
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Applications of bioluminescence in biotechnology and beyond†
Aisha J.
Syed‡
 * and
James C.
Anderson
* and
James C.
Anderson
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: Aisha_asrar@hotmail.com
First published on 18th March 2021
Abstract
Bioluminescence is the fascinating natural phenomenon by which living creatures produce light. Bioluminescence occurs when the oxidation of a small-molecule luciferin is catalysed by an enzyme luciferase to form an excited-state species that emits light. There are over 30 known bioluminescent systems but the luciferin–luciferase pairs of only 11 systems have been characterised to-date, whilst other novel systems are currently under investigation. The different luciferin–luciferase pairs have different light emission wavelengths and hence are suitable for various applications. The last decade or so has seen great advances in protein engineering, synthetic chemistry, and physics which have allowed luciferins and luciferases to reach previously uncharted applications. The bioluminescence reaction is now routinely used for gene assays, the detection of protein–protein interactions, high-throughput screening (HTS) in drug discovery, hygiene control, analysis of pollution in ecosystems and in vivo imaging in small mammals. Moving away from sensing and imaging, the more recent highlights of the applications of bioluminescence in biomedicine include the bioluminescence-induced photo-uncaging of small-molecules, bioluminescence based photodynamic therapy (PDT) and the use of bioluminescence to control neurons. There has also been an increase in blue-sky research such as the engineering of various light emitting plants. This has led to lots of exciting multidisciplinary science across various disciplines. This review focuses on the past, present, and future applications of bioluminescence. We aim to make this review accessible to all chemists to understand how these applications were developed and what they rely upon, in simple understandable terms for a graduate chemist.
 Aisha J. Syed | Aisha J. Syed received her BSc in Chemistry from the University of Birmingham in 2015. She then studied for a PhD in Organic Chemistry and Chemical Biology at University College London with Prof. Jim Anderson on the synthesis and bioluminescence properties of infraluciferins which she received in 2019. She is currently a Research Fellow in the labs of Prof. Andrew Wilson and Prof Adam Nelson at the University of Leeds. Her research interests include using synthesis and related chemical tools for visualising and manipulating biological processes including molecular imaging and protein–protein interactions. |
 James C. Anderson | Jim Anderson studied at Imperial College, receiving his PhD in 1990 working with Professor S. V. Ley FRS. After postdoctoral study with Professor D. A. Evans at Harvard University, he started his independent career at the University of Sheffield in 1993. He moved to the University of Nottingham in 1999 and to his current position at University College London in 2009. His work has been mainly concerned with asymmetric reaction methodology and total synthesis. At UCL these have been applied to the rational design of red shifted luciferins, which has led to the development of infraluciferin. |
1. Introduction
Bioluminescence is the fascinating natural phenomenon by which living creatures produce and emit light. In nature this has been evolutionarily conserved primarily in marine organisms, some species of bacteria, fungi and terrestrial insects for various purposes such as to hunt prey, ward-off predators, and attract mates.1–3 Since 1667 when one of the earliest scientific records of the study of bioluminescence was made by Robert Boyle, many researchers have contributed to our understanding of the mechanisms that govern bioluminescence and how this natural phenomenon can be tailored for targeted use as a powerful tool in biotechnology for the visualisation, imaging, and control of biochemical processes.4Naturally occurring light originates from two main kinds of systems – bioluminescent systems which comprise of distinct luciferase enzymes and luciferin moieties, and photoproteins in which the light-emitting chromophore is part of the protein itself and light emission is triggered by changes in the protein's environment. The discovery of the mechanisms underpinning photoproteins such as the green fluorescent protein (GFP), aequorin, kaede and pholasin and their subsequent applications have been previously reviewed and will not be discussed in this review.5–8
Whilst there are more than 40 known bioluminescent systems, the structures of the luciferin and luciferase have only been elucidated for 11 of them. The quest for the mechanistic characterisation of luciferin and luciferase pairs is an active area of research as is the search for new pairs.9–11 The bioluminescent reaction generally requires a luciferase enzyme, its luciferin substrate and an oxidant which is often molecular oxygen. Some systems require energy in the form of ATP or NADH as well. One of the earliest luciferin structures to be elucidated were those of D-luciferin found in fireflies, reported in the mid 1900s.12 About twenty years later the luciferin coelenterazine and its luciferase were discovered from the deep-sea shrimp Oplophorus gracilirostris.13
Since then, due to the low toxicity, high quantum yield and high sensitivity of both of these reactions, these molecules and their luciferase enzyme partners have found wide use as in vitro reporters of analytes and metabolites – for example the firefly luciferase (FLuc) and D-luciferin system are widely used in biochemical assays to determine ATP levels. The last decade or so has seen luciferins and luciferases to reach previously uncharted applications fuelled by great advances in protein engineering, synthetic chemistry, physics and light capture technology. We will first cover some of the basic qualities and characteristics of the most widely used natural luciferin/luciferase pairs, with a brief mention of their uses as well as their shortcomings that make them less than ideal candidates for certain applications. This then leads up to the developments in synthetic luciferins and engineered luciferases and their improved properties and uses. The applications of bioluminescence are then extensively discussed including ATP sensing, hygiene control in the fish and milk industries, mapping pollution in ecosystems using bioluminescence based assays, culture and heritage in the form of art work preservation, the sensing of pH, metal ions, membrane potential, drug molecules, other metabolites, gene assays, the detection of protein–protein interactions, high-throughput screening in drug discovery and in vivo imaging of tumours as well as infections. Apart from sensing applications, the discussion will then move to how new applications are now trying to make use of the light from bioluminescence for various purposes such as to effect healing in the form of bioluminescence based photodynamic therapy (PDT)14 or using the light for bioluminescence-induced photo-uncaging of small molecules,15,16 and the use of bioluminescence to control neurons.17 Exciting recent blue-sky research is also discussed such as the engineering of a light emitting plants of various types.18 We aim to make this review accessible to all chemists to understand how these applications were developed and what they rely upon, in simple understandable terms for a graduate chemist. Hence, a stepwise journey across the various applications of bioluminescence has been taken, and how protein-engineering, synthetic chemistry and chemical biology tools have fed into the development of novel, state-of-the-art applications.
Whilst bioluminescence has found utility in various fields including medicine, biology, physics and engineering and led to exciting multidisciplinary science across all of them, we also discuss the current limitations in the state of the art, as well as prospects and future directions for the research community for the future development of applications of the luciferin–luciferase reaction from a chemical biology tool to something more widely used in industry, daily life or in a clinic.
2. Natural luciferin, their luciferases and mechanisms
The structures of D-luciferin and coelenterazine, and their respective luciferases were some of the earliest luciferin–luciferase pairs to be elucidated and their enzymes synthetically expressed.12,19–25 Hence, these bioluminescence systems are used in the vast majority of applications of bioluminescence by virtue of our greater understanding of their mechanisms of action and ease in preparing the reagents needed for their assays.In this section the mechanisms of bioluminescence with respect to D-luciferin, coelenterazine and bacterial luciferin have been looked at in greater detail as these luciferins and their respective luciferases have found the most utility in various applications. The remaining eight known luciferin–luciferase pairs have been included and discussed briefly towards the end of the section, as their understanding and inclusion would help foster future applications.
2.1 D-Luciferin
Although each bioluminescent insect species has a distinct luciferase enzyme, they all share the same substrate D-luciferin which is found in around 40 different species of fireflies, click-beetles and the rail-road worms.12,26–33 In the bioluminescence reaction, the 62 kDa insect luciferase (Fluc)19 catalyses the oxidation of D-luciferin 1 in two distinct steps – the activation of the carboxyl group of 1 through adenylation, followed by the oxidation of the luciferyl-adenylate 2 to form oxyluciferin 4 as an excited state anionic species through a dioxetanone intermediate 3 (Fig. 1).34–36 The excited state oxyluciferin species 4 relaxes to its ground state 5 by giving off a photon of light. Around 20% of luciferyl adenylate undergoes a dark-side oxidation to form H2O2 and dehydro-luciferyladenylate 6 which is a strong inhibitor of luciferase (Ki = 3.8 ± 0.7 nM),37 and this limits the quantum yield of the bioluminescence reaction.38 Protonated 5, oxyluciferin itself is also an inhibitor of the luciferase enzyme (Ki = 500 ± 30 nM).37 Due to the dark-side reaction and other energy losses, firefly luciferase catalyses light emission from D-luciferin with a maximum quantum yield of 41% at pH = 8.5.39 | ||
| Fig. 1 The mechanism of firefly bioluminescence. | ||
The activation of D-luciferin by ATP prior to oxidation is a unique feature of D-luciferin bioluminescence that is not shared by other luciferins such as coelenterazine. This feature bestows a variety of unique benefits upon the D-luciferin/firefly luciferase system that are not enjoyed by other bioluminescent systems. For example, as D-luciferin requires activation in the form of adenylation, it is less susceptible to auto-oxidation and more stable in solution, leading to less background chemiluminescence.40 ATP is widely known as the ‘energy currency’ of a cell and found in varying amounts in virtually all eukaryotic and prokaryotic cells.41 As the D-luciferin/Fluc assay produces ATP dependent light output, this assay has been widely adapted to measure ATP concentration in the double digit nanomolar region in various systems for both quality control and hygiene, often to determine the extent of bacterial contamination.42,43 It is also used to monitor both ATP-forming reactions44–47 and ATP-degrading reactions.48–50 With the advent of more sensitive luminometers and improved assay techniques, ATP levels as low as attomole levels – corresponding to a single bacterial cell – can be routinely detected now using commercially available ATP bioluminescence reagents and kits.42 More details on these assays are discussed in Section 4.1.
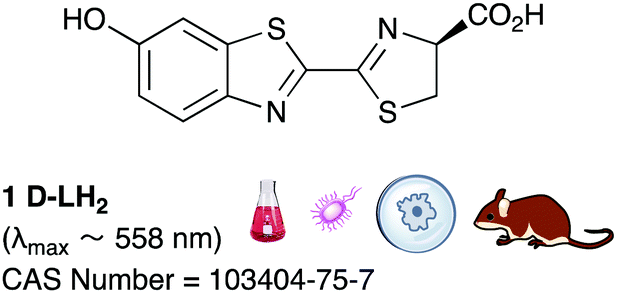
It is known that D-luciferin has greater aqueous solubility51 and lower toxicity than coelenterazine.52 Moreover D-luciferin has the most impressive quantum yield amongst all known luciferin/luciferase systems (41.0 ± 7.4% compared to 15–30% for most luciferase/luciferin pairs)39,53 and the longest emission wavelength amongst them (λmax = 558 nm39), it is more ideal for imaging applications where blood is involved. Haemoglobin and tissues absorb light most strongly of wavelength <500 nm.54 Mammalian cells and other biological entities of interest such as bacteria, fungi, protozoa and viruses can be genetically encoded with the luciferase gene of interest55,56 and introduced in to a small mammal for proliferation, tracking and imaging. From naturally occurring luciferins and luciferases, D-luciferin and the firefly luciferase from the North American firefly, Photinus pyralis are the most common pair used for in vivo imaging (Table 1).57
D-Luciferin is commercially available in its carboxylic acid form (CAS Number: 2591-17-5) as well as its sodium salt (CAS Number: 103404-75-7). It can also be readily prepared in up to >50 mg scale batches using multi-step organic synthesis – preparations up to 50 mg scale have been reported although it can be envisaged that larger batches of >200 mg could be readily prepared too.58,59 The plasmids and vectors used to express firefly luciferase are also readily available.
| PpyLuc | |
|---|---|
| Wavelength | 558 nm |
| Optimum pH | 7.8 |
| Optimum temperature | 28 °C |
| Quantum yield39 | 41 ± 7% at pH 8.5 |
Although the light from D-luciferin bioluminescence is red-shifted compared to that from other naturally occurring luciferins, it is still strongly absorbed by blood and tissue. Near infra-red light (650–900 nm) has better penetration through blood and tissue.60 Whilst there is a portion of light in the broad emission spectrum of D-luciferin within this desirable range of wavelengths, more red-shifted light would allow more sensitive imaging.54
There are other factors as well that make D-luciferin a less than ideal candidate for bioluminescence imaging. For example, D-luciferin has modest cell permeability61 and hence has to be dosed in large amounts for in vivo experiments.62 Studies in which the 14C-labelled radioactive D-luciferin substrate has been used have also demonstrated the inhomogeneous bio-distribution of the substrate in rodents.63 Moreover, there is poor uptake of the probe in some organs of interest such as the brain.64 Whilst D-luciferin is capable of emitting light of different wavelengths on interaction with various mutants of Fluc;65 the range of wavelengths emitted does not render it suitable for in vivo multi-parametric imaging particularly in deep tissue imaging because the most red-shifted λmax obtained by D-luciferin and Fluc mutants is 620 nm.66,67 The light at this wavelength suffers from absorption, attenuation and scatter in tissues making spectral unmixing from different luciferases more challenging.68 Some of these set-backs have been overcome with the design, synthesis and testing of novel D-luciferin analogues, which have led to brighter, red-shifted emission in some cases.69 Details on these can be found in Section 3.1.
There has also been a strong case for engineering the firefly luciferase enzyme to obtain better properties more suited for various applications. For example, in order to detect a variable ATP concentration, it is essential that the luciferase concentration remains constant during the duration of the measurement and luciferase is not inactivated by other factors such as pH, temperature, the concentration of metal ions, detergents and other soluble proteins such as bovine serum albumin, in the assay mixture. The optimum pH for wild-type luciferase activity is pH 7.8.70,71 A workable pH of 6–8 can be used for analytical applications using the wildtype enzyme.46 However, the colour of emission from some wild-type firefly luciferases such as P. pyralis and H. parvula changes from yellow-green to red when the pH is lowered below optimum.72–74 The reason for this has been proposed to be the disruption of a key hydrogen-bonding network involving key water molecules and amino acid residues in the enzyme's active site around the oxyluciferin emitter. It is believed that disruption at lower pH enhances the delocalisation of the phenolate negative charge that increases red shifted emission.75 This rendered these wild-type luciferases not as useful for comparative bio-analytical applications where often photons of a specific wavelength of light are being quantified. Moreover, most wild-type luciferases are thermolabile at even room temperature, whilst work on mammalian cells often requires temperatures of around 37 °C.76 Indeed, wild-type luciferase has a half-life of only 3–4 h in mammalian cells due to both thermal inactivation and proteolysis.77 Consequently, there has been significant work done in protein engineering to create mutant firefly luciferases of greater pH stability, thermostability and proteolytic stability. Indeed, a small change in the sequence of the enzyme can lead to changes significant changes in the emission and properties of the enzyme. For example, the mutant S286N of the Japanese Firefly Luciferase has red-shifted emission at λmax = 605 nm compared to the wild-type at λmax = 560 nm (Fig. 2A and B). This is because Ser 286 is involved in a key hydrogen-bonding network that is disrupted when it is mutated to an asparagine residue. Moreover, this also causes a change in the conformation of Ile 288 close to the emitter making the hydrophobic pocket more flexible (Fig. 2C and D). For further details on engineered Fluc enzymes please see Section 3.1.
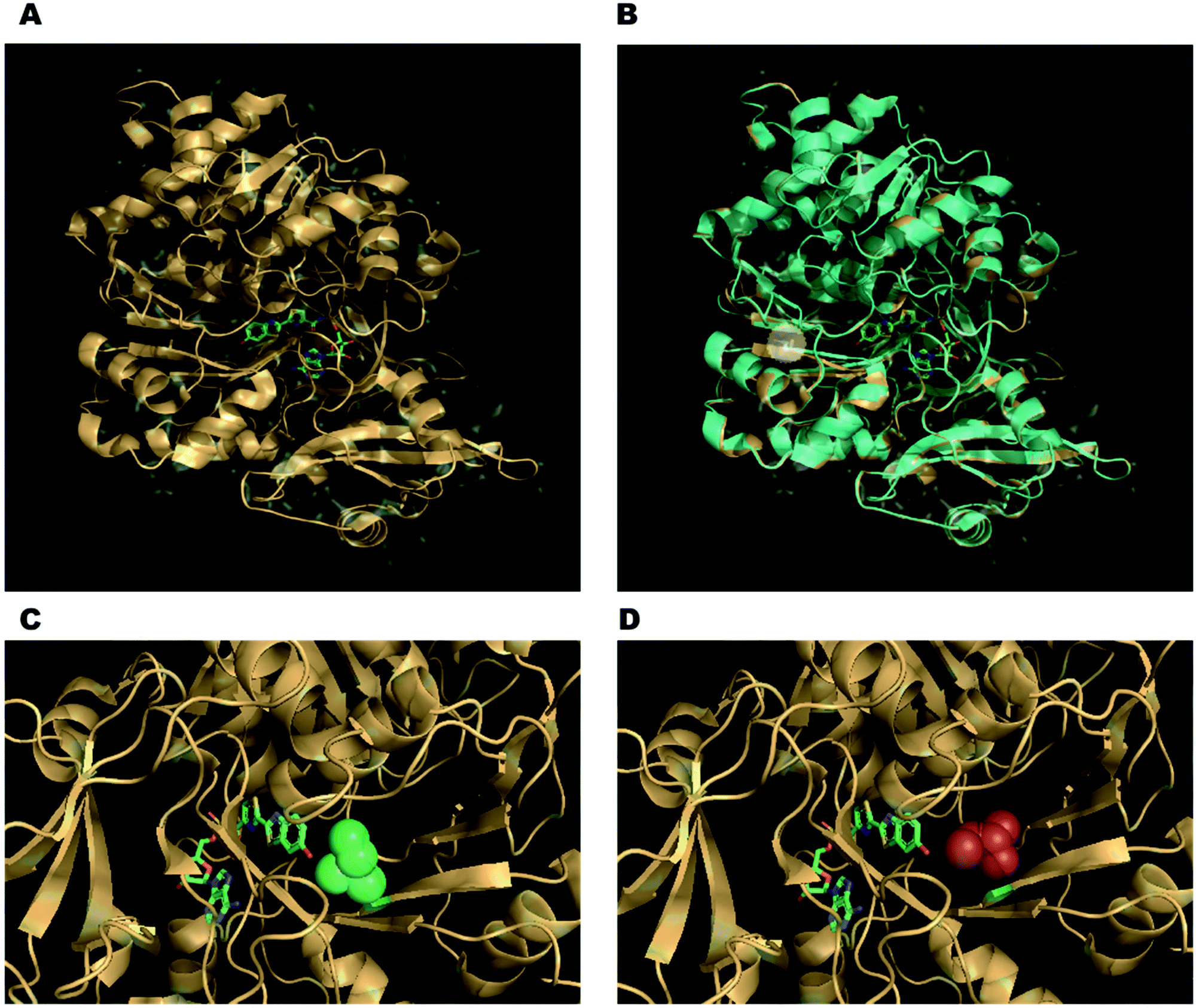 | ||
| Fig. 2 (A) The co-crystal structure of the Japanese firefly luciferase (Luciola cruciata) with a high-energy intermediate analogue, 5′-O-[N-(dehydroluciferyl)-sulfamoyl]adenosine (DLSA) – PDB code: 2D1S. (B) Overlay of complex of Luciola cruciata luciferase with DLSA (PDB code: 2D1S – coloured in light orange) with mutant S286N complexed with DLSA (PDB code: 2D1T – coloured in cyan). The side-chains of the residues of the key mutation are shown as sticks and highlighted in the white circle. (C) The conformation of Ile 288 (green spheres) in wild-type Luciola cruciata. (D) The changed conformation of Ile 288 in the S286N mutant (red spheres), leading to a more flexible hydrophobic pocket. | ||
2.2 Coelenterazine
Coelenterazine is found in several marine creatures including copepods and the deep-water shrimp, and is the other most commonly used luciferin.13,78 Coelenterazine is the substrate for around 15 different naturally occurring luciferases.24,25,79–84 From these, the luciferases from Renilla luciferase (Rluc), Gaussia luciferase (Gluc) and Metridia longa luciferase (Mluc) were one of the earliest to be cloned and have found most utility in biotechnological applications. The bioluminescent reaction of coelenterazine also involves an enzymatically catalysed oxidation. Coelenterazine 7 is converted to an excited state coelenteramide oxyluciferin 10 through a dioxetanone intermediate 9. The excited state oxyluciferin relaxes to its ground state to emit a photon of blue light of wavelength 454–493 nm, dependent upon the enzyme (Fig. 3). This bioluminescent reaction is not dependent on ATP. | ||
| Fig. 3 The mechanism of coelenterazine bioluminescence. | ||
Coelenterazine is commercially available (CAS Number: 55779-48-1) and can be synthesised in up to >200 mg scale batches using multi-step synthesis.85 However, coelenterazine is a larger molecule than D-luciferin, has poor water solubility, greater toxicity and is also susceptible to auto-oxidation leading to chemiluminescence in solution as it does not need activation in the form of adenylation.40 Moreover, coelenterazine has also been reported to be transported into cells through other mechanisms. For example, the multidrug resistance P-glycoprotein (MDR1 Pgp) was reported to mediate the transport of coelenterazine into cell lines. This led to greater amounts of coelenterazine being transported into cancer cells expressing greater quantities of MDR1 Pgp. This could lead to an inaccurate representation of tumours in small mammal in vivo imaging i.e. tumours that do not express MDR1 PGp would not be detected.86 Coelenterazine gives out blue light which is strongly absorbed by blood and tissue, making it a poor candidate for in vivo imaging when used alone without the red-shifting effects of BRET.54 To overcome these short-comings, several synthetic coelenterazine analogues have been prepared, of which some are commercially available. Details of these analogues are presented in Section 3.2.
In terms of size, both Rluc (∼34 kDa), Gluc and Mluc (both ∼20 kDa) are smaller than Fluc (∼62 kDa). This makes them more suitable for applications involving small vectors and/or proteins. In the sea pansy, Renilla reformis the luciferase Rluc is closely associated with a green fluorescent protein (GFP) and the blue light emitted by the luciferase is coupled through resonance energy transfer to the fluorophore of the GFP allowing it to form an excited-state species which emits a photon of green light (λmax 510 nm).87 This principle of resonance energy transfer led to the development of bioluminescence resonance energy transfer (BRET) and several associated applications where the BRET light emission has be used as a measure for the spatial proximity of two proteins.88
Most coelenterazine utilising luciferases possess several disulphide bonds in the protein structure which often help in protein-folding and confer these enzymes with greater thermal stability than firefly luciferases. However, these disulfide bonds also make the enzymes sensitive to any reducing agents in buffer solutions as it is vital for the cysteine residues to be in the correct oxidation state for native protein folding. Moreover, the optimum conditions for activity of these luciferases often mimic their natural marine environment. So, most of these luciferases are halophilic reflecting the saltiness of seawater and some forms – for example the Mluc2 isoform of Metridia longa luciferase – is psychrophilic i.e. has a very low optimum temperature reflecting the low temperatures at the bottom of the ocean (Table 2).89 Copepod luciferases Gluc and Mluc are the only known luciferases that are naturally secreted from eukaryotic cells.90 These unique properties make them potentially useful for a unique set of applications such as high throughput studies as cell-lysis is not required.
2.3 Bacterial luciferin
Besides firefly bioluminescence and coelenterazine based marine bioluminescence, bacterial bioluminescence has also received considerable attention in terms of applications over the past half century or so.57,94,95 All known bioluminescent bacteria are Gram-negative, facultative anaerobic94 and most are symbiotic and found in their host organisms such as the Hawaiian Bobtail squid (Aliivibrio fischeri)96 or terrestrial roundworms (Photorhabdus luminescens).97 Although some are free-living species such as Vibrio harveyi98 and Alteromonas hanedai.99 Although bioluminescent bacteria belong to various species and genre, the biochemical machinery of bacterial light emission is globally conserved between species. In bacteria, all the enzymes required for bioluminescence are completely genetically encoded for in the luxCDABEG operon.100 An operon is a functioning unit of DNA containing a cluster of genes under the control of a single promoter.101 A promoter is a sequence of DNA to which proteins bind that initiate transcription of a single RNA from the DNA downstream of it.102 The luxA and luxB genes encode for the 40 kDa α subunit and 37 kDa β subunit of the bacterial luciferase respectively. The luxC, luxD and luxE genes encode enzymes involved in the synthesis of the aldehyde co-factor, and luxG encodes for flavin reductase which participates in flavin mononucleotide (FMN) turnover.94,103The mechanism of bacterial bioluminescence is well understood (Fig. 4).104 Flavin mononucleotide (FMN) reductase reduces oxidised FMN 12 to form reduced FMN 13 which reacts with molecular oxygen to form an FMN-hydroperoxide species 14, possibly through a single electron transfer (SET) process as suggested for adenylated D-luciferin and oxygen.35,105,106 In the absence of long-chain aldehyde 15, intermediate 14 is fairly stable and was characterised by UV-vis absorption and NMR spectroscopy.107,108 It has been proposed that the long-chain aldehyde 15 adds to 14 to form the FMN-4a-peroxyhemiacetal species 16 which collapses to form the carboxylic acid 17 and FMN-4a-hydroxide 18 in an excited state.106 The exact mechanism of formation of the excited-state species is still under debate. A few different mechanisms have been proposed for the breakdown of FMN-4a-peroxyhemiacetal species 16, including the proposed formation of a dioxirane intermediate109 or a flavin-mediated intra-molecular electron transfer mechanism initiating the collapse of 16. There is greater computational110 and experimental data111 to support the intra-molecular flavin-mediated electron transfer from the 5-N of the isoalloxazine ring to the distal oxygen atom of FMN-4a-peroxyhemiacetal species 16. Light emission occurs when the flavin mononucleotide species FMN-4a-hydroxide 18 relaxes to its ground state 19 and emits a photon of bluish-green light (λmax 490 nm) (Table 3). The long-chain aldehyde is oxidised to the corresponding carboxylic acid as part of the cycle.
In essence, the entire light emission machinery including luciferase production, luciferin biosynthesis and recycling machinery is genetically encoded for together in a single operon, so transgenic auto-luminescence is possible. Exogenous administration of luciferin is not required in any imaging applications, unlike what is observed in firefly bioluminescence imaging and coelenterazine bioluminescence imaging. This is of great value in synthetic biology, especially as the administration of expensive and unstable luciferins in some hosts such as plants is difficult.112,113 However, considerable work and optimisation of the genetic make-up was required to make bacterial operons useful in eukaryotic cells and machinery. Although in early studies only luxA and luxB were expressed in plant cells such as tobacco and carrots to form functional luciferase in them, autoluminescent tobacco plants were later designed using the operons form Photobacterium leighognathi lux operon.114,115 Although previously, the bacterial lux operon had been used to produce a weakly autoluminescent human cell line (HEK293 cells), more recently codon-optimised lux sequences have been developed that produce bright, autonomous bioluminescence in mammalian cells.116,117 Further details of the codon-optimisation and enzyme engineering involved are presented in Section 3.3.
Bioluminescent bacteria are often used as biosensors in ecotoxicological studies as these can often be tuned to detect concentrations of a variety of different organic (alcohol, carboxylic acids, aromatic compounds) and inorganic substances (heavy metal ions).118–120 Moreover, bioluminescent bacteria are also being used to create unique glowing artwork and proposals have been suggested to use them in indoor aquariums as part of aesthetic architecture in skyscrapers.121
As both bacterial luciferase and luciferin are encoded for in the lux operon, this has led to limited mutations in the native luciferase structure and no mutations to the luciferin, although shorter-chain aldehydes are also tolerated and as expected result in no change in wavelength of the light emitted.105
2.4 Other known luciferins
There are over 30 known bioluminescent systems but the luciferin–luciferase pairs of only 11 systems have been characterised to-date with a handful of systems only partially characterised.10 Of these D-luciferin, coelenterazine and the bacterial bioluminescent systems have found most utility in biotechnological applications, by virtue of a more comprehensive understanding of them. The table below (Table 4) details succinctly the other known bioluminescence systems and some of their applications.| Luciferin | Luciferase size | Wavelength (λmax) | Required components | Key facts |
|---|---|---|---|---|
| a This is the proposed structure for the luciferin although other proposals suggest that the light emitting species could be a flavin moiety as is the case in the bacterial bioluminescent system. | ||||
|
∼61 kDa22 | 452 nm22 | Luciferin, luciferase and O2 | • Bioluminescence mechanism is very similar to that of coelenterazine |
| • It is a secreted luciferase | ||||
| • Several applications exist which will be discussed in depth in later sections. | ||||
| • Quantum yield = 28%125 | ||||
|
∼29 kDa126 | 520 nm126 | Luciferin,127 luciferase and O2 | • The only eukaryotic auto-luminescent system. |
| • So far used to create GM glowing plants.128 | ||||
| • Quantum yield not determined. | ||||

|
∼135 kDa129 3 distinct domains that are each catalytically active | 476 nm129 | Luciferin,130 luciferase and O2 at pH 6.3 | • No chemical synthesis of the luciferin reported to-date |
| • Fewer than half a dozen known applications of the luciferase exist. Examples include use as a reporter enzyme for in vitro gene expression,131 measuring quantities of cell-surface expressed membrane proteins,132 and pregnancy-specific glycoproteins in HeLa cells to investigate cell senescence.133 | ||||
| • Quantum yield not determined. | ||||

|
∼600 kDa134 | 468 nm135 | Luciferin,138 luciferase and O2 | • No chemical synthesis of the luciferin reported to-date and a pure sample of luciferin not isolated either – structure elucidated through crude luciferin sample and oxyluciferin sample |
| • Cross-reactivity is observed between krill luciferin and dinoflagellate luciferase and dinoflagellate luciferin and krill luciferase136 | ||||
| • No known applications to-date | ||||
| • Quantum yield not determined. | ||||
|
∼173 kDa137 | 536 nm138 | Luciferin,130 luciferase, purple protein and O2 | • The structure of the light-emitting species is still unknowna |
| • Synthetic analogues of the luciferin have been reported that are less active in bioluminescent emission139–141 | ||||
| • Quantum yield = 17%142 | ||||
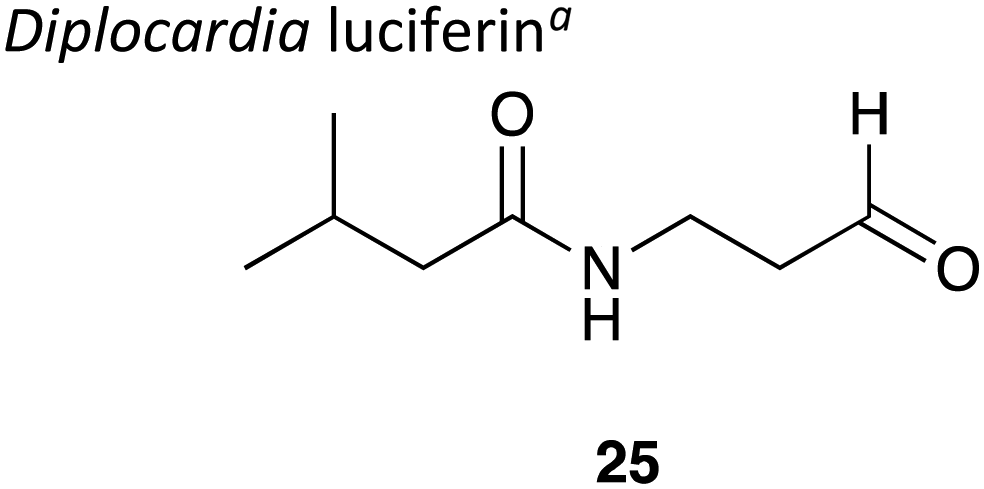
|
∼300 kDa143 copper dependent | 500–530 nm144 | Luciferin,144 luciferase and H2O2 | • Used in analytical tools to detect H2O2 levels and peroxidase activity.145 |
| • A major limitation on its use is that the luciferase has not been cloned so needs to be sourced directly from the earthworms themselves.146 | ||||
• Quantum yield = 3%a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 146 146 |
||||
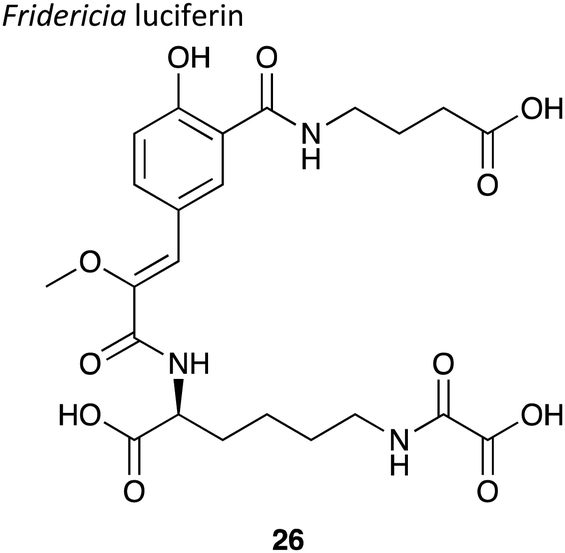
|
∼35 kDa147 | 478 nm147 | Luciferin,148,149 luciferase, Mg2+ ions, O2 and ATP | • A major limitation on its use is that the luciferase has not been cloned so needs to be sourced directly from the earthworms themselves.146 |
| • Quantum yield not determined. | ||||
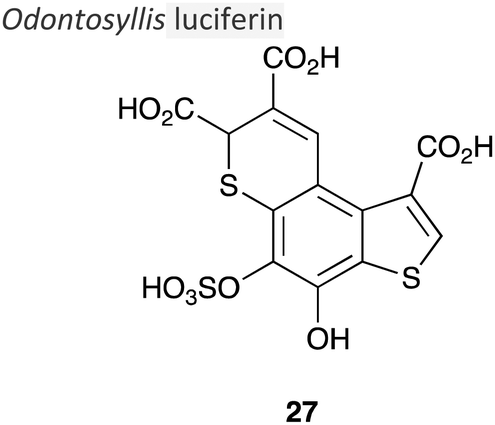
|
∼35 kDa150 | 510 nm150 | Luciferin, luciferase and O2 | • Luciferin structure was reported in late 2019.11 |
| • Quantum yield not determined. | ||||
| • No known applications to-date. | ||||
The discovery and characterisation of yet-unknown luciferins and luciferases is an area of active research and recent highlights include reports on the luciferin–luciferase systems of the New Zealand glow-worm Arachnocampa luminosa and the diptera Orfelia fultoni from the United States.151,152 Both systems emit blue light and although structures of the luciferins have been proposed based on NMR spectroscopy, mass spectrometry and isotopic labelling studies although, the proposed structures need to be validated through chemical synthesis.
3. Designer luciferins and their luciferases
Advances in synthetic chemistry and protein engineering have been crucial to take the known natural luciferin and luciferase systems to unchartered territories and use them for a wide range of applications. Significant work has been done in both fields to prepare optimum luciferin–luciferase pairs that give access to previously inaccessible applications through bioluminescence imaging (BLI). Although significant research has been done in accessing ‘caged’ luciferins and ‘tagged’ luciferins as well as ‘split’ luciferases and ‘tagged’ luciferases – this particular portion of the review will focus on the basic structural modifications of both luciferins and luciferases that have bestowed them with beneficial properties, whereas as ‘tagged’ and ‘caged’ luciferins and ‘split’ luciferases will be discussed where relevant and have been discussed in greater detail in Sections 4.5 and 4.7. This section will focus on some of the key developments in the area of novel luciferins and luciferases.3.1 Novel D-luciferin analogues and luciferase mutants
As the D-luciferin/Fluc assay is the most red-shifted and bright natural luciferin to date, it has emerged as the assay of choice for in vivo imaging in small mammals. However, it does have some drawbacks that limits quantitative deep-tissue imaging in small mammals (Section 2.1). In order to address these, the quest for bright, non-toxic, red-shifted luciferin analogues that have optimum biochemical properties such as solubility, biodistribution and the ability to be useful in quantitative, multi-parametric imaging continues.The chemical synthetic routes towards luciferin analogues were succinctly compiled by Podsiadly et al. in 2019.69 Herein, we briefly highlight synthetic luciferin analogues, particularly those that have found use in applications, or have useful and interesting properties such as brightness that could potentially make them useful in applications. As the primary use of red-shifted D-luciferin analogues is in in vivo applications, we particularly highlight analogues that emit in the near infra-red region (>650 nm), and of those analogues, specifically those that have been successfully tested in vivo (Table 5). It is important to note that although the Fluc/D-luciferin combination emits at λmax = 558 nm in vitro, there is a spectral red-shift observed in in vivo imaging, with λmax ∼ 610 nm due to attenuation through tissue.153
| λ max < 550 nm | Natural substrate | λ max ∼ 550–600 nm |
|
|
|
| λ max ∼ 600–650 nm | ||
|
||
| λ max ∼ 650–700 nm | ||
|
||
| λ max > 700 nm | ||
|
||
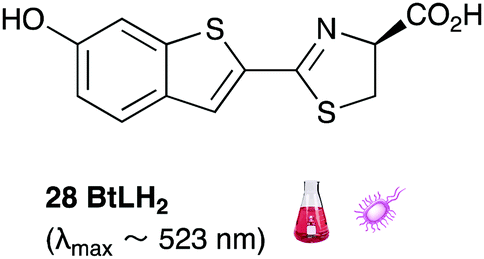
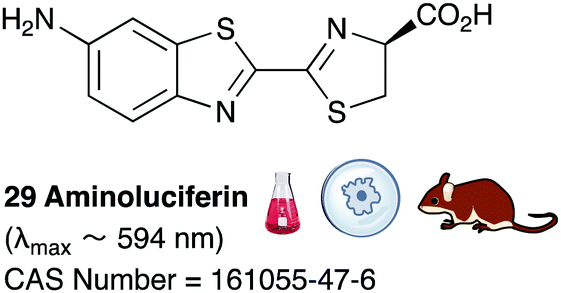
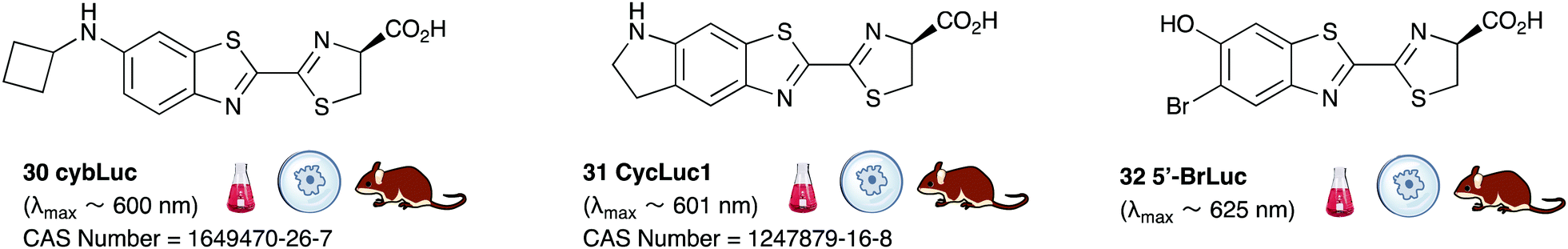
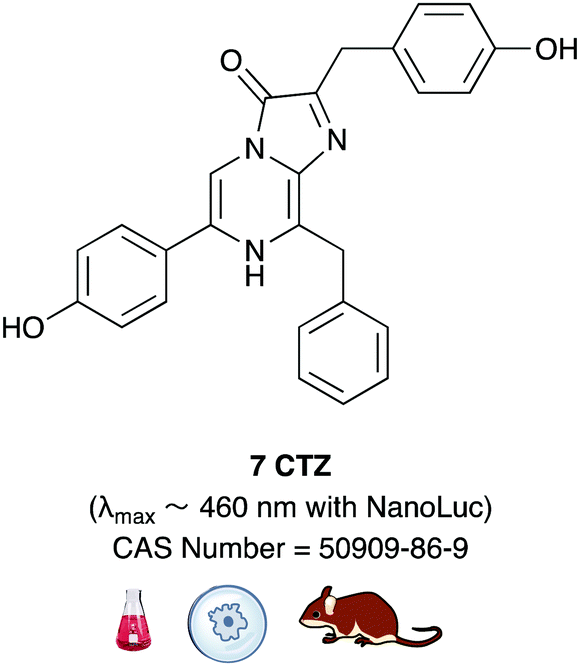
Of all the synthetic luciferin analogues reported to-date, BtLH228 was reported to have the highest bioluminescence quantum yield (70%) relative to that of D-luciferin 1 with wildtype Fluc. Although BtLH228, had a bioluminescence λmax of 523 nm which was around 20 nm blue-shifted, it was also reported to have a longer-lasting and sustained bioluminescence signal compared to that of D-luciferin.154 This could possibly make it a better candidate for applications that require blue-shifted and longer-lasting light emission. One of the earliest reported synthetic luciferins was the aminoluciferin 29 by White et al. This synthetic luciferin was red-shifted (λmax = 594 nm) compared to D-luciferin (λmax = 558 nm) but only about 10% as bright in vitro.61,155 Since then, several other synthetic aminoluciferin analogues were reported, and although all of them are dimmer than D-luciferin 1, with the wild-type Fluc, some have useful properties for in vivo imaging. For example, aminoluciferin analogues 30 and 31 have emission around λmax ∼ 600 nm. Both analogues 30 and 31 was reported to have better penetration than D-luciferin through the blood–brain barrier in mice.156,157 Moreover, analogue 31 CycLuc1 was reported to have brighter bioluminescence output from cells at lower substrate concentrations than D-luciferin 1, indicating that it had better cell-permeability than D-luciferin 1.62 Both analogues 30 and 31 are commercially available. The Prescher group reported a brominated luciferin analogue, that was red-shifted (λmax = 625 nm) and had an appreciable relative bioluminescence quantum yield of 46% compared to D-luciferin 1 at 100 μM substrate concentration and 1 μg of Fluc.158 At these concentrations they reported aminoluciferin 29 to be 61% as bright as D-luciferin 1.
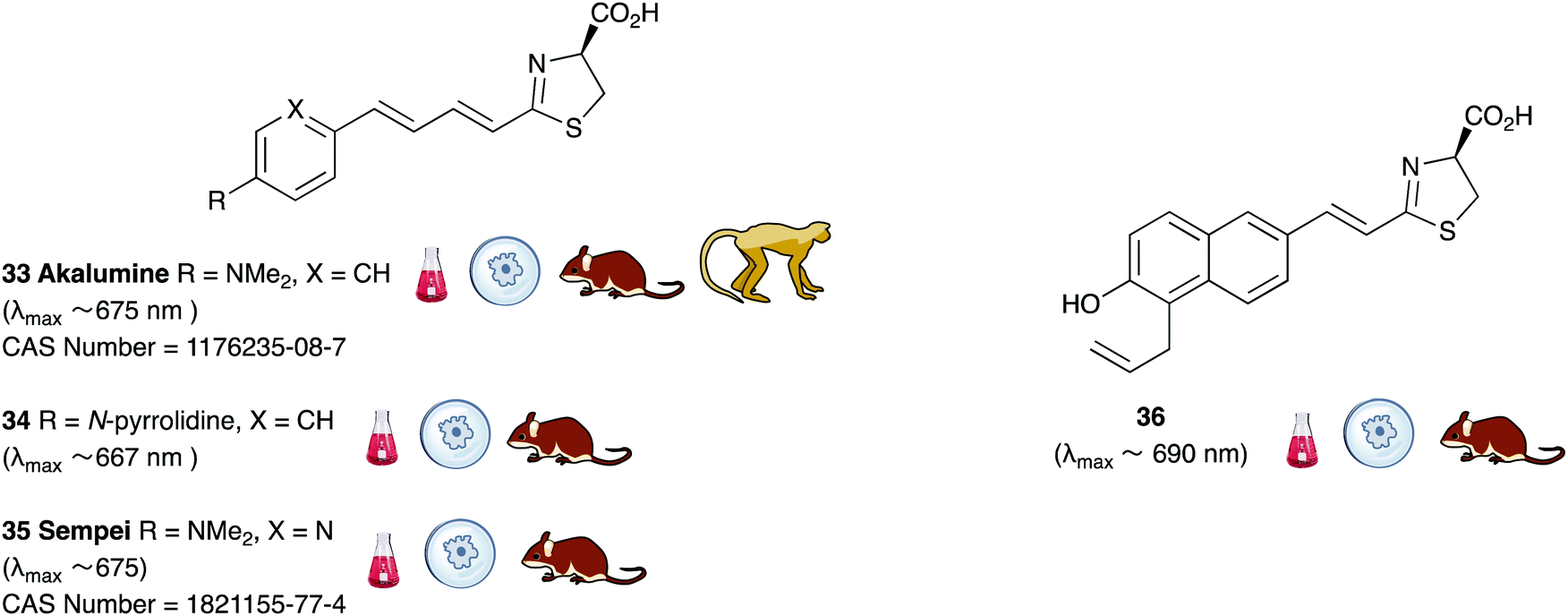
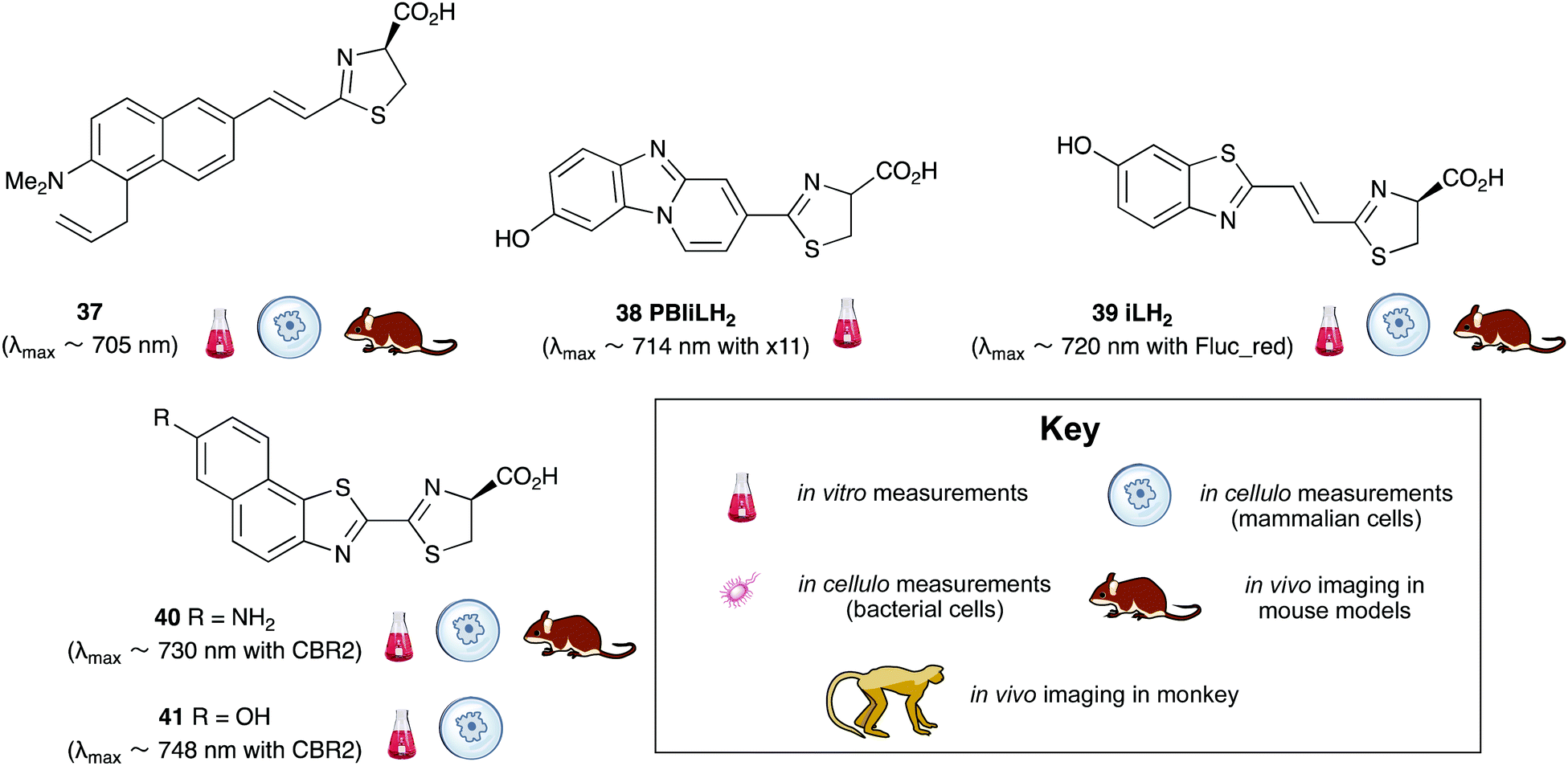
A handful of synthetic luciferin analogues truly emit in the nrIR region (>650 nm). The analogues 32–34 were reported by Maki et al. and have been successfully taken on to in vivo detection of tumours in mice.159–161 In particular, the hydrochloride salt of Akalumine 32 was used with the engineered firefly luciferase Akaluc in single-cell bioluminescence imaging (BLI) of deep-tissue in the lungs of live, freely-moving mice and to image small numbers of neurones in the brains of live marmosets.64 The analogues Akalumine 32 and Sempei 34 are now commercially available from Merck. The analogues 34 and 36 were also reported by Maki et al. as significantly dimmer substrates than Akalumine 32 but whose emission was ∼700 nm.162 Anderson et al. reported the analogue iLH238 as a significantly red-shifted luciferin that showed enhanced tumour burden in deep-seated tumours such as liver metastasis in mice due to less scattering and greater penetration of the near-infrared (nrIR) light.66 The luciferin iLH238 was designed to emit different colours of light with different Fluc mutants through retention of the phenol group which is deemed necessary to modulate the colour emission of D-luciferin analogues. This ability of iLH238 to emit different colours of light with different Fluc mutants made it unique, and it was proposed that iLH238 was a suitable analogue for multiparametric imaging and tomography. Recently, the suitability of iLH238 also demonstrated in a report by Anderson and co-workers in which racemic iLH238 together with stabilised colour mutants of firefly luciferase (Fluc_green ∼ 680 nm and Fluc_red ∼ 720 nm) were shown to be a suitable system for nrIR dual in vivo bioluminescence imaging in mouse models where they simultaneously monitored both tumour burden and CAR T cell therapy within a systemically induced mouse tumour model.68 The Anderson lab also reported the analogue PBIiLH237 as a racemic compound, which was prepared as a conformationally restrained infra-luciferin analogue.163 Although PBIiLH237 was only tested in in vitro assays and found to be less bright than racemic iLH238, it did demonstrate an increased bimodal emission with increasing pH. A primary bioluminescence peak at 608 nm was observed with Fluc x11 and a secondary peak at 714 nm of increasing intensity. This emission pattern could potentially be used to monitor pH, although the work does not build on this possibility.164 In 2018, Mezzanote et al. reported the most red-shifted luciferin analogue 40 and sister compound 39 with mutant CBR2opt luciferases without the use of resonance transfer.165 Both analogues 39 and 40 were tested in HEK-293 cell lines expressing CBR2opt, and as the analogue 39 gave higher light output than the analogue 40, the analogue 39 was tested in in vivo mouse studies. The most useful output from this study appeared to be the development of the mutant luciferase CBR2opt as the D-luciferin 1 and CBR2opt combination was demonstrated to be the brightest and most useful in all the experiments reported in the work, whilst both analogues 39 and 40 were dimmer than D-luciferin with CBR2opt.
In the area of luciferase engineering, several firefly and beetle luciferase mutants have been reported with improved properties such as increased stability, increased substrate affinity, and increased brightness over the years and these were comprehensively reviewed in 2016 by Yampolsky et al.10 Some highlights since then include engineered luciferases for improved light output of specific synthetic substrates such as Akaluc for Akalumine 32 and CBR2opt for the analogue 39.165,166 Other highlights include work by Miller et al. on mutants that have a significantly higher Km for D-luciferin and ATP than for cyclic amino-luciferins. The rate of reaction when the enzyme is saturated with substrate is the maximum rate of reaction, Vmax. For practical purposes, Km is the concentration of substrate which permits the enzyme to achieve half Vmax. An enzyme with a high Km for a particular substrate has a low affinity for that substrate and requires a greater concentration of the substrate to achieve Vmax. Hence the mutants developed by Miller et al. are more selective for cyclic aminoluciferins than for D-luciferin and this allowed substrate-selective BLI in mouse-brain.167 The Prescher group reported an elegant piece of work in which they prepared a library of 159 mutant luciferases by mutations of 23 key residues near the active site of the enzyme.168 These were then screened against 12 synthetic luciferins to identify orthogonal luciferin–luciferase pairs. Three of the ‘hit’ pairs from this analysis were taken up for in vivo mouse studies of mammary carcinoma.169 Imaging conditions are sensitive to a variety of factors, such as concentration of the imaging agent, the type of cell line used and the type of mouse model and tumour or infection studied. Although a number of synthetic luciferin analogues and mutant luciferases have been reported, there are very few studies reported that compare these against each other to match the best luciferin analogue and its complementary luciferase for a particular application in one study.
3.2 Coelenterazine analogues and luciferase mutants
Coelenterazine 7 (CTZ) is utilised by both photoproteins and luciferases. Coelenterazine utilising photoproteins such as aequorin, are often activated by Ca2+ ions and hence these are routinely used to detect intracellular Ca2+ ion concentration in biological studies. These photoproteins and the synthetic coelenterazine analogues with improved properties have been reviewed elsewhere.8,10A host of luciferases including Renilla luciferase (Rluc) from the sea pansy, Gaussia luciferase (Gluc) from the marine copepod and Oplophorus gracilirostris luciferase (Oluc), from the deep-sea shrimp use coelenterazine 7 as their substrate.24,25,79–84
A number of key developments using protein engineering were carried out on these coelenterazine utilising luciferases, which resulted in useful imaging and visualisation tools. For example, Nagai and co-workers developed the Nano-lantern, which was the brightest luminescent protein reported at the time in 2012. This was a chimera of Rluc8 (a brighter and more stable Rluc mutant)93 and a fluorescent protein called Venus which has high BRET efficiency.170 They then further developed this work by using different fluorescent proteins as BRET acceptors of Rluc8 and other Rluc mutants to develop a suite of Nano-lanterns that emit light of different colours, including the most red-shifted Nano-lantern ReNL (λmax 585 nm).171–173 The Nano-lantern series was shown to have broad applicability in both in vitro and in vivo imaging, as well as in the detection of Ca2+ ions.
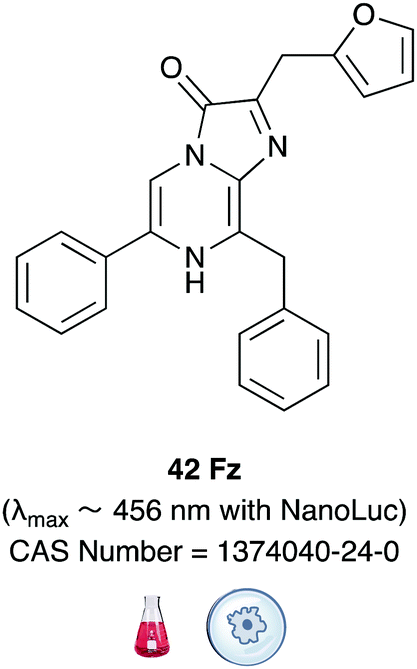
In another ground-breaking development, the catalytically active portion of Oluc was identified and mutated using a combination of both rational mutagenesis and random mutagenesis for enhanced thermal stability and light output by Promega.174 This small 19 kDa mutant enzyme was called Nanoluc and it was optimised to perform best with the synthetic substrate furimazine (Fz) 42Table 6.175 Both Nanoluc and furimazine are now commercially available. However, the Nanoluc-furimazine combination emits blue light (λmax ∼ 456 nm) which makes it unsuitable for in vivo applications, although the fact that this system has ATP-independent emission has led to possible advantages in some applications over the firefly bioluminescence system.176
Following the development of Nluc, there were reports of chimeric proteins that use Nluc as the BRET donor together with a fluorescent protein as the BRET acceptor. For example, the LumiFluor series was developed by creating chimeras of Nluc with bright, fluorescent proteins such as eGFP to get emission of around ∼460–508 nm or with an orange light emitting GFP variant LSSmOrange for emission ∼572 nm.177 The LumiFluor series was shown to be useful in the in vivo imaging of tumours as well. In a complimentary approach to the development of Nano-lanterns, small-molecule fluorophores could also be appended to Nluc through the development of Nluc-Halotag fusion proteins.178,179 BRET then occurs from the furimazine oxyluciferin to the fluorophore.
| λ max < 460 nm | Natural substrate | λ max ∼ 460–550 nm |
|
|
|
| λ max ∼ 550–600 nm | ||

|
||
| λ max ∼ 600–650 nm | ||
|
||
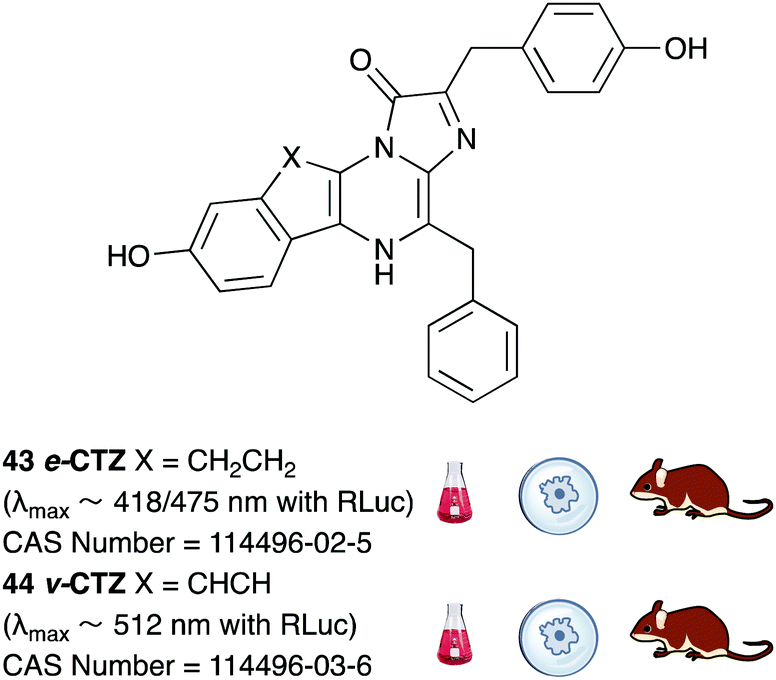
A number of coelenterazine analogues including 43 and 44 were reported by Shimomura et al. and these were tested against the wild-type luciferases that naturally utilise coelenterazine.180 The analogue e-CTZ 43 was reported to be ∼1.4 times brighter than CTZ 7 with Rluc and had a 7.5 times higher initial peak intensity than CTZ 7. This could potentially be due to the fact that this analogue is a conformationally restrained analogue of CTZ 7. The analogue v-CTZ 45 was reported to be 0.73 times brighter than CTZ 7 with Rluc but had a 6.4 times higher initial peak intensity than CTZ 7. It was also unsurprisingly more red-shifted than CTZ 7, possibly due to extended conjugation of the π-electon system. This led to an emission of λmax ∼ 512 nm. Both these analogues have been used with modified and optimised Rluc luciferase systems in the in vivo imaging of small mammals and are commercially available.181,182
Recently, some furimazine analogues such as 45 and 47 have been reported which have red-shifted emission with Nanoluc in the absence of any resonance transfer fluorophore.183 However, these analogues were about 102–104 times dimmer in in vitro assays than the Fz-Nanoluc combination. Consequently, Nanoluc was mutated further in attempts towards red-shifted emission. These attempts had limited success, with light emission being red-shifted only up to 509 nm which is still blueish-green light.184 In an attempt to create bright and red-shifted reporters, Nanoluc was fused with CyOFP1, a bright, engineered, orange-red fluorescent protein that is excitable by cyan light (497–523 nm), to develop a BRET-based genetically encoded reporter called Antares. The Fz-Antares combination was reported to be the brightest in vitro and in vivo when compared with D-luciferin-Fluc, Fz-Nanoluc and Fz-Orange Nanolantern combinations.185
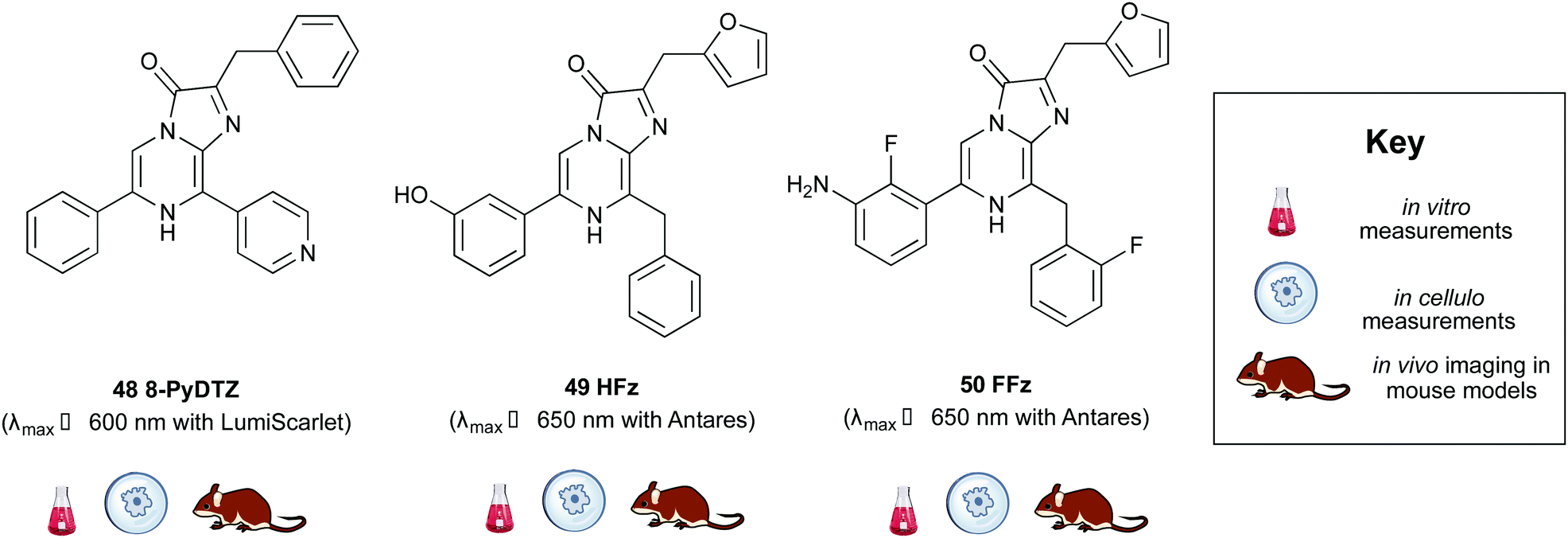
Building on from this work, Ai et al. created another fusion protein Antares2 in which random mutations were introduced in NanoLuc across the gene using error-prone PCR.184 From this, they identified a Nanoluc mutant (Nanoluc-D19S/D85N/C164H) with a 5.7-fold enhancement of DTZ bioluminescence and named it teLuc as it gave teal coloured emission (λmax ∼ 502 nm). The teLuc fusion with CyOFP1 was named Antares2. This BRET based reporter utilised DTZ 46 rather than Fz 42 and emitted 3.8 times more photons above 600 nm than Antares. Although this analogue is reported to be brighter than D-luciferin 1 and Akalumine 33in vitro, it suffers from poor solubility in aqueous media and poor stability as like all coelenterazine analogues it is prone to auto-oxidation. This makes in vivo studies particularly challenging. In order to address these challenges, Ai et al. reported a number of pyridyl analogues of DTZ 46 including 8-pyDTZ 48 that had ∼13 times better aqueous solubility and bioavailability than DTZ 46.186 In their work they reported the development of a teLuc mutant called LumiLuc which was through a series of error-prone PCR experiments on teLuc resulting in a total of 12 mutations, The emission from 8-pyDTZ/LumiLuc was ∼5 times brighter than 8-pyDTZ/teLuc and had emission around λmax ∼ 525 nm. LumiLuc was then fused to a fluorescent protein mSCarlet-I to form LumiScarlet which was useful in BRET based BLI and had emission around λmax ∼ 600 nm. The emission from 8-pyDTZ/LumiScarlet in in vivo imaging was reported to be comparable to that by Akalumine/Akaluc.
Two new substrates hydrofurimazine 49 (HFz) and fluorofurimazine 50 (FFz) were also reported recently to address the challenges of solubility and bioavailability in coelenterazine analogues. The analogue HFz 49 exhibited similar brightness to AkaLuc with its substrate Akalumine 33, whilst a second substrate, FFz 50 with even higher brightness in vivo. The FFz-Antares combination was used to track tumour size in vivo whilst Akalumine-AkaLuc combination was used to visualise CAR-T cells within the same mice.187
3.3 Bacterial bioluminescence
Several site-directed mutagenesis studies and some random-mutagenesis studies have been carried out on the bacterial luciferase system. However, most of these mutations have resulted in reduced activity, poorer quantum yield and have primarily served the purpose of improving our understanding of the nature of and the key residues in bacterial luciferase.188–192 A select few of these studies have resulted in altered properties of the bioluminescent system that could potentially be of use in applications. For example, an E175G mutation to the α-subunit of X. luminescens luciferase using random mutagenesis, resulted in faster kinetics and a faster decline in peak height, which could be useful in certain applications.193 Moreover, a number of red-shifted, mutant bacterial luciferases from Vibrio harveyi were reported.194 The mutant αA75G/C106V/V173A was reported to emit at 505 nm whilst the mutant αA75G/C106V/V173S emitted at 510 nm. However, both these systems were 80–90% dimmer than the wildtype system. Hence, they are too dim to be useful in in vivo imaging and would need substantial optimisation to make them more useful.A breakthrough in this area was reported by Gregor, Hell and co-workers, who engineered the ilux operon.195 Prior to their work, bacterial bioluminescence resulted in a weakly autoluminescent mammalian cell line.116 Hell and co-workers chose the luxCDABE operon from P. luminescens due to its thermostability and systematically carried out studies to identify the cause of poor luminescence in mammalian cells. This led to codon optimisation and enzyme engineering, including supplementing the FMN reductase in P. luminescens with that from V. campbellii followed by error-prone PCR to select brighter mutants. The final result ilux contains a total of at least 15 mutations and around 8 times brighter than the original construct allowing single-cell imaging of bacterial cells for extended periods of time in vitro.195 This work was then further developed by codon optimisation of ilux for mammalian cells, which led to brighter emission by around 3 orders of magnitude compared to previous approaches. The light output was also reported to be comparable to that of a D-luciferin/Fluc system in HeLa cells.
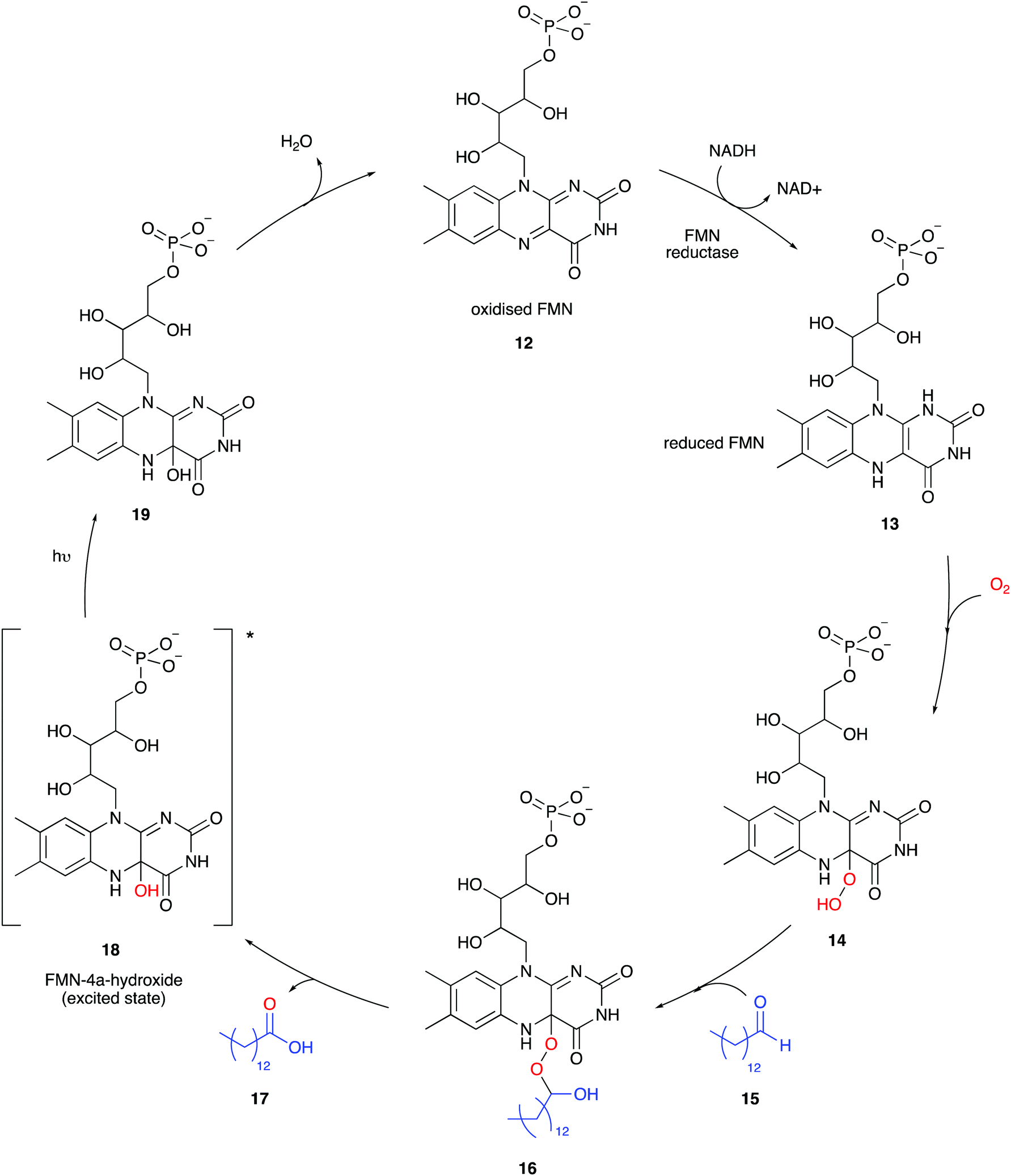 | ||
| Fig. 4 The mechanism of bacterial bioluminescence. | ||
4. Applications
4.1 ATP sensing
ATP is the energy currency in living cells and found in virtually all prokaryotic and eukaryotic cells. As Mg-ATP is a necessary co-factor in the mechanism of firefly bioluminescence, the firefly luciferase and D-luciferin system has been adapted into a variety of assays for ATP detection with different detection limit levels for ATP. Luciferase reagent preparations and their delivery devices for ATP detection vary from supplier to supplier and are optimised for each system. Each reagent system is a balanced cocktail of enzyme, co-factors, buffer and extractants.196Once the reagents are mixed at 25 °C, there is a 0.25 ms time lag until light emission is observed after which light output peaks at around 300 ms. This is followed by a rapid decay in light output and then finally slow, sustained light emission.197 This phenomenon is known as ‘burst kinetics’ and the decay in light output is thought to be due to inactivation of the enzyme or if a significant proportion of the substrates D-luciferin and ATP are consumed per minute in the reaction, when their concentration is low compared to that of the luciferase.42 Inactivation of the luciferase can occur if the luciferase is bound to surfaces or there is a significant concentration of an inhibitor such as oxyluciferin – the product of the reaction, or contaminants in the D-luciferin preparation such as L-luciferin and dehydroluciferin. Inactivation of the enzyme can be counteracted by using a highly pure D-luciferin sample to avoid contaminants, and by the addition of stabilising substances such as bovine serum albumin (BSA), neutral detergents and osmolytes for the protein, so that stable light output is obtained.198 In most ATP sensing assays, a fixed amount of D-luciferin is added to the assay mixture, which is in excess of ATP levels. At high luciferase concentrations, the peak light output is proportional to the amount of luciferase, as ATP is depleted in a first-order reaction. When luciferase concentration is low, ATP is slowly depleted and so light emission is stable and proportional to the ATP concentration, when the ATP concentration is significantly below the Km of the enzyme i.e. ATP < 0.1 mM L−1, as the rate of reaction and hence light output are proportional to the ATP concentration below Km.48 This is useful to monitor ATP forming and ATP degrading reactions including kinetic and end-point assays of enzymes and metabolites.42,48,199
Through the careful manipulation of all of these factors, a series of ATP-bioluminescence reagents have been made commercially available as simple-to-use kits which include the luciferin and luciferase preparations. These reagents are of two main types based on the intensity and duration of light-emission. Constant light emitting reagents have moderate sensitivity towards ATP (working range: 10−6 to 10−11 M ATP). The constant light signal is useful for kinetic studies of enzymes and metabolic studies, or if coupled enzymatic assays are applied. Such assays have been used to determine the amount of ATP in various diseased and healthy cell lines using both lysed human HeLa cells, mouse MEF cells and in worms such as the round worm, as well as intact cells or isolated mitochondria.200–203 This type of reagent can also be used to determine the activity of enzymes such as the activity of H+-ATP synthase from live isolated mitochondria.204
The second type of ATP-bioluminescence reagents are high sensitivity light emitting reagents. These have a higher concentration of luciferase and exploit the ‘burst kinetics’ phenomenon where the peak height is proportional to the amount of ATP in the sample and dependent on the concentration of luciferase. These reagent combinations have higher sensitivity towards ATP (working range: 10−5 to 10−12 M ATP), although reagents that report even lower detection limits are available in the market. These reagents are often sold packaged with cell lysis reagents, and are suitable for use in luminometers where automatic injection of the reagents is possible such as in tube luminometers and microplate luminometers (Table S1, ESI†).48
It is important to note that different types of cells have varying levels of ATP. For example, bacteria have lower levels of ATP compared to fungi or mammalian cells.48 It is important to pre-treat the sample effectively, and to use aseptic conditions to ensure that ATP levels from the correct desired source are detected. For example, a clinical urine sample may contain 3 different pools of ATP – extracellular ATP, ATP in mammalian cells, and ATP in bacterial cells and the purpose of an ATP bioluminescence measurement might be to estimate bacterial levels. The level of ATP from all 3 sources can be detected by using an appropriate kit containing ATP-degrading enzyme, neutral detergent, strong extractant, ATP reagent, and ATP standard (Fig. 5 and Table S1, ESI†).48
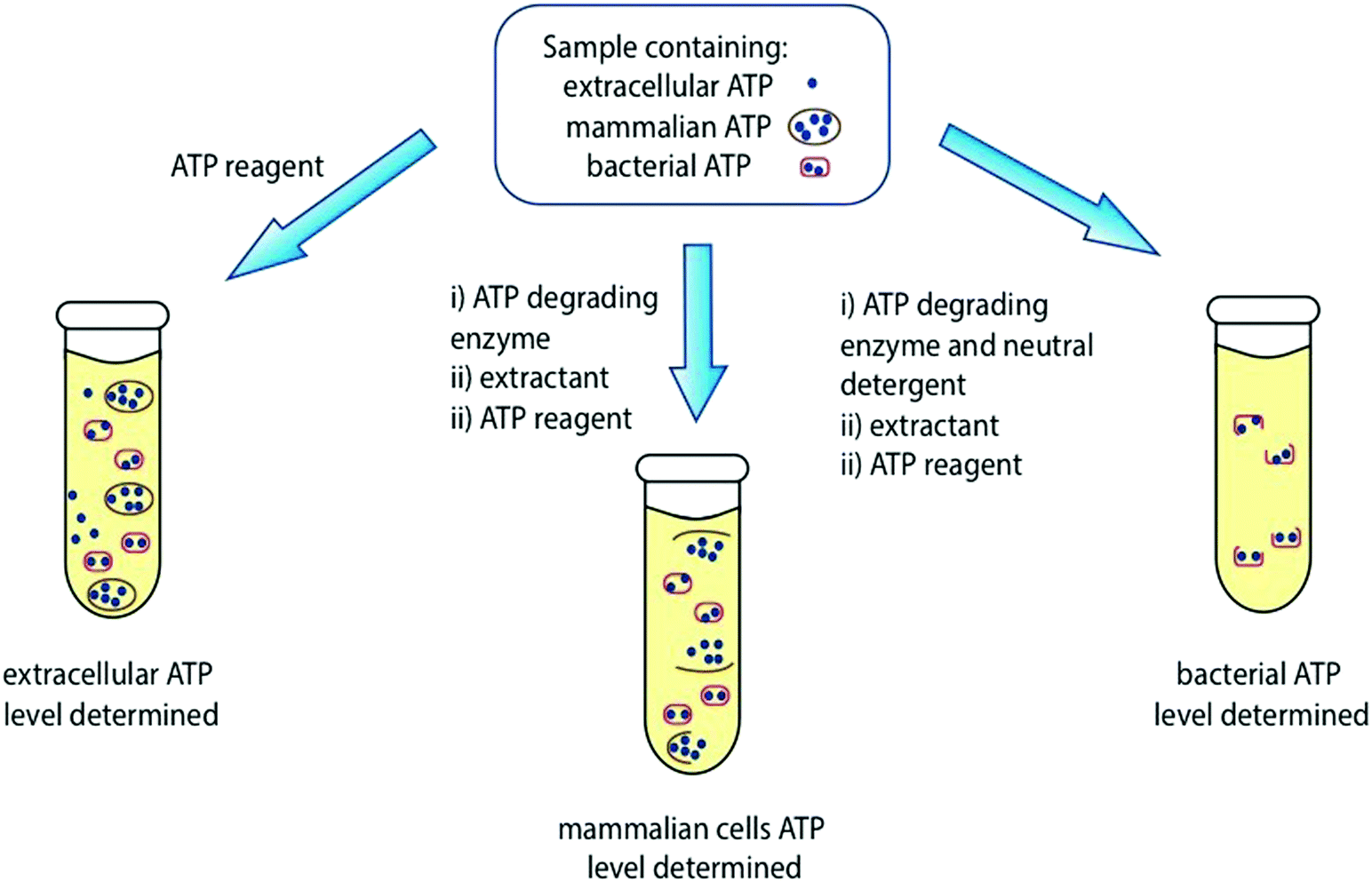 | ||
| Fig. 5 Determination of ATP levels in a sample containing extracellular ATP, mammalian cell ATP and bacterial ATP. Table S1 in the ESI† contains a list of commercially available ATP reagents and their details. | ||
Recently, there have been developments in luciferase engineering that allow ATP bioluminescence technology to reach unchartered territories. For example, Branchini et al. reported a red-emitting chimeric firefly luciferase that has a low Km for ATP and D-luciferin and would reach half of the maximum rate of the bioluminescence reaction at lower levels of ATP and D-luciferin than the wild-type enzyme making it suitable for in vivo imaging in low ATP cellular environments.205 Moreover Pinton et al reported protocols for the in cellulo and in vivo the use of chimeric luciferases that ensure the specific cellular localisation of the luciferase in a cell i.e. in the mitochondrial matrix and the outer surface of the plasma membrane to determine the ATP concentration in those areas.206 Viviani and co-workers also reported a blue-shifted luciferase that has the lowest reported Km for ATP, highest catalytic efficiency, and thermal stability among beetle luciferases that was suitable for ratiometric ATP, metal and pH biosensing assays.207 Metal sensing bioluminescence assays are covered in greater detail in Section 4.3, while pH sensing assays are covered in Section 4.5. There have also been advances in the development of more sensitive and portable luminometers and sensing devices.208,209 More recently, Roda and co-workers reported a low-cost wax-printed nitrocellulose paper biosensor that immobilised luciferase/luciferin reagents, and whose light signal could be detected and analysed by a smart phone to detect E. coli levels in urine samples.210 This low-cost, readily available technology would be ideal for use in developing countries where access to a luminometer and other specialist kit would be limited.
4.2 Hygiene control
Bioluminescence based sensing technology has been of great use in hygiene control for several decades now. In particular the ATP-bioluminescence assay based on the firefly bioluminescence system is routinely used to monitor the cleanliness of surfaces in healthcare facilities such as hospitals and clinics and in the dairy and meat processing industries.211,212 This is the technique of choice when the speed and ease of analysis are of vital importance as alternative methods such as culturing or microorganisms usually take days to offer results, whilst techniques based on fluorescence need an external light source for excitation and do not discriminate between living and dead cells.213 Bacterial bioluminescence is also used in the food industry to monitor the behaviour of Lux-tagged bacteria in situ in complex food systems, for problem-solving and for the development of modified and improved processing and storage purposes as discussed further below (Fig. 6).214Several studies have been reported on the use of swab taking and ATP bioluminescence as a quick and objective way of monitoring the cleanliness of hospital surfaces, including those of large objects such as tables and benches and small pieces of equipment such as tweezers and other kit. Swabs that are impregnated with buffer are often commercially available as part of ATP-bioluminescence kits. These are used to sample surfaces, and then processed with the ATP-bioluminescence reagent in a portable luminometer. The swabs and portable luminometer are often sold from the same supplier and complement each other. However, this technique is still poorly standardised at an international level and the difference in kit and reagents used in different studies is one reason why significant differences in ATP levels are reported.215,216 Despite these limitations over the comparability of results, ATP bioluminescence remains a quick and cost-effective measure for surface cleanliness and hygiene control in hospitals and routinely informs the cleaning practices of housekeeping and healthcare staff.217 More recently these assays have been used to monitor the cleanliness of not just surfaces but surgical instruments and dentures as well.218,219
ATP bioluminescence measurements are routinely used to monitor quality control and hygiene in the food industry.212 In particular, fish processing plants have used it for decades to determine contamination levels.220 Recently a study was reported to determine the contamination levels in various fish processing environments i.e. different lines of production including different fish types such as trout and cod, different types of meat such as protein-rich loin meat or fat-rich belly meat and different levels of processing such as slaughtered or cooked products, and the results were compared against conventional culture and plating techniques.221 It was established that it is essential to set up critical limits after a period of validation and calibration that are specific to each processing plant, type of ATP-bioluminescence kit used, specific areas, types of fish and fish meat and different hygiene zones, to obtain more robust, consistent and meaningful results. The dairy industry also benefits greatly from ATP bioluminescence assays as these are used to determine the quality of milk by selectively measuring the ATP from somatic cells and milk spoilage by determining ATP levels from bacteria and other microorganisms both before and after UHT treatment to estimate shelf life.222
The lux operon which is responsible for bacterial bioluminescence has also found great utility in the food industry.214 The lux genes responsible for bioluminescence can be genetically encoded onto bacteria that are not naturally bioluminescent, and the localisation, population size and environment of these bacteria can be monitored in real time. As all known bioluminescent bacteria are Gram-negative, there were initial challenges in obtaining a good level of light output and gene expression in Gram positive bacteria. However, this was overcome by introducing translational signals optimized for Gram positive bacteria in front of luxA, luxC and luxE genes.223,224 To date bioluminescent E. coli and Listeria have been used to monitor the survival of these bacteria post-processing in yoghurt and cheese.225 A number of lux-tagged bacteria such as Campylobacter jejuni and Salmonella enteritidis have also been used to determine egg-shell penetration and colonisation.225,226Lux-tagged bacteria have also been used to monitor the development of bacterial infection in plant seedlings so interventions can be made at the appropriate time.227 The lux-based gene expression system has also been fused to genes of bacterial toxin production such as the promoter of the cereulide toxin gene ces in B. cereus to determine the ability of various foods to support toxin formation.228 Lux-tagged bacteria have been administered to mice for in vivo imaging of the resultant developing bacterial infection – for example lux-tagged L. monocytogenes were shown to grow and localise in the gallbladder of mice and cause re-infection in the intestines when bile was released.229 However, as bacterial bioluminescence emits predominantly blue light which is strongly absorbed by blood and tissue, it is important do ex vivo analysis of the organs as well to ensure bacterial colonies are not missed. Another use of lux-tagged bacteria has also been to detect biofilms and to develop cleaning methods against them, as well as to test the efficacy of hand sanitisers and disinfectants. As well as monitoring the growth and development of pathogenic bacteria, lux-tagged probiotic bacteria can also be monitored in foods that contain them as well as tracking the bacteria using in vivo imaging to understand their lifecycle and environment.230,231
4.3 Mapping pollution in ecosystems
The most widely used bioluminescence sensors in the toxicology monitoring of ecosystems are whole-cell bacterial bioluminescence sensors.232 Like most bioluminescence-based assays, these sensors provide a quick result to help assess toxicity levels. Other protocols for measuring environmental toxicity often involve exposing test organisms, such as fish, crustaceans, plants or bacteria to environmental samples and to monitor survivorship. The benefit of bioluminescent bacteria is that their light output can be used as a quick measure of survival. Moreover, their bioluminescence is directly linked to their respiratory chain and so any toxin that interferes with their respiratory chain, interferes with the light output.233 These sensors have been used to monitor a wide variety of contaminants including both heavy metal contaminants and organic compounds such as toluene and naphthalene.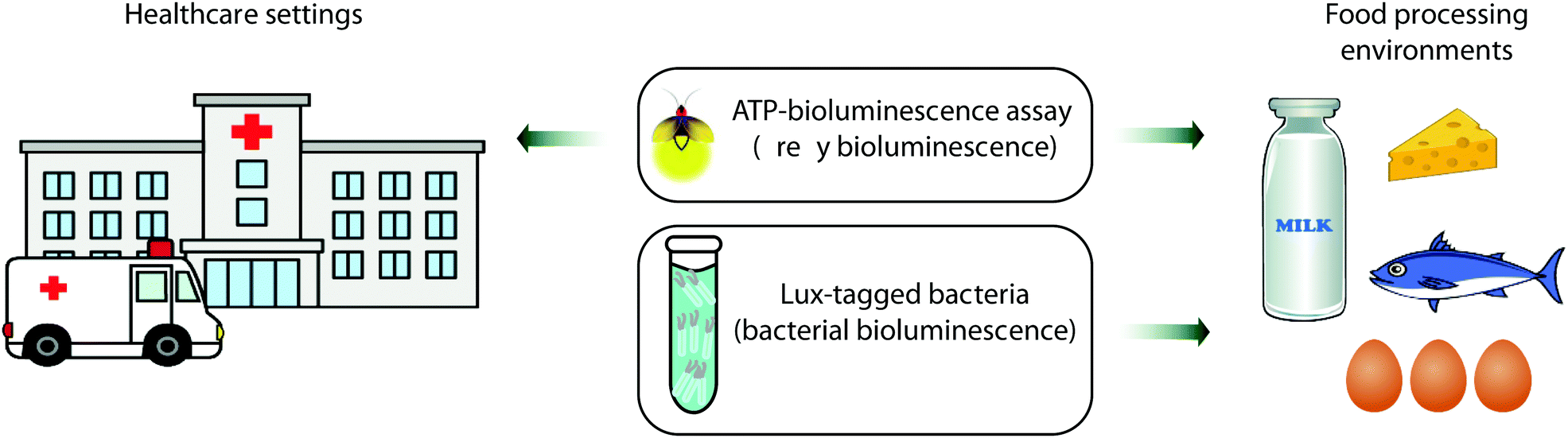 | ||
| Fig. 6 ATP-bioluminescence assays are used for hygiene control in several settings and industries including healthcare and food processing plants, whereas Lux-tagged bacterial bioluminescence is used to develop improved hygiene practices in the food industry. | ||
If the lux gene is expressed continuously, luciferase and luciferin will be formed continuously and the baseline light intensity would change on addition of the target analyte, depending on how well the bacterial cell survives. Alternatively, the lux gene can be controlled in an inducible manner wherein it would be fused to a promoter that is regulated by the compound of interest. In this case, the concentration of the compound can be quantitatively detected by measuring the bioluminescence intensity.118 Previously bacterial bioluminescence sensors were reported to analyse a variety of analytes including zinc, bioavailable toluene and uranium.234–236 Some recent examples in the development of bacterial bioluminescence biosensors to detect various analytes of ecotoxicology interest can be seen below (Table 7). Like most assays, careful pre-treatment of the sample to eliminate interference causing agents is essential to get meaningful results.
| Target | Microorganisms | Detection limit | Ref. |
|---|---|---|---|
| Common antibiotics | Bacillus WT and E. coli FhuAT | 0.043–324 mg L−1 | Jonkers et al.237 |
| Mercury | P. leiognathi | 9.87 mg L−1 | Kassim et al.238 |
| Chlorine | E. coli mutants | 1 mg L−1 | Borisover et al.239 |
| PyC12Phe (ionic liquid) | Vibrio fischeri | 4.17 mg L−1 | Kahru et al.240 |
| Sucralose (sweetener) | E. coli mutants | 1 g L−1 | Harpaz et al.241 |
| Terbutryn (herbicide) | Aliivibrio fischeri | 81 mg L−1 | Conrad et al.242 |
| Arsenite | E. coli | 39.6 mg L−1 | Ginet et al.243 |
The other common application of bioluminescence in ecotoxicology and pollutant monitoring is ATP quantification using the firefly bioluminescence ATP assay in both aquatic environments and bioaerosols in the atmosphere. ATP bioluminescence-based sensors have been used to determine the total ATP in water bodies including ocean environments and drinking water up to a detection limit of 1.1 × 10−11 M.244,245 This would include ATP from not just bacteria but also fungal cells and any parasitic protozoa. ATP bioluminescence-based sensors have also been used to detect the location and density of several air-borne bacteria in both artificially created and natural bioaerosols in indoor environments.246 The biosensors reported have either used fabricated paper disks immobilised with luciferase/D-luciferin or sensors microfluidic chips.247,248 Air was vented into and bubbled into a bio-sampler bottle containing 20 mL of deionised water to capture any cells found in the air. This solution was then concentrated and heated to lyse the cells. This lysate was then dripped on the fabricated paper disks immobilised with luciferase/D-luciferin. The fabricated paper disks with immobilised with luciferase/D-luciferin were reported to have up to 10 times longer shelf-life compared to the liquid assay reagents when stored at room temperature.247 Although this is a quick method to identify air-borne bacteria and their levels in studies where the identity of the bacterium is known i.e. artificially created bioaerosols, it is important to calibrate the assay effectively with known samples and use the ATP bioluminescence assay together with another assay to validate the results.249–251
4.4 Culture and heritage – preservation of art work
The ATP bioluminescence assay from the firefly has also been adapted to take surface measurements of ATP from old artwork to estimate bacterial, fungal, yeast, algae and lichen levels in antique work for the purposes of cultural and historic preservation.252 A bioluminescence low-light imaging technique was reported that was used on artwork consisting of paper, stone, fibre and wood.253,254 All reagents were applied directly to the samples and the conditions were optimised for sample geometry and surface conditions. More recently, the level of the microbial contamination of the seventeenth-century wall paintings in the nave of the old Church of the Holy Ascension (Veliki Krcimir, Serbia) was evaluated using the ATP bioluminescence method, and traditional cultivation-based method, using dip slides that were commercially available.255 It was established that ATP bioluminescence measurements can be a quick way to determine ‘hot spots’ of contamination on the art-work, allowing a quick assessment of areas that require greater concern.4.5 Sensing of pH, metal ions, reactive oxygen species (ROS), enzymes, drug molecules, and membrane potential including in cellulo applications
As the light output in bioluminescent reactions is dependent on the conditions of the assay in vitro or in vivo, it has been modified and adapted to be able sense various parameters such as pH, concentrations of metal ions, glucose, reactive oxygen species, enzymes and drug molecules. These sensing applications can use either modified luciferins such as caged luciferin structures or modified luciferases that are conjugated with sensing domains in a form of activity-based sensing (ABS) where the optical signal output is dependent on the intrinsic chemical activity of the bio-analyte in question with either the sensing domain of the luciferase or the caged-luciferin.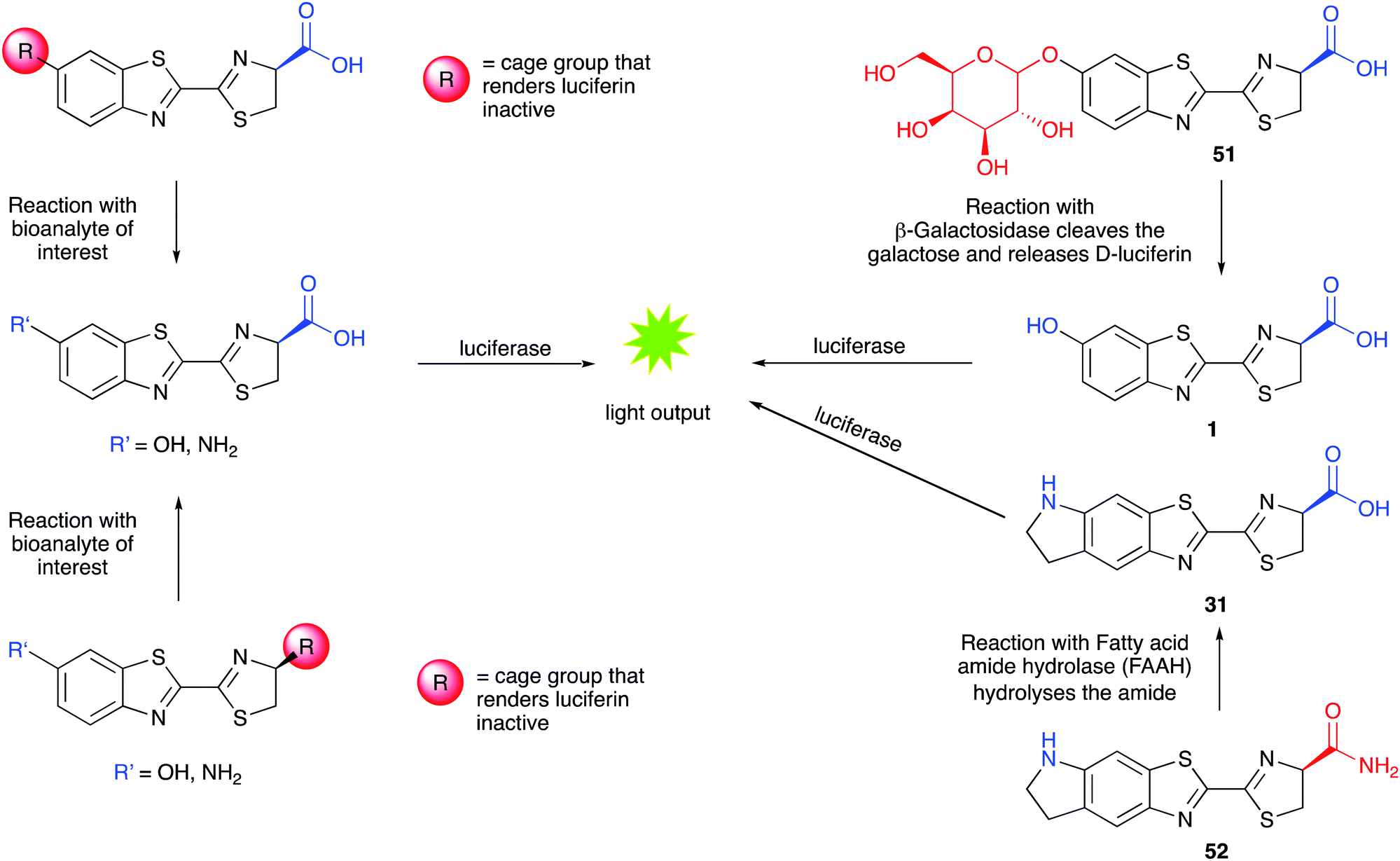 | ||
| Fig. 7 Caged luciferins – left hand side – a general scheme showing the 2 common sites of caging in red. Uncaging of the luciferin occurs in the presence of the bioanalyte of interest and the uncaged luciferin reacts with luciferase to emit light. A turn-on response is obtained in the presence of the analyte of interest. Right hand side – specific examples of caged-luciferin probes from the literature. Probe 51 was reported to measure β-galactosidase activity261 and probe 52 was reported to measure the activity of Fatty acid amide hydrolase.262 | ||
Caged D-luciferin probes have been used as sensors for enzyme activity,262–269 small molecule sensors for molecules such as glycans,270 hydrogen sulphide,271 hypochlorous acid,272 carbon monoxide273,274 and hydrogen peroxide,275 and sensors for metal ions such as copper,276 iron,277,278 and cobalt.279 The benefit of bioluminescence is that no incident light is needed and so the signal to noise ratio is higher and therefore more accurate. Although a potential drawback of these bioluminescence-based probes compared to similar fluorescent probes is that the bioluminescent probes are administered in much higher concentrations in cell-based assays than fluorescent probes, and this might affect the physiological conditions of the cells. Nonetheless, several caged-luciferins have been successfully used for in vivo imaging of mice and there has been an excellent recent review covering the advances in this area.280 Moreover, the first example of a bioluminescent probe that can measure mitochondrial membrane potential in a non-invasive manner in vivo has just been reported to be a caged luciferin probe that called a ‘mitochondria-activatable luciferin’ (MAL probe) (Fig. 8). The MAL probe is uncaged by a bioorthogonal Staudinger reaction with an organic azide (Azido-TPP1), to release a functional luciferin, which will emit light in the presence of luciferase. The triphenylphosphonium (TPP) groups on both the organic azide and the caged luciferin directs both reagents to the mitochondria. The rate of uncaging and hence rate of formation of active luciferin is proportional to the combined changes in mitochondrial membrane potential and plasma membrane potential.281
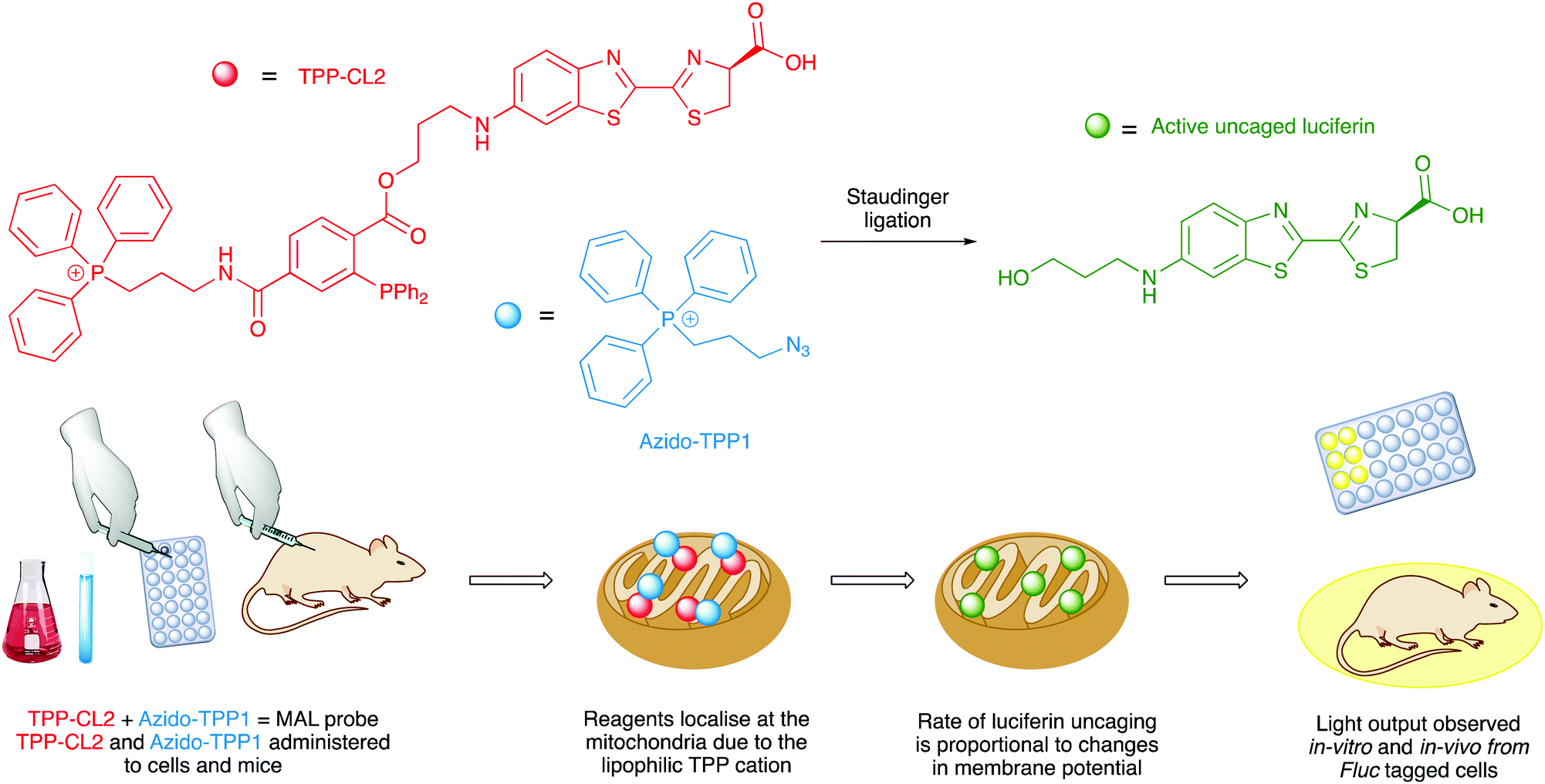 | ||
| Fig. 8 The design and functioning workflow of the mitochondria-activatable luciferin probe. | ||
The other type of luciferin probes that have been reported are designed to use the Bioluminescent Enzyme-Induced Electron Transfer (BioLeT) process to modify the light output generated. Bioluminescent Enzyme-Induced Electron Transfer (BioLeT) is analogous to photoinduced electron transfer (PeT) which has often been incorporated in the design of fluorescent probes.282,283 The design concept is that the singlet excited-state oxyluciferin species can be quenched by the electron transfer from the highest energy molecular orbital (HOMO) of an electron rich benzene moiety in close proximity. This was first reported in the design of a sensor for nitric oxide (NO), which is very dim in the absence of NO, due to BioLeT, but significantly brighter in the presence of NO, due to the absence of the electron-donating moiety (Fig. 9).284 This work also reported the successful use of this probe in vivo mice models. The authors have also reported another BioLet probe with turn-on luminescence that detect highly reactive oxygen species.285
 | ||
| Fig. 9 The use of a BioLeT probe to detect the presence of nitric oxide.285 The BioLeT donor has a high energy HOMO that is capable of donating an electron to the singlet excited state oxyluciferin. | ||
The Nano-lantern developed by Nagai et al. (Section 3.2) were also developed further in the same piece of work to detect ATP concentration.170 A chimeric fusion protein of the Nano-lantern with a subunit of bacterial FoF1-ATP synthase led to the development of Nano-lantern (ATP1) which exhibited an increase in light output on the addition of ATP with a Kd of 0.3 mM. This was then used to visualise ATP formation in chloroplasts.170 A number of Nano-lantern based GECIs were also developed by genetically engineering the Nano-lantern probe with a calcium-sensing domain from an established fluorescent Ca2+ sensor to form Nano-lantern (Ca2+) which gave comparable output and sensitivity to the fluorescent, genetically encoded Ca2+ sensor it was developed from.170
Another important class of luciferase-based sensors are the luciferase-based indicators of drugs (LUCIDs) developed by Johnsson and co-workers.288 These are semisynthetic bioluminescent sensor proteins that consist of three components: a receptor protein for the drug of interest covalently linked to a luciferase (Nluc), which is linked to a self-labelling protein such as SNAP-tag (Fig. 10A). The self-labelling protein was further linked to a synthetic molecule that consists of a fluorophore that can accept BRET from the luciferin–luciferase reaction and a ligand for the receptor protein. Binding of the protein with the ligand, lead to close proximity of the Nluc with the fluorophore and red-shifted emission due to BRET. In the presence of a drug molecule, this interaction is perturbed and hence a measure of the ratio of blue light/red light leads to a measure of drug concentration. LUCIDs were shown capable of detecting both small-molecule drugs and larger peptidic and macrocyclic drugs as well. The LUCIB and analytes were spotted on filter paper and the light output measured using a digital camera making them useful candidates for point-of-care diagnostics. Later Johnsson and co-workers reported the use of antibodies in place of the receptor protein to make the technology more easily accessible.289
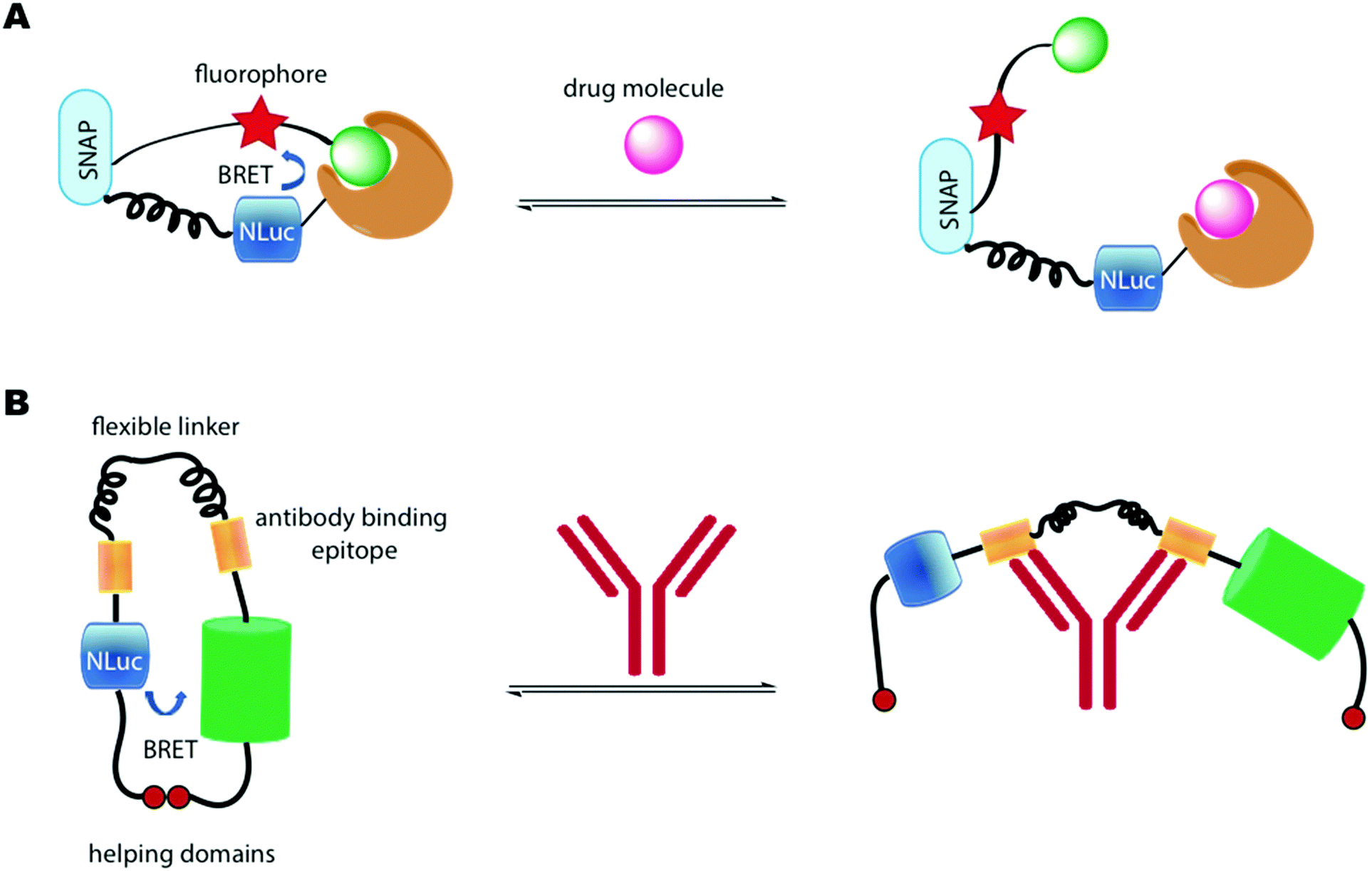 | ||
| Fig. 10 (A) LUCID developed by Johnsson and co-workers. BRET between NLuc and the fluorophore is disrupted in the presence of a drug molecule. (B) LUMABS developed by Merkx and co-workers. BRET between NLuc and mNeonGreen is disrupted in the presence of an antibody. | ||
A number of BRET-based antibody sensors have also been reported. Merkx and co-workers reported a luminescent antibody sensing (LUMABS) technology to detect antibodies in blood plasma.290 In these single protein sensors Nluc is connected to a green fluorescent protein mNeonGreen via a semiflexible linker and two antibody binding epitopes. A helper domain is found on each protein that keep both them in close contact to allow efficient BRET in the absence of an antibody (Fig. 10B). When an antibody binds to the sensing domain, the close proximity of the two light emitting proteins is disrupted leading to loss of BRET. A measure of this signal allows a ratiometric measure of antibody concentration. Initially this assay was optimised to a 384 well plate and the light output measured using a mobile phone. This technology has been further developed to enable identification of non-peptide epitopes,291 optimised to use as a microfluidic paper-based analytical device,292 and optimised further to require very small volumes of blood (∼5 μL) by depositing the biological machinery on cotton threads.293
As discussed earlier, whole cell bioluminescent bacterial biosensors are widely used to detect heavy metal concentrations such as mercury, zinc and chromium in ecotoxicology studies.294–296 For more details on this please refer to Section 4.3.
4.6 Gene assays
A number of gene and DNA based assays have been reported that use a bioluminescence-based readout to detect various analyte levels. For example, Christopoulos and co-workers reported some of the earliest work in this area, utilising either Aequorin as the photoprotein,297 or firefly luciferase.298 This expression immunoassay used a Fluc coding DNA fragment as a label, and reported the limit of detection of prostate specific antigen (PSA) as low as 1 pM ((Fig. 11).298 The antigen was isolated on polystyrene plates pre-coated with a suitable capture antibody. The captured antigen was then reacted with a biotinylated antibody. The biotin tag was then able to capture a streptavidin bound Fluc DNA tag. Transcription and translation of the Fluc DNA template resulted in 12–14 Fluc luciferase molecules per DNA tag bound to the plate. On addition of D-luciferin, the light output was proportional to the number of antigens bound. Recently, this limit of detection was improved to 0.007 pM by improving the number of enzyme molecules produced per DNA template by using a highly productive E. coli extract-based cell-free protein synthesis system.299 A DNA hybridization assay based on similar principles, but to detect DNA instead using Fluc bioluminescence was also reported by Christopoulos and co-workers.300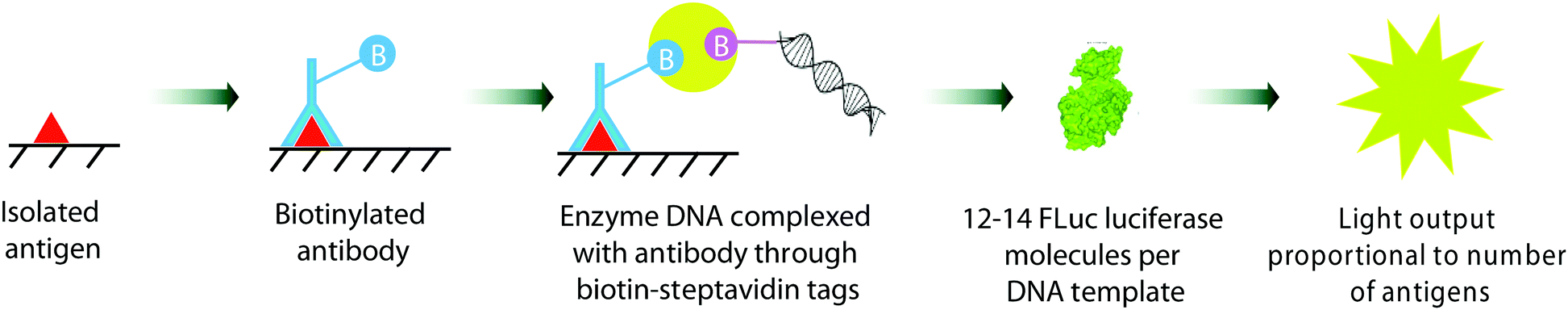 | ||
| Fig. 11 The gene expression immunoassay for detection of prostate specific antigen (PSA) reported by Christopoulos et al.264 | ||
One of the most common applications of Fluc and Rluc bioluminescence is their use as reporter genes for the study of gene expression in prokaryotic and eukaryotic cells and systems.301–304 A notable recent and relevant example is the use of luciferase based assays to determine the infectivity of viruses such as the various coronaviruses in different host cell types.305,306 Such an assay was also used to establish that SARS-Cov-2 coronavirus and a closely related RaTG13 coronavirus that was found in bats can both successful infect human cells to produce daughter viruses (Fig. 12).307 The interaction and binding of the spike glycoprotein in coronaviruses to the cell receptor Angiotensin-converting enzyme 2 (ACE2) in human cells is considered key in the entry of the viruses into human host cells.308 Plasmids containing the genes for Fluc and the genes for the spike protein from the coronavirus strain of interest were co-transfected into host cells and incubated for 72 h. The pseudoviruses formed after this period were collected and these pseudoviruses would have the desired spike-protein on their surface and the genetic material encoding for Fluc inside of them. These pseudoviruses were then incubated with ACE-2 expressing human cells for 60 h. If the viruses are successfully able to infect the human cells, daughter viruses and Fluc would be produced. On addition of D-luciferin, the light output would be a measure of infectivity.
There has also been a report of the blue light from a Nanoluc-furimazine reaction being used to activate a photoactive LOV protein that in-turn uncages a transcription factor – so in essence the light is used to regulate gene expression, although this assay can also be used as a protein–protein interaction assay.309 In their assay, protein A is linked to a light-activated LOV protein as well as a transcription factor through a protease cleavage site and protein B is linked to Nanoluc as well as a protease (tobacco etch virus protease TEVp). When proteins A and B come into contace with each other, and blue light is emitted from Nanoluc in the presence of Furimazine, this light activates the LOV protein, which changes conformation to present the protease cleavage site to the TEVp protease. The protease works on the protease cleavage site and releases the transcription factor, which then heads towards the nucleus for transcription. In the work, the transcription factor induces the transcription of the red fluorescent protein mCherry. The readout of the assay is the result of this transcription and hence the expression and fluorescence of mCherry (Fig. 13). This is one of the few examples of light from the bioluminescent reaction being used to control an effector function.
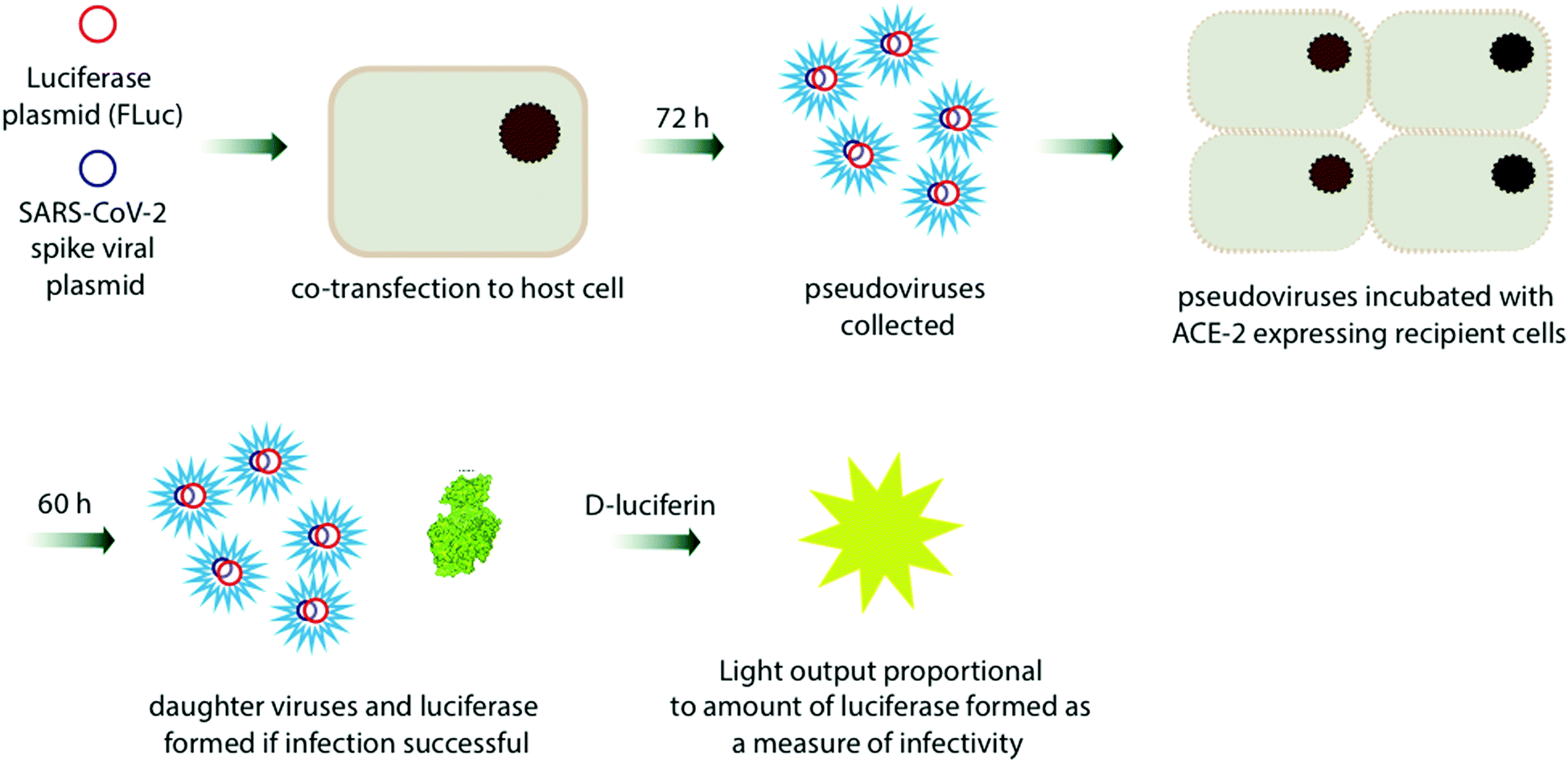 | ||
| Fig. 12 A Fluc/D-luciferin based assay used to determine the infectivity of coronaviruses in different host cells – image shows process used for SARS-CoV-2 in ref. 273. | ||
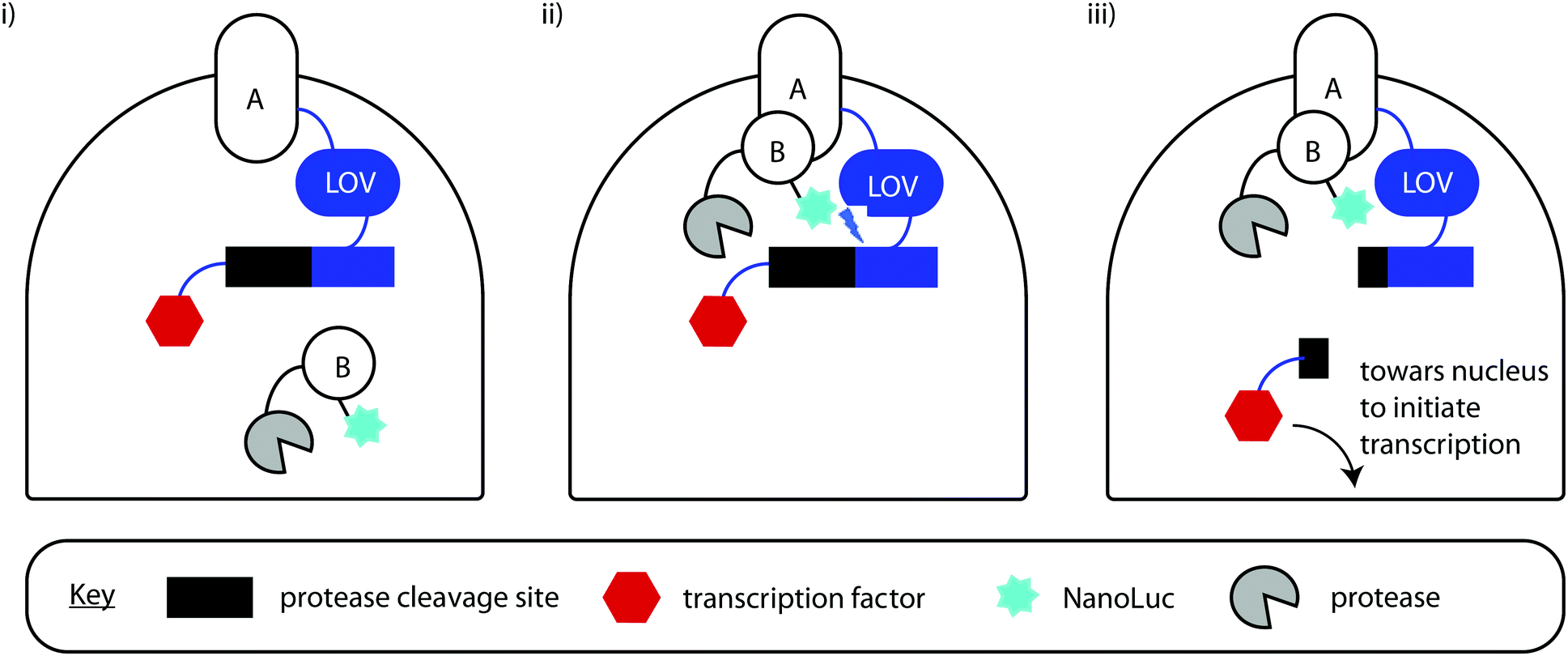 | ||
| Fig. 13 The use of bioluminescence to control gene expression – (i) When proteins A and B are distant, the LOV protein is deactivated and nothing happens. (ii) When A and B are in close proximity and interacting in the correct orientation AND Nanoluc reacts with furimazine to release light, the LOV protein gets activated and changes conformation to present the protease cleavage sit to the protease. (iii) After cleavage the transcription factor is free to move to the nucleus and initiate transcription of the fluorescent protein mCherry. | ||
4.7 Protein–protein interactions (PPIs)
One of the most established uses of bioluminescence-based assays is in the interrogation, analysis and determination of interactions between proteins. The two main types of technology used in this area are the bioluminescence resonance energy transfer assays (BRET) using the NanoBRET technology,175,310,311 and split-luciferase systems.181,312BRET is based on the concept of Förster resonance energy transfer, which is a non-radiative energy transfer between two luminescent molecules, an excited state donor that transfers its energy to an acceptor which then emits light. The efficiency of the energy transfer is dependent on the distance between the donor and acceptor and their respective dipoles, which means that for efficient energy transfer to occur the molecules must be in close proximity to each other (1–10 nm),313 and have the correct orientation.314 In BRET the donor is a photoprotein such as Aequorin or a luciferin molecule such as coelenterazine or furimazine, while the acceptor is often the green fluorescent protein GFP, which emits green light. This circumvents the problems with Förster resonance energy transfer (FRET) with fluorophores, such as the need for an external light source, photobleaching of the donor fluorescent protein, simultaneous excitation of both the donor and acceptor molecules and autofluorescence in cells or animal models (Fig. 14).
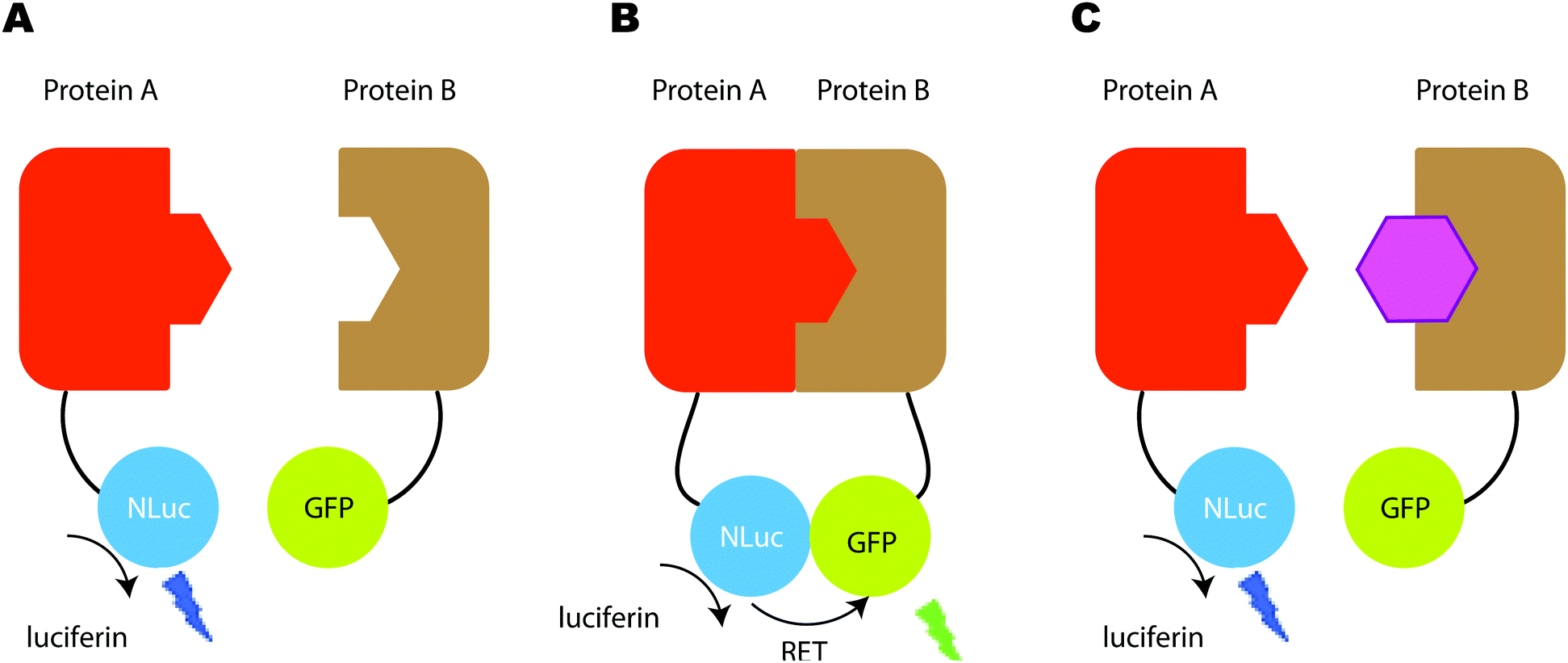 | ||
| Fig. 14 (A) When both protein partners are away from each other only blue luminescence is observed due to the NanoLuc reaction with furimazine. (B) When both protein partners are in close proximity, resonance energy transfer from NanoLuc to the acceptor GFP chromophore, results in the emission of yellow-green light from GFP. (C) In the presence of an inhibitor of the protein–protein interaction, both proteins are again at a distance and hence no BRET is observed. | ||
Previously, BRET based assays were well established to study various protein–protein interactions of interest such as oncology based targets including p53/hDM2 as well as G-coupled protein receptors both in vitro and in cellulo.315,316 More recently the inhibition or stabitisation of other PPIs of interest has also been successfully detected using the NanoBRET technology in which BRET from the bright Nanoluc to an acceptor chromophore allows the determination of the proximity and orientation of two proteins of interest. Examples of such analysis include the localisation and conformation of viral HCV NS5A protein,317 analysis of the interaction between the PRAS40 and hippo pathway,318 analysis of the CD26-ADA-A2AR trimeric complex in cells,319 the use of miniG proteins as probes for GPCRs,320 and analysis of the interaction between the H2 relaxin protein and the RXFP1 protein.321 With the development of BRET acceptors that emit red light, NanoBRET technology has also been used to measure protein–ligand interactions in vivo mouse models of breast cancer to determine target engagement.322 For a more detailed review on the developments in NanoBRET technology, please refer to the recently published review on the topic.311 The vast majority of BRET systems use the Nanoluc/Furimazine combination as the energy donor as it is significantly brighter than D-luciferin/Fluc and is also much a smaller enzyme than Fluc, which makes tagging it on to proteins of interest easier and more useful as it is unlikely to disturb the protein's natural state. However, this is of limited use for in vivo imaging studies. Consequently, some efforts have been made towards red-shifted BRET systems. In this regard, Fluc enzyme mutants such as Ppy RE10 (λmax 617 nm with D-luciferin) has been covalently labelled with nrIR fluorescent dyes such as Alexa-Fluor680. This resulted in BRET emission of λmax 705 nm with an acceptor to donor emission ratio of 34.0 (Fig. 15).323 Another interesting avenue has been BRET from a luciferase/luciferin combination to an appropriate quantum dot. Quantum dots are nanoparticles (diameters ∼ 2–10 nm) composed of a semiconducting material with diameters in the range of 2–10 nm. Due to their high surface-to-volume ratios they demonstrate a number of interesting properties such as fluorescence. For example, NanoLuc was covalently linked to a polymer-coated CdSe/ZnS core–shell quantum dot QD705 that emits at λmax 705 nm. BRET from the Nanoluc/Furimazine reaction to the quantum dot led to red-shifted emission at 705 nm and this was used to image a tumour in mouse.324 Nonetheless, the toxicity of quantum dots is a cause of concern for many, particularly for in vivo applications, leading to research into more biocompatible quantum dots.325
As brighter and red-shifted BRET systems are being engineered,184 and tagging technology is improving as well,326 it can be reasonably expected that increasing numbers of in vivo monitoring of protein–protein interactions and protein–ligand interactions will emerge in the near future.
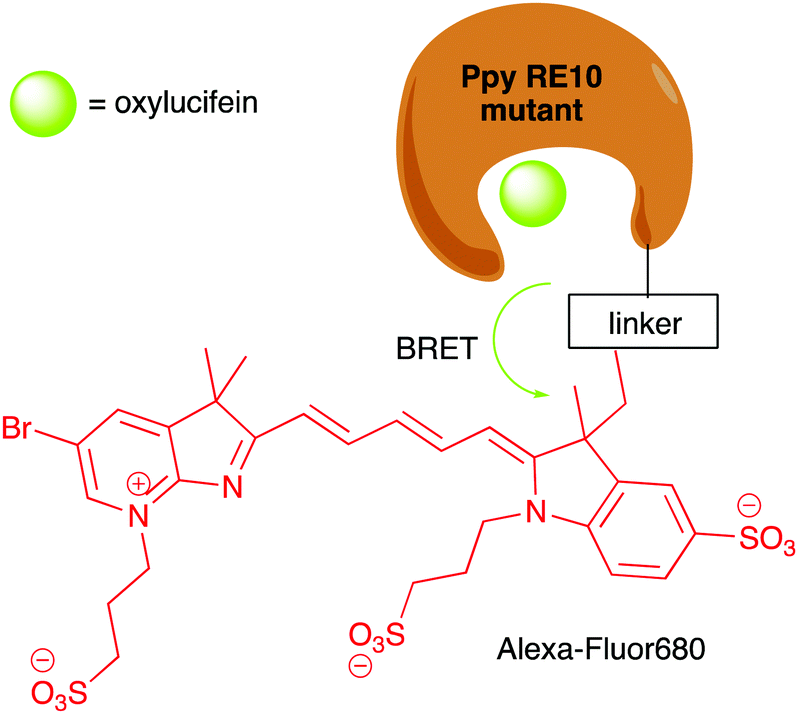 | ||
| Fig. 15 BRET from the oxyluciferin in the reaction of D-luciferin/PpyRE10 Fluc mutant to the covalently linked Alexa-Fluor680 resulted in emission at 705 nm with an excellent BRET ratio. | ||
Split luciferase assays are also widely used to detect and evaluate PPIs.327 These have been based on various luciferases including the firefly luciferase, click-beetle luciferase, Gaussia luciferase and Renilla luciferases, which have all been used to sensitively monitor dynamic PPIs with close to real-time kinetics both in vitro and in vivo.328,329 The luciferase is split into 2 portions with one consisting of the N-terminal and the other of the C-terminal domain. Each of these portions is tagged to proteins of interest. On addition of a ligand, the proteins of interest are brought together and both termini of the luciferase come in close proximity to each other, hence reconstituting the luciferase reporter function (Fig. 16).
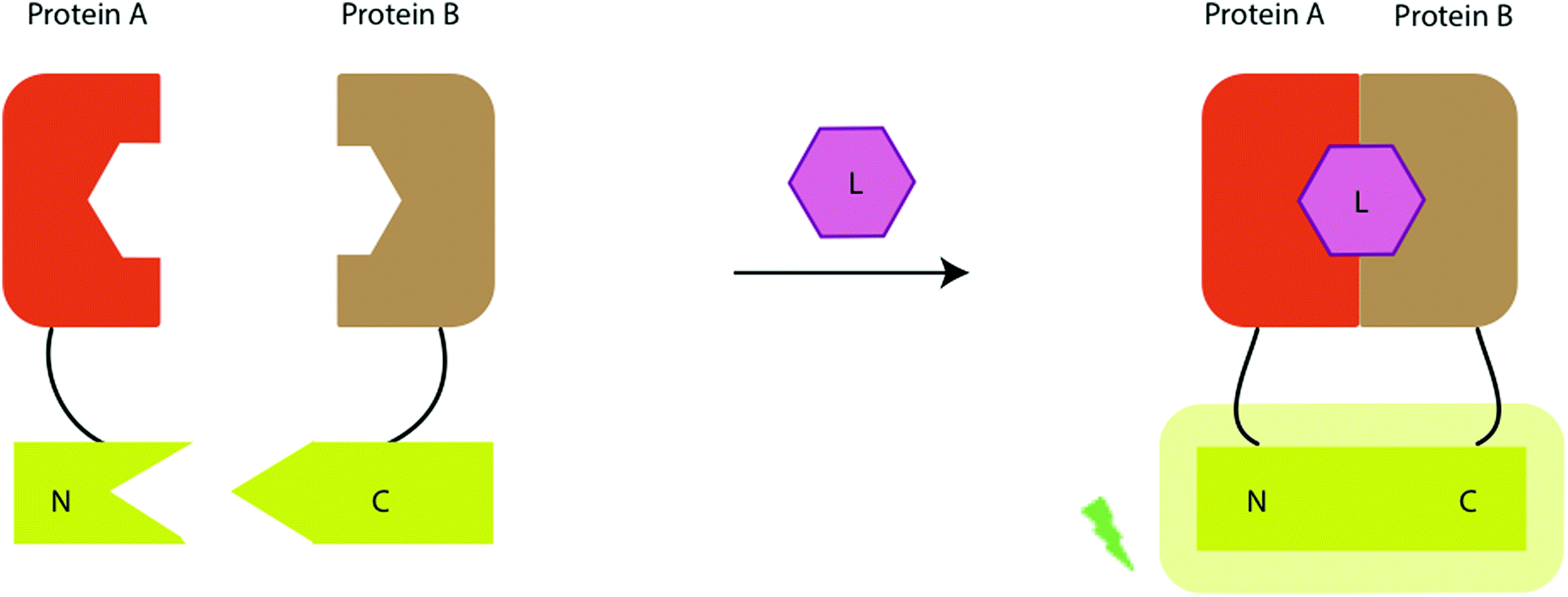 | ||
| Fig. 16 General principle behind split luciferases – on the addition of a ligand (L), the N and C terminals of the luciferase come together to give functional reporter luciferase and a glow and readout on administering of luciferin. | ||
Before the development of Nanoluc, Gaussia luciferase was arguably the most suited for protein-fragment complementation assays (PCA) as it is ATP independent, can be located in the extra-cellular space, is shown to be brighter than Rluc as part of these assays and the N-terminal and C-terminal enzyme fragments are small in size. The first split Gaussia luciferase assay was reported by Michnick et al. In their work, they interrogated the FRB/FKBP protein–protein interaction, using rapamycin as a ligand to induce complex formation, and FK506 as a competitive inhibitor. The PPI dynamics were visualised both in vitro and in vivo.330 Tao et al. reported the development of a split Gluc template which was adapted to visualise the protein–protein interaction of three different PPIs namely CaM/M13 with Ca2+ ions behaving as the ligand and the interactions of the ligand binding domains of (LBD) of representative steroid hormone receptors such as androgen receptor (AR), glucocorticoid receptor (GR), and oestrogen receptor (ER) with various petide or small-molecule ligands.331 Others have reported PCAs based on Gluc for visualisation of PPIs both in vitro and in vivo mouse models.332 Recently, split Nano luciferase has been used to determine protein–protein interactions in plant cells, wherein the PPI between receptor kinase flagellin-sensitive 2 (FLS2) and plant receptor kinase BAK1 (BRI1-associated receptor kinase 1) was found to be induced by bacterial flg22 peptide through a split Nanoluc system.333 Split luciferase assays have also been used to study viruses,334,335 as well as to detect protein–protein aggregation in human cells.336
4.8 High-throughput screening
Bioluminescence based assays are routinely used in drug-discovery programmes as part of high-throughput screens particularly against infectious pathogens such as bacteria, viruses or parasites. This is due to the ease in converting the assays to the commonly used 96 well or 384 well plate format and a quick, sensitive and straightforward readout of light output, that is often a measure of living cells. The gene, protein, enzyme or pathogen of interest are genetically encoded together with the luciferase gene and then pathogen replication along with simultaneous luciferase expression can be determined in the presence of luciferin through the light output of the sample both in the presence and absence of drug targets.For example, Tan and co-workers reported the development of high-throughput screening assay to identify inhibitors of coronaviruses.337 In particular, they replaced the ns2 accessory gene in the human coronanavirus strain HCoV-OC43 with the gene for Renilla luciferase (Rluc) to form a functional reporter virus strain rOC43-ns2DelRluc whose pathogenicity was unaltered. This mutant virus strain was then used to infect cells, and Rluc was expressed in the infected cells during viral replication. On addition of coelenterazine, the light output was proportional to the Rluc levels in the cells, which are a measure of viral replication. On addition, of small-molecule compounds that inhibit viral replication, a significant reduction in light-output was observed.338 This assay could be readily adapted in a 96-well format (Fig. 17).
 | ||
| Fig. 17 The high-throughput screening assay for coronavirus inhibitors that uses the coronavirus mutant strain rOC43-ns2DelRluc developed by Tan and co-workers.302 | ||
More recently, similar high-throughput assays have been developed for screening for antibiotics in aquatic samples,237 screening of antibacterial dental adhesives against mutants of streptococcus,339 and monitoring and inhibiting kinase activity.340,341
Both Fluc and Rluc have been extensively used in high-throughput screening campaigns against large compound libraries in both biochemical assays and cell-based assays. Whilst these assays are extremely useful and still widely used, it is important to note that they have some limitations. For example, both Fluc and Rluc can be inhibited by various small molecules. For example, Fluc in particular suffers from competitive inhibition from compound classes that have similar chemical structures to that of its substrate D-luciferin including benzothiazoles, benzimidazoles, benoxazoles and biaryl oxadiazoles. This can lead to false-positives in inhibitory assays for these compounds. Moreover, some compounds can also lead to increased trasnscription or translation of the reporter enzyme leading to a false-negative result. A critical discussion on the use of bioluminesce based assays is covered in seminal reviews written by Inglese and co-workers.342,343
4.9 In vivo imaging
The exceptional sensitivity and specificity afforded by bioluminescence imaging at the molecular level has rendered it particularly useful for in vivo imaging in small mammals.344 Whilst fluorescent probes suffer from photobleaching and background auto-luminescence and radioactive tracers have a short shelf-life and need a synchrotron source to be produced, bioluminescent probes do not suffer these disadvantages. Moreover, developments in charge couple devices allow the detection of low levels of light output. With the advent of engineered, brighter, substrate specific luciferases and red-shifted, synthetic luciferins, significant advances have been made in in vivo imaging, most of which have been discussed in Section 3. Most often, these new, engineered, bioluminesce systems are first trialled and used in the imaging of developing tumours in cell studies and in vivo in mice. Although initial studies described the monitoring of tumours and gene expression subcutaneously, more recent work has focused on the imaging of more-deep seated tumours and challenging targets such as imaging in the brain of moving animals with novel red-shifted and bright luciferin–luciferase pairs.345For bioluminescence in vivo imaging the cells of interest are genetically modified to include the gene for luciferase production. These cells can then be injected and tracked in the body of the small mammal, when the respective luciferin is added. Whilst, eukaryotic cells are often genetically tagged with Fluc, Rluc or Nluc mutants, bacterial cells are genetically modified to incorporate the lux codon responsible for bacterial bioluminescence. The light output is then recorded using a cooled charge-coupled device camera (Fig. 18). This allows the monitoring of in vivo processes in real time, without the need to sacrifice the animal.
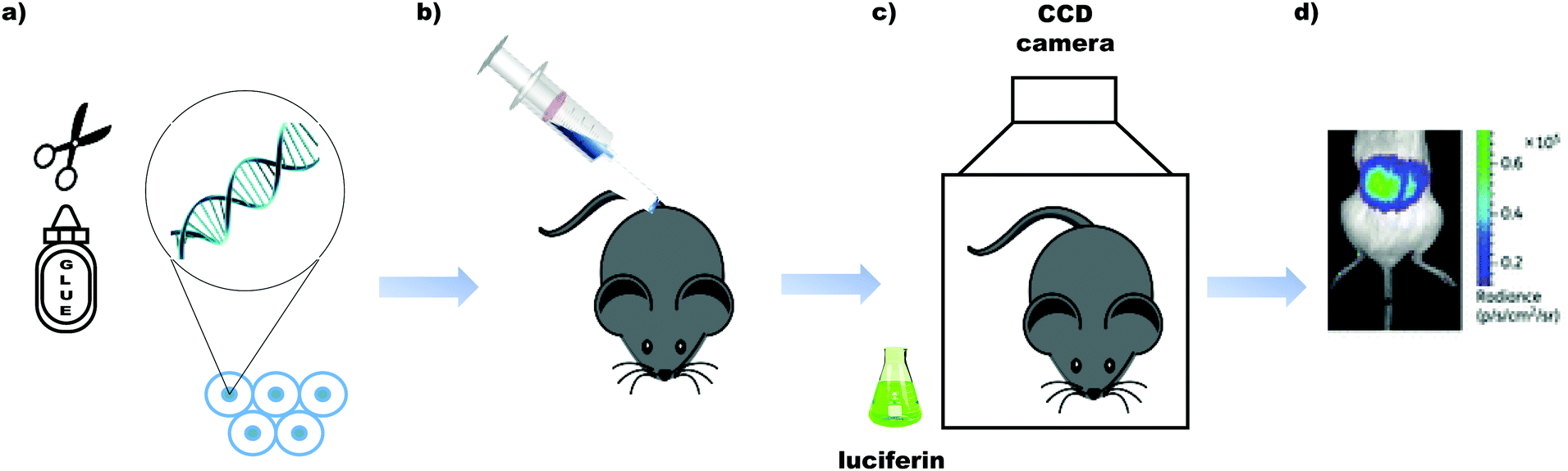 | ||
| Fig. 18 The process of in vivo imaging – (a) the cells of interest are genetically encoded with genes for expressing the luciferase of interest. (b) The luciferase expressing cells are injected in tot the mouse and incubated for a set period of time. (c) The respective luciferin is added, and the cells are tracked in real-time in vivo. Image acquisition is carried out using a camera with a cooled charged-coupled device. (d) An image of the tracked cells is obtained. This exemplar image is of the fungal cells of C. albicans, tagged with Fluc and imaged after the administration of D-luciferin. Image reused with permission from Oxford University Press on behalf of Brock et al.346 | ||
Bioluminescence imaging has also been effectively used in imaging the development of infectious diseases both in vitro and in vivo.347 Genetically modified bioluminescent pathogens, such as bacteria, parasites, viruses and fungi have been designed and monitored both in vivo and in vitro, in the presence and absence of therapies to test their effectiveness. The bioluminescent light output has been shown to correlate with the infection load.
Some notable examples include the first real time visualisation of the influenza virus in ferrets infected with A/California/04/2009 H1N1 virus (CA/09) encoding Nanoluc (Nluc) luciferase.348 The replication and development of human coronavirus strain HCoV-OC43 in the central nervous system of live mice was achieved using an Rluc reporter and coelenterazine,349 while another study reported the entry sites of encephalitis viruses in the central nervous system of mice, using the Fluc/D-luciferin system.350 An engineered firefly luciferase was also used with D-luciferin to monitor the development of a Candida albicans fungal infection real time in mice using in vivo imaging.351
In vivo BLI is also the modality of choice when monitoring the development of parasitic infections that cause neglected tropical diseases such as those from Toxoplasma gondii,352Trypanosoma cruzi,353,354 and Leishmania amazonesis.355,356 The gene for firefly luciferase can be readily encoded into these parasites and as bioluminescent output from the D-luciferin/Fluc system is ATP dependent, only living parasites are selectively imaged, which would not necessarily be the case in fluorescence imaging. Moreover, as the imaging technique is non-invasive, the diseased mice can be kept alive and monitored over time whilst being administered different treatments.
Bacterial infections such as Klebsiella pneumoniae, Citrobacter rodentium and antibiotic resistance to them is also monitored using BLI; however the bacterial lux operon is often used for this.357,358 The bacteria of interest are genetically encoded with the lux operon for bacterial bioluminescence and then injected into the mouse. The mouse is then treated with antibiotics and imaged over time to visualise the development of a disease. This technique has often been used to visualise the effectiveness of photodynamic therapy (PDT) on the treatment of bacterial infections such as surgical wounds, burns and lacerations as an alternative to treatment with antibiotics to combat antibiotic resistance. For example, dermal abrasions on mice infected with bioluminescent methicillin-resistant S. aureus (MRSA) were monitored while a treatment of PDT using a phthalocyanine derivative and toluidine blue with red light was administered by Hamblin and co-workers.359
Although now BLI reporters can emit light up to a wavelength of 750 nm, there is still much to be desired in terms of brightness to achieve desirable outcomes in in vivo imaging. Moreover, research into fluorescent probes has demonstrated that light output in the NIR-II window (1000–1700 nm) has significantly better penetration through blood and tissue than light in the NIR-I window.360 This is the next frontier in bioluminescence in vivo imaging.
4.10 Disease therapy using the light from bioluminescence
The light from bioluminescence has been used as a localised and controlled light source for the excitation of photosensitisers in photodynamic therapy. Photodynamic therapy makes use of photosensitiser molecules that are activated by light and generate reactive oxygen species (ROS) radicals, such as singlet oxygen that can destroy protein and tissue. This cytotoxicity can be advantageous in cases where cell death is required as a form of therapy such as in infection or cancer.Both the Fluc and Rluc systems have been used as genetically encoded sources of light for photosensitisers. For example, Theodossiou et al. reported that increased cell death was observed when both the photosensitiser Rose Bengal and D-luciferin were administered to a Fluc expressing cancer cell line, in the absence of ambient light.361 This work was contested by a later report that reported that the increase in cell death was insignificant when non-toxic levels of photosensitiser and luciferin were used, and that the quantum yield of the light output from the D-luciferin/Fluc reaction was the limiting factor.362 Another study by Lai et al. reported Rluc-immobilized quantum dots-655 (QD-Rluc8) for bioluminescence resonance energy transfer (BRET)-mediated PDT using the photosensitiser Foscan® to target cancer cells both in vitro and in vivo in mice.363 Although the PDT did not completely eradicate the tumour, it significantly delayed tumour growth, and it was proposed that this method of low-light dosage, compared to the use of external light, may cause lower inflammation and unnecessary death of healthy tissue.
Yun and co-workers reported Rluc and rose-bengal conjugates that generate singlet oxygen by bioluminescence resonance energy transfer (BRET). In their work, they used bovine serum albumin (BSA) as a central backbone and conjugated Rluc and rose-bengal to the BSA to achieve the desired distance between Rluc and Rose Bengal which allowed both moieties to be functional, without quenching the emission from either of them (Fig. 19). When coelenterazine was administered to this system, evidence of cytotoxicity and oxidative stress was observed in cells.14,364 Unsurprisingly, the uptake of the large and bulky conjugate into cells was poor and optimisation is required on that front.
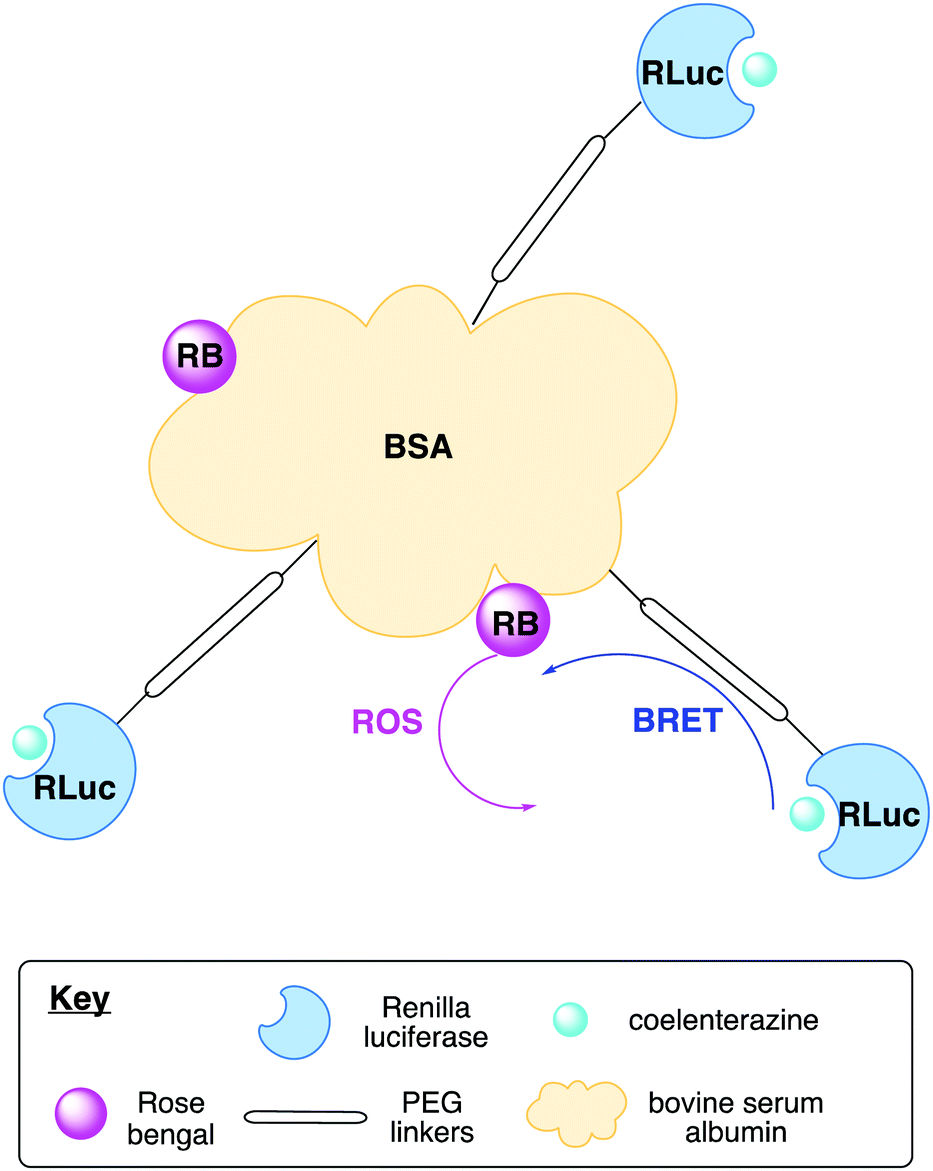 | ||
| Fig. 19 Figure showing the Rose-Bengal–BSA–Rluc conjugate. | ||
It is to be noted, that an improvement in the light output efficiency of the bioluminescent reaction, as well as the BRET efficiency between the coelenteramide and the photosensitiser would improve the generation of singlet oxygen.
An alternative approach that circumvents the need for cellular uptake of large and bulky biological conjugates could be to use a nanoparticle to bring the luciferase and photosensitiser in close proximity. This approach was taken by Wu et al. and in their work they encapsulated Rose-Bengal in biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles, which were then conjugated with Fluc. In the presence of D-luciferin, effective PDT and cancer cell death was observed both in vitro and in vivo in mice suffering from cancer of the liver.365
Although increasing numbers of studies with PDT activated by BRET from bioluminescence are being reported, the scope of the work is somewhat limited by amongst other factors, the brightness of the luciferin/luciferase reaction. As brighter luciferin/luciferase pairs are developed, it is hoped that this area will also expand into new territories.
4.11 Effector applications
Whilst traditionally the light from bioluminescence assays was primarily used for sensing applications such as the ones discussed previously, more recently bioluminescence-based technology has found avenues in effector technologies, wherein the light from the bioluminescent reaction can be used to affect or control other processes.366 This work was initially inspired by the relatively well-established field of optogenetics, where external light sources are used to control photoactive proteins and ion channels and hence cells such as neurones.367,368 Light from a luciferin–luciferase reaction provides an attractive possibility of localised and controlled light output of a wavelength of choice, compared to external light sources that are often harsh and can indiscriminately excite other endogenous fluorophores causing damage and autoluminescence.The earliest targets to be affected by bioluminescent light output were channelrhodopsins. Channelrhodopsins are light-sensitive ion channels that are naturally found in algae and are responsible for their movement in response to light i.e. phototaxis.369,370 When expressed in neurons, rhodopsins allow light to control the state of the ion-channel by either opening it or closing it and hence the ability of the neuron to fire action potentials.371 The channelrhodopsins absorb blue light (480 nm), so are ideal targets for Rluc, Gluc or Nluc.372 The gene for channelrhodopsins can be encoded together with the gene for luciferase leading to a fusion protein consisting of a channelrhodopsins with a luciferase. This led to the development of ‘luminopsins’ which are now a class of optogenetic reporters, in which the addition of coelenterazine luciferin and subsequent light output controls the state of the ion-channel, and hence the ability of neurone cells to fire action potentials.17,373–376 The use of bioluminescent light to control gene expression has been discussed in Section 4.6.
Bioluminescent light has also been used for novel photo-uncaging reactions by Winssinger and co-workers. In 2018, they reported a photo-uncaging using BRET from Nluc to a ruthenium photocatalyst to release pyridinium species. Further developments by this group in 2019 saw the first true ‘bioluminolysis’ wherein no photocatalyst was needed to uncage drugs and molecules of interest using BRET from Nanoluc-Halotag chimera protein (Hluc) to a coumarin photocage (Fig. 20).15,16
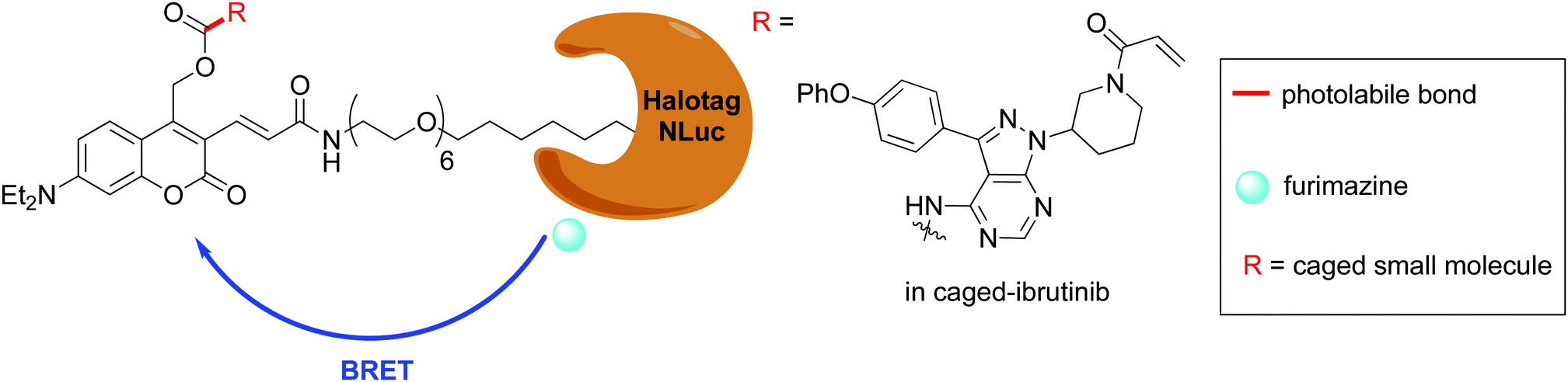 | ||
| Fig. 20 The use of bioluminolysis for the release of the photocaged small-molecule kinase inhibitor ibrutinib by BRET. | ||
Photopharmacology including photodynamic therapy, photouncaging and photoisomerism is an area of growing interest in medicinal chemistry to develop and herness new therapies.377 Bioluminescence has potential to be of great use here as a light source in vivo in close proximity to species of interest. The fact that luciferases are usually genetically encoded makes this a challenging endeavour in humans and complex on several fronts. However, with the advent of luciferase conjugated nanoparticles, this challenge in the delivery of the enzyme to its desired site of action might soon be getting addressed.
5. Blue-sky research and new horizons
5.1 Glowing plants
Bioluminescence imaging has found limited use in the imaging of plant metabolites, proteins and physiology. The fact that plants have chlorophyll rather than haemoglobin means that they have a different and distinct optical window compared to small mammals.378 The four types of chlorophyll each absorb light at two distinct wavelengths; the first λ ∼ 450 nm and the second λ ∼ 650 nm.379 Moreover, the circulation of water and nutrients in plants is not as rapid as in small mammals, and so the delivery of luciferin to the cells of interest is a challenge. This limited the use of BLI to small seedlings or cell cultures that could be grown in Petri dishes in the lab. Nonetheless, despite these limitations, noteworthy work was done to develop our understanding of the circadian clock using a fused, genetically encoded Fluc reporter in a pioneering study by Kay et al.380,381 In their seminal work, Kay et al. fused the Fluc gene with a gene for the promoter of the chlorophyll a-b binding protein (cab2), which is a membrane protein in plant cells and plays an important role in photosynthesis. The Arabidopsis plants were grown on sterile culture plates and sprayed with a solution of luciferin. Bioluminescence imaging was used to visualise the organ localisation of cab2 during the light and dark. In this study, based on the cyclical changes in cab2 expression levels, the length of the circadian clock cycle of the plants could be determined.380 After this work, the authors reported another study in which they used this methodology to identify Arabidopsis mutants which have aberrant circadian clock cycles, and tried to investigate which genes were responsible for these changes.381To circumvent the problem of delivery of D-luciferin to the cells of interest, it was thought that an autoluminescent plant would be of more use. To this end, genetically modified plants were produced, in which the bacterial lux operon would be expressed in the plastids, but this failed to produce sufficient light, partly due to the fact that bioluminescent bacteria emit blue light, whilst chlorophyll absorbs strongly in that region. Moreover, the expression of the bacterial bioluminescent system was found to be toxic to the plant.115 Since the recent discovery of the fully genetically encoded bioluminesce pathway of fungal bioluminescence in 2018,126 this year two different groups reported successfully creating genetically modified plants with the fungal bioluminescence pathway genetically encoded into them, making them autoluminescent.128,382 This opens up a new avenue for BLI in plant research as it has moved BLI in plants from the laboratory Petri-dish to more real life examples grown in soil. Moreover, it also opens the exciting avenue of using such plants for ‘green’ lighting purposes in the future. Notably, in their work Yampolsky and Sarkisyan et al. also reported an autoluminescent mammalian cell line using the genes responsible for fungal bioluminescence.128 However, no attempt was made to compare the brightness of the light output with that obtained from the bacterial ilux operon.
Apart from genetically encoded autoluminescent systems, a nanobionic light emitting plant has also been reported in the literature. In this system, Fluc was conjugated into silica nanoparticles (SNP-Luc) and D-luciferin was conjugated into poly(lactic-co-glycolic acid) (PLGA-LH2) nanoparticles. Both types of nanoparticles were slowly able to release their cargo inside plant cells to allow the bioluminescence reaction to take place and to give the plant a yellow-green glow.18 However, a pressurized bath infusion of nanoparticles was used to administer the mixture of nanoparticles to the plant, which makes this sort of a light emitting plant unsustainable.
5.2 Lifestyle
The natural phenomenon of bioluminescence has enthralled people for centuries and the advent of modern technology has allowed us to bring this natural beauty, from the wilderness to the lab and finally to our homes and places of recreation. Indeed, bioluminescent systems are now being considered as green alternatives to outdoor lighting in public spaces in architecture.383 Moreover, a simple search online would lead one to lamps based on bioluminescent dinoflagellate to decorate the home. The French start-up company Glowee was founded in 2014 and has used bioluminescent bacteria to develop lamps as a form of sustainable lighting. Since it's initiation, the company has managed to increase the lifespan of the bacteria, and hence the lamps from 3 days up to 1 month.As the use of bioluminescent systems become more widespread, they have inspired both scientists and artists alike towards innovation and novel applications. There are a number of examples of artists that are inspired by and actively use bioluminescence in their work. Novel and rare art work known as ‘living art’ has been produced using some bioluminescent systems – namely bioluminescent bacteria and the bioluminescence from single-celled dinoflagellate.384 The bacterial bioluminescence art work is done on Petri dishes and lasts up to 2 weeks, gradually dying off and depicting different aspects of the art as the light fades. Thus, these bioluminescent systems serve as tools for the artist's expression as well as starting points for science communication (Fig. 21).
 | ||
| Fig. 21 Art from bioluminescent creatures – (A) ‘flow visualisation’ by Iyvone Khoo made using bioluminescent dinoflagellate, http://www.iyvonekhoo.co.uk. (B) ‘Rabbit: stage 1’ photograph of drawing created with bioluminescent bacteria, Hunter Cole, http://www.HunterCole.org. | ||
5.3 Limitations of bioluminescent systems
Although the light from bioluminescence has quickly reached several unchartered territories, it is still limited from use in a wider-array of applications by the fact that most of the probes have poor quantum yields, with the quantum yield of D-luciferin/Fluc at 0.41, being one the highest photon outputs from a natural wild-type system. Although, engineered systems such as Nluc/coelenterazine have higher photon outputs, the wavelength of emission of these systems is not ideal for all potential applications and the more-redshifted variants of Nluc are not commercially available. Some of the natural and synthetic luciferins also suffer from issues arising due to poor stability, cell-compatibility and bioavailability. The development of complementary luciferin and luciferase pairs with improved properties is an active area of research, and with further advances, including the cloning and understanding of autoluminescent systems such as that of the fungi, will assuredly lead to more novel applications.6. Conclusions
Research in bioluminescence and bioluminescent organisms has made phenomenal progress over time. From early discovery research into undiscovered luciferins and luciferases, to new applications for known bioluminescent reporters, designer luciferins and luciferases, and other developing technology that assists these systems, a lot of great work has helped develop and progress the field. Moreover, enzyme engineering, biophotonics, computational chemistry and synthetic chemistry are rapidly evolving fields as well and it can be reasonably expected that developments in these areas will feed into the research and applications of bioluminescence in biotechnology. As bioluminescence has found utility in various fields including medicine, biology, physics and engineering and led to exciting multidisciplinary science across all of them, we hope this review, that briefly details the origins and mechanisms of bioluminescence, the currently available luciferin and luciferase systems and covers extensively the key and recent applications of bioluminescence in biotechnology across various fields and makes them accessible to chemists in particular will stimulate further exciting research and foster collaborations in the community.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the artists Iyvonne Khoo and Hunter Cole for sharing their work with us.Notes and references
- S. H. D. Haddock, M. A. Moline and J. F. Case, Ann. Rev. Mar. Sci., 2010, 2, 443–493 CrossRef PubMed.
- V. B. Meyer-Rochow, Luminescence, 2007, 22, 251–265 CrossRef.
- K. F. Stanger-Hall, J. E. Lloyd and D. M. Hillis, Mol. Phylogenet. Evol., 2007, 45, 33–49 CrossRef PubMed.
- R. Boyle, Philos. Trans. R. Soc., 1667, 2, 605–612 Search PubMed.
- O. Shimomura, J. Microsc., 2005, 217, 3–15 CrossRef PubMed.
- A. Chiesa, E. Rapizzi, V. Tosello, P. Pinton, M. De Virgilio, K. E. Fogarty and R. Rizzuto, Biochem. J., 2001, 355, 1–12 CrossRef.
- E. S. Vysotski and J. Lee, Acc. Chem. Res., 2004, 37, 405–415 CrossRef PubMed.
- L. P. Burakova and E. S. Vysotski, Appl. Microbiol. Biotechnol., 2019, 103, 5929–5946 CrossRef PubMed.
- A. Fleiss and K. S. Sarkisyan, Curr. Genet., 2019, 65, 877–882 CrossRef PubMed.
- Z. M. Kaskova, A. S. Tsarkova and I. V. Yampolsky, Chem. Soc. Rev., 2016, 45, 6048–6077 RSC.
- A. A. Kotlobay, M. A. Dubinnyi, K. V. Purtov, E. B. Guglya, N. S. Rodionova, V. N. Petushkov, Y. V. Bolt, V. S. Kublitski, Z. M. Kaskova, R. H. Ziganshin, Y. V. Nelyubina, P. V. Dorovatovskii, I. E. Eliseev, B. R. Branchini, G. Bourenkov, I. A. Ivanov, Y. Oba, I. V. Yampolsky and A. S. Tsarkova, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18911–18916 CrossRef CAS.
- E. H. White, F. McCapra, G. F. Field and W. D. McElroy, J. Am. Chem. Soc., 1961, 83, 2402–2403 CrossRef CAS.
- O. Shimomura, T. Masugi, F. H. Johnson and Y. Haneda, Biochemistry, 1978, 17, 994–998 CrossRef CAS.
- S. Kim, H. Jo, M. Jeon, M. G. Choi, S. K. Hahn and S. H. Yun, Chem. Commun., 2017, 53, 4569–4572 RSC.
- D. Chang, E. Lindberg, S. Feng, S. Angerani, H. Riezman and N. Winssinger, Angew. Chem., Int. Ed., 2019, 58, 16033–16037 CrossRef CAS.
- E. Lindberg, S. Angerani, M. Anzola and N. Winssinger, Nat. Commun., 2018, 9, 3539 CrossRef.
- S. Y. Park, S. H. Song, B. Palmateer, A. Pal, E. D. Petersen, G. P. Shall, R. M. Welchko, K. Ibata, A. Miyawaki, G. J. Augustine and U. Hochgeschwender, J. Neurosci. Res., 2020, 98, 410–421 CrossRef CAS.
- S. Y. Kwak, J. P. Giraldo, M. H. Wong, V. B. Koman, T. T. S. Lew, J. Ell, M. C. Weidman, R. M. Sinclair, M. P. Landry, W. A. Tisdale and M. S. Strano, Nano Lett., 2017, 17, 7951–7961 CrossRef CAS.
- E. Conti, N. P. Franks and P. Brick, Structure, 1996, 4, 287–298 CrossRef CAS.
- O. Shimomura, T. Goto and Y. Hirata, Bull. Chem. Soc. Jpn., 1957, 30, 929–933 CrossRef CAS.
- T. Kishi, T. Goto, Y. Hirata, O. Shimomura and F. H. Johnson, Tetrahedron Lett., 1966, 7, 3427–3436 CrossRef.
- E. M. Thompson, S. Nagata and F. I. Tsuji, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6567–6571 CrossRef.
- K. Hori, H. Charbonneau, R. C. Hart and M. J. Cormier, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4285–4287 CrossRef CAS.
- S. Inouye, K. Watanabe, H. Nakamura and O. Shimomura, FEBS Lett., 2000, 481, 19–25 CrossRef CAS.
- W. W. Lorenz, R. O. McCann, M. Longiaru and M. J. Cormier, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4438–4442 CrossRef CAS.
- J. R. de Wet, K. V. Wood, D. R. Helinski and M. DeLuca, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 7870–7873 CrossRef CAS PubMed.
- K. V. Wood, Y. A. Lam, H. H. Seliger and W. D. McElroy, Science, 1989, 244, 700–702 CrossRef CAS PubMed.
- J. H. Devine, G. D. Kutuzova, V. A. Green, N. N. Ugarova and T. O. Baldwin, Biochim. Biophys. Acta, Gene Struct. Expression, 1993, 1173, 121–132 CrossRef CAS.
- M. Tsutomu, T. Hiroki and N. Eiichi, Gene, 1989, 77, 265–270 CrossRef.
- N. Kajiyama, T. Masuda, H. Tatsumi and E. Nakano, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 228–232 CrossRef CAS.
- H. Tatsumi, N. Kajiyama and E. Nakano, Biochim. Biophys. Acta, Gene Struct. Expression, 1992, 1131, 161–165 CrossRef CAS.
- V. R. Viviani, A. C. R. Silva, G. L. O. Perez, R. V. Santelli, E. J. H. Bechara and F. C. Reinach, Photochem. Photobiol., 1999, 70, 254–260 CrossRef CAS PubMed.
- B. Said Alipour, S. Hosseinkhani, M. Nikkhah, H. Naderi-Manesh, M. J. Chaichi and S. K. Osaloo, Biochem. Biophys. Res. Commun., 2004, 325, 215–222 CrossRef CAS PubMed.
- P. S. F. McCapra, D. J. Gilfoyle, D. W. Young and N. J. Church, Bioluminescence and Chemiluminescence Fundamentals and Applied Aspects, Wiley-Blackwell, 1994 Search PubMed.
- B. R. Branchini, C. E. Behney, T. L. Southworth, D. M. Fontaine, A. M. Gulick, D. J. Vinyard and G. W. Brudvig, J. Am. Chem. Soc., 2015, 137, 7592–7595 CrossRef CAS PubMed.
- J. A. Koo, S. P. Schmidt and G. B. Schuster, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 30–33 CrossRef CAS.
- C. Ribeiro and J. C. G. Esteves da Silva, Photochem. Photobiol. Sci., 2008, 7, 1085–1090 CrossRef CAS.
- H. Fraga, D. Fernandes, J. Novotny, R. Fontes and J. C. G. Esteves da Silva, ChemBioChem, 2006, 7, 929–935 CrossRef CAS.
- Y. Ando, K. Niwa, N. Yamada, T. Enomoto, T. Irie, H. Kubota, Y. Ohmiya and H. Akiyama, Nat. Photonics, 2008, 2, 44–47 CrossRef CAS.
- D. K. Welsh and T. Noguchi, Cold Spring Harb. Protoc., 2012, 7, 852–866 Search PubMed.
- J. R. Knowles, Annu. Rev. Biochem., 1980, 49, 877–919 CrossRef CAS.
- A. Lundin, Methods Enzymol., 2000, 305, 346–370 CAS.
- B. R. Branchini, T. L. Southworth, D. M. Fontaine, D. Kohrt, M. Talukder, E. Michelini, L. Cevenini, A. Roda and M. J. Grossel, Anal. Biochem., 2015, 484, 148–153 CrossRef CAS.
- A. Lundin, B. Jaderlund and T. Lovgren, Clin. Chem., 1982, 28, 609–614 CrossRef CAS.
- G. Scheuerbrandt, A. Lundin, T. Lövgren and W. Mortier, Muscle Nerve, 1986, 9, 11–23 CrossRef CAS.
- A. Lundin, A. Rickardsson and A. Thore, Anal. Biochem., 1976, 75, 611–620 CrossRef CAS.
- A. Lundin, M. Hasenson, J. Persson and Å. Pousette, Methods Enzymol., 1986, 133, 27–42 CAS.
- A. Lundin, Adv. Biochem. Eng. Biotechnol., 2014, 145, 31–62 CrossRef CAS.
- A. Lundin, P. Arner and J. Hellmér, Anal. Biochem., 1989, 177, 125–131 CrossRef CAS.
- J. Hellmér, P. Arner and A. Lundin, Anal. Biochem., 1989, 177, 132–137 CrossRef.
- D. Morse and B. A. Tannous, Mol. Ther., 2012, 20, 692–693 CrossRef CAS.
- M. L. N. Dubuisson, B. De Wergifosse, A. Trouet, F. Baguet, J. Marchand-Brynaert and J. F. Rees, Biochem. Pharmacol., 2000, 60, 471–478 CrossRef CAS.
- A. Roda, M. Guardigli, E. Michelini and M. Mirasoli, TrAC, Trends Anal. Chem., 2009, 28, 307–322 CrossRef CAS.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS.
- G. L. Andreason and G. A. Evans, Anal. Biochem., 1989, 180, 269–275 CrossRef CAS.
- C. H. Contag, S. D. Spilman, P. R. Contag, M. Oshiro, B. Eames, P. Dennery, D. K. Stevenson and D. A. Benaron, Photochem. Photobiol., 1997, 66, 523–531 CrossRef CAS.
- C. H. Contag and M. H. Bachmann, Annu. Rev. Biomed. Eng., 2002, 4, 235–260 CrossRef CAS.
- D. C. McCutcheon, M. A. Paley, R. C. Steinhardt and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 7604–7607 CrossRef CAS.
- D. C. McCutcheon, W. B. Porterfield and J. A. Prescher, Org. Biomol. Chem., 2015, 13, 2117–2121 RSC.
- T. Vo-Dinh, Biomedical Photonics Handbook: Biomedical Diagnostics, CRC Press, 2014, 2nd edn, 2014 Search PubMed.
- R. Shinde, J. Perkins and C. H. Contag, Biochemistry, 2006, 45, 11103–11112 CrossRef CAS.
- M. S. Evans, J. P. Chaurette, S. T. Adams, G. R. Reddy, M. A. Paley, N. Aronin, J. A. Prescher and S. C. Miller, Nat. Methods, 2014, 11, 393–395 CrossRef CAS.
- F. Berger, R. Paulmurugan, S. Bhaumik and S. S. Gambhir, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 2275–2285 CrossRef.
- S. Iwano, M. Sugiyama, H. Hama, A. Watakabe, N. Hasegawa, T. Kuchimaru, K. Z. Tanaka, M. Takahashi, Y. Ishida, J. Hata, S. Shimozono, K. Namiki, T. Fukano, M. Kiyama, H. Okano, S. Kizaka-Kondoh, T. J. McHugh, T. Yamamori, H. Hioki, S. Maki and A. Miyawaki, Science, 2018, 359, 935–939 CrossRef CAS.
- G. Zomer, J. W. Hastings, F. Berthold, A. Lundin, A. M. Garcia Campana, R. Niessner, T. K. Christopolous, C. Lowik, B. Branchini, S. Daunert, L. Blum, L. J. Kricka and A. Roda, Chemiluminescence and Bioluminescence, The Royal Society of Chemistry, 2010 Search PubMed.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS.
- L. Mezzanotte, I. Que, E. Kaijzel, B. Branchini, A. Roda and C. Löwik, PLoS One, 2011, 6, 19277 CrossRef.
- C. L. Stowe, T. A. Burley, H. Allan, M. Vinci, G. Kramer-Marek, D. M. Ciobota, G. N. Parkinson, T. L. Southworth, G. Agliardi, A. Hotblack, M. F. Lythgoe, B. R. Branchini, T. L. Kalber, J. C. Anderson and M. A. Pule, eLife, 2019, 8, e45801 CrossRef.
- R. Podsiadły, A. Grzelakowska, J. Modrzejewska, P. Siarkiewicz, D. Słowiński, M. Szala and M. Świerczyńska, Dyes Pigm., 2019, 170 Search PubMed.
- W. D. McElroy and B. L. Strehler, Arch. Biochem., 1949, 22, 420–433 CAS.
- J. P. Steghens, K. L. Min and J. C. Bernengo, Biochem. J., 1998, 336, 109–113 CrossRef CAS.
- H. Caysa, R. Jacob, N. Müther, B. Branchini, M. Messerle and A. Söling, Photochem. Photobiol. Sci., 2009, 8, 52–56 CrossRef CAS.
- A. Kitayama, H. Yoshizaki, Y. Ohmiya, H. Ueda and T. Nagamune, Photochem. Photobiol., 2003, 77, 333 CrossRef CAS.
- H. H. Seliger and W. D. McElroy, Proc. Natl. Acad. Sci. U. S. A., 1964, 52, 75–81 CrossRef CAS.
- B. R. Branchini, T. L. Southworth, D. M. Fontaine, M. H. Murtiashaw, A. McGurk, M. H. Talukder, R. Qureshi, D. Yetil, J. A. Sundlov and A. M. Gulick, Photochem. Photobiol., 2017, 93, 479–485 CrossRef CAS PubMed.
- G. Oliveira and V. R. Viviani, Photochem. Photobiol. Sci., 2019, 18, 2682–2687 CrossRef CAS PubMed.
- J. F. Thompson, L. S. Hayes and D. B. Lloyd, Gene, 1991, 103, 171–177 CrossRef CAS.
- S. V. Markova, M. D. Larionova and E. S. Vysotski, Photochem. Photobiol., 2019, 95, 705–721 CrossRef CAS.
- G. A. Stepanyuk, Z. J. Liu, S. S. Markova, L. A. Frank, J. Lee, E. S. Vysotski and B. C. Wang, Photochem. Photobiol. Sci., 2008, 7, 442–447 CrossRef CAS PubMed.
- M. Verhaegent and T. K. Christopoulos, Anal. Chem., 2002, 74, 4378–4385 CrossRef.
- M. S. Titushin, S. V. Markova, L. A. Frank, N. P. Malikova, G. A. Stepanyuk, J. Lee and E. S. Vysotski, Photochem. Photobiol. Sci., 2008, 7, 189–196 CrossRef CAS.
- S. V. Markova, S. Golz, L. A. Frank, B. Kalthof and E. S. Vysotski, J. Biol. Chem., 2004, 279, 3212–3217 CrossRef CAS.
- Y. Takenaka, H. Masuda, A. Yamaguchi, S. Nishikawa, Y. Shigeri, Y. Yoshida and H. Mizuno, Gene, 2008, 425, 28–35 CrossRef CAS PubMed.
- Y. Takenaka, A. Yamaguchi, N. Tsuruoka, M. Torimura, T. Gojobori and Y. Shigeri, Mol. Biol. Evol., 2012, 29, 1669–1681 CrossRef CAS PubMed.
- T. B. Shrestha, D. L. Troyer and S. H. Bossmann, Synthesis, 2014, 646–652 CrossRef.
- A. Pichler, J. L. Prior and D. Piwnica-Worms, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1702–1707 CrossRef CAS.
- L. J. Kricka, Encyclopedia of Analytical Science, 2nd edn, 2005 Search PubMed.
- Y. Xu, D. W. Piston and C. H. Johnson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 151–156 CrossRef CAS.
- M. D. Larionova, S. V. Markova and E. S. Vysotski, Biochem. Biophys. Res. Commun., 2017, 483, 772–778 CrossRef CAS PubMed.
- S. Knappskog, H. Ravneberg, C. Gjerdrum, C. Tröße, B. Stern and I. F. Pryme, J. Biotechnol., 2007, 128, 705–715 CrossRef CAS PubMed.
- J. C. Matthews, K. Hori and M. J. Cormier, Biochemistry, 1977, 16, 85–91 CrossRef CAS PubMed.
- M. D. Larionova, S. V. Markova and E. S. Vysotski, J. Photochem. Photobiol., B, 2018, 183, 309–317 CrossRef CAS.
- A. M. Loening, T. D. Fenn, A. M. Wu and S. S. Gambhir, Protein Eng., Des. Sel., 2006, 19, 391–400 CrossRef CAS.
- E. A. Meighen, Microbiol. Rev., 1991, 55, 123–142 CrossRef CAS PubMed.
- J. Engebrecht, K. Nealson and M. Silverman, Cell, 1983, 32, 773–781 CrossRef CAS.
- J. L. Bose, C. S. Rosenberg and E. V. Stabb, Arch. Microbiol., 2008, 190, 169–183 CrossRef CAS PubMed.
- N. E. Boemare, R. J. Akhurst and R. G. Mourant, Int. J. Syst. Bacteriol., 1993, 43, 249–255 CrossRef CAS.
- B. Austin and X. H. Zhang, Lett. Appl. Microbiol., 2006, 43, 119–124 CrossRef CAS PubMed.
- M. J. Jensen, B. M. Tebo, P. Baumann, M. Mandel and K. H. Nealson, Curr. Microbiol., 1980, 3, 311–315 CrossRef.
- W. D. McElroy and A. A. Green, Arch. Biochem. Biophys., 1955, 56, 240–255 CrossRef CAS.
- E. P. C. Rocha, Annu. Rev. Genet., 2008, 42, 211–233 CrossRef CAS.
- H. Echols, Operators and Promoters: The Story of Molecular Biology and Its Creators, University of California Press, 2001 Search PubMed.
- S. Nijvipakul, J. Wongratana, C. Suadee, B. Entsch, D. P. Ballou and P. Chaiyen, J. Bacteriol., 2008, 190, 1531–1538 CrossRef CAS.
- E. Brodl, A. Winkler and P. Macheroux, Comput. Struct. Biotechnol. J., 2018, 16, 551–564 CrossRef CAS.
- T. O. Baldwin, M. Z. Nicoli, J. E. Becvar and J. W. Hastings, J. Biol. Chem., 1975, 250, 2763–2768 CrossRef CAS.
- M. Kurfurst, S. Ghisla and J. W. Hastings, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 2990–2994 CrossRef CAS PubMed.
- J. W. Hastings, C. Balny, C. L. Peuch and P. Douzou, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 3468–3472 CrossRef CAS PubMed.
- J. Vervoort, F. Muller, W. A. M. van den Berg, C. T. W. Moonen and J. Lee, Biochemistry, 1986, 25, 8062–8067 CrossRef CAS.
- F. M. Raushel and T. O. Baldwin, Biochem. Biophys. Res. Commun., 1989, 164, 1137–1142 CrossRef CAS.
- G. Merényi, J. Lind, H. I. X. Mager and S. C. Tu, J. Phys. Chem., 1992, 96, 10528–10533 CrossRef.
- J. W. Eckstein, J. W. Hastings and S. Ghisla, Biochemistry, 1993, 32, 404–411 CrossRef CAS.
- L. F. Greer and A. A. Szalay, Luminescence, 2002, 17, 43–74 CrossRef CAS PubMed.
- C. Koncz, W. H. R. Langridge, O. Olsson, J. Schell and A. A. Szalay, Dev. Genet., 1990, 11, 224–232 CrossRef CAS.
- C. Koncz, O. Olsson and W. H. R. Langridge, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 131–135 CrossRef CAS PubMed.
- A. Krichevsky, B. Meyers, A. Vainstein, P. Maliga and V. Citovsky, PLoS One, 2010, 5, e15461 CrossRef PubMed.
- D. M. Close, S. S. Patterson, S. Ripp, S. J. Baek, J. Sanseverino and G. S. Sayler, PLoS One, 2010, 5, e12441 CrossRef PubMed.
- C. Gregor, J. K. Pape, K. C. Gwosch, T. Gilat, S. J. Sahl and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26491–26496 CrossRef CAS PubMed.
- L. Su, W. Jia, C. Hou and Y. Lei, Biosens. Bioelectron., 2011, 26, 1788–1799 CrossRef CAS PubMed.
- F. Fernández-piñas, I. Rodea-palomares, F. Leganés, M. González-pleiter and M. Angeles MuñOz-MartíN, Adv. Biochem. Eng. Biotechnol., 2014, 145, 65–135 CrossRef.
- T. Xu, D. Close, A. Smartt, S. Ripp and G. Sayler, Adv. Biochem. Eng. Biotechnol., 2014, 144, 111–151 CrossRef CAS PubMed.
- Y. Aleksandrov, Bulg. J. Agric. Sci., 2020, 26, 332–338 Search PubMed.
- J. W. Hastings, K. Weber, J. Friedland, A. Eberhard, G. W. Mitchell and A. Gunsalus, Biochemistry, 1969, 8, 4681–4689 CrossRef CAS PubMed.
- J. G. Dorn, R. J. Frye and R. M. Maier, Appl. Environ. Microbiol., 2003, 69, 2209–2216 CrossRef CAS.
- J. W. Hastings and K. H. Nealson, Annu. Rev. Microbiol., 1977, 31, 549–595 CrossRef CAS.
- F. H. Johnson, O. Shimomura, Y. Saiga, L. C. Gershman, G. T. Reynolds and J. R. Waters, J. Cell. Comp. Physiol., 1962, 60, 85–103 CrossRef CAS.
- A. A. Kotlobay, K. S. Sarkisyan, Y. A. Mokrushina, M. Marcet-Houben, E. O. Serebrovskaya, N. M. Markina, L. G. Somermeyer, A. Y. Gorokhovatsky, A. Vvedensky, K. V. Purtov, V. N. Petushkov, N. S. Rodionova, T. V. Chepurnyh, L. I. Fakhranurova, E. B. Guglya, R. Ziganshin, A. S. Tsarkova, Z. M. Kaskova, V. Shender, M. Abakumov, T. O. Abakumova, I. S. Povolotskaya, F. M. Eroshkin, A. G. Zaraisky, A. S. Mishin, S. V. Dolgov, T. Y. Mitiouchkina, E. P. Kopantzev, H. E. Waldenmaier, A. G. Oliveira, Y. Oba, E. Barsova, E. A. Bogdanova, T. Gabaldón, C. V. Stevani, S. Lukyanov, I. V. Smirnov, J. I. Gitelson, F. A. Kondrashov and I. V. Yampolsky, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12728–12732 CrossRef CAS.
- Z. M. Kaskova, F. A. Dörr, V. N. Petushkov, K. V. Purtov, A. S. Tsarkova, N. S. Rodionova, K. S. Mineev, E. B. Guglya, A. Kotlobay, N. S. Baleeva, M. S. Baranov, A. S. Arseniev, J. I. Gitelson, S. Lukyanov, Y. Suzuki, S. Kanie, E. Pinto, P. Di Mascio, H. E. Waldenmaier, T. A. Pereira, R. P. Carvalho, A. G. Oliveira, Y. Oba, E. L. Bastos, C. V. Stevani and I. V. Yampolsky, Sci. Adv., 2017, 3, e1602847 CrossRef PubMed.
- T. Mitiouchkina, A. S. Mishin, L. G. Somermeyer, N. M. Markina, T. V. Chepurnyh, E. B. Guglya, T. A. Karataeva, K. A. Palkina, E. S. Shakhova, L. I. Fakhranurova, S. V. Chekova, A. S. Tsarkova, Y. V. Golubev, V. V. Negrebetsky, S. A. Dolgushin, P. V. Shalaev, D. Shlykov, O. A. Melnik, V. O. Shipunova, S. M. Deyev, A. I. Bubyrev, A. S. Pushin, V. V. Choob, S. V. Dolgov, F. A. Kondrashov, I. V. Yampolsky and K. S. Sarkisyan, Nat. Biotechnol., 2020, 38, 944–946 CrossRef CAS PubMed.
- L. Liu, T. Wilson and J. W. Hastings, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16555–16560 CrossRef CAS PubMed.
- D. Morse, A. M. Pappenheimer and J. W. Hastings, J. Biol. Chem., 1989, 264, 11822–11826 CrossRef CAS.
- C. Suzuki, Y. Nakajima, H. Akimoto, C. Wu and Y. Ohmiya, Gene, 2005, 344, 61–66 CrossRef CAS PubMed.
- M. Kato, T. Chiba, M. Li and Y. Hanyu, Assay Drug Dev. Technol., 2011, 9, 31–39 CrossRef CAS PubMed.
- M. Endoh, Y. Kobayashi, Y. Yamakami, R. Yonekura, M. Fujii and D. Ayusawa, Biogerontology, 2009, 10, 213–221 CrossRef CAS PubMed.
- O. Shimomura, J. Biolumin. Chemilumin., 1995, 10, 91–101 CrossRef CAS PubMed.
- R. Kay, Proc. R. Soc. London, Ser. B, 1965, 162, 365–386 CAS.
- H. Nakamura, B. Musicki, Y. Kishi and O. Shimomura, J. Am. Chem. Soc., 1988, 110, 2683–2685 CrossRef CAS.
- O. Shimomura and F. H. Johnson, Biochemistry, 1968, 7, 2574–2580 CrossRef CAS PubMed.
- O. Shimomura and F. H. Johnson, Biochemistry, 1968, 7, 1734–1738 CrossRef CAS PubMed.
- M. Nakamura, M. Mamino, M. Masaki, S. Maki, R. Matsui, S. Kojima, T. Hirano, Y. Ohmiya and H. Niwa, Tetrahedron Lett., 2005, 46, 53–56 CrossRef CAS.
- M. Nakamura, M. Masaki, S. Maki, R. Matsui, M. Hieda, M. Mamino, T. Hirano, Y. Ohmiya and H. Niwa, Tetrahedron Lett., 2004, 45, 2203–2205 CrossRef CAS.
- S. Kojima, S. Maki, T. Hirano, Y. Ohmiya and H. Niwa, Tetrahedron Lett., 2000, 41, 4409–4413 CrossRef CAS.
- O. Shimomura, F. H. Johnson and Y. Kohama, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2086–2089 CrossRef CAS.
- R. Bellisario, T. E. Spencer and M. J. Cormier, Biochemistry, 1972, 11, 2256–2266 CrossRef CAS PubMed.
- J. E. Wampler and B. G. M. Jamieson, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1980, 66, 43–50 CrossRef.
- J. E. Wampler, M. G. Mulkerrin and E. S. Rich Jr, Clin. Chem., 1979, 25, 1628–1634 CrossRef CAS.
- N. G. Rudie, M. G. Mulkerrin and J. E. Wampler, Biochemistry, 1981, 20, 344–350 CrossRef CAS PubMed.
- V. N. Petushkov and N. S. Rodionova, J. Photochem. Photobiol., B, 2007, 87, 130–136 CrossRef CAS.
- V. N. Petushkov and N. S. Rodionova, Dokl. Biochem. Biophys., 2005, 401, 115–118 CrossRef CAS PubMed.
- V. N. Petushkov, M. A. Dubinnyi, A. S. Tsarkova, N. S. Rodionova, M. S. Baranov, V. S. Kublitski, O. Shimomura and I. V. Yampolsky, Angew. Chem., Int. Ed., 2014, 53, 5566–5568 CrossRef CAS PubMed.
- Y. Mitani, R. Yasuno, M. Isaka, N. Mitsuda, R. Futahashi, Y. Kamagata and Y. Ohmiya, Sci. Rep., 2018, 8, 12789 CrossRef.
- O. C. Watkins, M. L. Sharpe, N. B. Perry and K. L. Krause, Sci. Rep., 2018, 8, 3278 CrossRef PubMed.
- V. R. Viviani, J. R. Silva, D. T. Amaral, V. R. Bevilaqua, F. C. Abdalla, B. R. Branchini and C. H. Johnson, Sci. Rep., 2020, 10, 9608 CrossRef PubMed.
- H. Zhao, T. C. Doyle, O. Coquoz, F. Kalish, B. W. Rice and C. H. Contag, J. Biomed. Opt., 2005, 10, 041210 CrossRef.
- C. C. Woodroofe, P. L. Meisenheimer, D. H. Klaubert, Y. Kovic, J. C. Rosenberg, C. E. Behney, T. L. Southworth and B. R. Branchini, Biochemistry, 2012, 51, 9807–9813 CrossRef CAS PubMed.
- E. H. White, H. Worther, H. H. Seliger and W. D. McElroy, J. Am. Chem. Soc., 1966, 88, 2015–2019 CrossRef CAS.
- W. Wu, J. Su, C. Tang, H. Bai, Z. Ma, T. Zhang, Z. Yuan, Z. Li, W. Zhou, H. Zhang, Z. Liu, Y. Wang, Y. Zhou, L. Du, L. Gu and M. Li, Anal. Chem., 2017, 89, 4808–4816 CrossRef CAS.
- H. Simonyan, C. Hurr and C. N. Young, Physiol. Genomics, 2016, 48, 762–770 CrossRef CAS PubMed.
- R. C. Steinhardt, C. M. Rathbun, B. T. Krull, J. M. Yu, Y. Yang, B. D. Nguyen, J. Kwon, D. C. McCutcheon, K. A. Jones, F. Furche and J. A. Prescher, ChemBioChem, 2017, 18, 96–100 CrossRef CAS PubMed.
- S. Iwano, R. Obata, C. Miura, M. Kiyama, K. Hama, M. Nakamura, Y. Amano, S. Kojima, T. Hirano, S. Maki and H. Niwa, Tetrahedron, 2013, 69, 3847–3856 CrossRef CAS.
- M. Kiyama, S. Iwano, S. Otsuka, S. W. Lu, R. Obata, A. Miyawaki, T. Hirano and S. A. Maki, Tetrahedron, 2018, 74, 652–660 CrossRef CAS.
- R. Saito, T. Kuchimaru, S. Higashi, S. W. Lu, M. Kiyama, S. Iwano, R. Obata, T. Hirano, S. Kizaka-Kondoh and S. A. Maki, Bull. Chem. Soc. Jpn., 2019, 608–618 CrossRef CAS.
- N. Kitada, R. Saito, R. Obata, S. Iwano, K. Karube, A. Miyawaki, T. Hirano and S. A. Maki, Chirality, 2020, 32, 922–931 CrossRef CAS PubMed.
- J. C. Anderson, C. H. Chang, A. P. Jathoul and A. J. Syed, Tetrahedron, 2019, 75, 347–356 CrossRef CAS.
- G. V. M. Gabriel and V. R. Viviani, Photochem. Photobiol. Sci., 2014, 13, 1661–1670 CrossRef CAS PubMed.
- M. P. Hall, C. C. Woodroofe, M. G. Wood, I. Que, M. Van’T Root, Y. Ridwan, C. Shi, T. A. Kirkland, L. P. Encell, K. V. Wood, C. Löwik and L. Mezzanotte, Nat. Commun., 2018, 9, 132 CrossRef.
- T. Kuchimaru, S. Iwano, M. Kiyama, S. Mitsumata, T. Kadonosono, H. Niwa, S. Maki and S. Kizaka-Kondoh, Nat. Commun., 2016, 7, 11856 CrossRef CAS PubMed.
- S. T. Adams, D. M. Mofford, G. S. K. K. Reddy and S. C. Miller, Angew. Chem., Int. Ed., 2016, 55, 4943–4946 CrossRef CAS PubMed.
- C. M. Rathbun, W. B. Porterfield, K. A. Jones, M. J. Sagoe, M. R. Reyes, C. T. Hua and J. A. Prescher, ACS Cent. Sci., 2017, 3, 1254–1261 CrossRef CAS PubMed.
- S. J. Williams and J. A. Prescher, Acc. Chem. Res., 2019, 52, 3039–3050 CrossRef CAS PubMed.
- K. Saito, Y. F. Chang, K. Horikawa, N. Hatsugai, Y. Higuchi, M. Hashida, Y. Yoshida, T. Matsuda, Y. Arai and T. Nagai, Nat. Commun., 2012, 3, 1262 CrossRef PubMed.
- A. Takai, R. Haruno, T. Nagai and Y. Okada, Mol. Biol. Cell, 2014, 25, 25 Search PubMed.
- A. Takai, M. Nakano, K. Saito, R. Haruno, T. M. Watanabe, T. Ohyanagi, T. Jin, Y. Okada and T. Nagai, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4352–4356 CrossRef CAS.
- K. Suzuki, T. Kimura, H. Shinoda, G. Bai, M. J. Daniels, Y. Arai, M. Nakano and T. Nagai, Nat. Commun., 2016, 7, 13718 CrossRef CAS.
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. MacHleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2012, 7, 1848–1857 CrossRef CAS.
- C. G. England, E. B. Ehlerding and W. Cai, Bioconjugate Chem., 2016, 27, 1175–1187 CrossRef CAS.
- H. W. Yeh, T. Wu, M. Chen and H. W. Ai, Biochemistry, 2019, 58, 1689–1697 CrossRef CAS.
- F. X. Schaub, M. S. Reza, C. A. Flaveny, W. Li, A. M. Musicant, S. Hoxha, M. Guo, J. L. Cleveland and A. L. Amelio, Cancer Res., 2015, 75, 5023–5033 CrossRef CAS PubMed.
- T. Machleidt, C. C. Woodroofe, M. K. Schwinn, J. Méndez, M. B. Robers, K. Zimmerman, P. Otto, D. L. Daniels, T. A. Kirkland and K. V. Wood, ACS Chem. Biol., 2015, 10, 1797–1804 CrossRef CAS PubMed.
- J. Hiblot, Q. Yu, M. D. B. Sabbadini, L. Reymond, L. Xue, A. Schena, O. Sallin, N. Hill, R. Griss and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 14556–14560 CrossRef CAS PubMed.
- S. Inouye and O. Shimomura, Biochem. Biophys. Res. Commun., 1997, 233, 349–353 CrossRef CAS PubMed.
- R. Paulmurugan, Y. Umezawa and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15608–15613 CrossRef CAS.
- A. M. Loening, A. M. Wu and S. S. Gambhir, Nat. Methods, 2007, 4, 641–643 CrossRef CAS PubMed.
- A. Shakhmin, M. P. Hall, T. Machleidt, J. R. Walker, K. V. Wood and T. A. Kirkland, Org. Biomol. Chem., 2017, 15, 8559–8567 RSC.
- H. W. Yeh, O. Karmach, A. Ji, D. Carter, M. M. Martins-Green and H. W. Ai, Nat. Methods, 2017, 14, 971–974 CrossRef CAS PubMed.
- J. Chu, Y. Oh, A. Sens, N. Ataie, H. Dana, J. J. Macklin, T. Laviv, E. S. Welf, K. M. Dean, F. Zhang, B. B. Kim, C. T. Tang, M. Hu, M. A. Baird, M. W. Davidson, M. A. Kay, R. Fiolka, R. Yasuda, D. S. Kim, H. L. Ng and M. Z. Lin, Nat. Biotechnol., 2016, 34, 760–767 CrossRef CAS.
- H. W. Yeh, Y. Xiong, T. Wu, M. Chen, A. Ji, X. Li and H. W. Ai, ACS Chem. Biol., 2019, 14, 959–965 CrossRef CAS.
- Y. Su, J. R. Walker, Y. Park, T. P. Smith, L. X. Liu, M. P. Hall, L. Labanieh, R. Hurst, D. C. Wang, L. P. Encell, N. Kim, F. Zhang, M. A. Kay, K. M. Casey, R. G. Majzner, J. R. Cochran, C. L. Mackall, T. A. Kirkland and M. Z. Lin, Nat. Methods, 2020, 17, 852–860 CrossRef CAS PubMed.
- C. H. Li and S. C. Tu, Biochemistry, 2005, 44, 13866–13873 CrossRef CAS.
- J. M. Sparks and T. O. Baldwin, Biochemistry, 2001, 40, 15436–15443 CrossRef CAS PubMed.
- C. H. Li and S. C. Tu, Biochemistry, 2005, 44, 12970–12977 CrossRef CAS PubMed.
- Z. T. Campbell, A. Weichsel, W. R. Montfort and T. O. Baldwin, Biochemistry, 2009, 48, 6085–6094 CrossRef CAS PubMed.
- Z. T. Campbell and T. O. Baldwin, J. Biol. Chem., 2009, 284, 32827–32834 CrossRef CAS PubMed.
- S. Hosseinkhani, R. Szittner and E. A. Meighen, Biochem. J., 2005, 385, 575–580 CrossRef CAS PubMed.
- L. Y. C. Lin, R. Szittner, R. Friedman and E. A. Meighen, Biochemistry, 2004, 43, 3183–3194 CrossRef CAS.
- C. Gregor, K. C. Gwosch, S. J. Sahl and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 962–967 CrossRef CAS PubMed.
- M. Easter, Food Sci. Technol., 2009, 23, 48–49 Search PubMed.
- M. DeLuca and W. D. McElroy, Biochemistry, 1974, 13, 921–925 CrossRef CAS PubMed.
- A. Lundin and J. Eriksson, Assay Drug Dev. Technol., 2008, 6, 531–541 CrossRef CAS.
- A. Lundin and I. Styrélius, Clin. Chim. Acta, 1978, 87, 199–209 CrossRef CAS.
- S. Lee, C. Zhang and X. Liu, J. Biol. Chem., 2015, 290, 904–917 CrossRef CAS PubMed.
- M. J. Bird, X. W. Wijeyeratne, J. C. Komen, A. Laskowski, M. T. Ryan, D. R. Thorburn and A. E. Frazier, Biosci. Rep., 2014, 34, 701–715 CrossRef CAS PubMed.
- N. Zhang, Z. Fu, S. Linke, J. Chicher, J. J. Gorman, D. Visk, G. G. Haddad, L. Poellinger, D. J. Peet, F. Powell and R. S. Johnson, Cell Metab., 2010, 11, 364–378 CrossRef CAS PubMed.
- K. Palikaras and N. Tavernarakis, Bio-Protoc., 2016, 6, e2048 Search PubMed.
- J. García-Bermúdez, C. Nuevo-Tapioles and J. M. Cuezva, Bio-Protoc., 2016, 6, e1905 Search PubMed.
- B. R. Branchini, T. L. Southworth, D. M. Fontaine, D. Kohrt, F. S. Welcome, C. M. Florentine, E. R. Henricks, D. B. DeBartolo, E. Michelini, L. Cevenini, A. Roda and M. J. Grossel, Anal. Biochem., 2017, 534, 36–39 CrossRef CAS PubMed.
- G. Morciano, A. C. Sarti, S. Marchi, S. Missiroli, S. Falzoni, L. Raffaghello, V. Pistoia, C. Giorgi, F. Di Virgilio and P. Pinton, Nat. Protoc., 2017, 12, 1542–1562 CrossRef CAS PubMed.
- G. F. Pelentir, V. R. Bevilaqua and V. R. Viviani, Photochem. Photobiol. Sci., 2019, 18, 2061–2070 CrossRef CAS PubMed.
- S. Kawabe and Y. Uchiho, Luminescence, 2020, 1–4, DOI:10.1002/bio.3828.
- M. F. Santangelo, S. Libertino, A. P. F. Turner, D. Filippini and W. C. Mak, Biosens. Bioelectron., 2018, 99, 464–470 CrossRef CAS.
- M. M. Calabretta, R. Álvarez-Diduk, E. Michelini, A. Roda and A. Merkoçi, Biosens. Bioelectron., 2020, 150, 1–8 CrossRef PubMed.
- E. Amodio and C. Dino, J. Infect. Public Health, 2014, 7, 92–98 CrossRef PubMed.
- P. Dostálek and T. Brányik, Czech J. Food Sci., 2005, 23, 85–92 CrossRef.
- A. A. Ali, A. B. Altemimi, N. Alhelfi and S. A. Ibrahim, Biosensors, 2020, 10, 58 CrossRef CAS PubMed.
- R. Morrissey, C. Hill and M. Begley, Trends Food Sci. Technol., 2013, 32, 4–15 CrossRef CAS.
- R. A. Cooper, C. J. Griffith, R. E. Malik, P. Obee and N. Looker, Am. J. Infect. Control, 2007, 35, 338–341 CrossRef PubMed.
- T. Lewis, C. Griffith, M. Gallo and M. Weinbren, J. Hosp. Infect., 2008, 69, 156–163 CrossRef CAS PubMed.
- B. G. Mitchell, A. McGhie, G. Whiteley, A. Farrington, L. Hall, K. Halton and N. M. White, Infect. Dis. Health, 2020, 25, 168–174 CrossRef PubMed.
- J. A. Rodriguez and G. Hooper, AORN J., 2019, 110, 596–604 CrossRef PubMed.
- Y. Baba, Y. Sato, G. Owada and S. Minakuchi, J. Prosthodont. Res., 2018, 62, 353–358 CrossRef.
- P. Howgate, Int. J. Food Sci. Technol., 2006, 41, 341–353 CrossRef CAS.
- T. Møretrø, M. A. Normann, H. R. Sæbø and S. Langsrud, J. Appl. Microbiol., 2019, 127, 186–195 CrossRef PubMed.
- J. W. F. Law, N. S. A. Mutalib, K. G. Chan and L. H. Lee, Front. Microbiol., 2015, 5, 1–19 Search PubMed.
- S. N. A. Qazi, E. Counil, J. Morrissey, C. E. D. Rees, A. Cockayne, K. Winzer, W. C. Chan, P. Williams and P. J. Hill, Infect. Immun., 2001, 69, 7074–7082 CrossRef CAS PubMed.
- M. F. Jacobs, S. Tynkkynen and M. Sibakov, Appl. Microbiol. Biotechnol., 1995, 44, 405–412 CrossRef CAS PubMed.
- H. Ramsaran, J. Chen, B. Brunke, A. Hill and M. W. Griffiths, J. Dairy Sci., 1998, 81, 1810–1817 CrossRef CAS PubMed.
- K. J. Allen and M. W. Griffiths, J. Food Prot., 2001, 64, 2058–2062 CrossRef CAS PubMed.
- X. Xu, S. A. Miller, F. Baysal-Gurel, K. H. Gartemann, R. Eichenlaub and G. Rajashekara, Appl. Environ. Microbiol., 2010, 76, 3978–3988 CrossRef CAS.
- M. K. Dommel, E. Frenzel, B. Strasser, C. Blöchinger, S. Scherer and M. Ehling-Schulz, Appl. Environ. Microbiol., 2010, 76, 1232–1240 CrossRef CAS PubMed.
- J. Hardy, K. P. Francis, M. DeBoer, P. Chu, K. Gibbs and C. H. Contag, Science, 2004, 303, 851–853 CrossRef CAS.
- H. Loessner, S. Leschner, A. Endmann, K. Westphal, K. Wolf, K. Kochruebe, T. Miloud, J. Altenbuchner and S. Weiss, Microbes Infect., 2009, 11, 1097–1105 CrossRef CAS PubMed.
- M. Cronin, R. D. Sleator, C. Hill, G. F. Fitzgerald and D. Van Sinderen, BMC Microbiol., 2008, 8, 1–12 CrossRef PubMed.
- S. H. A. Hassan, S. W. Van Ginkel, M. A. M. Hussein, R. Abskharon and S. E. Oh, Environ. Int., 2016, 92–93, 106–118 CrossRef CAS.
- E. A. Widder and B. Falls, IEEE J. Sel. Top. Quantum Electron., 2014, 20, 232–241 Search PubMed.
- N. C. Casavant, D. Thompson, G. A. Beattie, G. J. Phillips and L. J. Halverson, Environ. Microbiol., 2003, 5, 238–249 CrossRef CAS.
- A. Date, P. Pasini and S. Daunert, Anal. Chem., 2007, 79, 9391–9397 CrossRef CAS.
- N. J. Hillson, P. Hu, G. L. Andersen and L. Shapiro, Appl. Environ. Microbiol., 2007, 73, 7615–7621 CrossRef CAS PubMed.
- T. J. H. Jonkers, M. Steenhuis, L. Schalkwijk, J. Luirink, D. Bald, C. J. Houtman, J. Kool, M. H. Lamoree and T. Hamers, Sci. Total Environ., 2020, 729, 139028 CrossRef CAS PubMed.
- A. Kassim, M. I. E. Halmi, S. S. A. Gani, U. H. Zaidan, R. Othman, K. Mahmud and M. Y. A. Shukor, Ecotoxicol. Environ. Saf., 2020, 196, 1–13 CrossRef PubMed.
- M. Bhuvaneshwari, E. Eltzov, B. Veltman, O. Shapiro, G. Sadhasivam and M. Borisover, Water Res., 2019, 164, 1–12 CrossRef PubMed.
- D. K. A. Kusumahastuti, M. Sihtmäe, I. V. Kapitanov, Y. Karpichev, N. Gathergood and A. Kahru, Ecotoxicol. Environ. Saf., 2019, 172, 556–565 CrossRef CAS PubMed.
- D. Harpaz, L. P. Yeo, F. Cecchini, T. H. P. Koon, A. Kushmaro, A. I. Y. Tok, R. S. Marks and E. Eltzov, Molecules, 2018, 23, 2454 CrossRef PubMed.
- E. L. M. Vermeirssen, S. Campiche, C. Dietschweiler, I. Werner and M. Burkhardt, Environ. Toxicol. Chem., 2018, 37, 2246–2256 CrossRef CAS PubMed.
- S. Prévéral, C. Brutesco, E. C. T. Descamps, C. Escoffier, D. Pignol, N. Ginet and D. Garcia, Environ. Sci. Pollut. Res., 2017, 24, 25–32 CrossRef.
- T. Fukuba, Y. Aoki, N. Fukuzawa, T. Yamamoto, M. Kyo and T. Fujii, Lab Chip, 2011, 11, 3508–3515 RSC.
- C. B. Hansen, A. Kerrouche, K. Tatari, A. Rasmussen, T. Ryan, P. Summersgill, M. P. Y. Desmulliez, H. Bridle and H. J. Albrechtsen, J. Microbiol. Methods, 2019, 165, 105713 CrossRef CAS PubMed.
- E. Kabir, A. Azzouz, N. Raza, S. K. Bhardwaj, K. H. Kim, M. Tabatabaei and D. Kukkar, ACS Sens., 2020, 5, 1254–1267 CrossRef CAS PubMed.
- D. T. Nguyen, H. R. Kim, J. H. Jung, K. B. Lee and B. C. Kim, Sens. Actuators, B, 2018, 260, 274–281 CrossRef CAS.
- S. J. Lee, J. S. Park, H. T. Im and H. Il Jung, Sens. Actuators, B, 2008, 132, 443–448 CrossRef CAS.
- T. Han, M. Wren, K. DuBois, J. Therkorn and G. Mainelis, J. Aerosol Sci., 2015, 90, 114–123 CrossRef CAS PubMed.
- C. W. Park, J. W. Park, S. H. Lee and J. Hwang, Biosens. Bioelectron., 2014, 52, 379–383 CrossRef CAS.
- J. W. Park, H. R. Kim and J. Hwang, Anal. Chim. Acta, 2016, 941, 101–107 CrossRef CAS.
- P. Fernandes, Appl. Microbiol. Biotechnol., 2006, 73, 291–296 CrossRef CAS.
- G. Ranalli, P. Pasini and A. Roda, Proceedings of the 9th International Congress on Deterioration and Conservation of Stone, 2000.
- G. Ranalli, E. Zanardini, P. Pasini and A. Roda, Ann. Microbiol., 2003, 53, 1 CAS.
- N. Unković, M. Ljaljević Grbić, M. Stupar, J. Vukojević, G. Subakov-Simić, A. Jelikić and D. Stanojević, Nat. Prod. Res., 2019, 33, 1061–1069 CrossRef.
- J. Li, L. Chen, L. Du and M. Li, Chem. Soc. Rev., 2013, 42, 662–676 RSC.
- E. Lindberg, S. Mizukami, K. Ibata, A. Miyawaki and K. Kikuchi, Chem. – Eur. J., 2013, 19, 14970–14976 CrossRef CAS PubMed.
- M. Yuan, X. Ma, T. Jiang, C. Zhang, H. Chen, Y. Gao, X. Yang, L. Du and M. Li, Org. Biomol. Chem., 2016, 14, 10267–10274 RSC.
- N. Nomura, R. Nishihara, T. Nakajima, S. B. Kim, N. Iwasawa, Y. Hiruta, S. Nishiyama, M. Sato, D. Citterio and K. Suzuki, Anal. Chem., 2019, 91, 9546–9553 CrossRef CAS PubMed.
- D. H. Klaubert, P. Meisenheimer, J. Unch and W. Zhou, Eur. Pat. Search PubMed WO2012/061477, 2012.
- M. A. Sellmyer, L. Bronsart, H. Imoto, C. H. Contag, T. J. Wandless and J. A. Prescher, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8567–8572 CrossRef CAS PubMed.
- D. M. Mofford, S. T. Adams, G. S. K. K. Reddy, G. R. Reddy and S. C. Miller, J. Am. Chem. Soc., 2015, 137, 8684–8687 CrossRef CAS PubMed.
- R. Geigera, E. Schneider, K. Wallenfels and W. Miska, Biol. Chem. Hoppe-Seyler, 1992, 373, 1187–1192 CrossRef PubMed.
- A. Dragulescu-Andrasi, G. Liang and J. Rao, Bioconjugate Chem., 2009, 20, 1660–1666 CrossRef CAS PubMed.
- H. Yao, M. K. So and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 7031–7034 CrossRef CAS PubMed.
- T. Monsees, W. Miska and R. Geiger, Anal. Biochem., 1994, 221, 329–334 CrossRef CAS PubMed.
- W. Zhou, J. W. Shultz, N. Murphy, E. M. Hawkins, L. Bernad, T. Good, L. Moothart, S. Frackman, D. H. Klaubert, R. F. Bulleit and K. V. Wood, Chem. Commun., 2006, 4620–4622 RSC.
- A. G. Vorobyeva, M. Stanton, A. Godinat, K. B. Lund, G. G. Karateev, K. P. Francis, E. Allen, J. G. Gelovani, E. McCormack, M. Tangney and E. A. Dubikovskaya, PLoS One, 2015, 10, e0131037 CrossRef PubMed.
- S. J. Duellman, M. P. Valley, V. Kotraiah, J. Vidugiriene, W. Zhou, L. Bernad, J. Osterman, J. J. Kimball, P. Meisenheimer and J. J. Cali, Anal. Biochem., 2013, 434, 226–232 CrossRef CAS PubMed.
- A. S. Cohen, E. A. Dubikovskaya, J. S. Rush and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 8563–8565 CrossRef CAS PubMed.
- X. Tian, Z. Li, C. Lau and J. Lu, Anal. Chem., 2015, 87, 11325–11331 CrossRef CAS PubMed.
- G. C. Van De Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed.
- X. Tian, X. Liu, A. Wang, C. Lau and J. Lu, Anal. Chem., 2018, 90, 5951–5958 CrossRef CAS PubMed.
- A. Wang, X. Li, Y. Ju, D. Chen and J. Lu, Analyst, 2020, 145, 550–556 RSC.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- M. C. Heffern, H. M. Park, H. Y. Au-Yeung, G. C. Van De Bittner, C. M. Ackerman, A. Stahl and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14219–14224 CrossRef CAS PubMed.
- A. T. Aron, M. C. Heffern, Z. R. Lonergan, M. N. Vander Wal, B. R. Blank, B. Spangler, Y. Zhang, H. M. Park, A. Stahl, A. R. Renslo, E. P. Skaar and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12669–12674 CrossRef CAS.
- P. Feng, L. Ma, F. Xu, X. Gou, L. Du, B. Ke and M. Li, Talanta, 2019, 203, 29–33 CrossRef CAS PubMed.
- B. Ke, L. Ma, T. Kang, W. He, X. Gou, D. Gong, L. Du and M. Li, Anal. Chem., 2018, 90, 4946–4950 CrossRef CAS PubMed.
- T. A. Su, K. J. Bruemmer and C. J. Chang, Curr. Opin. Biotechnol., 2019, 60, 198–204 CrossRef CAS PubMed.
- A. A. Bazhin, R. Sinisi, U. De Marchi, A. Hermant, N. Sambiagio, T. Maric, G. Budin and E. A. Goun, Nat. Chem. Biol., 2020, 16, 1385–1393 CrossRef CAS PubMed.
- A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- H. Takakura, R. Kojima, M. Kamiya, E. Kobayashi, T. Komatsu, T. Ueno, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4010–4013 CrossRef CAS.
- R. Kojima, H. Takakura, M. Kamiya, E. Kobayashi, T. Komatsu, T. Ueno, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, Angew. Chem., Int. Ed., 2015, 54, 14768–14771 CrossRef CAS PubMed.
- T. T. Ong, Z. Ang, R. Verma, R. Koean, J. K. C. Tam and J. L. Ding, Front. Bioeng. Biotechnol., 2020, 8, 412 CrossRef PubMed.
- Y. Qian, V. Rancic, J. Wu, K. Ballanyi and R. E. Campbell, ChemBioChem, 2019, 20, 516–520 CrossRef CAS PubMed.
- R. Griss, A. Schena, L. Reymond, L. Patiny, D. Werner, C. E. Tinberg, D. Baker and K. Johnsson, Nat. Chem. Biol., 2014, 10, 598–603 CrossRef CAS PubMed.
- L. Xue, Q. Yu, R. Griss, A. Schena and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 7112–7116 CrossRef CAS PubMed.
- R. Arts, I. Den Hartog, S. E. Zijlema, V. Thijssen, S. H. E. Van Der Beelen and M. Merkx, Anal. Chem., 2016, 88, 4525–4532 CrossRef CAS PubMed.
- R. Arts, S. K. J. Ludwig, B. C. B. Van Gerven, E. M. Estirado, L. G. Milroy and M. Merkx, ACS Sens., 2017, 2, 1730–1736 CrossRef CAS.
- K. Tenda, B. van Gerven, R. Arts, Y. Hiruta, M. Merkx and D. Citterio, Angew. Chem., Int. Ed., 2018, 57, 15369–15373 CrossRef CAS PubMed.
- K. Tomimuro, K. Tenda, Y. Ni, Y. Hiruta, M. Merkx and D. Citterio, ACS Sens., 2020, 5, 1786–1794 CrossRef CAS PubMed.
- I. Brányiková, S. Lucáková, G. Kuncová, J. Trögl, V. Synek, J. Rohovec and T. Navrátil, Sensors, 2020, 20, 3138 CrossRef PubMed.
- R. Hajimohammadi, M. Johari-Ahar and S. Ahmadpour, Int. J. Environ. Anal. Chem., 2020 DOI:10.1080/03067319.2020.1760858.
- E. L. Sciuto, M. A. Coniglio, D. Corso, J. R. van der Meer, F. Acerbi, A. Gola and S. Libertino, Water, 2019, 11, 1986 CrossRef CAS.
- S. R. White and T. K. Christopoulos, Nucleic Acids Res., 1999, 27, e25–32 CrossRef CAS PubMed.
- T. K. Christopoulos and N. H. L. Chiu, Anal. Chem., 1995, 67, 4290–4294 CrossRef PubMed.
- J. Y. Byun, K. H. Lee, Y. B. Shin and D. M. Kim, ACS Sens., 2019, 4, 93–99 CrossRef CAS PubMed.
- N. H. L. Chiu and T. K. Christopoulos, Anal. Chem., 1996, 68, 2304–2308 CrossRef CAS PubMed.
- F. A. Iannotti, E. Pagano, O. Guardiola, S. Adinolfi, V. Saccone, S. Consalvi, F. Piscitelli, E. Gazzerro, G. Busetto, D. Carrella, R. Capasso, P. L. Puri, G. Minchiotti and V. Di Marzo, Nat. Commun., 2018, 9, 3950 CrossRef PubMed.
- E. Rincón, T. Cejalvo, D. Kanojia, A. Alfranca, M. Á. Rodríguez-Milla, R. A. G. Hoyos, Y. Han, L. Zhang, R. Alemany, M. S. Lesniak and J. García-Castro, Oncotarget, 2017, 8, 45415–45431 CrossRef PubMed.
- A. Taminiau, A. Draime, J. Tys, B. Lambert, J. Vandeputte, N. Nguyen, P. Renard, D. Geerts and R. Rezsöhazy, Nucleic Acids Res., 2016, 44, 7331–7349 CAS.
- S. Bhaumik and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 377–382 CrossRef CAS.
- G. Zhao, L. Du, C. Ma, Y. Li, L. Li, V. K. Poon, L. Wang, F. Yu, B. J. Zheng, S. Jiang and Y. Zhou, Virol. J., 2013, 10, 266 CrossRef PubMed.
- Y. Yang, L. Du, C. Liu, L. Wang, C. Ma, J. Tang, R. S. Baric, S. Jiang and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12516–12521 CrossRef CAS PubMed.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 581, 221–224 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS.
- C. K. Kim, K. F. Cho, M. W. Kim and A. Y. Ting, eLife, 2019, 8, e43826 CrossRef PubMed.
- K. D. G. Pfleger and K. A. Eidne, Nat. Methods, 2006, 3, 165–174 CrossRef CAS.
- N. C. Dale, E. K. M. Johnstone, C. W. White and K. D. G. Pfleger, Front. Bioeng. Biotechnol., 2019, 7, 56 CrossRef PubMed.
- T. Azad, A. Tashakor and S. Hosseinkhani, Anal. Bioanal. Chem., 2014, 406, 5541–5560 CrossRef PubMed.
- D. C. Harris, Quantitative Chemical Analysis, 8th edn, 2010, pp. 419–444 Search PubMed.
- J. Zheng, Methods Mol. Biol., 2006, 337, 65–77 CAS.
- P. A. Johnston, D. D. Dudgeon, S. N. Shinde, T. Y. Shun, J. S. Lazo, C. J. Strock, K. A. Giuliano, D. L. Taylor and P. A. Johnston, Assay Drug Dev. Technol., 2010, 8, 437–458 CrossRef.
- L. Comps-Agrar, D. Maurel, P. Rondard, J. P. Pin, E. Trinquet and L. Prézeau, Methods Mol. Biol., 2011, 756, 201–214 CrossRef CAS PubMed.
- N. S. Eyre, A. L. Aloia, M. A. Joyce, M. Chulanetra, D. L. Tyrrell and M. R. Beard, Virology, 2017, 507, 20–31 CrossRef CAS PubMed.
- X. L. Mo, Y. Luo, A. A. Ivanov, R. Su, J. J. Havel, Z. Li, F. R. Khuri, Y. Du and H. Fu, J. Mol. Cell Biol., 2016, 8, 271–281 CrossRef CAS PubMed.
- E. Moreno, J. Canet, E. Gracia, C. Lluís, J. Mallol, E. I. Canela, A. Cortés and V. Casadó, Front. Pharmacol., 2018, 9, 106 CrossRef.
- Q. Wan, N. Okashah, A. Inoue, R. Nehme, B. Carpenter, C. G. Tate and N. A. Lambert, J. Biol. Chem., 2018, 293, 7466–7473 CrossRef.
- B. L. Hoare, S. Bruell, A. Sethi, P. R. Gooley, M. J. Lew, M. A. Hossain, A. Inoue, D. J. Scott and R. A. D. Bathgate, iScience, 2019, 11, 93–113 CrossRef PubMed.
- D. C. Alcobia, A. I. Ziegler, A. Kondrashov, E. Comeo, S. Mistry, B. Kellam, A. Chang, J. Woolard, S. J. Hill and E. K. Sloan, iScience, 2018, 6, 280–288 CrossRef CAS.
- B. R. Branchini, D. M. Ablamsky and J. C. Rosenberg, Bioconjugate Chem., 2010, 21, 2023–2030 CrossRef CAS.
- A. Kamkaew, H. Sun, C. G. England, L. Cheng, Z. Liu and W. Cai, Chem. Commun., 2016, 52, 6997–7000 RSC.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS PubMed.
- O. M. Thirukkumaran, C. Wang, N. J. Asouzu, E. Fron, S. Rocha, J. Hofkens, L. D. Lavis and H. Mizuno, Front. Chem., 2020, 7, 938 CrossRef PubMed.
- M. C. Wehr and M. J. Rossner, Drug Discovery Today, 2016, 21, 415–429 CrossRef CAS.
- K. E. Luker, M. C. P. Smith, G. D. Luker, S. T. Gammon, H. Piwnica-Worms and D. Piwnica-Worms, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12288–12293 CrossRef CAS PubMed.
- E. Stefan, S. Aquin, N. Berger, C. R. Landry, B. Nyfeler, M. Bouvier and S. W. Michnick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16916–16921 CrossRef CAS PubMed.
- I. Remy and S. W. Michnick, Nat. Methods, 2006, 3, 977–979 CrossRef CAS.
- B. K. Sung, M. Sato and H. Tao, Anal. Chem., 2009, 81, 67–74 CrossRef.
- K. E. Luker and G. D. Luker, Methods Mol. Biol., 2014, 1098, 59–69 CrossRef CAS.
- F. Z. Wang, N. Zhang, Y. J. Guo, B. Q. Gong and J. F. Li, J. Integr. Plant Biol., 2020, 62, 1065–1079 CrossRef CAS.
- Y. Kanai, S. Komoto, T. Kawagishi, R. Nouda, N. Nagasawa, M. Onishi, Y. Matsuura, K. Taniguchi and T. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2343–2348 CrossRef PubMed.
- B. Mistry, J. S. Long, J. Schreyer, E. Staller, R. Y. Sanchez-David and W. S. Barclay, J. Virol., 2019, 94, e01353–19 Search PubMed.
- J. Zhao, T. J. Nelson, Q. Vu, T. Truong and C. I. Stains, ACS Chem. Biol., 2016, 11, 132–138 CrossRef CAS PubMed.
- L. Shen, Y. Yang, F. Ye, G. Liu, M. Desforges, P. J. Talbot and W. Tan, Antimicrob. Agents Chemother., 2016, 60, 5492–5503 CrossRef CAS PubMed.
- L. Shen, J. Niu, C. Wang, B. Huang, W. Wang, N. Zhu, Y. Deng, H. Wang, F. Ye, S. Cen and W. Tan, J. Virol., 2019, 93, e00023–19 CrossRef CAS.
- F. L. Esteban Florez, R. D. Hiers, Y. Zhao, J. Merritt, A. J. Rondinone and S. S. Khajotia, Dent. Mater., 2020, 36, 353–365 CrossRef.
- N. Sengupta, M. Jović, E. Barnaeva, D. W. Kim, X. Hu, N. Southall, M. Dejmek, I. Mejdrova, R. Nencka, A. Baumlova, D. Chalupska, E. Boura, M. Ferrer, J. Marugan and T. Balla, J. Lipid Res., 2019, 60, 683–693 CrossRef CAS.
- H. Y. Jin, Y. Tudor, K. Choi, Z. Shao, B. A. Sparling, J. G. McGivern and A. Symons, SLAS Discovery, 2020, 25, 215–222 CAS.
- N. Thorne, D. S. Auld and J. Inglese, Curr. Opin. Chem. Biol., 2010, 14, 315–324 CrossRef CAS PubMed.
- N. Thorne, J. Inglese and D. S. Auld, Chem. Biol., 2010, 17, 646–657 CrossRef CAS.
- L. Mezzanotte, M. van‘t Root, H. Karatas, E. A. Goun and C. W. G. M. Löwik, Trends Biotechnol., 2017, 35, 640–652 CrossRef CAS.
- M. Endo and T. Ozawa, Int. J. Mol. Sci., 2020, 21, 1–19 Search PubMed.
- I. D. Jacobsen, A. Lüttich, O. Kurzai, B. Hube and M. Brock, J. Antimicrob. Chemother., 2014, 69, 2785–2796 CrossRef CAS.
- M. Hutchens and G. D. Luker, Cell. Microbiol., 2007, 9, 2315–2322 CrossRef CAS PubMed.
- E. A. Karlsson, V. A. Meliopoulos, C. Savage, B. Livingston, A. Mehle and S. Schultz-Cherry, Nat. Commun., 2015, 6, 6738 CrossRef.
- J. Niu, L. Shen, B. Huang, F. Ye, L. Zhao, H. Wang, Y. Deng and W. Tan, Antiviral Res., 2020, 173, 104646 CrossRef CAS.
- A. T. Phillips, A. B. Rico, C. B. Stauft, S. L. Hammond, T. A. Aboellail, R. B. Tjalkens and K. E. Olson, J. Virol., 2016, 90, 5785–5796 CrossRef.
- S. Dorsaz, A. T. Coste and D. Sanglard, Front. Microbiol., 2017, 8, 1478 CrossRef.
- J. P. J. Saeij, J. P. Boyle, M. E. Grigg, G. Arrizabalaga and J. C. Boothroyd, Infect. Immun., 2005, 73, 695–702 CrossRef CAS.
- M. D. Lewis, A. Fortes Francisco, M. C. Taylor, H. Burrell-Saward, A. P. Mclatchie, M. A. Miles and J. M. Kelly, Cell. Microbiol., 2014, 16, 1285–1300 CrossRef.
- A. F. Francisco, M. D. Lewis, S. Jayawardhana, M. C. Taylor, E. Chatelain and J. M. Kelly, Antimicrob. Agents Chemother., 2015, 59, 4653–4661 CrossRef PubMed.
- J. Q. Reimão, C. T. Trinconi, J. K. Yokoyama-Yasunaka, D. C. Miguel, S. P. Kalil and S. R. B. Uliana, J. Microbiol. Methods, 2013, 93, 95–101 CrossRef.
- E. Calvo-Alvarez, C. Cren-Travaillé, A. Crouzols and B. Rotureau, Infect., Genet. Evol., 2018, 63, 391–403 CrossRef PubMed.
- J. L. C. Wong, M. Romano, L. E. Kerry, H. S. Kwong, W. W. Low, S. J. Brett, A. Clements, K. Beis and G. Frankel, Nat. Commun., 2019, 10, 3957 CrossRef.
- C. N. Berger, V. F. Crepin, T. I. Roumeliotis, J. C. Wright, D. Carson, M. Pevsner-Fischer, R. C. D. Furniss, G. Dougan, M. Dori-Bachash, L. Yu, A. Clements, J. W. Collins, E. Elinav, G. J. Larrouy-Maumus, J. S. Choudhary and G. Frankel, Cell Metab., 2017, 26, 738–752.e6 CrossRef.
- D. Vecchio, T. Dai, L. Huang, L. Fantetti, G. Roncucci and M. R. Hamblin, J. Biophotonics, 2013, 6, 733–742 CrossRef.
- L. Lu, B. Li, S. Ding, Y. Fan, S. Wang, C. Sun, M. Zhao, C. X. Zhao and F. Zhang, Nat. Commun., 2020, 11, 4192 CrossRef.
- T. Theodossiou, J. S. Hothersall, E. A. Woods, K. Okkenhaug, J. Jacobson and A. J. MacRobert, Cancer Res., 2003, 63, 1818–1821 CAS.
- M. L. Schipper, M. R. Patel and S. S. Gambhir, Mol. Imaging Biol., 2006, 8, 218–225 CrossRef PubMed.
- C. Y. Hsu, C. W. Chen, H. P. Yu, Y. F. Lin and P. S. Lai, Biomaterials, 2013, 34, 1204–1212 CrossRef.
- C. M. Magalhães, J. C. G. Esteves da Silva and L. Pinto da Silva, ChemPhysChem, 2016, 2286–2294 CrossRef.
- Y. Yang, W. Hou, S. Liu, K. Sun, M. Li and C. Wu, Biomacromolecules, 2018, 19, 201–208 CrossRef CAS.
- A. C. Love and J. A. Prescher, Cell Chem. Biol., 2020, 27, 904–920 CrossRef CAS PubMed.
- L. Fenno, O. Yizhar and K. Deisseroth, Annu. Rev. Neurosci., 2011, 34, 389–412 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS.
- G. Nagel, D. Ollig, M. Fuhrmann, S. Kateriya, A. M. Musti, E. Bamberg and P. Hegemann, Science, 2002, 296, 2395–2398 CrossRef CAS.
- O. A. Sineshchekov, K. H. Jung and J. L. Spudich, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8689–8694 CrossRef CAS PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263–1268 CrossRef CAS.
- C. Bamann, T. Kirsch, G. Nagel and E. Bamberg, J. Mol. Biol., 2008, 375, 686–694 CrossRef CAS.
- K. Berglund, E. Birkner, G. J. Augustine and U. Hochgeschwender, PLoS One, 2013, 8, e59759 CrossRef CAS PubMed.
- K. Berglund, K. Clissold, H. E. Li, L. Wen, S. Y. Park, J. Gleixner, M. E. Klein, D. Lu, J. W. Barter, M. A. Rossi, G. J. Augustine, H. H. Yin and U. Hochgeschwender, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E358–E367 CrossRef CAS.
- K. Berglund, A. M. Fernandez, C. A. N. Gutekunst, U. Hochgeschwender and R. E. Gross, J. Neurosci. Res., 2020, 98, 422–436 CrossRef CAS.
- J. R. Zenchak, B. Palmateer, N. Dorka, T. M. Brown, L. M. Wagner, W. E. Medendorp, E. D. Petersen, M. Prakash and U. Hochgeschwender, J. Neurosci. Res., 2020, 98, 458–468 CrossRef CAS.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS.
- Y. Furuhata, A. Sakai, T. Murakami, A. Nagasaki and Y. Kato, PLoS One, 2020, 15, e0227477 CrossRef CAS.
- R. J. Porra, W. A. Thompson and P. E. Kriedemann, Biochim. Biophys. Acta, Bioenerg., 1989, 975, 384–394 CrossRef CAS.
- A. J. Millar, S. R. Short, N. H. Chua and S. A. Kay, Plant Cell, 1992, 4, 1075–1087 CAS.
- A. J. Millar, I. A. Carré, C. A. Strayer, N. H. Chua and S. A. Kay, Science, 1995, 267, 1161–1163 CrossRef CAS.
- A. Khakhar, C. G. Starker, J. C. Chamness, N. Lee, S. Stokke, C. Wang, R. Swanson, F. Rizvi, T. Imaizumi and D. F. Voytas, eLife, 2020, 9, e52786 CrossRef CAS.
- R. Narboni, Light Eng., 2020, 28, 4–16 Search PubMed.
- M. I. Latz, Mater. Today: Proc., 2017, 4, 4959–4968 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs01492c |
| ‡ Present address: School of Chemistry, University of Leeds, Woodhouse Lane, LS2 9JT, Leeds, UK. |
| This journal is © The Royal Society of Chemistry 2021 |