
Biochemical and artificial pathways for the reduction of carbon dioxide, nitrite and the competing proton reduction: effect of 2nd sphere interactions in catalysis
Sk
Amanullah
 ,
Paramita
Saha
,
Abhijit
Nayek
,
Md Estak
Ahmed
and
Abhishek
Dey
*
,
Paramita
Saha
,
Abhijit
Nayek
,
Md Estak
Ahmed
and
Abhishek
Dey
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, 2A & 2B Raja SC Mullick Road, Kolkata, 700032, India. E-mail: icad@iacs.res.in
First published on 29th January 2021
Abstract
Reduction of oxides and oxoanions of carbon and nitrogen are of great contemporary importance as they are crucial for a sustainable environment. Substantial research has been dedicated to these areas in the last few decades. These reductions require both electrons and protons and their thermodynamic potentials often make them compete with hydrogen evolution reaction i.e., the reaction of protons and electrons to generate H2. These reactions are abundant in the environment in microorganisms and are facilitated by naturally occurring enzymes. This review brings together the state-of-the-art knowledge in the area of enzymatic reduction of CO2, NO2− and H+ with those of artificial molecular electrocatalysis. A simple ligand field theory-based design principle for electrocatalysts is first described. The electronic structure considerations developed automatically yield the basic geometry required and the 2nd sphere interactions which can potentially aid the activation and the further reduction of these small molecules. A systematic review of the enzymatic reaction followed by those reported in artificial molecular electrocatalysts is presented for the reduction of CO2, NO2− and H+. The review is focused on mechanism of action of these metalloenzymes and artificial electrocatalysts and discusses general principles that guide the rates and product selectivity of these reactions. The importance of the 2nd sphere interactions in both enzymatic and artificial molecular catalysis is discussed in detail.
 Sk Amanullah | Sk Amanullah received his BSc (2012) from Presidency College, Kolkata and MSc (2014) from Indian Institute of Technology, Kharagpur. He joined Prof. Dey's group as a PhD scholar in 2014. His thesis is focused to understand nature's choice of macrocyclic porphyrinoids for diverse reactivity. |
 Paramita Saha | Paramita Saha received her BSc (2015) and MSc (2017) from Jadavpur University. She joined Prof. Dey's group as a PhD scholar in 2017. Her thesis is focused to the synthesis of metallo-clusters for catalysis relevant to sustainable energy and environment. |
 Abhijit Nayek | Abhijit Nayek received his BSc (2015) from Midnapore College and MSc (2017) from Ramakrishna Mission Residential College, Narendrapur. He joined Prof. Dey's group as a PhD scholar in 2017. His current research focuses on understanding synthesis of active site mimics of [FeFe]-hydrogenases and the mechanism of H2–H+ interconversion. |
 Md Estak Ahmed | Md Estak Ahmed received his BSc (2012) from Ramakrishna Mission Residential College, Narendrapur and MSc (2014) from Indian Institute of Technology, Kanpur. He joined Prof. Dey's group as a PhD scholar in 2014. His thesis was focused to bio-inspired models of metalloenzymes involved in low-valent transformations. Currently he is a postdoctoral researcher at Georgetown University, USA. |
 Abhishek Dey | Abhishek Dey received his BSc (1999) from Presidency College, Kolkata and MSc (2001) from Indian Institute of Technology, Kanpur and PhD (2007) from Stanford University, CA, USA. After postdoctoral stay at Stanford University, he moved to Indian Association for the Cultivation of Science (IACS), Kolkata as an Assistant professor (2009). Currently, he is a professor at the same department. His primary research interests include inorganic reaction mechanisms relevant to renewable energy and clean environment. |
Introduction
Human activity over the last century has resulted in an increased distribution of oxidized forms of carbon, nitrogen and sulfur in the environment.1 Occam's razor would suggest that much of this is derived from the use of fossil fuel for energy to drive industrial and technological developments.2 Obtention of the energy stored in the C–H and C–C bonds of carbon-based fuels by oxidizing them with O2 has, expectedly, released large amounts of CO2 in the atmosphere; much beyond the capacity of the natural carbon fixation mechanism.3 This has led to rising concerns regarding the sustanibility of our current lifestyles and industrial activities. Logically, this has spurred research activites to find new methods of fixing these oxides and oxoanions of carbon, nitrogen and sulfur. These include both chemical and electrochemical methods. And inevitably a switch to non-fossil energy sources, such as wind, solar, nuclear, geothermal, etc is being advocated.4Atmospheric CO2 could be captured and converted into value added chemicals such as formic acid, carbon monoxide, methanol, ethylene, etc.5–9 Over the last few decades, extensive research has been conducted resulting in a number of homogeneous10,11 and heterogeneous12–14 electrochemical and photochemical15,16 pathways to achieve the same. Alternatively, the reduction of CO2 to various C-based products is a part of the geochemical Carbon cycle which fixes 250 gigatonnes of CO2 every year. Several earth abundant metal-based metalloenzymes are involved in this process which occurs under ambient conditions. The reduction of CO2 follows fundamentally different mechanisms in these enzymes. In photosynthetic carbondioxide reduction, the CO2, post capture, is reduced by hydride, more specifically via NADPH, by two electrons.17 In the reductive acetyl CoA pathway, CO2 is reduced to CO using H+ and e− derived from H2. In methanogens, however, the pathway to produce CH4 from CO2 involves reduction with hydride, molecular H2 as well as H+ and e− derived from H2via hydrogenases.18
Similarly, reduction of oxides and oxoanions of nitrogen poses challenges and heralds promises. Emission of NOx and SOx have same sources as COxi.e., fossil fuels. The atmospheric concentration of these oxides has been steadily increasing over the last decade and their detrimental effects is not limited to acid rain anymore but include long term damage to human health as well as the ozone layer.19 Like CO2, there are different routes of biochemical nitrogen cycle involve interconversion of various oxides of nitrogen (NxOy) each of which has prominent roles in biology. Nitrite, in particular, is a pivotal entity in the nitrogen cycle, mediating conversion of inorganic nitrates to ammonia or nitric oxide (NO). The NO generated is eventually recycled back to biological nitrogen pool. Nitrite reduction may be an assimilatory, respiratory or dissimilatory process. Assimilatory nitrite reduction by siroheme containing nitrite reductase produces ammonia which participates in biosynthetic pathways utilised for cell growth in bacteria. Some bacteria also exploit respiratory nitrite ammonification, involving cytochrome c containing nitrite reductase, as a process where nitrite acts as the terminal electron acceptor of the respiratory chain. This is a dissimilatory mode of energy conservation in lower living organisms. A contrasting route of nitrite reduction is denitrification. Denitrification involves the anaerobic nitrate reduction to molecular dinitrogen. Dissimilatory nitrite reductases, a heme or a Cu-containing enzyme, in denitrifiers converts nitrite to nitric oxide.
Diminishing fossil fuel and increasing global energy demand has encouraged the investigation into means of storing energy in the form of value added chemicals which can be obtained by the reduction of CO2 as well as oxonions of nitrogen like NO2−.20,21 In nature, the reduction of CO2 and NO2− proceeds through electron transfer from proteins like ferredoxin which are also involved in the reduction of proton from H2O to H2i.e., the reduction potentials for CO2, NO2− and H+ reduction are similar. Hence, while protons, H+, are necessary to reduce both CO2 and NO2−, its reduction to H2 competes with reduction of CO2 and NO2− itself. At the same time H2 itself is a convenient carier of energy and this energy can be extracted from it either by combustion or electrochemically. This, along with the drive to generate fuel from CO2, NO2−etc., has automatically brought to focus the storage of energy in the form of hydrogen itself. Hydrogen can be generated via reduction of H+ by e− and these, including H2, are components essential to reduction of CO2 and NO2− discussed above.
In this review, we focus on the reduction of CO2, NO2− and H+. We present an electronic structure-based approach for the activation and reduction of these species using mononuclear 1st row transition metal species. Thereon an overview of the natural enzymes involved in these reductions will be used followed by a review of artificial catalysts reported in the literature. In particular, the mechanism of action of these enzymes and complexes and the roles played by 2nd sphere residues will be stressed upon to understand the structure function correlations necessary for the design of efficient and selective catalysts.
Electronic structure and catalyst design
Molecular orbital (MO) theory considerations can be crucial when designing catalysts for the activation of small molecules. An analysis of the MO of the small molecule and possible ligand fields around a metal can narrow down the search for a metal-based catalyst and its ligand. CO2, for example, is a linear triatomic molecule. While its HOMO is localized on the more electronegative oxygen atoms, the LUMO is a C–O σ* (Scheme 1) orbital. The LUMO+1 is the degenerate set of in-plane and out-of-plane C–O π* orbitals (Scheme 1). Reduction of CO2 will entail population of these orbitals and the electrons required must be obtained from the metal center(s) of the catalyst used; a mononuclear complex having axial symmetry (x = y) is considered here. For efficient activation, it is desirable that the metal donor orbitals are poised to overlap with the CO2 acceptor orbitals. The contours of the orbitals involved suggest that a dz2 orbital is suitable for generous overlap with the C–O σ* and the dxz/yz orbitals are suitable to overlap with the C–O π* orbitals. Thus, it would seem that a metal center with occupied dz2 and dxz/yz would provide ideal configurational interaction for the initial activation and eventual final reduction of CO2.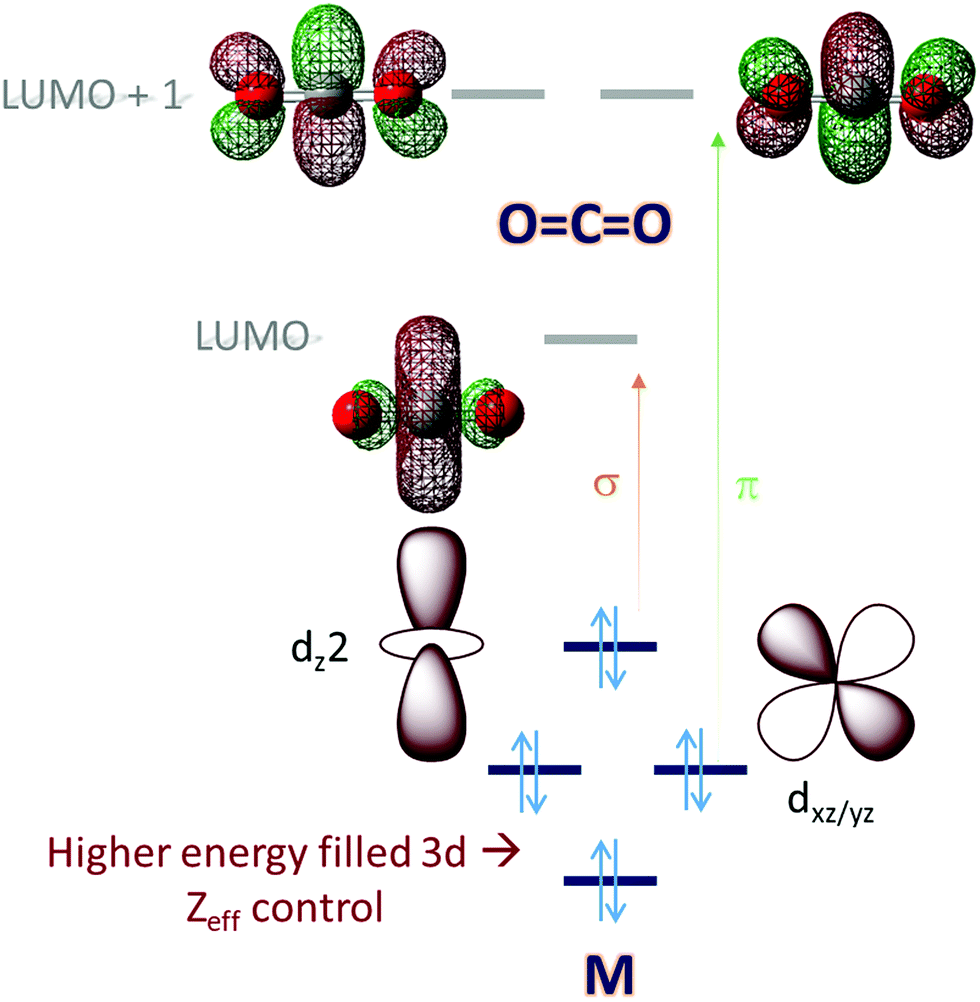 | ||
| Scheme 1 An electronic structure consideration of CO2 activation and reduction using a 3d metal active site. | ||
Considering the relative orbital energies of the d-orbitals in different trigonal and tetragonal ligand fields, two geometric dispositions of the metal catalyst appear attractive for CO2 activation i.e., occupied dz2 and dxz/yz orbitals (Scheme 2). A triaonal ligand field with no or weak ligand along the Z-axis can achieve the desired occupation of the dz2 and the dxz/yz orbitals with a d4, d5 and d6 (as demonstrated below) occupation to activate a CO2 molecule approaching along the Z-axis. For maximum activation of CO2, a low spin d6 cofiguration is required at least and further occupation of the higher energy dx2−y2 and dxy orbitals will not offer further advantage. This electronic structure automatically requires strong field ligands or 2nd or 3rd row transition metals to ensure low spin state at the metal as high spin configuration will lead to depopulation of the active dz2 and dxz/yz orbital and population of the inactive dx2−y2/dxy orbital. Similarly, a low spin d8 tetragonal ligand field is well equipped to activate a CO2 approaching along the Z-axis. Since the inactive dxy orbital is lower in energy in this ligand field than the active orbitals, additional two electrons will be required in this catalyst architecture than the trigonal architecture described earlier.
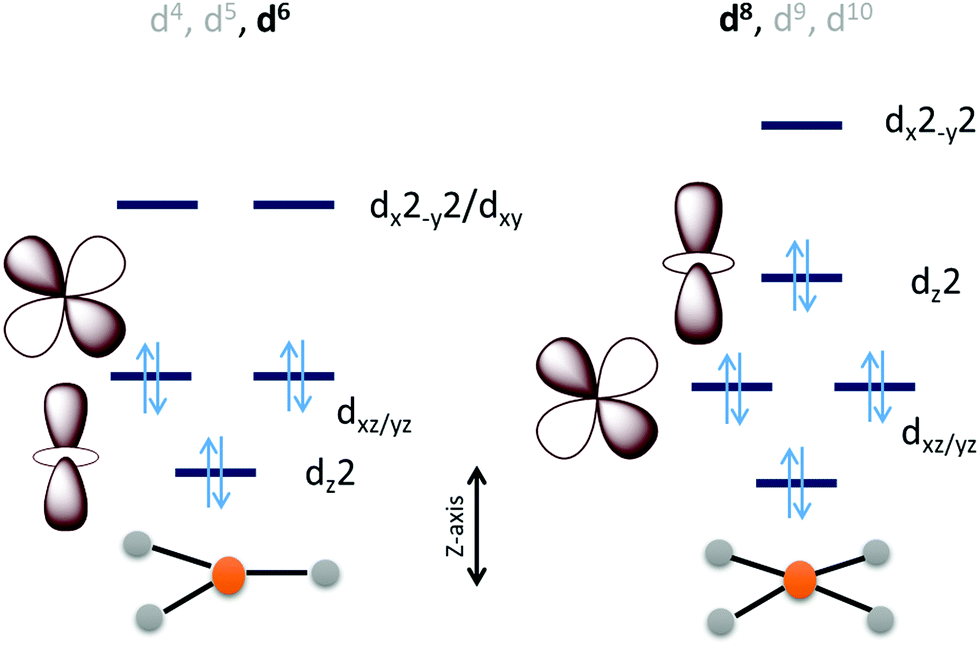 | ||
| Scheme 2 An electronic structure consideration of CO2 activation and reduction using a 3d metal active site having trigonal (left) and tetragonal (right) ligand field. | ||
The electronic structure considerations allow narrowing down the choices for metals and ligands required for CO2 activation as well as the oxidation states required which are eventually important for lowering the overpotential for the electrochemical reduction of CO2. For example, the analysis above suggests that for 2nd and 3rd row transition metal complexes, which tend to result in low spin states, the trigonal geometry can result in efficient CO2 activation and reduction with 6 d-electrons. Attempting to install low spin in a first-row metal in a trigonal geometry will require the use of π-acid ligands which would drain out the electron density from these metal donor orbitals compromising the activation of CO2. Obtention of stable three co-ordinate metal complexes (or with weak axial ligands) may be a synthetic challenge depending on the metal chosen. The second tetragonal option is more feasible with abundant examples in reported literature which inherently employed this design. A tetragonal ligand field will optimally require a low-spin d8 configuration. A reasonably strong σ donor ligand in the XY-plane will elevate the energy of the dx2−y2 orbital ensuring the occupation of the active dz2 orbital. As a result, metal centers like Fe0, CoI and NiII in tetragonal ligand field can, in principle, be able to activate CO2 efficiently. Of course, effective nuclear charge of Ni(II) may be prohibitive of the back donation from its occupied orbital and its further reduction to Ni(I) or Ni(0) states may be necessary. It is likely that CO2 activation can be achieved by a half-filled dz2 (e.g. FeI, CoII) in the tertragonal geometry but it should be less effective than a filled dz2 orbital. Complexes of the 2nd and 3rd row transition metal neighbours of Fe and Co, having tetragonal ligand field, should be active as well.
Finally, the activation of CO2 results in shift of electron density from the metal center to the bound CO2. The shift of electron density to the bound CO2 can be stabilized by suitably placed non-covalent 2nd sphere interactions. These may include secondary Lewis acidic metal center, hydrogen bonding and electrostatic interactions. Of these hydrogen bonding is particularly attractive as apart from stabilizing the charge transfer, it can facilitate proton transfer to the bound CO2 as well which is required for the reaction to proceed. Thus, the insight obtained from ligand field theory and MO theory (an electronic retrosynthesis of catalyst) suggests that Fe(0) and Co(I) square planar or square pyramidal complexes with weak axial ligands and 2nd sphere hydrogen bonding residues (which can also act as proton transfer residues) should be ideal for the activation and the ensuing reduction of CO2. The same approach can be used to design catalysts for NOx and SOx reduction as well.
In the case of NO2− the HOMO is in-plane and localized substantially on the nitrogen while the LUMO is a N–O π* (Scheme 3). To cleave the N–O bond one needs to bind the NO2− and populate the anti-bonding N–O π* orbital. The HOMO being delocalized over both N and O centers of NO2− can bind a metal center both η-N as well as η-O. An empty dz2 orbital can for a σ bond with the HOMO of the NO2− while a populated dxz/yz can assist in cleaving the N–O bond by back bonding into the N–O π*. Considering the MO of the HOMO and LUMO of a free NO2− it appears reasonable to assume that a η-N co-ordination to a metal center would lend it to better backbonding, essential for N–O bond cleavage, from the metal center as well. The electronic structure configurations that allow such bonding possibilities in trigonal and tetragonal ligand fields are d4 (Scheme 3) and d6 (Scheme 3), respectively. Considering the motivation of using a first-row transition metal complex with optimal Zeff to actuate the balance between forming a σ bond with the NO2− and backbonding to it, a d6 low spin configuration would be suitable for the purpose i.e., FeII, CoIetc. Similar to the case of CO2 the electron density shift of the metal center to the bound NO2− needs stabilization and the reaction forward requires protons. Thus, a hydrogen bonding from a group capable of proton transfer in the 2nd sphere would assist the reaction. Additionally, H-bonding interaction with the O atoms will also aid binding the NO2−via the N atom by restricting the electron density on the O atoms from participating in bonding.
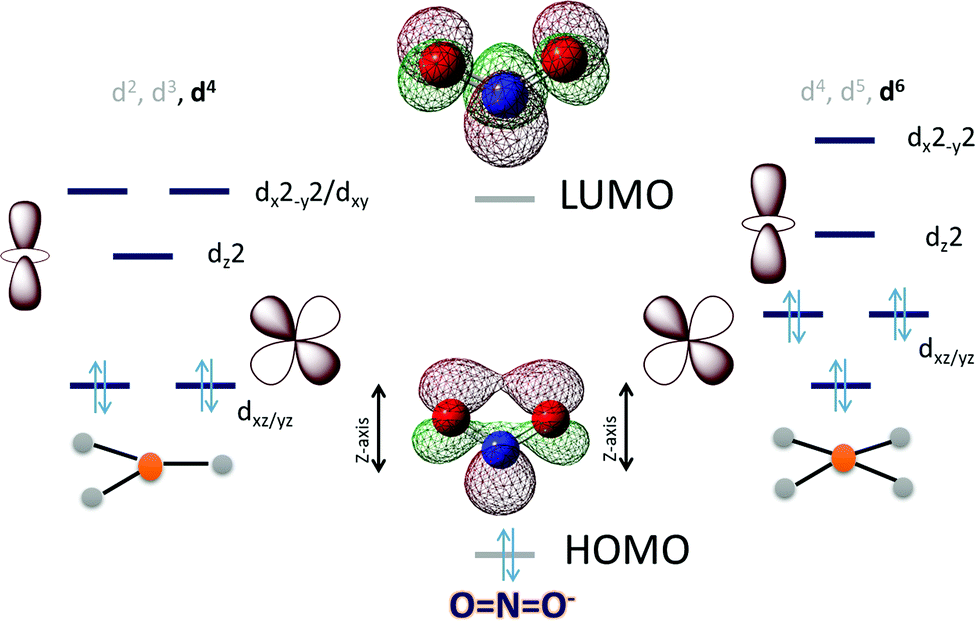 | ||
| Scheme 3 An electronic structure consideration of NO2− activation and reduction using a 3d metal active site having trigonal (left) and tetragonal (right) ligand field. | ||
Having considered a straight-forward and intuitive MO theory-based expectations for a monometallic molecular complex likely to be suitable for the reduction of CO2 and NO2−, the natural metalloenzymes doing the same is now discussed.
CO2 reduction
Introduction
CO2 is a linear molecule with a high C–O bond enthalpy of ∼191 kcal mol−1. The one electron reduction of CO2 to its corresponding anion radical (CO2−˙) is associated with a large re-organization energy due a change in geometry from linear to bent. This is reflected in the requirement of highly negative potential to reduce CO2 by one electron (−1.97 V and −1.90 V vs. NHE in an aprotic solvent, N,N-dimethylformamide (DMF)22 and in water,23 respectively). However, it is desirable to conduct these reactions involving multiple protons and electrons at relatively modest potentials (eqn (2)–(5)). Hence, catalytic strategies have been developed to avoid the high energy steps.3,24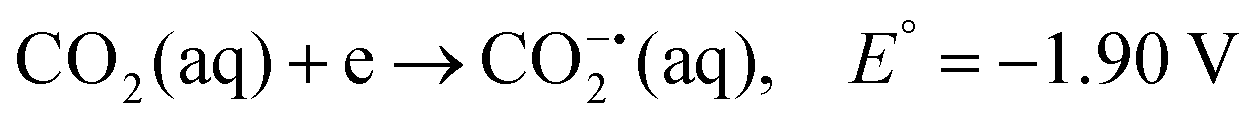 | (1) |
| CO2(g) + 2H+ + 2e → CO(g) + 2H2O, E° = −0.52 V | (2) |
| CO2(g) + H+ + 2e → HCO2−(aq), E° = −0.43 V | (3) |
| CO2(g) + 6H+ + 6e → CH3OH(aq) + H2O, E° = −0.38 V | (4) |
| CO2(g) + 8H+ + 8e → CH4(aq) + 2H2O, E° = −0.24 V | (5) |
In this section, we have attempted to summarize the conceptual development in the design of catalysts for electrocatalytic CO2 reduction. Several strategies have been devised considering the primary as well as the secondary coordination sphere and a wide variety of metal–ligand combinations. However, the central theme of this review is the contribution of the second sphere to catalysis. A bio-inspired approach is a good way of designing catalysts as metallo-enzymes have evolved over billions of years optimizing the rates and selectivity of specific reactions they catalyze using elegant 2nd sphere interactions. Enzymes have evolved to use amino acid residues, additional metal centres and well-defined substrate/product channels to regulate and optimize their reactivity. This allows catalyzing reactions with precise thermodynamic and kinetic control. Therefore, prior understanding about the enzymatic machinery is very important before designing molecular catalysts for the same purpose.
Lessons from nature
Six pathways are known for the fixation of inorganic carbon into organic materials to be incorporated into biomass: (i) reductive pentose phosphate (Calvin–Benson–Bassham cycle)25 (ii) reductive acetyl-CoA pathway (Wood–Ljungdahl pathway)26 (iii) reductive citric acid cycle (TCA and Arnon–Buchanan cycle)27,28 (iv) decarboxylate/4-hydroxybutyrate cycle (v) 3-hydroxypropionate/4-hydroxybutyrate cycle and (vi) 3-hydroxypropionate cycle. These processes have been discussed in detail in the previous reviews.3,29 In this review we will discuss about the second inorganic pathway which contains two CO2 reduction steps: (a) carbon monoxide dehydrogenase (CODHs) catalysed CO2 to CO interconversion and (b) formate dehydrogenase catalysed reduction of CO2 to formic acid.CO dehydrogenases (CODHs)
There are two types of CODHs participating in CO2 to CO interconversion. Oxygen-sensitive enzyme from obligate anaerobes like Moorella thermoacetica, Carboxythermus hydrogenoformas and Methanosarcina bakerii use the [NiFe]CODH active site for reversible conversion of CO2 to CO. These enzymes have turnover frequency (TOF) of ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 for CO oxidation (at 70 °C using methyl viologen) and 45 s−1 for CO2 reduction and operate with almost zero over-potential, i.e., close to the thermodynamic potential of the CO2/CO couple, at −0.52 V vs. SHE at pH 7.30 The second class of CODHs are air-stable [MoSCu]-containing enzymes which are found in aerobes such as Oligotropha carboxidovorans. These enzymes show lower CO oxidation activity (TOF ∼ 100 s−1) and their NiFe analogues and do not show any CO2 reduction activity. We will not discuss about this class of CODHs in this review which has been reviewed elsewhere.31,32
000 s−1 for CO oxidation (at 70 °C using methyl viologen) and 45 s−1 for CO2 reduction and operate with almost zero over-potential, i.e., close to the thermodynamic potential of the CO2/CO couple, at −0.52 V vs. SHE at pH 7.30 The second class of CODHs are air-stable [MoSCu]-containing enzymes which are found in aerobes such as Oligotropha carboxidovorans. These enzymes show lower CO oxidation activity (TOF ∼ 100 s−1) and their NiFe analogues and do not show any CO2 reduction activity. We will not discuss about this class of CODHs in this review which has been reviewed elsewhere.31,32
[NiFe]CO dehyrogenases
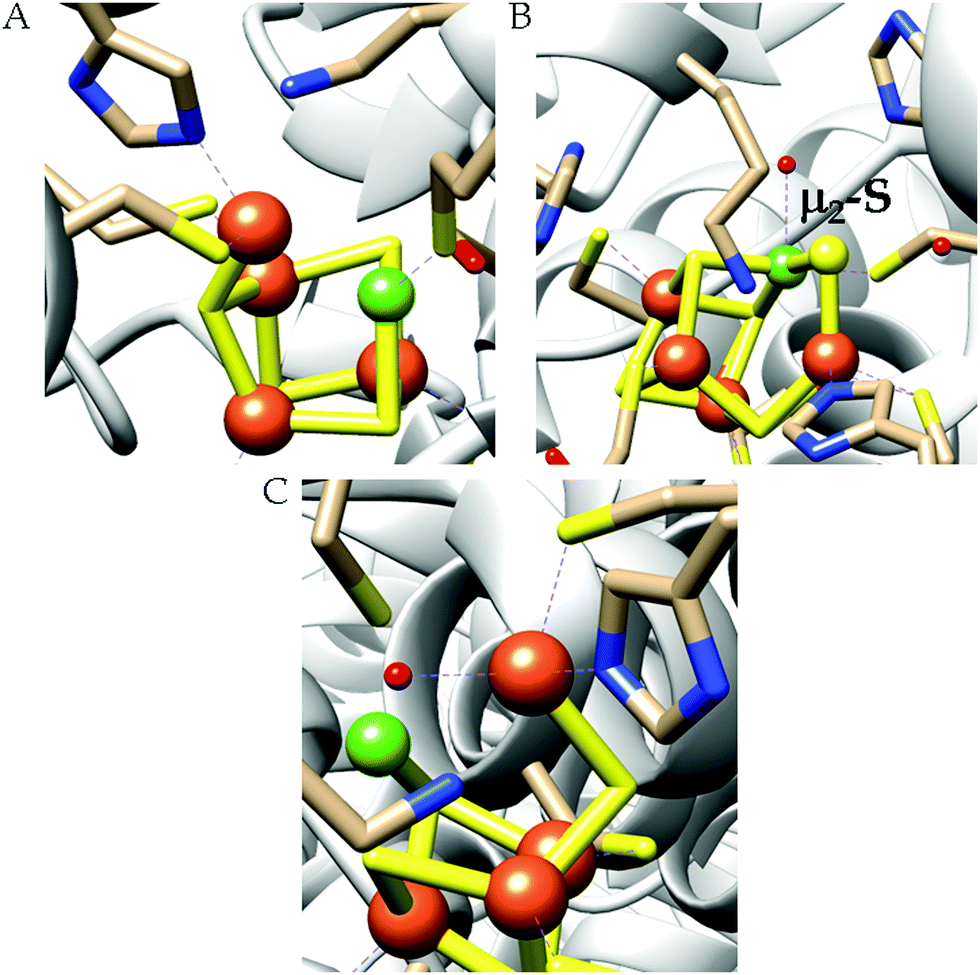
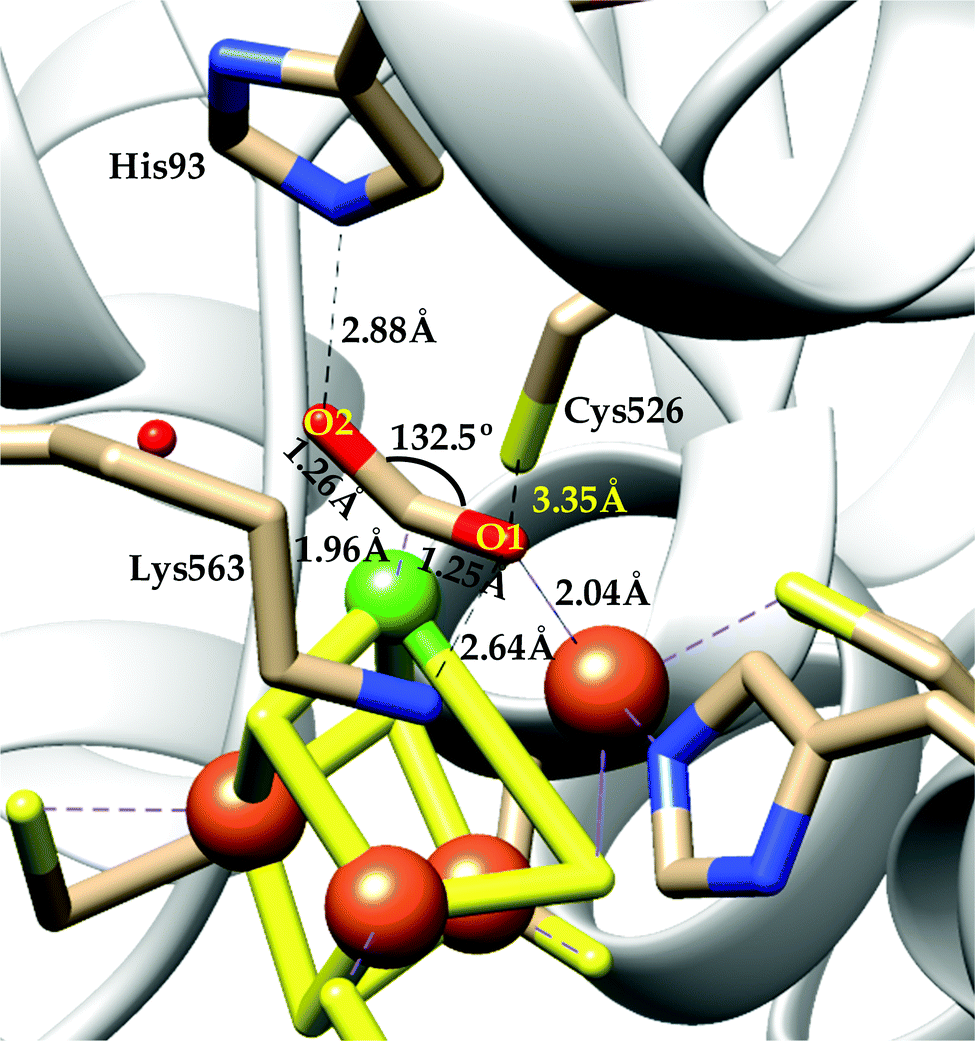
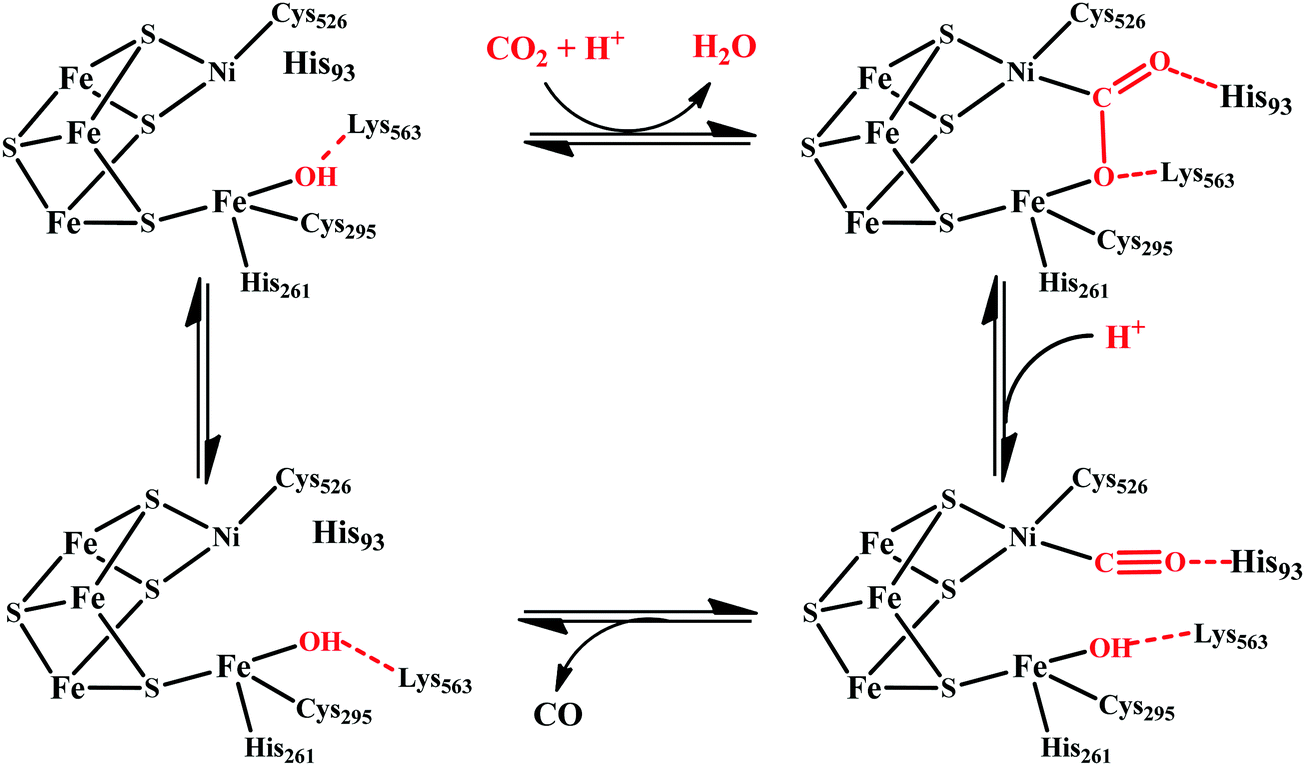 | ||
| Scheme 4 Proposed mechanistic cycle of CO2 reduction to CO by [NiFe]CODHs. | ||
Formate dehydrogenases (FDHs)
There are two types of formate dehydrogenases, metal independent NAD+ dependent and Mo/W containing formate dehydrogenases. The most abundant class of formate dehydrogenases are NAD(P)+ dependent. They are the major source of energy conversion in methylotrophic aerobic bacteria, fungi and plants.44,45 This class of enzymes have a characteristic property of transferring both proton and electron together in the form of a hydride, from one site to another. More specifically, from the formate to the C4-atom of the pyridine ring of NAD(P)+ for the oxidation of formate to CO2. It is difficult to drive the reaction in reverse direction, i.e., to reduce CO2 to formate, because the reduction potential of NAD(P)+ is more positive than that of CO2.46 Hence, this has not been included in this review. The Mo/W-containing formate dehydrogenases on the other hand, can catalyze the reduction of CO2 to formate (with rate ∼280 s−1) as well as formate oxidation (with rate ∼3400 s−1).47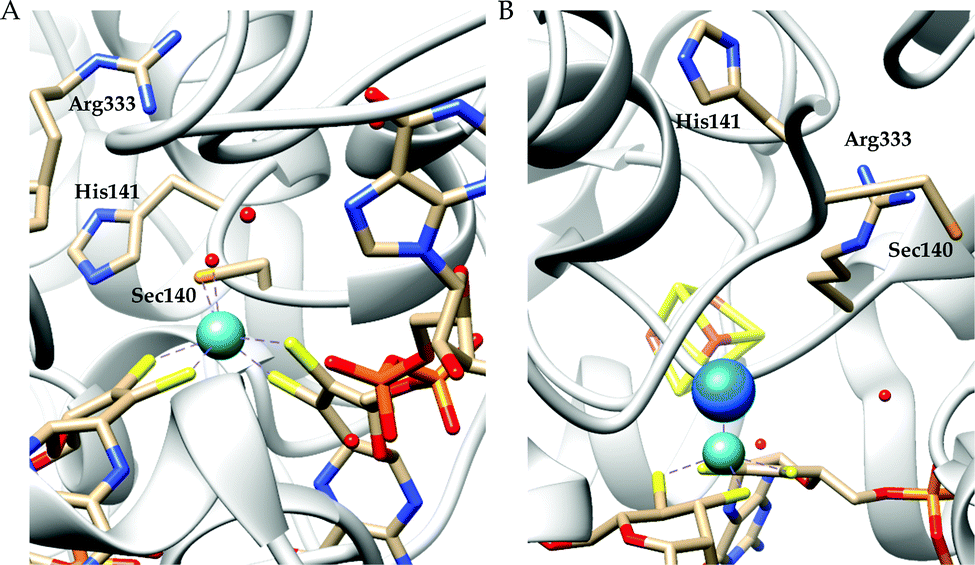 | ||
| Fig. 3 The active site structure of the formate dehydrogenase H from E. coli; (A) oxidized state (pdb: 1FDO);48 (B) reduced state (pdb: 2IV2).49 The figures are redrawn using the software package Chimera 1.12rc. | ||
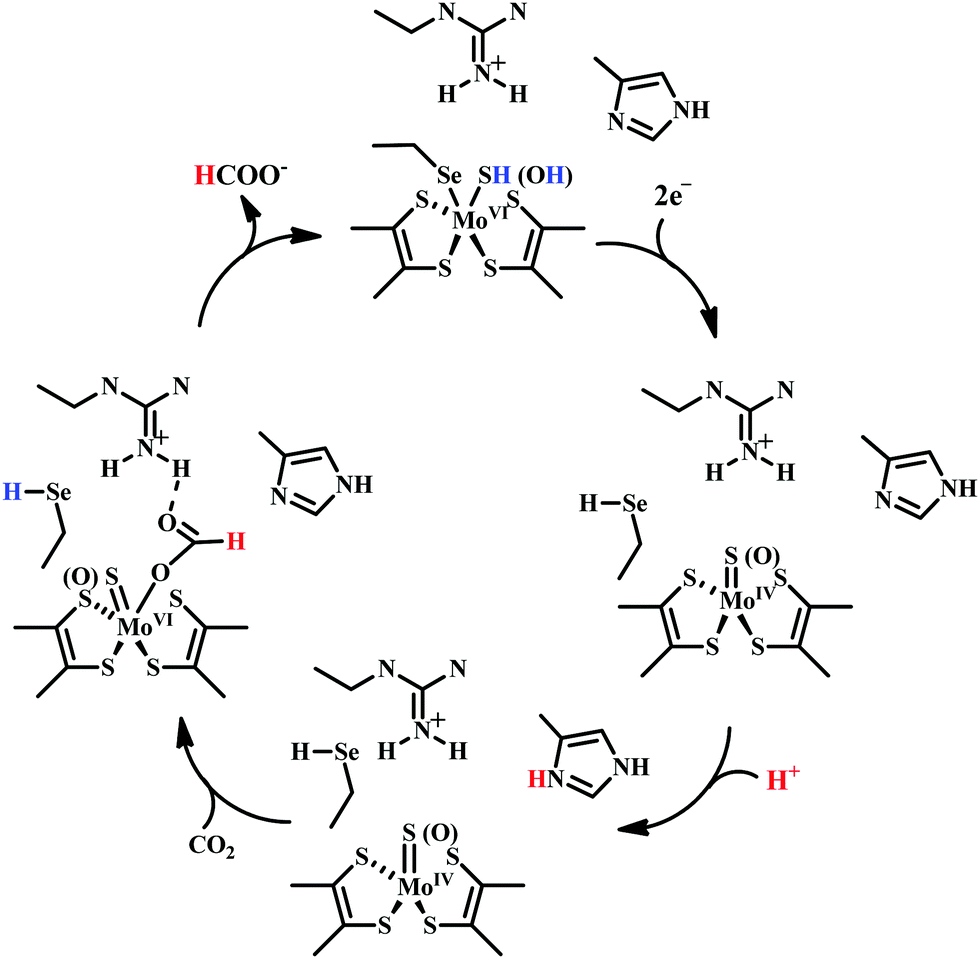 | ||
| Scheme 5 Proposed mechanistic cycle of CO2 reduction to formate by selenium and molybdenum dependent formate dehydrogenase. | ||
Challenges and general outlook
Efficient carbon dioxide reduction to useful molecules such as carbon monoxide, formic acid, methanol, methane etc., is an important challenge and provides a great opportunity to convert waste to valuable commodities. Although, the field of transition metal catalyzed CO2 reduction is relatively new, it has expanded substantially in the recent years. However, the major challenges associated with CO2 economy are: (i) capturing the pure CO2 from the atmosphere, which is quite difficult because the scarcity of CO2 in the atmosphere and (ii) reducing CO2 selectively as separation of mixture of products is undesirable. The reduction of CO2 competes wth the reduction of H+ which is another reactant in electrochemical CO2 reduction and strategies are needed to avoid competing H+ reduction. Similarly, practicable CO2 reduction systems need to be O2 tolerant or selective towards CO2 even in the presence of a strong oxidant as O2.In this section a review of the molecular electrocatalysts is presented with a focus on the effects of 2nd sphere interactions on the efficiency of ctalysis. These interations include the effect of hydrogen bonding, redox-active metal or ligands, additional metals, etc. A combination of suitable design or the primary coordination sphere and inclusion of 2nd sphere residues have been found to have a profound impact on CO2 reduction helping address some of the challenges discussed above.
Molecular catalysts
The idea that transition metals can activate CO2 dates back to the work by Aresta and Nobile, where a bent CO2 was attached in a η2-mode (through C and O atoms) with a Ni-centre.56 Meshitsuka reported electrochemical CO2 reduction with Co and Ni-phthalocyanines, but lacked product chracterization, TON, etc.57 Later in the 1980s, Eisenberg and co-workers reported electrochemical reduction of CO2 to CO (along with H2 production) with tetraaza-macrocycles of Co and Ni.58 It was likely one of the first report of electrochemical CO2 reduction with high current density and turn-over numbers (TON). In the following years, a number of electrocatalysts have been developed with high TONs and selectivity. We have divided this section in two major categories depending on the ligand type: (i) non-heme catalysts and (ii) heme or other macrocycle catalysts. It should be noted that in this review, we have discussed electrocatalytic reduction of carbon dioxide under homogeneous conditions only.Non-heme system
:1 DMF:water solution. It was later proposed that CO2 binds to the Re0 centre and after reductive decarbonylation generating a carboxylato-rhenium intermediate.60 Upon protonation it eliminates a molecule of water and generates [ReII(bpy)(CO)3Cl]+ complex which upon further reduction regenerates the starting molecule. Tanaka and co-workers did a remarkable study in electrocatalytic CO2 reduction with similar Ru-based catalysts ([Ru(bpy)2(CO)2]2+ or [Ru(bpy)2(CO)Cl]+).61 They showed a pH dependent product selectivity. Controlled potential electrolysis (at −1.25 V vs. NHE) at pH 6 generated a mixture of CO and H2. Alternatively, at pH 9 it generated CO and H2 along with HCOO−. The reaction was proposed to be initiated by the generation of an unstable penta-coordinated Ru0 intermediate ([Ru(bpy)2(CO)2]2+ or [Ru(bpy)2(CO)Cl]+). In the presence of CO2, both likely generated [Ru(bpy)2(CO)(COO−)]+ intermediate, which upon protonation should generate [Ru(bpy)2(CO)(COOH)]+ intermediate. Under acidic condition (pH 6), further protonation releases a molecule of water to regenerate the catalyst. Under weakly alkaline condition (pH 9), it may undergo a 2e−/1H+ process to regenerate the penta-coordinated Ru0 intermediate releasing HCOO−.
Meyer and co-workers reported electrocatalytic CO2 reduction by similar 2,2′-bipyridyl-complex of Rh and Ir.62 They reported selective reduction of CO2 to formate (without formation any CO during electrocatalytic condition) by cis-[Rh(bpy)2X2]+ (where, X is Cl− or OTf−) at −1.30 V vs. NHE. They also found [M(bpy)2(CO)H]+ (where M = Os, Ru) can reduce CO2 to CO as major product in anhydrous acetonitrile medium.63 However, up to 25% formate was observed upon addition of water into the solution. Later, Kubiak and co-workers developed a series of 2,2′-bipyridyl-complex of Re, Re(bpy-R)(CO)3Cl (where R = H/4,4′-dicarboxyl/4,4′-dimethyl/4,4′-di-tert-butyl/4,4′-dimethoxy) for electrocatalytic CO2 reduction (Fig. 4a).64 The Re(bpy-tBu)(CO)3Cl was found to be most effective to selectively reduce CO2 to CO at ∼100% faradaic efficiencies at −1.60 V vs. NHE. The observed high selectivity over competitive proton reduction was explained using extensive kinetic investigations, which suggests that under same condition, the reaction with CO2 is ∼25 times faster than that of water or methanol.65
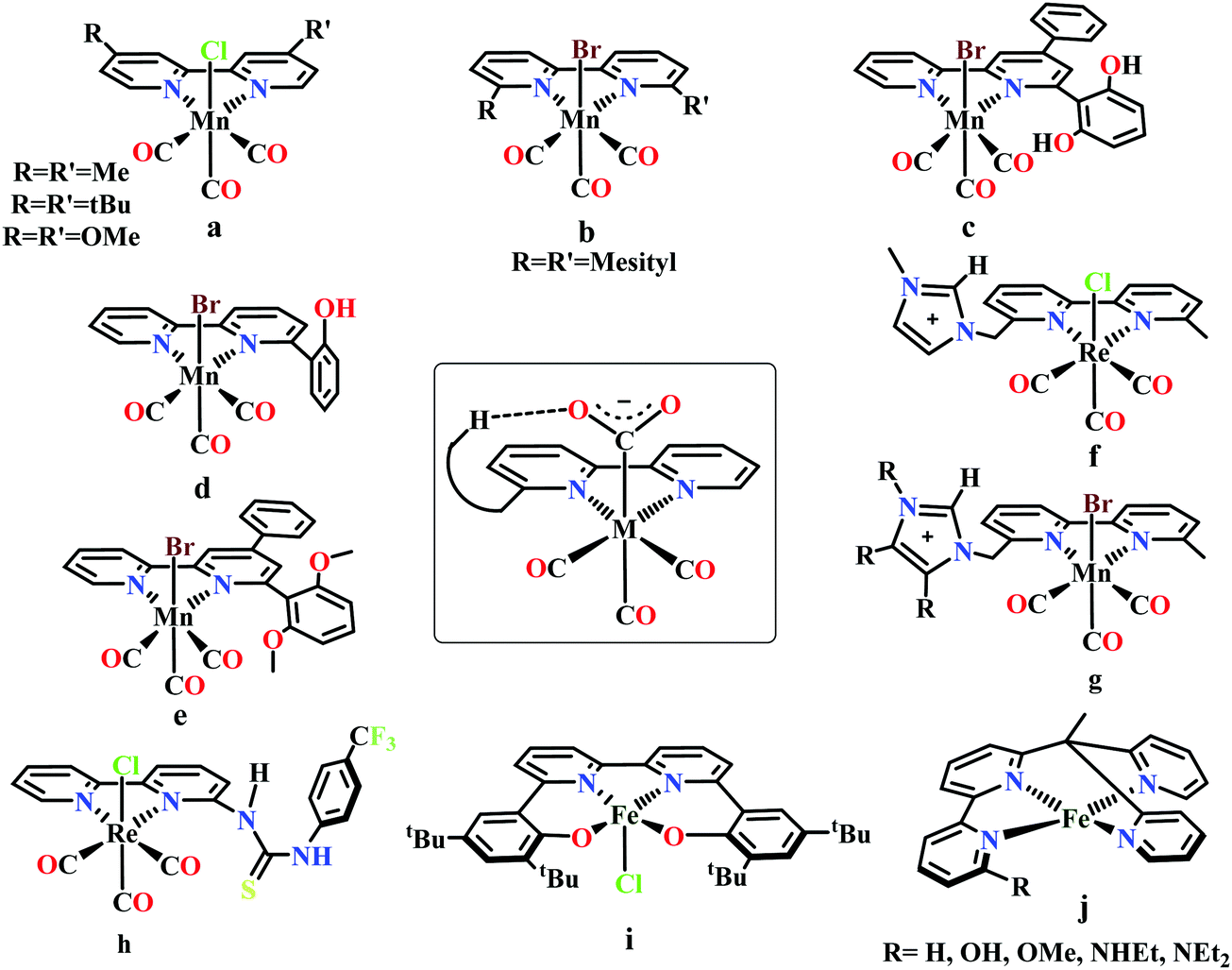 | ||
| Fig. 4 Representative molecules for bipyridine-based metal-complexes for CO2 reduction. | ||
Bourrez et al. reported earth abundant Mn-based catalysts, [Mn(L)(CO)3Br] complexes (where, L = 2,2′-bipyridine or 4,4′-dimethyl-2,2′-bipyridine, Fig. 4a), which can catalyze the electrochemical reduction of CO2 to CO at relatively lower overpotential (at −1.10 V vs. NHE).66 However, in comparison with the Re catalysts, there was one distinct difference in the corresponding Mn-catalysts, which is the tendency for dimerization after the first reduction. The irreversible loss of X (where, Cl−, Br− or OTf−) is responsible for such dimer formation (eqn (6)). This is in contrast to the Re counterparts, where the first reduction is reversible (no loss of X). Such a behaviour explains lower activity by Mn-bpy based electrocatalysts.67
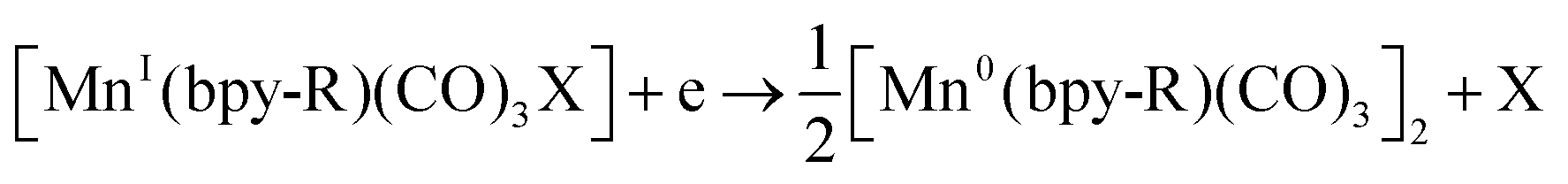 | (6) |
Tetracarbonyl Mo and W complex of bpy or bpy-tBu, M(bpy-R)(CO)4 (where, M = Mo/W; R = H/tBu in 4,4′-positions) were developed which has been shown to undergo electrocatalytic CO2 reduction.71 N-Heterocyclic carbene based Mn-complexes, MnBr(N-methyl-N′-2-pyridylbenzimidazol-2-ylidine)(CO)3 and MnBr(N-methyl-N′-2-pyridylimidazol-2-ylidine)(CO)3 were reported to catalyze electrochemical conversion of CO2 to CO.72
Gobetto and co-workers introduced a pendant 2,6-dihydroxyphenyl group in a [fac-Mn(dhbpy)(CO)3Br] (where, dhbpy = 4-phenyl-6-(1,3-dihydroxybenzen-2-yl)2,2′-bipyridine, Fig. 4c).73 The catalyst can undergo electrochemical CO2 reduction upon controlled potential electrolysis in anhydrous acetonitrile to CO (70%) and formic acid (22%), even in the absence of any external proton source. Later, Bocarsly and co-workers studied the effect of pendant phenol group in a similar model, Mn(hbpy)(CO)3Br (where, hbpy = 6-(2-hydroxyphenyl)-2,2′-bipyridine, Fig. 4d).74 The presence of phenol group in the second coordination sphere, the catalyst showed more than seven times higher current density with 86% faradaic efficiency for CO evolution in a 5% water–acetonitrile solution relative to Mn(bpy)(CO)3Br (Fig. 4b) at similar over-potential. An intramolecular proton-assisted dehydration of Mn(hbpy)(CO)3COOH with lower entropy of activation was proposed to be the reason for the enhanced catalytic activity. The effect of a H-bond acceptor anisole group instead of a phenolic –OH group was invesigated by Rochford and co-workers. They synthesized {fac-MnI([(MeO)2Ph]2bpy)(CO)3(CH3CN)}(OTf) (where, [(MeO)2Ph]2bpy = 6,6′-bis(2,6-dimethoxyphenyl)-2,2′-bipyridine), which contained four pendant methoxy groups attached with the benzene (Fig. 4e).75 In the presence of external proton source, the catalyst was shown to reduce CO2 to CO at remarkably lower over-potential. The [(MeO)2Ph]2bpy ligand exerts an additional electronic influence along with a weak allosteric hydrogen-bonding interaction which was proposed to be responsible for lowering down the activation barrier for C–OH bond cleavage from the metallocarboxylic acid intermediate. A schematic diagram depicting the influence of the second sphere residues with the bound CO2-adduct is shown in the inset in Fig. 4.
Nippe and co-workers have introduced a series of Lehn-type Re and Mn-complexes with pendant imidazolium moiety attached to the bipyridyl-ligand (Fig. 4f and g).76,77 The results obtained with Re complexes, {Re[bpyMe(ImMe)](CO)3Cl}PF6 and {Re[bpyMe(ImMe2)](CO)3Cl}PF6 suggested that the positively charged imidazolium group lowers the over-potential of CO2 reduction (Fig. 4f). The C2–H group from the imidazolium moiety facilitates the release of Cl− during reduction. Similarly, an extensive study was conducted on a series of Mn-complexes: {Mn[bpyMe(ImMe)](CO)3Br}PF6; {Mn[bpyMe(ImMe2)](CO)3Br}PF6; {Mn[bpyMe(ImMe4)](CO)3Br}PF6 and {Mn[bpyMe(ImtBu)](CO)3Br}PF6 (Fig. 4g).77 All of them could catalyze the electrochemical reduction of CO2 to CO in presence of water at much lower over-potential, suggesting a strong synergistic interaction between the imidazolium groups and the water molecules. This interaction resulted in local hydration and facilitate the CO2 reduction. Like the previous Re-system, here also the 2nd sphere C2–H of the imidazolium moiety was proposed to play a crucial role.
Neumann and co-workers modified the fac-[Re(bpy)(CO)3Cl] complex and tethered a thiourea-moiety in the second coordination sphere (Fig. 4h).78 The thiourea moiety may act both as hydrogen bond promoter and a proton donor. Hence, it not only stabilized the carboxylic acid intermediate but also act as local proton source to accelerate the C–O bond cleavage step of the catalytic cycle. This resulted the catalyst to undergo selective reduction of CO2 to CO at a TOF as high as 3040 h−1. Machan and co-workers have reported an iron(III) chloride (Fig. 4i), [FeIII(tbudhbpy)]Cl (where, tbudhbpy = 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine) which can catalyze the disproportionation of CO2 to CO and carbonate in anhydrous DMF.79 However, in the presence of an external acid source like phenol it was shown to generated formate as major product (FE ∼ 68%) with H2 (FE ∼ 30%) and CO (FE ∼ 1%) as minor products. Recently, Marinescu and co-workers have modified rhenium tricarbonyl bipyridine moiety with pendant secondary and tertiary amines in the 6- and 6′-positions of the bipyridine group.80 These catalysts have shown to reduce CO2 to CO with moderate faradaic efficiencies (51–73%).
Despite tremendous development in the CO2 reduction electrocatalysts with bipyridine-backbone, fewer studies have been carried out using first-row transition metals, except Mn. For example, no such report was present with iron, until recently, Long and co-workers have introduced a series of iron–bipyridine complexes with general formula, [(bpyRPY2Me)FeII]n+ (where, bpyRPY2Me = 6-(1,1-bis(pyridin-2-yl)ethyl)-2,2′-bipyridine) and R = H, OH, OMe, NHEt, NEt2, Fig. 4j.81 These complexes can undergo electrocatalytic CO2 reduction with different product selectivity depending upon the pendant protic functional groups of different acid strengths in the second coordination sphere. [(bpyNHEtPY2Me)FeII]2+ appeared to be the most efficient catalyst amongst them reducing CO2 to CO. The pendant NH group was proposed to facilitate the C–O bond cleavage by acting as a local proton source. In contrast, the more acidic OH group favors the generation of H2 over CO.
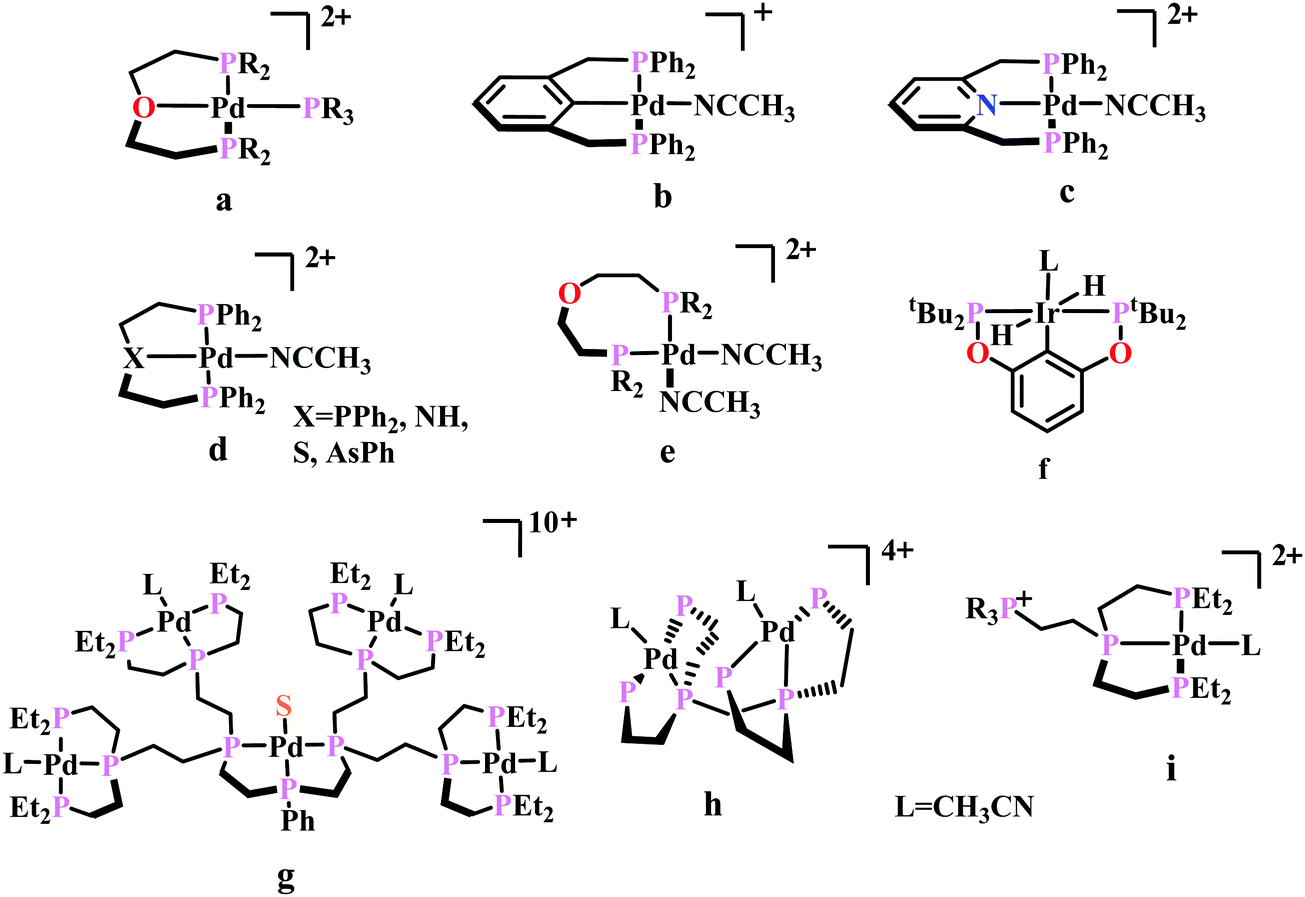 | ||
| Fig. 5 Representative molecules for phosphine-based metal-complexes for CO2 reduction. | ||
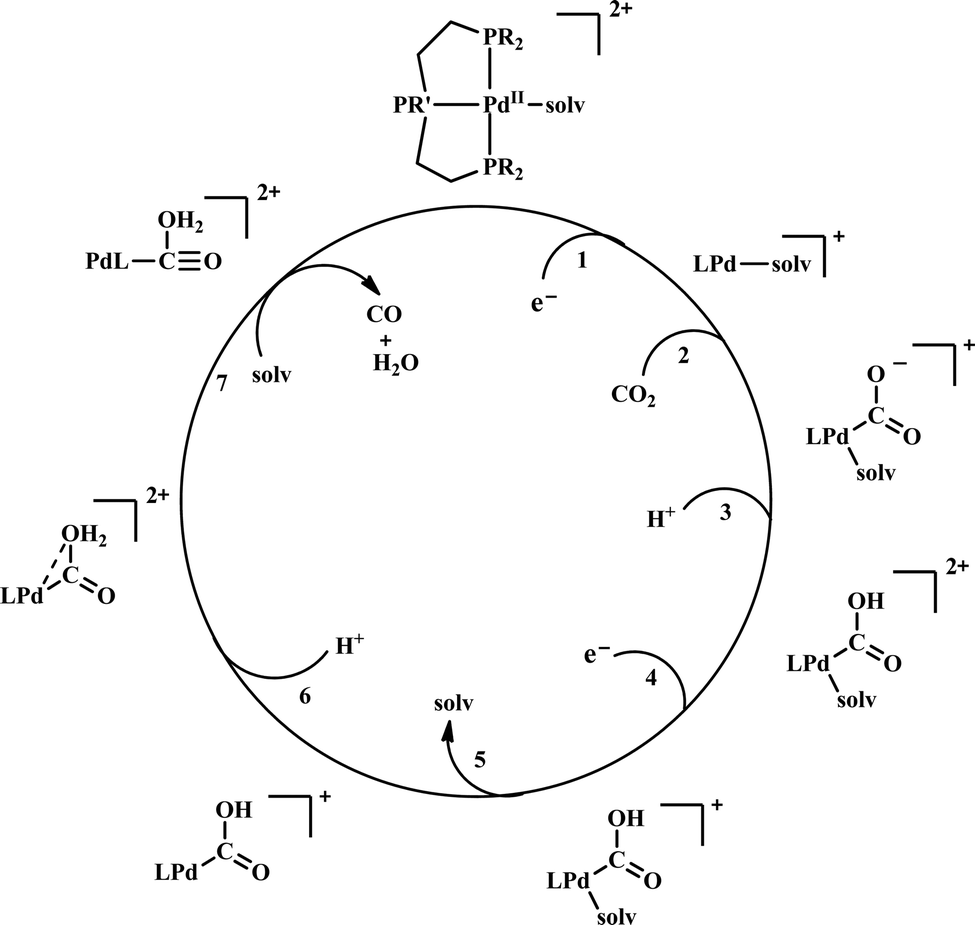 | ||
| Scheme 6 General mechanistic scheme for the electrocatalytic CO2 reduction by [Pd(triphosphine)(S)]2+ catalysts. | ||
To understand the effect of ligand donor strength on Pd, a series of square-planar palladium complexes containing tridentate ligands of type PXP (where X = C, N, O, S, and As) were synthesized. None of them were as effective as the triphosphine complex. During electrocatalysis, they were found to generate H2 instead of CO. The selectivity for producing H2 or CO appears to depend on the basicity or the redox potential of the complexes. Complexes with more negative redox potential, increases the basicity of the Pd-atom favouring the formation of Pd-hydride, which eventually generates H2. While, with less negative redox potentials or a less basic Pd-atom favours protonation of the “O”-atom of the coordinated CO2 to generate CO.86 An Ir-pincer complex, Ir(PCP)–H2(MeCN), was developed (Fig. 5f) which catalyzed electrochemical CO2 reduction to generate formate, selectively.87 Only a small amount of H2 and CO was detected.
A unique dendritic complex was synthesized (Fig. 5g) where the central-Pd is surrounded by four Pd-atoms linked to the terminal phosphorus via an ethylene linkage.88 The idea was to enhance the rate and/or decreasing the overpotential of CO2 reduction by co-operative binding of CO2. But the complex failed to show greater catalytic activity, instead it appeared to be a poor catalyst relative to its monomeric analogue. Later, a bimetallic complex was synthesized (Fig. 5h) which can be viewed as two [Pd(triphos)(solvent)]2+ units linked through a methylene bridge.89 This complex could reduce CO2 to CO at relatively lower potential (−1.05 V vs. NHE) with ∼1000 times enhancement in rate relative to the monomer. At higher acid concentration, the reaction was 1st order with respect to [catalyst] and 1st order in [CO2], indicating involvement of two Pd-atoms per one molecule of CO2. In contrast with the monomer (vide supra), at lower acid concentration, the reaction is 1st order with respect to [acid] instead of a 2nd order dependence. This clearly indicates the second Pd-atom binds with the “O-atom” of the bound CO2 at the transition state instead of a proton proton. However, formation of Pd–Pd bond deactivated the catalyst and only a TON of 10 could be obtained. As an extension of the logic flow, a pendant phosphonium ion was introduced (Fig. 5i) which can stabilize the negatively charged “O−”-atom of CO2 at the transition state via coulombic interaction.90 Although, the rate of CO2 binding was doubled, no catalytic rate enhancement was observed.
Previously, Kubiak and co-workers reported a binuclear “cradle”-type complex, [Ni2(μ-CNMe)(CNMe)2(dppm)2](PF6)2, (where dppm = bis(diphenylphosphino)methane, Fig. 6a) to catalyze the reduction of CO2 with quite low overpotential, −0.67 V vs. NHE. But, with time it ultimately undergoes complete carbonylation to afford Ni2(CO)3(dppm)2.91 A series of alkyl isocyanide bridged binuclear Ni0-clusters with the general formula [Ni2(μ-dppa)2(μ-CNR)(CNR)2] (where, dppa = bis(diphenylphosphine)amine; R = Me, n-Bu, 2,6-Me2C6H3, Fig. 6b) were reported to undergo reductive disproportionation of CO2 to CO and carbonate ion.92 Formate was produced in the presence of proton (from residual water). During catalysis the generated CO was found to be trapped by the catalyst. An interesting binuclear copper catalyst was developed (Fig. 6a), [Cu2(μ-PPh2bpy)2(MeCN)2][PF6]2 (where PPh2bpy = 6-(diphenylphosphino)-2,2′-bipyridyl, Fig. 6c).93 It was shown to undergo electrochemical CO2 reduction to CO and CO32− at −1.35 V vs. NHE. The unique bipyridine based ligand scaffold acts as an electron reservoir for these systems, where the electrons are localized in the π*-orbitals of the bipyridine-fragment. A turn-over frequency (TOF) of >2 h−1 was maintained throughout an experiment for 24 h and recovered quantitatively in its original form after the electrolysis.
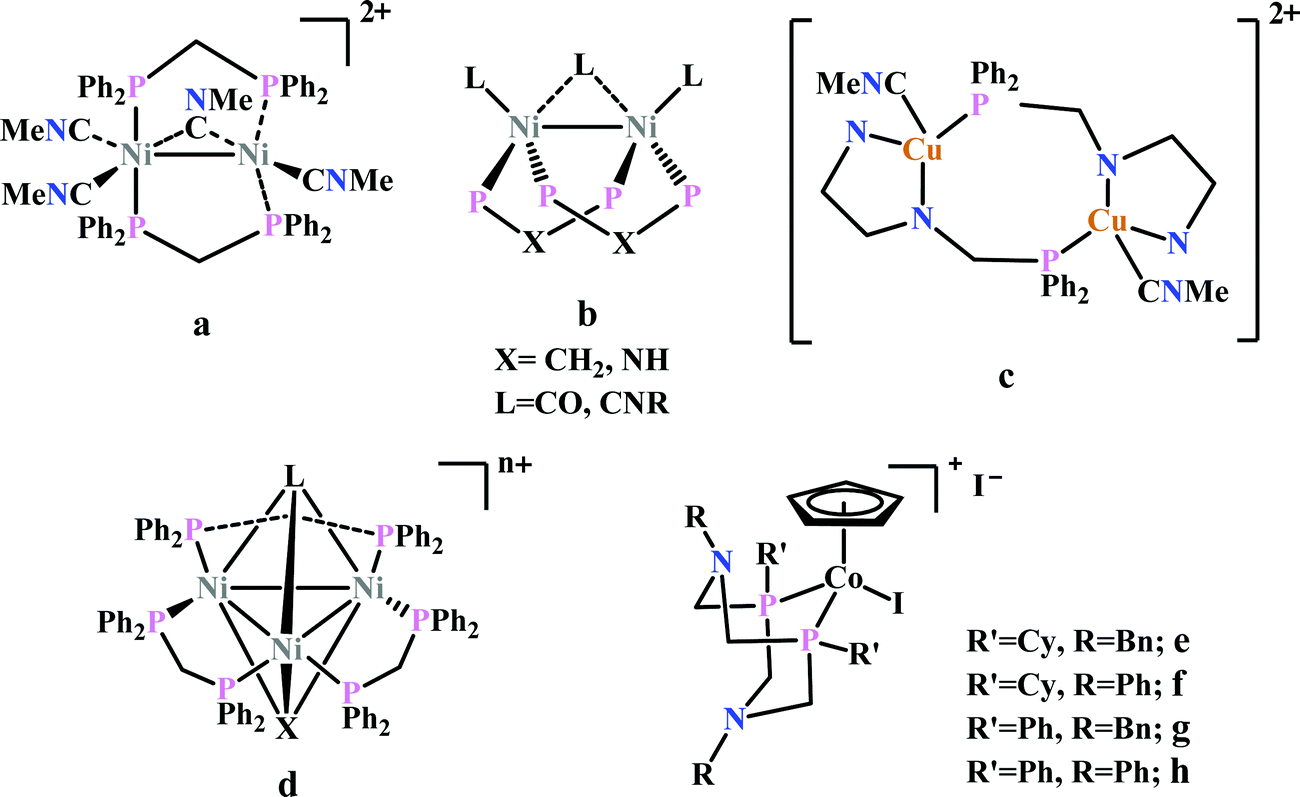 | ||
| Fig. 6 Representative non-heme molecules for electrocatalytic CO2 reduction with influence from second coordination sphere. | ||
As an extension of the concept of co-operative CO2 binding and reduction, a series of isocyanide and CO bound and halide capped trinuclear-nickel clusters were developed (Fig. 6d) with the general formula, [Ni3(μ3-L)(μ3-X)(μ2-dppm)3]n+ (where L = I−, Br−, CO, CNR; X = I−, Br−; n = 0, 1).94 They were reported to undergo reductive disproportionation of CO2 to CO and CO32− at relatively lower overpotential indicating co-operativity due to the presence of a trinuclear Nickel cluster. Although the reduction potentials of these molecules fall into a relatively narrow range of −0.84 V to −0.94 V vs. NHE, the slight difference in redox potentials affect the rate of CO2 reduction dramatically. This clearly invokes the influence of the substituents of the capping ligand.
Artero and co-workers reported a series of Co-diphosphine electrocatalysts (Fig. 6e–h) to convert selectively CO2 to formate in DMF–water system with high faradaic efficiency (>90%) and at moderate overpotential (500–700 mV in DMF).95 The catalysts had a general formula, [CpCo(PR2NR′2)I]I (where, PR2NR′2 = 1,5-diaza-3,7-diphosphacyclooctane). Cyclohexyl or phenyl substituents on P and benzyl or phenyl substituents on N were employed, to tune the donor ability of P and basicity of the N. Similar Ni-complexes were known for efficient formate oxidation.96–98 The high selectivity for formate (instead of CO or H2 formation) was postulated to be due to higher hydricity of the CpCoII(diphosphine)–H intermediate, they undergo hydride transfer to CO2. However, the pendant amine groups are not directly involved in proton transfer, but they stabilize the intermediates through H-bonding with the water molecules during hydride transfer. With the most electron-donating phosphine ligand and the most basic amine group, the catalyst could show an excellent TOF of >1000 s−1 which is much higher than other formate reducing catalysts.87,99–101
Heme and other macrocyclic system
The mode of activation relies on the ion-pairing with the negatively charged “O”-atoms of the Fe bound-CO2. This invokes a “push–pull” mechanism, i.e., an electron pair is pushed from the Fe0 into the CO2 molecule. This process is temperature dependent and at −40 °C, the reaction proceeds with the involvement of two CO2 molecules, while at room temperature only one CO2 was proposed to be present in the iron-coordination sphere. It required either one divalent cation or two mono-valent cations for the activation. This is an example of a bimetallic catalysis, where an electron-rich Fe0-center initiates the reaction with CO2 and an electron-deficient metal-centre assists the C–O bond cleavage to accelerate the reaction, mimicking the natural process catalyzed by enzyme, [NiFe]-CODH. Naruta and co-workers reported a series of bio-inspired cofacial iron-porphyrin dimer (o-Fe2DTPP, Fig. 7g and h) with the Fe⋯Fe separation of 3.4–4 Å, anticipating easy access and co-operative binding of the linear CO2 molecule (2.32 Å) in between.111 The high selectivity for CO (with 95% faradaic efficiency) at a considerably higher rate (TOF ∼ 4300 s−1) was attributed to the influence of second iron, which is absent in FeTPP or m-Fe2DTPP (Fig. 7h).
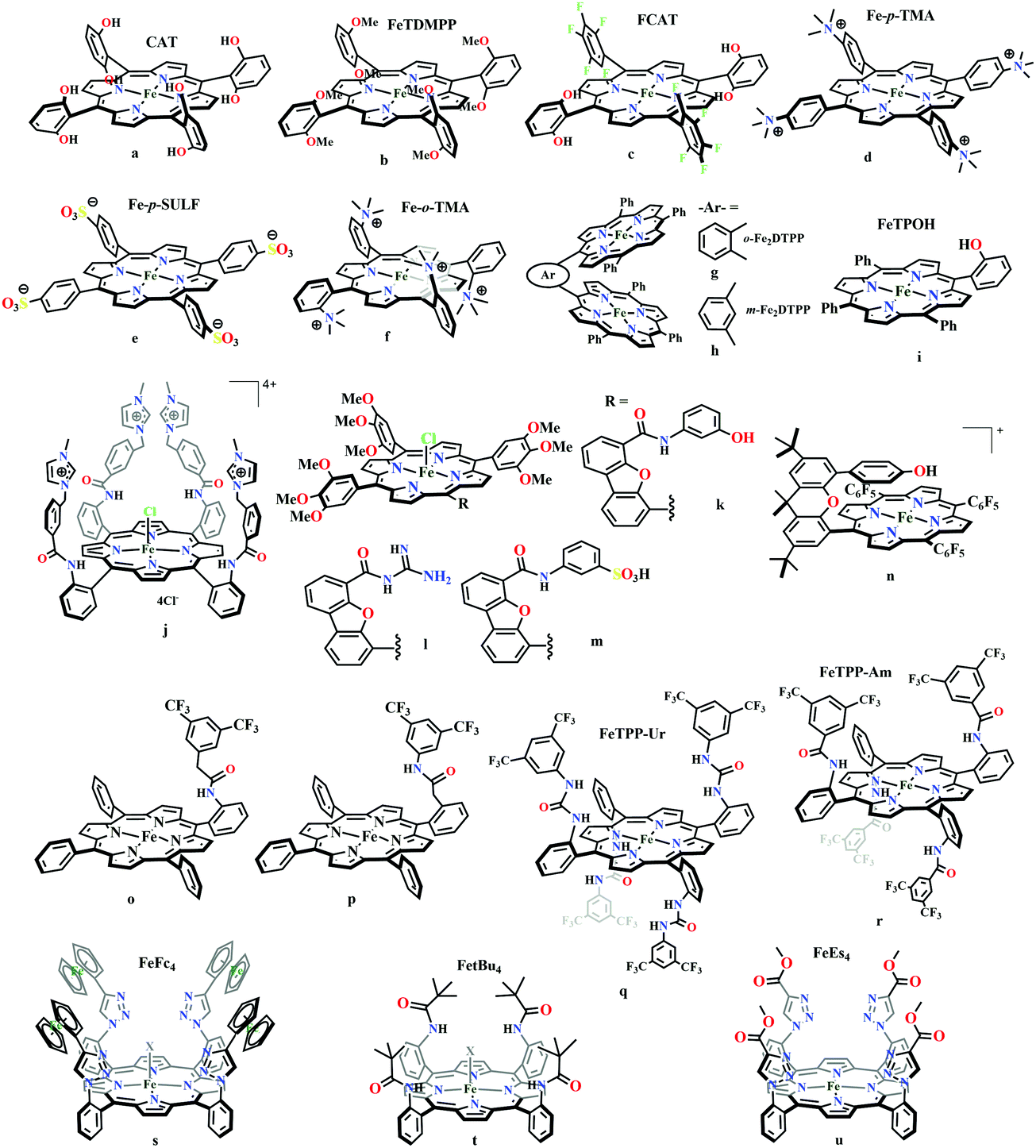 | ||
| Fig. 7 Representative molecules for electrocatalytic CO2 reduction by porphyrin-based catalysts. | ||
The reaction of CO2 with Fe0-porphyrin was proposed to be proceed through the formation of CO2 adduct (Scheme 7). The adduct is stabilized in presence of external acid source (AH). A second molecule of AH is involved in generation of a precursor complex, where a H-bond is being formed between one of the “O” of CO2 and the external proton source. Heterolysis of the C–O bond followed by reductive decarbonylation regenerates the catalyst. The rate-determining step is proposed to be the cleavage of the C–O bond, where an electron transfer (ET) from the central iron-atom is concerted with proton transfer (PT). Therefore, invoking an intramolecular concerted proton electron transfer with bond cleavage mechanism operated in the rate-determining step.112
 | ||
| Scheme 7 Proposed mechanistic pathway for the reduction of CO2 to CO by Fe-porphyrin electrocatalysts. | ||
Based on the observed role of proton donors, it was anticipated that tethering acid functionality with the catalyst should accelerate the reduction process by increasing the local proton concentration, which is otherwise impossible to attain in such high amount considering bimolecular reaction condition. A series of meso-phenyl substituted iron-porphyrins were synthesized with varying tethered phenol groups (Fig. 7a–c).113 The eight phenolic protons in CAT (Fig. 7a) was estimated to operate with phenol concentration as high as 150 M and it could efficiently converted CO2 to CO with high rate and faradaic efficiency. On the contrary, the octa-methoxy analogue, Fe0TDMPP (Fig. 7b) was a poor catalyst with large over-potential and low TOF, invoking the importance of the intramolecular proton source in catalysis.114 On an effort to lowering down the over-potential, a fluorinated analogue of CAT was considered, FCAT (Fig. 7c), anticipating the electron withdrawing inductive effect of the F-atoms. Despite lowering down the electron density over iron-centre, this catalyst was found to be one of the best electrocatalysts to convert CO2 to CO, selectively.115 The effect of solvent in electrocatalytic CO2 reduction was reported by Warren and co-workers using a 2-hydroxyphenyl substituted TPP (TPOH, simpler version of CAT, Fig. 7i).116 The Fe-TPOH complex was shown to reduce CO2 to CO (96% FE) at TOF ∼ 4500 s−1 in acetonitrile in presence of weak Brønsted acids (1 M water or 8 mM phenol). In contrast, the catalyst was found to be very poor in DMF. Based of Abraham's H-bond acidity117 and basicity118 as metrics, it has been suggested that DMF being a strong H-bond acceptor, plays a detrimental role in catalysis, where H-bonding is very crucial in accelerating the rate. Substituting the four meso-p-hydrogens of FeTPP by trimethylammonium groups (WSCAT) could afford formation of a water-soluble porphyrin (Fig. 7d). Under neutral pH, it catalyzes the reduction of CO2 to CO, selectively.119
The enhanced catalytic activity in CAT and FCAT was a direct proof of participation of pendant OH-functionalities installed in the catalysts. Appearance of a distinct pre-wave in front of the catalytic wave was suggestive of protonation of the Fe0–CO2 adduct prior to further reduction.120 Therefore, the stabilization of the Fe0–CO2 adduct through H-bonding with the pendant OH-groups is likely the main reason for such efficient catalysis (Scheme 8). The protonation is conducted from the pendant phenol groups, while the re-protonation of the phenoxide occurs from the externally added phenol. Hence, the role of the pre-positioned phenol groups was proposed to be as H-bond stabilizers and maintaining high concentration proton donor. The next step is concerted electron transfer and proton transfer with the cleavage of one of the two C–O bonds (CPETBC) of the bound CO2. However, the intermediates proposed were based on simulation of electochemical parameters, no such intermediate was isolated or spectroscopically characterized.
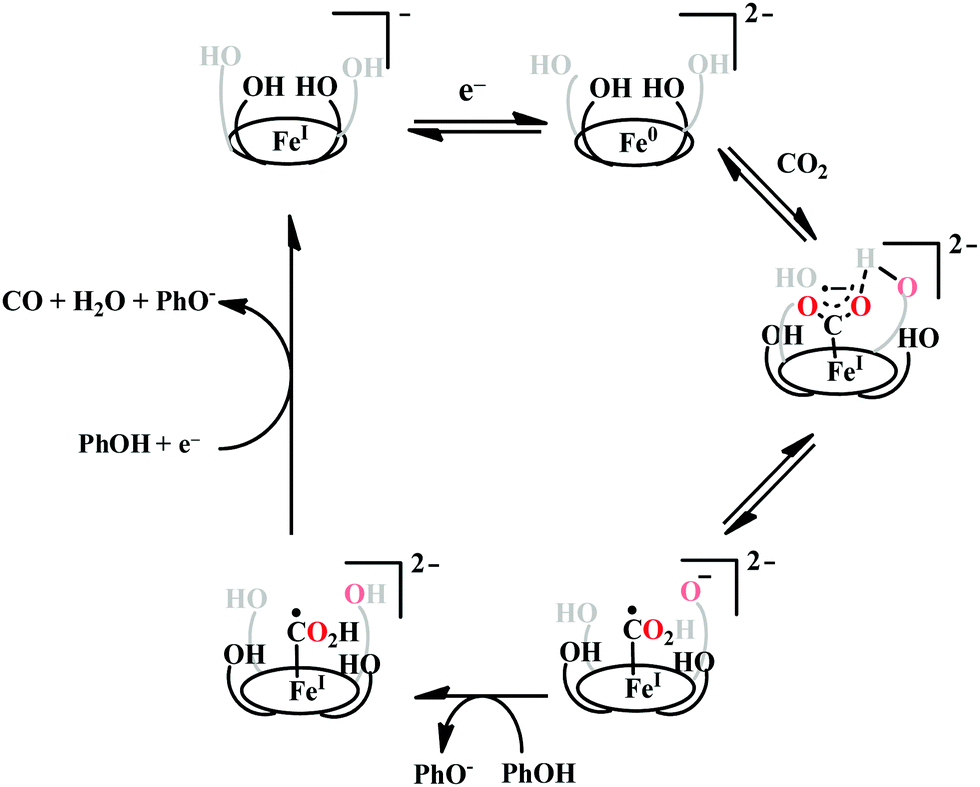 | ||
| Scheme 8 Proposed mechanistic pathway for the reduction of CO2 to CO by Fe-porphyrin electrocatalysts with pendant acid functionality, (OH)4. | ||
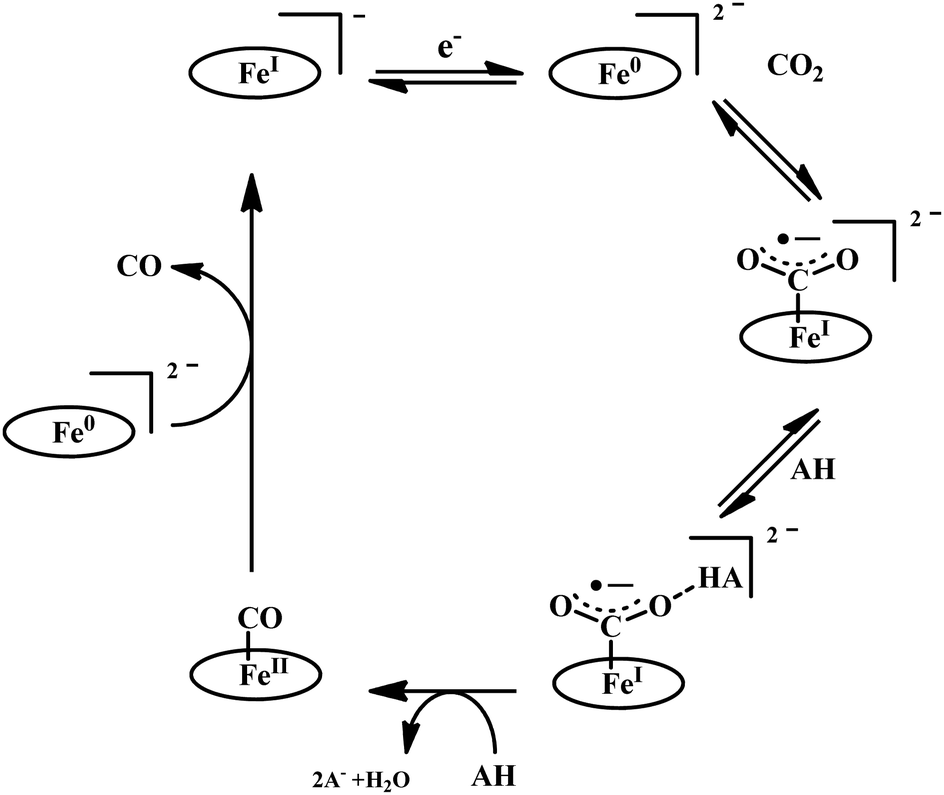
The reaction intermediate was probed by trapping reaction intermediates taking advantage of porphyrins bearing hydrogen bonding residues.121–123 At least two intermediates were observed using resonance Raman spectroscopy in an iron-porphyrin complex bearing a distal H-bonding pocket (Fig. 7u) involved in the homogeneous chemical reduction of CO2 (Scheme 9).124 The first intermediate (intermediate I), which could only be stabilized at −95 °C, was found to be a FeII–CO22− adduct. Warming up the solution to −80 °C lead to the protonation in presence of weak acid (methanol) to give rise to another intermediate; intermediate II. The intermediate II was established to be a FeII–COOH species (using labelled CO2 and proton). In the presence of phenol as acid source, only intermediate II could observed even at −95 °C and no intermediate I could be isolated. On the contrary, in presence of strong acid like p-toluenesulfonic acid the product, FeII–CO, species was generated rapidly at −95 °C. Intermediate I being very basic can abstract proton rapidly even from MeOH while intermediate II requires a strong acid to cleave the C–O bond to eliminate water. In the absence of strong acid, intermediate II gradually released formic acid. This investigation realized the definitive role of the M-COOH species in determining the selectivity of CO2 reduction; a C-protonation leads to HCOOH while an –OH protonation releases CO.
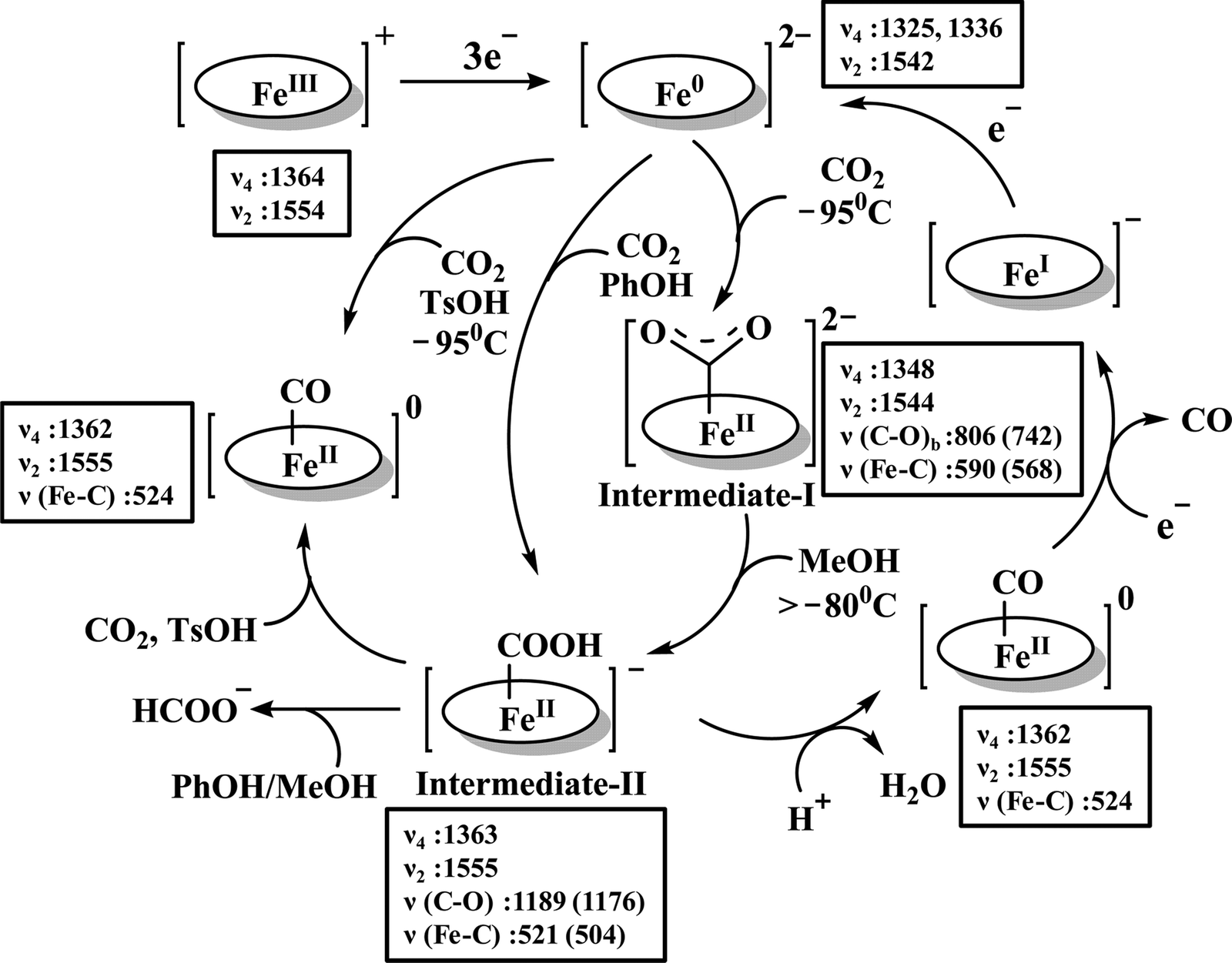 | ||
| Scheme 9 Proposed mechanistic cycle for the reduction of CO2 by a distal-triazole containing Fe-porphyrin. The vibrations (in cm−1) are given in the boxes. | ||
The pendant OH-groups can stabilize the negatively charged [porphyrin-Fe0–CO2]2− adduct through the formation of H-bond. However, this intermediate may also be stabilized by coulombic interactions with positively charged groups attached to the porphyrin. For example, the WSCAT (or Fe-p-TMA, Fig. 7d) can boost the catalysis, while replacing trimethylammonium with sulfonate (Fe-p-SULF, Fig. 7e) shows reduced catalytic effect. However, replacing four-ortho hydrogens by trimethylammonium group (Fe-o-TMA, Fig. 7f), can catalyze the reduction of CO2 to CO almost exclusively and that too with a TOF of 106 s−1 and very low over-potential (∼200 mV).125 Aukauloo and co-workers have attached methylimidazolium fragments as ionic liquid units, modifying the α4-tetraaminophenylporphyrin (Fig. 7j).126 The cationic pendant methylimidazolium was introduced to stabilize the negatively charged Fe–CO2 adduct. The catalyst was shown to be very active electrocatalyst to reduce CO2 to CO at only 418 mV over-potential in water with 91% faradaic efficiency and higher TOF, TON values of 14986 s−1 and 1.08 × 108, respectively. Nocera and co-workers have shown the proton-relay in iron hangman porphyrins with phenol (HPDFe-PhOH, Fig. 7k), guanidinium (HPDFe-GND, Fig. 7l), and sulfonic acid (HPDFe-3SA, Fig. 7m) proton donor groups.127 Selective reduction of CO2 to CO with >93% faradaic efficiencies were described where the CO2 binding within the hangman-cleft was proposed to determine the rate of the reaction. Deprotonation of the hanging sulfonate group was responsible for exhibiting lower TOF due to unfavourable electrostatic interaction with the negatively charged CO2-adduct. Despite tremendous rate acceleration by the hanging phenolic group, often improper alignment of the activating group may exert ill-defined results. For example, in the case of complex 7n (Fig. 7n), the axial Cl−, forming a H-bond with the phenolic-OH group renders an inefficient CO2 reduction, compared to the same complex with triflate ion.128 Therefore, distal H-bonding interaction plays a key role in determining the stability of the intermediates affecting the product selectivity as well as the rate of the reaction as suggested by the solution mechanistic investigation. Hence, a proper estimation about the role of distal residues was warranted.
Recalling the role of His and Lys in the activation of CO2 in [NiFe]CODHs, Chang and co-workers have synthesized a series of ortho- and para-substituted amide pendants at different positions from the porphyrin-plane (e.g., complex 7o–p in Fig. 7).129 The ortho-functionalized positional isomers (unlike para-functionalized) were found to engage in through-space interactions to enhance the rate of the electrochemical conversion of CO2 to CO. The complex with a distal amide, 7p was involved in greater enhancement over 7o, demonstrating the importance of precise geometric orientation of 2nd sphere pendants for designing effective catalyst. A series of iron-porphyrins with different distal environment spanning from amide (Fe picket-fence porphyrin) to triazole-with different substituents (complex 7t and 7u in Fig. 7).130 They exhibit TOFs ranging from 1–1000 s−1 without changing the pKa of the external acid source used (phenol). Theoretical calculations (DFT) suggested the intermediate-I (Fe–CO2−) is better stabilized by coulombic interactions, while the C–OH bond cleavage from the intermediate-II (Fe–COOH) is greatly influenced by the distal hydrogen bonding interactions. H-bonding alone could tune the rate of the CO2 reduction by as much as 1000-fold without change of overpotential or proton source. On a similar note, Aukauloo and co-workers pointed out the influence of multi-point H-bonding in the super-structured iron-porphyrin bearing urea (FeTPP-Ur, 7q) and amide (FeTPP-Am, 7r) functionalities in comparison with FeTPP.131 In comparison with FeTPP and FeTPP-Am (can make single-point H-bond), the pendant urea-functionalized FeTPP-Ur can reduce the over-potential of CO2 to CO electroreduction by ∼300 mV while retaining high TOFs. Also, the entrapped water molecules within the molecular clefts were found to be sufficient as a proton source.
Therefore, the Fe0-porphyrins are generally found to be biased for catalysing the reduction of CO2 to CO, where “pull-effect” from the secondary coordination sphere boosts up the rate of C–O bond cleavage up to 106 s−1. In absence of activators (such as external/internal acid source), the rate is relatively slow and formate is produced as a by-product likely via C-protonation of the FeII–COOH intermediate as demonstrated by Mondal et al.124 However, the reduction can be directed for the production of formate by the introduction of tertiary amines.132 Greater catalytic activity is attributed with the higher basicity of the tertiary amine. The axial amine appears to push the electron density over the bound “C”-atom (recalling trans-effect), thereby increasing the basicity on the C-atom of the Fe–COOH intermedate to allow the protonation and facilitate dissociation of formate (Scheme 10). The use of a weaker acid is warranted to avoid the formation of metal-hydride to undergo HER.
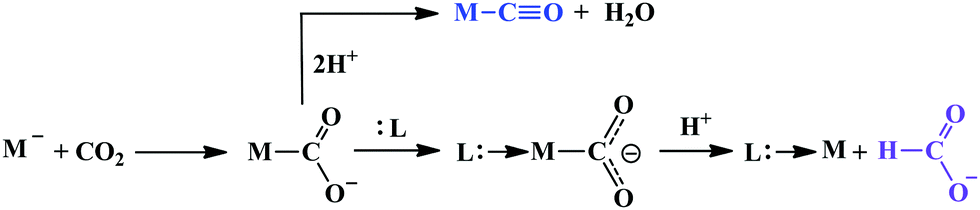 | ||
| Scheme 10 Contrasting pathways in CO2 reduction. | ||
Conceptual development of a suitable electrocatalysts for efficient CO2 reduction (in terms of over-potential, rate, stability, etc.) is quite fascinating for academic interests but its practical implementation is seriously deterred by the requirement of pure CO2.133 Considering the abundance of O2 over CO2 in the atmosphere and in flue gases (8–15% CO2), one of the major challenges in practical CO2 reduction is the O2-sensitivity of the low-valent metals. Note, O2 (0.83 V vs. NHE at pH 7)134 has much higher reduction potential in comparison with CO2. However, due to kinetic limitations, reduction of O2 to water is also very challenging, leading to the generation of partially reduced oxygenated species (PROS) such as superoxide, peroxide, etc., which are very reactive often responsible for irreversible catalyst degradation.135–137 Hence, an O2-tolarrant catalyst scheme is warranted. Successful technologies from hydrogen-evolving catalysts suggests three ways this can be achieved:138–145 (a) using a co-catalyst, which can scavenge any PROS; (b) the catalyst by itself can reduce O2 to water without releasing PROS and (c) choosing appropriate catalyst which can react selectively with CO2 over O2. The FeFc4 (Fig. 7s) complex has been known to catalyze the 4e/4H+ reduction of O2 to water over a wide range of pH.146 The three of the four appended ferrocene-moieties and the central FeII-atom give the required four electrons, while protons are derived from water.123 Hence this bi-functional catalyst was chosen to device an O2-tolarant CO2 catalyst, where the Fe0-state was activated for selective CO2 reduction (kinetic advantage) to CO, while FeII-state was activated for fast and selective O2 reduction to water (Scheme 11).147 Furthermore the Fe0-porphyrins was found to react with CO2 with ∼500 times faster rate relative to O2 offering selective CO2 reduction even in 3:1 mixture of O2:CO2. This was the first, and till date the only, report of any O2-tolarant low-valent transition metal-based CO2 reduction catalyst.
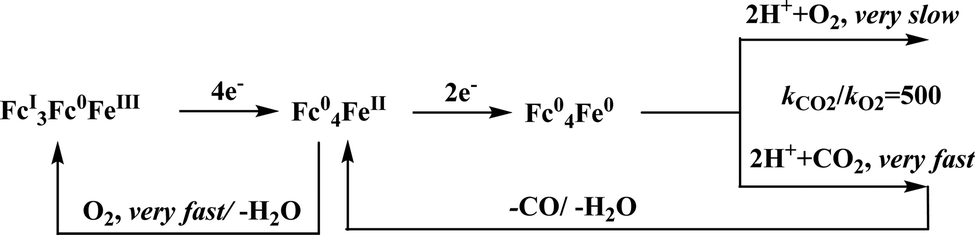 | ||
| Scheme 11 General reaction scheme of O2-tolerant electrocatalytic CO2 reduction by FeFc4. | ||
 | ||
| Fig. 8 Representative macrocyclic complexes for electrocatalytic CO2 reduction. | ||
Based on the initial work from Sauvage followed by Anson, Kubiak and Ye, a plausible mechanistic cycle has been proposed (Scheme 12).150,152,153,159–161 Initial electron-transfer to [NiII(cyclam)] generates the one electron reduced species, [NiI(cyclam)], which gets adsorbed on the electrode surface. In the presence of CO2, the adsorbed [NiI(cyclam)] likely forms an adduct with CO2, which gets stabilized by H-bonding to the adjacent N–H groups. Further, reduction and protonation lead to the formation of [NiI(cyclam)(COOH)] intermediate, which upon protonation generates [NiII(cyclam)(CO)] releasing a molecule of water. Reductive decarbonylation regenerates the active-catalyst, [NiI(cyclam)]. The facile and faster formation of [NiI(cyclam)(CO)] limits the rate of the process. Kubiak and co-workers have used an efficient CO-scavenger, [NiII(TMC)] (where, TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) to prevent from the catalyst deactivation.160 Shafaat and co-workers used a semi-enzymatic model by introducing [NiII(cyclam)] into a Cu-containing biological scaffold, azurin, which can provide a well-defined secondary coordination sphere to modulate catalytic efficiency and selectivity.161 Chang and co-workers have realized the effect of additives which can stabilize the CO2-adduct in [NiI(cyclam)] through H-bonding of different strength.162 A bis(aryl)urea additive could afford an enhanced current increment relative to isostructural amide additive. A co-operative multi-point H-bonding in urea-additive was proposed for such augmented catalytic activity.
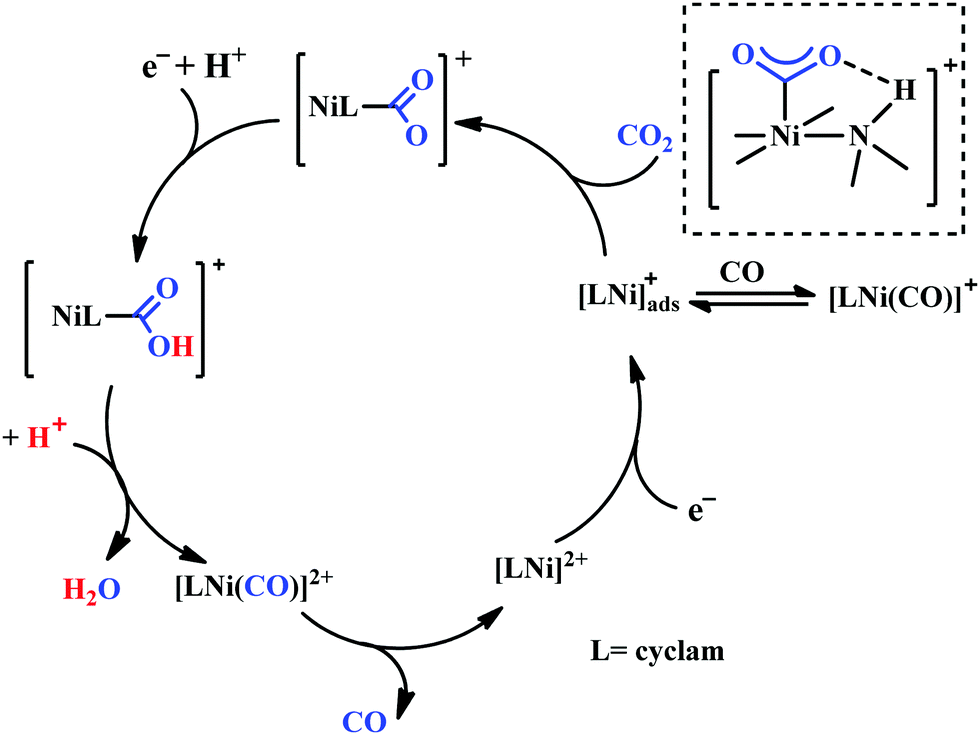 | ||
| Scheme 12 Proposed mechanistic pathway of electrocatalytic CO2 reduction to CO by [NiII(cyclam)]-based macrocycles. | ||
Marinescu and co-workers have introduced cobalt-bound azacalix[4](2,6)-pyridine framework with different substitutions on the pendant secondary and tertiary (methyl and allyl) amines (Fig. 8h–j).163 In the presence of weak Brønsted acid, the complex with N–H pendant can catalyze the reduction of CO2 to CO with ∼98% faradaic efficiency. While the other pendant N-alkyl variants were found to have poor catalytic rates which were lower by at least two order of magnitude. The pendant N–H groups were proposed to stabilize the Co0–CO2 adduct (through intramolecular H-bonding) as well as promote the C–OH bond cleavage to release CO (Scheme 13). Also, the N–H groups were proposed to decrease the over-potential by increasing the CoI/0 reduction potential. Unlike cyclam, this macrocycle is unique because here the pendant amines lie completely outside the primary coordination sphere of the metal-centre which allows discrete control over the number and configuration of the pendant proton donors in the outer sphere of the metal-centre without interfering in its primary coordination sphere.164 To discern the roles of the first and second coordination spheres in CO2 reduction catalysis, a series of cobalt complexes were synthesized with varying pendant secondary and tertiary amines. The theoretical calculations suggested that the N–H group was not transferring the proton directly to the bound CO2, in contrast, they bind acid molecules from the solution. Hence, in the rate-determining step each pendant amine was proposed to bind an acid molecule noncooperatively which eventually activate and enhance the local concentration of proton donors around the COOH adduct.
 | ||
| Scheme 13 Proposed mechanistic pathway for electrocatalytic CO2 reduction to CO by azacalix-pyridine based macrocycles. | ||
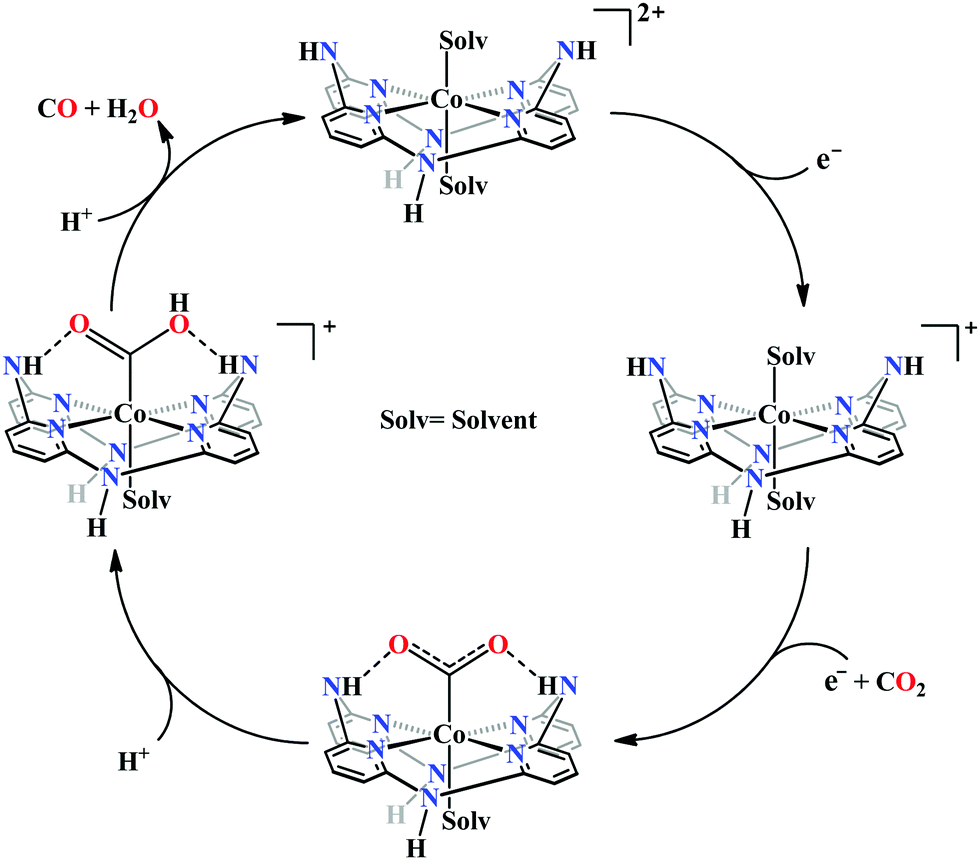
Peters and co-workers made a macrocyclic-cobalt complex with redox non-innocent ligand, [CoIIIN4H(Br)2]+ (where, N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca1(7),2,11,13,15-pentaene) (Fig. 8k).165 This complex was shown to undergo electrocatalytic CO2 reduction to CO (FE ∼ 45%) with concomitant generation of H2 (∼30%) in wet acetonitrile medium. In the same ligand framework, just by changing the metal, the CO2 reduction can be biased for either CO or formate. This phenomenon was shown by Robert and co-workers in a unique pentadentate ligand system, 2,13-dimethyl-3,6,9,12,18-pentaazabicyclo-[12.3.1]octadeca-1(18),2,12,14,16-pentaene (Fig. 8m).99 The CoII-complex is biased to generate CO with faradaic efficiency as high as 82%. In contrast, the FeIII-complex was shown to generate formate (FE ∼ 75–80%, with no detectable H2 or CO). This unusual change in product selectivity was explained considering the electronic structure of the M-COOH intermediate. In the case of Co-complex, there is greater π-back-bonding to the π*-orbitals of CO2 from the formal CoII-centre rendering the C–O bond weak to facilitate CO release upon C–O bond cleavage. On the contrary, the Fe-complex passes through a formal FeIII–COOH intermediate. Poor π-back-bonding from FeIII renders lower charge density over “O”, favouring the isomerization to Fe–OCOH intermediate, which leads to the generation of formate (Scheme 14). Jurss and co-workers have developed a series of nickel complexes with tetradentate chelating ligands comprised of a redox-active 2,2′-bipyridyl core and electron-rich N-heterocyclic carbene (NHC) donors (Fig. 8n–p).166 During electrocatalytic CO2 reduction, the complexes showed a drastic change in product selectivity from H2 to CO on transitioning from an open-chain to macrocyclic conformation with increasing linker size.
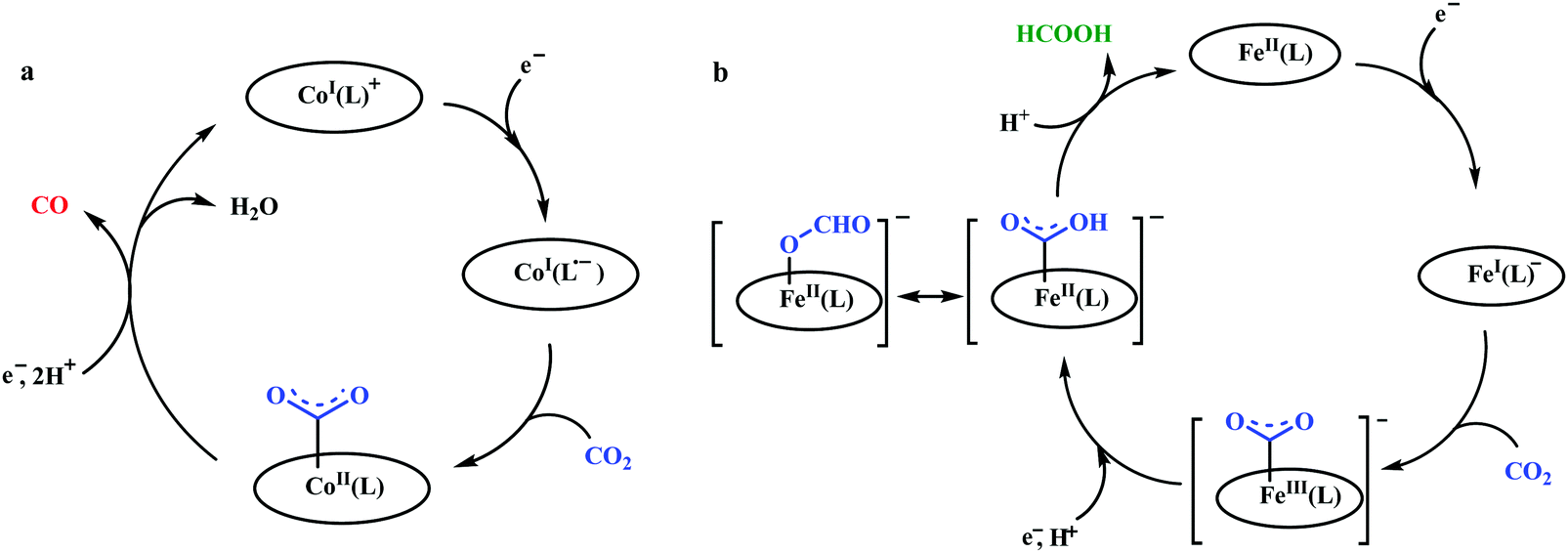 | ||
| Scheme 14 Proposed mechanistic pathway for electrocatalytic CO2 reduction to CO vs. formic acid; catalyzed by same ligand framework but with different metals: (a) cobalt and (b) iron. | ||
Over the years, several bio-inspired model of [NiFe]-CODHs have been developed involving several different ligand-framework, such as phosphine, bipyridine, porphyrin, cyclam, etc. It has been observed that second coordination sphere greatly influence the product selectivity as well as the tuning the rate of the reaction. Irrespective of the ligand frameworks, the initial CO2 activation was shown to be happen through the nucleophilic attack from the electron-rich metal centre. This implies, an electron-rich late transition metal should be more useful. The negatively charged metal–CO2 complex is stabilized either by protonated basic residues or any Lewis acidic metal. The C–O bond cleavage is proposed to be the rate-determining step, where an intramolecular basic residue (such as amines) are shown to play a pivotal role in shuttling the protons to the metal–COOH species. However, the role of thiols is not well explored. A nickel–tetrathiolate complex, mimicking molybdopterin (Fig. 9a), was reported to reduce CO2 to formate (with minor amounts of CO and H2) with 340 mV over-potential.167 To understand the effect of metal–thiolates in CO2 reduction, electron-rich late transition metal (cobalt) bound pyridine-dithiolate complex, 2,6-dithiomethylpyridinocobalt(dppe) (Fig. 9b) was synthesized.168 The catalyst was shown to reduce CO2 to CO (>95% faradaic efficiency) with an unprecedented <100 mV over-potential and that too with a TOF of 1559 ± 8 s−1. The electron-rich thiolates play a dual role. Firstly, on getting protonated under the experimental conditions (H2O + CO2), increases the formal potential of the CoII/I process, which in turn lowers down over-potential of CO2 reduction, and secondly, the other thiolate increases the electron density over CoI-state (through back-bonding) activating it to bind CO2 (Scheme 15A). The strong covalent bonding in a CoIII–COOH intermediate develops large negative charge-density over the “O”-atoms, preventing “C-protonation” and the generation formate. On the contrary, the thiolate mediates the proton relay to the “OH” group of CoIII–COOH intermediate in the rate-determining process of C–O bond cleavage leading to selective generation of CO. Hence, a dithiolate ligand framework can lead to the development of efficient CO2 reducing catalyst operative at low over-potential with high turnover rate. As an extension of the concept, a thio-pyridinato complex, cobaltIII-bis(2-thiopyridinato)-diphenylphosphenoethane chloride (Fig. 9c) was synthesized. This complex can also catalyze the reduction of CO2 to CO from the CoI-state.169 Unlike thiol-protonation, the hemilability of the pyridine arn on protonation mediates the proton-relay (Scheme 15B). Whereas, both thiolates being bound to the cobalt-centre, stabilizes the CoI-state for CO2 binding. Less energy demanding proton-transfer from the pyridine moiety selectively to the “OH” results in facile C–OH bond cleavage to form CO. Recently, Mougel and co-workers have reported a pyridinethiolate bound cobalt complex, bipyridine-bis-(2-pyridinethiolato)-cobalt(III)-hexaflurophosphate (9d).170 The catalyst can convert CO2 to formate selectively with a very low overpotential of110 mV and a TOF of 10 s−1.
 | ||
| Fig. 9 Representative metal–thiolate based electrocatalysts for CO2 reduction. | ||
 | ||
| Scheme 15 Proposed mechanistic cycle for the selective reduction of CO2 to CO through the influence of (a) a thiol-protonation; (b) a hemilabile-pyridine protonation. | ||
To provide a comprehensive overview, the catalysts which were found to convert CO2 to CO or formate with >90% product selectivity are summerized in Table 1. The TOF numbers were not included because they were determined under a variety of different experimental conditions using different mathematic equations. Each mathematical equation is derived considering different boundary conditions, hence benchmarking the catalysts in terms of TOF does not really help their comparability.
| Complex | Conditions | Overpotential | Product | TON | Ref. |
|---|---|---|---|---|---|
a Calculated from the equation: E = E° − 0.059pKa; assuming  172vs. Fc+/0 in CH3CN.
b Assuming 172vs. Fc+/0 in CH3CN.
b Assuming  vs. Fc+/0 in DMF, pKa of PhOH was considered to be 15.4.173 vs. Fc+/0 in DMF, pKa of PhOH was considered to be 15.4.173
|
|||||
| Re(bpy)(CO)3Cl | DMF (+ H2O), 0.1 M NEt4Cl, GC electrode | 0.56 V | 90% CO | 59 | |
| Re(bpy-tBu)(CO)3Cl | CH3CN (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.74 V | 100% CO | 64 | |
| Mn(bpy)(CO)3Br (4a) | CH3CN (+ H2O), 0.1 M NBu4ClO4, GC electrode | 0.24 V | 90% CO | 66 | |
| Re(bpy-thiourea)(CO)3Cl (4h) | CH3CN (+ H2O), 0.1 M NBu4PF6, GC electrode | ∼0.47 Va | 89% CO | 78 | |
| [(bpyNHEtPY2Me)FeII]2+ | CH3CN (+ H2O), 0.1 M NBu4PF6, GC electrode | ∼0.36 Va | 81 ± 11% CO | 81 | |
| o-Fe2DTPP (7g) | DMF (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.66 V | 95% CO | 1.58 × 108 | 111 |
| CAT (7a) | DMF (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.46 V | >90% CO | 5 × 107 | 113 |
| FCAT (7c) | DMF (+ PhOH), 0.1 M NBu4PF6, GC electrode | 0.39 V | ∼100% CO | 115 | |
| Fe-o-TMA (7f) | DMF (+ H2O + PhOH), 0.1 M NBu4PF6, GC/Hg electrode | 0.22 V | ∼100% CO | 125 | |
| FeTPOH (7i) | CH3CN (+ H2O), 0.1 M NBu4PF6, BPG electrode | 0.36 V | 96% CO | 4000 | 116 |
| (7j) | DMF (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.42 V | 91% CO | 1.08 × 108 | 126 |
| Fe-ortho-2-amide (7p) | DMF (+ PhOH), 0.1 M NBu4PF6, GC electrode | ∼0.54 Vb | 92% CO | 129 | |
| FeTPP-Ur (7q) | DMF (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.43 V | 91% CO | 3.28 × 106 | 131 |
| 6e–h | DMF (+ H2O), 0.1 M NBu4PF6, GC electrode | 0.50–0.70 V | >90% formate | 15–23 | 95 |
| [Pt(dmpe)2](PF6)2 | CH3CN (+ PhOH), 0.1 M NEt4PF6, GC electrode | <0.10 V | >90% formate | 171 | |
| (Ph4P)[NiIII(qpdt)2] (6m) | CH3CN (+ CF3CH2OH), 0.1 M NBu4ClO4, Hg/Au amalgam electrode | 0.34 V | ∼90% formate (trace CO, H2) | 167 | |
| Nickel cyclam (8a–b) | H2O (pH 4.1), 0.1 KNO3, electrode | ∼0.2 V | Up to 96% CO | ∼102 | 149–151 |
| [NiII(HTIM)] and [NiII(MTC)] | H2O (pH 5), 0.1 NaClO4, Hg electrode | ∼0.55 V | 88 ± 7% CO | 156 | |
| Cobalt aminopyridines (8h–j) | DMF (+ MeOH/CF3CH2OH), 0.1 M NBu4PF6, GC electrode | 0.35–0.68 V | ∼98% CO | 1.22 × 106 | 163 |
| 2,6-Dithiolatomethylpyridine-dppe cobalt(II) (9b) | CH3CN (+ H2O), 0.1 M NBu4ClO4, GC electrode | 0.05–0.07 V | 95% CO | 1 × 106 | 168 |
| [Co(dppe)(2-PyS)2]Cl (9c) | CH3CN (+ H2O), 0.1 M NBu4ClO4, GC electrode | 0.18 V | 92% CO | 1 × 105 | 169 |
Mechanistic consideration of CO2 reduction and rational design
A major challenge in CO2 reduction is control of competitive proton reduction process (hydrogen evolution reaction, HER, Scheme 16c), which is more favourable than CO2 reduction both thermodynamically and kinetically. Similarly, selectivity over the C-based product is desirable i.e. CO, HCOOH or CH4. The initial step of CO2 activation is the nucleophilic attack of the electron-rich metal centre on the electrophilic C-atom of CO2 (Scheme 16a). The CO2 reduction will result in a Mn+2–CO22− species which is very basic and will pick a proton easily to Mn+2–COOH species. Irrespective of the ligand framework used, the reaction generally proceeds via this metal–COOH species. The bound –COOH species bears a formal charge of −1. A C-protonation (g) will lead to the release of HCOOH while an O-protonation (f) will lead to CO (Scheme 16). The competing reaction involves the protonation of the metal to form the corresponding hydride (Scheme 16b). The resulting Mn+2–H species can either be protonated again (c) to release H2 or attack the CO2 as a nucleophile to produce formate (d).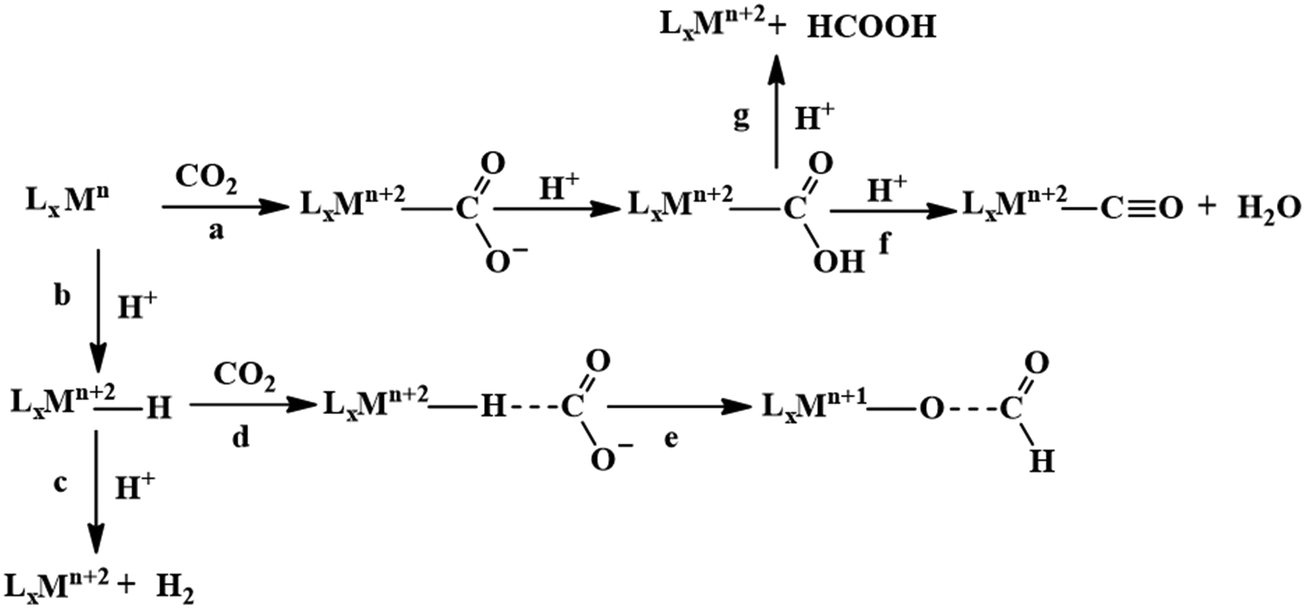 | ||
| Scheme 16 Controlling the competitive pathways in CO2 reduction. | ||
Production of CO from CO2 requires the reaction to move along a → f. Two key factors need to be controlled to achieve selectivity i. avoiding protonation of the metal centre (b) and ii. protonation of the O-centre of the Mn+2–COOH species (g).
i. Avoiding protonation of the metal centre:
The electronic structure of the metal centre may be tuned by judicious choice of ligands to restrain its basicity and avoid protonation. A good example of this is demonstrated in the Ni-cyclams. The CO2 binds the Ni-centre at its formal +1 state displacing a bound CO, the second electron required to reduce the CO2 further is provided to a [Ni–CO2]+ species.158,160,174 This avoids starting the reaction at a formal Ni(0) state which, apart from concerns of dissociation from the ligand, would be more prone to protonation and thereby loose selectivity.
Another approach to avoid metal protonation is the use of reductive disproportionation of CO2 to CO and CO32− which rescinds the requirement of proton all together and is inherently selective for CO by design. This is demonstrated in a series of copper and nickel-phosphine as well as manganese-bipyridine complexes.69,92–94
Finally, a bio-inspired approach is to use bound thiolate ligand or 2nd sphere pendant amine groups to act as the site of protonation keeping the metal centre free for CO2 reduction. This has been demonstrated in the cobalt thiolate complexes where the thiolate ligand is protonated.168 This protonation helps shift the reduction potential of the metal centre more positive reducing the over potential of the process and the protonated thiol ligand is later utilized to stabilize the Mn+2–CO22− intermediate and protonate it to Mn+2–COOH species.
ii. Protonation of the O-centre in the Mn+2–COOH species.
To selectively protonate the O-centre, several complexes have been developed with pendant groups which can help the proton transfer to this intermediate. Hydrogen bonding from these pendant groups helps in both facilitating the reduction of CO2 as well as the following proton transfer to the Mn+2–COO2− and Mn+2–COOH intermediate species. These include cyclam-NH,150,154,158 phenols,114 water,124,147 amide,129,130 thiols,168 azacalix-pyridine163 and pyridinium.169 The stabilization of the Mn+2–CO22− can also be attained by electrostatic field.125
Although the focus has been installing proton transfer and hydrogen bonding groups, an often-unappreciated fact is the possibility to control the charge density at the C-center by tuning the covalency of the metal carbon bond of the Mn+2–COOH species.169 This was suggested when analysing the selectivity for CO observed for a Cobalt dithiolate complex. The CoIII–COOH intermediate had a fairly covalent Co–C bond resulting in depletion of the negative charge on the C-centre of the –COOH− species and the negative charge was localized on the O-atom. This automatically biased the system for O protonation to release CO and no HCOOH could be thus be formed.
Nitrite reduction
Introduction
This section is focussed primarily on the heme-containing nitrite reductases. An attempt has been made to explicitly discuss different structural and functional aspects, focussing on enzymatic architecture and detailed mechanism emphasizing the roles of the basic residues within the active site of different enzymes involved in nitrite reduction. The later part of the review focusses on the recent investigations with ‘model systems’, directed towards understanding the mechanisms of these enzymes and the discussions concentrates on the factors which distinguishes the three non-identical nitrite reduction pathways. This review mainly concentrates on heme based enzymes and hence discussions on model systems will also be limited to heme related porphyrinoid complexes.Lesson from Nature
Nitrite reduction to ammonium
The conversion of nitrite to ammonium ion is accomplished in two different pathways, one involves assimilation of nitrite and the other is a dissimilatory process (Scheme 17). In each of the pathways, nitrite is reduced by six electrons to ammonium and the process is catalyzed using enzymes containing iron-based porphyrinoid cofactors, skipping the release of any detectable intermediates.175,176 The metal involved relates to the prebiotic ammonia formation from nitrite by reduced iron in the early earth.177 However, these two routes of nitrite reduction involve discrete cellular mechanism and occur in different cellular compartments: assimilation in cytoplasm/chloroplasts and dissimilation in periplasm. The enzymes involved in catalysis are structurally distinct with different physiological redox partners.176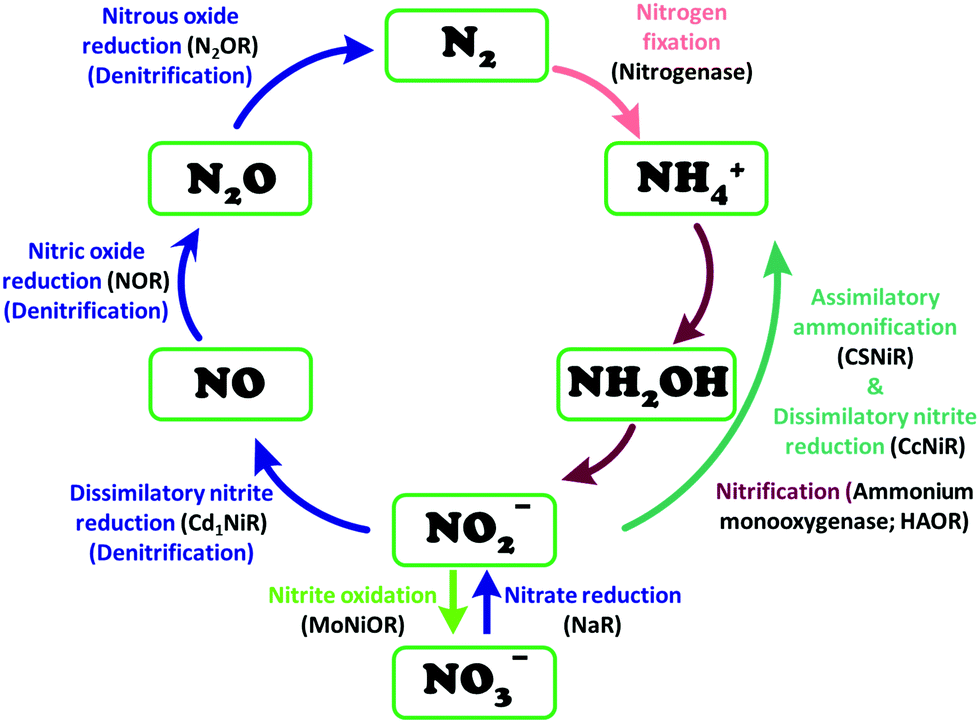 | ||
| Scheme 17 Biochemical nitrogen cycle. Different pathways of interconversion of various oxides of nitrogen, dinitrogen and ammonium are highlighted. The enzymes catalysing the conversions are written in black inside parenthesis. | ||
All cystallographically characterised enzymes (except T. nitratireducens) have homodimers of penta heme (labelled heme 1–5, Fig. 10b) subunits with extensive interaction between the monomers.189 Each monomer contains five heme c and arranged in near-parallel and near-perpendicular heme pairs (Fig. 10b). Hemes 2–4 are axially bis-histidine ligated with a CysXXCysHis binding motif and heme 1 constitute the enzyme active site coordinated by water/hydroxyl group in the distal position and lysine (Lys134) in the proximal position (belonging to a CysXXCysLys134 binding motif).178,191 The bis-histidine ligated hemes participate in electron relay during the catalysis.192,193 The active site is surrounded by conserved histidine (His277), tyrosine (Tyr218), arginine (Arg114) and glutamine (Gln276) residues (Fig. 10c, Wolinella succinogenes CcNiR sequence numbering). It also hosts two hexacoordinated calcium ions which have substantial structural and functional roles.178,194 Mutation studies revealed that Gln276 calcium ion pair stabilises the distal ligand binding to iron centre through a network of hydrogen bonding and in turn regulated the substrate affinity of the active site.195,196 The His277, Tyr218 and Arg114 residues play central roles in nitrite reduction through extensive hydrogen bonding to the substrate.175,197,198 Electron transfer from physiological redox partner to the active site is facilitated by adjacent hemes which is also evident from the redox potentials of the hemes which are compatible to the potential of NO2− reduction.192,198 In addition, an inlet channel with positive electrostatic surface potential assist approach of nitrite to the active site (also helps in proton supply) and an outlet channel with negative electrostatic surface potential facilitating ammonia release has also been identified.182,184,185 A novel octaheme cytochrome c nitrite reductase (TvNiR) from Thioalkalivibrio nitratireducens has also been structurally characterised. It exists as a homo-hexamer with eight hemes per monomer. Each monomer consists of two domains: in the N-terminal domain resides three hemes with a unique fold and C-terminal domain has five hemes which structurally and functionally conform with CcNiR.190
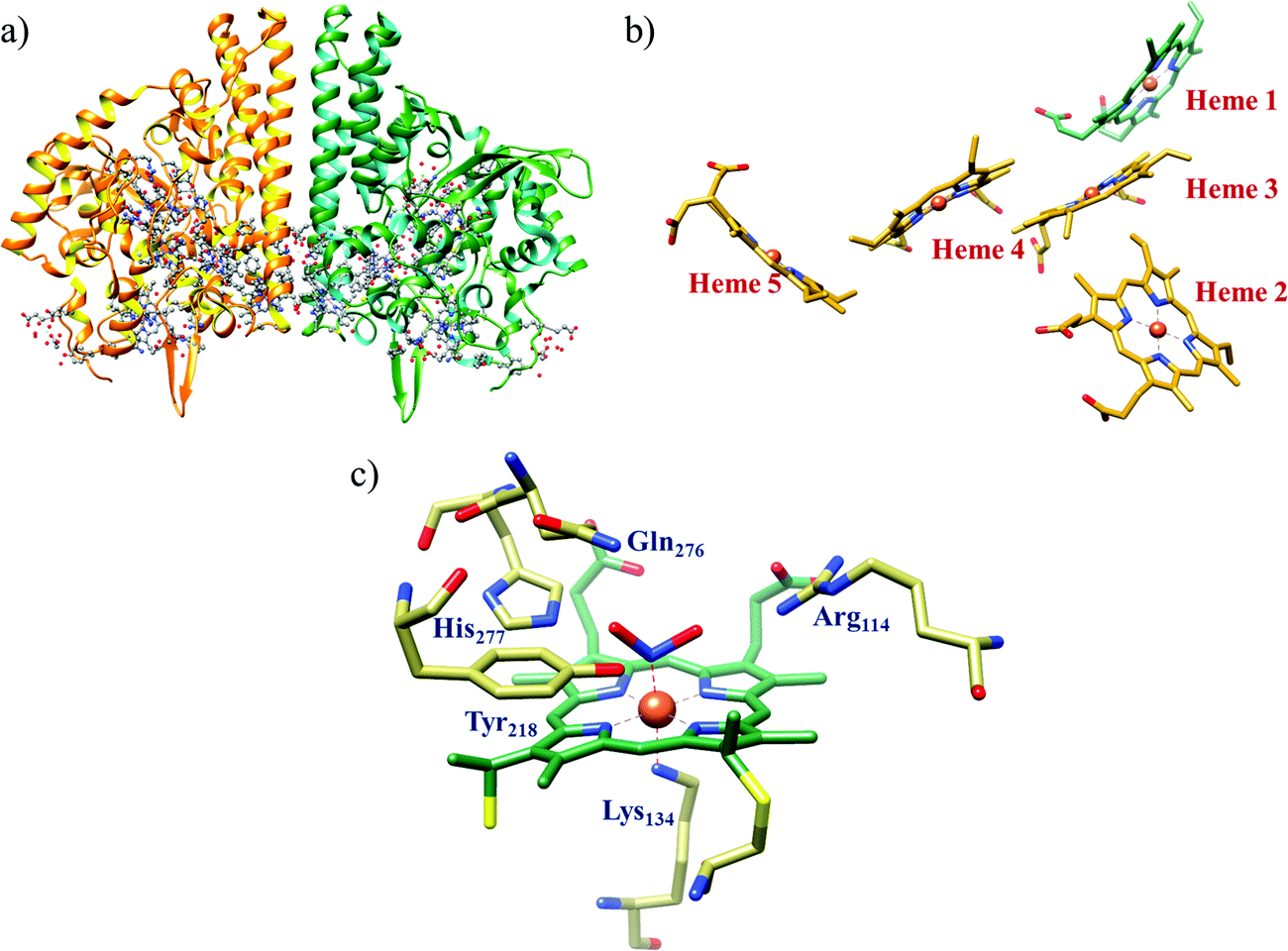 | ||
| Fig. 10 (a) Three-dimensional structure of the Wolinella succinogenes CcNiR homodimer, the momomers are shown in different colours (with yellow and green ribbon colour), (b) arrangement of hemes (1–5) in the monomer with green ribbons; the five heme c are numbered according to their attachment to the protein chain. Heme 1 contitutes the enzyme active site. (c) Nitrite bound active site structure. (a) and (b) are based on PDB file 1FS7182 and (c) is based on PDB file 2E80.175 | ||
The overall reaction can be split up into five episodes:
(a) The first episode deals with substrate binding to the active site and subsequent heterolytic N–O bond cleavage. The enzyme resting state is a high spin FeIII–OH2 (Scheme 18a), confirmed on the basis of Fe–O bond length of 2.1 Å.175 It undergoes reduction to FeII–OH2 which results in the weakening of Fe–O bond (2.39 Å) (Scheme 18b). The substrate nitrite then displaces the water molecule to bind to FeII in ‘nitro’ mode (through N-atom)175,197 and a low spin nitrite bound FeII is formed (Scheme 18c).205 The extensive back-bonding from FeII dxz orbital (HOMO) to nitrite π*-orbital (LUMO), results in strong Fe–N bond (1.9 Å) and a simultaneous weakening of N–O bond.175 Additionally, formation of hydrogen bonds from the protein residues to the oxygen atoms of the bound nitrite molecule contributes to N–O bond heterolysis.193,197 The structure of the nitrite bound states reveal that the two oxygen atoms of nitrite are at hydrogen bonding distances to the protonated His277 (2.6 Å) and positively charged Arg114 (2.8 Å).175 The N–O bond is cleaved through double protonation on one of the O-atom (Scheme 18d and e). At pH < 7, His277 is protonated and is found to facilitate initial protonation and the second protonation (after endothermic reprotonation). But at higher pH, the first proton transfer (PT) occurs from Tyr218 and the second PT is also mediated by the same residue after exothermic reprotonation.178,194 Hence, these two consecutive PT steps results in heterolytic N–O bond cleavage204 to form {FeNO}6 intermediate (Scheme 18f) and the first water molecule is released.197,206
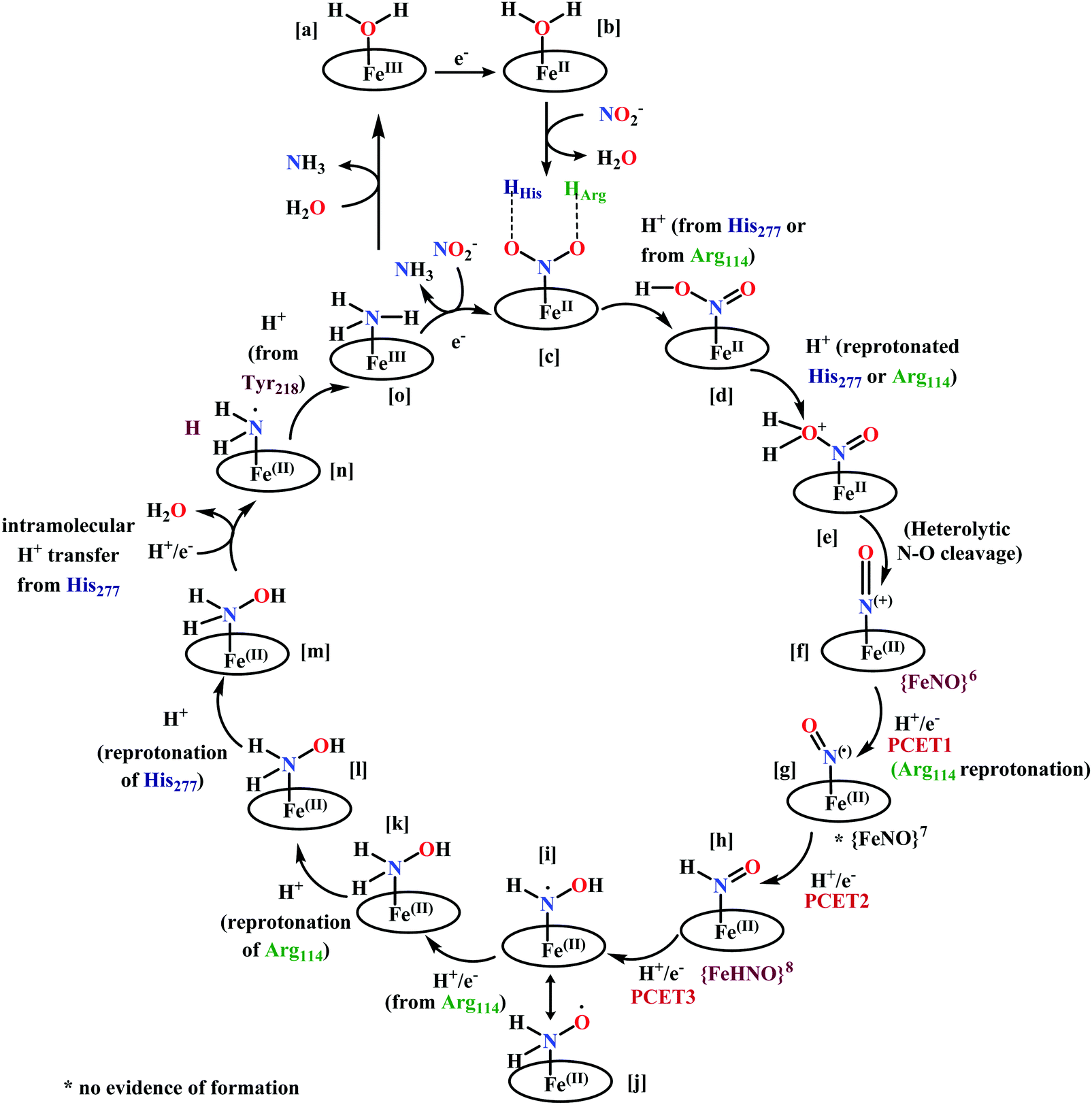 | ||
| Scheme 18 Mechanism of nitrite reduction to ammonia catalyzed by cytochrome c-containing nitrite reductase. | ||
All iron-nitrosyl intermediates in this review will be represented by Enemark–Feltham notation. In the Enemark–Feltham notation, {M(NO)x}n, the index x indicates the number of coordinated NO molecules to metal M, and the exponent n counts the number of valence electrons, which is the summation of the number of electrons in the metal(d) and NO(π*) orbitals.207
(b) The second episode of the mechanism comprises two proton-coupled electron-transfer (PCET) steps (PCET1 and PCET2), leading to an Fe-HNO intermediate. The electronic structure of {FeNO}6 is best described as a FeII metal centre bound to a NO+ ligand.208 {FeNO}6 are strong electrophiles209 while {FeNO}7 intermediate (Scheme 18g) is highly stable and represent a thermodynamic sink.210–212 So, to escape the formation of this intermediate two rapid consecutive PCET steps occur to the {FeNO}6 species to form an {Fe(HNO)}8 intermediate (Scheme 18h). Also, the reduction of {FeNO}6 to {FeNO}7 involves linear to bent transition of Fe–NO unit. The two consecutive electron transfer can be rationalised by the fact that {FeNO}8 is formed before this reorganisation of Fe–NO bond takes place.175 Conformational and electron structure studies reveal both the reductions to be ligand centred. Proton from PCET1 (f → g, Scheme 18) recharges the enzyme active site, with protonation of Arg114 and that from PCET2 (g → h, Scheme 18) does an electrophilic attack at the N-centre of NO forming {Fe(HNO)}8 (as shown in Scheme 18f–h).206
(c) In the third episode a hydroxylamine intermediate is formed and the necessary proton required is found to be supplied via Arg114. Initially a PCET step (PCET3) protonates His277 endothermically which then transfers the proton to {Fe(HNO)}8 with an electron forming two elusive isomeric intermediates (Scheme 18i and j). Next an intramolecular PT from Arg114 to this intermediate followed by exothermic 1-electron reduction leads to the hydroxylamine intermediate (Scheme 18k).202
(d) Second N–O bond cleavage occurs in the fourth episode of this catalytic cycle aided by a PT from His277. After the formation of hydroxylamine intermediate, the reaction proceeds through two protonation steps. This is the rate limiting step of the catalytic cycle.198,205 Two consecutive protonations of Arg114 and His277 occurs respectively (Scheme 18k–m). The fully protonated active site then undergoes an intramolecular PT from His277 resulting in another N–O bond cleavage with release of a water molecule (Scheme 18m and n).202,203
(e) In the fifth and final episode, the (H2N+)Fe intermediate formed previously undergoes reduction being highly electrophilic. The radical intermediate (H2N+˙)Fe (Scheme 18n) formed is also very reactive and theoretical and site-directed mutation investigations revealed that at this stage Tyr218 can participate in a PT step.198 The electronic structure of the radical intermediate explains stabilization by Tyr218 residue due to electron delocalisation from the radical to the aromatic ring.203 The issue of product dissociation was addressed by Neese by conducting a relaxed surface scans of the Fe–N(NH3) distance in two model complexes: (H3N+˙)HArg and (H3N+˙)HArg HTyr. A spin change likely occur during the dissociation of ammonia from (H3N+˙)HArg HTyr potential energy surface. Here, elongation of the Fe–N(NH3) bond results in switching from low to high spin surface. Due to this spin state change, dissociation step becomes much less energy demanding. Thus, ammonia is released from the active site in high spin FeIII state (Scheme 18o).203 The open coordination site is then occupied by another nitrite molecule available in the vicinity to initiate another cycle or by a water molecule resulting to the resting state.
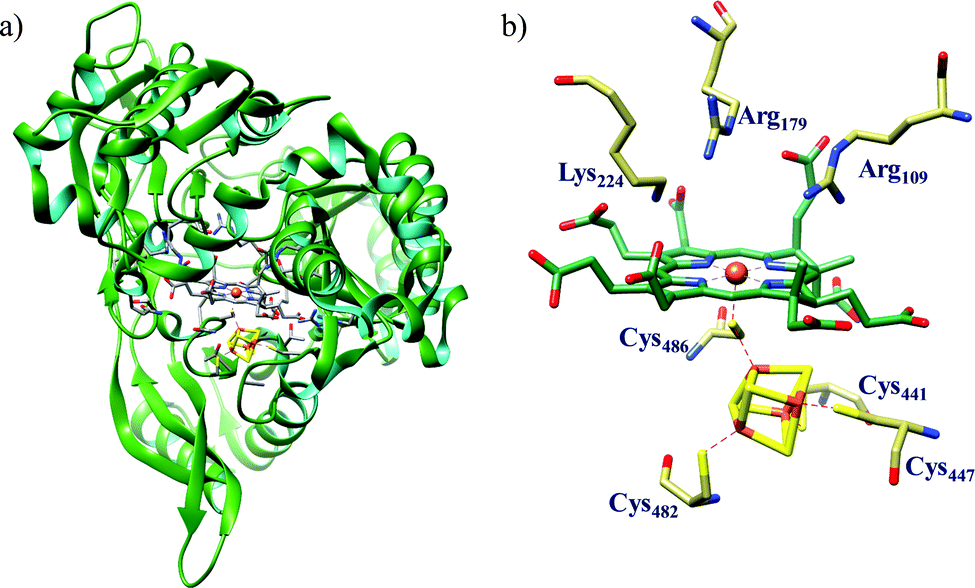 | ||
| Fig. 11 (a) Three dimensional structure of spinach nitrite reductase (ferredoxin dependent nitrite reductase) and (b) active site structure of CSNiR. Both structures are based on PDB 2AKJ.219 | ||
From crystallographic structure analysis (spinach chloroplast enzyme), it is quite evident that siroheme and Fe4S4 cluster are in very close proximity to one another and are directly linked through a bridging sulphur atom from the Cys486 residue. The cluster is coordinated to four Cys–S atoms (Cys441, Cys447, Cys482 and Cys486) which are essential for cofactor binding219 and also facilitates efficient electron transfer to the siroheme centre from reduced ferredoxin. The active site also contains conserved Arg109, Arg179 and Lys224 which forms a large pocket suitable for nitrite binding and reduction (Fig. 11b).219,222,225 There are several other Arg and Lys residues surrounding the active site which aids in the overall reduction process. These positively charged residues are also essential for stabilising the eight carboxylate groups present in the siroheme backbone. Electrostatic potential energy surface of NiR reveal the presence of a tapered tunnel (approx. diameter 8 Å) from the protein surface to the active site. Several positively charged basic residues surround the active site resulting in a positively charged electrostatic surface which promotes nitrite binding/stabilisation and induces expulsion of ammonium formed as the product (Fig. 12).219
 | ||
| Fig. 12 Electrostatic surface of CSNiR showing the presence of an open tunnel from the surface of the protein to the siroheme and iron–sulfur cluster. Positive regions are shown as blue, and negative regions are shown as red. Many Lys and Arg are found in this region as shown by blue colouration (based on PDB 2AKJ). | ||
NiR has only one ferredoxin (Fd) binding site.215 Modelling studies and site directed mutagenesis studies suggests electrostatically stabilised 1:1 complex formation between NiR and Fd wherein the acidic residues are supplied by Fd and NiR contributes the basic residues.225 From crystallographic data it was also revealed that the distance of Fe2S2 cluster of Fd to Fe4S4 cluster is shorter than distance between Fe2S2 cluster and siroheme. The shorter distance suggests that electron transfer (ET) should be significantly faster from Fd to Fe4S4 cluster than to siroheme. Using different electrochemical techniques, mid-point redox potential (Em) of the cofactors were determined. Em for siroheme (−290 mV) was found to be 75 mV more positive than Em Fe4S4 cluster (−370 mV).226 This result also agrees with the previous observation that Fe4S4 cluster is reduced prior to siroheme Fe-center.226
The enzyme nitrite reductase in its resting state is fully oxidised with the cluster in (Fe4S4)2+ state and siroheme iron in high spin FeIII state (Scheme 19a). Conversion of nitrite to ammonia by aNiR also involves five steps and the mechanism is almost same as that of CcNiR. The exact role of each of the active site residues are not clear yet and require more extensive investigations. Till date the proposed reaction mechanism involves assumption of the proton donating sites infered from ligand bound crystal structural data. The enzyme initially undergoes one electron reduction, and upon substrate binding, resulting in nitrite bound low spin FeII coordinated siroheme.176,219 Alternatively, several spectroscopic and kinetic data revealed that nitrite binds to FeIII–siroheme forming a low spin FeIII–siroheme–nitrite complex (Scheme 19b). Several Nitrite bound crystal structure of the active site reveals that nitrite is bound in ‘nitro’ mode and the oxygen atoms are at hydrogen bond forming distances to Lys224, Arg179 and Arg109.219,223 The shortest possible hydrogen bond around NO2 was with Lys224 and thus O(NO2) may be protonated. The Arg179 is assumed to be the second proton donor from structual data. The first N–O bond is cleaved with proton from Lys224 side conforming a similar S–O bond cleavage in siroheme containing dissimilatory sulphite reductases.227 After the N–O bond cleavage, a second electron is transferred and a NO-bound ferrosiroheme species with (Fe4S4)2+ state (Scheme 19c). The candidates for proton donor to this intermediate were Arg179 and Arg109 as elucidated from structural data.223 Addition of two more electrons coupled with the proton transfer results in reduction of NO to result in a hydroxylamine-bound siroheme (Scheme 19d). Crystal structures of Nii3–NO and Nii3–NH2OH were solved.223 Finally, two more electrons donated resulted in an ammonia bound siroheme (Scheme 19e), from where the ammonium ion was readily released out through the positively charged substrate channel. The protonations were assisted by the active site residues, the ultimate sources of these protons being the solvent within the channel. The enzyme receives the six electrons required in single steps from the reduced ferredoxin, which is quite challenging. Although recent efforts attempted to understand the complex mechanism of nitrite reduction by aNiR,225,228 there remains considerable uncertainity about exact pathways of reaction where an enzyme capable of storing two electrons at a time is involved. It is quite likely that the six-electron reduction proceeds by a series of three 2-electron steps.218
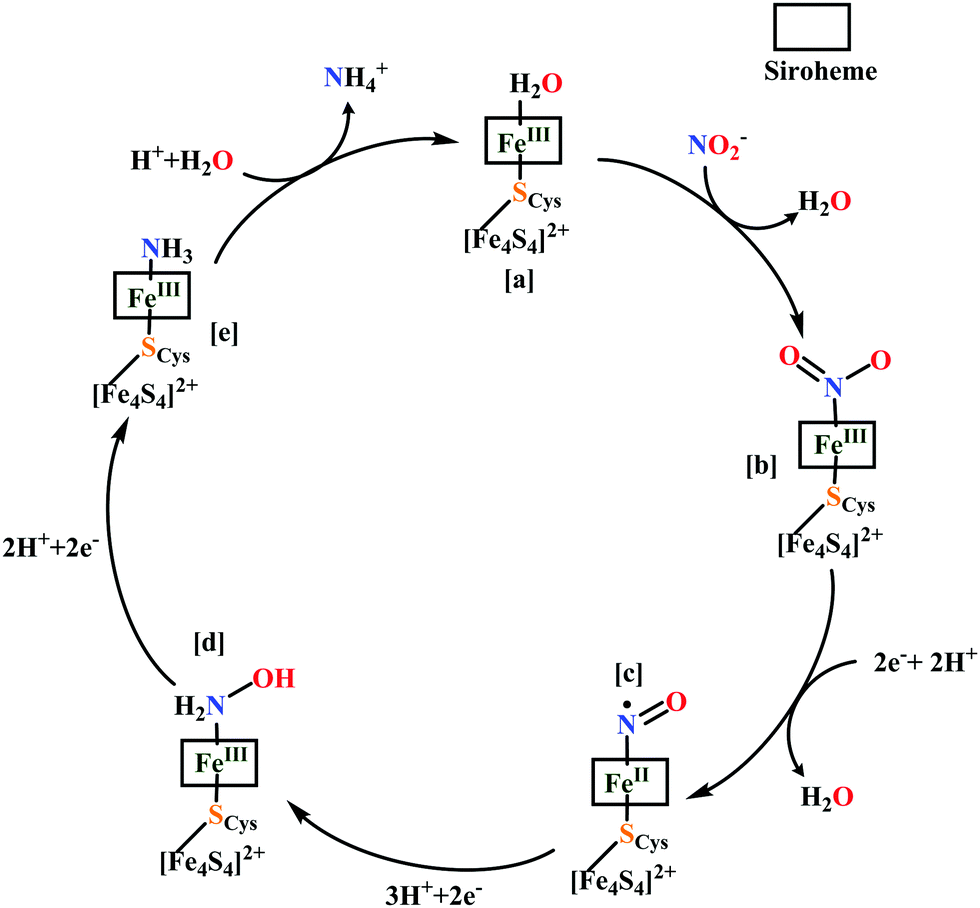 | ||
| Scheme 19 Proposed reaction mechanism of aNiR. Since the basic residues involved in the protonation steps are not extensively proved yet, informations about the residues are not included. | ||
After elucidation of reaction mechanism in both these type of nitrite reductases, an obvious question that comes to mind is that why nature implemented two very different heme cofactors varying widely in structure for catalysing the same reaction. A very recent theoretical investigation on electron transfer pathway of siroheme vs. heme in sulphite reductases suggested that siroheme prefers electron transfer via a bridging cysteine–S atom from the proximal Fe4S4 cluster more than heme. In contrast, heme prefers direct mode of electron transfer via porphyrin ring. This may lead to unwanted side reactions involving attack of radicals on heme. In siroheme, there is an interrupted π-conjugated system due to presence of sp3-carbons direct mode of electron transfer is disfavoured. Hence, nature might have implemented such a unique macrocycle for this reaction to protect the cofactor from undesired radical attack.229
Nitrite reduction to nitric oxide
Denitrification is the major metabolic process in facultative anaerobes under low oxygen tension where N-oxides serves as terminal electron acceptor. Molybdenum containing nitrate reductase (NaR) catalyses the first step, i.e., nitrate reduction to nitrite. Nitrite reductase (NiR) catalyse the reduction of nitrite to gaseous product nitric oxide (NO). This is thus considered as a major source of NO in bacteria. Purification and characterisation of NiR from several bacterial sources reveal that there are two distinct classes dissimilatory nitrite reductase which yield NO as the sole product. These could be either have copper (CuNiR) or heme (Cd1NiR) as cofactor; later being more abundant232 will be discussed here in details.
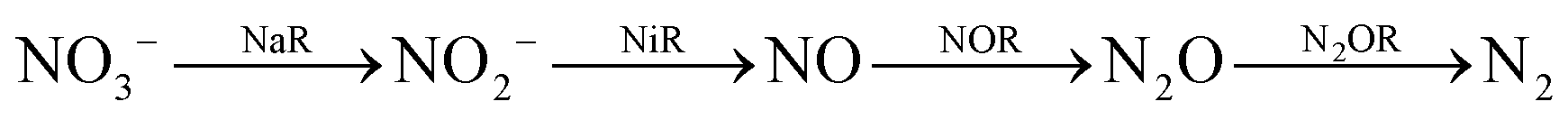
Enzyme architecture. Cd1NiR (expressed from nirS genes)176 is a homodimeric enzyme of the bacterial periplasm with each monomer being folded into two domains: (a) one α-helical N-terminal domain with a covalently attached heme c, and (b) one eight bladed β-propellar C-terminal domain containing heme d1 in its core. Structures of Cd1NiR from Pseudomonas aeruginosa (Fig. 13a)233–235 and Paracoccus pantotrophus236–239 have been extensively studied and their catalytic mechanism have also been probed using different theoretical and spectroscopic techniques. The heme-c moiety functions as electron acceptor from soluble electron carriers such as c-type cytochromes (cyt c550, cyt c551 and cyt c554) or copper proteins such as azurin or pseudoazurin240–243 and transfers the electron to the heme d1 which constitutes the enzyme active site (site of nitrite binding and reduction).233,239 Heme d1 (3,8-dioxo-17-acrylateporphyrindione) is unique to this class of enzyme distinguished from usual heme moieties by the presence of a partially saturated macrocycle and a set of oxo and acrylate substituents.244
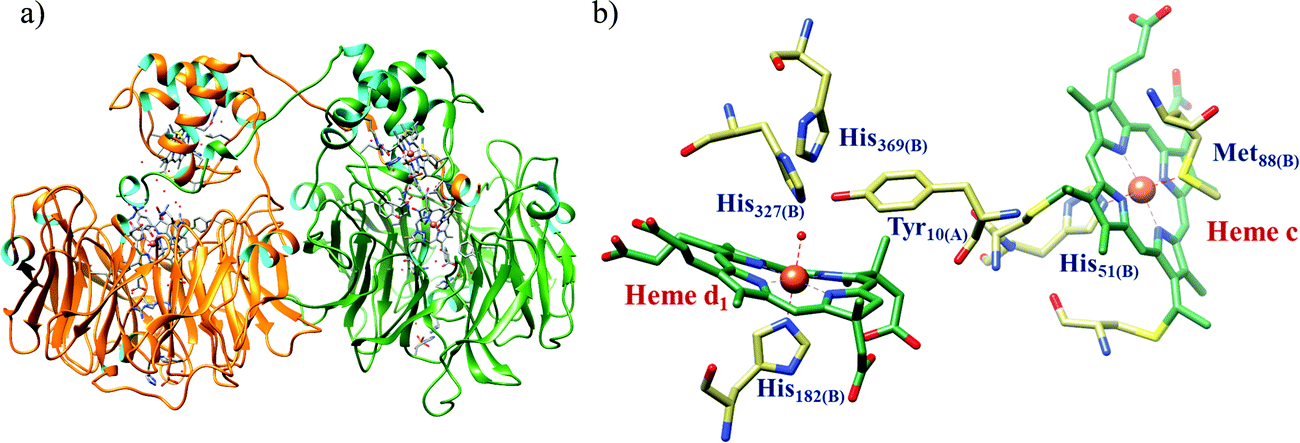 | ||
| Fig. 13 (a) 3D ribbon representation of Cd1NiR from Pseudomonas aeruginosa showing the homodimers and (b) active site structure of P. aeruginosa Cd1NiR in oxidised form, heme c is also included participating in electron transfer to the active site. Both (a) and (b) are based on PDB 1NIR.245 | ||
P. aeruginosa Cd1NiR (Pa-Cd1NiR) in oxidised form (resting state) has the heme-c axially coordinated by His51/Met88, whereas heme d1 is coordinated by His182 in the proximal position and by a hydroxyl group in the distal position. Distal site of the active site pocket constitutes residues such as Tyr10 (from the N-terminal arm of the adjacent partner monomer), two conserved histidines-His327 and His369. Crystal structure of the oxidised form revealed that Tyr10 remains hydrogen bonded to the distal hydroxyl ligand of heme d1. These distal residues presumably are involved in substrate binding and/or protonation.234,246 Reduction of heme d1 iron centre stimulates hydroxyl ligand release245 which further induces a series of conformational changes. Displacement of a loop in heme c domain is followed by rotation of Tyr10 away from heme d1 distal site and hydrogen bond formation between Thr59 and Gln11.233 All these changes in heme c domain of adjacent partner monomer exposes the heme d1 active site for nitrite binding.233,246
In P. pantotrophus Cd1NiR (formerly known as Thisophaera pantotropha) (Pp-Cd1NiR), the oxidised state (as isolated form) has bis-histidine coordinated heme c (His17/His69) and heme d1 is coordinated by His200 in the proximal position and by Tyr25 (from the same domain) at the distal site. Thus, the active site has closed coordination which implies that this as-isolated form is the ‘inactive form’ of the enzyme and is catalytically inert. Reduction of this enzyme results in movement of the N-domain loop causing structural rearrangement at both the heme centres. At heme c, His17 ligand is replaced by Met106 and the Tyr25 dissociates from the heme d1 thus leaving the active site open for nitrite binding. Conserved His345 and His388 completes the active site.236,239,247,248 Hence, redox energy driven conformational changes regulates the catalytic process. These conformational changes are very essential for the enzyme to transform into its ‘active form’ to perform its reductase activity. Thus, there is an ‘on/off’ mechanism for the regulation of the enzyme activity. Pa-Cd1NiR Tyr10Phe mutant is spectroscopically and functionally indistinguishable from the wild type enzyme.249,250 The Pp-Cd1NiR Tyr25Ser mutant, has a coordination environment of the active site similar to that in steady-state catalytic condition, i.e., its ‘active form’.240,251 These experimental findings suggest that the tyrosine residue, though do not have any role in catalysis may have been an intentional inclusion to regulate the ‘on/off’ mechanism.176 The structural changes were also governed by the protonation states of the conserved histidine residues in the enzyme active site. It was found that the structure of reduced Pp-Cd1NiR grown at pH 9.0251 where the histidines are likely to be deprotonated were electrostatically analogous to the structure of oxidised Pa-Cd1NiR with His–Ala mutants at pH 5.5–6.5.234 This gave rise to another hypothesis that the structural conformation of the enzyme is guided by the overall charge in the active site pocket rather than the oxidation states of the heme centres only.251
Mechanism of nitrite reduction. The catalytic cycle of nitrite reduction by Cd1NiR initiates with the reductive activation of the ‘inactive form’ (Scheme 20a and b). Initially, heme c of cFeIII–d1FeIII form (Scheme 20a) is rapidly reduced by external electron donors (such as cyt c551 or azurin) to cFeII–d1FeIII form (Scheme 20b). In Pp-Cd1NiR potentiometric titration potential in the reductive as well as oxidative directions (+60 mV and +210 mV, respectively) revealed hysteresis and cooperativity between the two heme centres.248 The external electron donors have redox titration potential in the range of +250 mV,252 hence electron transfer process in vivo is not clear. It was proposed that there may be some activation factor in bacterial periplasm or alternatively, the enzyme may function only when the ratio of reduced to oxidized electron donor proteins is very high.253 Electron transfer from heme c to heme d1 is coupled with conformational changes, generating the ‘active form’ of the enzyme. Heme c is replenished with electron from external electron donors resulting in fully reduced form: cFeII–d1FeII (Scheme 20c) which binds to the substrate.254
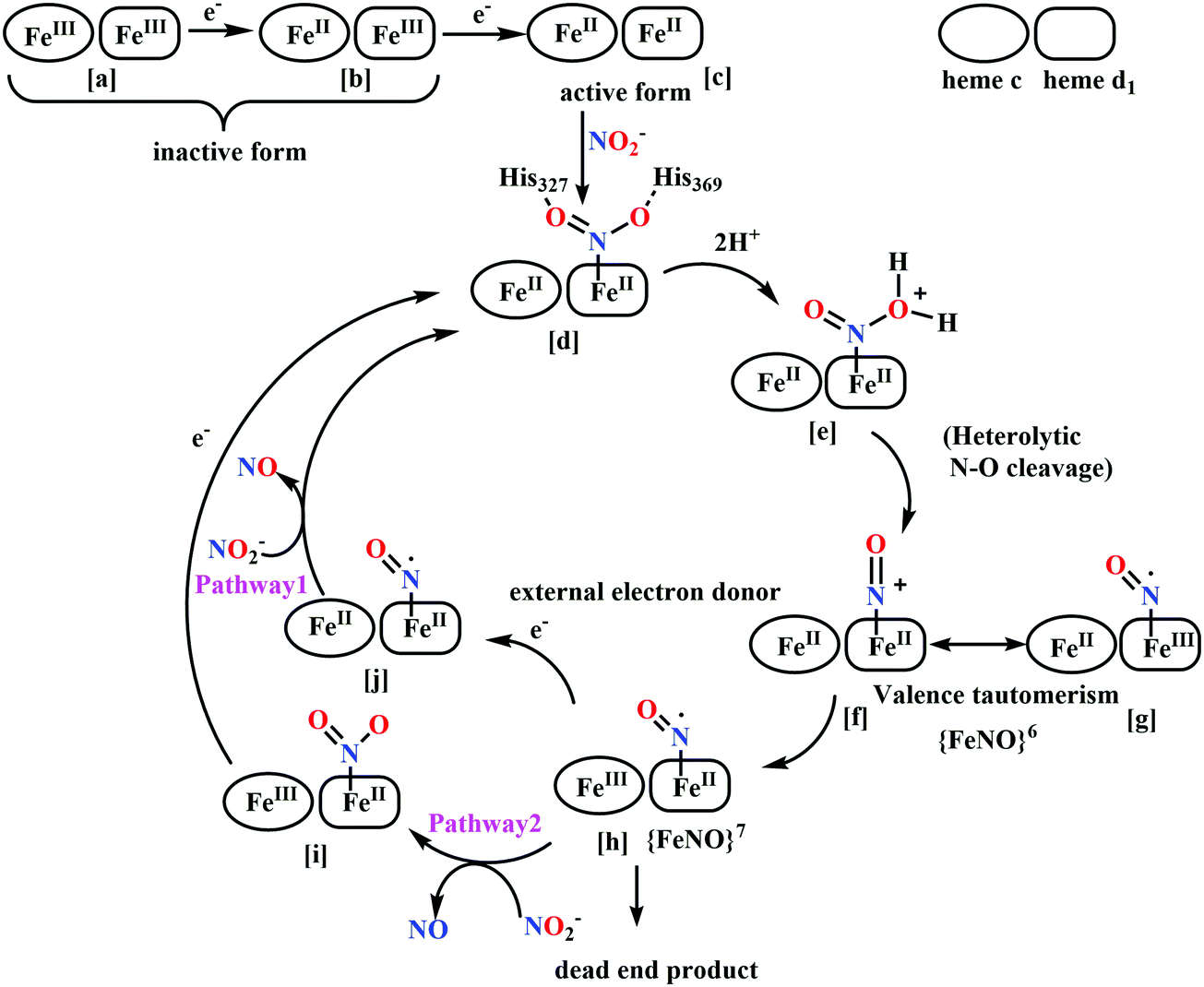 | ||
| Scheme 20 Proposed reaction mechanism of Cd1NiR. | ||
In Cd1NiR, nitrite binds to the d1FeII with high affinity.234 Although nitrite shows ‘nitrito’ mode of binding in haemoglobin and myoglobin,255–257 it exhibits a ‘nitro’ mode of binding in heme d1 (Scheme 20d).239 This mode of binding is also supported by proposed mechanism responsible for NO released. High affinity for nitrite can be explained by two factors (a) presence of two electron withdrawing oxo substituent in heme d1 ring moiety and (b) electrostatic interactions with two protonated conserved histidine residues in the active site pocket.246 Cyanide (an inhibitor ion) binding has been useful as a probe of the affinity of ferrous heme d1 for anions and role of the protein residues in controlling ligand binding. It has been observed that in Pa-Cd1NiR His369Ala mutant, affinity for cyanide binding is 10-fold reduced. Due to reduced steric hindrance in the active site, CO binds to the mutant much faster than the wild type (WT) enzyme than the His369Ala mutant.235 Thus, overall positive electrostatic potential of the distal site is the dominant factor affecting the affinity of anionic ligand (CN−, NO2−) which compensates for relatively weak affinity of ferrous heme iron towards anionic ligands.
The next step involves protonation at the oxygen atom and heterolytic N–O bond cleavage, resulting in release of a water molecule and d1{FeNO}6 formation (Scheme 20f and g). The two protonated histidine residues at the active site are well positioned to serve as the proton donors during catalysis233–239,258,259 and their role has been verified by site directed mutational studies234 and theoretical calculations.260 It has been observed that both Pa-Cd1NiR His369Ala mutant and His327Ala mutant lost nitrite reductase activity. However, effect of two residues are not equivalent. The His369 being at a shorter distance to both the oxygen atoms, play a more important role in stabilising the nitrite bound complex through hydrogen bonding.234,235,260
The last step of the catalytic cycle is NO dissociation which has been the subject of much debate over several decades. Earlier, the d1{FeNO}6 complex was considered to be a ‘dead end’ product supporting the fact that NO has very high association constants for binding to ferrous hemoproteins such as myoglobin and haemoglobin (Ka ∼ 1011–1012 M−1)261 and cyt aa3 oxidase (Ka ∼ 1010 M−1).262 So, NO was thought to be released from d1FeIII–˙NO complex formed by intramolecular electron transfer from FeII to π*-orbital of NO (valence tautomerism) (Scheme 20f and g).233,234,263 However, this hypothesis was never proven. Indeed, spectroscopic evidence of a long lived Pp-Cd1NiR d1FeIII–NO exists in absence of excess reducing equivalents. Also, this NO dissociation from this adduct is not observed.263,264 In addition, it was shown that NO is dissociated rapidly from reduced Pa-Cd1NiR (koff = 71 s−1),259,265 thus suggesting a reduced affinity of the ferrous heme d1 towards NO (Ka ∼ 107 M−1) in contrary to other hemoproteins. These conclusions on Pa-Cd1NiR are also supported by the results of ultra-fast kinetic studies on Pp-Cd1NiR which showed that intramolecular electron transfer from heme c to heme d1 triggers NO release (Scheme 20, pathway 1).254 Investigation of the reduction process of Pa-Cd1NiR by pulse radiolysis technique provided kinetic evidence for negative cooperativity between two d1heme sites of the dimeric enzyme and also suggested that intramolecular electron transfer from heme c to heme d1 is highly regulated by an allosteric mechanism.258,266,267 This can explain the requirement for two redox centres for Cd1NiR functioning.268 Hence, NO most likely is released from d1{FeNO}7. It is also established experimentally that nitrite reacts with reduced NO-bound Pa-Cd1NiR and has NO dissociation at rates much faster than the catalytic turnover (Scheme 20, pathway 2).265 Hence it can be concluded that Cd1NiR efficiently catalyses nitrite reduction and NO release only in presence of substrate and electron donors on the vicinity of the active site. In absence of reductants, NO release is sluggish.248,269–272
A combination of very high field electron-nuclear double resonance (ENDOR) techniques and theoretical calculations on Pa-Cd1NiR suggested that a dynamic and co-operative network of hydrogen bonds in the active site is also responsible for NO release. The structure obtained theoretically in best agreement with experimental findings reveal that the conserved His327 has no hydrogen bonds to the NO. Thus, the role of this residue is limited to maintaining positive electrostatic potential in the distal side of the active site. The d1Fe–NO complex is hydrogen bonded to OH from Tyr10 and the protonated His369 residues. In Tyr10Phe mutant-NO complex, the His369 residue moves closer to the NO, whereas mutation of both distal histidine residues with alanine displaces Tyr10 preventing hydrogen bond formation. This suggests cooperative behaviour for Tyr10 and His369. Hence, these observations suggest that hydrogen bonding network in the distal active site is dynamic and the position and strength of the hydrogen bonds are closely dependent on each other.273 All these factors act cooperatively along with the crucial contribution from unique electronic properties of heme d1 cofactor.237,274 Sperm whale apomyoglobin reconstituted with heme d1 also releases NO at 4 orders of magnitude greater rate than common heme b myoglobin (4 s−1vs. 2 × 10−4 s−1).268 Hence, the fast NO release is likely controlled by properties of heme d1. Two spectroscopic evidences point to this. The magnetic data revealed that ferric heme d1 has unusual low spin (dxz,yz)4(dxy)1 electronic ground state, completely different from the usual (dxy)2(dxz,dyz)3 protoheme ground state. This peculiar ground state results in the unpaired electron (HOMO) residing in the heme plane, whereas the filled orbitals (dxy and dyz) residing at the axial planes resulting in very weak π-interaction with NO π*-orbital forming weak Fe–NO bond as compared to NO bond with heme b.237 Secondly, presence of the two electron withdrawing keto substituent and saturation at two pyrrolic centres, contributes to NO dissociation and tunes the reduction potential of iron in heme d1 such that NO formed is not reduced further differentiating it from CcNiR. An important insight of the catalytic mechanism is the assignment of the rate determining step. In steady state turnover of nitrite, catalytic rate constant (kcat) for Pp-Cd1NiR is 72 s−1,269 which shows a higher value compared to 6 s−1 in Pa-Cd1NiR259 at pH 7.0. Therate of intramolecular electron transfer, which triggers NO dissociation from heme c to heme d1, is ∼1000 s−1 in Pp-Cd1NiR263,275 and 3–6 s−1 in Pa-Cd1NiR.266 Dissociation rate constant of NO (koff) in Pp-Cd1NiR is 65–200 s−1 (ref. 268) and 6–35 s−1 in Pa-Cd1NiR.259 Analysis of these kinetic parameters clearly indicate that in Pp-Cd1NiR, kcat is comparable to koff of {[FeNO]}7, hence probably NO dissociation is rate limiting step, whereas in Pa-Cd1NiR kcat corresponds to intramolecular electron transfer rate. Thus, in Pa-Cd1NiR, d1Fe(II) formation step is rate limiting followed by fast NO release.
An alternative possible reduction pathway through O-bound complex was first explored by Radu Silaghi–Dumitrescu in Cd1NiR.279 Theoretical calculations suggested that this reaction proceeded through a protonation from distal histidine residue leading to formation of hydroxo–Fe(III) complex and NO. Similar mechanism is also suggested for nitrite reduction by MbII and HbII (Scheme 21).276 Several other instances of metalloprotein-based nitrite reduction to NO have been reported in recent years.278,280–287
| MbII + NO2− + H+ → NO + MbIII + OH− | (7) |
| MbII + NO → MbII–NO | (8) |
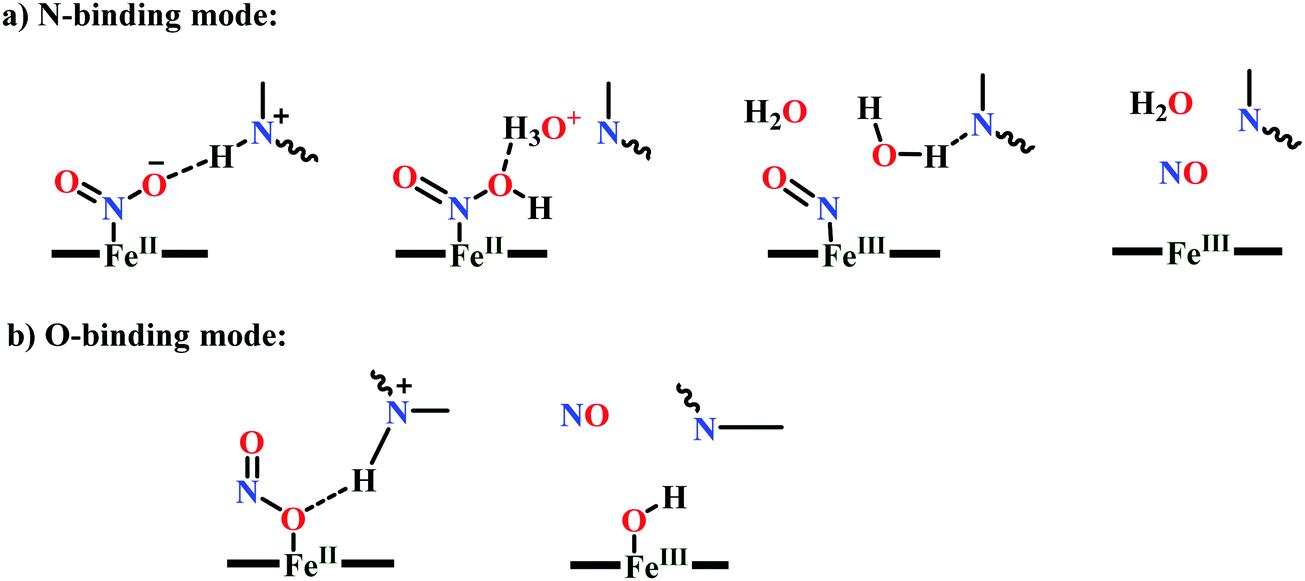 | ||
| Scheme 21 Proposed NO generation mechanisms catalysed by Hb. | ||
Challenges and general outlook. A single reaction (nitrite reduction) has been handled in nature by three different pathways utilising three completely different heme architectures. The chemistry behind the functioning of any enzyme active site is governed by both the primary coordination environment of the cofactors as well as the protein amino acid residues constituting the secondary environment. This chemistry is difficult to mimic in model complexes as it is difficult to design the exact protein ambiance outside a protein matrix. While selective roles of some of the residues can be probed, recreating the overall catalytically relevant protein architecture in a synthetic construct is a dauntic task. Detailed investigation of the synthetic analogues mimicking the cofactor structure, can however be equally important in predicting and/or reproducing the fundamental aspects of structure, spectroscopy, magnetic and electronic structure and chemical reactivity of the biological systems. These are discussed in he following sections.
Nitrite reductase model complexes
First generation models comprise iron-porphyrin complexes which could rationally explain oxidation states, coordination nature, identity of donor ligands, the ligation alternatives at the active site. Structural, spectroscopic and electrochemical characterisation of these led to logical designing of second-generation models to incorporate some feature of the enzyme, as has been understood from enzymatic studies. Blueprint of a third-generation model systems must include the consideration of the roles of basic residues residing in the enzyme active site.Solution chemistry of nitrite
Nitrite is the conjugate base of nitrous acid having pKa of 3.16 at 25 °C.288 This implies that at physiological pH, nitrite exists completely in its ionic form. The aqueous chemistry of nitrite is highly pH dependent, same has been observed in enzyme systems. One electron reduction of nitrite to NO requires two equivalents of acid, hence nitrous acid is oxidising in nature at very low pH, whereas reduction potential of nitrite to NO drops to 0.37 V at pH 7.0 and becomes energetically uphill at higher pH.289| HNO2 + H+ + e− → NO + H2O (at pH < 3) E° = 0.99 V (vs. NHE) | (9) |
| NO2− + 2H+ + e− → NO + H2O (at pH = 7) E° = 0.37 V (vs. NHE) | (10) |
| NO2− + H2O + e− → NO + 2OH− (at pH > 7) E° = 0.46 V (vs. NHE) | (11) |
NiR model complexes
Several metalloporphyrin complexes having coordinated nitrite ion have been characterised. We addressed the model complexes developed and characterised in a chronological order and based on their relevance in the catalytic cycle of nitrite reduction reaction.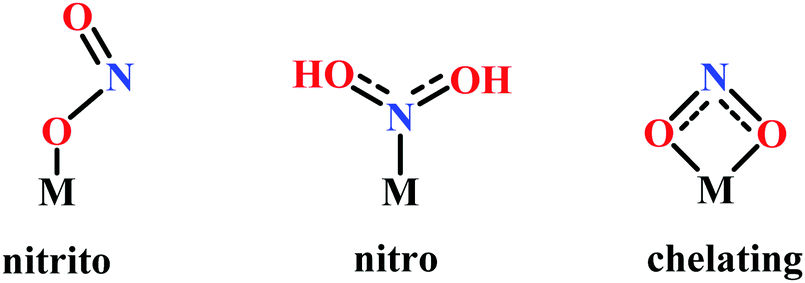 | ||
| Fig. 14 Nitrite ligand binding modes in monomeric complexes. | ||
The structures reported for heme containing nitrite reductases have been limited to ‘nitro’ binding mode despite the differences in proximal ligation to heme (e.g., Lys for CcNiR, μ-SCys for CSNiR and His for cyt cd1NiR), nature of macrocycle (heme c vs. siroheme vs. heme d1) and different basic residues in distal heme pocket.175,239,291 It is also interesting to find from the crystal structures of nitrite ligated synthetic iron porphyrin complexes that both FeII (ref. 292) and FeIII (ref. 293–295) binds via N-atom (Fig. 15).209,296 [(TpivPP)Fe(NO2)(NO)]− has been an exceptional complex where both nitro and nitrito mode of binding have been observed in same crystal due to disorder in nitrito group.297
 | ||
| Fig. 15 Molecular structure [(TpivPP)Fe(NO2)]−. | ||
Photoirradiation of (TPP)Fe(NO)(NO2) resulted in linkage isomerism of nitrite to its nitrito form.298,299 Such nitrito binding has also been observed in crystal structures of other metalloporphyrin nitrite complexes of Mn,300 Ru301,302 and Os.303,304 First instance of nitrito mode of binding to a heme protein was for horse heart FeIII–Mb (MbIII) and its derivatives256,257,305 and FeIII–Hb or deoxy–Hb(HbIII).255 Once bound to nitrite, all these complexes form low spin iron centres. When a crystal structure is not available, differentiation of the binding modes can be easily done by infrared vibrational spectroscopy. For nitro complexes, the antisymmetric and symmetric stretching modes are νa (NO2) = 1470–1370 cm−1 and νs (NO2) = 1340–1290 cm−1, respectively and for nitrito mode, νa (NO2) = 1510–1400 cm−1 and νs (NO2) = 1100–900 cm−1.290,306
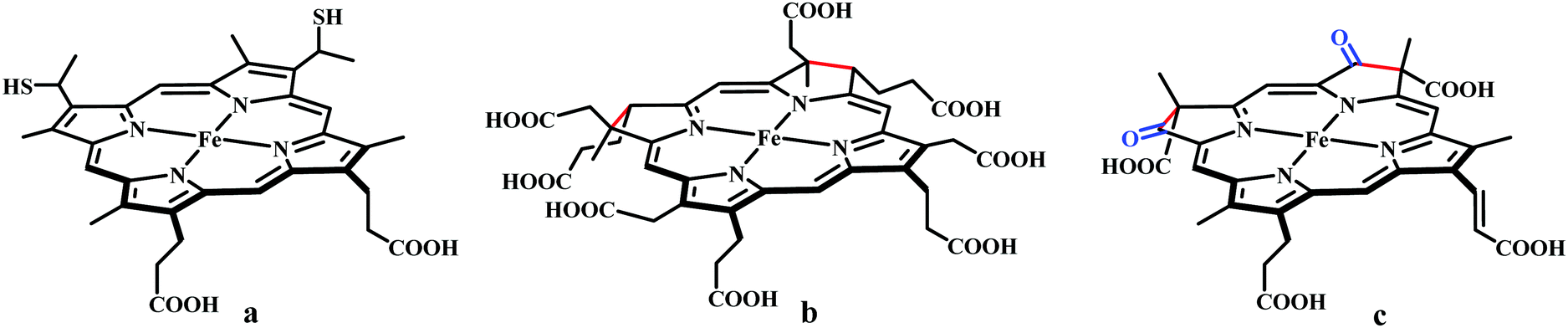 | ||
| Fig. 16 Structure of (a) heme c in CcNiR, (b) siroheme in CSNiR and (c) heme d1 in Cd1NiR. The saturated pyrrolic centres and electron withdrawing groups are highlighted in red and blue respectively. | ||
Several iron porphyrin-based nitrite reductase model complexes have been synthesised and their structural, spectroscopic, magnetic and electronic properties have been explored. Electrochemical investigations also gave some interesting results. The first area of focus was the differences in the heme backbone structure (Fig. 16) i.e., (a) heme c in CcNiR which is a substituted porphyrin macrocycle (Fig. 16a), (b) siroheme in CSNiR which has isobacteriochlorin unit with two saturated β-pyrroles (four sp3 hybridized peripheral carbons)224 (Fig. 16b) and (c) heme d1 in Cd1NiR which has an isobacteriochlorin macrocycle with two electron withdrawing oxo substituent and two saturated β-pyrroles (two sp3 hybridized peripheral carbons)307 (Fig. 16b). Hence, the divergent reactivity may stem from differences in structures of Fe-porphyrinoid macrocycles. To explore this possibility, several model complexes were synthesized (Fig. 17), characterized and electrochemistry of those complexes were investigated by several groups.296,308–316
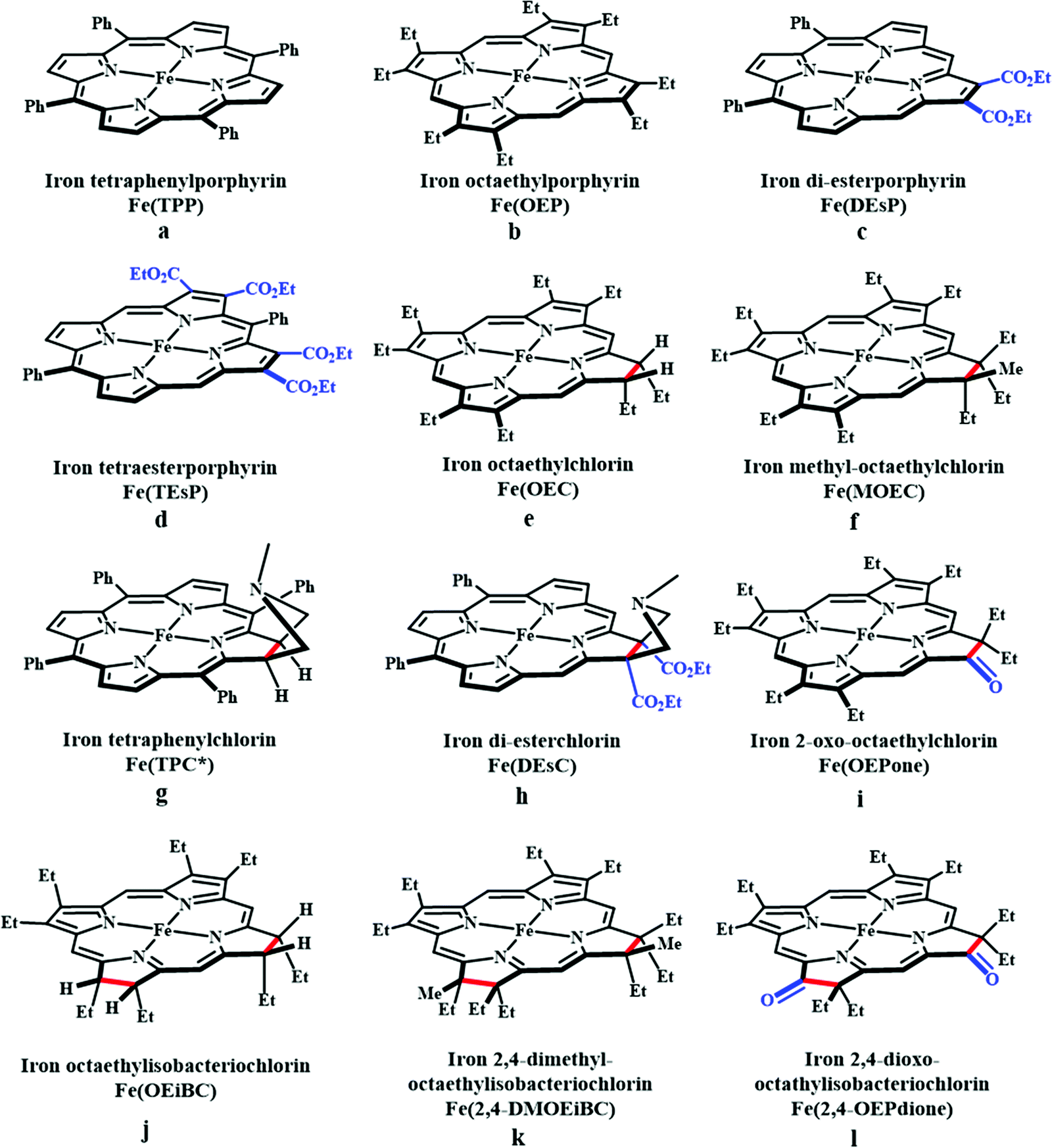 | ||
| Fig. 17 Molecular structures of heme based nitrite reductase model complexes. The saturated pyrrolic centres and electron withdrawing groups are highlighted in red and blue respectively. (j) and (k) Closely resembles siroheme structure and (l) resembles heme d1 structure. | ||
It was observed that nitrite bound complexes of these models were unstable to examine its transformation to {FeNO}6.292,294 But ferrous-nitrosyl adducts {FeNO}7 of these complexes were stable and thus {FeNO}6 could be prepared by one electron oxidation of {FeNO}7. Hence, the electrochemistry of nitrosyl adducts have also been explored as well (Table 2).210,211,317–320
| Complex | Solvent | E{FeNO}6/7 (V) vs. SCE | E{FeNO}7/8 (V) vs. SCE | Ref. |
|---|---|---|---|---|
| Fe(TPP)(NO) | DCM | 0.74 | −0.93 | 211 |
| Fe(OEP)(NO) | DCM | 0.71 | −1.02 | 320 |
| THF | 0.66 | −1.07 | 320 | |
| Butyronitrile | 0.63 | −1.08 | 317 | |
| Fe(DEsP)(NO) | DCM | 0.83 | −0.75 | 329 |
| Fe(TEsP)(NO) | DCM | 0.87 | −0.65 | 329 |
| Fe(TPC*)(NO) | DCM | 0.45 | −1.02 | 329 |
| Fe(OEC)(NO) | Butyronitrile | 0.48 | −1.08 | 317 |
| Fe(MOEC)(NO) | DCM | 0.59 | −1.02 | 320 |
| THF | 0.64 | −1.06 | ||
| Fe(OEPone)(NO) | DCM | 0.76 | −0.81 | 319 and 320 |
| THF | 0.76 | −0.86 | 320 | |
| Fe(DEsC)(NO) | DCM | 0.59 | −0.93 | 329 |
| Fe(OEiBC)(NO) | Butyronitrile | 0.22 | −1.11 | 317 |
| Fe(2,4-DMOEiBC)(NO) | DCM | 0.34 | −1.17 | 320 |
| THF | 0.38 | −1.16 | ||
| Fe(2,4-OEPdione)(NO) | DCM | 0.74 | −0.69 | 319 and 320 |
| THF | 0.80 | −0.80 |
Under coulometric conditions, reduction of nitrosyl adducts of most iron porphyrins proceeded exclusively to ammonia, mediated by hydroxylamine bound species.212,321–327 This was verified from reduction reaction of bis(hydroxylamine) complexes which yield ammonia in presence of excess hydroxylamine (eqn (16)). The rate of hydroxylamine formation was related directly to the basicity of {FeNO}8 complexes (eqn (12) and (13)). Changes in macrocycle moiety had a vast influence on the rate and was accelerated in hydroporphyrins and were slower in oxoporphyrins. Fe(P)(NH2O+) (eqn (14)) was readily formed in siroheme model Fe(2,4 DMOEiBC) (Fig. 17k) whereas its formation was least favourable in heme d1 model Fe(2,4-dioxoOEiBC) (Fig. 17l). It produces NO/N2O rather than ammonia.327
| Fe(P)(NO) + e− → Fe(P)(NO)−, where [Fe(P)(NO) = {FeNO}7] | (12) |
| Fe(P)(NO)− + H+ → Fe(P)(HNO) | (13) |
| Fe(P)(HNO) + H+ → Fe(P)(H2NO+) | (14) |
| Fe(P)(H2NO+) + H+ → Fe(P)(NH2OH) | (15) |
| 2Fe(P)(NH2OH)2 + NH2OH → Fe(P)(NO) + 3NH3 + 3H2O | (16) |
Reaction pathways of {FeHNO}8 hence formed may also include disproportionation to {FeNO}7 and H2 (eqn (17)) observed for FeTPP212 (Fig. 17a) or a cross reaction involving reduction of {FeHNO}8 (eqn (18)). Reduction of FeII(TFPPBr8)NO (TFPPBr8 = 2,3,7,8,12,13,17,18-octabromo-5,10,15,2-tetrakis-(pentafluorophenyl)porphyrin, Fig. 18) with cobaltocene yields a stable {FeNO}8 complex [Co(C5H5)2]+[Fe(TFPPBr8)NO]−. This complex was isolated and characterised, giving an exclusive example of stabilised {FeNO}8 intermediate, which is otherwise elusive. In the presence of strong acid such as triflic acid, it disproportionated to {FeNO}7 and H2, in a pathway observed earlier, without any direct evidence of mediation by a {FeHNO}8 species (eqn (19)).328 Electrochemistry of the reported heme-nitrosyl adducts also helped in understanding the electronic structure of the various heme model complexes.
| 2Fe(P)(HNO) → 2Fe(P)(NO) + H2 | (17) |
| Fe(P)(HNO) + Fe(P)(NO)− → Fe(P)(HNO)− + Fe(P)(NO) | (18) |
| 2[Co(C5H5)2]+[Fe(TFPPBr8)(NO)]− + 2CF3SO3H → 2[Fe(TFPPBr8)(NO)] + H2 + 2CF3SO3− + 2Co(Cp)3+ | (19) |
 | ||
| Fig. 18 Molecular structure of Fe(TFPPBr8). | ||
A closer look at Table 2 reveal a clear trend observed in reduction potential of both the oxidation process ({FeNO}6/7 couple) and reduction process ({FeNO}7/8 couple). Insertion of EWGs to the β-pyrrolic position shifted the redox couples to higher potential (for Fig. 17c and d). Conversely, saturating the pyrroles, i.e., making β-pyrrolic carbons sp3-hybridised had a reverse effect, shifting the redox couples to lower potentials (for Fig. 17e–h, j and k). Oxidation of {FeNO}7 complexes of oxoOEC and dioxoOEiBC resultes in a porphyrin π-cation radical generation whereas in OEP, the oxidation is metal centered.319 A very interesting work was done by Meyer and co-workers, who employed a water soluble porphyrin [Fe(III)(H2O)(TPPS)]3− (TPPS = (meso-tetrakis(p-sulfonatophenyl)porphyrin)), Fig. 19 in mildly aqueous acidic solution to electro-reduce nitrite to ammonia (eqn (20)–(22)), with N2O and hydroxylamine as side product. This reaction was highly pH dependent. Although, comprehensive mechanism of the reaction is not understood clearly, this may be considered as a structural and functional model of CcNiR.321,330
| [Fe(H2O)(TPPS)]4− + HONO + H+ → [Fe(TPPS)(NO+)]3− + 2H2O | (20) |
| [Fe(TPPS)(NO+)]3− + 2e− → [Fe(TPPS)(NO−)]5− | (21) |
| [Fe(TPPS)(NO−)]5− + 4e− + 5H+ → [Fe(TPPS)(NH3)]4− + H2O | (22) |
 | ||
| Fig. 19 Molecular structure of [Fe(III)(TPPS)]3−. | ||
Fe(III)-porphyrin complexes with NH3 as a ligand has been reported but not structurally characterised, magnetic studies reveal FeIII low spin state. Ammonia as is weakly bound and have a strong tendency to hydrolyse. This observation is relevant with release of NH3 during the turnover of nitrite reductase catalytic cycle.332
It has been recently reported that presence of EWGs on the periphery of porphyrin macrocycle (Fig. 17c, d and h) lowers the energy of porphyrin π*-orbitals. This results in competitive back-bonding from filled dπ-orbitals of iron between porphyrin π*-orbitals and NO π*-orbitals, weakening the Fe–NO adduct formation.333 This observation is evident from the analyses of the NO stretching frequencies (Table 4) (νNO) reported for {FeNO}7 complexes reported till date.
| Complex | {FeNO}6 (6C) | {FeNO}7 (6C) | {FeNO}8 | Ref. |
|---|---|---|---|---|
| Fe(TPP)(NO) | 1844 (1914) | 1676 (1626) | 1496 | 329 and 338 |
| Fe(OEP)(NO) | 1862 | 1670 | 1441 | 338 |
| Fe(DEsP)(NO) | (1923) | 1686 (1641) | 329 | |
| Fe(TEsP)(NO) | (1927) | 1688 (1646) | 1550 | 329 |
| Fe(TPC*)(NO) | — | 1680 (1635) | — | 329 |
| Fe(OEC)(NO) | — | 1670 | — | 317 |
| Fe(OEPone)(NO) | 1720 | 1662 (1916) | 1442 | 318, 319 and 338 |
| Fe(DEsC)(NO) | — | 1691 (1633) | 1537 | 329 |
| Fe(OEiBC)(NO) | — | 1670 | — | 317 |
| Fe(2,4-OEPdione) | 1700 | 1665 (1916) | 1442 | 318, 319 and 338 |
| Fe(TFPPBr8)(NO) | — | 1726 | 1547 | 328 |
Complexes having higher νNO for {FeNO}7 systems tend to release NO easily owing to weaker Fe–N bond (Fig. 20 and 21). This can be rationalised by the fact that stronger backbonding with porphyrin π* will reduce backbonding to NO π*. This effect also tunes the pKa and coherently affect the thermodynamics of the next reaction; NO release or a PCET step to {FeHNO}8. In heme c, νNO for {FeNO}7 is lower (1651–1671 cm−1), suggestive of disfavoured NO release, as has been the case in CcNiR.339 Aspects of facile PCET at this step has been recently reported for a series of model complexes where the effect of saturating the pyrroles and role of EGW were systematically investigated.329 It has been discussed that for PCET to be facile, the bond dissociation free energy (BDFE) of the N–H bond on {FeHNO}8 should be high. This report was first to evaluate the N–H BDFE involved in the PCET to {FeNO}7 species.340 The BDFE of N–H bond in {FeHNO}8 can be calculated using the following equation:341
| BDFENH = 1.38pKa + 23.06E° + C | (23) |
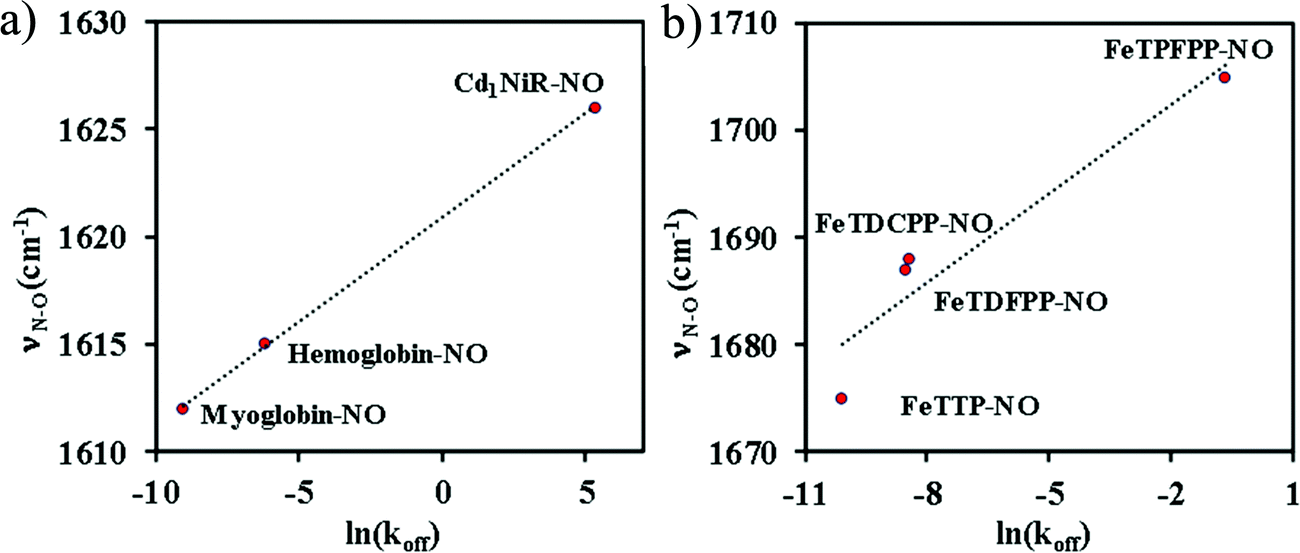 | ||
| Fig. 20 Correlation between N–O stretching frequency and rate of NO dissociation: (a) in six-coordinate heme nitrosyls in enzyme systems;334–336 (b) in synthetic Fe-porphyrin nitrosyl complexes, TTP (p-tolyl), TDFPP (2,6-difluorophenyl), TDCPP (2,6-dichlorophenyl), TPFPP (pentafluorophenyl).337 | ||
 | ||
| Fig. 21 Correlation between experimentally measured N–O stretching frequency and NO dissociation constant in model complexes. | ||
The ΔG° of protonation was computed using DFT calculations. The calculated BDFENH values revealed that the protonation of {FeNO}8 species gradually became less favorable from FeOEP to FeOEPone to Fe(2,4-OEPdione). This was consistent with the previously reported trend for nitrite reduction to ammonia, using moderately strong acids like phenols, under controlled potential electrolysis, i.e., the rate of the reaction: FeOEP > FeOEPone > Fe(2,4-OEPdione), where the protonation of {FeNO}8 species was proposed to be the rate-limiting step.327
Hence, it can be concluded that the E° and pKa of the {FeNO}7 species determine the thermodynamics of the PCET step or those of the competitive NO dissociation step. The presence of EWGs along with saturated pyrrolic centres result in weak BDFENH; probably explaining the role of heme d1 in Cd1NiR. On the contrary, higher values of BDFENH resulting from balance between E° and pKa is responsible for facile PCET to {FeNO}7 producing {FeHNO}8 which eventually proceeds to NH4+ formation and release relevant to CcNiR mechanism.
In this section, heme containing nitrite reductase enzymes and their structural and mechanistic aspects were discussed. The role of basic residues was highlighted, which has already been explored in details in the enzymes. Next, the different results based on synthetic model complexes were discussed. It is observed from the literature that main focus till now has been on the understanding of the role played by the different macrocyles employed by nature for nitrite reduction reaction. In the future more studies with reconstituted enzyme active site needs to be done. This may give a holistic view of the observed differences in the mechanistic pathways. Also, synthesis of tailor-made model complexes with a secondary structure is necessary for a deeper understanding of the roles played by proton transfer and electron transfer residues in nitrite reduction reaction to get more control of the fate of this reaction in an artificial setting. While the large structural changes responsible for the on–off switch may be difficult (or impossible) to mimic in artificial systems, the roles played by tyrosine, arginine, lysine and hstidines can definitely be understood better.
H2 and H+ interconversion
Introduction
Hydrogen is the most abundant element in the Universe but in Earth it is in the tenth position, as molecular H2 being a light gas escapes easily into space.342 Most of the hydrogen is fixed in the form of strong bonds with other elements like C, N and O. For example, the reaction of H2 with O2 releases substantial energy.| 2H2(g) + O2(g) → 2H2O(l), ΔH° = −286 kJ mol−1 | (24) |
Lessons from Nature
In nature, many lower microorganisms e.g. algae and bacteria354–357 have the capability either to pull out electrons from hydrogen molecules to carry out their metabolic processes or to reduce protons to hydrogen by removing electrons. In these microorganisms, the inter-conversion between hydrogen uptake and release is efficiently mediated by a class of enzymes, known as hydrogenases. Hydrogenases are class of a metalloenzyme354,355,358 that can efficiently convert proton to hydrogen reversibly with minimum overpotential with a turnover rates of 102–104 mol s−1 for per mole of the enzyme.345,359–362 Hydrogenases are widespread in nature and they occur in bacteria, archaea, and some eukarya.354–357 These enzymes catalyse the interconversion of H2 to H+ and participate in various physiological processes e.g. sulfate reduction, nitrate reduction, carbon dioxide reduction, etc.363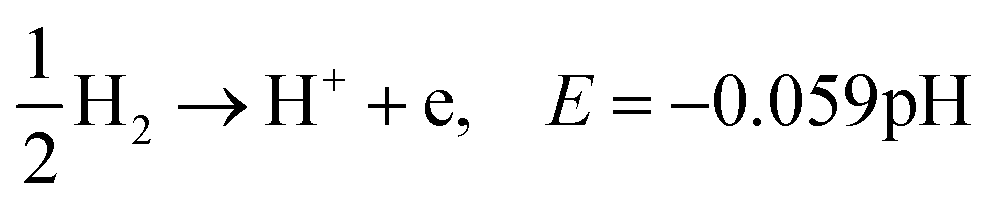 | (25) |
Types of hydrogenases
Based on their metal content, hydrogenases are classified into three classes namely [FeFe]-hydrogenase (Fig. 22), [NiFe]-hydrogenase (Fig. 23), and [Fe]-hydrogenase (Fig. 24).345,367–369 Among them [NiFe]-hydrogenases are the most abundant in nature and biased towards hydrogen oxidation whereas [FeFe]-hydrogenases are most selective towards proton reduction.355,363,368 The third class, [Fe]-hydrogenase or 5,10-methenyltetrahydromethanopterin dehydrogenase (Hmd) is mainly found in methanogens. Only in the presence of a hydride acceptor/donor substrate it reacts with H2 or produce H2.367,370,371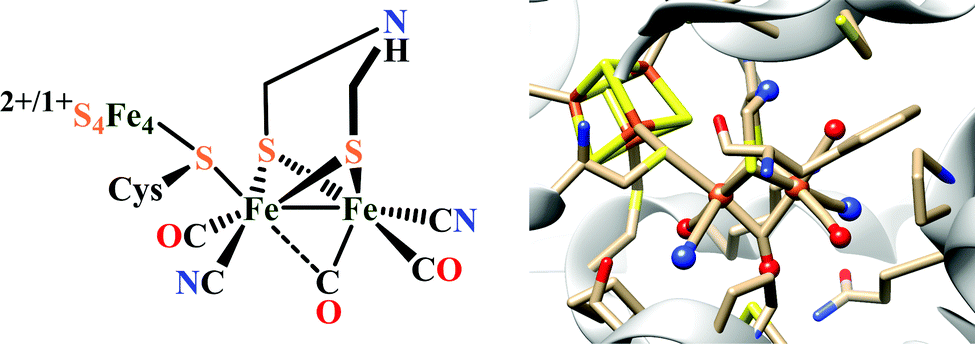 | ||
| Fig. 22 Schematic representation (left) and X-ray crystal structure (right) of the‘H-cluster’ in the Hox state, found in [FeFe]-hydrogenase (PDB ID: 3C8Y386 with modification of the bridgehead atom from oxygen to nitrogen). | ||
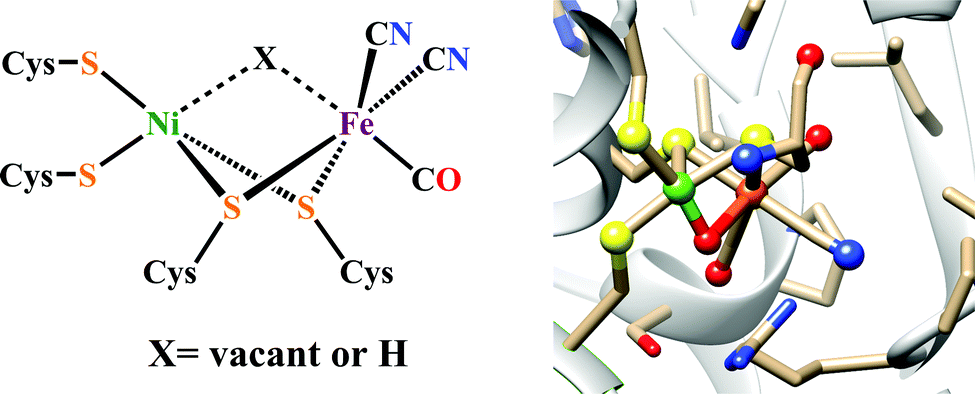 | ||
| Fig. 23 Schematic representation (left) and X-ray crystal structure (right) of the [NiFe]-hydrogenase (PDB ID: 1WUJ402) active site. | ||
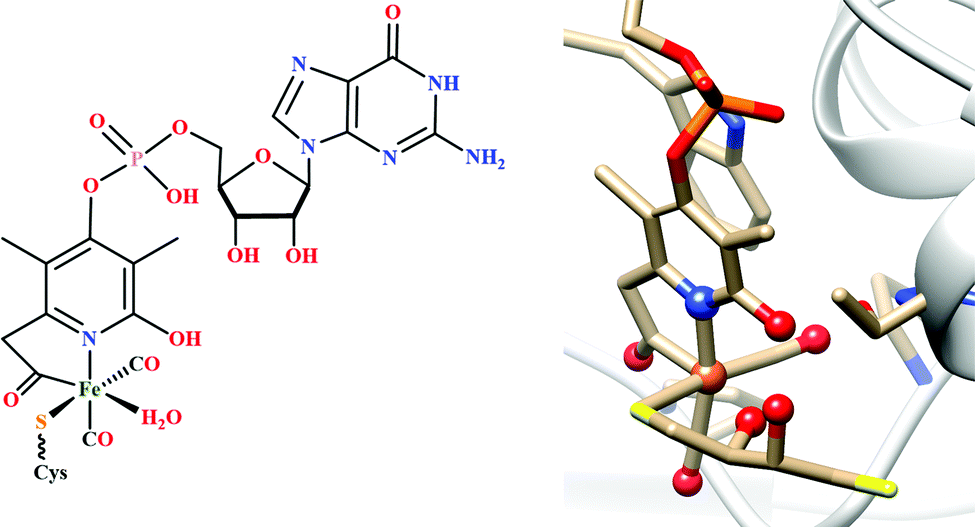 | ||
| Fig. 24 Schematic representation (left) of the [Fe]-hydrogenase active site with the Hmd cofactor, and the X-ray crystal structure (right) of the mono-iron complex obtained from the cysteine 176 mutant (PDB ID: 3H65406). | ||
Structure and activity of [FeFe]-Hydrogenases
The X-ray crystal structures of the [FeFe]-hydrogenases, particularly from Clostridium pasteurianum and Desulfovibrio desulfuricans, reveal the structure of this class of enzymes.372,373 The active site of [FeFe]-hydrogenase contains a unique dinuclear [2Fe] sub-cluster which is covalently linked by a cysteine sulphur to a [4Fe–4S]-cluster, thus forming the H-cluster. The Fe4S4 cluster acts as a source of electrons required for H+ reduction. Both X-ray crystallographic372,373 and infrared spectroscopic374,375 investigations confirmed that each of the two Fe centre contains one CN− and one CO ligand, both in a terminal binding mode. Whereas, an additional CO ligand is found to be bridging between both Fe atoms, in the oxidised Hox state of the H-cluster. The Hox state can be described as low spin Fe(II)Fe(I) state with spin state S = 1/2376,377 and a Fe4S4 cluster in the oxidized and diamagnetic [4Fe–4S]2+ state.378 The strong π acidic CO and CN− ligand stabilizes the low oxidation states of Fe by metal to ligand backbonding. The two diiron cores bridged by a dithiolate ligand, whose composition is matter of debate. From X-ray crystallographic data it is proposed to be –SCH2XCH2S–, where X can be nitrogen (–NH, adt), oxygen (O, odt), carbon (C, pdt) and a azadithiolate bridge is considered as the most likely.379 Furthermore,14N quadruple analysis and EPR spectroscopy support this hypothesis.380–382 The proximal iron (Fep) is directly bound to the [4Fe–4S] cluster and is coordinatively saturated. Whereas the distal iron (Fed) has a vacant coordination site where the substrate (H2 or H+) may bind as well as inhibitors like O2 and CO. Recent spectroelectrochemical investigations reveal that the Fe(I)Fe(I) state of the enzyme binds the first proton at the bridgehead nitrogen atom.383,384 Then successive reduction and proton rearrangement results in the formation of Fe(II)Fe(II)–H− species.385 At the bridgehead nitrogen atom, the second protonation occurs, which is well oriented to couple with the hydride, to release H2 (Scheme 22). | ||
| Scheme 22 Proposed catalytic cycles for reversible H+ reduction by [FeFe]-hydrogenases.387–390 | ||
Structure and activity of [NiFe]-Hydrogenases
Structural characterization of the [NiFe]-hydrogenases disclosed that these enzymes consist of a large subunit containing the [NiFe] active site and a small subunit containing three FeS clusters,391,392 which connects the active site of the enzyme to the surface of the protein and acts as an electron transport chain from the [NiFe] site to the electron acceptor cytochrome c3.393 Crystal structures of the hydrogenase show a [3Fe–4S] cluster in the centre of the chain, a [4Fe–4S] cluster close to the surface of the protein and a [4Fe–4S] cluster located approximately 13 Å away from the active site.394 The dinuclear active site contains two metal ions, Ni and Fe, bridged by two cysteine thiolates. Ni has two additional thiolates as terminal ligands and Fe is coordinated by one CO and two CN− ligands.391,392,395 Electron paramagnetic resonance spectroscopy and infrared spectroscopic techniques has revealed different possible redox states of the active site.396 Previously it was believed that only three intermediate species are involved in the catalytic cycle. The resting diamagnetic Ni-SIa species (NiIIFeII) takes up one electron and one proton to give the paramagnetic Ni-C state (NiIIIμ(H)FeII).397,398 The diamagnetic Ni-R state (NiIIIμ(H)FeII)399,400 is generated from the Ni-C state upon one electron reduction. Ni-R undergoes protonation at the hydride to produce H2, which eliminates to regenerate the resting Ni-SIa state. But recent studies established that Ni-C state upon low temperature photolysis produces new EPR active states, jointly called as Ni-L state, that are assigned to migration of the bridging hydride as proton.367,401 In both Ni-C state and Ni-R state the hydride bridges the two metallic centres (Scheme 23). Throughout this whole catalytic cycle, the Fe centre remains in the low spin FeII state, a configuration favoured by the two strong field CO and CN− ligands, whereas the Ni centre changes its oxidation states between the NiIII (in Ni-C) and NiII (in Ni-R and Ni-SIa). | ||
| Scheme 23 Proposed catalytic mechanism for reversible H2 oxidation by [NiFe]-hydrogenases.367,400 | ||
Structure and activity of [Fe]-Hydrogenases
In some methanogenic archaea, there is a third type of hydrogenase, which had long been believed to be purely bio-organic and they were initially called metal free hydrogenases.403 But it is now a well-known fact that these enzymes contain a iron in their active site and do not contain any nickel or iron–sulfur clusters. So, they are now referred as iron–sulfur cluster free hydrogenase and abbreviated as [Fe]-hydrogenase or Hmd. [Fe]-hydrogenases heterolytically split hydrogen into a proton (H+) and a hydride (H−), where the H− transferred to the substrate containing the carbocation, methenyl-tetrahydromethanopterin (methenyl-H4MPT+). Thus, it catalyzes the reversible reduction of methenyl-H4MPT+ with H2 to methylene-H4MPT and a H+(Scheme 24).404,405 This reduction is a key intermediary step in the biological conversion of carbon dioxide to methane. | ||
| Scheme 24 Scheme of the reversible heterolytic splitting of H2 by Hmd, showing a hydride stereospecifically transferred into the pro-R site of methenyl-H4MPT+ to yield methylene-H4MPT and a proton. | ||
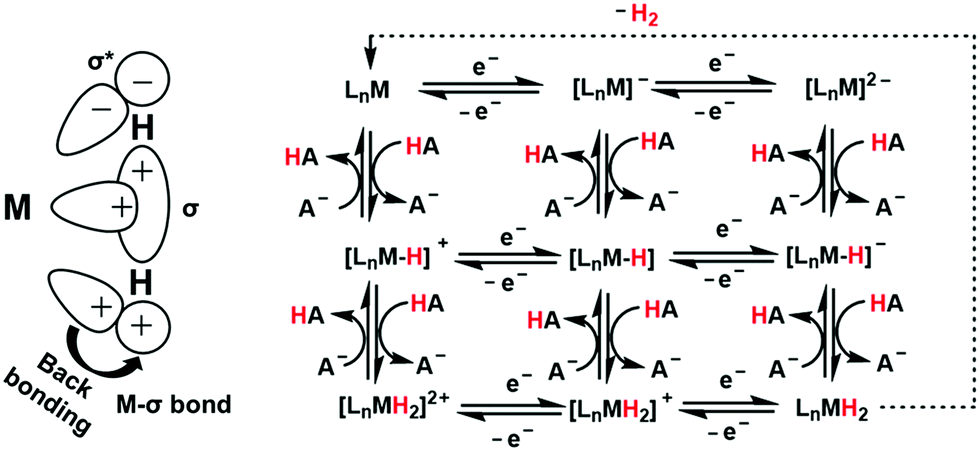 | ||
| Scheme 25 Schematic presentation of the formation of M–H2 σ bond and Mdπ → H2 backdonation (left). Possible mechanistic pathways for catalytic reduction of proton to hydrogen in the natural hydrogenases or synthetic catalysts (right). | ||
As pointed out by DuBois, the heterolytic cleavage of H2 should be at or near equilibrium to avoid high energy intermediates.416–420 This implies the hydride (H−) acceptor ability of the metal and the proton (H+) acceptor ability of the base (either external or internal) must be energetically matched to provide enough energy to drive the heterolysis of H2, but this reaction should not be strongly exergonic to offer reversibility in catalysis.
Synthetic model systems for HER
[FeFe]-Hydrogenase mimics
The number of structural and functional models of diiron dithiolate and related systems of the active site of the [FeFe]-hydrogenase has grown steadily since the first structures of the enzyme were reported some 20 years ago. Substantial progress has been made in producing stable mixed-valent species with bridging CO. Nonetheless many of the model systems that are similar to the enzyme in their oxidation state and valency of the iron and these consists of abiological phosphine or carbene ligands. On the functional side, mostly diiron hexacarbonyls have been explored. These systems generally reduce proton with large overpotentials and the catalytic cycle passes through the Fe(I)–Fe(I)/Fe(0)–Fe(0) levels as opposed to that of the natural FeII–FeI states in the enzymatic systems.Proton reduction by [FeFe]-Hydrogenase mimics in organic medium
In 1929, synthesis of [Fe2(CO)6(μ-SEt)2] was reported by Reihlen et al.423 When the protein crystal structure of [FeFe]-hydrogenase was available in 1998–1999,372,373 the striking resemblance of the diiron subsite of the H-cluster to the [(μ-pdt)Fe2(CO)6] (pdt = propanedithiolate), formerly described by Seyferth,424 became evident. The electrochemical reduction of proton to hydrogen has been investigated in substantial details using this diiron ditiolate bridged complexes by several groups previously.425–428 These complexes were classified based on the nature of the bridge, terminal ligands. In most cases the evolution of hydrogen has been confirmed by the use of gas chromatography (GC) and the faradaic yields are generally above 80%.In their resting state diiron model complexes are mainly in their FeI–FeI form and undergoes stepwise reversible or quasi-reversible reductions to produce Fe0–Fe0 state. In this section the focus is on the inclusion of 2nd here residues and their effect on proton reduction activity of diiron model complexes. Substituting the dithiolate bridge has little impact on the redox potentials of the model complexes. Replacing the CO ligands by electron donating ligands shifts the reduction towards more negative value. However, when the tereminal –CO ligands are replaced by electron donating ligands (e.g. phosphines, carbenes, CN−), the basicity of the iron centre increses and subsequently they are pronated by the proton source directly429–432 to produce the hydride intermediate. But when the diiron is not basic enough to undergo direct protonation they undergo one- or two-electron reduction to produce the FeIFe0 or the FeIFe0 state, which are enough basic to undergo protonation even in the presence of a weak proton source to produce the hydride intermediates.430,433–437 A second protonation of the hydride intermediates then produces the dihydride intermediates and which then releases H2 regenerating the starting species. Iron hexacarbonyl complexes, in presence of weak acids undergo reduction before protonation, due to their lower basicity. When a basic residue is present in the model complex, in presence of a suitable proton source, the electrocatalytic cycle is initiated by the protonation of the basic residue. Introduction of a redox active ligand in the model complex results a double reduction, one at the iron centre and other at the ligand. When a bidentate ligand is attached at the Fe centre, the chelating isomer is reported to be more efficient for H2 production than the symmetrically substituted bridging isomer both in terms of icat/ip value and turn over number (TON). Based electrochemical analysis various mechanisms (e.g., EECC, ECEC, ECCE, CECE, CEEC; E means an electron transfer step and C means a chemical step here protonation) are reported for the proton reduction activity in acidic medium.
 | ||
| Fig. 25 Diiron model complexes with nonbridging group with all carbon atoms. | ||
Diiron complexes of the type Fe2(CO)6(μ-R)2 were developed by Si et al., where [R = C6H4-o-OMe (Fig. 25b); C6H4-p-OMe (Fig. 25c); C6H4-p-Cl (Fig. 25d)].439 They showed that all these complexes reduces proton at their second reduction event in presence of AcOH. The complex b (Fig. 25) in presence of excess PMe3 was transformed to Fe2(CO)4(μ-SC6H4-o-CH3O)2(PMe3)2 (Fig. 25e), which was catalytically active at −2.08 V in CH3CN medium in presence of AcOH.
 | ||
| Fig. 26 Diiron model complexes with various –R groups in SCH2CH(R)CH2S bridge. | ||
Darensbourg and coworkers developed a series of diiron model complexes bearing carboxylic acid functionalities [(R1 = COOH, R2 = H; Fig. 26a and R1 = CONHPh, R2 = H Fig. 2c)].441 They reported that complex (μ-(SCH2)2CHCOOH)[Fe(CO)3]2 (Fig. 26a) and (μ-(SCH2)2CHCONHPh)[Fe(CO)3]2 (Fig. 26c) reduces proton in presence of AcOH in CH3CN at their Fe(I)–Fe(I)/Fe(I)–Fe(0) redox levels at −2.51, and −2.43 V vs. Fc+/Fc, respectively whereas the –PMe3 substituted complex (μ-(SCH2)2CHCOOH)[Fe(CO)2(PMe3)]2 (Fig. 26b) catalyzes H2 production from AcOH at its formal FeI–Fe0 state. Singleton et al. showed that when both R1 = R2 = Me and L = PPh3 (Fig. 26e) the Fe0–Fe0 species of the complex was active towards electrocatalytic H2 production, whereas the Fe0–FeI species is not.442 Apparently the weak donor ability of triphenylphosphine renders iron insufficiently electron rich to accommodate the oxidative addition of a proton.
Wang et al. reported electrocatlytic proton reduction by similar diiron model complexes (Here R1 = R2 = H) containing pendant nitrogen bases in phosphine ligands (μ-pdt)[Fe2(CO)5PPh2NH(CH2)2N(CH3)2] (Fig. 26f) and (μ-pdt)[Fe2(CO)5PPh2NH(2-NH2C6H4)] (Fig. 26g) in the presence of trifluoromethanesulfonic acid (CF3SO3H) in CH3CN medium.443 They reported that the secondary amine in complex (μ-pdt)[Fe2(CO)5PPh2NH(2-NH2C6H4)] (Fig. 26g) has a weak intramolecular hydrogen bonding with both the terminal nitrogen and sulfur atom. They suggested that a proton transfer pathway from amine in phosphine ligand to the sulfur atom of active site. Protonation of complexes (μ-pdt)[Fe2(CO)5PPh2NH(CH2)2N(CH3)2] (Fig. 26f) and (μ-pdt)[Fe2(CO)5PPh2NH(2-NH2C6H4)] (Fig. 26g) only occurred at the terminal nitrogen atom. Similarly Li et al. introduced pyridyl groups to the phosphine ligands of the diiron complex as a pendant basic site.444 The electrocatalytic proton reduction activity of (μ-pdt)[Fe2(CO)5(Ph2PCH2Py)] (Fig. 26h), (μ-pdt)[Fe2(CO)5(Ph2PPy)] (Fig. 26i) and (μ-pdt)[Fe(CO)2(PMe3)][Fe(CO)2(Ph2PCH2Py)] (Fig. 26j) was investigated in the prsesence of AcOH in CH3CN medium. The catalytic efficiency of complex (μ-pdt)[Fe2(CO)5(Ph2PCH2Py)] (Fig. 26h) and (μ-pdt)[Fe2(CO)5(Ph2PPy)] (Fig. 26i) was found to be relatively low. The overpotentials for the electrocatalytic reduction of protons from AcOH are 400 mV vs. Fc+/Fc for (μ-pdt)[Fe2(CO)5(Ph2PCH2Py)] (Fig. 26h) and 370 mV vs. Fc+/Fc for (μ-pdt)[Fe2(CO)5(Ph2PPy)] (Fig. 26i). The effect of water on the proton reduction activity of (μ-pdt)[Fe(CO)2(PMe3)][Fe(CO)2(Ph2PCH2Py)] (Fig. 26j) was probed in CH3CN and in CH3CN–H2O (50:1, v/v). The electrocatalytic current for proton reduction was considerably enhanced in CH3CN–H2O (50:1, v/v). The electrocatalytic efficiency for proton reduction by (μ-pdt)[Fe(CO)2(PMe3)][Fe(CO)2(Ph2PCH2Py)] (Fig. 26j) in CH3CN–H2O (50:1, v/v) increases 5 times than that of in pure CH3CN. Just like the secondary amine in the azadithiolate bridged diiron complexes, here the pyridyl group in the phosphine ligand of diiron complexes can serve as a proton relay in the presence of strong acid, resulting to the decrease of the overpotential for electrocatalytic proton reduction by 360–490 mV.444 Also, the hydrophilicity of the pyridyl group in these diiron complexes renders the electrocatalytic efficiency more susceptible to water.
On substituting the benzene ring with increasing number of electron withdrawing –Cl groups leads to an anodic shift in reduction potential and a associated lowering of the overpotential of hydrogen evolution was observed.445 A same ECEC mechanism (E corresponds to an electron transfer step, and C, to a chemical reaction, here protonation) is also suggested for the catalytic proton reduction from weak acids by compound b, c and d (Fig. 27).445 As the number of electrons withdrawing groups is increased in the complexes b to d (Fig. 27) 250 mV improvement of the overpotential of AcOH reduction was observed but compromises the catalytic efficiency in terms of TOF.445 The analogous methyl substituted complex [Fe2(CO)6{μ-SC6H3(CH3)S}] (Fig. 27e) produces H2 at −2.10 V in presence of AcOH with a overpotential of 0.64.445
 | ||
| Fig. 27 Diiron model complexes containing μ-arenedithiolato bridges. | ||
Chen et al. reported a series of Fe2S2 complexes containing phenolic –OH groups and –OMe groups adjacent to the sulfur centers.446 They showed that phenolic –OH groups adjacent to the sulfur centers in complexes f, h, i, j and k (Fig. 27) modestly render their reduction potentials less negative and hence lowered the catalytic overpotential compared to the unsubstituted complex [(μ-bdt)Fe2(CO)6] (Fig. 27a) due to internal hydrogen bonding. Spectroscopic investigations suggested the presence of intramolecular hydrogen bonding between the phenolic –OH groups and the adjacent sulfur atoms. However these hydroquinone complexes f, h, i, j and k (Fig. 27) is less effective catalyst for proton production from weak acids compared to [(μ-bdt)Fe2(CO)6] (Fig. 27a) because of the internal hydrogen bonding. The internal hydrogen bonding in these complexes lowered the basicity of the dianionic [Catalyst]2− species of the rate-determining step. Whereas the dimethoxy derivative g (Fig. 27), in which hydrogen bonding is absent, is a better catalyst compared to [(μ-bdt)Fe2(CO)6] (Fig. 27a) due to the increased basicity of the corresponding dianion. These results further indicate that the pKa value of the [CatalystH]− species is the most important factor for catalytic activity for this family of catalysts.446
Lin and coworkers reported phosphine derivatives (Fig. 27l–o) of [Fe2(CO)6{μ-SC6H3(CH3)S}] (Fig. 27e) complex and examined their proton reduction activites in CH3CN medium in presence of AcOH.447 The reduction potential of the parent complex [Fe2(CO)6{μ-SC6H3(CH3)S}] (Fig. 27e) is more positive than those of complexes l–o (Fig. 27) due to the fact that phosphine ligands are more electron-donating than CO.
Durgaprasad et al. reported a series of diiron model complexes by incorporating 1,2-ethylene-dithiolate on the benzenedithiolato bridgehead.448 They showed that compounds [Fe2{μ-diph-6,7-qdt}(CO)6] (Fig. 27p) and [Fe2{μ-btdt}(CO)6] (Fig. 27q) exhibit electrocatalytic proton reduction in presence of a strong acid TsOH. For the electrocatalytic proton reduction catalysed by compound [Fe2{μ-diph-6,7-qdt}(CO)6] (Fig. 27p), they proposed a CEEC mechanism. Whereas, the hexa carbonyl compound [Fe2{μ-btdt}(CO)6] (Fig. 27q) did not undergo protonation in presence of p-toluenesulfonic acid in its FeI–FeI oxidation state,may be due to the involvement or delocalization of lone pair of electrons of ring nitrogen with ring sulfur pi-electrons. Accordingly an EC electrocatalytic reduction mechanism is proposed for compound [Fe2{μ-btdt}(CO)6] (Fig. 27q).
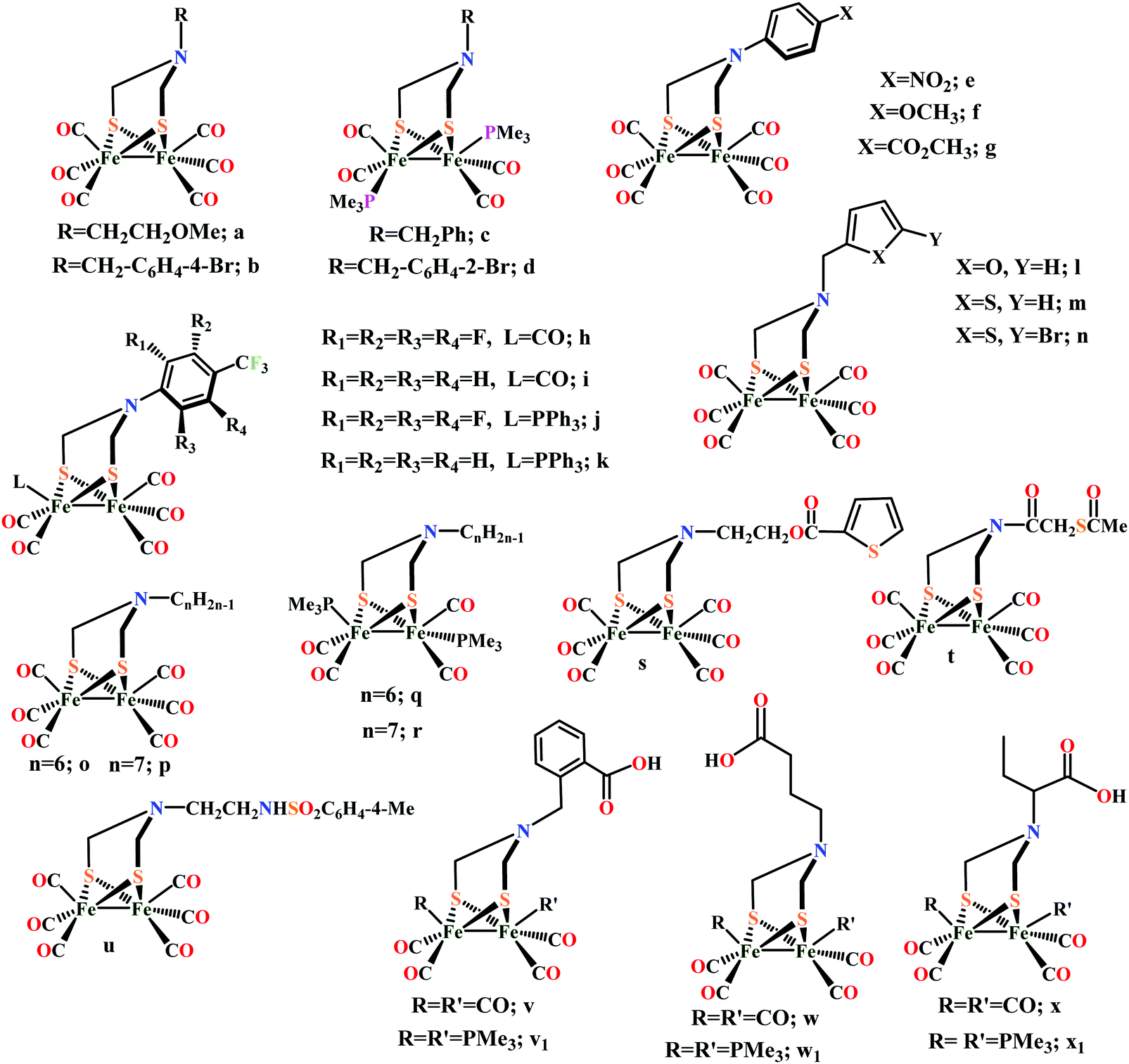 | ||
| Fig. 28 Diiron model complexes with azadithiolato bridges [SCH2N(R)CH2S]. | ||
 | ||
| Scheme 26 Proposed mechanisms of proton reduction catalysed by [Fe2(CO)6{μ-SCH2NH(R)CH2S}] complexes (R = CH2CH2OMe, Fig. 28a) in MeCN.426 | ||
Proton reduction by [{Fe(CO)2(PMe3)}2{μ-SCH2N(R)CH2S}] (Fig. 28c and d) was also monitored in the presence of a weak and of a strong acid.430,432,452 Weak acid like AcOH neither protonate the bridged-N atom nor the Fe–Fe site.432,452 In presence of a stronger acid (TfOH, pKa = 2.6 in CH3CN), catalysis takes place at around −1.5 V vs. Fc+/Fc, which is a potential assigned to the reduction of the protonated amino species (Fig. 28c, R = CH2Ph430) or of the bridging hydride (Fig. 28d, R = CH2C6H4-2-Br432). When R = CH2Ph (Fig. 28c), no catalysis was observed at −1.1 V vs. Fc+/Fc, at which the reduction of Fe(μ-H)Fe isomer takes place or at −1.0 V vs. Fc+/Fc at which the doubly protonated species get reduced.430 However on prolonged controlled potential electrolysis, slow catalysis is observed at these potentials.430 For the [{(μ-SCH2)2N(4-NO2C6H4)}Fe2(CO)6] (Fig. 28e) complex a CECE mechanism was suggested in presence of AcOH (0–10 mM) in CH3CN by Liu et al.453 But in case of the –OMe substituted complex f (Fig. 28) an EECC mechanism was proposed in presence of AcOH (0–10 mM) in CH3CN medium.454 Here the bridged N atom of f (Fig. 28) was not protonated by weak acid AcOH. From the Bulk electrolysis of a CH3CN solution of complex f (0.33 mM) with excess AcOH (6.6 mM) at −2.18 V showed hydrogen formation with ∼5 turnovers. Similarly for the complex g (Fig. 28) in presence of weak AcOH a EECC mechanism was also proposed.455 Bulk electrolysis experiment of the CH3CN solution of complex g (Fig. 28) (0.33 mM) with excess AcOH (6.6 mM) revealed that the initial rate of electrolysis was five times greater than that of in absence of the catalyst. In the presence of a strong acid like CF3CO2H an ECCE mechanism was proposed for the electrocatalytic proton reduction catalysed by complex g (Fig. 28).455
Wang et al. reported a set of adt-bridged fluorophenyl-substituted diiron active site models of [Fe–Fe] hydrogenase, [(μ-adt)C6F4CF3-p]Fe2(CO)6 (Fig. 28h), [(μ-adt)C6H4CF3-p]Fe2(CO)6 (Fig. 28i), [(μ-adt)C6F4CF3-p]Fe2(CO)5PPh3 (Fig. 28j), [(μ-adt)C6H4CF3-p]Fe2(CO)5PPh3 (Fig. 28k).456 They demonstrated that the electrocatalytic proton reduction by these complexes is mostly dependent on the strength of the proton source used. When weak AcOH is used as the proton source, the electrocatalytic proton reduction process starts off with two consecutive one-electron reduction processes to produce H2via the Fe0–Fe0 state whereas in the presence of strong acid, HBF4·Et2O, the catalytic process commenced by the protonation of the bridgehead N atom followed by reduction of the bridgehead protonated N atom around −1.29 V vs. Fc+/Fc, to generate H2via the Fe0–FeI state. Thus, by altering the strength of the acid triggers the initial electron-transfer step from the reduction of the bridgehead protonated N atom to the active site of [FeI–FeI].
Sun and coworkers prepared three heterocycle-containing diiron azadithiolate hexacarbonyl model complexes, [{(μ-SCH2)2NCH2(2-C4H3O)}](Fe2(CO)6) (Fig. 28l), [{(μ-SCH2)2NCH2(2-C4H3S)}](Fe2(CO)6) (Fig. 28m) and [{(μ-SCH2)2NCH2(5-Br-2-C4H2S)}Fe2(CO)6] (Fig. 28n).457 They proposed that the methyl group near the heterocycle might increase the electron density in the π system by donating electrons by hyperconjugation (σ conjugation), and slightly decrease the nuclear electron density of the Fe atom through the three atoms (N, C, S). Such an effect can be increased by the oxygen atom in furan and bromine atom on the thiophene, resulting in less negative reduction potentials. The catalytic proton reduction for these complexes were studied in CH3CN in presence of 0–8 mM HClO4 under CO atmosphere. They showed that the furan-containing model complex [{(μ-SCH2)2NCH2(2-C4H3O)}](Fe2(CO)6) (Fig. 28l), displayed electrocatalytic proton reduction at −1.13 V vs. Fc+/Fc whereas the thiophene-containing analogues m and n (Fig. 28) exhibit catalytic proton reduction at −1.20 and −1.09 V vs. Fc+/Fc, respectively in presence of HClO4. Along the same lines Si et al. reported a series of a novel class of (N–CnH2n−1)-1,3-azapropanedithiolate-bridged [Fe–Fe] hydrogenase model complexes [Fe2(CO)6(CH2S)2N–CnH2n−1] (n = 6, 7) and their PMe3-disubstituted complexes [Fe2(CO)4(PMe3)2(CH2S)2N–CnH2n−1] (n = 6, 7) and investigated their electrochemical behaviour in presence of AcOH.452 They proposed an ECEC mechanism for electrocatalytic proton reduction of the complexes o–r (Fig. 28) in presence of AcOH. From the bulk electrolysis experiment of 1 mM solution of complex [Fe2(CO)4(PMe3)2(CH2S)2N–CnH2n−1] (n = 6) (Fig. 28q) (5 ml, 5 mM) with excess AcOH (20 mM) at −2.3 V vs. Fc+/Fc it was observed that the initial rate of electrolysis was approximately 1.8 times that without the catalyst. 12 F charge per mole of q (Fig. 28) was passed over the course of 20 min, which corresponds to 6 turnovers.
Eletrocatalytic proton reduction by the [(μ-SCH2)2NCH2CH2O2CC4H3S-2]Fe2(CO)6 complex (Fig. 28s) in presence of AcOH in CH3CN medium was proposed to proceed via EECC mechanism.458 Similarly, Song et al. reported a N-acylated diiron azadithiolate complex [{(μ-SCH2)2-NC(O)CH2SC(O)Me}Fe2(CO)6] (Fig. 28t) which had the ability to to undergo proton reduction to generate hydrogen gas in the presence of weak acid AcOH via an EECC mechanism.459 The bulk electrolysis of AcOH (25 mM) catalyzed by t (Fig. 28t) (0.50 mM) in CH3CN medium at −2.34 V vs. Fc+/Fc indicated a total of 12.3 F per mol of t (Fig. 28) to be passed during half an hour. This corresponds to 6.1 turnovers. Complex u (Fig. 28) was reported to display proton reduction activity in presence of a strong acid CF3SO3H in CH3CN medium.460 Based on the electrochemical measurement a CECE mechanism was proposed for the proton reduction by u (Fig. 28).
A series of model complexes bearing aryl and alkyl carboxylic acid groups to the dithiolate bridge of diiron unit, [Fe2(CO)6{(μ-SCH2)2NCH2C6H4-2-COOH}] (Fig. 28v), [Fe2(CO)6{(μSCH2)2NCH2CH2CH2COOH}] (Fig. 28w) and [Fe2(CO)6{(μ-SCH2)2NCH(CH2CH3)COOH}] (Fig. 28x) have been synthesized with the aim of decreasing the potential for reduction of protons to hydrogen and their proton reduction activity in presence of TfOH is studied by Gao et al.461 They proposed that in the case of complex [Fe2(CO)6{(μSCH2)2NCH2CH2CH2COOH}] (Fig. 28w) the carbon chain is long enough to allow interaction between the carboxyl group and the diiron cluster. A CECE mechanism is suggested for the complex v, w and x (Fig. 28) for electrocatalytic proton reduction in accordance with a previous report.462 It was observed that the increase of catalytic current against amount of added acid (TfOH) for PMe3 substituted complex w1 and x1 is greater (200 μA for adding of 4 eq. of TfOH) than that for v1 (50 μA for adding of 4 eq. of TfOH). A CECE mechanism was proposed for the formation of hydrogen with v1, w1 and x1 (Fig. 28). In complex v1 (Fig. 28) the first protonation occurs at the bridging N atom whereas the in complex w1 and x1 (Fig. 28) the first protonation occurs at Fe–Fe bond. However, complexes w1 and x1 (Fig. 28) proved to be electrochemically active upon the addition of water alone, suggesting that the carboxyl group can provide H3O+ protons to the amino bridge thus catalyse proton reduction under neutral conditions (Scheme 27).
 | ||
| Scheme 27 Proposed mechanism for the electrocatalytic proton reduction mediated by [Fe2(CO)4(PMe3)2{(μ-SCH2)2NCH(CH2CH3)COOH}] (Fig. 28x1) in aqueous medium, explaining the role of the –COOH group as a proton transfer relay.461 | ||
Most of the above complexes have been investigated to test their ability to catalyze the reduction of acids to form hydrogen and the results are summarized in Table 5. The solvent used (mostly acetonotrile), the acid source, the peak potentials for the catalytic process, the standard potential for reduction of the acid in the used solvent, the overpotential and catalytic efficiency (C.E.) were tabulated. In a few instances, the overpotential is not reported either because the standard potential is not known in the solvent used (THF or CH2Cl2) or because the catalytic current may in fact not be due to reduction of the acid but to further reduction of the catalyst in the presence of acid. Potentials are referenced with respect to Fc+/Fc in acetonitrile and correspond to the peak of the catalytic process. Standard potentials are for the couple in acetonitrile. These were calculated as shown in previously reported literature.463 The pKa-values were used from the literature. Overpotentials are the catalytic peak potential minus the standard reduction potential of the acid used. The ratio of the catalytic current and the current for reduction of the catalyst in the absence of acid is examined. This ratio is divided by the ratio of the acid concentration, to the catalyst concentration to give catalytic efficiency (C.E.).
| Complex | Solvent used | Acid used | Catalytic peak potential (V) vs. Fc+/Fc | Standard reduction potential of the acid used (V) vs. Fc+/Fc | Overpotential (V) | Catalytic efficiency (CE) | Ref. |
|---|---|---|---|---|---|---|---|
| 25d | CH3CN | AcOH | −1.7 | −1.46 | −0.2 | 0.09 | 438 |
| 25i | CH3CN | AcOH | −2.08 | −1.46 | −0.62 | — | 439 |
| 26a | CH3CN | AcOH | −2.5 | −1.46 | −1.0 | 0.27 | 441 |
| 26b | CH3CN | AcOH | −2.3 | −1.46 | −0.8 | 0.28 | 441 |
| 26b | CH3CN | HCl | −1.45 | −0.67 | −0.78 | 0.06 | 441 |
| 26c | CH3CN | AcOH | −2.4 | −1.46 | −0.9 | 0.19 | 441 |
| 26e | CH3CN | AcOH | −2.3 | −1.46 | −0.8 | 0.40 | 442 |
| 26f | CH3CN | CF3SO3H | −1.75 | −0.29 | −1.46 | 1.0 | 443 |
| 26g | CH3CN | CF3SO3H | −1.7 | −0.29 | −1.4 | 0.88 | 443 |
| 27a | CH3CN | AcOH | −2.06 | −1.46 | −0.60 | 0.31 | 446 |
| 27a | CH2Cl2 | HBF4 | −1.4 | −0.28 | −1.1 | 0.38 | 464 |
| 27a | CH3CN | TsOH | −1.25 | −0.65 | −0.60 | 0.23 | 465 |
| 27b | CH3CN | AcOH | −1.97 | −1.46 | −0.51 | 0.32 | 445 |
| 27c | CH3CN | AcOH | −1.88 | −1.46 | −0.42 | 0.31 | 445 |
| 27d | CH3CN | AcOH | −1.85 | −1.46 | −0.39 | 0.23 | 445 |
| 27e | CH3CN | AcOH | −2.10 | −1.46 | −0.64 | 0.44 | 445 |
| 27f | CH3CN | AcOH | −2.05 | −1.46 | −0.59 | 0.12 | 446 |
| 27g | CH3CN | AcOH | −2.10 | −1.46 | −0.64 | 0.38 | 446 |
| 27h | CH3CN | AcOH | −2.05 | −1.46 | −0.59 | 0.12 | 446 |
| 27i | CH3CN | AcOH | −2.05 | −1.46 | −0.59 | 0.12 | 446 |
| 27j | CH3CN | AcOH | −2.10 | −1.46 | −0.6 | 0.09 | 446 |
| 27k | CH3CN | AcOH | −2.10 | −1.46 | −0.6 | 0.09 | 446 |
| 28a | CH3CN | HBF4 | −1.3 | −0.28 | −1.0 | 0.52 | 450 |
| 28a | CH3CN | TsOH | −1.3 | −0.65 | −0.6 | 0.29 | 450 |
| 28e | CH3CN | AcOH | −1.73 | −1.46 | −0.27 | 0.16 | 453 |
| 28f | CH3CN | AcOH | −2.20 | −1.46 | −0.7 | 0.19 | 454 |
| 28g | CH3CN | CF3CO2H | −1.60 | −0.89 | −0.7 | 0.34 | 455 |
| 28h | CH3CN | AcOH | −2.2 | −1.46 | −0.7 | 0.36 | 456 |
| 28h | CH3CN | HBF4 | −1.5 | −0.28 | −1.2 | 0.37 | 456 |
| 28i | CH3CN | AcOH | −2.1 | −1.46 | −0.6 | 0.23 | 456 |
| 28i | CH3CN | HBF4 | −1.5 | −0.28 | −1.2 | 0.22 | 456 |
| 28j | CH3CN | AcOH | −2.2 | −1.46 | −0.7 | 0.31 | 456 |
| 28j | CH3CN | HBF4 | −1.6 | −0.28 | −1.3 | 0.18 | 456 |
| 28k | CH3CN | AcOH | −2.3 | −1.46 | −0.8 | 0.60 | 456 |
| 28k | CH3CN | HBF4 | −1.6 | −0.28 | −1.3 | 0.80 | 456 |
| 28l | CH3CN | HClO4 | −1.2 | −0.26 | −0.9 | 0.32 | 466 |
| 28m | CH3CN | HClO4 | −1.45 | −0.26 | −1.19 | 0.50 | 457 |
| 28n | CH3CN | HClO4 | −1.25 | −0.26 | −0.99 | 0.43 | 457 |
| 28o | CH3CN | AcOH | −2.1 | −1.46 | −0.60 | — | 452 |
| 28p | CH3CN | AcOH | −2.2 | −1.46 | −0.70 | — | 452 |
| 28q | CH3CN | AcOH | −2.15 | −1.46 | −0.69 | — | 452 |
| 28r | CH3CN | AcOH | −2.1 | −1.46 | −0.60 | — | 452 |
| 28s | CH3CN | AcOH | −2.2 | −1.46 | −0.7 | 0.22 | 458 |
| 28t | CH3CN | AcOH | −2.2 | −1.46 | −0.7 | 0.50 | 459 |
The [(μ-bdt)Fe2(CO)6] (Fig. 27a) complex displays electrocatalytic proton reduction at −0.66 V vs. SHE in an aqueous sodium dodecyl sulphate (SDS) solution at pH 3.473 Bulk electrolysis experiment at −0.66 V vs. SHE of the complex at pH 3 confirms the production of H2 with faradaic efficiency close to 100%. In its catalytic cycle the complex first undergoes 2e− reduction to produce the active Fe0–Fe0 state followed by a PCET step to produce a hydride intermediate [Fe0–FeII(H)]− (Scheme 28), which then reacts with protons to produce H2 and regenerate the FeI–FeI form. However, in organic solvents, this [Fe0–FeII(H)]−(Scheme 28), intermediate is very hard to protonate by strong acids, which explains the poor activity of this for H2 production in acetonitrile.
 | ||
| Scheme 28 Proposed mechanism for the catalysis of electrochemical H2 production mediated by [(μ-bdt)Fe2(CO)6] (Fig. 27a). | ||
Several azadithiolate (ADT) bridged [FeFe]-hydrogenase model complexes are reported to produce H2 in aqueous medium.474,475 Complex BrC6H4N(CH2S)2Fe2(CO)6 (Fig. 29a) when physiadsorbed on edge plane graphite surface, has been demonstrated to display electrocatalytic proton reduction at pH < 3.474 While these catalysts are found to reduce proton at very negative potentials in organic medium, in aqueous medium the potential of proton reduction is shifted towards more positive value due to enhanced solvation of the anionic reduced states. The catalyst shows a TOF of 6400 s−1 at a potential of −0.5 V vs. SHE in 0.5 N H2SO4. Long term controlled potential electrolysis of the complex at −0.5 V indicates the TON of the catalyst is ≫108 and the faradaic efficiency >95%. The HER proceeds via the Fe(0)Fe(0) state under weakly acidic conditions and via the Fe(I)Fe(0) state in strongly acidic conditions. Complex BrC6H4N(CH2S)2Fe2(CO)6 (Fig. 29a) and some other molecular catalysts which efficiently produces H2 in water, are directly adsorbed or deposited on various electrode surface. However, a very few electrocatalysts are reported which are covalently attached onto the electrode surfaces in a site-specific manner.441,476–479 Covalent attachment of the catalyst solves the problem which appears in physiadsorption like excess catalyst loading, slower electron transfer between catalyst and the electrode, leaching of the catalyst.480 An azadithiolate bridged [Fe–Fe] hydrogenase mimic containing terminal alkyne group (Fig. 29b) is developed to covalently attach the complex on the modified electrode surface using click chemistry.475 The complex reduces proton electrocatalytically in presence of dil. H2SO4 medium with a high TON and TOF. The covalent attachment offers two orders of magnitude greater catalyst coverage without compromising the inherent catalytic properties of the complex. However, these catalysts need strongly acidic medium and substantial overpotential to be operative. Recently a series of N-aryl substituted azadithiolate (ADT) bridged [FeFe]-hydrogenase model complexes (Fig. 29c–e) have been reported in which the bridgehead N lone pair is fixed towards the one of the metal centres by ortho substitution of the aryl groups.145 Heterogeneous electrochemistry of these complexes physiadsorbed on edge plane graphite electrode reveals that the overpotential of proton reduction can be reduced to 160–180 mV in pH 5.5 water (Table 6).
 | ||
| Fig. 29 Proton reducing [FeFe]-hydrogenase model complexes in aqueous medium. | ||
| Complex | Medium | E (V) | TON | TOF (s−1) | Overpotential (V) |
|---|---|---|---|---|---|
| a | Aqueous medium (pH < 3) | −0.5 V vs. SHE | 108 | 6400 | 0.18 |
| c–e | Aqueous medium (pH 5.5) | −0.6 to −0.8 V vs. SHE | — | 11–50 | 0.16–0.18 |
| c | Organic medium (proton source TFA) | −1.8 V vs. Fc+/Fc | 0.15 ± 0.01 | 65 | 1.27 |
| d | Organic medium (proton source TFA) | −1.8 V vs. Fc+/Fc | 0.32 ± 0.05 | 32 | 1.31 |
| e | Organic medium (proton source TFA) | −1.8 V vs. Fc+/Fc | 0.35 ± 0.05 | 79 | 1.06 |
Complex (μ-pdt)[Fe(CO)2(PMe3)2]2 (Scheme 29a) on treatment with concentrated HCl produces a stable bridging hydride complex b483 (Scheme 29). Complex b (Scheme 29) mediates H/D exchange from H2/D2 and from H2/D2O. This reaction is inhibited by CO but promoted by sunlight suggesting a vacant coordination site for D2 binding. In the catalytic cycle D2 binding takes place prior to D–D cleavage. A series of similar complexes, (μ-L)2[Fe(CO)2(PMe3)]2, (L = SEt,SCH2CH2S, SCH2C6H4CH2S) and their protonated derivatives was also evaluated.484 The different thiolate ligands however has no effect on H/D exchange. Therefore, the sulphur ligands are unlikely proton acceptors in H2 splitting. Two mechanisms were proposed for the H/D exchange by b (Scheme 29).484 The (μ-pdt)[Fe(CO)2(PMe3)2]2 (Scheme 29a) complex also reacts with SMe+ to form c,485 which can take up H2 and catalyses the photolytic H/D exchange in D2/H2O. Complex (Scheme 29)c does not catalyse H2/D2 exchange under anhydrous conditions. Here the H2/D2 binding site is trans to the (μ-SMe) and H/D exchange is facilitated by H2O, the external base. So there is a need of a pentacoordinate Fe(II) Lewis acidic centre for (H/D)2 binding and an Brønsted base for deprotonation of the coordinated (H/D)2. Complexes b and c (Scheme 29) can catalytically cleavage H2 but cannot oxidize H2 into protons.
 | ||
| Scheme 29 Preparation of complex b and c. | ||
Initial attempts at activating and oxidizing H2 with synthetic [FeFe]-hydrogenase models utilized compounds in their Fe(I)Fe(I) or Fe(II)Fe(II) forms, neither of which is relevant to the biological mechanism, which proceeds through an Fe(I)Fe(II) state. The mixed-valent core of Hox exhibits a rotated Fed coordination geometry exposing a site on an electrophilic Fe center to bind H2 (or the inhibitor CO). Model complexes mimicking the Hox model with an azadithiolate ligand [Fe2[((SCH2)2N)(CO)3(PMe)3(dppv)]+] can react with H2, in a stoichiometric manner, to produce corresponding hydride complexes.486 But The related propanedithiolate complex [Fe2[((SCH2)2CH2)(CO)3(PMe)3(dppv)]+] and oxadithiolate complex [Fe2[((SCH2)2O)(CO)3(PMe)3(dppv)]+] analogues are inactive.487,488 These results suggest that heterolytic hydrogen activation is assisted by the azadithiolate ligand.
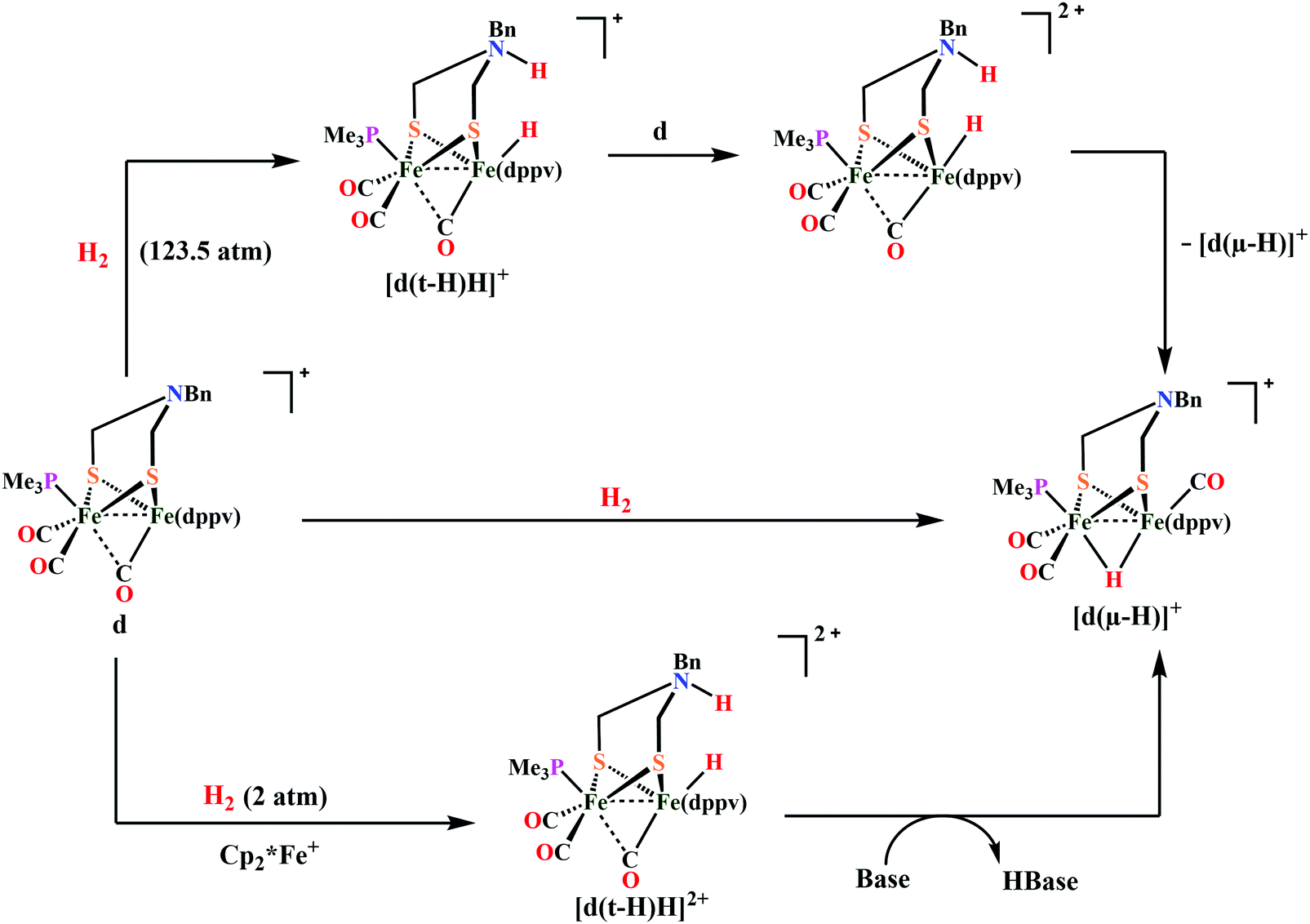 | ||
| Scheme 30 Stoichiometric activation of H2 with [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)] (Fig. 30d) in the presence489 and absence of an external oxidant.487 | ||
Complex [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)] (Fig. 30d) is the first model which slowly (>26 h, 25 °C) activates H2 and yields a bridging hydride at very high pressure (Scheme 30). But in presence of 1 eq. of a mild oxidant, [Fe(C5Me5)2]BArF4[ArF = 3.5-C6H3(CF3)2]([Fc]BArF4), and a weak base at 25 °C it reacts faster under 2 atm of H2 to produce [d(μ-H)]+ species (Scheme 30).489 However the more electrophilic model [Fe2[(SCH2)2NBn](CO)4(dppn)]+ [dppn = 1,8-bis(diphenylphosphino)naphthalene] (Fig. 30e), in presence of 1 eq. of [Fe(C5Me5)2]BArF4 reacts even more rapidly (t1/2 < 13 min at 20 °C) with 1 atm of H2 compared to [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)] (Fig. 30d). [Fe2[(SCH2)2NBn](CO)4(dppn)]+ (Fig. 30e) heterolyzes {in presence of [Fe(C5Me5)2]BArF4} H2 which is 20 times faster than [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)] (Fig. 30d) {in presence of [Fe(C5Me5)2]BArF4} and 104 times faster than that by complex [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)] (Fig. 30d) in the absence of an oxidant. The kinetic isotopic measurement studies indicate that H2 binding is the rate-determining step in these reactions.
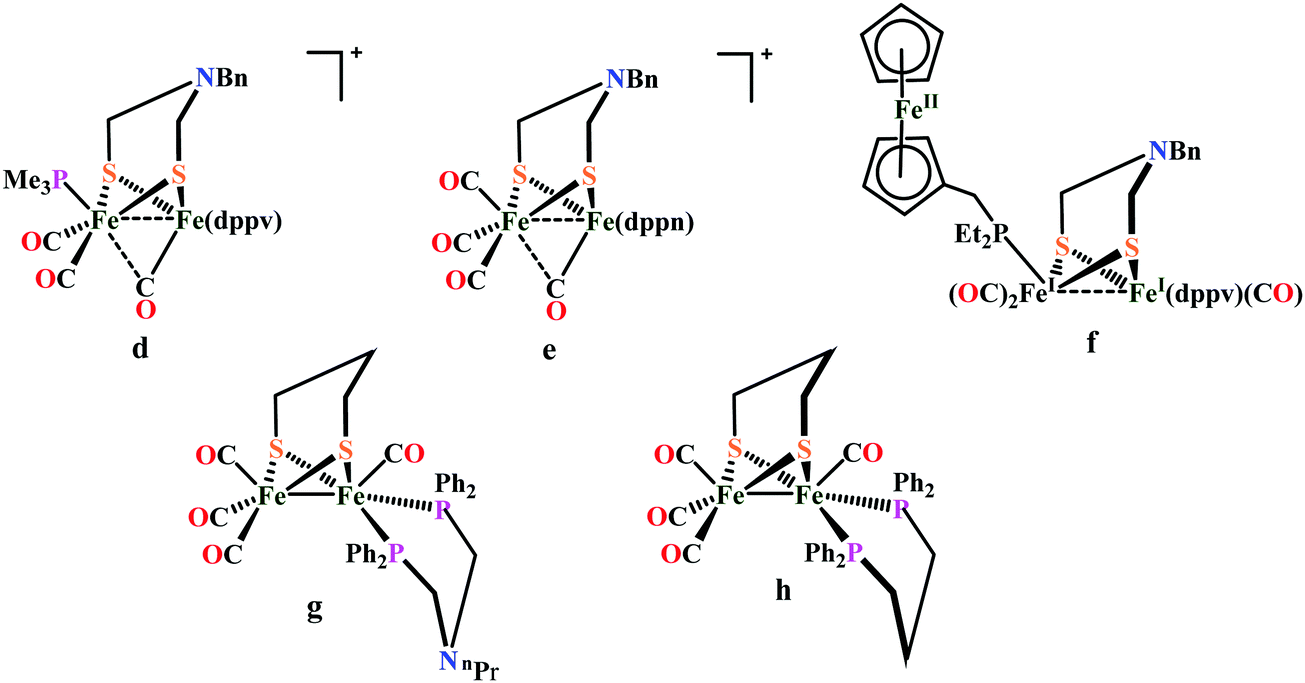 | ||
| Fig. 30 Hydrogen activating [FeFe]-hydrogenase model complexes d, e, f, g and the catalytically inactive complex h. | ||
 | ||
| Scheme 31 Proposed mechanism for O2 reduction by ortho substituted ADT bridged Fe–Fe mimics (Fig. 29c–e). | ||
Thus, for H2 activation the systems require a Lewis acidic Fe centre, a basic azadithiolate ligand and an electron sink. In the enzyme system these roles are beautifully served by the distal Fe atom, the azadithiolate group and the Fe4S4 cluster. To mimic the Fe4S4 cluster a redox active FcP* ligand is introduced to the complex [Me3P(CO)2Fe(NBn)Fe(CO)(dppv)]+ (Fig. 30d) by Rauchfuss and co-workers.490 The dicationic form of complex f(Fig. 30) (f2+) reacts with H2 in a PCET pathway to produce the bridging hydride complex. The FcP* ligand is reduced by one electron in a heterolytic H2 cleavage pathway. Only a 2-fold increase is observed here compared to d(Fig. 30) in presence of external oxidant. Complex d(Fig. 30) can only stoichiometrically activate H2, while the dication f2+ oxidises H2 in the presence of excess oxidant and excess base, but the rate is extremely slow (TOF = 0.4 h−1). Complex [(OC)3Fe(pdt)(μ-dppf)Fe(CO)] [dppf = 1,1-bis(diphenylphosphino)ferrocene]491,492 (Fig. 30g) catalyses H2 oxidation in presence pyridine at the surface of an electrode.493 It catalytically oxidises H2 in presence of excess oxidant FcBArF4 and base P(o-tolyl)3,494 demonstrating that the redox active group need not to be directly attached to the diiron core to attain the catalytic turnover.494 Analogous complex [(CO)3Fe(pdt)(μ-dppp)Fe(CO)] {dppp = 1,2-bis(diphenylphosphanyl)propane} (Fig. 30h) lacking an internal base moiety on the phosphine ligand does not react with 1 atm H2 under similar conditions, illustrating the necessity of proton relay.
To enhance this hydrogen bonding stability of the peroxide intermediate, to stall its hydrolysis, a series of [FeFe]-hydrogenase mimics with ortho substituted arenes attached to bridgehead N atoms is reported (Fig. 29c–e) where the phenyl ring is rotated off the nitrogen lone pair. These complexes reduce proton at very low overpotential in the presence of O2 near neutral pH (pH 5.5).145 This class of 2Fe subsite mimics produces almost no H2O2 during competitive O2 reduction; rather they reduce O2 to H2O. These complexes are the first examples of synthetic models of hydrogenases that can operate unabated in the presence of O2.
[NiFe]-Hydrogenase mimics
In the past two and half decades, after the first crystallographic structure of a [NiFe]-hydrogenase was reported in 1995, there has been attempts to structurally and functionally mimic the active site structures of [NiFe]-hydrogenases.392 For many years it has been a key challenge for synthetic chemists to reproducing the structure and function of the active site of [NiFe]-hydrogenase in artificial mimics motivated by its original dissymmetric structure and its atypical reactivity. Though there are several NiFe model complexes reported in the literature,394,468,469,504–507 only a few could accurately mimics the structure, function and reactivity of the active site of [NiFe]-hydrogenase.508 The major limitation of most active site mimics is, unlike enzyme, their chemistry is mainly centred on the Fe site505 or on M (M = Mn, Ru) in heterodinuclear NiM models.509–513 The NiFe model complexes reported so far show the hydride ligand is either bridging511,513–518 but displaced toward the Fe site, or terminaly bound to the Fe centre.519–521 Rauchfuss and co-workers have reported a NiFe mimic displaying redox activity at the Ni site with the formation of a NiIFeII species. However, DFT calculations showed that in the corresponding protonated species the hydride ligand is bound to iron.522 Recently, Artero, Duboc and co-workers reported a heterodinuclear NiFe complex, LN2S2NiIIFeII which shows nickel-centred proton reduction. They were also able to isolate two derivatives that are involved as intermediates in the catalytic cycle that reproduce the structure and spectroscopic signatures of the Ni-R and Ni-L states.508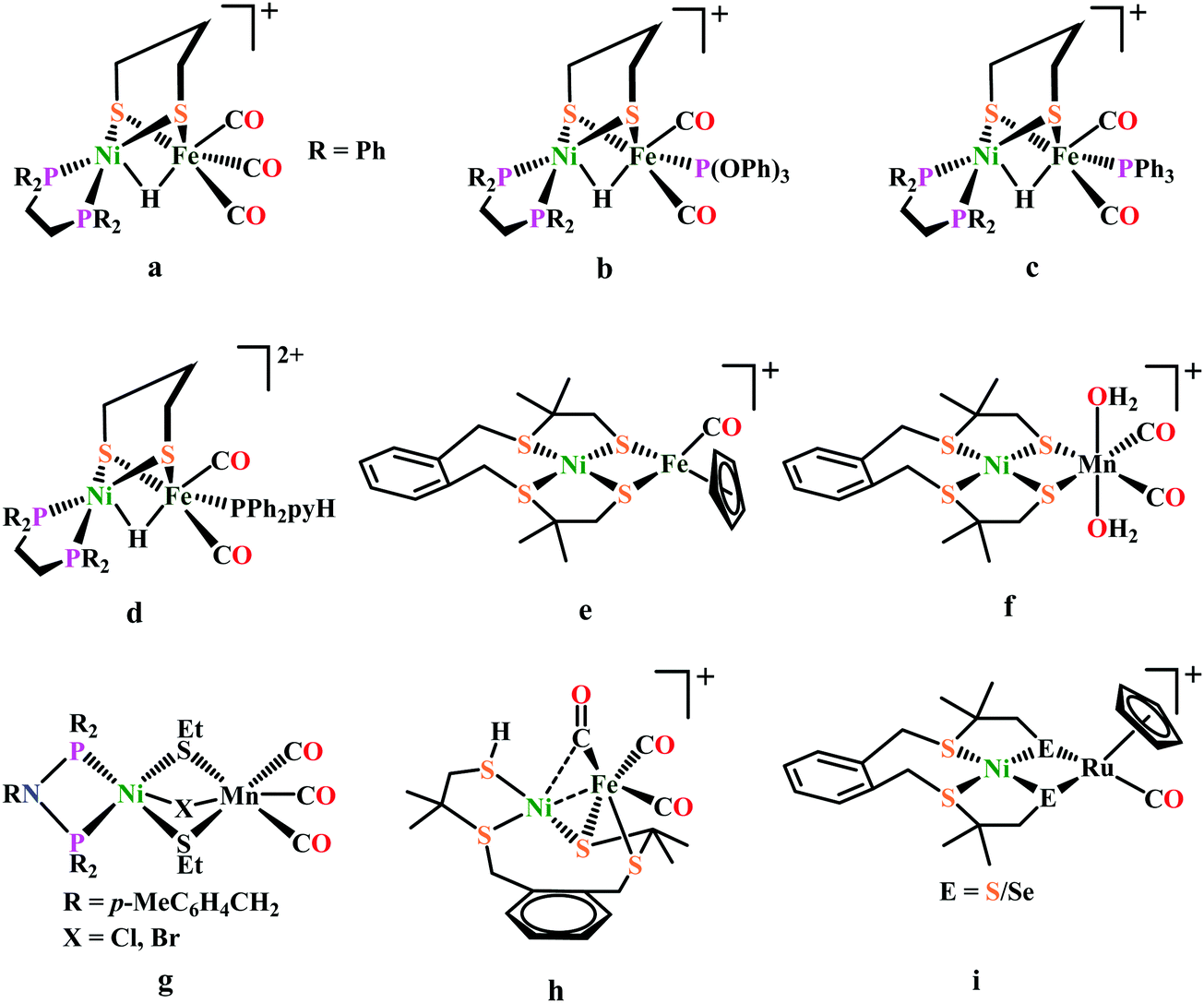 | ||
| Fig. 31 Structurally relevant, selected [NiFe]-hydrogenase mimics, active for electrocatalytic proton reduction. The only known [NiMn] systems (f and g) shows electrocatalytic proton reduction. | ||
In 2010, Fontecave, Artero and co-workers reported another functional model of [NiFe]-H2ase, with a S4 ligands framework around the nickel, like the enzyme and a cyclopentadienyl ligand on the iron center. The reaction of [Ni(xbsms)] with [CpFe(CO)2(thf)](BF4) in CH2Cl2 cleanly and rapidly yields [(xbsms)NiIIFeII(CO)2Cp]+. Upon exposure to light [(xbsms)NiIIFeII(CO)2Cp]+ loses one CO to form monocarbonyl species [(xbsms)NiIIFeII(CO)Cp]+ (Fig. 31e). Complex 31e was shown to catalyze H2 evolution from TFA in DMF with a 730 mV overpotential requirement. DFT calculations proposed that possible formation of a bridging hydride could occur through protonation at the CO ligand in the one-electron reduced state, followed by rearrangement and elimination of the CO ligand.523 A year later, the same group synthesized a dinuclear nickel–manganese complex [Ni(xbsms)Mn(CO)3(H2O)]+ (Fig. 31f) with the same ligand backbone around the nickel centre.51331f catalysed hydrogen evolution from TFA in DMF with an overpotential requirement of 860 mV, similar to complex 31e. Song et al. reported another series of Ni–Mn complexes, two of them (Fig. 31g) are active for H2 evolution from acetic acid in CH3CN512 with low catalytic current enhancement but reduced overpotential requirement (∼480 mV).
Weber et al. reported a structural and functional mimic of [NiFe]-H2ase with an unusual {S2Ni(μ-S)(μ-CO)Fe(CO)2S}-coordination environment around the metals (Fig. 31h). Interestingly, upon addition of 1 equivalent HBF4 to 31h, protonation took place at the terminal thiolate sulfur atom, that is coordinated to nickel, in a reversible way to produce protonated species [31H][BF4].523 This was the first experimental evidence of thiolate protonation, mimicking the postulated catalytic intermediates of [NiFe]-hydrogenases with a Cys-SH protonation.524–526 Both protonated and un-protonated species does proton reduction reaction from TFA in CH3CN at 540–570 mV overpotential.527 Using the analogous strategy, recently, Bouwman and co-workers reported two heterodinuclear NiRu complexes (Fig. 31i) one with bridging sulfur and another with bridging selenium. This complexes catalyze hydrogen evolution in the presence of acetic acid in acetonitrile solution with a overpotentials requirement of 810–830 mV.528 The same group also reported a NiFeSe complex with same ligand framework but here CO is ligated instead of PPh3. This complex was found to be active for hydrogen evolution from acetic acid in DMF solution.529 Ogo and co-workers reported a NiFe-mimic which was able to mediate both hydrogen evolution as well as oxidation in organic medium (CH3CN/CH3OH). This complex heterolytically activated H2 at atmospheric pressure and room temperature in presence of strong base, sodium methoxide, to form a hydride-bearing complex [NiII(X′)(μ-H)FeII{P(OEt)3}3]+ (Fig. 32a). The new hydride species thus formed is active for hydrogen evolution in presence of strong acid. So, this system shows promise as a catalyst for hydrogen evolution with a quite large overpotentials requirement.521
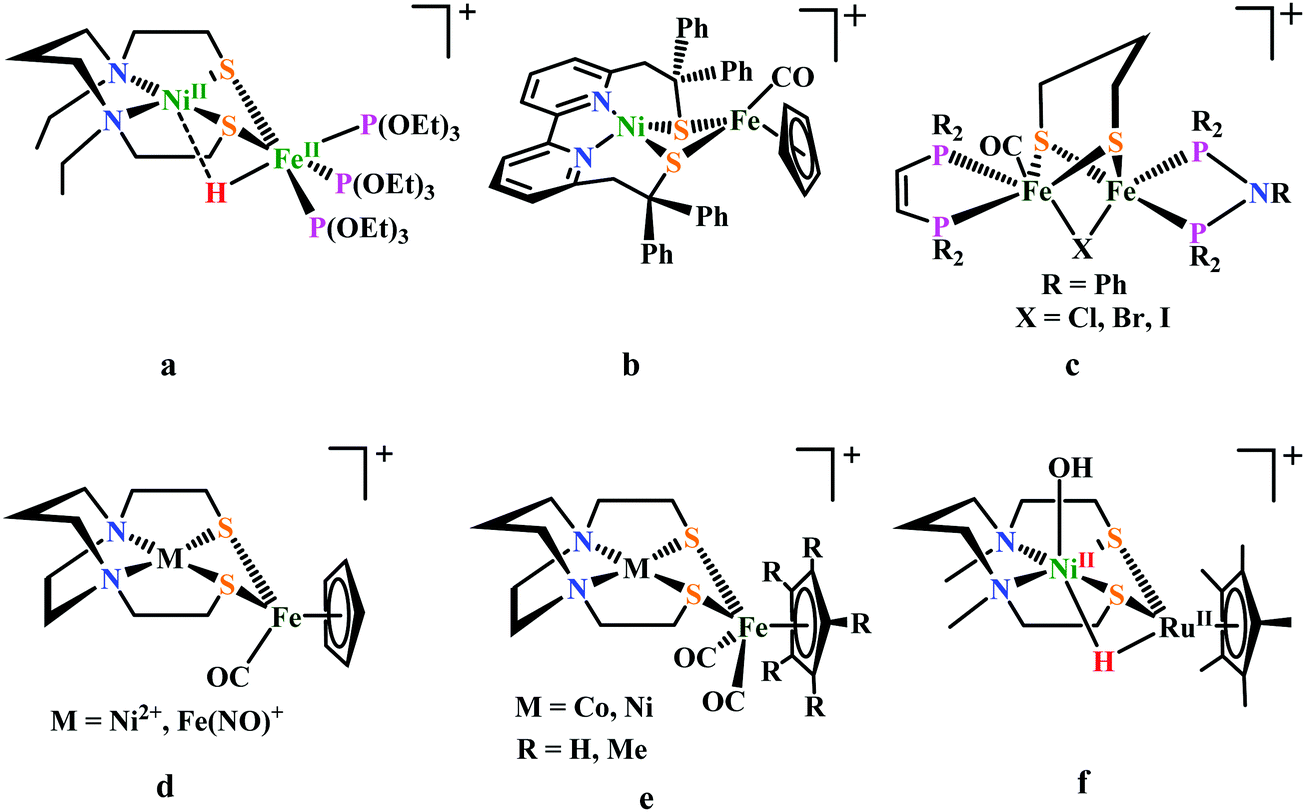 | ||
| Fig. 32 Selected NiFe-complexes as functional models of [NiFe]-hydrogenases (b–f); a is the NiFe-mimic active for the bidirectional conversion of protons and electrons into dihydrogen. | ||
Recently, Artero, Duboc and co-workers described a heterodinuclear NiFe complex, [LN2S2NiIIFeIIcp(CO)]+ (Fig. 32b) that shows ‘Ni’-centred proton reduction from [Et3NH][BF4] in CH3CN.508 In contrast to the NiFe complex containing terminal Fe(II)-hydride, reduction of 32b, with NaBH4 afforded a hydride complex, LN2S2NiII(H)FeII. Based on experimental evidences and theoretical calculations, it was proposed that LN2S2NiIIFeII first undergoes a one-electron reduction to generate LN2S2NiIFeII. This active species is further reduced to afford a bipyridine based redical anion, LN2S2·NiIFeII, which reacts with protons to yield a metal hydride intermediate akin to the metalloenzyme, LN2S2NiII(H)FeII. This metal hydride species further reacts with another equivalent of proton to produce H2 and regenerate the initial LN2S2NiIIFeII state (Scheme 32).40032b displays significant catalytic activity at a 500 mV overpotential from Et3NHBF4 in CH3CN solution. The second-order rate constant of proton reduction by this mimic is 2.5 × 104 M−1 s−1, which translates into a TOF of 250 s−1 at a concentration of 10 mM H+.508 Based on extensive DFT calculations, a year later, Tang et al. proposed that 32b does proton reduction following a much lower energy route via an E[ECEC] mechanism through an Fe-centred hydride intermediate.530 The same group in collaboration with others, synthesized a hybrid material NiFe@PCN-777 by incorporating the complex 32b into the Zr-based MOF PCN-777. FTO supported thin films of the NiFe@PCN-777 composite is active for proton reduction from Et3NHBF4 in CH3CN medium, inside the MOF cavities.531 The doubly reduced species of complex 32b is active for proton reduction in presence of weak acid like [Et3NH][BF4] while Ahmed et al. shown that in presence of strong acid like HBF4 singly reduced species is the active form for proton reduction in CH3CN medium.531
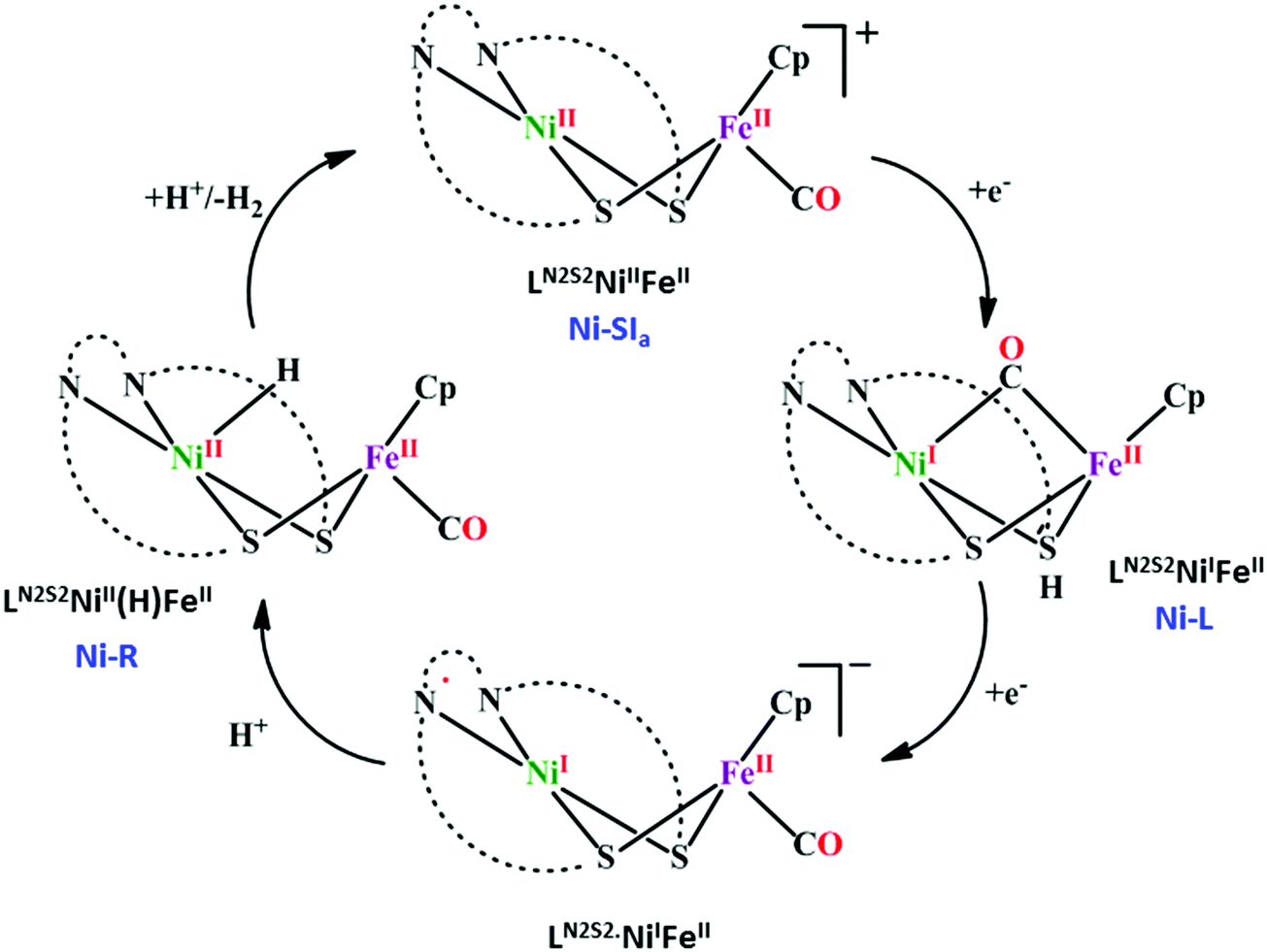 | ||
| Scheme 32 Proposed mechanistic cycle of proton reduction in complex [LN2S2NiIIFeIIcp(CO)]+ (Fig. 32b). | ||
To finely tune the reactivity of the [NiFe]-hydrogenase mimics Brazzolotto et al. modified the ligand environment, exclusively at the Fe site of complex 32b. Substitution of the Cp− (cyclopentadienyl) ligand by Cp*− (pentamethylcyclopentadienyl) to synthesize [LN2S2NiIIFeIIcp*(CO)]+ leads to remarkable modifications in its structural properties. Especially for the {Ni(μ-S)2Fe} core with a butterfly structure in [LN2S2NiIIFeIIcp(CO)]+ changes to a quasi-diamond structure in [LN2S2NiIIFeIIcp*(CO)]+. This new complex thus formed is also active for proton reduction in CH3CN medium in presence of [Et3NH][BF4] as a proton source. In the presence of CO, the electrocatalytic proton reduction activity of cp* species inhibits up to 90% while in case of complex 32b the inhibition is only 30%.532 A new series of the structural and functional models, [RN(PPh2)2Ni(μ-pdt)(μ-X)Fe(CO)(dppv)](BF4) (Fig. 32c) (R = p-MeC6H4CH2, EtO2CCH2 and X = Cl, Br, I) for the active site of [NiFe]-hydrogenases has been reported by Song and co-workers. These halogenido-bridged model complexes are active for H2 evolution from Cl2CHCO2H in CH3CN with an overpotential requirement of 530–570 mV. It was seen that the analogous chloro-bridged complexes are catalytically more active than bromo- or iodo-bridged.533
Recent electrochemical as well as theoretical studies by Hall, Darensbourg and co-workers of MN2S2 (M = Ni2+, Fe(NO)2+) bound to [(η5-C5H5)Fe(CO)]+ as electrocatalyst (Fig. 32d) demonstrated that in the electrochemical proton reduction process, reduction-induced hemi-lability of the bridging cis-dithiolates is a key step.534,535 For tuning the electronic character of the M(μ-S2)Fe core, the MN2S2·Fe(η5-C5R5)(CO) platform offers numerous possibilities. Modifying M within the metallodithiolate ligand, as well as replacing H by CH3 at the η5-C5R5 moiety increases the electron density at the Fe center, which might facilitate the reductive Fe–S bond cleavage. Although release of a free thiolate in these hemi-labile ligands creates a needed internal pendant base, this benefit might be countered by the increase in over-potential for addition of the first electron. Analogous study on MN2S2·[Fe(η5-C5R5)(CO)]+ (M = Ni2+, Co2+ and R = H, CH3) (Fig. 32e) complexes by Ghosh et al. shows that these catalysts are active for proton reduction in CH3CN medium in presence of TFA acid source. The overpotentials for the HER by these complexes are in the range of 1–1.3 V. The theoretical modelling suggests that the bridging thiol acts as a proton shuttle during catalysis.536 Different nitrosyl derivatives of NiFe complexes have also been synthesized to finely tune their catalytic hydrogen evolution activity in terms of TOF and overpotential.537
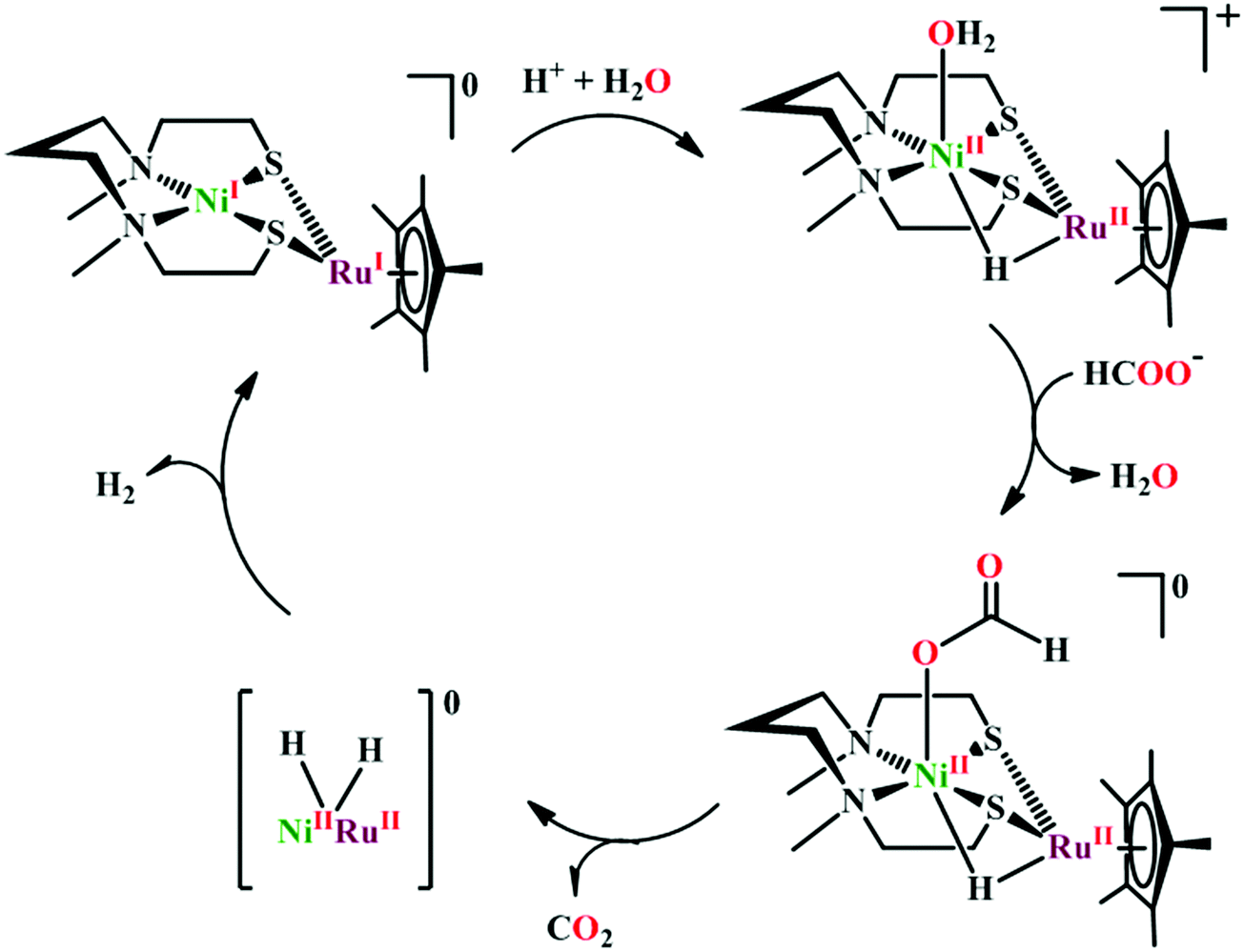 | ||
| Scheme 33 Proposed mechanism for proton reduction from formic acid catalyzed by NiRu complexes in acidic aqueous media. | ||
 | ||
| Scheme 34 Proposed mechanism of heterolytic H2 activation by complex 31a. | ||
 | ||
| Fig. 33 Selected examples of [NiFe]-hydrogenase mimics active for H2 activation. | ||
 | ||
| Fig. 34 Only known NiFe-complexes studied as a O2-tolerarent [NiFe]-hydrogenase mimics. | ||
Under aerobic conditions, the affinity of oxygen for sulfur and selenium, in [NiFeS]- and [NiFeSe]-hydrogenase, yields oxygenated chalcogens, and delays an inevitable and irreversible oxygen damage at the metals by maintaining the NiFe core structures. To identify the controlling features of S-site oxygen uptake, recently Darensbourg et al. synthesized a series of Ni(μ-EPhX)(μ-S′N2)Fe (E = S or Se, Fe = (η5-C5H5)FeII(CO)) (Fig. 34b) complexes and tuned them electronically by changing the para-substituent on μ-EPhX (X = CF3, Cl, H, OMe, NMe2). DFT analysis suggests that the more electron-rich sulfurs are more O2 responsive in the SPhX series; the selenium analogues were even more reactive with O2.545 In another study the same group described a biomimetic study for S/Se oxygenation in Ni(μ-EPh)(μ-SN2)Fe, (E = S or Se); Fe = (η5-C5H5)FeII(CO) complexes related to the oxygen-damaged active sites of [NiFeS]/[NiFeSe]-hydrogenases. Upon O2 exposure, it forms monooxygenated and dioxygenated species, and treatment with O-abstraction agents such as P(o-tolyl)3 or PMe3 reversed the oxygenation of the S/Se atoms. Although, NiEPhFe+ complex differs from the [NiFeSe]-hydrogenase active site as the selenium in the model is in a bridging position, rather than terminal as nature has adopted, the relative reactivities are consistent with what is found in nature.546 Very recently, Song and co-workers reported two S-oxygenated intermediate [(mmp-sulfeno-dach)Ni][Fe′X] (Fe′X = Fe(CO)4, Fe(CO)2cp) (Fig. 34c) of an O2-tolerant [NiFe]-hydrogenase mimics analogues to Driess's system. Reactivity study on both complexes indicates that they undergo the ligand dissociation reactions in the presence of Me3NO to give their precursor complex, while complex with cp ligand undergoes a complicated sequential reaction in the presence of organometallic oxidant FcBF4 to afford the trinuclear Ni(II) complex, unexpectedly.546
Ni(II) complexes. Inspired by the pendant azadithiolate ligand in the active site of [FeFe]-hydrogenase, a series of functional models of mononuclear nickel complexes based on bis diphosphine ligands with pendent amines [Ni(PR2NR′2)2]2+have been synthesized.547–549 This mononuclear Ni complexes having beneficial acid–base and redox qualities permits mediation of both H2 oxidation and production (Fig. 35).547–550
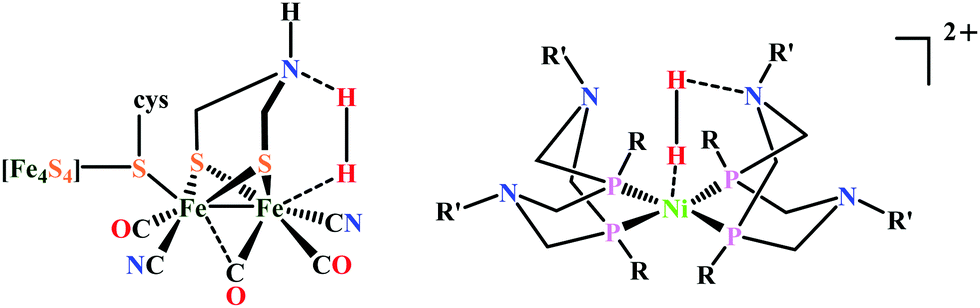 | ||
| Fig. 35 [FeFe]-hydrogenase active site (left) and [Ni(PR2NR′2)2]2+ molecular catalyst (right), H2 bound in each case in the active site. | ||
Catalytic oxidation of H2 by Ni complexes. A Ni complex without proton relays, [Ni(depp)2]2+ {depp = 1,2-bis(diethylphosphanyl) propane} (Fig. 36a) slowly catalyses the oxidation of H2 in the presence of Et3N.417 The NiII hydride complex [HNi(depp)2]2+, without proton relay, shows an irreversible oxidation peak at 0.0 V. But introducing a proton relay into the ligand by the form of a PNP (PNP = Et2PCH2NMeCH2PEt2) ligand [Ni(PNP)2]2+ (Fig. 36b) is obtained. The oxidation peak of [HNi(PNP)2]2+ is shifted to negative potential (−0.62 V). So, by introducing a PNP ligand as a proton relay leads to a much lower energy pathway for oxidation of H2. The [Ni(PNP)2]2+ complex has an overpotential of about 100 mV, that is 600 mV less than that observed with [Ni(depp)2]2+ for electrocatalytic H2 oxidation. But rate of catalysis was still slow. The incorporation of a pendant amine group into the PNP ligand triggers a notable decrease in the overpotential for the electrocatalytic H2 oxidation. Like the cyclohexane here the six-membered ring Ni–PNP go through chair/boat conformational transitions. But only in the boat conformation the pendant amine is well positioned to interact with a Ni–H bond. But from a crystal structure of [Ni(PNP)2]2+ derivative (with an nBu group on each N, Fig. 36c) it is seen that the both six-membered rings are in the chair form. So the energy of conversion of the chair form to the boat form contributes to the kinetic barrier for oxidation of H2 by [Ni(PNP)2]2+. At least one of the pendant amines adopts boat conformation when a second six-membered ring into the cyclic “P2N2” ligand structure of Ni(P2N2) complexes is added. By virtue of this the pendent amine becomes properly positioned to interact with a H− or H2 ligand on the metal.417 By varying the alkyl or aryl group bound to P or N atoms in Ni(P2N2) the steric and electronic properties can be tuned so that certain reactions are favoured. The calculated TOF of [Ni(PCy2NBn2)2]2+(Cy = cyclohexyl; Bn = benzyl) (Fig. 36d) complex for electrocatalytic H2 (1 atm) oxidation is about 10 s−1, which is much higher than that observed with [Ni(PNP)2]2+. A similar complex containing one P2N2 ligand, [Ni(PCy2NBn2)(dppp)]2+{dppp = 1,2-bis(diphenylphosphanyl)propane} catalyses the H2 oxidation of with a TOF of 0.4 s−1 which much less than that compared to the [Ni(PCy2NBn2)2]2+ due to with two well positioned pendant amines in later complex.417 The TOF increases with the number of well positioned pendant amines, indicating that the pendant amines facilitate the binding of H2. However theoretical studies on [Ni(PMe2NMe2)2]2+, reveals that only one pendant amine is directly associated in the heterolytic binding or cleavage of H2.417 The calculated TOF for the Oxidation of H2 (1 atm) by [Ni(PCy2NtBu2)2]2+ (tBu = tert-butyl) (Fig. 36e) Complex is 50 s−1 at −0.77V vs. Fc+/Fc in presence of Et3N.551,552 The higher TOF value for this complex compared to that for [Ni(PCy2NBn2)2]2+ is because of the higher basicity of nitrogen atoms attached with tert-butyl groups than those of benzyl groups.
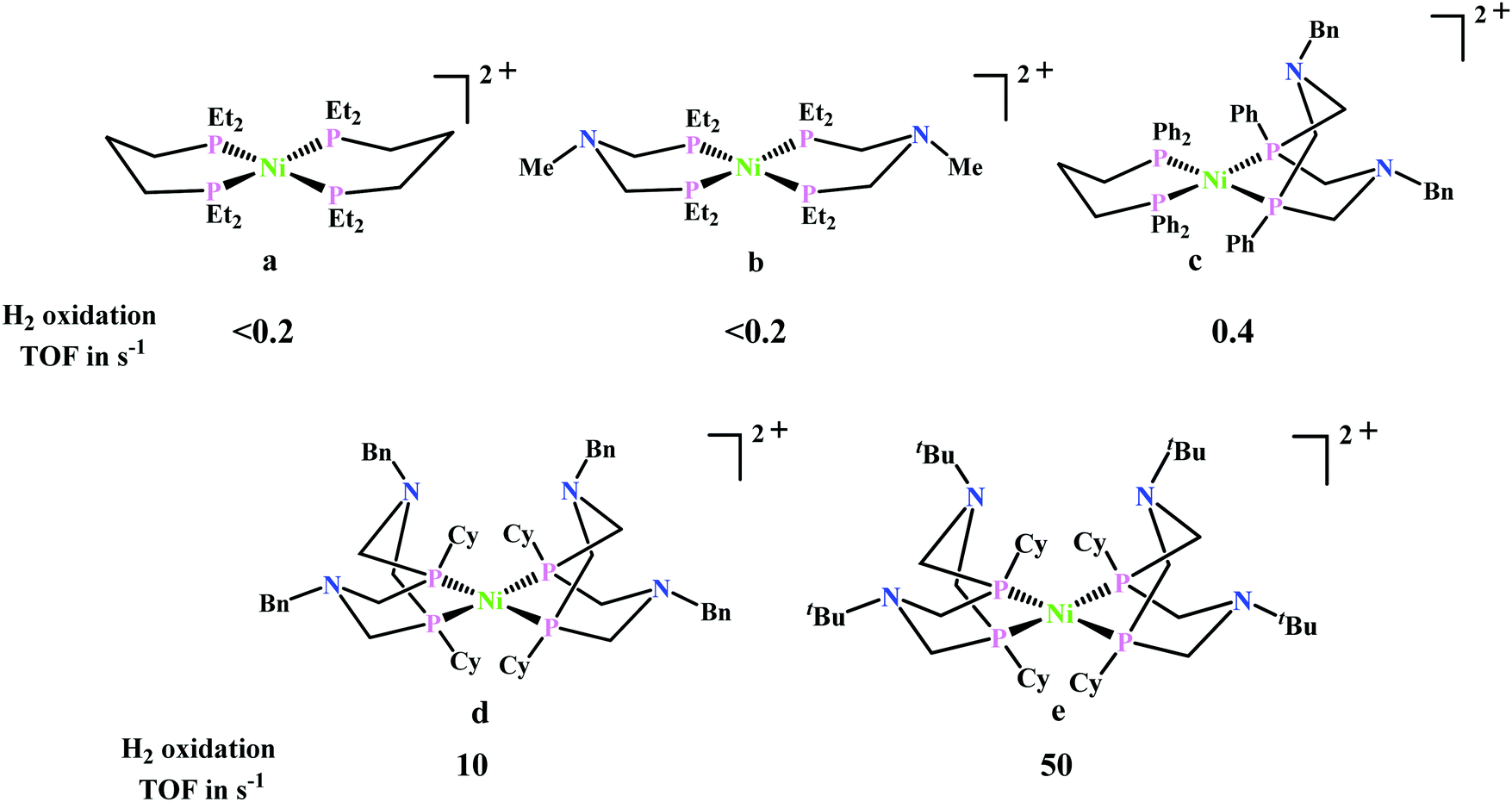 | ||
| Fig. 36 Ni(II) molecular complexes catalysing H2 oxidation. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OEt)2, phosphonate] the fastest rates are observed in presence of water. The rate of H2 production from H–DMF+ for this complex is 500 s−1 without added water. But in presence of H2O it displays a much higher rate of 1850 s−1. Due to the presence of four phosphonate groups, this complex encounter additional H-bonding interactions with H2O.553
O)(OEt)2, phosphonate] the fastest rates are observed in presence of water. The rate of H2 production from H–DMF+ for this complex is 500 s−1 without added water. But in presence of H2O it displays a much higher rate of 1850 s−1. Due to the presence of four phosphonate groups, this complex encounter additional H-bonding interactions with H2O.553
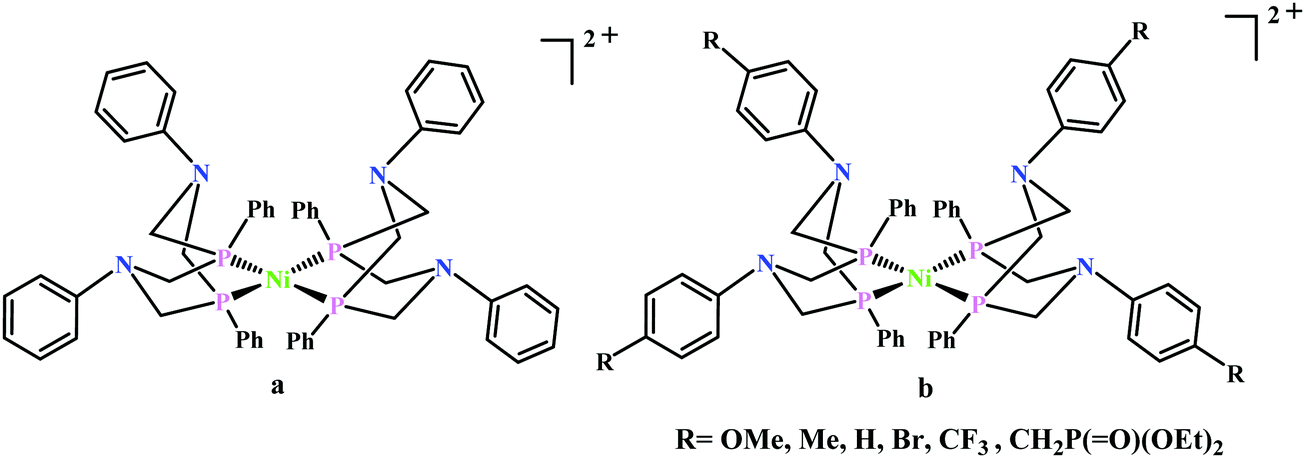 | ||
| Fig. 37 Proton reducing Ni(II) catalysts. | ||
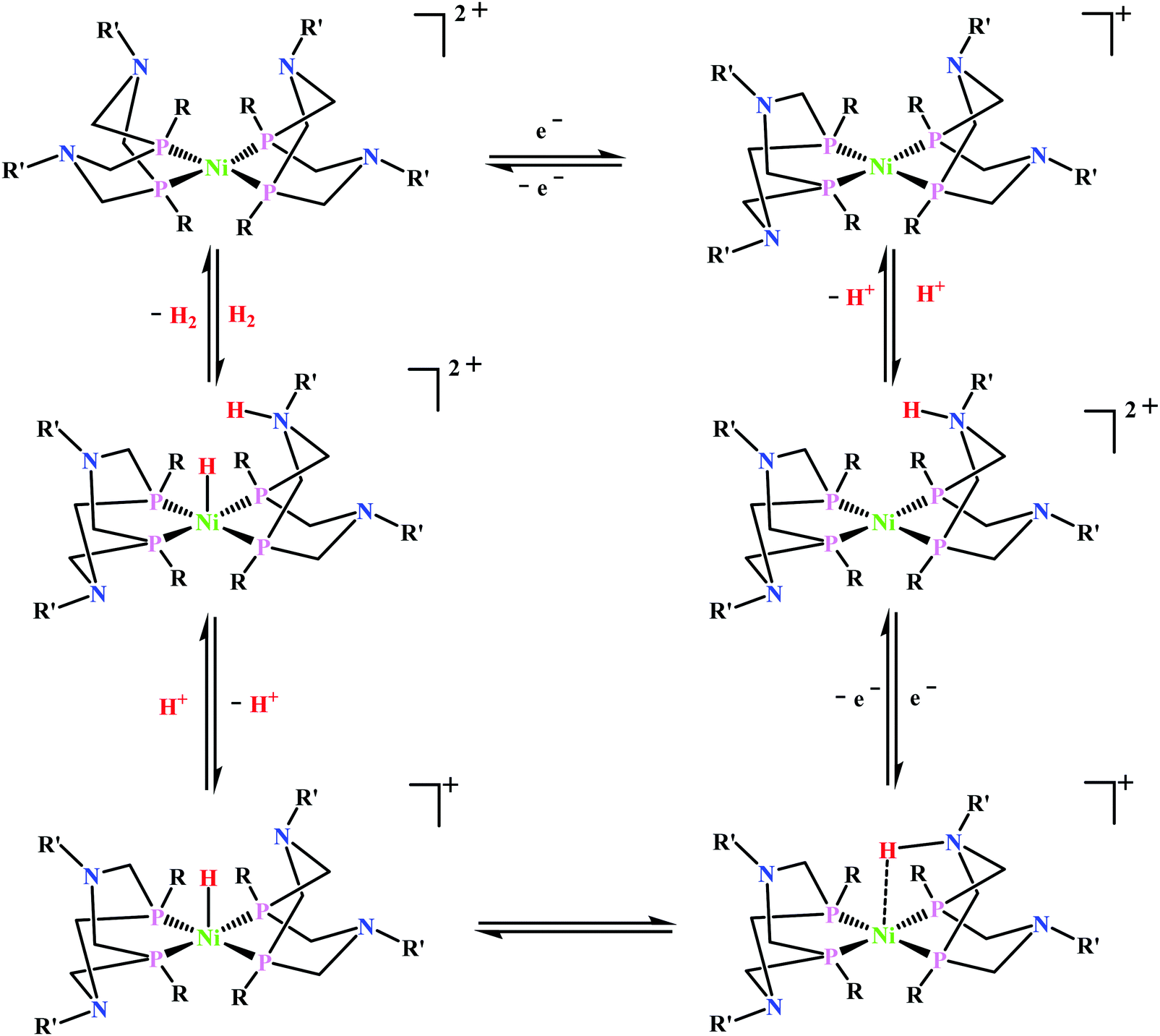 000 s−1 for complex 9. With the addition of water the catalytic current increases. In presence of 1.2 M water the TOF for the catalytic proton reduction catalysed by complex 9 is found to be 106000 s−1, which exceeds the value those reported for [Fe–Fe] hydrogenase enzyme (TOF = 9000 s−1 at 30 °C).557
000 s−1 for complex 9. With the addition of water the catalytic current increases. In presence of 1.2 M water the TOF for the catalytic proton reduction catalysed by complex 9 is found to be 106000 s−1, which exceeds the value those reported for [Fe–Fe] hydrogenase enzyme (TOF = 9000 s−1 at 30 °C).557
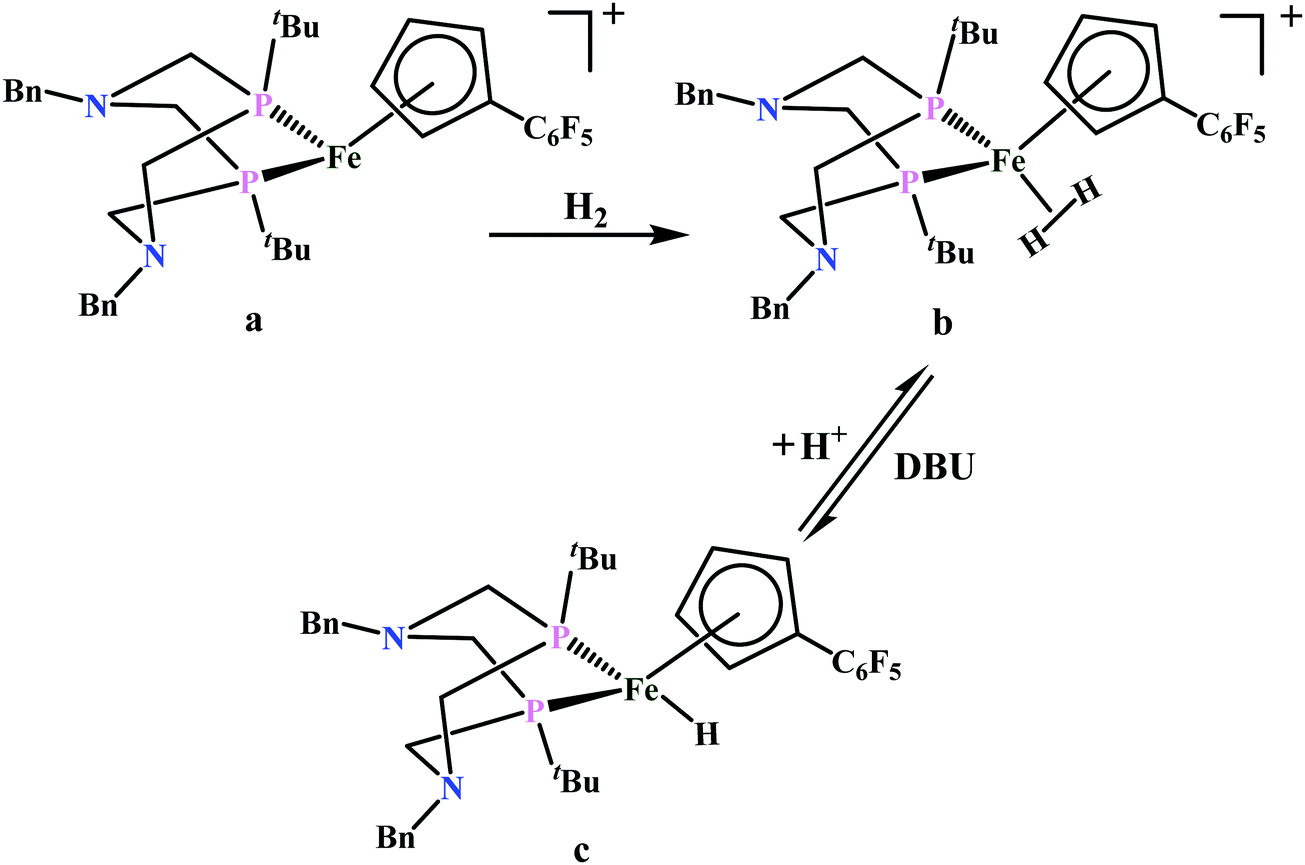 | ||
| Scheme 36 Reaction of [(CpC6F5)Fe(PtBu2NBn2)]+ with H2. | ||
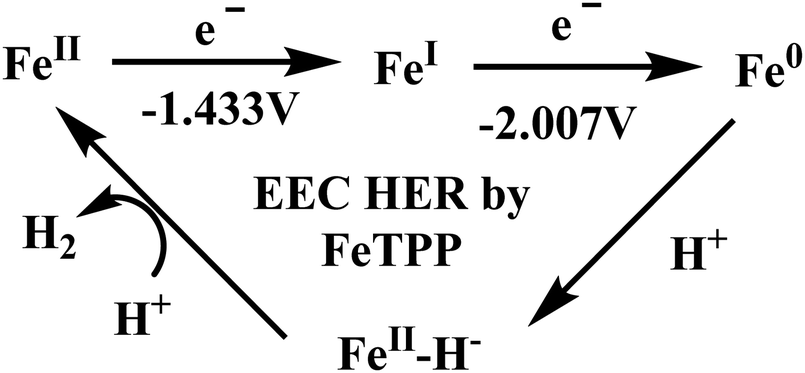 | ||
| Scheme 37 Probable mechanism of the HER catalysed by FeTPP. | ||
Three iron porphyrins namely FeFc4, FeFc1, and Fe(tBu)4 (where Fc and tBu represent bulky and hydrophobic ferrocene and tert-butyl substituents on the triazole ring, respectively, Fig. 38) with second sphere distal triazole residues are reported to show hydrogen evolution reaction in their Fe(I) state in both CH3CN medium in presence of TsOH (proton source) and in aqueous buffer solution when the catalyst is physiadsorbed on EPG and the pH of the buffer solution is below the pKa of triazole with the increase in the acid concentration the catalytic current at −1.44 V saturates.560 While Iron porphyrins without any such secondary structure (FeTPP) can catalyse HER only in their Fe(0) state in both organic and aqueous medium. But with increasing acid concentration the catalytic current increases instead of saturation. The Fe(I) state of FeTPP can catalyse HER under homogeneous condition only in presence of PPh3. The distal triazole residues of FeFc4, FeFc1, and Fe(tBu)4 complexes gets protonated in presence of TsOH. DFT calculations suggests that upon protonation the Fe(I) center produces an FeIII–H− species which gets stabilized by these protonated triazoles through dihydrogen bond formation. Thus, only after the protonation of the distal triazole(s) HER can be catalysed by the Fe(I) state. Based on these results it can be concluded that having a pendant base in the second sphere in iron porphyrins provide a thermodynamic advantage by increasing the pKa of the metal by enhancing the proton affinity by forming dihydrogen bonding (Scheme 38).560
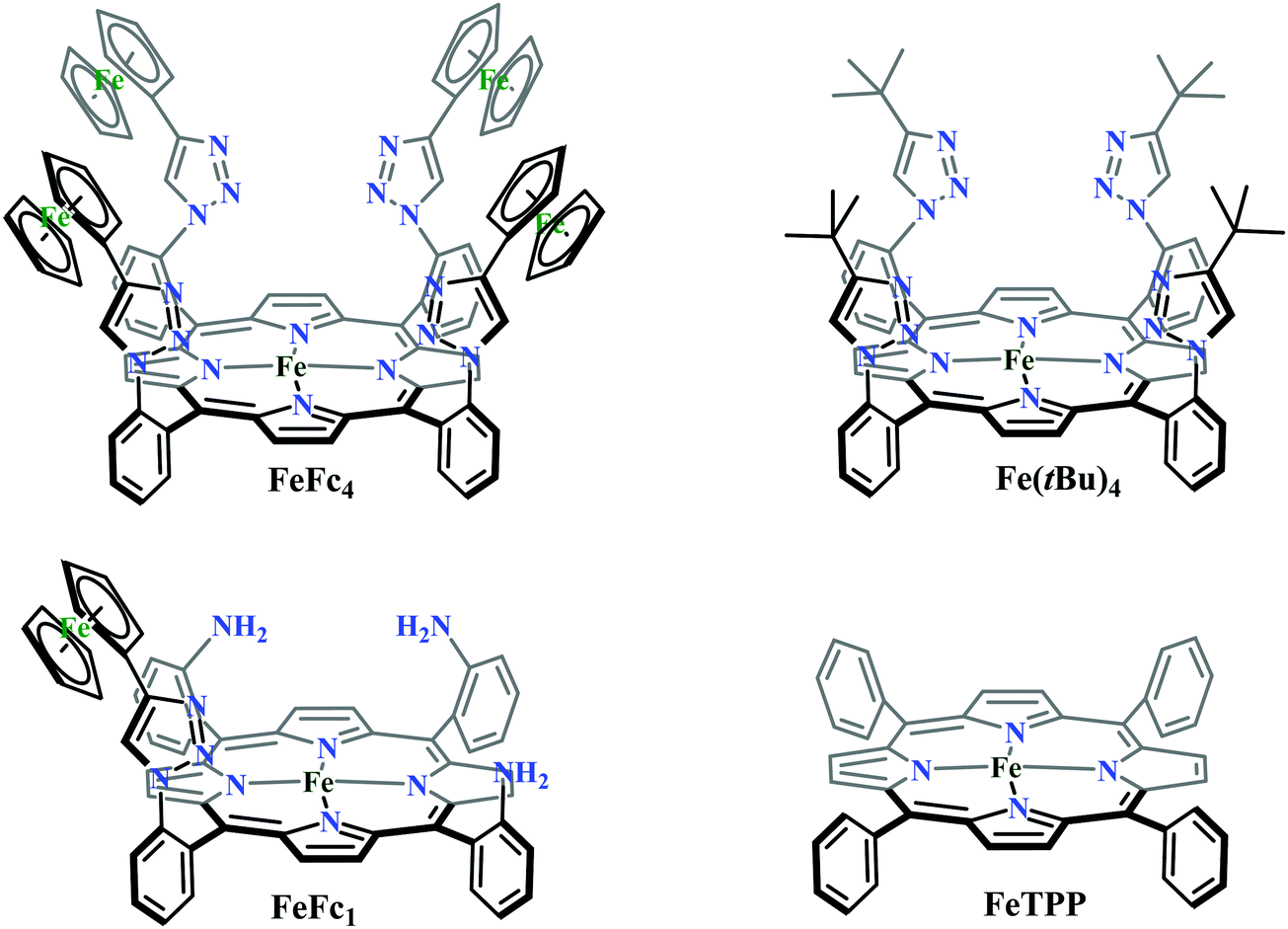 | ||
| Fig. 38 Structures of Fe porphyrins complexes showing HER. | ||
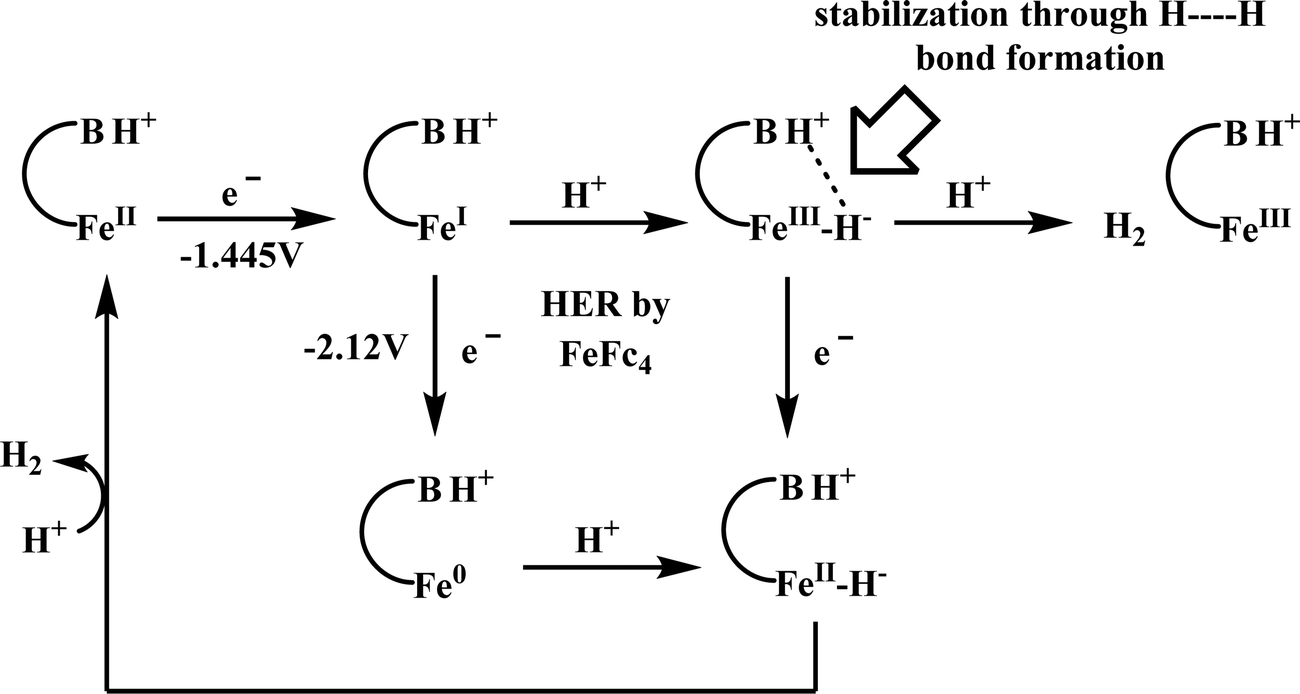 | ||
| Scheme 38 Probable mechanism of the HER catalysed by Fe porphyrins containing distal triazole basic residues. | ||
General conclusion and outlook
The natural systems have evolved to maximize efficiency for a given catalytic transformation and they exhibit high rates without compromising the selectivity. One needs to look deeper than the immediate structural disposition of a naturally occurring enzymatic active site, keeping in mind the function its primed for, to identify additional attributes that allow these metallo enzymes to access the efficiency they display. Often, for the small molecule reductions discussed here, this includes spatial and temporal control of delivery of protons and electrons to the active site. The metalloenzymes discussed here provide abundant examples of how the control over proton and electron transfer is crucial to maintain product selectivity in these transformations. The prevalent approach in developing small molecule catalysts for the activation of CO2 relies heavily on past accomplishments in organometallic chemistry.572 While this has resulted in several 2nd and 3rd row transition metal-based systems with encouraging rates and selectivity, recent developments have highlighted how high rates and selectivities can be accessed by being bio-inspired. Although there are several examples of establishing well-designed proton conduits in the catalysts, electron transfer rates are yet to be controlled to yield tangible advantages to the overall catalysis process.A thorough understanding of the mechanism of the metalloenzymes along with electronic structure function correlations can lead to the development of efficient 1st row transition metal catalysts for these tasks of contemporary importance. This requires detailed understanding and integration of the 2nd sphere effects in the design of the artificial catalysts. This is beautifully demonstrated in hydrogen evolution catalyzed by synthetic mimics and bio-inspired models of hydrogenases where bi-directional catalysis with practicable rates have been demonstrated.425 Gradually such approaches are increasingly being deployed to handle CO2 and NO2− reduction. The inclusion of hydrogen bonding residues in CO2 reduction has resulted not only in facile catalysis but also in much desired selectivity. This is demonstrated in the work by Saveant, Dey, Marinescu, Machan and others. Similarly, while there have been remarkable developments in the understanding of 2nd sphere residues in O2 reduction by iron porphyrins, these effects are yet to be realized in NO2− reduction.136,329 However, progress has been slow primarily because of the difficulty involved in designing the ligand. The electronic retrosynthesis approach discussed in this review may aid such design.
Selectivity is crucial for controlling the desired product from the reduction of these species. For example, CO2 can be reduced to CO, HCOOH, H2CO, CH3OH or CH4. Similarly, NO2− can be reduced to NO, HNO, N2, N2H4 and N2. Logically, selectivity over these processes are desirable. Here too, understanding the mechanism of the metalloenzymes can provide solutions as discussed in this review. Going forward one needs to address means to handle competing reactions. For example, the reduction of proton to hydrogen competes with the reduction of CO2. Similarly, the reduction of O2 competes with the reduction of protons, CO2 as well as NO2− as the reduction of O2 is thermodynamically more favourable than the reduction of the rest. While the competition between CO2 reduction and proton reduction has received some attention, the literature is scanty in the area of O2 tolerance, with a few rare instances of such selecitivity. Yet, practical application of any of these technologies will require maintaining catalytic performance in the presence of O2 which is abundant in the atmosphere and its removal will only increase the cost of any catalytic process. For instance, direct reduction of CO2 is severely limited by the cost of capturing CO2 from atmosphere. Alternatively, if CO2 present in the flue gas (8–15%) can be reduced, the cost of CO2 capture can be subverted. This will require the catalyst to be tolerant to O2. Unfortunately, the sensitivity to O2 is a limitation of the natural enzymes as well as many of them are inhibited by O2 albeit reversibly in some handful cases. Thus, this is an area that will require creative solutions above and beyond what exists and can be learnt from naturally occurring enzymes. The Dey group has established a few examples of O2 tolerant H+ and CO2 reduction catalysts.145,147,573 Finally, it is fair enough to assume that robust catalysts for these important multi-proton and multi-electron reactions are likely to be based on heterogeneous materials. Thus, a transition of the building knowledge in the area of bio-inspired molecular catalysts to smart materials is warranted.
Conflicts of interest
There are no conflicts to declare.References
- A. P. Ballantyne, C. B. Alden, J. B. Miller, P. P. Tans and J. W. C. White, Nature, 2012, 488, 70–72 CrossRef CAS PubMed.
- A. G. Chmielewski, Encyclopedia of Life Support Systems, 2008 Search PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- Bulletin of the American Meteorological Society, ed. J. Blunden and D. S. Arndt, 2020, vol. 101, pp. S1–S429 Search PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- K. S. Lackner, Annu. Rev. Energy Environ., 2002, 27, 193–232 CrossRef.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- H.-R. “Molly” Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC.
- Y. Hori, Handb. Fuel Cells, 2010 Search PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- M. P. Johnson, Essays Biochem., 2016, 60, 255–273 CrossRef PubMed.
- C. Welte and U. Deppenmeier, in Methods in Methane Metabolism, Part A, ed. A. C. Rosenzweig and S. W. B. T.-M. E. Ragsdale, Academic Press, 2011, vol. 494, pp. 257–280 Search PubMed.
- J. L. Peel, R. Haeuber, V. Garcia, A. G. Russell and L. Neas, Biogeochemistry, 2013, 114, 121–134 CrossRef CAS.
- M. Graetzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- E. Lamy, L. Nadjo and J. M. Saveant, J. Electroanal. Chem. Interfacial Electrochem., 1977, 78, 403–407 CrossRef CAS.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J. A. Bassham, A. A. Benson and M. Calvin, J. Biol. Chem., 1950, 185, 781–787 CrossRef CAS.
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1873–1898 CrossRef CAS PubMed.
- B. B. Buchanan and D. I. Arnon, Photosynth. Res., 1990, 24, 47–53 CrossRef CAS PubMed.
- M. C. Evans, B. B. Buchanan and D. I. Arnon, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 928–934 CrossRef CAS PubMed.
- G. Fuchs, Annu. Rev. Microbiol., 2011, 65, 631–658 CrossRef CAS PubMed.
- V. Svetlitchnyi, C. Peschel, G. Acker and O. Meyer, J. Bacteriol., 2001, 183, 5134–5144 CrossRef CAS PubMed.
- B. Zhang, C. F. Hemann and R. Hille, J. Biol. Chem., 2010, 285, 12571–12578 CrossRef CAS PubMed.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15971–15976 CrossRef CAS PubMed.
- S. W. Ragsdale and M. Kumar, Chem. Rev., 1996, 96, 2515–2540 CrossRef CAS PubMed.
- Z. Hu, N. J. Spangler, M. E. Anderson, J. Xia, P. W. Ludden, P. A. Lindahl and E. Münck, J. Am. Chem. Soc., 1996, 118, 830–845 CrossRef CAS.
- Y. Kung and C. L. Drennan, Curr. Opin. Chem. Biol., 2011, 15, 276–283 CrossRef CAS PubMed.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567–572 CrossRef CAS PubMed.
- H. Dobbek, V. Svetlitchnyi, J. Liss and O. Meyer, J. Am. Chem. Soc., 2004, 126, 5382–5387 CrossRef CAS PubMed.
- Y. Kung, T. I. Doukov, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Biochemistry, 2009, 48, 7432–7440 CrossRef CAS.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- V. J. DeRose, J. Telser, M. E. Anderson, P. A. Lindahl and B. M. Hoffman, J. Am. Chem. Soc., 1998, 120, 8767–8776 CrossRef CAS.
- E. J. Kim, J. Feng, M. R. Bramlett and P. A. Lindahl, Biochemistry, 2004, 43, 5728–5734 CrossRef CAS PubMed.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11973–11978 CrossRef CAS PubMed.
- B. Mondal, J. Song, F. Neese and S. Ye, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS PubMed.
- I. G. Shabalin, K. M. Polyakov, V. I. Tishkov and V. O. Popov, Acta Nat., 2009, 1, 89–93 CrossRef CAS.
- E. V. Filippova, K. M. Polyakov, T. V. Tikhonova, T. N. Stekhanova, K. M. Boiko and V. O. Popov, Crystallogr. Rep., 2005, 50, 796–800 CrossRef CAS.
- R. K. Thauer, FEBS Lett., 1972, 27, 111–115 CrossRef CAS.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- J. C. Boyington, V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman and P. D. Sun, Science, 1997, 275, 1305–1308 CrossRef CAS PubMed.
- H. C. A. Raaijmakers and M. J. Romão, JBIC, J. Biol. Inorg. Chem., 2006, 11, 849–854 CrossRef CAS PubMed.
- V. N. Gladyshev, J. C. Boyington, S. V. Khangulov, D. A. Grahame, T. C. Stadtman and P. D. Sun, J. Biol. Chem., 1996, 271, 8095–8100 CrossRef CAS.
- H. Dobbek, Coord. Chem. Rev., 2011, 255, 1104–1116 CrossRef CAS.
- H. Raaijmakers, S. Macieira, J. M. Dias, S. Teixeira, S. Bursakov, R. Huber, J. J. G. Moura, I. Moura and M. J. Romão, Structure, 2002, 10, 1261–1272 CrossRef CAS.
- S. V. Khangulov, V. N. Gladyshev, G. C. Dismukes and T. C. Stadtman, Biochemistry, 1998, 37, 3518–3528 CrossRef CAS PubMed.
- G. N. George, C. M. Colangelo, J. Dong, R. A. Scott, S. V. Khangulov, V. N. Gladyshev and T. C. Stadtman, J. Am. Chem. Soc., 1998, 120, 1267–1273 CrossRef CAS.
- C. S. Mota, M. G. Rivas, C. D. Brondino, I. Moura, J. J. G. Moura, P. J. González and N. M. F. S. A. Cerqueira, JBIC, J. Biol. Inorg. Chem., 2011, 16, 1255–1268 CrossRef CAS PubMed.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636–637 RSC.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- B. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 3, 328–330 RSC.
- J. Hawecker, J. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- C. Mark Bolinger, B. Nicole Story, P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582–4587 CrossRef.
- M. R. Bruce, E. Megehee, B. P. Sullivan, H. Thorn, T. R. O’Toole, A. Downard and T. J. Meyer, Organometallics, 1988, 7, 238–240 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. M. Miller, J. M. Mayer and C. P. Kubiak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 CrossRef CAS PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 1386–1393 CrossRef CAS PubMed.
- A. Zhanaidarova, H. Steger, M. H. Reineke and C. P. Kubiak, Dalton Trans., 2017, 46, 12413–12416 RSC.
- M. L. Clark, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Chem. Sci., 2014, 5, 1894–1900 RSC.
- J. Agarwal, T. W. Shaw, C. J. Stanton, G. F. Majetich, A. B. Bocarsly and H. F. Schaefer, Angew. Chem., Int. Ed., 2014, 53, 5152–5155 CrossRef CAS.
- F. Franco, C. Cometto, F. F. Vallana, F. Sordello, E. Priola, C. Minero, C. Nervi and R. Gobetto, Chem. Commun., 2014, 50, 14670–14673 RSC.
- N. P. Liyanage, H. A. Dulaney, A. J. Huckaba, J. W. Jurss and J. H. Delcamp, Inorg. Chem., 2016, 55, 6085–6094 CrossRef CAS PubMed.
- K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, D. C. Grills, M. Z. Ertem and J. Rochford, J. Am. Chem. Soc., 2017, 139, 2604–2618 CrossRef CAS PubMed.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- S. Sung, X. Li, L. M. Wolf, J. R. Meeder, N. S. Bhuvanesh, K. A. Grice, J. A. Panetier and M. Nippe, J. Am. Chem. Soc., 2019, 141, 6569–6582 CrossRef CAS PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451–12456 CrossRef CAS PubMed.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. MacHan, Inorg. Chem., 2018, 57, 2111–2121 CrossRef CAS PubMed.
- A. N. Hellman, R. Haiges and S. C. Marinescu, Dalton Trans., 2019, 48, 14251–14255 RSC.
- D. Z. Zee, M. Nippe, A. E. King, C. J. Chang and J. R. Long, Inorg. Chem., 2020, 59, 5206–5217 CrossRef CAS PubMed.
- S. Slater and J. H. Wagenknecht, J. Am. Chem. Soc., 1984, 106, 5367–5368 CrossRef CAS.
- A. Szymaszek and F. P. Pruchnik, J. Organomet. Chem., 1989, 376, 133–140 CrossRef CAS.
- D. L. Dubois, Comments Inorg. Chem., 1997, 19, 307–325 CrossRef CAS.
- A. M. Brun and A. Harriman, J. Am. Chem. Soc., 1991, 113, 8153–8159 CrossRef CAS.
- B. D. Steffey, A. Miedaner, M. L. Maciejewski-Farmer, P. R. Bernatis, A. M. Herring, V. S. Allured, V. Carperos and D. L. DuBois, Organometallics, 1994, 13, 4844–4855 Search PubMed.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed.
- A. Miedaner, C. J. Curtis, R. M. Barkley and D. L. DuBois, Inorg. Chem., 1994, 33, 5482–5490 CrossRef CAS.
- B. D. Steffey, C. J. Curtis and D. L. DuBois, Organometallics, 1995, 14, 4937–4943 CrossRef CAS.
- A. Miedaner, B. C. Noll and D. L. DuBois, Organometallics, 1997, 16, 5779–5791 CrossRef CAS.
- D. L. Delaet, R. Del Rosario, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1987, 109, 754–758 CrossRef CAS.
- E. Simón-Manso and C. P. Kubiak, Organometallics, 2005, 24, 96–102 CrossRef.
- R. J. Haines, R. E. Wittrig and C. P. Kubiak, Inorg. Chem., 1994, 33, 4723–4728 CrossRef CAS.
- D. A. Morgenstern, G. M. Ferrence, J. Washington, J. I. Henderson, L. Rosenhein, J. D. Heise, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1996, 118, 2198–2207 CrossRef CAS.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- C. S. Seu, A. M. Appel, M. D. Doud, D. L. Dubois and C. P. Kubiak, Energy Environ. Sci., 2012, 5, 6480–6490 RSC.
- B. R. Galan, M. L. Reback, A. Jain, A. M. Appel and W. J. Shaw, Eur. J. Inorg. Chem., 2013, 5366–5371 CrossRef CAS.
- B. R. Galan, J. Schöffel, J. C. Linehan, C. Seu, A. M. Appel, J. A. S. Roberts, M. L. Helm, U. J. Kilgore, J. Y. Yang, D. L. Dubois and C. P. Kubiak, J. Am. Chem. Soc., 2011, 133, 12767–12779 CrossRef CAS.
- L. Chen, Z. Guo, X. G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K. C. Lau, T. C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 CrossRef CAS.
- A. Taheri and L. A. Berben, Inorg. Chem., 2016, 55, 378–385 CrossRef CAS PubMed.
- D. Lexa, J. M. Savéant and D. L. Wang, Organometallics, 1986, 5, 1428–1434 CrossRef CAS.
- D. Lexa, J. M. Saveant, K. B. Su and D. L. Wang, J. Am. Chem. Soc., 1988, 110, 7617–7625 CrossRef CAS.
- C. Gueutin, D. Lexa, J. M. Savéant and D. L. Wang, Organometallics, 1989, 8, 1607–1613 CrossRef CAS.
- D. Lexa, J. M. Savéant, K. B. Su and D. L. Wang, J. Am. Chem. Soc., 1987, 109, 6464–6470 CrossRef CAS.
- D. Lexa, J. M. Savéant, K. B. Su, D. L. Wang, H. J. Schäfer and B. Vering, J. Am. Chem. Soc., 1990, 112, 6162–6177 CrossRef CAS.
- M. Hammouche, D. Lexa, J. M. Savéant and M. Momenteau, J. Electroanal. Chem., 1988, 249, 347–351 CrossRef CAS.
- M. Hammouche, D. Lexa, J. M. Savêant and M. Momenteau, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- I. Bhugun, D. Lexa and J. M. Savéant, J. Phys. Chem., 1996, 100, 19981–19985 CrossRef CAS.
- I. Bhugun, D. Lexa and J. M. Savéant, J. Am. Chem. Soc., 1994, 116, 5015–5016 CrossRef CAS.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- C. Costentin, S. Drouet, G. Passard, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2013, 135, 9023–9031 CrossRef CAS PubMed.
- C. Costentin, M. Robert, J. M. Savéant and C. Tard, Acc. Chem. Res., 2014, 47, 271–280 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J. M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed.
- S. Sinha and J. J. Warren, Inorg. Chem., 2018, 57, 12650–12656 CrossRef CAS PubMed.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1990, 521–529 RSC.
- M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 699–711 RSC.
- C. Costentin, M. Robert, J. M. Savéant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- P. Kumar Das, K. Mittra and A. Dey, Chem. Commun., 2014, 50, 5218–5220 RSC.
- K. Mittra, S. Chatterjee, S. Samanta, K. Sengupta, H. Bhattacharjee and A. Dey, Chem. Commun., 2012, 48, 10535–10537 RSC.
- K. Mittra, S. Chatterjee, S. Samanta and A. Dey, Inorg. Chem., 2013, 52, 14317–14325 CrossRef CAS PubMed.
- B. Mondal, A. Rana, P. Sen and A. Dey, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- C. G. Margarit, C. Schnedermann, N. G. Asimow and D. G. Nocera, Organometallics, 2019, 38, 1219–1223 CrossRef CAS.
- K. Guo, X. Li, H. Lei, W. Zhang and R. Cao, ChemCatChem, 2020, 12, 1591–1595 CrossRef CAS.
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952–2960 RSC.
- P. Sen, B. Mondal, D. Saha, A. Rana and A. Dey, Dalton Trans., 2019, 48, 5965–5977 RSC.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 CrossRef CAS PubMed.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- R. K. Parsapur, S. Chatterjee and K.-W. Huang, ACS Energy Lett., 2020, 2881–2885 CrossRef CAS.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 98 CrossRef CAS.
- S. Amanullah, A. Singha and A. Dey, Coord. Chem. Rev., 2019, 386, 183–208 CrossRef CAS.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313–12316 CrossRef CAS.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- B. Kandemir, L. Kubie, Y. Guo, B. Sheldon and K. L. Bren, Inorg. Chem., 2016, 55, 1355–1357 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2015, 8, 2283–2295 RSC.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015, 119, 13707–13713 CrossRef CAS PubMed.
- B. Mondal and A. Dey, Chem. Commun., 2017, 53, 7707–7715 RSC.
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 12457–12468 CrossRef CAS PubMed.
- S. Samanta, K. Sengupta, K. Mittra, S. Bandyopadhyay and A. Dey, Chem. Commun., 2012, 48, 7631–7633 RSC.
- B. Mondal, P. Sen, A. Rana, D. Saha, P. Das and A. Dey, ACS Catal., 2019, 9, 3895–3899 CrossRef CAS.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 2, 1315–1316 RSC.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1992, 322, 325–345 CrossRef CAS.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1993, 361, 149–157 CrossRef CAS.
- K. Bujno, R. Bilewicz, L. Siegfried and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS.
- E. Fujita, J. Haff, R. Sanzenbacher and H. Elias, Inorg. Chem., 1994, 33, 4627–4628 CrossRef CAS.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- P. J. Connolly and E. J. Billo, Inorg. Chem., 1987, 26, 3224–3226 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS.
- J. Song, E. L. Klein, F. Neese and S. Ye, Inorg. Chem., 2014, 53, 7500–7507 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef CAS PubMed.
- C. R. Schneider and H. S. Shafaat, Chem. Commun., 2016, 52, 9889–9892 RSC.
- E. M. Nichols and C. J. Chang, Organometallics, 2019, 38, 1213–1218 CrossRef CAS.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- E. X. Zhang, D. X. Wang, Z. T. Huang and M. X. Wang, J. Org. Chem., 2009, 74, 8595–8603 CrossRef CAS PubMed.
- D. C. Lacy, C. C. L. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980–4988 CrossRef CAS PubMed.
- X. Su, K. M. McCardle, J. A. Panetier and J. W. Jurss, Chem. Commun., 2018, 54, 3351–3354 RSC.
- T. Fogeron, T. K. Todorova, J. P. Porcher, M. Gomez-Mingot, L. M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, ACS Catal., 2018, 8, 2030–2038 CrossRef CAS.
- S. Dey, M. E. Ahmed and A. Dey, Inorg. Chem., 2018, 57, 5939–5947 CrossRef CAS.
- M. E. Ahmed, A. Rana, R. Saha, S. Dey and A. Dey, Inorg. Chem., 2020, 59, 5292–5302 CrossRef CAS.
- S. Dey, T. K. Todorova, M. Fontecave and V. Mougel, Angew. Chem., Int. Ed., 2020, 59, 15726–15733 CrossRef CAS PubMed.
- B. M. Ceballos and J. Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12686–12691 CrossRef CAS PubMed.
- Y. Matsubara, ACS Energy Lett., 2017, 2, 1886–1891 CrossRef CAS.
- K. Roy and P. L. A. Popelier, J. Phys. Org. Chem., 2009, 22, 186–196 CrossRef CAS.
- J. Song, E. L. Klein, F. Neese and S. Ye, Inorg. Chem., 2014, 53, 7500–7507 CrossRef CAS PubMed.
- O. Einsle, A. Messerschmidt, R. Huber, P. M. H. Kroneck, F. Neese and A. Strukturforschung, J. Am. Chem. Soc., 2002, 124, 11737–11745 CrossRef CAS PubMed.
- L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273–5357 CrossRef CAS PubMed.
- D. P. Summers and S. Chang, Nature, 1993, 365, 630–633 CrossRef CAS PubMed.
- B. Burlat, J. D. Gwyer, S. Poock, T. Clarke, J. A. Cole, A. M. Hemmings, M. R. Cheesman and J. N. Butt, Biochem. Soc. Trans., 2005, 33, 137–140 CrossRef CAS.
- M. Kern, Biochem. Soc. Trans., 2008, 36, 1011–1016 CrossRef.
- B. C. Berks, S. J. Ferguson, J. W. B. Moir and D. J. Richardson, Biochim. Biophys. Acta, Bioenerg., 1995, 1232, 97–173 CrossRef.
- V. A. Bamford, H. C. Angove, H. E. Seward, A. J. Thomson, J. A. Cole, J. N. Butt, A. M. Hemmings and D. J. Richardson, Biochemistry, 2002, 41, 2921–2931 CrossRef CAS PubMed.
- S. A. T. Å. Resolution, I. Binding, H. Motifs, O. Einsle, P. Stach, A. Messerschmidt, A. Kro, R. Huber and P. M. H. Kroneck, J. Biol. Chem., 2000, 275, 39608–39616 CrossRef.
- O. Einsle, P. Stach, A. Messerschmidt and È. Simon, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 341–342 CrossRef PubMed.
- O. Einsle, A. Messerschmidt, P. Stach, G. P. Bourenkov, H. D. Bartunik, R. Huber and P. M. H. Kroneck, Nature, 1999, 400, 476–480 CrossRef CAS PubMed.
- C. A. Cunha, S. Macieira, M. Dias, G. Almeida, L. L. Gonc, C. Costa, J. Lampreia, R. Huber, J. G. Moura, I. Moura and M. Joa, J. Biol. Chem., 2003, 278, 17455–17465 CrossRef CAS PubMed.
- M. G. Almeida, S. Macieira, L. L. Gonc, R. Huber, C. A. Cunha, C. Costa, J. Lampreia and M. Joa, Eur. J. Biochem., 2003, 270, 3904–3915 CrossRef CAS PubMed.
- F. Oliveira and M. L. Rodrigues, EMBO J., 2006, 25, 5951–5960 CrossRef PubMed.
- M. Youngblut, E. T. Judd, S. J. Elliott, M. Schmidt and A. A. Pacheco, J. Biol. Inorg. Chem., 2012, 17, 647–662 CrossRef CAS.
- E. T. Judd, M. Youngblut, A. Andrew Pacheco and S. J. Elliott, Biochemistry, 2012, 51, 10175–10185 CrossRef CAS PubMed.
- K. M. Polyakov, K. M. Boyko, T. V. Tikhonova, A. Slutsky, A. N. Antipov, R. A. Zvyagilskaya, A. N. Popov, G. P. Bourenkov, V. S. Lamzin and V. O. Popov, J. Mol. Biol., 2009, 389, 846–862 CrossRef CAS.
- P. Rydberg, E. Sigfridsson and U. Ryde, J. Biol. Inorg. Chem., 2004, 9, 203–223 CrossRef CAS PubMed.
- N. Stein, D. Love, E. T. Judd, S. J. Elliott, B. Bennett and A. A. Pacheco, Biochemistry, 2015, 54, 3749–3758 CrossRef CAS PubMed.
- C. W. J. Lockwood, M. R. Cheesman, M. Kern, J. Simon, T. A. Clarke, D. J. Richardson and J. N. Butt, J. Am. Chem. Soc., 2015, 137, 3059–3068 CrossRef CAS PubMed.
- P. Stach, O. Einsle, W. Schumacher, E. Kurun and P. M. H. Kroneck, J. Inorg. Biochem., 2000, 79, 381–385 CrossRef CAS.
- P. Lukat, M. Rudolf, P. Stach, A. Messerschmidt, P. M. H. Kroneck and O. Einsle, Biochemistry, 2008, 47, 2080–2086 CrossRef CAS PubMed.
- M. Sjo, Phys. Chem. Chem. Phys., 2004, 6, 4851–4858 RSC.
- D. Bykov and F. Neese, J. Biol. Inorg. Chem., 2011, 16, 417–430 CrossRef CAS PubMed.
- E. T. Judd, N. Stein, A. A. Pacheco and S. J. Elliott, Biochemistry, 2014, 53, 5638–5646 CrossRef CAS PubMed.
- Bruker, Apex Ii, 2013, Bruker AXS Inc., Madison, Wisconsin, USA.
- M. G. Almeida, C. M. Silveira, B. Guigliarelli, P. Bertrand, J. J. G. Moura, I. Moura and C. Léger, FEBS Lett., 2007, 581, 284–288 CrossRef CAS PubMed.
- T. A. Clarke, G. L. Kemp, J. H. Van Wonderen, R. A. S. Doyle, J. A. Cole, N. Tovell, M. R. Cheesman, J. N. Butt, D. J. Richardson and A. M. Hemmings, Biochemistry, 2008, 47, 3789–3799 CrossRef CAS PubMed.
- D. Bykov and F. Neese, Inorg. Chem., 2015, 54, 9303–9316 CrossRef CAS PubMed.
- D. Bykov, M. Plog and F. Neese, J. Biol. Inorg. Chem., 2014, 19, 97–112 CrossRef CAS PubMed.
- G. Martins, L. Rodrigues, F. M. Cunha, D. Matos, P. Hildebrandt, D. H. Murgida, A. C. Pereira and S. Todorovic, J. Phys. Chem. B, 2010, 114, 5563–5566 CrossRef CAS PubMed.
- M. Ali, N. Stein, Y. Mao, S. Shahid, M. Schmidt, B. Bennett and A. A. Pacheco, J. Am. Chem. Soc., 2019, 141, 13358–13371 CrossRef CAS PubMed.
- D. Bykov and F. Neese, J. Biol. Inorg. Chem., 2012, 741–760 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- L. E. Goodrich, F. Paulat, V. K. K. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293–6316 CrossRef CAS PubMed.
- G. R. A. Wyllie and W. R. Scheidt, Chem. Rev., 2002, 102, 1067–1089 CrossRef CAS PubMed.
- L. W. Olson, D. Schaeper, D. Lançon and K. M. Kadish, J. Am. Chem. Soc., 1982, 104, 2042–2044 CrossRef CAS.
- D. Lançon and K. M. Kadish, J. Am. Chem. Soc., 1983, 105, 5610–5617 CrossRef.
- I. K. Choi, Y. Liu, D. W. Feng, K. J. Paeng and M. D. Ryan, Inorg. Chem., 1991, 30, 1832–1839 CrossRef CAS.
- K. Fischer, G. G. Barbier, H. J. Hecht, R. R. Mendel, W. H. Campbell and G. Schwarz, Plant Cell, 2005, 17, 1167–1179 CrossRef CAS PubMed.
- E. Frı, L. M. Rubio, A. Herrero and E. Flores, Photosynth. Res., 2005, 83, 117–133 CrossRef.
- D. B. Knaff and M. Hirasawa, Biochim. Biophys. Acta, 1991, 1056, 93–125 CrossRef CAS.
- M. Hirasawa, K. Fukushima, G. Tamuna and D. B. Knaff, Biochim. Biophys. Acta, 1984, 791, 145–154 CrossRef CAS.
- C. Pino, F. Olmo-Mira, P. Cabello, M. Martínez-Luque, F. Castillo, M. D. Roldán and C. Moreno-Vivián, Biochem. Soc. Trans., 2006, 34, 127–129 CrossRef CAS PubMed.
- S. S. Merchant, D. B. Knaff and J. P. Allen, Photosynth. Res., 2010, 103, 67–77 CrossRef PubMed.
- U. Swamy, M. Wang, J. N. Tripathy, S. K. Kim, M. Hirasawa, D. B. Knaff and J. P. Allen, Biochemistry, 2005, 44, 16054–16063 CrossRef CAS PubMed.
- A. J. Gates, V. M. Luque-Almagro, A. D. Goddard, S. J. Ferguson, M. D. Roldán and D. J. Richardson, Biochem. J., 2011, 435, 743–753 CrossRef CAS PubMed.
- N. R. Harborne, L. Griffiths, S. J. W. Busby and J. A. Cole, Mol. Microbiol., 1992, 6, 2805–2813 CrossRef CAS PubMed.
- S. Nakano, M. Takahashi, A. Sakamoto, H. Morikawa and K. Katayanagi, Protein Sci., 2012, 21, 383–395 CrossRef CAS PubMed.
- S. Nakano, M. Takahashi, A. Sakamoto, H. Morikawa and K. Katayanagi, Proteins: Struct., Funct., Bioinf., 2012, 80, 2035–2045 CAS.
- M. J. Murphy, L. M. Siegel, S. R. Tove and H. Kamin, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 612–616 CrossRef CAS PubMed.
- P. Sétif, M. Hirasawa, N. Cassan, B. Lagoutte, J. N. Tripathy and D. B. Knaff, Biochemistry, 2009, 48, 2828–2838 CrossRef PubMed.
- M. Hirasawa, G. Tollin, Z. Salamon and D. B. Knaff, Biochim. Biophys. Acta, 1994, 1185, 336–345 CrossRef CAS.
- K. Parey, E. Warkentin, P. M. H. Kroneck and U. Ermler, Biochemistry, 2010, 49, 8912–8921 CrossRef CAS PubMed.
- S. Kuznetsova, D. B. Knaff, M. Hirasawa, P. Sétif and T. A. Mattioli, Biochemistry, 2004, 43, 10765–10774 CrossRef CAS PubMed.
- A. M. V. Brânzanic, U. Ryde and R. Silaghi-Dumitrescu, Chem. Commun., 2019, 55, 14047–14049 RSC.
- S. Rinaldo, G. Giardina, N. Castiglione, V. Stelitano and F. Cutruzzolà, Biochem. Soc. Trans., 2011, 39, 195–200 CrossRef CAS PubMed.
- W. G. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- F. Cutruzzolà, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 231–249 CrossRef.
- D. Nurizzo, F. Cutruzzolà, M. Arese, D. Bourgeois, M. Brunori, C. Cambillau and M. Tegoni, Biochemistry, 1998, 37, 13987–13996 CrossRef CAS PubMed.
- F. Cutruzzolà, K. Brown, E. K. Wilson, A. Bellelli, M. Arese, M. Tegoni, C. Cambillau and M. Brunori, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2232–2237 CrossRef PubMed.
- W. Sun, M. Arese, M. Brunori, F. Cutruzzolà, D. Nurizzo, K. Brown, C. Cambillau and M. Tegoni, Biochem. Biophys. Res. Commun., 2002, 291, 1–7 CrossRef CAS PubMed.
- S. C. Baker, N. F. W. Saunders, A. C. Willis, S. J. Ferguson, J. Hajdu and V. Fülöp, J. Mol. Biol., 1997, 269, 440–455 CrossRef CAS PubMed.
- M. R. Cheesman, S. J. Ferguson, J. W. B. Moir, D. J. Richardson, W. G. Zumft and A. J. Thomson, Biochemistry, 1997, 36, 16267–16276 CrossRef CAS PubMed.
- A. Jafferji, J. W. A. Allen, S. J. Ferguson and V. Fulop, J. Biol. Chem., 2000, 275, 25089–25094 CrossRef CAS.
- P. A. Williams, V. Fülöp, E. F. Carman, N. F. W. Saunders, S. J. Ferguson and J. Hajdu, Nature, 1997, 389, 406–412 CrossRef CAS PubMed.
- I. V. Pearson, M. D. Page, R. J. M. Van Spanning and S. J. Ferguson, J. Bacteriol., 2003, 185, 6308–6315 CrossRef CAS PubMed.
- T. Hard, H. J. Barnes, C. Larsson, J.-A. Gustafsson and J. Lund, Nature, 1995, 2, 983–989 CAS.
- E. Vijgenboom, J. E. Busch and G. W. Canters, Microbiology, 1997, 143, 2853–2863 CrossRef CAS PubMed.
- F. Rinaldo and S. Cutruzzola, Biology of the nitrogen cycle, Elsevier, Amsterdam, 2007 Search PubMed.
- W. Wu and C. K. Chang, J. Am. Chem. Soc., 1987, 109, 3149–3150 CrossRef CAS.
- D. Nurizzo, F. Cutruzzolà, M. Arese, D. Bourgeois, M. Brunori, C. Cambillau and M. Tegoni, J. Biol. Chem., 1999, 274, 14997–15004 CrossRef CAS.
- D. Nurizzo, M. C. Silvestrini, M. Mathieu, F. Cutruzzolà, D. Bourgeois, V. Fülöp, J. Hajdu, M. Brunori, M. Tegoni and C. Cambillau, Structure, 1997, 5, 1157–1171 CrossRef CAS PubMed.
- V. Fülöp, J. W. B. Moir, S. J. Ferguson and J. Hajdu, Cell, 1995, 81, 369–377 CrossRef.
- R. S. Zajicek, M. L. Cartron and S. J. Ferguson, Biochemistry, 2006, 45, 11208–11216 CrossRef CAS PubMed.
- F. Cutruzzolà, M. Arese, S. Grasso, A. Bellelli and M. Brunori, FEBS Lett., 1997, 412, 365–369 CrossRef.
- M. Radoul, F. Centola, S. Rinaldo, F. Cutruzzolá, I. Pecht and D. Goldfarb, Inorg. Chem., 2009, 48, 3913–3915 Search PubMed.
- T. Sjögren and J. Hajdu, J. Biol. Chem., 2001, 276, 29450–29455 CrossRef PubMed.
- B. Samyn, B. C. Berks, M. D. Page, S. J. Ferguson and J. J. Van Beeumen, Eur. J. Biochem., 1994, 219, 585–594 CrossRef CAS PubMed.
- A. Koppenhöfer, K. L. Turner, J. W. A. Allen, S. K. Chapman and S. J. Ferguson, Biochemistry, 2000, 39, 4243–4249 CrossRef PubMed.
- K. A. Sam, M. J. F. Strampraad, S. De Vries and S. J. Ferguson, J. Biol. Chem., 2008, 283, 27403–27409 CrossRef CAS PubMed.
- J. Yi, M. K. Safo and G. B. Richter-Addo, Biochemistry, 2008, 47, 8247–8249 CrossRef CAS.
- J. Yi, A. M. Orville, J. M. Skinner, M. J. Skinner and G. B. Richter-Addo, Biochemistry, 2010, 49, 5969–5971 CrossRef CAS PubMed.
- D. M. Copeland, A. S. Soares, A. H. West and G. B. Richter-Addo, J. Inorg. Biochem., 2006, 100, 1413–1425 CrossRef CAS PubMed.
- K. Brown, V. Roig-Zamboni, F. Cutruzzola’, M. Arese, W. Sun, M. Brunori, C. Cambillau and M. Tegoni, J. Mol. Biol., 2001, 312, 541–554 CrossRef CAS PubMed.
- S. Rinaldo, A. Arcovito, M. Brunori and F. Cutruzzolà, J. Biol. Chem., 2007, 282, 14761–14767 CrossRef CAS.
- G. Ranghino, E. Scorza, T. Sjogren, P. A. Williams, M. Ricci and J. Hajdu, Biochemistry, 2000, 39, 10958–10966 CrossRef CAS PubMed.
- E. G. Moore and Q. H. Gibson, J. Biol. Chem., 1976, 251, 2788–2794 CrossRef CAS.
- P. Sarti, A. Giuffré, E. Forte, D. Mastronicola, M. C. Barone and M. Brunori, Biochem. Biophys. Res. Commun., 2000, 274, 183–187 CrossRef CAS PubMed.
- S. J. George, J. W. A. Allen, S. J. Ferguson and R. N. F. Thorneley, J. Biol. Chem., 2000, 275, 33231–33237 CrossRef CAS PubMed.
- Y. Wang and B. A. Averill, J. Am. Chem. Soc., 1996, 118, 3972–3973 CrossRef CAS.
- S. Rinaldo, M. Brunori and F. Cutruzzolà, Biochem. Biophys. Res. Commun., 2007, 363, 662–666 CrossRef CAS PubMed.
- O. Farver, M. Brunori, F. Cutruzzolà, S. Rinaldo, S. Wherland and I. Pecht, Biophys. J., 2009, 96, 2849–2856 CrossRef CAS PubMed.
- O. Farver, P. M. H. Kroneck, I. Pecht and W. G. Zumft, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7622–7625 CrossRef CAS PubMed.
- S. Rinaldo, K. A. Sam, N. Castiglione, V. Stelitano, A. Arcovito, M. Brunori, J. W. A. Allen, S. J. Ferguson and F. Cutruzzolà, Biochem. J., 2011, 435, 217–225 CrossRef CAS PubMed.
- C. D. Richter, J. W. A. Allen, C. W. Higham, A. Koppenhöfer, R. S. Zajicek, N. J. Watmough and S. J. Ferguson, J. Biol. Chem., 2002, 277, 3093–3100 CrossRef CAS PubMed.
- K. A. Sam, J. D. Tolland, S. A. Fairhurst, C. W. Higham, D. J. Lowe, R. N. F. Thorneley, J. W. A. Allen and S. J. Ferguson, Biochem. Biophys. Res. Commun., 2008, 371, 719–723 CrossRef CAS PubMed.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- K. A. Sam, S. A. Fairhurst, R. N. F. Thorneley, J. W. A. Allen and S. J. Ferguson, J. Biol. Chem., 2008, 283, 12555–12563 CrossRef CAS PubMed.
- M. Radoul, D. Bykov, S. Rinaldo, F. Cutruzzolà, F. Neese and D. Goldfarb, J. Am. Chem. Soc., 2011, 133, 3043–3055 CrossRef CAS PubMed.
- J. W. A. Allen, P. D. Barker, O. Daltrop, J. M. Stevens, E. J. Tomlinson, N. Sinha and S. J. Ferguson, Dalton Trans., 2005, 3410–3418 RSC.
- K. Kobayashi, A. Koppenhöfer, S. J. Ferguson and S. Tagawa, Biochemistry, 1997, 36, 13611–13616 CrossRef CAS PubMed.
- L. L. Perissinotti, M. A. Marti, F. Doctorovich, F. J. Luque and D. A. Estrin, Biochemistry, 2008, 47, 9793–9802 CrossRef CAS PubMed.
- B. Wang, Y. Shi, J. Tejero, S. M. Powell, L. M. Thomas, M. T. Gladwin, S. Shiva, Y. Zhang and G. B. Richter-Addo, Biochemistry, 2018, 57, 4788–4802 CrossRef CAS PubMed.
- M. Tiso, J. Tejero, S. Basu, I. Azarov, X. Wang, V. Simplaceanu, S. Frizzell, T. Jayaraman, L. Geary, C. Shapiro, C. Ho, S. Shiva, D. B. Kim-Shapiro and M. T. Gladwin, J. Biol. Chem., 2011, 286, 18277–18289 CrossRef CAS PubMed.
- R. Silaghi-Dumitrescu, Inorg. Chem., 2004, 43, 3715–3718 CrossRef CAS PubMed.
- H. Li, A. Samouilov, X. Liu and J. L. Zweier, Biochemistry, 2003, 42, 1150–1159 CrossRef CAS PubMed.
- H. Li, C. Hemann, T. M. Abdelghany, M. A. El-Mahdy and J. L. Zweier, J. Biol. Chem., 2012, 287, 36623–36633 CrossRef CAS.
- P. Corti, M. Ieraci and J. Tejero, Nitric Oxide, 2016, 53, 22–34 CrossRef CAS PubMed.
- C. Gautier, E. Van Faassen, I. Mikula, P. Martasek and A. Slama-Schwok, Biochem. Biophys. Res. Commun., 2006, 341, 816–821 CrossRef CAS PubMed.
- J. Tejero, C. E. Sparacino-Watkins, V. Ragireddy, S. Frizzell and M. T. Gladwin, Biochemistry, 2015, 54, 722–733 CrossRef CAS PubMed.
- D. B. Kim-Shapiro and M. T. Gladwin, Nitric Oxide, 2014, 38, 58–68 CrossRef CAS PubMed.
- A. F. Vanin, L. M. Bevers, A. Slama-Schwok and E. E. Van Faassen, Cell. Mol. Life Sci., 2007, 64, 96–103 CrossRef CAS PubMed.
- B. J. Reeder and J. Ukeri, Nitric Oxide, 2018, 72, 16–23 CrossRef CAS PubMed.
- G. Da Silva, E. M. Kennedy and B. Z. Dlugogorski, J. Phys. Chem. A, 2006, 110, 11371–11376 CrossRef CAS PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, Dekker, New York, 1985 Search PubMed.
- J. Heinecke and P. C. Ford, Coord. Chem. Rev., 2010, 254, 235–247 CrossRef CAS.
- B. R. Crane, L. M. Siegel and E. D. Getzoff, Biochemistry, 1997, 36, 12120–12137 CrossRef CAS PubMed.
- H. Nasri, Y. Wang, B. H. Huynh and W. R. Scheldt, J. Am. Chem. Soc., 1991, 113, 717–719 CrossRef CAS.
- J. B. Fernandes, D. W. Feng, A. Chang, A. Keyser and M. D. Ryan, Inorg. Chem., 1986, 25, 2606–2610 CrossRef CAS.
- M. G. Finnegan, A. G. Lappin and W. R. Scheidt, Inorg. Chem., 1990, 29, 181–185 CrossRef CAS.
- H. Nasri, W. R. Scheidt, Y. Wang, B. H. Huynh and F. A. Walker, Inorg. Chem., 1991, 30, 1483–1489 CrossRef CAS.
- Z. Wei and M. D. Ryan, Inorg. Chim. Acta, 2001, 314, 49–57 CrossRef CAS.
- H. Nasri, M. K. Ellison, S. Chen, B. H. Huynh and W. R. Scheldt, J. Am. Chem. Soc., 1997, 119, 6274–6283 CrossRef CAS.
- J. Lee, A. Y. Kovalevsky, I. V. Novozhilova, K. A. Bagley, P. Coppens and G. B. Richter-Addo, J. Am. Chem. Soc., 2004, 126, 7180–7181 CrossRef CAS PubMed.
- I. V. Novozhilova, P. Coppens, J. Lee, G. B. Richter-Addo and K. A. Bagley, J. Am. Chem. Soc., 2006, 128, 2093–2104 CrossRef CAS.
- K. S. Suslick and R. A. Watson, Inorg. Chem., 1991, 30, 912–919 CrossRef CAS.
- K. M. Kadish, V. A. Adamian, E. Van Caemelbecke, Z. Tan, P. Tagliatesta, P. Bianco, T. Boschi, G.-B. Yi, M. A. Khan and G. B. Richter-Addo, Inorg. Chem., 1996, 35, 1343–1348 CrossRef CAS PubMed.
- K. M. Miranda, X. Bu, I. Lorković and P. C. Ford, Inorg. Chem., 1997, 36, 4838–4848 CrossRef CAS PubMed.
- F. A. Leal, I. M. Lorkovic, P. C. Ford, J. Lee, L. Chen, L. Torres, M. A. Khan and G. B. Richter-Addo, Can. J. Chem., 2003, 81, 872–881 CrossRef CAS.
- D. Awasabisah and G. B. Richter-Addo, NOx linkage isomerization in metal complexes, Elsevier Inc., 1st edn, 2015, vol. 67 Search PubMed.
- J. L. Heinecke, J. Yi, J. C. M. Pereira, G. B. Richter-Addo and P. C. Ford, J. Inorg. Biochem., 2012, 107, 47–53 CrossRef CAS PubMed.
- T. S. Kurtikyan and P. C. Ford, Coord. Chem. Rev., 2008, 252, 1486–1496 CrossRef CAS.
- C. K. Chang, J. Biol. Chem., 1985, 9520–9522 CrossRef CAS.
- D. Feng, Y. S. Ting and M. D. Ryan, Inorg. Chem., 1985, 24, 612–617 CrossRef CAS.
- Y. Liu and M. D. Ryan, Inorg. Chim. Acta, 1994, 225, 57–66 CrossRef CAS.
- A. M. Stolzenberg, S. H. Strauss and R. H. Holm, J. Am. Chem. Soc., 1981, 103, 4763–4778 CrossRef CAS.
- A. M. Stolzenberg, L. O. Spreer and R. H. Holm, J. Am. Chem. Soc., 1980, 102, 364–370 CrossRef CAS.
- A. M. Stolzenberg, L. O. Spreer and R. H. Holm, J. Chem. Soc., Chem. Commun., 1979, 1077–1078 RSC.
- C. K. Chang and J. Fajer, J. Am. Chem. Soc., 1980, 102, 848–851 CrossRef CAS.
- C. K. Chang, K. M. Barkigia, L. K. Hanson and J. Fajer, J. Am. Chem. Soc., 1986, 108, 1352–1354 CrossRef CAS.
- P. F. Richardson, C. K. Chang, L. K. Hanson, L. D. Spaulding and J. Fajer, J. Phys. Chem., 1979, 83, 3420–3424 CrossRef CAS.
- C. K. Chang, L. K. Hanson, P. F. Richardson, R. Young and J. Fajer, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2652–2656 CrossRef CAS PubMed.
- E. Fujita and J. Fajer, J. Am. Chem. Soc., 1983, 105, 6743–6745 CrossRef CAS.
- S. Ozawa, E. Sakamoto, Y. Watanabe and I. Morishima, J. Chem. Soc., Chem. Commun., 1994, 3, 935–936 RSC.
- S. Ozawa, E. Sakamoto, T. Ichikawa, Y. Watanabe and I. Morishima, Inorg. Chem., 1995, 34, 6362–6370 CrossRef CAS.
- Y. Liu, C. DeSilva and M. D. Ryan, Inorg. Chim. Acta, 1997, 258, 247–255 CrossRef CAS.
- M. H. Barley, K. Takeuchi, W. R. Murphy and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 507–508 RSC.
- M. H. Barley, K. J. Takeuchi and T. J. Meyer, J. Am. Chem. Soc., 1986, 108, 5876–5885 CrossRef CAS PubMed.
- M. R. Kumar, D. Pervitsky, L. Chen, T. Poulos, S. Kundu, M. S. Hargrove, E. J. Rivera, A. Diaz, J. L. Colón and P. J. Farmer, Biochemistry, 2009, 48, 5018–5025 CrossRef CAS PubMed.
- R. Lin and P. J. Farmer, J. Am. Chem. Soc., 2000, 122, 2393–2394 CrossRef CAS.
- L. E. Goodrich, S. Roy, E. E. Alp, J. Zhao, M. Y. Hu and N. Lehnert, Inorg. Chem., 2013, 52, 7766–7780 CrossRef CAS PubMed.
- M. H. Rahman and M. D. Ryan, Inorg. Chem., 2017, 56, 3302–3309 CrossRef CAS PubMed.
- Y. Liu and M. D. Ryan, J. Electroanal. Chem., 1994, 368, 209–219 CrossRef CAS.
- J. Pellegrino, S. E. Bari, D. E. Bikiel and F. Doctorovich, J. Am. Chem. Soc., 2010, 132, 989–995 CrossRef CAS PubMed.
- S. Amanullah and A. Dey, Chem. Sci., 2020, 11, 5909–5921 RSC.
- A. J. Timmons and M. D. Symes, Chem. Soc. Rev., 2015, 44, 6708–6722 RSC.
- I. K. Choi and M. D. Ryan, Inorg. Chim. Acta, 1988, 153, 25–30 CrossRef CAS.
- Y. O. Kim and H. M. Goff, Inorg. Chem., 1990, 29, 3907–3908 CrossRef CAS.
- S. Amanullah, P. Saha, R. Saha and A. Dey, Inorg. Chem., 2019, 58, 152–164 CrossRef CAS PubMed.
- T. K. Das, E. K. Wilson, F. Cutruzzolà, M. Brunori and D. L. Rousseau, Biochemistry, 2001, 40, 10774–10781 CrossRef CAS PubMed.
- X.-J. Zhao, V. Sampath and W. S. Caughey, Biochem. Biophys. Res. Commun., 1994, 537–543 CrossRef CAS PubMed.
- J. C. Maxwell and W. S. Caughey, Biochemistry, 1976, 15, 388–396 CrossRef CAS.
- D. S. Bohle and C.-H. Hung, J. Am. Chem. Soc., 1995, 117, 9584–9585 CrossRef CAS.
- Z. Wei and M. D. Ryan, Inorg. Chem., 2010, 49, 6948–6954 CrossRef CAS.
- A. E. Servid, A. L. McKay, C. A. Davis, E. M. Garton, A. Manole, P. S. Dobbin, M. A. Hough and C. R. Andrew, Biochemistry, 2015, 54, 3320–3327 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- D. D. M. Wayner and V. D. Parker, Acc. Chem. Res., 1993, 26, 287–294 CrossRef CAS.
- E. Wiberg, N. Wiberg and A. F. Holleman, Inorganic chemistry, Academic Press, De Gruyter, San Diego; Berlin; New York, 2001 Search PubMed.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- C. Mealli and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2007, 46, 8942–8944 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC.
- D. V. Esposito, S. T. Hunt, Y. C. Kimmel and J. G. Chen, J. Am. Chem. Soc., 2012, 134, 3025–3033 CrossRef CAS PubMed.
- R. B. Gordon, M. Bertram and T. E. Graedel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1209–1214 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- P. Tamagnini, R. Axelsson, P. Lindberg, F. Oxelfelt, R. Wünschiers and P. Lindblad, Microbiol. Mol. Biol. Rev., 2002, 66, 1–20 CrossRef CAS PubMed.
- R. K. Thauer, A.-K. Kaster, M. Goenrich, M. Schick, T. Hiromoto and S. Shima, Annu. Rev. Biochem., 2010, 79, 507–536 CrossRef CAS PubMed.
- R. Cammack, M. Frey and R. Robson, Hydrogen as a Fuel Learning from Nature, Taylor & Francis, 11 New Fetter Lane, London EC4P 4EE, 2001 Search PubMed.
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS PubMed.
- H. R. Pershad, J. L. C. Duff, H. A. Heering, E. C. Duin, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 1999, 38, 8992–8999 CrossRef CAS PubMed.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–275 RSC.
- S. Dey, P. K. Das and A. Dey, Coord. Chem. Rev., 2013, 257, 42–63 CrossRef CAS.
- K. Schütz, T. Happe, O. Troshina, P. Lindblad, E. Leitão, P. Oliveira and P. Tamagnini, Planta, 2004, 218, 350–359 CrossRef PubMed.
- A. Melis, L. Zhang, M. Forestier, M. L. Ghirardi and M. Seibert, Plant Physiol., 2000, 122, 127–136 CrossRef CAS PubMed.
- M. L. Ghirardi, Indian J. Biochem. Biophys., 2006, 43, 201–210 CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814–822 CrossRef CAS PubMed.
- S. Shima and R. K. Thauer, Chem. Rec., 2007, 7, 37–46 CrossRef CAS PubMed.
- S. Rospert, J. Breitung, K. Ma, B. Schwörer, C. Zirngibl, R. K. Thauer, D. Linder, R. Huber and K. O. Stetter, Arch. Microbiol., 1991, 156, 49–55 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, JBIC, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- Z. Chen, B. J. Lemon, S. Huang, D. J. Swartz, J. W. Peters and K. A. Bagley, Biochemistry, 2002, 41, 2036–2043 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414–4435 CrossRef CAS PubMed.
- A. L. de Lacey, E. C. Hatchikian, A. Volbeda, M. Frey, J. C. Fontecilla-Camps and V. M. Fernandez, J. Am. Chem. Soc., 1997, 119, 7181–7189 CrossRef CAS.
- C. V. Popescu and E. Münck, J. Am. Chem. Soc., 1999, 121, 7877–7884 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- O. F. Erdem, L. Schwartz, M. Stein, A. Silakov, S. Kaur-Ghumaan, P. Huang, S. Ott, E. J. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2011, 50, 1439–1443 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS PubMed.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- P. Rodríguez-Maciá, K. Pawlak, O. Rüdiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- J. H. Artz, D. W. Mulder, M. W. Ratzloff, C. E. Lubner, O. A. Zadvornyy, A. X. LeVan, S. G. Williams, M. W. W. Adams, A. K. Jones, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2017, 139, 9544–9550 CrossRef CAS PubMed.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, J. Am. Chem. Soc., 2008, 130, 4533–4540 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- M. L. K. Sanchez, C. Sommer, E. Reijerse, J. A. Birrell, W. Lubitz and R. B. Dyer, J. Am. Chem. Soc., 2019, 141, 16064–16070 CrossRef CAS PubMed.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577–7587 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- R. P. Happe, W. Roseboom, A. J. Pierik, S. P. J. Albracht and K. A. Bagley, Nature, 1997, 385, 126 CrossRef CAS PubMed.
- H. S. Shafaat, O. Rüdiger, H. Ogata and W. Lubitz, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 986–1002 CrossRef CAS PubMed.
- S. Foerster, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83–93 CrossRef CAS PubMed.
- M. Brecht, M. van Gastel, T. Buhrke, B. Friedrich and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 13075–13083 CrossRef CAS PubMed.
- S. J. George, S. Kurkin, R. N. F. Thorneley and S. P. J. Albracht, Biochemistry, 2004, 43, 6808–6819 CrossRef CAS PubMed.
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef PubMed.
- M. Kampa, M.-E. Pandelia, W. Lubitz, M. van Gastel and F. Neese, J. Am. Chem. Soc., 2013, 135, 3915–3925 CrossRef CAS PubMed.
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS PubMed.
- R. K. Thauer, A. R. Klein and G. C. Hartmann, Chem. Rev., 1996, 96, 3031–3042 CrossRef CAS PubMed.
- C. Zirngibl, R. Hedderich and R. K. Thauer, FEBS Lett., 1990, 261, 112–116 CrossRef CAS.
- C. Zirngibl, W. Van Dongen, B. Schwörer, R. Von Bünau, M. Richter, A. Klein and R. K. Thauer, Eur. J. Biochem., 1992, 208, 511–520 CrossRef CAS PubMed.
- T. Hiromoto, E. Warkentin, J. Moll, U. Ermler and S. Shima, Angew. Chem., Int. Ed., 2009, 48, 6457–6460 CrossRef CAS PubMed.
- A. Dey, J. Am. Chem. Soc., 2010, 132, 13892–13901 CrossRef CAS.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS PubMed.
- G. J. Kubas, R. R. Ryan, B. I. Swanson, P. J. Vergamini and H. J. Wasserman, J. Am. Chem. Soc., 1984, 106, 451–452 CrossRef CAS.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37–68 CrossRef CAS.
- R. H. Crabtree, Angew. Chem., Int. Ed. Engl., 1993, 32, 789–805 CrossRef.
- G. J. Kubas, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6901–6907 CrossRef CAS PubMed.
- R. H. Morris, Coord. Chem. Rev., 2008, 252, 2381–2394 CrossRef CAS.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71 Search PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS PubMed.
- A. D. Wilson, K. Fraze, B. Twamley, S. M. Miller, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2008, 130, 1061–1068 CrossRef CAS PubMed.
- W. Zhu, A. C. Marr, Q. Wang, F. Neese, D. J. E. Spencer, A. J. Blake, P. A. Cooke, C. Wilson and M. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18280–18285 CrossRef CAS PubMed.
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. R. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6951–6956 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 5, 7470–7490 RSC.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- H. Reihlen, A. Gruhl and G. v. Hessling, Justus Liebigs Ann. Chem., 1929, 472, 268–287 CrossRef CAS.
- D. Seyferth, G. B. Womack, M. K. Gallagher, M. Cowie, B. W. Hames, J. P. Fackler and A. M. Mazany, Organometallics, 1987, 6, 283–294 CrossRef CAS.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, Advance Article, 10.1039/D0CS01089H.
- J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2009, 253, 1476–1494 CrossRef CAS.
- G. A. N. Felton, C. A. Mebi, B. J. Petro, A. K. Vannucci, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Organomet. Chem., 2009, 694, 2681–2699 CrossRef CAS.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS PubMed.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Chem. – Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed.
- B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2008, 47, 2261–2263 CrossRef CAS PubMed.
- F. Wang, M. Wang, X. Liu, K. Jin, W. Dong and L. Sun, Dalton Trans., 2007, 3812–3819 RSC.
- U.-P. Apfel, C. R. Kowol, F. Kloss, H. Görls, B. K. Keppler and W. Weigand, J. Organomet. Chem., 2011, 696, 1084–1088 CrossRef CAS.
- H.-G. Cui, M. Wang, W.-B. Dong, L.-L. Duan, P. Li and L.-C. Sun, Polyhedron, 2007, 26, 904–910 CrossRef CAS.
- R. J. Wright, C. Lim and T. D. Tilley, Chem. – Eur. J., 2009, 15, 8518–8525 CrossRef CAS PubMed.
- G. Qian, W. Zhong, Z. Wei, H. Wang, Z. Xiao, L. Long and X. Liu, New J. Chem., 2015, 39, 9752–9760 RSC.
- D. Chong, I. P. Georgakaki, R. Mejia-Rodriguez, J. Sanabria-Chinchilla, M. P. Soriaga and M. Y. Darensbourg, Dalton Trans., 2003, 4158–4163 RSC.
- Z. Yu, M. Wang, P. Li, W. Dong, F. Wang and L. Sun, Dalton Trans., 2008, 2400–2406 RSC.
- Y. Si, M. Hu and C. Chen, C. R. Chim., 2008, 11, 932–937 CrossRef CAS.
- Y. Na, M. Wang, J. Pan, P. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2008, 47, 2805–2810 CrossRef CAS PubMed.
- C. M. Thomas, O. Rüdiger, T. Liu, C. E. Carson, M. B. Hall and M. Y. Darensbourg, Organometallics, 2007, 26, 3976–3984 CrossRef CAS.
- M. L. Singleton, R. M. Jenkins, C. L. Klemashevich and M. Y. Darensbourg, C. R. Chim., 2008, 11, 861–874 CrossRef CAS.
- Z. Wang, W. Jiang, J. Liu, W. Jiang, Y. Wang, B. Åkermark and L. Sun, J. Organomet. Chem., 2008, 693, 2828–2834 CrossRef CAS.
- P. Li, M. Wang, L. Chen, J. Liu, Z. Zhao and L. Sun, Dalton Trans., 2009, 1919–1926 RSC.
- E. S. Donovan, J. J. McCormick, G. S. Nichol and G. A. N. Felton, Organometallics, 2012, 31, 8067–8070 CrossRef CAS.
- J. Chen, A. K. Vannucci, C. A. Mebi, N. Okumura, S. C. Borowski, M. Swenson, L. T. Lockett, D. H. Evans, R. S. Glass and D. L. Lichtenberger, Organometallics, 2010, 29, 5330–5340 CrossRef CAS.
- H.-M. Lin, C. Mu, A. Li, X.-F. Liu, Y.-L. Li, Z.-Q. Jiang and H.-K. Wu, Transition Met. Chem., 2019, 44, 491–498 CrossRef CAS.
- G. Durgaprasad, R. Bolligarla and S. K. Das, J. Organomet. Chem., 2012, 706–707, 37–45 CrossRef CAS.
- R. M. Bullock and M. L. Helm, Acc. Chem. Res., 2015, 48, 2017–2026 CrossRef CAS PubMed.
- J.-F. Capon, S. Ezzaher, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Chem. – Eur. J., 2008, 14, 1954–1964 CrossRef CAS PubMed.
- S. Ott, M. Kritikos, B. Åkermark, L. Sun and R. Lomoth, Angew. Chem., Int. Ed., 2004, 43, 1006–1009 CrossRef CAS PubMed.
- Y. Si, C. Ma, M. Hu, H. Chen, C. Chen and Q. Liu, New J. Chem., 2007, 31, 1448–1454 RSC.
- T. Liu, M. Wang, Z. Shi, H. Cui, W. Dong, J. Chen, B. Åkermark and L. Sun, Chem. – Eur. J., 2004, 10, 4474–4479 CrossRef CAS PubMed.
- L.-C. Song, J.-H. Ge, X.-G. Zhang, Y. Liu and Q.-M. Hu, Eur. J. Inorg. Chem., 2006, 3204–3210 CrossRef CAS.
- L.-C. Song, J.-H. Ge, X.-F. Liu, L.-Q. Zhao and Q.-M. Hu, J. Organomet. Chem., 2006, 691, 5701–5709 CrossRef CAS.
- W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, Dalton Trans., 2009, 2712–2720 RSC.
- S. Jiang, J. Liu, Y. Shi, Z. Wang, B. Åkermark and L. Sun, Dalton Trans., 2007, 896–902 RSC.
- L.-C. Song, B.-S. Yin, Y.-L. Li, L.-Q. Zhao, J.-H. Ge, Z.-Y. Yang and Q.-M. Hu, Organometallics, 2007, 26, 4921–4929 CrossRef CAS.
- L.-C. Song, L.-X. Wang, B.-S. Yin, Y.-L. Li, X.-G. Zhang, Y.-W. Zhang, X. Luo and Q.-M. Hu, Eur. J. Inorg. Chem., 2008, 291–297 CrossRef CAS.
- W. Gao, J. Liu, C. Ma, L. Weng, K. Jin, C. Chen, B. Åkermark and L. Sun, Inorg. Chim. Acta, 2006, 359, 1071–1080 CrossRef CAS.
- W. Gao, J. Sun, T. Åkermark, M. Li, L. Eriksson, L. Sun and B. Åkermark, Chem. – Eur. J., 2010, 16, 2537–2546 CrossRef CAS PubMed.
- C. Costentin, Chem. Rev., 2008, 108, 2145–2179 CrossRef CAS PubMed.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2004, 566, 241–247 CrossRef CAS.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, J. Electroanal. Chem., 2006, 595, 47–52 CrossRef CAS.
- S. Jiang, J. Liu and L. Sun, Inorg. Chem. Commun., 2006, 9, 290–292 CrossRef CAS.
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, Coord. Chem. Rev., 2000, 206–207, 533–561 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- M. E. Ahmed and A. Dey, Curr. Opin. Electrochem., 2019, 15, 155–164 CrossRef CAS.
- J.-X. Jian, C. Ye, X.-Z. Wang, M. Wen, Z.-J. Li, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2016, 9, 2083–2089 RSC.
- M. Cheng, M. Wang, S. Zhang, F. Liu, Y. Yang, B. Wan and L. Sun, Faraday Discuss., 2017, 198, 197–209 RSC.
- R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS.
- F. Quentel, G. Passard and F. Gloaguen, Energy Environ. Sci., 2012, 5, 7757–7761 RSC.
- S. Dey, A. Rana, S. G. Dey and A. Dey, ACS Catal., 2013, 3, 429–436 CrossRef CAS.
- M. E. Ahmed, S. Dey, B. Mondal and A. Dey, Chem. Commun., 2017, 53, 8188–8191 RSC.
- V. Vijaikanth, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, J. Organomet. Chem., 2007, 692, 4177–4181 CrossRef CAS.
- A. Le Goff, V. Artero, R. Metayé, F. Moggia, B. Jousselme, M. Razavet, P. D. Tran, S. Palacin and M. Fontecave, Int. J. Hydrogen Energy, 2010, 35, 10790–10796 CrossRef CAS.
- E. S. Andreiadis, P.-A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- C. Caix, S. Chardon-Noblat, A. Deronzier and R. Ziessel, J. Electroanal. Chem., 1996, 403, 189–202 CrossRef.
- I. Sádaba, M. López Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS PubMed.
- K. Abdur-Rashid, T. P. Fong, B. Greaves, D. G. Gusev, J. G. Hinman, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2000, 122, 9155–9171 CrossRef CAS.
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710–9711 CrossRef CAS PubMed.
- X. Zhao, I. P. Georgakaki, M. L. Miller, R. Mejia-Rodriguez, C.-Y. Chiang and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 3917–3928 CrossRef CAS PubMed.
- I. P. Georgakaki, M. L. Miller and M. Y. Darensbourg, Inorg. Chem., 2003, 42, 2489–2494 CrossRef CAS PubMed.
- T. Xu, D. Chen and X. Hu, Coord. Chem. Rev., 2015, 303, 32–41 CrossRef CAS.
- M. T. Olsen, B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2009, 48, 7507–7509 CrossRef CAS PubMed.
- M. T. Olsen, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2010, 132, 17733–17740 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- N. Wang, M. Wang, T. Zhang, P. Li, J. Liu and L. Sun, Chem. Commun., 2008, 5800–5802 RSC.
- N. Wang, M. Wang, J. Liu, K. Jin, L. Chen and L. Sun, Inorg. Chem., 2009, 48, 11551–11558 CrossRef CAS PubMed.
- S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, S. E. Kabir and B. E. Sanchez, Chem. Commun., 2014, 50, 945–947 RSC.
- N. Wang, M. Wang, Y. Wang, D. Zheng, H. Han, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2013, 135, 13688–13691 CrossRef CAS PubMed.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- S. Dey, A. Rana, D. Crouthers, B. Mondal, P. K. Das, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2014, 136, 8847–8850 CrossRef CAS PubMed.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS PubMed.
- M. K. Bruska, M. T. Stiebritz and M. Reiher, J. Am. Chem. Soc., 2011, 133, 20588–20603 CrossRef CAS PubMed.
- M. T. Stiebritz and M. Reiher, Inorg. Chem., 2009, 48, 7127–7140 CrossRef CAS PubMed.
- C. Lambertz, N. Leidel, K. G. V. Havelius, J. Noth, P. Chernev, M. Winkler, T. Happe and M. Haumann, J. Biol. Chem., 2011, 286, 40614–40623 CrossRef CAS PubMed.
- C. Orain, L. Saujet, C. Gauquelin, P. Soucaille, I. Meynial-Salles, C. Baffert, V. Fourmond, H. Bottin and C. Léger, J. Am. Chem. Soc., 2015, 137, 12580–12587 CrossRef CAS PubMed.
- A. Kubas, D. De Sancho, R. B. Best and J. Blumberger, Angew. Chem., Int. Ed., 2014, 53, 4081–4084 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef CAS.
- S. Kaur-Ghumaan and M. Stein, Dalton Trans., 2014, 43, 9392–9405 RSC.
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem. – Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- Y. Ohki and K. Tatsumi, Eur. J. Inorg. Chem., 2011, 973–985 CrossRef CAS.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pécaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- Y. Oudart, V. Artero, J. Pécaut, C. Lebrun and M. Fontecave, Eur. J. Inorg. Chem., 2007, 2613–2626 CrossRef CAS.
- S. Canaguier, M. Fontecave and V. Artero, Eur. J. Inorg. Chem., 2011, 1094–1099 CrossRef CAS.
- S. Ogo, R. Kabe, K. Uehara, B. Kure, T. Nishimura, S. C. Menon, R. Harada, S. Fukuzumi, Y. Higuchi, T. Ohhara, T. Tamada and R. Kuroki, Science, 2007, 316, 585–587 CrossRef CAS PubMed.
- L.-C. Song, J.-P. Li, Z.-J. Xie and H.-B. Song, Inorg. Chem., 2013, 52, 11618–11626 CrossRef CAS PubMed.
- V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417–2427 RSC.
- B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877–14885 CrossRef CAS PubMed.
- B. E. Barton, C. M. Whaley, T. B. Rauchfuss and D. L. Gray, J. Am. Chem. Soc., 2009, 131, 6942–6943 CrossRef CAS PubMed.
- L. Vaccaro, V. Artero, S. Canaguier, M. Fontecave and M. J. Field, Dalton Trans., 2010, 39, 3043–3049 RSC.
- L.-C. Song, Y. Lu, L. Zhu and Q.-L. Li, Organometallics, 2017, 36, 750–760 CrossRef CAS.
- O. A. Ulloa, M. T. Huynh, C. P. Richers, J. A. Bertke, M. J. Nilges, S. Hammes-Schiffer and T. B. Rauchfuss, J. Am. Chem. Soc., 2016, 138, 9234–9245 CrossRef CAS PubMed.
- L.-C. Song, X.-Y. Yang, M. Cao, X.-Y. Gao, B.-B. Liu, L. Zhu and F. Jiang, Chem. Commun., 2017, 53, 3818–3821 RSC.
- T. R. Simmons and V. Artero, Angew. Chem., Int. Ed., 2013, 52, 6143–6145 CrossRef CAS PubMed.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- G. M. Chambers, M. T. Huynh, Y. Li, S. Hammes-Schiffer, T. B. Rauchfuss, E. Reijerse and W. Lubitz, Inorg. Chem., 2016, 55, 419–431 CrossRef CAS PubMed.
- S. Canaguier, M. Field, Y. Oudart, J. Pécaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876–5878 RSC.
- F. Dole, A. Fournel, V. Magro, E. C. Hatchikian, P. Bertrand and B. Guigliarelli, Biochemistry, 1997, 36, 7847–7854 CrossRef CAS PubMed.
- S. Niu, L. M. Thomson and M. B. Hall, J. Am. Chem. Soc., 1999, 121, 4000–4007 CrossRef CAS.
- L. De Gioia, P. Fantucci, B. Guigliarelli and P. Bertrand, Inorg. Chem., 1999, 38, 2658–2662 CrossRef CAS.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745–20755 CrossRef CAS PubMed.
- G. Gezer, S. Verbeek, M. A. Siegler and E. Bouwman, Dalton Trans., 2017, 46, 13590–13596 RSC.
- G. Gezer, D. Durán Jiménez, M. A. Siegler and E. Bouwman, Dalton Trans., 2017, 46, 7506–7514 RSC.
- H. Tang and M. B. Hall, J. Am. Chem. Soc., 2017, 139, 18065–18070 CrossRef CAS PubMed.
- D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò and M. Gennari, Inorg. Chem., 2017, 56, 14801–14808 CrossRef CAS PubMed.
- D. Brazzolotto, L. Wang, H. Tang, M. Gennari, N. Queyriaux, C. Philouze, S. Demeshko, F. Meyer, M. Orio, V. Artero, M. B. Hall and C. Duboc, ACS Catal., 2018, 8, 10658–10667 CrossRef CAS.
- L.-C. Song, X.-F. Han, W. Chen, J.-P. Li and X.-Y. Wang, Dalton Trans., 2017, 46, 10003–10013 RSC.
- S. Ding, P. Ghosh, M. Y. Darensbourg and M. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9775–E9782 CrossRef CAS PubMed.
- S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920–12927 CrossRef CAS PubMed.
- P. Ghosh, M. Quiroz, N. Wang, N. Bhuvanesh and M. Y. Darensbourg, Dalton Trans., 2017, 46, 5617–5624 RSC.
- P. Ghosh, S. Ding, R. B. Chupik, M. Quiroz, C.-H. Hsieh, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2017, 8, 8291–8300 RSC.
- M. E. Ahmed, S. Chattopadhyay, L. Wang, D. Brazzolotto, D. Pramanik, D. Aldakov, J. Fize, A. Morozan, M. Gennari, C. Duboc, A. Dey and V. Artero, Angew. Chem., Int. Ed., 2018, 57, 16001–16004 CrossRef CAS PubMed.
- N. T. Nguyen, Y. Mori, T. Matsumoto, T. Yatabe, R. Kabe, H. Nakai, K.-S. Yoon and S. Ogo, Chem. Commun., 2014, 50, 13385–13387 RSC.
- T. Matsumoto, B. Kure and S. Ogo, Chem. Lett., 2008, 37, 970–971 CrossRef CAS.
- G. M. Chambers, J. Mitra, T. B. Rauchfuss and M. Stein, Inorg. Chem., 2014, 53, 4243–4249 CrossRef CAS PubMed.
- B. Kure, T. Matsumoto, K. Ichikawa, S. Fukuzumi, Y. Higuchi, T. Yagi and S. Ogo, Dalton Trans., 2008, 4747–4755 RSC.
- M. Isegawa, A. K. Sharma, S. Ogo and K. Morokuma, ACS Catal., 2018, 8, 10419–10429 CrossRef CAS.
- J. Kalms, A. Schmidt, S. Frielingsdorf, T. Utesch, G. Gotthard, D. von Stetten, P. van der Linden, A. Royant, M. A. Mroginski, P. Carpentier, O. Lenz and P. Scheerer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2229–E2237 CrossRef CAS PubMed.
- N. J. Lindenmaier, S. Wahlefeld, E. Bill, T. Szilvási, C. Eberle, S. Yao, P. Hildebrandt, M. Horch, I. Zebger and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 2208–2211 CrossRef CAS PubMed.
- X. Yang, L. C. Elrod, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2019, 10, 1368–1373 RSC.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS PubMed.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- W. J. Shaw, M. L. Helm and D. L. DuBois, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1123–1139 CrossRef CAS PubMed.
- J. Y. Yang, S. Chen, W. G. Dougherty, W. S. Kassel, R. M. Bullock, D. L. DuBois, S. Raugei, R. Rousseau, M. Dupuis and M. R. DuBois, Chem. Commun., 2010, 46, 8618–8620 RSC.
- J. Y. Yang, R. M. Bullock, M. R. DuBois and D. L. DuBois, MRS Bull., 2011, 36, 39–47 CrossRef CAS.
- D. L. DuBois and R. M. Bullock, Eur. J. Inorg. Chem., 2011, 1017–1027 CrossRef CAS.
- U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861–5872 CrossRef CAS PubMed.
- S. Raugei, M. L. Helm, S. Hammes-Schiffer, A. M. Appel, M. O’Hagan, E. S. Wiedner and R. M. Bullock, Inorg. Chem., 2016, 55, 445–460 CrossRef CAS PubMed.
- S. Horvath, L. E. Fernandez, A. V. Soudackov and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15663–15668 CrossRef CAS PubMed.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS PubMed.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
- T. Liu, S. Chen, M. J. O’Hagan, M. Rakowski DuBois, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 134, 6257–6272 CrossRef CAS PubMed.
- N. Wang, H. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2018, 39, 228–244 CrossRef CAS.
- H. Lei, X. Li, J. Meng, H. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS.
- T. Nakae, M. Hirotsu and I. Kinoshita, Organometallics, 2015, 34, 3988–3997 CrossRef CAS.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed.
- A. Kochem, M. O’Hagan, E. S. Wiedner and M. van Gastel, Chem. – Eur. J., 2015, 21, 10338–10347 CrossRef CAS PubMed.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS PubMed.
- D. J. Graham and D. G. Nocera, Organometallics, 2014, 33, 4994–5001 CrossRef CAS.
- A. Dutta, J. A. S. Roberts and W. J. Shaw, Angew. Chem., Int. Ed., 2014, 53, 6487–6491 CrossRef CAS PubMed.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed.
- P. Das, M.-H. Ho, M. O’Hagan, W. J. Shaw, R. Morris Bullock, S. Raugei and M. L. Helm, Dalton Trans., 2014, 43, 2744–2754 RSC.
- A. Rana, B. Mondal, P. Sen, S. Dey and A. Dey, Inorg. Chem., 2017, 56, 1783–1793 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- S. Chatterjee, K. Sengupta, S. Dey and A. Dey, Inorg. Chem., 2013, 52, 14168–14177 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |