
Harnessing the power of transition metals in solid-phase peptide synthesis and key steps in the (semi)synthesis of proteins
Shay
Laps†
 ,
Gandhesiri
Satish†
and
Ashraf
Brik
*
,
Gandhesiri
Satish†
and
Ashraf
Brik
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa, 3200008, Israel. E-mail: abrik@technion.ac.il
First published on 12th January 2021
Abstract
Peptides and proteins can be either synthesized using solid-phase peptide synthesis (SPPS) or by applying a combination of SPPS and ligation approaches to address fundamental questions related to human health and disease, among others. The demand for their production either by chemical or biological methods continues to raise significant interests from the synthetic community. In this context, transition metals such as Pd, Ag, Hg, Tl, Au, Zn, Ni, and Cu have also contributed to the field of peptide and protein synthesis such as in peptide conjugation, extending native chemical ligation (NCL), and for regioselective disulfide bonds formation. In this review, we highlight, summarize, and evaluate the use of various transition metals in the chemical synthesis of peptides and proteins with emphasis on recent developments in this exciting research area.
 Shay Laps | Shay Laps received his teaching certificate and BSc degree in Chemistry from the Hebrew University of Jerusalem in 2011 and MA in Science and Technology Education from Ben-Gurion University of the Negev in 2014. He completed his MSc in Chemistry in the Technion-Israel Institute of Technology in 2018, under the supervision of Prof. Brik. He is currently a PhD student under the Jacobs excellence program also in Prof. Brik's laboratory at the Schulich Faculty of Chemistry in the Technion, working on the development of Pd chemistries for the synthesis of challenging and inaccessible posttranslationally modified peptides and proteins. |
 Gandhesiri Satish | Gandhesiri Satish received his BSc in Chemistry and MSc in Organic Chemistry from Kakatiya University, India. He obtained his PhD under Prof. Andivelu Ilangovan in 2017 from Bharathidasan University, India. Currently he is a postdoctoral fellow in Prof. Brik's laboratory at the Schulich Faculty of Chemistry, Technion – Israel Institute of Technology. His research work is related to the development of ubiquitin activity-based probes for studying and targeting deubiquitinases and proteolysis targeting chimeras (PROTACs) for protein degradation studies. |
 Ashraf Brik | Ashraf Brik is a professor at the Schulich Faculty of Chemistry in the Technion, Israel Institute of Technology, holding the Jordan and Irene Tark Academic Chair. He received his BSc in Chemistry from the Ben-Gurion University of the Negev and MSc in Chemistry from the Technion. He obtained his PhD in 2001 from the Faculty of Chemistry in the Technion. From 2002 to 2006, he was a Research Associate in the Scripps Research Institute. In 2007, he joined the Department of Chemistry in the Ben-Gurion University as an assistant professor and was promoted to a full professor in 2012. In 2015, Prof. Brik moved to Technion. |
1. Introduction
1.1 Peptide and protein synthesis
Proteins are often made using molecular biology approaches relying on various strategies that are based on the expression of proteins in yeasts, bacteria, or human cell lines, and cell free expression systems. These strategies, despite being useful in many systems, are still limited with the ability to produce proteins in high homogeneity and large quantities, which contain a diversity of posttranslational modifications (PTMs) at desired sites and numbers, unnatural functionalities, fluorophores, as well as other unique chemical moieties.1–3 Such complex proteins, in principle, could be obtained using total or semi chemical synthesis.4–6 In this approach, peptides derived from the protein sequence are synthesized using SPPS and ligated together by chemoselective reactions. The ligation is based on a chemoselective reaction between two unique functional groups that are introduced into C- and/or the N-termini of peptides.To simplify peptide synthesis and overcome the limitations of the solution phase synthesis, Bruce Merrifield developed SPPS in 1963.7 In SPPS, the first amino acid is attached to an insoluble solid support (resin) via a special linker (Fig. 1a). Then by repetitive cycles of α-amine deprotection and coupling of the next amino acid, the desired peptide is assembled on the resin. Fluorenylmethyloxycarbonyl (Fmoc)-SPPS,8 which is the most used strategy relies on α-amine protection with the Fmoc group and can be removed via treatment with a base (e.g., piperidine). Protected side chains with acid labile protecting groups (PGs, e.g., trityl (Trt)), are removed during the cleavage from the resin by treatment with trifluoroacetic acid (TFA) and scavengers.8
 | ||
| Fig. 1 Chemical synthesis of peptides and proteins: (a) peptide assembly on a solid support (SPPS), (b) general ligation of peptide segments via a chemoselective reaction between two functional groups. Examples of chemoselective reactions between two peptides based on: (c) NCL, (d) KAHA ligation, and (e) Ser/Thr ligation, R = H, CH3. | ||
Several chemoselective ligation reactions of peptides have been developed over the years (Fig. 1b) including: (1) NCL, which up to date is the most widely used approach in chemical protein synthesis (Fig. 1c).4,5,9 In this approach, a peptide bearing an N-terminal Cys reacts in a transthioesterification reaction with another peptide bearing a C-terminal thioester, followed by an N-to-S acyl shift to furnish the amide bond at the ligation junction; (2) the α-keto acid hydroxylamine (KAHA) ligation, which is based on the reaction between an hydroxylamine functionality and α-keto acid to give a native amide bond at the ligation junction (Fig. 1d);10 (3) Ser or Thr ligation, which is based on the reaction between a peptide with an N-terminal Ser/Thr and a peptide with a C-terminal O-salicylaldehyde ester to form an amide bond (Fig. 1e).11
In the process of the peptide synthesis and ligation steps, transition metals have found great utility due to their unique reactivity. This is in addition to the great knowledge we have about their known catalytic nature in a wide scope of organic transformations. Notably, in peptide and protein synthesis, transition metals have been found to be applicable both in organic and aqueous media for various synthetic targets (Fig. 2). This review covers these developments without covering their use in protein modifications (i.e., modification of an expressed form) as some excellent reviews in the literature have already covered this aspect.12–19
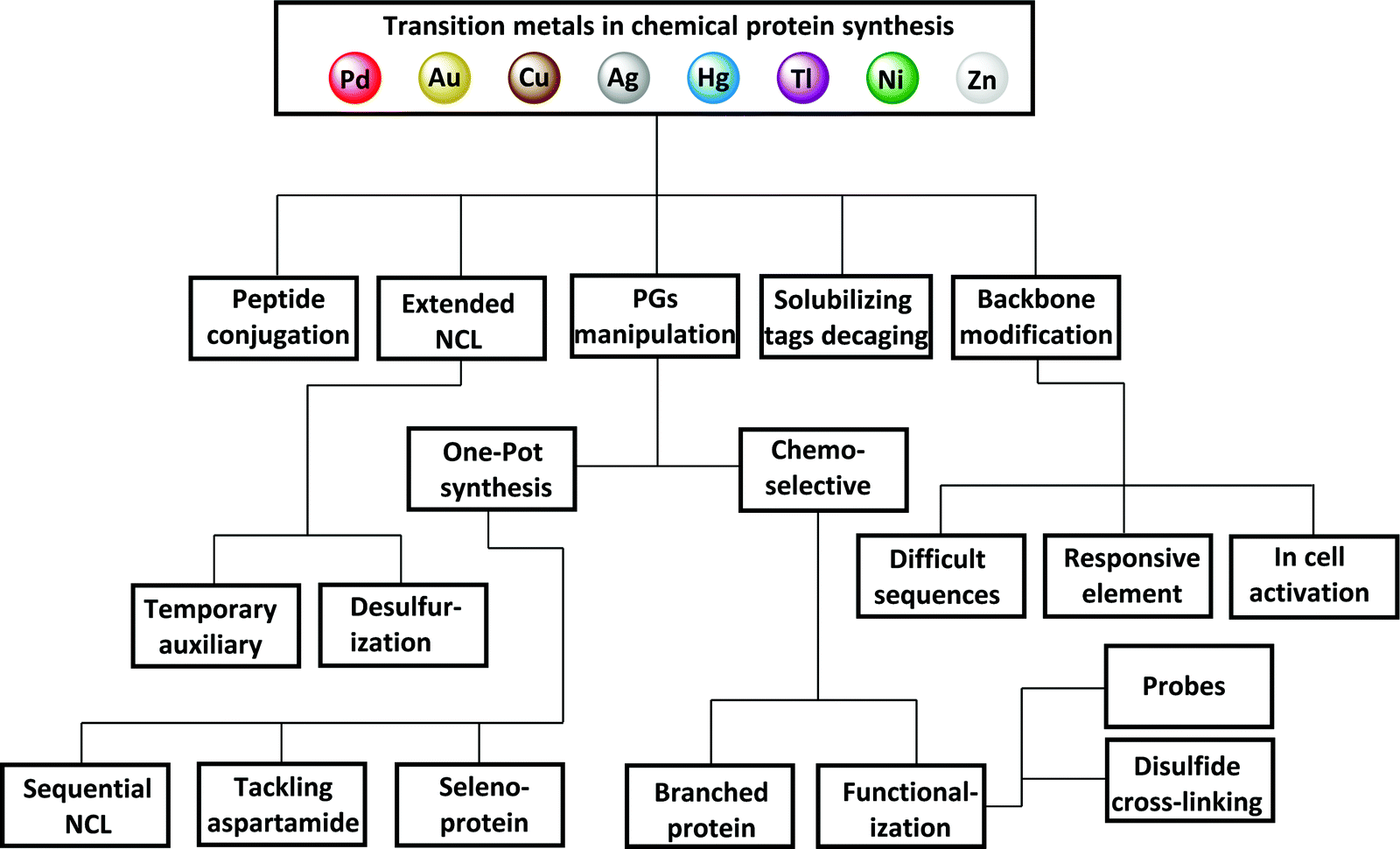 | ||
| Fig. 2 Transition metals in chemical protein synthesis. PG refers to protecting group and NCL refers to native chemical ligation. | ||
1.2 Protecting groups in peptide and protein synthesis
Protecting the α-amino group or the side chain of amino acids is crucial for performing SPPS (Fig. 3). In certain cases, protection is necessary also after SPPS and the cleavage from the resin. For example, when NCL is performed between two unprotected peptides, in several cases orthogonal protection of one or more amino acids during the ligation reaction is needed, as is the case in the combination of NCL with desulfurization.20 Here, native Cys residues have to be protected with orthogonal PGs that are stable to SPPS, cleavage, NCL, and desulfurization conditions, e.g., acetamidomethyl PG (Acm).21,22 Another example for the requirement of orthogonal protection is when sequential NCL between three peptides or more is desired. In this case, the middle peptide must bear both an N-terminal Cys and C-terminal thioester. In such peptides, the N-terminal Cys has to be protected in order to avoid polymerization or cyclization.4–6 Apart from this, the applicability of using orthogonal Cys protection was demonstrated in disulfide bond formation and the chemical synthesis of branched proteins, which are discussed in detail in the coming sections. | ||
| Fig. 3 Applications of PGs in peptide/protein synthesis: (a) cyclization, (b) direct single disulfide bond formation, (c) desulfurization, and (d) ligation. | ||
2. Protein synthesis
2.1 Silver-mediated peptide ligation
Silver-ion-mediated coupling of peptide bearing a free amine with other peptide bearing a C-terminal thioacid in aqueous DMF was originally reported by Blake in 1981 (Fig. 4a).23 Later on, Aimoto and co-workers expanded the scope and utility of this method by combining Ag(I) treatment with various activating agents, such as HOBt, to facilitate ligation with the N-terminal amine (Fig. 4a).24 This interesting strategy, in which the activation of the peptide thioester enables the condensation with an N-terminal amine from the peptide acceptor, was exploited by Hojo and co-workers in their impressive synthesis of a 23 kDa mucin-type glycoprotein, bearing multiple copies of the TN antigen (Fig. 4b).25 The synthesis involved the initial preparation of a 23-amino acid glycopeptide thioester bearing seven copies of the TN antigen. After being assembled on the solid support, the glycopeptide thioester was cleaved from the resin and then condensed with the trimer peptide (TQT-NH2) by the thioester method, which then underwent repetitive ligation cycles via silver-mediated activation to form the original repeat sequence.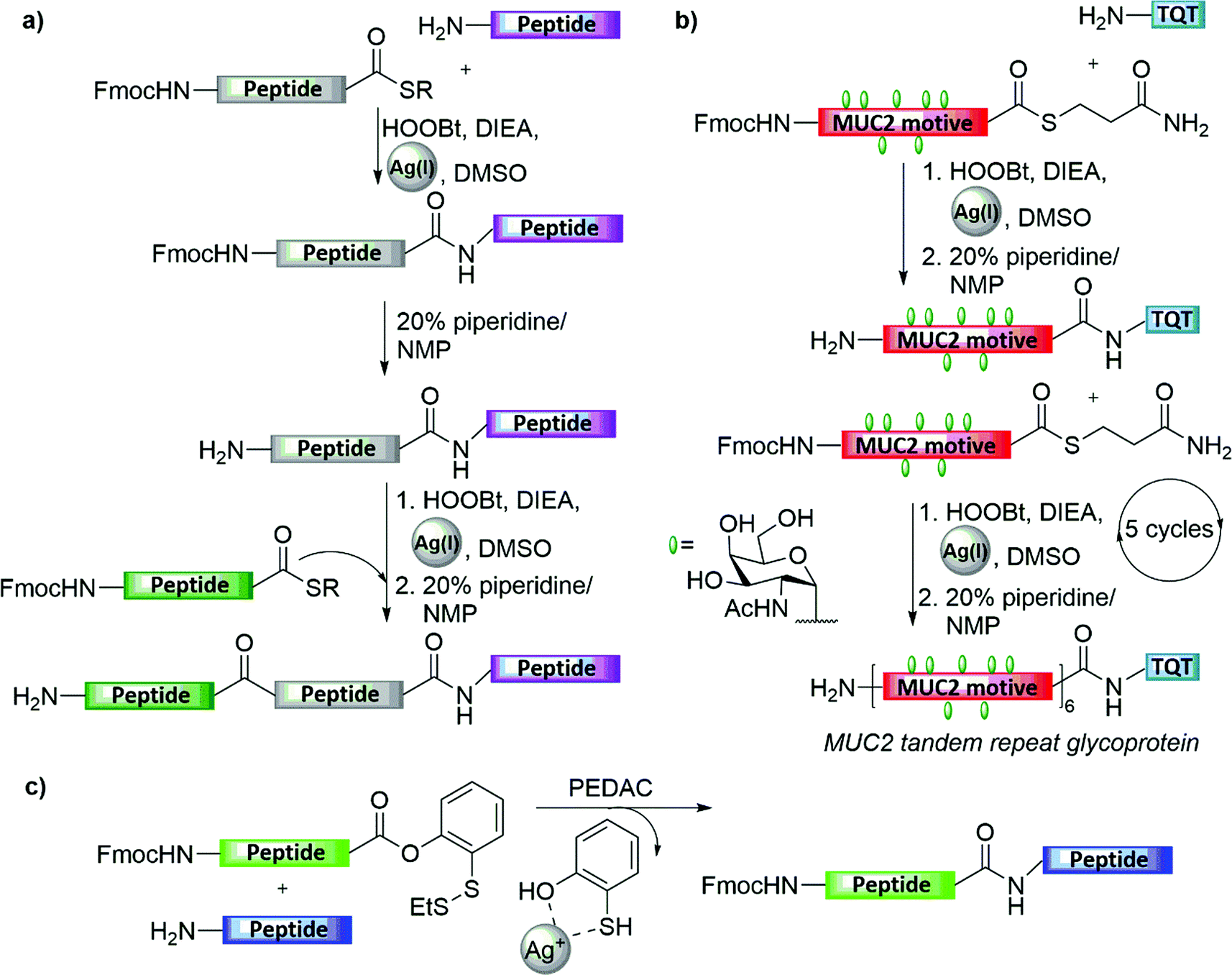 | ||
| Fig. 4 Silver-mediated peptide ligation: (a) general scheme for silver-mediated sequential ligation, (b) silver-mediated sequential ligation for the synthesis of MUC2, (c) AgCl-assisted phenolic ester-directed amide coupling (PEDAC AgCl). | ||
Later on, Danishefsky and co-workers reported AgCl-assisted phenolic ester-directed amide coupling (PEDAC AgCl) as an extension of this approach using C-terminal o-disulfide phenolic ester (Fig. 4c).26 In this strategy, peptides bearing this moiety on their C-terminus were shown to react rapidly with another peptide bearing an unprotected N-terminal amine under. In this reaction, the authors suspected a dual role of Ag(I), in which silver ions also enhanced disulfide reduction, and therefore they examined the use of a stronger reducing agent, namely tris(2-carboxyethyl)phosphine (TCEP), in place of silver(I) ions to obtain a faster ligation.26 Nevertheless, the epimerization of C-terminal residues other than Gly and Pro were obtained in both approaches. Another limitation of this approach is the necessity to protect the Lys and Cys side chains during the ligation step, to prevent unwanted side reactions.26
2.2 Desulfurization
A synthetic scheme was also developed for various protein targets that also included a thiol elimination step once the NCL between two or more segments has been accomplished (Fig. 5a).4–6,27 This combination of NCL and desulfurization extended the utility of protein synthesis for various ligation junctions.27 The synthesis based on these tools requires the introduction of a thiol functionality at the N-terminal peptide (e.g., N-terminal Cys) to enable the transthioesterification step followed by an S-to-N acyl shift. Upon polypeptide assembly, the thiol moiety then can be eliminated by employing metal or metal-free desulfurization reactions. Despite an important development emerging from Danishefsky's lab, where a radical-based approach was introduced to achieve rapid desulfurization under homogenous aqueous conditions,28 transition metals have found great utility in the chemical synthesis of many proteins via NCL coupled with desulfurization. | ||
| Fig. 5 (a) Strategy for NCL followed by desulfurization to recover native Ala. (b) Synthesis of Microcin J25 cyclic peptide. (c) Selected examples of non-native thiolated amino acids. | ||
Dawson and co-worker demonstrated the first total synthesis of Microcin J25, determined at that time as a 21 AA cyclic antibiotic peptide, which is naturally produced by a strain of E. coli, and which lacks a Cys residue (Fig. 5b).20 Further structural investigations revealed, however, that Microcin J25 comprises an unusual N-terminus to a Glu8 side-chain amide bond, resulting in an N-terminal ring in a conformation that has been described as a “lasso” type structure.29–31 In the original synthesis, the linear analogue of microcin J25 was prepared via Boc-SPPS. An N-terminal Cys was introduced, replacing natural Ala within the peptide sequence, while the C-terminal of the same peptide was functionalized with a thioester. Cyclization via NCL followed by metal-mediated desulfurization furnished the native microcin J25 (as was known before the structure was further revised).20 For the desulfurization step, the cyclized product was treated with Pd/Al2O3 (10%) in 20% aqueous acetic acid (AcOH) and exposed to H2 gas to eliminate the thiol functionality and convert it to the native Ala. In addition, the authors performed a comparison study between various conditions (e.g., Pd/Al2O3, (10%), Pd/C (10%), Pd/BaSO4 (10%), PdO and RANEY® nickel). The comparison revealed a similar efficiency of Pd/Al2O3 and Pd/C (10%) in 20% aqueous AcOH (>99% yield), while Pd/BaSO4 and PdO resulted in lowered yields (90% and 30%). RANEY® nickel was shown to be as efficient as Pd/Al2O3; however, the demethylthiolation of Met residues has also been observed.20
The Pd-mediated desulfurization enabled the development of sugar-assisted glycopeptide ligation by Brik et al., in which a sugar moiety of a glycopeptide modified with a thiol handle at the C-2 position to form an acetamido thiol handle facilitated the ligation of a Cys-free glycopeptide with a thioester peptide via an S-to-N-acyl shift followed by desulfurization of the ligation product to the corresponding native acetamide moiety on the sugar part.32,33 The same group developed a strategy for selective Cys desulfurization in the presence of a thiol moiety protected with Acm.21
In parallel, Pentelute et al. reported a selective desulfurization of Cys in the presence of Cys(Acm) in polypeptides obtained by NCL.21,22 In this study, the authors aimed to extend the use of NCL followed by desulfurization to enable a more versatile synthesis of Cys-rich peptide and protein targets. The unique and challenging knotted trypsin inhibitor EETI-II protein was chosen as a proof of concept.22 EETI-II contains 28 AA and six Cys residues that form a highly stable framework via three disulfide bonds. The synthetic route involved NCL between a segment bearing a C-terminal thioester and another segment in which Ala17 was mutated to Cys. All the natural six Cys residues were protected with Acm. After NCL of the two segments, selective desulfurization of residue Cys17 to give Ala17 was performed via RANEY® nickel-mediated reduction, in the presence of the Cys(Acm) residues that remained intact. Following HPLC purification, the Acm PGs were removed and the purified product was subjected to oxidative folding to form the native EETI-II.22
These early developments inspired several groups to exploit this concept to develop additional ligation junctions utilizing various artificial AAs bearing temporary thiol handles (such as Lys, Asp, Glu, Val, Trp, and Pro) (Fig. 5c).4–6,27 These further developments have added considerable flexibility in choosing the ligation junctions when attempting the synthesis of a particular protein.
2.3 Zn-Mediated removal of oxyalkyl auxiliary in peptide ligation
Kent and co-workers have sought to extend the applicability of NCL to more junctions beside Cys since the early days of its development. His group reported the development of a cleavable oxyethanethiol-based auxiliary to assist ligation, which was designed to be introduced at the backbone amide of the first AA of the peptide sequence (Fig. 6).34 In this approach, a C-terminal thioester peptide reacts via thiol exchange with another peptide containing the auxiliary, which through a rearrangement involving a six-membered ring gives the amide-linked product.34 The auxiliary showed stability under HF cleavage conditions yet could be simply removed under treatment with zinc dust to give the native backbone structure of the ligation product. Although this strategy was applied for the syntheses of a few proteins,35 it has been not widely used mainly due to the slow ligation rates (∼12 h for the ligation at Gly junction compared to ∼1 h via Cys-based NCL in the presence of 4-mercaptophenylacetic acid (MPAA) and TCEP).36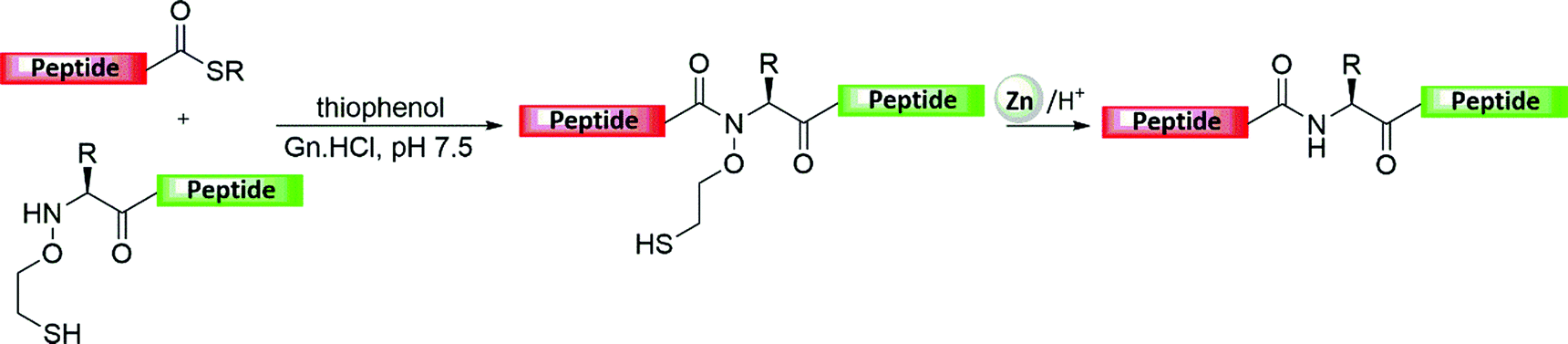 | ||
| Fig. 6 Representative depiction of a temporary auxiliary-based NCL followed by Zn-mediated removal. | ||
3. Removal of the protecting group with transition metals in organic solvents
3.1 Acm PG deprotection
The stability of the Acm group to acidic conditions allows its orthogonal use with other PGs (Fig. 7a and Table 1). It is also stable to the applied conditions for the generation of hydrazides from esters and azides from hydrazides.37 The main advantage of the Acm group comes from its stability under the conditions commonly used in peptide synthesis (Fmoc or Boc) in solution or in the solid-phase method. Acm can be used as a PG during segment condensation,38 sequential ligation,21,39–41 desulfurization,21,22,42 and disulfide bond formation.43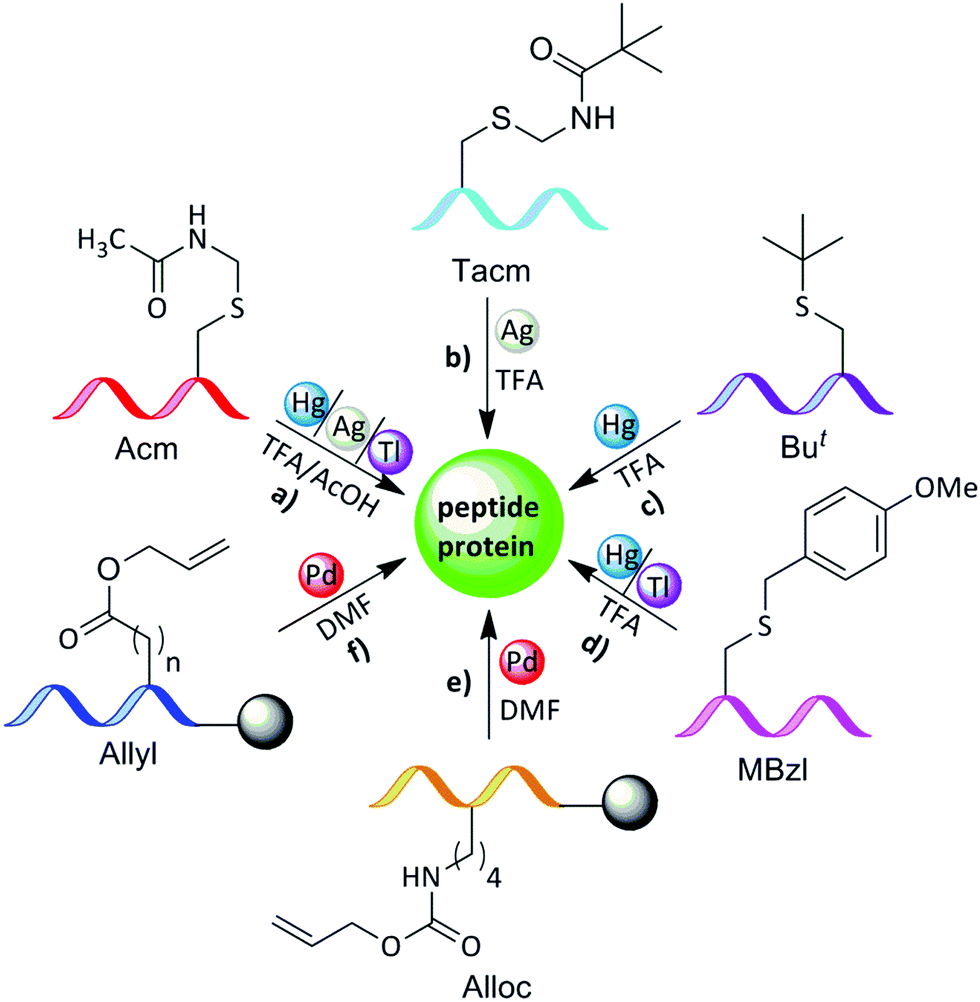 | ||
| Fig. 7 Transition metal-mediated removal of the PGs in organic solvents for peptide and protein synthesis: (a) removal of Acm PG with Hg, Ag or Tl in TFA or AcOH; (b) removal of S-trimethylacetamidelmethyl (Tacm) PG with Ag in TFA; (c) removal of tert-butyl PG with Hg in TFA; (d) removal of p-methoxybenzyl (MBzl) PG with Hg or Tl in TFA; (e) removal of Alloc PG with Pd in DMF; (f) removal of allyl PG with Pd in DMF. | ||
| Amino acid | Protecting group | Compatibility | Transition metal | Solvent |
|---|---|---|---|---|
| a Except for repetitive treatments. | ||||
| Cys | Acm | Boc, Fmoc | Hg(II) | AcOH |
| Ag(I) | TFA, TFE | |||
| Ag(II) | AcOH | |||
| Tl(III) | TFA + CH3CN | |||
| TFA | ||||
| TAcm | Boc, Fmoc | Hg(II) | TFA | |
| Ag(I) | TFA | |||
| But | Boc, Fmoc | Hg(II) | TFA, AcOH | |
| MBzl | Boc,a Fmoc | Hg(II) | TFA, AcOH | |
| Tl(III) | TFA | |||
| Lys | Alloc | Boc, Fmoc | Pd(0) | DCM |
| Asp/Glu | Allyl | Boc, Fmoc | Pd(0) | DCM |
The protection of thiol group of Cys by the Acm group was introduced by Hirshmann.37 In this report, mercury(II) acetate [Hg(OAc)2] in AcOH was used for the deprotection of Acm in octa-5-Acm-octahydro-5-protein to give S-protein, with further air oxidation to enable the synthesis of ribonuclease-S. The mercuric ion was removed by treatment with mercaptoethanol for a long time (19 h) by producing acetamido methanol as a byproduct.
Yajima and co-workers utilized Acm-protected Cys residues to form a single disulfide bond in oxytocin peptide using thalium(III)trifluoroacetate [Tl(OTf)3] in combination with TFA for the removal step.44 However, under these conditions, Trp and Met were found to be unstable and formed some unidentified side products. Hence, these functionalities must be protected during the Tl(OTf)3 treatment. To overcome these issues, another catalytic system involving silver trifluoromethanesufonate (AgOTf) in TFA and anisole was developed.45 Under these conditions, Trp, Met, and Tyr were less prone to undergo side reactions, and other Cys PGs, like S-t-butyl (But) and 4-methylbenzyl, were not affected.
Surprisingly, when these catalytic systems [Tl(OTf)3 and Hg(OAc)2] were employed for the synthesis of an amino terminal loop of the snake α-neurotoxin, the Acm group was shifted from the Cys residue to the Ser or Thr residue.46 However, the addition of glycerol (trivalent alcohol) as a scavenger in the presence of both metal complexes prevented this shift.
As discussed in Section 2.1,24 Aimoto and co-workers selectively achieved peptide ligation and Acm removal in the presence of AgNO3 by changing the nucleophile. Here, the nucleophilic character of the additive HOOBt was enough to attack the thioester activated with a silver ion, but was too weak to attack the thioether of the Acm, eventually leading to a coupling reaction. In another study, however, the same conditions (AgNO3, HOOBt) led to the removal of the Acm group when the synthesis of a reaper polypeptide was attempted.47 The reason for this could be explained by the influence of the amino acid residue in the immediate vicinity of the Cys(Acm), which greatly influences the stability of the Acm group toward different silver ions. Hence, changing the silver salt, such as by using AgCl instead of AgNO3, for the condensation step provided the reaper as well as the molt-inhibiting hormone (Prc-MIH).
Kent and co-workers reported the synthesis of crambin by three peptide segments, which gave a 10-fold increase in yield compared to from two segments (Fig. 8).48 Here, the N-terminal Cys of the middle segment was temporally protected with the Acm group to prevent undesired cyclization or polymerization. Following the ligation step, the Acm group was subjected to removal with Hg(OAc)2; however, the reaction was less efficient. Changing the metal to Ag(OAc)2, however, enabled the efficient removal of Acm to facilitate the subsequent ligation. Similarly, the total synthesis of a range of proteins, such as erythropoietin (EPO),38 sortase A,49 interferon-β-1a (IFN-β-1a),50 glycosylated granulocyte-macrophage colony-stimulating factor (GMCSF),43 designed ankyrin repeat protein (DARPin) PE59,41 bacillus amyloliquefaciens RNase (Barnase),41 and HIV-1 Rev protein,51 were achieved employing Acm as a PG.
 | ||
| Fig. 8 Protection of the middle fragment with an Acm group to facilitate sequential ligation for the synthesis of crambin. | ||
Our group utilized Acm as an N-terminal Cys PG of the middle fragment during isopeptide chemical ligation (ICL) for site-specific ubiquitination at the lysine 34 position of H2B as well as the δ-mercaptolysine of the α-synuclein-thioester fragment to enable expressed protein ligation (EPL) with the recombinant Cys fragment (Fig. 9),52 and later, the same target was prepared by a sequential strategy.40 Similarly, other ubiquitinated proteins, like ubiquitinated α-synuclein,53 H3K4me3,54 and H2K120Ub,42 were also obtained.
 | ||
| Fig. 9 Protection of the N-terminal Cys with Acm for ICL in the synthesis of ubiquitinated proteins. | ||
Kiso and co-workers developed the S-trimethylacetamidelmethyl (Tacm) group as a thiol PG of Cys (Fig. 7b).55 This PG has similar chemical properties as Acm, but is less susceptible to air oxidation. The utility of this group was demonstrated by the synthesis of porcine brain natriuretic peptide and somatostatin.55
3.2 Tertiary-butyl (But) PG deprotection
The Cys t-butyl (But) group is completely stable under the regular conditions of Fmoc-SPPS and its deprotection has been less explored (Fig. 7c and Table 1). Fujino and co-workers reported the removal of the S-But group under Hg(OAc)2 in TFA or Hg(OTf)2 in AcOH using anisole as a scavenger.56 In the subsequent step, the free thiol was regenerated quantitatively by treatment with hydrogen sulfide. Other heavy metal ions, including Ag+, Cu2+, Zn2+, Ni2+, and Pb2+, were completely ineffective for the removal of the S-But group.56 The same cleavage conditions for S-But were successfully applied for the removal of another thiol-PG, namely p-methoxybenzyl (MBzl),56 with an even faster rate and higher yields.3.3 p-Methoxybenzyl (MBzl) PG deprotection
Yazima and co-workers found that the treatment of Cys (MBzl) with Tl(OTf)3 as a soft Lewis acid under acidic conditions resulted in effective MBzl cleavage (Fig. 7d and Table 1).44 However, under these conditions, Trp gave several unidentified products and Met was partially oxidized. Hence, unprotected Trp and Met are not compatible with Tl(OTf)3 treatment. The utility of this method was demonstrated in the syntheses of a urotensin II peptide bearing a single disulfide bond and a Trp, which was temporally protected and upon formation of the disulfide bond, was then removed with TMSOTf/TFA in the presence of diphenyl sulfide.3.4 Allyloxycarbonyl (Alloc) PG deprotection
The Alloc group was first utilized by Stevens and Watanabe for the protection of the N-terminal group of D,L-amino acids (Fig. 7e, f and Table 1).57 This PG is widely used mainly due to its compatibility with either Boc- or Fmoc-SPPS, and as it can be cleaved efficiently under mild and neutral conditions. The cleavage of the Alloc group was demonstrated by catalytic hydrogenolysis either with Pt or Pd catalysts. Later, Kunz and Unverzagt developed effective conditions for the removal of the Alloc group by Pd(PPh3)4 in tetrahydrofuran via an allyl group transfer.58 In these reactions, dimedone and others, such as phenysilane, were used as scavengers to capture the released allyl group.59 Several research groups employed Alloc/allyl as PGs to the side chains of Lys/Glu residues for the synthesis of cyclic peptides on resin through a coupling of the head to tail/side chain to side chains.60,61 Apart from this, the Alloc group served as an orthogonal PG for the thiazolidine (Thz) on resin in the total synthesis of ubiquitinated α-globin.624. Removal of the protecting group with transition metals in aqueous solvents
4.1 N-terminal Cys protection
Thz is a widely used N-terminal-protected form of Cys when the sequential ligation of three or more fragments is applied (Table 2).63 Nevertheless, the unmasking step with methoxylamine takes a long time, which slows down the entire synthetic process of the target protein. In addition, thioester peptide with C-terminal Gly has been reported to be modified irreversibly with the methoxylamine.12,64–66 In addition, Thz was found to be unstable during the oxidative switching of hydrazide peptide.67 Our laboratory addressed these challenges and found that Pd(II) complexes can effectively open the Thz ring to give the free unprotected N-terminal Cys, within minutes, under NCL conditions (Fig. 10a).68 This approach has been applied for the synthesis of Lys34-ubiquitinated H2B among other proteins.69 We also reported that the propargyloxycarbonyl group (Proc) at the N-terminus Cys undergoes deprotection in an effective manner via exposure to PdCl2 under NCL conditions, as demonstrated in the first synthesis of neddlyated peptides (Fig. 10b).68 In this study, the addition of Mg2+ was found to facilitate the efficient one-pot Pd(II)-mediated Thz/Proc deprotection under NCL conditions.68| Amino acid | Protecting group | Transition metal | Solvent | Introduction to the sequence |
|---|---|---|---|---|
| Cys | Acm | Pd(II) | 6 M Gn·HCl | Fmoc-SPPS |
| Cu(II) | 6 M Gn·HCl | Fmoc-SPPS | ||
| Thz | Pd(II) | 6 M Gn·HCl | Fmoc-SPPS | |
| Cu(II) | 6 M Gn·HCl | Fmoc-SPPS | ||
| Proc | Pd(II) | 6 M Gn·HCl | Fmoc-SPPS | |
| Alloc | Pd(0) | 6 M Gn·HCl | Fmoc-SPPS | |
| But | Pd(II) | Urea/Tris buffer | Fmoc-SPPS | |
| Succinimde | Pd(II) | 6 M Gn·HCl | Solution phase | |
| Lys | Proc | Au(I) | 6 M Gn·HCl | Fmoc-SPPS |
| Asp/Glu | Allyl | Pd(II) | 6 M Gn·HCl | Fmoc-SPPS |
| Asp | Allyl | Pd(0) | 6 M Gn·HCl | Fmoc-SPPS |
| Sec | Sez | Cu(II) | 6 M Gn·HCl | Fmoc-SPPS |
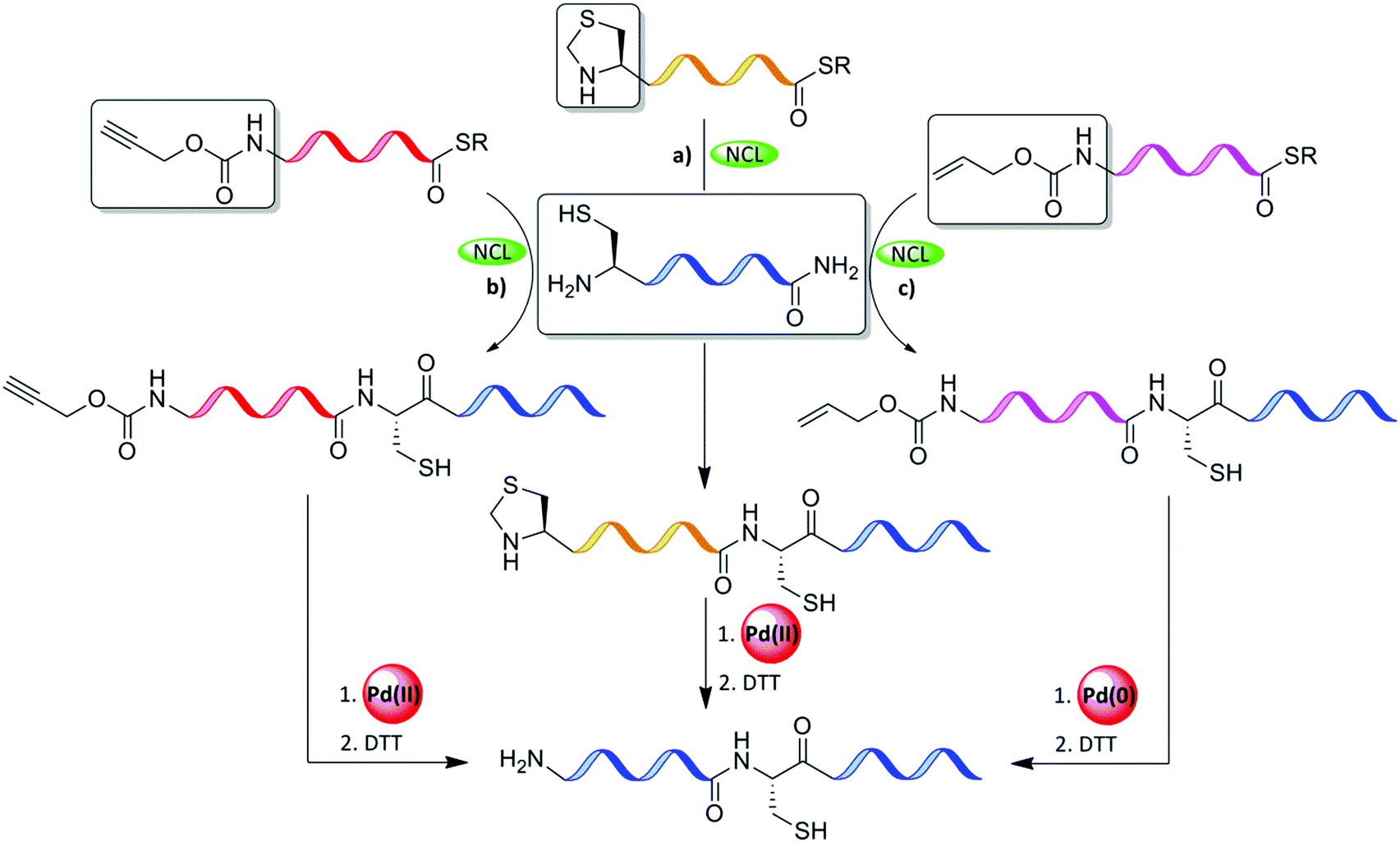 | ||
| Fig. 10 N-terminal Cys protection of the middle fragments (a) Thz, (b) Proc, and (c) Alloc during sequential ligation and removal with Pd complexes. | ||
Inspired by our findings, Okamoto's laboratory applied Pd(0) complexes to remove the Alloc PG from N-terminal Cys (Fig. 10c and Table 2). In this study, MPAA was used as an enhancement additive for ligation and as a scavenger for the π-allyl complexes, and for quenching Pd.70 This approach was demonstrated in the chemical synthesis of histone H2AX.
4.2 Internal Cys protection
The harsh removal conditions of the Acm PG, which were discussed in Section 3.1, often requires long reaction times, prevents one-pot ligation and/or desulfurization processes, and necessitates additional purification steps before and after its removal. Our laboratory discovered that Pd(II) complexes can effectively unmask Cys(Acm) in peptides and proteins, within 5 min, under aqueous NCL/desulfurization conditions (Fig. 11a and Table 2).71 Notably, PdCl2 dissolved in 6 M Gn·HCl more efficiently than in water to form the corresponding complexes, probably due to the coordination of the guanidinium group to Pd (II).72 We applied these mild Acm removal conditions to construct proteins containing native Cys residues by NCL and desulfurization.12,71,73 As a proof of concept, the ubiquitin-like protein (UBL-5) was synthesized by applying this chemistry.71 UBL-5 is a 73 AAs protein containing two Cys residues, which are not located in strategical positions suitable for NCL (6, 18). Consequently, the sequence was dissected into two segments: UBL-5N(1–32) bearing two Cys protected with Acm and a C-terminal thioester, and a second segment of UBL-5C(33–73), while a mutation of Ala33 into Cys was devised (Fig. 11b). A one-pot operation of NCL followed by desulfurization and Acm removal was successfully conducted, demonstrating the power of the developed tools for efficient protein synthesis.69,71,74 | ||
| Fig. 11 Transition metal-mediated removal of Acm in aqueous medium for peptide and protein syntheses. (a) General scheme for the removal of Acm with Pd(II). (b) One-pot synthesis of Ubl5 employing Acm. | ||
Inspired by our work, Wang and co-workers reported a method for the site-specific incorporation of selenocysteine (Sec) into proteins in E. coli and mammalian cells.75
The Sec incorporation required idiosyncratic translational machinery, which makes it challenging to recombinantly prepare selenoproteins with high Sec specificity. Here, the authors reported an orthogonal tRNApyl-ASecRS to site-specifically incorporate allyl-protected Sec. Subsequently, the allyl group was removed using Pd chemistry under mild conditions. This method provides the opportunity to introduce site-specific Sec to proteins and enables their biochemical study.75
Very recently, Otaka and co-workers reported the removal of Acm from Cys (Table 2).76 The deprotection was established via treatment with Cu(II) in the presence of free N-terminal Cys or an external source of 1,2-aminothiols, which was required in order to avoid severe side reactions. The reaction took 3 h compared to 5 min when using Pd, highlighting the differences in the used transition metal in these transformations. In their proposed mechanism, Cu was involved in the removal of Acm and the formation of Cys to the S–OH intermediate, which could then be reacted with EDTA or DTT to give the oxidized or reduced peptide, respectively.76
4.3 C-terminal Asp protection
As discussed in Section 1.1, NCL based on XX-Cys (XX represents any of the 20 canonical amino acids) sites has been shown to proceed with different kinetics.77 However, the carboxylate group side-chain of Asp and Glu are known to form the cyclic C-terminal anhydride through intramolecular cyclization, which can further be attacked by the Cys-peptide to form the β- or γ-linked product as well as the native product. (Fig. 12).78,79 When Boc chemistry79 or N-acylbenzimidazolinone (Nbz)-based methods80 were employed for the generation of C-terminal Asp thioesters, the formation of carboxylic anhydride was observed in 30% of the overall reaction products. However, in the case of the C-terminal hydrazide, the side-chain carboxyl group of Asp is prone to C-terminal cyclization upon oxidative switching, rendering it completely inactive in the ligation reaction.81 Therefore, the carboxylate side-chain of Asp and Glu must be protected during ligation to avoid undesired product formation.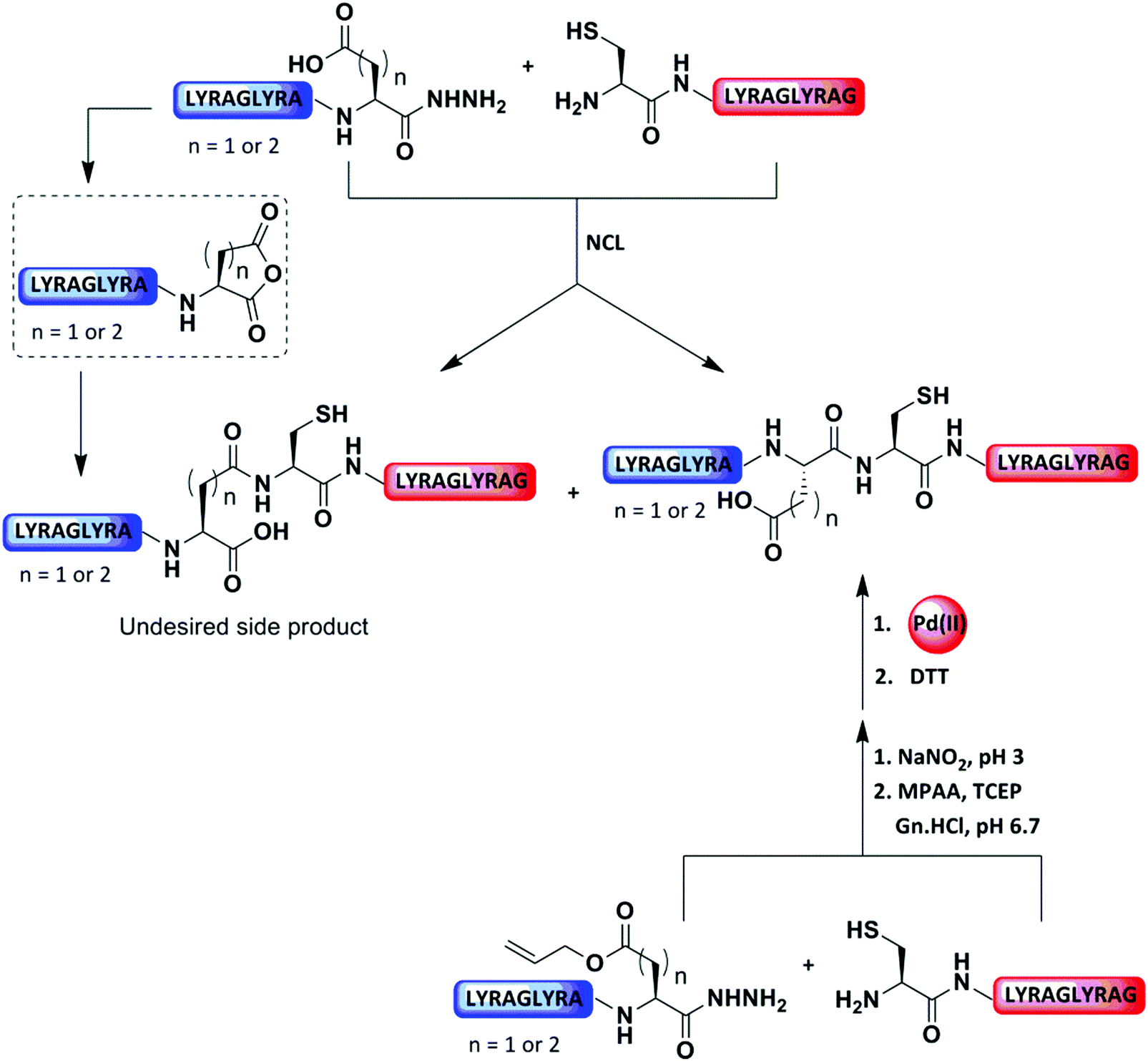 | ||
| Fig. 12 Pd(II)-Mediated removal of allyl PG after the ligation at Asp/Glu for peptide synthesis. | ||
Botti and co-workers developed the PG 1-methyl-2-oxo-2-phenylethyl ester (OMop) for the side chain of Asp and Glu, which was introduced during SPPS.78 This PG was found to be completely stable during HF cleavage and the ligation reaction. After ligation, the PG was removed by Zn in AcOH. In another report, Hojo's group protected the side chain of C-terminal Asp with a 2-(4-pyridyl)-2-propyl (PyP) group to prevent side reactions during NCL. Upon ligation, this PG could be selectively removed after 30 min by Zn in the presence of 3-mercaptopropionic acid (MPA).82 This method was applied to the synthesis of human histone H4.
Our group recently reported that the protection of the side chain of the Asp and Glu with allyl functionality could serve as a temporal PG to prevent undesired cyclization of the C-terminal hydrazide (Fig. 12).83 Upon ligation at these sites, the allyl group was rapidly (10 min) removed by [Pd(allyl)Cl]2 and GSH under aqueous conditions. In this case, GSH was found to enhance the Pd reactivity and acted as scavenger for the released allyl group (Fig. 12 and Table 2).83
Parallel to our study, Okamoto and co-workers demonstrated NCL at the Asp–Cys junction, while protecting the Asp residues with an allylic group. (Fig. 13 and Table 2).84 The removal step of the allylic moiety required less than 10 min upon the addition of Pd/TPPTS complex under the ligation conditions. Here, an excess amount of MPAA was used as a scavenger for the π-allyl complex and Mg2+ was added to speed up the deprotection reaction. This approach was demonstrated by the preparation of a fluorescein-labeled polypeptide, PEP-19.
 | ||
| Fig. 13 Pd(II)-Mediated removal of allyl PG to facilitate PEP-19 synthesis. | ||
5. Transition metal-mediated removal of reversible tags in protein synthesis and manipulations
5.1 Side-chain solubilizing tags
Chemists often encounter difficult sequences derived from proteins, which are problematic to make (e.g., β-amyloid peptides) and to handle at the purification step or in ligation reactions (e.g., membrane proteins).85 Dissecting a polypeptide into segments to enable protein synthesis could lead to the formation of insoluble peptides in aqueous media.4,8 To overcome these challenges, the concept of solubilizing tags has emerged, where a highly soluble tag is installed into the peptide fragments through a cleavable linker.86 The linker should be stable to SPPS, compatible to ligation approaches, and capable of being removed in full aqueous media.Based on the Acm functionality, our group designed the synthesis of an Alloc–Phacm linker and introduced this to SPPS to aid the solubility of histone H4 C-terminal fragment (Fig. 14a).87 This linker allowed the addition of several Arg (RRR-Phacm) as a solubilizing tag. Following the ligation step with the histone H4 N-terminal fragment, the reversible RRR-Phacm tag was completely removed using PdCl2, within 20 min, from the crude peptide under aqueous conditions. A final desulfurization step enabled the synthesis of the full-length histone protein H4 in a one-pot manner.74,87
 | ||
| Fig. 14 Pd(II)-Mediated removal of Acm-derived solubilizing tags to facilitate protein synthesis. (a) Histone H4 total synthesis. (b) ISGylated ubiquitin total synthesis (B-Arg6 refers to biotinylated six Arg). | ||
In another work, our group attempted to synthesize ISGylated-Ub, which turned out to be extremely challenging due to its hydrophobic nature and high tendency for its aggregation as well as it's fragments during SPPS (Fig. 14b).88 We envisioned that the introduction of a cleavable solubilizing tag could aid the solubility. Consequently, we designed a new reversible Acm solubilizing tag in which the α-carbon of the Acm was modified with a dimethylated amine moiety (Acm-NMe2).88 The key benefits of this tag were the positively charged amino groups lowered the aggregation tendency and there was less steric hindrance cause by the Acm-NMe2 group compared to the Ph-Acm-based tag. This solubilizing tag efficiently assisted the SPPS, assembly of the peptide segments by ligation, and the HPLC purification throughout the synthetic process. Upon the ISGylation of Ub through Lys 29, this temporal tag could be liberated by applying PdCl2 under aqueous conditions. Finally, a desulfurization step followed by folding afforded the target protein.88
5.2 Backbone modifications
Interest in the introduction of a responsive element to an external stimulus in peptides and proteins for various purposes has led to the use of different strategies, such as the expression of unnatural amino acids89,90 and the (semi)chemical synthesis of proteins.4,91,92We recently reported the backbone modification of a synthetic protein, namely ubiquitin (Ub)-aldehyde attached with a cell-penetrating peptide (CPP) through the Thz cleavable linker as a backbone (Fig. 15).93,94 We then delivered the engineered protein into live cells (DU-145 cells) with the help of CPP, and subsequently the Thz was cleaved by Pd as an external stimulus. Using this approach, we were able to generate the Ub C-terminal aldehyde as an electrophile, which resulted in an enhanced inhibition of the cellular deubiquitinating enzyme (DUB), USP2a. The inhibition of USP2a in the cellular environment was supported by western blot, using the antibody against MDM2, which is a known substrate for USP2a. The fluorescence quenching pair sulfo-cyanine 3/5 (Cy3/Cy5) were used to monitor the entire process.
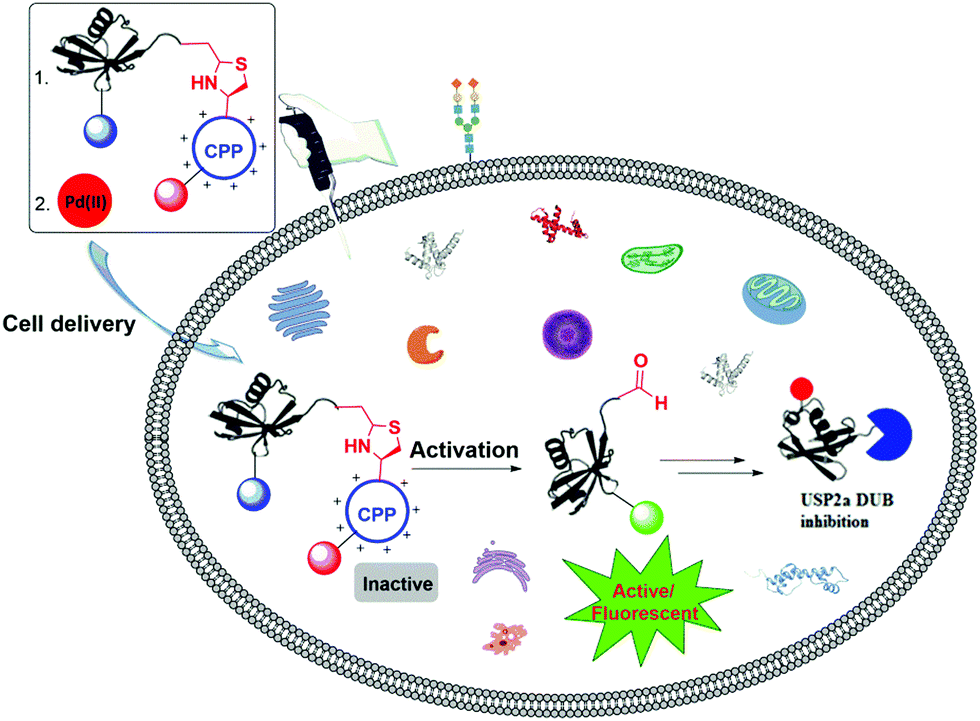 | ||
| Fig. 15 Pd(II)-Mediated cleavage of a Ub activity-based probe with a Thz-modified backbone in live cells. | ||
The incorporation of reversible backbone modification is a useful strategy to interrupt the secondary structures forming during SPPS or after cleavage, therefore assisting in avoiding peptide aggregation on the resin or in solution.95,96 The pseudoproline dipeptide introduced by Mutter and co-workers is restricted to particular amino acids (e.g., Ser) and often cleaved using TFA.97 Our group recently reported the depropargylation of amides in aqueous media utilizing gold [Au(I)] (Fig. 16 and Table 2).98 The incorporation of temporary propargylated amides in the sequence of NEDD8 protein enabled its efficient synthesis due to the effect of backbone modification on the peptide solubility. We also discovered a rapid site-selective amide bond cleavage, which depended on the position of the propargylation site (Gly-XX).98
 | ||
| Fig. 16 Au(I)-Mediated removal of the backbone facilitated the chemical synthesis of the protein and the cleavage reaction when reversing the residue order. | ||
6. Orthogonal removal of different PGs with transition metals
6.1 Orthogonal removal of Acm vs. Thz
The incorporation of orthogonal PGs is extremely important for the synthetic scheme of branched proteins and/or to guide the synthetic direction (e.g., in the case of sequential NCL and late-stage functionalization). Recently, we reported the Pd-mediated on-demand deprotection of Thz and Acm groups in a fully aqueous medium (Fig. 17a).99 We also showed the efficient removal of t-Bu PG using Pd and demonstrated its stability under the removal conditions of Thz and Acm (Fig. 17b).99 This chemistry has been applied for multiple selective functionalizations of peptides containing Cys residues and in the challenging total chemical synthesis of the copper storage protein 1 (Fig. 17c).99 Utilizing these synthetic tools, we also accomplished the chemical synthesis of an activity-based probe (ABP). The ABP was composed of histone H2A, which was specifically ubiquitinated at Lys119 and contained dehydroalanine (DHA), at the junction between H2A and Ub, as a Cys proteases trap. This ABP reacted selectively with the Calypso/ASX heterodimer DUB, via a Michael addition reaction, to furnish a nucleosome–enzyme complex stabilized by a covalent C–S bond.99 | ||
| Fig. 17 Cys deprotection on demand: (a) Pd(II)-mediated selective removal of Acm and Thz groups, (b) Pd(II)-mediated selective removal of Acm, Thz, and t-butyl groups, (c) Pd(II)-mediated site-specific and differential modification of a peptide. | ||
6.2 Orthogonal removal of Acm vs. Alloc
Okamoto's group developed another selective strategy relying on the different properties of Pd complexes for the selective removal of the Alloc PG for N-terminal Cys, in the presence of internal Cys protected with Acm, (Fig. 18).100 In this approach, the Alloc PG was cleaved via a Pd(0)-mediated reaction, while the Acm was effectively unmasked with Pd(II).100 The applicability of this chemistry has been demonstrated in the total chemical synthesis of histone H3 bearing a trimethylated Lys9 (K9me3) through the orthogonal removal of Alloc and Acm groups.100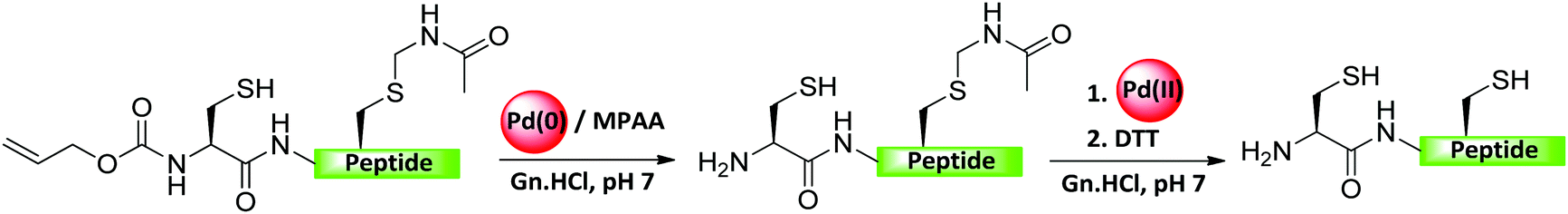 | ||
| Fig. 18 Pd-Mediated chemoselective removal of Alloc and Acm groups. | ||
6.3 Orthogonal removal of Thz vs. maleimide
As discussed in Sections 6.1 and 6.2, most of the PGs (e.g., Acm) employed in peptide and protein synthesis need to be installed during the synthesis of peptide fragments on SPPS. Our group recently developed maleimide as a PG, which could be attached or detached on demand to the Cys residues of unprotected peptides or proteins in aqueous conditions (Fig. 19).101 The maleimide group was efficiently removed by employing PdCl2/MgCl2 and dithiothreitol (DTT). To demonstrate the synthetic applicability, the maleimide group was employed as a stable linker during solid-phase chemical ligation for the synthesis of Ub-ABPs. To elongate the polypeptide on the solid support, the N-terminal-protected Thz was removed selectively against the maleimide group by using [Pd(allyl)Cl]2/GSH.101 After multiple ligations and desulfurization, the elongated polypeptide was then liberated efficiently from the solid support by cleaving the maleimide handle using PdCl2/MgCl2 (Fig. 19 and Table 2). In the final step, the liberated polypeptide was subjected with bisamide to form the corresponding Ub-ABPs.101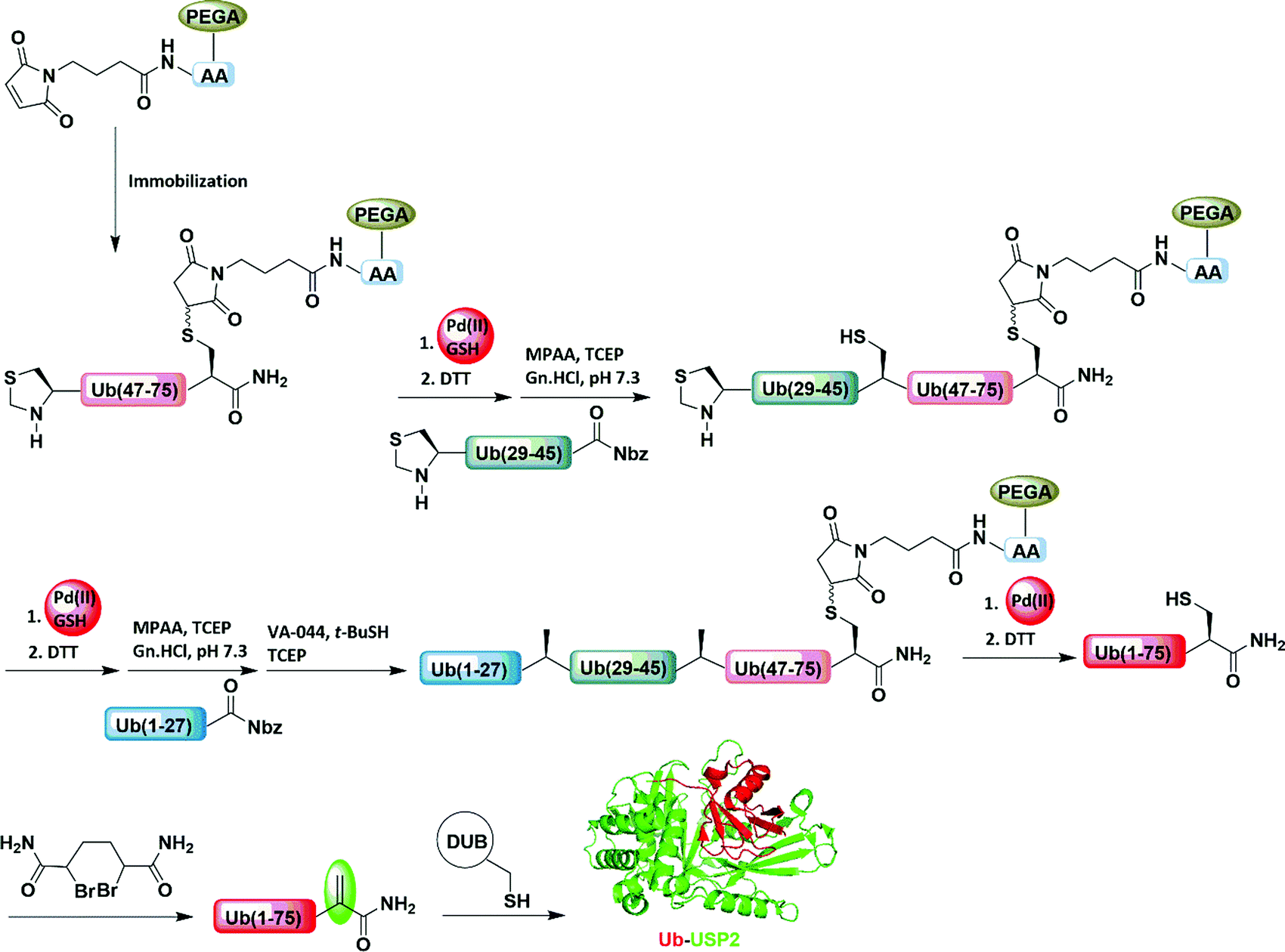 | ||
| Fig. 19 Pd(II)/GSH-mediated chemoselective removal of Thz against maleimide. | ||
7. Pd in the synthesis of peptides and proteins bearing disulfide bonds
For over six decades, tremendous efforts have been invested to synthesize peptides and proteins bearing multiple disulfide bonds, yet no effective and/or general synthetic scheme has been devised to date, despite the great potential of these products for various applications.102 Unfortunately, performing studies with isolated or biologically expressed peptides and proteins with disulfide bonds is very challenging for various targets, which has prompted the search for efficient synthetic approaches for their production. Two main strategies were adopted along this long journey: (1) the metal-free approach, which is based on oxidative folding and depends on the folding pathway of the particular target. However, this strategy suffers mainly from long reaction times (up to several days), low yields, and a lack of success in many cases; (2) a strategy based on forming disulfide bonds, which builds on the stepwise thiol deprotection and oxidation. In this approach, orthogonal PGs are applied for the side-chain Cys, such as of S-Trt, Acm, and t-Bu groups.102,103 Nevertheless, the removal conditions of these PGs and the requirement for purification before and after these steps reduce the practicability of this approach due to the long processing time, significant side reactions, and extremely low yields, meaning millions of potentially therapeutic active targets still remain chemically inaccessible.104,1057.1 Pd-Mediated single disulfide bond formation
Recently, our group developed a method for accomplishing single disulfide bond formation between two Cys(Acm) in a one-pot process using NCL, Pd chemistry, and diethyldithiocarbamate (DTC) (Fig. 20a).106 The crucial role of using DTC over DTT in this reaction was attributed to its Pd scavenging property and not its reducing property, like for DTT, which cleaves the disulfide bond. The applicability of this method was demonstrated by the synthesis of oxytocin and the first total chemical synthesis of thioredoxin-1 (Trx-1) enzyme via NCL, desulfurization, and a one-pot disulfide bond formation.106 Nevertheless, under these conditions, all attempts to create more than one disulfide bond in a regioselective manner failed and resulted in significant reshuffling, probably due to the formation of a Pd–S intermediate that destabilizes the already existing disulfide bonds via a yet unknown pathway. | ||
| Fig. 20 Pd(II)-Mediated one-pot disulfide bond formation for the total (semi) synthesis of Trx-1 employing Cys (Acm) (a) and Cys (maleimide) (b). | ||
Similarly, the maleimide group was utilized as a PG of Cys residues for intein-based expressed protein fragment [Trx-1(2–93)-Mesna] to enable (EPL)/desulfurization and subsequent direct single disulfide bond formation to give the full sequence of Trx-1 (Fig. 20b).101 Here, PdCl2/MgCl2 assisted the removal of the maleimide groups and DTC was used to efficiently recover the final product.
7.2 Ultrafast regioselective formation of multiple disulfide bonds in one pot
Very recently, we reported a novel synthetic strategy for the ultrafast, high-yielding formation of multiple disulfide bonds in peptides and proteins.107 Analysis of the current approaches and their main limitations prompted us to design a synthetic pathway in which: (1) the total process will be completed as fast as possible to avoid reshuffling and side reactions on sensitive AAs (e.g., Met and Trp); (2) the solvent will desaturate the peptide/protein (preferably aqueous) to overcome kinetically trapped folded intermediates and/or aggregations; and (3) purifications/isolations during the steps will be avoided (one-pot manner) to increase the synthesis yield. To achieve this, we designed an approach based on a combination of the small molecule and extremely active disulfiram (DSF), UV-light at 350 nm, and Pd(II) under pH tuning for the chemo- and regioselective activation of multiple Cys residues.107 Applying these conditions enabled the efficient one-pot syntheses of representative examples (in only 5 min for two- and 13 min for three disulfide bonds) from various peptide/protein families (Fig. 21), tackling a decades old standing problem in peptide and protein chemistry.107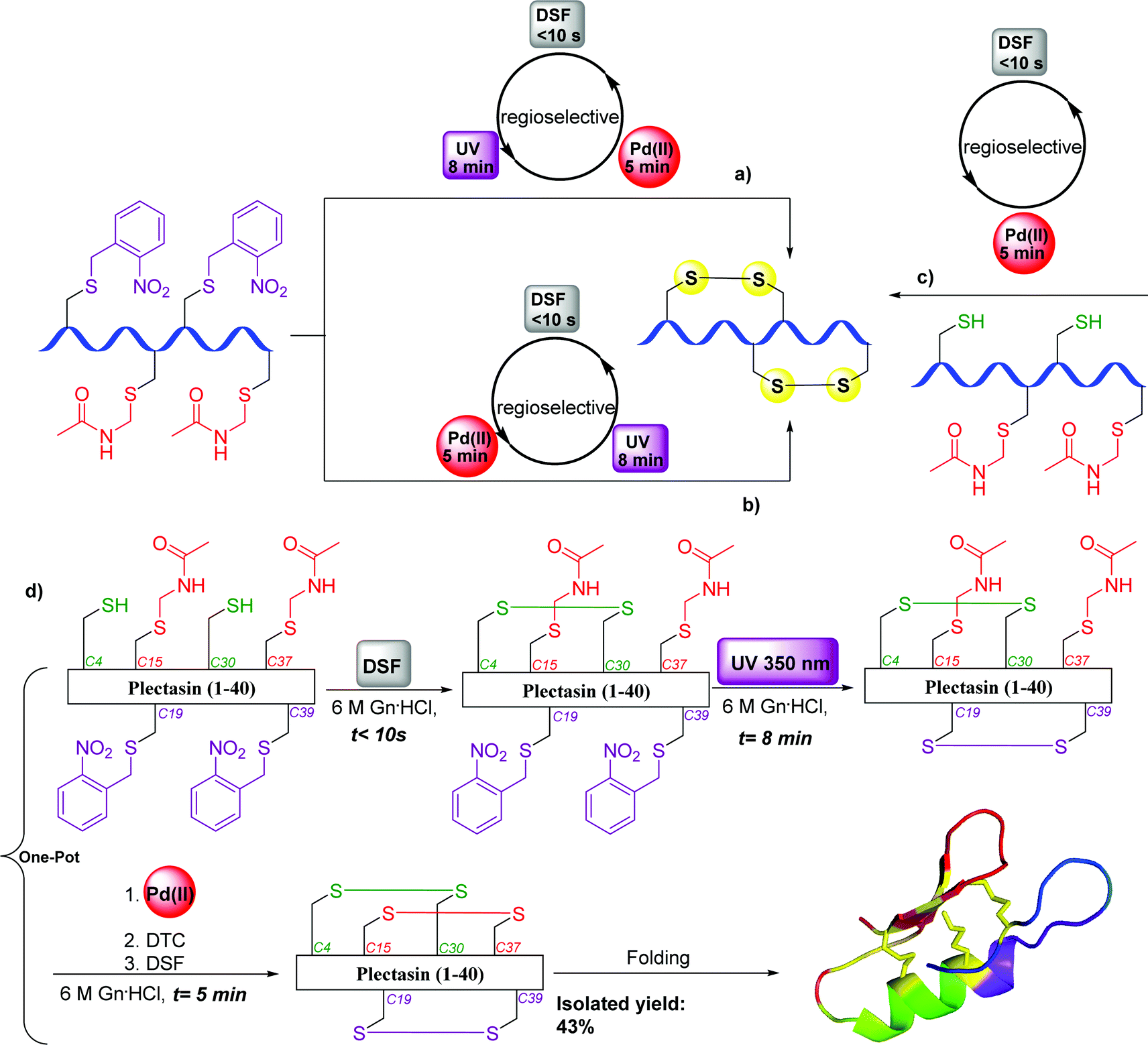 | ||
| Fig. 21 One-pot formation of disulfide bonds: (a) general scheme for the regioselective formation of two disulfide bonds via chemoselective UV-light irradiation in the presence of DSF followed by Pd(II) treatment in one pot; (b) regioselective formation of two disulfide bonds via chemoselective Pd(II) treatment in the presence of DSF followed by UV-light irradiation in one pot; (c) regioselective formation of two disulfide bonds via exposure to DSF followed by Pd(II) treatment in one pot; (d) regioselective synthesis of the antimicrobial plectasin peptide bearing three disulfide bonds. | ||
8. Synthesis of selenoprotein
The biological function of many Sec-containing proteins remains unclear or poorly studied.108 The incorporation of Sec into selenoproteins was achieved co-translationally by the suppression of the UGA stop codon.109,110 However, this requires unusual tRNA, a Sec insertion sequence element, a Sec-specific elongation factor, and other factors.111–113 Therefore it is highly difficult to obtain the desired amounts of wild-type selenoproteins by the recombinant expression method, hence chemical (semi)synthesis has emerged as an elegant approach to solve this issues.114The protection of Sec is highly necessary when aiming to perform the sequential ligations to enable selenoprotein synthesis. Recently, Metanis’ group developed selenazolidine (Sez) as an easily accessible N-terminal-protected form of Sec for the chemical synthesis of proteins.115 However, the generally used methoxylamine for the conversion of Sez-to-Sec requires a long reaction time similar to Thz-to-Cys conversion, and results in unwanted side products after reacting with the C-terminal thioester.115 To address these issues, Metanis’ group developed a Cu-mediated Sez deprotection for the synthesis of challenging proteins (Fig. 22a and Table 2).116 In this study, the group established the chemical synthesis of peptides and proteins using Cu(II)-mediated Sez deprotection, followed by NCL and deselenization, in a one-pot manner.116 The group also reported that among other metals the most efficient additive for Sez unmasking was Cu(II), which could complete the process within 30 min. The utility of this method was demonstrated in the syntheses of human thiosulfate:glutathione sulfurtransferase (TSTD1) and an analogue bearing Sec. In addition, this chemistry was extended for the preparation of cyclic peptides by sequential Sez deprotection using Cu and intramolecular cyclization in a one-pot manner (Fig. 22b).117 The usefulness of this method was also demonstrated by the synthesis of cyclotide Se-Kalata S, in which the Sez-linear peptide-MPAA was synthesized using Fmoc-SPPS followed by the deprotection of Sez by CuCl2.117
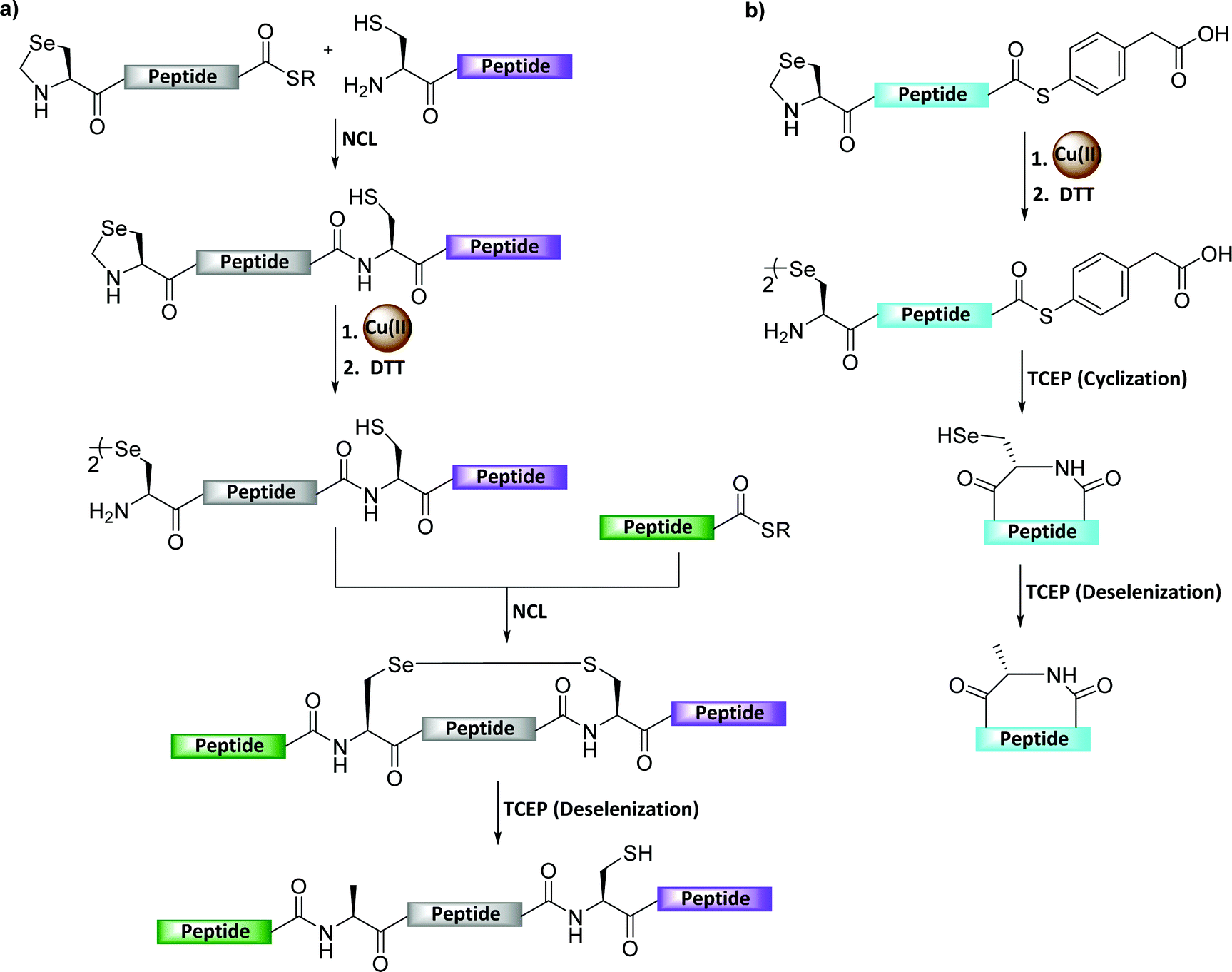 | ||
| Fig. 22 (a) Cu-Mediated Sez deprotection in the chemical synthesis of seleno-proteins. (b) Cu-mediated Sez deprotection in the chemical synthesis of cyclic peptides. | ||
In a closely related study by Otaka's group, they investigated the application of the Cu-mediated ring opening of the Thz to practical peptide synthesis. Their study revealed that the opening of Thz with CuSO4 in the presence of ascorbate proceeded within a few hours. In addition, they observed the opening of Sez under similar conditions.118 These tools have been applied for the chemical synthesis of 77-mer CXCL14 protein. In this study, the opening of Sez in a model peptide was also achieved with the use of a lower amount of Cu salt than for the Thz opening and therefore this process was applicable for one-pot/sequential reactions.118
9. Summary and outlook
In this review, we discussed the chemical synthesis of peptides and proteins assisted by transition metals as effective reagents for resolving various issues that arise with conventional methods during their syntheses. Using transition metals in peptide and protein synthesis enabled the reactions to be performed under mild conditions, with high selectivity and in a short time and as a one-pot process, and gave the products in high yields.With the increasing demand on the synthesis of complex peptides (e.g., with disulfide bonds) and proteins (ubiquitinated proteins),119 it became clear that the synthetic scheme requires PGs.4 On the other hand, these PGs must be removed under mild conditions, in a selective and orthogonal manner while being unreactive to other functional groups present in the sequence. In this regard, transition metals, especially Pd, chemistry offer the chance for the one-pot synthesis of complex protein targets and orthogonal cleavage of different PGs.69,99 This chemistry was also extended to the efficient formation of single disulfide bonds directly from the protected form of the Cys, e.g., Cys(Acm).106 Recently, Cu chemistry was also found to assist the removal of selenazolidine during the synthesis of selenoproteins.116,118
Pd chemistry played a crucial role in achieving the cleavage of temporally incorporated solubilizing tags, which were applied to aid the solubility of hydrophobic peptide fragments. Using this approach, several proteins with difficult sequences have been synthesized.74,87,88,99 In another direction, Au assisted also in the removal of backbone modifications of peptides and proteins that were applied to improve their synthesis.98 It is worth mentioning that transition metal chemistry has been extended to the cellular environment, where it helps to decage the temporal PGs or cleave the backbone elements of engineered peptides and proteins to aid studying the biochemical properties in the native environment.93,94 Despite the relative toxicity of some of the transition metals (e.g., Tl, Hg) being well established in various examples,120 the use of appropriate metal scavengers could afford highly purified and active proteins. For example, inductively coupled plasma (ICP) measurements for Pd traces after scavenging with DTT showed that the amount of Pd detected was below 0.07% of the protein.87,98,101,106 Moreover, metals such as Pd, Cu, and others have been shown to be fully compatible with the live cell environment.94,121–125
Despite possessing several advantages, many aspects remain to be addressed regarding the use of transition metals in peptide and protein chemistry. Can highly abundant and low-cost metals such as Fe, Ni be used instead of expensive or toxic metals? The newly evolved chemistry must be selective, must work under mild conditions, and ideally should work in catalytic amounts and with the possibility to work in the cellular environment. Such new inventions could further bring forward the field of the chemical synthesis of peptides and proteins and their modifications.
Future directions in this exciting research area include the employment of transition metal nanoparticles126 and metal-loaded water-compatible resin for peptide and protein synthesis,127 which have been proved to be excellent catalytic systems in modern organic synthesis. In addition, further studies should be performed in order to understand the mechanism of these transformations under aqueous media and in the presence of the various additives that are normally used in ligation chemistry, such as MPAA, TCEP, and guanidine. Such an understanding could have great implications in the design of new complexes or catalytic reactions for peptides and proteins as well as for other biomolecules (e.g., DNA). These developments might also inspire the scientific community to perform small molecule reactions in aqueous conditions or to use the newly discovered complexes in new chemical transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Brik holds The Jordan and Irene Tark Academic Chair. We would like to thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program for funding (grant agreement no. 831783). S. L. thanks the Technion for the Jacobs fellowship for excellence.References
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS.
- Y. Zhang, J. L. Ptacin, E. C. Fischer, H. R. Aerni, C. E. Caffaro, K. San Jose, A. W. Feldman, C. R. Turner and F. E. Romesberg, Nature, 2017, 551, 644–647 CAS.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS.
- S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CAS.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem., 2018, 2, 0122 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS.
- S. B. H. Kent, P. E. Dawson, T. W. Muir and I. Clark-Lewis, Science, 1994, 266, 776–779 CrossRef.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS.
- M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644–10655 CrossRef CAS.
- M. Jbara, Pure Appl. Chem., 2021 DOI:10.1515/pac-2020-0504.
- P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005–8013 CAS.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS.
- A. Brik, Adv. Synth. Catal., 2008, 350, 1661–1675 CrossRef CAS.
- E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 7710–7729 RSC.
- J. L. Mascarenas, P. Destito, C. Vidal and F. López, Chem. Eur. J., 2021 DOI:10.1002/chem.202003927.
- W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CAS.
- Y. Y. Yang, S. Ficht, A. Brik and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 7690–7701 CrossRef CAS.
- B. L. Pentelute and S. B. H. Kent, Org. Lett., 2007, 9, 687–690 CAS.
- J. Blake, Int. J. Pept. Protein Res., 1981, 17, 273–274 CrossRef CAS.
- S. Aimoto, N. Mizoguchi, H. Hojo and S. Yoshimura, Bull. Chem. Soc. Jpn., 1989, 62, 524–531 CrossRef CAS.
- H. Hojo, E. Haginoya, Y. Matsumoto, Y. Nakahara, K. Nabeshima, B. P. Toole and Y. Watanabe, Tetrahedron Lett., 2003, 44, 2961–2964 CrossRef CAS.
- G. Chen, Q. Wan, Z. Tan, C. Kan, Z. Hua, K. Ranganathan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 7383–7387 CrossRef CAS.
- P. E. Dawson, Isr. J. Chem., 2011, 51, 862–867 CrossRef CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS.
- K. J. Rosengren, R. J. Clark, N. L. Daly, U. Göransson, A. Jones and D. J. Craik, J. Am. Chem. Soc., 2003, 125, 12464–12474 CrossRef CAS.
- K. A. Wilson, M. Kalkum, J. Ottesen, J. Yuzenkova, B. T. Chait, R. Landick, T. Muir, K. Severinov and S. A. Darst, J. Am. Chem. Soc., 2003, 125, 12475–12483 CrossRef CAS.
- M. J. Bayro, J. Mukhopadhyay, G. V. T. Swapna, J. Y. Huang, L. C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382–12383 CrossRef CAS.
- A. Brik, Y. Y. Yang, S. Ficht and C. H. Wong, J. Am. Chem. Soc., 2006, 128, 5626–5627 CrossRef CAS.
- A. Brik, S. Ficht, Y. Y. Yang, C. S. Bennett and C. H. Wong, J. Am. Chem. Soc., 2006, 128, 15026–15033 CrossRef CAS.
- L. E. Canne, S. J. Bark and S. B. H. Kent, J. Am. Chem. Soc., 1996, 118, 5891–5896 CrossRef CAS.
- C. E. Weller, A. Dhall, F. Ding, E. Linares, S. D. Whedon, N. A. Senger, E. L. Tyson, J. D. Bagert, X. Li, O. Augusto and C. Chatterjee, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS.
- D. F. Veber, J. D. Milkowski, S. L. Varga, R. G. Denkewalter and R. Hirschmann, J. Am. Chem. Soc., 1972, 94, 5456–5461 CrossRef CAS.
- S. Liu, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2012, 51, 993–999 CrossRef CAS.
- M. T. Weinstock, M. T. Jacobsen and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11679–11684 CrossRef CAS.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS.
- S. K. Mong, A. A. Vinogradov, M. D. Simon and B. L. Pentelute, ChemBioChem, 2014, 15, 721–733 CrossRef CAS.
- Y. David, M. Vila-Perelló, S. Verma and T. W. Muir, Nat. Chem., 2015, 7, 394–402 CrossRef CAS.
- Q. Zhang, E. V. Johnston, J. H. Shieh, M. A. S. Moore and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2885–2890 CrossRef CAS.
- N. Fuji, A. Otaka, S. Funakoshi, K. Bessho, T. Watanabe, K. Akaji and H. Yajima, Chem. Pharm. Bull., 1987, 35, 2339–2347 CrossRef.
- N. Fujii, A. Otaka, T. Watanabe, A. Okamachi, H. Tamamura, H. Yajima, Y. Inagaki, M. Nomizu and K. Asano, J. Chem. Soc., Chem. Commun., 1989, 283–284 RSC.
- H. Lamthanh, C. Roumestand, C. Deprun and A. Ménez, Int. J. Pept. Protein Res., 1993, 41, 85–95 CrossRef CAS.
- T. Kawakami, S. Yoshimura and S. Aimoto, Tetrahedron Lett., 1998, 39, 7901–7904 CrossRef CAS.
- S. B. H. Kent, J. Am. Chem. Soc., 2004, 126, 1377–1383 CrossRef.
- F. K. Deng, L. Zhang, Y. T. Wang, O. Schneewind and S. B. H. Kent, Angew. Chem., Int. Ed., 2014, 53, 4662–4666 CrossRef CAS.
- I. Sakamoto, K. Tezuka, K. Fukae, K. Ishii, K. Taduru, M. Maeda, M. Ouchi, K. Yoshida, Y. Nambu, J. Igarashi, N. Hayashi, T. Tsuji and Y. Kajihara, J. Am. Chem. Soc., 2012, 134, 5428–5431 CrossRef CAS.
- P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS.
- P. Siman, S. V. Karthikeyan, M. Nikolov, W. Fischle and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 8059–8063 CrossRef CAS.
- M. Hejjaoui, M. Haj-Yahya, K. S. A. Kumar, A. Brik and H. A. Lashuel, Angew. Chem., Int. Ed., 2011, 50, 405–409 CrossRef CAS.
- J. Li, Y. Li, L. Liu, Y. Li, Q. He and H. Li, Org. Biomol. Chem., 2014, 12, 5435–5441 RSC.
- Y. K. M. Yoshida, T. Tatsumi, Y. Fujiwara, S. Iinuma, T. Kimura and K. Akaji, Chem. Pharm. Bull., 1990, 38, 1551–1557 CrossRef.
- O. Nishimura, C. Kitada and M. Fujino, Chem. Pharm. Bull., 1978, 26, 1576–1585 CrossRef CAS.
- C. M. Stevens and R. Watanabe, J. Am. Chem. Soc., 1950, 72, 725–727 CrossRef CAS.
- H. Kunz and C. Unverzagt, Angew. Chem., Int. Ed. Engl., 1984, 23, 436–437 CrossRef.
- G. B. F. Giube and O. Dangles, Tetrahedron Lett., 1986, 27, 2365–2368 CrossRef.
- J. Alsina, F. Rabanal, E. Giralt and F. Albericio, Tetrahedron Lett., 1994, 35, 9633–9636 CrossRef CAS.
- P. Grieco, P. M. Gitu and V. J. Hruby, J. Pept. Res., 2001, 57, 250–256 CrossRef CAS.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS.
- D. Bang and S. B. H. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS.
- M. Seenaiah, M. Jbara, S. M. Mali and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 12374–12378 CrossRef CAS.
- E. C. B. Johnson, E. Malito, Y. Shen, D. Rich, W. J. Tang and S. B. H. Kent, J. Am. Chem. Soc., 2007, 129, 11480–11490 CrossRef CAS.
- C. Piontek, D. Varón Silva, C. Heinlein, C. Pöhner, S. Mezzato, P. Ring, A. Martin, F. X. Schmid and C. Unverzagt, Angew. Chem., Int. Ed., 2009, 48, 1941–1945 CrossRef CAS.
- G.-M. Fang, J.-X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS.
- S. K. Maity, M. Jbara, G. Mann, G. Kamnesky and A. Brik, Nat. Protoc., 2017, 12, 2293–2322 CrossRef CAS.
- N. Kamo, G. Hayashi and A. Okamoto, Angew. Chem., Int. Ed., 2018, 57, 16533–16537 CrossRef CAS.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS.
- S. Li, Y. Lin, J. Cao and S. Zhang, J. Org. Chem., 2007, 72, 4067–4072 CrossRef CAS.
- M. Jbara, N. Guttmann-Raviv, S. K. Maity, N. Ayoub and A. Brik, Bioorg. Med. Chem., 2017, 25, 4966–4970 CrossRef CAS.
- S. Bondalapati, E. Eid, S. M. Mali, C. Wolberger and A. Brik, Chem. Sci., 2017, 8, 4027–4034 RSC.
- J. Liu, F. Zheng, R. Cheng, S. Li, S. Rozovsky, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 8807–8816 CrossRef CAS.
- D. Kobayashi, N. Naruse, M. Denda, A. Shigenaga and A. Otaka, Org. Biomol. Chem., 2020, 18, 8638–8645 RSC.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- M. Villain, H. Gaertner and P. Botti, Eur. J. Org. Chem., 2003, 3267–3272 CrossRef CAS.
- B. Dang, T. Kubota, K. Mandal, F. Bezanilla and S. B. H. Kent, J. Am. Chem. Soc., 2013, 135, 11911–11919 CrossRef CAS.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. Bin Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS.
- Y. Asahina, T. Kawakami and H. Hojo, Chem. Commun., 2017, 53, 2114–2117 RSC.
- M. Jbara, E. Eid and A. Brik, Org. Biomol. Chem., 2018, 16, 4061–4064 RSC.
- N. Kamo, G. Hayashi and A. Okamoto, Chem. Commun., 2018, 54, 4337–4340 RSC.
- T. Kobori, S. Iwamoto, K. Takeyasu and T. Ohtani, Biopolymers, 2007, 85, 392–406 CrossRef.
- J. Bin, Li, S. Tang, J. S. Zheng, C. L. Tian and L. Liu, Acc. Chem. Res., 2017, 50, 1143–1153 CrossRef.
- S. K. Maity, G. Mann, M. Jbara, S. Laps, G. Kamnesky and A. Brik, Org. Lett., 2016, 18, 3026–3029 CrossRef CAS.
- E. Eid, G. N. Boross, H. Sun, M. Msallam, S. K. Singh and A. Brik, Bioconjugate Chem., 2020, 31, 889–894 CrossRef CAS.
- C. H. Kim, J. Y. Axup and P. G. Schultz, Curr. Opin. Chem. Biol., 2013, 17, 412–419 CrossRef CAS.
- J. W. Chin, Annu. Rev. Biochem., 2014, 83, 379–408 CrossRef CAS.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS.
- M. Jbara, S. Laps, S. K. Maity and A. Brik, Chem. – Eur. J., 2016, 22, 14851–14855 CrossRef.
- G. Mann, G. Satish, R. Meledin, G. B. Vamisetti and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 13540–13549 CrossRef CAS.
- A. Isidro-Llobet, X. Just-Baringo, M. Álvarez and F. Albericio, Biopolymers, 2008, 90, 444–449 CrossRef CAS.
- J. S. Zheng, Y. He, C. Zuo, X. Y. Cai, S. Tang, Z. A. Wang, L. H. Zhang, C. L. Tian and L. Liu, J. Am. Chem. Soc., 2016, 138, 3553–3561 CrossRef CAS.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- M. Jbara, E. Eid and A. Brik, J. Am. Chem. Soc., 2020, 142, 8203–8210 CrossRef CAS.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 3154 CrossRef.
- N. Kamo, G. Hayashi and A. Okamoto, Org. Lett., 2019, 21, 8378–8382 CrossRef CAS.
- G. B. Vamisetti, G. Satish, S. Prasad, G. Mann, M. Glickman and A. Brik, J. Am. Chem. Soc., 2020, 142, 19558–19569 CrossRef CAS.
- M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS.
- S. J. De Veer, M. W. Kan and D. J. Craik, Chem. Rev., 2019, 119, 12375–12421 CrossRef CAS.
- A. H. Jin, M. Muttenthaler, S. Dutertre, S. W. A. Himaya, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Conotoxins: Chemistry and Biology, American Chemical Society, 2019, vol. 119 Search PubMed.
- S. Laps, H. Sun, G. Kamnesky and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 5729–5733 CrossRef CAS.
- S. Laps, F. Atamleh, G. Kamnesky, H. Sun and A. Brik, ChemRxiv, 2020 DOI:10.26434/chemrxiv.13065278.v1.
- R. Mousa, R. Notis Dardashti and N. Metanis, Angew. Chem., Int. Ed., 2017, 56, 15818–15827 CrossRef CAS.
- L. Flohé, Biochim. Biophys. Acta, 2009, 1790, 1389–1403 CrossRef.
- S. Yoshizawa and A. Böck, Biochim. Biophys. Acta, 2009, 1790, 1404–1414 CrossRef CAS.
- S. Chiba, Y. Itoh, S. ichi Sekine and S. Yokoyama, Mol. Cell, 2010, 39, 410–420 CrossRef CAS.
- S. Y. Y. Itoh, M. J. Brocker, S. Sekine, G. Hammond, S. Suetsugu and D. Söil, Science, 2013, 340, 75–79 CrossRef.
- K. Caban and P. R. Copeland, Selenium, 2012, 5, 61–72 Search PubMed.
- L. Dery, P. S. Reddy, S. Dery, R. Mousa, O. Ktorza, A. Talhami and N. Metanis, Chem. Sci., 2017, 8, 1922–1926 RSC.
- P. S. Reddy, S. Dery and N. Metanis, Angew. Chem., Int. Ed., 2016, 55, 992–995 CrossRef CAS.
- Z. Zhao and N. Metanis, Angew. Chem., Int. Ed., 2019, 58, 14610–14614 CrossRef CAS.
- Z. Zhao and N. Metanis, J. Org. Chem., 2020, 85, 1731–1739 CrossRef CAS.
- N. Naruse, D. Kobayashi, K. Ohkawachi, A. Shigenaga and A. Otaka, J. Org. Chem., 2020, 85, 1425–1433 CrossRef CAS.
- H. Sun and A. Brik, Acc. Chem. Res., 2019, 52, 3361–3371 CrossRef CAS.
- R. R. Crichton, RSC Metallobiol., 2017, 1–23 CAS.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS.
- S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Chem. Sci., 2017, 8, 2107–2114 RSC.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS.
- B. Van Vaerenbergh, J. Lauwaert, J. W. Thybaut, P. Vermeir and J. De Clercq, Chem. Eng. J., 2019, 374, 576–588 CrossRef CAS.
- P. Mastrorilli, M. M. Dell’anna, A. Rizzuti, M. Mali, M. Zapparoli and C. Leonelli, Molecules, 2015, 20, 18661–18684 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |