
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of chemical synthesis in developing RiPP antibiotics
Sam M.
Rowe
 and
David R.
Spring
*
and
David R.
Spring
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
First published on 26th February 2021
Abstract
The growing antimicrobial resistance crisis necessitates the discovery and development of novel classes of antibiotics if a ‘postantibiotic era’ is to be avoided. Ribosomally synthesised and post-translationally modified peptides, or RiPPs, are becoming increasingly recognised as a potential source of antimicrobial drugs. This is due to a combination of their potent antimicrobial activity and their high stability relative to unmodified linear peptides. However, as peptide drugs, their clinical development is often perturbed by issues such as low solubility and poor bioavailability. Chemical synthesis has the potential to overcome some of these challenges. Furthermore, the structural complexity of RiPPs makes them interesting synthetic targets in their own right, with the total synthesis of some structural classes having only been recently realised. This review focusses on the use of RiPPs as antimicrobial agents and will highlight various strategies that have been employed to chemically synthesise three major classes of RiPPs: lasso peptides, cyclotides, and lanthipeptides.
 Sam M. Rowe | Sam Rowe received his MSci in Natural Sciences from the University of Cambridge in 2017, specialising in organic and biological chemistry. He is currently a PhD student under the supervision of Professor David Spring, also at the University of Cambridge, and is working on the development of new peptide therapeutics. |
 David R. Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
Key learning points(1) Ribosomally-synthesised and post-translationally modified peptides (RiPPs) can display potent antimicrobial activity and high stability relative to unmodified linear peptides, making them attractive antimicrobial compounds for combatting the growing antimicrobial resistance crisis.(2) There are currently no antimicrobial RiPPs approved for clinical use in humans due to issues such as poor solubility and bioavailability. However, several are in clinical trials, and chemical synthesis has the potential to access RiPP analogues with improved therapeutic profiles. (3) The first total synthesis of a correctly folded lasso peptide was reported in 2019 and required a support with three linkage sites to direct folding. (4) Cyclotide chemical synthesis typically involves head-to-tail native chemical ligation (NCL)-mediated cyclisation followed by oxidative folding. (5) Lanthionine bridges in lanthipeptides are typically synthesised using orthogonally protected bisamino acids during solid-phase peptide synthesis (SPPS), or by Michael addition between cysteine and a dehydro amino acid after the linear peptide sequence has been synthesised. |
Introduction
Diseases caused by pathogens exhibiting antimicrobial resistance (AMR) are currently responsible for 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths a year globally. In a worst-case scenario this figure is set to rise to 10 million a year by 2050.1 This is in part because there is an inconsistency between the current pipeline of antibiotics and those that the world needs. Many of the antibiotics being researched target easier-to-treat infections of less overall concern in the context of AMR. Very few represent breakthroughs as first-in-class products, which is concerning because resistance to one antibiotic often leads to resistance against multiple antibiotics within the same class. The rate of antibiotic discovery and development needs to be greatly improved if a ‘postantibiotic era’ is to be avoided, where common infections are once again associated with high mortality.
000 deaths a year globally. In a worst-case scenario this figure is set to rise to 10 million a year by 2050.1 This is in part because there is an inconsistency between the current pipeline of antibiotics and those that the world needs. Many of the antibiotics being researched target easier-to-treat infections of less overall concern in the context of AMR. Very few represent breakthroughs as first-in-class products, which is concerning because resistance to one antibiotic often leads to resistance against multiple antibiotics within the same class. The rate of antibiotic discovery and development needs to be greatly improved if a ‘postantibiotic era’ is to be avoided, where common infections are once again associated with high mortality.
Many of our most familiar antibiotics are produced by non-ribosomal machinery (e.g., β-lactams, tetracyclines, macrolides, and glycopeptides), while their ribosomally synthesised counterparts have been comparatively overlooked. Genome sequencing efforts of the 21st century have highlighted the potential of ribosomally synthesised and post-translationally modified peptides (RiPPs) as an untapped source of antimicrobial drugs.2 In September 2020, a review was published by a community of scientists working on the biosynthesis of RiPPs, which outlines the current state of the field.3 It updates the work published in a seminal 2013 review where the term ‘RiPP’ was coined, and an upper size limit of 10 kDa was imposed to distinguish RiPPs from post-translationally modified proteins. The 2020 review highlights how rapidly the field is evolving, with 17 new classes of RiPPs having been identified since 2013, bringing the total number of classes to 41. This is likely to expand further in the coming years, and with the realisation that some RiPPs are made by hybrid biosynthetic pathways, this umbrella term may eventually outgrow its use.
RiPPs have been identified in all three domains of life and display impressive structural and functional diversity. The post-translational modifications afforded to these molecules enables them access to chemical space that is not typically explored by ribosomally-synthesised peptides. This is often achieved by conformational restriction, which has the dual benefit of enabling better target recognition while also significantly improving stability to both chemical and metabolic degradation relative to unmodified linear peptides. This enhanced stability makes RiPPs particularly attractive as peptide drugs, which are often rapidly degraded in vivo.
RiPPs have several other characteristics that make them attractive antimicrobial agents.4 Firstly, antimicrobial RiPPs typically display narrow spectrum activity, which can limit off-target effects that might otherwise disrupt normal flora. This in turn reduces the risk of the emergence of secondary infections, particularly by resistant antimicrobial strains. Secondly, many RiPPs have multiple mechanisms of action that can act simultaneously, reducing the risk of resistance development. Finally, antimicrobial RiPP targets are typically highly conserved, again reducing the risk of resistance development.
To the best of our knowledge, there are currently no antimicrobial RiPPs approved for clinical use in humans; however, there are several undergoing clinical trials. NVB302, a derivate of the lanthipeptide actagardine, became the first antibacterial lanthipeptide to enter clinical trials in November 2011. Despite a promising profile for C. difficile treatment, no further clinical trials have been reported to date.5 More recently, LFF571, a derivative of thiopeptide GE2270-A, completed a phase II clinical trial for treatment of moderate C. difficile infections, where it displayed similar safety and efficacy to vancomycin.5 Duramycin, a lanthipeptide, has also completed phase II clinical trials for the treatment of cystic fibrosis.6
Despite these counterexamples, clinical development of RiPPs is often hindered by issues such as poor solubility and bioavailability. However, the biosynthetic pathways of RiPPs display impressive malleability, which may be harnessed by engineering to overcome these challenges.3 Furthermore, chemical synthesis of these peptides is becoming ever more tractable and lends itself to the introduction of modifications that can enhance their therapeutic profiles. Finally, genome mining approaches developed in the new millennium are increasingly facilitating the identification of novel RiPPs, and so it is possible that those with favourable pharmacokinetic properties may yet be identified. Together, these strategies will facilitate the proper investigation of an entirely new class of antimicrobial compounds for clinical use.
This review will focus on three major classes of RiPPs: lasso peptides, cyclotides and lanthipeptides. It will highlight their structural characteristics and functions, drawing particular attention to members of each class that have been shown to display antimicrobial activity. The various routes used to chemically synthesise each of these classes will then be described.
Lasso peptides
Structure
Lasso peptides are structurally characterised by their knot topology, in which the C-terminal peptide ‘tail’ threads through an N-terminal lactam-bridged macrocycle in a right-handed conformation. This threading leads to the eponymous lasso structure (Fig. 1).7 The tail is often held in place by bulky ‘plug’ residues both above and below the plane of the ring, sometimes with the assistance of disulfide bridges. During biosynthesis, the peptide is pre-folded such that macrolactamisation locks the tail inside the ring.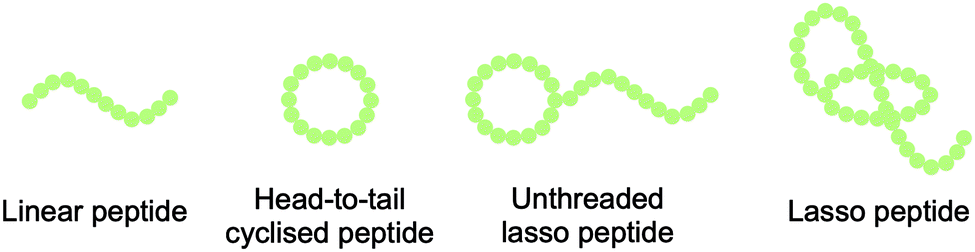 | ||
| Fig. 1 Linear, head-to-tail cyclic, unthreaded, and lasso peptide topologies. | ||
The peptides are typically 15–26 residues long, with the N-terminal macrolactam consisting of 7–9 residues. Cyclisation is achieved by isopeptide bond formation between the N-terminal α-amino group (typically belonging to Gly, Ala, Ser, or Cys) and the side chain of an Asp or Glu residue at position 7, 8, or 9.
Lasso peptides can be divided into four classes on the basis of the presence and number of disulfide bridges (Table 1). Around 55 lasso peptides have been reported to date, and of these Class II are the most common. Classes III and IV each have only one reported example with the Class IV peptide, LP2006, only having been identified in 2017.8 Although originally discovered by chance through activity-driven compound isolations, since 2008, lasso peptides have mostly been identified by genome mining approaches, which have revolutionised the field.9 Through genome mining, the rate of discovery of lasso peptides has increased drastically and it is therefore possible that more classes will be discovered in the coming years.
| Class | Description | Example | Structure |
|---|---|---|---|
| I | Two disulfide bonds. One from an N-terminal Cys to a loop Cys and another from a macrocycle Cys to a tail Cys. | Sviceucin |
|
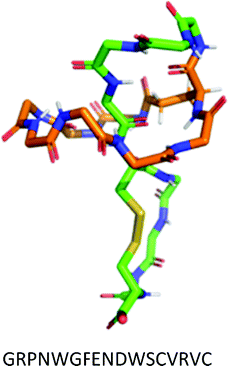
| II | No disulfide bonds. | Microcin J25 (MccJ25) |
|
| III | One disulfide bond from a macrocycle Cys to a tail Cys. | BI-32169 |
|
| IV | One disulfide bond between two tail Cys residues. | LP2006 |
|
Function
Lasso peptides display a range of biological functions, among which antimicrobial activity is common. There are currently 14 lasso peptides that are known to display antibacterial activity (against both Gram-positive and Gram-negative bacteria), and this knowledge has recently been summarised by Tan et al.7 Eight of these antibacterial lasso peptides have had their targets identified, although the mechanisms of action remain generally obscure. The three targets identified are: RNA polymerase; ClpC1, a component of the ClpC1P1P2 protease complex; and lipid II, a key precursor of the cell wall component peptidoglycan. The complex topology of lasso peptides is thought to be essential for their function, with ‘unthreading’ having been shown to lead to inactivity. (Fig. 1).9The compact structure of lasso peptides can also impart high stability to physical, chemical, and proteolytic degradation relative to unmodified linear peptides.7 This high stability has led to lasso peptides being used as molecular scaffolds for grafting of biologically active peptide epitopes (Scheme 1).10
 | ||
| Scheme 1 Lasso peptide-like [1]rotaxane (L1) synthesis from a [2]rotaxane core (4). In this example, a peptide sequence derived from octreotide (blue) was grafted into the structure by sequential α-ketoacid-hydroxylamine (KAHA) ligation and native chemical ligation (NCL) to afford 9. An unthreaded variant (B1) was also prepared separately. Adapted from F. Saito and J. W. Bode, Chem. Sci., 2017, 8, 2878–2884. | ||
Synthesis
Microcin J25 (MccJ25) is a 21-mer antimicrobial lasso peptide that shows activity against a range of Gram-negative pathogens through inhibition of bacterial RNA polymerase (RNAP).13 It is the most studied of the lasso peptides, and several approaches to its solid-phase chemical synthesis have been attempted; however, a synthesis leading to the properly folded lasso structure remains elusive. A four-year Horizon 2020 project (Grant agreement ID: 656999) with the goal of synthesising correctly folded MccJ25 ended in July 2019 and was ultimately unsuccessful.
Early attempted syntheses of MccJ25 included: head-to-tail macrocylisation of the full peptide sequence using native-chemical ligation; an unthreaded variant of the peptide with the correct macrolactam ring structure formed by an isopeptide bond between the α-amino group of Gly1 and the γ-carboxyl of Glu8 (Fig. 2a); and a similar unthreaded variant in which the Gly1 and Glu8 residues were replaced with isosteric azido acetic acid and propargylglycine residues for subsequent copper-catalysed azide–alkyne cycloaddition (CuAAC) ring-closing (Fig. 2b).9 All of these methods afforded variants of MccJ25 that showed no antimicrobial activity.
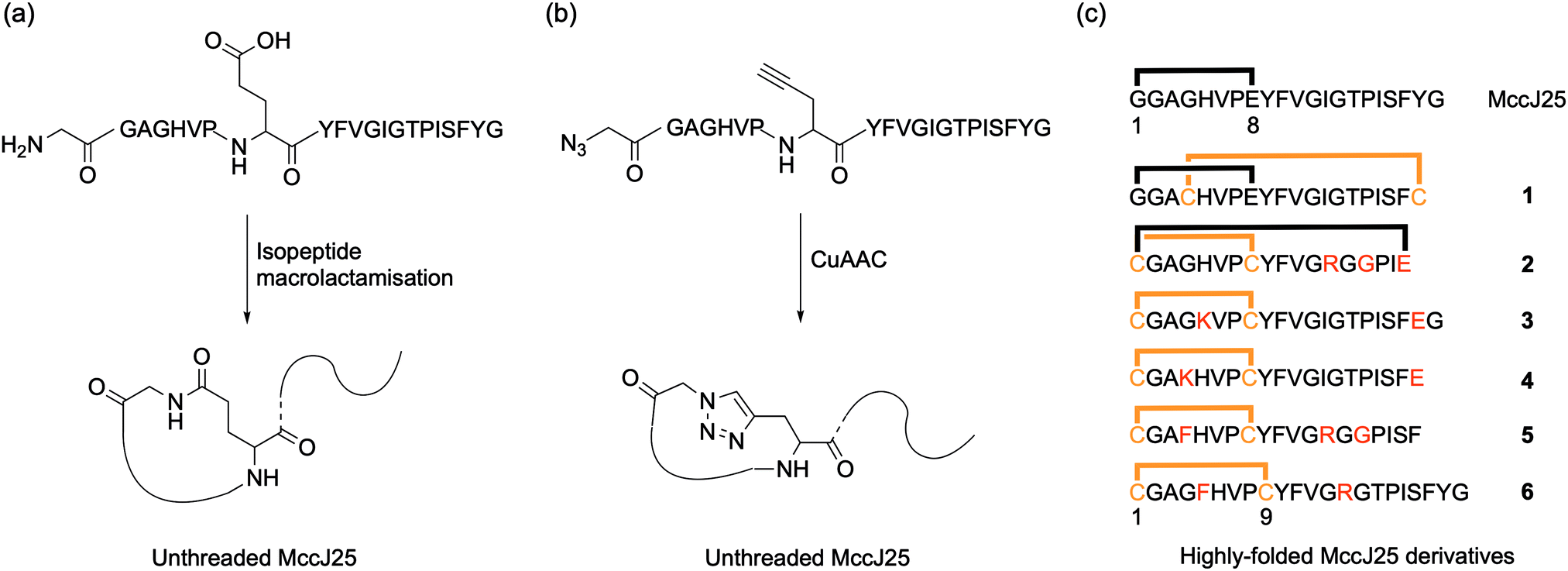 | ||
| Fig. 2 Chemical syntheses of incorrectly folded MccJ25. (a) Unthreaded MccJ25 formed by an isopeptide bond between the α-amino group of Gly1 and the γ-carboxyl of Glu8. (b) Unthreaded MccJ25 in which the Gly1 and Glu8 residues were replaced with isosteric azido acetic acid and propargylglycine residues for subsequent copper-catalysed azide–alkyne cycloaddition (CuAAC) ring-closing. (c) Amino acid sequences of MccJ25 and the highly-folded derivative peptides 1–6 produced by Kaur and co-workers. Substitutions of amino acids in the peptide derivatives are shown in orange for cysteine and red for others. (c) is adapted from Soudy, R., Wang, L., Kaur, K. Bioorganic Med. Chem. 2012, 20, 1794–1800 with permission from Elsevier. | ||
Kaur and co-workers were the first to report synthetic derivatives of MccJ25 that displayed antibacterial activity.14 In order to be active, the researchers proposed that a more folded conformation of the MccJ25 sequence was required, whereby the flexibility of the tail was reduced through interaction with the macrocycle. They reasoned that this could be achieved by a combination of intra-peptide disulfide bonds as well as electrostatic and hydrophobic interactions. A series of six peptides with various permutations of these constraints were synthesised and characterised, and peptides 1 and 6 displayed antimicrobial activity (Fig. 2c). Although both peptides were generally less potent than native MccJ25, peptide 1 showed antibacterial activity against two MccJ25-resistant Salmonella strains. Despite this activity, CD analysis confirmed that neither active peptide had the stabilising lasso structure of MccJ25, instead adopting an unthreaded conformation. Consequently, both peptides were significantly more susceptible to proteolytic degradation by chymotrypsin and pepsin than the native peptide. Fliss and co-workers have reported similarly ‘folded’ but non-lasso derivates of MccJ25 that retain some bactericidal activity.15 Tulla-Puche and co-workers have also reported synthesis of a lasso-inspired peptide in which the tail was covalently attached to the macrolactam ring but does not pass through it.16 The synthesis required the use of 10 different protecting groups, ultimately affording an unthreaded variant of chaxapeptin that displayed good thermal stability and protease resistance. Preliminary biological assays suggested that the synthetic peptide displays similar cytotoxicity and invasion activity to natural chaxapeptin.
 | ||
| Scheme 2 The cryptand-imidazolium supported synthesis of BI-32169 reported by Chen and co-workers. The imidazolium moiety is coloured red and the cryptand moiety blue. The residues forming the N-terminal ring are coloured green and those forming the C-terminal tail are coloured orange. All the protecting groups are indicated in italics. Reproduced from ref. 17 with permission from The Royal Society of Chemistry. | ||
The dipeptide 8 was extended until Ser8 of the N-terminal macrocycle was reached. The acid-sensitive protecting groups of the Ser8 side chain (triphenylmethyl, Trt) and the second cryptand linkage site (PhiPr) were removed with dilute trifluoroacetic acid (TFA), and the newly exposed functional groups were coupled to form the second linkage, affording 10. The peptide was then extended to the third linking residue, Trp4. Trp itself does not have a side chain easily amenable to chemical linkage, and so an Fmoc-protected 2H,3H-1-p-nitrobenzylcarboxyl tryptophan was used in its place. This residue has a carbamate handle that could be used to attach the residue to the third linkage site but can also easily be removed under final cleavage conditions to reveal unmodified Trp. The p-nitrobenzyl (pNB) group of the modified Trp and the tert-butyldiphenylsilyl (TBDPS) group of the third cryptand linkage site were removed simultaneously with tetra-n-butylammonium fluoride (TBAF), and the deprotected functional groups were coupled, giving 12. The peptide was extended with the final three residues such that the cryptand-imidazolium support was loaded with the complete BI-32169 sequence (13).
In order to form the macrolactam, the N-terminal Fmoc group and the 4-(N-[1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl]amino)benzyl (Dmab) group of Asp9 were both removed with 2% hydrazine, and the resulting amine and acid were coupled to afford the required isopeptide bond. With this coupling, the threaded lasso peptide structure 14 was realised. To release the structure from the support, the ortho-nitrobenzyl ester groups of linkages two and three were photolytically cleaved to give 15. The remaining protecting groups were subsequently removed with a standard peptide cleavage mixture of TFA, phenol, water, and triisopropylsilane (TIPS) in a ratio of 88:5:5:2, and the final support linkage was removed with sodium hydroxide. Oxidation in air led to formation of the desired disulfide bridge between Cys6 and Cys19, affording BI-32169. HPLC analysis revealed that the crude product was achieved in high purity, with the only by-product being unthreaded BI-32169. HPLC purification gave the 98.7% pure product (HPLC analysis) in 2.5% yield, based on the starting amount of imidazolium salt. The threaded structure of BI-32169 was confirmed by comparison to natural BI-32169 using HPLC, NMR, tandem MS, and CD analysis.
One clear advantage of chemical synthesis of BI-32169 is that the researchers were also able to synthesise the D-enantiomer of the bicyclic structure by using D-amino acids and swapping the chirality of the cryptand. The D-enantiomer showed improved proteolytic stability relative to the L-enantiomer, while maintaining potent antagonist activity.
The ability to engineer lasso peptides is highly desirable. Despite its remarkable resistance to multiple proteases, MccJ25 has been shown to be rapidly degraded in the human duodenum by pancreatic protease elastase.18 On the one hand, this suggests it is safe for food applications. On the other, it suggests that MccJ25 requires encapsulation or engineering in order to stabilise the loop region against degradation if it is to be used as an antibacterial agent in the gastrointestinal tract of humans or animals. Chemical synthesis is well situated to realise this engineering due to the comparative ease with which unnatural residues may be inserted into sequences, reducing protease recognition. By applying the tri-linkage support methodology of Chen and co-workers, the synthesis and enhancement of properly folded antimicrobial lasso peptides such as MccJ25 may be achieved.
Cyclotides
Structure
Cyclotides are head-to-tail macrocyclised peptides (28–37 residues long) found in several plant families including Rubiaceae, Violaceae, Cucurbitaceae, Solanaceae, and Fabaceae.19 They are characterised by the presence of a cyclic cystine knot (CCK) motif resulting from six conserved Cys residues, which form three intramolecular disulfide bonds (Fig. 3). Two of the disulfide bonds (CysI–CysIV and CysII–CysV) and their connecting backbone loops form a ring that is then threaded by the third disulfide bond (CysIII–CysVI). The CCK motif braces the structure, providing cyclotides with exceptional thermal, chemical, and proteolytic stability. CyBase (http://www.cybase.org.au), a database dedicated to cataloguing cyclotide sequences, structures, and activities, currently contains approximately 500 different cyclotide sequences, with some reports suggesting that the number of cyclotides in nature exceeds 10000.
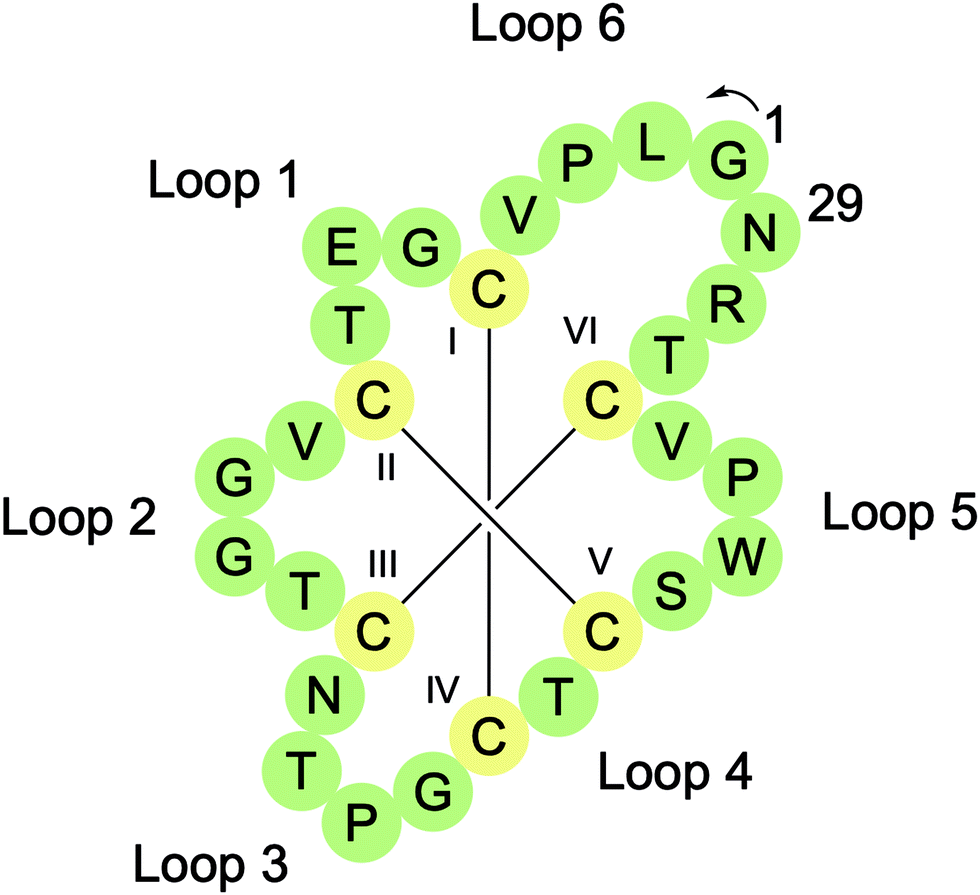 | ||
| Fig. 3 Sequence and connectivity of the prototypical cyclotide kalata B1. Adapted with permission from S. J. De Veer, M. W. Kan and D. J. Craik, Chem. Rev. 2019, 119, 12375–12421. Copyright (2019) American Chemical Society. | ||
Cyclotides can be organised into three classes based on their 3D structures.19 Approximately two-thirds of cyclotides have a bracelet structure, one-third have a Möbius structure, and a handful are trypsin inhibitors. Möbius cyclotides, such as the prototypical cyclotide kalata B1, typically have a Trp-Pro or Tyr-Pro motif in loop 5 that promotes a cis amide bond between the two residues. This introduces a 180° twist in the peptide backbone that has been likened to a Möbius strip. Bracelet cyclotides, such as cycloviolacin O2, are named for their circular topology and trypsin inhibitor cyclotides, such as MCoTI-II, are so named for their potent inhibition of the serine protease trypsin. The types and number of residues in different loops can also be used to distinguish between these three cyclotide classes.
Function
Cyclotides play a natural role as defense peptides in plants, but a diverse range of biological activities have been identified by screening plant extracts for medicinal properties.19 To date, approximately eight cyclotides have been shown to exhibit antimicrobial activity: kalata B1, kalata B2, circulin A, circulin B, cyclopsychotride, cycloviolacin O2, HB10, and HB11. While this is a limited number of examples, there is still a vast pool of potential candidates to explore and more with antimicrobial activity may be identified in the future. Furthermore, while few in number, some of these examples are active against Gram-negative bacteria, which are of particular concern in the context of AMR. For example, cycloviolacin O2 shows potent activity against the Gram-negative ESKAPE pathogens K. pneumoniae and P. aeruginosa in time-kill assays.20 Recently, the first in vivo assessment of cyclotide antimicrobial activity was reported in which cycloviolacin O2 and kalata B2 displayed anti-staphylococcal activity in a mouse subcutaneous infection model.21 Membrane insertion and pore formation appear to be the primary mechanism of action of cyclotides, although more work is required to confirm this in vivo.Much like lasso peptides, the exceptional stability of cyclotides makes them ideal molecular scaffolds for epitope grafting of biologically active peptides, and both natural and engineered cyclotides are being evaluated as drug leads.22 [T20K]kalata B1 (‘T20K’), an engineered kalata B1 derivative with a single point mutation, was the first cyclotide therapeutic to enter clinical trials, successfully completing phase I safety and tolerability studies in August 2019 for the treatment of multiple sclerosis.23
Synthesis
Cyclotides can be purified in high yields from plant material; however, chemical synthesis presents the opportunity to explore structure–activity relationships and to develop derivatives with new physical properties and biological activities. The chemical synthesis of cyclotides typically involves solid-phase peptide synthesis (SPPS) of the linear sequence, native chemical ligation (NCL)-mediated cyclisation, and finally oxidative folding of the cyclised peptide (Scheme 3). While in principle folding could be achieved either before or after backbone cyclisation, higher yields and greater purities have been observed when it occurs after ring closure.24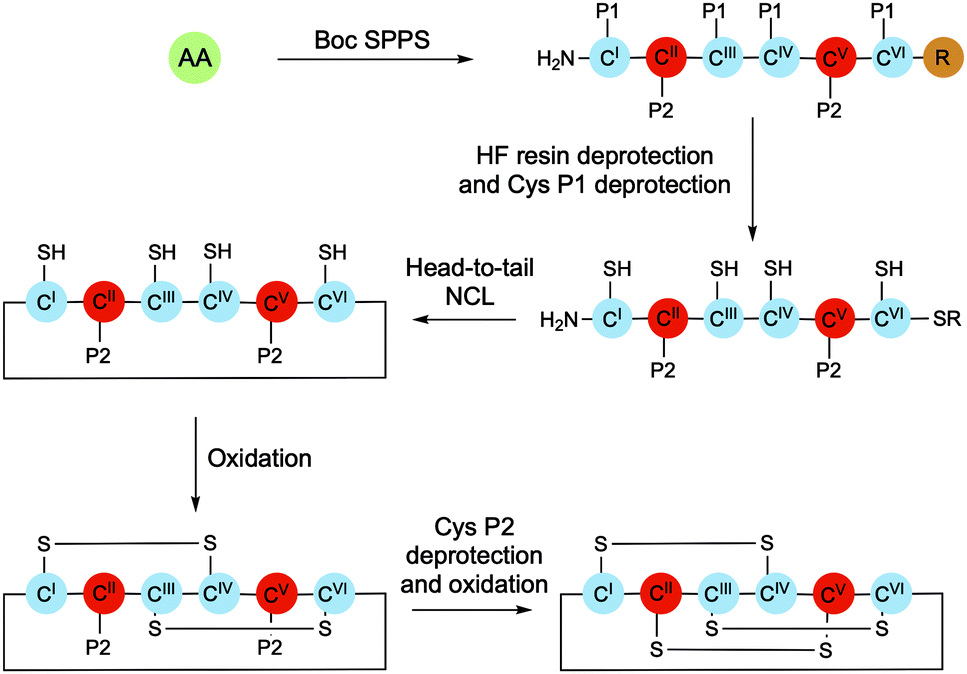 | ||
| Scheme 3 Typical route for the chemical synthesis of cyclotides. Differentially protected Cys residues are coloured blue (Cys I, III, IV, VI) and red (Cys II, V) respectively. The resin is coloured brown. AA, amino acid; P1, protecting group 1; P2 protecting group 2. | ||
Fmoc/Boc solid-phase peptide synthesis (SPPS)
Fluorenylmethyloxycarbonyl (Fmoc) SPPS is generally the preferred method of peptide synthesis as it requires less corrosive reagents and is more amenable to automation. However, tert-butyloxycarbonyl (Boc) SPPS is still commonly used for cyclotide synthesis due to its compatibility with NCL reaction conditions, which remain the most common method of cyclotide macrocyclisation.In order to achieve head-to-tail ligation, NCL requires nucleophilic attack by the thiol group of an N-terminal Cys residue onto a C-terminal thioester of the same peptide sequence (Scheme 4). As such, thioester bonds are often used to attach cyclotide peptide sequences to the solid support. Piperidine, which is used for iterative Fmoc deprotections during Fmoc-SPPS, is able to cleave thioester bonds, which can lead to impaired yields and thus makes Boc-SPPS more appealing.
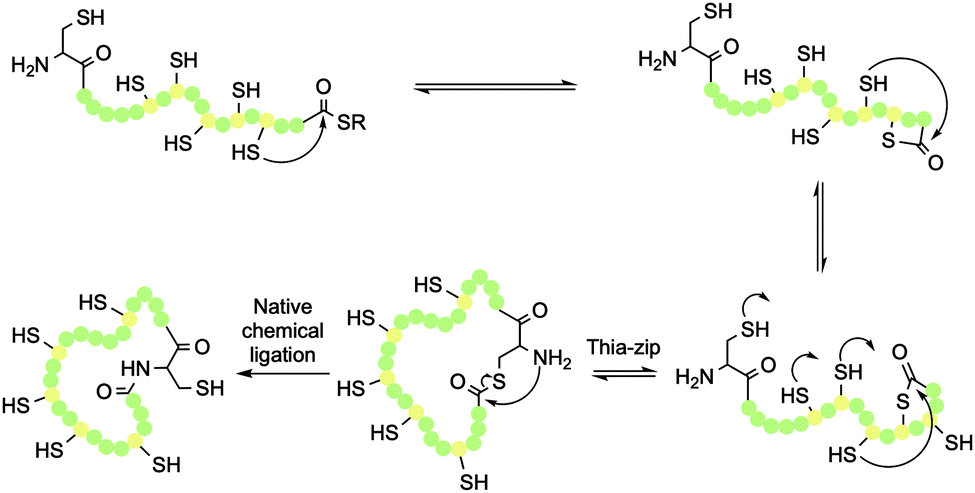 | ||
| Scheme 4 Thia-zip ring expansion and resulting head-to-tail native chemical ligation (NCL) in cyclotide synthesis. | ||
A significant advancement in Boc-SPPS of cyclotide sequences was the development of trityl-associated mercaptopropionic acid−leucine (TAMPAL) resin, which enables the use of all proteinogenic amino acids as the C-terminal thioester residue.25 The development of automated microwave-assisted methods of Boc-SPPS was another significant step forward for the field, improving synthetic yields and purity while also reducing assembly times to overnight reactions.26 Previous manual syntheses typically took 10–14 days for chain assembly, cleavage, and purification.
Nevertheless, methods for the Fmoc-SPPS of cyclotides have been developed, and multiple examples of both natural and engineered cyclotides synthesised with this methodology have been reported. For cyclotide synthesis methodologies that rely on NCL-cyclisation, the thioester-piperidine incompatability of Fmoc-SPPS is typically avoided by generating the C-terminal thioester in solution after the peptide has been cleaved from the resin. Recently, an alternative approach has been explored whereby peptides were synthesised by Fmoc-SPPS on 2-chlorotrityl chloride (2-CTC) resin.27 Peptide release from the resin can then be achieved under mild conditions (1% TFA), leaving side chain protecting groups intact and enabling head-to-tail macrocyclisation to be completed via standard amide bond formation conditions. A benefit of this strategy is that it is very flexible, enabling traceless macrocyclisation between any two residues in the sequence.
Cyclisation
Once the peptide has been deprotected, either in solution or on resin, NCL can be initiated by addition of a suitable buffer. This is typically phosphate buffer in the presence of excess tricarboxyethylphosphine (TCEP), which maintains the thiols in their reduced state. Once a thioester has formed between the N-terminal Cys thiol and the C-terminal thioester, an S,N-acyl transfer of the thioester intermediate results in formation of a peptide bond, while also regenerating the original Cys thiol (Scheme 4). This traceless cyclisation strategy can be achieved with any of the six Cys residues in the sequence by designing the SPPS to place the chosen Cys residue in the N-terminal position.The cyclisation process is believed to occur via a thia-zip mechanism, which is initiated by transthioesterification of the C-terminal thioester with an internal thiol. Successive ring expansions through reversible thiol-thiolactone exchanges in the direction of the N-terminus ultimately afford thiolactone formation with the N-terminal Cys residue thiol (Scheme 4). At this point, an irreversible S,N-acyl transfer occurs, giving the desired head-to-tail macrolactam. For a 31-mer cyclotide, there are 100 atoms in the backbone ring. As such, one step head-to-tail cyclisation is expected to be slow. Kinetic studies have shown that the zip-assisted cyclisation can be completed in 6 h and is 100 times faster than the unassisted one-step cyclisation.28
Chemoenzymatic cyclisation of cyclotides is a growing area of research, with current interest sparked by the work of Tam and co-workers who identified butelase 1, the fastest peptide ligase known, in 2014.29 Typically, linear sequences produced by SPPS need only be extended by a few residues in order to introduce the processing site for ligase enzymes. These cyclisations are often highly selective due to the very specific nature of ligase recognition sequences. Recently, an asparaginyl endopeptidase, MCoAEP2, was identified from Momordica cochinchinensis. It is one of the fastest cyclases reported to date and was used to efficiently cyclise the backbone of an engineered MCoTI-II cyclotide derivative with anti-angiogenic activity.30
Oxidation
Cyclotide synthesis ultimately requires oxidation of Cys residues in the correct pairings, and with six thiols present, 15 different arrangements are possible. Some chemical syntheses have limited this connectivity challenge by enabling the selective deprotection of different Cys residues. In the syntheses of cyclopsychotride A, circulin A, and circulin B, four Cys residues were S-methylbenzyl (Mbzl)-protected, while the remaining two Cys residues were S-acetamidomethyl (Acm)-protected.31 The peptides were cleaved from the resin using HF, which also removed the Mbzl protecting groups, leaving the Acm groups intact. The cleaved peptides were then extracted into 8 M urea containing TCEP, which maintained the free thiol groups in a reduced state. After dialysis to reduce the urea concentration and promote peptide folding, cyclisation was achieved by NCL and the formation of the first two disulfide bonds was promoted by the addition of DMSO. By only revealing four of the six thiols, the number of possible disulfide bond isomers was reduced from 15 to three. The isomers could then be separated by HPLC (∼30% yield), and the isomer with the correct disulfide connectivity could be confirmed by partial acid hydrolysis. Subsequent treatment of the resulting peptide with iodine led to removal of the two Acm groups and subsequent oxidation.In some cases, this two-step process is not necessary because the target cyclotide has a strong tendency to adopt the native CCK conformation. In these instances, NCL cyclisation and oxidation can be achieved in a one-pot procedure. The trypsin inhibitor cyclotide MCoTI-II is one such example, where it was shown that one-pot NCL-cyclisation and refolding could be achieved by dissolving the unprotected peptide and glutathione in carbonate buffer at pH 7.5, resulting in excellent conversion to MCoTI-II (>95% by HPLC).32
With their well-defined structure, resistance to proteolytic degradation, and ability to cross membranes, cyclotides are attractive molecular scaffolds for drug design. Increasingly sophisticated engineering approaches may be used to augment the natural antimicrobial properties of cyclotides, which appear to arise from membrane binding and disruption. Further work is required to outline the mechanism of their antimicrobial activity in vivo.
Lanthipeptides
Structure
Lanthipeptides are highly constrained polycyclic peptides produced by a wide variety of bacteria.33 They are among the best studied of the RiPPs and are characterised by the presence of the side chain thioether-linked bisamino acids lanthionine (Lan) and/or 3-methyllanthionine (MeLan) (Fig. 4a). In addition to Lan/MeLan bridges, other non-proteinogenic amino acids such as dehydroalanine (Dha), dehydrobutyrine (Dhb), S-[(Z)-2-aminovinyl]-(3S)-3-methyl-D-cysteine (AviMeCys) and labionin (Lab) are also often present (Fig. 4a). The Lan/MeLan bridges can be sequential, as in type A lanthipeptides such as nisin and epidermin, or highly overlapping, as in type B lanthipeptides such as duramycin and cinnamycin (Fig. 4b and c). Consequently, type A lanthipeptides have a relatively elongated and flexible conformation, whereas the type B lanthipeptides are more compact and globular. During biosynthesis, the Lan/MeLan bridges are formed post-translationally by the Michael addition of cysteine side chain thiols with Dha or Dhb residues respectively, which in turn are generated by dehydration of serine or threonine residues (Fig. 4d).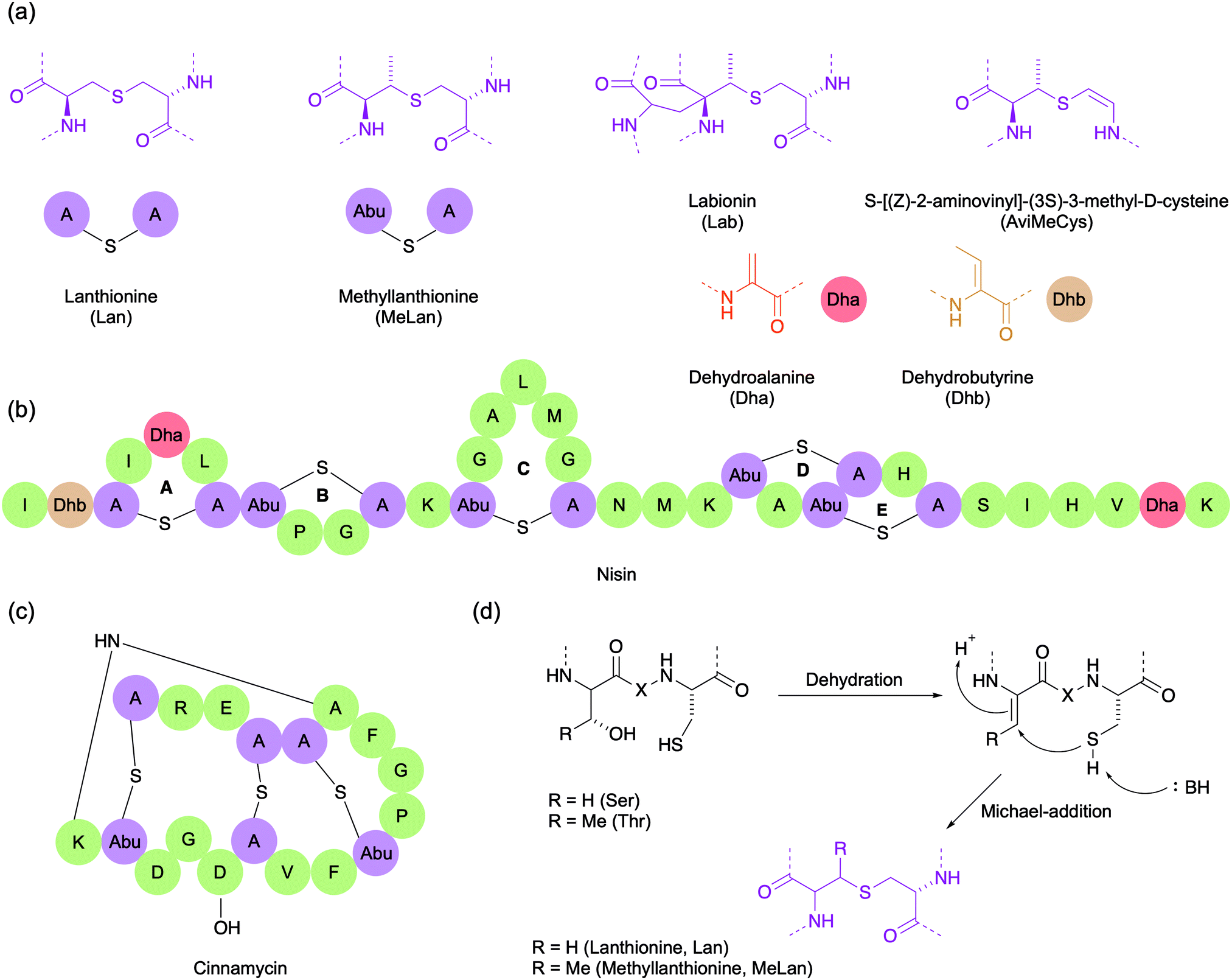 | ||
| Fig. 4 (a) The structures of some amino acids that are characteristically present in lanthipeptides. (b) The structure of the type A lanthipeptide nisin. (c) The structure of the type B lanthipeptide cinnamycin. (d) The mechanism of Lan/MeLan biosynthesis. X represents an intervening peptide sequence. | ||
Function
Many lanthipeptides display antibiotic activity against a range of Gram-positive bacteria, including multidrug resistant strains. These antimicrobial lanthipeptides are commonly referred to as lantibiotics. Two key targets of lantibiotics that have been identified are lipid II, a key precursor in bacterial cell wall biosynthesis, and phosphatidylethanolamine, a phospholipid and the substrate of phospholipase A2. Those that bind lipid II cause cell lysis by either preventing cell wall biosynthesis through its sequestration, or by forming of lipid II–peptide pores in the membrane.34,35 These mechanisms of action are unique to the lantibiotics, and because they have seen limited use in the clinic, many pathogenic bacteria have yet to develop resistance. Furthermore, the dual mechanisms of action associated with lipid II binding are likely responsible for the lack of resistance observed to the prototypical lantibiotic nisin, which has been used as a preservative in food for over 40 years. However, there are still many lantibiotics whose target is unknown. Like lasso peptides and cyclotides, the constrained conformation of lantibiotics typically results in good resistance to proteolytic degradation and high target specificity, and also makes them attractive for epitope grafting.For all these reasons, lantibiotics are promising candidates for the next generation of antimicrobial therapeutics. However, like many peptide drugs, they require engineering to overcome poor pharmacological properties. For example, despite its often nanomolar activity against Gram-positive bacteria, nisin is not suitable as a therapeutic agent in humans. This is due to its low stability in neutral or basic pH and its reactivity to nucleophiles such as water and thiols. Nevertheless, there are several lantibiotics in clinical or preclinical development including NVB302 (a derivative of actagardine), duramycin, and NAI-107 (microbisporicin).5,36,37 Recently, mutacin 1140 (MU1140) and its analogues have been extensively investigated by Oragenics, Inc. as lead compounds for the treatment of C. difficile-associated disease (CDAD). In 2019, the company began preclinical evaluation of enteric-coated capsules of OG253, a point-mutated analogue of MU1140 (Phe1Ile), where it displayed a favourable safety profile in rats.38
Synthesis
It can be difficult to isolate lanthipeptides from natural sources in sufficient quantities for in-depth study or therapeutic use in humans. Despite advances in genome mining enabling the identification of many lanthipeptide biosynthetic clusters, a robust heterologous expression system for large-scale production remains elusive. Alternatively, SPPS has been successfully used for the total synthesis of several lanthipeptides, though large-scale application currently appears economically infeasible. We direct the reader to an excellent review by Ongey and Neubauer, and the references therein, for further discussion of this issue.33Desulfurisation
The first total synthesis of a lanthipeptide, nisin A, was achieved in 1988 by Shiba and co-workers (Fig. 5a).39 The researchers synthesised disulfide-bridged analogues of all five rings of nisin (A–E), at which point desulfurisation was used to convert these cyclic peptides to their lanthionine equivalents. The rings were then coupled to afford the full lanthipeptide structure. The tedious nature of the procedure and its low overall yield means that it is not especially practical for the general synthesis of lanthipeptides.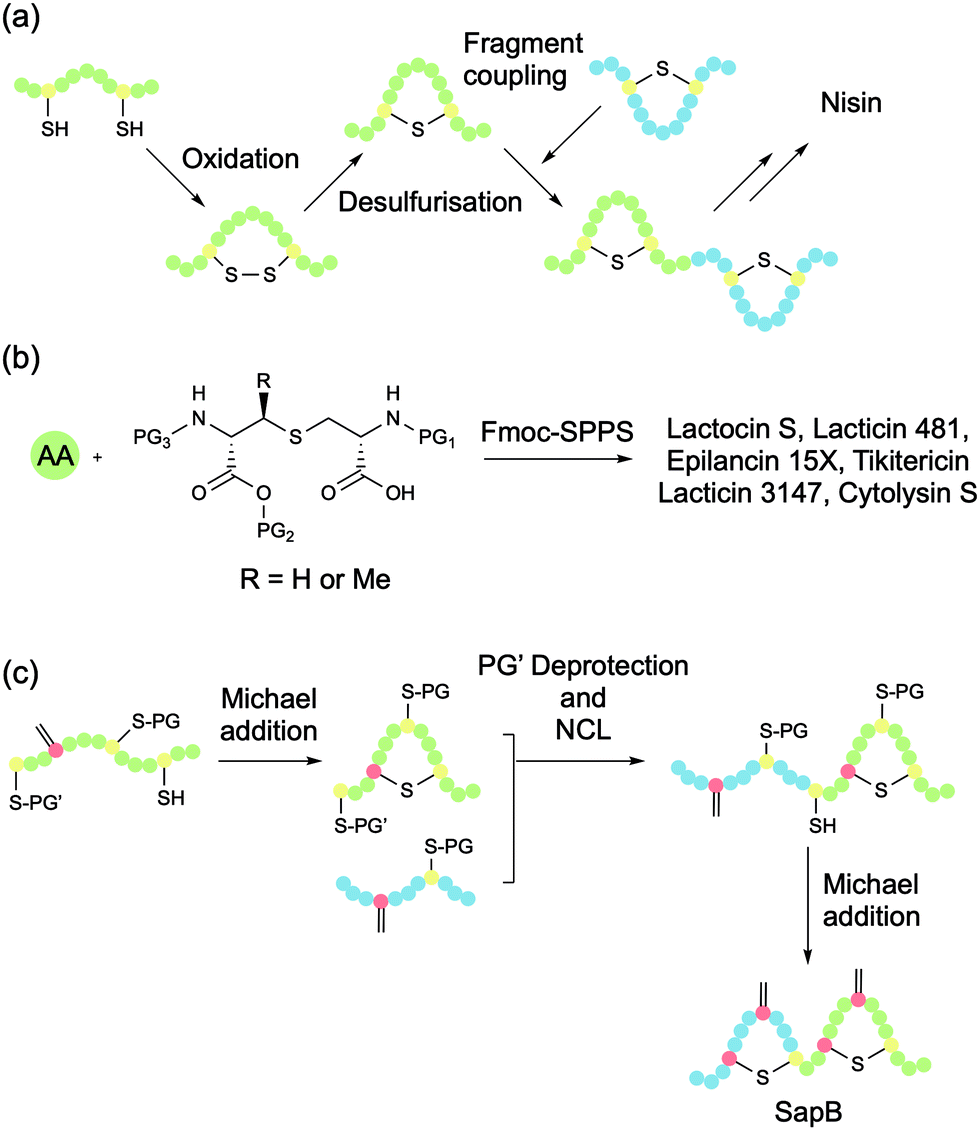 | ||
| Fig. 5 Strategies for the total synthesis of lanthipeptides. (a) Total synthesis of nisin by the Shiba group using a desulfurisation approach to generate Lan bridges and then condensing peptide fragments. (b) Using orthogonally protected bisamino acid building blocks to introduce Lan bridges in the total synthesis of several antimicrobial lanthipeptides. (c) Total synthesis of SapB by Chen et al. using an orthogonally protected cysteine strategy to enable in situ dehydro amino acid formation and regioselective Lan bridge formation. Adapted with permission from H. Chen, Y. Zhang, Q. Q. Li, Y. F. Zhao, Y. X. Chen and Y. M. Li, J. Org. Chem. 2018, 83, 7528–7533. Copyright (2020) American Chemical Society. | ||
Spatola and co-workers proposed that the mechanism of the desulfurisation approach involves the generation of a Dha residue followed by Michael addition that can be initiated by slightly basic conditions (pH 8).40 The general utility of this methodology is restricted by the lack of regio- and stereocontrol that can occur during the Michael addition step. Knerr and van der Donk demonstrated the importance of lanthionine stereochemistry in the lantibiotic lacticin 481 by chemically synthesising and biologically evaluating its stereoisomers.41 These investigations showed that substitution of any of the three DL-Lan/MeLan residues with the corresponding LL stereoisomers completely abolished biological activity. Nevertheless, several groups have used the explicit generation of Dha and subsequent Michael addition with Cys thiols to form Lan/MeLan bridges. In most of these examples, the products were formed as a single diastereoisomer, likely due to the particular conformation of the peptide sequences in question. However, all of these examples contained only a single Lan bridge. Regio- and stereoselective formation of multiple and overlapping Lan bridges presents a more significant challenge.
Orthogonally protected bisamino acids
Tabor and co-workers developed a general method for the solid-phase synthesis of side chain bridged peptides using orthogonally protected bisamino acid building blocks (Fig. 5b).42 The side chain linked bisamino acid is synthesised such that one amino acid is protected for typical Fmoc-synthesis (i.e., with an Nα-Fmoc group and a free acid), while the other is protected by an allyl and an allyloxycarbonyl (Alloc) group. This second set of protecting groups can both be removed using Pd(0), leaving acid- and base-labile peptide protecting groups intact. The bisamino acid is incorporated into the linear chain using standard Fmoc-SPPS conditions, and the chain is extended until the second residue requires incorporation. At this point, the allyl and Alloc group are selectively removed and the residue is coupled, which simultaneously affords the desired cyclisation. A key advantage of this methodology is the ability to generate the desired bridge as a single regio- and diastereoisomer. The lanthionine bisamino acid can be synthesised using a Mitsunobo reaction to generate N-trityl-β-iodo serine, and then reacting it with the side chain of Fmoc-Cys-OtBu. This orthogonal protecting groups strategy has been extended to facilitate the synthesis of overlapping lanthionine bridges by using lanthionine residues protected with Alloc/allyl-, β-(trimethylsilyl)ethoxycarbonyl (Teoc)/acid trimethylsilylethyl (TMSE)-, and p-nitrobenzyloxycarbonyl (pNz)/p-nitrobenzyl (pNb) groups. These methods have been excellently summarised in a 2014 review by Tabor.43The orthogonally protected Lan/MeLan strategy has become the methodology of choice for the total synthesis of several lantibiotics including lactocin S, lacticin 3147 (consisting of two peptides called A1 and A2), epilancin 15×, lacticin 481, cytolysin S, and tikitericin (antimicrobial activity not yet investigated), as well as some analogues.43–45 VanNieuwenhze and co-workers have also used a similar orthogonal protection strategy to synthesise the D-ring of mersacidin, a methicillin-resistant S. aureus (MRSA) active lantibiotic, which contains a sensitive AviMeCys bridge (Fig. 4a), showing that the methodology can be extended beyond the typical Lan/MeLan bridge.46
Oragenics, Inc. have applied their differentially protected orthogonal lanthionine technology (DPOLT) to the development of a synthetic equivalent of the lantibiotic mutacin 1140, MU1140-S. The analogue entered pre-clinical development against MRSA, C. difficile, M. tuberculosis, vancomycin-resistant Enterococci (VRE), and anthrax around a decade ago. In 2019, the researchers reported the first chemical synthesis of an analogue of the fused C/D ring of mutacin 1140 using an orthogonally protected lanthionine strategy (Fig. 6).47 In this way, a cysteamine (Cya) was incorporated into the rings, rather than the natural (S)-aminovinyl-D-cysteine (AviCys). The unsaturated double bond of AviCys had been shown to be non-essential for biological activity and its replacement with Cya simplifies the synthetic strategy.
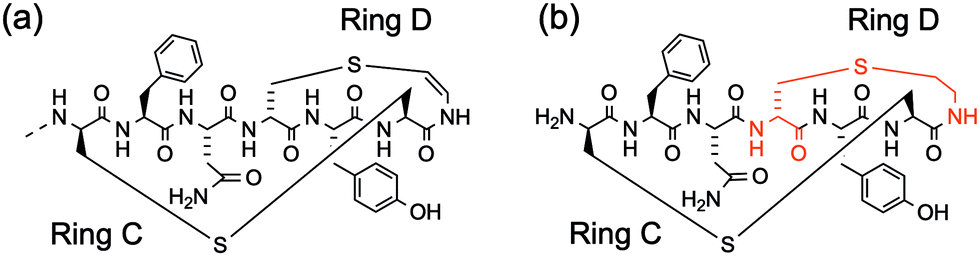 | ||
| Fig. 6 (a) The structure of the fused C/D rings of mutacin 1140. (b) The cysteamine analogue of the C/D rings synthesised by Park and co-workers using an orthogonally protected lanthionine strategy. | ||
Michael addition
While orthogonally protected Lan/MeLan strategies have dominated total syntheses of lanthipeptides in recent years, there is still merit in the Michael addition approach. In 2018, Chen et al. reported a novel Michael addition methodology for constructing lanthipeptides that utilises several commercially available and orthogonally protected Cys building blocks (Fig. 5c).48 With selective deprotections, the regioselectivity of Lan bridge formation can be controlled. The Lan bridges are formed by Michael addition reactions with Dha residues, which are generated in situ by bisalkylation and elimination of selectively deprotected Cys residues, either on the solid support or in solution. The researchers used this strategy to synthesise the lanthipeptide SapB, which is important for aerial mycelium formation in Streptomyces coelicolor.The synthesis of SapB was conducted by splitting the 21-mer into two sequences, SapB (1–9) and SapB (10–21), both of which contain one Lan bridge (Scheme 5). SapB (10–21) was synthesised on 2-chlorotrityl by standard Fmoc-SPPS, introducing a Boc-L-thiazolidine-4-carboxylic acid (Boc-Thz-OH) residue at the N-terminal position. Three Cys residues were introduced to the sequence, each with a different protecting group: Trt, Acm, and StBu (Scheme 5a). With the sequence assembled on the resin (2), the StBu protected Cys at position 13 was deprotected, and the resulting free thiol was activated with 2,5-dibromoadipamide (DBAA).49 Elimination of the alkylated product afforded the desired Dha. Global deprotection (including thiol Trt) and resin cleavage with TFA cleavage cocktail afforded the free peptide 3, with only the Acm and Thz protecting groups remaining intact. After HPLC purification, Michael addition between the newly revealed Cys thiol and Dha was effected by addition of alkaline aqueous buffer (50 mM Na2HPO4, pH 8.5) at room temperature, shaking for 1.5 h. Treatment of the resulting cyclised peptide 4 with MeONH2·HCl and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) at pH 4.0 deprotected the N-terminal Thz to reveal a Cys residue, which could then be used for native chemical ligation (5).
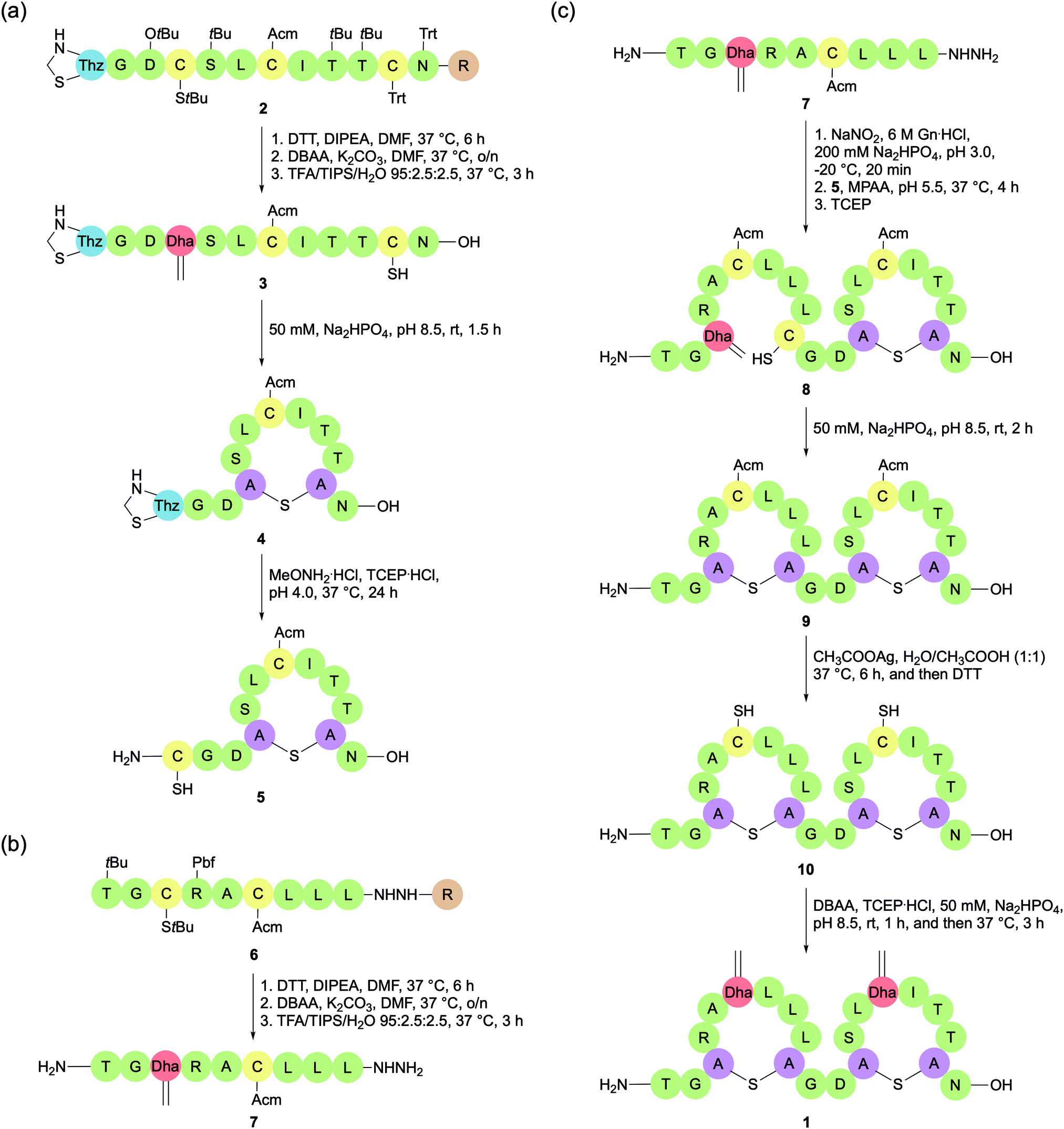 | ||
| Scheme 5 Synthesis of SapB by an orthogonal Cys deprotection strategy. (a) Synthesis of SapB fragment 10–21. (b) Synthesis of SapB fragment 1–9 hydrazide. (c) Native chemical ligation of fragments 5 and 7. Thz, L-thiazolidine; Dha, dehydroalanine; Dhb, dehyrobutyrine, Adapted with permission from H. Chen, Y. Zhang, Q. Q. Li, Y. F. Zhao, Y. X. Chen and Y. M. Li, J. Org. Chem. 2018, 83, 7528–7533. Copyright (2020) American Chemical Society. | ||
SapB (1–9) was separately prepared on 2-chlorotrityl resin by standard Fmoc-SPPS, including two Cys residues protected with Acm and StBu respectively. The peptide was attached to the resin via a hydrazide linkage (Scheme 5b, 6). As previously, a Dha residue was revealed from the StBu protected Cys, and the resulting peptide was globally deprotected and cleaved from the resin with a TFA cleavage cocktail. The resulting peptide hydrazide was purified by HPLC to afford 7. With the two peptide fragments in hand, NCL was used to connect them via a traceless linkage (Scheme 5c, 8). The second Lan bridge was then formed between the free Cys and Dha residues by addition of the same alkaline aqueous buffer as used previously, shaking for 2 h (9). Removal of the two remaining Acm protecting groups and subsequent conversion of the newly revealed Cys residues to Dha afforded the final product SapB (1). The synthesis was achieved in 9.8% yield after HPLC purification based on peptide 3 over six steps.
The main flaw in this methodology is poor stereoselectivity of the Michael addition reactions. In this instance, the second addition gave a mixture of epimers, which were then separated by HPLC. It is also important to note that the stereochemistry of natural SapB has not been determined. As such, it cannot be confirmed that the product synthesised by Chem et al. was in fact SapB without further investigation. Another issue is that the methodology does not yet demonstrate the introduction of Dhb residues, and consequently the formation of the commonly occurring MeLan bridge. However, with each step achieving reasonable yields and by using commercially available amino acids, lanthipeptides may be produced on a milligram scale. This is important if these compounds are to be fully tested for their biological properties. Both these attributes make this an attractive methodology for lanthipeptide synthesis, but it has yet to be applied to the synthesis of a lantibiotic.
Given the natural antimicrobial activity of lanthipeptides, there is great interest in their modification to improve their potency, solubility, and stability, such that they can be used as novel therapeutics in the clinic. While generation of analogues by mutagenesis has been used extensively, these methods require isolation and structural elucidation of the resulting active compounds to confirm their identities. Chemical synthesis provides a complementary platform for making analogues with improved pharmacochemical properties, some of which would not be accessible by biological means.
The characteristic Lan/MeLan bridges of lanthipeptides are susceptible to oxidation, which can eliminate their biological activity. In order to improve oxidative stability, Vederas and co-workers reported the SPPS of analogues of lactocin S, where the sulfur atom of its Lan bridges had been systematically replaced by a methylene unit.50
One of the analogues retained the full activity of the native peptide, while also displaying a marked increase in oxidative stability. This result was impressive as previous attempts to replace thioether bridges in lantibiotics had resulted in reduced activity. Replacing the sulfur atoms of the Lan/MeLan bridges in lacticin 3147 with either an oxygen atom or a two-carbon linker reduced its antimicrobial activity or abolished it respectively.33 The same group has also reported the solid-phase synthesis of a bis(desmethyl) analogue of the lacticin 3147 A2 peptide in which the two MeLan bridges were replaced by Lan bridges.33 Interestingly, the analogue maintains potent synergistic activity with the lacticin 3147 A1 peptide, but loses its inherent independent antimicrobial activity. This study demonstrates that the A2 peptide has two independent mechanisms of action and makes clear the role that chemical synthesis can play in understanding mechanisms of antimicrobial activity.
Conclusions
Widespread antimicrobial resistance risks plunging the world into a ‘postantibiotic era’. In order to avoid this outcome, it is essential that we investigate new classes of antibiotics, in particular those that have a propensity to overcome traditional resistance mechanisms. Ribosomally-synthesised and post-translationally modified peptides (RiPPs) are poised to fill this niche due to their potent antimicrobial activity, high stability, and multiple mechanisms of action. The increasing sophistication of chemical synthesis is making these structurally complex natural products ever more tractable, while also providing a facile means by which to introduce structural modifications that may improve their drug-like properties. Alongside bioengineering, chemical synthesis may yet see RiPPs reach the clinic as novel antimicrobial agents for human use.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Engineering and Physical Sciences Research Council and Royal Society (Wolfson Research Merit Award). We also thank B. H. Gan, J. Gaynord, and T. Deingruber for their critical proofreading of the manuscript.References
- J. O’Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, 2016 Search PubMed.
- G. A. Hudson and D. A. Mitchell, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- N. Poorinmohammad, R. Bagheban-Shemirani, J. Hamedi and A. van Leeuwenhoek, Int. J. Gen. Mol. Microbiol., 2019, 112, 1477–1499 CAS.
- N. Petrosillo, G. Granata and M. A. Cataldo, Front. Med., 2018, 5, 96 CrossRef.
- H. Grasemann, F. Stehling, H. Brunar, R. Widmann, T. W. Laliberte, L. Molina, G. Doring and F. Ratjen, Chest, 2007, 131, 1461–1466 CrossRef CAS.
- S. Tan, G. Moore and J. Nodwell, Antibiotics, 2019, 8, 117 CrossRef CAS.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H. C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS.
- H. Martin-Gómez and J. Tulla-Puche, Org. Biomol. Chem., 2018, 16, 5065–5080 RSC.
- J. D. Hegemann, M. De Simone, M. Zimmermann, T. A. Knappe, X. Xie, F. S. Di Leva, L. Marinelli, E. Novellino, S. Zahler, H. Kessler and M. A. Marahiel, J. Med. Chem., 2014, 57, 5829–5834 CrossRef CAS.
- F. Saito and J. W. Bode, Chem. Sci., 2017, 8, 2878–2884 RSC.
- C. Clavel, K. Fournel-Marotte and F. Coutrot, Molecules, 2013, 18, 11553–11575 CrossRef CAS.
- N. R. Braffman, F. J. Piscotta, J. Hauver, E. A. Campbell, A. James Link and S. A. Darst, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1273–1278 CrossRef CAS.
- R. Soudy, L. Wang and K. Kaur, Bioorg. Med. Chem., 2012, 20, 1794–1800 CrossRef CAS.
- R. Hammami, F. Bédard, A. Gomaa, M. Subirade, E. Biron and I. Fliss, Amino Acids, 2015, 47, 417–428 CrossRef CAS.
- H. Martin-Gómez, F. Albericio and J. Tulla-Puche, Chem. – Eur. J., 2018, 24, 19250–19257 CrossRef.
- M. Chen, S. Wang and X. Yu, Chem. Commun., 2019, 55, 3323–3326 RSC.
- S. Naimi, S. Zirah, R. Hammami, B. Fernandez, S. Rebuffat and I. Fliss, Front. Microbiol., 2018, 9, 1–13 CrossRef.
- S. J. De Veer, M. W. Kan and D. J. Craik, Chem. Rev., 2019, 119, 12375–12421 CrossRef CAS.
- M. Pränting, C. Lööv, R. Burman, U. Göransson and D. I. Andersson, J. Antimicrob. Chemother., 2010, 65, 1964–1971 CrossRef.
- I. C. M. Fensterseifer, O. N. Silva, U. Malik, A. S. Ravipati, N. R. F. Novaes, P. R. R. Miranda, E. A. Rodrigues, S. E. Moreno, D. J. Craik and O. L. Franco, Peptides, 2015, 63, 38–42 CrossRef CAS.
- J. A. Camarero and M. J. Campbell, Biomedicines, 2019, 7, 31 CrossRef CAS.
- C. Gründemann, K. G. Stenberg and C. W. Gruber, Int. J. Pept. Res. Ther., 2019, 25, 9–13 CrossRef.
- N. L. Daly, S. Love, P. F. Alewood and D. J. Craik, Biochemistry, 1999, 38, 10606–10614 CrossRef CAS.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- M. Cemazar and D. J. Craik, J. Pept. Sci., 2008, 14, 683–689 CrossRef CAS.
- C. A. Álvarez, P. A. Santana, O. Luna, C. Cárdenas, F. Albericio, M. S. Romero and F. Guzmán, Molecules, 2018, 23, 1–15 Search PubMed.
- J. P. Tam, Y. A. Lu and Q. Yu, J. Am. Chem. Soc., 1999, 121, 4316–4324 CrossRef CAS.
- G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS.
- J. Du, K. Yap, L. Y. Chan, F. B. H. Rehm, F. Y. Looi, A. G. Poth, E. K. Gilding, Q. Kaas, T. Durek and D. J. Craik, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- J. P. Tam, Y. A. Lu, J. L. Yang and K. W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8913–8918 CrossRef CAS.
- P. Thongyoo, E. W. Tate and R. J. Leatherbarrow, Chem. Commun., 2006, 2848–2850 RSC.
- E. L. Ongey and P. Neubauer, Microb. Cell Fact., 2016, 15, 1–16 CrossRef.
- R. Pokhrel, N. Bhattarai, P. Baral, B. S. Gerstman, J. H. Park, M. Handfield and P. P. Chapagain, Phys. Chem. Chem. Phys., 2019, 21, 12530–12539 RSC.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discovery, 2006, 5, 321–323 CrossRef CAS.
- Lancovutide (Moli1901) Inhalation Solution Study in Adolescents and Adults With Cystic Fibrosis, https://clinicaltrials.gov/ct2/show/NCT00671736, accessed 4 February 2021.
- C. Brunati, T. T. Thomsen, E. Gaspari, S. Maffioli, M. Sosio, D. Jabes, A. Løbner-Olesen and S. Donadio, J. Antimicrob. Chemother., 2018, 73, 414–424 CrossRef CAS.
- N. V. Rajeshkumar, J. A. Kers, S. Moncrief, A. W. Defusco, J. H. Park and M. Handfield, Toxicol. Appl. Pharmacol., 2019, 374, 32–40 CrossRef CAS.
- K. Fukase, M. Kitazawa, A. Sano, K. Shimbo, H. Fujita, S. Horimoto, T. Wakamiya and T. Shiba, Tetrahedron Lett., 1988, 29, 795–798 CrossRef CAS.
- A. K. Galande, J. O. Trent and A. F. Spatola, Pept. Sci., 2003, 71, 534–551 CrossRef CAS.
- P. J. Knerr and W. A. Van Der Donk, J. Am. Chem. Soc., 2013, 135, 7094–7097 CrossRef CAS.
- S. Bregant and A. B. Tabor, J. Org. Chem., 2005, 70, 2430–2438 CrossRef CAS.
- A. B. Tabor, Bioorg. Chem., 2014, 55, 39–50 CrossRef CAS.
- S. Mukherjee, L. Huo, G. N. Thibodeaux and W. A. Van Der Donk, Org. Lett., 2016, 18, 6188–6191 CrossRef CAS.
- B. Xu, E. J. Aitken, B. P. Baker, C. A. Turner, J. E. Harvey, M. B. Stott, J. F. Power, P. W. R. Harris, R. A. Keyzers and M. A. Brimble, Chem. Sci., 2018, 9, 7311–7317 RSC.
- P. García-Reynaga, A. K. Carrillo and M. S. VanNieuwenhze, Org. Lett., 2012, 14, 1030–1033 CrossRef.
- K. Kirichenko, J. D. Hillman, M. Handfield and J. H. Park, J. Pept. Sci., 2019, 25, e3214 CrossRef CAS.
- H. Chen, Y. Zhang, Q. Q. Li, Y. F. Zhao, Y. X. Chen and Y. M. Li, J. Org. Chem., 2018, 83, 7528–7533 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- A. C. Ross, S. M. K. McKinnie and J. C. Vederas, J. Am. Chem. Soc., 2012, 134, 2008–2011 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |