
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular and heterogeneous water oxidation catalysts: recent progress and joint perspectives
J.
Li
 ,
C. A.
Triana
,
W.
Wan
,
D. P.
Adiyeri Saseendran
,
Y.
Zhao
,
S. E.
Balaghi
,
S.
Heidari
and
G. R.
Patzke
*
,
C. A.
Triana
,
W.
Wan
,
D. P.
Adiyeri Saseendran
,
Y.
Zhao
,
S. E.
Balaghi
,
S.
Heidari
and
G. R.
Patzke
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: greta.patzke@chem.uzh.ch
First published on 6th January 2021
Abstract
The development of reliable water oxidation catalysts (WOCs) is essential for implementing artificial photosynthesis on a large technological scale. WOC research has evolved into two major branches, namely molecular and heterogeneous catalysts. Manifold design principles and plenty of mechanistic insights have been developed in these individual fields after decades of investigations. Over the past years, a growing need for knowledge transfer between both sides has emerged in order to expedite the development and optimization of next-generation WOCs. In this review, we first provide selected recent highlights in the area of molecular WOCs with different nuclearities, together with current mechanistic insight. WOCs offering molecular integrity under operational conditions are ideal platforms for elucidating reaction mechanisms and well-defined structure–function correlations at the atomic level. Next, recent mechanistic advances and design strategies for heterogeneous WOCs are illustrated for representative examples, together with a discussion of their inherent limitations in mechanistic studies. Finally, illustrative cases of knowledge transfer between molecular and heterogeneous WOCs are discussed to highlight the advantages of combining the best of both catalyst types. For the sake of conciseness, this review focuses primarily on WOCs based on the first-row transition metals, which are attracting increasing attention for both fundamental studies and economic applications.
 J. Li | Jingguo Li is a PhD student at the University of Zurich in the group of Prof. Greta R. Patzke. He obtained his BSc Degree from Henan University in 2010, and his MSc Degree from the University of Science and Technology of China in 2013. Subsequently, he worked as a Research Engineer at the National University of Singapore, developing polymeric thin films for water purification applications. Then he moved to UZH in 2016 and is currently focusing on photo(electrochemical) water oxidation research. |
 C. A. Triana | Carlos A. Triana received his BSc degree in Physics in 2010 and MSc in Science-Physics in 2013 from the National University of Colombia. In the same year, he started his studies in Solid State Physics at Uppsala University, Sweden, and received his Lic. Phil. degree in 2015 and his PhD in 2017. Currently, he is a postdoctoral researcher at the Department of Chemistry, University of Zurich. His research interests focus on the implementation of computational methods, theoretical and experimental approaches to understand the structure, transport, magnetic, optical, electronic properties and application of solid-state nanostructures, metal oxides, thin films and hybrid organic–inorganic systems. |
 W. Wan | Wenchao Wan received his MSc in Materials Science and Engineering from Southwest Petroleum University in 2016. Then he joined Prof. Greta R. Patzke's group to pursue his PhD degree at the University of Zurich. His current research focuses on the design, synthesis and characterization of new types of graphene-based catalysts for energy storage and conversion with special emphasis on single-atom catalysts for electrocatalytic applications. |
 D. P. Adiyeri Saseendran | Devi Prasad received his BS-MS Dual Degree in Chemistry from the Indian Institute of Science Education and Research, Bhopal in 2018, working on solid state materials and crystallography. Then he moved to Prof. Greta R. Patzke's research group at the University of Zurich for his PhD studies. His present research focuses on the synthesis, structural tuning and in situ mechanistic investigations of multi-nuclear molecular water oxidation catalysts using combined synchrotron X-ray and conventional spectroscopic techniques, together with their immobilization on metal oxide surfaces for photo/electrocatalytic water oxidation. |
 Y. Zhao | Yonggui Zhao received his BSc in Electrical Engineering from Northeastern University (2014) and his MSc in Solid-state Electronics from Tianjin University (2017). After working as a Junior Researcher in experimental physics at the University of Amsterdam, he joined the group of Prof. Greta R. Patzke as a PhD candidate in University of Zurich (December 2017). He is currently working on nanostructured materials in electrocatalysis. |
 S. E. Balaghi | S. Esmael Balaghi received his BSc in Chemistry from Shiraz University and MSc in Inorganic Chemistry from the Institute for Advanced Studies in Basic Sciences (IASBS). In 2016, he joined Prof. Greta R. Patzke's research group as a PhD candidate at the University of Zurich. He is currently working on in situ electrochemical studies of water splitting catalysts with different operando techniques such as X-ray absorption spectroscopy and in situ electrochemical liquid-cell electron microscopy methods. |
 S. Heidari | Sima Heidari is a Postdoctoral Researcher at the University of Zurich in the group of Prof. Greta R. Patzke. She received her MSc from the Institute for Advanced Studies in Basic Sciences (IASBS) in 2012, and her PhD degree from the University of Tabriz in 2017. Her current research is focused on developing earth-abundant transition metal-based electrocatalysts for the water oxidation reaction. |
 G. R. Patzke | Greta R. Patzke received her PhD from the University of Hannover and her venia legendi at ETH Zurich. She then moved to the University of Zurich, where she was promoted to Full Professor in 2016. Her research interests include the hydrothermal synthesis and monitoring of nanomaterials for environmental applications, together with the catalytic properties and bio-medical applications of polyoxometalates. She is a Board Member of the UZH Research Priority Program “Light to Chemical Energy Conversion”, where her group investigates a wide range of molecular, nanostructured and solid-state transition metal-based catalysts for water oxidation. |
The quest for reliable water oxidation catalysts (WOCs)
Finding efficient ways to capture and store renewable solar energy in chemical bonds through artificial photosynthesis (AP), e.g. water splitting, is currently one of the most competitive and intensely researched topics.1,2 To date, the large-scale production of renewable hydrogen via AP is significantly constrained by the lack of efficient, low cost and durable WOCs.3,4 This is due to the many unresolved fundamental issues of the energetically and kinetically demanding four-electron transfer oxygen evolution reaction (OER):| 2H2O → 4H+ + 4e− + O2 E0 = 1.23 V vs. NHE |
Further, limiting factors in both molecular and heterogeneous WOC research include the lack of operational durability on the molecular side and insufficient molecular-level understanding of heterogeneous catalysts on the other side.13 Thus, to promote progress on both fronts, innovative ways to solve these inherent problems are required, together with new strategies for reciprocal knowledge transfer. A common starting point is the involvement of equivalent reactive moieties in both molecular and heterogeneous WOCs at the atomic level.14 Specifically, the surface of a finite particle offers localized molecular centers for catalytic reactions, while some key molecular motifs are essentially surface cut-outs of solids. These structural similarities clearly link both fields for a mutual transfer of insight, which is beneficial and indispensable for optimizing the performance and durability of WOCs in general.
In this review, we discuss new strategies for knowledge transfer between both molecular and heterogeneous WOC research after a survey of their recent individual research progress (with main focus on first-row transition metal (TM)-based systems of application-oriented and fundamental interest). In Part I, the advantages of molecular WOCs are highlighted in terms of their nuclearity, mechanistic understanding and clear structure–activity correlations. This enables the targeted design of molecular WOCs with desired functionality, such as mimics of Nature's photosystem II (PSII). The spectrum of current optimization strategies such as engineering the metal nuclearity, tuning the electronic properties of the surrounding ligands, and introducing intramolecular proton regulation sites is concisely summarized. In addition, the importance of molecular integrity is addressed. Part II starts with advances in the mechanistic exploration of heterogeneous WOCs, taking Co- and Fe-oxides as representative and deeply explored examples of transition metal-based WOCs. We highlight the dynamic reaction pathways that are associated with material properties and experimental conditions. Further, emerging heterogeneous optimization strategies such as substitution of secondary elements, vacancies, lattice strain and crystal planes are discussed. Additionally, we also acknowledge the controversial topics of surface amorphization and real active centers of some classic heterogeneous WOCs. Finally, in Part III, the trends in the mutual knowledge transfer between both sides are reviewed. We begin with exploring the optimization of heterogeneous WOCs as tools for the molecular realm, such as the translation of reactive site motifs and application of the scaling relations. Furthermore, several molecular strategies emerging from heterogenized single-atom catalyst systems as models are introduced, such as structural features of the support and catalyst, coordination engineering and cooperative metal centers. Finally, emerging joint molecular and heterogeneous systems are discussed to highlight the advantages of combining both fields directly.
Part I: recent developments and mechanistic studies of molecular WOCs
1.1. Molecular WOCs as platforms for accessing reaction mechanisms
Recently, X-ray-free electron laser (XFEL) diffraction was used to solve the structure of metastable intermediates in the Mn4CaO5 complex of Nature's PSII at increased atomic resolutions of 1.85–1.87 Å.19 These low-dose XFEL experiments showed the presence of two environments around two monomers with different hydrogen bonding in the dimeric Mn4CaO5 moiety (Fig. 1a). In another representative study, the high-energy X-ray pair distribution function (PDF) was used to resolve the structure of an amorphous iridium-oxide WOC. The authors suggested that the peak at 3.07 Å in the PDF (Fig. 1b)20 resembles the local structures found in Cp*Ir(μ-O)3IrCp*, Ir2Cl2(1,5-cyclooctadiene)2(μ-OH)2(μ-O), and alkoxy derivatives featuring non-planar (μ-O(R))2 geometry. According to these results, the authors proposed that the local structure of amorphous Ir-oxide WOCs comprises distorted Ir(μ-O)3Ir/Ir(μ-O)2Ir substructures. A similar study on active Cp*IrIII complexes (Cp* = pentamethyl-cyclopentadienyl)20,21 showed that the local structure of these complexes features IrIV-dimers of mono-μ-O cores with terminal anionic ligands (Fig. 1c).21 Recently, some of us applied reverse Monte Carlo (RMC) modeling of PDF data to unravel the atomic range structure of a disordered 1D cobalt coordination polymer catalyst (Co-dppeO2).22 Our results revealed that the structure of Co-dppeO2 consists of hydrated edge-site {H2O–Co2(OH)2–OH2} motifs, nested within an organic matrix of bridging hydrophobic dppeO2 ligands (Fig. 1e).
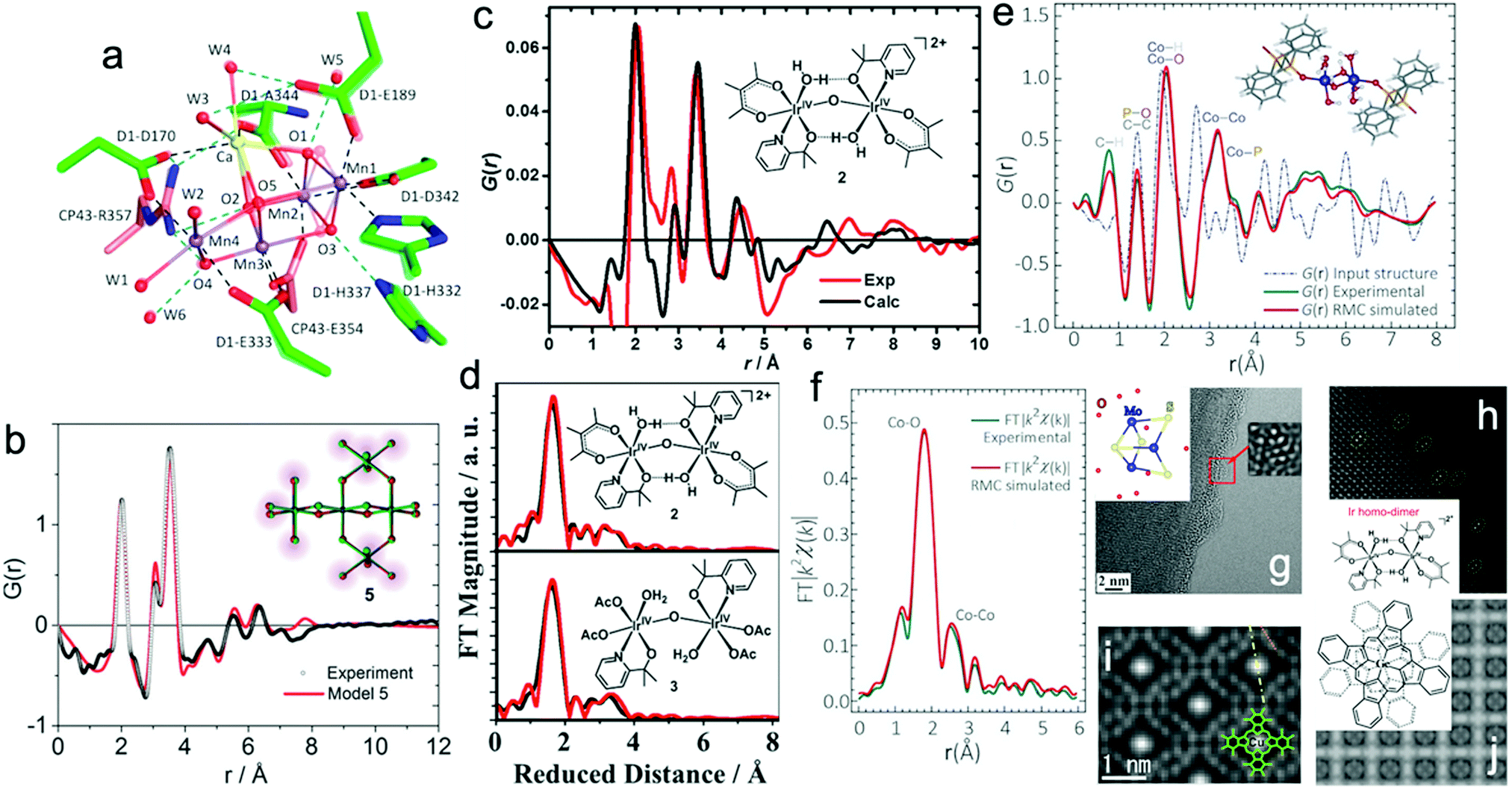 | ||
| Fig. 1 (a) Superposition of two different structures around the Mn4CaO5 cluster derived from low-dose XRD-XFEL experiments. Reproduced with permission from ref. 19. Copyright 2017 The American Chemical Society. (b) High-energy G(r) function (PDF) for the structure of an amorphous Ir-based WOC. Reproduced with permission from ref. 20. Copyright 2014 The Royal Society of Chemistry. (c) and (d) Experimental and calculated G(r) function and EXAFS spectra of a Cp*IrIII WOC for chemical and electrochemical activation. Reproduced with permission from ref. 21. Copyright 2016 The American Chemical Society. (e) and (f) Experimental and calculated G(r) function and EXAFS spectra of Co-dppeO2. Reproduced with permission from ref. 22. Copyright 2019 Elsevier. (g) Low dose-rate HRTEM image for the incomplete cubane-cluster [Mo3S4(H2O)9]Cl4. Reproduced with permission from ref. 32. Copyright 2020 Elsevier. (h) HAADF-STEM image of an immobilized molecular dinuclear Ir WOC on an α-Fe2O3 support. Reproduced with permission from ref. 31. Copyright 2018 National Academy of Sciences. (i) LAADF-STEM image of Cl16-CuPc. Reproduced with permission from ref. 29. Copyright 2008 Elsevier. (j) LAADF-STEM image of (GeOPc)n. Reproduced with permission from ref. 30. Copyright 2008 Taylor & Francis. | ||
Synchrotron X-ray absorption spectroscopy (XAS) also provides access to the electronic structure and coordination environment of active sites in molecular WOCs.23 XAS has been broadly used to study the electronic and coordination structure of metal centers, such as in {CaMn4O5},24 Cp*IrIII,21 [Cp*Ir(pyalc)Cl],25 {Co4O4}/{CoxNi4−xO4} cubanes,26 Co-dppeO2,22 polyoxometalates (POMs)27 and Ru-bda (bda; 2,2′-bipyridine-6,6′-dicarboxylate),28 among other molecular WOCs.
Since XAS is sensitive to local orders up to interatomic distances of ∼4 Å around the absorbing atom, XAS data can be well complemented with PDF data, which offers structural information up to 12–15 Å.
In this case, the structural correlations in the nearest coordination shells around the active metal sites are obtained from XAS data, and the extended structures of molecular ligands are extracted from the PDF data. This allows the domain size and structure of molecular WOCs to be elucidated, which are not accessible for conventional single crystal X-ray diffraction, including the first shell, outer sphere and long-range distances that are not accessible from XAS data alone.23 Recent studies on molecular Cp*IrIII (Fig. 1c and d)21 and our work on the disordered coordination polymer Co-dppeO2 (dppe = 1,2-bis(diphenylphosphino)ethane; Fig. 1e and f)22 combine high-energy PDF and XAS data with molecular dynamics (MD), density functional theory (DFT), and RMC modeling to solve the respective molecular structures. These unified methods allow the seamless correlation of experimental data at multiple atomic-length scales. This approach provides more realistic and extended 3D model structures of complex molecular WOCs, which cannot be achieved from conventional crystallographic analyses.
Imaging techniques such as transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and scanning tunneling microscopy (STM) offer access to structural investigations with atomic resolution at the sub-nanometer scale.16 This is especially of interest for molecules on supports or electrode surfaces (see Part III). However, the short mean-free path of electrons limits standard TEM methods to ultra-high vacuum (UHV) conditions and the aberration of magnetic lenses limits the application range of STEM/STM imaging.16,23 In STEM, a focused convergent electron beam scans a specific sample area to produce signals, which are recorded spot-by-spot for imaging.16,23 Depending on the type of detector used to obtain STEM images, they are classified as (i) bright field (BF-STEM), which detects the intensity in the direct beam after passing a point on the sample, (ii) annular dark field (ADF-STEM), which uses scattered/diffracted electrons through small angles to create an image, and (iii) high-angle annular dark field (HAADF-STEM), which detects scattered electrons at high angles arising mainly from incoherent scattering.16,23 ADF-STEM is sensitive to the atomic number Z, and thus the presence of heavier elements in a specimen increases the intensity of the image. This renders ADF-STEM a useful technique to image metal atoms embedded in a matrix of lower atomic numbers. To gain insight into the elemental composition, the STEM setup is often combined with electron energy loss spectroscopy (EELS) and energy dispersive X-ray spectroscopy (EDS).16,23
ADF-STEM was used to obtain Z-contrast images of the organic molecular structure in hexadecachloro-Cu-phthalocyanine (Cl16-CuPc), polyhalogenated CuPc-derivatives (CuPc-Br/-Cl) (Fig. 1i),29 hexadecachloro-Cu-phthalocyanine (Cl16CuPc),29 and poly-Ge-oxy-phthalocyanine (GeOPc)n (Fig. 1j).30 Recently, HAADF-STEM was used to investigate the immobilization of a molecular dinuclear Ir-based WOC after photochemical removal of organic ligands by bridged H2O/OH groups, which prevents detachment and aggregation (Fig. 1h).31
Advances in aberration-corrected STEM (AC-STEM) have further increased the atomic resolution to the sub-Ångström scale. This allows isolated molecules to be directly imaged on supporting layers through proper sample preparation.16 In a recent representative work, the molecular structure of a trimeric, incomplete aqua ion cubane-cluster ([Mo3S4(H2O)9]Cl4) was directly derived from AC-STEM imaging (Fig. 1g).32 The results showed that e-beam-sample interaction is a key factor to obtain one-to-one correspondence between the acquired images and atomic structure. Moreover, low-dose techniques must be further developed for increasing the resolution of the molecular structure.
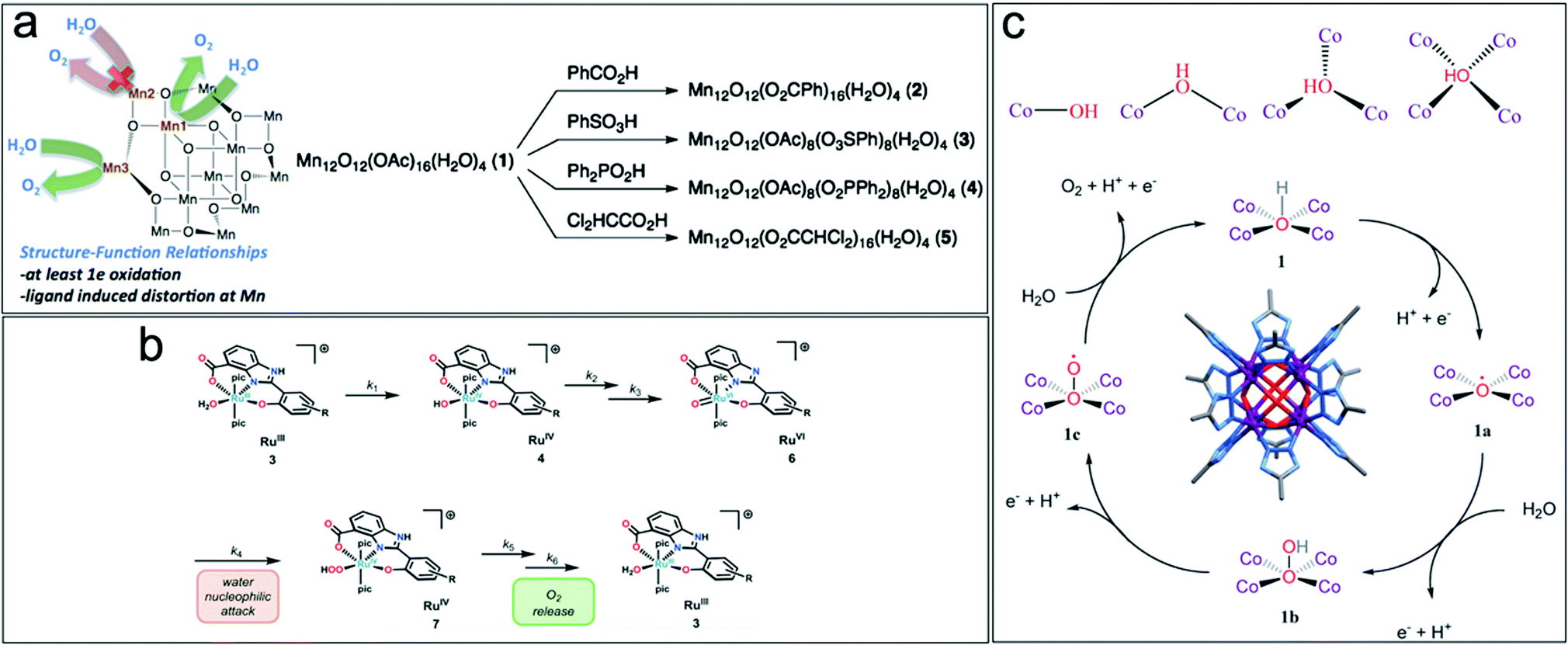 | ||
| Fig. 2 (a) Structure–function relationship and ligand exchange synthesis of Mn12O12(OAc)16−xLx(H2O)4 WOC. Reproduced with permission from ref. 35. Copyright 2015 The American Chemical Society. (b) Steps for the release of O2 from Ru-aqua complexes on RuIII complexes containing negatively charged hpbc3− ligands. Reproduced with permission from ref. 34. Copyright 2016 The Royal Society of Chemistry. (c) Reactant hydroxyl ion coordinated to cobalt ions during the OER, Co8(m4-OH)6(Rtrz)12 cluster and reaction mechanism for [Co8(–OH)6(bdt)4(Hbdt)2] WOC for photodriven water oxidation. Reproduced with permission from ref. 36. Copyright 2019 The Royal Society of Chemistry. | ||
Tuning the local structure of metal complexes by leaving coordination sites labile or open to act as active catalytic centers is another strategy towards highly active molecular WOCs. In octacobalt-cluster-MOFs, [Co8(–OH)6(bdt)4(Hbdt)2], featuring a network of linked Co8(m4-OH)6(Rtrz)12 (Rtrz = 1,2,3-triazolate) clusters and bdt2− ligands (bdt2− = 1,2-benzenedithiolate), it was shown that at each face of {Co8(μ4-OH)6}, the hydroxyl anion is linked to four coplanar CoII centers in a μ4-coordination to form a {Co4(μ4-OH)} unit.36 The μ4-OH group contributes to the activity by delivering an oxygen vacancy site with the optimal OH− adsorption energy. The OH− species are stabilized by four coplanar Co ions, becoming more stable with an increase in oxygen coordination, which further enhances the catalytic performance (Fig. 2c).36 Multi-nuclear molecular structures are often well suited for facilitated multi-electron transfer, i.e. highly active molecular WOCs.37 However, for this WOC design, it must be considered that the number of d-electrons of the transition metal drives the stability/reactivity of the resulting metal–oxo species. Transition metals with a moderate number of d-electrons (MnV-d2, FeV-d3, RuV-d3, FeIV-d4) efficiently produce reactive oxo species since they offer suitable empty d-orbitals to accept the 2px/2py electrons from the oxo ligand. Metals with low numbers of d-electrons (VV-d0, TiIV-d0, and ZrIV-d0) are inactive in O–O bond formation. Filling of the e(dxz;dyz) orbitals in Co, Ni, and Cu furthermore renders the formation of multiple metal–oxo species difficult.33
Overall, these trends pave the way for the development of highly efficient molecular WOCs by exploiting the wide combinatorial space of various metal atoms with negatively charged ligands (pyrrole, hpbc3−, carboxylate, sulfonic and phenolic groups) to efficiently regulate the stability/reactivity of high-valent metal–oxo species, which are considered key intermediates in water oxidation catalysis.
1.2. Recent progress in designing molecular WOCs
In this part, our discussion will primarily focus on recent progress on first-row TM-based (Fe, Co, Ni and Cu) molecular WOCs. For the widely studied Ru and Ir-based molecular WOCs, we refer to some excellent topical reviews.8,33,38,39 Here, instead of comparing their catalytic activity, we summarize their mechanistic aspects, namely (i) experimental trapping of high-valent metal centers, (ii) reaction mechanisms for O–O bond formation and the involvement of non-innocent ligands, (iii) electronic and structural tuning of ligand substitutions, and (iv) non-negligible role of reaction electrolytes. | ||
| Fig. 3 Molecular structures of the recently developed TM-based mononuclear WOCs discussed in Section 1.2.1. | ||
Second, we notice that the reaction mechanism of these first-row TM-based mononuclear WOCs is predominantly proposed as water nucleophilic attack (WNA) to the above discussed high-valent metal–oxo species or metal–oxyl radicals, where O–O bonds are formed.52,55–57 In this case, first-order reaction kinetics is normally observed for the mononuclear WOCs,58 which means that the rate-determining step (RDS) involves only one metal center. Meanwhile, intra- (via the formation of binuclear WOCs) and inter-molecular oxo coupling (I2M) pathways for O–O bond formation are also proposed as alternative reaction mechanisms.43,59,60 These reaction pathways are particularly validated for Fe- and Co-based mononuclear WOCs, where high-valent intermediate species were experimentally trapped.61 However, for Ni- and Cu-based systems, the validation of these classic mechanisms faces significant challenges since the existence of high-valent species is under debate.62–64 Instead, reaction mechanisms involving non-innocent ligands are widely proposed in this regard.65,66 For instance, the carbonate and phenolate ligands in Ni-based mononuclear WOCs 13 and 14 were believed to be redox-active centers in addition to the metal sites.67,68 Accordingly, high-valent metal centers can be circumvented for charge accumulation. The theory of non-innocent ligands was also employed for the Cu-based mononuclear WOC 19, where the redox active aryl oxime ligand was used to store oxidizing equivalents.69 Another important feature of these non-innocent ligands is that they can act as intramolecular bases to facilitate the proton removal for water activation, for example the carbonate and hydroxyl groups in Cu-based mononuclear WOCs 20–22.65,70,71 Therefore, the cooperative effect between the proton acceptor ligand and the catalytic metal center can help to minimize the energy barrier for O–O bond formation.
Here, it should be noted that the detailed reaction mechanism is not a stationary concept. On the contrary, it can be dynamic depending on the reaction conditions. For example, two labile ligands in the cis configuration were involved for Fe-based mononuclear WOC 1 when water oxidation proceeded in acidic conditions. However, in near-neutral electrolyte, a single labile site was sufficient for the reaction to occur.43 Another example is the Cu-based WOC 23, where reversible reaction pathways between mono and bi-nuclear (antiferromagnetically coupled) species were identified as a function of electrolyte pH.72
Third, many recent studies exhibited significant molecular activity and integrity changes upon a small variation in the ligand substitutions.73 We observed that it is hard to obtain unified observations for the ligand substitutions. For example, for the Fe- and Co-based mononuclear WOCs 6, 11, and 12, it was concluded that substituents with higher electronegativity favored higher catalytic activity.74–76 Instead, this positive promotion was supported with electron-donating ligands for Ni-based mononuclear WOC 15,77 which is consistent with the observations for the axial ligand R (R designates pyridine and its derivatives) on WOC 9.52 Nevertheless, it has been widely proposed that the activity promotion originates from a lower energy barrier for O–O bond formation. In addition, the steric hindrance of the substituents is another option to fine-tune the catalytic activity, while maintaining the required molecular stability.78 Ligand engineering still holds the key for the development of efficient and robust mononuclear WOCs. Finally, the nature of the electrolyte has also been proposed to play an important role during the catalytic reaction (complex 24). In another example, the presence of acetate anions could efficiently scavenge protons during the RDS of O–O bond formation for the mononuclear Ni-WOC 16.59 Similarly, phosphate anions were also explored as proton acceptors for mononuclear WOCs 17 and 18.62,79
Overall, while mononuclear WOCs are widely perceived as straightforward models for mechanistic studies, the notable complexity of these single-metal-site systems is often underestimated. Comprehensive studies that monitor not only the metal centers, but also the protecting ligands and the employed electrolytes are essential for their complete understanding. This insight into mononuclear systems is also important for designing higher nuclear first-row TM-based WOCs and single-atom catalysts (SACs), as discussed in the following parts.
Aiming towards the development of functional synthetic models for the oxygen evolution complex (OEC), the O2 evolution reaction of an Mn–terpy dimeric complex (terpy: 2,2′:6,2′′-terpyridine) was investigated by Brudvig et al., representing one of the first WOCs based on earth abundant metals.81,94 Although the initial studies focused mainly on Mn and Co, the statistics from the past six to seven years clearly show an enormous increase in the development of Fe- and Cu-based binuclear WOCs.15,37,95
We have summarized some model binuclear WOCs in Fig. 4. In 2011, Åkermark et al. developed Mn-based dimer 25, which catalyzes water oxidation under photocatalytic and chemical oxidative conditions, employing [Ru(bpy)3](PF6)2 as a photosensitizer and [Ru(bpy)3](PF6)3 as an oxidant.96 Standard isotopic labelling experiments using H218O were performed, which revealed that both oxygen atoms in the evolved O2 were contributed by solvent water. Further investigations on the correlation of the electronic effects in these Mn dimer catalysts towards their activity were carried out by synthetically modifying the catalyst structure and employing DFT-based computational calculations.97 Among the library of synthetically modified structures, the representatives with an aliphatic terminal carboxylate chain showed the highest rate of O2 evolution, which was attributed to facilitation of PCET processes by the involvement of probable hydrogen bonding interactions between water, carboxylate and a hydroxy Mn unit. In 2019, the Mn-dimer WOC 26 based on the bipyalkH ligand (bipyalkH: (2-([2,2′-bipyridin]-6-yl)propan-2-ol)) was reported by Brudvig et al., containing an anionic donor site capable of stabilizing high oxidation states on the metal.87 The Mn–bipyalkH dimer catalyses water oxidation in the presence of the sacrificial oxidant KHSO5. The possible catalytic pathways and intermediates were suggested based on EPR and spectroelectrochemical (SEC) investigations. In situ EPR spectroscopic analysis of the catalytic mixture (catalyst + KHSO5), following a serial freezing-thawing and re-freezing protocol, depicted a drastic change from a weak signal to an intense 16-line EPR spectral pattern, which suggested the probable involvement of an unusually highly oxidized MnV(μ-O)2MnV intermediate. The stabilization of this high-valent Mn species was associated with the high donicity of the ligand alkoxide group, which was also found to contribute to the redox levelling of the high-valent species Mn(II/III), Mn(III/IV) via successive deprotonations, as observed from SEC investigations. This also revealed that the Mn-dimer can be produced in situ from the corresponding Mn-monomers, and that dimer 26 remains dominant with a minor (around 20%) [MnIV(bipyalkH)(bipyalkH)](PF6)2 monomer content above an applied potential of 900 mV vs. NHE.
 | ||
| Fig. 4 Selected recently reported bi- and trinuclear WOCs discussed in Sections 1.2.2 and 1.2.3. | ||
Proceeding to Co-based binuclear WOCs, their representatives are still limited, but are well characterized with respect to their mechanistic propagations. A representative example is the Co–Hbpp complex, which was initially reported for the four-electron reduction of O2, leading to the formation of binuclear μ-peroxo species 27 as a result of PCET processes.98 More recently, combined spectroscopic analysis of 27 and isolation of the corresponding Co superoxido end-on ((H2O)–CoIII–Hbpp–CoIII–OO˙) species revealed its potential to catalyse water oxidation through the Co superoxido end-on intermediate following a number of sequential oxidations.99 Complex 27 containing oxygen isotopologues upon oxidation clearly displayed the resonance Raman (rR) vibrational features of the superoxido group at 1121 cm−1, 1088 cm−1 and 1055 cm−1, which correspond to 16O–16O, 17O–17O and 18O–18O, respectively. An EPR signal was observed for ((H2O)–CoIII–Hbpp–CoIII–OO˙) owing to the presence of an unpaired electron from the superoxido group. Furthermore, both of the above observations were in good accordance with the XANES spectra of the superoxido complex, which showed a positive edge shift of 0.4 eV and multiplet feature at the pre-edge region compared to that of 27, suggesting a change in the local geometry and electron density around the Co metal center. The above experimental observations in combination with DFT calculations were useful in identifying the catalytic pathways of 27. Similarly, the CoIII-(μ-peroxo-μ-hydroxo)-CoIII diastereomers 28 and 29 reported by Kojima et al. depict another example of Co-based binuclear WOCs.100 At pH 7, cis diastereomer 29 showed higher activity compared to 28, indicating the importance of structural symmetry in catalytic intermediates. The conclusions from the Pourbaix diagram suggested that the isomer-based activity originated from the slightly negative oxidation potential of the one-electron oxidised 29 compared to 28. Characterization of the one-electron oxidised species of 28 and 29 by EPR and Raman spectroscopy indicated the generation of a CoIII-(μ-superoxo)-CoIII dimer, which was confirmed by the presence of a 15-line EPR superhyperfine splitting and superoxo O–O vibrational mode, respectively. This suggested the plausible involvement of 28 and 29 as intermediates for the water oxidation catalyzed by the earlier reported Co–TPA dimer (TPA: tris(2-pyridylmethyl)-amine).101 However, later reports indicated that the formation of CoOx can drive heterogeneous water oxidation instead of 29.102,103 Recently, Goswami et al. synthesized mixed valence CoIICoIII binuclear complex 30, which was shown to be an active water oxidation catalyst owing to the presence of aqua ligands on one Co center.104 Preliminary catalytic studies based on cyclic voltammetry indicated an increase in catalytic current with pH over the range of 7–11, which was attributed to the conversion of the aqua to a hydroxide ligand at higher pH, as validated by the d–d-band changes in the UV-vis spectra.
Regarding Fe, which is the most abundant transition metal, the spectrum of Fe-based binuclear WOCs has witnessed great progress in the past five years.95,105 Experimental evidence from many Fe-based WOCs suggests that they generally involve FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/FeVO high-valent species as intermediates.106,107 As reported by Ma et al., the μ-(oxo)-μ-(acetato) form of di-iron complex 31 is the active species responsible for water oxidation compared to its monomer counterpart [Fe(TPA)(Cl2)] at pH 4.5.108 A high-valent FeIVO intermediate was assumed to be involved in the catalytic cycle, as confirmed from the evolution of a short-lived UV-vis peak at 726 nm upon the addition of oxone. By the targeted synthesis of 33, Åkermark et al. demonstrated the need for a proper ligand framework and nuclearity effects for Fe-based systems to function as efficient catalyts.109 Fe-μ-(oxo) dimers based on polypyridyl macrocyclic ligand frameworks have recently been shown to serve as enhanced WOCs.110,111 Among them, the binuclear Fe2-μ-(oxo)(ppq)2 complex 32 (ppq: 2-(pyrid-2′-yl)-8-(1′′,10′′-phenanthrolin-2′′-yl)-quinoline) drives water oxidation in the presence of ceric ammonium nitrate.110 Electrochemical and UV-vis absorption studies suggest that water oxidation by 32 involves a simultaneous two-electron oxidation process, producing an FeIIIFeVO intermediate owing to the presence of multiple redox peaks and an absorption band at 675 nm. Interestingly, the Fe2-μ-(oxo)(Hbbpya)2 complex 34 showed the influence of the electrode surface on the reaction mechanisms of homogenous water oxidation electrocatalysts.111 The mechanistic insight for 34 suggested the regeneration of the Fe2-μ-(oxo)(Hbbpya)2 dimer from two monomeric FeIV/FeV moieties over the catalytic turnover, thus confirming the μ-(oxo) dimer 34 to be the real catalyst. Recently, metallocorroles have been well established as promising candidates for water oxidation due to the electronic stability offered by the corrole framework.52,112 The binuclear Fe corroles 36 and 37 exhibit OER activity, with 37 displaying higher performance. This was attributed to the presence of a β–β′ linkage in the latter, which modulates the electronic factors between the two Fe centers by promoting their simultaneous participation compared to the μ-(oxo)-linked 36. Even though the majority of Fe-based WOCs involve high-valent FeIV/FeV species in their catalytic turnovers, a recent report from the Das group suggested O2 evolution from dimeric Fe complex 35 as a consequence of the replacement of labile chlorides by hydroxide ions, leading to the formation of a peroxo-bridged FeIII species.113
O/FeVO high-valent species as intermediates.106,107 As reported by Ma et al., the μ-(oxo)-μ-(acetato) form of di-iron complex 31 is the active species responsible for water oxidation compared to its monomer counterpart [Fe(TPA)(Cl2)] at pH 4.5.108 A high-valent FeIVO intermediate was assumed to be involved in the catalytic cycle, as confirmed from the evolution of a short-lived UV-vis peak at 726 nm upon the addition of oxone. By the targeted synthesis of 33, Åkermark et al. demonstrated the need for a proper ligand framework and nuclearity effects for Fe-based systems to function as efficient catalyts.109 Fe-μ-(oxo) dimers based on polypyridyl macrocyclic ligand frameworks have recently been shown to serve as enhanced WOCs.110,111 Among them, the binuclear Fe2-μ-(oxo)(ppq)2 complex 32 (ppq: 2-(pyrid-2′-yl)-8-(1′′,10′′-phenanthrolin-2′′-yl)-quinoline) drives water oxidation in the presence of ceric ammonium nitrate.110 Electrochemical and UV-vis absorption studies suggest that water oxidation by 32 involves a simultaneous two-electron oxidation process, producing an FeIIIFeVO intermediate owing to the presence of multiple redox peaks and an absorption band at 675 nm. Interestingly, the Fe2-μ-(oxo)(Hbbpya)2 complex 34 showed the influence of the electrode surface on the reaction mechanisms of homogenous water oxidation electrocatalysts.111 The mechanistic insight for 34 suggested the regeneration of the Fe2-μ-(oxo)(Hbbpya)2 dimer from two monomeric FeIV/FeV moieties over the catalytic turnover, thus confirming the μ-(oxo) dimer 34 to be the real catalyst. Recently, metallocorroles have been well established as promising candidates for water oxidation due to the electronic stability offered by the corrole framework.52,112 The binuclear Fe corroles 36 and 37 exhibit OER activity, with 37 displaying higher performance. This was attributed to the presence of a β–β′ linkage in the latter, which modulates the electronic factors between the two Fe centers by promoting their simultaneous participation compared to the μ-(oxo)-linked 36. Even though the majority of Fe-based WOCs involve high-valent FeIV/FeV species in their catalytic turnovers, a recent report from the Das group suggested O2 evolution from dimeric Fe complex 35 as a consequence of the replacement of labile chlorides by hydroxide ions, leading to the formation of a peroxo-bridged FeIII species.113
In accordance with the fact that Cu–oxo dimers play an active role in several redox processes in nature, the development of Cu-based WOCs has been a topic of great interest over the past decade due to the well-defined redox behaviour and relative earth abundance of Cu.85,114 With the report of 38 in 2015, a totally new pathway for water oxidation catalyzed by binuclear Cu systems was proposed, which was otherwise believed to involve high-valent CuIVO, as in the case of monomers.115 The Cu dimer 38 catalyses water oxidation at neutral pH, following an intramolecular proton transfer mechanism between the two CuIII centers by passing through a Cu2II,II superoxide intermediate. Subsequently, an unsymmetrical binuclear Cu complex 39 based on the earlier WOC demonstrated the key role of the intramolecular proton acceptor in the catalytic mechanism of 38 since 39 showed a buffer concentration-dependent catalysis, indicating the requirement of buffer to mediate the proton transfer process.116 In 2017, Emmons et al. proposed a new pathway for catalytic water oxidation by 41. This redox isomerization (RI) mechanism, which was supported with the identification of a CuIII2-(μ-O2) moiety, clearly suggested that single sites were disfavoured during the corresponding catalytic pathway.60 Furthermore, calculations revealed that both the WNA and RI mechanisms are energetically probable pathways for O–O bond formation. Very recently, Wang et al. succeeded in synthesizing a stable binuclear Cu core in WOC 42 using a suitable flexible linker group for the two metal centers.117 The electrochemical OER performance of 42 at pH 12 displayed a 30-fold higher activity compared to its monomeric analogue. The preference of a flexible linker over a rigid aromatic linker was demonstrated to promote the intramolecular cooperative effect between the metal centers, thereby reducing the energy barrier for the RDS, as validated by DFT calculations.
Earlier investigations in the development of cubane-type WOCs were solely concentrated on Mn-based systems owing to their presence in OEC and high natural abundance.122 Additionally, several cubane WOCs based on Co and Cu have been designed and investigated in the past decade. One of the most widely investigated Co cubane WOCs is Co4O4(Py)4(OAc)4, whose mechanistic and electronic features during water oxidation have been well determined recently via quantum chemical modelling and advanced spectroscopic techniques. Chen et al. characterized the possible high-valent intermediates in the OER catalyzed by Co4O4(Py)4(OAc)4 cubane using XAS and Kβ RIXS coupled with DFT calculations.123 The formation of high-valent Co(IV) species during the OER has been previously identified in Co4O4 cubane complexes and Co–Pi via multifrequency EPR spectroscopy.124,125 Owing to the faster timescale of the electronic transitions in X-ray spectroscopic methods compared to EPR, XAS/RIXS can be used as a valuable technique to probe the localized high-valent Co(IV) state. The combined XAS/RIXS analysis confirmed that the hole equivalents were stored within the cubane core, leading to the evolution of a Co(IV) state. Furthermore, in contrast to the previous EPR-based conclusions, these recent X-ray spectroscopy investigations indicated that the hole equivalents were localized rather than delocalized within the cubane cluster.123 Furthermore, these observations were supported by DFT calculations, which suggested the futility of electron hole delocalization for the formation of Co(IV) in clusters. Additionally, calculations based on hypothetical CoM3O4 cubanes formed from the substitution of redox-inactive metals (M) indicated the facilitated formation of localized Co(IV). Further studies on modelling the Co–Pi active sites based on the cofacial Co(IV) sites of Co4O4(Py)4(OAc)4 cubane have also been carried out recently to understand and correlate the formation of high-valent Co species in Co-based WOCs.126 This has been helpful in concluding the need for adjacent Co(IV) sites in oxidic cobalt WOCs to drive O–O bond formation. The in situ rapid-scan FT-IR spectroscopic investigations by Frei and co-workers indicated that the photo-driven water oxidation by Co4O4(Py)4(OAc)4 cubane involves a CoIII–(μ-peroxide)–CoIII species at the O–O bond formation step, which is preceded by a CoIIICoIV intermediate.127 In 2017, our group reported a highly intact and stable Co-dpk (dpk: di(2-pyridyl)ketone) cubane WOC 45 (Fig. 5), displaying the characteristic {H2O–Co2(OR)2–OH2} edge-site motif that catalyses photo-driven water oxidation.26In situ XANES investigations of 45 suggested the oxidation of the Co(II) centers to Co(III) or higher valent states during the catalytic turnover. DFT calculations indicated the role of the abovementioned edge-site aqua ligands in mediating the proton transfer process. Another work from our group involved the incorporation of Ln3+ as redox-inactive centers within the cubane core of a previously reported Co cubane WOC.128 The role of Ln3+ in 46, as a redox-inactive Ca2+ analogue of OEC, was depicted as an alternative strategy in the design of active cubane WOCs. Furthermore, the Ln3+ center also served as a catalytic promoter site by offering a flexible ligand-binding motif. Carrying forward the investigations on derivatives of Co4O4(Py)4(OAc)4, Tilley et al. established the role of heterometal incorporation and dangling metal sites on cubane cores in tuning the catalytic activity.129,130 Insertion of Mn into the cubane core led to the formation of MnCo3O4(Py)3(OAc)547, which was introduced as a molecular analogue for Mn-doped Co oxidic WOCs. The spectroscopic and electrochemical investigations suggested that the redox chemistry occurs at the Co sites, whose redox behaviours are tuned by the ligand attached on Mn, thus establishing a cooperative effect between Mn and Co.129 This in fact serves as a valuable strategy towards the understanding and design of heterobimetallic catalysts. Furthermore, attachment of a dangling Co site to the MnCo3O4(Py)3(OAc)5 cubane moiety resulted in the formation of 48. This synthetic analogue of the OEC indicated the role of a dangling metal center in modulating the structural rigidity and redox behaviour of the cubane core. Crystallographic information supported by computational data clearly suggested the distortion of the cubane core upon the addition of the dangling center. Furthermore, 48 shows an increased number of redox events compared to the parent cubane 47, thus indicating the productive role of the dangling metal center in facilitating multielectron processes.130 Wu et al. for the first time reported the Cu cubane 49, which serves as a water oxidation electrocatalyst. Mechanistic investigations based on in situ EPR spectroscopy suggested the formation of an EPR silent low-spin CuIII species, which is involved in the catalytic turnover during bulk electrolysis.131 This was further supported by the observation of μ4-peroxido O–O vibrational features in the Raman spectra. Therefore, it was concluded that the mechanistic intermediates involved were low-spin CuIII and CuIIIO species formed as a result of the successive two-electron oxidation of the initial CuII4 species.131 Another tetranuclear Cu cubane WOC 50 was reported by Wang et al., which serves as a stable electrocatalyst over a broad pH range of 8.92–11.78 with a relatively low overpotential of 400–665 mV.132 The mechanistic pathway was suggested to follow a PCET mechanism, as supported by the linear dependence of the catalytic water oxidation potentials on the pH.132 In 2018, Ding et al. reported an octanuclear Cu-based WOC comprising two Cu4O4 cubane units 51 supported by dpk ligands. This WOC served as an efficient catalyst for the photo-driven oxidation of water in the presence of the [Ru(bpy)3]2+ photosensitizer.133 The plausible mechanism was suggested to involve the pre-organization of solvent water molecules via hydrogen bonding from the dpk–OH− ligands, thus favouring the nucleophilic attack of water on CuII. Besides the tetranuclear cubane-type WOCs, trinuclear metal complexes based on Cu 43 and Co 44 have recently been reported as efficient WOCs.105,134
 | ||
| Fig. 5 Selected recent polynuclear WOCs discussed in Section 1.2.3. | ||
Besides tetranuclear cubanes and trinuclear WOCs, very few metal complexes of higher nuclearity have been reported as efficient WOCs. A representative well-investigated polynuclear WOC is the pentanuclear Fe complex 52, consisting of a triangular [Fe3(μ3-O)] core surrounded by two [Fe(μ-L)3] units, thus possessing open coordination sites.135 Earlier mechanistic insights proposed that the O–O bond formation proceeds through the coupling of two FeIVO units within the FeII2FeIIIFeIV2 cluster. However, a recent investigation based on DFT calculations on the same Fe system suggested a different and more energetically plausible pathway. The new mechanism is based on the involvement of a superoxide FeIII5 intermediate prior to O–O bond formation rather than the participation of ferrous sites.136 Another polynuclear Mn cluster with 12 Mn atoms was recently reported by the Christou group. The Mn12–dhbH (dhbH: 3,5-dihydroxybenzoic acid) cluster serves as a water oxidation electrocatalyst at pH 6.137 The specific ligand design with carboxylate groups endows the Mn12 cluster with water solubility and stability. The multinuclear metal sites contribute to the catalytic activity of the cluster, which is also supported by its multiple redox behaviour. Moreover, it was suggested that the hydroxyl groups serve as hydrogen bonding sites, thus enabling a shuttle pathway for H2O and H+ to and from the Mn active sites.
As mentioned earlier, the design of polyoxometalates with multinuclear TM metal cores is also important for the development of WOCs. POMs as structurally rigid and stable molecular metal oxo clusters have been well recognized as efficient WOCs owing to their capability to drive fast and reversible multi-electron transfer transformations, while preserving their structural motifs.138–140 POMs incorporating tetranuclear Co and trinuclear Cu metal sites have been identified as potential water oxidation photo- and electrocatalysts, respectively. Besides these smaller representatives, giant POMs with higher nuclearity have also been identified as stable WOCs. One such POM is the [Mo72Fe30O252(CH3COO)12{Mo2O7(H2O)}2{H2Mo2O8(H2O)}(H2O)91] oxo-cluster incorporating 30 Fe cores, which catalyses water oxidation under photocatalytic conditions at pH 9.141 For a more detailed discussion of POM-based water splitting catalysts, we would like to recommend recent representative reviews.142,143
As evident from the above discussion, molecular compounds offer a wide variety of options in the strategic design of efficient WOCs. The choice of a suitable ligand framework is essential, while targeting a multinuclear WOC favours the synergistic effects of metal sites and other electronic modulations via the ligand to enhance efficient hydrogen bonding interactions. Although combined spectroscopic techniques serve as a toolbox for the mechanistic investigations of molecular WOCs, they are sometimes unable to fully capture the chemical dynamics of molecules in solutions. Thus, future research strategies on molecular WOCs will move towards their immobilization on heterogeneous surfaces to avoid the limitations of conventional solution-based approaches.144–146
1.3. The importance of molecular integrity
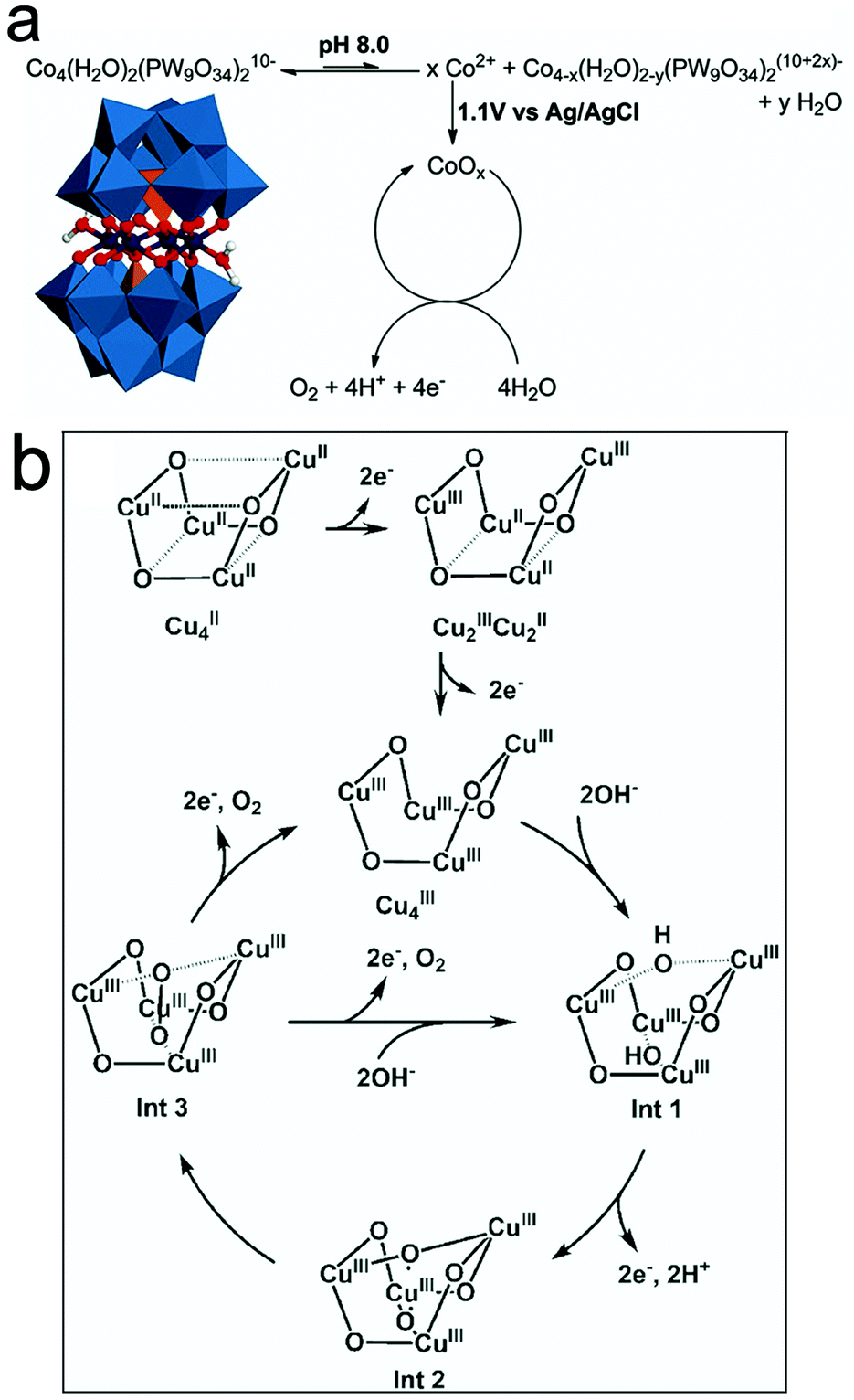 | ||
| Fig. 6 (a) Heterogeneous CoOx formation from homogeneous Co4POM. Reproduced with permission from ref. 148. Copyright 2011, The American Chemical Society. (b) Mechanism for the water oxidation of the Cu4O4 core for [(LGly-Cu)4]/[(LGlu-Cu)4] in alkaline solution. Reproduced with permission from ref. 131. Copyright 2018, Wiley VCH. | ||
However, more recent studies confirmed the molecular integrity of many molecular WOCs. Particularly, studies on the structural integrity of Cu-containing [V2Mo6O26]6− POM-WOCs152 showed that these POMs do not exhibit nanoparticle growth (0.04 M borate buffer, pH 8) based on DLS experiments. Regarding the Cu-sites, their high activity and low overpotential are controlled by their coordination environment. The first one-electron oxidation leads to the formation of CuIII–OH species followed by CuIVO oxidation, which assists in nucleophilic attack, while the aqua-oxo interaction favour the formation of intramolecular O–O bonds.152 Other works on the molecular integrity of Cu-cubane [(LGly-Cu)4]/[(LGlu-Cu)4] in alkaline solution131 indicated that the structure of the Cu4O4 core remains intact, with no oxide formation during the OER. The two-electron transfer processes involved in the tetranuclear Cu4O4-cubane centers yield high-valent CuIII/CuIIIO˙ intermediates during the OER (Fig. 6b). Other recent studies on the true catalyst in the Mn-containing [Mn3(H2O)3(SbW9O33)2]12− POM showed that no Mn2+/Mn-oxide species are formed under water oxidation conditions.153 The Sb heteroatoms in the polyoxoanions were found to play a central role in their stability during the water oxidation process.
However, the loss of structural integrity under harsh chemical oxidants or in the presence of sacrificial reagents remains a problem for molecular WOCs. Also, electrode fabrication from molecular WOCs by drop casting can lead to catalyst leakage, high aggregation, and inefficient activation of metal centers at the interfacial diffusion layer.15 Thus, to overcome these issues, novel strategies for the heterogeneous stabilization of molecular WOCs on solid surfaces have been developed.155 The main goals include (i) preserving the intrinsic activity and selectivity, (ii) stabilization of intermediate species, and lowering the energy barrier while facilitating O–O bond formation, (iii) tuning the catalyst loading to lower the water oxidation overpotentials, (iv) attaining facile electron transfer kinetics at the interface, and (v) promoting long-term resilience against leaching and highly oxidative media.15,23,38 All this must be complemented with the optimization of surface area/morphology, hydrophobicity and hydrophilicity, density of active metal sites, and mass/charge transfer properties. Particularly, the stabilization of molecular WOCs by tuning the electronic/steric properties of the ligand requires meticulous screening since the nature of the ligand can affect their activity and selectivity.155,156 Recently, N,N′-bidentate electronically asymmetric ligands were used to stabilize low-valent transient CoI species.157 The results showed that electron-deficient ligands removed electron density from the Co sites, thereby preventing deactivation of the catalyst, and stabilizing the complex (Fig. 7a). Similarly, mononuclear [Co(TPA-R)]2+ (TPA = tris(2-pyridylmethyl)amine, R = tri-αF; tri-αOMe and mono-αF) was stabilized through electron-withdrawing substituents on the TPA ligand.76 The oxidative resistance of the ligand on Co-intermediates was found to be a key factor to achieve oxidative stability. Mn-porphyrin dimer WOCs fabricated with mono/hexaphosphonic acid groups (Mn2DP-PO3H2 and Mn2DP-(PO3H2)6) were recently immobilized through covalent assembly on ITO electrodes.158 The immobilized Mn2DP-(PO3H2)6|ITO assists electron transfer from the WOC to the ITO-electrode, allowing efficient OER at a low overpotential with high turnover. Similarly, Co-complexes have been stabilized through the covalent anchoring of 2,2-bipyridine to graphene-modified ITO electrodes (Fig. 7b).159 The heterogenized system yields a surface-bound WOC with only covalently attached active catalytic species, displaying high catalytic activity. Thus, functionalization of graphene-modified electrodes offers a viable route for loading metal atoms on conductive surfaces with appropriate metal binding moieties. In a similar study, a molecular [(Lpy)CuII]2− complex (Lpy = 4-pyrenyl-1,2-phenylenebis(oxamidate) ligand) was stabilized on graphene electrodes.160 Anchoring promotes π-stacking interactions with the graphene layers, which provide π-delocalization to notably enhance the catalytic performance. The molecular [(Lpy)CuII]2− WOCs remained intact under catalytic turnover without any sign of CuO formation. Recently, cobalt porphyrin CoTCPP and Co4O4 cubane WOCs were stabilized on BiVO4 photoanodes (Fig. 7c and d; also see Part III).161,162 The results revealed that CoTCPP suppresses electron–hole recombination on the BiVO4 surface when using H2O2 as a hole scavenger, while improving both activity and stability. Loading Co4O4 cubane featuring alkoxycarboxylato bridging ligands on porous BiVO4 electrodes also resulted in highly efficient surface charge separation.
 | ||
| Fig. 7 (a) Stabilization of a low-valent transient CoI complex by using N,N′-bidentate electronically asymmetric ligands. Reproduced with permission from ref. 76. Copyright 2017 Elsevier. (b) TM complexes (M = Fe3+, Co2+, Ni2+, Cu2+, Zn2+) immobilized by covalent anchoring of 2,2-bipyridine to Gr-modified ITO electrodes. Reproduced with permission from ref. 159. Copyright 2014 The American Chemical Society. (c and d) Stabilization of molecular porphyrin complexes CoTCPP, and molecular Co4O4 cubane type WOCs on BiVO4 photoanodes. Reproduced with permission from ref. 161 and 162. Copyright 2016 The American Chemical Society and copyright 2017 Wiley VCH, respectively. (e). Stabilization of an Al2O3-coated [{RuIV4(OH)2(H2O)4}(γ-SiW10O34)2]10− POM on hematite photoelectrodes. Reproduced with permission from ref. 164. Copyright 2017 The American Chemical Society. (f) Stabilization of molecular Co-WOCs enclosing tris((2-benzimidazolymethyl)amine) ligands, Co-(BimC3F8)3, on the surface of an FTO electrode. Reproduced with permission from ref. 165. Copyright 2016 The American Chemical Society. (g) Stabilization of [Co4(H2O)2(PW9O34)2]10− polyoxometalate into the hexagonal channels of the Zr(IV) porphyrinic MOF-545 hybrid framework. Reproduced with permission from ref. 166. Copyright 2018 The American Chemical Society. | ||
Stabilization through covalent functionalization, namely modification of electrode surfaces with functional groups, has been used to stabilize tetranuclear [RuIV4O5(OH)(H2O)4(γ-PW10O36)2]9− and [{RuIV4(OH)2(H2O)4}(γ-SiW10O34)2]10− POM-WOCs on porous TiO2 electrodes.163 Treatment of the TiO2 electrode with cationic silylating agents resulted in better stabilization of the POM-WOC on the surface of the TiO2 electrodes, where it remained intact after photo-driven water oxidation. Stabilization of [{RuIV4(OH)2(H2O)4}(γ-SiW10O34)2]10− on Fe2O3 photoelectrodes was also reported.164 However, the attached POM on the Fe2O3 photoanode surface remained intact after water oxidation only if a protective Al2O3 layer was deposited on top the POM (Fig. 7e). Moreover, the thickness of the Al2O3 layer also drastically affected the catalytic performance of the attached POM-WOC. Another study reported the stabilization of Co-encapsulating C24H21N7 ligands Co-(BimC3F8)3 on FTO electrodes (Fig. 7f).165 The stability of this immobilized molecular Co-WOC was found to depend on the tailored hydrophobicity of the coordinating ligand. The Co-WOC, which was immobilized by physisorption, remained stable during extended electrolysis.
Stabilization of molecular WOCs through physical confinement was used for the encapsulation of the [Co4(H2O)2(PW9O34)2]10− POM in the hexagonal channels of a Zr(IV) porphyrinic MOF-545 (Fig. 7g).166 This approach showed that the POM-MOF interface delivers –OH species and labile H2O molecules, which clearly enhance the OER performance.166 Similarly, encapsulation of Co-substituted polyoxometalates (Co-POMs), was performed via an impregnation route using mesoporous Cr(III)terephthalate MIL-101(Cr) MOFs by stirring a solution of Co-POM and MIL-101(Cr).167 The results from 31P NMR and elemental analyses indicated that the [Co4(H2O)2(PW9O34)2]10− and [CoO39(H2O)PW11]5− POMs remained intact inside the MOF framework. A similar study encapsulated Ni4P2-POM [Ni4(H2O)2(PW9O34)2]10− into the pores of MOFs obtained from [Ir(ppy)2(bpy)]+ and [Ru(bpy)3]2+-derived dicarboxylate ligands.168 The results revealed that the proximity of Ni4P2 to the photosensitizer in Ni4P2@MOF assists multi-electron transfer for efficient catalysis. Hence, the hierarchically ordered POM@MOF assembly provides a tunable platform for achieving synergistic functionalization (also see Section 3.3.2).
Developing stabilization strategies for molecular WOCs remains a main challenge for large-scale applications. Accordingly, effective heterogenization of molecular WOCs is a promising method to achieve catalysts with enhanced atom-efficiency and intrinsic activity for large-scale artificial photosynthesis. Hence, further studies are needed for efficient device assembly with extended molecular stability, robustness and superior catalytic performance.
Part II: development and mechanistic studies of heterogeneous WOCs
2.1. Mechanistic advances for heterogeneous WOCs
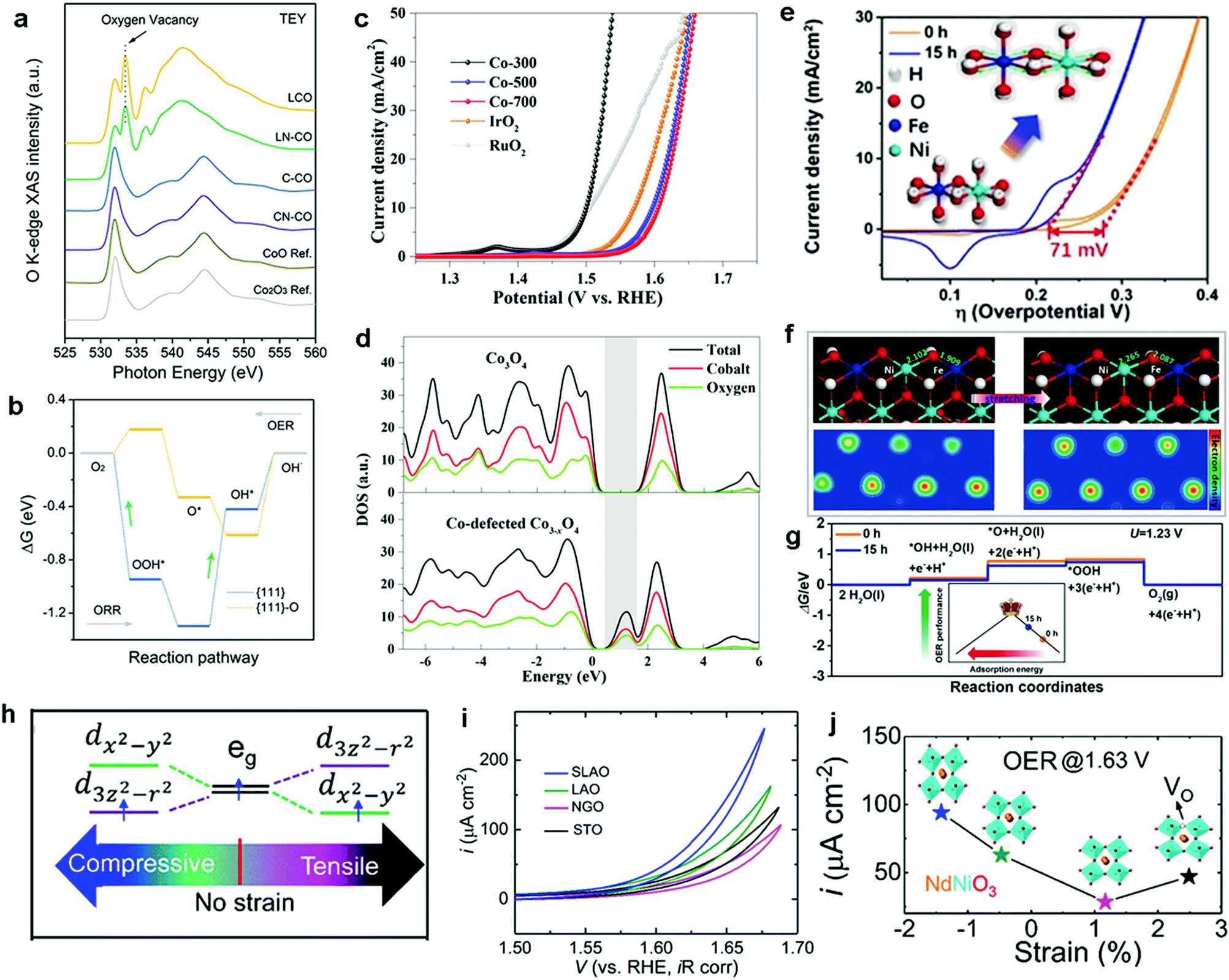
Over the past decade, considerable efforts in the heterogeneous WOC community were devoted to the design of efficient catalysts with a higher TOF at minimized overpotentials.3,169 Consequently, many first-row bi- and multi-TM-based systems have already surpassed noble metal oxides such as RuOx and IrOx in terms of TOF.5,170–173 However, the ongoing debate on the involved active sites and the pathways of the four-electron-transfer reaction still demand in-depth mechanistic investigations.174–177 In the following section, we highlight recent selected insights into the mechanisms of first-row TM-based heterogeneous WOCs. To render the discussion more concise, widely studied unary oxides, such as CoOx and α-Fe2O3 (hematite), are selected as model systems.To derive a model for the catalytic cycle, it is necessary to probe the chemical nature of the intermediate species on the surface. Accordingly, attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy was successfully implemented for probing the surface peroxide intermediates on a TiO2 photoanode film.189 This technique was also established for colloidal Co3O4 NPs, where two reaction intermediates with different temporal behaviors were resolved.190 Using isotope labelling experiments, the first one was confirmed to be a surface superoxide species, which was specified as the fast catalytic site where two adjacent metal centers are involved. Thus, the O–O bond formation was described as WNA at one of the CoIV–oxo sites with the assistance of another CoIV–oxo site as a proton acceptor (Fig. 8a). The second reaction intermediate was assigned to isolated CoIV–oxo sites and found to be catalytically slow (Fig. 8b). Compared to the electronically coupled dual-site centers, these structurally segregated CoIV–oxo sites had to overcome a significantly higher energy barrier for the WNA process to proceed. To understand the formation and the exceptional activity of adjacent CoIV–oxo centers, DFT calculations were performed on the Co3O4(001) surface.191 Surprisingly, it only took 1.42 meV to activate the separated CoIV–oxo species for their dimerization. Moreover, the kinetic barrier for the proton exchange between a CoIV–oxo site and its neighboring CoIII–OH site was merely 0.27 eV. Therefore, these CoIV–oxo species were quite mobile on the surface. In this case, when sufficient CoIV–oxo species are available, the chance of their encounter and formation of CoIV–oxo dimers through random walk was found to be very high. According to the calculations, the formed CoIV–oxo species could further assist the following steps (deprotonation of hydroperoxide and CoIII–H2O species) after WNA for O–O bond formation. This means that only the first step (CoIII–OH to CoIV–oxo) demands significant energy input, while the remaining steps in the catalytic cycle can proceed spontaneously if sufficient CoIV–oxo species are present on the surface. For the oxygen-bridged {H2O–Co2(OH)2–OH2} edge-site on Co3O4, it is important to note that the O–O bond formation proceeds exclusively through the WNA pathway since in H218O labeling experiments, both the unlabeled superoxide (16O16O) FTIR band and 32O2 product were absent.
 | ||
| Fig. 8 Proposed reaction mechanism for the fast (a) and slow (b) catalytic sites on the Co3O4 surface. Reproduced with permission from ref. 190. Copyright 2014 Springer Nature. (c) Five consecutive CVs were performed for 18O labelled Co-WOC (Co3O4) in unlabeled electrolyte, DEMS data indicate that 36O2 deplete fast as cycle number increases. (d) Proposed reaction mechanism for the Co-WOC. Reproduced with permission from ref. 193. Copyright 2016 The American Chemical Society. | ||
Direct coupling of the adjacent CoIV–oxo species as alternative O–O bond formation pathway was later confirmed with other Co-based heterogeneous analogues, which featured the key {H2O–Co2(OH)2–OH2} edge-sites. For example, the first turnover analysis on Co/methylenediphosphonate oxide NPs by time-resolved O-labelling isotope-ratio membrane-inlet mass spectroscopy indicated that the O–O bond was formed predominantly through the coupling between CoIV–O˙ and CoIV–OH.192 Unfortunately, it was not possible to completely exclude the contributions from the WNA pathway due to the limited temporal resolution (3 s) of this method, where exchange of the labelled O could not be avoided. To gain knowledge of the O exchange rate at the edge-site, Nocera et al. employed an oxygen-bridged bi-cobalt molecular model and found that the exchange of the terminal OH or OH2 groups with syn geometry was in the order of hours.193 This extremely slow isotope exchange rate would be in principle applicable for the edge-site of heterogeneous Co-WOCs due to their structural similarities at the molecular level. Differential electrochemical mass spectroscopy (DEMS) experiments with 18O-labeled Co-WOC in unlabeled electrolyte delivered much less 36O2 at higher cycling numbers (Fig. 8c). This indicates that the reaction proceeds primarily via the CoIV–oxo coupling mechanism (Fig. 8d) and no significant structural reorganization between the terminal and bridged oxygen occurs during the reaction. Thus far, we observed that terminal hydroxide groups are generally assumed as the resting state in most catalytic cycles. However, these OER reactions were carried out under near-neutral conditions. This implies that water molecules were the actual terminal ligands, and thus water dissociation was necessary in the first step. To visualize this process, STM experiments with atomic-level resolution were carried out on CoOx nanoislands.194 STM video monitoring revealed that water molecules first dissociated into protons and hydroxyls at the edge-sites before the basal plane took over the in following steps. The edge-site activity in water dissociation was further verified by CoOx nano-islands with different edge to basal plane ratios. Under the same water pressure, the density of hydroxyls was reversely proportional to the radius of the nano-islands. This observation confirmed the edge activity. It was also found that the presence of additional water molecules could significantly lower the energy barrier for both water dissociation and proton diffusion, and the results were supported by DFT calculations. Overall, this study highlighted the importance of edge-sites and their higher activity for water oxidation. To maximize the Co edge-site activity, ultra-small 2 nm size nanocubanes were prepared to take full advantage of the edge-site activity.195 However, the TOF of these nanocubanes (0.023 s−1 Co−1) was much lower than the fast catalytic site (3 s−1) reported by the Frei group.190 Therefore, a WNA on single CoIV-oxo sites was proposed, despite the presence of a large fraction of edge-sites. To account for this discrepancy, further research into the mechanisms for generating cooperative CoIV–oxo sites is required.
We observed that the catalytic activity of the above-discussed heterogeneous Co-WOCs was primarily assigned to the CoIII centers in the CoO6 octahedral geometry instead of the CoII centers in the tetrahedral geometry in the spinel structure of Co3O4. To distinguish between the roles of the two Co centers in different coordination environments, a cation-substitution strategy was systematically employed by Liu et al. to prepare spinel-type ZnCo2O4 and CoAl2O4.196 With the help of operando electrochemical impedance spectroscopy (EIS) and XAS analysis, the catalytic activity (normalized current density based on the electrocatalytic active surface area) revealed that the CoII centers in tetrahedral geometry were more active than the CoIII centers in the octahedral geometry (Fig. 9a). Operando Raman studies by the same group further identified the newly evolved 1068 cm−1 band as the superoxide species on the pristine Co3O4 surface.197 This characteristic reaction intermediate band was largely suppressed for ZnCo2O4 (Fig. 9b), while present in CoAl2O4 (Fig. 9c), thus indicating the crucial role of the tetrahedrally coordinated CoII centers for the formation of this intermediate species. This observation is in contrast with previous hypotheses that octahedrally coordinated CoIII edge-sites are the dominant catalytically active species.190,193 One possible explanation for these opposite conclusions is that the nature of the catalytic centers may have changed even for related starting materials considering that many previous studies have reported substantial material transformations during catalytic reactions. High-resolution transmission electron microscopy (HRTEM), for example, revealed surface reorganizations of crystalline oxides and the in situ formation of amorphous overlayers.9,198,199 Dau et al. made pioneering contributions on this key issue of the structural transformations of heterogeneous catalysts under operational conditions.199 To illustrate the reconstruction of CoPi, they utilized a variety of techniques ranging from in-house spectroscopic characterizations (UV-vis and FTIR) to synchrotron-based structural analyses (XANES in Fig. 9d and EXAFS in Fig. 9e). The restructuring of the crystalline solids was actually under kinetic control. In a representative study by Jaegermann et al., CoOx electrocatalysts with different preparative histories exhibited distinct material transformation routes, and finally displayed very different catalytic activities.200 This highlights the fact that electronic state differences will lead to divergent reaction pathways and apparent performance differences. Moreover, instead of the traditionally proposed surface activity that prevails for crystalline materials, the concept of volume activity was proposed for the newly formed amorphous materials.199 By applying operando XAS analyses, it was found that even amorphous Co-WOCs undergo interconversion between three different local molecular motifs (Fig. 9f).12 These molecular-scale motifs were actually found to be in dynamic equilibrium during the OER. Furthermore, a reduction of the activity by 10% was observed when 18O isotope tests were performed, which served as evidence for the direct coupling of CoIV–oxo species during the RDS.14
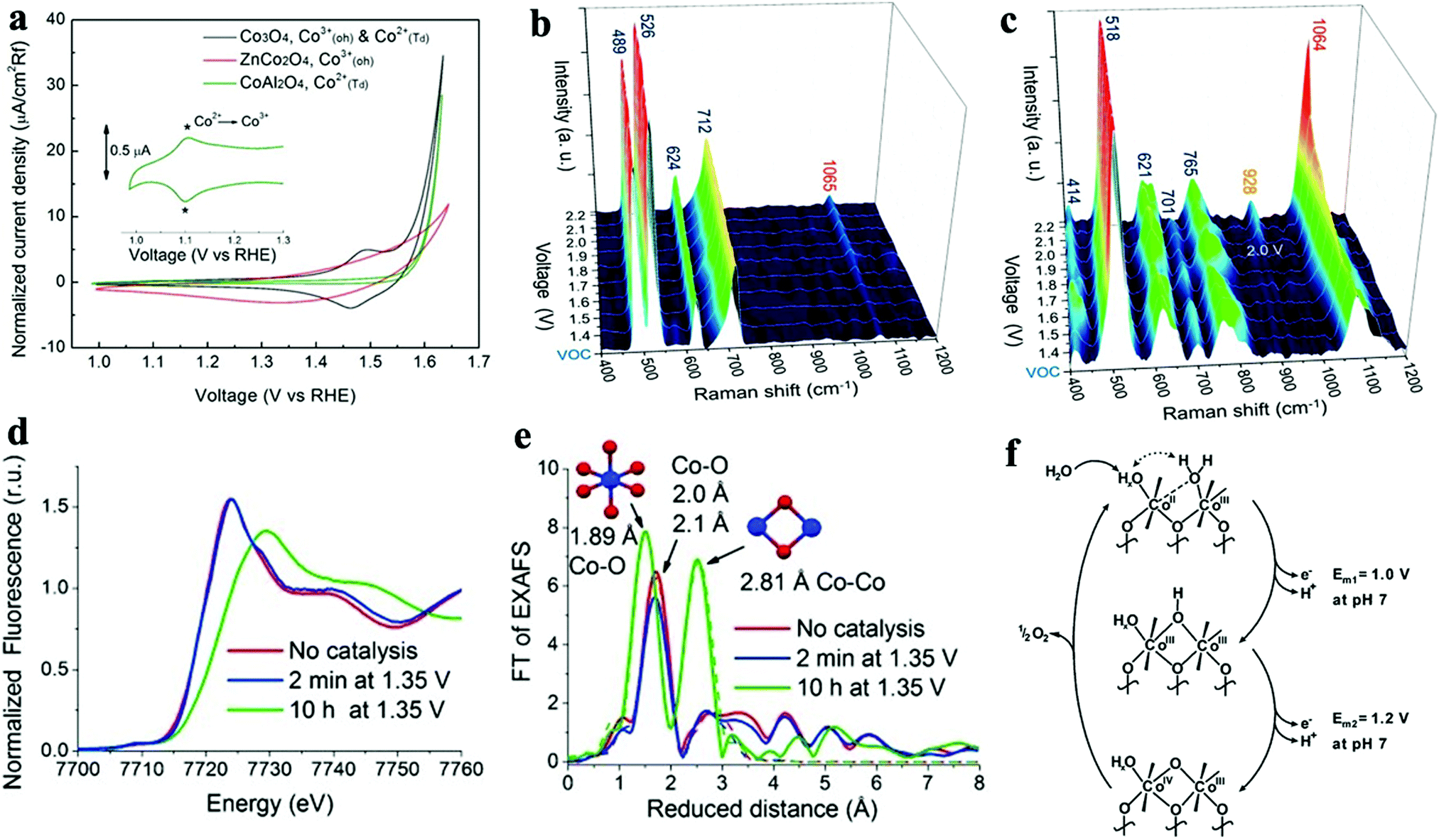 | ||
| Fig. 9 (a) Normalized electrochemical activity comparison of Co3O4, ZnCo2O4, and CoAl2O4. In situ Raman spectra of (b) ZnCo2O4 and (c) CoAl2O4 as a function of applied potential. Reproduced with permission from ref. 196 and 197. Copyright 2016, The American Chemical Society. XANES (d) and Fourier-transformed EXAFS (e) spectra of crystalline CoPi before and after anodic operation (1.76 V vs. RHE) for 2 min and 10 h. Reproduced with permission from ref. 199. Copyright 2015, Wiley VCH. (f) Schematic illustration of the three structural motifs during the OER. Reproduced with permission from ref. 12. Copyright 2015, The Royal Society of Chemistry. | ||
 | ||
| Fig. 10 Operando ATR-FTIR spectra of hematite scanned in the dark (a) and under illumination (b), with the reaction intermediate band at 898 cm−1 emerging at 1.83 V vs. RHE in the dark and 1.43 V vs. RHE under illumination. Reproduced with permission from ref. 202. Copyright 2016, Springer Nature. (c) Rate law analysis of surface hole density, where the surface hole density was derived from the intensity of the PIA signal at 650 nm at a steady state (1.5 V vs. RHE, 1 M NaOH). Reproduced with permission from ref. 204. Copyright 2015, The American Chemical Society. (d) Proposed mechanism for the third-order reaction: the equilibrium between triply oxidized edge-sites and three surface holes is followed by the RDS for O–O bond formation. Reproduced with permission from ref. 205. Copyright 2020, Springer Nature. | ||
With these and many other recent advances in mechanistic insights into unary first-row TM-based heterogeneous WOCs, it is clear that we are much closer to a full picture of the water oxidation cycle. The progress discussed here is inspirational and sometimes transferable for the rapidly growing number of first-row bi- or multi-metallic TM-based heterogeneous systems. It should be emphasized that the combination of many different techniques for multi-dimensional surface and structural information is required for the establishment of reliable and complete catalytic cycles.23 For example, time-resolved FTIR can provide molecular information of the active sites in action,208,209 while operando optical and XAS spectroscopy can uncover the electronic structure of the involved metal centres.210
2.2. Mechanistically driven design of heterogeneous WOCs
Advanced DFT simulations are an alternative approach to access the underlying OER mechanisms of heterogeneous WOCs. Nørskov et al. first studied the OER activity of a series of oxides and proposed that the binding energies of different reaction intermediates (HO*, O*, and HOO*) were responsible for the origin of the OER activity of the investigated catalysts.211 A universal scaling relationship between the adsorption energies of HOO* and O*, which are generally applied for the investigation of the reaction free energy diagrams of all the oxides, was discovered. This scaling relation gives rise to an activity volcano plot as a function of specific intermediates (Fig. 11a). Moreover, it suggests that the RDS of the free energy diagram plays a vital role in the OER activity. For instance, if the oxygen binding energy is too high, the OER overpotential is limited by the formation of HOO* species, otherwise, the energy barrier of HO* species formation is dominant when the oxygen binding energy is low. Therefore, to optimize the OER activity, methods to stabilize HOO* species compared to HO* species are desirable, and some general strategies are discussed below.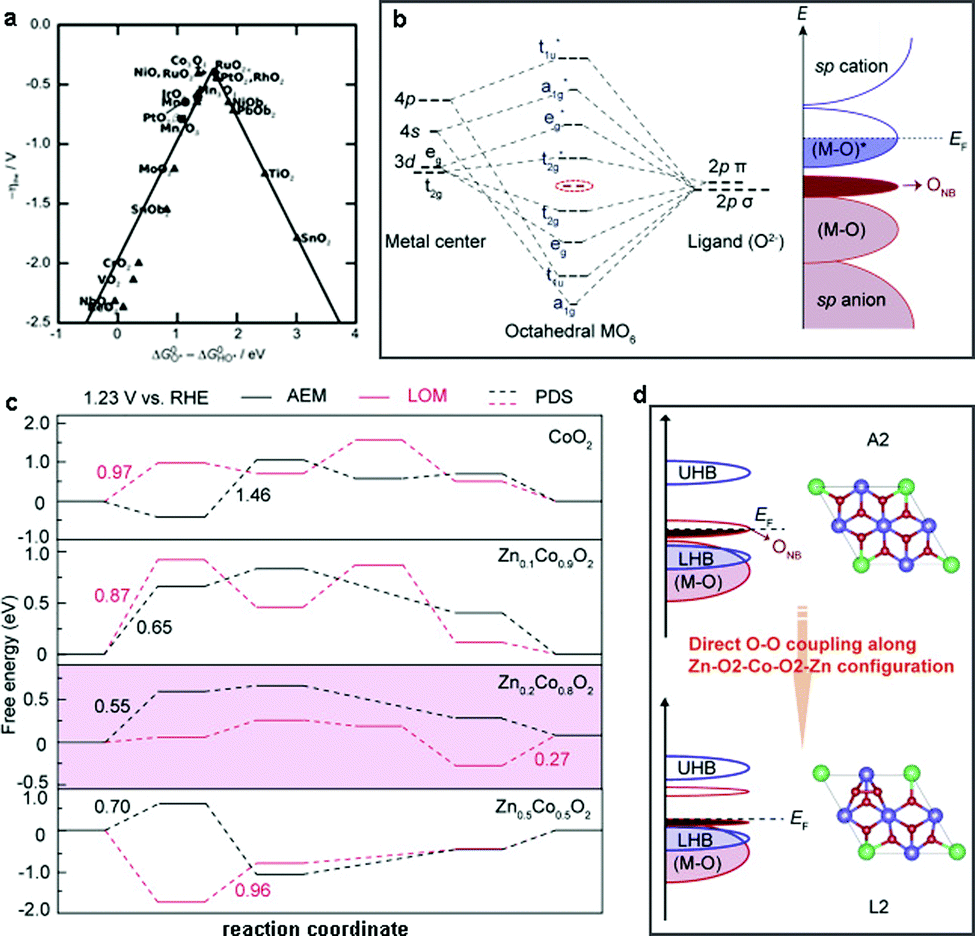 | ||
| Fig. 11 (a) OER activity trends vs. standard free energy of the ΔGHO*–ΔGO* step. Reproduced with permission from ref. 211. Copyright 2011, Wiley VCH. (b) Theoretical models for the formation of ONB with molecular orbital energy diagram for MO6 (EF = Fermi level). (c) Calculated free energies for the OER via AEM and LOM mechanisms over CoO2 and ZnxCo1−xO2. (d) Transformation of the OER mechanism from AEM to the LOM with the absence of unoccupied oxygen states in Zn0.2Co0.8O2. Reproduced with permission from ref. 224. Copyright 2019, Springer Nature. | ||
Recently, Wang et al. reported that the OER activity and mechanism of CoOOH could be appropriately modulated by the incorporation of redox-inert Zn2+ ions. Combined experimental and theoretical investigations showed that the insertion of Zn2+ into CoOOH could promote the generation of additional oxygen non-bonding states (ONB) with different local configurations (Fig. 11b).224 The presence of ONB can stabilize the peroxo- or superoxo-related O–O intermediates, thereby regulating the OER mechanism.226–228 Furthermore, DFT calculations confirmed that the less directional Zn–O bonds enhanced the electrical conductivities (σ) by overlapping with the neighboring O(2p) orbitals. As expected, the generated oxygen holes in the ONB states together with the Zn–O2–Co2–O2–Zn configuration tuned the oxygen binding energies and further promoted the change from a conventional adsorption evolution mechanism (AEM) to a lattice oxygen oxidation mechanism (LOM) (Fig. 11c and d). Consequently, the elaborately designed electrocatalyst Zn0.2Co0.8OOH with rich Zn–O2–Co2–O2–Zn configurations exhibited the best OER activity with the highest intrinsic mass activity of 0.38 mA cmox−2 (331.1 A g−1) at 1.5 V vs. RHE, which was 38(61) and 19(75) times better than that of CoOOH and commercial IrO2, respectively. However, lower intrinsic activities were found in Zn0.3Co0.7OOH and Zn0.4Co0.6OOH, which was mainly due to the lower content of Zn–O2–Co2–O2–Zn motifs. Therefore, high amounts of Zn substitution (over 20%) would decrease the OER activities and cause severe structural degradation of the catalysts due to the excessive amounts of oxygen vacancies. These observations were also confirmed by the investigation of the OER mechanism in spinel-type ZnCo2O4, which showed a decrease in OER activity compared to Co3O4 spinel due to the absence of Zn–O2–Co2–O2–Zn motifs.196,224 In conclusion, we suggest the following guidelines for the selection of secondary metal centers: (i) the charge conductivity of the catalyst matrix should be increased by regulation of its electronic structure, (ii) the adsorption energies of the reaction intermediates should be optimized by proper redistribution of the electron densities, and (iii) the reaction pathways should be accelerated by alteration of the energy barrier of certain steps.
 | ||
| Fig. 12 (a) O K-edge soft XAS spectra of L-CO, LN-CO, C-CO, and CN-CO and CoO and Co2O3 as references. (b) Calculated free energy profiles for the OER/ORR on {111} planes before and after the insertion of oxygen vacancies (green arrow = RDS). Reproduced with permission from ref. 236. Copyright 2019 Wiley VCH. (c) LSV curves of three catalysts and IrO2 and RuO2 references for the OER. (d) Computational DOS for Co3O4 and Co-deficient Co3−xO4. Reproduced with permission from ref. 234. Copyright 2018, The American Chemical Society. (e) OER performance of pristine NiFe-LDH and NiFe-LDH after 15 h of ball-milling treatment. (f) Theoretical slab model of NiFe-LDH with and without ball-milling treatment. (g) Computed free energy profiles for (f). Reproduced with permission from ref. 243. Copyright 2019, Wiley VCH. (h) Schematic plots for the strained NNO with eg orbital degeneracy. (i) OER performance for the NNO films grown on different substrates in O2-saturated 0.1 M KOH. (j) Plots of current density recorded at 1.63 V of NNO films against the induced strain. Reproduced with permission from ref. 244. Copyright 2019, The American Chemical Society. | ||
Other recent studies have further demonstrated that the introduction of metal cation vacancies (VM) is an effective alternative strategy to modulate the electronic structure and to tune the oxygen binding energy toward highly active OER catalysts.212,234,237,238 For example, Zou et al. synthesized Co-deficient Co3−xO4 and identified it as an efficient OER electrocatalyst.234 DFT calculations were conducted to investigate the electronic structure of Co3O4 before and after the introduction of VCo. The simulated results showed that the formation energy of VCo2+ was lower than that of VCo3+, indicating that the preferential type of vacancies in Co3O4 is VCo2+. Moreover, the presence of high VCo2+ concentrations can modulate the electron density of the Co centers in Co3−xO4, resulting in the electronic delocalization of the neighboring atoms and further increase of the overlap of the electron wavefunctions between the Co and O atoms. Consequently, the electronic delocalization effect enhanced the charge densities around the conduction band edge and narrowed the band gap in Co-deficient Co3−xO4 (Fig. 12d). In fact, the insertion of VCo2+ is accompanied by structural distortions, which decreased the surface energy and enhanced the structural stability during the OER. Consequently, the modulated Co-deficient Co3−xO4 displayed remarkable OER activity with a much lower overpotential of 268 mV at a current density of 10 mA cm−2 in comparison with that of normal Co3O4 (376 mV) (Fig. 12c). Overall, we noticed that the strategy of vacancy introduction is attracting increasing attention as an effective way to significantly improved catalyst properties, from exposing more surface sites to enhanced electrical properties.239 However, fundamental questions, such as ‘the more vacancies, the better?’ and ‘are vacancies stationary or mobile?’ remain to be answered for fully understanding the role of these structural defects.
 | ||
| Fig. 13 (a) LSV curves for the investigated cobalt oxide OER catalysts. (b) Surface atomic configurations of Co2+/Co3+ on different crystal planes. Reproduced with permission from ref. 251. Copyright 2018, Wiley VCH. (c) TEM and HRTEM images of NiFe–N–CNT–rGO. (d) LSV curves of as-prepared NiFe, NiFe–N–CNT–rGO, NiFe–N–CNT, and rGO, with blank GC as a reference. (e) Schematic structures of OER intermediates adsorbed on NiFe–CNT. (f) Calculated free energy for OER over pure CNT and NiFe–CNT at zero potential (U = 0), equilibrium potential (U = 1.23 V), and minimal potential where all steps become downhill. Reproduced with permission from ref. 171. Copyright 2019, The Royal Society of Chemistry. | ||
Recently, Khani et al. reported a convenient two-step sol–gel and carbothermal reduction strategy and prepared a series of graphitic-shell encapsulated metal catalysts (denoted as G–Ni, G–Co, G–Fe, G–Ni0.5Fe0.5, G–Ni0.5Co0.5, G–Co0.5Fe0.5, and G–Ni1/3Co1/3Fe1/3).256 Various ex situ techniques including powder XRD, Raman, and XPS spectroscopy confirmed that some metallic nanoparticles without the protection of graphitic shells were converted into metal oxide species after the OER. The graphitic-shell encapsulation of the metal catalysts not only preserved their pristine crystal structure, but also maintained their high electrocatalytic activity for a long time under the harsh electrochemical conditions. The results showed that the as-prepared G–Ni0.5Fe0.5 showed a low overpotential of 280 mV at 10 mA cm−2. All the above studies demonstrate that the rational utilization of a protective matrix material is a promising strategy for maintaining the structural integrity and high OER activity of electrocatalysts. It should be noted that many electrocatalysts generally underwent surface structural reconstruction under harsh oxidative conditions, which could affect their electrocatalytically active species and surface areas. Since this makes mechanistic investigations more complex, researchers should focus on the exploration of the real active species and aim to understand the dynamic processes under the operational reaction conditions. These challenges for the investigation of heterogeneous WOCs are summarized in the next sections.
2.3. Challenges in the mechanistic investigation of heterogeneous WOCs
The influence of many different parameters on the water oxidation performance, such as catalyst surface change, redox chemistry, defect chemistry, electrolyte, gas–liquid–solid interface, and adsorption/desorption energy as a function of the mentioned parameters, render a deep understanding of the catalytic process challenging. Therefore, the assignment of a distinct catalytic mechanism to a given heterogeneous water oxidation catalyst requires considerable analytical efforts to address all the issues discussed in the following sections.Perovskites have attracted particular interest among heterogeneous catalysts recently.263–267 Advances in this field have shown that the apparently stable perovskites can indeed transform into other phases when exposed to the corrosive conditions of water splitting.268–272 Suntivich et al. showed that in the initial stages of the catalysis, the crystalline Ba0.5Sr0.5Co0.8Fe0.2O3−δ mixed oxide (BSCF) changes and forms an amorphous mixed oxide shell on the surface, which acts as the true catalyst in the OER (Fig. 14a–d).273 In the operando electrochemical XAS study by Schmidt et al., the structural reconstruction and the dynamic surface amorphization of BSCF were resolved in detail.274 In this case, the Co atoms reached higher oxidation states and acted as the major catalytic centers in the BSCF structure.
 | ||
Fig. 14 HRTEM images and corresponding fast Fourier transforms (FFT) of (a) pre-catalytic Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF82) perovskite and BSCF82 samples after (b) 5 cycles and (c) 100 cycles. (d) Post-catalytic BSCF82 sample after 2 h chronopotentiometry at 1.7 V vs. RHE in 0.1 M KOH electrolyte at 10 mV s−1. Reproduced with permission from ref. 273. Copyright 2012, The American Chemical Society. (e) Fourier transform (FT) k3-weighted Co K-edge EXAFS spectra of BSCF-FS sample at different applied potentials in 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M KOH. The shoulder at ∼3.5 Å and the peak at ∼4 Å correspond to Co–Ba/Sr and Co–Co/Fe in corner-sharing octahedra in the perovskite structure, respectively. (f) Schematic representation of amorphization and lattice OER processes in BSCF-FS, which leads to leaching of Ba2+ and Sr2+ cations and dissolution of Co and Fe cations, respectively. Reproduced with permission from ref. 274. Copyright 2017, Springer Nature. M KOH. The shoulder at ∼3.5 Å and the peak at ∼4 Å correspond to Co–Ba/Sr and Co–Co/Fe in corner-sharing octahedra in the perovskite structure, respectively. (f) Schematic representation of amorphization and lattice OER processes in BSCF-FS, which leads to leaching of Ba2+ and Sr2+ cations and dissolution of Co and Fe cations, respectively. Reproduced with permission from ref. 274. Copyright 2017, Springer Nature. | ||
Additionally, in situ FT-EXAFS spectra further clarified that the growth of an amorphous Co/FeO(OH) shell on the electrode–electrolyte interface was induced by the lattice OER process (Fig. 14e and f).274 Surface reconstruction of these mixed oxides is usually accompanied by changes in the distribution of short-range order, the formation of local defects in the lattice structure, and oxygen vacancy changes.275,276 All these key factors must be considered in fundamental investigations of the OER mechanism.
The influence of surface corrosion on the lattice counter-anions makes the mechanistic understanding of heterogeneous OER catalysts even more complicated, giving rise to in-depth investigations.277,278 Despite the considerable OER activity of non-oxide first-row TM compounds, such as sulfides, selenides, and nitrides, these compounds are not thermodynamically stable under the highly oxidative OER conditions and are mostly converted to metal oxide hydroxide phases. Recently, various characterizations have been performed to screen the surface evolution and phase transition of non-oxide compounds during their OER performance.277–280 Fan et al. applied in situ HRTEM to study the structural changes of an amorphous CoSx chalcogenide during the OER. Their study showed that in the first step, the CoSx pre-catalyst is transformed into a Co(OH)2 intermediate. Next, the intermediate undergoes irreversible conversion into a crystalline CoOOH phase on the surface.279
In selected cases, the transformation of the crystalline pre-catalyst into an amorphous phase as the true active catalyst has been identified as a reversible process with different in situ X-ray techniques.9,281 The possibility of these reversible phenomena makes the precise identification of catalytically active sites via operando methods for the mechanistic understanding of OER highly important.
When trying to assess the various performance-related factors of solid-state WOCs, such as the degree of crystallinity, surface corrosion, particle size and morphology, the thickness of the electrocatalyst, and oxidation state of the active sites, the combination of different in situ methods can provide further fundamental understanding of the catalytic performance and the reaction mechanism. Recently, several in situ experiments have been undertaken to monitor the reaction intermediates and to specify redox state and local atomic structure of the active sites. Among the different applied in situ techniques, XAS,282–285 TEM,279,286 Raman,287 and Mössbauer spectroscopy175,288 are referred to here.
Impurities from the electrolyte are another crucial factor that can affect the OER activity of heterogeneous catalysts and the real active species.287,289,290 Bell et al. reinvestigated the role of Fe impurities in the OER activity of Ni(OH)2/NiOOH. They showed that aging an Ni-(oxy)hydroxide film in unpurified KOH electrolyte with Fe traces led to the formation of an Fe-doped Ni-oxy/hydroxide species. The newly formed phase displayed OER activity comparable to optimized Ni1−xFexOOH catalysts.287 Najafpour et al. revealed that the presence of Fe impurities in KOH electrolyte plays a significant role in the OER activity of NiV-LDH. Using operando UV-vis spectrophotometry and EXAFS techniques, it was shown that during water oxidation in impure KOH, the NiV-LDH catalyst is converted to a partially oxidized α-Ni(OH)2/NiOOH phase with Fe traces and this structure acts as the true catalyst.290
Regarding the dependence of different reports on the applied techniques and respective conditions, the obtained results should be interpreted carefully. Consequently, the results of case studies should not be considered as general concepts for reliably predicting key performance parameters and active species. Additionally, considering the critical role of the topmost layers of the electrocatalysts in the catalytic performance of the material and the kinetics and mechanism of the reaction, comprehensive monitoring of heterogeneous catalysts with atomic level operando techniques becomes mandatory. Although many reports add information on the bulk characteristics of catalysts, information based on in situ spectroscopic methods at the atomic scale, such as environmental TEM, are now required to derive optimization concepts for solid-state WOCs. Furthermore, these data can be supported by atomic-level computational modelling and simulation techniques on the path to predictive design.
Accordingly, surface reconstruction in the gas–liquid–solid interface merits more in-depth investigations with suitable operando techniques. This approach holds the key to a more comprehensive understanding of true water oxidation catalysts. Despite the rich body of literature on true catalyst studies in water oxidation, there is an obvious and increasing need for systematic studies of catalytic interfaces at the atomic scale by operando methods. Furthermore, this is an incentive to make them more accessible for everyday lab practice and to combine the results with advanced computational methods. Accordingly, artificial intelligence and different types of machine learning can speed up the exploration of reaction intermediates and the knowledge-based discovery of stable and highly efficient WOCs.291–293
Part III: perspectives for knowledge transfer between molecular and heterogeneous WOCs
3.1. Lessons learned from heterogeneous WOCs
Over the past decades, progress in heterogeneous WOCs was driven by both mechanistic understanding and new catalyst design concepts, as outlined in Part II of this review. We proceed with new options to transfer this rich knowledge to the field of molecular WOC development. In the following, we present some case studies and highlight key overlap between both research areas. | ||
| Fig. 15 Surface structure of Co3O4 electrocatalysts at non-catalytic (a, crystalline) and OER potentials (b, amorphous surface shell). Reproduced with permission from ref. 9. Copyright 2015 Springer Nature. (c) Molecular structure of the disordered moieties of Co-dppeO2 and the distorted edge-site motif {H2O–Co2(OH)2–OH2}. Reproduced with permission from ref. 22. Copyright 2019, Elsevier. (d) TOF evolution as a function of different amounts of cobalt centers (CoOx), where the near-zero slopes at lower operational potentials indicate that all the cobalt sites contribute equally across the deposited film. Reproduced with permission from ref. 304. Copyright 2014, Wiley VCH. | ||
However, this re-crystallization phenomenon is not a common observation for most electrocatalysts, which frequently display an ultrafine nano-structure or relatively low crystallinity.297–299 In this case, the ‘surface only’ model of catalytic heterogeneous WOCs requires adjustments since the amorphization can propagate towards the bulk region.199 The extension and rate of this irreversible structural reconstruction depends on many parameters such as the preparative history and nanostructure of heterogeneous WOCs, and on relevant reaction conditions, such as the anodic potential. Correspondingly, to derive convincing structure–activity relations for heterogeneous WOCs, confirmation of the molecular structure of newly developed amorphous materials is of great importance.12 In this regard, we employed the new disordered 1D coordination polymer WOC Co-dppeO2 as a model system and identified the edge-site {H2O–Co2(OH)2–OH2} motif as the active center, which was embedded into a ligand matrix (see Section 1.1.1), in close analogy to molecular cubane WOCs and Co3O4.22,26,210 The atomically resolved molecular motif within the highly disordered material was further refined with the coupling of in situ spectroscopic data and computational modeling (Fig. 15c, cf. Section 1.1.1). In addition, the direct utilization of amorphous WOCs promotes the participation of a higher fraction of metal centers in the catalytic reaction.300–303 For example, in a representative study, amorphous CoOx was found to be composed of nanoscale fragments, which were inter-connected by edge-sharing CoO6 octahedral dimers as a molecular motif.304,305 These dimers were randomly organized into polymeric networks. When exposed to electrolyte, they were saturated with water molecules and cations and anions from the reaction medium. This semi-permeability enabled the equal contribution of exposed metal centers when the mass diffusion was sufficient (Fig. 15d). An increasing number of studies have revealed that the real active motifs of most heterogeneous WOCs indeed bear close resemblance to molecular moieties, which is an prerequisite for mutual knowledge transfer.12
 | ||
| Fig. 16 (a) Structure of CoII4O4-dpk cubane representing a surface cut-out of the μ-oxo-bridged CoOx catalyst. Reproduced with permission from ref. 26. Copyright 2017, The American Chemical Society. (b) OER volcano plot summarized for a wide range of metal oxide surfaces employing ΔGO–ΔGOH as a single descriptor. (c) Comparison of the theoretical OER overpotentials on pure WO3(001), CoOOH(0112), FeOOH(010) and Fe,W-doped CoOOH, where the FeCoW oxyhydroxide shows the lowest overpotential. Reproduced with permission from ref. 315 and 324. Copyright 2017 and 2016, AAAS. | ||
Meanwhile, it is noteworthy that Mn cubanes behave less straightforward as molecular WOCs (see Section 1.2.3).122 Along the lines of cooperative molecular nuclearity motifs, an innovative pentanuclear iron complex (shown in Fig. 5) was designed and found to exhibit a high TOF of 1900 s−1, which outperformed previous lower nuclear iron-based molecular WOCs by at least three orders of magnitude.135 The superior activity was ascribed to (i) the presence of adjacent metal centers that significantly decreased the thermodynamic barrier for intramolecular O–O bond formation,99,101 and (ii) the large redox flexibility of the pentanuclear core, which could accommodate multiple oxidation equivalents. Therefore, we believe that the edge-site feature of heterogeneous WOCs can inspire future molecular design in the long term thanks to its remarkable flexibility in many aspects such as ligand and metal selection, along with tunability of electronic and protonation states. In addition, the efficient delocalization of oxidation equivalents over the polymeric metal centers on heterogeneous WOCs can be applied as a reciprocal guideline for designing molecular systems with higher nuclearity.118
Overall, the fast-growing field of heterogeneous WOCs has created many design principles and will continue to introduce more in the near future. With the discovery of shared mechanistic principles, the knowledge transfer from the pool of heterogeneous to molecular WOCs will greatly promote the design of the latter. Meanwhile, progress in molecular design can be inspiration for heterogeneous catalyst optimization, as we point out in the next section.
3.2. Lessons from molecular studies for heterogeneous WOC design
According to the mechanistic analyses presented in Part I and II, it is clear that molecular WOCs excel mainly through well-defined atomic structures, which is essential for an in-depth understanding of reaction intermediates. However, their isolation for heterogeneous WOCs remains a significant challenge, despite select success on some oxide surfaces such as CoOx and Fe2O3 (cf. Section 2.1). Therefore, identifying the structural similarities between molecular and heterogeneous WOCs at the atomic level can facilitate the transfer of mechanistic understanding between both catalyst types. In principle, this knowledge transfer can start by translating salient structural features of molecular WOCs to the surface or edge-sites of their heterogeneous counterparts. To date, the number of these examples in the heterogeneous field is still too limited to elaborate widely here. Accordingly, here we focus on the frontier research area of single-atom catalysts (SACs). SACs are presently the closest to the emerging bridging concept between molecular and heterogeneous WOCs because they feature the atomic dispersion of metal atoms on or in specific substrates as robust active centers for catalytic reactions.329 Therefore, SACs are actively connecting heterogeneous and molecular design concepts, and they are ideal platforms for bilateral knowledge transfer. In the following, we summarize recent research trends on SACs, with special emphasis on their molecular aspects of synthesis and structural–performance relationships, and on the debate on single vs. dual/multi-site systems.Support-coordinated SACs. Many materials have been reported as supports for SACs. Two types of supports are classified with respect to their contributions to the OER, namely non-catalytic supports and catalytic supports. In the case of non-catalytic supports, they adopt the role of “ligands”, which stabilize the metal centers and regulate their charge distribution. To implant single-metal atoms into the supports, their pretreatment by inducing defects or vacancies is necessary. One of the typical examples is the immobilization of single atoms in the lattice of defective graphene oxide (GO). Yao et al. reported on a Ni-SAC,331 where they first created increasingly defective GO sheets through thermally annealing a mixture of graphene and melamine under an inert atmosphere, and then anchored Ni ions onto the surface of this pretreated GO through an electrostatic adsorption process in solution. The final Ni SACs coordinated on GO were obtained via a pyrolysis process, leading to the formation of Ni–C bonds in the GO sheets.
In addition to the direct carbon coordination, most graphene-based SACs were coordinated by N atoms to form the typical M–N4 moieties on the graphene lattice. For instance, Huang et al. obtained Ni-, Co- and Fe-SACs coordinated by M–N4 moieties by subjecting defective graphene to a pyrolysis reaction under an NH3 atmosphere (Fig. 17a).332 The coordination geometries and the dispersion of single atoms were directly detected by HRTEM, as shown in Fig. 17b–d. N-coordination can not only modify the electronic structure of the isolated metal atoms, but also improve the adsorption of the reactants.333 In a recent study on Mn-SACs (Mn–N4–G), Li et al. evidenced that the four-fold N-coordination favors the formation of nucleophilic MnIV–oxo species during the OER, which leads to further attack of another water molecule in the electrolyte for O–O bond formation.334 However, the reaction mechanism in this study was solely supported by DFT calculations, and thus further experimental studies on the identification of the real functionality of N atoms are still expected. Other carbon-based SACs, such as those supported on CNT or C3N4, were also reported as efficient WOCs emerging from similar fabrication processes.335,336
 | ||
| Fig. 17 (a) Typical synthetic protocol of SACs coordinated by defective N-doped graphene. (b–d) High-resolution TEM images of graphene-coordinated Ni, Co and Fe SACs; scale bars = 0.5 nm. (e) Structural model showing Ru single atoms on PtCu alloy (blue, grey and purple represent Pt, Cu and Ru, respectively). (f) FT-EXAFS spectra of the Ru K-edge and reference samples. Reproduced with permission from ref. 332 and 337. Copyright 2018 and 2019, Springer Nature. (g) Cu molecules stabilized by Gr. Reproduced with permission from ref. 160. Copyright 2017, The American Chemical Society. | ||
Recently, alloys were introduced as alternative supports for SACs. Li et al. reported single Ru atoms supported by a PtCu alloy for the OER, with a minimized overpotential of 90 mV at a current density of 10 mA cm−2 (Fig. 17e and f).337 They found that the compressive strain from the PtCu lattice engineers the electronic structure of the isolated Ru atoms through the chemical bonds formed with Pt and Cu atoms in the support. Further investigations showed that the bulk alloy could also act as an electron reservoir to donate electrons to reaction intermediates, resulting in the prevention of the over-oxidation and dissolution of Ru atoms, which enhances their stability towards the OER.337 Although non-catalytic supports do not directly participate in the OER process, they can influence the properties of single atoms by modifying the electronic structures and optimizing the adsorption energies of the reactants.
Complementing the non-catalytic supports, catalytic supports have been also widely reported in water oxidation.338 Studies on an Au/NiFe-LDH SAC system reported by Zhang et al. demonstrated that the hybridization of Au atoms after coordinating with O atoms in NiFe-LDH promoted the charge redistribution of the active Fe and Ni atoms, resulting in enhanced OER activity of the Fe sites.339 A similar result was reported by Lin et al. for Pt/NiO-SAC, where the Pt single atoms promote the phase transformation from NiO to γ-NiOOH rather than acting as active sites.340 On the other hand, there are also many examples of active single atoms coordinated with catalytic supports. In a representative study, Sun et al. found that the Ru atoms in Ru/CoFe-LDH systems are more likely to represent the active sites compared to the Fe sites. However, the authors did not state whether Co and Fe also participate as active sites in the OER.341 A similar conclusion was also reported by Ma et al. for a W/α-Ni(OH)2 SAC.342 However, in the more recently developed Ir/NiO SAC system, Gu et al. found that the single Ir atoms not only serve as the active sites for the OER but also improve the reactivity of NiO. Therein, the formed *OH species on Ni sites could easily relocate to the surrounding Ir sites to create a joint reaction pathway.343 The discussion of the real active site in a catalytic support should consider both the electronic structure effects and multi-site reaction mechanisms. Current studies mainly rely on DFT calculations, and thus more experimental evidence based on recent advanced techniques, such as ATR-FTIR and operando XAS, as mentioned in Parts I and II, is required to unambiguously identify the active sites and their reaction mechanisms in future investigations.
The discussed examples clearly show that SACs provide a platform for understanding the behavior and functionalities of isolated reaction centers and their surrounding atoms during the OER. Therefore, the design and synthesis of SACs coordinated by non-catalytic supports (such as graphene) is one of the most convenient ways to exclude the influence of the support. However, their current OER activities do not yet meet application-oriented standards. SACs coordinated by catalytic supports are much more promising in terms of their performance; however, the nature of their catalytic supports, such as oxides, alloys and hydroxides and LDHs, brings us back to the above question: what are the conceptual differences between these SACs and the traditional heterogeneous catalysts? Therefore, when investigating SACs coordinated by catalytic supports, more emphasis should be placed on understanding their coordination–performance relationships, which can bring forward future design guidelines for more active SACs in applied OER.
Support-stabilized SACs. Although the development of support-coordinated SACs has turned into an important branch, the precise control of the coordination environment with the current widely used pyrolysis strategy for anchoring single metal atoms is still a massive preparative challenge. Researchers with expertise in molecular catalysis developed a molecule-support stabilization strategy, which paves another way to achieve both recyclable and well-coordinated SACs. However, in this protocol, establishing a strong chemical connection between molecules and solid supports remains difficult. Thus, many oxide supports have been reported to address this issue31 based on the hypothesis that molecules can be stabilized by the hydroxyl termination of the oxides, which connects most of these studies to oxide-based photochemical water oxidation using molecules as cocatalysts.4 We implemented an independent part in Section 3.3.1 to discuss this issue. Therefore, the focus in this section is on inert supports for stabilizing molecular catalysts.
Classic examples are graphene-like supports. Llobet et al. proposed a π-stacking strategy to stabilize Cu molecules on graphene sheets. It has been shown that the pyrene group in the molecule acts as the anchoring unit connecting the molecules to the graphene surface via π-conjugation (Fig. 17g).160 The OER performance was notably improved due to the extended π-delocalization arising from graphene. In a related study, N-doped defective graphene was used for supporting cobalt phthalocyanine (CoPc) molecules.344 The investigations showed that the Co center in the molecule was further coordinated by another N atom from the graphene support to form a Co–N5 moiety, acting as the active site for the OER. In addition to graphene, other carbon-based supports such as CNTs have been employed to stabilize Ru-based molecular catalysts.345,346 There are two reasons why carbon-based supports are widely developed to bridge homogeneous and heterogeneous catalysis. Firstly, their large surface area provides abundant anchoring sites to stabilize molecules; secondly, C–C sp2 hybridization provides extended π-delocalization, which offers the possibility of forming chemical bonds and electron transfer tunnels.347,348 The support-stabilized strategy can preserve the coordination environment of the original molecules to a large extent, thereby providing ample opportunities for the chemical design and optimization of molecules with high OER activities.
More recently, a key study of Ni-based SACs was reported by Feng et al., where one N atom was replaced with an S atom in the Ni–N4 moiety and the OER activity outperformed most of the current TM- and/or heteroatom-doped carbon-based electrocatalysts (Fig. 18a and b).353 It is well known that N coordinates more strongly to metal centers than S. The N/S replacement increased the distortion of the SAC, which rendered the M–N3S moiety chemically more active. In addition, the optimization of the coordinating atoms can also regulate the density-of-states distribution and the adsorption of OH groups and enhance the electron transfer ability of the metal centers (Fig. 18c). Accordingly, Wu et al. further constructed an M–O4 moiety, which exhibited an extremely low overpotential of 224 mV at a current density of 10 mA cm−2 for the OER (Fig. 18d).354 Theoretical studies showed that the stronger electronegativity of the O atoms is favorable for OH− adsorption, thus leading to the formation of an Ni–O4(OH)2 configuration with a high oxidation state for the Ni atoms (Fig. 18e and f). Similar studies on coordination–performance relationships have been also reported for SACs with other applications.355
 | ||
| Fig. 18 (a) Fourier-transformed magnitudes of the experimental K-edge EXAFS signals of Ni–N3S SAC. (b) Structural geometry of Ni–N3S SAC with O–O bond. (c) OER volcano plot of the overpotential η vs. the difference between the adsorption free energy of *O and *OH for the N–S, Ni–S, Ni–N4, and Ni–N3S models. Reproduced with permission from ref. 353. Copyright 2019, Springer Nature. (d) Atomic-scale HAADF-STEM image of Ni–O4–G SACs. (e) FT-EXAFS spectra of Ni–O4–G SACs with NiO and Ni foil references. (d) FT-EXAFS data of the proposed Ni–O4(OH)2 architecture compared to experimental Ni–O4. Reproduced with permission from ref. 354. Copyright 2020, Wiley VCH. | ||
The regulation of the coordination environment is an increasingly important research trend for improving the OER activity of SACs.356 However, the widely reported SACs with M–N4 moieties still show limited activity towards the four-electron transfer process of the OER because the strong coordination ability of N obstructs the electron transfer and adsorption of oxygen groups (e.g., H2O and *OH). Therefore, partial replacement of N atoms with other non-metal atoms, such as S, O and P, may provide an alternative option for improving the OER activity. However, the fact that the weak chemical bonding between the metal atoms and O/S atoms may reduce the stability should also be considered for these optimization strategies. Finally, we want to highlight that a well-defined coordination environment is of great importance for the interpretation of coordination–activity relationships. Thus, we recently introduced a soft-landing molecular strategy to generate well-defined model SAC targets.357 The function of M–N4 moieties could therein be unambiguously investigated in a near-perfect coordination environment.
Recently, Wang et al. found that the introduction of Fe3+ into graphene-conjugated molecular Ni2+ (Fig. 19a) could significantly increase the OER activity and stability of the active Ni–Fe sites of the catalyst with a distance of 2.7 Å between the two metal centers. It was observed that both the Ni and Fe sites could couple with the OH− ions through terminal and bridging geometries (Fig. 19b and c). Ni–Fe dual sites for water oxidation have been widely studied in traditional heterogeneous WOCs, as discussed in Section 2.2.1; however, this is the first experimental evidence reported to explain the synergistic effects of Ni and Fe at the atomic level.360 Another important example of a dual site system is the Co–Fe SACs reported by the Hu group. Their study showed that the high activity of Co–Fe SACs arises from a series of structural and coordination evolution processes during the OER in electrolyte containing a certain amount of Fe3+ ions (Fig. 19d). The Co atoms coordinated to the N-doped carbon support first interacted with OH−/OH2 species in the electrolyte, and then slowly bonded with trace amounts of Fe3+ ions in solution to form the Co–Fe site (Fig. 19e). The enhanced activity was ascribed to the joint functionalities of the Co–Fe–N–C motif.361 Although the authors did not elaborate on the detailed reaction mechanism, the in situ XAS data for the formation process of the Co–Fe–N–C site is of great significance to understand the reaction mechanism. Surprisingly, we could not find further studies on dual or multi-site SACs for the OER beyond the two works mentioned above at the time of writing this review. This leaves plenty of room for new explorations because multi-site SACs do not only hold the potential for high performance, but also provide a unique platform for investigating the role of each different metal center in the synergistic interaction of two metal atoms at the atomic level. This holds the key to the much sought-after general structure–performance relationships of WOCs. However, this research is still in its infancy, and the design and synthesis of these SACs are expected to greatly benefit from bi-/poly-nuclear molecular catalyst design concepts, as discussed in Part I.
 | ||
| Fig. 19 (a) DFT calculation-derived model of HG-Ni SAC. (b) ks of metal redox in HG-Ni, HG-NiFex, and HG-NiFe. (c) Redox potentials of Ni–Fe sites versus pH value of KOH solution. The inset shows the proposed configuration for the adsorption of HO− ions onto Ni–Fe sites for OER. Reproduced with permission from ref. 360. Copyright 2018 AAAS. (d) LSVs of the Co–N–C (single atom Co dispersed on a N-doped carbon support) with different qualities of KOH (and different concentrations of Fe3+). (e) Fourier transform of Co K-edge EXAFS spectra for as-prepared Co–N–C and the structure changes in KOH solution with different concentrations of Fe3+ under OER conditions. Reproduced with permission from ref. 361. Copyright 2019, The American Chemical Society. | ||
3.3. Combining the advantages of molecular and heterogeneous WOCs
While the previous sections were focused on mutual knowledge transfer between molecular and heterogeneous catalysts, we finally cover emerging hybrid strategies based on the direct coupling of both molecular and heterogeneous WOCs.362,363 In addition to the benefits of stabilizing molecular WOCs, as discussed in Section 1.3.2, hybrid systems also provide the opportunity to preserve their inherent advantages, such as synthetic flexibility, atomic-scale monitoring/modeling and structure-function correlations, as elaborated in Part I. They can be combined with the robustness, synthetically convenient tailoring, and favorable application-oriented features of heterogeneous WOCs (cf. Part II).345,364,365 More importantly, hybrid systems offer the possibility to overcome the limitations of both catalyst types.366,367 In the following, we highlight some key strategies such as hybrid molecular-heterogeneous (photo)electrodes or encapsulation strategies and briefly analyze their major advantages and remaining challenges.To facilitate the design and engineering of efficient hybrid devices, one important question has to be answered: what are the individual functions of the heterogeneous substrate and of the immobilized molecular species? For electrocatalytic devices, it is obvious that molecular WOCs are the dominant catalytic centers, especially when an inert substrate is employed. Concerning photoelectrochemical devices, the strong interplay of immobilized molecular cocatalysts and semiconductor substrate demands a stronger separation of their individual functions.378 The OER process of photoelectrochemical devices is deeply entangled with charge carrier separation and transportation processes. Therefore, the role of surface-loaded molecular WOCs is expected to be evolutionary rather than stationary. To clarify these complex relationships, our group employed a model system consisting of molecular Co4O4 cubanes immobilized on a hematite photoanode.379 For the first time, we identified the dynamic functionality of the molecular species as a function of the applied potential (Fig. 20a). Briefly, surface cubanes were activated first around the onset potential, and at moderate potentials they started to accumulate large amounts of oxidative equivalents. However, they were unable to transfer them efficiently to the electrolyte at moderate potentials since the competing hematite surface states were more catalytically active. At elevated potentials, the surface molecular WOCs finally started to contribute significantly to the OER. This study highlighted that the role of molecular WOCs in hybrid systems is not necessarily limited to cocatalysts. Instead, their functionality depends on the operational conditions and is closely linked to the properties of the heterogeneous substrate. We believe that understanding the interactive relationship between the immobilized molecular species and the underlying heterogeneous materials is crucial for the selection of optimal operation conditions and deserves to be at the center of future studies.
 | ||
| Fig. 20 (a) Dynamic role of molecular cocatalysts on hematite photoanodes upon the modulation of applied potentials. Reproduced with permission from ref. 379. Copyright 2019, The American Chemical Society. (b) Building unit of MOFs with programmable linkers that also function as a protecting ligand for molecular WOCs. Reproduced with permission from ref. 275. Copyright 2019, the National Academy of Sciences. | ||
Meanwhile, we have to point out that the development of hybrid systems also faces several major analytical challenges. Just to mention one, it is very demanding to probe their molecular structure after their immobilization.380 Similarly, their molecular integrity under operational conditions is another critical issue since both properties are essential for drawing any meaningful mechanistic conclusions, as detailed in Section 1.3. In this context, advanced surface-sensitive and atomic-level resolution techniques, such as HAADF-STEM, AC-TEM and XAS, are important to clarify these questions, as outlined in Section 1.1.1.
Overall, molecular-heterogeneous hybrid systems keep attracting interdisciplinary attention. Their advancement is not only of interest to promote industrial-scale AP with new components and building blocks, but also to explore fundamental aspects of catalyst design and stability. More importantly, these “crossover” studies are pushing the conventional boundaries between molecular and heterogeneous catalysts, which will considerably expedite the knowledge transfer and progress in catalyst optimization.
3.4. Enhanced knowledge transfer with advanced characterization techniques
Since the transfer of knowledge between molecular and heterogeneous WOCs relies on structural similarities at the atomic level, detailed atomic resolution models of both catalyst types are indispensable. Generally, this poses no special challenges for molecular WOCs due to the widely applicable crystallization and X-ray crystallography methods. However, for heterogeneous WOCs, this task is more problematic since most catalytically active solids are not perfectly crystalline materials, but contain many structural defects up to bulk disorder or amorphization. In this case, advanced characteristic techniques such as XAS, XFEL diffraction and PDF analysis are crucial for investigating the coordination environment and electronic structure of solid catalysts. Detailed structural insight into the surfaces or edges of heterogeneous WOCs is decisive for knowledge transfer to the molecular field, especially for the selective tailoring of molecular WOCs mimicking the motifs of efficient heterogeneous WOCs. Consequently, the as-constructed molecular WOCs can serve as important models for an in-depth understanding of the detailed reaction mechanisms of the original heterogeneous WOCs. Here, we can benefit from the wide accessibility of lab-scale spectroscopy for molecular WOCs, such as UV-Vis, Raman, EPS, and FTIR spectroscopy. Furthermore, the abovementioned methodological repertoire is also vital to verify the successful transfer of specially designed molecular features to the surfaces of a solid catalyst. For more experimental and theoretical details of these advanced characterization techniques, we kindly refer readers to our previous review.23As we pointed out above, operando measurements are at the center of identifying active intermediates and of ultimately uncovering the reaction mechanism. To date, we have a wide arsenal for operando tests, ranging from in-house spectroscopy (ATR-FTIR, Raman, EPR, etc.) and advanced microscopy (HRTEM, STEM, and STM) to synchrotron-based high-energy spectroscopic techniques. In addition to the identification of intermediates, most of these operando measurements can provide time-resolved information about the active intermediates on the way to understanding the full reaction kinetics. More importantly, some operando techniques, e.g. XAS and HRTEM, directly monitor detailed structural changes, and thus enabled exciting breakthroughs in recent years.274,276 The observed dynamic catalyst transformations illustrate the need for new, evolutionary concepts to point out parallels between molecular and heterogeneous WOCs. Here, it seems that growing insight into the mechanisms on both sides strengthens the bridges that connect the molecular and heterogeneous realms, especially in water oxidation catalysis, which finally, is a process revolving around the transformation of small molecules at specific catalytic centers. However, we have to make it clear that even with so many advanced operando techniques at our disposal, we need more studies to strengthen the inspirational continuous knowledge exchange between both catalytic research areas. In parallel, another knowledge gap is opening up at the interfaces of molecular and heterogeneous assemblies. These challenges will remain until we can unambiguously monitor, predict and explain the dynamic nature of catalytic processes.
Conclusions
Since the increasing worldwide need for clean energy has turned AP into a research hotspot, tremendous efforts have been devoted to the optimization of WOCs as a major bottleneck. Over the past decades, molecular and heterogeneous strategies in parallel have brought forward enormous progress in the design of new respective WOCs. Since they greatly facilitate mechanistic and structural tracking at the atomic level, molecular WOCs serve as model systems to investigate the versatile interplay and influence of both metal centers and ligands on the reaction pathways and catalytic activity. This line of research is now being intensified for molecular WOCs with higher nuclearities to benefit from their specific advantages and similarities to heterogeneous surface motifs. Meanwhile, rapid progress has been made in the mechanistic study of heterogeneous WOCs despite the analytical challenges still associated with the operando monitoring of their active sites. Now that increasing insight into the separate advantages and drawbacks of molecular and heterogeneous WOCs arising from their respective dimensionalities is available, at least four strategic approaches for knowledge transfer and their direct combination into hybrid systems are emerging. (1) The discovery of joint structural motifs of both molecular WOCs and active heterogeneous surfaces greatly promotes unification of design principles and mechanistic models from both sides in the search for next-generation WOCs. For instance, the translation of the most active surface structural features of heterogeneous WOCs into molecular analogues will facilitate their mechanistic investigation on the atomic scale with convenient analytical methods. (2) In turn, applying structure–activity relationships of molecular WOCs to the construction of heterogeneous systems can expedite the direct clarification of open questions. This is particularly applicable for optimizing the interactions of active metal sites and supports/ligands, as in the emerging field of SACs. Their in-depth study holds the key to fully understand the coordinative needs and reaction pathways of single (and dual) metal sites in diverse heterogeneous environments. (3) Furthermore, this crossover research can help to draw or erase the lines between heterogenized molecular systems, such as SACs, and low-degree doping of solids. (4) Moreover, the targeted construction of hybrid and encapsulated catalyst systems holds the key to combine the analytical accessibility and synthetic flexibility of molecular WOCs with the economic robustness of heterogeneous supports.The investigation of the dynamic roles of both components in these hybrids, e.g. the transitions of molecular cocatalysts on photoelectrodes or the participation of support atoms in single and dual-site SAC reactions, has just begun and will deliver further crucial insights. We want to highlight that this line of research requires rather demanding cutting-edge operando measurements. These investigations are indispensable for crucial snapshots of the catalytic process not only for understanding the reaction mechanism, but also to accelerate the dynamic mutual evolution of both molecular and heterogeneous WOCs. Ultimately, understanding these complex processes will help us combine the best of both realms and to circumvent the individual drawbacks of both catalyst types. Since the quest for applied AP is ongoing, crossing the borders between the different catalytic divisions is a key step to expedite the search for durable and technologically relevant catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University Research Priority Program (URPP) for Solar Light to Chemical Energy Conversion (LightChEC) and by the Swiss National Science Foundation (Sinergia Grant No. CRSII2_160801/1). W. Wan thanks the China Scholarship Council (CSC) for his PhD fellowship. S. Heidari thanks the University of Zurich for support of her postdoctoral research with a Forschungskredit grant (Grant No. FK-18-106).References
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421 RSC.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120 CrossRef CAS.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607 CrossRef CAS.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247 RSC.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331 CrossRef CAS.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661 RSC.
- B. Li, F. Li, S. Bai, Z. Wang, L. Sun, Q. Yang and C. Li, Energy Environ. Sci., 2012, 5, 8229 RSC.
- C. Pasquini, I. Zaharieva, D. González-Flores, P. Chernev, M. R. Mohammadi, L. Guidoni, R. D. L. Smith and H. Dau, J. Am. Chem. Soc., 2019, 141, 2938 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216 RSC.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215 RSC.
- M. A. Newton, Catal., Struct. React., 2017, 3, 2 CrossRef CAS.
- E. de La Mora, N. Coquelle, C. S. Bury, M. Rosenthal, J. M. Holton, I. Carmichael, E. F. Garman, M. Burghammer, J.-P. Colletier and M. Weik, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4142 CrossRef CAS.
- A. Tanaka, Y. Fukushima and N. Kamiya, J. Am. Chem. Soc., 2017, 139, 1718 CrossRef CAS.
- J. Huang, J. D. Blakemore, D. Fazi, O. Kokhan, N. D. Schley, R. H. Crabtree, G. W. Brudvig and D. M. Tiede, Phys. Chem. Chem. Phys., 2014, 16, 1814 RSC.
- K. R. Yang, A. J. Matula, G. Kwon, J. Hong, S. W. Sheehan, J. M. Thomsen, G. W. Brudvig, R. H. Crabtree, D. M. Tiede, L. X. Chen and V. S. Batista, J. Am. Chem. Soc., 2016, 138, 5511 CrossRef CAS.
- C. A. Triana, R. Moré, A. J. Bloomfield, P. V. Petrović, S. G. Ferrón, G. Stanley, S. D. Zarić, T. Fox, E. N. Brothers, S. W. Sheehan, P. T. Anastas and G. R. Patzke, Matter, 2019, 1, 1354 CrossRef.
- J. Li, R. Güttinger, R. Moré, F. Song, W. Wan and G. R. Patzke, Chem. Soc. Rev., 2017, 46, 6124 RSC.
- C. Glöckner, J. Kern, M. Broser, A. Zouni, V. Yachandra and J. Yano, J. Biol. Chem., 2013, 288, 22607 CrossRef.
- S. A. Bartlett, E. V. Sackville, E. K. Gibson, V. Celorrio, P. P. Wells, M. Nachtegaal, S. W. Sheehan and U. Hintermair, Chem. Commun., 2019, 55, 7832 RSC.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198 CrossRef CAS.
- R. Schiwon, K. Klingan, H. Dau and C. Limberg, Chem. Commun., 2014, 50, 100 RSC.
- D. Lebedev, Y. Pineda-Galvan, Y. Tokimaru, A. Fedorov, N. Kaeffer, C. Copéret and Y. Pushkar, J. Am. Chem. Soc., 2018, 140, 451 CrossRef CAS.
- M. Haruta, K. Yoshida, H. Kurata and S. Isoda, Ultramicroscopy, 2008, 108, 545 CrossRef CAS.
- M. Haruta, K. Yoshida, H. Kurata and S. Isoda, Mol. Cryst. Liq. Cryst., 2008, 492, 200 CAS.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902 CrossRef CAS.
- Y. Yu, Catal. Today, 2020, 350, 192 CrossRef CAS.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565 CrossRef CAS.
- M. D. Kärkäs, R.-Z. Liao, T. M. Laine, T. Åkermark, S. Ghanem, P. E. M. Siegbahn and B. Åkermark, Catal. Sci. Technol., 2016, 6, 1306 RSC.
- Y. Yan, J. S. Lee and D. A. Ruddy, Inorg. Chem., 2015, 54, 4550 CrossRef CAS.
- N.-Y. Huang, J.-Q. Shen, Z.-M. Ye, W.-X. Zhang, P.-Q. Liao and X.-M. Chen, Chem. Sci., 2019, 10, 9859 RSC.
- M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088 RSC.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef CAS.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS.
- Z. Codolà, L. Gómez, S. T. Kleespies, L. Que, M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 5865 CrossRef.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178 CrossRef CAS.
- G. Panchbhai, W. M. Singh, B. Das, R. T. Jane and A. Thapper, Eur. J. Inorg. Chem., 2016, 3262 CrossRef CAS.
- B. Yang, Q.-Q. Yang, X. Jiang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2017, 53, 9063 RSC.
- Z. Codolà, I. Gamba, F. Acuña-Parés, C. Casadevall, M. Clémancey, J.-M. Latour, J. M. Luis, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2019, 141, 323 CrossRef.
- S. I. Shylin, M. V. Pavliuk, L. D'Amario, F. Mamedov, J. Sá, G. Berggren and I. O. Fritsky, Chem. Commun., 2019, 55, 3335 RSC.
- D. Das, S. Pattanayak, K. K. Singh, B. Garai and S. Sen Gupta, Chem. Commun., 2016, 52, 11787 RSC.
- S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 10403 CrossRef CAS.
- B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2017, 8, 14839 CrossRef CAS.
- W. Sinha, A. Mizrahi, A. Mahammed, B. Tumanskii and Z. Gross, Inorg. Chem., 2018, 57, 478 CrossRef CAS.
- L. Xu, H. Lei, Z. Zhang, Z. Yao, J. Li, Z. Yu and R. Cao, Phys. Chem. Chem. Phys., 2017, 19, 9755 RSC.
- J.-W. Wang, W. Liu, D. Zhong and T. Lu, Coord. Chem. Rev., 2019, 378, 237 CrossRef CAS.
- J. Lin, Q. Han and Y. Ding, Chem. Rec., 2018, 18, 1531 CrossRef CAS.
- Y.-Y. Li, L.-P. Tong and R.-Z. Liao, Inorg. Chem., 2018, 57, 4590 CrossRef CAS.
- A. Q. Mir, G. Joshi, P. Ghosh, S. Khandelwal, A. Kar, R. Hegde, S. Khatua and A. Dutta, ACS Energy Lett., 2019, 4, 2428 CrossRef CAS.
- C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77 CrossRef CAS.
- R.-J. Xiang, H.-Y. Wang, Z.-J. Xin, C.-B. Li, Y.-X. Lu, X.-W. Gao, H.-M. Sun and R. Cao, Chem. – Eur. J., 2016, 22, 1602 CrossRef CAS.
- G.-Y. Luo, H.-H. Huang, J.-W. Wang and T.-B. Lu, ChemSusChem, 2016, 9, 485 CrossRef CAS.
- S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586 CrossRef CAS.
- M. Schilling, G. R. Patzke, J. Hutter and S. Luber, J. Phys. Chem. C, 2016, 120, 7966 CrossRef CAS.
- H. Pan, L. Duan and R.-Z. Liao, ChemCatChem, 2020, 12, 219 CrossRef CAS.
- Ł. Szyrwiel, D. Lukács, D. F. Srankó, Z. Kerner, A. Kotynia, J. Brasuń, B. Setner, Z. Szewczuk, K. Malec and J. S. Pap, RSC Adv., 2017, 7, 24657 RSC.
- J. Wang, H. Huang and T. Lu, Chin. J. Chem., 2017, 35, 586 CrossRef CAS.
- F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368 CrossRef CAS.
- Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613 RSC.
- B. Ariela, W. Yaniv, S. Dror, K. Haya, A. Yael, M. Eric and M. Dan, Dalton Trans., 2017, 46, 10774 RSC.
- D. Wang and C. O. Bruner, Inorg. Chem., 2017, 56, 13638 CrossRef CAS.
- H. Kuilya, N. Alam, D. Sarma, D. Choudhury and A. Kalita, Chem. Commun., 2019, 55, 5483 RSC.
- T. Ghosh, P. Ghosh and G. Maayan, ACS Catal., 2018, 8, 10631 CrossRef CAS.
- Q.-Y. Mao, Y.-J. Pang, X.-C. Li, G.-J. Chen and H.-W. Tan, ACS Catal., 2019, 9, 8798 CrossRef CAS.
- M.-C. Kafentzi, R. Papadakis, F. Gennarini, A. Kochem, O. Iranzo, Y. Le Mest, N. Le Poul, T. Tron, B. Faure, A. J. Simaan and M. Réglier, Chem. – Eur. J., 2018, 24, 5213 CrossRef CAS.
- M. Yoshida, M. Kondo, M. Okamura, M. Kanaike, S. Haesuwannakij, H. Sakurai and S. Masaoka, Faraday Discuss., 2017, 198, 181 RSC.
- P. Xu, S. Hu, H.-D. Zhang and X. Zheng, Phys. Chem. Chem. Phys., 2018, 20, 14919 RSC.
- H. Wang, Z. Xin, R. Xiang, S. Liu, X. Gao and C. Li, Chin. J. Chem., 2016, 34, 757 CrossRef CAS.
- S. Liu, Y.-J. Lei, Z.-J. Xin, R.-J. Xiang, S. Styring, A. Thapper and H.-Y. Wang, Int. J. Hydrogen Energy, 2017, 42, 29716 CrossRef CAS.
- P. Garrido-Barros, S. Grau, S. Drouet, J. Benet-Buchholz, C. Gimbert-Suriñach and A. Llobet, ACS Catal., 2019, 9, 3936 CrossRef CAS.
- A. Annunziata, R. Esposito, G. Gatto, M. E. Cucciolito, A. Tuzi, A. Macchioni and F. Ruffo, Eur. J. Inorg. Chem., 2018, 3304 CrossRef CAS.
- J.-W. Wang, X.-Q. Zhang, H.-H. Huang and T.-B. Lu, ChemCatChem, 2016, 8, 3287 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524 CrossRef CAS.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313 CrossRef.
- J.-W. Wang, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2018, 377, 225 CrossRef CAS.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059 CrossRef CAS.
- R. Dhiman and C. M. Nagaraja, Eur. J. Inorg. Chem., 2018, 2826 CrossRef CAS.
- H. M. C. Lant, T. K. Michaelos, L. S. Sharninghausen, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, Eur. J. Inorg. Chem., 2019, 2115 CrossRef CAS.
- S. Neudeck, S. Maji, I. López, S. Meyer, F. Meyer and A. Llobet, J. Am. Chem. Soc., 2014, 136, 24 CrossRef CAS.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170 RSC.
- C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798 CrossRef CAS.
- F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodríguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176 CrossRef CAS.
- S. Romain, F. Bozoglian, X. Sala and A. Llobet, J. Am. Chem. Soc., 2009, 131, 2768 CrossRef CAS.
- M. L. Rigsby, S. Mandal, W. Nam, L. C. Spencer, A. Llobet and S. S. Stahl, Chem. Sci., 2012, 3, 3058 RSC.
- J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423 CrossRef CAS.
- T. Liu, B. Zhang and L. Sun, Chem. – Asian J., 2019, 14, 31 CrossRef CAS.
- E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715 CrossRef CAS.
- W. A. A. Arafa, M. D. Kärkäs, B.-L. Lee, T. Åkermark, R.-Z. Liao, H.-M. Berends, J. Messinger, P. E. M. Siegbahn and B. Åkermark, Phys. Chem. Chem. Phys., 2014, 16, 11950 RSC.
- S. Fukuzumi, S. Mandal, K. Mase, K. Ohkubo, H. Park, J. Benet-Buchholz, W. Nam and A. Llobet, J. Am. Chem. Soc., 2012, 134, 9906 CrossRef CAS.
- C. Gimbert-Suriñach, D. Moonshiram, L. Francàs, N. Planas, V. Bernales, F. Bozoglian, A. Guda, L. Mognon, I. López, M. A. Hoque, L. Gagliardi, C. J. Cramer and A. Llobet, J. Am. Chem. Soc., 2016, 138, 15291 CrossRef.
- H. Kotani, D. Hong, K. Satonaka, T. Ishizuka and T. Kojima, Inorg. Chem., 2019, 58, 3676 CrossRef CAS.
- T. Ishizuka, A. Watanabe, H. Kotani, D. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154 CrossRef CAS.
- J.-W. Wang, P. Sahoo and T.-B. Lu, ACS Catal., 2016, 6, 5062 CrossRef CAS.
- H.-Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499 CrossRef CAS.
- R. Modak, B. Mondal, Y. Sikdar, J. Banerjee, E. Colacio, I. Oyarzabal, J. Cano and S. Goswami, Dalton Trans., 2020, 49, 6328 RSC.
- D. Wang and L. Que, Chem. Commun., 2013, 49, 10682 RSC.
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273 CrossRef CAS.
- W.-P. To, T. Wai-Shan Chow, C.-W. Tse, X. Guan, J.-S. Huang and C.-M. Che, Chem. Sci., 2015, 6, 5891 RSC.
- Y. Liu, R. Xiang, X. Du, Y. Ding and B. Ma, Chem. Commun., 2014, 50, 12779 RSC.
- B. Das, B.-L. Lee, E. A. Karlsson, T. Åkermark, A. Shatskiy, S. Demeshko, R.-Z. Liao, T. M. Laine, M. Haukka, E. Zeglio, A. F. Abdel-Magied, P. E. M. Siegbahn, F. Meyer, M. D. Kärkäs, E. V. Johnston, E. Nordlander and B. Åkermark, Dalton Trans., 2016, 45, 13289 RSC.
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260 CrossRef CAS.
- K. G. Kottrup, S. D'Agostini, P. H. van Langevelde, M. A. Siegler and D. G. H. Hetterscheid, ACS Catal., 2018, 8, 1052 CrossRef CAS.
- C. M. Lemon, M. Huynh, A. G. Maher, B. L. Anderson, E. D. Bloch, D. C. Powers and D. G. Nocera, Angew. Chem., Int. Ed., 2016, 55, 2176 CrossRef CAS.
- S. Karim, A. Chakraborty, D. Samanta, E. Zangrando, T. Ghosh and D. Das, Catal. Sci. Technol., 2020, 10, 2830 RSC.
- H. Lee, X. Wu and L. Sun, Nanoscale, 2020, 12, 4187 RSC.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909 CrossRef CAS.
- Q.-Q. Hu, X.-J. Su and M.-T. Zhang, Inorg. Chem., 2018, 57, 10481 CrossRef CAS.
- X. Zhang, Y.-Y. Li, J. Jiang, R. Zhang, R.-Z. Liao and M. Wang, Inorg. Chem., 2020, 59, 5424 CrossRef CAS.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262 RSC.
- H. B. Lee, D. A. Marchiori, R. Chatterjee, P. H. Oyala, J. Yano, R. D. Britt and T. Agapie, J. Am. Chem. Soc., 2020, 142, 3753 CrossRef CAS.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804 CrossRef CAS.
- M. Askerka, G. W. Brudvig and V. S. Batista, Acc. Chem. Res., 2017, 50, 41 CrossRef CAS.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935 CrossRef CAS.
- R. G. Hadt, D. Hayes, C. N. Brodsky, A. M. Ullman, D. M. Casa, M. H. Upton, D. G. Nocera and L. X. Chen, J. Am. Chem. Soc., 2016, 138, 11017 CrossRef CAS.
- J. G. McAlpin, Y. Surendranath, M. Dinca, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882 CrossRef CAS.
- J. G. McAlpin, T. A. Stich, C. A. Ohlin, Y. Surendranath, D. G. Nocera, W. H. Casey and R. D. Britt, J. Am. Chem. Soc., 2011, 133, 15444 CrossRef CAS.
- C. N. Brodsky, R. G. Hadt, D. Hayes, B. J. Reinhart, N. Li, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3855 CrossRef CAS.
- H. Liu and H. Frei, ACS Catal., 2020, 10, 2138 CrossRef CAS.
- F. Evangelisti, R. Moré, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076 CrossRef CAS.
- A. I. Nguyen, D. L. M. Suess, L. E. Darago, P. H. Oyala, D. S. Levine, M. S. Ziegler, R. D. Britt and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 5579 CrossRef CAS.
- A. I. Nguyen, L. E. Darago, D. Balcells and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 9030 CrossRef CAS.
- X. Jiang, J. Li, B. Yang, X.-Z. Wei, B.-W. Dong, Y. Kao, M.-Y. Huang, C.-H. Tung and L.-Z. Wu, Angew. Chem., 2018, 57, 7850 CrossRef CAS.
- W.-S. Gao, J.-M. Wang, N.-N. Shi, C.-N. Chen, Y.-H. Fan and M. Wang, New J. Chem., 2019, 43, 4640 RSC.
- J. Lin, X. Liang, X. Cao, N. Wei and Y. Ding, Chem. Commun., 2018, 54, 12515 RSC.
- J.-M. Wang, Y.-R. Liu, X.-Y. Mao, N.-N. Shi, X. Zhang, H.-S. Wang, Y.-H. Fan and M. Wang, Chem. – Asian J., 2019, 14, 2685 CrossRef CAS.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465 CrossRef CAS.
- R.-Z. Liao, S. Masaoka and P. E. M. Siegbahn, ACS Catal., 2018, 8, 11671 CrossRef CAS.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48 CrossRef CAS.
- Q. Lan, Z.-M. Zhang, C. Qin, X.-L. Wang, Y.-G. Li, H.-Q. Tan and E.-B. Wang, Chem. – Eur. J., 2016, 22, 15513 CrossRef CAS.
- J. Soriano-López, D. G. Musaev, C. L. Hill, J. R. Galán-Mascarós, J. J. Carbó and J. M. Poblet, J. Catal., 2017, 350, 56 CrossRef.
- X.-F. Su, W. Guan, L.-K. Yan and Z.-M. Su, J. Catal., 2020, 381, 402 CrossRef CAS.
- R. Kaushik, R. Sakla, D. Amilan Jose and A. Ghosh, New J. Chem., 2020, 44, 3764 RSC.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC.
- S. M. Lauinger, Q. Yin, Y. V. Geletii and C. L. Hill, in Advances in Inorganic Chemistry Polyoxometalate Chemistry, ed. R. van Eldik and L. Cronin, Academic Press, 2017, p. 117 Search PubMed.
- B. Chakraborty, G. Gan-Or, M. Raula, E. Gadot and I. A. Weinstock, Nat. Commun., 2018, 9, 4896 CrossRef.
- S. Bae, J.-E. Jang, H.-W. Lee and J. Ryu, Eur. J. Inorg. Chem., 2019, 2040 CrossRef CAS.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio, S. Caramori, P. Galloni and A. Sartorel, Chem. Commun., 2020, 56, 2248 RSC.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872 CrossRef CAS.
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209 CrossRef CAS.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 79 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110 CrossRef CAS.
- H. I. Buvailo, V. G. Makhankova, V. N. Kokozay, I. V. Omelchenko, S. V. Shishkina, J. Jezierska, M. V. Pavliuk and S. I. Shylin, Inorg. Chem. Front., 2019, 6, 1813 RSC.
- L. Yu, Y. Ding and M. Zheng, Appl. Catal., B, 2017, 209, 45 CrossRef CAS.
- M. Navarro, M. Li, H. Müller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2016, 22, 6740 CrossRef CAS.
- M. D. Bairagya, R. J. Bujol and N. Elgrishi, Chem. – Eur. J., 2019, 26, 3991 Search PubMed.
- B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159 RSC.
- D. P. Hickey, C. Sandford, Z. Rhodes, T. Gensch, L. R. Fries, M. S. Sigman and S. D. Minteer, J. Am. Chem. Soc., 2019, 141, 1382 CrossRef CAS.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, J. Catal., 2017, 352, 293 CrossRef CAS.
- X. Zhou, T. Zhang, C. W. Abney, Z. Li and W. Lin, ACS Appl. Mater. Interfaces, 2014, 6, 18475 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Suriñach, D. Moonshiram, A. Picón, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907 CrossRef CAS.
- B. Liu, J. Li, H.-L. Wu, W.-Q. Liu, X. Jiang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 18577 CrossRef CAS.
- Y. Wang, F. Li, X. Zhou, F. Yu, J. Du, L. Bai and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 6911 CrossRef CAS.
- S. M. Lauinger, J. M. Sumliner, Q. Yin, Z. Xu, G. Liang, E. N. Glass, T. Lian and C. L. Hill, Chem. Mater., 2015, 27, 5886 CrossRef CAS.
- S. M. Lauinger, B. D. Piercy, W. Li, Q. Yin, D. L. Collins-Wildman, E. N. Glass, M. D. Losego, D. Wang, Y. V. Geletii and C. L. Hill, ACS Appl. Mater. Interfaces, 2017, 9, 35048 CrossRef CAS.
- B.-T. Chen, N. Morlanés, E. Adogla, K. Takanabe and V. O. Rodionov, ACS Catal., 2016, 6, 4647 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613 CrossRef CAS.
- W. Salomon, F.-J. Yazigi, C. Roch-Marchal, P. Mialane, P. Horcajada, C. Serre, M. Haouas, F. Taulelle and A. Dolbecq, Dalton Trans., 2014, 43, 12698 RSC.
- X.-J. Kong, Z. Lin, Z.-M. Zhang, T. Zhang and W. Lin, Angew. Chem., 2016, 128, 6521 CrossRef.
- J. Wang, J. Liu, B. Zhang, H. Wan, Z. Li, X. Ji, K. Xu, C. Chen, D. Zha, L. Miao and J. Jiang, Nano Energy, 2017, 42, 98 CrossRef CAS.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef.
- W. Wan, S. Wei, J. Li, C. A. Triana, Y. Zhou and G. R. Patzke, J. Mater. Chem. A, 2019, 7, 15145 RSC.
- Y. Zhao, C. K. Mavrokefalos, P. Zhang, R. Erni, J. Li, C. A. Triana and G. R. Patzke, Chem. Mater., 2020, 32, 1371 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196 RSC.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019 CrossRef CAS.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090 CrossRef CAS.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Nat. Commun., 2019, 10, 5208 CrossRef.
- S. Lee, L. Bai and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 8072 CrossRef CAS.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886 CrossRef CAS.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692 CrossRef CAS.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431 CrossRef CAS.
- S. R. Pendlebury, M. Barroso, A. J. Cowan, K. Sivula, J. Tang, M. Grätzel, D. Klug and J. R. Durrant, Chem. Commun., 2011, 47, 716 RSC.
- C. Y. Cummings, F. Marken, L. M. Peter, A. A. Tahir and K. G. U. Wijayantha, Chem. Commun., 2012, 48, 2027 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626 RSC.
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724 RSC.
- K. M. H. Young, B. M. Klahr, O. Zandi and T. W. Hamann, Catal. Sci. Technol., 2013, 3, 1660 RSC.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 612 CrossRef CAS.
- J. Rodríguez-López, M. A. Alpuche-Avilés and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16985 CrossRef.
- C. Costentin, T. R. Porter and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 5615 CrossRef CAS.
- R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2004, 126, 1290 CrossRef CAS.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362 CrossRef CAS.
- H. H. Pham, M.-J. Cheng, H. Frei and L.-W. Wang, ACS Catal., 2016, 6, 5610 CrossRef CAS.
- S. Koroidov, M. F. Anderlund, S. Styring, A. Thapper and J. Messinger, Energy Environ. Sci., 2015, 8, 2492 RSC.
- A. M. Ullman, C. N. Brodsky, N. Li, S.-L. Zheng and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 4229 CrossRef CAS.
- J. Fester, M. García-Melchor, A. S. Walton, M. Bajdich, Z. Li, L. Lammich, A. Vojvodic and J. V. Lauritsen, Nat. Commun., 2017, 8, 14169 CrossRef CAS.
- G. S. Hutchings, Y. Zhang, J. Li, B. T. Yonemoto, X. Zhou, K. Zhu and F. Jiao, J. Am. Chem. Soc., 2015, 137, 4223 CrossRef CAS.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36 CrossRef CAS.
- H.-Y. Wang, S.-F. Hung, Y.-Y. Hsu, L. Zhang, J. Miao, T.-S. Chan, Q. Xiong and B. Liu, J. Phys. Chem. Lett., 2016, 7, 4847 CrossRef CAS.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959 CrossRef CAS.
- D. González-Flores, I. Sánchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Angew. Chem., Int. Ed., 2015, 54, 2472 CrossRef.
- N. Weidler, S. Paulus, J. Schuch, J. Klett, S. Hoch, P. Stenner, A. Maljusch, J. Brötz, C. Wittich, B. Kaiser and W. Jaegermann, Phys. Chem. Chem. Phys., 2016, 18, 10708 RSC.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432 CrossRef CAS.
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778 CrossRef CAS.
- S. Haschke, M. Mader, S. Schlicht, A. M. Roberts, A. M. Angeles-Boza, J. A. C. Barth and J. Bachmann, Nat. Commun., 2018, 9, 4565 CrossRef.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629 CrossRef CAS.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Nat. Chem., 2020, 12, 82 CrossRef CAS.
- Y. Zhang, H. Zhang, A. Liu, C. Chen, W. Song and J. Zhao, J. Am. Chem. Soc., 2018, 140, 3264 CrossRef CAS.
- Y. Zhang, H. Zhang, H. Ji, W. Ma, C. Chen and J. Zhao, J. Am. Chem. Soc., 2016, 138, 2705 CrossRef CAS.
- D. M. Herlihy, M. M. Waegele, X. Chen, C. D. Pemmaraju, D. Prendergast and T. Cuk, Nat. Chem., 2016, 8, 549 CrossRef CAS.
- X. Chen, D. J. Aschaffenburg and T. Cuk, Nat. Catal., 2019, 2, 820 CrossRef CAS.
- M. Zhang and H. Frei, Catal. Lett., 2015, 145, 420 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159 CrossRef CAS.
- M. Kuang, P. Han, L. Huang, N. Cao, L. Qian and G. Zheng, Adv. Funct. Mater., 2018, 28, 1804886 CrossRef.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603 CrossRef.
- Z. Li, W. Niu, L. Zhou and Y. Yang, ACS Energy Lett., 2018, 3, 892 CrossRef CAS.
- H. Hajiyani and R. Pentcheva, ACS Catal., 2018, 8, 11773 CrossRef CAS.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070 CrossRef.
- Y. Hu, Z. Wang, W. Liu, L. Xu, M. Guan, Y. Huang, Y. Zhao, J. Bao and H.-M. Li, ACS Sustainable Chem. Eng., 2019, 7, 16828 CrossRef CAS.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS.
- J. Bao, Z. Wang, J. Xie, L. Xu, F. Lei, M. Guan, Y. Zhao, Y. Huang and H. Li, Chem. Commun., 2019, 55, 3521 RSC.
- Y. Wang, Y. Sun, F. Yan, C. Zhu, P. Gao, X. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 8479 RSC.
- S. Sun, Y. Sun, Y. Zhou, J. Shen, D. Mandler, R. Neumann and Z. J. Xu, Chem. Mater., 2019, 31, 8106 CrossRef CAS.
- T. Tian, H. Gao, X. Zhou, L. Zheng, J. Wu, K. Li and Y. Ding, ACS Energy Lett., 2018, 3, 2150 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329 CrossRef CAS.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763 CrossRef CAS.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692 CrossRef CAS.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373 CrossRef CAS.
- J. Lee, D. A. Kitchaev, D.-H. Kwon, C.-W. Lee, J. K. Papp, Y.-S. Liu, Z. Lun, R. J. Clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196 RSC.
- M.-Q. Yang, J. Wang, H. Wu and G. W. Ho, Small, 2018, 14, 1703323 CrossRef.
- T. Ling, D.-Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X.-W. Du, Z. Hu, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef CAS.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277 CrossRef CAS.
- X. Zhang, Y. Zhao, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2019, 9, 1900881 CrossRef.
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J.-J. Zou, ACS Catal., 2018, 8, 3803 CrossRef CAS.
- D. Ji, L. Fan, L. Tao, Y. Sun, M. Li, G. Yang, T. Q. Tran, S. Ramakrishna and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 13840 CrossRef CAS.
- Z. Li, Y. Zhang, Y. Feng, C.-Q. Cheng, K.-W. Qiu, C.-K. Dong, H. Liu and X.-W. Du, Adv. Funct. Mater., 2019, 29, 1903444 CrossRef.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867 CrossRef CAS.
- Q. He, Y. Wan, H. Jiang, Z. Pan, C. Wu, M. Wang, X. Wu, B. Ye, P. M. Ajayan and L. Song, ACS Energy Lett., 2018, 3, 1373 CrossRef CAS.
- Y. Zhao, W. Wan, Y. Chen, R. Erni, C. A. Triana, J. Li, C. K. Mavrokefalos, Y. Zhou and G. R. Patzke, Adv. Energy Mater., 2020, 10, 2002228 CrossRef CAS.
- X. Du, J. Huang, J. Zhang, Y. Yan, C. Wu, Y. Hu, C. Yan, T. Lei, W. Chen, C. Fan and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 4484 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48 CrossRef CAS.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115 CrossRef CAS.
- D. Zhou, S. Wang, Y. Jia, X. Xiong, H. Yang, S. Liu, J. Tang, J. Zhang, D. Liu, L. Zheng, Y. Kuang, X. Sun and B. Liu, Angew. Chem., Int. Ed., 2019, 58, 736 CrossRef CAS.
- L. Wang, K. A. Stoerzinger, L. Chang, X. Yin, Y. Li, C. S. Tang, E. Jia, M. E. Bowden, Z. Yang, A. Abdelsamie, L. You, R. Guo, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 12941 CrossRef CAS.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870 CrossRef CAS.
- T. Ling, D.-Y. Yan, H. Wang, Y. Jiao, Z. Hu, Y. Zheng, L. Zheng, J. Mao, H. Liu, X.-W. Du, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2017, 8, 1509 CrossRef.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488 CrossRef CAS.
- Z. Chen, C. X. Kronawitter and B. E. Koel, Phys. Chem. Chem. Phys., 2015, 17, 29387 RSC.
- R. Gao, J. Zhu, X. Xiao, Z. Hu, J. Liu and X. Liu, J. Phys. Chem. C, 2015, 119, 4516 CrossRef CAS.
- L. Liu, Z. Jiang, L. Fang, H. Xu, H. Zhang, X. Gu and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 27736 CrossRef CAS.
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T.-Y. Ma, Adv. Energy Mater., 2018, 8, 1702222 CrossRef.
- X. Zhou, Z. Liu, Y. Wang and Y. Ding, Appl. Catal., B, 2018, 237, 74 CrossRef CAS.
- A. Govind Rajan, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2020, 142, 3600 CrossRef CAS.
- J. Deng, D. Deng and X. Bao, Adv. Mater., 2017, 29, 1606967 CrossRef.
- Q. Lu, J. Yu, X. Zou, K. Liao, P. Tan, W. Zhou, M. Ni and Z. Shao, Adv. Funct. Mater., 2019, 29, 1904481 CrossRef.
- H. Khani, N. S. Grundish, D. O. Wipf and J. B. Goodenough, Adv. Energy Mater., 2020, 10, 1903215 CrossRef CAS.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123 RSC.
- X. Han, W. Zhang, X. Ma, C. Zhong, N. Zhao, W. Hu and Y. Deng, Adv. Mater., 2019, 31, 1808281 CrossRef.
- J. Zhang, B. Qian, S. Sun, S. Tao, W. Chu, D. Wu and L. Song, Small, 2019, 15, 1904260 CrossRef CAS.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855 CrossRef CAS.
- Y. Li, X. Du, J. Huang, C. Wu, Y. Sun, G. Zou, C. Yang and J. Xiong, Small, 2019, 15, e1901980 CrossRef.
- M. M. Najafpour, M. Abbasi Isaloo, M. Abasi and M. Hołyńska, New J. Chem., 2014, 38, 852 RSC.
- J. O’M. Bockris and T. Otagawa, J. Electrochem. Soc., 2019, 131, 290 CrossRef.
- W. G. Hardin, J. T. Mefford, D. A. Slanac, B. B. Patel, X. Wang, S. Dai, X. Zhao, R. S. Ruoff, K. P. Johnston and K. J. Stevenson, Chem. Mater., 2014, 26, 3368 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404 RSC.
- R. A. Rincón, E. Ventosa, F. Tietz, J. Masa, S. Seisel, V. Kuznetsov and W. Schuhmann, ChemPhysChem, 2014, 15, 2810 CrossRef.
- W. Zhou, M. Zhao, F. Liang, S. C. Smith and Z. Zhu, Mater. Horiz., 2015, 2, 495 RSC.
- C. Lawley, M. Nachtegaal, J. Stahn, V. Roddatis, M. Döbeli, T. J. Schmidt, D. Pergolesi and T. Lippert, Nat. Commun., 2020, 11, 1728 CrossRef CAS.
- B.-J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231 CrossRef CAS.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 1 Search PubMed.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153 CrossRef CAS.
- B. Han, M. Risch, Y.-L. Lee, C. Ling, H. Jia and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2015, 17, 22576 RSC.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925 CrossRef CAS.
- A. I. Nguyen, K. M. van Allsburg, M. W. Terban, M. Bajdich, J. Oktawiec, J. Amtawong, M. S. Ziegler, J. P. Dombrowski, K. V. Lakshmi, W. S. Drisdell, J. Yano, S. J. L. Billinge and T. D. Tilley, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11630 CAS.
- D. Mierwaldt, V. Roddatis, M. Risch, J. Scholz, J. Geppert, M. E. Abrishami and C. Jooss, Adv. Sustainable Syst., 2017, 1, 1700109 CrossRef.
- S. Heidari, J. P. Singh, H. Feizi, R. Bagheri, K. H. Chae, Z. Song, M. Khatamian and M. M. Najafpour, Sci. Rep., 2019, 9, 7749 CrossRef.
- S. Heidari, M. M. Najafpour, M. Hołyńska, J. P. Singh, K. H. Chae and M. Khatamian, Dalton Trans., 2018, 47, 1557 RSC.
- K. Fan, H. Zou, Y. Lu, H. Chen, F. Li, J. Liu, L. Sun, L. Tong, M. F. Toney, M. Sui and J. Yu, ACS Nano, 2018, 12, 12369 CrossRef CAS.
- Y.-J. Tang, Y. Wang and K. Zhou, J. Mater. Chem. A, 2020, 8, 7925 RSC.
- X. Zhang, Y.-S. Chen, P. V. Kamat and S. Ptasinska, J. Phys. Chem. C, 2018, 122, 13894 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305 CrossRef CAS.
- T. Morikawa, S. Gul, Y. F. Nishimura, T. M. Suzuki and J. Yano, Chem. Commun., 2020, 56, 5158 RSC.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. González-Flores and H. Dau, Energy Environ. Sci., 2018, 11, 2476 RSC.
- D. González-Flores, K. Klingan, P. Chernev, S. Loos, M. R. Mohammadi, C. Pasquini, P. Kubella, I. Zaharieva, R. D. L. Smith and H. Dau, Sustainable Energy Fuels, 2018, 2, 1986 RSC.
- N. Ortiz Peña, D. Ihiawakrim, M. Han, B. Lassalle-Kaiser, S. Carenco, C. Sanchez, C. Laberty-Robert, D. Portehault and O. Ersen, ACS Nano, 2019, 13, 11372 CrossRef.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243 CrossRef CAS.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744 CrossRef CAS.
- Z. Zand, P. Salimi, M. R. Mohammadi, R. Bagheri, P. Chernev, Z. Song, H. Dau, M. Görlin and M. M. Najafpour, ACS Sustainable Chem. Eng., 2019, 7, 17252 CrossRef CAS.
- S. Back, K. Tran and Z. W. Ulissi, ACS Catal., 2019, 9, 7651 CrossRef CAS.
- A. Nandy, J. Zhu, J. P. Janet, C. Duan, R. B. Getman and H. J. Kulik, ACS Catal., 2019, 9, 8243 CrossRef CAS.
- R. Palkovits and S. Palkovits, ACS Catal., 2019, 9, 8383 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638 CrossRef CAS.
- P. W. Menezes, A. Indra, D. González-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017 CrossRef CAS.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Nat. Commun., 2017, 8, 2022 CrossRef.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628 CrossRef CAS.
- A. Indra, P. W. Menezes, C. Das, C. Göbel, M. Tallarida, D. Schmeiβer and M. Driess, J. Mater. Chem. A, 2017, 5, 5171 RSC.
- R. Zhang, Z. Wang, S. Hao, R. Ge, X. Ren, F. Qu, G. Du, A. M. Asiri, B. Zheng and X. Sun, ACS Sustainable Chem. Eng., 2017, 5, 8518 CrossRef CAS.
- Z. Chen, Z. Duan, Z. Wang, X. Liu, L. Gu, F. Zhang, M. Dupuis and C. Li, ChemCatChem, 2017, 9, 3641 CrossRef CAS.
- J. S. Kim, I. Park, E.-S. Jeong, K. Jin, W. M. Seong, G. Yoon, H. Kim, B. Kim, K. T. Nam and K. Kang, Adv. Mater., 2017, 29, 1606893 CrossRef.
- L. Liardet and X. Hu, ACS Catal., 2018, 8, 644 CrossRef CAS.
- X. Wang, W. Ma, C. Ding, Z. Xu, H. Wang, X. Zong and C. Li, ACS Catal., 2018, 8, 9926 CrossRef CAS.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301 CrossRef CAS.
- P. Du, O. Kokhan, K. W. Chapman, P. J. Chupas and D. M. Tiede, J. Am. Chem. Soc., 2012, 134, 11096 CrossRef CAS.
- J.-H. Xu, L.-Y. Guo, H.-F. Su, X. Gao, X.-F. Wu, W.-G. Wang, C.-H. Tung and D. Sun, Inorg. Chem., 2017, 56, 1591 CrossRef CAS.
- D. Y. Shopov, L. S. Sharninghausen, S. B. Sinha, B. Q. Mercado, D. Balcells, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 2018, 57, 5684 CrossRef CAS.
- M. Schilling and S. Luber, Front. Chem., 2018, 6, 100 CrossRef.
- I. D. Young, M. Ibrahim, R. Chatterjee, S. Gul, F. Fuller, S. Koroidov, A. S. Brewster, R. Tran, R. Alonso-Mori, T. Kroll, T. Michels-Clark, H. Laksmono, R. G. Sierra, C. A. Stan, R. Hussein, M. Zhang, L. Douthit, M. Kubin, C. de Lichtenberg, P. Long Vo, H. Nilsson, M. H. Cheah, D. Shevela, C. Saracini, M. A. Bean, I. Seuffert, D. Sokaras, T.-C. Weng, E. Pastor, C. Weninger, T. Fransson, L. Lassalle, P. Bräuer, P. Aller, P. T. Docker, B. Andi, A. M. Orville, J. M. Glownia, S. Nelson, M. Sikorski, D. Zhu, M. S. Hunter, T. J. Lane, A. Aquila, J. E. Koglin, J. Robinson, M. Liang, S. Boutet, A. Y. Lyubimov, M. Uervirojnangkoorn, N. W. Moriarty, D. Liebschner, P. V. Afonine, D. G. Waterman, G. Evans, P. Wernet, H. Dobbek, W. I. Weis, A. T. Brunger, P. H. Zwart, P. D. Adams, A. Zouni, J. Messinger, U. Bergmann, N. K. Sauter, J. Kern, V. K. Yachandra and J. Yano, Nature, 2016, 540, 453 CrossRef CAS.
- D. J. Vinyard and G. W. Brudvig, Annu. Rev. Phys. Chem., 2017, 68, 101 CrossRef CAS.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838 CrossRef CAS.
- Y. Zhao, J. Lin, Y. Liu, B. Ma, Y. Ding and M. Chen, Chem. Commun., 2015, 51, 17309 RSC.
- P. F. Smith, C. Kaplan, J. E. Sheats, D. M. Robinson, N. S. McCool, N. Mezle and G. C. Dismukes, Inorg. Chem., 2014, 53, 2113 CrossRef CAS.
- F. Song, K. Al-Ameed, M. Schilling, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 8846 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 CrossRef.
- J. Greeley, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 605 CrossRef.
- M. Busch, A. Fabrizio, S. Luber, J. Hutter and C. Corminboeuf, J. Phys. Chem. C, 2018, 122, 12404 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- J. Rossmeisl, E. Skúlason, M. E. Björketun, V. Tripkovic and J. K. Nørskov, Chem. Phys. Lett., 2008, 466, 68 CrossRef CAS.
- M. Busch, Curr. Opin. Electrochem., 2018, 9, 278 CrossRef CAS.
- J. Hessels, R. J. Detz, M. T. M. Koper and J. N. H. Reek, Chem. – Eur. J., 2017, 23, 16413 CrossRef CAS.
- K. Fan, Y. Ji, H. Zou, J. Zhang, B. Zhu, H. Chen, Q. Daniel, Y. Luo, J. Yu and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 3289 CrossRef CAS.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494 CrossRef.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. de Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333 CrossRef CAS.
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt and M. García-Melchor, Nat. Commun., 2019, 10, 4993 CrossRef.
- B. Schwarz, J. Forster, M. K. Goetz, D. Yücel, C. Berger, T. Jacob and C. Streb, Angew. Chem., Int. Ed., 2016, 55, 6329 CrossRef CAS.
- R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182 CrossRef CAS.
- M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242 CrossRef CAS.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451 CrossRef CAS.
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285 CAS.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63 CrossRef CAS.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef.
- J. Guan, Z. Duan, F. Zhang, S. D. Kelly, R. Si, M. Dupuis, Q. Huang, J. Q. Chen, C. Tang and C. Li, Nat. Catal., 2018, 1, 870 CrossRef CAS.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304 CrossRef CAS.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312 CrossRef CAS.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876 CrossRef CAS.
- C. Lin, Y. Zhao, H. Zhang, S. Xie, Y.-F. Li, X. Li, Z. Jiang and Z.-P. Liu, Chem. Sci., 2018, 9, 6803 RSC.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S.-T. Lee and T. Ma, Nat. Commun., 2019, 10, 2149 CrossRef.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218 CrossRef CAS.
- S. Zhan and M. S. G. Ahlquist, J. Am. Chem. Soc., 2018, 140, 7498 CrossRef CAS.
- F. Li, B. Zhang, X. Li, Y. Jiang, L. Chen, Y. Li and L. Sun, Angew. Chem., Int. Ed., 2011, 50, 12276 CrossRef CAS.
- T. Xue, B. Peng, M. Xue, X. Zhong, C.-Y. Chiu, S. Yang, Y. Qu, L. Ruan, S. Jiang, S. Dubin, R. B. Kaner, J. I. Zink, M. E. Meyerhoff, X. Duan and Y. Huang, Nat. Commun., 2014, 5, 3200 CrossRef.
- F. Li, H. Yang, W. Li and L. Sun, Joule, 2018, 2, 36 CrossRef CAS.
- N. Kuwamura, Y. Kurioka, N. Yoshinari and T. Konno, Chem. Commun., 2018, 54, 10766 RSC.
- P. Christopher, ACS Energy Lett., 2019, 4, 2249 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339 CrossRef CAS.
- G. Gao, S. Bottle and A. Du, Catal. Sci. Technol., 2018, 8, 996 RSC.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho, M. Chen, C. Yuan, L. Lei and X. Feng, Nat. Commun., 2019, 10, 1392 CrossRef.
- Y. Li, Z.-S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118 CrossRef CAS.
- B. Lu, Q. Liu and S. Chen, ACS Catal., 2020, 10, 7584 CrossRef CAS.
- W. Wan, C. A. Triana, J. Lan, J. Li, C. S. Allen, Y. Zhao, M. Iannuzzi and G. R. Patzke, ACS Nano, 2020, 14, 13279 CrossRef CAS.
- B. Wurster, D. Grumelli, D. Hötger, R. Gutzler and K. Kern, J. Am. Chem. Soc., 2016, 138, 3623 CrossRef CAS.
- H. Xiao, H. Shin and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5872 CrossRef CAS.
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, 7970 CrossRef.
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemCatChem, 2018, 10, 1686 CrossRef CAS.
- Y. Zhao, X. Yan, K. R. Yang, S. Cao, Q. Dong, J. E. Thorne, K. L. Materna, S. Zhu, X. Pan, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, ACS Cent. Sci., 2018, 4, 1166 CrossRef CAS.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS.
- Z. N. Zahran, Y. Tsubonouchi, E. A. Mohamed and M. Yagi, ChemSusChem, 2019, 12, 1775 CrossRef CAS.
- Z. Chen, J. J. Concepcion, J. F. Hull, P. G. Hoertz and T. J. Meyer, Dalton Trans., 2010, 39, 6950 RSC.
- G. Pastori, K. Wahab, A. Bucci, G. Bellachioma, C. Zuccaccia, J. Llorca, H. Idriss and A. Macchioni, Chem. – Eur. J., 2016, 22, 13459 CrossRef CAS.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918 CrossRef CAS.
- H. Baydoun, J. Burdick, B. Thapa, L. Wickramasinghe, D. Li, J. Niklas, O. G. Poluektov, H. B. Schlegel and C. N. Verani, Inorg. Chem., 2018, 57, 9748 CrossRef CAS.
- L. Wu, M. Eberhart, A. Nayak, M. K. Brennaman, B. Shan and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 15062 CrossRef CAS.
- R. M. Bullock, A. K. Das and A. M. Appel, Chem. – Eur. J., 2017, 23, 7626 CrossRef CAS.
- M. Yamamoto and K. Tanaka, ChemPlusChem, 2016, 81, 1028 CrossRef CAS.
- W. Jiang, X. Yang, F. Li, Q. Zhang, S. Li, H. Tong, Y. Jiang and L. Xia, Chem. Commun., 2019, 55, 1414 RSC.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296 RSC.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924 CrossRef CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099 RSC.
- L. Wu, A. Nayak, J. Shao and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11153 CAS.
- T. Morikawa, S. Sato, K. Sekizawa, T. Arai and T. M. Suzuki, ChemSusChem, 2019, 12, 1807 CAS.
- J. Li, W. Wan, C. A. Triana, Z. Novotny, J. Osterwalder, R. Erni and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 12839 CrossRef CAS.
- L. Xie, X. Li, B. Wang, J. Meng, H. Lei, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2019, 58, 18883 CrossRef CAS.
- P. Manna, J. Debgupta, S. Bose and S. K. Das, Angew. Chem., Int. Ed., 2016, 55, 2425 CrossRef CAS.
- B. A. Johnson, A. Bhunia and S. Ott, Dalton Trans., 2017, 46, 1382 RSC.
- X. Liu, Y. Maegawa, Y. Goto, K. Hara and S. Inagaki, Angew. Chem., Int. Ed., 2016, 55, 7943 CrossRef CAS.
- X. Song, C. Peng and H. Fei, ACS Appl. Energy Mater., 2018, 1, 2446 CrossRef CAS.
- S. Gutiérrez-Tarriño, J. L. Olloqui-Sariego, J. J. Calvente, M. Palomino, G. Mínguez Espallargas, J. L. Jordá, F. Rey and P. Oña-Burgos, ACS Appl. Mater. Interfaces, 2019, 11, 46658 CrossRef.
- N. Heidary, T. G. A. A. Harris, K. H. Ly and N. Kornienko, Physiol. Plant., 2019, 166, 460 CrossRef CAS.
- B. Wurster, D. Grumelli, D. Hötger, R. Gutzler and K. Kern, J. Am. Chem. Soc., 2016, 138, 3623 CrossRef CAS.
- Q. Shao, J. Yang and X. Huang, Chem. – Eur. J., 2018, 24, 15143 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |