
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine
Mauro
Sousa de Almeida†
 a,
Eva
Susnik†
a,
Barbara
Drasler
a,
Patricia
Taladriz-Blanco
a,
Alke
Petri-Fink
ab and
Barbara
Rothen-Rutishauser
*a
a,
Eva
Susnik†
a,
Barbara
Drasler
a,
Patricia
Taladriz-Blanco
a,
Alke
Petri-Fink
ab and
Barbara
Rothen-Rutishauser
*a
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: barbara.rothen@unifr.ch
bDepartment of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland
First published on 5th March 2021
Abstract
Nanoparticles (NPs) have attracted considerable attention in various fields, such as cosmetics, the food industry, material design, and nanomedicine. In particular, the fast-moving field of nanomedicine takes advantage of features of NPs for the detection and treatment of different types of cancer, fibrosis, inflammation, arthritis as well as neurodegenerative and gastrointestinal diseases. To this end, a detailed understanding of the NP uptake mechanisms by cells and intracellular localization is essential for safe and efficient therapeutic applications. In the first part of this review, we describe the several endocytic pathways involved in the internalization of NPs and we discuss the impact of the physicochemical properties of NPs on this process. In addition, the potential challenges of using various inhibitors, endocytic markers and genetic approaches to study endocytosis are addressed along with the principal (semi) quantification methods of NP uptake. The second part focuses on synthetic and bio-inspired substances, which can stimulate or decrease the cellular uptake of NPs. This approach could be interesting in nanomedicine where a high accumulation of drugs in the target cells is desirable and clearance by immune cells is to be avoided. This review contributes to an improved understanding of NP endocytic pathways and reveals potential substances, which can be used in nanomedicine to improve NP delivery.
 Mauro Sousa de Almeida | Mauro Sousa de Almeida obtained his Master's degree in medicine technologies from the Faculty of Pharmacy at the University of Coimbra (Portugal) in 2017. Currently he is a PhD student at the Adolphe Merkle Institute – Fribourg (Switzerland) under the supervision of Prof. Rothen-Rutishauser and Prof. Petri-Fink. His research interests focus on the cellular uptake mechanisms of nanoparticles. |
 Eva Susnik | Eva Susnik received her Master's degree in molecular and functional biology at the University of Ljubljana (Slovenia) in 2016. She then moved to Fribourg to start a PhD at the Adolphe Merkle Institute under supervision of Prof. Rothen-Rutishauser and Prof. Petri-Fink, where she is studying mechanisms of cellular endocytosis to enhance nanoparticle uptake. |
 Barbara Drasler | Barbara Drasler studied biology at the University of Ljubljana in Slovenia. She continued her studies there in the Nanobiology and Nanotoxicology research group and obtained a PhD in nanoscience and nanobiology in 2014. Barbara joined the BioNanomaterials research group at the Adolphe Merkle Institute as a post-doctoral research fellow in 2016. Her main research interest has been directed towards the use of nanomaterials in medical field, particularly regarding their inherent safety when intentionally used within such applications, as well as concerning the occupational hazard of nanomaterials. |
 Patricia Taladriz-Blanco | Patricia Taladriz-Blanco received her PhD degree in chemistry from the University of Vigo, Spain in 2012. She worked as a postdoctoral fellow at the University of Campinas (Brazil) and the University of Cambridge (UK) before joining the BioNanomaterials group at the Adolphe Merkle Institute in Switzerland as a senior postdoctoral researcher. Her research interests are synthesis, surface modifications, self-assembly and characterization of nanoparticles, thermoplasmonics and microfluidics. |
 Alke Petri-Fink | Prof. Alke Petri-Fink received her PhD in chemistry from the University of Ulm, Germany in 1999. After a post-doctoral stay at the University of Gainesville, Florida, she joined the Institute of Materials Science at the École Polytechnique Fédérale de Lausanne (EPFL), first as a post-doctoral researcher, then as a senior scientist. She became an Associate Swiss National Science Foundation Professor in the Department of Chemistry at the University of Fribourg in 2009, and a Full Professor in 2011 at the Adolphe Merkle Institute, Switzerland. Her research focuses on inorganic nanoparticles, their synthesis, surfaces, and interactions with biological cells. |
 Barbara Rothen-Rutishauser | Prof. Dr Barbara Rothen-Rutishauser received her PhD in 1996 in cell biology from the Swiss Federal Institute of Technology in Zurich (ETHZ). She worked as postdoc and group leader at ETHZ and University of Bern, Switzerland. She is an expert in the field of cell–nanoparticle interactions, with a special focus on human 3D tissue models. Since 2011 she is the Chair in BioNanomaterials at the Adolphe Merkle Institute, University of Fribourg, Switzerland; the position is shared equally with Prof. Alke Fink. Prof. Rothen-Rutishauser has published more than 290 peer-reviewed papers and is an associate editor of “Particle and Fibre Toxicology”. |
1. Introduction
Nanotechnology is a multidisciplinary field comprising, among others, chemistry, physics, biology, and medicine, which focuses on the design, production, and application of nanosized systems (e.g., devices, paints, food additives, and drug delivery systems).1 First discussions about nanotechnology date back to 1959, when Richard Feynman mentioned the opportunity of controlling atom by atom the production of miniaturized machines.2,3The term “nanotechnology” was introduced only a couple of years later, in 1974, by Norio Taniguchi.4 The possibility to manipulate materials at nanoscale provided a boost in the development of novel materials with different performance in comparison to the bulk material.5 To date, there are different definitions of nanomaterials, depending on the application field.6,7
The International Organization for Standardization (ISO)8 defines a nanomaterial as a “material with any external dimension in the nanoscale or having an internal structure or surface structure in the nanoscale (1–100 nm)”. Similarly, in 2011, the European Commission9 adopted a definition for a nanomaterial: “A natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1–100 nm”. The United States Food and Drug administration (US FDA)6 states that nanomaterials are “materials up to one micron if these ones exhibit properties or phenomena that are attributable to its dimensions”. In this review, a size range between 1–1000 nm is considered for nanomaterials and nanoparticles (NPs). The NP dimensions mentioned throughout the manuscript are expressed as a diameter, unless otherwise stated.
Pharmaceutical, medical, and device manufacturing industries are developing new nanomaterials for biomedical applications, such as drug delivery systems, biosensors and medical nanodevices.9,10 For drug delivery applications, cellular uptake, intracellular fate, and accumulation of NPs loaded with therapeutics play an important role in successful disease treatment.11 Several factors should be considered in the development of safe and efficient NP systems for medical purposes (i.e., nanomedicine): (i) NPs physico-chemical properties (e.g., size, shape, surface charge, and stiffness); (ii) colloidal stability; (iii) degradation rate, i.e. solubility; (iv) biocompatibility; (v) bioaccumulation; (vi) route of administration (e.g., intravenous, oral, inhalation and dermal) and (vii) target cell/tissue. The principal process used by cells to internalize NPs is endocytosis.12 Endocytosis is an important mechanism for cellular uptake of nutrients, regulation of cell surface receptors, control of cell polarity, motility, and signaling cascades.12,13
A thorough understanding of endocytosis is essential to optimize the safe-by-design, cellular targeting and uptake of NPs. It is still challenging to generalize the current findings, due to the variation on the endocytic mechanisms dependent on cell types and for different NPs.14–17 In addition, when NPs interact with physiological fluids such as mucus, lymph fluid, or blood, they can interact with different biomolecules including opsonins that promote cellular recognition and clearance by the mononuclear phagocyte system (MPS).18 Also the presence of efflux pumps, overexpression of specific transporters on the cell membrane, as well as mitosis can reduce NP accumulation in the target cells.19,20 Consequently, a very low number of NPs reach the target cells and might not be sufficient to treat the disease.21 For this reason, it becomes important to enhance NP-based targeted delivery and, at the same time, avoid internalization by MPS if this system is not the targeted one. Various stimuli, such as inflammatory cytokines,22 multiple NP co-exposure,23,24 or functionalization with ligands25–27 were described to increase NP uptake in target cells. Several other substances have also been found to decrease NP internalization.28,29 This effect is desired to avoid NP accumulation in non-target cells or organs, where they could cause unintended acute or chronic toxicity.21
This review contributes to the current understanding of NP cellular uptake and gives an overview about molecules, which can enhance or decrease cellular internalization of NPs. A description of the different endocytic mechanisms is included together with the approaches for NP quantification.
2. Cellular uptake mechanisms of NPs
NPs may enter the body via inhalation, oral ingestion, dermal and ocular penetration, and injection (intravenous, intramuscular and subcutaneous).21,30 Upon contact with physiological fluids or physical barriers, via different administration routes, NPs may undergo a process of aggregation and/or dissolution.31,32 In addition, the different constitution of physiological fluids (e.g., proteins, lipids and electrolytes) including blood, respiratory and gastrointestinal mucus, and tear fluid might affect NP properties and thus cell–NP interactions.33–35Inhalation has been used for the administration of nanomaterials to treat lung diseases or for systemic delivery.36 The lung possesses a large internal surface area (around 150 m2) and NPs can deposit in alveoli as opposed to their bulk counterparts, which is an advantage for pulmonary delivery.37,38 Furthermore, they might reach the peripheral gas exchange region and pass the air-blood tissue barrier to enter systemic circulation.39 Only a small amount of inhaled NPs reach systemic circulation because a large fraction of NPs is cleared by macrophages and the translocation of NPs inversely depends on particle size.40,41 In intravenous injection, NPs tend to penetrate and accumulate within the leaky tumor vasculature, which is designated as an enhanced permeation and retention (EPR) effect.30 A large retention of NPs by the reticuloendothelial system (RES), such as the liver, kidneys and spleen is observed when NPs are administered intravenously.42 The skin is the largest human organ, acting as an effective first barrier against external factors, such as pathogens.43 In dermal applications NPs usually remain on the skin surface, however for transdermal applications NPs may be able to penetrate into the stratum corneum via intercellular pathways, e.g. hair follicles or glandular tissue, or permeate the whole stratum corneum into deeper skin layers.44,45 The topical ocular drug delivery, usually based on eye drops, is used to treat ocular disorders.46 This approach usually requires the interaction of the drug with the sclera and the different tissues of the anterior segment (e.g., cornea, iris and conjunctiva).46 NPs have been studied as delivery systems for ophthalmic drugs and have revealed increased corneal permeability.47 The drug bioavailability in the target tissue may be affected by precorneal factors and anatomical barriers such as nasolacrimal drainage, tear turnover, and blinking.46
Following this first interplay with human chemical and physical barriers, NPs come in contact with the outer cell membrane. Herein, they may interact with the components of the outer plasma membrane and be subsequently internalized.48 The main cellular process of NP internalization is endocytosis and involves the invagination/ruffling of the cell membrane followed by the formation of intracellular/endocytic vesicles (Fig. 1).17 Depending on the cell–NP interaction, different signaling cascades can occur leading to various structural changes at the cell surface.49 These processes, together with the vast diversity of molecules (e.g., surface receptors, membrane lipids, cargo and adaptor proteins) needed for efficient endocytic trafficking of NPs, enable endocytosis to be categorized into two main types: phagocytosis and pinocytosis.50,51 Phagocytosis encompasses the uptake of large particles (≥0.5 μm) and is only performed by specialized cells, as described below (Section 2.1).52 Pinocytosis is associated with fluid-phase uptake and, based on the majority of the literature, includes macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis and clathrin/caveolae-independent endocytosis.50,53 These mechanisms occur in almost every eukaryotic cell.13
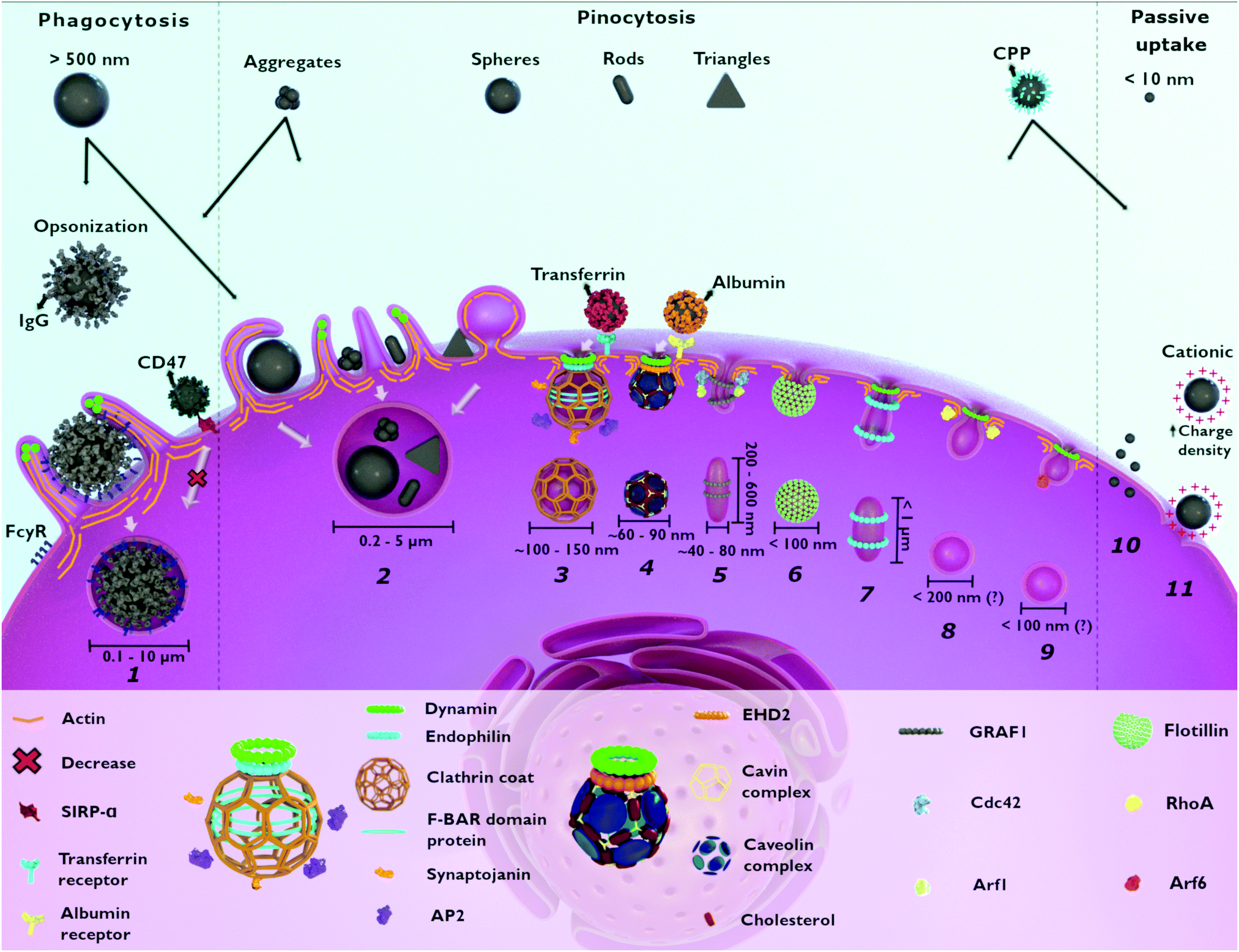 | ||
| Fig. 1 Possible entry mechanisms for nanoparticles (NPs). Large NPs (>500 nm) and aggregates enter the cell through phagocytosis (1) and macropinocytosis (2). NP opsonization via IgG leads to cellular recognition (FcγR) in phagocytes. Pinocytosis includes different mechanisms: macropinocytosis (2), clathrin-mediated endocytosis (3), caveolae-dependent endocytosis (4), CLIC–GEEC (5), flotillin-assisted endocytosis (6), fast-endophilin-mediated endocytosis (7), RhoA-dependent endocytosis (8) and Arf-6-associated endocytosis (9). As a non-selective endocytic process, macropinocytosis (2) is associated with the internalization of different NPs. Smaller NPs (<10 nm) and cationic NPs, with high charge density, enter the cell via direct penetration (10) and pore formation (11), respectively. NPs surface functionalization with different molecules has an impact on cellular uptake. NPs functionalized with transferrin and albumin are taken up through clathrin-mediated (3) and caveolae-dependent endocytosis (4), respectively. NPs functionalized with CPP can be internalized passively (10 and 11) as well as via other endocytic pathways. The functionalization of NPs with CD47 reduces phagocytosis (1). Flotillin-assisted endocytosis (6) and Arf-6-associated endocytosis (9) can occur both, in the presence or absence of dynamin. CPP: cell penetrating peptide. IgG: immunoglobulin G. CD47: cluster of differentiation 47. FcγR: Fc gamma receptor. SIRPα: signal regulatory protein alpha. AP2: adaptor protein 2. EHD2: Eps15-homology domain containing protein 2. GRAF1: GTPase regulator associated with focal adhesion kinase-1. CDC42: cell division cycle 42. Arf1: ADP-ribosylation factor 1. RhoA: Ras homolog family member A. Arf6: ADP-ribosylation factor 6. (?) – vesicle size still unclear. | ||
In this section, the mechanisms and the influence of the physicochemical properties of NPs on the subsequent uptake will be discussed. A common feature in endocytosis is the localization of NPs into endocytic vesicles after internalization. In addition, the occurrence of other internalization routes that do not involve vesicle formation, such as passive diffusion and pore formation by cell membrane disruption, will be discussed as well.11
2.1 Phagocytosis
Phagocytosis is an endocytic route carried out by professional phagocytes i.e., macrophages, monocytes, dendritic cells, osteoclasts, eosinophils and neutrophils where foreign bodies such as bacteria or fungi and cell debris are ingested and eliminated.54 In addition, it was shown that nonprofessional phagocytes such as fibroblasts, epithelial and endothelial cells, also possess phagocytic activity, but only to a limited extent, not being able to completely eliminate microorganisms.55,56Phagocytosis is usually associated with the uptake of large particles. In contrast, several studies also reported the phagocytosis of nanometer-sized particles, such as gold,57,58 silver,59 and polymeric NPs.60 It is hypothesized that this occurs mainly for aggregated NPs after an opsonization process (Fig. 1).61,62 Once NPs are dispersed in a physiological fluid such as mucosal, lymph fluid, or blood, they will interact with different proteins that will adsorb at their surface, creating the so-called “protein corona”.34,63 The immunoglobulins and complement proteins present in the protein corona, namely as opsonins, are recognized by opsonic receptors, i.e., Fc receptors (FcR) and complement receptors leading to NP internalization.54,64 Nevertheless, phagocytes also express non-opsonic receptors (e.g., mannose and scavenger receptors) that are able to interact directly with the molecular groups on the NP surface.64,389 For efficient recognition of the NPs, cooperation between multiple phagocyte receptors can occur depending on the density of both the molecules on the NPs surface and the receptors at the cellular membrane.56 The relative mobility of the receptors on the cell membrane and their affinity for the NPs influence phagocytosis efficiency.65
The protein composition and conformation on the NPs surface influence the interaction and recognition by the phagocyte surface receptors.33 The receptors involved in this recognition dictate the subsequent signaling cascade and may potentially initiate inflammatory events (e.g., FcR).62 Silica (SiO2) NPs of 50 and 100 nm triggered inflammation in THP-1 macrophage-like cells by the activation of the scavenger receptor A1, while 10 and 1000 nm did not.66 In a different context, a pre-coating of SiO2 NPs and single-carbon nanotubes coated with the surfactant Pluronic F127 reduced the adsorption of serum proteins and inhibited the anti-inflammatory effect on murine macrophages (RAW 264.7 cells).67 Therefore, the proteins adsorbed on the NP surface and the following receptor activation affect both the uptake of NPs and cell reactivity (i.e. inflammatory response).
Receptor-mediated phagocytosis is initiated upon cell–NP interaction, which leads to a signaling cascade resulting in the polymerization of actin filaments, membrane cup-shaped extensions and subsequent internalization of NPs.56 The formed phagosome containing NPs matures by a series of changes in its membrane composition and content. At the end it fuses with a lysosome, an acidic vesicle, that, depending on the NP material, is able to digest the ingested NPs.68
2.2 Macropinocytosis
Similar to phagocytosis, macropinocytosis is an actin-dependent process involved in the engulfment of fluids and micron-sized particles.69 This mechanism is a non-selective process where plasma membrane ruffles engulf high amounts of an external fluid, including particles and dissolved molecules, into large vesicles called macropinosomes (0.2–5 μm).70 Macropinocytosis allows the internalization of larger macromolecules by cells that do not possess phagocytic activity, which would not be possible through other endocytic mechanisms such as clathrin- and caveolae-mediated endocytosis.69 Depending on the cell type, macropinocytosis can occur in a constitutive or inducible way.71 In response to the stimulation by growth factors (e.g., epidermal and platelet-derived growth factors) and other molecules, actin-rich extensions of the plasma membrane, referred to as ruffles, can retreat back into the cell membrane or curve into circular ruffles that undergo membrane fission to form macropinosomes.72 This process, depending on the activation stimulus, may involve many molecules important to actin polymerization, cytoskeleton organization, macropinosome formation and closure. Here are encompassed several small GTPases from the Ras superfamily, kinases (p21-activated kinase 1 and protein kinase C) and lipids (phosphoinositides and diacylglycerol).73 More details about the molecular machinery involved in this process are available in other reviews.71,72,74The uptake of NPs through macropinocytosis occurs in a non-specific way, meaning that NPs are internalized due to the close contact with the plasma membrane where the ruffle formation starts.75 Therefore, the uptake through this mechanism is usually not dependent on specific NP properties such as size or shape. It is possible that adsorbed proteins on the NP surface or NPs functionalized with specific molecules are able to stimulate macropinocytosis, as will be discussed later in this review.
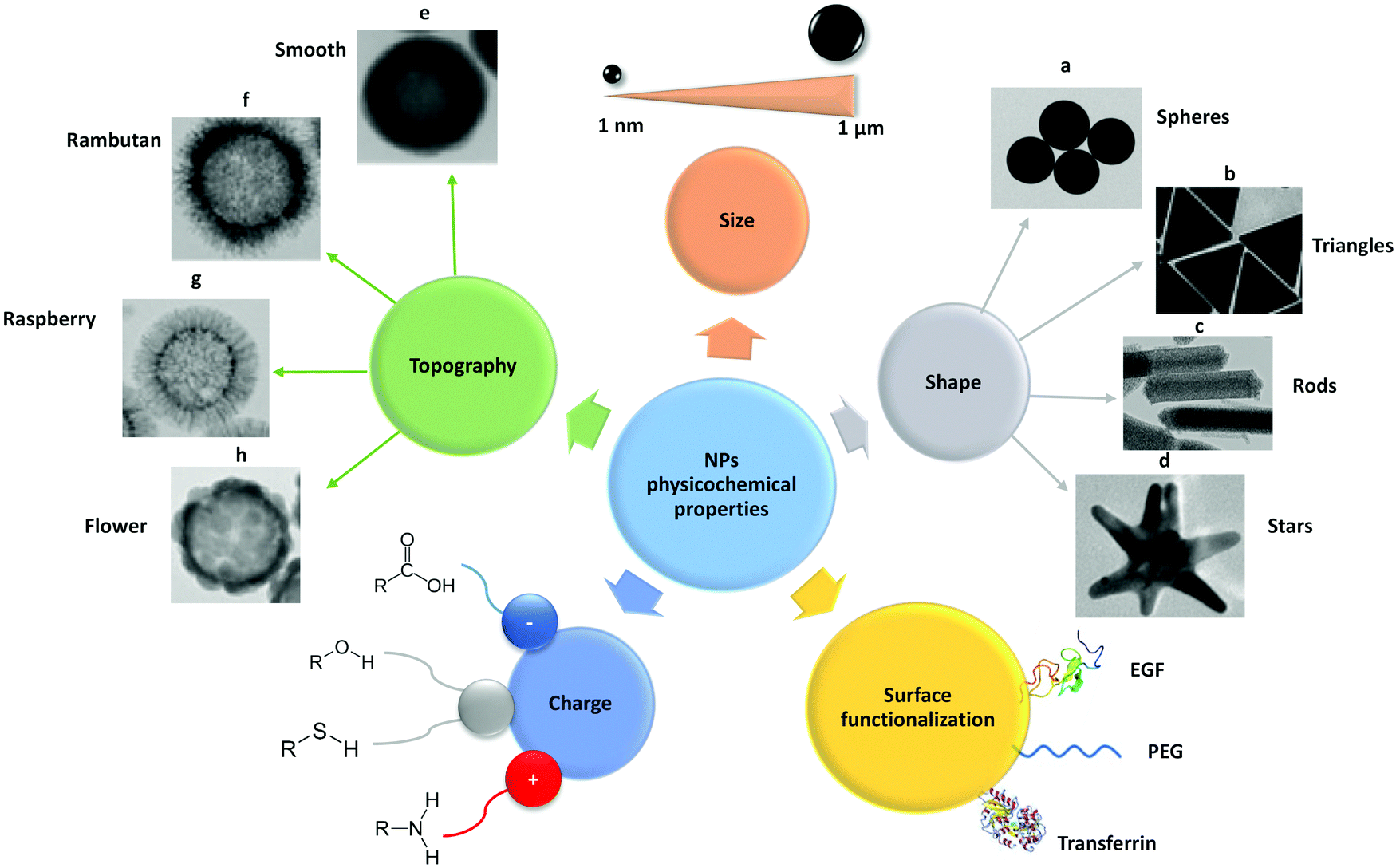 | ||
| Fig. 2 Schematic representation of the main physico-chemical properties of NPs, such as size, shape, surface functionalization, charge and topography. These properties need to be considered when employing NPs as carriers for drug delivery in nanomedicine. EGF – epidermal growth factor; PEG – polyethylene glycol. Adapted with permission from (a) ref. 421, (b) ref. 422, (c) ref. 423, (d) ref. 424 and (e–h) ref. 154. Copyright (a) Susnik et al. 2020, (b) Kuttner et al. 2018, (c) Madathiparambil Visalakshan et al. 2020, (d) Liu et al. 2015 and (e–h) Singh et al. 2019. | ||
2.3 Clathrin-mediated endocytosis (CME)
CME is the most studied endocytic mechanism and all the molecular details and cargo specificity are well described in the literature.50,76,77 It is considered as one of the most important mechanisms for NP uptake17 but is also related to many other functions such as regulation of expression of surface proteins and uptake of nutrients including iron via the transferrin receptor.11 The designation “clathrin-mediated endocytosis” is associated with the most abundant protein in the process, the triskelion clathrin that assembles in hexagons and pentagons to form a lattice-like coat around the endocytic vesicles.78 The process involving the formation of clathrin-coated vesicles (CCVs) can be divided into five different steps: initiation, cargo selection, coat assembly, scission, and un-coating.76 All these processes involve a set of proteins that localize intracellularly and are recruited to the plasma membrane in a coordinated manner.77 The protein machinery includes: (i) F-BAR domain-containing proteins important to initialize membrane curvature; (ii) clathrin adaptor proteins such as AP2, synaptojanin, and other accessory proteins – AP180, epsin and SNX9 – associated with cargo recognition, coat assembly and stabilization of membrane curvature; (iii) GTPase dynamin and endophilin for vesicle scission.76,79 For several years this mechanism was also designated as receptor-mediated endocytosis (RME), because it was believed that only specific interactions between the cargo and the receptor could lead to CME.11,17 Internalization of non-specific cargoes can also occur via non-specific interactions, such as hydrophobic and/or electrostatic interactions.11 Transferrin and epidermal growth factor (EGF) are ligands that bind to their specific receptors leading to RME via clathrin.80The CCV can have different sizes in different cell types depending on the cargo.13,77 During chemical synapses where fast processes are required, CCVs are usually smaller (∼50 nm), compared to CCVs in epithelial cells with a size of ∼120–150 nm.13 Nevertheless, the maximum reported size for CCVs was 200 nm.76 Therefore, the size range of CCVs and the occurrence of this process in all eukaryotic cells are the reason why CME is one of the principal mechanisms for NP uptake.
Several studies investigated the uptake of NPs in different cell types via CME. The route of internalization of 40 nm polystyrene (PS) NPs was studied in different cell types, cervical epithelial (HeLa), lung epithelial (A549), brain astrocytoma and macrophage (J774A.1), revealing the involvement of CME in all cells in combination with other mechanisms.60,81 In addition, several other NPs have been shown to be taken up by cells through CME, including fluorescein isothiocyanate (FITC)-incorporated silica-coated core-shell superparamagnetic iron oxide (SPION@SiO2),82 AuNPs83 and poly(ethylene glycol)-D,L-polylactide (PEG-PLA) NPs.84 NPs taken up via CME usually end up in the acidic environment of lysosomes.50
2.4 Caveolae-dependent endocytosis
Together with CME caveolae-dependent endocytosis is a common route of internalization of NPs.16 Caveola is the designation for the flask-shaped invaginations with 50–100 nm that can be found in the plasma membrane of specific mammalian cells.85 These structures are abundant in smooth muscle, endothelia and adipocytes, covering approximately a third of the plasma membrane area.86 Two important components are essential for caveolae formation: caveolin-1, or caveolin-3 in striated muscle cells, and cavin 1.87 Other components such as caveolin-2, and cavins 2, 3 and 4, and different accessory proteins (e.g., Eps-15 homology domain 2 (EHD2), PACSIN2 and dynamin 2) are also important for the formation, stabilization and scission of caveolae.87–89 Different cargoes have been linked to this pathway, including shiga and cholera bacterial toxins, non-enveloped viruses polyomavirus and simian virus 40, albumin, folic acid and plasma membrane components such as glycosylphosphatidylinositol (GPI)-anchored proteins.87,88 A commercially available NP conjugate of paclitaxel to human albumin (Nab-paclitaxel/Abraxane®) is successfully internalized by the cells through caveolae-mediated endocytosis.90 Similarly, lipid NPs with poly(styrene sulfonate) surface were revealed to be internalized via caveolae in HeLa and human endothelial cells (HUVEC).91Caveolae also play other important roles including their involvement in transcytosis across endothelial cells allowing the transport of solutes between the blood and the peripheral tissues (e.g., heart and lungs).92,93 An efficient transcytosis mechanism was observed for albumin-coated polymeric NPs of 20, 40 and 100 nm in lung endothelial cells (BLMVEC).94
2.5 Clathrin/caveolae-independent endocytosis
Besides the clathrin and caveolae-mediated endocytosis, which involve specific coated vesicles, NPs and other cargos such as cellular fluids, growth hormones and toxins, can be internalized via clathrin/caveolae-independent endocytosis.95 These mechanisms are usually cholesterol-dependent and require specific lipid composition.11 Despite the study of these processes, their contribution to endocytic uptake in mammalian cells is still not fully explained. Therefore, several ways of categorizing clathrin-/caveolae-independent endocytosis mechanisms are proposed in the literature.96–98 Herein, we have decided to categorize them according to the GTPases and associated proteins that are involved in the cellular entry pathway. In this regard, the clathrin/caveolae-independent endocytosis can be subdivided to: (i) clathrin-independent carriers/GPI-AP enriched early endosomal compartment (CLIC–GEEC), (ii) RhoA-dependent, (iii) Arf6-associated, (iv) flotillin-assisted and (v) fast endophilin-mediated endocytosis (for specific details see other publications95,97,99–101).(i) The CLIC–GEEC pathway is a dynamin-independent process leading to the formation of tubular/ring-like invaginations of the plasma membrane of around 200–600 nm in length and 40–80 nm in width.102 This process principally involves two small GTPases, Cdc42 and Arf1, the multidomain protein GTPase regulator associated with focal adhesion kinase (GRAF1), cholesterol and actin.95 The principal cargoes internalized through this pathway are extracellular fluid, GPI-anchored proteins and cholera toxin B.99
(ii) Contrary to CLIC–GEEC, RhoA-dependent endocytosis is a dynamin-dependent process responsible for the internalization of the interleukin-2 receptor and Clostridium botulinum C2 toxin.61 RhoA, one of the Ras homologous (Rho) protein family of GTPases, is the principal small GTPase protein involved in this process.103 Additionally, several other molecules, including small GTPase Rac1, its downstream p21-activated kinases (i.e., PAK-1 and PAK-2), and phosphatidylinositol 3-kinase are important regulators in this pathway.104
(iii) Although Arf6 was thought to be involved in macropinocytosis, the internalization of the major histocompatibility complex class I, β1-integrin and E-cadherin has been associated with Arf6-associated endocytosis in a clathrin- and dynamin-independent process.101 Uptake of other cargoes, including coxsackievirus A9 and the green fluorescent protein fused with the herpes simplex virus protein VP22, were shown to be dynamin-dependent.105,106 Arf6's key role is not in the process of endocytosis, but in recycling, and is thus referred to as Arf6-associated endocytosis.107
(iv) Flotillin-assisted endocytosis is another process that encompasses flotillin-1 (i.e., reggie-2) and flotillin-2 (i.e., reggie-1).95 They both associate with specific membrane microdomains that are important for the induction of membrane invaginations.108 It has been suggested that flotillin-1 is involved in sequestration of different cargoes, such as the GPI-anchored protein CD59, cholera toxin B subunit (CTxB), cationic molecules, proteoglycans and the Niemann–Pick C1-like 1 protein.109,110 Depending on the cargo, flotillin-assisted endocytosis can be dynamin-dependent (e.g., epidermal growth factor) or dynamin-independent (e.g., CTxB).108 Flotillins regulate several membrane trafficking events, although there is still no evidence that these proteins are essential elements of a specific endocytic pathway. With this in mind, Meister et al. proposed the term “flotillin-assisted endocytosis” and assumes an endocytic process that is facilitated by flotillins.110
(v) Fast-endophilin-mediated endocytosis (FEME) was recently discovered and is a non-constitutive process that is triggered upon activation of specific receptors including β1 adrenergic receptor, EGFR and interleukin-2 receptor.100 FEME occurs in the leading edges of the cell membrane where endophilin is recruited by lamellipodin.111 This pathway only takes place if pre-enriched endophilin (endophilin-Ptdlns(4,5)P2-lamellipodin) is already available at the plasma membrane to bind to the activated-receptor.112 FEME is characterized by the rapid formation, usually seconds, of tubulo-vesicular structures of <1 μm at the cell membrane.100 To date, the uptake of different NPs through clathrin/caveolae-independent endocytosis has been reported. SPIONs and silica-coated iron oxide NPs (Fe3O4@SiO2) with negative surface charge and a primary diameter of around 17 to 30 nm were shown to be internalized via CDC42 (CLIC–GEEC pathway) and caveolae in HeLa cells.113 Similarly, Arf6-associated endocytosis together with caveolae-dependent endocytosis and macropinocytosis were associated with the uptake of ∼130 nm polydopamine-coated mesoporous silica NPs (polydopamine@MSNs).114 In addition it has been demonstrated that flotillin-1 and -2 are involved in the uptake of 30 nm amorphous SiO2 NPs.115 The involvement of all the mechanisms in NP internalization is still unclear. There are three possible reasons: (i) clathrin/caveolae-independent endocytosis is not the principal mechanism for NP uptake; (ii) difficulties in distinguishing from other mechanisms, such as clathrin- and caveolae-dependent endocytosis; (iii) lack of knowledge about the process.
2.6 Passive uptake
The different entry routes discussed so far are known to be the principal mechanisms for NP uptake. Other processes such as passive diffusion across the outer cell membrane by van der Waals forces or steric interactions (subsumed as adhesive interactions) and pore formation may also be involved as well.116 For the study of passive uptake, red blood cells (RBCs) are usually used as models, once they are deprived of most cellular organelles and endocytosis machinery.117,118 Quantum dots (QDs) coated with the zwitterionic thiol ligand D-penicillamine (DPA-QDs) of 4 nm radius entered in RBC via membrane penetration without pore formation.119 Similarly, passive uptake was observed in lung cells after exposure of titanium dioxide (TiO2) NPs of 22 nm to rats via inhalation.120 The same findings were observed when different NP types were exposed to human RBCs.117 It was demonstrated that surface charge and material of the particles did not influence their uptake and that internalized particles were not membrane-bound. Also, several positively charged (cationic) NPs have been shown to lead to membrane disruption and formation of nanoscale holes.121 This finding is mostly related to experiments where serum-free cell culture medium was used. As an example, cationic AuNPs with high surface charge densities were able to create hydrophilic pores and diffuse into the plasma membrane.1223. Influence of NP physicochemical properties on endocytosis
Several factors can affect the internalization of NPs, including NP properties such as composition, size, shape, stiffness, and surface chemistry. These properties are, first and foremost, important for NP stability in the biological environment and, secondly, they can influence the cell–NP interactions and subsequent uptake. The principal parameters to take into consideration for NP stability are complex and have not been considered as a part of the review. For detailed information on this topic several published articles are available.18,31,123,124 Herein, the focus is in the influence of different NP physicochemical properties on endocytic mechanisms (Fig. 2).3.1 Size
In general, the size of a substance/particle is considered as one of the most important parameters in endocytosis. Large particles (>500 nm) are known to be internalized only via phagocytosis and/or macropinocytosis, while the other endocytic mechanisms are limited in terms of cargo size (maximum size of 200–300 nm).61 Several other uptake mechanisms can also be involved in the internalization of NPs. We assume that NP size is not a critical parameter influencing phagocytosis and macropinocytosis mechanisms.61 Firstly, because phagocytosis is mostly dependent on protein opsonization and secondly, because macropinocytosis is a non-specific cargo uptake mechanism.11Via macropinocytosis, cells can engulf NPs of various sizes at the same time and it usually occurs in combination with other mechanisms.61,73 In contrast, clathrin- and caveolae-mediated endocytosis seem to be dependent on NPs size.125,126 Caveolae-based vesicles are usually smaller (average size ∼60 nm) in comparison with clathrin-based vesicles (average size ∼120 nm), so it is expected that larger NPs preferentially are taken up by the cells via clathrin. These vesicle sizes are an average among different cell types, although it is possible to find larger or smaller vesicles in specific cells. Ho et al. revealed that 20 and 40 nm PS NPs are more dependent on caveolae-mediated endocytosis than 100 nm PS NPs, as seen in HUVEC cells.125 Similar observations were reported in hepatocytes (HepG2) where the uptake of 20 nm AuNPs showed a higher dependence of caveolae in comparison to 40 nm AuNPs.126 In addition, studies on BLMVEC revealed that a single caveolae vesicle was able to engulf up to three 20 nm or two 40 nm albumin-coated polymeric NPs.94An important size-related parameter that can affect NP internalization is the aggregation state, once NPs in an aggregated form or as individual particles interact differently with cells.124 Opsonization is another factor that can promote NP aggregation, change the surface properties and contributes to NP phagocytosis.62,127 The NP aggregation contributes to an overall increase in NP size and may affect their uptake96 and intracellular distribution.128 For example, Halamoda-Kenzaoui et al. showed that well-dispersed SiO2 NPs were internalized principally through caveolae-mediated endocytosis, but an increase in the NP agglomeration state shifted to a combination of endocytosis pathways with a predominant role of macropinocytosis.129
Size is not only related to the route of internalization but also affects the uptake rate. It has been reported that the internalization of SiO2 NPs in A549 lung epithelial cells becomes slower with increasing particle size130 and this might be related to the concept of dosimetry as size can determine the behaviour in cell culture medium and thus the delivered dose.131 The same observation was reported by Rejman et al. where the uptake of fluorescent carboxylate nano-/micro-spheres (50, 100, 200, 500, and 1000 nm), by melanoma cells, revealed to be size-dependent.132 A decreased internalization was related to increased microsphere size. In contrast, an increase in the uptake of larger NPs was observed in a different study where AuNPs (13 and 45 nm) were exposed to human dermal fibroblasts.133 They showed that 45 nm AuNPs enter the cell more efficiently than 13 nm AuNPs. Yet Li et al. demonstrated the interplay between different-sized SiO2 NPs (50, 100 and 150 nm) on their uptake in HeLa cells in a co-exposure scenario. When NPs were administered simultaneously to the cells, a competition between different-sized NPs in their cellular uptake was observed. Interestingly, the bigger NPs stimulated the uptake of smaller ones and vice versa.134 It is important to note that the comparisons between different studies cannot be made without considering the differences between NP types, cells, and experimental conditions. In order to investigate the size effect on NP uptake, the reductionist approach is proposed to minimize all influencing factors except the particle size.
3.2 Shape
Shape is a physical property that can also influence the uptake of NPs (for a review please refer to a previous publication).135 Xie et al. synthesized different gold nanostars, nanorods, and nanotriangles coated with methylpolyethylene glycol and exposed them to macrophage (RAW264.7).136 They showed that all AuNPs enter cells through clathrin-mediated uptake. Rods can also be internalized via caveolae/lipid raft-mediated endocytosis and triangles via additional actin and dynamin-dependent pathways (possibly phagocytosis and/or macropinocytosis). Shape can also have an impact on the increased or decreased uptake of NPs. Vàcha et al. described a simulation approach for investigation of the passive uptake of differently shaped NPs.137 They suggested that the sphero-cylinders are more efficiently endocytosed compared to spheres of the same diameter. Even though both shapes have the same kinetic barrier for uptake across a membrane, the sphero-cylinders possess the larger volume. For an idealized membrane, it is thermodynamically more convenient to encapsulate a sphero-cylinder than a sphere of the same radius, due to the smaller curvature of the cylindrical part. It has been shown that endocytosis was suppressed for particles with sharp edges (regions with high curvature).137 In support of this study, Huang et al. evaluated the cellular uptake of mesoporous silica NPs of similar diameter but different aspect ratios (AR): sphere-shaped (AR of ∼1), short rod-shaped (AR of ∼2) and a long rod-shaped (AR of ∼4) in human melanoma cells (A375). The uptake rate of long-rod particles was faster compared to those of short-rod and spherical particles. The most likely explanation for such behaviour could be the larger contact area of rod-shaped NPs with the cell membrane.138 Chithrani et al. investigated the uptake of differently shaped AuNPs in HeLa cells and found a lower amount of rod-shaped AuNPs within the cells compared to spherical ones.139 There are many speculations for this outcome, such as (i) differences in the membrane curvature, (ii) reduction of the available receptor binding sites, (iii) surfactant molecules, which prevent serum proteins from binding onto the NPs surface efficiently, and (iv) non-homogenous protein coating and thus lack of multivalent binding to the receptors.139The interaction between different NPs and cells depends not only on the shape of the NPs, but also on the cell type. Kinnear et al. synthesized different gold rods of varied aspect ratios and studied their interaction with macrophages and epithelial cells.140 Rods of ∼10–90 nm in length and with small aspect ratios (<5), revealed similar uptake efficiencies in both cells. In contrast, nanorods with higher aspect ratio (>5) were preferentially internalized by epithelial cells, whereas the uptake in macrophages revealed to be shape independent.
3.3 Surface charge
The surface charge of NPs influences their behaviour in biological environments due to the presence of biomolecules with various charges.17 Several factors can affect the surface charge of NPs, including adsorbed biomolecules, and pH. Herein, besides the design of cationic (positively charged) or anionic (negatively charged) NPs, it is crucial to understand how the NP surface charge changes in such complex environments. Based on the studies published so far, the internalization of cationic NPs is more efficient in comparison to neutral and anionic NPs.141–144 9 nm SPIONs functionalized with different polymers: polyvinyl alcohol (PVA), carboxylate-functionalized PVA (negative charge), thiol-functionalized PVA (neutral charge), and amino-functionalized PVA (positive charge) (amino-PVA) were synthesized, and a higher uptake for amino-PVA-SPIONs in melanoma cells in comparison with carboxy-PVA-SPIONs and thiol-PVA-SPIONs was shown.145 He et al. investigated the effects of surface charge on cellular uptake of polymeric particles with various sizes (150–500 nm) and zeta potentials between −40 mV and +35 mV in murine macrophages.146 They demonstrated more efficient uptake of large particles with higher positive surface charges. Uptake of negatively and positively charged NPs was cell-type-dependent. One of the explanations for the toxicity is that once cationic NPs reach the lysosomes they can lead to their swelling and subsequent membrane rupture.18 In contrast, Lunov et al. have shown uptake of anionic PS particles by monocyte-derived macrophages (MDMs) mainly via clathrin- and dynamin-dependent endocytosis, whereas the cationic ones were taken up via macropinocytosis. THP-1 cells did not differentiate between the particle charge and internalized both by macropinocytosis, and clathrin- and dynamin-dependent endocytosis.147 The uptake mechanism of charged particles thus strongly depends on the cell- and particle-types.3.4 Surface functionalization
The functionalization of NPs with specific moieties to target specific cellular receptors has been increasing over the years especially as a concept for targeted drug delivery. These receptors usually relate to certain endocytic mechanisms and can be specific for certain cell types or being upregulated in cancer cells.148,149 This includes, for example, NP functionalization with EGF150 and transferrin151 known to be internalized via clathrin-mediated endocytosis, but also with albumin90 that is associated with caveolae-mediated endocytosis. Besides cell targeting, NPs are functionalized with other molecules in order to increase NP stability and to prolong circulation time. Brandenberger et al. have compared the uptake of two different AuNPs, citrate-coated and polyethylene glycol (PEG)-coated, in A549 cells.128 The particles were aerosolized on the lung cell surface with an air–liquid cell exposure system to avoid exposure of the particles in the cell culture medium. Citrate-coated AuNPs were taken up by macropinocytosis and by clathrin- and caveolae-mediated endocytosis, whereas PEG-coated AuNPs were internalized via clathrin- and caveolae-mediated endocytosis, but not by macropinocytosis.3.5 Topography
Surface nanoscale topography of NPs has recently received particular attention, being considered an important surface property that influences cell–NP interaction.152 NPs with different surface topography have been synthesized, including smooth,153 rough,154 rambutan,154 raspberry,155 flower,156 and virus-like157 surfaces. Wang et al. investigated the internalization of three different SiO2 NPs: solid, mesoporous, and virus-like (spiky tubular rough surface) in HeLa cells.158 They showed that virus-like mesoporous SiO2 NPs had the fastest internalization rate among the three SiO2 NP types. Regarding the entry mechanisms, virus-like mesoporous SiO2 NPs were revealed to be internalized mainly via caveolae-mediated endocytosis and macropinocytosis, while clathrin-mediated endocytosis was the principal route for the other two NPs.4. Methods to study endocytosis
Our current understanding of cell–NP interactions is primarily due to recent developments in imaging and biochemical analysis. The answers to questions like “Are NPs inside the cell?” and “How many NPs are inside the cell?” are important, but even more important is the answer to “How are NPs internalized?”. Understanding NPs endocytosis mechanisms and its regulation allow the optimization of NP uptake by designing specific NPs and reduction of possible toxic effects. For the research community, in vitro and in vivo studies of the different endocytic processes require simple and reliable methods, whereas the research focus in this area is on the in vitro approach at the single cell level. Different approaches have been used, including chemical and pharmacological inhibitors, genetic approaches, protein and gene expression levels, specific biomarkers, and different microscopy techniques (fluorescence, electron, and atomic force). An overview of these methods will be given, and the pros and cons will be discussed in this section.4.1 Classical chemical and pharmacological inhibitors
The first possibility to evaluate endocytosis of NPs is the use of inhibitors to block the process. The use of classical chemical and pharmacological inhibitors is the most common approach to study NP uptake by cells. These inhibitors are the preferred choice due to: (i) fast action in blocking the uptake route; (ii) inhibition covers the overall cell population; (iii) efficient and cost-effective process.159 Nevertheless, when blocking a cellular process, there is always a possibility of side effects (e.g., cytotoxicity) and upregulation of compensatory uptake mechanisms. The major limitations are their lack of specificity and their efficacy variation between cell types, which may require dose-titration studies and the use of different inhibitors.160Several inhibitors have been introduced over recent years, including the recently discovered pharmacological inhibitors itstop 2 and dyngo compounds that inhibit CME and dynamin-dependent endocytosis, respectively (Table 1). Depending on the target pathway, inhibitors can be grouped into: (i) CME; (ii) lipid rafts and caveolae-mediated endocytosis; (iii) macropinocytosis and phagocytosis; (iv) dynamin-dependent endocytosis inhibitors. Compounds that block specifically each type of endocytic mechanism only are not yet available. In this regard, it is suggested to use the combination of different inhibitors in order to avoid ambiguous conclusions. In addition the use of appropriate controls to confirm blocking of a certain mechanism is highly recommended to evaluate the optimal and non-toxic time frame for cell experiments.161 The controls include molecules that are known to be internalized via the mechanism in study. Transferrin80 and lactosylceramide162 can be used as controls for CME and caveolae-mediated endocytosis, respectively, whereas dextran 70 kDa and lucifer yellow are controls for macropinocytosis.163,164 Several studies in the literature reported the use of other molecules such as cholera toxin B as a control for caveolae-mediated endocytosis. The issue with using cholera toxin B lies in its non-specificity, since this molecule can be internalized via different pathways.165 Included in CME inhibitors are the classical chemicals such as hypertonic sucrose and potassium depletion (Table 1). Singh et al. investigated the effect of both inhibitors on the uptake of cerium oxide NPs.166 They revealed that the NP uptake was inhibited in both approaches, which suggested internalization through CME. Even though hypertonic sucrose and potassium depletion have been related to CME, it was also proved that both interfere with other endocytic mechanisms.167,168 In a recent study, pitstop 2 was also included to evaluate the internalization of QDs conjugated with single-stranded oligonucleotide-based aptamers (APTs) in A549 cells.169 The target of pitstop 2 is CME, and they showed that in the presence of the chemical, APT-QDs were not able to enter into the cell, confirming CME as the predominant mechanism. Even if the mode of action of pitstop 2 is more selective, its non-specificity was also demonstrated, once it was able to block endocytic mechanisms independent of clathrin.170,171 Non-specificity is not the only issue with the use of inhibitors, given that they can also affect cellular processes, and cause unusual structural changes and cytotoxicity. Kuhn et al. tested four different inhibitors: chlorpromazine and monodansylcadaverine (both CME inhibitors), methyl-β-cyclodextrin (a caveolin-mediated endocytosis inhibitor), and cytochalasin D (a phagocytosis/macropinocytosis inhibitor), in the uptake of PS NPs in A549 and J774A.1 cells.60 Cellular damage was observed for A549 cells in the presence of monodansylcadaverine and cytochalasin D, and for J774A.1 cells in the presence of chlorpromazine. Vercauteren et al. also verified the cytotoxic effects of chlorpromazine and methyl-β-cyclodextrin in different cell types.172 They concluded that the cytotoxic effects caused by the inhibitors were cell-and concentration-dependent.
| Endocytosis inhibitors | Pathway(s) involved | Mechanism | Limitation | Conc. rangea | Ref. |
|---|---|---|---|---|---|
| Conc. – concentration; Ref. – reference(s); CPZ – chlorpromazine; MDC – monodansylcadaverine; CytD – cytochalasin D; CME – clathrin-mediated endocytosis; CCPs – clathrin-coated pits; CCVs – clathrin-coated vesicles; ES – endosidin; CHC – clathrin heavy chain; EIPA – ethyl-isopropyl amiloride; HOE-694 – 3-methylsulphonyl-4-piperidinobenzoyl, guanidine hydrochloride. n/a – not applicable.a The concentration of inhibitor is cell- and time-dependent. | |||||
| Hypertonic sucrose | CME | Traps clathrin in microcages | Interferes with macropinocytosis | 0.4–0.5 M | 167 |
| Potassium depletion | Removes clathrin lattices that associate with cell membrane | Affects actin cytoskeleton | n/a | 167 and 173 | |
| Cytosol acidification | Inhibits the scission of CCPs from the cell membrane | Affects actin cytoskeleton and interferes with macropinocytosis | 10–30 mM NH4Cl | 174 and 175 | |
| CPZ | AP2 inhibitor; blocks endosome recycling | Affects biogenesis of large intracellular vesicles such as phagosomes and macropinosomes | 50–100 μM | 176 | |
| MDC | Stabilizes CCVs | Global changes in the dynamics of the actin cytoskeleton | 100–300 μM | 177 and 178 | |
| Phenylarsine oxide | Inhibits protein tyrosine phosphatases; exact mechanism remains unknown | Inhibits phagocytosis and macropinocytosis | 1–20 μM | 178 | |
| Chloroquine | Reduces the expression of phosphatidylinositol binding clathrin assembly protein (PICALM) | May only affect phagocytic cells and interfere with other endocytic mechanisms | 20–400 μM | 179 | |
| Monensin | Dissipation of a proton gradient | Interferes with endocytic trafficking such as receptor recycling | 5–40 μM | 180 and 181 | |
| ES9-17 | Inhibitor of the CHC function; cytosol acidification | Still unknown (discovered recently) | 30 μM | 182 | |
| Pitstop 2 | Interferes with binding of proteins to the N-terminal domain of clathrin | Affects most endocytic pathways | 10–100 μM | 170 and 174 | |
| Methyl-β-cyclodextrin | Caveolae-mediated endocytosis/lipid rafts | Cholesterol depletion from the cell membrane | Interferes with macropinocytosis and CME | 5–10 mM | 183 |
| Filipin | Interacts with cholesterol at the cell membrane | Inhibits CME; unstable and toxic at high concentrations | 1–5 μg mL−1 | 184 | |
| Statins | Blocks cholesterol synthesis | Affects most endocytic mechanisms | 10–100 μM | 185 | |
| Genistein | Inhibits several tyrosine kinases | Interferes with dynamin and may affect other endocytic processes | 200–400 μM | 81, 172 and 186 | |
| CytD and lantruculins | Phagocytosis and macropinocytosis | Block actin polymerization | Affect most endocytic mechanisms | 1–10 μM | 60 and 187 |
| Amiloride or its derivatives (EIPA and HOE-694) | Na+/H+ exchanger pump inhibitor; blocks Rac1 and Cdc42 signaling | Inhibit CME | 1 mM for amiloride and 50–100 μM for its derivatives | 188 and 189 | |
| Imipramine | Inhibits plasma membrane ruffle formation | Still unknown (the mechanism of inhibition is still not fully understood) | 5 μM | 190 and 191 | |
| Wortmannin and LY294002 | Inhibit the activity of phosphatidylinositol 3-kinase | Affect most endocytic mechanisms | 10 nM–10 μM for Wortmannin and 20 μg mL−1 for LY294002 | 164, 192 and 193 | |
| Rottlerin | Inhibits the activity of protein kinase C delta | Affects most endocytic mechanisms | 1–3 μM | 194 and 195 | |
| Dynasore, dynole and dyngo compounds | Dynamin-dependent endocytosis | Inhibit the GTPase activity of dynamin1 and 2 | May interfere with cholesterol homeostasis and actin | 1–500 μM | 196 and 197 |
In short, the choice of inhibitors is cell- and experiment-dependent and once applied they should not cause significant side effects. As most of the inhibitors simultaneously affect different endocytic mechanisms, some caution should be taken when interpreting results and drawing conclusions.
4.2 Genetic approaches
In order to overcome the non-specificity of chemical inhibitors, genetic approaches were implemented to change the expression of specific proteins. Included in the genetic approaches are the use of interference RNA (RNAi) to silence specific genes, the use of knockout models, targeted genome editing and mutant proteins.198 RNAi refers to small noncoding RNAs (around 20 to 30 nucleotides), which includes micro RNA (miRNA), short interfering RNA (siRNA), short hairpin RNA (shRNA) and piwi interacting RNA (piRNA). Their principal role is the control of gene expression.199 siRNA are exogenous synthetic double stranded RNA, and have been used to interfere with specific genes related to the endocytosis process.160 The silencing mechanisms of siRNA occurs via degradation of the target mRNA, which ends up with the knockdown of the respective protein.200 Guggenheim et al. investigated the uptake of SPIONs in an epithelial breast cancer cell line (MDA-MB-231), A549, HeLa and THP-1 macrophage-like cells.201 They transfected the cells with different siRNA constructs: α-adaptin (AP2 inhibition) siRNA to block CME; caveolin-1 siRNA to block caveolae-mediated endocytosis; PAK-1 siRNA to block macropinocytosis. The uptake of SPIONs was inhibited in different ways by the various siRNA, revealing the involvement of different endocytic mechanisms, which confirms the findings with pharmaceutical inhibitors as described in the previous section.Knockout animal models can also be used to investigate the impact of specific proteins in a determined process. Sago et al. investigated the in vivo delivery of lipid NPs in a caveolin-1 knockout mouse model.202 They revealed that NP bio-distribution was affected by caveolin-1 in a cell type-specific manner. An additional genetic approach that is scarcely used in the study of NPs endocytosis, and that might be considered in future studies, is the use of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems.203 The CRISPR–Cas system is part of the RNA-mediated adaptive defense of prokaryotes and acts by cleaving the nucleic acids of invading viruses.204 These systems have been used to modify, regulate, or label specific genes in several cells and organisms.205 CRISPR-Cas9 is a novel efficient method for gene knockout and involves the Cas9 protein, an enzyme responsible for DNA cutting and a single guided RNA (sgRNA), which guides Cas9 to a specific location in the DNA sequence.204,205 This approach has been used to prepare AP2M1,206 CAV1,207 dynamin208 and Rac209 knockout cells. Patel et al., studied the intracellular delivery of mRNA from lipid NPs.210 They introduced CRISPR-based genetic perturbations on the lysosomal pathway of haploid cells (HAP1) via knockout of Rab5A, Rab4A, or Rab7A. These proteins can be found on early, recycling, and late endosomes, respectively. They concluded that late endosome/lysosome formation is essential for functional delivery of exogenously presented mRNA. The methods described above involve the inhibition of the expression of a protein of interest. There is still the possibility of using mutant proteins, where mutations (e.g., deletions) are introduced into the gene (i.e., DNA sequence) leading to the expression of dysfunctional proteins.211 Dominant negative mutations are the most common mutations to investigate the involvement of specific proteins in endocytosis. The overexpression of these mutated proteins in comparison to the endogenous wild-type proteins may culminate with non-functional proteins, inactive or hyperactive proteins [e.g., GTPases (Rho and Rab) and kinases (PAK and PKC)].198 A mutant form of dynamin, K44A, was one of the first mutated proteins used to study the importance of dynamin in the endocytosis of transferrin via CME.212 It was shown that internalization of transferrin in mammalian cells is blocked in the presence of dysfunctional dynamin. In a different study, Smith et al. investigated the effect of two mutant proteins, eps15 (EH29) and caveolin1 (Y14F), on the uptake of 20 nm carboxylate-modified PS NPs in HeLa cells (Fig. 3).213 They observed that the expression of the inhibitor of clathrin-mediated endocytosis eps15 (EH29), but not the dominant negative caveolin-1 (Y14F), significantly reduced NP uptake.
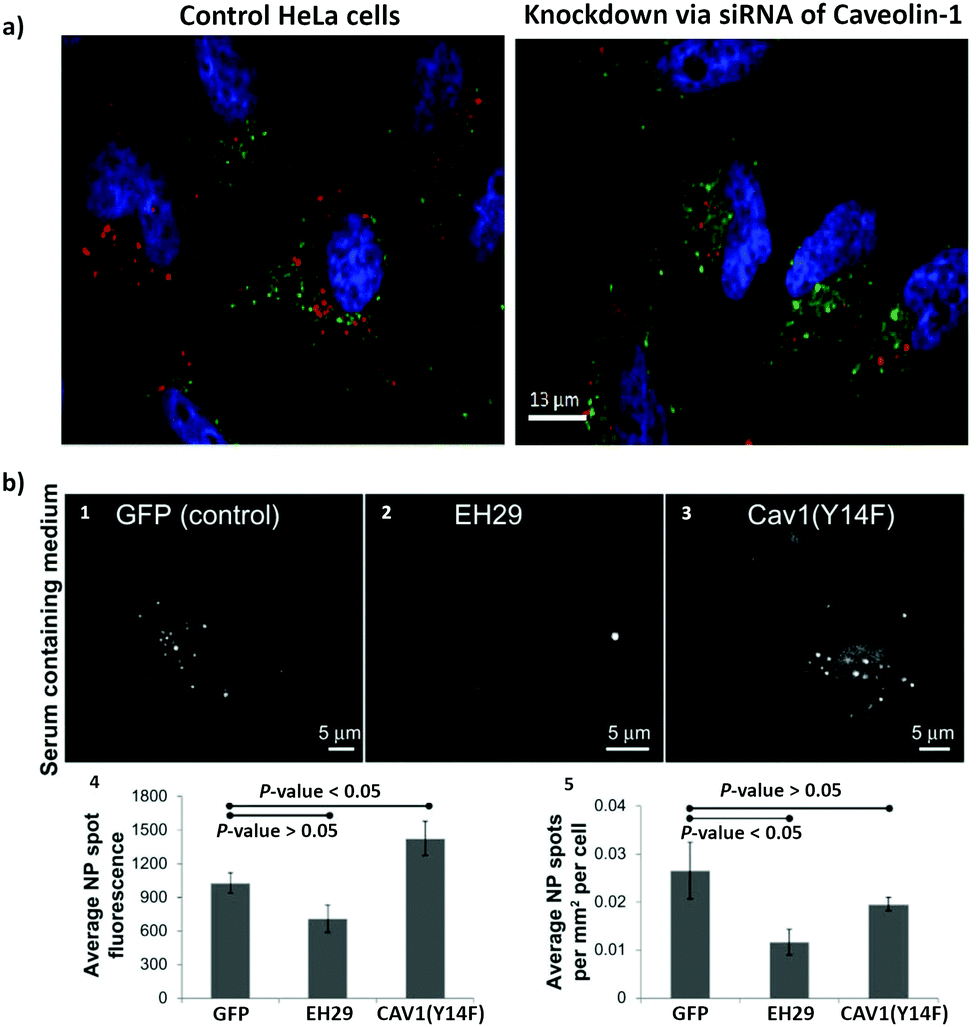 | ||
| Fig. 3 The use of siRNA and mutant proteins to investigate nanoparticle endocytosis. (a) Micrographs of HeLa cells, which were incubated with silica-coated iron oxide nanoparticles (Fe3O4@SiO2) for 4 h; on the left image control cells without siRNA treatment and on the right cells with knockdown of caveolin-1; blue = DAPI (nuclei), green = Transferrin Alexa Fluor® 488 conjugate (cytosol), red = Alexa Fluor® 555 (Fe3O4@SiO2). (b) Representative images of a HeLa cell expressing green fluorescent protein (1), EH29 (mutant form of eps15) (2), and Y14F (mutant form of caveolin 1) (3) incubated with carboxylate-modified polystyrene nanoparticles in serum-containing media. Average fluorescence based on nanoparticle spot (4) and average nanoparticle spot density per cell area (5) for HeLa cells expressing green fluorescent protein, EH29, and Y14F incubated with nanoparticles in serum-containing media. Adapted with permission from (a) ref. 113 and (b)ref. 213. Copyright (a) 2016 Bohmer et al., and (b) 2012 Smith et al., licensed by Dove Medical Press Limited. | ||
The genetic approaches discussed in this section present the advantage to be more specific than the chemical inhibitors, but they also have some limitations. Inhibition of protein expression may lead to compensatory mechanisms in the cell.160 Depending on the required time to silence a gene the cell may adapt and change gene expression.198 When transfecting cells with mutant proteins it is important to know if those proteins can be involved in more than one endocytic pathway.104 In addition, to get a dominant effect of the mutant protein, a certain level of overexpression (in comparison with the endogenous wild-type) should be achieved. Therefore, combining different approaches and the inclusion of appropriate positive and negative controls are crucial to avoid misinterpretations.
4.3 Protein and gene expression levels
The study of protein and gene expression levels, including proteomic and transcriptomic analysis enables the identification of the different proteins and transcripts associated with the process of endocytosis (Fig. 4). For example, it is possible to isolate endocytic vesicles and investigate the associated proteins by mass spectrometry (MS)-based proteomics.214 This approach, besides allowing the identification of proteins involved in endocytic uptake processes, also offers the possibility to quantify the protein levels. The two most widely used methods for high-throughput quantitative proteomic analysis in complex samples are gas chromatography MS (GC-MS) and liquid chromatography MS (LC-MS).215 LC-MS has been used as a bottom-up proteomics approach to analyze clathrin-coated vesicles,216,217 lipid rafts218 and phagosomes.219 Hofmann et al. included proteomic analysis, based on LC-MS, to investigate the uptake of superparamagnetic iron oxide PS NPs in HeLa cells (Fig. 4b).220 The identified proteins, including Arf1, together with other approaches, i.e. colocalization studies and the use of inhibitors, suggested the involvement of macropinocytosis in the internalization of NPs. Instead of using high-throughput analysis, there is the possibility to look directly at a few specific proteins. As a simple and more direct approach, protein levels can be determined via immunoassays [e.g., enzyme-linked immunosorbent assay (ELISA) and western blot]. Chaves et al. studied the expression levels of two proteins, clathrin heavy chain and caveolin 1, upon exposure to maghemite–rhodium citrate NPs in different cell types (breast cancer cell lines, MCF7 and MDA-MB-231, and primary human non-tumor mesenchymal cells) (Fig. 4a).221 For all cells, an increase in the clathrin heavy chain protein levels but not in caveolin-1 was observed, suggesting CME as the principal mechanism for internalization of maghemite–rhodium citrate NPs.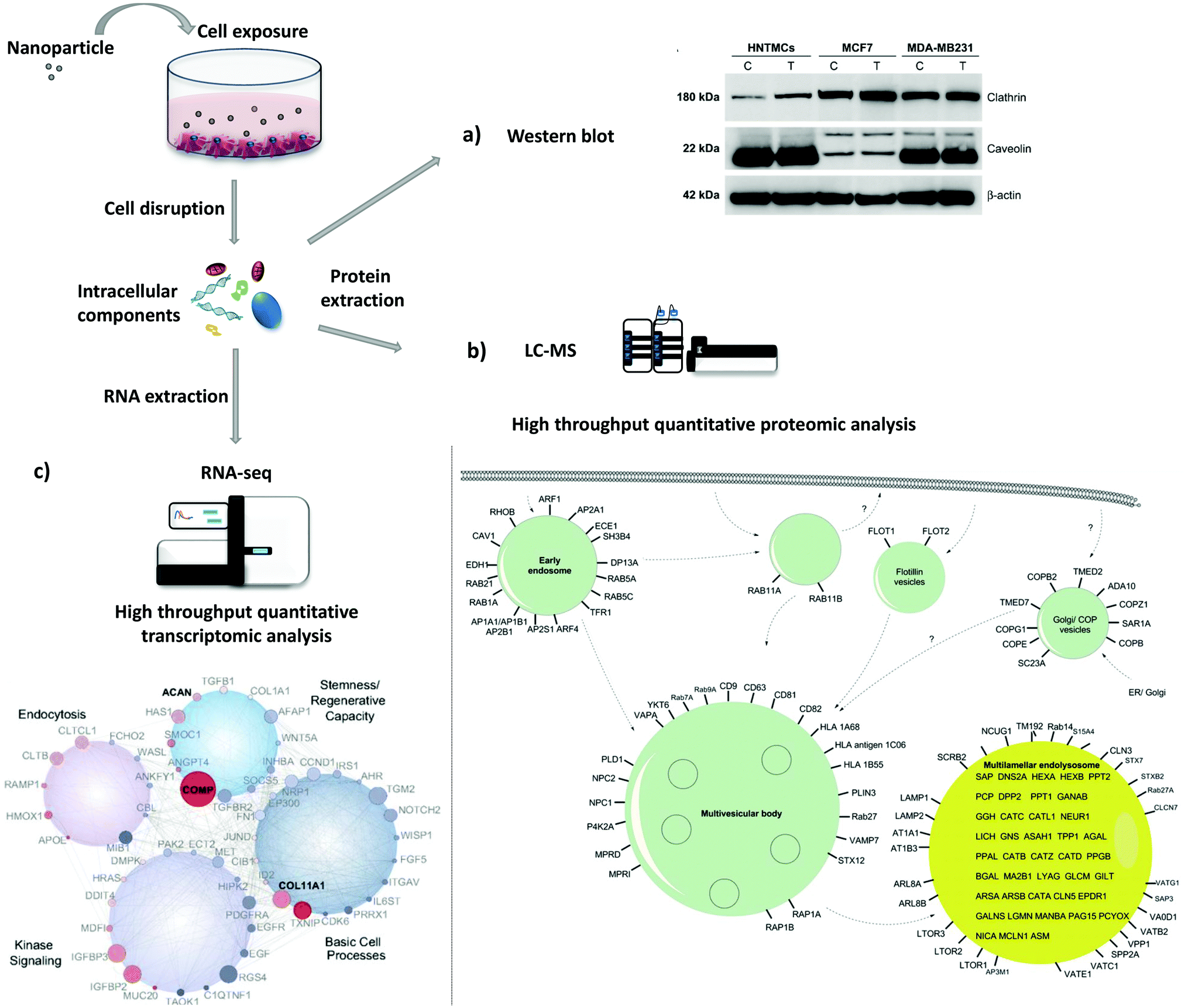 | ||
| Fig. 4 Evaluation of protein and gene expression upon nanoparticle exposure. Following nanoparticle exposure, cells can be disrupted and protein and RNA can be extracted. (a)–(c) are examples of possible downstream applications. (a) Western blot analysis of expression levels of clathrin and caveolin in MDA-MB231, MCF7, and human nontumor mesenchymal cells (HNTMCs). Control (C) and treated (T) cells with maghemite–rhodium citrate (MRC) nanoparticles (NPs) for 6 h. (b) Schematic illustration of intracellular nanoparticle trafficking based on gene ontology (GO) terms vesicle and lysosome. HeLa cells were exposed to superparamagnetic iron oxide polystyrene nanoparticles for 20 h and vesicles were magnetically separated. Label-free quantitative liquid chromatography-mass spectrometry (LC-MS) was performed followed by bioinformatic data analysis (DAVID ontology analysis). Only the proteins magnetically enriched by a factor of >2-fold are shown. (c) Analysis of the whole-transcriptome level by high-throughput sequencing (RNA-seq). Human mesenchymal stem cells (hMSCs) were exposed to nanosilicates for 7 days. At the bottom, scheme of the gene network, which comprises genes with different degrees of expression and high statistical significance (red, up-regulated; blue, down-regulated; size increases with significance). Reproduced with permission from (a)ref. 221 and (c)ref. 222. Adapted with permission from (b)ref. 220. Copyright (a) 2017 Chaves et al., licensed by Dove Medical Press Limited, (b) 2016 American Chemical Society and (c) 2018 Carrow et al., licensed by Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND). | ||
Instead of looking directly at protein levels, it is possible to evaluate the expression of particular genes (transcripts). This approach can be used to complement protein analysis. Here, the expression of certain genes or the analysis of the whole transcriptome (e.g., DNA microarrays and RNA-seq) can be performed. It is important to consider that transcript (mRNA) levels may not correlate with protein levels due to post-transcriptional modifications. Carrow et al. investigated the changes in the transcriptome profile of human mesenchymal stem cells induced by two-dimensional nanosilicates (Fig. 4c).222 They revealed significant changes in the expression level of 4.068 genes, where a large part has been found to be involved with endocytosis. In addition, genes related with CME were affected as well, revealing the involvement of CME on the uptake of nanosilicates.
Transcriptomic and proteomic analyses are powerful techniques that provide a large amount of information regarding endocytic processes and other biological processes (e.g., cell proliferation, inflammation, and apoptosis).223 These are expensive, time-consuming and laborious approaches, requiring fundamental support of bioinformatics and biostatistics.224 Investigation of a small group of transcripts and/or proteins are simpler and faster; however some proteins may be connected with different endocytic mechanisms and their expression may not change considerably.215 A crucial aspect to consider when looking for transcript and protein levels is the selection of the most appropriate time points. Depending on the experiment (i.e., cell type, NPs properties, administered dose, etc.) the expression of proteins involved in endocytosis can change. Therefore, it is recommended that a time-course analysis is carried out, involving different time points, to be able to see possible significant effects.
4.4 Endocytic markers and microscopy analysis
Recent advances in imaging, including electron, atomic force and super resolution light microscopy techniques, contribute to the understanding of NP uptake by cells and intracellular trafficking (Fig. 5).11 Microscopy comprises a variety of electron and light microscopes to visualize smaller scale structures in a sample by presenting a magnified image.225 Importantly, not all microscopes are able to resolve nanoscale structures such as NPs and subcellular compartments (e.g., clathrin and caveolae vesicles). In light microscopy, the resolution is limited due to the diffraction limit of light.226 In this sense, new advances in this field, with a focus on fluorescence techniques, improved the resolution by increasing spatial resolution [e.g., confocal, total internal reflection fluorescence (TIRF) and structure illumination microscopy (SIM)] or by bypassing the diffraction limit using super resolution fluorescence microscopy [e.g., stimulated emission depletion (STED), expansion microscopy (ExM) and single molecule localization microscopy (SMLM)].225 The importance of digital image restoration should be noted, including de-convolution algorithms that may increase the resolution by 2–3 fold.227 Super-resolution approaches were developed based on pre-existent microscope setups, wide-field, TIRF and confocal, to overcome the diffraction limit by taking advantage of particular fluorophore properties.228 In order to apply these techniques in biological systems, specific cellular structures or molecules have to be labeled with fluorophores. Fluorophores can be grouped into organic dyes [e.g., FITC], and biological fluorophores [e.g., green fluorescent protein (GFP)].229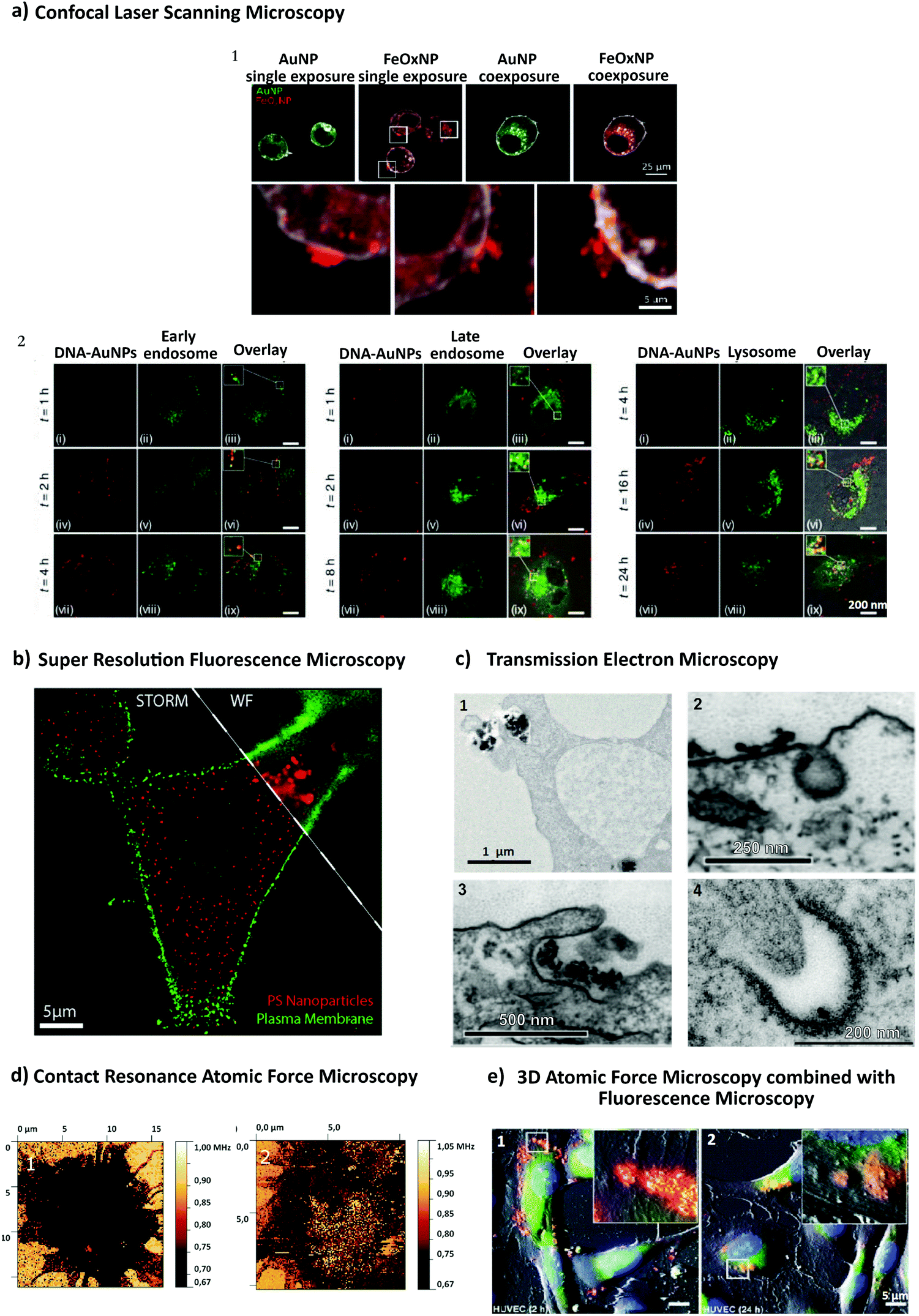 | ||
| Fig. 5 Different approaches to study NP endocytosis. (a) Confocal laser scanning microscopy reveals the effect of AuNPs and FeOx NPs uptake after monodansylcadaverine treatment of macrophage cells; the uptake of FeOx NPs is partially blocked in the presence of the inhibitor as shown by the aggregation of NPs in the outer side of cell membrane (1). Fluorescence microscopy images showing co-localization of DNA-decorated AuNPs (fPlas-gold) (red) with early endosomes (green), late endosomes (green) and lysosomes (green) over time (2). (b) Super-resolution fluorescence microscopy, stochastic optical reconstruction microscopy (STORM), of the uptake of 80 nm polystyrene NPs (red) in HeLa cervical carcinoma cells (plasma membrane, green) in comparison with conventional wide-field microscopic techniques. (c) Transmission electron microscopy (TEM) images of silica NPs being internalized via phagocytosis (1). TEM micrographs of clathrin-mediated endocytosis (CME) (2) and macropinocytosis (3) of magnetic cobalt ferrite NPs coated with polyacrylic acid (PAA). Electron microscopy image shows two nanoparticles (22 nm) located within a caveolin of a lung epithelial cell (A549) (4). (d) Contact resonance frequency (CRF) map, based on contact resonance atomic force microscopy (CR-AFM), of an empty microglia cell (1) and of a microglia engulfed with Fe3O4 NPs (2). (e) Three-dimensional AFM (3D-AFM) combined with fluorescence microscopy; perylene-labeled SiO2 NPs were visible on the outside of HUVEC cell membranes after exposure for 2 h (1); after 24 h (2) of incubation time, the cell surface is characterized by homogeneous distribution of only small humps, indicating intracellular localization of applied NPs. Adapted with permission from (a1) ref. 24, (a2) ref. 425, (b) ref. 426, (c1) ref. 427, (c2 and c3) ref. 428, (c4) ref. 429 (d) ref. 430 and (e) ref. 431. Copyright (a1) 2017 Vanhecke et al., (a2) 2017 Liu et al., (b) 2016 American Chemical Society, (c1) 2019 Martin et al., (c2 and c3) 2015 Lojk et al., (c4) 2011 John Wiley and Sons, (d) 2017 Royal Society of Chemistry and (e) 2013 John Wiley and Sons. | ||
Endocytic mechanisms can be differentiated based on the involvement of particular molecules called markers, such as clathrin in CME and caveolin-1 in caveolae-mediated endocytosis. Three main approaches are used to label these endocytic markers: (i) direct labeling via immunofluorescence, which is limited to fixed samples i.e., dead cells; (ii) cells expressing GFP-fusion proteins, where GFP-transcripts can be integrated (stable transfection) or not (transiently transfection) into the genome;230 or (iii) fluorescently labeled proteins, which are taken up by cells through specific mechanisms such as transferrin (clathrin-mediated endocytosis) and are finally located in the targeted vesicle. Nevertheless, besides the labeling of endocytic markers, NPs should also be tagged with fluorophores or some NPs can inherently emit specific optical signals (e.g., QDs and AuNPs).231,232 The identification of the internalization process of NPs is usually performed via fluorescence co-localization analysis of endocytic biological markers and NPs.233 This approach allows determining whether both, i.e. marker and NPs, localize within the same sub-cellular structures, such as NP co-localization with clathrin.234 In this case, it is possible to determine the extent of spatial overlap of both fluorophores (co-occurrence) and the proportional relation with each other (correlation).235 Peñaloza et al. studied the internalization mechanism of poly(3-hydroxybutyric acid-co-hydroxyvaleric acid) (PHBV) NPs in HeLa and human ovarian adenocarcinoma SKOV-3 cells.236 For that, NPs were functionalized with a fluorophore, Nile Red, and detection of caveolin-1 and early endosome antigen 1 (EEA1) was performed via immunofluorescence. Co-localization fluorescence microscopy in HeLa cells revealed that after 15 minutes NPs co-localize with caveolin-1 and EEA1, which suggests their cellular entry through caveolae-coated vesicles. In SKOV-3 cells, NPs did not co-localize with caveolin-1, indicating that PHBV NPs are internalized via a mechanism independent of caveolae. In addition, instead of NP co-localization with specific cell compartments, some researchers opt for co-localizing them with specific cargoes (e.g., fluorescently labeled cholera toxin B, transferrin and dextran).237,238 Dynamics and intracellular trafficking of NPs can also be studied in living cells. Li et al. applied a super-resolution photoactivated localization microscopy (PALM) with single particle tracking (SPT), which allows them to study the dynamics of PS NP interaction with cells with high spatial and temporal resolution.239 They concluded that NPs were internalized in two different ways via CME: (i) predominantly, NPs first attached to the cell membrane and clathrin-coated pits (CCP) formed at that site; (ii) NPs diffused on the membrane and translocated through a preformed CCP. Additionally, they revealed that 20 and 40 nm PS NPs were readily internalized via CCPs, whereas large PS particles (200 nm) were taken up by cells through a different mechanism.
Electron microscopy (EM) represents a different technique where a beam of electrons takes the place of light rays. EM is used to access the ultrastructural information of biological and non-biological specimens, providing a resolution in the nanometer range.240 In particular, transmission electron microscopy (TEM) provides valuable insight into the localization of NPs in the cellular compartments.241 Several drawbacks are associated with TEM, including the laborious sample preparation techniques, which includes fixation, dehydration, embedding, sectioning (<100 nm) and staining,240–242 thus does not allow one to visualize dynamic events. TEM is regularly used as a characterization method for NPs and it is also applied to visualize the localization and distribution of NPs within tissues, cells and subcellular structures.243 When investigating the cellular uptake mechanisms of NPs it is important to differentiate between cellular compartments. However, merely visualization of morphological appearance (e.g. clathrin vesicles, and early and late endosomes) can be challenging.240 To overcome these limitations, immunogold labelling of subcellular compartments can be performed.244 It is important to consider that organic NPs, such as polymer NPs, are composed by elements with a lower atomic number that are commonly present in the cell (e.g., carbon, hydrogen, nitrogen, and oxygen), which remarkably increases the difficulty to analyze them by EM techniques.243 Inorganic NPs such as gold and heavy metal NPs are easily identified inside or outside the cell due to their high electron density that enhances imaging contrast.240 Dobay et al. investigated the subcellular distribution of AuNPs in lung epithelial cells by TEM.245 They showed that AuNP uptake by cells occurs through different mechanisms including caveolae-dependent endocytosis. TEM has been applied in combination with different endocytic inhibitors to analyze the uptake route of polymer-coated AuNPs in A549 cells.161 It has been demonstrated that caveolae-mediated endocytosis was the main uptake mechanism for AuNPs.
Atomic force microscopy (AFM) is a powerful imaging tool that enables visualization and manipulation of biological samples, from single molecules to living cells.246 It can operate in liquid environments and allows the analysis of biomolecules and cells with resolution comparable to that of EM techniques (sub-nanometer).247 An AFM unit is composed of a flexible cantilever where an ultra-sharp tip is mounted. Topographical and mechanical information is obtained based on a piezoelectricity-driven scanning cantilever that measures the force between the sample and the sharp scanning probe tip.248 The principal drawbacks of imaging living cells are associated with tip contamination and possible interactions between the tip and the cell, which may lead to remodeling of events.247 Even if AFM provides three dimensional (x, y, z) information on tip position, cellular membrane deformations may occur due to the challenge in identifying the exact contact point between the tip and sample.246 In order to increase its potential, fluorescence microscopy techniques such as confocal and TIRF can be combined with AFM that allows multi-parameter characterization of single biomolecule structures and dynamic interactions.246 Hor et al. were able to study CME in human melanoma skin cells (SKMEL) by using AFM combined with fluorescence microscopy.249 In order to access the intracellular compartments, the upper membrane of the cells was removed using sonication (unroofing technique). By using genetically modified SKMEL cells that express a turquoise fluorescent protein (Tq2) along the clathrin light chain, they were able to correlate between the fluorescence and the topographical images of the clathrin-coated pit.
5. Methods to (semi)quantify NP uptake
Due to high interest in NPs applications for therapeutics delivery, accurate estimation of internalized NPs in cells is crucial. As an example, Dai et al. quantified the cancer cell-targeting efficiencies of the anti-cancer drug trastuzumab (Herceptin) and folic acid-coated gold and silica NPs in mouse tumor models.250 It has been shown that only 2% of cancer cells associated with the intravenously administrated NPs as assessed by flow cytometry. Less than 0.0014% of intravenously administrated NPs were delivered to targeted cancer cells as shown by inductively coupled plasma-mass spectrometry analysis. The majority of the NPs were either trapped in the extracellular matrix or taken up by perivascular tumor-associated macrophages. For this reason, it is crucial to develop methods to quantitatively assess the number of delivered drug-loaded NPs and to improve therapeutic efficacy. NP detection within single cells is challenging due to their small size.241 The development of highly sensitive methods and imaging techniques offers more reliable and reproducible results. There are several direct microscopic and analytical methods251 to quantify NP uptake by using fluorescently-labeled NPs,252 QDs,253 and magnetic NPs.254 Fluorescence microscopy may provide spatial information (e.g. subcellular localization) on NP uptake,255 but it is not optimal to estimate the total number of intracellular NPs. Also NP aggregation affects the estimation as it may cause difficulties to distinguish between intracellular and cell-associated NPs. A fluorescence signal depends on the excitation source, the number of fluorophores per NPs, the quantum yield of fluorophores or the NP, and the detector sensitivity. Therefore, the signals have to be compared with a control sample and are given as relative values, providing semi-quantitative outcomes.256In contrast, indirect methods such as inductively coupled plasma (ICP) techniques including ICP-optical emission spectroscopy (OES) and ICP-mass spectrometry (MS) can measure total mass of the NP elements.257,258 Quantification of NPs uptake using ICP-MS looks promising, but it requires sample destruction and does not provide any information about NP distribution within the cell.259 It is hard to draw a conclusion on which method is the most suitable for NP quantification, as it strongly depends on NP characteristics and therefore it is recommended to combine different methods, e.g. flow cytometry and confocal laser scanning microscopy260 or stereological sampling techniques and TEM.259 Recently, many new techniques have been described for NP quantification in cells, and only some of them will be described here.
5.1 Flow cytometry
Flow cytometry (FC) is a semi-quantitative fluorescence-based method for single-cell analysis,261 where a sheath fluid containing cells, loaded with fluorescent NPs, travels in a laminar flow regime. At the interrogation point, cells are sequentially excited by multiple lasers. Photomultiplier detectors capture the fluorescence emission signals. Every single cell is analyzed for visible light scatter to obtain information about the cell size (forward scatter or FSC) and granularity (side scatter or SSC), independently of the fluorescence signal.261 Further on, multiple lasers can obtain information about cell viability (according to live/dead staining) as well as integrated fluorescent signals coming from intracellular/associated NPs, such as QDs,262 TiO2263 or fluorescently labeled SiO2.264 FC is a rapid and sensitive method, providing information about thousands of cells (events) in one sample. Studies show that it is feasible to quantify in flow cytometry after determining the average fluorescence intensity per particle.264 However, this technique does not distinguish between intracellular and membrane-associated NPs.130,263 In this regard, flow cytometry can be used to complement confocal microscopy to obtain both, semi-quantitative and qualitative, information about NPs–cell interactions.130,252,265 The advantage of this technique is the ability to analyze numerous cells and fluorophores simultaneously, as well as to correct the spectral overlap between fluorophores (i.e. compensation). The latter is highly important in co-exposure studies. Finally, untreated and unstained control samples need to be included to achieve an optimized analysis of cell–NPs associations and to prevent autofluorescence detection.241 Another interesting aspect to consider is the possible NP interference with the fluorescent dyes used in FC. Two case studies addressed SiO2 NP and AuNP interference in the FC Annexin V/propidium iodide assay (quantifies apoptotic and necrotic cells). Both NPs caused distinct interference reactions. In the absence of serum proteins, SiO2 NPs induced false-positive signals, whereas gold NPs provoked fluorescence quenching effects.266For the design of NPs used in biomedical applications, it is important to obtain quantitative data about NPs uptake and at the same time information about spatial distribution of the NPs in live target cells. For this reason, imaging FC has been developed recently, which enables wide field imaging combined with high-throughput. It has been shown to be an efficient method for the quantification of nanoliposomes uptake in lymphocyte cells.267
5.2 Fluorescence microscopy combined with digital analysis
Confocal laser scanning microscopy (CLSM) is a commonly used imaging method to assess NPs–cell interactions for various biological systems.251 It provides insight into the uptake and intracellular fate of NPs in fixed and living cells,268,269 thus reflecting their spatial distribution. Discovery and development of fluorescent dyes offered new possibilities for CLSM application, such as use of NPs–antibody conjugates,270 particles loaded with dyes271 or NP functionalization.272Resolution limit of CLSM and the small NPs size prevent distinguishing between single particles and NP agglomerates. Alternatively, fluorescent particle events can be counted, where one event represents an individual compartment (e.g. endosome or lysosome) loaded with NPs.241 Therefore, only relative differences in NPs uptake per cell can be quantified by comparing different concentrations or time points.273
5.3 Elemental analysis
Elemental analysis techniques are available to quantitatively assess the elemental mass of NPs due to a very sensitive detection range (detection limit is less than 1 μg L−1). The most common techniques are ICP-MS, atomic emission spectrometry (AES) and OES.274Several research groups reported ICP-MS as a valuable technique to quantify NPs in various biological samples.139,275–279 For example, Sadauskas et al. quantified the gold content in mice liver and lungs by ICP-MS.278 ICP-MS/AES/OES has been used to investigate mouse brain samples for the presence of ceric oxide,277 uptake of the metal oxide NPs by human aortic endothelial cells (HAECs)276 and internalization of SV40-modified AuNPs in HeLa cells.275
Elemental analysis techniques have received considerable attention due to their sensitivity and precise quantification of various elements, especially metal- and metalloid-based NPs. Another advantage is the ability to quantify elements intracellularly. Unfortunately, sample digestion prior to analysis is inevitable and thus, distinguishing between internalized and membrane-associated NPs is not possible.241,251 In addition, ICP is not an optimal technique for quantification of silica, titania, and polymeric NPs due to intense polyatomic interferences.280
In order to obtain high-quality data, ICP techniques can be combined with optical and electron imaging methods such as contrast microscopy,275 TEM and CLSM281 that also provide spatial information.
5.4 Focused ion beam-scanning electron microscopy (FIB-SEM)
When paired with scanning electron microscopy (SEM), focused ion beam (FIB) provides high-resolution 3D images of the NP distribution at the single-cell level. The principle of FIB-SEM is to create an image of the surface layer by using a beam of charged gallium ions (Ga+), with a diameter of ∼10 nm. This technique enables both milling (with the FIB) and imaging (with the SEM) a specific region of interest in a fixed sample without moving the stage. By collecting many 2D-image stacks (i.e. parallel sections of constant thickness), we can reconstruct a 3D image of a sample.282Recently, FIB-SEM has become popular in nanoscience to visualize 3D nanostructures in a variety of biological specimens.283,284 James et al. describe FIB-SEM as a promising 3D approach for imaging and quantification of ZnO distribution and dissolution within human macrophages.285 FIB-SEM has also been used in combination with fluorescence microscopy and flow cytometry to study uptake and cellular distribution of silver NPs in mesenchymal stem cells.286
FIB-SEM automatically generates 3D images and has superior z-axis resolution (<10 nm). FIB operates at various angles and acts as a precise blade, thus creating a sample free of artifacts. The weakness of this technique is the time-consuming sample preparation and data collection steps, which can last up to several days.282 Furthermore, the process itself is destructive and does not allow re-imaging.287 In the future, FIB-SEM could be paired with other imaging techniques, which could open up new areas in nanoscience.282
5.5 Other methods
In addition to the above-described methods, there have been several other methods developed to assess NP uptake. Developments in environmental electron microscopy provide the possibility to study dynamic NP uptake at the subcellular resolution.288 Another approach is to combine stereology (i.e., estimation of three-dimensional structures from two-dimensional sections) and electron microscopy, allowing accurate assessment of the number of internalized NPs per cell.259 Brandenberger et al. applied this method to analyze the uptake of plain (i.e., citrate-stabilized) and PEG-coated AuNPs in A549 cells.128 The results showed the intracellular presence of ∼2500 and ∼3500 plain AuNPs per cell after 1 h and 4 h of exposure, respectively. In contrast, only ∼500 (1 h) and ∼1000 (4 h) PEG-coated AuNPs per cell were found inside the cells. This approach has been shown to be beneficial for estimating the relative distribution of NPs inside specific compartments (i.e., nucleus, mitochondria, endoplasmic reticulum and Golgi) and to follow the NP intracellular trafficking.Furthermore, surface-enhanced Raman spectroscopy (SERS) provides a detailed image of NPs–cell interactions.289 Next, correlative light and electron microscopy (CLEM) has become a powerful tool in studying NPs uptake.290 A combination of different approaches should be considered in order to correctly evaluate the results. The limitations of each system need to be considered for the correct interpretation of the results.
6. Stimulating NP endocytosis
In order to achieve high treatment efficacy, the enhanced uptake and accumulation of NPs in target cells is crucial. NPs uptake can be stimulated by their functionalization with targeting ligands on the surface or by co-exposure with other NP types. Besides, various bio-inspired molecules have been reported to significantly affect NPs cellular uptake. Table 2 represents the summary of molecules, which can stimulate NPs uptake.| Stimulus | Particles | Cell types | Summary of effects | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Concentration | Exp. time | Material | Size | Concentration | Exp. time | ||||
| ↑ increase; Exp. – exposure; Ref. – reference(s); LPS – lipopolysaccharide; IFN – interferon; IL – interleukin; MDM – monocyte-derived macrophages; BMDM – bone marrow-derived macrophages; TAM – tumor-associated macrophages; AM – alveolar macrophages; HDL/LDL – high/low density lipoprotein; PS – polystyrene; HS – human serum; BSA – bovine serum albumin; MSN – mesoporous silica; CB – carbon black; TAT – Trans-activator of transcription peptide; As – arsenic; IONPs – iron oxide nanoparticles; QDs – quantum dots; PLGA – poly lactic-co-glycolic acid; SPIONs – superparamagnetic iron oxide nanoparticles; PEG – polyethylene glycol; FA – folic acid; Cys –cysteine; SR9 – nona-arginine; R11 – polyarginine; PSL – polystyrene latex spheres; YGTM - Fluoresbrite microspheres; HA – hyaluronic acid; MET-H – Metformin hydrochloride; O/N – overnight; n/a – not applicable. | ||||||||||
| Inflammatory molecules | ||||||||||
| Cytokines | LPS, IFN-γ, IL-10 | 20–1000 ng mL−1 | 40 h | Silicon oxide | 26, 41, 1750 nm | NPs: 50 μg mL−1 | 1–3 h | MDM, AM, TAM, THP-1 | IL-10 ↑ uptake of 26 and 41 nm NPs in MDMs and THP-1 | 22 |
| Microparticles: 100 per cell | ||||||||||
| IL-10 | 20 ng mL−1 | 7 days | HDL, LDL | n/a | 40–50 μg mL−1 | 4 h | RAW264.7, MDM, Kupffer cells | IL-10 ↑ HDL and LDL uptake in RAW264.7 | 298 | |
| LPS, IFN-γ, IL-10, IL-4 | 10–100 ng mL−1 | 24–48 h | PS | 30, 50, 100 nm | 1013–1015 per mL | 8–24 h | BMDM | LPS and IFN-γ ↑ uptake of 100 nm NPs by M1 macrophages | 292 | |
| Serum | HS | 10% | 0.5–6 h | Silica | 50, 100, 200, 500, 1000 nm | 25 μg mL−1 | 0.5–6 h | THP-1 | HS ↑ uptake of NPs by M2 macrophages | 295 |
| BSA | n/a | n/a | MSN | 50 nm | 50–500 μg mL−1 | 2–4 h | RAW264.7 | BSA ↑ uptake of 100 nm NPs by M2 macrophages | 296 | |
| Immuno-globulins | CD200 | 30 molecules per μm2 | n/a | PLGA | 7 μm | 10 per cell | 12 h | BMDM, monocytes | CD200 ↑ phagocytosis of PLGA NPs | 355 |
| Co-exposure with different nano-/micro-particles | ||||||||||
| NPs | SiO2 | 10 μg mL−1 | 0.5 h, 4 h | TiO2 | <50 nm | 10 μg mL−1 | 0.5–4 h | BMDM | SiO2 and TiO2 NPs synergistically ↑ macrophage inflammatory response | 301 |
| Au | 38.6 μg mL−1 | 24 h | FeOx | 28 nm | 54.8 μg mL−1 | 24 h | J774A.1 | ↑ uptake and colocalisation of Au and FeOx NPs in co-exposure | 24 | |
| Carbon black | 0.5 μg mL−1 | 25 h | Fe2O3 | 45 nm | 0.5 μg mL−1 | 25 h | A549 | ↑ ROS; intracellular redox reaction between CB and Fe3+ | 23 | |
| Ag | 0.3 μg mL−1, 3.5 μg mL−1 | 4 h | HgCl2, CdCl2 | n/a | 2.8–28 μM HgCl2 | 4 h | HepG2 | ↑ uptake of HgCl2 in co-exposure with Ag NPs | 304 | |
| 0.15–1.5 μM CdCl2 | ||||||||||
| SiO2 | 10 μg mL−1 | 24 h | As | n/a | 1 μg mL−1 | 24 h | HepG2, HT1080 | ↑ intracellular level of As in the co-exposure with SiO2 | 303 | |
| Micro-particles | PSL | 1–10 per cell | 2 h, 4 h | R-PLGA | 3 μm | 1–10 per cell | 2–4 h | NR8383 | ↑ uptake of PSL in the presence of R-PLGA | 302 |
| Bio-inspired molecules | ||||||||||
| Cell-penetrating peptides | TAT | 10 μM | n/a | Ag, Au, IONPs, QDs, BSA, dextran | 20–50 nm | Ag/AuNPs: 0.27–0.79 nM; IONPs, QDs: 50 μg mL−1 | 1 h | CHO, H1975 | TAT-Ag ↑ the uptake of bystander Ag NPs | 195 |
| n/a | n/a | MSN | 50 nm | 100 μg mL−1 | 2–4 h | RAW264.7 | Cys-TAT ↑ uptake of MSN NPs | 296 | ||
| n/a | 4 h | PLGA | 189 nm | n/a | 4 h | Caco-2 | TAT-PLGA conjugation ↑ PLGA uptake | 327 | ||
| Pep TGN | n/a | n/a | PEG–PLGA | 100 nm | 50 μg mL−1 | 0.5–2 h | bEnd.3 | Conjugation with Pep TGN ↑ NPs cellular uptake | 322 | |
| SR9 | 6 μM | n/a | QDs | n/a | 100 nM | 1 h | A549 | SR9 ↑ QDs uptake | 324 | |
| R11 | n/a | n/a | SPIONs | 38 nm | 50 μg mL−1 | 4 h | T24, SV-HUC | ↑ uptake of SPIO-R11 | 325 | |
| Sta-R8 | n/a | n/a | FA–PLGA/PK3 | 137 nm | n/a | 2 h | RAW264.7 | FA and Sta-R8 ↑ uptake of PLGA NPs | 326 | |
| Bacteria | Listeria | n/a | n/a | n/a | 40, 200 nm | 109 particles | 0.5–72 h | KB | ↑ cargo uptake in the presence of bacteria | 335 |
| Vitamins | Folic acid | n/a | n/a | MSN | 110–168 nm | 0.01–20 μg mL−1 | 2 h | Y79 | ↑ uptake of FA-conjugated MSN via receptor-mediated endocytosis | 341 |
| n/a | n/a | ZnO | 20 nm | 1.25–10 mg mL−1 | 12–48 h | U87MG | ↑ uptake of FA-conjugated ZnO via receptor-mediated endocytosis | 340 | ||
| n/a | n/a | Au@SiO2 | 105 nm | n/a | 0.5–3 h | MCF-7, MDA-MB-231 | ↑ uptake of Au@SiO2 in MDA-MB-231 via receptor-mediated endocytosis | 339 | ||
| n/a | n/a | Pentablock copolymer | 21 nm | n/a | 4–24 h | HeLa, HEK293 | ↑ uptake of FA-conjugated NPs via receptor-mediated endocytosis | 338 | ||
| Vitamin B12 | n/a | n/a | YG™ | 50, 100, 200 nm | n/a | O/N | Caco-2 | Modification with vitamin B12 ↑ YG uptake; highest uptake for 50 nm YG | 351 | |
| Vitamin C | 300 mg mL−1 | n/a | ZnO | 112–196 nm | 15 mg L−1 | 2–24 h | GES-1 | Vitamin C ↑ endocytosis of ZnO and ↑ cytotoxicity | 352 | |
| Vitamin E | n/a | n/a | PLGA, PS | 200, 500, 1000 nm | n/a | 2 h | Caco-2 | Vitamin E ↑ endocytosis of PLGA | 353 | |
| Polysaccharides | Hyaluronic acid | n/a | n/a | Au | 5, 10, 20, 50, 100 nm | 0.01–0.25 mg mL−1 | 24 h | ARPE-19, NIH 3T3, CHO | ↑ uptake of 50 and 100 nm Au NPs functionalized with HA | 343 |
| n/a | n/a | Au | 30–50 nm | 0–18 μg mL−1 | n/a | HepG2, NIH 3T3 | MET-H-AuNPs ↑ apoptosis in HepG2; no impact on NPs uptake | 344 | ||
| n/a | n/a | siRNA | 50–80 nm | 100 nM | 12 h | A549, H69, MDA-MB468, Hep3B, B16F10 | ↑ siRNA silencing activity in presence of HA | 346 | ||
| n/a | n/a | PEG–PLGA | 250–350 nm | n/a | 12 h | L929, MDA-MB-231, MCF-7, HepG2 | ↑ endocytosis of HA–PEG–PLGA micelles in MDA-MB-231 and MCF-7 | 345 | ||
| Mannose | n/a | n/a | Liposomes | 1000 nm | 1 mM | 2 h | NR8383 | ↑ uptake of liposomes in the presence of mannose | 348 | |
6.1 Cytokines and other inflammatory molecules
Much attention has been given in recent years to investigate interactions between phagocytic cells and NPs.22 This is beneficial to better understand the behaviour of NPs in the human body in the presence of inflammatory molecules and their impact on phagocytic activity. Phagocytes are specialized cells of the innate immune system, which include monocytes, macrophages (mature monocytes), granulocytes (neutrophils and eosinophils) and dendritic cells. Their primary role is to eliminate foreign macromolecules/particles via macro- and microscale endocytic pathways and to degrade them by specific enzymes in lysosomes. As a response, phagocytes are able to secrete different inflammatory molecules, interferons, interleukins, and growth factors called cytokines, which stimulate a specific immune response by recruiting lymphocytes. Additionally, phagocytes are able to respond to different inflammatory stimuli from their local microenvironment.291Inflammatory stimuli not only reprogram (polarize) macrophages into pro-inflammatory M1 phenotype or anti-inflammatory M2 phenotype but can also enhance NPs uptake. Hoppstädter et al.22 investigated endocytosis of SiO2 NPs (26 and 41 nm) and SiO2 microparticles (1.75 μm) by different macrophage sub-populations, while stimulating them with various molecules: lipopolysaccharide (LPS)/interferon (IFN)-γ to generate M1 phenotype and interleukin (IL)-10 to differentiate cells into M2 macrophages. NPs uptake was higher in M2-polarized macrophages compared to M1 cells in all macrophage subpopulations. Higher endocytic capacity in M2-polarized cells has been explained due to higher expression of endocytosis-facilitating scavenger and lectin receptors on the surface. Interestingly, the uptake of microparticles did not differ between M1 and M2 phenotypes. In contrast, Qie et al.292 observed increased uptake of differently sized PS NPs (30, 50, and 100 nm) by M1 primary bone-marrow derived macrophages, stimulated with LPS (Fig. 6) or LPS and IFN-γ.
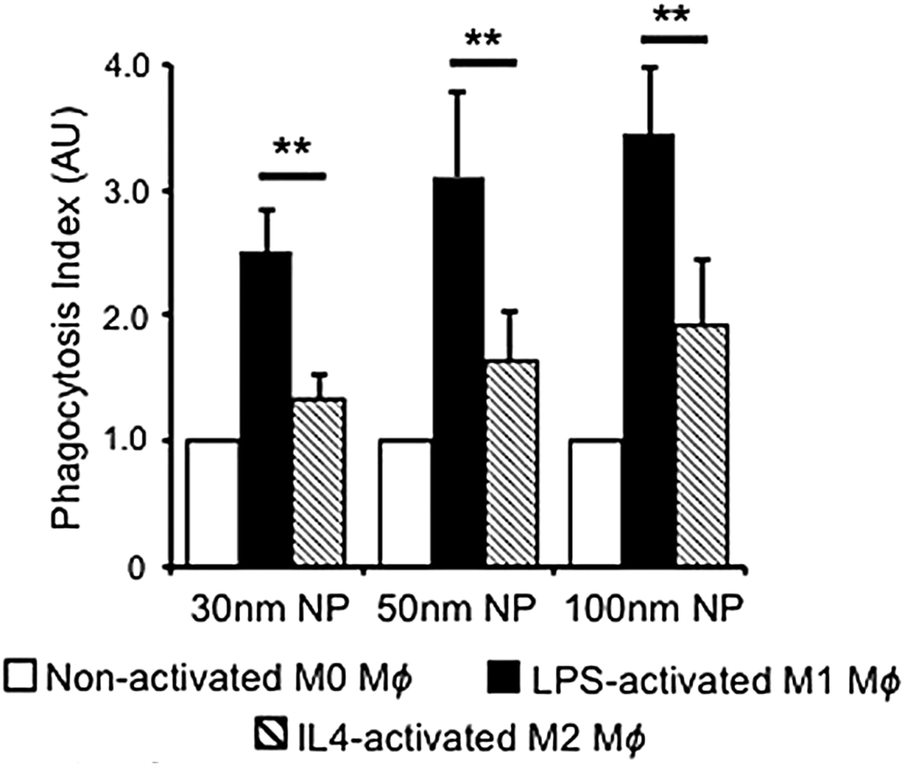 | ||
| Fig. 6 Increased uptake of different sized PS NPs in lipopolysaccharide-activated macrophages analyzed by flow cytometry and expressed as phagocytosis index (AU). Adapted with permission from Qie et al.292 Copyright 2016 Qie et al. | ||
According to the Food and Drug Administration (FDA), a novel nanomedicine, CYT-6091 (Aurimune™), constructed by binding recombinant human tumor necrosis factor alpha (rhTNF-α) to colloidal gold NPs, was tested in Phase 1 of the clinical trials in cancer patients.293 It has been demonstrated that colloidal gold-bound rhTNF-α successfully targets the tumors and can be administered at doses of rhTNF-α that were previously shown to be toxic to the patients. In the future, clinical studies will focus on combining CYT-6091 with approved chemotherapies for the systemic treatment of non-resectable cancers.294
Cytokines are not the only stimuli that can enhance NPs uptake by phagocytes. When NPs are exposed to biological fluids, formation of protein corona on NPs surface strongly influences their interaction with cells. For example, several serum proteins (immunoglobulins, albumin and fibrinogen) can act as ligands for macrophage receptors.295,296 Human monocytes (THP-1) were treated with the following cytokines to generate specific phenotypes: IFN-γ/LPS for M1 and IL-4/IL-13 for M2. Cellular uptake of SiO2 NPs (50, 100, 200, 500 and 1000 nm) was investigated with or without the presence of human serum in the cell culture medium. Uptake of 200–1000 nm NPs in the presence of serum was higher by M2 compared with M1 macrophages. The reason could be due to higher expression of receptors against protein corona ligands (fibrinogen, IgG and HDL) on M2 macrophages.295 In addition, higher uptake of various cargos other than NPs (e.g. HDL, LDL, and bacteria) in phagocytic cells when exposed to inflammatory stimuli (cytokines) has also been observed.297–299
There is no rule about which stimuli/NPs combination generally causes the highest uptake rate in phagocytes. We conclude that the uptake highly depends on cell type and NP physicochemical properties. A deeper knowledge of the interaction between NPs and phagocytes in the presence of inflammatory stimuli is of a great importance in nanomedicine as such NPs/drugs are or will be mainly applied in diseased patients. For instance, inflammatory stimuli can act as a NPs uptake accelerant to increase the delivery of a pharmaceutical substance to cancer cells.300
6.2 Co-exposure with different nanoparticles
With the development of nanotechnology, NPs have emerged as potential carriers for therapeutics. Several studies in recent years have pointed out that NPs can be internalized faster, while co-exposed due to activation of multiple parallel endocytic pathways, i.e., synergistic effect.23,24,134,301–304 This suggests NP co-exposure as a potential approach to increase the intracellular accumulation of drug-loaded NPs. When exposing cells to multiple particles, cytotoxic effects should also be considered.Vanhecke et al. revealed synergistic effects of Au and Fe3O4 NPs co-exposure on murine macrophage cells (J774A.1).24 Live cell imaging with confocal microscopy, environmental scanning electron microscopy (SEM) and ICP-OES data showed that co-exposure accelerates uptake due to activation and crosstalk between different endocytic pathways (Fig. 7).
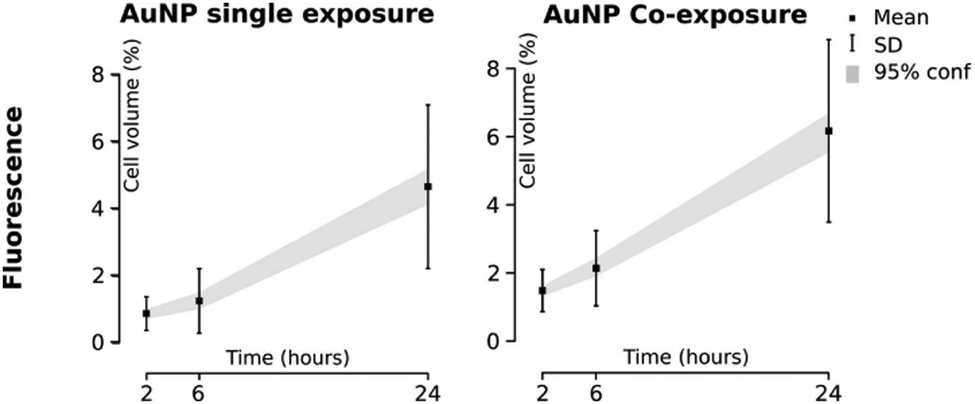 | ||
| Fig. 7 Quantification studies based on confocal laser scanning microscopy (CLSM) show a time-dependent increase in uptake of Au NPs in J774A.1 macrophages. The highest increase was observed in Au NPs co-exposure with Fe3O4 NPs. Adapted with permission from Vanhecke et al.24 Copyright 2017 Vanhecke et al. | ||
Tsugita et al. examined the effect of combined exposure of SiO2 and TiO2 NPs on murine bone-marrow-derived macrophages.301 This type of co-exposure synergistically induced the macrophage inflammatory response by use of distinct cellular mechanisms to cause cellular stress but does not reveal any effects on cellular NPs uptake.
Guo et al. examined synergistic oxidative effects of carbon black and Fe3O4 NPs co-exposure on human lung epithelial cells (A549) indicating intracellular redox reaction between both NPs.23 No significant difference was found in Fe3O4 NPs uptake between single- and co-exposure.
The synergistic effect of combined NPs exposure, such as silver and cadmium,304 SiO2 and arsenic,303 PS latex and poly-lactic-co-glycolic acid302 has also been investigated. These studies have focused mainly on synergistic cytotoxic effect, but no information is mentioned about their effect on cellular uptake.
6.3 Bio-inspired molecules
Nature has developed structurally and molecularly diverse substances, which can be used in combination with NPs (e.g. as a surface coating or co-exposure approaches) to achieve higher cellular uptake. Bio-inspired molecules have become increasingly interesting in science due to their ability to serve as alternative biocompatible drug delivery systems.305 In this section, various bio-inspired molecules found in the literature are discussed.NPs conjugates with CPPs could enhance NPs uptake and therapeutic effectiveness without causing significant cell damage. One of the first discovered CPPs was the cationic trans-activator of transcription peptide (TAT) from human immunodeficiency virus 1 (HIV-1).310 It has been demonstrated that TAT peptide is highly efficient in translocating different small molecules across the cell membrane.311–313 TAT peptide enhances the uptake of multiple bystander cargo (e.g. AgNPs, AuNPs, QDs, IONPs, BSA and dextran) through the macropinocytosis pathway in different cancer cell types (Fig. 8).195,313
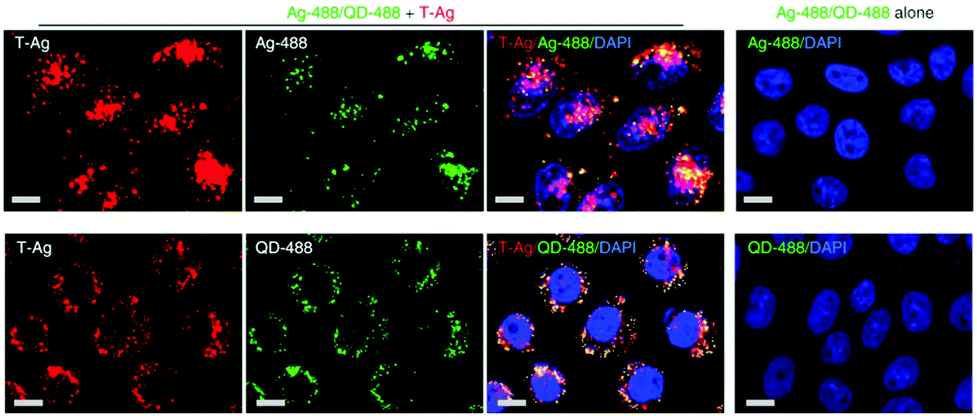 | ||
| Fig. 8 Ag NPs functionalized with cell-penetrating peptide TAT (T-Ag; red) enhanced cellular uptake of bystander NPs (Ag-488 and QD-488; green) in CHO cells. Representative CLSM images for Ag-488 (top panels) or QD-488 (bottom panels). Scale bar: 10 μm. Reproduced with permission from Wei et al.195 Copyright 2019 Wei et al. | ||
Shortly after TAT-peptide discovery, a homeoprotein penetratin of fruit fly Drosophila melanogaster has also been shown to have the ability for transporting therapeutic molecules across the cell membrane.314 More than 100 peptides have been used since then to accelerate therapeutics delivery, such as siRNA,315 antisense oligomers,316 enzymes,317etc. into eukaryotic cells in preclinical and clinical trials.307
Several studies exhibited the potential of NPs–CPP conjugates to enhance therapeutics delivery to various cancer cells both in vitro and in vivo, such as brain glioma,318 breast cancer,319 hepatocarcinoma,320 lung cancer321 and others. Conjugation of PEG–poly(lactic-co-glycolic acid) (PEG–PGLA) NPs with Pep–TGN peptides resulted in 3.6-fold higher delivery of coumarin-6 into brain cells compared to unmodified NPs.322 Cellular uptake of different NPs–CPP conjugates into drug resistant cancer cells showed that CPPs uptake is cell type-dependent.323
CPPs in combination with NPs can also be used as imaging agents to follow the pharmacokinetics and pharmacodynamics of the therapeutics. Liu et al. demonstrated that arginine-rich CPPs significantly increase cellular uptake of QDs.324 Higher accumulation of SPIONs functionalized with (CPP)–polyarginine peptides in comparison to unconjugated SPIONs in bladder cancer cells has also been described.325
The potential of CPPs to increase cellular NPs uptake has been applied for inflammatory disease treatment in mice, e.g. delivery of anti-rheumatic drug methotrexate using lipid polymeric hybrid nanoparticles (LNPs) conjugated with stearic acid–octarginine, folate–PEG–PLGA and polyketal (PK3).326 Finally, yet importantly, Yan et al. demonstrated the potential of NPs–CPP conjugates in neurodegenerative disease treatment, e.g. TAT functionalized PLGA NPs showed 4.5-fold increase in intracellular accumulation of insulin in the brain.327
Although there is an increasing number of CPPs reported in the literature, none of the CPP-based treatments have been approved yet by the FDA. The reasons could be a lack of proteolytic stability, selectivity, and the issues with the endosomal release. However, many CPPs are used in (pre)clinical studies, showing promising results in treating diseases, such as cancer.328,329
In conclusion, NP functionalization with CPPs has a great potential to enhance delivery of therapeutics into cells without provoking any cell damage. Structural characteristics such as sequence length, amino acid composition, and chirality play an important role in CPPs uptake and should be investigated for future improvements of efficiency of NPs as drug delivery carriers. Furthermore, development towards protease stable and water soluble CPPs is highly desirable.
By co-exposing cells to NPs and bacteria or their toxins and thus stimulating more endocytic pathways simultaneously, NPs uptake and therapeutics delivery can potentially be increased. Akin et al. co-exposed NPs and bacteria to deliver DNA-based therapeutics. A fluorescent or bioluminescent gene was loaded as a cargo on the NPs, which were carried on the bacteria. After exposure to different tumor cell lines, the cargo-carrying bacteria were phagocytosed. Bacterial toxins caused the disintegration of endosomal compartments and the cargo was released intracellularly from the NPs. This approach aimed to use bacteria as a carrier for various therapeutic peptides, antibodies or small molecule drugs.335
Folic acid (FA) is the synthetic form of folate, a water-soluble B9 vitamin. It is a natural product in foods such as green vegetables (lettuce, spinach, and broccoli), legumes (lentils, peas, and beans), and fruits (oranges, and mangoes).337 Many cancer cells overexpress FA receptors on their surface. Functionalization of NPs with FA could enhance NPs uptake in cancer cells as a specific targeting approach.338–340 FA was conjugated to MSNs loaded with the chemotherapeutic drug topotecan and delivered to retinoblastoma cells. The FA-conjugated NPs exhibited higher uptake in retinoblastoma cells compared to that of non-conjugated NPs. The higher uptake was attributed to FA receptor-mediated endocytosis.341
Hyaluronic acid (HA) is a carbohydrate, naturally present in the extracellular matrix in all living organisms. It is one of the most hydrophilic molecules found in nature, responsible for tissue hydration and water transport.342 HA serves as a ligand for many cell-surface receptors and has been investigated as a drug delivery agent in combination with various NPs. Karakocak et al. demonstrated facilitated uptake of 5–100 nm AuNPs, coated with end-thiolated hyaluronate (HS–HA) compared to that of uncoated NPs (Fig. 9).343 They also illustrated that the presence of CD44 receptors on the cell surface can facilitate HS–HA–AuNPs uptake. HA-conjugated NPs have potential as drug delivery vectors for cancer cells overexpressing CD44 receptors. The increased uptake of other NPs, such as AuNPs,344 PEG–PLGA345 and siRNA346 functionalized with HA has also been demonstrated.
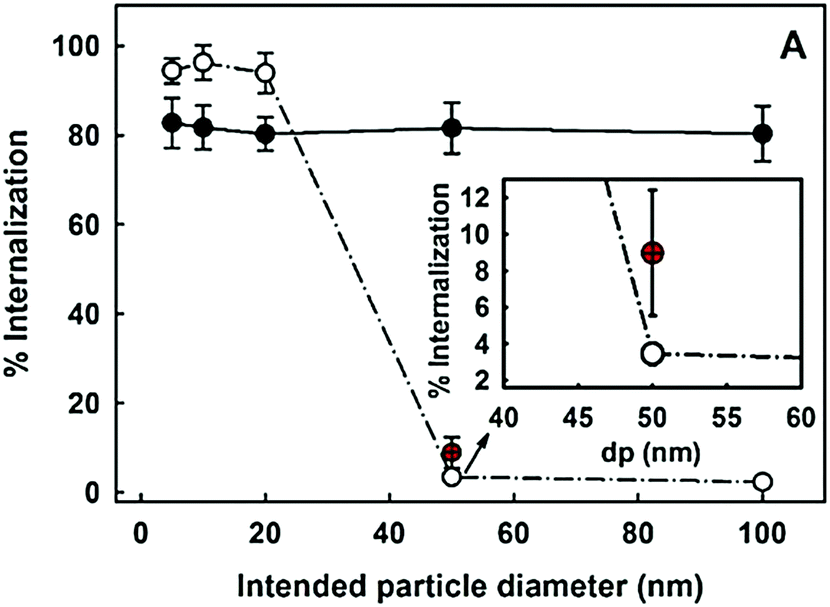 | ||
| Fig. 9 ICP-MS data showing increased uptake of Au NPs coated with (●) end-thiolated hyaluronic acid (HS–HA) and compared to (○) bare Au NPs in human retinal pigment epithelial cells. Adapted with permission from Elsevier.343 Copyright 2018 Elsevier. | ||
Mannose is a sugar monomer and a natural component of some plants (e.g. cranberries). Mannose-conjugated NPs can be used for targeted NPs delivery.347 Chono et al. exposed alveolar macrophages and NR8383 cells to liposomes, mannosylated with 4-aminophenyl-α-D-mannopyranoside. The differences in cellular uptake between mannosylated and non-mannosylated NPs were evaluated in vitro and in vivo. The uptake of mannosylated liposomes by both cell types was significantly greater compared to non-modified liposomes. The uptake mechanism of mannosylated liposomes by alveolar macrophages is believed to be mannose receptor-mediated.348
Vitamins are a group of essential organic compounds, which are required in nutrition for maintaining health, development and metabolic integrity. They function as co-enzymes, hormones, antioxidants, cell signaling mediators, growth regulators etc.349 In addition, some vitamins can enhance cellular NPs uptake. One of such vitamins is water-soluble vitamin B12 (i.e. cobalamin). Dietary products rich in vitamin B12 are fish, poultry, eggs and milk. B12 serves as a co-factor for enzymes, involved in amino acid synthesis. Its adequate supply is essential for red blood cell generation and optimal function of central nervous system.350 Russell-Jones et al. showed that conjugation of NPs (50 nm, 100 nm and 200 nm) with vitamin B12 highly enhanced their uptake into Caco-2 cells, an intestine cell line, compared to uncoated NPs.351 Wang et al. investigated the endocytosis and cytotoxicity of zinc oxide (ZnO) NPs and vitamin C co-exposure in gastric epithelial and neural stem cells.352 The presence of vitamin C increased intracellular concentration of Zn2+ ions and ZnO-associated cytotoxicity. Vitamin C created an acidic environment and caused the dissociation of ZnO into Zn2+ ions. By intracellular Zn2+ elevation, zinc homeostasis was disrupted, resulting in lysosomal and mitochondria damage and apoptosis.
Yin Win et al. showed that surface coating of PLGA NPs with vitamin E greatly improved adhesion to and cellular uptake into intestinal Caco-2 cells.353 In addition, it increased the ability of NPs to escape the cells via multi-drug resistance pumps. Since coating with vitamin E could increase NPs cellular uptake and inhibit P-glycoprotein-mediated drug transport, this mechanism could potentially improve the efficacy of oral NPs delivery.
A NPs-based therapeutic ND L02-s0201 (Nitto BioPharma Inc.) has been developed for the treatment of fibrosis of liver and other organs. It specifically targets hepatic stellate cells through the use of liposomes that are conjugated to vitamin A.354 Hepatic stellate cells will specifically take up vitamin A, so the drug is delivered to the stellate cells preferentially, but not to other liver cells. The specific targeting of stellate cells together with delivery of a siRNA that can downregulate collagen production has resulted in the reversal of cirrhosis and liver regeneration. The nanoformulation has already entered Phase 2 of clinical trials.
7. Decreasing NP endocytosis
The majority of studies investigated drug-loading capability of functionalized NPs in combination with different stimuli to improve NPs uptake into target cells. Another very important aspect is to investigate specific interactions of NPs with the microenvironment resulting in reduced cellular uptake. For instance, internalization of NPs loaded with therapeutics by cells of a mononuclear phagocyte system (MPS) decreases the treatment efficacy for other target cells/tissues. It is important to explore ways to make NPs invisible for the MPS, but still ensure highly efficient delivery and internalization of NPs into target cells.356 It has been shown that some pathogens are able to survive long periods in hosts without being detected and eliminated by the immune system.357 Therefore, a possibility exists to coat NPs with specific biomolecules that protect them from being phagocytosed by MPS and to prolong its circulation. The concept could also be used to decrease the uptake of unintentionally ingested or inhaled NPs. Table 3 represents an overview of the substances related to decreasing NPs uptake.| Stimuli | Particles | Cell types | Summary of effects | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Types | Concentration | Exp. time | Material | Size | Concentration | Exp. time | ||||
| ↓ – decrease; Exp. – exposure; Ref. – reference(s); IL – interleukin; HSA – human serum albumin; FBS – fetal bovine serum; YG™ – Fluoresbrite microspheres; BM – bone marrow; Tf – transferrin; TfR – transferrin receptor; ROS – reactive oxygen species; DHLA – dihydrolipoic acid; BMDC – bone marrow-derived dendritic cells; QDs – quantum dots; PLGA – poly lactic-co-glycolic acid; SPIO – superparamagnetic iron oxide; PEG – polyethylene glycol; MDM – monocyte-derived macrophages; PMN – polymorphonuclear leucocytes; PS – polystyrene; FFA – free fatty acids; Yop – Yersinia outer protein; Sic – Streptococcal inhibitor of complement; PICALM – phosphatidylinositol binding clathrin assembly protein; n/a – not applicable. | ||||||||||
| Inflammatory molecules | ||||||||||
| Cytokines | IL-4, IL-13 | 2–6 ng mL−1 | 48 h | Neisseria, E.coli, zymosan | n/a | 100 per cell | 2 h | Mouse peritoneal macrophages | IL-4 and IL-13 ↓ uptake of bacteria and zymosan via IL-4/IL-13 receptors | 362 |
| IL-4 | 10 ng mL−1 | 48 h | YG™ | 1 μm | 108 mL−1 | 0.5 h | Primary BM mononuclear cells | IL-4 ↓ phagocytosis of YG beads in macrophages | 364 | |
| 20 ng mL−1 | 48 h | B-lymphoma cells | n/a | 106 mL−1 | n/a | Human macrophages | IL-4 ↓ phagocytosis of rituximab opsonized lymphoma cells by macrophages | 365 | ||
| Co-exposure with different nanoparticles | ||||||||||
| Nano-particles | SiO2 | 10–250 μg mL−1 | 24–72 h | Fe3O4 | 20–30 nm | 10–250 μg mL−1 | 24–72 h | A549 | Effect on NPs uptake unknown; ↓ ROS and toxicity in co-exposure | 367 |
| Surface modification | ||||||||||
| Proteins | HSA | 100 μM | n/a | DHLA-QDs | 5 nm | 10 nM | 2 h | HeLa | HSA ↓ DHLA-QDs endocytosis | 418 |
| FBS | 0, 10, 55% | n/a | SiO2–PEG–Tf | 50 nm | 50 μg mL−1 | 1–8 h | A549 | FBS proteins ↓ TfR targeting for Tf-conjugated NPs | 272 | |
| Polyethylene-glycol | Amino-PEG | 10.000 g mol−1 | n/a | PS | 30, 50, 100 nm | 1013–1015 mL−1 | 8–24 h | Primary BMDM | Amino-PEG coating ↓ PS NPs uptake | 292 |
| PEG | n/a | n/a | FePt, Fe3O4 | 4–11 nm | 5–10 nM | 12 h | NIH/3T3 | PEGylation ↓ uptake of the NPs for 10% | 375 | |
| Thiolated-PEG | 50 PEG per nm2 Au | n/a | Au | 15 nm | 20 nM | 1–24 h | A549 | Thiolated-PEG ↓ uptake of Au NPs | 128 | |
| Surface markers | CD47 | n/a | n/a | PS | 30, 50, 100 nm | 1013–1015 mL−1 | 8–24 h | BMDM | ↓ phagocytosis of PS NPs, coated with CD47 | 292 |
| 21 μm−2 bead | n/a | Streptavidin–PS | 100, 160, 1100, 3500 nm | n/a | 3/4 h | THP-1 | CD47 ↓ phagocytosis of streptavidin–PS by signalling through the phagocyte receptor SIRPα | 398 | ||
| Bio-inspired molecules | ||||||||||
| Bacterial proteins | Yersinia (Yop) | 400 μg mL−1 | n/a | n/a | n/a | n/a | n/a | HeLa | YopE ↓ internalization of bacteria by ↓ PI3K/AKT pathway | 419 |
| n/a | 1 h | n/a | n/a | n/a | n/a | Murine BMDC | YopE, YopH and YopT ↓ uptake of bacteria by actin fibers destruction | 408 | ||
| n/a | 1 h | n/a | n/a | n/a | n/a | J774A.1 | YopH ↓ internalization of bacteria via ↓ PI3K/AKT pathway | 407 | ||
| Streptococcus (Sic) | 10 μg mL−1 | 1 h | n/a | n/a | n/a | n/a | PMN | Sic ↓ phagocytosis of bacteria | 409 | |
| Viral proteins | Herpes virus (K14) | n/a | n/a | n/a | n/a | n/a | n/a | MDM | K14 ↓ macrophage activation by interacting with human CD200R; unknown effect on uptake | 420 |
| Fatty acids | Oleic, stearic, α-linoleic | 500 μM | n/a | ZnO | 22 nm | 4 μg mL−1 | 6 h | THP-1 | FFA ↓ the NPs uptake and alter NPs–cell interactions | 411 |
| Others | ||||||||||
| Chloroquine | n/a | 20–400 μM | n/a | Albumin, lyposome, PS, silicon | 14–264 nm | Albumin: 20 μg mL−1; liposomes: 200–400 μM; PS: 3 × 109 mL−1; silicon: 6 × 107 mL−1 | 3–6 h | J774A.1, RAW264.7, Kupffer cells MDA-MB-231, MIA PaCa-2, H358 lung cancer cells | Chloroquine ↓ NPs uptake via clathrin-mediated endocytosis by ↓ PICALM expression | 179 |
7.1 Cytokines and other inflammatory molecules
Cytokine stimulation of macrophages,297 dendritic cells,358,359 fibroblasts360 and endothelial cells361 can regulate cellular endocytic mechanisms. An in vitro study investigated the uptake of the pathogenic bacteria Neisseria meningitidis in mouse peritoneal macrophages after IL-4 and IL-13 pretreatment.362 These cytokines are responsible for alternative activation of macrophages towards M2 anti-inflammatory phenotype.363 After 48 hours of IL-4 pretreatment, a remarkable reduction in Neisseria meningitidis uptake was reported. Reduction of uptake was also observed with E. coli and zymosan. Coincidently, IL-4 activation stimulated the secretion of pro-inflammatory cytokines by macrophages. Reduced phagocytosis of bacteria was due to inhibition of phagosome formation by down-regulation of PI3K activity.362 The inhibitory effect of IL-4 on uptake of 1 μm fluorescent beads364 and rituximab-opsonized lymphoma cells365 in macrophages have also been shown. We assume that decreased cellular uptake caused by IL-4 or IL-13 not only impacts uptake of bigger particles in the micrometer size but could also apply for NPs. Decreased uptake after cytokine stimulation has also been observed for dendritic cells. Patente et al. induced dendritic cell maturation by stimulation with the cytokines TNF-α and IFN-γ.359 While the immature cells expressed high endocytic capacity, the cytokine-stimulated mature dendritic cell showed a decrease in endocytic activity.7.2 Co-exposure with different nanoparticles
In addition to many synergistic effects,23,301,366 few studies have demonstrated the antagonistic effect of NPs co-exposure. Rafieepour et al. investigated the in vitro toxicological effects of single and combined exposure of magnetite (Fe3O4) NPs and polymorphous silica NPs on human epithelial cell line A549.367 The cells were exposed to four different NP concentrations (10, 50, 100, and 250 μg mL−1) of both NP types simultaneously for 24 h and 72 h. The data obtained in this study showed that increasing the concentration and exposure time to Fe3O4 and SiO2 NPs individually increased toxic effects. In contrast, the effect of combined exposure to Fe3O4 and SiO2 NPs led to antagonistic interactions. Combined exposure resulted in decreased reactive oxygen species (ROS) production and reduced toxicity (i.e. antagonistic effect) in comparison to single exposures. The reason for antagonistic effect in combined exposures has been explained by the accumulation of intracellular proteins on the SiO2 surface, forming a protein corona. Fe3O4 could provoke the synthesis of cellular proteins, causing the formation of protein corona on the surface of silica NPs and thereby reducing its cytotoxic effect.368,369 This phenomenon needs to be investigated further and we assume that another mechanism might be involved in the antagonistic effect of the two NPs. Unfortunately, this study did not investigate the antagonistic effect of combined exposure on intracellular uptake of NPs.7.3 Protein corona
NPs exposure to complex biological environment results in non-specific protein coating of particles, known as a “protein corona”.370 The corona consists of two components: (i) the inner firmly bound high-affinity proteins, known as “hard corona” and (ii) the outer loosely associated low-affinity proteins described as “soft corona”.124,371 “Hard” corona changes over time in terms of the amount of bound proteins, but not in composition.372 The majority of in vitro studies focus on the “hard” corona, as these proteins are strongly attached on the NPs and do not detach upon extensive washing.373 Phagocytes are able to recognize the proteins on the NPs surface and target them for internalization. The formation of a corona can reduce cellular uptake of functionalized NPs by shielding the ligands from binding to their receptors. As an example, the attachment of serum proteins on the surface of transferrin-functionalized SiO2 NPs resulted in a loss of its targeting capability.272 This “shielding” activity covers the active binding sites on the NPs and prevents the recognition by transferrin receptors. The composition of a protein corona is an important determinant of NPs fate and their cellular internalization. Various proteins, such as human serum albumin,374,375 fibrinogen,375 apolipoprotein,376 transferrin,377 and complement proteins378 were observed to adsorb onto the NPs.379 While some serum proteins contribute to decreased NPs endocytosis by shielding surface modification molecules on NPs, the binding of others may enhance the NPs uptake or modulate the endocytic mechanisms.379,380 This observation is supported by a multivariate model that used the protein corona fingerprint to predict NPs–cell association.380 Out of 64 tested serum proteins, 39 were classified as promoters of cell associations and 25 as inhibitors. The corona composition also varies depending on the nature of the biological fluids in which NPs are dispersed, such as human serum.381 Lesniak et al. demonstrated that protein corona of a very different nature is formed in the absence or presence of serum.369 In medium, supplemented with serum, the principal proteins that adsorb on NPs surface are immunoglobulins, complement proteins, and apolipoproteins, while under serum-free conditions mainly cytosolic proteins, components of the cytoskeleton, and membrane-associated proteins are adsorbed.369 When comparing in vivo and in vitro conditions, differences in NPs uptake should be considered due to variations in serum protein concentrations (in vivo, NPs in blood encounter much higher serum concentrations).382 Francia et al. reported lower uptake of 50 nm silica NPs into HeLa cells in the presence of a high amount of serum (62 mg mL−1) compared to that of NPs incubated with a five-times lower amount. The lower uptake could be related to differences in the corona composition. Additionally, since the corona biomolecules can mediate the uptake of NPs through recognition by specific receptors, it is likely that the free serum proteins compete for the same receptors and reduce the uptake of the corona–NPs complexes.383 When applying NPs in nanomedicine, one should consider the parameters that are affecting the protein corona formation, such as higher serum content, blood flow and higher serum complexity.372 A dynamic in vivo environment provides a continuous source of new proteins, resulting in more complex “hard” and “soft” coronae.3847.4 Surface functionalization
Exposure of macrophages to NPs coated with CD47 showed a significant reduction in NPs uptake across all macrophage sub-populations (Fig. 10). Uptake reduction was mediated through CD47-SIRPα interaction. The most pronounced inhibition in NPs uptake was observed in M1 macrophages, probably due to their higher expression of thrombospondin-1 (TSP-1), which serves to strengthen SIRPα and CD47 interaction.292,399 Interestingly, co-exposure of macrophages to bare NPs and free CD47 also reduced NPs uptake, but to a lesser extent than CD47-conjugated NPs.292
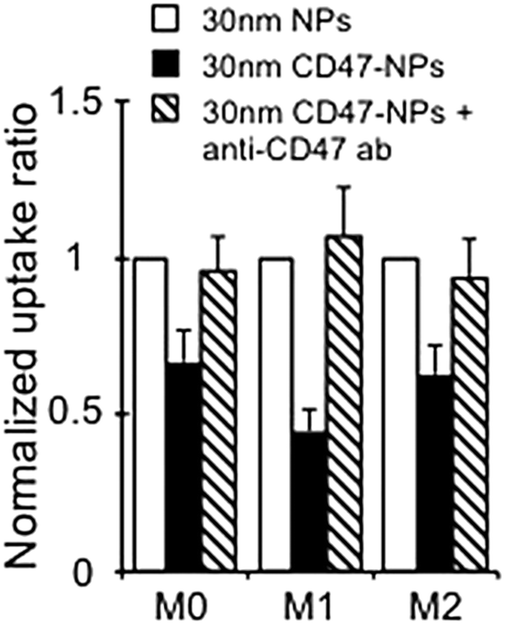 | ||
| Fig. 10 Surface modification of 30 nm PS NPs with CD47 protein reduced NPs uptake in macrophages. Adapted with permission from Qie et al.292 Copyright 2016 Qie et al. | ||
7.5 Bio-inspired molecules
Pathogenic food-borne Yersinia is able to resist phagocytosis in macrophages. The reason for this is plasmid-encoded Yop proteins (i.e. Yersinia outer proteins).400 Expression of Yop effector proteins is induced upon contact with eukaryotic target cells (e.g., macrophages), followed by channel opening and Yops injection into the cytosol.401,402 Several Yop proteins have been identified so far (YopH, YopE, YopM, YopK, and YpkA)403–405,419 and some share homology with eukaryotic proteins.401 Tyrosine phosphatase YopH is highly involved in the disruption of Fc receptor-mediated phagocytosis. It removes phosphate groups from the tyrosine-phosphorylated proteins, which are required for phagocytic invagination.406 Thus, YopH reduces the activation of the Akt pathway by blocking the integrin receptor-mediated activation of the PI3K signalling.407 The anti-phagocytic effect of Yersinia on macrophages and dendritic cells has also been demonstrated by other studies.408
Other pathogens, such as Streptococcus spp. are able to avoid host immune system by secreting specific molecules, which affect cytoskeleton function and inhibit phagocytosis. Many distinct Streptococcus serotypes have been identified, which differ in anti-phagocytic surface proteins (M proteins). Streptococcal inhibitor of complement (Sic) is a protein secreted by serotype M1. It has been shown that Sic binds with ezrin and moesin, two human proteins that functionally link the cytoskeleton to the plasma membrane. This data suggest that Sic together with other proteins decreases bacterial internalization and enhances its survival by enabling the pathogen to escape phagocytosis.409 Our idea in this context is to explore the usage of such bio-inspired proteins from bacteria in combination with various NPs to reduce their uptake.
7.6 Food supplements
Some food supplements can have an inhibitory (i.e., antagonistic) effect on NPs uptake and subsequent cellular response.Free fatty acids (FFA) are ubiquitous biological molecules, present in vegetable oils, avocado, various nuts, meat and milk products.410 After ingestion, the consumed FFA can interact with NPs. Jiang et al. investigated the interactions between ZnO NPs and saturated or unsaturated FFA. They observed significant reduction of intracellular Zn ions in THP-1 macrophages in the presence of stearic acid, oleic acid and α-linoleic acid. The reduced Zn uptake could be related to the altered NPs–cell interactions in the presence of FFA.411
Due to continuous exposure to multiple NPs and food supplements in our daily life via inhalation and ingestion, it is important to consider the potential interference of food supplements when assessing the biological effects of NPs.
7.7 Others
8. Conclusion
The use of NPs in nanomedicine for drug delivery and imaging is rapidly increasing. There is a high interest in development of novel delivery strategies, new technologies, new treatment approaches and research of novel NPs-based drug candidates. For biomedical applications, it is crucial to understand their interactions with different cell types as well as their behaviour in the in vivo system in the presence of various molecules. In recent years the knowledge about cell–NPs interaction, endocytosis and intracellular trafficking has largely increased. It is well known that the uptake of smaller NPs is mainly mediated via clathrin- and caveolae-mediated endocytosis, whereas bigger NPs and agglomerates enter the cells via macropinocytosis and phagocytosis. Based on the reviewed literature, there is still a lack of information regarding the NP type-specific endocytic mechanisms. In addition, the importance of clathrin-/caveolae-independent endocytosis needs to be explored in more detail. The main issue is the inconsistency between the existing results, which is a reflection of the complex nano–bio interactions as well as of the very different physico-chemical properties of different NP types. The choice of the optimal experiment is relevant, including the choice of the cell type, the introduction of more selective endocytic inhibitors such as dynasore, siRNAs and mutant proteins, as well as precise quantification of intracellular NP numbers and their subcellular localization. In an experimental set-up, a combination of different approaches for the assessment of NPs uptake and activation of specific endocytic pathways is highly recommended as most of inhibitors affect simultaneously different endocytic processes and may be cytotoxic. In addition, the advances in imaging techniques for sub-cellular analysis can provide new insights on the nano–bio interactions that occur at the cell surface and in subcellular compartments. Furthermore, CLEM and elemental analysis, flow cytometry and stereology became important tools for the accurate estimation of the number of internalized NPs (cellular dose). For the assessment of NPs–cell association as well as reliable quantification of the intracellular NP number it is recommended to combine different methods yielding complementary outcomes.Despite many successful approaches used in vitro for delivery of NPs-based drugs into the cells, the in vivo trials have not yet proven to be successful, due to the insufficient amount/number of NPs reaching targeted cells. The major hurdles that NPs face upon in vivo administration are the protein opsonization, uptake by the MPS system and/or the failure of delivery and penetration into the target tissue. In order to improve their delivery, NP co-exposure or functionalization with various molecules has been proposed as a tool to overcome the issue of low targetability. For example, pro-inflammatory stimuli have great potential to increase uptake of many different NPs but they mainly work specifically on immune cells. Other bio-inspired molecules, such as CPPs, bacterial toxins, and food supplements have shown that it is possible to increase NPs uptake in vitro and have a potential to be explored further in vivo. In the last ten years, more than 400 nano-formulations have already entered the clinical phases. Most of them are currently in the clinical Phases 1 and 2 for the treatment of various types of cancer, proving the growing interest of the biomedical community in using NPs-based approaches for targeted drug delivery. Overall, the literature on molecules that decrease NPs uptake is, as of now, very scarce. Reduction of NPs clearance by MPS upon their in vivo administration is an important aspect in nanomedicine with the goal of improving targeted drug delivery. For example, NPs surface coating with PEG or CD47 can prevent NPs from being recognized by cell receptors. Thus, NPs avoid phagocytic clearance, and their circulation time within the human body can be prolonged. Additionally, some bacterial pathogens are capable of using natural mechanisms (e.g., toxin release) to escape the endocytosis and survive longer in the host. Such approaches can be exploited further in combination with NPs. It is worth noting that when in contact with biological fluids, biomolecules present in the in vivo environment adsorb onto the NPs and form a protein corona. The corona formation alters the native properties and desired functionality of the NPs and thus modifies NPs uptake. Finally, food supplements and synthetic medications can interfere with NPs and reduce their uptake. Biological interactions are generally overlooked when designing NPs-based drug delivery systems. For this reason, it is important to consider the possible interactions between these substances and NPs to achieve efficient delivery to the target side. The increasing research interest in endocytic mechanisms can be merged with the chemical synthesis field to develop new NPs, which can be combined with nature-inspired substances, to improve physico-biochemical features for NPs delivery.
9. Future perspectives
The future perspectives in the field of improved targeting strategies in nanomedicine rely on the emergence of new tools capable of overcoming remaining knowledge gaps associated with the complex nano–bio interactions and the endocytic processes. The newly developed advanced tools should enable accurate discrimination between different endocytic pathways and an efficient quantification and intracellular localization of NPs. The introduction of CRISPR–Cas technology to specifically target key components of endocytic machinery presents a powerful tool that can be used to gain deeper insight into NPs uptake mechanisms and regulation of the pathways/processes. When applying NPs for biomedical purposes in vivo, the limitations lie in the fact that several factors are frequently overlooked in in vitro studies. Careful consideration of potential interactions of NPs with substances present in a realistic physiological microenvironment is essential for enhancing in vivo NPs delivery and for understanding the underlying mechanisms. Additionally, it would be of a great value to create a library of all potential molecules (natural and synthetic) that may have an impact, either stimulative or debilitating on NPs uptake. Examples of substances that could have a positive effect on the NPs uptake are inflammatory molecules, vitamins, polysaccharides, cell penetrating peptides and toxins. Encouraging preliminary results call for further investigation of these molecules. Ultimately, awareness and profound understanding of nano–bio interactions and how they drive cellular internalization is critical for successful implementation of NPs in nanomedical applications.Abbreviations
| Au | Gold |
| BM | Bone marrow |
| CCP | Clathrin coated pit |
| CCVs | Clathrin coated vesicles |
| CLICs | Clathrin-independent carriers |
| CLSM | Confocal laser scanning microscopy |
| CME | Clathrin-mediated endocytosis |
| CPP | Cell-penentrating peptide |
| CTxB | Cholera toxin B subunit |
| EEA1 | Early endosome antigen 1 |
| EGF | Epidermal growth factor |
| FA | Folic acid |
| FcR | Fc receptor |
| Fe3O4 | Iron oxide |
| FEME | Fast-endophilin-mediated endocytosis |
| FITC | Fluorescein isothiocyanate |
| GEEC | Glycosylphosphatidylinositol-anchored proteins-enriched early endosomal compartment |
| GPI | Glycosylphosphatidylinositol |
| GTP | Guanosine triphosphate |
| HA | Hyaluronic acid |
| HS | Human serum |
| ICP | Inductively coupled plasma |
| IFN | Interferon |
| IL | Interleukin |
| IONPs | Iron oxide nanoparticles |
| LDL | Low-density lipoprotein |
| LNP | Lipid nanoparticle |
| LPS | Lipopolysaccharide |
| MPS | Mononuclear phagocyte system |
| MSN | Mesoporous silica nanoparticles |
| NP | Nanoparticle |
| PEG | Poly(ethylene glycol) |
| PI3K | Phosphoinositide 3-kinase |
| PICALM | Phosphatidylinositol binding clathrin assembly protein |
| PLGA | Poly(lactic-co-glycolic acid) |
| PS | Polystyrene |
| PVA | Polyvinyl alcohol |
| QD | Quantum dot |
| RME | Receptor-mediated endocytosis |
| SiO2 | Silica |
| SPIO | Superparamagnetic iron oxide |
| TiO2 | Titanium dioxide |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swiss National Science Foundation (310030_192056/1), the Swiss National Science Foundation through the National Center of Competence in Research Bio-Inspired Materials, and the Adolphe Merkle Foundation. We kindly thank Dr Miguel Spuch-Calvar of mspuchdesign for the graphical design of Fig. 1.Notes and references
- J. Hulla, S. Sahu and A. Hayes, Hum. Exp. Toxicol., 2015, 34, 1318–1321 CrossRef CAS PubMed.
- R. Feynman, Eng. Sci., 1960, 22–36 Search PubMed.
- P. Mulvaney, ACS Nano, 2015, 9, 2215–2217 CrossRef CAS PubMed.
- N. Taniguchi, On the basic concept of ‘Nanotechnology’, Proceedings of the International Conference on Production Engineering, Japan Society of Precision Engineering, 1974.
- D. Nunes, A. Pimentel, L. Santos, P. Barquinha, L. Pereira, E. Fortunato and R. Martins, Metal Oxide Nanostructures, Elsevier, 2019, pp. 1–19 Search PubMed.
- D. R. Boverhof, C. M. Bramante, J. H. Butala, S. F. Clancy, W. M. Lafranconi, J. West and S. C. Gordon, Regul. Toxicol. Pharmacol., 2015, 73, 137–150 CrossRef CAS PubMed.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 Search PubMed.
- Nanotechnologies—Vocab.—Part 1 Core terms, 2015.
- EC, Commission recommendation of 18 October 2011 on the definition of nanomaterial (2011/696/EU), 2011.
- FDA, Considering whether an FDA-regulated product involves the application of nanotechnology, 2014.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- D. Dutta and J. G. Donaldson, Cell. Logist., 2012, 2, 203–208 CrossRef PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012.
- V. Mailänder and K. Landfester, Biomacromolecules, 2009, 10, 2379–2400 CrossRef.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- J. Zhao and M. H. Stenzel, Polym. Chem., 2018, 9, 259–272 RSC.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- A. Sangtani, E. Petryayeva, K. Susumu, E. Oh, A. L. Huston, G. Lasarte-Aragones, I. L. Medintz, W. R. Algar and J. B. Delehanty, Bioconjugate Chem., 2019, 30, 525–530 CrossRef CAS.
- J. Bourquin, D. Septiadi, D. Vanhecke, S. Balog, L. Steinmetz, M. Spuch-Calvar, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, ACS Nano, 2019, 13, 7759–7770 CrossRef CAS PubMed.
- J. Bourquin, A. Milosevic, D. Hauser, R. Lehner, F. Blank, A. Petri-Fink and B. Rothen-Rutishauser, Adv. Mater., 2018, 30, 1–31 CrossRef.
- J. Hoppstädter, M. Seif, A. Dembek, C. Cavelius, H. Huwer, A. Kraegeloh and A. K. Kiemer, Front. Pharmacol., 2015, 6, 1–12 Search PubMed.
- B. Guo, R. Zebda, S. J. Drake and C. M. Sayes, Part. Fibre Toxicol., 2009, 6, 1–13 CrossRef PubMed.
- D. Vanhecke, D. A. Kuhn, D. J. de Aberasturi, S. Balog, A. Milosevic, D. Urban, D. Peckys, N. de Jonge, W. J. Parak, A. Petri-Fink and B. Rothen-Rutishauser, Beilstein J. Nanotechnol., 2017, 8, 2396–2409 CrossRef CAS PubMed.
- S. Silva, A. Almeida and N. Vale, Biomolecules, 2019, 9, 22 CrossRef PubMed.
- W. Qu, B. Meng, Y. Yu and S. Wang, Int. J. Nanomed., 2018, 13, 4379–4389 CrossRef CAS PubMed.
- G. Grasso, S. Mercuri, A. Danani and M. A. Deriu, J. Phys. Chem. B, 2019, 123, 10622–10630 CrossRef CAS PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- S. Jaiswal, C. H. M. Jamieson, W. W. Pang, C. Y. Park, M. P. Chao, R. Majeti, D. Traver, N. van Rooijen and I. L. Weissman, Cell, 2009, 138, 271–285 CrossRef CAS PubMed.
- L. Yildirimer, N. T. K. Thanh, M. Loizidou and A. M. Seifalian, Nano Today, 2011, 6, 585–607 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- L. Steinmetz, C. Geers, S. Balog, M. Bonmarin, L. Rodriguez-Lorenzo, P. Taladriz-Blanco, B. Rothen-Rutishauser and A. Petri-Fink, Nanoscale Adv., 2020, 2, 5760–5768 RSC.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- D. A. Urban, L. Rodriguez-Lorenzo, S. Balog, C. Kinnear, B. Rothen-Rutishauser and A. Petri-Fink, Colloids Surf., B, 2016, 137, 39–49 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270.
- S. Mangal, W. Gao, T. Li and Q. Zhou, Acta Pharmacol. Sin., 2017, 38, 782–797 CrossRef CAS PubMed.
- J. Heyder, J. Gebhart, G. Rudolf, C. F. Schiller and W. Stahlhofen, J. Aerosol Sci., 1986, 17, 811–825 CrossRef.
- M. Geiser and W. G. Kreyling, Part. Fibre Toxicol., 2010, 7, 2 CrossRef PubMed.
- J. S. Patton and P. R. Byron, Nat. Rev. Drug Discovery, 2007, 6, 67–74 CrossRef CAS PubMed.
- W. G. Kreyling, S. Hirn, W. Möller, C. Schleh, A. Wenk, G. Celik, J. Lipka, M. Schäffler, N. Haberl, B. D. Johnston, R. Sperling, G. Schmid, U. Simon, W. J. Parak and M. Semmler-Behnke, ACS Nano, 2014, 8, 222–233 CrossRef CAS PubMed.
- M. Geiser, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, 207–217 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- A. L. Byrd, Y. Belkaid and J. A. Segre, Nat. Rev. Microbiol., 2018, 16, 143–155 CrossRef CAS PubMed.
- H. I. Labouta and M. Schneider, Nanomedicine, 2013, 9, 39–54 CrossRef CAS PubMed.
- A. Patzelt and J. Lademann, Expert Opin. Drug Delivery, 2013, 10, 787–797 CrossRef CAS PubMed.
- V. Agrahari, A. Mandal, V. Agrahari, H. M. Trinh, M. Joseph, A. Ray, H. Hadji, R. Mitra, D. Pal and A. K. Mitra, Drug Delivery Transl. Res., 2016, 6, 735–754 CrossRef CAS PubMed.
- A. Patel, K. Cholkar, V. Agrahari and A. K. Mitra, World J. Pharmacol., 2013, 2, 47–64 CrossRef PubMed.
- J. H. Park and N. Oh, Int. J. Nanomed., 2014, 9(Suppl. 1), 51 CrossRef PubMed.
- A. Sorkin and M. Von Zastrow, Nat. Rev. Mol. Cell Biol., 2010, 10, 609–622 CrossRef PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed.
- P. P. Di Fiore and M. von Zastrow, Cold Spring Harbor Perspect. Biol., 2014, 6, a016865 CrossRef PubMed.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- S. Kumari, S. Mg and S. Mayor, Cell Res., 2010, 20, 256–275 CrossRef CAS PubMed.
- S. Gordon, Immunity, 2016, 44, 463–475 CrossRef CAS PubMed.
- J. A. Swanson, Nat. Rev. Mol. Cell Biol., 2008, 9, 639–649 CrossRef CAS PubMed.
- C. Rosales and E. Uribe-Querol, BioMed Res. Int., 2017, 2017, 1–18 CrossRef PubMed.
- Z. Krpetić, F. Porta, E. Caneva, V. Dal Santo and G. Scarì, Langmuir, 2010, 26, 14799–14805 CrossRef PubMed.
- A. França, P. Aggarwal, E. V. Barsov, S. V. Kozlov, M. A. Dobrovolskaia and Á. González-Fernández, Nanomedicine, 2011, 6, 1175–1188 CrossRef PubMed.
- M. V. D. Z. Park, A. M. Neigh, J. P. Vermeulen, L. J. J. de la Fonteyne, H. W. Verharen, J. J. Briedé, H. van Loveren and W. H. de Jong, Biomaterials, 2011, 32, 9810–9817 CrossRef CAS PubMed.
- D. A. Kuhn, D. Vanhecke, B. Michen, F. Blank, P. Gehr, A. Petri-Fink and B. Rothen-Rutishauser, Beilstein J. Nanotechnol., 2014, 5, 1625–1636 CrossRef PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718 RSC.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- C. Gunawan, M. Lim, C. P. Marquis and R. Amal, J. Mater. Chem. B, 2014, 2, 2060 RSC.
- R. S. Flannagan, V. Jaumouillé and S. Grinstein, Annu. Rev. Pathol.: Mech. Dis., 2012, 7, 61–98 CrossRef CAS PubMed.
- V. Jaumouillé and S. Grinstein, Curr. Opin. Cell Biol., 2011, 23, 22–29 CrossRef PubMed.
- N. Nishijima, T. Hirai, K. Misato, M. Aoyama, E. Kuroda, K. J. Ishii, K. Higashisaka, Y. Yoshioka and Y. Tsutsumi, Front. Immunol., 2017, 8, 379 Search PubMed.
- D. Dutta, S. K. Sundaram, J. G. Teeguarden, B. J. Riley, L. S. Fifield, J. M. Jacobs, S. R. Addleman, G. A. Kaysen, B. M. Moudgil and T. J. Weber, Toxicol. Sci, 2007, 100, 303–315 CrossRef CAS PubMed.
- J. M. Kinchen and K. S. Ravichandran, Nat. Rev. Mol. Cell Biol., 2008, 9, 781–795 CrossRef CAS PubMed.
- J. A. Swanson and C. Watts, Trends Cell Biol., 1995, 5, 424–428 CrossRef CAS PubMed.
- J. A. Swanson, Nat. Rev. Mol. Cell Biol., 2008, 9, 639–649 CrossRef CAS PubMed.
- J. A. Swanson and S. Yoshida, Encyclopedia of Cell Biology, 2016 Search PubMed.
- J. A. Swanson and S. Yoshida, Philos. Trans. R. Soc., B, 2019, 374, 20180157 CrossRef CAS PubMed.
- G. Bloomfield and R. R. Kay, J. Cell Sci., 2016, 129, 2697–2705 CAS.
- J. Mercer and A. Helenius, Nat. Cell Biol., 2009, 11, 510–520 CrossRef CAS PubMed.
- M. Reifarth, S. Hoeppener and U. S. Schubert, Adv. Mater., 2018, 30, 1703704 CrossRef PubMed.
- H. T. McMahon and E. Boucrot, Nat. Rev. Mol. Cell Biol., 2011, 12, 517–533 CrossRef CAS PubMed.
- M. Kaksonen and A. Roux, Nat. Rev. Mol. Cell Biol., 2018, 19, 313–326 CrossRef CAS.
- L. M. Traub and J. S. Bonifacino, Cold Spring Harbor Perspect. Biol., 2013, 5, a016790 CrossRef PubMed.
- M. Ehrlich, W. Boll, A. van Oijen, R. Hariharan, K. Chandran, M. L. Nibert and T. Kirchhausen, Cell, 2004, 118, 591–605 CrossRef CAS PubMed.
- C. R. Hopkins, K. Miller and J. M. Beardmore, J. Cell Sci., 1985, 1985, 173–186 CrossRef PubMed.
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, PLoS One, 2011, 6, e24438 CrossRef CAS.
- C. W. Lu, Y. Hung, J. K. Hsiao, M. Yao, T. H. Chung, Y. S. Lin, S. H. Wu, S. C. Hsu, H. M. Liu, C. Y. Mou, C. S. Yang, D. M. Huang and Y. C. Chen, Nano Lett., 2007, 7, 149–154 CrossRef CAS.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- C. M. Luz, M. S. P. Boyles, P. Falagan-Lotsch, M. R. Pereira, H. R. Tutumi, E. Oliveira Santos, N. B. Martins, M. Himly, A. Sommer, I. Foissner, A. Duschl, J. M. Granjeiro and P. E. C. Leite, J. Nanobiotechnol., 2017, 15, 11 CrossRef.
- O. Kovtun, V. A. Tillu, N. Ariotti, R. G. Parton and B. M. Collins, J. Cell Sci., 2015, 128, 1269–1278 CrossRef CAS PubMed.
- A. L. Kiss and E. Botos, J. Cell. Mol. Med., 2009, 13, 1228–1237 CrossRef PubMed.
- J. P. X. Cheng and B. J. Nichols, Trends Cell Biol., 2016, 26, 177–189 CrossRef CAS PubMed.
- L. Pelkmans and A. Helenius, Traffic, 2002.
- R. G. Parton, V. A. Tillu and B. M. Collins, Curr. Biol., 2018, 28, R402–R405 CrossRef CAS PubMed.
- M. Chatterjee, E. Ben-Josef, R. Robb, M. Vedaie, S. Seum, K. Thirumoorthy, K. Palanichamy, M. Harbrecht, A. Chakravarti and T. M. Williams, Cancer Res., 2017, 77, 5925–5937 CrossRef CAS PubMed.
- J. Voigt, J. Christensen and V. P. Shastri, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2942–2947.
- B. J. Andreone, B. W. Chow, A. Tata, B. Lacoste, A. Ben-Zvi, K. Bullock, A. A. Deik, D. D. Ginty, C. B. Clish and C. Gu, Neuron, 2017, 94, 581–594.e5 CrossRef CAS PubMed.
- P. G. Frank, S. Pavlides and M. P. Lisanti, Cell Tissue Res., 2009, 335, 41–47 CrossRef CAS PubMed.
- Z. Wang, C. Tiruppathi, R. D. Minshall and A. B. Malik, ACS Nano, 2009, 3, 4110–4116 CrossRef CAS.
- S. Mayor, R. G. Parton and J. G. Donaldson, Cold Spring Harbor Perspect. Biol., 2014, 6, a016758 CrossRef PubMed.
- B. J. Nichols and J. Lippincott-Schwartz, Trends Cell Biol., 2001, 11, 406–412 CrossRef CAS.
- L. Johannes, R. G. Parton, P. Bassereau and S. Mayor, Nat. Rev. Mol. Cell Biol., 2015.
- V. Bitsikas, I. R. Corrêa and B. J. Nichols, eLife, 2014, 3, e05256 CrossRef.
- K. Sandvig, S. Pust, T. Skotland and B. van Deurs, Curr. Opin. Cell Biol., 2011, 23, 413–420 CrossRef CAS PubMed.
- E. Boucrot, A. P. A. Ferreira, L. Almeida-Souza, S. Debard, Y. Vallis, G. Howard, L. Bertot, N. Sauvonnet and H. T. McMahon, Nature, 2015, 517, 460–465 CrossRef CAS PubMed.
- B. D. Grant and J. G. Donaldson, Nat. Rev. Mol. Cell Biol., 2009, 10, 597–608 CrossRef CAS PubMed.
- M. Kirkham, A. Fujita, R. Chadda, S. J. Nixon, T. V. Kurzchalia, D. K. Sharma, R. E. Pagano, J. F. Hancock, S. Mayor and R. G. Parton, J. Cell Biol., 2005, 168, 465–476 CrossRef CAS PubMed.
- X. Chi, S. Wang, Y. Huang, M. Stamnes and J.-L. Chen, Int. J. Mol. Sci., 2013, 14, 7089–7108 CrossRef CAS PubMed.
- K. Sandvig, S. Kavaliauskiene and T. Skotland, Histochem. Cell Biol., 2018, 150, 107–118 CrossRef CAS.
- O. Heikkilä, P. Susi, T. Tevaluoto, H. Härmä, V. Marjomäki, T. Hyypiä and S. Kiljunen, J. Virol., 2010, 84, 3666–3681 CrossRef.
- K. Nishi and K. Saigo, J. Biol. Chem., 2007, 282, 27503–27517 CrossRef CAS.
- H. Radhakrishna and J. G. Donaldson, J. Cell Biol., 1997, 139, 49–61 CrossRef CAS PubMed.
- G. P. Otto and B. J. Nichols, J. Cell Sci., 2011, 124, 3933–3940 CrossRef CAS PubMed.
- O. O. Glebov, N. A. Bright and B. J. Nichols, Nat. Cell Biol., 2006, 8, 46–54 CrossRef CAS PubMed.
- M. Meister and R. Tikkanen, Membranes, 2014, 4, 356–371 CrossRef PubMed.
- L. Chan Wah Hak, S. Khan, I. Di Meglio, A.-L. Law, S. Lucken-Ardjomande Häsler, L. M. Quintaneiro, A. P. A. Ferreira, M. Krause, H. T. McMahon and E. Boucrot, Nat. Cell Biol., 2018, 20, 1023–1031 CrossRef CAS PubMed.
- A. P. A. Ferreira and E. Boucrot, Trends Cell Biol., 2018, 28, 188–200 CrossRef CAS PubMed.
- N. Bohmer and A. Jordan, Beilstein J. Nanotechnol., 2015, 6, 167–176 CrossRef PubMed.
- L. Ding, X. Zhu, Y. Wang, B. Shi, X. Ling, H. Chen, W. Nan, A. Barrett, Z. Guo, W. Tao, J. Wu and X. Shi, Nano Lett., 2017, 17, 6790–6801 CrossRef CAS PubMed.
- J. Kasper, M. I. Hermanns, C. Bantz, S. Utech, O. Koshkina, M. Maskos, C. Brochhausen, C. Pohl, S. Fuchs, R. E. Unger and C. James Kirkpatrick, Eur. J. Pharm. Biopharm., 2013, 84, 275–287 CrossRef CAS PubMed.
- D. Rimai, D. Quesnel and A. Busnaina, Colloids Surf., A, 2000, 165, 3–10 CrossRef CAS.
- B. M. Rothen-Rutishauser, S. Schürch, B. Haenni, N. Kapp and P. Gehr, Environ. Sci. Technol., 2006, 40, 4353–4359 CrossRef CAS PubMed.
- W. B. Gratzer, Biochem. J., 1981, 198, 1–8 CrossRef CAS PubMed.
- T. Wang, J. Bai, X. Jiang and G. U. Nienhaus, ACS Nano, 2012, 6, 1251–1259 CrossRef CAS PubMed.
- M. Geiser, B. Rothen-Rutishauser, N. Kapp, S. Schürch, W. Kreyling, H. Schulz, M. Semmler, V. I. Hof, J. Heyder and P. Gehr, Environ. Health Perspect., 2005, 113, 1555–1560 CrossRef PubMed.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS PubMed.
- L. Treuel, K. A. Eslahian, D. Docter, T. Lang, R. Zellner, K. Nienhaus, G. U. Nienhaus, R. H. Stauber and M. Maskos, Phys. Chem. Chem. Phys., 2014, 16, 15053–15067 RSC.
- W.-K. K. Fong, T. L. Moore, S. Balog, D. Vanhecke, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, NanoScience and Technology, 2019, pp. 101–150 Search PubMed.
- Y. T. Ho, R. D. Kamm and J. C. Y. Kah, Nanoscale, 2018, 10, 12386–12397 RSC.
- X. Cheng, X. Tian, A. Wu, J. Li, J. Tian, Y. Chong, Z. Chai, Y. Zhao, C. Chen and C. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 20568–20575 CrossRef CAS PubMed.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- C. Brandenberger, C. Mühlfeld, Z. Ali, A.-G. Lenz, O. Schmid, W. J. Parak, P. Gehr and B. Rothen-Rutishauser, Small, 2010, 6, 1669–1678 CrossRef CAS PubMed.
- B. Halamoda-Kenzaoui, M. Ceridono, P. Urbán, A. Bogni, J. Ponti, S. Gioria and A. Kinsner-Ovaskainen, J. Nanobiotechnol., 2017, 15, 48 CrossRef PubMed.
- K. Shapero, F. Fenaroli, I. Lynch, D. C. Cottell, A. Salvati and K. A. Dawson, Mol. BioSyst., 2011, 7, 371–378 RSC.
- O. Schmid and F. R. Cassee, Part. Fibre Toxicol., 2017, 14, 52 CrossRef PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- T. Mironava, M. Hadjiargyrou, M. Simon, V. Jurukovski and M. H. Rafailovich, Nanotoxicology, 2010, 4, 120–137 CrossRef CAS PubMed.
- L. Li, W. S. Xi, Q. Su, Y. Li, G. H. Yan, Y. Liu, H. Wang and A. Cao, Small, 2019, 15, 1–8 Search PubMed.
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- X. Xie, J. Liao, X. Shao, Q. Li and Y. Lin, Sci. Rep., 2017, 7, 3827 CrossRef PubMed.
- R. Vácha, F. J. Martinez-Veracoechea and D. Frenkel, Nano Lett., 2011, 11, 5391–5395 CrossRef PubMed.
- X. Huang, X. Teng, D. Chen, F. Tang and J. He, Biomaterials, 2010, 31, 438–448 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- C. Kinnear, L. Rodriguez-Lorenzo, M. J. D. Clift, B. Goris, S. Bals, B. Rothen-Rutishauser and A. Petri-Fink, Nanoscale, 2016, 8, 16416–16426 RSC.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577 CrossRef PubMed.
- A. Akinc and G. Battaglia, Cold Spring Harbor Perspect. Biol., 2013, 5, a016980–a016980 CrossRef PubMed.
- L. Chen, J. M. Mccrate, J. C.-M. Lee and H. Li, Nanotechnology, 2011, 22, 105708 CrossRef.
- S. Jambhrunkar, Z. Qu, A. Popat, J. Yang, O. Noonan, L. Acauan, Y. Ahmad Nor, C. Yu and S. Karmakar, Mol. Pharmaceutics, 2014, 11, 3642–3655 CrossRef CAS.
- A. Petri-Fink, M. Chastellain, L. Juillerat-Jeanneret, A. Ferrari and H. Hofmann, Biomaterials, 2005, 26, 2685–2694 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- T. Wileman, C. Harding and P. Stahl, Biochem. J., 1985, 232, 1–14 CrossRef CAS PubMed.
- B. Yu, H. C. Tai, W. Xue, L. J. Lee and R. J. Lee, Mol. Membr. Biol., 2010, 27, 286–298 CrossRef CAS PubMed.
- W. Kim, K.-Y. Na, K.-H. Lee, H. W. Lee, J. K. Lee and K.-T. Kim, Sci. Rep., 2017, 7, 10971 CrossRef PubMed.
- D. Hauser, M. Estermann, A. Milosevic, L. Steinmetz, D. Vanhecke, D. Septiadi, B. Drasler, A. Petri-Fink, V. Ball and B. Rothen-Rutishauser, Nanomaterials, 2018, 8, 1065 CrossRef PubMed.
- R. K. Singh, J. C. Knowles and H.-W. Kim, J. Tissue Eng., 2019, 10, 204173141987752 CrossRef PubMed.
- C. Xu, Y. Niu, A. Popat, S. Jambhrunkar, S. Karmakar and C. Yu, J. Mater. Chem. B, 2014, 2, 253–256 RSC.
- R. K. Singh, J. C. Knowles and H.-W. Kim, J. Tissue Eng., 2019, 10, 204173141987752 CrossRef PubMed.
- S.-H. Cho, J. H. Hong, Y.-W. Noh, E. Lee, C.-S. Lee and Y. T. Lim, Int. J. Nanomed., 2016, 11, 5621–5632 CrossRef CAS PubMed.
- S. Del Sol-Fernández, Y. Portilla-Tundidor, L. Gutiérrez, O. F. Odio, E. Reguera, D. F. Barber and M. P. Morales, ACS Appl. Mater. Interfaces, 2019, 11, 26648–26663 CrossRef PubMed.
- A. Parodi, R. Molinaro, M. Sushnitha, M. Evangelopoulos, J. O. Martinez, N. Arrighetti, C. Corbo and E. Tasciotti, Biomaterials, 2017, 147, 155–168 CrossRef CAS.
- W. Wang, P. Wang, X. Tang, A. A. Elzatahry, S. Wang, D. Al-Dahyan, M. Zhao, C. Yao, C.-T. Hung, X. Zhu, T. Zhao, X. Li, F. Zhang and D. Zhao, ACS Cent. Sci., 2017, 3, 839–846 CrossRef CAS.
- A. I. Ivanov, Methods in Molecular Biology, 2008, pp. 15–33 Search PubMed.
- D. Dutta and J. G. Donaldson, Cell. Logist., 2012, 2, 203–208 CrossRef PubMed.
- B. Rothen-Rutishauser, D. A. Kuhn, Z. Ali, M. Gasser, F. Amin, W. J. Parak, D. Vanhecke, A. Fink, P. Gehr and C. Brandenberger, Nanomedicine, 2014, 9, 607–621 CrossRef CAS PubMed.
- R. D. Singh, V. Puri, J. T. Valiyaveettil, D. L. Marks, R. Bittman and R. E. Pagano, Mol. Biol. Cell, 2003, 14, 3254–3265 CrossRef CAS.
- C. Commisso, R. J. Flinn and D. Bar-Sagi, Nat. Protoc., 2014, 9, 182–192 CrossRef CAS.
- N. Araki, M. T. Johnson and J. A. Swanson, J. Cell Biol., 1996, 135, 1249–1260 CrossRef CAS PubMed.
- M. L. Torgersen, G. Skretting, B. van Deurs and K. Sandvig, J. Cell Sci., 2001, 114, 3737–3747 CrossRef CAS.
- S. Singh, A. Kumar, A. Karakoti, S. Seal and W. T. Self, Mol. BioSyst., 2010, 6, 1813 RSC.
- J. L. Carpentier, F. Sawano, D. Geiger, P. Gorden, A. Perrelet and L. Orci, J. Cell. Physiol., 1989, 138, 519–526 CrossRef CAS PubMed.
- G. Altankov and F. Grinnell, J. Cell Biol., 1993, 120, 1449–1459 CrossRef CAS PubMed.
- S. Engelberg, J. Modrejewski, J. G. Walter, Y. D. Livney and Y. G. Assaraf, Oncotarget, 2018, 9, 20993–21006 CrossRef PubMed.
- A. K. Willox, Y. M. E. Sahraoui and S. J. Royle, Biol. Open, 2014, 3, 326–331 CrossRef CAS PubMed.
- D. Dutta, C. D. Williamson, N. B. Cole and J. G. Donaldson, PLoS One, 2012, 7, e45799 CrossRef CAS PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- G. Altankov and F. Grinnell, J. Cell Biol., 1993, 120, 1449–1459 CrossRef CAS PubMed.
- P. Cosson, I. de Curtis, J. Pouysségur, G. Griffiths and J. Davoust, J. Cell Biol., 1989, 108, 377–387 CrossRef CAS PubMed.
- K. Sandvig, S. Olsnes, O. W. Petersen and B. van Deurs, J. Cell Biol., 1987, 105, 679–689 CrossRef CAS.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS PubMed.
- P. J. A. Davies, D. R. Davies, A. Levitzki, F. R. Maxfield, P. Milhaud, M. C. Willingham and I. H. Pastan, Nature, 1980, 283, 162–167 CrossRef CAS.
- J. R. Bradley, D. R. Johnson and J. S. Pober, J. Immunol., 1993, 150, 5544–5555 CAS.
- J. Wolfram, S. Nizzero, H. Liu, F. Li, G. Zhang, Z. Li, H. Shen, E. Blanco and M. Ferrari, Sci. Rep., 2017, 7, 13738 CrossRef.
- R. B. Dickson, M. C. Willingham and I. H. Pastan, Ann. N. Y. Acad. Sci., 1982, 401, 38–49 CrossRef CAS PubMed.
- C.-L. Chen, W.-H. Hou, I.-H. Liu, G. Hsiao, S. S. Huang and J. S. Huang, J. Cell Sci., 2009, 122, 1863–1871 CrossRef CAS PubMed.
- W. Dejonghe, I. Sharma, B. Denoo, S. De Munck, Q. Lu, K. Mishev, H. Bulut, E. Mylle, R. De Rycke, M. Vasileva, D. V. Savatin, W. Nerinckx, A. Staes, A. Drozdzecki, D. Audenaert, K. Yperman, A. Madder, J. Friml, D. Van Damme, K. Gevaert, V. Haucke, S. N. Savvides, J. Winne and E. Russinova, Nat. Chem. Biol., 2019, 15, 641–649 CrossRef CAS PubMed.
- S. K. Rodal, G. Skretting, Ø. Garred, F. Vilhardt, B. van Deurs and K. Sandvig, Mol. Biol. Cell, 1999, 10, 961–974 CrossRef CAS PubMed.
- J. E. Schnitzer, P. Oh, E. Pinney and J. Allard, J. Cell Biol., 1994, 127, 1217–1232 CrossRef CAS PubMed.
- J. E. Sidaway, J. Am. Soc. Nephrol., 2004, 15, 2258–2265 CrossRef CAS PubMed.
- T. Akiyama, J. Ishida, S. Nakagawa, H. Ogawara, S. Watanabe, N. Itoh, M. Shibuya and Y. Fukami, J. Biol. Chem., 1987, 62, 5592–5595 CrossRef.
- L. M. Fujimoto, R. Roth, J. E. Heuser and S. L. Schmid, Traffic, 2000, 1, 161–171 CrossRef CAS.
- M. Koivusalo, C. Welch, H. Hayashi, C. C. Scott, M. Kim, T. Alexander, N. Touret, K. M. Hahn and S. Grinstein, J. Cell Biol., 2010, 188, 547–563 CrossRef CAS.
- V. Francia, C. Reker-Smit, G. Boel and A. Salvati, Nanomedicine, 2019, 14, 1533–1549 CrossRef CAS PubMed.
- H.-P. Lin, B. Singla, P. Ghoshal, J. L. Faulkner, M. Cherian-Shaw, P. M. O’Connor, J.-X. She, E. J. Belin de Chantemele and G. Csányi, Br. J. Pharmacol., 2018, 175, 3640–3655 CrossRef CAS PubMed.
- D. van der Hoeven, K. Cho, Y. Zhou, X. Ma, W. Chen, A. Naji, D. Montufar-Solis, Y. Zuo, S. E. Kovar, K. R. Levental, J. A. Frost, R. van der Hoeven and J. F. Hancock, Mol. Cell. Biol., 2017, 38, e300373-17 Search PubMed.
- L. P. Fernando, P. K. Kandel, J. Yu, J. McNeill, P. C. Ackroyd and K. A. Christensen, Biomacromolecules, 2010, 11, 2675–2682 CrossRef CAS PubMed.
- H. Herd, N. Daum, A. T. Jones, H. Huwer, H. Ghandehari and C.-M. Lehr, ACS Nano, 2013, 7, 1961–1973 CrossRef CAS PubMed.
- K. Sarkar, M. J. Kruhlak, S. L. Erlandsen and S. Shaw, Immunology, 2005, 116, 513–524 CAS.
- Y. Wei, T. Tang and H.-B. Pang, Nat. Commun., 2019, 10, 3646 CrossRef PubMed.
- E. Macia, M. Ehrlich, R. Massol, E. Boucrot, C. Brunner and T. Kirchhausen, Dev. Cell, 2006, 10, 839–850 CrossRef CAS.
- G. Preta, J. G. Cronin and I. M. Sheldon, Cell Commun. Signaling, 2015, 13, 24 CrossRef PubMed.
- L. Kühling and M. Schelhaas, Exocytosis and Endocytosis, 2014, pp. 19–46 Search PubMed.
- R. C. Wilson and J. A. Doudna, Annu. Rev. Biophys., 2013, 42, 217–239 CrossRef CAS PubMed.
- N. M. Snead and J. J. Rossi, Wiley Interdiscip. Rev.: RNA, 2010, 1, 117–131 CrossRef CAS PubMed.
- E. J. Guggenheim, J. Z. Rappoport and I. Lynch, Nanotoxicology, 2020, 14, 504–532 CrossRef CAS PubMed.
- C. D. Sago, M. P. Lokugamage, G. N. Lando, N. Djeddar, N. N. Shah, C. Syed, A. V. Bryksin and J. E. Dahlman, Nano Lett., 2018, 18, 7590–7600 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- B. Wiedenheft, S. H. Sternberg and J. A. Doudna, Nature, 2012, 482, 331–338 CrossRef CAS PubMed.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed.
- M. Maksymowicz, M. Miączyńska and M. Banach-Orłowska, Cell Commun. Signaling, 2020, 18, 176 CrossRef CAS PubMed.
- C. Mendoza-Topaz, G. Nelson, G. Howard, S. Hafner, P. Rademacher, M. Frick and B. J. Nichols, PLoS One, 2018, 13, e0205306 CrossRef CAS PubMed.
- S. Srinivasan, C. J. Burckhardt, M. Bhave, Z. Chen, P.-H. Chen, X. Wang, G. Danuser and S. L. Schmid, PLoS Biol., 2018, 16, e2005377 CrossRef PubMed.
- M. Schaks, S. P. Singh, F. Kage, P. Thomason, T. Klünemann, A. Steffen, W. Blankenfeldt, T. E. Stradal, R. H. Insall and K. Rottner, Curr. Biol., 2018, 28, 3674–3684.e6 CrossRef CAS PubMed.
- S. Patel, N. Ashwanikumar, E. Robinson, A. DuRoss, C. Sun, K. E. Murphy-Benenato, C. Mihai, Ö. Almarsson and G. Sahay, Nano Lett., 2017, 17, 5711–5718 CrossRef CAS PubMed.
- I. Herskowitz, Nature, 1987, 329, 219–222 CrossRef CAS.
- A. van der Bliek, T. Redelmeier, H. Damke, E. Tisdale, E. Meyerowitz and S. Schmid, J. Cell Biol., 1993, 122, 553–563 CrossRef CAS PubMed.
- J. Rappoport, P. J. Smith, M. Giroud, H. L. Wiggins, J. Mazzolin and R. J. Dyson, Int. J. Nanomed., 2012, 2045 CrossRef PubMed.
- A. Mukhopadhyay, E. Nieves, F.-Y. Che, J. Wang, L. Jin, J. W. Murray, K. Gordon, R. H. Angeletti and A. W. Wolkoff, J. Cell Sci., 2011, 124, 765–775 CrossRef CAS.
- Y. V. Karpievitch, A. D. Polpitiya, G. A. Anderson, R. D. Smith and A. R. Dabney, Ann. Appl. Stat., 2010, 4, 1797–1823 Search PubMed.
- G. H. H. Borner, M. Harbour, S. Hester, K. S. Lilley and M. S. Robinson, J. Cell Biol., 2006, 175, 571–578 CrossRef CAS PubMed.
- M. Girard, P. D. Allaire, P. S. McPherson and F. Blondeau, Mol. Cell. Proteomics, 2005, 4, 1145–1154 CrossRef CAS.
- A. Dávalos, C. Fernández-Hernando, G. Sowa, B. Derakhshan, M. I. Lin, J. Y. Lee, H. Zhao, R. Luo, C. Colangelo and W. C. Sessa, Mol. Cell. Proteomics, 2010, 9, 2109–2124 CrossRef PubMed.
- W. Shui, L. Sheu, J. Liu, B. Smart, C. J. Petzold, T.-Y. Hsieh, A. Pitcher, J. D. Keasling and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16952–16957.
- D. Hofmann, S. Tenzer, M. B. Bannwarth, C. Messerschmidt, S.-F. Glaser, H. Schild, K. Landfester and V. Mailänder, ACS Nano, 2014, 8, 10077–10088 CrossRef CAS PubMed.
- N. Chaves, I. Estrela-Lopis, J. Böttner, C. A. P. Lopes, B. Cândido Guido, A. Souza and S. Bao, Int. J. Nanomed., 2017, 12, 5511–5523 CrossRef CAS PubMed.
- J. K. Carrow, L. M. Cross, R. W. Reese, M. K. Jaiswal, C. A. Gregory, R. Kaunas, I. Singh and A. K. Gaharwar, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3905–E3913.
- X. Cai, J. Dong, J. Liu, H. Zheng, C. Kaweeteerawat, F. Wang, Z. Ji and R. Li, Nat. Commun., 2018, 9, 4416 CrossRef PubMed.
- S. Haider and R. Pal, Curr. Genomics, 2013, 14, 91–110 CrossRef CAS.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- A. Descloux, K. S. Grußmayer and A. Radenovic, Nat. Methods, 2019, 16, 918–924 CrossRef CAS.
- J. B. Pawley, Handbook Of Biological Confocal Microscopy, Springer US, Boston, MA, 2006, pp. 20–42 Search PubMed.
- L. Schermelleh, A. Ferrand, T. Huser, C. Eggeling, M. Sauer, O. Biehlmaier and G. P. C. Drummen, Nat. Cell Biol., 2019, 21, 72–84 CrossRef CAS.
- I. D. Johnson, in Handbook Of Biological Confocal Microscopy, ed. J. B. Pawley, Springer US, Boston, MA, 2006, pp. 353–367 Search PubMed.
- D. L. Spector and R. D. Goldman, Cold Spring Harbor Perspect. Biol., 2010, 35–42 CAS.
- F. W. Pratiwi, C. W. Kuo, B.-C. Chen and P. Chen, Nanomedicine, 2019, 14, 1759–1769 CrossRef CAS PubMed.
- L. Rodriguez-Lorenzo, K. Fytianos, F. Blank, C. von Garnier, B. Rothen-Rutishauser and A. Petri-Fink, Small, 2014, 10, 1341–1350 CrossRef CAS PubMed.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol. Physiol., 2011, 300, C723–C742 CrossRef CAS PubMed.
- D. Vercauteren, H. Deschout, K. Remaut, J. F. J. Engbersen, A. T. Jones, J. Demeester, S. C. De Smedt and K. Braeckmans, ACS Nano, 2011, 5, 7874–7884 CrossRef CAS PubMed.
- J. S. Aaron, A. B. Taylor and T.-L. Chew, J. Cell Sci., 2018, 131, jsc211847 CrossRef PubMed.
- J. P. Peñaloza, V. Márquez-Miranda, M. Cabaña-Brunod, R. Reyes-Ramírez, F. M. Llancalahuen, C. Vilos, F. Maldonado-Biermann, L. A. Velásquez, J. A. Fuentes, F. D. González-Nilo, M. Rodríguez-Díaz and C. Otero, J. Nanobiotechnol., 2017, 15, 1 CrossRef PubMed.
- X. Jiang, C. Röcker, M. Hafner, S. Brandholt, R. M. Dörlich and G. U. Nienhaus, ACS Nano, 2010, 4, 6787–6797 CrossRef CAS PubMed.
- K. C. Partlow, G. M. Lanza and S. A. Wickline, Biomaterials, 2008, 29, 3367–3375 CrossRef CAS.
- Y. Li, L. Shang and G. U. Nienhaus, Nanoscale, 2016, 8, 7423–7429 RSC.
- M. Reifarth, U. S. Schubert and S. Hoeppener, Adv. Biosyst., 2018, 2, 1700254 CrossRef.
- D. Vanhecke, L. Rodriguez-Lorenzo, M. J. D. Clift, F. Blank, A. Petri-Fink and B. Rothen-Rutishauser, Nanomedicine, 2014, 9, 1885–1900 CrossRef CAS.
- L. E. Franken, K. Grünewald, E. J. Boekema and M. C. A. Stuart, Small, 2020, 16, 1906198 CrossRef CAS PubMed.
- C. Mühlfeld, B. Rothen-Rutishauser, D. Vanhecke, F. Blank, P. Gehr and M. Ochs, Part. Fibre Toxicol., 2007, 4, 11 CrossRef PubMed.
- T. M. Mayhew, C. Mühlfeld, D. Vanhecke and M. Ochs, Ann. Anat., 2009, 191, 153–170 CrossRef CAS.
- M. P. D. Dobay, A. P. Alberola, E. R. Mendoza and J. O. Rädler, J. Nanopart. Res., 2012, 14, 821 CrossRef.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Müller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef PubMed.
- K. Haase and A. E. Pelling, J. R. Soc., Interface, 2015, 12, 20140970 CrossRef PubMed.
- S.-H. Jung, D. Park, J. H. Park, Y.-M. Kim and K.-S. Ha, Exp. Mol. Med., 2010, 42, 597 CrossRef CAS PubMed.
- A. Hor, A. Luu, L. Kang, B. Scott, E. Bailey, A. Hoppe and S. Smith, SPIE Rev., 2017, 1007101 DOI:10.1117/12.2256683.
- Q. Dai, S. Wilhelm, D. Ding, A. M. Syed, S. Sindhwani, Y. Zhang, Y. Y. Chen, P. Macmillan and W. C. W. Chan, ACS Nano, 2018, 12, 8423–8435 CrossRef CAS PubMed.
- A. Elsaesser, A. Taylor, G. S. de Yanés, G. McKerr, E.-M. Kim, E. O’Hare and C. V. Howard, Nanomedicine, 2010, 5, 1447–1457 CrossRef CAS PubMed.
- M. J. D. Clift, B. Rothen-Rutishauser, D. M. Brown, R. Duffin, K. Donaldson, L. Proudfoot, K. Guy and V. Stone, Toxicol. Appl. Pharmacol., 2008, 232, 418–427 CrossRef CAS PubMed.
- Y. Williams, A. Sukhanova, M. Nowostawska, A. M. Davies, S. Mitchell, V. Oleinikov, Y. Gun’ko, I. Nabiev, D. Kelleher and Y. Volkov, Small, 2009, 5, 2581–2588 CrossRef CAS PubMed.
- H. Paysen, N. Loewa, A. Stach, J. Wells, O. Kosch, S. Twamley, M. R. Makowski, T. Schaeffter, A. Ludwig and F. Wiekhorst, Sci. Rep., 2020, 10, 1922 CrossRef CAS PubMed.
- J. Davda and V. Labhasetwar, Int. J. Pharm., 2002, 233, 51–59 CrossRef CAS.
- B. Drasler, D. Vanhecke, L. Rodriguez-Lorenzo, A. Petri-Fink and B. Rothen-Rutishauser, Nanomedicine, 2017, 12, 1095–1099 CrossRef CAS PubMed.
- G. D. W. Montaser, Inductively coupled plasmas in analytical atomic spectrometry, Wiley, 1992 Search PubMed.
- H. E. Taylor, Inductively Coupled Plasma-Mass Spectrometry: Practices and Techniques, Academic Press Inc., 1st edn, 2000 Search PubMed.
- A. Elsaesser, C. A. Barnes, G. McKerr, A. Salvati, I. Lynch, K. A. Dawson and C. V. Howard, Nanomedicine, 2011, 6, 1189–1198 CrossRef CAS PubMed.
- C. Gottstein, G. Wu, B. J. Wong and J. A. Zasadzinski, ACS Nano, 2013, 7, 4933–4945 CrossRef CAS.
- K. M. McKinnon, Curr. Protoc. Immunol., 2018, 120, 5.1.1–5.1.11 Search PubMed.
- M. Chen, X. He, K. Wang, D. He, X. Yang and H. Shi, TrAC, Trends Anal. Chem., 2014, 58, 120–129 CrossRef CAS.
- R. M. Zucker, E. J. Massaro, K. M. Sanders, L. L. Degn and W. K. Boyes, Cytometry, Part A, 2010, 77, 677–685 CrossRef CAS.
- I. L. Hsiao, A. M. Gramatke, R. Joksimovic, M. Sokolowski, M. Gradzielski and A. J. Haase, J. Nanomed. Nanotechnol., 2014, 5, 248 Search PubMed.
- S. Vranic, N. Boggetto, V. Contremoulins, S. Mornet, N. Reinhardt, F. Marano, A. Baeza-Squiban and S. Boland, Part. Fibre Toxicol., 2013, 10, 2 CrossRef CAS PubMed.
- N. Bohmer, A. Rippl, S. May, A. Walter, M. B. Heo, M. Kwak, M. Roesslein, N. W. Song, P. Wick and C. Hirsch, Colloids Surf., B, 2018, 172, 635–645 CrossRef CAS PubMed.
- L. E. Weiss, Y. Shalev Ezra, S. Goldberg, B. Ferdman, O. Adir, A. Schroeder, O. Alalouf and Y. Shechtman, Nat. Nanotechnol., 2020, 15, 500–506 CrossRef CAS PubMed.
- A. D. Lehmann, W. J. Parak, F. Zhang, Z. Ali, C. Röcker, G. U. Nienhaus, P. Gehr and B. Rothen-Rutishauser, Small, 2010, 6, 753–762 CrossRef CAS PubMed.
- B. Rothen-Rutishauser, D. A. Kuhn, Z. Ali, M. Gasser, F. Amin, W. J. Parak, D. Vanhecke, A. Fink, P. Gehr and C. Brandenberger, Nanomedicine, 2014, 9, 607–621 CrossRef CAS PubMed.
- F. Oltolina, D. Colangelo, I. Miletto, N. Clemente, M. Miola, E. Verné, M. Prat and A. Follenzi, Nanomaterials, 2019, 9, 1575 CrossRef CAS PubMed.
- M. Huang, Z. Ma, E. Khor and L. Y. Lim, Pharm. Res., 2002, 19, 1488–1494 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- B. Rothen-Rutishauser, C. Mühlfeld, F. Blank, C. Musso and P. Gehr, Part. Fibre Toxicol., 2007, 4, 9 CrossRef.
- G. A. Jenner, H. P. Longerich, S. E. Jackson and B. J. Fryer, Chem. Geol., 1990, 83, 133–148 CrossRef CAS.
- J. A. Ryan, K. W. Overton, M. E. Speight, C. N. Oldenburg, L. Loo, W. Robarge, S. Franzen and D. L. Feldheim, Anal. Chem., 2007, 79, 9150–9159 CrossRef CAS PubMed.
- A. Gojova, B. Guo, R. S. Kota, J. C. Rutledge, I. M. Kennedy and A. I. Barakat, Environ. Health Perspect., 2007, 115, 403–409 CrossRef CAS.
- S. S. Hardas, D. A. Butterfield, R. Sultana, M. T. Tseng, M. Dan, R. L. Florence, J. M. Unrine, U. M. Graham, P. Wu, E. A. Grulke and R. A. Yokel, Toxicol. Sci, 2010, 116, 562–576 CrossRef CAS.
- E. Sadauskas, N. R. Jacobsen, G. Danscher, M. Stoltenberg, U. Vogel, A. Larsen, W. Kreyling and H. Wallin, Chem. Cent. J., 2009, 3, 16 CrossRef PubMed.
- A. López-Serrano Oliver, S. Baumgart, W. Bremser, S. Flemig, D. Wittke, A. Grützkau, A. Luch, A. Haase and N. Jakubowski, J. Anal. At. Spectrom., 2018, 33, 1256–1263 RSC.
- T. W. May and R. H. Wiedmeyer, At. Spectrosc., 1998, 19, 150–155 CAS.
- C. M. Ackerman, P. K. Weber, T. Xiao, B. Thai, T. J. Kuo, E. Zhang, J. Pett-Ridge and C. J. Chang, Metallomics, 2018, 10, 474–485 CrossRef CAS PubMed.
- K. Narayan and S. Subramaniam, Nat. Methods, 2015, 12, 1021–1031 CrossRef CAS PubMed.
- E. Guehrs, M. Schneider, C. M. Günther, P. Hessing, K. Heitz, D. Wittke, A. López-Serrano Oliver, N. Jakubowski, J. Plendl, S. Eisebitt and A. Haase, J. Nanobiotechnol., 2017, 15, 21 CrossRef PubMed.
- V. Raffa, C. Riggio, M. P. Calatayud, C. Hoskins, J. Pinkernelle, G. F. Goya, L. Wang, G. Keilhoff and A. Cuschieri, Int. J. Nanomed., 2012, 3155 CrossRef PubMed.
- S. A. James, B. N. Feltis, M. D. De Jonge, M. Sridhar, J. A. Kimpton, M. Altissimo, S. Mayo, C. Zheng, A. Hastings, D. L. Howard, D. J. Paterson, P. F. A. Wright, G. F. Moorhead, T. W. Turney and J. Fu, ACS Nano, 2013, 7, 10621–10635 CrossRef CAS PubMed.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Acta Biomater., 2011, 7, 347–354 CrossRef CAS PubMed.
- C. S. Xu, K. J. Hayworth, Z. Lu, P. Grob, A. M. Hassan, J. G. García-Cerdán, K. K. Niyogi, E. Nogales, R. J. Weinberg and H. F. Hess, eLife, 2017, 6, 25916 CrossRef PubMed.
- R. Sharma and P. A. Crozier, Handbook of Microscopy for Nanotechnology, Kluwer Academic Publishers, Boston, 2005, pp. 531–565 Search PubMed.
- J. Taylor, A. Huefner, L. Li, J. Wingfield and S. Mahajan, Analyst, 2016, 141, 5037–5055 RSC.
- S. Watanabe, M. Lehmann, E. Hujber, R. D. Fetter, J. Richards, B. Söhl-kielczynski, A. Felies, J. Schmoranzer and E. M. Jorgensen, Electron Microscopy, Humana Press, Totowa, NJ, 2014, vol. 1117 Search PubMed.
- T. D. Helth, Basic immunology, 1978, vol. 272 Search PubMed.
- Y. Qie, H. Yuan, C. A. Von Roemeling, Y. Chen, X. Liu, K. D. Shih, J. A. Knight, H. W. Tun, R. E. Wharen, W. Jiang and B. Y. S. Kim, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- L. Tamarkin, L. Myer, R. Haynes and G. Paciotti, Nanomedicine, 2006, 2, 273–274 CrossRef.
- S. K. Libutti, G. F. Paciotti, A. A. Byrnes, H. R. Alexander, W. E. Gannon, M. Walker, G. D. Seidel, N. Yuldasheva and L. Tamarkin, Clin. Cancer Res., 2010, 16, 6139–6149 CrossRef CAS PubMed.
- K. A. Binnemars-Postma, H. W. Ten Hoopen, G. Storm and J. Prakash, Nanomedicine, 2016, 11, 2889–2902 CrossRef CAS PubMed.
- Y.-P. Chen, S.-H. Wu, I.-C. Chen and C.-T. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 10254–10265 CrossRef CAS PubMed.
- S. BoseDasgupta and J. Pieters, PLoS Pathog., 2014, 10, e1003879 CrossRef PubMed.
- D. Lucero, P. Islam, L. A. Freeman, X. Jin, M. Pryor, J. Tang, H. S. Kruth and A. T. Remaley, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2020, 1865, 158537 CrossRef CAS PubMed.
- N. H. Trivedi, J. J. Yu, C. Y. Hung, R. P. Doelger, C. S. Navara, L. Y. Armitige, J. Seshu, A. P. Sinai, J. P. Chambers, M. N. Guentzel and B. P. Arulanandam, Innate Immun., 2018, 24, 152–162 CrossRef CAS PubMed.
- D. Reichel, M. Tripathi and J. M. Perez, Nanotheranostics, 2019, 3, 66–88 CrossRef PubMed.
- M. Tsugita, N. Morimoto and M. Nakayama, Part. Fibre Toxicol., 2017, 14, 1–9 CrossRef PubMed.
- K. Hirota, T. Hasegawa, T. Nakajima, K. Makino and H. Terada, Colloids Surf., B, 2011, 87, 293–298 CrossRef CAS PubMed.
- M. Ahamed, M. J. Akhtar and H. A. Alhadlaq, Int. J. Environ. Res. Public Health, 2019, 16, 3199 CrossRef CAS PubMed.
- R. R. Miranda, V. Gorshkov, B. Korzeniowska, S. J. Kempf, F. F. Neto and F. Kjeldsen, Nanotoxicology, 2018, 12, 781–795 CrossRef CAS PubMed.
- G. M. Ngandeu Neubi, Y. Opoku-Damoah, X. Gu, Y. Han, J. Zhou and Y. Ding, Biomater. Sci., 2018, 6, 958–973 RSC.
- T. M. Allen, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed.
- K. Kardani, A. Milani, S. H. Shabani and A. Bolhassani, Expert Opin. Drug Delivery, 2019, 16, 1227–1258 CrossRef CAS PubMed.
- J. R. Maiolo, M. Ferrer and E. A. Ottinger, Biochim. Biophys. Acta, Biomembr., 2005, 1712, 161–172 CrossRef CAS PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS.
- G. Cao, W. Pei, H. Ge, Q. Liang, Y. Luo, F. R. Sharp, A. Lu, R. Ran, S. H. Graham and J. Chen, J. Neurosci., 2002, 22, 5423–5431 CrossRef CAS PubMed.
- H. D. Herce and A. E. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20805–20810 CrossRef CAS PubMed.
- I. M. Kaplan, J. S. Wadia and S. F. Dowdy, J. Controlled Release, 2005, 102, 247–253 CrossRef CAS PubMed.
- A. Joliot, C. Pernelle, H. Deagostini-Bazin and A. Prochiantz, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 1864–1868 CrossRef CAS.
- L. Crombez, M. C. Morris, S. Dufort, G. Aldrian-Herrada, Q. Nguyen, G. Mc Master, J.-L. Coll, F. Heitz and G. Divita, Nucleic Acids Res., 2009, 37, 4559–4569 CrossRef CAS.
- A. Hosseini, F. A. Lattanzio, S. S. Samudre, G. DiSandro, J. D. Sheppard and P. B. Williams, J. Ocul. Pharmacol. Ther., 2012, 28, 194–201 CrossRef CAS PubMed.
- G.-Q. Huang, J.-N. Wang, J.-M. Tang, L. Zhang, F. Zheng, J.-Y. Yang, L.-Y. Guo, X. Kong, Y.-Z. Huang, Y. Liu and S.-Y. Chen, J. Transl. Med., 2011, 9, 73 CrossRef CAS PubMed.
- H. Xin, X. Jiang, J. Gu, X. Sha, L. Chen, K. Law, Y. Chen, X. Wang, Y. Jiang and X. Fang, Biomaterials, 2011, 32, 4293–4305 CrossRef CAS PubMed.
- R. A. Morshed, M. E. Muroski, Q. Dai, M. L. Wegscheid, B. Auffinger, D. Yu, Y. Han, L. Zhang, M. Wu, Y. Cheng and M. S. Lesniak, Mol. Pharmaceutics, 2016, 13, 1843–1854 CrossRef CAS PubMed.
- L. Zhang, F. Liu, G. Li, Y. Zhou and Y. Yang, J. Pharm. Sci., 2015, 104, 4185–4196 CrossRef CAS PubMed.
- H. Gao, Q. Zhang, Y. Yang, X. Jiang and Q. He, Int. J. Pharm., 2015, 478, 240–250 CrossRef CAS PubMed.
- J. Li, L. Feng, L. Fan, Y. Zha, L. Guo, Q. Zhang, J. Chen, Z. Pang, Y. Wang, X. Jiang, V. C. Yang and L. Wen, Biomaterials, 2011, 32, 4943–4950 CrossRef CAS PubMed.
- K. J. F. Carnevale, M. E. Muroski, P. N. Vakil, M. E. Foley, G. Laufersky, R. Kenworthy, D. A. R. Zorio, T. J. Morgan, C. W. Levenson and G. F. Strouse, Bioconjugate Chem., 2018, 29, 3273–3284 CrossRef CAS PubMed.
- C. Hendrix, A. Abernethy, R. Sloane and J. Misuraca, Bone, 2013, 23, 1–7 Search PubMed.
- C. Ding, K. Wu, W. Wang, Z. Guan, L. Wang, X. Wang, R. Wang, L. Liu and J. Fan, Oncotarget, 2017, 8, 4718–4729 CrossRef PubMed.
- J. Zhao, X. Zhang, X. Sun, M. Zhao, C. Yu, R. J. Lee, F. Sun, Y. Zhou, Y. Li and L. Teng, Eur. J. Pharm. Biopharm., 2018, 130, 39–47 CrossRef CAS PubMed.
- L. Yan, H. Wang, Y. Jiang, J. Liu, Z. Wang, Y. Yang, S. Huang and Y. Huang, Macromol. Res., 2013, 21, 435–441 CrossRef CAS.
- K. Kurrikoff and Ü. Langel, Expert Opin. Drug Delivery, 2019, 16, 1183–1191 CrossRef CAS PubMed.
- I. Gessner and I. Neundorf, Int. J. Mol. Sci., 2020, 21, 1–21 Search PubMed.
- M. L. Torgersen, S. U. Lauvrak and K. Sandvig, FEBS J., 2005, 272, 4103–4113 CrossRef CAS PubMed.
- J. S. Lim, H. S. Na, H. C. Lee, H. E. Choy, S. C. Park, J. M. Han and K. A. Cho, Biochem. Biophys. Res. Commun., 2009, 390, 1322–1327 CrossRef CAS PubMed.
- P. A. Orlandi and P. H. Fishman, J. Cell Biol., 1998, 141, 905–915 CrossRef CAS PubMed.
- E. M. Damm, L. Pelkmans, J. Kartenbeck, A. Mezzacasa, T. Kurzchalia and A. Helenius, J. Cell Biol., 2005, 168, 477–488 CrossRef CAS PubMed.
- J. S. Shin and S. N. Abraham, Microbes Infect., 2001, 3, 755–761 CrossRef CAS.
- D. Akin, J. Sturgis, K. Ragheb, D. Sherman, K. Burkholder, J. P. Robinson, A. K. Bhunia, S. Mohammed and R. Bashir, Nat. Nanotechnol., 2007, 2, 441–449 CrossRef CAS PubMed.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015, 44, 6094–6121 RSC.
- K. Bibbins-Domingo, D. C. Grossman, S. J. Curry, K. W. Davidson, J. W. Epling, F. A. R. Garcia, A. R. Kemper, A. H. Krist, A. E. Kurth, C. S. Landefeld, C. M. Mangione, W. R. Phillips, M. G. Phipps, M. P. Pignone, M. Silverstein and C. W. Tseng, JAMA, J. Am. Med. Assoc., 2017, 317, 183–189 CrossRef PubMed.
- R. Lehner, K. Liu, X. Wang and P. Hunziker, Biomacromolecules, 2017, 18, 2654–2662 CrossRef CAS PubMed.
- R. Agabeigi, S. H. Rasta, M. Rahmati-Yamchi, R. Salehi and E. Alizadeh, Nanoscale Res. Lett., 2020, 15, 1–14 CrossRef PubMed.
- Z. H. Marfavi, M. Farhadi, S. B. Jameie, M. Zahmatkeshan, V. Pirhajati and M. Jameie, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 2783–2790 CrossRef CAS PubMed.
- W. Qu, Int. J. Nanomed., 2018, 4379–4389 CrossRef CAS PubMed.
- J. Necas, L. Bartosikova, P. Brauner and J. Kolar, Vet. Med., 2008, 53, 397–411 CrossRef CAS.
- B. B. Karakocak, J. Liang, P. Biswas and N. Ravi, Carbohydr. Polym., 2018, 186, 243–251 CrossRef CAS.
- C. S. Kumar, M. D. Raja, D. S. Sundar, M. Gover Antoniraj and K. Ruckmani, Carbohydr. Polym., 2015, 128, 63–74 CrossRef CAS.
- W. J. Lin, W. C. Lee and M. J. Shieh, Carbohydr. Polym., 2017, 155, 101–108 CrossRef CAS PubMed.
- S. Ganesh, A. K. Iyer, D. V. Morrissey and M. M. Amiji, Biomaterials, 2013, 34, 3489–3502 CrossRef CAS PubMed.
- R. Narayanaswamy, S. Kanagesan, A. Pandurangan and P. Padmanabhan, Nanobiomaterials in Medical Imaging, Elsevier, 2016, pp. 101–129 Search PubMed.
- S. Chono, T. Tanino, T. Seki and K. Morimoto, J. Pharm. Pharmacol., 2007, 59, 75–80 CrossRef CAS PubMed.
- D. A. Bender, Nutritional Biochemistry of the Vitamins, Cambridge University Press, 2003 Search PubMed.
- P. M. Finglas, Trends Food Sci. Technol., 2000, 11, 296–297 CrossRef CAS.
- G. J. Russell-Jones, L. Arthur and H. Walker, Int. J. Pharm., 1999, 179, 247–255 CrossRef CAS PubMed.
- Y. Wang, L. Yuan, C. Yao, L. Ding, C. Li, J. Fang, K. Sui, Y. Liu and M. Wu, Nanoscale, 2014, 6, 15333–15342 RSC.
- K. Yin Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef PubMed.
- N. Sakamoto, K. Ogawa, G. Suda, K. Morikawa, T. Sho, M. Nakai, H. Suzuki, N. Yamagata, Y. Tanaka, W. Ying, Y. Tamura, Y. Niitsu and K. Maruyama, J. Hepatol., 2018, 68, S242 CrossRef.
- E. Y. Chen, S. H. Chu, L. Gov, Y. K. Kim, M. B. Lodoen, A. J. Tenner and W. F. Liu, J. Mater. Chem. B, 2017, 5, 1574–1584 RSC.
- M. Cavadas, Á. González-Fernández and R. Franco, Nanomedicine, 2011, 7, 730–743 CrossRef CAS PubMed.
- J. Pieters, Curr. Opin. Immunol., 2001, 13, 37–44 CrossRef CAS.
- D. Longoni, L. Piemonti, S. Bernasconi, A. Mantovani and P. Allavena, Int. J. Clin. Lab. Res., 1998, 28, 162–169 CrossRef CAS PubMed.
- T. A. Patente, M. P. Pinho, A. A. Oliveira, G. C. M. Evangelista, P. C. Bergami-Santos and J. A. M. Barbuto, Front. Immunol., 2019, 9, 3176 CrossRef PubMed.
- J. F. Telfer and J. H. Brock, Med. Sci. Monit., 2004, 10, 91–96 Search PubMed.
- I. Martinez, B. Sveinbjornsson, F. Vidalvanaclocha, A. Asumendi and B. Smedsrod, Biochem. Biophys. Res. Commun., 1995, 212, 235–241 CrossRef CAS PubMed.
- A. Varin, S. Mukhopadhyay, G. Herbein and S. Gordon, Blood, 2010, 115, 353–362 CrossRef CAS PubMed.
- D. M. Mosser and X. Zhang, Curr. Protoc. Immunol., 2008, 83, 14 Search PubMed.
- J. L. Moreno, I. Mikhailenko, M. M. Tondravi and A. D. Keegan, J. Leukocyte Biol., 2007, 82, 1542–1553 CrossRef CAS PubMed.
- M. Leidi, E. Gotti, L. Bologna, E. Miranda, M. Rimoldi, A. Sica, M. Roncalli, G. A. Palumbo, M. Introna and J. Golay, J. Immunol., 2009, 182, 4415–4422 CrossRef CAS PubMed.
- B.-A. Chen, Y.-Y. Dai, X.-M. Wang, R.-Y. Zhang, W.-L. Xu, H.-L. Shen, F. Gao, Q. Sun, X.-J. Deng, J.-H. Ding, C. Gao, Y.-Y. Sun, J. Cheng, J. Wang, G. Zhao and N.-N. Chen, Int. J. Nanomed., 2008, 3, 343–350 CrossRef CAS.
- A. Rafieepour, M. R. Azari, F. Khodagholi, J. P. Jaktaji, Y. Mehrabi and H. Peirovi, Environ. Sci. Pollut. Res., 2019, 26, 31752–31762 CrossRef CAS PubMed.
- O. Okoturo-Evans, A. Dybowska, E. Valsami-Jones, J. Cupitt, M. Gierula, A. R. Boobis and R. J. Edwards, PLoS One, 2013, 8, e72363 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- L. A. Lane, X. Qian, A. M. Smith and S. Nie, Annu. Rev. Phys. Chem., 2015, 66, 521–547 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- V. H. Nguyen and B.-J. Lee, Int. J. Nanomed., 2017, 12, 3137–3151 CrossRef CAS PubMed.
- P. Maffre, S. Brandholt, K. Nienhaus, L. Shang, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2014, 5, 2036–2047 CrossRef PubMed.
- B. Pelaz, P. Del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. De La Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- P. Maffre, K. Nienhaus, F. Amin, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2011, 2, 374–383 CrossRef CAS PubMed.
- X. Jiang, S. Weise, M. Hafner, C. Röcker, F. Zhang, W. J. Parak and G. U. Nienhaus, J. R. Soc. Interface, 2010, 7, S5–S13 CAS.
- Y. Klapper, P. Maffre, L. Shang, K. N. Ekdahl, B. Nilsson, S. Hettler, M. Dries, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2015, 7, 9980–9984 RSC.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo, D. W. H. Olsen, Y. Cohen, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 2439–2455 CrossRef CAS PubMed.
- V. Mirshafiee, R. Kim, M. Mahmoudi and M. L. Kraft, Int. J. Biochem. Cell Biol., 2016, 75, 188–195 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- V. Francia, K. Yang, S. Deville, C. Reker-Smit, I. Nelissen and A. Salvati, ACS Nano, 2019, 13, 11107–11121 CrossRef CAS PubMed.
- A. C. G. Weiss, K. Kempe, S. Förster and F. Caruso, Biomacromolecules, 2018, 19, 2580–2594 CrossRef CAS PubMed.
- J.-W. Yoo, E. Chambers and S. Mitragotri, Curr. Pharm. Des., 2010, 16, 2298–2307 CrossRef CAS PubMed.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–3166 CrossRef CAS.
- S. Hak, E. Helgesen, H. H. Hektoen, E. M. Huuse, P. A. Jarzyna, W. J. M. Mulder, O. Haraldseth and C. de L. Davies, ACS Nano, 2012, 6, 5648–5658 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS.
- J. Canton, D. Neculai and S. Grinstein, Nat. Rev. Immunol., 2013, 13, 621–634 CrossRef CAS PubMed.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- E. Tahover, R. Bar-Shalom, E. Sapir, R. Pfeffer, I. Nemirovsky, Y. Turner, M. Gips, P. Ohana, B. W. Corn, A. Z. Wang and A. A. Gabizon, Front. Oncol., 2018, 8, 544 CrossRef PubMed.
- P. Munster, I. E. Krop, P. LoRusso, C. Ma, B. A. Siegel, A. F. Shields, I. Molnár, T. J. Wickham, J. Reynolds, K. Campbell, B. S. Hendriks, B. S. Adiwijaya, E. Geretti, V. Moyo and K. D. Miller, Br. J. Cancer, 2018, 119, 1086–1093 CrossRef CAS PubMed.
- K. A. Autio, J. A. Garcia, A. S. Alva, L. L. Hart, M. I. Milowsky, E. M. Posadas, C. J. Ryan, J. M. Summa, H. Youssoufian, H. I. Scher and R. Dreicer, J. Clin. Oncol., 2016, 34, 233 Search PubMed.
- P. Jiang, C. F. Lagenaur and V. Narayanan, J. Biol. Chem., 1999, 274, 559–562 CrossRef CAS PubMed.
- E. Brown, Trends Cell Biol., 2001, 11, 130–135 CrossRef CAS PubMed.
- S. B. Willingham, J. P. Volkmer, A. J. Gentles, D. Sahoo, P. Dalerba, S. S. Mitra, J. Wang, H. Contreras-Trujillo, R. Martin, J. D. Cohen, P. Lovelace, F. A. Scheeren, M. P. Chao, K. Weiskopf, C. Tang, A. K. Volkmer, T. J. Naik, T. A. Storm, A. R. Mosley, B. Edris, S. M. Schmid, C. K. Sun, M. S. Chua, O. Murillo, P. Rajendran, A. C. Cha, R. K. Chin, D. Kim, M. Adorno, T. Raveh, D. Tseng, S. Jaiswal, P. Ø. Enger, G. K. Steinberg, G. Li, S. K. So, R. Majeti, G. R. Harsh, M. De Van Rijn, N. N. H. Teng, J. B. Sunwoo, A. A. Alizadeh, M. F. Clarke and I. L. Weissman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6662–6667 CrossRef CAS PubMed.
- P. L. Rodriguez, T. Harada, D. A. Christian, D. A. Pantano, R. K. Tsai and D. E. Discher, Science, 2013, 339, 971–975 CrossRef CAS.
- P. Grimbert, S. Bouguermouh, N. Baba, T. Nakajima, Z. Allakhverdi, D. Braun, H. Saito, M. Rubio, G. Delespesse and M. Sarfati, J. Immunol., 2006, 177, 3534–3541 CrossRef CAS PubMed.
- G. R. Cornelis, J. Cell Biol., 2002, 158, 401–408 CrossRef CAS.
- K. Guan and J. Dixon, Science, 1990, 249, 553–556 CrossRef CAS PubMed.
- M. Fallman, K. Andersson, S. Hakansson, K. E. Magnusson, O. Stendahl and H. Wolf-Watz, Infect. Immun., 1995, 63, 3117–3124 CrossRef CAS.
- I. Bolin, A. Forsberg, L. Norlander, M. Skurnik and H. Wolf-Watz, Infect. Immun., 1988, 56, 343–348 CrossRef CAS PubMed.
- A. Forsberg and H. Wolf-Watz, Mol. Microbiol., 1988, 2, 121–133 CrossRef CAS PubMed.
- K. Y. Leung, B. S. Reisner and S. C. Straley, Infect. Immun., 1990, 58, 3262–3271 CrossRef CAS PubMed.
- S. Greenberg, P. Chang and S. C. Silverstein, J. Exp. Med., 1993, 177, 529–534 CrossRef CAS.
- N. Sauvonnet, I. Lambermont, P. van der Bruggen and G. R. Cornelis, Mol. Microbiol., 2002, 45, 805–815 CrossRef CAS PubMed.
- I. Adkins, M. Köberle, S. Gröbner, E. Bohn, I. B. Autenrieth and S. Borgmann, Int. J. Med. Microbiol., 2007, 297, 235–244 CrossRef CAS.
- N. P. Hoe, R. M. Ireland, F. R. DeLeo, B. B. Gowen, D. W. Dorward, J. M. Voyich, M. Liu, E. H. Burns, D. M. Culnan, A. Bretscher and J. M. Musser, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7646–7651 CrossRef CAS PubMed.
- B. White, Am. Fam. Physician, 2009, 80, 345–350 Search PubMed.
- M. Jiang, B. Wu, Y. Sun, Y. Ding, Y. Xie, L. Liu and Y. Cao, Toxicol. Mech. Methods, 2019, 29, 291–299 CrossRef CAS.
- T. E. Wellems and C. V. Plowe, J. Infect. Dis., 2001, 184, 770–776 CrossRef CAS PubMed.
- T. Y. Hu, M. Frieman and J. Wolfram, Nat. Nanotechnol., 2020, 15, 247–249 CrossRef CAS.
- M. Hoffmann, K. Mösbauer, H. Hofmann-Winkler, A. Kaul, H. Kleine-Weber, N. Krüger, N. C. Gassen, M. A. Müller, C. Drosten and S. Pöhlmann, Nature, 2020, 585, 588–590 CrossRef CAS PubMed.
- J. Pelt, S. Busatto, M. Ferrari, E. A. Thompson, K. Mody and J. Wolfram, Pharmacol. Ther., 2018, 191, 43–49 CrossRef CAS PubMed.
- S. E. Miller, S. Mathiasen, N. A. Bright, F. Pierre, B. T. Kelly, N. Kladt, A. Schauss, C. J. Merrifield, D. Stamou, S. Höning and D. J. Owen, Dev. Cell, 2015, 33, 163–175 CrossRef CAS PubMed.
- E. Haładyj, M. Sikora, A. Felis-Giemza and M. Olesińska, Reumatologia, 2018, 56, 164–173 CrossRef PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- R. Rosqvist, A. Forsberg and H. Wolf-Watz, Biochem. Soc. Trans., 1991, 19, 1131–1132 CrossRef CAS PubMed.
- M. Foster-Cuevas, G. J. Wright, M. J. Puklavec, M. H. Brown and A. N. Barclay, J. Virol., 2004, 78, 7667–7676 CrossRef CAS PubMed.
- E. Susnik, P. Taladriz-Blanco, B. Drasler, S. Balog, A. Petri-Fink and B. Rothen-Rutishauser, Cells, 2020, 9, 2099 CrossRef CAS PubMed.
- C. Kuttner, M. Mayer, M. Dulle, A. Moscoso, J. M. López-Romero, S. Förster, A. Fery, J. Pérez-Juste and R. Contreras-Cáceres, ACS Appl. Mater. Interfaces, 2018, 10, 11152–11163 CrossRef CAS.
- R. Madathiparambil Visalakshan, L. E. González García, M. R. Benzigar, A. Ghazaryan, J. Simon, A. Mierczynska-Vasilev, T. D. Michl, A. Vinu, V. Mailänder, S. Morsbach, K. Landfester and K. Vasilev, Small, 2020, 16, 2000285 CrossRef CAS PubMed.
- Y. Liu, H. Yuan, F. R. Kersey, J. K. Register, M. C. Parrott and T. Vo-Dinh, Sensors, 2015, 15, 3706–3720 CrossRef CAS PubMed.
- M. Liu, Q. Li, L. Liang, J. Li, K. Wang, J. Li, M. Lv, N. Chen, H. Song, J. Lee, J. Shi, L. Wang, R. Lal and C. Fan, Nat. Commun., 2017, 8, 15646 CrossRef CAS PubMed.
- D. van der Zwaag, N. Vanparijs, S. Wijnands, R. De Rycke, B. G. De Geest and L. Albertazzi, ACS Appl. Mater. Interfaces, 2016, 8, 6391–6399 CrossRef CAS PubMed.
- M. Wiemann, A. Vennemann, M. Stintz, R. Retamal Marín, F. Babick, G.-G. Lindner, T. Schuster, U. Brinkmann and N. Krueger, Nanomaterials, 2018, 9, 11 CrossRef.
- M. Pavlin, J. Lojk, V. B. Bregar, M. Rajh, K. Mis, M. E. Kreft, S. Pirkmajer and P. Veranic, Int. J. Nanomed., 2015, 1449 CrossRef.
- H. F. Krug and P. Wick, Angew. Chem., Int. Ed., 2011, 50, 1260–1278 CrossRef CAS PubMed.
- M. Reggente, D. Passeri, L. Angeloni, F. A. Scaramuzzo, M. Barteri, F. De Angelis, I. Persiconi, M. E. De Stefano and M. Rossi, Nanoscale, 2017, 9, 5671–5676 RSC.
- J. Blechinger, A. T. Bauer, A. A. Torrano, C. Gorzelanny, C. Bräuchle and S. W. Schneider, Small, 2013, 9, 3970–3980 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |