
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApproaches for the inhibition and elimination of microbial biofilms using macromolecular agents
Lewis D.
Blackman
 *a,
Yue
Qu
bc,
Peter
Cass
a and
Katherine E. S.
Locock
*a
*a,
Yue
Qu
bc,
Peter
Cass
a and
Katherine E. S.
Locock
*a
aCSIRO Manufacturing, Research Way, Clayton, VIC 3168, Australia. E-mail: Lewis.Blackman@csiro.au; Katherine.Locock@csiro.au
bInfection and Immunity Program, Department of Microbiology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia
cDepartment of Infectious Diseases, The Alfred Hospital and Central Clinical School, Monash University, Melbourne, VIC 3004, Australia
First published on 6th January 2021
Abstract
Biofilms are complex three-dimensional structures formed at interfaces by the vast majority of bacteria and fungi. These robust communities have an important detrimental impact on a wide range of industries and other facets of our daily lives, yet their removal is challenging owing to the high tolerance of biofilms towards conventional antimicrobial agents. This key issue has driven an urgent search for new innovative antibiofilm materials. Amongst these emerging approaches are highly promising materials that employ aqueous-soluble macromolecules, including peptides, proteins, synthetic polymers, and nanomaterials thereof, which exhibit a range of functionalities that can inhibit biofilm formation or detach and destroy organisms residing within established biofilms. In this Review, we outline the progress made in inhibiting and removing biofilms using macromolecular approaches, including a spotlight on cutting-edge materials that respond to environmental stimuli for “on-demand” antibiofilm activity, as well as synergistic multi-action antibiofilm materials. We also highlight materials that imitate and harness naturally derived species to achieve new and improved biomimetic and biohybrid antibiofilm materials. Finally, we share some speculative insights into possible future directions for this exciting and highly significant field of research.
 Lewis D. Blackman | Dr Lewis Blackman is a Research Scientist within the Biomedical Manufacturing Program at the CSIRO in Melbourne, Australia. Lewis was awarded an MChem from the University of Southampton (2012), and a PhD focused on block copolymer self-assembly, under the supervision of Prof. Rachel O’Reilly and Prof. Matthew Gibson, from the University of Warwick (2018). In 2017, he joined the Polymeric Biomaterials Team at CSIRO as a CERC Postdoctoral Research Fellow, where he was recently appointed as a Research Scientist. Lewis’ interests include antimicrobial materials, stimuli-responsive systems, polymer self-assembly, and biohybrid materials for biomedical applications, as well as commercial research activities. |
 Yue Qu | Dr Yue Qu is a microbiologist whose expertise is in biofilm-related infections. He received a MBBS from Wenzhou Medical University in 2000, a PhD from RMIT University in 2010 and an Australian Research Council (ARC) Super Science Fellowship at Monash University in 2012. He is a rare example of fundamental researcher equipped with medical knowledge and experience. Dr Qu has contributed significantly to the field of biofilm-related infections, from disease pathogenesis to prevention and treatment. His research has a broad coverage of bacterial and fungal pathogens, and different disease models, including bloodstream infections, vaginal candidiasis, and VAD driveline infections. |
 Peter Cass | Dr Peter Cass received his PhD in polymer chemistry from Swinburne University, Australia in 2000. Peter undertook a Postdoctoral Fellowship at Melbourne University, Australia, from 2001 to 2003. He is currently working as a Principle Research Consultant at the Commonwealth Scientific and Industrial Research Organisation in Melbourne, Australia. His research interests lie in polymer chemistry including industrial polymers, composites, drug delivery and biomaterials. |
 Katherine E. S. Locock | Dr Katherine Locock is a Senior Research Scientist in the Manufacturing Business Unit of the CSIRO in Melbourne, Australia. Her research focuses on the development of biologically active polymers, with a particular focus on antimicrobial therapeutics. Prior to the CSIRO, Katherine held a position as an Associate Lecturer in Pharmacology at the University of Sydney, where she focused on rational drug design to develop analogues to target memory and mood. She received the Victorian Tall Poppy of the Year and Julius Career Development Awards in 2016, and has had an active role in driving diversity in the chemical sciences. |
Introduction
Biofilms are dynamic surface-attached communities of bacteria and/or fungi surrounded by a robust self-secreted polymeric extracellular matrix.1–3 Biofilms are ubiquitous in nature, with the vast majority of bacteria being either surface-adhered or existing within a biofilm in natural environments.4 The wide-spread presence of biofilms has significant implications for a range of applications including in healthcare, agriculture and food processing and storage, industrial processes, and transportation. Whilst biofilms can be harnessed for good to provide innovative biotechnological solutions, such as their use as biofertilizers,5 in biodiesel production,6 and in the degradation of pollutants for water remediation,7 the prevalence of biofilms in other sectors costs the global economy billions of dollars annually, as well as having significant social implications for our daily lives. In the healthcare sector, up to an estimated 80% of all microbial infections are associated with biofilm formation.8 These include chronic bacterial and fungal infections that have great significance in cystic fibrosis-related pulmonary infections, in chronic wounds, such as those associated with diabetes, and in chronic middle ear infections.9,10 Additionally, biofilms strongly enable nosocomial infections, including those associated with medical devices, such as catheter-associated urinary tract infections (CAUTI), central line-associated bloodstream infections (CLABI), and ventilator-associated pneumonia.11,12 Such hospital-acquired infections are solely estimated to cost between $28–45 billion annually in the USA alone.13 Biofilms also have a significant negative impact on dental care,14 as well as in the food industry, with biofilms being the dominant mode of growth for food-borne pathogens.15,16 Furthermore, biofilms can be highly detrimental to steel and other alloys through biofouling and biocorrosion,17,18 placing a substantial economic burden on industries that are reliant on marine applications, such as maritime transportation and underwater mining.Bacteria and fungi within biofilms are typically highly resistant to conventional antimicrobials, making their eradication and treatment exceptionally challenging.19 As such, there has been extensive investigation into the development of new materials for the elimination of established biofilms using small molecule agents,20–22 carbon nanomaterials,23 as well as metal, metal oxide, and other inorganic nanoparticles.24–26 These materials have been widely reviewed in the cited texts and will not be covered in this Review. Aside from these newly emerging materials, aqueous-soluble macromolecular species, such as peptides, enzymes, synthetic polymers, and polymer nanoparticles, have also shown great promise as antibiofilm agents. The focus of this article is to give an overview of the approaches for inhibiting and eliminating biofilms using either natural or synthetic macromolecular antibiofilm agents. Note that surface-based approaches, such as the development of new antiadhesive and biocidal surface coatings and surface modification strategies, as well as antiadhesive materials (e.g. hydrogels and other biomaterials), will not be included in this article and are reviewed elsewhere.27–33 Similarly, this Review does not intend to detail the chemistry and synthetic approaches for achieving such macromolecular agents. For more information on this aspect of the field, the reader is referred to the following texts.34–36 In this Review, an overview of biofilm formation will be given, including a brief description of biofilm structure and function, which gives rise to their resistance and tolerance towards conventional antibiotic treatments. This will be followed by a review of strategies that exploit antibiofilm peptides and proteins, as well as their synthetic polymer counterparts, and nanoparticles thereof. A detailed account of cutting-edge emerging strategies for tackling biofilms will be also given, with a focus on multi-mechanistic approaches and other highly novel unconventional approaches. Finally, we share our outlook and offer some speculative views on the future directions of this fascinating and vital field of research.
Overview of biofilm formation, regulation, and resistance
Whilst bacteria and fungi have been widely studied in their planktonic form, such microorganisms seldom exist in an independent, free-swimming planktonic state in natural systems.4 Instead, bacteria and fungi typically exist in interdependent multicellular surface-adhered communities, surrounded by a self-secreted extracellular matrix, known as biofilms.2,37 Biofilms can form at the interface of both biotic and abiotic surfaces, as well as at liquid–solid and liquid–air interfaces,37 and can comprise either a single species (monomicrobial biofilms), or multiple diverse species (polymicrobial biofilms), occasionally spanning different biological kingdoms.38 It is typically observed that polymicrobial biofilms can exhibit even higher tolerance to antimicrobial treatment, particularly in the case of cross-kingdom polymicrobial biofilms.39Biofilm formation and proliferation is a complex, multistep process (Scheme 1).1,2,40 Prior to initial microbial attachment, the surface is typically fouled through non-specific adsorption of proteins or other such species present in the surrounding medium, providing a conditioning layer for attachment.40 This conditioning layer alters the surface energy, electrostatic charge, topography of the substrate surface, and surface elemental composition, and typically aids microbial attachment.41 Microbes are typically transported to the surface through passive sedimentation and mass transport mechanisms, but they can also be propelled through active chemotaxis and flagella-driven motion in some instances.40 Individual cells then attach to the surface in a dual-phase process involving reversible electrostatic and hydrophobic interactions, followed by relatively irreversible covalent and hydrogen bonding interactions.40 In certain cases, this initial attachment is aided by the presence of wall teichoic acids or other adhesive units present on the microbial surface.42 Bacterial pilli and pillus-like adhesins are particularly important for host–microbe interaction and attachment to living host tissue,43 whereas non-specific interactions are generally dominant in the case of biofilm formation onto abiotic surfaces.1
 | ||
| Scheme 1 Schematic illustrating the various biofilm stages of a monomicrobial biofilm's formation. | ||
Following initial attachment and the generation of surface-adhered microcolonies, the biofilm enters the proliferation phase. During this period, the attached microbes proliferate, whilst also secreting extracellular polymeric substances (EPS) to form a biofilm matrix, comprised of a complex mixture of exopolysaccharides, proteins, nucleic acids, and lipids.3,44 This provides mechanical stability and cohesion amongst the individual microbes, as well as promoting further “anchoring” interactions with the surface.42 In the subsequent maturation phase, the biofilm restructures itself into a tall three-dimensional morphology, often described as a mushroom-like or tower-like structure.2,42 This structure is highly hydrated and typically contains water channels between the tower-like structures.2 To facilitate this restructuring, enzymes such as proteases and nucleases that can degrade the EPS are secreted by the biofilm population.42 Finally, partial dispersal of the biofilm occurs to allow the release of microorganisms from the biofilm into the planktonic form, in turn facilitating the spread of the species to colonize new surface regions.45
Biofilm formation, maturation and dispersal is often driven largely by cell–cell signalling pathways, known collectively as quorum sensing.45,46 Quorum sensing is a mechanism for regulating gene expression in response to changes in local microbial population density and is driven by the secretion and recognition of signalling molecules called autoinducers or quorum sensing molecules.47,48 Recognition of extracellular quorum sensing autoinducers allows individual organisms to monitor the population density of other microbes in their local environment, and respond if above or below a certain autoinducer threshold. Their response is achieved by regulating gene expression, thereby promoting phenotypic changes to drive an appropriate biological process. Such processes include regulation of virulence, sporulation, swarming motility, and biofilm formation and dispersal.46,47 Autoinducer signalling molecules in bacterial pathways include acyl homoserine lactones in Gram-negative bacteria, oligopeptides in Gram-positive bacteria, and the somewhat universal boron-containing autoinducer, autoinducer-2.47 In fungi, quorum sensing molecules include the terpene-based alcohol, farnesol, as well as alcohols derived from aromatic amino acids, such as tyrosine (tyrosol), phenylalanine (phenylethanol) and tryptophan (tryptophol).48
The EPS secreted by organisms within the biofilm form the hydrated crosslinked network of the biofilm matrix.3 The role of the biofilm matrix is highly multifaceted and includes facilitating bacterial aggregation and providing biofilm cohesion, allowing horizontal gene transfer and cell–cell communication amongst the biofilm population, allowing sorption of inorganic ions and organic small molecules and providing a source of nutrients, allowing water retention, and exhibiting enzymatic activity, amongst others.3 The biofilm matrix also provides extreme protection against both mechanical stresses and antibiotic treatments. Bacteria and fungi within a biofilm are typically able to resist up to the order of one thousand-fold concentrations of organic small molecule antibiotics or antimicrobial heavy metals, relative to their planktonic form.49,50 Note that the mechanisms responsible for biofilm-associated resistance are typically distinct from the genetic mutations or gene transfer mechanisms observed in conventional antimicrobial resistance pathways, with biofilm resistance mechanisms being structural and environmental in nature.49 Though debated,49 possible mechanistic pathways proposed for the enhanced resistance of biofilm-dwelling microorganisms include the retarded diffusion and limited penetration of antibiotics through the biofilm matrix, which hampers the antibiotic's activity over its lifetime prior to degradation or clearance. Additionally, the biofilm has an altered chemical environment relative to the surrounding medium, for example the formation of anaerobic regions and significant changes in pH are common, which can alter the effectiveness and stability of certain antibiotics.
Further to physicochemical resistance mechanisms, biofilms can also include a discrete sub-population of phenotypically distinct bacteria known as persister cells.51,52 These persister cells comprise up to 1% of the total population and exist in a protected, dormant state, and exhibit high multidrug tolerance. Contrary to antibiotic resistance, whereby the antibiotic is degraded or prevented from reaching its intended target, antibiotic tolerance occurs when persister cells shut down the target itself, thereby circumventing the antibiotic's mode of action. As such, persister cells typically exhibit non-metabolic behaviour consistent with a dormant phenotype that is resistant to antibiotics that target metabolic pathways.51,52 Fungal biofilms have also been shown to contain a sub-population of persister cells with inherent antifungal tolerance,53 suggesting that persister cells may be ubiquitous in providing antimicrobial tolerance to biofilms across a diverse range of species.
Macromolecular antibiofilm agents
Owing to the high resistance and tolerance of biofilms towards conventional small molecule antibiotics, there has been significant attention given to less conventional treatments, such as those that employ macromolecular species. These can directly target biofilm-dwelling bacteria or fungi through alternative non-metabolic pathways, or otherwise can inhibit the formation of the biofilm matrix. Additionally, those that can degrade the biofilm matrix components or interfere with cell–cell signalling can act as biofilm dispersants, breathing new life into existing conventional antibiotics, or can act synergistically with lethal macromolecular antibiofilm species or functionalities. Macromolecular approaches for the inhibition, dispersal and killing of biofilms using natural, synthetic and semi-synthetic materials are discussed in the following sections and are summarized in Table 1. In the following sections, numerous techniques for assessing antibiofilm agents will be highlighted throughout the discussion, however the techniques themselves will not be explicitly described in this focused Review; for details of the assessment of antibiofilm agents, the reader is referred to the following dedicated text.54| Material class | Examples | Mode of activity | References |
|---|---|---|---|
| Antibiofilm peptides | LL-37, IDR-1018, HD6 | Multiple (see Fig. 1C) | 56 and 58–61 |
| Nucleases | DNase I | Degradation of extracellular DNA to destabilize the biofilm matrix | 62–64 |
| Polysaccharide depolymerases | Dispersin B, lysozyme | Degradation of exopolysaccahrides and/or peptidoglycan | 64–66 |
| Oxidoreductases | Glucose oxidase, lactoperoxidase | Production of reactive oxygen species (ROS) and other reactive species | 67–69 |
| Proteases | Proteinase K, lysostaphin | Degradation of protein/peptide components of biofilm matrix and/or peptidoglycan | 70–73 |
| Quorum quenching enzymes | Acylases, lactonases | Interference with microbial cell–cell signalling | 74 and 75 |
| Antibodies | Anti-(β-1,6-poly-N-acetylglucosamine) | Binding and subsequent deactivation or disruption of biofilm components | 76 |
| (Semi-)synthetic polycationic materials | Poly(guanidine)s, poly(amine)s, chitosan | Predominantly rupturing of bacterial membranes | 34 and 77–83 |
| Peptidomimetics | Peptoids, β-peptides | Predominantly rupturing of bacterial membranes, among others | 84–86 |
| Nitric oxide-releasing polymers | N-Diazeniumdiolate-functional polymers | Release of nitric oxide in addition to subsequent formation of related reactive species | 87–89 |
| Photodynamic polymers | Conjugated polymers, porphyrin-functional polymers | Production of ROS upon illumination | 90–92 |
| Quorum quenching synthetic polymers | Poly(diol)s, poly(catechol)s, polyanions | Interference with microbial cell–cell signalling | 93 and 94 |
| Biohybrid systems | Bioconjugates, protein-loaded nanoparticles | Numerous, dependent on biologic and synthetic material employed | 95–98 |
Antibiofilm strategies employing peptides and proteins
Antibiofilm peptides
Antimicrobial peptides are a promising class of biocidal materials that are widely found in nature as part of various organisms’ innate immune systems.55–57 Typically, these short peptides (around 12–50 residues) contain both hydrophobic and cationic residues and are capable of penetration and rupture of bacterial membranes, giving rise to their biocidal properties.55 Such peptides are typically easily cleared in vivo and are poorly bioavailable, ensuring that they do not persist in the body or in the environment for extended periods of time.58 The nature of antimicrobial peptides’ membrane-rupturing biocidal mechanism, along with their low bioavailability and rapid in vivo proteolytic degradation lead to low levels of acquired resistance development towards such treatments.58 The structure of one natural (LL-37) and one synthetic (IDR-1018) peptide are shown in Fig. 1A and B, which illustrate the helical secondary structure typically observed in such antimicrobial peptides. The use of antimicrobial peptides for tackling biofilms has been reviewed in detail elsewhere,20,56,58,99 however these will be briefly discussed in this section owing to their similarity and overlap with other antibiofilm approaches. As will be discussed, antimicrobial peptides show numerous modes of action and can inhibit biofilm formation as well as being able to weaken the matrix of established biofilms to directly kill microbes residing within. | ||
| Fig. 1 Structure and function of the antibiofilm peptides LL-37 and IDR-1018. (A) Helical wheel structure of LL-37 reproduced with permission from ref. 59. Published by the Royal Society of Chemistry. (B) Primary structure of IDR-1018 along with its secondary helical structure calculated from NMR spectroscopy of the peptide in dodecylphosphocholine micelles. Reproduced with permission from ref. 104. Copyright©2010 Elsevier Ltd. All rights reserved. (C) Various mechanisms of biofilm disruption and elimination demonstrated for antibiofilm peptides. (D) CLSM micrographs of established biofilms with either no treatment or treatment with the IDR-1018 peptide, a model antibiotic as indicated, or a combination of IDR-1018 and antibiotic, after staining with a BacLight LIVE/DEAD viability stain. Adapted with permission from the American Society for Microbiology, ref. 105. | ||
Okuda and Mizunoe studied the antibiofilm activity of various ribosomally-synthesized bacteria-derived antimicrobial peptides (nisin A, lacticin Q, and nukacin ISK-1) against biofilms formed by methicillin resistant Staphylococcus aureus (MRSA) clinical isolates.100 They found that whilst all three showed effective anti-planktonic activity, nisin showed the greatest antibiofilm activity, with lacticin Q having significantly reduced antibiofilm activity. Nukacin ISK-1 was ineffective at dispersing or eradicating biofilms despite its activity against the planktonic form, which was similar to vancomycin, a control small molecule antibiotic with ineffective antibiofilm properties. Mechanistic studies showed that peptides like nisin A could form stable pores in the bacterial membranes and lead to efflux of adenosine triphosphate (ATP), which was important for bactericidal activity against biofilm cells.100
Whilst peptides that show activity against planktonic cells and those that exhibit antibiofilm activity are structurally similar with regards to their cationic and hydrophobic balance, they show distinct structure–property relationships for each process. For example, the human cathelicidin peptide LL-37 showed inhibition of Pseudomonas aeruginosa biofilm formation at 1/16th of its minimum inhibitory concentration (MIC) obtained against the planktonic form of this bacterium.101 Furthermore, it was able to significantly reduce the thickness of established biofilms at this same sub-lethal concentration in a flow cell assay.101 This observation highlights that aside from direct killing of bacteria through membrane rupture, antimicrobial peptides have other distinct mechanisms for the eradication of biofilms (Fig. 1C),102 for example the interruption of bacterial signalling pathways. In the case of LL-37, the peptide was shown to interfere with two major quorum sensing pathways required for biofilm formation, as well as stimulating twitching motility.101
Certain antimicrobial peptides show an immunomodulatory effect both in vitro and in vivo, including natural peptides such as LL-37, and synthetic peptides such as the innate defense regulator peptide, IDR-1018.103 Whilst both peptides show only limited direct antimicrobial activity under physiological conditions, they can trigger a host immune response in eukaryotic cells. For IDR-1018, this includes influencing macrophage differentiation to release anti-inflammatory mediators and upregulating wound healing genes, amongst other host immunomodulatory effects.60 Furthermore, there is also evidence to suggest that IDR-1018 can induce a bacterial cellular stress response by suppressing guanosine pentaphosphate ((p)ppGpp), an important signalling nucleotide for biofilm development.106 Many of these modulatory mechanisms require translocation across eukaryotic or prokaryotic membranes and typically such peptides exhibit high arginine functionality, which is known to also feature heavily in other cell penetrating peptides.107
Whilst charge is important for antimicrobial activity and translocation, too many cationic residues can hamper the peptide's ability to penetrate into the biofilm's EPS. The extracellular DNA and exopolysaccharides within this matrix are typically highly charged and enact strong attractive or repulsive interactions towards cationic peptides, thus significantly reducing the peptide's effectiveness and mobility through the biofilm.102 However, some synthetic peptides have been postulated to suppress EPS secretion or limit the interactions between key extracellular polymers in the biofilm matrix, thus resulting in poorly adhered cellular aggregates that are easily detached, rather than strongly adhered biofilm networks in the untreated control.108 Finally, it is important to note that biofilm dispersal alone may be inappropriate in a clinical setting as the dispersed pathogens remain a threat to other organs or regions in the body and septic shock can also occur upon rapid dispersal.56 Therefore, peptides that are able to disperse biofilms as well as actively destroy the resultant planktonic bacteria are highly desirable. Whilst the design of such peptides is complex, many have been demonstrated to show synergistic activity with conventional small molecule antibiotics for combination therapy, for example work by Hancock et al. using IDR-1018 (Fig. 1D).105 Many of the previously discussed biofilm-dispersing peptides are therefore also highly promising adjuvant materials for use in combination with other conventional approaches.
One notable non-lethal antibiofilm peptide that exhibits strong anti-adhesion activity, particularly towards inhibiting fungal biofilm formation, is human α-defensin 6 (HD6).61 This short 32 residue peptide is naturally expressed as part of the human innate immune system and like many other defensins is rich in cysteine-disulfide bridges, which stabilize a three-stranded β-sheet tertiary structure, thereby providing the peptide strong resistance towards proteolysis. However, in contrast to typical defensins, HD6 shows negligible lethal antimicrobial activity against Gram-positive or Gram-negative bacteria,109 nor antifungal activity against fungal species.110 Nevertheless, the peptide undergoes unique self-assembly behavior to form oligomeric fibrils, observable by electron microscopy, which can act as “nanonets” by entrapping bacteria, causing non-lethal aggregation and sedimentation. In the case of the opportunistic fungal pathogen Candida albicans, HD6 was shown to prevent the yeast cell from adhering to human intestinal epithelial cells, thereby preventing subsequent fungal invasion and systemic dissemination.110 Furthermore, HD6 was found to significantly suppress the formation of fungal biofilms but was ineffective at disrupting established biofilms, thereby providing mechanistic insight through the “nanonet” entrapment and entanglement of these fungal cells, which inhibited initial attachment. The non-lethal nature of HD6 was proposed to allow C. albicans to reside commensally within the intestine, whilst preventing systemic invasion events and suppressing other virulence traits.110
Antimicrobial peptides remain amongst the most promising antibiofilm materials for clinical translation111 and form the basis of numerous antibiofilm strategies employed by other material classes. Their well-defined sequence and multiple possible modes of action make them highly promising therapeutic agents for a range of antibiofilm clinical applications, yet their instability in the environment poses a challenge for certain indications. Other synthetic materials discussed in later sections aim to address this significant limitation, however the relatively low environmental persistence of peptides is advantageous to ensure the development of acquired antimicrobial resistance towards such agents is minimized.
Antimicrobial proteins
Antimicrobial functional proteins such as enzymes and antibodies are another promising class of naturally occurring antibiofilm materials. The use of certain antimicrobial enzymes for general antimicrobial applications, including a brief overview of some of their antibiofilm properties, has been reviewed in the following texts.64,99,112 In this section, we aim to highlight the main classes of enzymes and other functional proteins that exhibit antibiofilm activity. Enzymes are the broadest class of antibiofilm proteins and these can be further divided into various subclasses, including proteolytic enzymes, which degrade essential microbial proteins and peptides, oxidoreductases, which produce reactive oxygen and (pseudo)halide species, polysaccharide depolymerases and nucleases, which degrade polysaccharides or extracellular DNA respectively, and quorum quenching enzymes, which interfere with cell–cell signalling. We will briefly outline these subclasses using a few selected examples.As well as non-biocidal destruction of the biofilm matrix, other proteases directly degrade the bacterial cell wall to enact bactericidal properties. The peptidoglycan is a major structural component of the cell walls of both Gram-positive and Gram-negative bacteria and typically contains β-(1,4)-linked N-acetylglucosamine and N-acetylmuramic acid crosslinked into a 3D network through pendant tri-, tetra- and pentapeptide bridges, which link the polysaccharides together.115 The 27 kDa glycylglycine endopeptidase bacteriolysin, lysostaphin, is a classic example of such a cell wall-degrading enzyme, which acts through selective cleavage of pentaglycine crosslinking bridges found in the peptidoglycan of certain staphylococci.70 For example, recombinant lysostaphin was shown to effectively eradicate established S. aureus and S. epidermidis biofilms, including those of drug-resistant strains, whereas small molecule antibiotics such as oxacillin and vancomycin were ineffective.71 Whilst lysostaphin is highly selective for staphylococcal strains, and therefore does not show broad-spectrum antibiofilm activity, it shows an advantage in its ability to both degrade the biofilm matrix as well as enact a biocidal bacteriolysis mechanism.
 | ||
| Fig. 2 Typical enzymatic processes of various classes of antibiofilm enzymes. The proposed pyocyanine demethylase mechanism (bottom) was adapted with permission from ref. 116. Copyright©2017 American Association for the Advancement of Science. The red dashed oval indicates an unfavorable coulombic interaction between the substrate intermediate and the enzyme, which likely leads to release of this intermediate from the active site, followed by subsequent spontaneous hydrolysis to yield the reduced and inactivated 1-hydroxyphenazine and formaldehyde byproduct. | ||
PGN is present in a range of biofilms formed by bacterial pathogens, however it is absent in P. aeruginosa biofilms. The EPS of biofilms formed by this bacterium are comprised of at least three biosynthetic exopolysaccharides; Psl, a pentasaccharide containing D-mannose, L-rhamnose, and D-glucose units; Pel, a cationic polysaccharide containing partially deacetylated N-acetyl-D-glucosamine and N-acetyl-D-galactosamine; and the anionic polysaccharide, alginate.118 Howell and co-workers identified enzymes, PslG and PelA, which showed glycoside hydrolase activity against Psl and Pel, using bioinformatics analysis.118 They purified and characterized corresponding enzyme sequence segments (constructs), PslGh and PelAh, which contained the catalytic and binding domains but unlike the parent enzymes were water-soluble. Addition of these rationally designed enzyme constructs resulted in the removal of up to 99% biomass of P. aueruginosa biofilms, whereas variants with site-specific mutations in the catalytic domain were inactive, supporting the importance of the enzyme's active sites to their biofilm dispersing activities.118
Aside from the degradation of exopolysaccharides in the biofilm matrix, other enzymes such as lysozyme attack bacteria directly by degrading the polysaccharides in the peptidoglycan of the bacterial cell wall. Lysozyme is a 14 kDa glycoside hydrolase that is expressed as part of various animals’ innate immune systems, and cleaves the 1,4-β-linkages between the N-acetylmuramic acid and N-acetyl-D-glucosamine residues in the peptidoglycan, thus destabilizing the bacterial cell wall and leading to bacterial lysis.66 In one study, Nagarsenker and co-workers developed an inhalable spray-dried formulation comprised of a small molecule antibiotic encapsulated within liposomes, levofloxacin, supplemented with additional lysozyme.119 The formulation removed 85% of S. aureus biofilms at sub-MIC concentrations of the antibiotic in vitro. They further demonstrated that the formulation resulted in up to around a 2-log reduction in bacterial burden in vivo, whilst not inducing inflammatory markers in a rat model of S. aureus pulmonary infection after administration by inhalation.119
Other oxidoreductases such as hexose oxidase and glucose oxidase facilitate the oxidation of monosaccharides to their corresponding lactones, whilst similarly producing the reactive oxygen species, hydrogen peroxide (Fig. 2(III)). Glucose oxidase (GOx) is a 160 kDa homodimer that is commercially isolated from filamentous fungi Aspergillus species and shows relatively high specificity for its substrate D-glucose.68 By contrast, hexose oxidase is a genetically modified enzyme produced by the yeast Hansenula polymorpha, which shows promiscuity in its substrate reactivity and can oxidise a range of monosaccharides, including glucose, maltose, lactose and galactose.122 Both are used extensively in the food technology industry122 and in toothpaste formulations,123 typically as part of a cocktail with other enzymes.
GOx also contributes to the antimicrobial properties of natural honeys.124,125 In one study, Hammer and co-workers investigated the antibiofilm effect of various honeys derived from Australian flora, including Eucalyptus marginata and Corymbia calophylla.126 The authors showed that treatment of established S. aureus, E. faecalis and P. aeruginosa biofilms with 20–50% honey formulations resulted in a significant reduction in biofilm viability.126 The authors proposed that the antibiofilm activity of the honeys resulted from a complex interplay between GOx-produced hydrogen peroxide and other honey components, such as bee defensin and plant-derived compounds, as well as osmotic stress and low pH, which likely acted synergistically, however further studies of these honeys are necessary to fully elucidate their various modes of activity.126 More recently, phenoloxidases and prophenoloxidases are of growing interest as antibiofilm oxidoreductases.127,128 These are expressed as part of the immune system of various insects and crustaceans and produce reactive oxygen and nitrogen species from phenols.
Peroxidases such as lactoperoxidase (LPO) and myleoperoxidase,112 which also belong to the subclass of haloperoxidases, generate reactive antimicrobial hypothiocyanite, hypochlorite, hypobromite and hypoiodite from hydrogen peroxide and endogenous (pseudo)halides (Fig. 2(IV) and (V)), thereby showing excellent applicability for use in combination with oxidative enzymes such as GOx. For example, Johansen and co-workers investigated a cocktail of oxidoreductases, glycoside hydrolases and proteases in their antibiofilm activity against S. aureus, S. epidermidis, Pseudomonas fluorescens, and P. aeruginosa biofilms formed on steel and polypropylene substrates, as well as Streptococcus mutans, Actinomyces viscosus, and Fusobacterium nucleatum biofilms grown on saliva-coated hydroxyapatite.67 They found that whilst a combination of GOx and LPO showed bactericidal activity against the biofilms, they did not effectively remove the adhered biofilms, leaving a non-viable surface-adhered biomass. In the same study, Pectinex Ultra SP (a complex mixture of polysaccharide depolymerases and proteases) showed effective biofilm removal, however it was non-bactericidal. Therefore, combination of Pectinex Ultra SP with GOx and LPO was positioned to likely exhibit ideal biofilm-removal and bactericidal activities, employing multiple modes of action.67
DNases have also been shown to be effective at increasing the susceptibility of Gram-positive biofilms towards small molecule biocides. For example, Kaplan and co-workers evaluated the antibiofilm activity of a recombinant human DNase (dornase alpha) and found that pre-treatment of a range of pre-formed S. aureus biofilms with the DNase rendered them much more susceptible to small molecule antibiotics.62 The DNase-pretreated biofilms showed a 6-log reduction in viability when exposed to small molecule biocides such as chlorhexidine gluconate, whereas either the biocide or DNase alone were ineffective. The authors further demonstrated that use of DNase in conjunction with tobramycin resulted in better in vivo survival rate for the nematode Caenorhabditis elegans infected with S. aureus, compared to use of either tobramycin or DNase alone. In the same study, a cocktail of the glycoside hydrolase dispersin B with DNase was found to also greatly improve the effectiveness of vancomycin and tigecycline against S. epidermidis biofilms.62
One indirect mechanism for targeting eDNA is through the degradation of small signalling molecules that drive the extracellular release of DNA, or those that bind DNA. Pyocyanine is one such bacterium-derived small signalling molecule and DNA intercalator, which has recently been used as an enzymatic target for inhibiting P. aeruginosa biofilm formation.116 Newman and co-workers characterized the structure of a tautomerizing pyocyanine demethylase, PodA, which oxidizes the methyl group of pyocyanine, thereby reducing the pyrazine ring structure through a spontaneous, hydrolysis-driven demethylation reaction, ultimately releasing formaldehyde as a byproduct (Fig. 2(IX)).116 Upon addition of this 45.6 kDa trimeric enzyme to P. aeruginosa, biofilm formation was partially inhibited after 5 h. A control group where DNase I was used in place of PodA showed identical biofilm inhibition, whereas the use of a combination of DNase I and PodA did not show an additive antibiofilm effect. This is consistent with the mechanism that PodA influenced DNA release through degradation of pyocyanine, thereby hampering the structural role of eDNA during early biofilm formation by P. aeruginosa.116
As can be seen in this section, enzymatic and non-enzymatic proteins are a richly diverse class of antibiofilm agents with numerous mechanisms for biofilm eradication and dispersal. As with peptides, a major challenge in using proteins for biofilm removal is their low in vivo residence time, high relative cost and somewhat difficult synthetic modification. However, advances in scale-up technology for protein manufacture can rapidly decrease the cost of identified candidates, which we believe will be pivotal for the wide adoption of protein-based antibiofilm agents.
Antibiofilm strategies employing solution-based polymer materials
Cationic polymers and polymer nanoparticles
Whilst peptides are highly effective agents for eliminating biofilms, they suffer from low stability and early clearance in vivo. Additionally, their ease of preparation is complicated by multi-step solid phase synthesis, which limits their scope and significantly increases production costs. Many researchers have turned to investigating synthetic mimics of antimicrobial peptides (sometimes abbreviated to SMAMPs, or similar acronyms) to remove or inhibit biofilms, which combine the benefits of low production costs and high proteolytic stability, with the benefit of antimicrobial bacterial membrane rupturing properties.34,77,79–83,141 These polymeric materials are chemically similar to antimicrobial peptides in that they contain repeating units with cationic and lipophilic functionality and are typically low molar mass and soluble in aqueous solutions. There are four main types of cationic moieties that give rise to antimicrobial activity in synthetic polymers: amine groups that mimic the structure of lysine residues, guanidine groups that mimic the structure of arginine residues, as well as quaternary ammonium moieties and phosphonium functionalities, neither of which typically exist in naturally occurring peptides. Whilst these materials have been extensively investigated as antimicrobial agents against planktonic bacteria, fewer studies have focused on the interaction of such polymers with established biofilms.Qu and Locock et al. investigated the ability for guanidine-functional methacrylate polymers to disperse and eradicate both monomicrobial and polymicrobial biofilms in vitro.78 Copolymers of poly(2-guanidinoethyl methacrylate hydrochloride-co-methyl methacrylate) (P(GMA-co-MMA)) with a GMA content of 50 mol% and 68 mol% were tested as they had previously shown optimal antimicrobial activity against a range of planktonic species, whilst exhibiting low hemolytic properties (shown in Fig. 3A).142 It was observed that these polymers, when employed at concentrations of up to 16 times their planktonic MIC, outperformed state-of-the-art clinical small molecule antibiotics, antifungals, and combinations thereof, when these small molecule agents were tested at their highest serum-achievable concentrations.78 The polymers were effective against both S. aureus and C. albicans monomicrobial biofilms, as well as a cross-kingdom polymicrobial biofilm comprised of both species, as shown in Fig. 3A. It was demonstrated that the polymers were somewhat hampered by the presence of the extracellular matrix, with the planktonic MICs increasing by up to 4-fold in the presence of the isolated biofilm matrix, compared with a 16-fold increase in the case of fluconazole.78 Building from this in vitro study, the optimized guanidine copolymers were investigated as a topical treatment against epithelial-associated C. albicans biofilms in an in vivo murine vulvovaginal candidiasis (VVC) model.143 The results from this study showed that the polymers displayed negligible local toxicity and resistance to vaginal acidity, and that this formulation outperformed conventional antifungal agents at concentrations 50-fold lower than the over-the-counter concentration of the conventional antifungal drugs. Furthermore, topical treatment using these polymers resulted in fewer persister cell survivors in the fungal biofilms in comparison with conventional antifungals.143 This highlights the numerous advantages of using guanidine-functional polymeric materials over conventional small molecule drugs in the treatment of biofilm-related local infections. Furthermore, they are able to be used as a sole therapeutic agent for cross-kingdom polymicrobial infections, replacing conventional combination therapies.
 | ||
| Fig. 3 Polycationic antibiofilm agents. (A) Guanidine-functional polymethacrylates are effective single therapeutic agents against polymicrobial biofilms. Biofilms treated with various agents as indicated are shown after LIVE/DEAD viability staining. Adapted with permission from ref. 78. Copyright©2015 Oxford University Press. (B) Combination therapy using mixtures of cationic copolymers with conventional small molecule antibiotics. Adapted with permission from ref. 144. Copyright©2019 American Chemical Society. (C) Oil-in-water emulsions of various antimicrobial essential oils stabilized by cationic copolymers show synergistic efficacy against P. aeruginosa biofilms. Adapted with permission from ref. 145. Copyright©2020 American Chemical Society. | ||
Hedrick and Yang et al. developed biodegradable antimicrobial polycarbonates with quaternary ammonium functionality.146 These polymers were found to reduce the cell viability of both S. aureus and E. coli monomicrobial biofilms upon treatment at 8 times the MIC, resulting in around 10% viability relative to a non-treated control. Furthermore, the adhered biomass was reduced to around 25% under the same conditions, highlighting the polymers’ ability to both disrupt the biofilm structure and kill embedded bacteria. Owing to their biodegradability, the polymers showed significantly higher lethal dose concentrations (LD50 = 99 mg kg−1) compared with a small molecule antiseptic (chlorhexidine, LD50 = 12.5 mg kg−1) and a peptide antibiotic (polymyxin B, LD50 = 5.4 mg kg−1) in a mouse model. Furthermore, liver and kidney function and polyelectrolyte balance remained normal after two intravenous administrations of 30 mg kg−1 polymer, supporting the biocompatibility of these polymers. The polymers were shown to significantly reduce the bacterial viability in a S. aureus mouse sepsis model and that unlike the control PBS treatment, the mice in the polymer-treated cohort showed an 80% survival rate 48 h post-infection and did not show symptoms associated with sepsis such as lethargy, dehydration and weight loss.146
Kuroda and co-workers developed polycationic poly(4-aminobutyl methacrylate-co-ethyl methacrylate) copolymers for eliminating Streptococcus mutans biofilms commonly found in dental cavities.147 Whilst both the small molecule disinfectants, chlorhexidine, and the copolymers were both shown to inhibit biofilm formation, the polymers led to 85% reduction in the biomass of established biofilms, whereas chlorhexidine was ineffective at biofilm removal. It was speculated that the interaction of the cationic polymer with the EPS weakened the integrity of the biofilm matrix, facilitating biofilm removal and effective killing. Furthermore, the copolymers were found to be effective at removing S. mutans biofilms in an in vitro “swishing” simulation assay, designed to mimic the mechanical/chemical nature of mouthwash treatments.147
Antimicrobial polymers have also been investigated in combination with conventional antibiotics to eradicate established biofilms. Our aforementioned study using a mouse VVC model showed that antimicrobial polymers could even further eradicate persister cells pre-selected by conventional antifungal agents.143 Francolini and co-workers investigated the synergistic effects of using an antimicrobial tertiary amine-functional polymer, poly(2-dimethylamino acrylamide), with either daptomycin or moxifloxacin against biofilms formed by a range of methicillin-susceptible and methicillin-resistant S. aureus.148 Here, the authors found that the synergy was dependent on the antibiotic's mechanism of action, with synergy being observed in the case of the membrane disrupting lipopeptide, daptomycin, but no additional benefit being observed for the topoisomerase inhibitor, moxifloxacin.148 Similarly, Haldar and co-workers showed that the addition of amino acid conjugates of poly(maleic anhydride-alt-isobutylene) with a quaternary ammonium-functional linker in the side chain (shown in Fig. 3B) facilitated a 4-log reduction in multi-drug resistant P. aeruginosa and E. coli monomicrobial biofilms when used in combination with rifampicin, a bacterial RNA polymerase inhibitor.144 The polymer alone showed no antibiofilm activity and the free drug only resulted in one-log viability reduction. More recently, Wong and Boyer et al. showed that water-in-oil emulsions of antimicrobial essential oils stabilized with amphiphilic primary amine-functional copolymers and block copolymers showed synergistic antibiofilm activity against P. aeruginosa biofilms (Fig. 3C).145
Aside from unimer polycations, cationic nanoparticles formed by polymer self-assembly have also been investigated as antibiofilm agents. Tzanov and co-workers developed electrostatic alternating layer-by-layer (LbL) coated nanoparticles using two polysaccharides, anionic hyaluronic acid and cationic aminocellulose.149 The nanoparticles were able to inhibit the formation of S. aureus and E. coli biofilms up to 99% and 62% respectively. However, interestingly when the nanoparticles with anionic hyaluronic acid on their outer layers were employed, the best biofilm inhibition resulted, the reason for which was not investigated further.149 One possible mechanism for this counter-intuitive behaviour is exemplified by Yang and Park et al., who developed block copolymer core–shell nanoparticles comprised of dextran-block-poly((3-acrylamidopropyl) trimethylammonium chloride-co-butyl methacrylate).150 Here, the nanoparticles had a cationic, lipophilic core-forming block and a neutral dextran polysaccharide corona-forming block. These were investigated as antibiofilm agents against biofilms of either MRSA, vancomycin-resistant Enterococci (VRE), or Enterococcus faecalis. Whilst the particles had excellent antimicrobial activity against planktonic MRSA, they were ineffective against VRE or E. faecalis. However, the authors showed that the micelles were able to attach to biofilm-embedded bacteria, without inducing bactericidal effects, and disrupt established biofilms through biofilm debridement. The authors explained that binding of the nanoparticles to the bacterial membranes altered the surface properties and increased the microorganisms’ solubility in the medium, leading to destabilization of the biofilm.150 This new non-lethal mechanism for tackling biofilms may be important for reducing the evolutionary pressure for the development of acquired antimicrobial resistance towards such agents.
Aside from synthetic polymers, polymers derived from natural materials also show promise as effective antibiofilm agents. Chitosan is a polysaccharide material, prepared from treatment of natural chitin obtained from the exoskeleton of shellfish and crustaceans.151 Chitosan's structure consists of randomly distributed β-linked D-glucosamine and N-acetyl-D-glucosamine repeating units. Being a polycationic material, many researchers have shown chitosan materials exhibit effective antibiofilm properties.152–155 For example, Pintado and co-workers developed chitosan nanoparticles held together electrostatically using the anionic binding agent tripolyphosphate.153 These nanoparticles were effective at inhibiting planktonic growth of methicillin-resistant and methicillin-susceptible strains of S. aureus (MRSA and MSSA, respectively), as well as a methicillin-resistant strain of S. epidermidis (MRSE). The nanoparticles showed moderate activity in disrupting MRSE's ability to bind onto a protein-coated polystyrene substrate, whilst minimal activity was observed for S. aureus strains. However, the nanoparticles were able to strongly inhibit biofilm formation of both S. aureus strains in microwell plates at sub-MIC concentrations but showed only moderate inhibitory activity against biofilm formation by MRSE. This was explained by the distinct mechanisms for biofilm formation between the two staphylococcal species, with S. epidermidis relying more heavily on hydrophobic interactions for initial attachment, whilst S. aureus exhibited greater reliance on an active adhesin-dependent binding mechanism.153
Chitosan is also amenable to further modification in order to impart additional antimicrobial functionality. Sahariah and co-workers developed a range of chitosan materials functionalized with various quaternary ammoniumyl derivatives, as well as lipophilic groups.155 They demonstrated that the presence of cationic charge and short alkyl chains resulted in enhanced activity towards planktonic S. aureus, while longer alkyl chains resulted in reduction in the antibacterial activity. However, in the case of S. aureus biofilms, the ideal candidate had a combination of trimethylamine and acetyl moieties, as well as long, lipophilic stearoyl functionality, which allowed for deep penetration into the biofilm network.155 This work highlights the importance of identifying structure–activity relationships in polymer materials using application-specific assays. Similarly, Guo and co-workers developed chitosan functionalized with N-phosphonuim cationic groups.154 They showed that at 3% and 13% phosphonium substitution, these materials showed superior inhibitory activity against S. aureus and E. coli biofilms than comparable chitosan derivates functionalized with quaternary ammonium species.154
Cationic polymer materials are excellent alternatives to antimicrobial peptides for the eradication of biofilms and show greatly improved in vivo stability, ease of tunability and low cost of production. However, as with peptides, the poor penetration of highly charged species through the biofilm matrix, as well as their hemotoxicity, must be carefully considered when designing such systems. Studies from our group and others have revealed the influence of hydrophobicity, molar mass, end group, and monomer sequence, which play a significant role in determining selectivity against planktonic bacteria.77,83,142,156–161 Through detailed structure–property relationship studies such as these, we believe that ideal antibiofilm candidates from within this class of materials can be developed for a range of applications.
Drug-releasing polymer systems
Whilst antibiofilm polycations have been the focus of significant research attention, others have focused on polymer systems that can better deliver conventional antibiotics. These often show advantages over the use of free antibiotics alone because they can often circumvent traditional resistance mechanisms and better penetrate into the biofilm matrix, as well as having other pharmacokinetic advantages owing to the additional size and stability that the polymer material imparts. For example, Benoit and Koo et al. developed antibiotic-loaded pH-responsive polymeric micelles comprised of poly(2-(dimethylamino)ethyl methacrylate)-block-(2-(dimethylamino)ethyl methacrylate-co-butyl methacrylate-co-propyl acrylic acid) (pDMAEMA-b-(DMAEMA-co-BMA-co-PAA)) for the treatment of oral biofilms associated with dental cavities.162 Here, the cationic pDMAEMA corona could effectively target anionic surfaces resulting in adherence to the hydroxyapatite tooth surface, including saliva-coated hydroxyapatite and exopolysaccharide-coated hydroxyapatite. Upon exposure to a reduced pH environment, a virulence factor for cavity-producing biofilms, the core of the micelles became destabilized owing to core ionization, which led to the release of the loaded antimicrobial, farnesol. Note that as previously mentioned farnesol is a fungal quorum sensing molecule, but it also displays antimicrobial properties against S. mutans. The loaded micelles were 4-fold more effective at disrupting S. mutans biofilms than the free antibiotic and it was further demonstrated in a rodent model that the loaded micelles reduced the occurrence and severity of carious legions, whilst the free drug showed no effect.162Rotello and co-workers developed drug-loaded biodegradable nanocomposites from guanidine-functional polynorbornenes with additional poly(ethylene glycol) and maleimide moieties in the polymer's side chain.163 Upon crosslinking an oil-in-water emulsion containing the naturally occurring membrane disrupting antibiotic, carvacrol, using a disulfide-based crosslinker, drug-loaded nanocomposites were prepared, which could readily degrade in the presence of either glutathione or esterases to release the antimicrobial payload. The nanocomposites were shown to readily penetrate into E. coli biofilms and were found to be highly effective at killing biofilms formed by four clinical isolates, including multi-drug resistant strains. This was in stark contrast to treatment with the carvacrol alone, or the non-crosslinked guanidine polymer, which were both ineffective. The authors demonstrated the nanocomposites could selectively kill bacteria in a co-culture in vitro model using P. aeruginosa biofilms grown on top of mammalian fibroblasts, and that unlike the conventional antibiotic, the development of resistance to such treatment was negligible after 20 serial passages at subinhibitory concentrations.163
Liu and co-workers developed antibiotic delivery vehicles based on chitosan nanoparticles surface-functionalised with various compositions of zwitterionic 2-methacryloyloxyethyl phosphorylcholine (MPC) and cationic N-(3-aminopropyl) methacrylamide.152 These could be loaded with the small molecule antibiotic triclosan and were shown to rapidly release this cargo upon acidification from pH 7.4 to pH 5.0. It was demonstrated that greater zwitterionic functionality present on the particle surface led to greater particle penetration and accumulation within S. aureus biofilms, thus leading to better antimicrobial efficacy of the released cargo.152 This highlights the importance of electrostatic interactions between the biofilm's extracellular matrix and the drug delivery vehicle, with highly charged cationic species generally being hampered by the anionic matrix.
Cavallaro and co-workers prepared hierarchical drug delivery vehicles for superior biofilm penetration for the treatment of cystic fibrosis pulmonary infections.164 An anionic polymer was synthesized by grafting D-(+)-glucuronic acid γ-lactone onto a α,β-poly-(N-2-hydroxyethyl)-D,L-aspartamide and used the resultant anionic graft copolymer to formulate a polyion complex with the cationic antibiotic tobramycin at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge ratio. These nanosized polyion complexes were formulated into microparticles of a mucus-disrupting sugar, mannitol, using a spray drying technique to produce nano into micro formulations (NiMs). These formulations were shown to sustainably release the polyion complexes and were amenable to pulmonary administration as an inhalable dry powder. The NiMs interrupted the mucoadhesive properties of an artificial cystic fibrosis mucus and it was demonstrated that the NiMs led to faster initial penetration through the artificial mucus but slow and sustained release after longer time periods owing to the polyion complexation of the tobramycin. After loading with other optimising agents such as cysteamine, these hierarchical drug delivery vehicles significantly outperformed the free antibiotic, as well as a market leading porous inhalable formulation for cystic fibrosis treatment, at removing P. aeruginosa biofilms in the presence of the artificial mucus.164
:1 charge ratio. These nanosized polyion complexes were formulated into microparticles of a mucus-disrupting sugar, mannitol, using a spray drying technique to produce nano into micro formulations (NiMs). These formulations were shown to sustainably release the polyion complexes and were amenable to pulmonary administration as an inhalable dry powder. The NiMs interrupted the mucoadhesive properties of an artificial cystic fibrosis mucus and it was demonstrated that the NiMs led to faster initial penetration through the artificial mucus but slow and sustained release after longer time periods owing to the polyion complexation of the tobramycin. After loading with other optimising agents such as cysteamine, these hierarchical drug delivery vehicles significantly outperformed the free antibiotic, as well as a market leading porous inhalable formulation for cystic fibrosis treatment, at removing P. aeruginosa biofilms in the presence of the artificial mucus.164
Outside of polymer self-assemblies, Zhang and co-workers developed unimolecular core-crosslinked micelles prepared from quaternary ammonium-functional lipids employing an acid and/or lipase labile linker.167 These crosslinked micelles were shown to degrade and disassemble in under low pH conditions or in the presence of lipase and showed greater penetration into established S. aureus biofilms, resulting in significantly lower biofilm biomass and viability at equal concentration to the non-crosslinked lipid micelles. Furthermore, owing to their crosslinked nature, which permanently shielded the lipid hydrophobic tails from solution, the crosslinked micelles were far less hemolytic than the non-crosslinked micelles under non-acidic, physiological pH conditions, demonstrating the importance of site-directed toxicity or antimicrobial activity.167
Delivery of small molecule and peptide agents using polymeric nanoparticles can greatly improve their antibiofilm properties and can allow for complementary functionality to be installed, such as biofilm targeting agents. Furthermore, these systems can exploit environmental stimuli to release their cargo more exclusively at the infection site. However, such systems do not alleviate the issues surrounding acquired antimicrobial resistance to their cargo. Whilst these drug delivery systems effectively repurpose existing antibiofilm agents, new strategies must be developed that can overcome the growing threat of antimicrobial resistance through employing both unconventional killing mechanisms, as well as non-lethal biofilm removal strategies.
New directions in antibiofilm materials
Peptidomimetics
Whilst both synthetic and natural antimicrobial peptides with well-defined primary sequence and secondary structure have been used extensively for antibiofilm applications, others have developed biomimetic antibiofilm materials, known collectively as peptidomimetics (Fig. 4).84,86 For the purpose of this Review, we also include poly(peptide)s under the bracket of peptidomimetics but it is worth clarifying at the start of this section that there is some ambiguity in the nomenclature of such materials. IUPAC describe “polypeptides” as any peptide containing 10 or more amino acids, however for certain synthetic poly(peptide)s, often one is referring to synthetic polymers prepared by the polymerization of amino acids or their precursor monomers to form (co)polymer materials with a distribution of molar masses and an ill-defined sequence, as opposed to a mass-defined and sequence-defined peptide. In this regard, we refer to poly(peptide) peptidomimetics as synthetic materials that have the same peptide backbones as naturally occurring peptides, but are prepared through the ring opening polymerization of strained cyclic monomers, such as N-carboxyanhydrides, or through polycondensation of amino acids, as opposed to iterative solid phase synthesis in the case of synthetic peptides. As such by this definition, whilst poly(peptide)s possess similar properties to antimicrobial peptides, they do not exhibit the same level of control over their primary sequence, nor length homogeneity. However, these materials show an advantage over peptides through their ease of preparation, often in a one-pot methodology from the starting monomers, which significantly reduces the production cost of these materials.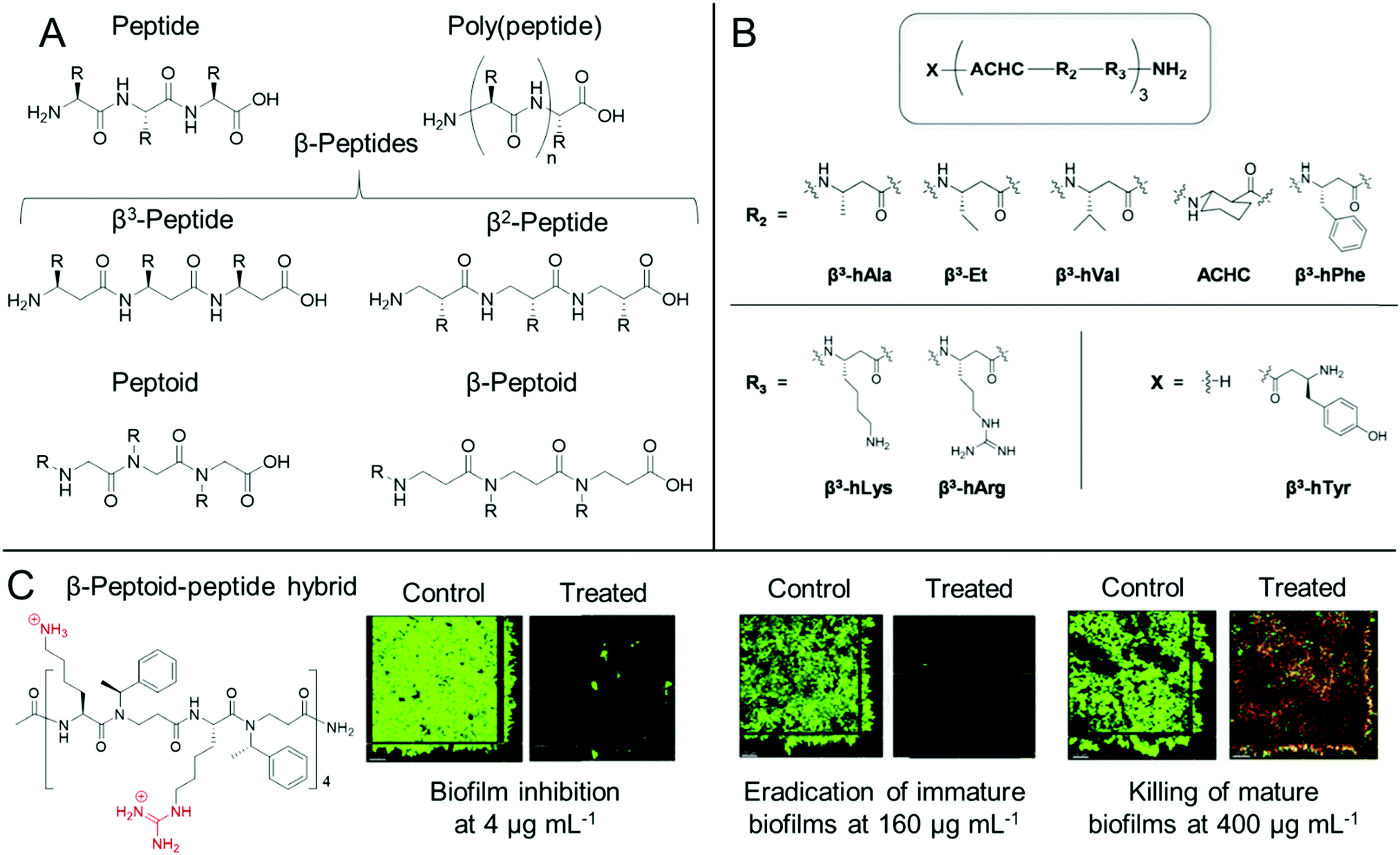 | ||
| Fig. 4 Antibiofilm peptidomimetics. (A) Generic structures of peptides, poly(peptide)s, peptoids, β-peptides and β-peptoids. (B) Modular 14-helical β-peptide materials with the general structure X-(ACHC-R2-R3)-NH2 investigated by Palecek and co-workers. Reproduced with permission from ref. 165. (C) Structure and antibiofilm activity of the β-peptoid–peptide hybrid oligomers investigated by Folkesson and co-workers. Adapted from “High in vitro antimicrobial activity of β-peptoid–peptide hybrid oligomers against planktonic and biofilm cultures of Staphylococcus epidermidis”, Y. Liu, K. M. Knapp, L. Yang, S. Molin, H. Franzyk and A. Folkesson, Int. J. Antimicrob. Agents, 2013, 41, 20–27,166 with permission from Elsevier. | ||
For poly(peptide)s, the side chain residue is present on the carbon adjacent to the peptide bond to give an identical backbone to peptides. Conversely, in a distinct class of peptidomimetics known as peptoids, the side chain functionality is N-substituted to give a secondary amide peptide backbone (each structure shown for comparison in Fig. 4A).84 This results in stark differences in the self-assembly and in vivo behaviour of peptides and peptoids. Whilst peptoids can be prepared by either iterative solid phase methods or through ring opening polymerization, contrary to both peptides and poly(peptide)s, peptoids show no backbone chirality or inter-backbone hydrogen bonding, whilst showing significant proteolytic stability.84 As previously discussed, the persistence of non-degradable materials in the environment is cause for some concern over antimicrobial resistance development. However, the enhanced proteolytic stability of peptoids may be important for certain therapeutic indications.
The design principles for peptidomimetics are similar to that of antibiofilm peptides, in that those with cationic and hydrophobic side chains typically show good antibiofilm properties, with poly(L-lysine) being commonly used in the case of poly(peptide)s. Whilst bacterial membrane rupture is thought to be a major mechanism, similarly to antibiofilm peptides, peptidomimetics can show multiple modes of therapeutic action.86 For example, Hervé and co-workers showed that a commercial poly(L-lysine) compacted DNA in cystic fibrosis sputum clinical isolates, exerting mucolytic activity.168 The compacting of DNA was important for allowing natural and exogenous protease inhibitors, which are hampered by the presence of DNA in cystic fibrosis patients, to control neutrophil serine protease activity in the cystic fibrosis sputum. In such a way, treatment with poly(L-lysine) provided a fluid phase in the sputum that improved ciliary beating frequency in an in vivo mouse model after aerosol administration, whilst also destroying opportunistic bacteria such as P. aeruginosa and S. aureus.168 In a later study, the authors utilized poly(L-lysine) to treat ex vivo P. aeruginosa biofilms formed on endotracheal tubes collected from mechanically ventilated clinical patients.169 They showed that the poly(peptide) removed 90% of the endotracheal tube biofilm after a single 2 min administration and was also well tolerated by the lung tissue of mechanically ventilated pigs in an in vivo porcine model.169 These studies demonstrate the potential of a simple poly(peptide) for the treatment of biofilms accompanying both cystic fibrosis and ventilator-associated pneumonia.
Cobb and Lundy et al. investigated the in vitro antibiofilm activity of 18 sequence defined peptoids, containing N-substituted side chains with cationic primary amine groups as well as hydrophobic aromatic functionalities, against established polymicrobial cross-kingdom bacterial/fungal biofilms comprised of either S. aureus/C. albicans or E. coli/C. albicans species.170 Optimal peptoid candidates that could facilitate up to a 2-log reduction in bacterial cell counts, as well as up to a 3-log reduction in fungal cell counts were identified, whilst it was also demonstrated that the peptoids exhibited a membrane-rupturing mechanism of action against all three species.170 The authors further explained that the modified polymerase chain reaction quantification technique employed for assessing biofilm viability was a powerful tool for including species that are viable but non-culturable in the viability quantification, as opposed to traditional colony forming unit counting assays.170 Similarly, Jenssen and co-workers identified a synthetic peptide GN-2 through quantitative structure activity relationship (QSAR) in silico methods and prepared a range of sequence-defined synthetic peptides and their corresponding peptoids, which mimicked the GN-2 structure, as well as the GN-2 peptide itself.171 Interestingly, they found that structures that showed high planktonic antimicrobial activity showed poorer antibiofilm activity and vice versa. Peptoid materials generally showed superior E. coli antibiofilm activity, compared to the corresponding peptides, despite showing lower activity against planktonic bacteria. The authors explained this through consideration of the differences in backbone rigidity, different spatial arrangements of the charged and hydrophobic residues, as well as differences in hydrogen bonding capability, which were competing factors in determining potency against planktonic bacteria and biofilms.171
One other important class of peptidomimetics are β-peptides.85 Whilst typical α-peptides have both the amino and carboxylic acid functionality bound to the same carbon (the α-carbon), β-peptides are bound through the β-carbon (Fig. 4A), present on the side chain of each natural amino acid except glycine. As such, whilst all β-peptide residues, except for β-alanine, retain their chirality in a similar manner to peptides, they are also proteolytically stable, as observed with peptoids. β-Peptides are also emerging materials as anti-biofilm agents, particularly in the case of fungal biofilms. Palecek and co-workers investigated the antibiofilm effect of a range of amphiphilic 14-helical β-peptides, which form helical secondary structures in aqueous solution.165,172,173 In one study, the authors identified a β-peptide, Y-(ACHC-V-K)3, that inhibited the further growth of C. albicans established biofilms.172 Here, ACHC is a helix stabilizing residue containing trans-2-aminocyclohexanecarboxylic acid, linking the β3 and β2 positions (see Fig. 4B), whilst Y, V and K are residues containing tyrosine, valine and lysine mimicking groups in the β3 position.172 In a later study, they developed a range of 14-helical β-peptides with the structure X-(ACHC-R2-R3)-NH2 (Fig. 4B), which inhibited the formation of fungal biofilms by C. albicans, C. glabrata, C. parapsilosis and C. tropicalis.165 However, it was demonstrated that the β-peptides did not show potency against pre-formed biofilms from these species.165 More recently, small molecule modulators for fungal morphogenesis, which inhibit the hyphal elongation of C. albicans, were shown to markedly improve the biofilm inhibitory activity of these β-peptides, demonstrating their applicability to be used in combination with complementary antibiofilm agents.173 One further related structural class, β-peptoids, which are bound through the β-carbon, similar to β-peptides, but have their side chain functionality present on the nitrogen atom, similar to peptoids (structure shown in Fig. 4A), also show antibiofilm properties. For example, Folkesson and co-workers prepared β-peptoid–peptide hybrid oligomers and demonstrated their in vitro activity against established S. epidermidis biofilms (an example structure is shown in Fig. 4C).166 It was demonstrated that the hybrid oligomers could inhibit biofilm formation at concentrations equal to the MIC, whereas much higher concentrations were required to tackle established immature biofilms grown for 6 h. For established mature biofilms grown for 24 h, even higher concentrations were required and after treatment, a layer of dead bacteria was still present, unlike in the case of the immature biofilms (Fig. 4C).166
Peptidomimetics can also be utilized to form hybrid structures with non-peptidomimetic polymers. Recently, Pethe and Chan-Park reported a convenient one pot, two-step protocol for preparing diblock copolymers based on a glycopolymer block and a β-peptide block, poly(amido-D-glucose)-block-poly(β-L-lysine).178 Here, the authors utilized the anionic ring opening polymerization of 4-membered ring precursor monomers, followed by a global deprotection step to afford the cationic hybrid polymer. Owing to the vastly different homopolymerization rates of the two monomers, a block-like structure was formed. It was demonstrated that these hybrid polymers at just 2 × MIC showed high efficacy against S. aureus persister cells pre-selected by antibiotics from planktonic cultures and that they could disrupt and kill preformed MRSA biofilms, reducing the biofilm viability by 99.9%, and significantly reducing the adhered biomass. It was theorized that the polymer underwent a coil-to-helix transition in response to bacterial membranes, which upon insertion of the β-peptide block, created a surface layer of the glycopolymer block. This low-fouling layer promoted detachment of the bacteria from the surface, which was not observed for the β-peptide alone.178
Peptidomimetic materials share many of the strengths of antimicrobial peptides but offer additional advantages such as low production costs in certain cases, such as poly(peptide)s, or higher proteolytic stability in the case of peptoids and β-peptides. Whilst antibiofilm peptidomimetics have been known for longer than other synthetic antibiofilm materials detailed in this Review, none have currently been granted clinical approval, although the poly(peptide) ε-polylysine has received “Generally Regarded As Safe” (GRAS) status by the Food and Drug Administration (FDA) and can be used as a food additive. As with peptides, their translation is broadly hampered by off-target effects, toxicity (particularly hemotoxicity), and a high cost-benefit factor for development and clinical trials. For non-sequence defined peptidomimetics, additional challenges exist pertaining to their ill-defined structure, which are discussed in more detail towards the end of this Review.
Nitric oxide-releasing polymers and polymer nanoparticles
Nitric oxide-releasing polymers are a relatively new class of antibiofilm materials that have gained significant attention in recent years (Fig. 5).87–89 Nitric oxide (NO) itself is an endogenous small molecule gasotransmitter, which is involved in a number of normal physiological processes. As a free radical, nitric oxide shows inherent reactivity towards proteins, metabolic enzymes, DNA, and cell surfaces, and as such exhibits broad-spectrum antimicrobial activity through both oxidative and nitrosative processes.87,179 Furthermore, upon reaction with molecular oxygen and other reactive oxygen species, nitric oxide readily converts into further reactive species such as nitrogen dioxide (NO2), dinitrogen trioxide (N2O3) and peroxynitrite (ONOO−), which also exhibit separate antimicrobial modes of activity.87,179 Whilst nitric oxide itself is a promising antimicrobial agent, it has a very short half-life in vivo and is challenging to handle and deliver owing to its gaseous nature. Therefore, materials that can slowly release nitric oxide from a polymeric scaffold provide a convenient means for local delivery of nitric oxide to biofilms.87–89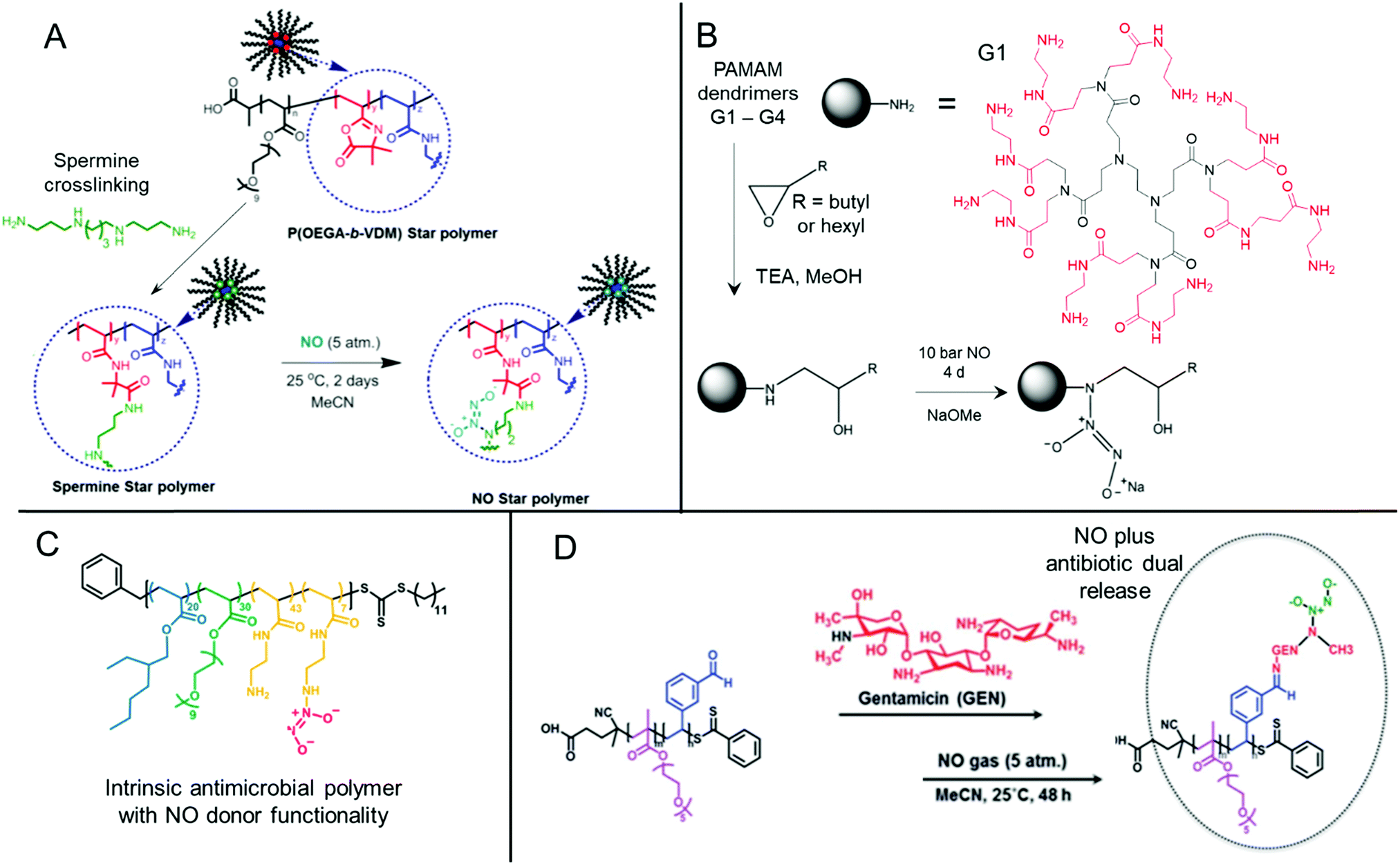 | ||
| Fig. 5 Chemical structures of NONOate-functional NO-releasing polymer materials. (A) Preparation of NONOate-functional star-like polymers. Adapted with permission from ref. 174. Copyright©2014 American Chemical Society. (B) Functionalization of PAMAM dendrimers with NONOate groups. The structure of a typical PAMAM first generation (G1) dendrimer is also shown with core (black) and 1st generation (red) units indicated. Adapted with permission from ref. 175. Copyright©2015 American Chemical Society. (C) Structure of NO-releasing antimicrobial polycations. Reprinted with permission from ref. 176. Copyright©2018 American Chemical Society. (D) Preparation of dual-releasing NO and gentamicin-loaded micelles. Adapted with permission from ref. 177. Published by the Royal Society of Chemistry. | ||
Two major classes of nitric oxide donor moieties are typically employed; these include N-diazeniumdiolates (NONOates) and S-nitrosothiols (RSNOs, which are discussed in a later section), both of which release nitric oxide under certain physiological conditions. NONOates are formed through the reaction of NO with secondary amine species and readily degrade back to the parent amine to release two NO molecules in a passive manner under physiological conditions. Boyer and co-workers developed poly(oligo(ethylene glycol) acrylate monomethyl ether) based star-like polymers crosslinked in their core through a secondary amine containing spermine linker (Fig. 5A). Upon reaction with gaseous NO, star polymers containing NONOate-functional cores were achieved. In contrast to the spermine NONOate conjugate alone, which rapidly released NO with t1/2 = 40 min, the star-like polymer was shown to steadily release NO over a 70 h period. This slow, sustained release enabled inhibition of P. aeruginosa biofilm formation at non-biocidal polymer concentrations. This was consistent with the notion that the star-like NONOate polymers prevented the bacterial behavioral switch from planktonic free-swimming, to biofilm-forming behavior, which was further confirmed using mutant strains.174 More recently, work from the same group investigated amphiphilic block copolymer self-assembled nanoparticles formed by polymerization-induced self-assembly (PISA) as precursors for NONOate functionalization, through employing a reactive epoxide-containing core-forming monomer.180 By ring-opening of the reactive epoxide group with benzylamine to form a secondary amine, followed by reaction with gaseous NO, NONOate-functional nanoparticles were obtained. Interestingly, the authors showed that two self-assembled nanoparticle morphologies, spherical micelles and non-spherical worm-like micelles, exhibited markedly different NO release kinetics. Slow releasing worm-like micelles were less effective against established P. aeruginosa biofilms than the fast releasing spherical micelles after 30 min treatment. Conversely, the opposite was true when a 60 min nanoparticle treatment regime was employed, whereby the worm-like micelles after a 60 min treatment outperformed the spherical micelles’ antibiofilm activity after either 30 min or 60 min treatment.180 These studies highlight the importance of slow, sustained NO-releasing polymer systems for the effective inhibition and killing of biofilms.
Synthetically enhanced biomaterials have also been used as NO-releasing materials. For example, Schoenfisch and co-workers developed chitosan oligosaccharides functionalized with NONOates and demonstrated their antibiofilm activity against established P. aeruginosa biofilms.181 Though chitosan itself is typically biocidal, the short oligomers used in this study did not exhibit biocidal activity. However, the NONOate-functional chitosan oligomers were able to disperse and kill established P. aeruginosa biofilms.181 As an aside, under aerobic conditions, oxygen, superoxide and hydrogen peroxide play a complex role in determining NO biocidal activity. These reactive oxygen species facilitate the conversion of NO into the previously mentioned higher oxidation biocidal species, however they also play a role in converting NO into relatively benign species such as nitrate (NO3−) and nitrite (NO2−).179 Therefore, Schoenfisch and co-workers used the NO-releasing chitosan oligomers to study the influence of oxygen availability on the antimicrobial and antibiofilm activity of NO donors in the aforementioned study.181 Under anaerobic conditions, the NO-releasing chitosan was shown to release more bioavailable NO than under aerobic conditions, which typically led to lower MICs of the NO-releasing chitosan under anaerobic conditions against planktonic P. aeruginosa bacteria. However, this corresponded to higher concentrations of bioavailable NO. The NO-releasing chitosan was much more effective at killing both mucoid and non-mucoid P. aeruginosa biofilms under anaerobic conditions, in stark contrast to the small molecule antibiotic tobramycin, which showed up to an 8-fold reduction in efficacy under anaerobic conditions. Furthermore, the NO-releasing chitosan was shown to kill both phenotypes (mucoid and non-mucoid biofilms) with equal efficacy, whereas tobramycin showed phenotypic dependency, with mucoid biofilms exhibiting 4-fold lower susceptibility.181 More recently, work from the same group demonstrated that NONOate-functional alginate polysaccharides showed superior antibiofilm activity against Gram-positive biofilms formed by S. aureus including MRSA under anaerobic conditions, compared to tobramycin.182 These studies demonstrate the influence of environmental factors, such as oxygen availability, on the effectiveness of antibiotics against established biofilms and showcase how NO-releasing materials can use these conventionally challenging conditions to their advantage.
NO-releasing materials show excellent promise in their own right through their relative ease of preparation from both vinyl commodity monomers and natural polymers. However, as discussed in the next section, their true strength is in their compatibility with other antibiofilm moieties to produce multi-action materials with a broad arsenal of antibiofilm functionalities.
Multi-action antibiofilm materials
An emerging trend in antibiofilm materials research includes those that combine the synergistic benefits of two or more distinct antibiofilm mechanisms. Unlike the materials with single modalities discussed previously, multi-action materials couple both biocidal functionality and biofilm dispersal moieties, or can undergo staged release of multiple synergistic biocidal drugs. For example, Schoenfisch and co-workers investigated modified antimicrobial amine-terminal poly(amidoamine) (PAMAM) dendrimers of different molar masses (different dendritic generations, G1 through to G4) as dual-action biocides/NO donors (Fig. 5B).175 Following functionalization of the PAMAM amine groups through epoxide ring opening using alkyl monoepoxides and subsequent reaction of the resulting secondary amines with NO, dual-action NONOate-functional dendrimers were achieved. Note that dendrimers with greater generations (i.e. G4 as opposed to G1), have more terminal amine groups per dendrimer molecule and as such had a higher NO loading capacity onto these terminal units. Both NO-loaded and non-loaded modified dendrimers showed antimicrobial activity against a range of planktonic bacteria, demonstrating the dual-action capability of the NONOate-functional materials. Whilst each dendrimer in the series showed similar NO release kinetics, the terminal group chemistry and dendrimer generation had a marked effect on the species-dependent antibiofilm activity. The higher molar mass G3 and G4 dendrimers that were functionalized with hexyl groups exhibited the best broad-spectrum in vitro antibiofilm activity against established mature P. aeruginosa, drug-susceptible S. aureus, and MRSA biofilms grown for 24 h. This was shown to be a result of the improved penetration of these species into the biofilm matrix, owing to their greater functional group density and greater hydrophobicity, which enabled better association with and disruption of exopolysaccharides and bacterial cell membranes. However, the butyl-functional dendrimers from this study favorably showed reduced cytotoxicity profiles against mouse fibroblasts, demonstrating a trade-off between antibiofilm activity and mammalian toxicity.175Boyer and co-workers developed dual-action NO-releasing antimicrobial polycations formed by reversible addition–fragmentation chain transfer (RAFT) polymerization.176 To this end, optimized antimicrobial acrylate/acrylamide copolymers, containing hydrophobic ethylhexyl groups, hydrophilic oligo(ethylene glycol) groups, and primary amine groups in their side chains, were partially loaded with NO to form NONOates (Fig. 5C). These could passively release NO in vitro, and subsequently restore the antimicrobial bacteriolytic primary amine functionality. The NO-loaded polymer outperformed both the non-loaded antimicrobial polymer, the small molecule spermine-NO conjugate, or a blended mixture of the two, against established P. aeruginosa biofilms pre-grown for 6.5 h, resulting in 80% biomass reduction and over 5-log reduction in biofilm viability, relative to the untreated control.176 The same group investigated block copolymer micelles with oligo(ethylene glycol) shells and benzaldehyde-functional cores, which could be core-loaded with the small molecule antibiotic, gentamycin, through a pH-cleavable imine bond.177 The attached gentamycin, which contains multiple primary and secondary amines, could be further loaded with NO to form polymer–gentamycin–NONOate conjugates (Fig. 5D). Similarly, these antibiotic/NO dual-releasing micelles outperformed the antibiofilm activity of free gentamycin, a spermine–NO conjugate, or the polymer–gentamycin conjugate, all of which showed <20% reduction in P. aeruginosa biofilm viability, whereas the dual-action system resulted in 90% reduction in biofilm viability relative to the untreated control.177 These studies demonstrate the importance of multi-action synergistic materials, which show superior antibiofilm activity compared to their individual components, or mixtures thereof.
Recently, Du and co-workers developed dual corona vesicles comprised of a co-assembled mixture of poly(ε-caprolactone)-block-poly(lysine-stat-phenylalanine) (PCL-b-P(Lys-stat-Phe)) and poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL), as shown in Fig. 6A.183 These showed intrinsic broad-spectrum antimicrobial activity imparted by the polypeptide P(Lys-stat-Phe) corona and stealth-like protein repellence imparted by the PEO corona, as well as biodegradability imparted by the hydrolytic properties of the PCL core. The latter property allowed these nanoparticles to penetrate deeper into S. aureus and E. coli biofilms. These particles could be further loaded with the small molecule antibiotic ciprofloxacin. The combined antimicrobial properties of the dual-corona vesicles, along with their enhanced stealth properties, allowed for superior delivery of the small molecule antibiotic relative to the single corona vesicle controls, resulting in greater biomass removal. Additionally, the release of lipases and other hydrolytic enzymes upon lysis of the bacterial membranes were thought to enhance the degradation of the particles, thus leading to faster burst release of the loaded antibiotic (Fig. 6B). Topical treatment with the antibiotic-loaded vesicles was shown to significantly reduce bacterial plaque formation, gingival bleeding and periodontal inflammation in a rat periodontitis model, whilst requiring just 50% of the loaded antibiotic to achieve similar bactericidal effects.183 Similarly, van der Mei and Shi et al. developed surface adaptive mixed corona micelles comprised of a PCL core and a mixed corona of both PEO and a pH-responsive poly(β-amino ester).185 PEO provided stealth properties until acidification in the S. aureus biofilm environment, which led to cationization of the poly(β-amino ester) block. This resulted in lysis of anionic bacterial membranes, subsequent release of secretory bacterial lipases, enzyme-mediated degradation of the PCL core, and release of the loaded antibiotic cargo, triclosan (similar to the schematic mechanism shown in Fig. 6B).185 Materials that exploit such “kill-degrade-release” cascades are promising candidates for combination treatment of biofilms through their multiple mechanisms of action, which subverts biofilm defences against conventional antibiotics, as well as reducing antimicrobial resistance often observed in single mechanism approaches.
 | ||
| Fig. 6 Self-assembled multi-action antibiofilm materials. (A) Dual corona vesicles with both stealth-like and antimicrobial coronas and a pH- and lipase-degradable core, loaded with antibiotic. (B) Schematic of the “kill-degrade-release” cascade mechanism. Both (A) and (B) adapted with permission from ref. 183. Copyright©2019 American Chemical Society. (C) Preparation of the poly(aspartamide)-based copolymer self-assembly with azithromycin (Az) covalently bound and the D-tyrosine prodrug electrostatically sequestered (shown in black). A schematic for the multi-stage drug release is given, including charge reversal upon D-tyrosine prodrug release, followed by subsequent lipase-mediated micelle degradation and azithromycin release. Adapted from ref. 184, with permission from the Royal Society of Chemistry. | ||
One intricate example of a dual-antibiotic-loaded nanoparticle that exploits a similar multi-stage release mechanism was given by Li and co-workers, who developed a mixed corona micelle drug delivery system based on functionalized poly(aspartamide) copolymers.184 The authors prepared a cationic homopolymer of poly(aspartamide) grafted with the hydrophobic antibiotic azithromycin through diethylene triamine spacers and an ester linkage (Fig. 6C, structure shown in green). This was combined with an ABA triblock copolymer, poly(diisopropylaspartamide)-block-poly(ethylene oxide)-block-poly(diisopropylaspartamide) (Fig. 6C, blue and red block copolymer), in aqueous solution to form self-assembled micelles. These micelles had cationic poly(ethylene oxide)/poly(aspartamine-diethylenetriamine) shells and a poly(diisopropylaspartamide) core, stabilizing the grafted hydrophobic azithromycin (Fig. 6C). An anionic small molecule biofilm dispersing prodrug, cis-aconityl D-tyrosine, could be electrostatically sequestered to the cationic shell, resulting in the formation of anionic nanoparticles. Upon acidification in the biofilm environment, the acid-labile cis-aconityl group of the sequestered small molecule prodrug was cleaved releasing the now zwitterionic D-tyrosine, which was able to disperse the biofilm matrix. Release of D-tyrosine resulted in charge switching of the micelles from anionic back to cationic, resulting in the subsequent shrinkage of the micelles from 79 nm to 24 nm. In the presence of the resulting secretory bacterial lipases following biofilm disruption, the ester linkages between the poly(peptide) and the antibiotic azithromycin were cleaved, resulting in local drug release within the biofilm to destroy the dispersed P. aeruginosa bacteria. The mixed poly(peptide) micelles were highly efficient at killing biofilms present on implanted catheters in an in vivo rat model. Close to 3-log reduction in viable counts of P. aeruginosa biofilms was observed upon treatment, relative to the negative control group.184 This study demonstrates the design of highly complex delivery vehicles with multiple controlled release mechanisms that can effectively exploit the biofilm microenvironment for stepwise payload delivery, more examples of which will be described in a later section.
Combining synergistic mechanisms of action through the use of antimicrobial enzyme/peptide/antibiotic cocktails has recently been systematically analyzed by Jorge and co-workers in the treatment of S. aureus and P. aeruginosa biofilms.187 This has led to the curation of an available online resource, the Antimicrobial Enzyme Combinations Database (https://www.ceb.uminho.pt/aecd), which details enzyme combination therapies and their effectiveness against these microbial biofilm systems. Such databases will be key to identifying knowledge gaps and new research opportunities, with the authors identifying the study of polymicrobial biofilms as a future focus area.187 In a similar initiative, the recently updated Data Repository of Antimicrobial Peptides (DRAMP; http://dramp.cpu-bioinfor.org) holds information on the structure and antimicrobial properties of over 20000 peptides and peptide sequence entries.188,189
“On-demand” stimuli-responsive materials
The ability of highly active solution-based macromolecular antimicrobials to penetrate biofilms is often severely hampered by the size of the antimicrobial and through virtue of attractive and repulsive coulombic interactions with the biofilm matrix. Additionally, the robustness of synthetic polymer materials over that of peptides enables their prolonged environmental persistence at lethal and sub-lethal concentrations, which may eventually lead to the emergence of acquired antimicrobial resistance to such polymeric agents. Emerging strategies to tackle these newly found issues have led to the design of stimuli-responsive materials with “on-demand” antibiofilm activity. Whilst endogenous factors that stimulate the release of small molecule antibiotics has been discussed in previous sections, here we highlight a few examples where external stimuli have been employed, or where a stimuli-responsive change in the polymer chemistry results in an intrinsic change in antibiofilm activity. These include photodynamic polymer materials that respond to light, materials that employ dynamic covalent chemistries or host–guest complexation, and materials with stimuli-responsive “on-demand” NO release.Wang and Liu et al. prepared conjugated polymers with cationic amine-functional side chains, which showed photo-responsive antibiofilm activity.190 Through zeta-potential measurements, the polymers were shown to adhere to the surface of S. aureus, increasing the concentration of the polymer on the bacterial surface. Upon illumination with white light, the conjugated polymer produced reactive oxygen species, which led to bacterial death in a process known as photodynamic therapy. These polymers were shown to effectively inhibit S. aureus biofilm formation, as well as killing established mature biofilms.190 In a more elaborate example, Xu and co-workers designed dual stimuli-responsive supramolecular polymers held together through cucurbit[8]uril-mediated host–guest and metal coordination interactions.90 A cationic porphyrin moiety (Por) was non-covalently attached to an intermediate linker (NpDi) through metal coordination interaction with the porphyrin guest Sn(IV) ion. The linker allowed the porphyrin to be non-covalently bound to the side chain of a cationic polymer (BPB) using cucurbit[8]uril host–guest interactions, forming the polymer complexation product (SPP) (Fig. 7A). The porphyrin moiety could be used as a photosensitizer, producing antimicrobial reactive oxygen species upon illumination with white light (Fig. 7B and C). The cationic supramolecular polymer adhered to negative bacterial membranes, thereby increasing the local concentration of porphyrin moieties around these pathogens. Therefore, this polymer showed higher biofilm-killing activity than the free porphyrin after illumination, whilst the mammalian toxicity was significantly reduced. Upon addition of a competitive binding agent, cucurbit[7]uril, the supramolecular polymer could be controllably disassembled, thus reducing their antibiofilm activity to match that of the free porphyrin.90 This study highlights the potential of dynamic non-covalent interactions in designing antibiofilm agents that controllably degrade, so as not to persist in the environment. For more information on photodynamic therapy in the treatment of biofilms (including inorganic and carbon nanomaterials), the reader is referred to the following text.92
 | ||
| Fig. 7 Examples of stimuli-responsive antibiofilm polymers based on non-covalent host–guest interactions and dynamic covalent interactions. (A) Preparation and disassembly behaviour of the non-covalent porphyrin-functional polymer SPP. (B) Schematic of photoresponsive reactive oxygen species generation upon illumination. (C) CLSM images of S. aureus biofilms stained with PI and SYTO9 LIVE/DEAD stain after various treatments. (A–C) Adapted with permission from ref. 90. Copyright©2019 American Chemical Society. (D) Structures of the PONI-GAT stabilizer and ATA crosslinker used in ref. 186 and schematic showing their pH-responsive dynamic covalent interactions at the carvacrol oil–water interface of the formed nanocomposites. | ||
Another example of dynamic interactions exploited in antibiofilm materials is in the use of dynamic covalent Schiff base chemistry.186 Such imine-functional materials are highly pH-sensitive, with the equilibrium strongly favoring the hydrolysis degradation products (amine plus carbonyl) at low pH, whilst the imine bond remains intact under physiological conditions. Rotello and Guo reported the preparation of dynamic covalent nanocomposites of an oil-in-water emulsion of the antibiotic oil, carvacrol, in aqueous solution.186 The emulsion was stabilized through use of a cationic poly(oxonorbornene) copolymer present in the aqueous phase (PONI-GAT), which contained guanidine, amine and tetra(ethylene glycol) functionality in its side chains, in combination with a tetra functional aldehyde, adamantyl-core tetrakisaldehyde (ATA) present in the carvacrol oil phase (Fig. 7D). Upon emulsification, the amine side chain groups of PONI-GAT formed Schiff base dynamic covalent bonds with the aldehyde functionality of ATA at the oil–water interface. These nanocomposites remained stable under physiological conditions but became destabilized under acidic conditions (pH 5.0–6.5), leading to release of the antimicrobials PONI-GAT and carvacrol. Unlike the individual components, the nanocomposites could penetrate deep within biofilms and resulted in near complete loss of viability in biofilms formed by P. aeruginosa, E. coli, S. aureus and Enterobacter cloacae drug-resistant clinical isolates respectively, whereas PONI-GAT and carvacrol alone were ineffective.186
Aside from stimuli-responsive polymer materials, Zhou and co-workers have reported the use of stimuli-responsive moieties built into synthetic antimicrobial peptides to modulate their antibiofilm properties.191 Here, the authors synthesized isomeric tripeptides based on either Gly–Gly–Ala or Ala–Gly–Gly with a butyl azobenzene moiety present at the N terminus of each peptide. These were shown to self-assemble into twisted nanowires or untwisted nanofibers in their native form. It is noteworthy that in these tripeptides, the entire sequence, including the trans-azobenzene moiety, is hydrophobic and that the peptides are amphiphilic only through the charge in their terminal carboxylate group. Upon complexation with β-cyclodextrin, an amphiphilic host molecule for the hydrophobic guest trans-azobenzene moiety, circular dichroism spectroscopy and electron microscopy indicated disassembly of the chiral self-assemblies, owing to an increase in the overall hydrophilicity. Upon further addition of a competitive guest molecule, adamantane, to disrupt the β-cyclodextrin-azobenzene complexation, the self-assembly could be reformed. Similarly, photoirradiation of the native tripeptides with UV light could invoke a trans-to-cis isomerisation in the azobenzene unit, thus greatly increasing its hydrophilicity and resulting in disassembly. Again, this process was reversible by further irradiating with visible light to afford the trans isomer. By varying the host–guest complexation, or photoirradiation conditions, the biofilm inhibitory activity against a range of Gram-negative and Gram-positive pathogens could be effectively modulated.191 The activation/deactivation of antibiofilm activity in response to external triggers is a promising strategy for controlling antimicrobial environmental persistence, as well as potentially providing a future platform for spatiotemporal antibiofilm control using photoirradiation.
Aside from the passive release of NO from NONOates discussed previously, other materials based on S-nitrosothiols show on-demand release of NO in response to stimuli such as enzymes, heat, light, metal ions or reduction.87–89 Small molecule S-nitrosothiols such as S-nitroso-N-acetyl-D-penicillamine (SNAP) employ such NO-release mechanisms, as well as the tripeptide S-nitrosoglutathione. For example, S-nitrosoglutathione has been shown to be an effective antibiofilm agent against P. aeruginosa biofilms grown on medical grade polyurethane.192 Whilst numerous reports of S-nitrosothiol-functional polymers and polymer nanoparticle delivery of S-nitrosothiol small molecules exist for the treatment of planktonic bacteria, reports of their use against established biofilms are rarer, thereby presenting excellent opportunities for stimuli-responsive delivery of NO to treat biofilms using such materials. A recent report by Hu and Yu et al. described a new, distinct type of NO donor based on a photoresponsive coumarin-containing monomer, whose secondary amine moiety could be loaded with NO upon treatment with the gaseous biocide.193 The monomer could be directly polymerized in a RAFT chain extension process, starting from a poly(ethylene glycol) macro chain transfer agent, to form amphiphilic block copolymers. These formed self-assembled micelles with NO-loaded hydrophobic cores in aqueous solution. Upon photoirradiation at 410 nm, NO could be controllably released from the micelles, which was also associated with restoration of the fluorescence of the coumarin unit, thereby providing an in situ probe for monitoring NO release at the administration site. Furthermore, light-mediated NO release was associated with a reduction in the micelle core hydrophobicity, which was exploited as a mechanism for the dual release of NO and the sequestered small molecule antibiotic, ciprofloxacin. After light irradiation, these dual-loaded micelles outperformed micelles loaded with the individual antibiotic or NO alone, against established P. aeruginosa biofilms, but showed no activity under dark conditions, demonstrating their controlled temporal release.193
The ability to externally control antibiofilm activity will likely give rise to the next generation of biofilm removal strategies. This control will be particularly important for overcoming challenges related to the overuse and persistence of biocidal agents. Additionally, the on-demand antibiofilm properties are expected show fewer off-target effects and lower toxicity in vivo, which could potentially show synergy with other strategies employed in nanomedicine, such as site-specific targeting.
Polymers for quorum sensing modulation
Whilst quorum quenching enzymes are promising antibiofilm agents through their effective elimination of quorum sensing autoinducers, others have more recently designed polymers that are able to modulate quorum sensing in order to influence biofilm formation. For example, Duarte and co-workers prepared linear anionic copolymers poly(methyl methacrylate-co-methacrylic acid) and poly(methyl methacrylate-co-itaconic acid) and demonstrated their ability to interact and interfere with quorum sensing AHL autoinducers. The copolymers reduced the quorum sensing-moderated bioluminescence in Vibrio fischeri, as well as reducing biofilm formation of A. hydrophila. The phenotypical effects reverted after subsequent addition of additional AHLs, indicating an autoinducer sequestration mechanism. Shepherd and co-workers designed a highly branched non-biocidal polymer with its multiple end groups functionalized with AHLs.194 Through the use of colorimetric biosensor assays, it was demonstrated that this polymer could block AHL-mediated quorum sensing in Chromobacterium violaceum. The authors explained that these polymers could show potential as antibiofilm materials owing to their quorum quenching activity, but no further antibiofilm experiments were performed in this very recent study.194Another interesting approach to passive quorum quenching is through the use of molecular imprinted polymer materials (MIPs). These materials are prepared through the polymerization of a monomer and crosslinker in the presence of a non-polymerizable template molecule, which is subsequently removed after curing. These MIPs selectively bind and sequester the template molecule of interest. Piletska and co-workers used molecular dynamics simulations to investigate in silico the suitability of a range of monomers to interact with the template quorum sensing autoinducer, N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-AHL).195 After identifying itaconic acid as an ideal monomer for interacting with the autoinducer, MIPs were prepared by polymerizing this monomer in the presence of 3-oxo-C12-AHL and a dimethacrylate crosslinker. MIP microparticles sized 38–125 μm were shown to sequester 3-oxo-C12-AHL and inhibit the formation of P. aeruginosa biofilms, which relied on the 3-oxo-C12-AHL quorum sensing pathway for upregulation of this phenotype.195
Whilst some polymers are able to reduce quorum sensing, other polymers such as polycations have been shown to cluster bacteria, which in turn enhances quorum sensing.196,197 Whilst this effect is advantageous for certain applications, this inadvertently promotes quorum sensing-induced processes like biofilm formation.196,197 Alexander, Krasnogor and Fernadez-Trillo et al. investigated the interaction with bacteria of a tertiary amine-functional polycation, which was shown to cluster bacteria, and a polyol (poly(vinyl alcohol)), which was shown to competitively bind to the boron species present in the quorum sensing autoinducer, autoinducer-2, leading to autoinducer inactivation.93 Additionally, a copolymer containing both catechol units and cationic units was included in the study, which could facilitate both bacterial clustering through the cationic functionality and AI-2 inactivation through the diol present in the catechol unit. It was shown that the polycation induced quorum sensing-mediated bioluminescence in Vibrio harveyi, whereas the polyol reduced this phenotype. By contrast, the copolymer showed concentration dependency and could facilitate both induction and suppression of bioluminescence under certain concentration regimes.93 Krachler and Fernandez-Trillo et al. in a later study showed that polycations induced the formation of biofilms in Vibrio cholerae but downregulated the expression of virulence genes.197
These studies highlight the complex processes involved in quorum sensing modulation, which occasionally lead to non-intuitive biological outcomes. The design of polymers that interfere with quorum sensing for the treatment and dispersal of biofilms is non-trivial and therefore presents excellent opportunities for future research; more in-depth investigation of these promising materials is required to fully understand their properties and take advantage of their therapeutic potential. Indeed, the ability to remove biofilms from surfaces without inducing microbial destruction is an attractive approach for overcoming some of the challenges associated with antimicrobial resistance.
Enzyme-mimicking materials
One highly novel approach towards eliminating biofilms is through the use of macromolecular structures that can mimic antibiofilm enzymes. These materials can show an advantage over biosynthesized enzymes in their greater stability in vivo and more cost-effective preparation. This approach is at the very forefront of antibiofilm technology and we anticipate that such materials will form a new class of therapeutic agents with great future potential. One example is given by Ellis, Locock and Qu et al., who prepared antibiofilm materials from guanine-rich DNA sequences that can assemble into three-dimensional complexes known as G-quadraplexes.202 These DNA sequences could be coupled to a β-lactam antibiotic, oxacillin, and complexed with the iron-containing protoporphyrin IX (hemin) to form a drug conjugated oxacillin–G-quadraplex–hemin complex. Given the oxidoreductase capability of the iron present in hemin, this species can mimic the enzymatic activity of peroxidases through the production of reactive, oxidized species such as hypothiocyanite ([OSCN]−) in the presence of hydrogen peroxide. Against established S. aureus biofilms and in the presence of 0.5% H2O2, the G-quadraplex–hemin complex (without oxacillin) showed a 50% reduction in biofilm viability at the concentration as hemin alone, which showed reduced activity (30% reduction), demonstrating that complexation with the G-quadraplex improved penetration of hemin through the biofilm. In the absence of H2O2 (where hemin is inactive), the oxacillin–G-quadraplexes, with or without hemin, showed around a 25% reduction in viability, where the oxacillin alone showed only 11% reduction. Whilst more mechanistic investigation is required to fully elucidate the various modes of action, taken together, these findings demonstrate how the catalytic nature of hemin, along with the enhanced biofilm penetration of the DNA–drug conjugate, could be used to synergistically eliminate established biofilms.202 For more information on non-macromolecular enzyme mimics for biofilm eradication, such as inorganic nanoparticles, the reader is referred to the following text.203Biohybrid materials
Biohybrid materials are those that incorporate both synthetic materials, such as polymers and nanoparticles, with biologically derived species. As with the biomimetic structures discussed above, biohybrid materials can possess the beneficial properties of both classes; they can exhibit high specificity and selectivity associated with peptides and proteins, combined with greater stability, tunability and cost-effectiveness associated with synthetic materials. For example, encapsulation of enzymes inside semi-permeable self-assembled polymersomes to form therapeutic nanoreactors has been shown by Blackman, Gibson and O’Reilly et al.,198,204 which was shown to significantly reduce the immunogenicity and proteolytic susceptibility of the encapsulated species, relative to both the native free enzyme and a PEGylated conjugate (Fig. 8A, LHS).198 This polymerization-induced self-assembly (PISA) encapsulation technique has recently been exploited by Blackman and Locock et al. to encapsulate the antimicrobial enzyme GOx to form antimicrobial nanoreactors (Fig. 8A, RHS).95 The subsequent nanoreactors were permeable to the small molecule substrates D-glucose and molecular oxygen, as well as the products δ-glucono-1,5-lactone and the antimicrobial reactive oxygen species, hydrogen peroxide, yet retained the active enzyme inside the polymersome lumen. Subsequently, the encapsulated enzyme was highly effective against planktonic Gram-positive staphylococcal bacteria and showed glucose-dependent activity against planktonic Gram-negative bacteria. Meanwhile, the nanoreactors retained their antibiofilm activity against an established MRSA clinical isolate biofilm, relative to the native free enzyme.95 This PISA approach offers a simple encapsulation strategy for antibiofilm agents and could potentially be used as a future platform for modulating antibiofilm activity, for example through the use of further complementary antibiofilm functionalities or biofilm targeting groups. | ||
| Fig. 8 Biohybrid systems. (A) Schematic of the protective nature of semi-permeable enzyme-loaded nanoreactors formed by PISA (LHS) and the antimicrobial and antibiofilm activity of GOx-loaded nanoreactors (RHS). LHS adapted with permission from ref. 198 (https://pubs.acs.org/doi/10.1021/acscentsci.8b00168) Copyright©2018 American Chemical Society. Further permissions related to this excerpted material should be directed to the American Chemical Society. RHS adapted from ref. 95. Copyright©2020 American Chemical Society. (B) Preparation and antibiofilm activity of GOx-functionalized magnetic chitosan nanocomposites. The insets show established S. aureus biofilms treated with the chitosan particles alone (I) and the nanocomposites (II). Adapted with permission from ref. 199. Copyright©2019 American Chemical Society. (C) Schematic of DNase I-functional ciprofloxacin-loaded poly(lactic-co-glycolic acid) nanoparticles (blue)200 and DNase I/cellobiose dehydrogenase (CDH) co-immobilized onto chitosan nanoparticles (red).97 (D) Enzyme-responsive activation of the cyclic peptide colistin from the biohybrid colistin-dextrin conjugate used in antibiofilm PUMPT treatments. Adapted with permission from ref. 201 (https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.9b00393). Copyright©2019 American Chemical Society. Further permissions related to this excerpted material should be directed to the American Chemical Society. | ||
Using the same GOx active species, Kim and Dordick et al. developed a strategy for GOx immobilization onto the surface of chitosan nanoparticles.199 In one formulation, the enzyme was immobilized onto the chitosan surface via precipitation, along with magnetic iron oxide nanoparticles to produce biohybrid nanocomposites with both oxidative activity and magnetic properties for simple extraction from solution (Fig. 8B). These nanocomposites showed significantly enhanced anti-biofilm activity against established S. aureus biofilms, relative to treatment with the chitosan nanoparticles alone, demonstrating the importance of the additional catalytic functionality (Fig. 8B, insets I and II).199
Other antimicrobial enzymes have been incorporated into biohybrid systems. For example, Han and co-workers immobilized dispersin B within hydrogel nanoparticles formed by linoleic acid-modified carboxymethyl chitosan through a sonication method.98 These nanoparticles showed enhanced thermal and storage stability compared with the native free enzyme, as well as improved dispersin B reusability. Owing to the synergistic action of dispersin B with the chitosan material, which also independently showed antibiofilm properties, the loaded nanoparticles showed superior inhibitory and killing activity against A. actinomycetemcomitans, S. aureus and S. epidermidis biofilms.98
Biohybrid materials provide a convenient platform for co-localization of synergistic antibiofilm or therapeutic components (Fig. 8C). For example, Torrents and co-workers developed poly(lactic-co-glycolic acid) nanoparticles, which were loaded with the small molecule antibiotic ciprofloxacin, and surface decorated with immobilized DNase I from bovine pancreas.200 These nanoparticles were shown to passively release the antibiotic and showed biocidal properties against planktonic S. aureus and P. aeruginosa. The nanoparticles outperformed each of the individual components, as well as a cocktail of the individual components, which had not been covalently immobilized to the nanoparticles, in inhibition of P. aeruginosa biofilm formation, as well as killing 99.8% of 48 h mature biofilms upon repeated exposure over a 3-day period.200 Similarly, Tan and co-workers co-immobilized two antibiofilm enzymes, DNase I and cellobiose dehydrogenase, onto the surface of chitosan nanoparticles.97 In the presence of cellobiose, the co-immobilized biohybrid nanoparticles outperformed nanoparticles decorated with just a single enzyme and were found to penetrate into cross-kingdom polymicrobial biofilms comprised of C. albicans and S. aureus. The co-immobilized enzymes could effectively inhibit mono- and polymicrobial biofilms, as well as removing established mono- and polymicrobial biofilms formed on silicone.97 These examples highlight the power of biohybrid nanoparticles to co-localize various antibiofilm components, leading to local synergistic modes of action, which surpass the activity of the individual components.
The conjugation of polymers with controllable degradability to therapeutic peptides and proteins, forms the basis of polymer masked–unmasked protein therapy (PUMPT).201,205–207 Upon a stimulus such as enzymatic hydrolysis, the polymer component of the relatively inactive polymer–protein or polymer–peptide conjugate degrades, releasing the active peptide or protein component to enact its therapeutic function at the disease site (Fig. 8D). This strategy has been investigated for a range of applications, including cancer therapy and wound healing, as well as the effective treatment of planktonic bacteria.205–207 Recently, Ferguson and co-workers demonstrated that dextrin–colistin conjugates could be used as effective amylase-responsive PUMPT agents against established biofilms.201 Colistin (also known as polymyxin E) is a biosynthesized cyclic cationic peptide that enacts a membrane-rupturing mechanism of action against bacterial membranes; dextrin is a semi-synthetic polysaccharide prepared from the hydrolysis of starch. After treatment of the dextrin-colistin conjugate with α-amylase from human saliva, which could selectively degrade the dextrin, the unmasked peptide was purified by size exclusion chromatography and its ability to inhibit E. coli biofilm formation was confirmed.201 In previous studies, the authors demonstrated that such bioconjugation resulted in significantly lower toxicity and an extended in vivo plasma half-life, compared with free colistin sulfate.205 Aside from peptide–polymer conjugates used in PUMPT approaches, more information about the general antimicrobial applications of peptide–polymer conjugates, including biohybrid antibiofilm coatings and hydrogel materials, can be found in the following recent Review.96
Biohybrid materials aim to combine the advantages of natural and synthetic materials, whilst minimizing the inherent limitations of each material class. The term captures a wide range of materials from highly complex multicomponent systems, down to simple bioconjugates. The barriers to their clinical translation increase along with the complexity of the antibiofilm system and whilst these materials show numerous advantages over their natural or synthetic components, significant challenges exist in obtaining approval for clinical applications. Nevertheless, we anticipate that further investigation into new biohybrid materials could lead to revolutionary advances in biofilm removal and eradication.
Challenges for the translation of macromolecular antibiofilm agents
Whilst macromolecules and polymers hold significant promise for the inhibition and elimination of biofilms, challenges remain for their translation into real-world applications, particularly for clinical applications. To date, natural species such as peptides and enzymes lead the way in obtaining clinical approval, with numerous species approved by the FDA for antimicrobial indications, either through topical or systemic administration.55,111 These include recombinant human DNase I (Pulmozyme®) for cystic fibrosis treatments, the topical antimicrobial peptide bacitracin for bacterial skin infections, and members of the non-ribosomal antimicrobial peptide family of polymyxins; polymyxin B and polymyxin E. Meanwhile, the antimicrobial peptide nisin, the poly(peptide) ε-polylysine, and the enzymes, glucose oxidase and lysozyme, amongst others, have received “Generally Regarded As Safe” (GRAS) status and are approved for used as food additives. However, particularly when designing new bacteriolytic peptides, a major challenge is in reducing hemotoxicity whilst still ensuring high antibiofilm and antimicrobial efficacy against a broad spectrum of clinically relevant pathogens. As with any new therapeutic agent, ensuring low cytotoxicity of these agents towards tissue in the local environment is important for external topical indications, whereas material pharmacokinetics and biodistribution play a much larger additional role in determining suitability for systemic indications. Finally, other factors such as the immunogenicity of the species in question, as well as the biocompatibility of any metabolites, must be understood in detail to ensure off-target effects lie within acceptable tolerances.Whilst peptides and enzymes have been successful in obtaining approval for use, their stability in vivo is still limited, making them non-optimal candidates for certain indications where long-acting therapeutics are required, for instance in chronic or recurring infections. In this respect, synthetic materials, including biomimetic and biohybrid materials, can show a significant advantage. However, these synthetic materials face additional hurdles for entering the clinic, which pertain to their relatively ill-defined sequence, molar mass and chemical composition, along with a host of other challenges.208 Furthermore, batch-to-batch variability and reproducibility of processing techniques are particularly important for polymer nanoparticle formulations. Indeed, detailed reporting of therapeutic nanoparticles, using a range of complementary characterization techniques, will be important for widespread translation of these nanomedicines into clinical applications.209 Despite these challenges, hope remains that such polymeric materials will be able to overcome these hurdles to obtain approval for clinical use in antibiofilm indications. Furthermore, these materials face significantly lower barriers for adoption in non-clinical antibiofilm applications.
As mentioned throughout this Review, the long-term use of biocidal antibiofilm agents is cause for concern over resistance development. Strict stewardship over the use of both existing and newly approved biocidal agents is vital for ensuring that the development of acquired resistance towards such agents is minimized and that newly resistant strains are rapidly identified.210 This stewardship includes the reservation of certain antimicrobials as last-resort treatments in the clinic, or employing agents that do not persist in the environment. Here, we believe that materials with on-demand activity, including those with spatiotemporal control, will be important for addressing such challenges, in addition to materials that have programmable degradation for on-demand deactivation after use.
Conclusions and outlook
Macromolecular approaches for the inhibition and killing of biofilms are promising alternatives to the use of small molecule antibiotics and inorganic materials. These approaches employ a range of material classes, including naturally occurring biological species refined through evolutionary processes, or synthetic materials with expansive scope in their design space and ease of tunability. With the threat of antimicrobial resistance already constraining global healthcare systems, and with its expanding and impending impact just on the horizon, it is imperative that the scientific community find novel, innovative solutions for the effective treatment of biofilms. In this article, we have highlighted the composition of biofilms and their importance in a wide range of biomedical and industrial fields. The use of naturally occurring and synthetic peptides and proteins were emphasized as exceptional candidates for biofilm inhibition, destruction and/or dispersal. Modern strategies using synthetic and semi-synthetic polymer materials were also discussed, including those with intrinsic antibiofilm properties, or those that deliver conventional small molecule antibiotics. Finally, emerging areas in the treatment of biofilms were highlighted, which we envisage could lead to greater advancements in this important field. These include materials that deliver reactive oxygen and nitrogen species, or that interfere with bacterial communication pathways, those with multiple modes of action and “smart” stimuli-responsive systems. Furthermore, materials that combine the benefits of both synthetic and natural materials, either through biomimicry or through uniting both classes to form biohybrid systems, were highlighted as promising emerging approaches. In our future outlook for this field, we expect to see further advanced strategies, such as those that combine biofilm-targeting functionality and mucoadhesion with multi-modal dispersing/killing activity and sensing properties as anti-infective theranostic agents. It is envisioned that detailed studies investigating the prevalence of antimicrobial resistance upon exposure to these new materials will become more numerous, as well as strategies to controllably switch on/off antibiofilm activity, and if necessary, degrade these antibiofilm agents after their intended use. We anticipate that the study of polymicrobial growth models, in vivo and ex vivo models, and the design of materials that can eradicate persister cells, also represent great challenges and research opportunities. Furthermore, we expect that the curation of databases and the use of advanced statistical analysis of material libraries to provide more comprehensive structure–activity relationships in antibiofilm materials will also be key for identifying new candidate systems. We hope that this article highlights the importance of identifying new antibiofilm strategies for improving a vast array of aspects in our daily lives, and that this Review gives further insights into the design of new advanced materials for the inhibition and killing of microbial biofilms.Conflicts of interest
Please note that Y. Q. and K. E. S. L. are listed as co-inventors of a family of patents related to antibiofilm polymers (Worldwide Patent Application No.: WO2016138558A1). No specific commercial activities in relation to these patents were underway at the time of writing. No funding from any external agencies, other than those listed in the acknowledgements section, has been obtained in relation to the preparation of this article.Acknowledgements
This work was supported by a CSIRO Early Research Career Postdoctoral Fellowship (L. D. B.), a CSIRO Julius Career Award (K. E. S. L.), and the National Natural Science Foundation of China, grant number 81772241 (Y. Q.). Dr Pathiraja Gunatillake is gratefully acknowledged for helpful discussions.Notes and references
- W. M. Dunne Jr, Clin. Microbiol. Rev., 2002, 15, 155–166 CrossRef.
- G. O'Toole, H. B. Kaplan and R. Kolter, Annu. Rev. Microbiol., 2000, 54, 49–79 CrossRef.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS.
- H. M. Dalton and P. E. March, Curr. Opin. Biotechnol, 1998, 9, 252–255 CrossRef CAS.
- T. Parween, P. Bhandari, Z. H. Siddiqui, S. Jan, T. Fatma and P. K. Patanjali, in Mycoremediation and Environmental Sustainability, ed. R. Prasad, Springer International Publishing, Cham, 2017, vol. 1, pp. 39–51 DOI:10.1007/978-3-319-68957-9_3.
- Z.-W. Wang and S. Chen, Appl. Microbiol. Biotechnol., 2009, 83, 1–18 CrossRef CAS.
- S. J. Edwards and B. V. Kjellerup, Appl. Microbiol. Biotechnol., 2013, 97, 9909–9921 CAS.
- H.-S. Joo and M. Otto, Chem. Biol., 2012, 19, 1503–1513 CrossRef CAS.
- M. Burmølle, T. R. Thomsen, M. Fazli, I. Dige, L. Christensen, P. Homøe, M. Tvede, B. Nyvad, T. Tolker-Nielsen, M. Givskov, C. Moser, K. Kirketerp-Møller, H. K. Johansen, N. Høiby, P. Ø. Jensen, S. J. Sørensen and T. Bjarnsholt, FEMS Immunol. Med. Microbiol., 2010, 59, 324–336 CrossRef.
- S. Furukawa, S. L. Kuchma and G. A. Toole, J. Bacteriol., 2006, 188, 1211 CAS.
- R. M. Donlan, Emerging Infect. Dis., 2001, 7, 277–281 CrossRef CAS.
- S. L. Percival, L. Suleman, C. Vuotto and G. Donelli, J. Med. Microbiol., 2015, 64, 323–334 CrossRef.
- P. W. Stone, Expert Rev. Pharmacoecon. Outcomes Res., 2009, 9, 417–422 CrossRef.
- P. D. Marsh, BMC Oral Health, 2006, 6, S14 CrossRef.
- A. Bridier, P. Sanchez-Vizuete, M. Guilbaud, J. C. Piard, M. Naïtali and R. Briandet, Food Microbiol., 2015, 45, 167–178 CrossRef CAS.
- S. Galié, C. García-Gutiérrez, E. M. Miguélez, C. J. Villar and F. Lombó, Front. Microbiol., 2018, 9, 898 CrossRef.
- I. B. Beech and J. Sunner, Curr. Opin. Biotechnol, 2004, 15, 181–186 CrossRef CAS.
- G. Caruso, J. Mar. Sci. Eng., 2020, 8, 78 CrossRef.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Front. Chem., 2019, 7, 824 CrossRef CAS.
- B. Parrino, D. Schillaci, I. Carnevale, E. Giovannetti, P. Diana, G. Cirrincione and S. Cascioferro, Eur. J. Med. Chem., 2019, 161, 154–178 CrossRef CAS.
- K. Qvortrup, L. D. Hultqvist, M. Nilsson, T. H. Jakobsen, C. U. Jansen, J. Uhd, J. B. Andersen, T. E. Nielsen, M. Givskov and T. Tolker-Nielsen, Front. Chem., 2019, 7, 742 CrossRef CAS.
- A. Al-Jumaili, S. Alancherry, K. Bazaka and M. V. Jacob, Materials, 2017, 10, 1066 CrossRef.
- M. Ramasamy and J. Lee, BioMed Res. Int., 2016, 2016, 1851242 Search PubMed.
- M.-H. Kim, IEEE Trans Nanobioscience, 2016, 15, 294–304 Search PubMed.
- K. P. Miller, L. Wang, B. C. Benicewicz and A. W. Decho, Chem. Soc. Rev., 2015, 44, 7787–7807 RSC.
- M. Salwiczek, Y. Qu, J. Gardiner, R. A. Strugnell, T. Lithgow, K. M. McLean and H. Thissen, Trends Biotechnol., 2014, 32, 82–90 CrossRef CAS.
- Z. K. Zander and M. L. Becker, ACS Macro Lett., 2018, 7, 16–25 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS.
- P. Singha, J. Locklin and H. Handa, Acta Biomater., 2017, 50, 20–40 CrossRef CAS.
- T. Murosaki, N. Ahmed and J. Ping-Gong, Sci. Technol. Adv. Mater., 2011, 12, 064706 CrossRef.
- Z. Wang, L. Scheres, H. Xia and H. Zuilhof, Adv. Funct. Mater., 2020, 30, 1908098 CrossRef CAS.
- V. B. Damodaran and N. S. Murthy, Biomater. Res., 2016, 20, 18 CrossRef.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- N. F. Kamaruzzaman, L. P. Tan, R. H. Hamdan, S. S. Choong, W. K. Wong, A. J. Gibson, A. Chivu and M. D. F. Pina, Int. J. Mol. Sci., 2019, 20, 2747 CrossRef CAS.
- Y. L. Qin, Int. J. Mol. Sci., 2020, 21, 499 CrossRef CAS.
- M. E. Davey and G. A. O'Toole, Microbiol. Mol. Biol. Rev., 2000, 64, 847–867 CrossRef CAS.
- B. M. Peters, M. A. Jabra-Rizk, G. A. O'May, J. W. Costerton and M. E. Shirtliff, Clin. Microbiol. Rev., 2012, 25, 193–213 CrossRef.
- M. M. Harriott and M. C. Noverr, Trends Microbiol., 2011, 19, 557–563 CrossRef CAS.
- J. Palmer, S. Flint and J. Brooks, J. Ind. Microbiol. Biotechnol., 2007, 34, 577–588 CrossRef CAS.
- Y. Qu, Y. Li, D. R. Cameron, C. D. Easton, X. Zhu, M. Zhu, M. Salwiczek, B. W. Muir, H. Thissen, A. Daley, J. S. Forsythe, A. Y. Peleg and T. Lithgow, Front. Microbiol., 2020, 11, 920 CrossRef.
- M. Otto, Gram-Positive Pathogens, 2019, pp. 699–711 DOI:10.1128/9781683670131.ch43.
- K. A. Kline, S. Fälker, S. Dahlberg, S. Normark and B. Henriques-Normark, Cell Host Microbe, 2009, 5, 580–592 CrossRef CAS.
- I. W. Sutherland, Microbiology, 2001, 147, 3–9 CrossRef CAS.
- C. Solano, M. Echeverz and I. Lasa, Curr. Opin. Microbiol., 2014, 18, 96–104 CrossRef CAS.
- M. R. Parsek and E. P. Greenberg, Trends Microbiol., 2005, 13, 27–33 CrossRef CAS.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef CAS.
- P. Albuquerque and A. Casadevall, Med. Mycol., 2012, 50, 337–345 CrossRef CAS.
- P. S. Stewart and J. William-Costerton, Lancet, 2001, 358, 135–138 CAS.
- G. M. Teitzel and M. R. Parsek, Appl. Environ. Microbiol., 2003, 69, 2313 CrossRef CAS.
- K. Lewis, Biochemistry, 2005, 70, 267–274 CAS.
- K. Lewis, in Bacterial Biofilms, ed. T. Romeo, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 107–131 DOI:10.1007/978-3-540-75418-3_6.
- M. D. LaFleur, C. A. Kumamoto and K. Lewis, Antimicrob. Agents Chemother., 2006, 50, 3839–3846 CrossRef CAS.
- E. F. Haney, M. J. Trimble, J. T. Cheng, Q. Vallé and R. E. W. Hancock, Biomolecules, 2018, 8, 29 CrossRef.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- D. Pletzer and R. E. W. Hancock, J. Bacteriol., 2016, 198, 2572 CrossRef CAS.
- L.-j. Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS.
- E. Galdiero, L. Lombardi, A. Falanga, G. Libralato, M. Guida and R. Carotenuto, Pharmaceutics, 2019, 11, 322 CrossRef CAS.
- M. F. Burton and P. G. Steel, Nat. Prod. Rep., 2009, 26, 1572–1584 RSC.
- S. C. Mansour, C. de la Fuente-Núñez and R. E. W. Hancock, J. Pept. Sci., 2015, 21, 323–329 CrossRef CAS.
- P. Chairatana and E. M. Nolan, Acc. Chem. Res., 2017, 50, 960–967 CrossRef CAS.
- J. B. Kaplan, K. LoVetri, S. T. Cardona, S. Madhyastha, I. Sadovskaya, S. Jabbouri and E. A. Izano, J. Antibiot., 2012, 65, 73–77 CrossRef CAS.
- K. Sharma and A. Pagedar Singh, Foods, 2018, 7, 42 CrossRef.
- J. B. Kaplan, Int. J. Artif. Organs, 2009, 32, 545–554 CrossRef CAS.
- N. Ramasubbu, L. M. Thomas, C. Ragunath and J. B. Kaplan, J. Mol. Biol., 2005, 349, 475–486 CrossRef CAS.
- S. A. C. Ragland and A. K. Criss, PLoS Pathog., 2017, 13, e1006512 CrossRef.
- C. Johansen, P. Falholt and L. Gram, Appl. Environ. Microbiol., 1997, 63, 3724–3728 CrossRef CAS.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS.
- G. Henriksson, G. Johansson and G. Pettersson, J. Biotechnol., 2000, 78, 93–113 CrossRef CAS.
- M. D. C. de Freire Bastos, B. G. Coutinho and M. L. V. Coelho, Pharmaceuticals, 2010, 3, 1139–1161 CrossRef.
- J. A. Wu, C. Kusuma, J. J. Mond and J. F. Kokai-Kun, Antimicrob. Agents Chemother., 2003, 47, 3407–3414 CrossRef CAS.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS.
- S. Kumar Shukla and T. S. Rao, J. Antibiot., 2013, 66, 55–60 CrossRef CAS.
- D. M. Roche, J. T. Byers, D. S. Smith, F. G. Glansdorp, D. R. Spring and M. Welch, Microbiology, 2004, 150, 2023–2028 CrossRef CAS.
- E. Paluch, J. Rewak-Soroczyńska, I. Jędrusik, E. Mazurkiewicz and K. Jermakow, Appl. Microbiol. Biotechnol., 2020, 104, 1871–1881 CrossRef CAS.
- D. Raafat, M. Otto, K. Reppschläger, J. Iqbal and S. Holtfreter, Trends Microbiol., 2019, 27, 303–322 CrossRef CAS.
- K. E. S. Locock, Aust. J. Chem., 2016, 69, 717–724 CrossRef CAS.
- Y. Qu, K. Locock, J. Verma-Gaur, I. D. Hay, L. Meagher and A. Traven, J. Antimicrob. Chemother., 2015, 71, 413–421 CrossRef.
- A. Al-Ahmad, D. Laird, P. Zou, P. Tomakidi, T. Steinberg and K. Lienkamp, PLoS One, 2013, 8, e73812 CrossRef CAS.
- M. Álvarez-Paino, A. Muñoz-Bonilla and M. Fernández-García, Nanomaterials, 2017, 7, 48 CrossRef.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., 2019, 131, 3728–3731 Search PubMed.
- B. A. Chan, S. Xuan, A. Li, J. M. Simpson, G. L. Sternhagen, T. Yu, O. A. Darvish, N. Jiang and D. Zhang, Biopolymers, 2018, 109, e23070 CrossRef.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS.
- R. Gomes Von Borowski, S. C. B. Gnoatto, A. J. Macedo and R. Gillet, Front. Microbiol., 2018, 9, 2157 CrossRef.
- F. Rong, Y. Tang, T. Wang, T. Feng, J. Song, P. Li and W. Huang, Antioxidants, 2019, 8, 556 CrossRef CAS.
- Z. Sadrearhami, T.-K. Nguyen, R. Namivandi-Zangeneh, K. Jung, E. H. H. Wong and C. Boyer, J. Mater. Chem. B, 2018, 6, 2945–2959 RSC.
- L. Yang, E. S. Feura, M. J. R. Ahonen and M. H. Schoenfisch, Adv. Healthcare Mater., 2018, 7, e1800155 CrossRef.
- L. Chen, Y. Yang, P. Zhang, S. Wang, J.-F. Xu and X. Zhang, ACS Appl. Bio Mater., 2019, 2, 2920–2926 CrossRef CAS.
- B. S. T. Peddinti, F. Scholle, R. A. Ghiladi and R. J. Spontak, ACS Appl. Mater. Interfaces, 2018, 10, 25955–25959 CrossRef CAS.
- Y. Wang, Y. Jin, W. Chen, J. Wang, H. Chen, L. Sun, X. Li, J. Ji, Q. Yu, L. Shen and B. Wang, Chem. Eng. J., 2019, 358, 74–90 CrossRef CAS.
- L. T. Lui, X. Xue, C. Sui, A. Brown, D. I. Pritchard, N. Halliday, K. Winzer, S. M. Howdle, F. Fernandez-Trillo, N. Krasnogor and C. Alexander, Nat. Chem., 2013, 5, 1058–1065 CrossRef CAS.
- E. Cavaleiro, A. S. Duarte, A. C. Esteves, A. Correia, M. J. Whitcombe, E. V. Piletska, S. A. Piletsky and I. Chianella, Macromol. Biosci., 2015, 15, 647–656 CrossRef CAS.
- L. D. Blackman, Z. Y. Oo, Y. Qu, P. A. Gunatillake, P. Cass and K. E. S. Locock, ACS Appl. Mater. Interfaces, 2020, 12, 11353–11362 CrossRef CAS.
- H. Sun, Y. Hong, Y. Xi, Y. Zou, J. Gao and J. Du, Biomacromolecules, 2018, 19, 1701–1720 CrossRef CAS.
- Y. Tan, S. Ma, M. Leonhard, D. Moser, R. Ludwig and B. Schneider-Stickler, Mater. Sci. Eng., C, 2020, 108, 110499 CrossRef CAS.
- Y. Tan, S. Ma, C. Liu, W. Yu and F. Han, Microbiol. Res., 2015, 178, 35–41 CrossRef CAS.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS.
- K.-i. Okuda, T. Zendo, S. Sugimoto, T. Iwase, A. Tajima, S. Yamada, K. Sonomoto and Y. Mizunoe, Antimicrob. Agents Chemother., 2013, 57, 5572 CrossRef CAS.
- J. Overhage, A. Campisano, M. Bains, E. C. W. Torfs, B. H. A. Rehm and R. E. W. Hancock, Infect. Immun., 2008, 76, 4176 CrossRef CAS.
- M. Yasir, D. M. Willcox and D. Dutta, Materials, 2018, 11, 2468 CrossRef.
- E. F. Haney and R. E. W. Hancock, Biopolymers, 2013, 100, 572–583 CrossRef CAS.
- M. Wieczorek, H. Jenssen, J. Kindrachuk, W. R. P. Scott, M. Elliott, K. Hilpert, J. T. J. Cheng, R. E. W. Hancock and S. K. Straus, Chem. Biol., 2010, 17, 970–980 CrossRef CAS.
- F. Reffuveille, C. de la Fuente-Núñez, S. Mansour and R. E. W. Hancock, Antimicrob. Agents Chemother., 2014, 58, 5363–5371 CrossRef.
- C. de la Fuente-Núñez, F. Reffuveille, E. F. Haney, S. K. Straus and R. E. W. Hancock, PLoS Pathog., 2014, 10, e1004152 CrossRef.
- M. Vazdar, J. Heyda, P. E. Mason, G. Tesei, C. Allolio, M. Lund and P. Jungwirth, Acc. Chem. Res., 2018, 51, 1455–1464 CrossRef CAS.
- J. M. Ansari, N. M. Abraham, J. Massaro, K. Murphy, J. Smith-Carpenter and E. Fikrig, Front. Microbiol., 2017, 8, 488 Search PubMed.
- P. Chairatana and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 13267–13276 CrossRef CAS.
- P. Chairatana, I. L. Chiang and E. M. Nolan, Biochemistry, 2017, 56, 1033–1041 CrossRef CAS.
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, Am. J. Transl. Res., 2019, 11, 3919–3931 CAS.
- B. Thallinger, E. N. Prasetyo, G. S. Nyanhongo and G. M. Guebitz, Biotechnol. J., 2013, 8, 97–109 CrossRef CAS.
- T. Iwase, Y. Uehara, H. Shinji, A. Tajima, H. Seo, K. Takada, T. Agata and Y. Mizunoe, Nature, 2010, 465, 346–349 CrossRef CAS.
- J.-H. Park, J.-H. Lee, M. H. Cho, M. Herzberg and J. Lee, FEMS Microbiol. Lett., 2012, 335, 31–38 CrossRef CAS.
- W. Vollmer, D. Blanot and M. A. De Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS.
- K. C. Costa, N. R. Glasser, S. J. Conway and D. K. Newman, Science, 2017, 355, 170 CrossRef CAS.
- J. B. Kaplan, C. Ragunath, K. Velliyagounder, D. H. Fine and N. Ramasubbu, Antimicrob. Agents Chemother., 2004, 48, 2633–2636 CrossRef CAS.
- P. Baker, P. J. Hill, B. D. Snarr, N. Alnabelseya, M. J. Pestrak, M. J. Lee, L. K. Jennings, J. Tam, R. A. Melnyk, M. R. Parsek, D. C. Sheppard, D. J. Wozniak and P. L. Howell, Sci. Adv., 2016, 2, e1501632 CrossRef.
- P. V. Gupta, A. M. Nirwane and M. S. Nagarsenker, AAPS PharmSciTech, 2018, 19, 1454–1467 CrossRef CAS.
- B. Thallinger, M. Argirova, M. Lesseva, R. Ludwig, C. Sygmund, A. Schlick, G. S. Nyanhongo and G. M. Guebitz, Int. J. Antimicrob. Agents, 2014, 44, 402–408 CrossRef CAS.
- G. S. Nyanhongo, B. Thallinger and G. M. Guebitz, Process Biochem., 2017, 59, 37–45 CrossRef CAS.
- K. Hellmuth and J. M. van den Brink, in Microbial Production of Food Ingredients, Enzymes and Nutraceuticals, ed. B. McNeil, D. Archer, I. Giavasis and L. Harvey, Woodhead Publishing, 2013, pp. 262–287 DOI:10.1533/9780857093547.2.262.
- S. E. Adams, D. Arnold, B. Murphy, P. Carroll, A. K. Green, A. M. Smith, P. D. Marsh, T. Chen, R. E. Marriott and M. G. Brading, Sci. Rep., 2017, 7, 43344 CrossRef CAS.
- M. Bucekova, I. Valachova, L. Kohutova, E. Prochazka, J. Klaudiny and J. Majtan, Naturwissenschaften, 2014, 101, 661–670 CrossRef CAS.
- Z. H. Israili, Am. J. Ther., 2014, 21, 304–323 CrossRef.
- A. Sindi, M. V. B. Chawn, M. E. Hernandez, K. Green, M. K. Islam, C. Locher and K. Hammer, Sci. Rep., 2019, 9, 17666 CrossRef.
- M. Divya, B. Vaseeharan, M. Anjugam, A. Iswarya, S. Karthikeyan, P. Velusamy, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, J. M. Khaled and C. Vágvölgyi, Int. J. Biol. Macromol., 2018, 114, 864–873 CrossRef CAS.
- S. Jayanthi, B. Vaseeharan, R. Ishwarya, S. Karthikeyan, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, J. M. Khaled and C. Vágvölgyi, Int. J. Biol. Macromol., 2018, 113, 996–1007 CrossRef CAS.
- C. B. Whitchurch, T. Tolker-Nielsen, P. C. Ragas and J. S. Mattick, Science, 2002, 295, 1487 CrossRef CAS.
- J. S. Kavanaugh, C. E. Flack, J. Lister, E. B. Ricker, C. B. Ibberson, C. Jenul, D. E. Moormeier, E. A. Delmain, K. W. Bayles and A. R. Horswill, mBio, 2019, 10, e01137 CrossRef CAS.
- E. S. Gloag, L. Turnbull, A. Huang, P. Vallotton, H. Wang, L. M. Nolan, L. Mililli, C. Hunt, J. Lu, S. R. Osvath, L. G. Monahan, R. Cavaliere, I. G. Charles, M. P. Wand, M. L. Gee, R. Prabhakar and C. B. Whitchurch, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11541–11546 CrossRef CAS.
- E. A. Izano, M. A. Amarante, W. B. Kher and J. B. Kaplan, Appl. Environ. Microbiol., 2008, 74, 470 CrossRef CAS.
- S. Molin and T. Tolker-Nielsen, Curr. Opin. Biotechnol, 2003, 14, 255–261 CrossRef CAS.
- C. J. H. von Wintersdorff, J. Penders, J. M. van Niekerk, N. D. Mills, S. Majumder, L. B. van Alphen, P. H. M. Savelkoul and P. F. G. Wolffs, Front. Microbiol., 2016, 7, 173 Search PubMed.
- J. B. Kaplan, K. LoVetri, S. T. Cardona, S. Madhyastha, I. Sadovskaya, S. Jabbouri and E. A. Izano, J. Antibiot., 2012, 65, 73–77 CrossRef CAS.
- R. Pei and G. R. Lamas-Samanamud, Appl. Environ. Microbiol., 2014, 80, 5340 CrossRef.
- D. R. Harper, H. M. R. T. Parracho, J. Walker, R. Sharp, G. Hughes, M. Werthén, S. Lehman and S. Morales, Antibiotics, 2014, 3, 270–284 CrossRef.
- D. Paul, Y. S. Kim, K. Ponnusamy and J. H. Kweon, Environ. Eng. Sci., 2009, 26, 1319–1324 CrossRef CAS.
- B. LaSarre and M. J. Federle, Microbiol. Mol. Biol. Rev., 2013, 77, 73–111 CrossRef CAS.
- S. G. Dashper, Y. Pan, P. D. Veith, Y. Y. Chen, E. C. Toh, S. W. Liu, K. J. Cross and E. C. Reynolds, Antimicrob. Agents Chemother., 2012, 56, 1548–1556 CrossRef CAS.
- S. J. Lam, E. H. H. Wong, C. Boyer and G. G. Qiao, Prog. Polym. Sci., 2018, 76, 40–64 CrossRef CAS.
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS.
- X. Wu, S. Zhang, X. Xu, L. Shen, B. Xu, W. Qu, W. Zhuang, K. Locock, M. Deighton and Y. Qu, Front. Microbiol., 2019, 10, 2592 CrossRef.
- S. Barman, S. Mukherjee, S. Ghosh and J. Haldar, ACS Appl. Bio Mater., 2019, 2, 5404–5414 CrossRef CAS.
- R. Namivandi-Zangeneh, Y. Yang, S. Xu, E. H. H. Wong and C. Boyer, Biomacromolecules, 2020, 21, 262–272 CrossRef CAS.
- J. P. K. Tan, D. J. Coady, H. Sardon, A. Yuen, S. Gao, S. W. Lim, Z. C. Liang, E. W. Tan, S. Venkataraman, A. C. Engler, M. Fevre, R. Ono, Y. Y. Yang and J. L. Hedrick, Adv. Healthcare Mater., 2017, 6, 1601420 CrossRef.
- H. Takahashi, E. T. Nadres and K. Kuroda, Biomacromolecules, 2017, 18, 257–265 CrossRef CAS.
- W. Siala, F. Â. o. Van Bambeke, V. Taresco, A. Piozzi and I. Francolini, Pathog. Dis., 2016, 74, ftw042 CrossRef.
- A. Ivanova, K. Ivanova, J. Hoyo, T. Heinze, S. Sanchez-Gomez and T. Tzanov, ACS Appl. Mater. Interfaces, 2018, 10, 3314–3323 CrossRef CAS.
- J. Li, K. Zhang, L. Ruan, S. F. Chin, N. Wickramasinghe, H. Liu, V. Ravikumar, J. Ren, H. Duan, L. Yang and M. B. Chan-Park, Nano Lett., 2018, 18, 4180–4187 CrossRef CAS.
- M. N. V. Ravi Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- J. Cao, Y. Zhao, Y. Liu, S. Tian, C. Zheng, C. Liu, Y. Zhai, Y. An, H. J. Busscher, L. Shi and Y. Liu, ACS Macro Lett., 2019, 651–657, DOI:10.1021/acsmacrolett.9b00142.
- E. M. Costa, S. Silva, S. Vicente, C. Neto, P. M. Castro, M. Veiga, R. Madureira, F. Tavaria and M. M. Pintado, Mater. Sci. Eng., C, 2017, 79, 221–226 CrossRef CAS.
- A. Guo, F. Wang, W. Lin, X. Xu, T. Tang, Y. Shen and S. Guo, Int. J. Biol. Macromol., 2014, 67, 163–171 CrossRef CAS.
- P. Sahariah, M. Másson and R. L. Meyer, Biomacromolecules, 2018, 19, 3649–3658 CrossRef CAS.
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem., 2014, 5, 5813–5822 RSC.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS.
- P. R. Judzewitsch, T. K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559–4564 CrossRef CAS.
- J. L. Grace, J. X. Huang, S.-E. Cheah, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, RSC Adv., 2016, 6, 15469–15477 RSC.
- E. F. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS.
- S.-J. Richards, A. Jones, R. M. F. Tomás and M. I. Gibson, Chem. – Eur. J., 2018, 24, 13758–13761 CrossRef CAS.
- B. Horev, M. I. Klein, G. Hwang, Y. Li, D. Kim, H. Koo and D. S. W. Benoit, ACS Nano, 2015, 9, 2390–2404 CrossRef CAS.
- R. F. Landis, C.-H. Li, A. Gupta, Y.-W. Lee, M. Yazdani, N. Ngernyuang, I. Altinbasak, S. Mansoor, M. A. S. Khichi, A. Sanyal and V. M. Rotello, J. Am. Chem. Soc., 2018, 140, 6176–6182 CrossRef CAS.
- B. Porsio, M. G. Cusimano, D. Schillaci, E. F. Craparo, G. Giammona and G. Cavallaro, Biomacromolecules, 2017, 18, 3924–3935 CrossRef CAS.
- N. Raman, M.-R. Lee, D. M. Lynn and S. P. Palecek, Pharmaceuticals, 2015, 8, 403–583 CrossRef.
- Y. Liu, K. M. Knapp, L. Yang, S. Molin, H. Franzyk and A. Folkesson, Int. J. Antimicrob. Agents, 2013, 41, 20–27 CrossRef.
- F. Liu, D. He, Y. Yu, L. Cheng and S. Zhang, Bioconjugate Chem., 2019, 30, 541–546 CrossRef CAS.
- A. V. Dubois, P. Midoux, D. Gras, M. Si-Tahar, D. Bréa, S. Attucci, M.-K. Khelloufi, R. Ramphal, P. Diot, F. Gauthier and V. Hervé, Am. J. Respir. Crit. Care Med., 2013, 188, 703–709 CrossRef CAS.
- A. Guillon, D. Fouquenet, E. Morello, C. Henry, S. Georgeault, M. Si-Tahar and V. Hervé, Antimicrob. Agents Chemother., 2018, 62, e00564 CrossRef.
- Y. Luo, H. L. Bolt, G. A. Eggimann, D. F. McAuley, R. McMullan, T. Curran, M. Zhou, P. C. A. B. Jahoda, S. L. Cobb and F. T. Lundy, ChemBioChem, 2017, 18, 111–118 CrossRef CAS.
- P. Saporito, B. Mojsoska, A. Løbner Olesen and H. Jenssen, Biopolymers, 2019, 110, e23275 CrossRef.
- A. J. Karlsson, W. C. Pomerantz, K. J. Neilsen, S. H. Gellman and S. P. Palecek, ACS Chem. Biol., 2009, 4, 567–579 CrossRef CAS.
- A. d. L. Rodríguez López, M.-R. Lee, N. B. Wang, K. K. Dunn, H. Sanchez, N. Raman, D. R. Andes, D. M. Lynn and S. P. Palecek, Antimicrob. Agents Chemother., 2019, 63, e02653 CrossRef.
- H. T. T. Duong, K. Jung, S. K. Kutty, S. Agustina, N. N. M. Adnan, J. S. Basuki, N. Kumar, T. P. Davis, N. Barraud and C. Boyer, Biomacromolecules, 2014, 15, 2583–2589 CrossRef CAS.
- B. V. Worley, K. M. Schilly and M. H. Schoenfisch, Mol. Pharmaceutics, 2015, 12, 1573–1583 CrossRef CAS.
- R. Namivandi-Zangeneh, Z. Sadrearhami, A. Bagheri, M. Sauvage-Nguyen, K. K. K. Ho, N. Kumar, E. H. H. Wong and C. Boyer, ACS Macro Lett., 2018, 7, 592–597 CrossRef CAS.
- T.-K. Nguyen, R. Selvanayagam, K. K. K. Ho, R. Chen, S. K. Kutty, S. A. Rice, N. Kumar, N. Barraud, H. T. T. Duong and C. Boyer, Chem. Sci., 2016, 7, 1016–1027 RSC.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy, S. Reghu, K. Marimuthu, P. P. De, O. T. Ng, J. R. Mediavilla, B. N. Kreiswirth, Y. R. Chi, J. Ren, K. C. Tam, X.-W. Liu, H. Duan, Y. Zhu, Y. Mu, P. T. Hammond, G. C. Bazan, K. Pethe and M. B. Chan-Park, Nat. Commun., 2019, 10, 4792 CrossRef.
- F. C. Fang, J. Clin. Invest., 1997, 99, 2818–2825 CrossRef CAS.
- Z. Sadrearhami, J. Yeow, T.-K. Nguyen, K. K. K. Ho, N. Kumar and C. Boyer, Chem. Commun., 2017, 53, 12894–12897 RSC.
- K. P. Reighard and M. H. Schoenfisch, Antimicrob. Agents Chemother., 2015, 59, 6506–6513 CrossRef CAS.
- M. J. R. Ahonen, J. M. Dorrier and M. H. Schoenfisch, ACS Infect. Dis., 2019, 5, 1327–1335 CrossRef CAS.
- Y. Xi, Y. Wang, J. Gao, Y. Xiao and J. Du, ACS Nano, 2019, 13, 13645–13657 CrossRef CAS.
- M. Chen, J. Wei, S. Xie, X. Tao, Z. Zhang, P. Ran and X. Li, Nanoscale, 2019, 11, 1410–1422 RSC.
- Y. Liu, H. J. Busscher, B. Zhao, Y. Li, Z. Zhang, H. C. van der Mei, Y. Ren and L. Shi, ACS Nano, 2016, 10, 4779–4789 CrossRef CAS.
- D. Y. Zhu, R. F. Landis, C.-H. Li, A. Gupta, L.-S. Wang, Y. Geng, S. Gopalakrishnan, J. W. Guo and V. M. Rotello, Nanoscale, 2018, 10, 18651–18656 RSC.
- P. Jorge, D. Alves and M. O. Pereira, Int. J. Antimicrob. Agents, 2019, 53, 598–605 CrossRef CAS.
- L. Fan, J. Sun, M. Zhou, J. Zhou, X. Lao, H. Zheng and H. Xu, Sci. Rep., 2016, 6, 24482 CrossRef CAS.
- X. Kang, F. Dong, C. Shi, S. Liu, J. Sun, J. Chen, H. Li, H. Xu, X. Lao and H. Zheng, Sci. Data, 2019, 6, 148 CrossRef.
- P. Zhang, S. Li, H. Chen, X. Wang, L. Liu, F. Lv and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16933–16938 CrossRef CAS.
- L. Chen, D. Yang, J. Feng, M. Zhang, Q. Qian and Y. Zhou, J. Mater. Chem. B, 2019, 7, 6420–6427 RSC.
- B. H. Neufeld and M. M. Reynolds, Biointerphases, 2016, 11, 031012 CrossRef.
- Z. Shen, K. He, Z. Ding, M. Zhang, Y. Yu and J. Hu, Macromolecules, 2019, 52, 7668–7677 CrossRef CAS.
- J. Shepherd, T. Swift, C.-Y. Chang, J. R. Boyne, S. Rimmer and W. H. C. Martin, Chem. Commun., 2019, 55, 9765–9768 RSC.
- E. V. Piletska, G. Stavroulakis, L. D. Larcombe, M. J. Whitcombe, A. Sharma, S. Primrose, G. K. Robinson and S. A. Piletsky, Biomacromolecules, 2011, 12, 1067–1071 CrossRef CAS.
- N. Perez-Soto, O. Creese, F. Fernandez-Trillo and A.-M. Krachler, ACS Chem. Biol., 2018, 13, 3021–3029 CrossRef CAS.
- N. Perez-Soto, L. Moule, D. N. Crisan, I. Insua, L. M. Taylor-Smith, K. Voelz, F. Fernandez-Trillo and A. M. Krachler, Chem. Sci., 2017, 8, 5291–5298 RSC.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O’Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS.
- K.-M. Yeon, J. You, M. D. Adhikari, S.-G. Hong, I. Lee, H. S. Kim, L. N. Kim, J. Nam, S.-J. Kwon, M. I. Kim, W. Sajomsang, J. S. Dordick and J. Kim, Biomacromolecules, 2019, 20, 2477–2485 CrossRef CAS.
- A. Baelo, R. Levato, E. Julián, A. Crespo, J. Astola, J. Gavaldà, E. Engel, M. A. Mateos-Timoneda and E. Torrents, J. Controlled Release, 2015, 209, 150–158 CrossRef CAS.
- M. Varache, L. C. Powell, O. A. Aarstad, T. L. Williams, M. N. Wenzel, D. W. Thomas and E. L. Ferguson, Mol. Pharmaceutics, 2019, 16, 3199–3207 CrossRef CAS.
- R. A. Fenati, K. Locock, Y. Qu and A. V. Ellis, ACS Appl. Bio Mater., 2019, 2, 3002–3008 CrossRef CAS.
- Z. Chen, Z. Wang, J. Ren and X. Qu, Acc. Chem. Res., 2018, 51, 789–799 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O’Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS.
- E. L. Ferguson, E. Azzopardi, J. L. Roberts, T. R. Walsh and D. W. Thomas, Mol. Pharmaceutics, 2014, 11, 4437–4447 CrossRef CAS.
- E. L. Ferguson and R. Duncan, Biomacromolecules, 2009, 10, 1358–1364 CrossRef CAS.
- J. Hardwicke, E. L. Ferguson, R. Moseley, P. Stephens, D. W. Thomas and R. Duncan, J. Controlled Release, 2008, 130, 275–283 CrossRef CAS.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS.
- A. Chokshi, Z. Sifri, D. Cennimo and H. Horng, J. Global Infect. Dis., 2019, 11, 36–42 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |