
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoft materials as biological and artificial membranes
Shukun
Tang†
a,
Zahra
Davoudi†
b,
Guangtian
Wang†
a,
Zihao
Xu
c,
Tanzeel
Rehman
c,
Aleksander
Prominski
d,
Bozhi
Tian
 d,
Kaitlin M.
Bratlie
bc,
Haisheng
Peng
*a and
Qun
Wang
*b
d,
Kaitlin M.
Bratlie
bc,
Haisheng
Peng
*a and
Qun
Wang
*b
aDepartment of Pharmaceutics, Daqing Branch, Harbin Medical University, Research and Development of Natural Products Key Laboratory of Harbin Medical University, 39 Xin Yang Road, Daqing, 163319, China. E-mail: fisher1688@163.com; Fax: +86-459-8977 588; Tel: +86-459-8153 003
bDepartment of Chemical and Biological Engineering, Iowa State University, 1014 Sweeney Hall, Ames, IA 50011, USA. E-mail: qunwang@iastate.edu; Fax: +1-515-294-8216; Tel: +1-515-294-4218
cDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011, USA
dThe James Franck Institute, Department of Chemistry, The Institute for Biophysical Dynamics, The University of Chicago, Chicago, Illinois 60637, USA
First published on 12th October 2021
Abstract
The past few decades have seen emerging growth in the field of soft materials for synthetic biology. This review focuses on soft materials involved in biological and artificial membranes. The biological membranes discussed here are mainly those involved in the structure and function of cells and organelles. As building blocks in medicine, non-native membranes including nanocarriers (NCs), especially liposomes and DQAsomes, and polymeric membranes for scaffolds are constructed from amphiphilic combinations of lipids, proteins, and carbohydrates. Artificial membranes can be prepared using synthetic, soft materials and molecules and then incorporated into structures through self-organization to form micelles or niosomes. The modification of artificial membranes can be realized using traditional chemical methods such as click reactions to target the delivery of NCs and control the release of therapeutics. The biomembrane, a lamellar structure inlaid with ion channels, receptors, lipid rafts, enzymes, and other functional units, separates cells and organelles from the environment. An active domain inserted into the membrane and organelles for energy conversion and cellular communication can target disease by changing the membrane's composition, structure, and fluidity and affecting the on/off status of the membrane gates. The biological membrane targets analyzing pathological mechanisms and curing complex diseases, which inspires us to create NCs with artificial membranes.
 Bozhi Tian | Professor Bozhi Tian received his BSc and MSc degrees in Chemistry from Fudan University Shanghai, China, and his A. M. and PhD degrees in Physical Chemistry from Harvard University in 2010. As an Associate Professor in the Department of Chemistry at the University of Chicago, his current research focuses on the semiconductor-enabled understanding of subcellular biophysics and studies of dynamics at soft–hard interfaces. The Tian group is interested in probing the nanoscopic or molecular interface between biological and inorganic systems, placing emphasis on novel material synthesis and device conception. |
 Kaitlin M. Bratlie | Professor Katie Bratlie earned her BS degree from the University of Minnesota in 2003 and her PhD degree from the University of California, Berkeley, under Prof. Gabor Somorjai. She then conducted post-doctoral research at MIT as an NIH fellow in Prof. Robert Langer's lab. Subsequently, She joined the Department of Materials Science & Engineering and the Department of Chemical & Biological Engineering at Iowa State University as an Assistant Professor in 2011. Her current research thrusts include developing biomaterials for medical applications and evaluating these materials in vitro and in vivo for drug delivery and regenerative medicine. She received the NSF BRIGE Award in 2012, the ISU Honors Mentor Award in 2014, was nominated as the “Outstanding Faculty Member” by the Interfraternity Council in 2015 and won both the Akinc Excellence in Research and Teaching Awards in 2015. |
 Haisheng Peng | Professor Haisheng Peng is the Dean of the College of Pharmacy in the Department of Pharmaceutics at Harbin Medical University (Daqing). He gained his PhD in Neural Biology from Harbin Medical University in 2008. He was a visiting scholar in the Department of Chemical & Biological Engineering at Iowa State University. Professor Peng's areas of interest include the synthesis of polymers, and the design and construction of nanocarriers. He has guided 22 projects, including NSFC. He has published 90 papers, and has won 13 awards, the Natural Science Achievement Award of Heilongjiang Provincial Government and the Natural Science Achievement Award of Heilongjiang Provincial Health Commission; meanwhile, he has received five authorized patents. |
 Qun Wang | Professor Qun Wang currently works in the Department of Chemical and Biological Engineering at Iowa State University. He gained his PhD in Chemical and Petroleum Engineering from the University of Kansas in 2010. Dr Wang's areas of interest include biomaterials, organoids, nanotechnology, and drug delivery. At Iowa State University, Dr Wang BONDs his research in these areas to produce innovative solutions and products for improving human health. Specifically, the current research in the Wang Laboratory focuses on tissue engineering and regenerative medicine using organoids, biomaterial-mediated organoids and stem cell reprogramming, and organoids and cell-mediated drug delivery. |
1. Introduction to soft materials
Soft materials refer to materials that become easily deformed upon thermal stress or thermal fluctuation at room temperature, including human-made polymeric molecules, colloidal vesicles, granular materials, and biological materials, such as the membranes of cells and organelles. There are close membrane connections from simple vesicles to complicated cells, including the compositions, structures, and functions of materials synthesized by nature or humans. As is known, we are struggling to clarify those connections. This review focuses on the compositions, structures, and applications of soft materials as biological and artificial membranes. Deepening our understanding of the biological membrane may help us to analyze the pathogenesis, and to diagnose and cure disease. Clarifying the connections between biological and artificial membranes and mimicking and utilizing biological membranes is beneficial for designing nanocarriers (NCs). A conceptual illustration of soft materials related to the membrane is illustrated in Fig. 1. This research area is an emerging convergence of material physics, physical chemistry, biophysics, and biochemistry. We are exploiting artificial membranes as building blocks that have become a potential medicine method, especially liposomes, DQAsomes (liposome-like vesicles containing dequalinium chloride), and polymeric membranes.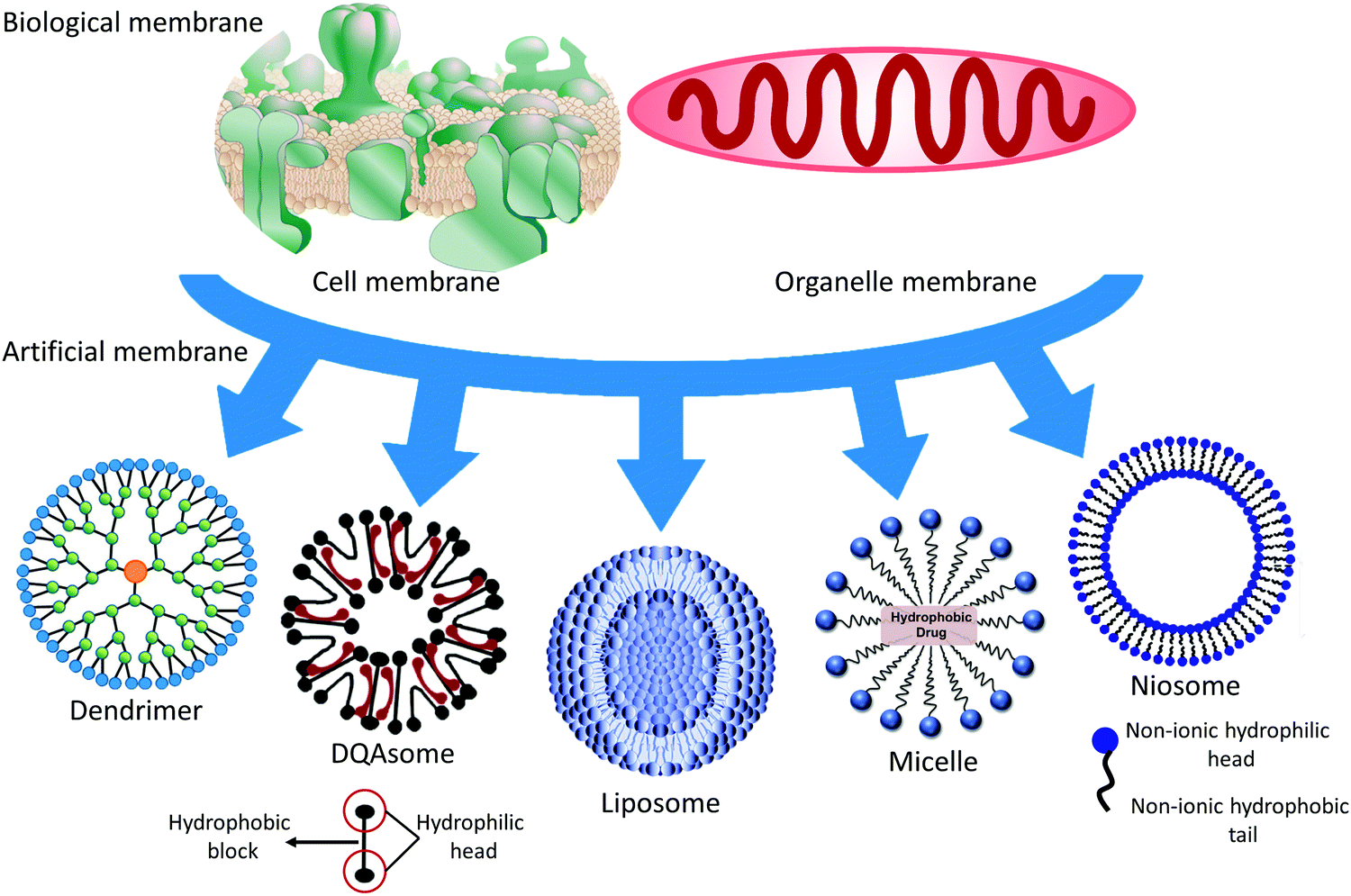 | ||
| Fig. 1 Conceptual illustration of soft materials, including human-made polymeric molecules, colloidal vesicles, granular materials, and biological materials such as organelle and cell membranes. Top left to right: Cell membrane and organelle membrane. Bottom left to right: dendrimer, DQAsome, liposome, micelle, and niosome. | ||
A cell is a container that is separated from the outside environment by membrane envelopes and needs to obtain essential nutrients and metabolize waste via the membrane barrier to maintain life's physiological activities. Also, cells need to respond to alterations in the exterior surroundings and produce signals across their biomembranes, which is particularly vital for multicellular beings. To continuously carry out these essential physiological functions under changing conditions, cells must modulate the shape of their membranes to reflect the cell's requirements in a particular environment during cellular activities, including migration, endocytosis, cell division, exocytosis, and cytoskeletal modulation.1 Electron microscopy has been used to visualize the microstructures of biomembranes to enrich our knowledge of membrane structures.
The cellular membrane encloses a limited space that is necessary for organizing the cell. Analogously, the cytoskeleton, which is composed of proteins, decreases the membrane's diffusion limits and carries out mechanical work. Spatial complexity and temporal regulation enable cells to have more complicated features and more efficiently respond to environmental changes in the evolution of multicellular life.2 Secreted organelles are transported by microtubules and motor-associated proteins, one of the most critical processes being investigated.3 When nanoscale carriers reach the target organelle, such as lysosomes, mitochondria, the endoplasmic reticulum (ER), or nuclei, they can repair the damaged functions of the cell.4 Gene therapy can influence the transcription of DNA in the nucleus and RNA translation in the cytoplasm.5 Enzymes in the lysosomal compartment catalyze the reaction of their substrates under specific conditions. The mitochondria are organelles that can induce anti-apoptotic or proapoptotic therapy. The transport of therapeutics across a soft material biomembrane is a challenge for drug delivery. After the safe delivery of NCs to the cytoplasm, therapeutics still need to find their way to the targeted organellar compartment, where their therapeutic efficacy is enhanced.6 The half-life of the payload in the NCs is prolonged due to the avoidance of premature degradation in the blood and cells, resulting in enhancement of the bioavailability. The intelligent design of NCs will solve many of the problems faced by therapeutic agents.
In the following sections, we review the native membranes of cells and organelles and non-native membranes of the following: liposomes, which are nanoscale cell-like vesicles with a bilayer structure of phospholipids; niosomes, which are liposome-like vesicles with a membrane containing non-ionic surfactants; polymeric membranes, which are molecular films of polymeric materials existing on the surface or at the interface of particles or the scaffold; and micelles, which are colloidal particles composed of amphiphilic molecular aggregates in water. The compositions of different NCs lead to a change of the surface features. The surface properties of NCs may influence drug molecules, cells, and tissues between the vesicles. They may further affect the molecular diffusion, cellular uptake, intracellular traffic, and targeting efficiency of the drug in the body. In the following sections, we will discuss these effects in detail. Finally, the affected membrane may give us a target or a vital clue to treating pathological cells or organelles; we also briefly introduce the applications of biological and artificial membranes.
2. Biological membranes
2.1 Cell membrane
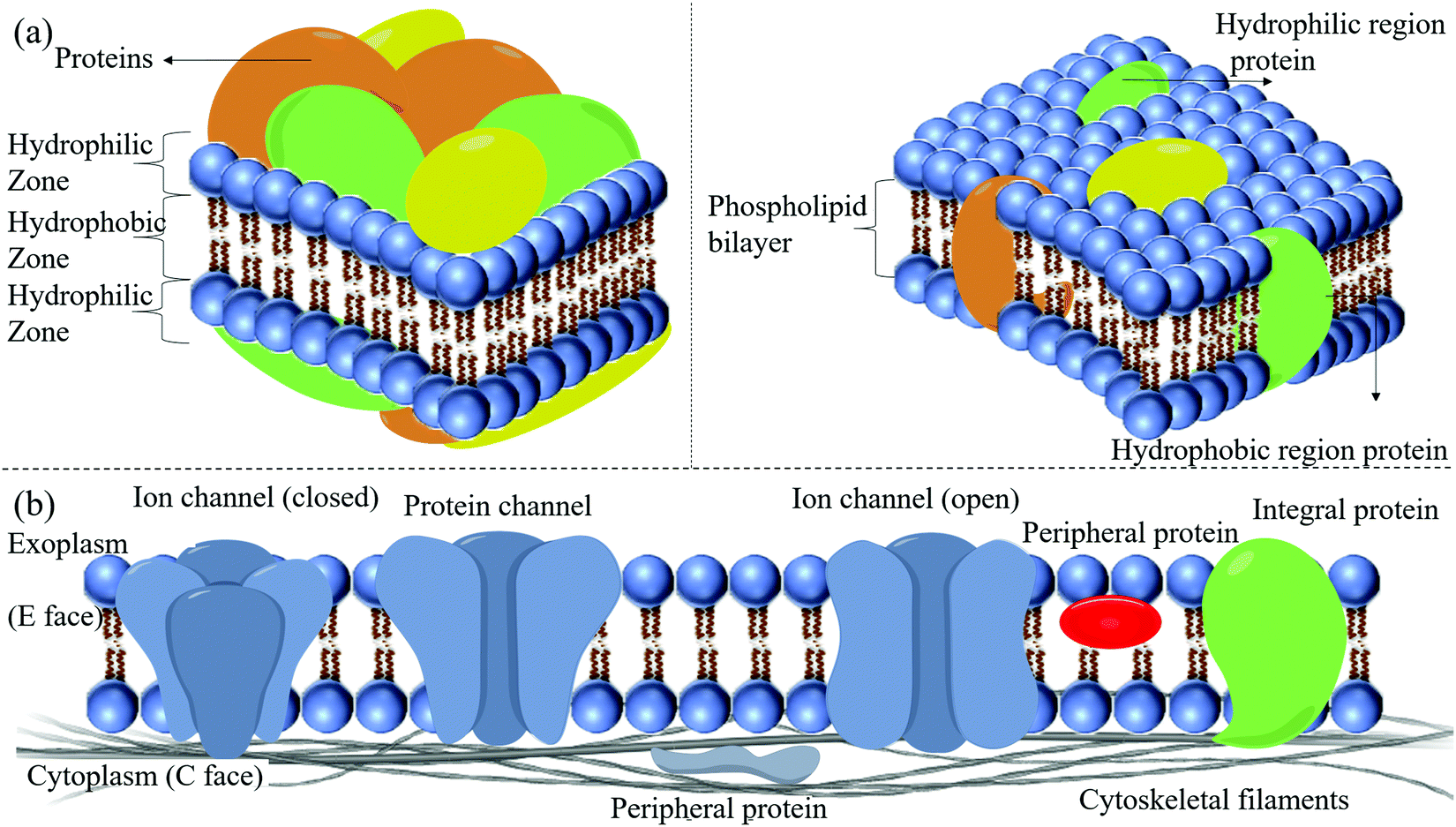 | ||
| Fig. 2 (a) Comparison between the old fluid-mosaic membrane model (F-MMM) model (the Davson–Danielli model, 1935) (left) and the new F-MMM (the Singer–Nicolson model, 1972) (right). The old F-MMM model proposed that proteins form a distinct layer on the top and bottom of the bilayer, making a sandwich-like structure. The new F-MMM model suggests that the proteins are embedded within the bilayer, forming a fluid mosaic. (b) Detailed schematic of the current F-MMM showing various components of the F-MMM embedded in the phospholipid bilayer. | ||
2.1.3.1 The skeleton of the phospholipid bilayer. The phospholipid bilayer constitutes the basic skeleton of the membrane, and it is not stationary. The mobility of the lipid bilayer resembles that of oil. Phospholipids are insoluble in water and self-emulsify to form a bilayer structure with the hydrophilic units exposed to water. By contrast, the long hydrophobic tails are inserted into the lipophilic part of the bilayer. The functional zone of the phospholipid bilayer is relatively poorly understood. The interplay amongst cholesterol, fatty acid residues, and the hydrophobic heads of the phospholipids regulates the cellular membrane's basic structure and function. Integral proteins are embedded in the membrane or membrane microdomains. This membrane domain is composed of phospholipid protein complexes with a delicate architecture for the protein's optimized function. Fatty acid residues play a vital role in the micro-modulation of the bilayer's physical–chemical properties. The bilayer's disturbance around a functional protein will affect its integration and structure, and function.11
Eukaryotic cells have intracellular membranes, whereas most non-eukaryotic cells have no intracellular membranes but a plasma membrane (PM). The internal membranes of eukaryotic cells usually include many bends, leading to a complicated membrane curvature. The PM is a larger, flatter structure but includes many delicate micro-membrane designs, adding complexity to the curvature of the flat PM. The PM is primarily made up of phospholipids. Various phospholipids can be incorporated into the bilayer of the membrane. Variation of the lipid composition may affect the physiological functions of the membrane. In eukaryotic membranes, the four significant phospholipids are sphingomyelin (SM), phosphatidylethanolamine (PE), phosphatidylcholine (PC), and phosphatidylserine (PS). It has also been confirmed that small amounts of phosphatidylinositol (PI) and phosphatidic acid (PA) can be detected in the membrane. Trace amounts of other phospholipids may even exist, but their functions need to be further characterized.12 Besides, PI is only present as meager amounts of phosphorylated inositol, known as the signal substance.13 Although most phosphoinositide in the bilayers structure is even more scarce, it can still activate the signal transduction of receptors in a signal-dependent manner. These lipids are the target of NCs. Pathologically, the 'flip-flopping' of PS from the inner side of the membrane to the layer parts is a typical pathological phenomenon at an early stage of cell apoptosis, and may possibly be utilized to design a targeting delivery system modified with annexin V protein.
2.1.3.2 Receptors. Receptors are macromolecular substances that can identify and selectively bind ligands. They have at least two functional regions: a region associated with ligand binding and a part that produces the biological effect. A series of processes can be activated when the receptor binds a ligand; the receptor's conformation changes and produces the activity, leading to the natural effect. The features that affect the interaction of the receptor with the ligand are specificity, saturation, and affinity. According to the receptor's site on the target cell, the receptor is classified as an intracellular receptor or a surface receptor.
Proteins preferentially bind to phosphoinositide rather than PS, probably because of their higher charge. Thus, phosphoinositide has a crucial role in controlling the subtle changes in the membrane structure that occur depending on the various cell functions, such as membrane trafficking, cell proliferation, and cell migration. Signaling clusters, or large-scale molecular assemblies, are essential regulators of cell signaling in the cell membrane. The T cell receptor (TCR) groups act as a classic model. Within the past decades, receptor oligomerization at a small scale has been studied to explain signal transduction across the cell membrane. However, unique spatial patterns at a large scale, for example, signal transmission in the immunological synapse (IS), have been identified more recently.14–16 Intercellular junctions between immunocytes and effector cells comprise the IS. Upon binding, diverse receptor–ligand couples divide into different spatial patterns through the interface, extending to ∼1 mm in size. Many spatial components are called ISs; TCR microclusters and other signaling molecular units, named signalosomes, can actively transduce signaling17 (Fig. 3). Recently, apparent advances in the comprehension of TCR microclusters have been achieved, and these novel findings could prove highly consequential in biology.
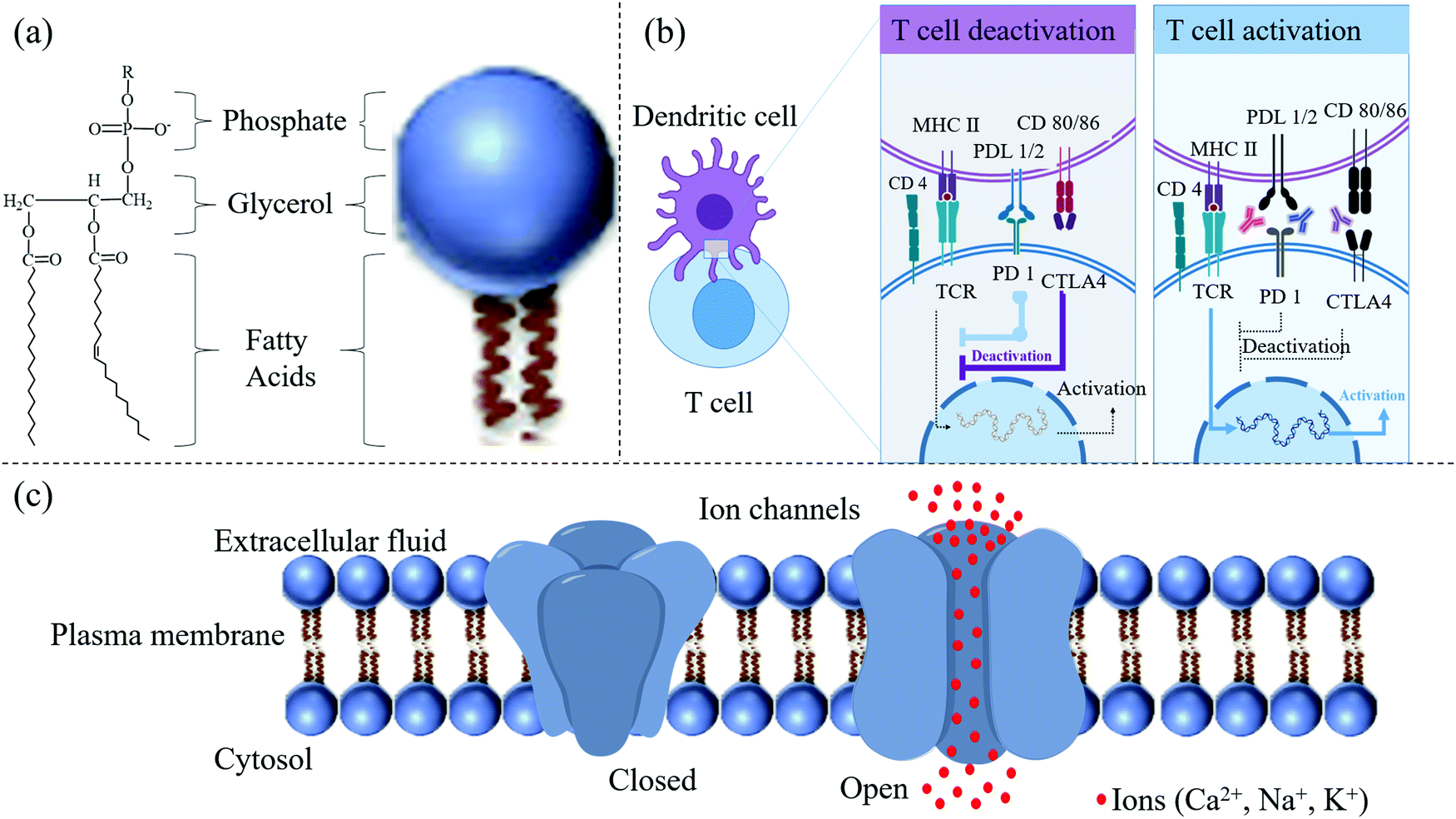 | ||
| Fig. 3 (a) Detailed structure of a phospholipid. (b) T-cell activation results in a series of events culminating in producing antibodies needed for the dendritic cell. (c) Schematic of ion channels (closed vs. open). Various types of ions can pass through the ion channels inserted into the bilayer membrane. | ||
Dendritic cells (DCs) primed by pathogens interact with T cells through TCRs and promote the cells to transform into protective effector T cells (Fig. 3b).18 Three significant signals provided by dendritic cells determine the activation of the cells. Stimulatory signal 1, after stimulation by the specific antigen, is mediated by TCRs. In this mode, pathogens are internalized by DCs through pattern recognition receptors (PRRs) and generate major histocompatibility complex (MHC) II peptides on the surface of the DC membrane. The MHC peptides help DCs present the antigen to helper T cells to activate them via interaction with TCRs.18 However, the antigen interaction with the receptor, called the stimulatory signal 1, cannot initiate the protective immunity alone but requires co-stimulation, signal 2. In this mode, CD80 and CD 86 produced by DCs trigger a co-stimulator (CD28) after the interaction of PRRs with pathogenic antigen, for example, Toll-like receptors (TLRs). Signal 3 is triggered by hydrophilic or membrane-bound factors, for instance, a secreted cytokine in the microenvironment of interleukin-12 (IL-12) or CC-chemokine ligand 2 (CCL2) polarizing signals, to help the maturation of the helper T cells TH1 or TH2, respectively.
Interestingly, cytokines released from DCs can also inhibit T cell activation or can induce apoptosis. Programmed death-ligand 1 (PDL-1) and PDL-2 expressed on DCs share the same T cell receptor inducing the programmed cell death protein 1 (PD-1). PD-1 can down-regulate T cell activation.19,20 There is negative feedback in the process of T cell activation. The binding of CD80 and CD86 on the DC membrane to CD28 on T cells leads to the production of cytotoxic lymphocyte-associated antigen-4 (CTLA4), which has a higher affinity for CD80/86 and thus interrupts the activation signal21 (Fig. 3b). The interaction between receptor and ligand would influence the selective accumulation of particles if the ligand were conjugated on the surface of NCs. However, the influence of the ligand's signaling activation on the NCs is always ignored when developing the particles. We paid more attention to the selectivity and targeting features of particles than cascade reactions in the cells. All factors, from simple materials to biological events, should be considered to acquire the best NCs.
2.1.3.3 Ion channels. Living cells must continuously exchange substances with their surrounding environment. Ion channels in the cell membrane provide a vital way to exchange substances. The most crucial substances, such as various ions and sugars, are soluble in water. Ion channels in the cell membrane can control the entry of ions and sugars or the excretion of soluble waste. Ion channels are inherent membrane proteins that control ion movement between aqueous environments through a hydrocarbon barrier of low dielectric constant. In higher eukaryotes, ion channels play primary roles in cellular function, specifically in intercellular signaling, especially in electrically excitable cells.1 Reconstitution experiments with specific lipids and their specific combinations are usually necessary for ion channels to show native properties.22 These preferences may reflect the protein structure's optimization for the specific lipid environment in which the protein is found. They may also be related to nascent protein folding, targeting the protein to a membrane microdomain, or regulating the protein function.
Additionally, one specific preference is produced by the particular binding of the lipid to the protein because it changes the membrane's physical properties. The challenge is to understand the effect of lipids on the ion channel. Any change of lipid composition in the membrane may alter its physical properties. Hence it is not easy to define the potential molecular mechanisms underlying the observed effect. Thus, clarifying the physicochemical effects of these properties on the membrane-associated protein structure is necessary to understand the targeting and regulation of the ion channels. These regulatory lipids interact with a wide variety of proteins.23 Their action on ion channels and synaptic function have been extensively documented.24 Lipid metabolites have also been reported on ion channels; for example, free fatty acids have modulated secretory chloride channels.25
Like the immunological synapse, the structure and function of many ion channels may be sensitively regulated by the unstable features of the membrane where they exist. Cholesterol and sphingomyelins (SPHs) are the main contents of synaptic membranes. Recently, researchers have found that these constituents have modulatory effects26 and lipid rafts on the membrane have several specific functions in electrically excitable cells. For instance, lipid rafts can sort and modulate voltage-gated K+ channels.27 Concerning ionotropic receptors, the effects of the bilayer on K+ channels and nicotinicoid receptors have been discussed. These superfamilies of receptors were chosen to broadly represent voltage-gated channels and ligand-gated channels, respectively. K+ channels are members of the more extended family of tetrameric cation channels that contain sodium ion-, calcium ion-, and cyclic nucleotide-gated channels. Generally, ion channels are formed with a central pore surrounded by four membrane-spanning domains or subunits. All these ion channels have four membrane-spanning domains. Nicotinicoid receptors, along with glutamate receptors, are the largest class of ligand-gated ion channels.
2.2 Organelle membrane
Eukaryotic cells have evolved to provide complicated membrane-bound organelles to perform specific biochemical reactions.28 Organelles are regarded as independent membrane-bound biochemical units. Several organelles such as lysosomes, peroxisomes, the endoplasmic reticulum (ER), or mitochondria are connected to perform different cellular activities such as protein transport and degradation, ATP production, and lipid breakdown.29In the past few years, a combination of ultrastructural studies, molecular cell biology methods, proteomics approaches, and fluorescence-marked live-cell imaging techniques has dramatically changed the view that subcellular compartments provide physical separation to an understanding that they offer a dynamic, collaborative and sophisticated network, and can interact and communicate with each other.30 Intracellular compartments exchange materials and deliver signals to each other to balance and maintain the cellular behavior. Collaborative roles of organelle networks include (1) intracellular signaling, (2) metabolic interaction, (3) modulation of cell apoptosis/necrosis, (4) pathogen defense, and (5) cellular maintenance.31,32 The functional interaction of organelles can be investigated by exchanging diffused signaling molecules or metabolites or by direct physical contact with specialized membrane sites. Molecular motors and the cytoskeleton are not the only players that organize the cellular architecture.
Furthermore, membrane contacts can change the motility and position of the organelles. Organelle interaction is also influenced by the number of organelles and is modulated by organelle formation and the biomembrane dynamic, as well as the process of autophagy. Many cellular functions are achieved through the intracellular transport of organelles. Most movements of organelles are controlled by actin filaments and the microtubules of motor proteins. Some fascinating proteins realize the conversion of chemical energy from nucleotide hydrolysis to action. These behaviors influence cell-shape changes, such as elongating the spindle, dividing the cytoplasm, or promoting cell migration during muscle contraction. Alternatively, motors can be used to transport cargo intracellularly through processes including centrosome positioning, chromosome separation, mRNA transport, the trafficking of membranous organelles, and the placement of polarity determinants.33,34
The autophagy-dependent cell death is mediated due to BECLIN-1 upregulation on a non-return point after reperfusion in the ischemic myocardium.42 During the autophagy phase, autophagosomes with double-membrane bubbles are continuously formed to eat intracellular materials and deliver this cargo to the ‘stomach’ of the cell, the lysosome, for subsequent degradation.43 Meanwhile, this ‘stomach’ of the cell controls the metabolism and signal hub of intracellular activities.44 Therefore, the reformed, activated, or damaged lysosomes are a target to regulate degradation during autophagy.45 Protective autophagy is precisely controlled to degrade the protein and reutilize damaged organelles.46
Furthermore, the OMM also includes proteins that master the hereditary pattern and shape of the organelle and members of the Bcl-2 family.50 Simultaneously, the IMM contains a boundary membrane and cristae with many crosstalk functional units to orchestrate activities in the mitochondrion, including the respiratory chain. Pro-apoptotic proteins, such as BCL-2-associated protein X (Bax) and BCL-2 antagonist/killer (Bak), cooperate with lipids to open the OMM to trigger programmed cell death. BCL-2 on the IMM prevents the cell from inducing apoptosis.51,52 The perturbations or reconstruction of the membrane integrity are caused by proteins, such as P53,53 caspase in the aging and trauma brain,54 GTPase,55 and estradiol.56 The negatively charged mitochondrial membrane is utilized to design targeting drug conjugates to deliver anticancer drugs, antioxidants, and sensor molecules for the diagnosis and treatment of diseases.57
Researchers have demonstrated that mitochondria are involved in respiratory chain abnormalities in patients with encephalomyopathy.58 The IMM protein encoded with the mutant SLC25A42 gene loses the capability to import coenzyme A into the mitochondrial lumen. Transmembrane protein 65 (TMEM65) within the IMM leads to severe neurological manifestations.59 Except for the central nervous system, mitochondrial diseases (MD) with respiratory chain defects may be multisystemic, including the heart, visual and auditory pathways, the gastrointestinal system, and myopathy.60 Alzheimer's disease (AD) is a type of neurodegenerative disorder. The accumulation of Aβ protein in the mitochondria improperly opens the mitochondrial permeability transition pore (mPTP), leading to breakdown of the OMM and the abnormal binding of IMM with the release of a cascade incorrect signals.61 Mitochondrial bioenergetics, mitochondrial dysfunction, transcriptional dysregulation, mitochondrial fusion/fission, and mitophagy have been confirmed as critical pathogenic factors of neurodegenerative disorders62 (Fig. 4).
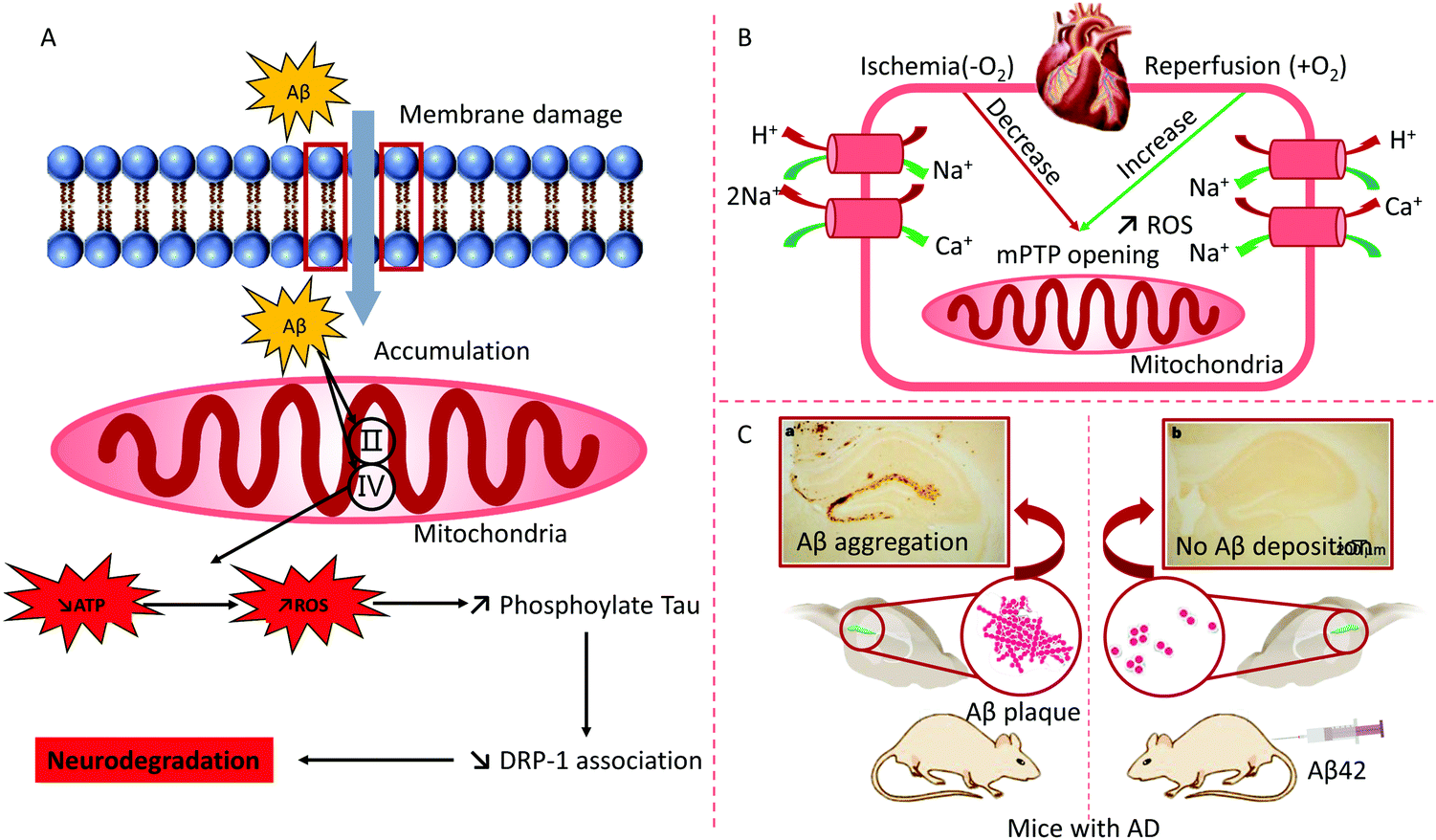 | ||
| Fig. 4 (A) Abnormal function of mitochondria and treatment for AD: the over-expression of amyloid-β (Aβ) destroys the mitochondrial function of AD animals. Aβ hampers protein complex II and IV on the mitochondrion, which inhibits ATP production and enhances ROS. Reproduced from ref. 62 with permission from Elsevier, copyright 2013. (B) The major mechanisms of myocardial ischemia–reperfusion injury. mPTP, mitochondrial permeability transition pore. (C) Immunization with Aβ attenuates AD-like pathology in mice. Reproduced from ref. 63 with permission from Springer Nature, copyright 1999. | ||
During cell division, the biogenesis of the GA is a complex process of deformation and reformation. The process can be divided into three phases: the disassembly and reassembly of the Golgi cisternae, stacking, and formation of the ribbon-like structure.67,68 The Golgi Reassembly Stacking Proteins (GRASPs) are not solely responsible for Golgi stacking. In the fruit fly Drosophila melanogaster, loss of a single GRASP influences the Golgi stacking slightly.69 Thus, in the budding phase of Saccharomyces cerevisiae, largely unstacked cisternae are found even with the existence of a GRASP protein such as Grh1p.70 These results suggest that other factors are also involved in the stacking process of Golgi cisternae. Besides cisternal stacking, GRASPs participate in the lateral coupling of Golgi cisternae and the formation of ribbon-like structures in vertebrates.71 These proteins may also influence the anchoring of transport vesicles, probably through GM130 binding.71 The physiological function of the GA is based on its unique cisternal-stacking structure.72 In neurodegenerative diseases, the Golgi structure is damaged, indicating the contribution of abnormal GAs to these diseases. It was recently shown in Alzheimer's disease (AD) that phosphorylation of GRASP65 destroyed its ability to form the Golgi morphology, leading to fragmentation of the GA. Inhibition of GRASP65 phosphorylation restored the Golgi morphology from Aβ-induced fragments and reduced Aβ formation. Disturbance of the Golgi morphology and function in nerve cells may directly impact the transport of substances and proteins indispensable for the structural integrity of synapses and dendrites.73
Glycosylation is a modification of lipids and proteins, and most glycosylation takes place in the GA. Although the transfer of initial carbohydrates into glycolipids or glycoproteins is carried out in the ER membrane or the ER, the subsequent reaction of many carbohydrates that comprise glycan maturation is completed in the GA. Golgi membranes are inlaid with glycosidases, glycosyltransferases, and transporters positioned in a highly organized way. Each active component in the membrane can act on a specific substrate produced at the early stage of the pathway.
The types and activities of glycosyltransferases impact the glycosylation process and the final structure of glycoconjugates in various cell types. Also, glycan synthesis is impacted by the protein integrity in the peripheral membrane within the GA, the pH in the GA, growth factors, cellular stress, and the membrane dynamics of the GA. Glycosylation in the GA can be used to develop assay protocols to observe the process of intracellular vesicle transport and help with the synthesis of recombinant glycoproteins.74 Earlier results confirmed that the trafficking and dynamics of the Golgi membrane were protein–centric.75 Data have recently demonstrated that lipid metabolism in the GA is also involved in vesicle transport and function maintenance.
Since the earliest confirmation of this mechanism in yeast and permeabilized cell systems,76 researchers have maintained that there is complicated interface trafficking of the membrane in the process of lipid metabolism. This interface plays a vital role in regulating the morphology and dynamics of the GA.77 Many proteins and enzymes of interest are as follows: lipid-transfer proteins, phosphatases, and lipid kinases, phospholipases D and A2,78,79 phospholipid acyl-transferases, and amino-phospholipid flippases,80 which regulate the catalysis capacity of ATPase to control the lipid position within the bilayer leaflets.81
Many studies have shown that nucleophagy (micronucleophagy) is related to partial and specific nucleus degradation. Autophagy locally changes the composition and dynamics of membrane tissue. Under various pathological and physiological conditions, the nucleophile plays a role in ‘housecleaning’. In eukaryotic cells, the nucleus is the largest membrane-enclosed and indisputably the most detailed organelle. Two primary units constitute the metazoan nucleus. First, the nuclear envelope (NE) provides the boundary between the contents of the nucleus and the cytoplasm.83,84 Second, the nuclear lamina consists of a crowded but fenestrated network comprised of intermediate filaments (IFs). The IFs contain lamin-associated proteins, and lamins underlie the inner side of the NE. The NE is necessary to maintain the space for nuclear pore complexes (NPCs), the nuclear shape, DNA synthesis and the modulation of transcription factors, and the organization of heterochromatin.85–87 For instance, some living beings, plants, or fungi do not express lamins, resulting in a lack of lamina.88 The nucleoskeleton components of herbs include coiled-coil proteins, which are not related to lamins.88 In non-metazoans, particularly Trypanosoma brucei and yeast, coiled-coil proteins are present in the nucleoskeleton.88,89
Historically, the NE has been regarded as a physical barrier, like the wall of a medieval castle.90 A moat usually protects such walls. Similarly, the NE structure comprises two isolated phospholipid bilayers, the inner (INM) and outer (ONM) layers. This structure is different from the single phospholipid bilayer of the cytoplasmic membrane. The NE has two protecting layers to guard the genome against hazards. The space between the two layers is called the lumen. It has been proposed that the primary evolution of the NE was an accidental result of excess growth of tubular ER and invaginations of the cytoplasmic membrane. By suppressing and precisely separating larger genomes in mitosis, the NE enabled increased complexity of the genome.90 The INM and ONM fusion at a specific domain of the NE is where NPCs exist. Composed of multiple copies of 30 subunits, NPCs, known as gatekeepers, regulate macromolecules to enter and exit the nucleus in eukaryotes.91 Topological and sequence analyses have confirmed that this gatekeeper has a central channel of phenylalanine-glycine (FG)-rich repeat-containing nucleoporins (FG-Nups). The FG-Nup structure plays a vital role in restricting the entry or exit of molecules (Stokes radius: ≳2.63 nm or MW: ∼40–60 kDa).92 This gatekeeper also has many peripheral channels, which regulate the transport of INM proteins.93,94
3. Artificial membranes
3.1 Liposomes
Liposomes are nano- or microscale cell-like vesicles with bilayer membranes that are ∼25–1000 nm in size and are composed of natural or artificial phospholipids. Liposomes can encapsulate hydrophilic drugs in their inner compartment and hydrophobic molecules within their membrane layers. Over the past few decades, liposomes have been considered to be promising drug carriers.95,96 They are biocompatible and have little to no toxicity or antigen response, and drugs encapsulated into liposomes are protected from external degrading reactions. Liposomes have obvious pharmacokinetic advantages, such as delayed drug absorption, limitation of drug distribution in vivo, decreased drug removal, and sustained drug metabolism.97Liposomes in the bloodstream can be rapidly cleared and pass through the reticuloendothelial system, especially the liver and kidneys. Most liposomes are eaten by phagocytic cells and are eventually degraded in lysosomes.98 Modified liposomes can selectively accumulate in specific tissues or organs and may also enhance the efficacy of drugs. Many methods obtain targeted liposomes by connecting the corresponding targeted moieties to the surface of vesicles. For instance, modifying the antibody on a liposome's surface without damaging the antibody's targeting property is a case in point.99 Also, the limitation of immunoliposomes may be due to the short lifetime of vesicles in blood vessels.100 The primary organ that accumulates antibody-modified liposomes is the liver, which prevents the drug from gathering in the target tissue. If the lifetime of the liposomes in the blood is prolonged, the higher is the drug concentration at the target site and the more opportunities there are for interaction with the targeted antigens. This is why stealth liposomes have become the most researched NCs over the past ten years.101 The results indicate that the combination of properties of targeted and long-circulating liposomes is valued in these carriers. Therefore, several specific binding molecules, including antibodies, on the surfaces of carriers have been linked to the polyethylene glycol (PEG) chain with water-exposed fragments.102 The chain length, the grafted density of the PEG, and ligands conjugated onto the PEG termini change the folding of the PEG chain. The surface properties of liposomes and components cause them to adhere to the vesicles, forming what is called the protein corona. The protein corona is responsible for the off-target effects of NCs and is closely related to the distribution of particles in vivo. Moreover, the addition of targeting molecules also changes the conformation of PEG and negative feedback regulation of the position of targeting molecules, which decrease the targeting efficiency of NCs modified with ligands to some extent. The components of the protein corona in blood may also fluctuate according to the type and stage of a disease, further increasing the complexity of the design of NCs.
3.2 Niosomes
Genetic engineering and biotechnology are developing specific drug-delivery systems that mimic cell membranes, and these have been widely emphasized.1,100,103 Vesicles are widely used to mimic cell membranes and deliver drugs to the affected foci and viruses.104 The characterization of vesicles, such as via the size and zeta potential, is essential in the pharmacokinetics of vesicle-loaded therapeutics. The delivery efficiency of a particle decreases from 0.7% to 0.4% when the particle diameter changes from 100 nm to 200 nm. Also, delivery efficiency of neutral particles is more significant than negatively or positively charged particles (0.7% vs. 0.5% or 0.6%).105 The vesicles can improve drug stability, enhance the treatment effects, prolong the circulation time, promote absorption at the target site, and reduce drug toxicity. Vesicles containing non-ionic surfactants are termed niosomes. Niosomes are biodegradable, stable, relatively non-toxic, and an inexpensive alternative to liposomes. Interest in these vesicles is deepening and is expanding to many scientific disciplines, particularly in medicine.104Generally, niosomes containing non-ionic surfactants or amphipathic molecules have a structure and features like liposomes; their structure and morphology are illustrated in Fig. 5A. The materials used to prepare niosomes are relatively inexpensive, making niosomes more attractive than liposomes for pharmaceutical and cosmetic applications. Also, liposomes are limited by the breakdown of phospholipids in aqueous media.106 Another advantage of niosomes is that their industrial fabrication is free from toxic solid solvents. A novel niosome was formulated using a cationic lipid mixed with squalene and Tween 80 to increase the transfection efficiency of a specific gene fragment to rat retinas. The researchers used a solvent emulsification evaporation process to obtain the niosomes. Then, blank vesicles were incubated with the pCMSEGFP plasmid to obtain lipoplexes. The characteristics of the niosomes were evaluated by observing the size, shape, loading efficiency, degradation and release of DNA, and the surface charge. In vitro experiments using ARPE-19 and HEK-293 cells were aimed at estimating the transfection efficiency and the mechanism of internalization.107 The formulation's efficacy was assessed in rat eyes by checking the level of EGFP protein after subretinal and intravitreal injections. Successful internalization of lipoplexes in niosomes by HEK-293 and ARPE-19 cells was realized without decreasing their cellular viability. Meanwhile, lipoplexes were internalized by retinal cells through a clathrin-mediated endocytosis mechanism. On the other hand, the lipoplexes entered the HEK-293 cells through a routine of caveolae-dependent uptake. FITC was used as a fluorescent probe to investigate the in vivo skin permeation of niosomes. Niosomes demonstrated continuous permeation and could diffuse far into the deep dermis, as shown in Fig. 5A.
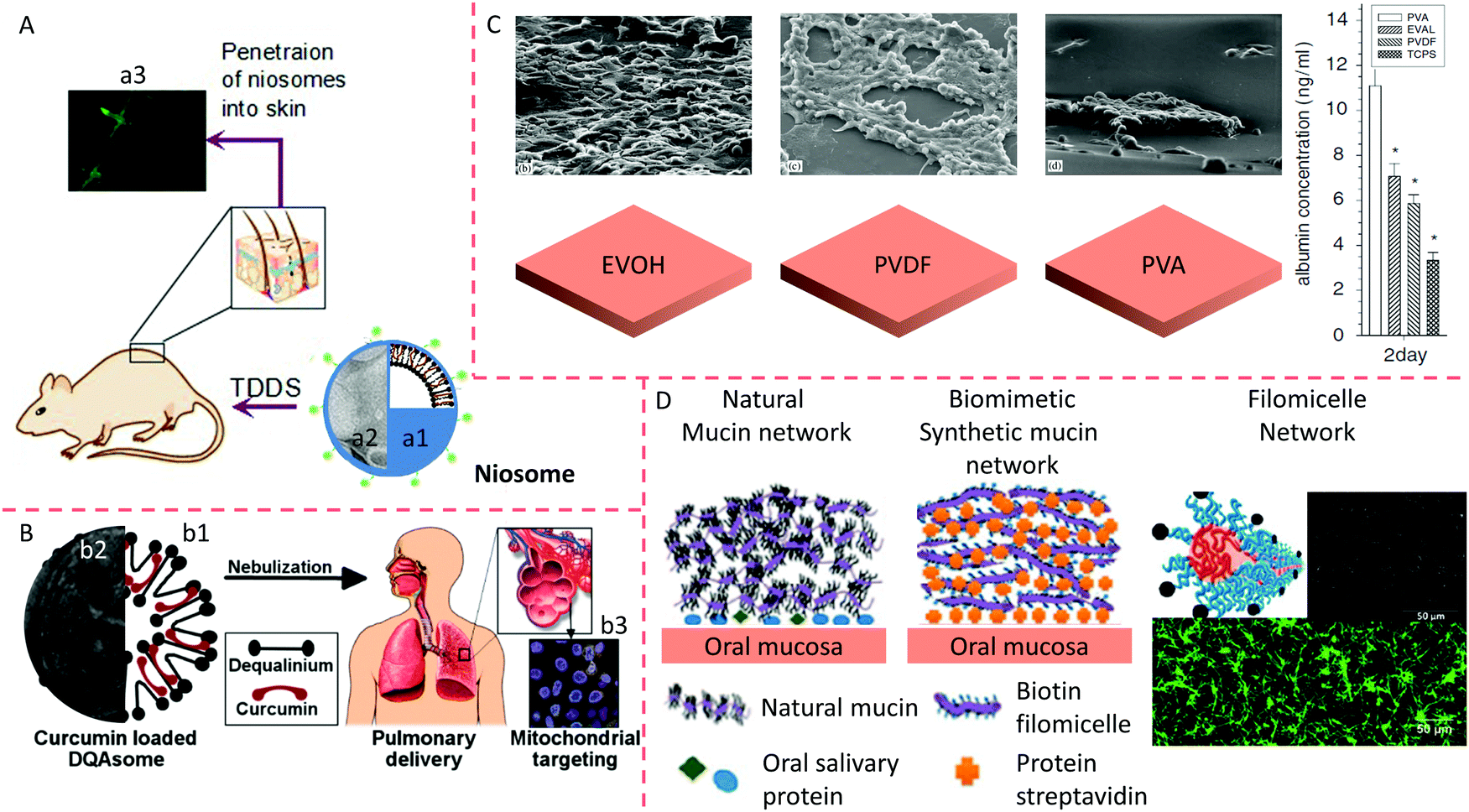 | ||
| Fig. 5 Penetration of niosomes into mice skin. (A) Schematic (a1) and TEM image (a2) of a niosome and penetration of vesicles into mouse skin (a3). Reproduced from ref. 107 with permission from Elsevier, copyright 2014. (B) Schematic (b1) and TEM image (b2) of DQAsomes and targeting of nanocarriers to the mitochondria of cells in the lung after administration of aerosol (b3). Reproduced from ref.111 with permission from Elsevier, copyright 2003. (C) HepG2 cells are cultured on different polymeric membrane substrates, and their corresponding albumin adsorption. Reproduced from ref. 119 with permission from Elsevier, copyright 2007. (D) Comparison between a synthetic oral mucin mimic containing a polymeric micelle layer-by-layer (LBL) network and a natural oral mucin counterpart. Reproduced from ref. 118 with permission from the American Chemical Society, copyright 2014. | ||
3.3 DQAsomes
Weissig et al. (1998) prepared cationic vesicles mixed with dequalinium chloride (DQA), called DQAsomes.108 The data revealed that DQAsomes could attract DNA, avoiding degradation by DNase.109 DQA preferentially accumulates in mitochondria due to the difference in electrochemical gradient between the mitochondrial membrane and other organelle membranes. Thus, DQAsomes might be used as carriers to deliver DNA fragments to mitochondria in live cells. This research group also confirmed that DQAsomes could selectively release DNA fragments from cardiolipin-rich liposomes, a mitochondrial membrane model. They reported that DNA remained in the complex with DQAsomes containing superfluous anionic lipids that are different from cardiolipin.110 DQAsomes, which are mitochondriotropic cationic vesicles, have been used to transport DNA or drugs to mitochondria in live cells (Fig. 5B);111 the morphology of a DQAsome is shown in Fig. 5B. The entry of DNA fragments into the mitochondria is based on the conjugation of DNA within DQAsomes with a signal peptide of mitochondria. The specific downgrade of this DNA from DQAsomes before the entry of the mitochondria and the internalization of DNA fragments by mitochondrial protein.The DNA fragments within DQAplexes selectively released pDNA when they met mitochondria-like membranes. A novel protocol was used to selectively stain uncoupled pDNA in the plasma of living cells using confocal fluorescence microscopy. The data showed that DQAsomes were not degraded after endosomes and continuously loaded their pDNA and transported it to the mitochondria, where the pDNA was downloaded.109 Confocal microscopy could be used to detect pDNA in the mitochondria, but it was not detected at other organelle membranes of transfected cells, indicating the selective delivery of DNA to mitochondria by DQAsomes.108,111 In a novel mitochondrial transfection protocol, Lyrawat et al. (2011) utilized the reporter gene of green fluorescent protein (GFP) to design and construct an artificial mitochondrial genome. From the jellyfish Aequorea victoria, the GFP gene is conditionally expressed to report a specific gene level since GFP expression can be easily observed in the mitochondria using fluorescence microscopy.112 Lyrawat concluded that a DQAsome-based transfection protocol was successfully developed using the mitochondrial construct pmtGFP. The imaging showed that GFP was expressed in the mitochondria. The toxicity of DQAsomes to nude mice depends on the administration route of the vesicles. DQAsome loading of anti-cancer drugs caused the senseless death of mice after vein injection of the vesicles. DQA may be responsible for the unexpected death of the mice (unpublished data).
3.4 Polymeric membrane
The polymeric membrane is a multiple-layer molecular film composed of polymeric materials, coated on the surface of particles or at the interface of a tissue engineering scaffold to tune the physicochemical properties, biocompatibility, release or loading pattern, and the targeting efficiency as well as to optimize the maintenance, growth, differentiation, and migration of cells to eventually form an artificial tissue or organ. With the development of tissue engineering, incorporating biological cells into biomaterials forms an essential part in biological materials.113 In many studies, biomaterials in a serum medium have been co-cultured with cells. The medium adsorbs proteins onto the biomaterial's surface layer, regulating the interaction of the biomaterial with the cells.114,115 The physicochemical characterization of the biomaterial surface is affected by the type, conformation, and quantity of adsorbed proteins.116 These absorbed proteins include extracellular matrix (ECM) proteins, for instance, fibronectin and vitronectin. These proteins recognize the cell-binding domain through integrin receptors on the cell surface. It is well known that integrins induce focal adhesion formation, regulate signal transduction pathways, and modulate several vital activities such as cytoskeletal reorganization, spreading, and long-term events.117 Hence, clarifying these mechanisms will enable biomaterials to enhance their cell function and compatibility.The HepG2 cell line was used as an in vitro model of human hepatoma. The interactions of polymer materials with HepG2 cells have been widely explored in many studies, including poly(ethylene vinyl alcohol) (EVOH), polyvinylidene fluoride (PVDF), and polyvinyl alcohol (PVA). The mobility of cells, co-cultured in serum medium after the addition of PVA, was significantly more robust than that of cells treated with PVDF or EVOH (Fig. 5C). In the absence of a serum medium, the PVA effect was entirely inhibited by cycloheximide (CHX). Taken together, the cell-surface charge can be changed after growth on various polymer matrix surfaces. The exact reasons for these changes in the electrophoretic mobility of these cells are still unknown. Still, researchers believe that the surface properties of polymer matrices may be changed by the absorbed substances, leading to electrophoretic mobility changes and regulating the adhesion, growth, and function of the cells.116 In the oral cavity, mucin networks are formed by complexing salivary proteins with glycoproteins on the surface of mucin to yield a hydrated lubricating layer. The physiological function of the hydrated layer is related to the physicochemical and mechanical properties of its components. However, it is not easy to clarify their relative importance owing to their interdependent properties. Authimoolam et al. (2014) showed that a worm-like fibrous network could be deposited to form a three-dimensional (3D) porous network by multiple washing of polymeric worm-like micelles118,119 (Fig. 5). The structure, the microbial-capturing capabilities, and the stability were proposed to affect the network's functions and properties. The results showed that increasing the structure's order, even by adding adhesion-resistant PEG, helped to capture bacteria. This artificial network offers an outstanding and effortless means of independent evaluation of mucin network properties (Fig. 5D).
3.5 Micelles
Micelles are molecular aggregates from the self-assembly of surfactants in water with hydrophilic segments pointing outward and the hydrophobic parts tucked inside, and these are also promising nanoparticles for drug delivery.120 Generally, the particle size of a micelle is not larger than 100 nm. One of the vital properties of these particles is that they can improve the bioavailability of hydrophobic drugs by increasing the solubility of poorly soluble molecules. The building blocks of micelles are amphiphilic molecules composed of segments of hydrophobic and hydrophilic chains, prolonging the half-life of carriers. Because of the nanoscale size of micelles (<100 nm), they show spontaneous penetration into vascular leakage (tumor, infarction); this selective drug delivery is called passive targeting.121 Micellar entrapment of anticancer drugs has been repeatedly used in this way. For example, Doxil accumulates more in tumors than in other organs, minimizing an adverse reaction to the drug in normal tissue.122 It has been reported that the accumulation and diffusion parameters of drug carriers might depend on the permeability of the vessels. Still, the vessel permeability is mainly regulated by the pore size of the tumor vessel wall.123 However, the statistical data based on ten-year literature studies show that only less than 1% (median) of the administrated dose is detected in the tumor tissue. Proof-of-concept data did cure mice loaded with a tumor xenograft, but the clinical results frustrated us when translated to clinical application.105 Possibly, when the tumor is too small, the lack of blood vessels limits the diffusion of nanoparticles into the tumor tissue without the enhanced permeability and retention (EPR) effects. Comparably, when the tumor is big enough, it may be removed by surgical operation, and the residual tumor tissue also lost the blood supply with the EPR effects. Indeed, there might be significant histological differences in blood vessels between humans and animals such as mice and rats.Understanding the structure and features of micelles is vital in fundamental science and its application – generally, the dimensions of micelles increase with the aggregation number of surfactants. The size and shape of micelles will change with the type of surfactant molecule and their aggregation number. Enhancement of the charge on the ether oxygen within the skeleton of ethylene oxide group will enhance the interaction of micelles. Regulation of the surface charge will be a benefit to improving the colloidal stability. Self-assembled mixed micelles with multiple surfactants have some features that are different from those of individual micelles. Mixed micelles could reduce the critical micelle concentration, increase foaming, and hold beneficial rheological properties.124 Ligands, including some small molecules or antibodies, can be connected to the free termini of the hydrophilic segment of polymers. Using targeting micelles and separating drugs from their carriers at the correct location increases drug utilization, improving the therapeutic efficacy, and reducing the toxicity of drugs.125 The stable structure of micelles is determined by the proportion of hydrophilic fragments and the hydrophobic counterpart. Some bigger ligands, such as proteins and antibodies, may enhance the part of the hydrophilic segment too much, destroying the system balance of the structure, and damaging stable micelles. Therefore, the modification of micelles aims to improve the above-mentioned features of the micelle, and the dynamic stability of the colloidal form should be taken into account.
4. Biophysical insights into membranes
Lipid membranes can display different deformation classes, such as bending, stretching/pulling, twisting, and coating of various curvilinear substrates. They are fluidic in the lateral direction but show elastic behavior upon stretching or bending. Lipid bilayers can also establish charge separation and use it for biological signal transduction. The most remarkable bioelectric behavior of the lipid bilayer is the elicitation of action potentials in excitable cells, such as neurons and muscle cells. The mechanical, topographical, and bioelectric states of membranes can all produce downstream biochemical responses in cells, leading to various changes in cell states and fates, such as directed cell migration and electrical signal propagation in a neural network. Here, we only focus on some latest advances in our biophysical understanding of lipid membranes.4.1 Membrane tension
Membrane tension is generalized as the force per unit length of an imaginary boundary that defines a membrane patch. The biological or biointerface processes that yield membrane tension include (1) interactions between the cytoskeletal components such as the actin–myosin bundles and the plasma membrane, (2) dynamic focal adhesions established during the cell attachment or migration, (3) the osmotic pressure generated across a cell or an organelle, and (4) the mechanical impact upon the device or material applications onto single cells or tissues. Different biophysical tools have been developed for mechanical property characterizations. For example, micropipette aspiration experiments have frequently been used to measure the area stretch modulus (Ka) of a giant unilamellar vesicle or other membrane elastic properties. While the mechanical properties of natural and synthetic membranes have been studied extensively, our understanding of the membrane tension in biological systems is minimal (e.g., how does the membrane tension control cell morphogenesis or force transduction in tissues? And how does the tension mediate the biomechanical or even bioelectrical signal transduction at an implant surface?). A recent breakthrough discovery from micropipette-assisted experiments suggests that cortical cytoskeletal filaments apply significant resistance against the propagation of tension across the plasma membranes (Fig. 6A).126 The cytoskeletal filaments connect to the plasma membranes through the membrane proteins, forming a ‘composite’.127 This biological composite confines the tension and helps the membranes to reach mechanical equilibrium more rapidly (compared to synthetic membrane systems) when the geometry of the membranes is locally perturbed, without causing an invasive impact on the rest of the cellular parts. The confined tension can also yield a highly localized and strong tension gradient, triggering membrane protein activities (e.g., mechanosensitive ion channels) and cellular functions (e.g., vesicle fusion).126 Specifically, the authors found that cell long-range membrane tension transmission did not occur throughout ∼10 min. The diffusion coefficient was ∼0.024 mm2 s−1, which is slow enough to make the local tension ‘static’ for most characterizations or other biological events. | ||
| Fig. 6 Localized membrane tension and induced membrane curvature. (A) The membrane tension is localized at the plasma membrane; the membrane proteins and the cytoskeletal filaments form a composite. This yields membrane tethers when pulled from the body of a HeLa cell expressing GPI-eGFP. The schematics (left) and the fluorescence image (right) show that membrane tension transmission is only limited to a very local area. The left panel is reproduced from ref. 126 with permission from Cell Press, copyright 2018. (B) The localized membrane tension also yielded a successful intracellular electrical recording from a U-shaped silicon nanowire FET device, where a curvature-dependent device entry was observed. The right panel is a zoom-in view, highlighting the curved biointerfaces where the lipid bilayer from the nanowire backbone and the cell's plasma membrane can fuse upon contact. Reproduced from ref. 129 with permission from Springer Nature, copyright 2019. (C) A recent study showed that the membrane curvature accumulated Arp2/3 and related activator proteins when establishing an extracellular nano–bio interface, leading to branched actin network formation. Reproduced from ref. 132 with permission from the American Association for the Advancement of Science, copyright 2019. | ||
This discovery is a significant advance in the field, and its implications go far beyond molecular and cell biology. For example, the confined tension distribution in the cell membrane is consistent with a recent report in the field of nano-bioelectronics,128,129 which shows that U-shaped nanowire-based intracellular penetration is dependent on the tip curvature (Fig. 6B). The nanowire was configured into a field-effect-transistor configuration for intracellular electrical recording from neurons and cardiomyocytes. The nanowire surface was modified with a phospholipid bilayer that could potentially fuse with the plasma membrane, exposing the nanowire tip to the cytosol for recording. As long as the membrane tension was not confined in the contacting areas but instead quickly relaxed to other cell regions, there would be significantly smaller curvature dependence for penetration of the nanowire device. This confined membrane tension also suggests that nano–bio interfaces established between synthetic materials and cell membranes may yield a local tension gradient that is strong enough to drive biomolecular diffusion, liquid/gel phase changes, and various biological signaling processes.127 Future studies in this area can benefit from multiple in situ characterization tools130 that capture the correlation between the bioelectrical activities of cells and the dynamics of membrane tension, cytoskeletal organization, extracellular matrix viscoelasticity, and membrane protein organization.
4.2. Substrate-induced curvature
Lipid membranes can self-assemble into various forms such as vesicles, tubes, sheets, and liquid crystals. In recent years, membranes coated over synthetic substrates have gained much attention as they offer new drug-delivery opportunities and minimally invasive biomedical devices for recording and modulation.Recent studies have demonstrated that when plasma membranes are deformed against nano-pillar or nano-bar substrates, the membrane elicited responses in curvature-sensing membrane proteins and triggered clathrin-mediated endocytosis at the biointerfaces.137,138 Notably, a very recent study has discovered a mechanism by which plasma membrane curvature promotes the actin polymerization into a branched network around curved biointerfaces (Fig. 6C).132,139 The actin accumulation is more pronounced for nanostructures with smaller widths and larger local curvatures. Notably, the curvature sensing was found to be related to Arp2/3 and its activator proteins instead of formins; this would yield the branched actin meshwork around the biointerfaces. Such an accumulation of actin is also found to accompany reduced expression of the mechanotransducer YAP,140 suggesting a lower probability of cellular migration and proliferation.141 These recent studies highlight a membrane curvature engineering approach for cellular activity controls.
4.3. Bioelectric properties
 , where Q is the charge flow to/from the capacitor and V is the voltage across the membrane. The increase of the electrical capacitance can be understood by the fact that heating can expand the membrane surface area while reducing the membrane thickness. Upon ‘injection’ into the cells, the capacitive current elevates the intracellular potential (i.e., depolarization) to a level that excitable cells such as neurons or cardiomyocytes can elicit single action potentials or the spike trains through a coordinated process among different ion channels. In traditional intracellular neuronal or cardiac excitation, depolarization is achieved by injecting currents through a glass micropipette electrode. However, the bulky electrode and associated electronics could be invasive to cells and are less scalable.
, where Q is the charge flow to/from the capacitor and V is the voltage across the membrane. The increase of the electrical capacitance can be understood by the fact that heating can expand the membrane surface area while reducing the membrane thickness. Upon ‘injection’ into the cells, the capacitive current elevates the intracellular potential (i.e., depolarization) to a level that excitable cells such as neurons or cardiomyocytes can elicit single action potentials or the spike trains through a coordinated process among different ion channels. In traditional intracellular neuronal or cardiac excitation, depolarization is achieved by injecting currents through a glass micropipette electrode. However, the bulky electrode and associated electronics could be invasive to cells and are less scalable.
Various synthetic materials, such as gold or nanoporous silicon (Si) particles (Fig. 7A)147,148 can enhance this process as they produce heat even with visible or near-infrared (NIR) light. Additionally, the surfaces of these nanomaterials can be modified with high-affinity ligands or antibodies for cellular targeting.149 In this regard, light-sensitive and heating-producing nanomaterials enable the highly localized and selective optical stimulation of cells and tissues. Light sources with wavelengths accessible in most research and clinical settings can be used.149
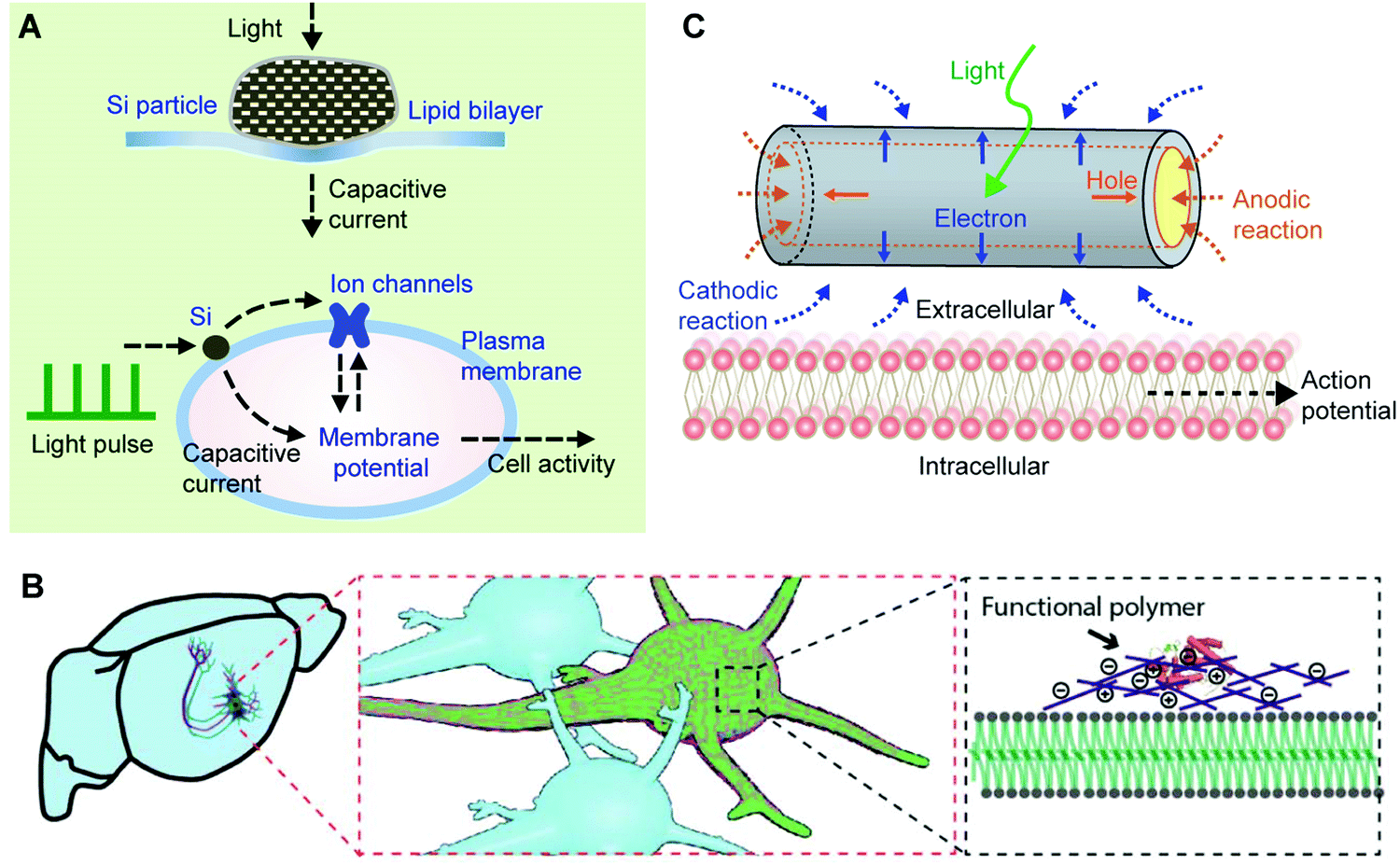 | ||
| Fig. 7 The bioelectric properties of membranes can be modulated. (A) Modulation of a lipid bilayer electrical capacitance with nanoporous Si particles. The light-induced heating produced a transient capacitive current, which can be used for neuromodulation through an interplay among light pulses, Si particles, the plasma membrane, and ion channels. Reproduced from ref. 147 with permission from Springer Nature, copyright 2016. (B) The plasma membrane capacitance can also be modulated long-term through a genetically targeted chemical assembly of functional polymers. Reproduced from ref. 150 with permission from the American Association for the Advancement of Science, copyright 2020. (C) The local electrochemical potential near a plasma membrane can be controlled through a photoelectrochemical process from a coaxial Si nanowire. The photocathodic effect from the nanowire can depolarize the cell membrane, triggering action potentials in neurons. Reproduced from ref. 154 with permission from Springer Nature, copyright 2018. | ||
While local heating can induce a membrane capacitance change, this effect is transient. Recently, a long-term variation of the membrane capacitance has been demonstrated through engineered protein-induced polymerization of synthetic materials over cell surfaces.150 Liu et al. showed both in vitro and in vivo that neuronal spiking activities decreased when the neurons were coated with conducting polymers (Fig. 7B). By contrast, the activities increased with an insulating polymer coating.150 The authors have attributed such an observation to the membrane capacitance effect. Specifically, when conducting polymers were used, their large electric permittivity151–153 contributes to increasing the membrane ‘composite’ electrical capacitance, following C = εoA/d (where A and d are the surface area and the thickness of the membrane capacitor, respectively). For an insulating polymer coating over neurons, the increase of d tends to reduce the size of C, yielding a faster neuronal spiking frequency. Decreasing the C value would potentially speed up the action potential propagation in axons, in a similar way that myelinated nerves conduct action potentials faster than that in unmyelinated fibers.
5. Internalization of the artificial membrane by cells
The endocytic pathway is characterized as pinocytosis for liquids and phagocytosis for solids. The NCs are internalized by cells through different pathways, including clathrin-mediated, caveolin-mediated, clathrin- and caveolin-independent pathways, and macropinocytosis.155 The physicochemical properties of NCs most commonly emphasize the internalization pathway of particles. The properties include the size, zeta potential, shape, surface, rigidity, and surface molecules of the particles.156 The size of the NCs is a significant factor for trafficking in the cellular compartments. However, an infinitely small particle cannot be captured and shrouded due to the limitation of the radius of curvature of membrane folding. Therefore, it is vital to optimize the size of NCs to realize effective endocytosis. The stiffness and softness of NCs change the cellular uptake and consequently the tissue distribution. The Young's modulus can be used to assess the stiffness based on the relationship between stress and strain. The contact angle, the contact surface area with the biomembrane, and the receptor-mediated diffusion all affect the uptake of NCs.157 To achieve the recycling of materials, maintain a reasonable cell size, or transmit signals and materials to neighboring cells, after entry of the NCs into the cells, one part of the endosome will be fused with the lysosome to acquire the raw materials. At the same time, the rest will be released through an exosome pathway to the environment as well as helping newly-formed NCs penetrate deeper tissue. Alternatively, the microvesicles that are larger than the exosome leave the cell in a budding manner. There are significant differences in proteins and lipids between exosomes and microvesicles.158Understanding the exact mechanisms of cellular uptake, intracellular trafficking, and the design of gene vectors has become a growing demand to optimize targeting carriers for clinical applications. Integrins such as αvβ3 and αvβ5 have become promising targets for cancer therapy; the levels of integrins are increased in tumor vessels and malignant cells and modulate the growth and metastasis of tumors.159 Many researchers have used peptides with the Arg-Gly-Asp (RGD) recognition sequence to modify nanoparticles to target integrins on the tumor endothelium and to penetrate tumor tissues. This targeted nano-drug delivery system has been shown to have specific and curative effects on tumor cells and tumor vessels in vitro and in vivo. The polyplex micelle is a synthetic gene vector that is a DNA plasmid complexed with thiolated PEG-block-poly(lysine) copolymers, targeting various integrins by modifying the RGD peptides. The polyplex micelle has become a potential formulation for application in clinical treatment.160 The effects of RGD ligand modification on the uptake of micelles were analyzed using an flow cytometer and advanced fluorescent microscope. By changing the chain length of the PEG molecules (PEG12, MW: 12 kDa and PEG17, MW: 17 kDa) on the shell layers of the micelles, the impact of surface shielding on the uptake of carriers by the cells was observed (Fig. 8). The RGD peptide promoted the uptake of micelles by HeLa cells highly-expressing integrin but did not affect the entry pathway of the particles.161
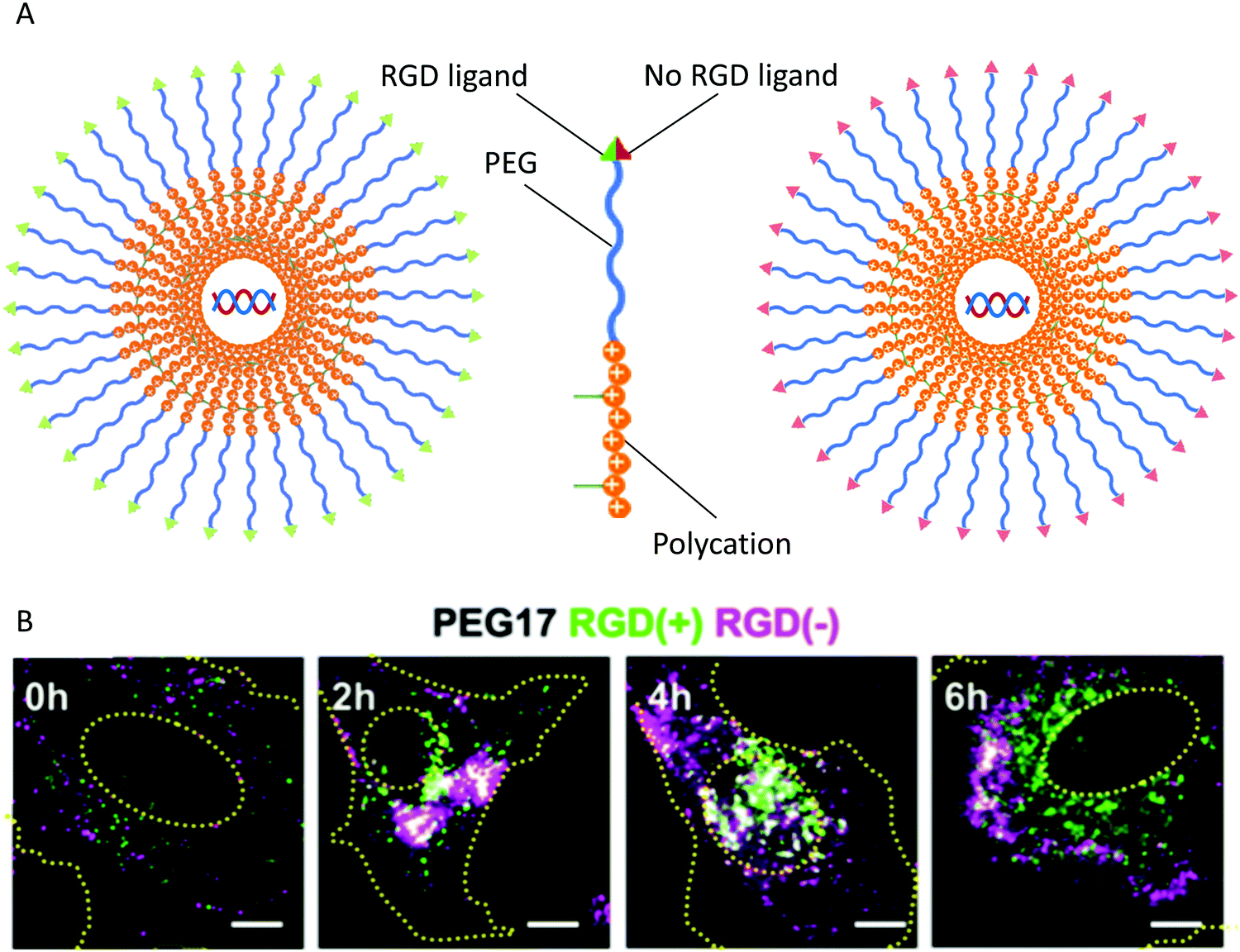 | ||
| Fig. 8 Cellular localization. (A) HeLa cells were co-incubated with micelles modified with PEG shielding and modified with RGD(+) and RGD(−). (B) Distribution of RGD(+) and RGD(−) modified micelles inside HeLa cells. Reproduced from ref. 161 with permission from Elsevier, copyright 2011. | ||
In clinical practice, the effectiveness of chemotherapeutic drugs in the multidrug resistance (MDR) of cancer cells can decrease significantly. In doxorubicin-resistant breast cancer cells, fluorescent therapeutics accumulated only in various organelles, and both in vitro and in vivo treatment effects were lost. As part of photodynamic therapy (PDT), photochemical internalization (PCI) is an approach for specific site-controlled drug and gene release.162,163 The objective of nanoparticle-based PDT/PCI was used to reverse drug resistance. Dendrimer fluorescence-loaded polymeric micelle (DF/m)-mediated PCI was used to control the release of doxorubicin, and the efficacy of DF/m was evaluated in drug-resistant MCF-7 cells and a xenograft model. Photoirradiation promoted the release of doxorubicin from the lysosomes into the nucleus, demonstrating the specific properties of PCI inside the cells. Moreover, the untriggered PCI had the most robust antitumor ability and penetrated deeply into the tumor tissue. After DF/m-regulated PDT, therapy with doxorubicin was improved, indicating that the drawback of PDT, the power of penetration, may be enhanced by PCI.164
The extent and rate of the internalization of drugs into cells is a significant concern for treatment. Hydrophilic drugs tend to internalize slowly and poorly, whereas the internalization of highly lipophilic drugs is too fast. A block copolymer can be optionally adapted to specific drugs to adjust their internalization parameters of incorporation into micelles. A non-cytotoxic and highly lipophilic fluorescent probe are used widely in biology. Researchers compared the uptake of the fluorescent probe CM-DiI (DiI) of micelle-incorporated states with non-micelle-encapsulated states. DiI was effectively incorporated into the spherical micelles of polycaprolactone-b-polyethylene oxide block copolymers of around 25–50 nm in diameter.165 The internalization of DiI by glial cells was observed using confocal microscopy. Encapsulation of DiI into particles significantly reduced the extent and rate of its uptake. The block copolymer's structure and the chemical component, and the solvent applied to prepare the micelles statistically impacted the incorporated probe's loading efficiency into the carriers.
6. Modification of the artificial membrane
The artificial membrane is a vital interface to regulate the interaction of NCs with environmental substances such as macromolecules of peptide, protein, lipid, and DNA, small molecules of theranostics, live cells, or NCs themselves. The interface of NCs can be modified to change the delivery efficiency of NCs and internalized speed by the cells and release behavior of drugs from the particles due to the altered interaction. Targeting delivery of NCs can be improved due to the enhanced selectivity of vesicles based on the interaction of ligand and receptor, antigen and antibody, etc. The uptake of vesicles by the cell can also be accelerated due to the existence of the interaction strength between NCs and cells. Additionally, the charge, composition, and structure of the NC membrane can be changed to tune the release mechanism of loaded molecules from NCs to meet the need for disease treatment.6.1 Targeting delivery
A significant drawback of microscale artificial biologicals is their non-selective interaction with macromolecules, including proteins, lipids, carbohydrates, and cellular surfaces, resulting in their premature removal from circulation. Macrophages of the mononuclear system (MPS) can rapidly clear conventional liposomes, especially Kupffer cells in the liver and spleen macrophages. By shielding the water-exposed surface of the particles with hydrophilic macromolecules, such as PEG, this shortcoming of liposomes can be overcome. Coating PEG on the surface of liposomes creates a stealth barrier.166 The delivery of liposomes can be targeted to specific sites by connecting small molecules, monoclonal antibodies (mAbs), and peptide sequences to PEG on the liposomal surface.167 Horseradish peroxidase (HRP)-loaded liposomes were modified with transferrin (Tf) using PEG as a linker to overcome the blood–brain barrier. The post-insertion process was used to prepare targeting liposomes.168 Although the specific entry routine of the particles into the cells and the intracellular trafficking of Tf-liposomes could not be determined, it was still feasible for enhancing the intracellular concentration of active agents using Tf-modified vesicles. The internalization and intracellular fate of a protein can be determined. The conjugates of drug molecules in particles were protected after injection to avoid degradation in the serum. Moreover, particles can circumvent the efflux transporter of P-gp to increase drug entry into the brain.169 The lipid composition of particles impacts intracellular transport. Also, the size of particles determines the pathway of internalization.170,171Over the past few years, several new technologies have allowed the liposomal content to be released into the cytoplasm but not degraded by the microenvironment of the lysosomes. For instance, liposomes contain several pH-sensitive materials, avoiding the breakdown of the therapeutic molecules in the endosome and lysosome. Soon after internalization, the endosomal pH decreases, and these liposomes disintegrate and release their contents. By changing the ratio of pH-sensitive fusogenic peptides, such as peptizing with multiple Glu-Ala-Leu-Ala (GALA) units, in the particles, endosomal escape might be induced.172 By contrast, targeting liposomes with peptides or proteins to the TfR on brain capillary endothelial cells (BCEC) may fail due to the intracellular inactivity of the therapeutic proteins or peptides. Some active substances that are not sensitive to the lysosome microenvironment and can live under rigorous conditions may be able to treat lysosome diseases using specific carriers.
The folate receptor is another target used to design the targeting liposomes of tumors based on the folate receptor's expression. Low levels of folate receptors are observed in astrogliomas, while little or no folate receptors are usually expressed in normal cells. However, HeLa and kB cells express folate receptors highly. It is still unclear whether it is possible to deliver the drug to tumor cells without or with little expression of folate receptors. Optimizing the targeting ligands by folic acid can enhance the doxorubicin concentration in C6 glioma cells but not in normal cortical cells. Conjugating liposome surfaces can produce immunoliposomes with antibodies to deliver drugs targeted at specific organs or tissues. The high selectivity of mAbs with their particular antigens can further enhance the site-specific targeting delivery of the carriers.173
A non-covalent complexing protocol for the formulation of stealth immunoliposomes is available, which simplifies the adherence of targeting molecules to surface stealth liposomes. A biotinylated PEG phospholipid was employed to bridge the streptavidin-coupled anti-transferrin receptor monoclonal antibody (OX26 mAb) and 150 nm liposomes. OX26-streptavidin has 2–3 biotin-binding sites. Immunostaining observation of the OX26 mAb using fluorescent confocal microscopy demonstrated fluorescent markers of the transferrin receptor on skeletal muscle. The surface modification of micelles can increase the circulation time in the blood. The most used polymer in the surface modification of micelles is the hydrophilic polymer. PEG is the most commonly used of all hydrophilic polymers and can increase persistence inside the blood due to an external hydrated layer on the polymeric micellar surface.174 The hydrophilic PEG hydrated layer is essential for preventing opsonin adherence and removal by the mononuclear phagocytic system.175 The half-life and stealth properties of acetaldehyde-functionalized PEG-b-poly(D,L-lactic acid) (PEG-b-PDLLA) micelles were strengthened by anchoring a peptide ligand with a negative charge on the particles.176
Although the modification of NCs can improve the delivery efficiency of particles to some extent, the meta-analysis results are not ideal for developing an active targeting drug-delivery system. Many factors need to be taken into account when designing and evaluating the features of NCs. Currently, researchers always find some targets in the physiological or pathological tissues and utilize these findings for scanning some ligand or antigen to modify the NCs, which is expected to improve the delivery efficiency of the particles. However, it is impossible to find one target that only exists in the affected cells by far. Additionally, our design of NCs always ignores the influences of dynamic and complicate life behavior of ligand and receptor, or antigen and antibody. Therefore, there is a long way to go to develop an active targeting drug-delivery system.
6.2 Enhanced internalization
As the development of new drugs and molecular targets has become promising for cancer therapy, asparagine–glycine–arginine peptide (NGR)-modified liposomal doxorubicin (TVT-DOX) has been studied.177 The liposome surface conjugated with NGR peptide can target CD13 to inhibit tumor angiogenesis and thus is a promising strategy.178,179 TVT-DOX is associated explicitly with CD13+ cells, and the intracellular release and nuclear uptake of TVT-DOX were enhanced, as shown by fluorescence microscopy in various CD13+ cells.177 Neutral nucleolipids containing carbohydrate components are glycosylated nucleolipids (GNLs) with the addition of the carbohydrate moiety to obtain supplementary molecular recognition capabilities.180 Many glycosylated proteins are markers on the surface of stem cells.181 A novel synthesis of glycosylated nucleolipids with lipidic molecules with two stearic acid molecules was used to observe the interaction of GNL-based liposomes with adipose-derived stem cells (ADSC). Double-stranded GNL could promote the uptake of vesicles by stem cells. Internalization of GNL-decorated liposomes by cells was enhanced over non-nucleoside-decorated vesicles containing glycosylated lipids or common liposomes.182Gene therapy can be used to treat hereditary and acquired diseases. Although viral vectors are incredibly efficient for the insertion of genes, their use may lead to immunogenicity and substantial inherited toxicity. Due to their low toxicity and weak immunogenicity, synthetic or non-viral vectors have become compelling alternatives to viral vectors. The liposome will be available in the clinical setting after the challenge of cellular uptake of conventional or non-specific liposomes has been overcome.183 Researchers have focused on short peptides in drug development because they can be used as a shuttle to transport biologically active macromolecules to cells.184 The primary importance of these peptides is that they can transport large payloads that are several times larger than their size into the cell. Other advantages of these peptides include large inertia, low immunogenicity, and weak toxicity.185 Surprisingly, these peptides can efficiently (∼100% of exposed cells) and quickly (within a few minutes) deliver different payloads into cells and even into the nucleus.186
Common characteristics of these peptides include membrane translocation (MTSs), membrane penetration (CPS), and protein or peptide transduction, such as by protein transduction domains (PTDs). The peptide of polyarginine shows the “Trojan horse” feature of the PTDs and can efficiently and safely deliver drugs when modified on the surfaces of liposomal carriers. The modification of liposomes with the polyarginine peptide enhances cellular internalization and protects the drug's intracellular activities by avoiding degradation by active agents in the lysosome.183
After internalization, the NCs may be wrapped by the cellular membrane, and the new bigger endosomes quickly infused with the lysosome. The fate of the NCs depends on whether lysosome escape can occur or not. The escaped vesicles partly coated with cellular or organelle membrane are translocated into deeper tissue. Before NCs reach the affected cell in the foci, the lost targeting molecule on the surface membrane of the NCs will decrease the internalization of the particles and the delivery efficiency of the carriers to the desired cells. We have no idea how many layers of cells the NCs must penetrate, and we do not know how fast the surface component of NCs is lost. It is a considerable challenge to clarify that the process changes from initial internalization by the outermost cells to the terminal affected cells.
6.3 Controlled release
Artificial cells are water-insoluble artificial vesicles, which can be utilized to identify a specific biological function without inducing the body's defense system. Polymersomes are simple artificial cells with a large compartment to encapsulate active macromolecules, tuning the features, functionality, and membrane stability.187 Biofunctionality refers to the ability to change the biological activity of compounds in the artificial cell in situ. The biofunctionality of artificial cells can be obtained by encapsulating or anchoring a biofunctional component, such as an antibody, drug, enzyme, DNA, or peptide, in or on the cell. The encapsulation of biologically active compounds in these simple cell bodies can be achieved during the formation process.188–190Carboxyfluorescein is a water-soluble biofunctional component that is easily merged into polymersome membrane by dissolving it in the water during formulation. The liberation of fluorescent substances from polymeric materials can be realized by regulating the copolymer composition, especially the hydrophobic block type of the copolymer and the molecular weight. The plasma protein complexities are greater than that of albumin, which can inhibit the release of fluorescent substances. Carboxyfluorescein in the polymer system can go into phosphate buffered saline (PBS) at room temperature or ∼60–80 °C through first-order kinetics, a type of membrane control system. These artificial cells can target specific tissues by accurately complexing antibodies on the surfaces of artificial cells with an antigen-decorated sensor surface.187 The release mechanism can be modulated by combining lipid chains, targeting molecules, and a bridging polymer in membrane constituents under specific conditions.
The four release mechanisms of active molecules from carriers are as follows. First, opposite charges from various termini of biomembrane constituents can neutralize each other with a change in pH.166 The second mechanism involves the pH-dependent hydrolysis of uncharged components integrated into membranes.191 The third mechanism involves the thiolysis of disulfide bonds present in membrane lipids. Thiolysis changes the surrounding environment's redox potential, such as moving from outside the cell surface to the cytoplasm.166 The fourth mechanism is the temperature-regulating release used in thermosensitive liposomes to modulate the release of a drug.192 These mechanisms are summarized in Fig. 9.
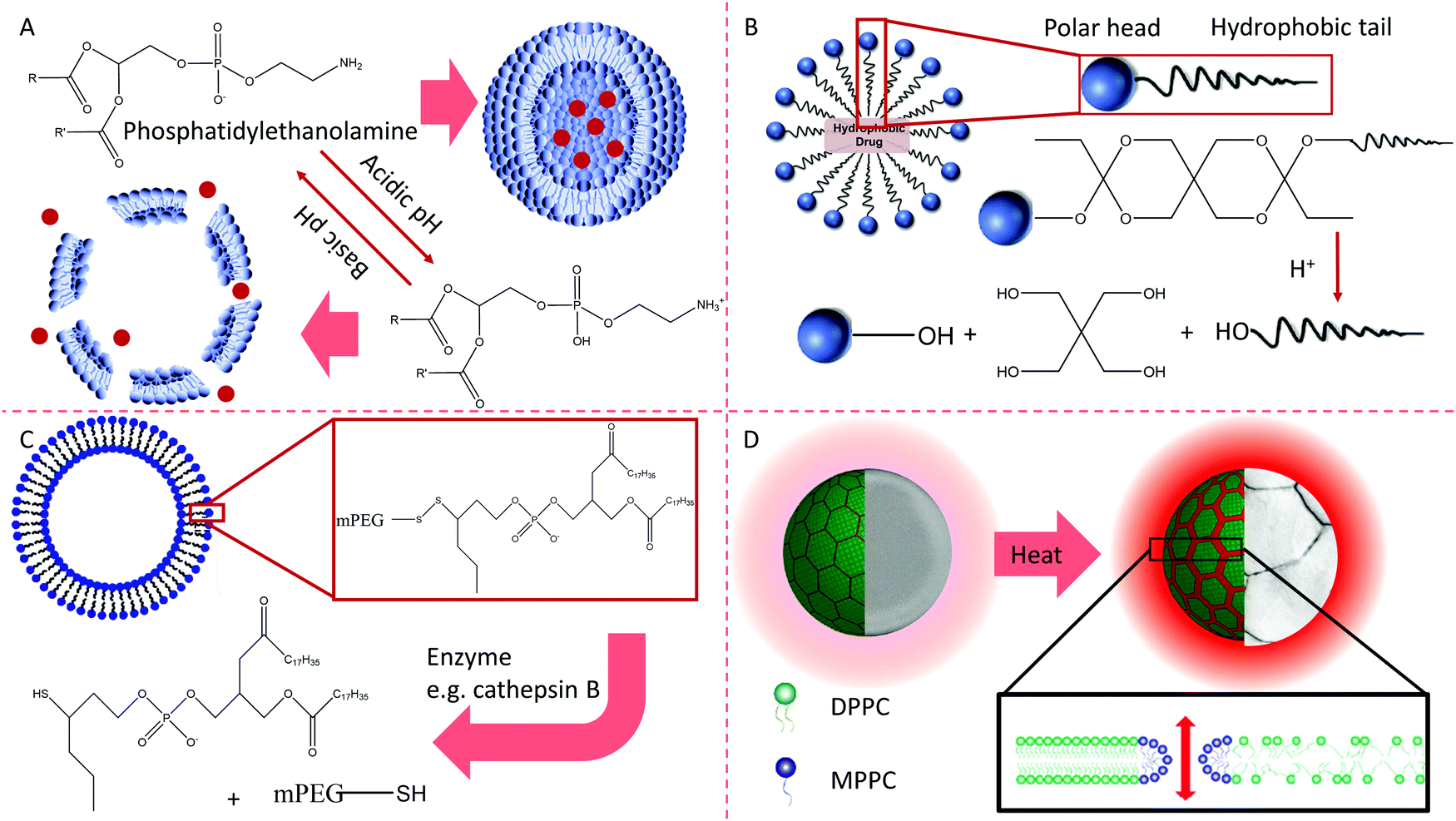 | ||
| Fig. 9 Mechanisms of active molecule release from liposomes. (A) Disassembly of liposome induced by neutralization of oppositely charged biomembrane constituents. (B) pH-dependent hydrolysis of uncharged components integrated into membrane lipids. (C) Liposome disassembly by thiolysis of disulfide bonds on the membrane. (D) Cracking of thermosensitive liposomes by increasing the temperature. | ||
Active molecules can physically insert into or chemically combine with the membrane of polymeric micelles.174 These molecules can also enter the inner compartment of micelles with organic solvents through an oil-in-water emulsion, solid dispersion, and dialysis. Many factors impact how the active molecules are released from particles, such as the loading efficiency, the thickness of the hydrophobic core of micelles, the interaction between the active molecules and the core components.193 The release of the physically entrapped active molecules is regulated by the mechanisms of diffusion and the partition coefficient.166
The responses of NCs to the microenvironment are utilized for designing NCs due to the breakage of the chemical bond between the surface payload and the NC. Although acidic conditions and specific enzymes in the microenvironment of the foci are commonly used triggers to promote the release of drugs from NCs, all of these conditions are met in the lysosome after internalization by the first cell. Avoiding the premature release of drugs in the NCs is tough to produce intelligent NCs. Surface modification of NCs changes the component, structure, charge, thickness, octanol–water partition coefficients, and accessibility of acid, enzyme, and further influencing the release behavior of NCs.
7. Conclusion
In conclusion, the biomembrane, a lamellar structure inlaid with ion channels, receptors, lipid rafts, enzymes, and other functional units, can separate cells and organelles from the environment. The functional units inserted into the membrane and the organelles responsible for phospholipid bilayer inlaid may be influenced by the membrane fluid. The amount, composition, and position of functional units also impact the regional membrane fluid. Many diseases are caused by functional abnormalities of biomembranes, such as MD, neurodegeneration, and ischemia/reperfusion damage. Artificial membranes are also constructed using surfactants, amphipathic substances, modified macromolecules, and polymeric materials. They are loaded with active agents to influence the activities of cells in the body. To improve efficacy of these therapeutics and decrease the adverse effects of drugs, modification with ligands, PEG, and other molecules on the surface of artificial membranes of nanoscale particles is performed. After such amendment, the fate of the nanovesicles changes after interacting with cells, including the efficiency of targeting, internalization, trafficking, and removal from the body. The surface properties of the polymeric membrane are changed to regulate the hydrophilicity/hydrophobicity balance for cell proliferation to facilitate cell expansion during scaffold tissue engineering. Surely, restoring the structure and function of the biological membrane can cure a disease by changing the composition, design, membrane fluid, and the on/off operation of signals. The biological membrane may be a mentor of the artificial membrane, which teaches us how to understand the differences and connections between them and bravely face diseases with more robust tools of diagnosis and treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81671814), the Heilongjiang Provincial Natural Science Foundation (D201031 and ZD2016013), the Scientific Research Fund of Harbin Medical University-Daqing (No. 2018XN-24), the Postgraduate Tutor Foundation of Harbin Medical University-Daqing (DSJJ2015004), the Weihan Yu Outstanding Youth Training Foundation of Harbin Medical University, and the Innovative Fund of Harbin Medical University Graduate Student (YJSCX2015-60HYD). Dr Wang is grateful for the support from the Crohn's & Colitis Foundation (CCF) (Career Award No. 348137), the PhRMA Foundation Research Starter Award (No. RSGTMT17) and the McGee-Wagner Interdisciplinary Research Foundation.References
- Q. Wang, H. Cheng, H. Peng, H. Zhou, P. Y. Li and R. Langer, Adv. Drug Delivery Rev., 2015, 91, 125–140 CrossRef CAS PubMed.
- Y. Pichugin, H. J. Park and A. Traulsen, J. R. Soc., Interface, 2019, 16, 20190054 CrossRef PubMed.
- F. Brandizzi and G. O. Wasteneys, Plant J., 2013, 75, 339–349 CrossRef CAS PubMed.
- Z. Davoudi, N. Peroutka-Bigus, B. Bellaire, M. Wannemuehler, T. A. Barrett, B. Narasimhan and Q. Wang, J. Biomed. Mater. Res., Part A, 2018, 106(4), 876–886 CrossRef CAS PubMed.
- H. Peng, C. Wang, X. Xu, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4354–4360 RSC.
- J. Di, J. Yu, Q. Wang, S. Yao, D. Suo, Y. Ye, M. Pless, Y. Zhu, Y. Jing and Z. Gu, Nano Res., 2017, 10, 1393–1402 CrossRef CAS.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CrossRef CAS PubMed.
- J. M. Sanderson, Mol. Membr. Biol., 2012, 29, 118–143 CrossRef CAS PubMed.
- G. L. Nicolson, Biochim. Biophys. Acta, 1976, 457, 57–108 CrossRef CAS.
- G. L. Nicolson, Biochim. Biophys. Acta, Gen. Subj., 2014, 1838, 1451–1466 CrossRef CAS PubMed.
- L.-L. Huang, W. Nie, J. Zhang and H.-Y. Xie, Acc. Chem. Res., 2020, 53, 276–287 CrossRef CAS PubMed.
- A. Panevska, M. Skočaj, I. Križaj, P. Maček and K. Sepčić, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1284–1292 CrossRef CAS PubMed.
- J. Liang, J. Lyu, M. Zhao, D. Li, M. Zheng, Y. Fang, F. Zhao, J. Lou, C. Guo and L. Wang, Nat. Commun., 2017, 8, 1–13 CrossRef PubMed.
- C. R. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky and A. Kupfer, Nature, 1998, 395, 82–86 CrossRef CAS PubMed.
- G. M. Redpath, M. Ecker, N. Kapoor-Kaushik, H. Vartoukian, M. Carnell, D. Kempe, M. Biro, N. Ariotti and J. Rossy, Nat. Commun., 2019, 10, 1–14 CrossRef CAS PubMed.
- J. Veldman, L. Visser, M. Huberts-Kregel, N. Muller, B. Hepkema, A. Van den Berg and A. Diepstra, Blood, 2020, 136(21), 2437–2441 CrossRef CAS PubMed.
- N. C. Hartman and J. T. Groves, Curr. Opin. Cell Biol., 2011, 23, 370–376 CrossRef CAS PubMed.
- M. L. Kapsenberg, Nat. Rev. Immunol., 2003, 3, 984–993 CrossRef CAS PubMed.
- S. Chen, L. Fang, W. Guo, Y. Zhou, G. Yu, W. Li, K. Dong, J. Liu, Y. Luo and B. Wang, Nat. Commun., 2018, 9, 1–16 CrossRef PubMed.
- H. Hassannia, M. Ghasemi Chaleshtari, F. Atyabi, M. Nosouhian, A. Masjedi, M. Hojjat-Farsangi, A. Namdar, G. Azizi, H. Mohammadi and G. Ghalamfarsa, Immunology, 2020, 159, 75–87 CrossRef CAS PubMed.
- A. Vasaturo, S. Di Blasio, D. G. Peeters, C. C. De Koning, J. De Vries, C. Figdor and S. V. Hato, Front. Immunol., 2013, 4, 417 Search PubMed.
- M. le Maire, P. Champeil and J. V. Moller, Biochim. Biophys. Acta, Gen. Subj., 2000, 1508, 86–111 CrossRef CAS.
- M. G. Ciardo and A. Ferrer-Montiel, Biochim. Biophys. Acta, Biomembr., 2017, 1859(9), 1615–1628 CrossRef CAS PubMed.
- T. Nakagawa, Science, 2019, 366, 1259–1263 CrossRef CAS PubMed.
- T. C. Hwang, S. E. Guggino and W. B. Guggino, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5706–5709 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- A. Alday, H. Ahyayauch, V. Fernández-López, L. Echeazarra, J. Urrutia, O. Casis and M. Gallego, Cell. Physiol. Biochem., 2020, 54, 27–39 CAS.
- H. Peng, N. Poovaiah, M. Forrester, E. Cochran and Q. Wang, ACS Biomater. Sci. Eng., 2014, 1, 37–42 CrossRef PubMed.
- S. Minami, T. Yamamoto, Y. Takabatake, A. Takahashi, T. Namba, J. Matsuda, T. Kimura, J.-Y. Kaimori, I. Matsui and T. Hamano, Autophagy, 2017, 13, 1629–1647 CrossRef CAS PubMed.
- R. Kalluri and V. S. LeBleu, Science, 2020, 367, eaau6977 CrossRef CAS PubMed.
- N. Pfanner, B. Warscheid and N. Wiedemann, Nat. Rev. Mol. Cell Biol., 2019, 20, 267–284 CrossRef CAS PubMed.
- J. Moretti, S. Roy, D. Bozec, J. Martinez, J. R. Chapman, B. Ueberheide, D. W. Lamming, Z. J. Chen, T. Horng and G. Yeretssian, Cell, 2017, 171(809–823), e813 Search PubMed.
- C. I. Dix, H. C. Soundararajan, N. S. Dzhindzhev, F. Begum, B. Suter, H. Ohkura, E. Stephens and S. L. Bullock, J. Cell Biol., 2013, 202, 479–494 CrossRef CAS PubMed.
- S. Doxsey, Nat. Rev. Mol. Cell Biol., 2001, 2, 688–698 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283–296 CrossRef CAS PubMed.
- M. Madureira, N. Connor-Robson and R. Wade-Martins, Front. Neurosci., 2020, 14, 498 CrossRef PubMed.
- Q. Song, B. Meng, H. Xu and Z. Mao, Transl. Neurodegener., 2020, 9, 1–14 CrossRef PubMed.
- Y. Zhan, W. Zeng, G. Jiang, Q. Wang, X. Shi, Z. Zhou, H. Deng and Y. Du, J. Appl. Polym. Sci., 2015, 132(8), 41496–41505 CrossRef.
- W. Li, X. Li, Q. Wang, Y. Pan, T. Wang, H. Wang, R. Song and H. Deng, Carbohydr. Polym., 2014, 99, 218–225 CrossRef CAS PubMed.
- S. Kaushik and A. M. Cuervo, Trends Cell Biol., 2012, 22, 407–417 CrossRef CAS PubMed.
- J. Kaur and J. Debnath, Nat. Rev. Mol. Cell Biol., 2015, 16, 461–472 CrossRef CAS PubMed.
- X. Ma, H. Liu, S. R. Foyil, R. J. Godar, C. J. Weinheimer, J. A. Hill and A. Diwan, Circulation, 2012, 125, 3170–3181 CrossRef CAS PubMed.
- H. Nakatogawa, Nat. Rev. Mol. Cell Biol., 2020, 1–20 Search PubMed.
- L. K. Holland, I. Ø. Nielsen, K. Maeda and M. Jäättelä, Cell, 2020, 181(748–748), e741 Search PubMed.
- L. Yu, Cell Res., 2020, 30, 553 CrossRef PubMed.
- S. Yitzhaki, C. Huang, W. Liu, Y. Lee, Å. B. Gustafsson, R. M. Mentzer and R. A. Gottlieb, Basic Res. Cardiol., 2009, 104, 157–167 CrossRef CAS PubMed.
- J. Farrant, R. Robertson and M. J. Wilkins, Nature, 1953, 171, 401–402 CrossRef CAS PubMed.
- M. Giacomello, A. Pyakurel, C. Glytsou and L. Scorrano, Nat. Rev. Mol. Cell Biol., 2020, 1–21 Search PubMed.
- M. Sukumar, J. Liu, G. U. Mehta, S. J. Patel, R. Roychoudhuri, J. G. Crompton, C. A. Klebanoff, Y. Ji, P. Li and Z. Yu, Cell Metab., 2016, 23, 63–76 CrossRef CAS PubMed.
- D. Rapaport, EMBO Rep., 2003, 4, 948–952 CrossRef CAS PubMed.
- T. Kuwana, M. R. Mackey, G. Perkins, M. H. Ellisman, M. Latterich, R. Schneiter, D. R. Green and D. D. Newmeyer, Cell, 2002, 111, 331–342 CrossRef CAS PubMed.
- D. Hockenbery, G. Nuñez, C. Milliman, R. D. Schreiber and S. J. Korsmeyer, Nature, 1990, 348, 334–336 CrossRef CAS PubMed.
- E. Morselli, L. Galluzzi and G. Kroemer, Cell Res., 2008, 18, 708–710 CrossRef CAS PubMed.
- L. Galluzzi, K. Blomgren and G. Kroemer, Nat. Rev. Neurosci., 2009, 10, 481–494 CrossRef CAS PubMed.
- K. Faelber, L. Dietrich, J. K. Noel, F. Wollweber, A.-K. Pfitzner, A. Mühleip, R. Sánchez, M. Kudryashev, N. Chiaruttini and H. Lilie, Nature, 2019, 571, 429–433 CrossRef CAS PubMed.
- M. J. Torres, K. A. Kew, T. E. Ryan, E. R. Pennington, C.-T. Lin, K. A. Buddo, A. M. Fix, C. A. Smith, L. A. Gilliam and S. Karvinen, Cell Metab., 2018, 27(167–179), e167 CrossRef PubMed.
- G. Battogtokh, Y. S. Choi, D. S. Kang, S. J. Park, M. S. Shim, K. M. Huh, Y.-Y. Cho, J. Y. Lee, H. S. Lee and H. C. Kang, Acta Pharm. Sin. B, 2018, 8, 862–880 CrossRef PubMed.
- D. Piekutowska-Abramczuk, Z. Assouline, L. Mataković, R. G. Feichtinger, E. Koňařiková, E. Jurkiewicz, P. Stawiński, M. Gusic, A. Koller and A. Pollak, Am. J. Hum. Genet., 2018, 102, 460–467 CrossRef CAS PubMed.
- M. Almannai, A. Alasmari, A. Alqasmi, E. Faqeih, F. Al Mutairi, M. Alotaibi, M. M. Samman, W. Eyaid, Y. I. Aljadhai and H. E. Shamseldin, Clin. Genet., 2018, 93, 1097–1102 CrossRef CAS PubMed.
- Y. Yang, H. Wu, X. Kang, Y. Liang, T. Lan, T. Li, T. Tan, J. Peng, Q. Zhang and G. An, Protein cell, 2018, 9, 283–297 CrossRef CAS PubMed.
- A. A. Starkov and F. M. Beal, Nat. Med., 2008, 14, 1020–1021 CrossRef CAS PubMed.
- R. K. Chaturvedi and M. Flint Beal, Free Radical Biol. Med., 2013, 63, 1–29 CrossRef CAS PubMed.
- D. Schenk, R. Barbour, W. Dunn, G. Gordon, H. Grajeda, T. Guido, K. Hu, J. Huang, K. Johnson-Wood and K. Khan, Nature, 1999, 400, 173–177 CrossRef CAS PubMed.
- J. G. Carlton, H. Jones and U. S. Eggert, Nat. Rev. Mol. Cell Biol., 2020, 1–16 Search PubMed.
- J. L. Parker and S. Newstead, Curr. Opin. Struct. Biol., 2019, 57, 127–134 CrossRef CAS PubMed.
- S. Emr, B. S. Glick, A. D. Linstedt, J. Lippincott-Schwartz, A. Luini, V. Malhotra, B. J. Marsh, A. Nakano, S. R. Pfeffer, C. Rabouille, J. E. Rothman, G. Warren and F. T. Wieland, J. Cell Biol., 2009, 187, 449–453 CrossRef CAS PubMed.
- I. Ayala, F. Mascanzoni and A. Colanzi, Biochem. Soc. Trans., 2020, 48, 245–256 CrossRef CAS PubMed.
- M. Tachikawa and A. Mochizuki, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5177–5182 CrossRef CAS PubMed.
- J. Li, D. Tang, S. C. Ireland and Y. Wang, Mol. Biol. Cell, 2019, 30, 478–490 CrossRef CAS PubMed.
- S. K. Levi, D. Bhattacharyya, R. L. Strack, J. R. Austin, 2nd and B. S. Glick, Traffic, 2010, 11, 1168–1179 CrossRef CAS PubMed.
- M. Lowe, Curr. Opin. Cell Biol., 2011, 23, 85–93 CrossRef CAS PubMed.
- J. Klumperman, Cold Spring Harbor Perspect. Biol., 2011, 3, a0051 Search PubMed.
- G. Joshi, M. E. Bekier, 2nd and Y. Wang, Front. Neurosci., 2015, 9, 340 Search PubMed.
- P. Stanley, Cold Spring Harbor Perspect. Biol., 2011, 3, a005199 Search PubMed.
- J. E. Rothman, Protein Sci., 1996, 5, 185–194 CrossRef CAS PubMed.
- V. A. Bankaitis, J. R. Aitken, A. E. Cleves and W. Dowhan, Nature, 1990, 347, 561–562 CrossRef CAS PubMed.
- J. T. Best, P. Xu and T. R. Graham, Curr. Opin. Cell Biol., 2019, 59, 8–15 CrossRef CAS PubMed.
- P. Romani, I. Brian, G. Santinon, A. Pocaterra, M. Audano, S. Pedretti, S. Mathieu, M. Forcato, S. Bicciato and J.-B. Manneville, Nat. Cell Biol., 2019, 21, 338–347 CrossRef CAS PubMed.
- D. Xu, Z. Wang, Y. Xia, F. Shao, W. Xia, Y. Wei, X. Li, X. Qian, J.-H. Lee and L. Du, Nature, 2020, 580, 530–535 CrossRef CAS PubMed.
- A. Pagliuso, C. Valente, L. L. Giordano, A. Filograna, G. Li, D. Circolo, G. Turacchio, V. M. Marzullo, L. Mandrich and M. A. Zhukovsky, Nat. Commun., 2016, 7, 12148 CrossRef CAS PubMed.
- I. J. McGough, R. E. De Groot, A. P. Jellett, M. C. Betist, K. C. Varandas, C. M. Danson, K. J. Heesom, H. C. Korswagen and P. J. Cullen, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- K. Rippe, Curr. Opin. Genet. Dev., 2007, 17, 373–380 CrossRef CAS PubMed.
- F.-Y. Chu, S. C. Haley and A. Zidovska, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10338–10343 CrossRef CAS PubMed.
- P. De Magistris and W. Antonin, Curr. Biol., 2018, 28, R487–R497 CrossRef CAS PubMed.
- M. Borsos, S. M. Perricone, T. Schauer, J. Pontabry, K. L. de Luca, S. S. de Vries, E. R. Ruiz-Morales, M.-E. Torres-Padilla and J. Kind, Nature, 2019, 569, 729–733 CrossRef CAS PubMed.
- M. Falk, Y. Feodorova, N. Naumova, M. Imakaev, B. R. Lajoie, H. Leonhardt, B. Joffe, J. Dekker, G. Fudenberg and I. Solovei, Nature, 2019, 570, 395–399 CrossRef CAS PubMed.
- A. Poleshko, P. P. Shah, M. Gupta, A. Babu, M. P. Morley, L. J. Manderfield, J. L. Ifkovits, D. Calderon, H. Aghajanian and J. E. Sierra-Pagán, Cell, 2017, 171, 573. e514–587. e514 CrossRef PubMed.
- B. J. Mans, V. Anantharaman, L. Aravind and E. V. Koonin, Cell Cycle, 2004, 3, 1612–1637 CrossRef CAS PubMed.
- K. Graumann, E. Vanrobays, S. Tutois, A. V. Probst, D. E. Evans and C. Tatout, J. Exp. Bot., 2014, 65, 6499–6512 CrossRef CAS PubMed.
- J. I. de Las Heras, P. Meinke, D. G. Batrakou, V. Srsen, N. Zuleger, A. R. Kerr and E. C. Schirmer, Nucleus, 2013, 4, 460–477 CrossRef PubMed.
- T. Schlichthaerle, M. T. Strauss, F. Schueder, A. Auer, B. Nijmeijer, M. Kueblbeck, V. Jimenez Sabinina, J. V. Thevathasan, J. Ries and J. Ellenberg, Angew. Chem., Int. Ed., 2019, 58, 13004–13008 CrossRef CAS PubMed.
- L. J. Terry and S. R. Wente, Eukaryotic Cell, 2009, 8, 1814–1827 CrossRef CAS PubMed.
- F. Alber, S. Dokudovskaya, L. M. Veenhoff, W. Zhang, J. Kipper, D. Devos, A. Suprapto, O. Karni-Schmidt, R. Williams, B. T. Chait, M. P. Rout and A. Sali, Nature, 2007, 450, 683–694 CrossRef CAS PubMed.
- F. Alber, S. Dokudovskaya, L. M. Veenhoff, W. Zhang, J. Kipper, D. Devos, A. Suprapto, O. Karni-Schmidt, R. Williams and B. T. Chait, Nature, 2007, 450, 695 CrossRef CAS PubMed.
- T. X. Nguyen, L. Huang, M. Gauthier, G. Yang and Q. Wang, Nanomedicine, 2016, 11, 1169–1185 CrossRef CAS PubMed.
- M. Li, H. Yu, T. Wang, N. Chang, J. Zhang, D. Du, M. Liu, S. Sun, R. Wang and H. Tao, J. Mater. Chem. B, 2014, 2, 1619–1625 RSC.
- J. Wang, P. Li, Y. Yu, Y. Fu, H. Jiang, M. Lu, Z. Sun, S. Jiang, L. Lu and M. X. Wu, Science, 2020, 367(6480), eaau0810 CrossRef CAS PubMed.
- J. H. Senior, Crit. Rev. Ther. Drug Carrier Syst., 1987, 3, 123–193 CAS.
- S. Luli, D. Di Paolo, P. Perri, C. Brignole, S. J. Hill, H. Brown, J. Leslie, H. Marshall, M. C. Wright and D. A. Mann, J. Hepatol., 2016, 65, 75–83 CrossRef CAS PubMed.
- F. Jia, X. Liu, L. Li, S. Mallapragada, B. Narasimhan and Q. Wang, J. Controlled Release, 2013, 172, 1020–1034 CrossRef CAS PubMed.
- H. Peng, X. Liu, R. Wang, F. Jia, L. Dong and Q. Wang, J. Mater. Chem. B, 2014, 2, 6435–6461 RSC.
- N. Wang, W. Zhang, D. Hu, L. Jiang, X. Liu, S. Tang, X. Zhou, T. Liu, X. Tang and Y. Chai, J. Pharm. Sci., 2020, 109(9), 2861–2873 CrossRef CAS PubMed.
- J. Ye, Q. Wang, X. Zhou and N. Zhang, Int. J. Pharm., 2008, 352, 273–279 CrossRef CAS PubMed.
- K. M. Kazi, A. S. Mandal, N. Biswas, A. Guha, S. Chatterjee, M. Behera and K. Kuotsu, J. Adv. Pharm. Technol. Res., 2010, 1, 374–380 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- I. A. Alsarra, A. A. Bosela, S. M. Ahmed and G. M. Mahrous, Eur. J. Pharm. Biopharm., 2005, 59, 485–490 CrossRef CAS PubMed.
- G. Puras, M. Mashal, J. Zarate, M. Agirre, E. Ojeda, S. Grijalvo, R. Eritja, A. Diaz-Tahoces, G. Martinez Navarrete, M. Aviles-Trigueros, E. Fernandez and J. L. Pedraz, J. Controlled Release, 2014, 174, 27–36 CrossRef CAS PubMed.
- V. Weissig, J. Lasch, G. Erdos, H. W. Meyer, T. C. Rowe and J. Hughes, Pharm. Res., 1998, 15, 334–337 CrossRef CAS PubMed.
- J. Lasch, A. Meye, H. Taubert, R. Koelsch, J. Mansa-ard and V. Weissig, Biol. Chem., 1999, 380, 647–652 CAS.
- V. Weissig, C. Lizano and V. P. Torchilin, Drug Delivery, 2000, 7, 1–5 CrossRef CAS PubMed.
- G. G. D'Souza, R. Rammohan, S. M. Cheng, V. P. Torchilin and V. Weissig, J. Controlled Release, 2003, 92, 189–197 CrossRef.
- D. Lyrawati, A. Trounson and D. Cram, Pharm. Res., 2011, 28, 2848–2862 CrossRef CAS PubMed.
- Q. Wang, S. Jamal, M. S. Detamore and C. Berkland, J. Biomed. Mater. Res., Part A, 2011, 96, 520–527 CrossRef PubMed.
- S. J. Williams, Q. Wang, R. R. MacGregor, T. J. Siahaan, L. Stehno-Bittel and C. Berkland, Biopolymers, 2009, 91, 676–685 CrossRef CAS PubMed.
- Q. Wang, Y.-M. Du, L.-H. Fan, H. Liu and X.-H. Wang, J. Wuhan Univ. (Nat. Sci. Ed.), 2003, 49(6), 725–730 CAS.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387–393 CrossRef CAS PubMed.
- S. Miyamoto, H. Teramoto, O. A. Coso, J. S. Gutkind, P. D. Burbelo, S. K. Akiyama and K. M. Yamada, J. Cell Biol., 1995, 131, 791–805 CrossRef CAS PubMed.
- S. P. Authimoolam, A. L. Vasilakes, N. M. Shah, D. A. Puleo and T. D. Dziubla, Biomacromolecules, 2014, 15, 3099–3111 CrossRef CAS PubMed.
- C.-C. Wang, J.-N. Lu and T.-H. Young, Biomaterials, 2007, 28, 625–631 CrossRef CAS PubMed.
- X. Han, Y. Lu, J. Xie, E. Zhang, H. Zhu, H. Du, K. Wang, B. Song, C. Yang and Y. Shi, Nat. Nanotechnol., 2020, 1–10 Search PubMed.
- H. Yin, H. Wang, Z. Li, D. Shu and P. Guo, ACS Nano, 2018, 13, 706–717 CrossRef PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- M. Ludwig, R. Geisler, S. Prévost and R. von Klitzing, Molecules, 2021, 26, 4136 CrossRef PubMed.
- V. P. Torchilin, Annu. Rev. Biomed. Eng., 2006, 8, 343–375 CrossRef CAS PubMed.
- Z. Shi, Z. T. Graber, T. Baumgart, H. A. Stone and A. E. Cohen, Cell, 2018, 175(1769–1779), e1713 Search PubMed.
- J. T. Groves, Dev. Cell, 2019, 48, 15–16 CrossRef CAS PubMed.
- Y. Zhao, S. S. You, A. Zhang, J.-H. Lee, J. Huang and C. M. Lieber, Nat. Nanotechnol., 2019, 14, 783–790 CrossRef CAS PubMed.
- Y. Fang and B. Tian, Nat. Nanotechnol., 2019, 14, 733–735 CrossRef CAS PubMed.
- A. Diz-Muñoz, O. D. Weiner and D. A. Fletcher, Nat. Phys., 2018, 14, 648–652 Search PubMed.
- B. Tian, T. Cohen-Karni, Q. Qing, X. Duan, P. Xie and C. M. Lieber, Science, 2010, 329, 830–834 CrossRef CAS PubMed.
- E. N. Schaumann and B. Tian, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 22897–22898 CrossRef CAS PubMed.
- K.-J. Chang, S. A. Redmond and J. R. Chan, Nat. Neurosci., 2016, 19, 190–197 CrossRef CAS PubMed.
- M. Doktorova, J. L. Symons and I. Levental, Nat. Chem. Biol., 2020, 16, 1321–1330 CrossRef CAS PubMed.
- H. T. McMahon and E. Boucrot, Nat. Rev. Mol. Cell Biol., 2011, 12, 517–533 CrossRef CAS PubMed.
- J. C. Stachowiak, F. M. Brodsky and E. A. Miller, Nat. Cell Biol., 2013, 15, 1019–1027 CrossRef CAS PubMed.
- W. Zhao, L. Hanson, H.-Y. Lou, M. Akamatsu, P. D. Chowdary, F. Santoro, J. R. Marks, A. Grassart, D. G. Drubin and Y. Cui, Nat. Nanotechnol., 2017, 12, 750 CrossRef CAS PubMed.
- X. Li, L. Matino, W. Zhang, L. Klausen, A. F. McGuire, C. Lubrano, W. Zhao, F. Santoro and B. Cui, Nat. Protoc., 2019, 14, 1772–1802 CrossRef CAS PubMed.
- H.-Y. Lou, W. Zhao, X. Li, L. Duan, A. Powers, M. Akamatsu, F. Santoro, A. F. McGuire, Y. Cui and D. G. Drubin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23143–23151 CrossRef CAS PubMed.
- C. S. Hansel, S. W. Crowder, S. Cooper, S. Gopal, M. João Pardelha da Cruz, L. de Oliveira Martins, D. Keller, S. Rothery, M. Becce and A. E. Cass, ACS Nano, 2019, 13, 2913–2926 CrossRef CAS PubMed.
- M. Aragona, T. Panciera, A. Manfrin, S. Giulitti, F. Michielin, N. Elvassore, S. Dupont and S. Piccolo, Cell, 2013, 154, 1047–1059 CrossRef CAS PubMed.
- A. L. Efros, J. B. Delehanty, A. L. Huston, I. L. Medintz, M. Barbic and T. D. Harris, Nat. Nanotechnol., 2018, 13, 278–288 CrossRef CAS PubMed.
- D. S. Peterka, H. Takahashi and R. Yuste, Neuron, 2011, 69, 9–21 CrossRef CAS PubMed.
- J. D. Marshall and M. J. Schnitzer, ACS Nano, 2013, 7, 4601–4609 CrossRef CAS PubMed.
- M. G. Shapiro, K. Homma, S. Villarreal, C.-P. Richter and F. Bezanilla, Nat. Commun., 2012, 3, 1–11 Search PubMed.
- J. L. Carvalho-de-Souza, B. I. Pinto, D. R. Pepperberg and F. Bezanilla, Biophys. J., 2018, 114, 283–288 CrossRef CAS PubMed.
- Y. Jiang, J. L. Carvalho-de-Souza, R. C. Wong, Z. Luo, D. Isheim, X. Zuo, A. W. Nicholls, I. W. Jung, J. Yue and D.-J. Liu, Nat. Mater., 2016, 15, 1023–1030 CrossRef CAS PubMed.
- Y. Fang, Y. Jiang, H. Acaron Ledesma, J. Yi, X. Gao, D. E. Weiss, F. Shi and B. Tian, Nano Lett., 2018, 18, 4487–4492 CrossRef CAS PubMed.
- J. L. Carvalho-de-Souza, J. S. Treger, B. Dang, S. B. Kent, D. R. Pepperberg and F. Bezanilla, Neuron, 2015, 86, 207–217 CrossRef CAS PubMed.
- J. Liu, Y. S. Kim, C. E. Richardson, A. Tom, C. Ramakrishnan, F. Birey, T. Katsumata, S. Chen, C. Wang and X. Wang, Science, 2020, 367, 1372–1376 CrossRef CAS PubMed.
- I. Bekri-Abbes and E. Srasra, J. Nanomater., 2015, 16, 428–435 Search PubMed.
- D. Mezdour, Spectrosc. Lett., 2017, 50, 214–219 CrossRef CAS.
- S. M. Chauhan, M. Jayveer and B. Chakarabarty, J. Nano Adv. Mater., 2016, 4, 9–17 Search PubMed.
- R. Parameswaran, J. L. Carvalho-de-Souza, Y. Jiang, M. J. Burke, J. F. Zimmerman, K. Koehler, A. W. Phillips, J. Yi, E. J. Adams and F. Bezanilla, Nat. Nanotechnol., 2018, 13, 260–266 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- S. E. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- A. Madni, S. Rehman, H. Sultan, M. M. Khan, F. Ahmad, M. R. Raza, N. Rai and F. Parveen, AAPS PharmSciTech, 2021, 22, 1–17 CrossRef PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- M. Oba, S. Fukushima, N. Kanayama, K. Aoyagi, N. Nishiyama, H. Koyama and K. Kataoka, Bioconjugate Chem., 2007, 18, 1415–1423 CrossRef CAS PubMed.
- A. Dirisala, K. Osada, Q. Chen, T. A. Tockary, K. Machitani, S. Osawa, X. Liu, T. Ishii, K. Miyata and M. Oba, Biomaterials, 2014, 35, 5359–5368 CrossRef CAS PubMed.
- F. Mickler, Y. Vachutinsky, M. Oba, K. Miyata, N. Nishiyama, K. Kataoka, C. Bräuchle and N. Ruthardt, J. Controlled Release, 2011, 156, 364–373 CrossRef CAS PubMed.
- S. Kwon, H. Ko, D. G. You, K. Kataoka and J. H. Park, Acc. Chem. Res., 2019, 52, 1771–1782 CrossRef CAS PubMed.
- V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC.
- H. L. Lu, W. J. Syu, N. Nishiyama, K. Kataoka and P. S. Lai, J. Controlled Release, 2011, 155, 458–464 CrossRef CAS PubMed.
- D. Maysinger, O. Berezovska, R. Savic, P. L. Soo and A. Eisenberg, Biochim. Biophys. Acta, Mol. Cell Res., 2001, 1539, 205–217 CrossRef CAS.
- A. Kumari, R. Singla, A. Guliani and S. K. Yadav, EXCLI J., 2014, 13, 265–286 Search PubMed.
- X. Zhang, C. Lin, W. Chan, K. Liu, A. Lu, G. Lin, R. Hu, H. Shi, H. Zhang and Z. Yang, Molecules, 2019, 24, 3332 CrossRef CAS PubMed.
- X. Zhang, J. Xie, S. Li, X. Wang and X. Hou, J. Drug Targeting, 2003, 11, 117–122 CrossRef CAS PubMed.
- K. AboulFotouh, A. A. Allam, M. El-Badry and A. M. El-Sayed, Eur. J. Pharm. Sci., 2017, 109, 503–513 CrossRef CAS PubMed.
- K. He, R. Marsland III, S. Upadhyayula, E. Song, S. Dang, B. R. Capraro, W. Wang, W. Skillern, R. Gaudin and M. Ma, Nature, 2017, 552, 410–414 CrossRef CAS PubMed.
- Y. Jiang, S. Huo, T. Mizuhara, R. Das, Y.-W. Lee, S. Hou, D. F. Moyano, B. Duncan, X.-J. Liang and V. M. Rotello, ACS Nano, 2015, 9, 9986–9993 CrossRef CAS PubMed.
- T. Kakudo, S. Chaki, S. Futaki, I. Nakase, K. Akaji, T. Kawakami, K. Maruyama, H. Kamiya and H. Harashima, Biochemistry, 2004, 43, 5618–5628 CrossRef CAS PubMed.
- A. Schnyder, S. Krahenbuhl, M. Torok, J. Drewe and J. Huwyler, Biochem. J., 2004, 377, 61–67 CrossRef CAS PubMed.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- L. Viitala, S. Pajari, L. Gentile, J. Määttä, M. Gubitosi, J. Deska, M. Sammalkorpi, U. Olsson and L. Murtomäki, Langmuir, 2019, 35, 3999–4010 CrossRef CAS PubMed.
- Y. Yamamoto, Y. Nagasaki, Y. Kato, Y. Sugiyama and K. Kataoka, J. Controlled Release, 2001, 77, 27–38 CrossRef CAS PubMed.
- N.-Q. Shi, Y. Li, Y. Zhang, Z.-Q. Li and X.-R. Qi, Int. J. Nanomed., 2018, 13, 5537 CrossRef CAS PubMed.
- F. Curnis, G. Arrigoni, A. Sacchi, L. Fischetti, W. Arap, R. Pasqualini and A. Corti, Cancer Res., 2002, 62, 867–874 CAS.
- A. Corti, A. M. Gasparri, M. Ghitti, A. Sacchi, F. Sudati, M. Fiocchi, V. Buttiglione, L. Perani, A. Gori and S. Valtorta, Adv. Funct. Mater., 2017, 27, 1701245 CrossRef PubMed.
- G. Godeau and P. Barthelemy, Langmuir, 2009, 25, 8447–8450 CrossRef CAS PubMed.
- J. Stadlmann, J. Taubenschmid, D. Wenzel, A. Gattinger, G. Dürnberger, F. Dusberger, U. Elling, L. Mach, K. Mechtler and J. M. Penninger, Nature, 2017, 549, 538–542 CrossRef PubMed.
- L. Latxague, S. Ziane, O. Chassande, A. Patwa, M. J. Dalila and P. Barthelemy, Chem. Commun., 2011, 47, 12598–12600 RSC.
- I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, Int. J. Pharm., 2008, 354, 39–48 CrossRef CAS PubMed.
- J.-L. Li, Y.-J. Cheng, C. Zhang, H. Cheng, J. Feng, R.-X. Zhuo, X. Zeng and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 5287–5295 CrossRef CAS PubMed.
- B. Oller-Salvia, M. Sánchez-Navarro, S. Ciudad, M. Guiu, P. Arranz-Gibert, C. Garcia, R. R. Gomis, R. Cecchelli, J. García and E. Giralt, Angew. Chem., 2016, 128, 582–585 CrossRef.
- T. Barka, E. W. Gresik and H. van Der Noen, J. Histochem. Cytochem., 2000, 48, 1453–1460 CrossRef CAS PubMed.
- C. Zhang, Z. Chen, W. Li, X. Liu, S. Tang, L. Jiang, M. Li, H. Peng and M. Lian, J. Drug Targeting, 2020, 1–12 Search PubMed.
- M. Hadjidemetriou, S. McAdam, G. Garner, C. Thackeray, D. Knight, D. Smith, Z. Al-Ahmady, M. Mazza, J. Rogan and A. Clamp, Adv. Mater., 2019, 31, 1803335 CrossRef PubMed.
- W.-C. Huang, B. Deng, C. Lin, K. A. Carter, J. Geng, A. Razi, X. He, U. Chitgupi, J. Federizon and B. Sun, Nat. Nanotechnol., 2018, 13, 1174–1181 CrossRef CAS PubMed.
- P. C. Lyon, M. D. Gray, C. Mannaris, L. K. Folkes, M. Stratford, L. Campo, D. Y. Chung, S. Scott, M. Anderson and R. Goldin, Lancet Oncol., 2018, 19, 1027–1039 CrossRef CAS PubMed.
- C. Liu, K. K. Ewert, N. Wang, Y. Li, C. R. Safinya and W. Qiao, Biomaterials, 2019, 221, 119412 CrossRef CAS PubMed.
- D. Needham and M. W. Dewhirst, Adv. Drug Delivery Rev., 2001, 53, 285–305 CrossRef CAS PubMed.
- K. M. Huh, S. C. Lee, Y. W. Cho, J. Lee, J. H. Jeong and K. Park, J. Controlled Release, 2005, 101, 59–68 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |