
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cysteine protecting groups: applications in peptide and protein science
Richard J.
Spears
*,
Clíona
McMahon
and
Vijay
Chudasama
 *
*
Department of Chemistry, University College London, London, UK. E-mail: r.spears@ucl.ac.uk; v.chudasama@ucl.ac.uk
First published on 17th August 2021
Abstract
Protecting group chemistry for the cysteine thiol group has enabled a vast array of peptide and protein chemistry over the last several decades. Increasingly sophisticated strategies for the protection, and subsequent deprotection, of cysteine have been developed, facilitating synthesis of complex disulfide-rich peptides, semisynthesis of proteins, and peptide/protein labelling in vitro and in vivo. In this review, we analyse and discuss the 60+ individual protecting groups reported for cysteine, highlighting their applications in peptide synthesis and protein science.
 Richard J. Spears | Richard J. Spears received his MChem degree in Chemistry from The University of York in 2014. He then obtained his PhD in Chemistry from The University of York in 2018 under the supervision of Dr Martin A. Fascione, where his work focused on developing new methodologies for site-selective protein modification. In 2018 he joined Prof. Vijay Chudasama's lab as a postdoctoral researcher within University College London, where he currently works on developing novel methodology for use in peptide chemistry and protein bioconjugation. |
 Clíona McMahon | Clíona McMahon obtained her MSci degree in Natural Sciences at UCL, majoring in synthetic organic chemistry and minoring in cell and molecular biology. Her master's research project focused on the development of a cysteine protecting group for use in solid phase peptide synthesis. She will begin a PhD in Chemical Biology in September 2021 in the Chudasama Group, working in the areas of protein and peptide modification. |
 Vijay Chudasama | Prof. Vijay Chudasama obtained his MSci degree and PhD from University College London (UCL) in 2008 and 2011, respectively. Following post-doctoral studies under the supervision of Prof. Stephen Caddick, Vijay obtained a Ramsay Memorial Fellowship. During this time, he was made Technical Director of a biotechnology spin-out (ThioLogics). In April 2015, he was appointed as a Lecturer at UCL Department of Chemistry, before being promoted to Reader (2017) and then Professor (2019) at the same institution. Vijay's research has been highlighted by Forbes, Scientific American, CNN News, Nature Chemistry and the Royal Society of Chemistry. |
1. Introduction
The concept of protecting, and subsequently deprotecting, functional groups is of paramount importance in synthetic chemistry.1–3 Protecting group strategies in particular feature extensively in the synthesis of peptides, whereby amino acid building blocks are coupled to one another via amide bonds. This is unsurprising, given the array of functional groups that are found within the 20 proteinogenic amino acids, including: amines, alcohols, thioethers, imidazole rings, and carboxylic acids.4 Of the proteinogenic amino acids, cysteine (Cys) has garnered enormous interest within the field of peptide (and protein) chemistry and biology, owing to the unique properties of its thiol side chain. For example, disulfide bonds, which can be formed via the covalent linkage of two Cys thiol side chains, are of utmost importance in defining certain peptides and proteins conformational, proteolytic, chemical, and biophysical properties.5,6 Disulfide-containing peptides have also gained huge interest as therapeutic agents.7–9 In 1953 Vigneaud, Ressler, Swan, Roberts, Katsoyannis, and Gordon, demonstrated the chemical synthesis of the hormone oxytocin, a disulfide containing peptide that is the focus of a great deal of therapeutic research (Fig. 1a).10,11 Vigneaud was later awarded the Nobel prize in Chemistry in 1955. The hormone insulin, which is produced in the pancreas and contains one intrachain and two interchain disulfide bonds, has been a critical component of treating Type I diabetes over the last 100 years (Fig. 1b)12 Disulfide-containing peptides also feature in a number of Food and Drug Administration (FDA) approved therapeutics, such as Ziconotide (Prialt™, a disulfide-rich peptide for treating severe chronic pain, Fig. 1c),13 Linaclotide (Linzess™, a disulfide-rich peptide for treating severe irritable bowel syndrome),14 -and 68Ga-DOTATOC (Fig. 1d, an octreotide-based tumour imaging agent).15 In biology, Cys often finds itself of critical importance in redox processes,16 and in the active sites of enzymes such as cysteine proteases.17 Cys is also often the site of choice when it comes to the site-specific modification of proteins, also known as bioconjugation, owing to its favourable properties (nucleophilic profile of the thiol at neutral/near-neutral pH, low natural abundance, general ease of incorporation into proteins via site-directed mutagenesis).18 This growing field in chemical biology has led to the development of a range of therapeutically relevant bioconjugates, most notable of which include antibody–drug conjugates (ADCs);19 these include FDA approved Adcetris™ (Fig. 1e, brentuximab vedotin, used to treat lymphoma), Polivy™ (polatuzumab vedotin, used to treat lymphoma), and Padcev™ (enfortumab vedotin, used to treat urothelial cancer), all of which have their therapeutic payload covalently attached to the antibody via Cys conjugation.20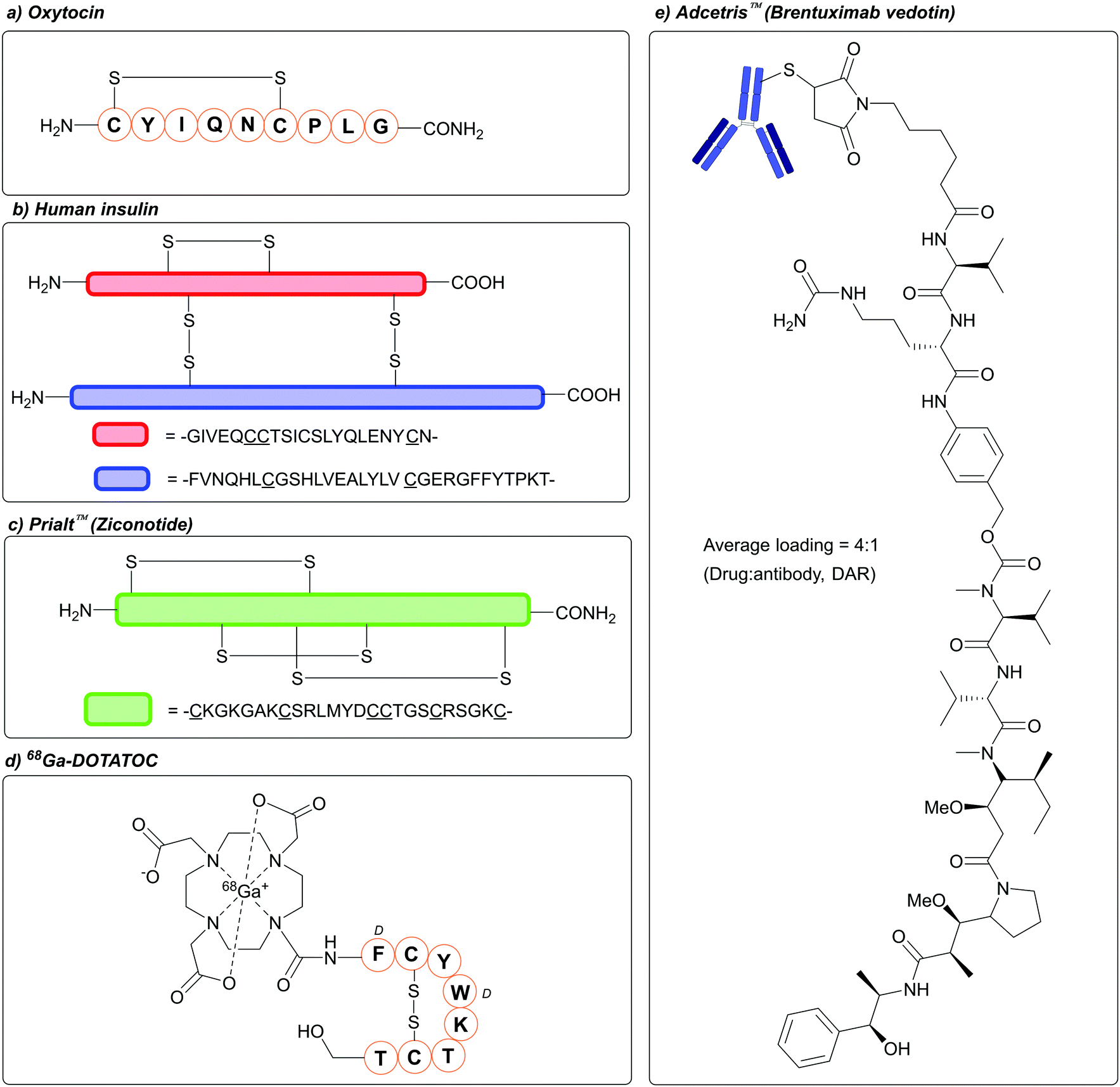 | ||
| Fig. 1 Examples of Cys-containing biomolecules of therapeutic interest. (a) Oxytocin, (b) Human insulin, (c) Prialt™/Ziconotide, (d) 68Ga-DOTATOC, (e) Adcetris™/Brentuximab vedotin. | ||
The reactivity profile of Cys enables a plethora of applications in chemistry and biology related to peptides and proteins. However, this reactivity also leads to significant challenges when employing the residue for peptide chemistry in particular. The reactive nature of the thiol side chain makes it prone to side reactions such as alkylation and oxidation.21 Particularly for disulfide-rich peptides, ensuring the regioselective formation/correct connectivity of disulfides presents a significant challenge in peptide synthesis.5,6 Ensuring racemisation of Cys at the α-carbon stereocenter does not occur during peptide synthesis also presents a notable challenge; this is especially true for synthesising peptides containing a C-terminal Cys.22 Other side reactions related to Fmoc SPPS include formation of 3-(1-piperidinyl)alanine by-products at the C-terminal Cys position formed by an elimination–addition reaction involving piperidine.23,24 Unsurprisingly, this has led to significant research into strategies to protect (and subsequently deprotect) the thiol side chain to overcome these challenges, and to further expand upon the synthesis of biomolecules. Indeed, protection of Cys during standard peptide synthesis is therefore often critical to avoid undesired alkylation/oxidation, allowing for synthesis of the desired peptide. In the case of disulfide-containing peptide synthesis, the development of “orthogonal” Cys protecting groups (which can be selectively deprotected in the presence of one another) has proved paramount to ensuring regioselective disulfide formation.6
Herein we review the different thiol protecting groups reported for Cys throughout the last 70+ years. We analyse and discuss each of the individual 60+ protecting groups that have been reported for Cys protection (and deprotection). For each group (and where applicable), this review will focus on (a) the conditions used for its deprotection, (b) use in Boc and Fmoc peptide synthesis, (c) its comparison to other related Cys protecting groups, and (d) notable applications of the Cys protecting group in peptide and protein chemistry.
2. Organisation of review
This review begins with a brief introduction to key concepts relevant to the content covered within the review and Cys protecting group chemistry as a whole. This is then followed by a table (Table 1) and a series of discussions about each individual protecting group. Taking inspiration from previously reported literature,4–6 the protecting groups within this review have been divided into separate categories based on the conditions that have primarily been used for their deprotection. We note that, in cases where multiple methods of deprotection are available, certain protecting groups do not sit rigidly within these categories. The categories are as follows: Acid labile, oxidatively labile, base labile, enzyme-labile, hydrazine-labile, palladium-labile, and reductively labile. The “benzyl” protecting group, protecting groups specific to N-terminal Cys, and the “safety catch” protecting group, have also been assigned to their own separate categories. Additionally, in order to give historical context, the groups in each category are broadly arranged in chronological order. For additional reading, we direct the reader to extensive reviews on general amino acid protecting groups by Isidrio-Llobet, Álveraz, and Albericio,4 along with reviews that focus on regioselective disulfide formation by Postma, and Albericio,6 and more recently by He, Pan, Mayer, and Li.5 We also wish to highlight a recent review by Laps, Satish, and Brik discussing recent advances in transition metal chemistry relating to peptide/protein synthesis.253. Key concepts in cysteine protecting group chemistry
3.1 Solid phase peptide synthesis
Briefly, chemical peptide synthesis dates back well over 100 years ago, when Curtius synthesised benzoylglycylglycine, an N-protected dipeptide, in 1882 using benzoyl chloride and the silver salt of glycine.17 In the early 1900s, Fischer and Fourneau reported the synthesis of the unprotected dipeptide via hydrolysis of glycine ester to give the diketopiperazine, which subsequently hydrolysed to give glycylglycine.26 Later, in 1932, the concept of using protecting groups to temporarily mask functional groups in the context of peptide synthesis was established when Bergmann and Zervas introduced the carbobenzoxy group (Cbz or Z) for the protection of amines.26 Another key concept was then introduced in 1955 by Sheehan and Hess, where N,N-dicyclohexylcarbodiimide (DCC) was established as an amide coupling reagent for peptide synthesis.27 This would later be followed by the introduction of the acid-labile tert-butyloxycarbonyl (Boc) protecting group in 1957 by Carpino,28 Mckay, and Albertson.29 Peptide synthesis would later be revolutionised by the advent of solid phase peptide synthesis (SPPS) reported in 1963 by Merrifield.30 Here, peptides are synthesised through sequential coupling of protected amino acids on a solid support (typically a polystyrene resin); impurities and excess reagents can then be removed by simple wash and filtration of the solid support (Fig. 2). Specific cocktails of reagents can then be used to cleave and isolate the peptide from the resin; different resins have since been reported over the last few decades, which yield peptides with different functional groups at the C-terminus once cleaved e.g. acid, amide, thioester.31 Carpino, along with Han, would then introduce the base-labile 9-fluorenylmethoxycarbonyl (Fmoc) protecting group in 1970.32 The combination of these concepts has led to development of two solid phase strategies for peptide synthesis: the Boc/Benzyl (Bn) strategy, and the Fmoc/tert-butyl (tBu) strategy. The Fmoc/tBu strategy is now the more routinely used, as it avoids both the repeated acidic treatments of the peptide, and the use of hydrogen fluoride (HF) (which requires specialist equipment and is extremely toxic) that is required in Boc/Bn peptide synthesis.33 For a more extensive breakdown of the history of peptide chemistry and related key concepts, we direct the reader's attention to discussions in recently published reviews.33,34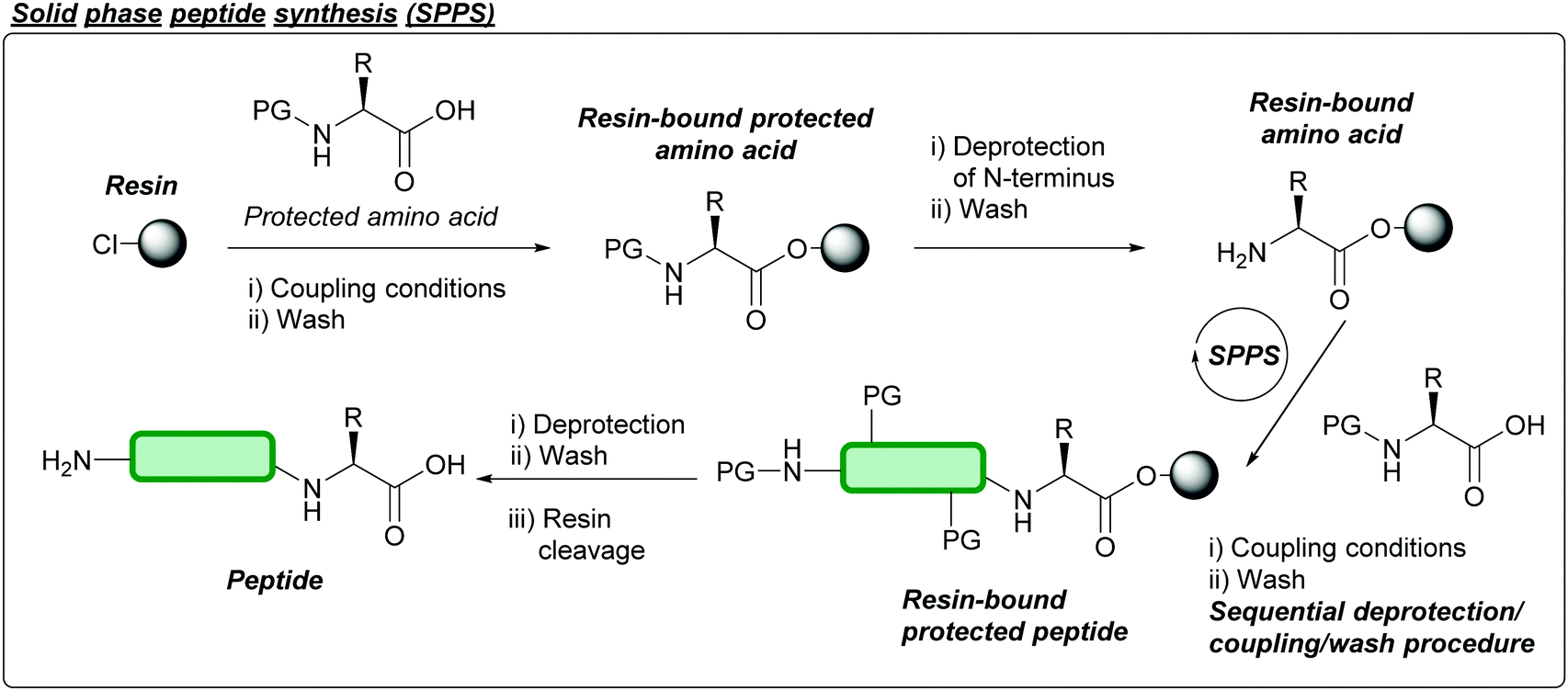 | ||
| Fig. 2 Outline of solid phase peptide synthesis (SPPS) procedure. Following loading of a desired amino acid onto a functionalised polystyrene resin, sequential deprotections of the N-terminal protecting group and amide coupling reactions yield a resin-bound, protected peptide of a desired sequence. Excess reagents/impurities can be removed by washing and filtration at each step. Removal of protecting groups and cleavage of the peptide off the resin with specific cocktails of reagents yields the final desired peptide. | ||
3.2 Native chemical ligation and related ligations strategies
SPPS is now the standard for the chemical peptide synthesis; however, the process is generally limited to peptides of ca. 50 amino acids long.35 As such, ligation reactions are usually employed when synthesising larger peptides,36 the most common of which is the native chemical ligation (NCL) strategy. Introduced in 1994 by Dawson, Muir, Clark-Lewis, and Kent,37 NCL is a ligation reaction between a C-terminal peptide thioester and an N-terminal cysteinyl peptide. An initial exchange reaction occurs between the thiol and the thioester linking the two peptides via a thioester bond. The molecule then spontaneously rearranges via an intramolecular S,N-acyl transfer, resulting in the formation of a native peptide bond (Fig. 3a).38 A notable example of NCL is in the one-pot synthesis of Crambin reported in 2004, whereby three peptide fragments were ligated together using NCL, followed by folding to yield the desired protein.39 Peptide fragments for NCL can be synthesised chemically via SPPS or, when significantly longer peptide fragments are desired, can be produced recombinantly in bacterial expression systems such as in Escherichia coli (E. coli) cells. In some cases, these recombinantly produced peptides contain an intein segment fused to the C-terminus, which can then undergo N,S-acyl shift and, following addition of an exogenous thiol such as sodium 2-mercaptoethanesulfonate (MESNa), cleavage to yield a thioester-containing peptide fragment that can subsequently participate in NCL; this process is known as expressed protein ligation (EPL, Fig. 3b).40,41 The resulting Cys residue post-NCL can then be converted to alanine through metal-based desulfurisation,42 or metal-free-based desulfurisation using radical initiators such as 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) in combination with tris(2-carboxyethyl)phosphine (TCEP, Fig. 3c).43 As a result, it is therefore common for alanine within a target peptide sequence to be the ligation site of choice.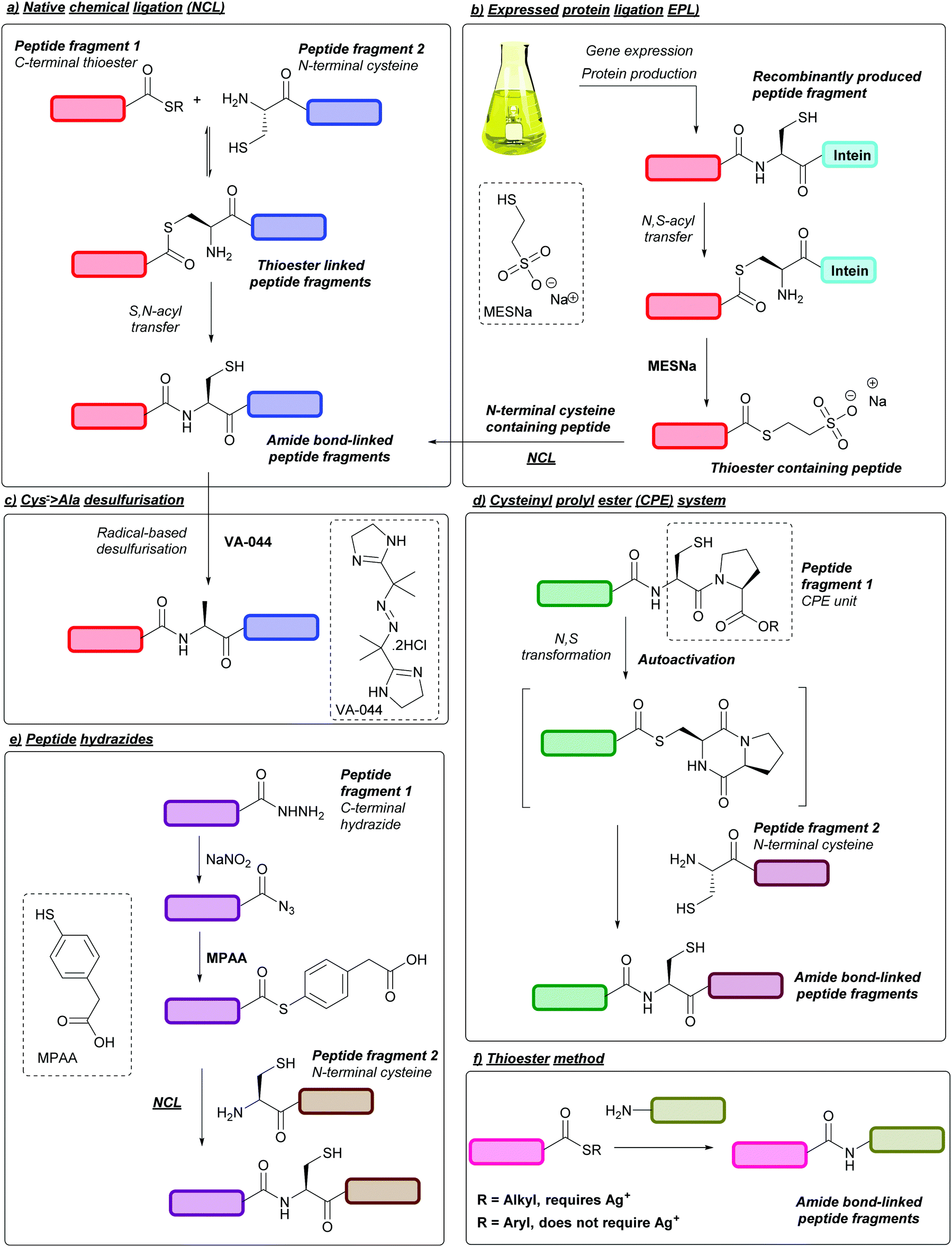 | ||
| Fig. 3 Strategies related to peptide ligation. (a) Native chemical ligation (NCL), (b) expressed protein ligation (EPL), (c) radical-based desulfurisation of Cys to yield alanine using 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044), (d) cysteinyl prolyl ester(CPE) system, (e) ligation of peptides using peptide hydrazides, (f) thioester method for ligation of peptides. | ||
Other common systems used for ligation of large peptides are N,S-acyl shift systems, one example of which is the cysteinylprolyl ester (CPE) system.44 Here, peptides containing a CPE unit at the C-terminus can undergo spontaneous N- > S transformations into diketopiperazine thioesters. The resulting thioester can then undergo NCL in a one-pot process (Fig. 3d). A related system that uses cysteinylprolyl imide (CPI) peptides has more recently been described and used in the synthesis of proteins such as ZHER2 affibody.45 In addition, peptides containing C-terminal hydrazides can be used as more chemically stable peptide thioester precursors.46 In this strategy the hydrazide group can be chemically converted to a C-terminal azide or acyl pyrazole using nitrous acid or acetyl acetone, followed by conversion to a thioester through addition of 4-mercaptophenylacetic acid (MPAA, Fig. 3(e)).47 This methodology has since been used in the synthesis of multiple proteins, including Centruroides suffusus suffusus toxin II protein (CssII)46 and α-synuclein.48 Peptide hydrazides can also be prepared through Cys cyanylation using 2-nitro-5-thiocyanatobenzoic acid (NTCB) followed by addition of hydrazine, as shown in the recently described activated Cys-directed protein ligation (APCL).49 Another ligation strategy is the thioester method, whereby a silver (Ag+) activated C-terminal alkyl thioester is directly displaced by an N-terminal amine to yield the desired amide bond.50 A silver-free protocol has also been developed for the thioester method, which involves the use of aryl, as opposed to alkyl, thioesters (Fig. 3f).51 To ensure a successful peptide synthesis both thioester methods require peptide fragments with protected amino groups, such as Fmoc protection of N-terminal α-amino groups, or Boc/Z protection of Lys side chain ε-amino groups; alternatively, Lys side chains can be protected by using an azido-lysine analogue during peptide synthesis, followed by azide reduction to give the desired ε-amino group post-thioester ligation.52 Thioester methods have since been utilised for the synthesis of proteins such as chemokine CCL2751and various glycoproteins.53 For further reading on ligation methods, we direct the reader to extensive reviews on the subject.53–57
3.3 Orthogonality and bioorthogonality
In 1977, Barany and Merrifield described the concept of “orthogonality”, which (as mentioned previously) describes protecting groups that can be chemoselectively removed in the presence of one another without affecting/removing each other under certain sets of conditions.58 This concept has proved especially important in peptide synthesis when considering disulfide-containing peptides, where the use of protecting groups can avoid off-target and undesired disulfide formation (Fig. 4a).5,6 The concept of orthogonality would later be extended to chemical biology when Bertozzi coined the term “bioorthogonal”. First referenced by Bertozzi in 2003, along with Hang, Yu, and Kato,59 bioorthogonal/bioorthogonality refers to chemical reactions that can be performed in biological systems without interfering with the native environment (Fig. 4b).60,61 This typically relies on incorporation of non-natural functionalities, which can be used as “handles” for selective, biooorthogonal reactions. The field of bioorthogonal chemistry has grown rapidly over the last two decades, with in vivo applications such as metabolic labelling of developing zebrafish embryos.62,63 On the related subject of protein modification, advances in synthetic biology and genetic code expansion have enabled the incorporation of non-natural functionalities into proteins via unnatural amino acid mutagenesis. This typically involves expression of a gene (encoding for a protein of interest) containing an amber STOP codon (in E. coli cells), along with expression of an orthogonal tRNA/tRNA synthetase in the presence of an unnatural amino acid, to produce the unnatural amino acid-containing protein of interest (Fig. 4c). Functionalities introduced into proteins in this manner include azides, alkynes, aldehydes, and tetrazines. Similar to the thiol side chain of Cys, these groups can subsequently be used as handles for site-selective bioconjugation to yield conjugates such as ADCs.64 Alternatively, amino acids with modified or protected side chains can also be introduced; these include analogues that mimic post-translational modifications (PTMs) in proteins e.g. histones,65 or photocaged analogues of inactive enzymes which can subsequently be activated via deprotection e.g. photocaged Tyr in β-galactosidase.66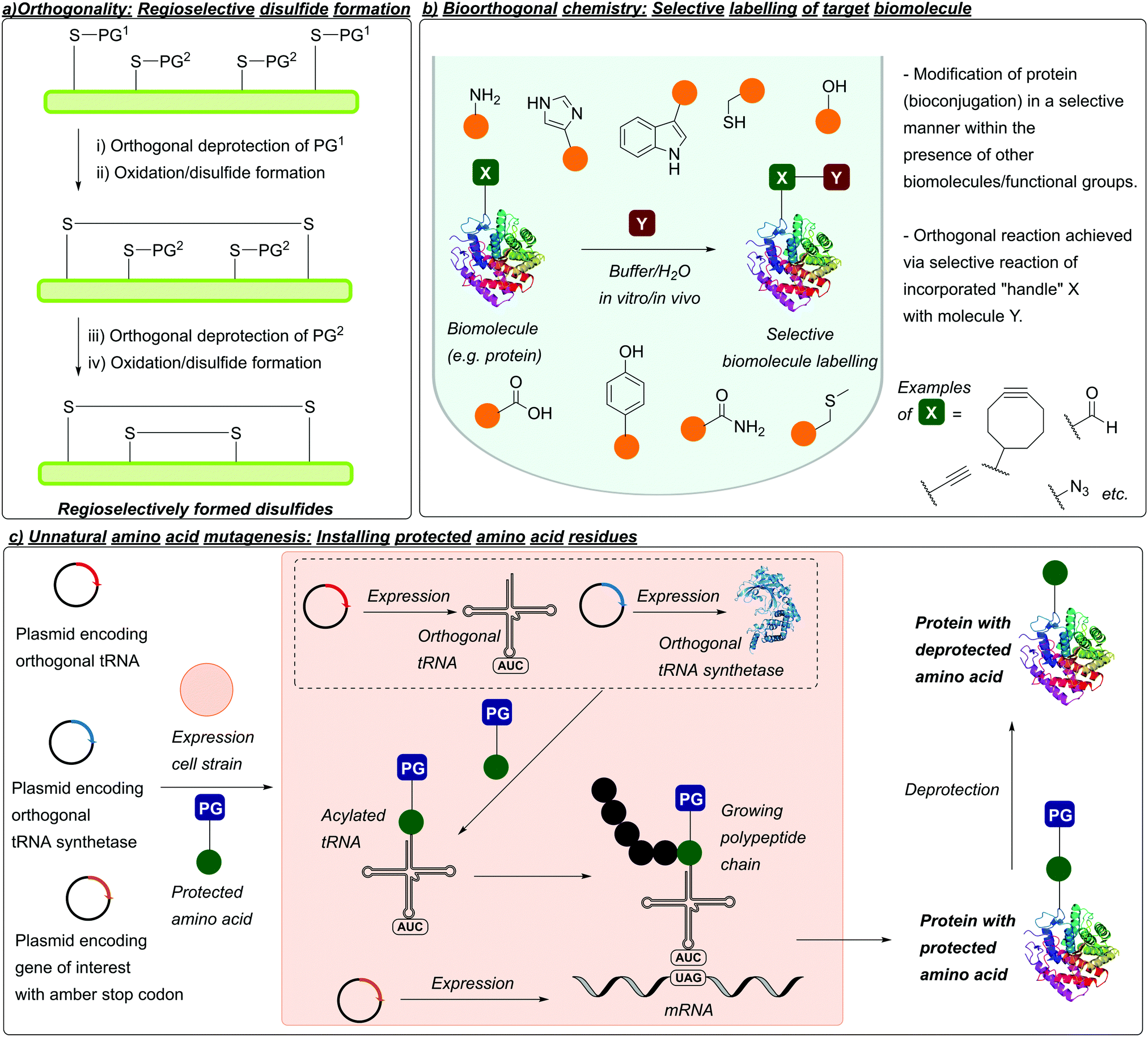 | ||
| Fig. 4 (a) Concept of orthogonality and its relation to protecting group chemistry and disulfide formation. Using combinations of protecting groups that are orthogonal i.e. can be removed in the presence of one another, followed by disulfide bond formation in a sequential manner allows for controlled, regioselective disulfide bond formation. (b) Outline of bioorthogonal chemistry. Selective modification of a given biomolecule is carried out without off-target modification/perturbation of the native environment. (c) Outline of unnatural amino acid mutagenesis. Using a suitable cell strain, expression of a plasmid encoding for the desired protein (which contains an amber STOP codon) alongside an orthogonal tRNA synthetase/tRNA pair and unnatural amino acid allows for incorporation of said unnatural amino acid at the aforementioned STOP position. | ||
4. Table of cysteine protecting groups
| Protecting group, abbreviations and location within review | Lability | Deprotection conditions | Stable to/compatible with | Structure |
|---|---|---|---|---|
| Benzyl (Bzl, Bn); Section 5.1 | Acid, reducing agents | Na/NH3 (liq.)67 | Standard Boc SPPS reagents4 |
|
| HF (25 °C)68 | ||||
| TMSBr/TFA/thioanisole69 | ||||
| Trityl (Trt); Section 5.2.1 | Acid, oxidising agents | Ag(I)70 | Standard Fmoc SPPS reagents74 |
|
| Hg(II)70 | ||||
| HBr/AcOH70 | ||||
TFA/TIS (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10)71 :10)71 |
||||
| HBF4/scavengers72 | ||||
| I24 | ||||
| CuSO4-cysteamine, Gdn·HCl/HEPPS buffer73 | ||||
| Diphenylmethyl (Dpm, Bzh, Bh); Section 5.2.2 | Acid | TFA/TIS/H2O/DCM (90:2.5:2.5:5)75 |
Cocktails of <25% TFA75 |
|
| Standard Fmoc SPPS reagents75 | ||||
| Tetrahydropyranyl (Thp); Section 5.2.3 | Acid | Silver nitrate (aq., 0 °C)76 | Na/NH3 (liq.)76 |
|
| TFA/TIS/DCM (95:2.5:7.5)77 |
1% TFA in DCM77 | |||
| Standard Fmoc SPPS reagents77 | ||||
| tert-Butyl (tBu); Section 5.2.4 | Acid | NpsCl (2 h, RT), then NaBH478 | I272 |
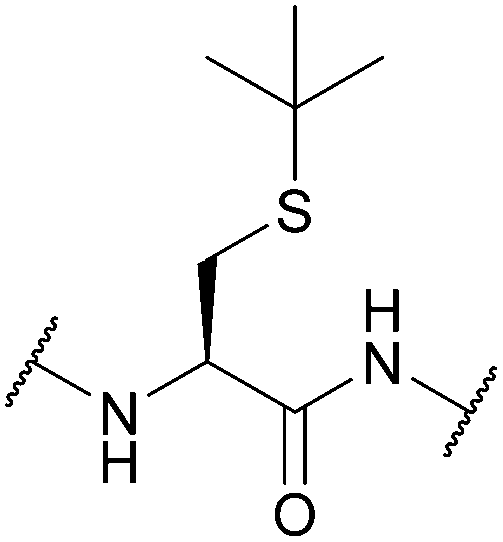
|
| HF (20 °C)79 | TFA86 | |||
| DTNP/TFA80 | AgOTf/TFA87 | |||
| Hg(OAc)2/TFA/anisole81 | Na/NH3 (liq.)78 | |||
| Silyl chloride-sulfoxide/TFA82 | Hydrazine78 | |||
| Tl(TFA)383 | Standard Fmoc SPPS reagents88 | |||
| DMSO/TFA84 | ||||
| PdCl2, 50 mM Tris or urea buffer (37 °C)85 | ||||
| 4-Methoxybenzyl (Mob, MBzl); Section 5.2.5 | Acid | TFA (100 °C)89 | HBr89 |
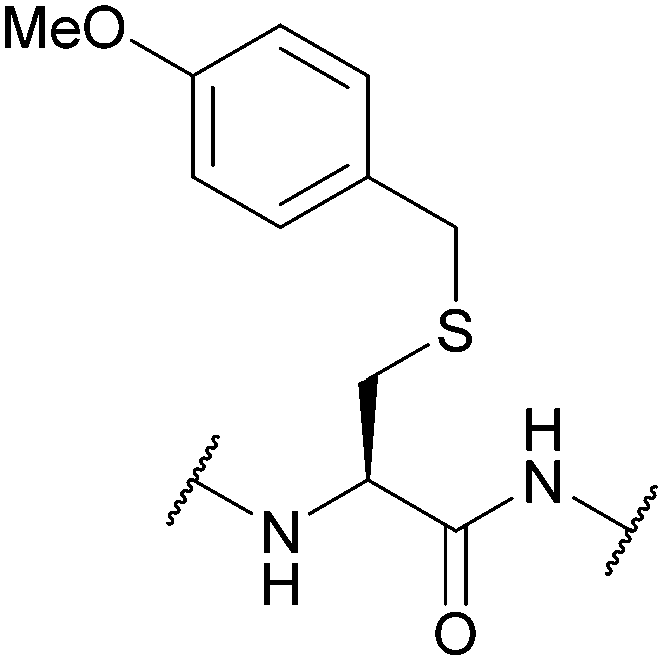
|
| HF79 | TFA (without scavengers)86 | |||
| DTNP or DTP/TFA/thioanisole90 | Standard Fmoc SPPS reagents75 | |||
| TFA/TIS (12 h, 37 °C)86 | Standard Boc SPPS reagents (for small peptides)4,91 | |||
| Hg(TFA)281 | ||||
| Tl(TFA)383 | ||||
| AgOTf/TFA/thioanisole87 | ||||
| 3,4-Dimethylbenzyl (DMB); Section 5.2.6 | Acid | HF (10 min, 0 °C)92 | 50% TFA in DCM (23 h, 24 °C)92 |
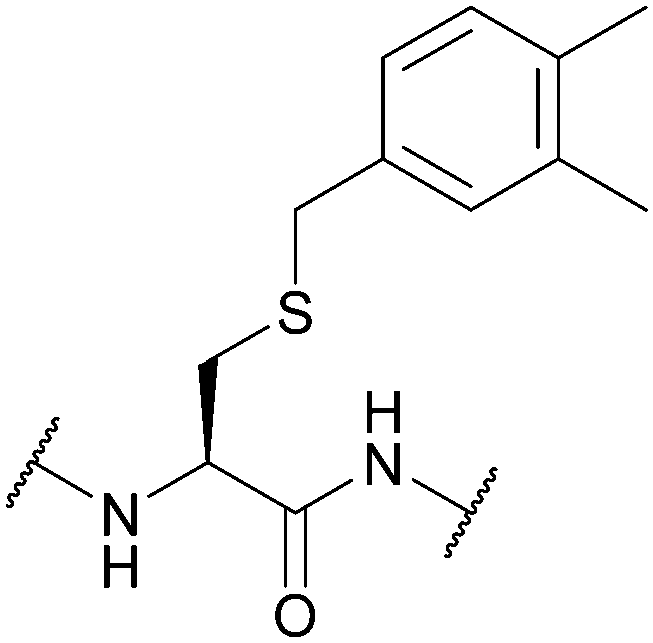
|
| Standard Boc SPPS reagents92 | ||||
| Methylbenzyl (Meb, 4-MeBn, 4-MeBzl); Section 5.2.7 | Acid | HF/anisole (1 h, 0 °C)91 | AgOTf87 |
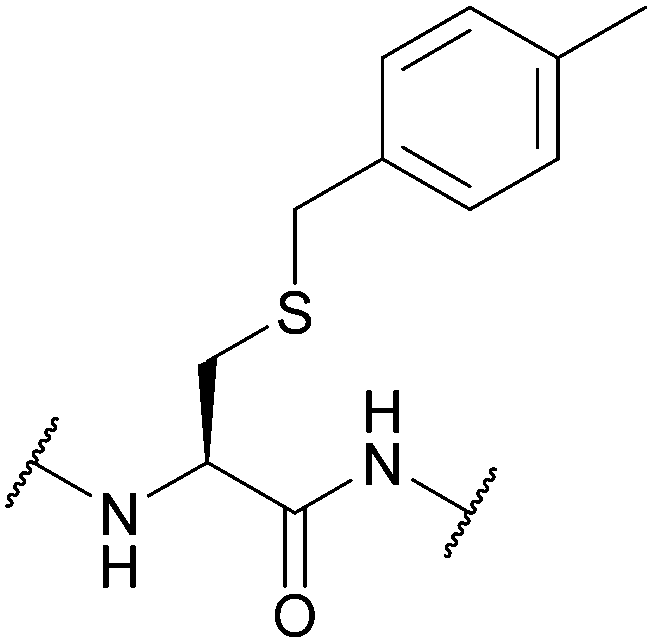
|
| Tl(TFA)383 | Standard Fmoc SPPS reagents74 | |||
| DMSO/TFA (45 °C)74 | Standard Boc SPPS reagents91 | |||
| 1-Adamantyl (Ad, 1-Ada); Section 5.2.8 | Acid | Hg(OAc)2/TFA81 | TFA (2.5 h, 0 °C)93 |
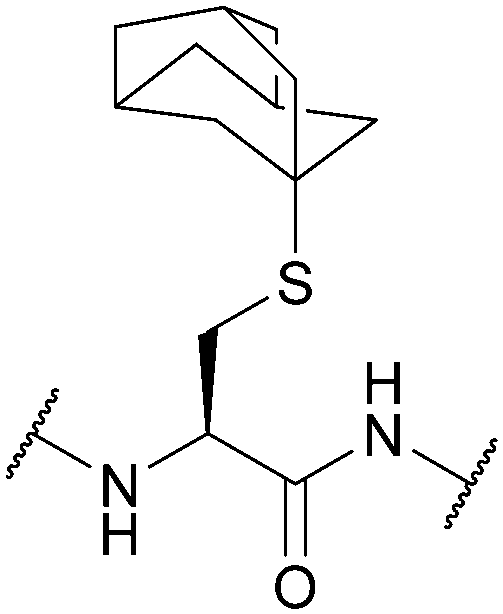
|
| 1 M TFMSA/anisole/TFA93 | AgOTf/TFA87 | |||
| Tl(TFA)393 | Standard Boc SPPS reagents87 | |||
| Standard Fmoc SPPS reagents4 | ||||
| Benzyloxymethyl (Bom); Section 5.2.9 | Acid | AgOTf/anisole/TFA (1 h, 0 °C)94 | TFA (4 h, 0 °C)94 |
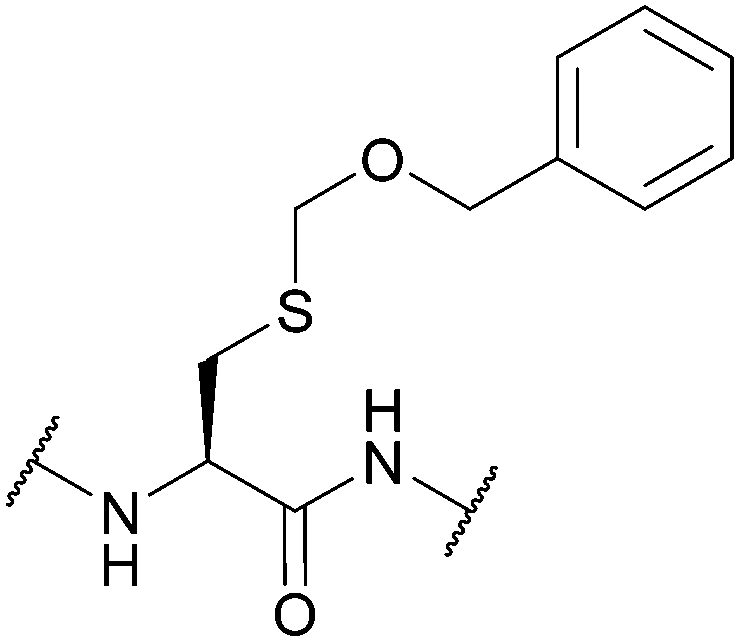
|
| 1 M TMSOTf/thioanisole/TFA (1 h, 0 °C)94 | Hydrazine94 | |||
| Tl(TFA)394 | Piperidine/DMF94 | |||
| NaBO394 | ||||
| Standard Fmoc SPPS reagents94 | ||||
| 2,4,6-Trimethoxybenzyl (Tmob); Section 5.2.10 | Acid | ≥6% TFA with TES or TIS (0.5%) in DCM (5 min, 25 °C)95 | Standard Fmoc SPPS reagents96 |
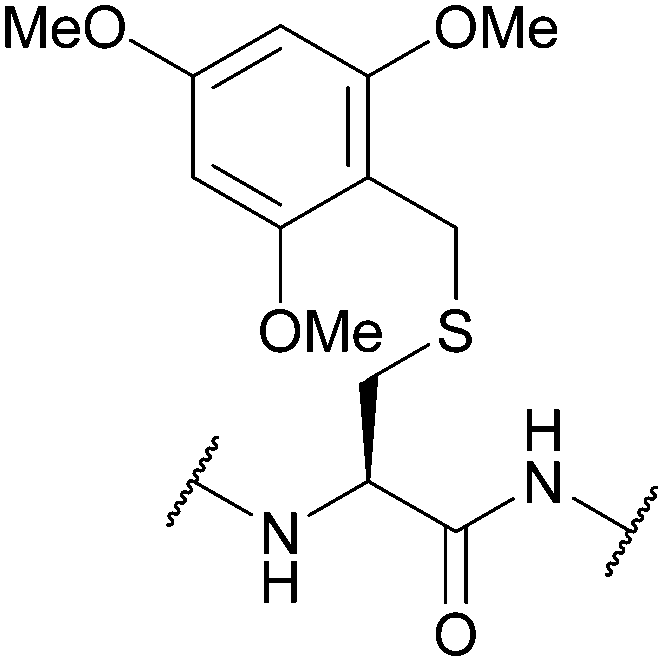
|
| ≥30% TFA in DCM with phenol/thioanisole/H2O (5% each)95 | ||||
| I2/DMF (0 °C)95 | ||||
| Tl(TFA)3/DMF/anisole (0 °C)95 | ||||
| 4,4′,4′′,-Trimethoxy-triphenylmethyl (TMTr); Section 5.2.11 | Acid | 1% TFA in DCM96 | Standard Fmoc SPPS reagents96 |
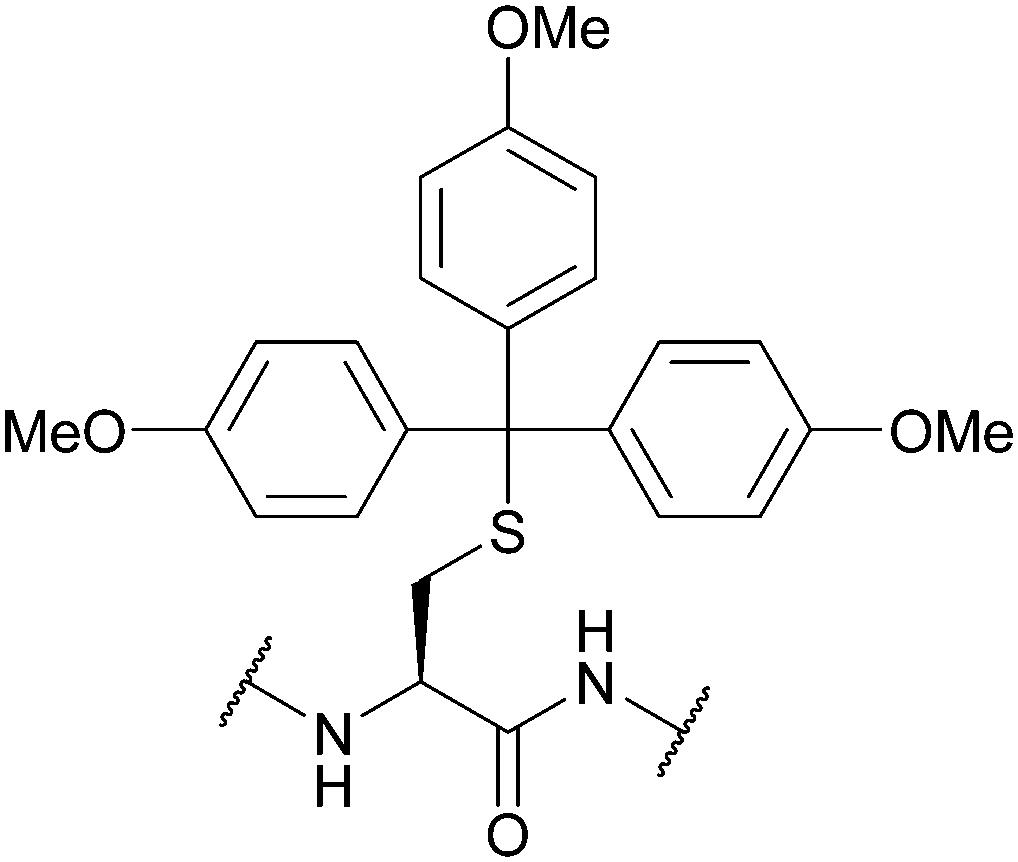
|
| Pseudoprolines (ΨPro); Section 5.2.12 | Acid | TFA (ΨMe,Mepro, 32–36 h, sequence dependant)97 | Standard Fmoc SPPS reagents97 |
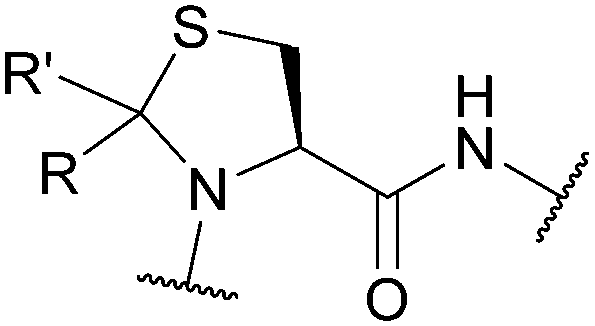
|
| TFMSA (15 min, 0 °C)98 | TFA (ΨH,Hpro)97 | |||
| TFA (ΨMe,Mepro, 1–6 h, sequence dependant)99 | TFA/MeOH/TIS/H2O (80:15:2.5:2.5, ΨMe,Mepro, 1 h, cyclic peptides)101 |
|||
| TFA (ΨH,Dmppro, minutes)97 | ||||
| TFA (ΨH,Dmppro, C-terminal resin-bound, 1.5 h)100 | ||||
| 4-Methyltrityl (Mtt); Section 5.2.13 | Acid | 1% TFA/scavengers75 | Standard Fmoc SPPS reagents75 |
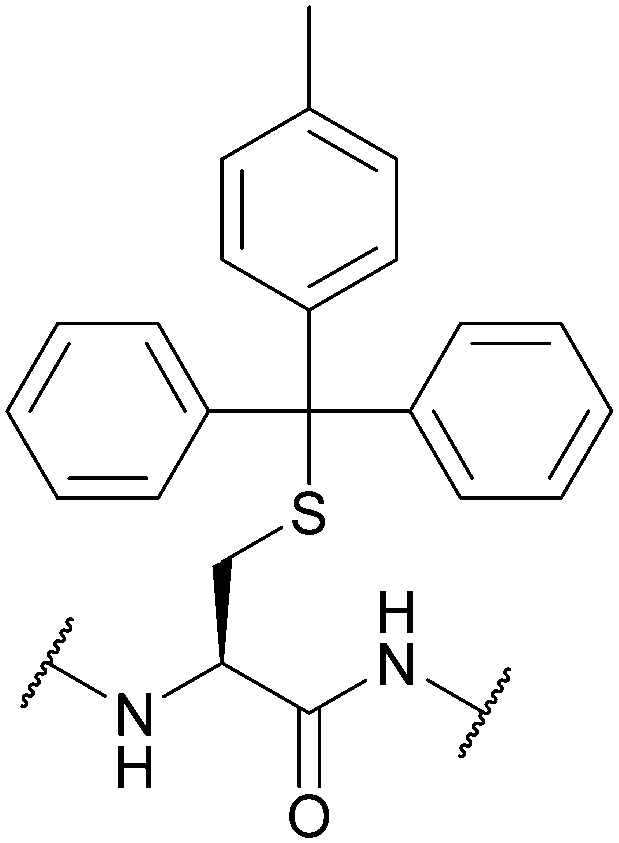
|
| 4-Methoxytrityl (Mmt); Section 5.2.13 | Acid | 1–3% TFA in DCM/TES (95:5)71 |
Bases, e.g. 30% piperidine in DMF (24 h, 22 °C)71 |
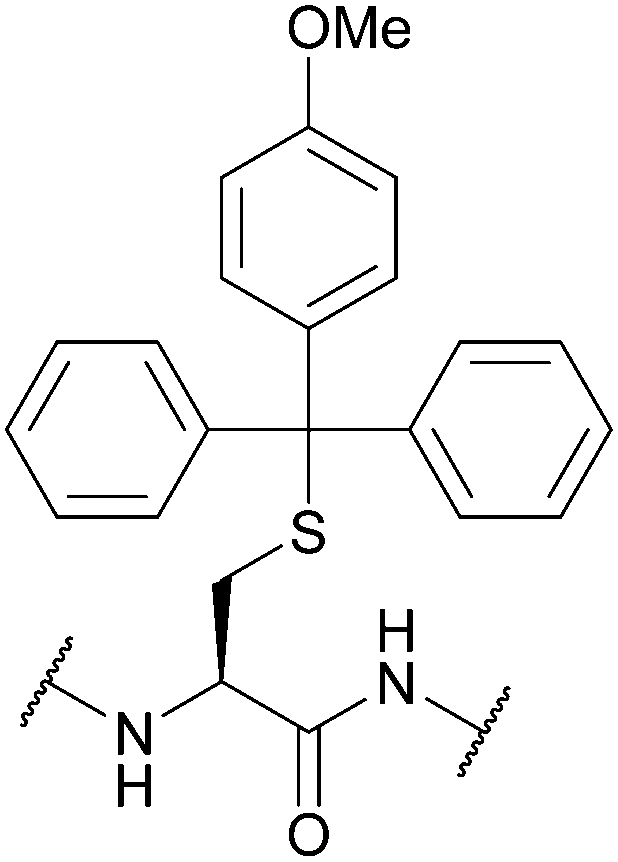
|
| I271 | Very weak acids, e.g. AcOH/TFE/DCM (1:2:7, 30 min)71 |
|||
| Standard Fmoc SPPS reagents71 | ||||
| 9H-Xanthen-9-yl (Xan); Section 5.2.15 | Acid | TFA:DCM:TES (0.1:99.4:0.5)102 |
Piperidine/DMF (≥24 h, 25 °C)102 |
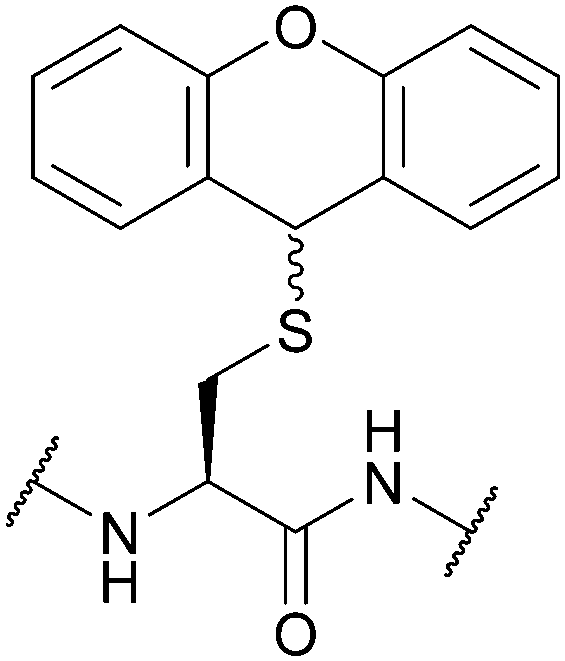
|
| TFA:DCM:BME (10:85:5)102 |
HOBt/DMF (24 h, 25 °C); | |||
| TFA:DCM:TES (1:98.5:0.5) solid phase (25 C, 2 h)102 |
AcOH102 | |||
| I2/MeOH102 | Standard Fmoc SPPS reagents102 | |||
| Tl(TFA)3102 | ||||
| 2-Methoxy-9H-xanthen-9-yl (2-Moxan); Section 5.2.15 | Acid | TFA:DCM:TES (0.1:99.4:0.5)102 |
Piperidine/DMF (≥24 h, 25 °C)102 |
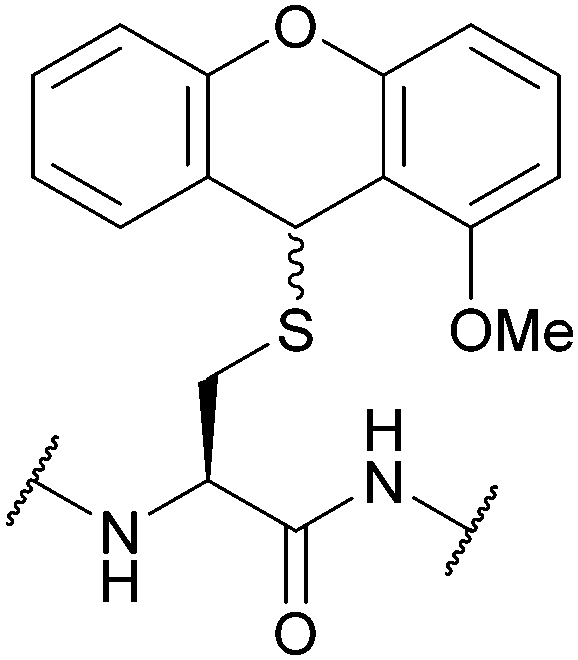
|
| TFA:DCM:BME (10:85:5)102 |
HOBt/DMF (24 h, 25 °C); | |||
| TFA:DCM:TES (1:98.5:0.5) solid phase (25 C, 2 h)102 |
AcOH102 | |||
| I2/MeOH102 | Standard Fmoc SPPS reagents102 | |||
| Tl(TFA)3102 | ||||
| 4,5,6-Trimethoxy-2,2-dimethyl-2,3-dihydrobenzofuran-7-methyl (Tmbm); Section 5.2.16 | Acid | TFA/TES/DCM (1:5:94)103 |
Standard Fmoc SPPS reagents103 |
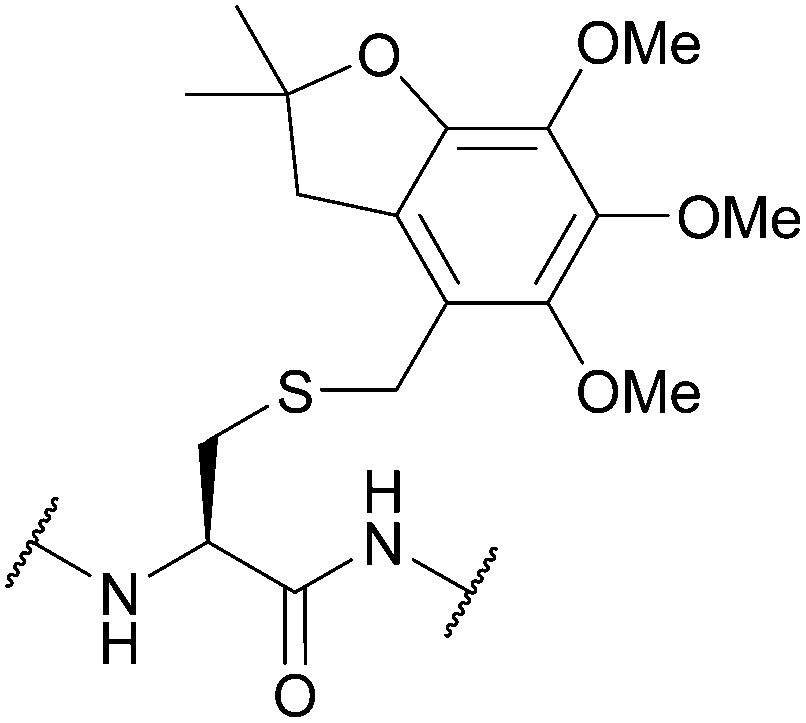
|
| 2,2,5,7,8-Pentamethylchroman-6-methyl (Pmcm); Section 5.2.16 | Acid | TFA/TES/DCM (1:5:94)103 |
Standard Fmoc SPPS reagents103 |
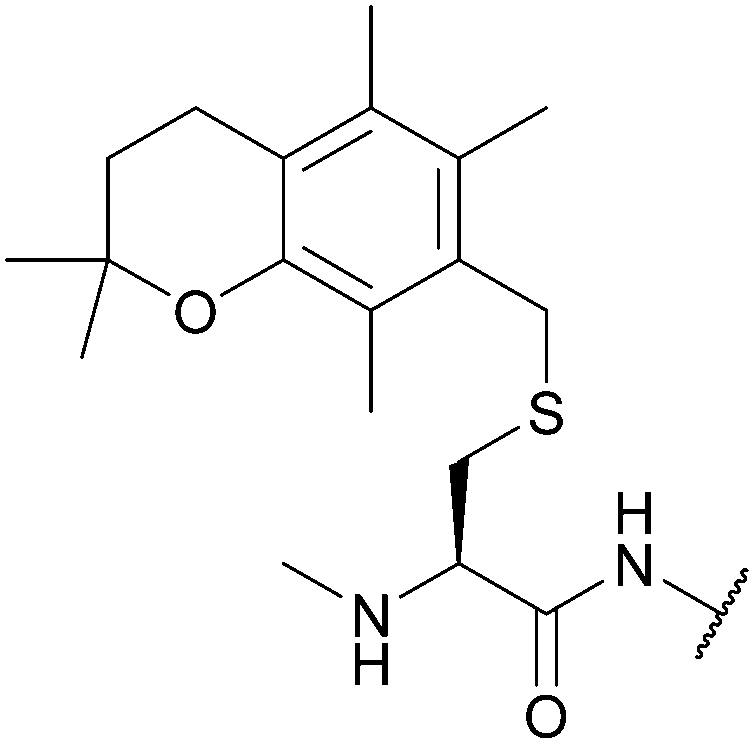
|
| 2,2,4,6,7-Pentamethyl-2,3-dihydrobenzofuran-5-methyl (Pbfm); Section 5.2.16 | Acid | TFA/DCM/TIS (1:5:94)103 |
Standard Fmoc SPPS reagents103 |
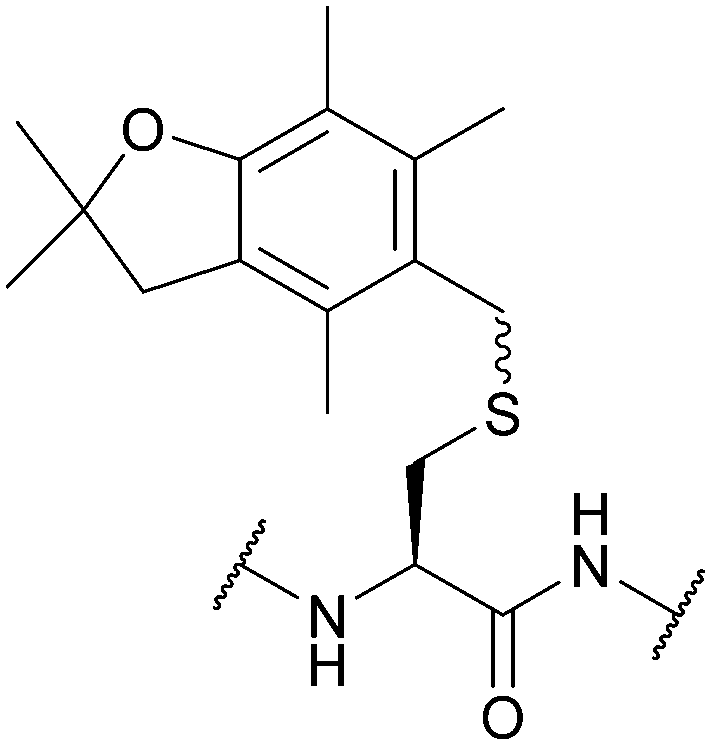
|
| I2103 | ||||
| HFIP or TFE in DMF (15 min)103 | ||||
| 4-Methoxybenzyloxymethyl (Mbom); Section 5.2.17 | Acid | Reagent K/MeONH2·HCl104 | Standard Fmoc SPPS reagents104 |
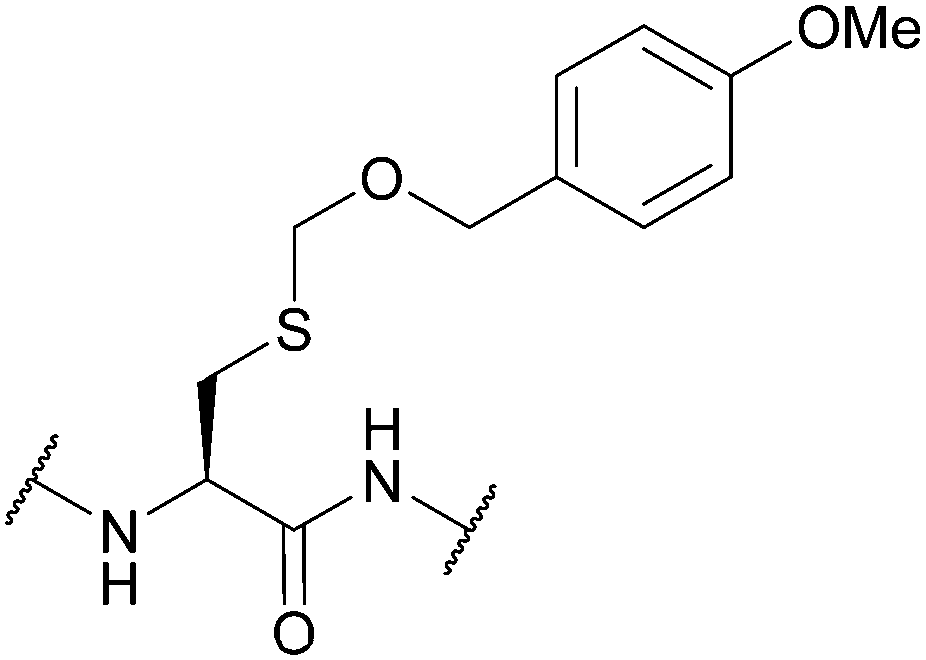
|
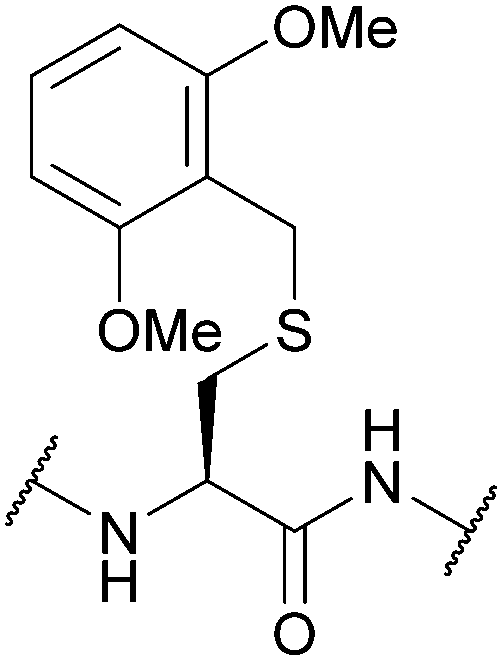
| 2,6-Dimethoxybenzyl (2,6-diMeOBn); Section 5.2.18 | Acid | TFA:DCM:TIS:H2O (50:45:2.5:2.5, 1 h, 25 °C)105 |
Standard Fmoc SPPS reagents75 |
|
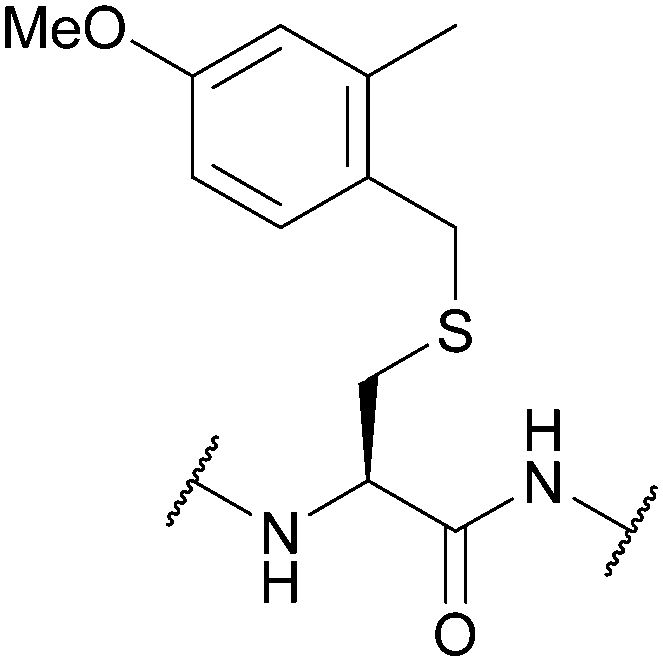
| 4-Methoxy-2-methylbenzyl (4-MeO-2MeBn); Section 5.2.18 | Acid | TFA:DCM:TIS:H2O (50:45:2.5:2.5, 1 h, 25 °C)105 |
Standard Fmoc SPPS reagents75 |
|
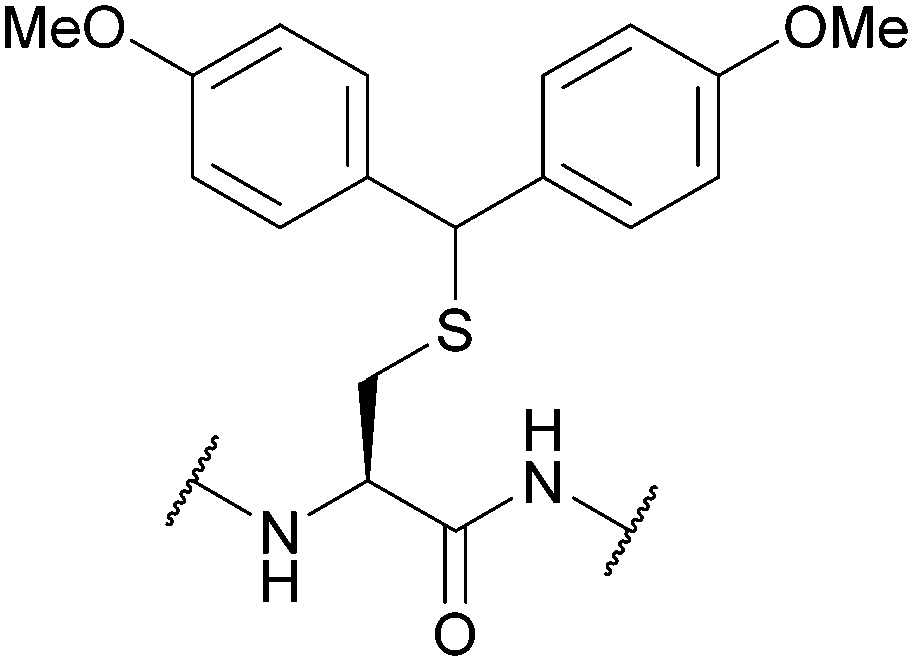
| 4,4′-Dimethoxydiphenylmethyl (Ddm); Section 5.2.19 | Acid | TFA:DCM:TIS:H2O (10:85:2.5:2.5, 1 h, 25 °C)105 |
Standard Fmoc SPPS reagents106 |
|
| Hmbon/off; Section 5.2.20 | Acid | TFA/TIS/H2O (95:2.5:2.5, 2 h, 25 °C) in Hmbon form107 |
TFA/TIS/H2O (95:2.5:2.5, 2 h, 25 °C) in Hmboff form107 |
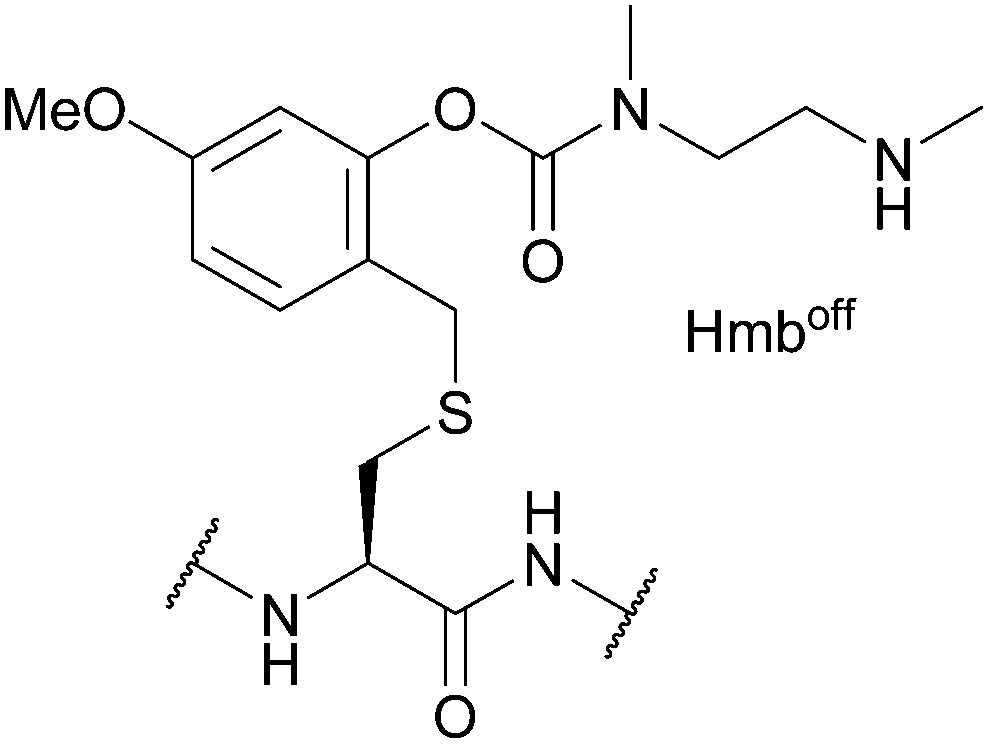
|
| Standard Fmoc SPPS reagents in Hmboff form107 | ||||
| Standard NCL/desulfurisation/HPLC reagents in Hmbon form107 | ||||
| Acetamidomethyl (Acm); Section 5.3.1 | Oxidising agents | NpsCl (2 h, RT), then NaBH478 | TFA (25 °C)110 |
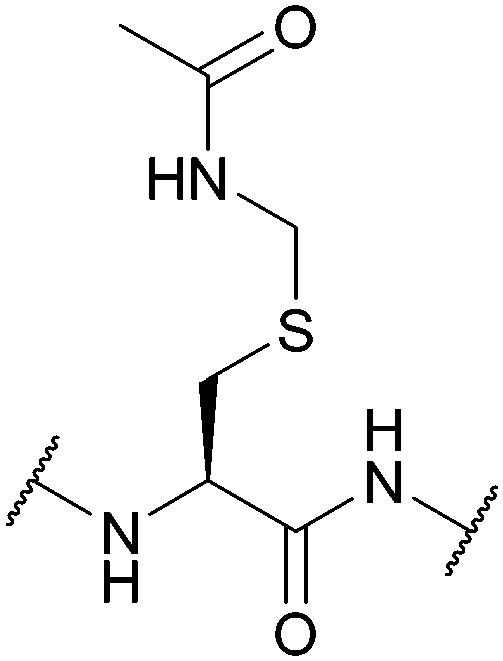
|
| Hg(II)108 Ag(I)87 Pd(II)109 | HBr/AcOH (25 °C)110 | |||
| 6 M HCl (20 h, 110 °C)110 | HCl/EtOH (25 °C)110 | |||
| 15 eq. DTNP in 97.5% TFA/thioanisole78,90 | HF (0 °C)110 | |||
| 98% TFA with scavengers86 | Pd(0)113 | |||
| I2111 | TCEP114 | |||
| Tl(TFA)3111 | Standard Fmoc SPPS reagents74 | |||
| Silyl chloride-sulfoxide/TFA111 | ||||
| PdCl2, aqueous/buffered conditions112 | ||||
| CuSO4, aqueous/buffered conditions with aminothiol source73 | ||||
| 5-Dibenzosuberyl (Dbs, Sub); Section 5.3.2 | Oxidising agents | Hg(OAc)2115 | TFA115 |
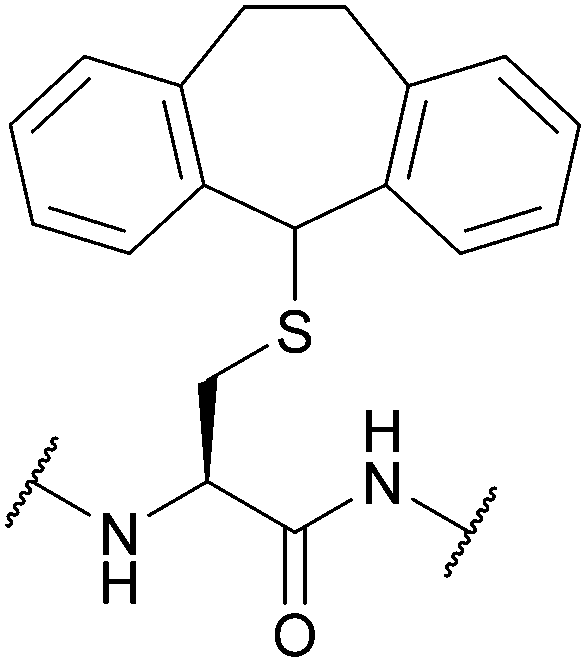
|
| I2/MeOH or AcOH115 | ||||
| Tl(TFA)383 | ||||
| Benzamidomethyl (Bam); Section 5.3.3 | Oxidising agents | Hg(OAc)2 (1 h, RT)116 | 1 M HCl (25 °C)116 |
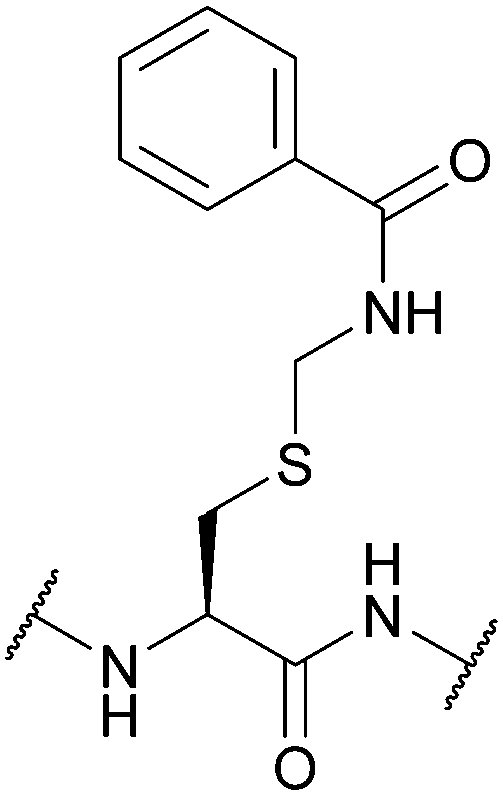
|
| Ag(OTf)/TFA/anisole (1 h, 0 °C)87 | 1 M NaOH (25 °C)116 | |||
| Silyl chloride-sulfoxide/TFA82 | TFA (25 °C)116 | |||
| 6 M HCl (24 h, 110 °C)116 | 90% Zn/AcOH (0 °C)116 | |||
| Standard Boc SPPS reagents116 | ||||
| (2-Oxo-1-pyrrolidinyl)methyl (Pym); Section 5.3.4 | Oxidising agents | HF117 | Solution phase Boc reagents117 |
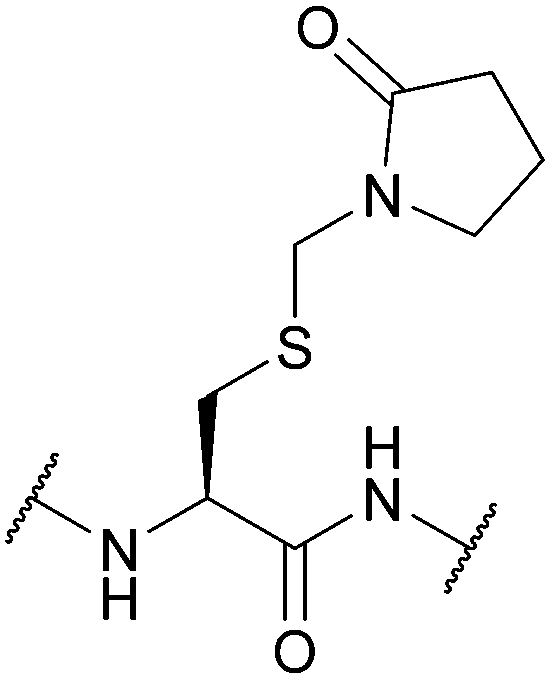
|
| NaBO3117 | ||||
| Dimethylphosphinothioyl (Mpt); Section 5.3.5 | Oxidising agents | AgNO3 (2–4 equiv., 20 min, 1 h, 0 °C) in H2O118 | 2 M HCl/EtOAc118 |
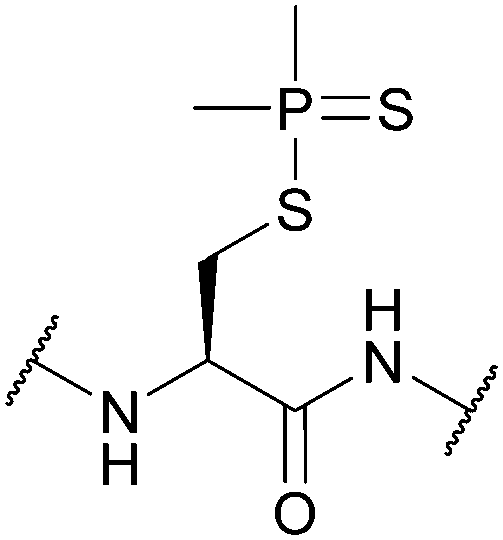
|
| Hg(OAc)2 (1–3 equiv., 1 h, 0 °C) in H2O118 | 1 M HCl/MeOH118 | |||
| TBAF in THF (free thiol)119 | 2 M HCl/AcOH118 | |||
| TBAF in DMF (disulfide)119 | 1 M HCl/H2O118 | |||
| TBAF in DCM (–S–CH2–S– formation)119 | TFA118 | |||
| Solution phase Boc reagents118 | ||||
| Trimethyl-acetamidomethyl (Tacm); Section 5.3.6 | Oxidising agents | Hg(OAc)2120 | HF (1 h, 0 °C)117 |
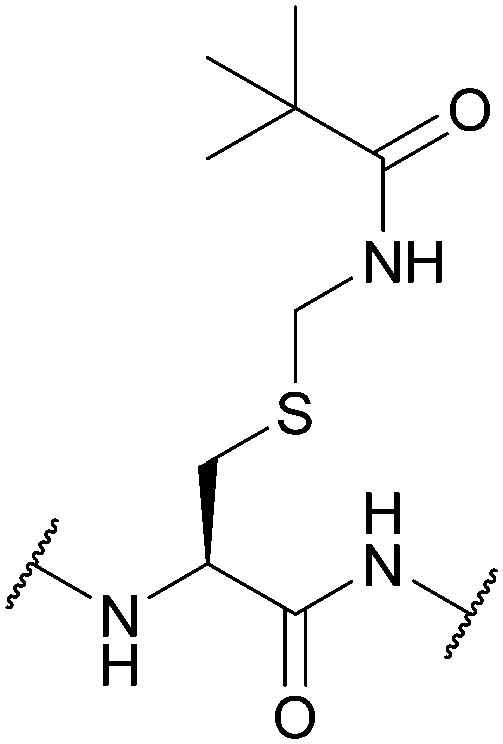
|
| AgBF4/TFA/thioanisole121 | TFMSA/thioanisole/TFA (2 h, 0 °C)117 | |||
| I2/EtOH in AcOH120 | 0.05 M NaOH in MeOH (aq., 1 h, 0 °C)117 | |||
| Hydrazine/MeOH (24 h, RT)117 | ||||
| Zn in 90% AcOH (1 h, 25 °C)117 | ||||
| Standard Boc SPPS reagents93 | ||||
| 9-Fluorenylmethyl (Fm); Section 5.4.1 | Base | NH3/MeOH122 | TFA93 |
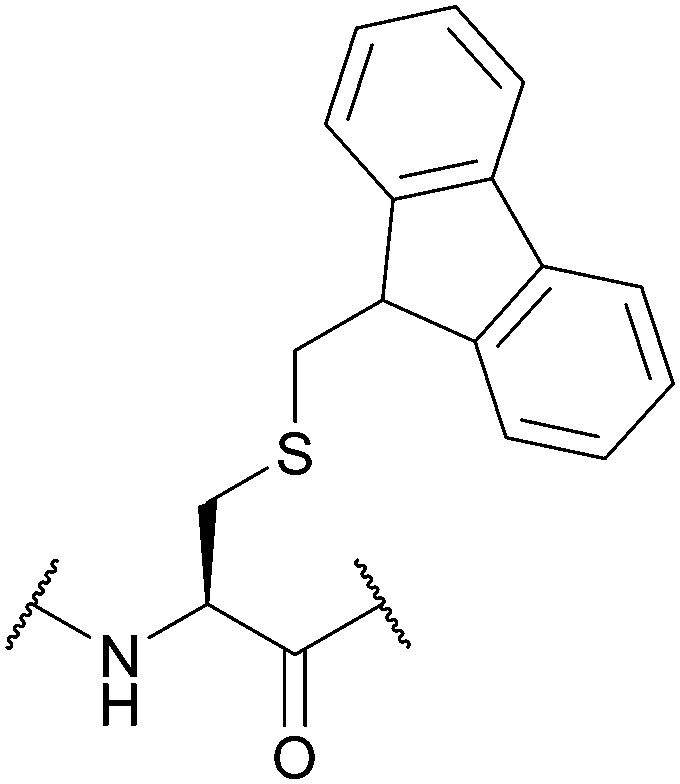
|
| 50% Piperidine in DMF (2 h, RT)123 | TFMSA/TFA93 | |||
| HCl (110 °C)93 | ||||
| HF/anisole (95:5, 1 h, 0 °C)93 |
||||
| 0.1 M I2/DMF123 | ||||
| H2, Pd/C122 | ||||
| Standard Boc SPPS reagents123 | ||||
| 2-(2,4-Dinitrophenyl)ethyl (Dnpe); Section 5.4.2 | Base | 50% Piperidine in DMF (30 min)124 | 5% DIEA in DCM (2 h)124 |
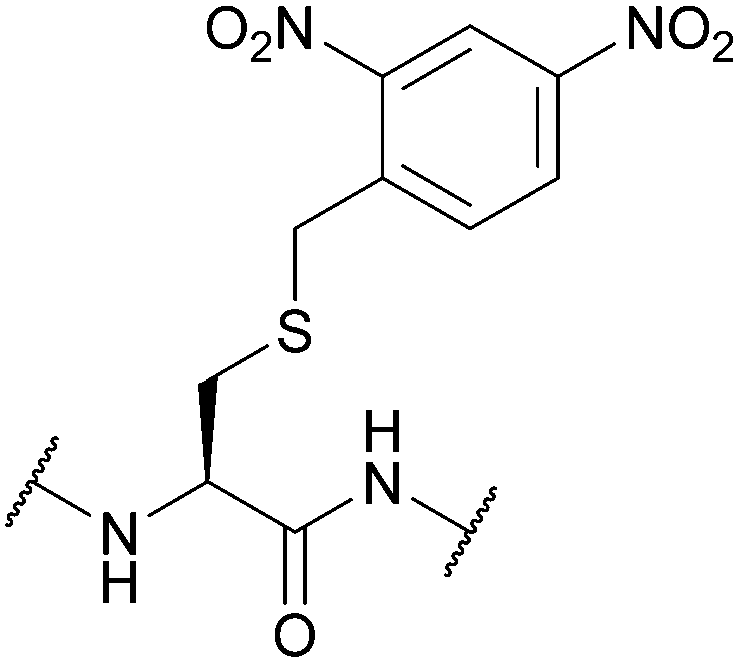
|
| Dilute DBU68 | 40% TFA in DCM (24 h)124 | |||
| 90% HF/p-cresol or anisole (1 h, 0 °C)124 | ||||
| Tl(TFA)3/TFA124 | ||||
| I2 in 80% AcOH (aq.)124 | ||||
| Standard Boc SPPS reagents124 | ||||
| 9-Fluorenylmethyl-oxycarbonyl (Fmoc); Section 5.4.3 | Base | Et3N, then NH3/MeOH or | 4 M HCl in dioxane125 |
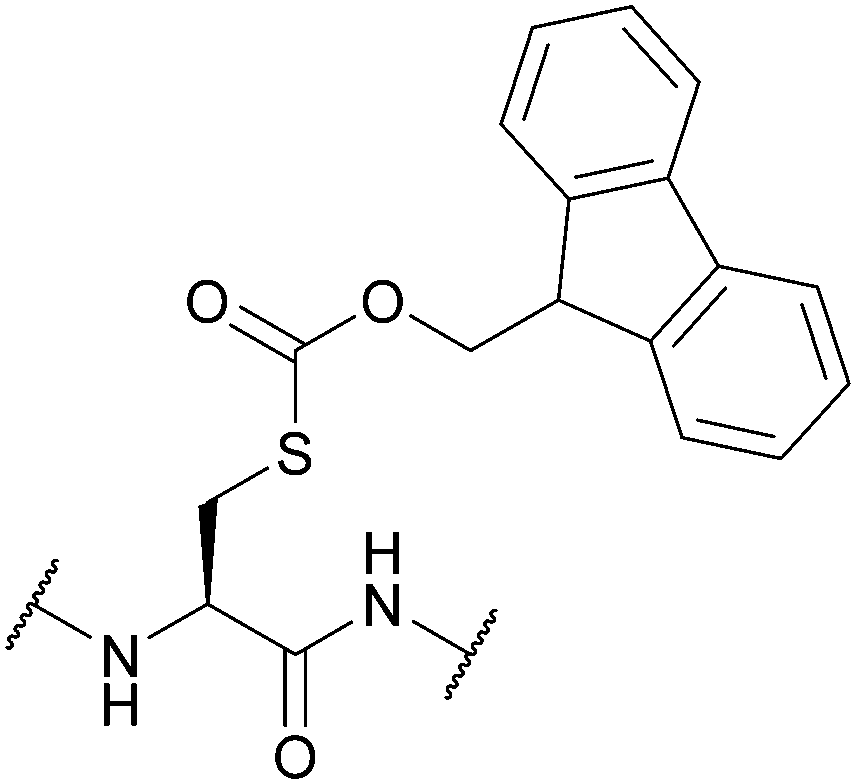
|
| 50% Piperidine in DMF (2 h, RT)125 | ||||
| Phenyl-acetamidomethyl (Phacm); Section 5.5.1 | Enzyme, oxidising agents | Penicillin G acylase (pH 7.9 buffer)126 | 5% DIEA in DCM (24 h, 25 °C)126 |
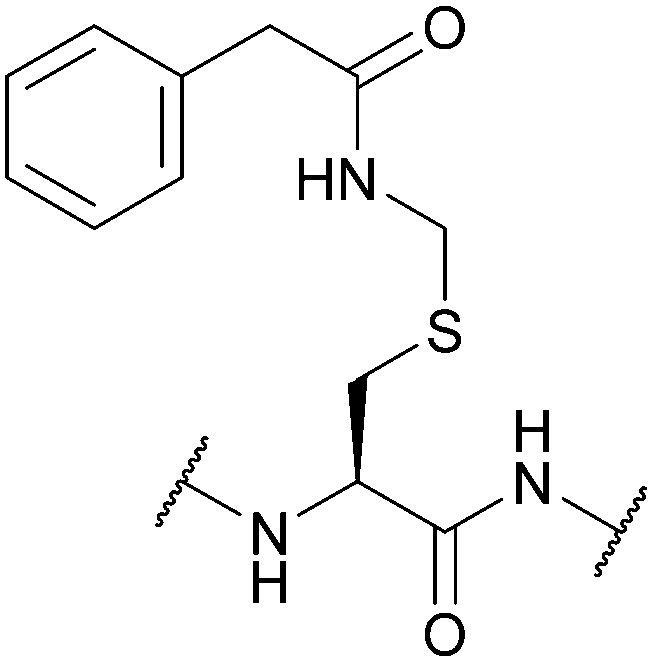
|
| Hg(II)127 Ag(II)127 | 40% TFA in DCM (24 h, 25 °C)126 | |||
| I2127 | 25% piperidine in DMF (24 h, 25 °C)126 | |||
| Tl(TFA)3127 | 0.1 M TBAF in DMF (24 h, 25 °C)126 | |||
| 5% DBU in DMF (24 h, 25 °C)126 | ||||
| 90% HF/anisole or p-cresol (1 h, 0 °C)126 | ||||
| 90% TFA/scavengers (2 h, 25 °C)126 | ||||
| Standard Boc SPPS reagents126 | ||||
| Standard Fmoc SPPS reagents126 | ||||
| Hydroxyglycine-Acm (Hgm); Section 5.6.1 | Hydrazine | 5% hydrazine in H2O (pH 8.5, 3 days, 37 °C)128 | Standard Fmoc SPPS reagents128 |
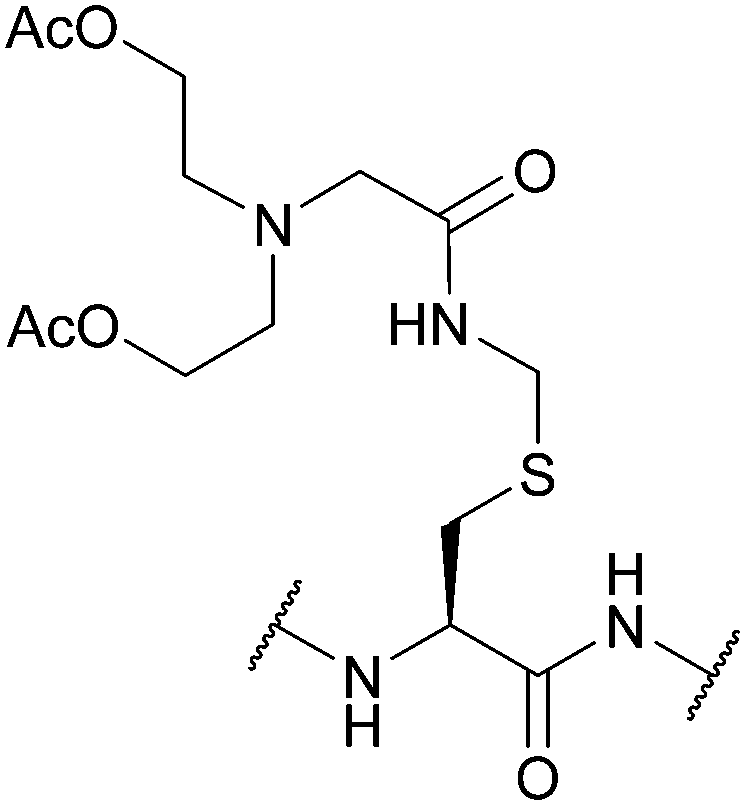
|
| Standard Boc SPPS reagents128 | ||||
| Hydroxyquinoline-Acm (Hqm); Section 5.6.1 | Hydrazine | 5% hydrazine in H2O (pH 8.5, 8 h, 37 °C)128 | Standard Fmoc SPPS reagents128 |
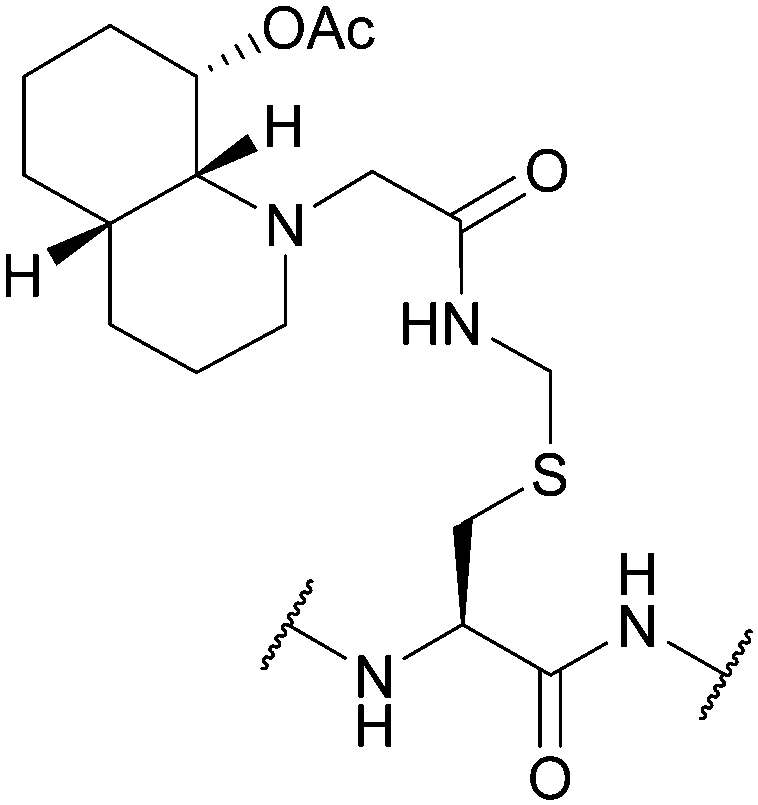
|
| I2 (30 min)128 | Standard Boc SPPS reagents128 | |||
| AgOAc (30 min)128 | ||||
| Allyloxycarbonyl (Alloc); Section 5.7.1 | Pd, base | Pd(0) cat./Bu3SnH/AcOH126 | TFA/DCM (24 h, 50 °C)129 |
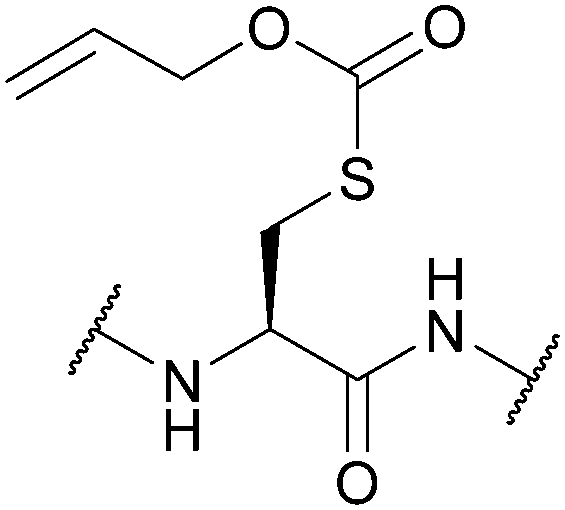
|
| Piperidine (3 h, 30 °C)129 | Standard Boc SPPS reagents129 | |||
| Allyloxy-carbonylaminomethyl (Allocam); Section 5.7.2 | Pd | Pd(0) cat./Bu3SnH/AcOH (10 min, RT)130 | Piperidine130 |
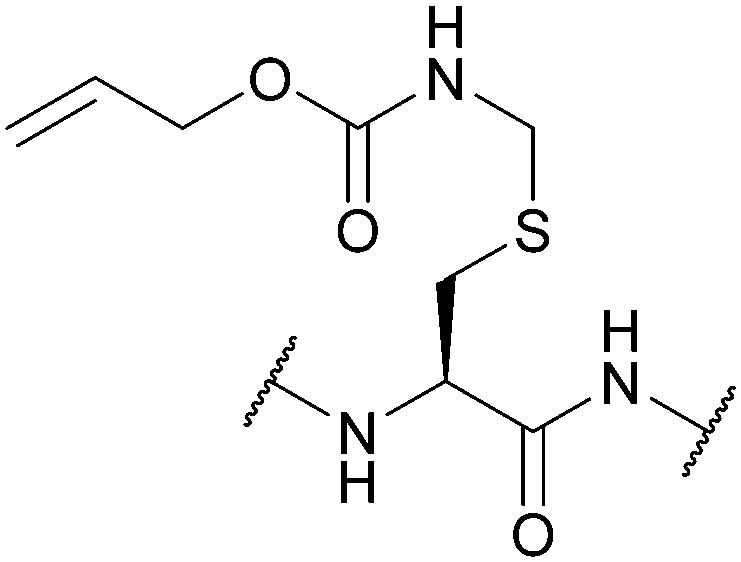
|
| Pd(OAc)2/NMM/AcOH in DMSO (2 h, disulfide)131 | Acids (partially)130 | |||
| Standard Fmoc SPPS reagents130 | ||||
| [N-[2,3,5,6-Tetrafluoro-4-(N′-piperidino)-phenyl], N-allyloxycarbonyl]-aminomethyl (Fnam); Section 5.7.3 | Pd, oxidising agents | Pd(0) cat./allyl scavenger then AcOH/BME132 | Acid132 |
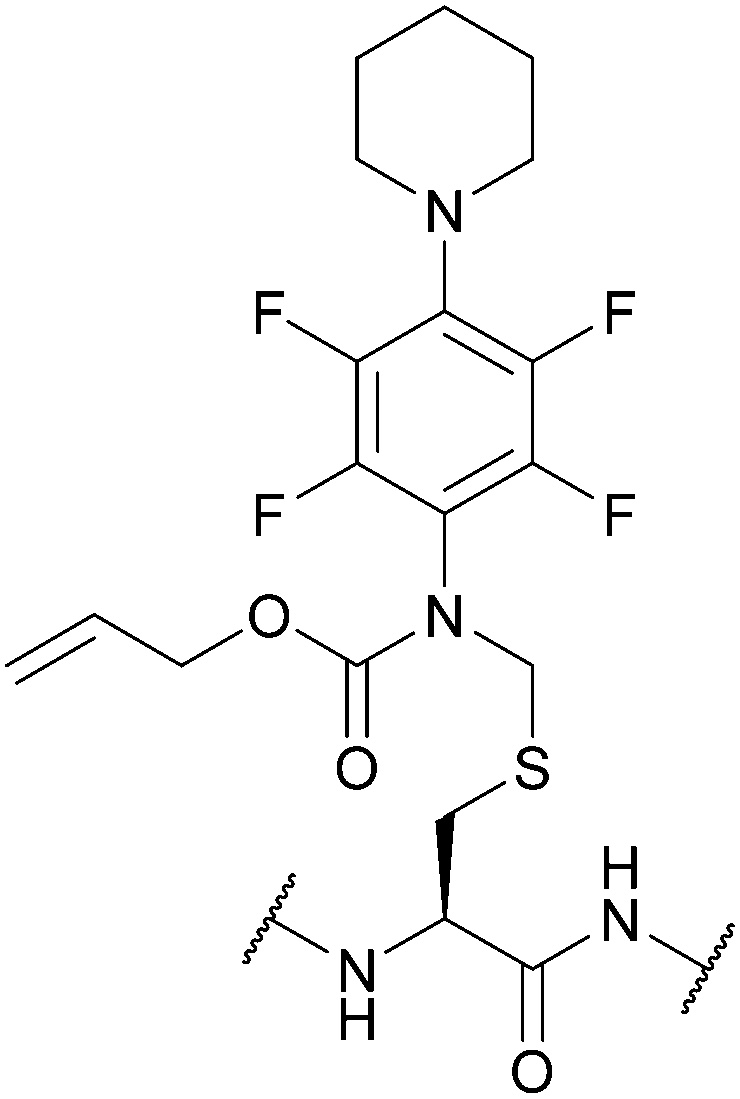
|
| Heavy metal salts132 | Base132 | |||
| Tl(TFA)3132 | Standard Boc SPPS reagents132 | |||
| Standard Fmoc SPPS reagents132 | ||||
| S-[N-[2,3,5,6-Tetrafluoro-4-(phenylthio)-phenyl], N-allyloxycarbonyl]-aminomethyl (Fsam); Section 5.7.4 | Pd, oxidising agents | Pd(0) cat./allyl scavenger then AcOH/BME133 | Acid133 |
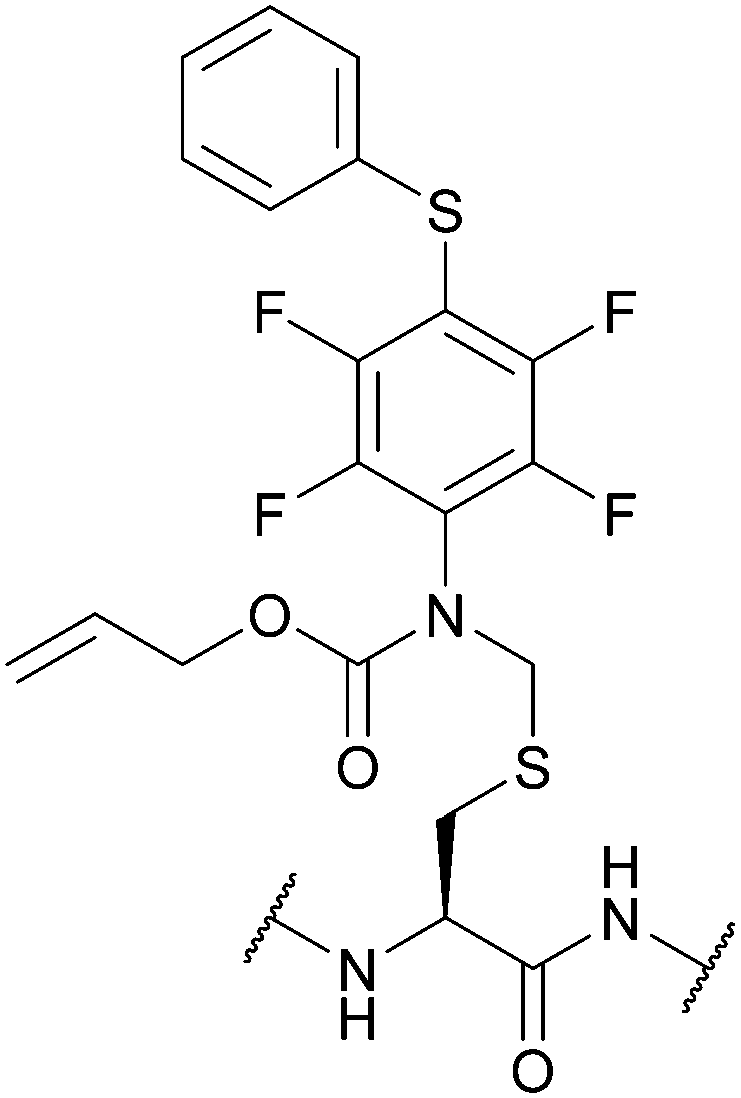
|
| I2 and other oxidants133 | Base133 | |||
| Standard Boc SPPS reagents133 | ||||
| Standard Fmoc SPPS reagents133 | ||||
| Allyl (Sac); Section 5.7.5 | Pd | Pd(tppts)4134 | Unnatural amino acid mutagenesis134 |
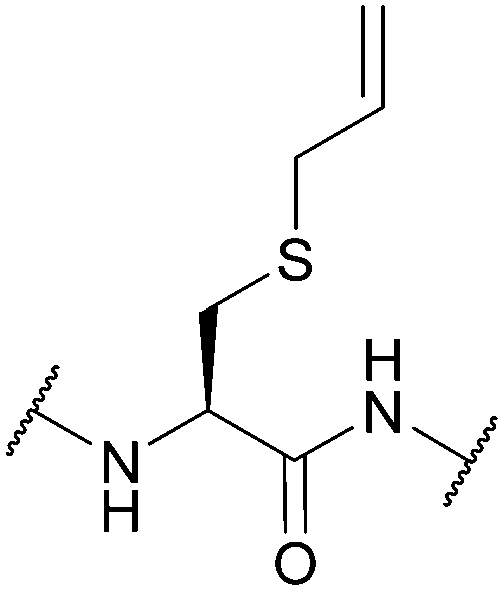
|
| S-Propargyl-cysteine (SprC); Section 5.7.6 | Pd | Pd(tppts)4135 | Unnatural amino acid mutagenesis |
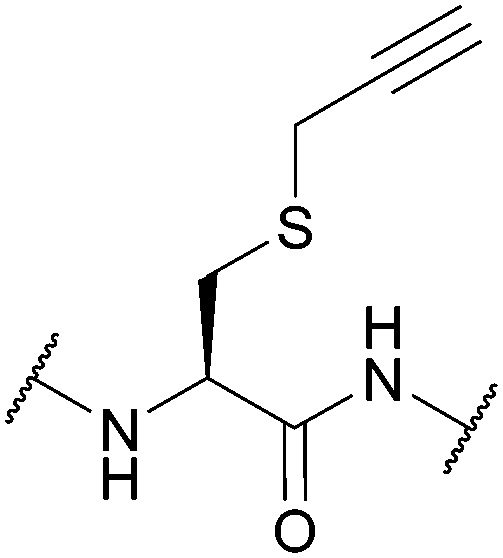
|
| CuAAC, Sonagashira coupling (alkyne functionality can be used for conjugation without cleavage)135 | ||||
| Succinimide (Suc); Section 5.7.7 | Pd | PdCl2 and MgCl2, 6 M Gdn.HCl/0.2 M phosphate buffer pH 5.5, 37 °C then DTT136 | Aqueous solution phase conditions136 |
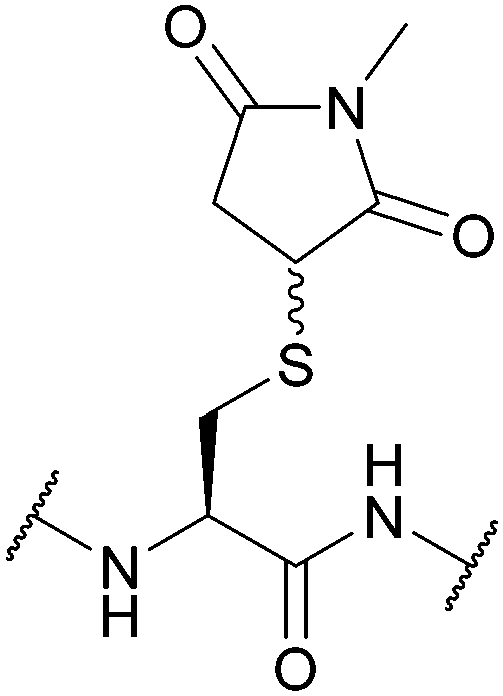
|
| Desulfurisation conditions136 | ||||
| Thiazolidine (Thz); Section 5.8.1 | N-terminal | H2O2137 I2137 | PdCl2/H2O85 |
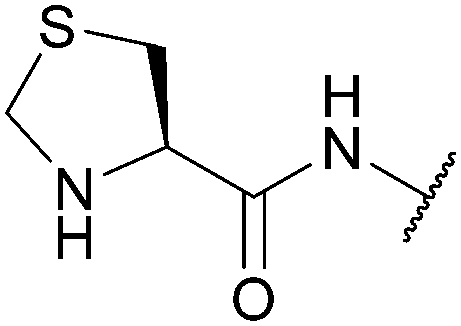
|
| Pd, oxidising agents, Cu | Iodoacetic acid/benzyl chloride (pH 10-11, RT)137 | Standard NCL reagents139 | ||
| Air/ferric chloride (trace, pH 10)137 | ||||
| Excess methoxyamine at pH 4 (8 h)138 | ||||
| Pd(II) and MPAA/TCEP or GSH/6 M Gdn·HCl (pH ∼6.5, 37 °C, 45 min)85,138 | ||||
| CuSO4/sodium ascorbate/5 M Gdn·HCl/HEPPS buffer then DTT (pH 7.0, 1 h, 37 °C)139 | ||||
| 20 mM DPDS, 50% MeCN (0.1% TFA)140 | ||||
| Ninhydrin (Nin); Section 5.8.2 | N-terminal | Excess Cys (pH 7.7, 30 min, 23 °C)141 | Standard Boc SPPS reagents141 |
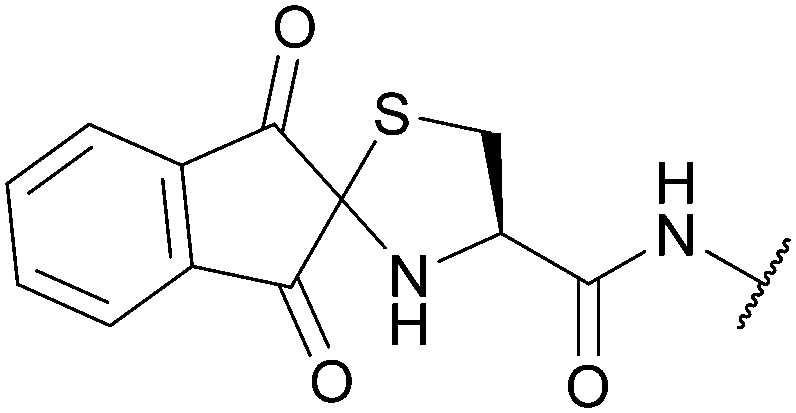
|
| Reducing agents | Cysteine O-methylester/DMF/DIEA (on resin)141 | |||
| Excess MPS, pH 7141 | ||||
| 10% TFA/H2O/Zn dust, 1 h141 | ||||
| 2-Nitrobenzyl (oNB); Section 5.9.1 | Light | hν ≥ 350 nm142 | Standard Fmoc SPPS reagents143 |
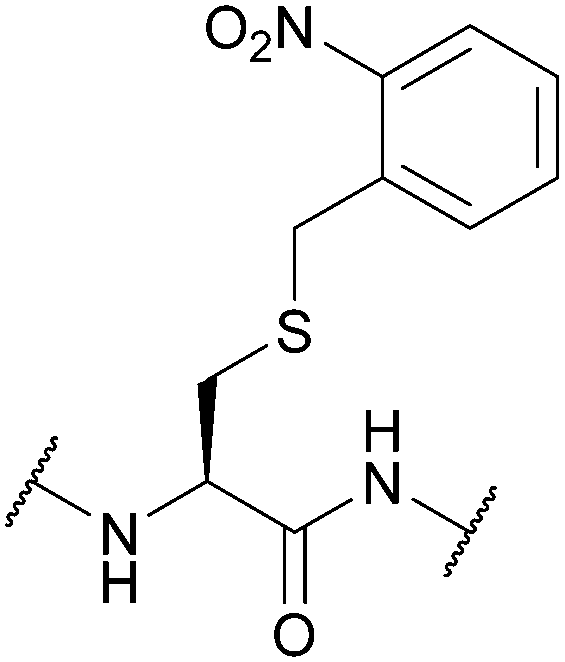
|
| Standard Boc SPPS reagents143 | ||||
| [7,8-Bis(carboxymethoxy)coumarin-4-yl]methoxycarbonyl (7,8-BCMCMOC); Section 5.9.2 | Light | hν ≥ 325 nm144 | TFA144 |
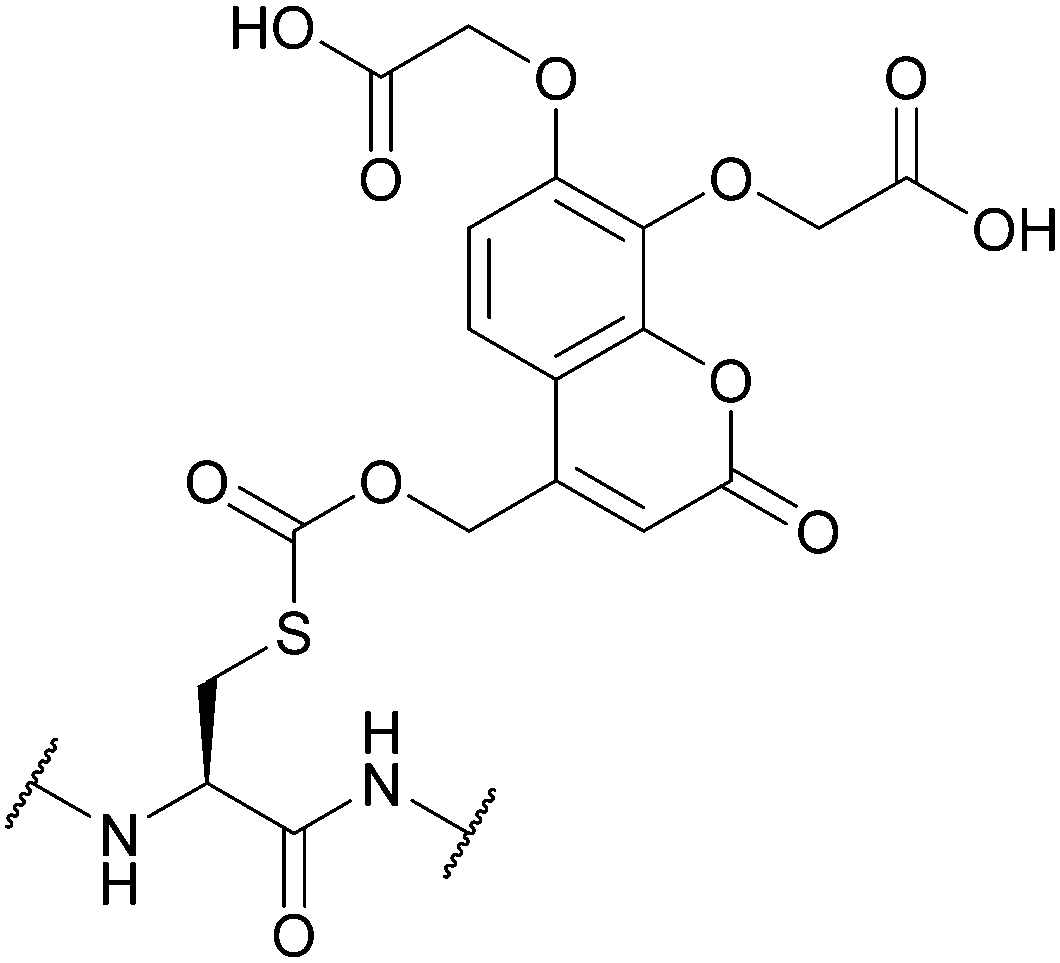
|
| Thiolysis144 | ||||
| [7-Bis(carboxymethyl)-amino-coumarin-4-yl]methoxycarbonyl (BCMACMOC); Section 5.9.2 | Light | hν ≥ 402 nm144 | TFA144 |
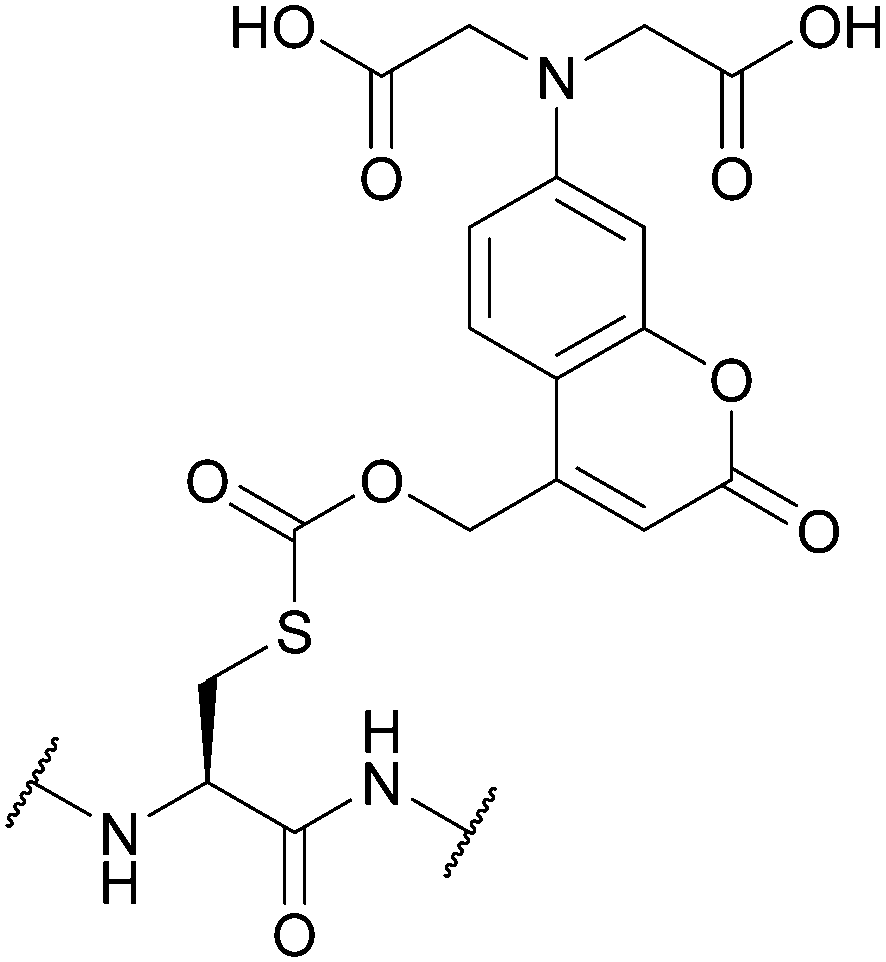
|
| Thiolysis144 | ||||
| α-Carboxy-4-methoxy- 2-nitrobenzyl (CDMNB); Section 5.9.2 | Light | hν ≥ 325 nm144 | TFA144 |
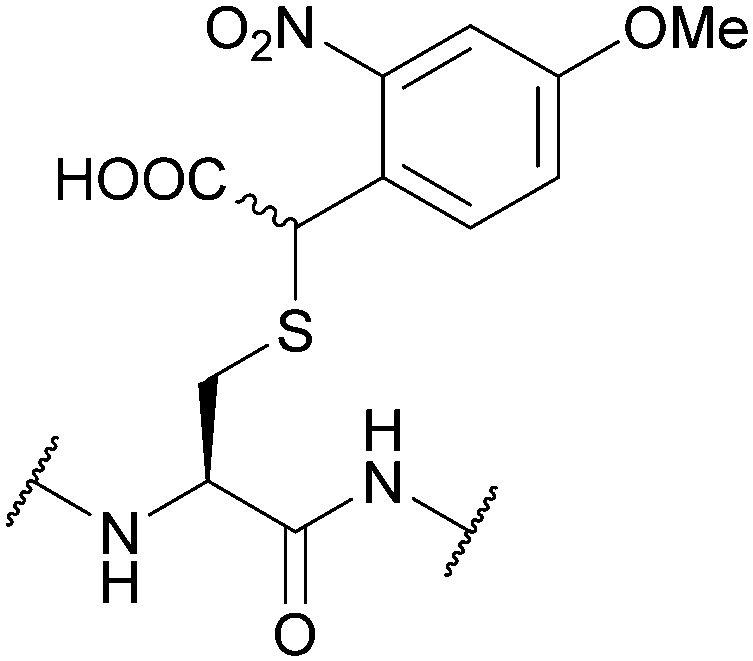
|
| Thiolysis144 | ||||
| Piperidine144 | ||||
| 2-Nitroveratryl | Light | hν = 350 nm (30 min)142 | 10% TFMSA/TFA/excess dipyridine disulfide142 |
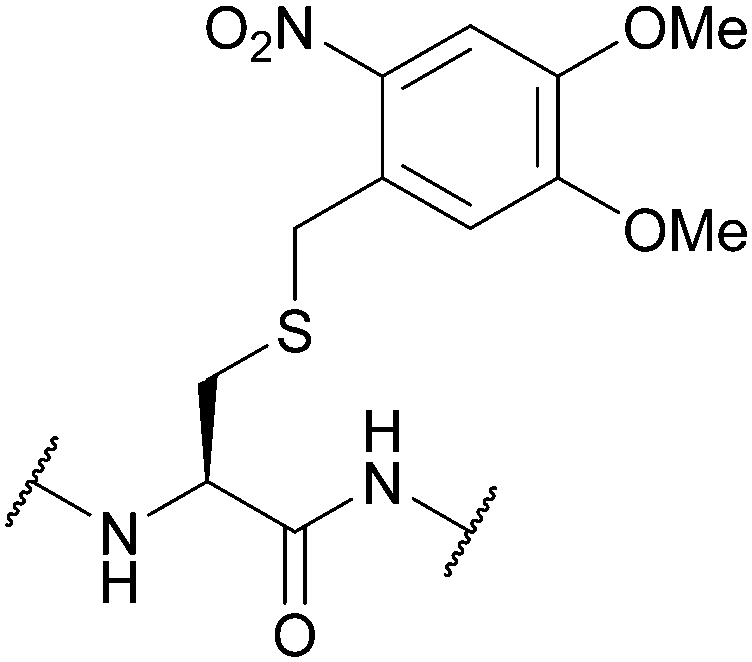
|
| 6-Nitroveratryl | Standard Fmoc SPPS reagents142 | |||
| 4,5-Dimethoxy-2-nitrobenzyl (oNV, DMNB); Section 5.9.3 | ||||
| 6-Bromo-7-hydroxycoumarin (Bhc); Section 5.9.4 | Light | hν = 365 nm in photolysis buffer (1 mM DTT in 50 mM PB, pH 7.2)145 | Standard Fmoc SPPS reagents145 |
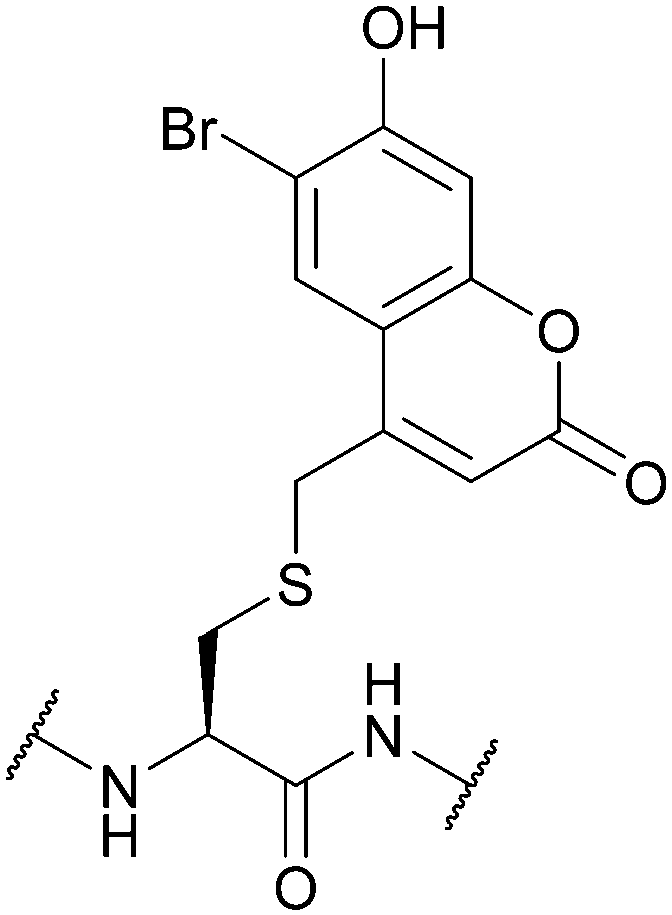
|
| Nitrodibenzofuran (NDBF); Section 5.9.5 | Light | hν = 365 nm145 | Standard Fmoc SPPS reagents145 |
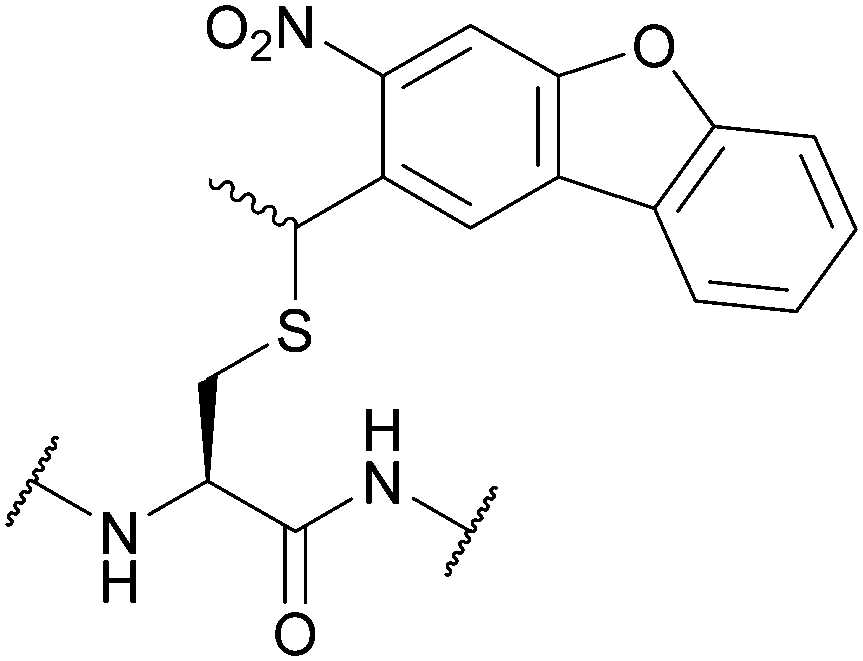
|
| 6-Bromo-7-hydroxy-3-methylcoumarin (mBhc); Section 5.9.6 | Light | hν = 365 nm146 | Standard Fmoc SPPS reagents146 |
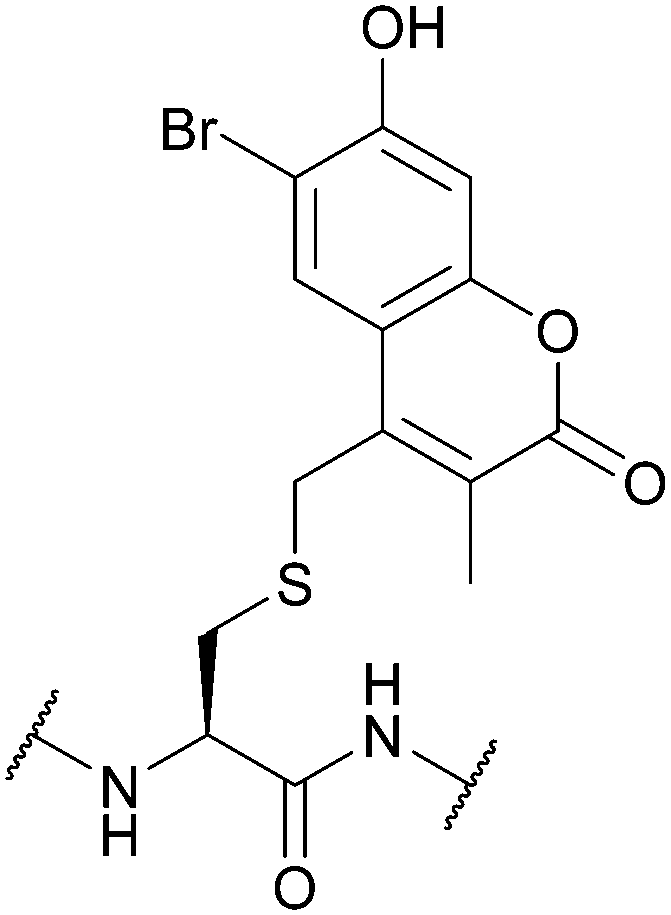
|
| Methoxy-nitrodibenzofuran (OMe-NDBF); Section 5.9.7 | Light | hν ≥ 350 nm147 | Standard Fmoc SPPS reagents147 |
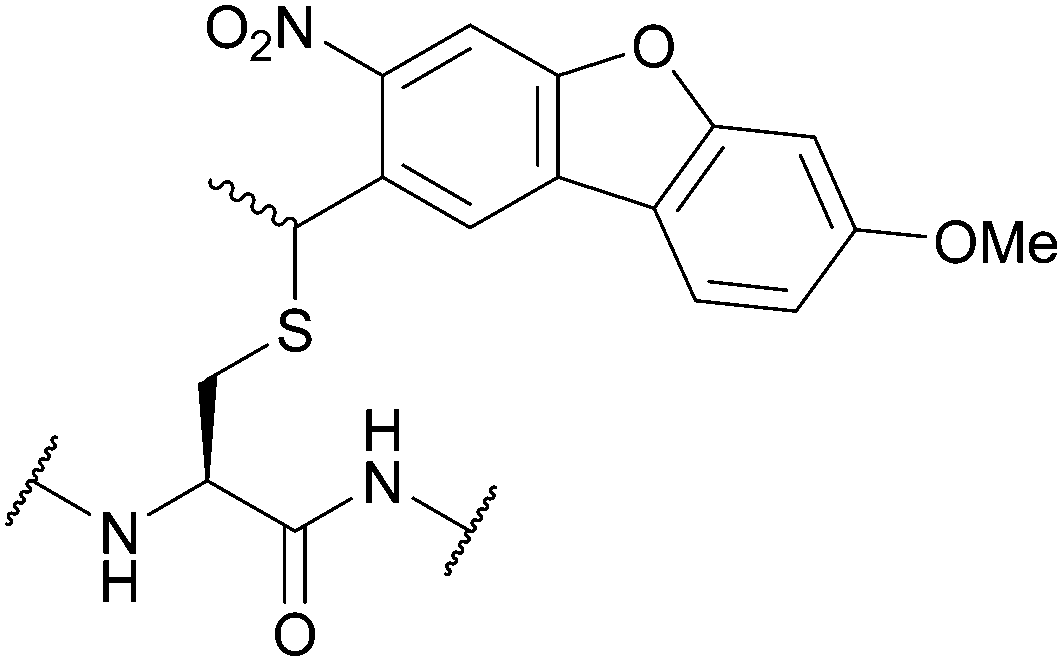
|
| para-Nitrobenzyl (pNB); Section 5.10.1 | Reducing agents | Zn/AcOH, then I2 (in solution)111 | HF/p-cresol (9:1, 1 h, 0 °C)111 |
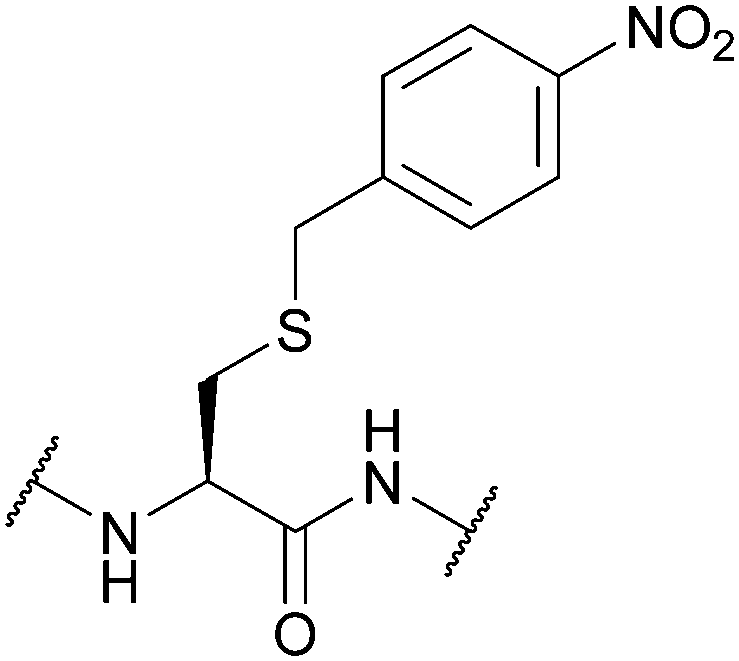
|
| SnCl2/HCl, then I2 (on resin)111 | TFA111 | |||
| Excess CAN/Hopkins reagent111 | I2/AcOH/2 M HCl111 | |||
| H2, Pd/C, then oxidant111 | Standard Boc SPPS reagents111 | |||
| Carbomethoxysulfenyl (Scm); Section 5.10.2 | Reducing agents | DTT148 | Strong acids – anhydrous HF, TFMSA149 |
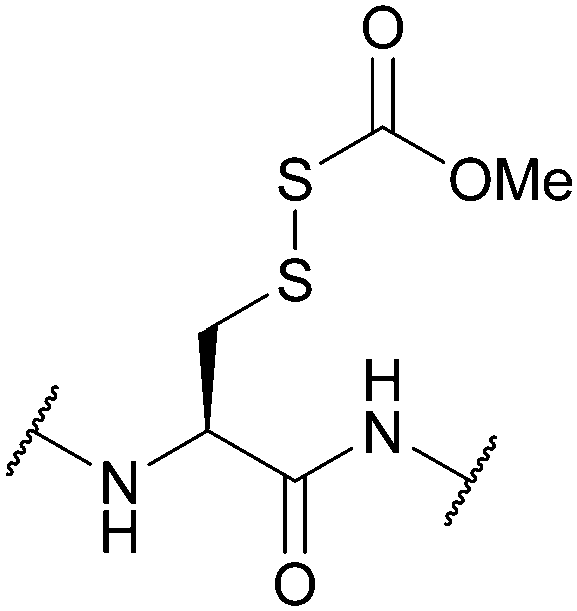
|
| Standard Boc SPPS reagents149 | ||||
| (N′-Methyl-N′-phenylcarbamoyl)sulfenyl (Snm); Section 5.10.3 | Reducing agents | DTT/NMM/CDCl3149 | Strong acids – anhydrous HF, TFMSA149 |
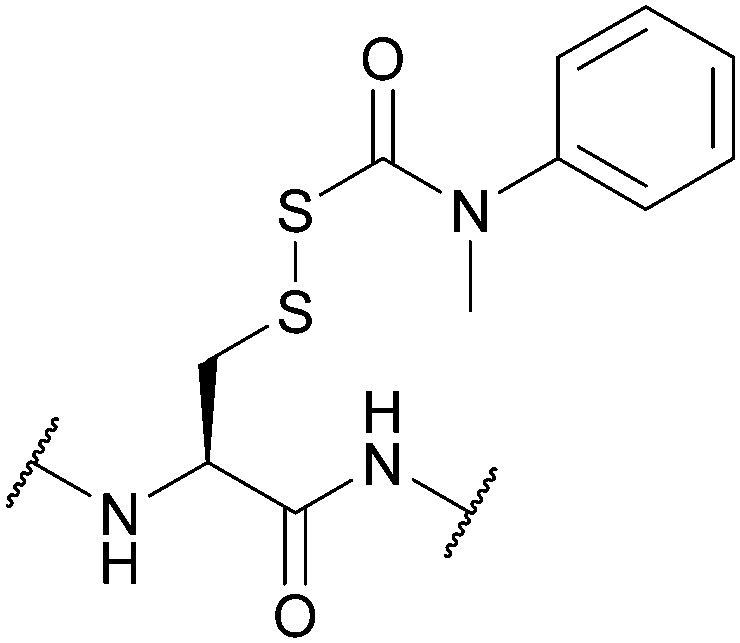
|
| Standard Boc SPPS reagents149 | ||||
| 4-Picolyl; Section 5.10.4 | Reducing agents | Zn/AcOH150 | TFA151 |
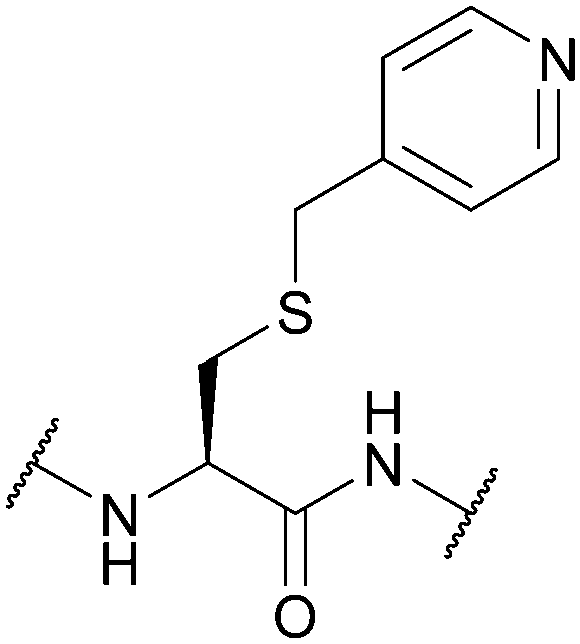
|
| Electrolytic reduction in 0.5 M H2SO4151 | 32% HBr/AcOH (1 week, RT)151 | |||
| Standard Boc SPPS reagents151 | ||||
| Sulfonic acid/sulfonyl (SO3H/SO2R); Section 5.10.5 | Reducing agents | Thiols152 | Standard Fmoc SPPS reagents153 |
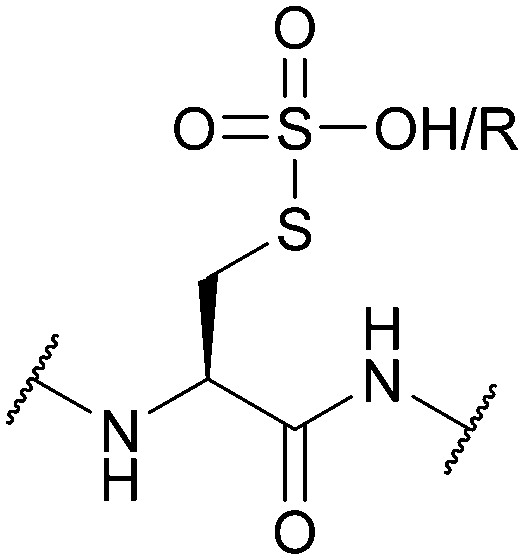
|
| PBu3153 | Standard Boc SPPS reagents153 | |||
| 3-Nitro-2-pyridinesulfenyl (Npys); Section 5.10.6 | Reducing agents | Aliphatic thiols154 | TFA (24 h, RT)155 |
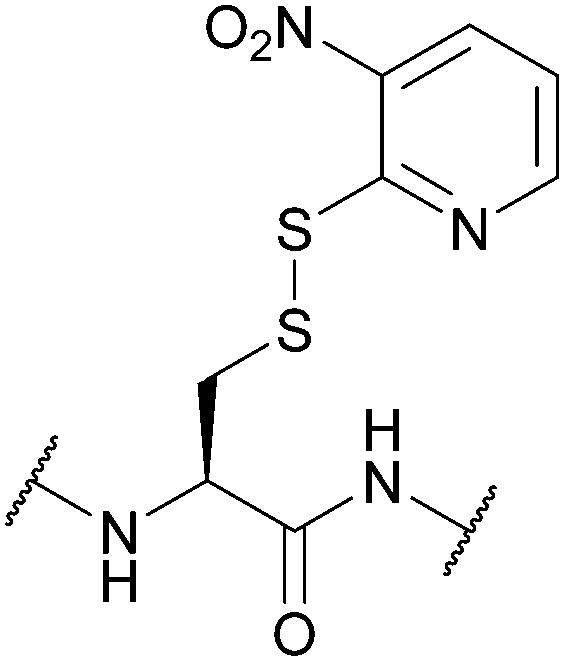
|
| Tertiary phosphine/H2O155,156 | HF (1 h, RT)155 | |||
| 4 M HCl/dioxane (24 h)155 | ||||
| DCM157 DMF157 MeOH157 | ||||
| N,N-Dimethylacetaminde157 | ||||
| N-Methylpyrrolidone157 Trifluoroethanol157 | ||||
| Pentafluorophenol157 | ||||
| Standard Boc SPPS reagents155 | ||||
| 5-Nitro-2-pyridinesulfenyl (5-Nyps); Section 5.10.7 | Reducing agents | Thiols158 | Standard Boc SPPS reagents (see Nyps)158 |
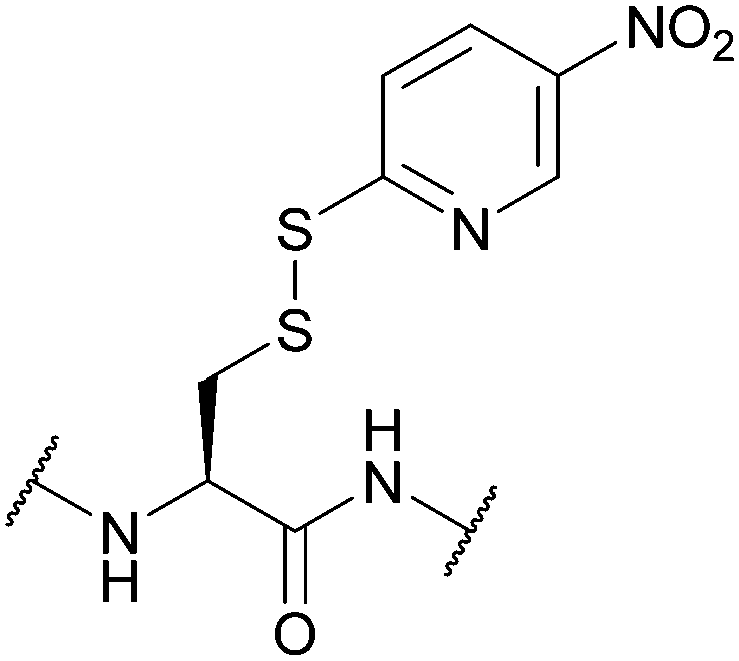
|
| tert-Butylsulphenyl (StBu); Section 5.10.8 | Reducing agents | Thiols159 | Acid159 |
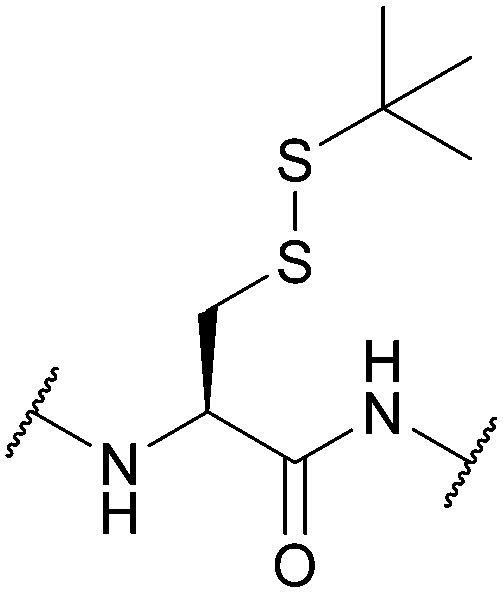
|
| Phosphines114 | Base159 | |||
| TFA/DTNP/thioanisole80 | TFA/DNTP80 | |||
| Standard Fmoc SPPS reagents74 | ||||
| N-Methyl-phenacyloxy-carbamidomethyl (Pocam); Section 5.10.9 | Reducing agents, acid | Zn/AcOH (aq.)160 | TFA (4 h, 4 °C)160 |
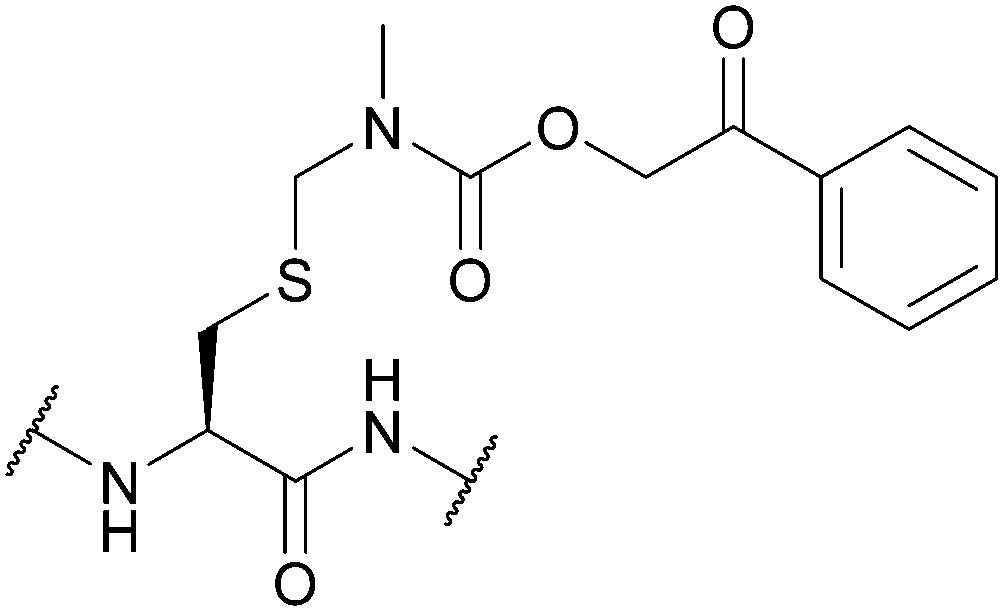
|
| TFA (1 h, 50 °C)160 | Standard Fmoc SPPS reagents160 | |||
| Phenacyl (Pac); Section 5.10.10 | Reducing agents | Zn powder, AcOH161 | Mild acid161 |
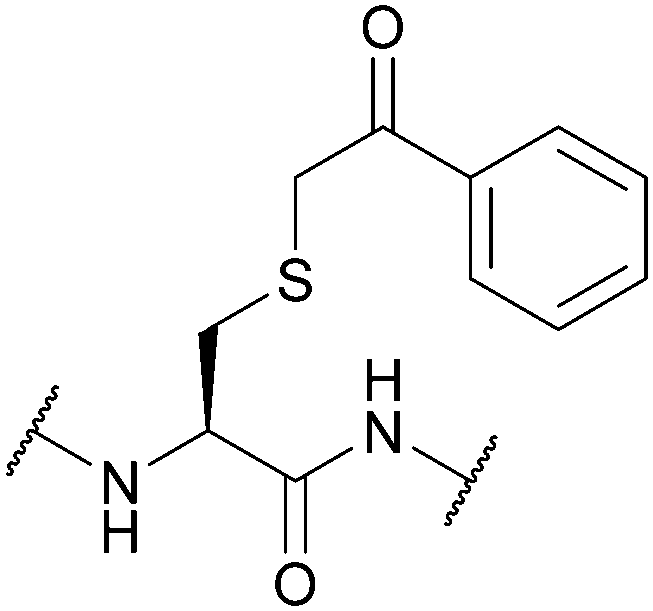
|
| Standard Fmoc SPPS reagents161 | ||||
| Standard NCL reagents162 | ||||
| S-Iso-Propyl (SiPr); Section 5.10.11 | Reducing agents | TCEP in PBS pH 7.4 | Standard Fmoc SPPS reagents114 |
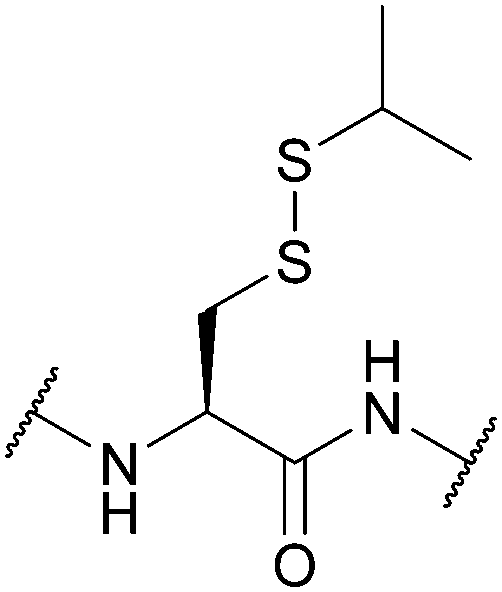
|
| 37 °C114 | ||||
| Dimethoxyphenylthio (S-Dmp); Section 5.10.12 | Reducing agents | NMM (0.1 M) then either 20% BME/DMF or 5% DTT/DMF (5 min)163 | 20% piperidine in DMF (4 h)163 |
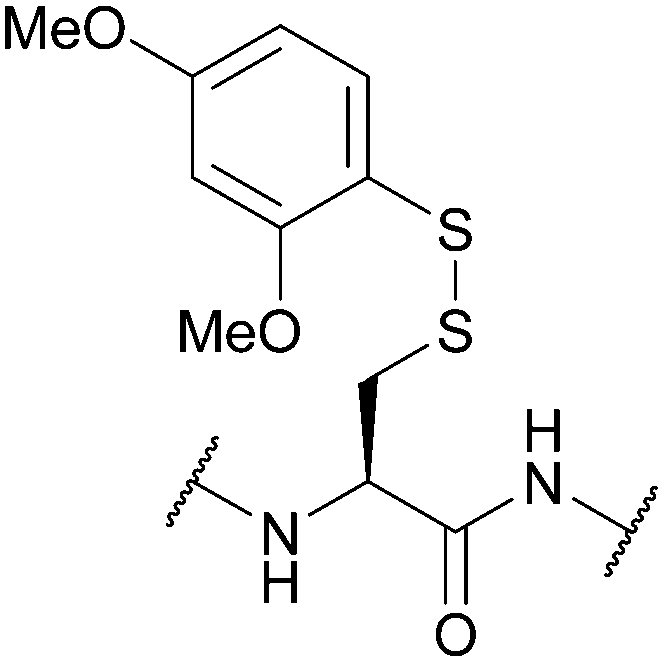
|
| DABDT, DIEA/H2O/MeCN (3:3:94)164 |
95% TFA (1 h, RT)163 | |||
| Standard Fmoc SPPS reagents163 | ||||
| 2,4,6-Trimethoxyphenylthio (S-Tmp); Section 5.10.12 | Reducing agents | NMM (0.1 M) then either 20% BME/DMF or 5% DTT/DMF (5 min)163 | 20% piperidine in DMF (4 h)163 |
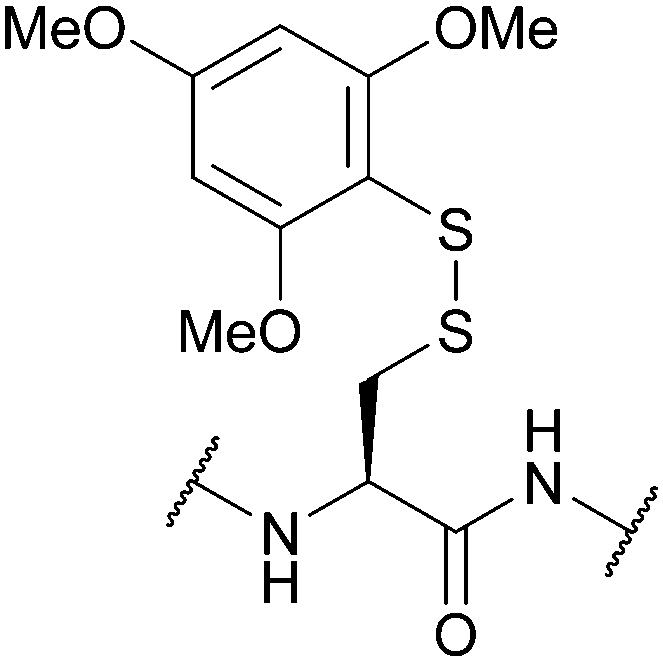
|
| 95% TFA (1 h, RT)163 | ||||
| Standard Fmoc SPPS reagents163 | ||||
| Sec-isoamyl mercaptan 3-methyl-2-butanethiol (SIT); Section 5.10.13 | Reducing agents | BME in DMF (1:4), 0.1 M DIEA165 |
Standard Fmoc SPPS reagents165 |
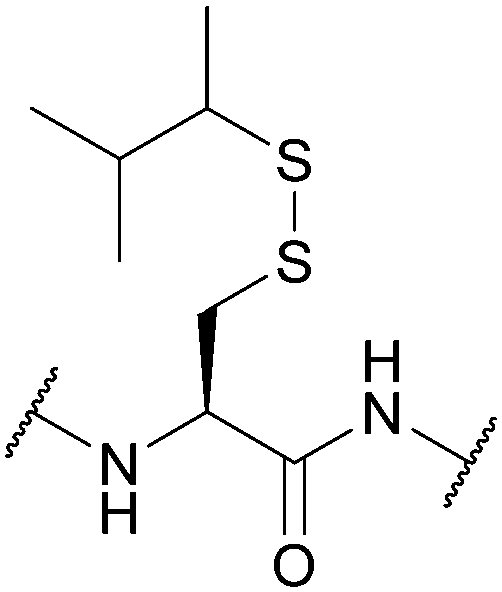
|
| 20 equiv. DTT, MeCN/DIEA/H2O (90:5:5)165 |
||||
| 5 equiv. DTT × 3, DMF/DIEA/H2O (95:2.5:2.5)165 |
||||
| 2-Methyloxolane-3-thiol (MOT); Section 5.10.13 | Reducing agents | BME in DMF (1:4), 0.1 M DIEA165 |
Standard Fmoc SPPS reagents165 |
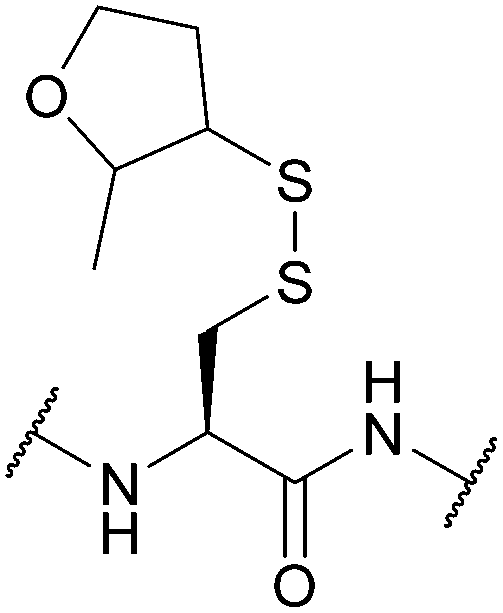
|
| 20 equiv. DTT, MeCN/DIEA/H2O (90:5:5)165 |
||||
| 5 equiv. DTT × 3, DMF/DIEA/H2O (95:2.5:2.5)165 |
||||
| 2-Pyridinesulfenyl (S-Pyr); Section 5.10.14 | Reducing agents | Thiols4 | 1 M TFMSA in TFA-anisole (10:1, 2 h, 0 °C)4 |
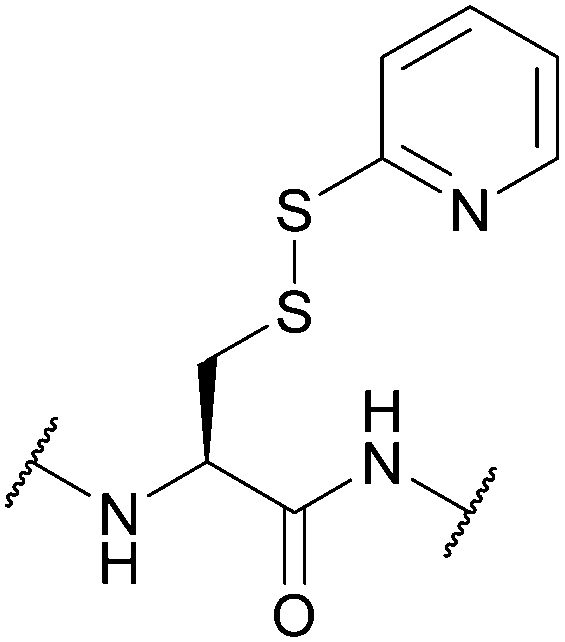
|
| Standard Boc SPPS reagents4 | ||||
| 4,4-Bis(dimethylsulfinyl)benzhydryl (Msbh); Section 5.11.1 | Safety-catch | NH4I/DMS/TFA166 | Acid (TFA, HF)166 |
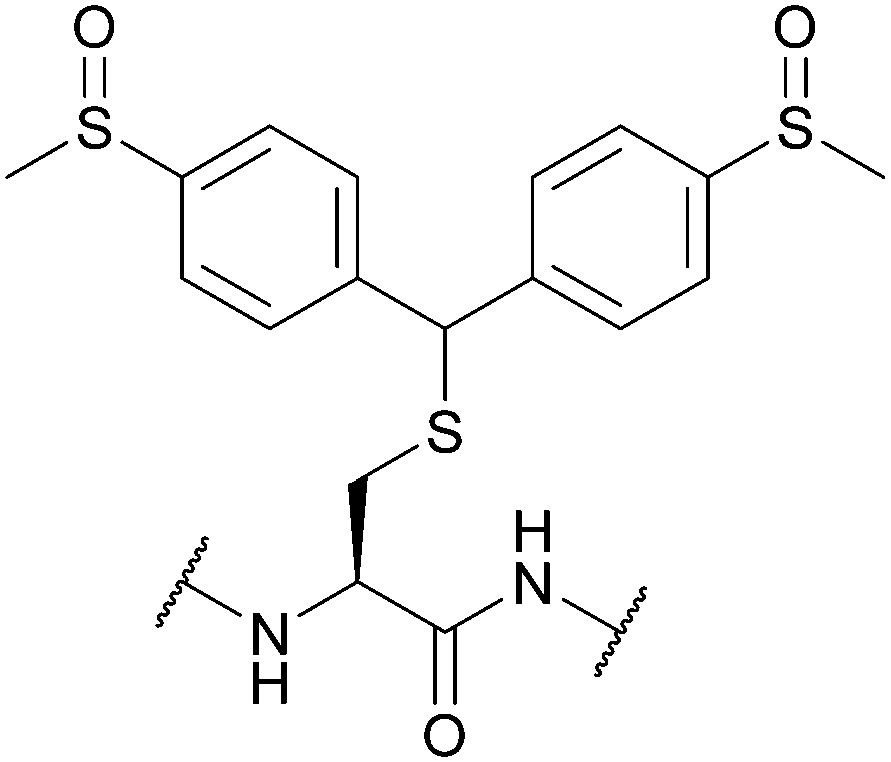
|
| Oxidants166 | ||||
| Reductants166 | ||||
| Standard Boc SPPS reagents166 |
5. Cysteine protecting groups in peptide and protein science
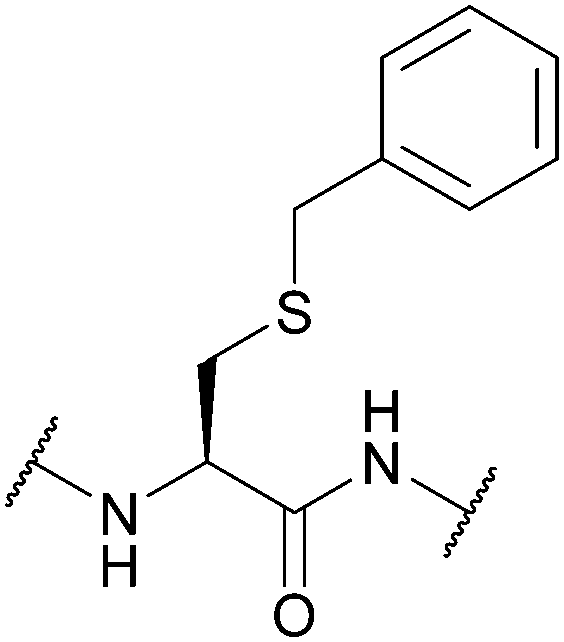
5.1 Benzyl (Bzl)
The benzyl (Bzl, Bn) protecting group (Fig. 5a) has been used for many years.67,167 One of the most noteworthy examples where the Cys(Bzl) protecting group has featured is in the landmark chemical synthesis of the hormone oxytocin in 1953 (Fig. 5b); this was the first example of a polypeptide hormone synthesis, and contributed towards Vincent du Vigneaud being awarded the Nobel Prize in Chemistry in 1955. Synthesis of other Cys-containing oxytocin analogues have also employed the Bzl protecting group.168 Additionally, Cys(Bzl) protected tripeptides have been shown to display apoptotic activity.169 Protocols for removing Bzl with Na/NH3 (liq.) have been described as far back as 1930,67 and Bzl can also be removed by treatment with HF at 25 °C,68 or with strong Lewis acids (e.g. TMSBr) in TFA-thioanisole.69 However, these removal methods have a number of significant issues associated with them. For example, using Na/NH3 (liq.) cleaves existing disulfide bonds alongside Bzl removal.70 HF gas, in particular, is expensive, not widely available, and highly toxic. TMSBr been reported to undergo side reactions with peptides and has a relatively poor solubility in diethyl ether (Et2O, a commonly used solvent for precipitation of a synthesised peptide once cleaved from the resin). TMSBr can thus precipitate with the peptide instead of being removed resulting in issues with handling following lyophilisation.90,170 Considering this, it is likely that Bzl will see diminishing use as a Cys protecting group in routine peptide synthesis, as it is superseded by newer protecting groups, benzyl-based or otherwise, that do not require such harsh conditions for removal.4,78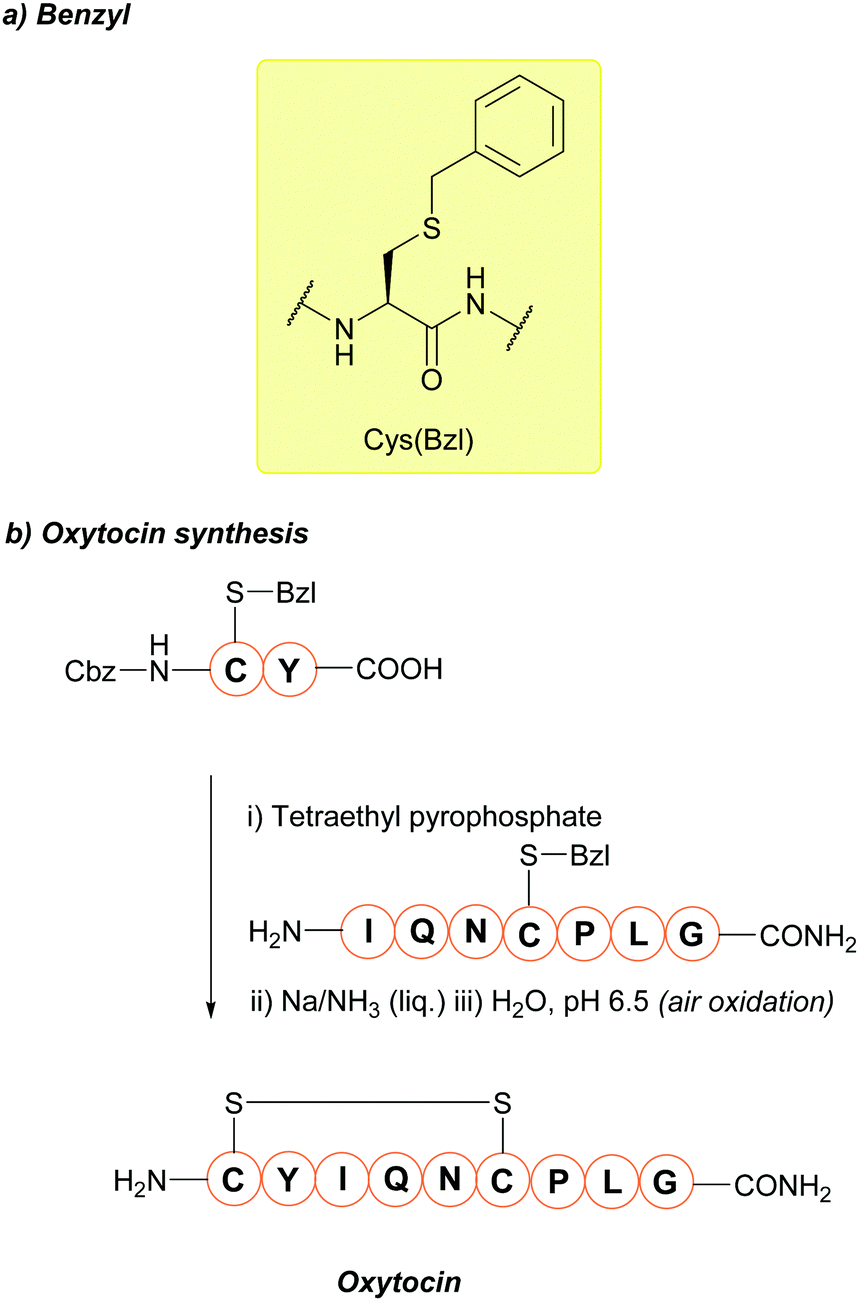 | ||
| Fig. 5 (a) Cys thiol protection with the benzyl (Bn/Bzl) protecting group (b) the synthesis of oxytocin using Cys(Bzl). | ||
5.2 Acid-labile protecting groups
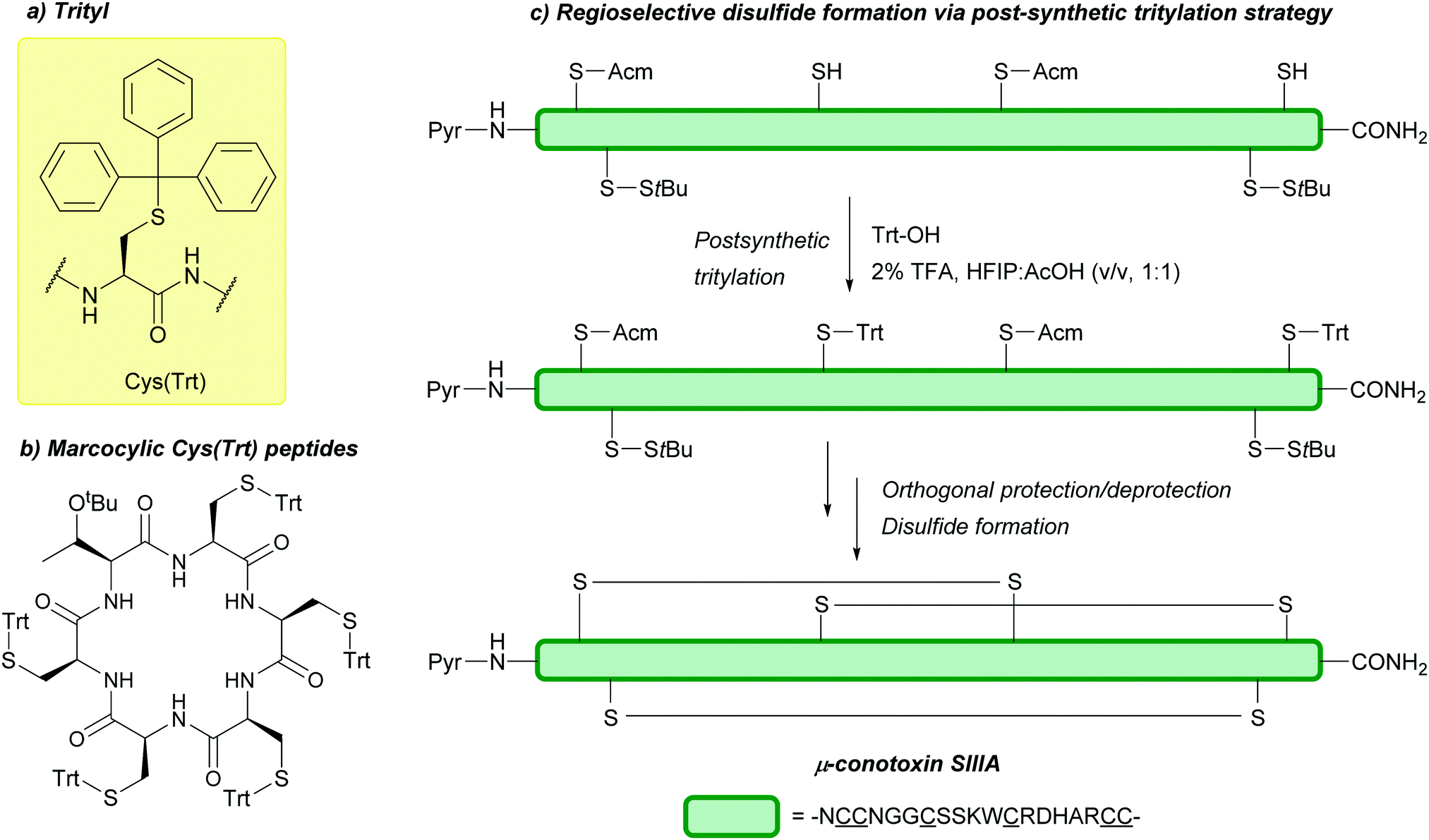 | ||
| Fig. 6 (a) Cys thiol protection with the trityl (Trt) protecting group. Examples of Cys(Trt) use include (b) macrocyclic peptide synthesis and (c) regioselective disulfide formation in μ-conotoxin SIIIA via a post-synthetic tritylation strategy. | ||
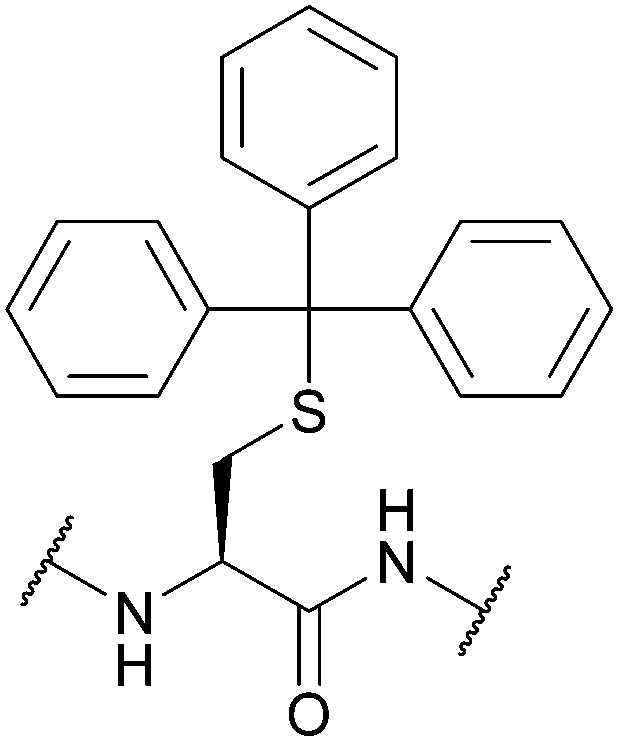
Should the desired product of Trt cleavage be a new disulfide bond, oxidising agents such as I2 may be used.4 The rate of Trt removal and subsequent disulfide formation by I2 has previously been to shown to depend on the choice of solvent.171 It is common to remove Trt using weak acids (e.g. HBF4 or TFA) in the presence of scavengers such as triisopropylsilane (TIS) or triethylsilane (TES); these scavengers prevent the released Trt cations from adding back onto the synthesised peptide during cleavage and isolation.172 This method of Trt removal renders Trt orthogonal to other common protecting groups such as Acm or tBu, as shown in the regioselective synthesis of human insulin, via sequential disulfide formation.72 Other applications of Cys(Trt) in SPPS include synthesis of human relaxin,173 and α-melanocyte stimulating hormone (α-MSH) analogues.174 Additionally, macrocyclic peptides displaying in vitro anti-malarial properties have also been synthesised using Cys(Trt); in this example, the Trt protecting group is retained rather than removed in the final bioactive product (Fig. 6b).175 Cys(Trt) has also very recently been reported to undergo complete deprotection in model peptides when treated with CuSO4 and cysteamine in aqueous buffered conditions.73
Cys(Trt) is routinely used in Fmoc SPPS. It is worth noting that Trt is highly hydrophobic and that the Trt cation-scavenger adducts formed during deprotection may not be removed completely during final TFA cleavage however. The presence of Trt can then mask the quality of the crude peptide due to its high UV absorbance.103 To avoid incomplete detritylation, typical cleavage cocktails of >90% TFA with TIS/TES (typically ca. 5%) can be used. These conditions can, however, cause the reduction of the indole ring of Trp.71,172 Due to its lability to TFA, Trt is removed during cleavage of acid-labile resins (as commonly seen in Fmoc SPPS). It is still possible to use Trt as a protecting group when forming disulfide bonds in solution post-cleavage from the resin, however, the deprotected residues must be re-tritylated post-synthetically. This was exemplified in the regioselective syntheses of μ-conotoxin SIIIA (Fig. 6c) and human hepcidin, using combinations of StBu, Trt, Meb/Mob and Acm.74
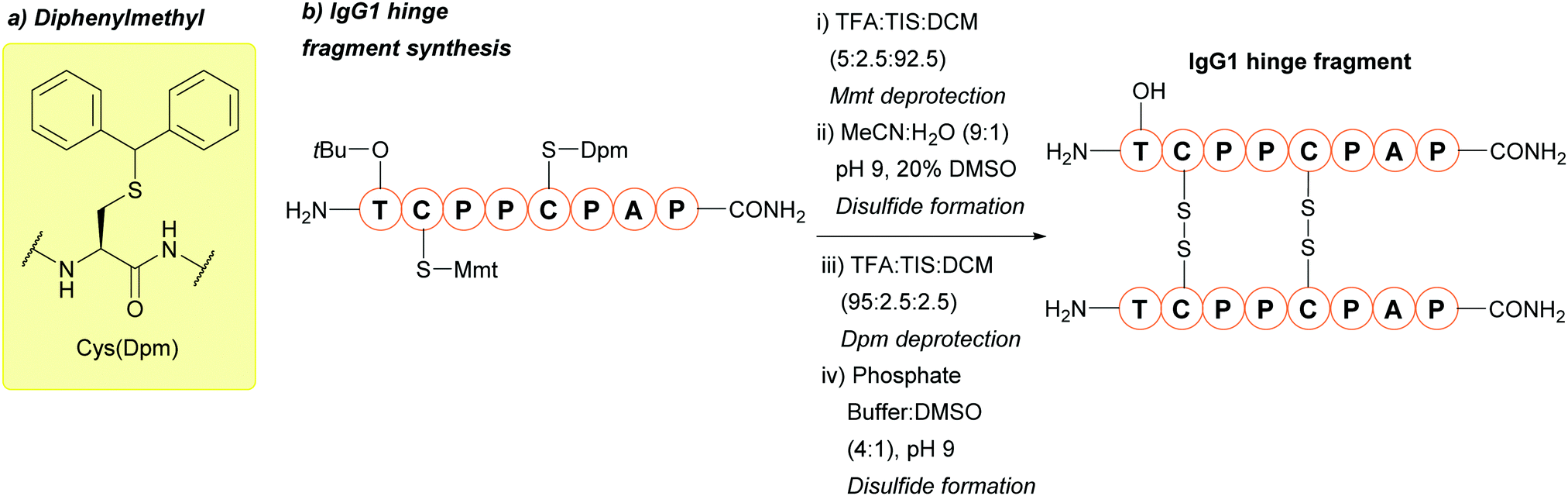 | ||
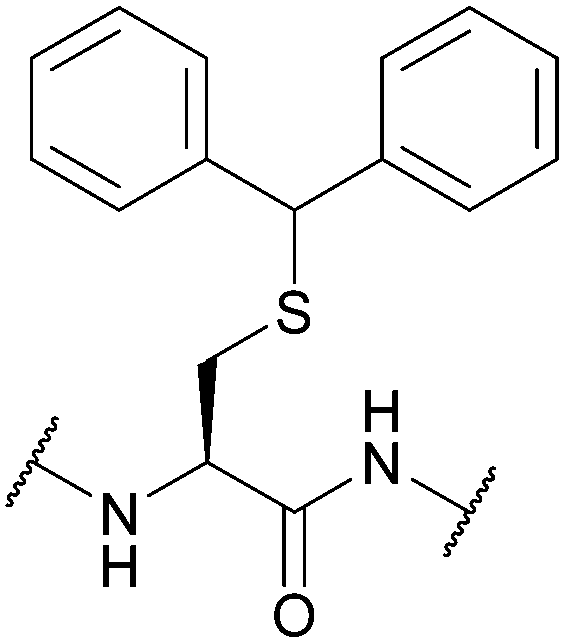
| Fig. 7 (a) Cys thiol protection with the diphenylmethyl (Dpm) protecting group (b) synthesis of the IgG1 hinge region fragment using Cys(Dpm). | ||
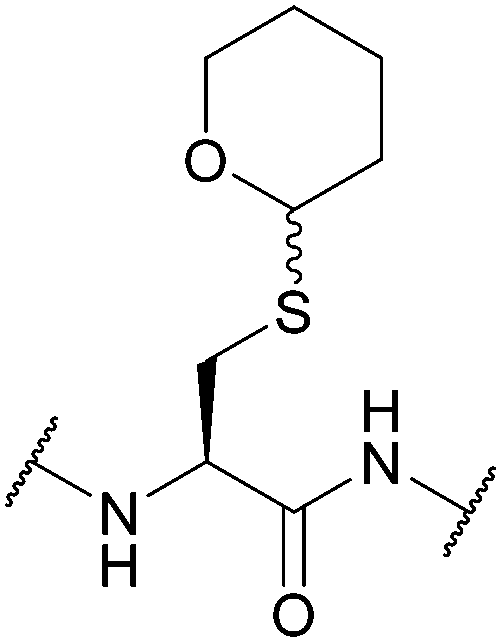
:1). Cys(Thp)-protected peptides exist as a diastereomeric mixture; however, once cleaved in concentrated TFA with a scavenger (e.g. 95% TFA, 2.5% TIS in DCM), a single pure product is obtained.77 Thp protected tripeptides displayed greater solubility in H2O/MeCN solvent systems (as assessed by reverse-phase HPLC) than either Trt or Dpm protected tripeptides. Racemisation is also decreased compared to Trt, Dpm or StBu, and fewer side products, e.g. C-terminal 3-(1-piperidinyl)alanine adducts, are observed.77 More recently, Thp has been explored as a protecting group for additional amino acid residues such as Ser and Thr.179
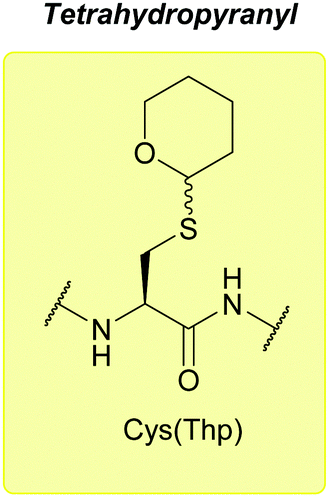 | ||
| Fig. 8 Cys thiol protection with the tetrahydropyranyl (Thp) protecting group. | ||
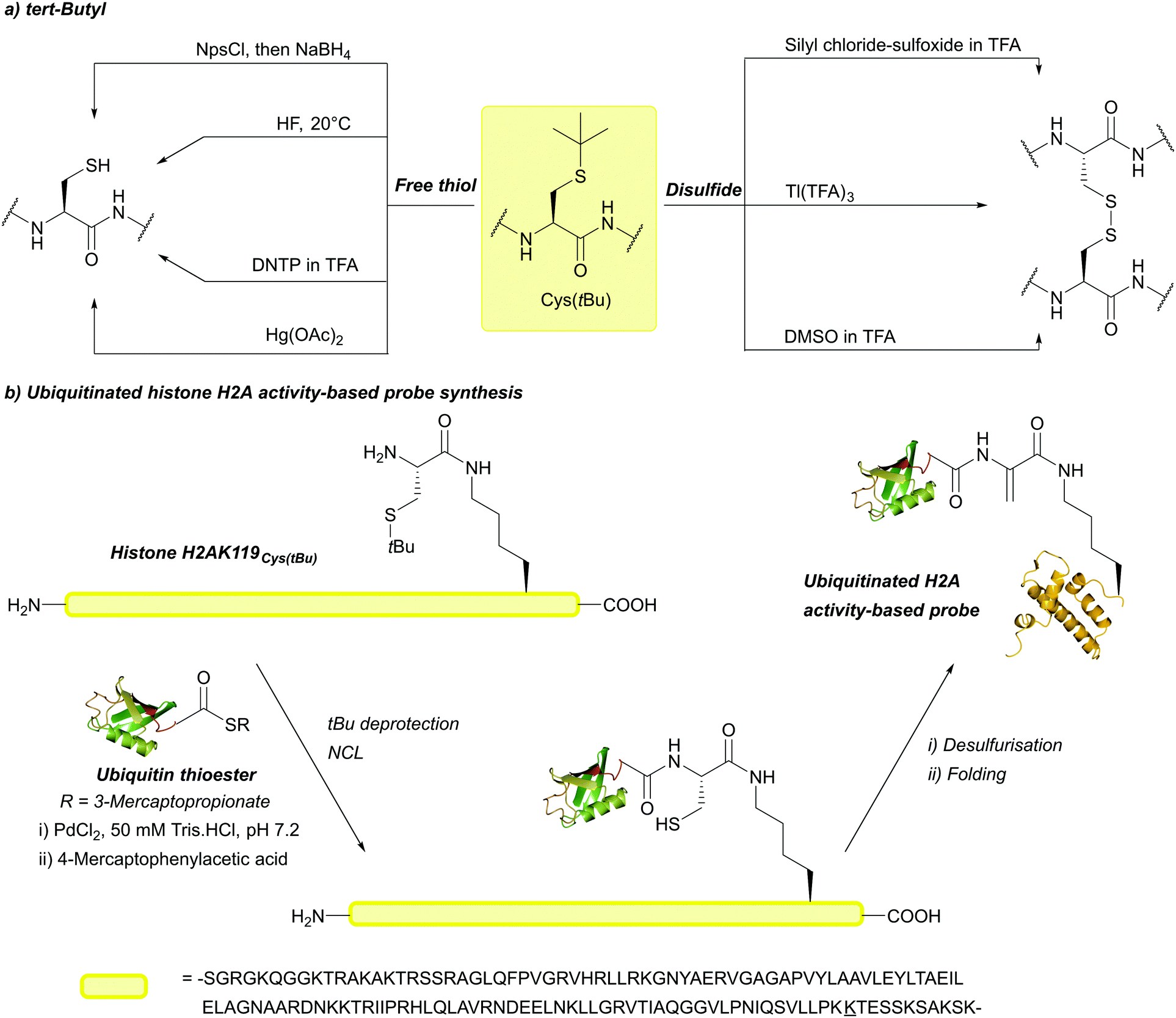 | ||
| Fig. 9 (a) Cys thiol protection with the tert-butyl (tBu) protecting group and different methods of deprotection to give the corresponding Cys or cystine. (b) Synthesis of a ubiquitinated histone H2A activity-based probe using Cys(tBu) and PdCl2 for deprotection under aqueous conditions. | ||
The above methods of deprotection are harsh,128 and often result in the formation of side products and low yields.166 Additionally, incubation in neat TFA causes ∼20% deprotection.86 In 2018, PdCl2 in a 50 mM Tris or urea buffer at 37 °C was shown to cleave tBu, providing a much milder way to remove the protecting group.85 This is significant for tBu, as the harsh conditions required for deprotection can hinder its practical use. tBu is not removed by [Pd(allyl)Cl]2, making it orthogonal to Thz and Acm under those conditions. This method of removal was successfully used to synthesise an activity-based probe of ubiquitinated histone H2A (Fig. 9b).85
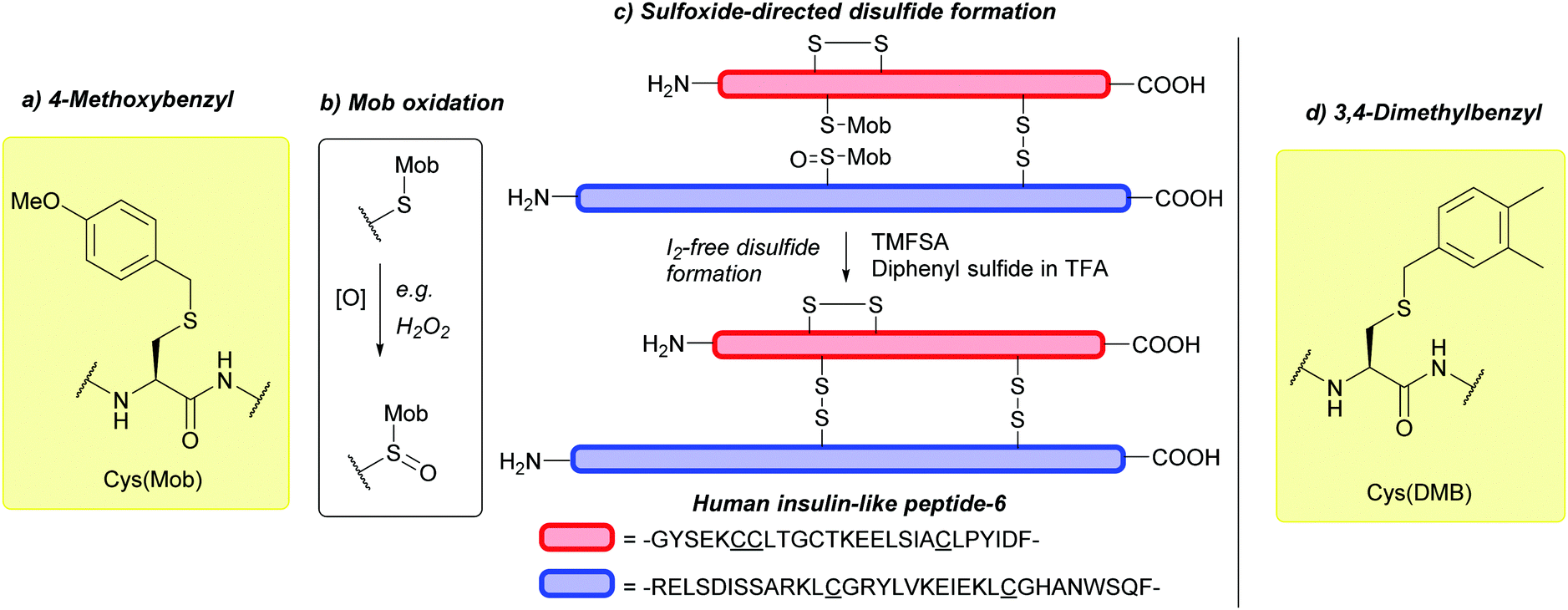 | ||
| Fig. 10 (a) Cys thiol protection with the 4-methoxybenzyl (Mob) protecting group. (b) Oxidation of Cys(Mob) gives the corresponding Mob-protected sulfone, Cys(Mob(O)). (c) Synthesis of human insulin-like peptide-6 (INSL-6) using Cys(Mob) and Cys(Mob(O)) for sulfoxide-directed disulfide formation. (d) Cys thiol protection with the 3,4-dimethylbenzyl (DMB) protecting group. | ||
Cys(Mob) can undergo oxidation to give the corresponding Mob-protected sulfone, Cys(Mob(O)) (Fig. 10b) with either NaBO3185 or H2O2.186 Cys(Mob(O)) can in turn be incorporated into peptides via Fmoc-SPPS. Deprotection with TfOH:TFA:H2O (50:45:5) leads to removal of the Mob protecting group, generating peptides bearing Cys sulfinic acid, a known PTM of Cys-containing proteins that can regulate protein function.186 Additionally, Cys(Mob(O)) can be used for disulfide formation via sulfoxide-directed disulfide reactions187 in an I2-free manner (I2 can otherwise oxidise amino acids such as Trp).188 Example syntheses include oxytocin,189 chicken calcitonin-gene-related peptide (cCGRP),190 and human insulin-like peptide-6 (INSL-6) (Fig. 10c).188
We note to the reader that the DMB protecting group is different to that of the similarly abbreviated 2,4-dimethoxybenzyl (Dmb) group, which is used as an amide backbone protecting group to prevent peptide aggregation and off-target reactions during peptide synthesis.4
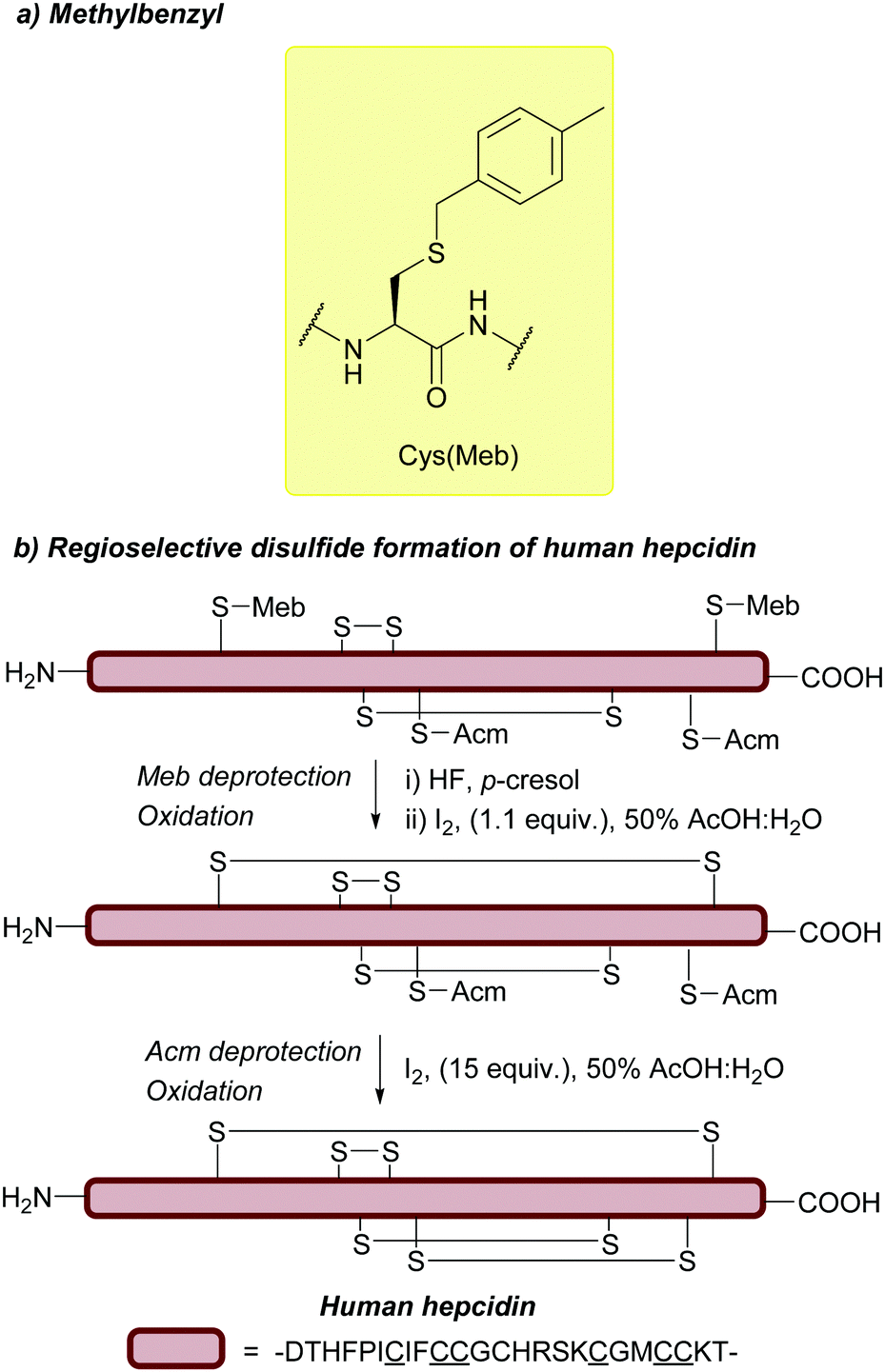 | ||
| Fig. 11 (a) Cys thiol protection with the methylbenzyl (Meb) protecting group. (b) Synthesis of human hepcidin using Cys(Meb). | ||
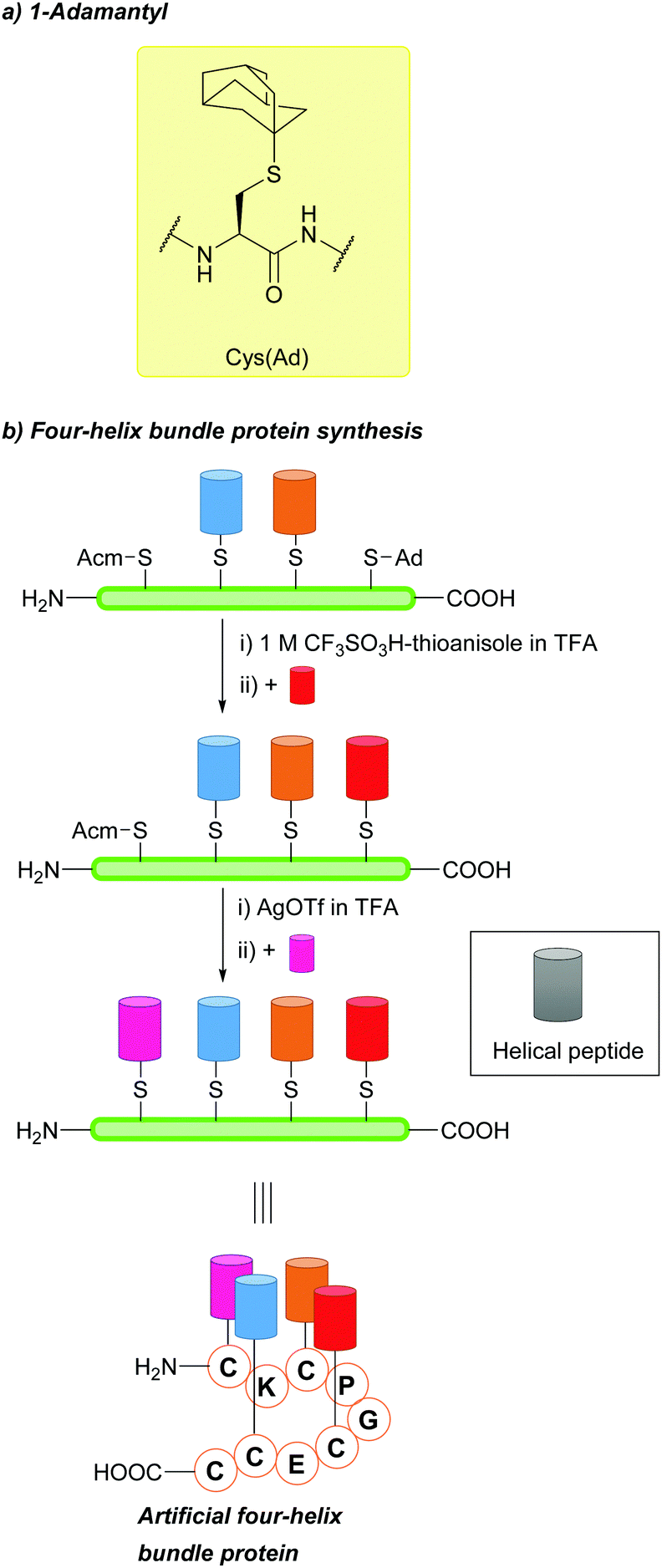 | ||
| Fig. 12 (a) Cys thiol protection with the 1-adamantyl (Ad) protecting group. (b) Synthesis of four-helix bundle proteins using Cys(Ad). | ||
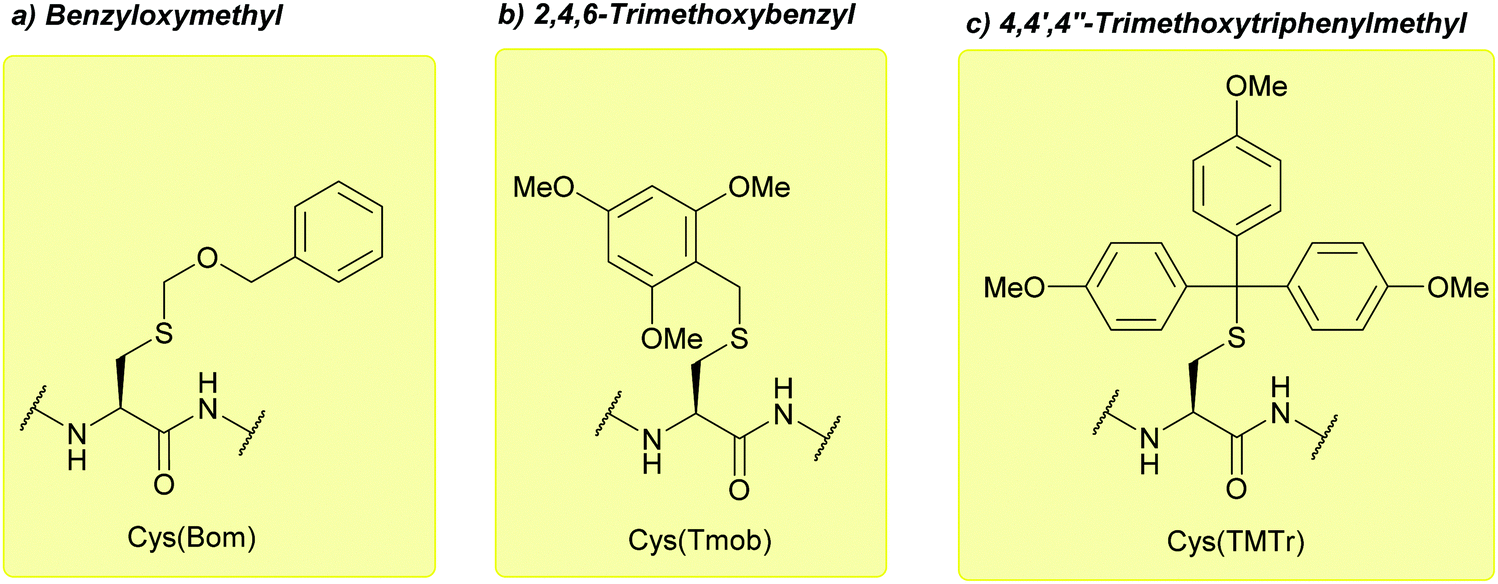 | ||
| Fig. 13 (a) Cys thiol protection with the benzyloxymethyl (Bom) protecting group. (b) Cys thiol protection with the 2,4,6-trimethoxybenzyl (Tmob) protecting group. (c) Cys thiol protection with the 4,4′,4′′-trimethoxytriphenylmethyl (TMTr). | ||
:2.5:2.5) for 1 h.100
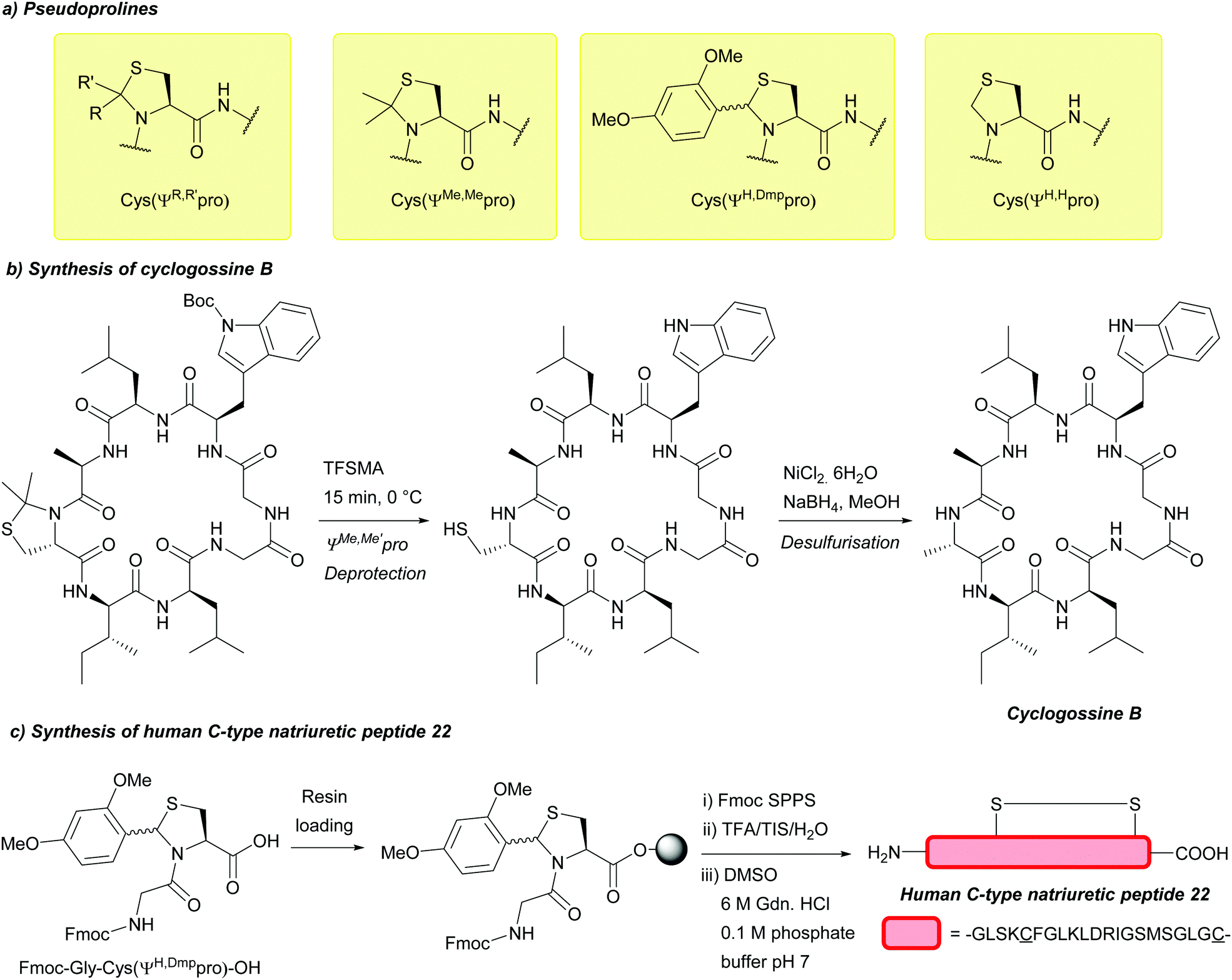 | ||
| Fig. 14 (a) Cys thiol protection with pseudoprolines bearing different substituents. (b) Synthesis of cyclogossine B using Cys(ΨMe,Mepro). (c) Synthesis of human C-type natriuretic peptide 22 using a resin-bound C-terminal protected Gly-Cys(ΨH,Dmppro) dipeptide. | ||
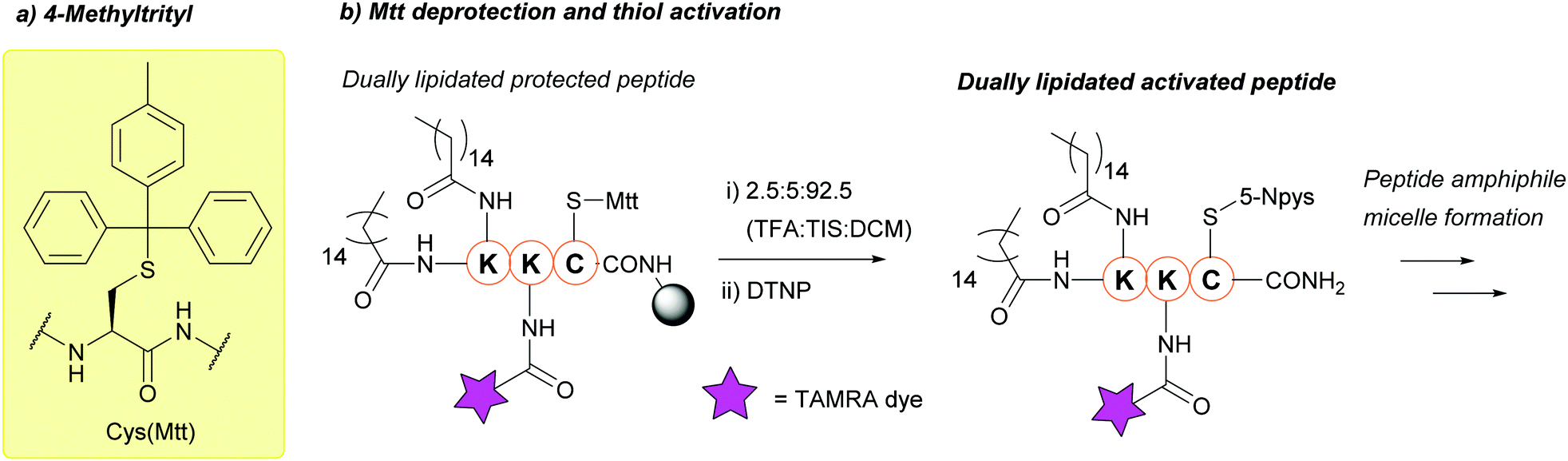 | ||
| Fig. 15 (a) Cys thiol protection with the 4-methyltrityl (Mtt) protecting group. (b) Deprotection of Cys(Mtt) and activation with DTNP in preparation for peptide amphiphile micelle formation. | ||
:2:7, 30 min). Most of its other properties, such as coupling rates, stability to piperidine, oxidative removal using I2, are identical to those of Trt. However, no modification of Trp residues is seen when using thiols or silanes as Mmt cation scavengers, which is advantageous when compared to using Tmob or Trt.71 The Mmt group displays orthogonality to multiple Cys protecting groups, including tBu,84 Dpm,75 oNv,142 StBu,84 and Acm.203 As a result, Cys(Mmt) has been frequently used in combination with other Cys protecting groups to synthesise disulfide containing peptides, such as α-conotoxin MII (Fig. 16b).203 In addition, as it is more acid-labile than Trt, the two groups are technically orthogonal to each other; care, however, must be taken if using Mmt and Trt together to avoid an overlap in deprotection conditions (4–5% Trt deprotection is observed in 1% TFA for 30 min at 22 °C).71,75 Cys(Mmt) has also been used in the synthesis of cyclic tumour-homing peptides,204 and conopeptides in combination with Cys(Allocam).205 The protecting group also features in the synthesis of palmitoylated peptide fragments; these peptides can be subsequently used as building blocks in Ser/Thr ligation for the chemical synthesis of complex, palmitoylated membrane proteins, including S-palmitoylated rabbit sarcolipin (S-palm SLN),206S-palmitoylated interferon-induced transmembrane protein 3 (S-palm IFITM3),207 and S-palmitoylated matrix-2 ion channel from influenza A virus (S-palm M2).206,207
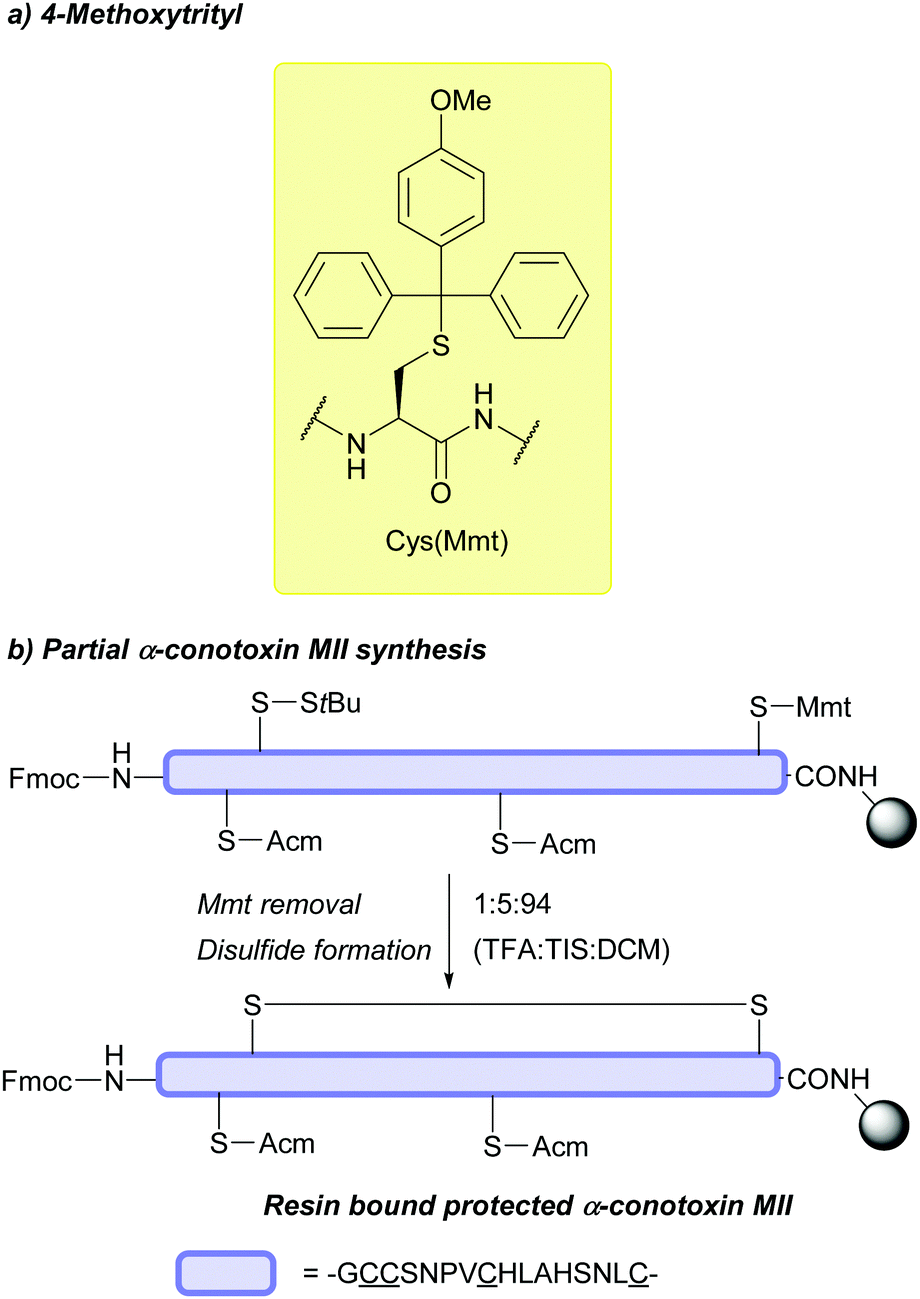 | ||
| Fig. 16 (a) Cys thiol protection with the 4-methoxyltrityl (Mmt) protecting group. (b) Cys(Mmt) deprotection and subsequent disulfide formation through displacement of StBu in the synthesis of α-conotoxin MII. | ||
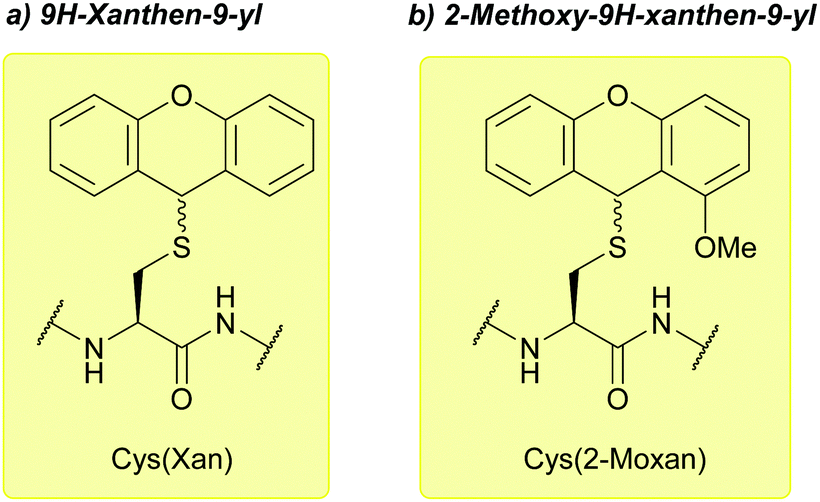 | ||
| Fig. 17 (a) Cys thiol protection with the 9H-xanthen-9-yl (Xan) protecting group. (b) Cys thiol protection with the 2-methoxy-9H-xanthen-9-yl (2-Moxan). | ||
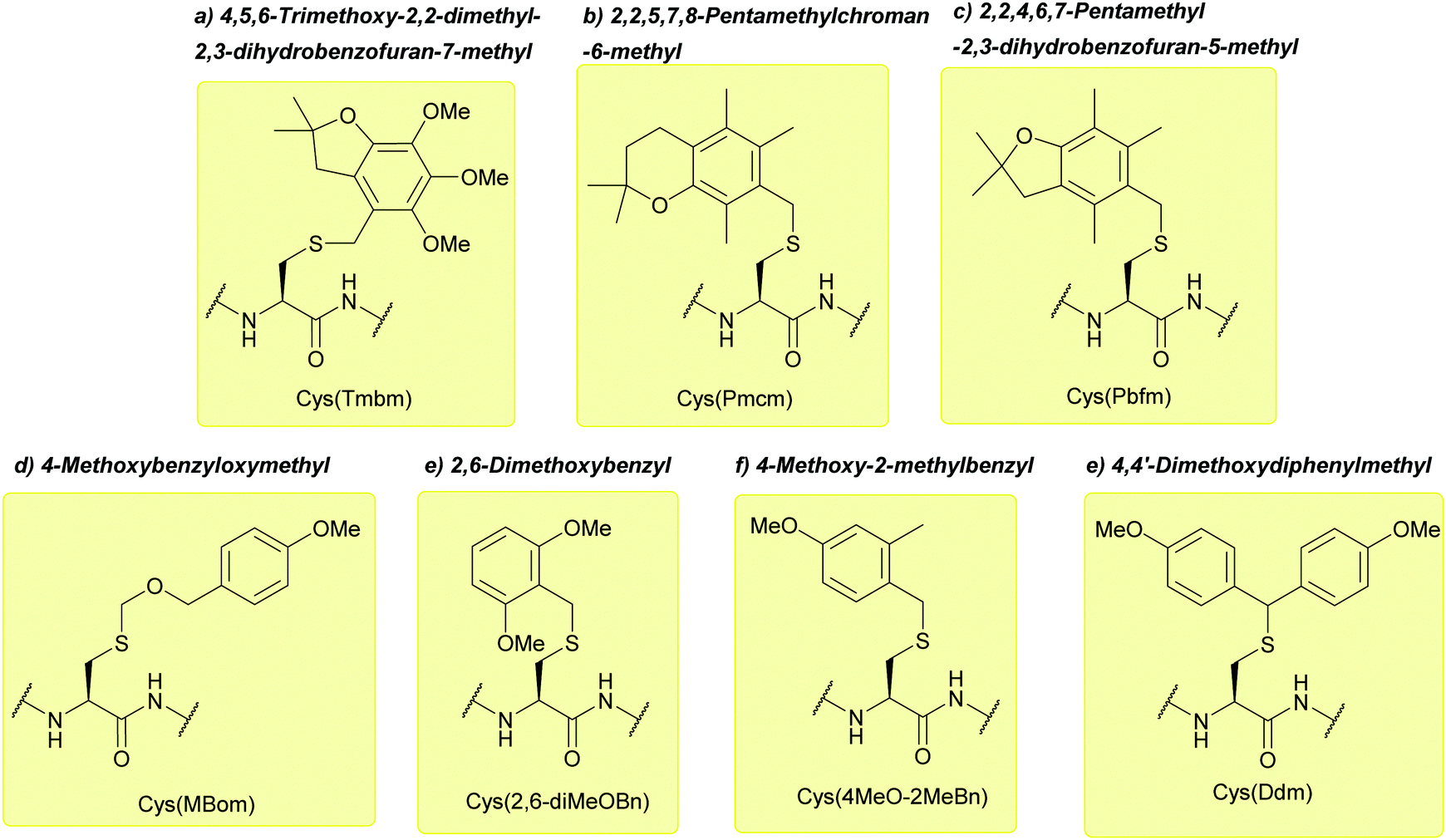 | ||
| Fig. 18 (a) Cys thiol protection with the 4,5,6-trimethoxy-2,2-dimethyl-2,3-dihydrobenzofuran-7-methyl (Tmbm) protecting group. (b) Cys thiol protection with the 2,2,5,7,8-pentamethylchroman-6-methyl (Pmcm) protecting group. (c) Cys thiol protection with the 2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-methyl (Pbfm) protecting group. (d) Cys thiol protection with the 4-methoxybenzyloxymethyl (Mbom). (e) Cys thiol protection with the 2,6-dimethoxybenzyl (2,6-diMeOBn) protecting group. (f) Cys thiol protection with the 4-methoxy-2-methylbenzyl (4MeO-2MeBn). (g) Cys thiol protection with the 4,4'-dimethoxydiphenylmethyl (Ddm) protecting group. | ||
:5:5:5:2.5. Both methoxybenzyl cations and formaldehyde are generated during TFA cleavage, which can undergo side reactions with the peptide; the methoxybenzyl cations can alkylate Cys residues, whereas formaldehyde can lead to hydroxymethylation, and react with N-terminal Cys residues producing Thz-capped peptides. Such side reactions are prevented by using a thiol as a methoxybenzyl cation scavenger, and MeONH2·HCl as a formaldehyde scavenger.104
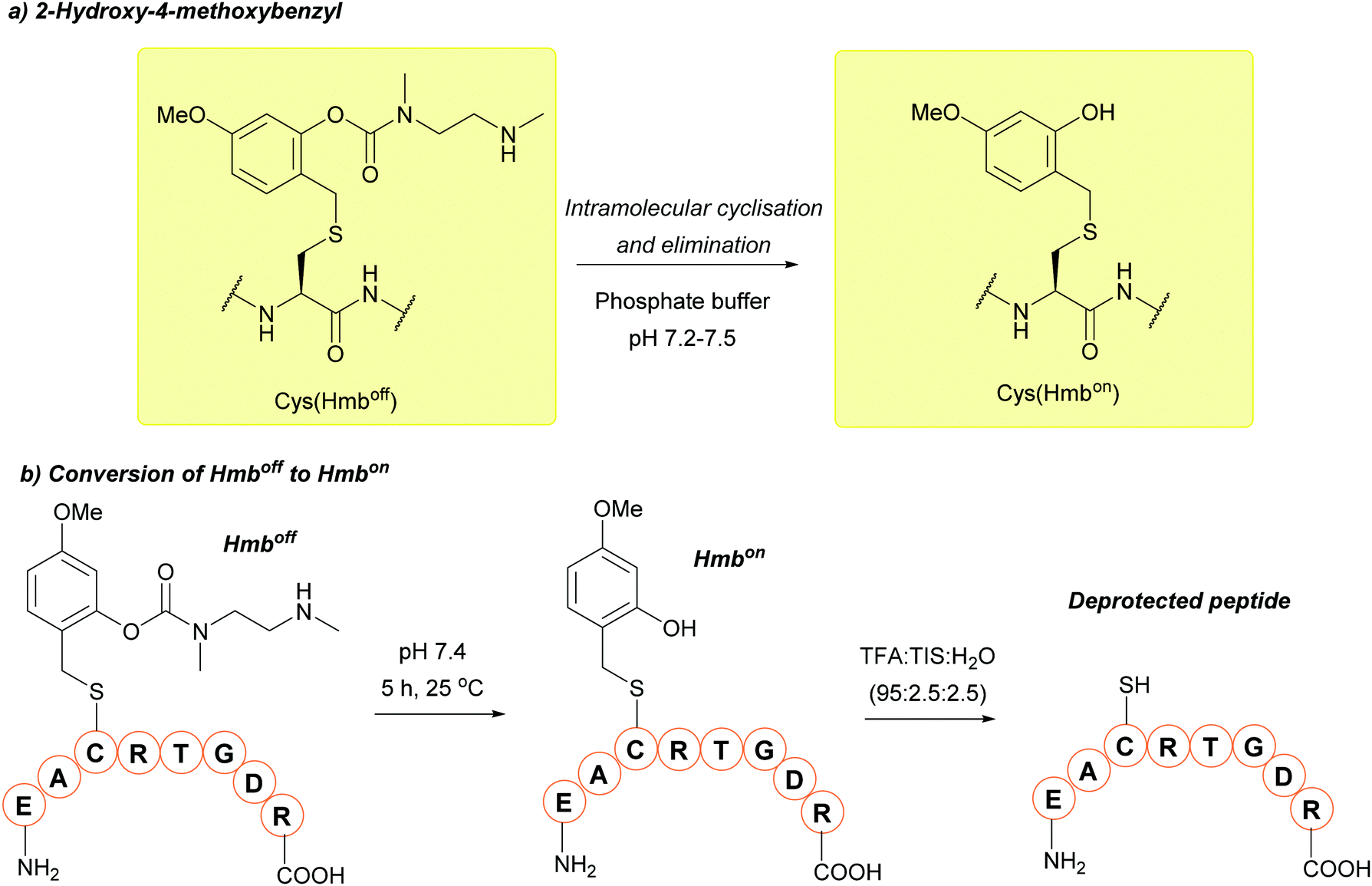 | ||
| Fig. 19 (a) Cys thiol protection with the Hmboff/on protecting group. (b) Conversion of Cys(Hmboff) to Cys(Hmbon) and subsequent deprotection of a model peptide. | ||
5.3 Oxidatively-labile protecting groups
:1) as a scavenger with Hg(II) or Tl(III) deprotection methods.211 I2 can cause back-alkylation, resulting in the formation of residual products, which can be difficult to remove.127 Other potential side-reactions include the iodination of Trp and Tyr residues128 or over-oxidation to the sulfonic acid.109 Although mostly acid-stable, premature cleavage of Acm occasionally occurs in mildly acidic conditions in the presence of free thiol and thioanisole scavengers. Irreversible alkylation of Tyr residues with Acm groups can then occur. This reaction appears to be suppressed with scavengers (e.g. phenol) at comparatively high peptide dilution and may be sequence-specific.212 The preparation of Acm can also be problematic, as thiazolidine-2-carboxylic acid can be formed as a byproduct. Activation using DCC/HOBt can also result in side reactions. The Bam and Tacm protecting groups have been reported in an attempt to address these issues.120 Due to its partial instability to HF, Acm is not particularly suitable for use in Boc SPPS.111 In addition, Acm reacts with strong electrophiles, such as the tBu cation (which is generated during Boc removal), causing S-alkylation – although this reaction can be suppressed by the addition of dimethyl sulfide.110
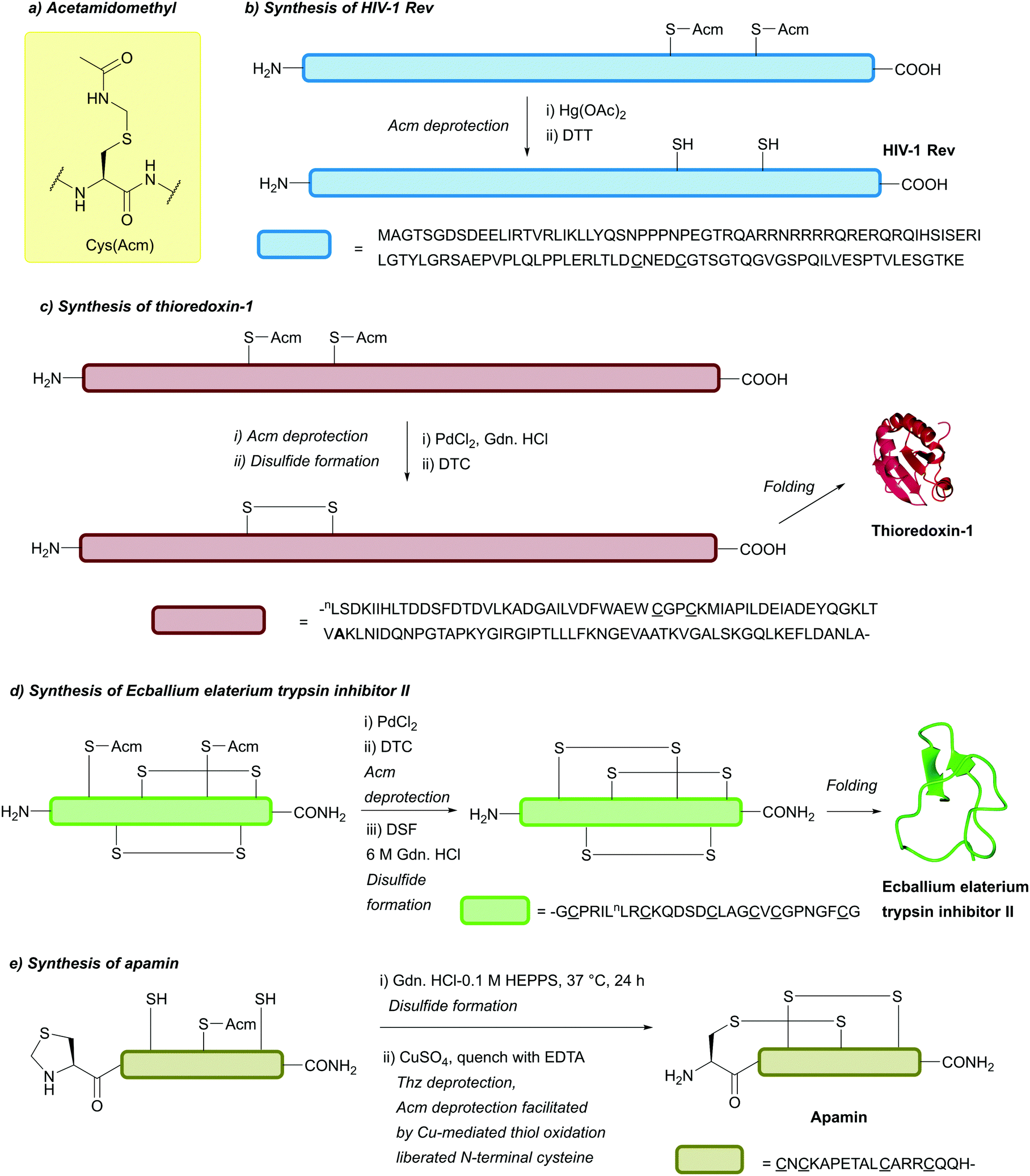 | ||
| Fig. 20 (a) Cys thiol protection with the acetamidomethyl (Acm) protecting group. (b) Synthesis of HIV-1 Rev using Cys(Acm) and Hg-mediated deprotection. (c) Synthesis of thioredoxin-1 using Cys(Acm) and Pd-mediated deprotection. DTC is used for Pd quenching. (d) Synthesis of Ecballium elaterium trypsin inhibitor II (EETI-II) using Cys(Acm) and Pd-mediated deprotection. DSF is used for disulfide formation post-Acm removal. (e) Synthesis of apamin using Cys(Acm) and a Cu-mediated deprotection process. | ||
More recently, it has been reported that Acm can be removed under significantly milder conditions using transition metal catalysts, such as Pd(II) complexes.109,112For example, the synthesis of the two-Cys containing ubiquitin-like protein 5 (UBL5) could be achieved in a one-pot manner, with deprotection of Cys(Acm) in the final step performed using 30 equiv. PdCl2 within 15 min (followed by DTT addition).112 Treatment of Cys(Acm)-containing peptides with PdCl2 in H2O or guanidinium chloride (Gdn·HCl) pH 7 (30 min, 37 °C) followed by DTT addition led to total deprotection to yield the free thiol-containing peptide. Alternatively, using 100 equiv. of diethyldithiocarbamate (DTC) after Pd-mediated deprotection of multi-Cys(Acm)-containing peptides allows for oxidation to form disulfide bonds. Total synthesis of E. Coli thioredoxin (Trx-1) in a one pot manner has been reported using this strategy. First NCL was performed between an N-terminal fragment (bearing two Cys(Acm) residues and a C-terminal thioester, Trx(1-56)) and a C-terminal fragment (bearing an N-terminal Cys (Trx58-109)). This was then followed by desulfurisation, and finally sequential Acm deprotection and disulfide formation using PdCl2 and DTC respectively to yield the target protein (Fig. 20c).109 Moreover, whilst Acm and Thz are both labile to Pd(II) complexes, Acm is stable to the lower concentrations of [Pd(allyl)Cl]2 compared to Thz, giving the two groups some orthogonality to one another. Following Thz deprotection, Acm can be deprotected within 5 h following the addition of an extra 10 equiv. of [Pd(allyl)Cl]2. Additionally, Acm is orthogonal to tBu under these conditions.85 Acm is also stable to Pd(0)-mediated removal of N-terminal Alloc-protected α-amino groups. This, in tandem with a β-thiolactone-mediated NCL procedure,213,214 was demonstrated in the full chemical synthesis of histone H3 bearing an Nε trimethylated Lys residue ([Lys(Me3)9]H3.1).113 The use of PdCl2 for Cys(Acm) deprotection, with DTC for Pd quenching and disulfiram (DSF) for disulfide formation, has very recently been described in combination with photolabile Cys protecting groups for rapid deprotection/disulfide formation in multiple disulfide-rich peptides, including Linaclotide and Ecballium elaterium trypsin inhibitor II (EETI-II, Fig. 20d).215
In addition to Pd(II) chemistry, deprotection of Cys(Acm) using CuSO4 has been reported.73 Initially, it was noted that CuSO4-mediated deprotection of Cys(Thz) would also lead to deprotection of Cys(Acm) in the same peptide scaffold if ascorbate was not added, but would leave Acm intact if ascorbate was added. In model peptides, CuSO4 would only deprotect Cys(Acm) if the peptide contained an N-terminal Cys that was either unprotected or protected with Thz (which could undergo CuSO4-mediated deprotection to reveal an N-terminal Cys). This instance of double deprotection has been hypothesised to result from Cu-mediated oxidation of the deprotected/unprotected N-terminal thiol to sulfenic acid, which then undergoes nucleophilic attack from the Cys(Acm) sulfur, leading to disulfide formation and loss of Acm. Excess Cu can then be quenched with DTT (leading to the free thiol-containing peptide) or ethylenediaminetetraacetic acid (EDTA, leading to the disulfide containing peptide). This protocol was demonstrated in the synthesis of the two-disulfide containing peptide apamin. Following formation of the first disulfide bond via Cys(Trt) deprotection and air oxidation, sequential deprotection of Cys(Thz) and Cys(Acm) with CuSO4 (100 mM, 6 M Gdn.HCl, 0.1 M HEPPS) followed by quenching with EDTA (100 mM), could be used to engineer the second disulfide bond in a regioselective manner (Fig. 20e). Oxidation by-products were formed, however, if the order of disulfide bond formation in this procedure was reversed (with Cys(Mob) used as opposed to Cys(Trt)). In the case of peptides which lack an unprotected N-terminal Cys, Cys(Acm) can be deprotected by addition of CuSO4 in the presence of an equimolar amount of aminothiol additive, such as cysteamine.73
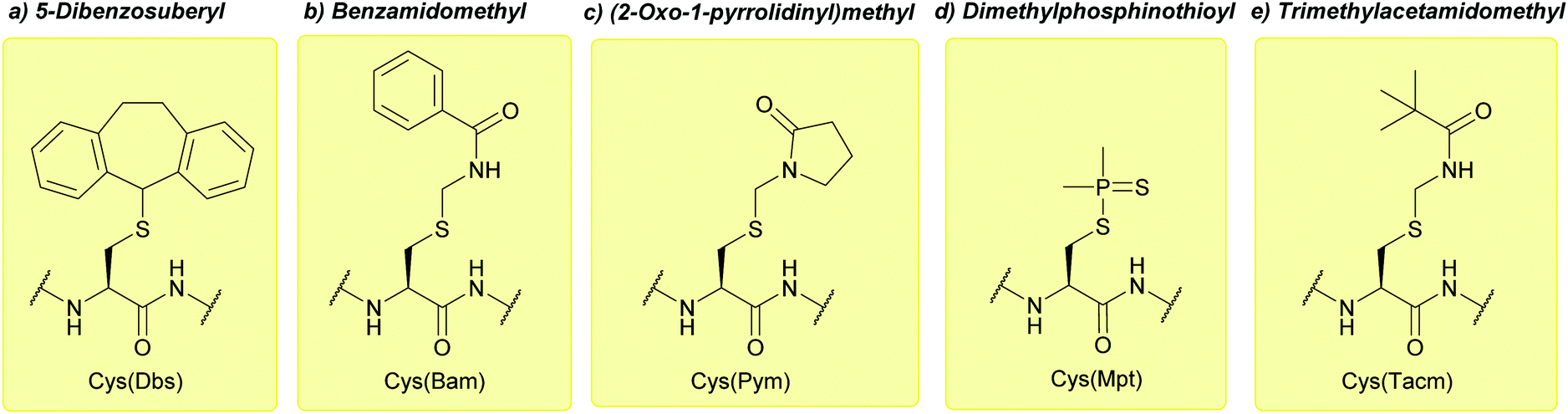 | ||
| Fig. 21 (a) Cys thiol protection with the 5-dibenzosuberyl (Dbs) protecting group. (b) Cys thiol protection with the benzamidomethyl (Bam) protecting group. (c) Cys thiol protection with the (2-oxo-1-pyrrolidinyl)methyl (Pym) protecting group. (d) Cys thiol protection with the dimethylphosphinothioyl (Mpt) protecting group. (e) Cys thiol protection with the trimethylacetamidomethyl (Tacm) protecting group. | ||
Care must be taken when using I2 in AcOH, as partial oxidation of methionine (Met) residues to methionine sulfoxide (Met(O)) can occur. Modification of Trp residues has also been observed.121 Despite its higher resistance to acid than Bam, Tacm can still show instability in HF and TFA/heat. It is therefore not particularly suitable for Boc SPPS,111 although it has been used successfully in Boc solution phase synthesis of pBNP using this strategy.120 Furthermore, Cys(Tacm) has been used in Fmoc SPPS to synthesise oxytocin in combination with the silyl chloride-sulfoxide system.220
5.4 Base-labile protecting groups
:5, 1 h, 0 °C). It is also stable to 0.1 M I2 in DMF,123 and catalytic hydrogenation.122 As with all of the base-labile protecting groups described here, Fm is incompatible with Fmoc SPPS.123 Some Fm-protected peptides can display poor solubility in polar media, and a low peptide yield is observed upon final HF cleavage from the resin when the C-terminal amino acid is Cys(Fm). Dnpe is an alternative protecting group that may be used to avoid these complications.124
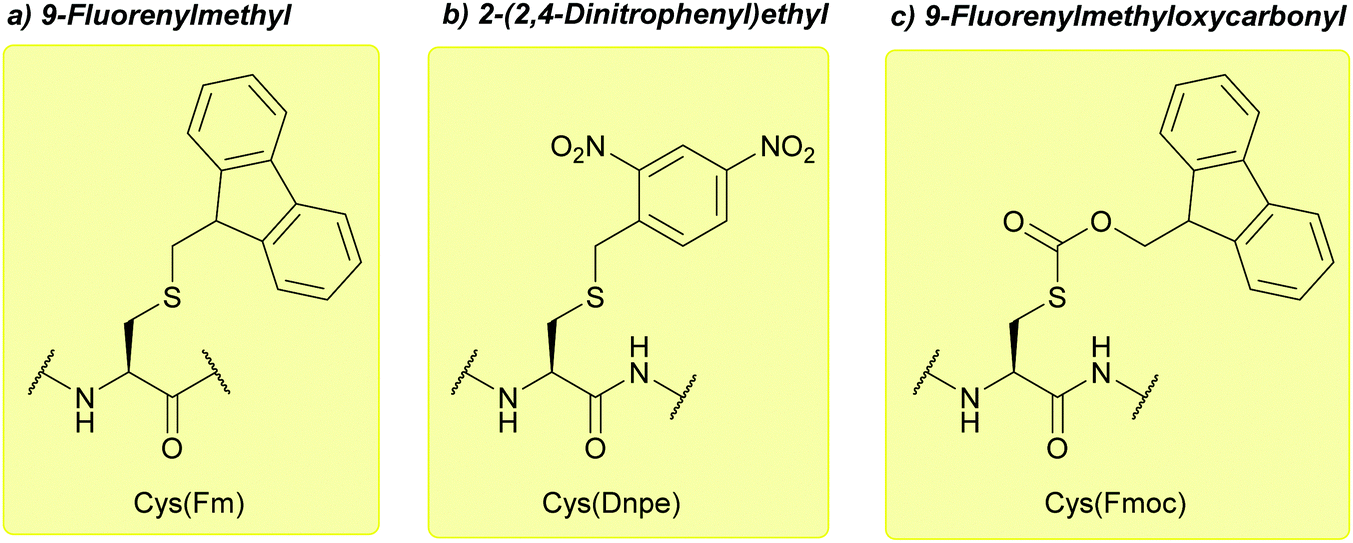 | ||
| Fig. 22 (a) Cys thiol protection with the 9-fluorenylmethyl (Fm) protecting group. (b) Cys thiol protection with the 2-(2,4-dinitrophenyl)ethyl (Dnpe). (c) Cys thiol protection with the 9-fluorenylmethyloxycarbonyl (Fmoc) protecting groups. | ||
Dnpe is stable to a wide variety of reagents, both basic and acidic. These include: 5% DIEA in DCM (2 h), 40% TFA in DCM (24 h), 90% HF in the presence of p-cresol or anisole (1 h, 0 °C), and TFMSA-p-cresol in TFA (1:3:10, 2 h, 25 °C). It is also stable to oxidative conditions, e.g. Tl(TFA)3 in TFA, I2 in 80% AcOH (aq.) and is thus orthogonal to protecting groups including Meb, Acm and StBu.124
5.5 Enzyme-labile protecting groups
:1), at 37 °C. DMSO is the best-tolerated solvent, but enzymatic activity remains almost completely intact when DMSO is replaced by any of a variety of organic co-solvents: MeCN, DMF, EtOH, various other alcohols and Et2O. The pH range under which deprotection can occur is also broad, with clean removal still observed at pH 5.9.127 If enzymatic hydrolysis is performed in the presence of BME, the free thiol is obtained, otherwise, the Cys is oxidised directly to the disulfide.126 The efficiency of iPGA can depend on the peptide sequence containing Cys(Phacm), as some peptide sequences appear to tolerate a broader range of conditions than others. Care must therefore be taken to choose the optimal conditions for a given sequence.127 Phacm may also be removed using Hg(II) or Ag(II) salts, or under conventional oxidative conditions, e.g. using I2 or Tl(TFA)3. However, removal under enzymatic conditions circumvents the problems with such reagents, as there is no need to use toxic heavy metals or reagents (e.g. I2) which can favour peptide chain modifications (e.g. back alkylation), have difficult to remove residual products and are damaging to the environment.127 Phacm is stable to acidic and basic conditions, including 5% DIEA in DCM, 40% TFA in DCM, 25% piperidine in DMF, 0.1 M TBAF in DMF and 5% DBU in DMF (all for 24 h at 25 °C). Phacm is also stable to 90% HF in the presence of anisole or p-cresol (1 h, 0 °C) and 90% TFA in the presence of a number of scavengers (i.e. phenol, ethane-1,2-dithiol (EDT), p-cresol, anisole, 2 h, 25 °C). This makes Phacm suitable for both Fmoc and Boc SPPS. However, it is more useful for Fmoc SPPS, as it is partially cleaved (<20%) by TFMSA-TFA-p-cresol (1:10:0.3, 2 h, 25 °C).126 Phacm is orthogonal to a wide variety of protecting groups, including Fm, Dnpe, Meb, Trt and Tmob.126 In addition to those mentioned, Phacm is orthogonal to many acid-labile protecting groups.127 It is also partially orthogonal to Acm, provided Phacm is removed enzymatically prior to Acm deprotection.126 This has been demonstrated on model tripeptides containing Cys(Phacm) and Cys(Acm) (Fig. 23c).127 Cys(Phacm) derivatives, which have been used as protecting groups/solubilising tags in the chemical synthesis of proteins such as histone H4, can also be removed using PdCl2 under aqueous conditions.222 Additionally, the orthogonality of Cys(Phacm) can be used to facilitate multi-interchain disulfide formation in the final step of three-disulfide containing peptides synthesis, such as in the I2 free synthesis of a porcine insulin analogue (Fig. 23d).223 Here, the A chain (containing four c) can be synthesised using a combination of Cys(S-Tmp), Cys(Mmt), Cys(Trt) and Cys(Phacm). Selective deprotection, followed by activation, yields the Cys(Trt), Cys(Phacm), disulfide containing resin bound A chain. Simultaneous Trt removal and resin cleavage could then be achieved with a cleavage cocktail of TFA:TIS:H2O (92.5:5:2.5). Incubation of this A chain peptide with a Cys(Phacm), Cys(5-Npys) activated B chain peptide in ligation buffer (0.2 M NH4OAc, 6 M urea, pH 4.5) lead to formation of one of the interchain disulfides. Finally, selective removal of the Phacm protecting group with immobilised PGA and activation with 3 equiv. 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) lead to formation of the second interchain disulfide to give the desired product. Optimisation of activation with DTNB proved critical to minimise misfolding and disulfide scrambling; oxidation with no DTNB or in 10% DMSO led to only scrambled side products, whereas double-DTNB side products were observed when using >4 equiv. DTNB.223 A similar strategy was adopted for the synthesis of human relaxin-2 using Cys(Phacm) and Cys(tBu); here, DTNB was replaced with activating agent bis(5-(2-methoxyethoxy)-2-pyrimidinyl disulfide (BMPD) post-Phacm-removal to give an improved peptide yield.224
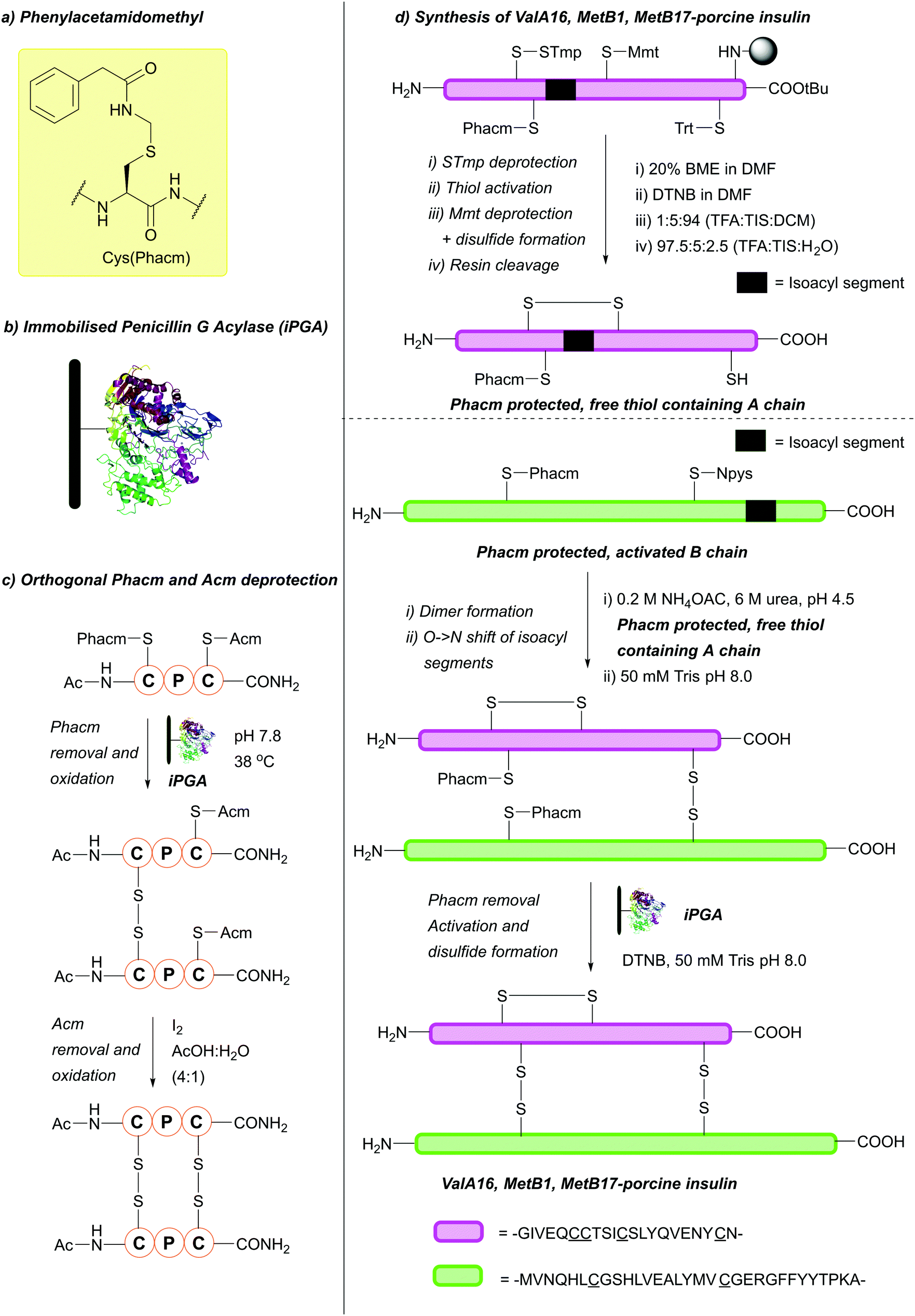 | ||
| Fig. 23 (a) Cys thiol protection with the phenylacetamidomethyl (Phacm) protecting group. (b) Schematic of immobilised Penicillin G Acylase (iPGA) used for Cys(Phacm) deprotection. (c) Orthogonal deprotection of Cys(Phacm) and Cys(Acm) in a model tripeptide. (d) Synthesis of a porcine insulin analogue using Cys(Phacm). | ||
5.6 Hydrazine-labile protecting groups
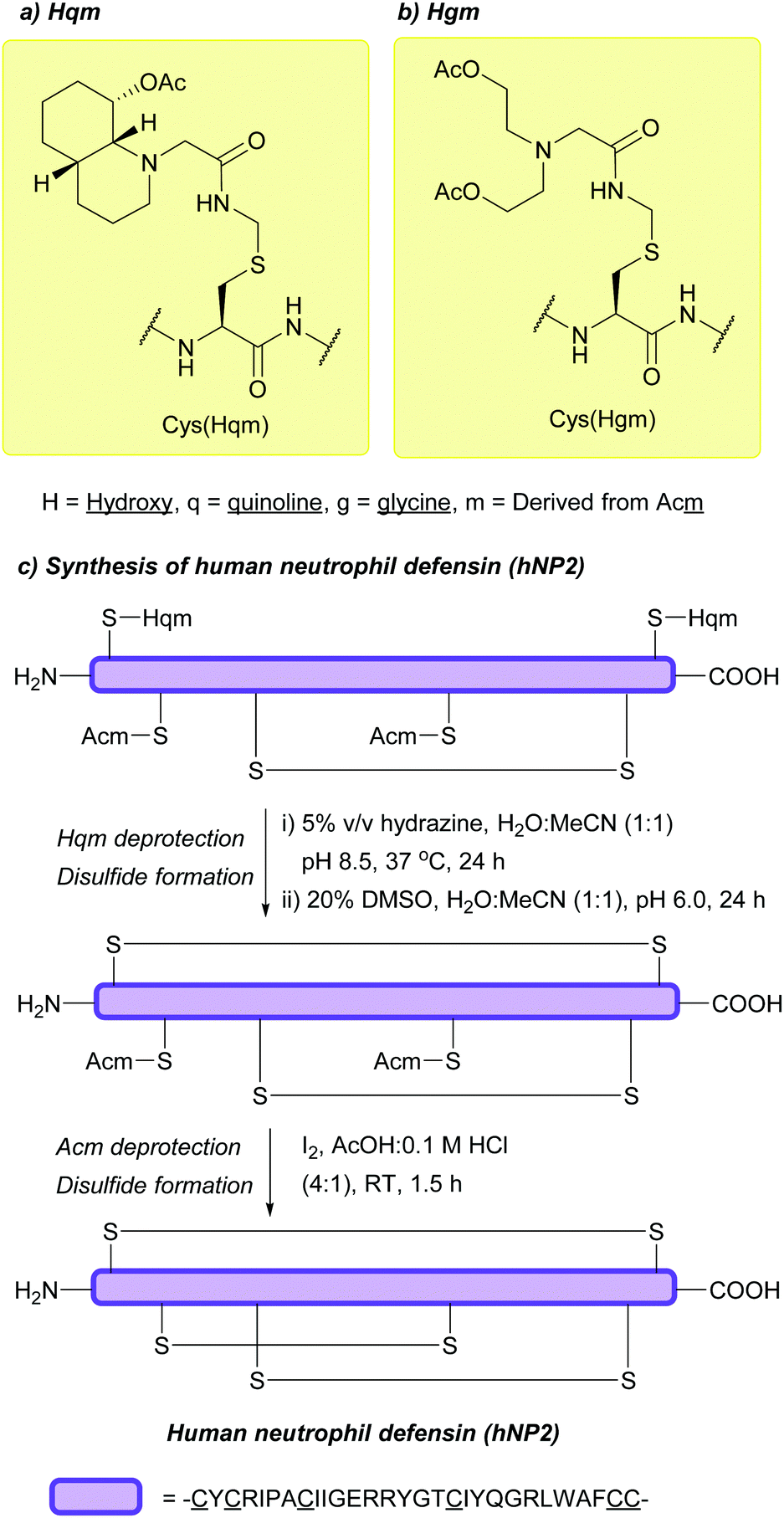 | ||
| Fig. 24 (a) Cys thiol protection with the Hqm protecting group. (b) Cys thiol protection with the Hgm protecting group. (c) Synthesis of human neutrophil defensin (hNP2) using Cys(Phacm). | ||
5.7 Palladium-labile protecting groups
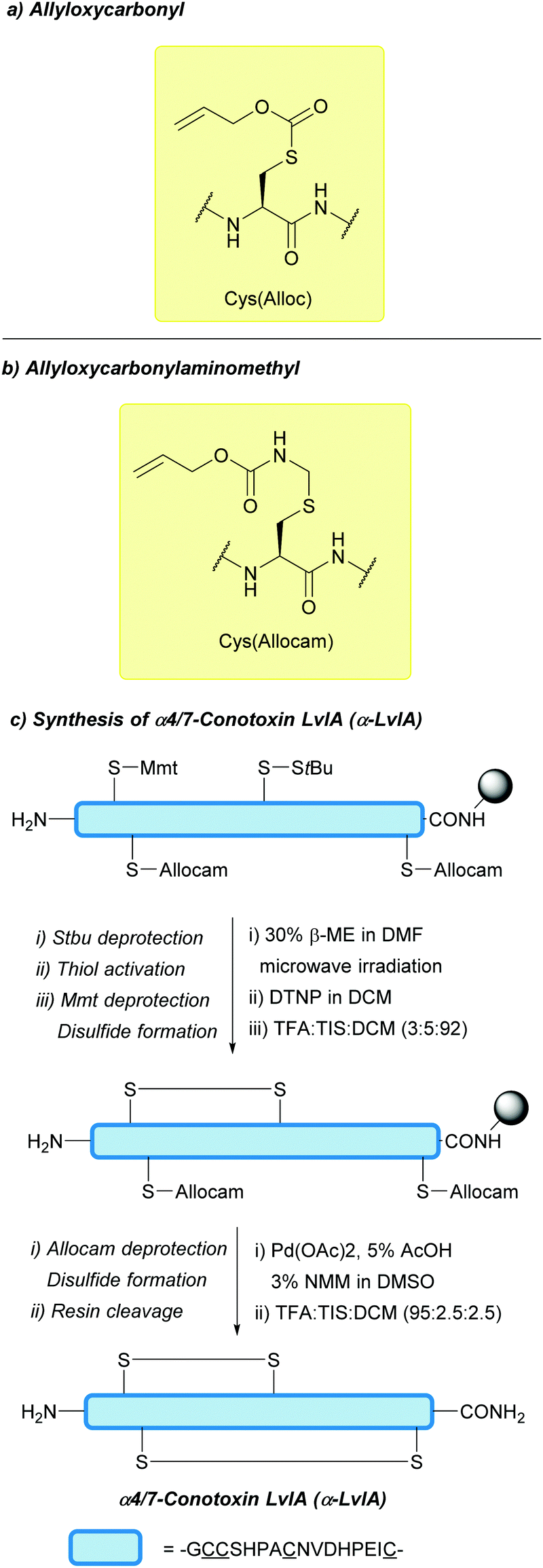 | ||
| Fig. 25 (a) Cys thiol protection with the allyloxycarbonyl (Alloc) protecting group. (b) Cys thiol protection with the allyloxycarbonylaminomethyl (Allocam) protecting group. (c) Synthesis of α4/7 Conotoxin LvlA (α-LvlA) using Cys(Allocam). | ||
After its initial documentation, the Allocam group saw little use as a protecting group for Cys in the literature. More recently, however, it has seen a revival as an orthogonal Cys protecting group. Here, alternative deprotection protocols that lead directly to the disulfide formation on-resin have been developed: Pd(OAc)2 (1.5 equiv.), 3% NMM and 5% AcOH in DMSO for 2 h. Under these conditions, complete removal of Allocam could be achieved to yield the disulfide-containing resin-bound peptide.131 The use of Cys(Allocam) has since been expanded towards higher-yielding, on-resin synthesis of α4/7-Conotoxin LvIA (α-LvIA, Fig. 25c).205 Orthogonality of Cys(Allocam) to other protecting groups was also further established. For example, treatment of peptides containing Cys(Trt) and Cys(tBu) with Pd(OAc)2 (1.5 equiv.), 3% NMM and 5% AcOH in DMSO for 2 h, followed by resin cleavage, led to no removal of either protecting group, whereas treatment of Cys(Mmt) with the same conditions gave a mixture of products. Peptides containing two Cys(Allocam) residues could be fully deprotected to give the corresponding disulfide when using I2, whereas the Allocam protecting groups remained intact when using conditions typically used for StBu/Mmt removal (20% BME, DTNP, 1% TFA, 5% TIS). Depending on the combination of Cys protecting groups used, Cys(Allocam) could be orthogonally deprotected first or last in a given sequence to perform regioselective synthesis of disulfide-containing α-LvIA on-resin.205
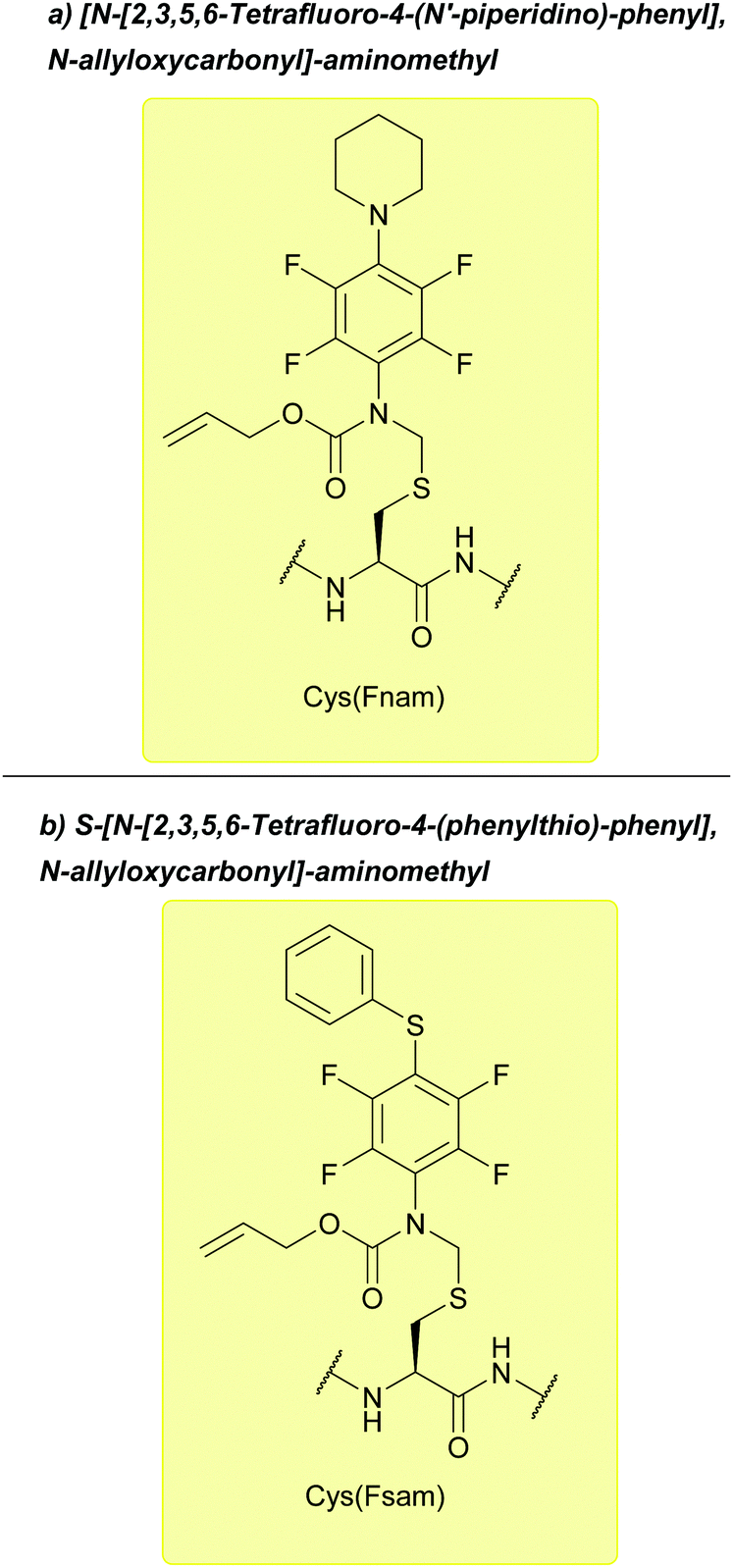 | ||
| Fig. 26 (a) Cys thiol protection with the [N-[2,3,5,6-tetrafluoro-4-(N′-piperidino)-phenyl], N-allyloxycarbonyl]-aminomethyl (Fnam) (b) Cys thiol protection with the S-[N-[2,3,5,6-tetrafluoro-4-(phenylthio)-phenyl], N-allyloxycarbonyl]-aminomethyl (Fsam) protecting group. | ||
Fsam is stable to acidic and basic conditions, and is compatible with both Boc and Fmoc SPPS. Fsam is labile to similar oxidative conditions to Acm and Phacm. A side reaction was observed during SPPS, which was suggested to be due to N-allyl peptides forming via nucleophilic attack of the neighbouring α-amino function at the allyl group. This reaction was proposed to occur under the basic conditions present during Fmoc removal or during the coupling step. This phenomenon has also been observed with the Alloc group.133
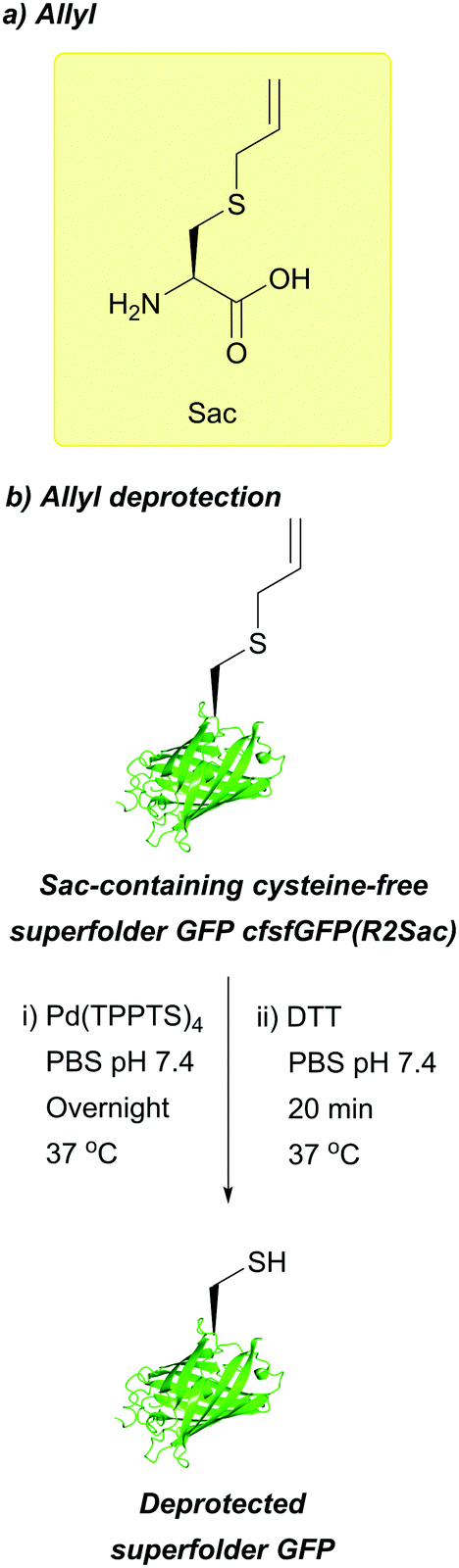 | ||
| Fig. 27 (a) S-allyl cysteine (Sac). (b) Pd-mediated deprotection of Sac-containing cysteine-free superfolder GFP (cfsfGFP(R2Sac)). | ||
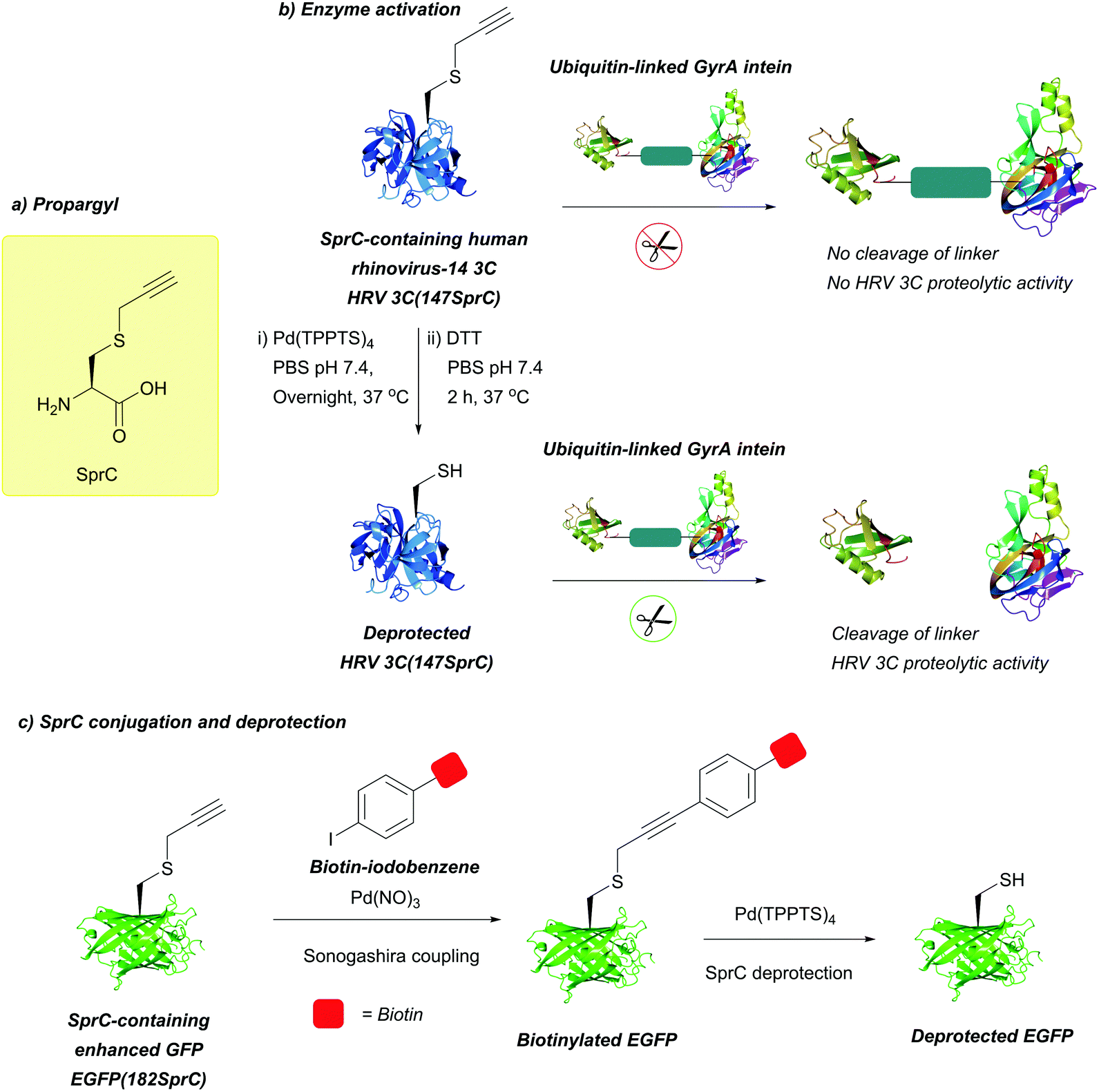 | ||
| Fig. 28 (a) S-Propargyl-cysteine (SprC). (b) Pd-Mediated deprotection of SprC-containing human rhinovirus-14 3C (HRV 3C(147SprC)). Upon SprC deprotection, HRV 3C proteolytic activity is restored. (c) Sonogashira coupling of SprC-containing enhanced GFP (EGFP(182SprC)), followed by subsequent Pd-mediated deprotection to yield the free-thiol containing EGFP. | ||
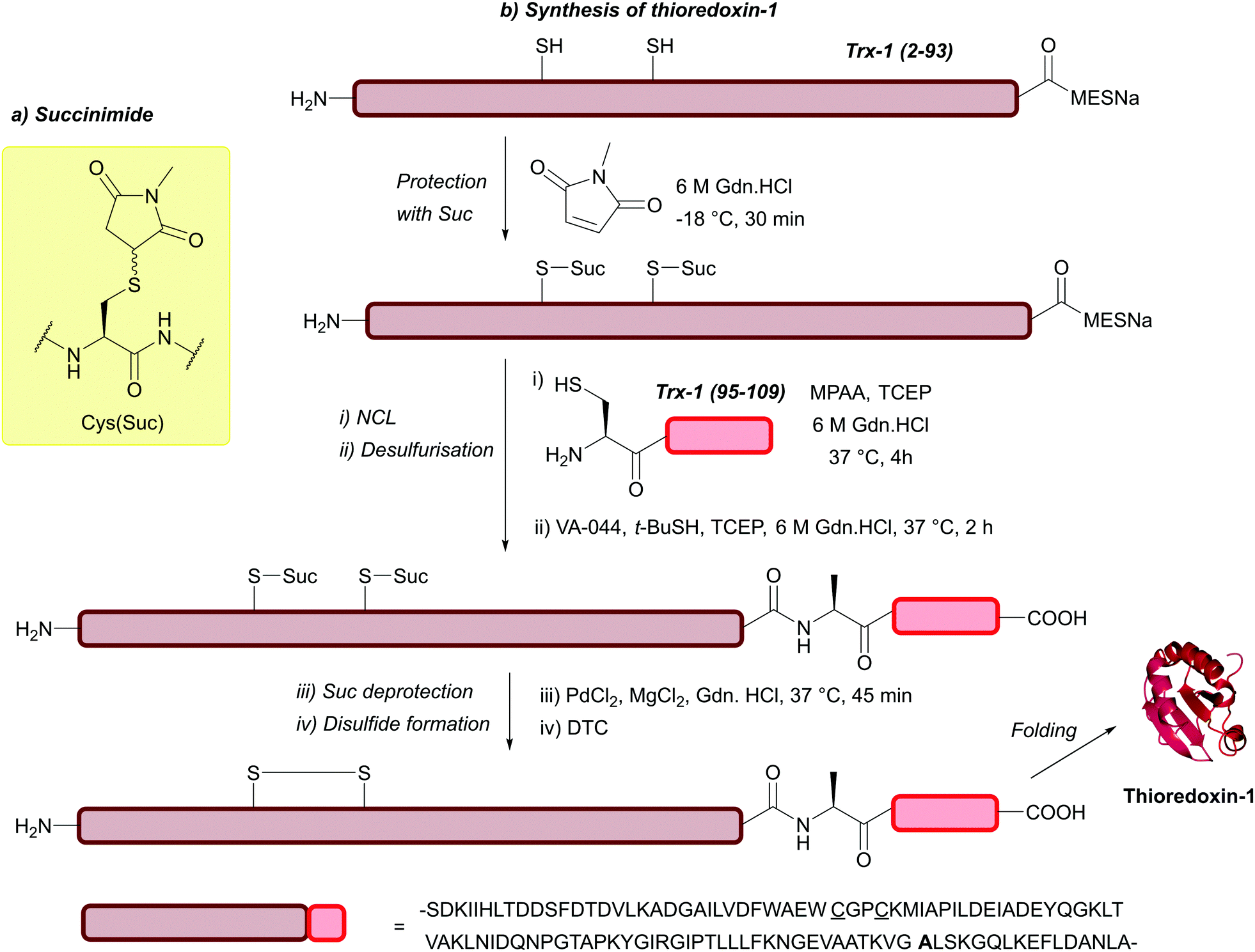 | ||
| Fig. 29 (a) Cys thiol protection with N-methyl maleimide yielding a thiol-succinimide conjugate (Suc). (b) Synthesis of thioredoxin-1 using Cys(Suc). | ||
5.8 N-terminal cysteine protecting groups
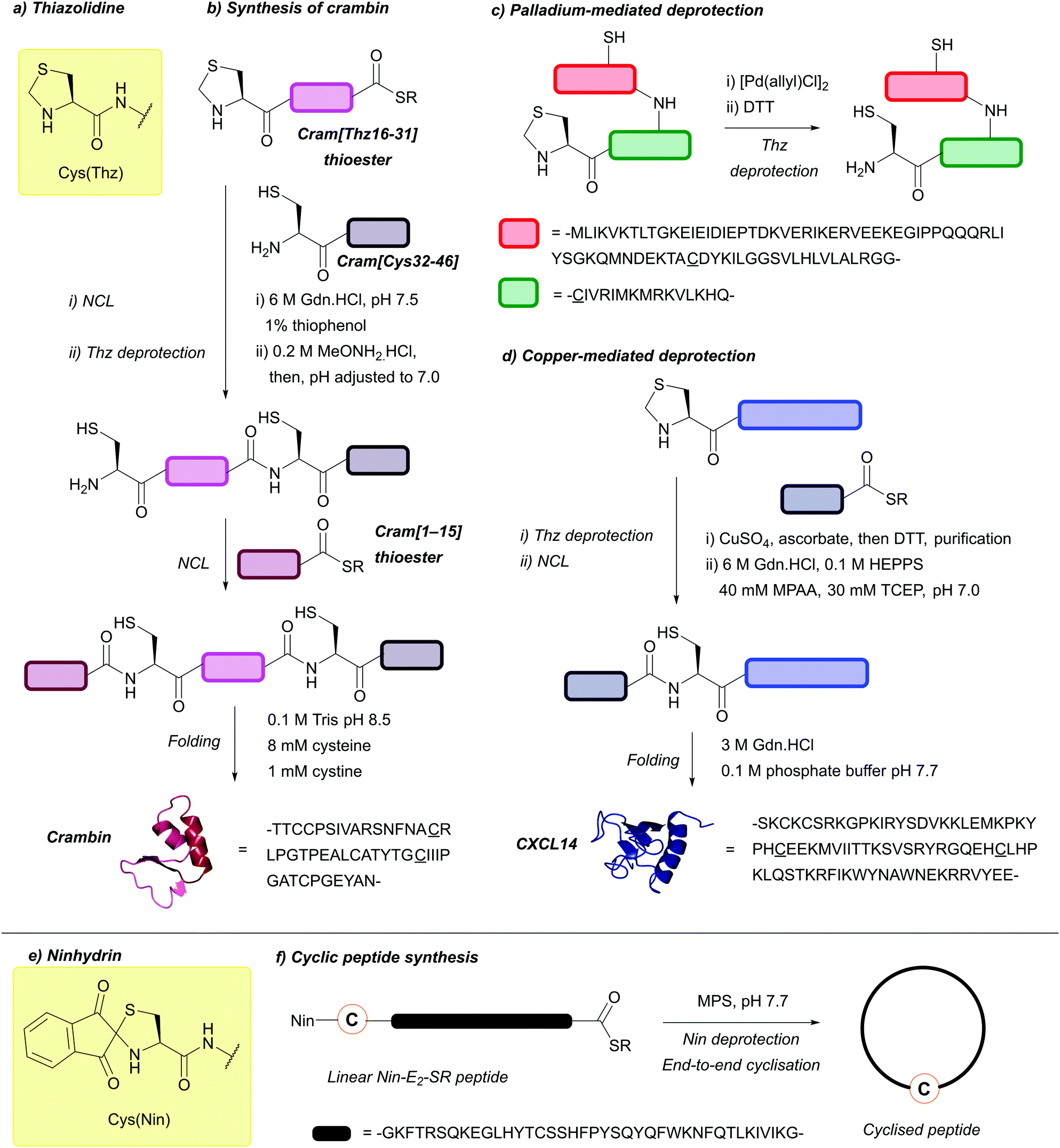 | ||
| Fig. 30 (a) N-terminal Cys protection with the thiazolidine (Thz) protecting group. (b) Synthesis of Crambin using Cys(Thz) with methoxyamine-mediated deprotection. (c) Peptide synthesis using Cys(Thz) with Pd-mediated deprotection. (d) Synthesis of CXCL14 using Cys(Thz) with Cu-mediated deprotection. (e) N-terminal Cys protection with the ninhydrin (Nin) protecting group. (f) Cyclic peptide synthesis using Cys(Nin). | ||
More recently, alternative strategies for the deprotection of Thz have been described; these have primarily been transition-metal based.235 For example, deprotection of Thz can be achieved using water-soluble Pd(II) complexes, such as [Pd(allyl)Cl]2 followed by treatment with DTT to both obtain the free thiol and quench remaining Pd species (Fig. 30c). This is performed under native chemical ligation (NCL) conditions and in the presence of MPAA and TCEP, with complete removal obtained within 15 min.138 MPAA and TCEP appear to be crucial for efficient removal (100% removal in 15 min vs. 40% removal after 4 h). It has been hypothesised that this is due to MPAA and TCEP chelating to Pd(II), or possibly reducing it to Pd(0). To demonstrate the usage of this deprotection method, Lys34-ubiquitinated H2B and several other sample peptides have been synthesised using Pd(II) to deprotect Thz.138 In further work, it was found that [Pd(allyl)Cl]2 and GSH (1:1) in 6 M Gdn·HCl (pH 6.5, 37 °C, 45 min) were sufficient for full removal, along with its orthogonality demonstrated to both Acm and tBu.85 Pd-mediated Thz deprotection has also been successfully applied towards in vivo systems for triggered release/chemical activation of peptides/proteins.236 Furthermore, Thz analogues have also been employed in within the field of protein bioconjugation; Pd-mediated237 (or Ag-mediated)238 “unmasking” of unnatural Thz side chains leads to generation of an α-oxo aldehyde side chain, which can be used for downstream site-selective protein modification.239 Aside from Pd, Cu complexes can also be used to deprotect Thz.139,240 Thz is removed by CuSO4 in the presence of sodium ascorbate (critical for avoiding oxidation by-products), in 5 M Gdn·HCl in HEPPS buffer (pH 7.0, 1 h, 37 °C). This was demonstrated in the synthesis of CXCL14 (Fig. 30d).139 As discussed previously, the reaction can then be quenched with DTT or EDTA.73 No epimerisation or side products are observed using this method of deprotection, and the reaction can be performed under standard NCL conditions.139 Outside of transition-metal based strategies, 2,2′-dipyridyl disulfide (DPDS) in 50% MeCN (0.1% TFA) as a reagent for Thz deprotection has also been described.140,241
Protection using the Nin group avoids the formylation side reactions normally found during TFA and HF cleavage when His(Bom) groups are present, without the need for additional scavengers. Additionally, in thioester-containing peptides, the products will cyclise under deprotection conditions, enabling a one-step process. As proof of concept, a Nin-E2-SR peptide was synthesised and cyclised in one-step (Fig. 30f), using an excess of MPS (where E2 refers to a 40-residue long sequence of the chemokine receptor CCR5's second extracellular loop).141
5.9 Photo-labile protecting groups
:1 at pH 6, in the presence of semicarbazide and L-(−)-ascorbic acid.143 The oNB protecting group also shows a high one-photon efficiency, and a high yield of the free thiol is obtained upon photolysis.145
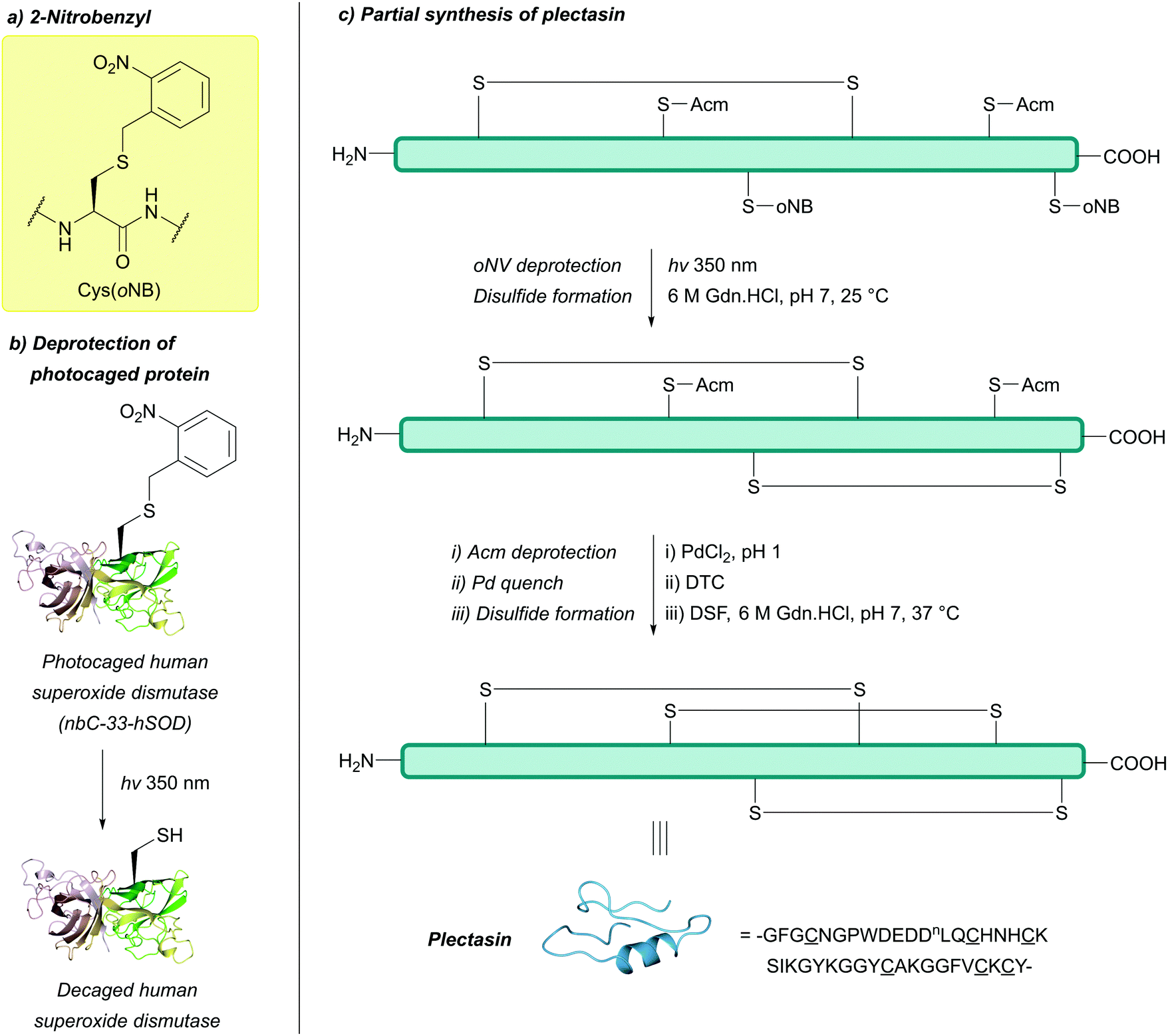 | ||
| Fig. 31 (a) Cys thiol protection with the 2-nitrobenzyl (oNB) protecting group. (b) Photodecaging of a photocaged human superoxide dismutase containing Cys(oNB) as an unnatural amino acid (nbC-33-hSOD). (c) Partial synthesis of plectasin using Cys(oNB). | ||
The protecting group has since featured in the synthesis of a thioester-containing HCDLP pentapeptide; this peptide can then undergo NCL to semisynthesise a variant of a nickel-dependant superoxide dismutase (NiSOD). Critically, Cys(oNB) remained intact after acidic deprotection of other amino acid residues (95% TFA) of the pentapeptide, and subsequent NCL to a recombinant Streptomyces coelicolor NiSOD bearing an N-terminal Cys. Photochemical deprotection of oNB could then be achieved by irradiation at 365 nm (100 mM NaOAc, 20 mM TCEP, 10 mM semicarbazide, pH 5.8).244 Cys(oNB) can also be incorporated into proteins via unnatural amino acid mutagenesis245,246 as demonstrated with a photocaged human superoxide dismutase (nbC-33-hSOD, Fig. 31b),246 along with being used to study protein activity,247,248 As discussed previously, Cys(oNB) is also orthogonal to Pd-mediated deprotection of Acm, as recently displayed in the synthesis of Linaclotide and plectasin (Fig. 31c).215 The oNB protecting group is, however, a poor chromophore and displays low two-photon sensitivity. Coumarin-based protecting groups have since been described in an attempt to address some of these issues.145
:MeCN (95:5, pH 7.2). The 7,8-BCMCMOC and BCMACMOC groups were, however, unstable when treated with piperidine, resulting in intramolecular S- > N acyl shifts in the case of N-terminal Cys; an alternative Fmoc removal protocol (1% DBU in DMF, acidic washing) and acetylation of the N-terminus was therefore utilised. The BCMACMOC group can be removed by irradiation at ≥402 nm whilst the 7,8-BCMCMOC and C4MNB groups can be removed by irradiation at ≥325 nm, allowing for orthogonal deprotection (provided BCMACMOC is photolysed using long-wavelength irradiation first). These groups were subsequently used to synthesise an N-acetylated resact peptide (Ac0-resact).144
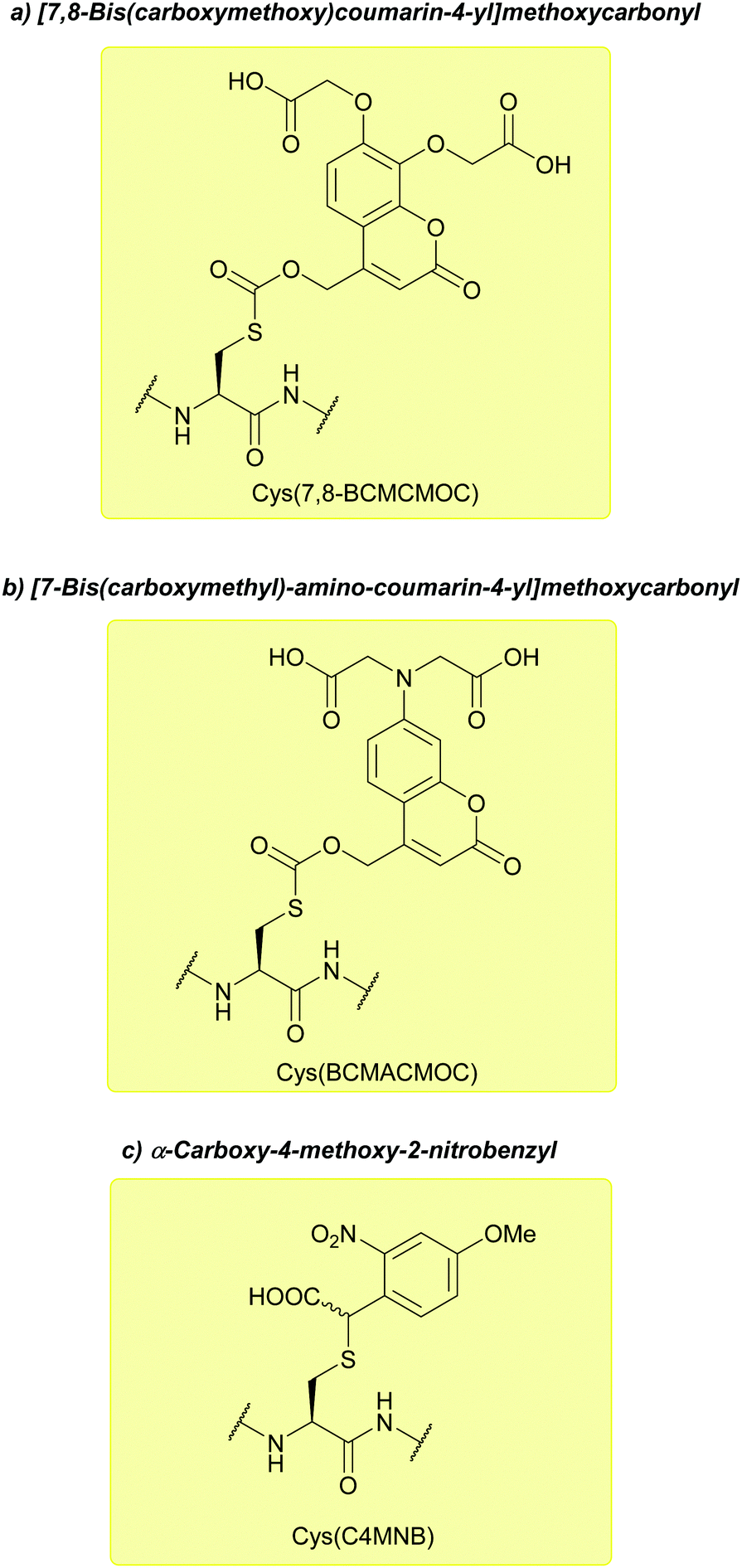 | ||
| Fig. 32 (a) Cys thiol protection with the [7,8-bis(carboxymethoxy)coumarin-4-yl]methoxycarbonyl (7,8-BCMCMOC) protecting group. (b) Cys thiol protection with the [7-bis(carboxymethyl)-amino-coumarin-4-yl]methoxycarbonyl (BCMACMOC) protecting group. (c) Cys thiol protection with the α-carboxy-4-methoxy-2-nitrobenzyl (C4MNB) protecting group. | ||
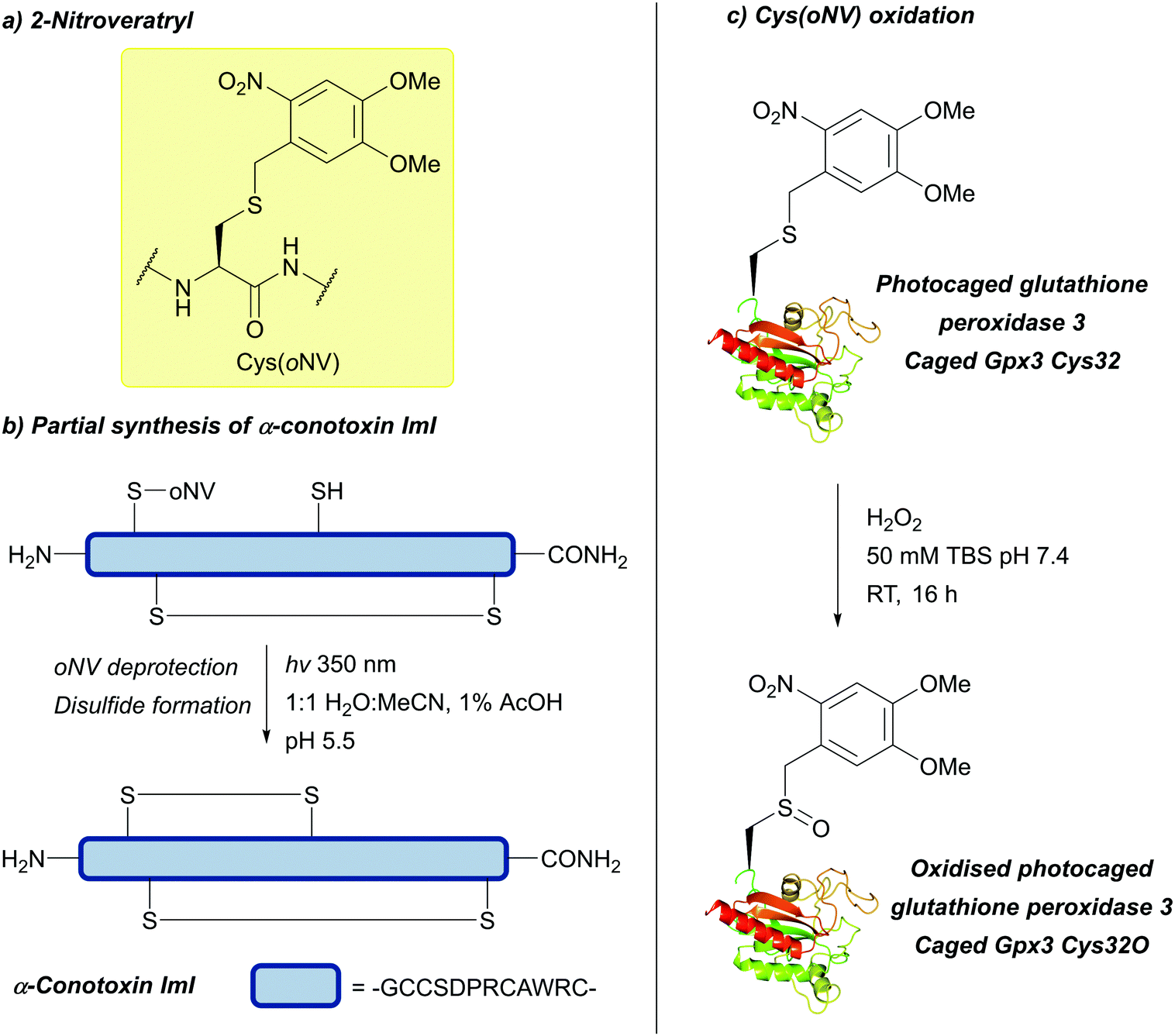 | ||
| Fig. 33 (a) Cys thiol protection with the 2-nitroveratryl (oNV) protecting group. (b) Partial synthesis of α-conotoxin ImI using Cys(oNV). (c) Photocaged glutathione peroxidase 3 (caged Gpx3 Cys32) and subsequent oxidation to yield the the sulfenic acid analogue (caged Gpx3 Cys32O). | ||
oNV is fully compatible with Fmoc SPPS, including stability to 10% TFMSA/TFA in the presence of excess dipyridine disulfide, the conditions necessary to convert the tBu protecting group to the activated S-Pyr group. The two groups may, therefore, be used together – photolysis of the oNV is followed by thiolysis in the presence of Cys(S-Pyr), selectively forming a disulfide bond. This strategy has been used for the solid-phase synthesis of several Cys-rich peptides, including human insulin and α-conotoxin ImI (Fig. 33b).142 oNv photocaged Cys has since been utilised in the synthesis of multi-Cys-containing peptide fragments.252 The protected amino acid has also been incorporated into proteins via unnatural amino acid mutagenesis to yield photocaged eGFP (EGFPTyr39DMNB-Cys),253 and photocaged glutathione peroxidase 3 (caged Gpx3 Cys32); the latter can then be oxidised with H2O2 to generate the sulfenic acid analogue (caged Gpx3 Cys32O, Fig. 33c).254
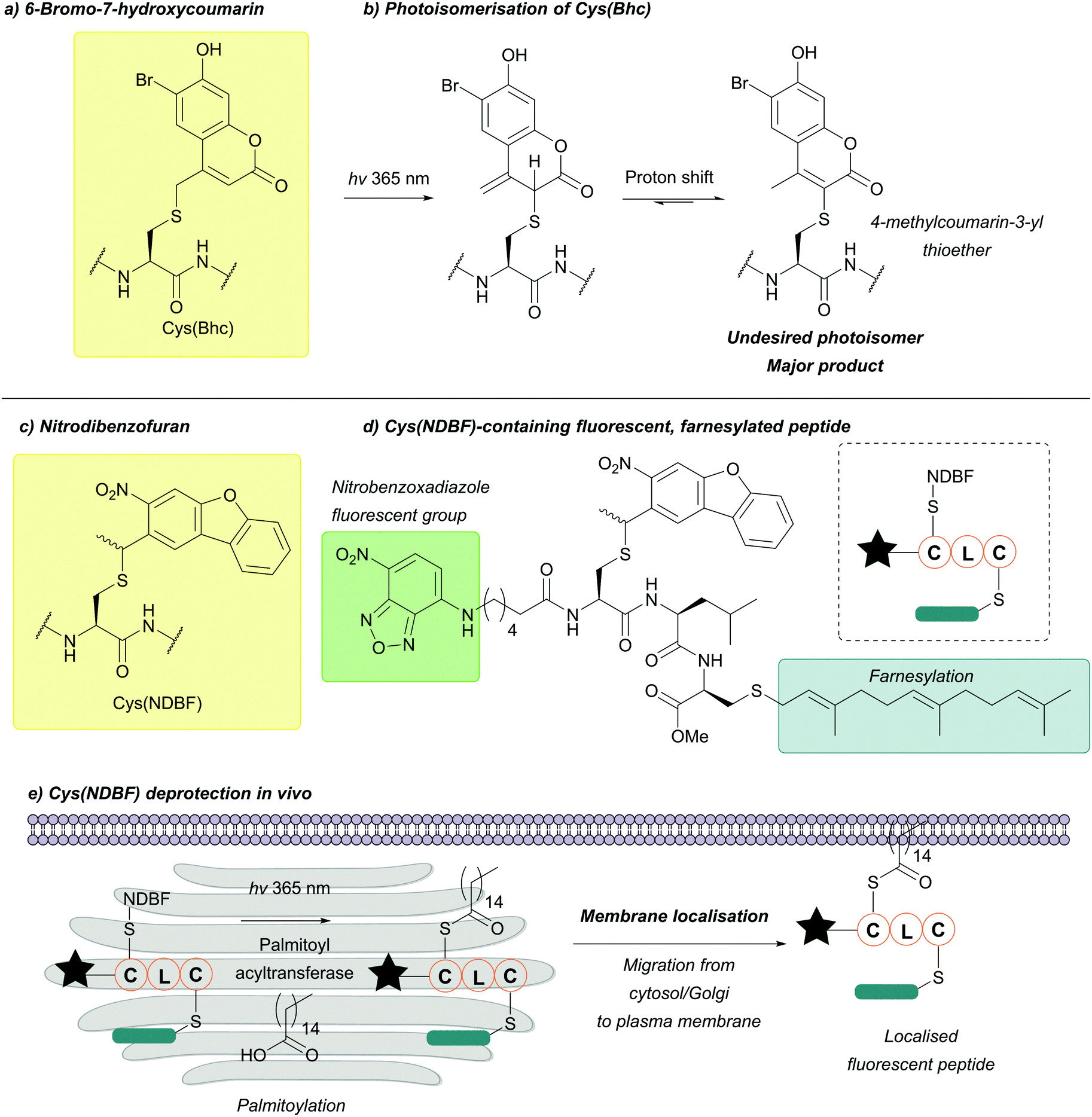 | ||
| Fig. 34 (a) Cys thiol protection with the 6-bromo-7-hydroxycoumarin (Bhc) protecting group. (b) Irradiation of Cys(Bhc) and subsequent formation of a 4-methylcoumarin-3-yl thioether photoisomer. (c) Cys thiol protection with the nitrodibenzofuran (NDBF) protecting group. (d) Fluorescent, farnesylated peptide containing Cys(NDBF). (e) Incubation and subsequent localisation in vivo of the aforementioned peptide within the cytosol and Golgi apparatus. Irradiation of the cells leads to deprotection of Cys(NDBF) which can then under enzymatic palmitoylation, leading to membrane localisation of the peptide. | ||
Although incorporation into a peptide using Fmoc SPPS is not an issue, the photocleavage efficiency of Bhc-protected thiols is context-dependent. This is because the major product of irradiation is typically an unwanted photoisomer (a 4-methylcoumarin-3-yl thioether, Fig. 34b) instead of the free thiol, which limits the applications of Bhc as a Cys protecting group.
The NDBF protecting group has been additionally used for live cell applications. A fluorescent, farnesylated peptide which can undergo enzymatic palmitoylation by palmitoyl acyltransferase was synthesised and protected as a NDBF thioether (Fig. 34d).145 Incubation of this peptide with human ovarian carcinoma SKOV3 cells led to localisation of the peptide to the cytosol and the Golgi apparatus. Upon irradiation, the peptide migrated to the plasma membrane, indicating that enzymatic palmitoylation had occurred (Fig. 34e).
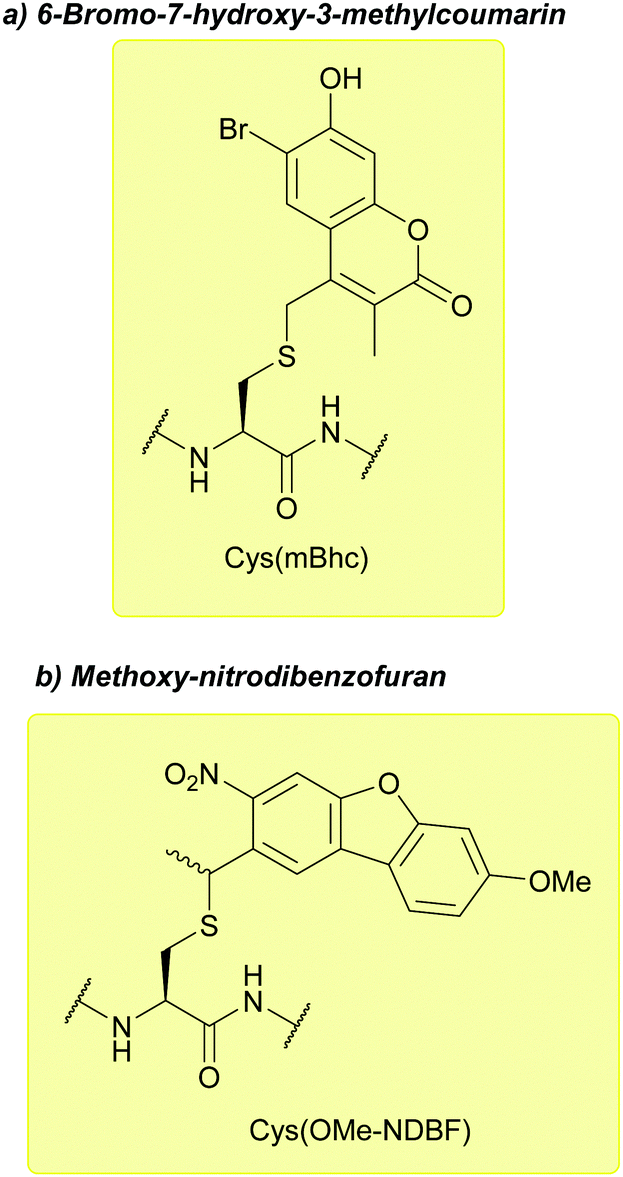 | ||
| Fig. 35 (a) Cys thiol protection with the 6-bromo-7-hydroxy-3-methylcoumarin (mBhc) protecting group. (b) Cys thiol protection with the methoxy-nitrodibenzofuran (OMe-NDBF) protecting group. | ||
5.10 Reducing agent-labile protecting groups
:1, 1 h, 0 °C), TFA and I2/AcOH/2 M HCl. No side products are observed during cleavage. pNB has been proposed as an alternative to Acm for use in Boc SPPS due to its greater stability to acid.111
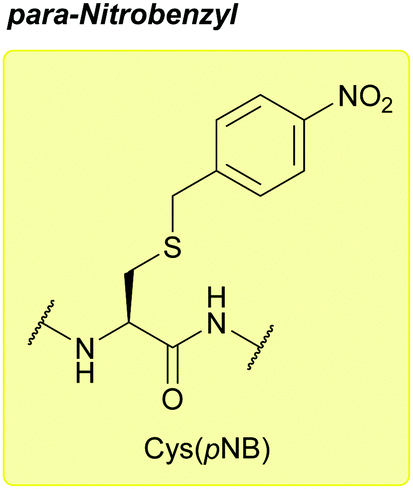 | ||
| Fig. 36 Cys thiol protection with the para-nitrobenzyl (pNB) protecting group. | ||
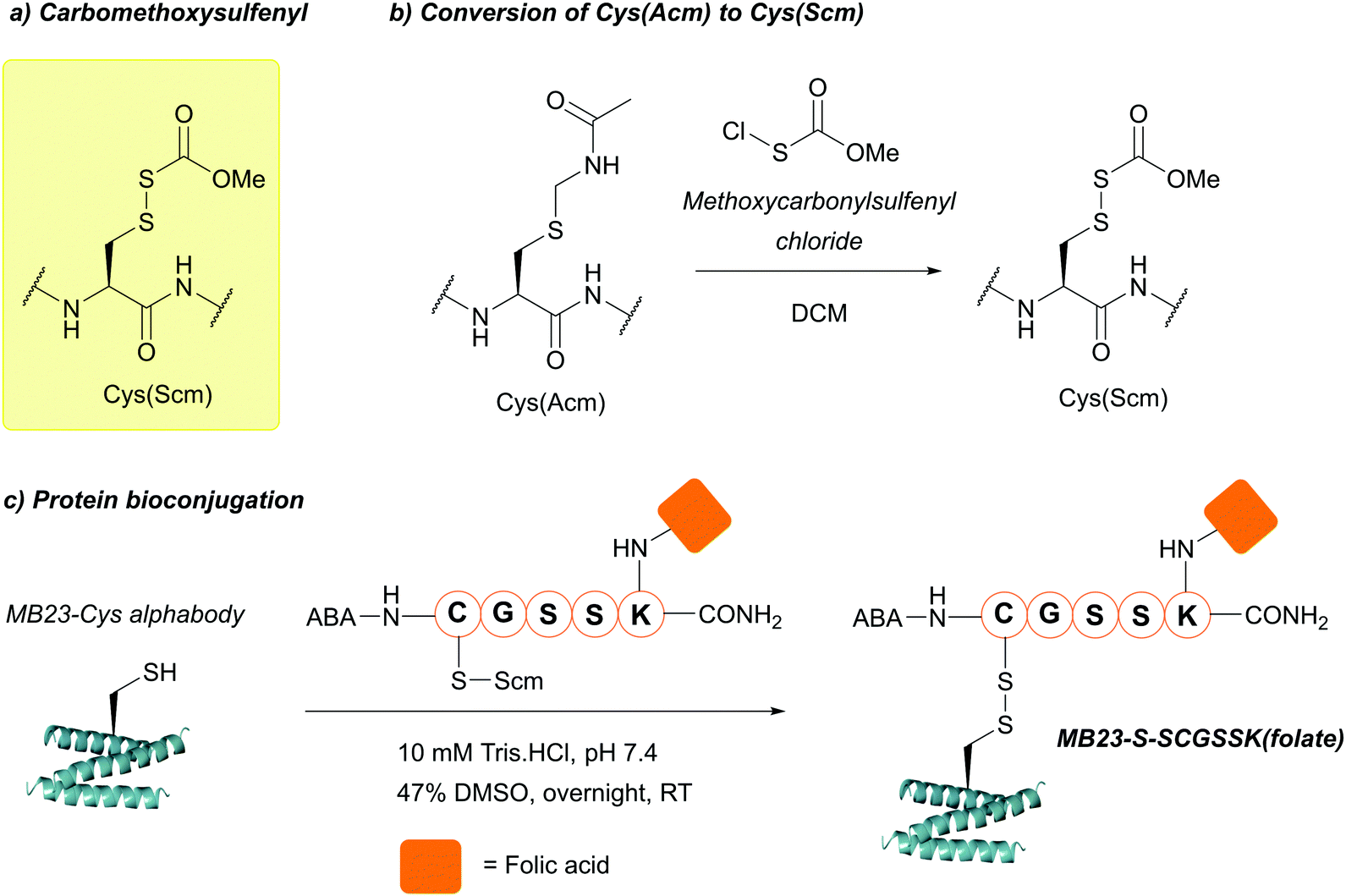 | ||
| Fig. 37 (a) Cys thiol protection with the carbomethoxysulfenyl (Scm) protecting group. (b) Conversion of Cys(Acm) to Cys(Scm) using methoxycarbonylsulfenyl chloride. (c) Protein bioconjugation with Cys(Scm)-containing peptides. | ||
Cys(Scm) has recently used in protein bioconjugation to facilitate the modification of MB23-Cys (an Alphabody – a trihelical peptide with potential as an anti-cancer therapeutic) via disulfide linkages. MB23-Cys was first reduced using DTT (to remove any protein dimer), then modified with a folic acid, Cys(Scm)-containing peptide in 10 mM Tris–HCl, pH 7.4, 37 °C (Fig. 37c).148
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Piperidine), referred to as the Snip protecting group (Fig. 38a). These derivatives can be used for intramolecular disulfide formation both on and off resin akin to peptides containing Cys(Snm).259
O)Piperidine), referred to as the Snip protecting group (Fig. 38a). These derivatives can be used for intramolecular disulfide formation both on and off resin akin to peptides containing Cys(Snm).259
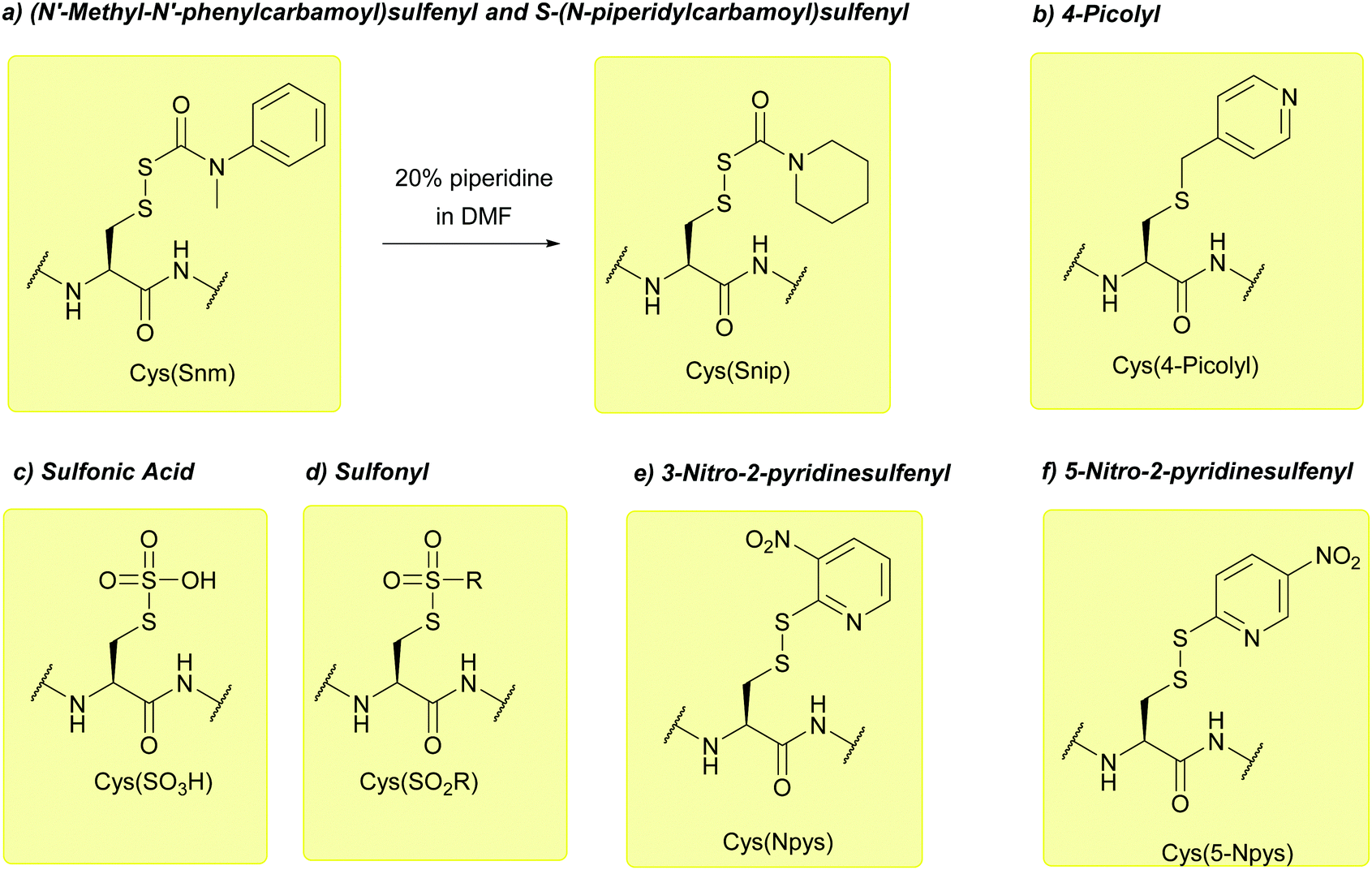 | ||
| Fig. 38 (a) Cys thiol protection with the (N′-methyl-N′-phenylcarbamoyl)sulfenyl (Snm) protecting group. Conversion of Cys(Snm) to Cys(Snip) occurs upon treatment with piperidine (b) Cys thiol protection with the 4-picolyl protecting group. (c) Cys thiol protection with the sulfonic acid (SO3H) protecting group. (d) Cys thiol protection with the sulfonyl (SO2R) protecting group. (e) Cys thiol protection with the 3-nitro-2-pyridinesulfenyl (Npys) protecting group. (f) Cys thiol protection with the 5-nitro-2-pyridinesulfenyl (5-Npys) protecting group. | ||
Sulfonyl (SO2R) protecting groups (Fig. 38d) have recently found some use in the synthesis of enantiomerically-enriched α-hydroxy and α-chloro acid building blocks. The sulfonyl group is first introduced to the Cys residue, and diazotisation is subsequently used to generate the α-hydroxy or α-chloro acid (depending on whether H2SO4 or HCl is used).260
Npys is not compatible with Fmoc SPPS, as it is unstable to piperidine (81% decomposition following treatment with 50% piperidine in DCM for 10 min). It is also unstable to TBAF−, an alternative to piperidine for Fmoc deprotection. Additionally, while treatment with HF in the presence of anisole or p-cresol leaves Npys unaltered, HF in the presence of p-thiocresol or DMS (HF/DMS/p-cresol, 25:65:10 or HF/DMS/p-cresol/p-thiocresol, 25:65:5:5), has been shown to cause significant loss of the protecting group.157 Npys has been used in many syntheses.154 An early example of this is the synthesis of [Lys]8vasopressin, which was chosen as a model peptide to display the use of Npys in Boc SPPS.156 More recently, the Npys protecting group has been the focus of a solid phase disulfide ligation (SPDSL) system.262 Here, Npys-Bn is loaded onto a solid support and converted to resin-bound Npys-Cl through chlorosulfenylation. Cys(tBu)-containing peptides can then be loaded onto the resin via Npys-mediated displacement of the tBu protecting group. Peptide release, along with disulfide bond formation, can then be achieved through addition of a Cys-containing peptide. Subsequent intramolecular amide formation then yields a disulfide containing peptide, as demonstrated in the synthesis of oxytocin.262 A similar platform that replaces the Npys-Cl group for a more stable Npys-OPh(para-fluoro) group has very recently been reported in the literature.263
:2.5:2.5 v/v) provided no thiol scavenger is added. In cases such as for tryptophan-containing peptides, 2-methylindole and anisole can be used as alternative scavengers to avoid StBu deprotection.159 The group is also compatible with basic conditions for Fmoc SPPS; extended incubations of resin-bound C-terminal Cys(StBu)-containing peptides in 20% piperidine in DMF can lead to 3-(1-piperidinyl)alanine by-products however.77 StBu can also be removed by TFA in the presence of DTNP-thioanisole. Interestingly, StBu exhibits no lability to TFA/DTNP without the addition of thioanisole to the mixture, making it orthogonal to groups which are removed by TFA/DTNP, such as tBu.80 Partial removal in HF has also been reported.159 The StBu protecting group is orthogonal to other protecting groups, such as Trt, Acm, Meb/Mob, tBu,74,163 and Allocam.205 Protected Cys(StBu) has featured in the synthesis of somatostatin,268 coumarin-based probes,270 and cryptophane-based probes.271 Additionally, peptides bearing Cys(StBu) and perfluoroaryl-modified Cys residues can undergo cyclisation to form macrocyclic peptides; this was achieved with in situ reduction of Cys(StBu) with TCEP in the presence of glutathione S-transferase (GST, which facilitates conjugation of the newly released thiol and perfluoroaryl group, Fig. 39b).272 In the case of N-terminal Cys protected with StBu, in situ reduction/protecting group removal followed by NCL can be achieved in the presence of a suitable peptide thioester substrate.273,274 Alternatively, the N-terminal Cys can react with 2-cyano-6-aminobenzothiazoles (CBTs), as demonstrated in the synthesis of imaging agents for positron emission tomography (PET).269,275 For formation of disulfide bonds using an orthogonal protecting group strategy, StBu must be removed before other protecting groups to avoid cleaving or scrambling existing disulfides (as would occur upon treatment with a reducing agent). This is typically done on-resin prior to TFA acidolysis, which can result in side products and low yields. As discussed previously, one solution to this problem that has been reported was to perform post-synthetic tritylation; multiple disulfide bonds could then be regioselectively formed in solution using combinations of StBu, Trt, Acm and Meb/Mob, all of which survive the final cleavage step in Fmoc SPPS, bar Trt.74
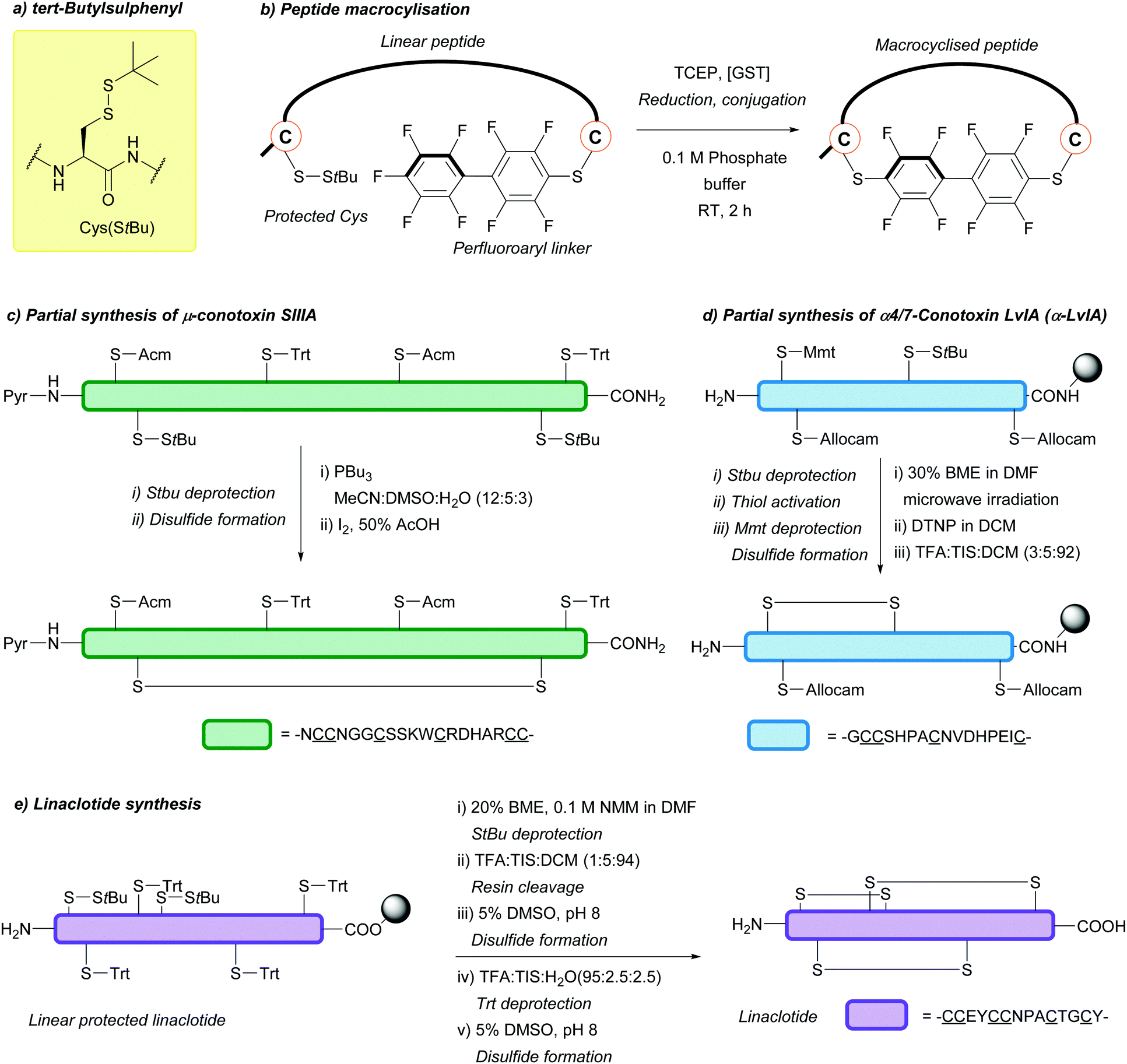 | ||
| Fig. 39 (a) Cys thiol protection with the tert-butylsulphenyl (StBu) protecting group. (b) Reduction of Cys(StBu) and perfluoroaryl linker-containing peptide with TCEP in the presence of glutathione S-transferase (GST). Deprotection of Cys(StBu) yields the free thiol, which can then undergo macrocyclisation with the perfluoroaryl linker facilitated by GST. (c) Partial synthesis of μ-conotoxin SIIIA using Cys(StBu). (d) Partial synthesis of α-LvIA using Cys(StBu). (e) Synthesis of Linaclotide using Cys(StBu). | ||
Cys(StBu) has been used in the synthesis of a range of different peptides, including μ-SIIIA (Fig. 39c),74 α-LvIA (Fig. 39d),205 and Linaclotide (Fig. 39e).276 The removal of StBu with reducing agents has previously been shown to be sequence-dependent and challenging;166 additionally, the lengthy times required for deprotection are undesirable when using the group in routine SPPS.163 Alternative disulfide-based protecting groups such as dimethoxyphenylthio (S-Dmp) and 2,4,6-trimethoxyphenylthio (S-Tmp) have since been reported as replacements for the StBu protecting group.163
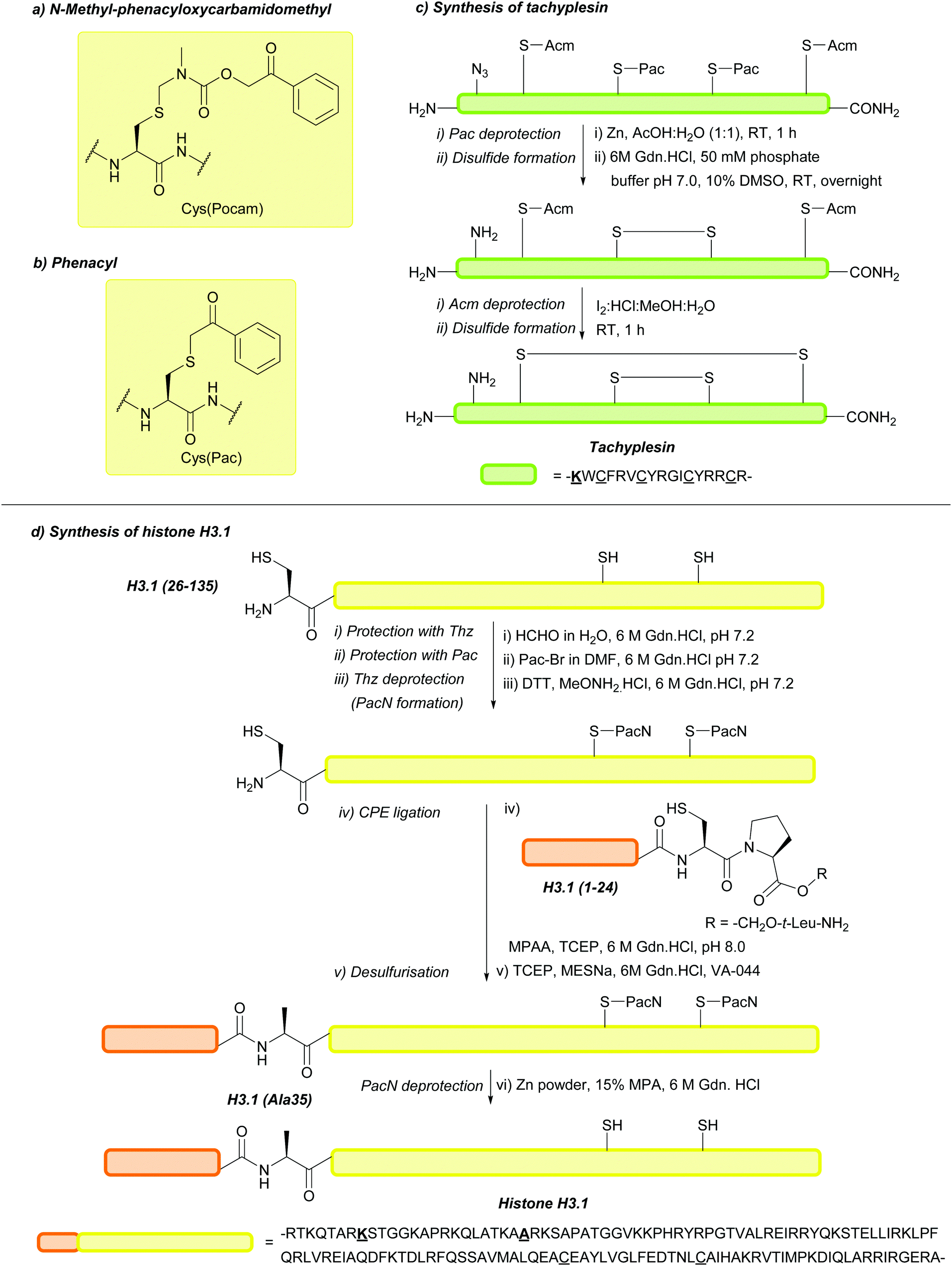 | ||
| Fig. 40 (a) Cys thiol protection with the N-methyl-phenacyloxycarbamidomethyl (Pocam) protecting group. (b) Cys thiol protection with the phenacyl (Pac) protecting group. (c) Synthesis of tachyplesin using Cys(Pac). (d) Semi-synthesis of histone H3.1 using Cys(Pac). | ||
The Pac protecting group has been employed in the semi-synthesis of histone H3 bearing an Nε trimethylated Lys residue ([Lys(Me3)9]H3.1, Fig. 40d).278 First, the C-terminal fragment of the peptide containing an N-terminal Cys and two internal Cys residues (H3.1(26-135)) was recombinantly produced by gene expression and subsequent peptide production in E. coli. The N-terminal Cys residue was then orthogonally protected with Thz, whereas both the internal Cys were protected with Pac. Conditions for deprotection of N-terminal Cys(Thz) using methoxyamine also lead to oxime ether formation at the ketone position of Pac, converting Pac to “PacN”; this, however, does not impact on downstream deprotection as PacN also shows lability to Zn/AcOH similar to Pac. Deprotection of the N-terminal Cys(Thz), followed by CPE ligation with a suitable H3 N-terminal fragment and subsequent desulfurisation gave the dual Cys(PacN) protected peptide. Critically, the Cys(PacN) groups remained resistant to desulfurisation. Deprotection with Zn powder and 15% 3-mercaptopropionic acid (MPA) in H2O with 6 M Gdn yielded the product [Lys(Me3)9]H3.1 histone.278 Similarly, it has been shown that Cys(Pac) can be used in conjunction with recombinantly generated protein segments for the traceless semisynthesis of human small heat shock protein (Hsp27) and a lipidated variant of murine prion protein (Prp).162 Pac proved compatible both with installation into thioester-containing peptides, and in radical desulfurisation steps. Cys(Trt) was also investigated; however, in this case, desulfurisation of Cys(Trt) containing Hsp27 proved lead to a mixture of products.162
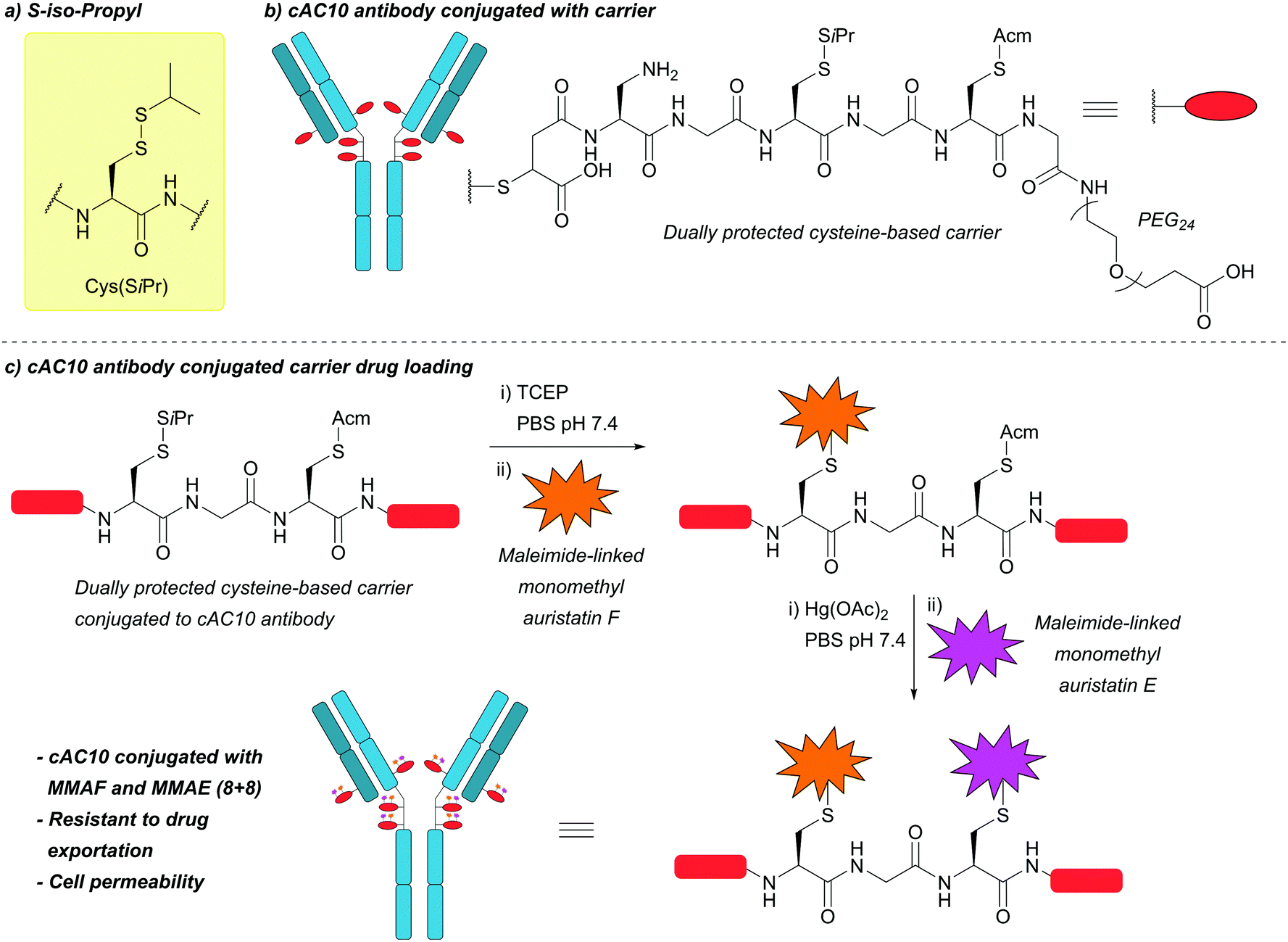 | ||
| Fig. 41 (a) Cys thiol protection with the S-iso-propyl (SiPr) protecting group. (b) CD30-directed antibody cAC10 bearing a dual protected cysteine-based carrier containing Cys(SiPr) and Cys(Acm). (c) Construction of cAC10 conjugated with MMAF and MMAE (8 + 8) using Cys(SiPr). | ||
:2.5:2.5) at RT when applied to oxytocin synthesis. This could be avoided by using a more acid-labile resin, avoiding the requirement for high TFA concentrations. As with StBu, S-Dmp and S-Tmp should be deprotected first in orthogonal protecting group strategies (which usually results in deprotection prior to TFA cleavage from the resin). The use of S-Dmp and S-Tmp has been demonstrated practically, with sample syntheses of oxytocin on-resin, and T22 carried out.163 Both groups produced better yields than the corresponding peptides synthesised using StBu. Out of the two groups, S-Tmp showed higher stability and produced purer peptides, and is therefore recommended for use.163 In addition to using DTT, deprotection of the S-Dmp group has also been demonstrated using a novel reducing agent 2-(dibenzylamino)butane-1,4-dithiol (DABDT).164 The S-Tmp protecting group has since been used in conjunction with N-chlorosuccinimide in the on-resin, regioselective synthesis of α-conotoxin SI (Fig. 42c),280 and very recently in the synthesis of a disulfide containing, truncated neuropeptide Y (NPY) analogue.281
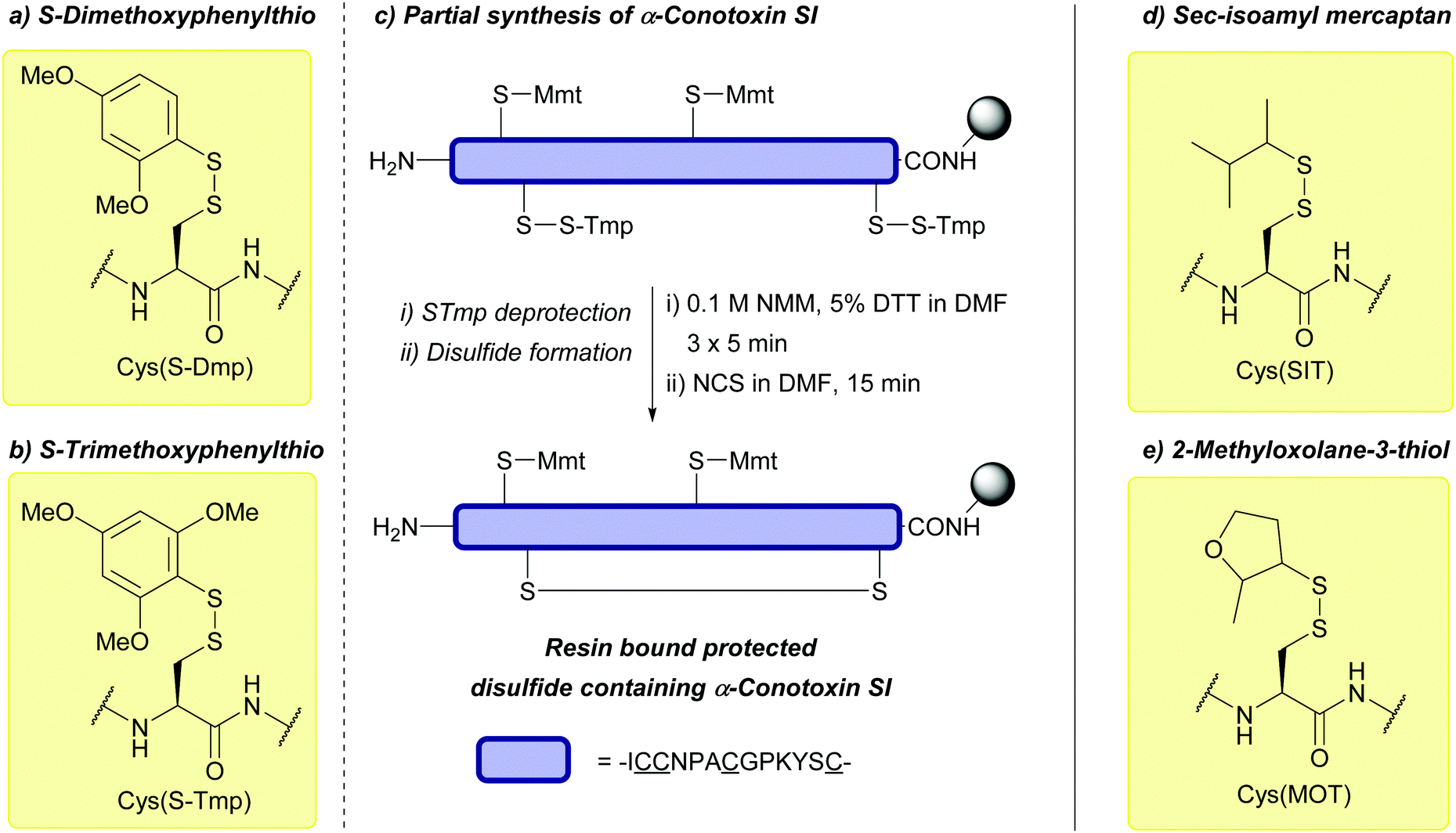 | ||
| Fig. 42 (a) Cys thiol protection with the dimethoxyphenylthio (S-Dmp) protecting group. (b) Cys thiol protection with the 2,4,6-trimethoxyphenylthio (S-Tmp) protecting group. (c) Partial synthesis of synthesis of α-conotoxin SI using Cys(S-Tmp). (d) Cys thiol protection with the sec-isoamyl mercaptan (SIT) protecting group. (e) Cys thiol protection with the 2-methyloxolane-3-thiol (MOT) protecting group. | ||
:4) and 0.1 M DIEA. Alternatively, 20 equiv. DTT in MeCN with 5% DIEA could also be used to remove both groups; the rate of deprotection was further enhanced by addition of 5% H2O, with SIT and MOT fully removed within 40 and 20 min respectively. Treatments of 5 equiv. DTT in DMF:DIEA:H2O (95:2.5:2.5, three treatments, 10 min) was also sufficient for deprotection of Cys(SIT) and Cys(MOT) located within Asn(Trt)-Cys(SIT/MOT)-Asn(Trt) tripeptides. Both of these groups have been successfully used in the synthesis of vasopressin via Fmoc SPPS, although minor thiol deprotection (ca. 2%) of protected vasopressin during SPPS was noted for the more labile MOT group. Furthermore, the SIT protecting group could be used in microwave-assisted synthesis of vasopressin.165
:1, 2 h, 0 °C).4S-Pyr can also act as an activating group; free thiols can attack the activated sulfur atom, displacing S-Pyr to generate disulfide containing peptides.142,284 This has recently been shown in the synthesis of sialic acid-containing insulin analogues, referred to as “Sialic-Ins”.285
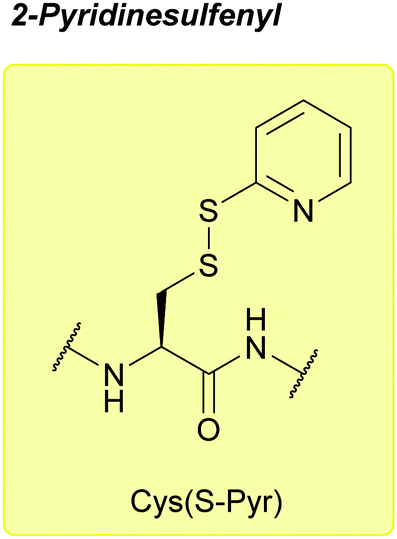 | ||
| Fig. 43 Cys thiol protection with the 2-pyridinesulfenyl (S-Pyr) protecting group. | ||
5.11 Safety-catch protecting groups
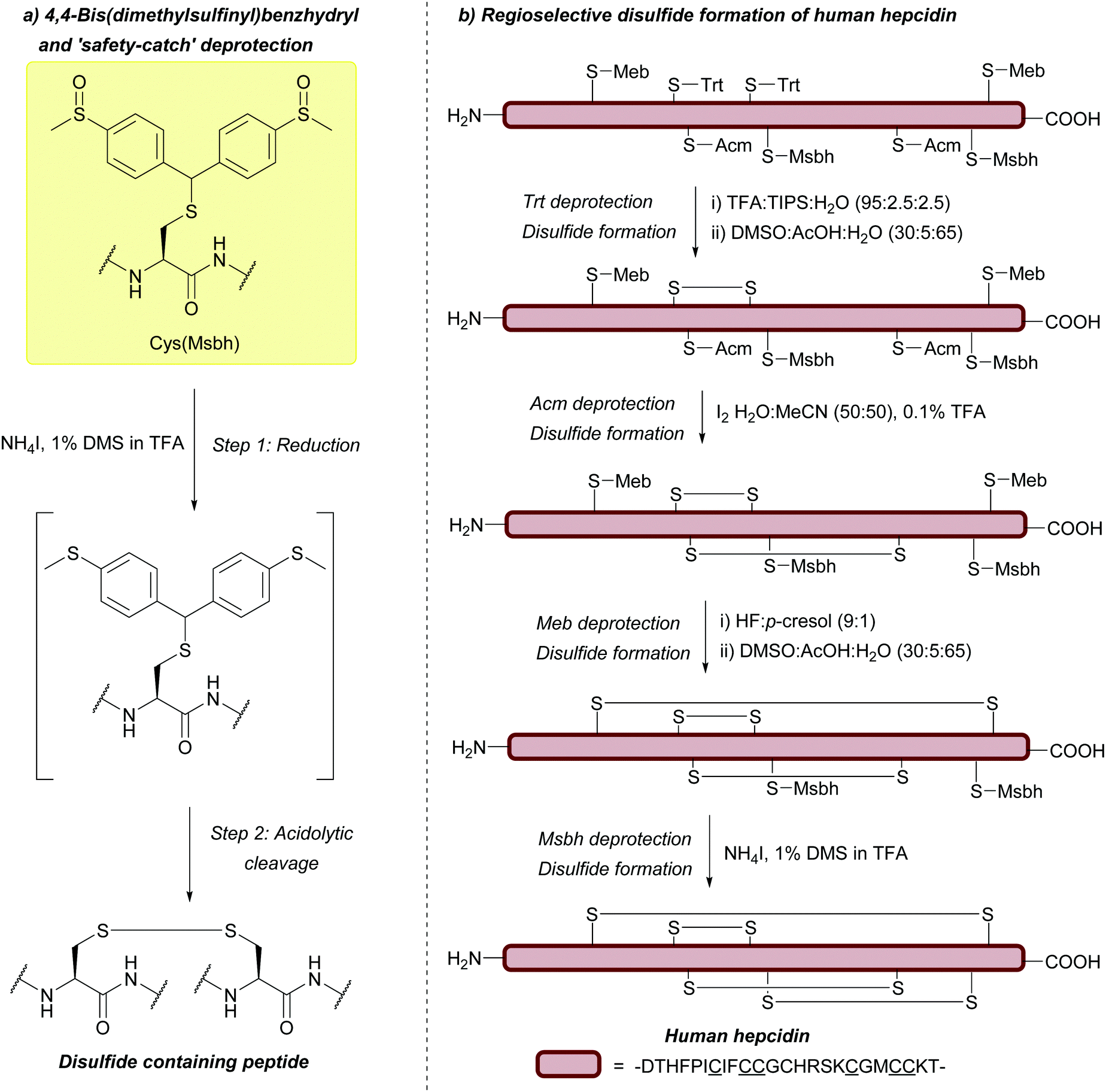 | ||
| Fig. 44 (a) Cys thiol protection with the 4,4-bis(dimethylsulfinyl)benzhydryl (Msbh) protecting group. Treatment with a cocktail of NH4I/DMS/TFA results in initial reduction of the Msbh protecting group, followed by acid-mediated cleavage and subsequent iodine-mediated oxidation to yield disulfide containing peptides. (b) Partial synthesis and regioselective disulfide formation of human hepcidin using Cys(Msbh). | ||
NH4I/TFA treatment is incompatible with Trp-containing peptides, as Trp undergoes a number of side reactions (due to the presence of I2) unless the indole nitrogen is protected with a formyl group. This is standard in Boc SPPS and thus should not present a significant issue in this case. The formyl group is, however, removed by piperidine treatment so if Fmoc SPPS is being used alternative sulfoxide reduction methods or Trp protecting groups are needed.166 Msbh has been used in the regioselective synthesis of human hepcidin (Fig. 44b), providing an orthogonal strategy for the synthesise of peptides containing four disulfide bonds.166 It has also been theorised to be key to the regiospecific construction of peptides containing five or more disulfide bonds, a feat that is yet to be accomplished.5
6. Conclusions
Methods for the protection, and subsequent deprotection, of the thiol side chain of Cys has enabled a vast array of peptide and protein chemistry over the last 70 years. The development of protecting groups that are orthogonal to one another in particular has proved critical to facilitating the synthesis of complex products such as disulfide rich peptides. Although there are now numerous protecting strategies that have been reported for Cys, it is likely that research and development on the subject will continue over the course of the coming years. This will undoubtedly involve taking well-established Cys protecting groups, and developing new deprotection conditions that offer advantages over established methods. For example, protecting groups originally reported decades ago for use in Boc peptide synthesis such as Dpm, Thp, and SiPr, have greatly benefitted from being revisited for use in Fmoc peptide synthesis, both from the perspective of developing new deprotection protocols, and the application of such groups. Transition metal chemistry applied to Cys protecting group chemistry in recent years has also yielded new, milder conditions to deprotect protecting groups such as Trt, tBu, Acm, and Thz, or improved deprotection protocols in the case of Allocam. Alternatively, the development of new protecting groups with their own set of deprotection conditions will also likely prove paramount to furthering Cys-based peptide and protein chemistry. Protecting groups such as Mbom and Ddm offer C-terminal Cys-containing peptides with reduced racemisation side-reactions. Redesigning previously reported protecting group scaffolds has also led to the development of new protecting groups that are significantly easier to deprotect, as seen with S-Dmp/S-Tmp and SIT/MOT. Furthermore, newer protecting groups with unique “turn on/off” deprotection conditions, such as Hmboff/on and Msbh, will likely see further development in challenging peptide synthesis. For example, regioselective synthesis of peptides with greater than four disulfide bonds has not yet been reported; novel protecting groups will likely prove critical to achieving this.Finally, there is an increasing desire to make the process of SPPS more “green”, which currently uses toxic and environmentally unfriendly reagents such as piperidine, DMF, and DCM.286 This will require protecting group chemistry that is compatible with greener reagents, such as water/aqueous-based solvent systems. Enzymatically labile protecting groups such as Phacm, or photolabile groups such as NDBF, have already been successfully utilised in this context; future protecting groups with similar properties will likely prove important in the “greening” of SPPS. We additionally anticipate the field of peptide chemistry and growing field of protein bioconjugation will likely benefit each other in the coming years. In contrast to conventional peptide synthesis, protein bioconjugation/deconjugation is, by design, carried out in benign/environmentally friendly aqueous systems in a site-specific manner. Additionally, there is a continuing search for new methodology for use in bioconjugation; although these must yield stable conjugates, strategies whereby the bioconjugate can be released in a controlled manner offers huge potential in applications such as controlled drug release. This interplay has already been demonstrated with Cys protecting groups that have since been applied to bioconjugation, such as Acm, Scm, Thz, and SiPr. Similarly, Suc, which is routinely used in bioconjugation, has very recently been demonstrated as a Cys protecting group in peptide synthesis. It is likely other bioconjugation strategies will also be applied to peptide synthesis in the coming years.
We have reviewed, analysed, and discussed over 60 individual protecting groups for the thiol group of Cys. We hope that this review provides a useful resource for peptide and protein chemists research, and encourages further research into both old and new Cys protecting groups.
Abbreviations
| 2-Moxan | 2-methoxy-9H-xanthen-9-yl |
| 2,6-diMeOBn | 2,6-dimethoxylbenzyl |
| 4MeO-2MeBn | 4-methoxy-2-methylbenzyl |
| 5-Npys | 5-nitro-2-pyridinesulfenyl |
| 7,8BCMCMOC | [7,8-bis(carboxymethoxy)coumarin-4-yl]methoxycarbonyl |
| Acm | acetamidomethyl |
| AcOH | acetic acid |
| Ad/1-Ada | 1-adamantyl |
| ADC | antibody drug conjugate |
| Alloc | allyloxycarbonyl |
| Allocam | allyloxycarbonylaminomethyl |
| AgOAc | silver acetate |
| AgOTf | silver trifluoromethanesulfonate |
| Arg | arginine |
| Bam | benzamidomethyl |
| BCMACMOC | [7-bis(carboxymethyl)-amino-coumarin-4-yl]methoxycarbonyl |
| Bhc | 6-bromo-7-hydroxycoumarin |
| BME | 2-mercaptoethanol |
| Boc | tert-butyloxycarbonyl |
| Bom | benzyloxymethyl |
| BSA | bovine serum albumin |
| Bu3SnH | tributyltin hydride |
| Bzl/Bn | benzyl |
| C4MNB | α-carboxy-4-methoxy-2-nitrobenzyl |
| CAN | ceric ammonium nitrate |
| CDMNB | α-carboxy-4-methoxy-2-nitrobenzyl |
| CuAAC | copper(I)-catalysed alkyne-azide cycloaddition |
| CPE | cysteinylprolyl ester |
| CPI | cysteinylprolyl imide |
| Cys | cysteine |
| Dbs/Sub | 5-dibenzosuberyl |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DCC | dicyclohexylcarbodiimide |
| DCM | dichloromethane |
| Ddm/4,4′-diMeODpm | 4,4′-dimethoxydiphenylmethyl |
| DIEA | N,N-diisopropylethylamine |
| DMB | 3,4-dimethylbenzyl |
| Dmbm | (4,6-dimethoxy-2,2-dimethyl-2,3-dihydrobenzofuran-7-yl)methanol |
| DMF | dimethylformamide |
| DMS | dimethyl sulfide |
| DMSO | dimethylsulfoxide |
| Dnpe | 2-(2,4-dinitrophenyl)ethyl |
| DPDS | 2,2′-dipyridyl disulfide |
| Dpm | diphenylmethyl |
| DSF | disulfiram |
| DTC | diethyldithiocarbamate |
| DTNB | 5,5′-dithiobis-(2-nitrobenzoic acid) |
| DTNP | 2,2′-dithiobis(5-nitropyridine) |
| DTP | 2,2′-dithiodipyridine |
| DTT | dithiothreitol |
| EDT | ethane-1,2-dithiol |
| EDTA | ethylenediaminetetraacetic acid |
| EETI-II | Ecballium elaterium trypsin inhibitor II |
| EGFP | enhanced green fluorescent protein |
| EtOH | ethanol |
| Fm | 9-fluorenylmethyl |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| Fnam | [N-[2,3,5,6-tetrafluoro-4-(N′-piperidino)-phenyl], N-allyloxycarbonyl]-aminomethyl |
| Fsam | S-[N-[2,3,5,6-tetrafluoro-4-(phenylthio)-phenyl], N-allyloxycarbonyl]-aminomethyl |
| GBP | growth-blocking peptide |
| Gdn·HCl | guanidinium chloride |
| GFP | green fluorescent protein |
| GSH | glutathione |
| H2O | water |
| HEPES | (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) |
| HEPPS | 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid |
| HFIP | hexafluoro-2-propanol |
| Hgm | hydroxyglycine-Acm |
| Hg(OAc)2 | mercury(II) acetate |
| His | histidine |
| Hmb | 2-hydroxy-4-methoxy benzyl |
| hNP2 | defensin human neutrophil peptide-2 |
| HOBt | 1-hydroxybenzotriazole |
| Hqm | hydroxyquinoline-Acm |
| Lys | lysine |
| MAA | 3-mercaptoacetic acid |
| mBhc | 6-bromo-7-hydroxy-3-methylcoumarin |
| Mbom | 4-methoxybenzyloxymethyl |
| Meb/4-MeBn/4-MeBzl | 4-methylbenzyl |
| MeCN | acetonitrile |
| MeOH | methanol |
| MeONH2·HCl | O-methylhydroxylamine |
| MESNA | sodium 2-mercaptoethanesulfonate |
| Met | methionine |
| Mmt | 4-methoxytrityl |
| Mob/MBzl | 4-methoxybenzyl |
| MOT | 2-methyloxolane-3-thiol |
| MPAA | 4-mercaptophenylacetic acid |
| MPS | mercaptopropiosulfonic acid |
| Mpt | dimethylphosphinothioyl |
| Msbh | 4,4-bis(dimethylsulfinyl)benzhydryl |
| Mtt | 4-methyltrityl |
| NCL | native chemical ligation |
| NDBF | nitrodibenzofuran |
| NDMBA | 1,3-dimethylbarbituric acid |
| Nin | ninhydrin |
| NMM | N-methylmorpholine |
| NpsCl | 2-nitrophenylsulfenyl chloride |
| Npys | 3-nitro-2-pyridinesulfenyl |
| OMe-NDBF | methoxy-nitrodibenzofuran |
| oNB | 2-nitrobenzyl |
| oNV | 2-nitroveratryl |
| pAB | para-aminobenzyl |
| Pac | phenacyl |
| PAL | peptide-amide-linker |
| PB | phosphate buffer |
| Pbfm/Pmbf | 2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-methyl |
| pBNP | porcine brain natriuretic peptide |
| PBS | phosphate-buffered saline |
| [Pd(allyl)Cl]2 | allylpalladium(II) chloride dimer |
| PdCl2(PPh3)2 | bis(triphenylphosphine)palladium(II) dichloride |
| Pd(OAc)2 | palladium(II) acetate |
| Pd(PPh3)4 | tetrakis(triphenylphosphine)palladium(0) |
| PFTase | protein farnesyltransferase |
| PGA | penicillin G acylase |
| Phacm | phenylacetamidomethyl |
| PhSiH3 | phenylsilane |
| Pmcm | 2,2,5,7,8-pentamethylchroman-6-methyl |
| pNB | para-nitrobenzyl |
| Pocam | N-methyl-phenacyloxycarbamidomethyl |
| PTM | post-translational modification |
| Pym | 2-oxo-1-pyrrolidinyl)methyl |
| RP-HPLC | reversed phase high-performance liquid chromatography |
| Sac | S-allyl cysteine |
| Scm | carbomethoxysulfenyl |
| S-Dmp | dimethoxyphenylthio |
| SIT | sec-isoamyl mercaptan/3-methyl-2-butanethiol |
| SiPr | S-iso-propyl |
| Snm | (N′-methyl-N′-phenylcarbamoyl)sulfenyl |
| SO2R | sulfonyl |
| SO3H | sulfonic acid |
| SPPS | solid phase peptide synthesis |
| SprC | S-propargyl-cysteine |
| S-Pyr | 2-pyridinesulfenyl |
| StBu | tert-butylsulphenyl |
| S-Tmp | 2,4,6-trimethoxyphenylthio |
| Suc | succinimide |
| Tacm | trimethylacetamidomethyl |
| TBAF | tetrabutylammonium fluoride |
| tBu | tert-butyl |
| TCEP | tris(2-carboxyethyl)phosphine |
| TES | triethylsilane |
| TFA | trifluoroacetic acid |
| TFE | tetrafluoroethylene |
| TFMSA | trifluoromethanesulfonic acid |
| THF | tetrahydrofuran |
| Thp | tetrahydropyranyl |
| Thz | thiazolidine |
| TIS | triisopropylsilane |
| Tmbm | 4,5,6-trimethoxy-2,2-dimethyl-2,3-dihydrobenzofuran-7-methyl |
| Tmob | 2,4,6-trimethoxybenzyl |
| TMSBr | bromotrimethylsilane |
| TMSOTf | trimethylsilyl trifluoromethanesulfonate |
| TMTr | 4,4′,4′′-trimethoxytriphenylmethyl |
| Tppts | 3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt |
| Trp | tryptophan |
| Trt | trityl |
| Trx | thioredoxin |
| Tyr | tyrosine |
| VA-044 | 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride |
| UBL5 | ubiquitin-like protein 5 |
| Xan | 9H-xanthen-9-yl |
| ΨPro | pseudoproline |
| μ-SIIA | μ-conotoxin SIIIA |
Author contributions
R. J. S., C. M. and V. C. co-wrote the review and co-analysed the literature with R. J. S. and V. C. leading in writing and analysing.Conflicts of interest
V. C. is a co-founder and director of the company ThioLogics.Acknowledgements
We gratefully acknowledge the Leverhulme Trust (RPG-2017-288, 176274) for funding R. J. S.References
- G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS PubMed.
- B. Ghosh and S. S. Kulkarni, Chem. – Asian J., 2020, 15, 450–462 CrossRef CAS PubMed.
- M. Schelhaas and H. Waldmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2056–2083 CrossRef CAS.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- R. He, J. Pan, J. P. Mayer and F. Liu, ChemBioChem, 2020, 21, 1101–1111 CrossRef CAS PubMed.
- T. M. Postma and F. Albericio, Eur. J. Org. Chem., 2014, 3519–3530 CrossRef CAS.
- B. J. Tombling, C. K. Wang and D. J. Craik, Angew. Chem., Int. Ed., 2020, 59, 11218–11232 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef.
- D. Imhof, D. Roy and F. Albericio, Front. Chem., 2020, 8, 841 Search PubMed.
- V. du Vigneaud, C. Ressler, J. M. Swan, C. W. Roberts and P. G. Katsoyannis, J. Am. Chem. Soc., 1954, 76, 3115–3121 CrossRef CAS.
- V. du Vigneaud, C. Ressler, C. J. M. Swan, C. W. Roberts, P. G. Katsoyannis and S. Gordon, J. Am. Chem. Soc., 1953, 75, 4879–4880 CrossRef CAS.
- I. Vecchio, C. Tornali, N. L. Bragazzi and M. Martini, Front. Endocrinol., 2018, 9, 613 CrossRef.
- J. G. McGivern, Neuropsychiatr. Dis. Treat., 2007, 3, 69–85 CrossRef CAS PubMed.
- M. Corsetti and J. Tack, United Eur. Gastroenterol. J., 2013, 1, 7–20 CrossRef.
- U. Hennrich and M. Benešová, Pharmaceuticals, 2020, 13(3), 38 CrossRef CAS PubMed.
- A. Fra, E. D. Yoboue and R. Sitia, Front. Mol. Neurosci., 2017, 10, 167 CrossRef.
- S. Verma, R. Dixit and K. C. Pandey, Front. Pharmacol., 2016, 7, 107 Search PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- N. Joubert, A. Beck, C. Dumontet and C. Denevault-Sabourin, Pharmaceuticals, 2020, 13, 245 CrossRef CAS PubMed.
- P. Stathopoulos, S. Papas, C. Pappas, V. Mousis, N. Sayyad, V. Theodorou, A. G. Tzakos and V. Tsikaris, Amino Acids, 2013, 44, 1357–1363 CrossRef CAS.
- Y. Fujiwara, K. Akaji and Y. Kiso, Chem. Pharm. Bull., 1994, 42, 724–726 CrossRef CAS PubMed.
- D. Lelièvre, V. P. Terrier, A. F. Delmas and V. Aucagne, Org. Lett., 2016, 18, 920–923 CrossRef PubMed.
- J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- S. Laps, G. Satish and A. Brik, Chem. Soc. Rev., 2021, 50, 2367–2387 RSC.
- E. Fischer and E. Fourneau, Ber. Dtsch. Chem. Ges., 1901, 34, 2868–2877 CrossRef.
- J. C. Sheehan and G. P. Hess, J. Am. Chem. Soc., 1955, 77, 1067–1068 CrossRef CAS.
- L. A. Carpino, J. Am. Chem. Soc., 1957, 79, 4427–4431 CrossRef CAS.
- F. C. McKay and N. F. Albertson, J. Am. Chem. Soc., 1957, 79, 4686–4690 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- L. A. Carpino and G. Y. Han, J. Am. Chem. Soc., 1970, 92, 5748–5749 CrossRef CAS.
- D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 CrossRef CAS.
- M. Ahangarpour, I. Kavianinia, P. W. R. Harris and M. A. Brimble, Chem. Soc. Rev., 2021, 50, 898–944 RSC.
- P. R. Hansen and A. Oddo, in Fmoc Solid-Phase Peptide Synthesis, ed. G. Houen, Springer New York, New York, NY, 2015, pp. 33–50 Search PubMed.
- V. Agouridas, O. El Mahdi, M. Cargoët and O. Melnyk, Bioorg. Med. Chem., 2017, 25, 4938–4945 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed.
- D. Bang and S. B. H. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS PubMed.
- T. C. Evans Jr., J. Benner and M.-Q. Xu, Protein Sci., 1998, 7, 2256–2264 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, LP6705–LP6710 CrossRef PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- T. Kawakami and S. Aimoto, Chem. Lett., 2006, 76–77 Search PubMed.
- M. Yanase, K. Nakatsu, C. J. Cardos, Y. Konda, G. Hayashi and A. Okamoto, Chem. Sci., 2019, 10, 5967–5975 RSC.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- G.-M. Fang, J.-X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- J.-S. Zheng, S. Tang, Y.-K. Qi, Z.-P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- Y. Qiao, G. Yu, K. C. Kratch, X. A. Wang, W. W. Wang, S. Z. Leeuwon, S. Xu, J. S. Morse and W. R. Liu, J. Am. Chem. Soc., 2020, 142, 7047–7054 CrossRef CAS PubMed.
- H. Hojo and S. Aimoto, Bull. Chem. Soc. Jpn., 1991, 64, 111–117 CrossRef CAS.
- H. Hojo, Y. Murasawa, H. Katayama, T. Ohira, Y. Nakahara and Y. Nakahara, Org. Biomol. Chem., 2008, 6, 1808–1813 RSC.
- H. Katayama, H. Hojo, T. Ohira and Y. Nakahara, Tetrahedron Lett., 2008, 49, 5492–5494 CrossRef CAS.
- H. Hojo, Org. Biomol. Chem., 2016, 14, 6368–6374 RSC.
- W. Hou, X. Zhang and C.-F. Liu, Trans. Tianjin Univ., 2017, 23, 401–419 CrossRef.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem., 2018, 2, 122 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- G. Barany and R. B. Merrifield, J. Am. Chem. Soc., 1977, 99, 7363–7365 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, LP14846–LP14851 CrossRef PubMed.
- S. S. Nguyen and J. A. Prescher, Nat. Rev. Chem., 2020, 4, 476–489 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- P. Agarwal, B. J. Beahm, P. Shieh and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 11504–11510 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- R. Brabham and M. A. Fascione, ChemBioChem, 2017, 18, 1973–1983 CrossRef CAS PubMed.
- C. H. Kim, M. Kang, H. J. Kim, A. Chatterjee and P. G. Schultz, Angew. Chem., Int. Ed., 2012, 51, 7246–7249 CrossRef CAS PubMed.
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728–2731 CrossRef CAS PubMed.
- V. du Vigneaud, L. F. Audrieth and H. S. Loring, J. Am. Chem. Soc., 1930, 52, 4500–4504 CrossRef CAS.
- D. Andreu, F. Albericio, N. A. Solé, M. C. Munson and G. Barany, in Methods in Molecular Biology, Peptide Synthesis Protocols, ed. M. W. Pennington and B. M. Dunn, Human Press Inc., Totowa, NJ, 1994, vol. 35, pp. 91–169 Search PubMed.
- H. Yajima, N. Fujii, S. Funakoshi, T. Watanabe, E. Murayama and A. Otaka, Tetrahedron, 1988, 44, 805–819 CrossRef CAS.
- L. Zervas and I. Photaki, J. Am. Chem. Soc., 1962, 84, 3887–3897 CrossRef CAS.
- K. Barlos, D. Gatos, O. Hatzi, N. Koch and S. Koutsogianni, Int. J. Pept. Protein Res., 1996, 47, 148–153 CrossRef CAS PubMed.
- K. Akaji, K. Fujino, T. Tatsumi and Y. Kiso, J. Am. Chem. Soc., 1993, 115, 11384–11392 CrossRef CAS.
- D. Kobayashi, N. Naruse, M. Denda, A. Shigenaga and A. Otaka, Org. Biomol. Chem., 2020, 18, 8638–8645 RSC.
- M. Mochizuki, S. Tsuda, K. Tanimura and Y. Nishiuchi, Org. Lett., 2015, 17, 2202–2205 CrossRef CAS PubMed.
- M. Góngora-Benítez, L. Mendive-Tapia, I. Ramos-Tomillero, A. C. Breman, J. Tulla-Puche and F. Albericio, Org. Lett., 2012, 14, 5472–5475 CrossRef PubMed.
- G. F. Holland and L. A. Cohen, J. Am. Chem. Soc., 1958, 80, 3765–3769 CrossRef CAS.
- I. Ramos-Tomillero, H. Rodriguez and F. Albericio, Org. Lett., 2015, 17, 1680–1683 CrossRef CAS PubMed.
- J. J. Pastuszak and A. Chimiak, J. Org. Chem., 1981, 46, 1868–1873 CrossRef CAS.
- S. Sakakibara, Y. Shimonishi, Y. Kishida, M. Okada and H. Sugihara, Bull. Chem. Soc. Jpn., 1967, 40, 2164–2167 CrossRef CAS PubMed.
- A. L. Schroll, R. J. Hondal and S. Flemer, J. Pept. Sci., 2012, 18, 1–9 CrossRef CAS PubMed.
- O. Nishimura, C. Kitada and M. Fujino, Chem. Pharm. Bull., 1978, 26, 1576–1585 CrossRef CAS.
- K. Akaji, T. Tatsumi, M. Yoshida, T. Kimura, Y. Fujiwara and Y. Kiso, J. Chem. Soc., Chem. Commun., 1991, 167–168 RSC.
- N. Fujii, A. Otaka, S. Funakoshi, K. Bessho, T. Watanabe, K. Akaji and H. Yajima, Chem. Pharm. Bull., 1987, 35, 2339–2347 CrossRef CAS PubMed.
- F. Wu, J. P. Mayer, V. M. Gelfanov, F. Liu and R. D. Dimarchi, J. Org. Chem., 2017, 82, 3506–3512 CrossRef CAS PubMed.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 3154 CrossRef PubMed.
- E. J. Ste Marie and R. J. Hondal, J. Pept. Sci., 2018, 24, e3130 CrossRef PubMed.
- N. Fujii, A. Otaka, T. Watanabe, A. Okamachi, H. Tamamura, H. Yajima, Y. Inagaki, M. Nomizu and K. Asano, J. Chem. Soc., Chem. Commun., 1989, 283 RSC.
- A. Cuthbertson and B. Indrevoll, Org. Lett., 2003, 5, 2955–2957 CrossRef CAS PubMed.
- S. Akabori, S. Sakakibara, Y. Shimonishi and Y. Nobuhara, Bull. Chem. Soc. Jpn., 1964, 37, 433–434 CrossRef CAS.
- K. M. Harris, S. Flemer and R. J. Hondal, J. Pept. Sci., 2007, 13, 81–93 CrossRef CAS PubMed.
- B. W. Erickson and R. B. Merrifield, J. Am. Chem. Soc., 1973, 95, 3750–3756 CrossRef CAS PubMed.
- D. Yamashiro, R. L. Noble and C. H. Li, J. Org. Chem., 1973, 38, 3561–3565 CrossRef CAS PubMed.
- N. Fujii, A. Otaka, S. Funakoshi, H. Yajima, O. Nishimura and M. Fujino, Chem. Pharm. Bull., 1986, 34, 869–872 CrossRef CAS.
- A. Otaka, H. Morimoto, N. Fujii, T. Koide, S. Funakoshi and H. Yajima, Chem. Pharm. Bull., 1989, 37, 526–528 CrossRef CAS.
- M. C. Munson, C. Garcia-Echeverria, F. Albericio and G. Barany, J. Org. Chem., 1992, 57, 3013–3018 CrossRef CAS.
- M. C. Munson, C. Garcia-Echeverria, F. Albericio and G. Barany, in Peptides Chemistry and Biology (Proceedings of the Twelfth American Peptide Symposium), ed. J. A. Smith and J. E. Rivier, Leiden, 1992, pp. 605–606 Search PubMed.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- M. S. Y. Wong and K. A. Jolliffe, Aust. J. Chem., 2010, 63, 797–801 CrossRef CAS.
- T. M. Postma and F. Albericio, Org. Lett., 2014, 16, 1772–1775 CrossRef CAS PubMed.
- S. Tsuda, S. Masuda and T. Yoshiya, J. Org. Chem., 2020, 85, 1674–1679 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Meli, J. Tulla-Puche and F. Albericio, J. Med. Chem., 2013, 56, 9780–9788 CrossRef CAS PubMed.
- Y. Han and G. Barany, J. Org. Chem., 1997, 62, 3841–3848 CrossRef CAS.
- O. Garcia, J. M. Bofill, E. Nicolas and F. Albericio, Eur. J. Org. Chem., 2010, 3631–3640 CrossRef CAS.
- H. Hibino and Y. Nishiuchi, Org. Lett., 2012, 14, 1926–1929 CrossRef CAS PubMed.
- I. Ramos-Tomillero, L. Mendive-Tapia, M. Góngora-Benítez, E. Nicolás, J. Tulla-Puche and F. Albericio, Molecules, 2013, 18, 5155–5162 CrossRef CAS PubMed.
- H. Hibino, Y. Miki and Y. Nishiuchi, J. Pept. Sci., 2014, 20, 30–35 CrossRef CAS PubMed.
- Y.-K. Qi, S. Tang, Y.-C. Huang, M. Pan, J.-S. Zheng and L. Liu, Org. Biomol. Chem., 2016, 14, 4194–4198 RSC.
- D. F. Veber, J. D. Milkowski, R. G. Denkewalter and R. Hirschmann, Tetrahedron Lett., 1968, 9, 3057–3058 CrossRef.
- S. Laps, H. Sun, G. Kamnesky and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 5729–5733 CrossRef CAS PubMed.
- D. F. Veber, J. D. Milkowski, S. L. Varga, R. G. Denkewalter and R. Hirschmann, J. Am. Chem. Soc., 1972, 94, 5456–5461 CrossRef CAS PubMed.
- M. Muttenthaler, Y. G. Ramos, D. Feytens, A. D. de Araujo and P. F. Alewood, Biopolymers, 2010, 94, 423–432 CrossRef CAS PubMed.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
- N. Kamo, G. Hayashi and A. Okamoto, Org. Lett., 2019, 21, 8378–8382 CrossRef CAS PubMed.
- M. R. Levengood, X. Zhang, J. H. Hunter, K. K. Emmerton, J. B. Miyamoto, T. S. Lewis and P. D. Senter, Angew. Chem., Int. Ed., 2017, 56, 733–737 CrossRef CAS PubMed.
- J. Pless, Helv. Chim. Acta, 1976, 59, 499–512 CrossRef CAS.
- P. K. Chakravarty and R. K. Olsen, J. Org. Chem., 1978, 43, 1270–1271 CrossRef CAS.
- Y. Kiso, M. Yoshida, Y. Fujiwara, T. Koimura, M. Shimokura and K. Akaji, Chem. Pharm. Bull., 1990, 38, 673–675 CrossRef CAS.
- M. Ueki and K. Shinozaki, Bull. Chem. Soc. Jpn., 1983, 56, 1187–1191 CrossRef CAS.
- M. Ueki, T. Ikeo, K. Hokari, K. Nakamura, A. Saeki and H. Komatsu, Bull. Chem. Soc. Jpn., 1999, 72, 829–838 CrossRef CAS.
- Y. Kiso, M. Yoshida, T. Kimura, Y. Fujiwara and M. Shimokura, Tetrahedron Lett., 1989, 30, 1979–1982 CrossRef CAS.
- M. Yoshida, T. Tatsumi, Y. Fujiwara, S. Iinuma, T. Kimura, K. Akaji and Y. Kiso, Chem. Pharm. Bull., 1990, 38, 1551–1557 CrossRef CAS PubMed.
- M. Bodanszky and M. A. Bednarek, Int. J. Pept. Protein Res., 1982, 20, 434–437 CrossRef CAS PubMed.
- M. Ruiz-Gayo, F. Albericio, E. Pedroso and E. Giralt, J. Chem. Soc., Chem. Commun., 1986, 1501 RSC.
- M. Royo, C. García-Echeverría, E. Giralt, R. Eritja and F. Albericio, Tetrahedron Lett., 1992, 33, 2391–2394 CrossRef CAS.
- C. W. West, M. A. Estiarte and D. H. Rich, Org. Lett., 2001, 3, 1205–1208 CrossRef CAS PubMed.
- M. Royo, J. Alsina, E. Giralt, U. Slomcyznska and F. Albericio, J. Chem. Soc., Perkin Trans. 1, 1995, 1095–1102 RSC.
- M. Góngora-Benítez, A. Basso, T. Bruckdorfer, M. Royo, J. Tulla-Puche and F. Albericio, Chem. – Eur. J., 2012, 18, 16166–16176 CrossRef PubMed.
- F. Shen, Z. P. Zhang, J. Bin Li, Y. Lin and L. Liu, Org. Lett., 2011, 13, 568–571 CrossRef CAS PubMed.
- A. Loffet and H. X. Zhang, Int. J. Pept. Protein Res., 1993, 42, 346–351 CrossRef CAS PubMed.
- A. M. Kimbonguila, A. Merzouk, F. Guibé and A. Loffet, Tetrahedron Lett., 1994, 35, 9035–9038 CrossRef CAS.
- T. D. Kondasinghe, H. Y. Saraha, S. B. Odeesho and J. L. Stockdill, Org. Biomol. Chem., 2017, 15, 2914–2918 RSC.
- P. Gomez-Martinez, A. Malanda Kimbonguila and F. Guibé, Tetrahedron, 1999, 55, 6945–6960 CrossRef CAS.
- P. Gomez-Martinez, F. Guibé and F. Albericio, Lett. Pept. Sci., 2000, 7, 187–194 CAS.
- M. P. Exner, T. Kuenzl, T. M. T. To, Z. Ouyang, S. Schwagerus, M. G. Hoesl, C. P. R. Hackenberger, M. C. Lensen, S. Panke and N. Budisa, ChemBioChem, 2017, 18, 85–90 CrossRef CAS PubMed.
- J. Liu, R. Cheng, H. Wu, S. Li, P. G. Wang, W. F. DeGrado, S. Rozovsky and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 12702–12706 CrossRef CAS PubMed.
- G. B. Vamisetti, G. Satish, P. Sulkshane, G. Mann, M. H. Glickman and A. Brik, J. Am. Chem. Soc., 2020, 142, 19558–19569 CrossRef CAS PubMed.
- S. Rather and H. T. Clarke, J. Am. Chem. Soc., 1937, 59, 200–206 CrossRef.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- N. Naruse, D. Kobayashi, K. Ohkawachi, A. Shigenaga and A. Otaka, J. Org. Chem., 2020, 85, 1425–1433 CrossRef CAS PubMed.
- H. Katayama and S. Morisue, Tetrahedron, 2017, 73, 3541–3547 CrossRef CAS.
- C. T. Pool, J. G. Boyd and J. P. Tam, J. Pept. Res., 2004, 63, 223–234 CrossRef CAS PubMed.
- J. A. Karas, D. B. Scanlon, B. E. Forbes, I. Vetter, R. J. Lewis, J. Gardiner, F. Separovic, J. D. Wade and M. A. Hossain, Chem. – Eur. J., 2014, 20, 9549–9552 CrossRef CAS PubMed.
- A. B. Smith, S. N. Savinov, U. V. Manjappara and I. M. Chaiken, Org. Lett., 2002, 4, 4041–4044 CrossRef CAS PubMed.
- N. Kotzur, B. Briand, M. Beyermann and V. Hagen, J. Am. Chem. Soc., 2009, 131, 16927–16931 CrossRef CAS PubMed.
- M. M. Mahmoodi, D. Abate-Pella, T. J. Pundsack, C. C. Palsuledesai, P. C. Goff, D. A. Blank and M. D. Distefano, J. Am. Chem. Soc., 2016, 138, 5848–5859 CrossRef CAS PubMed.
- M. M. Mahmoodi, S. A. Fisher, R. Y. Tam, P. C. Goff, R. B. Anderson, J. E. Wissinger, D. A. Blank, M. S. Shoichet and M. D. Distefano, Org. Biomol. Chem., 2016, 14, 8289–8300 RSC.
- T. K. Bader, F. Xu, M. H. Hodny, D. A. Blank and M. D. Distefano, J. Org. Chem., 2020, 85, 1614–1625 CrossRef CAS PubMed.
- S. B. Gunnoo, A. Iyer, W. Vannecke, K. W. Decoene, T. Hebbrecht, J. Gettemans, M. Laga, S. Loverix, I. Lasters and A. Madder, Chem. Commun., 2018, 54, 11929–11932 RSC.
- A. L. Schroll and G. Barany, J. Org. Chem., 1989, 54, 244–247 CrossRef CAS.
- W. Chan and P. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, Oxford, 2000, p. 87 Search PubMed.
- A. Gosden, D. Stevenson and G. T. Young, J. Chem. Soc., Chem. Commun., 1972, 1123 RSC.
- R. B. Merrifield, The Chemistry of Polypeptides, Springer US, Boston, MA, 1973 Search PubMed.
- I. Maugras, J. Gosteli, E. Rapp and R. Nyfeler, Int. J. Pept. Protein Res., 1995, 45, 152–156 CrossRef CAS PubMed.
- C. Rentier, K. Fukumoto, A. Taguchi and Y. Hayashi, J. Pept. Sci., 2017, 23, 496–504 CrossRef CAS PubMed.
- R. Matsueda, T. Kimura, E. T. Kaiser and G. R. Matsueda, Chem. Lett., 1981, 737–740 CrossRef CAS.
- R. J. Ridge, G. R. Matsueda, E. Haber and R. Matsueda, Int. J. Pept. Protein Res., 1982, 19, 490–498 CrossRef CAS PubMed.
- F. Albericio, D. Andreu, E. Giralt, C. Navalpotro, E. Pedroso, B. Ponsati and M. Rue-Gayo, Int. J. Pept. Protein Res., 1989, 34, 124–128 CrossRef CAS PubMed.
- F. Rabanal, W. F. DeGrado and P. L. Dutton, Tetrahedron Lett., 1996, 37, 1347–1350 CrossRef CAS.
- R. Eritja, J. P. Ziehler-Martin, P. A. Walker, T. D. Lee, K. Legesse, F. Albericio and B. E. Kaplan, Tetrahedron, 1987, 43, 2675–2680 CrossRef CAS.
- H. Katayama, Y. Nakahara and H. Hojo, Org. Biomol. Chem., 2011, 9, 4653 RSC.
- H. Katayama and H. Hojo, Org. Biomol. Chem., 2013, 11, 4405–4413 RSC.
- M. Matveenko, S. Hackl and C. F. W. Becker, ChemistryOpen, 2018, 7, 106–110 CrossRef CAS PubMed.
- T. M. Postma, M. Giraud and F. Albericio, Org. Lett., 2012, 14, 5468–5471 CrossRef CAS PubMed.
- S. N. Mthembu, A. Sharma, F. Albericio and B. G. de la Torre, Org. Lett., 2019, 21, 10111–10114 CrossRef CAS PubMed.
- A. Chakraborty, A. Sharma, F. Albericio and B. G. de la Torre, Org. Lett., 2020, 22, 9644–9647 CrossRef CAS PubMed.
- Z. Dekan, M. Mobli, M. W. Pennington, E. Fung, E. Nemeth and P. F. Alewood, Angew. Chem., Int. Ed., 2014, 53, 2931–2934 CrossRef CAS PubMed.
- R. Deslauriers, R. Walter and I. C. P. Smith, Biochem. Biophys. Res. Commun., 1972, 48, 854–859 CrossRef CAS PubMed.
- G. Flouret, W. Brieher, K. Mahan and L. Wilson, J. Med. Chem., 1991, 34, 642–646 CrossRef CAS PubMed.
- B. Banerji, M. Chatterjee, C. Prodhan and K. Chaudhuri, RSC Adv., 2016, 6, 112667–112676 RSC.
- M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed.
- B. Kamber, A. Hartmann, K. Eisler, B. Riniker, H. Rink, P. Sieber and W. Rittel, Helv. Chim. Acta, 1980, 63, 899–915 CrossRef CAS.
- D. A. Pearson, M. Blanchette, M. Lou Baker and C. A. Guindon, Tetrahedron Lett., 1989, 30, 2739–2742 CrossRef CAS.
- E. E. Büllesbach and C. Schwabe, J. Biol. Chem., 1991, 266, 10754–10761 CrossRef.
- J. Ying, X. Gu, M. Cai, M. Dedek, J. Vagner, D. B. Trivedi and V. J. Hruby, J. Med. Chem., 2006, 49, 6888–6896 CrossRef CAS PubMed.
- C. Fagundez, D. Sellanes and G. Serra, ACS Comb. Sci., 2018, 20, 212–219 CrossRef CAS PubMed.
- G. P. Schwartz and P. G. Katsoyannis, J. Chem. Soc., Perkin Trans, 1973, 1, 2894 RSC.
- R. G. Hiskey and J. B. Adams, J. Org. Chem., 1966, 31, 2178–2183 CrossRef CAS.
- S. A. Khan and K. M. Sivanandaiah, Int. J. Pept. Protein Res., 1978, 12, 164–169 CrossRef CAS PubMed.
- A. Sharma, I. Ramos-Tomillero, A. El-Faham, E. Nicolas, H. Rodriguez, B. G. de la Torre and F. Albericio, ChemistryOpen, 2017, 6, 168–177 CrossRef CAS PubMed.
- A. Chimiak, in Proceedings of 5th European Peptide Symposium, 1962, p. 37 Search PubMed.
- X. Zhang, Y. Yang, M. Zhao, L. Liu, M. Zheng, Y. Wang, J. Wu and S. Peng, Eur. J. Med. Chem., 2011, 46, 3410–3419 CrossRef CAS PubMed.
- N. Zheng, P. Karra, M. A. Vandenberg, J. H. Kim, M. J. Webber, W. L. Holland and D. H. C. Chou, J. Med. Chem., 2019, 62, 11437–11443 CrossRef CAS PubMed.
- J. Tytgat, K. G. Chandy, M. L. Garcia, G. A. Gutman, M.-F. Martin-Eauclaire, J. J. van der Walt and L. D. Possani, Trends Pharmacol. Sci., 1999, 20, 444–447 CrossRef CAS PubMed.
- H. Juichi, R. Ando, T. Ishido, M. Miyashita, Y. Nakagawa and H. Miyagawa, J. Pept. Sci., 2018, 24, e3133 CrossRef PubMed.
- H. Yajima, S. Funakoshi, N. Fujii, K. Akaji and H. Irie, Chem. Pharm. Bull., 1979, 27, 1060–1061 CrossRef CAS.
- A. R. Urmey and N. J. Zondlo, Pept. Sci., 2020, 112, e24137 CAS.
- N. Fujii, A. Otaka, T. Watanabe, H. Arai, S. Funakoshi, K. Bessho and H. Yajima, J. Chem. Soc., Chem. Commun., 1987, 1676–1678 RSC.
- F. Wu, J. P. Mayer, A. N. Zaykov, F. Zhang, F. Liu and R. D. DiMarchi, Chem. – Eur. J., 2016, 22, 9777–9783 CrossRef CAS PubMed.
- N. Fujii, A. Otaka, S. Funakoshi, T. Watanabe, H. Arai, K. Bessho and H. Yajima, J. Protein Chem., 1988, 7, 151–156 CrossRef CAS PubMed.
- N. Fujii, T. Watanabe, T. Aotake, A. Otaka, I. Yamamoto, J. Konishe and H. Yajima, Chem. Pharm. Bull., 1988, 36, 3304–3311 CrossRef CAS PubMed.
- V. J. Hruby, D. A. Upson and N. S. Agarwal, J. Org. Chem., 1977, 42, 3552–3556 CrossRef CAS.
- J. J. Knittel, T. K. Sawyer, V. J. Hruby and M. E. Hadley, J. Med. Chem., 1983, 26, 125–129 CrossRef CAS PubMed.
- S. Tsuda, T. Yoshiya, M. Mochizuki and Y. Nishiuchi, Org. Lett., 2015, 17, 1806–1809 CrossRef CAS PubMed.
- N. Fujii, H. Yajima, A. Otaka, S. Funakoshi, M. Nomizu, K. Akaji, I. Yamamoto, K. Torizuka, K. Kitagawa, T. Akita, K. Ando, T. Kawamoto, Y. Shimonishi and T. Takao, J. Chem. Soc., Chem. Commun., 1985, 602–603 RSC.
- S. Futaki, M. Aoki, T. Ishikawa, F. Kondo, T. Asahara, M. Niwa, Y. Nakaya, T. Yagami and K. Kitagawa, Bioorg. Med. Chem., 1999, 7, 187–192 CrossRef CAS PubMed.
- T. Brown, J. H. Jones and J. D. Richards, J. Chem. Soc., Perkin Trans. 1, 1982, 1553–1561 RSC.
- T. Haack and M. Mutter, Tetrahedron Lett., 1992, 33, 1589–1592 CrossRef CAS.
- A. Kreżel, J. Wójcik, M. Maciejczyk and W. Bal, Inorg. Chem., 2011, 50, 72–85 CrossRef PubMed.
- A. Krężel and W. Bal, Org. Biomol. Chem., 2003, 1, 3885–3890 RSC.
- V. Martos, P. Castreño, M. Royo, F. Albericio and J. de Mendoza, J. Comb. Chem., 2009, 11, 410–421 CrossRef CAS PubMed.
- J. D. Smith, L. N. Cardwell, D. Porciani, A. Nolla, B. T. Cornelison, M. C. Schulte, F. Gallazzi, D. H. Burke, M. A. Daniels and B. D. Ulery, Mol. Syst. Des. Eng., 2020, 5, 269–283 RSC.
- A. Aletras, K. Barlos, D. Gatos, S. Koutsogianni and P. Mamos, Int. J. Pept. Protein Res., 1995, 45, 488–496 CrossRef CAS PubMed.
- A. S. Galanis, F. Albericio and M. Grøtli, Pept. Sci., 2009, 92, 23–34 CrossRef CAS PubMed.
- A. Graziadio, M. Zanda, S. Frau, I. N. Fleming, M. Musolino, S. Dall’Angelo, M. Baldassarre and M. Piras, Bioconjugate Chem., 2016, 27, 1332–1340 CrossRef CAS PubMed.
- T. D. Kondasinghe, H. Y. Saraha, S. T. Jackowski and J. L. Stockdill, Tetrahedron Lett., 2019, 60, 23–28 CrossRef CAS PubMed.
- D.-L. Huang, C. Montigny, Y. Zheng, V. Beswick, Y. Li, X.-X. Cao, T. Barbot, C. Jaxel, J. Liang, M. Xue, C.-L. Tian, N. Jamin and J.-S. Zheng, Angew. Chem., Int. Ed., 2020, 59, 5178–5184 CrossRef CAS PubMed.
- D.-L. Huang, Y. Li, J. Liang, L. Yu, M. Xue, X.-X. Cao, B. Xiao, C.-L. Tian, L. Liu and J.-S. Zheng, J. Am. Chem. Soc., 2020, 142, 8790–8799 CrossRef PubMed.
- J. Halstrøm, K. Brunfeldt and K. Kovács, Acta Chem. Scand., 1979, 33b, 685–689 CrossRef.
- J. Tulla-Puche and G. Barany, J. Org. Chem., 2004, 69, 4101–4107 CrossRef CAS PubMed.
- S. Liu, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2012, 51, 993–999 CrossRef CAS PubMed.
- H. Lamthanh, C. Roumestand, C. Deprun and A. Ménez, Int. J. Pept. Protein Res., 1993, 41, 85–95 CrossRef CAS PubMed.
- M. A. Y. Engebretsen, E. Agner, J. Sandosham and P. M. Fischer, J. Pept. Res., 1997, 49, 341–346 CrossRef CAS PubMed.
- Y. Wang, L. Han, N. Yuan, H. Wang, H. Li, J. Liu, H. Chen, Q. Zhang and S. Dong, Chem. Sci., 2018, 9, 1940–1946 RSC.
- H. Chen, Y. Xiao, N. Yuan, J. Weng, P. Gao, L. Breindel, A. Shekhtman and Q. Zhang, Chem. Sci., 2018, 9, 1982–1988 RSC.
- S. Laps, F. Atamleh, G. Kamnesky, H. Sun and A. Brik, Nat. Commun., 2021, 12, 870 CrossRef CAS PubMed.
- N. Fujii, A. Otaka, S. Funakoshi, K. Bessho and H. Yajima, J. Chem. Soc., Chem. Commun., 1987, 163–164 RSC.
- M. Noda and M. Kiffe, J. Pept. Res., 1997, 50, 329–335 CrossRef CAS PubMed.
- A. K. Ivanov, E. É. Grigor’eva, A. A. Antonov and I. A. Donetskii, Chem. Nat. Compd., 1993, 29, 97–103 CrossRef.
- M. Ueki, T. Inazu and S. Ikeda, Bull. Chem. Soc. Jpn., 1979, 52, 2424–2427 CrossRef CAS.
- K. Akaji, T. Tatsumi, M. Yoshida, T. Kimura, Y. Fujiwara and Y. Kiso, J. Am. Chem. Soc., 1992, 114, 4137–4143 CrossRef CAS.
- G. Greiner and P. Hermann, in Peptides 1990, Proceedings of the Twenty-First European Peptide Symposium, ed. E. Giralt and D. Andreu, ESCOM Science Publishers B.V., Leiden, 1991, pp. 227–278 Search PubMed.
- S. K. Maity, G. Mann, M. Jbara, S. Laps, G. Kamnesky and A. Brik, Org. Lett., 2016, 18, 3026–3029 CrossRef CAS PubMed.
- F. Liu, Q. Liu and A. R. Mezo, Org. Lett., 2014, 16, 3126–3129 CrossRef CAS PubMed.
- X. Yang, V. Gelfanov, F. Liu and R. DiMarchi, Org. Lett., 2016, 18, 5516–5519 CrossRef CAS PubMed.
- O. Dangles, F. Guibe, G. Balavoine, S. Lavielle and A. Marquet, J. Org. Chem., 1987, 52, 4984–4993 CrossRef CAS.
- F. Guibé, Tetrahedron, 1998, 54, 2967–3042 CrossRef.
- A. Malanda Kimbonguila, A. Merzouk, F. Guibé and A. Loffet, Tetrahedron, 1999, 55, 6931–6944 CrossRef.
- J. Borlinghaus, F. Albrecht, M. C. H. Gruhlke, I. D. Nwachukwu and A. J. Slusarenko, Molecules, 2014, 19, 12591–12618 CrossRef PubMed.
- N. D. J. Yates, M. A. Fascione and A. Parkin, Chem. – Eur. J., 2018, 24, 12164–12182 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- K. Renault, J. W. Fredy, P.-Y. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS PubMed.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS PubMed.
- V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2007, 46, 1667–1670 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644–10655 CrossRef CAS PubMed.
- G. Mann, G. Satish, R. Meledin, G. B. Vamisetti and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 13540–13549 CrossRef CAS PubMed.
- R. L. Brabham, R. J. Spears, J. Walton, S. Tyagi, E. A. Lemke and M. A. Fascione, Chem. Commun., 2018, 54, 1501–1504 RSC.
- X. Bi, K. K. Pasunooti, J. Lescar and C.-F. Liu, Bioconjugate Chem., 2017, 28, 325–329 CrossRef CAS PubMed.
- R. J. Spears, R. L. Brabham, D. Budhadev, T. Keenan, S. McKenna, J. Walton, J. A. Brannigan, A. M. Brzozowski, A. J. Wilkinson, M. Plevin and M. A. Fascione, Chem. Sci., 2018, 9, 5585–5593 RSC.
- K. Ohkawachi, D. Kobayashi, K. Morimoto, A. Shigenaga, M. Denda, K. Yamatsugu, M. Kanai and A. Otaka, Org. Lett., 2020, 22, 5289–5293 CrossRef CAS PubMed.
- H. Katayama and K. Nagata, J. Pept. Sci., 2021, 27, e3290 CrossRef CAS PubMed.
- G. Prota and E. Ponsiglione, Tetrahedron, 1973, 29, 4271–4274 CrossRef CAS.
- G. J. Hoogerheidem, in Peptides 1980 (Proceedings of the sixteenth European peptide symposium), ed. K. Brunfeldt, Copenhagen, 1981 Search PubMed.
- J. O. Campeciño, L. W. Dudycz, D. Tumelty, V. Berg, D. E. Cabelli and M. J. Maroney, J. Am. Chem. Soc., 2015, 137, 9044–9052 CrossRef PubMed.
- D. P. Nguyen, M. Mahesh, S. J. Elsässer, S. M. Hancock, C. Uttamapinant and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 2240–2243 CrossRef CAS PubMed.
- N. Wu, A. Deiters, T. A. Cropp, D. King and P. G. Schultz, J. Am. Chem. Soc., 2004, 126, 14306–14307 CrossRef CAS PubMed.
- K. D. Philipson, J. P. Gallivan, G. S. Brandt, D. A. Dougherty and H. A. Lester, Am. J. Physiol. – Cell Physiol., 2001, 281, C195–C206 CrossRef CAS PubMed.
- G. Marriott and M. Heidecker, Biochemistry, 1996, 35, 3170–3174 CrossRef CAS PubMed.
- A. G. Russell, M.-E. Ragoussi, R. Ramalho, C. W. Wharton, D. Carteau, D. M. Bassani and J. S. Snaith, J. Org. Chem., 2010, 75, 4648–4651 CrossRef CAS PubMed.
- S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu and D. Solas, Science, 1991, 251, LP767–LP773 CrossRef PubMed.
- A. Patchornik, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- R. Miyajima, Y. Tsuda, T. Inokuma, A. Shigenaga, M. Imanishi, S. Futaki and A. Otaka, Biopolymers, 2016, 531–546 CrossRef CAS PubMed.
- R. Rakauskaitė, G. Urbanavičiūtė, A. Rukšėnaitė, Z. Liutkevičiūtė, R. Juškėnas, V. Masevičius and S. Klimašauskas, Chem. Commun., 2015, 51, 8245–8248 RSC.
- J. Pan and K. S. Carroll, Org. Lett., 2015, 17, 6014–6017 CrossRef CAS PubMed.
- J. H. Wosnick and M. S. Shoichet, Chem. Mater., 2008, 20, 55–60 CrossRef CAS.
- C. Berse, R. Boucher and L. Piché, J. Org. Chem., 1957, 22, 805–808 CrossRef CAS.
- M. D. Bachi and K. J. Ross-Petersen, J. Org. Chem., 1972, 37, 3550–3551 CrossRef CAS.
- S. J. Brois, J. F. Pilot and H. W. Barnum, J. Am. Chem. Soc., 1970, 92, 7629–7631 CrossRef CAS.
- L. Chen, I. Zoulíková, J. Slaninová and G. Barany, J. Med. Chem., 1997, 40, 864–876 CrossRef CAS PubMed.
- S. J. Mear and T. F. Jamison, J. Org. Chem., 2019, 84, 15001–15007 CrossRef CAS PubMed.
- R. Matsueda and K. Aiba, Chem. Lett., 1978, 951–952 CrossRef CAS.
- A. Taguchi, K. Fukumoto, Y. Asahina, A. Kajiyama, S. Shimura, K. Hamada, K. Takayama, F. Yakushiji, H. Hojo and Y. Hayashi, Org. Biomol. Chem., 2015, 13, 3186–3189 RSC.
- Y. Cui, A. Taguchi, K. Kobayashi, H. Shida, K. Takayama, A. Taniguchi and Y. Hayashi, Org. Biomol. Chem., 2020, 18, 7094–7097 RSC.
- E. J. Ste Marie, E. L. Ruggles and R. J. Hondal, J. Pept. Sci., 2016, 22, 571–576 CrossRef CAS PubMed.
- U. Weber and P. Hartter, Hoppe. Seylers. Z. Physiol. Chem, 1970, 351, 1384–1388 CrossRef CAS PubMed.
- P. S. Herradura, K. A. Pendola and R. K. Guy, Org. Lett., 2000, 2, 2019–2022 CrossRef CAS PubMed.
- S. S. More and R. Vince, Bioorg. Med. Chem. Lett., 2006, 16, 6039–6042 CrossRef CAS PubMed.
- L. Moroder, M. Gemeiner, W. Goehring, E. Jaeger, P. Thamm and E. Wünsch, Biopolymers, 1981, 20, 17–37 CrossRef CAS PubMed.
- S. Wang, D. Gao, K. Li, S. Ye, Q. Liu, Y. Peng, G. Lv, L. Qiu and J. Lin, Org. Biomol. Chem., 2020, 18, 3512–3521 RSC.
- B. Roubinet, A. Chevalier, P.-Y. Renard and A. Romieu, Eur. J. Org. Chem., 2015, 166–182 CrossRef CAS.
- N. S. Khan, B. A. Riggle, G. K. Seward, Y. Bai and I. J. Dmochowski, Bioconjugate Chem., 2015, 26, 101–109 CrossRef CAS PubMed.
- C. Zhang, P. Dai, A. M. Spokoyny and B. L. Pentelute, Org. Lett., 2014, 16, 3652–3655 CrossRef CAS PubMed.
- J. A. Brailsford, J. L. Stockdill, A. J. Axelrod, M. T. Peterson, P. A. Vadola, E. V. Johnston and S. J. Danishefsky, Tetrahedron, 2018, 74, 1951–1956 CrossRef CAS PubMed.
- B. J. Umlauf, K. A. Mix, V. A. Grosskopf, R. T. Raines and E. V. Shusta, Bioconjugate Chem., 2018, 29, 1605–1613 CrossRef CAS PubMed.
- X. Zhao, G. Lv, K. Li, Y. Peng, Q. Liu, L. Qiu and J. Lin, Nucl. Med. Biol., 2020, 82–83, 72–79 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche, M. Paradís-Bas, O. Werbitzky, M. Giraud and F. Albericio, Biopolymers, 2011, 96, 69–80 CrossRef PubMed.
- G. Tang, T. Ji, A. F. Hu and Y. F. Zhao, Synlett, 2008, 1907–1909 CrossRef CAS.
- T. Kawakami, R. Yoshikawa, Y. Fujiyoshi, Y. Mishima, H. Hojo, S. Tajima and I. Suetake, J. Biochem., 2015, 158, 403–411 CrossRef CAS PubMed.
- P. Hartter, Biol. Chem., 1976, 357, 1683–1694 CrossRef CAS PubMed.
- T. M. Postma and F. Albericio, Org. Lett., 2013, 15, 616–619 CrossRef CAS PubMed.
- K. Krieger, B. Wängler, R. Schirrmacher and C. Wängler, ChemMedChem, 2020, 15, 1652–1660 CrossRef CAS PubMed.
- M. Ruiz-Gayo, F. Albericio, M. Pons, M. Royo, E. Pedroso and E. Giralt, Tetrahedron Lett., 1988, 29, 3845–3848 CrossRef CAS.
- H. Huang and R. I. Carey, J. Pept. Res., 1998, 51, 290–296 CrossRef CAS PubMed.
- R. A. D. Bathgate, F. Lin, N. F. Hanson, L. Otvos, A. Guidolin, C. Giannakis, S. Bastiras, S. L. Layfield, T. Ferraro, S. Ma, C. Zhao, A. L. Gundlach, C. S. Samuel, G. W. Tregear and J. D. Wade, Biochemistry, 2006, 45, 1043–1053 CrossRef CAS PubMed.
- D. E. K. Kabotso, D. Smiley, J. P. Mayer, V. M. Gelfanov, D. Perez-Tilve, R. D. DiMarchi, N. L. B. Pohl and F. Liu, J. Med. Chem., 2020, 63, 6134–6143 CrossRef CAS PubMed.
- O. Al Musaimi, B. G. de la Torre and F. Albericio, Green Chem., 2020, 22, 996–1018 RSC.
| This journal is © The Royal Society of Chemistry 2021 |