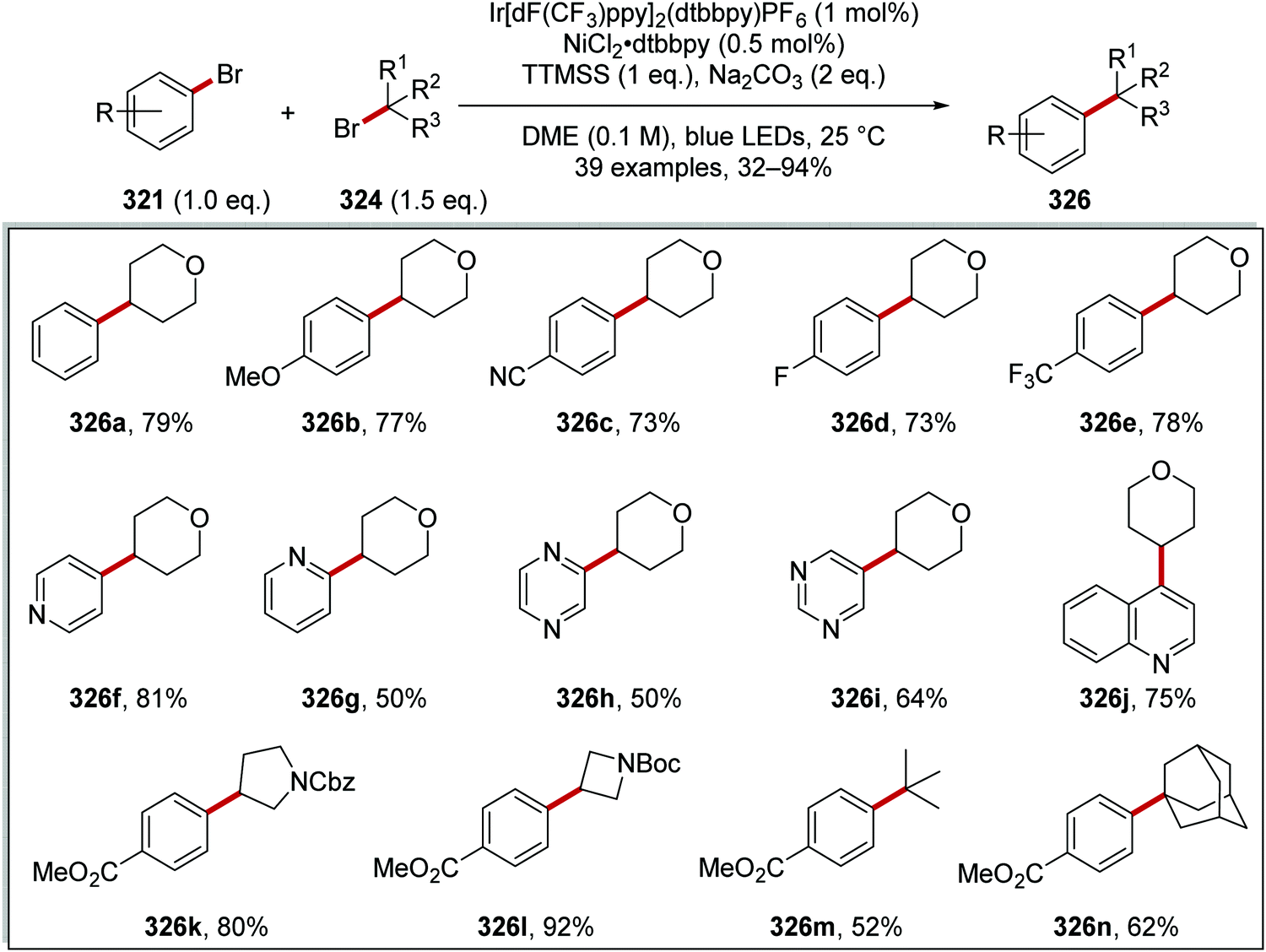
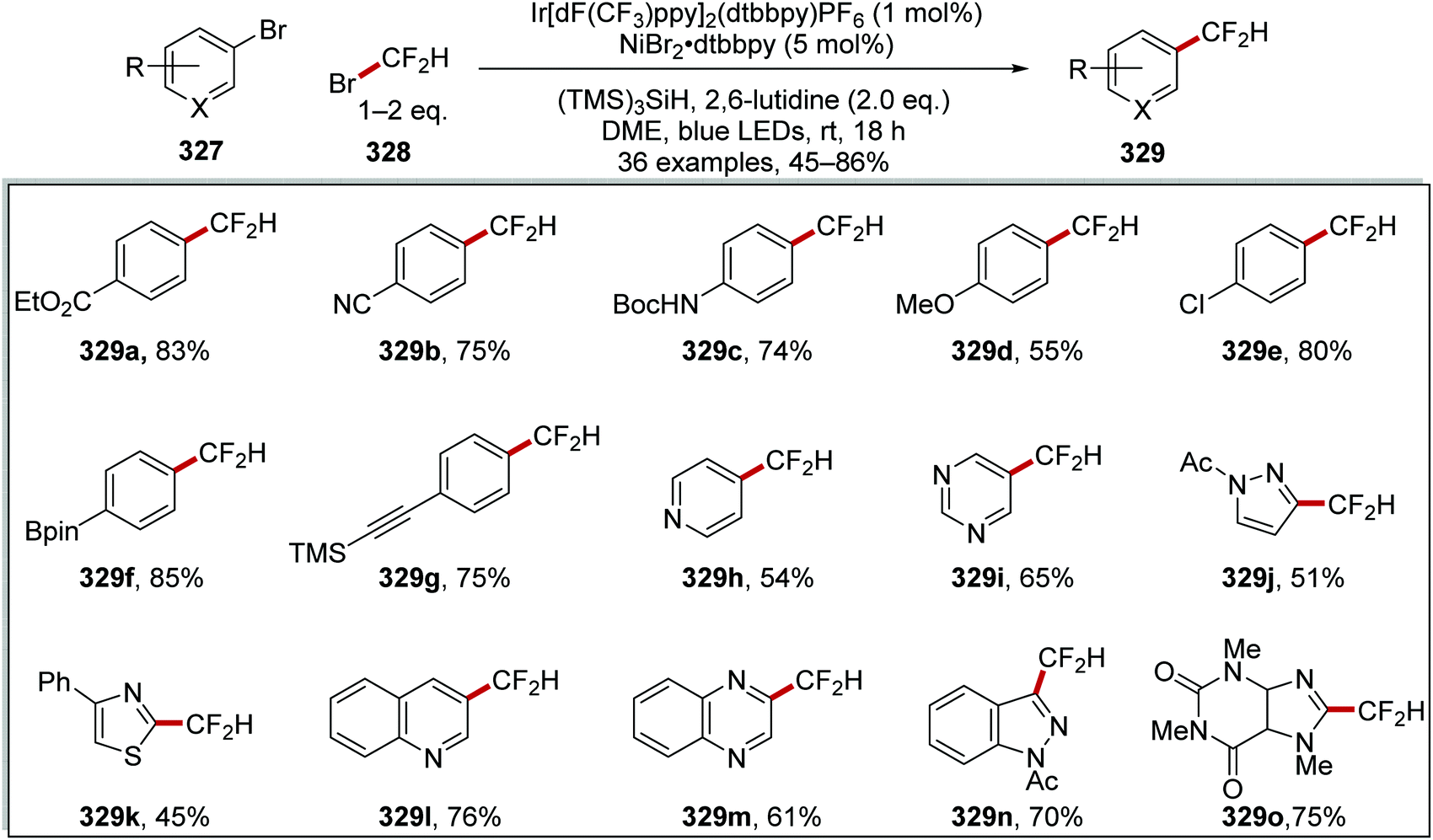
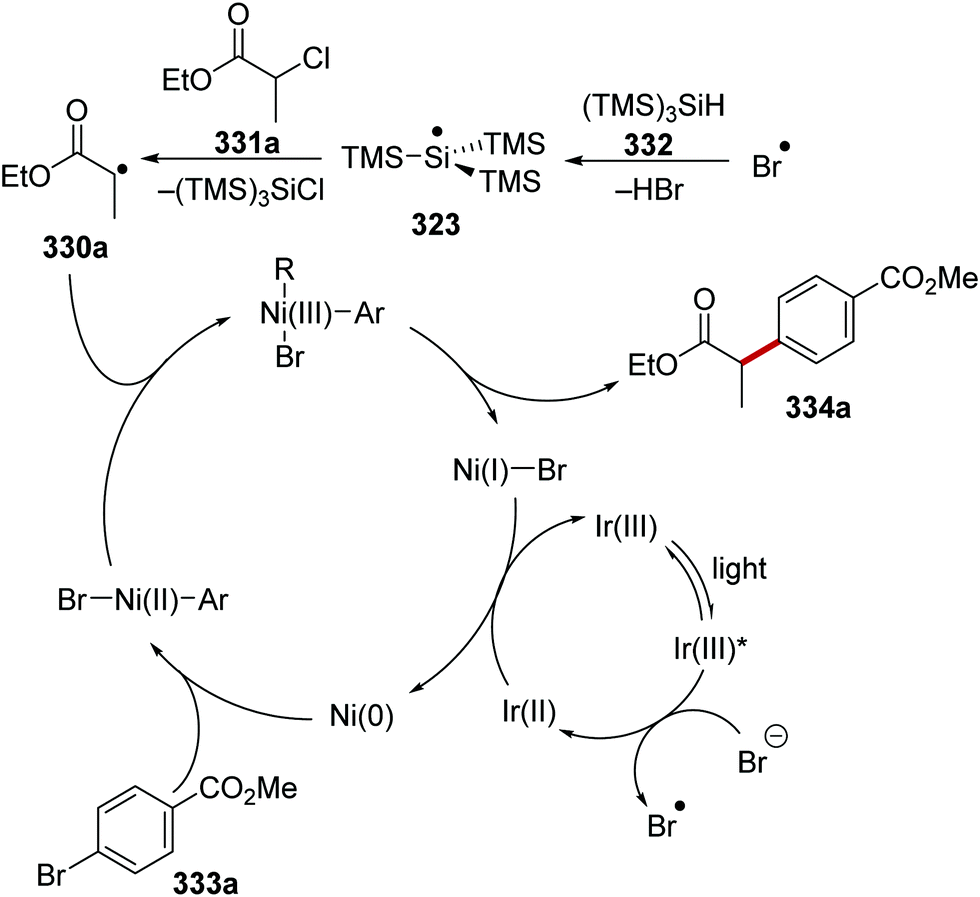
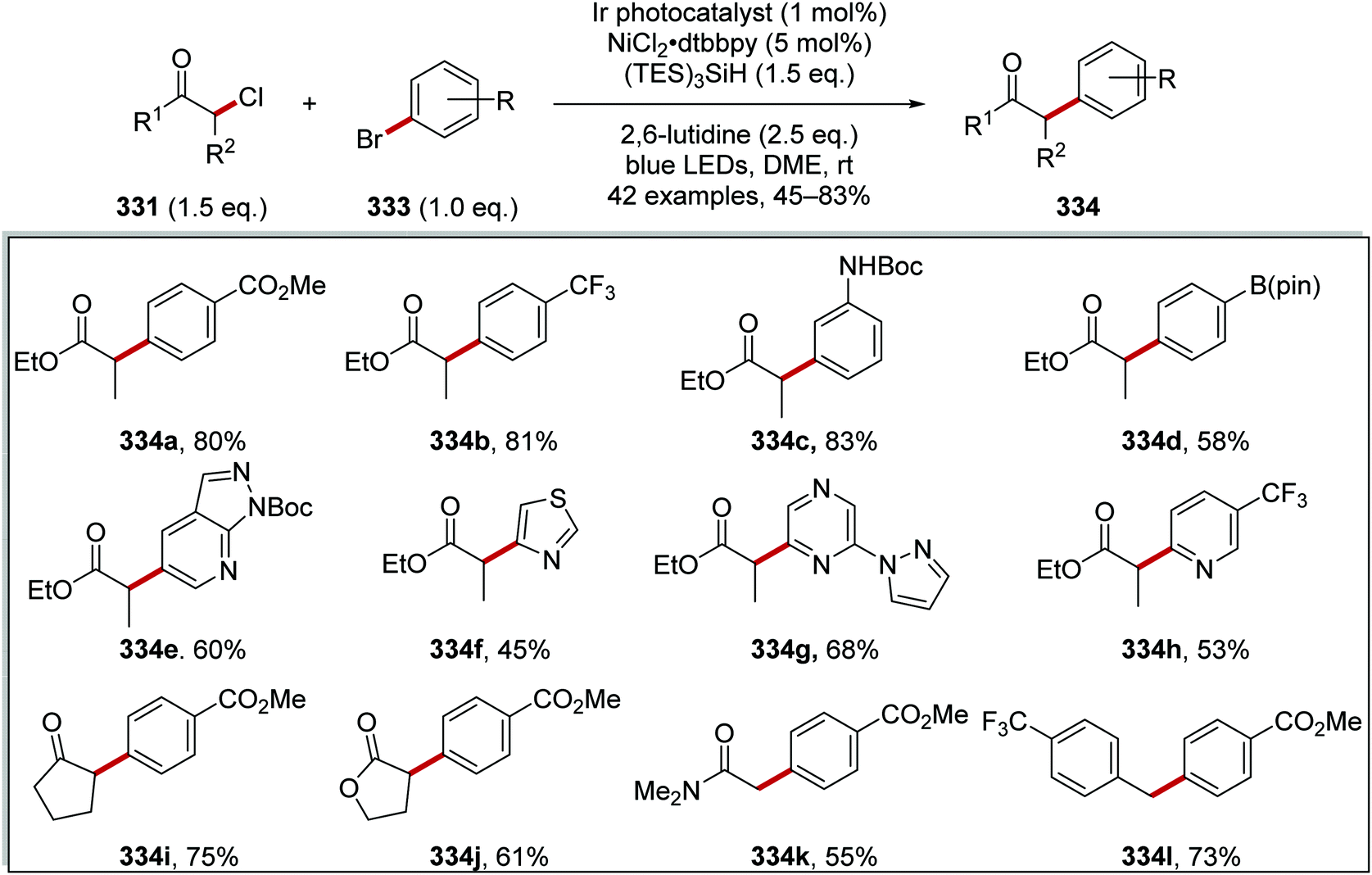
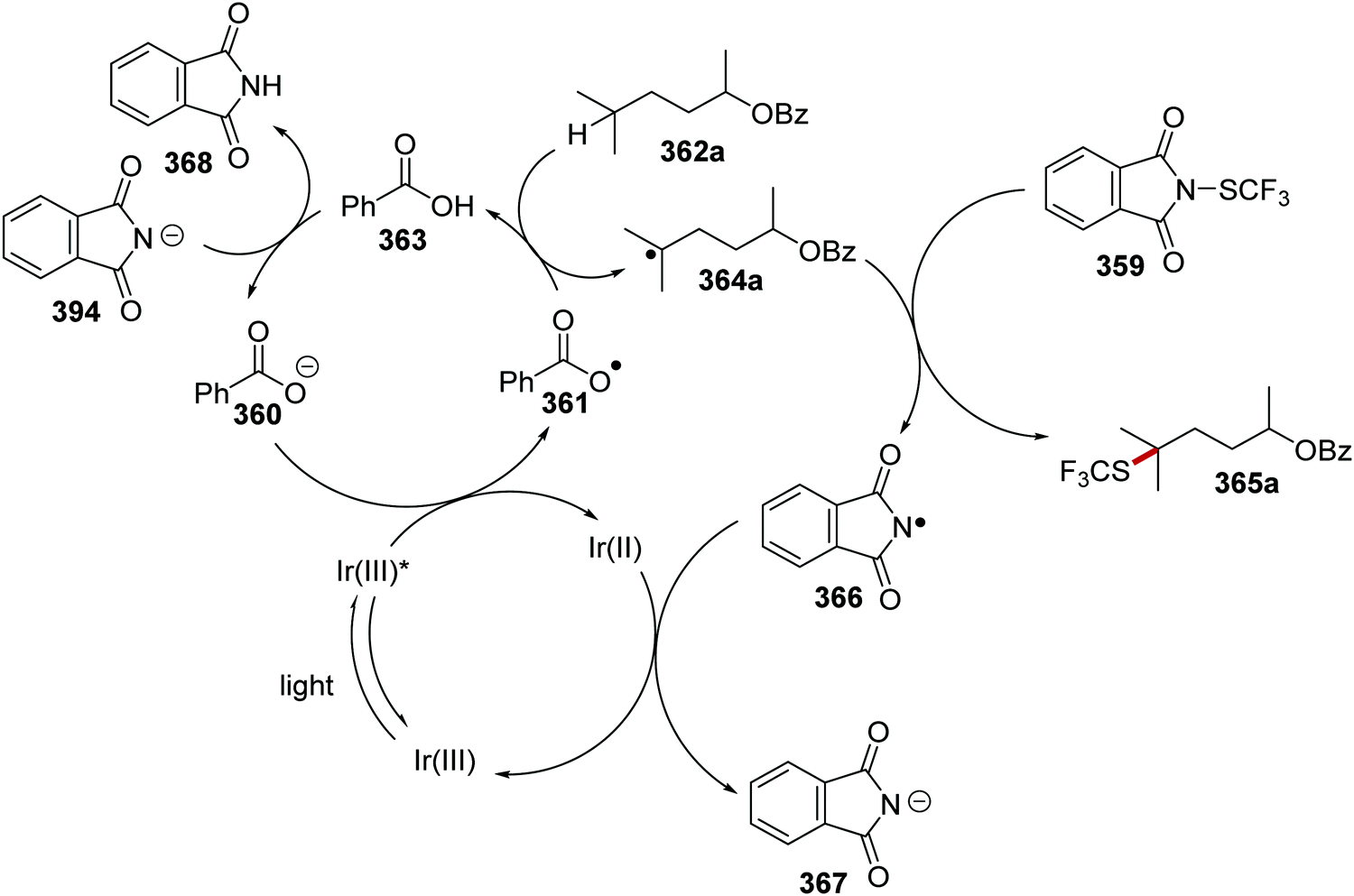
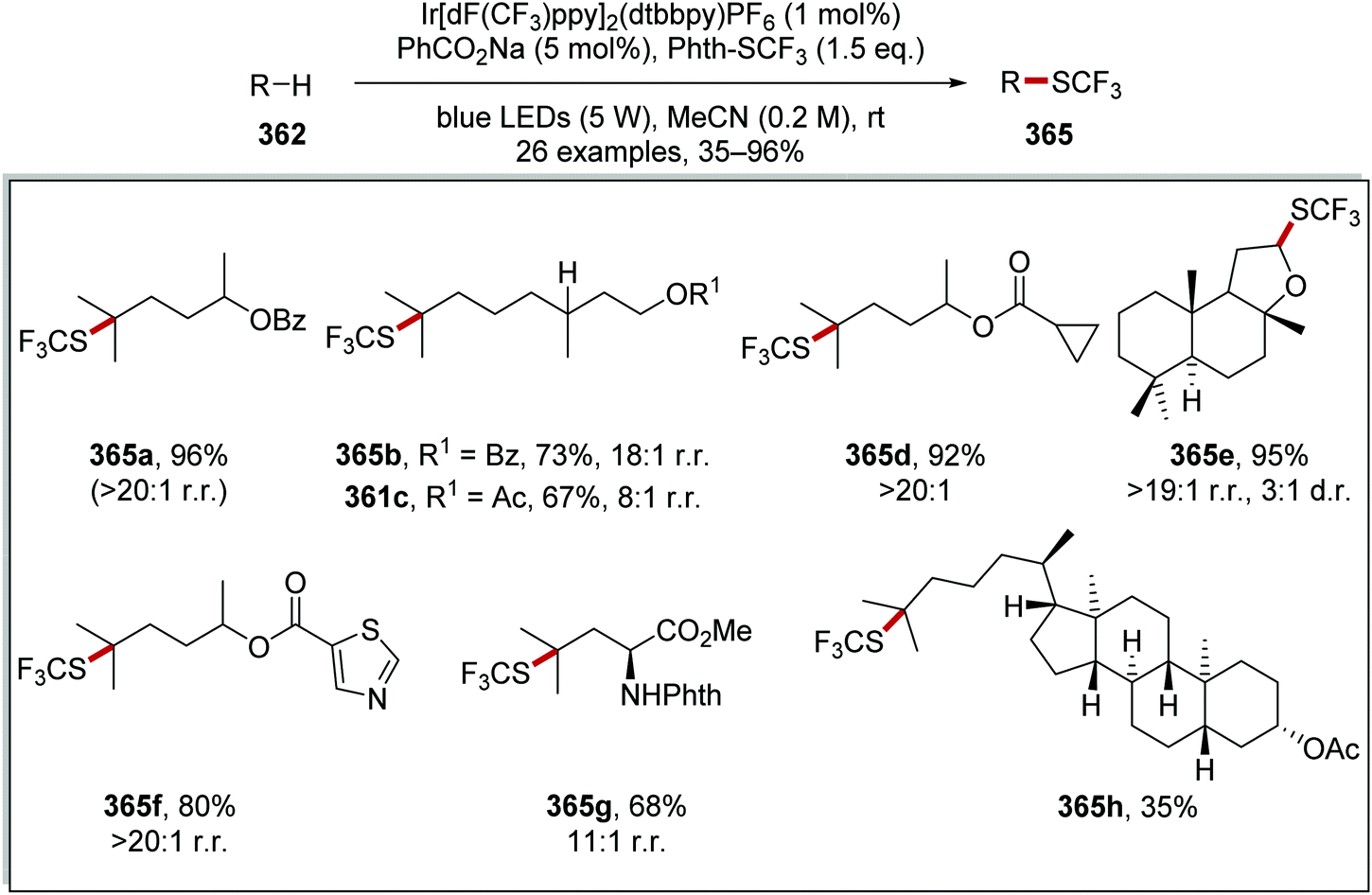
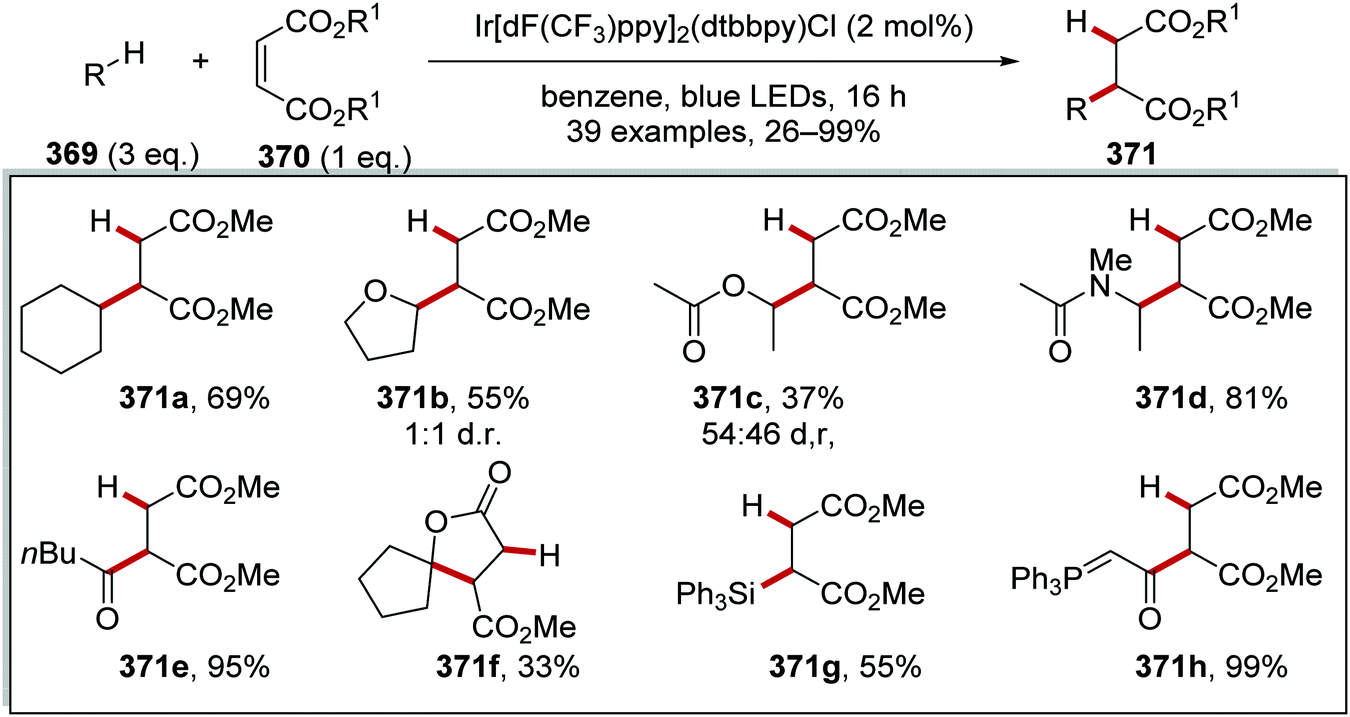
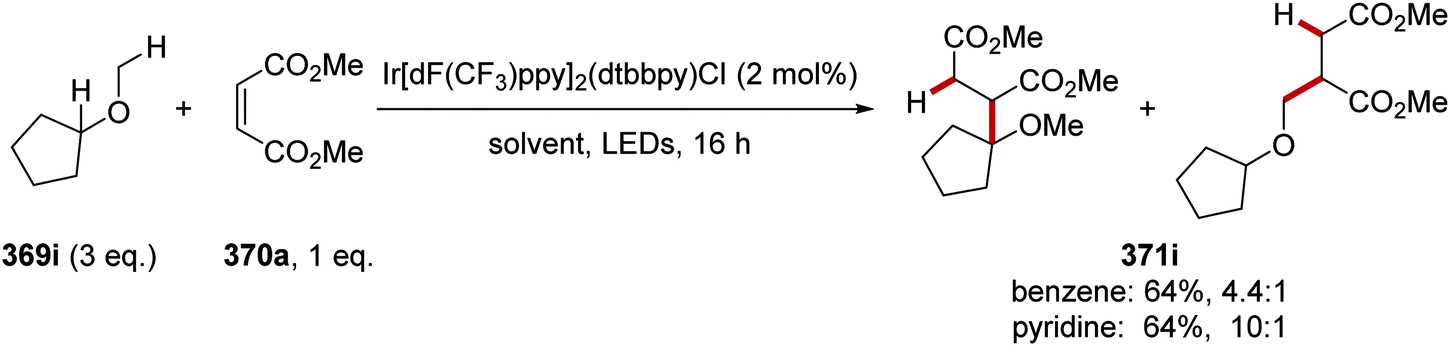
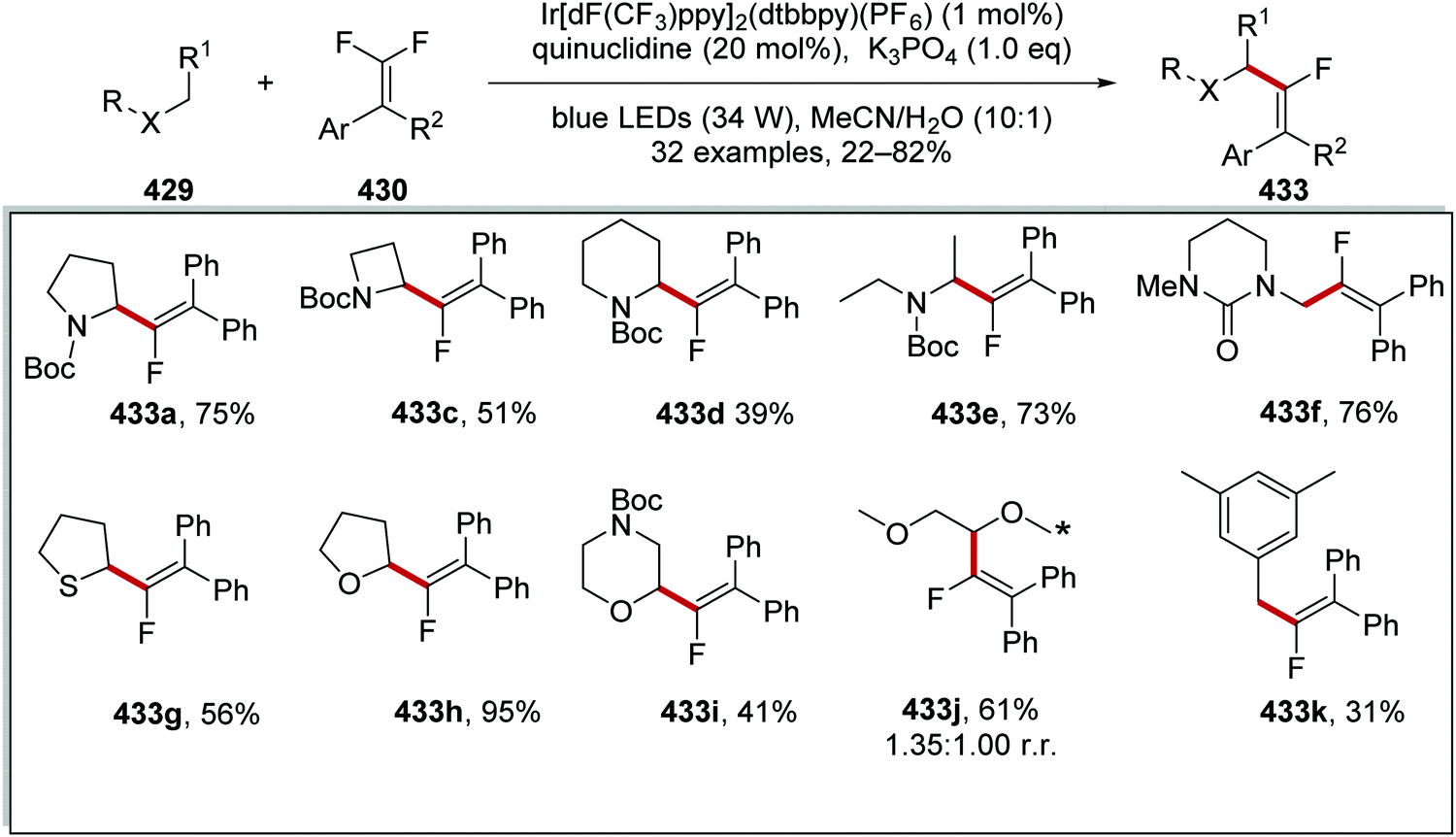
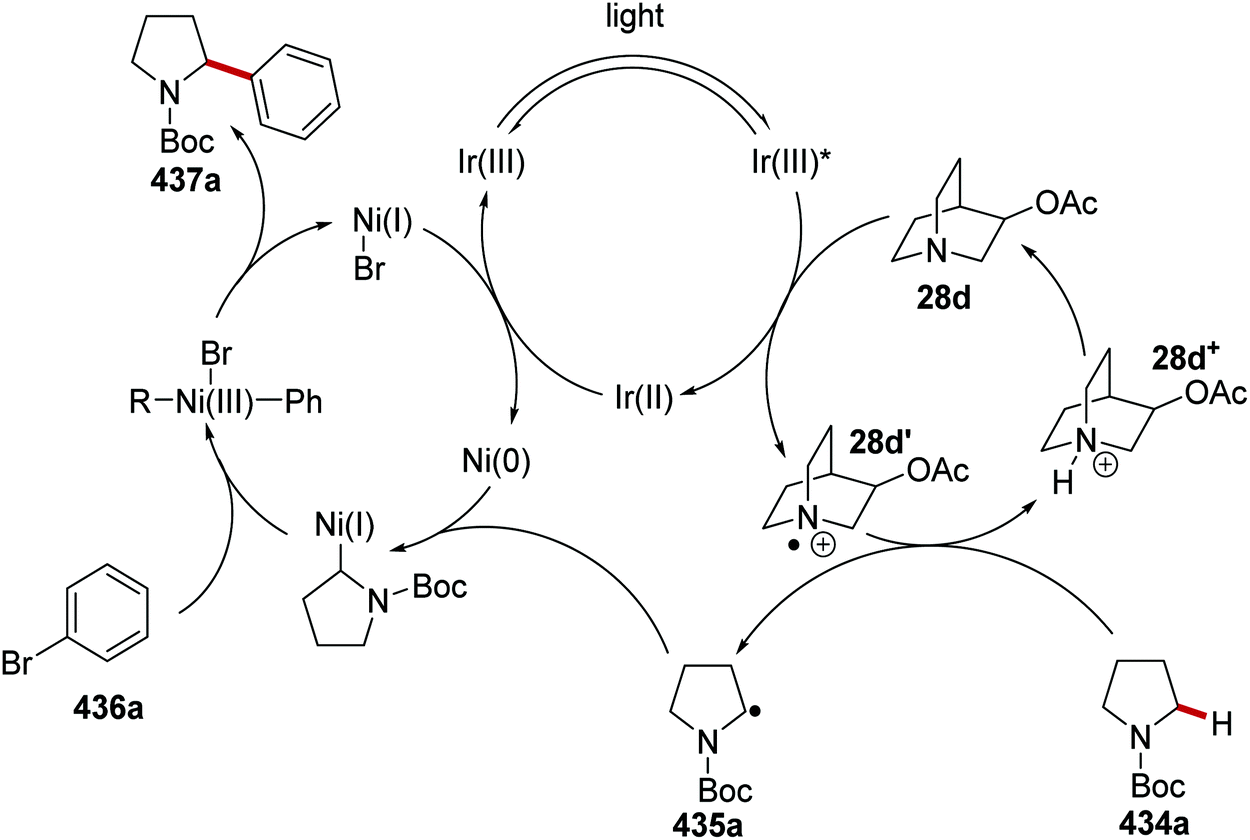
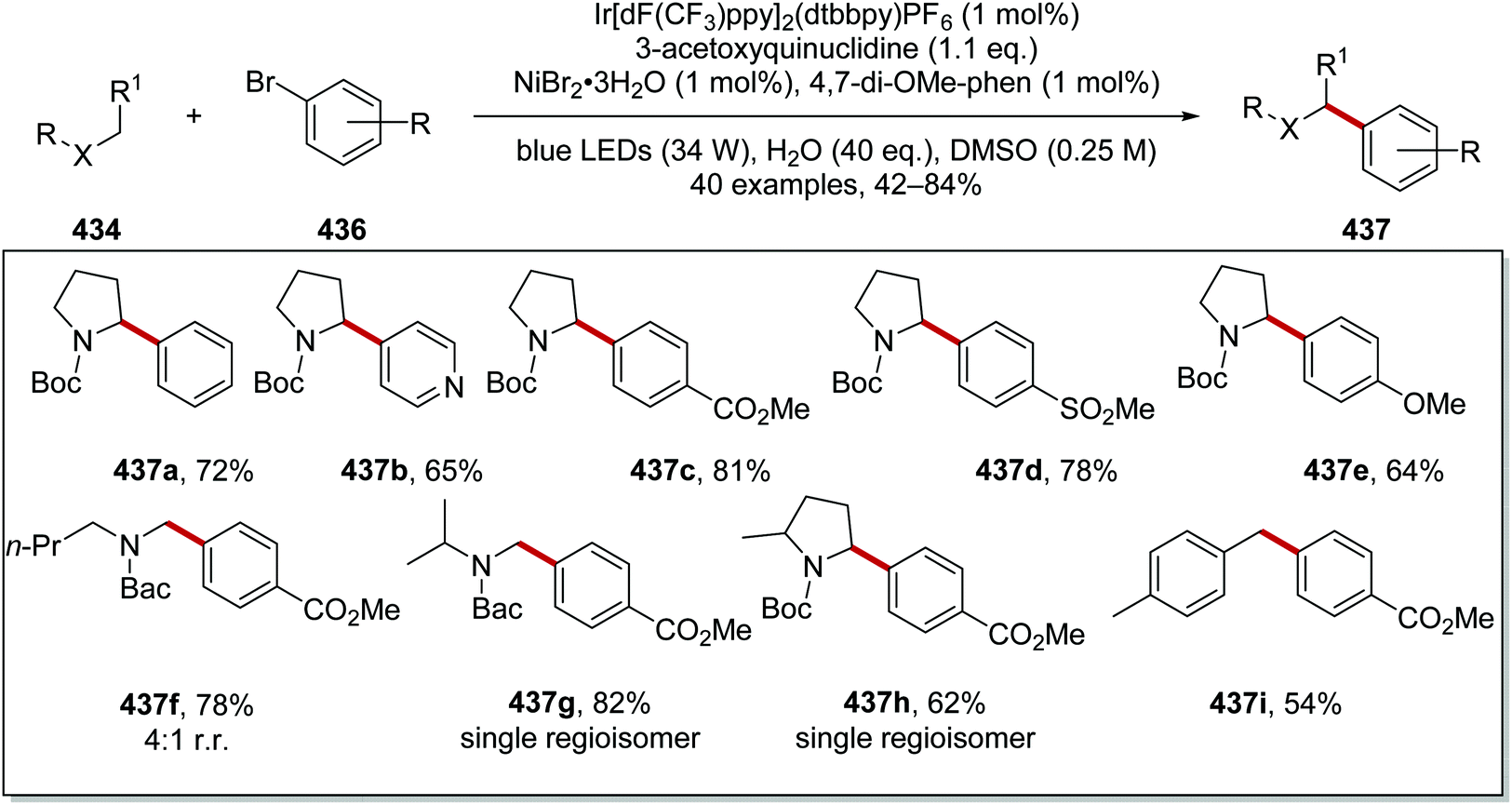
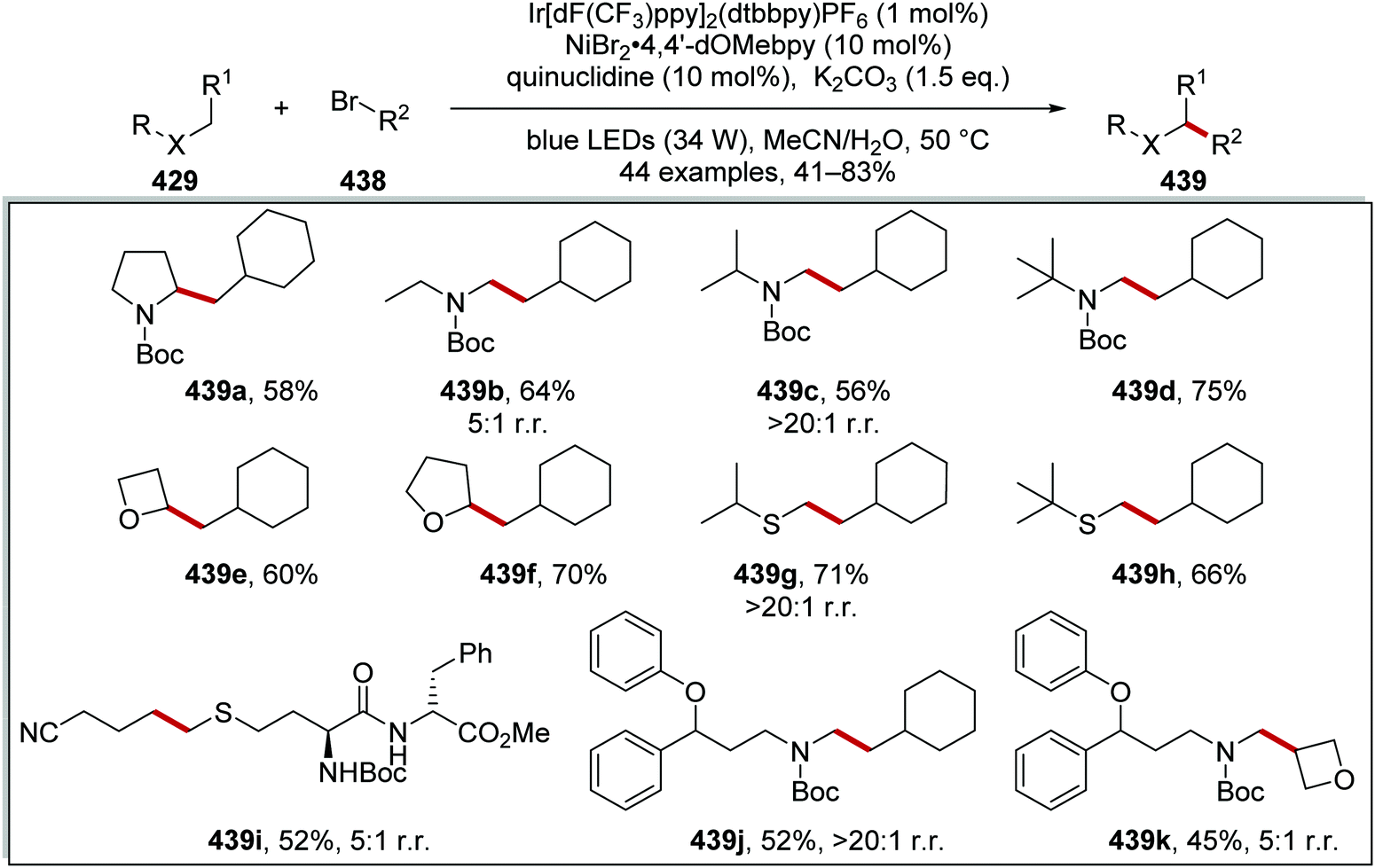
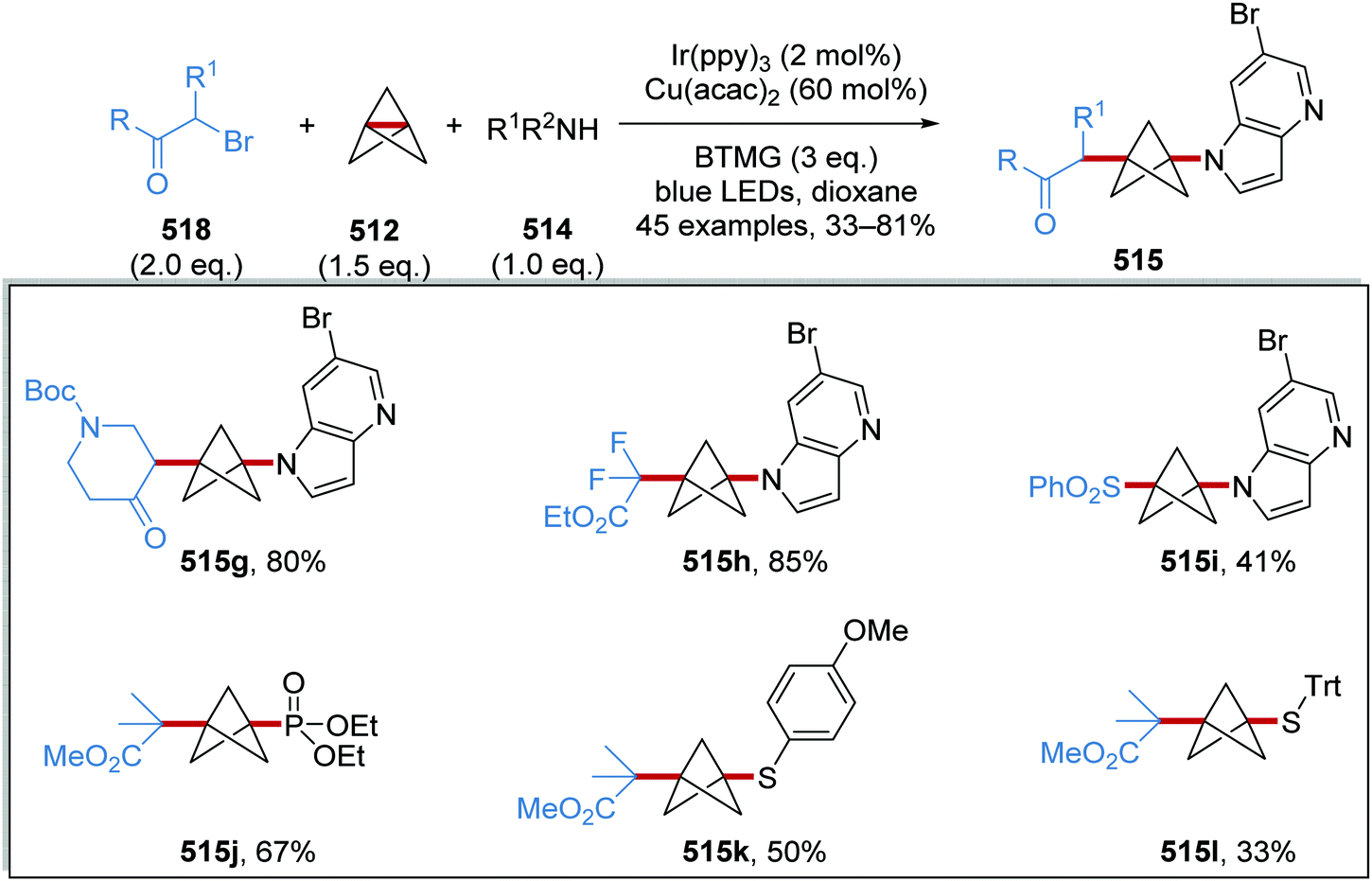
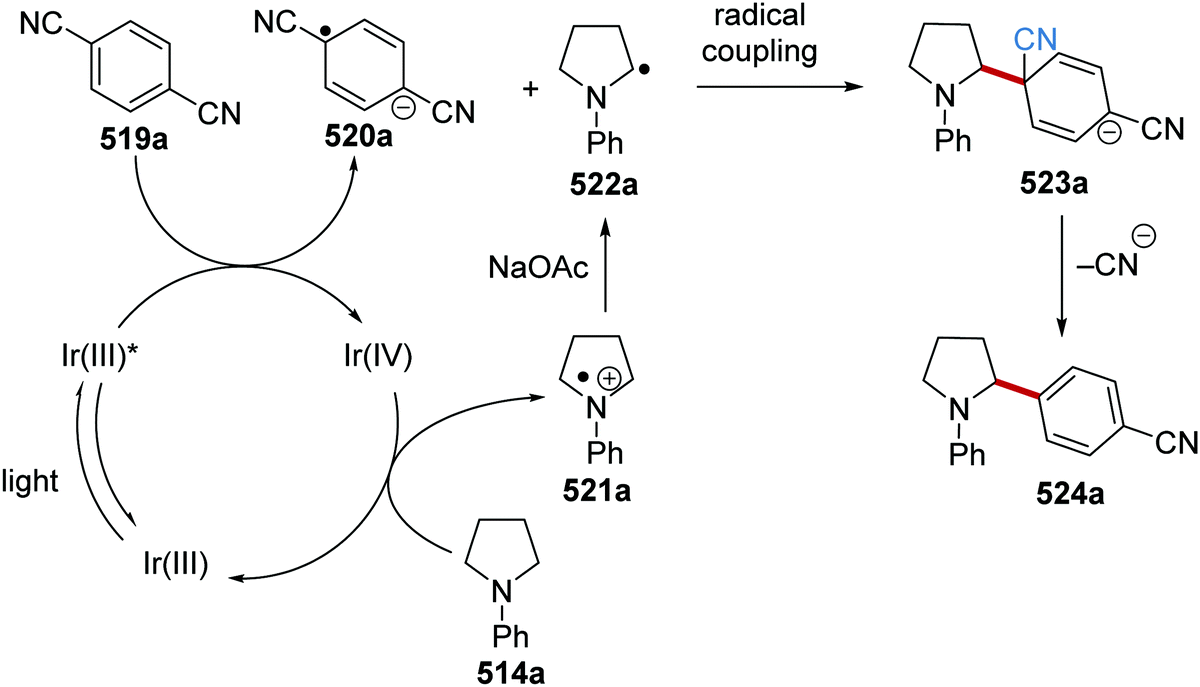
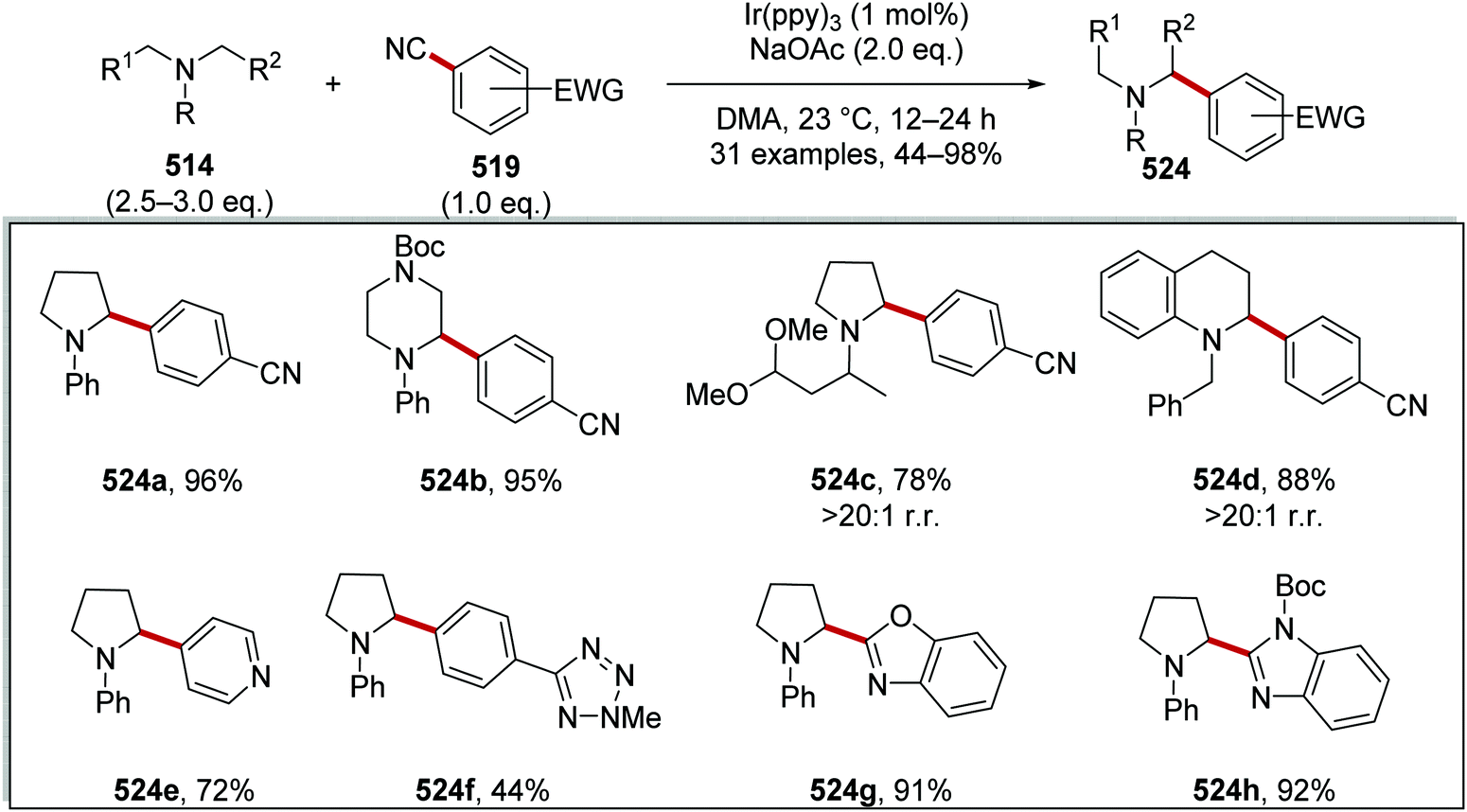
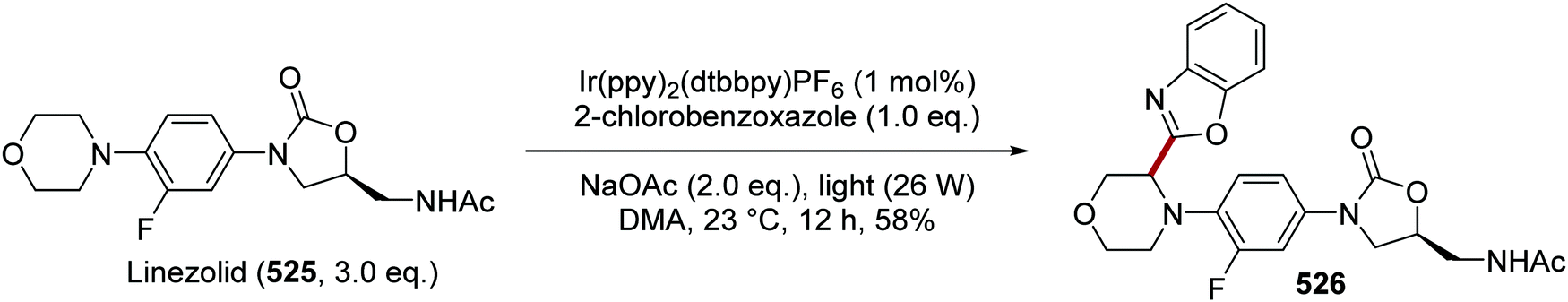
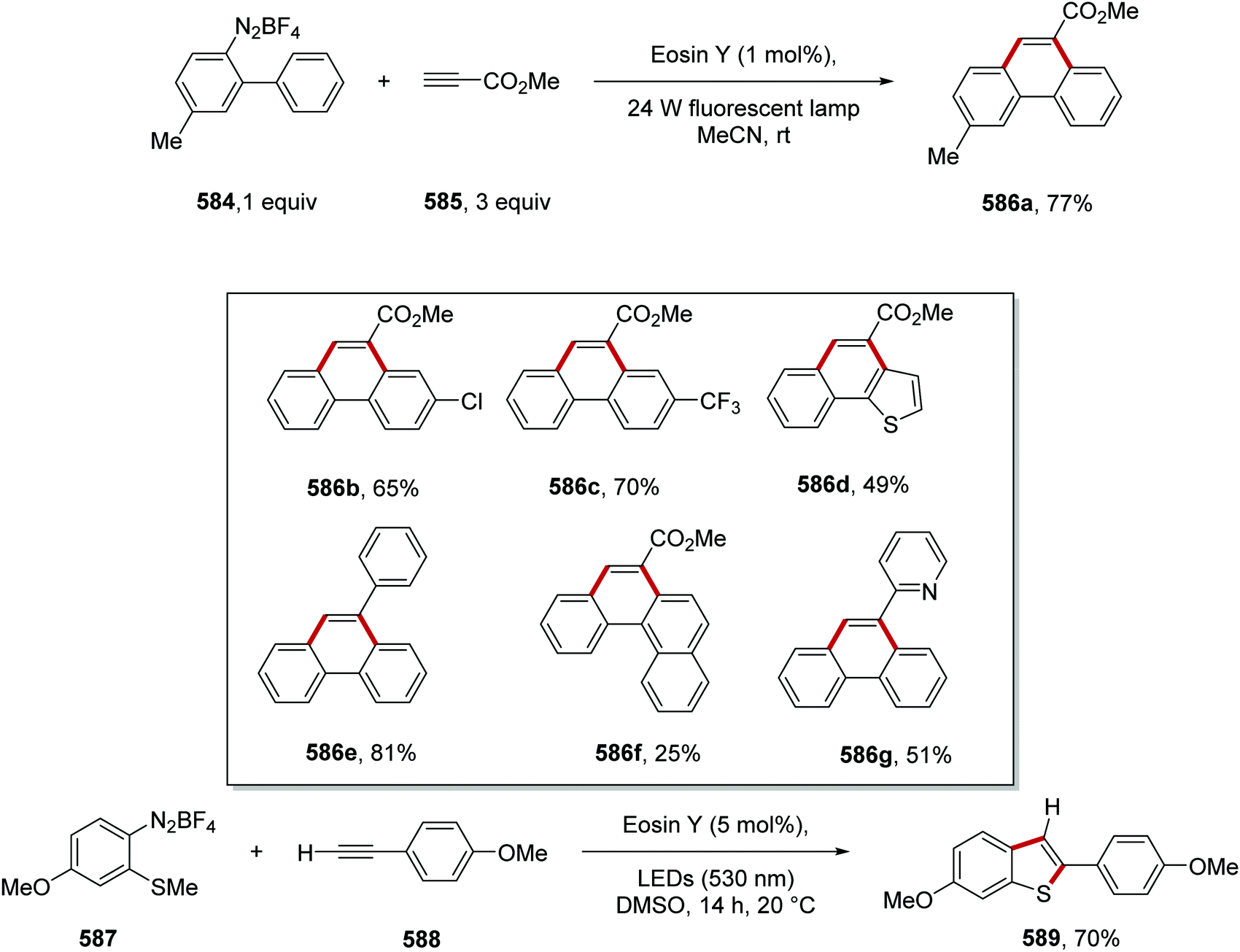
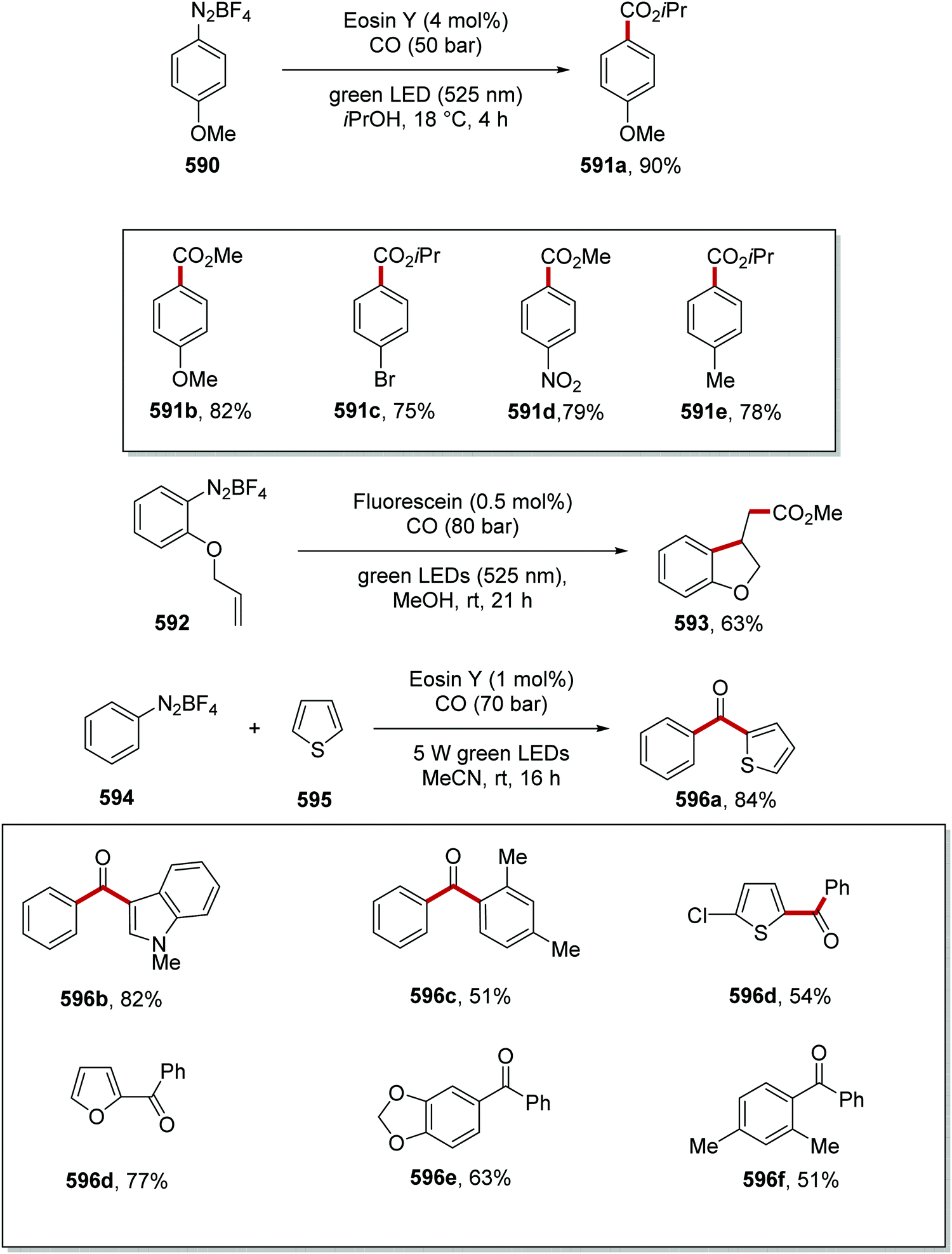
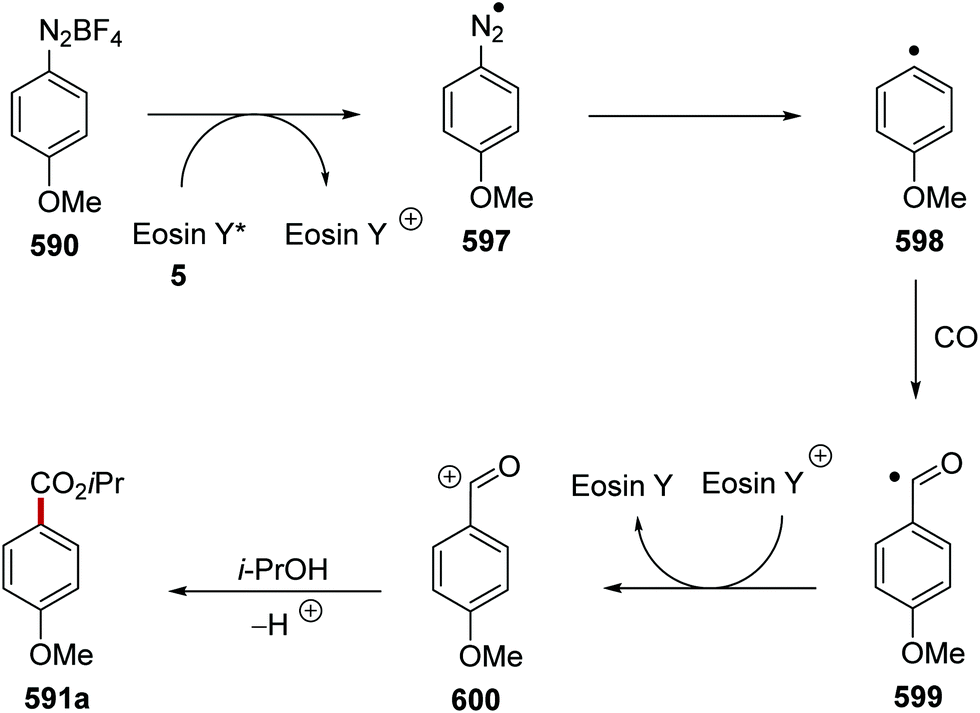
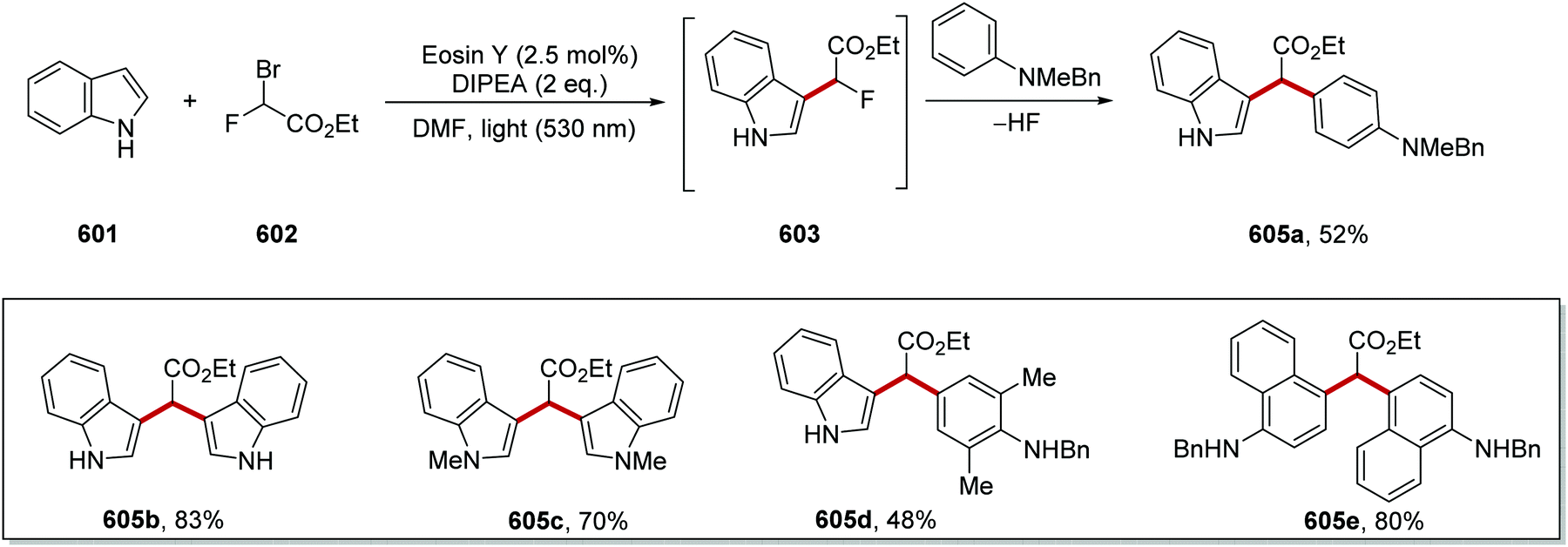
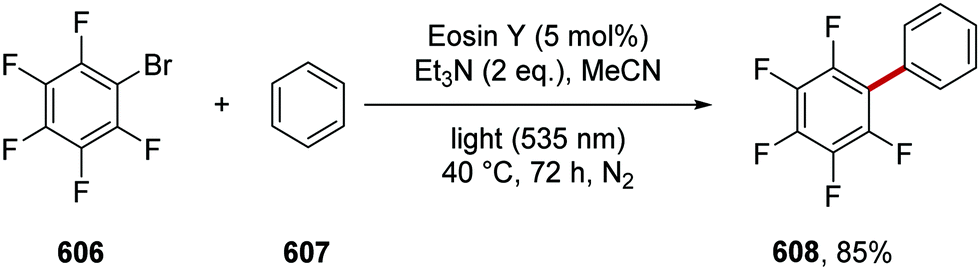
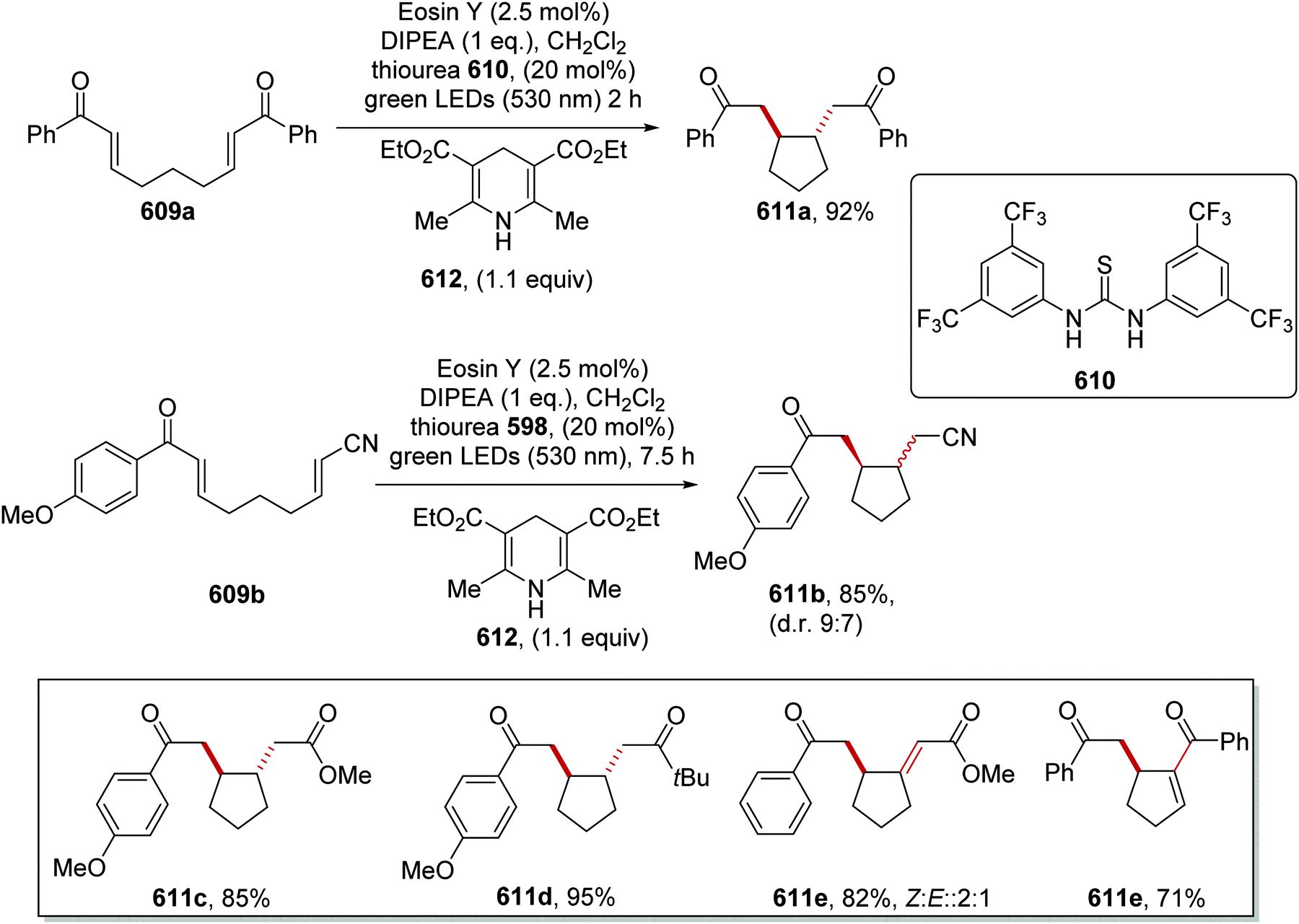
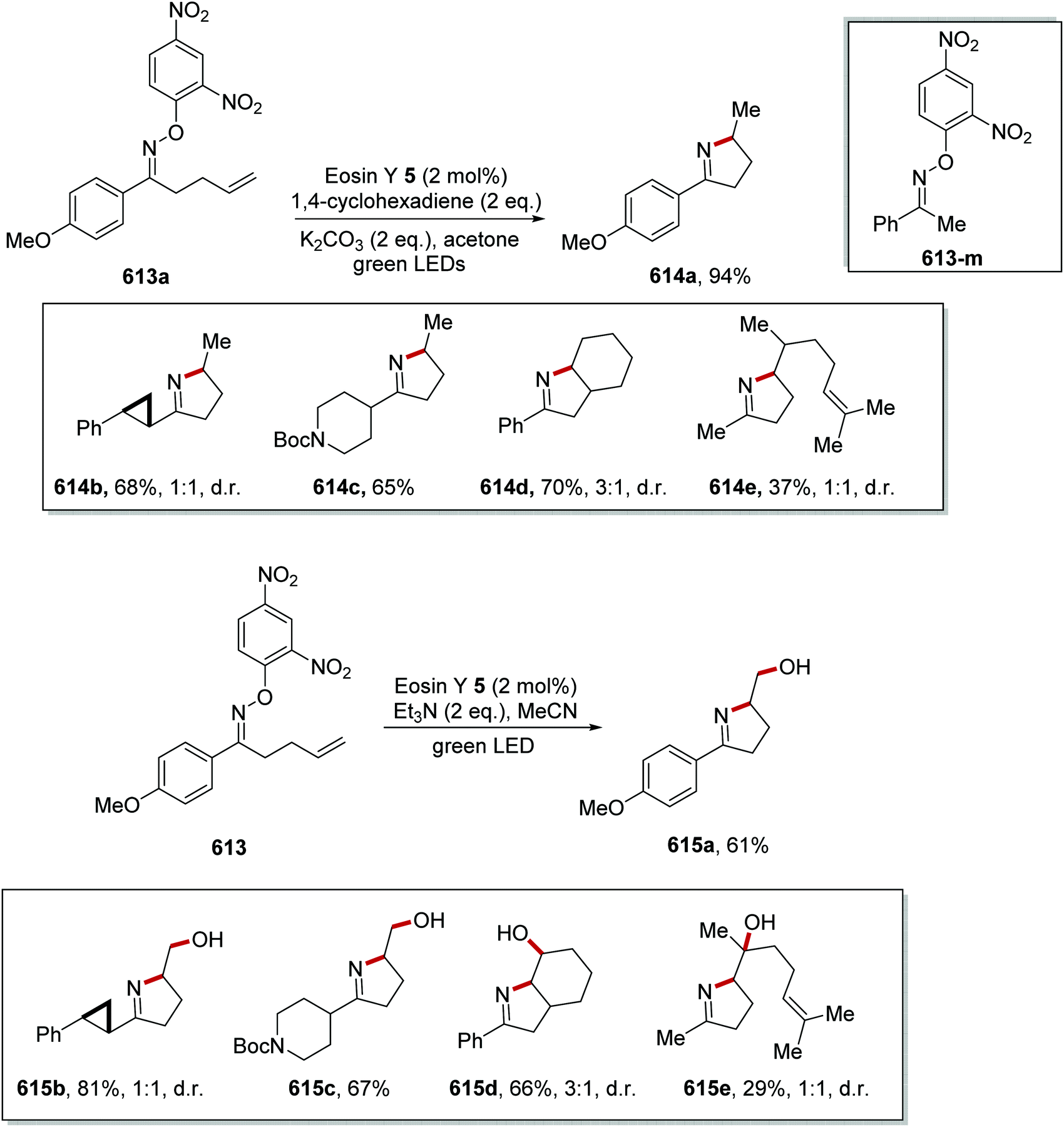
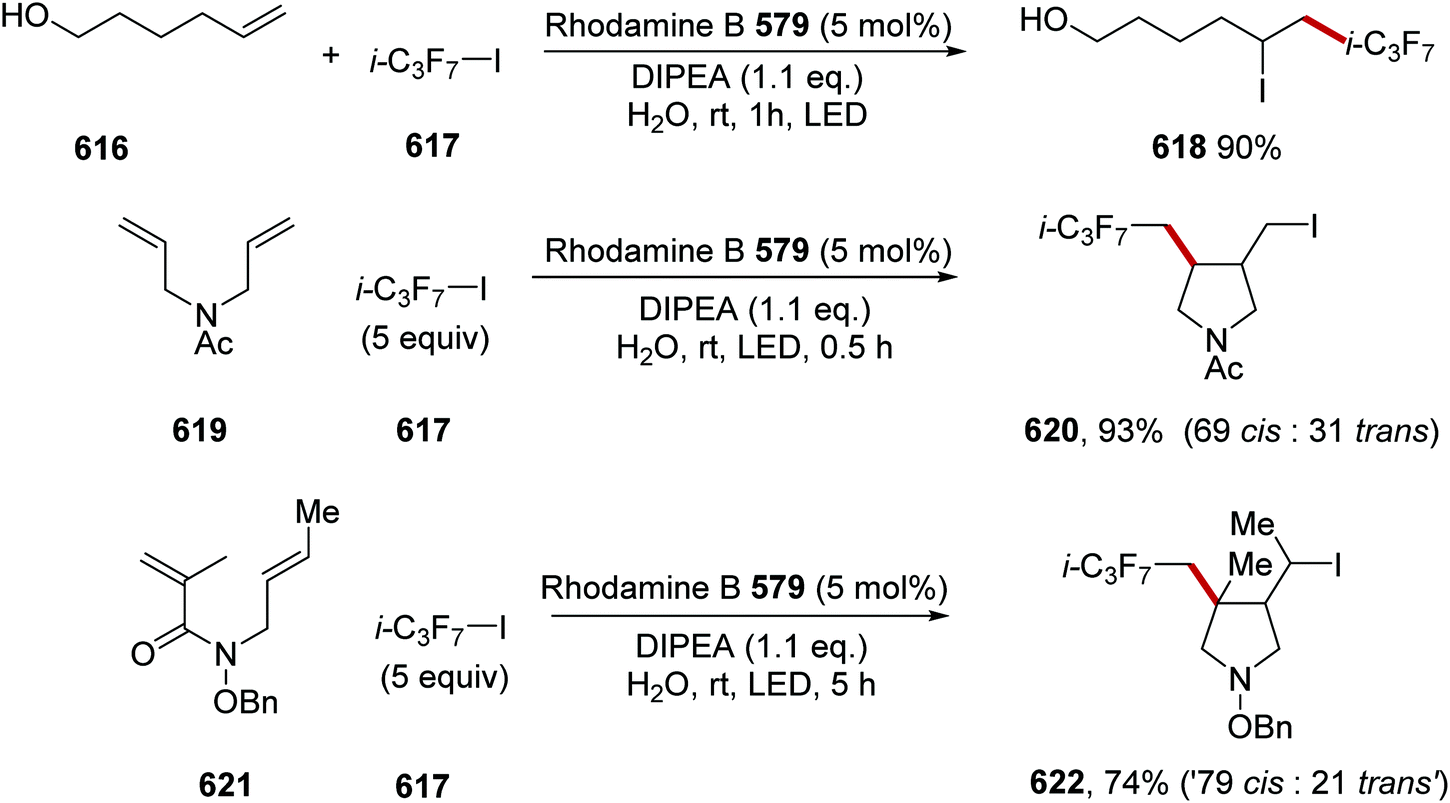
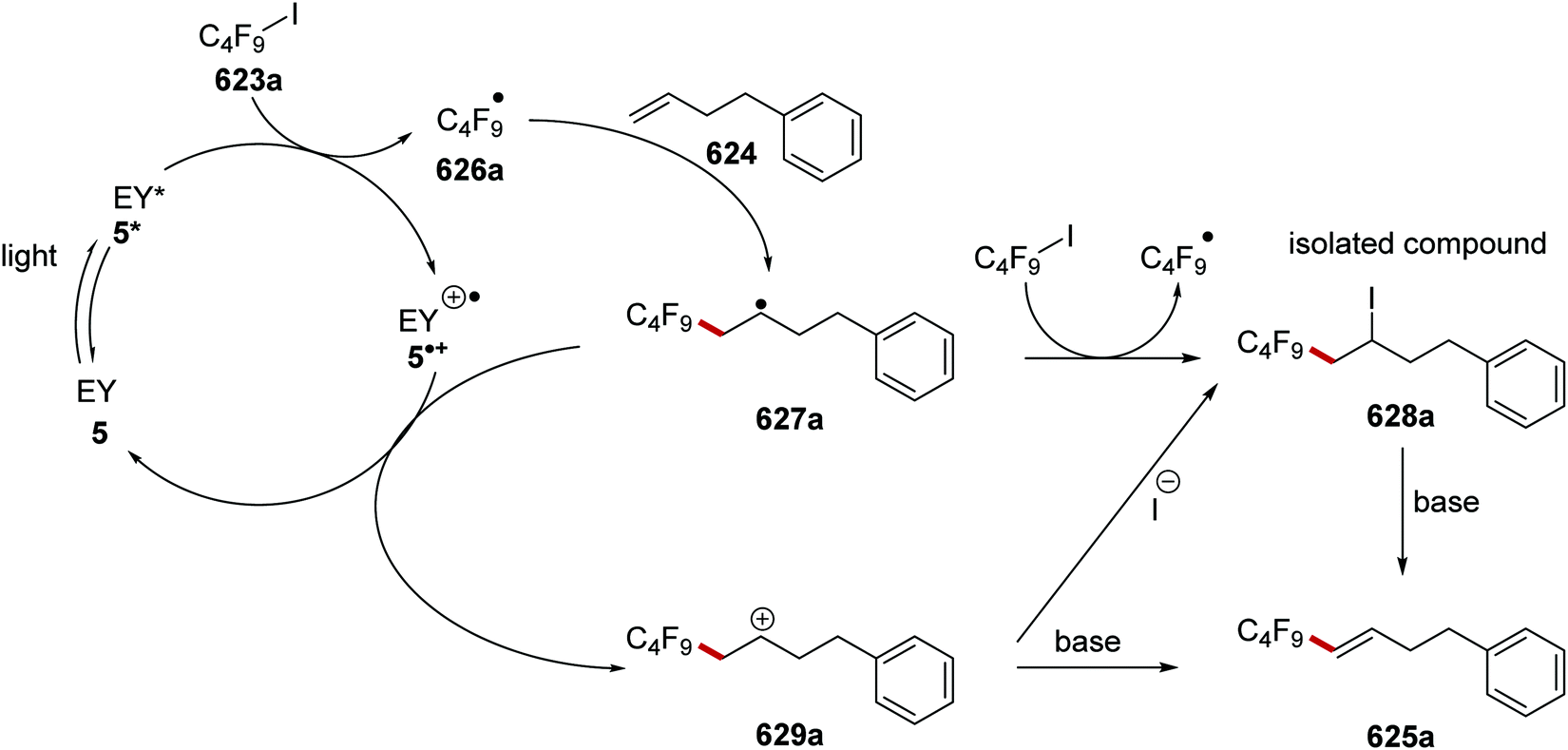
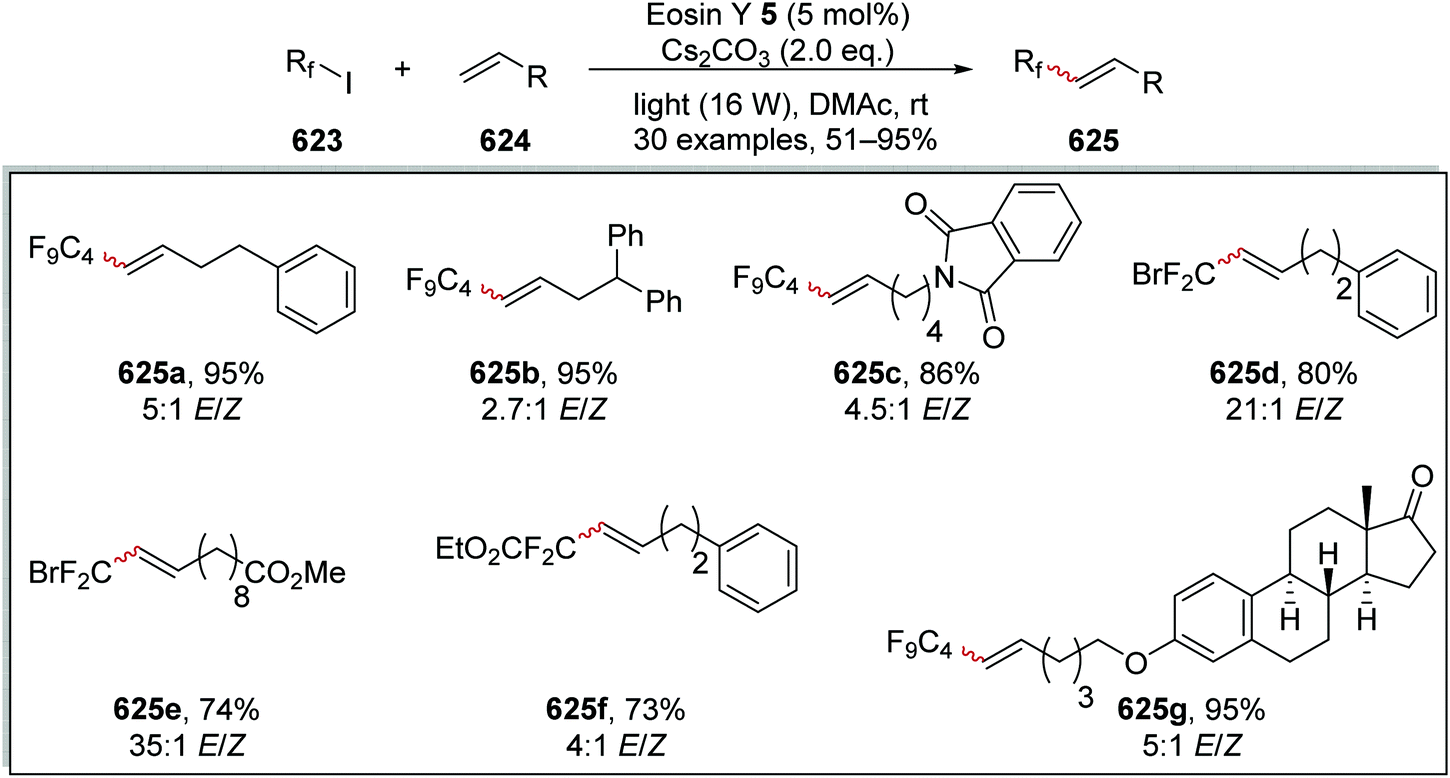
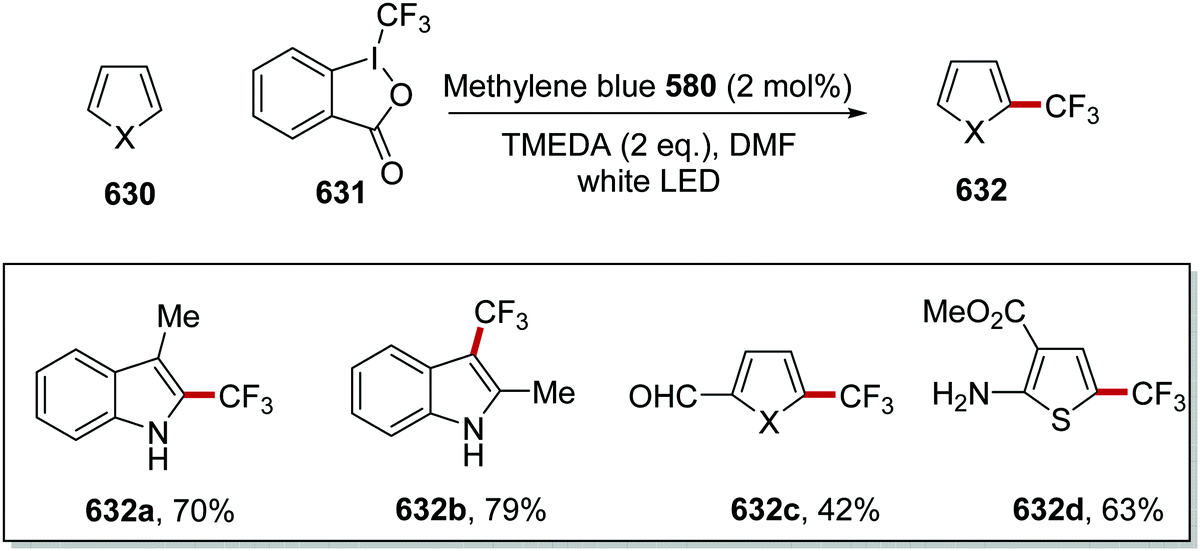
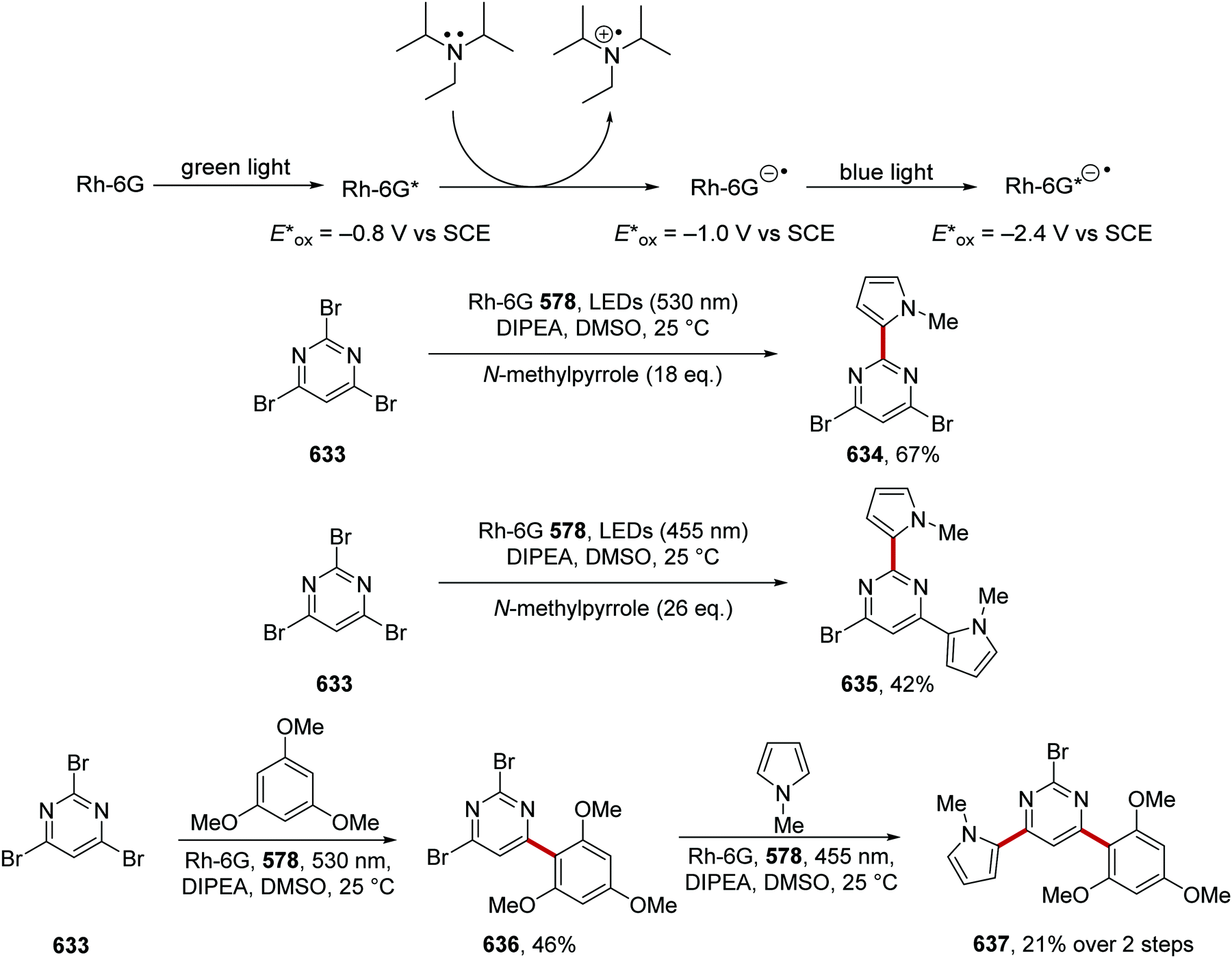
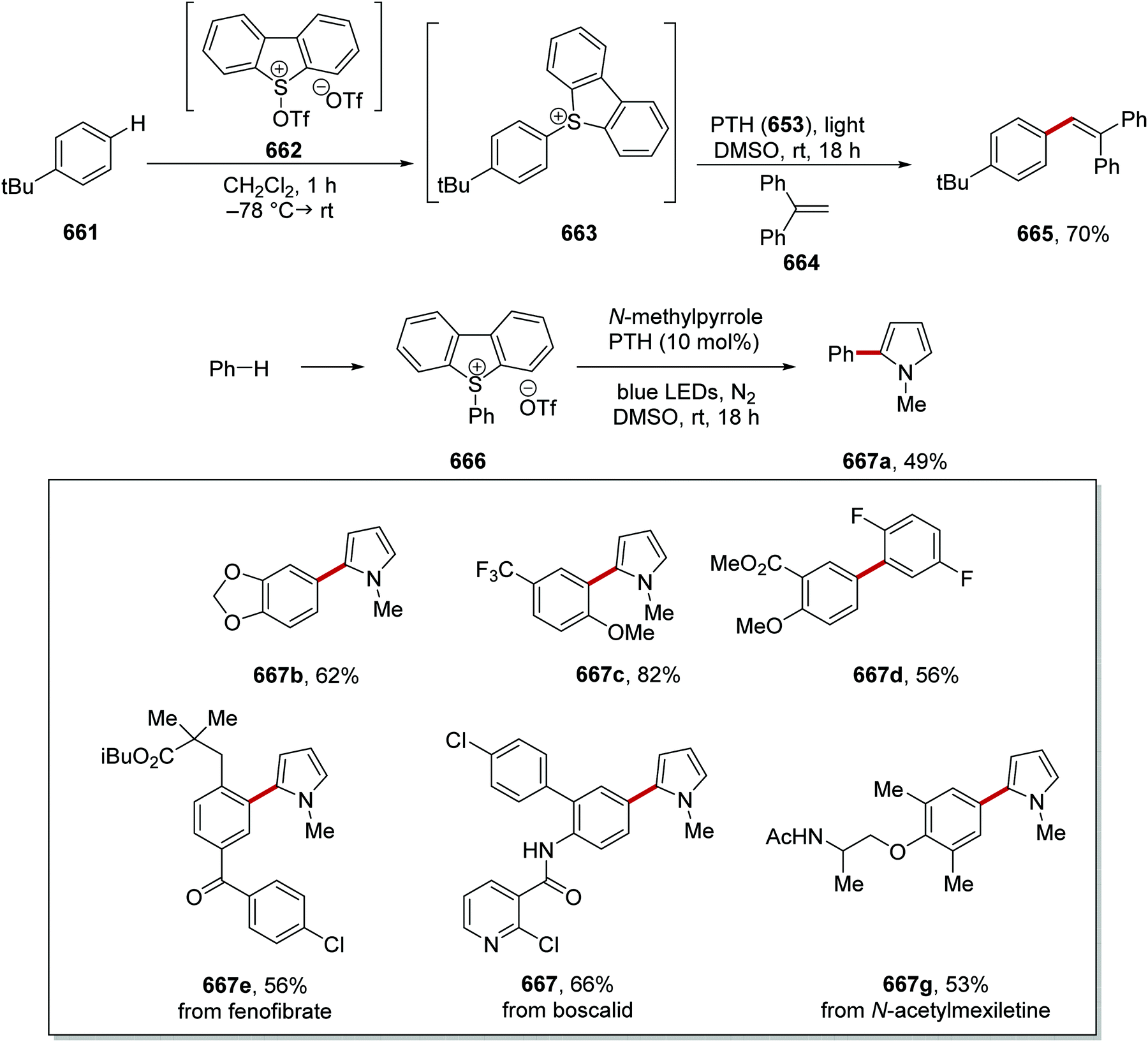
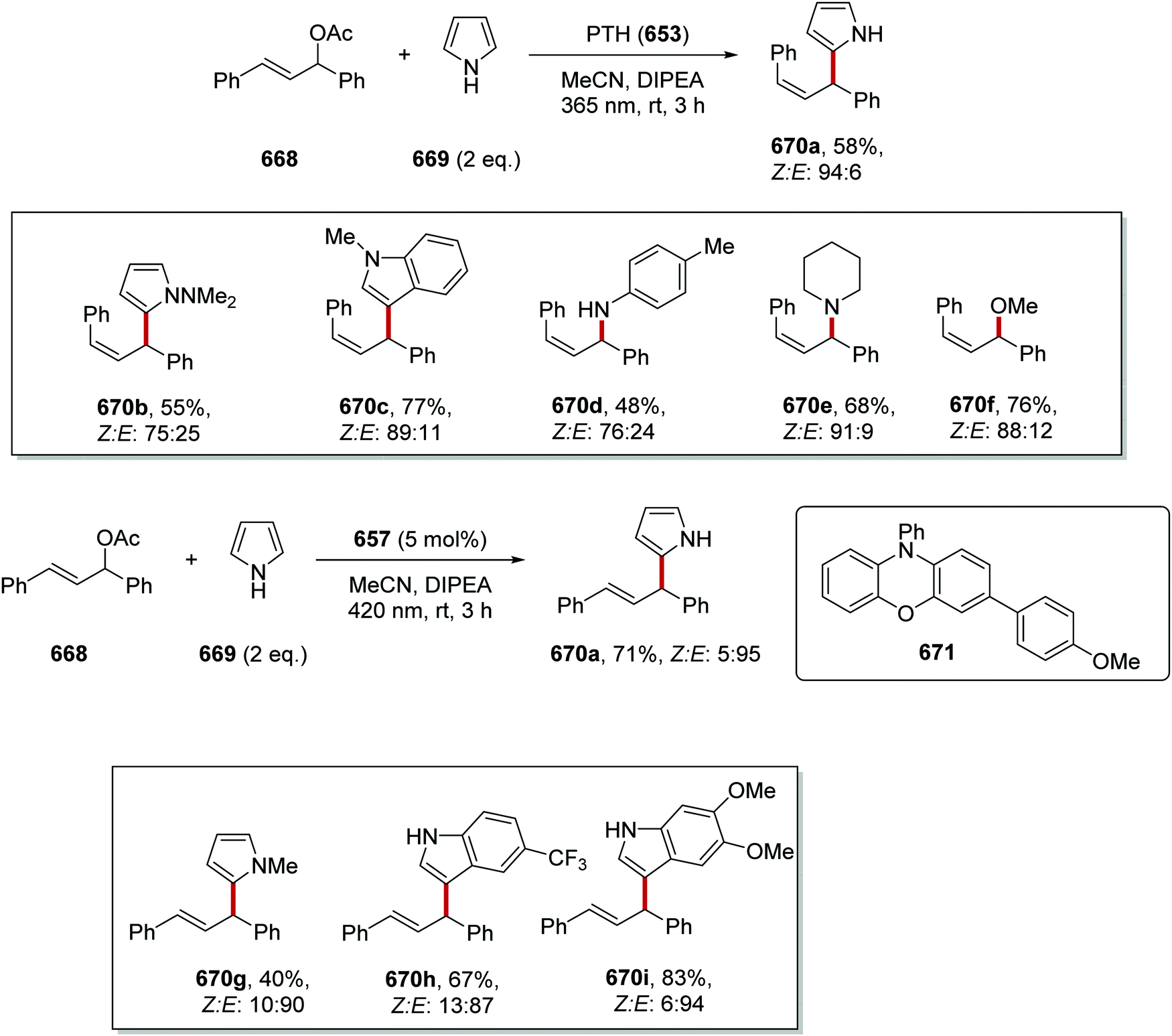
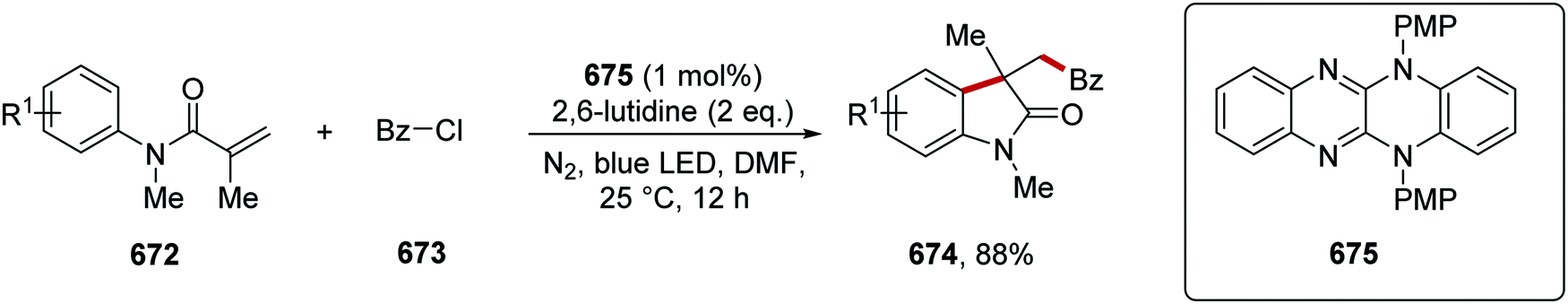
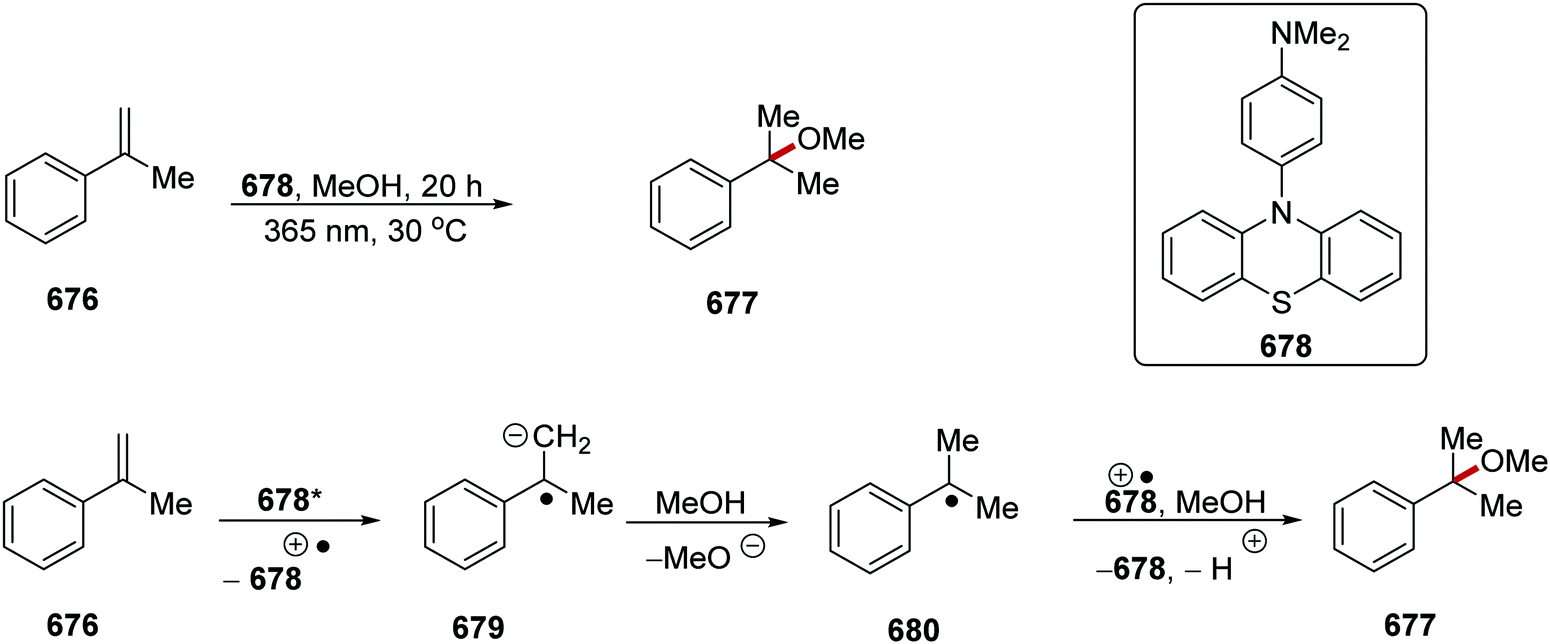
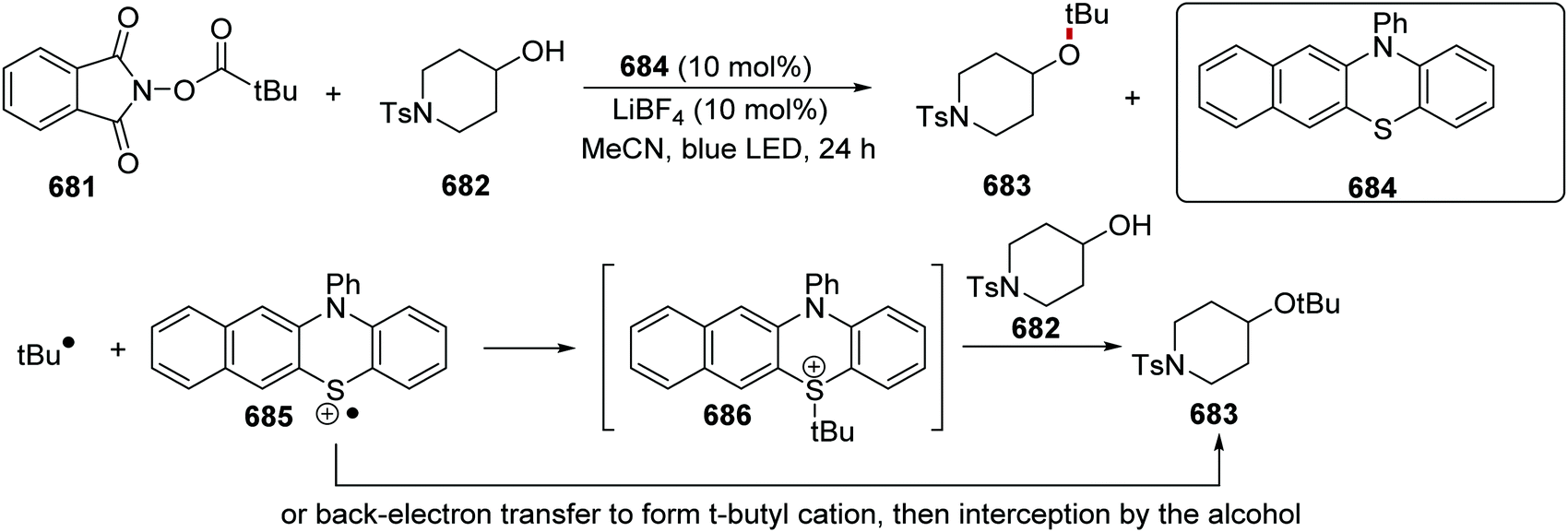
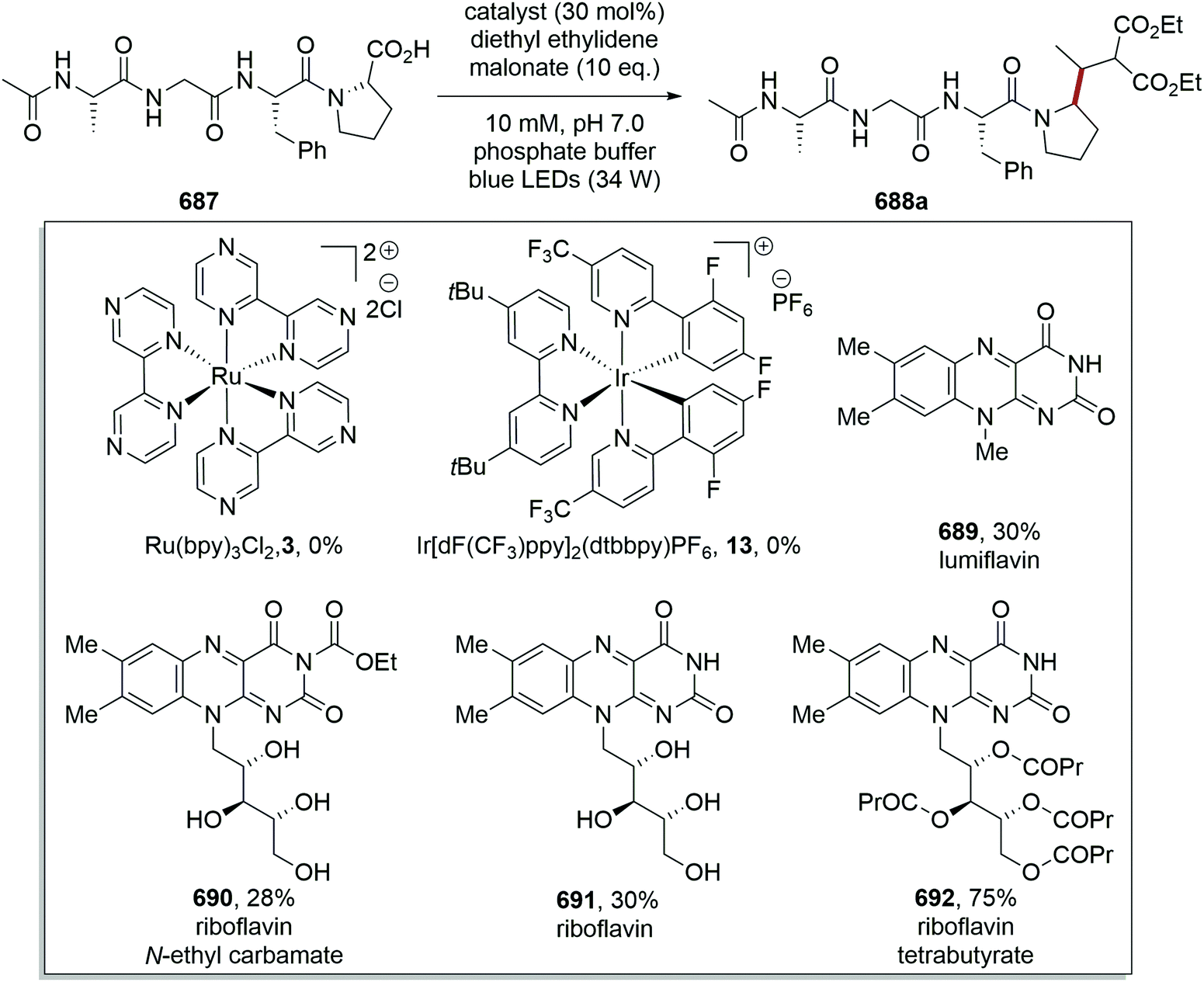
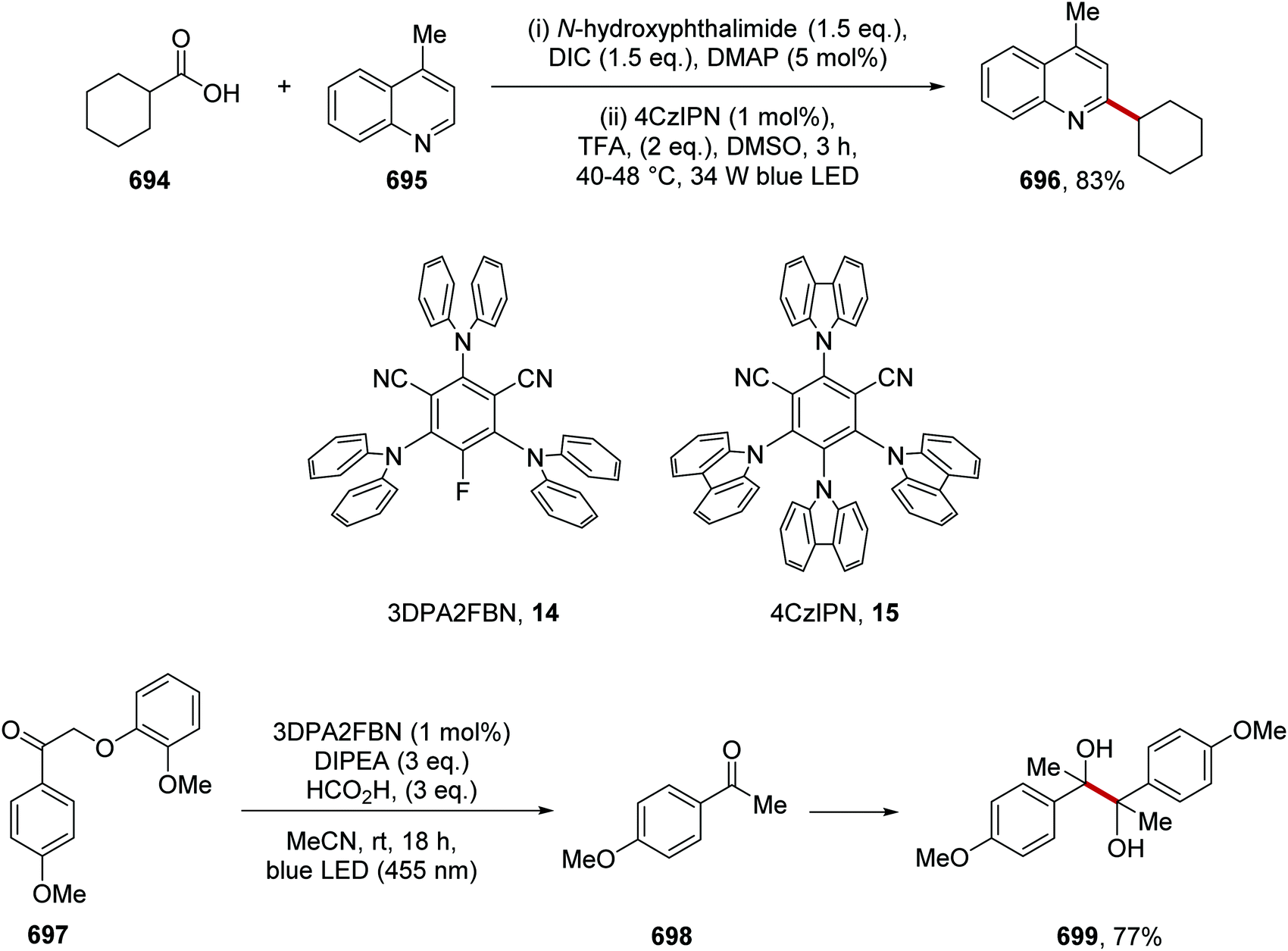
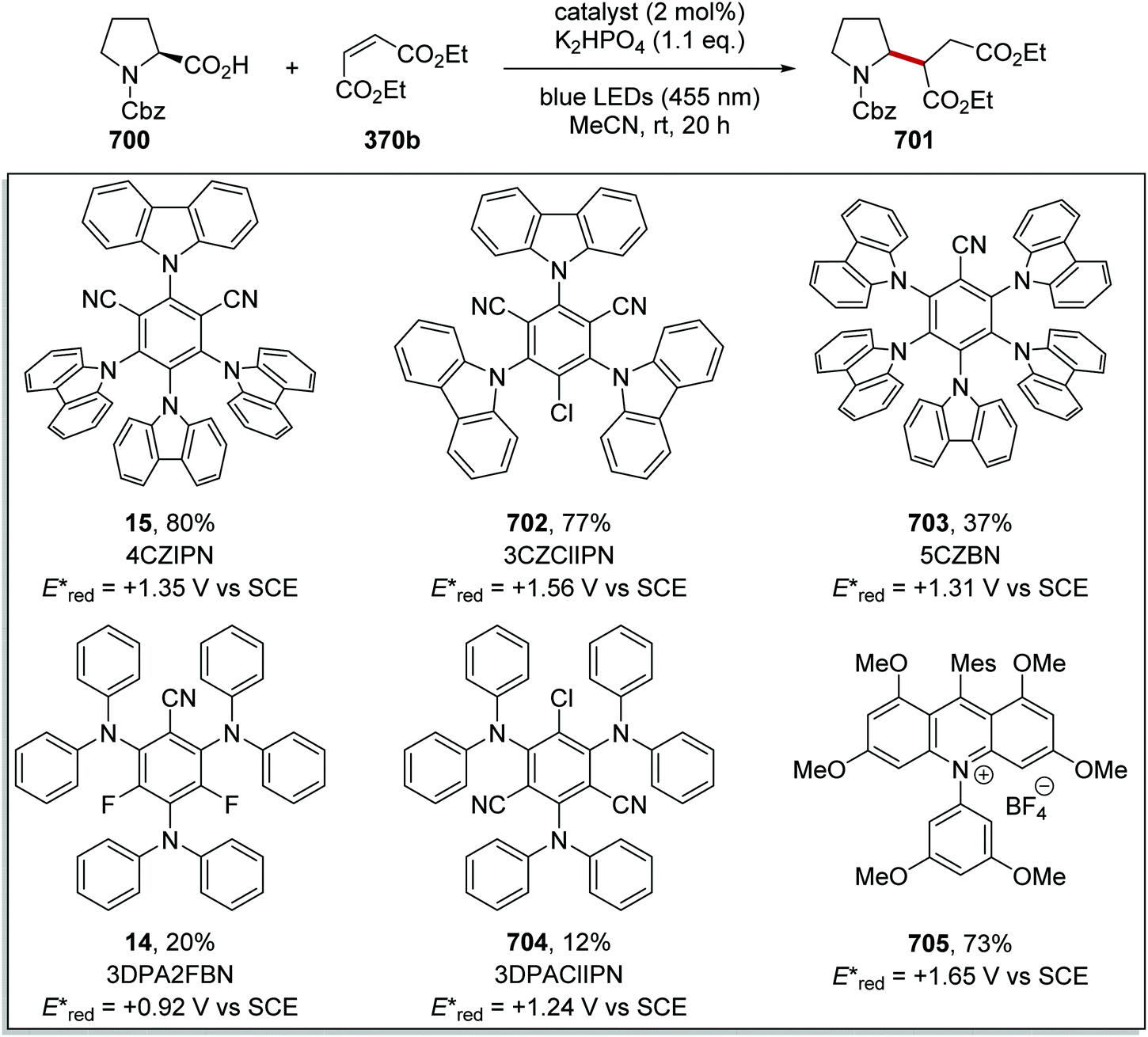
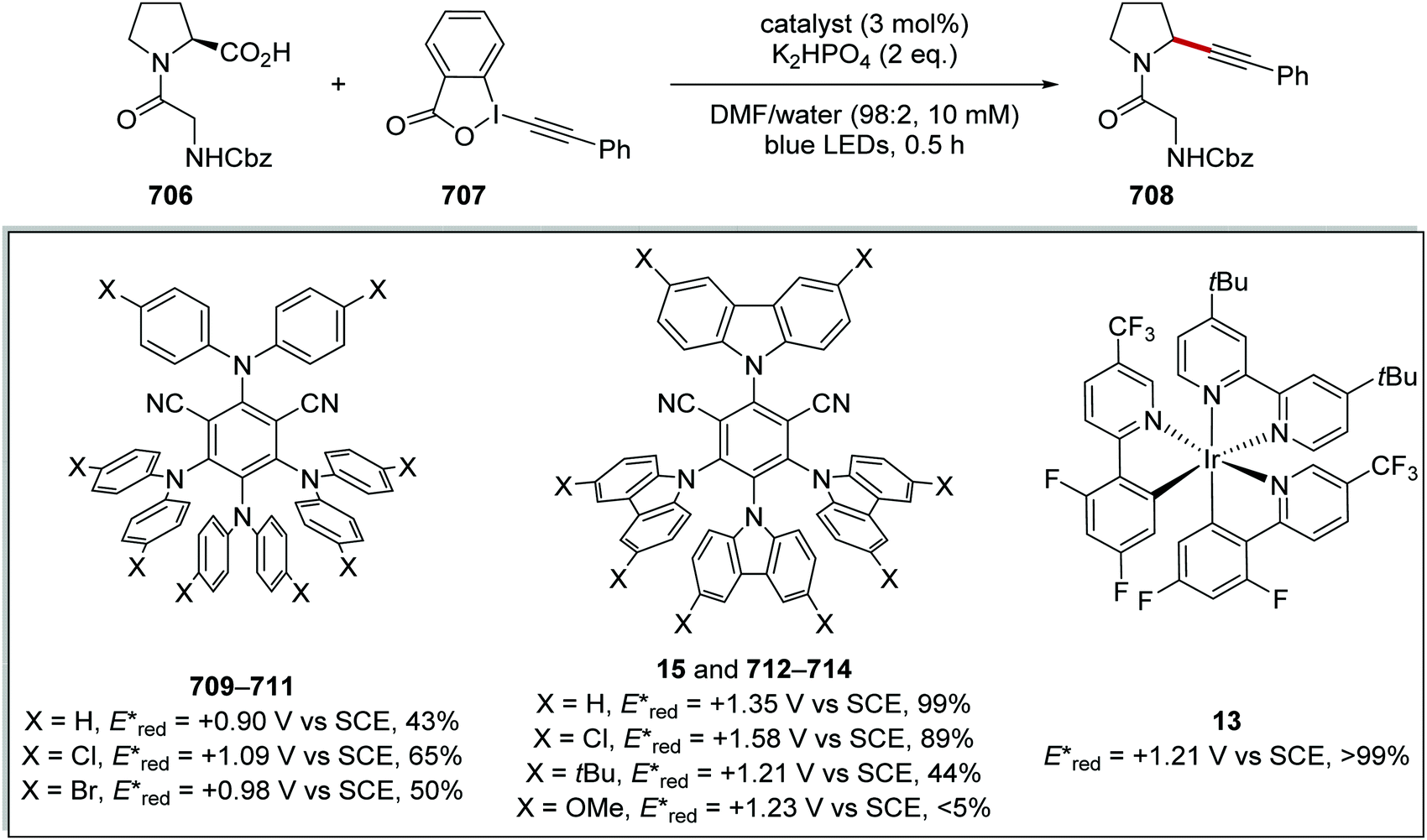
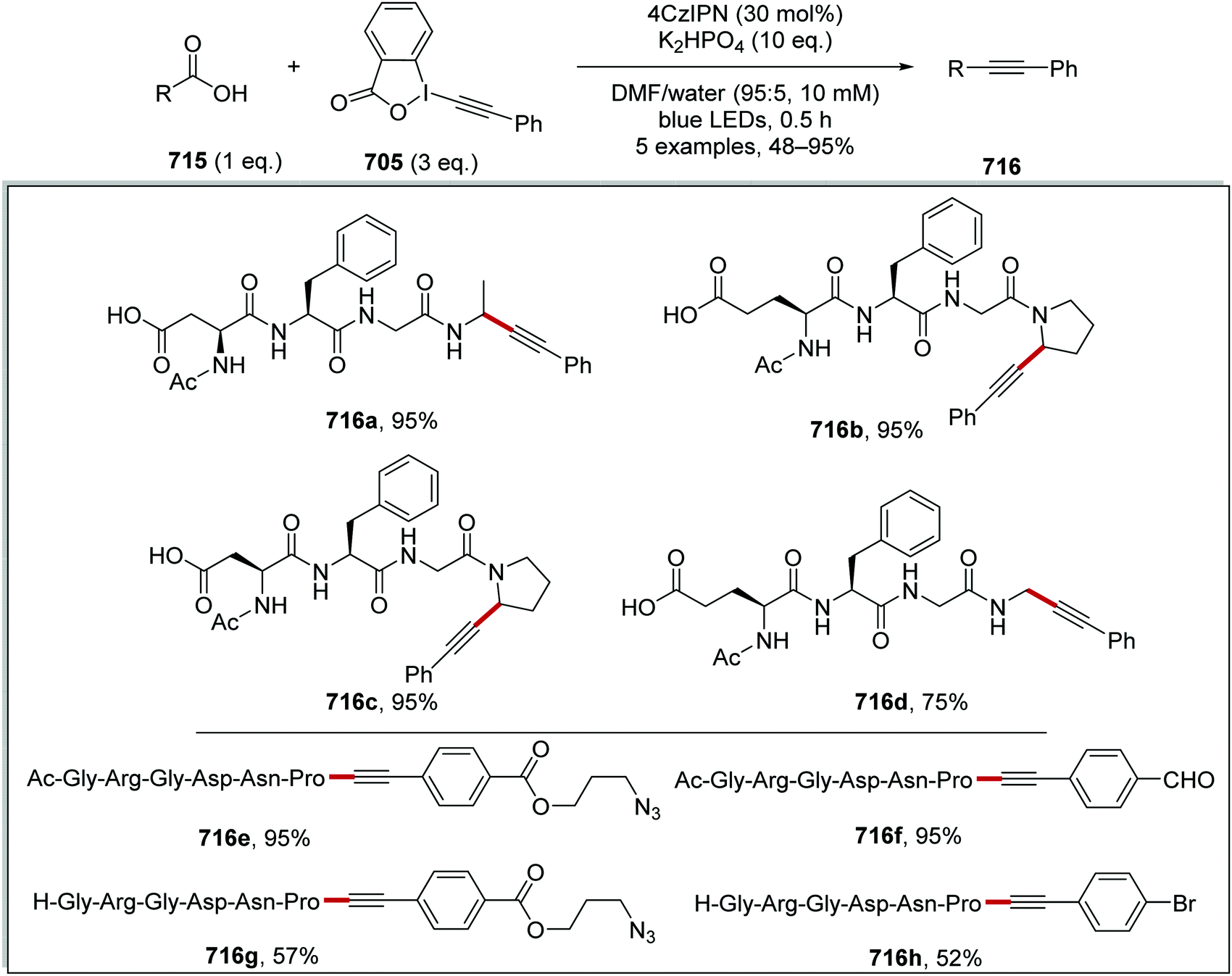
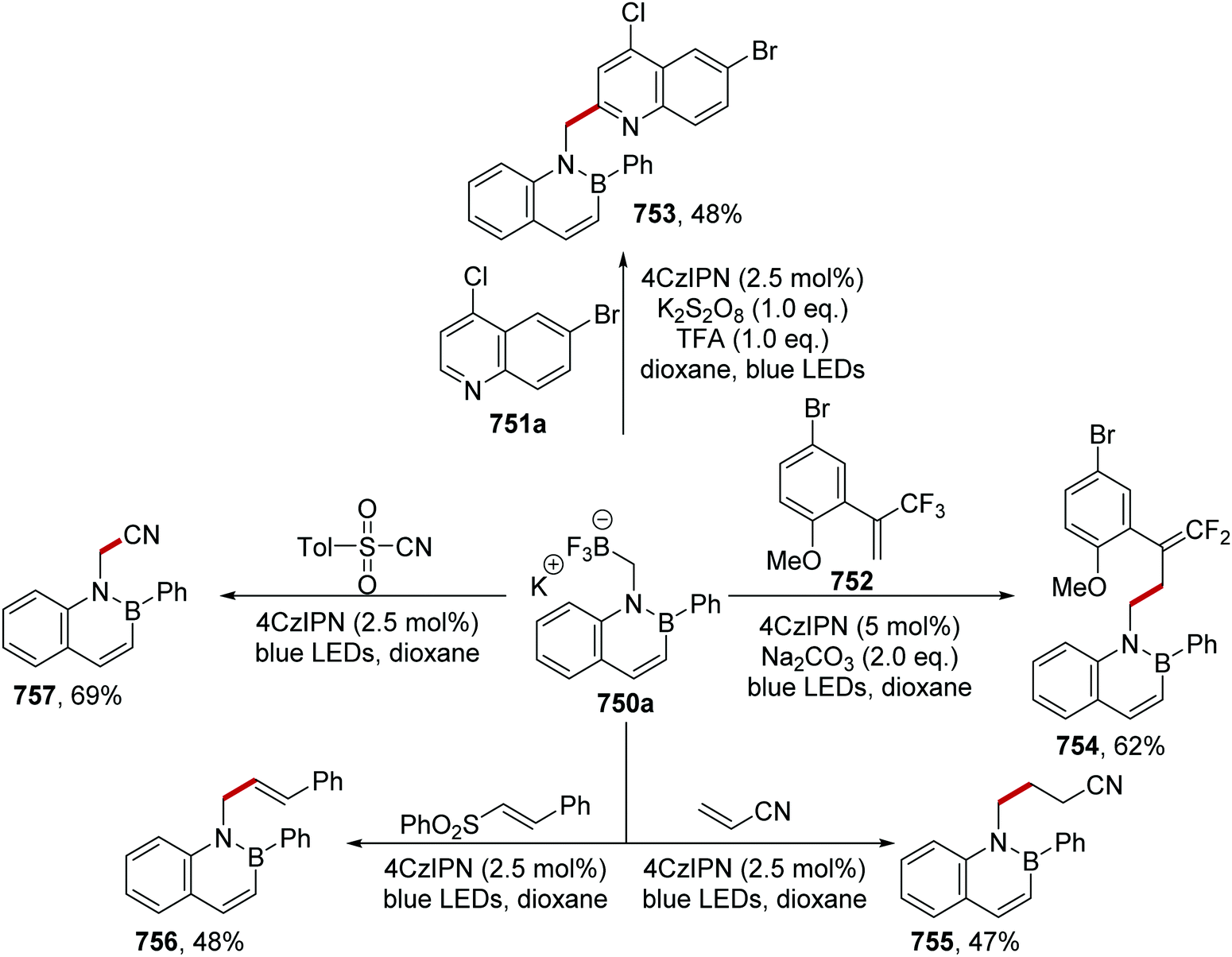
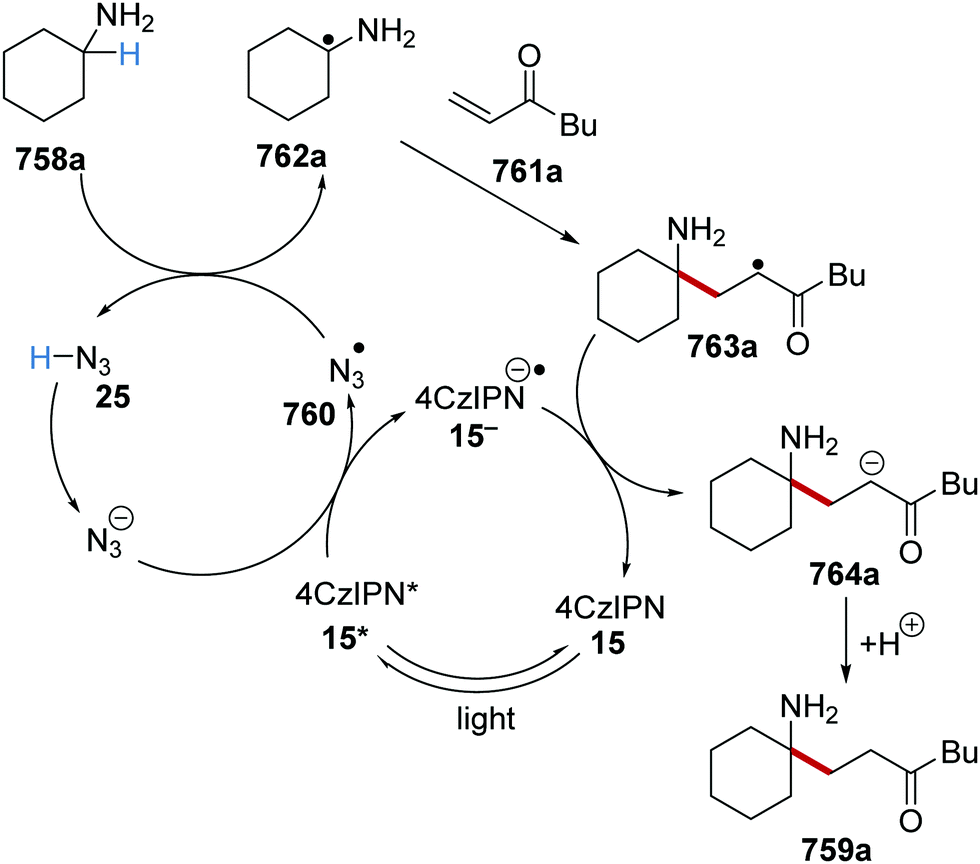
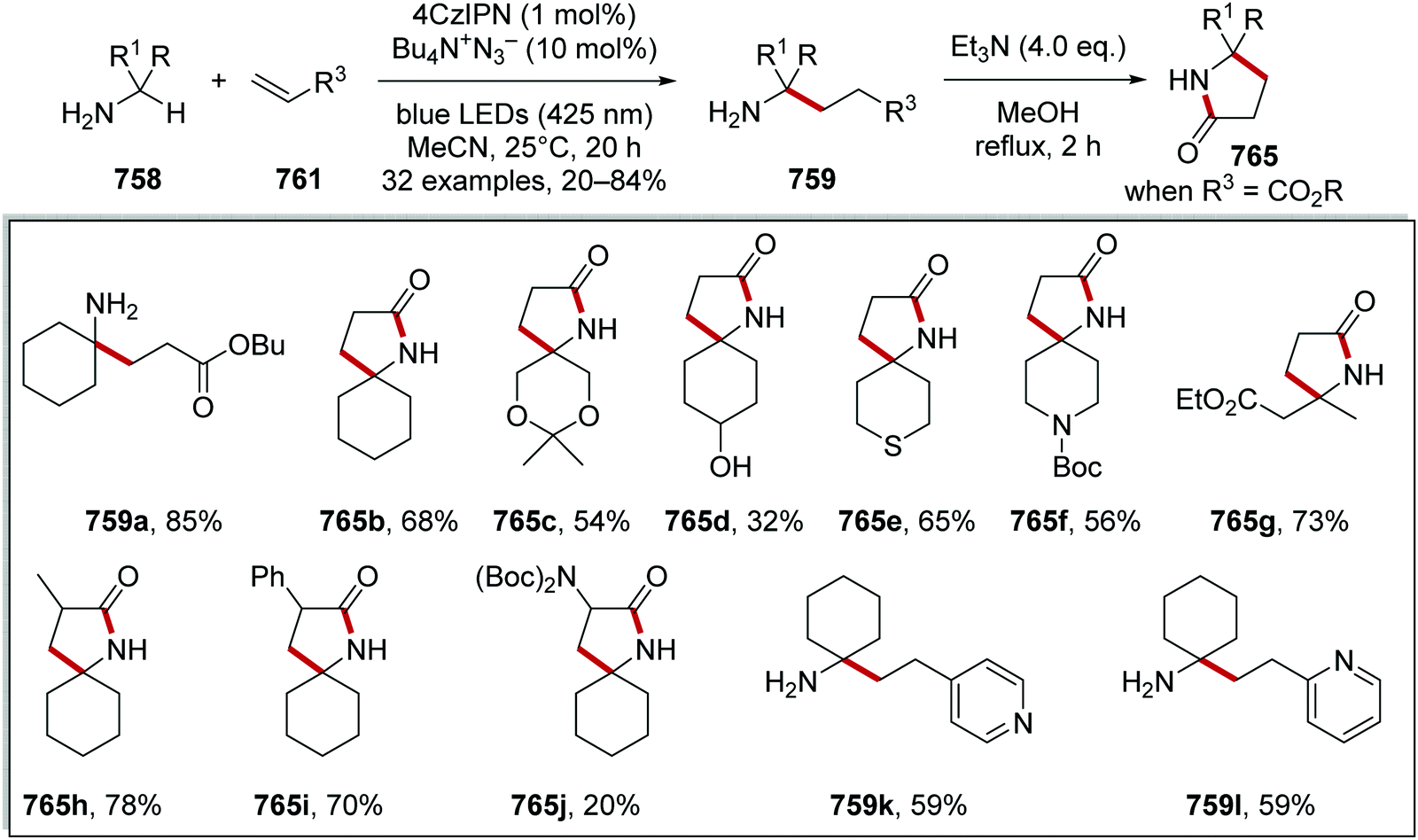
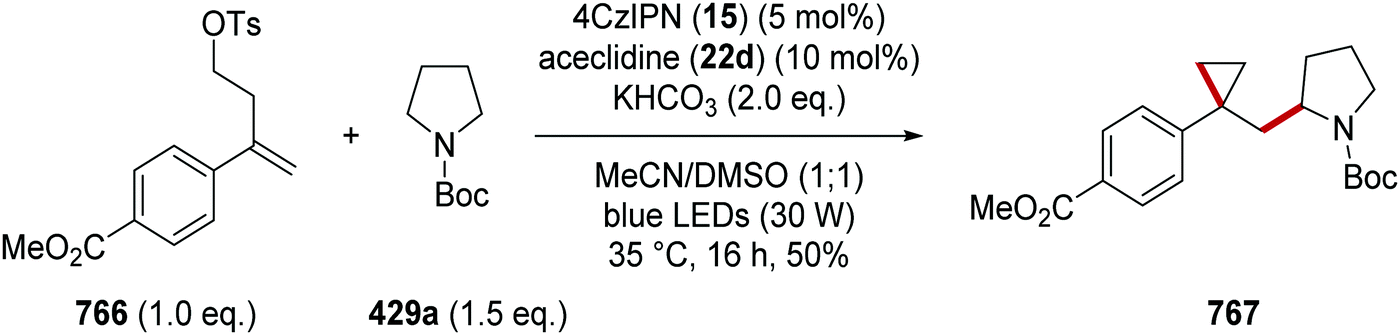
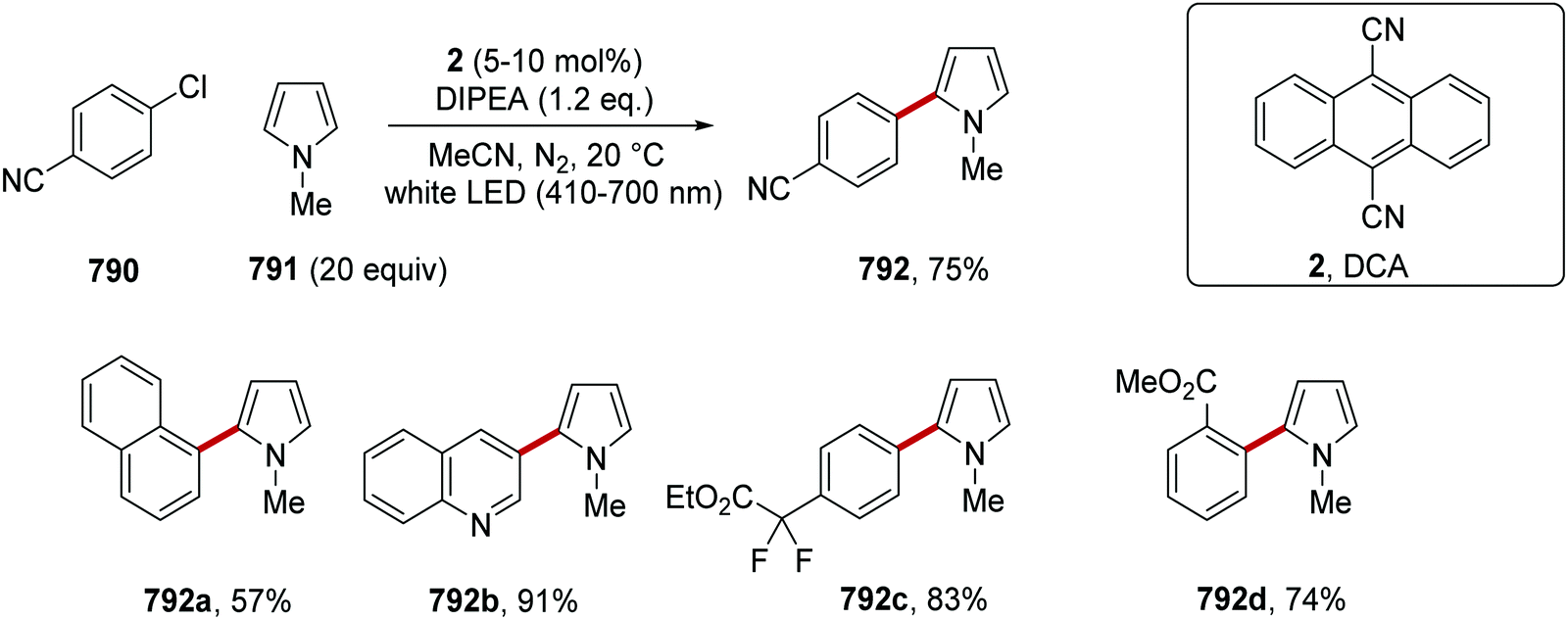
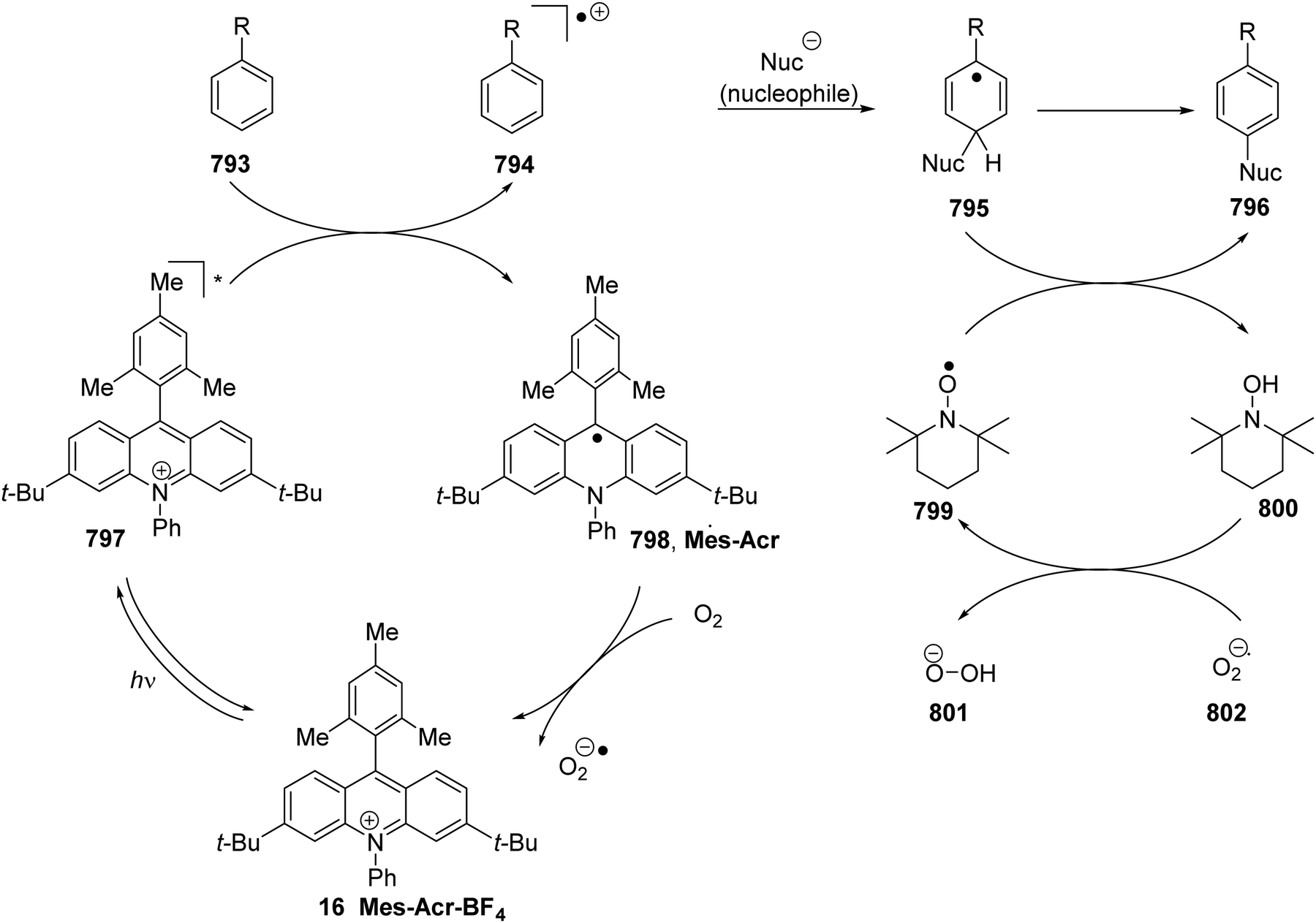
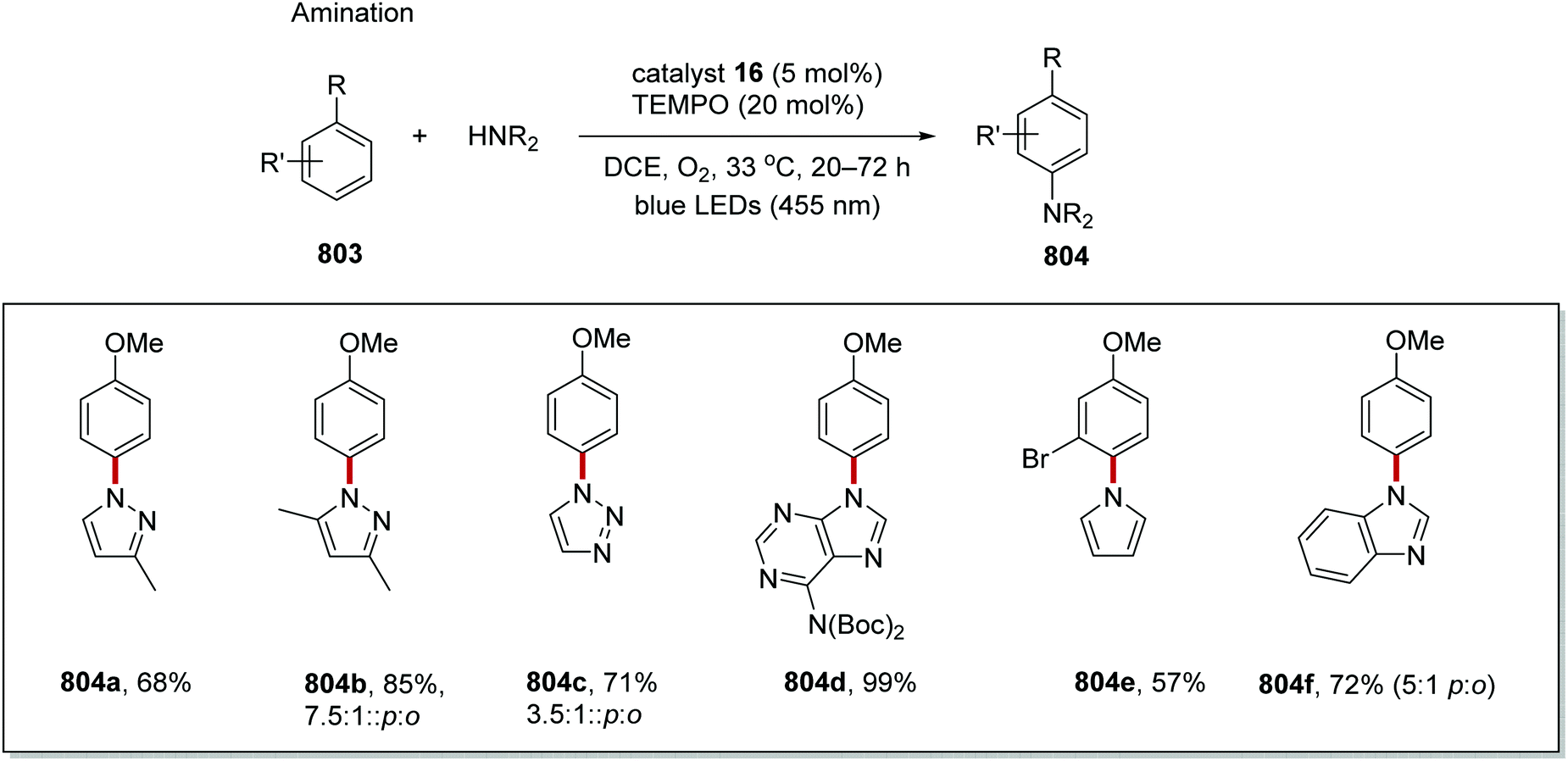
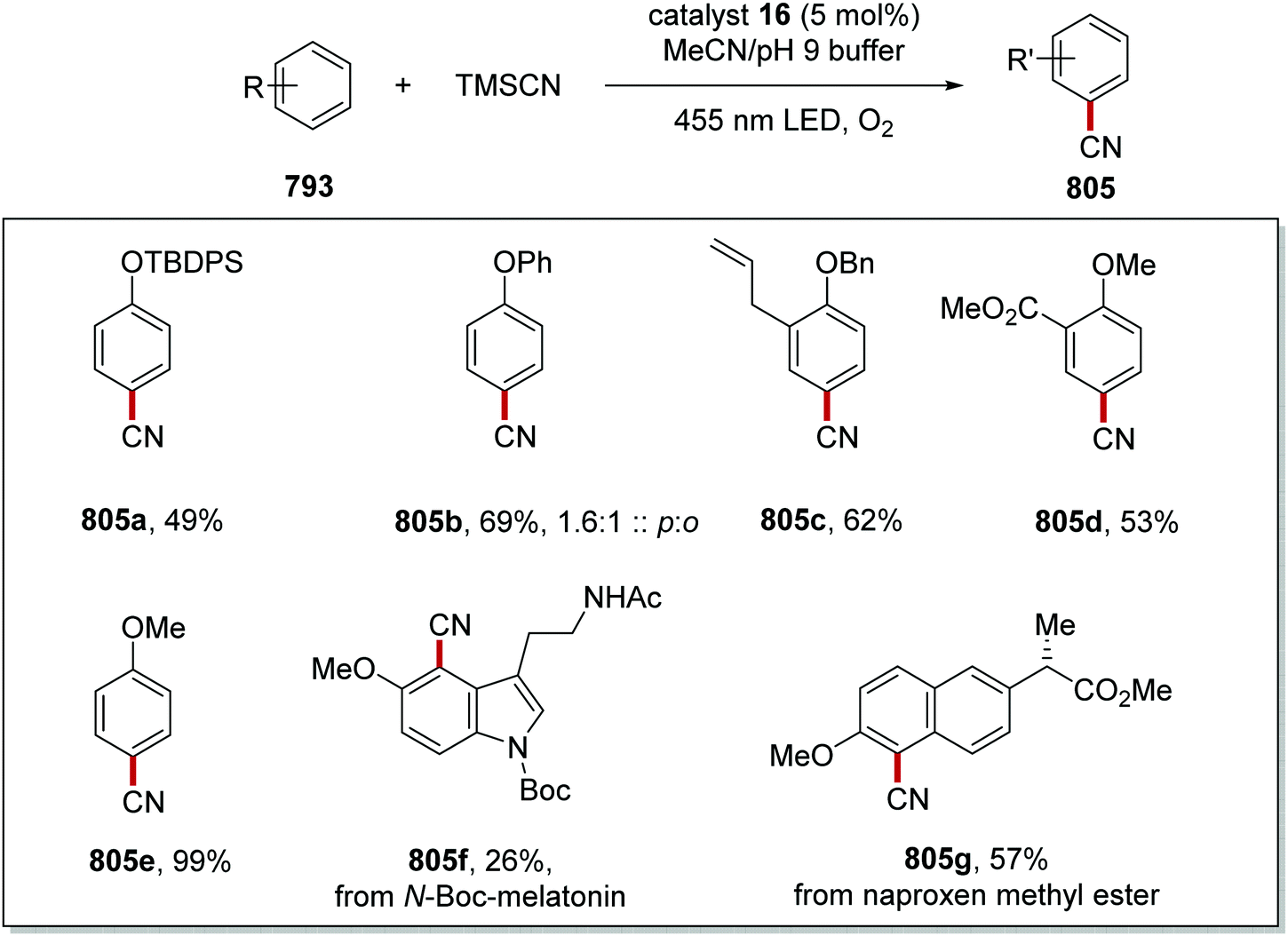
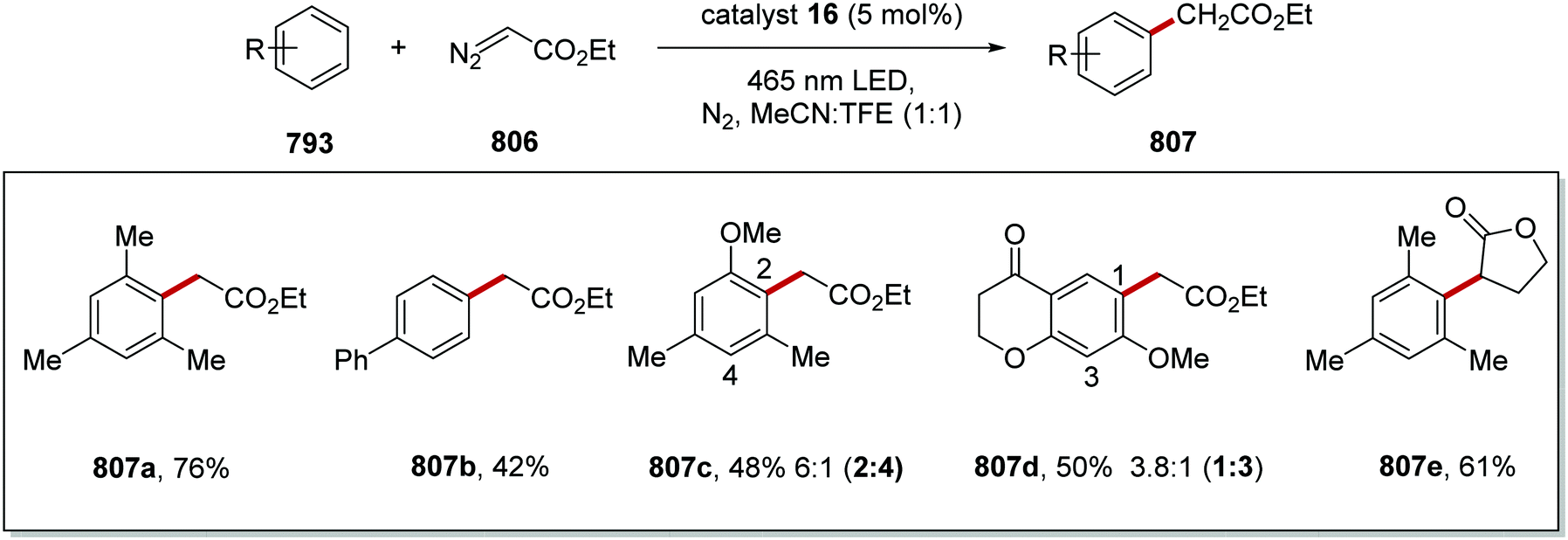
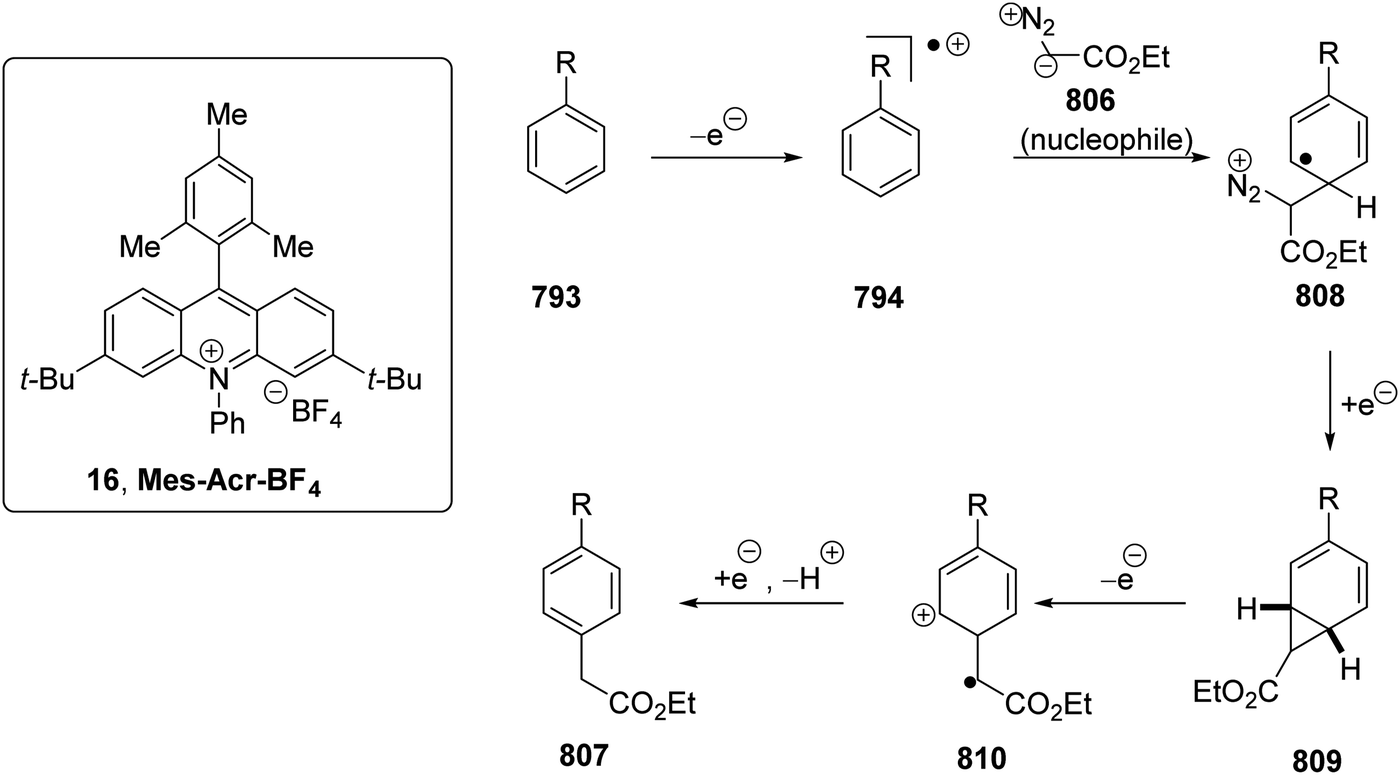
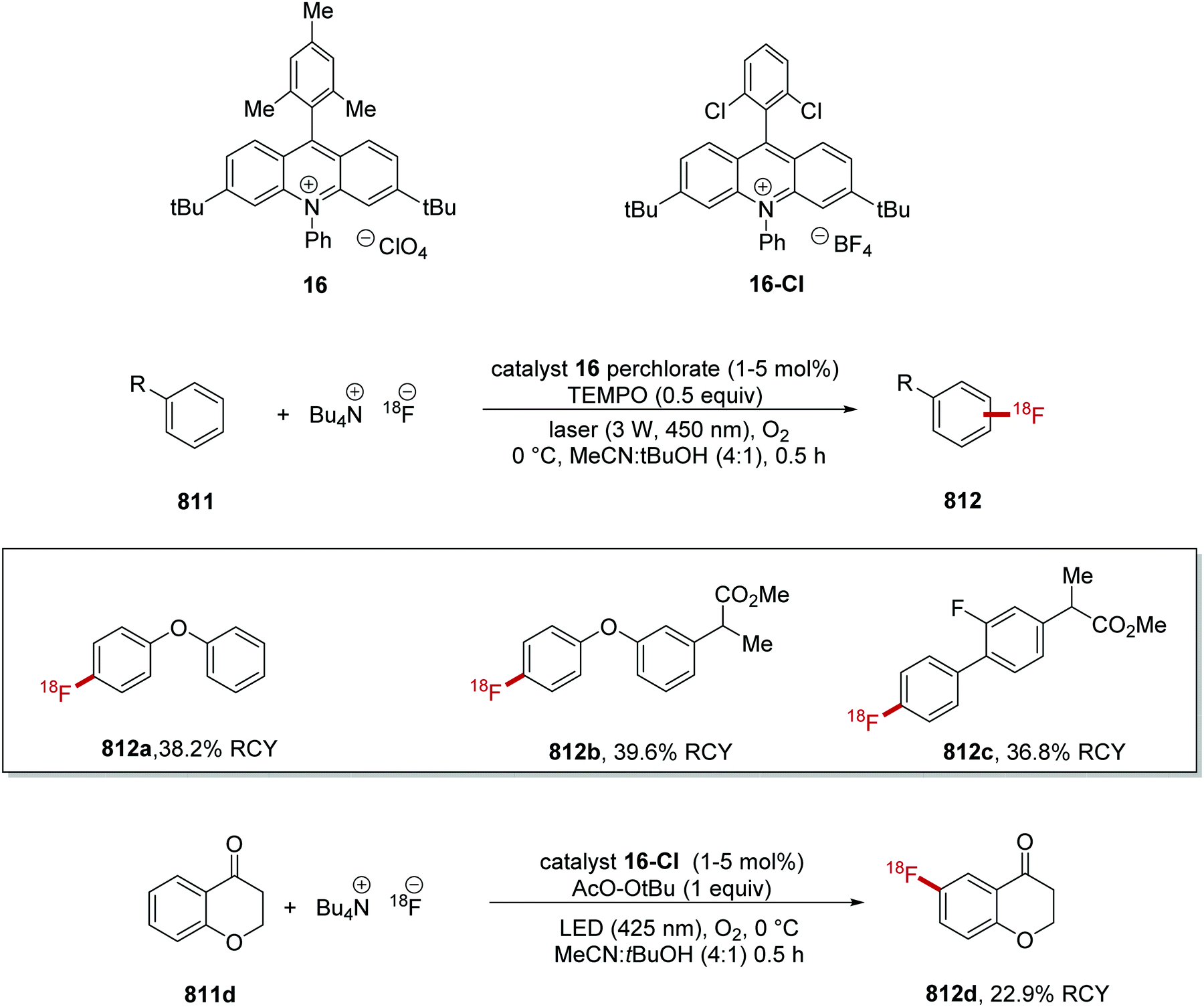
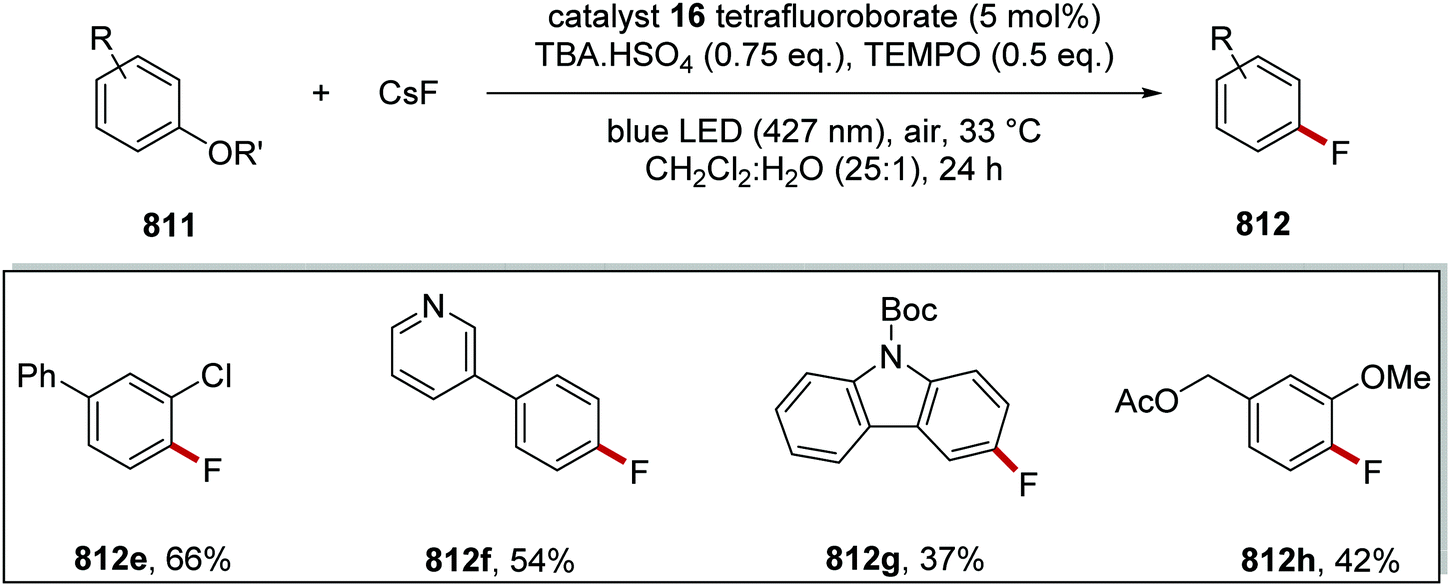
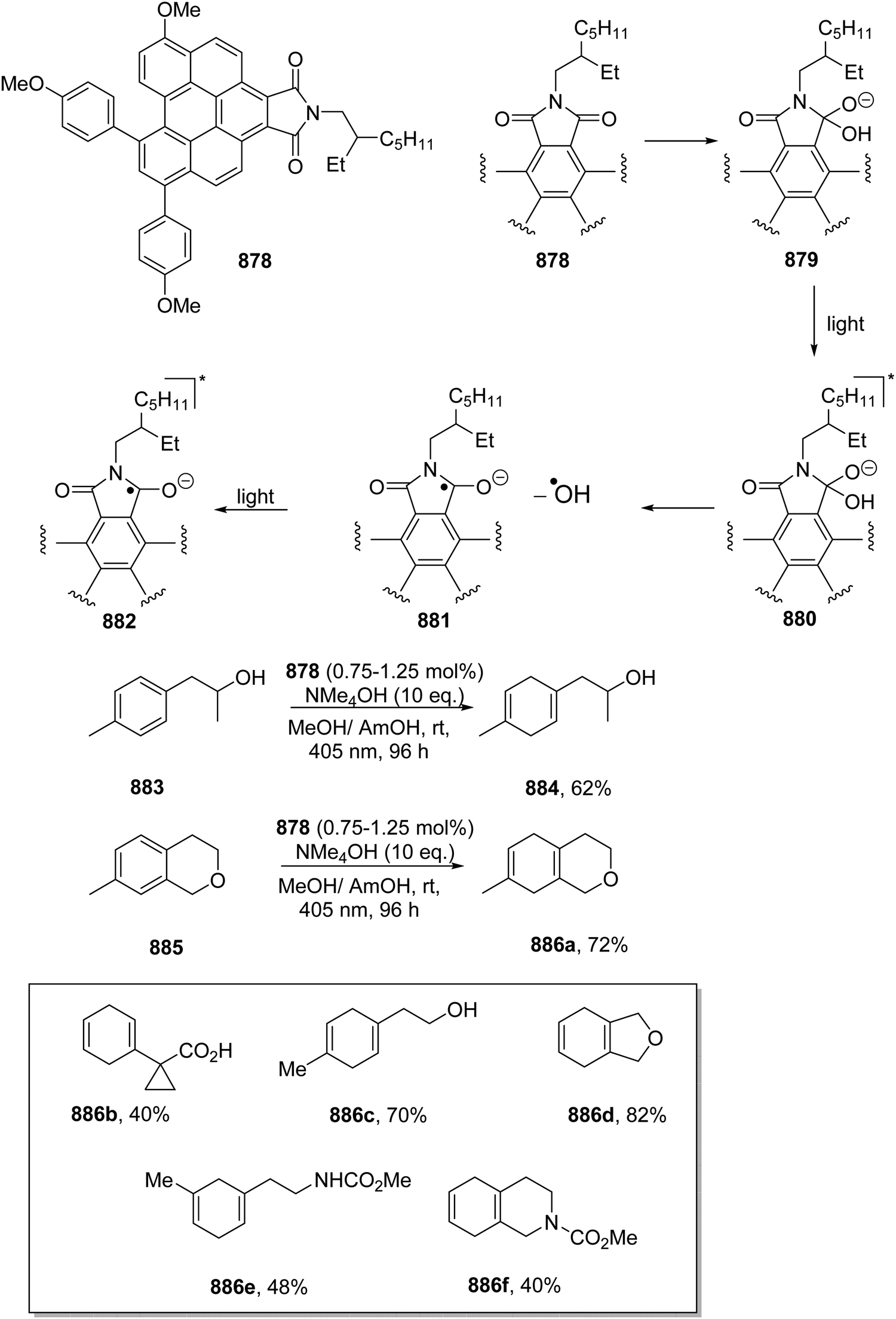
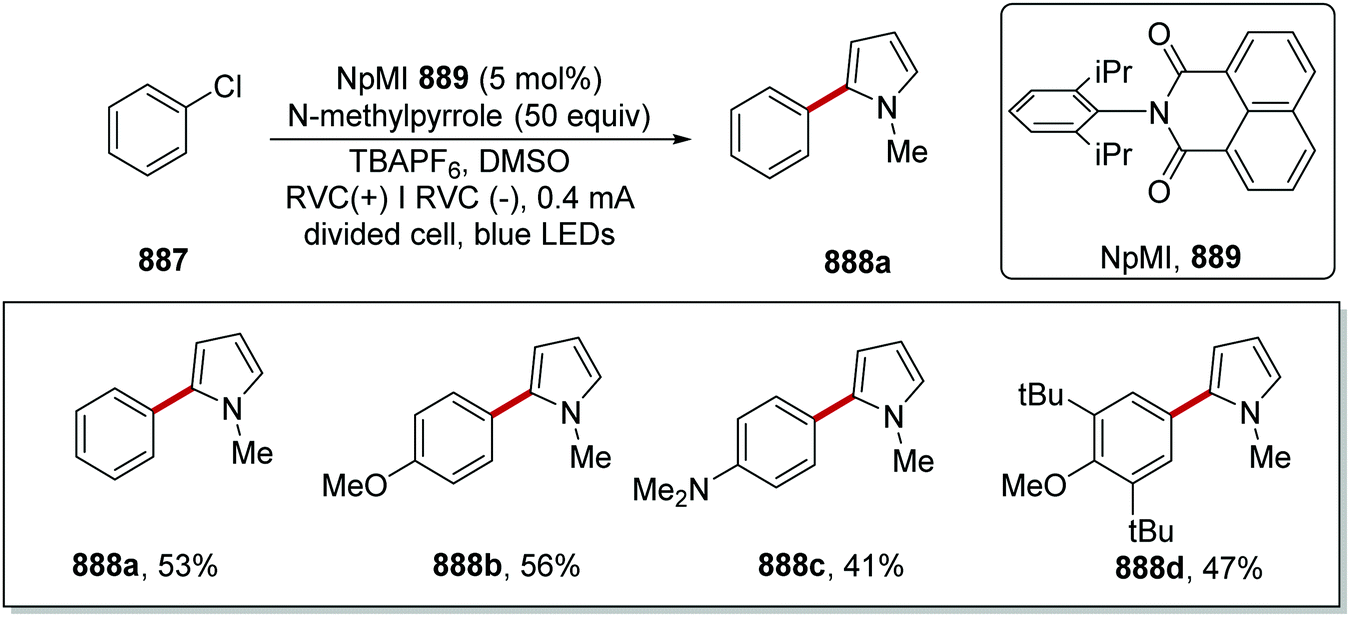
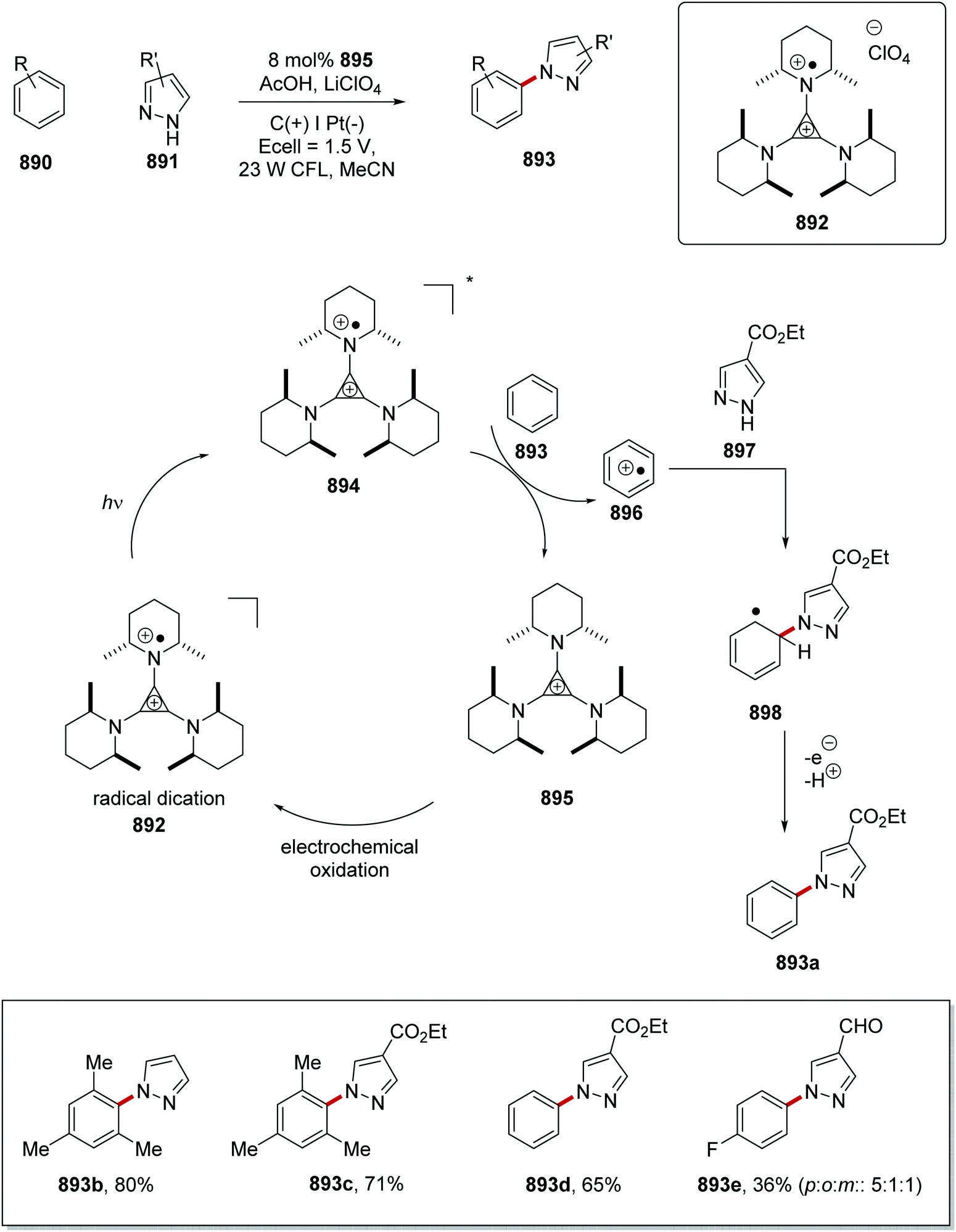
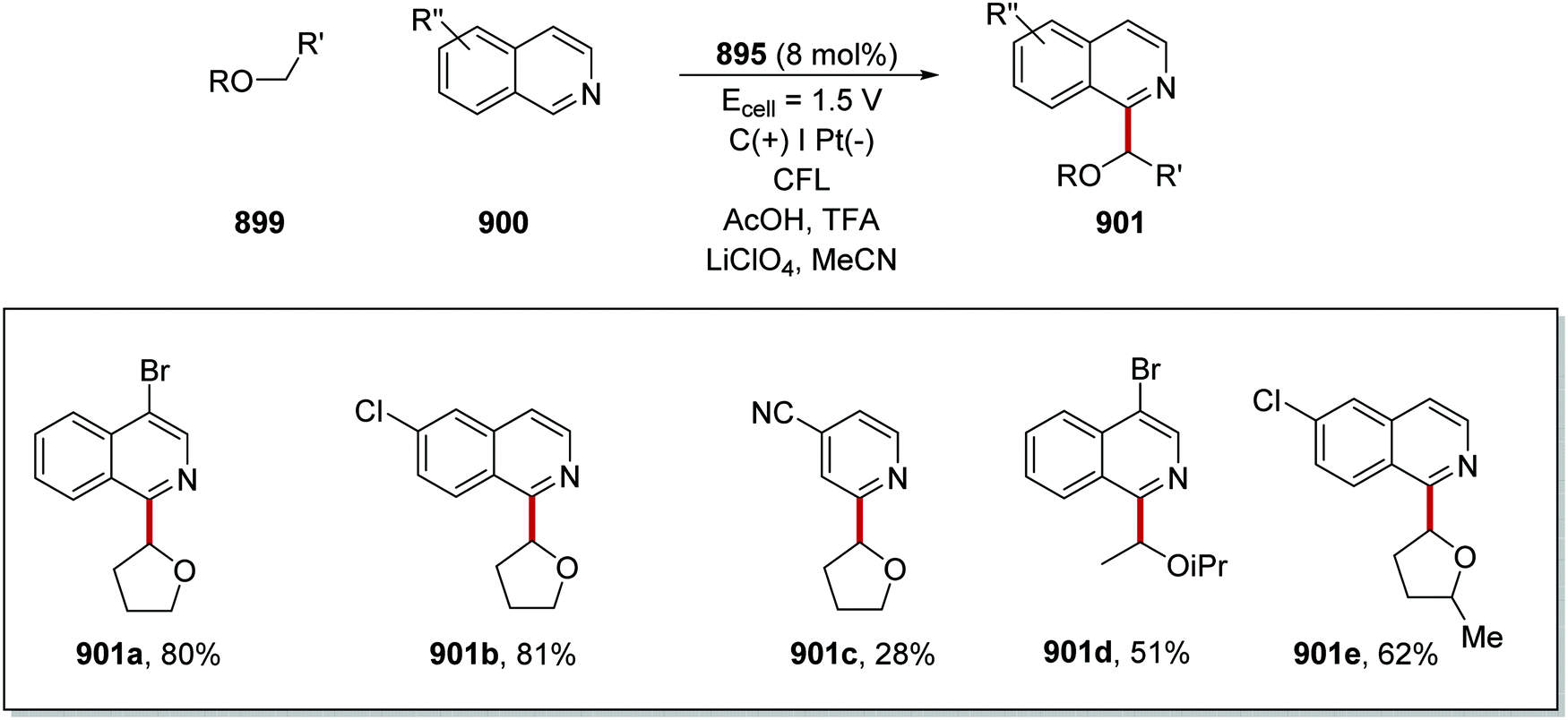
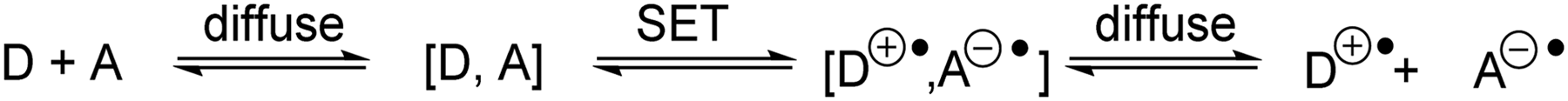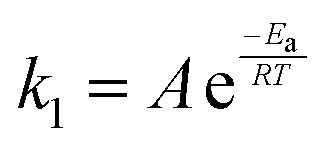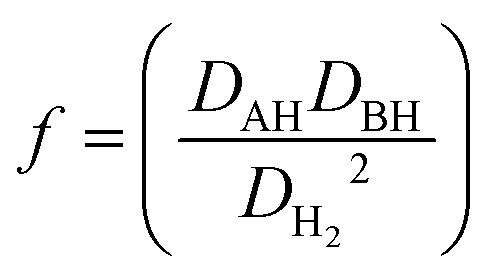DOI:
10.1039/D1CS00311A
(Review Article)
Chem. Soc. Rev., 2021,
50, 9540-9685
Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents
Received
29th March 2021
First published on 26th July 2021
Abstract
Photoredox chemistry with organic or transition metal agents has been reviewed in earlier years, but such is the pace of progress that we will overlap very little with earlier comprehensive reviews. This review first presents an overview of the area of research and then examines recent examples of C–C, C–N, C–O and C–S bond formations via radical intermediates with transition metal and organic radical promoters. Recent successes with Birch reductions are also included. The transition metal chemistry will be restricted to photocatalysts based on the most widely used metals, Ru and Ir, but includes coupling chemistries that take advantage of low-valent nickel, or occasionally copper, complexes to process the radicals that are formed. Our focus is on developments in the past 10 years (2011–2021). This period has also seen great advances in the chemistry of organic photoredox reagents and the review covers this area. The review is intended to present highlights and is not comprehensive.

Jonathan D. Bell
| Dr Jonathan D. Bell graduated with a MChem honours from the University of St Andrews in 2014, which included a research project with Dr Euan Kay. He completed his PhD at the University of Glasgow under the supervision of Prof. Andrew Sutherland and Dr Chris Wellaway, where he investigated the stereoselective synthesis of fluorescent amino acids, natural products, and biomolecules. In 2019 he joined the Prof. John Murphy group at the University of Strathclyde for postdoctoral studies into C–H functionalisation of drug-like compounds. |

John A. Murphy
| John Murphy was educated at the University of Dublin (TCD) and the University of Cambridge. After Fellowships at Alberta and Oxford, he was appointed as Lecturer, then Reader, at the University of Nottingham. Since 1995, he has held the Merck–Pauson Professorship at the University of Strathclyde. His interests are in reactivity, reaction mechanisms and synthesis. |
1 Introduction
Radical coupling reactions have a rich history with many prominent examples such as the Meerwein arylation reaction,1 the Minisci reaction,2,3 and the Giese addition of radicals to unsaturated substrates.4 Recently, there has been a surge in interest in radical-mediated coupling reactions due to the emergence of new catalysts and reagents. New transformations have emerged that take advantage of photoinduced electron transfer (PET), hydrogen atom transfer (HAT) and energy transfer processes, and these three processes have their own selectivity criteria. PET selectivity is governed by redox potentials and electron transfer kinetics of the functional groups present upon the substrate.5 HAT processes are principally determined by bond energies,6 and energy transfers are regulated by triplet energy values of the species involved.7 The area of photoredox chemistry has become vast. The area has moved rapidly since landmark general reviews;8 many of the recent reviews represent very specific areas. Our aim here is to provide an overview of some of the areas of rapid development that deploy (i) transition metal photoredox reagents based on ruthenium and iridium complexes or (ii) organic photoredox catalysts.9
Photophysics
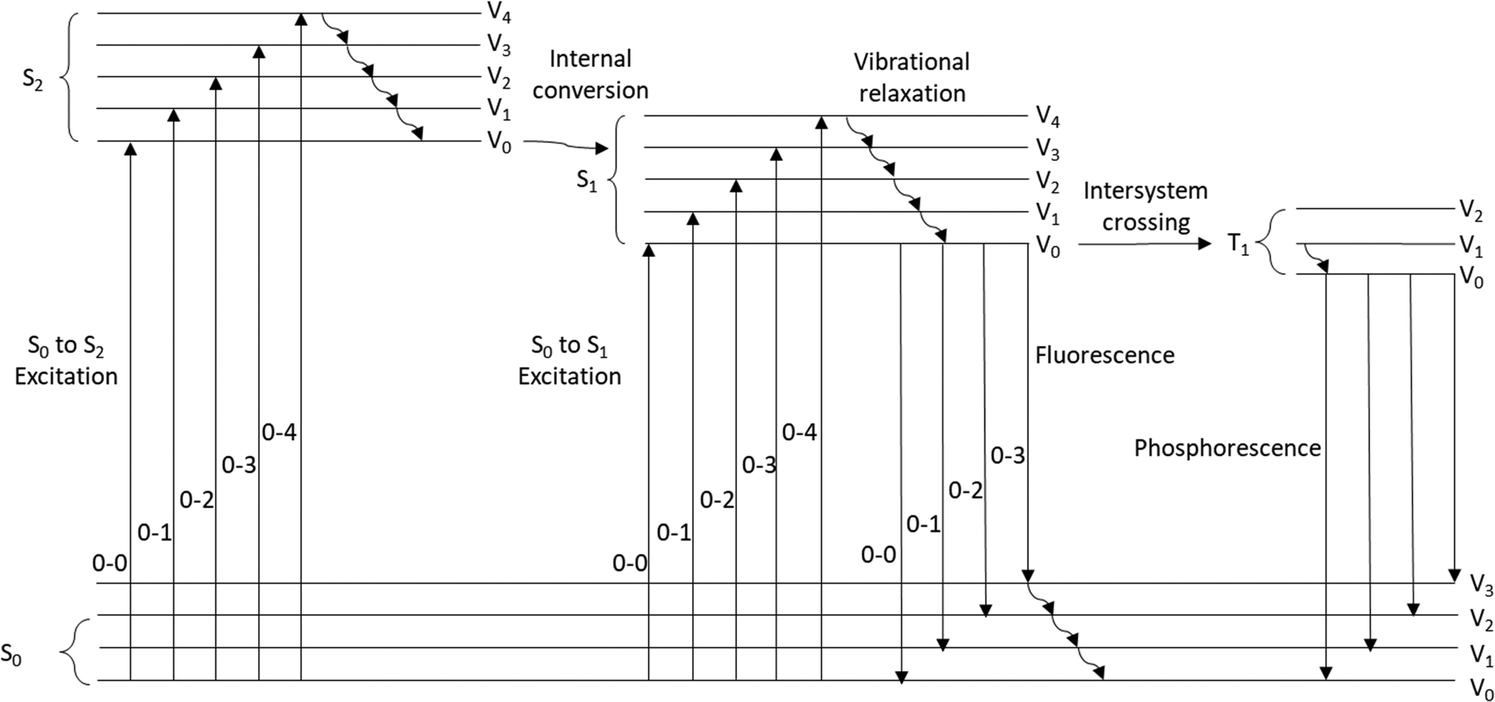
As a compound is excited with a photon of light, an electron is transferred from the ground state, (usually S0), to a higher energy orbital (Fig. 1). An indication that a compound is an effective photocatalyst is that it is strongly photoluminescent as it signifies that non-productive deactivation pathways are minimised.8a
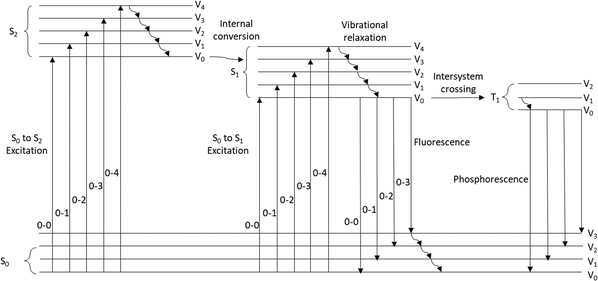 |
| | Fig. 1 Jablonski–Perrin diagram representing electron transitions between ground and excited states. | |
Kasha's rule states that in photoemissive processes, the emitting electronic level of a given multiplicity (usually singlet or triplet) is the lowest excited state of that multiplicity (in these cases S1 or T1). To date, azulene which has a large energy gap between the first and second singlet excited states (ΔES2→S1 = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1) was reported in 2019 as the only compound that breaks Kasha's rule.10 Recently it was suggested that “breaking” Kasha's rule may lead to more effective photochemistry.11
000 cm−1) was reported in 2019 as the only compound that breaks Kasha's rule.10 Recently it was suggested that “breaking” Kasha's rule may lead to more effective photochemistry.11
Intersystem crossing (ISC)
The multiplicity of an excited state has a major influence upon the photophysical properties of a compound. Triplet states are long-lived and this is due to the T1 → S0 electronic transition being spin–forbidden. Therefore, the excited state lifetimes of these triplet states are much greater than for the corresponding singlet states. It can be envisioned that longer lifetimes may result in more successful photochemical processes due to more favourable kinetics.5 However, it has been stated that efficient photoinduced SET only requires an excited state lifetime of 1 ns. Intersystem crossing (ISC) is the process through which a compound changes multiplicity, typically from the lowest excited singlet state to the lowest excited triplet state.12 This process is formally forbidden within nonrelativistic quantum theory, and therefore access to the long-lived triplet states is limited.8,13 However, efficient ISC is achieved with the inclusion of heavy atoms, paramagnetic atoms or the change of orbital character with the change in multiplicity.12–16
El-Sayed's rules
Processes where the total angular momentum is conserved are non-forbidden. Hence, if a change of electron spin coincides with a change in angular momentum, then efficient ISC is viable.13–16 Therefore, highly efficient ISC is found with compounds such as benzophenone,17 xanthone15 and benzaldehyde,18 with the ϕISC of benzaldehyde being close to unity in the vapour phase.19,20
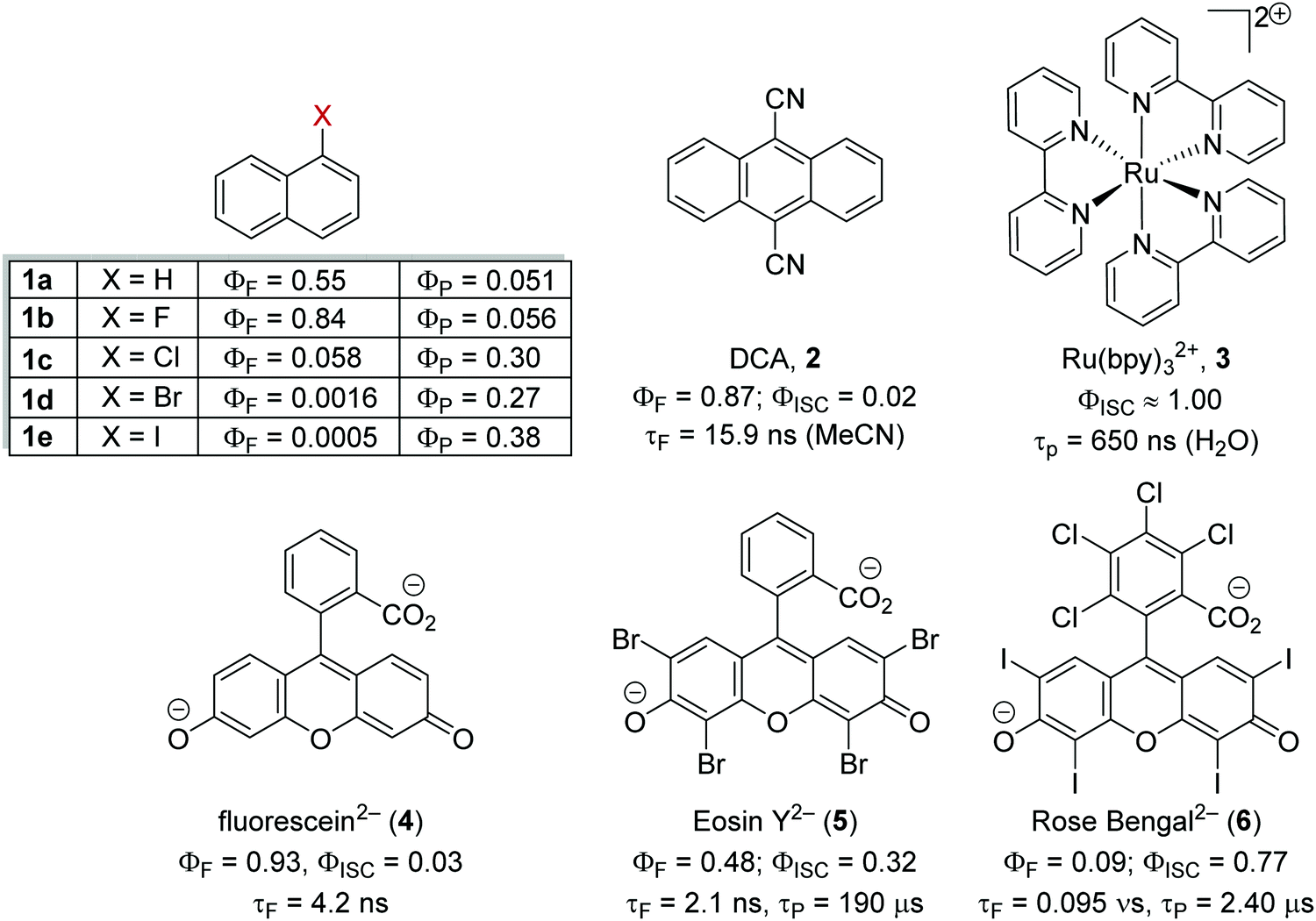
Heavy atom effect
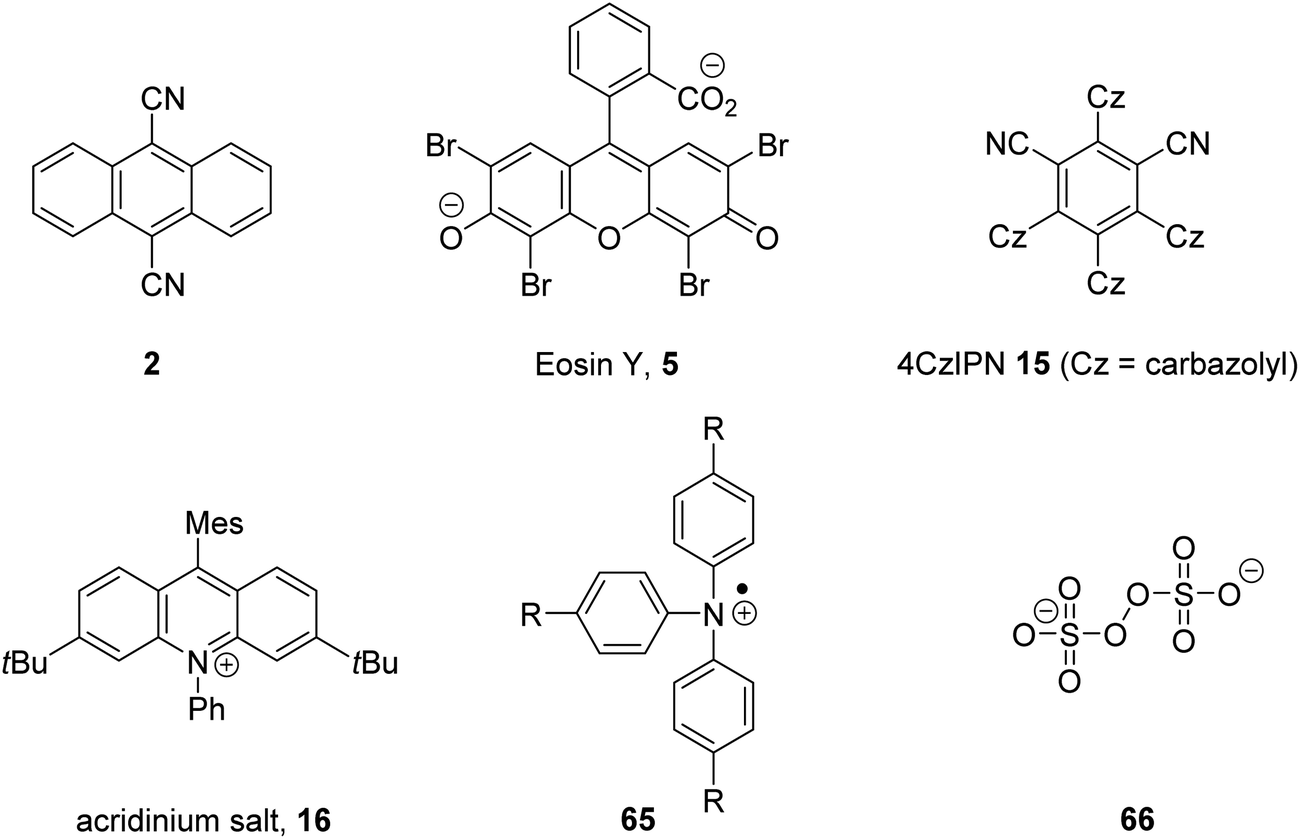
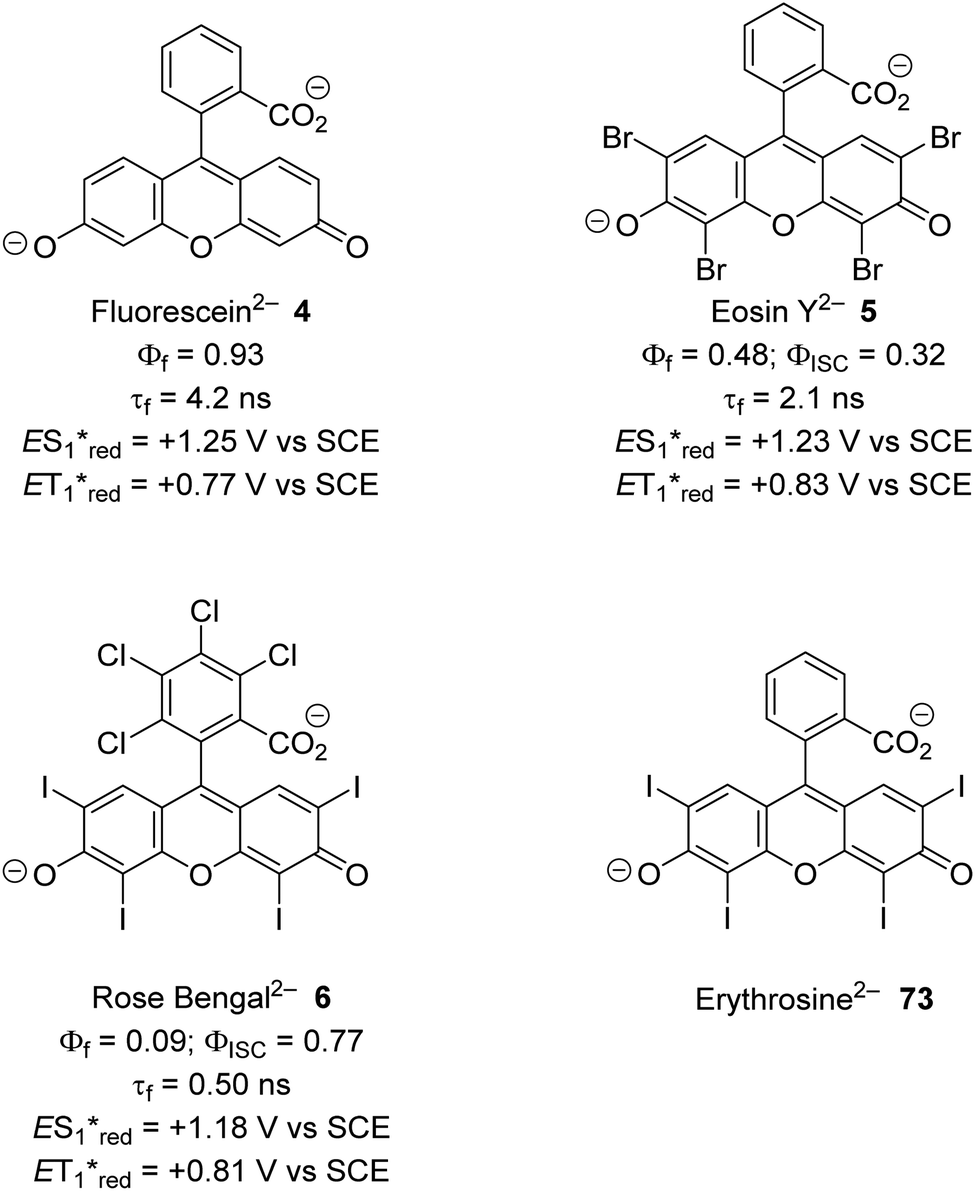
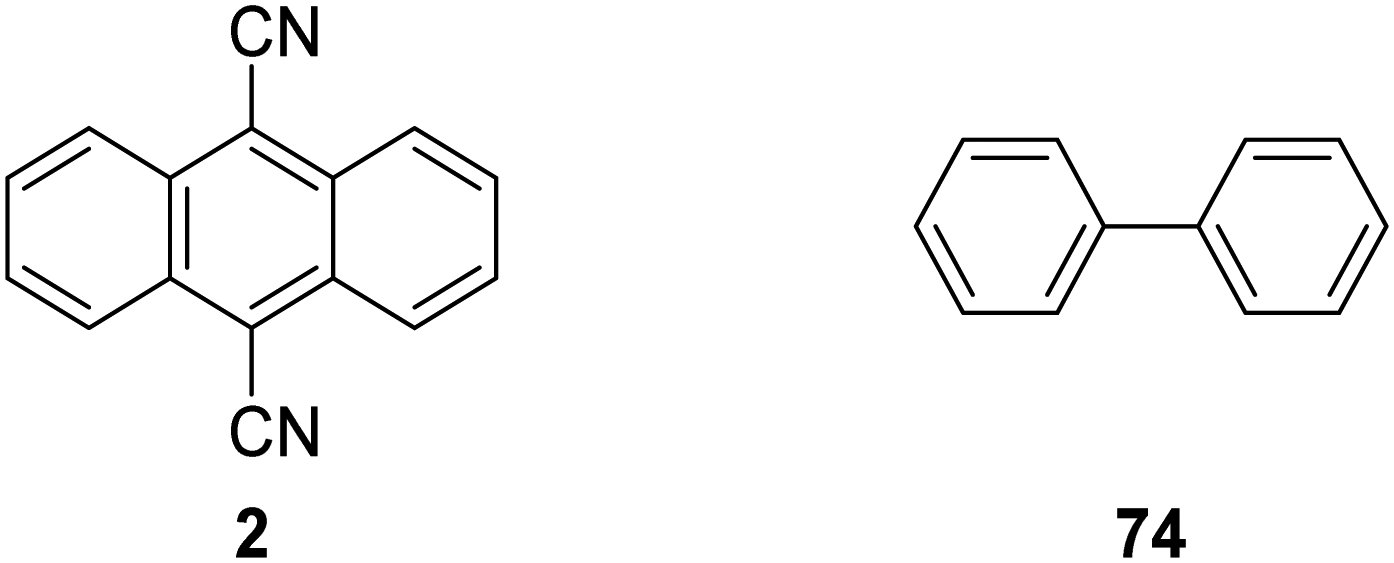
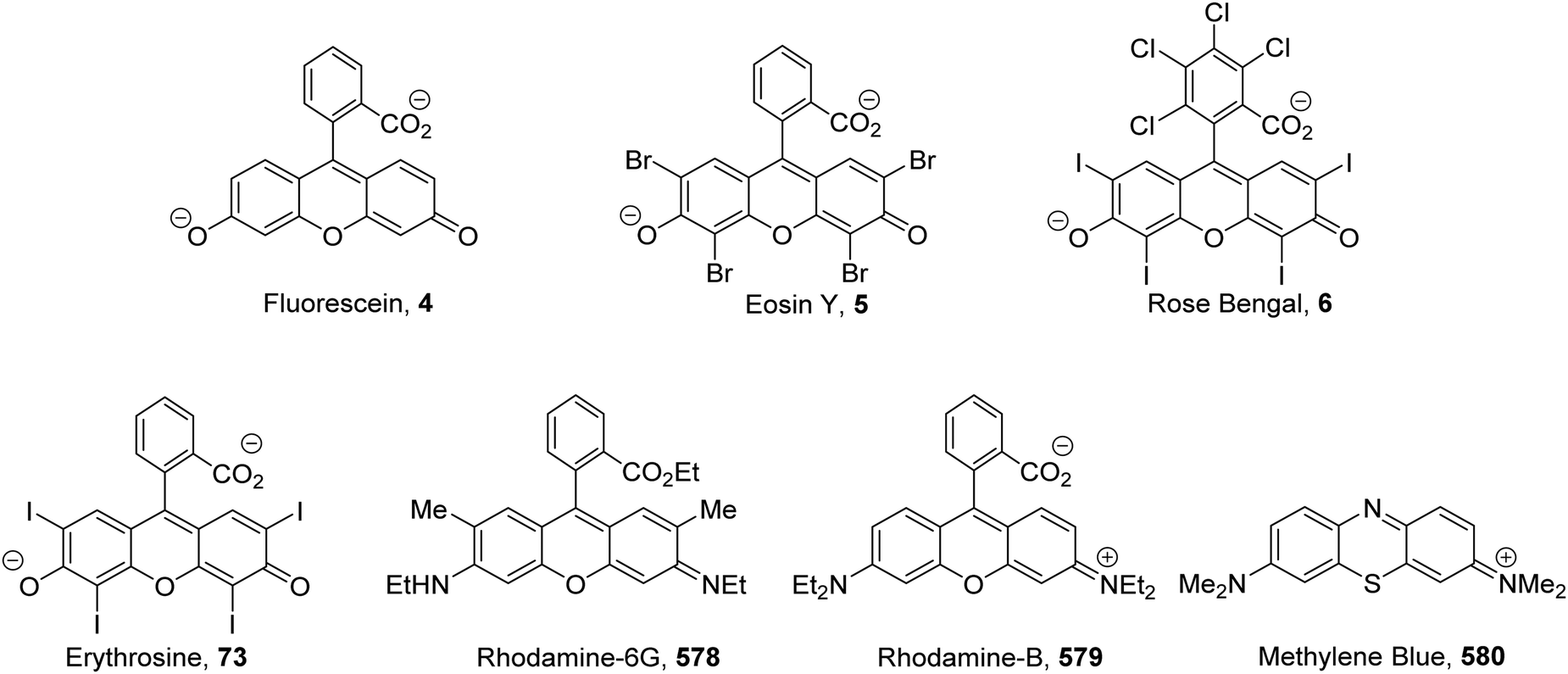
The inclusion of heavy atoms increases ISC efficiency and this is demonstrated with naphthalene analogues 1a–1e (Fig. 2); when successively heavier halogen atoms are added, the ϕISC increases. The heavy atom effect can operate intermolecularly and through the chemically impermeable walls of molecular cages.21 The presence of heavy atoms has a significant impact upon the characteristics of photocatalysts. For a photocatalyst without heavy atoms, such as 9,10-dicyanoanthracene DCA (2), low efficiency for ISC is observed (ϕISC = 0.02) and its photophysical behaviour is dominated by its S1 state, which has a lifetime of 15.9 ns.8a However, the photocatalyst Ru(bpy)32+ (3) has a heavy ruthenium atom and therefore its ISC is almost quantitative; its triplet state is long-lived.22 The effect of heavy atom inclusion is also displayed with the xanthene dyes, fluorescein (4), Eosin Y (5) and Rose Bengal (6). Fluorescein has no heavy halogen atoms and has a ϕISC of 0.03 but Eosin Y and Rose Bengal contain bromine and iodine atoms respectively and this results in higher ϕISC with values of 0.32 and 0.77 respectively.8a This results in 4 having a short excited state lifetime (4.2 ns) while catalyst 6 has a much longer excited lifetime (2.4 μs).8a,23
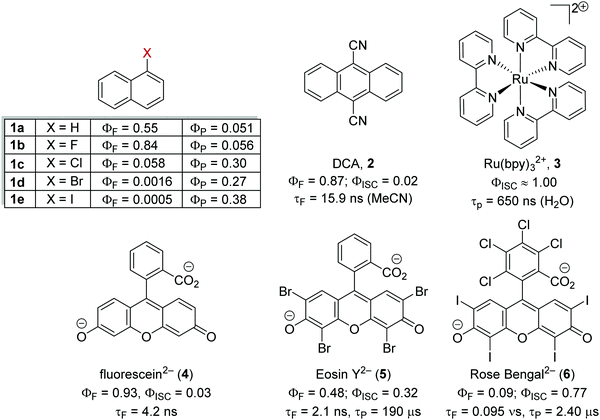 |
| | Fig. 2 The effect of heavy atoms upon photophysical properties. | |
Photochemical processes
Once a compound is promoted to an excited state, it can go through a number of different processes such as single electron transfer (SET, commonly described as photoinduced electron transfer, PET),24 HAT (hydrogen atom transfer),25 and energy transfer.7
Electron transfer
Single-electron transfer (SET) is a transfer of an electron from an electron donor (D) to an electron acceptor (A).26–28 SET can proceed via a transition state where the donor and acceptor molecules interact strongly (binding energy >4.8 kcal mol−1) and this is referred to as inner-sphere electron transfer (ISET). Electron transfer events between species that are weakly interacting (1.0–3.8 kcal mol−1) or non-bonded organic compounds are known as outer-sphere electron transfer (OSET).29 Marcus theory was developed so that rates of OSET between donor and acceptor species could be calculated (Scheme 1).
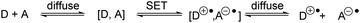 |
| | Scheme 1 Outer-sphere electron transfer depicted by Marcus theory. | |
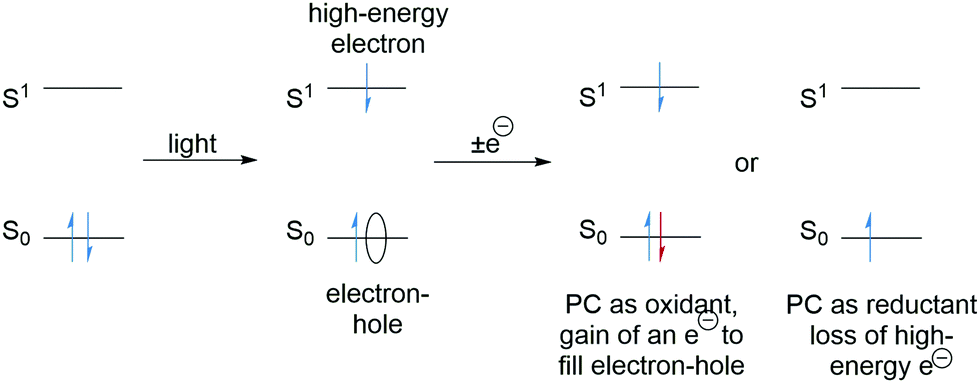
Photocatalysts are excellent species for SET as the excitation of an electron results in (i) a low energy electron–hole and (ii) a high-energy electron (Scheme 2). The electron–hole left by the transition can be filled with an electron from a donor molecule and thus the photocatalyst becomes a powerful SET oxidant. Alternatively, the high energy electron can be lost to an acceptor molecule and the photocatalyst is a strong SET reductant. The feasibility of SET processes can be evaluated with consideration of redox potentials and a comprehensive list of various functional groups has been reported.30
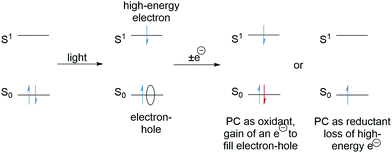 |
| | Scheme 2 Photoredox processes. | |
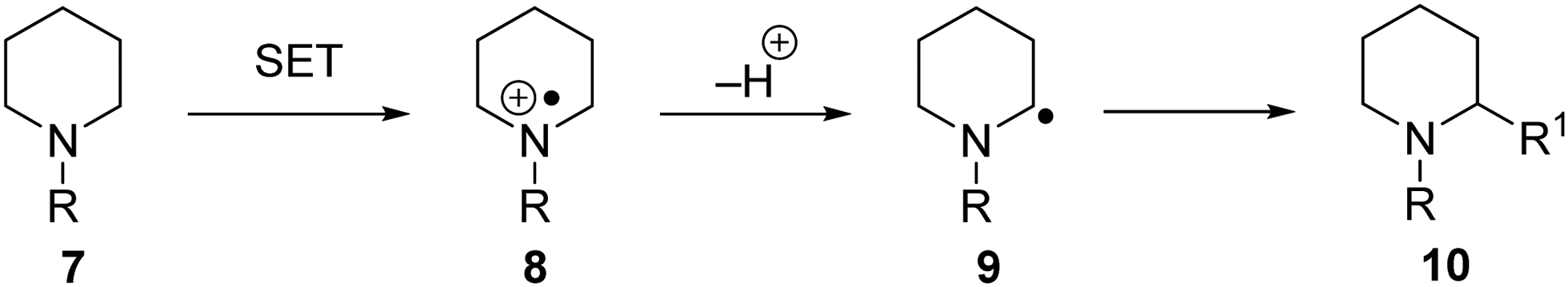
C–H functionalisation can be achieved via a SET process (Scheme 3). If an amine 7 is oxidised by electron transfer to the amine radical cation 8, this can result in proton loss and formation of carbon-centred radical 9. Numerous radical-based reactions can then take place and radical 9 is converted to functionalised product 10.
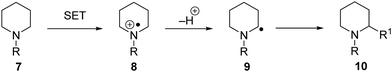 |
| | Scheme 3 C–H activation via SET. | |
Photoredox catalysts
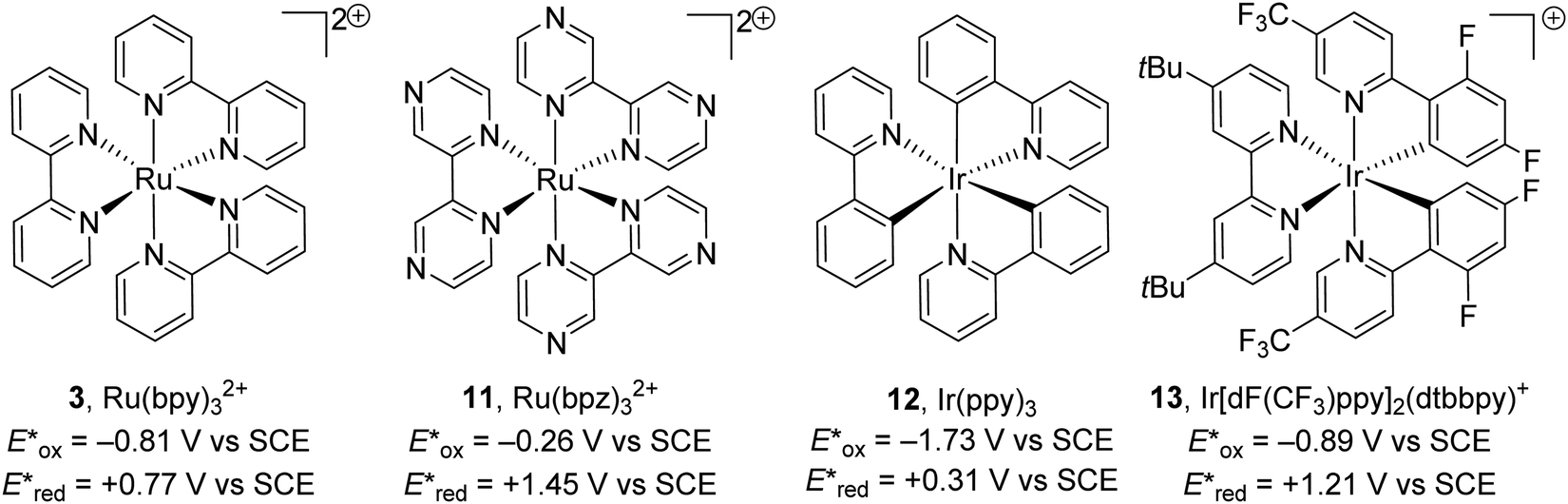
There is a vast array of metal9,31 and organic8 photoredox catalysts with different excited state redox potentials. The redox potential of metal catalysts can be altered with the selection of ligands around the metal centre, with more electron-poor ligands resulting in more oxidising catalysts and more electron-rich ligands giving better reducing catalysts. The photoexcited metal complex Ru(bpy)32+ (3) is a weak oxidant but a strong reductant. When the more electron-poor bipyrazine are used as ligands, the resulting catalyst 11 becomes a strong oxidant but weak reductant. A similar observation is found with iridium catalysts; the photoactivated catalyst Ir(ppy)312 is an excellent reductant but weak oxidant whereas 13 is a very strong oxidant but weak reductant (Fig. 3).
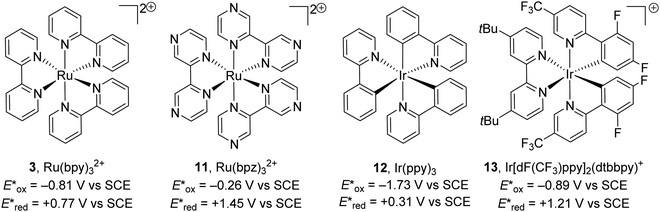 |
| | Fig. 3 Excited state redox potentials of metal photoredox catalysts. | |
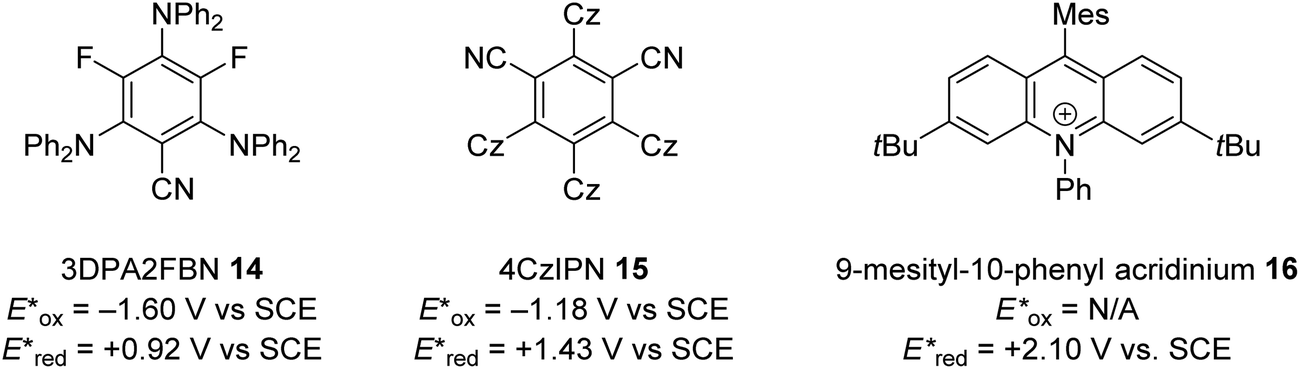
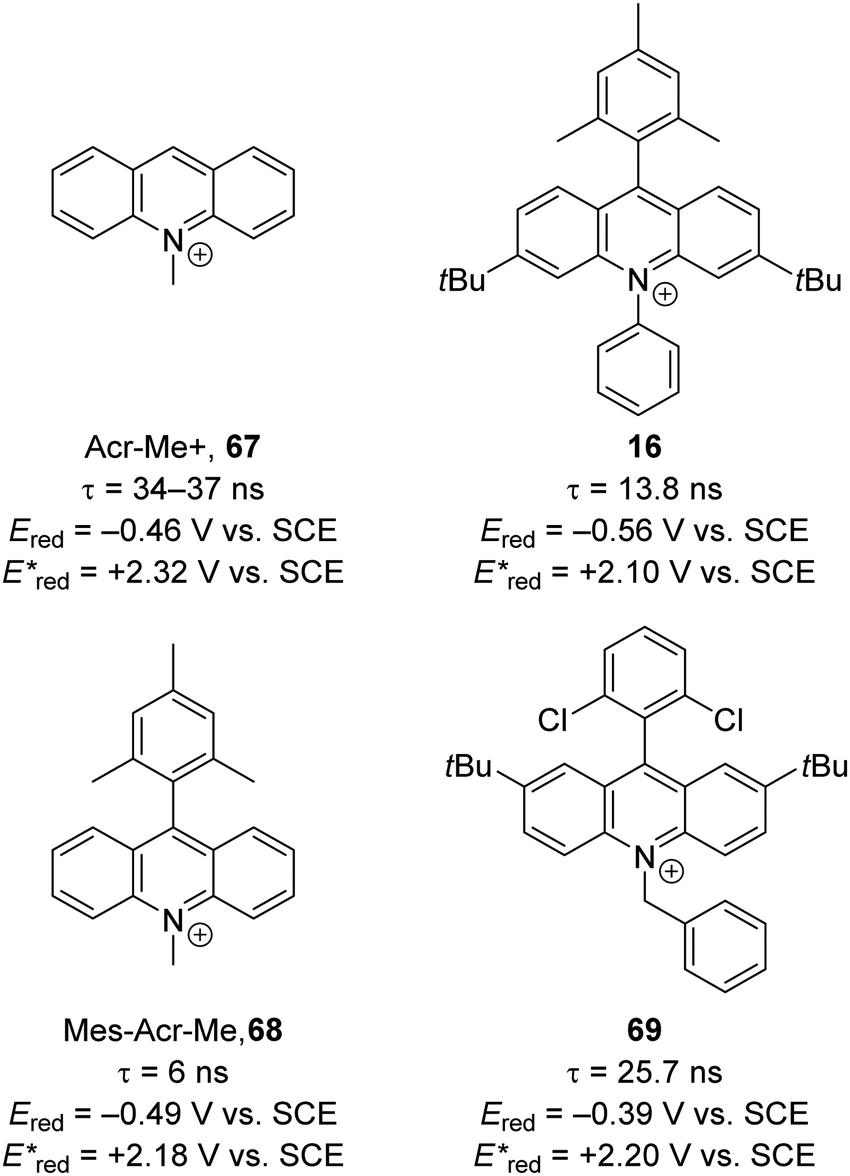
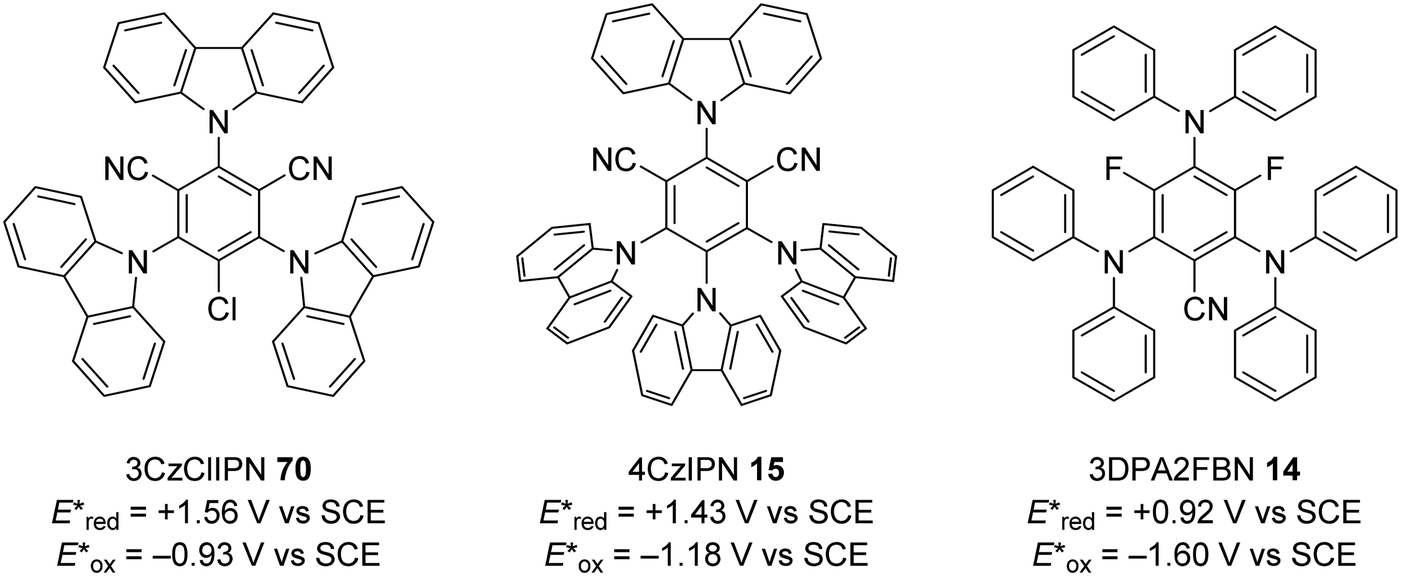
The excited state redox potentials of organic catalysts are determined by the electronic properties of the compound, with more electron-poor compounds being greater oxidants and more prone to reduction. The relationship between excited state reduction potentials and the electronic effects of substituents is demonstrated with the following compounds (Fig. 4). The most electron-rich compound 3PDA2FBN 14 has the lowest excited state reduction potential (+0.92 V vs. SCE). If the electron-rich diphenylamine groups of 14 are replaced with carbazole groups and an additional nitrile group is introduced, a more oxidising catalyst is obtained 4CzIPN 15 (+1.43 V vs. SCE).32,33 Due to the presence of a formal positive charge, acridinium salts (16) make extremely strong photooxidants (+2.10 V vs. SCE).32
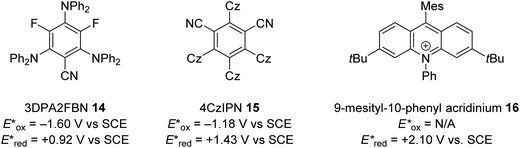 |
| | Fig. 4 Excited state redox potentials of non-metal photoredox catalysts. | |
Hydrogen atom transfer
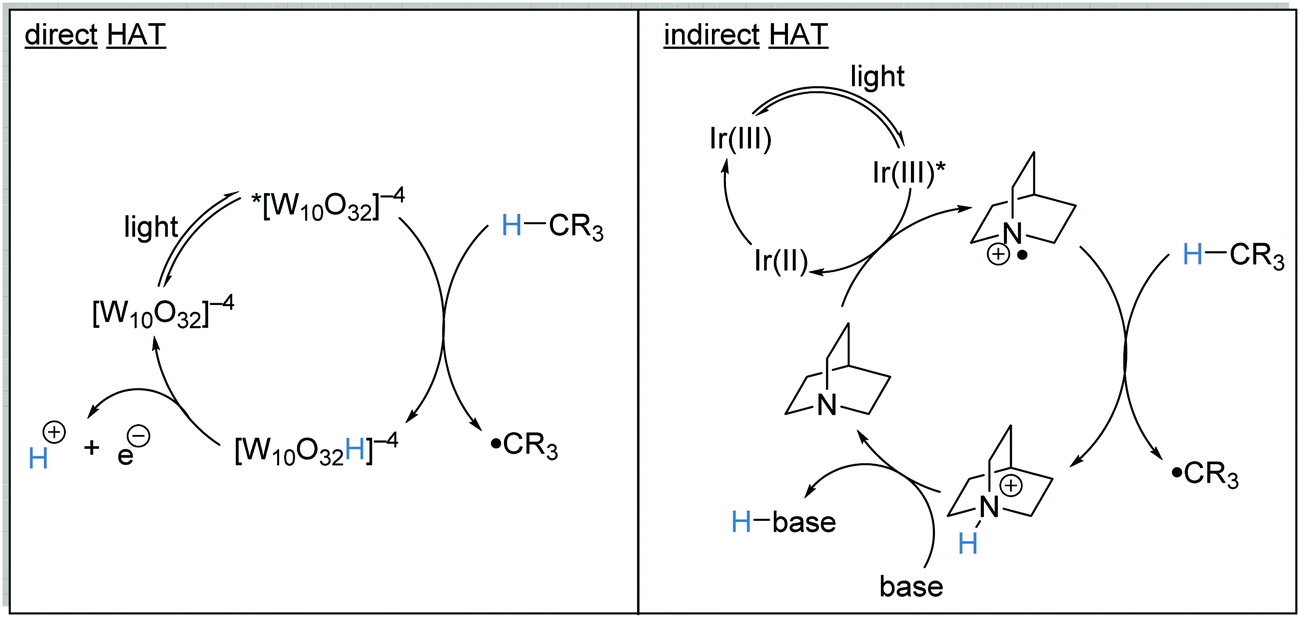
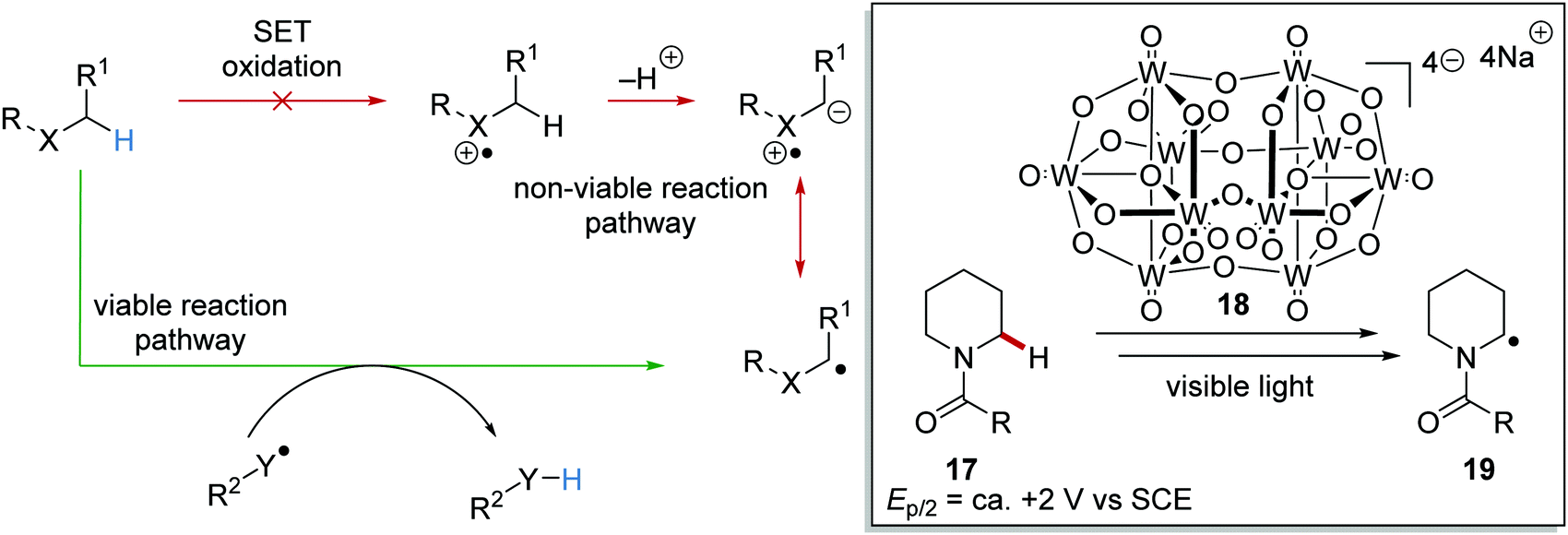
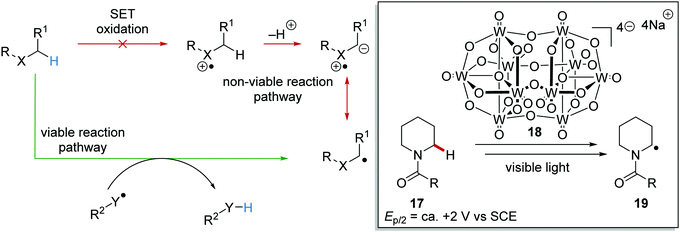
HAT is the transfer of an H-atom from one species to another. A wide range of HAT reagents (and catalysts) exists and they can be used in direct and indirect processes.34 A direct HAT process is where the excited photocatalyst can abstract a hydrogen atom from the substrate. Sodium decatungstate 18 (Schemes 4 and 5) is a prominent example of a direct HAT catalyst. Indirect processes involve the activation of a HAT catalyst with another catalyst or reagent; a topical example of this process is the combination of an iridium photocatalyst with quinuclidine (Scheme 4).
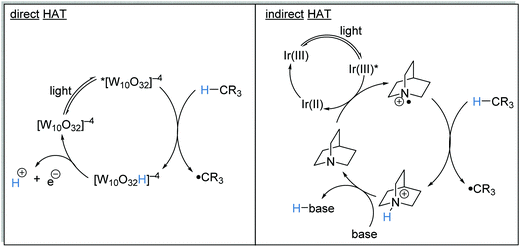 |
| | Scheme 4 Catalytic direct HAT vs. catalytic indirect HAT approaches. | |
 |
| | Scheme 5 HAT strategy vs. SET strategy. | |
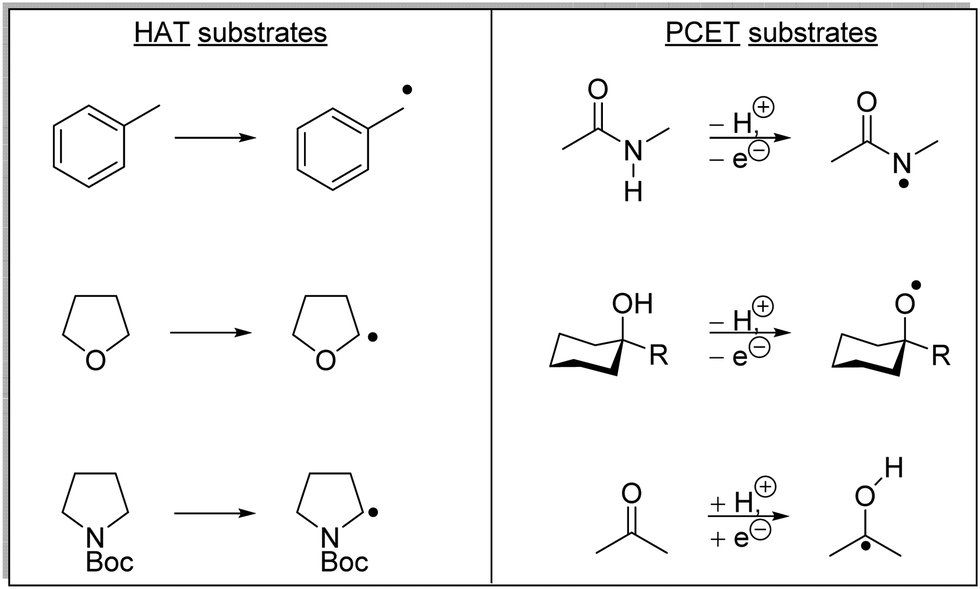
HAT has one distinct advantage over SET processes in radical coupling.34 Hard-to-oxidise functional groups (amides, ethers, alkanes) can be functionalised with a HAT approach, as HAT reagents have the advantage that the substrate does not need to be oxidised through loss of an electron for radical formation. Therefore, for hard-to-oxidise substrates, radical formation is achievable through an atom abstraction process for which a SET oxidation process is not feasible (Scheme 5). For example, N-Boc-piperidine 17, (R = OtBu) has an oxidation potential of +1.96 V vs. SCE so commercial photoredox catalysts such as Ru(bpy)3(PF6)2 (Ered* = +0.77 V vs. SCE) and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (Ered* = 1.21 V vs. SCE) are unable to oxidise this substrate.35 However, decatungstate HAT catalyst 18 was able to functionalise 17via α-amido hydrogen atom transfer after activation with visible light, and this gave radical 19.36 Hence, the use of HAT processes allows for radical formation on substrates that are unable to be easily oxidised with a photoredox catalyst.
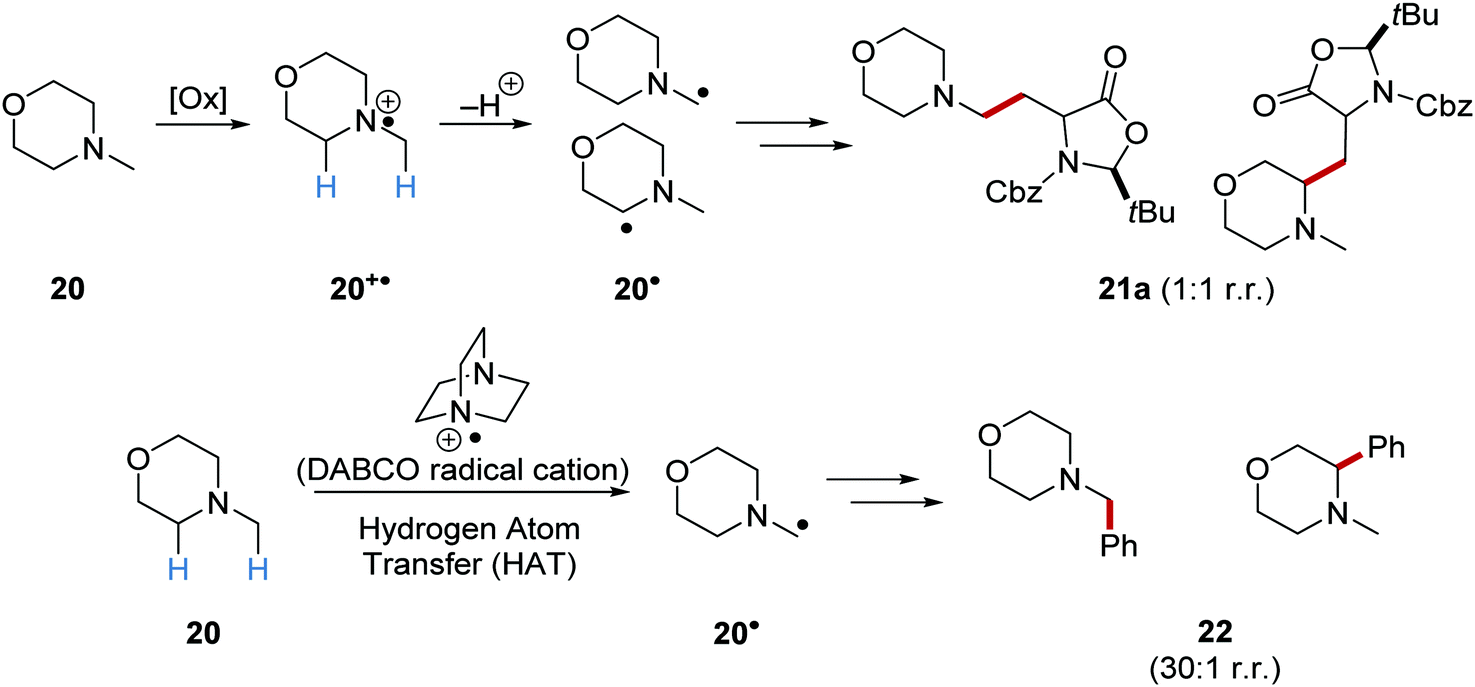
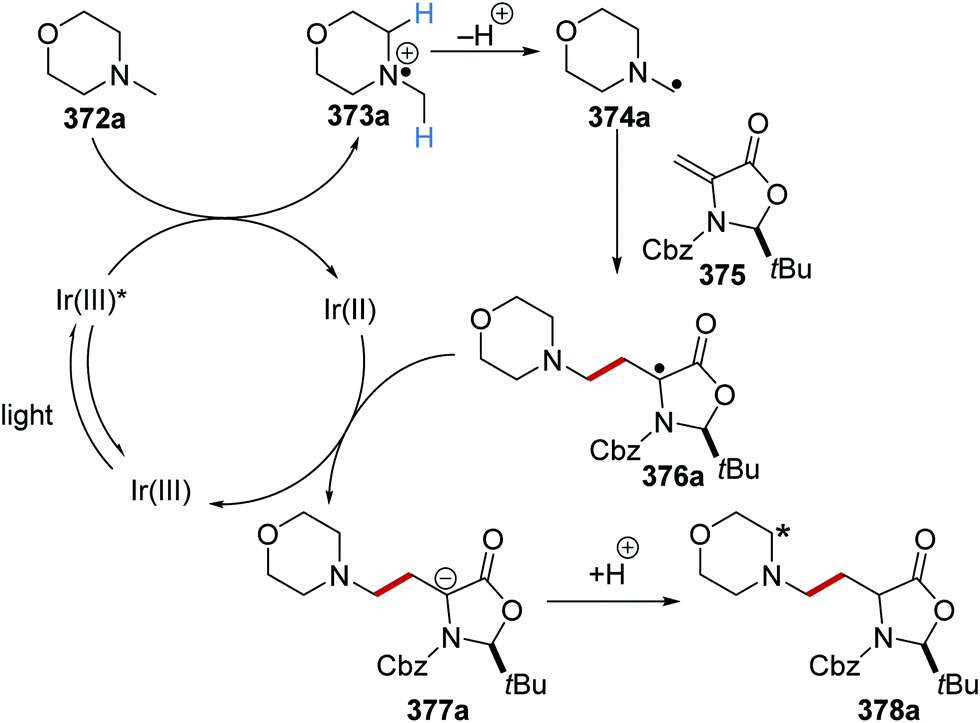
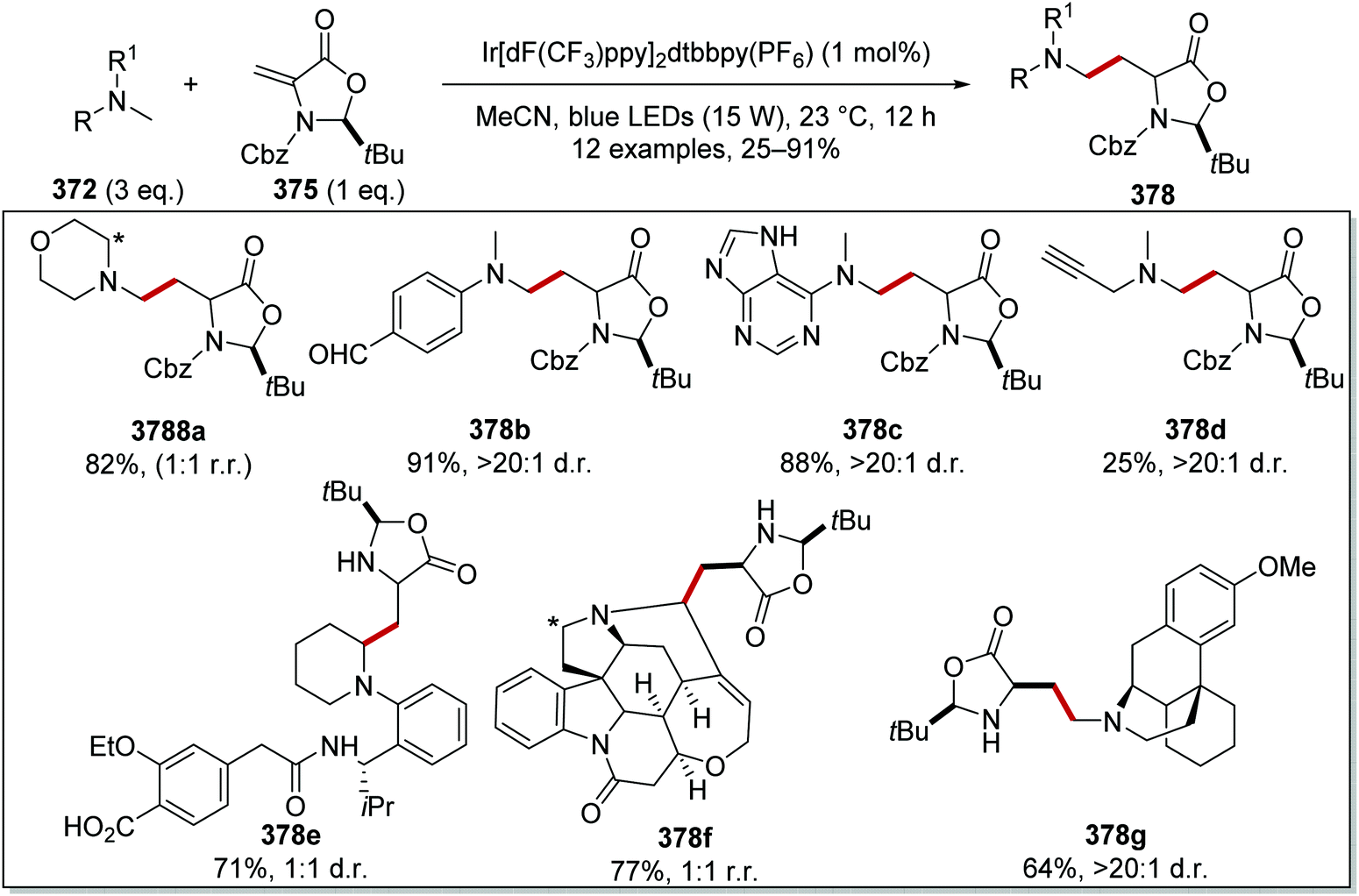
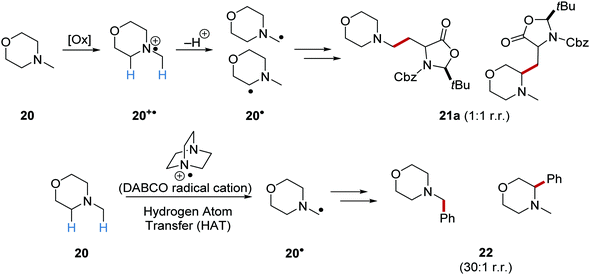
On occasion, HAT processes are more regioselective than SET processes in radical coupling reactions (Scheme 6). For example, it has been shown that amines, such as N-methylmorpholine 20, are oxidised via SET processes by an iridium photocatalyst and this generates amine radical cation 20+˙.37 Proton loss showed little or no selectivity in forming two regioisomeric carbon-centred radicals 20˙. Radical addition of 20˙ to an electron-poor alkene resulted in a 1:1 mixture of regioisomers of compound 21a. However, when a DABCO-based HAT reagent was used, due to steric factors, hydrogen abstraction took place from the N-CH3 position essentially exclusively and this gave a >30:1 mixture of regioisomers of 22.38
 |
| | Scheme 6 Regioselectivity of SET and HAT processes. | |
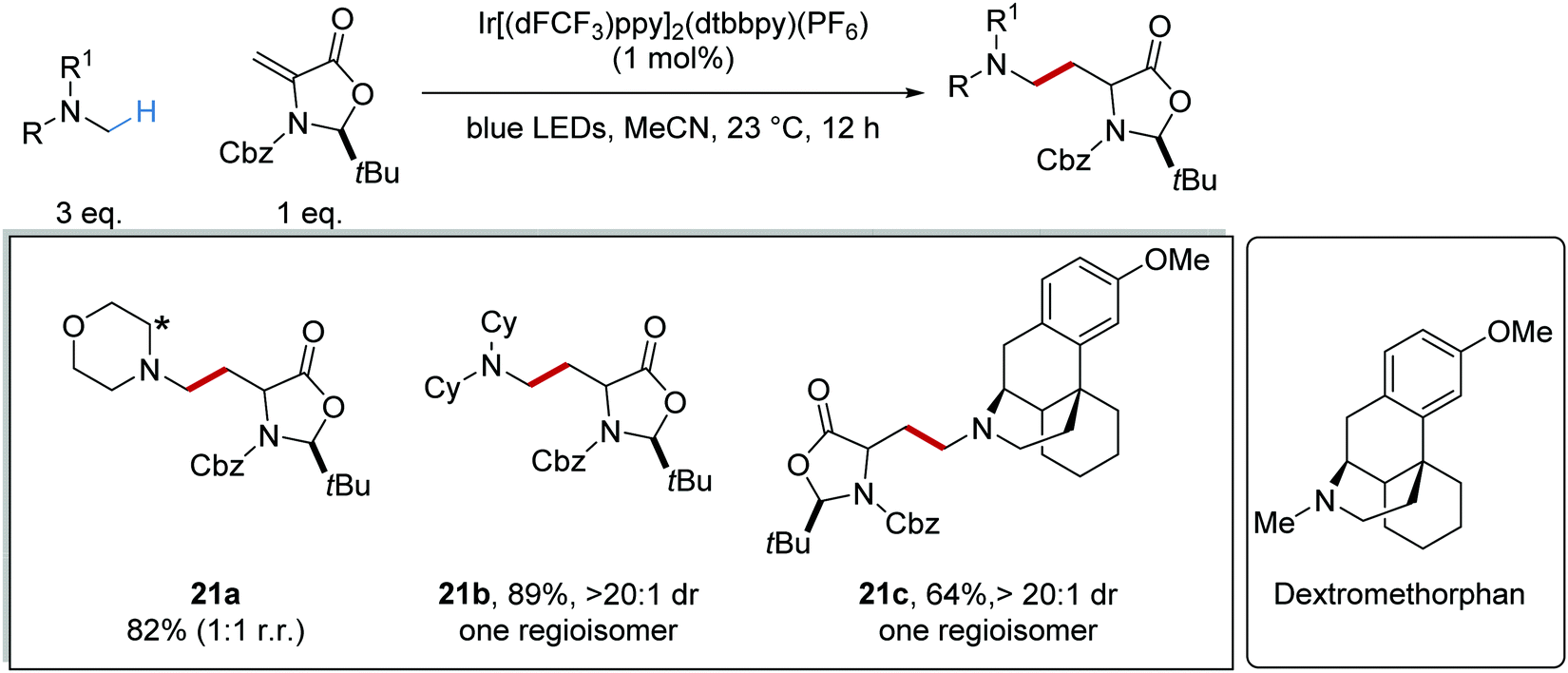
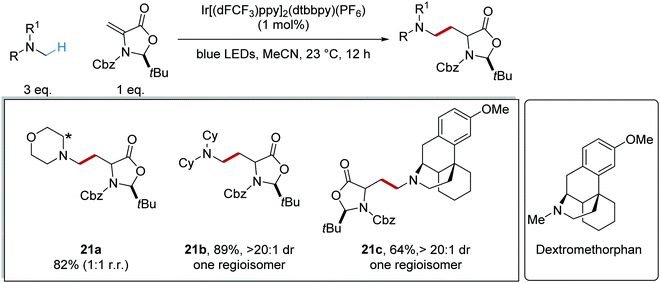
In some instances, substrate control results in regiocontrol, even with transformations that involve sequential oxidation and loss of a proton. This was demonstrated when dextromethorphan (inset, Scheme 7) and N-methyldicyclohexylamine were used as substrates; only single regioisomers of compounds 21b and 21c were produced, unlike for 21a.37
 |
| | Scheme 7 Substrate regiocontrol of SET processes. [Asterisk denotes alternative connectivity for another regioisomer.] | |
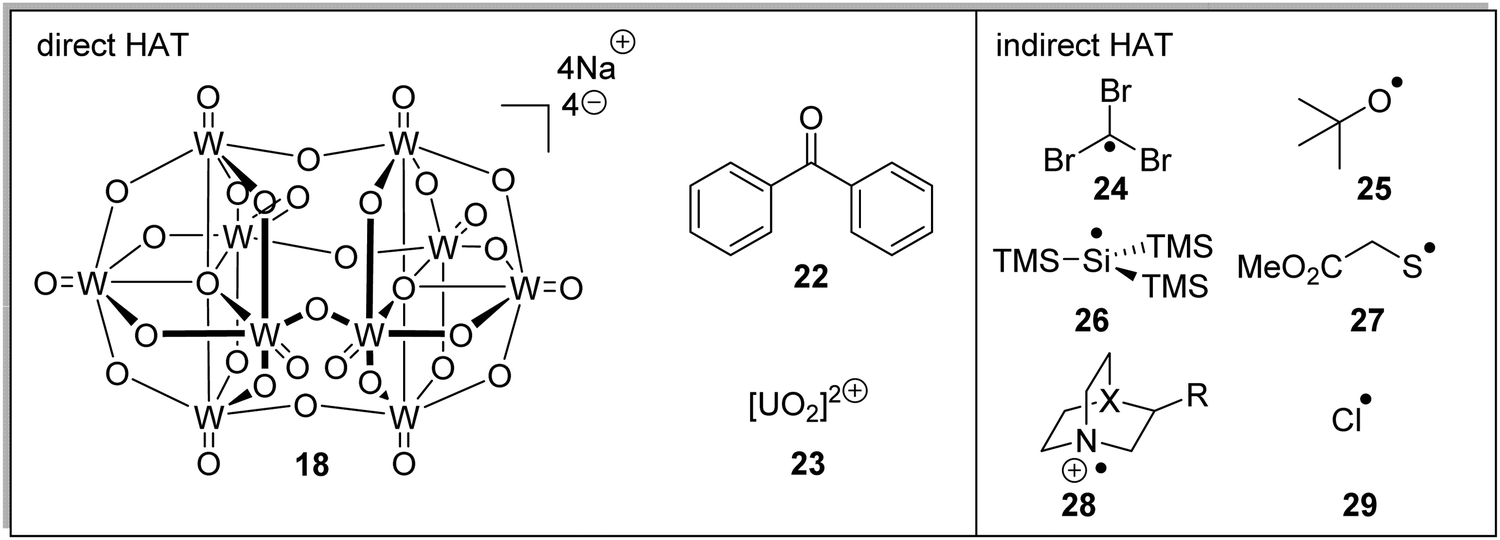
HAT reagents
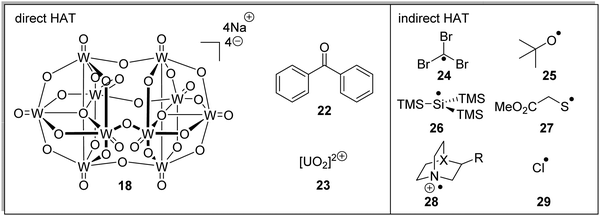
Both direct HAT and indirect HAT catalysts (and reagents) have been well-reviewed.34 Sodium decatungstate 18, benzophenone 22 and uranyl cation 23 are all direct HAT species as, upon their excitation, they can abstract a hydrogen atom from the substrate (Fig. 5). The compounds 24–29 are all indirect HAT compounds as they require an additional reagent or catalyst for their formation. As discussed below, the rate of hydrogen abstraction can be calculated with consideration of bond energies and polarities. Therefore, different hydrogen atoms can be targeted for abstraction, depending upon the nature of the HAT catalyst.
 |
| | Fig. 5 Direct and indirect HAT species. | |
Kinetics of HAhT
As mentioned above, the rate of hydrogen-atom abstraction can be predicted from the ground-state properties of the reactants and products.6 The rate constant (k1) for a reaction can be determined with the Arrhenius equation (eqn (1)) in terms of activation energy (Ea) and pre-exponential factor (A). Therefore, the rate of a reaction could be predicted if a value of Ea could be calculated.| | 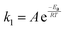 | (1) |
The Ea of a HAT process (eqn (2)) is determined with the following equation (eqn (3)).6 The term f is defined in eqn (4), where DAH, DBH and DH2 correspond to the bond dissociation energy (BDE) for AH, BH and H2 respectively. Data tables for BDE are abundant in the literature,39–43 but it has been suggested that free energies should be used instead.44 The term ΔχAB2 is the difference in electronegativity between the radicals A˙ and B˙. The S-factors (SA and SB) relate to structural parameters of the radicals A˙ and B˙ taking into account structural changes during the reaction. Small S factors are obtained if little structural changes occur during the reaction, for example if A˙ and B˙ both represent atoms. Large values are obtained if significant structural changes occur such as conversion of a tetrahedral structure to trigonal planar for the formation of the methyl radical from methane. To account for the non-linearity in stabilisation of the newly formed radical B˙ by conjugative delocalisation of the unpaired electron, the term d is included into the equation. The constants E0, α, β and γ are determined from multiple regression analysis of experimental data. This approach was successful in reproducing the activation energies for 65 reactions. Therefore, it can be concluded that bond energies, polarities, structural changes and radical stabilisation with delocalisation of the unpaired electron must all be considered in evaluating whether a HAT process is viable. However, most attention should be spent considering the strengths of the bonds involved in the process; as stated by Roberts “the activation energy for an endothermic reaction cannot be less than (ΔH° + RT), no matter how favourable are the polar factors!”45| | | Ea = E0f + αΔH°(1 − d) + βΔχAB2 + γ(SA + SB) | (3) |
| | 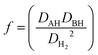 | (4) |
Bond energies of HAT catalysts and substrates
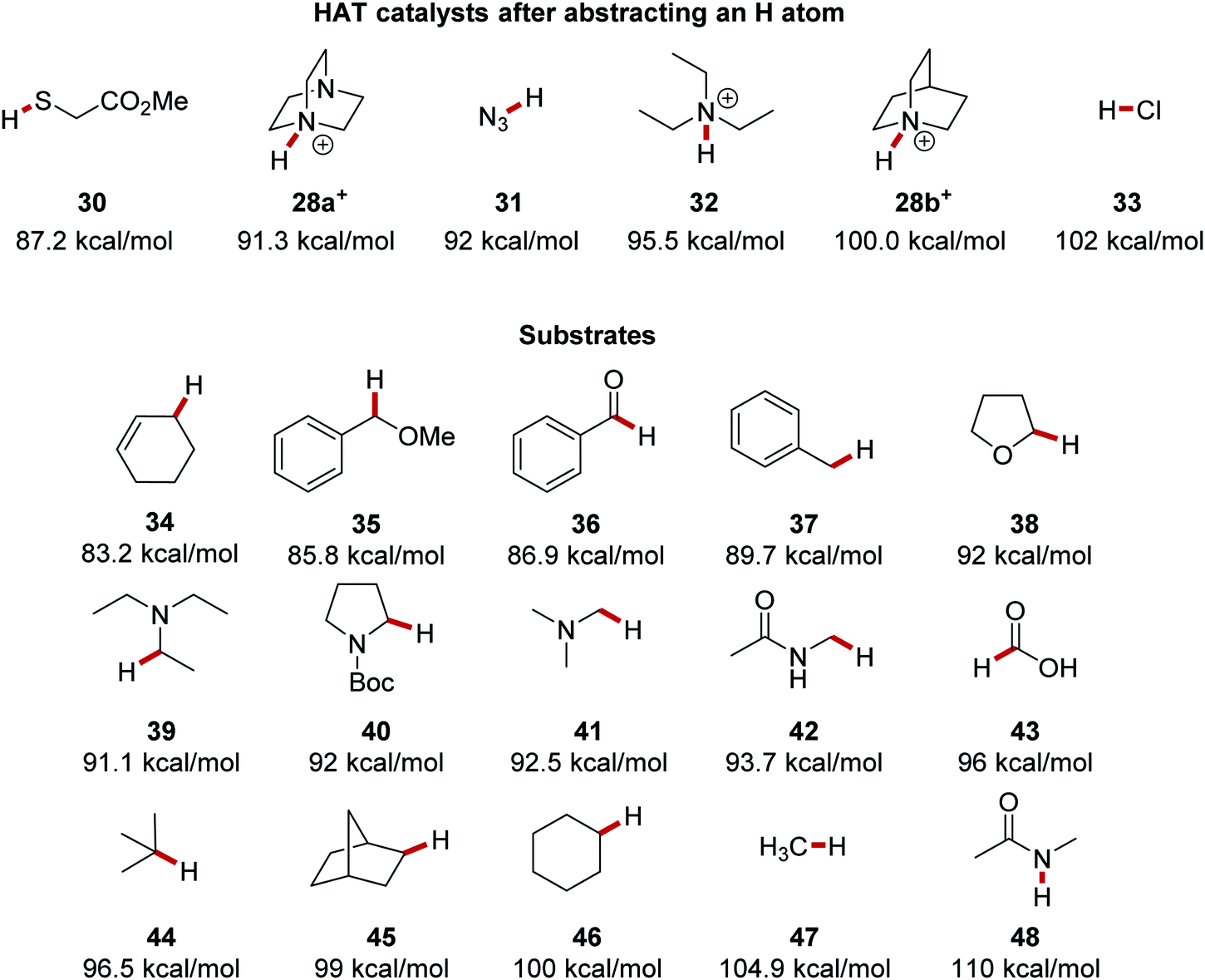
The resulting bond between the HAT catalyst and the sequestered hydrogen atom is a key driving force in propagating a HAT process. The bond strength between H atoms and HAT catalysts vary and the strength of this bond dictates which substrates a HAT catalyst can be used upon. For example, thiol 30 has an S–H bond energy of 87.2 kcal mol−1.46 Therefore, a thiyl radical is unsuitable for breaking strong C–H bonds but it could break weak C–H bonds of benzyl ethers, such as 35, which has a bond strength of 85.8 kcal mol−1 (Fig. 6). The chlorine atom forms a strong bond to a hydrogen atom with the formation of HCl 33 (BDE = 102 kcal mol−1) and thus it can abstract hydrogen atoms from stronger C–H bonds. A well-reported HAT catalyst, the quinuclidine radical cation 28b forms a strong N–H bond (BDE = 100 kcal mol−1) in HAT processes.43 Triethylamine radical cation forms a weaker bond to hydrogen in 32 than quinuclidine as cleavage of the N–H bond of 32 results in strain relief which is not possible due to the rigid structure of 28b. The additional nitrogen atom present in DABCO 28a stabilises the corresponding radical cation with a 1-spin-4-non-bonded-electron orbital interaction.47 Therefore, the N–H bond in 28a+ (91.3 kcal mol−1) is weaker than in 28b+ as homolytic N–H bond cleavage results in a more stable radical cation. Quinuclidine radical cation can functionalise α-amido C–H bonds (92 kcal mol−1) whereas its DABCO counterpart cannot due to its resulting weaker bond to the hydrogen atom.38,48 The BDE of C–H bonds are well reported in the literature and thus by comparing bond energies, feasible HAT processes can be designed. Methane has the strongest C–H bond of alkanes (BDE = 104.9 kcal mol−1) as there is no radical stabilisation for the methyl radical after H-atom abstraction. Carbon atoms with greater substitution have weaker C–H bonds, such as cyclohexane 46 (100 kcal mol−1) and norbornane 45 (99 kcal mol−1 for C–H in the methylene group shown) due to hyperconjugation effects.36 Due to the high stability of tertiary radicals, isobutane has a BDE of 96.5 kcal mol−1 for its methine C–H bond.40
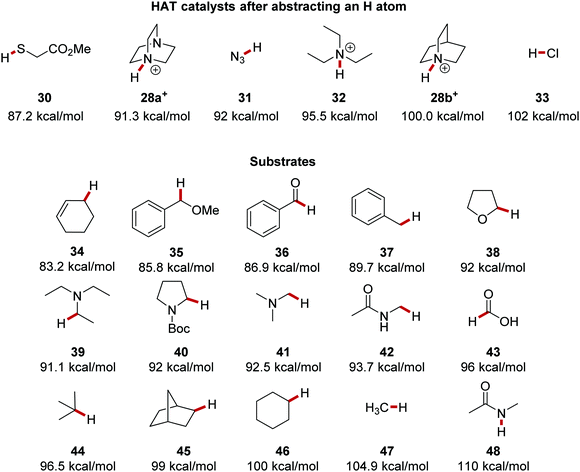 |
| | Fig. 6 BDE of N+–H bonds and BDE of selected organic substrates. | |
The C–H bonds adjacent to heteroatoms bearing one or more electron pairs are weak; for example, triethylamine has an α-amino C–H BDE of 91.1 kcal mol−1.39 The radical formed from triethylamine is stabilised with the sharing of π electrons between the C and N atoms. Benzylic and other α-heteroatom C–H bonds are suitable substrates for HAT due to their weak bonds.36,39,49,50 Other appropriate HAT substrates are aldehydes,40,51 benzylic ethers,52 and allylic C–H bonds.53 However, unlike C–H bonds, heteroatom–H bonds can be much stronger, and N-methyl acetamide (48) has a BDFE of 110 kcal mol−1 and is unable to be activated with HAT strategies.54
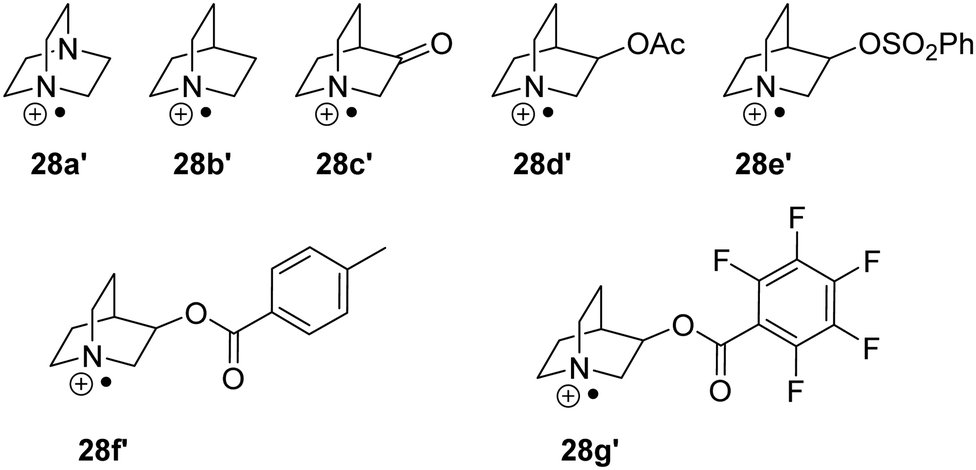
As mentioned above, polarity effects also have a significant effect on the feasibility of HAT reactions and thus various analogues of quinuclidine 28a′–28g′ have been used as HAT catalysts (Fig. 7). In one instance, it was demonstrated that the C–H abstraction of adamantane was only achievable with sulfonate HAT catalyst 28e′ due to its strong polarity.55
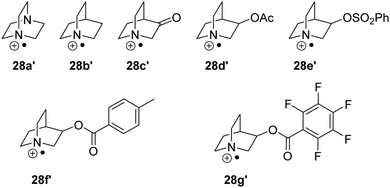 |
| | Fig. 7 Nitrogen-based HAT catalysts. | |
PCET
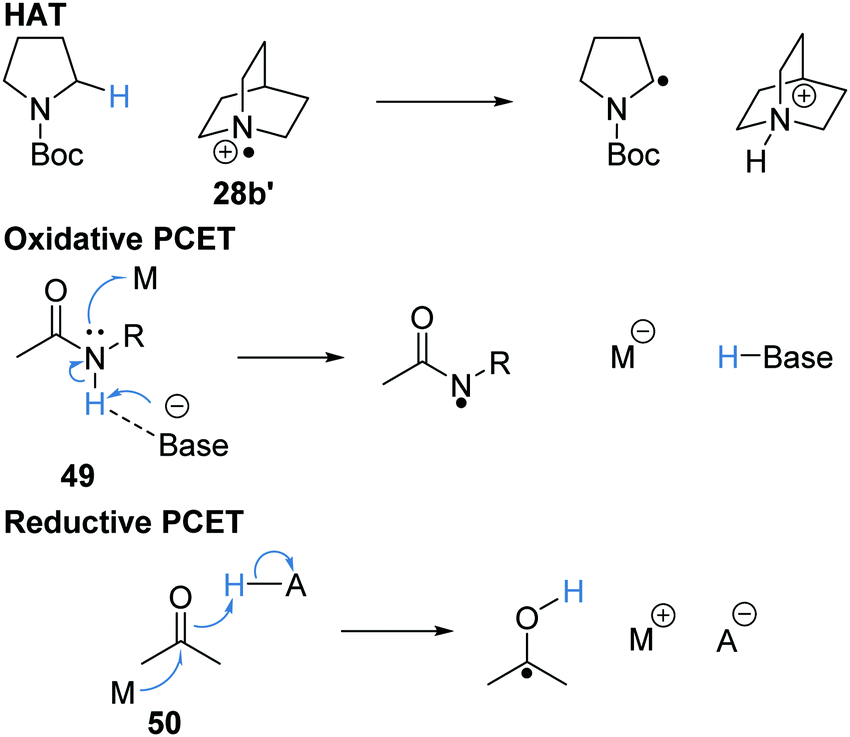
Proton-coupled electron transfer (PCET) is closely related to HAT and there have been many conflicting definitions regarding HAT and PCET.54–57 HAT is best defined as the concerted transfer of a proton and an electron from one species to another, while PCET is a concerted process, the proton and electron are transferred either to separate acceptors (oxidative PCET) or received from separate donors (reductive PCET). For example, for HAT, an electron and a proton from the α-amino C–H bond is transferred to the quinuclidine radical cation 28b′ and this gives the α-amido radical shown in Scheme 8. The oxidative PCET of amide 49 involves the simultaneous transfer of the N–H proton to a base and transfer of an electron to an electron acceptor, M. Reductive PCET of ketone 50 necessitates donation of a proton (from a suitable acid) and an electron from a donor M.
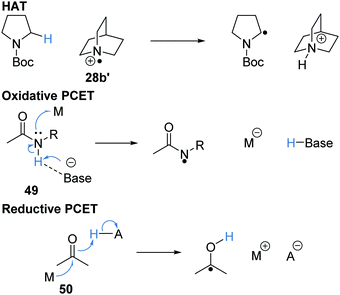 |
| | Scheme 8 HAT and PCET processes. | |
These are complementary transformations with PCET being able to abstract hydrogen atoms, typically from X–H bonds (where X = O and N), where HAT processes are not feasible due to the high bond energies. For example, the N–H bond present in N-methyl acetamide 48 has a bond dissociation free energy (BDFE) of 110 kcal mol−1.54 Common HAT catalysts such as quinuclidine radical cation and carbon-centred radicals resulted in the formation of N–H and C–H bonds that have BDE 98–100 kcal mol−1. Therefore, for most HAT catalysts the removal of a hydrogen atom from the amide functional group is thermodynamically unfavourable. However, it is routinely observed that the PCET of amides is feasible with the use of a weak Brønsted base and a photoredox catalyst. PCET has also been demonstrated upon ketones and alcohols (Scheme 9).54,58
 |
| | Scheme 9 HAT processes and PCET processes. | |
Energy transfer
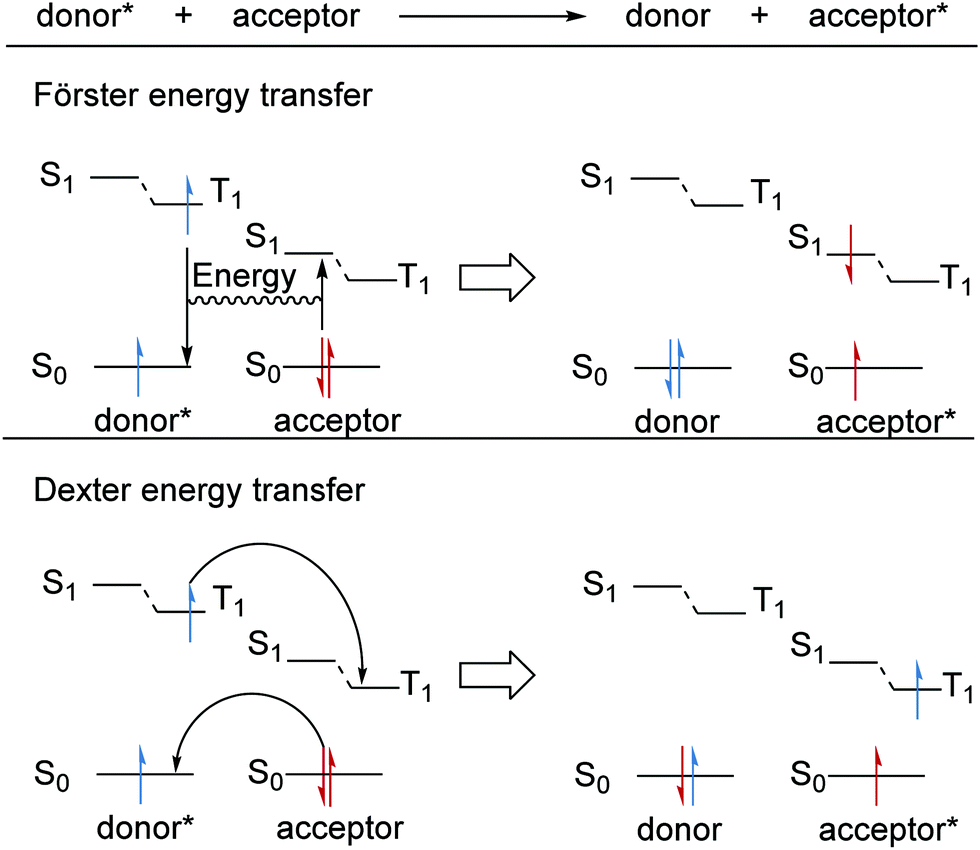
Alongside PET and HAT, energy transfer is a complementary method that allows reactions that are not feasible in the ground-state to take place in an excited state.59,60 For these transformations to take place, the substrate must access its excited triplet state. However, direct methods to access T1 commonly require high-energy light which may result in potential degradation of the organic substrate and the required ISC crossing can be inefficient.60 Therefore, photocatalysts (sensitisers) have been employed as energy-transfer reagents so that organic substrates can access their T1 state without the need for high-energy UV light. There are two energy transfer mechanisms, Förster resonance energy transfer (FRET) and Dexter energy transfer (Scheme 10).61 In both cases, an excited donor molecule transfers its energy to an acceptor molecule and this becomes excited with the quenching of the original donor compound. For FRET, coulombic interactions between the electronic transitions gives rise to the transfer of energy.61 After a FRET, the substrate molecule will be in its singlet excited state and will still require an ISC processes to populate its triplet state. On the other hand, Dexter energy transfer results from the exchange of electrons; for this, wavefunction overlap is required and thus Dexter energy transfer can only take place over short distances (<10 Å). Here, triplet to triplet (TT) energy transfer operates as a Dexter process with two simultaneous electron transfers.61
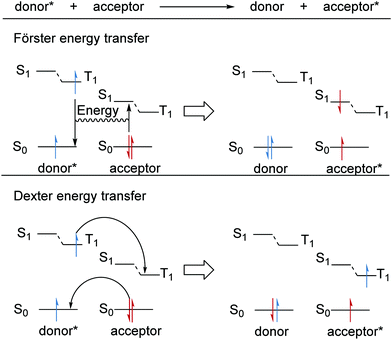 |
| | Scheme 10 Energy transfer processes. | |
Energy-transfer catalysts
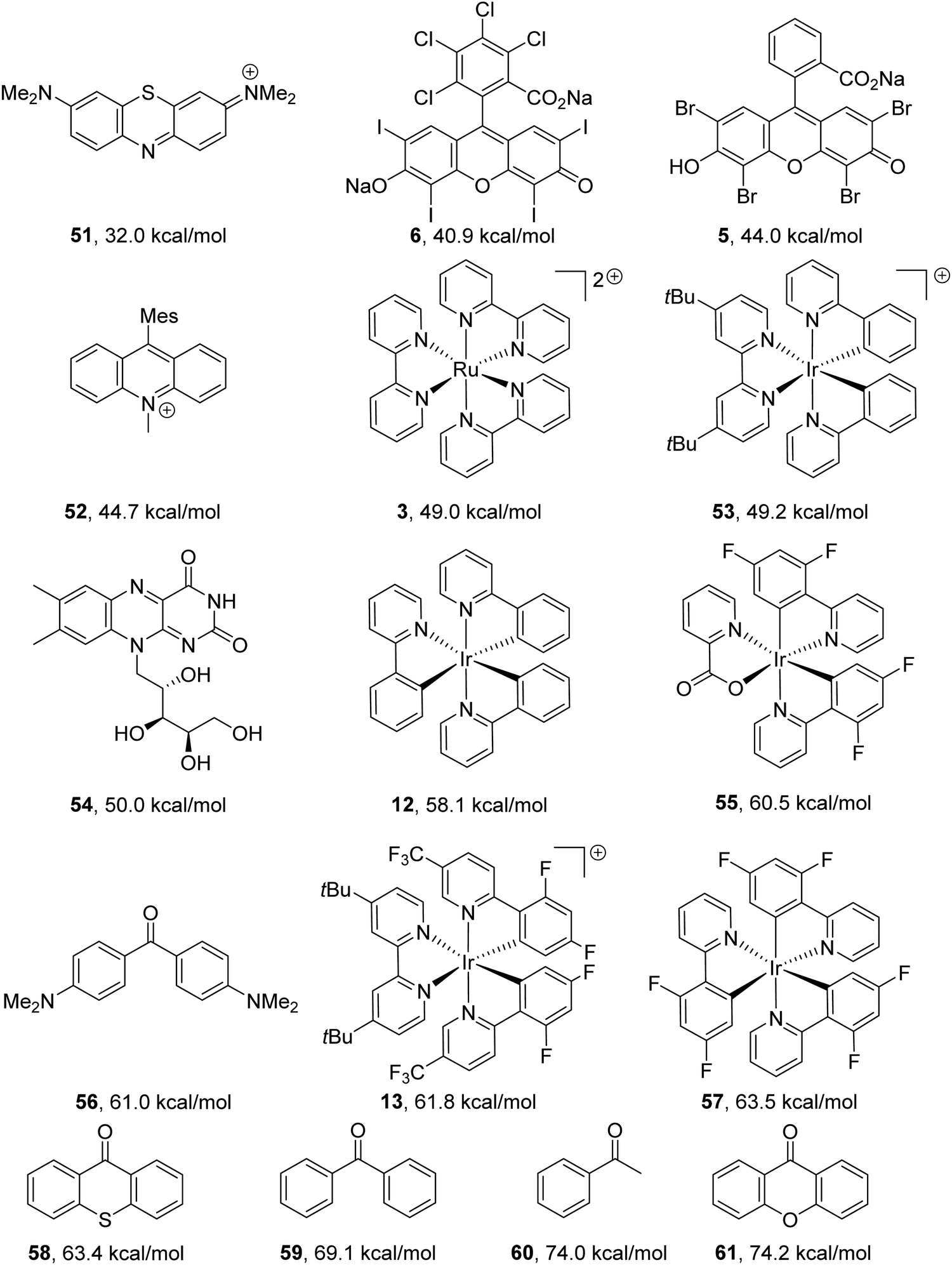
Previously mentioned photoredox catalysts can also be used as energy-transfer catalysts. However, for energy-transfer, the triplet energy of the donor must be greater than the triplet energy of the acceptor.7 Triplet energy values of catalysts can be found in the literature (Fig. 8).
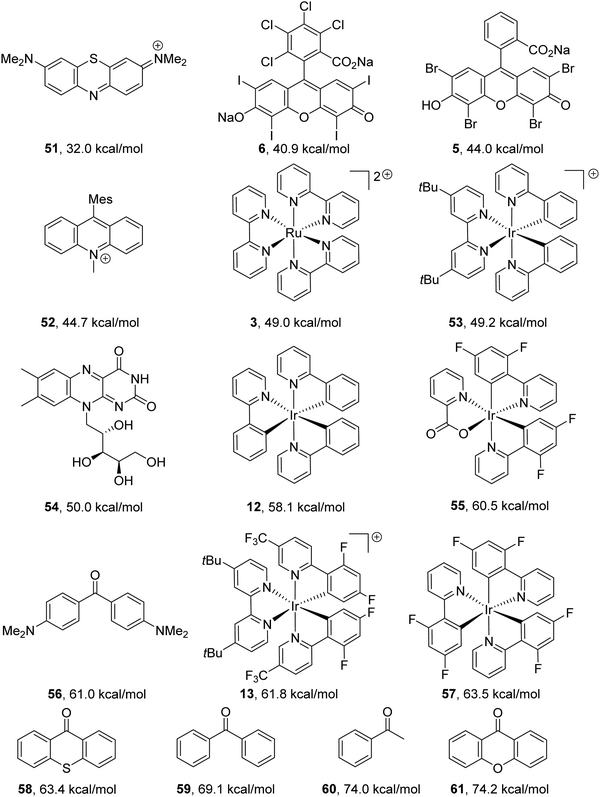 |
| | Fig. 8 Triplet energy values taken from the following sources.7,8,60,62,63 | |
Bicatalytic processes
Redox mediation.
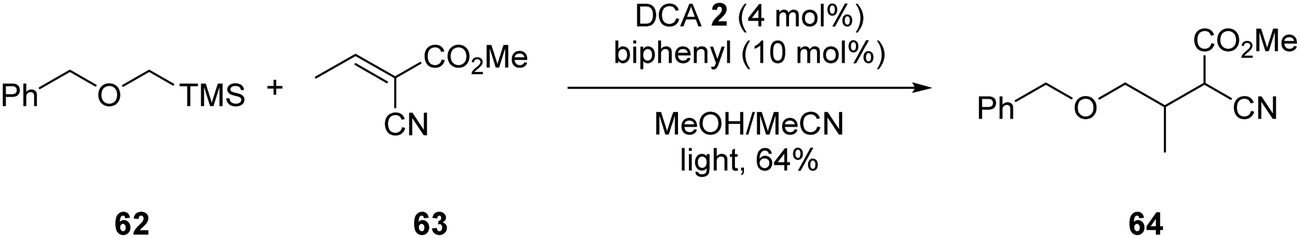
In some cases, a catalytic amount of redox mediator is required for PET processes which do not have favourable kinetics or suffer from rapid back-electron transfer (BET).5 An example of redox mediation was the coupling of α-silyl ether 62 with electron-poor alkene 63 with catalytic quantities of DCA 2 (Scheme 11). Successful formation of ester 64 required the addition of biphenyl (10 mol%) as a redox mediator to decrease BET, which has been reported as an issue with DCA.64 (See later for further discussion.)
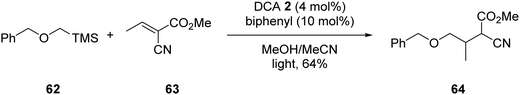 |
| | Scheme 11 DCA and BP mediated coupling of α-silyl ether 3 with electron-poor alkene 63. | |
Auxiliary metal catalysis.
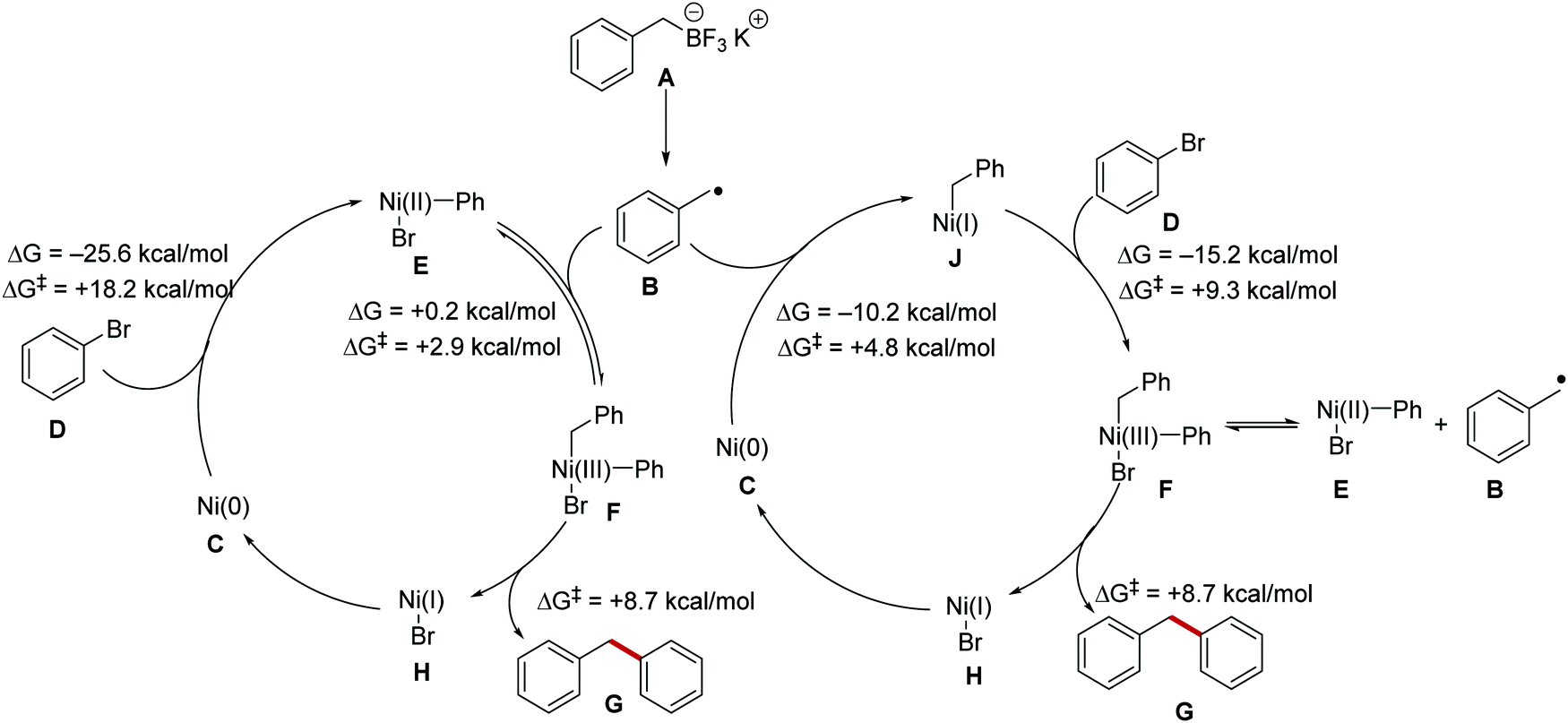
The addition of a transition metal catalyst has vastly increased the scope of functionalisation that was achieved with PET, HAT, or energy transfer reactions. Nickel, in particular, has found extensive use in radical coupling chemistry and computational investigations into the mechanistic pathways of these reactions have been carried out.65 Two mechanisms have been proposed for radical cross-coupling chemistry with a nickel catalyst (Scheme 12). It was thought that a radical could either add to a Ni(0) species or a Ni(II) complex and this was investigated computationally with the nickel cross-coupling reaction of benzylic radical B (derived from boronate salt A) with bromobenzene D and this found that both mechanisms were feasible. Radical addition of B to Ni(II) complex E was only slightly endergonic (ΔG = +0.2 kcal mol−1) and had a favourable transition state energy (ΔG‡ = +2.9 kcal mol−1). The addition of radical B to Ni(0) complex C was thermodynamically favoured (ΔG = −10.2 kcal mol−1) and had a low transition state energy (ΔG‡ = +4.8 kcal mol−1). The highest energy barrier found for both mechanisms was the oxidative addition of aryl bromide D to Ni(0) complex C forming Ni(II) complex E, this had a transition state energy of +18.2 kcal mol−1 but was exergonic by −25.6 kcal mol−1. Therefore, it can be concluded that both reaction pathways could be operative for radical nickel cross-coupling reactions. The transition state energy for reductive elimination from common complex F was +8.7 kcal mol−1, meaning that radical dissociation happens more rapidly than reductive elimination leading to a reversible process between complexes E and F.
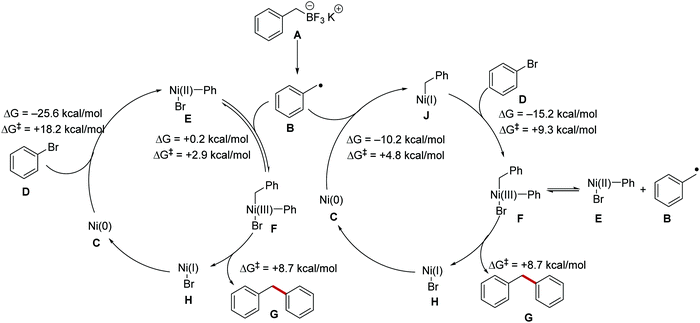 |
| | Scheme 12 Processing of radicals by nickel complexes. | |
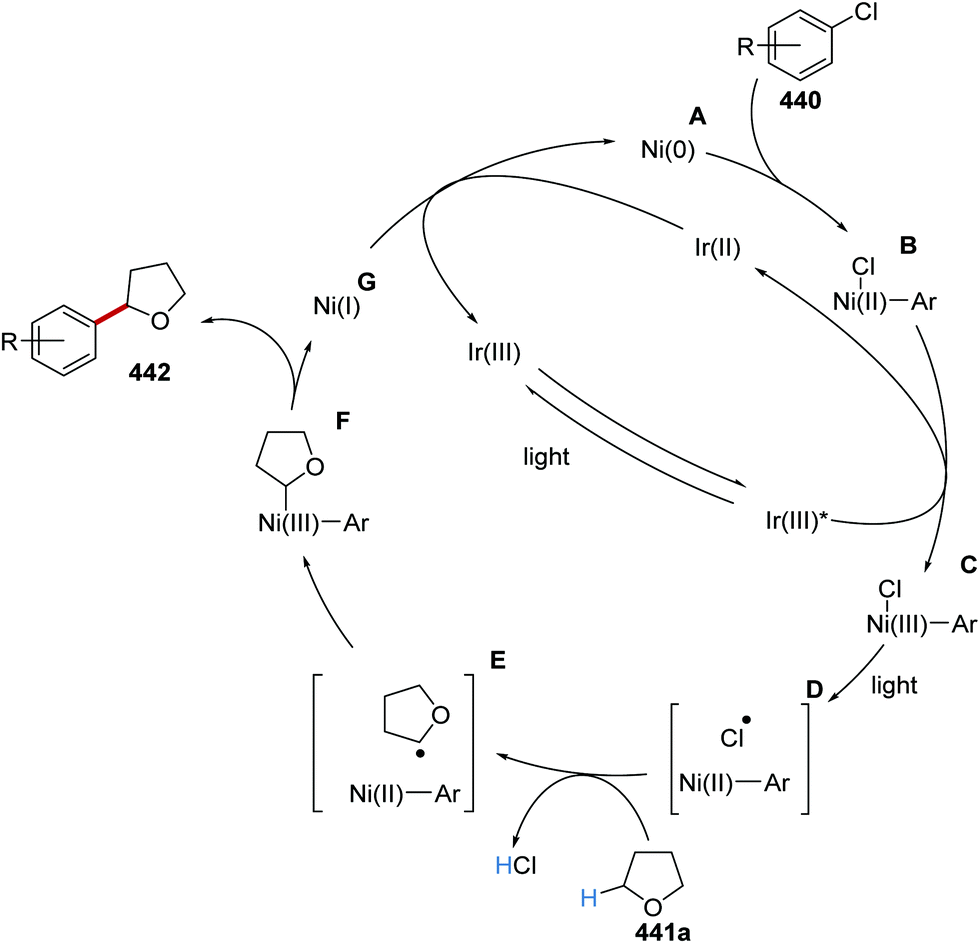
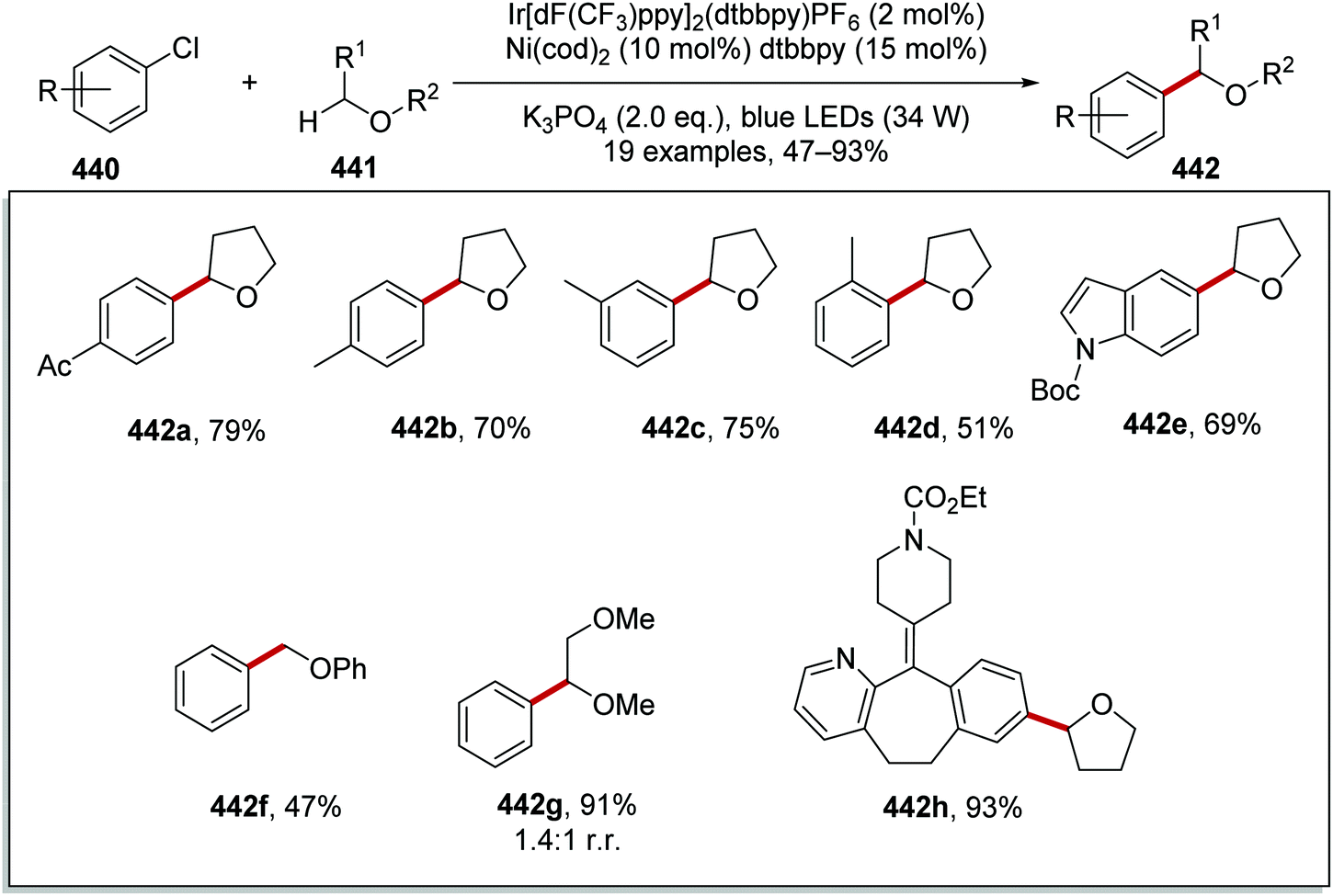
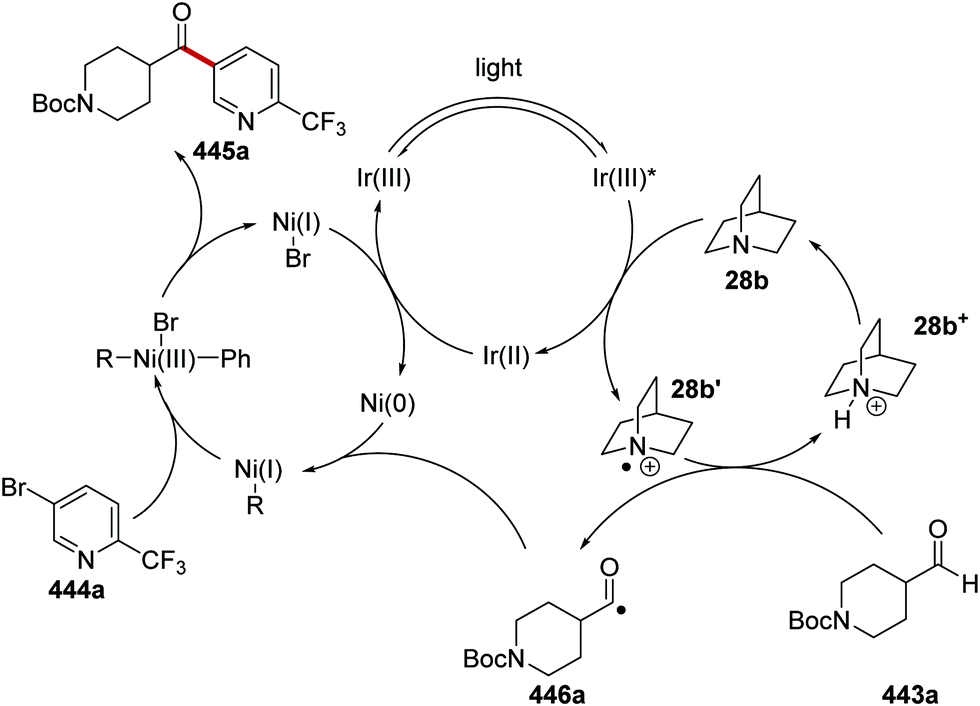
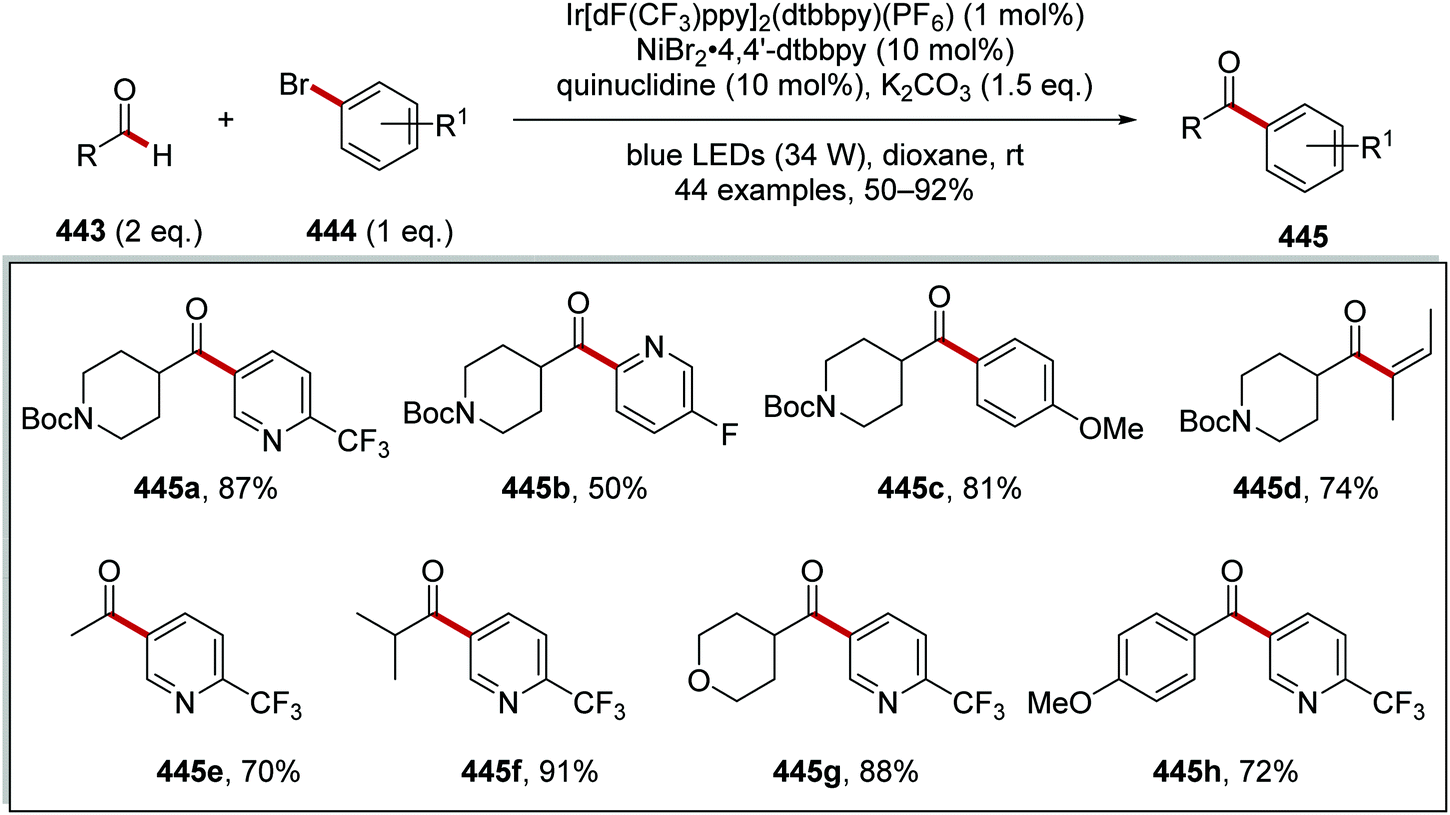
The intricacies of the role of nickel complexes in coupling reactions continue to be studied. In 2020, the MacMillan team studied66 the mechanism of C–N bond formation in the presence of iridium(III) photoredox catalysts and DABCO. DABCO reduced the photoactivated Ir(III) to Ir(II), and this in turn reduced Ni(II) to Ni(I). Ni(I) and Ni(III) then operated a catalytic cycle transforming aryl halide to arylamine.
Examples of catalysts and reagents
Metal-based photoredox agents.
It has been demonstrated that ruthenium and iridium complexes make effective photoredox and energy transfer catalysts.7,67 The inclusion of heavy metal atoms results in efficient ISC and long excited state lifetimes. Furthermore, excited state redox potentials and triplet energies can be finely tuned with the use of the right ligands, making them very attractive catalysts. However, there are issues regarding the sustainability of these metal catalysts.68
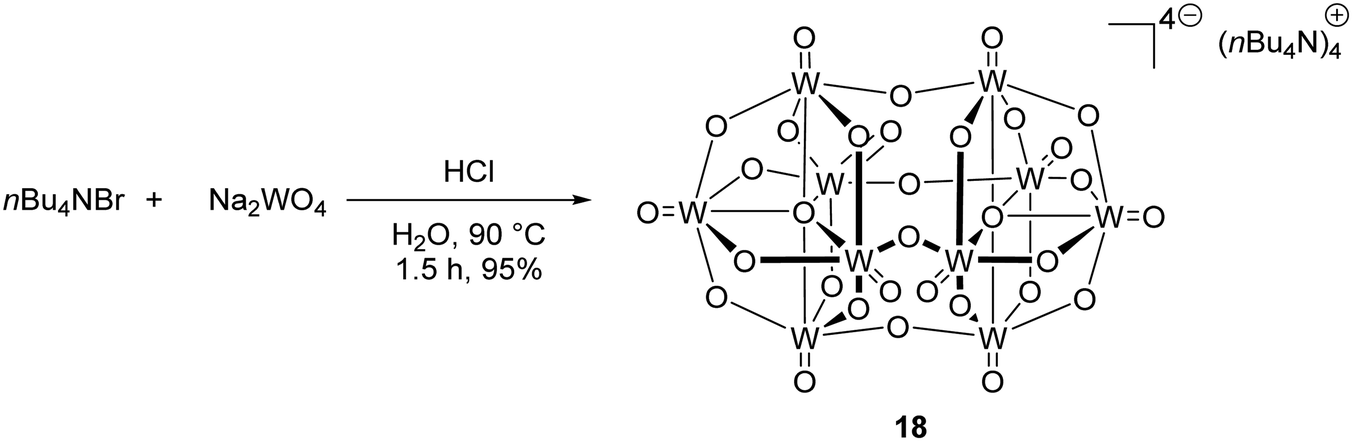
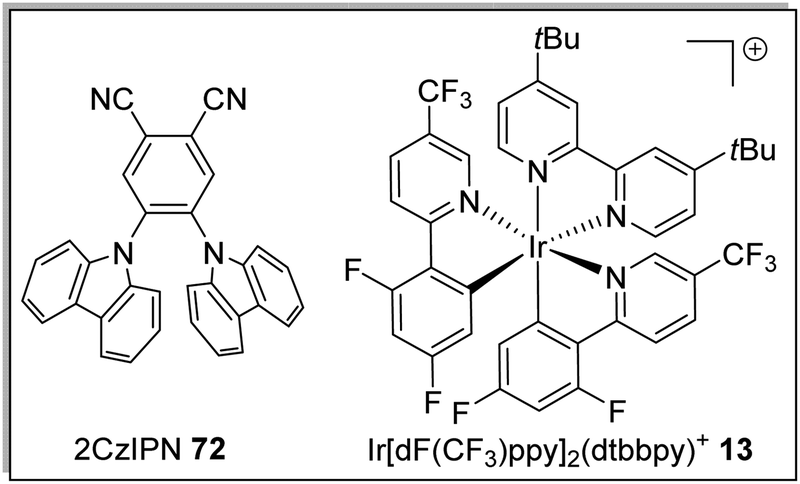
In the excited state, decatungstate salts (sodium and tetrabutylammonium salts have been reported)69,71 make highly reactive direct HAT catalysts, which have been shown to remove hydrogen atoms from the strong C–H bonds present in cyclohexane (BDE up to 100 kcal mol−1).36,70,71 Decatungstate salts are also strong PET catalysts (Ered* = +2.44 V vs. SCE) and have an excited triplet state lifetime of 51.5 ± 5 ns.72 Tetrabutylammonium decatungstate (TBADT, 18) can be prepared in one step from tetrabutylammonium bromide and sodium tungstate (Scheme 13).73 Alongside decatungstate catalyst, a manganese perchlorophthalocyanine catalyst has been used for C–H functionalisation.74 These compounds will not be discussed in detail in this review.
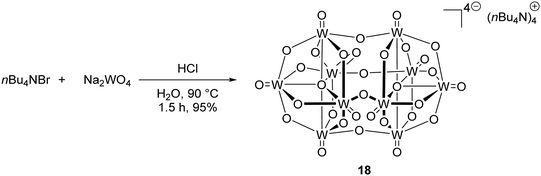 |
| | Scheme 13 Synthesis of tetrabutylammonium decatungstate (18) | |
Non-metal based photoredox agents.
Due to sustainability issues with some transition-metal catalysts, non-metal-based catalysts can be used instead (Fig. 9), including DCA 2, xanthene dyes (such as Eosin Y, 5), cyanobenzene-derived photocatalysts (such as 4CzIPN, 15), acridinium dyes 16, triarylaminium radical cations 65 and persulfate salts 66. An important advantage for non-metal based catalysts is the cost difference between TM and non-TM catalysts. In 2016, Ru(bpy)3Cl2 was $57.3 per g from Sigma-Aldrich, Ir(ppy)3 cost $1078 per g purchased from Sigma-Aldrich, Ir[dF(CF3)ppy]2(dtbbpy)PF6 cost $117.56 per g to synthesise, whereas non-TM were much less, for example, 4CzIPN cost $6.01 per g to synthesise.75 Another popular organic catalyst, 3,6-di-tert-butyl-9-mesityl-10-phenylacridin-10-ium tetrafluoroborate 16 (Fig. 11) can be purchased for $871 per g but can be easily synthesised in three steps from 3-tert-butylbromobenzene and 3-tert-butylphenol.32
 |
| | Fig. 9 Examples of sustainable radical promoters. | |
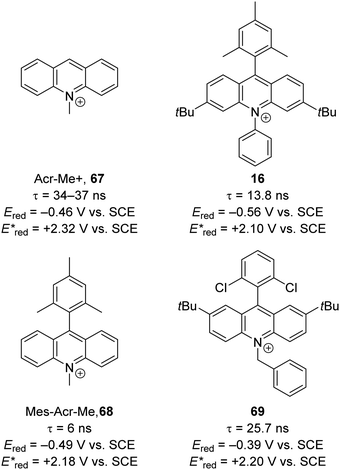 |
| | Fig. 10 Examples of acridinium catalysts. | |
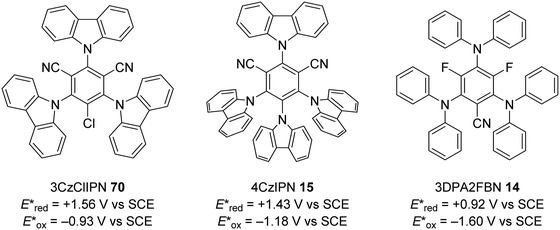 |
| | Fig. 11 Cyanobenzene derived photocatalysts and photoredox values. | |
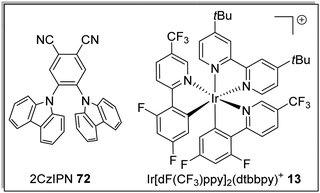
Table 1 Maximum solubility of catalysts 72 and 13 in various organic solvents (molar)
|
|
| Solvent |
2CzIPN 72 (M) |
13 (M) |
| Toluene |
1.3 × 10−2 |
7.0 × 10−5 |
| 1,4-Dioxane |
1.6 × 10−2 |
2.2 × 10−4 |
| Dichloromethane |
8.3 × 10−2 |
5.6 × 10−3 |
| Methyl tert-butyl ether |
9.2 × 10−4 |
7.0 × 10−5 |
| Dimethyl sulfoxide |
1.6 × 10−2 |
1.6 × 10−1 |
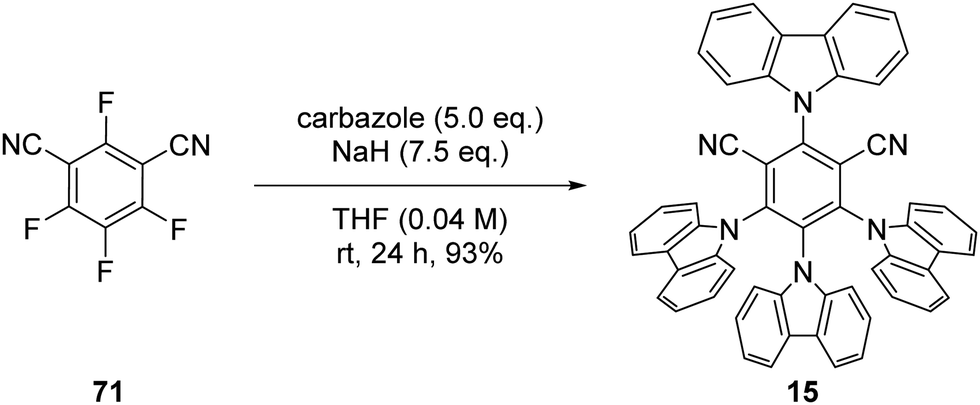
The photocatalyst 4CzIPN (15) is accessed in one step from tetrafluoroisophthalonitrile (71) and carbazole with sodium hydride in 93% yield, rapidly and economically without column chromatography (Scheme 14).78
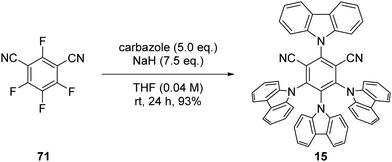 |
| | Scheme 14 Synthesis of 4CzIPN (15). | |
A key advantage of these cyanobenzene-derived photocatalysts over Ru and Ir photocatalysts is their high solubility in non-polar solvents.79 For example, the catalyst 2CzIPN (72) has a much greater solubility in non-polar solvents than Ir[dF(CF3)ppy2](dtbbpy)PF6, 13. This was demonstrated with 72 having a maximum solubility in toluene of 1.3 × 10−2 M while 13 had a maximum solubility of 7.0 × 10−5 M. It has been recently highlighted that the majority of iridium and ruthenium photocatalysts have low solubility in non-polar organic solvents.80
Xanthene dyes.
The xanthene dyes fluorescein (4), Eosin Y (5), rose Bengal (6) and erythrosine (73) have remarkably similar photoredox values but very different photophysical properties due to the presence or absence of heavy halogen atoms (Fig. 12; for discussion, see ‘Heavy atom effect’).8a
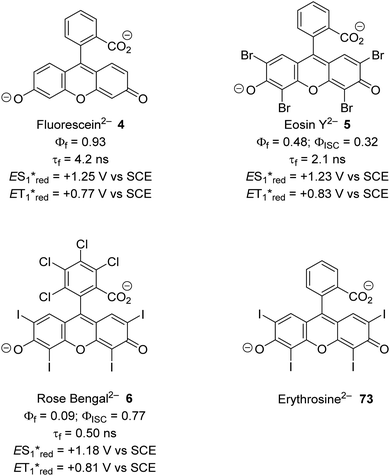 |
| | Fig. 12 Some photophysical properties of xanthene dyes.8a | |
 |
| | Fig. 13 Structures of DCA (2) and biphenyl (74). | |
 |
| | Fig. 14 Structures of triarylamine radical cations. | |
 |
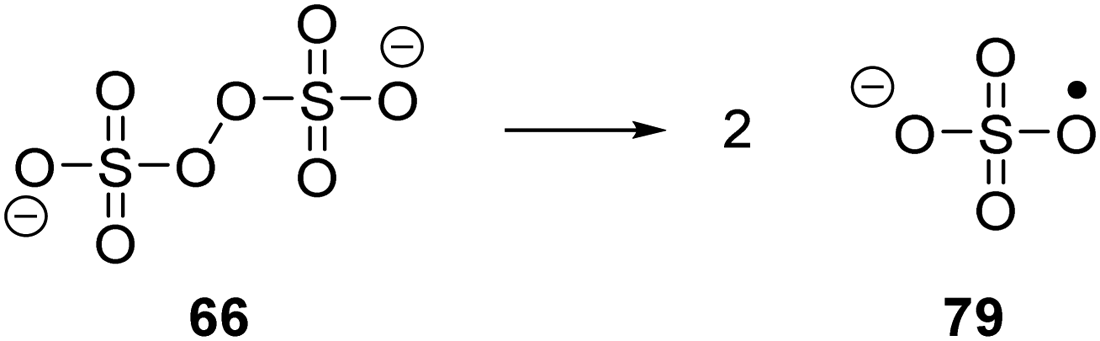
| | Scheme 15 Generation of sulfate radical anion 79. | |
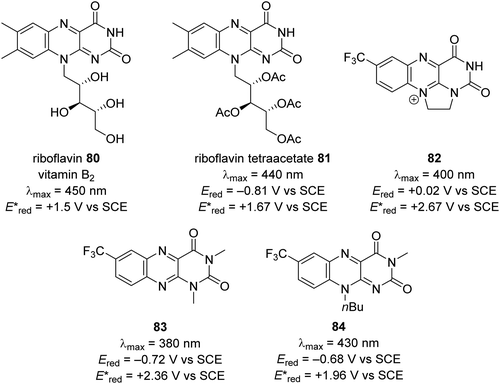 |
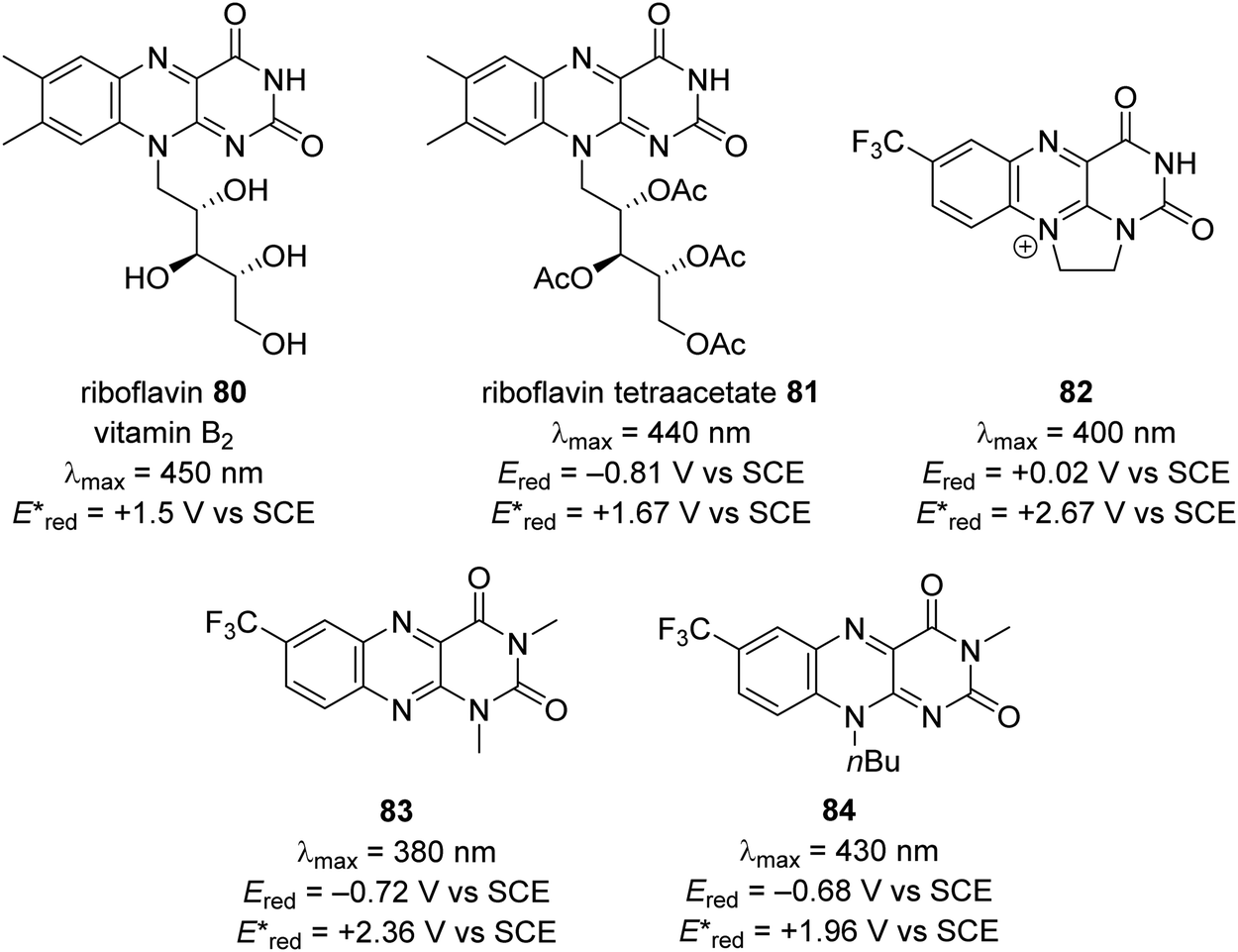
| | Fig. 15 Redox values for flavin-based compounds.95–99 | |
Summary of radical coupling reactions
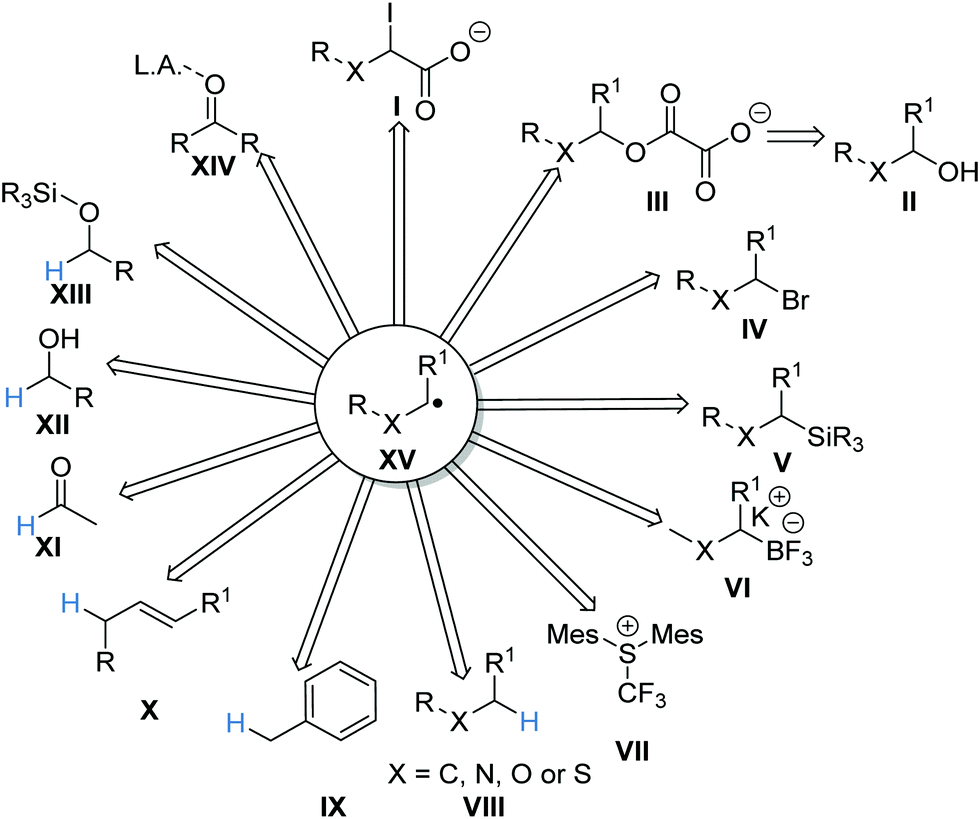
A summary of the reactions discussed in this review is presented here. The reactions have been categorised, differentiating between reactions proceeding via a carbon-centred radical, reactions proceeding via a heteroatom-centred radical and energy transfer. Carbon-centred radicals XV can be accessed from many different substrates (Scheme 16). For example, carboxylates I were oxidised via a SET process and this resulted in a carboxyl radical, which after radical decarboxylation gave radical XV. Similar approaches have been developed to convert alcohols II to carbon-centred radicals XVvia oxalate intermediates III. Carbon-centred radicals have also been prepared from alkyl halides IV, organosilanes V and boronates VI. The use of HAT allows carbon-centred radicals to be prepared from α-heteroatom C–H bonds VIII, benzylic C–H bonds IX, allylic C–H bonds X, aldehyde C–H bonds XI, hydroxy C–H bonds and C–H bonds of silyl protected alcoholic compounds XIII. The use of PCET allows for carbon-centred radical formation from carbonyl compounds XIV.
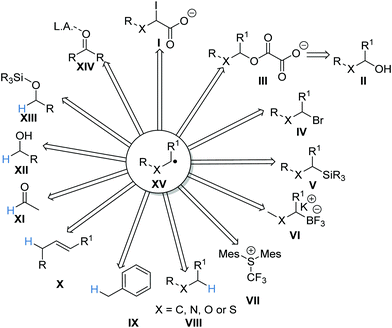 |
| | Scheme 16 Radical precursors. | |
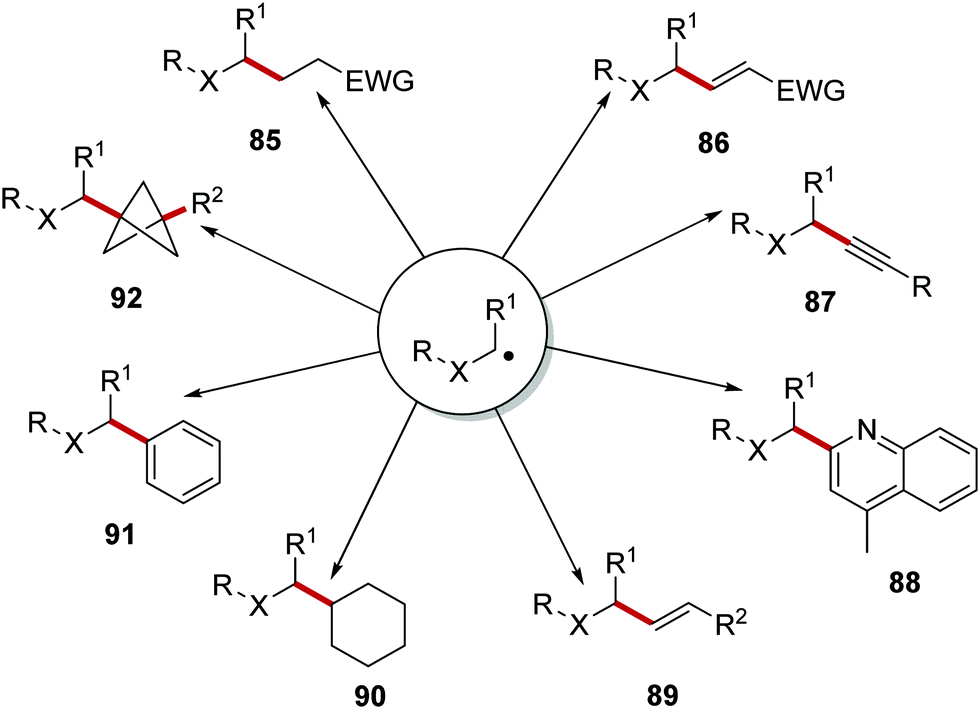
Once formed, the carbon-centred radical can take part in many different reactions (Scheme 17). A Giese reaction can be performed where the radical adds to an electron-poor alkene and this gives the alkylated compounds 85.99,100 The use of vinyl sulfones in combination with a carbon-centred radical led to the formation of vinyl compounds 86 (following loss of the sulfonyl radical)101,102 The use of ethynyl-benziodoxolone (EBX) reagents allowed for the preparation of alkyne compounds 87 with carbon-centred radicals under mild conditions.103 Carbon-centred radicals were used in the Minisci reaction, with functionalised heterocycles 88 being produced.2 Tandem photoredox and cross-coupling catalytic strategies have allowed for the formation of alkenes 89, alkyl derivatives 90 and aryl products 91. Reactions of [1.1.1]propellane with carbon-centred radicals have led to the formation of bicyclo[1.1.1]pentanes 92, as potential isosteres of aromatic rings.104
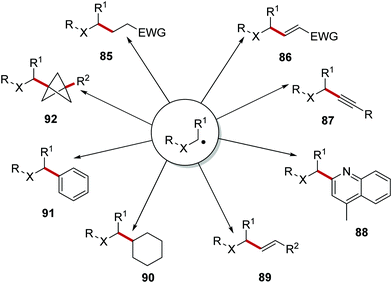 |
| | Scheme 17 Common radical reactions reported in the literature. | |
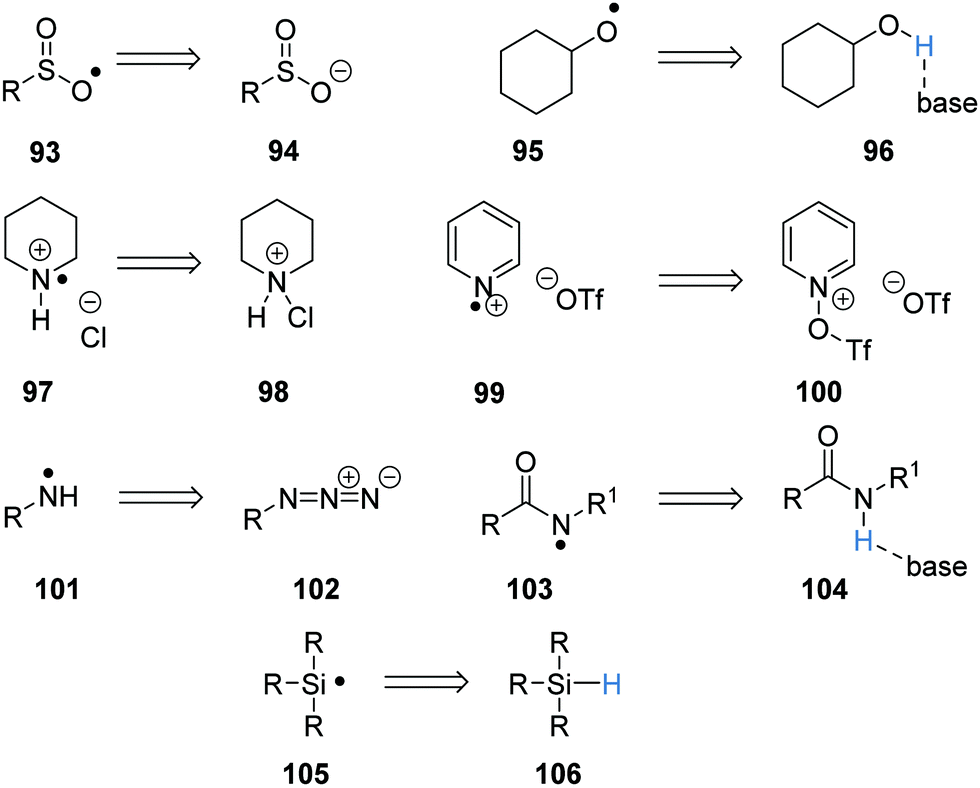
As well as carbon-centred radicals, heteroatom-centred radicals can also be formed (Scheme 18). Sulfonyl radicals 93 are formed via SET oxidation of sulfinate salts 94, whereas the oxygen-centred radical 95 is formed from a PCET from the alcohol 96. Nitrogen-centred radicals 97 and 99 are formed under a similar process, where the cations 98 and 100 gain a single electron and the loss of an anion gives these radical cations. Primary aminyl radicals 101 were formed from the single electron reduction of azides 102 in the presence of a proton source. PCET can give rise to amidyl radicals 103 from amides 104 and a HAT process can give silyl radicals 105 from silanes 106.
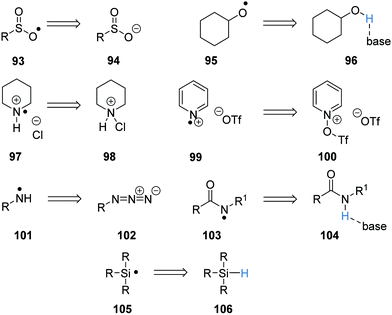 |
| | Scheme 18 Heteroatom radical precursors. | |
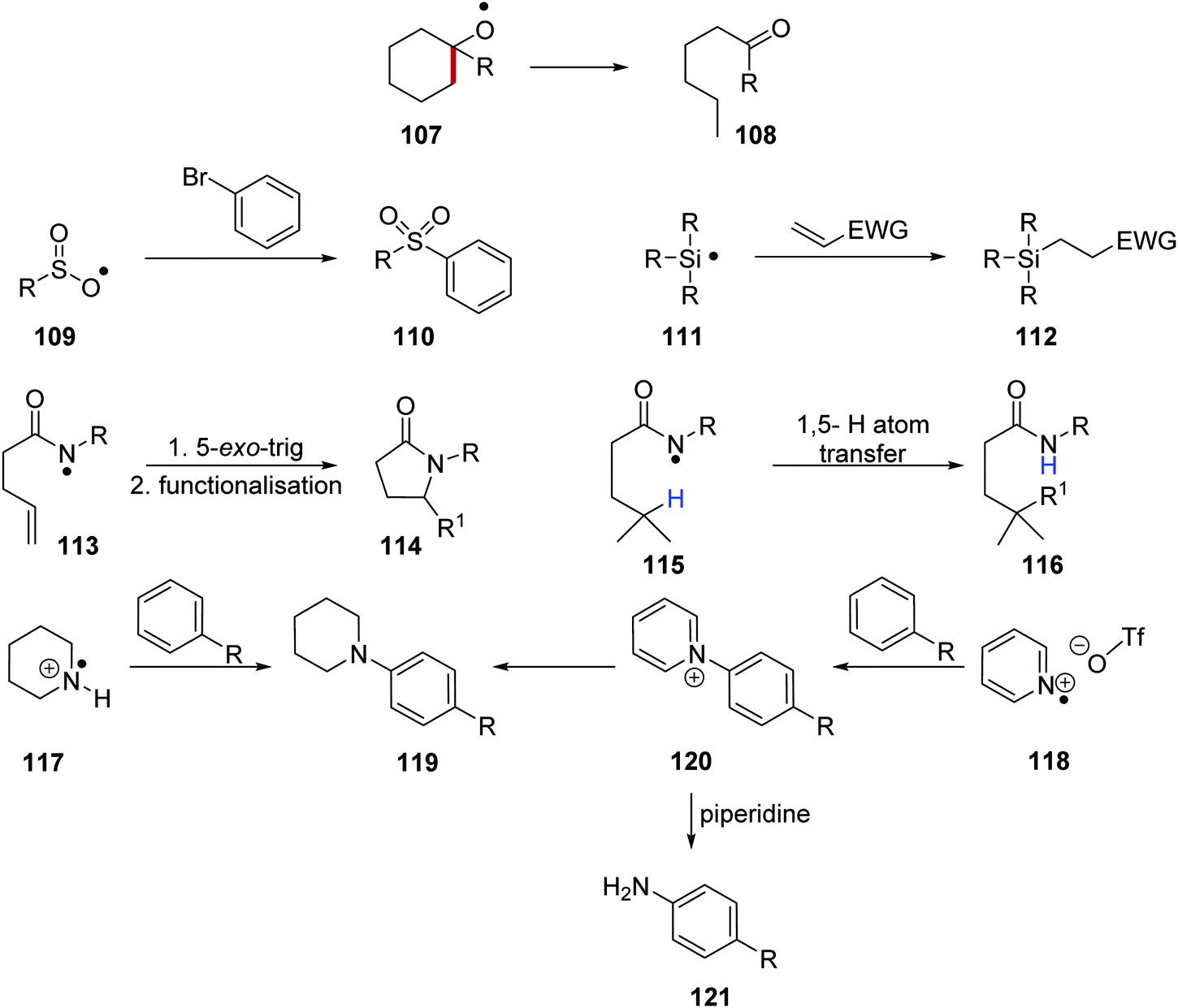
These heteroatom radicals can take part in a variety of reactions (Scheme 19). For example, the formation of oxygen-centred radical 107 results in ketone formation and C–C bond fragmentation leading to products such as 108.105 Incorporation of a nickel catalyst facilitated cross-coupling reactions between sulfonyl radicals 109 and bromobenzene and this led to coupled products such as 110.106 Amidyl radicals 113 can partake in 5-exo-trig cyclisation, followed by further radical functionalisation and this gave lactams such as 114. Additionally, amidyl radicals 115 also underwent 1,5-hydrogen atom transfer and this allowed for remote site-selective functionalisation giving amides such as 116. Secondary amine radical cations 117 and pyridine radical cations 118 have both been used to form substituted aniline products 119. It was also demonstrated that a Zincke reaction with piperidine could be performed on pyridinium adduct intermediate 120 and this generated unsubstituted anilines 121.
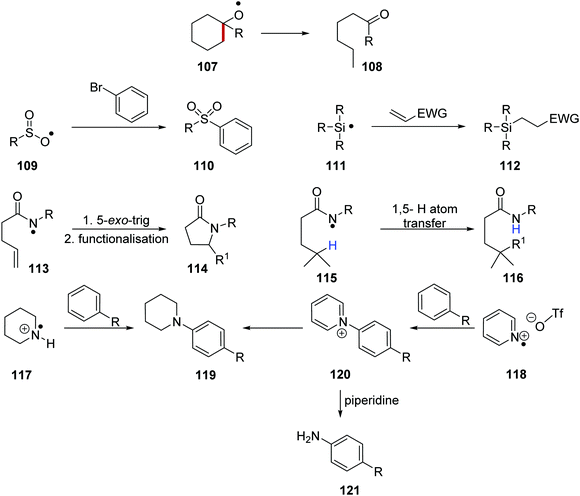 |
| | Scheme 19 Heteroatom radical reactions found in the literature. | |
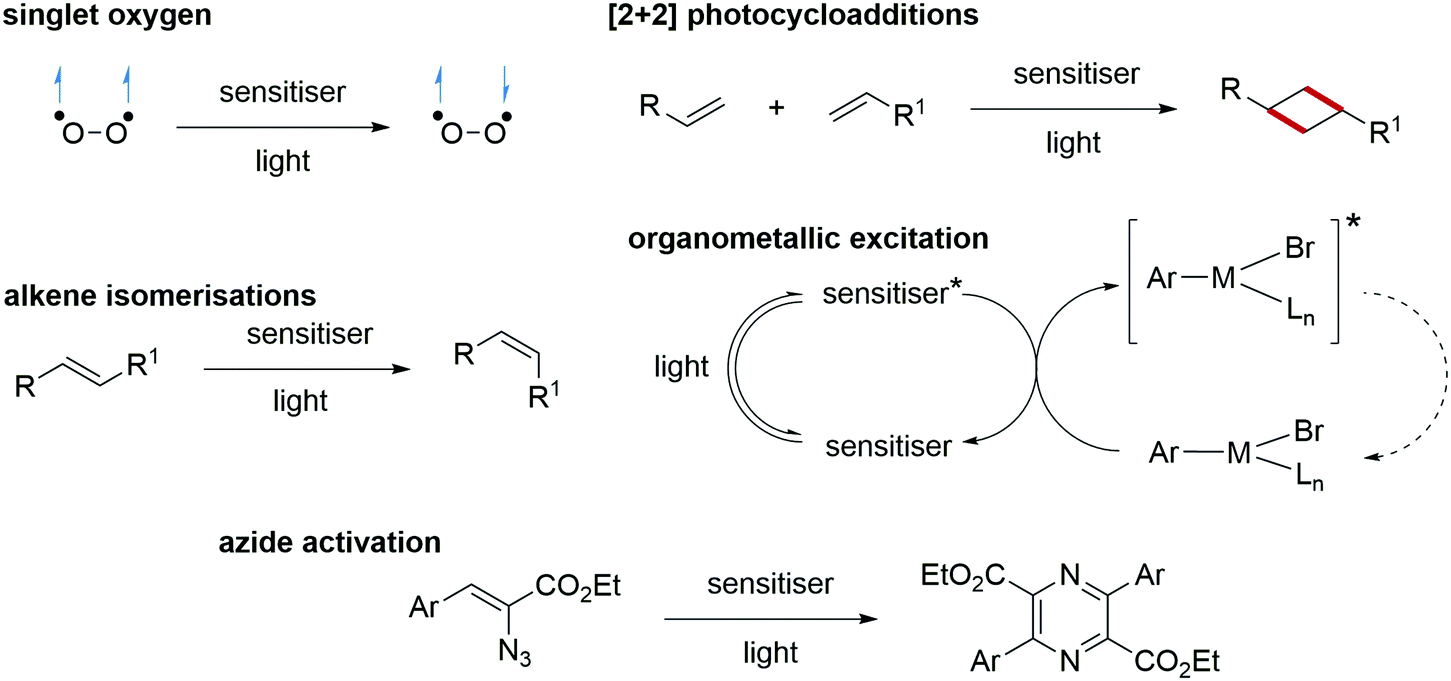
Energy transfer processes facilitated complementary transformations (Scheme 20). Singlet oxygen generation can be accessible through energy transfer processes.107,108 The [2+2] cycloadditions of alkenes are highly successful processes with energy transfer catalysis.109,110 The application of quantum dots as energy transfer catalysts in organometallic cross-coupling reactions has increased the efficiency and selectivity of these transformations.111 Energy transfer has also found use in the activation of azides and, from this, N-heterocycles have been formed, such as pyrazines.112
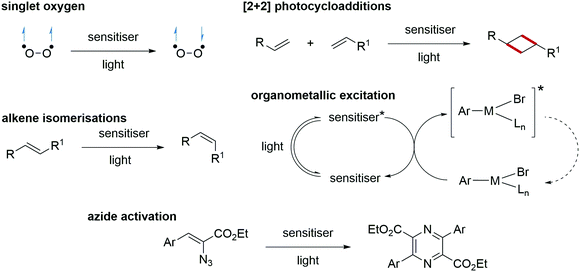 |
| | Scheme 20 Energy transfer reactions. | |
Summary of introduction
A considerable amount of progress in radical coupling has been achieved due to PET, HAT and energy transfer approaches. The reactivity and selectivity of these processes are dependent upon different factors. Photoredox approaches are dependent upon redox potentials, HAT approaches are dependent upon bond dissociation energies and energy transfer is dependent upon triplet energies. With these three different but complementary processes, a wide range of mild, non-hazardous transformations has been developed. Photocatalysts, in particular, have garnered considerable attention due to the enhanced reactivity of the excited state. The use of nickel and copper catalyst alongside a PET, HAT or energy transfer catalyst has resulted in a greatly expanded scope of feasible transformations.
2 Metal-mediated bond formations
Ruthenium
Photoactivated ruthenium(II)* complexes can act as electron donors or as acceptors. This means that catalytic cycles can either involve Ru(I) and Ru(II), or Ru(II) and Ru(III). In addition, photoactivated Ru(II)* complexes can engage in energy transfer reactions. Examples of these roles are now described, based on proposals for mechanisms described in the original research papers. This section builds on extensive earlier discoveries on visible light photoredox catalysis with transition metal complexes, and the reader is referred there for further reading.9
2.1 Catalytic cycles featuring Ru(II)/Ru(I)
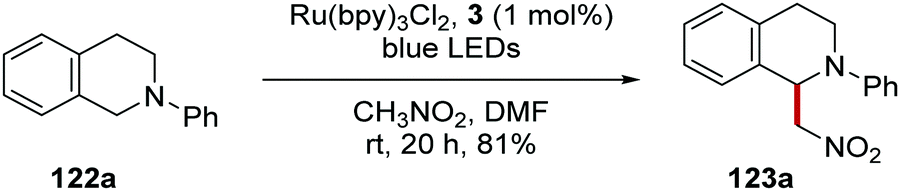
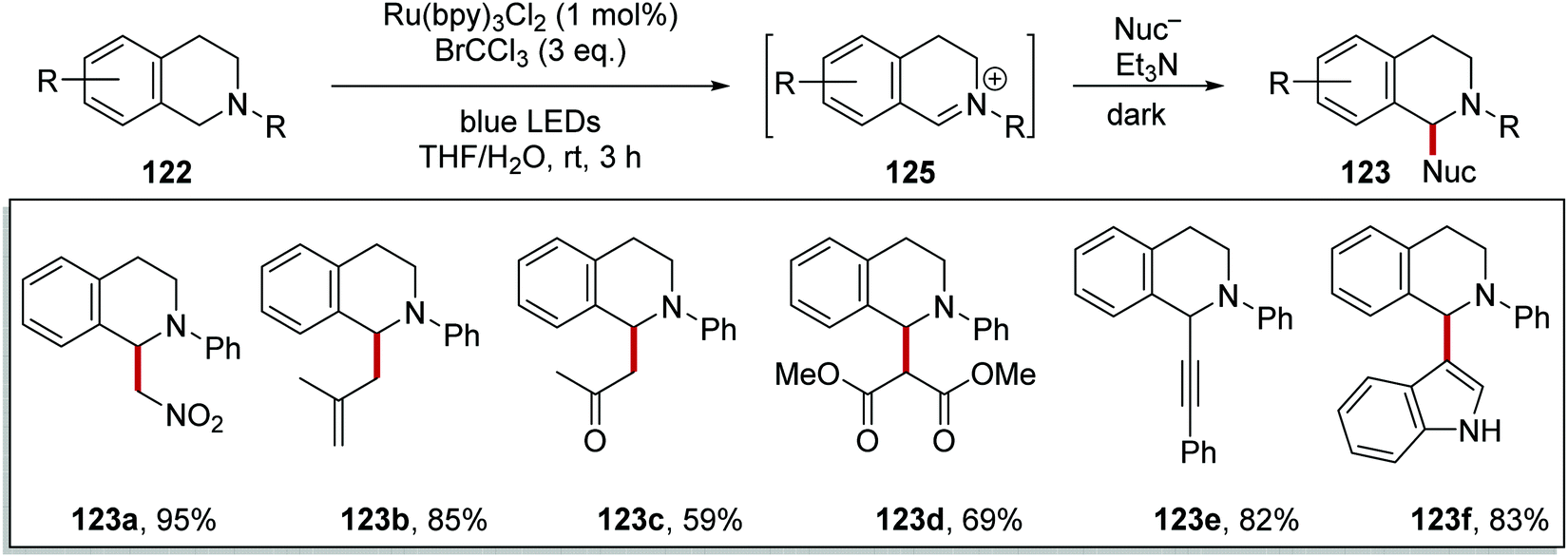
Stephenson's team converted tetrahydroisoquinoline 122a into the Henry product 123a in the presence of nitromethane using Ru(bpy)Cl2 in 81% yield or Ir(ppy)2(dtbbpy)PF6 in 92% yield (Scheme 21).113
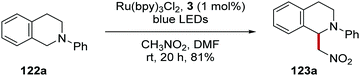 |
| | Scheme 21 C–H functionalisation of THIQ 122a with nitromethane. | |
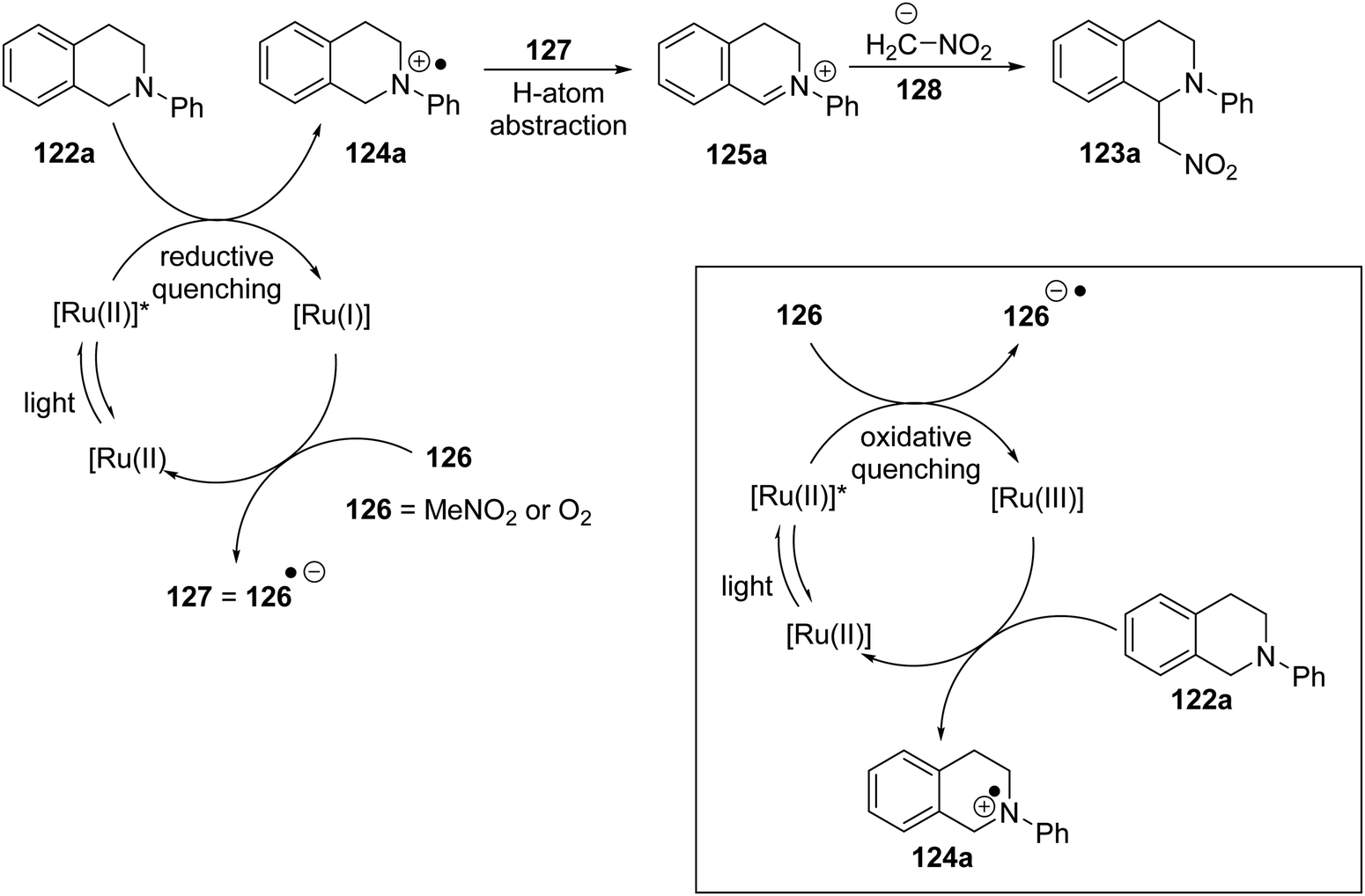
A specific mechanism is proposed for the Ir(III)-catalysed reaction, and may also be valid for the Ru(II) variant. Here, the photoactivated catalyst is reductively quenched by the amine substrate 122a to form the radical cation 124 (Scheme 22). The reduced form of the catalyst Ir(II) or Ru(I) is returned to its original oxidation state Ir(III)/Ru(II) through interaction with an oxidant 126 (MeNO2 or O2) which is converted to its radical anion 127. The radical component of this radical anion abstracts an H atom from the amine radical cation, to form an iminium salt 125, while the anionic component can deprotonate nitromethane to form the nitronate nucleophile 128.
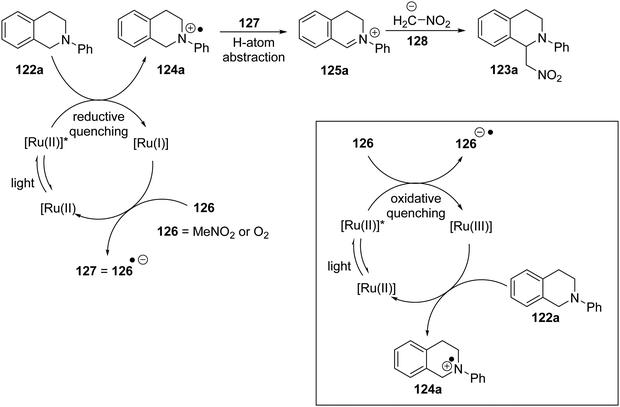 |
| | Scheme 22 | |
However, for the ruthenium pathway, an alternative mechanism may or may not be at play. It was reported that 122a (+0.88 V vs. SCE)113b cannot be oxidised with photoactivated [Ru(bpy)32+]* (+0.77 V vs. SCE113b) (+0.84 V is cited113a) but can be oxidised with Ru(bpy)33+ (+1.29 V vs. SCE). This seems also to have been considered by Stephenson et al. who noted that [Ru(bpy)32+]* can be oxidised by nitromethane (inset Scheme 22). The result would be that the same species, iminium salt 125 and nitronate anion 128, would be produced in solution, but would involve an oxidative quenching of the photoactivated Ru catalyst and therefore involve a Ru(II)/Ru(III) cycle. Subsequent papers from Stephenson promote the Ru(I)/Ru(II) cycle for these Henry reactions, so we proceed on that basis.
The slow conversion observed in Scheme 21, was due to the slow re-oxidation of Ru(I) to Ru(II). Therefore, it was thought that a terminal oxidant could be added to accelerate the reaction.114 It was found that bromotrichloromethane was the best terminal oxidant for this process. The inclusion of BrCCl3 led to the formation of iminium salts 125 from THIQs 122 within 3 h (Scheme 23). Afterwards, a variety of different nucleophiles gave the products corresponding to 123. Addition of the nucleophile was best performed in the dark with an excess of triethylamine and the reactions were complete within 3 h. Under these conditions the aza-Henry reaction was feasible, and this gave 123a in 95% yield. The use of methallyltrimethylsilane gave 123b in 85% yield, whilst 2-(trimethylsilyloxy)propene gave ketone 123c in 59% yield. When dimethyl malonate was used in the reaction with potassium carbonate, diester analogue 123d was given in 69% yield. Alkynylation of iminium salt 125 was also achieved with phenylacetylene when a catalytic amount of Cu(I)Br was added and this gave alkyne derivative 123e in 82% yield. Indole was another suitable nucleophile for this transformation when used with KOtBu, and this gave 123f in 83% yield.
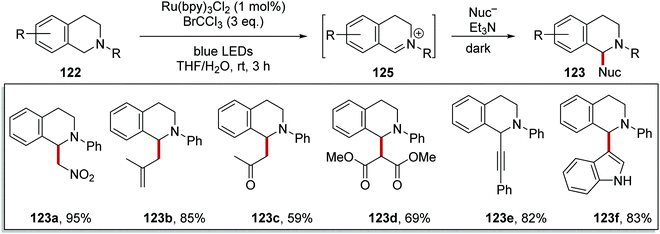 |
| | Scheme 23 Iminium salt formation and subsequent nucleophile addition upon ring system 122. | |
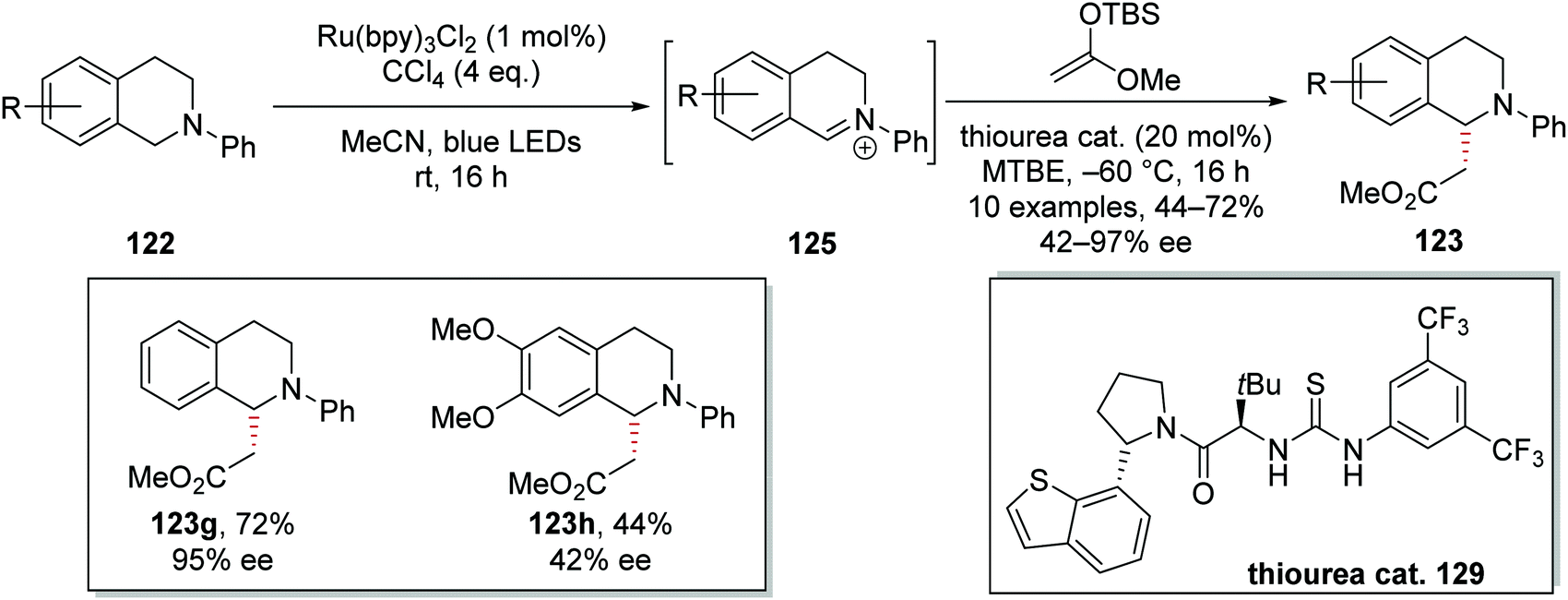
C–H functionalisation of THIQ was achieved asymmetrically (Scheme 24) with a thiourea catalyst 129.115 As previously discussed, iminium salt 124 was formed from 122. During optimisation of the reaction conditions, it was found that iminium salt formation was most efficient in MeCN, whereas the asymmetric addition of 1-(tert-butyldimethylsilyloxy)-1-methoxyethylene proceeded best in MTBE. Therefore, in preparing ester 123g, a solvent swap was carried out between formation of intermediate iminium salt 125 and addition of the nucleophile, and this achieved the highest yields and highest ee for this compound. In investigating different terminal oxidants for iminium salt formation, it was discovered that the photocatalyst counterion and halogen atom source had significant effects on enantioselectivity. Higher ee values were obtained when CCl4 was used as a terminal oxidant over BrCCl3. Furthermore, the common counterion PF6 also gave lower ee than the chloride counterion. Once optimisation of the reaction conditions was concluded, a small substrate scoping study was carried out. From this, it was observed that the enantioselectivity of the reaction was very sensitive to the electronic nature of the THIQ ring; electron-rich substituents gave lower yields and lower ee. For example, ester 123g was isolated in 72% yield and 95% ee, whereas the more electron-rich analogue 123h was isolated in 44% yield and 42% ee.
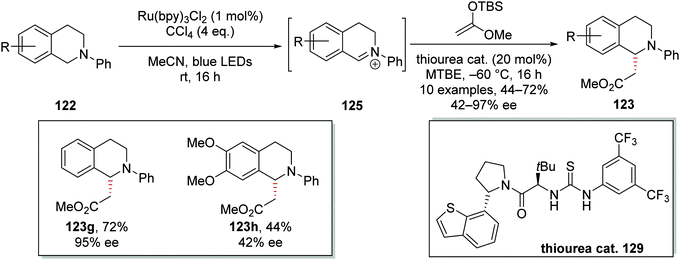 |
| | Scheme 24 Asymmetric alkylation of THIQs 122. | |
Silanes.
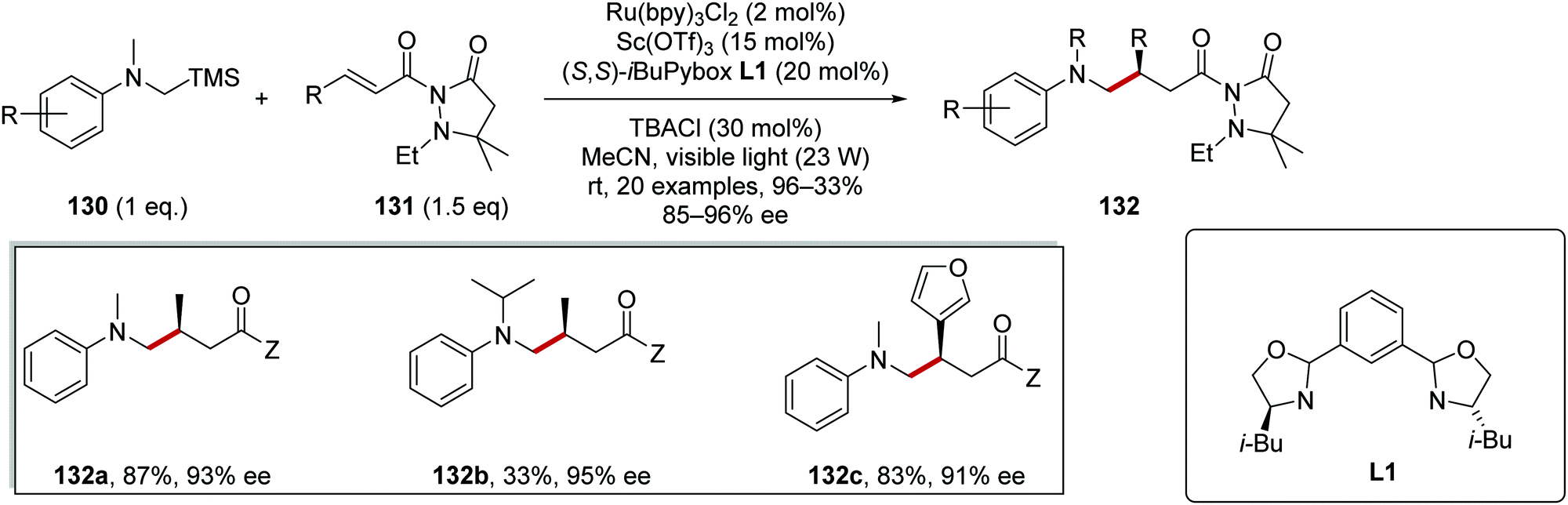
α-Silylamines 130 were successful coupling partners for radical coupling reactions to electron-poor alkenes 131 (Scheme 25).116 With consideration of redox potentials, SET between Ru(II)* and 130 was feasible (Rured* = +0.77 V vs. SCE, dimethylaniline has a redox potential of +0.74 V vs. SCE and the adjacent silyl group further lowers the oxidation potential)30,117 and this gave amine radical cation. Loss of +SiMe3 resulted in a carbon-centred α-aminoalkyl radical. Asymmetric radical addition to dicarbonyl compound 131 facilitated with scandium triflate as Lewis acid and chiral PyBox ligand L1 gave coupled alkylated anilines 132. These conditions allowed for the preparation of a library of 20 alkylated anilines 132; for example, analogues 132a–c were among a series of products all prepared with high ee (85–96%).
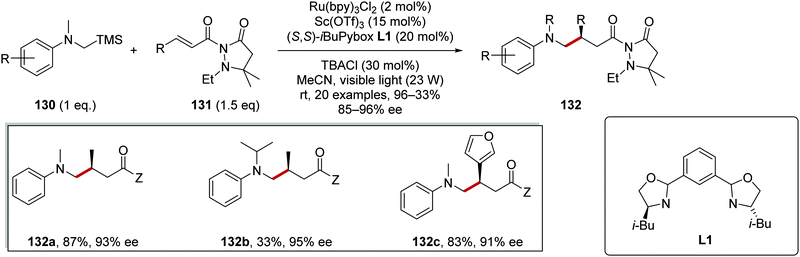 |
| | Scheme 25 Lewis acid-promoted coupling of α-silylamines to electron-poor alkenes. | |
Silicate chemistry.
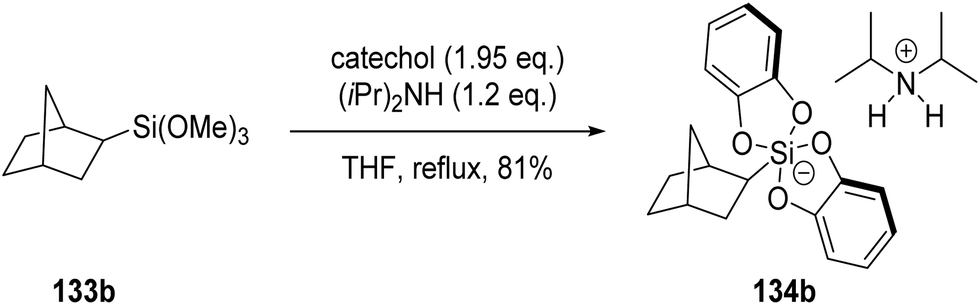
Organosilicates 134 are excellent radical precursors due to their low oxidation potentials (Ered = +0.4 → +0.7 V vs. SCE).118 These silicates were accessed by reacting the corresponding trimethoxysilane 133 with catechol and an amine base, such as diisopropylamine.119 For example, the use of silane 133b led to the formation of silicate 134b in 81% yield (Scheme 26).
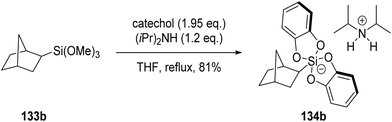 |
| | Scheme 26 Formation of silicate 134b. | |
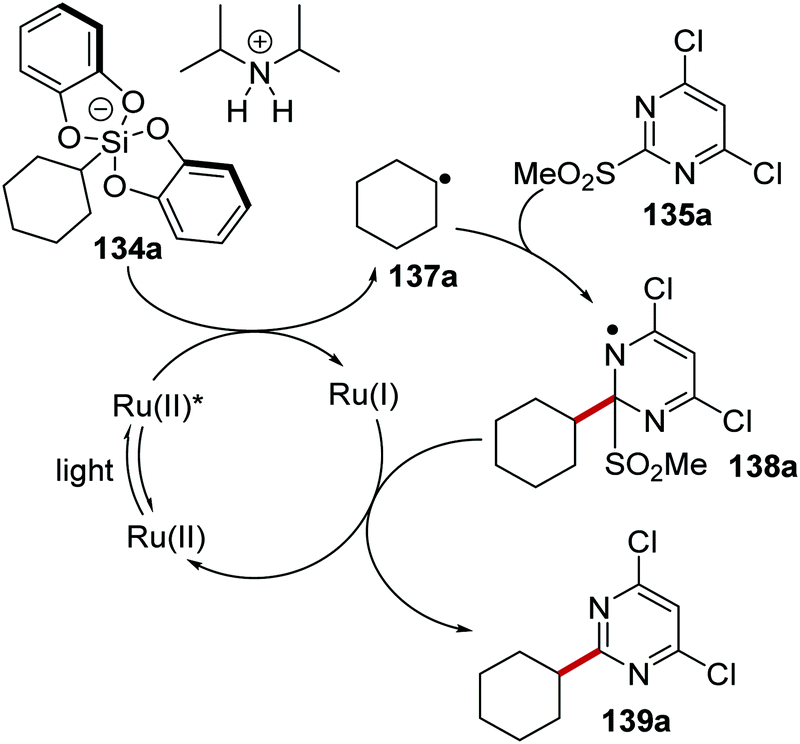
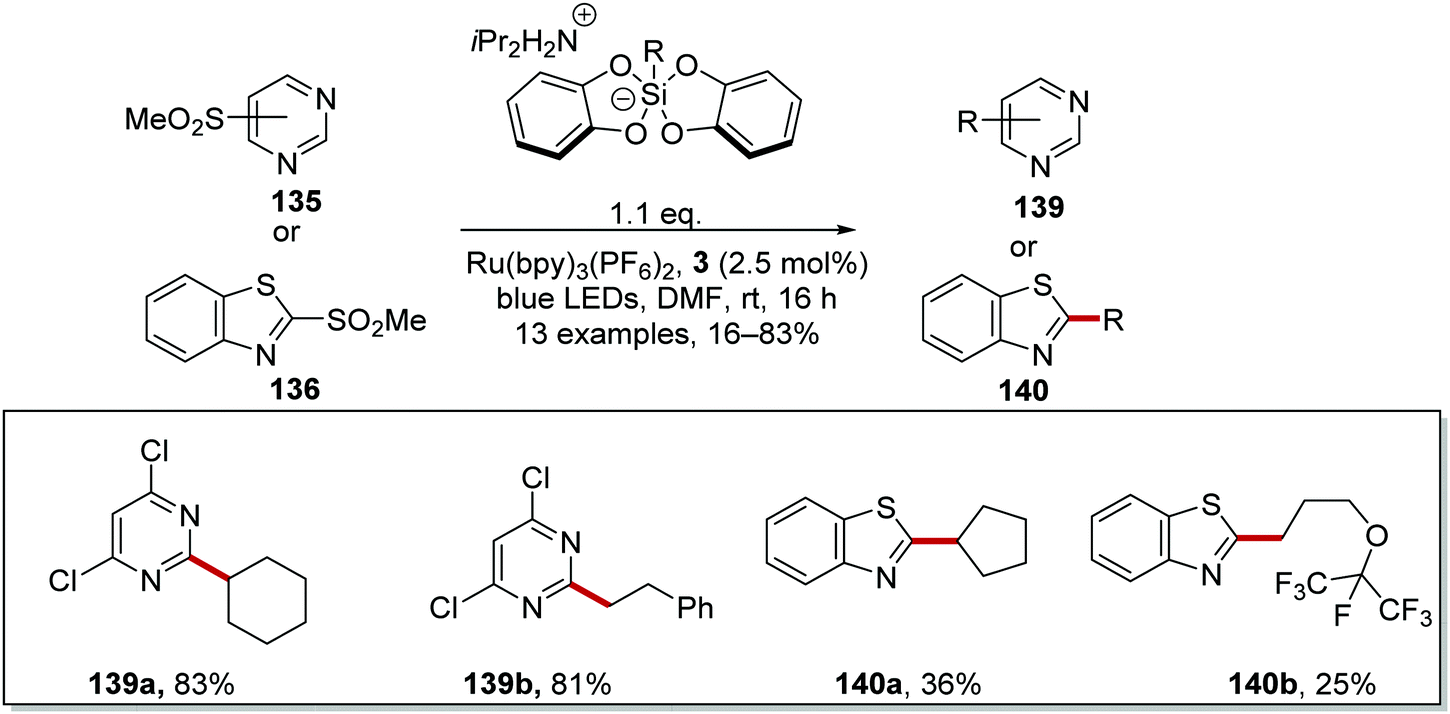
The coupling of organosilanes 133 with electron-poor arenes, such as pyrimidine sulfones 135 (Schemes 27 and 28) and benzothiazole-sulfones 136 was realised via a silicate intermediate 134 under blue light and a Ru(bpy)3(PF6)2 photoredox catalyst. Under the reaction conditions, the Ru(II) complex was excited to the Ru(II)* species (Scheme 27). SET to Ru(II)* complex (Ered* = +0.77 V vs. SCE) from 134a (ca. +0.40 V vs. SCE)119 resulted in the formation of alkyl radical 137a and Ru(I) complex. Addition of 137a to electron-poor pyrimidine 135a formed the key C–C bond with the generation of radical 138a. The ruthenium catalytic cycle was closed with a SET to 138a from Ru(I) species. This reformed the original Ru(II) complex and after the loss of sulfinate anion from 138a, gave coupled compound 139a.
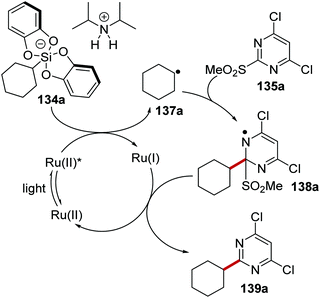 |
| | Scheme 27 Formation of 139a from 134a. | |
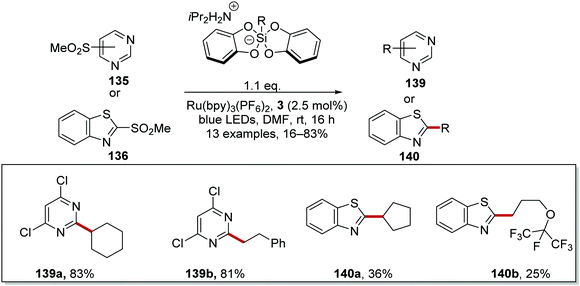 |
| | Scheme 28 Silicate coupling to pyrimidine sulfones 135 and benzothiazole sulfones 136 with Ru(bpy)3(PF6)2. | |
The reaction worked better for pyrimidines than benzothiazoles. Low yields were recorded for benzothiazoles 140a (36%)and 140b (25%) (Scheme 28), whereas high yields were found for pyrimidine compounds 139a (83%) and 139b (81%).
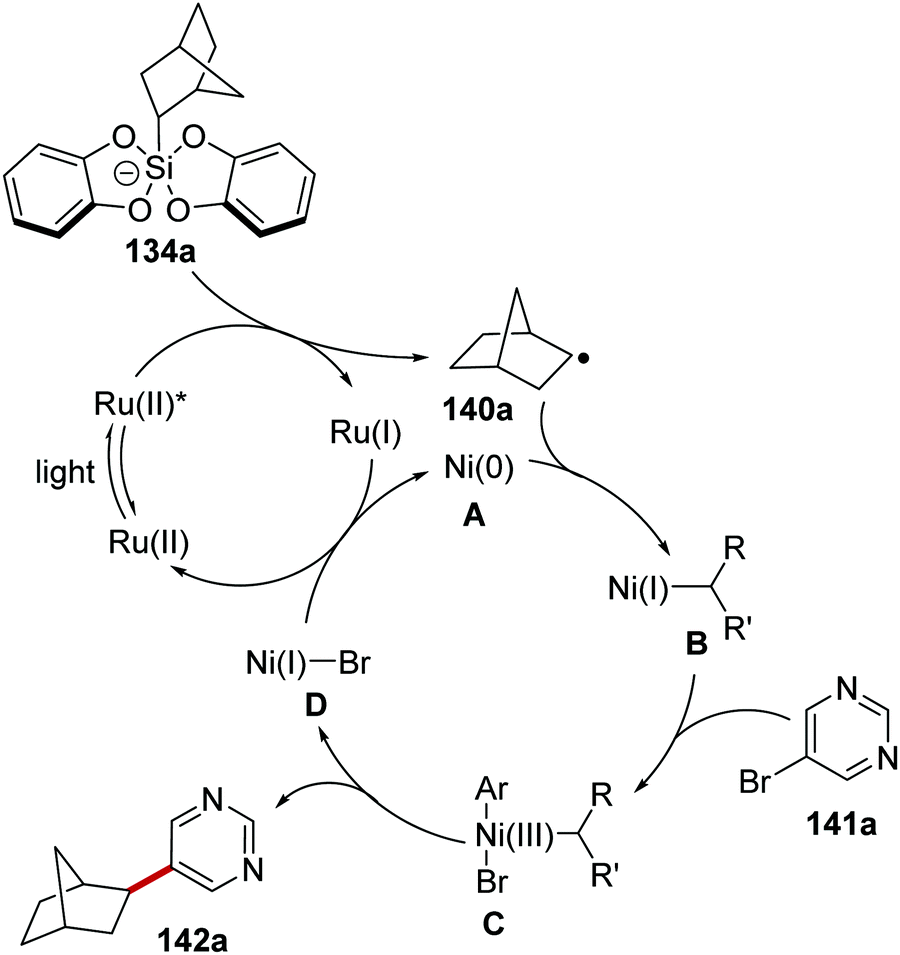
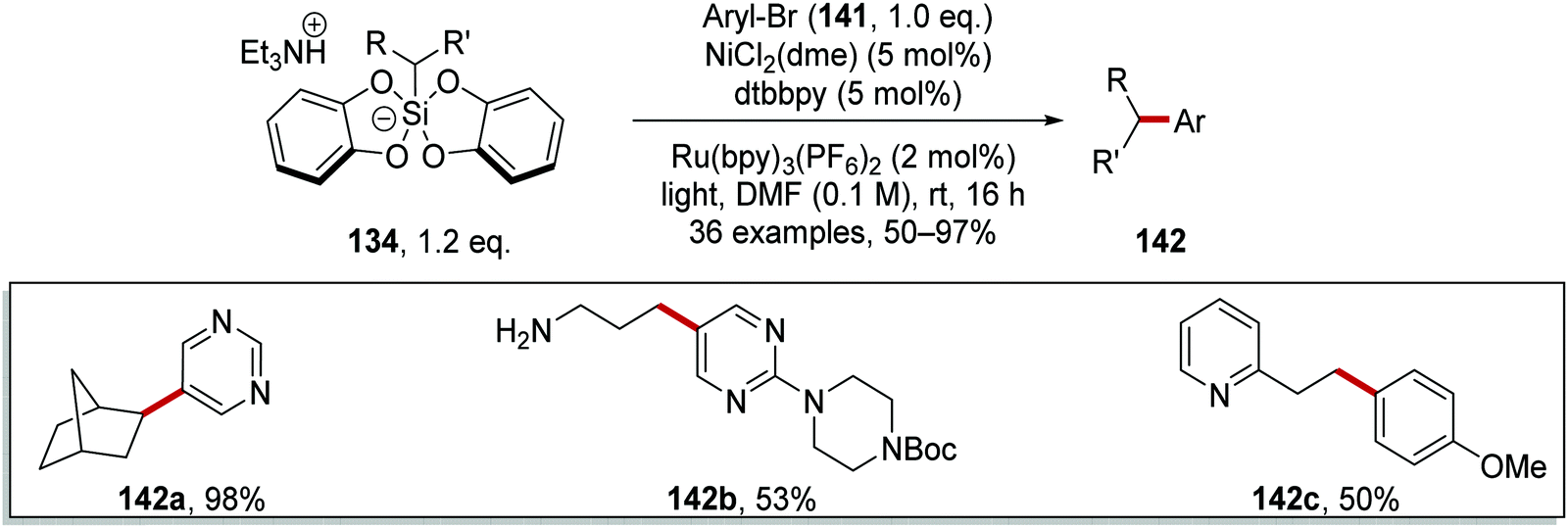
The inclusion of a nickel catalyst permitted coupling between organosilicates 134 and aryl bromides 141.119 Irradiation of the reaction mixture with blue light resulted in the formation of Ru(II)* species from Ru(II) complex (Scheme 29). SET to Ru(II)* from silicate 134a afforded Ru(I) complex and radical 140a. Interception of 140a with Ni(0) complex A gave Ni(I) complex B. Oxidative addition of complex B to aryl bromide 141a resulted in Ni(III) complex C, which underwent reductive elimination and this yielded coupled product 142a and Ni(I) complex D. The ruthenium and nickel catalytic cycles were closed with an outer-sphere SET between Ni(I) and Ru(I) complexes as both original Ni(0) and Ru(II) species were returned.
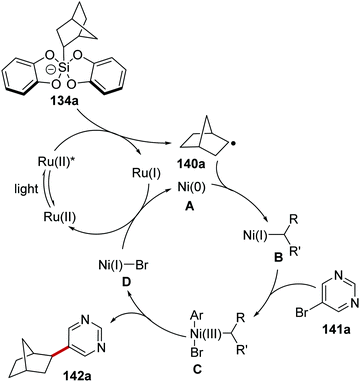 |
| | Scheme 29 Mechanism of silicate coupling to aryl bromides. | |
Coupled compound 142a was also produced when the Ir[dF(CF3)ppy](bpy)(PF6) photocatalyst was used but similar yields of 142a were obtained with the more economical Ru(bpy)3(PF6)2 photocatalyst. These optimised conditions afforded bicyclic 142a, N-Boc protected amine 142b (53%) and heterocyclic 142c (50%) (Scheme 30).
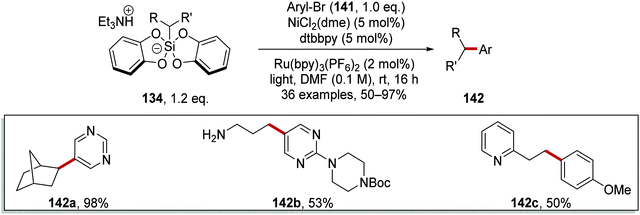 |
| | Scheme 30 Coupling of silicates to aryl bromides. | |
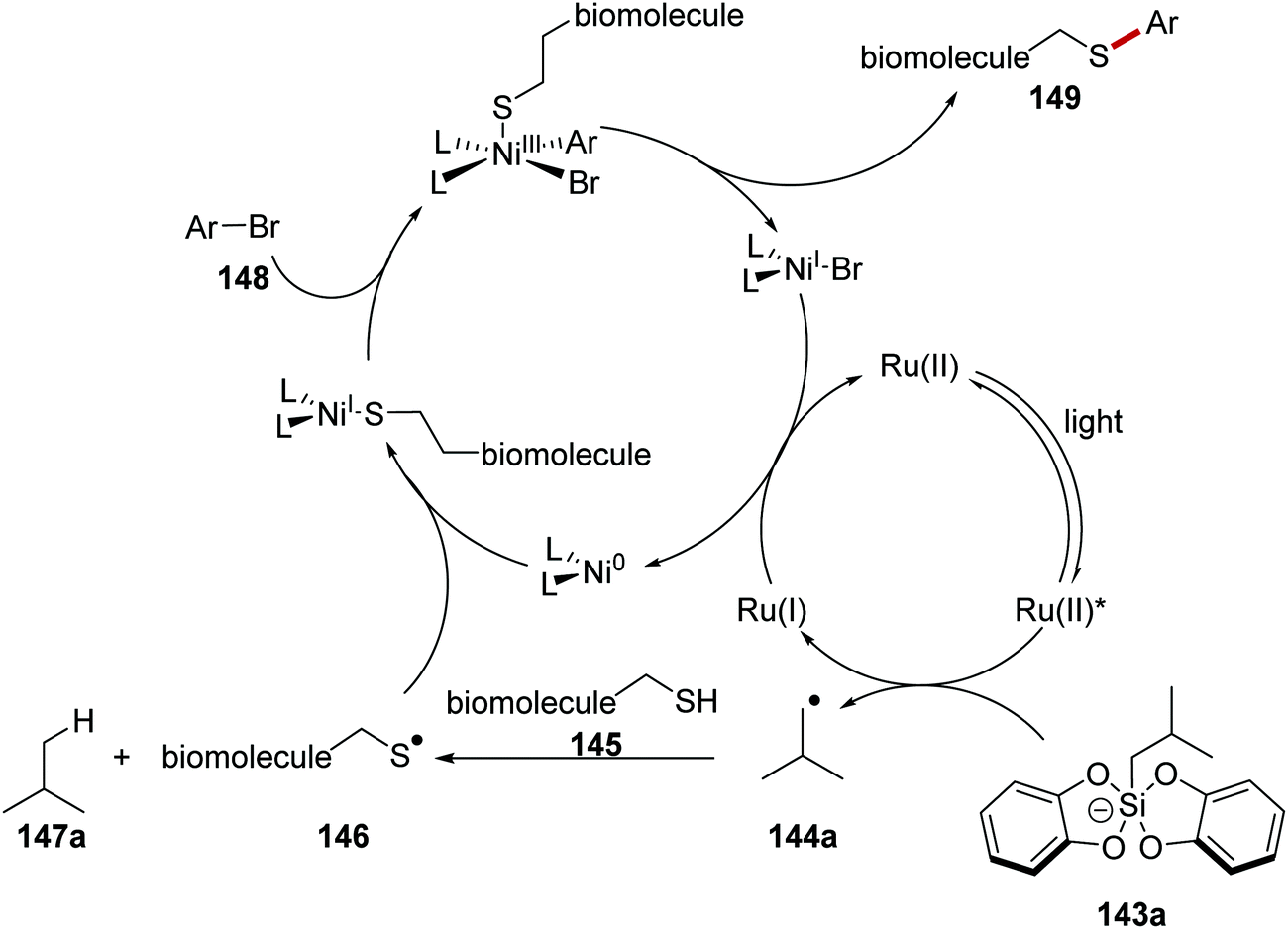
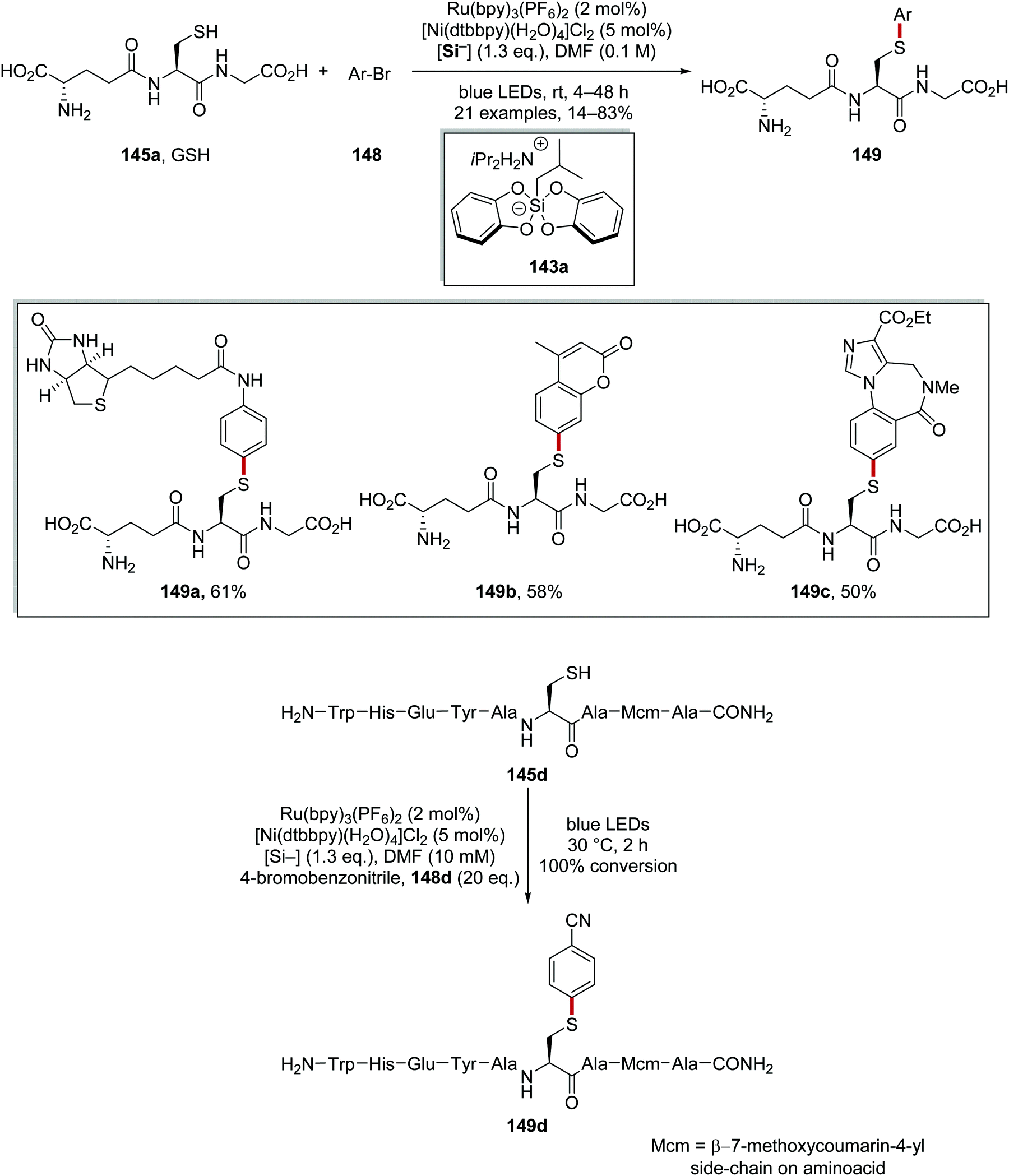
The arylation of biomolecules (containing cysteine residues) 145 (Schemes 31 and 32) with aryl bromides 148 was achieved with a ruthenium/nickel and silicate strategy, using silicate 143a as a HAT reagent precursor (Scheme 31).120 Irradiation of the reaction mixture led to Ru(II)* formation from Ru(II) complex. SET from silicate 143a to Ru(II)* afforded radical 144a, and the Ru(I) complex. Radical 144a abstracted a hydrogen atom from a cysteine residue present in 145 and this gave thiyl radical 146 and alkane 147a. Radical 146 was intercepted with Ni(0) complex and a Ni(I) complex was formed. Oxidative addition of aryl bromide 148 generated Ni(III) complex and this underwent reductive elimination forming biomolecule 149 and Ni(I) complex. Both ruthenium and nickel catalytic cycles were closed with an electron transfer to Ni(I) from Ru(I).
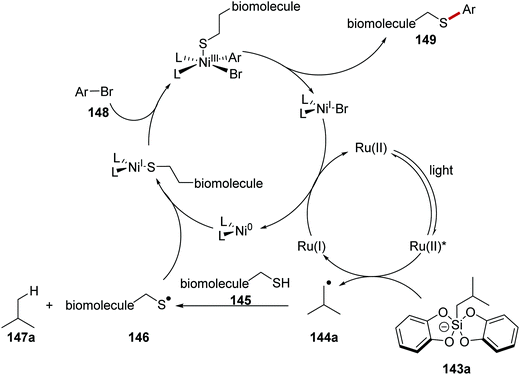 |
| | Scheme 31 Proposed mechanism for radical coupling to biomolecules with ammonium bis(catechol)silicate as a HAT reagent precursor. | |
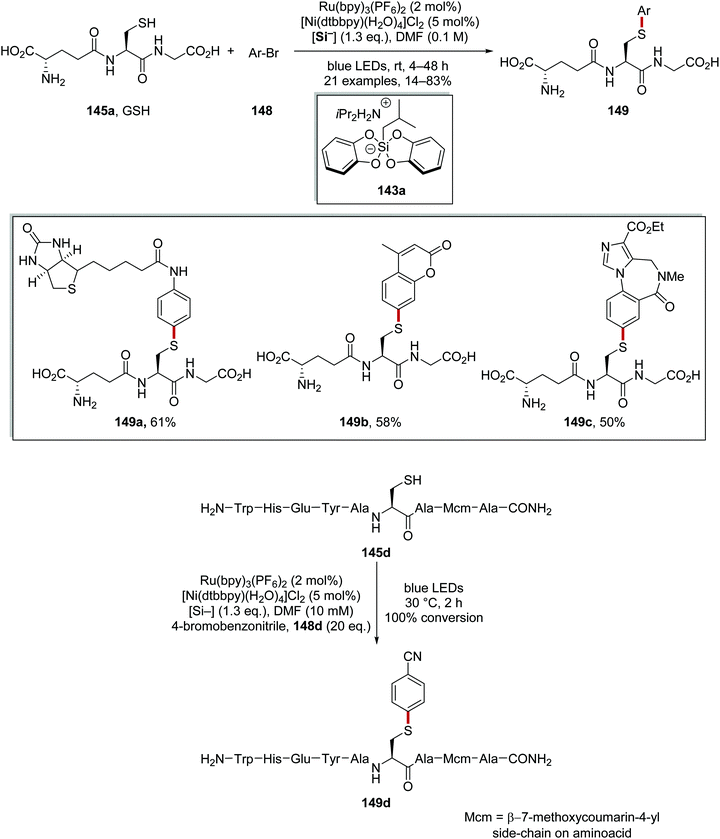 |
| | Scheme 32 Results of radical coupling to biomolecules. | |
This transformation allowed for the functionalisation of tripeptide L-glutathione (γ-Glu-Cys-Gly; GSH, 145a). Both a biotin analogue 149a and a fluorescent variant 149b of GSH were prepared in 61% and 58% yield, respectively, using low loadings of both Ru and Ni catalyst. The drug molecule flumazenil was also coupled with GSH and this gave 149c in 50% yield (Scheme 32). Finally, this methodology was used to functionalise nonapeptide 145d with 4-bromobenzonitrile 148d and this gave full conversion to 149d within 2 h.
Sulfinate oxidation to sulfonyl radicals.
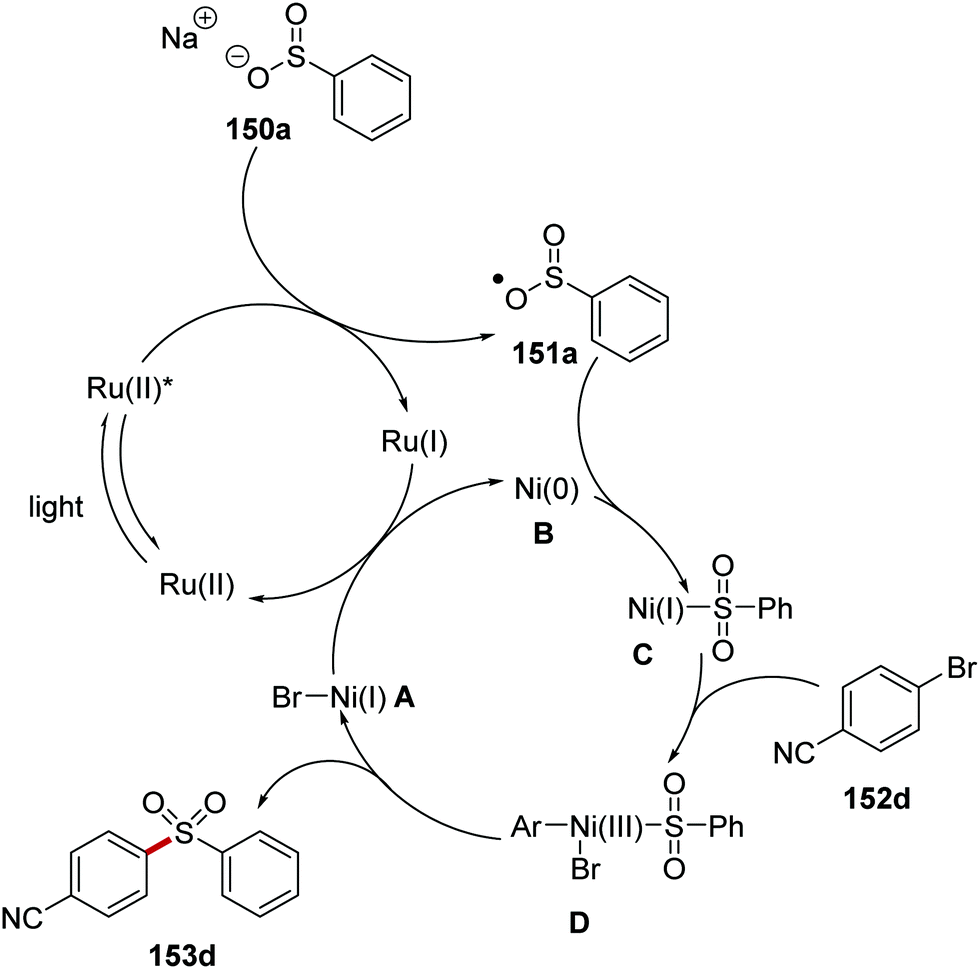
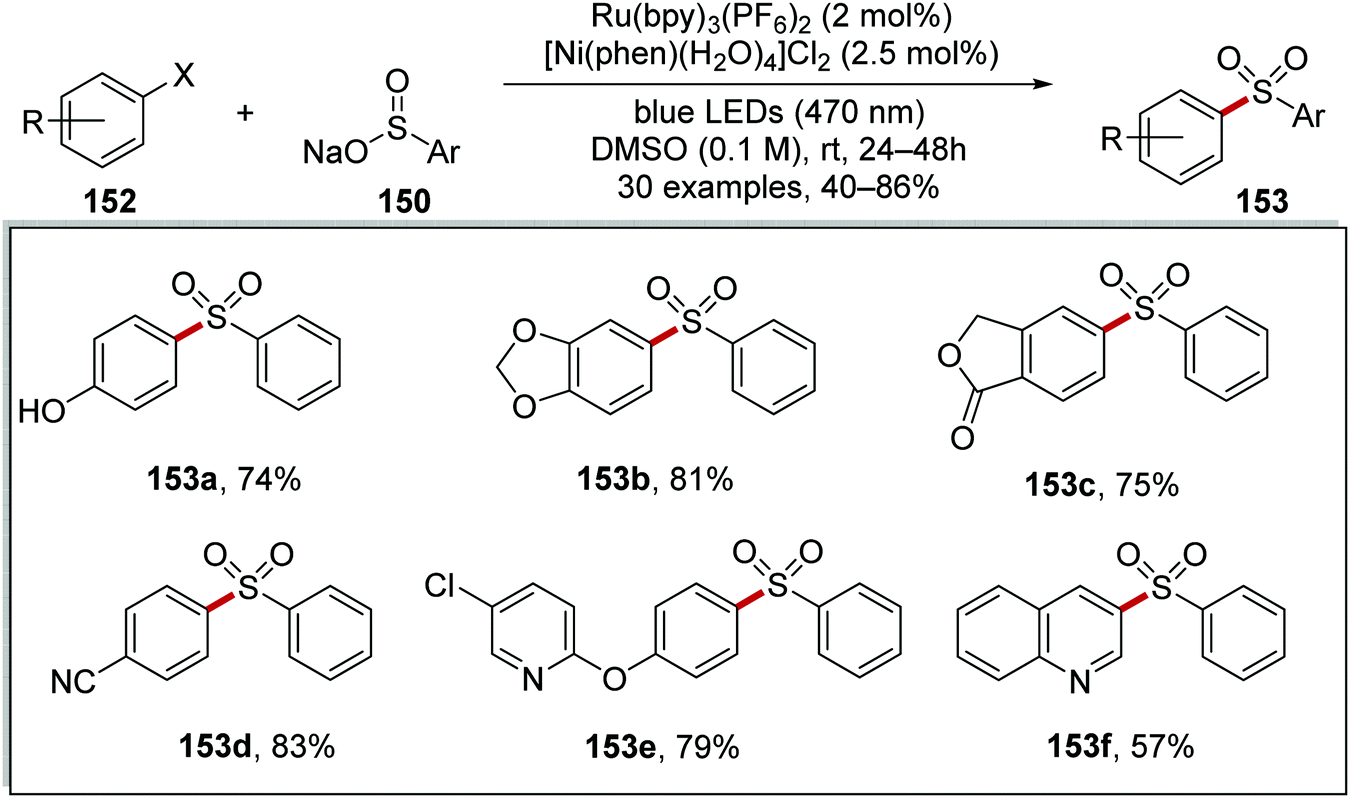
The formation of C–S bonds was achieved with a ruthenium/nickel catalytic strategy under blue light.106 Here, sodium sulfinate salts 150 and aryl bromides/iodides 152 were coupled in high yields and this gave aryl sulfones 153 (Scheme 33). Under the reaction conditions, Ru(bpy)3(PF6)2 catalyst is excited from exposure to light giving excited Ru(II)* complex. Electron transfer from sulfinate 150a (E1/2 = −0.37 V vs. SCE)106 to Ru(II)* (+0.77 V vs. SCE) gave sulfonyl radical 151a and Ru(I) complex. Ni(0) complex B intercepted the sulfonyl radical 151a and Ni(I) complex C was formed. Oxidative addition of aryl bromide 152d to Ni(I) complex C and then subsequent reductive elimination resulted in the generation of coupled product 153d and restoration of the original Ni(I) complex A. Electron transfer to Ni(I) complex A from Ru(I) complex resulted in the formation of Ni(0) complex B and the original Ru(II) complex.
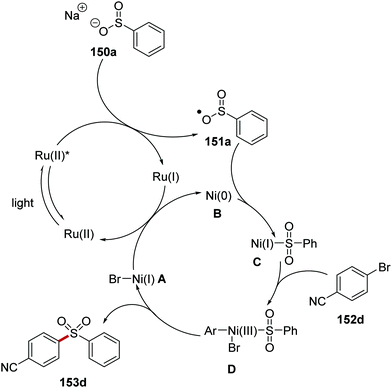 |
| | Scheme 33 Reaction mechanism of coupling between sulfonyl radicals with aryl halides. | |
Both aryl iodides and aryl bromides were used with this Ru/Ni catalytic system and this afforded 40 examples of the coupled sulfone products in 33–86% yields (Scheme 34 for selected examples). The reaction was functional group-tolerant; phenol 153a was isolated in 74% yield and 1,3-benzodioxole derivative 153b was given in 81% yield. Furthermore, lactone 153c was prepared in 75% yield from the corresponding aryl bromide. When 4-bromobenzonitrile was used in this reaction, 153d was formed in 83% yield. The presence of heterocycles did not impede this transformation with 153e and 153f isolated in 79% and 57% yield, respectively.
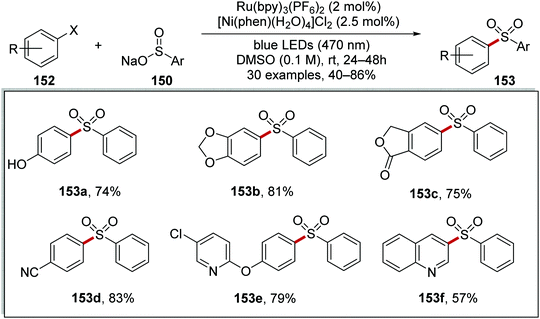 |
| | Scheme 34 Scope of the coupling reaction between sulfonyl radicals and aryl halides. | |
Lewis acid-activated ketones.
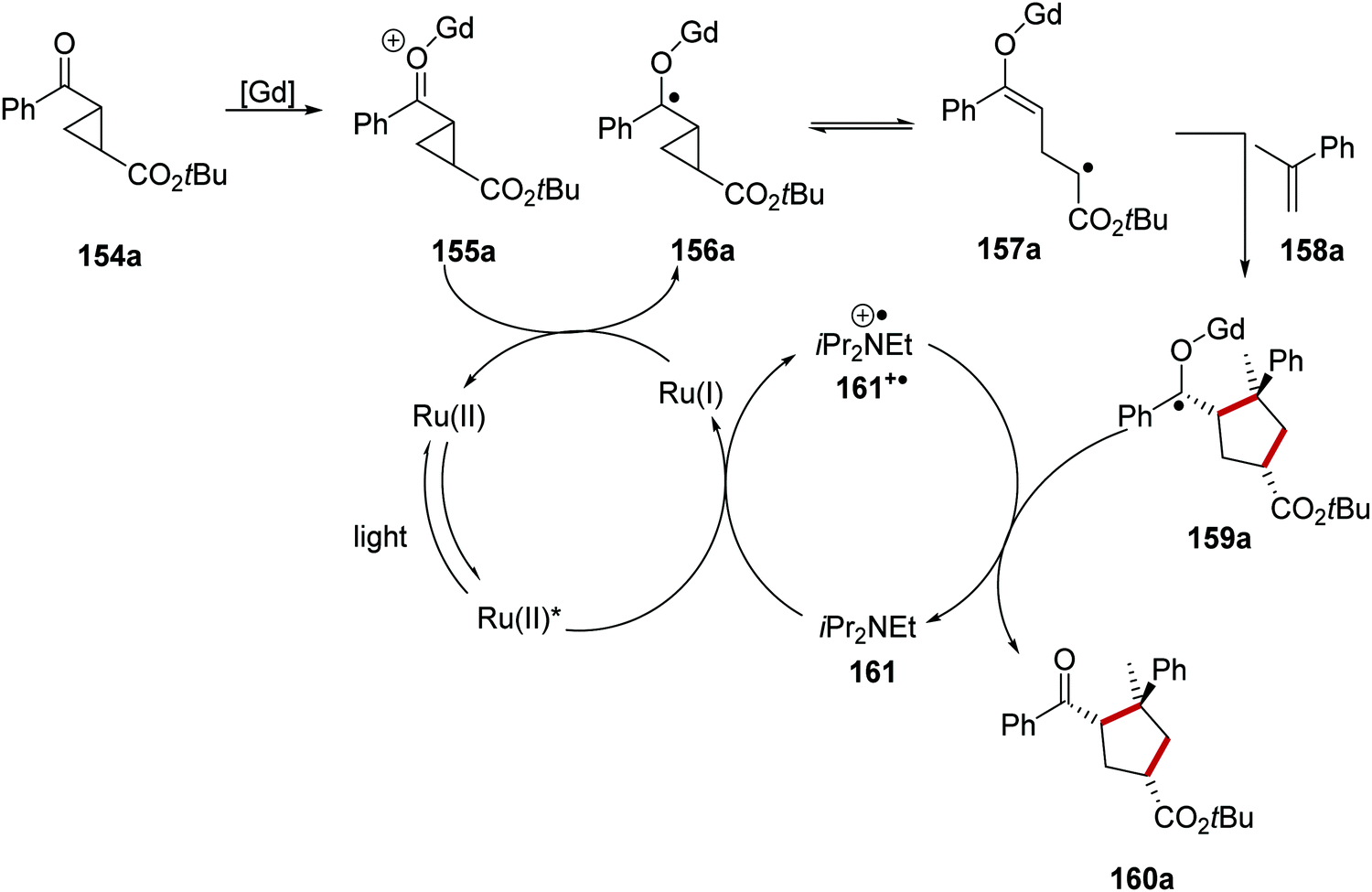
The combination of Lewis acid catalysis, chiral ligands and ruthenium photoredox was also employed in the asymmetric ring-expansion of cyclopropanes 154 with coupling to terminal alkenes 158 (Scheme 35).121 In this case, gadolinium(III) triflate was employed as the Lewis acid in combination with a PyBox ligand and a Ru(bpy)3X2 photocatalyst. SET to Ru(II)* from the amine base, DIPEA (161), resulted in the formation of a Ru(I) complex and the DIPEA radical cation 161+˙. Donation of an electron from Ru(I) to gadolinium adduct 155a resulted in carbon-centred radical 156a. Direct reduction of ketone 154a is not possible with the Ru(I) complex and this is supported by the reduction potential of acetophenone (−2.11 V vs. SCE)30 and the oxidation potential of Ru(bpy)3+ (−1.33 V vs. SCE).8b Electron donation from Ru(I) complex to the carbonyl group present in compound 155a, formed from 154a, generated cyclopropyl ketyl radical 156a. Subsequent reversible endergonic radical ring-opening of the cyclopropane moiety resulted in alkyl radical 157a.122 Slow step-wise cycloaddition with alkene 158a resulted in a [Gd]-ketyl radical 159a. Chain-propagating electron transfer to another equivalent of the substrate or chain-terminating reduction of the amine radical cation 161+˙ resulted in the formation of 160a.
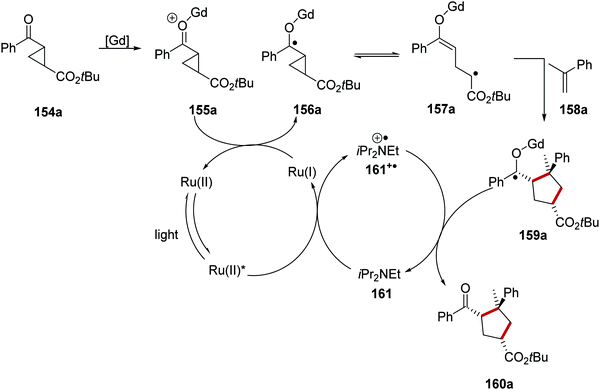 |
| | Scheme 35 Ruthenium and Lewis acid-promoted ring-expansion. | |
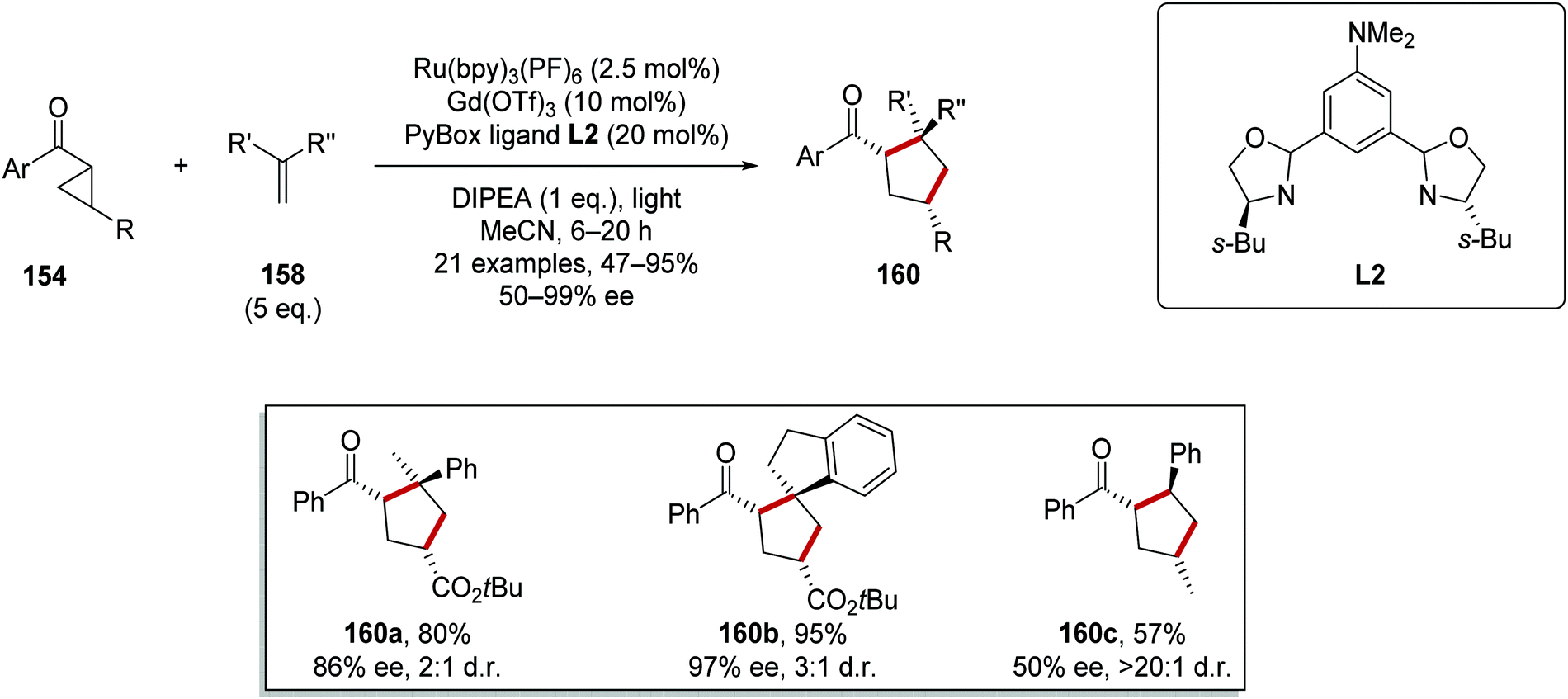
This reaction was applied to a wide range of different substrates generating a library of enantioenriched cyclopentanes 160 where the vast majority of yields and ee values were in the range 80–100% (Scheme 36). Terminal alkenes bearing two substituents were tolerated under the reaction conditions and this allowed for the preparation of compounds 160a and 160b in 80 and 95% yield, respectively. The use of a methyl-substituted cyclopropane substrate resulted in a lower 57% yield and only 50% ee for the corresponding cyclised product 160c.
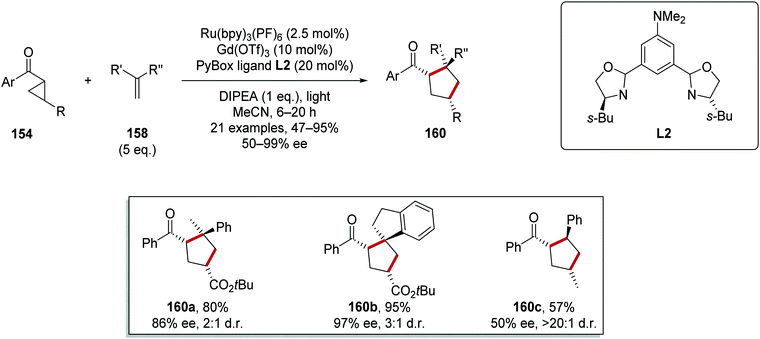 |
| | Scheme 36 Ru and Lewis acid-mediated cyclopropane ring expansion. | |
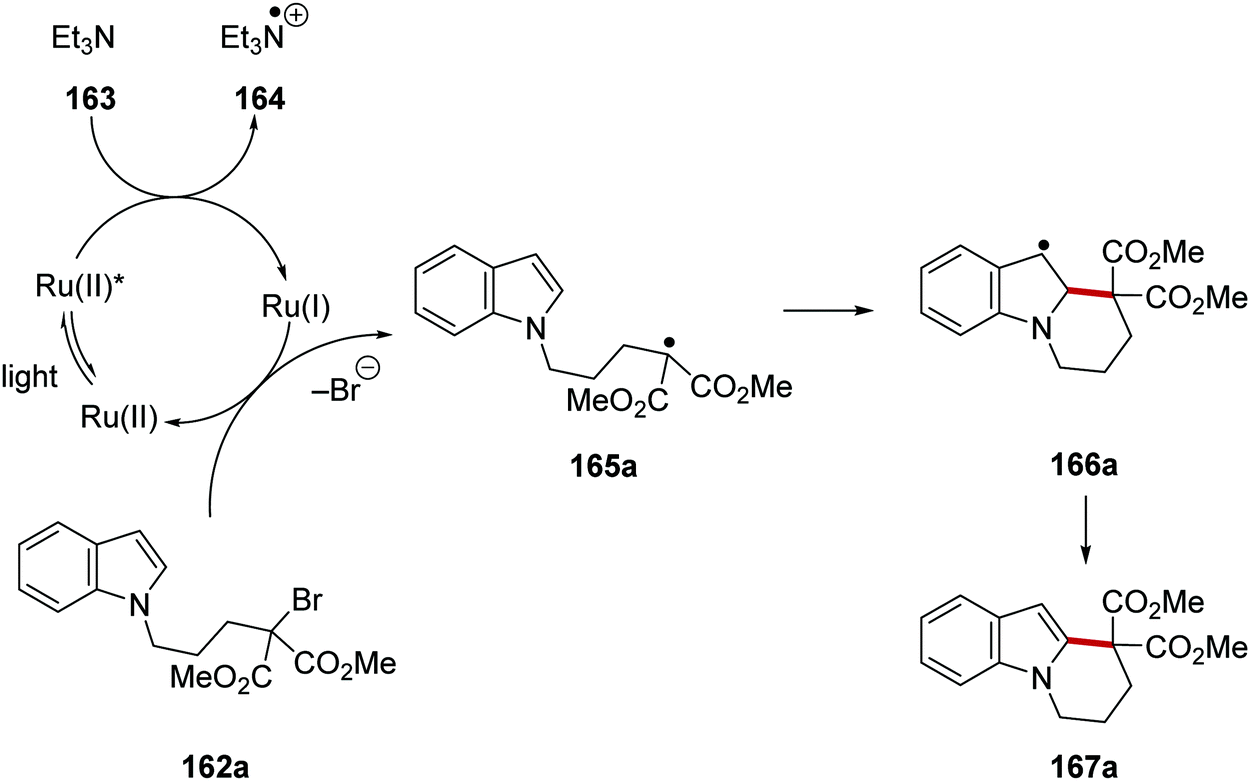
Ruthenium(I) behaved as an electron donor when it reduced alkyl C–Br bonds below and this led to cyclisation (Schemes 37 and 38).123 Under the reaction conditions, the excited Ru(II)* complex received an electron from triethylamine (163), resulting in the formation of a Ru(I) complex and the amine radical cation 160. Ru(I) then reduced the C–Br bond in 162a which formed radical 165a and the bromide anion. This SET was supported by the reduction potential of ethyl bromoacetate (E = −1.08 V vs. SCE)30 which should be comfortably reduced by Ru(bpy)+ (E = −1.33 V vs. SCE). Cyclisation of the electron-poor radical onto the electron-rich indole in 165a gave 166a. It was not determined whether the transformation of benzylic radical 166a to indole compound 167a operated via a radical chain mechanism (via bromine atom abstraction from 162a) or through electron transfer to Ru(II)* complex followed by deprotonation.
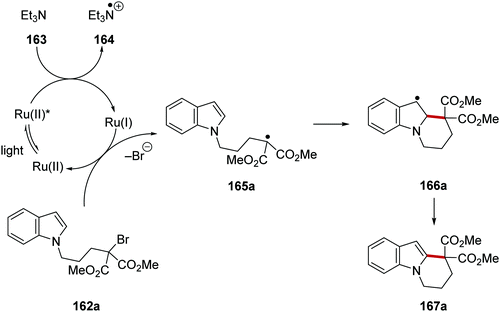 |
| | Scheme 37 Cyclisation onto indoles with Ru(bpy)3Cl2. | |
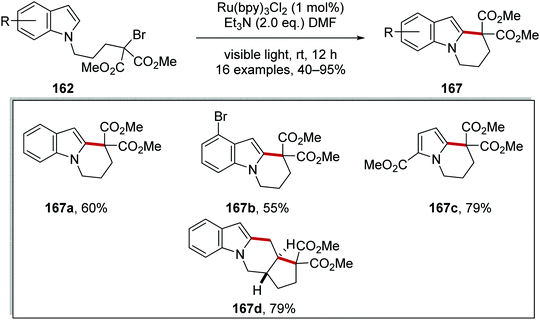 |
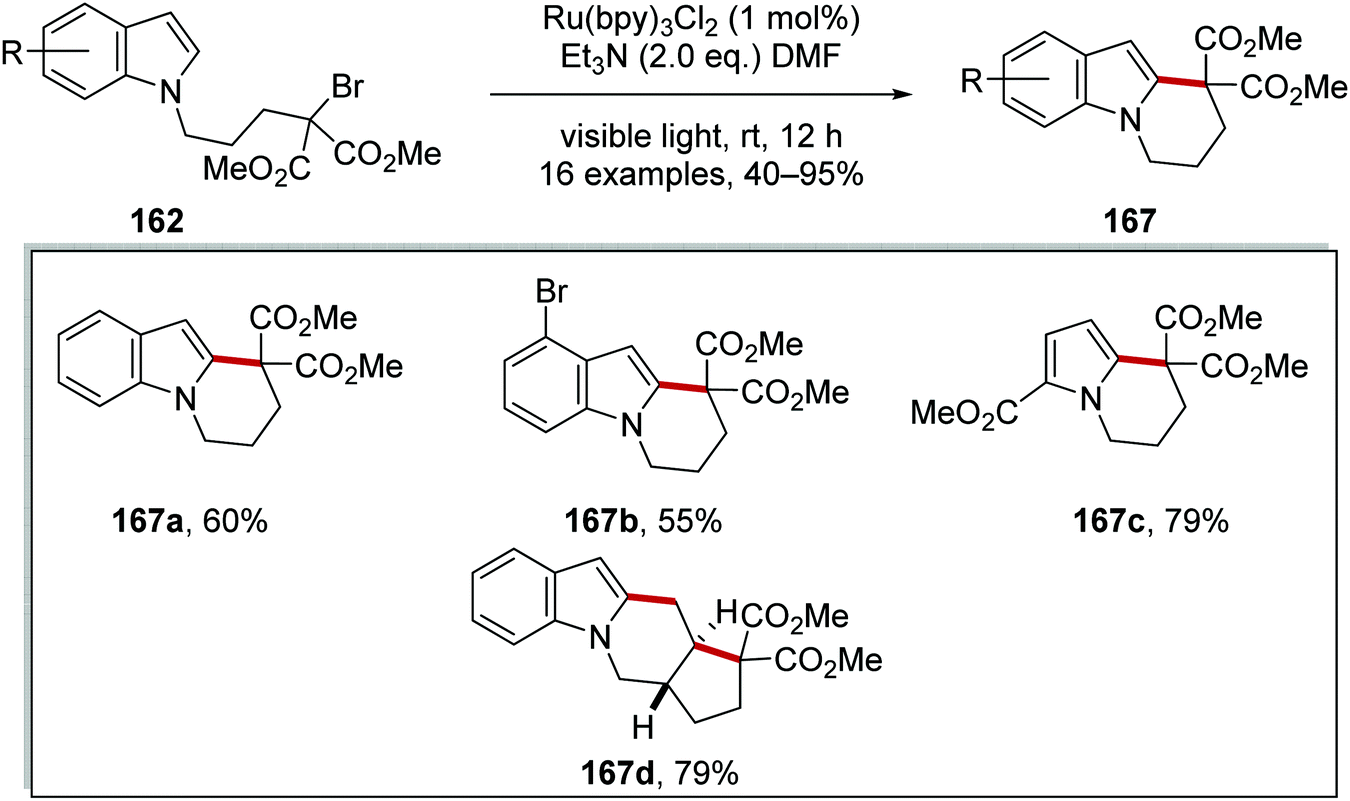
| | Scheme 38 Cyclisation of bromoalkylindoles and bromoalkylpyrroles. | |
An optimisation study was carried out where it was found that triethylamine was the optimal base to generate Ru(I) complex from Ru(II)*. Other bases tested such as DABCO and trimethylamine resulted in the loss of the bromine atom from the substrate but with no cyclisation. Under the optimised reaction conditions, tricycle 167a was isolated in 60% yield (Scheme 38). Thirteen other cyclised products were also prepared with yields ranging from 40% to 90%. These conditions were also applied to a radical cascade cyclisation giving tetracycle 167d in 79% yield.
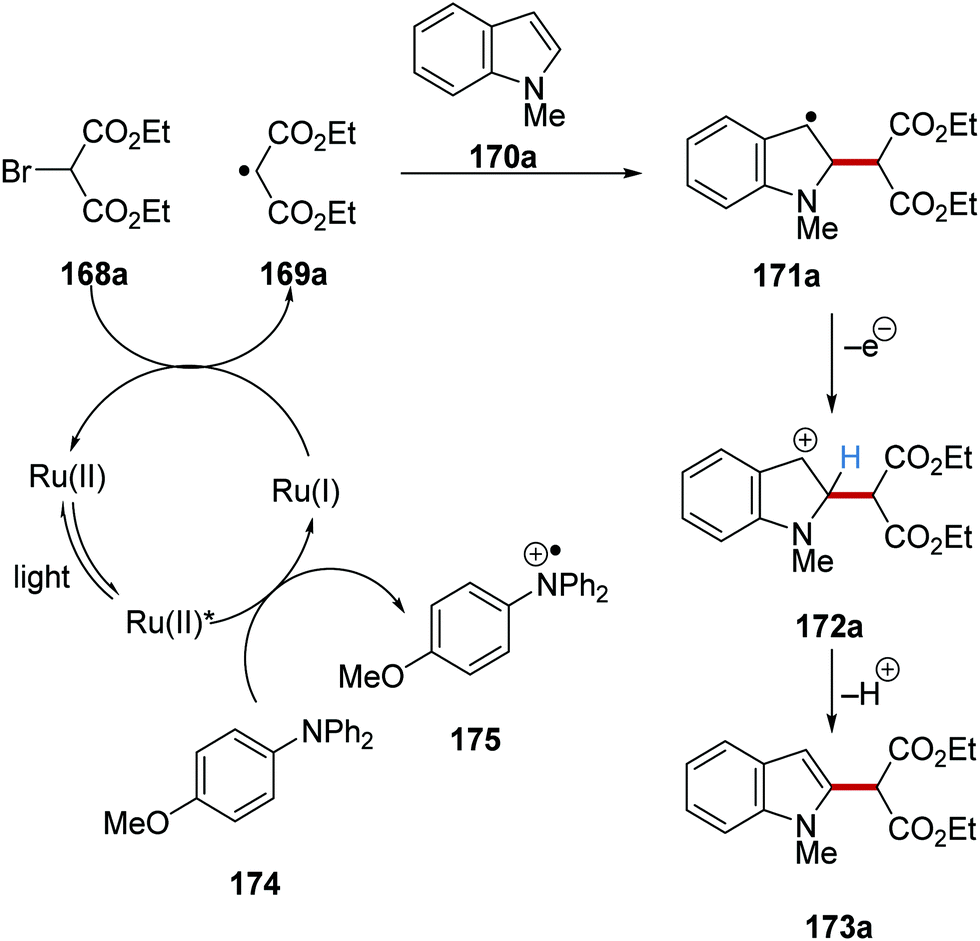
The C–H functionalisation of furan, pyrroles and indoles was achieved via a radical coupling strategy with diethyl bromomalonate (168a) as coupling partner.124N,N-Diphenyl-4-methoxyaniline (174) reduced the excited Ru(II)* complex to Ru(I) species with 175 being formed (Scheme 39). The strongly reducing Ru(I) reduced 168a, resulting in bromide loss and formation of alkyl radical 169a. This electron-deficient malonate radical then underwent coupling with electron-rich indole 170a, which provided benzylic radical 171a. The loss of an electron from 171a gave benzylic cation 172a, which, after proton loss, afforded the coupled product 173a.
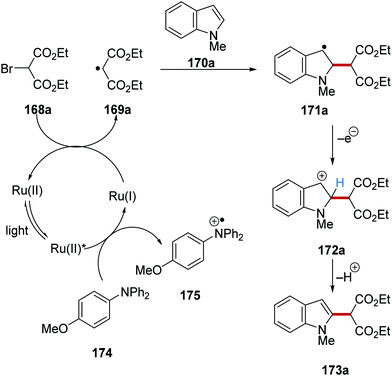 |
| | Scheme 39 Radical coupling of heterocycles with diethyl bromomalonate. | |
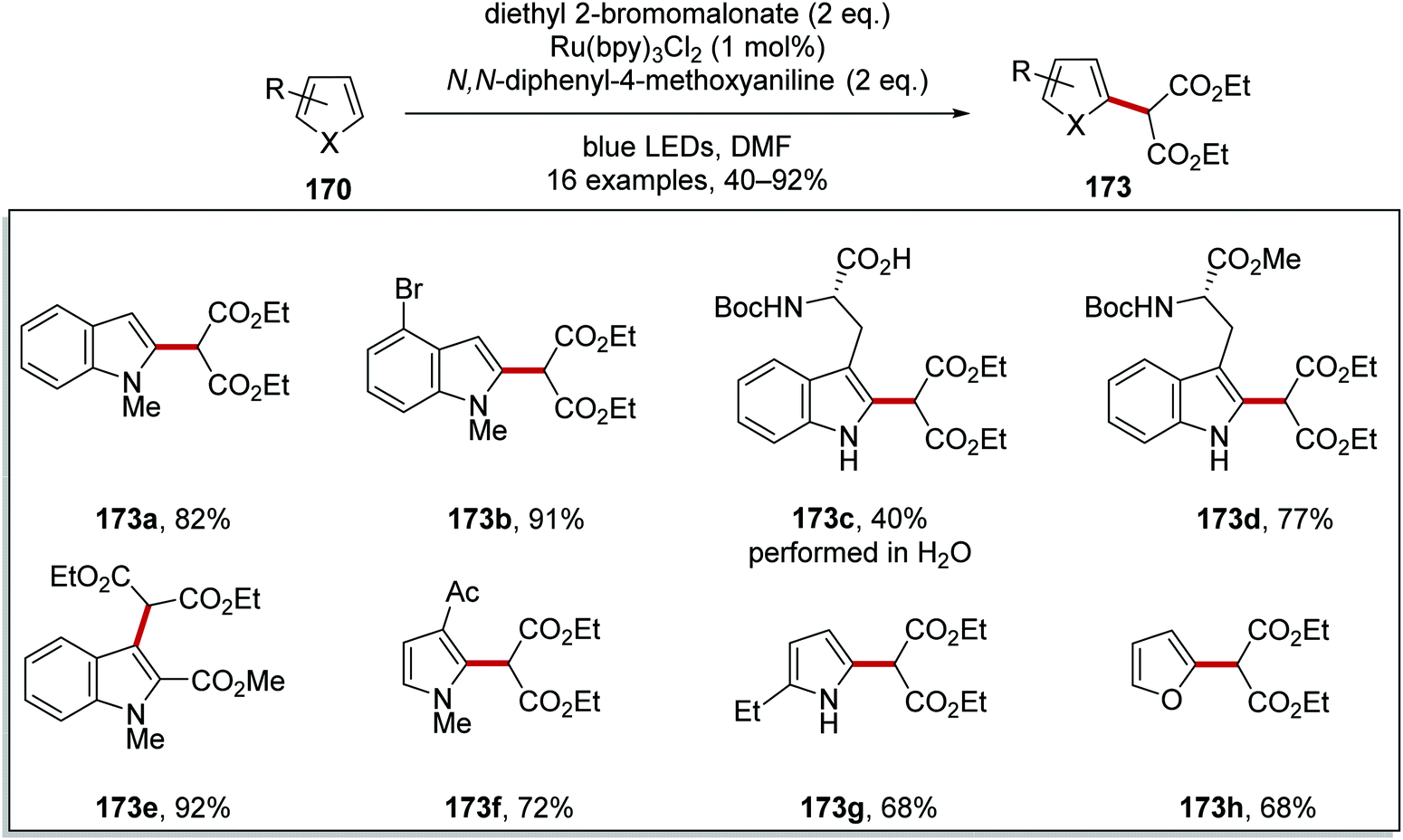
This reaction was used to prepare functionalised indoles 173a–e, pyrroles 173f and 173g and furan 173h analogues (Scheme 40). The reaction generally worked well although a low yield (40%) was obtained for tryptophan analogue 173c when the reaction was conducted in water.
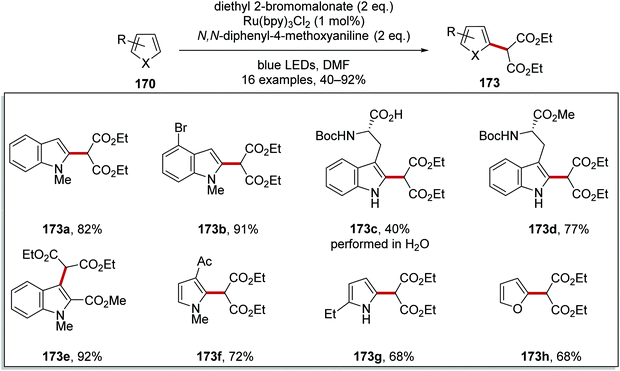 |
| | Scheme 40 Radical coupling of diethyl malonate to heteroarenes. | |
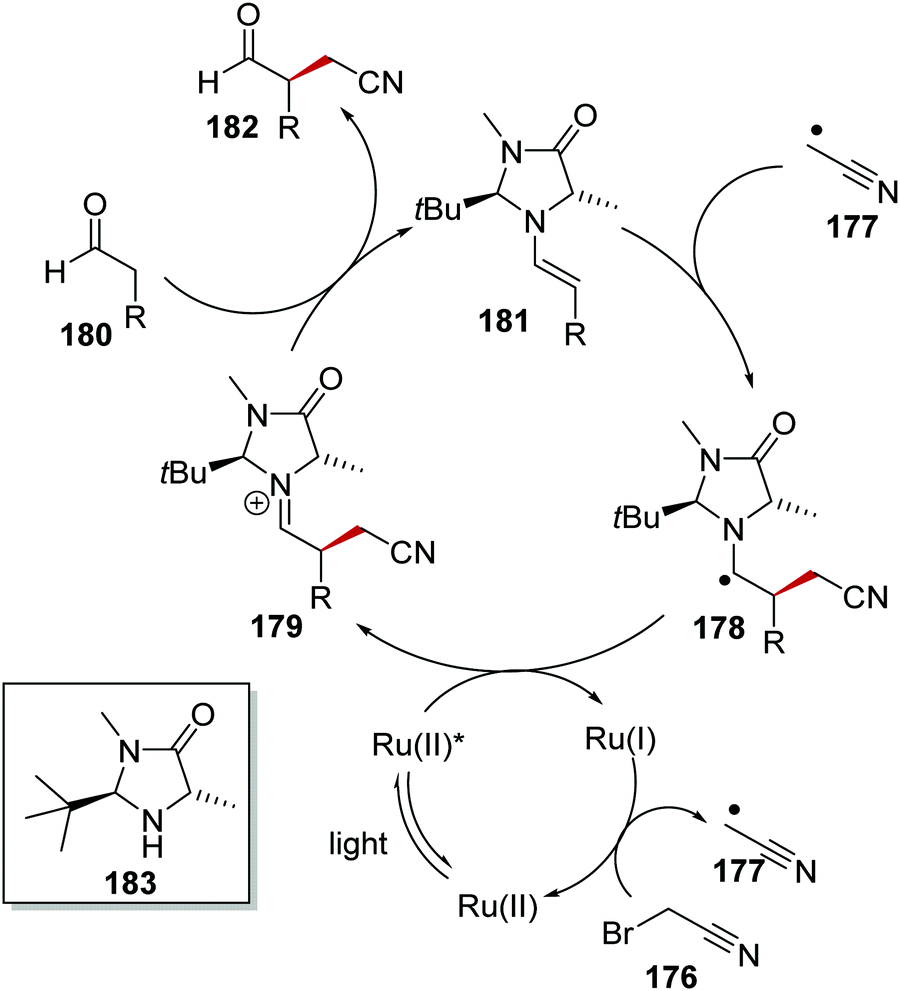
α-Functionalisation of aldehydes 180 with bromoacetonitrile (176) via C–H activation was an effective enantioselective transformation with a Ru photoredox and organocatalyst system.125 The reaction was proposed to go through the following reaction mechanism (Scheme 41). The reduced Ru(I) complex (E = −1.33 V vs. SCE) was able to donate an electron to nitrile 176 (Ered1/2 = −0.69 V vs. SCE)124 and this gave alkyl radical 177 and Ru(II) complex. Coincidentally, aldehyde 180 and organocatalyst 183 formed chiral electron-rich enamine adduct 181. Radical coupling between enamine 181 and radical 177 resulted in the formation of α-aminoalkyl radical 178. Subsequent single-electron oxidation with Ru(II)* resulted in the reformation of Ru(I) complex and iminium compound 179, hydrolysis of which gave enantioenriched product 182.
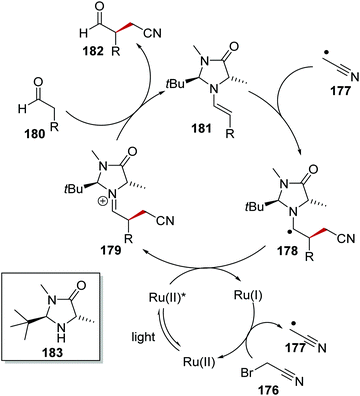 |
| | Scheme 41 Reaction mechanism between bromoacetonitrile (176) and aldehyde 180. | |
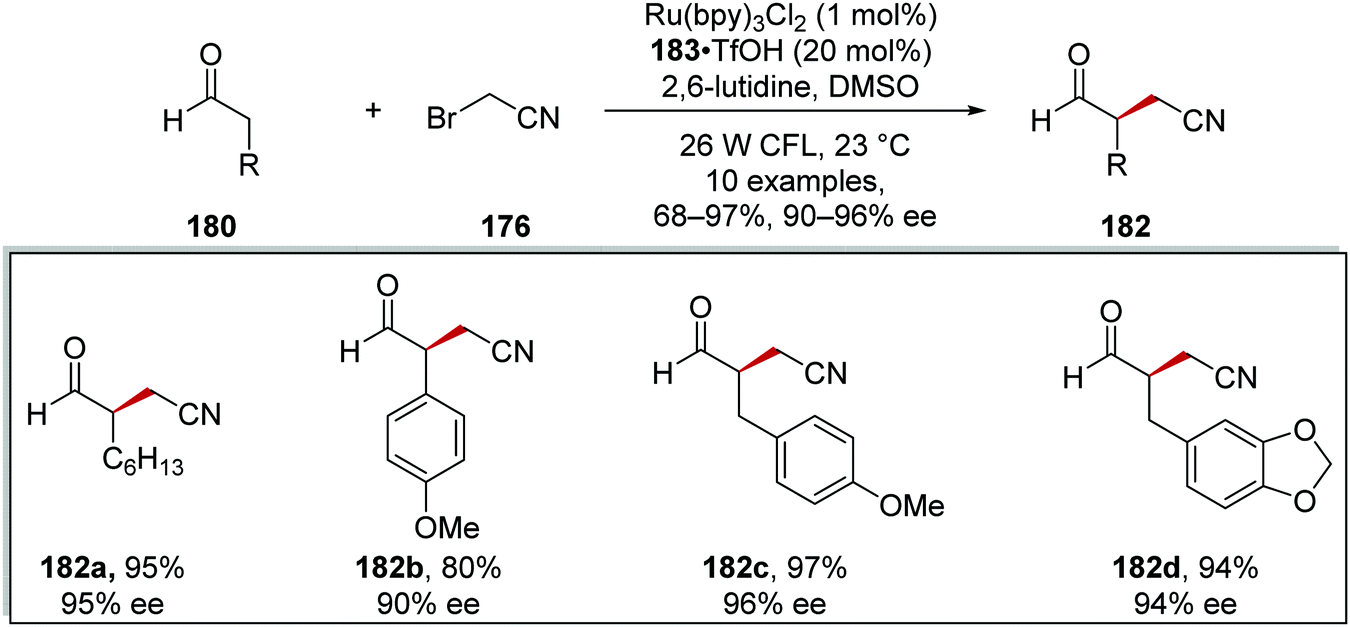
These conditions were used to generate a library of 10 compounds in 68–97% yields and with 90–97% ee. Aliphatic 182a, aromatic 182b and benzyl 182c–d substituents were all tolerated in the reaction and with a high yield and enantiomeric excess in each case, as shown in Scheme 42.
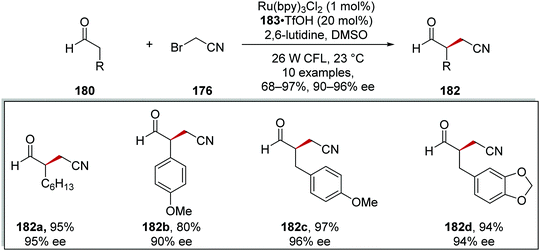 |
| | Scheme 42 Substrate scope for the coupling of aldehydes 180 and bromoacetonitrile (176). | |
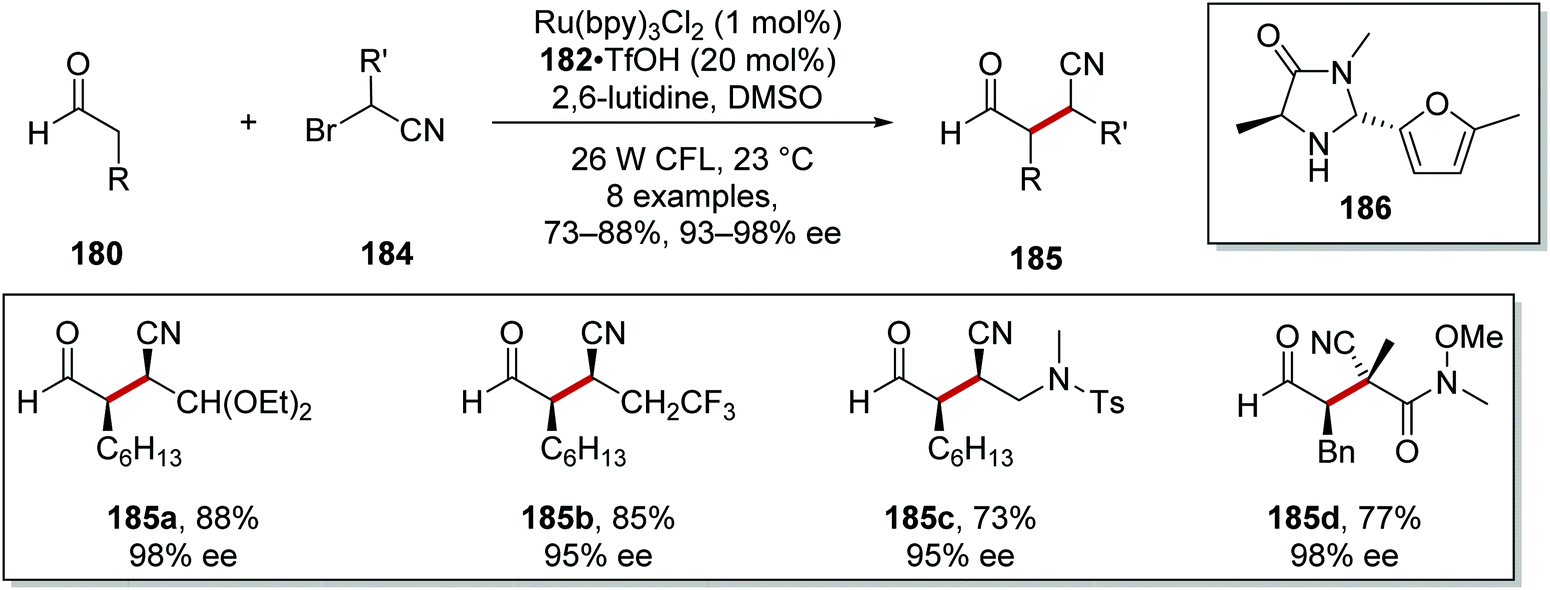
Substituted α-bromonitriles 184 were also employed under similar reaction conditions to afford nitrile products 185 with organocatalyst 186 (Scheme 43).125 Acetal 185a, trifluoromethyl derivative 185b and sulfonamide analogue 185c were all prepared under the optimised reaction conditions. All-carbon quaternary stereocentres with excellent enantiocontrol were also prepared, as seen in the formation of 185d with 98% ee.
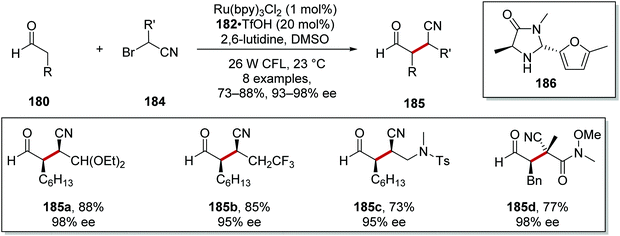 |
| | Scheme 43 Scope of the α-bromonitriles 184. | |
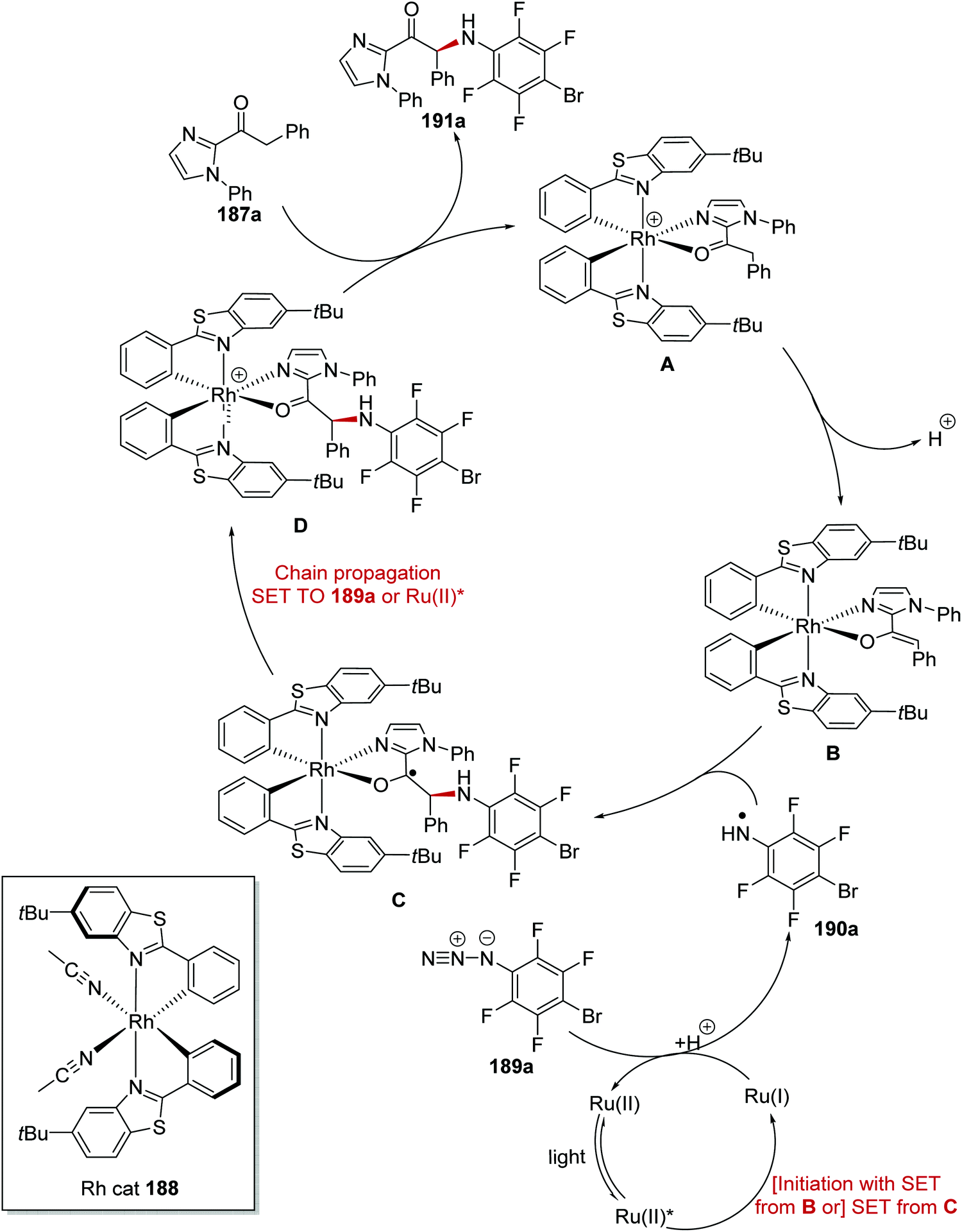
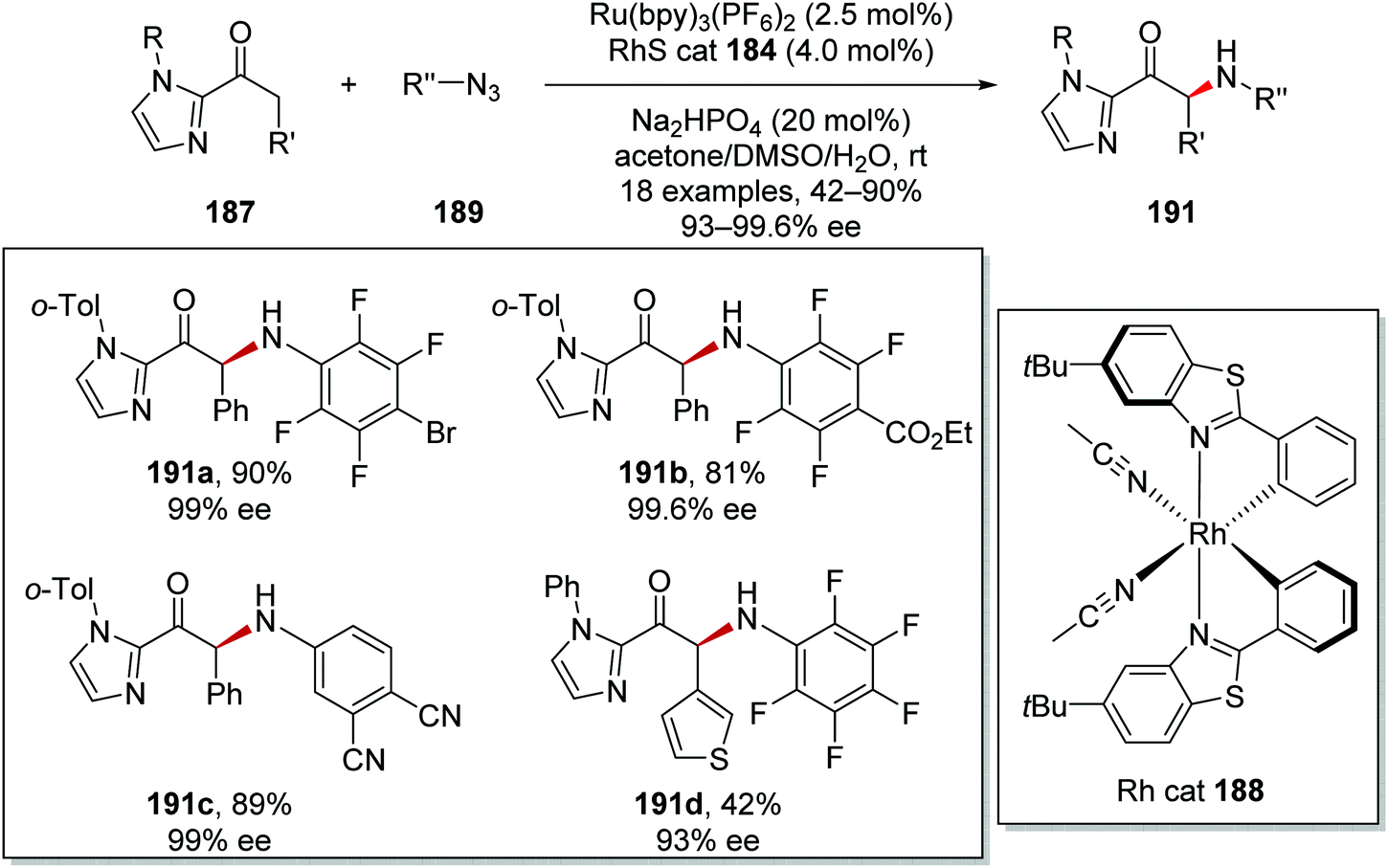
The asymmetric α-functionalisation of ketones 187 with azides 189 (Schemes 44 and 45) or diazoacetates 193 (Scheme 45) was achieved with a Ru/Rh catalytic system.126 Coordination of 187a to the Rh catalyst 188 gave Rh complex A and this allowed for base-promoted deprotonation, which gave complex B. Stern–Volmer studies it was shown that only the Rh-enolate complex could quench the excited Ru(II)* complex. Therefore, it was thought that an outer-sphere SET between B and Ru(II)* initiated the reaction with formation of the strongly reducing Ru(I) species. The Ru(I) complex reduced azide 189a giving anilinyl radical 190a after protonation. This mechanism was supported with cyclic voltammetry studies performed by the authors. Organic azide 189a has an irreversible reduction peak at −1.82 V vs. Fc/Fc+ which would not be reduced by the excited state of Ru(bpy)32+ (−0.81 V vs. SCE) but could feasibly be reduced by Ru(bpy)3+ (−1.33 V vs. SCE). Addition of 190a to electron-rich enolate B resulted in enantioselective C–N bond formation with ketyl radical C being formed. It was thought that the strongly reducing radical C could either participate in a radical-chain mechanism with reduction of 189a or SET with Ru(II)* complex. The loss of an electron from C resulted in cationic intermediate D. Dissociation of complex D gave the enantioenriched amine product 191a with the Rh catalyst being regenerated.
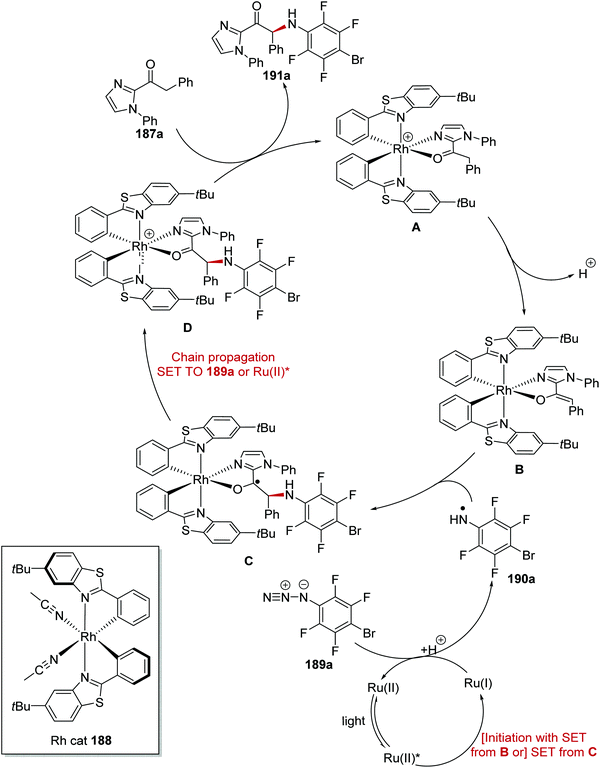 |
| | Scheme 44 Stereocontrolled coupling of azide 189a to ketone 187a. | |
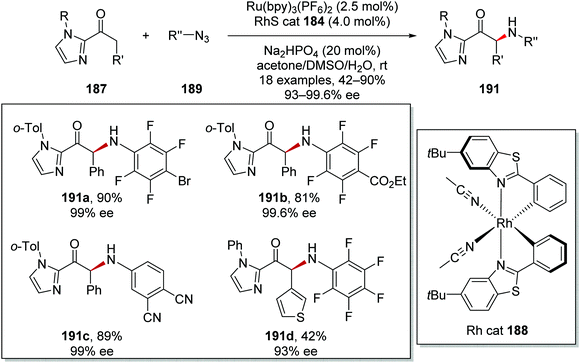 |
| | Scheme 45 Scope of products from Ru/Rh tandem catalytic coupling of ketones 187 with azides 189. | |
This methodology allowed for the preparation of 18 enantioenriched anilines 191 (Scheme 45). High yields and ee were recorded for bromo 191a, ester 191b and nitrile 191c analogues. The reaction was tolerant of heterocyclic motifs; for example, thiophene analogue 191d was isolated in 42% yield.
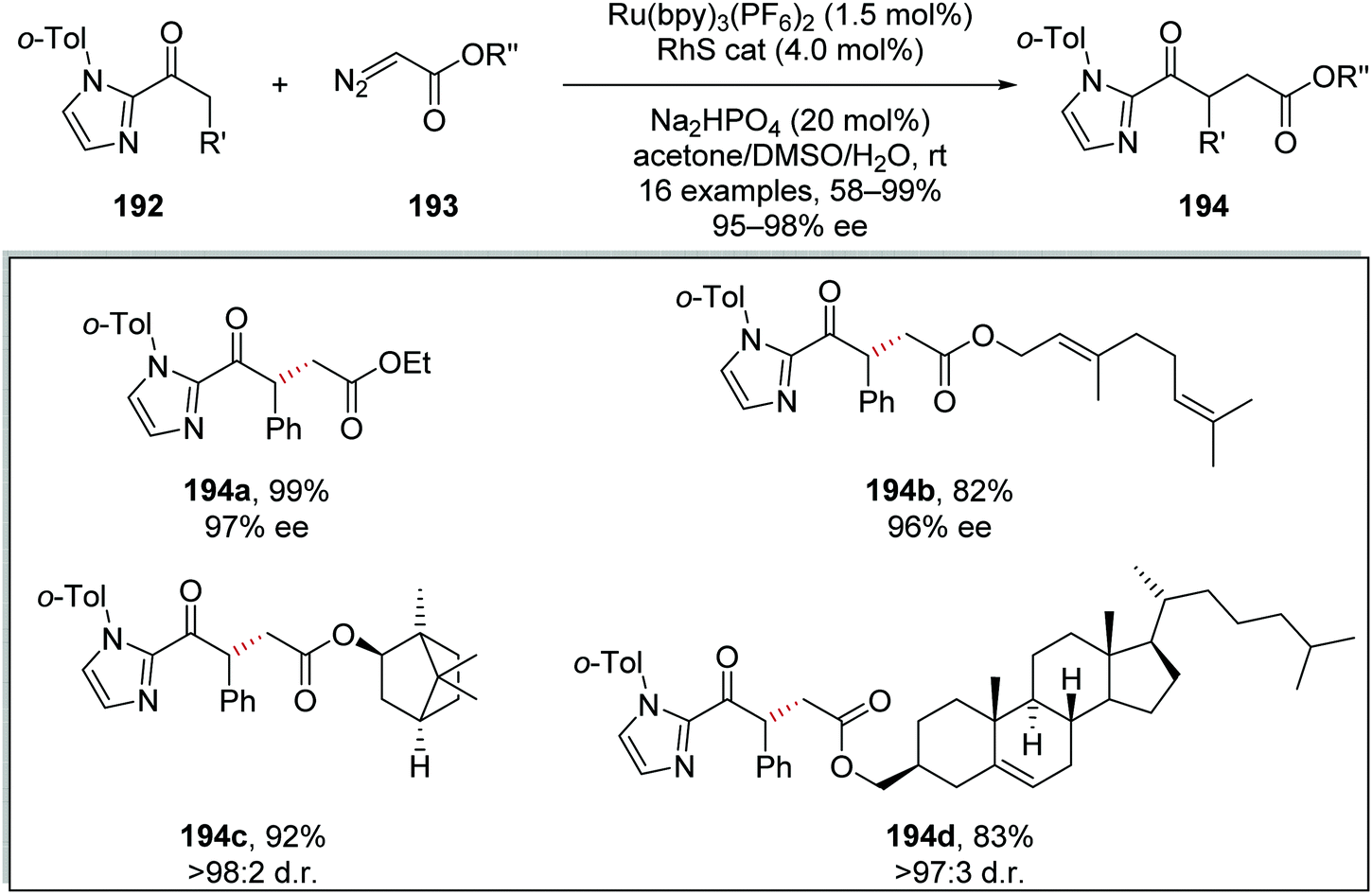
Diazoacetates 193 were coupled to 2-acylimidazoles 192 using the same Ru/Rh catalytic system (Scheme 46). This was exemplified by the preparation of 16 coupled products. Ethyl ester 194a was prepared in 99% yield and with 97% ee using this methodology. Additionally, a geraniol derivative 194b, L-(−)-borneol derivative 194c and a cholesterol derivative 194d were all prepared in the yields stereoselectivities shown in Scheme 45.
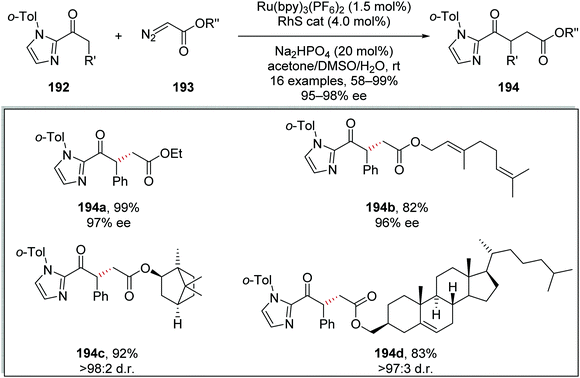 |
| | Scheme 46 Substrate scope of Ru/Rh tandem catalytic coupling of ketones 192 to diazoacetates 193. | |
2.2 Catalytic cycles featuring Ru(II)/Ru(III)
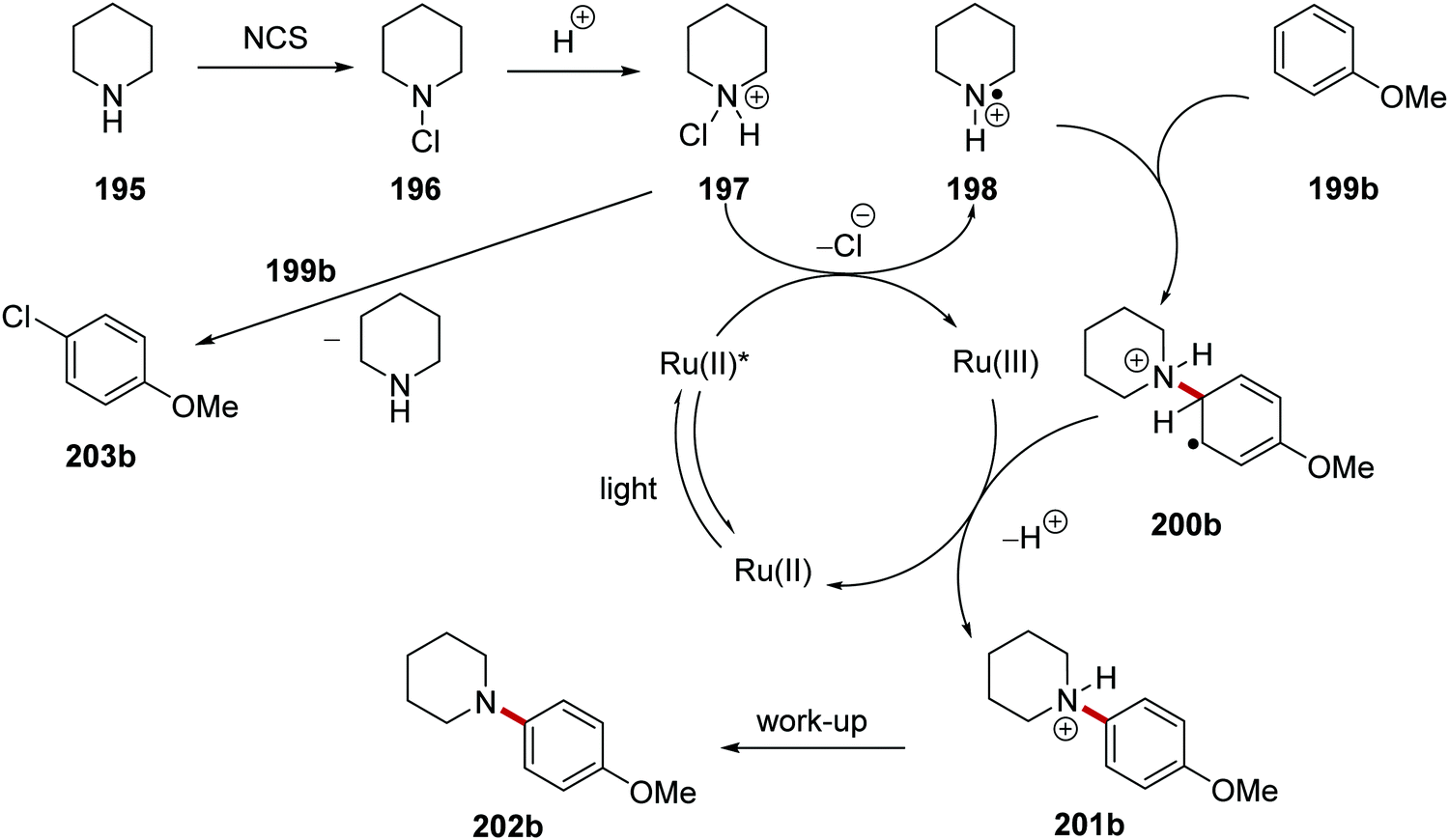
The coupling of secondary amines to arenes was achieved with the ruthenium photocatalyst in an HLF-inspired reaction (Scheme 47).127 The chlorination and protonation of amine 195 gave the compound 197. This salt received an electron from excited Ru(II)(bpy)3(PF6)2* complex and this gave amine radical cation 198 and Ru(III) complex. It was clearly demonstrated by the authors that SET would only occur with protonated salt 197 and not with N-chloropiperidine (196). Cyclic voltammetry studies found that 196 (Ered = −1.80 V vs. SCE, in MeCN) was unable to be reduced by Ru(II)* (Eox* = −0.81 vs. SCE) but SET was very feasible with 197 (Ered = +0.43 V vs. SCE). Addition of the electron-poor radical 198 to electron-rich arene 199b resulted in para-substituted radical 200b, which was oxidised by Ru(III) forming anilinium salt 201b following loss of one proton.
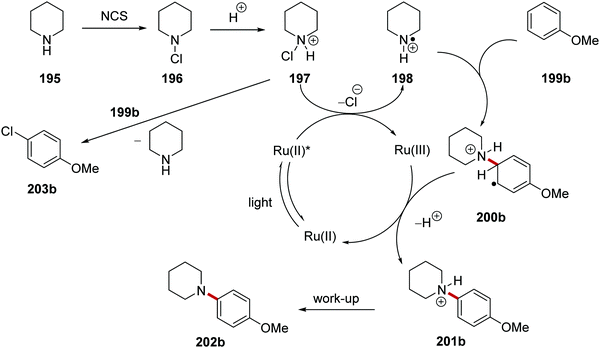 |
| | Scheme 47 Aryl coupling of secondary amines. | |
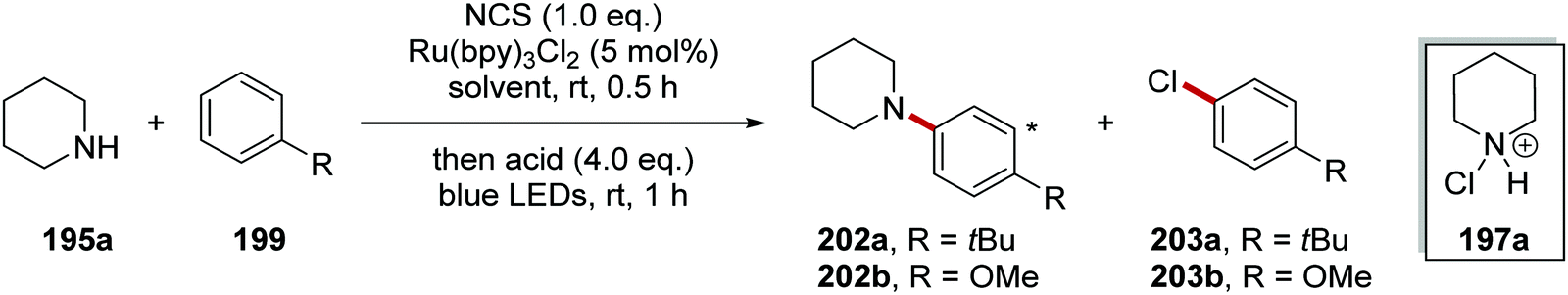
N-Chloroamine salt 197 is well known to chlorinate nucleophiles through SN2 attack at the Cl atom (i.e. a non-radical mechanism). Therefore, it was crucial to develop reaction conditions that would exclusively produce the desired C–N product 202 and not the unwanted C–Cl product 203 (Table 2 and Scheme 48). This would require that the species 197 is susceptible to rapid reduction to 198 by the photoredox agent and also that the radical 198 does not lose a proton to become a neutral aminyl radical (this would be far less reactive towards arenes). Therefore the acidity of the reaction medium is crucial. Successful amination also depends on the nucleophilicity of the arene. If this is too nucleophilic, then it may attack 197 before the transformation to 198 can occur.
Table 2 Reaction optimisation, n.d. = not detected (where relevant, asterisk * indicates the alternative site of attachment of the amine)
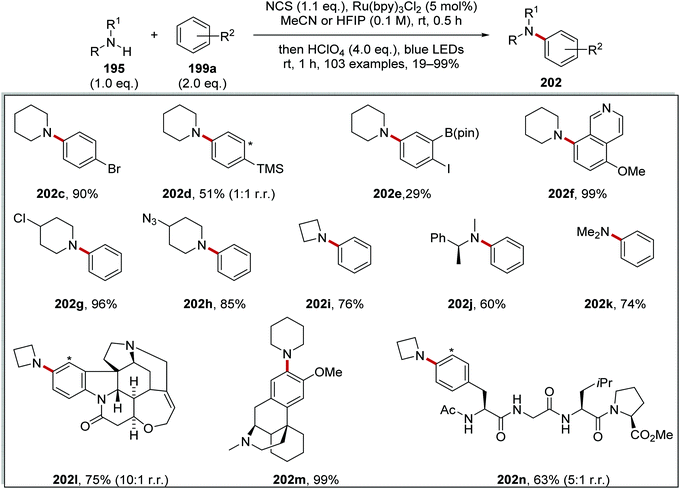 |
| | Scheme 48 Substrate scope for secondary amines coupling to arenes. (Asterisk * indicated alternative site for incoming group.) | |
To study the reaction, tert-butylbenzene 199a and anisole 199b were chosen as substrates of moderately and highly electron-rich arenes, respectively. A range of different acids and solvents was tested to ascertain the best conditions for the formation of 202. Initially, the use of acetic acid (entry 1, Table 2) gave no aniline or aryl chloride products for both substrates. The use of TFA (entry 2) gave no product formation for 202b but it did provide 4-chloroanisole 203b in 17% yield. p-TsOH (entry 3) gave unwanted product 203b in 90% yield. When the superacid, perchloric acid, was used (entry 4) this gave exclusive aniline formation for both substrates with 202a in 71% yield and 202b in 94% yield. Further investigation demonstrated that using HFIP as solvent (entry 5) showed interesting but complex results; thus it gave an increased yield of 98% for 202a relative to MeCN. Furthermore, HFIP allowed for the more convenient acid, TFA, to be used as the acid for moderately electron-rich arene substrates, with 202a being prepared in 88% yield.
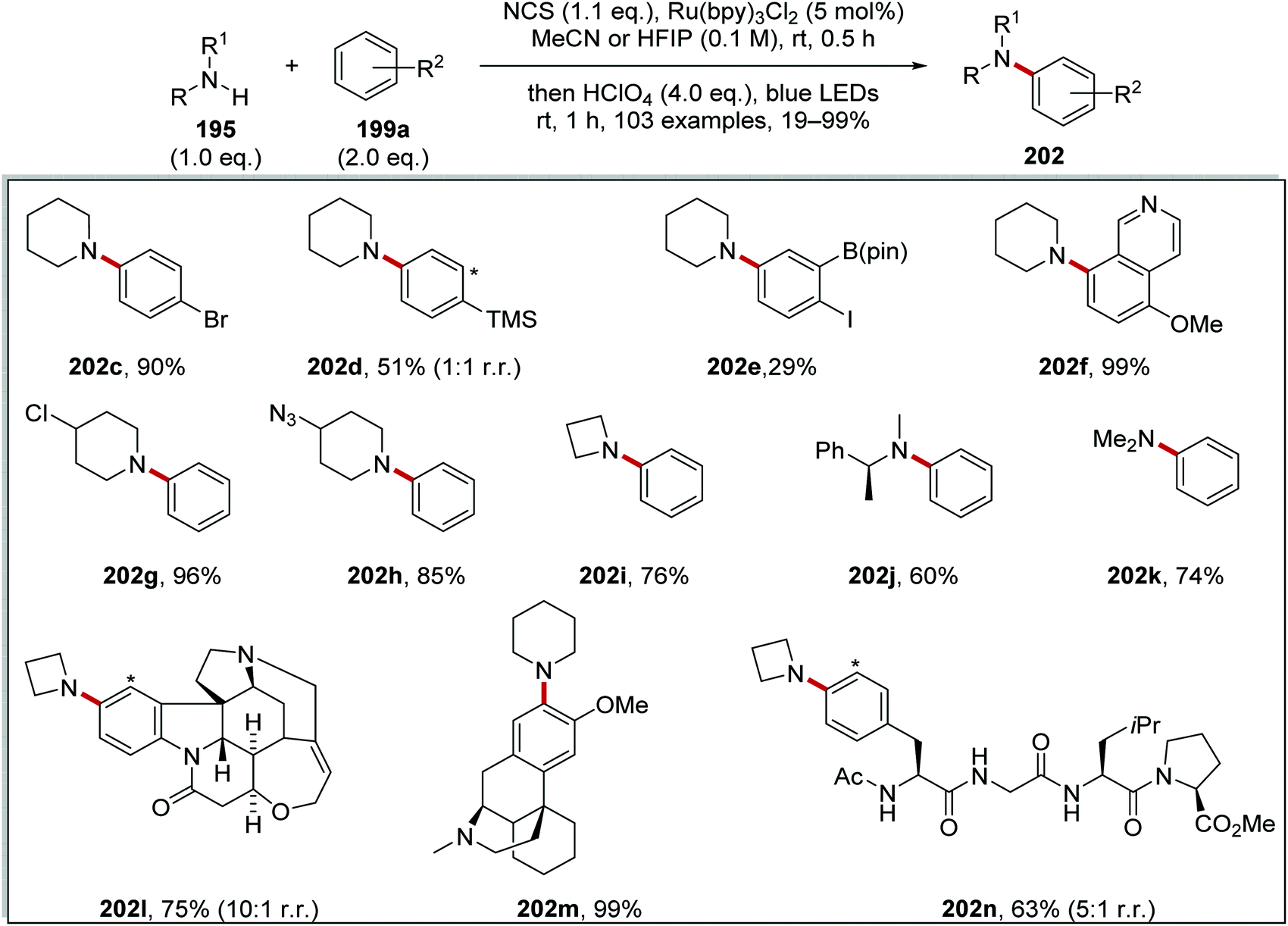
The optimised reaction conditions were used to investigate the scope of the reaction. The reaction accommodated a wide range of substrates, with the preparation of 103 coupled amine compounds (Scheme 48 for selected examples). With piperidine as the amine, various functionalised aryl compounds were employed giving coupled compounds in 21–99% yields, e.g.202c–f in Scheme 48. An assorted selection of secondary amines provided successful substrates for this reaction giving products in 40–99% yields, e.g.202g–k. The reaction also found a use in late-stage functionalisation of complex compounds; this resulted in the isolation of bioactive derivatives 202l–n in the yields shown.
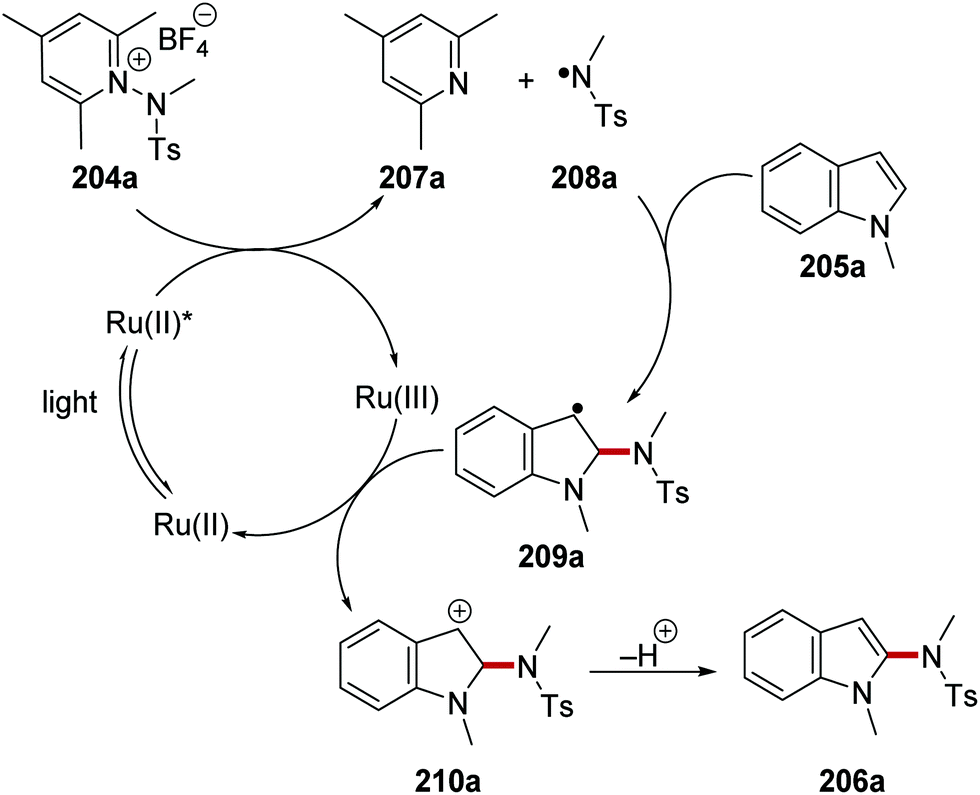
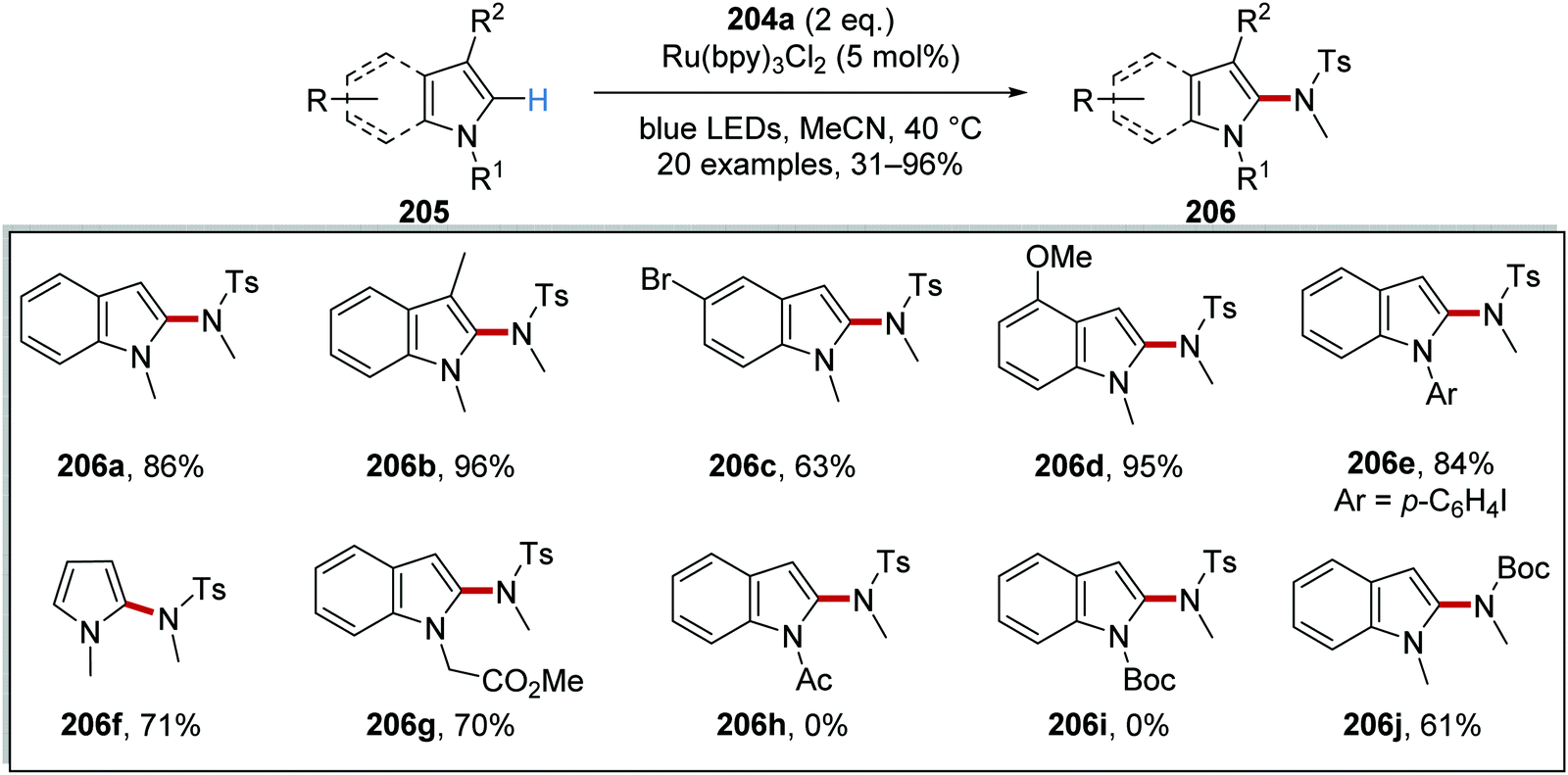
N-Centred radicals were formed from reaction of N-sulfonamidopyridinium salts with Ru(bpy)3Cl2 photocatalyst.128 These N-centred radicals were then used to functionalise arenes and heterocycles without any pre-functionalisation. For example, N-sulfonamidopyridinium 204a was used to functionalise N-methylindole 205a and this produced sulfonamide 206a (Scheme 49). Irradiation of the reaction mixture with blue light yielded excited Ru(II)* complex from the original Ru(bpy)3Cl2 complex. SET from Ru(II)* to 204a resulted in N–N bond breakage which led to 2,4,6-collidine (207a) and nitrogen-centred radical 208a. The excited photocatalyst Ru(bpy)32+ (Eox* = −0.81 V vs. SCE)8b was able to reduce pyridinium salt 204a (Ered = −0.70 V vs. Ag/Ag+).128 Regioselective addition of 208a to N-methylindole (205a) resulted in carbon-centred radical 209a. A second electron transfer to Ru(III) from 209a closed the ruthenium catalytic cycle while cation 210a was formed. Proton loss gave sulfonamide 206a as the observed product.
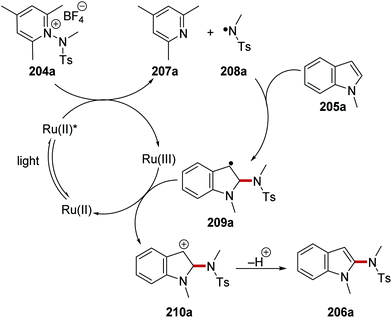 |
| | Scheme 49 Generation of sulfonamidyl radicals from pyridinium derivatives. | |
Alongside sulfonamide 206a which was isolated in 86% yield, 19 other functionalised compounds were prepared with this methodology (Scheme 50). A range of indole functionalised compounds was used as substrates and yielded sulfonamides 206b–d. N-Aryl indoles were amenable under these conditions and aryl iodide 206e was produced in 84% yield. When N-methylpyrrole was used in the reaction, compound 206f was isolated in 71% yield. However, the substituent upon the nitrogen atom of the indole heterocycle had a significant influence upon the efficiency of the reaction. While ester 206g was formed in 70% yield, the use of N-Boc and N-Ac indole gave no reaction, presumably due to ring deactivation. Finally, the synthetically useful N-Boc derivative 206j was prepared in 61% yield when a Boc analogue of 204a was used.
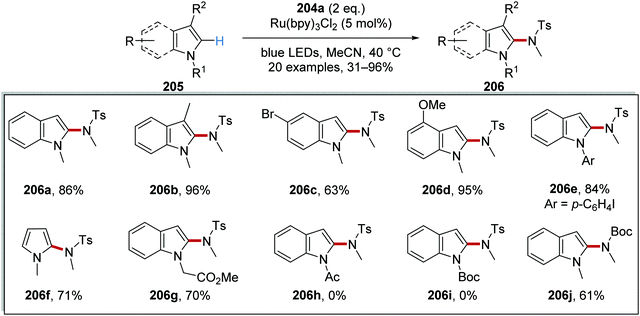 |
| | Scheme 50 Substitution of indoles and pyrroles. | |
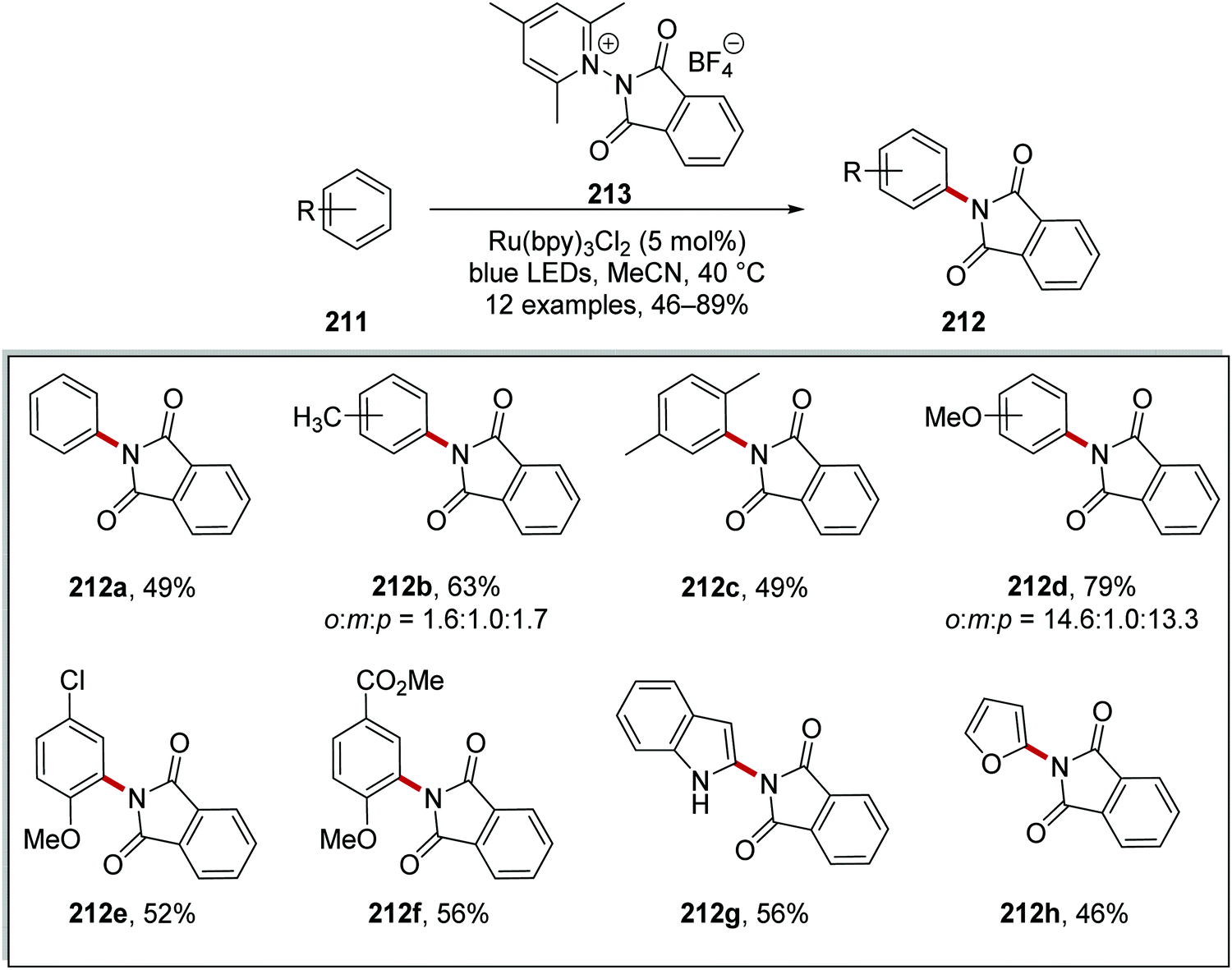
Furthermore, phthaloyl reagent 213 was used to prepare imides 212 from arenes 211 when used with Ru(bpy)3Cl2 (Scheme 51). The reaction was very successful with 12 imide compounds being prepared in 46–89% yield, although compounds 212b and 212d were isolated as a mixture of regioisomers. The reaction also tolerated heterocycles, and furan product 212h was prepared in 46% yield.
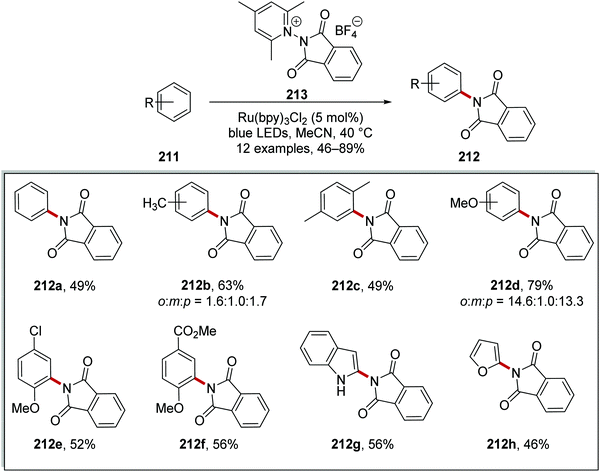 |
| | Scheme 51 Phthalimidation of arenes. | |
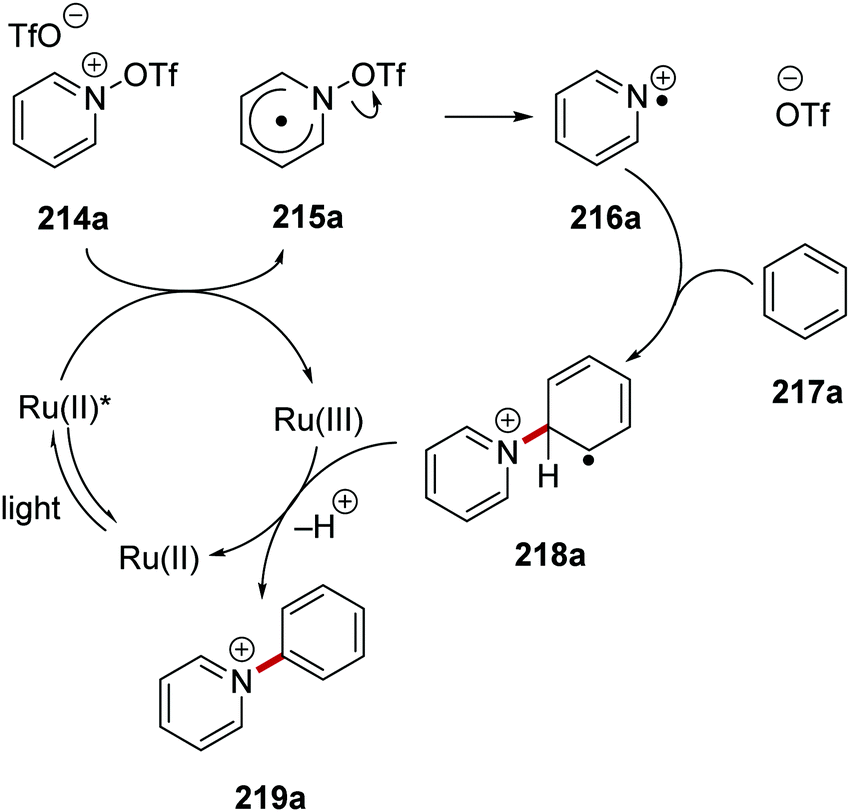
The catalyst Ru(bpy)3(PF6)2 was used as an electron donor in the generation of pyridyl radical cations 215 from N-(trifluoromethylsulfonyloxy)pyridinium salt 214.129,130 Irradiation of the reaction mixture with blue light led to excited Ru(II)* species (Scheme 52). SET gave pyridyl radical 215a (Ered = −0.14 V vs. SCE)130 and a Ru(III) complex from Ru(II)* (Eox* = −0.81 V vs. SCE)8b species and triflate salt 214a. Heterolytic N–O bond cleavage in 215a formed triflate anion and pyridyl radical cation 216a. Addition to arene 217a formed the key C–C bond giving radical 218a. The ruthenium catalytic cycle was closed with SET to Ru(III) complex from 218a, this gave the original Ru(II) complex and, following proton loss, pyridinium 219a.
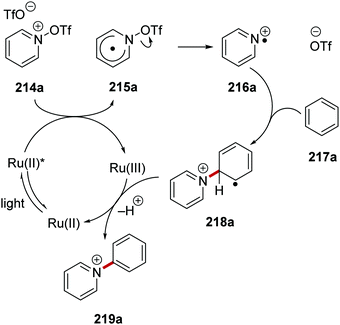 |
| | Scheme 52 Formation of pyridine radical cation from 214a, and subsequent coupling. | |
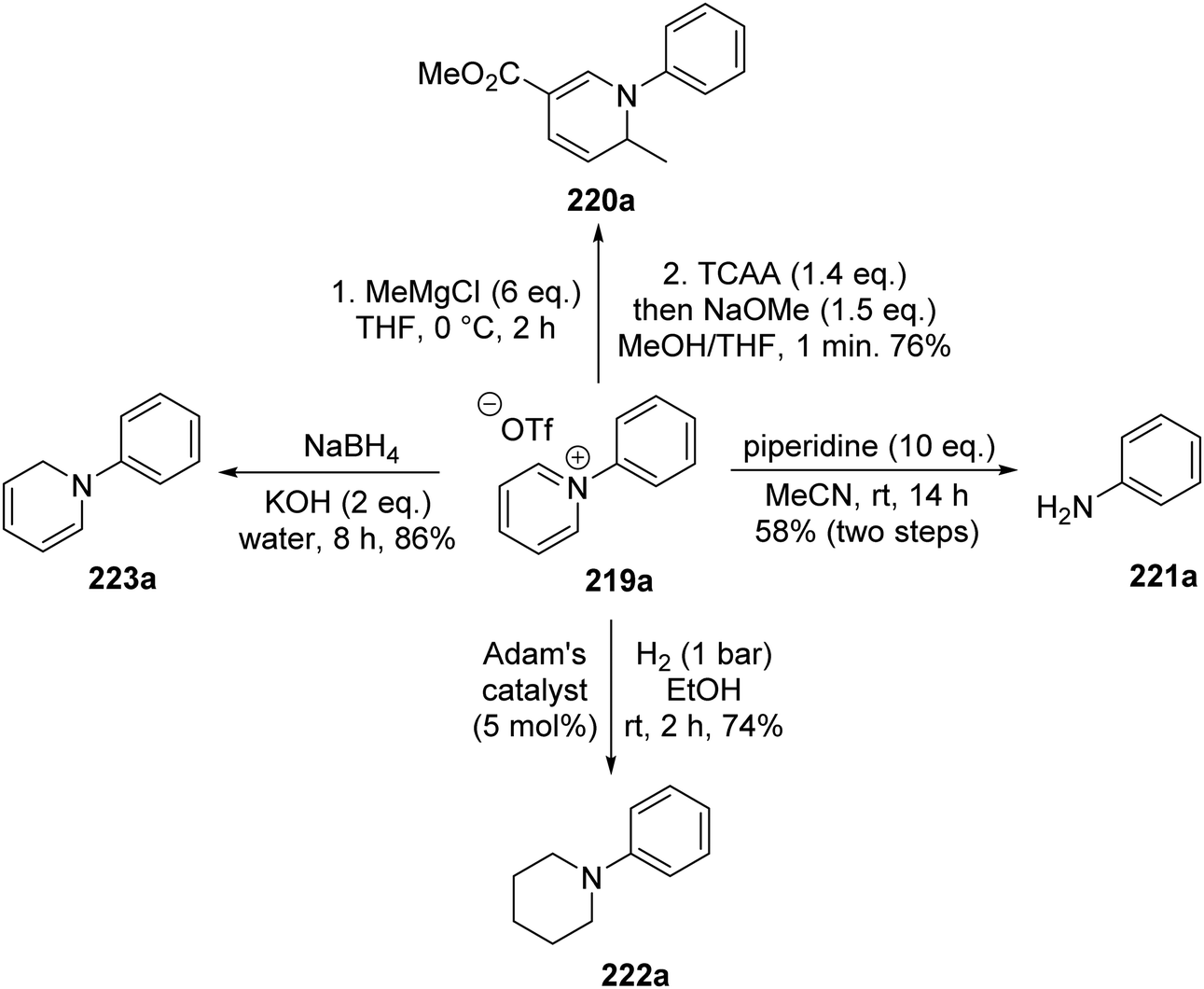
The pyridinium salts 219 were utilised in a range of different reactions (Scheme 53).129 Methylmagnesium chloride led to selective C2 alkylation and the combination of trichloroacetic anhydride and then sodium methoxide gave ester 220a in 76% yield. Treatment of 219a with piperidine resulted in aniline 221a being isolated in 58% yield from triflate 214avia a Zincke reaction. N-Arylated piperidine 222a was produced in 74% yield after 219a was exposed to hydrogenation conditions with Adam's catalyst (PtO2). Diene 223a was the product following treatment of 219a with sodium borohydride in 86% yield.
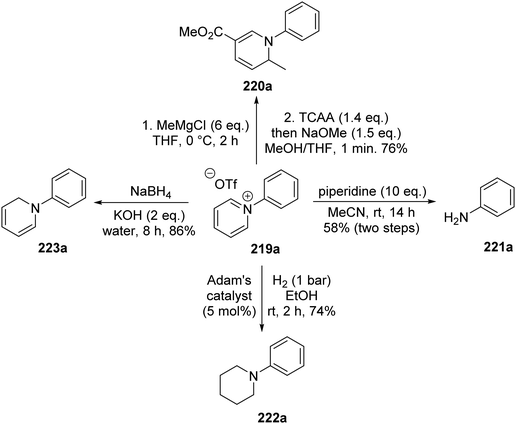 |
| | Scheme 53 Transformations of pyridinium salt 219a. | |
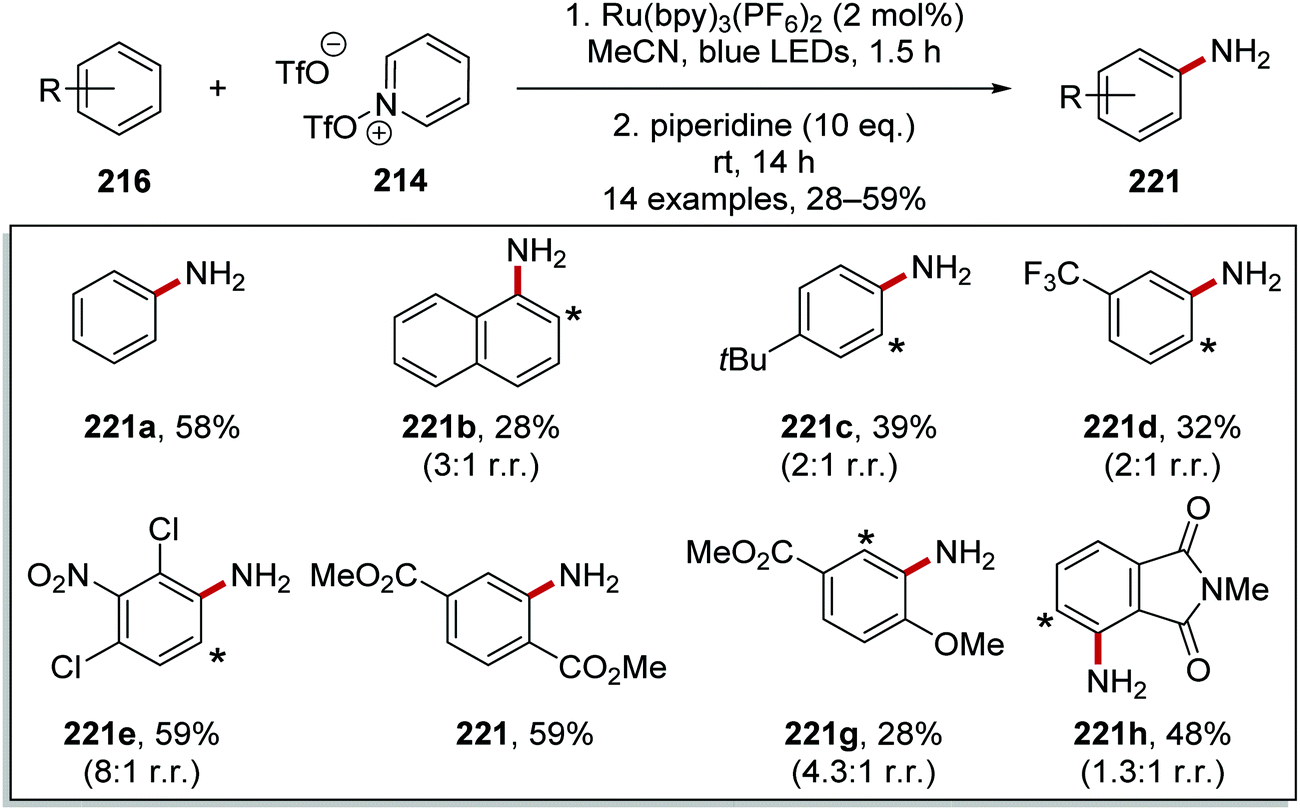
Typical conditions for this transformation included the use of Ru(bpy)3(PF6)2 (2 mol%) in MeCN and irradiation with blue light.129 The pyridinium products were converted in situ to anilines with use of piperidine (10 eq.) and this allowed for the preparation of 14 anilines 221 in yields ranging from 28% to 59% (Scheme 54).
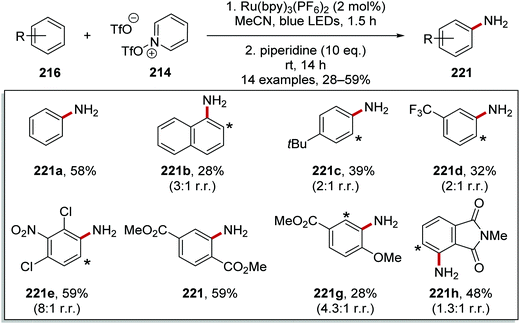 |
| | Scheme 54 Synthesis of anilines 221 from arenes 216. (Asterisk * marks alternative site of attachment.) | |
2.3 Ru energy transfer reactions
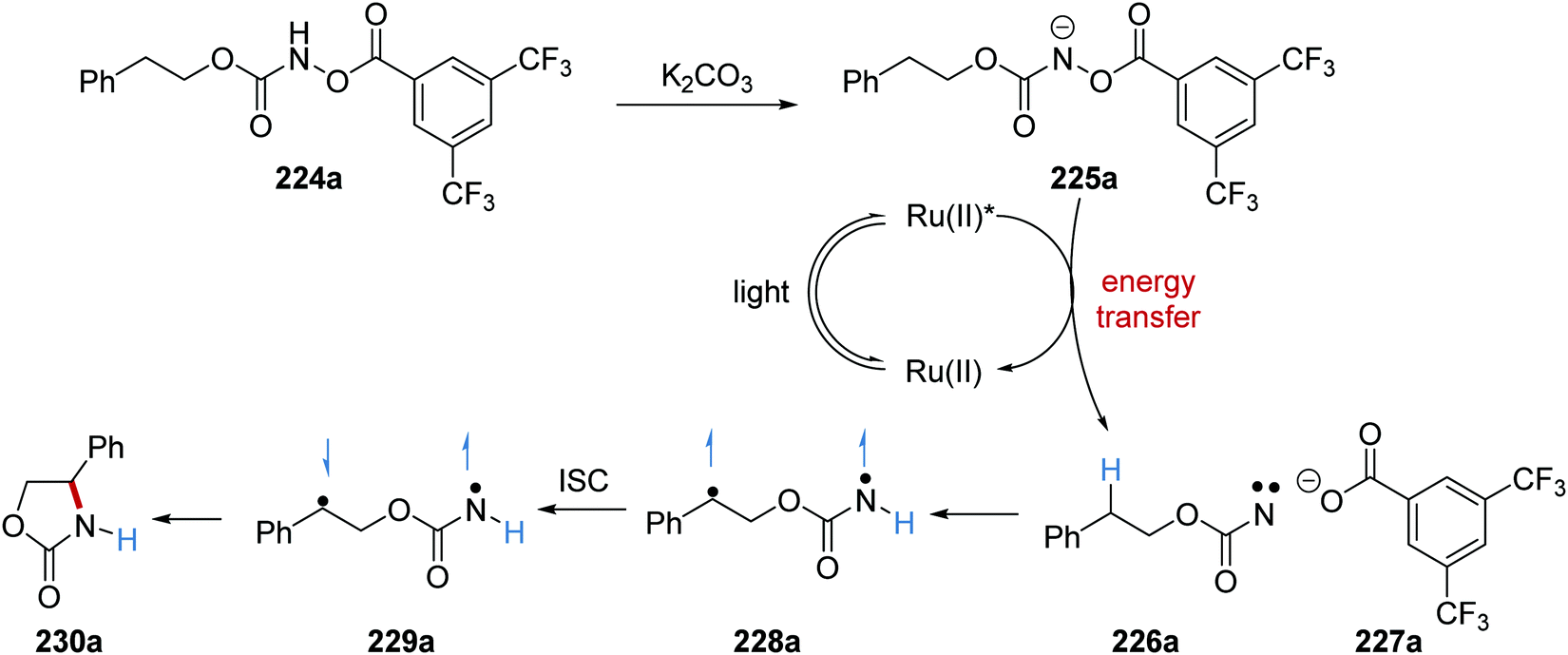
Formation of oxazolidinone 230 was achieved with an energy transfer strategy from carbamate 224via nitrene intermediate under basic conditions.131 Carbamate 224a was deprotonated with potassium carbonate and this gave anion 225a (Scheme 55). The Ru(bpy)3(PF6)2 catalyst was excited with blue light. An energy transfer to 225a (ET = 40.1 kcal mol−1) from Ru(II)* (ET = 47.7 kcal mol−1) resulted in N–O heterolytic bond cleavage and nitrene 226a and carboxylate 227a were formed. This bond cleavage was supported by DFT calculations. An intramolecular HAT to the nitrene in 226a resulted in triplet diradical 228a. ISC delivered singlet diradical 229a from 230a, and subsequent radical coupling gave lactam 230a.
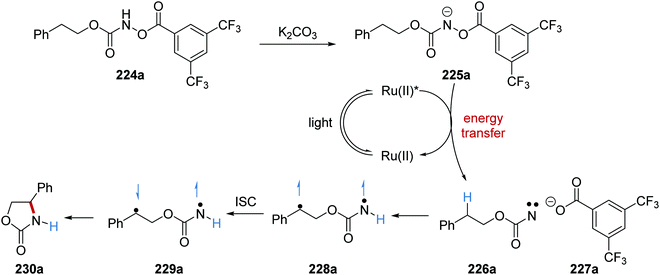 |
| | Scheme 55 Oxazolidinone ring formation from carbamate 224a. | |
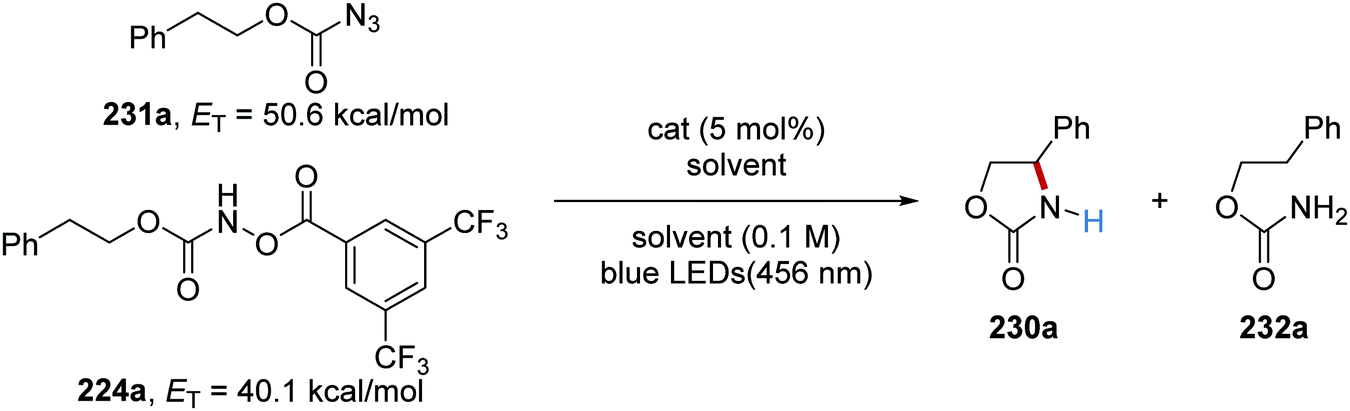
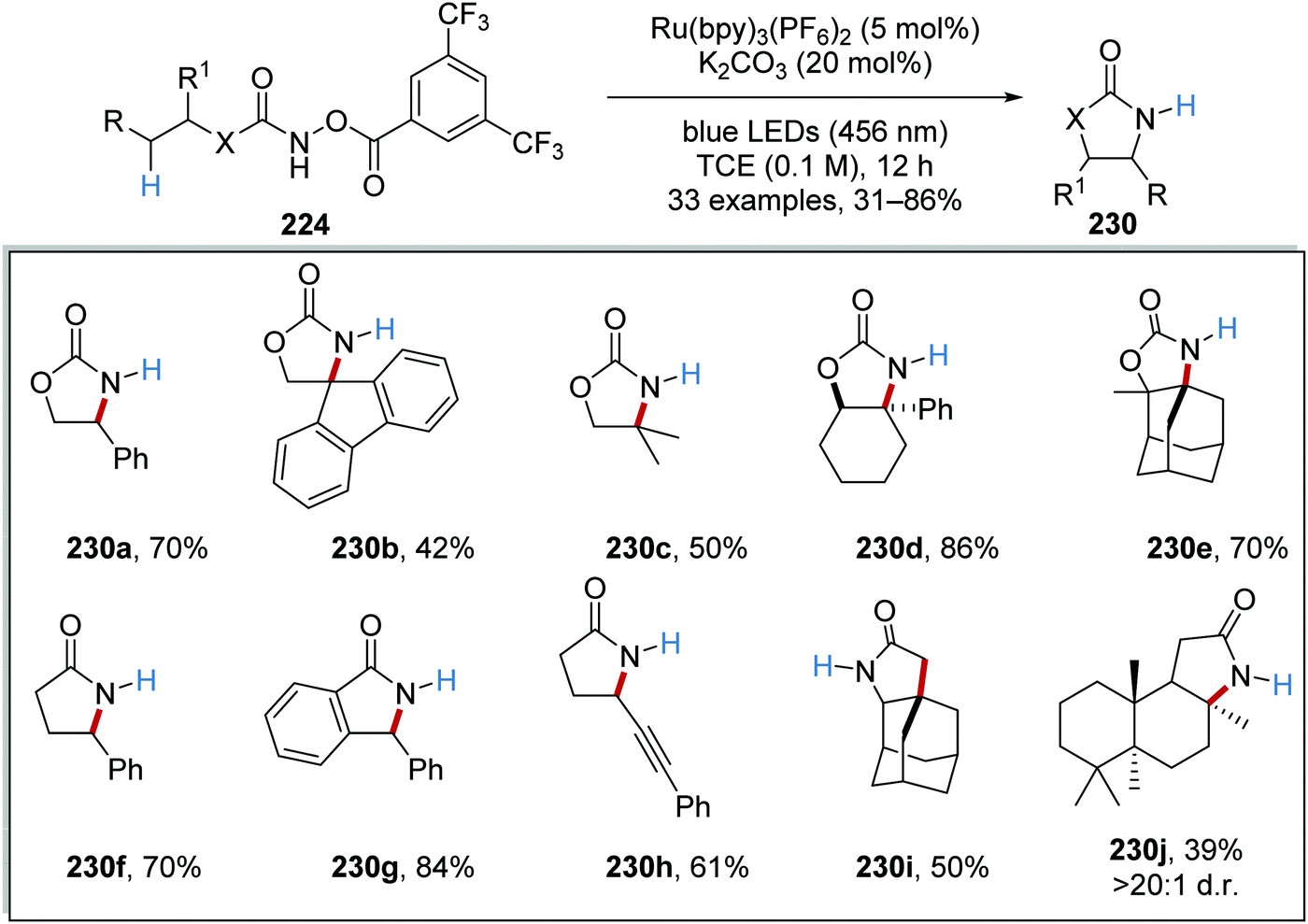
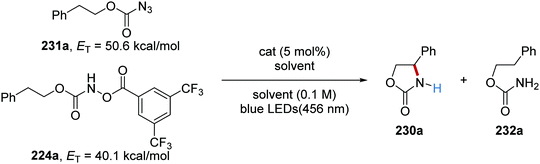
The reaction was initially trialled with azidoformate 231a which had a triplet energy of 50.6 kcal mol−1 (Table 3). However, the use of commercial iridium and ruthenium photoredox catalysts resulted in either no formation of oxazolidinone 230a or trace amounts (Table 3, entries 1–4), with carbamate 232a being formed instead. It was postulated that the poor efficiency of this reaction was due to incompatible triplet sensitisation. Therefore, other nitrene precursors were examined, such as 224a, which had triplet energy of 40.1 kcal mol−1 and a smaller singlet reorganisation energy. The combination of 224a and the Ru(bpy)3(PF6) photocatalyst gave desired oxazolidine 230a in 70% isolated yield.
Table 3 Optimisation of oxazolidinone formation
|
|
| Entry |
Substrate |
Catalyst |
E
T, kcal mol−1 |
Solvent |
Yield of 230a (%) |
Yield of 232a (%) |
| 1 |
231a
|
Ru(bpy)3(PF6)2 |
47.7 |
CH2Cl2 |
<5 |
<5 |
| 2 |
231a
|
Ir(ppy)2(dtbbpy)(PF6) |
51.0 |
CH2Cl2 |
<5 |
18 |
| 3 |
231a
|
Ir(ppy)3 |
55.4 |
CH2Cl2 |
5 |
16 |
| 4 |
231a
|
Ir[dF(CF3)ppy]2dtbbpy(PF6) |
61.0 |
CH2Cl2 |
<5 |
13 |
| 5 |
224a
|
Ru(bpy)3(PF6)2 |
47.7 |
(CHCl2)2 |
70 |
N/A |
With optimised conditions in hand, the substrate scope of the reaction was investigated. A range of 33 oxazolidines and lactams 230 was prepared from 224 (Scheme 56). Functional groups like alkynes were also accommodated under the reaction conditions as seen with product 224h and polycyclic compounds like 230e, 230i and 230j were also prepared.
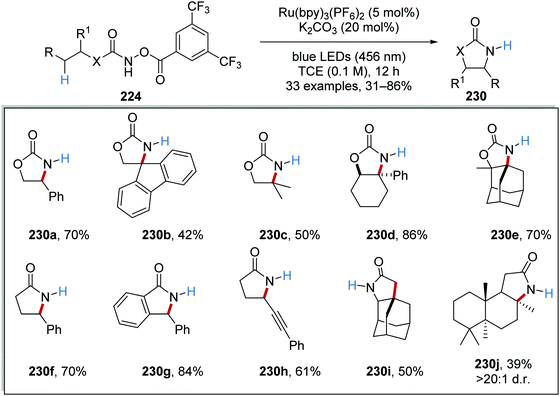 |
| | Scheme 56 Substrate scope for oxazolidinone synthesis. | |
Iridium.
As with ruthenium(II) complexes, photoexcited iridium(III) complexes can act as electron acceptors, in this case resulting in a catalytic cycle that shuttles between Ir(III) and Ir(II), or as electron donors, leading to a cycle that incorporates Ir(III) and Ir(IV).
2.4 Catalytic cycles featuring Ir(III)/Ir(II)
Decarboxylation of carboxylic acids.
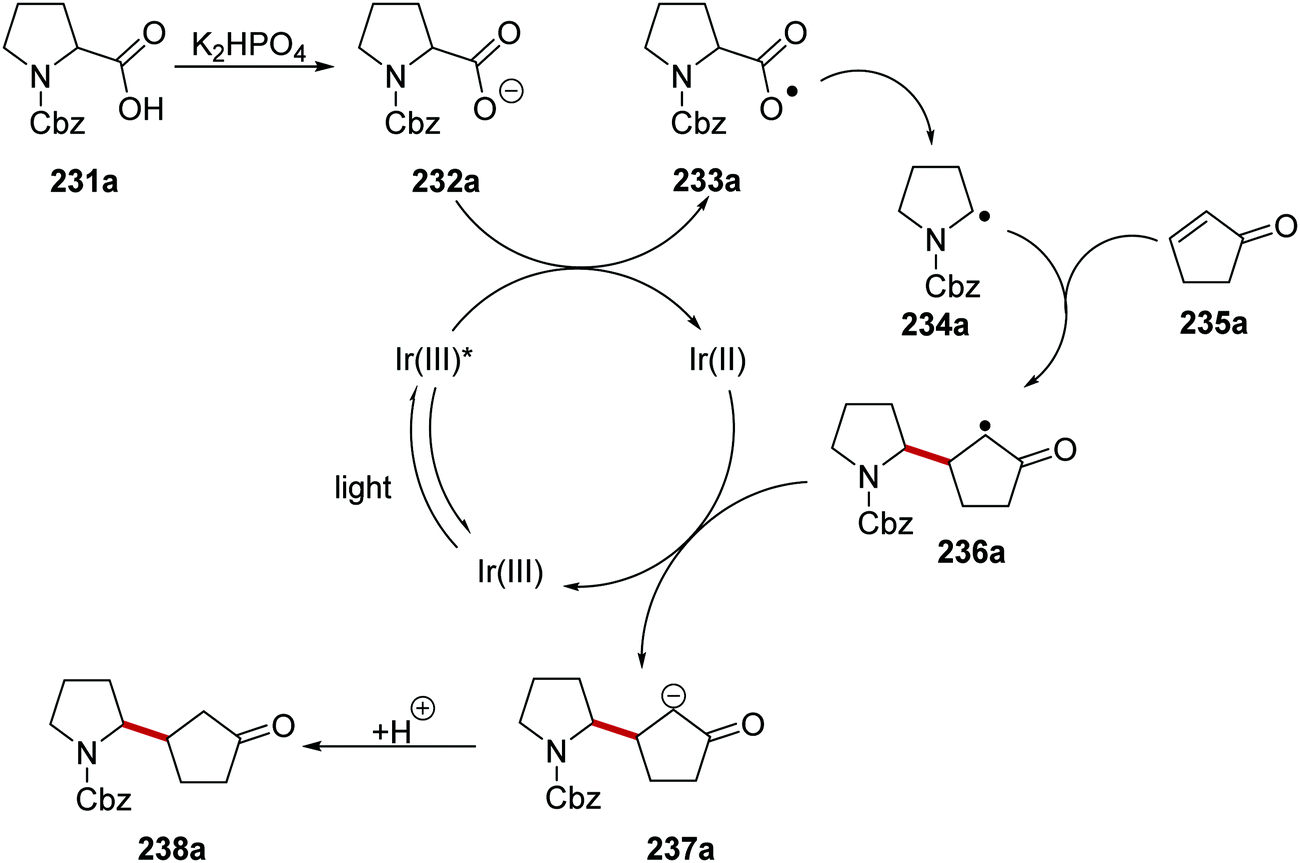
The decarboxylation of aliphatic carboxylic acids with iridium photoredox catalysis is a very successful strategy at generating carbon-centred radicals. For example, a Giese coupling reaction of carboxylate-derived radicals with electron-poor alkenes has been reported (Scheme 57).132 Under the reaction conditions, carboxylic acid 231a was deprotonated with the phosphate base and this gave carboxylate 232a. Carboxylates (Boc-Pro-OCs, E = +0.95 V vs. SCE)134 can be oxidised by the excited Ir(III)* complex (Ered* = +1.21 V vs. SCE) and thus carboxyl radical 233a and reduced Ir(II) species were formed SET. Decarboxylation of radical 233a gave the electron-rich radical 234a which added to the electron-poor alkene 235a and this gave α-keto radical 236a. The iridium catalytic cycle was closed with a SET between 236a and Ir(II) complex which regenerated the original Ir(III) complex as well as anion 237a. Protonation of 237a led to the ketone 238a.
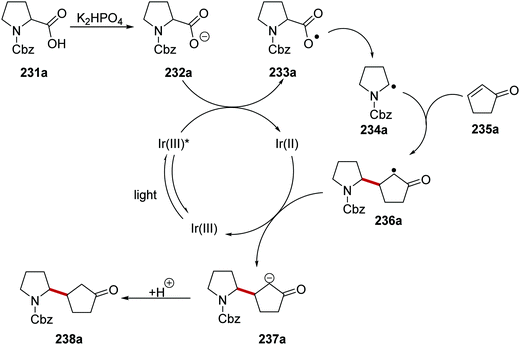 |
| | Scheme 57 Decarboxylative radical coupling to electron-poor alkenes. | |
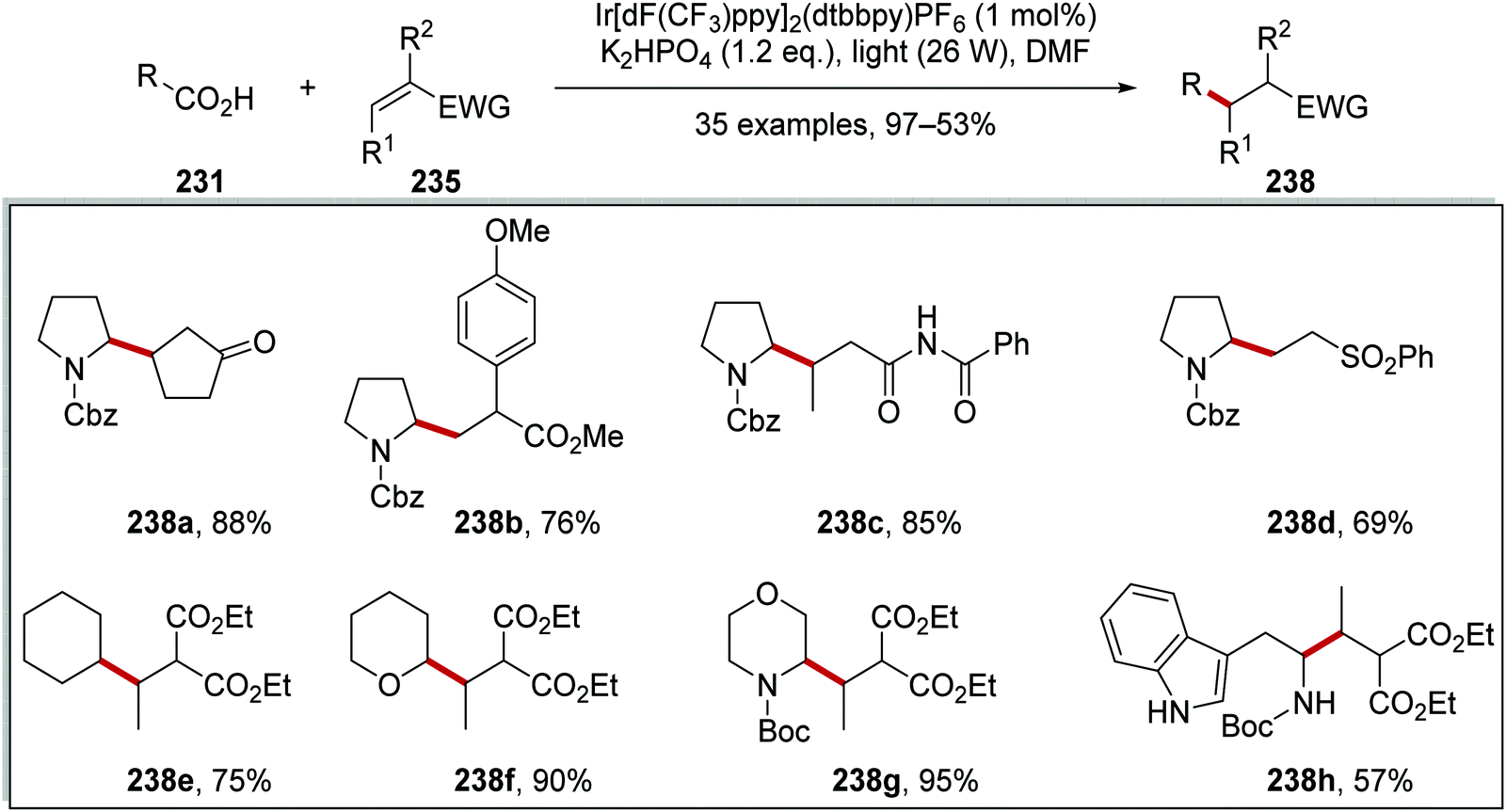
As stated, this reaction allowed for the coupling of carboxylic acids 231 to electron-poor alkenes 235 and this resulted in the formation of Csp3–Csp3 coupled products 238 (Scheme 58). The wide applicability of this reaction was proven with the synthesis of 35 compounds. Whilst the alkene had to be conjugated to an electron-withdrawing group, it was shown that ketones, esters, amides, and sulfones could all be utilised as electron-withdrawing groups (EWGs) with the preparations of 238a–d. Different aliphatic carboxylic acids were also used in this reaction, as seen in the preparation of 238e–h in 57–95% yields.
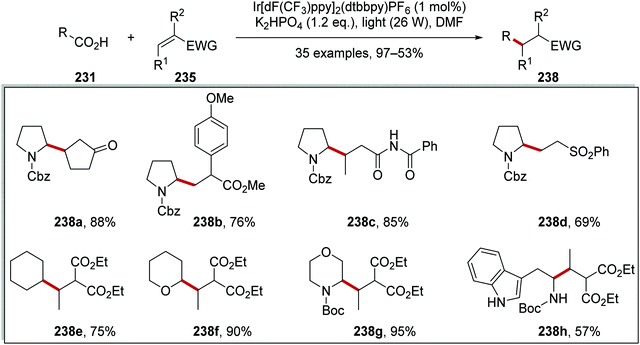 |
| | Scheme 58 Products of decarboxylative radical coupling to electron-poor alkenes. | |
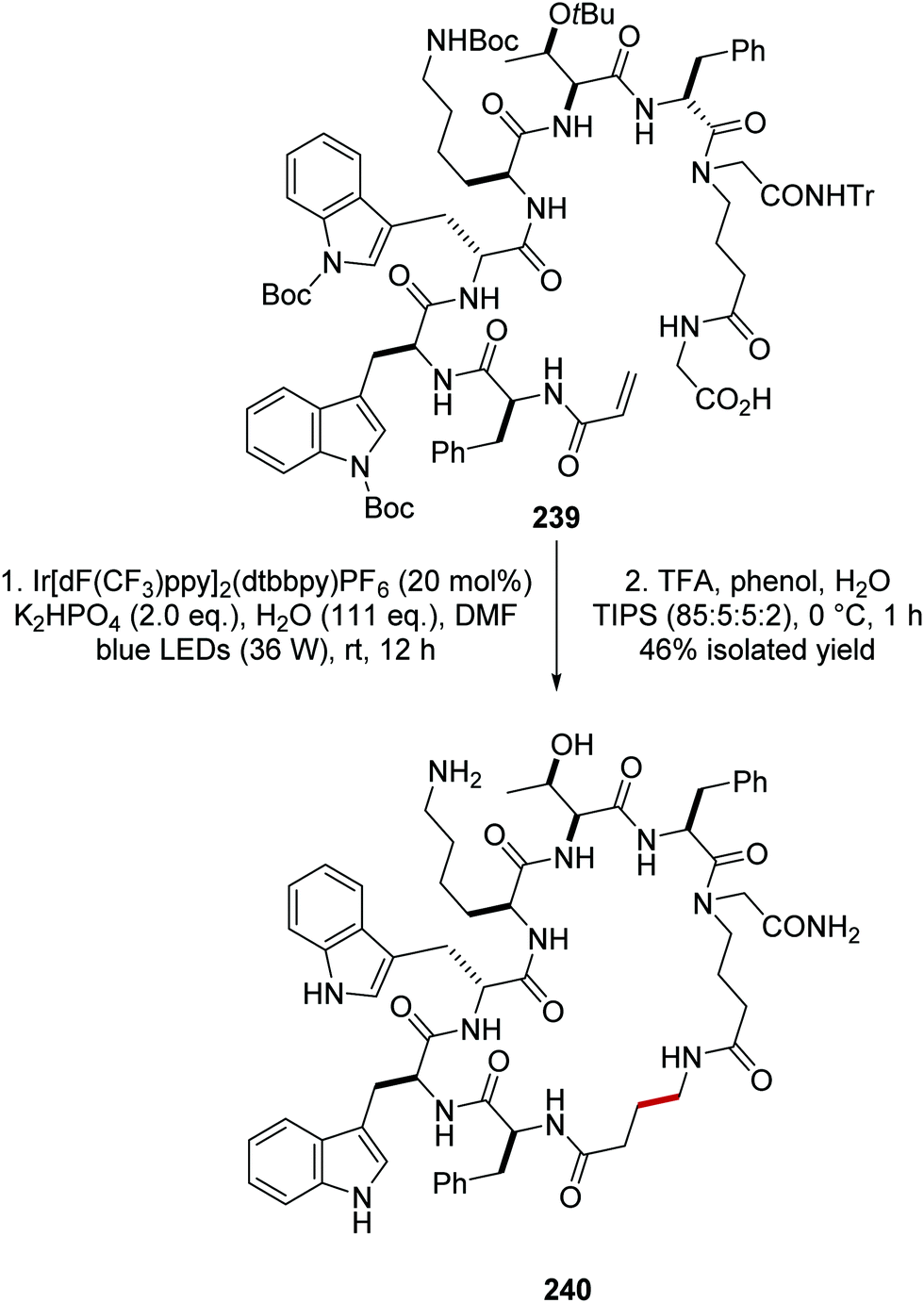
The synthetic utility of this reaction was demonstrated in the macrocyclisation of peptides (Scheme 59).133 Treatment of alkene-derived peptide 239 with the optimised reaction conditions led to cyclised peptide 240 in 46% yield, using a commercially available iridium photocatalyst with catalytic loadings (20 mol%).
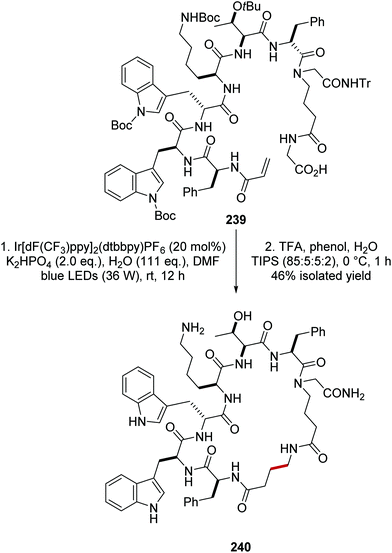 |
| | Scheme 59 Radical macrocyclisation of peptide 239. | |
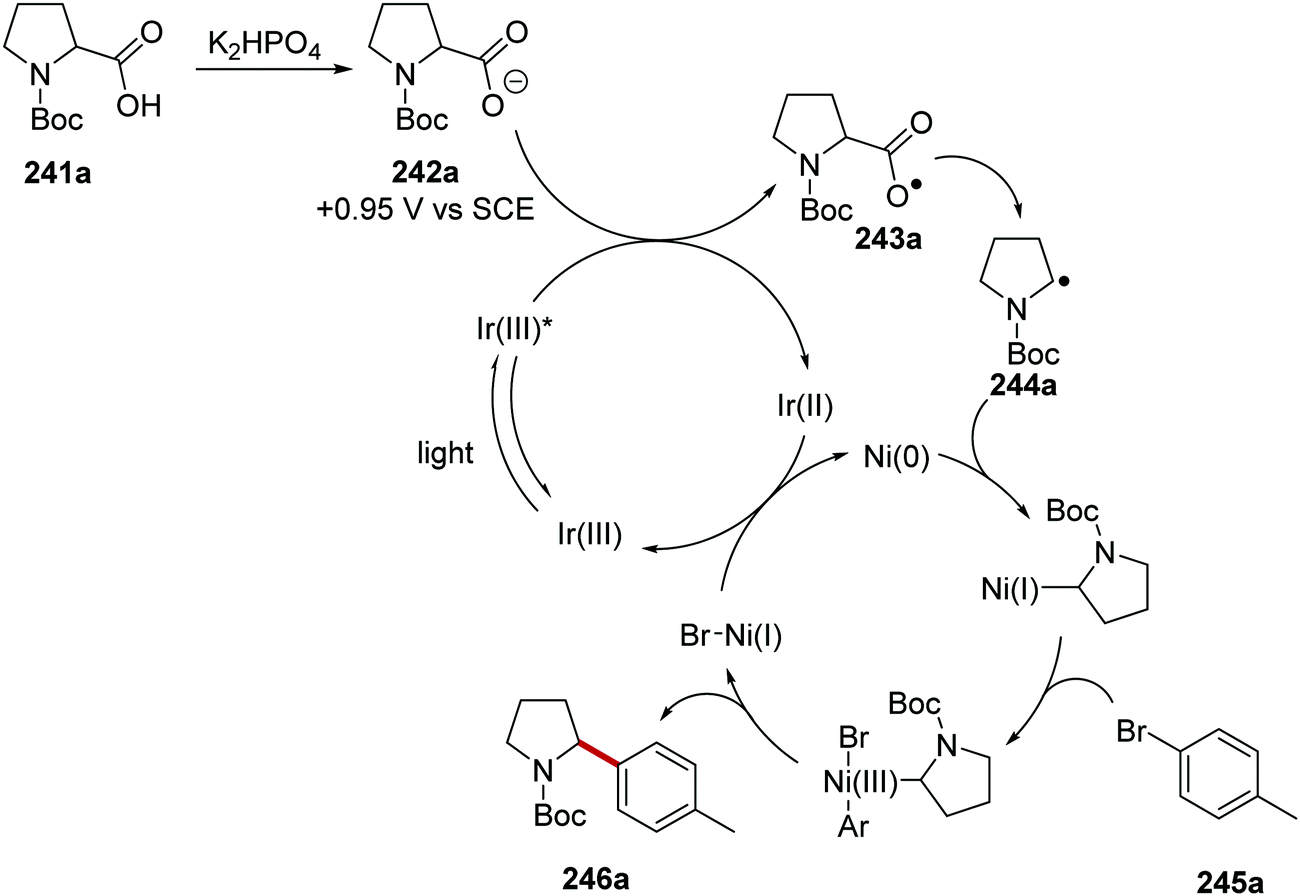
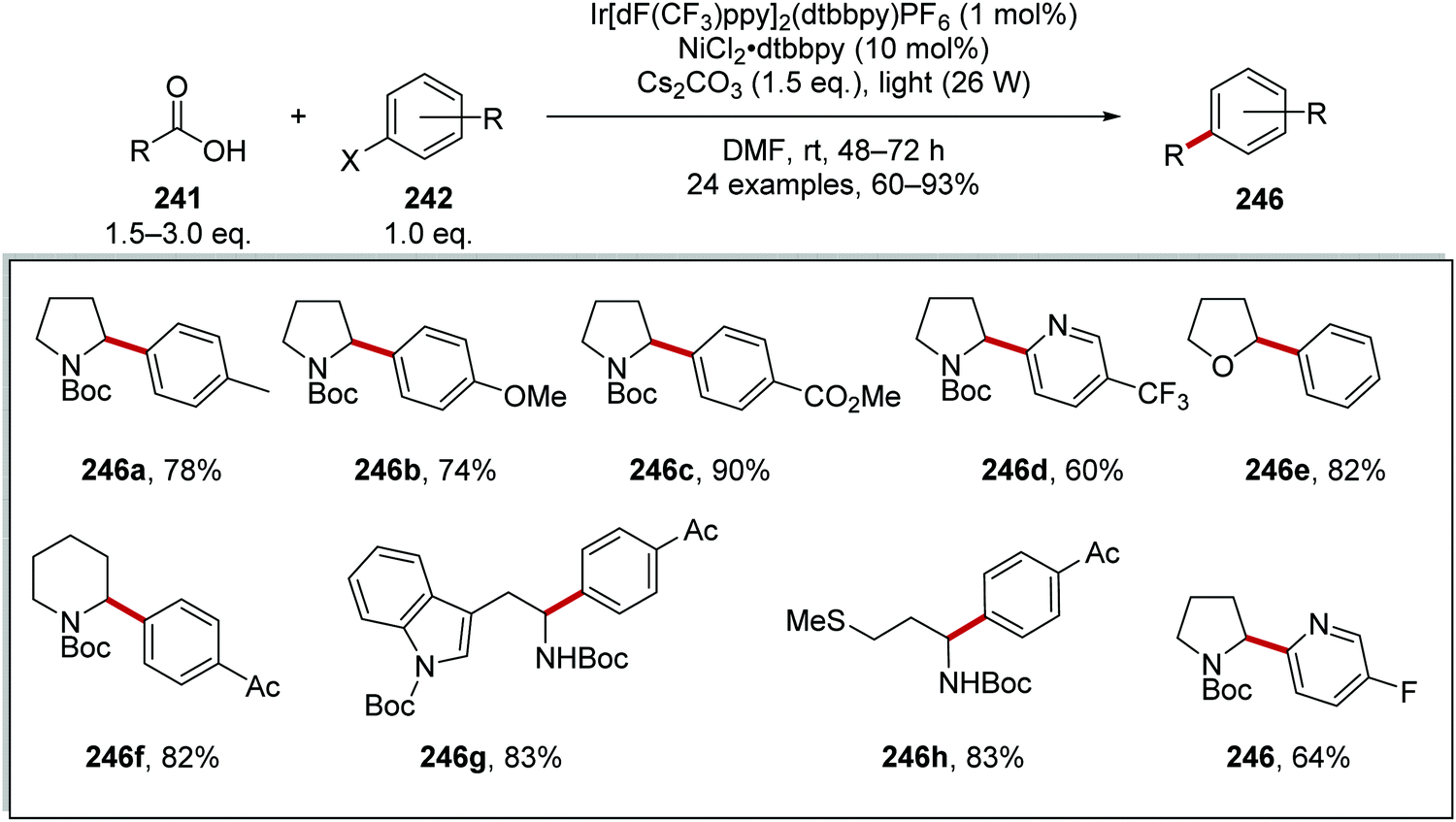
Carboxylic acids 241 were coupled to aryl halides 245 with use of an iridium and nickel catalytic system.134 Akin to the formation of radical 234 (Scheme 57), radical 244a was formed from N-Boc amine 242a (Scheme 60). A Ni(0) complex intercepted radical 244a and a Ni(I) complex was formed. Oxidative addition of the Ni(I) complex to 245a gave a Ni(III) complex and this underwent reductive elimination, which gave the functionalised amine product 246a. Both Ir and Ni catalytic cycles were closed with SET between Ir(II) and Ni(I) complexes.
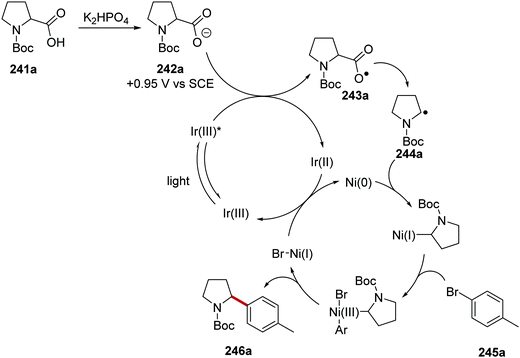 |
| | Scheme 60 Mechanism of radical coupling between carboxyl acid radical precursor 241a and aryl halide 245a. | |
From this reaction, a wide range of coupled compounds was prepared in 60–93% yields, (Scheme 61 for 246a-I as selected examples).134 It should be noted that compounds 246a and 246b were prepared from the aryl iodide, whereas compounds 246c–h were prepared from the aryl bromides and compound 246i was prepared from the aryl chloride.
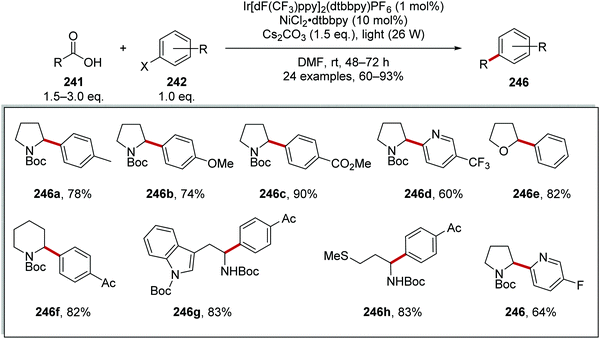 |
| | Scheme 61 Results of radical coupling between carboxyl acid 241 and aryl halide 242. | |
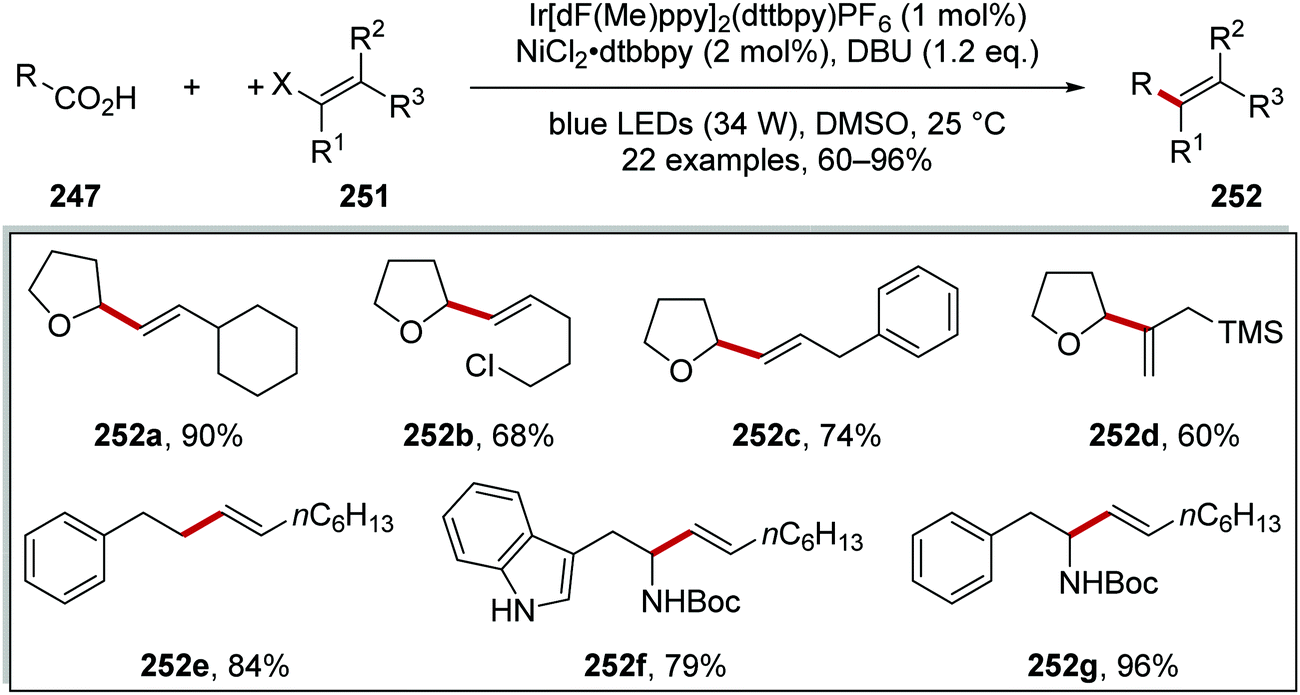
The coupling of carbon-centred radicals derived from carboxylic acids 247 with vinyl halides 251 was achieved with a similar nickel and iridium catalytic system and this gave alkene products 252 (Scheme 62).135 Deprotonation of the acid to its carboxylate salt 248 (not shown) was followed by oxidation to the carboxyl radical 249 and decarboxylation to aliphatic radical 250. The Ni-mediated coupling of vinyl halides 251 to these radicals was a very facile process and 22 alkenes 252 were isolated in 60–96% yields. A vinyl bromide was used in the preparation of compound 252d, whereas the other products were prepared from the vinyl iodide. This transformation was tolerant of functional groups 252b, silyl groups 252d and Boc-protected amines 252f and 252g.
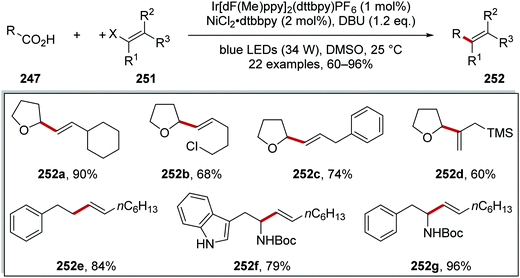 |
| | Scheme 62 Results of decarboxylative radical coupling to vinyl halides. | |
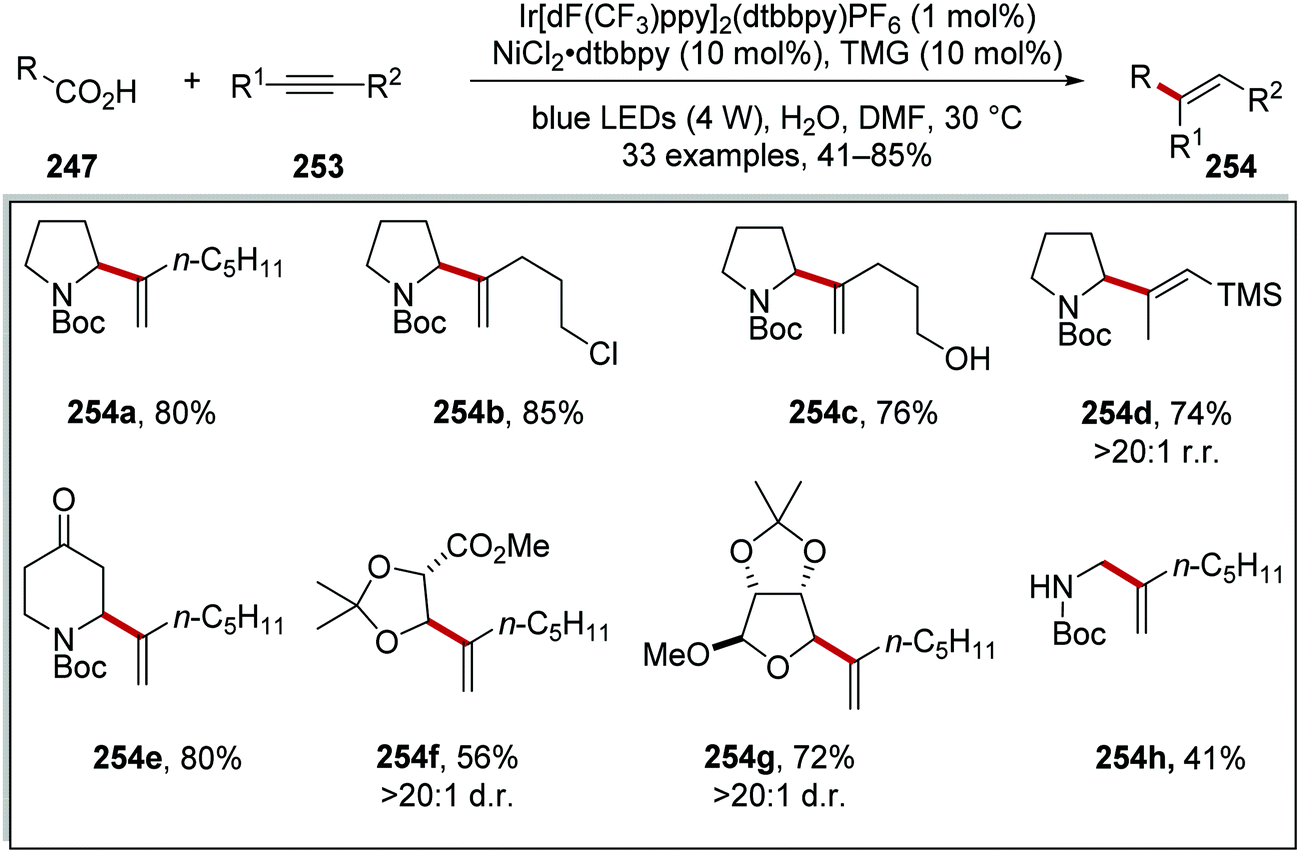
Radical coupling to alkynes 253 was achieved using a similar iridium and nickel catalytic system (Scheme 63).136 It was found that the use of 1,1,3,3-tetramethylguanidine (TMG) allowed for successful coupling and this produced alkenes 254. The reaction allowed for the preparation of chloro 254b, hydroxy 254c, silyl 254d, ketal 254g, ester 254f analogues. A diminished yield of secondary N-Boc amine was observed with 254h (41% yield).
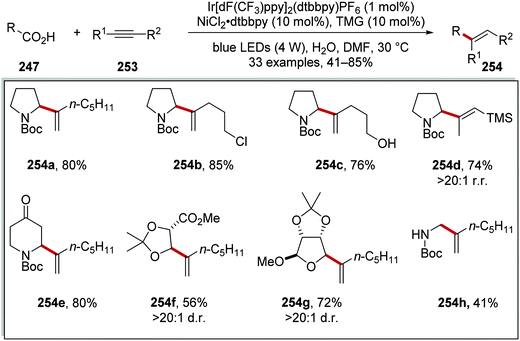 |
| | Scheme 63 Decarboxylative radical coupling to alkynes. | |
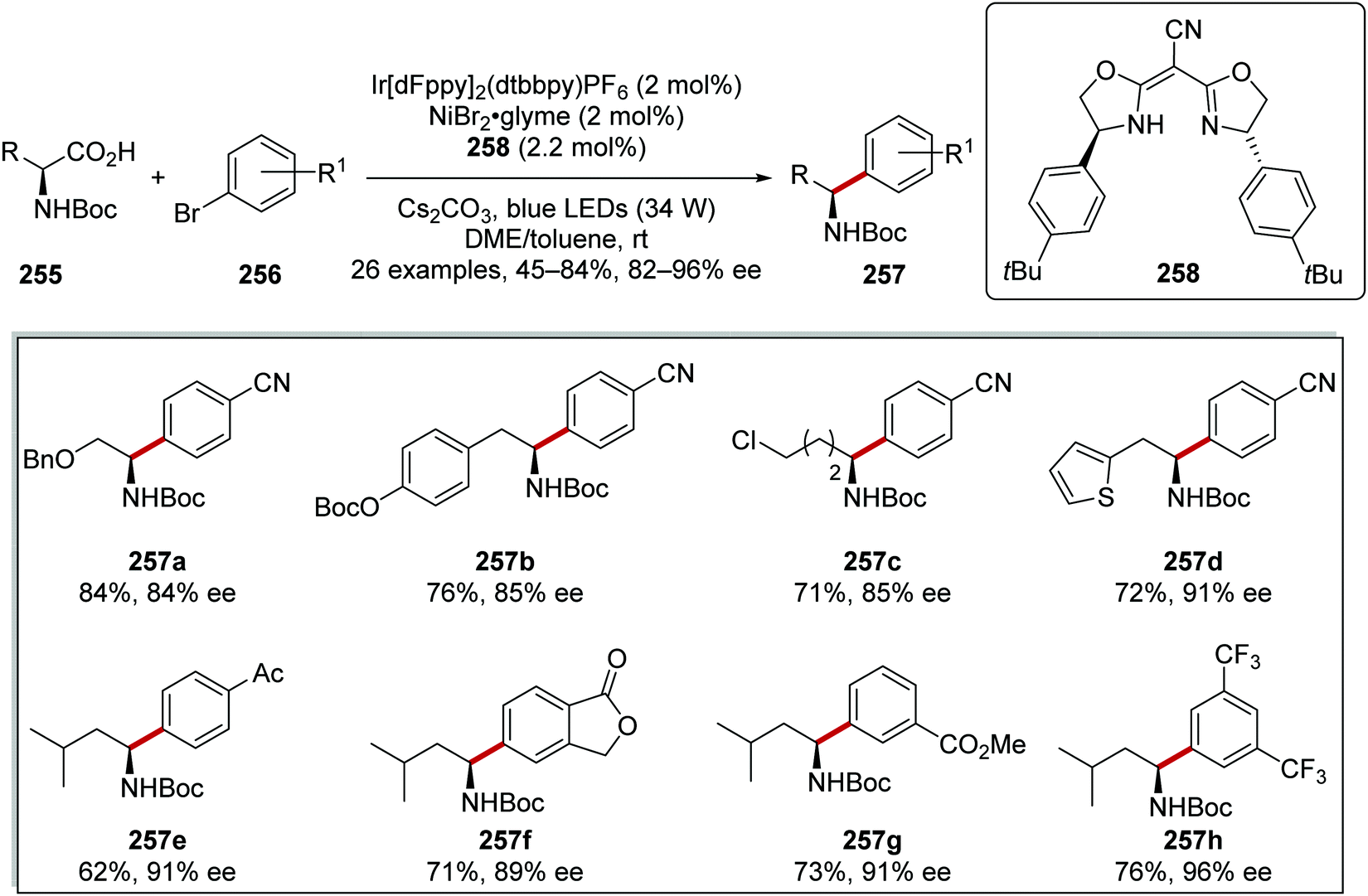
Enantioenriched amine derivatives 257 were prepared from the coupling of carboxylic acids 255 with aryl bromides 256 (Scheme 64).137 The use of a chiral ligand 258 gave high yields and high levels of enantiopurity as exemplified with products 257a–h.
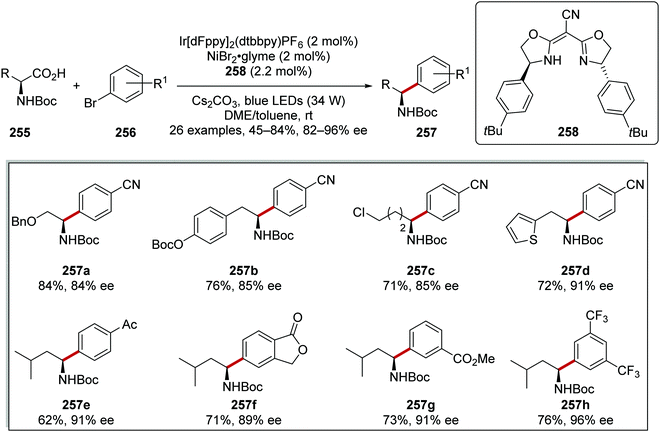 |
| | Scheme 64 Asymmetric C–C bond formation. | |
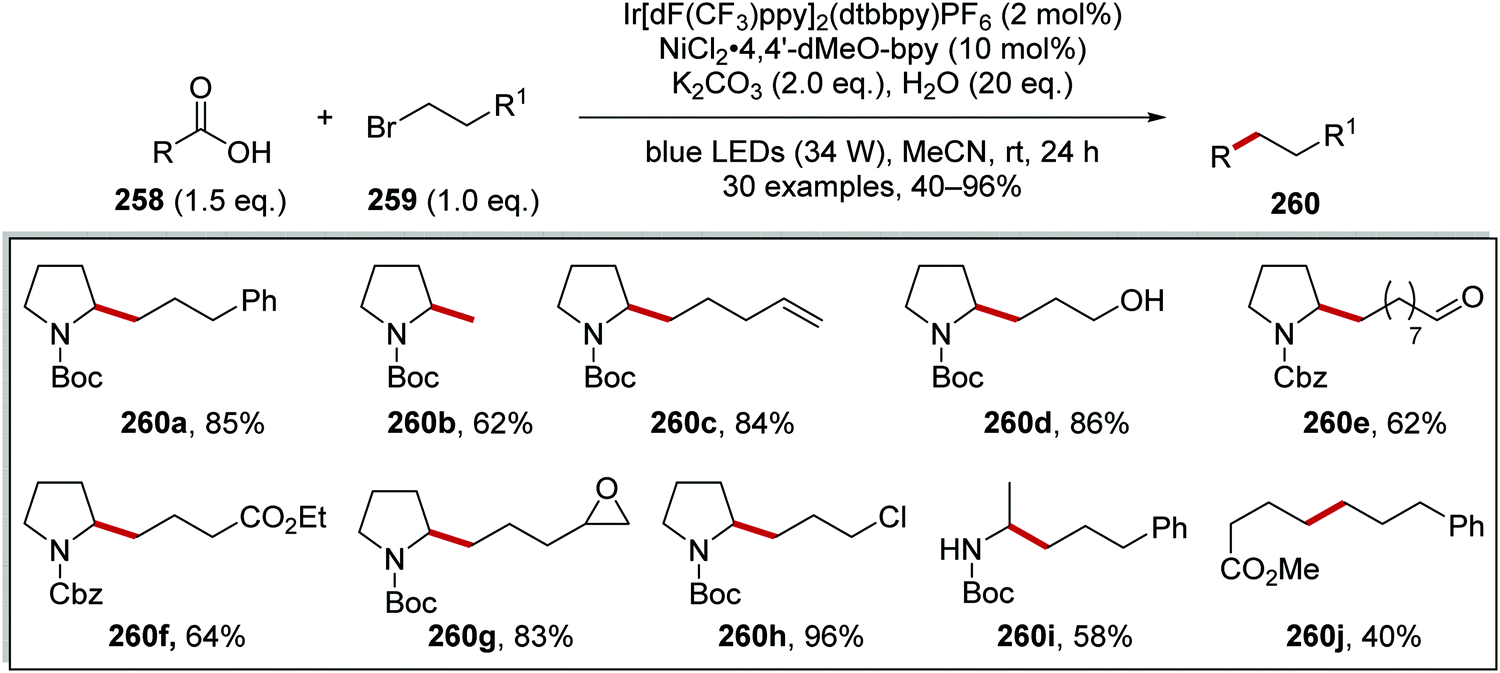
An iridium- and nickel-mediated Csp3–Csp3 coupling reaction between carboxylic acids 258 and alkyl bromides 259 was reported (Scheme 65).138 This reaction was based upon the transformation discussed in Scheme 61. However, it was found that the reaction conditions had to be modified. The use of Cs2CO3 as base in DMF led to ester formation between carboxylic acid 258 and alkyl bromide 259. After an extensive solvent screen, it was found that K2CO3 in MeCN allowed for the formation of 260a in 68% GC yield. The use of the more electron-rich ligand 4,4′-dimethoxy-2,2′-bipyridine (4,4′-dMeO-bpy) increased the GC yield to 74% over dtbbpy ligand. The inclusion of water (20 eq.) in the reaction mixture, further suppressed ester formation, giving 260a in 96% GC yield and 85% isolated yield. The synthetic utility of this reaction was seen with the preparation of alcohol 260d, aldehyde 260e, ester 260f and 260j, epoxide 260g and chloroalkane 260h analogues.
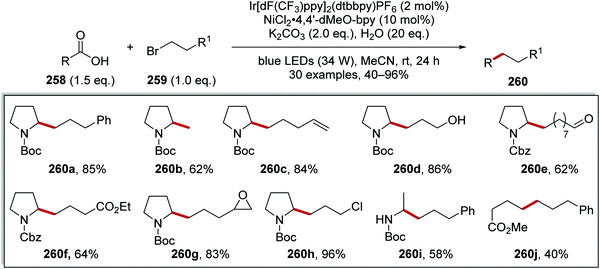 |
| | Scheme 65 Results of alkyl bromide 259 coupling to carboxylic acids 258 as radical precursors. | |
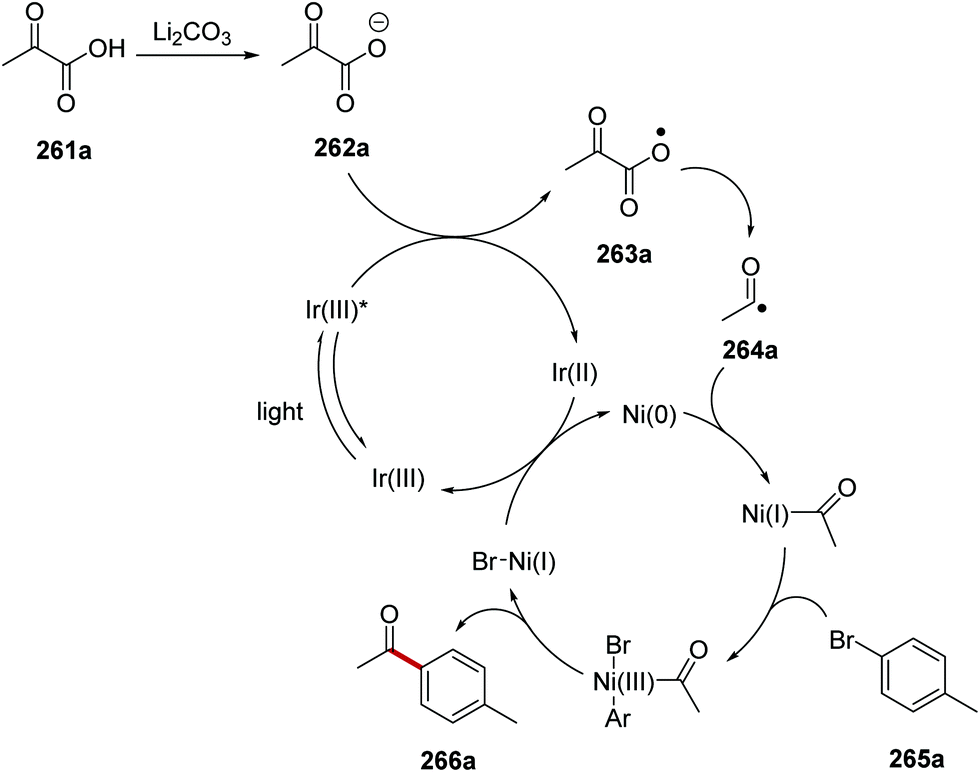
The acylation of aryl halides 265 with α-oxo carboxylic acids 261 under an iridium and nickel catalytic system was developed.139 Under the reaction conditions, the carboxylate 262a was formed from carboxylic acid 261a and lithium carbonate (Scheme 66). SET between excited Ir(III)* complex (Ered* = +1.21 V vs. SCE) and 262a (E1/2 = +1.03 V vs. SCE) resulted in carboxyl radical 263a and the Ir(II) species. Decarboxylation of 263a gave acyl radical 264a. The presence of the nickel catalyst allowed for cross-coupling between 264a and 4-bromotoluene (265a) and this gave ketone 266a.
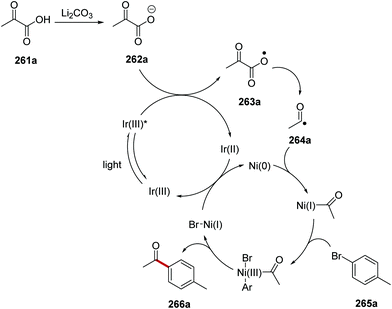 |
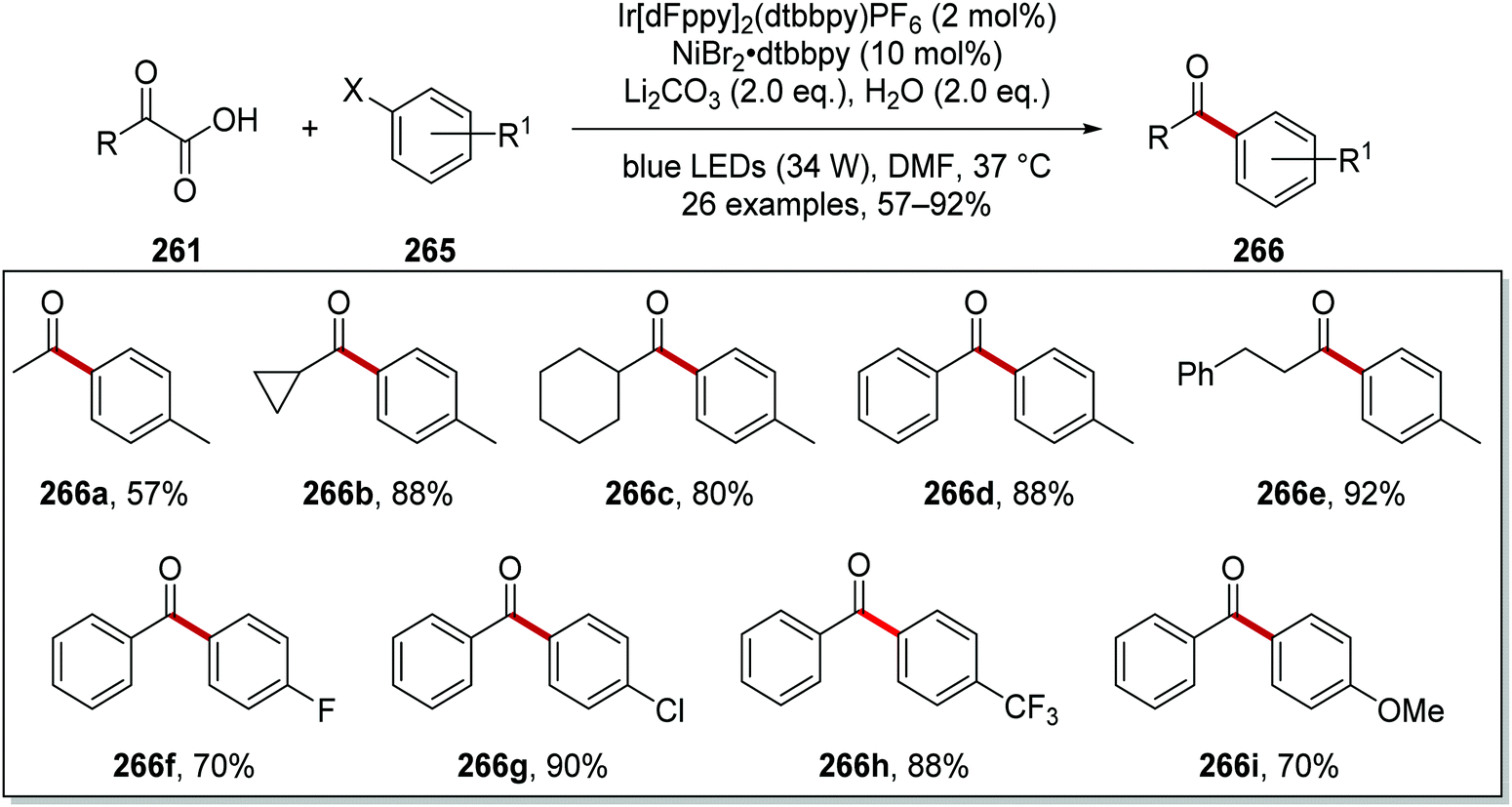
| | Scheme 66 Formation of ketone 266a from α-oxo acid 261a and aryl halide 265a. | |
From optimisation studies, it was found that improved yields of 266 were obtained when lithium carbonate, rather than caesium carbonate, was used as base. Increased yields were also isolated when a more powerful blue light source was used, and so it was inferred that the reaction was photon-limited. The 1H-NMR yield of 266c was increased to 84% from 74% when 2 eq. of water were added to the reaction mixture. Higher loading of Ir[dF(CF3)ppy]2dtbby from 1 mol% to 2 mol% gave the optimal 1H-NMR yield of 266c (88%) and it was isolated in 80% yield (Scheme 67). Various α-oxoacids 261 and aryl halides could be used in this reaction giving ketones 266a–i.
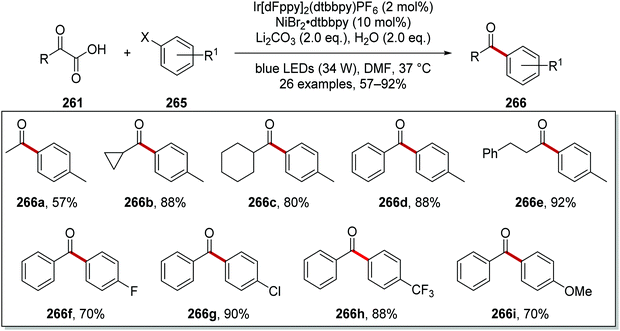 |
| | Scheme 67 Ketone formation from α-keto acids. | |
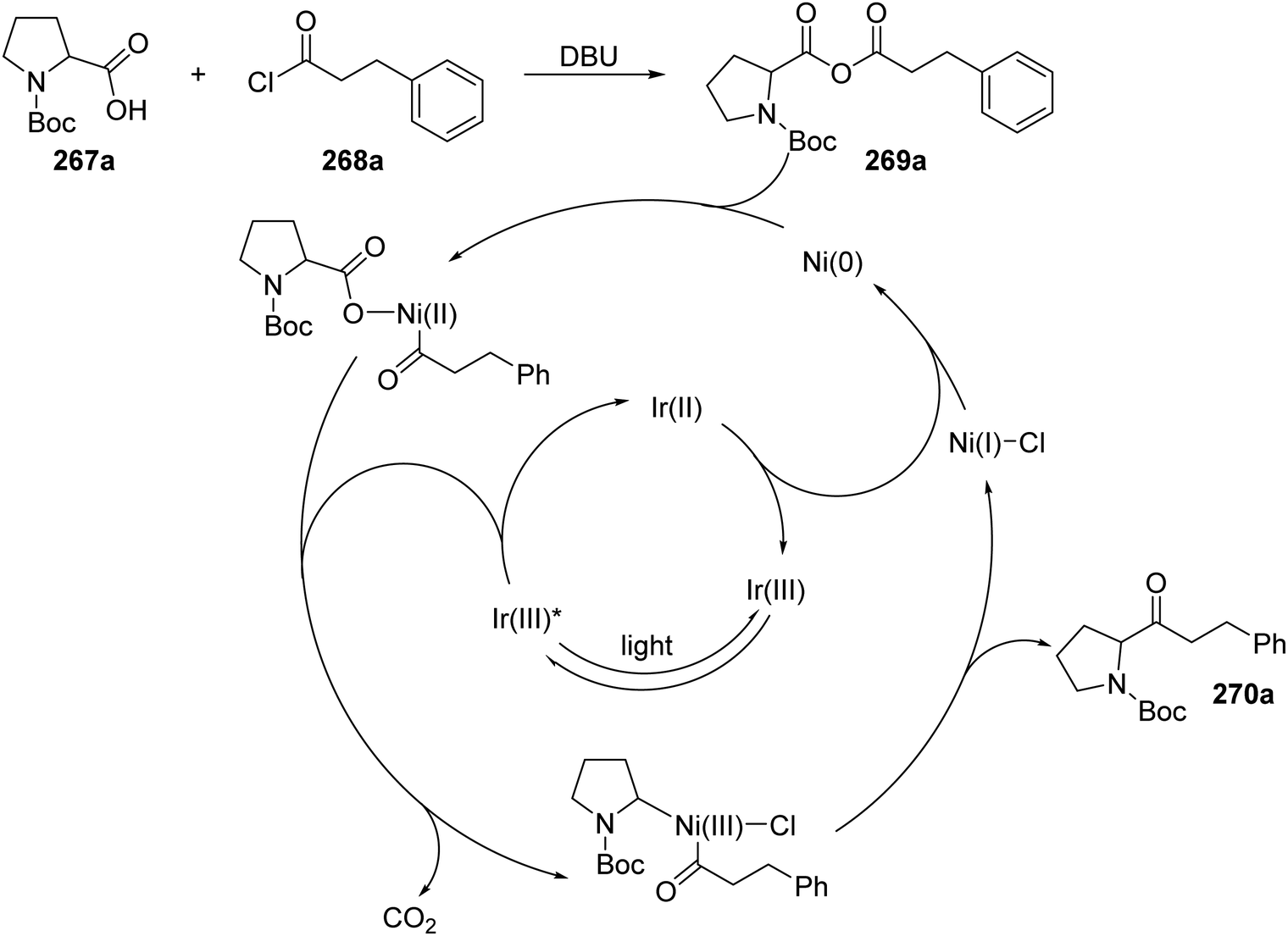
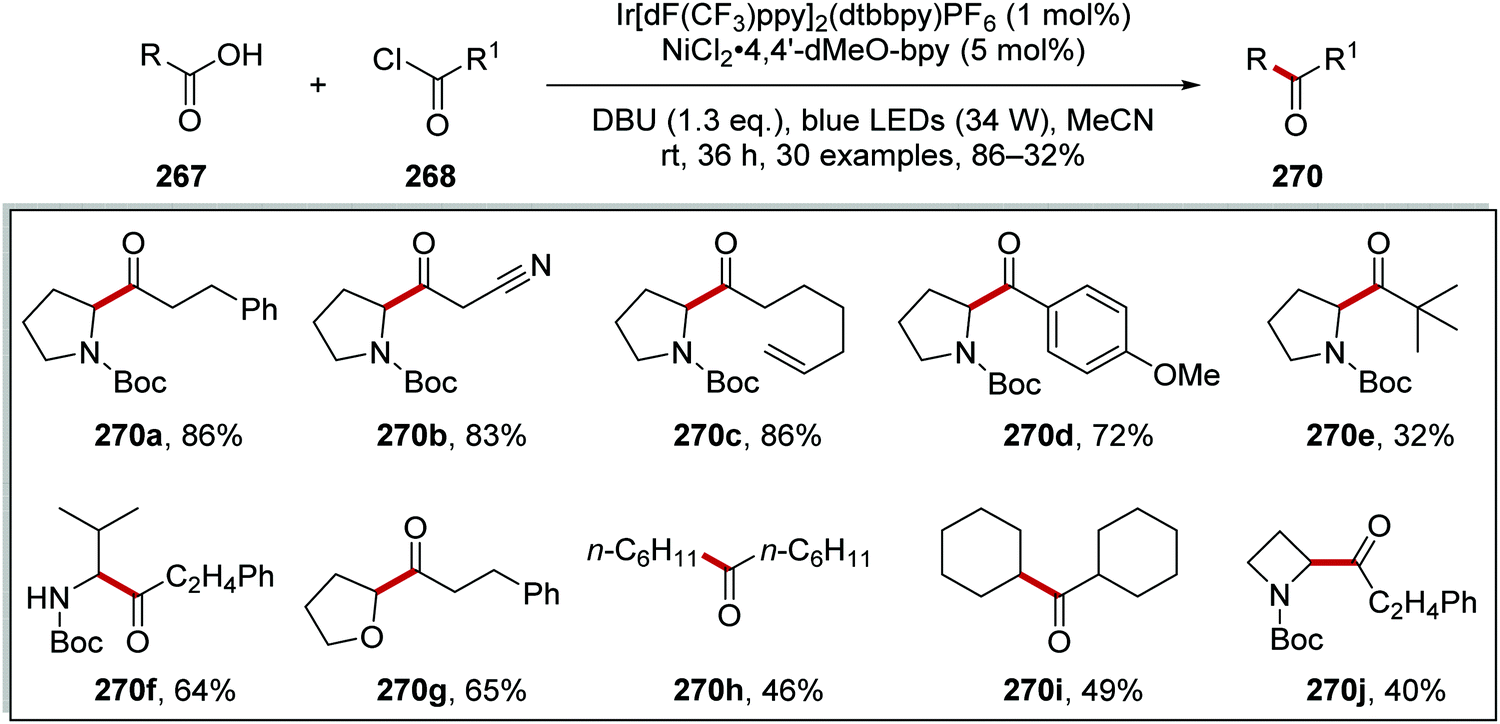
Ketones 270 were prepared with the combination of carboxylic acids 267, DBU and acyl chlorides 268, under an iridium and nickel catalytic system (Scheme 68).140 The combination of 267a and 268a under basic conditions gave anhydride 269a. Interception of 269a with a Ni(0) complex led to a Ni(II) complex. Loss of an electron, to an excited Ir(III)* complex, and carbon dioxide led to the formation of a Ni(II) complex. Reductive elimination of Ni complex led to ketone compound 270a and a Ni(I) complex. An outer-sphere electron transfer between Ir(II) species and Ni(I) species closed both catalytic cycles as both the original Ni(0) and Ir(III) complexes were reformed.
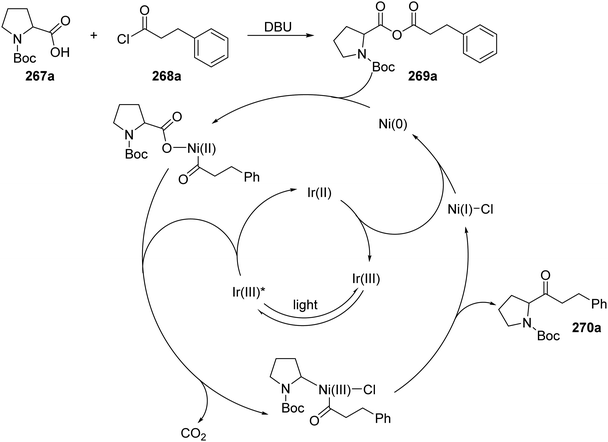 |
| | Scheme 68 Formation of ketone 270a from carboxylic acid 267a and acyl chloride 268a. | |
This transformation worked well as demonstrated with the preparation of 30 alkyl ketones 270 in yields ranging from 32% to 86%. (Scheme 69). The reaction was functional group-tolerant with nitrile 270b, alkene 270c, anisole 270d, and tert-butyl 270e analogues all being prepared. Various carboxyl radical precursors were used in this reaction and N-Boc tertiary amine 270j, N-Boc secondary amine 270f, tetrahydrofuran 270g, alkyl 270h and 270i derivatives were all prepared.
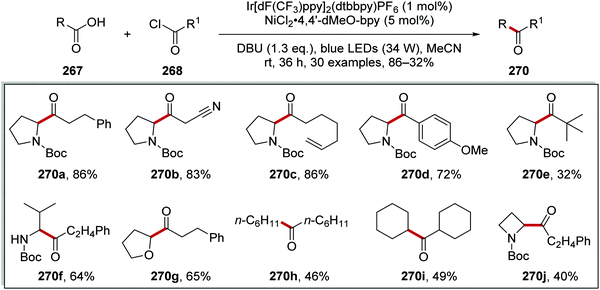 |
| | Scheme 69 Formation of ketone 270 from carboxylic acid 267 and acyl chloride 268. | |
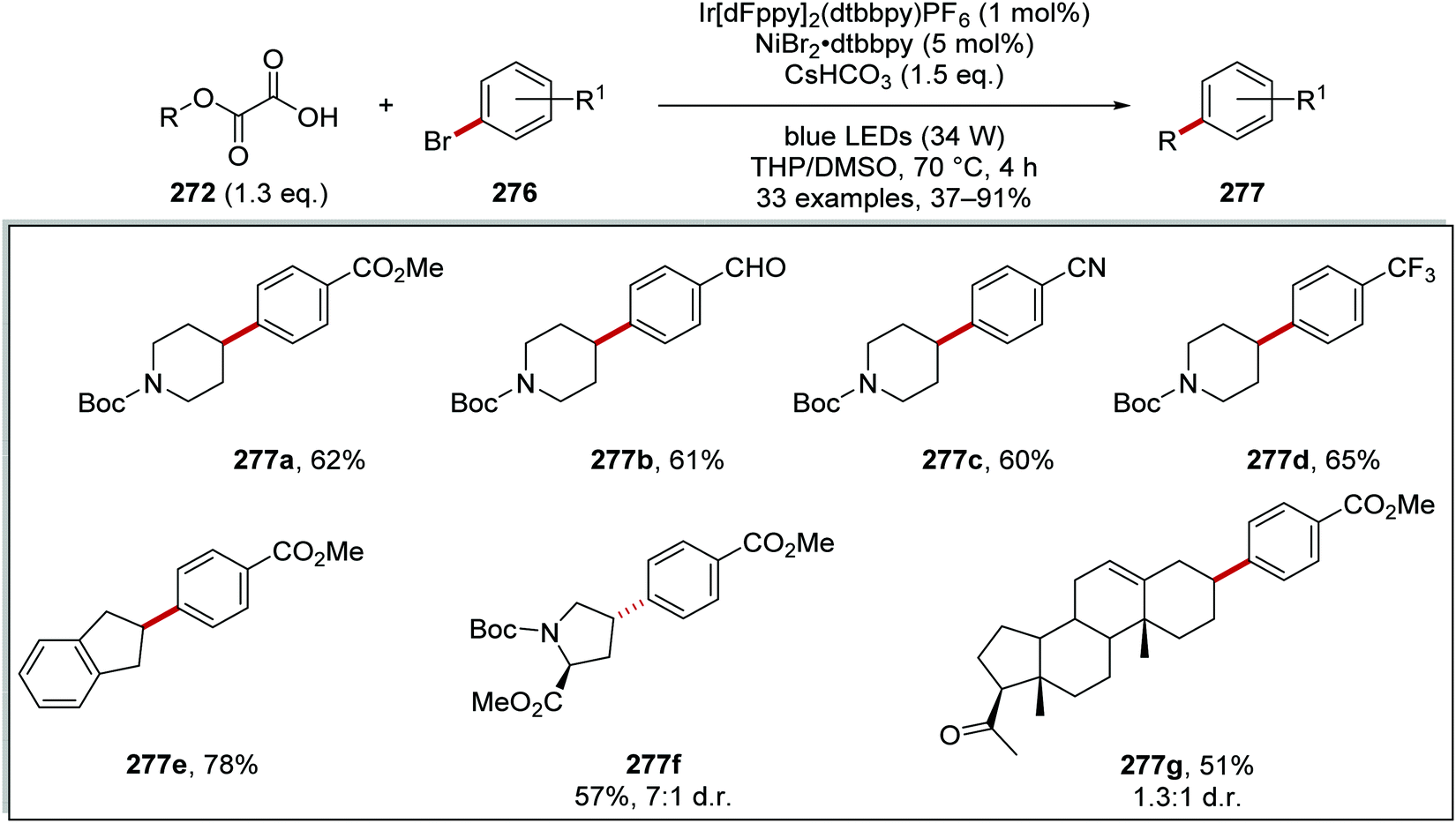
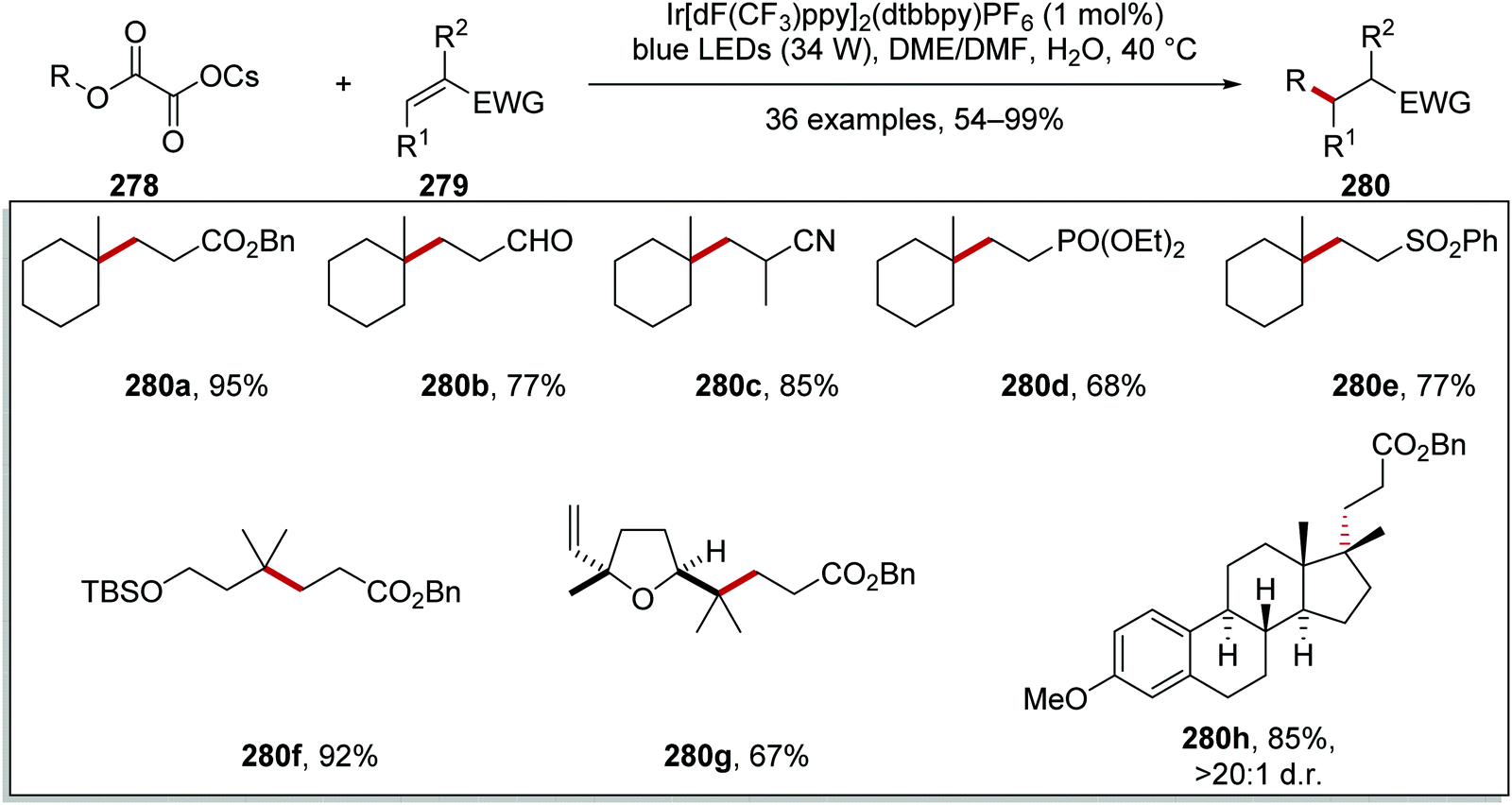
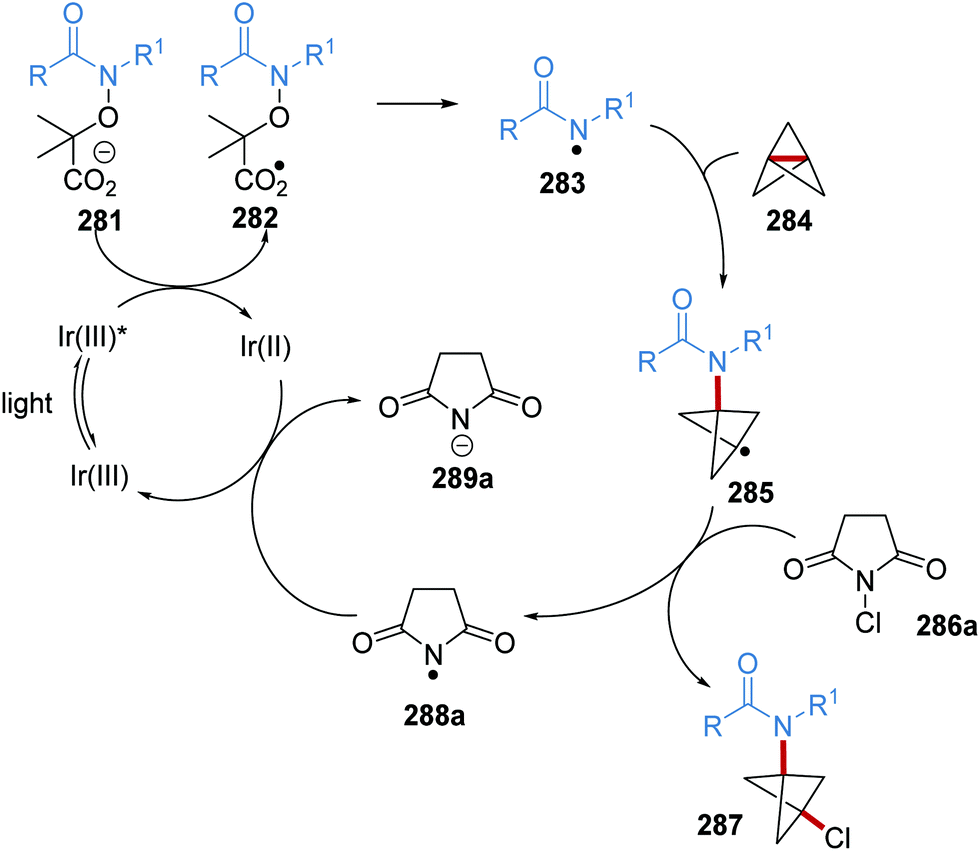
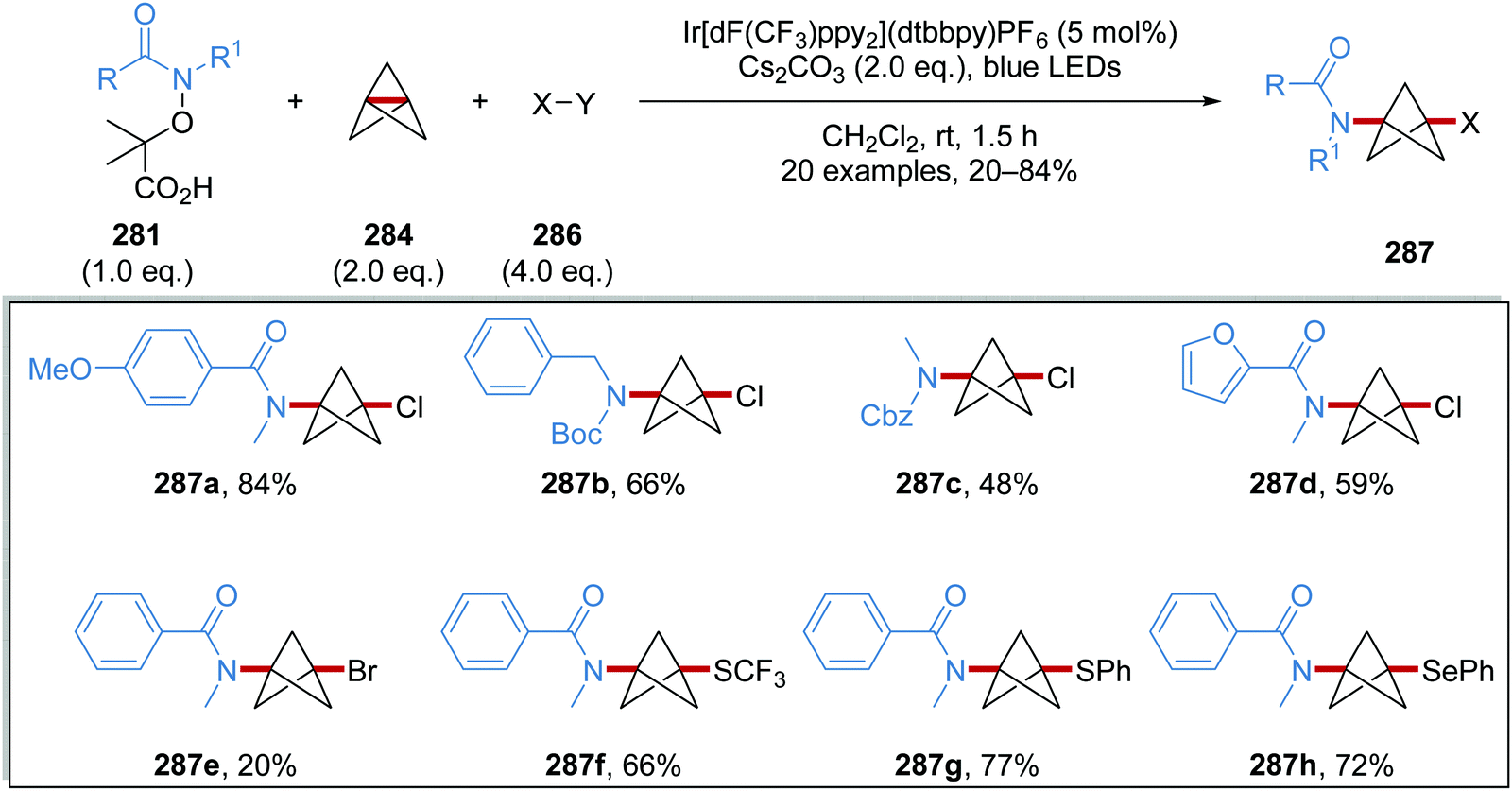
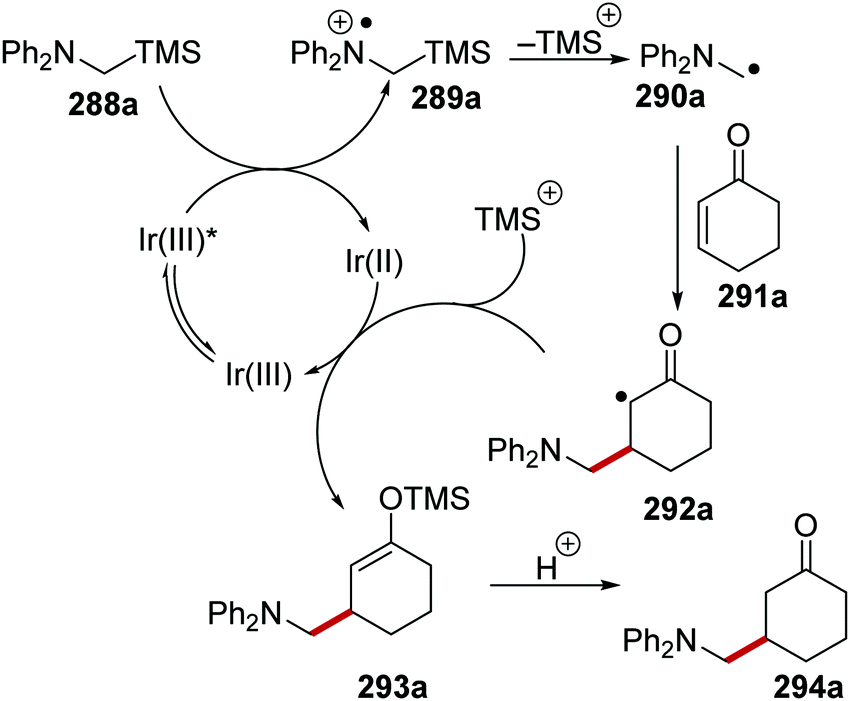
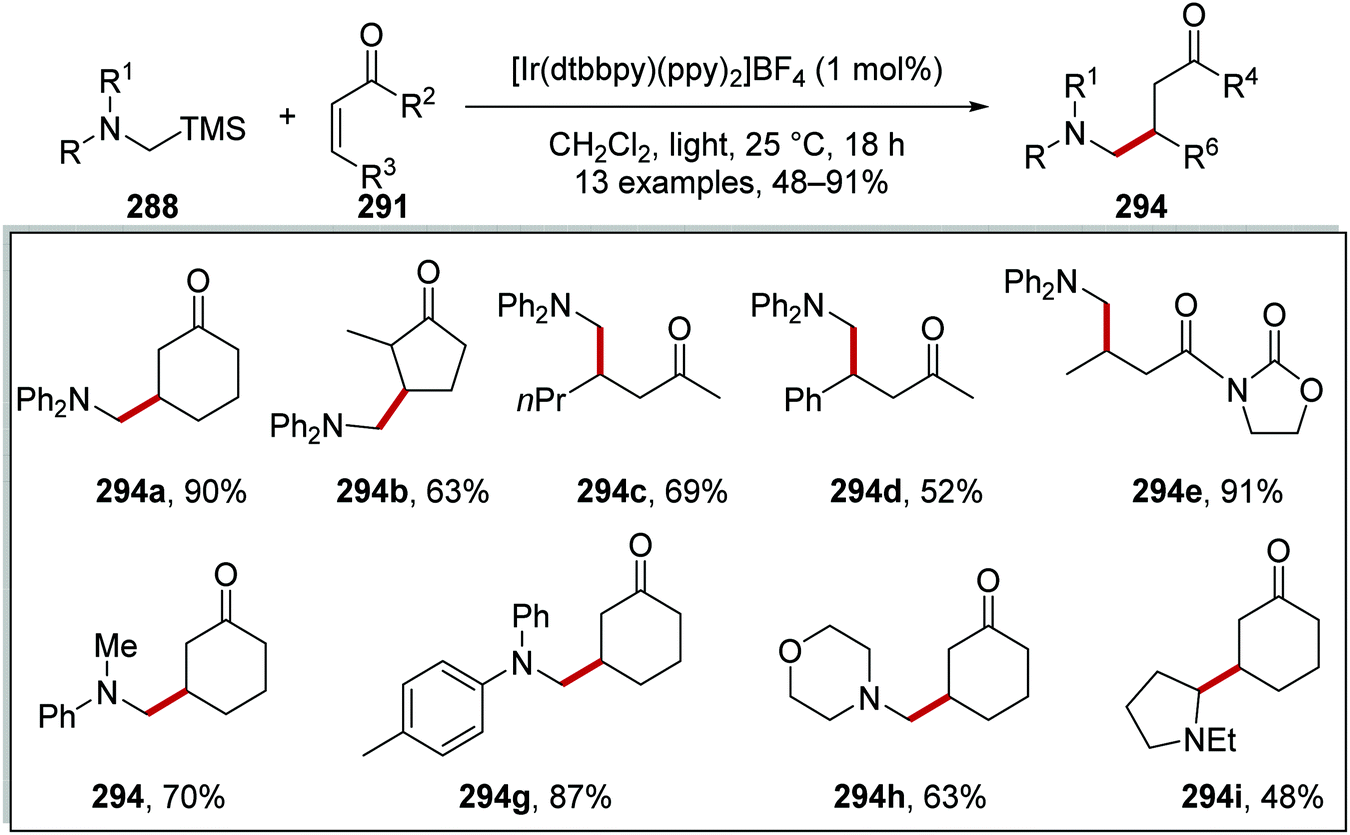
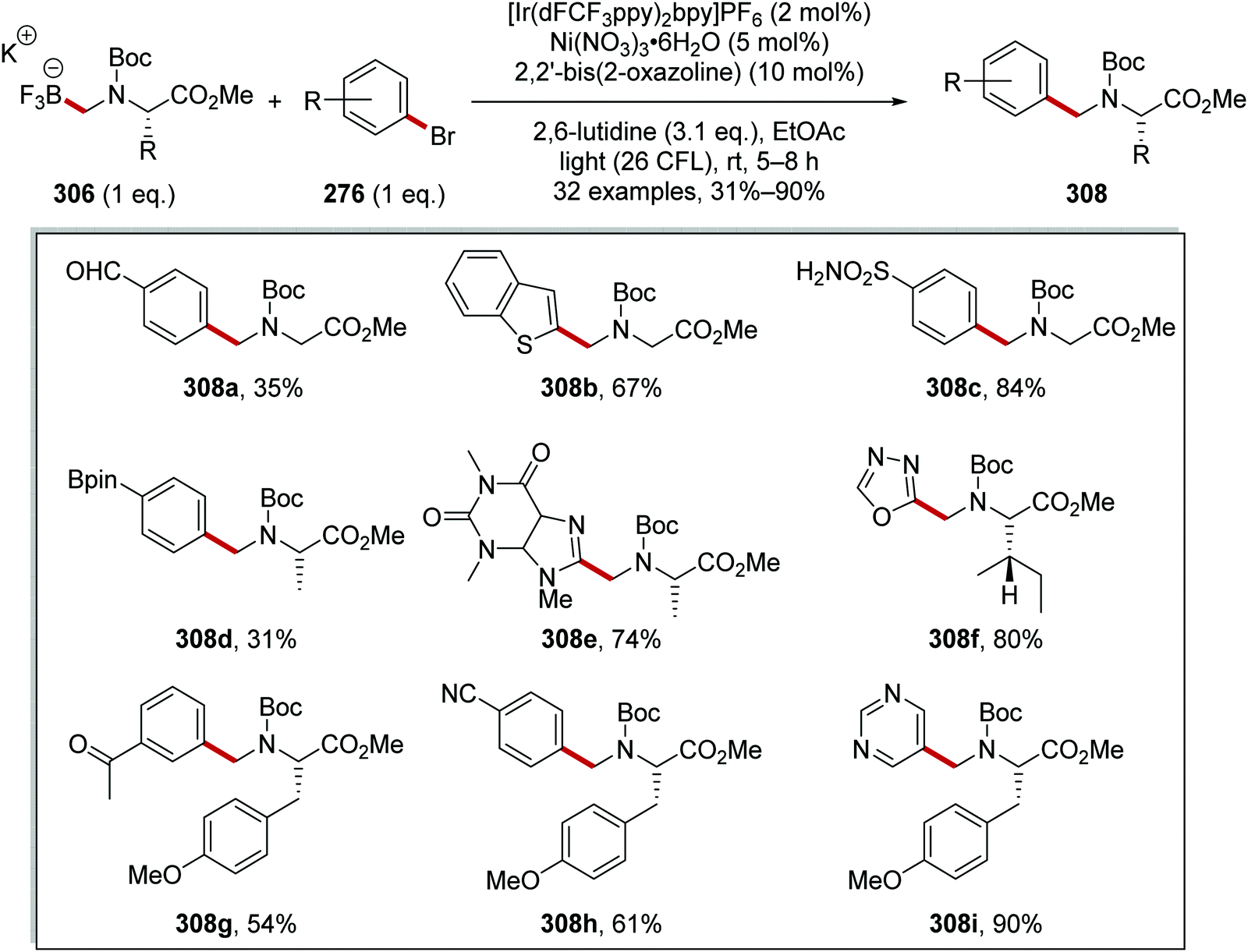
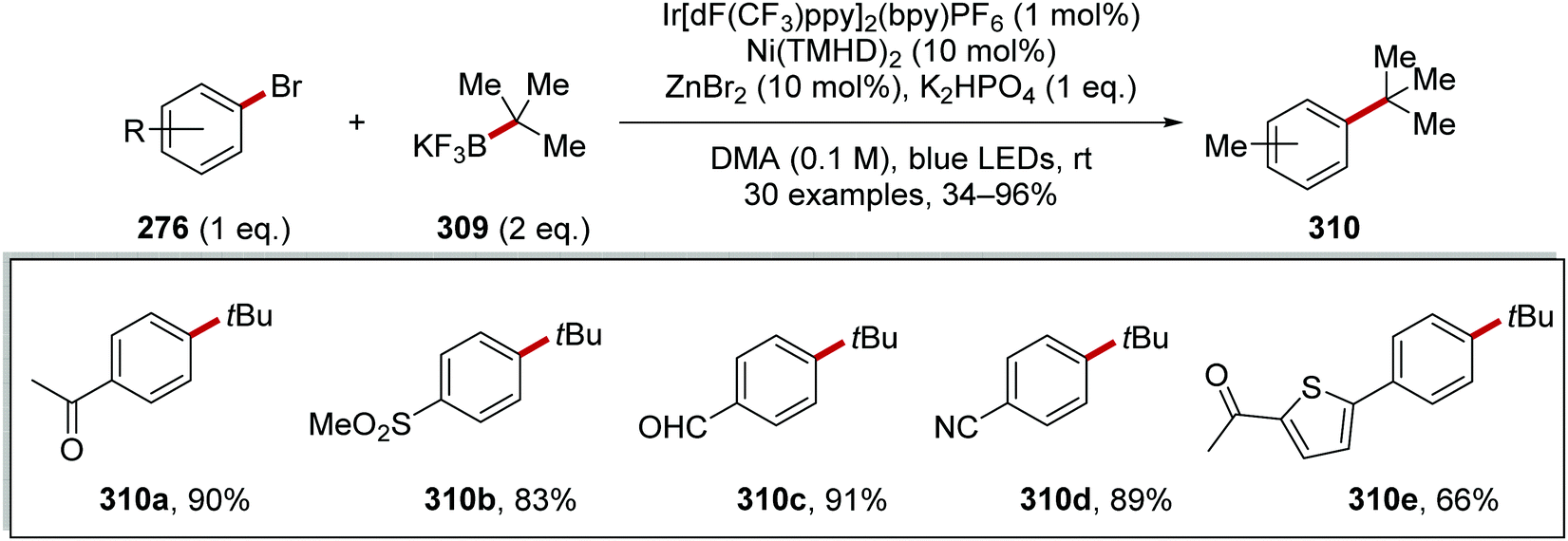
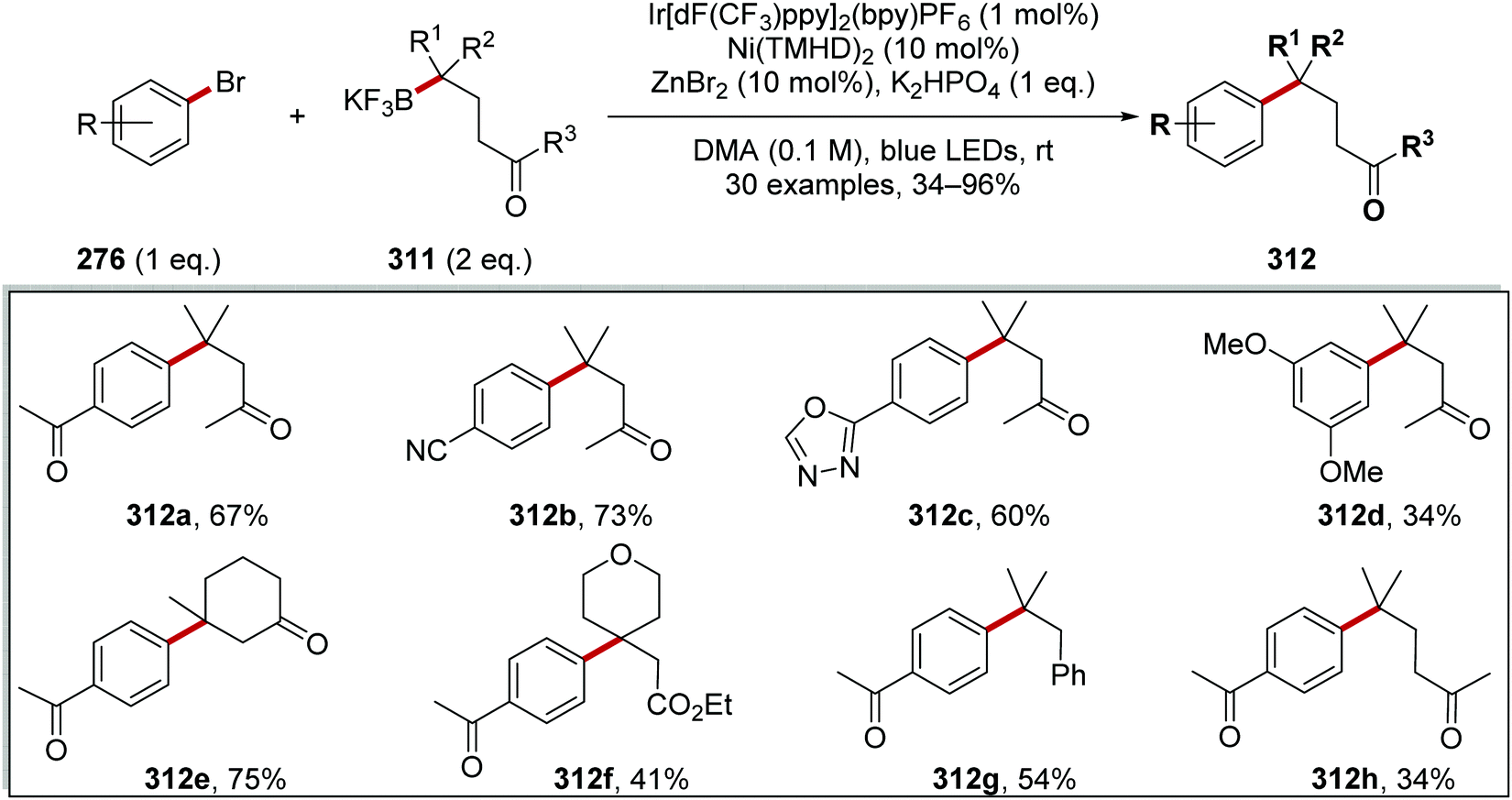
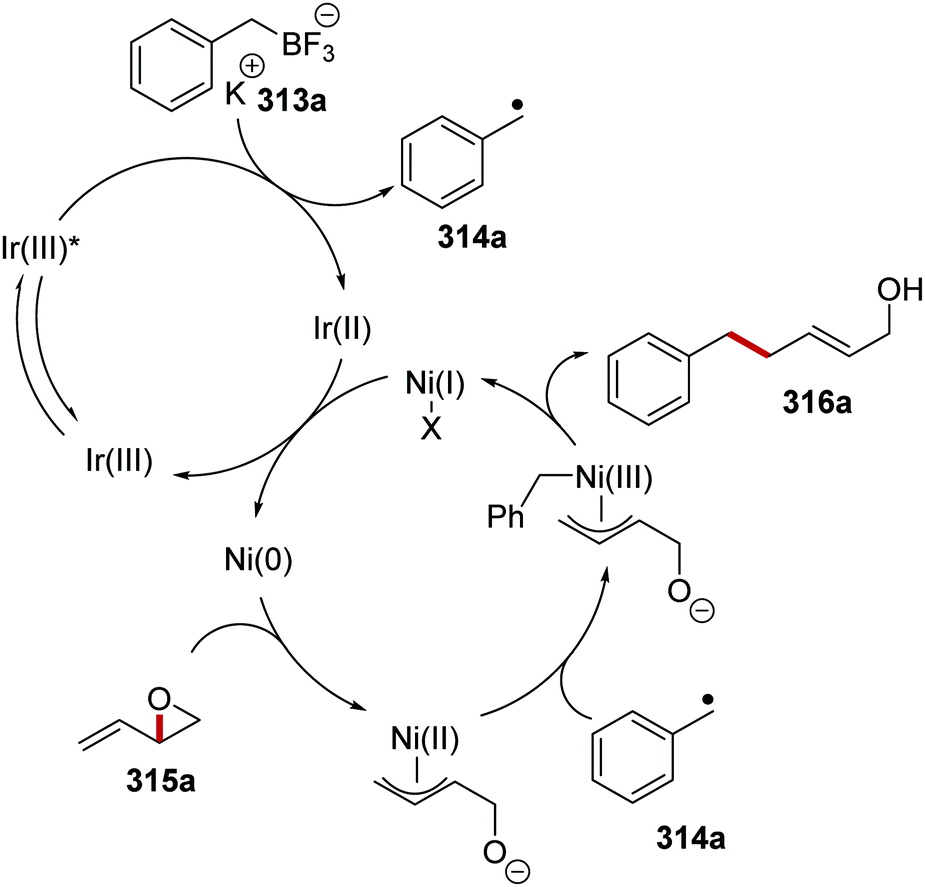
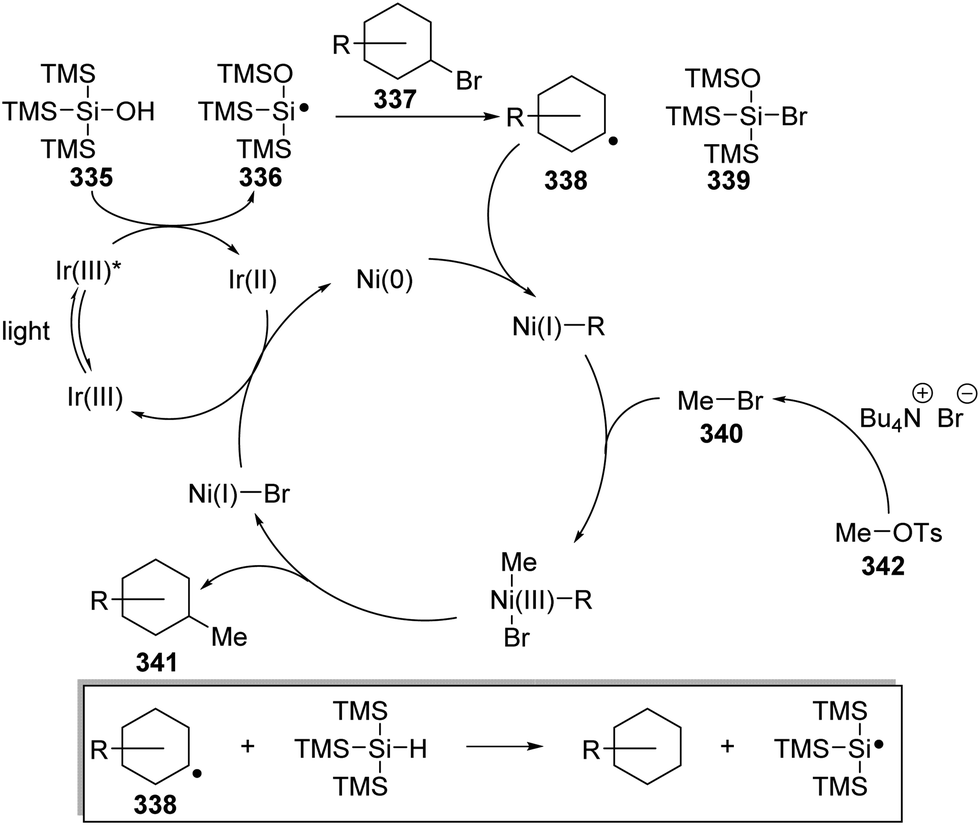
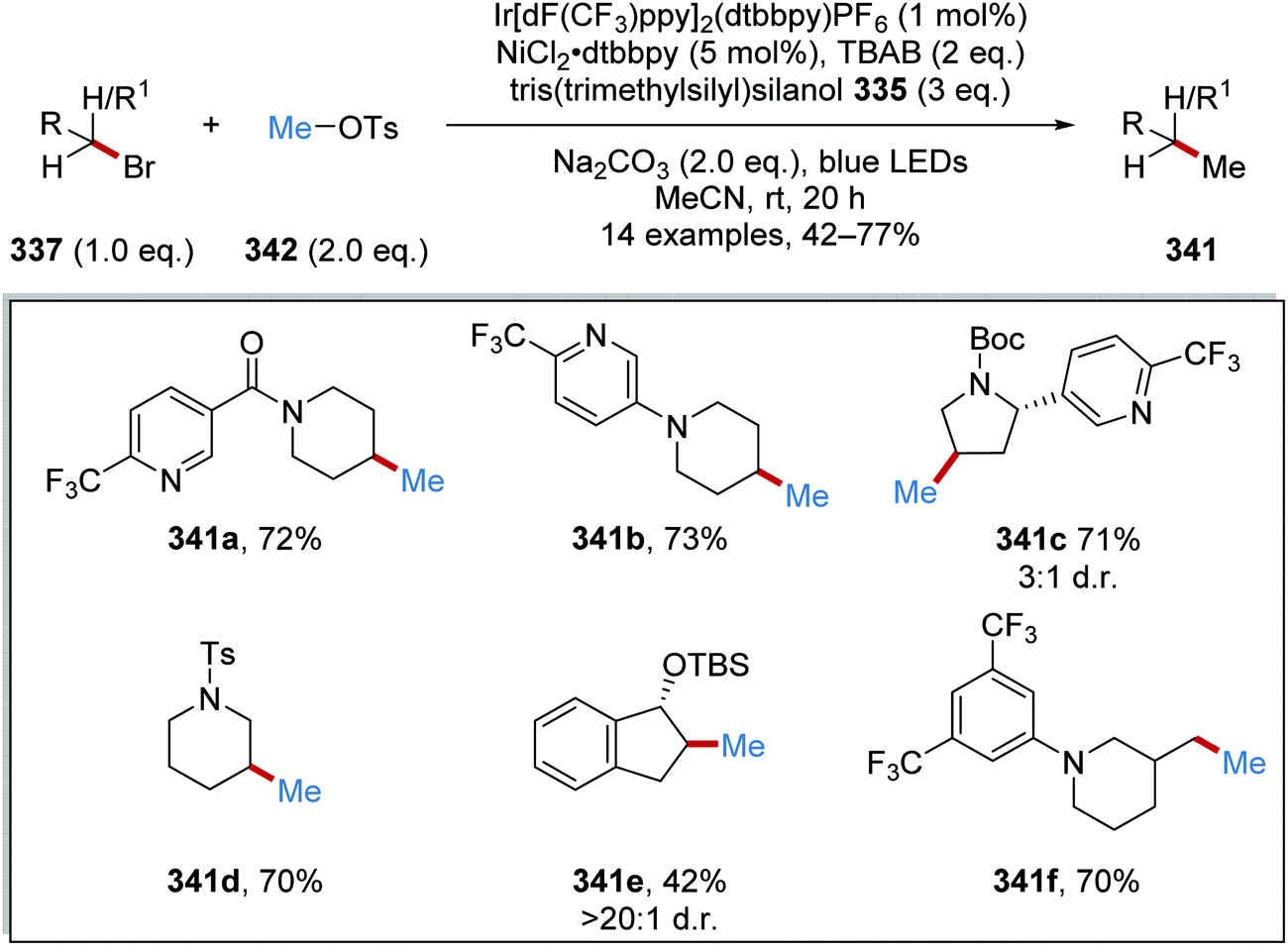
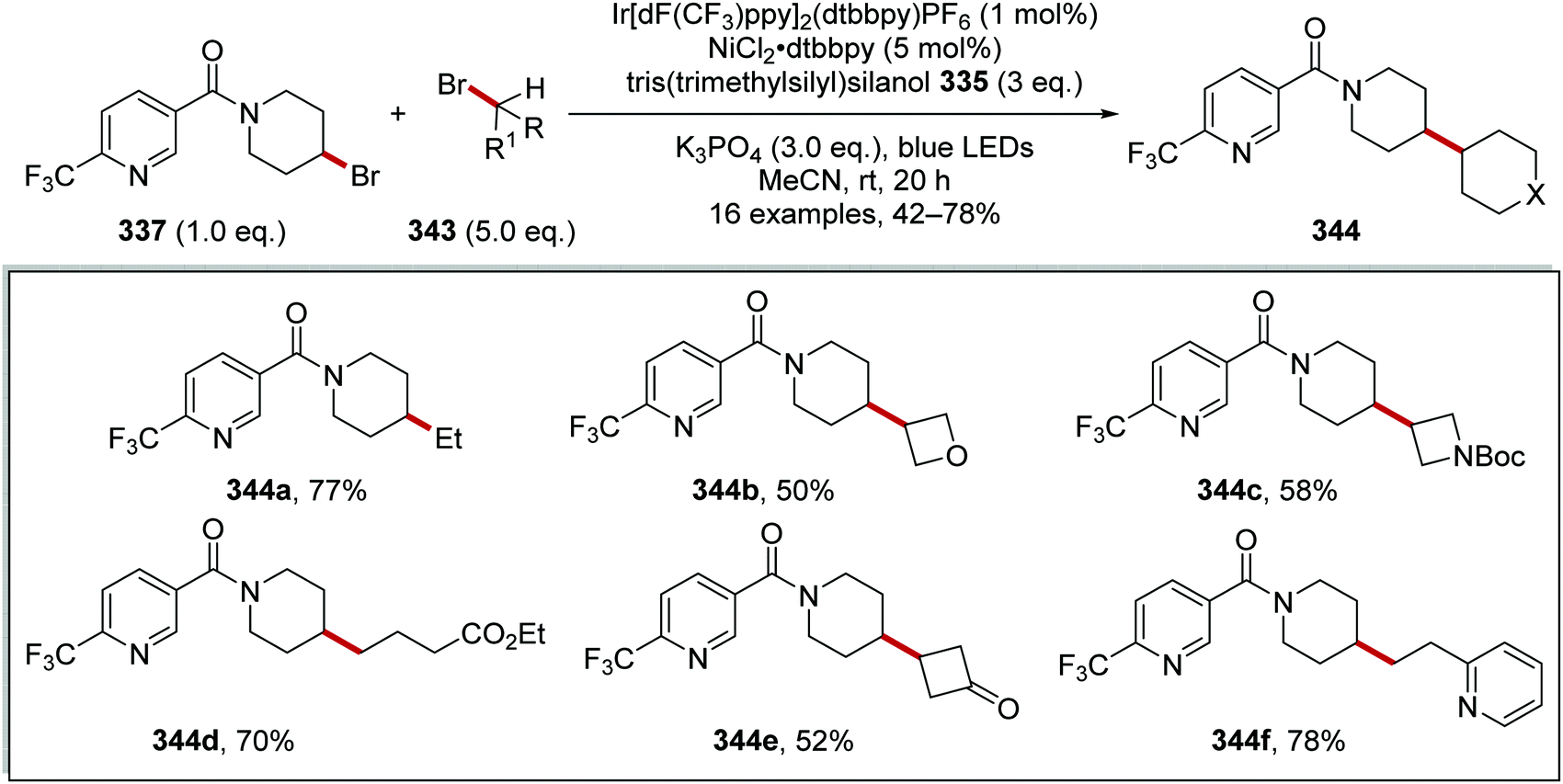
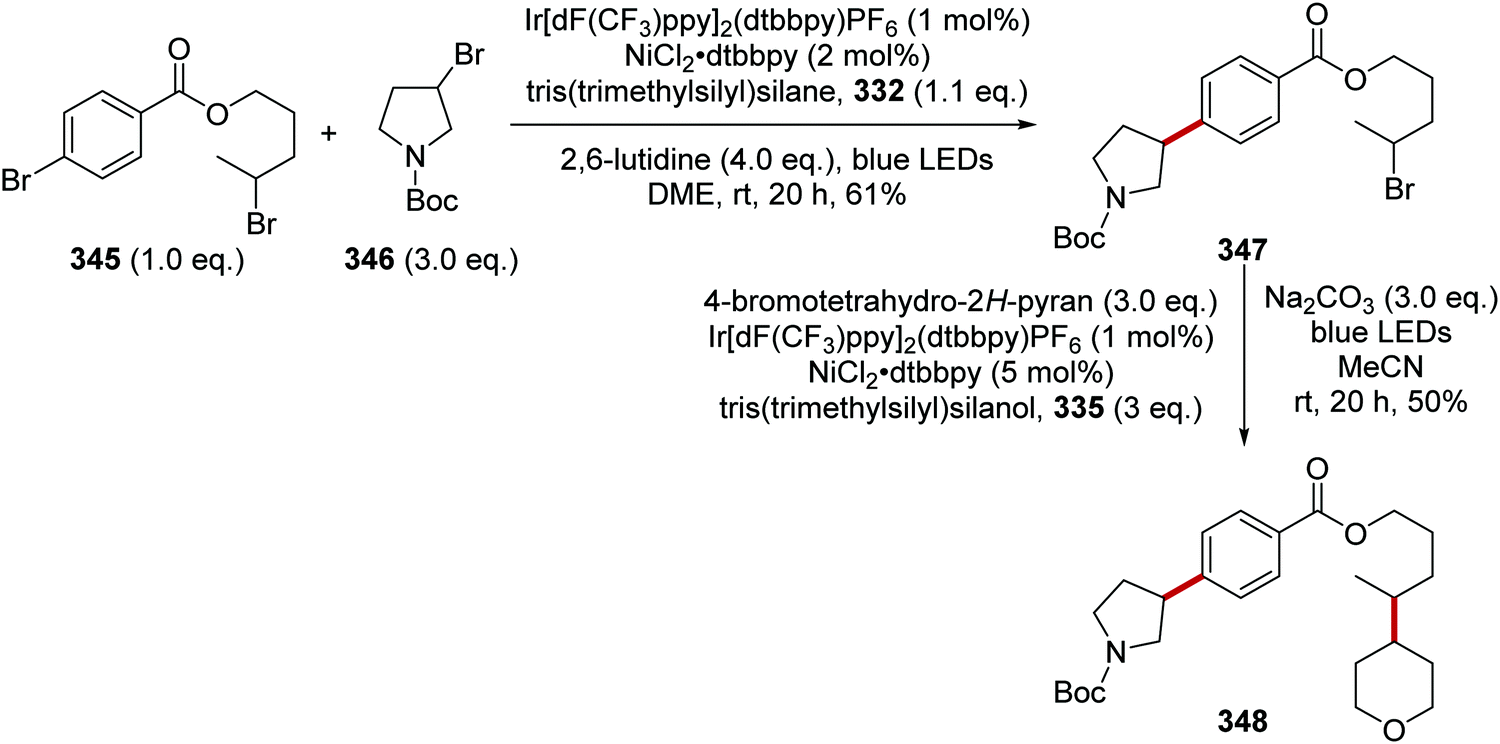
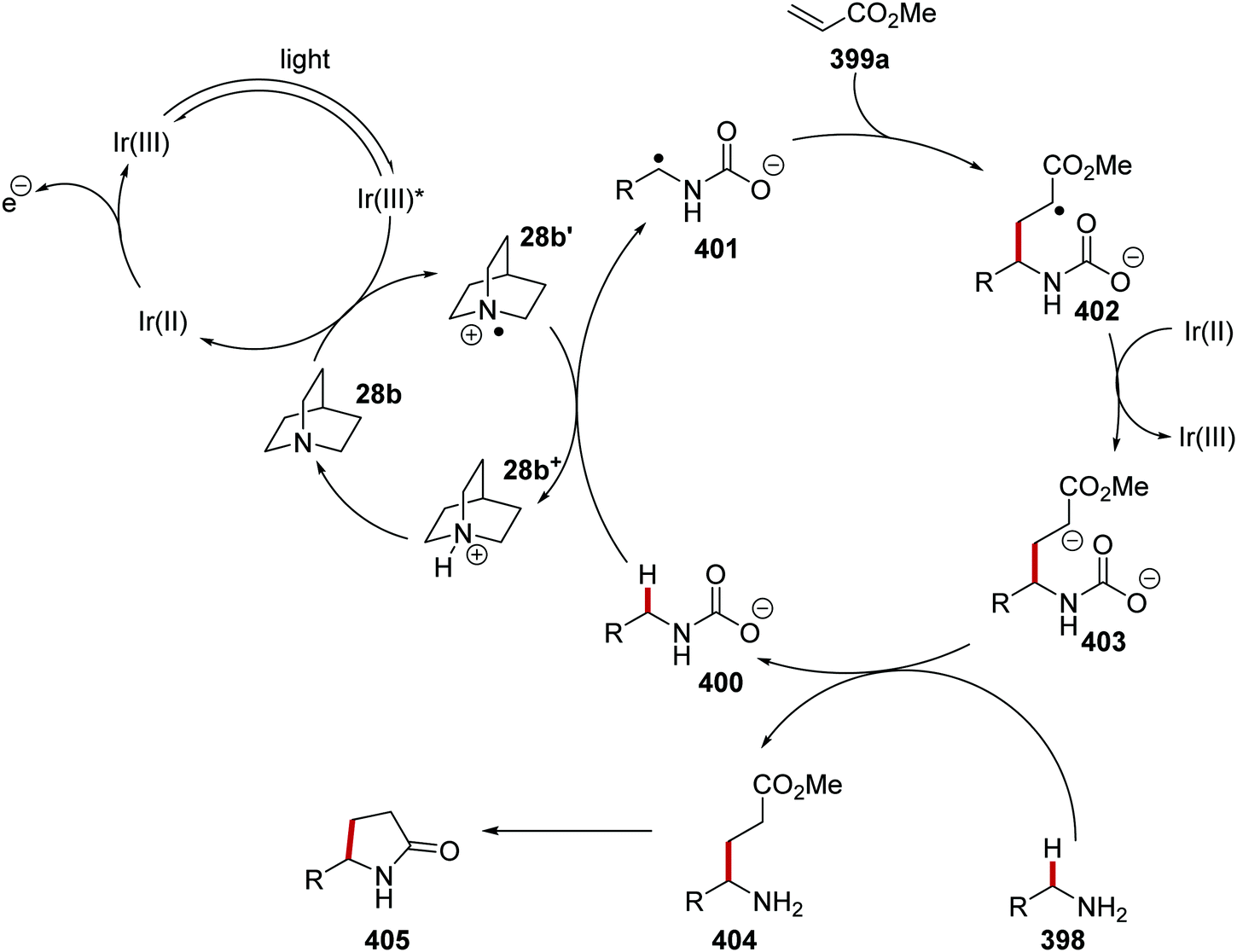
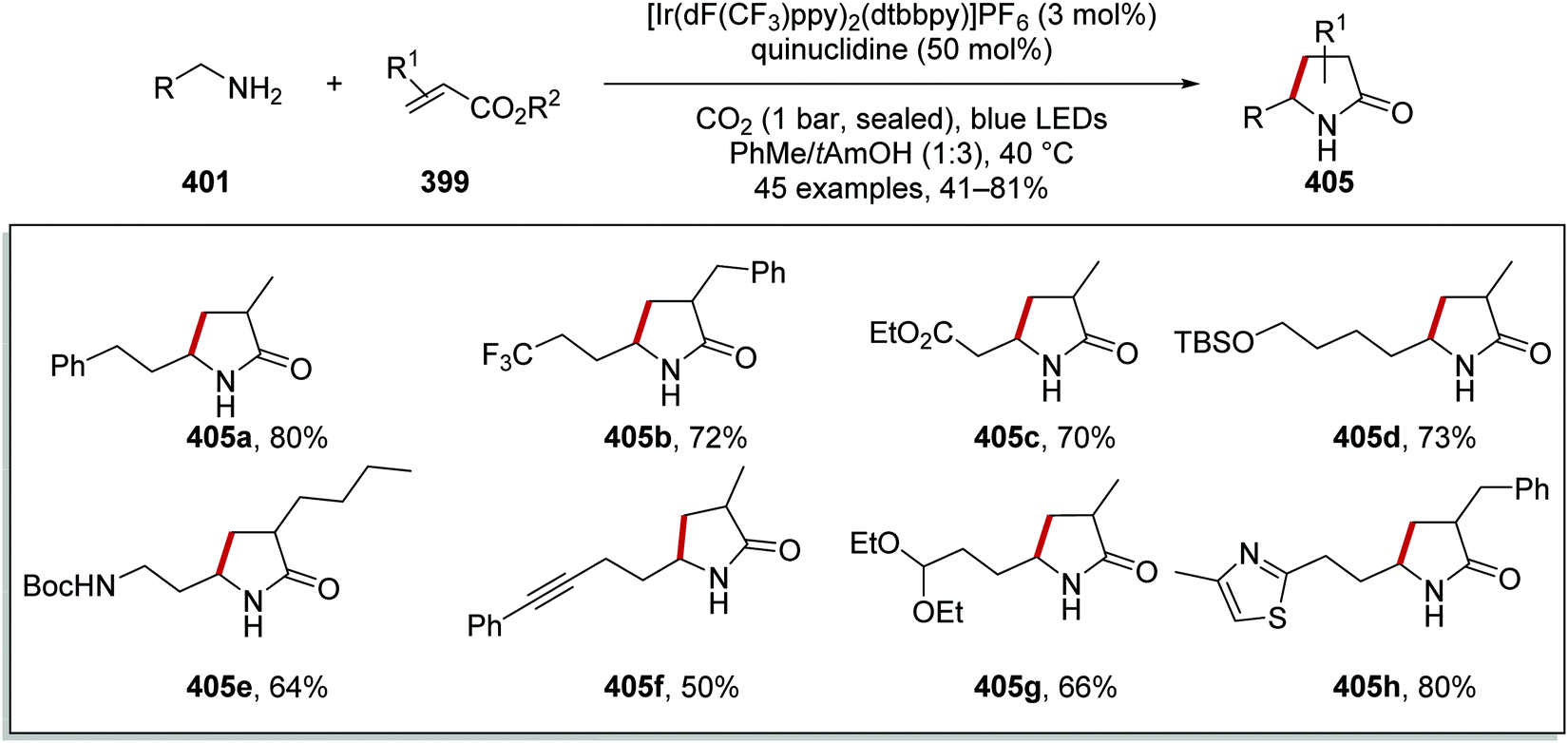
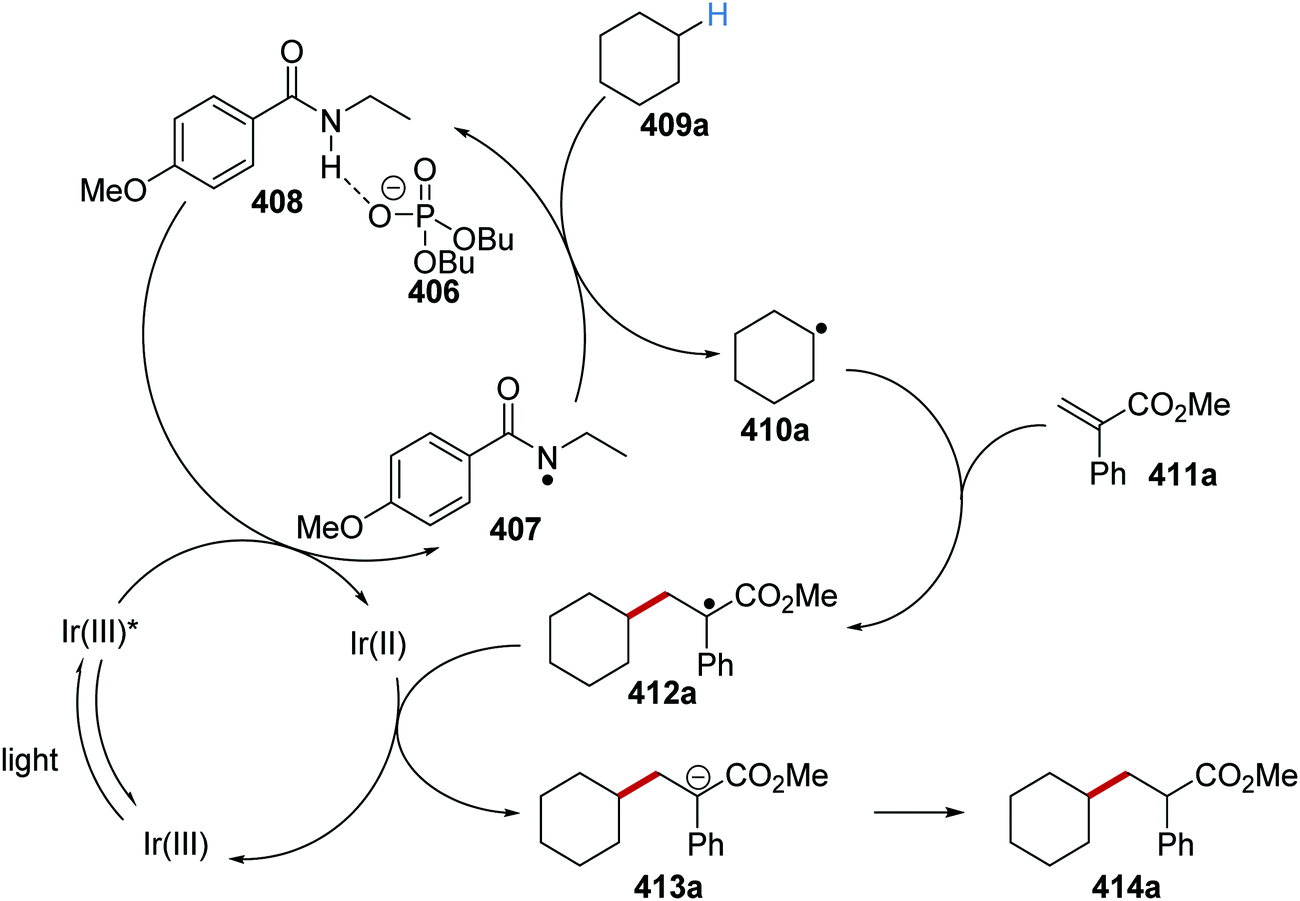
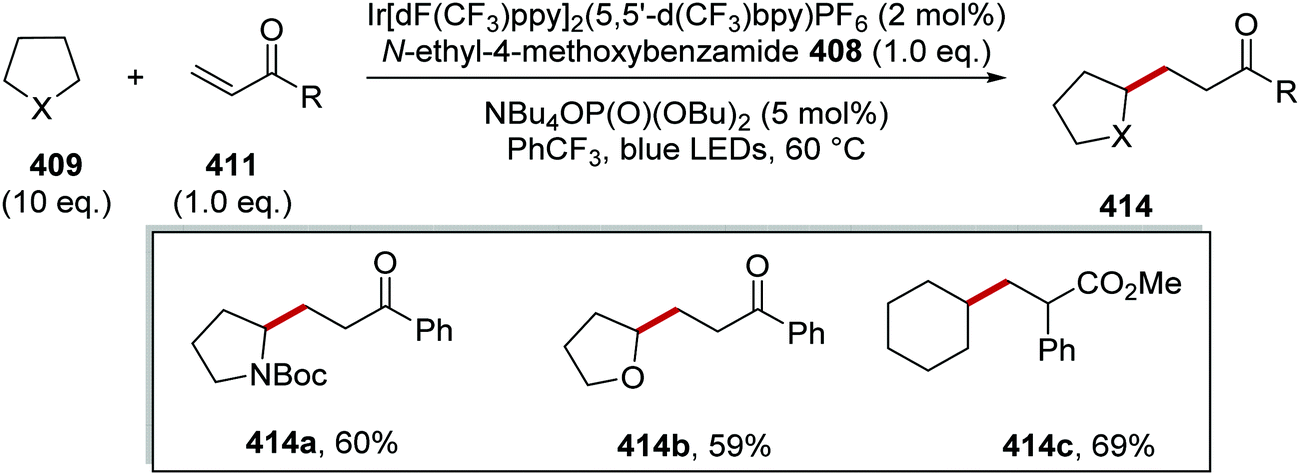
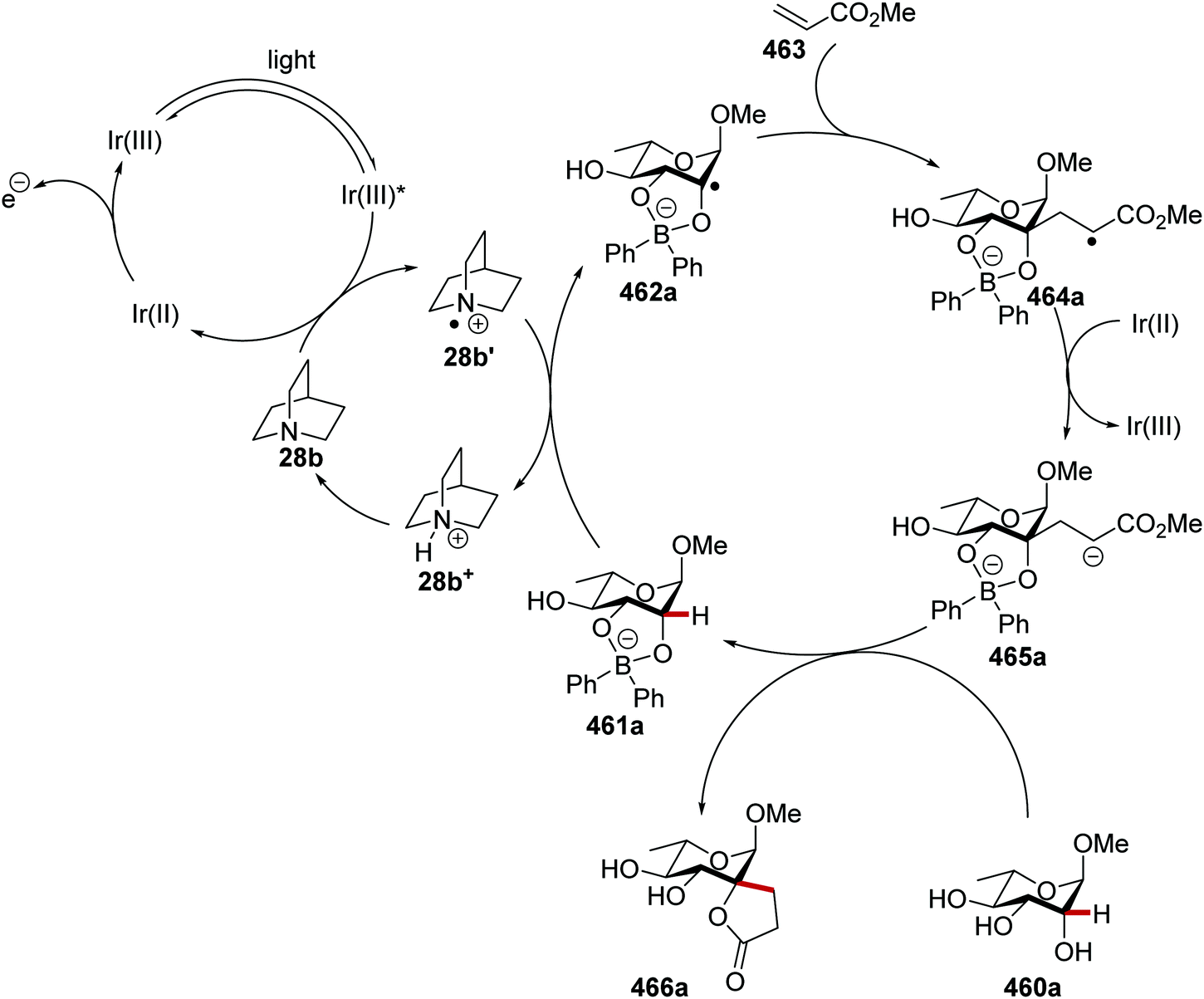
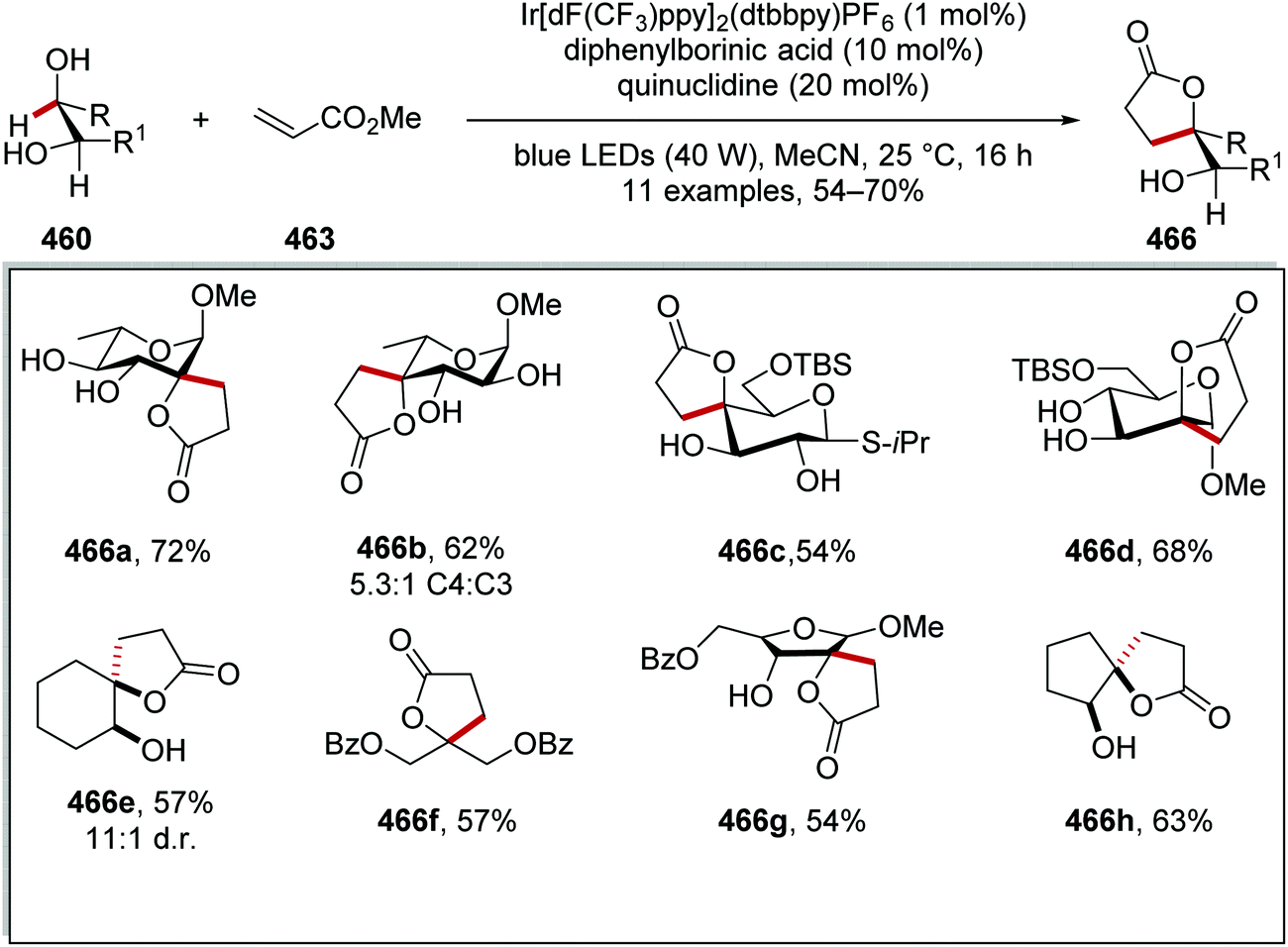
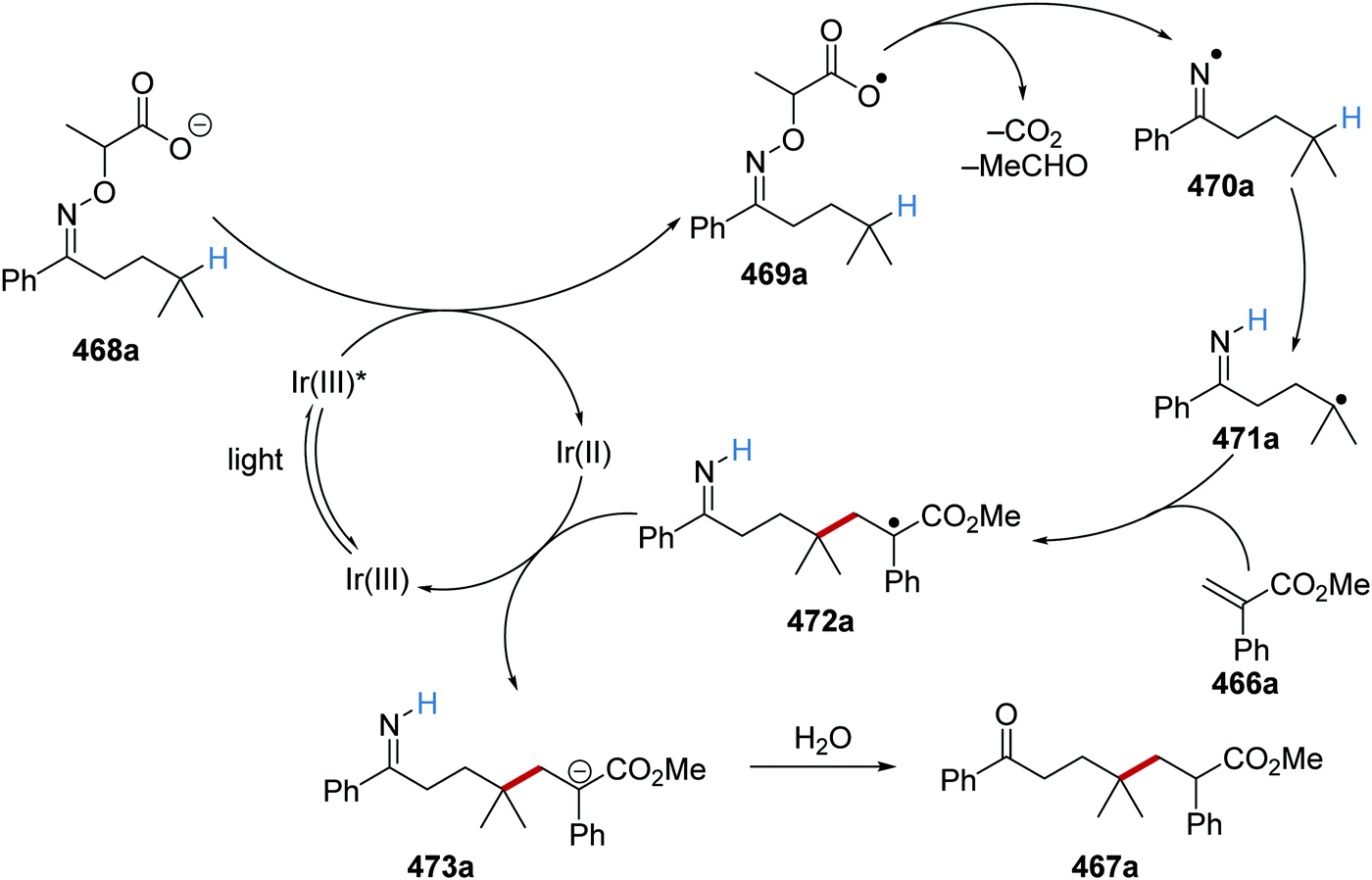
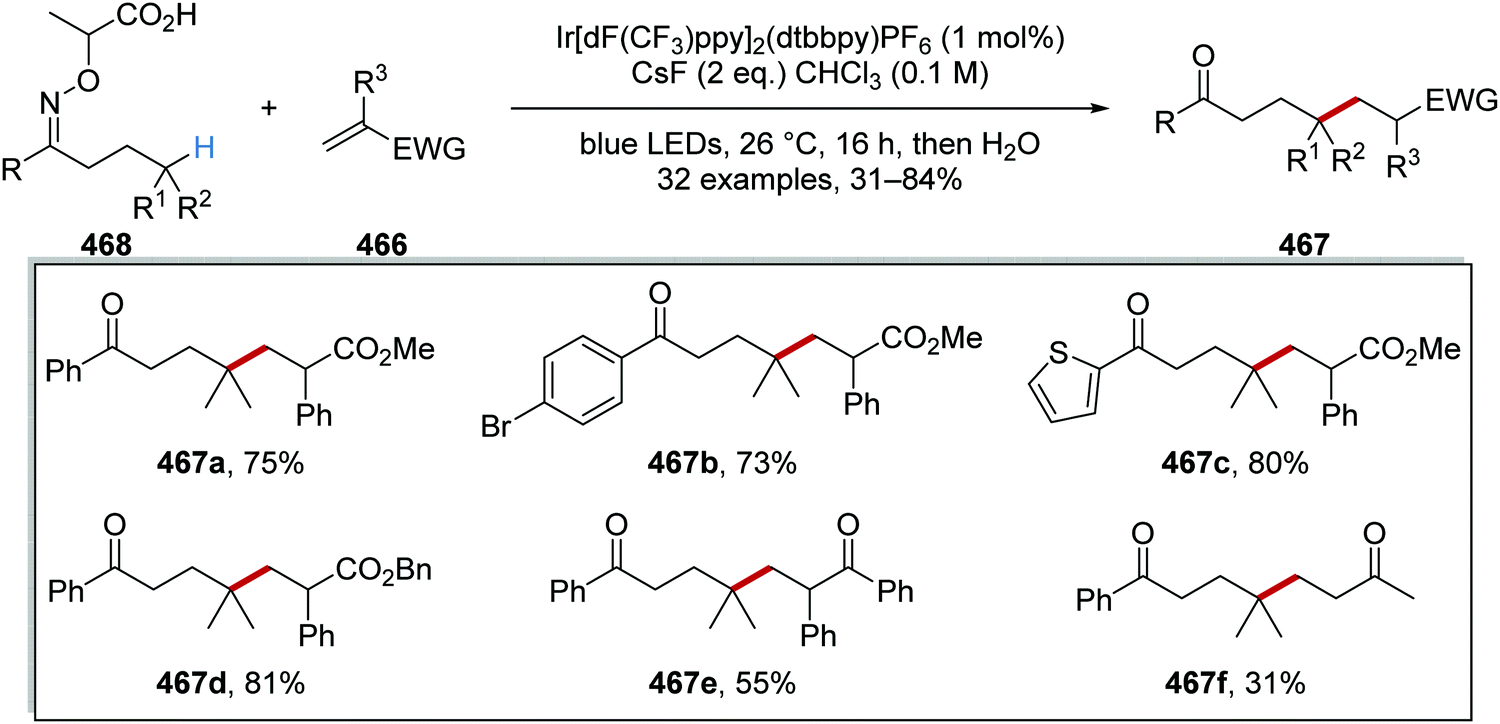
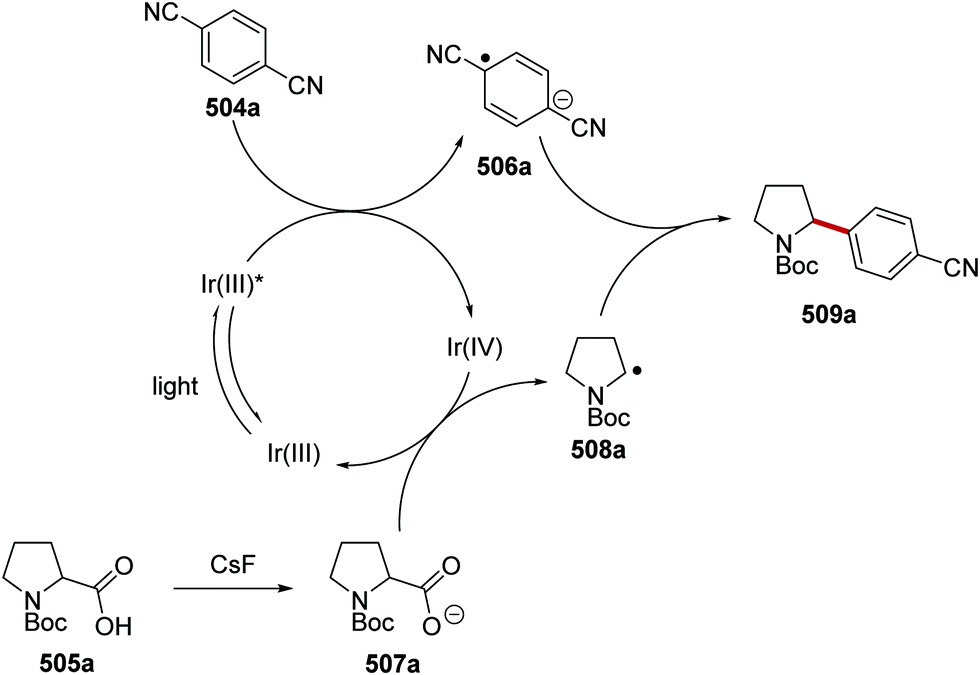
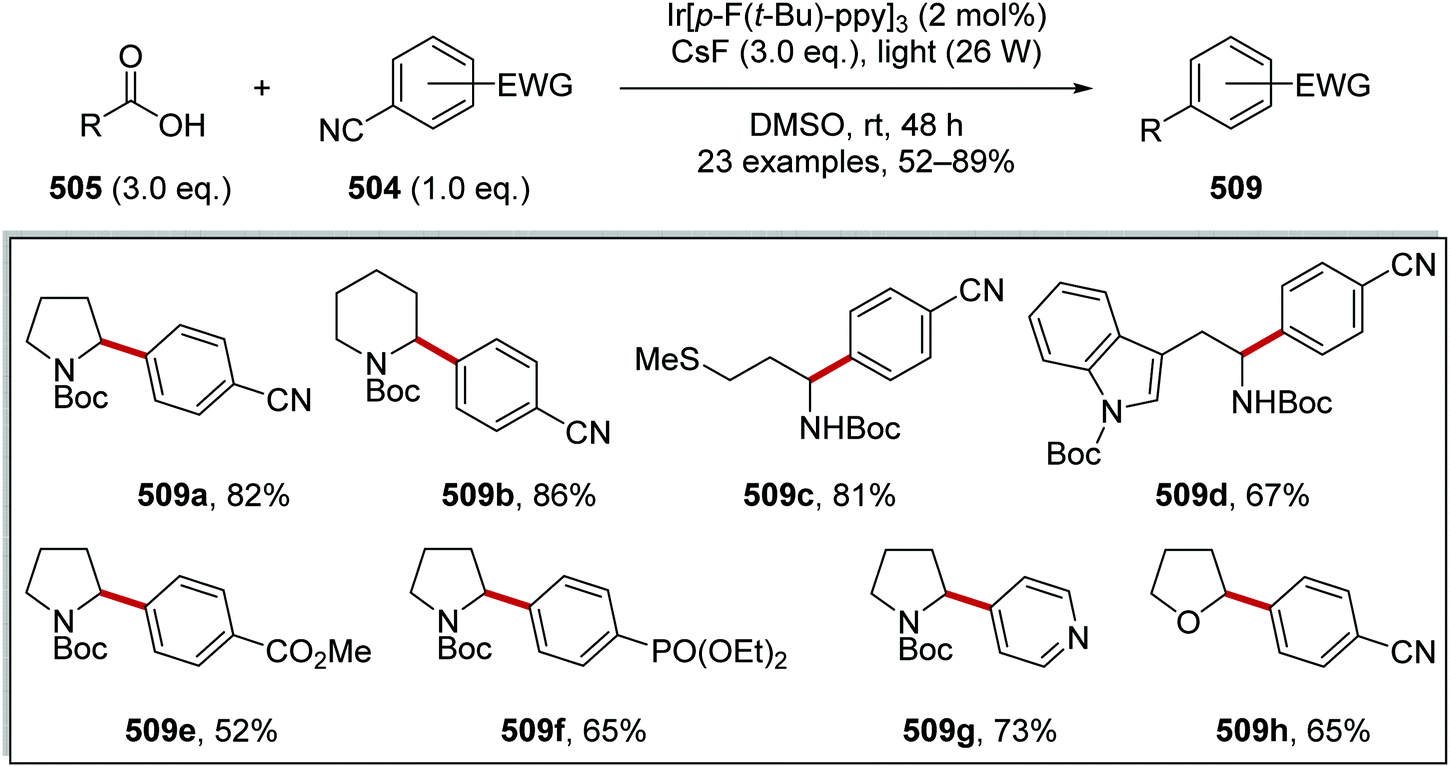
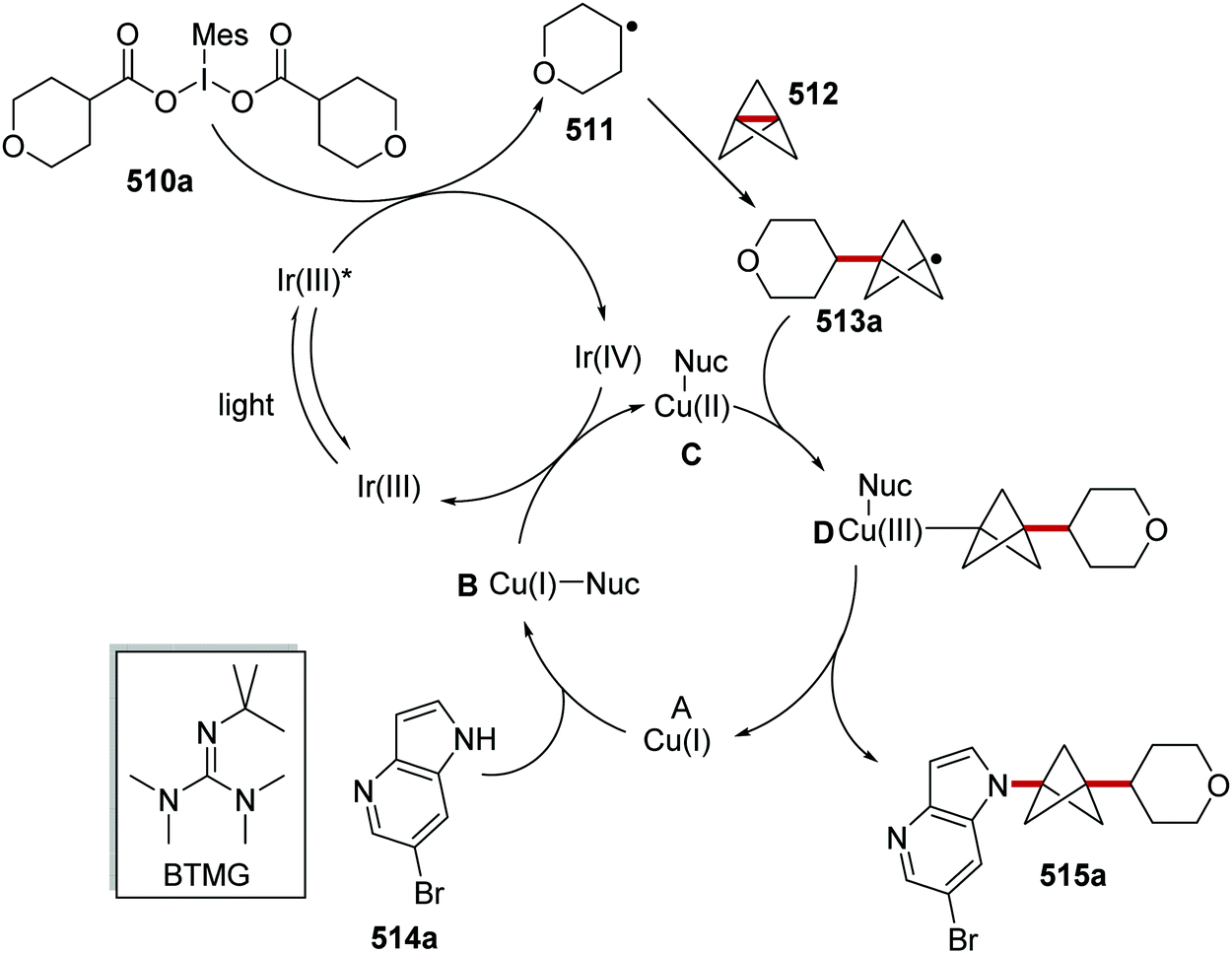
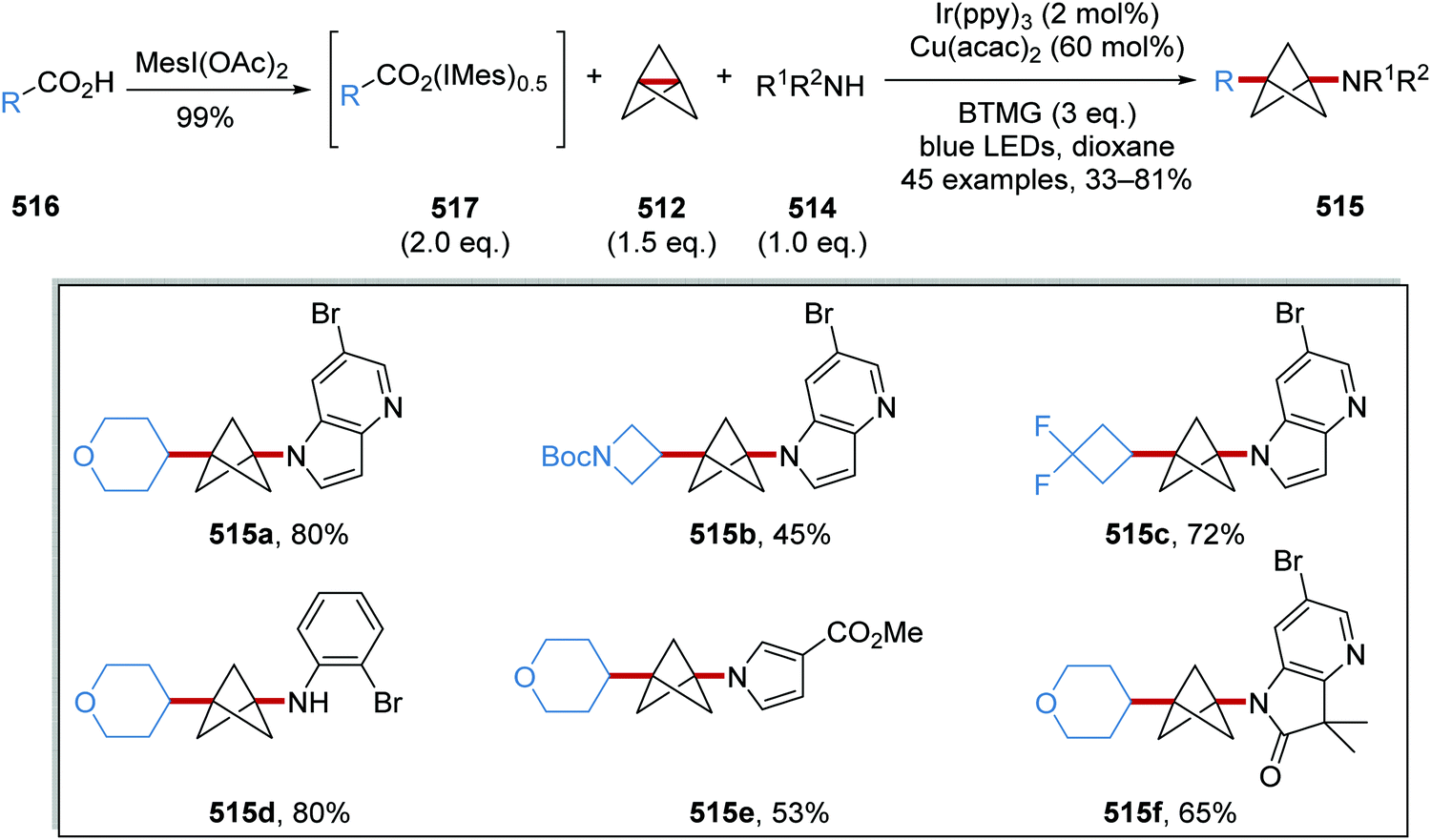
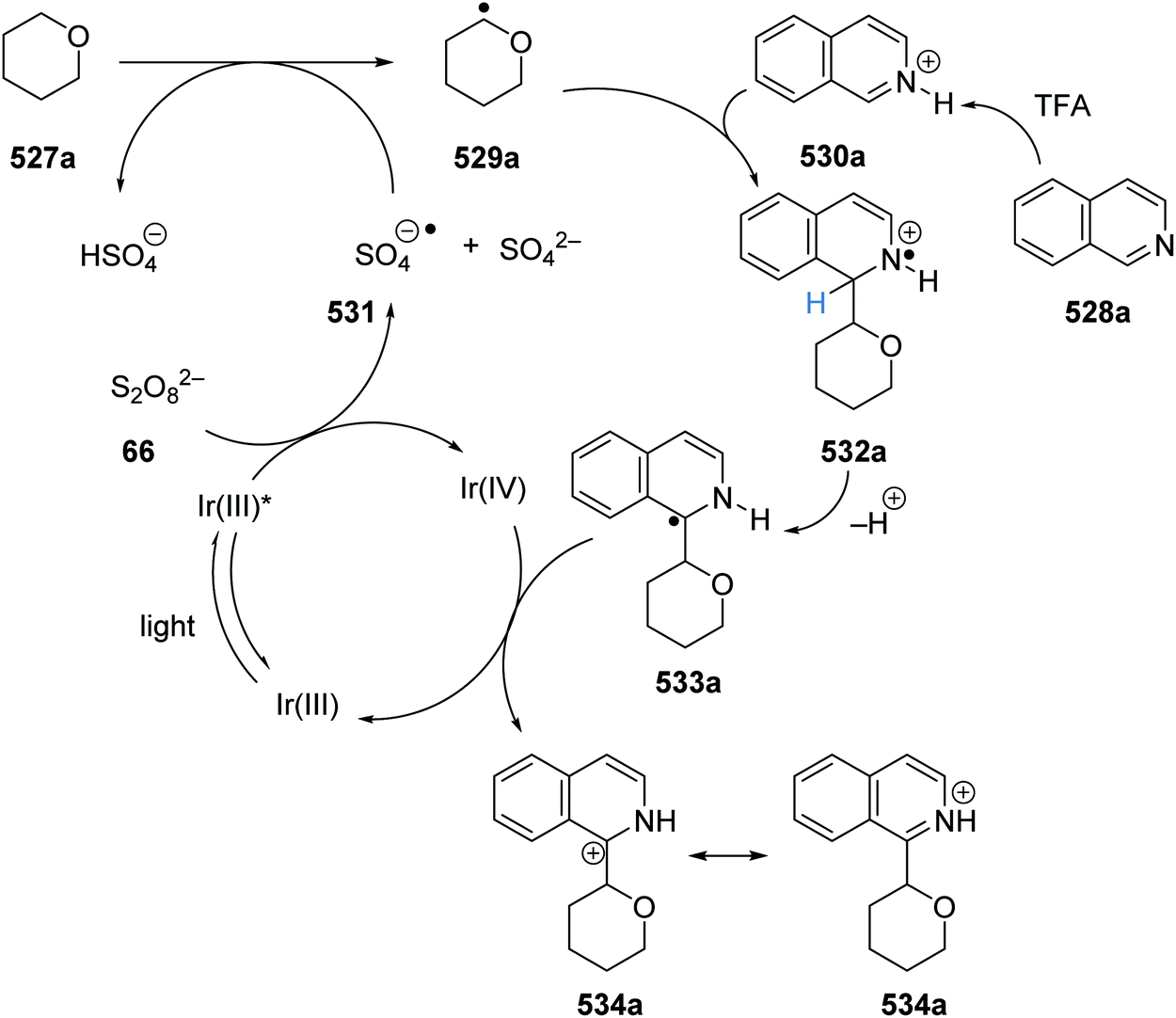
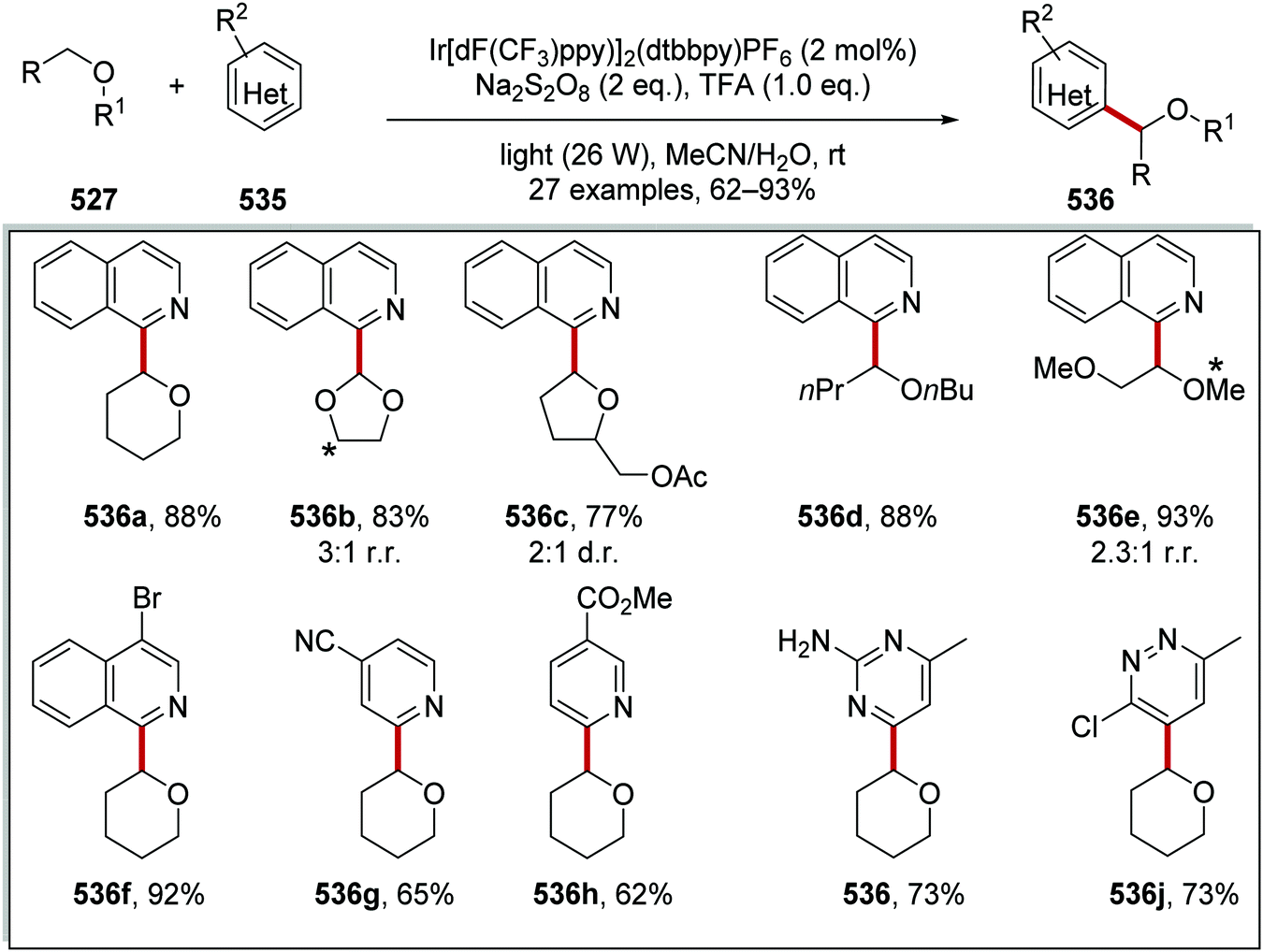
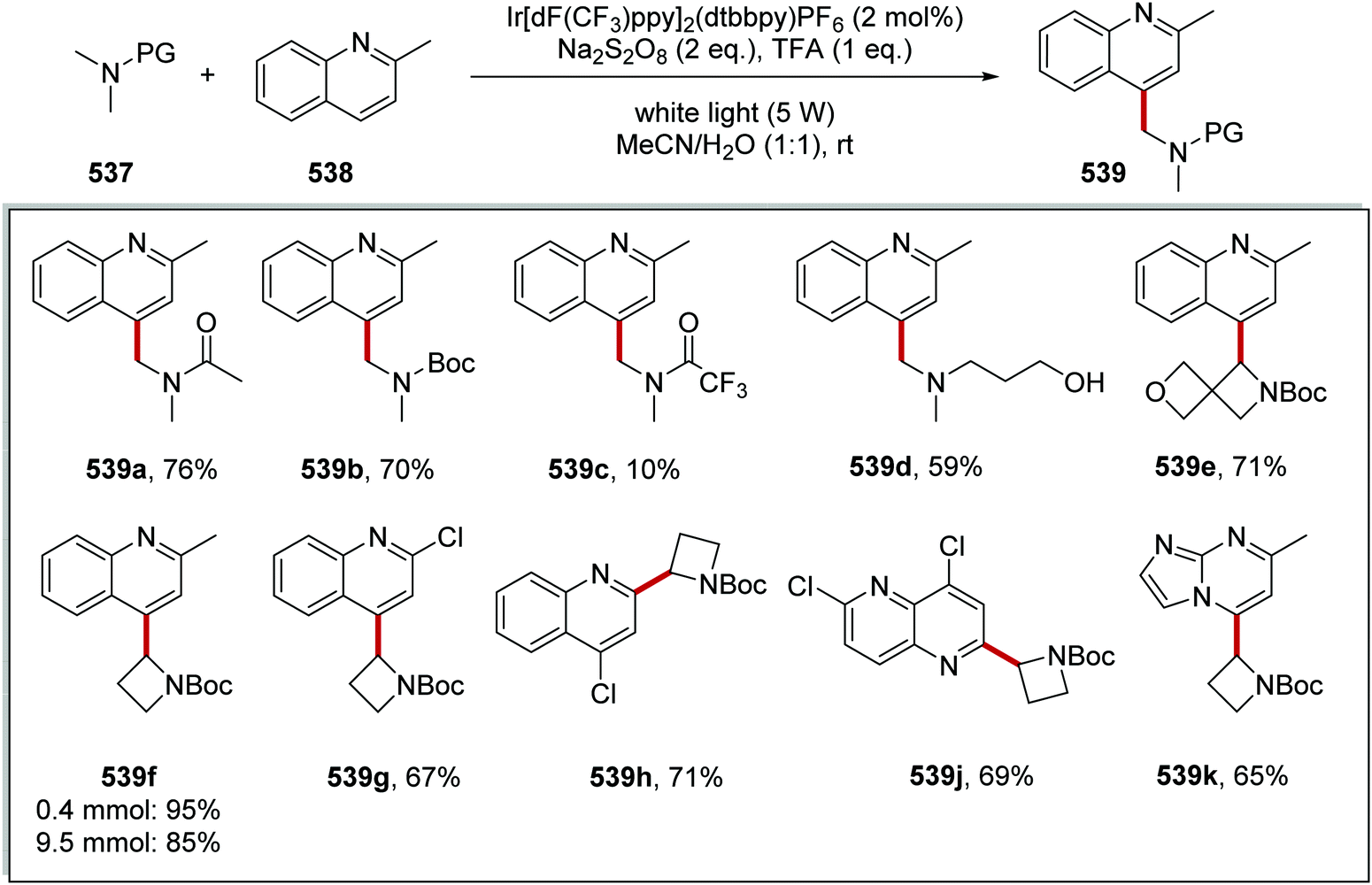
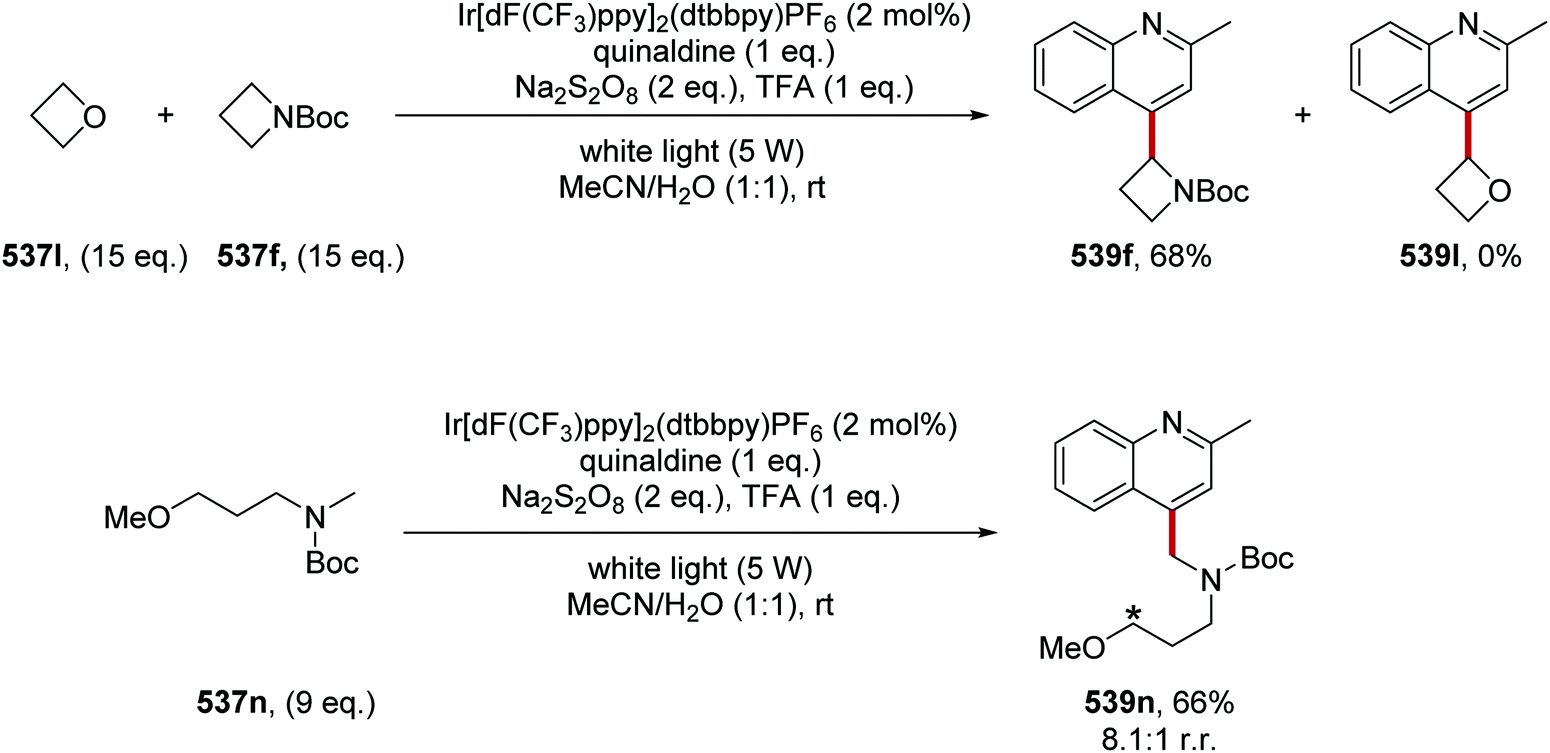
Oxalate decarboxylation.
Alcohols 271 were used as radical precursors via oxalate 272via a radical decarboxylation reaction.141,142 Oxalates such as 272a were prepared by reacting alcohol 271a, with oxalyl chloride and after quenching the reaction mixture with water, this gave compound 272a in 99% yield (Scheme 70).
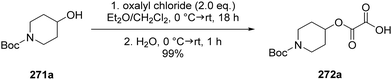 |
| | Scheme 70 Preparation of oxalate 272a from alcohol 271a. | |
Coupling of oxalates 272 with aryl bromides 276 was accomplished with an iridium and nickel catalytic system.142 Oxalate 272a was deprotonated with caesium bicarbonate and this gave anion 273a (Scheme 71). The iridium photocatalyst was excited with blue light and this afforded Ir(III)* species. A SET to Ir(III)* [Ir(dFppy)2(ptbbpy)PF6, Ered* = +1.1 V vs. SCE] from 273a (Ep/2 = +1.26 V vs. SCE) gave Ir(II) species and radical 274a, which underwent decarboxylation and alkyl radical 275a was formed. The nickel catalyst intercepted radical 275a and giving a Ni(I) complex. Oxidative addition of aryl bromide 276a gave a Ni(III) species, which underwent reductive elimination giving coupled product 277a.
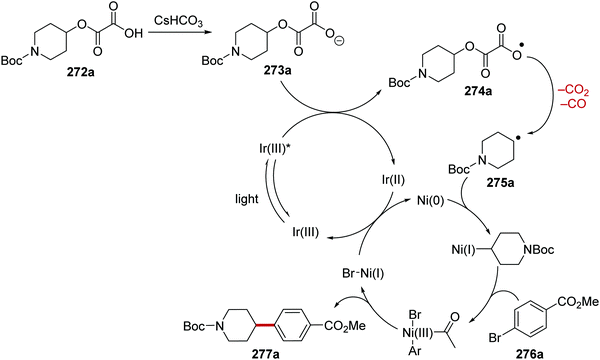 |
| | Scheme 71 Coupling of oxalate 272a with aryl bromide 276a. | |
The reaction of oxalates 272 with aryl bromides 276 was a highly facile reaction with the preparation of 33 coupled compounds 277 in yields of 37–91% (Scheme 72). A wide range of functional groups was tolerated, and this allowed for the preparation of functionalised derivatives 277a–f. The synthetic utility of this reaction was demonstrated with the preparation of the bioactive steroid adduct 277g in 51% yield.
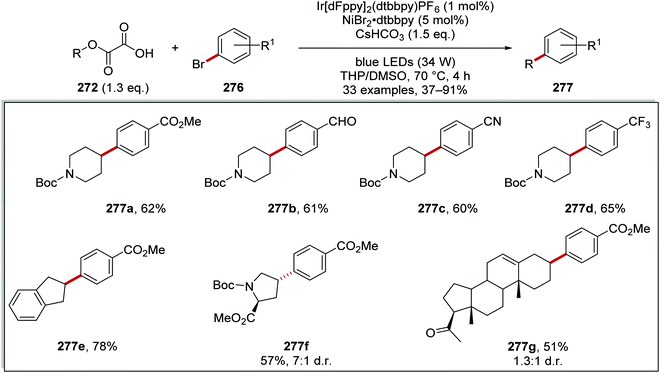 |
| | Scheme 72 Coupled products synthesised from corresponding alcohols. | |
The coupling of oxalates was also achieved with an iridium photocatalyst and radical decarboxylation strategy (Scheme 73).141 A range of different electron-poor alkenes gave the products 280a–g in yields of 67–95%. It was also shown that bioactive compounds were tolerated under the reaction conditions; for example, steroid-derived ester 280h was prepared in 85% yield.
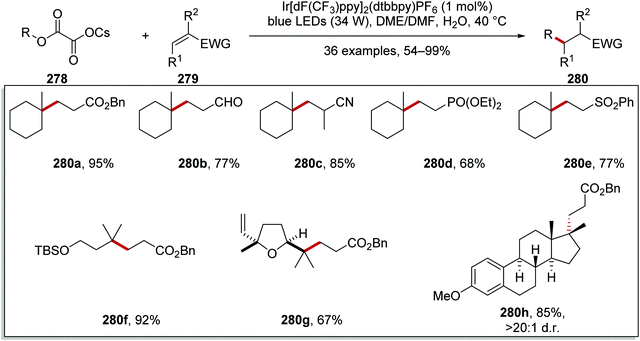 |
| | Scheme 73 Results of radical coupling between caesium carboxylates 278 and electron-poor alkenes 279. | |
Other decarboxylative substrates.
The inclusion of the bicyclopentane (BCP) bioisosteres into compounds was accomplished with a decarboxylative radical strategy.143 SET oxidation of carboxylate 281 with Ir(III)* complex resulted in carboxyl radical 282 and Ir(II) species (Scheme 74). The cyclohexanecarboxylate anion has an electrochemical potential of +1.16 V vs. SCE132 and therefore Ir[dF(CF3)ppy2]dtbpy(PF6) (Ered* = +1.21 V vs. SCE)8b is able to oxidise 281. Radical decarboxylation led to amidyl radical 283, which added to [1.1.1]propellane 284 and led to alkyl radical 285. The inclusion of NCS (286a) led to compound 287 and succinimidyl radical 288a. The iridium catalytic cycle was closed with a SET to 288a from Ir(II) and this gave anion 289a along with the original Ir(III) catalytic species.
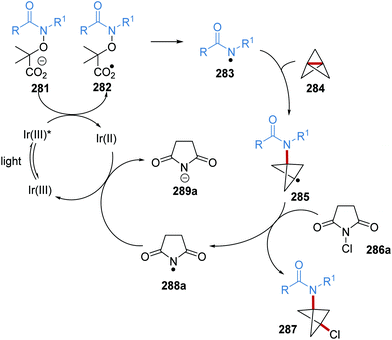 |
| | Scheme 74 Formation of bicyclopentane 287 from carboxylate 281. | |
This transformation worked well when N-chlorosuccinimide was used as 286 and this gave BCPs 287a–d (Scheme 75). When bromotrichloromethane was used in the reaction as 286, this gave 287e in 20% yield. Trifluoromethylthio 287f, thioether 287g and selenyl ether 287h derivatives were prepared from N-trifluoromethylthiophthalimide, N-phenylthiophthalimide and N-phenylselanylphthalimide.
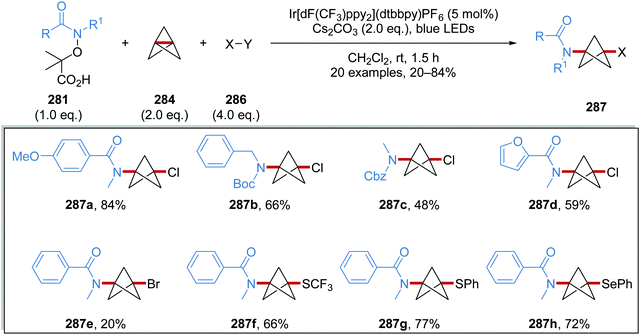 |
| | Scheme 75 Synthesis of amide-functionalised BCPs 287. | |
Silanes.
α-Aminosilanes 288 were used in a Giese coupling reaction with electron-poor alkenes 291 (Schemes 76 and 77).144 Control reactions gave evidence for the mechanism for this transformation. The use of N-methyldiphenylamine gave no Giese coupled product but trimethylsilyl analogue 288a with cyclohexenone gave coupled compound 294a in 90% yield. Furthermore, when the reaction was performed with no aqueous work-up, silyl enol ether 293a was isolated instead. Therefore, the following reaction mechanism was proposed. Irradiation of the reaction mixture with blue light gave the Ir(III)* complex from the iridium catalyst. Electron transfer from 288a to Ir(III)* resulted in amine radical cation 289a (Scheme 76). As previously mentioned, the presence the trimethylsilyl group decreases the oxidation potentials of substrates and thus 288a has a very low oxidation potential (+0.41 V vs. SCE;144bN-methyldiphenylamine Eox = 0.94 V vs. SCE).144c Loss of trimethylsilyl cation gave carbon-centred radical 290a which added to enone 291a, forming radical 292a. SET involving the Ir(II) complex, the TMS cation and 292a gave silyl ether 292a, which was detected in control experiments and this closed the iridium catalytic cycle.
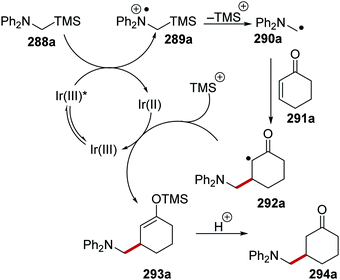 |
| | Scheme 76 Redox activation of silylamines. | |
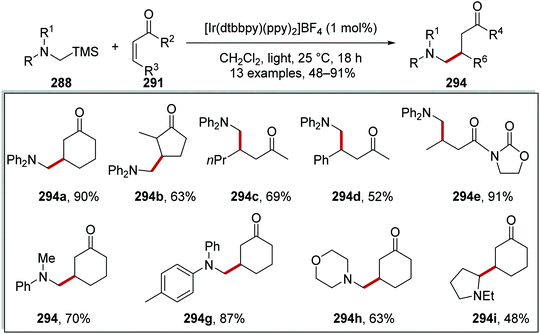 |
| | Scheme 77 Results of iridium-mediated coupling of α-silylamines 288 to electron-poor alkenes 291. | |
Optimisation studies found that use of the Ir(dtbbpy)(ppy)2BF4 catalyst gave coupled product 294a in 90% yield. However, the use of Ru(bpy)3(PF6)2 returned 294a in 40% yield. The optimised reaction conditions were used to prepare a library of 13 coupled amine compounds 294. Under the reaction conditions, silylaniline 288a was coupled to a range of electron-poor alkenes and this products 294a–e in 52–91% yield. Many different amines, including N-ethyl-2-trimethylsilylpyrrolidine, were compatible substrates and these gave products 294f–i.
Use of α-aminosilanes 295 with electron-poor alkenes 298 (not shown in Scheme 78) and an Ir/Yb catalytic system allowed for the formation of tricyclic compounds 302.145 The reaction progressed with single-electron oxidation of indole 295a (Eox = +1.16 V vs. Ag/AgCl) with Ir(III)* complex and this gave radical cation 296a and Ir(II) species (Scheme 78). The loss of TMS+ from 296a resulted in carbon-centred radical 297a. Addition of 297a to ytterbium-activated enone 299a, from alkene 298a, gave α-keto radical 300a. The electron-poor radical present in 300a then added to the electron-rich heterocycle and this formed 301a. Oxidation and proton loss from 301a resulted in the formation of 302a.
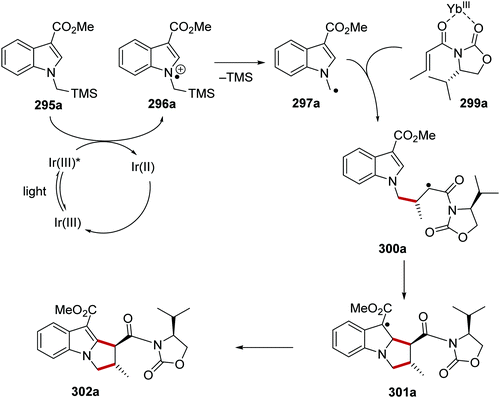 |
| | Scheme 78 Stereocontrolled coupling to activated alkene 299a. | |
Initially, the reaction was developed with a chiral oxazolidinone auxiliary 298 and this resulted in a highly diastereoselective transformation in combination with an ytterbium Lewis acid and a bipyridyl ligand (Scheme 79). Various indoles and electron-poor alkenes led to 302a–g being isolated. However, the use of pyrroles led to the preparation of 302h in 14% yield and a 93:3 d.r.
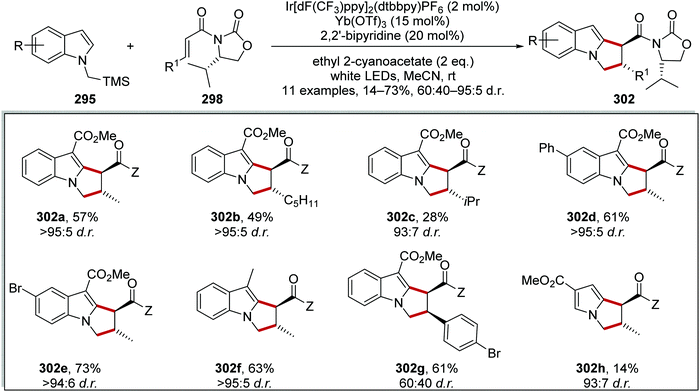 |
| | Scheme 79 Lewis acid-mediated diastereoselective radical ring formation of α-silyl amines and electron-poor alkenes. | |
An enantioselective version of this reaction was developed with pro-chiral alkene 303 (Scheme 80).145 The enantioenriched products 304a–d were formed with modest ee using a Lewis acid/chiral ligand strategy with a PyBox ligand 305.
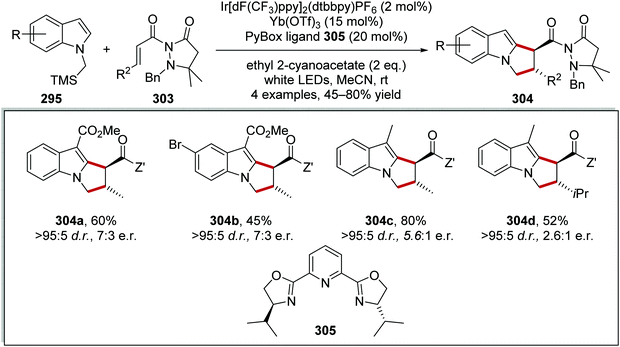 |
| | Scheme 80 Lewis acid-mediated enantioselective radical ring formation of α-silyl amines and electron-poor alkenes. | |
Boronates has radical precursors.
The coupling of boronate-containing amino esters 306 to aryl bromides 276 under an iridium/nickel catalytic system was reported.146 Under the reaction conditions, the Ir(III) complex was excited by blue light giving the Ir(III)* complex (Scheme 81). SET between Ir(III)* complex (Ered* = +1.21 V vs. SCE) and boronate 306 (Eox = +1.01 V vs. SCE)resulted in radical 307 and the Ir(II) complex. The iridium catalytic cycle was closed with a SET with Ni(I) complex D and this resulted in Ni(0) complex and the original Ir(III) species. Radical 307 was intercepted by Ni(0) complex A and this gave Ni(I) species B. Oxidative addition of aryl bromide 276 to B resulted in complex C, which underwent reductive elimination yielding coupled product 308 and Ni complex D.
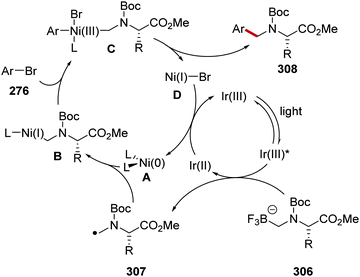 |
| | Scheme 81 Mechanism of coupling with boronate salts as radical precursors. | |
This transformation allowed for a wide-range of α-amino esters 308 to be prepared from α-amino boronic salts 306 and aryl bromides 276 (Scheme 82). Benzaldehyde 308a, benzothiophene 308b, sulfonamide 308c, boronic ester 308d, caffeine 308e, oxadiazole 308f, ketone 308g, benzonitrile 308h and pyrimidine 308i analogues were all prepared in the yields shown. From chiral HPLC of two of the synthesised amino esters, the stereointegrity of these compounds was not compromised.
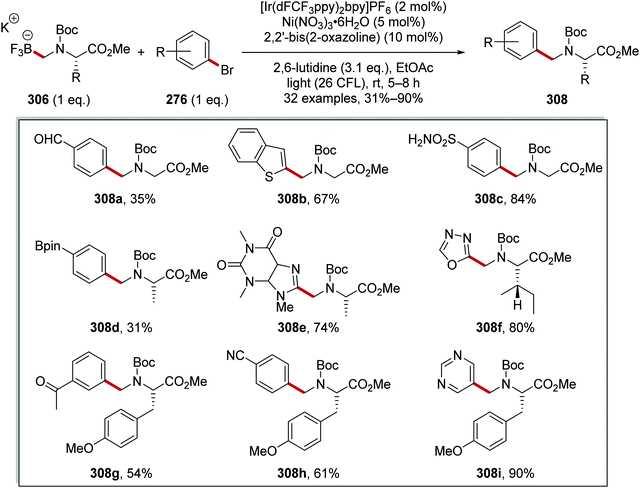 |
| | Scheme 82 Coupling of amino acid-derived boronic salts with aryl bromides. | |
All-carbon quaternary centres were prepared from organo-trifluoroboronate salts (Scheme 83).147 From optimisation studies, it was found that nickel-bipyridyl ligands were unsuitable for this transformation, as only starting material and protodehalogenation products were returned from the reaction mixture. Even with stoichiometric quantities of nickel catalyst and prolonged reaction times, no coupled compound was given with nickel bipyridyl complexes. Ligands for the nickel catalyst were screened, and it was found that only diketone-type ligands resulted in any significant product formation. The diketone, tetramethylheptanedione (TMHD) was the best ligand for the nickel complex for this transformation. The presence of inorganic bases, such as K2HPO4, decreased protodehalogenation and increased formation of 310a. The inclusion of Lewis acid ZnBr2 in the reaction mixture decreased the initial induction period and further increased the yield of 310a. Under these optimal conditions, coupled product 310a was prepared in a >95% HPLC yield and 90% isolated yield. The substrate scope of the reaction was examined with these optimised conditions. The tert-butyl group could be installed on electron-poor arene systems, such that ketone 310a, and 310e, aldehyde 310c, sulfone 310b and nitrile 310d analogues were all synthesised.
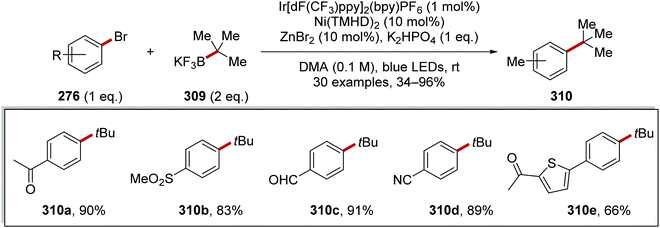 |
| | Scheme 83 Formation of all-carbon quaternary centres with boronates. | |
Alkyltrifluoroborates derived from enones were also used in this reaction (Scheme 84). Therefore, compounds such as ketone 312a, nitrile 312b and 1,3,4-oxadiazole 312c were all prepared. Electron-rich analogues such as dimethoxy 312d were isolated in diminished yields. Other alkyltrifluoroborates allowed for the preparation of 312e–h.
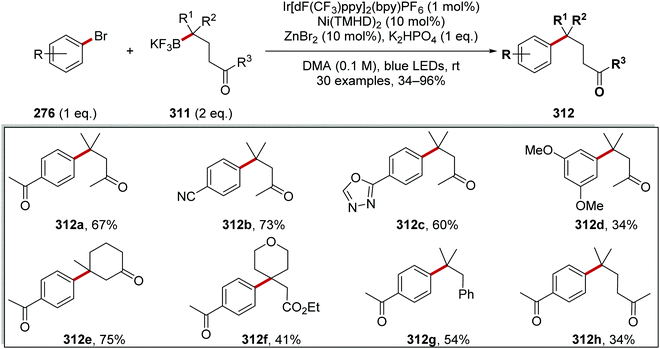 |
| | Scheme 84 Formation of all-carbon quaternary centres using a Ni/Ir mediated process. | |
The preparation of allylic alcohols 316 was achieved from alkyltrifluoroboronates 313via a radical Tsuji–Trost reaction under Ir photoredox conditions.148 The transformation was investigated with DFT calculations and from this, a plausible mechanism was suggested (Scheme 85). Under the reaction conditions, benzylic radical 314a was formed from boronate 313a (Eox = +0.93 V vs. SCE)148b and Ir(III)* complex (Ered* = +1.21 V vs. SCE). The iridium catalytic cycle was closed with electron transfer to Ni(I) from Ir(II) and this reformed the original Ir(III) catalytic species and a Ni(0) complex. It was calculated that complex formation between Ni(0) and vinyl epoxide 315a was favoured by 8.9 kcal mol−1 over the complexation between Ni(0) and benzyl radical 314a; the former led to a Ni(II) π-allyl complex. Addition of benzylic radical 314a to the Ni(II) complex resulted in a Ni(III) complex. Thereafter, C–C bond formation followed by reductive elimination was proposed to lead to the formation of allylic alcohol 316a and Ni(I) complex.
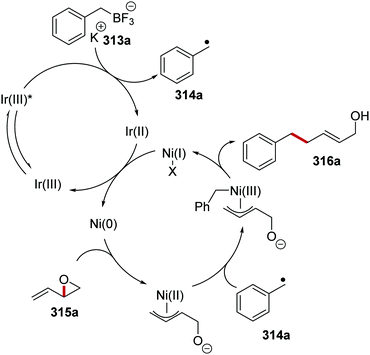 |
| | Scheme 85 Mechanism of ‘radical Tsuji–Trost’ reaction. | |
The reaction was tolerant of both electron-rich aryl rings, giving allylic alcohols 316a–c and electron-poor aryl rings affording 316d–f (Scheme 86). However, higher yields and greater stereoselectivity were found for the electron-poor analogues. Alkyl-substituted products 316g–i were also accessed.
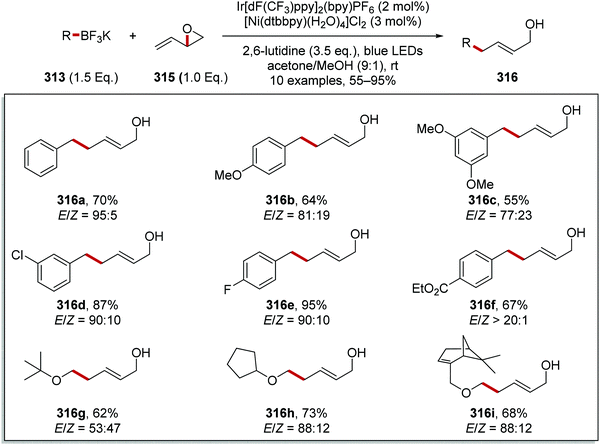 |
| | Scheme 86 Results of a Ni/Ir promoted Tsuji–Trost reaction. | |
A three-component reaction involving boronate 317, vinylboronic ester 319 and aryl bromide 321 under an Ir/Ni catalytic system resulted in saturated boronic ester products 322.149 Under the reaction conditions, boronate salt 317a (Eox = +1.26 V vs. SCE)149 was oxidised by Ir(III)* and this gave a tert-alkyl radical 318a (Scheme 87). Addition of radical 318a to alkene 319a resulted in α-boronic ester radical 320a. Radical 320a was stabilised via overlap with the p-orbital situated on the boron atom; it was intercepted by a Ni(0) complex and this resulted in a Ni(I) complex. Oxidative addition of aryl bromide 321a to the Ni(I) complex gave a Ni(III) complex. Reductive elimination of the Ni(III) complex resulted in boronate ester 322a and a Ni(I) species. Both iridium and nickel catalytic cycles were closed with an outer-sphere SET from the Ir(II) to the Ni(I) complex.
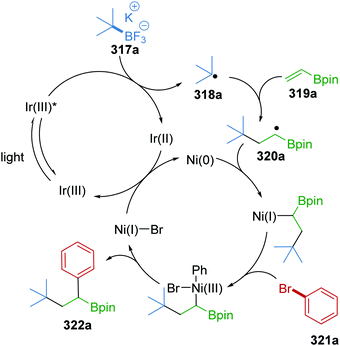 |
| | Scheme 87 Formation of boronic ester 322a from boronate salt 317a, alkene 319a and aryl bromide 321a. | |
This three-component reaction with an Ir/Ni catalytic system was highly facile, with the preparation of 57 analogues. As examples, analogues 322a–i were prepared in 62–85% yields (Scheme 88).
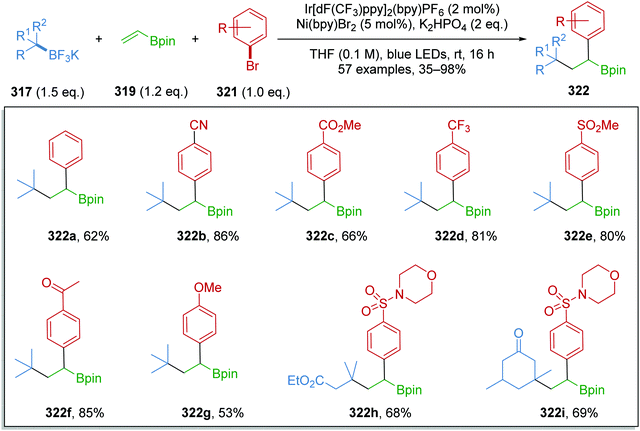 |
| | Scheme 88 Preparation of boronic esters 322via a Ni/Ir mediated three-component reaction. | |
Halide ions as radical precursors.
Alkyl halides 324 were used as radical precursors for C–C bond formation with an iridium, nickel, silane catalytic system (Schemes 89 and 90).150 Irradiation of the reaction mixture with blue light gave excited Ir(III)* complex from the Ir(III) complex (Scheme 89). Electron transfer from bromide ion (Eox = +0.80 V vs. SCE) to Ir(III)* (E*red = +1.21 V vs. SCE) resulted in a bromine atom and an Ir(II) complex. H-atom abstraction from tris(trimethylsilyl)silane (TTMSS) by the bromine atom resulted in the formation of silyl radical 323 and hydrogen bromide. Bromine abstraction from alkyl bromide 324a by silyl radical 323 gave alkyl radical 325a and silyl bromide byproduct. Nickel cross-coupling reaction between alkyl radical 325a and aryl bromide 321a resulted in coupled product 326a. The resulting Ni(I) complex accepted an electron from the Ir(II) complex to regenerate a Ni(0) complex and the original Ir(III) complex.
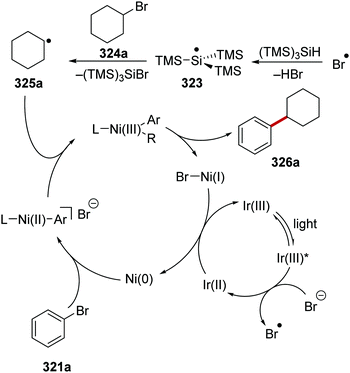 |
| | Scheme 89 Radical coupling of aryl bromides 321 with alkyl bromides 324. | |
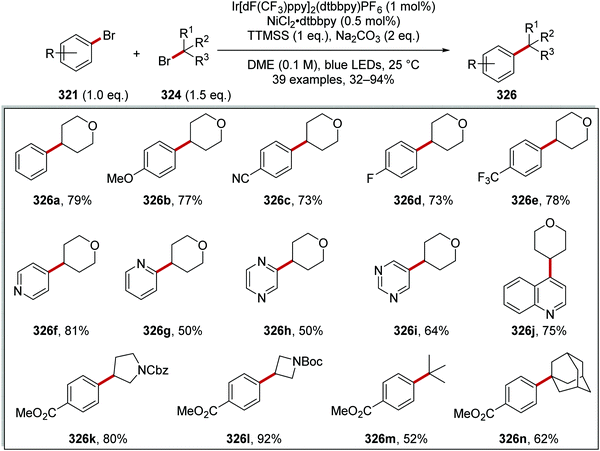 |
| | Scheme 90 Results of radical coupling between aryl bromides 321 and alkyl bromides 324. | |
The combination of aryl bromides 321 and alkyl bromides 324 with iridium and nickel complexes and a silane was successful for the preparation of 39 coupled compounds 326 (Scheme 90). This transformation was effective in preparing coupled compound 326a in 79% yield. Electron-rich anisole derivative 326b was prepared in 77% yield and electron-poor analogues 326c–e with yields ranging from 73% to 78%. Coupling to heterocycles as seen in 326f–j was achieved. The transformation also accommodated Cbz and Boc protecting groups with N-Cbz 326k and N-Boc 326l derivatives being prepared in 80% and 92% yield, respectively. Coupling of tertiary carbon centres was achieved with the formation of 326m and 326n.
The difluoromethylation of aryl bromides was also achieved with the same iridium, nickel catalytic system-mediated with supersilane TTMSS, [which is (TMS)3SiH] and 2,6-lutidine as base (Scheme 91).151 The substrate scope for this reaction was explored and, for 36 examples, the reaction was found to be very accommodating, with yields obtained ranging from 45% to 86%. Esters 329a, nitrile 329b, N-Boc amines 329c, methoxy 329d aryl chlorides 329e, boronic esters 329f and alkynes 329g analogues were all prepared. Furthermore, heterocycles were also functionalised, with pyridine 329h, pyrimidine 329i, pyrazole 329j, thiazole 329k, quinoline 329l, quinoxaline 329m, 1H-indazole 329n and caffeine 329o derivatives all being prepared. Four late-stage pharmaceutical agents also acted as substrates in this transformation.
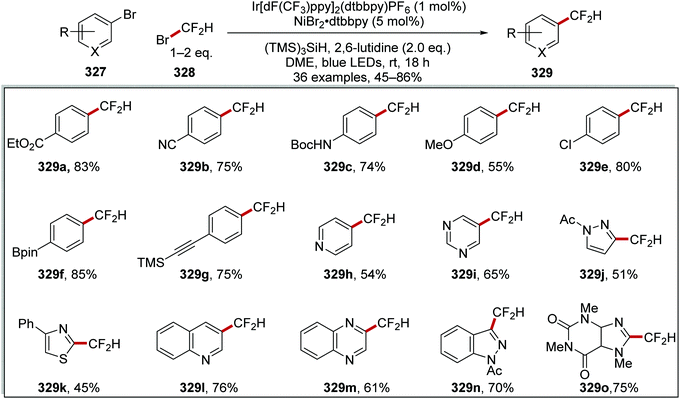 |
| | Scheme 91 Results of Ir/Ni catalysed difluoromethylation of aryl bromides 327. | |
The Ir/Ni/Si radical coupling strategy was then tested with the coupling of α-chlorocarbonyl compounds 331 with aryl bromides 333.152 Silyl radical 323 abstracted the chlorine atom present on 331a and this led to α-keto radical 330a (Scheme 92). Radical 330a was intercepted by the Ni(II) complex and this resulted in coupling to aryl bromide 333a with the formation of compound 334a.
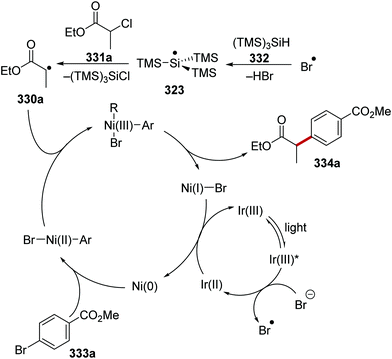 |
| | Scheme 92 Coupling of aryl bromide 333a to α-chloroester 331a. | |
The use of the bulkier silane, tris(triethylsilyl)silane over tris(trimethylsilyl)silane 332 gave an increased yield of 334a (from 65% to 80%) (Scheme 93). With these optimised conditions in hand, the substrate scope of the reaction was explored. Under the reaction conditions aryl esters 334a, trifluoromethyl 334b, Boc-protected amines 334c and boronic ester 334d derivatives were all prepared. It was also shown that 7-azaindazole 334e and thiazole 334f were accessed in 60% and in 45% yield, respectively. While the reaction worked well for aryl bromides 333, it was observed that activated aryl chlorides could also be utilised in this reaction and this gave coupled compounds 334g and 334h. Alternatively, α-chloro carbonyl compounds were also good substrates for this reaction and this allowed for the isolation of ketone 334i, lactone 334j and amide 334k derivatives. It was also shown that 4-(trifluoromethyl)benzyl chloride was a successful substrate for this reaction and this allowed for the preparation of diaryl compound 334l in 73% yield.
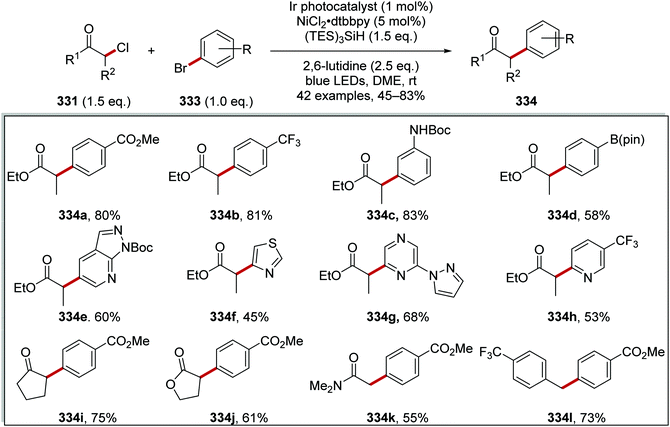 |
| | Scheme 93 Radical coupling between α-chlorocarbonyl 331 and aryl bromides 333. | |
The generation of Csp3–Csp3 bonds from alkyl-halogen compounds was achieved with the use of iridium, nickel catalysts and supersilanol 335 as a radical precursor (Scheme 94).153 The reaction occurred with the oxidation of silanol 335 (Eox = +1.54 V vs. SCE)154 with Ir(III)* complex (Ered* = +1.21 V vs. SCE)8b and after rearrangement, this gave silyl radical 336 and an Ir(II) species. Radical 336 abstracted a bromine atom from 337 and this gave silyl bromide 339 and alkyl radical 338. Interception of radical 338 with a Ni(0) complex resulted in a Ni(I) complex, which became a Ni(III) complex after oxidative addition of methyl bromide (340), formed from methyl tosylate (342) and tetrabutylammonium bromide. Reductive elimination from the Ni(III) complex gave methylated product 341 and a Ni(I) complex. Both catalytic cycles were closed with electron transfer to Ni(I) from the Ir(II) complex.
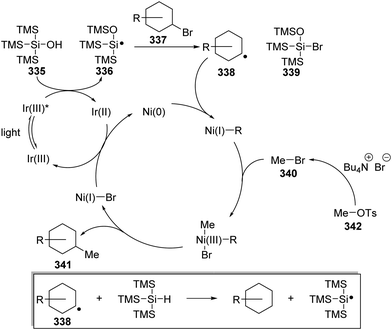 |
| | Scheme 94 Coupling of alkyl bromide 337 with methyl tosylate 342via methyl bromide 340. | |
Initially, it was investigated whether methylation of alkyl bromides was feasible with tris(trimethylsilyl)silane (TTMSS, 332). However, this resulted in a low yield (25%) of methylated product 341a and large amounts of dehalogenated alkane product were produced instead. The presence of a Si–H bond in the silane reagent 332 led to deleterious HAT which gave the dehalogenated compound (mechanism shown inset in Scheme 94). [This issue was solved by replacing TTMSS with tris(trimethylsilyl)silanol (335), as the chemistry of 335, shown in Scheme 94, never features an Si–H bond.] Finally, the addition of tetrabutylammonium bromide increased the efficiency of converting methyl tosylate (342) to methyl bromide 340in situ and this allowed for 341a to be prepared in 72% isolated yield (Scheme 95). When the optimised reaction conditions were trialled on a range of 14 substrates, this resulted in good-to-high yields of methylated analogues 341. The reaction tolerated functional groups and allowed for preparation of amide 341a and trifluoromethyl analogues 341b and 341f. Protecting groups were also accommodated under the reaction conditions affording N-Boc 341c (71%), and N-tosyl 341d (70%) analogues.
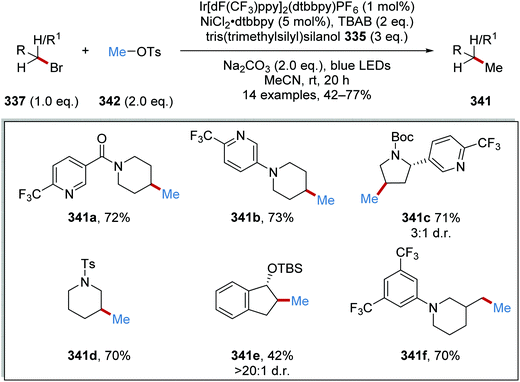 |
| | Scheme 95 Csp3–Csp3 radical cross-coupling of alkyl bromides 337 and methyl tosylate 342. | |
More complicated alkyl bromides were also successful coupling partners when one was used in large excess (Scheme 96). This allowed for two alkyl bromides (337 and 343) to be coupled together. A wide-range of coupled products was accessed (from diverse examples of 343) that included alkyl 344a, ethers 344b, N-Boc amine 344c, ester 344d, ketone 344e and pyridine 344f derivatives.
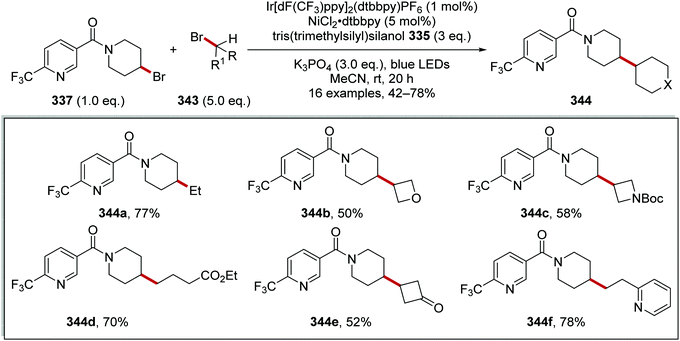 |
| | Scheme 96 Csp3–Csp3 radical coupling between two alkyl bromides 337 and 343. | |
An iterative coupling synthesis was then demonstrated using this chemistry with dibromide 345 (Scheme 97). Utilising TTMSS 332, the aryl bromide component of 345 was selectively functionalised over the alkyl bromide 346 and this gave 347 in 61% yield. With 347 in hand, reverting to tris(trimethylsilyl)silanol 335, the alkyl bromide was activated for a coupling reaction with 4-bromotetrahydro-2H-pyran and this gave pyran 348 in 50% yield.
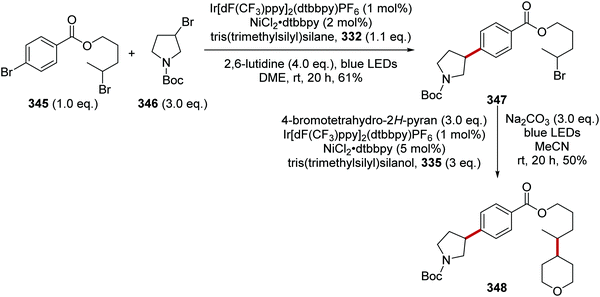 |
| | Scheme 97 Iterative coupling strategy. | |
Sulfonium salts as radical precursors.
The conversion of aryl bromides 333 to aryl trifluoromethyl compounds 352 was achieved with an iridium and copper catalytic strategy.154 The slow oxidative addition to Cu has limited its use in cross-coupling chemistry, even though it readily undergoes reductive elimination. Therefore, it was explored whether two successive radical additions to a Cu(I) complex would result in an efficient coupling reaction. The preparation of trifluoromethyl arenes 352 from bromoarenes 333 with the Mes-Umemoto reagent (350) was chosen to investigate this proposal. Under irradiation by blue light, the Ir(III) complex was excited to the Ir(III)* complex (Scheme 98). Silanol 335 (Eox = 1.54 V vs. SCE) was oxidised by Ir(III)* [Ir(dFFppy)2(4,4′-dCF3bpy)PF6Ered* = +1.55 V vs. SCE] giving, after rearrangement and proton loss, the silyl radical 336. Bromine abstraction from 333a led to aryl radical 349a. At the same time, the reduced iridium catalyst (Eox = −0.83 V vs. SCE) reduced sulfonium cation 350 (E = −0.52 V vs. SCE) and this gave trifluoromethyl radical 351 whilst reforming Ir(III) photocatalyst, closing the iridium catalytic cycle. Successive radical additions of 351 and 349a to Cu(I) complex resulted in the formation of a Cu(III) complex. Reductive elimination of Cu(III) complex reformed Cu(I) catalyst and gave trifluoromethyl arene 352a as product.
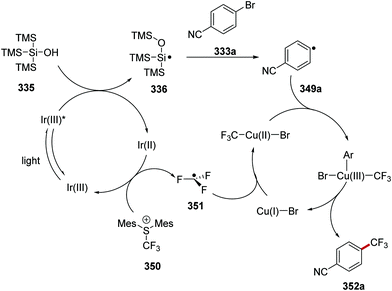 |
| | Scheme 98 Tandem catalytic process for the generation of aryl trifluoromethyl compound 352a from aryl bromide 333a. | |
Under an iridium and copper catalytic system, the desired trifluoromethyl compounds 352 were produced in yields ranging from 38% to 96% (Scheme 99). The optimal base and iridium catalyst varied, depending upon the substrate being used in the reaction. Nevertheless, a wide range of different functional groups was tolerated under the reaction conditions with 352a–e being formed in 80–96% yields. Heterocyclic products 352f–h were isolated in 38–64% yields. Furthermore, celecoxib (COX-2 inhibitor) derivative 352i was also isolated in 77% yield.
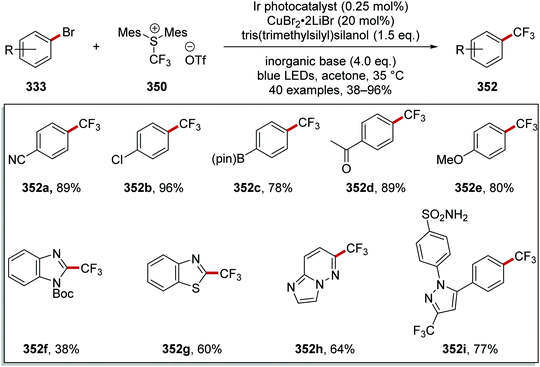 |
| | Scheme 99 Trifluoromethylation of aryl bromides. | |
Alkane C–H functionalisation.
The functionalisation of adamantane 353a with electron-poor alkenes 357 was achieved with the HAT catalyst quinuclidin-3-yl benzenesulfonate (28e) and an iridium complex.55 In competition experiments, it was found that HAT catalyst 28e was highly selective for adamantane compounds. In a reaction mixture with equal amounts of adamantane (353a) and n-octanal (354), preferred functionalisation of 353a was observed affording alkylated adamantane derivative 355a in 63% yield and aldehyde-derived alkylated compound 356a in 7% yield (Table 4, entry 1). Using quinuclidine (28b) as HAT catalyst in conjunction with an Ir photoredox agent, also favoured the formation of 355a over 356a but the reaction was less successful (entry 2). The use of an acridinium photocatalyst in conjunction with a phosphate base gave equal amounts of 355a and 356a (entry 3). The catalysts 5,7,12,14-pentacenetetrone 358 and 2-chloroanthraquinone 359 gave a slight preference for the aldehyde-derived product 356a instead of the desired alkylated compound 355a. Strong preference for aldehyde-derived analogue 356a was observed with the HAT catalysts tetrabutylammonium benzoate (oxidised by Ir(III)*) and tetrabutylammonium decatungstate (entries 6 and 7). These competition reactions demonstrated the exquisite selectivity of quinuclidin-3-yl benzenesulfonate (28e) as a HAT catalyst.
Table 4 Selectivity of different catalytic systems
As it was established that the electron-poor quinuclidine HAT catalyst 28e was optimal for activating the tertiary C–H bonds in adamantane, the reaction was optimised. When the reaction was performed with Ir[dF(CF3)ppy]2[d(CF3)bpy]PF6 (2 mol%) and quinuclidin-3-yl benzenesulfonate (20 mol%) an isolated yield of 72% was obtained for 355a. When the more electron-rich 3-acetoxyquinuclidine was employed as a HAT catalyst, a yield of 74% was attained for 355a. This trend was continued when quinuclidine was used as HAT catalyst with 355a being given in a 33% GC yield. Replacement of Ir[dF(CF3)ppy]2[d(CF3)bpy]PF6 with Ir[dF(CF3)ppy]2[d(tBu)bpy]PF6 also led to a diminished yield, affording 358a in 16% GC yield. This can be explained with the oxidation potentials of the iridium catalyst and the HAT catalyst. The catalyst Ir[dF(CF3)ppy]2[d(tBu)bpy]PF6 has an excited reduction potential of +1.21 V vs. SCE and Ir[dF(CF3)ppy]2[d(CF3)bpy]PF6 has an excited reduction potential of +1.68 V vs. SCE.55,155,156 As quinuclidin-3-yl benzenesulfonate has an Ered1/2 + 1.41 V vs. SCE only excited Ir[dF(CF3)ppy]2[d(CF3)bpy]PF6 is a sufficiently strong oxidant to oxidise quinuclidin-3-yl benzenesulfonate. With optimal conditions being found, adamantane was coupled to various electron-poor alkenes and this led to a range of coupled products 355a–h (Scheme 100).
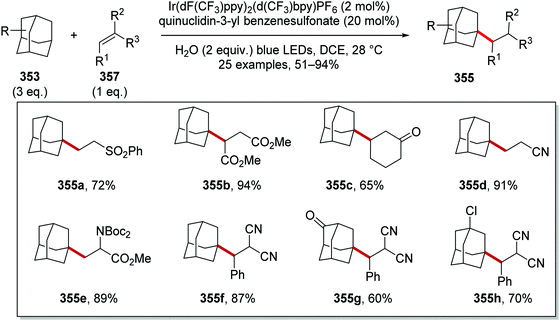 |
| | Scheme 100 Results of coupling to adamantanes. | |
From a high-throughput study, tetrabutylammonium benzoate was an effective quenching agent of Ir[dF(CF3)ppy](dtbbpy)6 complex, indicating electron transfer from benzoate 360 to Ir(III)* and this gave oxygen-centred radical 361 (Scheme 101).157 These radicals are slow to undergo radical decarboxylation and they participate in HAT reactions.158 Due to the great strength of O–H bonds (BDE of benzoic acid O–H bond = 111 kcal mol−1) it was proposed that 361 could activate strong non-activated C–H bonds.159 When benzoate ester 362a was in the presence of 361, hydrogen atom abstraction was observed from the most electron-rich C(sp3)–H bond and this gave tertiary radical 364a. The use of N-(trifluoromethylthio)phthalimide (359) resulted in formation of trifluoromethylsulfide 365a and phthalimidyl radical 366 from alkyl radical 364a. The iridium catalytic cycle was closed with SET to phthalimidyl radical 366 from Ir(II) complex with anion 367 being given. Benzoate anion 360 was regained with proton exchange between phthalimidyl anion 367 (pKa of phthalimide = 8.3, in water) and benzoic acid (363) (pKa of benzoic acid = 4.2, in water). Calculation of average radical chain length (ϕ/Q) supported this mechanism over a radical-chain mechanism with a value of 2.0 being determined. The average radical chain length was calculated from the quantum yield of the reaction (ϕ = 1.76) and the quenching fraction (Q) (86% of the photons absorbed by the Ir catalyst participated in productive electron transfer). Further evidence for this mechanism was the high selectivity obtained. The selectivity was found to be >20:1 for methine C–H bonds, which suggested abstraction by radical 361, as it has been previously shown that the phthalimidyl radical 366 is much less selective.160
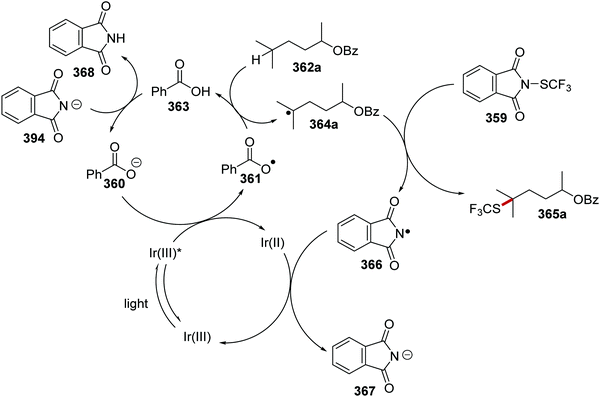 |
| | Scheme 101 Formation of trifluoromethylthio derivative 365a from 362a with 359. | |
Initially, the reaction was performed with tetrabutylammonium benzoate (5 mol%) and Ir[dF(CF3)ppy](dtbbpy)PF6 (2 mol%) and this gave 365a in 96% isolated yield and with high regioselectivity (>20:1). Further study showed that this transformation would also operate with 1 mol% of Ir catalyst and the use of the more accessible sodium benzoate also gave 365a in 96% yield. With the optimised reaction conditions, a small library of 26 compounds 365 was prepared (Scheme 102). Benzoate 365b was obtained in higher selectivity than acetate product 365c. The reaction conditions were also tolerant of a strained cyclopropyl ring and this gave 365d in 92% yield, with a >20:1 site-selectivity. Generally, methylene C–H bonds reacted slowly under these conditions. However, methylene C–H bonds neighbouring heteroatoms were excellent substrates, and this allowed for 365e to be isolated in 95% yield. Heterocycle-bearing substrates were tolerated in this process; for example, 365f was given in 80% yield. Bioactive molecules such as leucine and 5α-cholestan-3β-acetate were satisfactory substrates for this reaction and thus fluorinated derivatives 365g and 365h were prepared in 68% yield and 35% yield, respectively.
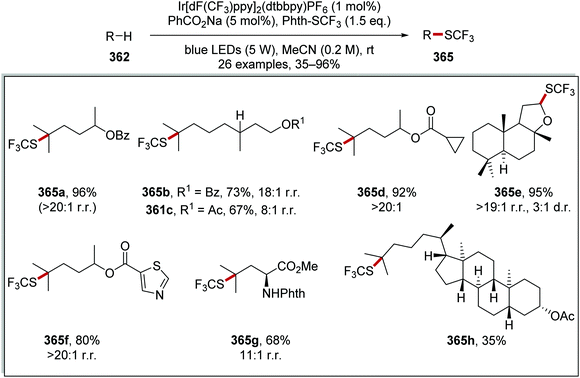 |
| | Scheme 102 Substrate scope of the trifluoromethylthiolation reaction. | |
Giese reaction between diesters 370 and organic compounds bearing C–H functionality, 369, was achieved with hydrogen atom abstraction by chlorine atoms.161 Originally, investigations began with Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%) photocatalyst and tetrabutylammonium chloride (10 mol%) to generate the chlorine atoms from chloride ions. However, it was then found by using the chloride salt of the iridium photocatalyst, the TBACl co-catalyst could be omitted and this gave 371a in a slightly improved yield. Therefore, investigations into the substrate scope were conducted with Ir[dF(CF3)ppy]2(dtbbpy)Cl catalyst (Scheme 103). These reaction conditions were applied to a wide range of substrates. Cyclohexane was converted to diester 371a in 69% yield. Tetrahydrofurans were suitable coupling partners; for example, THF was used in the preparation of 371b in 55% yield, as a single regioisomer. Both esters and amides were capable substrates, and this allowed for compounds 371c and 371d to be isolated. Aldehydes with weak C–H bonds were found to be suitable in this transformation – for example, n-pentanal gave ketone 371e in 95% yield after column chromatography. When cyclopentanol was used in the reaction, the cyclised lactone 371f was given as product in 33% yield. Silanes or phosphorus-containing compounds were successfully coupled to the electron-poor alkenes and this gave compounds such as 371g and 371h. During the determination of the substrate scope, it was found that benzylic C–H bonds were not suitable targets. Therefore, compounds such as toluene and mesitylene could not be coupled to alkenes 370.
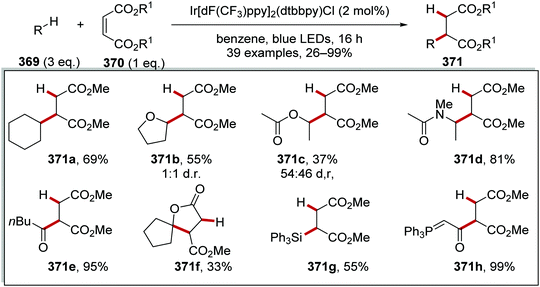 |
| | Scheme 103 Giese coupling with a Cl-atom HAT strategy. | |
The selectivity of the reaction was tempered by choice of the reaction solvent (Scheme 104).161 With cyclopentyl methyl ether (369i) as substrate reacting with dimethyl maleate, a preference for the reaction of the methine C–H bond over the methyl C–H bond was found. In benzene, using the Ir chloride photocatalyst, a yield of 64% with a 4.4:1 mixture of regioisomers was obtained for 371i. This was improved with the use of pyridine as the reaction solvent, with 371i being isolated in 64% yield with a 10:1 mixture of regioisomers.
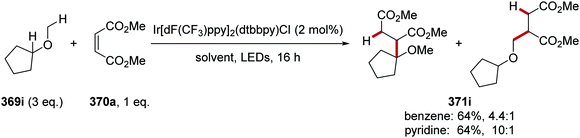 |
| | Scheme 104 Effect of solvent upon selectivity. | |
Carbon α-heteroatom C–H functionalisation.
Giese coupling.
The Giese coupling of tertiary amines was achieved with the use of an iridium photoredox catalyst.37 Irradiation with blue light gave excited Ir(III)* species from Ir(III) complex. Electron transfer from N-methylmorpholine (372a, Epox = +1.10 V)38 to Ir(III)* [Ir[(dFCF3)ppy]2dtbbpy(PF6)], (Ered* = +1.21 V vs. SCE)8b gave amine radical cation 373a and Ir(II) species (Scheme 105). Deprotonation of 373a led to the carbon-centred radical 374a. For some substrates, such as 373a, more than one deprotonation site exists (highlighted in blue) and this gave rise to products being isolated as mixtures of regioisomers. Nevertheless, radical 374a added to alkene 375 and this gave captodative radical 376a. The iridium catalytic cycle is closed with electron transfer from Ir(II) to 376a giving the original Ir(III) complex along with anion 377a. Protonation of 377a led to the formation of 378a, which was isolated from the reaction mixture.
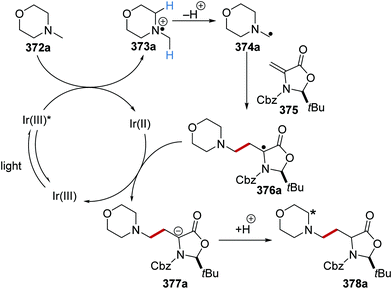 |
| | Scheme 105 Formation of 378a from 372a. [The asterisk * denotes alternative connectivity in formation of alternative regioisomers.] | |
Under these conditions, 12 different amines 372 were alkylated with oxazolidinone 375 with Ir[df(CF3)ppy]2dtbbpy(PF6) in MeCN, giving functionalised oxazolidinones 378 (Scheme 106). The yields of isolated 4-oxazolidinones 378 ranged from 25–91%, and alkyne 378d was isolated in 25% yield. As mentioned earlier, when compounds such as N-methylmorpholine and strychnine were used as substrate, this gave 378a and 378f as mixtures of regioisomers as proton loss occurred competitively at two different sites for these substrates.
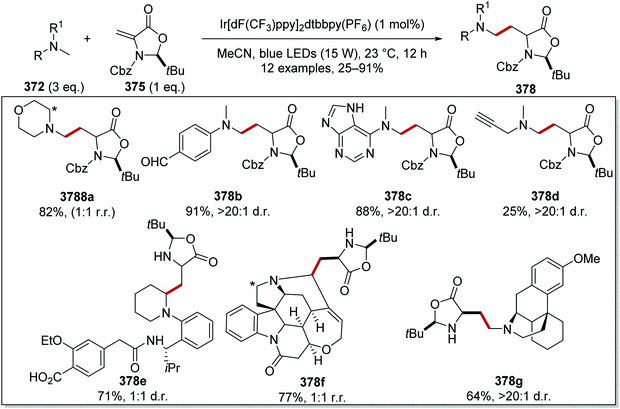 |
| | Scheme 106 Products from radical Michael addition to electron-poor alkenes. | |
The synthetic utility of this methodology was then demonstrated with the functionalisation of peptides (Scheme 107). There was no control of stereochemistry of the new chiral centre and therefore this gave a 1:1 mixture of diastereoisomers. The high degree of chemoselectivity in this process was remarkable. For example, while peptide 379 has several reactive functional groups, including primary amines, aniline peptide 380 was selectively prepared in 67% yield.
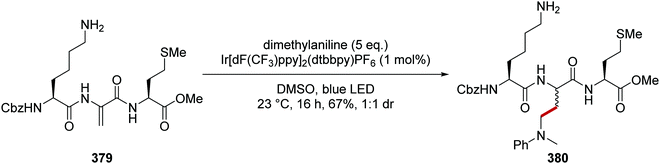 |
| | Scheme 107 Selective functionalisation of peptide 379. | |
Regioselective addition of benzylamines to electron-poor alkenes was investigated under photoredox conditions mediated by an iridium photocatalyst.162 When N-benzyl-N-methylaniline (381a) was reacted with methyl vinyl ketone (3 eq.) using Ir(ppy)2(dtbbpy)PF6 in dichloromethane, functionalisation at the benzylic position was observed giving 382a in 74% yield (Table 5, entry 1). When the reaction was conducted in MeCN, the site-selectivity of the reaction was completely reversed, there was no functionalisation at the benzylic position, and instead 383a was the sole-product in 70% yield (entry 2). Addition of TFA (0.4 eq.) to the reaction mixture gave 382a in 49% yield even when performed in MeCN (entry 3). From this optimisation study, it was observed that 383a gave a maximum yield with 1.1 eq. of methyl vinyl ketone, 0.3 eq. of DBU as additive and with the reaction performed in MeCN (entry 4). The use of higher loadings of methyl vinyl ketone led to more than one Giese addition giving 384a in 76% yield (entry 5). The inclusion of an inorganic base gave aldol product 385a in 70% isolated yield as the major product (entry 6).
Table 5 Site-selective functionalisation of aniline 381
|
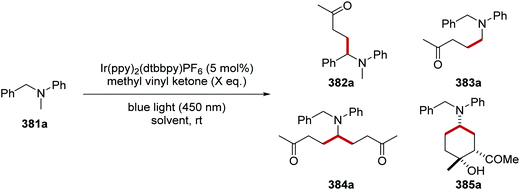
|
| Entry |
Methyl vinyl ketone, eq. |
Solvent |
Additive |
Isolated yield (%) |
|
382a
|
383a
|
384a
|
385a
|
| 1 |
3 |
CH2Cl2 |
|
74 |
|
|
|
| 2 |
3 |
MeCN |
|
|
70 |
|
|
| 3 |
3 |
MeCN |
TFA |
49 |
|
|
|
| 4 |
1.1 |
MeCN |
DBU |
|
84 |
|
|
| 5 |
3 |
MeCN |
DBU |
|
|
76 |
<5 |
| 6 |
3 |
MeCN |
K3PO4 |
|
|
<5 |
70 |
A mechanism was proposed for the observed regioselectivity in these reactions (Scheme 108). A PET to Ir(III)* from aniline 381a resulted in radical cation 386a. It was then suggested that reversible proton loss occurred at the more acidic benzylic position (pKa 2.4, methyl position pKa = 8.2) to give 387a and this explained some of the experimental observations. When the reaction was conducted in dichloromethane, very fast addition to MVK (389a) occurred and this resulted in ketone 382a being formed under the reaction conditions. However, when the reaction was conducted in MeCN, the reverse reaction from 387a to 386a was more significant, perhaps due to enhanced stabilisation of the charged species 386a in the more polar MeCN, and from this the N-methyl radical 388a formed. DFT calculations indicated that Giese addition of 388a to give 390a would have a lower transition state by 6.9 kcal mol−1 compared to addition of 387a, when the reaction was conducted in MeCN. The lower transition state energy in the reaction of 388a with 389a would result in a rapid and irreversible formation of radical 390a. Electron transfer from Ir(II) complex to 390a would result in formation of the corresponding enolate and, after proton transfer, the ketone 383a.163
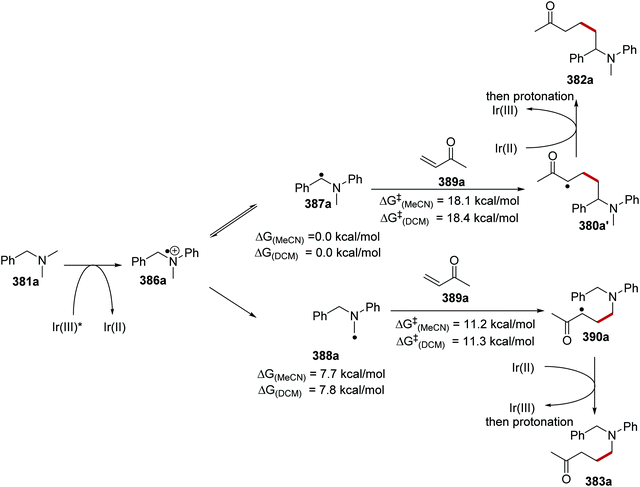 |
| | Scheme 108 Selectivity of products determined by solvent. | |
With four unique reaction conditions optimised, aniline 381 was converted into a wide range of different compounds 382–385. When aniline 381a was reacted with electron-poor alkenes 389 (2 eq.) in dichloromethane mediated by an iridium photocatalyst, the benzylic functionalised compounds 382a–c were returned in 74%, 77% and 40% yield, respectively. When the reaction was performed with lowered equivalents of electron-poor alkene (1.1 eq.) and in MeCN, the N-methyl functionalised compounds were produced. For example, ketone 383a and ester 383b were isolated in 85% and 82% yield. Sequential N-methyl functionalisation was performed, and this gave compounds such as 384b (71% yield) and 384c (82% yield) from 383a. This was accomplished from the reaction of 383a with electron-poor alkene (2.0 eq.) in MeCN mediated with the iridium photocatalyst. Cyclic aldol compounds, such as 385a and 385b were produced when 381 was treated with methyl vinyl ketone (3 eq.), in the presence of K3PO4 and the Ir photocatalyst in MeCN (Scheme 109).
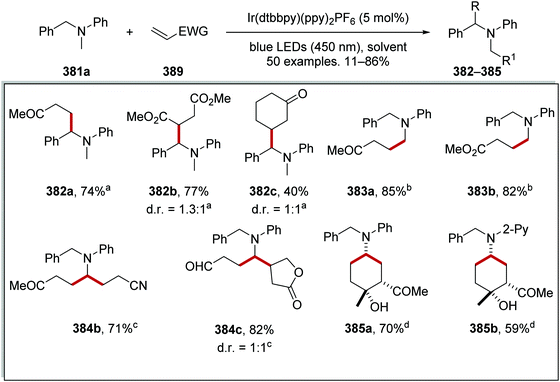 |
| | Scheme 109 Substrate-scope for site-elective coupling. a389 (2 eq.), CH2Cl2, rt, 24 h. b389 (1.1. eq.), DBU (0.3 eq.), MeCN, rt, 24 h. c389 (1.1 eq.), DBU (0.3 eq.), MeCN, rt, 24 h then 389′ (2.0 eq.), DBU (0.3 eq.), MeCN, rt, 24 h. d389 (3.0 eq.), K3PO4 (0.3 eq.), MeCN rt, 24 h. | |
The radical coupling of protected amines 391 with electron-deficient alkenes 392 was investigated with iridium photocatalysis.164 The use of N-propyl triflamide and benzyl acrylate as substrate with Ir[dF(CF3)ppy]2dtbbpy(PF6) and caesium carbonate as base gave ester 397 in 50% yield (Table 6, entry 1). Studies with other inorganic bases and other iridium catalysts determined that comparable yields of 397 were generated with Ir[dF(CH3)ppy]2dtbbpy(PF6) (entries 2–4). However, increased efficiency was discovered when quinuclidine was employed as base and tert-butyl acrylate as electron-poor alkene giving 397 in 77% yield (entry 5). This reaction was only feasible when the triflamide protecting group was used (entries 5–8).
Table 6 Reaction optimisation. Catalyst A = Ir[dF(CF3)ppy]2dtbbpy(PF6), catalyst B = Ir[dF(CH3)ppy]2dtbbpy(PF6). Yields determined by 1H NMR spectroscopy, asterisk denotes isolated yield
|

|
| Entry |
PG |
Photocatalyst |
R |
Base |
Yield (%) |
| 1 |
Tf |
A |
Bn |
Cs2CO3 |
50 |
| 2 |
Tf |
A |
Bn |
K2CO3 |
54 |
| 3 |
Tf |
A |
Bn |
K3PO4 |
58 |
| 4 |
Tf |
B |
Bn |
K3PO4 |
65 |
| 5 |
Tf |
B |
tBu |
Quinuclidine |
77* |
| 6 |
COCF3 |
B |
tBu |
Quinuclidine |
0 |
| 7 |
Ts |
B |
tBu |
Quinuclidine |
0 |
| 8 |
Ac |
B |
tBu |
Quinuclidine |
0 |
It was inferred that two feasible mechanisms could apply, depending upon the base employed in the reaction. When quinuclidine (28b) was used as base, it was thought, under the reaction conditions, triflamide 391a was deprotonated with an equivalent of 28b to give the anion 393a (Scheme 110). Deprotonation of 391a was supported by experimental pKa values, N-methyltrifluoromethanesulfonamide and phenyl trifluoromethanesulfonamide have pKa values of 7.56 and 4.45 respectively in water,165 and the pKa of quinuclidine conjugate acid is 11.3 in water.166 It was thought that the excited iridium photocatalyst [Ir[(dFCF3)ppy]2dtbbpy(PF6), (Ered* = +1.21 V vs. SCE) oxidised quinuclidine (Eox = +1.1 V vs. SCE).156 A selective HAT of the activated α-amino C–H bond of anion 393a with quinuclidine radical 28b′ resulted in the formation of radical anion 394a. Stern–Volmer experiments showed that effective quenching of Ir(III)* complex was seen with 28b. Electron-rich radical 394a was then trapped with an electron-poor alkene 392a and this gave coupled radical 395a. SET and protonation of 395a led to the formation of ester 397a and closure of the iridium catalytic cycle. When K3PO4 was used as base, a different mechanism was thought to be in operation (Scheme 110, inset). The combination of 391a, K3PO4 and excited Ir(III)* gave N-centred radical 393˙ either through a concerted PCET process or a sequential deprotonation and SET event. The mechanism was supported by Stern–Volmer studies that showed quenching of Ir(III)* was achievable with 393a. Radical 393˙ was then able to react with another molecule of 393a and this generated key intermediate 394a for C–C bond formation with alkene 392a.
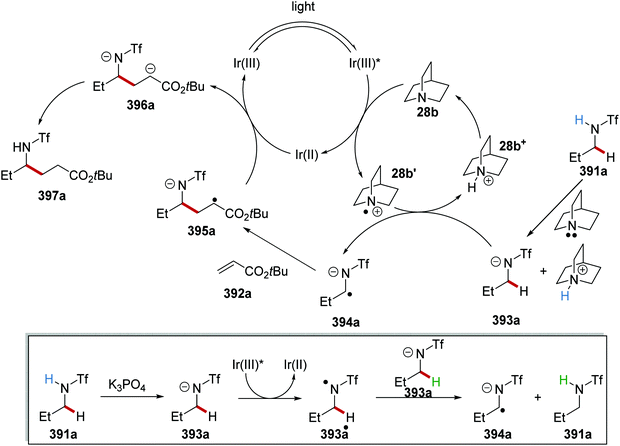 |
| | Scheme 110 α-C–H functionalisation of triflamides 391 with electron-poor alkenes 392. | |
When optimal reaction conditions were employed, the site-selectivity of this reaction was then demonstrated with the preparation of glucose 397g, N-Boc 397h and benzyl ether 397i derivatives. Finally, it was shown that a range of different electron-poor alkenes could be used in this transformation, for example, compounds 397b–e were prepared in yields of 32–73% (Scheme 111).
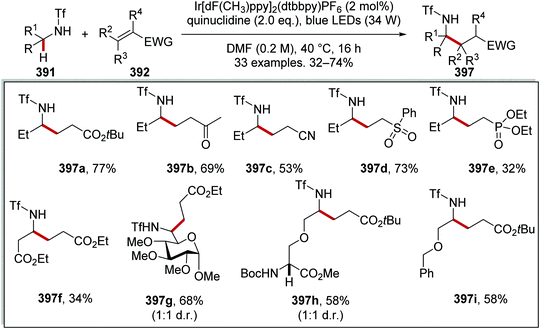 |
| | Scheme 111 α-C–H functionalisation of triflamides 387 with electron-poor alkenes 388. | |
Quinuclidine (28b) was used as a HAT catalyst alongside photoredox catalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 in the preparation of lactams 405 from primary amines 398 and methyl acrylate (399a) (Scheme 112).167 Quinuclidine radical cation 28b′ was generated with SET to excited Ir(III)* complex (Ir[dF(CF3)ppy]2dtbbpy(PF6) Ered* = +1.21 V vs. SCE) from quinuclidine (Eox = +1.1 V vs. SCE). As the reaction was performed under carbon dioxide, carbamate 400 was formed from amine 398 and carbon dioxide. The formation of carboxylate 400 assisted in the HAT event and formation of 401, due to the electrostatic attraction between the negatively charged carboxylate ion and the positively charged 28b′; this enhanced the selectivity with abstraction at the most hydridic C–H bond. Once alkyl radical 401 was delivered, it reacted with the electron-poor alkene 399a and this gave α-carbonyl radical 402. Electron transfer from the reduced Ir(II) complex to radical 402 resulted in the generation of the original Ir(III) species and enolate anion 403. Protonation of anion 403 and loss of carbon dioxide gave amine 404. Lactam 405 was formed from 404 with the loss of methanol.
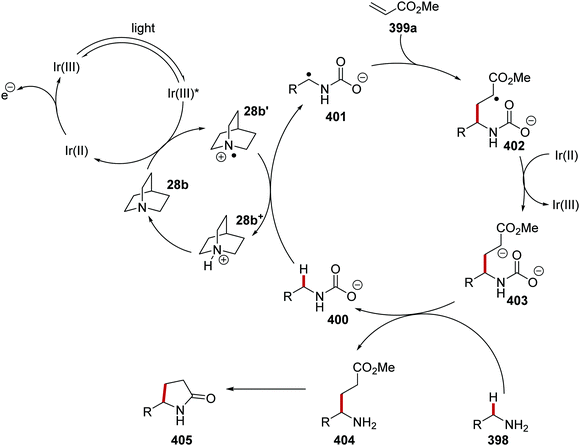 |
| | Scheme 112 Lactam formation from primary amines. | |
Optimisation of the reaction conditions found that quinuclidine was the best HAT catalyst. When 3-acetoxyquinuclidine (28d) was used instead of quinuclidine (28b), a decreased 1H-NMR yield of lactam 405a was obtained. Use of DABCO instead of quinuclidine led to no observable product formation. When the reaction was performed with no carbon dioxide, the 1H-NMR yield decreased from 85% to 25%. With optimal conditions being established, a library of 45 lactams 405 was prepared in 41–80% yields (Scheme 113 for selected examples). The reaction was tolerant of functional groups with trifluoromethyl 405b, ester 405c, silyl protected alcohols 405d, alkynes 405f, acetals 405g and heterocyclic 405h analogues all being synthesised. The directing effect of carboxylate intermediate 400 with quinuclidine radical 28b′ was demonstrated with the preparation of N-Boc amine compound 405e as there was no alkylation adjacent to the N-Boc group.
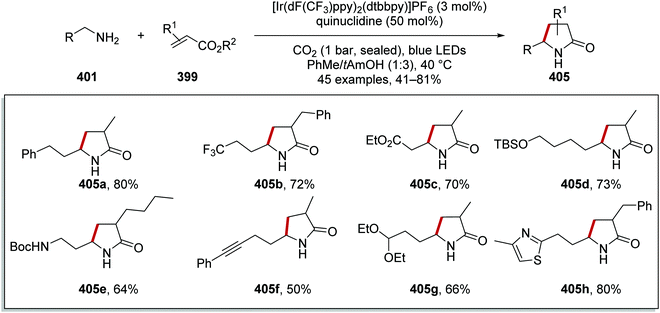 |
| | Scheme 113 Products from lactam formation from primary amines. | |
It was shown that amidyl radicals could be used as effective HAT catalysts.168 The combination of Ir[dF(CF3)ppy]2(5,5′-dCF3bpy)PF6 and dibutyl phosphate (406) gave amidyl radical 407 from N-ethyl-4-methoxybenzamide (408) through PCET when irradiated with blue LEDs (Scheme 114). Amide 408 (Ep = +1.48 V vs. Fc/Fc+) was unable to directly quench the Ir catalyst (Ir[dF(CF3)ppy]2(5,5′-dCF3bpy)PF6E1/2* = +1.30 V vs. Fc/Fc+) but the presence of tetrabutylammonium dibutyl phosphate resulted in large decrease in emission intensity. Radical 407 abstracted a hydrogen atom from cyclohexane (409a) and this gave alkyl radical 410a. A Giese coupling between 410a and electron-poor alkene 411a resulted in C–C bond formation giving α-ester radical 412a. The iridium catalytic cycle was closed with an outer-sphere electron transfer from Ir(II) to 412a and this gave anion 413a, which was converted to 414a with protonation.
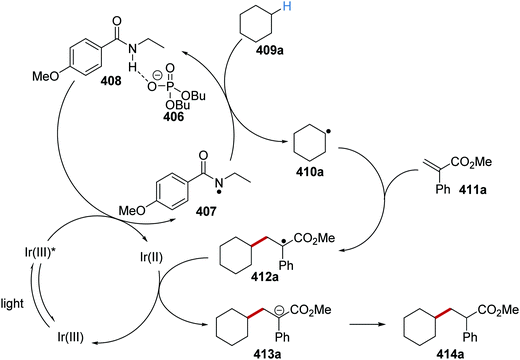 |
| | Scheme 114 Preparation of 414a with amidyl HAT catalyst. | |
The use of amidyl radicals as HAT reagents allowed for C–H activation of alkanes, ethers, and amines. Thus, with electron-poor alkenes, 414a, 414b and 414c were prepared (Scheme 115).
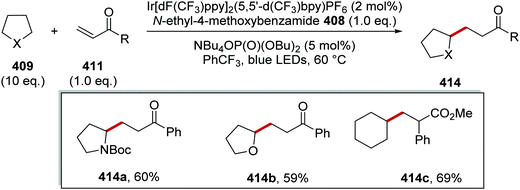 |
| | Scheme 115 Applications of amidyl HAT catalyst 408. | |
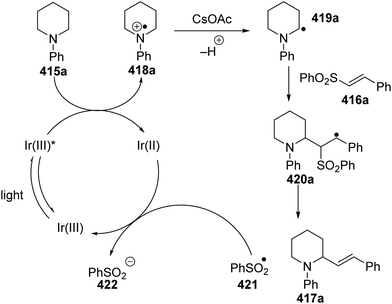 |
| | Scheme 116 C–H activation and radical coupling to electron-poor alkenes. | |
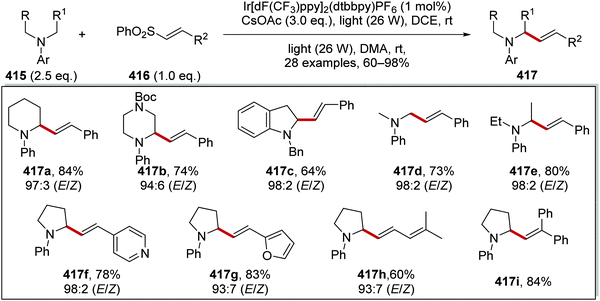 |
| | Scheme 117 Results of radical coupling between amines 415 and vinyl sulfones 416. | |
This transformation was then realised with the use of Ir(dF(CF3)ppy)]2(dtbbpy) photocatalyst and CsOAc as base (Scheme 117). This system allowed for the preparation of 28 alkenes 417 from the corresponding amines 415 and vinyl sulfones 416. In most cases, the E isomer was predominantly formed.
The coupling of heteroaryl chlorides 423 to dialkylanilines 415 was investigated and this gave amines 424 (Scheme 118).170 A wide range of coupled products 424a–424i, was prepared in 72–94% yields although only 22% yield was obtained for compound 424j.
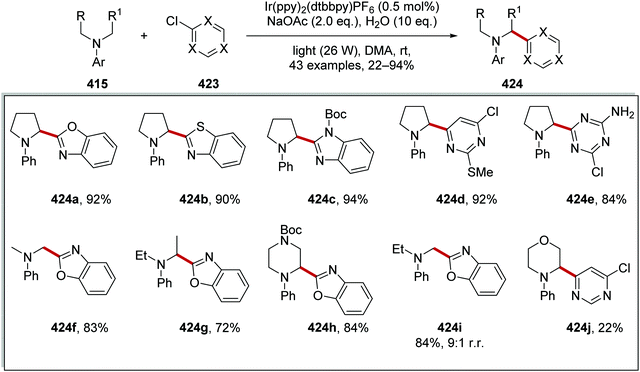 |
| | Scheme 118 Results of coupling between heteroaryl chlorides 423 and alkylanilines 415. | |
Gandotnib (428) is a selective inhibitor of JAK2-V617F and is currently being studied in phase 2 clinical trials in patients with myeloproliferative neoplasms (http://ClinicalTrials.gov Identifier: NCT01594723).171 Preparation of key intermediate 426 was carried out with C–H activation and radical coupling chemistry (Scheme 119). The coupling of unfunctionalised pyridazine 425 with N-methylmorpholine (20 eq.) was achieved with an Ir(ppy)3 photocatalyst (0.5 mol%). From extensive investigation, it was found that a solvent system of 10:1 DMA/H2O gave the highest selectivity for desired regioisomer 426. The reaction, performed in DMA as the sole solvent, led to a substantial amount of unwanted endo compound 427; however, the addition of water minimised this byproduct. Product 426 was isolated from the reaction mixture by the addition of 3:1 H2O:EtOH and this afforded the product in 56% isolated yield and with 90% purity. Highly pure 426 was obtained with an acid/base recrystallisation procedure which gave 426 in 98% UPLC purity.
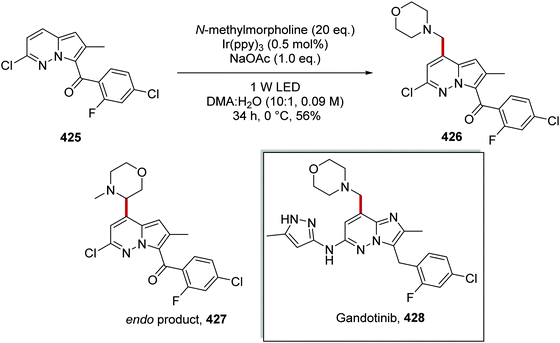 |
| | Scheme 119 Preparation of intermediate 426 for the synthesis of 428, a selective inhibitor of JAK2-V617F. | |
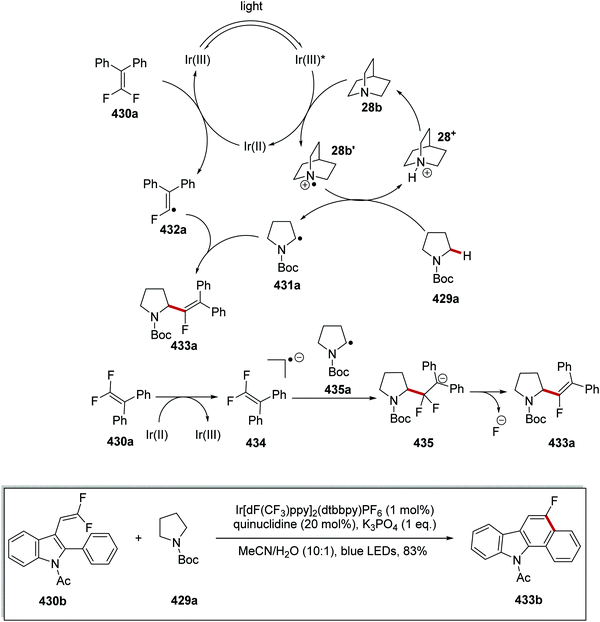 |
| | Scheme 120 HAT mediated coupling of N-Boc amine 429a with gem-difluoroalkene 430a. | |
The use of 1 mol% of Ir(dFCF3ppy)2(dtbbpy)(PF6) as photocatalyst and 20 mol% quinuclidine under basic conditions gave coupling between 429 and 430 after 24 h at room temperature. From optimisation studies, it was found that the solvent system MeCN:H2O (10:1 ratio) was optimal for the formation of 433a. When MeCN was used as the sole reaction solvent, the yield decreased from 79% to 43% for 433a. It was postulated that the inclusion of water in the reaction mixture increased the solubility of the base and this increased the efficiency of the reaction. The use of the HAT reagents aceclidine (3-acetoxyquinuclidine) or 3-quinuclidinol also led to a less successful reaction with 433a being isolated in 40% or 0% yield, respectively. The use of potassium carbonate as base also led to an inferior reaction, forming 433a in 60% yield. No reaction proceeded when photocatalyst, HAT catalyst, light or base was absent from the reaction mixture. Furthermore, there was no reaction when Ru(bpy)3(PF6)2 was used as the photocatalyst. This is understandable as the excited Ru complex is unable to oxidise quinuclidine. When the optimised conditions were applied, a wide range of products was prepared 433a–k (Scheme 121).
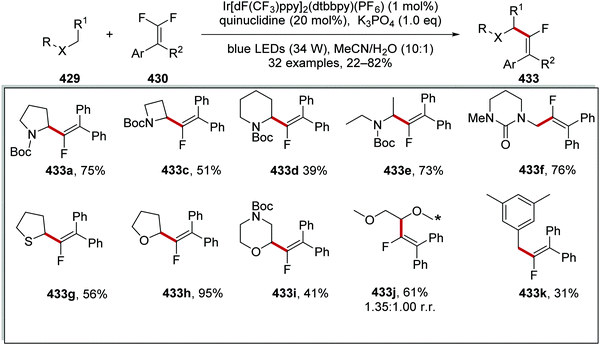 |
| | Scheme 121 Results of the coupling between N-Boc amines 429 and gem-difluoroalkenes 430. | |
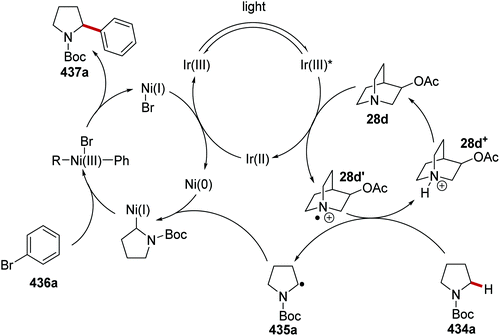 |
| | Scheme 122 HAT-mediated radical cross-coupling. | |
The combination of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), 3-acetoxyquinuclidine (1.1 eq.), NiBr2 (1 mol%), the ligand 4,7-dimethoxy-1,10-phenanthroline (1 mol%) gave 437c in 81% yield from N-Boc pyrrolidine and 4-bromobenzoate. The reaction was regioselective with only one regioisomer being detected. The compound 434a was functionalised at the most hydridic C–H bond. The optimised reaction conditions were trialled on a range of substrates and this produced 40 coupled compounds (Scheme 123). Functionalisation of α-carbamate C–H bonds was achieved and coupled compounds 437a–437h were achieved in yields of 62–81%. Benzylic C–H functionalisation was also achievable; for example, diaryl compound 437i was isolated in 54% yield. From the substrate scope, it was observed that this transformation preferred to functionalise the most sterically accessible site. For example, when N-Bac butylmethylamine was treated with these optimised conditions, a 4:1 mixture of regioisomers was obtained for 437f in favour of functionalisation on the methyl group over the methylene group. Additionally, only one regioisomer was obtained for starting amines possessing α-methyl or methylene and α-methine groups, see compounds 437g and 437h. Therefore, no functionalisation at α-amino methine groups was achievable under these conditions.
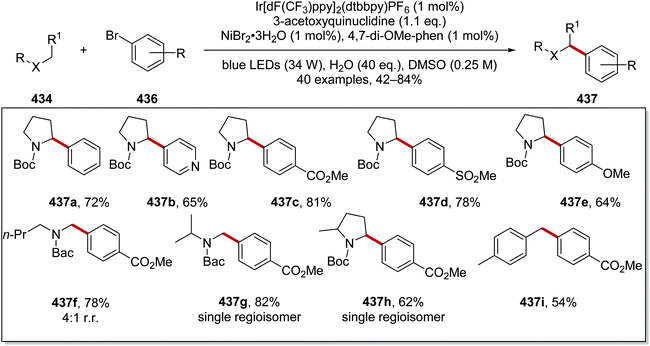 |
| | Scheme 123 Results of HAT mediated radical cross-coupling. | |
Carbon sp3–sp3 bonds were formed with functionalisation of α-amido C–H bonds with the use of alkyl bromides 438 with a similar iridium/nickel/quinuclidine catalytic system (Scheme 124).156 It was observed that C–H functionalisation occurred at the site of least steric hindrance. Therefore, the preparation of coupled compounds 439b, 439c, 439g, 439j and 439k was achieved with a high degree of regiocontrol. In addition, C–H bonds adjacent to oxygen atoms and sulfur atoms were susceptible to functionalisation and this gave coupled compounds 439e–439i.
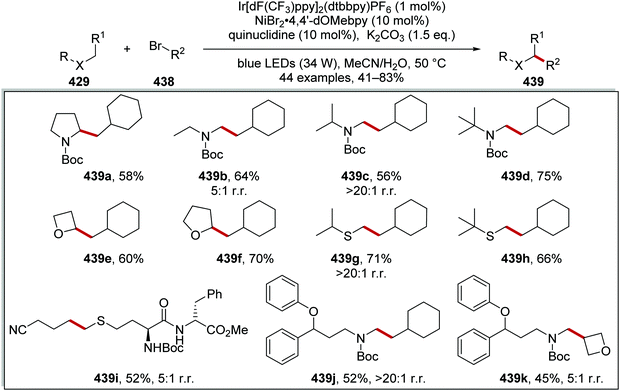 |
| | Scheme 124 HAT catalyst mediated sp3–sp3 coupling reaction. | |
The coupling of aryl chlorides 440 with ethers 441 was achieved with iridium photoredox conditions, blue-light irradiation and the use of chlorine atoms as HAT catalysts.49 The reaction occurred via oxidative addition of aryl chloride 440 to Ni(0) complex A and this gave Ni(II) complex B (Scheme 125). This was followed with a SET between B and excited Ir(III)* complex and gave Ir(II) complex and Ni(III) complexes C. From cyclic voltammetry, it was found that oxidation of Ni(II) complex B (Ep = +0.85 V vs. SCE) was achievable with Ir(III)* species (E1/2* = +1.21 V vs. SCE). Furthermore, from a Stern–Volmer quenching study, it was found that Ni(II) complex B significantly quenched the excited Ir(III)* complex. It was reported that a chlorine atom dissociated under irradiation with light.172 Therefore, it was thought that, under the reaction conditions, Ni(III) complex C liberates a chlorine atom. HAT from tetrahydrofuran substrate 441a (THF C–H BDE = 92 kcal mol−1) to a chlorine atom (H–Cl BDE = 102 kcal mol−1) resulted in the alkyl radical and this culminated in Ni complex F. Desired product 442 was formed with the reductive elimination from F. SET between Ni(I) complex G and Ir(II) complex closed both catalytic cycles.
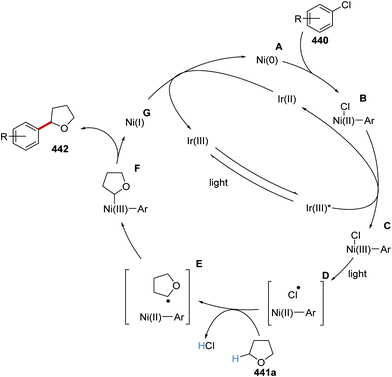 |
| | Scheme 125 Formation of tetrahydrofuran 442 with chlorine atom HAT catalysis. | |
From optimisation studies, it was observed that the combination of Ni(cod)2, dtbbpy, Ir[dF(CF3)ppy(dtbbpy)PF6 and K3PO4 allowed for the preparation of 442a in 92% 1H-NMR yield and 79% isolated yield (Scheme 126). The less oxidising photocatalyst, Ru(bpy)3(PF6), gave no product. When NiCl2·glyme was used as a Ni source, a diminished NMR yield of 61% of 442a was obtained. This transformation also operated without the use of base. However, this gave a less productive process affording 442a in 13% 1H-NMR yield. The source of light also had a crucial bearing upon the efficiency of this reaction. When a blue LED array (25 W) was used as a light source for the preparation of 442a, a 33% GC-FID yield was obtained, after 72 h. However, using a blue LED lamp (34 W) this yield was increased to 65%. When the optimised reaction conditions were established, the scope of this transformation was explored. It was found that a range of tolyl products could be prepared, 442b–442d, while carbamate-protected heterocyclic compound 442e was given in 69% yield. When methyl phenyl ether and dimethoxyethane were employed in the reaction, compounds 442f and 442g were given. For 442g a mixture of regioisomers was obtained with a ratio of 1.4:1 in favour of the branched isomer. Therefore, this indicated selectivity for the weaker C–H bond. Furthermore, this transformation could be carried out on bioactive molecules, for example, the medicinal compound loratadine and this gave compound 442h in 93% yield.
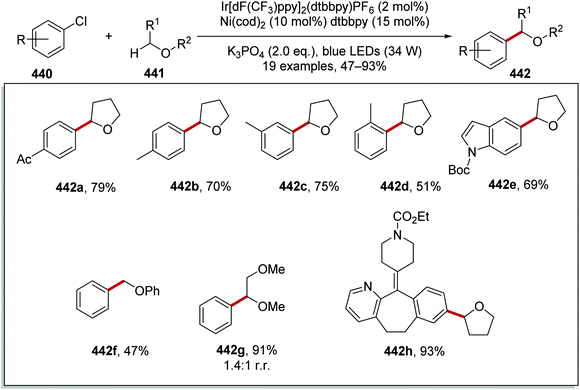 |
| | Scheme 126 Chlorine atom as HAT catalyst. | |
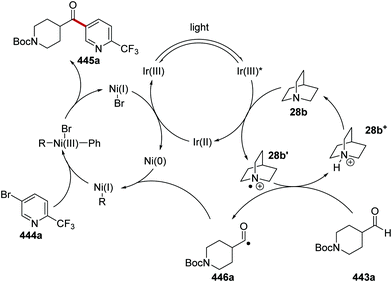 |
| | Scheme 127 Formation of 445a with a tricatalytic system. | |
Optimal reaction conditions were found in the preparation of ketone product 445a. Initially, the reaction was attempted in DMSO and MeCN, and this gave 445a in 2% and 8% yield, respectively. The low yields were due to a competing α-amido C–H abstraction mechanism. However, this was overcome when the reaction was performed in 1,4-dioxane, which gave ketone compound 445a in 87% isolated yield (Scheme 128). It was thought that the solvent dielectric constant was crucial in stabilising the ionic quinuclidinium radical cation, which influenced the roles of BDEs and bond polarization upon the kinetics of this transformation. With these optimised reaction conditions, 44 coupled compounds were prepared in yields ranging from 50% to 92%. Electron-poor 445b and electron-rich 445c arenes were prepared, as well as alkyl ketones 445d. The aldehyde coupling partner was also varied and this gave ketones 445e–445h.
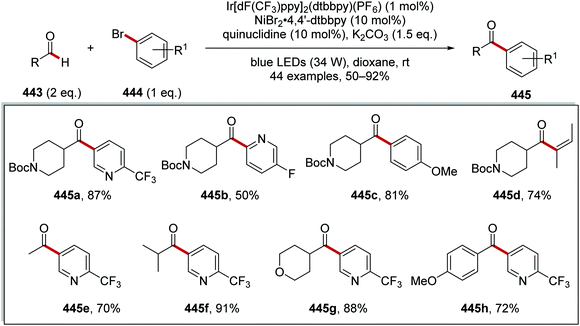 |
| | Scheme 128 Preparation of ketones 445 from aldehydes 443. | |
Aldehydes 447 were functionalised with benziodoxole-derived alkynes 448 mediated with sodium 2-iodobenzoate as a HAT catalyst and an iridium photocatalyst, and this gave alkyne products 449 (Scheme 129).174 From these conditions, 36 alkynes 449 were prepared in yields ranging from 46% to 90%. Silyl functional groups such as TIPS were accommodated under the reaction conditions and this gave 449d in a 70% yield. Aromatic and non-aromatic heterocycles were also suitable substrates, giving 449e and 449f in a 65% and a 77% yield, respectively. The reaction conditions also tolerated ester functionality and ester 449g was prepared in 56% yield.
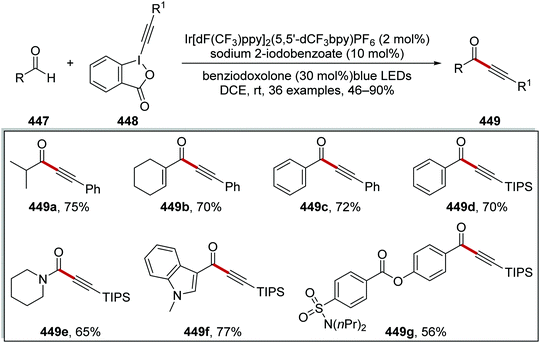 |
| | Scheme 129 Formation of alkyne compounds 449 from aldehydes 447. | |
The reaction mechanism for this reaction (Scheme 130) proposes that the photoactivated iridium catalyst was quenched by iodobenzoate 450 which was oxidised to the corresponding carboxyl radical 451. This then abstracted the formyl hydrogen from the aldehyde substrate 447. The resulting acyl radical 453 attacks the alkyne of the alkynyl benziodoxolone 448 displacing the benziodoxolonyl radical 454. This can either isomerise to the carboxyl radical 451 or can oxidise the Ir(II) complex back to Ir(III).
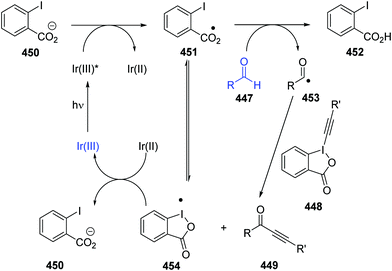 |
| | Scheme 130 Mechanism for coupling alkynes to aldehydes. | |
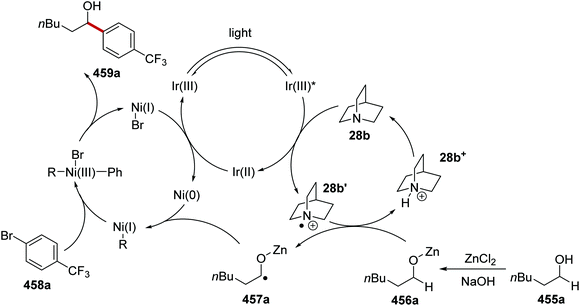 |
| | Scheme 131 Formation of benzylic alcohol 459a from n-hexanol (455a). | |
The reaction was optimised with the preparation of alcohol 459a (Schemes 131 and 132). Initially, when no Lewis acid was present, this resulted in exclusive Ar–O bond formation (yield of the ether product was 54%) and 459a was isolated in a 3% yield. From a survey of Lewis acids, it was found that the Lewis acids ZnCl2/ZnBr2 gave maximal yields of 459a. When ZnCl2 was employed as Lewis acid, with quinuclidine (3 eq., as base and HAT catalyst), this delivered 459a in 44% yield. When the reaction was performed with sodium hydroxide and a lower loading of quinuclidine (30 mol%), this gave 459a in 66% yield. When the reaction was performed with the base, potassium phosphate, and ZnCl2 (1.5 eq.), this afforded 459a in 75% yield and these were taken as optimal conditions (Scheme 132). With these conditions, the scope of the reaction was studied. From the substrate scope, it was observed that n-hexanol was coupled to both electron-rich and electron-poor aryl rings; this gave hydroxy compounds 459a–459d. Functionalisation was site-selective for α-hydroxy C–H bonds, despite when multiple “hydridic” C–H bonds were present. This was demonstrated with the preparation of silyl-protected alcohol 459e, ether 459f and N-benzoyl amide 459g analogues, all with pronounced site-selectivity. The synthetic utility of this process was demonstrated with the preparation of N-benzoyl amide compound 459h, an intermediate for the drug-molecule Prozac.
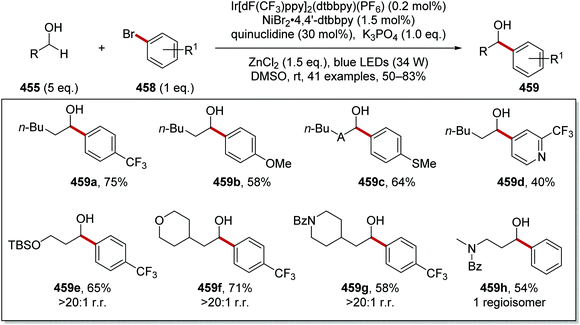 |
| | Scheme 132 Preparation of benzylic alcohols 459. | |
The stereo- and site-selective C–H alkylation of carbohydrate-based compounds was achieved with the implementation of a tri-catalytic reaction system.176 The functionalisation of sugar molecules was facilitated with Ir[dF(CF3)ppy](dtbbpy)2PF6 as photocatalyst, quinuclidine (28b) as HAT catalyst and diphenylboronic acid as cocatalyst. The tricatalytic reaction system allowed selective radical alkylation of C–H bonds of cis-1,2-diol moieties at the equatorial position. Under the reaction conditions, the iridium photocatalyst was excited with blue light and this gave Ir(III)* species (Scheme 133). Ir(III)* converted 28b to radical cation 28b′. At the same time, diphenylboronic acid coordinated to rhamnopyranoside (460a) and this gave boronate adduct 461a. This activated the less hindered equatorial C–H bond for HAT. The selectivity of the HAT process was due to the greater hydridic nature of the C–H bond and the electrostatic attractions between boronate 461a and the positively charged radical species 28b′. The HAT event between 28b′ and 461a resulted in radical 462a and protonated quinuclidinium (22+). Giese addition between carbon-centred radical 462a and electron-poor alkene 463 resulted in α-carbonyl radical 464a. The iridium catalytic cycle was closed with SET between reduced Ir(II) complex and radical 464a, which resulted in anion 465a and Ir(III) complex. Anionic compound 465a underwent protonation, dissociation of boronic acid and transesterification and this gave lactone compound 466a. From control reactions, it was found that the absence of diphenylboronic acid from the reaction mixture resulted in compound 466a being given in 5% yield and the C-2 epimer of 466a was isolated in 22% yield. When diphenylboronic acid was present in the reaction mixture (10 mol%) this gave 466a in 72% yield with no other regioisomers being formed. Therefore, this demonstrated the importance of diphenylboronic acid in the reaction mixture. To further understand this reaction, a computational study was performed, the BDEs and transition state energies of compounds on the methyl α-L-rhamnopyranoside (460a) route were calculated. It was determined that monosaccharide 460a had a weak C–H bond at the C4 position (BDE 87.4 kcal mol−1), which was weaker than the C–H bond at the C2 position (BDE = 89.6 kcal mol−1). Therefore, this explained the poor conversion and selectivity when diphenylboronic acid was omitted from the reaction mixture. For the boronate compound 461a, the C–H bond at the C2 position was much weaker (85.1 kcal mol−1). This made it the weakest C–H bond present in the compound and this increased the efficiency and selectivity of the reaction when diphenylboronic acid was in the reaction mixture.
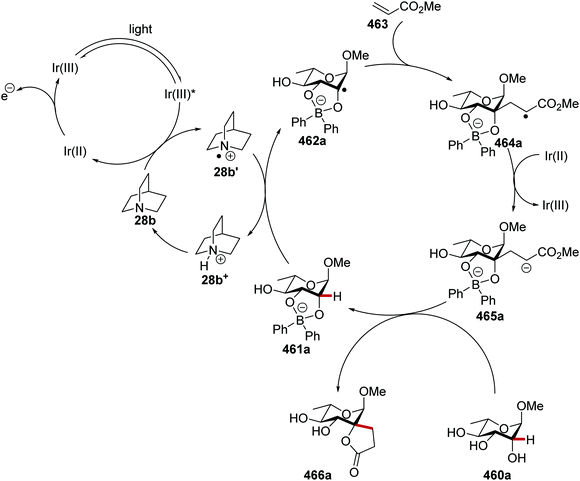 |
| | Scheme 133 Formation of lactone 466a from methyl α-L-rhamnopyranoside 460a. | |
From optimisation studies, it was found that diphenylboronic acid was the optimal boronic acid for this process as it gave 466a in the highest yield (72%) (Scheme 134). A series of 11 lactones 457 was prepared in yields of 54–70%, with notable regio- and stereoselectivity. Silyl protected alcohols were tolerated under the reaction conditions giving lactones 466c and 466d in 54% and 68% yield respectively.
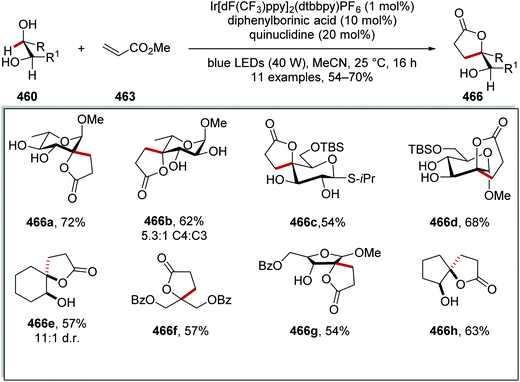 |
| | Scheme 134 Products from lactone formation. | |
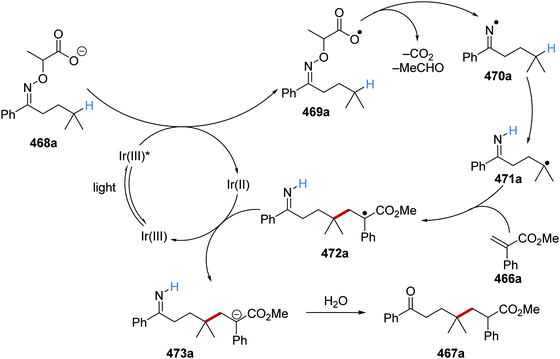 |
| | Scheme 135 Tandem reactions of iminyl radicals. | |
The conversion of 468a to ketone 467a was trialled with three different iridium photocatalysts. From a short optimisation study it was found that Ir(dF(CF3)ppy)]2(dtbbpy)PF6 gave optimal yields of 467a (75% isolated yield, Scheme 136). The optimal conditions were used for a substrate scope and this gave a library of 32 ketones 467 in yields of 31–81%. The reaction was applied to a wide range of substrates and aryl bromides, heterocycle, benzyl esters and ketone groups were all tolerated (467b–e). However, a low yield was obtained for 467f when methyl vinyl ketone was used. It was suggested that competitive polymerisation reactions resulted in this low yield.
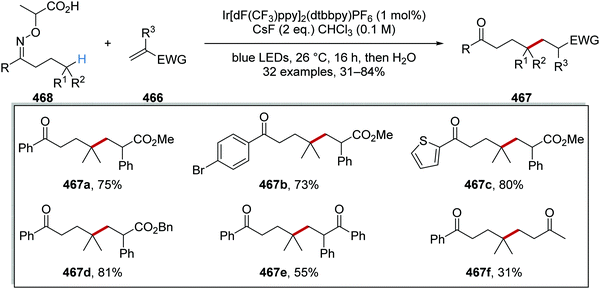 |
| | Scheme 136 Products arising from tandem reactions arising from iminyl radicals. | |
The similar reaction conditions were also used with unsaturated α-iminoxyacetic acid compounds 468 with electron-poor alkenes 466 and this led to dihydropyrroles 474 (Scheme 137).178 These reaction conditions were highly successful as 32 dihydropyrrole compounds were prepared in yields 42–91%. Various electron-withdrawing groups were suitable for this transformation as highlighted with compounds 474a–f. The reaction was amenable to heterocyclic substrates, and this allowed thiophene 474g to be accessed in 68% yield. Substrates where R = alkyl also performed well under the reaction conditions and thus compounds 474h and 474i were given in the yields shown.
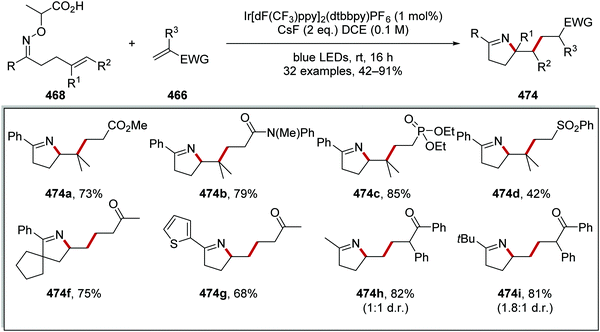 |
| | Scheme 137 Scope of products. | |
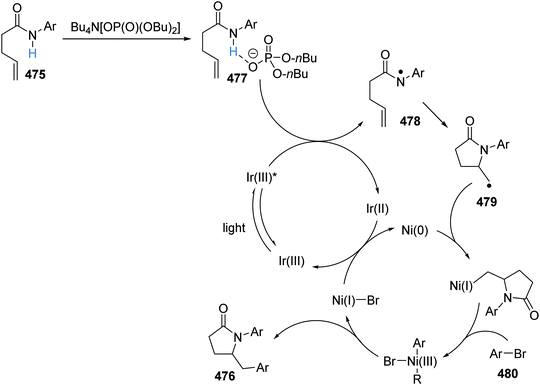 |
| | Scheme 138 PCET and radical cascade and coupling of 466. | |
This process was utilised in the preparation of 50 lactams, 476 (Scheme 139). This methodology allowed for the preparation of nitrile analogue 476a in 78% yield. Spirocyclic 476b, bicyclic 476c, dinitrile 476d, N-Boc amine 476f, and thiophene 476g derivatives were all prepared with this transformation.
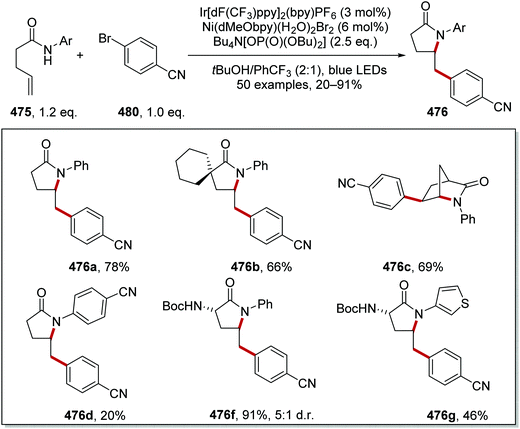 |
| | Scheme 139 Results of PCET and radical cascade and cross-coupling of 475. | |
A site-selective remote C–H activation, inspired by the Hofmann–Löffler–Freytag reaction, and involving a photocatalytic reaction with a PCET was reported.168 Under the reaction conditions, the combination of amide 481a and phosphate base resulted in hydrogen-bond interactions as in adduct 482a (Scheme 140). A concerted PCET with adduct 482a and Ir(III)* complex allowed for cleavage of the strong N–H bond (N-methylacetamide has an N–H BDE = 106.5 kcal mol−1, in DMSO)42 and formation of amidyl radical 483a and Ir(II) species. This PCET process was confirmed with Stern–Volmer experiments, where it was shown that efficient quenching of Ir(III)* only occurred when both amide and base were present. Additionally, as shown above (Scheme 138), the addition of base to an amide drastically facilitates the oxidation of amides. Amidyl radical 483a effected 1,5-HAT giving radical 484a. Giese addition between radical 484a and electron-poor alkene 485a gave α-keto radical 486a. The iridium catalytic cycle was closed with electron transfer from the Ir(II) complex to radical 486a. Protonation of anion 487a led to coupled compound 488a.
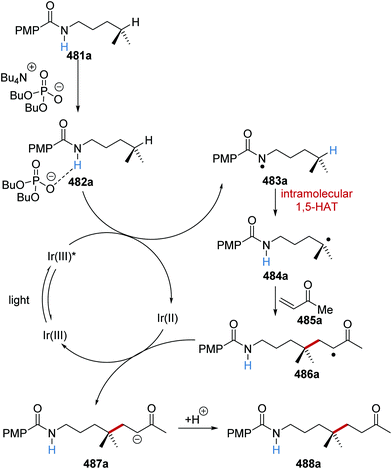 |
| | Scheme 140 Iridium-promoted HLF type reaction. | |
Ir[dF(CF3)ppy]2(5,5′-dCF3bpy)PF6 was the optimal catalyst for this process, as it gave 488a in the highest yield (78%, Scheme 141). Optimised reaction conditions allowed for the preparation of 27 alkylated compounds 488 in 29–87% yield. Silyl-protected hydroxy groups were tolerated and thus compound 488b was isolated in 66% yield. Furthermore, the site-selectivity of this process was maintained even with N-Boc groups being present giving product 488c in 45% yield. The high degree of selectivity of this process is due to the cyclic HAT transition state, and this ensured only one site of hydrogen atom abstraction was feasible. Both acrylate and acrylamide were poor coupling partners, providing only trace amounts of alkylated products. It was concluded that this was due to mismatching of the reduction potentials of the α-carbonyl radicals and Ir(II) complex, which led to sluggish electron transfer and thus diminished yields. However, when the more reactive dicarbonyl coupling partners were used, this resulted in formation of diester 488d in 87% yield. This PCET transformation was also applicable to sulfonamides and N-Boc carbamates, providing 488e and 488f in 74% and 29% yield, respectively.
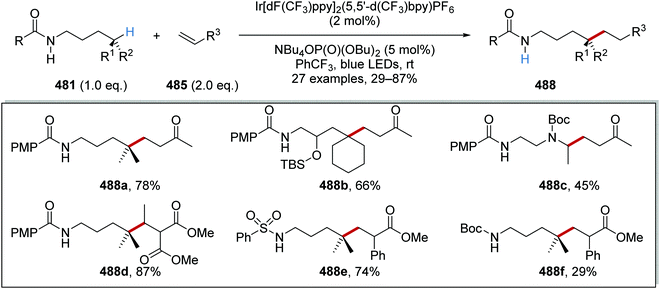 |
| | Scheme 141 HLF-inspired reaction with PCET. | |
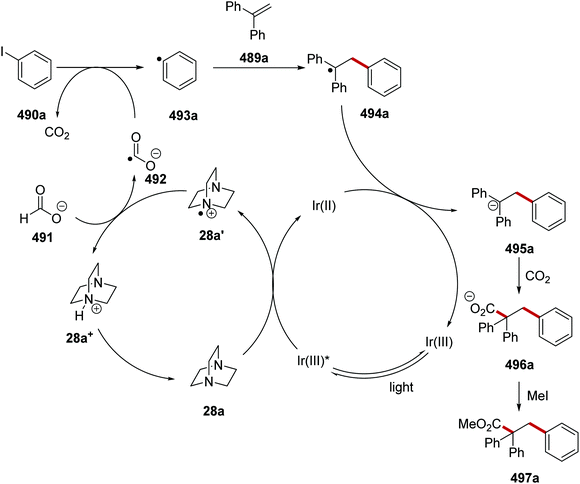 |
| | Scheme 142 DABCO-facilitated Meerwein reaction. | |
Optimal conditions for this reaction used Ir(ppy)2(dtbbpy)PF6 as photocatalyst, DABCO as HAT catalyst, potassium carbonate as base, DMSO as the solvent, and this gave 497a in 78% yield (Scheme 143). A library of 82 compounds was prepared with the use of these optimal conditions. Functional groups and heterocycles were accommodated under the reaction conditions and thus compounds 497b–f were prepared. It was found, alongside aryl iodides or bromides, that aryl chlorides could also be used in this transformation and 497e was prepared in 72% yield from the aryl chloride. The use of an α,β-substituted styrene allowed for the formation of compound 497f in 56% yield. Alkyl bromides and alkyl iodides under the reaction conditions gave 497g and 497h, in 59–71% yields.
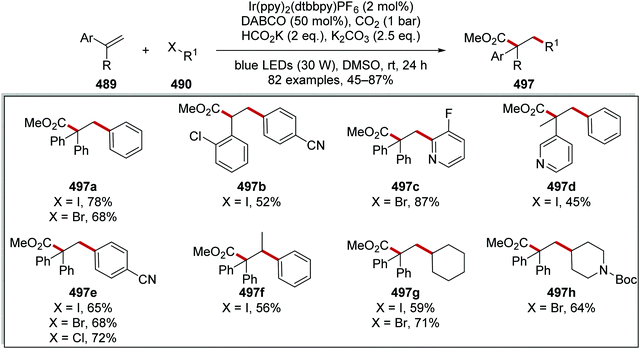 |
| | Scheme 143 Selected products from a DABCO ‘photo-Meerwein’ reaction. | |
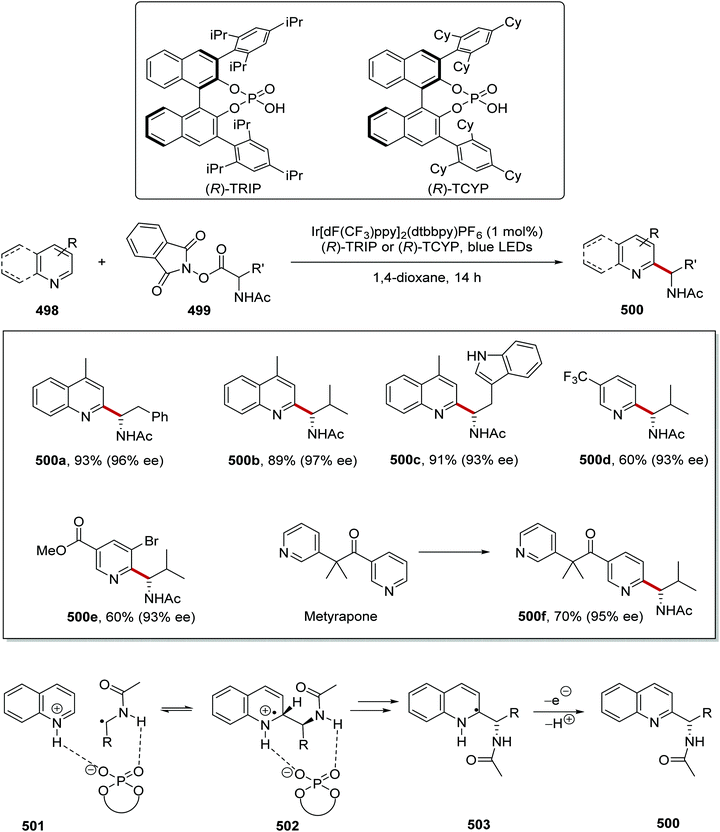 |
| | Scheme 144 Stereoselective Minisci reaction. | |
It might be imagined that the initial reduction of the substrates 499 occurred by the photoactivated Ir(III)* complex. However, investigations by the authors concluded that this is unlikely as Ir(III)* has insufficient reducing power. Instead, they propose that Ir(II) is produced off the main catalytic cycle, and perhaps by reduction of Ir(III)* by a TRIP anion. The resulting Ir(II) complex would then reduce substrates 499 as above. This proposal is supported by electrochemical potential values reported elsewhere. The compound N-hydroxyphthalimide benzoate (Ered = −1.4 V vs. SCE)184b can undergo irreversible SET with the reduced iridium catalyst (Ir(II) Eox = −1.37 V vs. SCE).8b The transformation of radical 503 to product 500 provides the electron that closes the redox catalytic cycle.
2.4 Catalytic cycles featuring Ir(III)/Ir(IV)
Carboxylates as radical precursors.
The radical coupling between benzonitriles 504 and carboxylic acid radical precursors 505 was reported.187 Irradiation of the reaction mixture with blue light gave Ir(III)* species from Ir(III) complex (Scheme 145). SET reduction of terephthalonitrile (504a, Ered = −1.61 V vs. SCE) with Ir(III)* [For the related (Ir(ppy)3 [Eox* = −1.73 V vs. SCE)] resulted in the formation of radical anion 506a and an Ir(IV) complex. The iridium catalytic cycle was closed with a SET transfer from carboxylate 507a to Ir(IV) complex. After decarboxylation, α-amino radical 508a added to 506a, and subsequent cyanide loss afforded coupled product 509a.
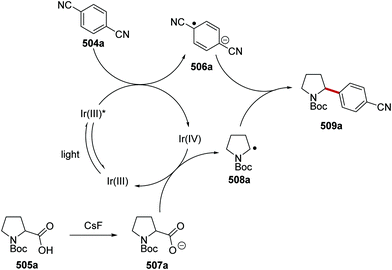 |
| | Scheme 145 Radical coupling between benzonitrile 504a and carboxylic acid radical precursor 505a. | |
The coupling of carboxylic acids 505 to electron-poor benzonitriles 504 succeeded and this gave a range of arylated compounds 509 in yields of 52–89% (Scheme 146).187 Many α-amino-acids provided acceptable substrates for this transformation and afforded arylated products 509a–d in high yields. A range of different electron-withdrawing groups was employed in this transformation as seen in electron-poor aromatic products 509e–h.
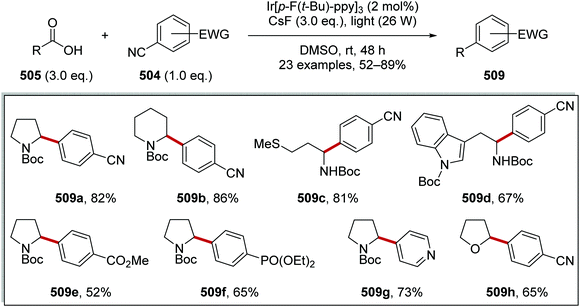 |
| | Scheme 146 Products of radical coupling between benzonitriles 504 and carboxylic acid radical precursors 505. | |
An Ir and Cu catalytic system was used to prepare disubstituted BCPs in one step from hypervalent-iodine species 510 and [1.1.1]propellane (512)104 (Scheme 147). SET from excited Ir(III)* complex to radical precursor 510a (Ered = −0.82 V vs. SCE) resulted in alkyl radical 502a and electron-deficient Ir(IV) complex. Radical addition of 511a to [1.1.1]propellane (512) broke the weak central C–C bond and formed BCP radical 513a. Coincidentally, Cu(I) complex underwent ligation with nucleophile 514a and this resulted in Cu(I) complex B. Oxidation of Cu(I) complex B with the aforementioned Ir(IV) complex, resulted in the closure of the iridium catalytic cycle and Cu(II) complex C. Addition of alkyl radical 513a to C gave Cu complex D. Reductive elimination of complex D resulted in the closure of the copper catalytic cycle and formation of BCP 515a.
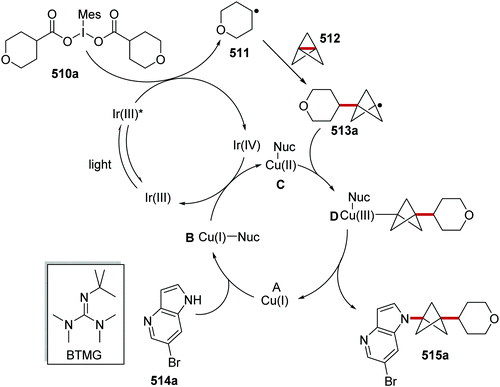 |
| | Scheme 147 Formation of disubstituted BCP analogue 515a. | |
This three-component reaction was successful as it allowed carboxylic acid precursors 516 to be converted to bioisostere 515via iodine intermediate 517 (Scheme 148). Variation of carboxylic acid precursors allowed for a range of different bioisosteres to be formed. For example, the formation of ether 515a, N-Boc amine 515b and gem-difluoroalkane 515c analogues were achieved. It was also observed that different nucleophiles made successful coupling partners and derivatives 515d–f were prepared in yields of 53–80%.
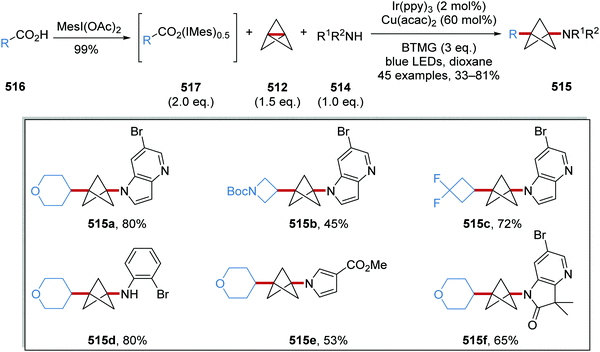 |
| | Scheme 148 Results of radical coupling to form disubstituted BCPs 515. | |
Activated alkyl bromides 518 were also substrates for this transformation (Scheme 149). Therefore, a range of BCP compounds 515g–l was prepared from the corresponding α-bromocarbonyl compounds.
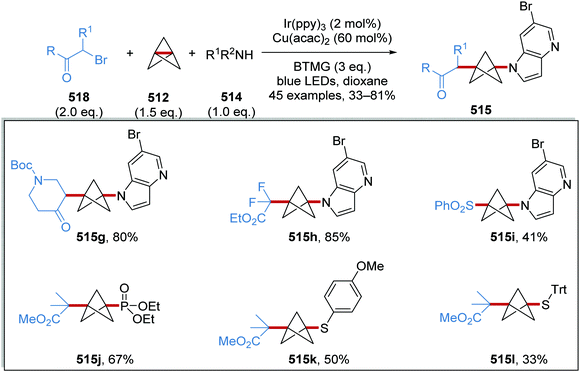 |
| | Scheme 149 Formation of disubstituted BCPs 515 from α-bromocarbonyl compounds 518. | |
Carbon α-heteroatom C–H functionalisation.
The coupling of amines 514 with dicyanobenzene 519 was achieved with the Ir(ppy)3 photocatalyst, and sodium acetate (Scheme 150).188 Ir(III)* (Eox* = −1.73 V vs. SCE)8b reduced 519a (Ered = −0.82 V vs. SCE) to its radical anion 520a. The iridium catalytic cycle was closed with SET from N-phenylpyrrolidine (514a, Eox = 0.82 V vs. SCE)188b to the Ir(IV) complex (Ered = +0.77 V vs. SCE). This gave amine radical cation 521 and after proton loss, carbon-centred radical 522a. Coupling between 520a and 522a formed anion 523a. Loss of the cyanide ion from 523a gave the coupled product 524a.
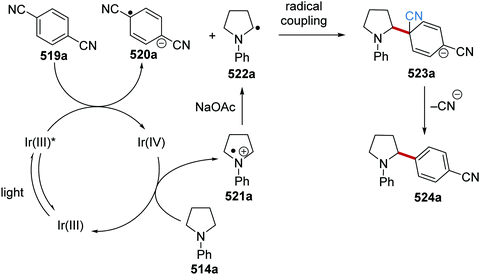 |
| | Scheme 150 α-Aminoalkyl radical 522a coupling to benzonitrile analogue 519a. | |
The photocatalyst tris[2-phenylpyridinato-C2,N]iridium(III) resulted in high yields of coupled compounds 524 from anilines 514 and electron-poor benzonitrile compounds 519 (Scheme 151). Further investigation showed that the reaction worked well when carried out in DMA with sodium acetate as base. Application of the optimised reaction conditions to the corresponding substrates gave acetal 524c, N-Boc amines 524b and 524h derivatives in yields of 78–95%. Heterocycles were compatible with the reaction conditions, and thus coupled compounds 524e–524h were prepared and isolated in yields of 44–92%. This reaction was regioselective; for example, coupled product 524d formed as a mixture of regioisomers (20:1 ratio), with arylation taking place upon the less sterically hindered α-amino C–H bond. The regioselectivity was observed for substrates with α-amino methylene and methine positions, with the methylene functionalised compound 524c being isolated as the only isomer.
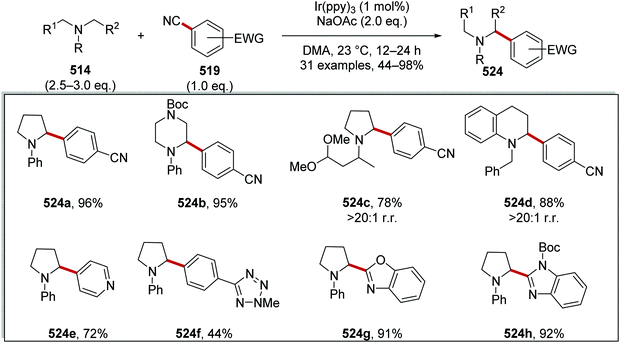 |
| | Scheme 151 Results of amines 514 coupling to benzonitriles 519. | |
The synthetic utility of this methodology was demonstrated with the preparation of derivative 526 from the antibiotic, linezolid (525, Scheme 152). With the use of Ir(ppy)2(dtbbpy) photocatalyst, linezolid was coupled to 2-chlorobenzoxazole and this gave linezolid derivative 520 in 58% yield.
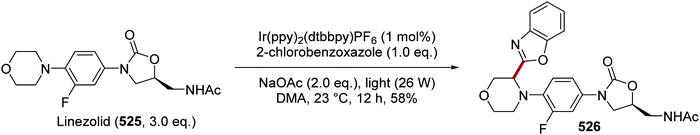 |
| | Scheme 152 C–H functionalisation of linezolid 525. | |
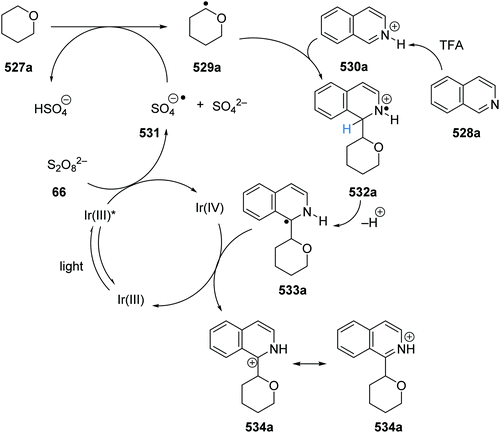 |
| | Scheme 153 Ether-derived radicals undergo Minisci reaction. | |
When the reaction was being optimised, it was found that the addition of water to the reaction mixture gave increased yields of 536a (Scheme 154). When water was added to the reaction mixture, this resulted in a biphasic mixture and it was inferred that this allowed for greater photon penetration leading to a more successful reaction. Higher yields of 536a were obtained when sodium persulfate was used in the reaction, over potassium persulfate and ammonium persulfate. Therefore, it was concluded that the optimal conditions for this transformation were: Ir[dF(CF3)ppy](dtbbpy)PF6 as photocatalyst (2 mol%), sodium persulfate (2 eq.), TFA (1 eq.), ether substrate (50 eq.) and N-based heterocycle (1 eq.) (Scheme 154). These optimised conditions gave an 88% yield of 536a. 1,3-Dioxolane gave 536b as the product in 83% yield with 3:1 ratio of regioisomers. Tetrahydrofurfuryl acetate as substrate gave tetrahydrofuran product 536c in 77% yield with a 2:1 ratio of diastereoisomers and only one regioisomer was isolated, highlighting the site-selectivity of this process. Ether 536d was isolated in 88% yield when dibutyl ether was employed as the substrate. A 2.3:1 mixture of regioisomers (with preferred functionalisation at the methylene position) was obtained, in 93% yield for coupled compound 536e when dimethoxyethane was used as substrate. It was shown that coupling to various N-based heterocycles was feasible with the preparation of coupled heterocycles 536f–j.
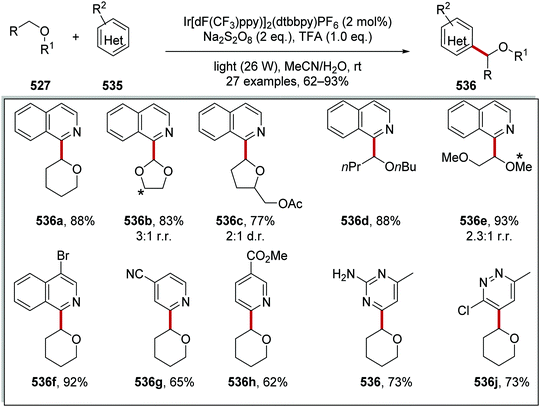 |
| | Scheme 154 Scope of products from Minisci reaction. (Asterisk * indicates an alternative site of attachment.) | |
The Minisci coupling of N-based heterocycles 538 with amine derivatives 537 was investigated, with particular attention paid to protecting group influences (Scheme 155).191 The photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6, TFA and sodium persulfate were used to facilitate the Minisci reaction; under these conditions acetamide, trifluoroacetamide and Boc-protected amines were investigated. The use of DMA and tert-butyl N,N-dimethylcarbamate (BDC) afforded coupled amine compounds 539a and 539b in 76% and 70% yields respectively. However, N,N-dimethyltrifluoroacetamide (DTA) gave coupled amine compound 539c in 10% yield after 13 days of reaction. To understand this phenomenon, the BDE of DMA, BDC and DTA were calculated, and it was found that all three compounds had similar α-amino C–H bond energies. The calculated α-amino C–H BDE for these compounds were: DMA = 97.7 kcal mol−1, BDC = 99.0 kcal mol−1 and DTA = 99.9 kcal mol−1. Therefore, this indicated that the HAT event had limited influence upon the success of this transformation. The SOMO orbitals energy levels of these substrates were then calculated and from this, it was found that the SOMO energy levels of the radicals derived from N-BocNMe2 and DMA (−134.2 and −138.4 kcal mol−1, respectively) were much higher than for DTA (−151.3 kcal mol−1). This implied that the corresponding carbon-centred radical of DTA is much less nucleophilic and more stable than the DMA and BDC radicals and this inhibited reactivity with the heterocycle. With the understanding gained from these control experiments and calculations, 34 Minisci coupled products 539 were prepared. Previous work reported that high loadings of ether coupling partner (50 eq.) were required for successful coupling.189 However, it was shown that a much lower number of equivalents was required for this process and, for some transformations, two equivalents of 537 were sufficient. These optimised conditions were mild, and thus sensitive functional groups were tolerated, and compounds like hydroxy-functionalised quinoline 539d and spiroether product 539e were prepared in 59% and 71% yield, respectively. In preparing analogue 539e, no reactivity was observed at the α-alkoxy position and only one regioisomer was given. 2- and 4-Chloroquinoline led to the formation of 539g and 539h in 67% and 71% yield, respectively. Furthermore, the use of 1,5-naphthyridine and imidazo[1,2-a]pyrimidine analogues as substrates gave 539j and 539k in 69% and 65% yield, highlighting the range of heterocycles that can be used in this transformation. The reaction was insensitive to scale and compound 539f was prepared on a 0.4 mmol scale in a yield of 95%. The preparation of 539f on a 9.5 mmol scale led to isolation of the material in 85% yield. The reaction was still operative without the use of light or Ir photocatalyst, as it was demonstrated that carbamate 539f was prepared in 47% yield simply by heating the reaction mixture at 50 °C.
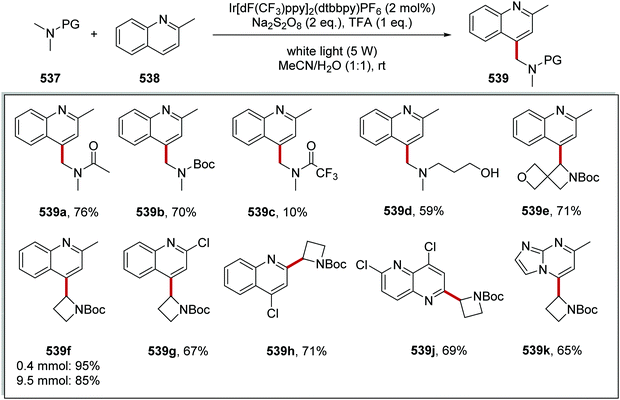 |
| | Scheme 155 Range of products from the sodium persulfate-initiated Minisci reaction. | |
The origin of the high site-selectivity observed for derivative 539e was investigated with competition reactions (Scheme 156). When a mixture of oxetane 537l (15 eq.) and N-Boc azetidine 537f (15 eq.) with quinaldine (538, 1 eq.) was reacted under standard reaction conditions, this gave carbamate 539f in 68% yield as sole-product, and no ether coupled compound 539l was detected (Scheme 156). However, when THF was the substrate, the reaction did yield some α-alkoxy functionalised compound, but the selectivity was biased for azetidine 539f. An intra-molecular competition experiment was also carried out with bifunctional compound 537n, which featured both ether and N-Boc amine functionalities. This gave 539n in 66% yield as an 8.1:1 mixture of regioisomers with the α-carbamate functionalised compound as the major regioisomer. The results of these competition experiments were not explained with the BDE or calculated SOMO energies from ab initio calculations. Therefore, it was postulated that the observed selectivity was due to polar effects with the HAT event taking place at the most hydridic C–H bond being attacked by the electrophilic sulfate radical anion.
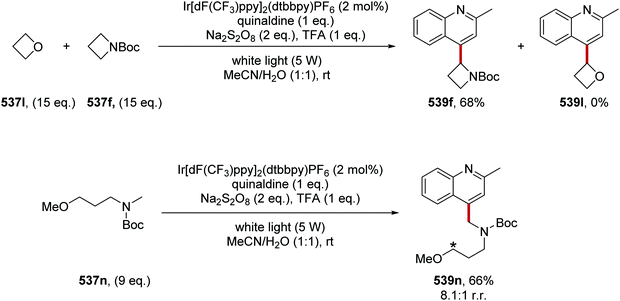 |
| | Scheme 156 α-Carbamate and α-alkoxy C–H competition experiments. | |
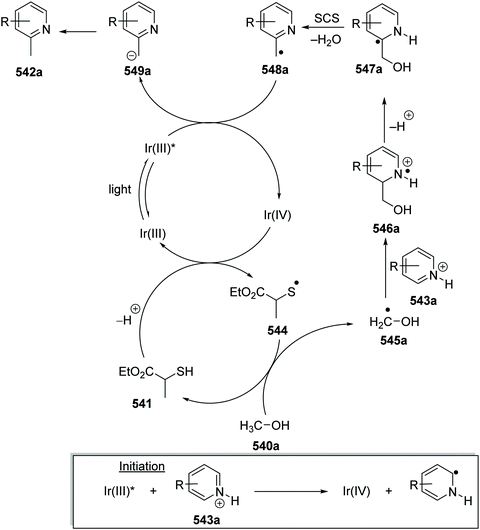 |
| | Scheme 157 Preparation of alkylated heterocycles via spin-centred-shift (SCS). | |
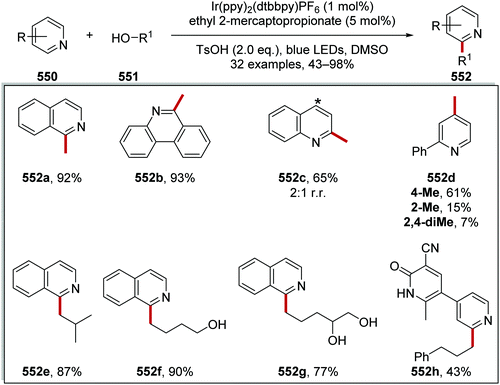 |
| | Scheme 158 Products from the dehydrative Minisci variant. | |
The combination of photoredox catalyst Ir(ppy)2(dtbbpy)PF6 (1 mol%), ethyl 2-mercaptopropionate (5 mol%) and p-toluenesulfonic acid (1 eq.) in DMSO with methanol (co-solvent) gave methylated heterocycles 552a–d. Isoquinoline and phenanthridine as substrates gave methylated products 552a and 552b, in a regioselective manner, in 92% and 93% yield, respectively. When quinoline was treated to the reaction conditions, this gave 552c as a 2:1 mixture of regioisomers (the site of the other regioisomer is denoted with an asterisk) in 65% combined yield. 2-Phenylpyridine as substrate led to the formation of three compounds 552d in 84% combined yield. Other alcohols were also used as alkylating agents. The coupling of isoquinoline and isobutanol (10 eq.) gave alkylated analogue 552e in 87% yield. Coupling of tetrahydrofuran (10 eq.) with isoquinoline resulted in the formation of alcohol 552f in 90% yield. Similar reactivity was observed when tetrahydrofurfuryl alcohol (10 eq.) was used as coupling partner and this gave diol 552g in 77% yield. Bioactive compounds were functionalised with this reaction protocol. For example, milrinone, a phosphodiesterase 3 inhibitor was selectively alkylated with 3-phenylpropan-1-ol (10 eq.) and this gave 552h in 43% yield.
The use of methyl 2-mercaptoacetate (553) alongside an iridium photoredox catalyst allowed for C–C coupling between benzylic ethers 554 and electron-poor benzonitriles 555, and this gave benzhydryl products 556.50 Under the reaction conditions, the Ir(ppy)3 complex was excited with blue light and this afforded excited Ir(III)* complex (Scheme 159). SET to 555a (Ered = −1.61 V vs. SCE in MeCN) and Ir(III)* (Eox* = −1.73 V vs. SCE in MeCN) resulted in the formation of Ir(IV) species and arene radical anion 557a. The iridium catalytic cycle was closed with SET from thiol 553 to Ir(IV) (Ered = +0.77 V vs. SCE) and this gave thiyl radical 558a and it was inferred that 558a was formed via a PCET process. The reaction was performed under weakly basic conditions with the base K2HPO4 being used and as mentioned above in the text accompanying Scheme 157, Ir(IV) might be unable to directly oxidise the thiol by HAT. Therefore, the pre-association of the S–H proton with the phosphate base allowed for a PCET and this avoided high-energy intermediates.57 Radical 558a abstracted an H atom from benzylic ether 554a, to give radical 559a. This reaction was thermodynamically favourable as the BDE for the S–H of 553 (BDE of S–H bond of the analogous methyl 2-mercaptoacetate = 86.8–87.2 kcal mol−1) is likely greater than the BDE for the C–H present in 554a (benzyl methyl ether has an αC–H BDE = 85.8 kcal mol−1). Coupling between arene radical anion 557a and benzylic radical 559a gave anion 560a and, after cyanide ion loss, this gave coupled product 556a.
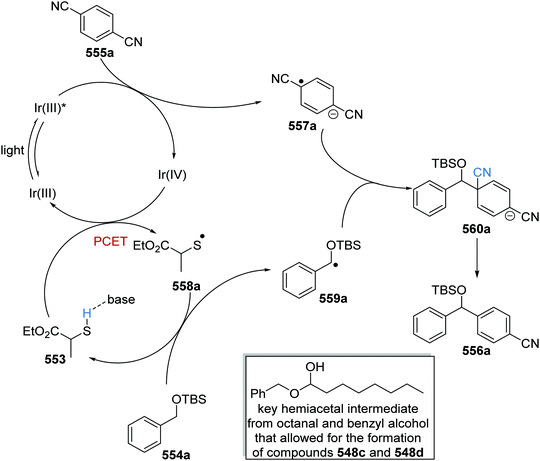 |
| | Scheme 159 Benzyl silyl ethers couple to 1,4-dicyanobenzene. | |
As the reaction was optimised it was found that the use of Ir(ppy)3 (1 mol%), cysteine (20 mol%) as HAT catalyst in MeCN with K2HPO4 gave a 14% yield of 556a. Investigations then revealed that the addition of an aldehyde (octanal) was critical in forming 556a in a higher (32%) yield. It was postulated that the presence of the aldehyde was beneficial as it sequestered the cyanide anion. When DMA was used as reaction solvent instead of MeCN and methyl 2-mercaptoacetate (553) was used as HAT catalyst instead of cysteine, this gave 556a in 77% yield. These optimised reaction conditions were then applied to various substrates resulting in the preparation of 28 coupled compounds 556 in yields of 41–86% (Scheme 160). Various benzylic ethers were suitable substrates and methoxy 556a and cyclic ether 556b derivatives were isolated. Benzylic ethers were substrates for this transformation and alcohols 556c and 556d were isolated in 77% and 72% yield, respectively. The use of silyl-protected benzylic alcohol allowed for the preparation of ester 556e, o-cyano 556f, pyridine 556g and azaindole 556h coupled analogues in yields ranging from 41–76%. During optimisation and substrate scope studies, there was no evidence of oxidation of the alcohol substrates to the corresponding aldehyde or ketone products nor of any O-functionalised products. Control reactions were performed to elucidate the importance of octanal with these alcohol compounds. When this reaction was conducted without octanal, this resulted in exclusive aldehyde formation and with no aryl coupling. Analysis by 1H-NMR spectroscopy of the reaction mixture revealed a transient hemiacetal intermediate (Scheme 159, inset), which was responsible for C–C bond formation and the prevention of benzylic oxidation.
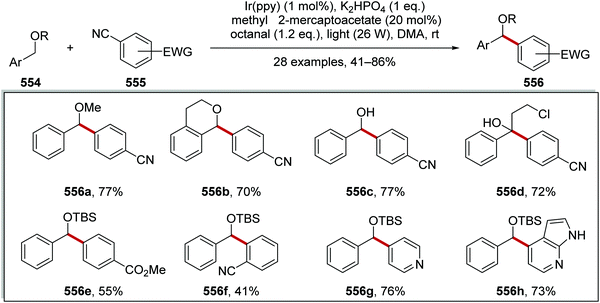 |
| | Scheme 160 Benzylic C–H activation and coupling of the resulting radicals. | |
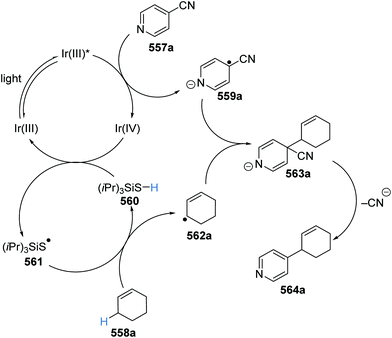 |
| | Scheme 161 HAT of allylic hydrogen atoms on sp3 carbon and coupling to electron-poor aryl systems. | |
The applicability of this methodology was then demonstrated with the preparation of 27 coupled products (Scheme 162). With regards to the cyanoarene, it was demonstrated that the EWG could either be in the para or ortho position with 564b being prepared in a 50% yield from 1,2-dicyanobenzene. When 1-(trimethylsiloxy)cyclohexene was used in this transformation, this resulted in ketone 564d in 83% yield. It was also found that the reaction was tolerant of ester functional groups and heterocycles with the preparation of compounds 564e and 564f.
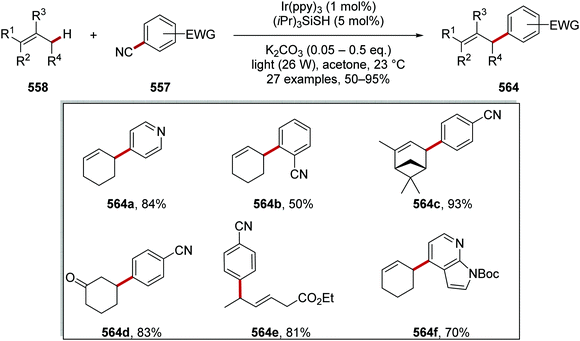 |
| | Scheme 162 Results of HAT of allylic hydrogen atoms on sp3 carbon and coupling to electron-poor aryl systems. | |
2.5 Ir(III) energy transfer reactions
Progress in radical aromatic decarboxylation reactions of aromatic carboxylic acids has been slow due to the inherent stability of these substrates.158,195 This problem was overcome with an energy-transfer-mediated concerted fragmentation/decarboxylation process from a redox active ester. The iridium catalyst Ir[dF(CF3)ppy]2dtbbpy(PF6) was promoted to its excited state Ir(III)*, which has a triplet energy of 60.8 kcal mol−1 and an excited state lifetime of 2.3 μs. Ir(III)* participated in an energy transfer with ester 565a due to its lower triplet energy (ET = 46.4 kcal mol−1) and excited species 566a* was produced (Scheme 163). Concerted N–O homolytic bond cleavage and decarboxylation of 566a* resulted in carbon dioxide, iminyl radical 567a and aryl radical 568a. Aryl radical 568a was trapped with a radical trapping reagent; chloroiodomethane gave aryl iodide 569a, but alternative trapping agents were also used as seen in products 573a–d. Concurrently, iminyl radical decomposed to byproducts 570–572, which were always detected in optimisation and substrate scope reactions.
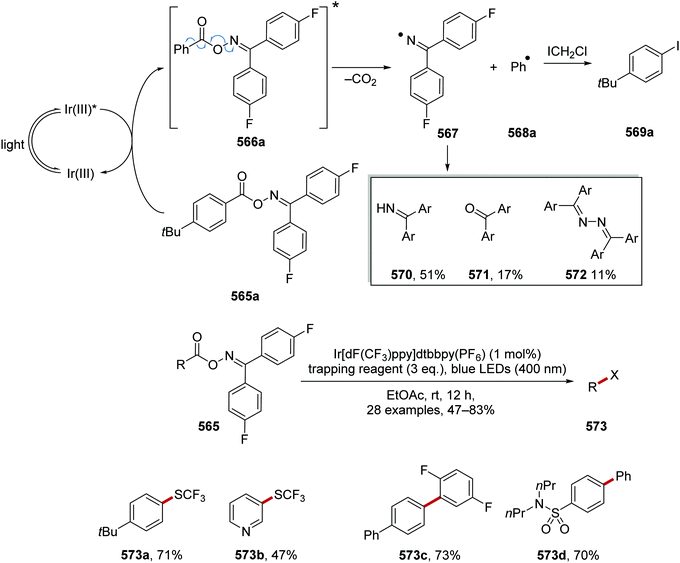 |
| | Scheme 163 Coupling of aryl radicals arising from decarboxylation. | |
An iridium-catalysed approach has been reported for the construction of azetidine compounds 574via an intramolecular aza Paternò–Büchi reaction.196 Under the reaction conditions, the iridium catalyst became excited with irradiation with blue light and this gave Ir(III)* species (Scheme 164). From Stern–Volmer studies it was shown that styrene moieties 575 quenched the excited catalyst, which returned the Ir(III) complex and triplet styrene 576a. Reversible C–C bond formation resulted in cyclic intermediate 577a. Free rotation around the C–N bond present in 577a led to the observed oxime E/Z scrambling as styrene 575a was returned from 577a after ring-opening and deexcitation pathways. Cyclic intermediate 577a was also able to partake in an ISC process and this delivered singlet biradical 578a, which allowed for C–N bond formation and giving azetidine 574a.
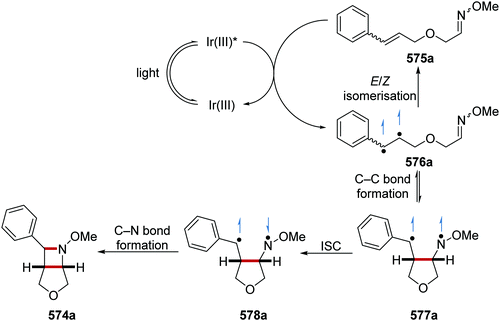 |
| | Scheme 164 Aza-Paternò–Büchi reactions. | |
A challenge for this transformation was the selective excitation of the alkene over the imine motif. It has been found that aza-Paternò–Büchi reactions work less well than Paternò–Büchi reactions, as imine excitation leads to non-productive radiationless decay processes. The addition of a sensitiser to the reaction mixture overcomes the problem of non-productive imine excitation.61 Oxime 575a was chosen as substrate to find optimal reaction conditions. Initially, it was found that direct excitation of 575a with UV-light led to the formation of 574a in 6% yield (Table 7, entry 1). The use of catalytic amounts of xanthone (ET = 74.1 kcal mol−1) gave 574a in 43% yield (entry 2).197 With complete consumption of starting material, it was postulated that the strong photoredox properties of xanthone led to decomposition of the substrate. When Ru(bpy)3(PF6) was used as catalyst, no conversion to product was observed (entry 3), and this was due to the triplet energy of this catalyst being too small to effectively transfer energy to 575a. Styrenes have a triplet energy of ca. 62 kcal mol−1; therefore an effective energy transfer catalyst has to have a triplet energy greater than this.198 Imines have greater triplet energy, and arylimines have triplet energy levels between 72–85 kcal mol−1.199 The Ir(ppy)3 photocatalyst has a triplet energy of 58 kcal mol−1 and thus this gave 574a in 39% yield. The use of Ir[dF(CF3)ppy]2dtbbpy(PF6) that has a triplet energy level of 62 kcal mol−1 efficiently generated 574a in 98% yield with only 0.5 mol% of catalyst.
Table 7 Optimisation of reaction conditions for Aza-Paternò–Büchi reactions
|
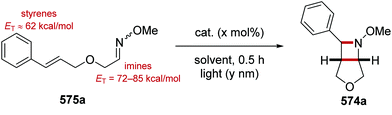
|
| Entry |
Catalyst (mol%) |
E
T (kcal mol−1) |
Wavelength (nm) |
Solvent |
Yield of 574aa (%) |
|
Yield determined by quantitative 1H NMR spectroscopy with an internal standard.
|
| 1 |
None |
— |
365 |
CH2Cl2 |
6 |
| 2 |
Xanthone (30) |
74 |
365 |
MeCN |
43 |
| 3 |
Ru(bpy)3(PF6)2 (2.5) |
49 |
427 |
THF |
0 |
| 4 |
Ir(ppy)3 (2.5) |
58 |
427 |
THF |
39 |
| 5 |
Ir[dF(CF3)ppy]2dtbbpy(PF6) (0.5) |
62 |
427 |
THF |
98 |
Optimised reaction conditions led to the generation of 21 azetidine compounds 574 from the corresponding alkene-imine substrates 575 (Scheme 165). This methodology allowed for the preparation of bicyclic fused tetrahydrofuran analogues 574a and 574b in 96% and 85% yield, respectively. The reaction proceeded smoothly even with the presence of ester, silyl-protected alcohol and alkene groups and thus compounds 574d–f were all prepared. The generation of heterocycle-containing products and preparation of polycyclic compounds was also achieved and 574g–j were all produced.
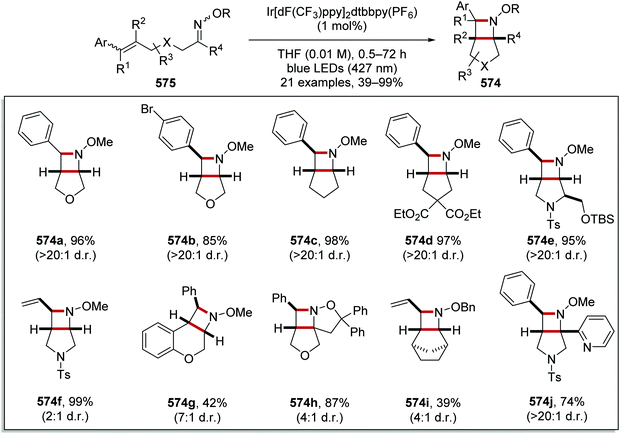 |
| | Scheme 165 Product scope for Ir-promoted 4-membered ring formation. | |
3 Radical coupling reactions triggered by organic photoredox agents
3.1 Xanthene and related dyes
Xanthene dyes undergo photoexcitation. The excited species can act as electron donors or electron acceptors or as energy transfer agents. Fluorescein 4, Eosin Y 5, Rose Bengal 6 and erythrosine 73 are all members of this family (Fig. 16). The molecules can have various protonation states but are represented above in their electron-rich ‘basic’ forms. Nitrogen-substituted analogues of this family are the rhodamines, represented here by rhodamine-6G 578 and rhodamine B 579, while another closely related structure is methylene blue 580.8,200 As mentioned previously, Eosin Y does have an efficient ISC (ϕISC = 0.33) and the strength of its oxidising/reducing abilities does vary between the excited singlet (Ered* = +1.23 V vs. SCE, Eox* = −1.58 V vs. SCE) and triplet state (Ered* = +0.83 V vs. SCE, Eox* = −1.15 V vs. SCE), with the excited singlet state being a stronger oxidant and reductant.
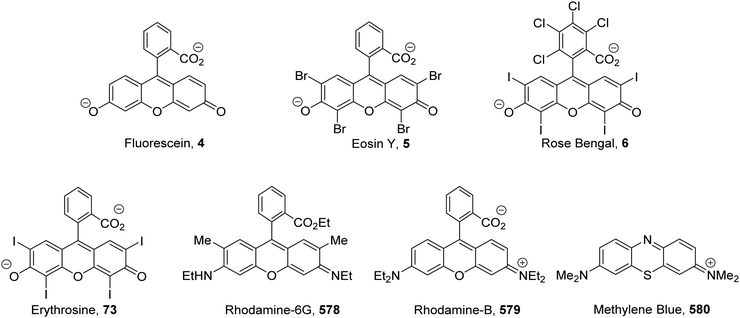 |
| | Fig. 16 Eosin Y (5) and structurally related dyes. | |
Eosin Y 5 is widely reported for a range of reaction types. The photoexcited forms of Eosin and related molecules are generally mild electron donors but they react well with good electron acceptors such as arenediazonium salts (Scheme 166). Single electron transfer from 5 (Eox* = −1.58 V vs. SCE)8a reduces the diazonium cation 581 (Ered = +0.33 V vs. Ag/AgCl and 4-methylbenzenediazonium tetrafluoroborate Ered = −0.18 V vs. SCE) to the corresponding radical, from which a molecule of N2 is lost to afford an aryl radical. This couples regioselectively to the 2-position of electron-rich heteroarenes like furans, thiophenes and N-Boc pyrroles as reported by König et al.202 The reaction works well for a wide range of substituted diazonium salts.
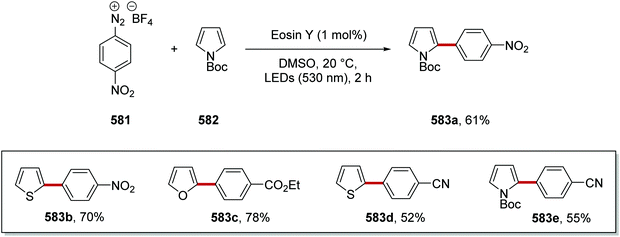 |
| | Scheme 166 Photoactivated Eosin Y reduced arenediazonium salts to aryl radicals. | |
Zhou‘s team has used diazonium salts in benzannulation reactions (Scheme 167).203 Diazonium salt 584, derived from a biaryl, illustrates the point. The salt was activated as above, and the aryl radical so created reacted with methyl propiolate 585. The resulting vinyl radical then cyclised onto the remaining aryl ring to give an intermediate cyclohexadienyl radical. The final product formed following electron transfer [likely to another molecule of diazonium salt or to the oxidised dye], followed by deprotonation. A range of phenanthrenes was prepared in similar manner as exemplified by 586a–g.
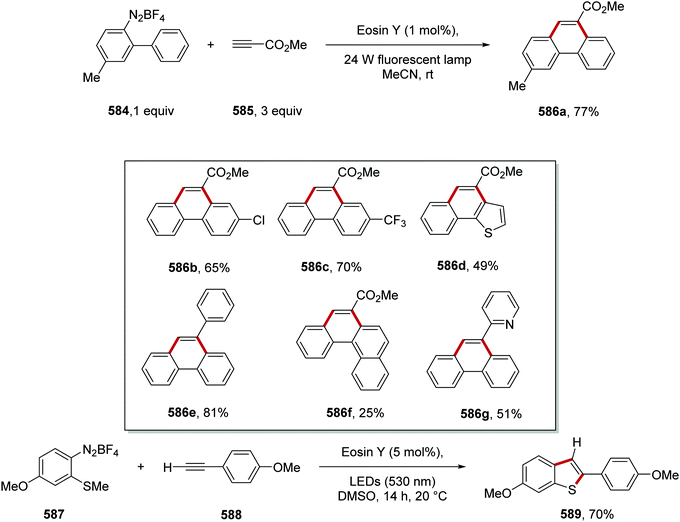 |
| | Scheme 167 Reductive annulation with photoexcited Eosin Y as electron donor. | |
Similarly, König prepared fused heterocycles such as the dibenzothiophene 589. In these cases, an aryl radical, formed from 587 as described above, reacted with an alkyne 588 to form a vinyl radical.204 Cyclisation of the vinyl radical onto sulfur afforded a hypervalent sulfur radical. Oxidation transformed this to a sulfonium salt, which was demethylated to give the benzothiophene product.
Diazonium salts have also been deployed in acylation chemistry (Scheme 168). Following SET to 590 (Ered = −0.22 V vs. Ag/AgCl)205b and loss of N2, the resulting aryl radical reacted with CO to form an aroyl radical (Scheme 169). In turn, this underwent electron transfer to the oxidised form of the redox dye to afford the corresponding acyl cation. This was then trapped by alcohols to form esters 591 by Jacobi von Wangelin205 and, separately, Xiao.206 Likewise, aroyl cations, generated from diazonium salts, have been subjected by Gu et al. to Friedel–Crafts chemistry by arene nucleophiles thereby forming diaryl ketones 596.207
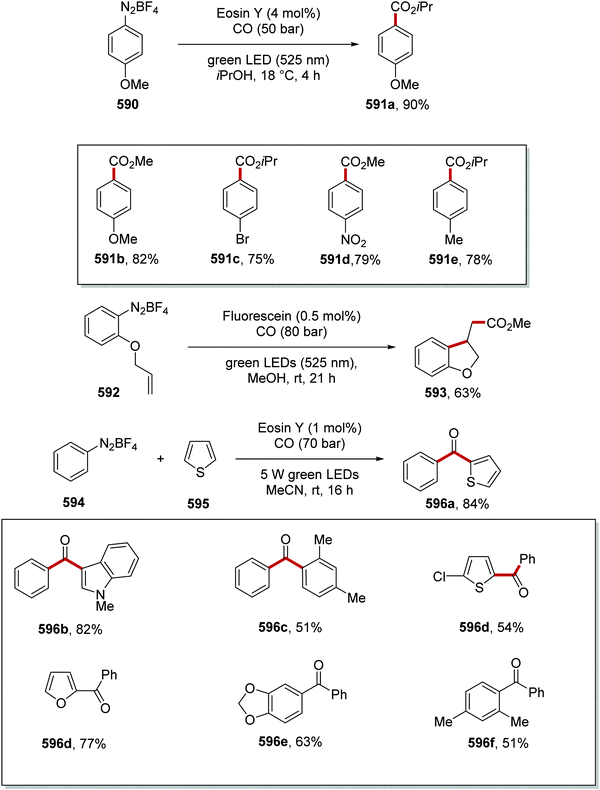 |
| | Scheme 168 Acylation of diazonium salts. | |
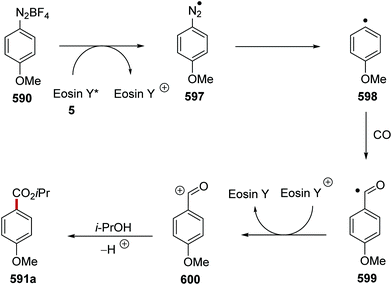 |
| | Scheme 169 Reaction pathway for acylation of arenediazonium salts. | |
Here the diazonium salt quenches (Ered = −0.22 V vs. Ag/AgCl)205b the excited state Eosin Y* by electron transfer giving the oxidised form of Eosin Y. Loss of dinitrogen from 597 leads to an aryl radical 598. This reacts with CO to form the acyl radical 599, and this radical then reduces the oxidised form of Eosin Y while being converted into an acyl cation 600, that is trapped by isopropanol. In principle, the cycle could be more direct if the acyl radical can act as electron donor to a fresh molecule of diazonium salt.
Eosin Y (5) (Eox* = −1.15 V vs. SCE)8a has been used to reduce other types of functional groups also. The excited state can lead to cleavage of labile C–halogen bonds. Thus, the electron-poor ethyl bromofluoroacetate 602 is a good electron acceptor leading to a fluoroacetate radical and a bromide anion (Scheme 170).208 The intermediate indole α-fluoroester 603 loses fluoride anion and the resulting conjugated iminium electrophile is attacked by nucleophilic arene, to yield the observed product. Symmetrical or unsymmetrical products (e.g.605d) can be targeted.
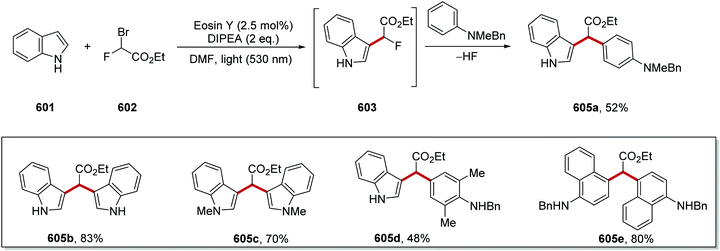 |
| | Scheme 170 Double arylation of ethyl bromofluoroacetate. | |
Electron-poor arenes such as bromopentafluorobenzene 606 (Ered = −1.39 V vs. SCE) can also be reduced by photoactivated Eosin209 (Eox* = −1.58 V vs. SCE) as shown by the team of König. Electron transfer forms the radical anion of the substrate that expels bromide ion, leaving a perfluoroaryl radical, which couples to arene partners such as benzene 607, to afford products 608 (Scheme 171).
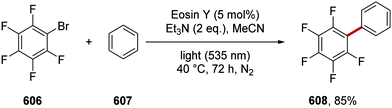 |
| | Scheme 171 Photoactivated Eosin Y reductively activates a reactive bromoarene. | |
Zeitler et al. used210 excited state Eosin Y 5 as an electron donor to enones and related systems that were activated through hydrogen bonding to appropriate thioureas e.g.610 (Scheme 172). Monocyclisation occurred to give products 611 in the presence of a Hantzsch ester 612 as an H-atom donor. Similar cyclisations can also be achieved,211 using alternative redox active reagents e.g. the radical anion of DCA or the chrysene radical anion and Ru redox reagents, in the presence or absence of acids.212 It was proposed that the excited catalyst underwent SET from the Hantzsch ester and it was the reduced catalyst that was responsible for electron-donation to the enone system that was activated by the thiourea. Both electron-rich and electron-poor aromatic enones underwent successful cyclisation with Eosin Y due to their low reduction potential (enone 609Ered = −1.20 V vs. SCE).210b However, it was observed that aliphatic enones could not be reduced by Eosin Y, due to their more negative reduction potential;210c in these cases an iridium catalyst [Ir(dtbbpy)(ppy)2PF6, (E(M/M−) = −1.51 V vs. SCE) was found to be very successful.
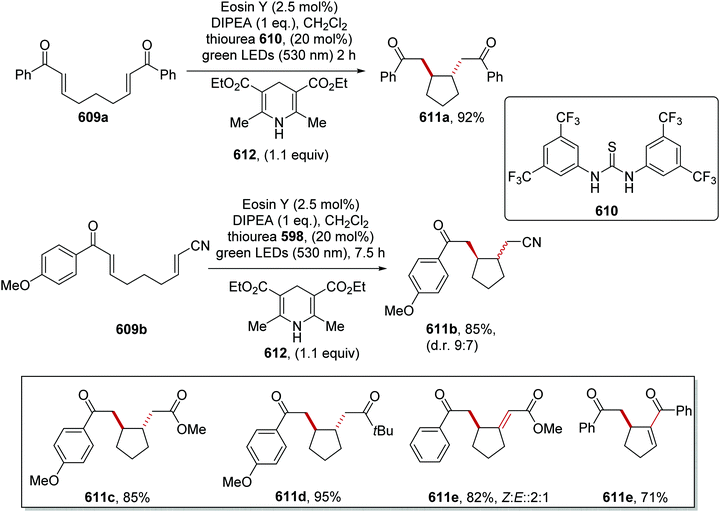 |
| | Scheme 172 Reductive cyclisation of activated bis-enones and related compounds mediated by Eosin Y and Hantzsch esters. | |
Leonori et al. reported photoactivated Eosin Y 5 as an electron donor to dinitroaryl oxime ethers such as 613.213 The resulting radical anions fragmented to dinitrophenolate anions and iminyl radicals; the radicals cyclised and the resulting carbon-centred radicals abstracted H from 1,4-cyclohexadiene to form products 614 and analogues (Scheme 173). When the cyclohexadiene was omitted, then the final radical instead attacked a nitro group, ultimately resulting in formation of alcohols 615. During the study, the reduction potential of the oxime was of critical importance in ensuring a successful reaction as it must match the oxidation potential of the Eosin Y catalyst. The model substrate 613-m (see inset, Scheme 173) was studied by cyclic voltammetry (Ered = −0.55 V vs. SCE) it was found it could be reduced by Eosin Y (Eox* = −1.15 V vs. SCE) and thus this provided an efficient reaction. However, when the aromatic ring only contained a single para-nitro group this gave a more negative reduction potential (Ered = −0.93 V vs. SCE) and this translated into a more challenging substrate for Eosin Y to reduce with SET. When an analogue containing just a single aromatic para-nitro group Eosin Y delivered the corresponding cyclised product in 7% isolated yield, the more strongly reducing Ir(ppy)3 (Eox* = −1.77 V vs. SCE) in 91% yield.
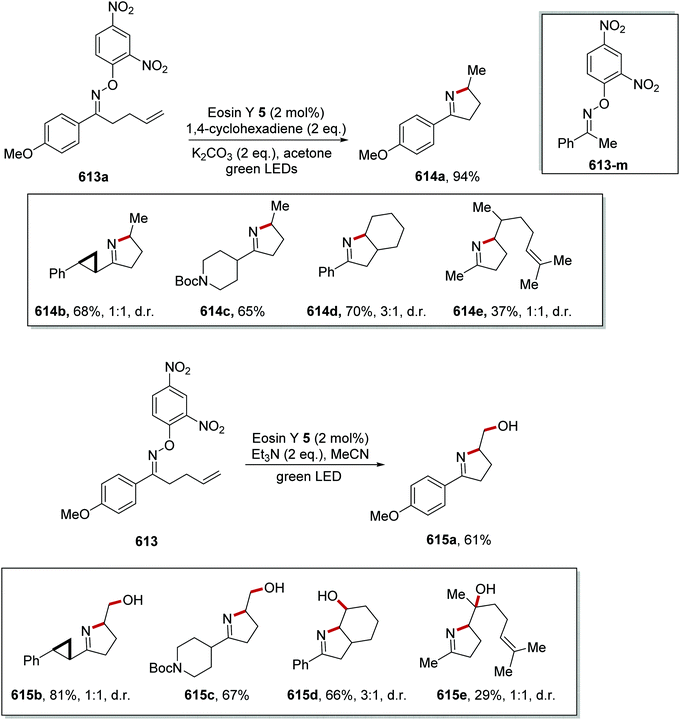 |
| | Scheme 173 Iminyl radicals are formed through reduction of oxime ethers. | |
Although Eosin Y 5 is the most commonly used of these dyes, other members of the family have also found applications. Rhodamine B (579) was found to be the best of a number of related dyes that were tested for the activation of perfluoroalkyl iodides e.g.617 and other electron-deficient alkyl iodides (Scheme 174).214,215 Electron transfer from 579 (Eox* = −1.3 V vs. SCE) to 617 (Ered = −1.00 V vs. SCE)214b leads to the perfluoroalkyl radical. This adds to the less hindered end of an alkene to form an alkyl radical that, in the simplest case, reacts by iodine atom transfer to afford the product, (e.g.618) and also to form another perfluoroalkyl radical, thereby sustaining a chain reaction. Many of the substrates e.g.619, 621 are 1,6-dienes which allow cyclisation of an intermediate carbon radical before iodine atom transfer.
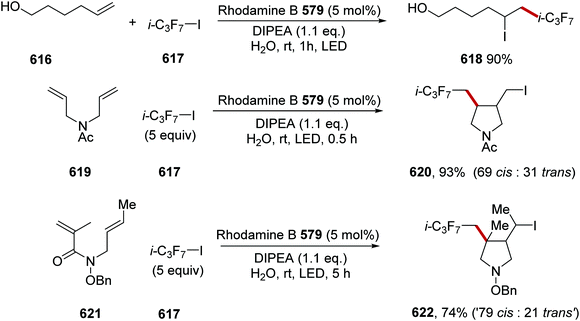 |
| | Scheme 174 Trifluoromethylations are readily achieved through photoactivated dyes acting as electron donors. | |
However, in a separate study, perfluoroalkyl halides 623 were coupled to terminal alkenes 624, which gave fluorinated alkene 625via an atom transfer radical addition elimination (ATRE) process, promoted by Eosin Y photocatalyst (Scheme 175).216 Alkene 625a was formed when the reaction mixture was irradiated with white light (16 W household fluorescent lamp) and this resulted in excited Eosin Y* (5*) from 5. SET from 5* (Eox* = −1.1 V vs. SCE)215 to halide 623a (Ered = −1.10 V vs. SCE)216b gave perfluoroalkyl radical 626a and oxidised catalyst 5˙+. Radical addition of 626a to alkene 624a gave the carbon-centred radical 627a. The exact reaction pathway to reach alkene 625a from 627a was not fully elucidated and three different reaction pathways were feasible. A radical-chain mechanism may have been operative with an abstraction of an iodine atom from 623a with radical 627a and this would have given 628a. The combination of 628a with base would have resulted in an E2 elimination and this would have produced alkene 625a. Alternatively, SET between radical 627a and oxidised catalyst 5˙+ would have resulted in the formation of cation 629a and closure of the Eosin Y catalytic cycle. Proton loss from 629a would have resulted in the formation of the alkene product. However, a charge combination between cation 629a and iodide was also possible this would have delivered alkyl iodide 628a, from which alkene 625a is accessed through an E2 elimination. During the optimisation of this reaction, alkyl iodide 628a was sometimes the sole product; therefore, it may be a key intermediate for this transformation.
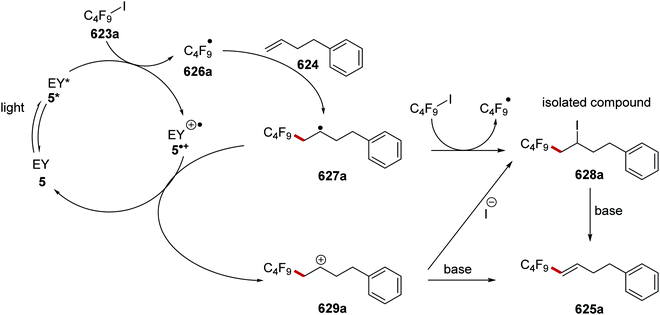 |
| | Scheme 175 Perfluoroalkyl radical addition to alkene 624. | |
From optimisation studies, it was found that Eosin Y performed better than the other organic dyes, Rose Bengal, Rhodamine B and dibenzylamine. Highest yields of 625a were obtained when caesium carbonate was used as the base. DBU, triazabicyclodecene, DIPEA and K2HPO4 were also tested. Product 625a was formed in the highest yields when DMA was used as the solvent. The use of THF or water as solvent gave alkyl iodide 628a as the sole product in 66% and 63% 1H-NMR yield, respectively. With these optimised reaction conditions, a range of 30 perfluoroalkylated alkene products 625 was prepared in 51–95% yields (Scheme 176). Synthetically useful functional groups were tolerated under the reaction conditions and N-alkylphthalimide analogue 625c was isolated in 86% yield, allowing for later conversion to an amine. Difluoroalkyl bromides were accessible under the reaction conditions when difluorodibromomethane was used in the reaction and this gave difluorobromomethyl compounds 625d–e in yields of 74–80%. When ethyl 2,2-difluoro-2-iodoacetate was used in the reaction, this gave ester derivatives 625f–g. The reaction was also amenable to complex substrates, for example, steroid compound 625g was prepared in 95% yield in a 5:1 mixture of stereoisomers.
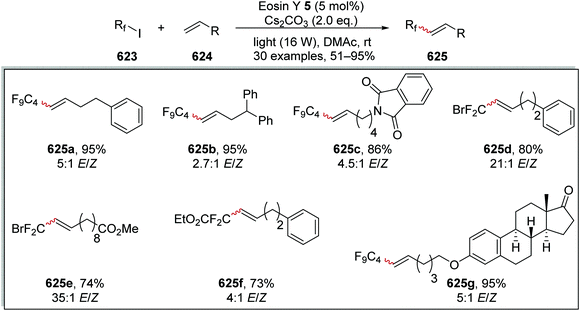 |
| | Scheme 176 Scope of alkene products. | |
Meanwhile, Scaiano et al. used photoactivated methylene blue 580 to activate Togni's reagent 631 towards the release of trifluoromethyl radicals that substituted onto pyrroles, indoles and thiazoles giving trifluoromethylated products (Scheme 177).217 Specific examples include 632a-d. In addition, products derived from addition to alkenes and alkynes were isolated. A probable mechanism would be SET from 3-methylindole (Eox = +1.12 V vs. SCE) to an excited (T1) molecule of methylene blue (Ered* = +1.60 V vs. SCE).8a The resulting reduced catalytic species should be able to provide an electron to reagent 631 (Ered = −1.34 V vs. SCE).
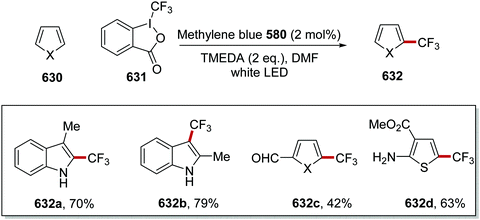 |
| | Scheme 177 Togni's reagent as a source of trifluoromethyl radicals. | |
A clever application is reported by König et al. for Rhodamine 6G, 578, where the effective reduction potential is dependent on the wavelength of radiation.218 2,4,6-Tribromopyrimidine, 633, nicely illustrates the point (Scheme 178). Excitation of rhodamine-6G, 578, with green light afforded the excited state (Eox* = ca. −0.8 V vs. SCE) and this was reduced by electron transfer from DIPEA to afford the radical anion (Eox−˙ = ca. −1.0 V vs. SCE), which is a stronger reducing reagent. This species has sufficient driving force to reduce the tribromopyrimidine 633 to its radical anion, which expels bromide ion to form the aryl radical. This was now used to couple to heteroaromatics; an example shows N-methylpyrrole reacting to yield the dibromoproduct 634. However, if the reaction was performed under blue light a different reaction pathway was feasible. When the reaction mixture was irradiated with blue light it was speculated that the radical anion became excited, and this created the strongly reducing reagent Rh-6G*−˙ (Eox* = −2.4 V vs. SCE), the exact electronic transition was not identified. It was proposed that it was this strongly reducing species that could reduce pyrimidine 634 and thus under blue light pyrimidine 633 was converted through 634 to pyrrole derivative 635. The reaction was also performed stepwise through the two activations, and this allowed for selective substitutions as in the conversion of 633 to 637. Alongside pyrimidine 633, 1,3,5-tribromobenzene (Ered = −1.61 V vs. SCE)218b was used extensively as substrate in this paper.
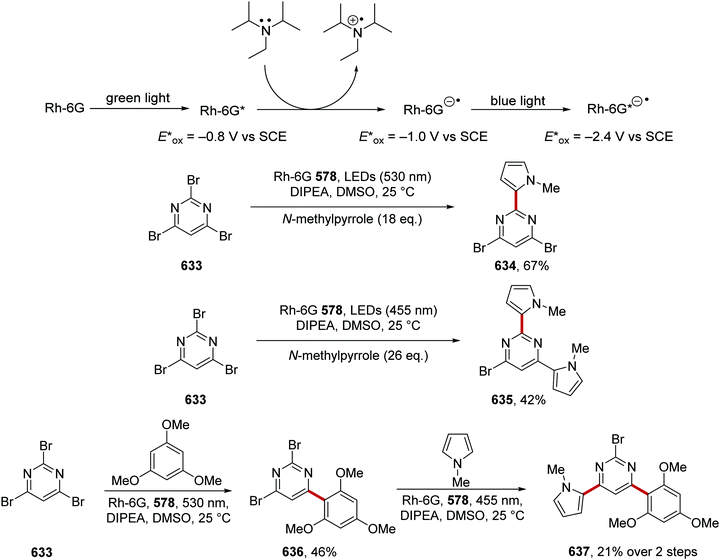 |
| | Scheme 178 Wavelength-dependent control of substitution of polyhaloarenes. | |
Boronates as radical precursors.
The preparation of alkenes 640 from trifluoroborates 638 and sulfones 639 with the photoredox catalyst Eosin Y was recently reported in the literature.219 The reaction was performed in DMF and 49–77% yields were obtained and high levels of stereocontrol were observed for the E-isomer (Scheme 179). Eosin Y (Ered* = +0.83 V) could oxidise trifluoroborates (638, Eox = +0.78 V vs. SCE).
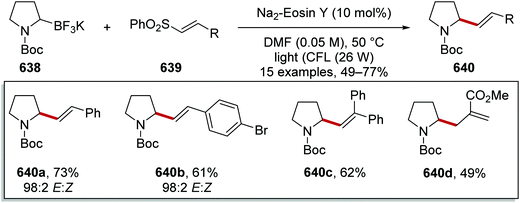 |
| | Scheme 179 Eosin-mediated coupling with α-aminoalkylboronates. | |
α-Heteroatom functionalisation.
The asymmetric alkylation of lactams 641 with ketones 642 was achieved with catalytic amounts of proline 645 and Eosin Y, 5.220 From Stern–Volmer analysis, it was established that lactam 641a was able to reductively quench the excited state of Eosin Y dye 5. Therefore, it was proposed that under the reaction conditions 641a (in terms of published anilines, N,N-dimethylaniline has Eox = +0.74 V vs. SCE) quenched 5* (Ered* = 0.83 V vs. SCE) and this gave amine radical cation 643a (Scheme 180) and the reduced form of the Eosin dye. This, in turn, passed an electron to dioxygen in air to form superoxide radical anion, and superoxide was proposed to convert 643a to 644a oxidised the amine radical cation to imine 644a. [The reaction was inhibited when it was conducted in the absence of oxygen.] Meanwhile, acetone 642a and L-proline 645 reacted to form enamine 646a, which reacted with the imine to give 648a in a Mannich reaction with stereochemistry being set by transition state 647a.
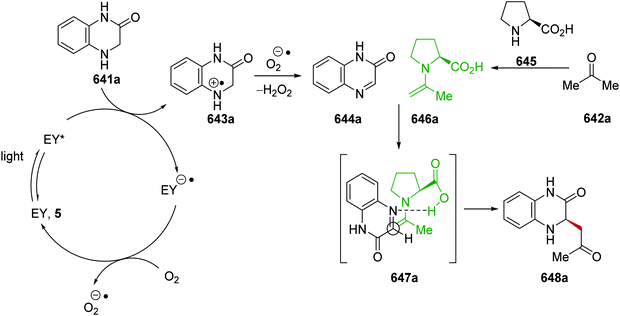 |
| | Scheme 180 Eosin Y and proline-dependent asymmetric Mannich reaction. | |
The transformation was successful when Ir(ppy)3, Ru(bpy)3Cl2, acridinium dye and Eosin Y were used as photoredox catalysts. However, to give a more sustainable and cost-effective transformation, further investigations were carried out with Eosin Y. MeCN, PhMe, CHCl3 and DMF were all screened as solvents for this reaction, and it was found that, when DMF was used as solvent, this gave the greatest yield for 648a.
From these optimised reaction conditions, a library of 22 coupled compounds was prepared. The use of various lactams gave coupled compounds 648a–d in high yields with high enantiomeric excess (Scheme 181). However, the use of assorted ketones resulted in greater variation in the optical purity of the coupled compounds 648f–h. The reaction was regioselective when unsymmetrical ketones were used. For example, when 2-butanone was used as the substrate, this was functionalised at the less sterically encumbered position (methyl vs. methylene) giving coupled compound 648e in 55% yield in a 11:1 ratio of regioisomers. Greater regioselectivity was observed between α-methyl and α-methine groups as demonstrated with the formation of 648f with only one regioisomer being detected. When prochiral ketones were employed as the substrates, no control of diastereoselectivity was observed with a 1:1 mixture of diastereoisomers being given, as seen for example, with coupled compounds 648g and 648h.
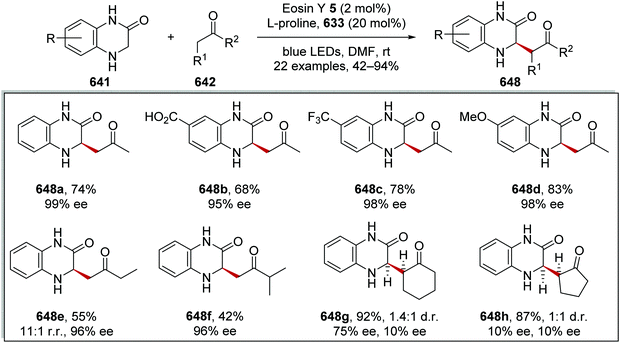 |
| | Scheme 181 Stereoselective coupling of ketones 642. | |
Aldehyde C–H functionalisation.
Additions of acyl radicals, derived from aldehydes 649, to electron-poor alkenes 650 were facilitated with an excited state HAT with Eosin Y photocatalyst.221 Under the reaction conditions, irradiation with blue light led to excited Eosin Y species 5* (Scheme 182). HAT to 5* from aldehyde 649a gave acyl radical 651a and protonated Eosin Y complex 5′. Concurrently, electron-poor alkene 650 added to Rh catalyst 213 and this gave Rh complex A. Acyl radical 651a was intercepted by Rh complex A and this gave Rh complex B, which formed the key C–C bond. A reverse hydrogen atom transfer (RHAT) between complex B and 5′ closed the Eosin Y catalytic cycle with formation of Rh complex C. The catalytic cycle was closed with the dissociation of dicarbonyl product 652 as product and association of another molecule 650.
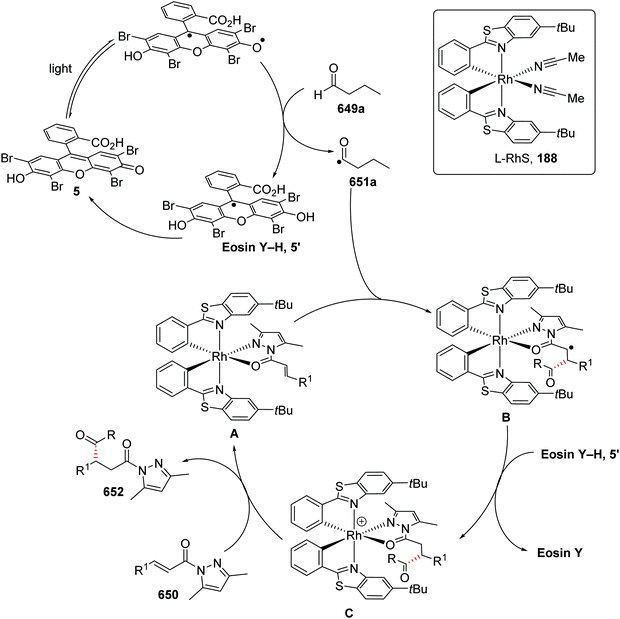 |
| | Scheme 182 Tandem Eosin Y and Rh-dependent Giese reaction. | |
A maximum 1H-NMR yield (52%) of compound 652a was obtained when water (20 eq.) was added to the reaction mixture. When water was absent from the reaction mixture this was diminished to 6% 1H-NMR yield. The reaction did not propagate when Ru(bpy)3Cl2 or Ru(phen)3Cl2 was used instead of Eosin Y, highlighting the HAT activity required to activate aldehyde 649a. A decreased yield of product was obtained when fluorescein was used instead of Eosin Y, with 652a being given in 11% 1H-NMR yield. When the reaction was performed at 30 °C instead of 10 °C, the enantiomeric excess decreased from 94% to 76%. It was also established that TBME was the optimal reaction solvent, with 652a being given in lower yields when acetone or benzene was used instead (Scheme 183). A range of dicarbonyl compounds 652 was produced with these optimised reaction conditions in 23–99% yields. The use of cyclopropanecarboxaldehyde as substrate under these conditions gave cyclopropane analogue 652b in 60% yield and with 81% ee. Aromatic aldehydes were also compatible with the reaction conditions and giving compound 652c in 44% yield and with 99% ee. Sensitive functional groups were tolerated under the reaction conditions and this allowed for acetals 652d and 652e to be prepared in 73% yield (99% ee) and in 99% yield (59% ee), respectively. The ethers, tetrahydrofuran and 2-methyltetrahydrofuran were successful coupling partners for this transformation and products 652f and 652g were isolated in high yields (81% and 78%) and high ee (96% and 70%). The reaction with 2-methyltetrahydrofuran led to the formation of the more sterically hindered compound 652g, via the more stable radical intermediate. The use of dimethylaniline as a coupling partner resulted in amine compound 652h in 48% yield and with 96% ee.
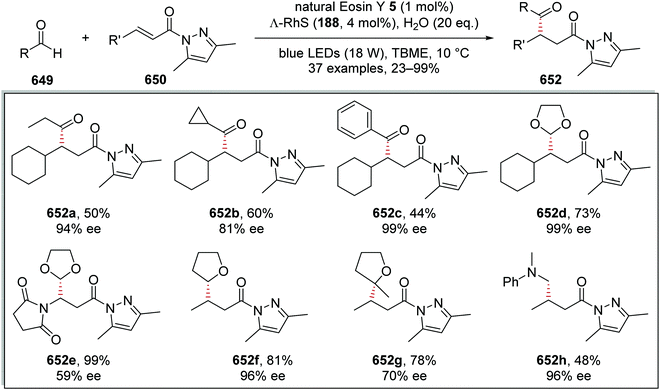 |
| | Scheme 183 Eosin Y and Rh Giese reaction. | |
3.2
N-Phenylphenothiazine and related heterocycles
N-Phenylphenothiazine 653 (PTH) was deployed by the teams of Hawker and Read de Alaniz222 for radical dehalogenations of aryl and alkyl halides (the reactions are not shown here). The work was eminently successful, leading to this and related photoactivated redox agents being adopted by a number of authors. Alemán et al. irradiated PTH at 385 nm (Eox* = −2.1 V vs. SCE) in the presence of iPrEt2N in methanol on aryl halides including 654 (Ered = −2.21 V vs. SCE for the related 4-iodoanisole) (Scheme 184).223 This resulted in formation of aryl radicals that then cyclised onto appropriate functional groups within the substrate to form C–S, C–P or C–Si bonds. Examples of C–S bond formation are shown below, featuring cyclisations onto sulfinate esters. The authors confirmed that the iPrEt2N did not quench the PTH* and hence that the initial SET occurs to the aryl halide.
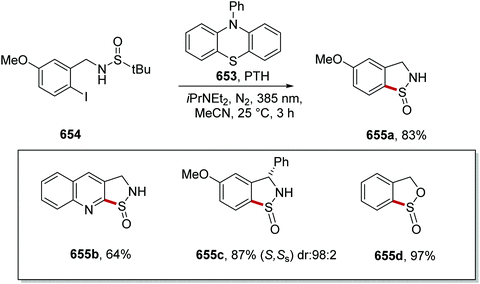 |
| | Scheme 184 Photoexcited N-phenyl phenothiazine as electron donor. | |
The aryl iodide 654 quenches the excited state PTH*. In so doing it forms an aryl radical 656 and iodide anion as well as the radical cation of PTH. The radical then cyclises onto sulfur. A hypervalent intermediate 657 may be present before loss of the tert-butyl radical (Scheme 185).
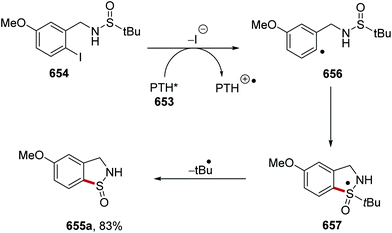 |
| | Scheme 185 | |
Jui et al. have exploited blue LEDs to activate PTH (653, Eox* = −2.10 V vs. SCE) for electron transfer to aryl iodides 658 (as an estimate, iodobenzene Ered = −2.30 V vs. SCE),224b allowing the aryl radical to add to enamides 659.224 The photoactivated PTH initiates the cycle. The nucleophilic radical resulting from the addition abstracts an H atom from a thiol, and the formed thiyl radical then abstracts H from formate ion to give the radical anion of carbon dioxide which propagates the chain by electron transfer (Scheme 186).
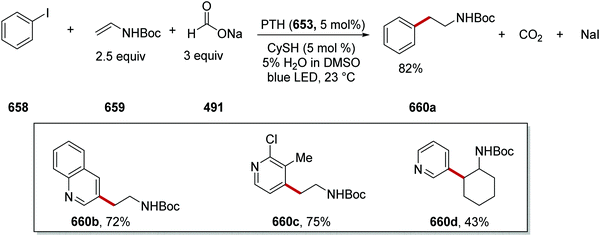 |
| | Scheme 186 Additions of aryl radical to enamides initiated by electron transfer from photoactivated PTH. | |
Procter et al. used excited state PTH as an electron donor to activate dibenzothiophenylium salt derivatives of arenes, e.g.663, prepared in situ by interrupted Pummerer reactions of arene 661 (Scheme 187).225 This afforded aryl radicals that were then used to couple to arenes and heteroarenes. SET between PTH* (Eox* = −2.1 V vs. SCE) and 663 (Ered = −1.10 V vs. SCE) was thermodynamically feasible. Excitation was performed with a blue Kessil lamp. The method was applied to the formation of simpler biaryls e.g.667a–d, but also to products arising from late-stage functionalisation of pharmaceutically relevant molecules 667e–g.
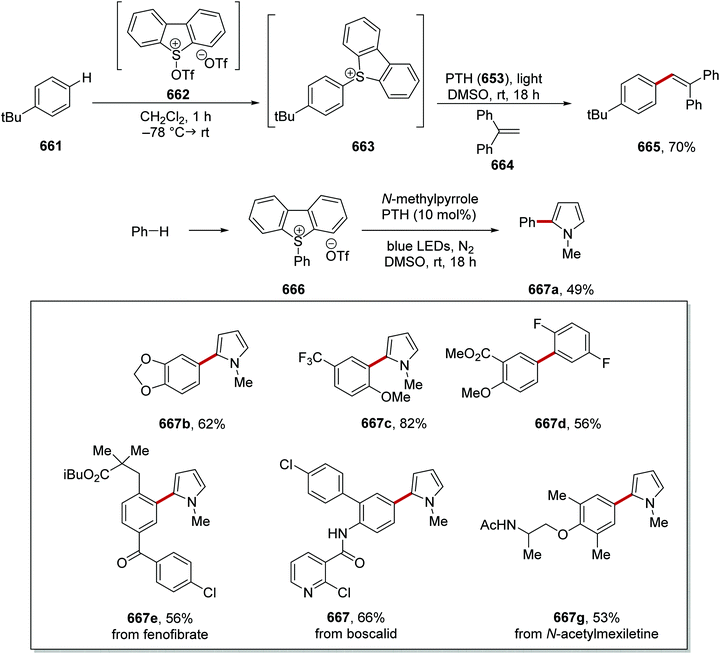 |
| | Scheme 187 Interrupted Pummerer reactions provide substrates for reductive cleavage by PTH*. | |
Aleman et al.226 developed wavelength-dependent transformations access to (E)- or (Z)-products in allylic substitution (Scheme 188). The allylic acetate 668 (Ered = −2.35 V vs. SCE) was substituted by indoles in their 3-position or by pyrroles in their 2-position when appropriate photocatalysts were used. The (Z)-isomers were the predominant products when PTH (653, Eox* = −2.1 V vs. SCE) was irradiated at 365 nm, while the (E)-isomers were selectively formed when heterocycle 671 was irradiated at 420 nm. A study of the mechanism of the substitution reaction showed electron transfer to the allylic acetate, with loss of acetate ion as a leaving group. The resulting allylic radical then transferred an electron back to the oxidised form of the electron donor to give an allylic cation, and this was intercepted by nucleophiles. Electron-rich arenes including pyrroles, indoles and anilines, as well as aliphatic amines and alcohols acted as suitable nucleophiles. Stern–Volmer studies again showed that DIPEA or pyrrole 669 did not quench the excited state of 653. However, acetate 668 was a strong quencher of 653* indicating that 668 was the acceptor in the initial electron transfer. To rationalise the stereoselectivity, studies indicated that the starting acetate 668 was selectively photoisomerised to its Z-isomer through irradiation at 365 nm, but not at 420 nm. On the other hand, catalyst 671 facilitated the preparation of E alkenes as it could be excited at 420 nm due to its extended conjugation.
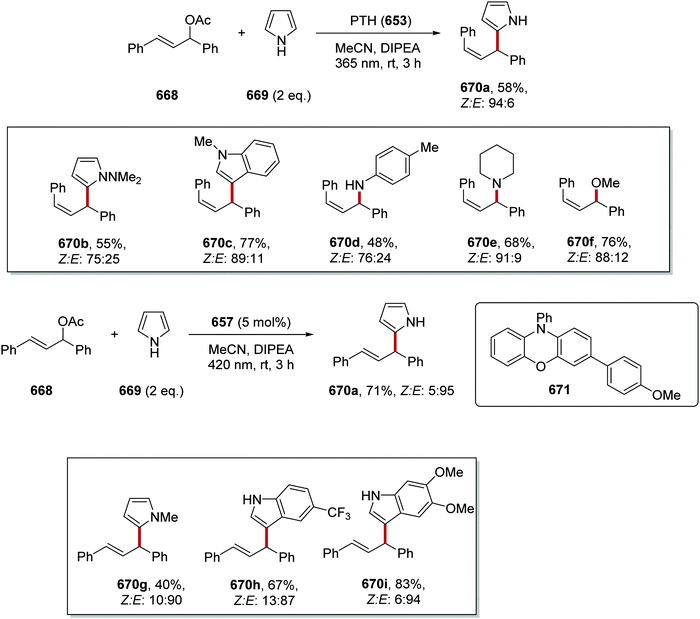 |
| | Scheme 188 Allylic acetate 668 undergoes photoredox allylic arylation, where the stereochemistry of the product is dependent on the wavelength used. | |
Xu et al. used a family of photoexcited heterocycles including 675 (PMP = p-methoxyphenyl) as electron donors to afford aroyl radicals from aroyl chlorides as shown for benzoyl chloride, 673 (Ered = −1.56 V vs. SCE). It was calculated from ground-state redox potentials and excited-state energy levels that 675 was capable of reducing 672 both in its S1 state (Eox* = −1.91 V vs. SCE) and its T1 state (Eox* = −1.65 V vs. SCE). Catalyst 675 was also used to accomplish conversion of activated aryl iodides, bromides and chlorides to aryl radicals for a range of reactions (Scheme 189).227
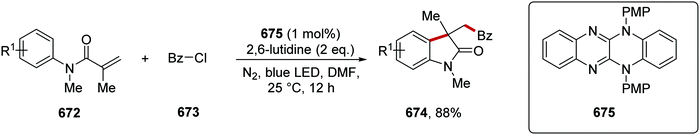 |
| | Scheme 189 Aroyl radicals are produced by aroyl chlorides. | |
Wagenknecht et al. harnessed forceful photoredox agents for the Markovnikov photoaddition of alcohols to styrenes.228 Here, the authors propose that electron transfer from photoexcited donor 678* (Eox* = −2.5 to −2.6 V vs. SCE) to the styrene 676 (Ered = −2.5 to −2.7 V vs. SCE) produced the corresponding radical anion 679 (Scheme 190). Protonation then left a benzylic radical 680 that was oxidised to the benzylic cation before product 677 formed as a result of interception by the alcohol.
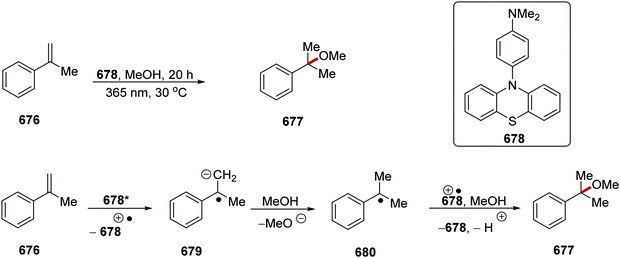 |
| | Scheme 190 Styrene activation through its radical anion and a benzylic cation leads to Markovnikov addition of alcohols. | |
Ohmiya et al. took activated N-oxyphthalimide esters of tertiary alkylcarboxylic acids 681 (Scheme 191).229 Electron transfer from PTH analogue 684 (Eox* = −1.97 V vs. SCE, for the S1 state) gave the phthalimide anion and the carboxyl radical, which rapidly lost carbon dioxide to give a tertiary alkyl radical. This radical was then converted to the corresponding tertiary allyl cation by back electron transfer to the oxidised form of the photocatalyst, before the ether product was formed as a result of attack by alcohol nucleophiles such as 682. When 684 was used as catalyst an isolated yield of 81% was recorded for 683 whereas PTH (Eox* = −2.1 V vs. SCE) gave 26% yield.
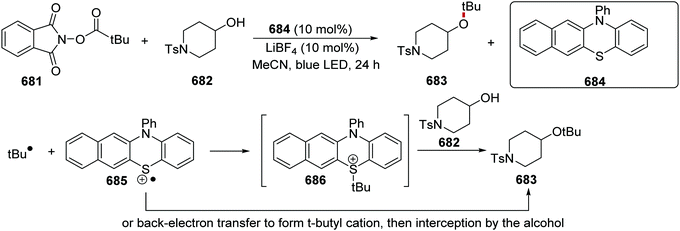 |
| | Scheme 191 Reduction of N-hydroxyphthalimide esters leading to ether formations. | |
3.3 Flavin photocatalysts
Carboxylic acids as radical precursors.
The site-selective alkylation of peptides and proteins was studied with a photoredox decarboxylative strategy with a flavin photocatalyst.95 Initial investigations probed Giese coupling between tetrapeptide 687 (Eox = +1.3 V vs. SCE) and diethyl ethylidene malonate with a range of photocatalysts (Scheme 192). From the initial screen of catalysts, it was found that the photocatalysts Ir[dF(CF3)ppy]2(dtbbpy)PF6 (Ered* = +1.21 V vs. SCE) and Ru(bpz)3Cl2 (Ered* = +1.45 V vs. SCE)95b resulted in no formation of coupled product 688. Due to their high aqueous solubility, a range of flavin photocatalysts was investigated for this reaction and riboflavin analogues 689–691 gave low yields for Giese reaction product 688. However, the use of riboflavin tetrabutyrate 688 (Ered* = +1.5 V vs. SCE) afforded peptide 688a in 75% yield.
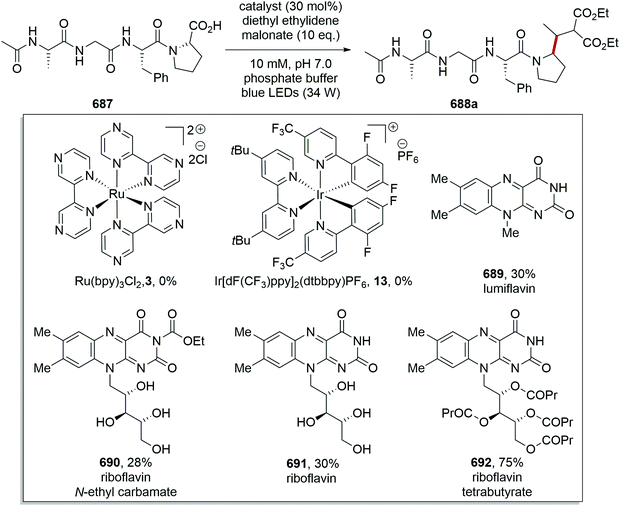 |
| | Scheme 192 | |
Optimised reaction conditions were applied to a range of tetramer peptides with the Giese-coupled peptides 688 being given as products. For the majority of peptides tested with these conditions, the reactions worked very well (Table 8, entries 1–11). However, in some cases, decreased yields were obtained (entries 12–14); this was due to deleterious oxidations of the sidechain (R) of these residues. The oxidation of Tyr, His and Lys was overcome with the reaction being performed at a lower pH (pH = 3.5) and this significantly increased the yields. The preparation of the tyrosine-derived peptide (entry 12) worked best when flavin 689 was used as photocatalyst and this gave the coupled peptide in 23% yield.
Table 8 Results of decarboxylative alkylation with 692
|
|
| Entry |
R = |
Yield at pH 7.0, 8 h (%) |
Yield at pH 3.5, 8 h (%) |
|
Indicates that flavin 689 was used as photocatalyst.
|
| 1 |
Ala |
79 |
92 |
| 2 |
Arg |
71 |
87 |
| 3 |
Asn |
76 |
91 |
| 4 |
Asp |
77 |
93 |
| 5 |
Gln |
71 |
94 |
| 6 |
Glu |
75 |
91 |
| 7 |
Ile |
77 |
90 |
| 8 |
Leu |
74 |
95 |
| 9 |
Ser |
75 |
87 |
| 10 |
Thr |
73 |
90 |
| 11 |
Val |
76 |
95 |
| 12 |
Tyra |
8 |
23 |
| 13 |
His |
11 |
70 |
| 14 |
Lys |
52 |
65 |
This methodology was demonstrated upon human insulin, and this was alkylated with electron-poor enone 693 on a 500 nmol scale (Fig. 17). This reaction gave the ligated protein in 41% conversion as a single monoalkylated product (functionalising the C-terminal amino acid on the A chain of insulin) after 8 h with irradiation by blue light (34 W), based upon reverse-phase HPLC.
 |
| | Fig. 17 Electron-poor enone. | |
3.4 Cyanobenzene-derived photocatalysts
The tetracarbazolylisophthalonitrile 4CzIPN 15 is an organoredox reagent that has principally been used for oxidative interactions with substrates. However, in a one-pot procedure, Sherwood et al.230 converted aliphatic carboxylic acids into their N-hydroxyphthalimide esters, and then, using 4CzIPN as reducing agent, transformed these into alkyl radicals which performed Minisci reactions on protonated derivatives of pyridines, quinolines e.g.694 and related heterocycles (Scheme 193). N-Hydroxyphthalimide esters (Ered = −1.26 to −1.39 V vs. SCE) have reduction potentials that can be targeted by photocatalysts (Eox* = −1.73 V vs. SCE) for Ir(ppy)3. It was speculated that the use of 4CzIPN that the reduced species (4CzIPN−, Eox = −1.21 V vs. SCE) might be the active species as it is a stronger reductant that the excited species (4CzIPN*, Eox* = −1.04 V vs. SCE).
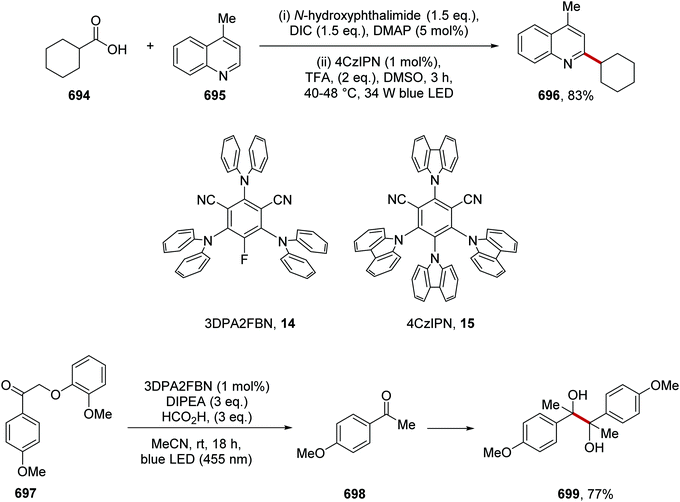 |
| | Scheme 193 Excited state 3DPA2FBN as a potent electron donor. | |
4CzIPN is an attractive redox reagent and is becoming more widely used; as new analogues become available, the potentials can be modulated. This was the thrust of the report by Zeitler et al.33a who made a wide investigation of the redox activity of derivatives of 4CzIPN, 15, notably the analogue 3DPA2FBN, 14.33b This compound in its photoexcited state performed a series of challenging reactions. With ketone 697, reduction led to expulsion of o-methoxyphenolate anion and formation of p-methoxyacetophenone 698. This compound was then reduced further to its ketyl, resulting in coupling to form pinacol 699. Consideration of the redox values for this reaction illustrates why the catalyst 3DPA2FBN is successful in this reaction. 2-Phenoxy-1-phenylethan-1-one, which can be used as a model compound for ketone 697 has a very reduction potential (Ered = −1.72 V vs. SCE).33c While the excited-state species of 14 (Eox* = −1.60 V vs. SCE) cannot reduce 697, the radical anion of 14 (Eox˙− = −1.92 V vs. SCE) can, and this can be formed with reductive quenching with DIPEA (Eox = +0.64 V vs. SCE).33d The reduction of acetophenone 698 (Ered = −2.17 V vs. SCE)33e is more challenging, it was suggested that a LUMO-lowering activation of the carbonyl group with the radical cation of DIPEA could be promoting SET, by analogy with a similar coupling observed by Rueping et al. using iridium photoredox catalysts.33f
Carboxylic acids as radical precursors.
The formation of diester 701via the coupling of proline 700 (Boc-Pro-OCs, E = +0.95 V vs. SCE)134 with diethyl maleate 370b was investigated with a range of different organic photoredox catalysts (Scheme 194).77 When the photocatalyst 4CzIPN (15) was used for this transformation, it gave 701 in 80% yield. A comparable yield was obtained when 3CzClIPN (702) was used as photocatalyst with 701 being given in 77% yield. Diminished yields of 701 were obtained when less oxidising photocatalysts were used. For example, the pentacarbazole catalyst 703 gave 701 in 37% yield. Benzonitrile catalyst 703 had a lower excited reduction potential (Ered* = +1.31 V vs. SCE) than 15 (Ered* = +1.35 V vs. SCE) and 702 (Ered* = +1.56 V vs. SCE) and this correlated with the decreased efficiency of this transformation. This trend was continued with the dyes 14 (Ered* = +0.92 V vs. SCE) and 704 (Ered* = +1.24 V vs. SCE), and these catalysts gave diester 701 in 20% and 12% yield, respectively. The acridinium dye 705 was also a good catalyst for this process and afforded diester 701 in 73% yield.
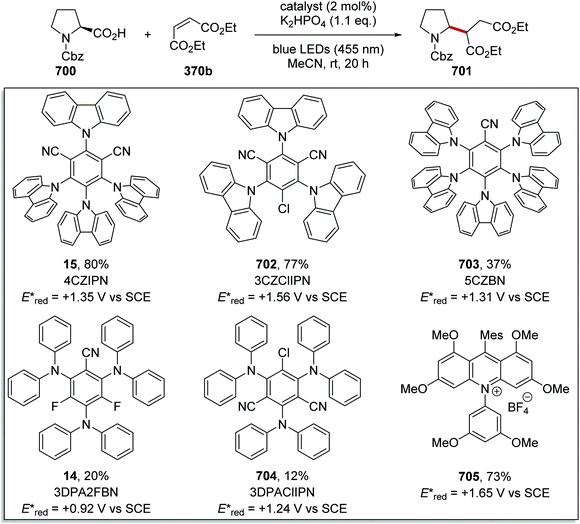 |
| | Scheme 194 Organic-photocatalyst mediated Giese-coupling. | |
The bioconjugation of peptide chains was achieved under metal-free conditions.231 The C-terminus of the peptide chain underwent radical decarboxylation and this gave the corresponding carbon-centred radical. The use of ethynylbenziodoxolone (EBX, 707) in the reaction mixture resulted in the formation of alkyne product 708 (Scheme 195). One challenge for this transformation was the site-selectivity when aspartic and glutamic acid residues were present within the peptide substrate. However, this was overcome by taking into account the different oxidation potentials of the carboxylic acids.95 To avoid the use of expensive transition metals this study focused on the use of organic dyes. Initially, the reaction of dipeptide Cbz-Gly-Pro-OH (706, Eox = +0.95 V vs. SCE) for proline carboxylate with Ph-EBX (707, 1.5 eq.) was examined. Commercially available iridium photocatalyst 13 gave alkyne derivative 708 in 99% isolated yield. A range of different organic dyes with various oxidation potentials (from +0.90 V to +1.58 V vs. SCE) was then tested. In general, the more oxidising organic dyes gave superior yields of the target compound. For example, when 4CzIPN (15) was used as catalyst, this gave 708 in 99% yield. From this finding, 4CzIPN was then used in the alkylation of di- and tetrapeptides. For the majority of amino acid combinations, smooth conversion to the alkylated product was achieved. Only substrates containing tryptophan and tyrosine residues were found to be problematic, due to their low oxidation potentials (tyrosine Eox = +1.1 to 1.27 V vs. SCE and tryptophan Eox = +0.77 to 1.66 V vs. SCE).
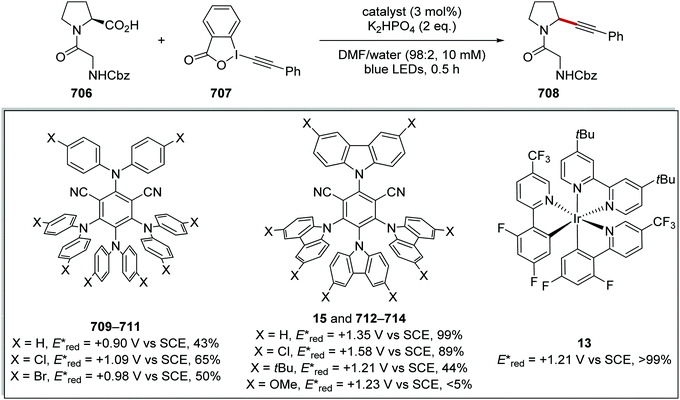 |
| | Scheme 195 Yields of 708 using a range of organic photoredox catalysts. | |
As stated, the selective oxidation of C-terminus carboxylic acids was achieved due to the differences in oxidation potentials. Therefore, the use of the right catalyst gave selective alkylation of the terminal acid over any internal carboxylate moieties. This was shown with 4CzIPN (15), as this resulted in the site-selective alkylated peptides 716a–h (Scheme 196). This transformation was achieved on biologically relevant hexapeptides such as GRGDNP, a potent inhibitor of cell attachment to fibronectin. Azide 716e and aldehyde 716f derivatives were prepared in near-quantitative yield from protected a protected analogue of GRGDNP. The use of unprotected hexamer peptide as substrate allowed for preparation of azide 716g in 57% yield and aryl bromide 716h in 52% yield.
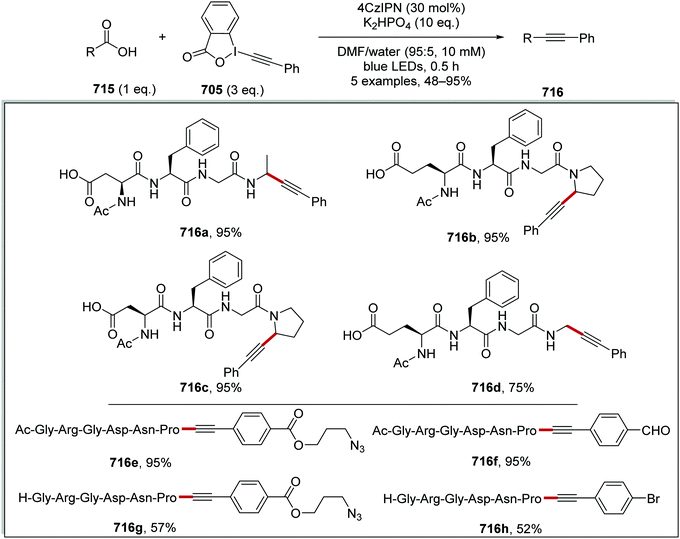 |
| | Scheme 196 Preparation of alkyne peptides 716. | |
An organic photoredox-mediated radical decarboxylation followed by anion formation and addition to aldehyde was reported.232 4CzIPN (15) allowed for coupling between arylacetic acid 717 and carbonyl compounds 722 and this led to the formation of alcohol 723 under blue light (Scheme 197). Mechanistic studies were carried out on this transformation to elucidate the reaction mechanism. From UV-vis spectroscopy, it was detected that photodecomposition of the photocatalyst was occurring under the reaction conditions. Isolation and X-ray crystallography determined the structure of this photodegraded product was 4CzBnBN (726). It was found that benzonitrile derivative 726 was formed from 4CzIPN (15) with phenylacetic acid 717a under basic conditions with irradiation with blue light in 64% isolated yield. Benzonitrile 726 was less reactive to substitution than 15 as further benzyl substitution could only be detected in trace quantities. It was demonstrated that 726 was capable of catalysing the reaction between 717a and 722a. Therefore, it was concluded that 726 was the active catalyst in this process. Therefore, a reaction mechanism for this process was proposed with 725 as the active catalytic species. Photocatalyst 726 was excited with blue light and this gave excited compound 725*. SET between carboxylate 718a (Eox = +1.27 V vs. SCE for the tetrabutylammonium salt of phenylacetate) with 725* (Ered* = +1.21 V vs. SCE) resulted in the formation of carboxyl radical 719a and reduced catalyst 724˙−. Decarboxylation of 719a resulted in benzylic radical 720a, which underwent a SET reduction by 724˙− and this gave benzyl anion 721a. Cyclic voltammetry indicated that 724˙− (Eox = −1.72 V vs. SCE) could only reaction with radical 720a (Ered = −1.43 V vs. SCE) and not with the alkyl aldehyde (Ered = −2.24 V vs. SCE for 3-methylbutanal). C–C bond formation is achieved with the reaction between the benzyl anion and aldehyde 722a and this gave alcohol 723a.
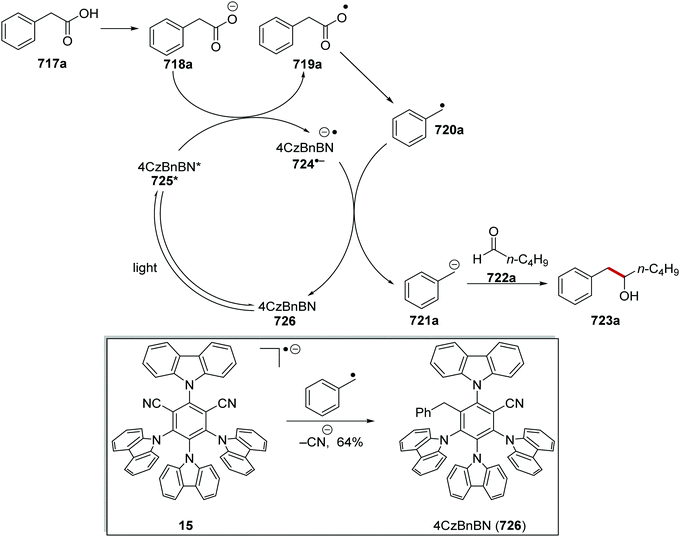 |
| | Scheme 197 Conversion of acid 717a to alcohol 723a with organic photocatalyst 726. | |
The reaction operated best when degassed DMA was used as solvent (Scheme 198). The yield of 723a did not vary between anhydrous DMA and undried DMA although the addition of water (3 eq.) did result in a lower yield of 723a. With the optimised reaction conditions, 723a was prepared in 63% yield. Aryl fluoride 723b and aryl meta-methoxy compound 723c analogues were prepared in 62% and 63% yield, respectively. When 4-methoxyphenylacetic acid was used as substrate, a poor yield of 723d was obtained, whilst 75% of starting material was returned after 16 h of reaction time, indicating poor conversion. Both heterocyclic 723e and N-Boc protected compounds 723f were prepared using this methodology. Acetone was also used as a coupling partner, although only moderate yields for 723h (32%) were obtained when the reaction was performed in a solvent mixture of acetone/DMA (1:1).
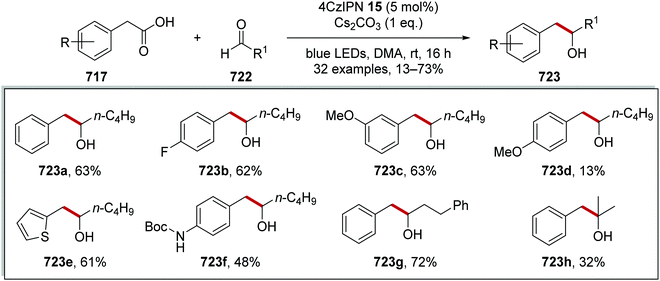 |
| | Scheme 198 Formation of alcohols 709. | |
An HLF-inspired reaction mediated by the organic photocatalyst 3CzClIPN 70 was reported and this allowed for the site-selective remote heteroarylation of amides, such as 727a (Scheme 199).233 It was proposed in the paper and supported by DFT calculations that the following mechanism took place. Excited catalyst 70* gave an electron to substrate 727a with simultaneous N–O heterolytic bond cleavage. This resulted in formation of amidyl radical 728a and oxidised catalyst 70˙+. An intra-molecular 1,5-HAT in 728a resulted in carbon-centred radical 729a. Under the basic reaction conditions (K2CO3), alkyl radical 729a attacked heterocycle 730a which afforded radical 731a. SET from 731a to oxidised catalyst 70˙+ closed the catalytic cycle and, after proton transfer, afforded the observed product 732a.
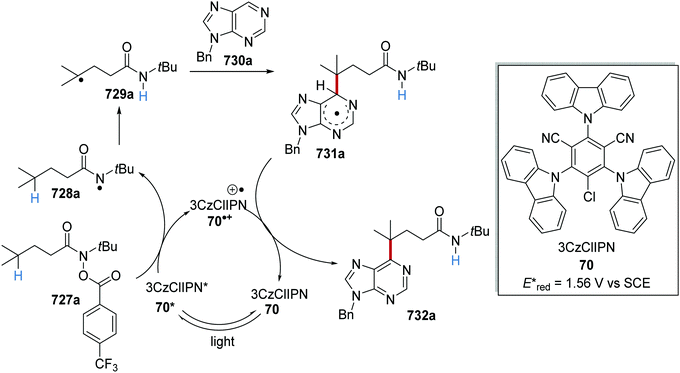 |
| | Scheme 199 Formation of alkyl radical 715a and its coupling to heteroarene 718a. | |
Under the optimised conditions of 3CzClIPN (2 mol%), K2CO3 (1.0 eq.), DMSO (0.2 M) and 90 W blue LEDs, compound 732a was produced in 89% isolated yield (Scheme 200). Other photocatalysts gave inferior yields, for example, Ir(ppy)3 gave 46% yield for 732a, Ru(bpy)3Cl2 gave a 27%, Eosin Y gave 54% yield and 4CzIPN gave 67% yield. [A control experiment was carried out where no photocatalyst was added and this gave 732a in 65% yield after 46 h. It was thought that an electron-donor–acceptor complex formed between 727a and 730a, and this facilitated the reaction.] With these optimal conditions using 3CzClIPN, the scope of the reaction was investigated. A range of different heterocycles was used in the reaction mixture, and this gave a range of products 732b–c. The use of an adamantane-derived amide allowed gave benzothiazole 732d in 67% yield. Ether functionalised compounds were suitable substrates for this transformation with 732e and 732f being isolated in 60% and 87% yield, respectively. This methodology was applied to late-stage functionalisation of bio-active compounds with the formation of 732g and 732h.
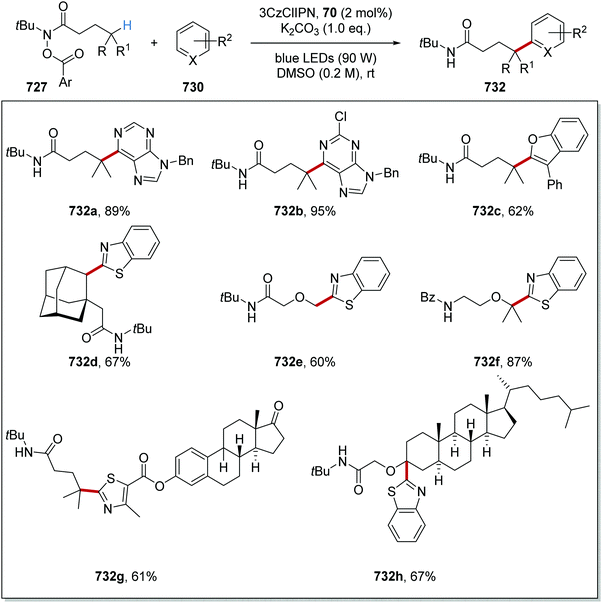 |
| | Scheme 200 Examples of coupling reactions arising from radical precursor 727. | |
Boronates and silicates as radical precursors.
Here, we only mention cases where the oxidation is achieved by an organic oxidant, but readers can refer to references for reactions with transition metal-based oxidants and coupling agents.234 The silicon-based reagents arise from studies by the team of Fensterbank, Ollivier et al.235 who oxidised alkyl silylcathecholates 733 (Eox = +0.3 to +0.9 V vs. SCE) in DMF with a range of organic oxidants, the most successful of which was photoexcited 4CzIPN, (15, Ered* = +1.35 V vs. SCE) (Scheme 201). The liberated alkyl radicals 734 were trapped by a number of species, including allylic sulfones, e.g.736, forming product 737. In this case, addition of the alkyl radical led to an intermediate, which expelled a sulfonyl radical 738. This was reduced by the reduced radical anion form of 4CzIPN 15 to a sulfinate anion 739, thereby regenerating 4CzIPN and rendering the process catalytic. Examples of products are 737a–c. Besides allylic sulfones, a range of other acceptors, including 740–742, proved successful.
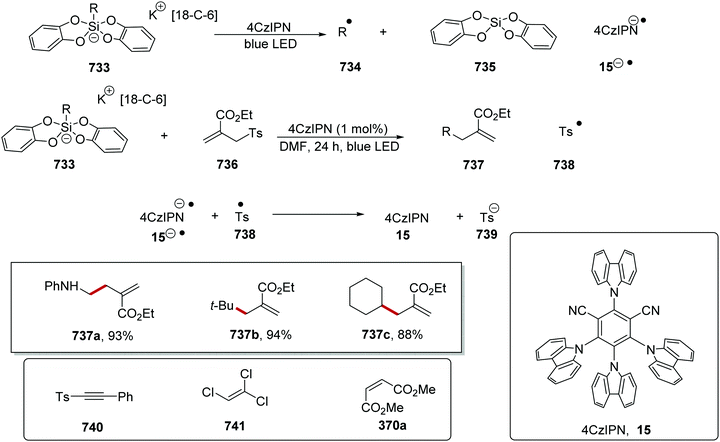 |
| | Scheme 201 Silanates act as electron donors to 4CzIPN 15. | |
Molander et al., using ammonium silane catecholates 743 and the photoactivated 4CzIPN as oxidant,236 showed that the liberated radicals add to imines 742 to afford benzylic amine products 744 (Scheme 202). On the other hand, Mancheño et al. used benzyltrialkylsilanes as sources of benzyl radicals, but a strong oxidant, a mesitylacridinium salt was needed to liberate the radical in this case and the chemistry was limited to benzyl radicals,237 while Bode et al. liberated alkoxyakyl radicals from trimethylsilyloxy groups using triphenylpyrylium salts as photoredox reagents for cyclisation reactions in flow-based chemistry (not shown here).238
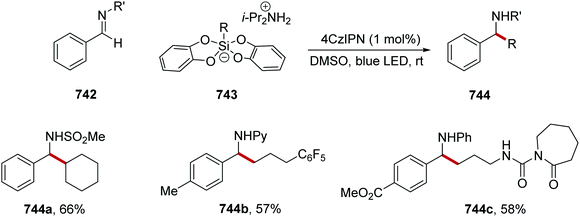 |
| | Scheme 202 Radicals are liberated from silanates and add to imines. | |
Liberated alkyl radicals were also used239 as precursors to acyl radicals 745 for addition to Michael acceptors yielding products 746. Specific examples are shown as 746a–d (Scheme 203).
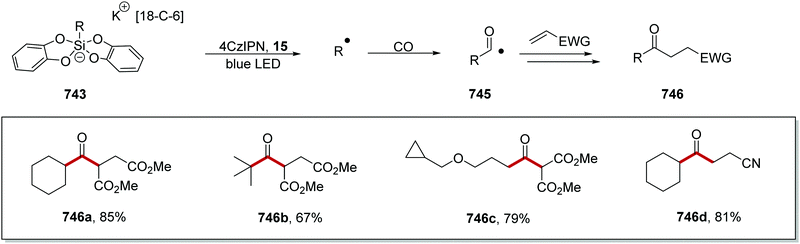 |
| | Scheme 203 Giese reactions, by acyl radicals, generated from carbonylation of radicals liberated from silanates. | |
4CzIPN (15, Ered* = +1.35 V vs. SCE) allowed for α-amino functionalisation of 2,1-borazaronaphthyltrifluoroborates 747 (Eox = +1.07 V vs. SCE for when R = H).240 The inclusion of a nickel catalyst resulted in a cross-coupling reaction between the intermediate α-aminoalkyl radical and aryl bromides 748 and this gave coupled azaborine compounds 749 as products (Scheme 204). Initially, the reaction was optimised with the preparation of nitrile derivative 749a. The reaction was first trialled with Ir[dF(CF3)ppy]2(bpy)PF6 photocatalyst, caesium carbonate as base and this gave a 3.21 product:internal standard (P:IS) ratio. Minimal formation of 749a (0.18 P/IS) was observed when Ru(bpy)3PF6 was used as photocatalyst. However, it was found that using 2,6-lutidine as base increased formation of 749a with a 5.46 P/IS ratio being achieved. The more affordable 4CzIPN photocatalyst gave 749a in a 4.98 P/IS ratio. Although 4CzIPN gave a lower yield for 749a than the iridium catalyst, it was taken forward due to its lower cost. The preparation of 749a with 4CzIPN was scaled up from a 0.100 mmol scale to a 0.500 mmol scale and this gave the azaborine compound in 80% yield. These optimised conditions were tried for a range of different coupling partners and this gave 1,3-benzodioxole 749b, 2-fluoropyridyl 735c, anethole 749d and ketyl 749e derivatives all in 50–88% yields. Analogously functionalised 2,1-borazaronaphthyl-trifluoroborates were used in this transformation as substrates and this gave benzothiophene 749f, trifluoromethyl 749g, benzofuran 749h and intramolecular coupled product 749i.
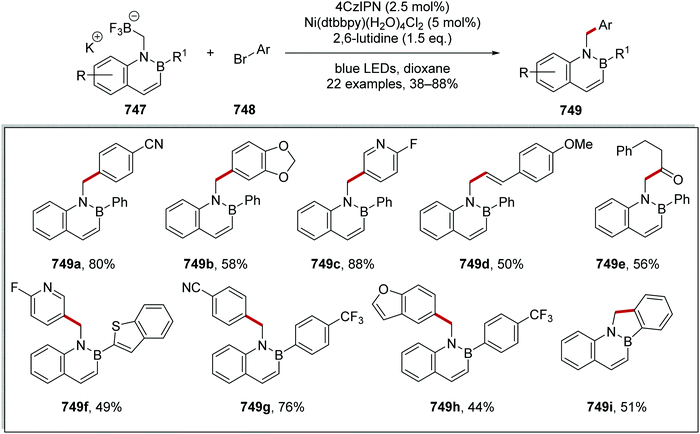 |
| | Scheme 204 Formation of azaborines 749. | |
The synthetic utility of these 2,1-borazaronaphthyltrifluoroborates was investigated further with compound 750a (Scheme 205). From this, it was found that a Minisci reaction with quinoline 751a afforded derivative 753 in 48% yield. A radical-polar crossover reaction of 750a in a defluorinative alkylation reaction gave gem-difluoroalkene 754, a carbonyl bioisostere in 62%. Radical addition to the electron-poor alkene present in acrylonitrile gave nitrile 755 in 47% yield. The use of phenyl styryl sulfone gave alkene 756 in 48% yield and arenesulfonyl cyanide afforded 757 in 69% yield.
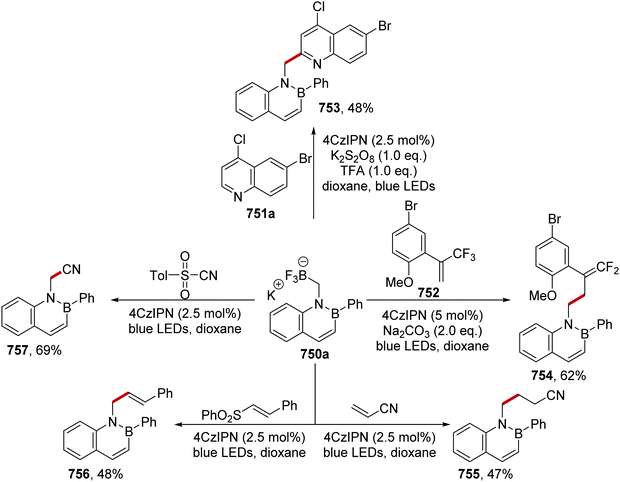 |
| | Scheme 205 Transformations of 2,1-borazaronaphthyltrifluoroborates 750a. | |
α-Heteroatom functionalisation.
The functionalisation of α-secondary amines 758 to α-tertiary amines 759 with electron-poor alkenes 761via a Giese reaction was achieved with the 4CzIPN photocatalyst and tetra-n-butylamine azide.241 (Schemes 206 and 207). Photoexcited dye 15* (Ered* = +1.35 V vs. SCE) received an electron from azide anion (Eox = +0.87 V vs. SCE for the tetrabutylammonium salt), giving reduced catalyst 15− and radical 760. Hydrogen atom abstraction from amine 758a by 760 gave α-aminoalkyl radical 762a and hydrazoic acid (25). The α-amino C–H bonds are quite weak (BDE = 89–91 ± 2 kcal mol−1). Due to its thermal instability, hydrazoic acid has not been extensively studied, although in 1981 the N–H BDE was experimentally found to be 92 ± 5 kcal mol−1.242 Therefore, the hydrogen abstraction from 758a was feasible with radical 760. Addition of radical 762a to alkene 761a resulted in α-carbonyl radical 763a. Electron transfer to 763a from 15− closed the 4CzIPN catalytic cycle and delivered enolate 764a, which was protonated to afford the product 759a. Due to relative BDE (α-amino C–H = ca. 90 kcal mol−1 and α-ester C–H = ca. 96 kcal mol−1) it was thought that a radical chain mechanism could be operative. However, the calculation of a quantum yield value of 0.04 makes this mechanism improbable.
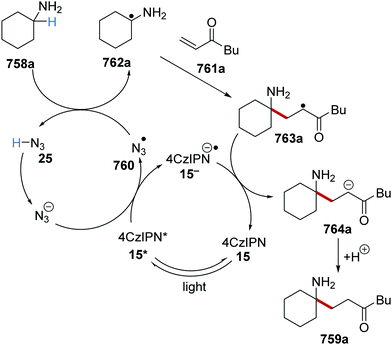 |
| | Scheme 206 Formation and reaction of α-aminoalkyl radicals. | |
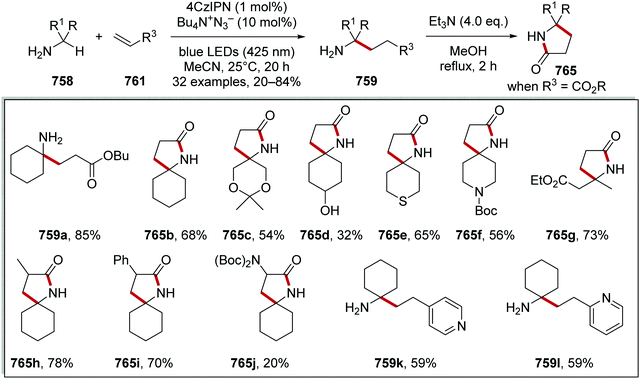 |
| | Scheme 207 Products from Giese reactions of α-aminoalkyl radicals. | |
From the result of optimisation studies, it was found that the formation of α-tertiary amines 759 proceeded best when 4CzIPN and Bu4N+N3− were used respectively as photocatalyst and precursor to the HAT catalyst and this gave 759a in 85% isolated yield (Scheme 207). Similar yields were obtained with Ir[dF(CF3)ppy]2(dtbbpy)PF6 as photocatalyst with 759a being given in 83% yield. The use of quinuclidine or (TMS)3SiH as HAT catalyst led to diminished yields of 759a, 42% and 70% respectively. The concentration of the reaction was also crucial for this transformation. When the reaction was performed at a 0.075 M concentration, this gave 759a in 85% yield. When the reaction was carried out at a 0.02 M concentration, 759a was given in 84% yield and with a 0.4 M concentration, it was isolated in 44% yield. With a good process for the formation of α-tertiary amines 759 established, it was then demonstrated that pharmacologically valuable γ-lactams 765 could be prepared from 759. For example, lactam 765b was prepared from cyclohexylamine and methyl acrylate in 68% yield, over the two steps. It was also demonstrated that ketal 765c, alcohol 765d, thioether 765e, N-Boc amine 765f and ester 765g analogues were prepared from cyclohexylamine. The use of other electron-poor alkenes led to the formation of lactams 765h–j and amines 759k–l.
N-Boc pyrrolidine 429a was coupled to alkene 766 using an 3-acetoxyquinuclidine (22d)/4CzIPN (15) catalytic system and this gave cyclopropane 767 in 50% yield via a radical/polar crossover reaction. From an optimisation study, it was found that the inclusion of the inorganic base potassium bicarbonate led to the highest yield of 767 over potassium carbonate and caesium carbonate. Furthermore, it was established that a 1:1 solvent mixture of MeCN and DMSO gave the highest yield of 767 (Scheme 208).
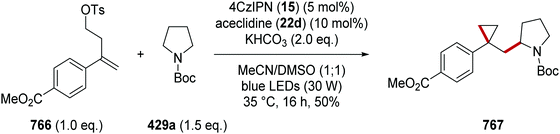 |
| | Scheme 208 Radical-polar crossover with N-Boc amine 429a. | |
The reaction propagated with irradiation by blue light and this gave excited compound 15* from 15 (Scheme 209). SET to 15*(Ered* = +1.35 V vs. SCE) from 3-acetoxyquinuclidine (22d, Eox = +1.22 V vs. SCE)48 resulted in the formation of 22d′. HAT from N-Boc pyrrolidine 429a to 22d′ gave α-amido radical 768. Addition of radical 768 to alkene 766 gave benzylic radical 769 which was reduced by 15− and this resulted in anion 770. An intra-molecular polar substitution reaction of 770 gave cyclopropane product 767.
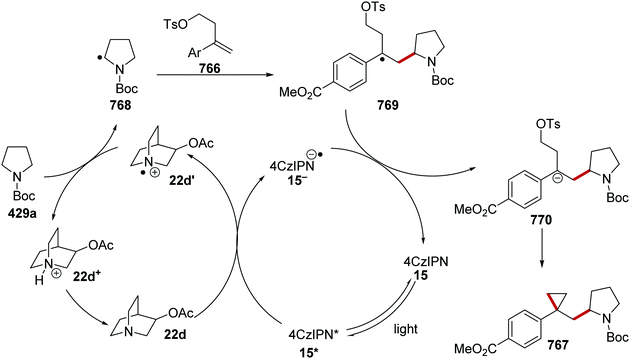 |
| | Scheme 209 4CzIPN mediated RPC with substrate 766. | |
Energy transfer.
Dearomative rearrangements of naphthols and indoles were achieved with an organic photocatalyst, 2CzIPN (72) (Scheme 210).79 Under irradiation with blue light (455 nm), the organic photocatalyst was excited and this gave 2CzIPN*, which has a triplet energy of 60.6 kcal mol−1. The triplet energy of biaryl compound 771a was calculated to be 59.8 kcal mol−1.243 Therefore, 2CzIPN* was able to transfer its energy to naphthol compound 771a and this gave triplet diradical 772a and the original photocatalyst. Diradical 772a underwent a dearomatizing [2+2] giving the 6/6/4/5-tetracycle 773a. From computational calculations, it was determined that this tetracycle 773a had a triplet energy of 55.9 kcal mol−1 and thus it was able to receive energy from the photocatalyst. Therefore, a second Dexter-energy transfer event between 773a and 2CzIPN* resulted in excited triplet species 774a. This diradical underwent excited homolytic ring opening of the strained four-membered ring and, after ISC, this gave the singlet diradical 775a. Carbon–carbon bond formation between the two radicals resulted in a 6/4/6/5-tetracycle 776a, which was isolated as product.
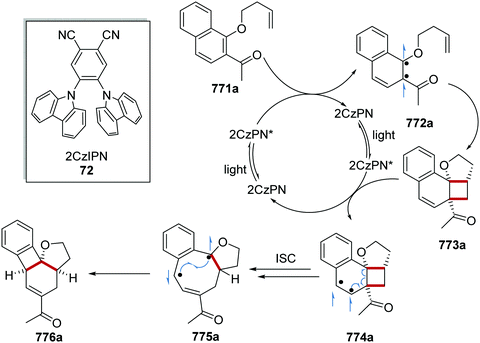 |
| | Scheme 210 Dearomatising rearrangements of naphthol derivative 771a. | |
In optimising the reaction, it was found that, dependent upon reaction conditions, the intermediate 773a or tetracyclic compound 776a was isolated, or a mixture of both. When the reaction was performed in 1,4-dioxane, full conversion of starting alkene 771a was achieved but poor selectivity was achieved, with 773a and 776a being formed in equal amounts (Table 9, entry 1). A modest increase in the selectivity was achieved when the reaction was performed in toluene (entry 2). When the reaction was performed in chloroform, high levels of selectivity for the desired compound 776a was obtained (16:1, entry 3). Fully optimised conditions employed low levels of catalyst (1 mol%) in chloroform and this returned 776a in 94% isolated yield (entry 4). The selectivity of the reaction was reversed with the addition of catalytic amounts of scandium triflate (entry 5).
Table 9 Optimisation of rearrangement of 771a
| Entry |
2CzIPN (x mol%) |
Solvent |
Time (h) |
Additive |
Ratio of 773a:7776a |
| 1 |
5 |
1,4-Dioxane |
17 |
|
1.2:1 |
| 2 |
5 |
PhMe |
17 |
|
1.8:1 |
| 3 |
5 |
CHCl3 |
17 |
|
16:1 |
| 4 |
1 |
CHCl3 |
14 |
|
16 (94%):1 |
| 5 |
1 |
CHCl3 |
17 |
Sc(OTf)3 (0.1 eq.) |
1:20 |
The optimised reaction conditions were applied to naphthol derivatives 771 (Scheme 211) and this resulted in the 6/4/6/5-tetracyclic compounds 776 as products. This allowed for the preparation of ketone 776a, 776d–e and ester 776b–c analogues all within 14 h. The reaction was scaled up to gram quantities as 2.04 g of 776c were isolated in 92% yield. Recycling of the photocatalyst was also feasible, as recycled 2CzIPN gave 776d in 88% isolated yield.
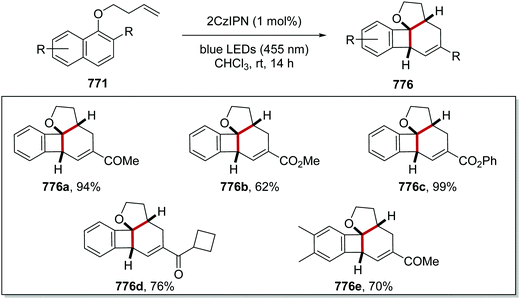 |
| | Scheme 211 Products arising from dearomatising rearrangements of naphthol 771. | |
The reaction conditions developed for the synthesis of tetracyclic compounds 776 were also applied to indoles 777 and this gave compounds 778, which had a 6/5/4/5-ring system (Scheme 212). Conversion of indoles 778 to tetracyclic compounds 778 worked better in toluene than CHCl3. It was postulated that the use of a less polar solvent inhibited electron transfer and thus energy transfer processes were more dominant. The use of toluene as reaction solvent allowed for the preparation of six tetracyclic compounds 778 in 51–99% yields.
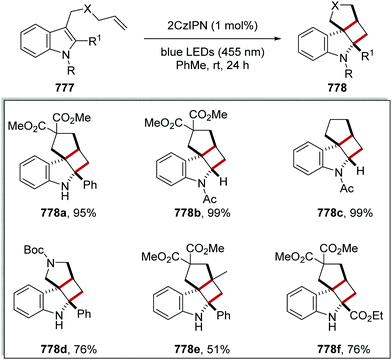 |
| | Scheme 212 Dearomatising rearrangements of indoles. | |
3.5 Dicyanoanthracene
Carboxylic acids as radical precursors.
The coupling of amino acids 779 to electron-poor alkenes 78066 (and also to alkynes244) was investigated with DCA (2) as photocatalyst. The reaction between phenylalanine (779a) and E-1,2-bis(phenylsulfonyl)ethylene (780a) was promoted with the DCA (20 mol%) and biphenyl (BP) catalytic system and this led to alkene product 781a in 67% yield, within 24 h (entry 1, Table 10). The loading of the DCA catalyst could be decreased down to 1 mol% (entries 2–4). The removal of biphenyl from the reaction mixture led to a diminished yield of 781a (entry 5). Consumption of the starting material was not achieved when BP was absent from the reaction even with a reaction time of 70 h (entry 6).
Table 10 Optimisation table
|
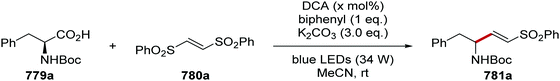
|
| Entry |
DCA (mol%) |
Biphenyl |
Time (h) |
Yield of alkene |
| 1 |
20 |
Yes |
24 |
67 |
| 2 |
10 |
Yes |
24 |
70 |
| 3 |
5 |
Yes |
24 |
70 |
| 4 |
1 |
Yes |
24 |
70 |
| 5 |
1 |
No |
24 |
37 |
| 6 |
1 |
No |
70 |
59 |
The coupling of 779a to 780a under DCA/BP mediated conditions was achieved through the following mechanism (Scheme 213). Upon excitation, with blue light, DCA was promoted to its excited singlet state (S1*, 2*). The S1* state of DCA is a very strong oxidant (Ered* = +1.99 V vs. SCE) and it can oxidise both biphenyl [E1/2(BP˙+/BP) = +1.95 V vs. SCE] and carboxylate (E1/2(RCO2˙/RCO2−) = +0.95 V vs. SCE). The S1* state of DCA has a lifetime of ca. 15 ns in nitrogen-saturated acetonitrile and it has an inefficient intersystem crossing (ISC) to its triplet state (ϕISC = 0.01 in MeCN). It has been communicated that a lifetime of 1 ns is sufficient for an effective SET process to occur.8a However, when the reaction is performed without BP, the reaction is slower and with compromised yields, even though 2* is sufficiently long-lived for a SET from 782a. Therefore, the role of BP could be suppression of back-electron transfer (BET) and thus higher rates of reaction, although more investigation is required. SET between BP radical cation 70′ and carboxylate 782a gave 783a and biphenyl (74). Carboxyl radical 783a underwent decarboxylation and this gave α-amido radical 784a, which was stabilised by the adjacent nitrogen atom. Radical coupling between this radical and alkene 780a resulted in α-sulfone radical 785a. The phenylsulfonyl radical 786 is a good radical leaving group and radical elimination of 786 resulted in alkene 781a. The DCA catalytic cycle was closed with SET from 2˙− to 786, giving anion 787 and DCA 2.
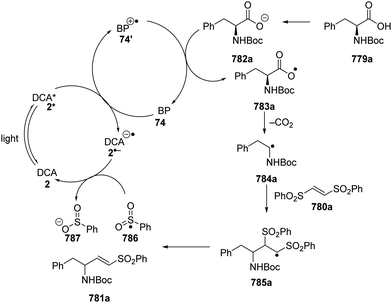 |
| | Scheme 213 DCA and BP-mediated Giese reaction. | |
The use of the DCA/BP catalytic system allowed for coupling of a range of carboxylic acids 779 with electron-poor alkenes 780 and this resulted in the preparation of 26 alkenes 781 in yields of 10–87% (Scheme 214). It was found that α-amino acids as substrates worked well in forming alkene products 781a–e, due to radical stabilisation. However, the absence of an α-amino group led to diminished yields as shown in the preparation of 781f (10%). A low yield of 30% was also obtained for 781g, due to the lack of a stabilising α-amino group.
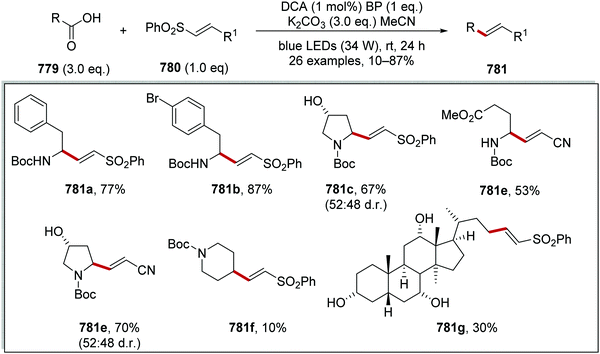 |
| | Scheme 214 DCA mediated coupling reaction with biphenyl (BP). | |
DCA* 2*, generated by irradiating the ground state parent with visible light had earlier been used as an oxidant by Pandey in the amidation of benzylic C–H bonds.245 On the other hand, the radical anion of DCA, generated by photoinduced electron transfer from PPh3, is a much more powerful reducing agent, and was used to generate the radical anion of conjugated enones (not shown here).246
However, in a different approach, the radical anion of DCA, produced electrochemically, was excited to its relatively stable excited state (lifetime of 13.5 ns), which then underwent SET, converting aryl chlorides 788, 790 to aryl radicals for coupling to arenes and pyrroles (Schemes 215 and 216).247 DCA (E1/2 = −0.82 V vs. SCE) was reduced and this radical anion was excited by blue light and this gave the excited radical anion species. From DFT calculations suggested that this involved an electronic transition from the HOMO to the SOMO resulting in a SOMO–HOMO level inversion. This radical species was calculated to be strongly reducing (Eox = −3.2 V vs. SCE, for comparison Eox = −3.3 V vs. SCE for lithium metal). A highlight of this methodology was the reduction of 4-chloroanisole (Ered = −2.90 V vs. SCE) in the formation of 789a.
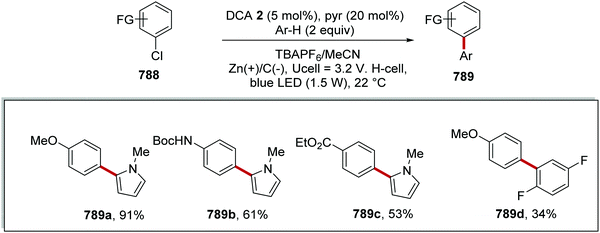 |
| | Scheme 215 Photoactivation of the radical anion of DCA, 2, produced by electrochemical reduction. | |
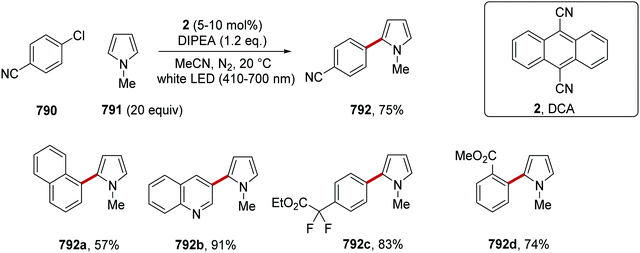 |
| | Scheme 216 Consecutive photoactivations of dicyanoanthracene (DCA, 2) and its radical anion afford a strong electron donor. | |
In 2018 von Wangelin, Peres-Ruiz et al. used 9,10-dicyanoanthracene (9,10-DCA) 2 for coupling similarly activated aryl halides 790 to heteroarenes 791 to form C–B, C–S and C–P bonds (Scheme 216).248 In this case, however, the DCA 2 was photoexcited, and received an electron from DIPEA. The resulting radical anion then received a second quantum to form a donor that activated aryl bromides and chlorides [usually bearing an additional electron-withdrawing substituent] for coupling to pyrroles to form products 792 or for formation of C–P, C–S or C–B bonds. This double quantum approach followed an earlier report of this conPET concept by König et al. in 2014 that will be discussed later in relation to Scheme 239.
3.6 Acridinium dyes
Among the most impressive organic redox agents are the acridinium salts. Initially championed by Fukuzumi et al., their application to synthetic organic redox chemistry has principally been led by the team of Nicewicz.8a They have principally been recognised for the oxidative prowess of their excited states, but a recent example shows that, in the presence of a sacrificial electron donor such as diisopropylethylamine, a metastable mesitylacridyl radical is produced. Excitation of this radical to its excited state is accomplished at about 350 nm, and that excited state converts aryl chlorides and bromides to aryl radicals for hydrodehalogenation reactions and undertakes removal of arenesulfonyl groups from arenesulfonamides.201 Until now, this has not been exploited for formation of the types of bonds discussed in this review. However, the more developed side of the chemistry of photoexcited acridinium salts is as strong oxidants. In particular, they oxidise many arene substrates to their radical cations. The radical cations are then subject to attack by nucleophiles, followed by the rearomatisation of the original arene. In these reactions, Ar–H or Ar–OMe (Ar–OR) or Ar–F are the usual candidates for substitution.
Examples of functionalisation reactions with Ar–H shown below and include C–H amination,249 C–H cyanation,250 and C–H alkylation,251 in addition to C–H fluorination.252Scheme 217 shows the likely general mechanism. The arene substrate 793 is converted into the corresponding radical cation 794 by electron transfer to the excited state redox agent, (797, Ered* = +2.15 V vs. SCE) which itself is transformed to the acridinyl radical 798. The radical cation derived from the substrate, 794, can then be attacked by a nucleophile, normally para to the substituent. The resulting radical 795 undergoes loss of an H atom to air or TEMPO to afford the product 796. Meanwhile the acridinyl radical can be reoxidised to 16e.g. by air, to complete its catalytic cycle.
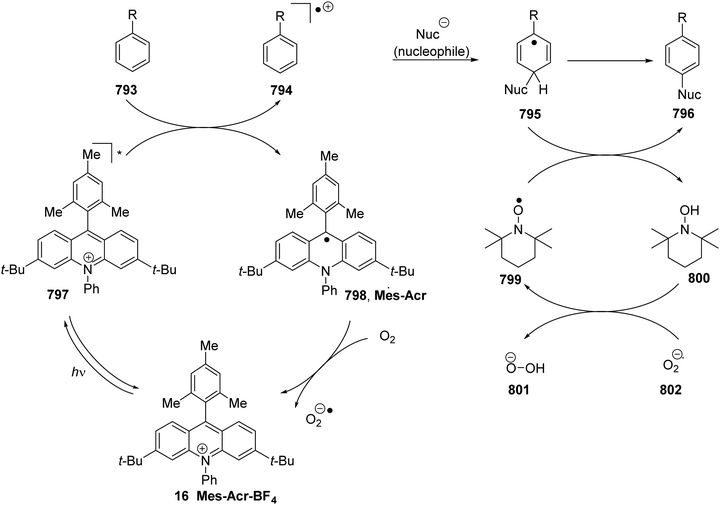 |
| | Scheme 217 General mechanism for C–H substitution of arene radical cations. | |
Examples of C–H amination are shown in Scheme 218.249 Azoles including pyrroles (e.g.804e), pyrazoles (e.g.804a, b) imidazoles, triazoles (804c) and tetrazoles and a number of ring-fused analogues (804d, 804f) formed the majority of the reported examples. Direct amination, Ar–H → Ar–NH2, was also achieved when ammonium carbamate (NH4+ H2N–CO2–) was used as the source of the nitrogen nucleophile.
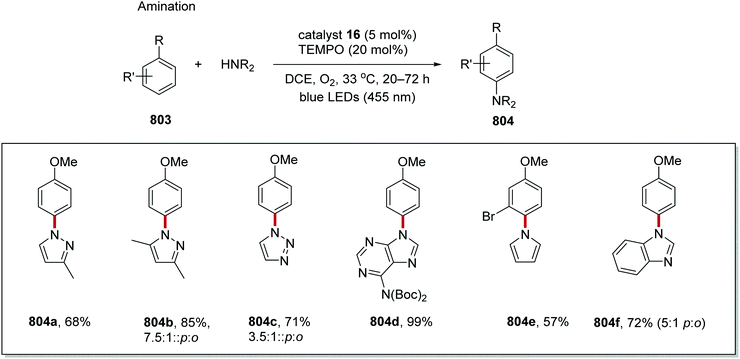 |
| | Scheme 218 C–H amination of arenes with azoles. | |
The cyanation protocol is shown in Scheme 219.250 TMSCN was the most successful source of cyanide for these reactions, which were carried out open to air and in the presence of the pH 9 phosphate buffer. The process worked well also for more complex substrates. Thus N-Boc-melatonin was selectively cyanated to afford 805f (26%), and naproxen methyl ester afforded a single regioisomer of cyanated product, i.e.805g (57%).
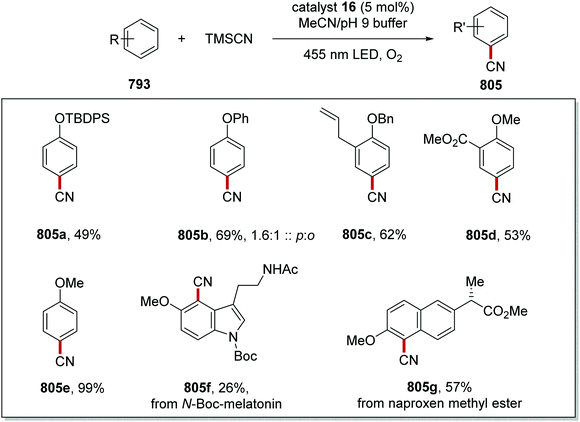 |
| | Scheme 219 C–H cyanation of arene radical cations. | |
Alkylation was achieved using diazoesters as the alkylating agents (Scheme 220).251 The mechanism for alkylation is a little different. Here, (Scheme 221) the arene is again proposed to be oxidised to its radical cation, 794, which is then attacked by the diazoester 806 as a nucleophile. This gives the diazonium intermediate 808 which then receives an electron from the reduced form of the MesAcrBF4 photocatalyst to give 809. Oxidation of this species opens the cyclopropane affording the distal radical cation 810. Reductive electron transfer and deprotonation then yields the final product.
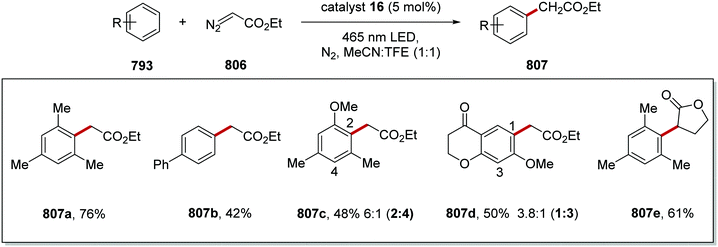 |
| | Scheme 220 C–H alkylation of arene radical cations. | |
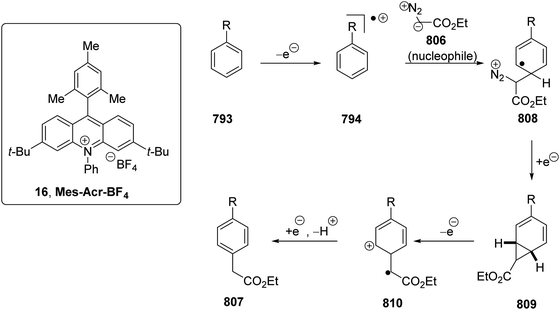 |
| | Scheme 221 Mechanism of alkylation of arene radical cations. | |
In addition to the reaction types just mentioned, direct Ar–H fluorination reactions have been extensively developed by the Nicewicz team.252 One of the most useful applications of this work is in the direct incorporation of radioactive 18F into organic molecules for positron emission tomography (PET) applications in medicine. An extensive range of substrates has been fluorinated in this way, and radiochemical yields (RCY) for some are presented in Scheme 222. Initially, the chemistry required 450 nm laser activation, but recent developments have allowed LED activation at 425 nm as well as the use of structurally optimised acridinium catalysts in a user-friendly manner and in flow. For these reactions, tert-butyl peracetate was used as the sacrificial oxidant. Labelling of xanthones had not been possible with the earlier technology but afforded a 22.9% RCY of product 812d with the new methodology with catalyst 16-Cl.252b
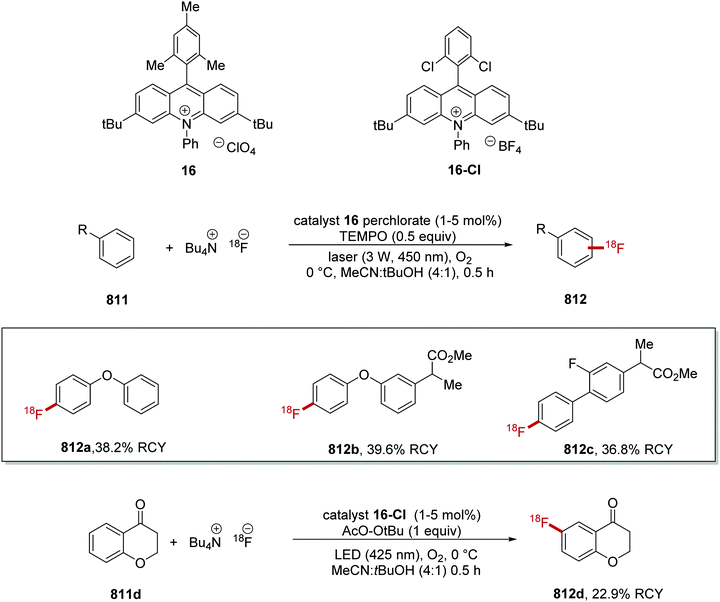 |
| | Scheme 222 C–H radiofluorination of arenes. | |
These reactions have all involved substitution of the incoming nucleophile onto an Ar–H position, with loss of the hydrogen atom as a proton. The alternative class of reactions involves displacement of an anionic leaving group, rather than a proton, from the arene. Initial reports featured deoxyaminations and deoxyfluorinations with loss of an alkoxide group from alkoxybenzenes. Deoxyfluorinations253 are useful for the same reasons as the fluorinations just described, i.e. in PET medical applications using 18F. A wide range of radiolabelled products have been produced in that way, but 19F products can also be prepared, and Scheme 223 shows a range of these.
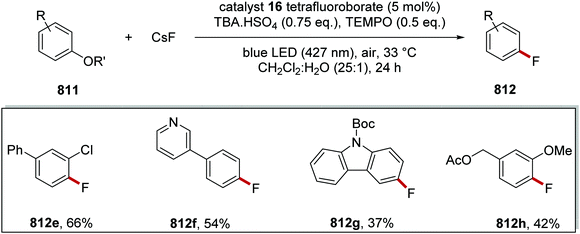 |
| | Scheme 223 Fluorodealkoxylation reactions of arene radical cations. | |
Deoxycyanation reactions254 and deoxyamination reactions255 have been developed. Scheme 224 below shows examples of the latter. For the deoxyamination reaction, questions of whether the arene or the amine was oxidised with the photoactivated acridinium salt were addressed through measurements of the rates of quenching of the catalyst fluorescence. Both amines and the arene substrates were effective quenching agents but, for the cases studied, the electron-rich arenes were more rapidly quenched than the amines. When (R)-phenethylamine was used to prepare 813d, the product was completely racemic. This likely arises due to oxidation of the product (but possibly also the amine starting material) to its radical cation, followed by reversible loss of the ArN–CH proton to afford an α-aminoalkyl radical at the original chiral centre.
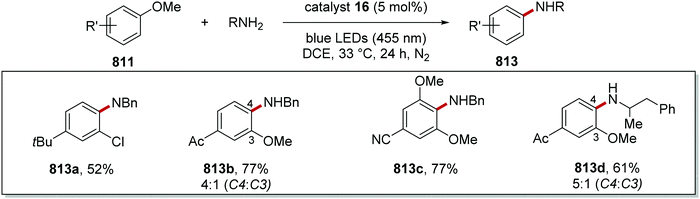 |
| | Scheme 224 Deoxyamination of anisole radical cations. | |
Intramolecular displacements of leaving groups in the radical cations of arenes have also been reported for a number of cases, e.g. Smiles rearrangements 814 → 815256 and amination reactions, 816a → 817a256 have also been observed to work well. In this case, the cyclising amines can also be azoles as exemplified in formation of product 817b (Scheme 225). The reactions are not limited to displacements by nitrogen nucleophiles, as seen in the Newman–Kwart reactions, using a pyrylium salt photoredox agent, exemplified by conversion 818 → 819.257
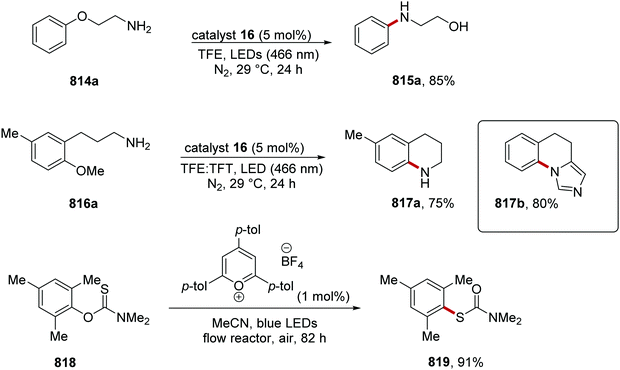 |
| | Scheme 225 Substitutions involving cleavage of Ar–O bonds by sulfur and nitrogen nucleophiles. | |
The reactions of arene radical cations discussed here so far, featuring anionic leaving groups, have focussed on alkoxide leaving groups. However, recently, reports of displacement of fluoride anions from aryl fluorides have emerged from the laboratories of Nicewicz258 and Lambert (see later, Scheme 244). Given that fluoroarenes 821 (Eox = +2.24 V vs. SCE for 4-fluorotoluene) are not generally easily oxidised, the conditions of the reactions are important. The acridinium salt 16 can mediate the desired oxidations to the arene radical cation provided that the oxidation potential of the arenes is less than +2.14 V vs. SCE (Scheme 226). Azoles, primary amines and carboxylates can then act as intermolecular nucleophiles affording products 822. With less easily oxidised arenes, a more reactive xanthylium photocatalyst 820 (Ered* = +2.57 V vs. SCE) is required. Here, pyrazoles and triazoles can act as intermolecular nucleophiles, in addition to carboxylates as successful [usually intramolecular] nucleophiles. With the acridinium salt reactions, different optimal conditions were devised for the three classes of nucleophile.
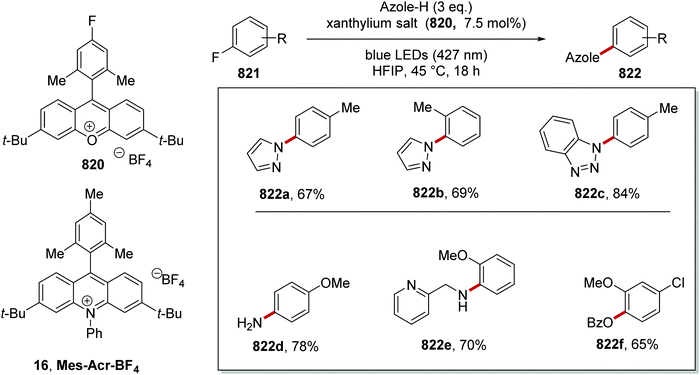 |
| | Scheme 226 Defluorinative amination of fluorobenzenes. | |
Although the majority of recent developments have been associated with the oxidation of arenes to their radical cations, it has also been possible to oxidise certain alkenes to their radical cations which are subsequently attacked by nucleophiles, either intermolecular or intramolecular (Scheme 227).259,260 This was achieved with acridinium catalyst 68 (Ered* = + 2.18 V vs. SCE)8a and alkenes such as 821 (Eox = +1.86 V vs. SCE, when R = H), 829 (Eox = + 1.69 V vs. SCE) and 831 (Eox = +1.61 V vs. SCE).
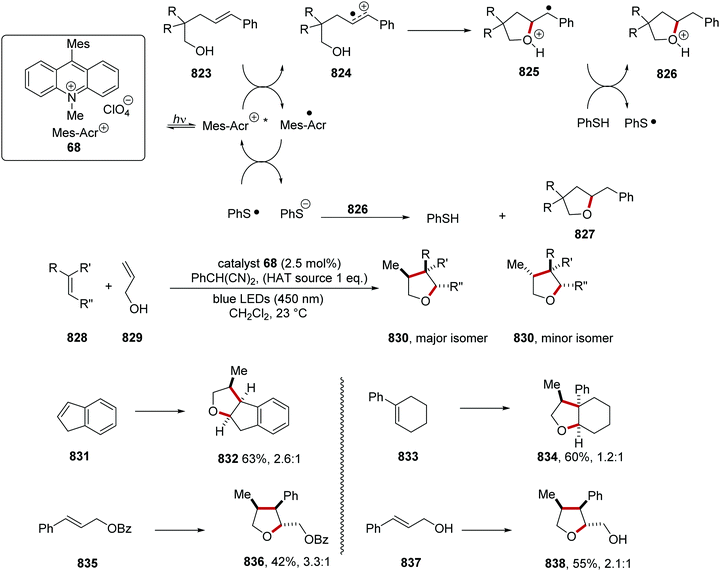 |
| | Scheme 227 Tetrahydrofuran formation via alkene radical cations. | |
Photoactivated acridinium salts have also been used agents to liberate radicals from boron-based precursors.261 Ley's team used acridinium salts as photoredox reagents. BPin esters 839 and boronic acids 843 in the presence of Lewis bases were studied under photoactivation of a mesitylacridinium salt 841 or 4CzIPN 15 and gave the desired products from reactions of radicals with Michael acceptors (Scheme 228).262 They demonstrated success with both alkyl and aryl radicals. The Lewis bases were required to make the boronic esters more suspectable to SET oxidation. In a previous report it was shown that 4-methoxybenzylboronic acid (Eox = +1.43 V vs. SCE) had a high oxidation potential but this was significantly decreased when it was complexed to DMAP (Eox = +0.81 V vs. SCE, for boronic acid/DMAP adduct).262b However, all these species listed should easily be oxidised by catalyst 841 (Ered* = +1.65 V vs. SCE).
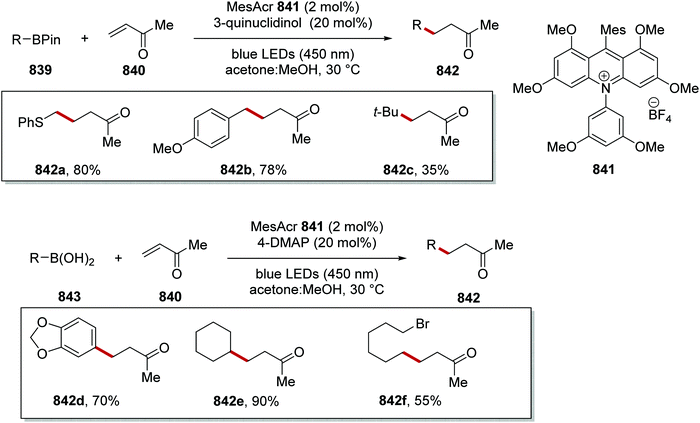 |
| | Scheme 228 Comparison of BPin esters and boronic acids as sources of radicals for Giese reactions. | |
The Molander group used alkyltrifluoroborate salts for cyanation and allylation reactions.263 Oxidation of the salts led to radicals which then reacted with tosyl cyanide displacing tosyl radicals, which are reduced to toluenesulfinate anions to complete the catalytic cycle of the photoredox agent (see Scheme 229).
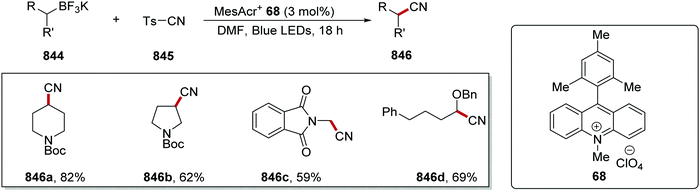 |
| | Scheme 229 Formation of nitriles using radicals liberated from oxidation of alkyl trifluoroborates. | |
Minisci reactions were also carried out by Molander's team using photoactivated acridinium chemistry. For example, reaction between 2-trifluoroborato-4-chromanones 847 and N-heterocycles 848 mediated with acridinium dye 16 and potassium persulfate 66 was reported, affording products 849 (Scheme 230).264 Irradiation with white light (26 W CFLs) gave excited dye 797* from 16. Electron transfer to 797* (Ered* = +2.20 V vs. SCE) from trifluoroborate 847a (Eox ≈ 1.11 V vs. SCE) gave α-alkoxyalkyl radical 850a, which added to protonated heterocycle 851a and this gave amine radical cation 852a. Coincidentally, electron transfer from reduced catalyst 798˙ (Eox = −0.57 V vs. SCE)266 to potassium persulfate 66 (Ered = ca. ≤0.35 V vs. SCE, see ref. 189) resulted in oxygen-centred radical 79 and sulfate ion. Hydrogen atom transfer to 79 from radical cation 852a gave the coupled product 853a, which was neutralised on workup to give 849a.
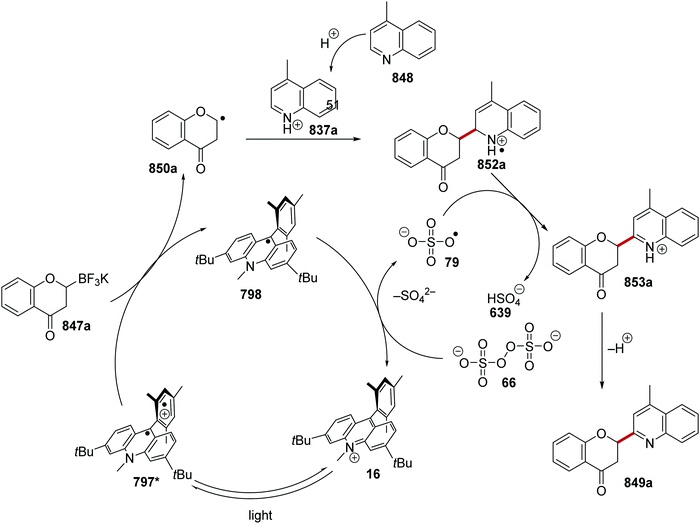 |
| | Scheme 230 Minisci reactions with trifluoroboronates mediated by acridinium salt 16 and persulfate. | |
Various photocatalysts were trialled for this transformation. Initially, Ir[dFCF3ppy]2(bpy)]PF6 (2 mol%) was tested with K2S2O8 (2 eq.) and this gave 849a in 43% yield. The use of the organic dye Eosin Y gave 849a in 40% yield. The yield of 849a was increased to 57% when MesAcr 16 (1 mol%) was used as photocatalyst. Fully optimised conditions were established when the reaction was performed with one equivalent of TFA and this gave 849a in 60% yield (Scheme 231). These optimised conditions allowed for the preparation of 20 alkylated heterocycles 849b–e, including a caffeine analogue 849f which was given in 44% yield.
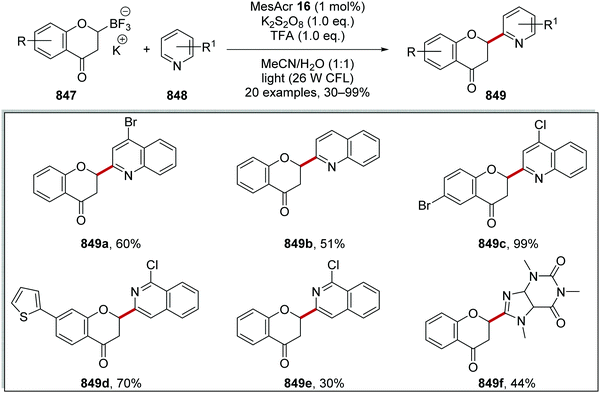 |
| | Scheme 231 Acridinium and persulfate-mediated Minisci reaction. | |
α-Heteroatom functionalisation.
Coupling of N-Boc protected amines 854a with electron-poor alkenes 855 was achieved with acridinium dye 16 and this resulted in γ-amidoketone compounds 856 (Scheme 232).35 When the reaction mixture was irradiated with blue light (455 nm), this resulted in the formation of 856a from N-Boc piperidine 854a and methyl vinyl ketone 855a. Acridinium dye 16 was excited by the blue light and this gave the excited species 797, which is a strong oxidant (Ered* = +2.15 V vs. SCE). N-Boc amine 854a (Ep/2 = 1.96 V vs. SCE) underwent SET with 797 and this resulted in amine radical cation 857a and reduced catalyst 798. Proton loss from 857a resulted in carbon-centred radical 858a, which added to alkene 855a and this gave α-keto radical 859a. The acridinium catalytic cycle was closed with SET to 859a from 798; this also gave anion 860a, which was protonated and this resulted in γ-aminoketone 856a.
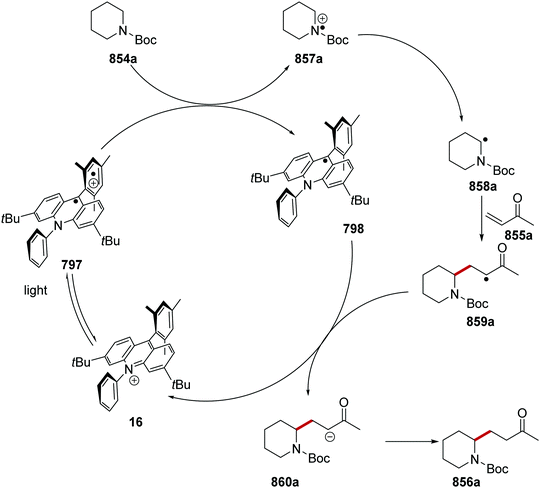 |
| | Scheme 232 Giese coupling of radicals derived from N-Boc piperidine 854. | |
Product 856a was formed in the highest yields when the reaction was conducted in dichloromethane, with a reaction time of 6 h and a concentration of 0.1 M. These optimised reaction conditions were then applied to the preparation of products 856 in yields of 20–100%. With the optimised reaction conditions, N-Boc amines were coupled to methyl vinyl ketone, and this gave compounds 856a and 856b in 83% and 39% yield, respectively (Scheme 233). When N-Boc morpholine was used as the substrate, functionalisation occurred exclusively at the α-amino position and this formed 856c in 62% yield. Furthermore, N-Boc protected tetrahydroisoquinoline resulted in exclusive reactivity at the more reactive benzylic position and this gave 856d in 99% yield. When (2S)-2-methyl N-Boc piperidine was treated with methyl vinyl ketone under these reaction conditions, 856e was given as a single regioisomer, in a 10:1 diastereomeric ratio (95:5, er). Various other enones were suitable coupling partners in this reaction and 856f and 856g were formed in 82% and 53% yield, respectively. The preparation of dinitrile 856h was achieved in 94% yield on a 0.3 mmol scale. The preparation of 856h was conducted on 15.0 mmol scale with a photochemical flow reactor and this gave the dinitrile compound in 94% yield (1.71 g).
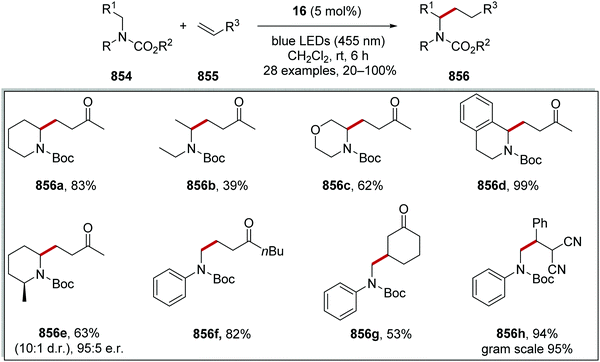 |
| | Scheme 233 Giese coupling with acridinium catalyst 16. | |
The site-selective C–H functionalisation of piperazines 861 with electron-poor alkenes 855 was achieved through use of the acridinium catalyst 16 and this gave products of conjugate addition 862 (Scheme 234).265 Initially, derivative 862a was prepared in 90% yield from 1,4-di-Boc-piperazine and methyl vinyl ketone. Replacement of a Boc group with the Fmoc protecting group resulted in preferential C–H functionalisation adjacent to the Boc protected amine and this gave 862b as a 2.2:1 mixture of regioisomers in 71% yield. Greater site-selectivity was attained when N-Boc-N′-benzoylpiperazine was used as substrate, with 862c being given in 81% yield with a 10:1 ratio of regioisomers at 29 °C. In the preparation of 862c, the site-selectivity obtained was enhanced when the transformation was performed at 0 °C with the compound being given as a 21:1 mixture of regioisomers. Strongly deactivating groups such as N-sulfonamide resulted in the exclusive formation of one regioisomer, for example, 862d albeit a low yield was obtained. A reversal of selectivity was obtained when arylamines were used as substrates with a preference for the α-aminoaryl position over the α-amido position. For example, when 4-Boc-1-(2-pyridyl)piperazine was used as substrate this gave 862e as a 19:1 mixture of regioisomers and the use of 4-Boc-1-(2-trifluoromethylphenyl)piperazine gave 862f as a 41:1 mixture of regioisomers in 58% yield. Enhancement in the regioselectivity was achieved for different electron-poor alkene coupling partners. For example, 1,1-bis(phenylsulfonyl)ethylene as coupling partner gave 862g as a single regioisomer in 90% yield. The functionalisation of bioactive molecules was achieved; the use of the commercial antibiotic, ciprofloxacin as substrate gave 862h in 47% yield and as a single regioisomer. From the course of the investigation, it was found that some substrates gave superior results when the less oxidising acridinium dye 841 (Ered* = +1.65 V vs. SCE) was used as catalyst, denoted with †.
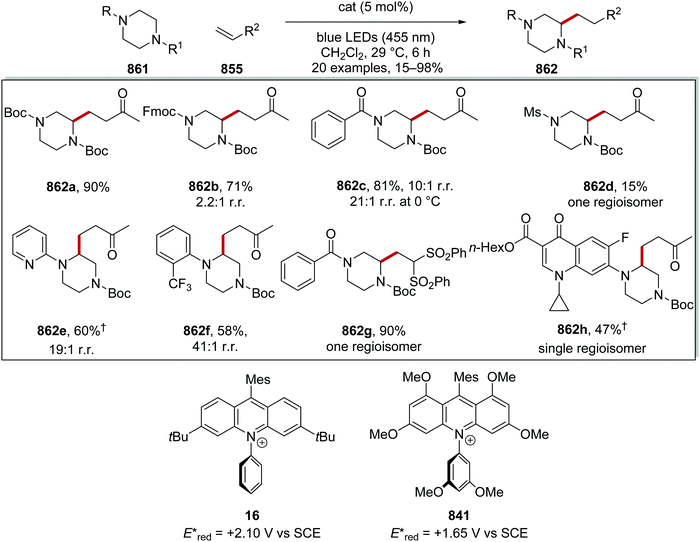 |
| | Scheme 234 Site-selective alkylation of piperazine, † denotes the use of 841. | |
Alkane functionalisation.
A dual catalytic system of acridinium dye 16 and hydrogen chloride worked well for the coupling of a range of functional groups, such as alkanes, to electron-poor alkenes under blue light (Scheme 235).266 Irradiation of the reaction mixture gave excited compound 797 from 16. SET from chloride ion (Eox = +2.03 V vs. SCE) to 797* (Ered* = +2.18 V vs. SCE) formed a chlorine radical and acridinyl radical 798. The abstraction of a hydrogen atom from cyclohexane 863a by the chlorine atom resulted in alkyl radical 864a and hydrogen chloride. C–C bond formation between electron-poor alkene 865a and 864a gave alkyl radical 866a, which received an electron from reduced catalyst 798 (E˙− = −0.57 V vs. SCE) forming anion 867a and this step closed the acridinium catalytic cycle. Protonation of 867a gave 868a.
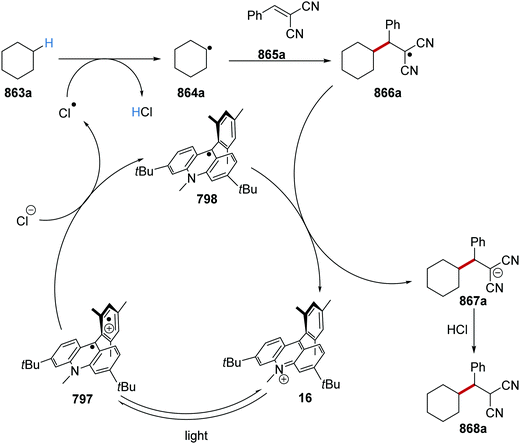 |
| | Scheme 235 C–H activation mediated with acridinium and chloride anion. | |
This transformation performed best when performed in a “stop-flow microtubing reactor” (SFMT) instead of a traditional batch reactor (Scheme 236). This allowed for the preparation of 34 dinitrile compounds 868. From the course of this investigation, it was found that alkanes gave coupled compounds 868a–e as products. Benzylic C–H bonds were activated, and this gave 868f in 69% yield. The use of ethers and alcohols led to compounds such as 868g and 868h in 75% and 90% yield, respectively. This reaction was also performed in a continuous-flow reactor, and this gave 868j in 95% yield with a 1.12 g h−1 rate.
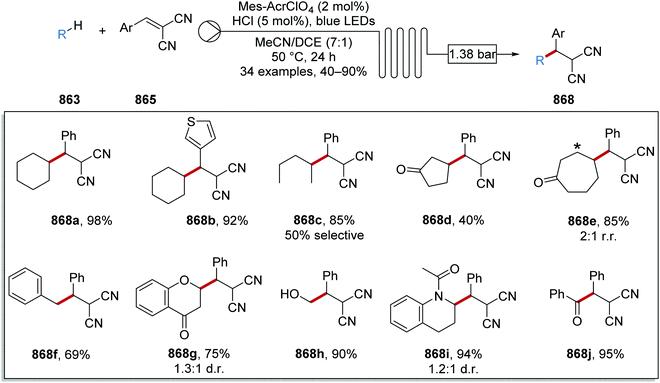 |
| | Scheme 236 Stop-flow Giese-type reaction with acridinium photocatalyst. | |
3.7 Excitation of radical cations and radical anions9a
Back in Scheme 216, the 2018 report of two quantum excitations was mentioned in relation to von Wangelin et al. This conPET (consecutive photoelectron transfers) was devised by König's team in 2014268 for reducing challenging substrates. Here the perylenediimide (PDI) 873 was first reduced to its relatively stabilised radical anion by a photo-induced reaction (Scheme 239). The radical anion was then photoexcited and, in its excited state, behaved as a powerful electron donor, reducing aryl iodides, bromides or chlorides that were slightly activated by an electron-withdrawing group in the para- or ortho-position, for example 2-chlorobenzonitrile (Ered = −1.91 V vs. SCE).222
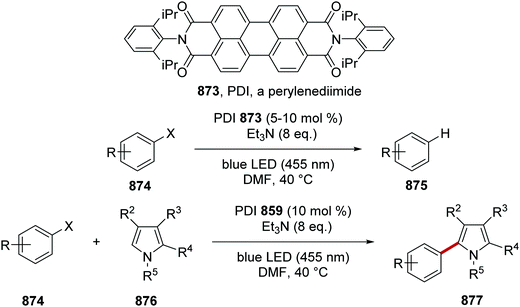 |
| | Scheme 239 ConPET (consecutive photoelectron transfers) of perylenediimide 859 in the presence of triethylamine generates a strong enough electron donor to convert aryl halides to aryl radicals. | |
Recently, the potency of such conPET donors has been extended by Miyake et al., using different dyes.269–271 Here the perylenediimide 873 was replaced by imide 878. In the presence of NMe4OH, this compound formed an adduct 879; photoexcitation produces 880 that underwent fragmentation to the somewhat stabilised radical anion 881 (Scheme 240). This radical anion was then photoexcited to afford a very strong donor, which has been used for metal-free Birch reductions of benzenes like 883 and 885 (Ered = −3.42 V vs. SCE for benzene). To investigate this reaction calculated and experimental redox potentials were found for the photocatalyst. The ground state reduced anion 881 was unable to reduce benzene (Ecalc˙− = −1.30 V vs. SCE and Eexp˙− = −1.24 V vs. SCE). Excited complex 882 had a range of different electrochemical potentials depending upon the excited state of the catalyst. Crucially, it was calculated that the first doublet state had an oxidation potential of Eox˙−* = −2.43 V vs. SCE, (calculated value, first doublet state). Therefore, these calculations could suggest that this catalyst exhibits anti-Kasha behaviour. As previously mentioned, anti-Kasha behaviour is very rare as internal conversion to the lowest excited state is extremely. However, alternatively, the formation of a solvated electron can also occur rapidly (11 ± 1 ps).269b Mechanistic studies to fully elucidate the mechanism are still in progress but it was suggested by the authors that a ground-state π-stacking complex or an exciplex could be critical for the transfer of the electron. Culmination in the ability to produce Birch reductions of benzenes in the absence of redox-active metals is a remarkable achievement for the organic electron transfer chemists. Other photoredox272a and non-photoredox272b approaches to Birch reduction have also recently been published.
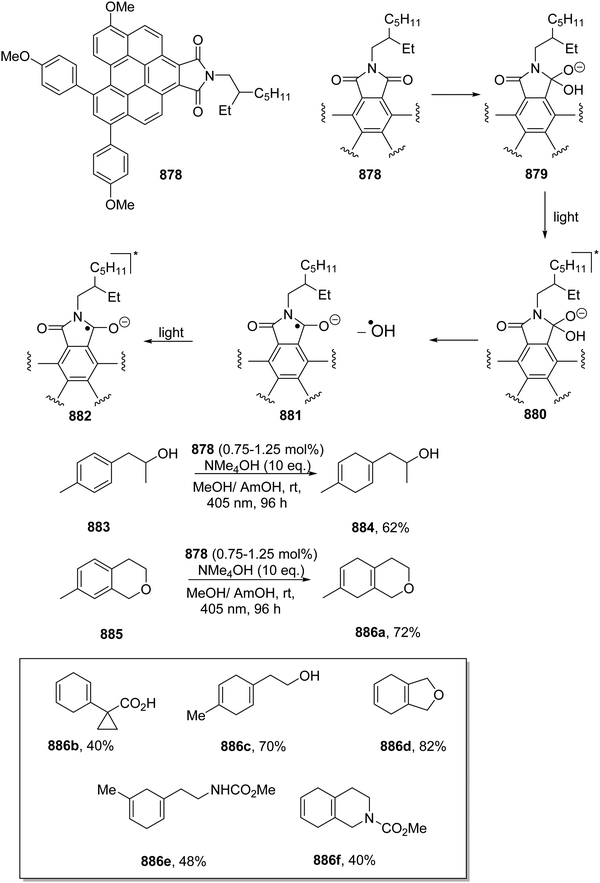 |
| | Scheme 240 Consecutive photoactivations of diimide 878 led to Birch reduction of substituted benzenes. | |
Inspired by the perylenes diimides work of König, alternative diimides were explored by the team of Wickens.273 They reported in 2020 that photoactivation of the radical anion of imide 889 usefully converts aryl chlorides, some with very negative reduction potentials (Ered = −2.8 V vs. SCE for chlorobenzene), to the corresponding aryl radicals and used these radicals in heteroarylation reactions (Scheme 241) and for coupling to trialkyl phosphites. In their case, the imide is reduced to its radical anion electrochemically and this species is then photoexcited by blue LEDs to afford electron transfer.
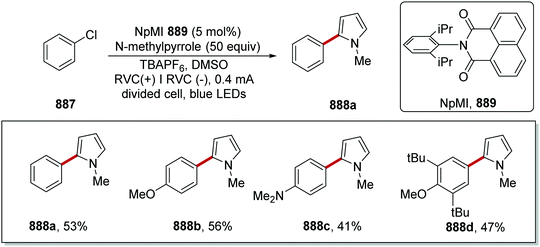 |
| | Scheme 241 Heteroarylation of aryl chlorides. | |
A combination of electrochemical activation followed by photoactivation was published in 2019 by Lambert's group.274 They prepared the trisaminocyclopropenium salt 895, and oxidised it electrochemically to the radical dication 892, which was then excited by a 23 W CFL (Schemes 242 and 243). When the diradical cation was excited, it became a very strong oxidant (Ered* = +3.33 V vs. SCE) and thus it was able to oxidise compounds such as benzene (Eox = +2.54 V vs. SCE). Oxidation of arenes afforded the radical cations that were then trapped by azole nucleophiles giving products 893a–e.
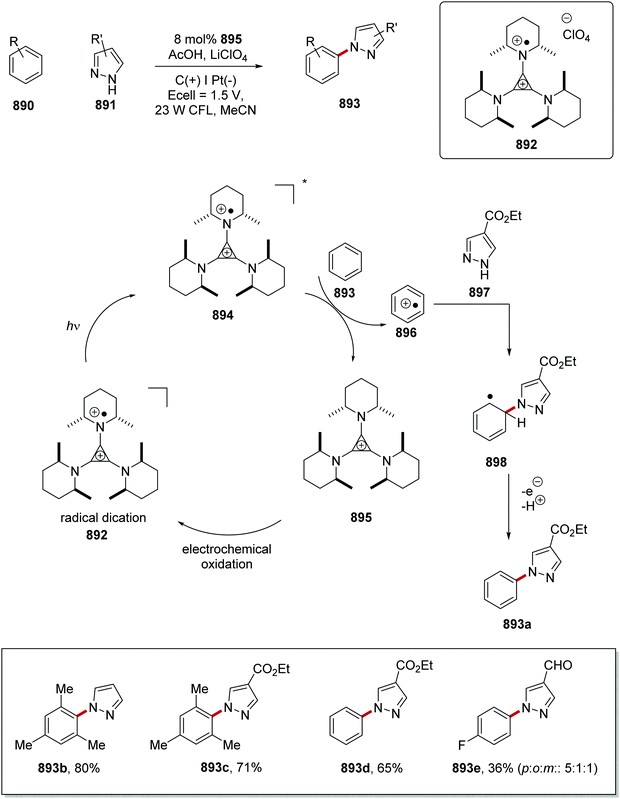 |
| | Scheme 242 C–H amination of arene radical cations with azoles. | |
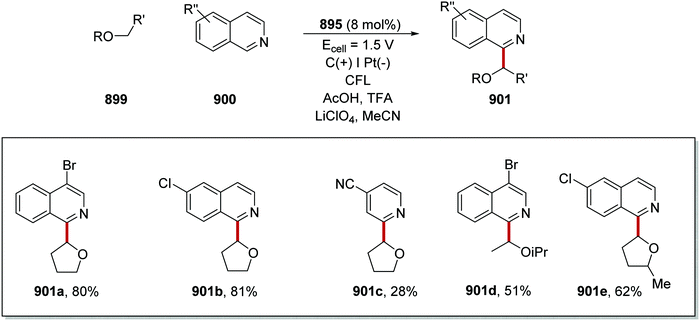 |
| | Scheme 243 Minisci reactions of ethers. | |
The strong oxidising power of the excited radical dication 894 was also deployed in the Minisci reactions of alkyl ethers (Scheme 243).275 Using the same cycle as shown above, it is thought that the photoexcited radical dication has HAT properties to abstract a hydrogen atom from an ether 899 to bring about Minisci reactions with protonated pyridines and quinolines *. Additionally, the ether-derived radicals undergo Giese reactions with electron-poor alkenes and C–N coupling to azoles (not shown here).
Expanding on the repertoire, the cyclopropenium reagent 895 has been used in a study of SNAr reactions of aryl fluoride radical cations.276 (See also the studies by Nicewicz et al. in this review, Scheme 226.) However, photoexcited DDQ was found to work better. This excitation of DDQ with blue LEDs afforded a highly oxidising DDQ* (Ered* = +3.18 V vs. SCE) that converted aryl fluorides into their radical cations. Even p-difluorobenzene (Eox = +2.35 V vs. SCE) was activated in this way to afford the derivative 904e (63%, Scheme 244).
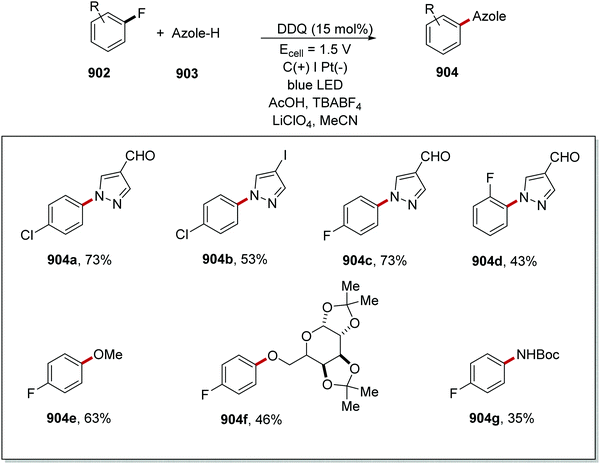 |
| | Scheme 244 Defluorinative amination of aryl fluorides. | |
Triarylamine radical cations are ground state oxidants and have been used e.g. to oxidise DABCO to its radical cation for highly selective HAT chemistry.38 More recently, the photoactivated radical cation of [(tri[1,1′]-biphenyl)-4-yl]aminetolylaminium salt (TCPBA) has been used to oxidise arenes to their radical cations by the Barham team. The examples shown in Scheme 245 feature C–H substitution by azoles.277 The formation of fluoroarene 904m from fluorobenzene shows that this is a highly powerful oxidising system for arenes and competitive with those discussed above [from the teams of Nicewicz and Lambert]. The best catalyst used in this transformation, TCBPA, was calculated to be a very strong oxidant (Ered* = +4.19 V vs. SCE).
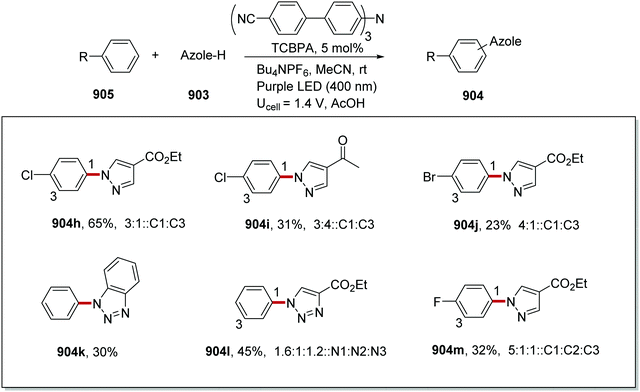 |
| | Scheme 245 Photoexcitation of triarylaminium salts affords powerful oxidants. | |
3.8 Selectfluor®
A Minisci coupling reaction using Selectfluor® (22h) as a HAT reagent precursor was reported (Schemes 246 and 247).278,279 It was proposed, with irradiation with blue light (427 nm, 40 W) that 22h was converted to HAT reagent, TEDA2+˙ (22h*) via homolytic bond cleavage. Hydrogen atom abstraction from ether substrate 905a with 22h* resulted in the formation of alkyl radical 906a and protonated species 22h+. Addition of alkyl radical 906a to protonated heterocycle 907a resulted in the formation of a new C–C bond giving 908a. Proton loss from 908a gave carbon-centred radical 909a. SET oxidation of 909a with another equivalent of 22h and aromatisation resulted in Minisci coupled product 910a. From light on/off experiments, it was concluded that a radical-chain mechanism was unlikely for this process.
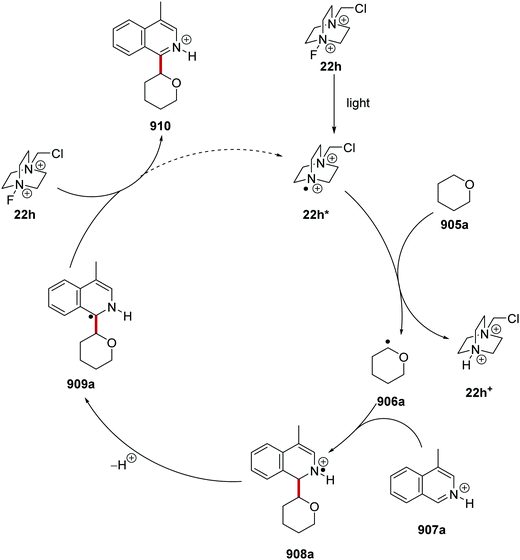 |
| | Scheme 246 Minisci reaction with Selectfluor. | |
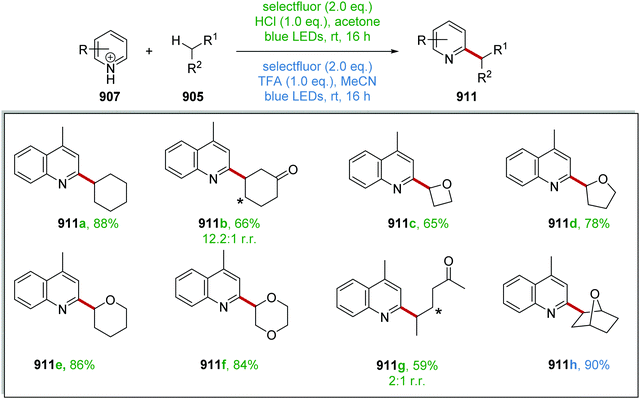 |
| | Scheme 247 Selectfluor Minisci reaction. | |
From the use of two different reaction conditions, a wide range of Minisci coupled products 911 was prepared (Scheme 247). It was found that alkanes, ketones, and ethers were all effective coupling partners for N-based heterocycles 907 and this allowed functionalised heterocyclic compounds 911 to be isolated efficiently. The use of cyclohexenone and 2-hexenone led to compounds 911b and 911g being isolated as mixtures of regioisomers. However, the use of ethers gave a highly regioselective reaction with 911c–f being formed as single regioisomers.
From competition experiments, it was shown that the reaction propagated with a higher rate of reaction for benzylic and α-hetero C–H bonds than alkane C–H bonds (Scheme 248).278 Functionalisation was quicker for benzylic C–H bonds than α-heteroatom C–H bonds.
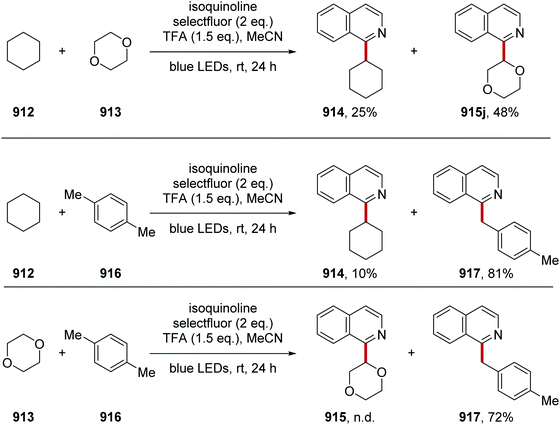 |
| | Scheme 248 Competition experiments. | |
3.9 Thiocarbamate catalyst
Benzylic C–H functionalisation.
Radical carbon–carbon bond formation via a Giese-type addition was achieved with a thiocarbamate nucleophilic catalyst 918 (Scheme 249).280 This reaction was unlike previous radical coupling strategies as it did not rely upon redox potentials of substrates, BDEs or triplet energies. The use of 918 as catalyst allowed for coupling of compounds with suitable leaving groups, such as benzyl chloride 919a with electron-poor alkenes such as dimethyl fumarate 920a and this gave diester 921a. Reaction between catalyst 918 and benzyl chloride 919a gave indole derivative 922a. The UV-vis absorbance of indole 922a showed strong absorbance (λmax = 344 nm, ε344 = 18400 M−1 cm−1) and could absorb light in the visible region. Irradiation of the reaction mixture with blue light (465 nm) LED led to homolytic bond cleavage of 922a and radicals 923a and 924 were produced. Addition of benzylic radical 923a to alkene 920a yielded α-ester radical 925a. γ-Terpinene (926) was used as a cost-effective replacement for 1,4-cyclohexadiene as hydrogen atom source and reductant. HAT to 925a from 926 gave the isolated diester 921a as product and radical 927. The nucleophilic catalytic cycle was closed with a SET from 927 to radical 924 and reformed original catalytic species 918 and cation 928, which, after proton loss, yielded aryl derivative 929.
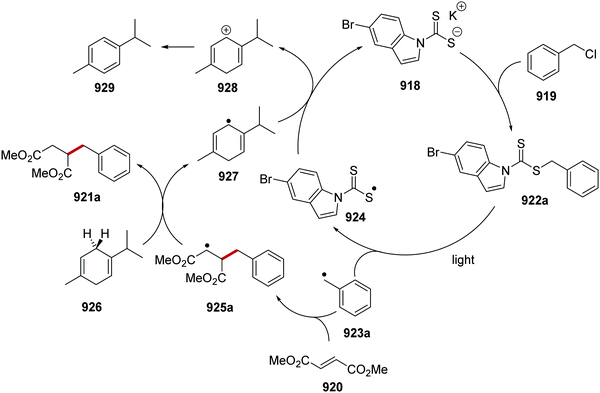 |
| | Scheme 249 The chemistry of nucleophilic catalyst 918. | |
The preparation of diester 921a from benzyl chloride 919a and dimethyl fumarate 920a was chosen as a model reaction in developing optimised conditions. As mentioned above, γ-terpinene (1.2 eq.) 926 was chosen as 1,4-CHD replacement and 2,6-lutidine was chosen as base. Originally, potassium ethyl xanthogenate (20 mol%) was used as a nucleophilic catalyst with irradiation at 400 nm but this delivered 921a in 19% NMR yield. When more red-shifted light was used (465 nm), no product formation was observed. It was thought that incorporation of a chromophore onto the nucleophilic catalyst would lead to greater conversion to product. Indole 922a had far superior absorbance than the ethyl xanthogenate analogue and this did lead to greater product formation. The combination of 918 with benzyl chloride (1.5 eq.) and 920 gave 921a in 43% NMR yield when conducted at room temperature and irradiation with 465 nm light. The yield of 921a was increased to 90% NMR yield (73% isolated yield, 8.6 g scale) when the temperature was increased to 60 °C (Scheme 250).
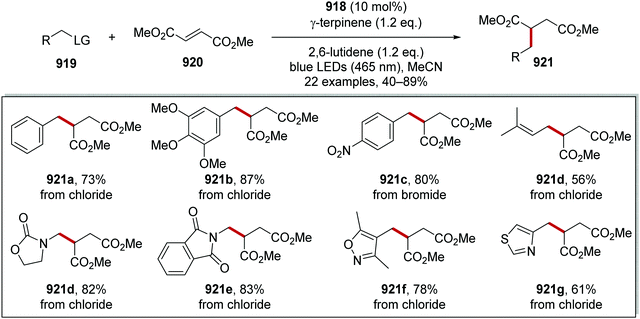 |
| | Scheme 250 Coupling products arising through use of catalyst 918. | |
An investigation into other leaving groups was carried out. It was found when benzyl bromide was used instead of benzyl chloride, a yield of 94% was achieved for 921a. Benzyl iodide gave 85% yield for 921a and benzyl mesylate gave 921a in 91% yield. Other benzylic halogens were successful substrates for this process and compounds 921b and 921c were prepared. When prenyl chloride was used in this transformation, this gave 921d in 56% yield. Both aliphatic and aromatic heterocycles were used in this transformation and this gave compounds 921d–g.
The use of stoichiometric sacrificial cyclohexadienyl radicals was avoided when the reactions were performed with acrylamides 930 and electron-rich heterocycles 931 (Scheme 251). When chloroacetonitrile was treated with various acrylamides under optimised reaction conditions, compounds 932a–c were prepared. Ethyl bromofluoroacetate was an excellent substrate for this transformation and ester compound 932d was prepared in 97% yield from it. Compound 921e was afforded from 2-chloroacetophenone in 68% yield. When 2-bromo-1-(3′-bromophenyl)ethan-1-one was treated with N-methylpyrrole under the reaction conditions this yielded ketone 933a in 65% yield. N-Methylpyrrole was also reacted with other alkyl halides including α-bromo-γ-butyrolactone (933b) and chloroacetonitrile (933d) and this afforded pyrrole products. Other heterocycles were also amenable to these reaction conditions; for example, furfuryl alcohol was transformed to 933e with chloroacetonitrile in 50% yield.
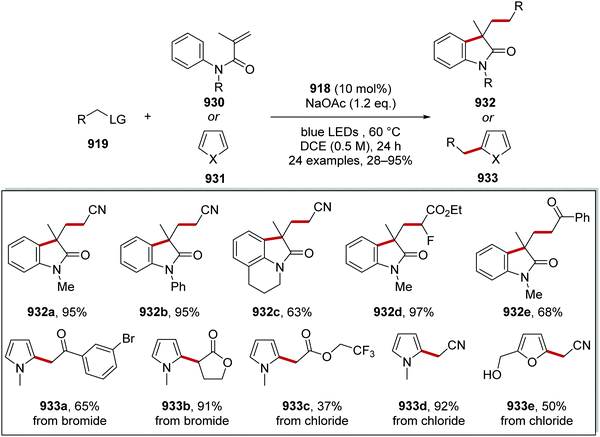 |
| | Scheme 251 Tandem reactions induced by catalyst 918. | |
3.10 Charge transfer complexes
Most of the reactions detailed so far have been accomplished by the addition of external photoredox catalysts to the reaction. However, a number of chemists, notably the group of Melchiorre, have focused on the formation of charge transfer complexes when the reactants are mixed. In these cases, photoactivation can lead to full electron transfer, thereby triggering a chemical reaction.
α-Aminosilane as radical precursor.
Organic chiral catalyst 937 allowed for the coupling of enones 934 to α-silyl amines 935 which gave ketones 936 without any photocatalyst (Scheme 252).281 This was achieved via an intramolecular iminium-ion-based electron donor–acceptor complex. Condensation between enones 934 and chiral catalyst 937 under acidic conditions resulted in a chiral iminium ion that contained an electron-rich carbazole moiety and an electron-poor alkene. A light-promoted electron transfer to the alkene from the carbazole gave an intermediate that featured both an allylic radical and a carbazole radical cation. Adjacent silyl groups lower the redox potential of amines. Therefore, the use of the right α-silylamine compound would participate in inter-molecular SET with the carbazole radical cation. Dimethyl(phenyl)silyl groups (Eox = +0.97 V vs. Ag/Ag+ in MeCN for α-dimethyl(phenyl)silyl carbazole) were successful at participating in SET with the carbazole radical cation (Ered = +1.09 V vs. Ag/Ag+ in MeCN), this allowed for α-aminoalkyl radical formation and propagation of the reaction. The reaction was more efficient when the catalyst had more electron-withdrawing groups present and thus, from a screen of five catalysts, catalyst 937 was the most effective. Employment of the optimal reaction conditions gave a library of nine aniline products 936a–i in yields of 57–83%.
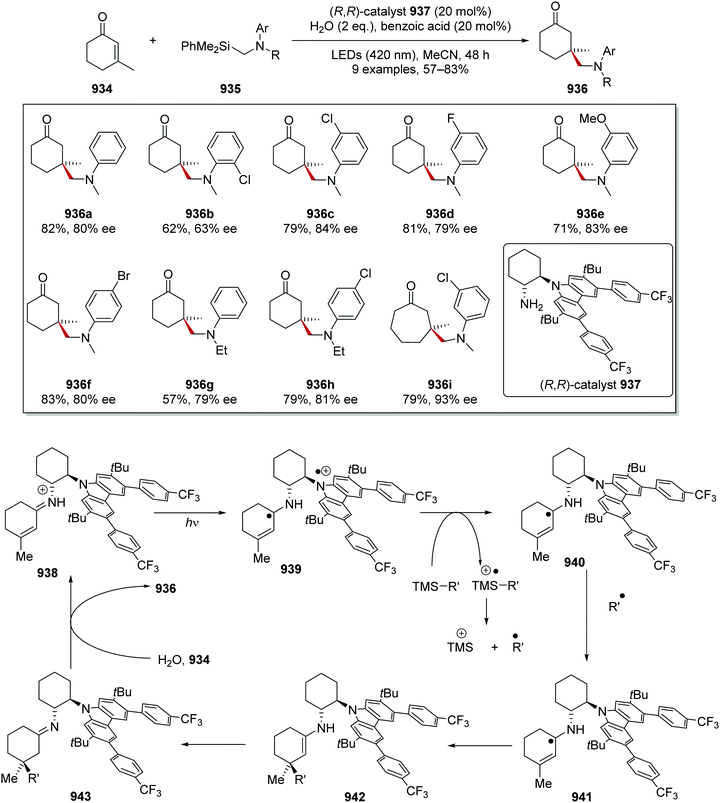 |
| | Scheme 252 Enantioselective radical addition reactions controlled by diamine 937. | |
Even one nitrogen atom attached to an alkene gives interesting electron donating properties to enamines, notably when photoexcited. Melchiorre et al.282 examined the enantioselective α-methylation and benzylation of aldehydes. (Scheme 253) Such reactions had been previously promoted by TM-based photoredox agents,283 but Melchiorre's contribution was to bring about the reactions in their absence. Taking butyraldehyde 944 as an example, reaction with an enantiomerically pure secondary amine 952 creates the corresponding enamine 947. When the enamine was excited, a strongly reducing species was formed (Eox* = ca. −2.0 V vs. Ag/AgCl) and this was able to reduce sulfone 945 (Ered = −1.49 V vs. Ag/AgCl) to form the radical 948. [The authors discuss the possibility of excitsation of the individual species, as well as excitation of a charge-transfer complex formed from the enamine and the iodosulfone.] The resulting radical 948 added to the enamine in a stereocontrolled manner to give α-aminoalkyl radical 949. The reducing power of 949 (Eox = −0.95 V vs. Ag/AgCl) was insufficient of reducing 945 therefore it was proposed that a halogen atom abstraction process was operative to continue the radical chain process (ϕ = 3.8). The iminium salt was hydrolysed in situ to provide product 951. For ease of isolation, the product was reduced with NaBH4 to the corresponding alcohol. Products 946a–d exemplify high yields and stereoselectivity.
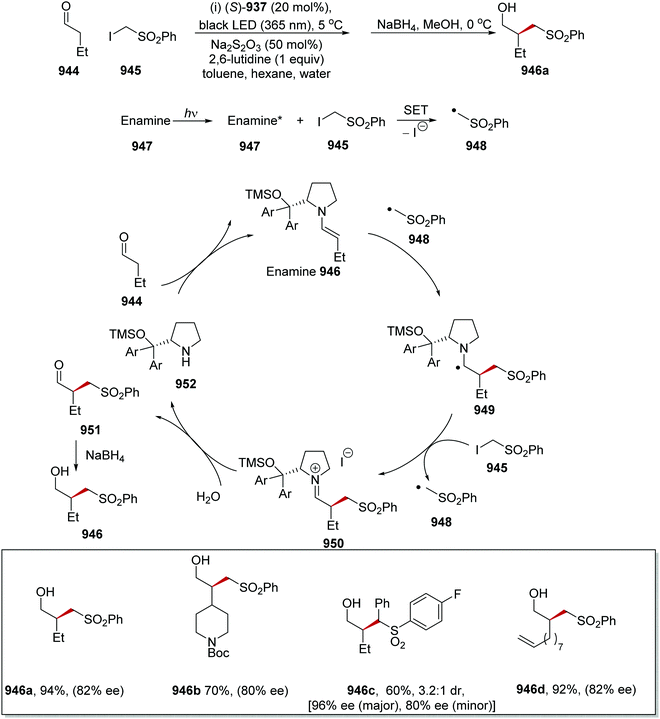 |
| | Scheme 253 Enantioselective alkylation of aldehydes. | |
Melchiorre also demonstrated electron transfer properties for the dienamine (dihydropyridine) 953 (Scheme 254). Here, the excited state chromophore 956 (Eox* = ca. −1.1 V vs. SCE) should not be able to transfer an electron to compound 957 (Ered = −1.32 V vs. SCE).284 Instead it was thought that electron transfer to pyridinium ion 961 (Ered = −1.01 V vs. SCE), which could be formed with photolysis from 953, was more feasible. The dihydropyridine radical cation 959 underwent bond fragmentation and acyl radical 960 was delivered. Radical attack of 960 to protonated heterocycle 957 resulted in C–C bond formation. The adduct 962 easily undergoes deprotonation on carbon to give radical 963. Protonation on oxygen affords ketyl radical 964, which can receive an electron and a proton to give 965, the protonated form of isolated product 955. Hence the process does not follow the normal Minisci pathway, providing an hydroxyalkyl substituted heterocycle, rather than the acyl-substituted counterpart. Products 955a–c illustrate examples of outcomes from analogous substrates.
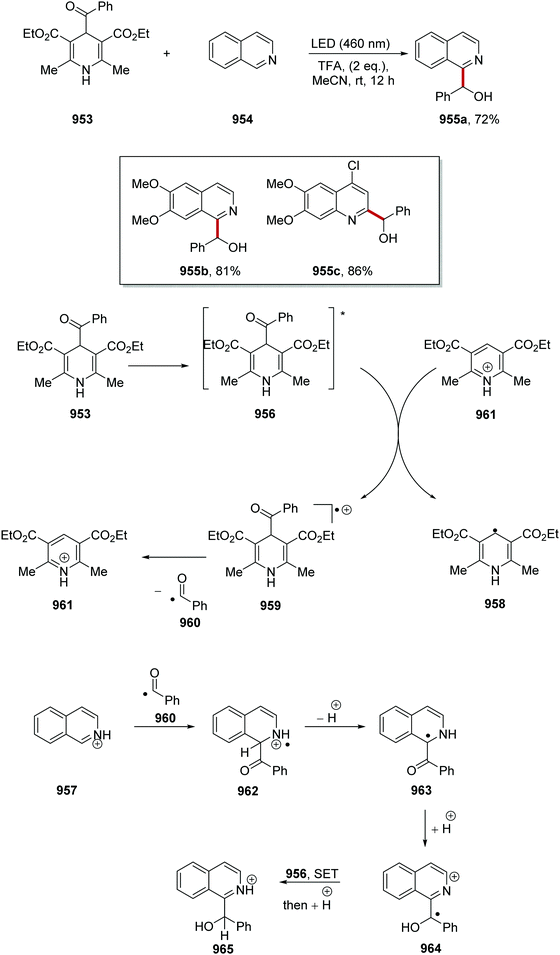 |
| | Scheme 254 A modified hydroxyalkylation Minisci process from scission of photoactivated 953. | |
A final example285 illustrates the use of charge-transfer complexes for the C2 alkylation of tryptophan residues 966 (Scheme 255). Here pyridinium salts 967 act as electron-poor π-systems that can complex with the electron-rich π-system of indoles, as in tryptophan. Visible light triggers electron transfer to the charge-transfer complex, giving an indole radical cation and a pyridinyl radical that cleaves to give the parent pyridine and an alkyl radical. Under appropriate conditions, the radical reacts selectively attacking the 2-position of the indole radical cation. Subsequent proton loss affords the substituted tryptophan product 968.
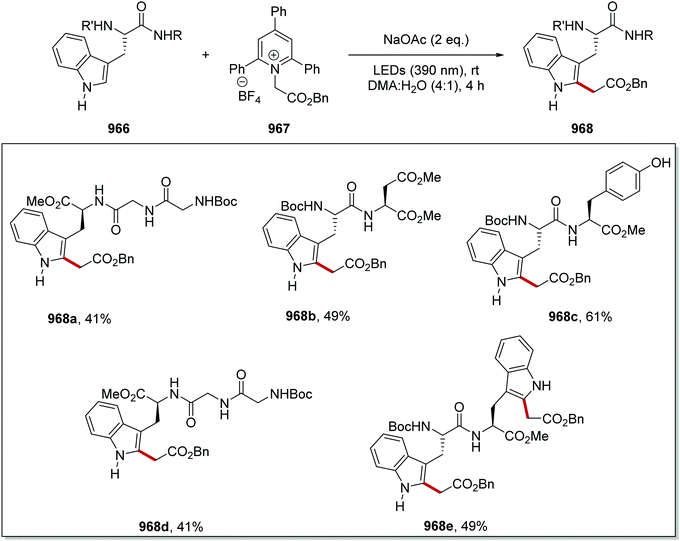 |
| | Scheme 255 Stereocontrolled alkylation at C-2 of indoles. | |
4 Conclusions
Over the past ten years considerable progress has been made in radical mediated coupling reactions exploiting PET, HAT and energy transfer processes. During that period, ruthenium and iridium photocatalysts have led the way for transition metal based redox reagents and have resulted in an astonishing number of research papers, addressing a wide variety of reactions. Perhaps because of concerns about the sustainability of supplies of some transition metals,65 but also about management of waste-streams in pharmaceutical chemistry, organic photoredox catalysts have come to the fore, and are proving capable of attaining very challenging potentials, both reductive and oxidative. Photoactivation of cationic and anionic organic pi-systems has produced really reactive redox active molecules, and coupling of photoactivation with other methodologies, e.g. electrochemistry, has begun to create diverse methods of access these. The study of reactions between reagents which, although each is individually unreactive to stimulus by visible light, form a charge-transfer complex that undergoes full electron transfer upon irradiation by visible light, is another area where development is accelerating. Finally, a number of cases of photoredox catalysis that provide excellent levels of asymmetry will simulate much interest in the coming decade. The pursuit of more sustainable reagents for radical promoted coupling reactions should allow further uptake of these strategies in particular late-stage functionalisation of pharmaceutical compounds.25
Abbreviations
|
A
| Pre-exponential factor |
| ATRE | Atom transfer radical addition–elimination |
| BCP | Bicyclopentane |
| BDC |
tert-Butyl N,N-dimethylcarbamate |
| BDE | Bond dissociation energy |
| BDFE | Bond dissociation free energy |
| BET | Back electron transfer |
| Boc |
tert-Butyloxycarbonyl |
| Bn | Benzyl |
| BP | Biphenyl |
| Bpy | Bipyridyl |
| Bpz | Bipyrazyl |
| Bz | Benzoyl |
| Cbz | Carboxybenzyl |
| ConPET | Consecutive photoelectron transfers |
| Cz | Carbazolyl |
| 4CzIPN | Tetracarbazolylisophthalonitrile |
| DABCO | Diazabicyclooctane |
| DCA | 9,10-Dicyanoanthracene |
| DCE | 1,2-Dichloroethane |
| DFT | Density functional theory |
| DMA |
N,N-Dimethylacetamide |
| DMF |
N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DIC |
N,N′-Diisopropylcarbodiimide |
| DTA |
N,N-Dimethyltrifluoroacetamide |
| Dtbbpy | Di-tert-butylbipyridyl |
| dr | Diastereomeric ratio |
| Ea | Activation energy |
| EDA | Electron-donor–acceptor |
| ee | Enantiomeric excess |
| er | Enantiomeric ratio |
| EWG | Electron-withdrawing group |
| FRET | Förster resonance energy transfer |
| Fmoc | Fluorenylmethoxycarbonyl |
| HAT | Hydrogen atom transfer |
| HFIP | Hexafluoroisopropanol |
| HPLC | High-performance liquid chromatography |
| IC | Internal conversion |
| ISC | Inter-system crossing |
| ISET | Inner-sphere electron transfer |
| HAT | Hydrogen atom transfer |
| HLF | Hofmann–Löffler–Freytag |
| KIE | Kinetic isotope effect |
| LED | Light emitting diode |
| MLCT | Metal–ligand charge transfer |
| MTBE | Methyl tert-butyl ether |
| NCS |
N-Chlorosuccinimide |
| OSET | Outer-sphere electron transfer |
| PDI | Perylenediimide |
| PET | Positron emission tomography or Photoelectron transfer |
| Ppy | Phenylpyridyl |
| SCE | Saturated calomel electrode |
| SCS | Spin-centred-shift |
| SET | Single electron transfer |
| SOMO | Singly occupied molecular orbital |
| TBACl | Tetra-n-butylammonium chloride |
| TCAA | Trichloroacetic acid |
| Tf | Trifluoromethanesulfonyl |
| THIQ | Tetrahydroisoquinolinyl |
| TM | Transition metal |
| TMG | 1,1,3,3-Tetramethylguanidine |
| TMHD | 2,2,6,6-Tetramethylheptanedione |
| TMS | Trimethylsilyl |
| PCET | Proton-coupled electron transfer |
| PET | Photoinduced electron transfer |
| RCY | Radiochemical yield |
| RDS | Rate determining step |
| rr | Ratio of regioisomers |
| UPLC | Ultra-performance liquid chromatography |
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank the EPSRC for funding via EPSRC Prosperity Partnership EP/S035990/1.
References
-
(a) M. R. Heinrich, Chem. – Eur. J., 2009, 15, 820–833 CrossRef CAS PubMed;
(b) S. Kindt and M. R. Heinrich, Synthesis, 2016, 1597–1606 CAS;
(c) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed;
(d) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- W. Buratti, G. P. Gardini, F. Minisci, F. Bertini, R. Galli and M. Perchinunno, Tetrahedron, 1971, 27, 3655–3668 CrossRef CAS.
- F. Minisci, A. Citterio, E. Vismara and C. Giordano, Tetrahedron, 1985, 41, 4157–4170 CrossRef CAS.
-
(a) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753–764 CrossRef;
(b) B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef;
(c) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef CAS;
(d) T. Kawamoto and I. Ryu, Org. Biomol. Chem., 2014, 12, 9733–9742 RSC;
(e) J. Streuff and A. Gansäuer, Angew. Chem., Int. Ed., 2015, 54, 14232–14242 CrossRef CAS PubMed;
(f) Q. Huang, S. R. Suravarapu and P. Renaud, Chem. Sci., 2021, 12, 2225–2230 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- B. P. Roberts and A. J. Steel, J. Chem. Soc., Perkin Trans. 2, 1994, 2155–2162 RSC.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- Selected reviews, general:
(a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed;
(b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed;
(c) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1(special issue), 111–129 CrossRef CAS;
(d) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- Selected recent reviews, more specific:
(a) M. Schmalzbauer, M. Marcon and B. König, Angew. Chem., Int. Ed., 2021, 60, 6270–6293 CrossRef CAS PubMed;
(b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed;
(c) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed;
(d) F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS;
(e) R. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed;
(f) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC;
(g) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC;
(h) A. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC;
(i) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed;
(j) N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC;
(k) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed.
- J. C. Del Valle and J. Catalán, Phys. Chem. Chem. Phys., 2019, 21, 10061–10069 RSC.
- A. P. Demchenko, V. I. Tomin and P. T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- B. Heinz, B. Schmidt, C. Root, H. Satzger, F. Milota, B. Fierz, T. Kiefhaber, W. Zinth and P. Gilch, Phys. Chem. Chem. Phys., 2006, 8, 3432–3439 RSC.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- S. Yamauchi and D. W. Pratt, Mol. Phys., 1979, 37, 541–569 CrossRef CAS.
- N. Hirota, M. Baba, Y. Hirata and S. Nagaoka, J. Phys. Chem., 1979, 83, 3350–3354 CrossRef CAS.
- T. Itoh, Chem. Phys. Lett., 1988, 151, 166 CrossRef CAS.
- M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833–857 CrossRef CAS PubMed.
- Z. S. Romanova, K. Deshayes and P. Piotrowiak, J. Am. Chem. Soc., 2001, 123, 2444–2445 CrossRef CAS PubMed.
- L. M. Hancock, E. Marchi, P. Ceroni and P. D. Beer, Chem. – Eur. J., 2012, 18, 11277–11283 CrossRef CAS PubMed.
- A. Seret, E. Gandin and A. Van De Vorst, Chem. Phys. Lett., 1987, 135, 427–431 CrossRef CAS.
- I. R. Gould and S. Farid, Acc. Chem. Res., 1996, 29, 522–528 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC.
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848–5958 CrossRef CAS PubMed.
- A. Pross, Acc. Chem. Res., 1985, 18, 212–219 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
-
IUPAC, Compendium of Chemical Terminology, 2nd edn (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford ( 1997), Online version (2019-) created by S. J. Chalk, ISBN 0-9678550-9-8, DOI:10.1351/goldbook.
- H. G. Roth, N. A. Romero and D. Nicewicz, Synlett, 2016, 714–723 CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 827–832 CAS.
-
(a) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed;
(b) J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed;
(c) K. Chen, J. Schwarz, T. A. Karl, A. Chatterjee and B. König, Chem. Commun., 2019, 55, 13144–13147 RSC;
(d) H. Cheng, T.-L. Lam, Y. Liu, Z. Tang and C.-M. Che, Angew. Chem., Int. Ed., 2021, 60, 1383–1389 CrossRef CAS PubMed;
(e) A. A. Isse and A. Gennaro, Collect. Czech. Chem. Commun., 2003, 68, 1379–1394 CrossRef CAS;
(f) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- R. A. Aycock, C. J. Pratt and N. T. Jui, ACS Catal., 2018, 8, 9115–9119 CrossRef CAS.
- J. P. Barham, M. P. John and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 15482–15487 CrossRef CAS PubMed.
- D. D. M. Wayner, K. B. Clark, A. Rauk, D. Yu and D. A. Armstrong, J. Am. Chem. Soc., 1997, 119, 8925–8932 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- J. Berkowitz, G. B. Ellison and D. Gutman, J. Phys. Chem., 1994, 98, 2744–2765 CrossRef CAS.
- F. G. Bordwell, S. Zhang, X.-M. Zhang and W.-Z. Liu, J. Am. Chem. Soc., 1995, 117, 7092–7096 CrossRef CAS.
- W.-Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778–4783 CrossRef CAS PubMed.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- D. Hager and D. W. C. Macmillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS PubMed.
- R. W. Alder, R. J. Arrowsmith, A. Casson, R. B. Sessions, E. Heilbronner, B. Kovač, H. Huber and M. Taagepera, J. Am. Chem. Soc., 1981, 103, 6137–6142 CrossRef CAS.
-
(a) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed;
(b) T. Toshili and N. Atsushi, Chem. Lett., 2009, 38, 160–161 CrossRef.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
-
S. Charaya, PhD thesis, New Jersey Institute of Technology, 2011.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed.
- K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed.
-
(a) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef CAS PubMed;
(b) C. Zhou, T. Lei, X.-Z. Wei, C. Ye, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142, 16805–16813 CrossRef CAS PubMed.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
- H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708–5715 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed.
- P. A. Tanner, L. Zhou, C. Duan and K.-L. Wong, Chem. Soc. Rev., 2018, 47, 5234–5265 RSC.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- Z. Q. You, C. P. Hsu and G. R. Fleming, J. Chem. Phys., 2006, 124, 044506 CrossRef PubMed.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, H. J. Van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 127, 16054–16064 CrossRef CAS PubMed.
- E. Kumarasamy, S. K. Kandappa, R. Raghunathan, S. Jockusch and J. Sivaguru, Angew. Chem., Int. Ed., 2017, 56, 7056–7061 CrossRef CAS PubMed.
- L. M. Kammer, B. Lipp and T. Opatz, J. Org. Chem., 2019, 84, 2379–2392 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830–15841 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803, DOI:10.1038/s41467-019-13887-8.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed.
- V. De Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef.
- T. Basile, L. Capaldo, D. Ravelli and P. Quadrelli, Eur. J. Org. Chem., 2020, 1443–1447 CrossRef CAS.
- J. R. Clark, K. Feng, A. Sookezian and M. C. White, Nat. Chem., 2018, 10, 583–591 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- S. S. Shah, A. Paul, M. Bera, Y. Venkatesh and N. D. P. Singh, Org. Lett., 2018, 20, 5533–5536 CrossRef CAS PubMed.
- A. B. Rolka and B. König, Org. Lett., 2020, 22, 5035–5040 CrossRef CAS PubMed.
- D. Jespersen, B. Keen, J. I. Day, A. Singh, J. Briles, D. Mullins and J. D. Weaver III, Org. Process Res. Dev., 2019, 23, 1087–1095 CrossRef CAS PubMed.
- S. Blanc, T. Pigot, C. Cugnet, R. Brown and S. Lacombe, Phys. Chem. Chem. Phys., 2010, 12, 11280–11290 RSC.
- F. A. Bell, A. Ledwith and D. C. Sherrington, J. Chem. Soc. C, 1969, 2719–2720 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. R. Talipov, M. M. Hossain, A. Boddeda, K. Thakur and R. Rathore, Org. Biomol. Chem., 2016, 14, 2961–2968 RSC.
- M. Schorpp, T. Heizmann, M. Schmucker, S. Rein, S. Weber and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 9453–9459 CrossRef CAS PubMed.
- S.-i. Kato, T. Matsuoka, S. Suzuki, M. S. Asano, T. Yoshihara, S. Tobita, T. Matsumoto and C. Kitamura, Org. Lett., 2020, 22, 734–738 CrossRef CAS PubMed.
- T. A. Schaub, T. Mekelburg, P. O. Dral, M. Miehlich, F. Hampel, K. Meyer and M. Kivala, Chem. – Eur. J., 2020, 26, 3264–3269 CrossRef CAS PubMed.
-
(a)
H. Jakob, S. Leininger, T. Lehmann, S. Jacobi and S. Gutewort, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, pp. 503–519 Search PubMed;
(b) S. Härtinger, J. Rosenmund, E. Savinova, S. Wasle and K. Doblhofer, J. Phys. Chem. B, 1997, 101, 2411–2414 CrossRef.
- E. J. Behrman, Beilstein J. Org. Chem., 2006, 2, 22 CAS.
- M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A. L. Lee, Org. Lett., 2019, 21, 7119–7123 CrossRef CAS PubMed.
- C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425–4431 CrossRef CAS PubMed.
- D. R. Sutherland, M. Veguillas, C. L. Oates and A.-L. Lee, Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed.
- V. Piano, B. A. Palfey and A. Mattevi, Trends Biochem. Sci., 2017, 42, 457–469 CrossRef CAS PubMed.
-
J. B. Metternich, R. J. Mudd and R. Gilmour, in Photocatalysis in Organic Synthesis, ed. B. Koenig, Georg Thieme Verlag, Stuttgart, 2019, vol. 6, pp. 391–404 Search PubMed.
-
(a) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed;
(b) D. M. Schultz, J. W. Sawicki and T. P. Yoon, Beilstein J. Org. Chem., 2015, 11, 61–65 CrossRef PubMed.
- A. H. Tolba, F. Vávra, J. Chudoba and R. Cibulka, Eur. J. Org. Chem., 2020, 1579–1585 CrossRef CAS.
- S. L. J. Tan and R. D. Webster, J. Am. Chem. Soc., 2012, 134, 5954–5964 CrossRef CAS PubMed.
- R. Martinez-Haya, M. A. Miranda and M. L. Marin, Eur. J. Org. Chem., 2017, 2164–2169 CrossRef CAS.
- B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef.
- D. B. Gerth and B. Giese, J. Org. Chem., 1986, 51, 3726–3729 CrossRef CAS.
- N. Miyamoto, D. Fukuoka, K. Utimoto and H. Nozaki, Bull. Chem. Soc. Jpn., 1974, 47, 503–504 CrossRef CAS.
- J. Xiang, W. Jiang, J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 4123–4129 CrossRef CAS.
- F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 CrossRef CAS PubMed.
- X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220–226 CrossRef CAS PubMed.
- E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC.
- M. J. Cabrera-Afonso, Z.-P. Lu, C. B. Kelly, S. B. Lang, R. Dykstra, O. Gutierrez and G. A. Molander, Chem. Sci., 2018, 9, 3186–3191 RSC.
- M. L. Agazzi, V. A. S. Almodovar, N. S. Gsponer, S. Bertolotti, A. C. Tomé and E. N. Durantini, Org. Biomol. Chem., 2020, 18, 1449–1461 RSC.
- J. Fischer, P. Nun and V. Coeffard, Synthesis, 2020, 1617–1624 CAS.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Nat. Chem., 2019, 11, 1034–1040 CrossRef CAS PubMed.
- E. M. Sherbrook, H. Jung, D. Cho, M. H. Baik and T. P. Yoon, Chem. Sci., 2020, 11, 856–861 RSC.
- Z. Zhang, C. R. Rogers and E. A. Weiss, J. Am. Chem. Soc., 2020, 142, 495–501 CrossRef CAS PubMed.
- A. Hossain, S. K. Pagire and O. Reiser, Synlett, 2017, 1707–1714 CAS.
-
(a) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed;
(b) Q. Yang, L. Zhang, C. Ye, S. Lou, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed.
- D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94–97 CrossRef CAS PubMed.
- G. Bergonzini, C. S. Schindler, C. J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC.
- L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef CAS PubMed.
- J. Yoshida, T. Maekawa, T. Murata, S. Matsunaga and S. Isoe, J. Am. Chem. Soc., 1990, 112, 1962–1970 CrossRef CAS.
- Z.-J. Wang, S. Zheng, J. K. Matsui, Z. Lu and G. A. Molander, Chem. Sci., 2019, 10, 4389–4393 RSC.
- M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed.
- B. A. Vara, X. Li, S. Berritt, C. R. Walters, E. J. Petersson and G. A. Molander, Chem. Sci., 2018, 9, 336–344 RSC.
- A. G. Amador, E. M. Sherbrook and T. P. Yoon, J. Am. Chem. Soc., 2016, 138, 4722–4725 CrossRef CAS PubMed.
- J. M. Tanko and R. E. Drumright, J. Am. Chem. Soc., 1992, 114, 1844–1854 CrossRef CAS.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS PubMed.
- L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS PubMed.
- E. R. Welin, A. A. Warkentin, J. C. Conrad and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 9668–9672 CrossRef CAS PubMed.
- X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
-
(a) A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11, 426–433 CrossRef CAS PubMed;
(b) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC;
(c) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254–257 CrossRef CAS PubMed.
- S. L. Rössler, B. J. Jelier, P. F. Tripet, A. Shemet, G. Jeschke, A. Togni and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 526–531 CrossRef PubMed.
- W. S. Ham, J. Hillenbrand, J. Jacq, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 532–536 CrossRef CAS PubMed.
- H. Jung, H. Keum, J. Kweon and S. Chang, J. Am. Chem. Soc., 2020, 142, 5811–5818 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- A. Noble, S. J. McCarver and D. W. C. Macmillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 CrossRef CAS PubMed.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. Macmillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed.
- C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 11938–11941 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 13862–13865 CrossRef CAS PubMed.
- J. H. Kim, A. Ruffoni, Y. S. S. Al-Faiyz, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2020, 59, 8225–8231 CrossRef CAS PubMed.
-
(a) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966–6968 RSC;
(b) B. E. Cooper and W. J. Owen, J. Organomet. Chem., 1971, 29, 33–40 CrossRef CAS;
(c) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed.
- A. Casado-Sánchez, P. Domingo-Legarda, S. Cabrera and J. Alemán, Chem. Commun., 2019, 55, 11303–11306 RSC.
- M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254–258 CrossRef CAS PubMed.
- D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef CAS PubMed.
-
(a) J. K. Matsui, Á. Gutiérrez-Bonet, M. Rotella, R. Alam, O. Gutierrez and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 15847–15851 CrossRef CAS PubMed;
(b) Y. Nishigaichi, T. Orimi and A. Takuwa, J. Organomet. Chem., 2009, 694, 3837–3839 CrossRef CAS.
- M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed.
- P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS PubMed.
- T. Q. Chen and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2019, 58, 14584–14588 CrossRef CAS PubMed.
- R. T. Smith, X. Zhang, J. A. Rincón, J. Agejas, C. Mateos, M. Barberis, S. García-Cerrada, O. de Frutos and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 17433–17438 CrossRef CAS PubMed.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
-
(a) H. Tian, Q. Xia, Q. Wang, J. Dong, Y. Liu and Q. Wang, Org. Lett., 2019, 21, 4585–4589 CrossRef CAS PubMed;
(b) J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS PubMed.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- M. N. Hopkinson, A. Gómez-Suárez, M. Teders, B. Sahoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 4361–4366 CrossRef CAS PubMed.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 2886–2893 CrossRef CAS.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed.
- J. C. Day, N. Govindaraj, D. S. McBain, P. S. Skell and J. M. Tanko, J. Org. Chem., 1986, 51, 4959–4963 CrossRef CAS.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS PubMed.
- L. Leng, Y. Fu, P. Liu and J. M. Ready, J. Am. Chem. Soc., 2020, 142, 11972–11977 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed.
- R. D. Trepka, J. W. Belisle and J. K. Harrington, J. Org. Chem., 1974, 39, 1094–1098 CrossRef CAS.
- V. K. Aggarwal, I. Emme and S. Y. Fulford, J. Org. Chem., 2003, 68, 692–700 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed.
- C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC.
- J. J. Douglas, K. P. Cole and C. R. J. Stephenson, J. Org. Chem., 2014, 79, 11631–11643 CrossRef CAS PubMed.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed.
- S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723–14726 CrossRef CAS PubMed.
- J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed.
- V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692–1696 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273–12276 CrossRef CAS PubMed.
- S. Zheng, Á. Gutiérrez-Bonet and G. A. Molander, Chem, 2019, 5, 339–352 CAS.
- G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed.
- H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed.
- A. R. Gregory, K. G. Kidd and G. W. Burton, THEOCHEM, 1983, 104, 9–22 CrossRef.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
-
(a) R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed;
(b) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS PubMed.
- K. Ermanis, A. C. Colgan, R. S. J. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 21091–21101 CrossRef CAS PubMed.
- Z. Zuo and D. W. C. Macmillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
-
(a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed;
(b) L. Wei, M. Yuan, Y. Yingwu and Z. Yufen, Bull. Chem. Soc. Jpn., 2006, 79, 577–579 CrossRef.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- E. A. Mayeda, L. L. Miller and J. F. Wolf, J. Am. Chem. Soc., 1972, 94, 6812–6816 CrossRef CAS.
- C. Bosset, H. Beucher, G. Bretel, E. Pasquier, L. Queguiner, C. Henry, A. Vos, J. P. Edwards, L. Meerpoel and D. Berthelot, Org. Lett., 2018, 20, 6003–6006 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- S. Escoubet, S. Gastaldi, N. Vanthuyne, G. Gil, D. Siri and M. P. Bertrand, J. Org. Chem., 2006, 71, 7288–7292 CrossRef CAS PubMed.
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219–2232 CrossRef CAS.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed.
- M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095, DOI:10.1038/s41467-019-13072-x.
- M. Yoshida, H. Sakuragi, T. Nishimura, S. Ishikawa and K. Tokumaru, Chem. Lett., 1975, 1125–1130 CrossRef CAS.
- T. Ni, R. A. Caldwell and L. A. Melton, J. Am. Chem. Soc., 1989, 111, 457–464 CrossRef CAS.
- A. Padwa, W. Bergmark and D. Pashayan, J. Am. Chem. Soc., 1969, 91, 2653–2660 CrossRef CAS.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- Our review has considered visible light activations of the acridinium skeleton to provide a strong oxidant. Intermediate 798 can be activated to give a powerful reductant on irradiation at 360 nm. I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- D. P. Hari, P. Schroll and B. Koenig, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed.
- T. Xiao, X. Dong, Y. Tang and L. Zhou, Adv. Synth. Catal., 2012, 354, 3195–3199 CrossRef CAS.
- D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334–5337 CrossRef CAS PubMed.
-
(a) M. Majek and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 2270–2274 CrossRef CAS PubMed;
(b) J. Liu, Y. Wei and M. Shi, Chin. J. Chem., 2021, 39, 295–300 CrossRef CAS.
- W. Guo, L.-Q. Lu, Y. Wang, Y.-N. Wang, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 2265–2269 CrossRef CAS PubMed.
- L. Gu, C. Jin and J. Liu, Green Chem., 2015, 54, 3733–3736 RSC.
- S. A. Jadhav, D. Bakshi and A. Singh, J. Org. Chem., 2015, 80, 10187–10196 CrossRef CAS PubMed.
- A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
-
(a) M. Neumann and K. Zeitler, Chem. – Eur. J., 2013, 19, 6950–6955 CrossRef CAS PubMed;
(b) Y. Roh, H.-Y. Jang, V. Lynch, N. L. Bauld and M. J. Krische, Org. Lett., 2002, 4, 611–613 CrossRef CAS PubMed;
(c) H. O. House, L. E. Huber and M. J. Umen, J. Am. Chem. Soc., 1972, 94, 8471–8475 CrossRef CAS.
-
(a) G. Pandey and S. Hajra, Angew. Chem., Int. Ed. Engl., 1994, 33, 1169–1171 CrossRef;
(b) J. Yang, G. A. N. Felton, N. L. Bauld and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 1634–1635 CrossRef CAS PubMed.
-
(a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed;
(b) J. Du, L. R. Espelt, I. A. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115–2119 RSC.
- J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed.
-
(a) E. Yoshioka, S. Kohtani, T. Jichu, T. Fukazawa, T. Nagai, A. Kawashima, Y. Takemoto and H. Miyabe, J. Org. Chem., 2016, 81, 7217–7229 CrossRef CAS PubMed;
(b) I. N. Rozhkov, S. M. Igumnov, G. Y. Bekker, S. I. Pletnev, G. D. Rempel and L. E. Deev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 2041–2043 CrossRef.
- E. Yoshioka, S. Kohtani, T. Jichu, T. Fukazawa, T. Nagai, Y. Takemoto and H. Miyabe, Synlett, 2015, 265–270 CAS.
-
(a) D. P. Tiwari, S. Dabral, J. Wen, J. Wiesenthal, S. Terhorst and C. Bolm, Org. Lett., 2017, 19, 4295–4298 CrossRef CAS PubMed;
(b) X.-Q. Chu, D. Ge, M.-L. Wang, W. Rao, T.-P. Loh and Z.-L. Shen, Adv. Synth. Catal., 2019, 361, 4082–4090 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan, H. Ismailoi and J. C. Scaiano, ACS Catal., 2014, 4, 2530–2535 CrossRef CAS.
-
(a) I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed;
(b) S. Nishimoto, T. Izukawa and T. Kagiya, J. Chem. Soc., Perkin Trans. 2, 1983, 1147–1152 RSC.
- D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed.
- J. Rostoll-Berenguer, G. Blay, M. C. Muñoz, J. R. Pedro and C. Vila, Org. Lett., 2019, 21, 6011–6015 CrossRef CAS PubMed.
- Y. Kuang, K. Wang, X. Shi, X. Huang, E. Meggers and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. C. Read de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- A. F. Garrido-Castro, N. Salaverri, M. C. Maestro and J. Alemán, Org. Lett., 2019, 21, 5295–5300 CrossRef CAS PubMed.
-
(a) A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. Jui, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed;
(b) S. M. Soria-Castro, D. M. Andrada, D. A. Caminos, J. E. Argüello, M. Robert and A. B. Peñéñory, J. Org. Chem., 2017, 82, 11464–11473 CrossRef CAS PubMed.
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS.
- A. M. Martinez-Gualda, R. Cano, L. Marzo, R. Perez-Ruiz, J. Luis-Barrera, R. Mas-Balleste, A. Fraile, V. A. de la Pena O’Shea and J. Aleman, Nat. Commun., 2019, 10, 2634, DOI:10.1038/s41467-019-10441-4.
- D. Liu, V. Jiao, Z.-T. Feng, X.-Z. Wang, G.-Q. Xu and P.-F. Xu, Org. Lett., 2018, 20, 5700–5704 CrossRef CAS PubMed.
- F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed.
- S. Shibutani, T. Kodo, M. Takeda, K. Nagao, N. Tokunaga, Y. Sasaki and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 1211–1216 CrossRef CAS PubMed.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. G. Murali Dhar, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 RSC.
-
(a) H. Chen, W. Fan, X.-A. Yuan and S. Yu, Nat. Commun., 2019, 10, 4743, DOI:10.1038/s41467-019-12722-4;
(b) H. Chen, W. Jin and S. Yu, Org. Lett., 2020, 22, 5910–5914 CrossRef CAS PubMed.
- Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966–6968 RSC.
- C. Lévêque, L. Cheneberg, V. Corcé, C. Ollivier and L. Fensterbank, Chem. Commun., 2016, 52, 9877–9880 RSC.
- N. R. Patel, C. B. Kelly, A. P. Sigenfeld and G. A. Molander, ACS Catal., 2017, 7, 1766–1770 CrossRef CAS PubMed.
- M. Uygur, T. Danelzik and O. G. Mancheño, Chem. Commun., 2019, 55, 2980–2983 RSC.
- M. K. Jackl, L. Legnani, B. Morandi and J. W. Bode, Org. Lett., 2017, 19, 4696–4699 CrossRef CAS PubMed.
- A. Cartier, E. Levernier, V. Corcé, T. Fukuyama, A.-L. Dhimane, C. Ollivier, I. Ryu and L. Fensterbank, Angew. Chem., Int. Ed., 2019, 58, 1789–1793 CrossRef CAS PubMed.
- X. Wang, G. H. M. Davies, A. Koschitzky, S. R. Wisniewski, C. B. Kelly and G. A. Molander, Org. Lett., 2019, 21, 2880–2884 CrossRef CAS PubMed.
- A. Ryder, W. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS PubMed.
- M. J. Pellerite, R. L. Jackson and J. I. Brauman, J. Phys. Chem., 1981, 85, 1624–1626 CrossRef CAS.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed.
- C. Yang, J.-D. Yang, Y.-H. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 CrossRef CAS PubMed.
- G. Pandey and R. Laha, Angew. Chem., Int. Ed., 2015, 54, 14875–14879 CrossRef CAS PubMed.
- G. Pandey and S. Hajra, Angew. Chem., Int. Ed. Engl., 1994, 33, 1169–1171 CrossRef.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Majek, V. A. de la Pena O’Shea, A. Jacobi von Wangelin and R. Peres-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- N. Holmberg-Douglas, N. P. R. Onuska and D. A. Nicewicz, Angew. Chem., Int. Ed., 2020, 59, 7425–7429 CrossRef CAS PubMed.
-
(a) W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Science, 2019, 364, 1170–1174 CrossRef CAS PubMed;
(b) L. Wang, A. R. White, W. Chen, Z. Wu, D. A. Nicewicz and Z. Li, Org. Lett., 2020, 22, 7971–7975 CrossRef CAS PubMed.
- N. E. S. Tay, W. Chen, A. Levens, V. A. Pistritto, Z. Huang, Z. Wu, Z. Li and D. A. Nicewicz, Nat. Catal., 2020, 3, 734–742 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Org. Lett., 2019, 21, 7114–7118 CrossRef CAS PubMed.
- N. J. Venditto and D. A. Nicewicz, Org. Lett., 2020, 22, 4817–4822 CrossRef CAS PubMed.
- C. A. Lawson, A. P. Dominey, G. D. Williams and J. A. Murphy, Chem. Commun., 2020, 56, 11445–11448 RSC.
- A. J. Perkowski, C. L. Cruz and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 15684–15687 CrossRef CAS PubMed.
- V. A. Pistritto, M. E. Schutzbach-Horton and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 17187–17194 CrossRef CAS PubMed.
- J.-M. M. Grandjean and D. A. Nicewicz, Angew. Chem., Int. Ed., 2013, 52, 3967–3971 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
-
(a) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed;
(b) L. Cheneberg, C. Lévêque, V. Corcé, A. Baralle, J. P. Goddard, C. Ollivier and L. Fensterbank, Synlett, 2016, 731–735 Search PubMed.
-
(a) F. Lima, L. Grunenberg, H. B. A. Rahman, R. Labes, J. Sedelmeier and S. V. Ley, Chem. Commun., 2018, 54, 5606–5609 RSC;
(b) F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed.
- D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed.
- J. K. Matsui and G. A. Molander, Org. Lett., 2017, 19, 950–953 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska, M. S. Jeffreys, N. C. Goodwin and D. A. Nicewicz, Org. Lett., 2020, 22, 679–683 CrossRef CAS PubMed.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed.
- L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. Koenig, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
-
(a) J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C. H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed;
(b) G. A. Kenny-Wallace and C. D. Jonah, J. Phys. Chem., 1982, 86, 2572–2586 CrossRef.
- D. A. Corbin, C.-H. Lim and G. M. Miyake, Aldrichimica Acta, 2019, 52, 7–21 Search PubMed.
- Y. Du, R. M. Pearson, C.-H. Lim, S.-M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed.
-
(a) A. Chatterjee and B. Koenig, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed;
(b) B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater and T. H. Lambert, J. Am. Chem. Soc., 2020, 142, 1698–1703 CrossRef CAS PubMed.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2020, 59, 658–662 CrossRef CAS PubMed.
- S. Wu, J. Zurauskas, M. Domanski, P. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- X. A. Liang, L. Niu, S. Wang, J. Liu and A. Lei, Org. Lett., 2019, 21, 2441–2444 CrossRef CAS PubMed.
- H. Zhao and J. Jin, Org. Lett., 2019, 21, 6179–6184 CrossRef CAS PubMed.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- Z. Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274, DOI:10.1038/s41467-018-05375-2.
-
(a) G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447–4451 CrossRef CAS PubMed;
(b) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed.
- A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017, 9, 1073–1077, DOI:10.1038/NCHEM.2797.
- B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878–16883 CrossRef CAS PubMed.
- B. Laroche, X. Tang, G. Archer, R. Di Sanza and P. Melchiorre, Org. Lett., 2021, 23, 285–289 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
John A.
Murphy
and
John A.
Murphy