
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli responsive dynamic transformations in supramolecular gels
Santanu
Panja
 and
Dave J.
Adams
*
and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 1st March 2021
Abstract
Supramolecular gels are formed by the self-assembly of small molecules under the influence of various non-covalent interactions. As the interactions are individually weak and reversible, it is possible to perturb the gels easily, which in turn enables fine tuning of their properties. Synthetic supramolecular gels are kinetically trapped and usually do not show time variable changes in material properties after formation. However, such materials potentially become switchable when exposed to external stimuli like temperature, pH, light, enzyme, redox, and chemical analytes resulting in reconfiguration of gel matrix into a different type of network. Such transformations allow gel-to-gel transitions while the changes in the molecular aggregation result in alteration of physical and chemical properties of the gel with time. Here, we discuss various methods that have been used to achieve gel-to-gel transitions by modifying a pre-formed gel material through external perturbation. We also describe methods that allow time-dependent autonomous switching of gels into different networks enabling synthesis of next generation functional materials. Dynamic modification of gels allows construction of an array of supramolecular gels with various properties from a single material which eventually extend the limit of applications of the gels. In some cases, gel-to-gel transitions lead to materials that cannot be accessed directly. Finally, we point out the necessity and possibility of further exploration of the field.
 Santanu Panja | Santanu Panja received his MSc degree with specialization in organic chemistry from the University of Calcutta, India in 2011. He obtained his PhD in 2017 from the University of Kalyani, India. After postdoctoral studies at the Université de Bordeaux, France, he joined the group of Prof. Dave J. Adams at the University of Glasgow in 2018 under the Newton International Fellowship program. Presently he is associated with the same group as a research associate. He is an Associate Editorial board member of Mini Reviews in Organic Chemistry (MROC) and Current Smart Materials (CSM). His current research focuses on developing methods to control hydrogel properties. |
 Dave J. Adams | Dave Adams received his DPhil from the University of York in 1999. After carrying out postdoctoral research at York, Leeds, and Leicester, he joined Unilever R&D for four years. In 2008, he joined the University of Liverpool before joining the University of Glasgow in 2016. His current research interests encompass many areas of materials chemistry including supramolecular polymers and gels. |
1. Introduction
One route for preparing interesting and useful functional soft materials is to use supramolecular gels. Such gels are formed by the self-assembly of small organic molecules into larger structures by non-covalent forces like π–π stacking, hydrogen bonding, van der Waals interactions, ion pairing, solvophobic forces and ion coordination;1,2 these interactions lead to fiber formation that immobilizes the solvent.3–5 This solvent entrapment ultimately results in the formation of a viscoelastic semi-solid. In contrast to their polymeric counterparts, supramolecular gels derived from low molecular weight gelators (LMWGs) involve discrete molecular components with well-defined chemical structures. As a consequence, tuning of the materials is conceptually possible at the molecular level.6–8 These gels are easy to make and if structural designs are appropriate, it is then possible to utilize these small molecules as functional materials by controlling the gel properties.9–12The design and synthesis of LMWGs is complicated because the structural requirements of the gelators that influence self-aggregation are often not well understood.13–15 In spite of reasonable progress in the field, it is still unclear why some molecules form gels and some do not. In particular, the correlation between precursor, assembly conditions and self-assembled structure is not fully understood.16–18 Furthermore, prediction of gel properties is difficult because of the pathway dependency of the gelation.19–22 The final gel properties are often influenced by the kinetics of self-assembly which means a number of different supramolecular structures can be constructed under similar final conditions from a single building block by varying the method used to carry out the self-assembly.19,20 However, typically once the self-assembly reaches the final set of conditions, the assembled structures usually do not change their properties with time. Most gels are typically devoid of self-regulating behavior which means time dependent changes in gel properties are not possible until the environmental conditions in which the gel system exists are changed. However, such materials potentially become switchable when exposed to external stimuli. As the gel structure is maintained by non-covalent interactions, supramolecular gels are highly stimuli responsive where the properties can be modulated through external perturbation, for example by exploiting chemical, thermal, optical, electrical, mechanical, or magnetic stimuli.23–25 On perturbation, the response can bring about microscopic changes like changes in aggregation and morphology. Such a transformation enables a gel-to-gel transition resulting in an alteration of physical and chemical properties of the gel. This is an extremely powerful approach to adapt material properties.
As most of the synthetic gels exist in a kinetically trapped state, autonomous behavior is largely absent, and so the modification of gel properties mostly relies on external triggers or stimuli. However, under certain conditions, a subset of supramolecular gels can evolve and reconfigure their properties over time (Fig. 1). In such systems, initially a gel matrix is formed which gradually transforms into a different type of network without any external input. Such self-regulating switching between different gel states is relatively a new concept in material research to prepare exciting materials involving biomimicry. In some cases, self-regulating gel-to-gel transitions lead to materials that cannot be obtained directly or through most assembly modifications. Here, we summarize the different methods that allow gel-to-gel transitions. We have categorized the work into two approaches: (i) gel-to-gel transitions induced by stimuli-responsive post-assembly fabrication and (ii) time-programmable self-regulating gel-to-gel transitions. Each section is further divided depending upon the nature of the conditions used for reconfiguring the gel states. We also address how such methods can be explored in controlling material properties during assembly transitions.
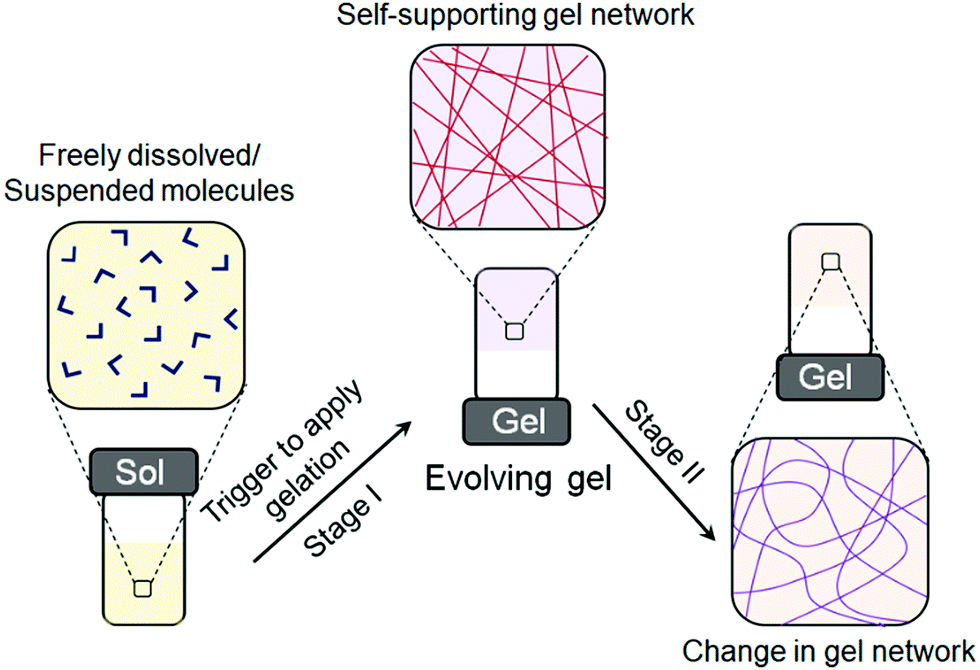 | ||
| Fig. 1 Cartoon representing formation of a self-supported gel network from initially discrete molecules on applying a trigger followed by a change in network structure with time. For traditional gels, a stimulus is applied externally at stage 2 to induce the changes. For systems that show self-regulating gel-to-gel transitions, no external stimulus is required at this stage. | ||
2. Gel to gel transitions and energy landscape
Supramolecular gels are formed by the self-assembly of an initially freely dissolved or colloidal dispersion of small building blocks (the LMWGs). The assembly process needs to be activated first by a trigger, after which one- or two-dimensional anisotropic structures are formed, followed by their entanglement into a 3D network. As a result, the solvent is retained and trapped inside the resulting self-supporting matrix through surface tension and capillary forces. Typically, only a low concentration of gelator is needed to trap the solvent and so even strong gels can have a final composition of less than 1 wt% of the LMWG.The self-assembly of LMWGs can be triggered by various techniques; a pH change, UV light irradiation, temperature change, addition of chemical analytes, or solvent switch have all been used to trigger the self-assembly. In order to form a gel, the environmental conditions need to be controlled such that the initially-freely dispersed molecules would prefer to associate with one another as opposed to remaining discrete. Ideally, when the conditions are changed to favour self-assembly, this leads to a situation where there is a significant amount of the free molecule present in solution whilst the self-assembly is occurring and the ‘on–off rate’ (the rate at which molecules add to and leave the assembled structure) is high (Fig. 2a).22,26–28 This means that the assembled structure can easily reach a low energy state, with errors in assembly being corrected during the process. In other words, if less favourable assembly happens, the fast exchange of building blocks facilitates a correction to a more energetically favourable state, ideally the state at a global thermodynamic minimum.26,27 For instance, da Silva et al. reported that, in case of relatively weak non-covalent bonds between peptide amphiphiles, the ongoing exchange of such molecules between the sample network and the medium is high, and the energy of the assembled structure is in equilibrium with the environment.29
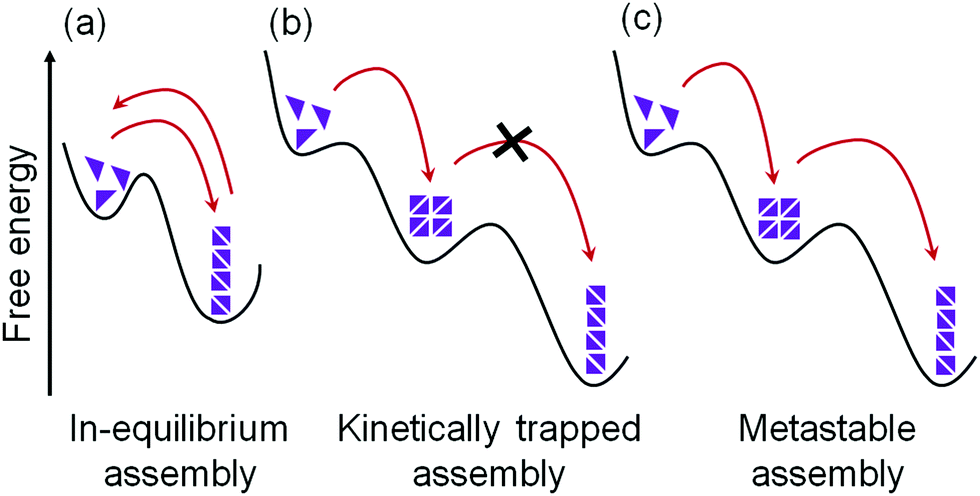 | ||
| Fig. 2 Schematic representation of possible energy landscapes during the self-assembly of supramolecular materials: (a) in-equilibrium assembly, (b) kinetically trapped assembly and (c) metastable assembly. The kinetically trapped state cannot overcome the energy barrier to reach the thermodynamic minimum unlike the metastable state. | ||
Unfortunately, this ideal case is not always possible. With stronger non-covalent interactions between building blocks, the dynamic exchange and consequently thermodynamic optimization become harder to achieve and sometimes may even be impossible.20,30,31 For traditional gel systems, the self-assembly process occurs at a high rate, or there are very ‘sticky’ molecules, meaning that errors are maintained and cannot be corrected during self-assembly.19,30 Hence, the self-assembled structures cannot reach the most stable state; instead, they exist as kinetically trapped structures (Fig. 2b).19,30–32 Although such a system is not at the thermodynamic minimum, a rearrangement into a more favourable state is not possible as there is little or no exchange of the molecules. The assembly is therefore trapped in a local energy minimum from which it cannot escape. This kinetically-trapped structure may be in a sufficiently deep energy well such that the gel states are stable, and their mechanical properties do not generally change over their useful lifetime. However, reconfiguration of such gel state is possible by inducing post-assembly fabrication (PAF) where exposure of the gel state towards some external stimuli such as heat can provide the system with enough energy to overcome the barrier and re-assemble into the more stable product.33,34 PAF typically allows transition between two different thermodynamically stable states on energy input (stimuli).
In some cases, matter is exchanged with the environment, and a slow transformation of a kinetically trapped state into the thermodynamically favoured state is possible.30,31,35,36 This kind of systems is referred to as metastable (Fig. 2c). For metastable gels, the formation and material properties are governed by both kinetics and thermodynamics where a kinetic product formed at first which slowly transforms into a thermodynamic one. Accessing a metastable state can allow autonomous programming of gel-to-gel transition. However, unlike PAF, no additional trigger/stimuli is required for rearrangement of the initially formed gel state. Initially, the gelation process can be controlled by kinetic factors that eventually dictates the final self-assembled structure and consequently physio-chemical properties of the material.
In all of this, a key part of the system is the solvent. Depending on the solvent, a wide number of parameters can be changed including polarity, hydrogen bond donor and acceptor ability, boiling point and viscosity. As such, it is normal for a gelator to only be able to gel a number of solvents, as opposed to all solvents. It can be hard to predict in advance which solvents will be gelled, although there has been recent progress using Hansen parameters.37,38 The solvent molecules cannot be ignored when considering the dynamic nature of such gels, but to the best of our knowledge there is little currently available data discussing the importance of solvent in gel-to-gel transitions. This is an area which could prove very fruitful in terms of increasing our understanding in the future.
3. Gel to gel transitions induced by stimuli responsive post-assembly fabrication
Typically, gels are used in their original form without any major changes in their mechanical properties. However, there are ways to adapt the material properties outside the range of the original gelator. One option is gel functionalisation after the assembly process has taken place. PAF aims to expand the range of applications for already known supramolecular structures without losing their self-assembly abilities.(i) Thermal annealing of supramolecular gels
Temperature is one of the most common stimuli applied in gel chemistry.39,40 When supramolecular gels are subjected to heating, they usually exhibit a gel-to-sol phase transformation driven by the weakening of the intermolecular interactions prevailing in gel system. On decreasing the temperature below the temperature of gelation (Tgel), reaggregation occurs due to the change in solubility followed by establishment of the hydrophilic/hydrophobic balance within the network. One specific example of thermo responsive supramolecular hydrogels was using a N-acetyl-galactosamine-appended amino acid gelator.41 Temperature induced gel–sol transition is commonly considered as a parameter of the strength of the intermolecular interactions existing in gel architectures.4,42,43 However, rare reports are also found where an increase in temperature stimulates aggregation.44–46 For example, Kimizuka et al. reported a metallogelator based on Co2+ coordinated-4-alkyl triazole complex that formed a gel upon heating.47 On lowering the temperature to 0 °C, the blue gel converted to a pink coloured solution, which formed a gel state again by increasing the temperature to room temperature. Such thermochromic behaviour accompanying a phase transformation occurs because of the reconfiguration of the coordination polymer system from tetrahedral complexes in the gel state to the metastable octahedral complexes in solution state.Typically, supramolecular gels are formed after cooling the gelator solution from an elevated temperature. Depending upon the thermal history, more specifically upon the cooling rate, the gel network may exist as a kinetically trapped state.32 For instance, Murata et al. showed that the microstructure of a cholesterol coupled azobenzene gel depends upon the cooling rate.48 While a rapidly cooling solution of the gelator favoured the formation of right-handed helices, slow cooling preferred left-handed helices formation. Interestingly, chirotopic switching from the right-handed to left-handed helix was achieved by slow heating but the reverse transition was not possible without dissolution of the gel. These results indicate that the right-handed helices network were the kinetically trapped metastable product, whilst the left-handed helical aggregates were the thermodynamically stable state. Furthermore, the metastable state persisted only if the cooling rate was higher than the rate of formation of the thermodynamic product. Hence, a heat-cool operation on a kinetically trapped state eventually drives the system towards the, or at least a, thermodynamic minimum state.48,49 This method is typically known as thermal annealing.
Annealing is applied for many kinetically trapped systems to achieve their local thermodynamic minimum.50–52 Although thermal annealing is a powerful strategy to avoid competition between self-assembly pathways, surprisingly little work has been carried out in this domain for supramolecular gels. In case of supramolecular gels, thermal annealing at a temperature above the melting temperature of the gel results in sol formation and re-gelling on cooling.53–56 Thermal annealing results in more homogeneous network formation56 by eliminating the errors in the initial assembly states and often causes substantial increase in robustness of the gels.57–59 Recently, thermal annealing has been shown to be also applicable to tune the material properties for multicomponent gel systems (Fig. 3).60 Two gelators were chosen and adjusted their concentration in such a way that the single component gels exhibit two distinctly different gel melting temperature (65 °C and 80 °C). On heating the multicomponent gel, sequential melting of the individual gels close to their respective gel melting temperature was recorded by temperature sweep rheology. The heat-cool operation led to a self-sorted network with a different microstructure to the initial gels, which resulted in a considerable increase in the storage modulus. Furthermore, heating the multicomponent gel to 65 °C followed by cooling subsequently allowed the selective melting and annealing of only one of the networks. However, heating a gel is not always desirable as some gelators containing specific functional groups can undergo bond cleavage,61,62 Diels–Alder reactions63–65etc. at the elevated temperature that causes permanent chemical changes on gelator backbone. Such limitations of thermal annealing demands new and sophisticated methods of annealing gel systems under mild conditions.
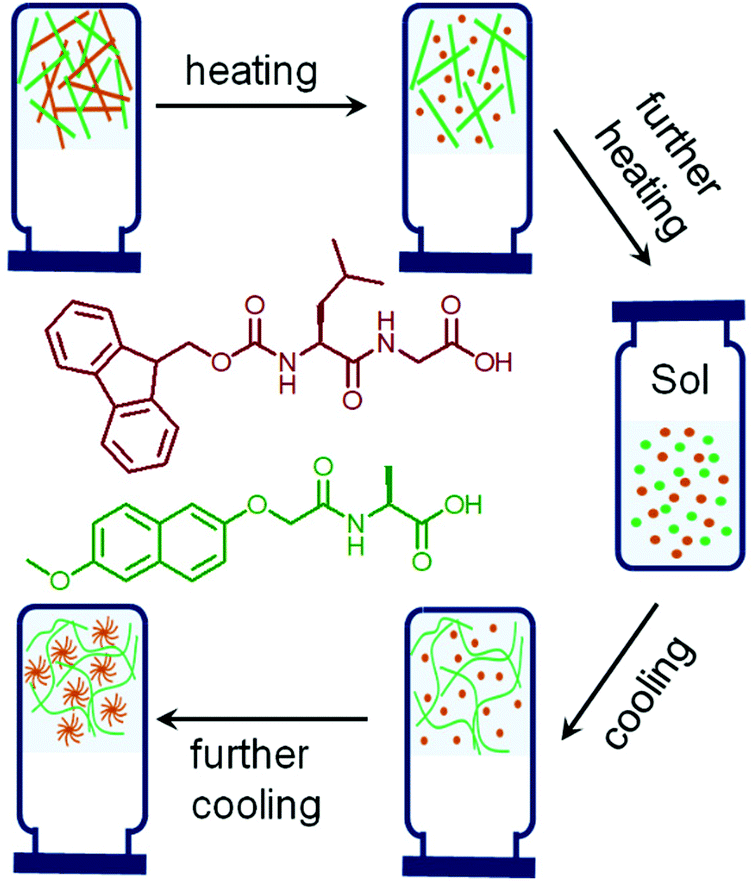 | ||
| Fig. 3 Cartoon representing thermal induced sequential disassembly and reassembly of networks in a multicomponent self-sorted hydrogel system. The chemical structures used to prepare the multicomponent hydrogel are shown and color coded.60 | ||
Recent research focuses on designing more complex systems where a gel state can be directly tailored to form a different gel state by heating without the formation of a sol state. These types of gels are referred to as “thermally bisignate” gels where reorganization of the hydrogen bonded networks, preferentially driven by solvent exchange allows temperature induced gel-to-gel transitions. A pioneering work was carried out by the Aida group where supramolecular polymerization was designed to occur upon both heating and cooling.66 They synthesized trialkyl substituted gallic acid-based metalloporphyrin monomer with eight H-bonding amide units on backbone. The resulting supramolecular aggregate obtained from hydrocarbon-based solvents at ambient temperature was exceptionally thermodynamically stable and hardly depolymerized upon heating to 110 °C. However, the presence of alcohols acted as competitive solvents (described as hydrogen-bond scavengers) and destroyed the existing H-bonded network leading to depolymerization. Whether on heating or cooling, the alcohol molecules detached from the amide groups of the monomers, making them eligible for polymerization.66,67 Xie et al. utilized this concept and reported temperature dependent accessing of two distinctly different gel states obtained from a zwitterionic amphiphile 3,3′-(octadecylazanediyl)dipropionic acid (Fig. 4).68 On heating, a turbid hydrogel at 25 °C gradually transformed to a translucent gel at 50 °C. The gel-to-gel transition temperature was determined to be 43 °C. The change in turbidity of the material was attributed to the conversion of a low-temperature crystalline branched-tubular gel into a high-temperature entangled worm-like micellar gel. Such “thermally bisignate” gels provide opportunity of accessing two fundamentally different gel networks just by successive cooling-heating cycles.68–70
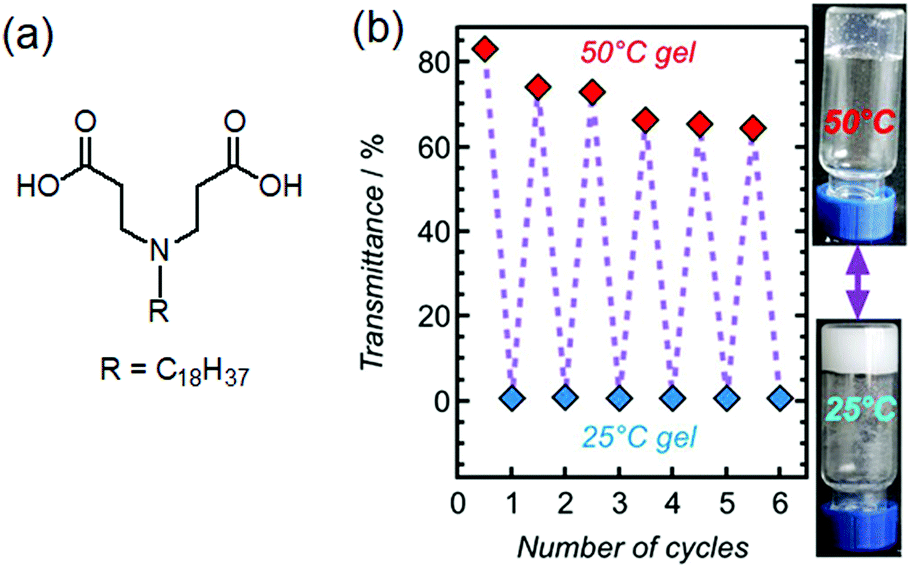 | ||
| Fig. 4 (a) Chemical structure of the gelator studied by Xie et al.68 (b) Pictures of 2% hydrogel at pH = 5.37 at 25 °C and 50 °C together with its transmittance data during successive cooling–heating cycles. Figure (b) is adapted from ref. 68 with permission from the Springer Nature, copyright 2017. | ||
(ii) Enzymatic reaction
Enzymes are abundant biomacromolecules that play catalytic roles in numerous physiological and metabolic processes.71,72 As enzymatic reactions are highly substrate specific as well as regio- and stereo-specific in nature, they are widely used for constructing various functional materials.73–75 Among various functional materials, supramolecular hydrogels are promising for biological applications. Enzyme-triggered assembly of small molecules provides a facile approach to study the properties of the assembled structures in a cellular environment which is important for exploring the possibilities of supramolecular gels in biomedical research.76–78Enzyme responsive assemblies are usually formed by enzyme-reactive centres on a gelator skeleton which leads to either formation or cleavage of a covalent bond upon enzymatic reaction. Such chemical transformations result in a change in chemical structure of the building blocks which in turn perturbs the existing non-covalent interactions, leading to self-assembly. For example, while phosphatases catalyze phospho–ester bond cleavage, thermolysin catalyzes the formation of an amide bond via reverse hydrolysis. Both these strategies have been effectively used to trigger gelation by converting a pro-gelator to an active gelator. Yang et al. reported alkaline phosphatase induced dephosphorylation of the pro-gelator Fmoc-tyrosine phosphate to generate Fmoc-tyrosine (the gelator) which self-assembles to form nanofibres and so a gel.79 Ulijn's group investigated hydrogelation of a tripeptide produced by a chemical reaction between diphenylalanine dipeptide and Fmoc-phenylalanine in the presence of thermolysin.80 Many enzymes such as β-lactamase,81 chymotrypsin,82 β-galactosidase,83 esterase,84 glucose oxidase,85,86 peroxidase87 phosphatase,79,88 and thermolysin80,89 have been explored to trigger and control molecular gelation. Abul-Haija and Ulijn illustrated that dynamic transformation of different nanostructures could be achieved by coassembly of sequence adaptive peptide obtained through dynamic combinatorial chemistry in presence of polysaccharides and thermolysin.90 When a mixture of anionic Fmoc-cysteic acid (CA), cationic Fmoc-lysine (K) and phenylalanine amide (F) coexist with thermolysin, the rate of amide bond formation and hydrolysis was comparable and no self-assembly occurred in absence of the polysaccharide template. In the presence of the polysaccharide templates, amide bond formation could be accelerated which in turn led to a dynamic peptide library consisting of two different Fmoc-dipeptides (CA–F and K–F). The presence of the cationic polysaccharide chitosan led to the formation CA–F peptide, whilst the anionic heparin resulted in formation of K–F selectively. The co-assemblies formed by CA–F and K–F were nanosheets and nanotubes in their respective gel states.
It is possible to induce a gel-to-gel transformation involving a solution state as an intermediate by using an enzyme in a hydrogel system. Yang et al. sequentially performed the tyrosine kinase catalyzed phosphorylation followed by the alkaline phosphatase induced dephosphorylation reaction on a pentapeptide hydrogelator and explored the kinase/phosphatase switch to regulate self-assembled structures (Fig. 5).91 When kinase was added to the hydrogel prepared in presence of adenosine triphosphate (ATP), phosphorylation of the corresponding peptide led to return to the solution state. The phosphorylated compound served as a precursor for phosphatase. Further treatment of the resulted solution with phosphatase regenerated the pentapeptide through dephosphorylation and caused reappearance of the gel with time.
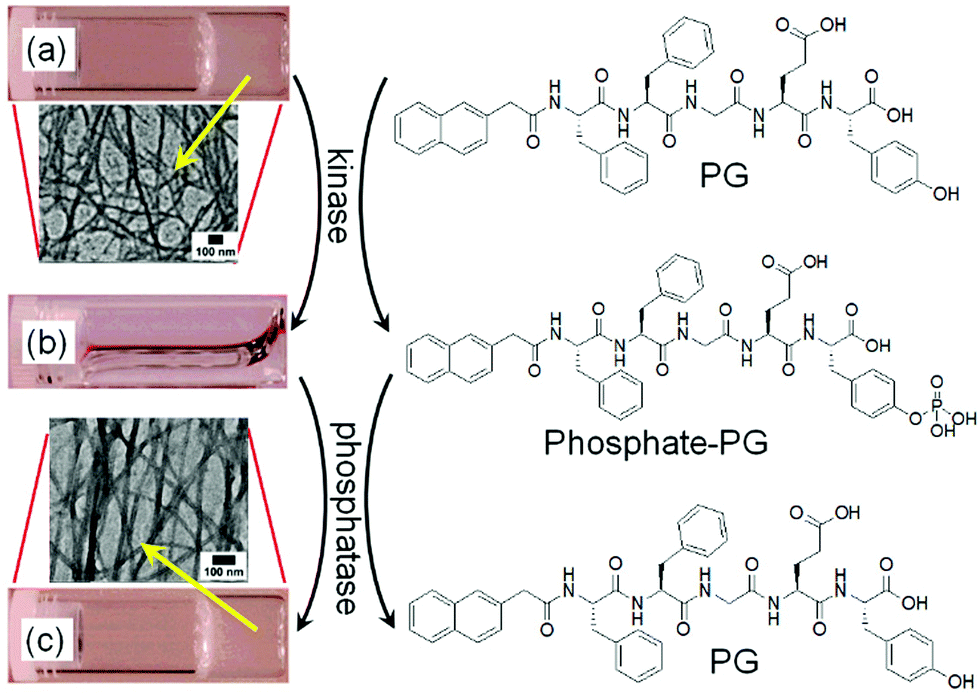 | ||
| Fig. 5 Change in chemical structures of the pentapeptide gelator (PG) and the phosphorylated compound (phosphate-PG) with corresponding phase transformations catalyzed by kinase and phosphatase. The hydrogel of PG (a) transformed into a solution (b) when treated with kinase and ATP. (c) the hydrogel of PG restored by adding phosphatase. Insets represent TEM images of the nanofibres of PG formed by adjusting the pH (a), and the restored nanofibres of PG by enzymatic dephosphorylation of phosphate-PG. Reproduced from ref. 91 with permission from the American Chemical Society, copyright 2006. | ||
(iii) UV light induced reaction
Photoinduced morphology transformation is an important and convenient method in gel chemistry.4,42,92 Unlike other stimuli, light offers the opportunity for ambient simulation of part of a system in a spatially controlled manner.93–96 Photoresponsive gels are extensively used in cell culture and optoelectronics.97–100 In these gels, usually the gelators are directly functionalized with photoresponsive chromophoric units, or noncovalently binds with the photoresponsive moieties. The photoresponsive groups, upon irradiation, absorb light of a specific wavelength and undergo photoreactions such as molecular switching, bond cleavage or bond formation leading to a change in physical and chemical properties like dipole moment, conjugation, geometric structures and electronic properties. Such changes alter the molecular level supramolecular interactions and result in transformations of the self-assembly. A number of chromophores including azobenzene, stilbene, diacetylene, spiropyran, coumarin, and salicyl-imine have been utilized to prepare photoresponsive gels. A common pattern of reactivity of the chromophores can be identified which includes trans–cis photoisomerization, photodimerization and photopolymerization, electrocyclic reactions, photoredox reaction etc.101–104 Such photochemical reactions often either lead to gelation or trigger gel-to-sol phase transformations. However, few structurally designed LMWGs are found that execute changes in material properties like viscosity, rigidity, morphology transformations, swelling, or electronic properties in response to light without phase transformations. Here, we discuss the different approaches that have been used to devise optically responsive gel-to-gel transitions.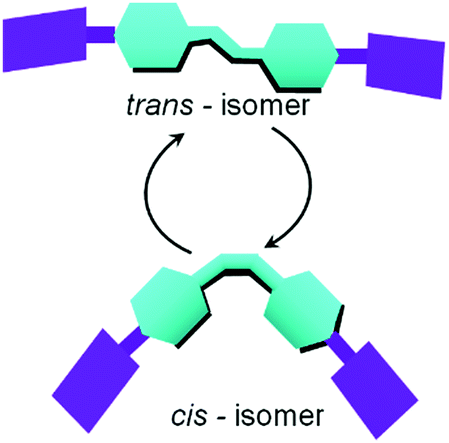 | ||
| Fig. 6 Schematic representation trans/cis (Z → E) photoisomerization in presence of light resulting in significant changes to the molecular shape. | ||
The vast majority of photo-isomerized gels belong to the azobenzene family. For azobenzene-based gelators, photoisomerization usually causes either formation or disruption of gels.105–110 Zhao et al. presented a fluoroazobenzene-based hydrogel, where E → Z isomerization of the azo group could disrupt the physical cross-links leading to less elastic hydrogel formation.111 Similar observations were documented by the Ravoo group, where the E-configuration of the hydrogelators produced stiff gels due to more stable and planar structure, whilst photoinduced conversion to the Z-isomer disrupted the planarity, resulting in a softer gel with a lower storage modulus.112 In order to maintain the assembled structure upon photoirradiation, multicomponent systems were proposed by Xie et al. who reported a hydrogel system prepared by mixing a L-glutamic acid-based amphiphile (OGAc) and an azobenzene containing pyridinium-salt (AzoPy) (Fig. 7).113 The hydrogel exhibited thermally reversible deswelling at ambient temperature (20 °C) but converted to swollen state upon UV-irradiation. On exposure of UV-light, the peak at 344 nm for the shrunken gel slowly decreased, while a new peak at 420 nm was generated over time indicating trans to cis-azobenzene formation. The photoirradiated swollen gel consisted of wider and twisted fibrous structure than the original gel. However, a drawback of azobenzene gels is that thermal re-isomerisation can be very fast that results in quick reversion to the trans-form. Because of this, the trans-to-cis conversion never reaches completion and photoisomerization typically leads to a photostationary state (the equilibrium state where the composition of both isomers remains unchanged on further irradiation)92 with a 30–50% conversation to the cis-isomer.42 Ajayaghosh and co-workers demonstrated UV-triggered gel-to-gel transformation of a metal–organic azobenzene gel with about 37.5% conversion to the cis isomer in the photostationary state.114
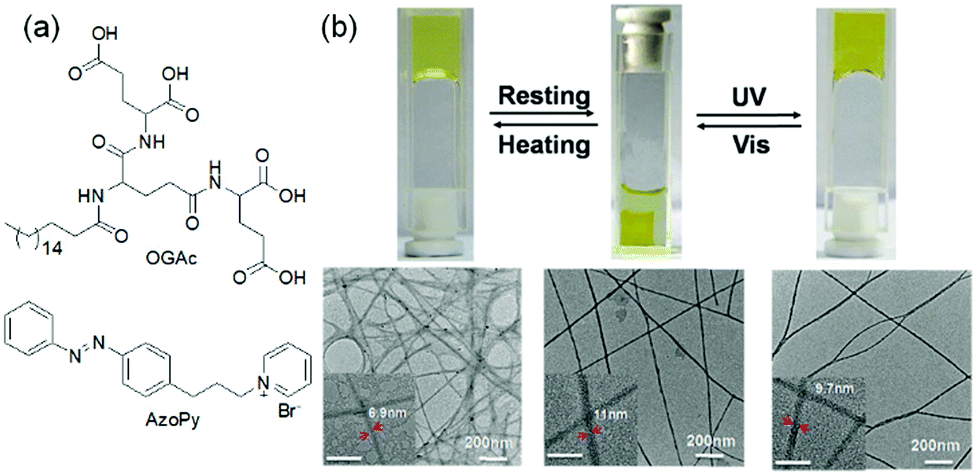 | ||
| Fig. 7 (a) Molecular structures of amphiphile OGAc and AzoPY. (b) Reversible deswelling and swelling transitions of a two-component hydrogel of OGAc and AzoPy on resting and after exposure to UV light. Reproduced from ref. 113 with permission from the Royal Society of Chemistry. | ||
Like azobenzenes, stilbenes undergo trans-to-cis isomeric switching in a similar way, but with relatively high conversion rate and quantum yields.115 Again here the trans-stilbenes are thermally more stable than cis-stilbenes and UV-illumination usually causes gel-to-sol transformations.115–117 A methodology to modify gel properties by using a stilbene gelator as one component in a two component self-sorted gel has been reported (Fig. 8).118 Importantly, the stilbene gelator alone could form a hydrogel and a gel-to-sol transition was achieved by irradiation in a single component system. The two-component hydrogel was prepared by a decrease in pH of the mixture of the stilbene gelator and a dipeptide-based gelator. Due to their different pKa, the slow pH decrease resulted in sequential self-assembly and formation of a self-sorted system. On irradiation, only the stilbene gelator underwent isomerization while the non-photoreactive gelator remained unchanged. The gel state maintained its integrity, however due to selective removal of stilbene assemblies through the gel-to-sol transition, the stiffness of the overall gel decreased. By using photomask, it was possible to convert selected regions of the two-component system into the single component gel, and hence prepare photopatterned multicomponent networks.
 | ||
| Fig. 8 (a) Chemical structures of the stilbene gelator and the dipeptide gelator. (b) Cartoon representing photo tuning of the multicomponent gel formed. From left to right: a slow pH decrease resulted in sequential fibre formation for both stilbene (blue segments) and dipeptide (red segments) leading to a multicomponent self-sorted gel. (blue and red lines for the fibres that consist of stilbene and dipeptide, respectively). The fibrous network formed by the stilbene gelator is then selectively removed by irradiation with light. Figure (b) is adapted from ref. 118 with permission from the Springer Nature, Copyright 2015. | ||
Recently, hydrazine-based Schiff bases have been used to synthesized photochromic gels. Such gelators exhibit E → Z photoswitching across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and produce remarkable changes in absorption and emission spectra. A naphthalimide-hydrazine Schiff base was reported to show emission colour changes from orange to blue under sunlight.119 Che et al. mixed a photoactive acylhydrazone Schiff base with a photo-inert gelator and found that the properties of the two-component gel could be tuned by visible light (350–570 nm).120 Light irradiation caused trans–cis photoisomerization across the CN bond of the photoactive component while the second component remained unaffected. Photoirradiation resulted in 30 nm hypsochromic shift during the gel–gel transition. Further, the irradiated gel showed weak emission as well as reduced mechanical strength. Interestingly, the single component gel of the photoactive Schiff base showed gel-to-sol transition upon photo exposure. Based on these results, it was proposed that the two component system was formed by the orthogonal self-assembly of two gelators. Elimination of one fibre from the mixed system under visible light was responsible for inferior mechanical properties of the irradiated gel. Use of a photo mask further allowed patterned two-component gel to be constructed by selectively removing one component from a specific area of the gel.
N bond and produce remarkable changes in absorption and emission spectra. A naphthalimide-hydrazine Schiff base was reported to show emission colour changes from orange to blue under sunlight.119 Che et al. mixed a photoactive acylhydrazone Schiff base with a photo-inert gelator and found that the properties of the two-component gel could be tuned by visible light (350–570 nm).120 Light irradiation caused trans–cis photoisomerization across the CN bond of the photoactive component while the second component remained unaffected. Photoirradiation resulted in 30 nm hypsochromic shift during the gel–gel transition. Further, the irradiated gel showed weak emission as well as reduced mechanical strength. Interestingly, the single component gel of the photoactive Schiff base showed gel-to-sol transition upon photo exposure. Based on these results, it was proposed that the two component system was formed by the orthogonal self-assembly of two gelators. Elimination of one fibre from the mixed system under visible light was responsible for inferior mechanical properties of the irradiated gel. Use of a photo mask further allowed patterned two-component gel to be constructed by selectively removing one component from a specific area of the gel.
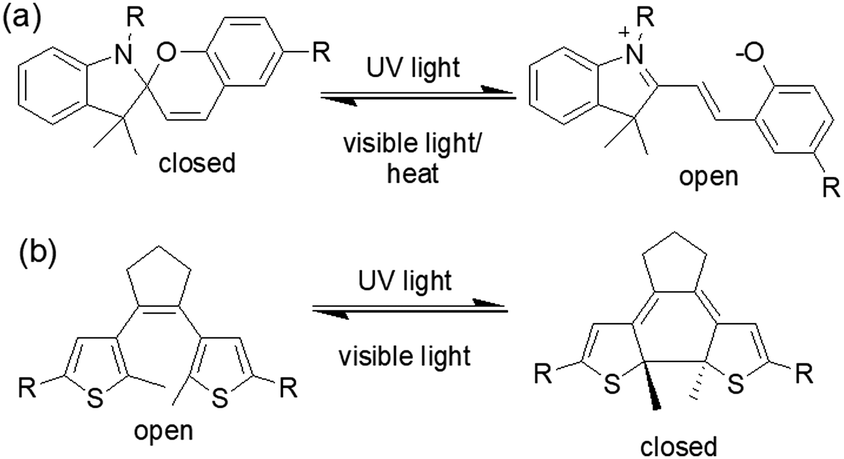 | ||
| Fig. 9 Photoresponsive ring closing and opening of (a) spiropyran and (b) dithienylethene molecules. | ||
An interesting example was carried out by Chen et al. showing photophysical properties of spiropyran gel can be altered by light (Fig. 10).125 In their design, aromatic stacking was used as the driving force in order to maintain the assembled structure after photoreaction. A spiropyran-functionalized dendron (DG) formed a yellow gel on cooling a toluene/benzene solution to 0 °C. Extensive π–π stacking exerted by the dendron was ascribed to the formation of long fibres of width 50 nm in the gel state. On UV-irradiation, the closed form of the spiropyran was converted to the open form, which produced a new absorption band in the region 500–700 nm. The gel did not collapse but showed a colour change from yellow to purple-blue. The presence of the open form of the spiropyran group allowed the purple-blue gel to emit relatively strong red fluorescence. Repetitive transition between these two gel states was possible by alternating UV and visible light irradiation. An opportunity afforded by spiropyrans is that they act as a proton source on ring closing. Using catalytic amounts of spiropyran, it is possible to achieve spatial control over gelation.134 However, a serious issue is the short lifetime of the open form of the spiropyrans in the excited state, and they return back to the closed form in the dark. Even the photoresponsive product in the gel state which has a longer stability shows a significant loss of product within minutes.124,135 In comparison, dithienylethenes (DTE) exhibit relatively photo-stable products.136,137 The closed-ring structure formed upon UV exposure shows high stability from months to years at ambient temperature, making such systems suitable for chemical information storage.138,139
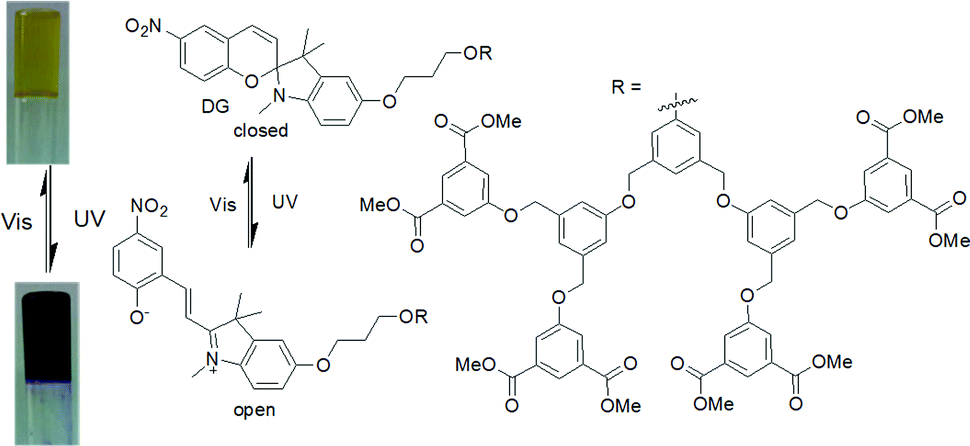 | ||
| Fig. 10 Photoresponsive structural change of the dendron organogelator DG responsible for the colour change during gel to gel transitions. Reproduced from ref. 125 with permission from the John Wiley and Sons, copyright 2009. | ||
The first photoswitching dithienylethene organogel was reported by Wang et al. in 2006 (Fig. 11).140 The gelator (BTE-NA-Chol) was synthesized by coupling of a diarylethene segment and a cholesteryl-naphthalimide with the intention that the cholesterol part aid to gelation through van der Waals interaction and the naphthalimide groups contribute to fluorescence of the molecules. The gel exhibited a photochromic response associated with a colour change from yellow to red when irradiated. The colour change was attributed to the cyclization of the dithienylethene functionality. The emission intensity of the irradiated gel significantly increased at around 460 nm than the original yellow gel. Similarly, Feringa and co-workers developed a lysine–glycine functionalised dithienylethene which showed a colour change in gel state on light exposure.141 The yellow hydrogel produced by the ring-open dithienylethene when irradiated at 312 nm turned to a red gel due to the formation of the more conjugated and rigid structure in the closed-ring isomer. Surprisingly, such a transition did not apparently influence the macroscopic appearance or the mechanical properties of the gel. The closed to open ring conversion was reversible using visible light (>500 nm) and the original yellow colour of the gel was recovered. Reversible switching between two gel states was achieved over several cycles without a loss of efficiency. Xiao et al. introduced a dithienylethene coupled pyridinium gelator suitable for non-destructive fluorescent storage and switching.142 In their study, isomerisation to the closed- and open-ring structures in acetonitrile was achieved by irradiation at 365 and 620 nm, respectively. The pale brown gel formed by the ring-open-ring state changed to blue because of increased π-electron delocalization in the closed form. Recently, Park and co-workers introduced a reversible and multistate addressable fluorescent organogel formed by the mixing of spiropyran and diarylethene gelators.143 In the co-assembled gel, while the spiropyran switching can be thermally activated, the diarylethene dopant exhibited selective photoinduced turn-on switching. A combinational logic gate circuit was established by applying these two orthogonal stimuli (heat and light) that allowed access to different gel states with different fluorescence colours. Such logic circuits based on optical memory are crucial for future optoelectronic applications.144,145 Another advantage of dithienylethenes is that, if the open-ring molecules are chiral, cyclization lead to diastereomeric products which can have different aggregation and optical properties.146
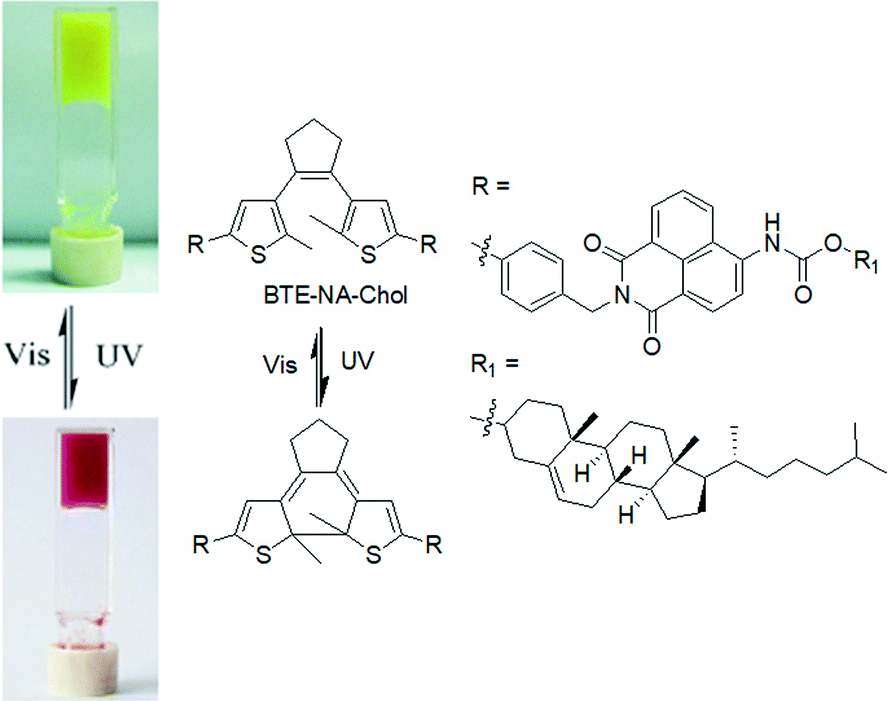 | ||
| Fig. 11 Photoresponsive switching of BTE-NA-Chol and the corresponding colour change of the organogels. Reproduced from ref. 140 with permission from the Royal Society of Chemistry. | ||
Coumarin-based gelators when irradiated with light >300 nm149 undergo a [2+2] dimerization that alters the intermolecular interactions (Fig. 12a). There are a number of possibilities for the product formation such as syn–syn, anti–syn, syn–anti and anti–anti cyclobutanes depending on the orientation of the molecules in the aggregated states. However, in gel chemistry, the effect of photodimerization on morphology transition and mechanical properties is most studied. For instance, dimerization of coumarin under 365 nm was found by Draper et al. to convert a peptide hydrogel into a stiffer material.153 Photoreaction resulted in increase in turbidity of the gel and a decrease in fibre diameter from 42 nm to 39 nm which indicates dimerization occurred within the fibres rather than cross-linking between fibres. Similarly, the Parquette group illustrated photodimerization of a lysine-functionalized 7-(diethylamino)-3-coumarin carboxylic acid-based hydrogelator within the fibre under 365 nm light that enhanced the mechanical properties.154 However, a drawback of these systems is that, although partial crosslinking enhances the mechanical properties of the gels, prolonged irradiation disrupts the network due to stiffening153 and can lead to precipitation in some cases.154 Therefore, the irradiation time is crucial in controlling the gel properties. Another issue is, as the generated cyclobutane ring is highly stressed, reverse ring opening is possible on further irradiation by 260–280 nm light. Hence, the choice of wavelength if light is vital for coumarin gelators. Yu et al. prepared an alkyl amide bearing a coumarin chromophore that showed reversible photodimerization in the gel phase without losing its mechanical integrity.155 The cyclohexane gel was subjected to photoirradiation successively by >300 nm and <280 nm light and the variation of material properties was investigated by fluorescence spectroscopy and scanning electron microscopy (SEM) studies. Irradiation with >300 nm light significantly decreased the emission of the gel at 390 nm due to dimerization of coumarin; further irradiation with <280 nm light stimulated bond cleavage and caused a slow recovery of the emission spectra. SEM revealed a morphological transformation of the fibres from helical fibres to lumps upon irradiation with >300 nm and then again changed to a sponge like aggregates when irradiated with <280 nm.
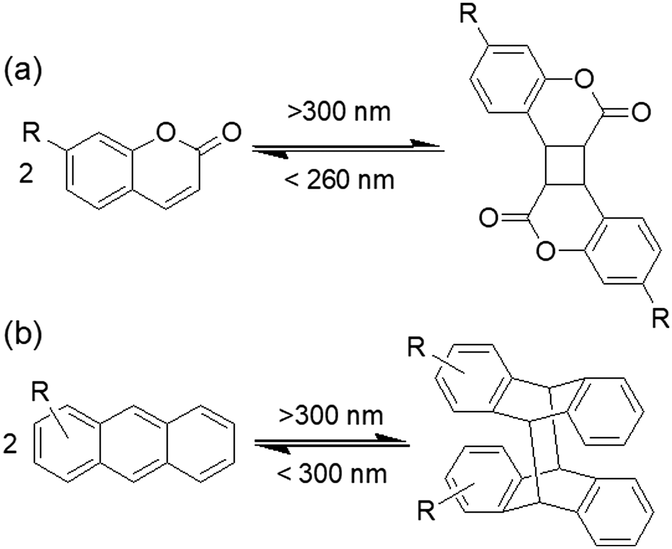 | ||
| Fig. 12 Reversible photodimerization of (a) coumarin and (b) anthracene. | ||
In a related fashion, anthracene-based systems can undergo a [4+4] dimerization when irradiated with light >300 nm and the reverse cycloaddition occurs in presence of light of <300 nm (Fig. 12b).156 Again, depending upon the relative position of the anthracenes during the cycloaddition reaction, syn-head to tail, anti-head to tail, syn-head to head and anti-head to head stereodimers can be formed. However, in the gel state, photodimerization can be highly selective and lead predominantly to a single photodimer.157,158 For example, Wang et al. designed an anthracene modified urea organogelator which showed head-to-tail cycloadduct formation upon on irradiation with >300 nm light without showing major deformation of the gel structure.159
Photocoupling of alkenes has also been used in photoresponsive gel-to-gel transitions. Jung et al. reported the gels obtained from Schiff base formation between a calixarene–acylhydrazone and a stilbene–aldehyde when exposed to UV light, the mechanical strength of the gel was enhanced (>1300-fold increase in rheological moduli) because of the formation of cyclobutane units that contributed to the tolerance against sheer stress.160 Furthermore, the stilbene units of the Schiff base exhibited strong blue emission due to H-aggregation in the gel state (Fig. 13). On heating the gel to 60 °C, solvent encroachment into the gel network modulated the stacking pattern of the molecules which resulted in partial conversion to J-aggregation and a change in fluorescence to green. The authors noted that, in presence of UV-light, while the H-aggregate underwent cycloaddition between the stilbene functionalities, the J-aggregate remained unperturbed. As a result, a partially cyclobutane conjugated gel could be obtained where the blue fluorescence was suppressed. The addition of DMSO led to destruction of the J-aggregates present in the cyclobutane gel and consequently the green emission was diminished. It was finally shown that, thermal induced tuning of molecular aggregation followed by photo cross-linking of the H-aggregates allowed controlled fluorescence photopatterning of the gels.
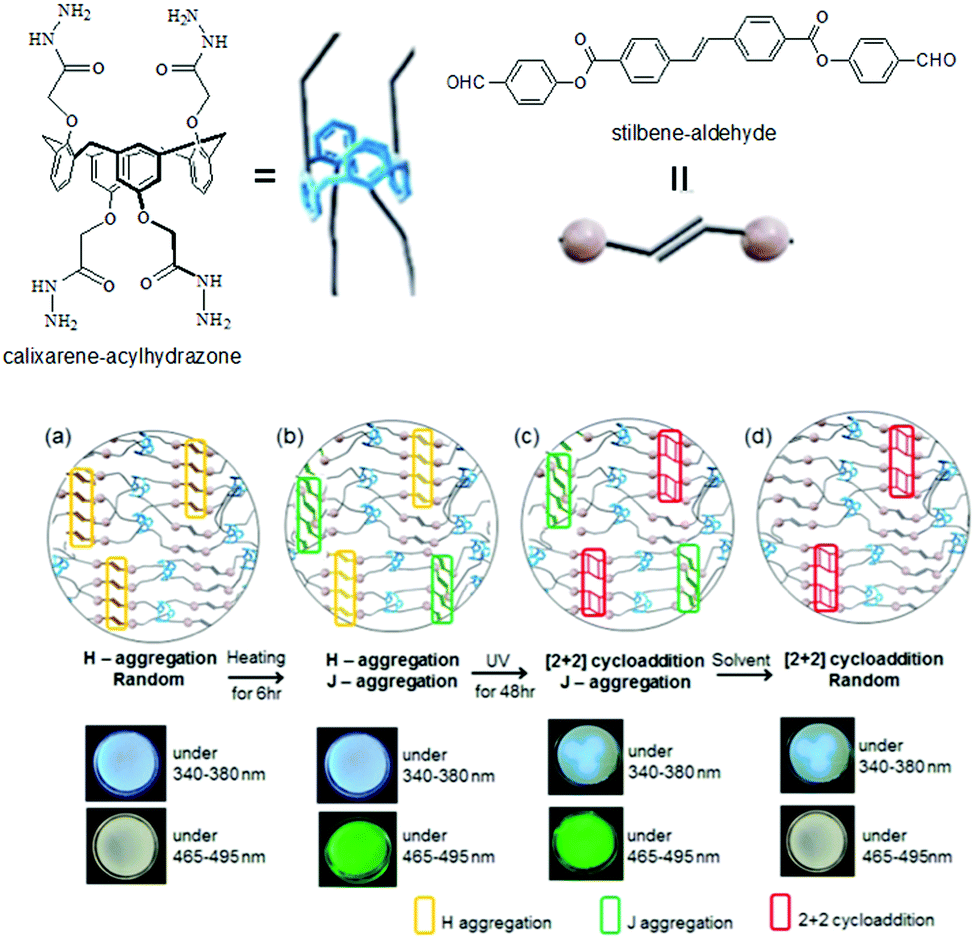 | ||
| Fig. 13 Illustration of gel formation by calixarene–acylhydrazone with stilbene–aldehyde: (a) before and (b) after heating at 60 °C; (c) after UV irradiation; (d) after immersion in DMSO. Reproduced from ref. 160 with permission from the American Chemical Society, copyright 2017. | ||
Wang et al. presented a new strategy, using the ruthenium-catalyzed photo-crosslinking of tyrosine to dityrosine to enhance the mechanical strength of a Fmoc-peptide hydrogel by 104-fold with a final storage modulus of around 100 kPa.161 Recently, photocoupling of 1,3,4-oxadiazole coupled styrenes was utilized by Dumur et al. to convert the blue fluorescence of the organogels into yellow by a tandem [4+2] and [3+2] cascade reaction.162
Another fascinating approach is photopolymerization of diacetylenes to create conductive arrays of conjugated π-bonds (Fig. 14).163,164 The final polydiacetylene gel shows a dramatic increase in rheological strength because of the formation of covalent cross-links into the network. An early report by Shinkai and co-workers showed polymerization resulted in significant increase in thermal stability (from 46 °C to 58 °C) and robustness of the material.165 Later, George and Weiss reported that photopolymerization of diacetylenes led to organogels that may lose thermoreversible properties unlike the primitive non-polymerized gels.166 However, because of increase in conjugated π-length, the polydiacetylene gels became highly coloured. Applying a similar strategy, a number of diacetylene gelators have been reported that showed remarkable stability as well as drastic colour change (usually colourless to blue) and morphology transformations during gel-to-gel transitions.167–178 However, structural modifications have been carried out keeping the basic diacetylene platform intact to achieve high control over optical and mechanical properties of the polymerized gel. For example, Stupp's group synthesized diacetylene containing linear peptide hydrogels and showed that the efficiency of photopolymerization decreased in branched architectures due to poor molecular packing within the fibre.167 In some cases, instead of long alkyl chain, cholesteryl segments have been introduced168 to uphold the polymerized network through extensive hydrophobic and van der Waals interactions.169 Moreover, incorporation of sugar or amino acids at the terminal positions provided H-bond assisted extra stability to the polymerized gels and increased material strengths.170–173 Metalloporphyrins have also been used to induce template-directed polymerization of diacetylene in gels, forming highly elongated networks with a unimolecularly stacked array.174,175 Shirakawa et al. utilized this concept and used diacetylene decorated Cu(II)-porphyrins as photopolymerization template. On photoirradiation, unimolecularly segregated polydiacetylene nanowires were obtained from columnar fibrils.176C3-symmetric diacetylenes also showed similar topochemical coupling upon photoirradiation, producing conjugated 1D nanowires made of three connected polydiacetylenes under solvent free conditions.177
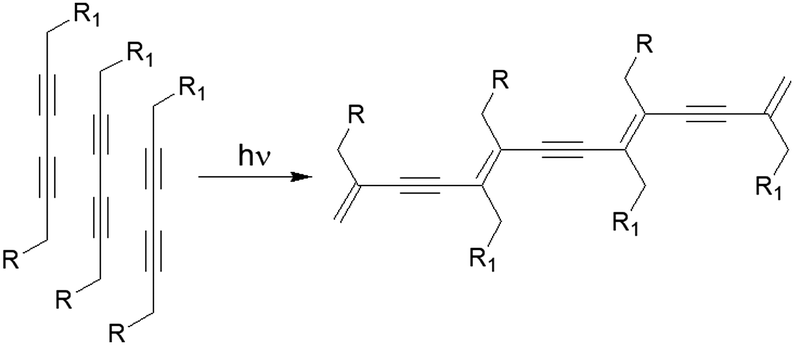 | ||
| Fig. 14 Photopolymerization of a diacetylene-containing LMWG leads to formation of an arrays of conjugated π-bonds. | ||
Study of such structure–property relationship is essential for exploring the possibilities of these gels because minor structural changes can significantly influence the optoelectronic properties of the polyacetylenes. For example, Aoki et al. studied photogelation of a series of diacetylene diastereomers (DD) and showed that a good gelation ability was found for enantiomers while for the racemic mixtures of diastereomers no gelation was observed (Fig. 15a and b).179 Additionally, a prominent odd–even effect in the alkyl chain length was found that regulated the properties of the photopolymerized gels. Upon photoirradiation, the cyclohexane gels with an odd number of carbon atoms exhibited thermal stability and a vivid colour change from orange (n = 3) to blue-purple (n = 7). Gels with even number of carbon spacers were non-photoreactive. A similar odd–even effect was observed by the Shinkai group for a series of diacetylene-gallic ester conjugates (DGE).180 The carbon atoms present on the aliphatic chains controlled the planarity during polymerization and produced various colours from red to dark blue (Fig. 15c). Apart from these, photopolymerization of methyl methacrylate also resulted in change in turbidity, thermal stability as well as morphology of gels.181
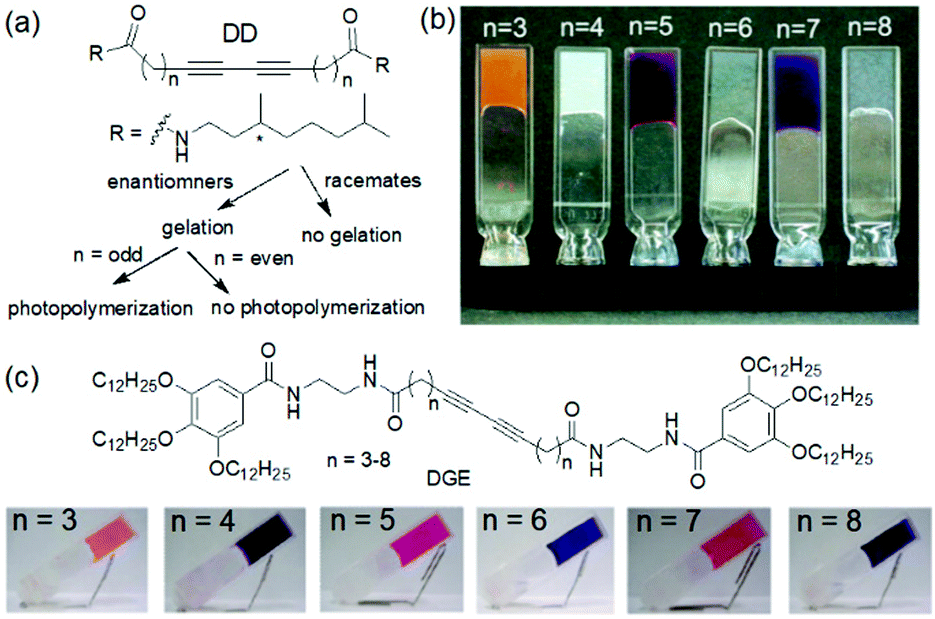 | ||
| Fig. 15 (a) Illustration of gelation ability of diacetylene diastereomers (DD).179 (b) Photographs of the cyclohexane gels of diacetylene diastereomers (DD) after photoirradiation. Reprinted from ref. 179 with permission from the American Chemical Society, copyright 2004. (c) Structures of the diacetylene–gallic ester conjugate (DGE) gelators and photographs of DGE gels after photoirradiation.180 Reproduced from ref. 180 with permission from the American Chemical Society, copyright 2007. | ||
Recently, the Jeong group illustrated thermal behaviour of a diacetylene-functionalized dendron in n-butanol.182 The dendron molecules contain amide functionalities for hydrogen bonding, long alkyl chains for van der Waals interactions and a diacetylene core as a photoresponsive unit. Initially, a turbid organogel was formed by the self-assembly of the molecules involving hydrogen bonding and hydrophobic interactions. After photoinduced polymerization of the diacetylene cores, the colorless turbid gel turned into purple. These two gels behaved differently when subjected to heating. While the non-polymerized gel underwent gel–sol transition on heating, the polydiacetylene gel exhibited wringing behavior at 85 °C with a color change to red. From wide-angle X-ray diffraction studies, they suggested that at 85 °C, the solvent was completely squeezed out of the gel network due to wringing behavior arises from the poor miscibility of the polydiacetylene fibers with n-butanol.
Based on the same principle, Fe(III)/Fe(II) redox conversion has been utilized extremely in fabricating dynamic gels. For example, Zhang et al. synthesized a redox tunable hydrogel system comprising of poly(acrylic acid) (PAAc), hexadecyl methacrylate and Fe(III)-citrate that evolved under UV-light.191 Photoreduction of Fe(III) in presence of citric acid led to significant decrease in mechanical strength of the corresponding Fe(II)-gel. The reaction could be reverted by oxygen and so forth the mechanical strength of the hydrogel can be controlled. Similar methodology was undertaken by Tong and other research groups for further developing iron-based dynamic gels.192,193
Photoreduction can be used to change the properties of perylene bisimide (PBI) based gels (Fig. 16). A red coloured self-supported hydrogel was formed by acidification of a PBI substituted with a valine at the imide positions. The fibre network was controlled primarily by stacking of the perylene cores.194 Photoreduction of the gelator by 365 nm UV-light lead to generation of a stable PBI-radical anion which was confirmed by electron paramagnetic resonance (EPR) studies. With time, the intensity at 710 and 810 nm in the UV-vis spectra of the gel progressively increased due to generation of the radial anion and the colour of the gel changed to dark purple. These radical anions altered the PBI stacking within the network resulted in a change in fibrous structures and improved the rheological properties. The reverse reaction occurred on keeping the gel in air for several hours and allowed recovery of the initial gel properties. Using a naphthalene diimide (NDI) core resulted in a transparent pale-yellow gel (NDIG) instead of the turbid red gel (Fig. 17).195 When this gel was exposed to sunlight or irradiated by 365 nm light, the gel darken due to the formation of the radical anion. Generation of NDI-radical anions that turned the transparent gel into black could also be achieved electrochemically. The reverse colour change could be observed by applying a low voltage of 0.6 V for 30 s. Such reversible and highly desirable transparent to dark colour change offered a great potential for smart windows.196 In addition to these attributes, use of photomask on PBI and NDI gels further allowed spatiotemporal control and switching of material properties that also extends the utility of such gels as organic electronics and semiconducting materials.197–202
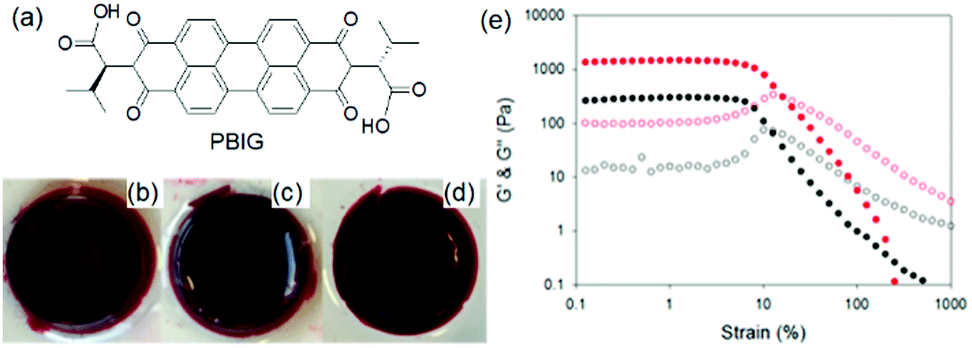 | ||
| Fig. 16 (a) Chemical structure of the gelator PBIG. (b) Photograph of a gel of PBIG prepared at a pH of 3.4 in a mould and then removed; (c) after irradiation with a 365 nm LED for 30 min and (d) after 24 h photographed under natural light. (e) Strain sweeps performed on gels of PBIG before (black data) and after photoirradiation with a 365 nm LED for 1 h (red data). The closed symbols represent G′, the open symbols G′′. Reproduced from ref. 203 with permission from the American Chemical society, copyright 2016. | ||
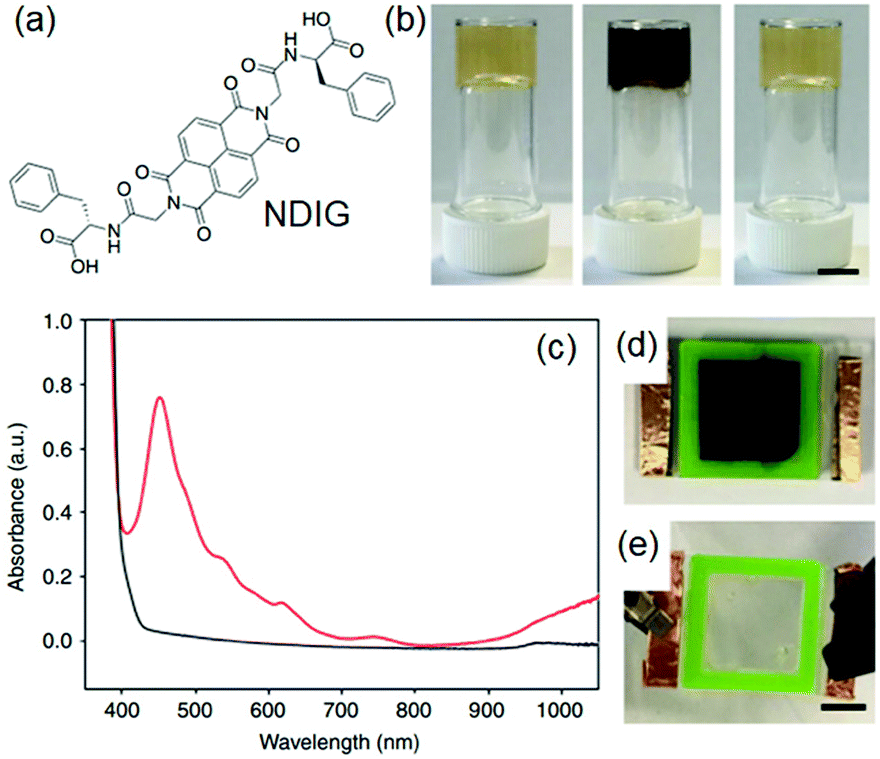 | ||
| Fig. 17 (a) Chemical structure of NDIG used to construct smart window. (b) (from left to right): transparent NDIG gel as prepared, and the same gel after irradiation and recovered. (c) UV-Vis absorption data of NDIG gel prepared in a cell irradiated with UV light (red data and d) and after applying 0.6 V for 60 s (black data and e). Reproduced from ref. 195 with permission from the Springer Nature, copyright 2018. | ||
(iv) pH changes
A change of pH is one of the most effective and convenient method used to trigger and control molecular gelation in water.204 With biological systems being highly pH dependent, pH responsive hydrogels are particularly of interest for biomedical research.205–207 A subtle change of pH brings about dramatic changes in gel properties. For example, Singh et al. reported hydrogelation of a 3-nitrotyrosine functionalized Fmoc-gelator at a wide pH range from pH 4.5–8.5 in 50 mM phosphate buffer solutions (Fig. 18).208 Field emission scanning electron microscopy (FESEM) studies showed an increase in nanofibres overlapping at high pH. Interestingly, the yellow colour of the gel gradually increased from lower pH to higher pH. The colour change was ascribed to the deprotonation of the –OH group at higher pH. The corresponding phenolate ion involved in electron conjugation with the nitro group that resulted in a red-shift in the absorption spectrum and produced intense yellow colouration.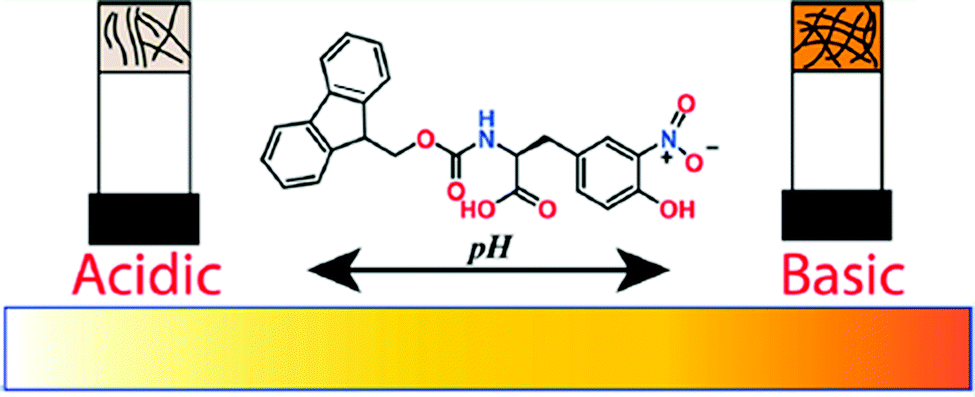 | ||
| Fig. 18 Cartoon representing pH dependent morphology transformation of the hydrogel formed from 3-nitrotyrosine functionalized Fmoc-gelator. Reproduced from ref. 208 with permission from the American Chemical society, copyright 2019. | ||
The key concept of preparing pH-triggered gels either includes the presence of ionizable pendant groups such as amine or carboxylic groups on the gelator backbone or incorporates active cleavable bonds like Schiff base bonds into the structure. As consequence, such gels typically exhibit reversible sol–gel transformations on reversal of the pH of the medium. For ionizable functionalities, the solubility of the molecules is controlled by the degree of protonation or deprotonation of the groups. For example, many peptide hydrogels are pH-triggered and exhibit hydrogel formation at a pH above or below the apparent pKa of the gelators. pH-controlled deprotonation of a carboxylic group results in increase in electrostatic repulsion and causes gel-to-sol transition.209–211 Similarly, amine or pyridine functionalized gels are stable at basic pH, while decreasing the pH below the pKa causes formation of ammonium or pyridinium ions that results in return to the solution state.212–216 Hydrogelation involving imine bond formation is typically favoured at neutral or slightly basic pH while at acidic pH the reverse reaction occurs that causes a gel-to-sol transition.217 In addition, gelators containing amphoteric functionality like imidazole or benzimidazole exhibit gelation near neutral pH while either an increase or decrease in pH of the medium results in weakening of the gels.218,219 Because of such limitations, gel properties have often been varied within a short pH window220 typically without crossing the pKa.221 Outside this pH region, sol formation occurs.
Variation of pH changes the nature of non-covalent interactions such as hydrogen bonding, electrostatic as well as hydrophobic interactions, chemical compositions etc. between the assembled building blocks. As a result, apart from phase transitions, pH induced morphological transformations or a change in molecular conformation is common in supramolecular self-assembled systems.222–225 pH triggered gel-to-gel transitions are limited in the literature. Several strategies have been undertaken to provide stability to the assembly structures of LMWGs upon pH change. A common method is to increase the hydrophobicity of the peptide gelators so that at high pH they behave as a surfactant and form micellar aggregates. This can be achieved by incorporating large π-surface or long alkyl chain into the structure.226–228 The micellar aggregates may be capable of sufficiently increasing the viscosity of the solution such that it can resist flow.229 The carboxylate anions formed upon deprotonation at high pH can bind the counter cation of the base to reduce the electrostatic repulsion and further crosslinking results in formation of a stable gels at high pH.230–233 The change in cation induces different packing in the micellar phase and thereby determines the material properties.234 A transition from the salt-induced gel to pH-triggered gel is possible by reducing the pH using hydrolysis agent like glucono-δ-lactone (GdL).234 This method further allowed to prepare gels with transient gradient in pH which can be visually observed by incorporating a pH responsive dye into the gel. The changes in material properties during the transition was further demonstrated by time sweep rheology.
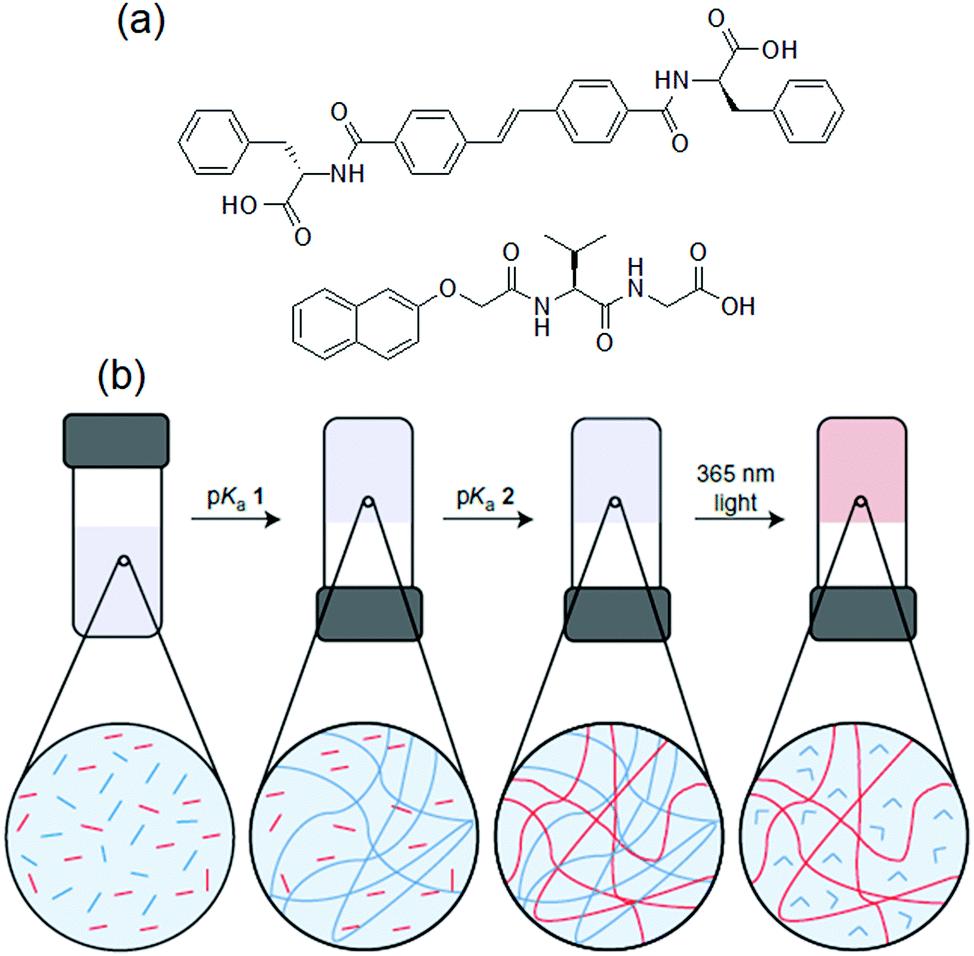
Another method involves incorporation of two opposite ionizable pendant groups on the same gelator backbone so that when the pH is changed, one group may ionise and so decrease its ability to sustain a self-assembled network, but the other loses charge and so can trigger self-assembly. For example, one can exploit the presence of a histidine functionality where the degree of solubility of the molecules can be determined by protonation/deprotonation of both the carboxylic acid and imidazole groups. Patra et al. synthesized a L-histidine-based amphiphiles containing an octyl chain.235 This compound exists mostly in three different forms: cationic, zwitterionic, and anionic and thereby exhibited hydrogelation over a broad pH range of pH 2–11. A dipeptide hydrogelator H2N–ADDAPhe–Phe–COOH (ADDA: 12-amino dodecanoic acid) containing an amine and a carboxylic acid functionality at two opposite terminals was introduced by the Banerjee group.236 In presence of either HCl or NaOH, the peptide formed self-supportive hydrogels, while it produced white aggregates under neutral conditions. The high pH gel was translucent in nature while the HCl triggered gel was quite transparent. The Escuder group modified this strategy and prepared pH-responsive gels through combination of two oppositely charged self-assembling peptides (Fig. 19).237 The gelators either contained acid sensitive amine functionality or a base sensitive acid group. The co-assembled gel was formed at neutral pH which turned into a single component gel either at acidic or basic pH due to the in situ dissolution of one component through protonation of amine or deprotonation of carboxylic acid group, respectively. Consequently, the morphology and mechanical properties varied depending on the pH of the medium.
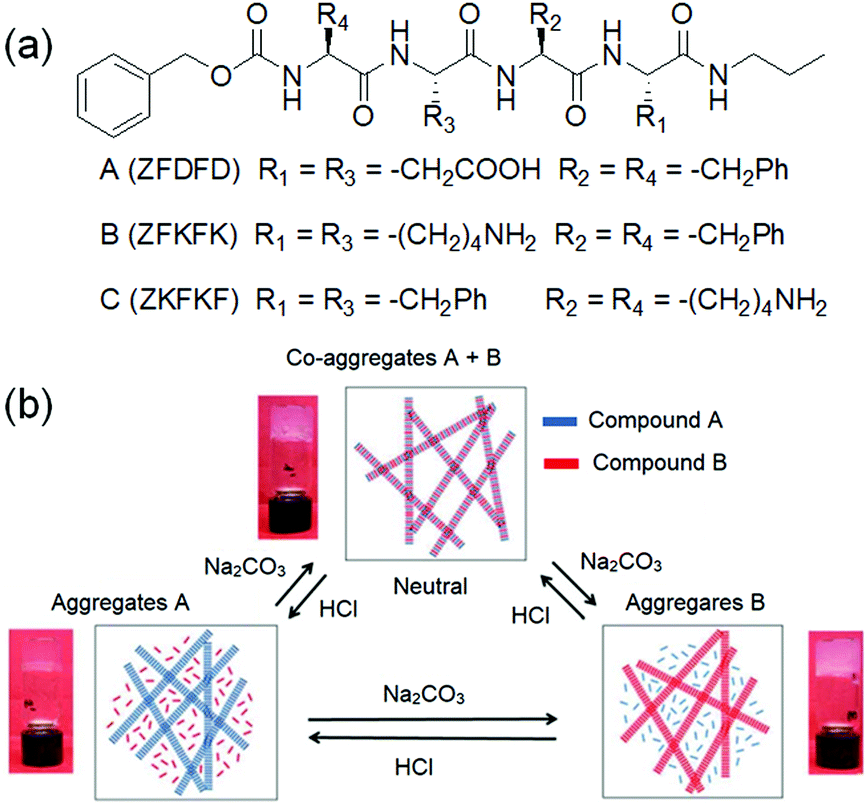 | ||
| Fig. 19 (a) Chemical structures of the gelators A–C. (b) Schematic representation of the pH-responsiveness of mixtures A and B. Insets demonstrate the macroscopic aspect of the hydrogels. Reproduced from ref. 237 with permission from the Royal Society of Chemistry. | ||
A change of pH can also be used to tune the degree of metal–ligand crosslinking in coordination polymer gels. Lee et al. reported a polymer hydrogel obtained through polymerization of dopaminemethacrylamide (DMA).238 The polymer chain contained a number of catechol units that formed strong complexes with Fe(III) ions. Depending on the pH on the medium, three different metal–ligand complex [mono(<pH 5.6), bis(pH 5.6–9.1) and cis(>pH 9.1)] formations was observed. The gels formed at lower pH were week because of a smaller number of cross-linking while stronger catechol–Fe(III) binding at high pH allowed deswelling and formation of relatively stiff gels. A similar strategy has been taken by various research groups to devise pH-responsive hydrogel-based actuators.239–242
Smith's group proposed a new idea to control material properties of multi-component gels by sequential implementation of two different pH triggers (Fig. 20).243 They synthesized two carboxylic acid functionalized gelators by derivatizing 1,3:2,4-dibenzyldene-D-sorbitol (DBS) that form self-sorted gels when the pH is lowered in a controlled way. The DBS gelators chosen had two distinctly different pKa meaning that in the multicomponent system, their self-assembly can be triggered at different pHs. They used base promoted hydrolysis of glucono-δ-lactone (GdL) as a proton source to lower the pH of the mixture of gelators which initially triggered self-assembly of DBS–CO2H (pKa 5.4), followed by DBS–Gly (pKa 4.3). By adjusting the initial GdL concentration, a high degree of kinetic control was achieved where almost all the DBS–CO2H was incorporated into the network while negligible amounts of DBS–Gly were immobilized. At this point they turned on the second pH-trigger i.e. the diphenyliodonium nitrate (DPIN), a well-known photoacid generator.244 Upon irradiation, further activation of DBS–Gly allowed second network formation. Hence, use of two different triggers allowed two step acidifications of the multicomponent gel system.
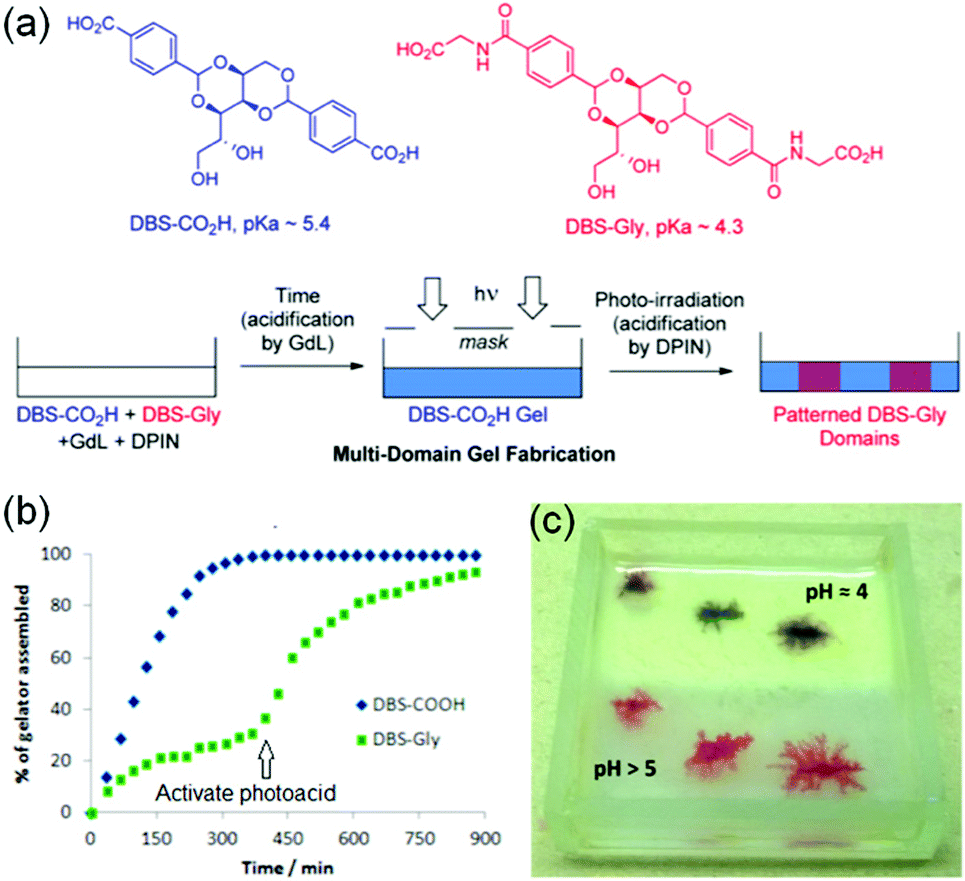 | ||
| Fig. 20 (a) Fabrication approach to multidomain hydrogels formed by DBS–CO2H and DBS–Gly. Initially, because of higher pKa, DBS–CO2H assembled in the presence of GdL before DBS-Gly. The DBS-Gly then assembled second which when activated by the photoacid DPIN. Photo-triggered network formation in presence of a photomask yielded a photopatterned hydrogel system. (b) NMR experiments demonstrating that DBS-CO2H assembled in the presence of GdL before DBS-Gly. (c) Photopatterned multicomponent gel of DBS-CO2H and DBS-Gly in which one-half was exposed to UV to activate DPIN (upper, opaque half, pH 4) while other-half was left unexposed (lower, translucent half, pH 5). The experiment was carried in presence of Congo Red indicator to monitor the pH change visually. The lower pH of the photopatterned region than the unexposed region indicated proton generation through DPIN activation in the patterned region. Reproduced from ref. 243 with permission from the American Chemical society, copyright 2015. | ||
The same research group later introduced photopolymerization to generate poly(ethylene glycol) dimethacrylate (PEGDM) polymer gel (PG) inside the mixture of DBS-CO2H and DBS-Gly.245 They successively employed three different triggers to fabricate step by step assembly of three gelators (two LMWGs and one polymer gel). Initially they prepared an alkaline mixture of six components: three gelators, GdL, DPIN and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (HMMP). HMMP was used as the photo-initiator for PEGDM polymerisation. On irradiation of the mixture solution, initially rapid photopolymerization resulted in formation of polymer gel while the rest of the LMWGs were nonresponding. Then GdL hydrolysis with time allowed second network formation involving DBS-CO2H at acidic pH. Finally, DPIN activation led to further decrease in pH of the medium and allowed DBS-Gly network assembly. They also performed same photopolymerization reaction inside a preformed DBS-hydrazide gel and showed that formation of polymer network resulted in significant increase in the mechanical stiffness of the final hybrid gel.246
(v) Dynamic covalent chemistry
Modification of gel networks can be achieved by exploiting dynamic covalent chemistry. Dynamic covalent bonds undergo formation and rupture reversibly under mild reaction condition.247–249 Dynamic covalent chemistry has emerged as a powerful tool to construct functional materials.250–253 In such materials, dynamic covalent bonds usually work alongside various non-covalent forces. Generally, only mild conditions are needed to reversibly make or break a dynamic covalent bond, however such systems still show resistance against small environmental fluctuations (for example temperature).247 Thermodynamic products that are stable with time are favoured which makes the kinetics of bond formation and cleavage slower than in case of kinetically controlled systems. Moreover, as the dynamic exchange generally occurs at equilibrium, this allows error correction of kinetically-trapped imperfections. Possible suitable reactions include hydrazone and acylhydrazone formation, olefin metathesis or Diels–Alder cycloaddition. Condensation of amino and carbonyl groups to generate CN double bond is very popular, especially imine formation from the reaction between a primary amine and aldehyde or ketone.254,255 Generally, this Schiff base condensation is acid catalysed, the resulting imine is stable at neutral or basic pH. On the other hand, hydrazines and acylhydrazines react with aldehydes or ketones under mild acidic conditions256–258 and produce hydrazone bonds that are stable under neutral, basic as well as acidic conditions.259 However, the imine system can be kinetically fixed to prevent the reversibility, e.g. reduction of dynamic CN bond to a non-reversible covalent C–N bond.260
There are numerous examples of supramolecular gelators formed from dynamic covalent chemistry that includes formation of imines, hydrazones, acylhydrazones, boronic esters, Diels–Alder reaction, disulfides exchange reaction. Such gelators form under equilibrium condition and often exhibit unidirectional responses on perturbation resulting in gel-to-sol transitions. A number of reviews have described the opportunities afforded by stimuli responsive dynamic covalent gels.250,251,261
Fabrication of dynamic covalent gels relies on multilevel self-assembly mechanism. Lehn and co-workers introduced a guanosine hydrazide-based supramolecular gelator which underwent G-quartet formation in the presence of template cations like K+ (Fig. 21).262 This G-quadruplex superstructure, stabilized by H-bonding, self-assembles to form fibres and gels. This superstructure was capable to undergoing dynamic decoration through construction of acylhydrazone bonds with aldehydes. Among the aldehydes tested, 1-formyl furan-3-sulfonic acid and pyridoxal-5-phosphate reacted to produce gels from the corresponding acylhydrazones. This is a unique example of cation-templated multilevel self-organization with component selection and amplification in dynamic hydrogels based on G-quadruplex superstructure formation. Later on, Arnal-Hérault et al. utilized similar strategy to synthesize G-quartet macroscopic hydrogen-bonded membrane films.263
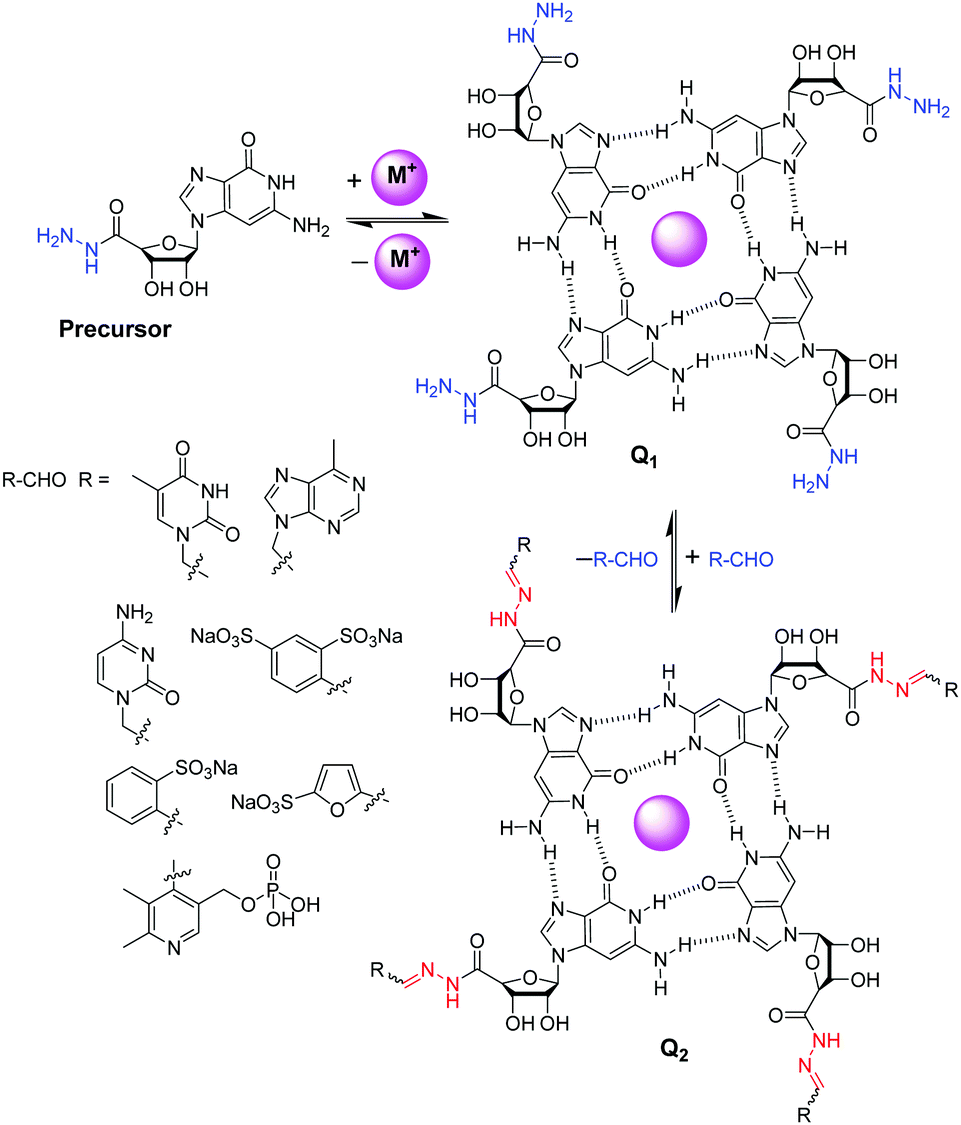 | ||
| Fig. 21 Reversible decoration of G-quartet assembly Q1 of guanosine hydrazide precursor through hydrazone formation with various aldehydes.262 | ||
Chirality transfer as well as chiroptical switching can be achieved through dynamic covalent bond formation and cleavage.264–266 The Liu group reported the synthesis of a chiral supramolecular organogel obtained from the co-assembly of an achiral gelator with N,N′-bis(octadecyl)-L-Boc-glutamic diamide.267 The achiral imine 2-((octadecylimino)methyl)phenol did not form a gel in many organic solvents tested; however, in the presence of the universal gelator N,N′-bis(octadecyl)-L-Boc-glutamic diamide,268,269 it produced a gel in ethanol. The co-assembled gel showed a left-handed helical morphology in scanning electron microscope. The supramolecular chirality was thought to arise because of chirality transfer from the chiral diamide through interchain interactions. Importantly, the chirality transfer stopped when the imine bond was ruptured on acidification and reappeared under alkaline condition. Such a pH-induced rupture and formation of imine bond enabled the gel to show significant change in circular dichroism (CD) signals. Additionally, based on pH-responsive imine chemistry, the morphological transformation of imine assemblies can also be controlled.265,270
Khalily et al. described post-modification of a hexapeptide-based gel involving dynamic imine bond formation.271 In their study, they synthesized a peptide gelator (Lauryl-VVAGKK-Am) which self-assembled into nanofibres (diameter of ca. 10 nm) directed by β-sheet formation at pH 7.5. The hydrophobic interactions exerted by the lauryl group as well as the hydrogen-bonding facilitated by β-sheet forming amino acids (VVA) drives the peptide to form a self-supported gel. When the free amines of the peptide were reacted with glutaraldehyde, the corresponding crosslinked gel was formed via imine bonding. Interestingly, no significant change of the nanofibre morphology or porosity was noticed during the gel-to-gel transition. However, dynamic imine bond formation resulted in considerable increase in viscoelastic properties compared to the initially formed gel. It was possible to tune the viscoelastic properties of the gels either by changing the ratio of glutaraldehyde or by adding aldehydes of different lengths. A similar methodology was undertaken by the Smith group for dynamic modification of acylhydrazide-functionalised 1,3:2,4-dibenzylidene sorbitol (DBS-CONHNH2) gels.272 Reaction of DBS-CONHNH2 with various aromatic and aliphatic aldehydes produced acylhydrazones that retained the aggregated structure. However, the thermal stability, morphology and rheological properties of the aldehyde modified gels differ from the initially prepared gel. Aldehyde modification resulted in decrease in fibre lengths, however the fibre width increases from around 50 nm to around 500 nm in the modified gel. The increased thermal stability and higher rheological properties were more prominent when aldehydes with aromatic groups were used compared to simple aliphatic aldehydes.
(vi) Redox reactions
A common strategy to achieve gel-to-gel transformations is to use redox reactions. Redox-responsive gels typically either contain redox active functionalities like tetrathiafulvalenes, ferrocenes, disulfides, thiophenes on the gelator backbone or they accumulate redox active metal ions into the gel fibres.273–275 Hence, such gels are intrinsically capable to respond to oxidation or reduction. While a redox-induced change in molecular structure dramatically influences the intermolecular interactions, a change in oxidation state of the metal ions in a metal ligand complex alters the coordination mode and potentially impacts the electrostatic interactions.Redox-responsive gels can be achieved either chemically or electrochemically. In chemically active gels, chemical reagents are used to activate the molecular components. Zhou et al. synthesized a yellow coloured coordination polymer supramolecular gel by interactions between 3,5-dinitrosalicylic acid (DNSA) and europium (Eu3+) ions in presence of TiO2 (Fig. 22).276 The gel exhibited the characteristic red fluorescence of Eu3+. The yellow colour of the gel turned to red on addition of NaBH4 due to reduction of DNSA to 3-amino-5-nitrosalicylic acid. The reduced gel became non-fluorescent. However, the presence of TiO2 made the gel photoactive that causes further oxidation to DNSA and restoration of the original yellow gel by UV-light. Panja and Ghosh illustrated a H2O2 induced transformation of a Fe(II)-metallogel into a Fe(III)-gel.277 Their study showed that a yellow coloured gel could be obtained from the self-assembly of 1-((pyridin-3-ylimino)methyl)naphthalen-2-ol, a well explored imine functionalized 2-naphthol.278 The gelation was proposed to be mostly driven by π-stacking interactions exerted by the naphthyl groups. The hydroxyl group of the gelator involved in intramolecular hydrogen bonding and locks the molecular conformation in a planar arrangement. Such a molecular arrangement resulted in extensive π–π stacking between the naphthyl rings and triggered gelation. The presence of either H2O2 or Fe(II) individually caused a marginal effect on photophysical properties of the gel. However, when H2O2 was added on the top of the Fe(II)-metallogel, a visual transformation to a deep brown coloured gel was observed with time. The colour change was associated to the oxidation of Fe(II) to Fe(III) ions followed by a change in the metal–ligand coordination. They further explored the Fe(II)/Fe(III) redox conversion in visual recognition of H2O2 from other reactive oxygen species (ROS) by performing Fenton reaction inside the Fe(II)-gel medium. The deep-brown Fe(III) gel underwent demetallation in presence of F− ions and regenerated the original yellow coloured gel. Based on such colour changes in presence of different analytes, paper-based test kits were prepared for further development of the gels as rewritable display materials. In a recent development, Zhu and co-workers exploited the redox properties of thiols to control swelling of gels.279 They synthesized a poly(acrylamide)-based disulfide cross-linked photonic crystal hydrogel containing monodisperse carbon-encapsulated Fe3O4 nanoparticles. The nanoparticles were regularly spaced within the polymeric network and exhibited a green structural color indicative of their lattice spacing. When the gel was exposed to glutathione solution, reduction of the disulfide bonds occurred which led to decrease in the cross-linking degree of the gel network, triggering the swelling of the hydrogel. As a result, an increase of the nanoparticle lattice spacing happened which causes a red-shift in the hydrogel's structural color. Increasing concentration of glutathione (1 to 10 mM) resulted in more reduction of the disulfide bonds, allowing the gel to expand in a greater degree. Consequently, the lattice spacing between the nanoparticles increases with higher concentration of glutathione, causing a larger red-shift in the hydrogel's structural color (from green to yellow and then red gradually).
 | ||
| Fig. 22 (a) Schematic representation of the reversible redox switching of fluorescence and visual colour change of DNSA/Eu3+/TiO2 coordination polymer gel. (b) Emission spectra of the DNSA/Eu3+/TiO2 gels in presence of different concentrations of reducing agent NaBH4 (0–6 mmol L−1) and the reduced DNSA/Eu3+/TiO2 gels with different UV light irradiation times (0–4 h). (c–e) represent SEM images of the initial DNSA/Eu3+/TiO2 gel (c), reduced DNSA/Eu3+/TiO2 gel by aqueous NaBH4 (d), and the restored oxidised DNSA/Eu3+/TiO2 gel after UV irradiation (e). Reproduced from ref. 276 with permission from the American Chemical society, copyright 2019. | ||
Electrochemically-responsive hydrogels change their properties on exposure to externally applied electrical field.280–282 In this case, the gelators bear redox-responsive functionalities such as catechol or anthraquinone which undergo reversible electro-mediated oxidation–reduction and exhibit changes in their physical and chemical properties. An anthraquinone functionalized glycine-based peptide was synthesized by Jones et al.283 The peptide formed a self-supported hydrogel driven by π–π stacking and hydrogen bonding. Electrochemical conversion of anthraquinone to anthrahydroxyquinone caused reversible variation in transmittance over the entire visible range. Similarly, Xue et al. coupled 3,4-dihydroxyphenylalanine (DOPA) with 2-naphthalenyl-glycine–phenylalanine–phenylalanine to synthesize an electrochemical active hydrogelator (Fig. 23).284 The presence of the DOPA segment made the hydrogel redox-responsive. Electrochemical oxidation of the corresponding hydrogel resulted in conversion of DOPA to dopaquinone and made the hydrogel more hydrophobic. This resulted in swelling of the hydrogel (up to 50%) as well as an increase in mechanical properties. In presence of an alternative redox potential, it was possible to constitute a hydrogel actuator that showed unidirectional expansion and shrinkage with a change in the redox states of the peptide. In some cases, redox active metal ions have been incorporated to fabricate the supramolecular gels which, upon electrical simulation, change the oxidation state of the metal ions and cause ion mobility between the electrodes.285 Such an uneven distribution of the ions inside the gel active led to reorganization of the counter anions and generated an osmotic pressure gradient leading to swelling or shrinking of the hydrogels.284,286 The Meyer group reported Cu(II)/Cu(I) based redox responsive polymer based actuators where conversion to Cu(I) resulted in a significant decrease in material strength.287 The polymer gel could reversibly switch between hard and soft states, where the mechanical properties were controlled by the oxidation states.
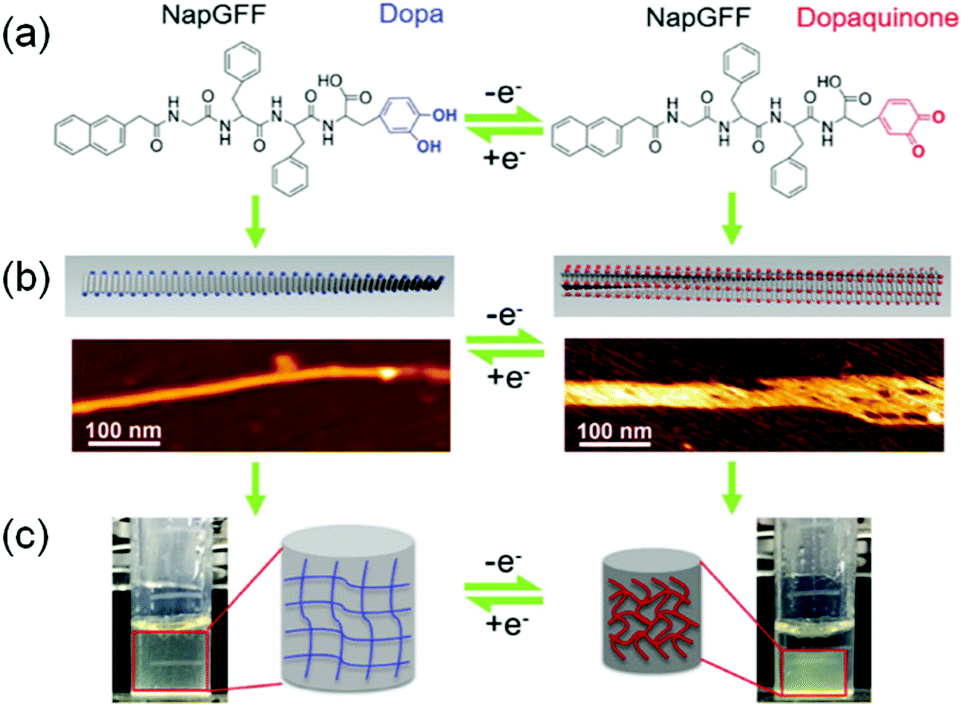 | ||
| Fig. 23 Schematic representation of the electro-responsive supramolecular peptide hydrogel actuator. Presence of DOPA segment on naphthyl gelator (a) enables the hydrogel to exhibit changes in morphology (AFM images) (b) and volume (c) at different potentials. Adapted from ref. 284 with permission from the John Wiley and Sons, copyright 2016. | ||
(vii) Post assembly fabrication of gels by chemical analytes
Gelators with specific binding sites for charged as well as neutral analytes provide interesting opportunities in gel chemistry.288–295 The key concept relies on binding of the analytes into specific sites present on the gelator structure, leading to changes in the aggregation and properties of gels. Common chemical analytes include metal ions, amino acids, carbohydrates, amines and carboxylic acids.296–301 The nature of the chemical analytes demands specific design of the gelators in order to exhibit changes upon interaction. Generally, gelators possessing heteroaromatic scaffolds (like pyridine, imidazole, thiazole etc.), crown ethers, triazole, hydrazone, and salicyl-imine segments are responsive towards cations whilst the presence of hydrogen bond donor groups like hydroxyl, urea and amides tend to interact with anions. While the ionic analytes interact electrostatically with the gelators, neutral guests mostly depend on hydrogen bonding and stacking interactions. The donor–acceptor charge transfer interaction is also a possibility to prepare co-assembled gels with remarkable photophysical properties.302,303 Although rare, cation–π and anion–π interactions are also found in gels.304,305 Chemical analyte responsive dosimetric gelators are another topic of interest in recent years.306–308 Generally, the binding sites are connected to some associating units on the gelator structure that assist self-assembly involving non-covalent interactions.18,309–311 Such associating units may contain hydrophobic moieties like cholesterol169,312–314 and aromatic surface,315 or hydrophilic functionalities like peptide,316 sugar317etc. Chemical analyte responsive gels have huge potential as molecular sensors, aggregation probes, antibacterial agents, conducting materials and water purifying agents.212,299,318–322The presence of chemical analytes can dramatically alter the gel properties.323–325 A specific example is the Tb3+ metallogel investigated by Ma et al.326 The hydrogel of 2-(2-butyl-1H-benzimidazol-1-yl)acethydrazide exhibited weak fluorescence at 480 nm which was significantly increased (∼4-fold increase) and red shifted to 498 nm in the presence of 0.3 equivalent of terbium nitrate. The enhanced and red shifted emission was attributed to the coordination effect between the gelator and Tb3+ ions which induced the morphology change of the gel from rod like nanostructure to highly entangled and densely bundled aggregate. As an extension, the authors used a similar strategy to convert a weakly fluorescent hydrogel of a benzimidazole functionalized acylhydrazone Schiff base to a strong blue-light emitting metal–organic gel by implanting Cd2+ into the gel network.327 In a seminal work, Maji and co-workers reported a terpyridine substituted tetraphenylethelene-based LMWG whose optical and fibre properties can be tuned by Eu3+.328 Insertion of metal ions into the LMWG converts the supramolecular gel into a 3D coordination polymer gel. It is further possible to incorporate two different metal ions simultaneously into the same gel network and the properties of the metallogels can be controlled by varying the stoichiometries of the metal ions.329–331 For example, Sutar et al. reported a series of Ln-coordination polymer organogels where the emission properties of the gels was tuned by varying the stoichiometry of Tb3+ and Eu3+ (Fig. 24).329 However, the metal ions were introduced at the beginning of gelation for these systems, and hence, the changes in gel properties can be ascribed to the different self-assembly pathway than the original gels.19,327,332,333 Furthermore, in some cases, a heat–cool operation was performed after addition of the guest analytes onto the gels.326,334–340 Here, we discuss the possibilities of modifying gel properties by various chemical analytes in a post-assembly pathway at room temperature.
 | ||
| Fig. 24 An example showing tuning of gel properties by varying concentration of chemical analyte. The ligand self-assembled in presence of Ln3+ ions. Different ratios of Tb3+ and Eu3+ produced a number of luminescent coordination polymer gels. Reproduced from ref. 329 with permission from the Royal Society of Chemistry. | ||
Yao et al. explored an acylhydrazone functionalized benzimidazole gelator in multianalyte sensing in a gel-to-gel fashion (Fig. 25).296 The LMWG formed a gel in ethylene glycol and glycerol exhibiting strong yellow-green fluorescence. Various chemical analytes were added on the top of the gel, and then allowed to diffuse through the gel. The organogel selectively recognized CN−, Al3+, Fe3+ and L-Cys via a fluorescence change. Depending upon the nature of the chemical guests, different modes of interaction were established in the recognition events. The presence of CN− ions resulted in deprotonation of both phenolic-OH and the acylhydrazone –NH because of the strong basicity. The yellow-green fluorescence of the gel changed to blue with enhanced intensity on interaction with CN− ions. In addition to anions, the gel state could selectively recognize both Al3+ and Fe3+ by exhibiting opposite responses in emission spectra. While addition of Al3+ caused aggregation induced blue shifted emission with colour change to blue, Fe3+ quenched the yellow-green emission of the gel. Demetallation of Fe3+ from the Fe-metallogel by L-cysteine resulted in restoration of the original yellow-green emission, and thereby the Fe-metallogel was inspected as a secondary sensing probe to detect L-cysteine from other amino acids. The fluorescence off–on switching by successive addition of Fe3+ and L-cysteine was found to be an effective strategy for constructing rewritable display materials. Many research groups adopted similar metalation and demetallation techniques to modify the optical properties of the gels and so to detect several anions by off–on switching of the emission property of the metallogels.341–344 In a recent study, Sebastian and Prasad synthesized a Cu-gel of a terpyridine linked poly(aryl ether) dendrone.345 The non-emissive spectra of the Cu-gel was selectively turned on in presence of CN− ion. As suggested, scavenging of Cu2+ from the gel fibres by CN− ions through the formation of [Cu(CN)x]n− complex led to attend the original gelation attributes of the poly(aryl ether) dendrone gelator which resulted in significant enhancement in the emission of the gel. In some cases, instead of demetallation, complexation of the incoming guest (or binding) with the metal centre can also occur. Mukhopadhyay and co-workers synthesized an organogelator N1,N3,N5-tris(4-cyanomethylphenyl)benzene-1,3,5-tricarboxamide. On addition of Co2+, the colourless organogel became pink.346 This Co-metallogel selectively senses L-tryptophan from other amino acids (Tyr, Leu, Lys, Phe, Trp, Ala, Glu, Asp, Ser, His, Arg, Pro, and Cys) through quenching of emission and a visible colour change to yellow.347 It was proposed that tryptophan binding with the Co(II) ion changes the coordination environment around the metal centre, which was responsible for the vivid colour change of the metallogel. Interestingly, the Co-metallogel could also sense L-tryptophan in a complex structure like bovine serum albumin protein by exhibiting similar colour change with a detection limit of protein of 8.7 × 10−9 M.
 | ||
| Fig. 25 (a) Schematic representation of the multianalyte responsive system. (b) Fluorescence responses of the multi-analyte sensor array gel (WJG) towards various chemical analytes. Adapted from ref. 296 with permission from the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS) and the RSC. | ||
Such post-assembly modification enables the use the gels as a catalyst and sensor for antibiotics. Huang et al. showed that diffusion of Pd2+ into a Fe3+ metallogel resulted in replacement of the iron ions.348 The corresponding Pd-gel behaved as an efficient recoverable catalyst for Suzuki–Miyaura coupling reaction under mild conditions. Similarly, diffusion of Tb3+ into an Al3+-gel of 1,3,5-benzenetricarboxylic acid produced the corresponding Pb-gel that exhibited an intense green emission due to energy-transfer from the ligand to the metal centre.349 Addition of sulfamide-antibiotics, such as sulfamethazine, sulfadiazine caused quenching of the emission of the Tb-gel by disturbing the ligand–metal energy transfer process.
Post-assembly fabrication can often give gels that can discriminate between basic anions like F− and AcO−. Xu et al. synthesized a π-gelator (PG) containing a pyrenefluorophore and urea-sulfonamide groups as anion binding sites (Fig. 26).350 Due to the strong basicity, both F− and AcO− ions (both as tetrabutylammonium salt) formed strong intermolecular hydrogen bonding with the urea –NH. However, the authors speculated that owing to its small size, F− ions can fit within the gel network. The gel structure was maintained by aromatic stacking and hydrophobic interactions, but the emission intensity was quenched significantly. On the other hand, AcO− ions altered the intermolecular interactions significantly and destroyed the gel structure. This was ascribed to the relatively larger size of the AcO− ions. Recently, Pang et al. introduced a cholesterol coupled naphthalimide (NC) that exhibited the opposite response in presence of the same anions (as tetrabutylammonium salt) (Fig. 26).351 While addition of F− ion resulted in a rapid gel-to-sol transition associated with a colour change from red to dark blue, addition of AcO− ion showed only a colour change of the gel from red to black without a phase transition. The colour changes under both conditions were attributed to the deprotonation of the acidic –NH that enhanced the π-electron conjugation. Proton NMR spectroscopy studies showed both hydrogen bonding and anion–π interaction were responsible for these observations. Due to strong hydrogen bonding and anion–π interaction between the ligand and F− caused destruction of the gel. In this context, it is mentionable that, while triggering gelation involving ion–ligand interactions, the nature of the counterion often play decisive role on material properties.289,352 However, the effect of counterions on the kinetics of gel to gel transitions is unexplored and hence demand future attention.
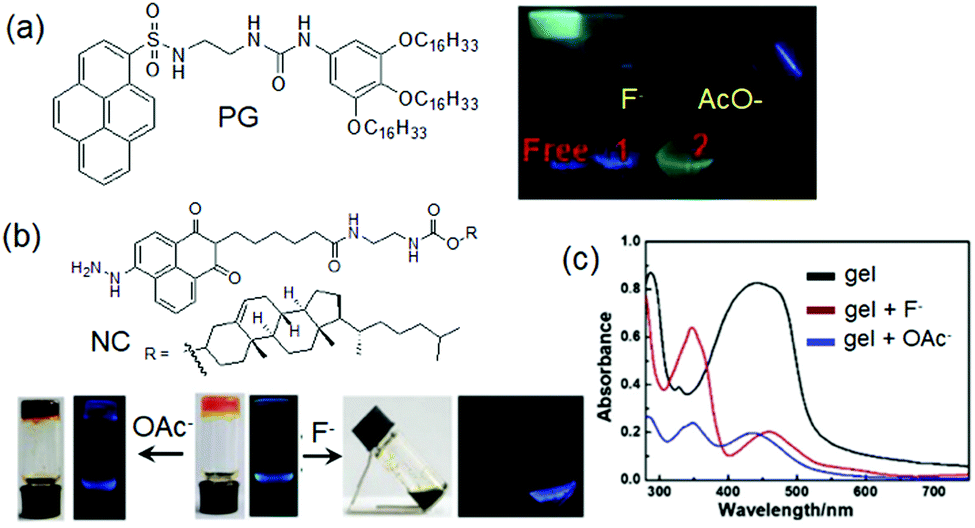 | ||
| Fig. 26 Photographs showing anion responsive behaviour of PG towards F− and AcO− ions. Reproduced from ref. 350 with permission from Elsevier, copyright 2017. In comparison to PG, the naphthalimide–cholesterol gelator NC exhibited opposite response for F− and AcO− (b). (c) UV-vis spectra of the organogel NC in presence of F− and AcO− ions. Figures (b) and (c) are reproduced from ref. 351 with permission from The Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS) and the RSC. | ||
Anions can also modify gel properties by forming charge transfer complexes. Ghosh et al. introduced bisurea gelator 1-pyridin-3-yl-3-[8-(3-pyridin-3-yl-ureido)-naphthalen-1-yl]-urea, that formed a colourless gel in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:2, v/v).353 Experimental evidence showed that extensive π–π stacking by the naphthyl rings along with the bifurcated hydrogen bonding involving the urea groups played pivotal role in fibre formation. The gel became reddish brown when treated with iodide ions due to formation of charge transfer complex. SEM images suggested an increase in fibre density in the iodide gel. It was proposed that cooperative hydrogen bonding interactions of the pyridyl ureas with iodide ion packed the gelators molecules effectively, reinforcing the stacking interaction between the naphthalene groups. As a result, the iodide gel showed a higher thermal stability than the initially formed gel. However, with time, I3− ions was generated in situ either through photooxidation or by DMSO induced oxidation of I−. A strong band at 370 nm in the absorption spectrum of the reddish-brown gel indicated the coexistence of I3− ions with I− in the gel network. No such colour change was observed with other anions such as HSO4−, AcO−, H2PO4− including halides F−, Cl− and Br−. Hence, the gel state can be used for the visual recognition of iodide ion over other anions. According to the authors, this was the first report of naked eye detection of iodide through a colour change involving no phase transformation.
:H2O (1:2, v/v).353 Experimental evidence showed that extensive π–π stacking by the naphthyl rings along with the bifurcated hydrogen bonding involving the urea groups played pivotal role in fibre formation. The gel became reddish brown when treated with iodide ions due to formation of charge transfer complex. SEM images suggested an increase in fibre density in the iodide gel. It was proposed that cooperative hydrogen bonding interactions of the pyridyl ureas with iodide ion packed the gelators molecules effectively, reinforcing the stacking interaction between the naphthalene groups. As a result, the iodide gel showed a higher thermal stability than the initially formed gel. However, with time, I3− ions was generated in situ either through photooxidation or by DMSO induced oxidation of I−. A strong band at 370 nm in the absorption spectrum of the reddish-brown gel indicated the coexistence of I3− ions with I− in the gel network. No such colour change was observed with other anions such as HSO4−, AcO−, H2PO4− including halides F−, Cl− and Br−. Hence, the gel state can be used for the visual recognition of iodide ion over other anions. According to the authors, this was the first report of naked eye detection of iodide through a colour change involving no phase transformation.
A recent report describes a chemodosimetric method to modify gel properties. In this case, the gelator backbone contains a reaction centre where instead of reversible interactions, a covalent bond formation or rupturing occurs. As a result, the gelator molecules undergo permanent chemical changes after reaction with the analyte. For example, nucleophilic addition of cyanide ion to the gelator 2-(hexadecylthio)oxazolo[4,5-b]phenazine resulted in a quenching in emission of the DMSO–water gel.354 In a recent study, Mandegania et al. presented a supramolecular gelator composed of indolin-2-one and quinoline moieties with an activated olefinic bond that exhibited gelation in DMSO/water.355 Among various anions, only CN− ion brought about a color change of the gel from orange to pink. The selectivity arises from high nucleophilicity of cyanide compared to other anions. The gelator first undergoes a pseudo-Michael attack by CN− ion followed by a ring closing. Subsequent proton shift and tautomerization finally led to generate corresponding conjugate anion which was responsible for the dark purple color. Although such chemodosimetric changes alter the optical and morphological properties of gels, very little information is available on the changes in the rheological properties.
A simple but efficient method was developed by the Smith group to modify gel properties by chiral selection within gels (Fig. 27).356 They synthesized an L-lysine dendron and demonstrated selective incorporation of one enantiomer of an amine over other into the fibrous network. When co-assembled gels were prepared by the dendron with chiral amines (R and S amines), the most stable gel was obtained from R-amine. They showed that addition of R-enantiomeric amine to the gel formed by the co-assembly of L-lysine dendron and S-amine allowed diffusion of R-amine into the system followed by displacement of the S-amine from the solid-like fibres. Chiral recognition allowed selection and incorporation of component (R-amine) leading to more thermodynamically stable gel. These results suggest that chirality of the molecules plays crucial role in determining thermodynamic stability and properties of chiral gels.2,264,357 Other biomolecules like sugars can also be used to tailor gel to gel transitions. Mahendar et al. synthesized a hydrazone coupled phenylboronic acid-based chiral gelator.358 The design principle underlines with the fact that the boronic acid moiety exhibits selective interaction with D-(+)-glucose at alkaline pH. Initially, a stiff metallogel was obtained from DMSO/water in presence of LiOH. The white Li+-gel was then converted to a red-colored metallogel by treating with D-(+)-glucose. Experimental evidence revealed that, while Li+ ions binds to the acyl carbonyls, the boronic acid moieties undergoes esterification with D-(+)-glucose. Glucose adduct formation resulted in weakening of the mechanical properties. This decrease in stiffness in the D-(+)-glucose bind gels led to a significant enhancement of the conductance of the metallogel (compared to the D-(+)-glucose free metallogel).
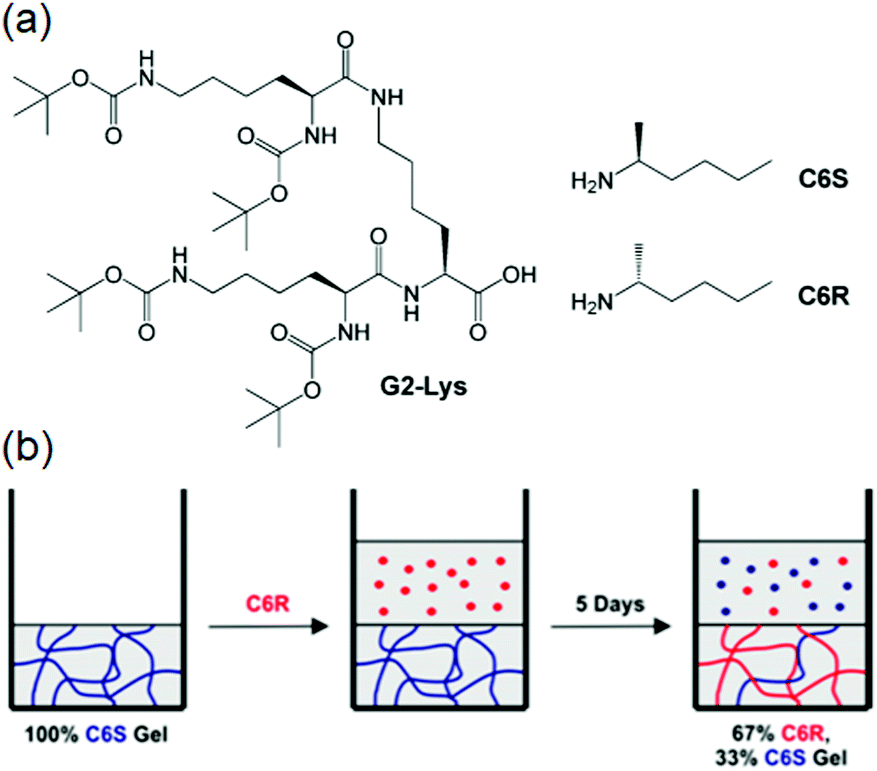 | ||
| Fig. 27 (a) Chemical structures of the chiral dendron (G2-Lys) and chiral amines (C6R/S). (b) Cartoon representing thermodynamically controlled gel evolution upon addition of C6R to the gel prepared from G2-Lys and C6S. Reprinted from ref. 356 with permission from the American Chemical Society, copyright 2014. | ||
Self-assembled gels can show successive disassembly and reassembly upon exposure to a chemical reagent followed by a complementary counter-trigger resulting in gel–sol–gel transitions.359–363 However, the changes in mechanical properties and morphology of the gels upon reappearance have been rarely investigated. Recent research includes implementation of a reaction cycle on a pre-formed gel to execute successive phase change autonomously. The Hermans group synthesized a hydrogel from an aldehyde saccharide hydrogelator (SachCHO) (Fig. 28).364 Addition of a chemical reaction cycle consisting of dithionite (DT), GdL and hexamethylenetetramine (HMTA) to the hydrogel gave a self-regulating gel–sol–gel transition involving chemical changes of the gelator. Initially, SachCHO was quantitatively converted to an α-hydroxy sulfonate (SachSO3−) that resulted in collapse of the hydrogel into a clear solution. The second chemical reaction between GdL and HMTA produced formaldehyde in situ, which consumed SachSO3− and regenerate the SachCHO gelator. The production of formaldehyde can be controlled by adjusting the initial GdL concentration, and thereby the onset of regelation could be programmed which allowed reappearance of the gel at different time. The final properties of the gels depend upon the initial concentration of GdL, and hence the rate of formaldehyde (fuel) production. While flower-like assemblies consisting of short fibres (60–100 μm) appeared during slow catalyzed ([GdL] = 234 mM) formation of SachCHO, fractal-like assemblies composed of long fibres (≥200 μm long) formed at fast catalyzed ([GdL] = 374 mM) formation of SachCHO. Consequently, the storage moduli of the chemically fuelled gels varied from ∼110 Pa ([GdL] = 46.8 mM) to ∼2100 Pa ([GdL] = 374 mM) depending upon the initial concentration of GdL. By employing the reaction cycle, they were able to achieve 13 gel–sol–gel cycle without significant loss of material properties.
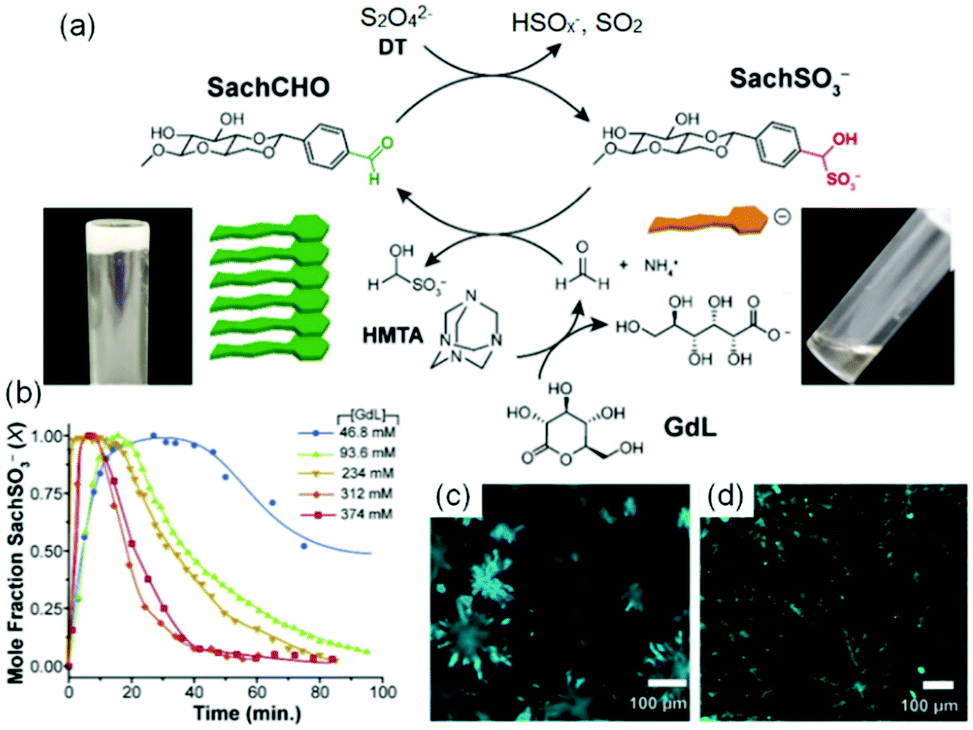 | ||
| Fig. 28 (a) Schematic representation of sequential disassembly and reassembly of SachCHO hydrogel in presence of a chemical reaction cycle. (b) Evolution of SachSO3− in the transient cycles as recorded by NMR at different [GdL]. Initial gel with [SachCHO] = 54 mM, and [HMTA] = 190 mM, [DT] = 191 mM. (c and d) represent confocal microscopy images of the SashCHO gels obtained from a slow catalyzed ([GdL] = 234 mM) (c) and a fast catalyzed ([GdL] = 374 mM) (d) production of SachCHO during the transient cycles. Reproduced from ref. 364 with permission from the American Chemical Society, copyright 2020. | ||
4. Time programmable self-regulating gel to gel transitions
Stimuli-triggered post assembly fabrication of supramolecular assemblies provides an effective strategy to prepare functional nanomaterials with well-defined architectures and properties. In living organisms, many biological processes are associated with stimuli responsive structural changes.365–369 Integration of biological processes with the state-of-the-art stimuli responsiveness of synthetic materials has helped understanding of the fundamental principles related to the biological processes.370–373 However, natural processes operate under temporal regulation and are always associated with self-monitoring and self-correcting properties.365–369,374–376 A true challenge emerges when considering dynamic transformations of synthetic gels by post assembly modification. Here, self-regulating and self-correcting usually demands an external input from the user which limits the scope of autonomous access of the two states. In this section, we discuss various methods that are documented in order to induce time programmable self-regulating gel-to-gel transitions. Integration of biomimicking with traditional gels not only allows temporal access of various properties, but also provides an opportunity to synthesize sophisticated materials which cannot be prepared under normal conditions. In particular, engineering of complexities of living organisms into synthetic gels has opened new avenues toward the construction of next generation functional materials.196,282,377–381(i) Aging
In some ways, gel-to-gel transition is the process of aging. In cases where the assembled network is formed under kinetic control and not necessarily in a thermodynamic minimum, it may undergo a transition over time where the initially formed aggregates rearrange or ‘self-correct’ to form a more stable assembly. This often leads to a change in microstructure as well as bulk properties like gel melting temperature, mechanical strength and visual appearance of the material. For example, Baral et al. reported an increase in turbidity of a peptide hydrogel over two days.382 This transition was driven by an increase in the helicity of the fibre network which resulted in a stiffer (higher G′) gel over time. The Pozzo group reported an amide and carboxylic acid functionalized dynamic system that showed entanglement of small, branched and needle-like fibres to produce unbranched and larger rod-like structures involving Ostwald ripening during aging.383 While aging simply involve spontaneous reorganization of assembled structures, it may be associated with conformational switching of the gelator molecules in the aggregated state. Xu and co-workers demonstrated a gel-to-gel transition of a cinnamoyl-based peptide gel with time.384 Initially, an opaque gel appeared that finally evolved into a transparent hydrogel after aging for ten days. They proposed that the reversible cis/trans-isomerization of the cinnamoyl group is responsible for rearrangement of the supramolecular structures. Aging is also common in multicomponent gel system. Nandi and co-workers synthesized a series of multicomponent hydrogels by varying the concentration of 6,7-dimethoxy-2,4[1H,3H]-quinazolinedione and melamine.385 They found that all the multicomponent gel showed aging induced optical as well as rheological changes. Recently, Gayen et al. reported aging induced tuning of thermal and mechanical properties of a two-component hydrogel system formed from a histidine-coupled naphthalenediimide gelator and tartaric acid.386 Initially (for the freshly prepared gels), the interaction between the imidazole moieties and the carboxylic groups was poor due to the weak dissociation constant of tartaric acid. With time, the imidazole ring of the gelator molecule was slowly protonated. Consequently, the electrostatic interaction set up between the oppositely charged imidazolium unit of the gelator and the carboxylate group of tartrate increases. As a result, the gel became more rigid which led to increase in thermal stability of the gel. The weak fluorescence of the fresh gel was also transformed into bright greenish fluorescence on aging. Importantly, the stiffness of the gel was enhanced by 146 times upon aging for 40 days.Although this natural evolution might be utilized in some cases, it is mostly an uncontrollable process. However, there are various factors that can influence aging. For the gels where gelation is triggered by a heat–cool cycle, aging is observed when the cooling rate is high.383 This is quite obvious as the aggregated structures can have sufficient time to form thermodynamically minimum network when the cooling rate is slow. If the cooling rate is sufficiently slow, then the system directly leads to more stable equilibrated state.387 Baral et al. in their system mentioned above, showed that while a fast cooling produced a kinetically-trapped gel capable of exhibiting aging phenomena, significantly slower cooling triggered the formation of turbid solution instead of any gelation.382 An unusual gel aging was reported which was driven by the carbonate concentration as a result of CO2 in the air dissolving into solution.388 The aging time for turbid to transparent gel transition could be tuned by treatment of the gelator solution either by gaseous CO2 or by directly with carbonate salt. Gel aging was ascribed purely due to morphological transformation from spherical aggregates to long continuous fibres structures (three days). The presence of carbonate ions affected the hydrophobicity of the fibres and so the amount of bound water molecules which resulted in reduction in the inter-fibre interaction leading to decrease in turbidity of the gel. Such solvent exchange between the fibres and medium is also possible in absence of ionic analytes. Mallia et al. described peptide-based organogels in CCl4 that showed thermally-induced opaque to transparent gel-to-gel transitions.389 They suggested that when the opaque gels were heated, reversible inclusion of further solvent molecules between the fibres occurred which caused decrease in turbidity.
The effect of aging is unpredictable, but however crucial for determining the final material properties and hence, for desired applications. The Gazit group reported a aging induced two-step assembly formation of a Fmoc-based dipeptide and successfully studied its use in cell scaffolding.390 Aging can allow self-correcting properties for the gels that can, for example, result in an increase in thermal stability of the network.391 Although aging is known to have an effect on thermal behaviour of gels, surprisingly little work is carried out in this domain. People typically use gels after the systems reach the (apparent) equilibrium conditions and do not change their properties further. Hence, there are real opportunities here to study time variable dynamic changes in thermal behaviour within supramolecular frameworks. The Smith group explored the possibility of increasing the thermal stability of a multicomponent gel system by using one gelator which showed aging-induced enhanced thermal stability.391 Later, Wang et al. utilized a similar concept in developing more thermodynamically stable hydrogels involving a multilevel self-sorting process.392,393 Using multicomponent gelators that spontaneously self-assemble into a stable heterogeneous network, they found that the age of gelator precursors plays an important role in the assembly process. One of the precursor molecules when subjected to an aging treatment produced so-called “tiny aggregates”. These age-induced aggregates then served as seeds forming a kinetically-controlled transparent homogeneous gel in the multicomponent system at a faster rate compared to the case with fresh precursor. After some time, this metastable state converted to the more stable heterogeneous turbid gel. Accessing aging-induced seeded metastable state also resulted in significant robustness of the final gel. Aging often leads to improvement in rheological properties of the gels.382,388 However, this is not always true. Saha et al. reported Zn2+ capped metallogels that showed an opaque-to-transparent transition associated with inferior mechanical properties both in terms of gel stiffness and gel strength.394 In some cases, aging can causes a considerable weakening of interlocked fibre networks leading to spontaneous evolution to crystallization.395–401 Furthermore, there are examples where the aged gel shows a shrinking behavior.402–404 Recently, Sugiura et al. constructed a hydrogel system involving hydrazone-oxime exchange reaction in response to hydroxylamine (Fig. 29).405 When the free-flowing solution of a hydrazine coupled peptide was treated with hydroxylamine in presence of catalytic amounts of aniline, the multicomponent system underwent dynamic covalent bond exchange reaction to produce hydrazide-containing peptide (with the removal of the aldehyde segment) and converted to a gel. With time, the gel underwent a morphological transformation from long fibres to bundled and aggregated clots of the fibrous that caused reduction in the volume of the gel. The aging time depends on various factors like initial concentration of hydroxylamine, aniline and the solution pH. As a result of shrinking, considerable amounts of solvent is released from the gel matrix. Such aging induced shrinking of gel was effectively utilized in removal of water polluting toxic metal ions and dyes as showed by the Banerjee group.406
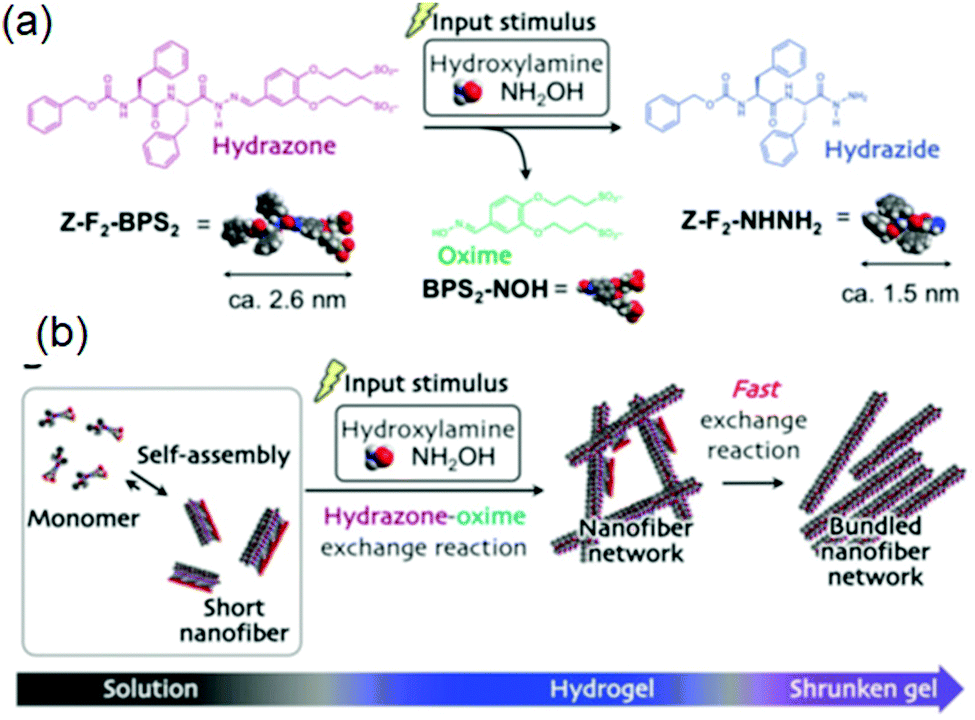 | ||
| Fig. 29 (a) Hydroxylamine induced hydrazone–oxime exchange reaction of progelator Z-F2-BPS2. (b) Schematic representation of aging induced transition of a supramolecular nanofibre network (hydrogel state) to subsequent bundled and aggregated nanofibre network (shrunken gel state). Reproduced from ref. 405 with permission from the Royal Society of Chemistry. | ||
(ii) pH regulated autonomous programming of gel-to-gel transformations
pH driven self-regulated gel-to-gel transitions involve switching between two pH-dependent energy states driven by the presence of a particular chemical reagent. The key concept relies on slow reactivity of the reagent at the early stages so that initially a self-supported gel network can be accessed. However, after a certain time, the chemical reaction is accelerated leading to a rapid pH change of the medium. As a result, the initially-formed gel state spontaneously evolves to a different type of material over time. Alternatively, two different scenarios can be considered. Firstly, the chemical reaction shows a unidirectional response leading to alter the pH of the medium. Another situation may arise; if the chemical reaction is coupled with a second reaction that resist the pH change with time, it is then possible to revert the medium to its initial pH. However, under both conditions, the pH changes need to be controlled to achieve time dependent accessing of different gel states.Typically, strong acids or bases are added externally to change the pH of the medium. However, this is not desirable for an autonomous gel-to-gel transition which demands a local change in the pH of the medium so that an initially formed gel state could be accessed before it evolves to a new state. A slow pH decrease may be effective to convert one component system into a two component gel networks through sequential assembly formation. For example, when an alkaline mixture of two gelators having distinctly different pKa was treated with GdL, slow hydrolysis of GdL to gluconic acid resulted in a gradual decrease in pH that enabled sequentially assembly of each gelator at their respective pKa values and allowed formation of a self-sorted system.407,408 Use of GdL provides an advantage over strong acids as it is possible to probe the kinetics of the pH change by the amount of added GdL.228 This method often leads to more stable homogeneous hydrogel formation as compared to the use of mineral acids and hence faster pH changes.211,409
Recently, we developed a method to achieve an unusual gel-to-gel transition for a single peptide system (Fig. 30). We chose a naphthyl-derived dipeptide (2NapFF) that can form a gel at low pH as well as exhibits Ca2+ induced hydrogelation at alkaline pH. We then employed the autocatalytic reaction between urease and urea to increase the pH of the medium. The urease-urea reaction was developed by Taylor et al.410,411 as an alternative of conventional bases like gaseous NH3, metal hydroxides or basic buffers, and then used by many other research groups.216,412–416 In aqueous medium, urea undergoes hydrolysis in presence of urease and produces ammonia. As a result, the pH of the medium increases. The reaction is clean and allows accessing of a broad pH range of pH 4 to pH 9. The reaction itself is highly pH dependent as below pH 4 urease loses its activity216,414,417 Therefore, the rate of the reaction can easily be controlled by adjusting the initial pH and the reagent concentrations. When we coupled the enzymatic reaction with our hydrogel system in presence of Ca2+, initially a self-supportive gel was formed exhibiting pH of around 4. With time, the pH increases slowly, deprotonation of carboxylic acid occurred, and the gel gradually returned to a free-flowing solution state under dissipative condition. Again, over time, Ca2+ ions binding to the carboxylates at high pH resulted in reappearance of the gel. When we performed the same reaction in the absence of Ca2+, a transient hydrogelation occurred (gel-to-sol transition) showing that the presence of Ca2+ was necessary to drive gelation at high pH. Time sweep rheology and confocal fluorescence microscopic studies confirmed the pH-dependent destruction of the primary assembled structures and salt induced gradual reconstruction of the assemblies with time. The lifetime of the initially formed gel significantly depends upon the rate of pH change and could be controlled by adjusting the initial concentrations of urease, urea and Ca2+ ions which eventually led to the formation of transparent, homogeneous and reproducible hydrogels with good control over the final material properties. Interestingly, unlike the gels formed by the dissipative process, direct hydrogelation by adding Ca2+ to the alkaline solution led to inhomogeneous turbid gels. Inhomogeneity can be a real issue when the mixing of the components competes with gelation and often results in gels with irreproducible properties.19,418 Hence our approach is effective in triggering homogeneous hydrogelation of peptides at high pH. Furthermore, unlike conventional gel-based dissipative assemblies217,419–423 where the limited lifetime of the gels has rare practical applications,424,425 our pH responsive out-of-equilibrium system permits pre-programming of a system to drive an unusual gel-to-sol-to-gel transition which allows access of interesting materials that cannot be directly prepared.
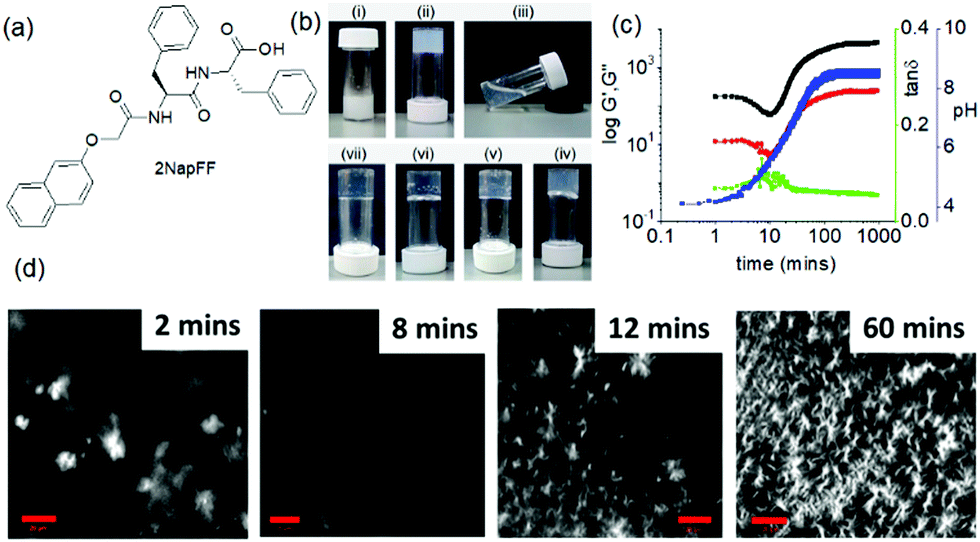 | ||
| Fig. 30 (a) Chemical structure of dipeptide 2NapFF. (b) Photographs showing autonomous gel to gel transition of the hydrogel of 2NapFF in presence of Ca2+ involving urease–urea reaction: after (i) immediate addition, (ii) 2 min, (iii) 8 min, (iv) 30 min, (v) 1 h, (vi) 3 h, (vii) 16 h. (c) Correlation of time sweep rheology with the pH change shows an initially formed gel slowly disintegrates into solution as the pH of the medium increases. Further increase in pH allowed Ca2+-ion binding by the carboxylate of 2NapFF. Consequently, there was an increase in rheological moduli values with time. Black data represents G′, red data G′′ and blue data pH. (d) Confocal microscopy images showed pH induced destruction of the primary assembled structures followed by Ca2+ induced reconstruction of aggregates at high pH. Reproduced from ref. 426 with permission from the Royal Society of Chemistry. | ||
We utilised the same enzymatic reaction to anneal pH-responsive hydrogels (Fig. 31).427 We employed a reaction relay on a peptide system (1ThNapFF) to anneal the structures locally through a uniform and control pH change. To construct the consecutive reaction relay, we coupled the autocatalytic urease-urea reaction with methyl formate, a hydrolysis agent that undergoes base promoted saponification to generate formic acid that results in reduction of the pH. We adjusted the initial conditions in such a way that at the early stages, production of the ammonia is faster than the hydrolysis of methyl formate while after certain time base promoted hydrolysis of methyl formate becomes predominant. Again, the urea-urease reaction initially drives the system to form an out-of-equilibrium gel, followed by increasing the pH of the medium to form a transient hydrogel. Subsequently, hydrolysis of methyl formate led to reassembly by reducing the pH over time. Our approach enabled us to prepare homogeneous and reproducible gels which are both stiffer and contain a different network structure than the original gel. We suggested that, annealing resulted in a change in the gel structure from spherulitic domains to a more uniform distribution of fibres, that ultimately led to the formation of homogeneous hydrogels with improved mechanical properties. Conventionally, a heat–cool operation is conduced to anneal gels.53–56,60 We observed that, our annealing approach is more effective in improving gel robustness than thermal annealing for the present system.
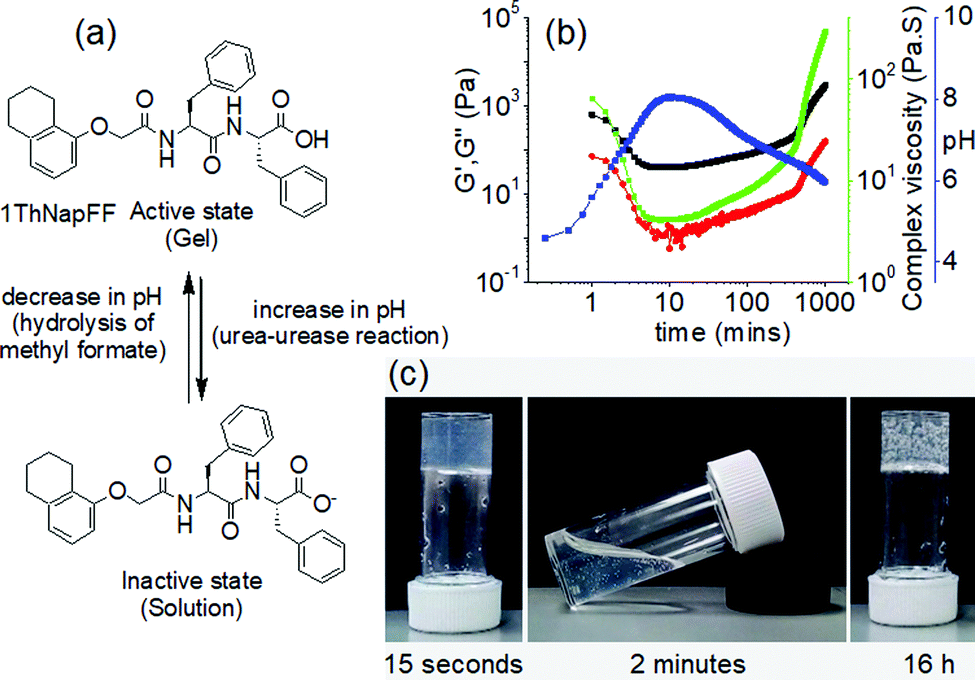 | ||
| Fig. 31 (a) Annealing peptide hydrogel of 1ThNapFF by a pH cycle. The pH cycle was constructed by coupling the urease–urea enzymatic reaction with methyl formate. (b) represents variation of rheological moduli with pH over time. Black data represents G′, red data G′′, green data complex viscosity and blue data pH. (c) A gel–sol–gel phase transition was observed visually during the annealing process. For (b) and (c), initial reaction condition: [1ThNapFF] = 2 mg mL−1, [urease] = 0.2 mg mL−1, [urea] = 0.02 M, volume of methyl formate = 100 μL. Reproduced from ref. 427 with permission from the American Chemical Society, copyright 2020. | ||
As we discussed earlier, thermal annealing has limitations in terms of thermal sensitivity of various functional groups present on the gelator skeleton.61–65,428,429 Furthermore, there may be issues related to solvent evaporation that leads to a change in the solvent composition of gels. During thermal annealing, typically the gel is prepared inside a vial or a container and then the whole system is subjected to heating from outside, which imposes restrictions in terms of the volume of gel that can be annealed. Moreover, during the heat–cool cycle, there may be temperature gradients within the gel as well as at the liquid-container junction that often prevents a homogeneous temperature change inside the system. Consequently, in true sense, homogeneous ‘moulding and casting’ (i.e. melting followed by flowing and then solidification) of gels is not possible using conventional annealing process. Our approach of annealing offers local changes in pH at ambient temperature and hence annealing of gels without any volume restrictions (Fig. 31). The rate of annealing can easily be controlled by adjusting initial reaction conditions which enabled us to produce a range of materials from a single starting gel. Furthermore, as it produced a free-flowing solution as an intermediate during annealing, it was possible to use our annealing method for autonomous programming of homogeneous ‘moulding and casting’ of the hydrogel assemblies in time. Additionally, this method can also be used to drive controlled mixing of encapsulated components within different gel environments.
The Wang group reported switching of a polymeric hydrogel between two different pH dependent states in the presence of the urea–urease reaction.430 The polymer gel was fabricated by in situ formation of dynamic acylhydrazone bonds. The chemical bond formation was efficient at a low initial pH of 4 yielding a red coloured gel in presence of neutral red (used as dye to generate the colour). They observed that the hydrogel behaved as a kinetically labile system at acidic pH where the activation of the acylhydrazone bonds through protonation led to faster gelation. On treatment with urea, production of ammonia drives the system to a yellow coloured kinetically stable gel with time. Interestingly, the hydrogel showed better healing ability under acidic condition. A damage-healing experiment was carried out where the kinetically stable yellow gel was harmed first, followed by feeding with acidic urea solution at the damaged site (Fig. 32). As a result, a transient acidic pH state was temporarily created that allowed recovery of the damage with red colouration. With time the enzymatic reaction again drove the system to the yellow coloured kinetically stable gel with complete recovery of the material properties.
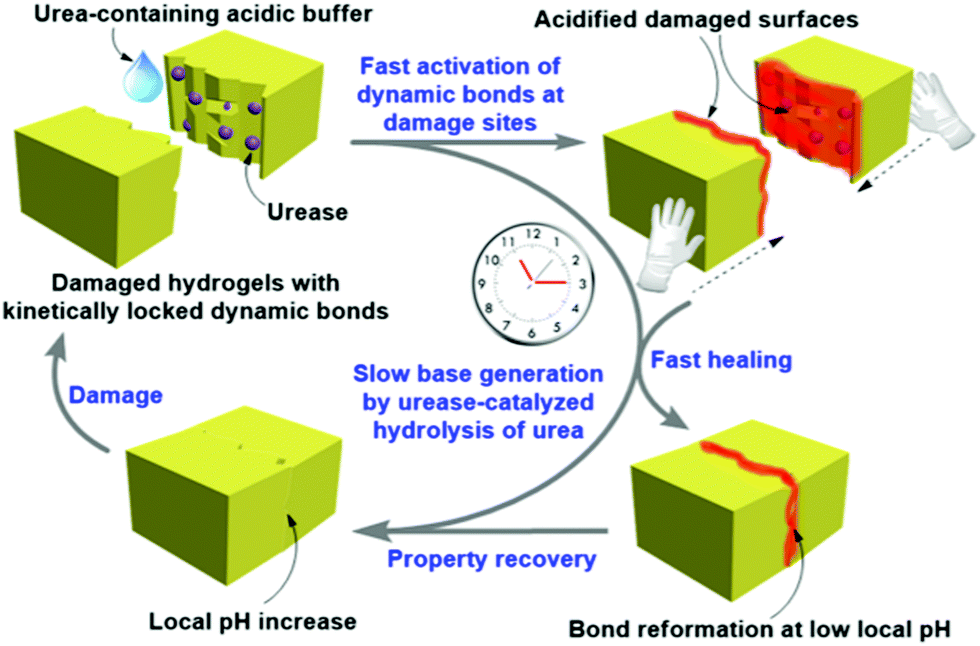 | ||
| Fig. 32 Schematic representation of kinetically controlled damage-healing experiment of hydrogel. A transient acidic pH state was temporarily created that allowed recovery of the damage with red colouration. The transient healability of damaged hydrogels was temporally programmed by combining a fast acidic activator (acylhydrazone activation) with the slow enzymatic generation of a base (urea–urease reaction). Adapted from ref. 430 with permission from the American Chemical Society, copyright 2020. | ||
We followed similar strategy to control properties and lifetime of metastable hydrogels.431 We devised a system by employing dynamic imine chemistry between 4-(dimethylamino)benzaldehyde and hydrazine monohydrochloride in DMSO/H2O (molar ratio of aldehyde and amine was 2:1). The system led to either a kinetically controlled transient orange gel at acidic pH or a thermodynamically stable yellow gel at high pH. Under both conditions, formation of the imine was responsible for gelation, however, the response of the systems was governed by the protonation/deprotonation of the in situ synthesized imine compound. The orange colour of the hydrogels was attributed to the protonation state of the imine product while the yellow colour was generated due to the unprotonated form of the imine at high pH. Protonation of the imine bond accelerates the nucleation of self-assembly by intermolecular hydrogen bonding resulting in rapid hydrogelation. By adjusting the initial pH, a metastable yellow gel was accessed as intermediate which initially evolved into an orange gel and then to an orange sol over time. Hence, transient aggregation was occurred by successive formation of two distinctly different kinetically controlled metastable states. Interestingly, unlike Wang's system mentioned above,430 exploitation of the urease-urea reaction did not allow conversion of the low pH-orange gel to the high pH yellow gel in our case. Instead, we observed a controlled switching from a yellow gel (high pH) to an orange gel (low pH) without any phase transformation. Exploitation of the enzymatic reaction enabled us to programming properties of a metastable assembly state which ultimately pushed the system to a new material exhibiting a gel-to-gel transition.
(iii) Redox responsive self-regulating gel to gel transitions
Redox responsive dynamic conformational changes are crucial for the human organism. In living system, many processes like Ca2+ regulation, muscle control, cell division, genetic expression, cell immunity, mitochondrial function, neurogenic events include redox responsiveness to “sense and react”.432 Unregulated redox mechanisms can lead to severe diseases like diabetes, fibrosis, cancer, cardiovascular and neurological diseases etc.433 From this perspective, there is significant interest in using a change in redox state as a trigger for constructing biomimetic hydrogels.377,434,435A well-accepted method of constructing gels that show self-regulated redox-triggered gel-to-gel transitions is to synthesize a metal–organic gel system followed by an in situ change in the oxidation state of the metal ion by adding an appropriate chemical reagent during the self-assembly process. This allows the formation of time-programmable redox-induced gels. Such gels have potential in designing biodegradable scaffolds, tissue engineering, sensors and actuators. However, most of the examples fall in this domain are based on polymeric hydrogels. Yoshida et al. developed a copolymer gel of N-isopropylacrylamide (IPAAm) and incorporated ruthenium(II)tris-(2,2′-bipyridine) (Ru(bpy)32+) as Belousov–Zhabotinsky (BZ) reaction catalyst within the gel.436 In the composite polymer, the BZ catalyst was directly attached to the polymer chain via covalently bond. The composite gel showed reversible shrinking and swelling of the hydrogel due to change in oxidation state between the reduced (Ru2+) and oxidized form (Ru3+). The BZ reaction in the gel caused periodic charge modification on the polymer backbone which resulting in mechanical oscillation of the polymer network. Based on same strategy, the Suzuki group further developed an oscillating microgel system which exhibited fast swelling/deswelling and dispersing/flocculating oscillation.437 They suggested that this system could be explored in fabricating autonomously oscillating micropumps similar to human heartbeat.
Ballav and co-workers undertook a similar strategy to convert a coordination polymer gel into a conducting polymer gel.438 A metal–organic gel initially comprised of Fe(III) and terephthalic acid was subjected to redox changes in presence of organic molecules like pyrrole, aniline and bithiophene. In situ oxidation by Fe(III) led to the formation of polypyrrole, polyaniline and polythiophene, respectively, and allowed transition of the Fe(III)-gel into hybrid Fe(II)-conducting polymer gels accompanied with drastic changes in morphology and luminescent properties. The redox reaction induced a remarkable change in morphology of the metal–organic gel from spindle-like structure to nanofibres. Additionally, the original nonfluorescent metallogel was converted into a highly photoluminescent hybrid gel. The fluorescence turn on response was attributed to the production of oxidized polymers inside the porous gel matrix.
Zhang et al. prepared a complex heterometallic coordination polymer gel by integrating Pd2+ and Cu2+ ions simultaneously with a pyridine functionalized isophthaloyl ligand (Fig. 33).439 The ligand 5-(pyridin-4-yl)isophthalic acid was consisting of two metal coordinating sites. When the ligand was mixed with Pd2+ and Cu2+ ions in a molar ratio of 2:2:1 (ligand:Cu2+:Pd2+) in a mixed solvent like e.g. DMF–MeOH–MeCN, a greenish yellow gel was obtained. In the gel state, while the Pd2+ ions coordinate to the pyridine group and acted as a bridging unit, Cu2+ ions coordinate to the carboxylate groups resulted in formation of a coordination polymers gel. With time, the colour of the gel changes and after ∼1 month it transformed into a blue coloured gel without showing any phase transformation. By X-ray photoelectron spectroscopic studies, it was suggested that the colour change was due to reduction of Cu(II) to Cu(I). However, since no Pd(0) was formed, the changes in Cu-oxidation state was explained by the nanofibres reducing Cu(II) to Cu(I) in a confined environment.
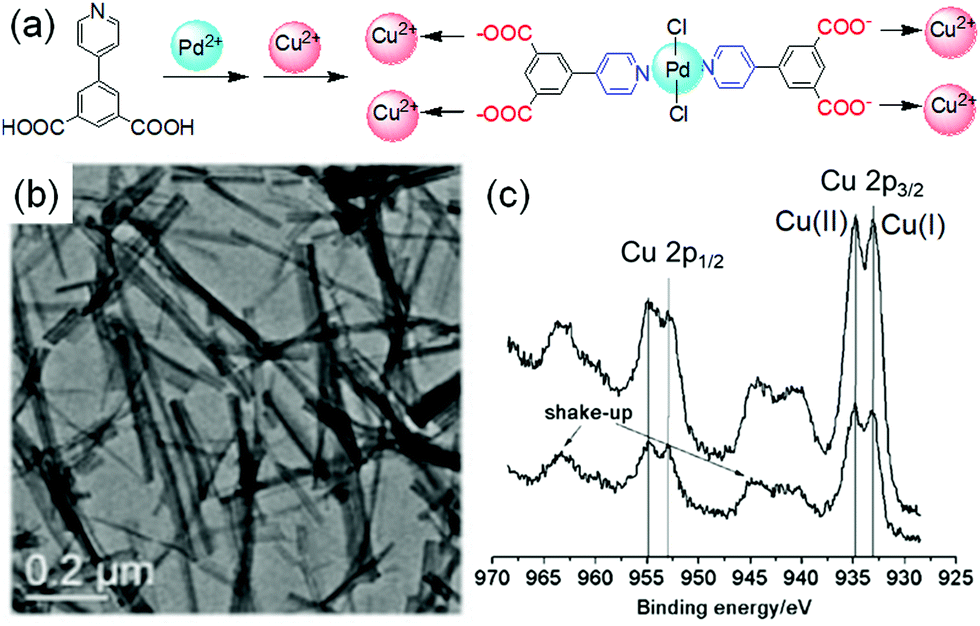 | ||
| Fig. 33 (a) Proposed coordination in the heterometallic gels of 5-(pyridin-4-yl)isophthalic acid. (b) TEM images of PdCu(NO3)2·H2O gel of the ligand. (c) Comparison XPS data over the Cu 2p regions for the xerogels of PdCu(NO3)2·MeOH (bottom), and Cu(I) containing PdCu(NO3)2·MeOH (top). The Cu 2p3/2 signal at 932.8 eV was due to Cu(I) and the signal at 934.8 eV was for Cu(II). Reproduced from ref. 439 with permission from the John Wiley and Sons, copyright 2011. | ||
Recently, we reported a redox responsive dynamic system and emphasized the importance of pathway dependency for redox-based supramolecular gels.440 We synthesized an iron-containing organic gelator by performing dynamic covalent bond formation reaction between 4-(dimethylamino)benzaldehyde and N,N-dimethyl-p-phenylenediamine in presence of Fe(II) salt. The presence of Fe(II) ions made the gel system redox responsive. In situ oxidation of the Fe(II) ions by NaNO2 resulted in conversion of the Fe(II)-gel into a Fe(III)-gel. We showed that the final properties of the Fe(III)-gel depend upon the rate of oxidation of Fe(II) which actually control the final gel properties like rheological and optical properties. As the rate of oxidation increased, the corresponding Fe(III) gel became weaker. Moreover, upon oxidation, the materials exhibited up to 38% increase in volume of the final material without any destruction of the gel architecture. The extent of the swelling could be controlled just by adjusting the initial NaNO2 concentration. The temporal changes in gel properties depended upon the assembled structures, and the rate of formation of Fe(III) changed the Fe-interactions with the fibres.
Another variant of the redox-responsive dynamic system is the synthesise of temporally regulated transient hydrogels where the assembled structures display a limited lifetime before it undergoes sol formation.423,441,442 There has been recent interest in constructing transient hydrogels based on redox reactions. Most of such systems are usually developed either from the intermolecular disulfide exchange reaction-based molecular probes,86,443,444 or from incorporating redox responsive metal ions onto the organic gelator backbone.445,446 However, a key issue is the short lifetime of the gels in their active state which limit their scope of application. In this regard, redox responsive time programmable gel-to-gel transitions are worthy of interest because of their widespread applications.
(iv) Chemically fuelled systems
A key issue for most of the self-regulating dynamic systems (except aging, reaction cycle) is that the environmental conditions (e.g. pH) of the final gel significantly differ from the initially formed material. Even in some cases, the applied method (e.g. covalent bond formation) results in changes in the chemical structure of the gelators. To overcome these issues, our group introduced a fuel-driven dynamic system that exhibited an automated gel-to-gel transformation at similar final conditions.447 Unlike conventional gel-to-gel transitions, our target was to modify the gelator structure at the beginning of gelation involving chemical reaction so that the initially formed gel state incorporates the modified molecules. With time, as the modified molecules started to regenerate the original precursor, a different gel state could be achieved at the end under similar final conditions.To achieve this, we utilized the carbodiimide-induced acid-anhydride reversible conversion as the key chemical reaction. We used 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as the chemical fuel to convert a C-terminal peptide into its anhydride. EDC has been extensively used for constructing fuel-driven transient hydrogel systems.421,448–453 Here, we exploited the same protocol to achieve a temporally programmed gel-to-gel transition. We deliberately chose the gelator as it could form self-supporting gel in 20/80 DMSO/water in absence of EDC exhibiting densely packed needle-like fibres in the aggregated state. However, when EDC was added to, the gelator molecules was converted into its anhydride that initially caused a transition from a solution to a gel state by co-assembly. Over time, the anhydride component started to hydrolyze and regenerated the precursor, although the network structure remained intact. As a consequence, the complete cycle resulted in an autonomous gel-to-gel transformation with the same gelator structure at the end. Although, no significant change in the visual appearance as well as microstructures was noticed during the gel evolution process, time sweep rheology and spectroscopic studies demonstrated the gel-to-gel transition through successive covalent bond formation and rupture. We also established that exploitation of fuel-driven pathway not only allowed access to the materials that cannot be obtained directly under similar final conditions, but also it provided opportunity to control the final properties of the gels just by varying the initial EDC concentrations. We proposed that when different concentrations of EDC were used, the co-assembled gels formed by the anhydride were kinetically trapped. Energy dissipation from these kinetically-trapped states through hydrolysis eventually governed the final properties of the gels. This method was effective in improving gel strength than the material prepared directly without using EDC.
There is a sharp difference between fuel-driven chemical bonds and dynamic covalent bonds. Dynamic covalent bonds are formed and broken reversibly under thermodynamic equilibrium.247,248 In particular, formation of a dynamic covalent bond follows energetically down-hill processes. As a result, the newly formed bond is stable unless the equilibrium is disturbed by means of chemical stimulants. In contrast, fuel-driven covalent bond formation is an energetically up-hill process and so a constant energy source is required to maintain the high energy chemical structure.32,454,455 On removal of the energy source, the system spontaneously drives to low-energy precursor structure. This difference probably makes fuel-driven pathways superior to constructing gel-to-gel transition under similar final conditions.447
5. Future perspective
Functional supramolecular gels are promising in the field of cell culturing, sensing, drug delivery, catalysis, tissue engineering, environmental remediation, and optoelectronics.9–12 In all of these cases, the gels need specific, but different properties. In order to explore these gels for task-specific applications, it is essential to adapt and control the material properties. This can be achieved by changing the underlying gel structure through gel evolution. Hence, the desired application apparently depends on the chemical structure of gelator. In this context, aromatic peptide gelators gain sever attention as they are easily functionalizing as well as biocompatible.11,210 Again, the choice of gelator is important as the functional group present on gelator backbone determines the suitability of the applied stimuli. The most common stimuli that are used to induce dynamic changes include temperature, pH, light, enzyme, redox, and chemical analytes. The desired application also determines the appropriateness of these stimuli.Stimuli-triggered post assembly fabrication (PAF) of gels is the most common strategy applied in gel chemistry to adapt material properties. One potential advantage of PAF is that it provides opportunity to create heterogeneous and hierarchical hydrogels with internal gradients in nanostructure, crosslink density, mechanical and responsive properties.456,457 Gradient hydrogels enable to integrate variable properties into a single gel material leading to many possible applications including tissue engineering and cell culturing.458,459 Different stimuli can be employed to generate hydrogels with intrinsic heterogeneous structures over a broad length scale.457 Photo-patterned hydrogels are the most common heterogeneous gels with a tunable gradients in mechanical and photophysical properties (discussed in Section 3(iii)). On the other hand, diffusion of metal ions, chemical analytes can also be used to fabricate to spatiotemporal gradient gels.456,460 For example, Wang et al. developed a simple method, called ion transfer printing, to devise gradient hydrogels.461 They used a solution of Fe3+ as an ink to print on a filter paper. When the filter paper printed with Fe3+ solution was placed in contact with a polymer hydrogel sheet, slow diffusion of Fe3+ ions into the hydrogel resulted in a change in local coordination and crosslinking of the fibres. A patterned gel with gradient in stiffness was obtained with a higher stiffness at the Fe3+ coordinated zones. Furthermore, the oxidation state of the metal ion in the gel can be controlled electrochemically or by redox reaction (discussed in Section 3(vi)) which enables to programme redox-responsive gradient gels with pre-defined properties. Similarly, a change of pH can also induce a diffusion controlled transient gradient in pH during gel-to-gel transitions and thereby integrate transient heterogeneous structures.234 Longo and co-workers utilized pH gradients to facilitate protein adsorption to a polymer hydrogel film from binary and ternary mixtures of myoglobin, lysozyme and cytochrome c.462 Change of pH can also be utilized to control the degree of ion–ligand coordination as well as distribution of metal ions in pH responsive metallogels (discussed in Section 3(iii)) and thereby enables to control inhomogeneity in crosslink density, rheological moduli, and responsive properties in gradient hydrogel networks. However, most of the chemical stimuli responsive gradient gels are composed of polymeric backbone. It will be interesting to investigate the effectiveness supramolecular low molecular weight gels in devising chemical stimuli-induced gradient gels. Furthermore, a major difficulty is to investigate the local changes in material properties in gradient gels by oscillatory rheology (as it measures the bulk properties). Modern techniques such as nanoindentation,463 cavitation rheology464etc. could be used to provide information about how the underlying changes in the network affect the local mechanical properties.
Dynamic transformations of supramolecular gels in presence of a cyclic energy input is getting more and more interest to reprogramme gel properties after they are formed (Fig. 28 and 31).364,427 However, the key challenge arises from how these dynamic systems evolve. In presence of an energy cycle, mostly the dynamic system incorporates a solution state as an intermediate (i.e. gel-to-sol-to-gel transition) where the gel loses its integrity and first falls apart completely before it reassembles again (Fig. 28 and 31).364,427 Hence, there is a possibility that after the gel structures are completely destroyed, the free molecules simply reassemble kinetically. To counter this, present research focuses on creating more complex systems in which the molecular dynamics can be controlled in such a way that the gel system would resist the sol formation during evolution. This leads to the development of ‘homeostatic’ hydrogels where a gel material evolves through a gel-to-gel-to-gel pathway.465
Typically, homeostasis is a biological concept that refers to the ability of a system to sustain its integrity in response to a change in the environment.466,467 Systems exhibiting homeostasis are called homeostatic systems. Recently we tried to mimic the concept of homeostasis by which a gel system can maintain its gel form during evolution in presence of a feedback loop.465 We consider the conservation of gel state (i.e phase integrity) throughout an energy cycle as the essential criterion of homeostasis. We deliberately chose the system described in Fig. 31 for this purpose. In Fig. 31, the gel fell apart completely or loses its integrity at high pH.427 For the system to function as a homeostatic module, instead of a gel–sol–gel transition, it needs to evolve through a gel-to-gel-to-gel transition. To prevent the sol formation, we incorporated Ca2+ into the system described in Fig. 31. At high pH, cross-linking of the micellar dispersion of the carboxylate anions by Ca2+ was possible which resulted in formation of a Ca2+-triggered gel at elevated pH and eventually allowed the system to evolve in a gel-to-gel-to-gel fashion in presence of a pH cycle.465 The on–off switching of the pH cycle resulted in different feedback from the gel and creates three mini-homeostatic modules with time. The value of this behavior is clear: the gelator molecules utilized the sources available in the medium to resist structural destruction under particular condition. Furthermore, the evolution of the mini-homeostatic modules induces self-correcting behaviour in the system leading to improvement in the mechanical properties of the gel.
Apart from biomimicry, attempts have also been made to explore homeostatic hydrogels for practical applications. He et al. demonstrated a hydrogel-supported bilayer-based homeostatic system that exhibits chemo-mechano-chemical self-regulation.468 They utilized this platform to perform several exothermic catalytic reactions. However, the study of homeostatic hydrogels is still in its infancy. Development of new models for homeostatic hydrogels is necessary to explore this field. The effect of other energy cycles (e.g. chemical reaction cycle, light, redox etc.) on the synthetic homeostatic system also needs to be investigated.
6. Conclusions and outlook
Supramolecular self-assembly is the heart of bio-organic chemistry. Among various self-assembled system, low molecular weight gelators are receiving much current attention. Typically, supramolecular gels exhibit responsive behaviour because of the existence of non-covalent interactions and undergo gel to gel transitions with a dramatic change in microscopic as well as macroscopic properties over time. Here, we have discussed various methods that have been used to achieve gel-to-gel transitions. While traditional gel systems mostly rely on an external input, self-regulating gels offer autonomous switching between two gel states. Stimuli-triggered gel-to-gel transitions allow construction of an array of supramolecular gels with various properties from a single material which extends the limit of applications of the gels. Application of gels in a particular field is determined by the bulk properties or specifically mechanical properties of the gels. The mechanical properties depend on the nature of the fibre formed during self-assembly. Reconfiguration of gel states provide further advantage in tuning and controlling the mechanical properties. In some cases, gel-to-gel transitions allow one to produce materials that cannot be accessed directly.The research has been carried out so far in this domain has encountered several drawbacks. One potential issue is limited understanding of changes in the underlying microstructure during gel evolution.469 Techniques such as scanning electron microscopy (SEM), transmission electron microscopy (TEM) or atomic force microscopy (AFM) have been used to compare the morphology changes after evolution. Drying of the sample is usually required for these experiments although drying of gels can lead to changes in the self-assembled network.470 Confocal laser scanning microscopy (CLSM),471 and super-resolution microscopy techniques such as Points Accumulation for Imaging in Nanoscale Topography (PAINT)472 demand attention in this context as they enable real time monitoring of changes in microstructure of gels under native conditions and thereby understanding their impact in material properties. Further, small angle scattering473 experiments are rarely carried out to probe the changes at the fibre level assembly, but could be used to provide information as to how the underlying changes are occurring. However, the major drawback is that rheological studies are rarely performed to understand the changes in mechanical properties of the gels during the transition process. Surprisingly, people mostly perform UV-vis and fluorescence studies to demonstrate the gel-to-gel transitions, which cannot inform as to the mechanical properties. Indeed, the topic stimuli responsive chemical and structural changes of gels is highly demanding to inform new models of supramolecular gelation.380,474–476 The rising popularity in the field certainly promises construction of bioinspired next generation functional materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP thanks the University of Glasgow for funding. DA thanks the EPSRC for funding (EP/S019472/1).References
- P. A. Kollman, Acc. Chem. Res., 1977, 10, 365–371 CrossRef CAS.
- G. Ouyang and M. Liu, Mater. Chem. Front., 2020, 4, 155–167 RSC.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS.
- J. Zhang, Y. Hu and Y. Li, Gel Chemistry: Interactions, Structures and Properties, Springer Singapore, Singapore, 2018, pp. 9–59 DOI:10.1007/978-981-10-6881-2_2.
- Y. Wang, J. Chou, Y. Sun, S. Wen, S. Vasilescu and H. Zhang, Mater. Sci. Eng., C, 2019, 101, 650–659 CrossRef CAS.
- R. K. Mishra, S. Das, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Molecular Gels: Structure and Dynamics, The Royal Society of Chemistry, 2018, pp. 190–226 10.1039/9781788013147-00190.
- G. R. Deen and X. J. Loh, Gels, 2018, 4, 13 CrossRef.
- W. Zhang and C. Gao, J. Mater. Chem. A, 2017, 5, 16059–16104 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- Y. Cai, W. Ran, Y. Zhai, J. Wang, C. Zheng, Y. Li and P. Zhang, Biomater. Sci., 2020, 8, 1045–1057 RSC.
- A. Dawn, Int. J. Mol. Sci., 2019, 20, 781 CrossRef CAS.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- D. M. Zurcher and A. J. McNeil, J. Org. Chem., 2015, 80, 2473–2478 CrossRef CAS.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- E. R. Triboni, T. B. F. Moraes and M. J. Politi, in Nano Design for Smart Gels, ed. R. Bacani, F. Trindade, M. J. Politi and E. R. Triboni, Elsevier, 2019, pp. 35–69 DOI:10.1016/B978-0-12-814825-9.00003-5.
- M. Liu, G. Ouyang, D. Niu and Y. Sang, Org. Chem. Front., 2018, 5, 2885–2900 RSC.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS.
- S. Panettieri and R. V. Ulijn, Curr. Opin. Struct. Biol., 2018, 51, 9–18 CrossRef CAS.
- D. Kuckling, Gels, 2018, 4, 60 CrossRef.
- M. A. Mohamed, A. Fallahi, A. M. A. El-Sokkary, S. Salehi, M. A. Akl, A. Jafari, A. Tamayol, H. Fenniri, A. Khademhosseini, S. T. Andreadis and C. Cheng, Prog. Polym. Sci., 2019, 98, 101147 CrossRef CAS.
- C. Echeverria, S. N. Fernandes, M. H. Godinho, J. P. Borges and P. I. P. Soares, Gels, 2018, 4, 54 CrossRef.
- A. Ghosh, M. Haverick, K. Stump, X. Yang, M. F. Tweedle and J. E. Goldberger, J. Am. Chem. Soc., 2012, 134, 3647–3650 CrossRef CAS.
- S. A. Deshmukh, L. A. Solomon, G. Kamath, H. C. Fry and S. K. R. S. Sankaranarayanan, Nat. Commun., 2016, 7, 12367 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS.
- R. M. P. da Silva, D. van der Zwaag, L. Albertazzi, S. S. Lee, E. W. Meijer and S. I. Stupp, Nat. Commun., 2016, 7, 11561 CrossRef CAS.
- R. K. Grötsch and J. Boekhoven, in Self-assembling Biomaterials, ed. H. S. Azevedo and R. M. P. da Silva, Woodhead Publishing, 2018, pp. 235–250 DOI:10.1016/B978-0-08-102015-9.00012-5.
- S. H. Jung, M. Takeuchi and K. Sugiyasu, in Kinetic Control in Synthesis and Self-Assembly, ed. M. Numata, S. Yagai and T. Hamura, Academic Press, 2019, pp. 205–229 DOI:10.1016/B978-0-12-812126-9.00010-9.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS.
- B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS.
- Y. Li and Y. Cao, Chinese J. Polym. Sci., 2018, 36, 366–378 CrossRef CAS.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- M. Raynal and L. Bouteiller, Chem. Commun., 2011, 47, 8271–8273 RSC.
- D. Rosa Nunes, M. Raynal, B. Isare, P.-A. Albouy and L. Bouteiller, Soft Matter, 2018, 14, 4805–4809 RSC.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- K. Godrikus, D. J. Young and X. J. Loh, Biodegradable Thermogels, The Royal Society of Chemistry, 2019, pp. 102–112 10.1039/9781788012676-00102.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- K. Ghosh, D. Kar, S. Panja and S. Bhattacharya, RSC Adv., 2014, 4, 3798–3803 CAS.
- Y. Hou, F. Xin, M. Yin, L. Kong, H. Zhang, T. Sun, P. Xing and A. Hao, Colloids Surf., A, 2012, 414, 160–167 CrossRef CAS.
- M. Yan, S. K. P. Velu, M. Maréchal, G. Royal, J. Galvez and P. Terech, Soft Matter, 2013, 9, 4428–4436 RSC.
- V. J. Nebot, J. J. Ojeda-Flores, J. Smets, S. Fernández-Prieto, B. Escuder and J. F. Miravet, Chem. – Eur. J., 2014, 20, 14465–14472 CrossRef CAS.
- K. Kuroiwa, T. Shibata, A. Takada, N. Nemoto and N. Kimizuka, J. Am. Chem. Soc., 2004, 126, 2016–2021 CrossRef CAS.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- J. Cui, A. Liu, Y. Guan, J. Zheng, Z. Shen and X. Wan, Langmuir, 2010, 26, 3615–3622 CrossRef CAS.
- R. Alberstein, Y. Suzuki, F. Paesani and F. A. Tezcan, Nat. Chem., 2018, 10, 732–739 CrossRef CAS.
- J. Jiang, A. G. Jacobs, B. Wenning, C. Liedel, M. O. Thompson and C. K. Ober, ACS Appl. Mater. Interfaces, 2017, 9, 31317–31324 CrossRef CAS.
- H.-L. Yip, H. Ma, Y. Tian, O. Acton, N. M. Tucker and A. K. Y. Jen, J. Mater. Res., 2011, 26, 311–321 CrossRef CAS.
- L. Wang, X. Shi and J. Wang, Soft Matter, 2018, 14, 3090–3095 RSC.
- J. Fujiwara, M. Takahashi, T. Hatakeyama and H. Hatakeyama, Polym. Int., 2000, 49, 1604–1608 CrossRef CAS.
- M. Llusar, B. Escuder, J. D. D. López-Castro, S. Trasobares and G. Monrós, Gels, 2017, 3, 23 CrossRef.
- C. M. Rubert Pérez, L. A. Rank and J. Chmielewski, Chem. Commun., 2014, 50, 8174–8176 RSC.
- H. Kumari, S. E. Armitage, S. R. Kline, K. K. Damodaran, S. R. Kennedy, J. L. Atwood and J. W. Steed, Soft Matter, 2015, 11, 8471–8478 RSC.
- E. C. Barker, A. D. Martin, C. J. Garvey, C. Y. Goh, F. Jones, M. Mocerino, B. W. Skelton, M. I. Ogden and T. Becker, Soft Matter, 2017, 13, 1006–1011 RSC.
- D. M. Murali and G. Shanmugam, New J. Chem., 2019, 43, 12396–12409 RSC.
- A. M. Fuentes-Caparrós, F. de Paula Gómez-Franco, B. Dietrich, C. Wilson, C. Brasnett, A. Seddon and D. J. Adams, Nanoscale, 2019, 11, 3275–3280 RSC.
- S. Höck, R. Marti, R. Riedl and M. Simeunovic, Chimia, 2010, 64, 200–202 CrossRef.
- A. M. Genaev, L. N. Shchegoleva, G. E. Salnikov, A. V. Shernyukov, L. A. Shundrin, I. K. Shundrina, Z. Zhu and K. Y. Koltunov, J. Org. Chem., 2019, 84, 7238–7243 CrossRef CAS.
- M. Ikeda, R. Ochi, Y.-S. Kurita, D. J. Pochan and I. Hamachi, Chem. – Eur. J., 2012, 18, 13091–13096 CrossRef CAS.
- R. Ochi, T. Nishida, M. Ikeda and I. Hamachi, J. Mater. Chem. B, 2014, 2, 1464–1469 RSC.
- K. C. Koehler, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS.
- K. Venkata Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133–1139 CrossRef CAS.
- K. V. Rao, M. F. J. Mabesoone, D. Miyajima, A. Nihonyanagi, E. W. Meijer and T. Aida, J. Am. Chem. Soc., 2020, 142, 598–605 CrossRef CAS.
- H. Xie, M. Asad Ayoubi, W. Lu, J. Wang, J. Huang and W. Wang, Sci. Rep., 2017, 7, 8459 CrossRef.
- M. Kuddushi, S. Rajput, A. Shah, J. Mata, V. K. Aswal, O. El Seoud, A. Kumar and N. I. Malek, ACS Appl. Mater. Interfaces, 2019, 11, 19572–19583 CrossRef CAS.
- M. Kuddushi, N. K. Patel, S. Rajput, A. Shah, O. A. El Seoud and N. I. Malek, ACS Omega, 2018, 3, 12068–12078 CrossRef CAS.
- P. K. Agarwal, Microb. Cell Fact., 2006, 5, 2 CrossRef.
- S. Martínez Cuesta, S. A. Rahman, N. Furnham and J. M. Thornton, Biophys. J., 2015, 109, 1082–1086 CrossRef.
- P. K. Robinson, Essays Biochem., 2015, 59, 1–41 CrossRef.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- V. Kumar, T. M. Koyasseril-Yehiya and S. Thayumanavan, Molecular Assemblies: Characterization and Applications, American Chemical Society, 2020, ch. 7, vol. 1355, pp. 95–107 DOI:10.1021/bk-2020-1355.ch007.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS.
- P. Li, Y. Zhong, X. Wang and J. Hao, ACS Cent. Sci., 2020, 6, 1507–1522 CrossRef CAS.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS.
- Z. Yang, P.-L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS.
- K. N. Plunkett, K. L. Berkowski and J. S. Moore, Biomacromolecules, 2005, 6, 632–637 CrossRef CAS.
- F. Zhao, C. S. Weitzel, Y. Gao, H. M. Browdy, J. Shi, H.-C. Lin, S. T. Lovett and B. Xu, Nanoscale, 2011, 3, 2859–2861 RSC.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 19, 3152–3156 CrossRef CAS.
- Y. Liu, V. Javvaji, S. R. Raghavan, W. E. Bentley and G. F. Payne, J. Agric. Food Chem., 2012, 60, 8963–8967 CrossRef CAS.
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461–15465 CrossRef CAS.
- S. Sakai and M. Nakahata, Chem. – Asian J., 2017, 12, 3098–3109 CrossRef CAS.
- J. Leckie, A. Hope, M. Hughes, S. Debnath, S. Fleming, A. W. Wark, R. V. Ulijn and M. D. Haw, ACS Nano, 2014, 8, 9580–9589 CrossRef CAS.
- L. Szkolar, J.-B. Guilbaud, A. F. Miller, J. E. Gough and A. Saiani, J. Pept. Sci., 2014, 20, 578–584 CrossRef CAS.
- Y. M. Abul-Haija and R. V. Ulijn, Biomacromolecules, 2015, 16, 3473–3479 CrossRef CAS.
- Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem. Commun., 2016, 52, 8196–8206 RSC.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS.
- S. Yagai, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2005, 11, 4054–4063 CrossRef CAS.
- P. R. A. Chivers, J. A. Kelly, M. J. S. Hill and D. K. Smith, React. Chem. Eng., 2020, 5, 1112–1117 RSC.
- J. J. D. de Jong, P. R. Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS.
- S. Khetan and J. A. Burdick, Soft Matter, 2011, 7, 830–838 RSC.
- F. Ercole, T. P. Davis and R. A. Evans, Polym. Chem., 2010, 1, 37–54 RSC.
- Y. Zhou, X. Dong, Y. Mi, F. Fan, Q. Xu, H. Zhao, S. Wang and Y. Long, J. Mater. Chem. A, 2020, 8, 10007–10025 RSC.
- M. Mauro, J. Mater. Chem. B, 2019, 7, 4234–4242 RSC.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS.
- S. Das, S. Varghese and N. S. S. Kumar, Langmuir, 2010, 26, 1598–1609 CrossRef CAS.
- A. Dey and K. Biradha, Isr. J. Chem., 2019, 59, 220–232 CrossRef CAS.
- S.-T. Zheng, H.-H. Yin, Z.-G. Ma, N.-L. Sheng, T.-G. Zhan, X.-Y. Yan, J. Cui, L.-J. Liu and K.-D. Zhang, Chin. Chem. Lett., 2019, 30, 707–709 CrossRef CAS.
- M. Salzano de Luna, V. Marturano, M. Manganelli, C. Santillo, V. Ambrogi, G. Filippone and P. Cerruti, J. Colloid Interface Sci., 2020, 568, 16–24 CrossRef CAS.
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 RSC.
- Z. Chen, Z. Lv, G. Qing and T. Sun, J. Mater. Chem. B, 2017, 5, 3163–3171 RSC.
- Y. Wu, S. Wu, X. Tian, X. Wang, W. Wu, G. Zou and Q. Zhang, Soft Matter, 2011, 7, 716–721 RSC.
- S. Lee, S. Oh, J. Lee, Y. Malpani, Y.-S. Jung, B. Kang, J. Y. Lee, K. Ozasa, T. Isoshima, S. Y. Lee, M. Hara, D. Hashizume and J.-M. Kim, Langmuir, 2013, 29, 5869–5877 CrossRef CAS.
- F. Zhao, A. Bonasera, U. Nöchel, M. Behl and D. Bléger, Macromol. Rapid Commun., 2018, 39, 1700527 CrossRef.
- C.-W. Chu, L. Stricker, T. M. Kirse, M. Hayduk and B. J. Ravoo, Chem. – Eur. J., 2019, 25, 6131–6140 CrossRef CAS.
- F. Xie, L. Qin and M. Liu, Chem. Commun., 2016, 52, 930–933 RSC.
- R. D. Mukhopadhyay, V. K. Praveen, A. Hazra, T. K. Maji and A. Ajayaghosh, Chem. Sci., 2015, 6, 6583–6591 RSC.
- J.-F. Xu, Y.-Z. Chen, D. Wu, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2013, 52, 9738–9742 CrossRef CAS.
- J. Eastoe, M. Sánchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609, 10.1039/B410158H.
- S. Miljanić, L. Frkanec, Z. Meić and M. Žinić, Eur. J. Org. Chem., 2006, 1323–1334 CrossRef.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS.
- X. Yu, D. Xie, Y. Li, L. Geng, J. Ren, T. Wang and X. Pang, Sens. Actuators, B, 2017, 251, 828–835 CrossRef CAS.
- X. Che, B. Bai, T. Zhang, C. Zhang, C. Zhang, P. Zhang, H. Wang and M. Li, New J. Chem., 2017, 41, 8614–8619 RSC.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS.
- H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- K. Zheng, H. Wang and H.-F. Chow, Org. Biomol. Chem., 2019, 17, 3285–3291 RSC.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS.
- Q. Chen, Y. Feng, D. Zhang, G. Zhang, Q. Fan, S. Sun and D. Zhu, Adv. Funct. Mater., 2010, 20, 36–42 CrossRef CAS.
- D. Moldenhauer and F. Gröhn, Chem. – Eur. J., 2017, 23, 3966–3978 CrossRef CAS.
- A. V. Metelitsa, C. Coudret, J. C. Micheau and N. A. Voloshin, RSC Adv., 2014, 4, 20974–20983 RSC.
- A. R. Katritzky, R. Sakhuja, L. Khelashvili and K. Shanab, J. Org. Chem., 2009, 74, 3062–3065 CrossRef CAS.
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS.
- S. Yagai, K. Ishiwatari, X. Lin, T. Karatsu, A. Kitamura and S. Uemura, Chem. – Eur. J., 2013, 19, 6971–6975 CrossRef CAS.
- K. Rameshbabu, L. Zou, C. Kim, A. Urbas and Q. Li, J. Mater. Chem., 2011, 21, 15673–15677 RSC.
- M. D. Johnstone, C.-W. Hsu, N. Hochbaum, J. Andréasson and H. Sundén, Chem. Commun., 2020, 56, 988–991 RSC.
- O. Tosic, K. Altenhöner and J. Mattay, Photochem. Photobiol. Sci., 2010, 9, 128–130 CrossRef CAS.
- C. Maity, W. E. Hendriksen, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998–1001 CrossRef CAS.
- V. I. Minkin, Russian Chem. Rev., 2013, 82, 1–26 CrossRef.
- M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS.
- Y. Nakayama, K. Hayashi and M. Irie, J. Org. Chem., 1990, 55, 2592–2596 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS.
- S. Wang, W. Shen, Y. Feng and H. Tian, Chem. Commun., 2006, 1497–1499, 10.1039/B515412J.
- J. T. van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 3077–3083 CrossRef CAS.
- S. Xiao, Y. Zou, M. Yu, T. Yi, Y. Zhou, F. Li and C. Huang, Chem. Commun., 2007, 4758–4760, 10.1039/B709409D.
- D. Kim, J. E. Kwon and S. Y. Park, Adv. Funct. Mater., 2018, 28, 1706213 CrossRef.
- T. Sarkar, K. Selvakumar, L. Motiei and D. Margulies, Nat. Commun., 2016, 7, 11374 CrossRef CAS.
- S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Chem. Soc. Rev., 2018, 47, 2228–2248 RSC.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS.
- V. Ramamurthy and B. Mondal, J. Photochem. Photobiol., C, 2015, 23, 68–102 CrossRef CAS.
- M. D’Auria and R. Racioppi, J. Photochem. Photobiol., A, 2004, 163, 557–559 CrossRef.
- D. S. Pal, H. Kar and S. Ghosh, Chem. – Eur. J., 2016, 22, 16872–16877 CrossRef CAS.
- H. Lai, J. Zhang, F. Xing and P. Xiao, Chem. Soc. Rev., 2020, 49, 1867–1886 RSC.
- X. Qian and B. Städler, Chem. Mater., 2019, 31, 1196–1222 CrossRef CAS.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 12827–12830 RSC.
- S. H. Kim, Y. Sun, J. A. Kaplan, M. W. Grinstaff and J. R. Parquette, New J. Chem., 2015, 39, 3225–3228 RSC.
- H. Yu, H. Mizufune, K. Uenaka, T. Moritoki and H. Koshima, Tetrahedron, 2005, 61, 8932–8938 CrossRef CAS.
- S. Dong, A. Ong and C. Chi, J. Photochem. Photobiol., C, 2019, 38, 27–46 CrossRef CAS.
- Y. Sako and Y. Takaguchi, Org. Biomol. Chem., 2008, 6, 3843–3847 RSC.
- A. Dawn, N. Fujita, S. Haraguchi, K. Sada and S. Shinkai, Chem. Commun., 2009, 2100–2102, 10.1039/B820565E.
- C. Wang, D. Zhang, J. Xiang and D. Zhu, Langmuir, 2007, 23, 9195–9200 CrossRef CAS.
- J. H. Lee, S. H. Jung, S. S. Lee, K.-Y. Kwon, K. Sakurai, J. Jaworski and J. H. Jung, ACS Nano, 2017, 11, 4155–4164 CrossRef CAS.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2013, 29, 13299–13306 CrossRef CAS.
- F. Dumur, E. Contal, G. Wantz, T. N. T. Phan, D. Bertin and D. Gigmes, Chem. – Eur. J., 2013, 19, 1373–1384 CrossRef CAS.
- S. R. Diegelmann and J. D. Tovar, Macromol. Rapid Commun., 2013, 34, 1343–1350 CrossRef CAS.
- M. A. Markowitz, A. Singh and E. L. Chang, Biochem. Biophys. Res. Commun., 1994, 203, 296–305 CrossRef CAS.
- I. Kazuhiko, O. Yoshiyuki, K. Yasumasa, H. Kenji and S. Seiji, Chem. Lett., 1999, 429–430 Search PubMed.
- M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879–2888 CrossRef CAS.
- L. Hsu, G. L. Cvetanovich and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 3892–3899 CrossRef CAS.
- N. Tamaoki, S. Shimada, Y. Okada, A. Belaissaoui, G. Kruk, K. Yase and H. Matsuda, Langmuir, 2000, 16, 7545–7547 CrossRef CAS.
- H. M. T. Albuquerque, C. M. M. Santos and A. M. S. Silva, Molecules, 2019, 24, 116 CrossRef.
- M. Masuda, T. Hanada, K. Yase and T. Shimizu, Macromolecules, 1998, 31, 9403–9405 CrossRef CAS.
- M. Masuda, T. Hanada, Y. Okada, K. Yase and T. Shimizu, Macromolecules, 2000, 33, 9233–9238 CrossRef CAS.
- S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3121–3132 CrossRef CAS.
- S. R. Diegelmann, N. Hartman, N. Markovic and J. D. Tovar, J. Am. Chem. Soc., 2012, 134, 2028–2031 CrossRef CAS.
- A. Ghosh, T. Selvamani, D. A. Jose, A. Das and I. Mukhopadhyay, J. Nanomater., 2007, 2007, 047234 Search PubMed.
- G. Magna, D. Monti, C. Di Natale, R. Paolesse and M. Stefanelli, Molecules, 2019, 24, 4307 CrossRef CAS.
- M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS.
- J. R. Néabo, S. Rondeau-Gagné, C. Vigier-Carrière and J.-F. Morin, Langmuir, 2013, 29, 3446–3452 CrossRef.
- Y. Meng, J. Jiang and M. Liu, Nanoscale, 2017, 9, 7199–7206 RSC.
- K. i. Aoki, M. Kudo and N. Tamaoki, Org. Lett., 2004, 6, 4009–4012 CrossRef CAS.
- N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134–4135 CrossRef CAS.
- M. de Loos, J. van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675–12676 CrossRef CAS.
- Y.-J. Choi, D. Jung, S.-I. Lim, W.-J. Yoon, D.-Y. Kim and K.-U. Jeong, ACS Appl. Mater. Interfaces, 2020, 12, 33239–33245 CrossRef CAS.
- G. K. Oster and G. Oster, J. Am. Chem. Soc., 1959, 81, 5543–5545 CrossRef CAS.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- R. O. Marcon and S. Brochsztain, J. Phys. Chem. A, 2009, 113, 1747–1752 CrossRef CAS.
- M. Cametti and Z. Džolić, Chem. Commun., 2014, 50, 8273–8286 RSC.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Soft Matter, 2011, 7, 2412–2418 RSC.
- S. Marimuthu, A. J. Antonisamy, S. Malayandi, K. Rajendran, P.-C. Tsai, A. Pugazhendhi and V. K. Ponnusamy, J. Photochem. Photobiol., B, 2020, 205, 111823 CrossRef CAS.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS.
- Y. Jiang, N. Krishnan, J. Heo, R. H. Fang and L. Zhang, J. Controlled Release, 2020, 324, 505–521 CrossRef CAS.
- Y. Zhang, Y. Li, Z. Gao, G. Gao and L. Duan, Mater. Chem. Phys., 2018, 207, 175–180 CrossRef CAS.
- F. Peng, G. Li, X. Liu, S. Wu and Z. Tong, J. Am. Chem. Soc., 2008, 130, 16166–16167 CrossRef CAS.
- P. Calvo-Marzal, M. P. Delaney, J. T. Auletta, T. Pan, N. M. Perri, L. M. Weiland, D. H. Waldeck, W. W. Clark and T. Y. Meyer, ACS Macro Lett., 2012, 1, 204–208 CrossRef CAS.
- E. Krieg, E. Shirman, H. Weissman, E. Shimoni, S. G. Wolf, I. Pinkas and B. Rybtchinski, J. Am. Chem. Soc., 2009, 131, 14365–14373 CrossRef CAS.
- L. Gonzalez, C. Liu, B. Dietrich, H. Su, S. Sproules, H. Cui, D. Honecker, D. J. Adams and E. R. Draper, Chem. Commun., 2018, 1, 77 CrossRef.
- Y. Zhou, X. Dong, Y. Mi, F. Fan, Q. Xu, H. Zhao, S. Wang and Y. Long, J. Mater. Chem. A, 2020, 8, 10007–10025 RSC.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef.
- H. Wang, C. N. Zhu, H. Zeng, X. Ji, T. Xie, X. Yan, Z. L. Wu and F. Huang, Adv. Mater., 2019, 31, 1807328 CrossRef.
- Z. Guo, X. Zhang, Y. Wang and Z. Li, Langmuir, 2019, 35, 342–358 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- V. Adams, J. Cameron, M. Wallace and E. R. Draper, Chem. – Eur. J., 2020, 26, 9879–9882 CrossRef CAS.
- E. R. Draper, R. Schweins, R. Akhtar, P. Groves, V. Chechik, M. A. Zwijnenburg and D. J. Adams, Chem. Mater., 2016, 28, 6336–6341 CrossRef CAS.
- D. S. Peterson, in Encyclopedia of Microfluidics and Nanofluidics, ed. D. Li, Springer US, Boston, MA, 2013, pp. 1–5 DOI:10.1007/978-3-642-27758-0_1230-2.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, Polymers, 2017, 9, 137 CrossRef.
- A. R. Hibbins, P. Kumar, Y. E. Choonara, P. P. D. Kondiah, T. Marimuthu, L. C. Du Toit and V. Pillay, Polymers, 2017, 9, 474 CrossRef.
- H. W. Ooi, S. Hafeez, C. A. van Blitterswijk, L. Moroni and M. B. Baker, Mater. Horiz., 2017, 4, 1020–1040 RSC.
- P. Singh, S. Misra, A. Das, S. Roy, P. Datta, G. Bhattacharjee, B. Satpati and J. Nanda, ACS Appl. Bio Mater., 2019, 2, 4881–4891 CrossRef CAS.
- A. M. Jonker, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Mater., 2012, 24, 759–773 CrossRef CAS.
- J. Chen and X. Zou, Bioact. Mater., 2019, 4, 120–131 CrossRef.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- A. Panja and K. Ghosh, New J. Chem., 2019, 43, 934–945 RSC.
- D. K. Kumar, D. A. Jose, P. Dastidar and A. Das, Langmuir, 2004, 20, 10413–10418 CrossRef CAS.
- L. Ma, H. Kang, R. Liu and Y. Huang, Langmuir, 2010, 26, 18519–18525 CrossRef CAS.
- G.-F. Liu, W. Ji and C.-L. Feng, Langmuir, 2015, 31, 7122–7128 CrossRef CAS.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 47–50 RSC.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS.
- S. Panja, S. Bhattacharya and K. Ghosh, Mater. Chem. Front., 2018, 2, 385–395 RSC.
- P. Santanu and G. Kumaresh, Mini Rev. Org. Chem., 2020, 17, 1042–1055 CrossRef.
- K. Samanta, M. Ehlers and C. Schmuck, Chem. – Eur. J., 2016, 22, 15242–15247 CrossRef.
- S. M. M. Reddy, G. Augustine, N. Ayyadurai and G. Shanmugam, ACS Appl. Bio Mater., 2018, 1, 1382–1388 CrossRef CAS.
- K. C.-F. Leung, C.-P. Chak, C.-M. Lo, W.-Y. Wong, S. Xuan and C. H. K. Cheng, Chem. – Asian J., 2009, 4, 364–381 CrossRef CAS.
- H.-Y. Zhou, Y. Han and C.-F. Chen, Mater. Chem. Front., 2020, 4, 12–28 RSC.
- H. Frisch and P. Besenius, Macromol. Rapid Commun., 2015, 36, 346–363 CrossRef CAS.
- H. Shao and J. R. Parquette, Angew. Chem., Int. Ed., 2009, 48, 2525–2528 CrossRef CAS.
- T. Kar, S. Debnath, D. Das, A. Shome and P. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS.
- E. R. Draper, H. Su, C. Brasnett, R. J. Poole, S. Rogers, H. Cui, A. Seddon and D. J. Adams, Angew. Chem., Int. Ed., 2017, 56, 10467–10470 CrossRef CAS.
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714–8720 RSC.
- C. Colquhoun, E. R. Draper, R. Schweins, M. Marcello, D. Vadukul, L. C. Serpell and D. J. Adams, Soft Matter, 2017, 13, 1914–1919 RSC.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Soft Matter, 2016, 12, 3612–3621 RSC.
- K. McAulay, P. A. Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Chem. Commun., 2020, 56, 4094–4097 RSC.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- T. Patra, A. Pal and J. Dey, Langmuir, 2010, 26, 7761–7767 CrossRef CAS.
- S. Paul, K. Basu, K. S. Das and A. Banerjee, ChemNanoMat, 2018, 4, 882–887 CrossRef CAS.
- M. Tena-Solsona, S. Alonso-de Castro, J. F. Miravet and B. Escuder, J. Mater. Chem. B, 2014, 2, 6192–6197 RSC.
- B. P. Lee, M.-H. Lin, A. Narkar, S. Konst and R. Wilharm, Sens. Actuators, B, 2015, 206, 456–462 CrossRef CAS.
- W.-Y. Quan, Z. Hu, H.-Z. Liu, Q.-Q. Ouyang, D.-Y. Zhang, S.-D. Li, P.-W. Li and Z.-M. Yang, Molecules, 2019, 24, 2586 CrossRef CAS.
- M. Krogsgaard, A. Andersen and H. Birkedal, Chem. Commun., 2014, 50, 13278–13281 RSC.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS.
- J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355–9357 RSC.
- D. J. Cornwell and D. K. Smith, Chem. Commun., 2020, 56, 7029–7032 RSC.
- P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- S. Kubik, Angew. Chem., Int. Ed., 2018, 57, 3005 CrossRef CAS.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- J. Zhang, Y. Hu and Y. Li, Gel Chemistry: Interactions, Structures and Properties, Springer Singapore, Singapore, 2018, pp. 119–151 DOI:10.1007/978-981-10-6881-2_4.
- J.-Y. Zhang, L.-H. Zeng and J. Feng, Chin. Chem. Lett., 2017, 28, 168–183 CrossRef CAS.
- Y. Chen, L. Yang, W. Zheng, P. Ouyang, H. Zhang, Y. Ruan, W. Weng, X. He and H. Xia, ACS Macro Lett., 2020, 9, 344–349 CrossRef CAS.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- T. Jiao, G. Wu, Y. Zhang, L. Shen, Y. Lei, C.-Y. Wang, A. C. Fahrenbach and H. Li, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef CAS.
- J. Xu, Y. Liu and S.-H. Hsu, Molecules, 2019, 24, 3005 CrossRef CAS.
- J. M. Poolman, C. Maity, J. Boekhoven, L. van der Mee, V. A. A. le Sage, G. J. M. Groenewold, S. I. van Kasteren, F. Versluis, J. H. van Esch and R. Eelkema, J. Mater. Chem. B, 2016, 4, 852–858 RSC.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433–437 CrossRef CAS.
- J. S. Foster, J. M. Żurek, N. M. S. Almeida, W. E. Hendriksen, V. A. A. le Sage, V. Lakshminarayanan, A. L. Thompson, R. Banerjee, R. Eelkema, H. Mulvana, M. J. Paterson, J. H. van Esch and G. O. Lloyd, J. Am. Chem. Soc., 2015, 137, 14236–14239 CrossRef CAS.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- F. Picchioni and H. Muljana, Gels, 2018, 4, 21 CrossRef.
- N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS.
- C. Arnal-Hérault, A. Pasc, M. Michau, D. Cot, E. Petit and M. Barboiu, Angew. Chem., Int. Ed., 2007, 46, 8409–8413 CrossRef.
- P. Duan, H. Cao, L. Zhang and M. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- P. Sun, S. Ren, A. Wu, N. Sun, L. Shi and L. Zheng, Chem. Commun., 2019, 55, 9861–9864 RSC.
- N. Mehwish, X. Dou, C. Zhao, C. Feng and Q. Fu, Adv. Fiber Mater., 2020, 2, 204–211 CrossRef.
- K. Lv, L. Qin, X. Wang, L. Zhang and M. Liu, Phys. Chem. Chem. Phys., 2013, 15, 20197–20202 RSC.
- Y. Li, T. Wang and M. Liu, Soft Matter, 2007, 3, 1312–1317 RSC.
- P. Duan, Y. Li, J. Jiang, T. Wang and M. Liu, Sci. China: Chem., 2011, 54, 1051–1063 CrossRef CAS.
- Y. Wang, P. Xing, S. Li, M. Ma, M. Yang, Y. Zhang, B. Wang and A. Hao, Langmuir, 2016, 32, 10705–10711 CrossRef CAS.
- M. A. Khalily, M. Goktas and M. O. Guler, Org. Biomol. Chem., 2015, 13, 1983–1987 RSC.
- S. J. Beckers, S. Parkinson, E. Wheeldon and D. K. Smith, Chem. Commun., 2019, 55, 1947–1950 RSC.
- X. Sui, X. Feng, M. A. Hempenius and G. J. Vancso, J. Mater. Chem. B, 2013, 1, 1658–1672 RSC.
- T. Fukino, H. Yamagishi and T. Aida, Adv. Mater., 2017, 29, 1603888 CrossRef.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- Q. Zhou, X. Dong, J. Yuan, B. Zhang, S. Lu, Q. Wang, Y. Liao, Y. Yang and H. Wang, Langmuir, 2019, 35, 15344–15351 CrossRef CAS.
- A. Panja and K. Ghosh, Mater. Chem. Front., 2018, 2, 1866–1875 RSC.
- G. R. Ferreira, B. L. Marcial, H. C. Garcia, F. R. L. Faulstich, H. F. Dos Santos and L. F. C. de Oliveira, Supramol. Chem., 2015, 27, 13–20 CrossRef CAS.
- X. Jia, T. Xiao, Z. Hou, L. Xiao, Y. Qi, Z. Hou and J. Zhu, ACS Omega, 2019, 4, 12043–12048 CrossRef CAS.
- S. Sun and A. F. T. Mak, J. Polym. Sci. B Polym. Phys., 2001, 39, 236–246 CrossRef CAS.
- S. Szunerits, F. Teodorescu and R. Boukherroub, Eur. Polym. J., 2016, 83, 467–477 CrossRef CAS.
- V. Lakshminarayanan, L. Poltorak, E. J. R. Sudhölter, E. Mendes and J. van Esch, Electrochim. Acta, 2020, 350, 136352 CrossRef CAS.
- S. Jones, K. Wong, P. Thordarson and F. Ladouceur, J. Phys.: Condens. Matter, 2010, 22, 494105 CrossRef.
- B. Xue, M. Qin, T. Wang, J. Wu, D. Luo, Q. Jiang, Y. Li, Y. Cao and W. Wang, Adv. Funct. Mater., 2016, 26, 9053–9062 CrossRef CAS.
- J. T. Auletta, G. J. LeDonne, K. C. Gronborg, C. D. Ladd, H. Liu, W. W. Clark and T. Y. Meyer, Macromolecules, 2015, 48, 1736–1747 CrossRef CAS.
- M. J. Bassetti, A. N. Chatterjee, N. R. Aluru and D. J. Beebe, J. Microelectromech. Syst., 2005, 14, 1198–1207 Search PubMed.
- R. D. Harris, J. T. Auletta, S. A. M. Motlagh, M. J. Lawless, N. M. Perri, S. Saxena, L. M. Weiland, D. H. Waldeck, W. W. Clark and T. Y. Meyer, ACS Macro Lett., 2013, 2, 1095–1099 CrossRef CAS.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS.
- H. Maeda, Chem. – Eur. J., 2008, 14, 11274–11282 CrossRef CAS.
- P. Byrne, G. O. Lloyd, L. Applegarth, K. M. Anderson, N. Clarke and J. W. Steed, New J. Chem., 2010, 34, 2261–2274 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Langmuir, 2009, 25, 8451–8456 CrossRef CAS.
- S. Ha, J. Lee, K.-S. Kim, E. J. Choi, P. Nhem and C. Song, Chem. Mater., 2019, 31, 5735–5741 CrossRef CAS.
- H. Yao, J. Wang, S.-S. Song, Y.-Q. Fan, X.-W. Guan, Q. Zhou, T.-B. Wei, Q. Lin and Y.-M. Zhang, New J. Chem., 2018, 42, 18059–18065 RSC.
- M. A. Ramin, K. R. Sindhu, A. Appavoo, K. Oumzil, M. W. Grinstaff, O. Chassande and P. Barthélémy, Adv. Mater., 2017, 29, 1605227 CrossRef.
- T. Tu, W. Fang and Z. Sun, Adv. Mater., 2013, 25, 5304–5313 CrossRef CAS.
- X. Li, Y. Gao and M. J. Serpe, Gels, 2016, 2, 8 CrossRef.
- W. P. Singh and R. S. Singh, Soft Mater., 2019, 17, 93–118 CrossRef.
- C. Mahendar, M. K. Dixit, Y. Kumar and M. Dubey, J. Mater. Chem. C, 2020, 8, 11008–11012 RSC.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS.
- A. Gainar, T.-L. Lai, C. Oliveras-González, F. Pop, M. Raynal, B. Isare, L. Bouteiller, M. Linares, D. Canevet, N. Avarvari and M. Sallé, Chem. – Eur. J., 2021, 27, 2410–2420 CrossRef CAS.
- H. Fan, H. Guo, J. Wang and J. P. Gong, Giant, 2020, 1, 100005 CrossRef.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS.
- A. Panja and K. Ghosh, ChemistrySelect, 2018, 3, 9448–9453 CrossRef CAS.
- L. Xiong, J. Feng, R. Hu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2013, 85, 4113–4119 CrossRef CAS.
- R. Raza, A. Panja, M. Mukherjee, P. Chattopadhyay and K. Ghosh, ACS Omega, 2018, 3, 17319–17325 CrossRef CAS.
- P. Dastidar, Gels, 2019, 5, 15 CrossRef CAS.
- P. Dastidar, R. Roy, R. Parveen and K. Sarkar, Adv. Ther., 2019, 2, 1800061 CrossRef.
- D. Ghosh, M. T. Mulvee and K. K. Damodaran, Molecules, 2019, 24, 3472 CrossRef CAS.
- R. Kuosmanen, K. Rissanen and E. Sievänen, Chem. Soc. Rev., 2020, 49, 1977–1998 RSC.
- R. Raza, A. Panja and K. Ghosh, New J. Chem., 2020, 44, 10275–10285 RSC.
- A. Panja and K. Ghosh, Mater. Chem. Front., 2018, 2, 2286–2296 RSC.
- S. Ghosh, V. K. Praveen and A. Ajayaghosh, Annu. Rev. Mater. Res., 2016, 46, 235–262 CrossRef CAS.
- T. Shao, N. Falcone and H.-B. Kraatz, ACS Omega, 2020, 5, 1312–1317 CrossRef CAS.
- N. Basu, A. Chakraborty and R. Ghosh, Gels, 2018, 4, 52 CrossRef.
- N. Falcone and H.-B. Kraatz, Chem. – Eur. J., 2018, 24, 14316–14328 CrossRef CAS.
- S. M. George, S. Tandon and B. Kandasubramanian, ACS Omega, 2020, 5, 2060–2068 CrossRef CAS.
- H. Wu, J. Zheng, A.-L. Kjøniksen, W. Wang, Y. Zhang and J. Ma, Adv. Mater., 2019, 31, 1806204 CrossRef.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- H. Wang, X. Ji, M. Ahmed, F. Huang and J. L. Sessler, J. Mater. Chem. A, 2019, 7, 1394–1403 RSC.
- Z. Sun, Q. Huang, T. He, Z. Li, Y. Zhang and L. Yi, ChemPhysChem, 2014, 15, 2421–2430 CrossRef CAS.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- A. Dawn, B. Roy and S. Shinkai, Chemoresponsive Materials: Stimulation by Chemical and Biological Signals, The Royal Society of Chemistry, 2015, pp. 67–97 10.1039/9781782622420-00067.
- X. Ma, D. Yu, N. Tang and J. Wu, Dalton Trans., 2014, 43, 9856–9859 RSC.
- X. Ma, J. Xie, N. Tang and J. Wu, New J. Chem., 2016, 40, 6584–6587 RSC.
- V. M. Suresh, A. De and T. K. Maji, Chem. Commun., 2015, 51, 14678–14681 RSC.
- P. Sutar, V. M. Suresh and T. K. Maji, Chem. Commun., 2015, 51, 9876–9879 RSC.
- M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef.
- P. Sutar and T. K. Maji, Dalton Trans., 2020, 49, 7658–7672 RSC.
- X. Ma, Z. Zhang, H. Xie, Y. Ma, C. Liu, S. Liu and M. Liu, Chem. Commun., 2018, 54, 13674–13677 RSC.
- S. Basak, I. Singh, A. Banerjee and H.-B. Kraatz, RSC Adv., 2017, 7, 14461–14465 RSC.
- H. Yao, H.-P. Wu, J. Chang, Q. Lin, T.-B. Wei and Y.-M. Zhang, New J. Chem., 2016, 40, 4940–4944 RSC.
- X. Ma, J. Zhang, N. Tang and J. Wu, Dalton Trans., 2014, 43, 17236–17239 RSC.
- L. Qin, P. Duan, F. Xie, L. Zhang and M. Liu, Chem. Commun., 2013, 49, 10823–10825 RSC.
- X. Ma, S. Liu, Z. Zhang, Y. Niu and J. Wu, Soft Matter, 2017, 13, 8882–8885 RSC.
- Q. Jin, L. Zhang, X. Zhu, P. Duan and M. Liu, Chem. – Eur. J., 2012, 18, 4916–4922 CrossRef CAS.
- P. Mukhopadhyay, Y. Iwashita, M. Shirakawa, S.-I. Kawano, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2006, 45, 1592–1595 CrossRef CAS.
- D. Bairagi, P. Biswas, K. Basu, S. Hazra, D. Hermida-Merino, D. K. Sinha, I. W. Hamley and A. Banerjee, ACS Appl. Bio Mater., 2019, 2, 5235–5244 CrossRef CAS.
- H. Mehdi, H. Pang, W. Gong, M. K. Dhinakaran, A. Wajahat, X. Kuang and G. Ning, Org. Biomol. Chem., 2016, 14, 5956–5964 RSC.
- N. Malviya, M. Das, P. Mandal and S. Mukhopadhyay, Soft Matter, 2017, 13, 6243–6249 RSC.
- D. Yuan, Y. D. Zhang, Z. W. Jiang, Z. W. Peng, C. Z. Huang and Y. F. Li, Mater. Lett., 2018, 211, 157–160 CrossRef CAS.
- Q. Lin, T.-T. Lu, X. Zhu, T.-B. Wei, H. Li and Y.-M. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC.
- A. Sebastian and E. Prasad, Langmuir, 2020, 36, 10537–10547 CrossRef CAS.
- N. Malviya, C. Sonkar, B. K. Kundu and S. Mukhopadhyay, Langmuir, 2018, 34, 11575–11585 CrossRef CAS.
- N. Malviya, C. Sonkar, R. Ganguly and S. Mukhopadhyay, Inorg. Chem., 2019, 58, 7324–7334 CrossRef CAS.
- J. Huang, L. He, J. Zhang, L. Chen and C.-Y. Su, J. Mol. Catal. A: Chem., 2010, 317, 97–103 CrossRef CAS.
- Z.-S. Qin, W.-W. Dong, J. Zhao, Y.-P. Wu, Q. Zhang and D.-S. Li, Inorg. Chem. Front., 2018, 5, 120–126 RSC.
- Y.-L. Xu, C.-T. Li, Q.-Y. Cao, B.-Y. Wang and Y. Xie, Dyes Pigm., 2017, 139, 681–687 CrossRef CAS.
- X. Pang, J. Ge, X. Yu, Y. Li, F. Shen, Y. Wang and J. Ren, New J. Chem., 2019, 43, 10554–10559 RSC.
- S. Sarkar, S. Dutta, P. Bairi and T. Pal, Langmuir, 2014, 30, 7833–7841 CrossRef CAS.
- K. Ghosh, S. Panja and S. Bhattacharya, RSC Adv., 2015, 5, 72772–72779 RSC.
- H. Fang, W.-J. Qu, H.-H. Yang, J.-X. He, H. Yao, Q. Lin, T.-B. Wei and Y.-M. Zhang, Dyes Pigm., 2020, 174, 108066 CrossRef CAS.
- F. Mandegani, H. Zali-Boeini, Z. Khayat and R. Scopelliti, Talanta, 2020, 219, 121237 CrossRef CAS.
- W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS.
- K. McAulay, B. Dietrich, H. Su, M. T. Scott, S. Rogers, Y. K. Al-Hilaly, H. Cui, L. C. Serpell, A. M. Seddon, E. R. Draper and D. J. Adams, Chem. Sci., 2019, 10, 7801–7806 RSC.
- C. Mahendar, M. K. Dixit, Y. Kumar and M. Dubey, J. Mater. Chem. C, 2020, 8, 11008–11012 RSC.
- M. Yamanaka, T. Nakamura, T. Nakagawa and H. Itagaki, Tetrahedron Lett., 2007, 48, 8990–8993 CrossRef CAS.
- K. Ghosh and S. Panja, RSC Adv., 2015, 5, 12094–12099 RSC.
- S. Panja, S. Ghosh and K. Ghosh, New J. Chem., 2018, 42, 6488–6497 RSC.
- L.-B. Xing, B. Yang, X.-J. Wang, J.-J. Wang, B. Chen, Q. Wu, H.-X. Peng, L.-P. Zhang, C.-H. Tung and L.-Z. Wu, Langmuir, 2013, 29, 2843–2848 CrossRef CAS.
- J.-W. Liu, Y. Yang, C.-F. Chen and J.-T. Ma, Langmuir, 2010, 26, 9040–9044 CrossRef CAS.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS.
- D. J. Kushner, Bacteriol. Rev., 1969, 33, 302–345 CrossRef CAS.
- P. S. Agutter and D. N. Wheatley, About Life: Concepts in Modern Biology, Springer Netherlands, Dordrecht, 2007, pp. 79–89 DOI:10.1007/978-1-4020-5418-1_9.
- F. Kremer, L. Santo, A. Poglitsch, C. Koschnitzke, H. Behrens and L. Genzel, in Biological Coherence and Response to External Stimuli, ed. H. Fröhlich, Springer Berlin Heidelberg, Berlin, Heidelberg, 1988, pp. 86–101 DOI:10.1007/978-3-642-73309-3_5.
- D. L. Bader and M. M. Knight, Med. Biol. Eng. Comput., 2008, 46, 951 CrossRef CAS.
- G. Bao, R. D. Kamm, W. Thomas, W. Hwang, D. A. Fletcher, A. J. Grodzinsky, C. Zhu and M. R. K. Mofrad, Cell. Mol. Bioeng., 2010, 3, 91–105 CrossRef.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913–18925 RSC.
- B. A. Grzybowski and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 585–592 CrossRef CAS.
- D. Bochicchio and G. M. Pavan, Adv. Phys. X, 2018, 3, 1436408 Search PubMed.
- F. Nabeel, T. Rasheed, M. Bilal, C. Li, C. Yu and H. M. N. Iqbal, Sep. Purif. Rev., 2020, 49, 20–36 CrossRef.
- D. D. Klug, D. E. Kranbuehl and W. E. Vaughan, J. Chem. Phys., 1969, 50, 3904–3905 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- J. B. Hasted, in Biological Coherence and Response to External Stimuli, ed. H. Fröhlich, Springer Berlin Heidelberg, Berlin, Heidelberg, 1988, pp. 102–113 DOI:10.1007/978-3-642-73309-3_6.
- O. Erol, A. Pantula, W. Liu and D. H. Gracias, Adv. Mater. Technol., 2019, 4, 1900043 CrossRef.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- P. Dorishetty, N. K. Dutta and N. R. Choudhury, Adv. Colloid Interface Sci., 2020, 281, 102163 CrossRef CAS.
- A. Walther, Adv. Mater., 2020, 32, 1905111 CrossRef CAS.
- J. Hoque, N. Sangaj and S. Varghese, Macromol. Biosci., 2019, 19, 1800259 CrossRef.
- A. Baral, S. Basak, K. Basu, A. Dehsorkhi, I. W. Hamley and A. Banerjee, Soft Matter, 2015, 11, 4944–4951 RSC.
- M. Lescanne, P. Grondin, A. d'Aléo, F. Fages, J. L. Pozzo, O. M. Monval, P. Reinheimer and A. Colin, Langmuir, 2004, 20, 3032–3041 CrossRef CAS.
- J. Shi, Y. Gao, Z. Yang and B. Xu, Beilstein J. Org. Chem., 2011, 7, 167–172 CrossRef CAS.
- B. Roy, A. Saha, A. Esterrani and A. K. Nandi, Soft Matter, 2010, 6, 3337–3345 RSC.
- K. Gayen, N. Nandi, K. S. Das, D. Hermida-Merino, I. W. Hamley and A. Banerjee, Soft Matter, 2020, 16, 10106–10114 RSC.
- F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 209–211, 10.1039/B816234D.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 6595–6597 RSC.
- V. A. Mallia, P. D. Butler, B. Sarkar, K. T. Holman and R. G. Weiss, J. Am. Chem. Soc., 2011, 133, 15045–15054 CrossRef CAS.
- P. Chakraborty, Y. Tang, T. Yamamoto, Y. Yao, T. Guterman, S. Zilberzwige-Tal, N. Adadi, W. Ji, T. Dvir, A. Ramamoorthy, G. Wei and E. Gazit, Adv. Mater., 2020, 32, 1906043 CrossRef CAS.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- Y. Wang, R. M. de Kruijff, M. Lovrak, X. Guo, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2019, 58, 3800–3803 CrossRef CAS.
- Y. Wang, T. K. Piskorz, M. Lovrak, E. Mendes, X. Guo, R. Eelkema and J. H. van Esch, Adv. Sci., 2020, 7, 1902487 CrossRef CAS.
- S. Saha, J. Bachl, T. Kundu, D. Díaz Díaz and R. Banerjee, Chem. Commun., 2014, 50, 7032–7035 RSC.
- Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884–889 CrossRef CAS.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 2248–2250, 10.1039/B801913D.
- D. Ghosh, I. Lebedytė, D. S. Yufit, K. K. Damodaran and J. W. Steed, CrystEngComm, 2015, 17, 8130–8138 RSC.
- X.-J. Jia, J. Wang, D.-C. Zhong, J. Wu, B. Zhao, D. D. Engelsen and X.-Z. Luo, RSC Adv., 2016, 6, 109425–109433 RSC.
- J. Cui, Z. Shen and X. Wan, Langmuir, 2010, 26, 97–103 CrossRef CAS.
- Y. Xu, C. Kang, Y. Chen, Z. Bian, X. Qiu, L. Gao and Q. Meng, Chem. – Eur. J., 2012, 18, 16955–16961 CrossRef CAS.
- T. Guterman, M. Levin, S. Kolusheva, D. Levy, N. Noor, Y. Roichman and E. Gazit, Angew. Chem., Int. Ed., 2019, 58, 15869–15875 CrossRef CAS.
- J. Chen, T. Wang and M. Liu, Inorg. Chem. Front., 2016, 3, 1559–1565 RSC.
- J. Chen, T. Wang and M. Liu, Chem. Commun., 2016, 52, 11277–11280 RSC.
- G. W. Scherer, J. Non-Cryst. Solids, 1988, 100, 77–92 CrossRef CAS.
- T. Sugiura, T. Kanada, D. Mori, H. Sakai, A. Shibata, Y. Kitamura and M. Ikeda, Soft Matter, 2020, 16, 899–906 RSC.
- S. Basak, N. Nandi, S. Paul, I. W. Hamley and A. Banerjee, Chem. Commun., 2017, 53, 5910–5913 RSC.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O’Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef.
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC.
- E. R. Draper, L. L. E. Mears, A. M. Castilla, S. M. King, T. O. McDonald, R. Akhtar and D. J. Adams, RSC Adv., 2015, 5, 95369–95378 RSC.
- G. Hu, J. A. Pojman, S. K. Scott, M. M. Wrobel and A. F. Taylor, J. Phys. Chem. B, 2010, 114, 14059–14063 CrossRef CAS.
- T. Bánsági and A. F. Taylor, J. Phys. Chem. B, 2014, 118, 6092–6097 CrossRef.
- C.-M. Chang, G. R. D. Prabhu, C.-M. Tseng and P. L. Urban, Anal. Chem., 2019, 91, 8814–8819 CrossRef CAS.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS.
- E. Jee, T. Bánsági Jr., A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS.
- H. E. Cingil, N. C. H. Meertens and I. K. Voets, Small, 2018, 14, 1802089 CrossRef.
- Y. Miele, Z. Medveczky, G. Holló, B. Tegze, I. Derényi, Z. Hórvölgyi, E. Altamura, I. Lagzi and F. Rossi, Chem. Sci., 2020, 11, 3228–3235 RSC.
- I. N. Bubanja, T. Bánsági and A. F. Taylor, React. Kinet. Mech., 2018, 123, 177–185 CrossRef CAS.
- W. Helen, P. de Leonardis, R. V. Ulijn, J. Gough and N. Tirelli, Soft Matter, 2011, 7, 1732–1740 RSC.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS.
- S. Panja, C. Patterson and D. J. Adams, Macromol. Rapid Commun., 2019, 40, 1900251 CrossRef.
- M. Cheng, C. Qian, Y. Ding, Y. Chen, T. Xiao, X. Lu, J. Jiang and L. Wang, ACS Mater. Lett., 2020, 2, 425–429 CrossRef CAS.
- N. Singh, G. J. M. Formon, S. De Piccoli and T. M. Hermans, Adv. Mater., 2020, 32, 1906834 CrossRef CAS.
- B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 Search PubMed.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- B. Rieß and J. Boekhoven, ChemNanoMat, 2018, 4, 710–719 CrossRef.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 10154–10157 RSC.
- S. Panja, A. M. Fuentes-Caparros, E. R. Cross, L. Cavalcanti and D. J. Adams, Chem. Mater., 2020, 32, 5264–5271 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS.
- A. M. Cok, H. Zhou and J. A. Johnson, Macromol. Symp., 2013, 329, 108–112 CrossRef CAS.
- Y. Zhong, P. Li, J. Hao and X. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 6471–6478 CrossRef CAS.
- S. Panja, K. Bohacova, B. Dietrich and D. Adams, Nanoscale, 2020, 12, 12840–12848 RSC.
- O. N. Oktyabrsky and G. V. Smirnova, Biochemistry, 2007, 72, 132–145 CAS.
- E. H. Sarsour, M. G. Kumar, L. Chaudhuri, A. L. Kalen and P. C. Goswami, Antioxid. Redox Signal., 2009, 11, 2985–3011 CrossRef CAS.
- W. Cheng and Y. Liu, in Biopolymer-Based Composites, ed. S. Jana, S. Maiti and S. Jana, Woodhead Publishing, 2017, pp. 31–60 DOI:10.1016/B978-0-08-101914-6.00002-8.
- P. Li, Y. Xia, J. Hao and X. Wang, Macromolecules, 2020, 53, 2856–2863 CrossRef CAS.
- R. Yoshida, T. Takahashi, T. Yamaguchi and H. Ichijo, J. Am. Chem. Soc., 1996, 118, 5134–5135 CrossRef CAS.
- S. Matsui, K. Inui, Y. Kumai, R. Yoshida and D. Suzuki, ACS Biomater. Sci. Eng., 2019, 5, 5615–5622 CrossRef CAS.
- B. Dhara, P. P. Patra, P. K. Jha, S. V. Jadhav, G. V. Pavan Kumar and N. Ballav, J. Phys. Chem. C, 2014, 118, 19287–19293 CrossRef CAS.
- J. Zhang, S. Chen, S. Xiang, J. Huang, L. Chen and C.-Y. Su, Chem. – Eur. J., 2011, 17, 2369–2372 CrossRef CAS.
- S. Panja and D. J. Adams, Chem. – Eur. J., 2020, 26, 6130–6135 CrossRef CAS.
- M. Tena-Solsona and J. Boekhoven, Isr. J. Chem., 2019, 59, 898–905 CrossRef CAS.
- S. Dhiman, K. Jalani and S. J. George, ACS Appl. Mater. Interfaces, 2020, 12, 5259–5264 CrossRef CAS.
- W. A. Ogden and Z. Guan, ChemSystChem, 2020, 2, e1900030 CAS.
- J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS.
- S. Ahmed, A. Chatterjee, K. Das and D. Das, Chem. Sci., 2019, 10, 7574–7578 RSC.
- N. J. Oldenhuis, K. P. Qin, S. Wang, H.-Z. Ye, E. A. Alt, A. P. Willard, T. Van Voorhis, S. L. Craig and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 2784–2792 CrossRef CAS.
- S. Panja, B. Dietrich and D. J. Adams, ChemSystChem, 2020, 2, e1900038 CAS.
- M. Tena-Solsona, C. Wanzke, B. Riess, A. R. Bausch and J. Boekhoven, Nat. Commun., 2018, 9, 2044 CrossRef.
- M. Tena-Solsona, B. Rieß, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS.
- B. Zhang, I. M. Jayalath, J. Ke, J. L. Sparks, C. S. Hartley and D. Konkolewicz, Chem. Commun., 2019, 55, 2086–2089 RSC.
- L. S. Kariyawasam and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 11949–11955 CrossRef CAS.
- B. Rieß, C. Wanzke, M. Tena-Solsona, R. K. Grötsch, C. Maity and J. Boekhoven, Soft Matter, 2018, 14, 4852–4859 RSC.
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef.
- F.-M. Cheng, H.-X. Chen and H.-D. Li, J. Mater. Chem. B, 2021 10.1039/D0TB02524K.
- L. M. Cross, K. Shah, S. Palani, C. W. Peak and A. K. Gaharwar, Nanomedicine, 2018, 14, 2465–2474 CrossRef CAS.
- L. Zhang, L. Fu, X. Zhang, L. Chen, Q. Cai and X. Yang, Biomater. Sci., 2021 10.1039/D0BM01595D.
- M. Lovrak, W. E. J. Hendriksen, C. Maity, S. Mytnyk, V. van Steijn, R. Eelkema and J. H. van Esch, Nat. Commun., 2017, 8, 15317 CrossRef CAS.
- X. Zhou, T. Li, J. Wang, F. Chen, D. Zhou, Q. Liu, B. Li, J. Cheng, X. Zhou and B. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 9077–9084 CrossRef CAS.
- A. Hagemann, J. M. Giussi and G. S. Longo, Macromolecules, 2018, 51, 8205–8216 CrossRef CAS.
- P. A. Panteli, C. S. Patrickios, M. Constantinou and G. Constantinides, Macromol. Symp., 2019, 385, 1800201 CrossRef.
- J. A. Zimberlin, N. Sanabria-DeLong, G. N. Tew and A. J. Crosby, Soft Matter, 2007, 3, 763–767 RSC.
- S. Panja and D. J. Adams, Giant, 2021, 5, 100041 CrossRef.
- W. B. Cannon, Physiol. Rev., 1929, 9, 399–431 CrossRef.
- M. M. Lerch, A. Grinthal and J. Aizenberg, Adv. Mater., 2020, 32, 1905554 CrossRef CAS.
- X. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214–218 CrossRef CAS.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS.
- R. Kubota, K. Nakamura, S. Torigoe and I. Hamachi, ChemistryOpen, 2020, 9, 67–79 CrossRef CAS.
- E. Fuentes, K. Boháčová, A. M. Fuentes-Caparrós, R. Schweins, E. R. Draper, D. J. Adams, S. Pujals and L. Albertazzi, Chem. – Eur. J., 2020, 26, 9869–9873 CrossRef CAS.
- E. R. Draper, B. Dietrich, K. McAulay, C. Brasnett, H. Abdizadeh, I. Patmanidis, S. J. Marrink, H. Su, H. Cui, R. Schweins, A. Seddon and D. J. Adams, Matter, 2020, 2, 764–778 CrossRef.
- M. M. Lerch, A. Grinthal and J. Aizenberg, Adv. Mater., 2020, 32, 2070159 CrossRef CAS.
- G. D. Cha, W. H. Lee, C. Lim, M. K. Choi and D.-H. Kim, Nanoscale, 2020, 12, 10456–10473 RSC.
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |