
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Installing the “magic methyl” – C–H methylation in synthesis
Daniya
Aynetdinova†
 ,
Mia C.
Callens†
,
Harry B.
Hicks†
,
Charmaine Y. X.
Poh†
,
Benjamin D. A.
Shennan†
,
Alistair M.
Boyd‡
,
Zhong Hui
Lim‡
,
Jamie A.
Leitch
* and
Darren J.
Dixon
*
,
Mia C.
Callens†
,
Harry B.
Hicks†
,
Charmaine Y. X.
Poh†
,
Benjamin D. A.
Shennan†
,
Alistair M.
Boyd‡
,
Zhong Hui
Lim‡
,
Jamie A.
Leitch
* and
Darren J.
Dixon
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: j.leitch@ucl.ac.uk; darren.dixon@chem.ox.ac.uk
First published on 10th March 2021
Abstract
The selective and efficient C–H methylation of sp2 and sp3 carbon centres has become a powerful transformation in the synthetic toolbox. Due to the potential for profound changes to physicochemical properties attributed to the installation of a “Magic Methyl” group at a strategic site in a lead compound, such techniques have become highly desirable in modern drug discovery and synthesis programmes. This review will cover the diverse techniques that have been employed to enable the selective installation of the C–Me bond in a wide range of chemical structures, from simple building blocks to complex drug-like architectures.
 (First row, left to right) Daniya Aynetdinova, Mia Callens, Harry Hicks; (Second row) Charmaine Poh, Benjamin Shennan, Alistair Boyd; (Third row) Zhong Hui Lim, Jamie Leitch, Darren Dixon | The following seven authors are pursuing their DPhil studies in the EPSRC Synthesis for Biology and Medicine Centre for Doctoral Training (SBM CDT) at the University of Oxford. Daniya Aynetdinova: Daniya obtained her MChem degree from the University of Oxford, completing her Master's project with Prof. Timothy Donohoe working on the total synthesis of lophotoxin using RCM based furan methodology. She then returned to the Donohoe group to work on forming complex carbocycles via cationic cyclisation reactions mediated by carbon-electrophiles. Mia C. Callens: Mia received her MChem in Medicinal Chemistry (Industrial, GSK) from the University of Leeds, where she carried out her final project with Prof. Adam Nelson using activity-directed synthesis in the discovery of kinase inhibitor scaffolds. She is now with Prof. Paul Brennan, where her work focuses on the development of chemical probes to investigate GEF/GTPase targets associated with neurodegenerative diseases. Harry B. Hicks: Harry completed his undergraduate studies at the University of Cambridge attaining an MSc in Natural Sciences, before working as a Medicinal Chemist at Charles River Laboratories for a year. He is now pursuing a DPhil at the University of Oxford, exploring oxonium ion chemistry in natural product synthesis under the supervision of Prof. Jonathan Burton. Charmaine Y. X. Poh: Charmaine is a recipient of the National Science Scholarship from A*STAR in Singapore, where she grew up. With this scholarship, she completed her BSc in Chemistry at Imperial College London and now works on bifunctional iminophosphorane organocatalysis with Prof. Darren Dixon. Benjamin D. A. Shennan: Ben received his MChem degree from the University of Oxford, completing his Master's project with Prof. Darren Dixon investigating new synthetic routes towards spirocyclic pyrrolidines. He now continues research with Prof. Dixon, focusing on the total synthesis of complex diamine natural products. Alistair M. Boyd: Alistair graduated with an MChem from the University of York and completed a research project on the use of vibrational spectroscopy to quantify impurities in pharmaceutical intermediates. He now conducts research in the group of Prof. Stuart Conway, investigating macrocyclic ligands for bromodomain containing proteins. Zhong Hui Lim: Zhong Hui received his MChem degree from the University of Oxford, working with Prof. Ed Anderson on atropselective [5+2] cycloisomerisations for his Master's thesis. Under the supervision of Prof. Hagan Bayley, his current research focuses on single molecule kinetics within protein nanopores. |
Jamie A. Leitch: Jamie obtained his MChem at the University of Bath in 2014 then pursued a PhD at the same institution in the group of Prof. Chris Frost, in a CASE collaboration with Syngenta. He specialised in the remote C–H functionalisation of arenes using ruthenium(II) catalysis, and completed his training in 2017. Following this, he took up a position as a Leverhulme Trust Postdoctoral Research Fellow in the Dixon group at the University of Oxford, as part of the photoredox team working on the reductive generation of α-amino and α-oxy radicals. |
Darren J. Dixon: studied at the University of Oxford where he received a Masters degree in 1993 and DPhil in 1997 under the supervision of Prof. Stephen Davies. After postdoctoral work with Prof. Steven Ley CBE, FRS, he joined the faculty at the Department of Chemistry in Cambridge in 2000. In 2004 he took a Senior Lecturership at The University of Manchester and in 2007 he was promoted to Reader. In 2008 he moved to his current post at the University of Oxford where he is Professor of Chemistry and the Knowles–Williams Fellow in Organic Chemistry at Wadham College. |
1. Introduction
The “Magic Methyl” effect has become coveted in medicinal chemistry due to the profound pharmacological effects that have been observed upon conversion of a C–H bond to the C–Me bond. As outlined in detail in a case-study-focused review by Cernak in 2013,1 the “Magic Methyl” effect2 can be attributed to a number of physical phenomena, including (but not limited to): favourable desolvation energetics,3 metabolic stability changes (both decrease and increase),4 tailored hydrophobic interactions,5 and induced conformational effects.6 The last of these has been attributed to the headline effects of the “Magic Methyl”, and potency jumps of 100–1000-fold have been suggested to be primarily due to such conformational effects,1 of which two remarkable examples are outlined in Fig. 1. | ||
| Fig. 1 Examples of profound conformational effects due to a “Magic Methyl”. | ||
The first of which, by GlaxoSmithKline's (GSK's) discovery team, demonstrated that the installation of a methyl group in the ortho position of a biaryl unit, led to a >200-fold increase in binding affinity (Ki) of p38α MAP3 kinase.7 Analysis of the protein–ligand complex revealed substantial torsional twist in the bound ligand, where a biaryl dihedral angle of 85° was observed. Computational analysis of the two compounds elucidated that the methylated species more closely mimicked the protein-bound structure (C–Me, 65° vs. C–H, 50°), which was hypothesised to cause the substantial binding and potency uplift observed.
In a second example, Pfizer discovered that installation of a cis-methyl group at the α-oxy position of a morpholine-containing lead compound, gave rise to a 45-fold potency increase as a mineralocorticoid receptor (MR) agonist.8 Computational and single crystal X-ray diffraction experiments revealed that the presence of a methyl group cis to the phenyl group locked the arene in the axial position (>5 kcal mol−1). This conformation was suggested to cause burial of both the phenyl and methyl groups into hydrophobic pockets within the active site, leading to the observed boost in potency.
The examples above are two in a wide library of documented “Magic Methyl” effects demonstrating the potential for marked improvements in drug properties via the strategic exchange of a C–H to a C–Me group.3–6 It is worth noting that a literature survey from 2012 found that 8% of all methyl installations led to a potency boost of 10-fold or more, increasing further to >100-fold in 0.4% of cases. Despite this statistically low chance of success, the potential for rapidly increasing potency upon formal C–H methylation cannot be understated.9 In addition, C–Me introduction is accompanied by negligible effects on the lipophilicity (Δc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) log
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ≈ 0.5) and molecular weight (ΔMW = 14 g mol−1) of a lead compound. This is especially pronounced when compared to the medicinally relevant trifluoromethyl group (ΔclogP ≈ 0.9 & ΔMW = 68 g mol−1), the installation of which leads to marked changes in the physical properties of a compound. When considering Lipinski's rules on small-molecule drug candidates,10 such increases can prove immensely costly, and even critical, if a lead compound is already lipophilic or of high molecular-weight.11
P ≈ 0.5) and molecular weight (ΔMW = 14 g mol−1) of a lead compound. This is especially pronounced when compared to the medicinally relevant trifluoromethyl group (ΔclogP ≈ 0.9 & ΔMW = 68 g mol−1), the installation of which leads to marked changes in the physical properties of a compound. When considering Lipinski's rules on small-molecule drug candidates,10 such increases can prove immensely costly, and even critical, if a lead compound is already lipophilic or of high molecular-weight.11
For these reasons, the ability to incorporate the Me group at specific points in a structure–activity-relationship (SAR) programme would be of high value to medicinal chemistry. Despite this, the C–H methylation of both sp2 and sp3 centres has been traditionally limited to deprotonation of acidic C–H bonds followed by alkylation with electrophilic methyl sources, such as methyl iodide.12 Accordingly, in the absence of such acidic C–H bonds at the target position (for example an enolisable carbonyl functional group), the methyl subunit must be incorporated at the early stages in the synthetic route before building further complexity (Fig. 2A). In one notable case, during a SAR investigation towards mGluR5 antagonists, the discovery team at GSK explored the exchange of C–H to C–Me at two separate positions of a lead compound. While an impressive >754-fold boost in potency was observed when the methylation pattern was optimal, all of the compounds studied required de novo synthetic routes from methylated feedstocks.13 Such approaches often lack divergence and, in turn, are time- and resource-consuming for drug discovery programmes, where route efficiency, sustainability, and atom economy are of critical importance.14
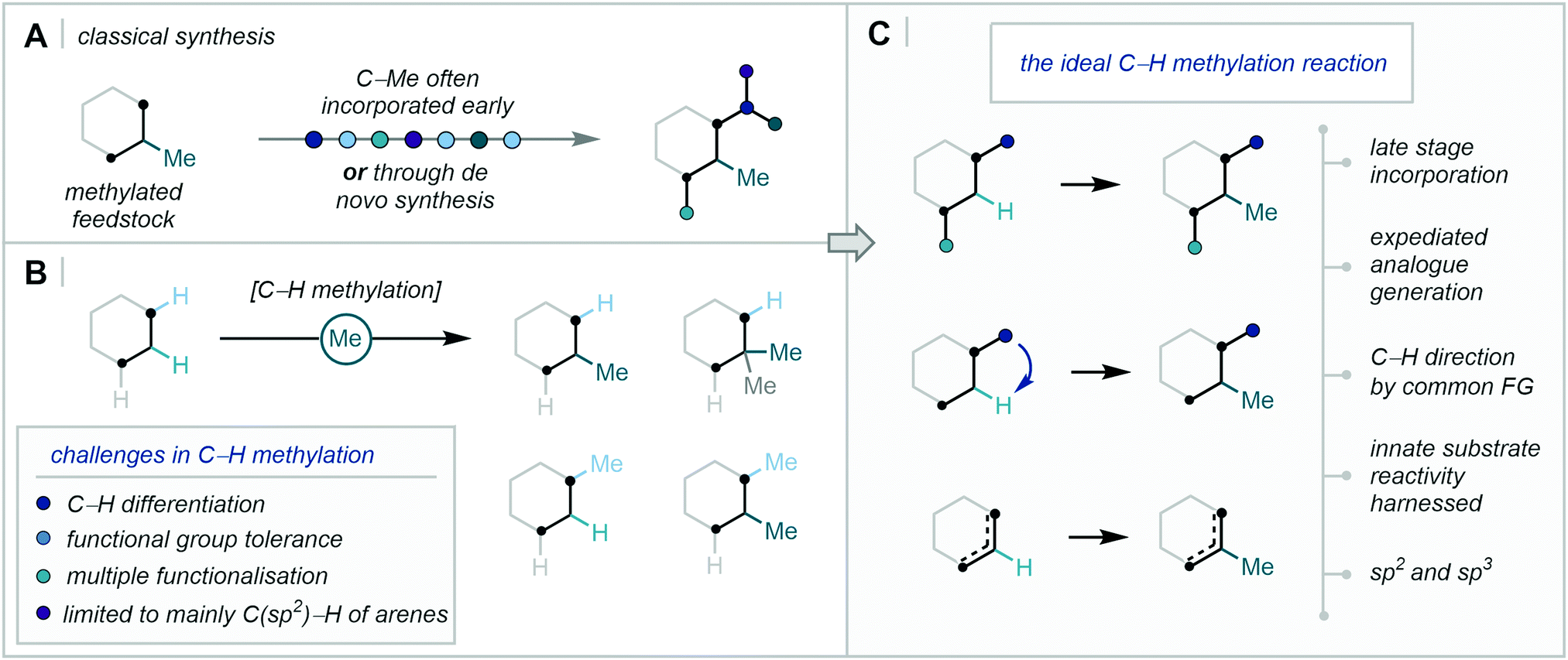 | ||
| Fig. 2 C–H methylation in synthesis – (A) Classical synthesis. (B) Challenges in C–H methylation. (C) The ideal C–H methylation reaction. | ||
To this end, the functionalisation of C–H bonds for downstream C–C bond formation has become an influential tool in streamlining complex synthetic routes.15 However, the challenge of differentiating sterically and electronically similar C–H bonds (such as in an aromatic ring or an alkyl chain), especially in complex biologically relevant molecules, remains ever-present.16 Consequently, a concerted effort in modern synthesis has sought to overcome these hurdles, enabling chemo-, regio-, and enantio-selective C–H functionalisation.17 Within the context of C–H methylation, however, the efficient and selective methylation of C–H bonds faces additional challenges often attributed to the small size of the methyl unit, such as heightened regioselectivity issues and in the undesired over-functionalisation of one or more sites (Fig. 2B).
Accordingly, technologies for the selective and efficient installation of a methyl group in a strategic position, especially in the late-stage functionalisation of drug-like compounds, have wide-ranging and potentially immediate applications in medicinal chemistry (Fig. 2C).18 This review will cover the diverse strategies and techniques that have achieved the C–H methylation of C(sp2)–H and C(sp3)–H bonds, with specific focus on the recent influx of research efforts that have answered Cernak's call for the discovery of new C–H methylation reactions in 2013.19 Transition-metal directed C–H activation, direct radical addition approaches, and complementary two-electron activation pathways will be covered. However, we will primarily turn our attention away from methods which have relied on traditional deprotonation then alkylation/aldol sequences, and for those reasons, elegant recent developments in transition-metal-catalysed hydrogen borrowing methodology will not be discussed.20
2. Directed C(sp2)–H methylation
2.1 4d transition metal-catalysed C(sp2)–H methylation
Transition metal-catalysis has undoubtedly become one of the most powerful tools in organic synthesis for C–C bond formation. Among the myriad of transformations that have been developed, 4d transition metals (namely Pd, Rh, and Ru) have continuously demonstrated their versatility in an impressive array of these reactions, particularly in the field of C–H activation.21Despite this, coupled with their renowned chemical inertness, the selective activation and functionalisation of specific aromatic C–H bonds remains a persistent challenge.21,22 One approach that has been employed to tackle this problem is the use of directing groups (DGs),23 which generally consist of a Lewis basic coordinating moiety that directs the metal centre to enable C–H activation at a specific site (Scheme 1).24
 | ||
| Scheme 1 General concept underlying the directed transition metal-catalysed C–H activation. | ||
A DG-facilitated C–H activation strategy in the context of methylation was reported in 1984 by Tremont and Rahman, who harnessed the ortho directing capability of acetanilides using stoichiometric Pd(OAc)2 and methyl iodide as the methylation reagent.25 The proposed mechanism involved a PdII/PdIV cycle, in which the oxidation of PdII to PdIV by methyl iodide was later supported by X-ray crystallography (Scheme 2).26 A comprehensive overview of the types of DGs used in recent metal-catalysed C–H functionalisation was described previously23b and, due to the vast array available, only those recently applied to C–H methylation will be discussed herein.
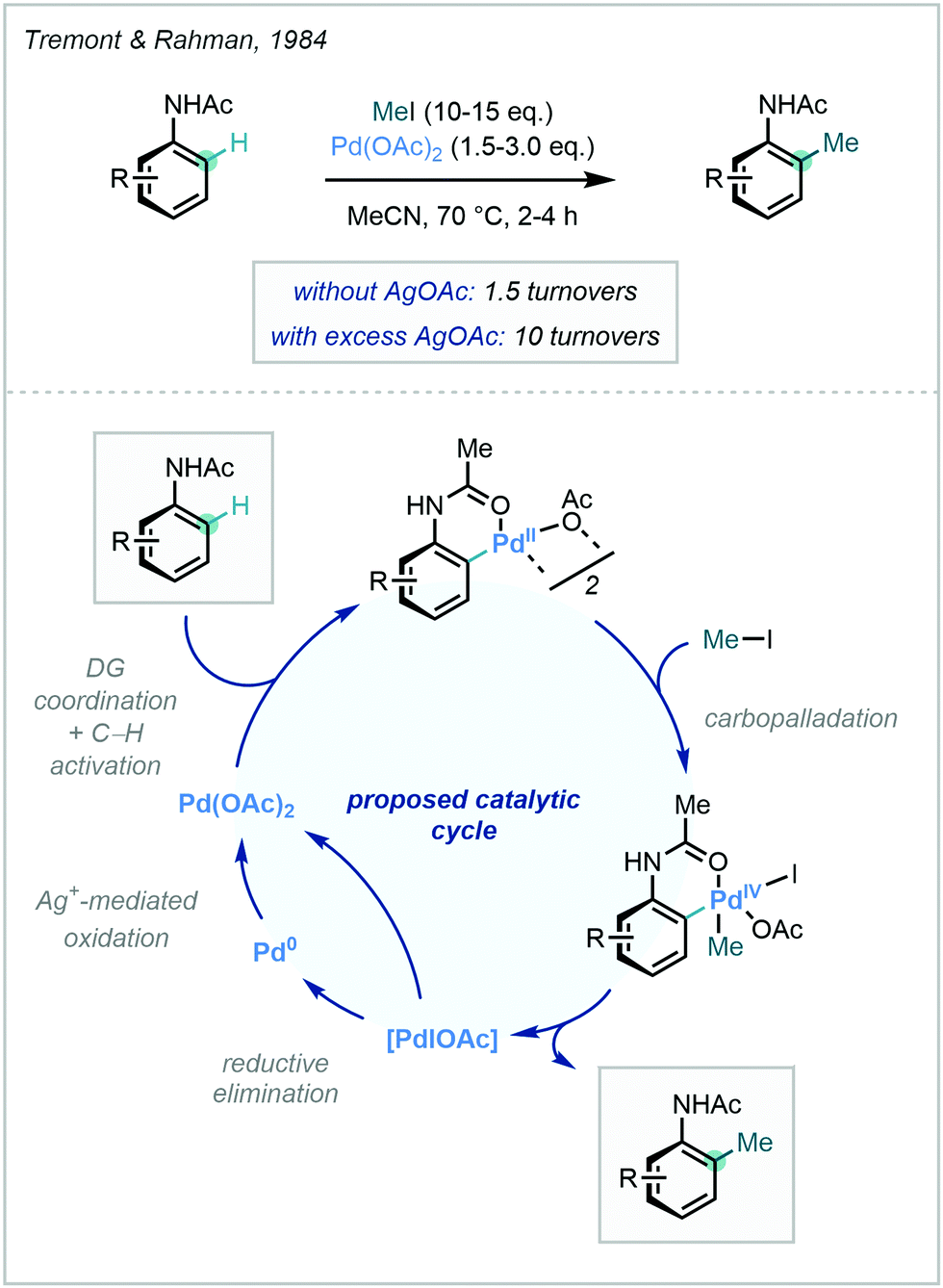 | ||
| Scheme 2 Pd-mediated directed ortho methylation of acetanilides with Me–I. | ||
Although the use of methyl halides in this transformation has remained popular, attractive alternatives have also been reported. A prominent class of alkylating agents which have been employed in directed C(sp2)–C(sp3) alkylations are boronic acid-derived reagents, disclosed by Yu in 2006.28 In this work, ortho C–H methylation of substituted arenes bearing a pyridyl or pyrazole DG was achieved with trimethylboroxine or methyl boronic acid (Scheme 3). This was reported as the first protocol for the Pd-catalysed alkylation of sp2 and sp3 C–H bonds with such methylating reagents.
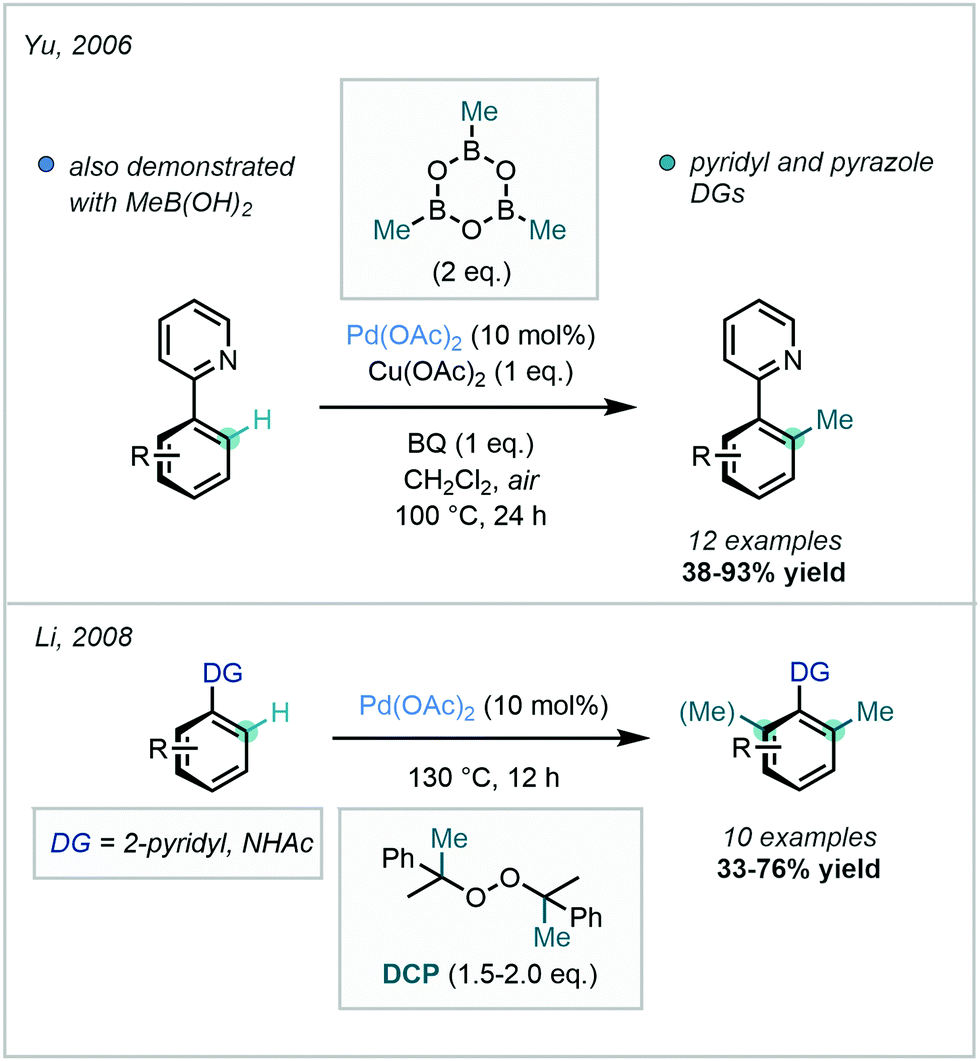 | ||
| Scheme 3 Directed C–H methylation of pyridyl arenes and acetanilides with trimethylboroxine, methylboronic acid and dicumyl peroxide. | ||
Peroxides have similarly been deployed as valuable methylating reagents. Their use in directed C–H methylation was described by Li in 2008, using dicumyl peroxide (DCP) to install a methyl group at the ortho position of arenes bearing a pyridyl or amidic DG (Scheme 3).29 The mechanism was subsequently investigated by Sunoj.30
In 2011, Youn revisited the Pd-catalysed ortho methylation of acetanilides to reveal the reaction could be performed at room temperature with methyl iodide (Scheme 4A).31 The use of pseudo alkyl halides was detailed more recently by Bach, where methyl mesylate and sodium bromide were utilised in the ortho alkylation of methoxyarenes (Scheme 4B).32 A cyano-pyrrole DG was employed, and methylation was demonstrated in good yield. Additionally, methyl iodide was used by Roy & Kundu in their ortho methylation of arenes bearing a 2-pyridone unit (Scheme 4C).33 In this instance, water was used as the solvent.
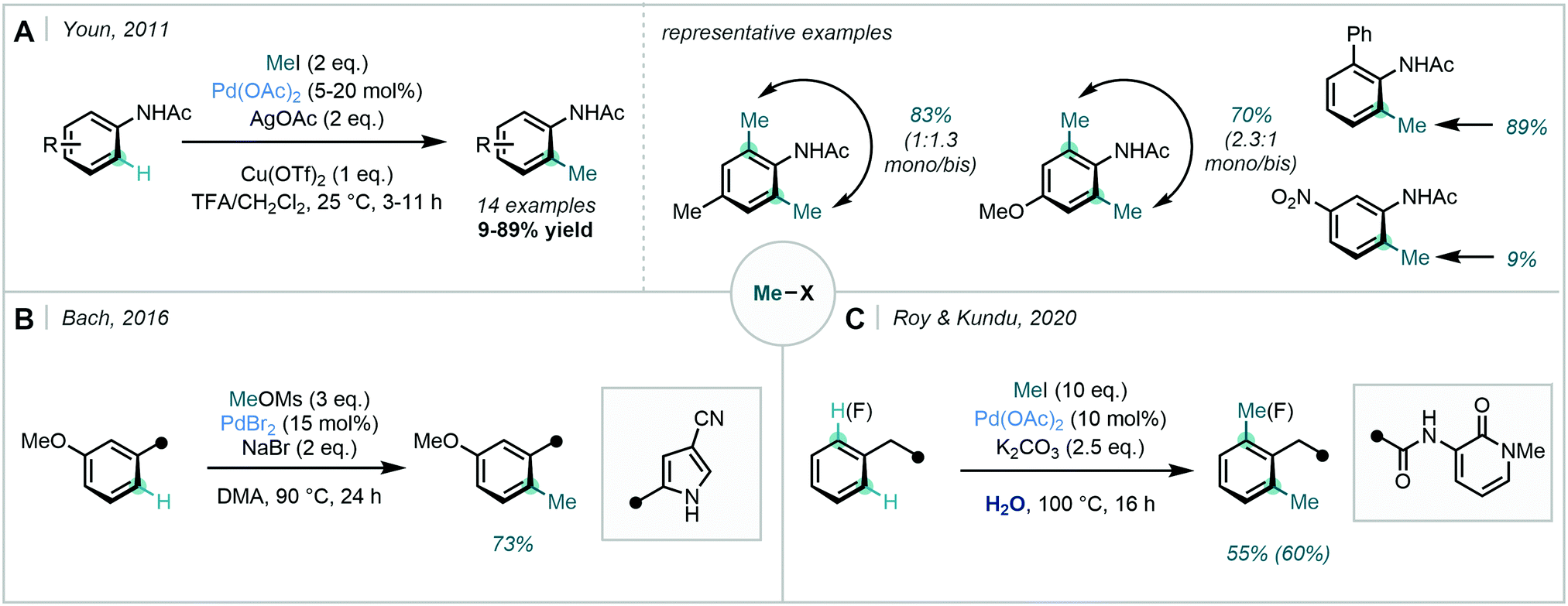 | ||
| Scheme 4 C–H methylation using nitrogen-containing DGs, with Me–X reagents as the methyl source. (A) ortho methylation of acetanilides at room temperature. (B) Use of a cyano-pyrrole DG. (C) C–H methylation of phenyl acetamide derivatives in water. | ||
An extensive array of both aromatic and vinylic C–H methylation examples was disclosed by Yu in 2014, where a ligand-promoted alkylation approach was demonstrated using methyl iodide (Scheme 5). 9-Methylacridine (L1) was selected after a rigorous ligand survey, facilitating effective methylation of sp2 centres bearing an amidic DG. The broad scope included methylation of substituted arenes, thiazoles, alkenes, and sp3 centres.34
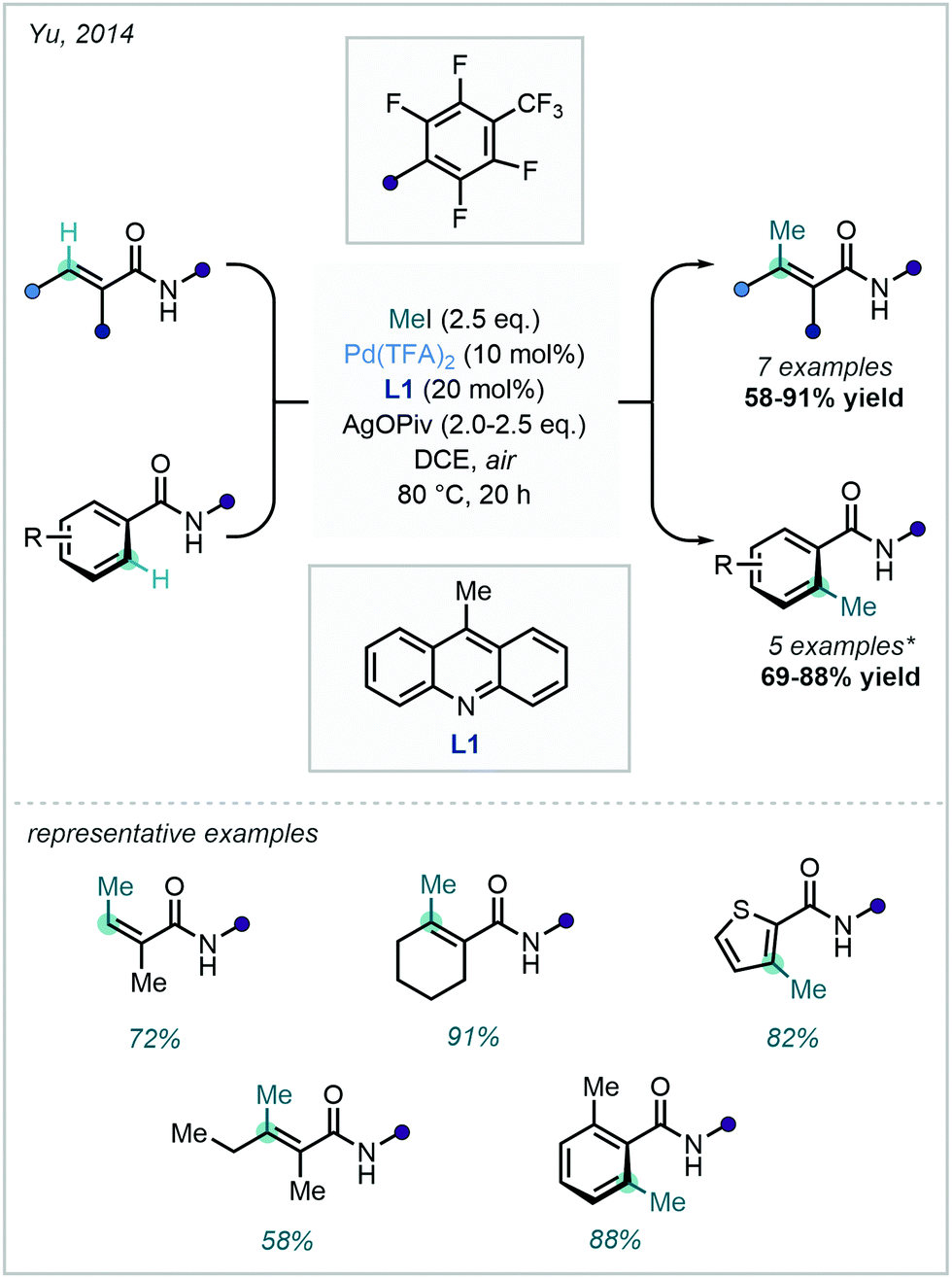 | ||
| Scheme 5 C–H methylation of (hetero)aryl and vinylic substrates with methyl iodide. *Pentafluorobenzoic acid (PFBA, 40 mol%) was required in the methylation of benzamides to suppress N-alkylation. | ||
Further developments to methyl boronic acid derived methylation reagents came in 2013, when Sanford described a methylation procedure with MeBF3K and used MnF3 as the oxidant (Scheme 6A).35 Two different mechanisms were proposed that notably bypassed more traditional PdII reductive eliminations, opting for operation of either a PdIII/PdI or PdIV/PdII manifold facilitated by MnF3 oxidation.
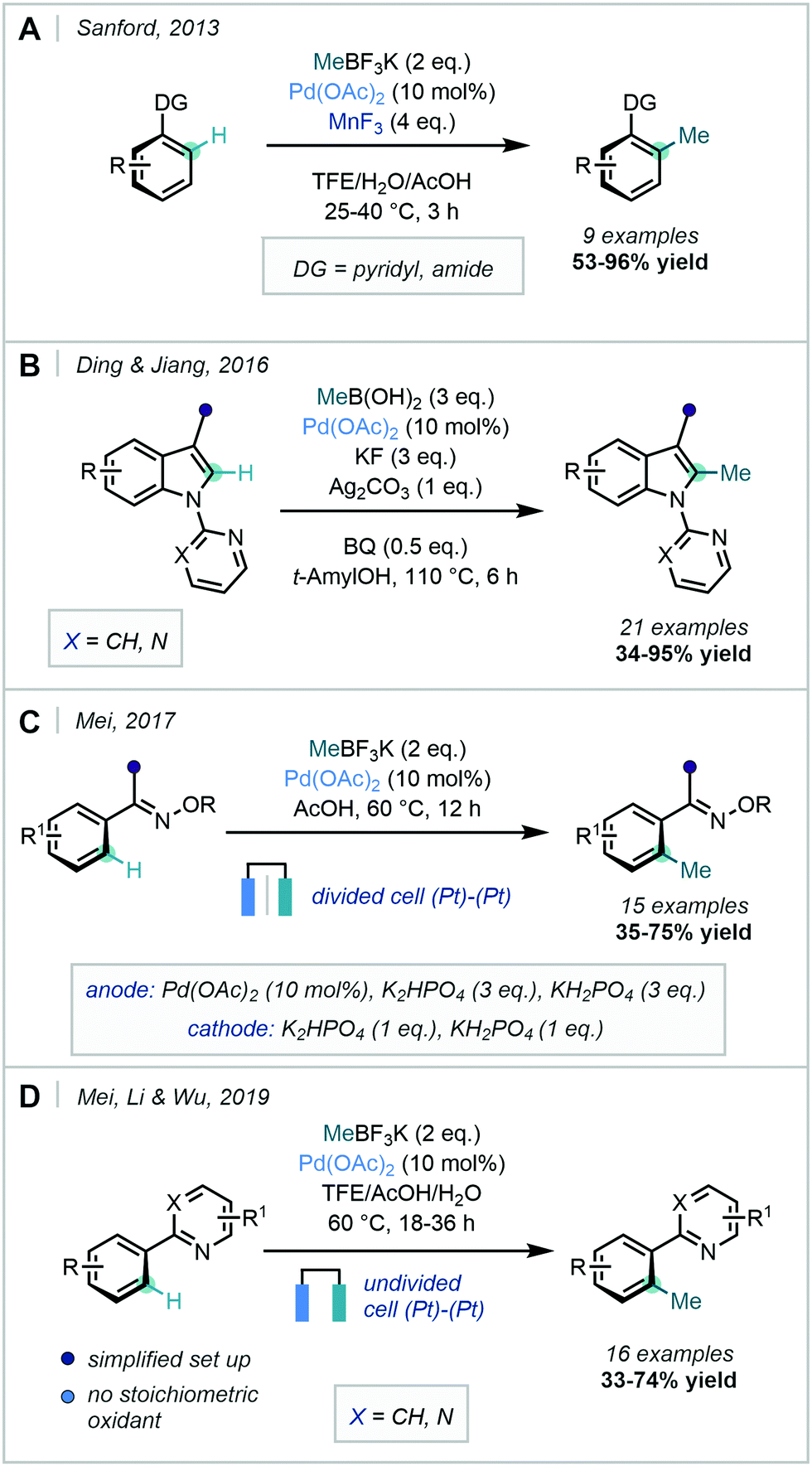 | ||
| Scheme 6 Application of MeBF3K and MeB(OH)2 in the methylation of substituted arenes and heterocycles using a variety of N-based directing groups. (A) Use of MnF3 as an alternative oxidant. (B) C-2 methylation of indoles. (C and D) Electrochemical approaches to Pd-catalysed directed C–H methylation. | ||
Ding & Jiang employed methylboronic acid as the methylating reagent in a pyridyl- and pyrimidyl-directed C2-methylation of indoles, which required high temperatures and 0.5 equivalents of benzoquinone (BQ) in order to promote the slow reductive elimination step (Scheme 6B).36
As the use of a stoichiometric oxidant is generally required in this type of transformation, Mei recognised that an electrochemical strategy could provide an atom-economic alternative. Accordingly, in 2017 the Pd-catalysed ortho-C–H methylation of ketoximes using MeBF3K was documented using electrochemical oxidation to facilitate catalyst turnover (Scheme 6C).37 This approach was applied to a range of functionalised arenes in moderate to good yields. Mei, Li & Wu more recently adapted this procedure to use pyridyl-DGs and an undivided cell, which improved the general accessibility and applicability of electrochemical synthetic set-ups (Scheme 6D).38
Through use of peroxide-based methylating reagents, Cai developed an interesting condition-dependent selective C–H/N–H methylation procedure using DCP or di-tert-butyl peroxide (DTBP) (Scheme 7).39 By altering the methylating agent and catalyst, the authors effectively controlled whether N–H or C–H methylation occurred, where the DGs were sulfonamides and methanamides.
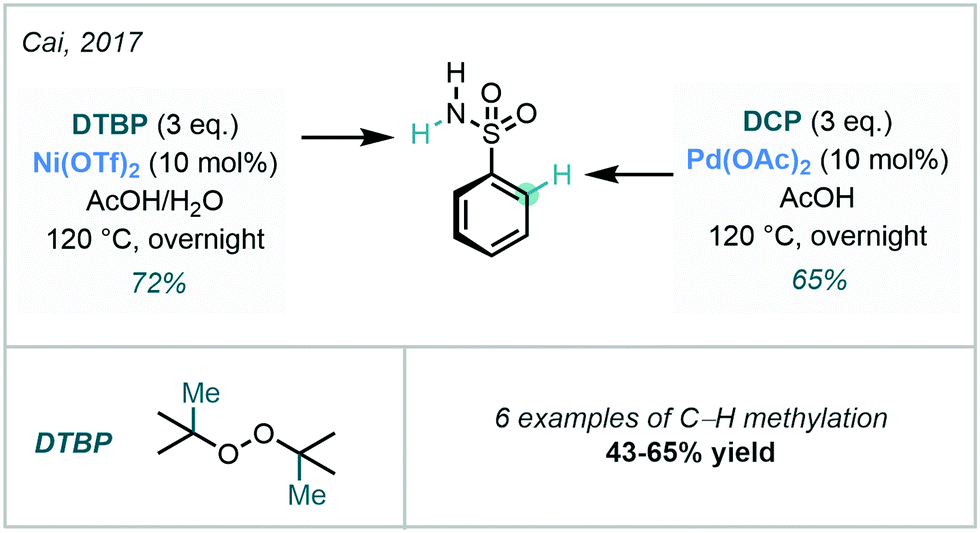 | ||
| Scheme 7 C–H/N–H site-selectivity via peroxide-mediated methylation. | ||
In 2018, Novák successfully prepared a number of S-alkyldibenzothiophenium salts that were effective electrophiles for Pd-catalysed C–H alkylation, further expanding the repertoire of methylating reagents available (Scheme 8). With these in hand, ortho methylation of simple acetanilides was achieved, and aromatic ureas were equally applicable.40 The thiophenium salts could be synthesised via the alkylation of dibenzothiophene with formate esters in strong acid.
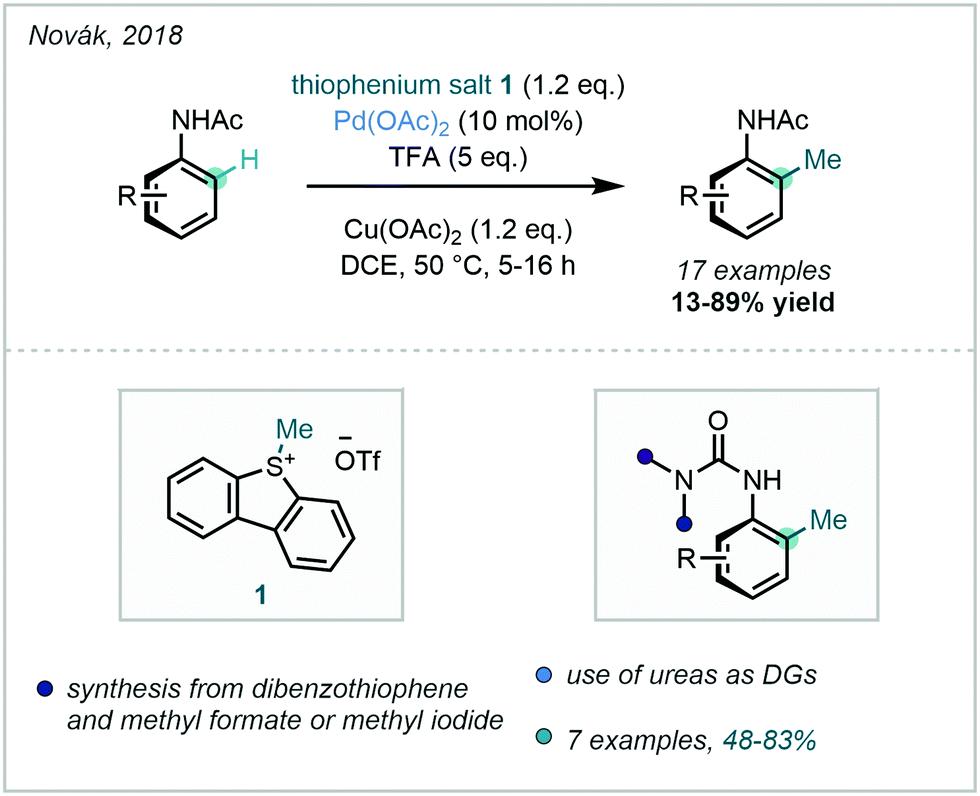 | ||
| Scheme 8 Application of thiophenium salts as methylating reagents. | ||
The use of 8-aminoquinoline (8-AQ) as a DG was pioneered by Daugulis in 2010,41 and has since received significant synthetic interest in both directed C(sp2)–H and C(sp3)–H functionalisation. Chen achieved condition-dependent mono-/di-selective methylation using substrates bearing an 8-AQ DG, enabled simply by adjusting the amount of NaHCO3 used (Scheme 9A).42 Qi identified that similar quinolinamide (QA) or picolinamide (PA) bidentate groups could enable the selective mono-methylation of the C8-position of 1-naphthylamine scaffolds, following their previous success with QA-directed C8-arylation of the same scaffold (Scheme 9B).43
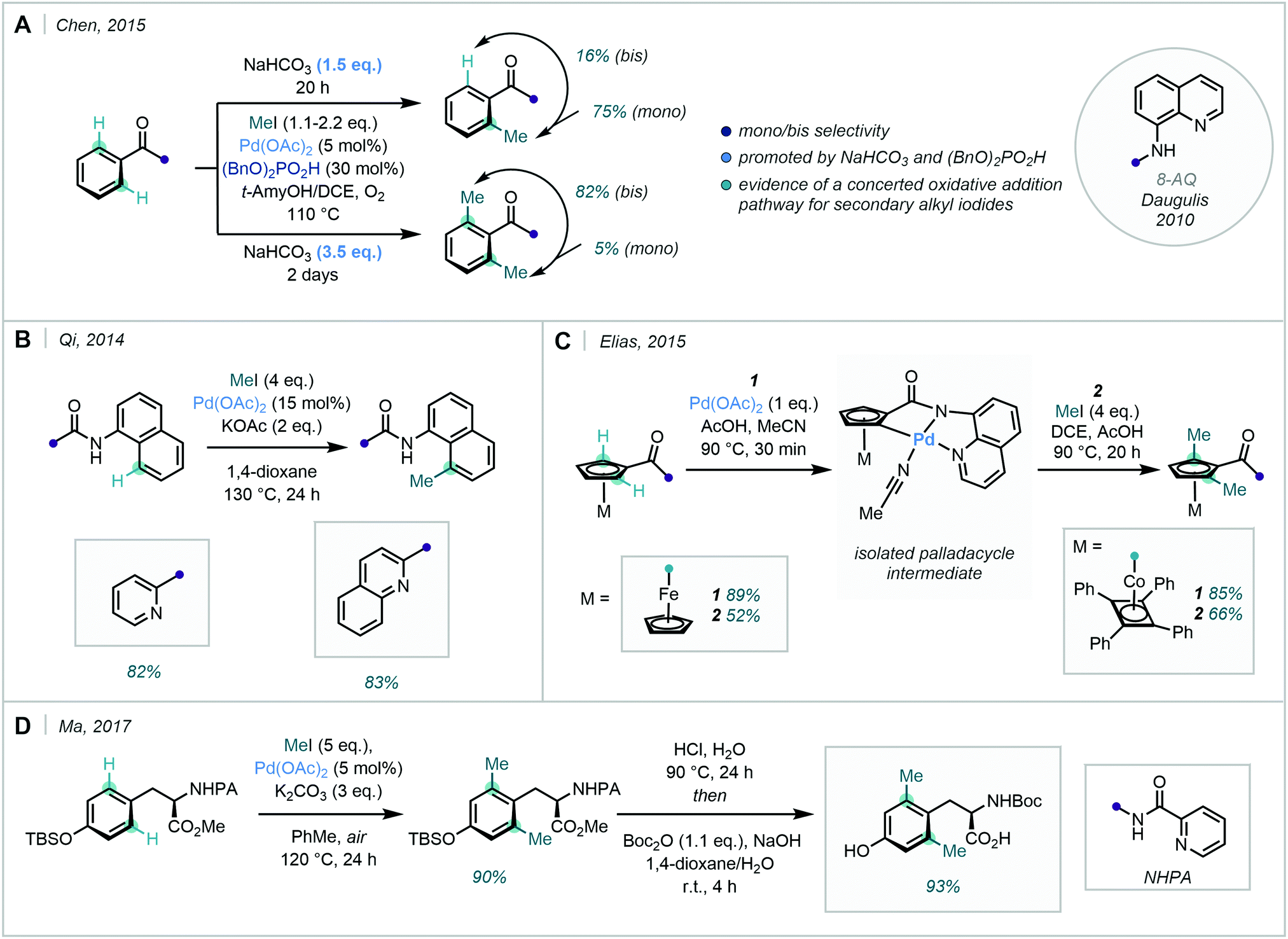 | ||
| Scheme 9 (A) NaHCO3 dependent mono/di-selective ortho methylation of substrates bearing an 8-AQ group. (B) Methylation of 1-naphthylamine scaffolds. (C) Application of the 8-AQ directing group in the ortho dimethylation of iron and cobalt sandwich complexes. (D) Synthesis of methylated tyrosine analogues. | ||
8-AQ was also applied in the ortho di-methylation of cobalt and iron sandwich complexes, where Elias was successful in forming novel palladacycles through the C–H activation of the cyclopentadienyl (Cp) rings (Scheme 9C).44 These examples showcased the continued use of N-based bidentate ligands in the C–H methylation of arenes and heteroarenes, highlighting their robustness and versatility.
Bidentate DGs have also been applied in the synthesis of bespoke amino acid analogues (Scheme 9D). Ma described an ortho-selective di-methylation of (S)-N-Boc-tyrosine, using picolinamide and other amide-based groups.45 Di-methylation occurred in excellent yield, offering an appealing means of acquiring methylated tyrosine analogues.
Expanding this chemistry, in 2013, Yu & Baran developed a Pd-catalysed benzoic acid-directed C–H methylation procedure using amino acid-derived ligands; these were shown to be crucial to reactivity, exhibiting a profound ligand-acceleration effect (Scheme 10A).47
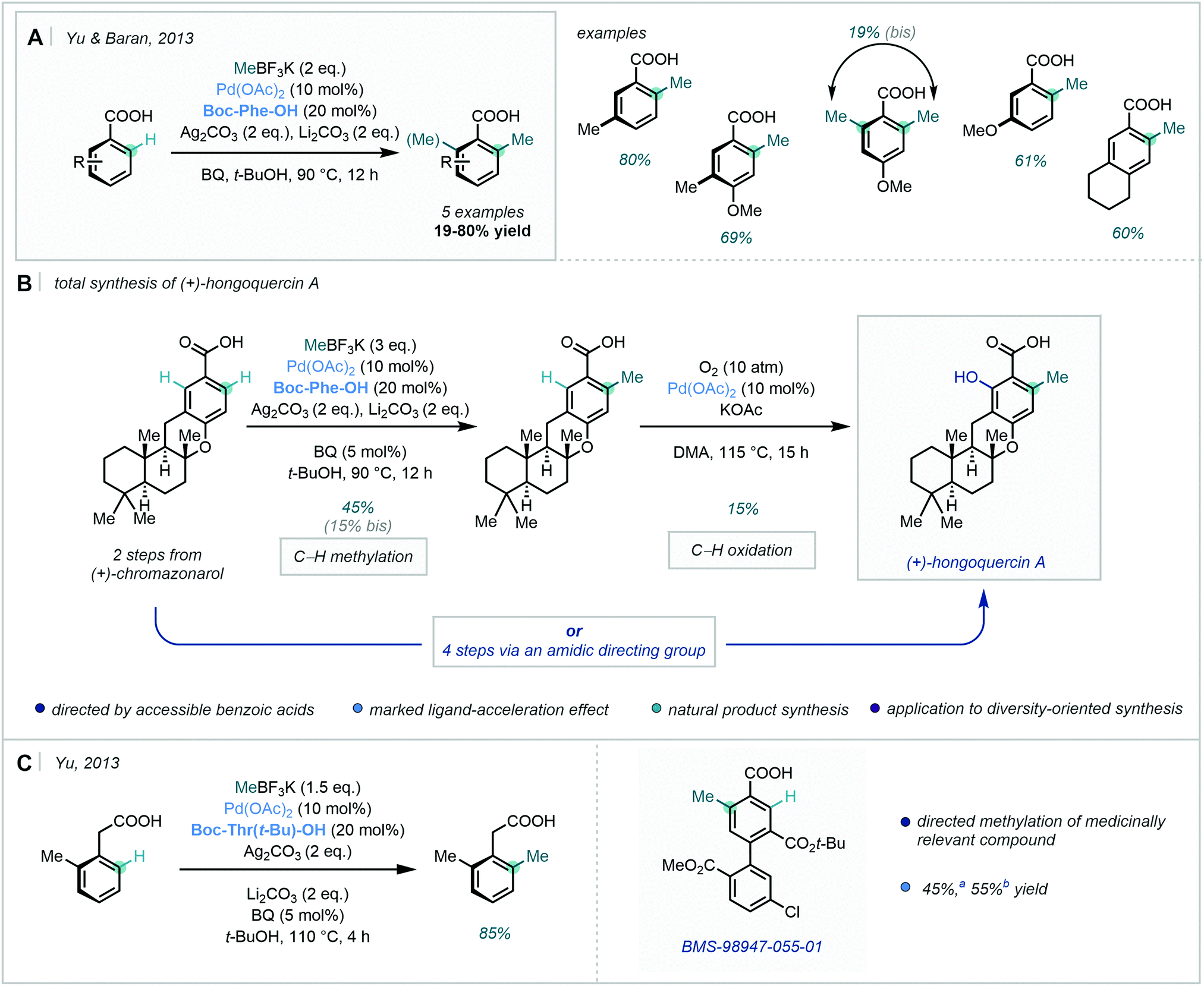 | ||
| Scheme 10 (A) ortho methylation of benzoic acids. (B) The total synthesis of (+)-hongoquercin A. (C) C–H methylation of arylacetic acid derivatives and medicinally relevant compound BMS-98947-055-01. a1.5 eq. MeBF3K, ligand = Boc-Thr(t-Bu)–OH, 110 °C, 2 h. b3.0 eq. MeBF3K, ligand = Boc-Phe–OH, 90 °C, 12 h. | ||
This C–H methylation strategy was successfully applied as the penultimate step in an elegant total synthesis of (+)-hongoquercin A (Scheme 10B). In this instance, the carboxylic acid directed two sequential site-selective C–H functionalisation events – C–H methylation followed by C–H oxidation at the second ortho position. An alternative route via an amidic-DG was equally applicable, and markedly increased the yield of the C–H oxidation step. The divergent design allowed for the generation of a small library of related analogues, showcasing its applicability to diversity-oriented synthesis.
In the same year, Yu described a similar approach for the ortho C–H alkylation of arylacetic acid derivatives and benzoic acids (Scheme 10C).48 After selecting amino acid-derived ligand Boc-Thr(t-Bu)–OH from a comprehensive ligand screen, electron-rich and electron-poor phenylacetic acids were shown to be compatible substrates in the C–H alkylation chemistry. Notably, C–H methylation occurred in almost quantitative conversion and high yield. Mechanistic studies supported the possibility of a radical process, and the rate-determining step appeared to be highly dependent on the electronics of the substrate. Furthermore, the methylation protocol was applied in the late-stage methylation of medicinally relevant compound BMS-98947-055-01.
A similar protocol was more recently reported by Cheng, who employed DTBP as both the methylating agent and oxidant. This protocol allowed the ortho C–H methylation of benzoic acids to occur under external oxidant-free and ligand-free conditions (Scheme 11).49
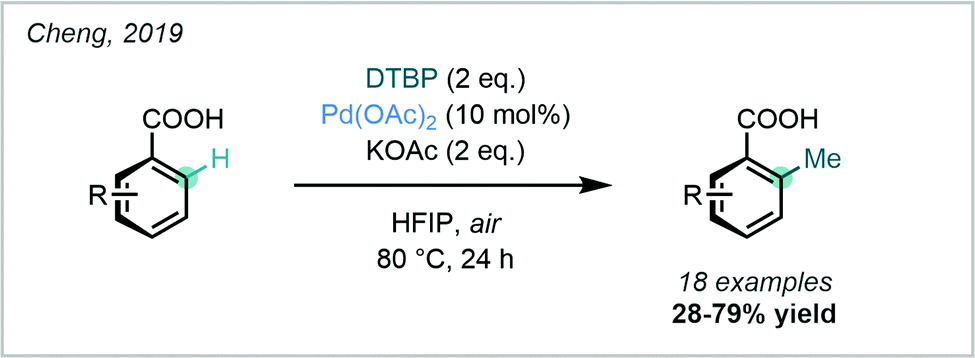 | ||
| Scheme 11 ortho methylation of benzoic acids employing DTBP as the methylating reagent. | ||
To this end, a variety of silane DGs, that are able to serve as downstream functional handles, have been developed.51 Gevorgyan has previously documented the removable and modifiable pyridyldiisopropylsilyl (PyDipSi) and pyrimidyldiisopropylsilyl (PyrDipSi) DGs, and in 2016 applied PyrDipSi in the ortho C–H alkylation of arenes. The example of C–H methylation was mono-selective, and occurred in good yield (Scheme 12).52 Such Si-tethered arenes possess impressive versatility and can be transformed into a number of functional groups including aryl halides, phenols, biaryls, and boronic ester derivatives, with the opportunity to enable a streamlined C–H methylation and diversification platform.
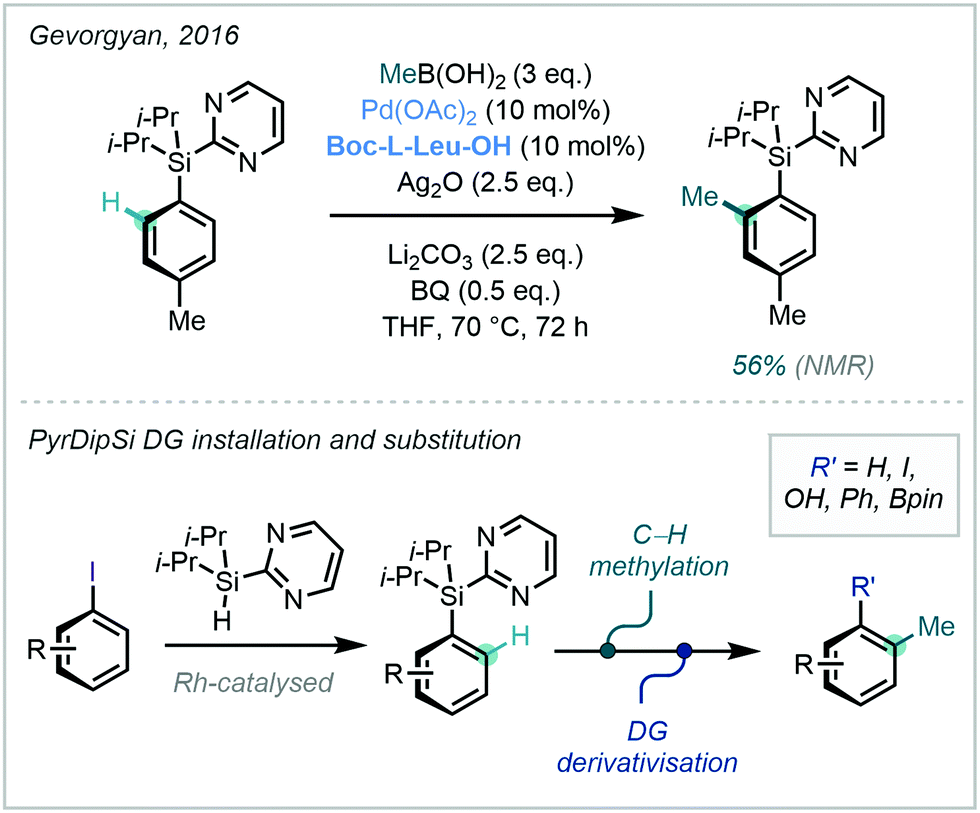 | ||
| Scheme 12 ortho methylation using the modifiable and removable directing group PyrDipSi. | ||
Alternatively, the use of transient directing groups (TDGs) has positioned itself as an extremely appealing method for expediting syntheses.53 An early report of this approach was described by Jun in 1997,54 and was notably exemplified by Yu in the identification of an amino acid transient DG for the functionalisation of C(sp3)–H bonds in 2016.55
Within the context of C–H methylation methodology, Chen & Sorensen harnessed this approach in 2018, where the Pd-catalysed ortho methylation (and fluorination) of benzaldehydes was enabled (Scheme 13).56
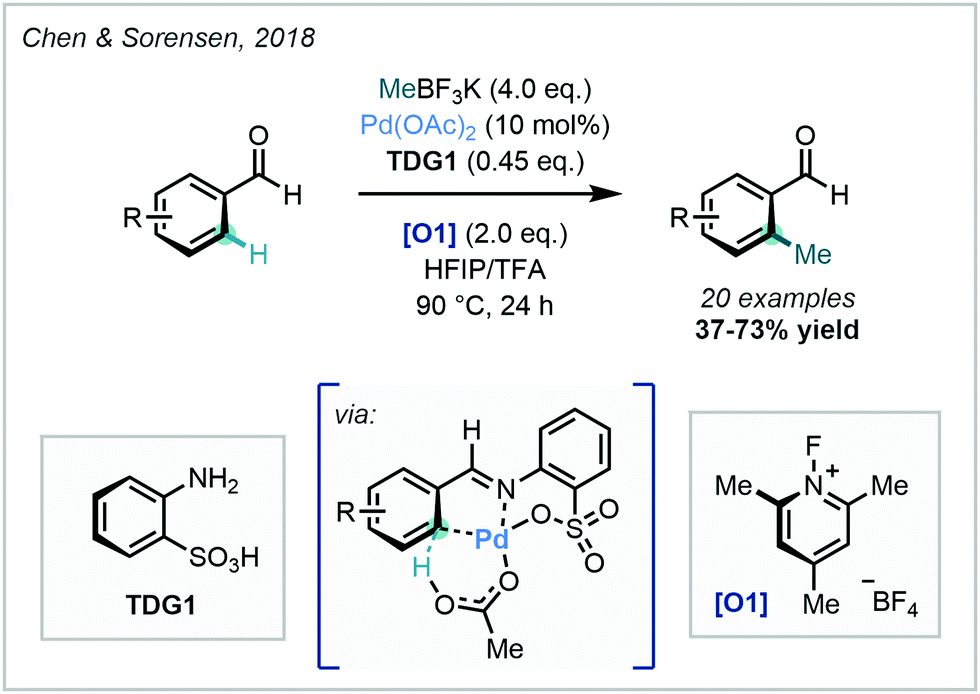 | ||
| Scheme 13 Identification of orthanilic acid as a transient directing group in the C–H methylation of benzaldehydes. | ||
In this work, ortho methylation was achieved using orthanilic acid-analogue TDG1, and the protocol was demonstrated on a variety of benzaldehydes in good yields. Crystallographic evidence was obtained of the TDG forming a multidentate cyclometalated species, supporting the proposed mechanism. This work not only offered a novel TDG, but also expanded the scope of catalytic C–H methylations of aldehydes. The approach was extended to other alkyl groups in an Ir-catalysed ortho-alkylation of heteroaromatic aldehydes, but in this instance, C–H methylation was only demonstrated in low yield.57
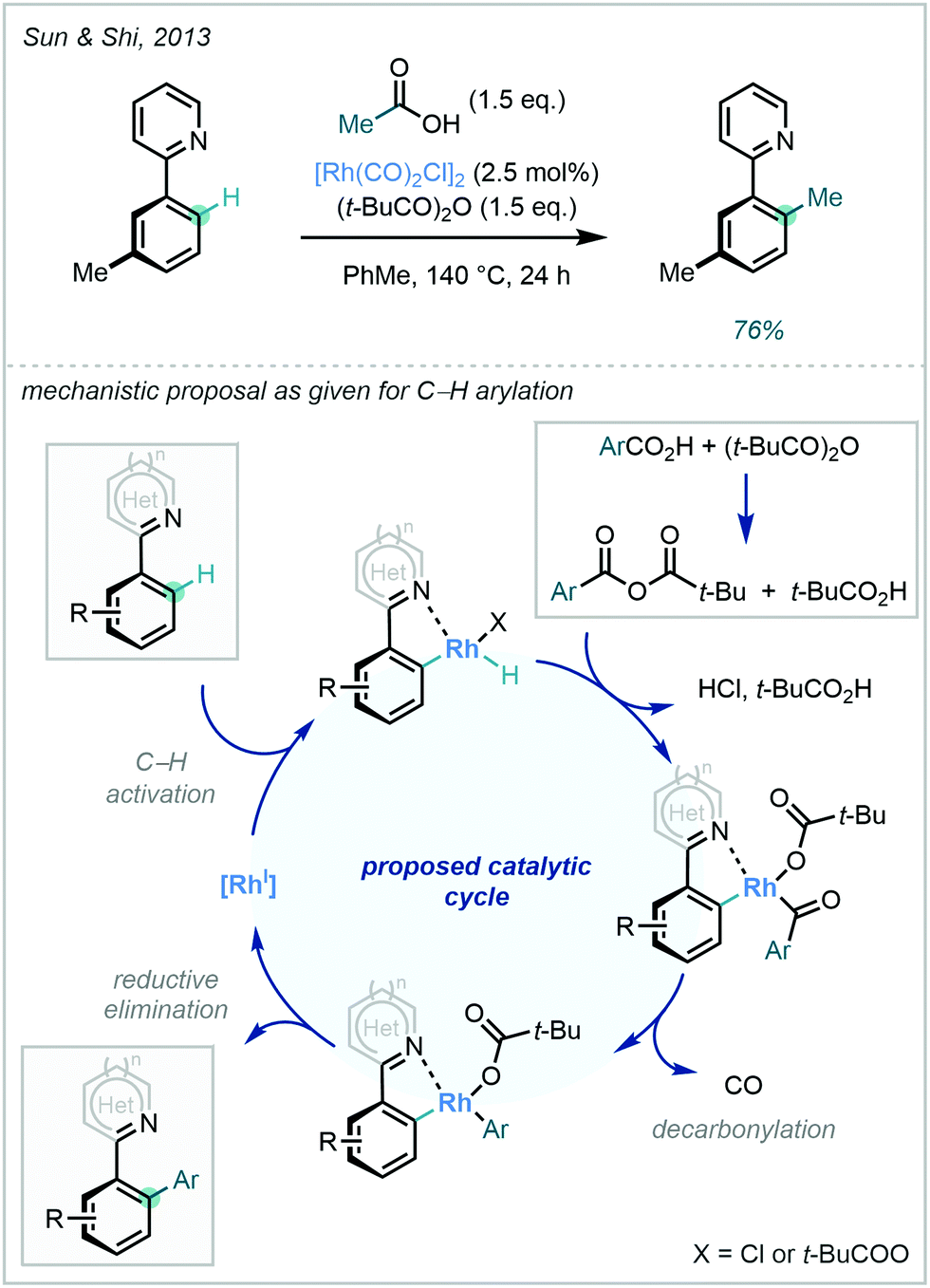 | ||
| Scheme 14 C–H methylation of a 2-arylpyridine using acetic acid as the methyl source. | ||
Many of the reports that followed this work adopted the MeBF3K methylating reagent discussed previously. In 2015, Li reported the Rh-catalysed ortho-selective C–H methylation directed by a suite of N-based DGs (Scheme 15).59 MeBF3K was used as the methyl source, resulting in the ortho methylation of a variety of arenes and the C2 methylation of indoles. Mechanistic studies suggested that a radical process was not operational, and thus transmetalation followed by C–C bond-forming reductive elimination was proposed as the likely mechanism (Scheme 15A).
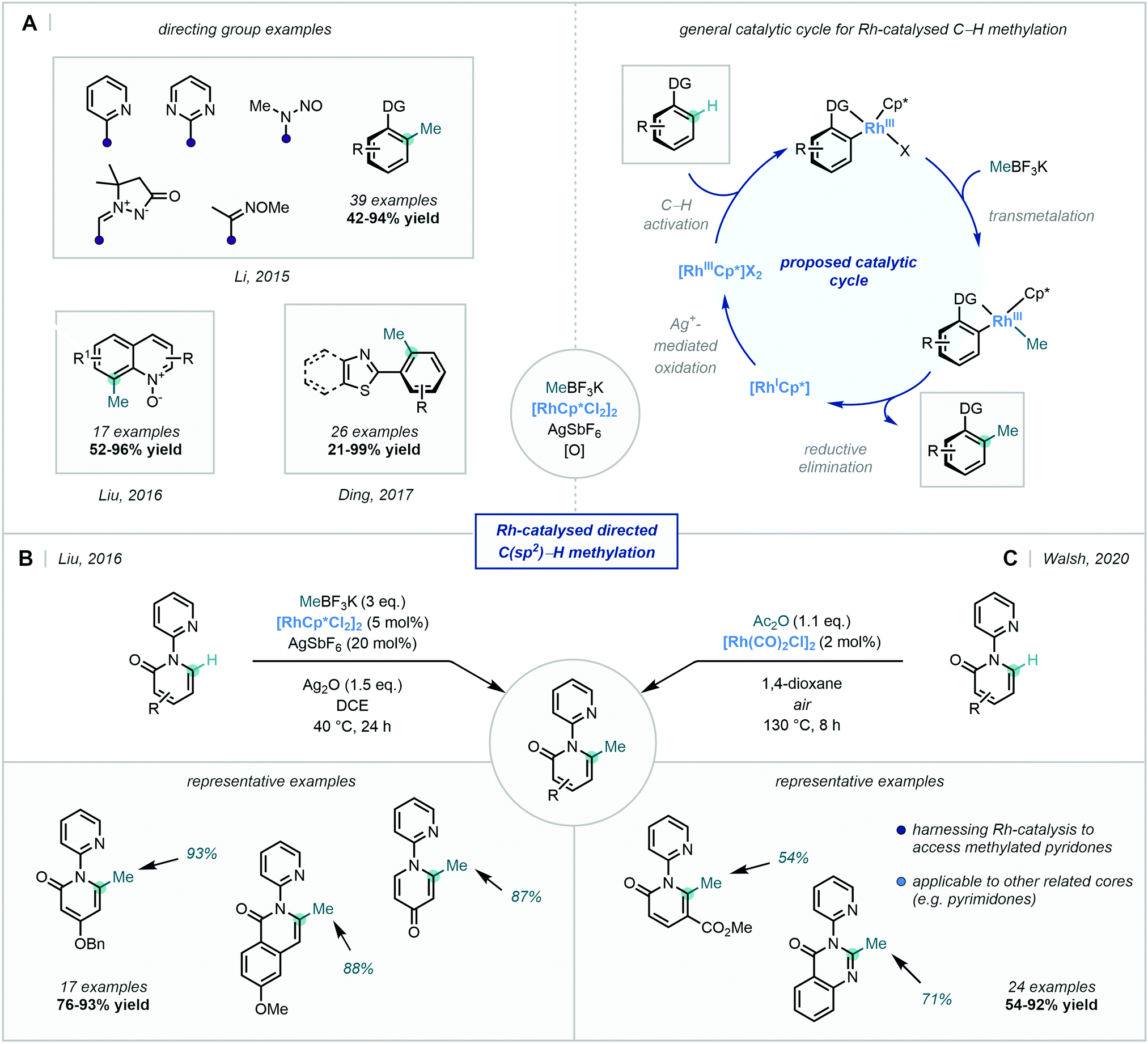 | ||
| Scheme 15 C(sp2)–H directed methylations harnessing Rh catalysis. (A) A selection of recently reported directing groups capable of directing C–H methylation using Rh-catalysis and the general catalytic cycle. (B and C) Two complementary procedures for the Rh-catalysed C–H methylation of pyridones. | ||
In 2017, Ding developed a general method for the ortho methylation of 2-aryl(benzo)thiazoles, using MeBF3K as the methyl source, albeit with limited selectivity between mono- and di-functionalisation (Scheme 15A).60 Similar conditions were also used in the regioselective C8 C–H methylation of quinoline N-oxides by Liu (Scheme 15A),61 as well as in the selective directed C–H methylation of 2,4-diarylquinazolines described by Peng.62
Liu also took advantage of the catalytic reactivity of Rh, in order to access the 6-position of a number of pyridones (Scheme 15B), facilitating C–H methylation enabled by a pyridyl directing group.63 A related transformation was more recently described by Walsh, using acetic anhydride as the methylation agent in a C6-selective decarbonylative alkylation of pyridones (Scheme 15C).64
An exciting recent development disclosed by Pilarski in 2020, was the ability to perform this Rh-catalysed transformation using mechanochemistry (Scheme 16).65 The ball-milling approach66 was not only solvent-free, but also highly regioselective. Interestingly, they developed a frequency-dependent regioselective C–H methylation protocol on an indoline substrate (C7 vs. C2 insertion), and explicitly highlighted the advantages of using this ball-milling approach in the preparation of rhodacycles. C–H methylation was also achieved on a number of biologically-relevant compounds, thus highlighting the applicability of this mechanochemical approach in late-stage functionalisation.
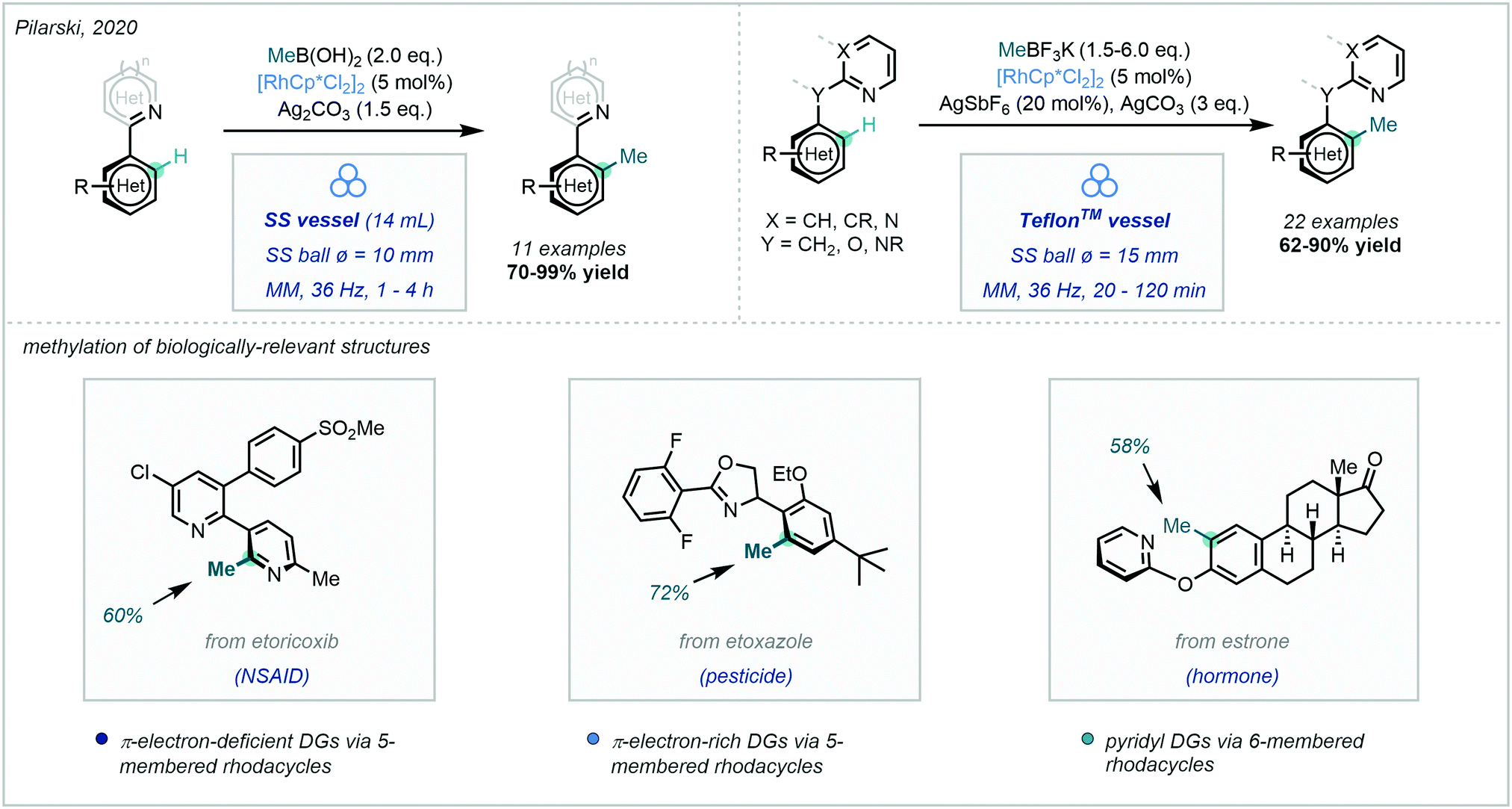 | ||
| Scheme 16 A mechanochemical approach to C–H methylation, including in the methylation of biologically-relevant compounds (MM = mixer mill). | ||
Despite this, Ackermann – a pioneer in Ru-catalysed C–H functionalisation67 – documented the Ru(II)-catalysed C–H methylation of both indoles and pyrroles using a pyridyl DG appended to the indole nitrogen (Scheme 17). In line with other reports, MeBF3K was applied as the methyl source and silver salts proved to be optimal as the terminal oxidant. The reaction also proved successful for the methylation of a tryptophan derivative, occurring in excellent yield.68
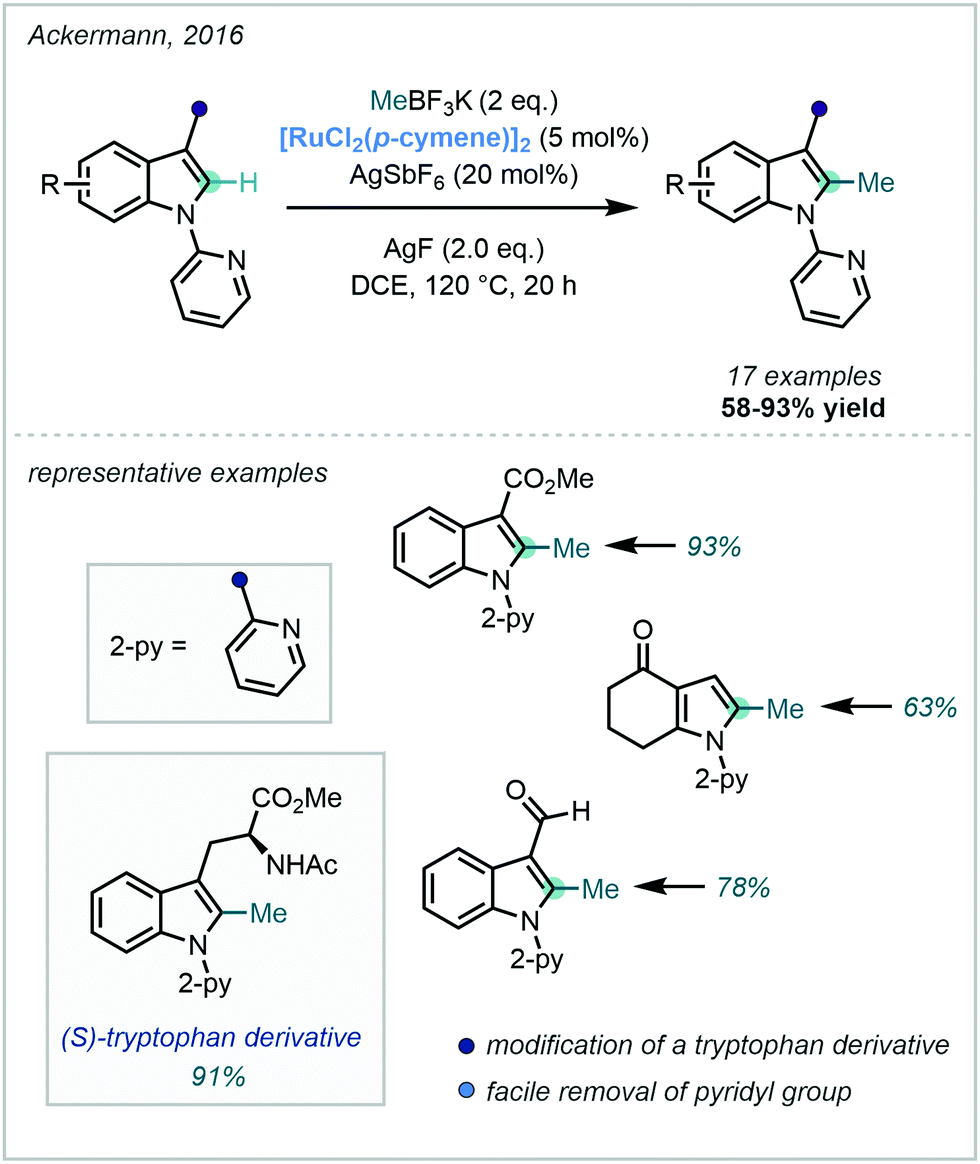 | ||
| Scheme 17 Methylation of indoles utilising Ru-catalysis, applied in the methylation of a tryptophan derivative. | ||
2.2 3d transition metal-catalysed C(sp2)–H methylation
In contrast to the transition metals discussed previously, the application of the earth-abundant 3d transition metals offers an attractive alternative. Such 3d transition metal catalysts, namely Co, Fe, Ni or Mn, are generally readily available, inexpensive, relatively non-toxic, and can also possess unique catalytic capabilities.15fAs with the previously discussed 4d transition metal systems, selective C–H activation using 3d transition metals is often facilitated by the use of an appropriate DG. Typically, such transformations rely on the use of a proximal DG to assist in the activation of C–H bonds, with the C–H bonds at the ortho position to the group most commonly activated.23 The directing moieties that allow this reactivity have evolved from highly privileged auxiliaries to commonplace functional groups, including primary amides and ketones; these advancements have considerably improved the practicality of this approach. The following section will highlight the most recent advances in directed C(sp2)–H methylation catalysed by the 3d transition metals.
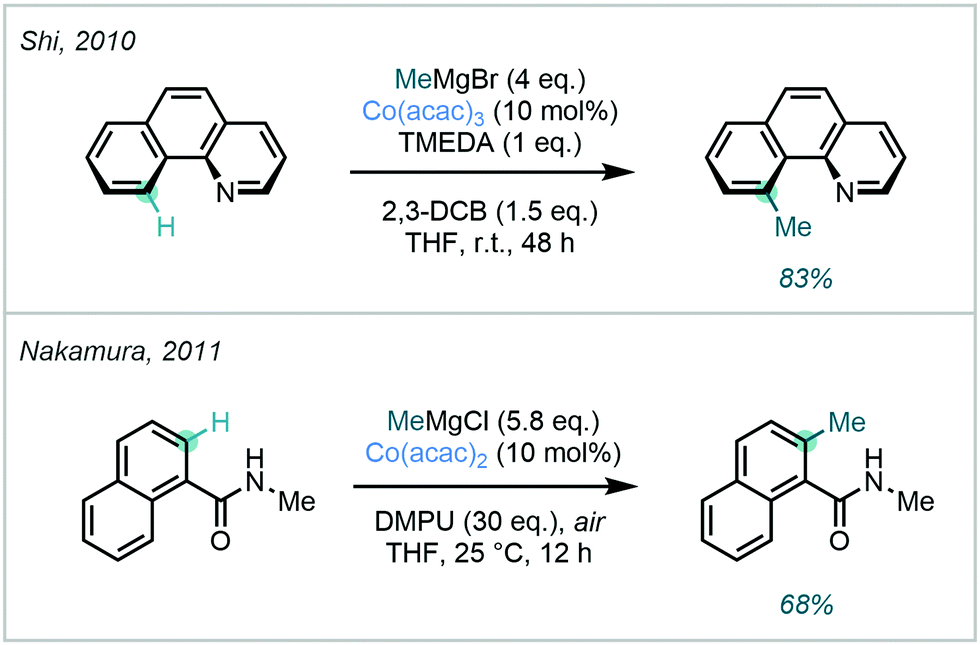 | ||
| Scheme 18 Early work featuring Co-catalysed C–H methylation, effected by superstoichiometric amounts of Grignard reagents. | ||
Soon after, Nakamura disclosed the Co-catalysed C–H alkylation of secondary benzamides, also using simple Grignard reagents (Scheme 18).70 In a singular example, the ortho methylation of N-methyl-1-naphthalenecarboxamide was demonstrated in good yield.
Co-catalysed C–H methylation reactions have more recently expanded to include methodologies featuring other nitrogen-centred DGs, together with a variety of electrophilic and nucleophilic methyl sources. In 2016, Xu demonstrated the use of AlMe3 as the methyl source for the 8-AQ-directed C–H methylation of a variety of substituted arenes (Scheme 19).71 This methylation procedure was selective for the less sterically-hindered ortho position on meta-substituted benzamides and exclusively displayed mono-selectivity. This was attributed to steric interactions in the key cobaltacycle complex with an already-present methyl moiety in the ortho position. The authors also reported the methylation of 8-AQ protected 1-methylcyclohexane-1-carboxamide at the terminal methyl group, demonstrating the applicability of the methodology in C(sp3)–H methylation.
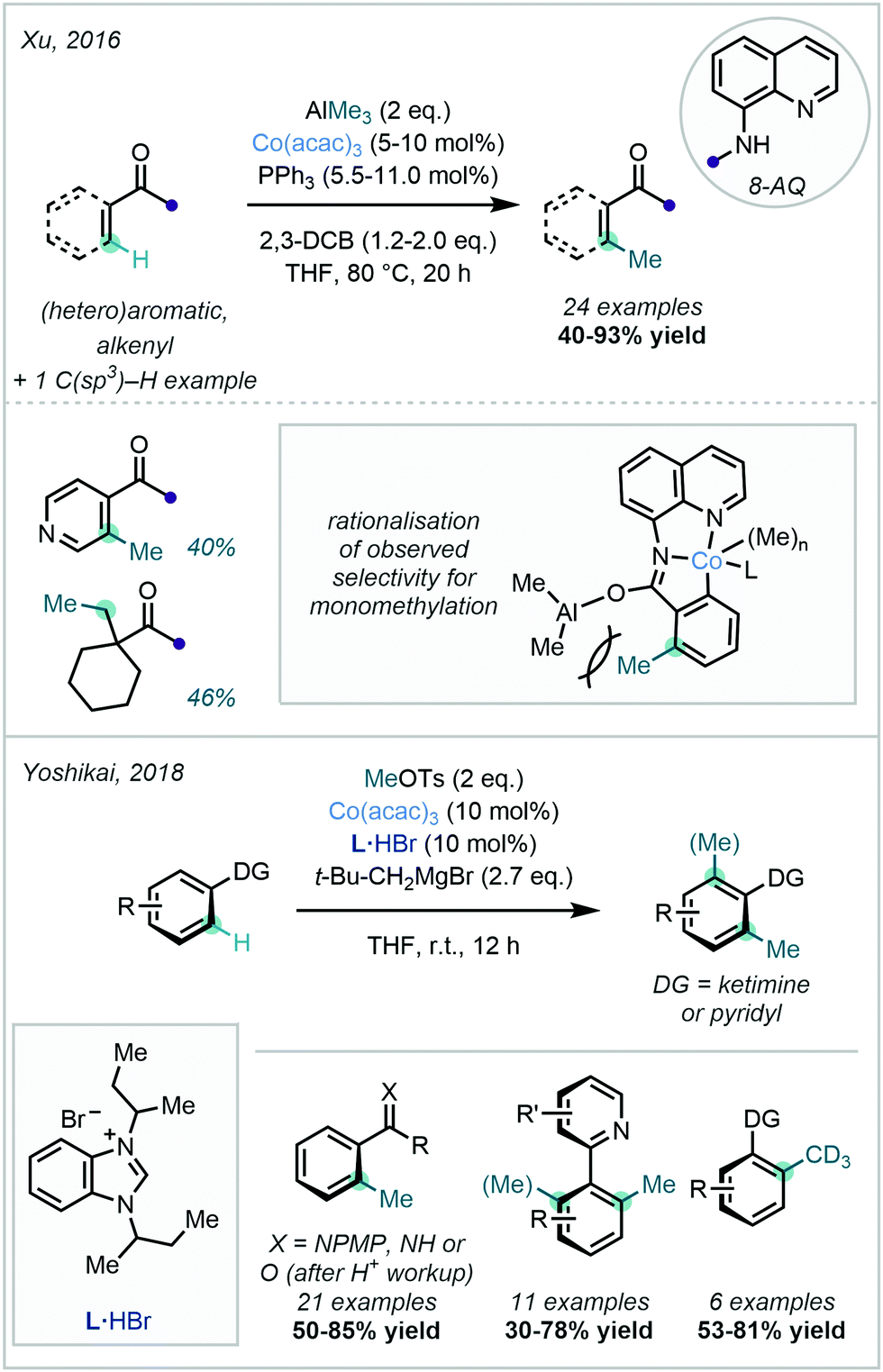 | ||
| Scheme 19 Co-Catalysed C–H methylation reactions employing alternative methylation reagents. | ||
Complementary to the use of nucleophilic methyl sources, Yoshikai successfully utilised methyl tosylate (MeOTs) for Co-catalysed C–H methylation (Scheme 19).72 The reaction was demonstrated with a variety of nitrogen-centred DGs, including N-aryl imines, N–H imines, and 2-pyridines. Conveniently, under suitably acidic conditions, imine hydrolysis could be conducted to recover the corresponding C–H methylated ketones. Neopentyl magnesium bromide was selected as a base in this procedure, and a mechanism invoking a single electron transfer (SET) for the activation of MeBr (formed in situ from MeOTs and L·HBr) was proposed. With MeOTs serving as the preliminary methyl source, high value CD3 groups were conveniently appended by the use of CD3OTs.
In 2017, Butenschön documented the Co-catalysed ortho methylation of metallocenes, constituting a welcome addition to the expanding field of Co-catalysed methylation strategies (Scheme 20).73 The reaction was amenable to ferrocenes bearing a variety of nitrogen-based DGs such as oxazolines, 8-AQ and triazolyldimethyl (TAM) carboxamides. Both mono- and di-methylated products were isolated, but only the di-methylated product was observed for substrates bearing an 8-AQ DG.
 | ||
| Scheme 20 The Co-catalysed methylation of ferrocene showcasing the use of multiple DGs. | ||
Peroxides have also been used as methylating reagents in Co-catalysed C–H methylation reactions. In 2016, Li & Lu reported the methylation of a wide variety of aryl amides bearing a 2-pyridinylisopropyl (PIP) directing group using DCP as both the methyl source and the oxidant (Scheme 21).74 The proposed mechanism involved a CoII/CoIII/CoIV catalytic cycle, where the Co centre coordinated to the amide and PIP DG nitrogen atoms. After substrate coordination to the cobalt complex, C–H activation led to formation of the cobaltacycle. This was then followed by a methyl transfer from the oxygen-centred cumyloxy radical generated from DCP and an accompanying oxidation of CoIII. Reductive elimination and ligand exchange subsequently liberated the methylated product. A similar catalytic cycle was suggested to hold true for a Co-catalysed methylation described by Cai in 2019,75 as well as in an 8-AQ-directed Ni-catalysed C–H methylation reported by Chatani using DCP as the methyl source (vide infra).76
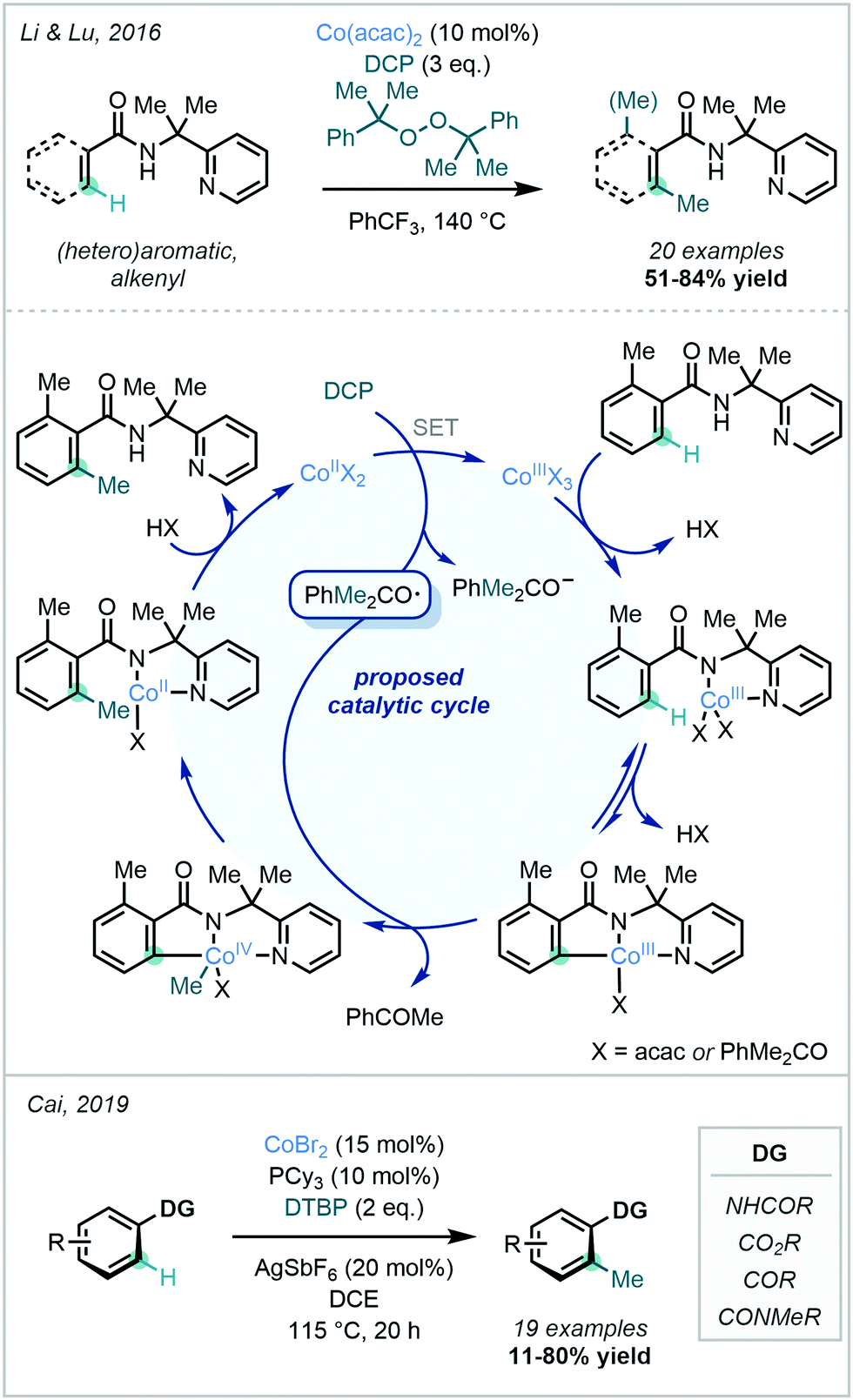 | ||
| Scheme 21 Catalytic cycle of Co-catalysed C–H methylation using organic peroxides as methylating agents. | ||
Cai disclosed a mechanistically-related Co-catalysed C–H methylation strategy for anilides using a CoBr2/PCy3 catalytic system together with DTBP as the methyl source (Scheme 21).75 Not only did the method circumvent the need for elaborate DGs and the use of precious metals, it also made use of inexpensive DTBP. The reaction was also amenable to the use of more commonplace functional groups such as aryl ketones, amides and esters as directing groups.
A major development in the field of arene C–H methylation, and 3d transition metal-catalysed C–H functionalisation in general, was achieved in 2020 when Johansson & Ackermann developed an impressive Co-catalysed late-stage C–H methylation protocol (Scheme 22).77 Me3B3O3 was employed as the methyl source, and the highly electrophilic Cp*Co(PhH)(PF6)2 was shown to be the optimal catalyst for this transformation. Using this combination, directed C–H methylation was shown to be achievable using a plethora of Lewis basic functional groups.
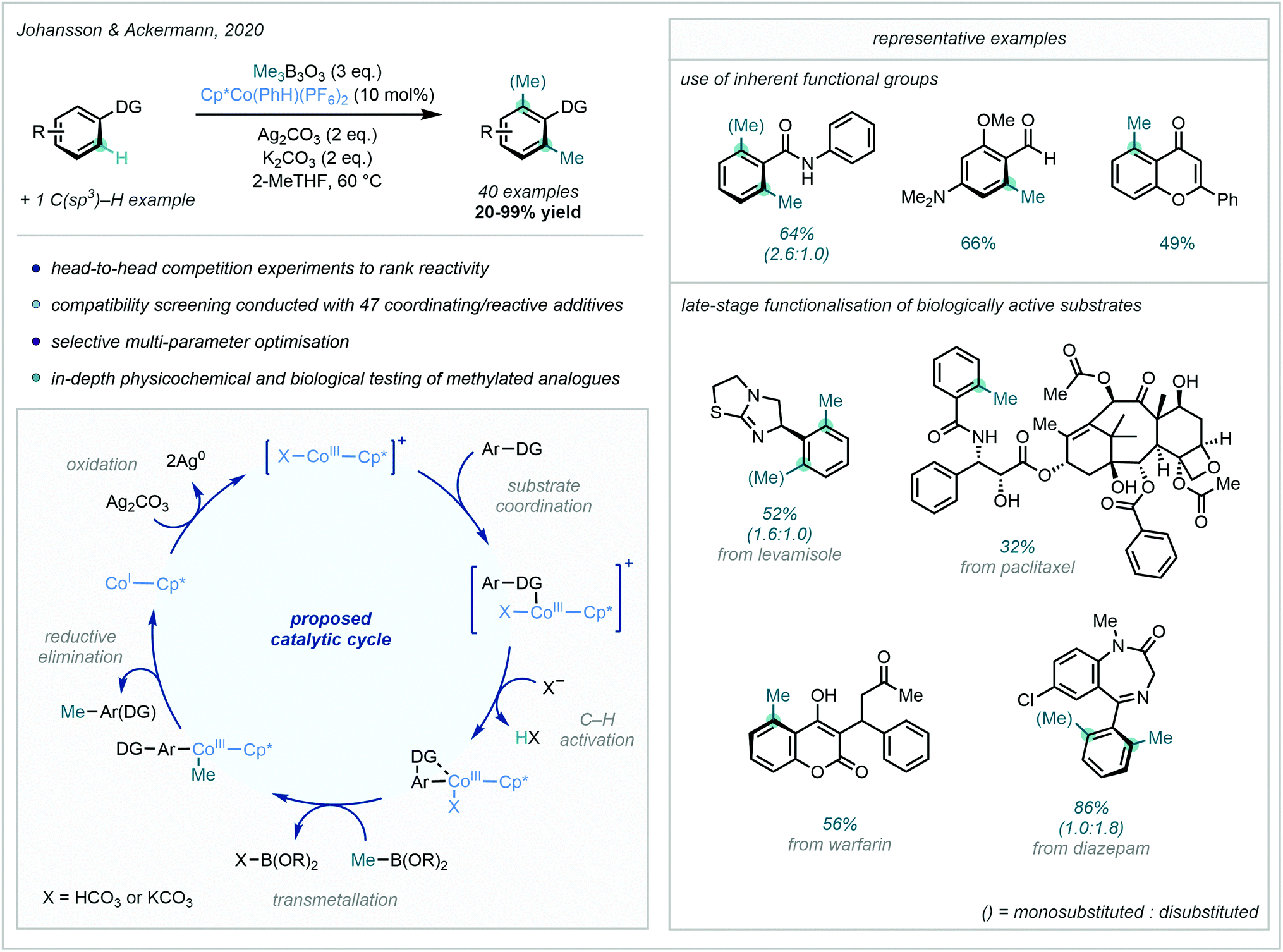 | ||
| Scheme 22 Co-Catalysed C–H methylation utilising a broad range of DGs and applied to the late-stage functionalisation of select drug compounds. | ||
This wide and versatile selection of DGs included nitrogen-containing heterocycles and primary amides, but also traditionally “weakly” coordinating groups such as ketones and aldehydes. To further demonstrate the power of the approach, the late-stage methylation of an array of complex drug molecules was achieved, enabling the potential investigation of the “Magic Methyl” effect in a convenient, late-stage manner.
The proposed mechanism for this reaction involved a CoI/CoIII cycle, with the heteroatom lone pair of the DG coordinating to the catalyst. Subsequent C–H cobaltation followed by transmetalation with Me3B3O3 introduced the methyl group to the metal centre. After reductive elimination liberated the methylated product, Ag2CO3 was able to regenerate the active CoIII catalyst.
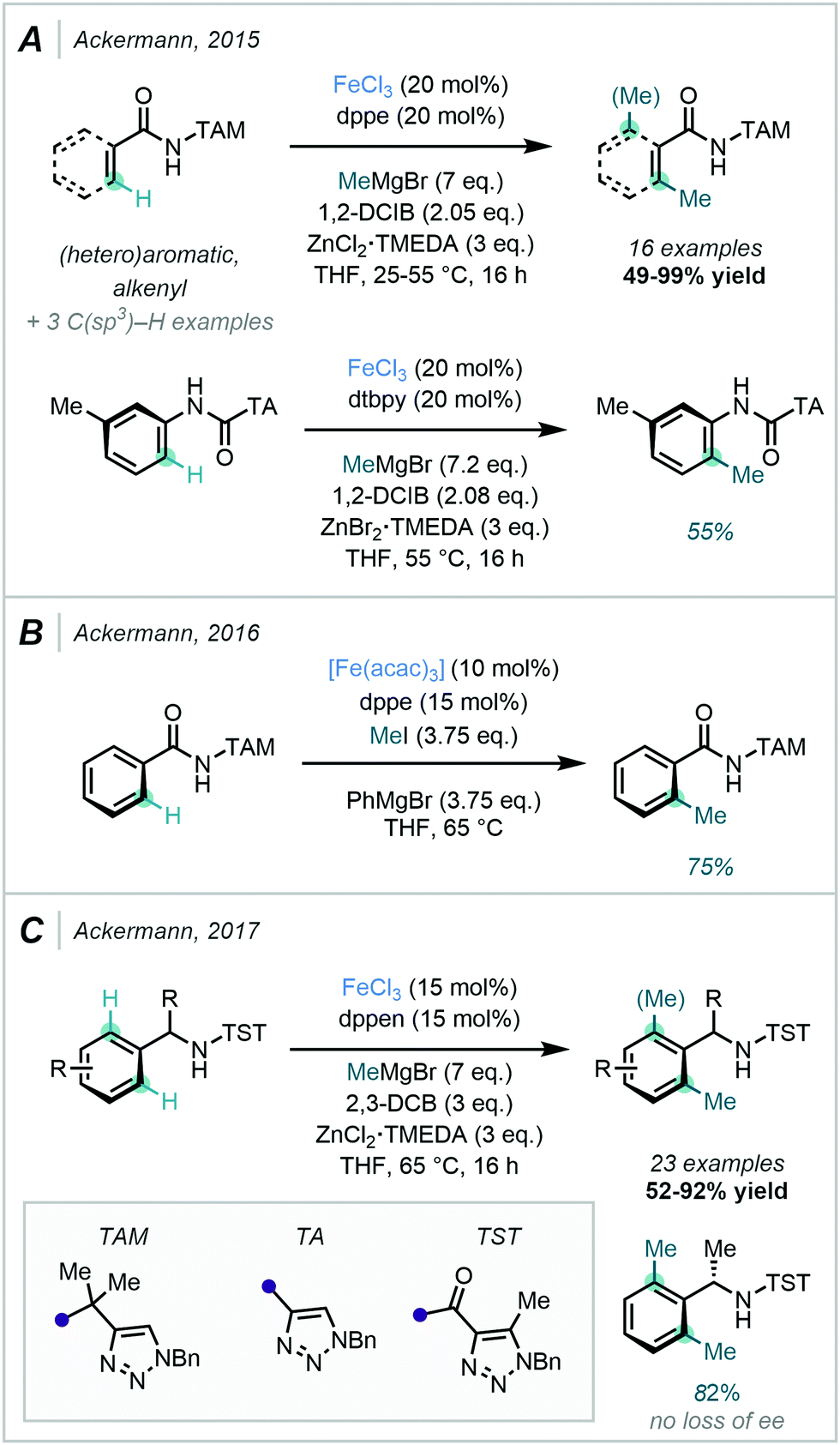 | ||
| Scheme 23 Fe-Catalysed C–H methylation using triazole directing groups. | ||
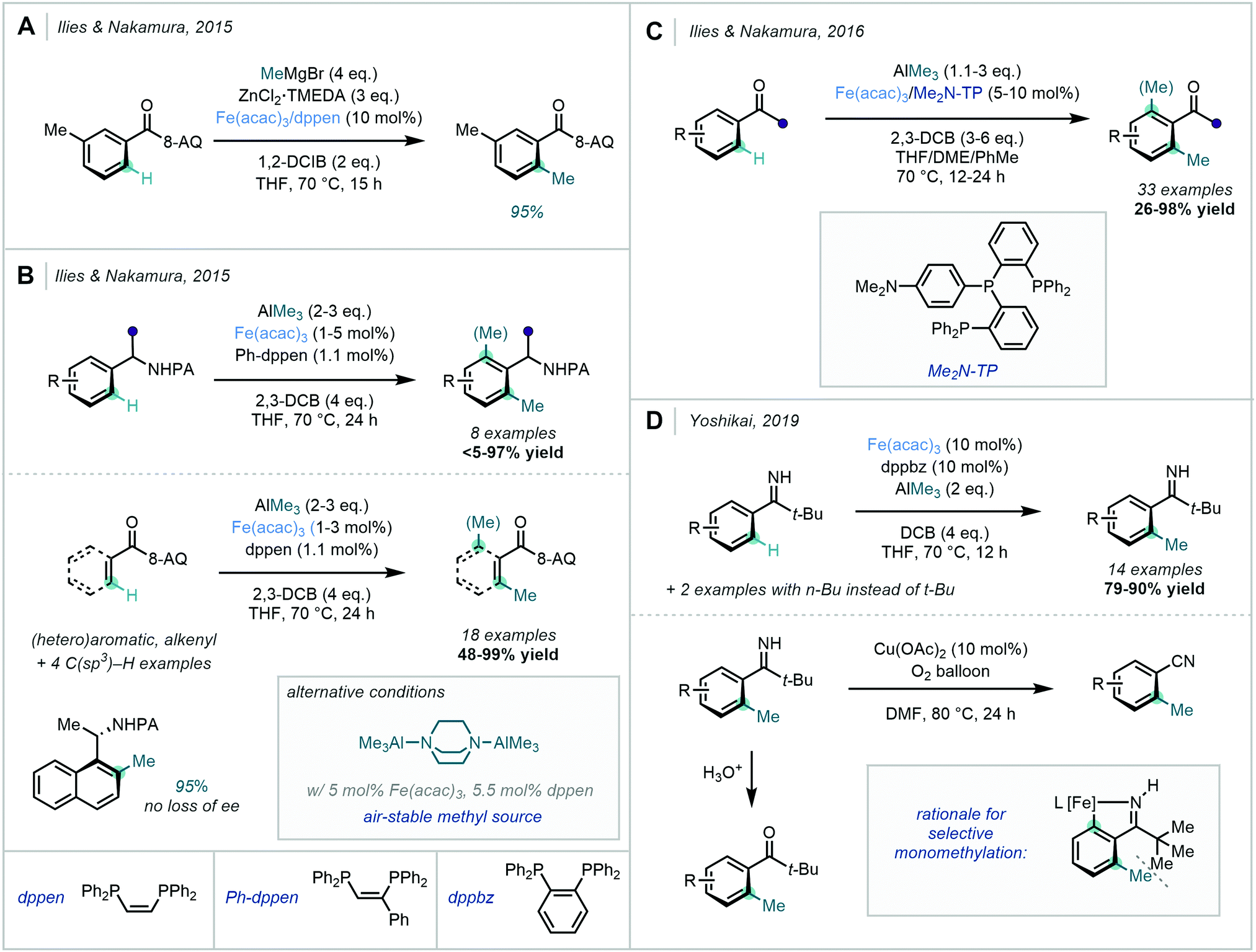 | ||
| Scheme 24 Fe-Catalysed C–H methylations (A) of benzamides using 8-AQ as a DG; (B) of benzylamines using PA as a DG as well as the application of an air-stable methyl source; (C) using carbonyl groups as directing elements; (D) using N–H imines as DGs. | ||
A key feature of Ackermann's C–H methylation strategy was the application of triazole-based auxiliaries. This moiety acted as the directing group during methylation, and was conveniently removed with aqueous HCl to recover the corresponding primary amide.78 The bidentate auxiliary (TAM) was shown to be an effective directing group for facilitating C–H methylation, and was also proposed to possess greater directing capability when compared to the more commonly used 8-AQ.
The related auxiliary triazolyl (TA) was also successfully applied in the ortho C–H methylation of anilides. In conjunction with the appropriate DGs, MeMgBr and 1,2-dichloroisobutane (1,2-DCIB) were utilised as the methyl source and oxidant respectively. ZnCl2·TMEDA was suggested to be involved in the facilitation of the transmetallation step via the in situ generation of the activated organozinc halide ZnMeCl (Scheme 23A). As an alternative to Grignard reagents, Ackermann also described the use of MeI as an electrophilic methyl source. Notably, when coupled with an Fe-catalysed TAM-directed reaction system, excellent mono-selectivity was achieved (Scheme 23B).79
In 2017, Ackermann further reported an alternative set of conditions for Fe-catalysed ortho di-methylation of benzylamines. In this methodology, tri-substituted 1,2,3-triazole (TST) acted as the DG, MeMgBr as the methyl source and 2,3-DCB as the oxidant (Scheme 23C).80 Like TAM, the TST directing group was easily removed post-methylation. Pleasingly, it was demonstrated that the presence of the TST group did not affect the stereochemical integrity of the product formed from an enantioenriched substrate.
Ilies & Nakamura's complementary approach from 2015 opted for the use of a benzamide bearing an 8-AQ DG under similar conditions (Scheme 24A).81 In agreement with Ackermann, ZnMeCl was proposed as the active methylating reagent. They also suggested that a catalytic quantity of ZnMeCl was used to generate the active organoiron(II) species responsible for C–H activation. Excellent mono-selectivity was observed at the less hindered ortho C–H site.
In the same year, Ilies & Nakamura described a related methylation of amine substrates bearing a PA DG, and of benzamides bearing an 8-AQ DG (Scheme 24B).82 In this case, AlMe3 was used as the methyl source with 2,3-DCB as the oxidant. An alternative protocol was also disclosed using the air-stable bis-(trimethylaluminum)·1,4-diazabicyclo[2.2.2]octane adduct (DABAL-Me3) as the methyl source, albeit requiring longer reaction times. The use of a milder Al-based methyl source – when compared to Grignard reagents – was proposed to prevent premature reduction of the reactive organoiron species in the catalytic cycle, allowing for catalyst turnover numbers as high as 6500. Exclusive mono-methylation was observed for substrates bearing a meta substituent on the aromatic ring, which suppressed methylation at the neighbouring ortho site. However, ortho di-methylated products were observed to be the major products for unsubstituted or para-substituted substrates. For acyclic alkenecarboxamides, Z to E isomerization occurred to generate a mixture of isomers of the methylated products. It was again noted that the presence of a PA group did not affect the stereochemical integrity of the product formed from an enantioenriched substrate.
This technique was later expanded by Ilies & Nakamura, in the carbonyl-directed ortho methylation of arenes using AlMe3 as the methyl source (Scheme 24C).83 DABAL-Me3 proved to be an effective alternative methyl source, albeit with lower reactivity. Employing a simple carbonyl group as the DG complemented the amidic DGs discussed previously, and allowed for the C–H methylation of readily available aromatic ketones, carboxylic acids, esters and amides. Furthermore, functional groups including boronates and enolisable ketones were tolerated. Tridentate phosphine ligand 4-(bis(2(diphenylphosphanyl)phenyl)phosphanyl)-N,N-dimethylaniline (Me2N-TP) was found to be key for reactivity in this transformation.
In 2019, Yoshikai further expanded the repertoire of Fe-catalysed ortho C–H methylations (Scheme 24D).84 Pivalophenone N–H imines were used as monodentate directing groups given their downstream value as intermediates towards ketones and nitriles. This method also exclusively displayed mono-methylation, supposedly due to steric repulsions between the t-Bu group and the initially introduced methyl group.
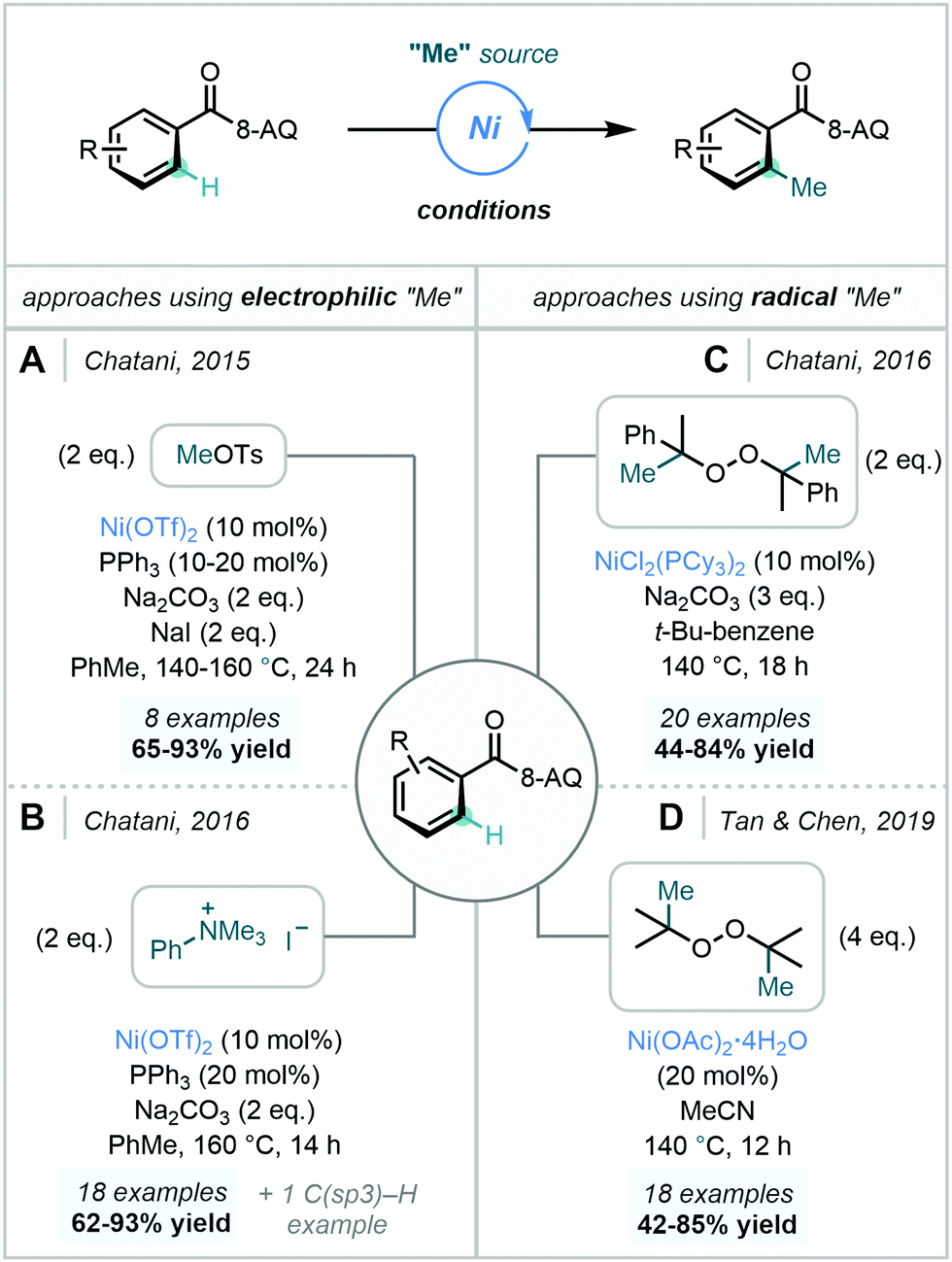 | ||
| Scheme 25 Ni-Catalysed C–H methylation processes utilising different electrophilic and radical methyl sources using the 8-AQ DG. | ||
Further expanding the repertoire of methyl sources applicable in Ni-catalysed C–H methylation reactions, Chatani later documented the use of DCP as a methyl source, in a proposed radical-based mechanism (vide supra) involving a NiII/NiIII/NiIV catalytic cycle (Scheme 25C).76 Similarly to the previously discussed reports, meta-substitution enforced methylation only at the less hindered ortho C–H bond. Mechanistic experiments suggested that C–H bond cleavage was reversible and that reductive elimination was likely to be rate-determining.
In 2019, Tan & Chen developed a robust Ni-catalysed ortho C–H methylation using DTBP. Conveniently, the methodology did not require the use of an external base or ligand, nor demanded moisture-free or inert atmospheric conditions (Scheme 25D).87 Additionally, the use of DTBP afforded acetone as a by-product, aiding the isolation of the methylated product. In agreement with Chatani, a radical-based mechanism involving a NiII/NiIII/NiIV catalytic cycle was proposed.
In 2017, Ackermann developed a TAM-directed MnCl2-catalysed C–H methylation procedure with MeMgBr (Scheme 26).88 Unlike the previously-reported triazole-directed Fe-catalysed approach,78–80 the transition to a Mn-catalysed system obviated the need for a phosphine ligand or a zinc additive. The suggested mechanism involved a MnII/MnIII/MnI catalytic cycle featuring two one-electron oxidation steps with C–H activation as the rate-determining step, and excellent mono-selectivity was observed.
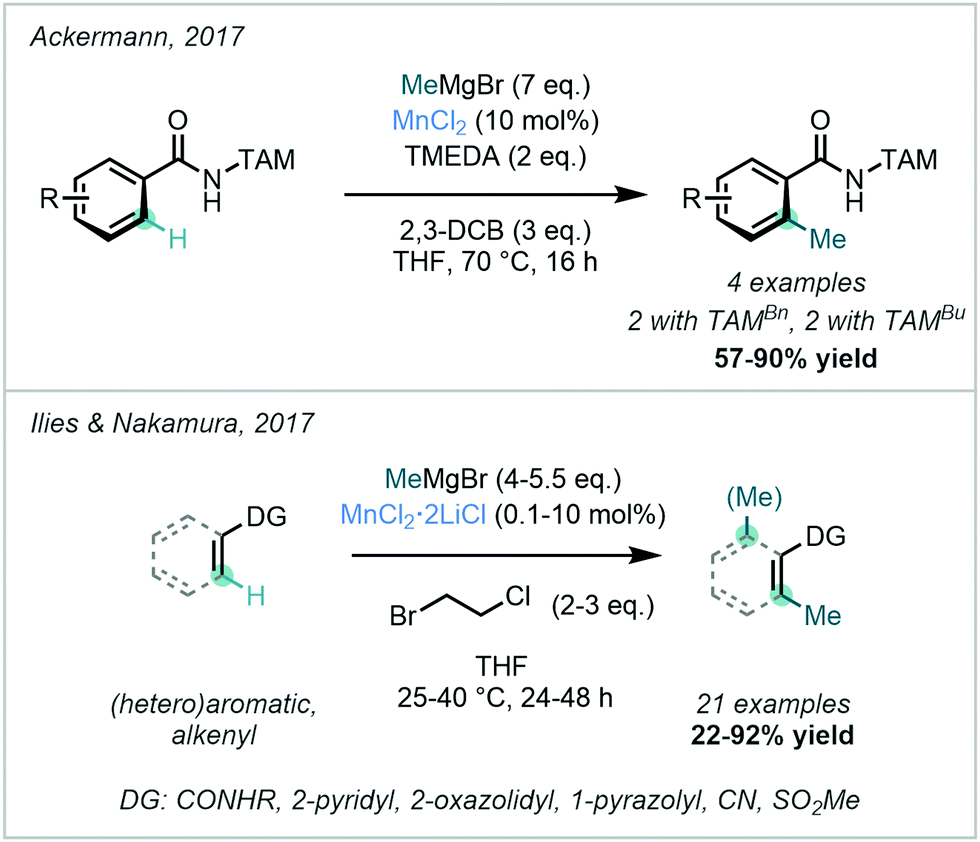 | ||
| Scheme 26 Mn-catalysed ortho C–H methylations of arenes and alkenyl substrates using diverse directing groups, including TAM and alternative amidic and heterocyclic species. | ||
Ilies & Nakamura described the use of MnCl2·2LiCl for C–H methylation. The reaction was shown to operate at room temperature with MeMgBr as the methyl source, 1-bromo-2-chloroethane as the oxidant, and without an external ligand (Scheme 26).89
This process boasted low catalyst loadings, with a catalytic turnover of up to 5900. A diverse range of DGs including amides, pyridines, oxazolines, pyrazoles, nitriles and methylsulfones were applicable, rendering this method a substantial addition to previously-reported directed C–H methylation methods. Unlike their previous Fe-catalysed methylation protocol with AlMe3,82 this Mn-catalysed reaction was not amenable to bidentate directing groups. The proposed mechanism for this reaction involved a MnII/MnIII/MnI catalytic cycle where in this case intermediary radical generation was suggested to be unlikely.
2.3 Catellani-type strategies for C(sp2)–H methylation
Building upon a wealth of established cross-coupling literature, the Catellani reaction has gained significant attention as a powerful difunctionalisation strategy.90 A necessary feature of the reaction is the norbornene (NBE) co-catalyst. After an initial oxidative addition to the C–X bond to furnish intermediate I, NBE acts as a transient mediator to direct Pd to the ortho position via carbopalladation and palladacycle formation. The resulting intermediate II can then undergo oxidative addition with a suitable methyl electrophile to generate intermediate III, which is capable of reductive elimination to position the methyl group at the ortho position to the original position of the C–X bond (Scheme 27).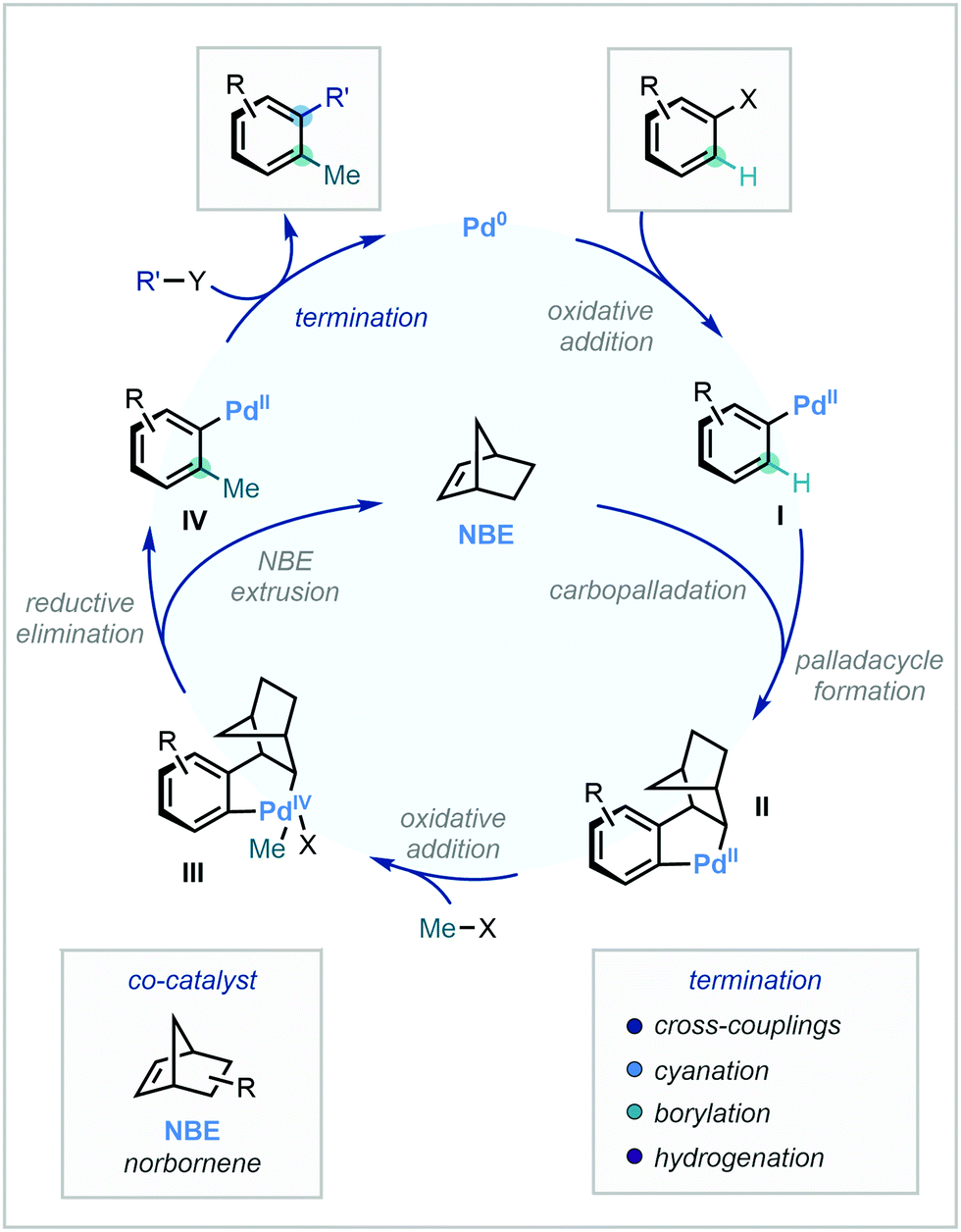 | ||
| Scheme 27 General mechanism of the Catellani reaction in the case of C–H methylation. | ||
Subsequent retro-carbopalladation and norbornene disassociation affords intermediate IV. Attesting to the high modularity of the Catellani reaction, various termination protocols can be exploited in the final ipso functionalisation. Di-methylation is also possible in the instance that both ortho positions are unsubstituted, allowing for tri-functionalisation in a singular step. The following section details recent developments in C(sp2)–H methylation that capitalise on Catellani-type Pd/NBE cooperative catalysis. While initial reports have focused solely on aryl substrates, recent advancements extending to vinyl substrates will also be discussed here.
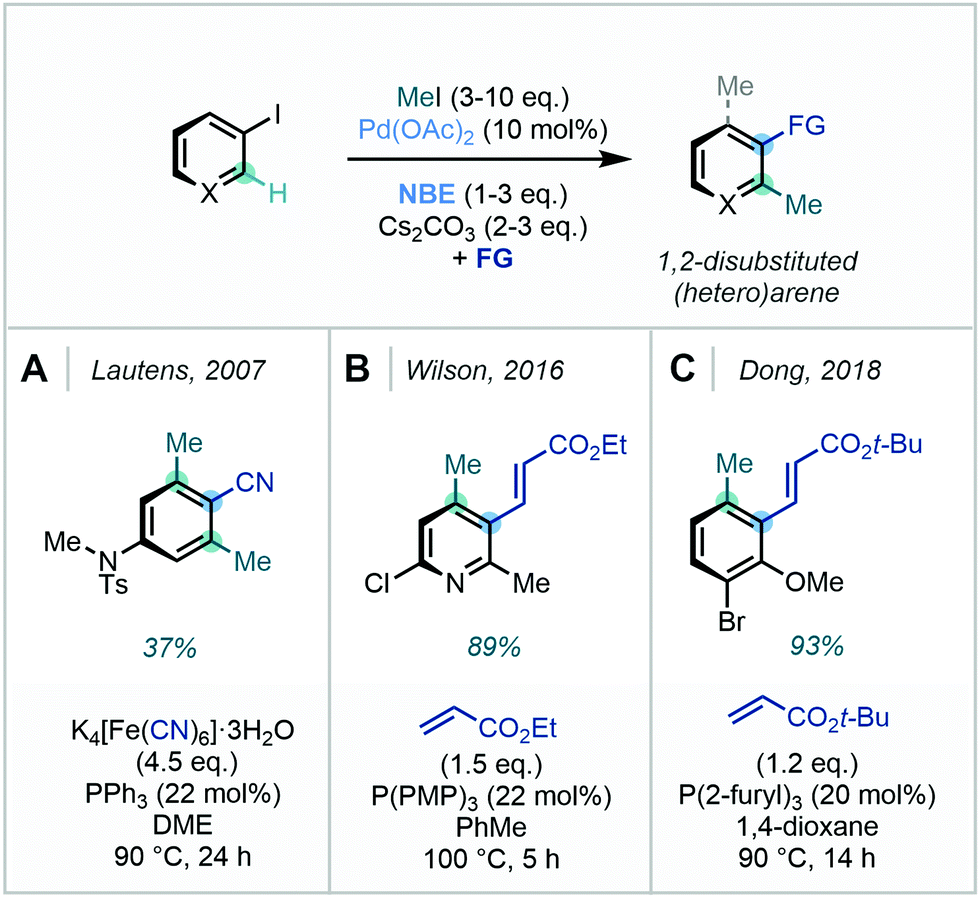 | ||
| Scheme 28 Catellani-type ortho C(sp2)–H methylations utilising methyl iodide. | ||
Noting the volatility of methyl iodide, Zhou identified MeOTs and trimethyl phosphate (PO(OMe)3) as effective methylating agents for Catellani reactivity (Scheme 29).90c Furthermore, the use of CD3OTs was equally effective in appending high-value CD3 groups. The authors extended the chemistry to a vast substrate scope, attesting to the robustness of the methodology. Impressively, this protocol was shown to be amenable to a host of termination strategies, where various cross-couplings, cyanation, borylation and hydrogenation were viable. Applicability to late-stage methylation was also demonstrated, resulting in the synthesis of methylated fenofibrate and ezetimibe analogues.
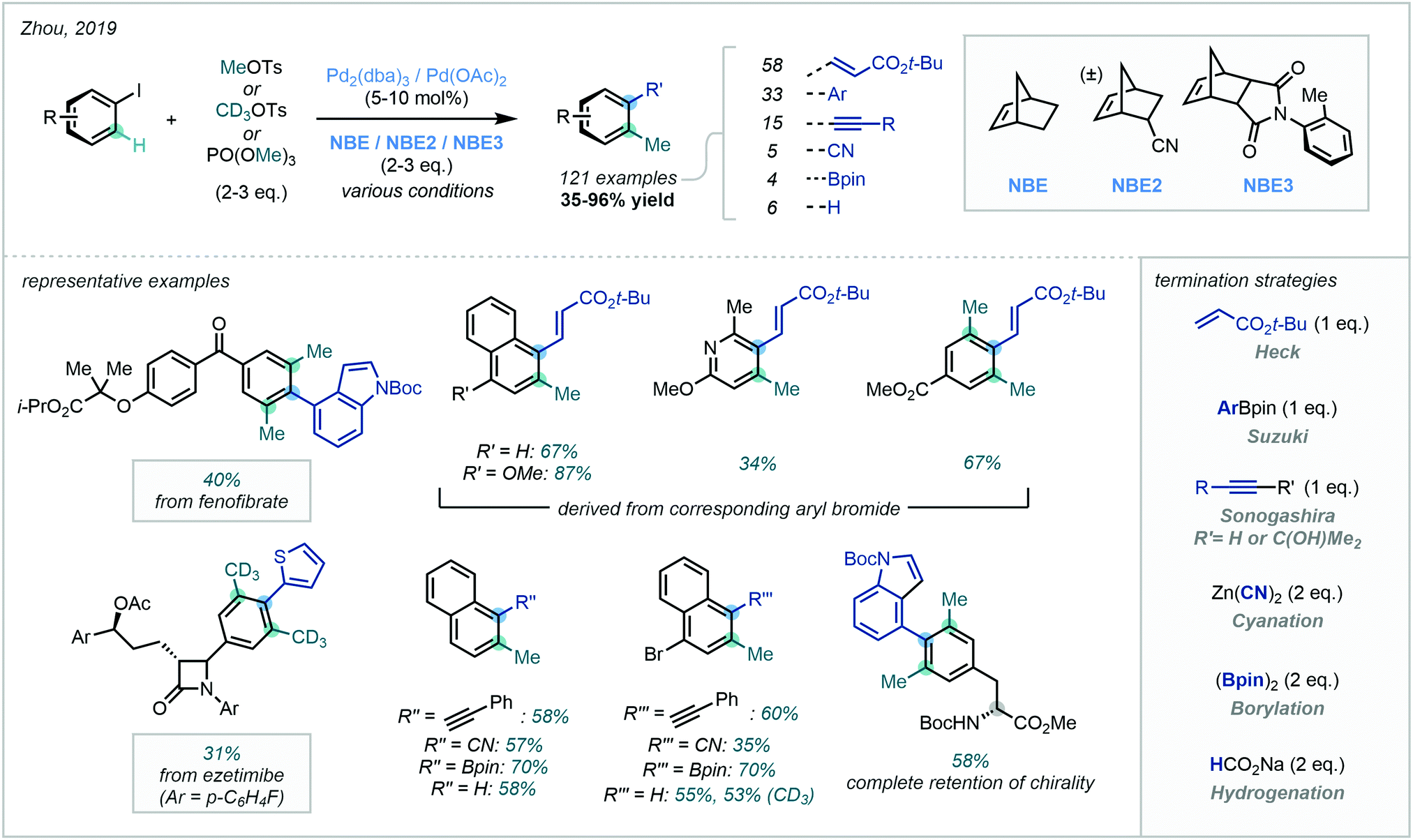 | ||
| Scheme 29 Pd-Catalysed Catellani-type reaction using MeOTs or PO(OMe)3, enabling C(sp2)–H methylation with diverse termination strategies. | ||
In a separate study, a complementary Catellani-type ortho methylation/ipso borylation strategy was demonstrated by Smith, where methyl iodide was employed to enable the C(sp2)–H methylation/borylation of 2-iodotoluene in 60% yield.94
The methodology reported by Zhou (vide supra) was also successfully performed on several aryl bromides, an uncommon but synthetically useful development. Dong similarly conducted a Catellani-type C(sp2)–H methylation of 2-bromoanisole, in which methyl 4-nitrobenzenesulfonate (MeONs) was utilised as the methylating agent (Scheme 30).93
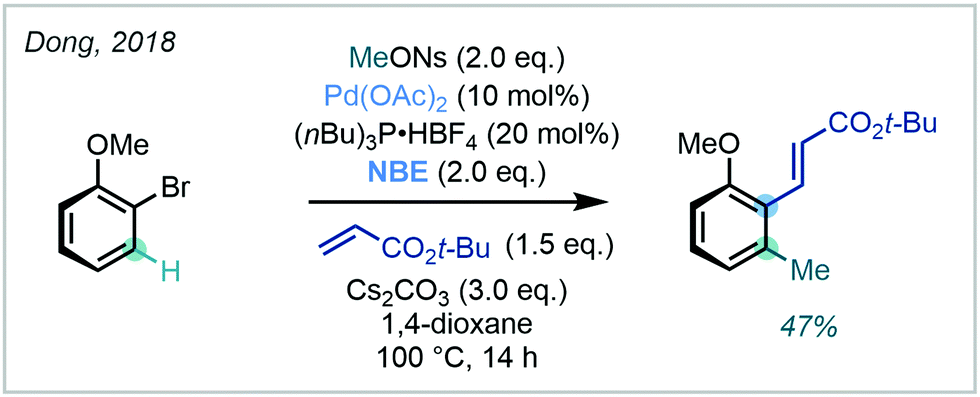 | ||
| Scheme 30 Use of an aryl bromide for a Catellani-type aryl C(sp2)–H methylation. | ||
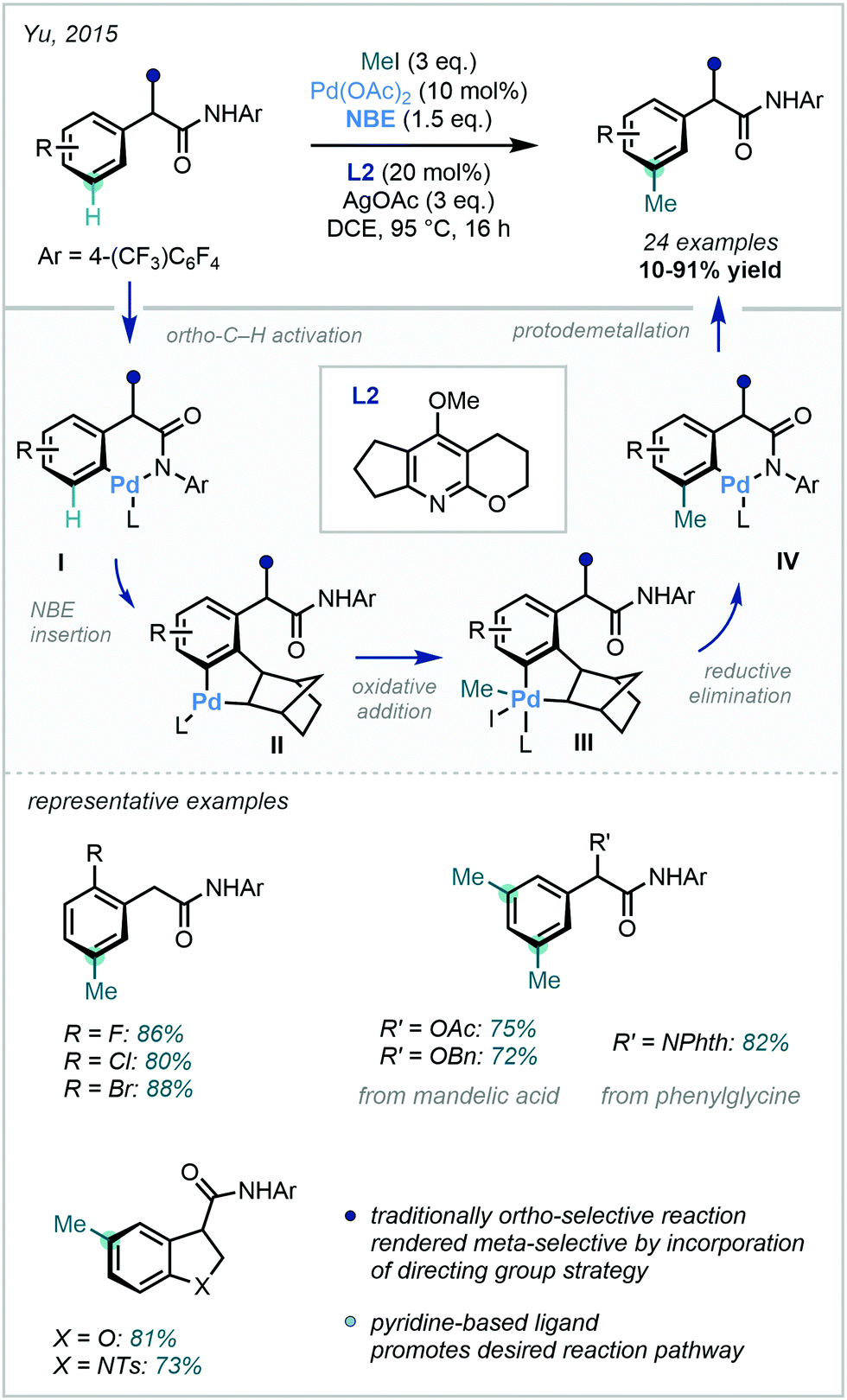 | ||
| Scheme 31 meta C–H methylation of aryl C(sp2)–H bonds, combining a directing group approach and Catellani-type Pd/NBE catalysis. | ||
In this protocol, the N-2,3,5,6-tetrafluoro-4-trifluoromethylphenyl amide directing group facilitates the initial ortho-C–H palladation to afford intermediate I. Subsequent 1,2-migratory insertion of norbornene was enabled through the use of pyridine-based ligands, with ligand L2 eventually chosen after a rigorous screening process. Following NBE insertion, intermediate II can then undergo methylation at the meta position. Intermediate II was noted to be potentially susceptible to an undesirable C–C reductive elimination which would generate the benzocyclobutane side product. A subsequent computational study conducted by Yang demonstrated the importance of ligand L2 in shifting the energetics of the reaction pathway to favour the observed oxidative addition to the alkyl iodide.96 Unlike traditional Catellani reactions, where a secondary termination event takes place, protodemetalation of intermediate IV occurred to deliver the meta-C–H methylated product. This methodology was also shown to be a general strategy for meta C(sp2)–H activation – the use of alternative coupling partners such as benzyl bromides and aryl iodides allowed for the generation of alternative alkylation and arylation products respectively. Via prudent selection of the aryl ipso handle and optimised reaction conditions, this innovative strategy introduced a powerful, new approach to meta functionalisation employing Catellani-type reactivity.
Using the same directing group strategy, Yu subsequently demonstrated that the benzylsulfonamide moiety, prevalent in pharmaceuticals, could also act as an ortho DG (Scheme 32). Ding later showed that nosyl-protected phenylalanines were also suitable substrates for similar meta-methylation (Scheme 32).97 With each directing group, a survey of suitable NBE derivatives, as well as pyridine- and quinoline-derived ligands, allowed for optimisation of reaction efficiency. The methylation protocol was successfully applied to an L-phenylalanine substrate, with no observed racemisation of the amino acid stereogenic centre.
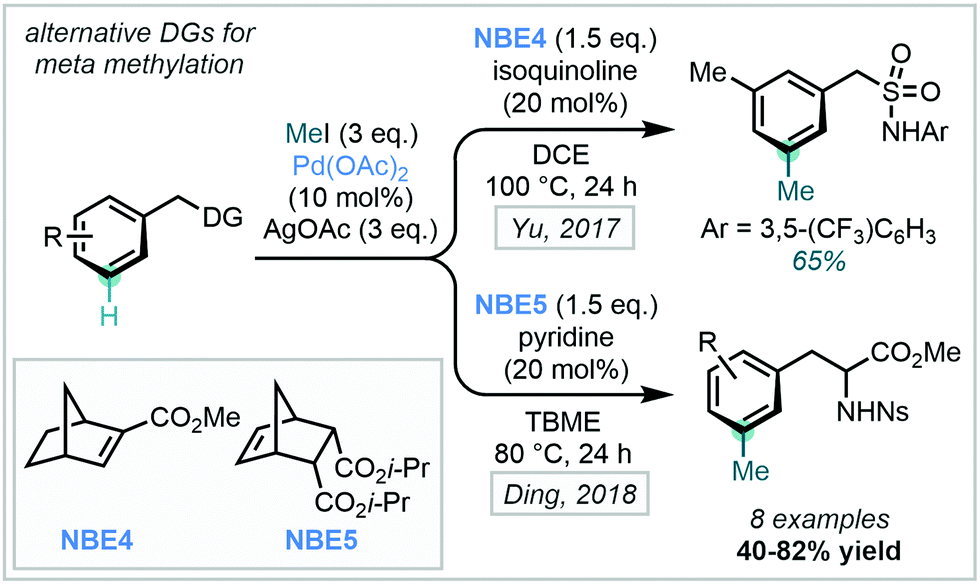 | ||
| Scheme 32 Alternative directing groups utilised to conduct meta-directed C(sp2)–H methylations. | ||
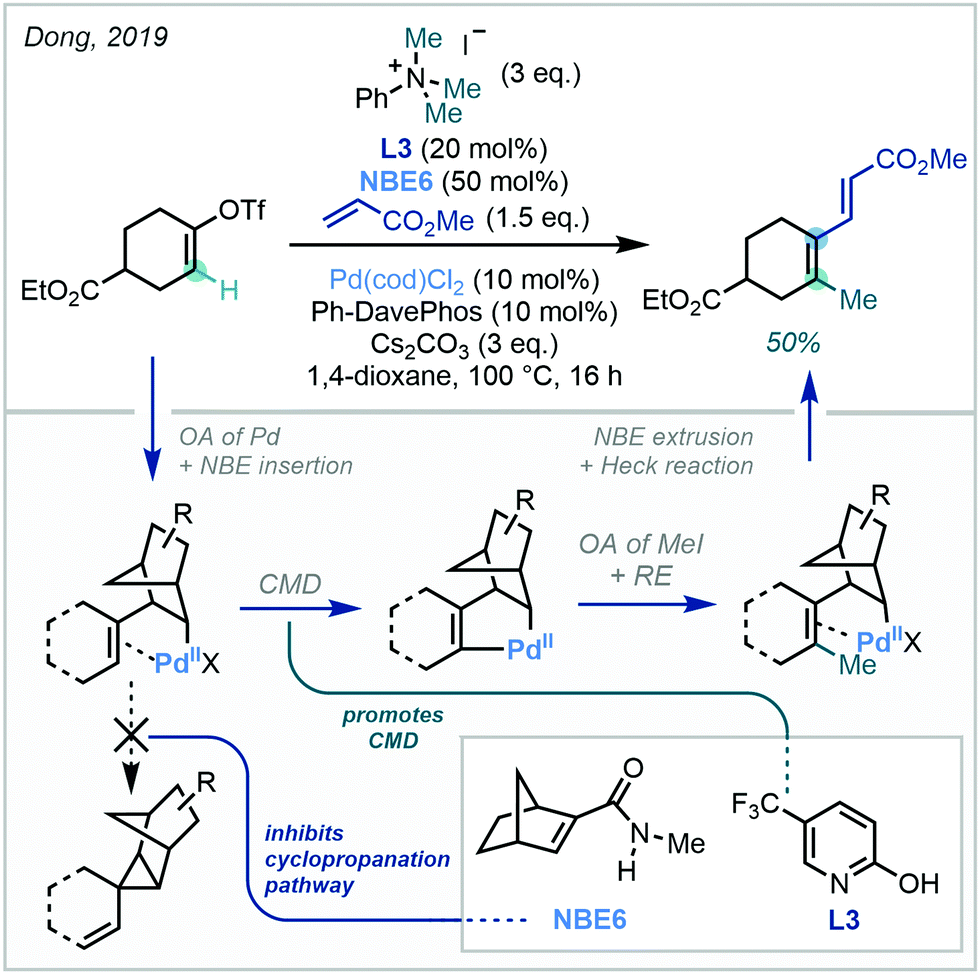 | ||
| Scheme 33 C(sp2)–H methylation of alkenyl substrates via Catellani-type Pd/NBE cooperative catalysis. OA = oxidative addition, RE = reductive elimination, CMD = concerted metalation-deprotonation. | ||
Expanding upon this vinylic functionalisation, Dong extended the approach to distal functionalisation via directed C–H activation and Pd/NBE cooperative catalysis (Scheme 34).99
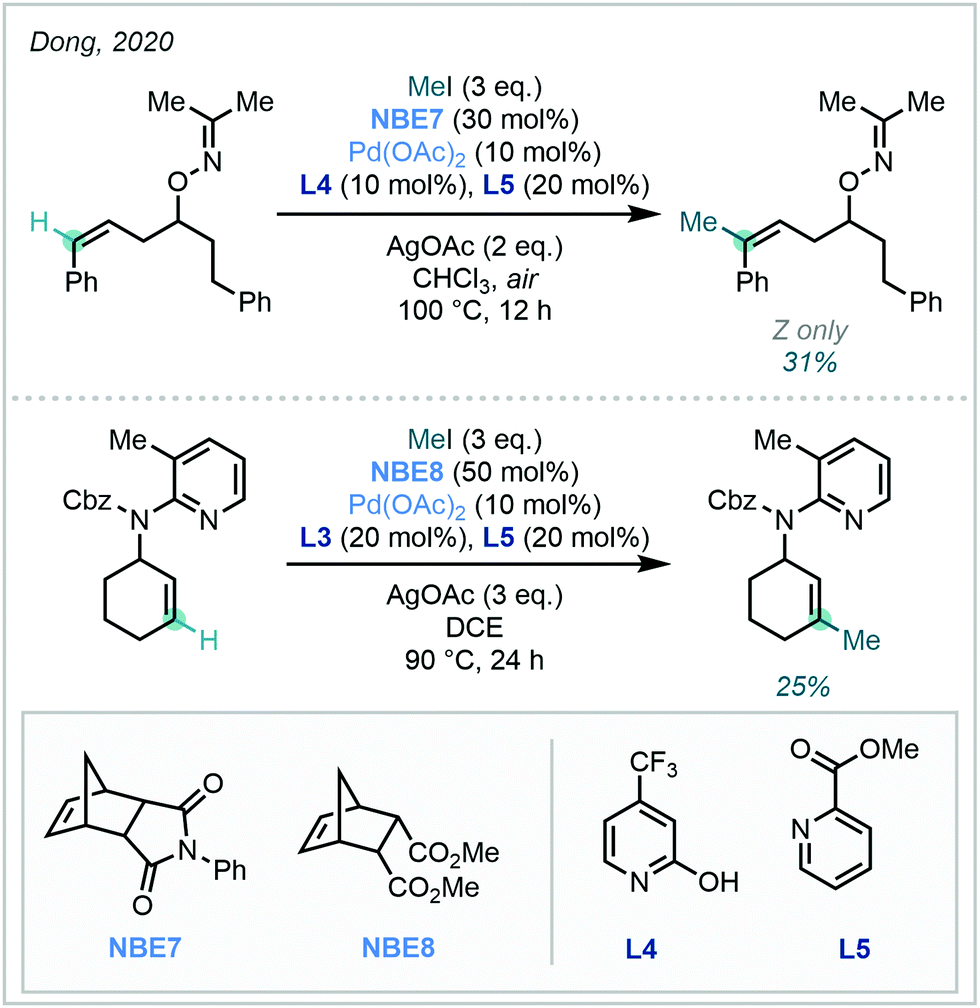 | ||
| Scheme 34 The use of directing groups to effect distal C(sp2)–H methylation on alkenyl substrates. For L3, see Scheme 33. | ||
While principally focussed upon arylation, C(sp2)–H methylation was achieved with both cyclic and conformationally flexible acyclic alkene systems in modest yields, under either oxime or 2-aminopyridine direction – a remarkable feat given the challenging nature of the transformation.
3. Innate/direct C(sp2)–H methylation
3.1 One-electron strategies
A profound development in the field came following pioneering work from Minisci, in which radicals generated via a Ag(II)-mediated oxidative decarboxylation were observed to add to nitrogen-containing aromatic bases followed by subsequent rearomatisation (Scheme 35).101 Bolstered by the development of new radical generation manifolds, the now-eponymous reaction – which notably complements well-established Friedel–Crafts reactivity – has become a powerful tool in the construction of C–C bonds in a plethora of heteroaromatic substrates.102
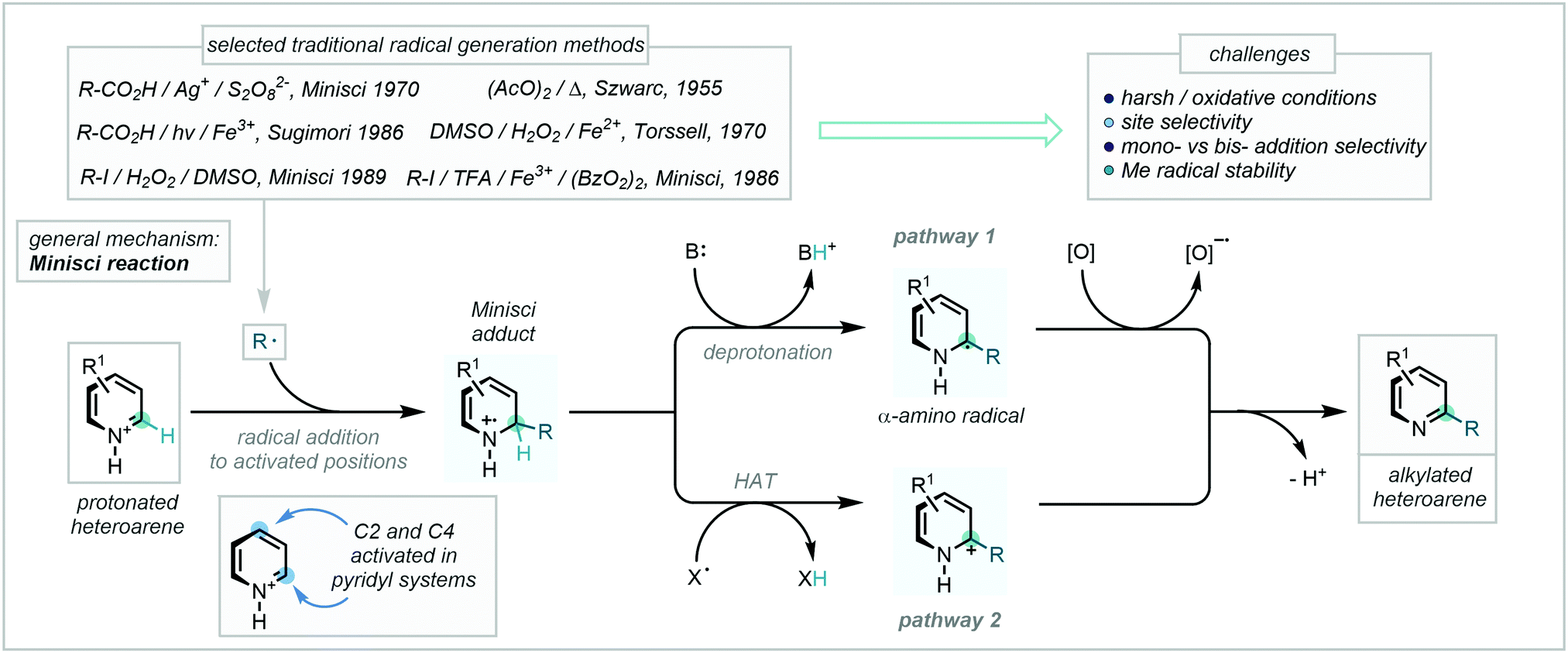 | ||
| Scheme 35 A mechanistic overview and examples of traditional radical generation methods for Minisci reactions. | ||
Despite initial challenges, such as the harsh conditions required for radical generation101,102c,g,103 and the generally unpredictable behaviour of the resulting radicals, the development of this mechanistic framework as a C–H methylation tool has enjoyed a recent expansion, with numerous exciting and valuable methodologies being reported. These will be the subject of the following section and have been categorised according to the nature of methyl radical/methyl radical surrogate generation.
 | ||
| Scheme 36 Peroxide decomposition as a source of methyl radicals. | ||
The power of this approach as a C–H methylation strategy was highlighted in a seminal report from DiRocco in 2014, in which a photocatalytic manifold was employed to initiate the decomposition of tert-butyl peracetate (Scheme 37A).106 A high-throughput screen was performed to establish a set of optimal conditions which were later applied to the C–H methylation of a number of complex biologically-active heterocycles. Despite modest yields in some cases, the methodology was developed in a manner that rendered it of great value to medicinal chemistry programmes.
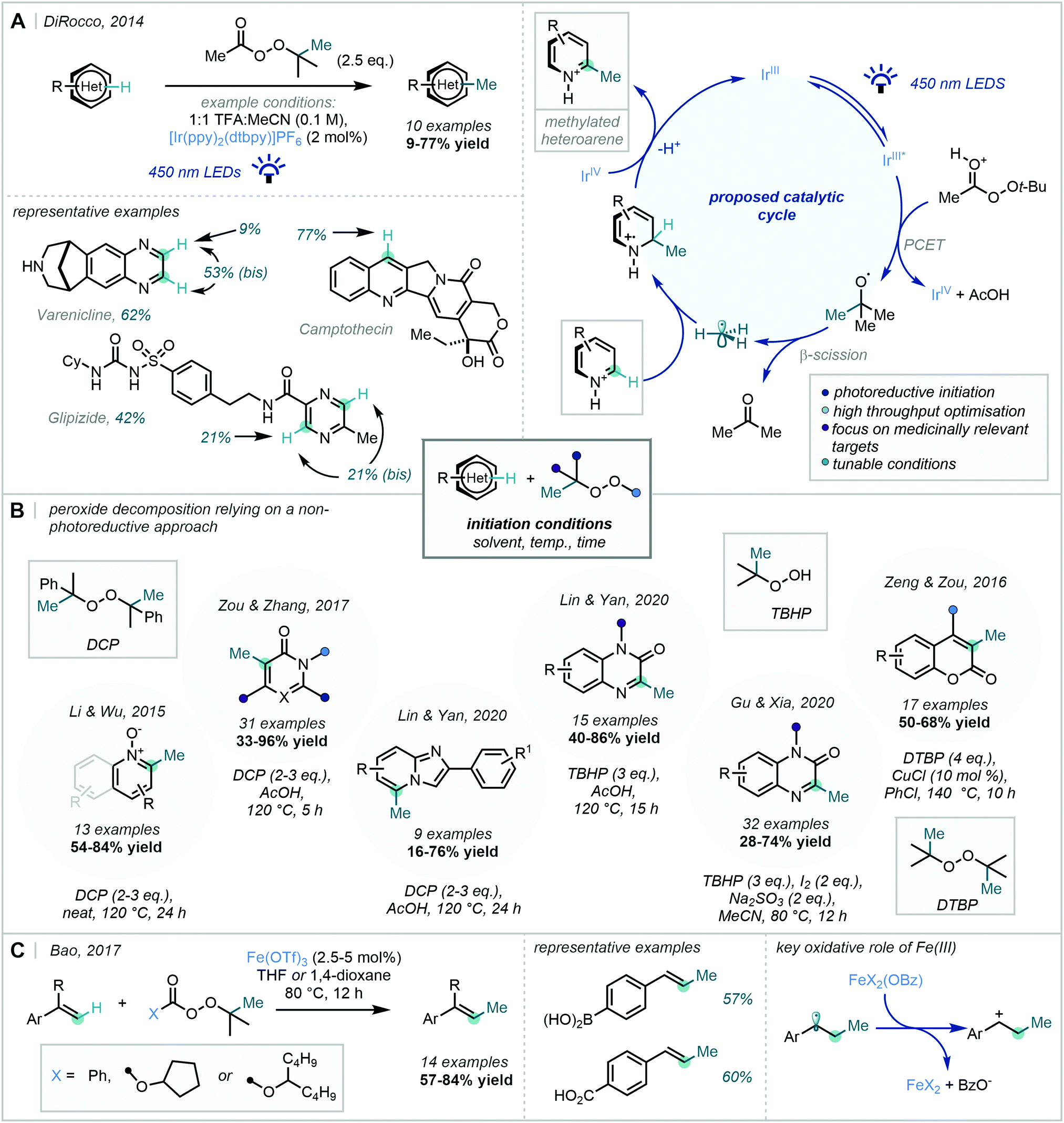 | ||
| Scheme 37 (A) Late-stage methylation of biologically-active heteroarenes via photoredox-initiated peroxide decomposition (B) Recent peroxide decomposition methods relying on a non-photoreductive approach. (C) Fe-catalysed styrenyl C–H methylation. | ||
While this chemistry enabled the generation of methyl radicals under mild conditions, an alternative approach focuses on the facile, thermolytic cleavage of organic peroxides. Such routes have been exemplified in the methylation of a number of privileged heterocyclic scaffolds such as pyridine N-oxides,107 pyrimidines108 and imidazo[1,2-a]pyridines109 (Scheme 37B). Lin & Yan further developed their methylation of imidazo[1,2-a]pyridines to enable the methylation of quinoxalin-2(1H)-ones by the use of tert-butyl hydroperoxide (TBHP) instead of DCP.109
Zeng & Zou demonstrated that the use of additives can further widen the scope of these methylation strategies. They employed a Cu(I)/Cu(II) catalytic cycle to enable the methylation of coumarins utilising DTBP as the methyl source.110 A distinct approach from Gu & Xia used a Na2SO3/I2 couple to act as an external oxidant, via the gradual release of I-radicals capable of triggering rearomatisation of the Minisci adduct.111
In a 2017 report from Bao, this approach has found further application in the methylation of styrene derivatives (Scheme 37C).112 The adoption of Fe(OTf)3 enabled both the fragmentation of a wide range of organic peroxides and the oxidation of the benzylic radical, resulting addition of the methyl radical, to enable reformation of the alkene π-bond.
A mechanistically-related approach from Ghosh in 2018 obviated the need to use organic peroxides via the in situ generation of an iodine(III)/tert-butanol adduct, which was shown to undergo fragmentation to generate methyl radicals, thus constituting a formal C–C activation (Scheme 38).113 This system was limited to the methylation of heteroarene N-oxides, with the oxide being invoked as a crucial directing and activating factor in the proposed mechanism.
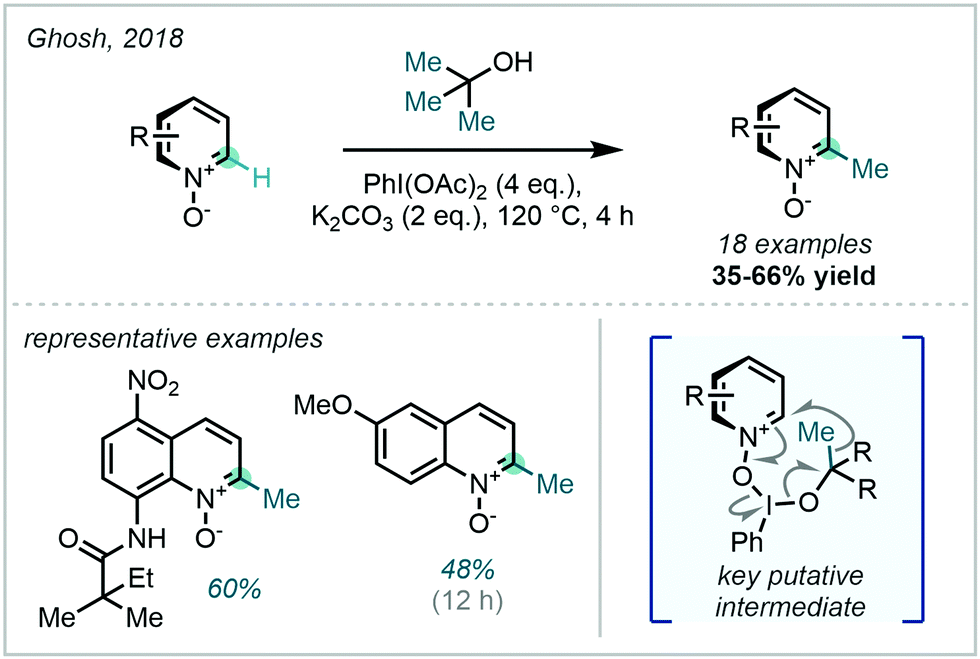 | ||
| Scheme 38 C–C activation of alcohols using hypervalent iodine reagents. | ||
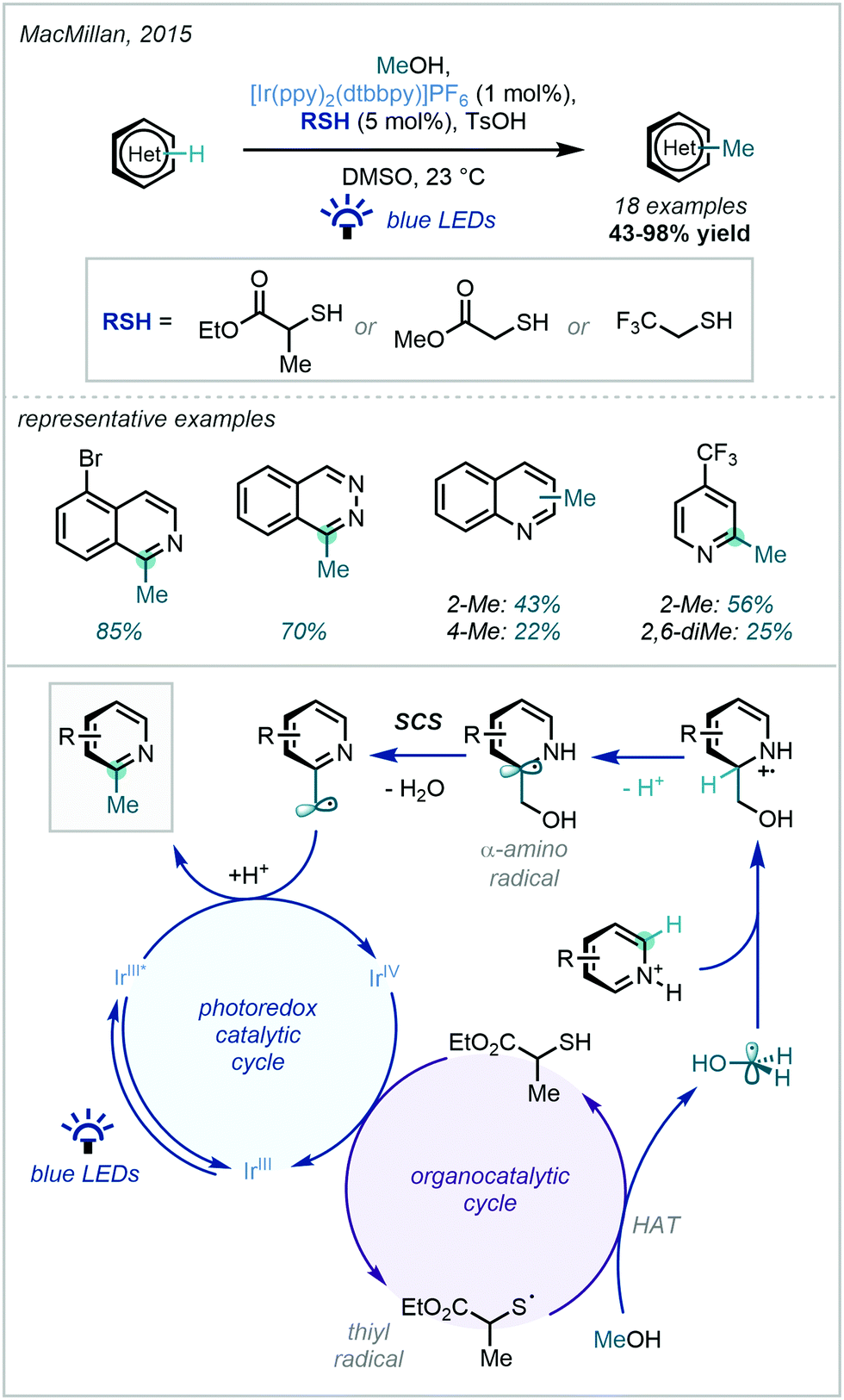 | ||
| Scheme 39 Dual catalytic activation of methanol for heteroarene C–H methylation. | ||
Following major developments in the fields of both hydrogen atom transfer (HAT) reactivity and photocatalysis, they elegantly entwined a photocatalytic cycle with a thiol-catalysed HAT cycle, via oxidative generation of a key thiyl radical by a photocatalytically generated IrIV oxidant. The critical dehydroxylation occurred via a spin-centre shift (SCS), a mechanistic principle describing the elimination of a leaving group adjacent to a radical which is accompanied by the transferral of the spin density to the adjacent atom.115
A further key development to this concept was made in 2017, when Li reported a catalyst-free photoactivation of methanol (Scheme 40).116 By engaging higher energy radiation, it was observed that hydroxymethyl radical generation was feasible without an external photocatalyst, and that the efficiency of this process could be further improved by the use of dichloromethane or benzophenone as additives. The authors postulated numerous viable mechanisms for this radical generation. The protocol demonstrated a broad scope, performing well on medicinally relevant pyridines, quinolines and isoquinolines.
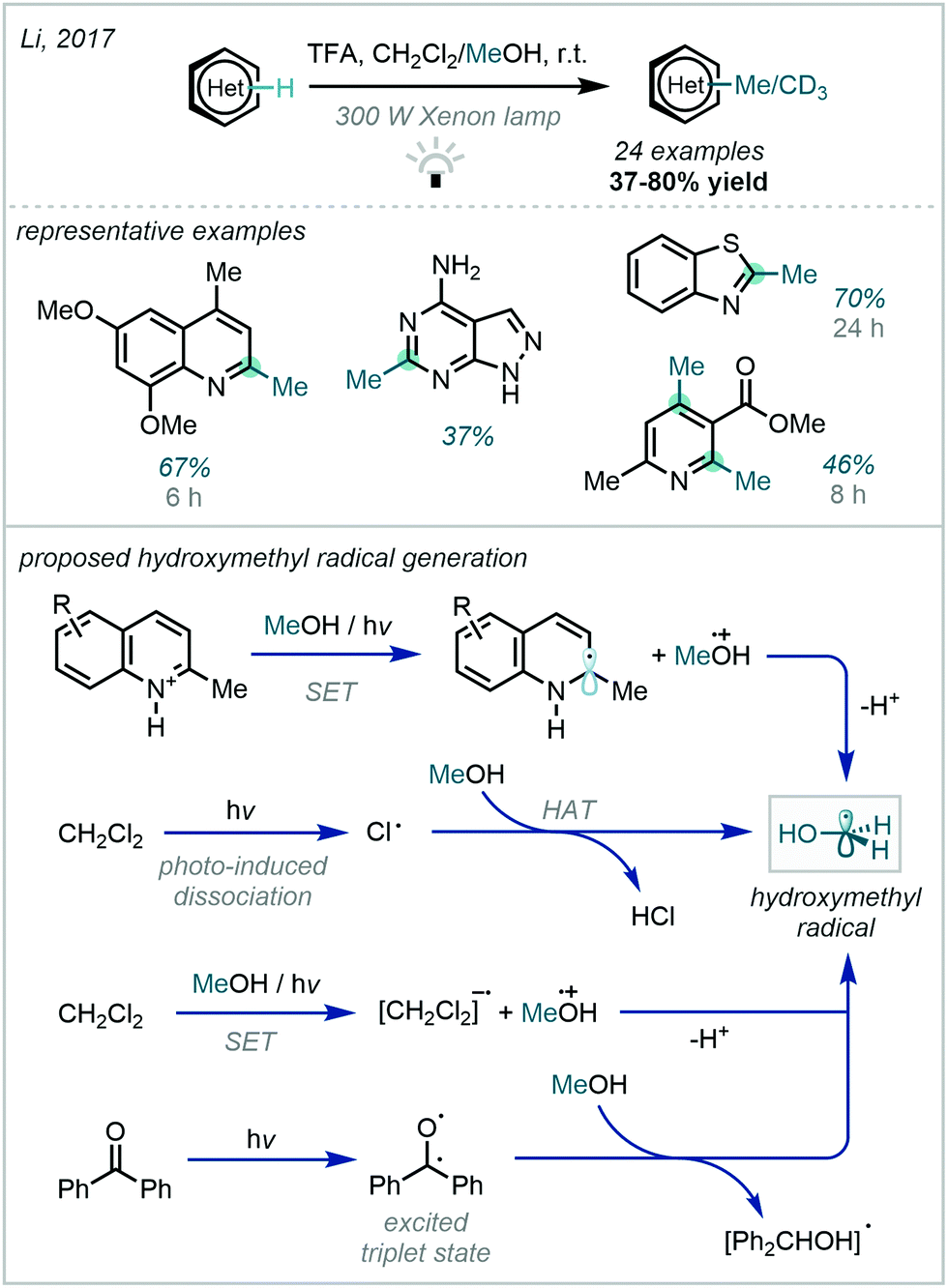 | ||
| Scheme 40 Activation of methanol via high energy photoexcitation to generate hydroxymethyl radicals capable of Minisci-type reactions. | ||
Furthermore, in 2017, Barriault & Scaiano demonstrated a related photo-activation of methanol (Scheme 41);117 by harnessing the power of UVA radiation the procedure avoided the use of an external photocatalytic cycle. Featuring subtly different mechanistic proposals, this work suggested the role of a sacrificial quantity of protonated substrate in forming a photo-catalytic cycle, both enabling generation of the hydroxymethyl radical and facilitating reduction of the Minisci adduct.
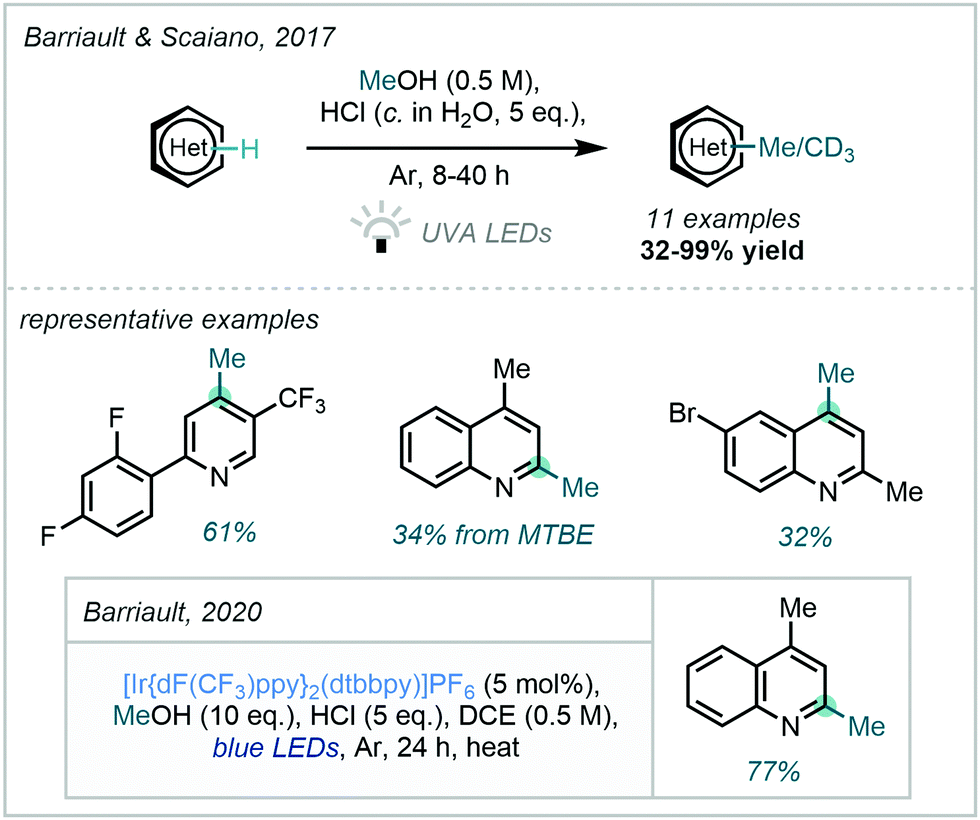 | ||
| Scheme 41 Activation of MeOH under high energy irradiation and subsequent visible light photocatalytic modification. | ||
Following this work, Barriault described a more general alkylation procedure from alcohols which employed an iridium photocatalyst to enable the use of lower energy radiation.118 The procedure was also found to be applicable for the C–H methylation of quinolines (Scheme 41). Intriguingly, following Stern–Volmer quenching studies, the chloride ion derived from the HCl acid promoter was implicated in the photocatalytic cycle and therefore a hydrogen abstraction by a chloride radical was proposed.
These approaches, while well-established for the generation of substituted alkyl radicals, have achieved limited success in the generation and the productive application of reactive methyl radicals. An initial achievement by Shang & Fu used an iridium photocatalyst ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6) in the presence of a Lewis acid co-catalyst (In(OTf)3) to selectively methylate phenanthridine in a modest 32% yield (Scheme 42).120
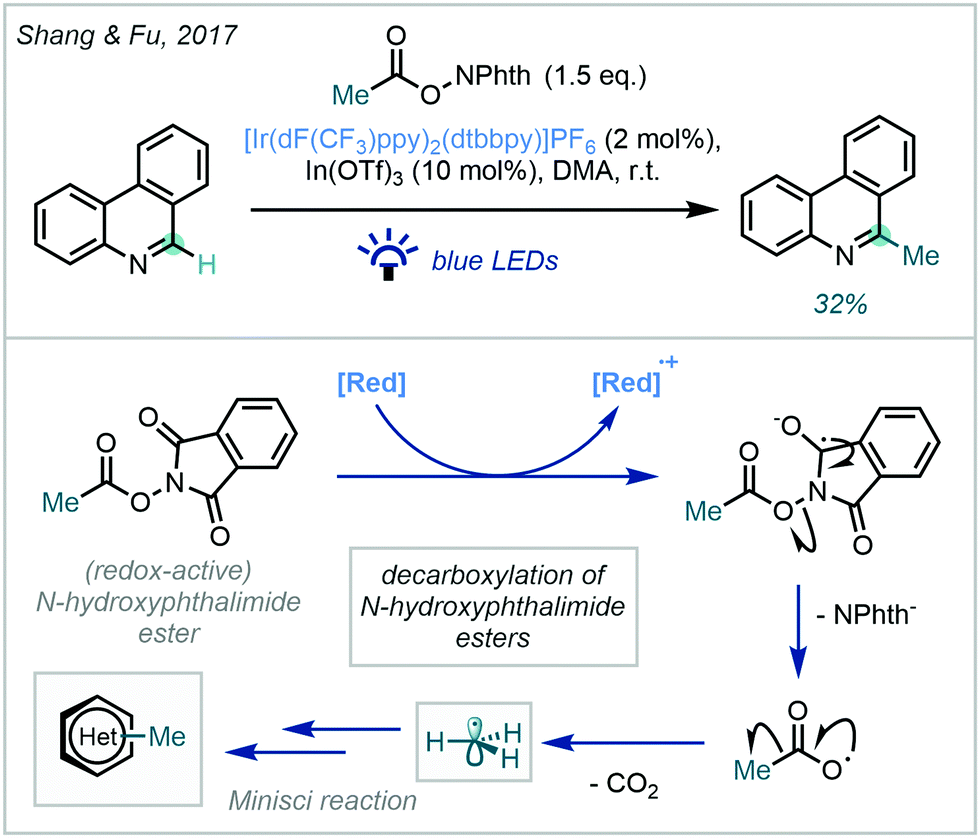 | ||
| Scheme 42 Photocatalytic decarboxylation of a N-hydroxyphthalimide ester for the methylation of phenanthridine. | ||
Genovino & Frenette opted for the photo-reductive fragmentation of an in situ-generated hypervalent iodine species, enabling methylation of lepidine in 43% yield, using acetic acid directly as the methyl source (conditions A, Scheme 43A).121 In contrast, Sherwood reported an in situ coupling to afford the redox-active ester which could undergo decarboxylation upon single electron reduction by an organophotocatalyst (4-CzIPN) and, subsequently, methylate lepidine in 26% yield (conditions B, Scheme 43A).122
 | ||
| Scheme 43 (A) Photodecarboxylation strategies for generation of methyl radicals directly employing acetic acid (B) Photocatalytic decarboxylation of hypervalent iodine reagents. | ||
While effectively demonstrating proof of concept, the aforementioned methodologies were arguably not developed as broadly applicable high-yielding heterocycle methylation platforms. Directly following Genovino & Frenette's report, Hu described the photo-induced reductive decarboxylation of hypervalent iodine dicarboxylates to afford methyl radicals which could participate in Minisci-type radical addition to quinoxalin-2-(1H)-ones (Scheme 43B).123 In contrast to the previous examples of decarboxylative methylation, Hu's work showed a wide scope in the quinoxalin-2(1H)-one substrate.
Furthermore, Xu & Song applied electrophotocatalysis to either methylate or trideuteromethylate lepidine, albeit in modest yields (conditions C, Scheme 43A).124 Key to this strategy was a dual catalytic cycle in which a cerium photocatalyst was proposed to effect both an oxidative fragmentation of the carboxylate and then the re-oxidation of the Minisci adduct; anodic oxidation then enabled catalytic turnover by recycling Ce(III) to Ce(IV).
Recently, Antonchick developed this work to enable the trideuteromethylation of a number of quinolines and isoquinolines, demonstrating good selectivity and yields (Scheme 44).127 The mechanistic feature common to these protocols is the generation of a hydroxyl radical from hydrogen peroxide which then adds to DMSO-d6 to facilitate β-scission that releases a (trideutero)methyl radical and a sulfinate salt/sulfinic acid.
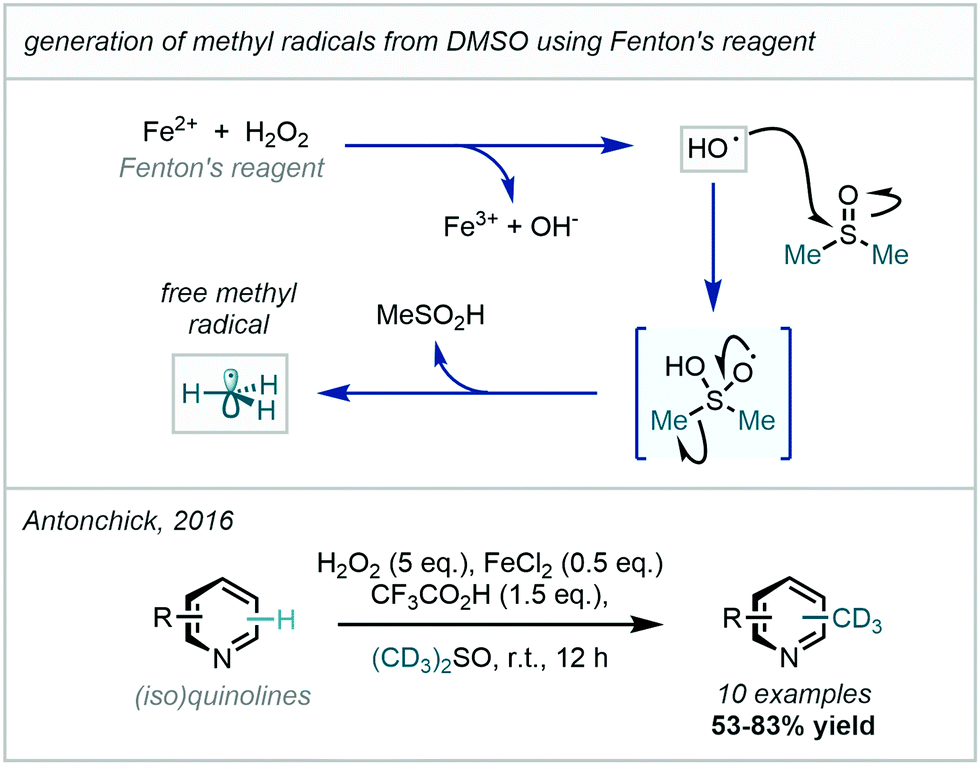 | ||
| Scheme 44 Generation of methyl radicals from DMSO using Fenton's reagent and H2O2; trideuteromethylation employing DMSO-d6 under related radical generation conditions. | ||
An alternative strategy, reported by Glorius in 2018, elegantly employed the photo-induced reduction of in situ-generated Me2SCl+.128 The key cationic intermediate was generated from the reaction of DMSO with an electrophilic activator (PhPOCl2) while an iridium photocatalyst was chosen to facilitate the SET required for both methyl radical generation and subsequent oxidation of the Minisci adduct (Scheme 45). The conditions were demonstrated to be broadly applicable for the methylation and trideuteromethylation of quinolines and isoquinolines and, notably, a moderate alteration in the conditions enabled methylthiomethylation as well.
 | ||
| Scheme 45 A photocatalytic approach to the activation of DMSO using PhPOCl2 as an electrophilic activator. | ||
Complementary to the Minisci-type approaches described above, a base-mediated system applicable to 1,8-naphthyridines was developed by Zhu & Chen in 2019 (Scheme 46).129 An unusual photo-induced SET from t-BuONa to DMSO was proposed to account for the methyl radical generation. On the back of numerous deuteration experiments, a Meerwein–Pondorff–Verley-type reduction (and subsequent tautomerisation) of the 1,8-naphthyridine substrates was proposed to precede the methylation event. Auto-oxidation then enabled regeneration of aromaticity.
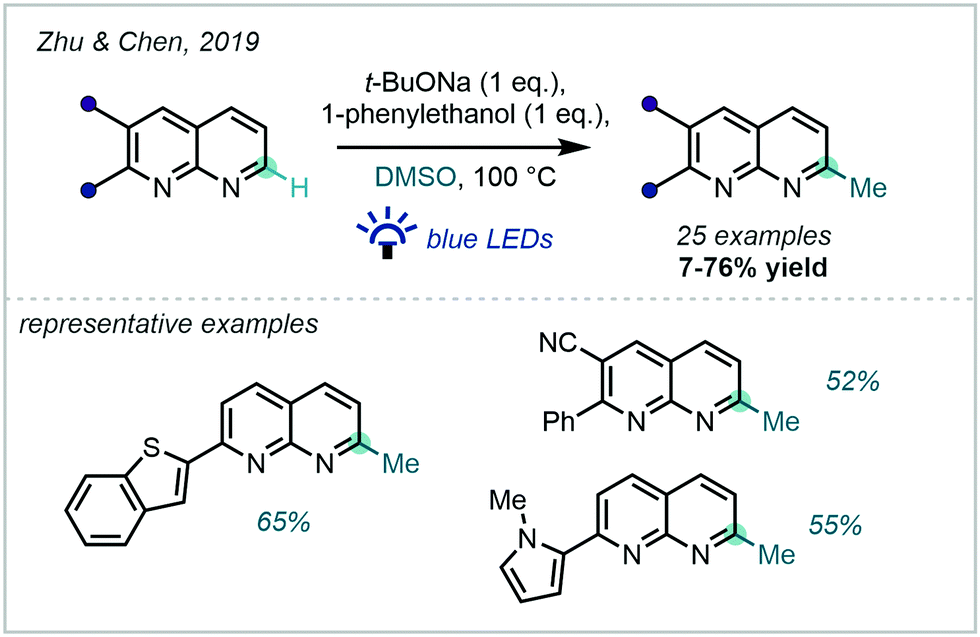 | ||
| Scheme 46 Methylation of 1,8-naphthyridines via DMSO activation. | ||
A major development in this field came in 2014 with Baran's report of using a novel zinc alkyl sulfinate to install a phenylsulfonylmethyl moiety, which could be smoothly converted to a methyl group under numerous reductive conditions (Scheme 47).130 This approach took inspiration from S-adenosylmethionine (SAM), the methylating “reagent” used by living organisms, which has been shown to methylate heteroarenes through what is likely to be an enzymatically generated methyl radical.131
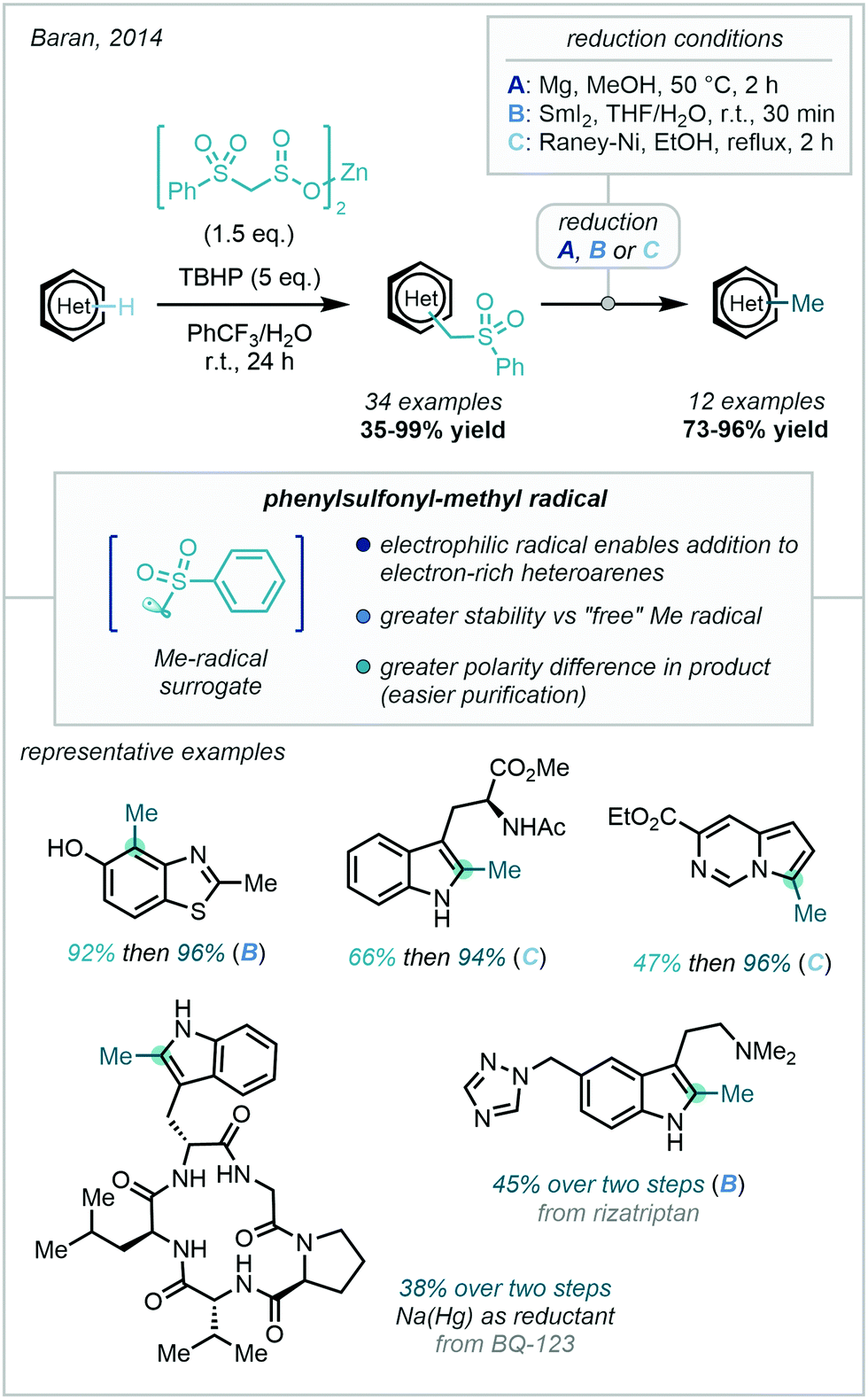 | ||
| Scheme 47 Two-step SAM-inspired methylation of heteroarenes. | ||
Zinc bis(phenylsulfonylmethanesulfinate), a free-flowing, bench-stable solid, was developed and employed in the presence of 5 equivalents of TBHP to install the phenylsulfonylmethyl group on a diverse range of heterocyclic scaffolds. The use of zinc sulfinates as radical precursors had been previously documented by Baran, where the putative mechanism involves single electron oxidation of the sulfinate by a tert-butoxy radical, generated from TBHP decomposition, which in turn triggers a desulfonylation reaction, releasing the desired radical.132 The greater stability of the radical in this work enabled a wider range of heteroarenes to undergo C–H methylation. In particular, the electrophilic nature of the phenylsulfonyl-methyl radical enable efficient reaction with electron-rich heteroarenes. A related system utilising sodium alkyl sulfinates and phenyliodine(III) diacetate (PIDA) as an oxidant has also been reported by Zhang & Zhang to methylate quinoxalinones (18 examples, 45–90% yield).133
A conceptually related approach was disclosed by Zard in 2018, in which a carboxylic xanthate was used to generate stabilised α-carboxymethyl radicals, capable of taking part in Minisci reactions (Scheme 48).134 The resulting carboxymethyl groups would then undergo a subsequent decarboxylation following heat and microwave treatment to afford the desired methyl appendage. The generation of the key α-carboxymethyl radical was initiated by the decomposition of dilauroyl peroxide (DLP). Similar to Baran's work, the greater stability of the active radical species enabled a broad substrate scope in the carboxymethylation, of which numerous examples were then subjected to in situ decarboxylation.
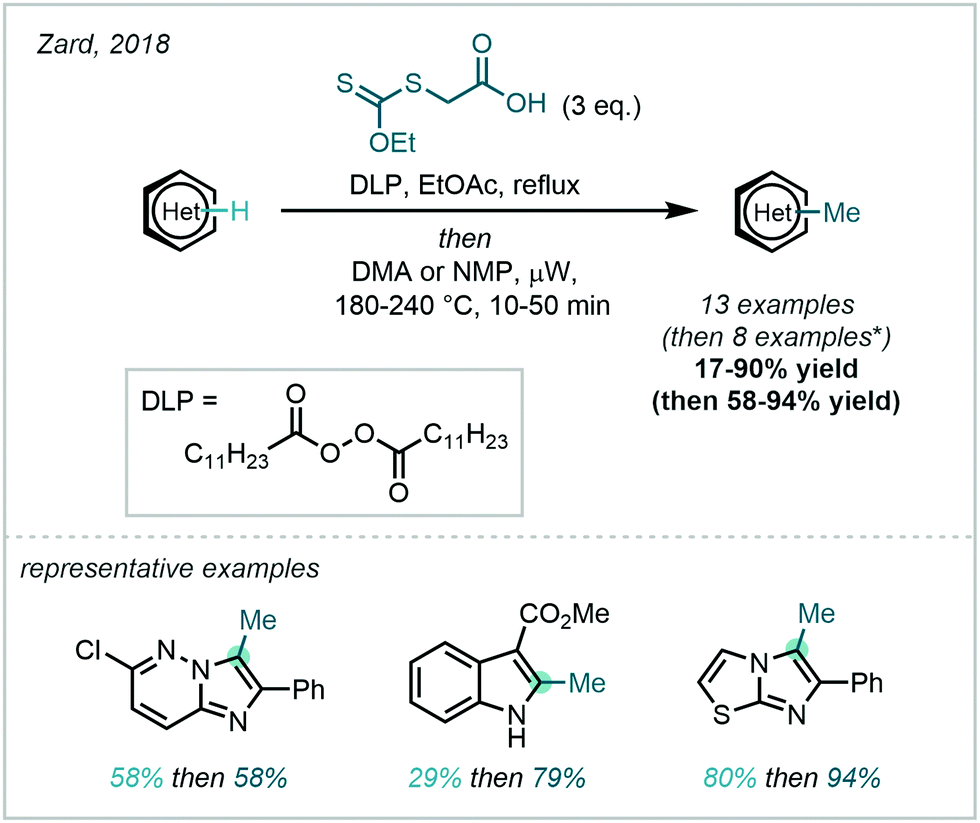 | ||
| Scheme 48 Use of a carboxylic xanthate for the methylation of heteroarenes. *Four additional examples decarboxylate spontaneously. | ||
From an alternative standpoint, building on the powerful recent applications of alkyl trifluoroborates as radical precursors,135 a study in 2016 from Chen & Liu highlighted the potential of boronic acids (and their derivatives) as methyl radical sources, following reaction with photocatalytically-generated benzoyloxy radicals (Scheme 49).136 In this work, numerous alkylations were described however only one example of a methylation was reported, in 46% yield on 4-chloroquinoline.
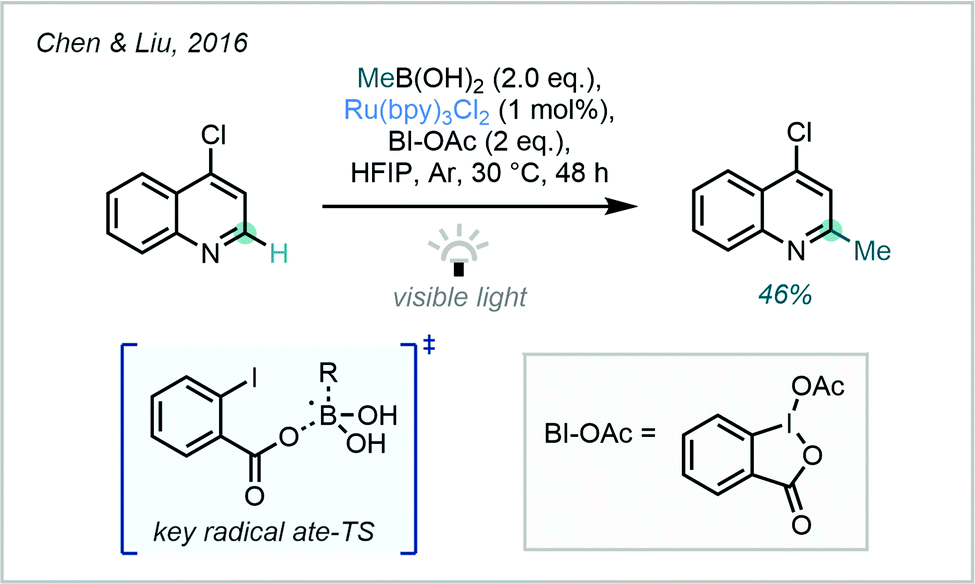 | ||
| Scheme 49 Application of boronic acids for the photocatalytic methylation of 4-chloroquinoline. | ||
In 2020 Wang demonstrated the use of PEG-400, under an O2 atmosphere in the presence of a Brønsted acid (TsOH·H2O), as a source of α-oxy radicals which could participate in Minisci-type reactions on 3-arylquinazolin-4(3H)-ones (Scheme 50). Subsequent tandem deoxygenation and rearomatisation generated the desired methylated product.137 This procedure enabled the methylation of a broad range of 3-phenylquinazolin-4(3H)-ones.
 | ||
| Scheme 50 Oxidative fragmentation of PEG-400 to methylate 3-arylquinazolin-4(3H)-ones. | ||
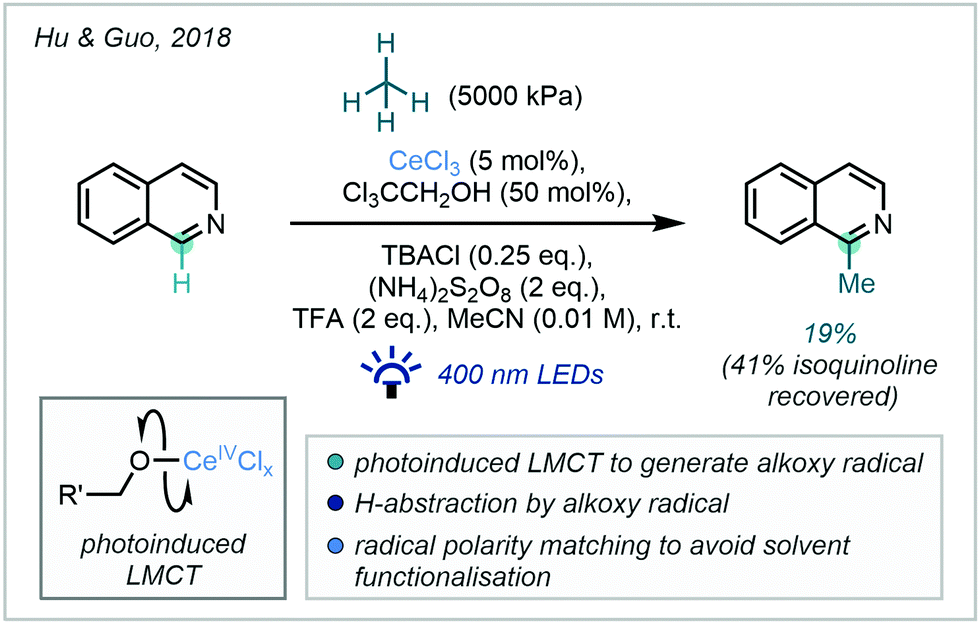 | ||
| Scheme 51 Direct application of methane under Ce-photocatalysis to methylate isoquinoline. | ||
3.2 Two-electron strategies for the C(sp2)–H methylation
One strategy that has been employed to raise the innate electrophilicity of the C2 and C4 positions is through N-activation. Azine-N-oxides have found extensive use as tandem activating and leaving groups for generating C–H methylated azines. These N-oxides are readily obtained from the parent N-heteroarenes under mild oxidative conditions and often obviate the considerable challenges presented by the formation of inseparable mixtures of starting material and product.139 An early example from Nicolaou demonstrated methylation through this sequence, utilising Tebbe's reagent to reductively methylate three structurally simple azine N-oxides at the C2-position (Scheme 52).140
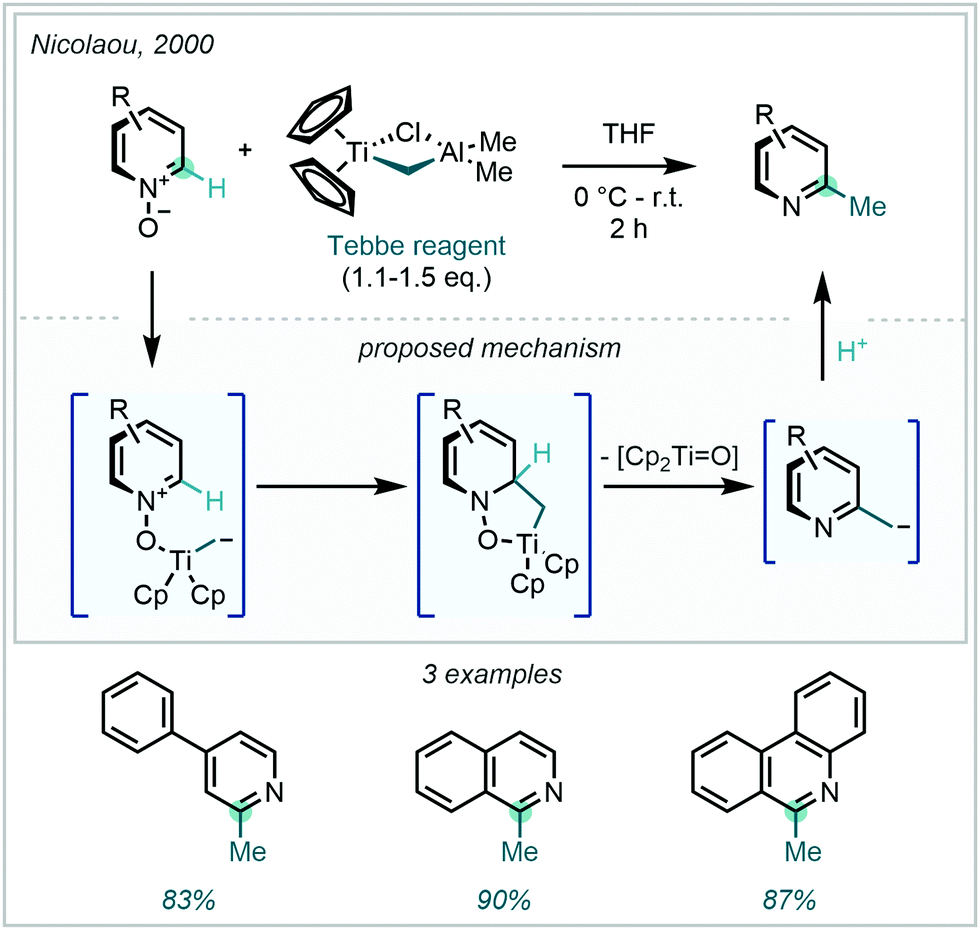 | ||
| Scheme 52 Reductive C–H methylation of azine-N-oxides using Tebbe's reagent. | ||
Following this, Almqvist and Olsson described a single low yielding example of the addition of MeMgCl into the C2-position of 4-benzyloxypyridine-N-oxide.141 Subsequent heating of the N-oxide with Ac2O enabled re-aromatisation.
A problem facing the use of alkyl Grignard reagents is their highly carbanionic nature. This has been postulated to facilitate the deleterious metalation of azines, outcompeting addition of the alkyl group at the same position.139,142 Duan found that MeMgBr could be successfully added to nitropyridine-N-oxides, with retainment of the N-oxide handle through 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) oxidation of the initial addition adducts.143 Interestingly in para-nitropyridine-N-oxides, alkyl Grignards were found to react ortho to the nitro substituent, as opposed to aryl Grignards which preferred to react ortho to the azine N. For meta-nitropyridine-N-oxides these two directional effects were able to combine, delivering methylation in high yield (Scheme 53A). Further to this, in 2014, Larionov discovered that incorporation of catalytic CuCl and stoichiometric LiF or MgCl2 enabled the reaction of MeMgBr with, less electronically activated, heteroarene N-oxides (Scheme 53B).144 These milder reaction conditions – through employment of the key additives – allowed for a wider scope of aryl functional groups, which was exemplified in the preparation of the phenolic antimicrobial reagent chlorquinaldol and a des-chloro analogue.
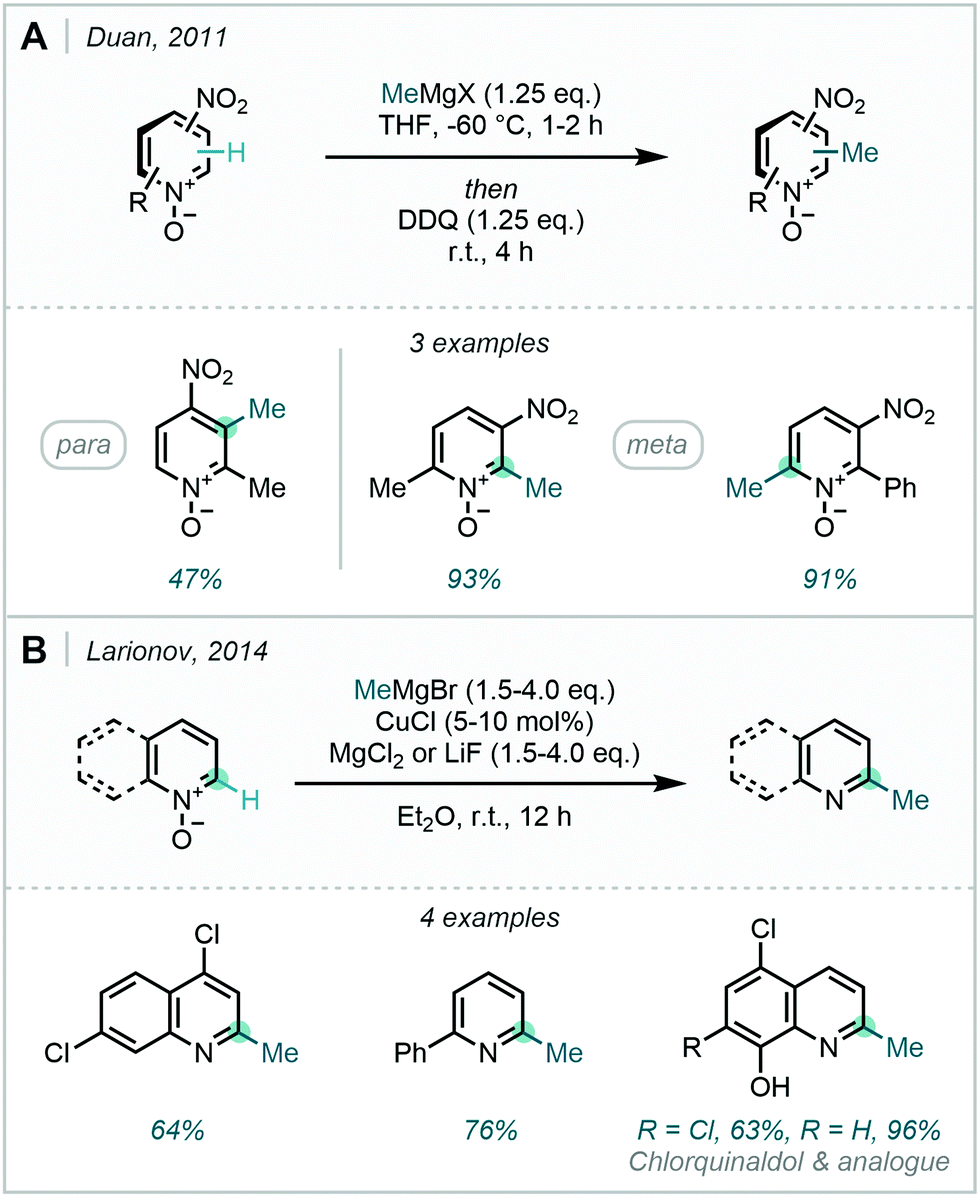 | ||
| Scheme 53 (A) Alkylation of pyridyl-N-oxides with retention of the N-oxide. (B) Cu-catalysed methylation of pyridyl-N-oxides to generate pyridines. | ||
In 2016, Cho disclosed that the α-borylcarbanion, derived from bis[(pinacolato)boryl]methane, was an excellent nucleophilic methyl group surrogate for the C2-selective methylation of a range of heteroarene-N-oxides (Scheme 54A).145
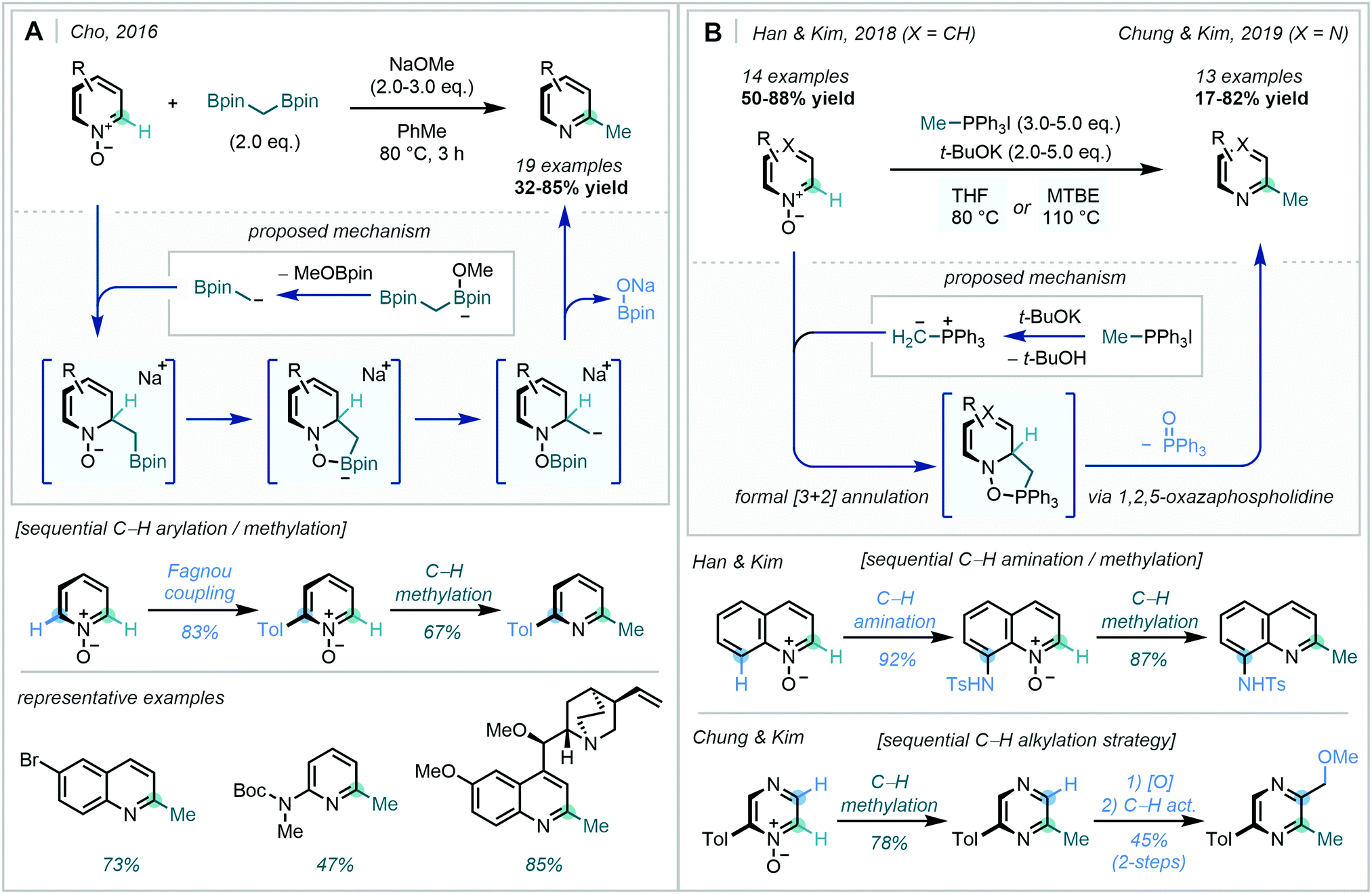 | ||
| Scheme 54 (A) Diborylmethane addition to azine-N-oxides. (B) Phosphonium ylide addition to azine-N-oxides. | ||
The transition metal-free, base-promoted approach was shown to be scalable and tolerant of a variety of functional groups, including carbamates, amides, cyclic acetals, tertiary amines and aryl halides, thus allowing the preparation of a diverse set of methylated azine fragments. A powerful example of this transformation was displayed by sequential Fagnou coupling and methylation to generate a 2,6-disubstituted pyridine, a highly valuable motif within drug discovery programs.146
In two recent reports, Han & Kim,147 followed by Chung & Kim,148 highlighted the use of methyltriphenylphosphonium salts for the C2-selective methylation of pyridine and diazine N-oxides, respectively (Scheme 54B). Believed to proceed via a formal [3+2] annulation between the azine-N-oxide and ylide, subsequent decomposition of the 1,2,5-oxazaphospholidine to extrude triphenylphosphine oxide was suggested to be the driving force for the reaction.
The initial methodology on pyridines and quinolines was found to readily translate across various diazines, with a particular focus on pyrazines. Tandem C–H activation sequences offered the opportunity for the rapid generation of value-added substrates, as showcased by the two-step amination and methylation of quinoline-N-oxide alongside the dialkylation of simple pyrazines. Furthermore, access to unsymmetric bipyridyl substrates and gram scale performance was also achieved.
The ideal C–H methylation of arenes is both direct (one-step) and, crucially, site-selective, a facet often lacking in radical approaches which often present undesirable regioselectivities. Until recently, a lone two-electron example had been reported by Sarpong,149 enabling direct C6 methylation of a pyridyl alcohol with MeLi. However, in 2020 Baik & Cho succeeded in developing a reliable protocol to overcome the energy barriers associated with unactivated azine C–H methylation.150 Building on previous developments in the area (Scheme 54A), the authors facilitated in situ activation-methylation of N-heteroarenes with ZnMe2 in combination with diborylalkanes (Scheme 55). A combination of NMR, deuterium labelling and DFT studies were used to delineate the complex mechanism. These investigations suggested that complexation of the diborylmethane-derived α-borylcarbanion to the Lewis acidic MeZnOt-Bu generated the reactive zincate intermediate, prior to a concerted C2-attack/N-activation of the azine. This zincate coordinated adduct was then believed to undergo decomposition, re-aromatising to the boromethylated azine. Subsequent protodeborylation yielded the C–H methylated product. This highly C2-selective procedure found great application on a range of quinolines, pyridines and bipyridines, tolerating a range of aryl substituents.
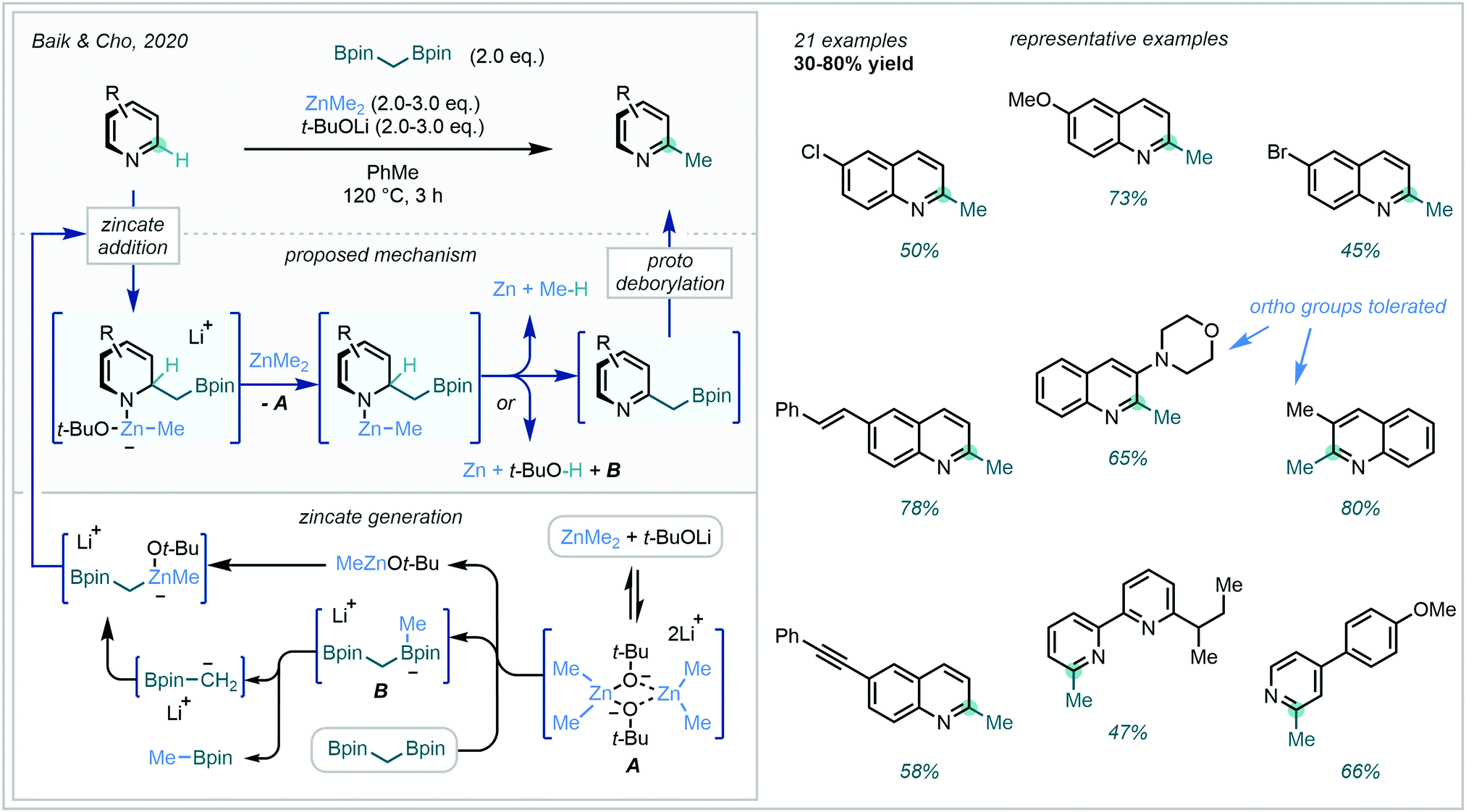 | ||
| Scheme 55 Mechanism and scope of ZnMe2-promoted direct C–H methylation of N-heteroarenes with diborylmethane. | ||
In 2020, Kim extended the range of ylides capable of achieving azine methylation, by using sulfoxonium ylides to directly methylate pyrazinone, quinoxalinone and azauracil scaffolds (Scheme 56).151 The latter of these scaffolds, the azauracils, are of high interest in the medicinal chemistry community due to their potential to display antiviral, antitumor, and antifungal activity. Formed through deprotonation in aqueous or alcoholic media, the sulfoxonium ylides were found to exhibit nucleophilic attack exclusively at the carbon centre formally making up the imino fragment of the iminoamido heteroarene. Elimination of the sulfoxide and protonation at N generates an exo-methylene enamine which undergoes tautomerisation to furnish the C–H methylated azine. Intriguingly, no aziridine containing by-products were observed, indicating a distinct difference in reactivity to Corey–Chaykovsky aziridinations. The wide substrate range reported highlights the great tolerance of multiple functionalities under the basic conditions, including N-THP and N-Bn protecting groups. Diverse downstream reactivity was achieved from the newly installed methyl group, with C–H alkylation and two-step oxidation-amination being exemplified amongst other transformations. Gram-scale reactivity coupled with the use of purely aqueous media seek to further showcase the utility of sulfoxonium ylides as methylating agents.
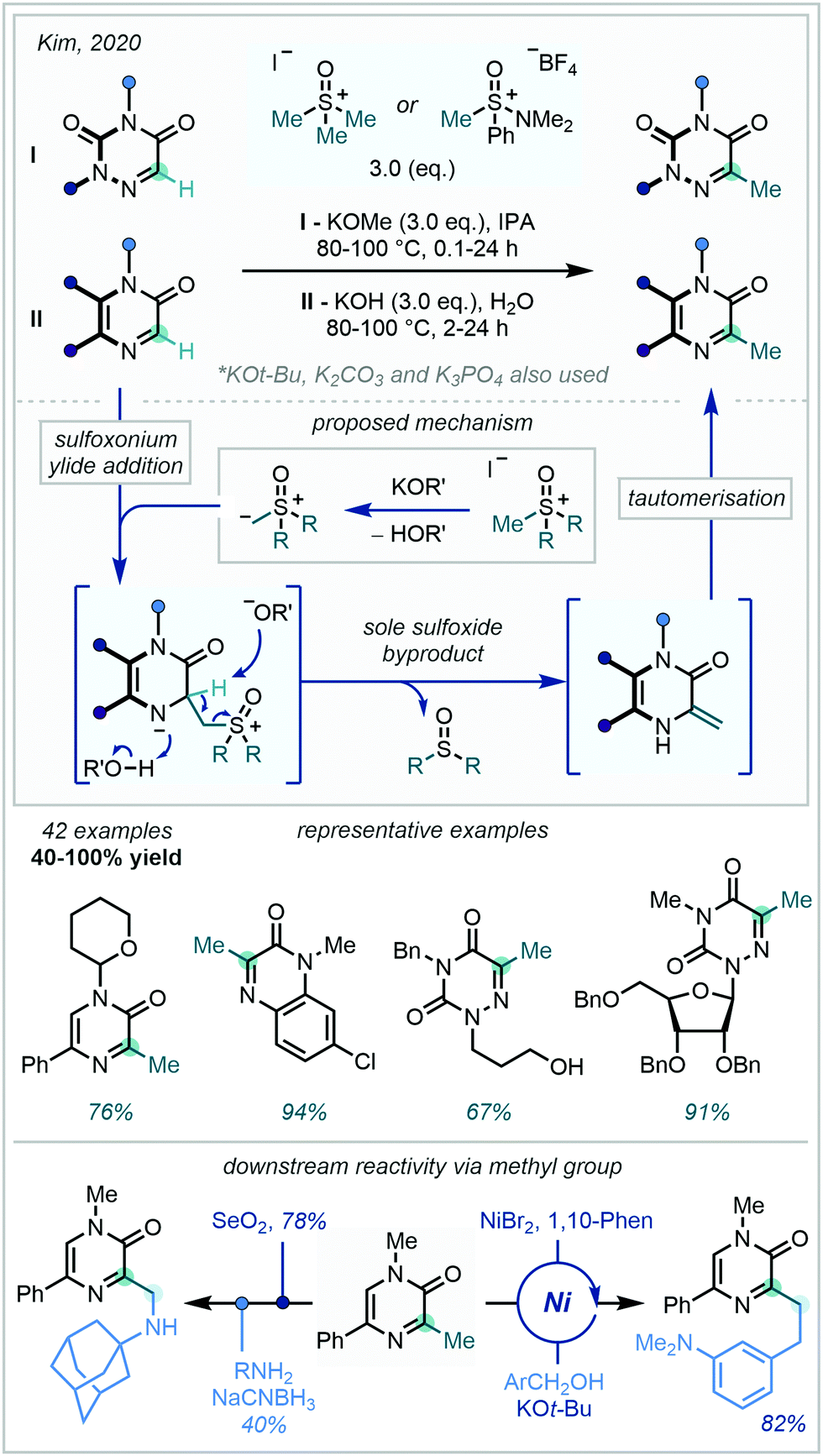 | ||
| Scheme 56 Sulfoxonium ylides as vicarious nucleophiles for the C–H methylation of iminoamido heterocycles. | ||
Very recently, Han, Hong & Kim documented an additional use for sulfur-based ylides in C–H methylation, employing trimethylsulfonium ylides for the redox neutral C2-selective methylation of azine-N-oxides with retention of the N-oxide (Scheme 57).152 Previous work from Han & Kim (Scheme 54B), focused on achieving a formal [3+2] cycloaddition between a phosphonium ylide and azine-N-oxide prior to extruding triphenylphosphine oxide to generate C2 methylated azines. In contrast, the trimethylsulfonium ylide avoids forming the analogous 1,2,5-oxathiazolidine [3+2] intermediate, the formation of which is proposed to be endergonic. Instead, the initial betaine adduct is stable and undergoes pyrrolidine-assisted E-2 elimination, as supported by DFT calculations, of dimethyl sulfide, in turn yielding C2 methylated azines with N-oxide retainment.
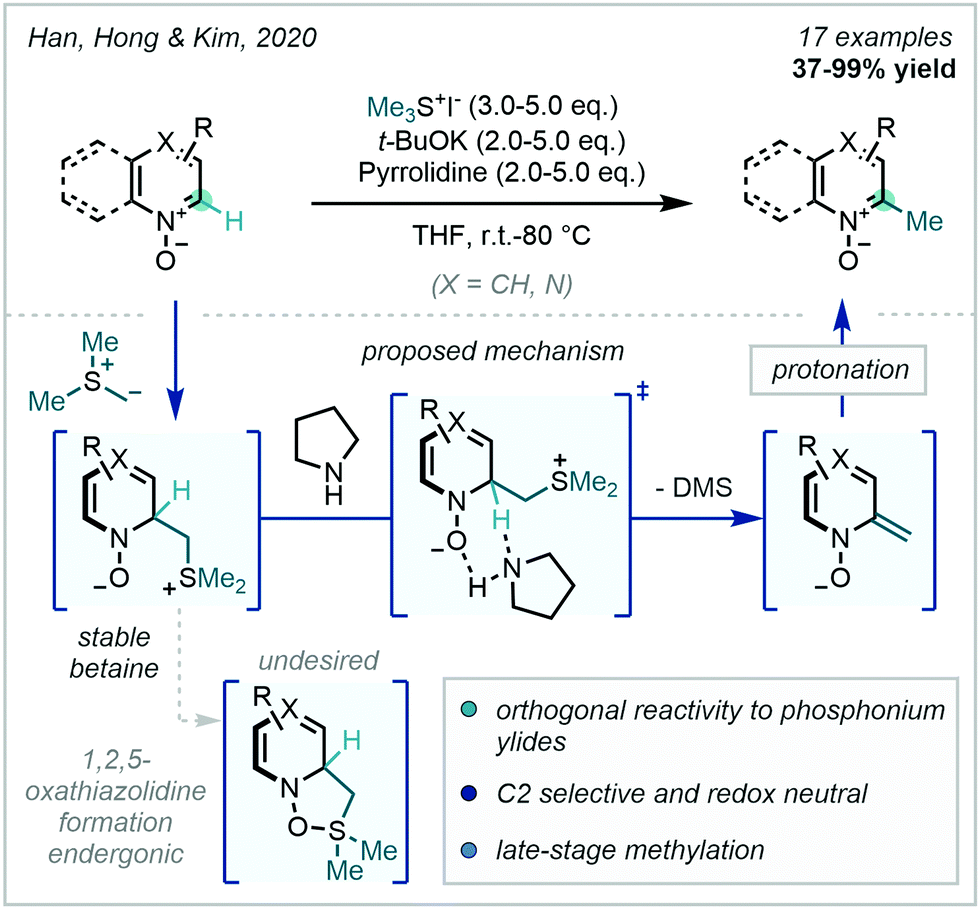 | ||
| Scheme 57 Trimethylsulfonium ylides for redox neutral methylation of azine-N-oxides. | ||
With sustainability rapidly becoming a priority in process design, the valorisation of simple and renewable feedstock chemicals is becoming ever more desirable. Building on their previous N-methylation protocol,154 Beller achieved the analogous and more challenging C–H methylation on pyrroles, indoles and other electron rich arenes utilising feedstock gases CO2 and H2 (Scheme 58).155 A catalytic reduction of CO2 and subsequent nucleophilic attack on a transient ruthenium formate complex was proposed to deliver an intermediary hemiacetal species. In situ reduction by ruthenium-hydride then delivered the methyl group, generating water as the sole by-product. Numerous indoles and pyrroles were successfully methylated at their innately reactive C3-positions, alongside trimethoxybenzene analogues, the latter previously having found use in the synthesis of flavanones.156
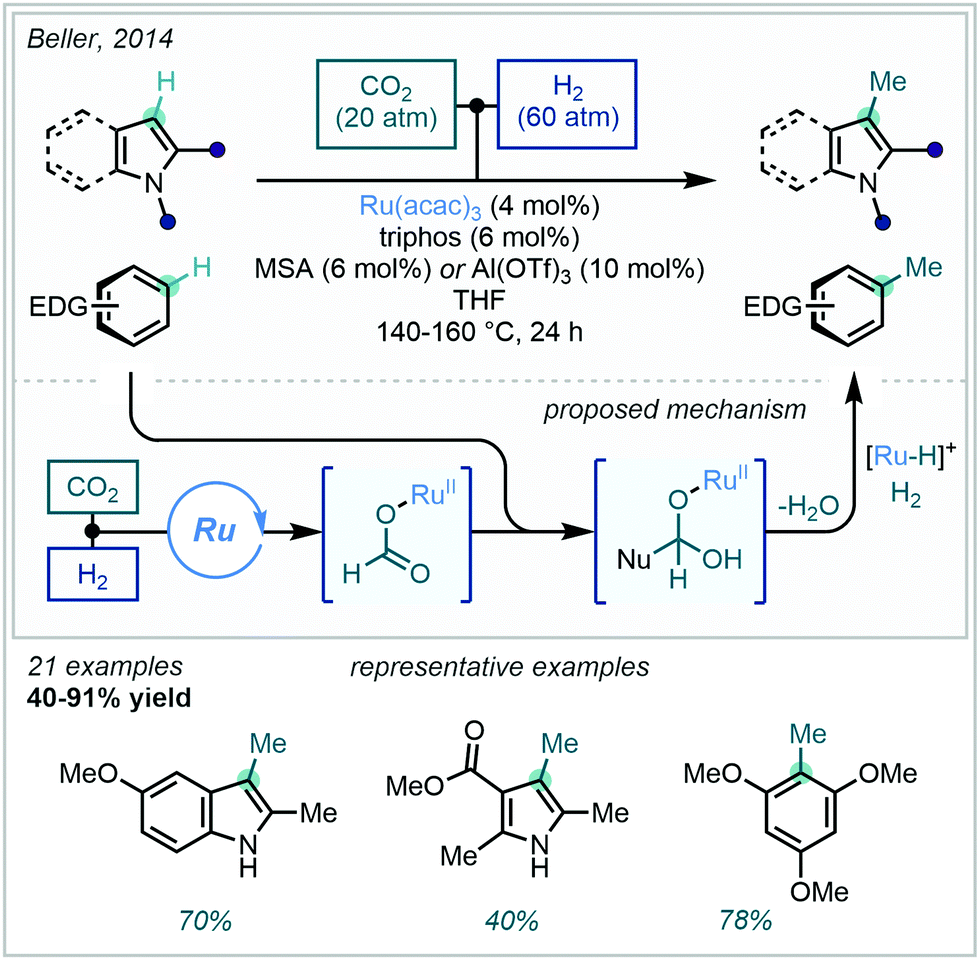 | ||
| Scheme 58 Ru-Catalysed C–H methylation of electron rich (hetero)arenes using CO2/H2 as a methyl source. | ||
In 2008, Brown disclosed the feasibility of Pd mediated methyl transfer from silanes to alkenes, in an olefinic Fujiwara–Moritani (or oxidative Heck) reaction (Scheme 59).158 Prepared in one step from 1,1-dimethyl-3-phenylurea, the bespoke disilylurea reagent readily underwent methyl transfer – via Pd – to a wide range of olefins including styrenes, acrylates and enones. A dative interaction between the proximal urea and TMS silicon centre was proposed to enable Si–Me activation, facilitating methyl transfer to PdII. Carbopalladation and subsequent β–hydride elimination then furnished the methylated-E-olefins with high geometric selectivity. E/Z-Selectivity could be reversed for olefins with adjacent and sterically hindered sp3 sites, giving access to methylated-Z-olefins.
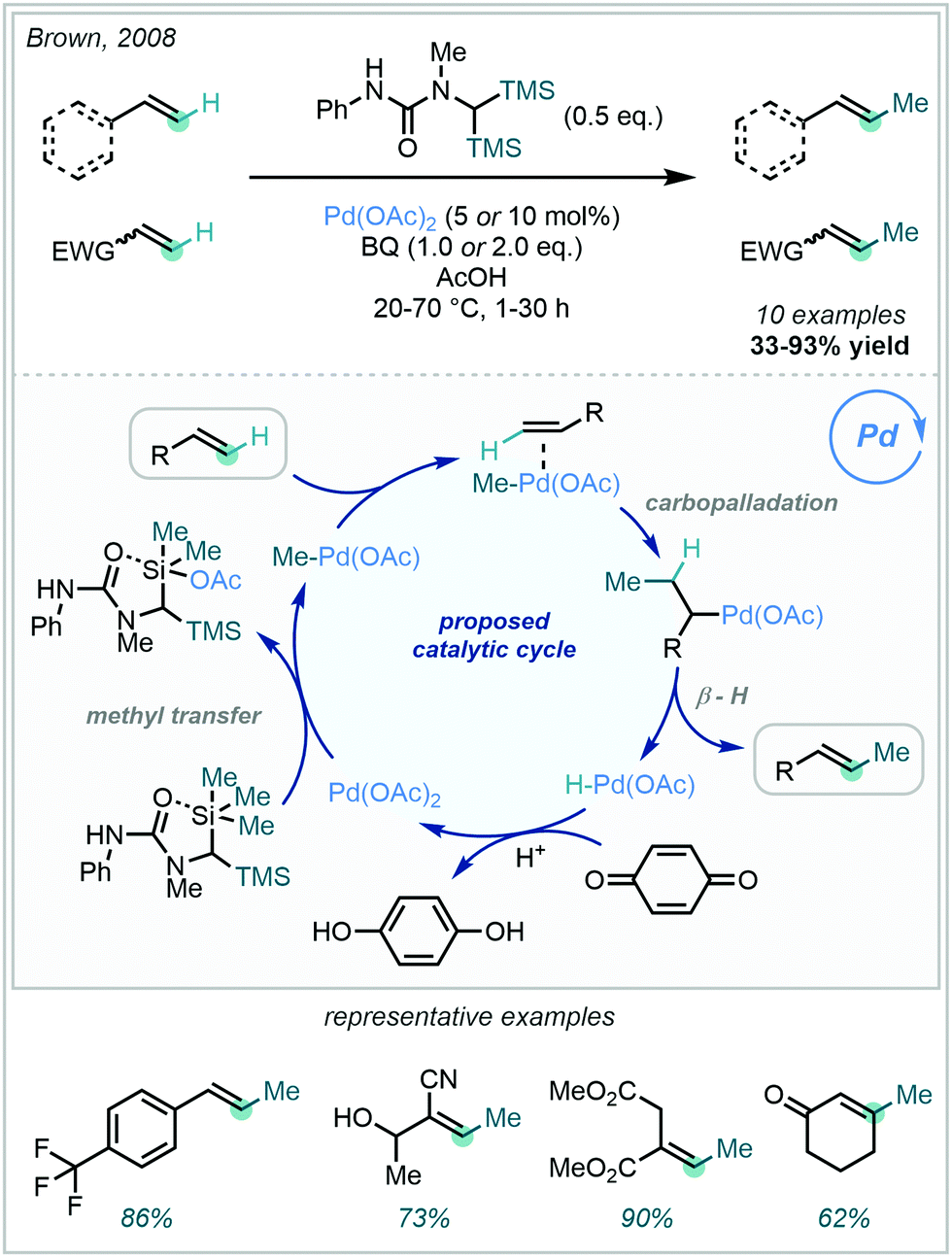 | ||
| Scheme 59 Vinyl C–H methylation via the Fujiwara–Moritani reaction. | ||
3.3 Tandem C–H functionalisation/methylation strategies
Pre-functionalised arenes with synthetically versatile functional groups have an esteemed history in the field of C–C bond formation. The potential for divergent synthesis enabled by these handles has stimulated intensive research into methods to access them from C–H bonds. By combining state-of-the-art C–H functionalisation methodology with robust cross coupling protocols, both precise C–H selectivity and efficient C–C bond formation can be realised (Fig. 3). | ||
| Fig. 3 Strategy for 2-step C–H functionalisation/methylation of arenes. | ||
For pyridyl arenes, a key challenge to address is selective C4 (or para) methylation. Both the C2 and C4 sites are innately electrophilic, and product distributions can often arise. However, C2 reactivity tends to dominate due to inductive/directional effects from the proximal N atom or N-bound activating group. Despite this, work from McNally, who has pioneered the use of azine derived arylphosphonium salts as functional handles in synthesis, presented an elegant solution to the problem at hand.159 Relying on activation of pyridine derivatives through initial N-triflation, triarylphospines have been shown to possess almost exclusive reactivity towards addition at the C4 position, and readily generate C4-substituted arylphosphonium salts after elimination of triflate. Intriguingly, the rationale for this exquisite regioselectivity is thought to stem not from steric effects, but from the enhanced orbital interaction between the pyridyl π-system at C4 and the incoming phosphorus lone pair.
As part of a wider research effort, McNally recently added alkyl groups to the increasing repertoire of nucleophiles capable of substitution with the versatile phosphonium handle, detailing four examples of methylation (Scheme 60).160 A Co-catalysed variant of the Negishi cross coupling was developed to achieve this, with catalytic i-PrMgCl proving to be essential for efficient methylation.160 In the presence of C4-substituents, C2-phosphorylation, and in turn C2-selective C–H methylation, was observed.
 | ||
| Scheme 60 Aryl phosphonium salts as cross-coupling handles for C–H methylation. | ||
C–H borylation chemistry to prepare versatile aryl-boron species has progressed rapidly in recent decades.15d,161 In two accounts, Hartwig exploited sequential Ir and Pd/Cu catalysis to methylate a variety of (hetero)arenes via intermediary aryl-Bpin species (Scheme 61A and B).162 These tandem operations could be carried out as one-pot processes, requiring only a simple solvent swap between reactions. Furthermore, they showcased the selectivity and efficiency of the well-established iridium-catalysed C–H borylation of arenes, including on drug-like scaffolds. The site selectivity of the Ir catalyst for borylation tended to be dominated by steric effects for carboarenes, in contrast to heteroarenes where electronic effects had the greatest influence. Methyl iodide proved essential as the reactive methyl source in both the Pd and Cu cross couplings, being elegantly generated in situ from PO(OMe)3 in the latter of the two methods.
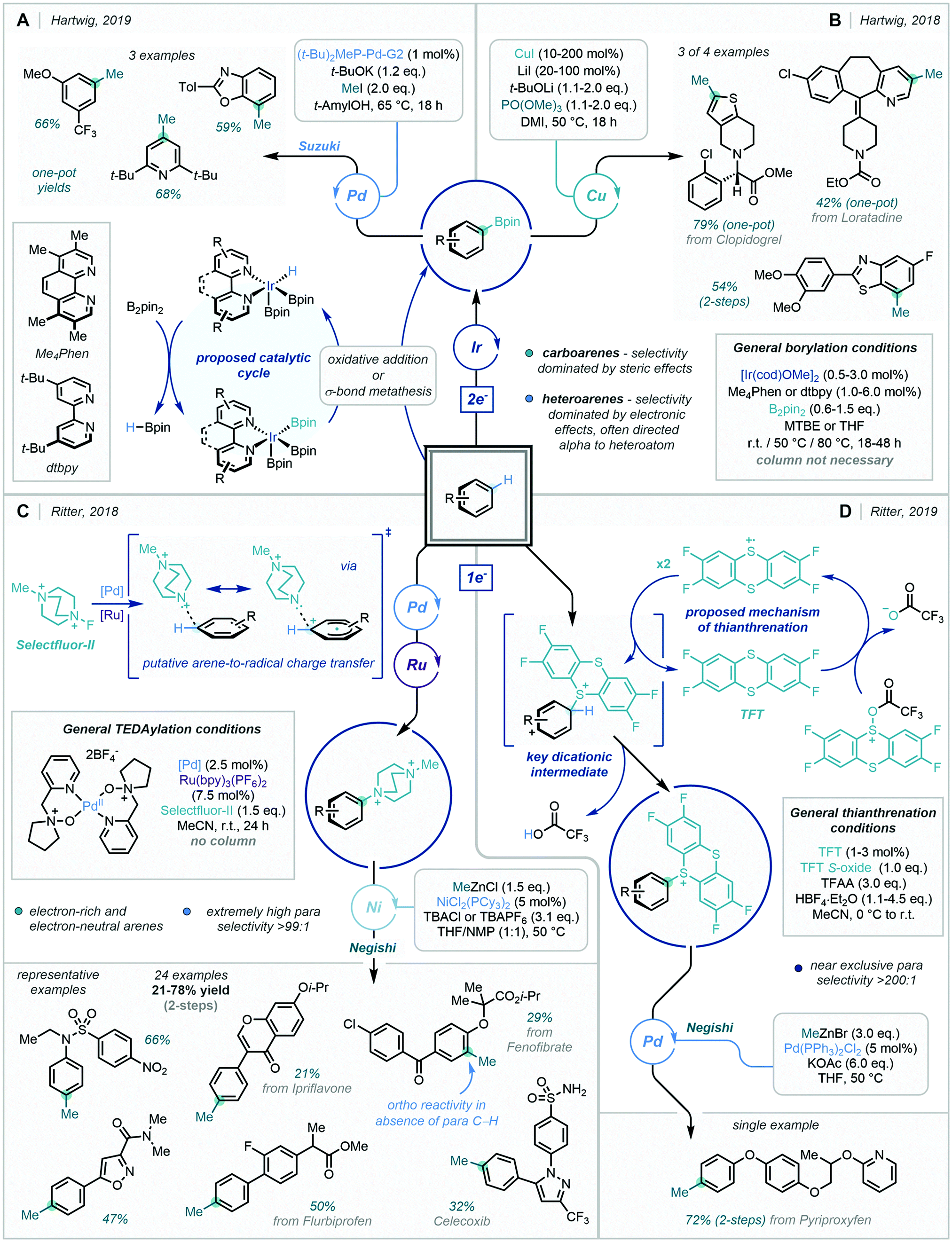 | ||
| Scheme 61 (A + B) Methylation via C–H Borylation. (C) Methylation via C–H TEDAylation (D). Methylation via C–H Thianthrenation. | ||
Positional selectivity in the innate C–H functionalisation of arenes has remained one of the greatest challenges in the field. Inherent limits exist within sterically controlled methodologies, owing to both the limited range of the effect and similar spatial volumes of many functional groups. Subtle positional disparities in electronic structure extend across the entire arene scaffold, and subsequently, reactivity contingent on electronic directing effects has the potential to confer site selectivity between distal C–H bonds. This principle has been impressively harnessed by Ritter, demonstrating charge-transfer directed radical aromatic substitution as a means of para-selective C–H functionalisation (Scheme 61C).17j,163 The highly electrophilic [triethylenediamine]2+ (TEDA2+) aminium radical generated from Selectfluor-II, was found to forge C–N bonds selectively at the para-position of numerous complex carboarenes and biologically active substrates with near-absolute selectivity. Arene-to-radical charge transfer in the transition state for radical addition was postulated as the primary reason for high positional selectivity, with substitution occurring at the site from which charge transfer is the greatest. Notably, the authors were previously able to use Fukui indices as a function to predict the site of C–H functionalisation.164 The dicationic nature of the aryl–TEDA complex also allows for silica-free purification of the intermediate, prior to methylation via a Ni-catalysed Negishi reaction. Superstoichiometric TBACl or TBAPF6 were found to be critical for enabling transmetalation of the methyl group. Applications of the protocol for gram-scale reactivity and the late stage methylation of pharmaceuticals further highlight the power of this two-step approach to C–H methylation.
Understanding the utility of strongly electrophilic radical cations for precise C–H functionalisation, Ritter developed C–H thianthrenation as a means of accessing diverse downstream reactivity (Scheme 61D).17q Akin to the C–H TEDAylation, near absolute site selectivity was attained on formation of the C–S bond to the thianthrenium handle. Electronic directing effects were found to dominate steric factors for site differentiation in the radical C–S bond formation. A plethora of substrates ranging from simple monosubstituted arenes, to complex natural products such as strychnine, were found to participate in the reaction delivering the bench stable thianthrenium salts. Among the myriad of downstream transformations enabled by the thianthrenium handle, methylation was shown to be possible through Ni-catalysed Negishi coupling with MeZnCl. The extreme applicability and efficacy of this transformation on both small molecular building blocks and pharmaceuticals, marks a significant milestone in arene C–H functionalisation.
4. C(sp3)–H methylation
As a consequence of the growing desire to explore 3D chemical space further, sp3-rich structures with multiple positional vectors are of ever-increasing importance in drug discovery programs.100 Spanning diverse structures such as saturated heterocyclic frameworks, peptidomimetics, steroidal and glycosyl fragments, these sp3-rich scaffolds readily appear within a host of medicinally relevant molecules. It has been demonstrated that increased C(sp3) character correlates with higher clinical success, through suppressed binding promiscuity (improved affinity into specific 3D binding sites) and greater stability of compounds under physiological conditions (decreased metabolite formation via cytochrome oxidation).165Accordingly, the desire for methodologies that enable the selective C(sp3)–H methylation of such substrates is of increasing significance. Although this transformation is synthetically challenging, several major advances in the field have been reported in recent years. The following section endeavours to cover these recent advances, and their applications in the elaboration of saturated architectures.
4.1 Directed C(sp3)–H methylation
The selective pin-point activation of C(sp3)–H bonds has proved more synthetically challenging than analogous C(sp2)–H centres, and accordingly functionalisation of saturated C–H bonds has relied heavily on the use of DGs. This inherent difficulty stems from the rotatable nature of sp3 hybridized bonds and the formation of weaker M–C(sp3) bonds, rendering C–H activation events more energetically challenging.166 This is often coupled with deleterious potential side reactions such as β-hydride elimination and undesired alkylations.167 Lewis basic directing groups have been found to not only reduce the entropy change associated with C–H insertion in systems of high free rotation, such as sp3-rich structures, but also aid in controlling the site selectivity of the C–H activation event.The prominent role of nitrogen-based directing groups in the site-selective C–H activation of sp2 centres has been mirrored in their application in C(sp3)–H methylation (see Section 2). An early report of C(sp3)–H methylation – disclosed by Yu in 2006 as part of a wider C–H methylation project – exploited a pendant pyridyl directing group to dictate site selective methylation at the β-position to the pyridyl moiety (Scheme 62A).28 Good yields were achieved for the C–H methylation of terminal methyl groups, with di-methylation occurring in some instances. The methodology was also applicable to the C(sp3)–H methylation of CH2 methylene units, albeit with reduced yields. A 5-membered palladacycle, formed via a pyridyl-directed C–H activation by Pd, was proposed to be key in achieving the site selectivity observed. Methylboroxine and methylboronic acid were both found to act as efficient transmetalating agents, and the authors suggested that the addition of benzoquinone was key for the reductive elimination step. This work signified a major step towards developing C–H methylation methodologies for medicinally relevant heteroarene scaffolds, and accordingly has acted as a springboard for further reaction development.
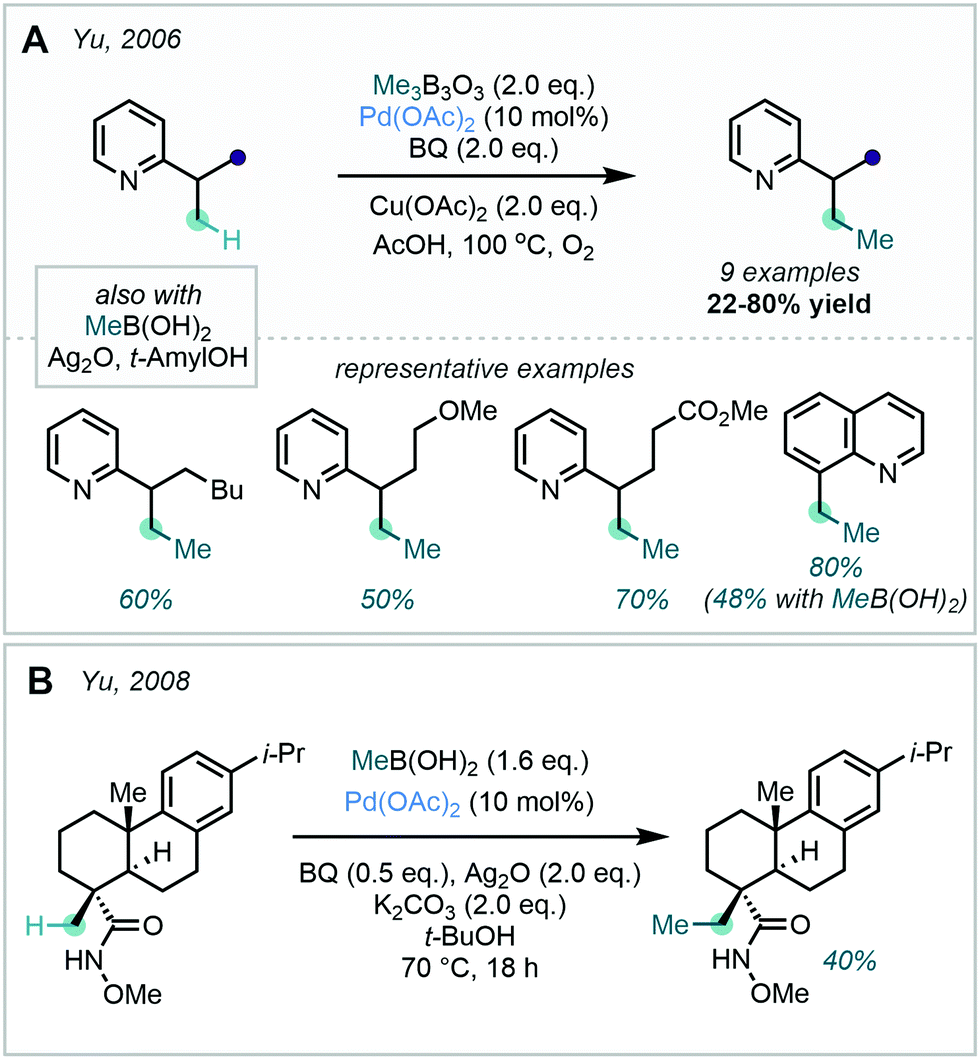 | ||
| Scheme 62 Early reports of Pd-catalysed C(sp3)–H methylation. | ||
In 2008, Yu reported a related system for the C(sp3)–H methylation of dehydroabietic acid derivatives (Scheme 62B).168 The use of O-methyl hydroxamic acids was demonstrated as an effective directing group in C–H methylation exclusively at the methyl appendage β to the DG. Methylation constituted a standalone example in a wider C–H alkylation study and occurred in moderate yield.
The selective activation and functionalisation of C(sp3)–H bonds saw further development when Chen described the use of a PA directing group in the C–H methylation of a diverse family of amine derivatives (Scheme 63).169 The methylation protocol displayed good to excellent selectivity for the functionalisation of unactivated C(sp3)–H bonds γ to the directing group. Interestingly, in conformationally rigid frameworks, the construction of higher order alkyl chains through sequential methylations was observed. This insight was showcased in a one-pot triple methylation of a norbornane derived substrate to install an isopropyl moiety. Following methylation, the PA directing group could be readily hydrolysed to the corresponding free amine under acidic conditions.
 | ||
| Scheme 63 Pd-Catalysed PA-directed C(sp3)–H methylation with methyl iodide. | ||
The 8-AQ directing group for C–H activation – which has been highlighted in the above sections – has proved equally powerful in saturated systems. In 2013, Shi170 and Chen167 contemporaneously reported the C(sp3)–H methylation of amino acid derivatives appended with the 8-AQ directing group, using methyl iodide as the methyl source (Scheme 64A), where cyclometalation proceeded via a CMD mechanism. Subsequent oxidative addition and reductive elimination forged the new carbon–carbon bond, and finally protonolysis released the C–H alkylated product, in turn regenerating the catalyst. It was hypothesised that Ag+ played crucial roles in the reaction mechanism: firstly, in facilitating reductive elimination and catalyst turnover by acting as an iodide scavenger, and secondly in aiding the oxidative addition through increasing the electrophilicity of the alkylating agent in an SN2-type oxidative addition mechanism. The acidic additive, dibenzyl phosphate, employed by both groups was shown to be a key factor in improving reaction efficiency, and this – in line with earlier mechanistic proposals from Chen (Scheme 63) – was attributed to the acid acting as a solid-to-solution phase transfer catalyst for Ag+. Chen also reported modest to excellent diastereoselectivity with respect to the methylation of secondary β-C(sp3)–H bonds (3–15:1 dr), and furthermore illustrated that both CH3 and CD3 groups could be incorporated with this method, allowing for access to isotope labelled β-methylated α-amino acids (Scheme 64B).
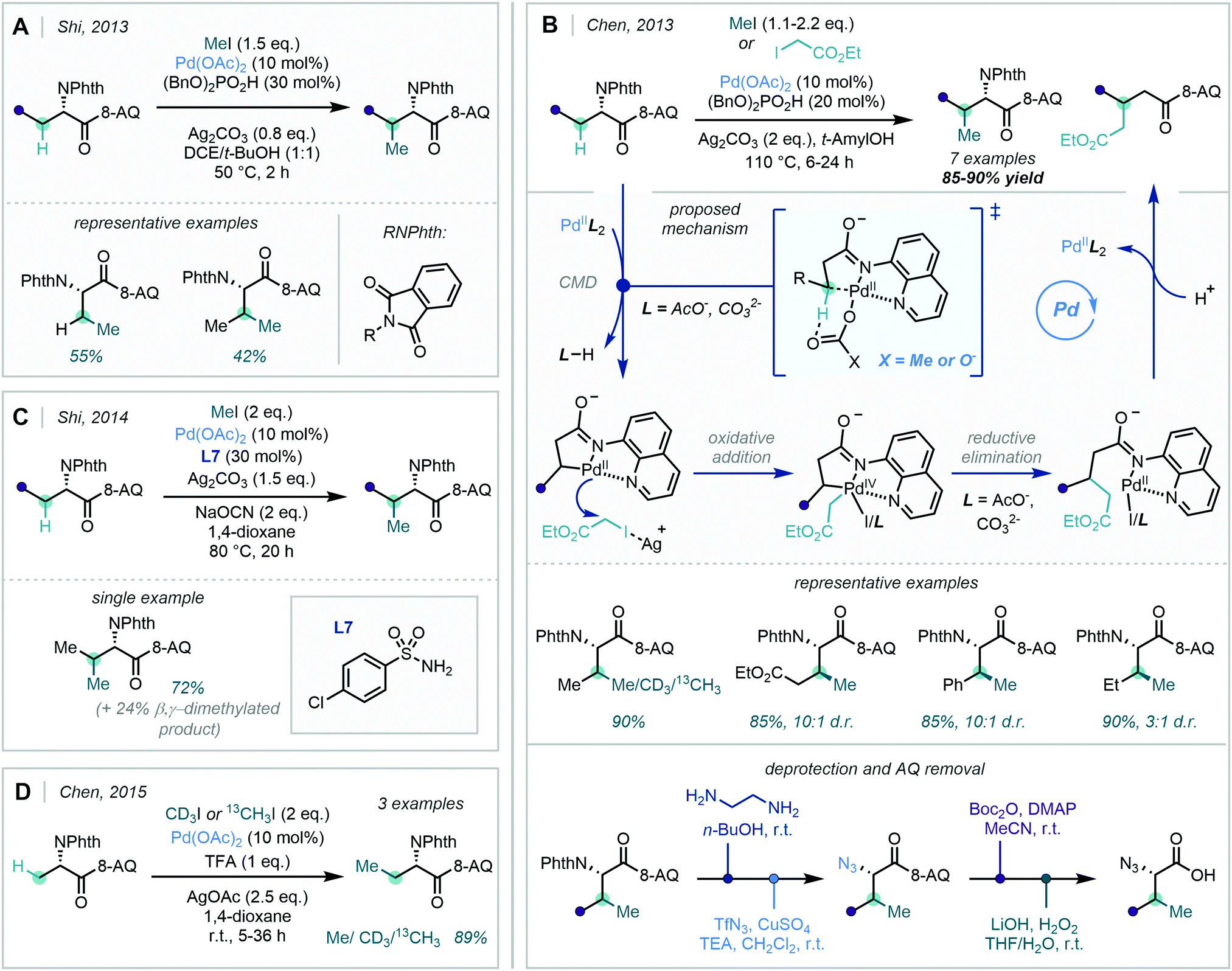 | ||
| Scheme 64 Developments in the C(sp3)–H methylation of amino acid derivatives using an 8-AQ directing group. | ||
Further work by Shi in 2014, introduced the use of a sulfonamide ligand L7 to promote the C–H alkylation amino acid derivatives (Scheme 64C).171 The addition of the ligand negated the use of an external acid additive. In 2015, Chen disclosed a complementary procedure that could be carried out at room temperature, opting to retain the acidic additive – in this case trifluoroacetic acid (Scheme 64D).172 These conditions aided in improving this method's applicability without detriment to the high yields (∼90%), showcasing the robustness of this C–H methylation protocol. The improvements also enabled the mono-methylation of primary C(sp3)–H bonds, a feature which was not demonstrated in their first report.
The application of finely-tuned secondary amide directing groups, discussed in Section 2 (Scheme 5), was demonstrated by Yu to be equally capable in the directed C–H methylation of sp3 centres (Scheme 65).34 Notably, N-heteroaromatic ligands were found to be crucial to reaction efficiency, where an acridine derivative was identified as optimal amongst an array of potential ligands.
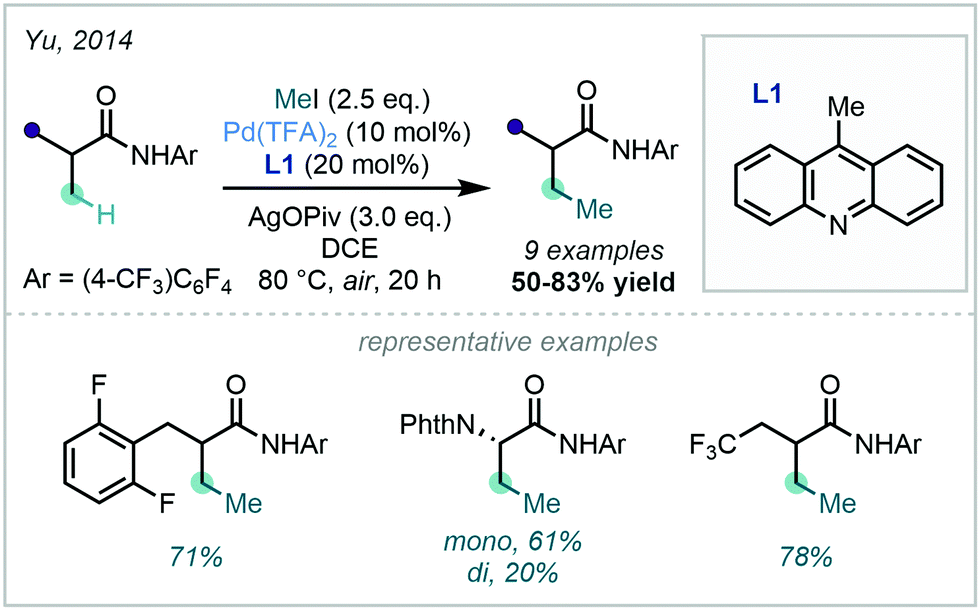 | ||
| Scheme 65 Ligand-promoted C(sp3)–H methylation with secondary amide directing groups. | ||
Of the methods discussed thus far, all have exploited palladium catalysis to enact C–H activation and subsequent functionalisation. However, in 2020, Sharma described the directed C(sp3)–H methylation of 8-methylquinoline structures using a Cp*RhIII catalyst system.173 This method also allowed for the use of MeBF3K as a nucleophilic methyl source. While the yields of this methylation protocol remained modest, the demonstrated quinoline scope was notably broad (Scheme 66). Mechanistically, the quinoline was proposed to direct C–H activation, resulting in the formation of a five-membered rhodacycle; this could subsequently undergo transmetalation, followed by reductive elimination to assemble the desired mono-methylated product.
 | ||
| Scheme 66 Directed C(sp3)–H methylation of 8-methylquinolines. | ||
Nitrogen-based directing groups have been shown to dominate the directed C–H methylation of sp3 centres, primarily due to the strong binding of the N-centred lone pair to transition metal species. Despite this, in 2020, Wang & Yang reported the use of a thioamide directing group to enable the C–H arylation of 3-pyrroline derivatives using boronic acid nucleophiles (Scheme 67).174 One example of methylation was exemplified, occurring in moderate yield. Following, downstream post-synthetic modifications, the protocol provided access to α-arylated or methylated pyrrolidines, a motif that appears in a variety of bioactive molecules.
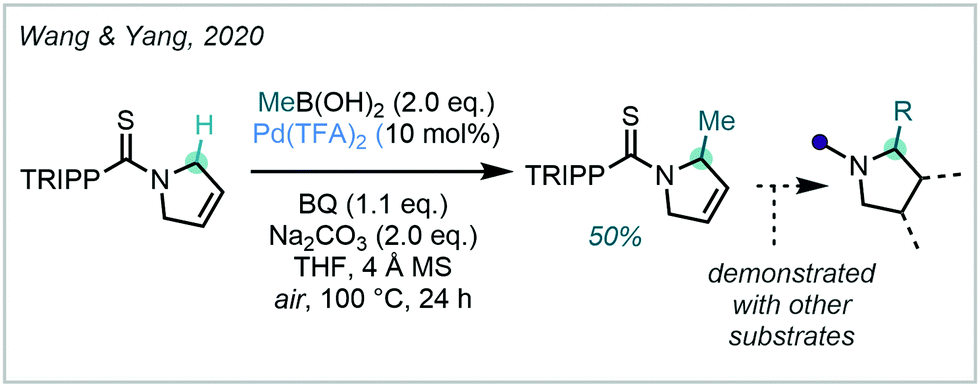 | ||
| Scheme 67 Thioamide-directed C(sp3)–H methylation of 3-pyrrolines. TRIPP = 2,4,6-triisopropylphenyl. | ||
4.2 Oxidative C(sp3)–H methylation with a functional group at the α-position
The conversion of a C–H to C–Me has been shown to elicit some of the most profound pharmacological effects when the methyl group is installed adjacent to a heteroatom in saturated heterocycles.1,2,175 Accordingly, the direct C(sp3)–H methylation of saturated C(sp3)–H bonds α to a heteroatom in cyclic and acyclic systems – without the necessity of a proximal directing group – is of great value to the synthetic community. Moreover, the development of reaction conditions that enable the late-stage C–H methylation of complex molecules would be of particularly high value to medicinal chemistry programmes, obviating the need for de novo introduction of the methyl group. Nevertheless, such transformations remain a challenging task owing to the low acidity of α-protons and numerous competing C–H oxidation pathways. Despite these challenges, the oxidative C(sp3)–H methylation at sites with α functionality has garnered recent attention and the notable advances in this approach will be discussed herein.In 2017, MacMillan disclosed a pioneering development in C–H alkylation chemistry in the polarity-match-based selective α-C(sp3)–H alkylation of various cyclic and acyclic amines, thiols and ethers, including two examples of C–H methylation on N-protected pyrrolidines.176 The authors combined photoredox, polarity-matched HAT, and nickel catalytic cycles, where the high positional selectivity is determined via polarity-matched HAT (Scheme 68). Oxidation of quinuclidine by excited Ir(III) generates an electrophilic nitrogen radical which then abstracts a hydrogen from the most electron rich (hydridic) C–H bond of the substrate, forming a nucleophilic α-aminoalkyl radical in this case. The Ir(II) species formed then reduces a Ni(I) intermediate to furnish a Ni(0) complex which can productively combine with an α-aminoalkyl radical to form a Ni(I)–alkyl complex. Subsequent oxidative addition to the alkylating agent forms a Ni(III) species, with reductive elimination delivering the α-alkylated heterocycle and regenerating Ni(I). With this approach, exclusive alkylation at the most electron rich C–H site of the substrate was achieved, notably even in the presence of activated benzylic C–H bonds.
 | ||
| Scheme 68 Polarity-match-based approach for selective C(sp3)–H alkylation; ayield calculated from a calibrated GC assay. | ||
Interestingly, for the C–H methylation examples, a modification to the reaction conditions was required, with MeBr prepared in situ from MeOTs and CsBr and quinuclidine proving to be detrimental to reaction efficiency. The authors postulated that this was due to undesired reactivity of the highly electrophilic MeOTs with quinuclidine. Without quinuclidine present, it was proposed that the HAT cycle is performed via an analogous catalytic cycle with the bromide anion, bromide radical and HBr as HAT cycle components. A similar mechanism was previously described by MacMillan for photoredox-enabled C(sp3)–C(sp2) coupling from alkyl and aryl halide precursors.177
The presence of a heteroatom can also grant access to new avenues of reactivity, which can enable selective activation of otherwise unreactive C(sp3)–H bonds in heterocyclic systems. For tetrahydroisoquinolines (THIQs) and related fused heterocycles, commonly used in drug discovery programs, one of the α positions is further activated by an adjacent aromatic ring. As a result, single electron oxidation of the amine can selectively lead to an iminium ion at the benzylic position via an intermediary α-amino radical. Accordingly, several groups have reported conditions to convert THIQs and related structures to the corresponding iminium ions through oxidative methods.178 These reactive intermediates have then been shown to react with an array of nucleophiles such as cyanide, electron-rich aromatics and boronic acids, furnishing valuable C–H functionalised products.
In 2017 Cheng detailed an oxidative approach to perform C–H methylation on 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidines (THPPs) (Scheme 69A).179 From an initial oxidant screen, it was found that I2, Co and Cu salts failed to deliver any desired products, however the use of catalytic RuCl3 and NaIO4 as a co-oxidant in THF/MeOH generated the desired THPP-derived hemiaminal intermediates in excellent yield. These in situ-generated hemiaminals were then activated by BF3·OEt2 to form the reactive iminium ions, which upon trapping with Me2Zn yielded the methylated products. The one-pot procedure was applied to a number of heterocyclic systems, furnishing benzylic α-C–H methylated analogues.
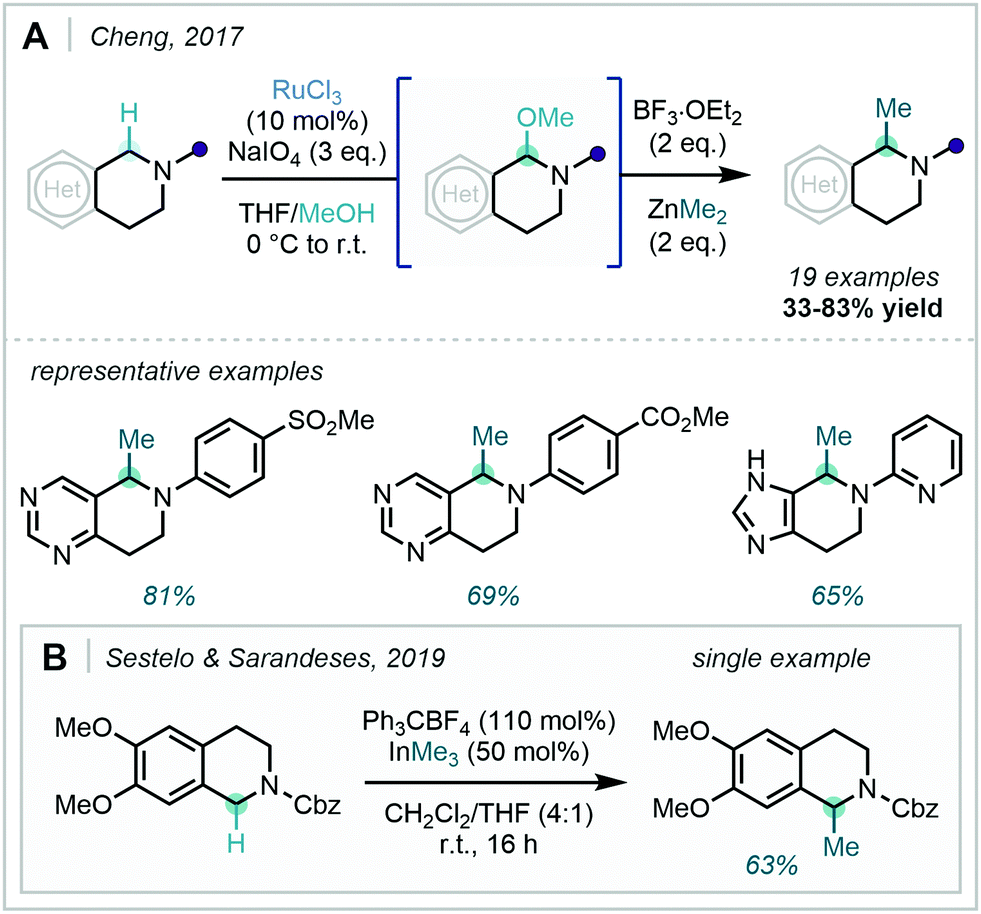 | ||
| Scheme 69 (A) α-C–H methylation of THPPs and related systems. (B) Trityl cation-mediated oxidative methylation of THIQs. | ||
In 2019, Sestelo & Sarandeses demonstrated a complementary oxidative approach employing organoindium compounds as mild reagents for the alkylation of electron-rich and electron-neutral THIQs (Scheme 69B).180 The oxidation was performed with triphenylcarbenium (trityl) salts via a putative hydride transfer mechanism to afford the analogous iminium ions. Amine oxidation took place exclusively at the benzylic position, furnishing transient iminium ions which were trapped by organoindium species in a one-pot procedure. Notably, the use of Me3In furnished the desired C–H methylated product in 63% yield. Electron-deficient THIQs showed no reactivity under the optimised conditions, accounted for by the deactivation of the benzylic position towards hydride transfer.
Although these methods enabled C–H methylation of C(sp3)–H bonds α to a nitrogen atom, they remain limited to benzylic systems. To widen the scope of this strategy, subtle modifications to the electronics of the amine, to facilitate more favourable hydride transfer, can allow application in non-benzylic systems.
This approach was exploited by Seidel in 2019 to facilitate the α-functionalisation of cyclic secondary amines, enabled by organolithium deprotonation of the N–H bond (Scheme 70).181 Transient imines were generated via selective hydride transfer from the α-position of the lithium amide to a carefully-selected hydride acceptor, through a proposed 6-membered transition state organised by lithium. These intermediary imines, in combination with a Lewis acid, were subsequently intercepted by a range of organolithium or Grignard reagents, in turn generating α-functionalised secondary amines in a one-pot procedure. Of note, an α-methylated 13-membered azacycle was obtained in 68% yield using MeMgBr and TMSOTf as the Lewis acid activator.
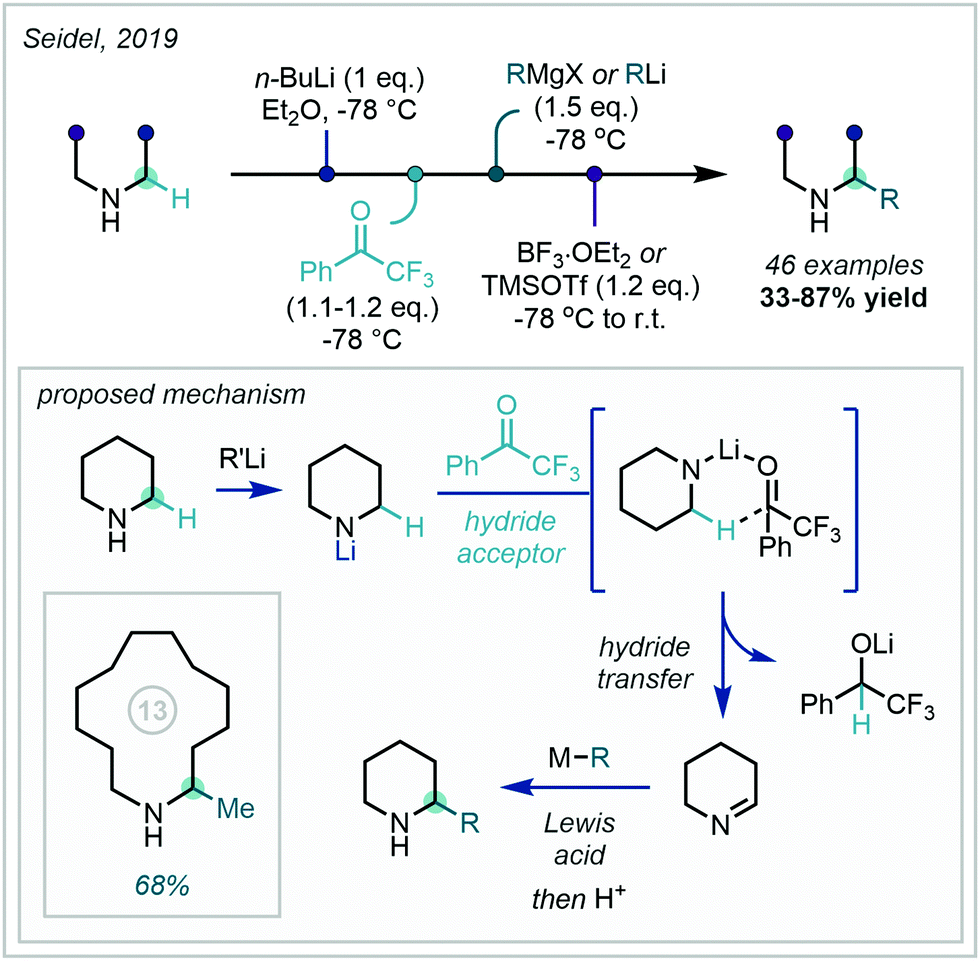 | ||
| Scheme 70 α-Functionalisation of cyclic secondary amines via hydride transfer. | ||
More recently, Seidel elegantly extended the methodology to the β-functionalisation of cyclic secondary amines (Scheme 71).182 In this study, the imines, formed in situ via hydride transfer, were further deprotonated at the β-position, generating key endocyclic 1-azaallyl anions. A rigorous optimisation of the trapping of this intermediate with electrophilic alkyl halides, led to the development of conditions for selective β-C-alkylation over N-alkylation. Subsequent trapping of the resulting imines with suitable nucleophiles, could then furnish α,β-disubstituted amines in a one-pot procedure, often with good-to-excellent diastereoselectivity. This procedure was applied to the 1-azaallyl anion derived from piperidine, using methyl iodide followed by 4-chlorophenyl lithium to furnish 63% of the desired methylated product.
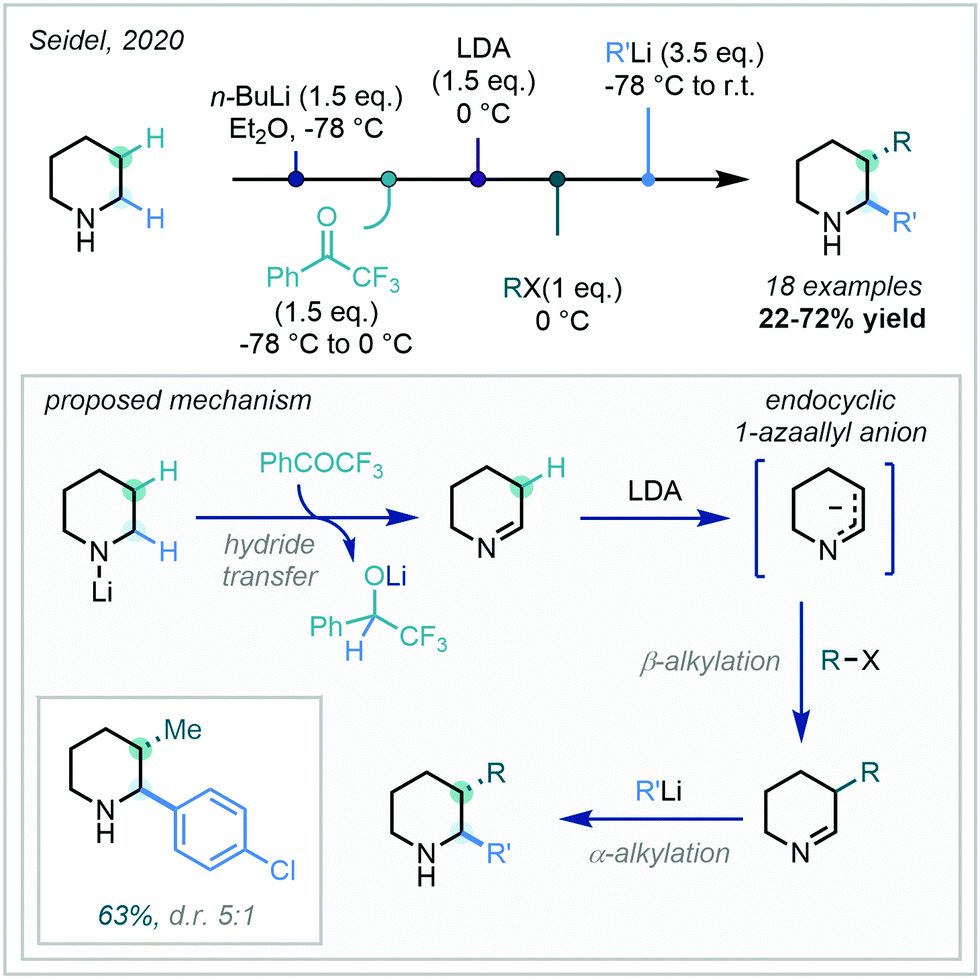 | ||
| Scheme 71 Tandem one-pot α,β-alkylation strategy on saturated azacycles. | ||
The C(sp3)–H methylation techniques described so far give access to substituted secondary and tertiary amines. The α-C–H methylation of primary unprotected amines, however, presents unique challenges, requiring an alternative approach. Less-hindered primary amines are typically more nucleophilic and are hence incompatible with the aforementioned hydride transfer methodologies, however, harnessing this inherent nucleophilicity, Dixon reported the use of reactive imines en route to the synthesis of primary α-tertiary amines.183 Inspired by the enzymatic process in which metalloenzymes selectively oxidise primary amines to aldehydes via a quinone-derived imine (Scheme 72),184 abundant α,α-disubstituted primary amines were condensed with quinones, generating Schiff base intermediates. These species underwent a [1,5]-H-shift, generating reactive ketimine intermediates which were then intercepted with organomagnesium and organolithium reagents, or cyanide nucleophiles. After oxidative hydrolytic work-up, the hydroxyarene was detached, furnishing α-tertiary primary amines in a three step one-pot procedure and up to 95% yield. Trapping the ketimine intermediates with MeLi furnished the desired product in excellent yield, demonstrating the applicability of the method for α-C–H methylation of α,α-disubstituted primary amines.
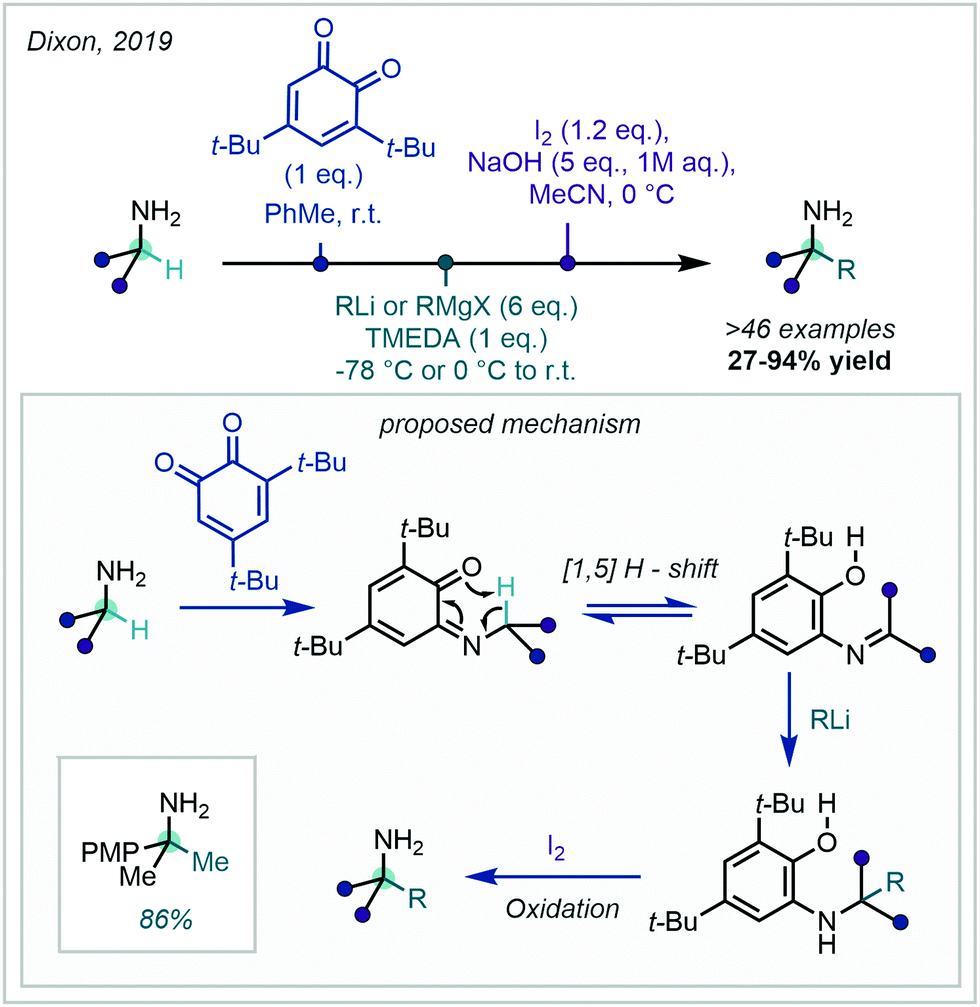 | ||
| Scheme 72 Primary α-tertiary amine synthesis via bioinspired oxidation. (PMP = p-methoxyphenyl). | ||
Despite the significant progress in α-amine C(sp3)–H alkylation achieved above, the application of these methods to C–H methylation in particular has remained limited. However, in 2020, White developed a general platform for C(sp3)–H methylation, methylating at the α-heteroatom site of numerous cyclic and acyclic systems, with the late-stage functionalisation of complex molecules also being exemplified. Similar to the approaches of Seidel and Dixon, the strategy centred on the oxidation of a substrate to a corresponding electrophilic species, which could be intercepted by a nucleophilic methyl source (Scheme 73A).175 Oxidation was performed via α-C–H hydroxylation to generate an intermediate hemiaminal or hemiacetal. This hydroxylation was enabled using hydrogen peroxide and a manganese catalyst [Mn(CF3PDP)(MeCN)2](SbF6)2, previously reported by White as a highly selective catalyst for C(sp3)–H methylene hydroxylation.185 The oxidative conditions do not involve the use of strong nucleophilic bases or the generation of an alkoxide by-product, making the substrate scope markedly tolerant of common functionalities. The resultant hemiaminals or hemiacetals could then be ionized by Lewis acids such as BF3·OEt2, diethylaminosulfur trifluoride (DAST) or in some cases by esterification with TFAA and subsequent activation with TFA, to furnish the reactive iminium or oxocarbenium intermediates. The use of AlMe3 proved to be essential in delivering C–H methylated products with high functional group tolerance.
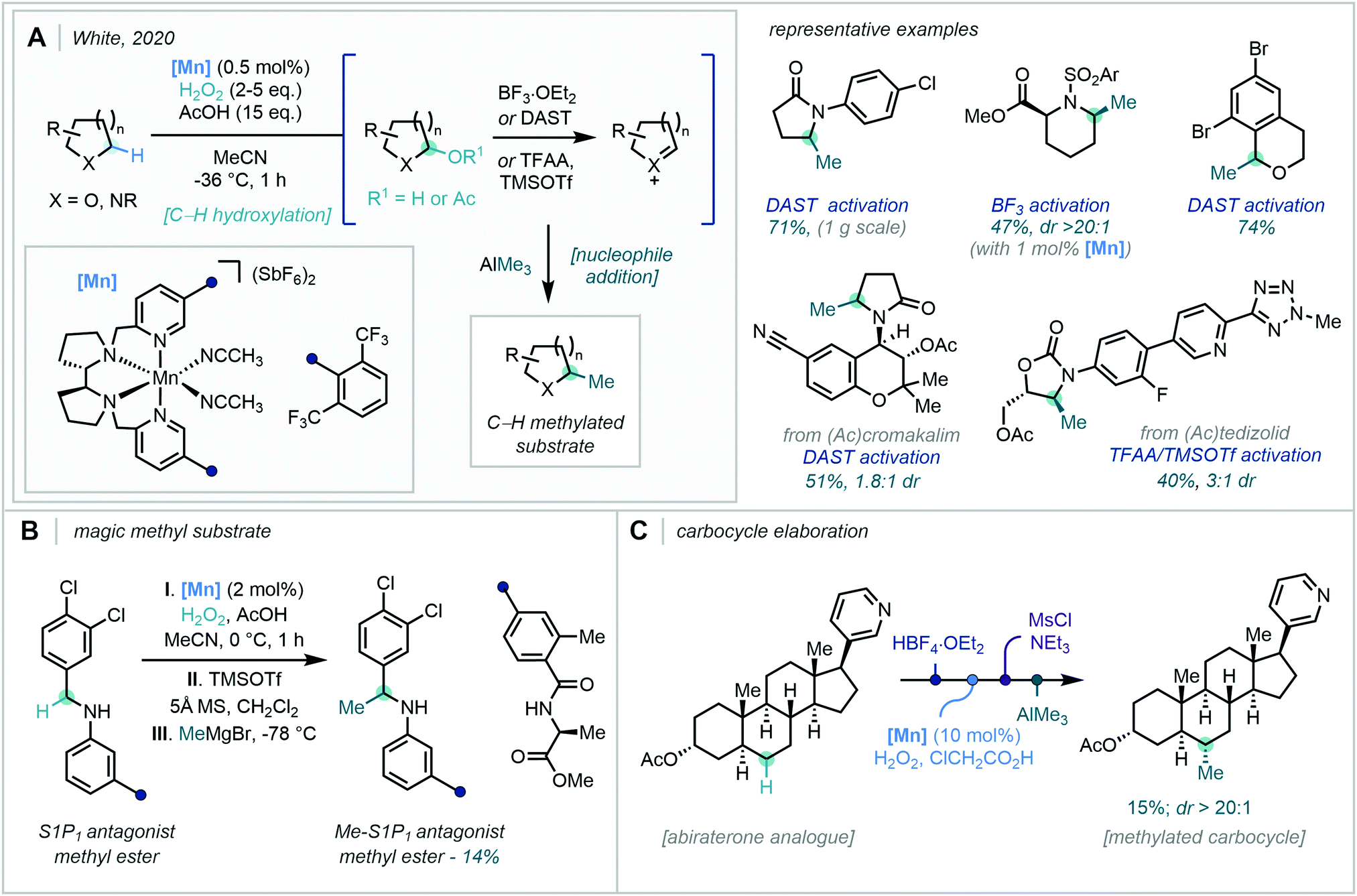 | ||
| Scheme 73 (A) One-pot C–H methylation via tandem hydroxylation-activation-methylation. (B) Access to a Magic Methyl substrate. (C) Remote C–H methylation. | ||
The broad scope covers a variety of complex saturated N- and O-heterocyclic scaffolds, with varying ring sizes, epimerisable stereocenters, and nucleophilic functional groups all being well-tolerated. In the case of unsymmetrical saturated heterocycles, the least hindered α-position was found to undergo C–H methylation, and high regioselectivity was proposed to arise from catalyst control in the C–H hydroxylation step. Notably, methylation of piperidines – the most common saturated N-heterocycle in small-molecule drugs100 – was achieved with high diastereoselectivity owing to a rigid half-chair conformation of the iminium ion intermediate. Furthermore, this methodology was found to be extremely applicable to late-stage functionalisation, as demonstrated by the successful methylation of numerous complex bioactive molecules, natural products and drugs such as, acetylated cromakalim and acetylated tedizolid alongside many other drug precursors and derivatives. The utility of this method in medicinal chemistry was further showcased by synthesis of a “Magic Methyl” substrate, Me-S1P1 antagonist methyl ester, which possessed a remarkable 2135-fold increase in potency when compared to the non-methylated analogue (Scheme 73B). This transformation required some modification to the standard procedure, where the transient imine was activated and methylated with TMSOTf and MeMgBr respectively, to account for the lower reactivity of imines compared to iminium ions. Moreover, it was demonstrated that oxidative C–H methylation could be extended beyond methylenic sites bound to heteroatoms, with the transformation being successfully applied to unactivated carbocycles (Scheme 73C). By increasing the catalyst loading, C(sp3)–H bond hydroxylation of an unactivated methylene on an abiraterone analogue, a prostate cancer drug, was achieved. Subsequent activation-methylation of the intermediate alcohol with MsCl and AlMe3 furnished a Me-abiraterone analogue as a single regioisomer. The unique selectivity in this transformation is determined in the C–H oxidation step and arises from multiple factors including the strong inductive electron-withdrawing effect of the protonated pyridine on the adjacent 5- and 6-membered rings. The same site selectivity for the remote C–H oxidation of abiraterone was also observed previously with a similar catalytic system.186
Heteroatoms are not the only functionality shown to enable selective α-C(sp3)–H methylation. In 2019, Tambar exploited the allylic ene reaction, to perform copper-catalysed allylic C–H alkylation (Scheme 74).187 In this work, sulfur diimide was used both as an electrophilic oxidant and as a leaving group in the subsequent copper-catalysed alkylation step, where regioselectivity was controlled by a sterically demanding phosphine ligand. The proposed reaction mechanism begins with an ene reaction between the sulphur diimide oxidant and the terminal allyl group, generating adduct (I), which is then activated by a Grignard reagent to furnish sulfimine (II). Following this, the sulfimine serves as an allylic leaving group, facilitating oxidative addition of the CuI complex bearing the alkyl coupling partner. This step is regiodetermining and results in the formation of CuIII adduct (IV) via TS (III), with the sterically demanding ligand located on the less substituted side of the π-allyl system. Bulky ligands t-BuXPhos or (R)-SITCP were found to be necessary to promote formation of organocopper intermediate (IV), which subsequently undergoes reductive elimination to yield the branched alkylated product. Alongside the generation of racemic branched allyl products, stereoinduction was also shown to be possible by employing (R)-SITCP as the ligand. Generally, good levels of e.e. (up to 88%) were achieved, however enantioinduction proved to be poor in the case of C–H methylation.
 | ||
| Scheme 74 Allylic one-pot C(sp3)–H methylation enabled by a sulfur diimide ene reaction and subsequent Cu-catalysed allylic alkylation. | ||
Almost all of the techniques discussed above rely on the presence of functionality at the α- or β-position to facilitate C–H oxidation and subsequent methylation. Barring select cases (Scheme 73C), the innate C–H methylation of unactivated C(sp3)–H bonds is much less developed owing to their innately low reactivity.
In 2020, Mi & Li disclosed a novel approach to enable methylation of unactivated C–H bonds by employing highly reactive methylene carbenes (Scheme 75). The carbenes were formed by photochemical activation of MeOH on a specifically designed p-type doped GaN nanowire (NW) deposited with Cu nanoparticles.188 Unfunctionalised hydrocarbon feedstock chemicals were methylated in modest yield for both sp2 and sp3 systems. Interestingly, with unsymmetrical systems there was an observed preference for insertion of the carbene into the C–H bond bound to the most substituted carbon atom. This selectivity is unusual for free methylene carbenes, which are highly reactive and are thus typically unselective in C–H insertions.189 The observed selectivity was proposed to be due to a strong adsorption of the methylene carbenes generated on the GaN surface.190 This interaction is believed to influence their reactivity, rendering them more selective for the weaker C–H bonds at the more substituted carbon centres. Indeed, when 2-methylpropane was subjected to the optimised reaction conditions at higher temperatures – suggested to promote desorption of methyl carbenes from the catalyst surface – the selectivity was reversed in favour of 1° C–H insertion in line with statistical predictions.
 | ||
| Scheme 75 GaN-mediated methyl carbene formation for the C–H methylation of feedstock hydrocarbons. | ||
To date, there remains vast chemical space to be explored for developing the C–H methylation of unactivated C(sp3)–H bonds. The challenges associated with this powerful and ambitious disconnection will no doubt be grappled with for years to come.
5. Enantioselective C–H methylation
Methodology capable of facilitating both C–H alkylation and stereoselectivity has long been one of the greatest outstanding challenges in organic synthesis. Progress within the subset of enantioselective C–H methylation remains in its embryonic stage, with few examples detailed to date.To this end, promise has been displayed in utilising directed transition metal catalysis coupled with chiral mediators to enable enantioselective C–H methylation. In 2016, Yu disclosed a lone example showcasing that the catalytic enantioselective methylation of saturated azacycles was possible, using BINOL-derived chiral phosphoric acid co-catalysts to achieve stereoinduction (Scheme 76A).191 The disconnection is akin to that of sparteine-mediated lithiation-methylation, an approach which remains largely limited to pyrrolidines, piperidines and piperazines with varying levels of enantioselectivity.192 The sterically congested TRIPP-substituted thioamide proved to be essential for both directing the ligated Pd species to the site of reactivity, and for achieving good enantioinduction. A strict exclusion of achiral anions in the Pd source was also hypothesised to remove any non-stereoselective background reactivity, with Pd2(dba)3 producing the highest levels of enantioselectivity. In a later account, Yu detailed another single example of asymmetric methylation, exemplifying meta C–H methylation as a means of kinetically resolving a naphthylalanine derivative (Scheme 76B).193 The impressive methodology relied on a chiral norbornene mediator transiently relaying an initial ortho C–H activation to the meta position, enabling the remote C–H methylation of one of the homobenzylamine enantiomers with marked enantiofacial selectivity (see 2.3 Catellani-type strategies for C(sp2)–H methylation).
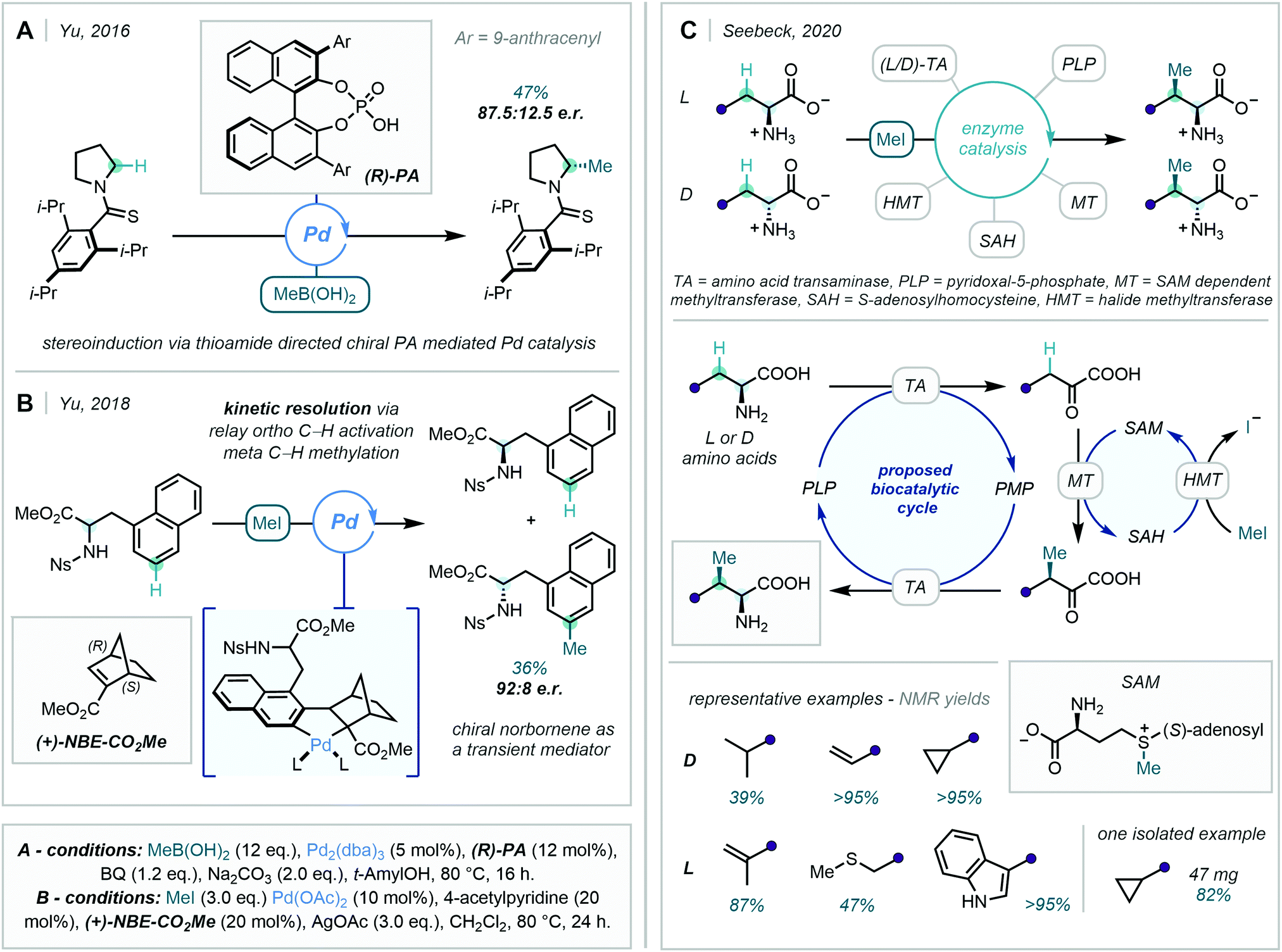 | ||
| Scheme 76 Emerging transition metal (A + B) and enzyme (C) mediated strategies for enantioselective methylation. | ||
Nature has developed highly evolved enzymatic cascades capable of forging complex natural products with near total stereochemical control. Due to both the myriad of biological species involved and the reliance on cellular metabolism to turn over reactivity, translation of in vivo reactivity to synthetically useful in vitro reactivity remains challenging. In 2020, Seebeck managed to assemble a synthetic replica of nature's methylation framework, allowing for the synthesis of β-methyl-α-amino acids, a prevalent motif in natural products (Scheme 76C).194
Functioning through a combination of bacterially grown enzymes and co-substrates to facilitate artificial metabolism, numerous L and D β-methylated amino acids were produced on small scale with high diastereoselectivity. The mechanistic sequence began with amino acid oxidation to the achiral α-keto acid by a transaminase (TA), which was then asymmetrically methylated by a SAM-dependent methyl transferase (MT). These α-keto acids are prone to racemisation but crucially, due to the lack of additional amine acceptors in the system, the steady state concentration of the α-keto acids was kept low, limited by the initial concentration of pyridoxal-5-phosphate (PLP). The β-methyl-α-keto acids were finally re-aminated by the transaminase, restoring the initial stereochemistry of the amino group. It was also shown that by switching from an L-TA to a D-TA, achiral α-keto acids could also be produced from D-amino acids, thus enabling efficient β-methylation of D-amino acids. A halide methyl transferase (HMT) was utilised to generate in situ S-adenosyl methionine (SAM), by combination of S-adenosyl-homocysteine (SAH) and methyl iodide, to feed the reactive methyl source into the biocatalytic cascade. Although restricted to small scale reactivity, the broad scope of β-methyl-α-amino acids generated clearly demonstrates the promise of cascade biocatalysis for the enantioselective C–H methylation of biologically relevant building blocks.
6. Conclusions
Whether as part of a wider C–H alkylation platform or in bespoke C–H methylation studies, the interest and research effort invested in C–H methylation methodology has grown rapidly in recent times. This is in part due to the expanding appreciation of the “Magic Methyl” effect in medicinal chemistry programmes, where the exchange of C–H to C–Me has been demonstrated to have profound effects on potency and other pharmacological properties when installed at a strategic position.In line with the remarkable recent advances in directed C–H functionalisation, the directed C–H methylation of both sp2 and sp3 centres has witnessed profound developments covering a panoply of bespoke directing groups and methyl sources, high power catalytic systems, and, more recently, the application of commonplace functional groups as the directing moiety. Complementary to this, a comprehensive suite of methods capable of harnessing the innate reactivity of molecules have arisen for the direct C–H methylation of sp2 centres. While one-electron approaches have advanced Minisci-type reactivity, largely fuelled by modern radical generation techniques, two-electron processes have exploited and developed a notably diverse set of methylating reagents and – often – highly varied chemistries. These approaches have found marked success when constituting two-step tandem processes, employing methyl surrogates or appropriate functional handles. Furthermore, given the numerous examples in which a pronounced “Magic Methyl” effect has been observed at the α-position of a heteroatom in a saturated ring system, the recent developments in oxidative α-C–H methylation have particularly powerful implications in drug development.
Finally, despite the pioneering reports discussed above, the enantioselective C–H methylation of both C(sp2)–H and C(sp3)–H bonds remains conspicuously underdeveloped. For direct applications in medicinal chemistry programmes, which are endeavouring to expand further into 3D chemical space, the ability to selectively install a methyl group with enantiocontrol is of great importance. To this end, there remains hope that the recent growth in methodology for C–H methylation will in turn foster new approaches towards enantioselective variants.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are supported by the Centre for Doctoral Training in Synthesis for Biology and Medicine, generously supported by GlaxoSmithKline, MSD, Syngenta and Vertex. D. A. also thanks the Clarendon Scholarship, M. C. thanks the Oxford-RE Jones Scholarship, H. H. thanks the Oxford Radcliffe Scholarship, C. Y. X. P. also thanks the A*STAR Singapore, B. S. thanks the Oxford-Leon E & Iris L Beghian Scholarship, A. B. thanks the Derek Calam Scholarship and Z. H. L. thanks the Oxford-Richards Scholarship for their individual studentships. J. A. L. thanks the Leverhulme Trust (RPG-2017-069) for a research fellowship.Notes and references
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- (a) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (b) S. Sun and J. Fu, Bioorg. Med. Chem. Lett., 2018, 28, 3283–3289 CrossRef CAS PubMed; (c) M. Egbertson, G. B. McGaughey, S. M. Pitzenberger, S. R. Stauffer, C. A. Coburn, S. J. Stachel, W. Yang, J. C. Barrow, L. A. Neilson, M. McWherter, D. Perlow, B. Fahr, S. Munshi, T. J. Allison, K. Holloway, H. G. Selnick, Z. Yang, J. Swestock, A. J. Simon, S. Sankaranarayanan, D. Colussi, K. Tugusheva, M. T. Lai, B. Pietrak, S. Haugabook, L. Jin, I. W. Chen, M. Holahan, M. Stranieri-Michener, J. J. Cook, J. Vacca and S. L. Graham, Bioorg. Med. Chem. Lett., 2015, 25, 4812–4819 CrossRef CAS PubMed; (d) M. K. Ameriks, H. Ao, N. I. Carruthers, B. Lord, S. Ravula, J. C. Rech, B. M. Savall, J. L. Wall, Q. Wang, A. Bhattacharya and M. A. Letavic, Bioorg. Med. Chem. Lett., 2016, 26, 257–261 CrossRef CAS PubMed; (e) W. Zhang, Acc. Chem. Res., 2017, 50, 2381–2388 CrossRef CAS PubMed.
- (a) G. Némethy, Angew. Chem., Int. Ed. Engl., 1967, 6, 195–206 CrossRef PubMed; (b) N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS; (c) A. Khawam and D. R. Flanagan, J. Pharm. Sci., 2006, 95, 472–498 CrossRef CAS PubMed; (d) G. Eugene Kellogg and D. J. Abraham, Eur. J. Med. Chem., 2000, 35, 651–661 CrossRef CAS PubMed; (e) P. R. Andrews, D. J. Craik and J. L. Martin, J. Med. Chem., 1984, 27, 1648–1657 CrossRef CAS PubMed.
- (a) A. Gomtsyan, E. K. Bayburt, R. Keddy, S. C. Turner, T. K. Jinkerson, S. Didomenico, R. J. Perner, J. R. Koenig, I. Drizin, H. A. McDonald, C. S. Surowy, P. Honore, J. Mikusa, K. C. Marsh, J. M. Wetter, C. R. Faltynek and C.-H. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 3894–3899 CrossRef CAS PubMed; (b) A. Bahl, P. Barton, K. Bowers, M. V. Caffrey, R. Denton, P. Gilmour, S. Hawley, T. Linannen, C. A. Luckhurst, T. Mochel, M. W. D. Perry, R. J. Riley, E. Roe, B. Springthorpe, L. Stein and P. Webborn, Bioorg. Med. Chem. Lett., 2012, 22, 6694–6699 CrossRef CAS PubMed; (c) R. W. Friesen, C. Brideau, C. C. Chan, S. Charleson, D. Deschênes, D. Dubé, D. Ethier, R. Fortin, J. Y. Gauthier, Y. Girard, R. Gordon, G. M. Greig, D. Riendeau, C. Savoie, Z. Wang, E. Wong, D. Visco, L. J. Xu and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 2777–2782 CrossRef CAS PubMed; (d) C. A. Brooks, L. S. Barton, D. J. Behm, H. S. Eidam, R. M. Fox, M. Hammond, T. H. Hoang, D. A. Holt, M. A. Hilfiker, B. G. Lawhorn, J. R. Patterson, P. Stoy, T. J. Roethke, G. Ye, S. Zhao, K. S. Thorneloe, K. B. Goodman and M. Cheung, ACS Med. Chem. Lett., 2019, 10, 1228–1233 CrossRef CAS PubMed.
- (a) M. Boehringer, H. Fischer, M. Hennig, D. Hunziker, J. Huwyler, B. Kuhn, B. M. Loeffler, T. Luebbers, P. Mattei, R. Narquizian, E. Sebokova, U. Sprecher and H. P. Wessel, Bioorg. Med. Chem. Lett., 2010, 20, 1106–1108 CrossRef CAS PubMed; (b) K. Fukunaga, F. Uehara, K. Aritomo, A. Shoda, S. Hiki, M. Okuyama, Y. Usui, K. Watanabe, K. Yamakoshi, T. Kohara, T. Hanano, H. Tanaka, S. Tsuchiya, S. Sunada, K.-I. Saito, J.-I. Eguchi, S. Yuki, S. Asano, S. Tanaka, A. Mori, K. Yamagami, H. Baba, T. Horikawa and M. Fujimura, Bioorg. Med. Chem. Lett., 2013, 23, 6933–6937 CrossRef CAS PubMed; (c) M. L. Vazquez, N. Kaila, J. W. Strohbach, J. D. Trzupek, M. F. Brown, M. E. Flanagan, M. J. Mitton-Fry, T. A. Johnson, R. E. TenBrink, E. P. Arnold, A. Basak, S. E. Heasley, S. Kwon, J. Langille, M. D. Parikh, S. H. Griffin, J. M. Casavant, B. A. Duclos, A. E. Fenwick, T. M. Harris, S. Han, N. Caspers, M. E. Dowty, X. Yang, M. E. Banker, M. Hegen, P. T. Symanowicz, L. Li, L. Wang, T. H. Lin, J. Jussif, J. D. Clark, J. B. Telliez, R. P. Robinson and R. Unwalla, J. Med. Chem., 2018, 61, 1130–1152 CrossRef CAS PubMed.
- (a) P. J. Coleman, J. D. Schreier, C. D. Cox, M. J. Breslin, D. B. Whitman, M. J. Bogusky, G. B. McGaughey, R. A. Bednar, W. Lemaire, S. M. Doran, S. V. Fox, S. L. Garson, A. L. Gotter, C. M. Harrell, D. R. Reiss, T. D. Cabalu, D. Cui, T. Prueksaritanont, J. Stevens, P. L. Tannenbaum, R. G. Ball, J. Stellabott, S. D. Young, G. D. Hartman, C. J. Winrow and J. J. Renger, ChemMedChem, 2012, 7, 415–424 CrossRef CAS PubMed; (b) C. D. Cox, G. B. McGaughey, M. J. Bogusky, D. B. Whitman, R. G. Ball, C. J. Winrow, J. J. Renger and P. J. Coleman, Bioorg. Med. Chem. Lett., 2009, 19, 2997–3001 CrossRef CAS PubMed; (c) W. R. Judd, P. M. Slattum, K. C. Hoang, L. Bhoite, L. Valppu, G. Alberts, B. Brown, B. Roth, K. Ostanin, L. Huang, D. Wettstein, B. Richards and J. A. Willardsen, J. Med. Chem., 2011, 54, 5031–5047 CrossRef CAS PubMed; (d) M. I. Lansdell, D. Hepworth, A. Calabrese, A. D. Brown, J. Blagg, D. J. Burring, P. Wilson, D. Fradet, T. B. Brown, F. Quinton, N. Mistry, K. Tang, N. Mount, P. Stacey, N. Edmunds, C. Adams, S. Gaboardi, S. Neal-Morgan, C. Wayman, S. Cole, J. Phipps, M. Lewis, H. Verrier, V. Gillon, N. Feeder, A. Heatherington, S. Sultana, S. Haughie, S. W. Martin, M. Sudworth and S. Tweedy, J. Med. Chem., 2010, 53, 3183–3197 CrossRef CAS PubMed; (e) K. W. Kuntz, J. E. Campbell, H. Keilhack, R. M. Pollock, S. K. Knutson, M. Porter-Scott, V. M. Richon, C. J. Sneeringer, T. J. Wigle, C. J. Allain, C. R. Majer, M. P. Moyer, R. A. Copeland and R. Chesworth, J. Med. Chem., 2016, 59, 1556–1564 CrossRef CAS PubMed; (f) R. K. Pavana, S. Choudhary, A. Bastian, M. A. Ihnat, R. Bai, E. Hamel and A. Gangjee, Bioorg. Med. Chem., 2017, 25, 545–556 CrossRef CAS PubMed.
- R. Angell, N. M. Aston, P. Bamborough, J. B. Buckton, S. Cockerill, S. J. deBoeck, C. D. Edwards, D. S. Holmes, K. L. Jones, D. I. Laine, S. Patel, P. A. Smee, K. J. Smith, D. O. Somers and A. L. Walker, Bioorg. Med. Chem. Lett., 2008, 18, 4428–4432 CrossRef CAS PubMed.
- D. W. Piotrowski, K. Futatsugi, A. Casimiro-Garcia, L. Wei, M. F. Sammons, M. Herr, W. Jiao, S. Y. Lavergne, S. B. Coffey, S. W. Wright, K. Song, P. M. Loria, M. E. Banker, D. N. Petersen and J. Bauman, J. Med. Chem., 2018, 61, 1086–1097 CrossRef CAS PubMed.
- C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489–4500 CrossRef CAS PubMed.
- C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS PubMed.
- (a) M. D. Engstrom and B. F. Pfleger, Synth. Syst. Biotechnol., 2017, 2, 176–191 CrossRef PubMed; (b) T. W. Johnson, R. A. Gallego and M. P. Edwards, J. Med. Chem., 2018, 61, 6401–6420 CrossRef CAS PubMed.
- Y. Chen, Chemistry, 2019, 25, 3405–3439 CrossRef CAS PubMed.
- M. Pilla, M. Andreoli, M. Tessari, S. Delle-Fratte, A. Roth, S. Butler, F. Brown, P. Shah, E. Bettini, P. Cavallini, R. Benedetti, D. Minick, P. Smith, B. Tehan, P. D'Alessandro, O. Lorthioir, C. Ball, V. Garzya, C. Goodacre and S. Watson, Bioorg. Med. Chem. Lett., 2010, 20, 7521–7524 CrossRef CAS PubMed.
- (a) B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed; (b) J. Yeston, Science, 2019, 363, 241–243 Search PubMed.
- (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (e) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (f) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (b) E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, Bioorg. Med. Chem. Lett., 2016, 26, 5378–5383 CrossRef CAS PubMed; (c) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS PubMed; (d) S. Sengupta and G. Mehta, Tetrahedron Lett., 2017, 58, 1357–1372 CrossRef CAS.
- (a) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed; (c) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed; (d) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (e) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538–542 CrossRef CAS PubMed; (f) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (g) Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed; (h) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed; (i) N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed; (j) G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Nat. Chem., 2016, 8, 810–815 CrossRef CAS PubMed; (k) B. F. Shi, N. Maugel, Y. H. Zhang and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed; (l) B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460–461 CrossRef CAS PubMed; (m) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed; (n) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598–19601 CrossRef CAS PubMed; (o) H. M. L. Davies and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075–9076 CrossRef CAS; (p) G. R. Genov, J. L. Douthwaite, A. S. K. Lahdenperä, D. C. Gibson and R. J. Phipps, Science, 2020, 367, 1246–1251 CrossRef CAS PubMed; (q) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed; (r) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (s) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149–171 RSC; (t) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC; (u) A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398–12414 RSC.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- (a) G. Yan, A. J. Borah, L. Wang and M. Yang, Adv. Synth. Catal., 2015, 357, 1333–1350 CrossRef CAS; (b) J. Kim and S. H. Cho, Synlett, 2016, 2525–2529 CAS; (c) L. Hu, Y. A. Liu and X. Liao, Synlett, 2018, 375–382 CAS.
- (a) L. K. Chan, D. L. Poole, D. Shen, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2014, 53, 761–765 CrossRef CAS PubMed; (b) D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642–1645 CrossRef CAS PubMed; (c) K. Polidano, J. M. J. Williams and L. C. Morrill, ACS Catal., 2019, 9, 8575–8580 CrossRef CAS PubMed; (d) A. Kaithal, P. van Bonn, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 215–220 CrossRef CAS PubMed; (e) Q. Liu, G. Xu, Z. Wang, X. Liu, X. Wang, L. Dong, X. Mu and H. Liu, ChemSusChem, 2017, 10, 4748–4755 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- S. J. Tremont and H. U. Rahman, J. Am. Chem. Soc., 1984, 106, 5759–5760 CrossRef CAS.
- A. J. Canty, M. C. Denney, G. van Koten, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 5432–5439 CrossRef CAS.
- M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC.
- X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634–12635 CrossRef CAS PubMed.
- Y. Zhang, J. Feng and C.-J. Li, J. Am. Chem. Soc., 2008, 130, 2900–2901 CrossRef CAS PubMed.
- A. K. Sharma, D. Roy and R. B. Sunoj, Dalton Trans., 2014, 43, 10183–10201 RSC.
- M.-J. Jang and S. W. Youn, Bull. Korean Chem. Soc., 2011, 32, 2865–2866 CrossRef CAS.
- J. M. Wiest, A. Pöthig and T. Bach, Org. Lett., 2016, 18, 852–855 CrossRef CAS.
- T. Mitra, M. Kundu and B. Roy, J. Org. Chem., 2020, 85, 345–359 CrossRef CAS PubMed.
- R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194–13197 CrossRef CAS PubMed.
- S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302–2305 CrossRef CAS.
- D. Tu, X. Cheng, Y. Gao, P. Yang, Y. Ding and C. Jiang, Org. Biomol. Chem., 2016, 14, 7443–7446 RSC.
- C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC.
- Q.-L. Yang, C.-Z. Li, L.-W. Zhang, Y.-Y. Li, X. Tong, X.-Y. Wu and T.-S. Mei, Organometallics, 2019, 38, 1208–1212 CrossRef CAS.
- Z.-l. Li and C. Cai, Org. Chem. Front., 2017, 4, 2207–2210 RSC.
- D. C. Simkó, P. Elekes, V. Pázmándi and Z. Novák, Org. Lett., 2018, 20, 676–679 CrossRef PubMed.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS.
- S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531–539 CrossRef CAS PubMed.
- (a) L. Huang, X. Sun, Q. Li and C. Qi, J. Org. Chem., 2014, 79, 6720–6725 CrossRef CAS PubMed; (b) L. Huang, Q. Li, C. Wang and C. Qi, J. Org. Chem., 2013, 78, 3030–3038 CrossRef CAS PubMed.
- J. Singh, M. Deb and A. J. Elias, Organometallics, 2015, 34, 4946–4951 CrossRef CAS.
- X. Wang, S. Niu, L. Xu, C. Zhang, L. Meng, X. Zhang and D. Ma, Org. Lett., 2017, 19, 246–249 CrossRef CAS PubMed.
- (a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (b) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef CAS PubMed; (c) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS PubMed; (d) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed; (e) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed; (f) L. Hu, P.-X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134–2138 CrossRef CAS PubMed; (g) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 CrossRef CAS PubMed.
- B. R. Rosen, L. R. Simke, P. S. Thuy-Boun, D. D. Dixon, J.-Q. Yu and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 7317–7320 CrossRef CAS PubMed.
- P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508–17513 CrossRef CAS PubMed.
- W. Lv, S. Wen, J. Liu and G. Cheng, J. Org. Chem., 2019, 84, 9786–9791 CrossRef CAS PubMed.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC.
- D. Sarkar and V. Gevorgyan, Chem. – Eur. J., 2016, 22, 11201–11204 CrossRef CAS PubMed.
- S. John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC.
- C.-H. Jun, H. Lee and J.-B. Hong, J. Org. Chem., 1997, 62, 1200–1201 CrossRef CAS.
- (a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (b) Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 4808–4826 CAS; (c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS.
- X.-Y. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS PubMed.
- X.-Y. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951–8956 RSC.
- F. Pan, Z.-Q. Lei, H. Wang, H. Li, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 2063–2067 CrossRef CAS PubMed.
- H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef CAS PubMed.
- Y. Ping, Z. Chen, Q. Ding and Y. Peng, Synthesis, 2017, 2015–2024 CAS.
- B. Wang, C. Li and H. Liu, Adv. Synth. Catal., 2017, 359, 3029–3034 CrossRef CAS.
- W. Gao, C. Gong, Q. Yang, J. Yuan, L. Xu and Y. Peng, Can. J. Chem., 2017, 95, 1052–1058 CrossRef CAS.
- P. Peng, J. Wang, H. Jiang and H. Liu, Org. Lett., 2016, 18, 5376–5379 CrossRef CAS PubMed.
- H. Zhao, X. Xu, H. Yu, B. Li, X. Xu, H. Li, L. Xu, Q. Fan and P. J. Walsh, Org. Lett., 2020, 22, 4228–4234 CrossRef CAS PubMed.
- S. Ni, M. Hribersek, S. K. Baddigam, F. J. L. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Angew. Chem., Int. Ed., 2020, 59, 2–9 CrossRef.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (b) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (c) Z. Ruan, S.-K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 2045–2049 CrossRef CAS PubMed; (d) I. Choi, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12534–12540 CrossRef CAS PubMed; (e) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS PubMed; (f) G. G. Dias, T. A. D. Nascimento, A. K. A. de Almeida, A. C. S. Bombaça, R. F. S. Menna-Barreto, C. Jacob, S. Warratz, E. N. da Silva Júnior and L. Ackermann, Eur. J. Org. Chem., 2019, 2344–2353 CrossRef CAS.
- M. D. L. Tonin, D. Zell, V. Müller and L. Ackermann, Synthesis, 2016, 127–134 Search PubMed.
- B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109–1113 CrossRef CAS PubMed.
- Q. Chen, L. Ilies, N. Yoshikai and E. Nakamura, Org. Lett., 2011, 13, 3232–3234 CrossRef CAS PubMed.
- H. Wang, S. Zhang, Z. Wang, M. He and K. Xu, Org. Lett., 2016, 18, 5628–5631 CrossRef CAS PubMed.
- Q. Sun and N. Yoshikai, Org. Chem. Front., 2018, 5, 2214–2218 RSC.
- D. Schmiel and H. Butenschön, Eur. J. Org. Chem., 2017, 3041–3048 CrossRef CAS.
- Q. Li, Y. Li, W. Hu, R. Hu, G. Li and H. Lu, Chem. – Eur. J., 2016, 22, 12286–12289 CrossRef CAS PubMed.
- Z.-l. Li, P.-Y. Wu and C. Cai, Org. Chem. Front., 2019, 6, 2043–2047 RSC.
- T. Kubo and N. Chatani, Org. Lett., 2016, 18, 1698–1701 CrossRef CAS PubMed.
- S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed.
- K. Graczyk, T. Haven and L. Ackermann, Chem. – Eur. J., 2015, 21, 8812–8815 CrossRef CAS PubMed.
- G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484–1488 CrossRef CAS PubMed.
- Z. Shen, G. Cera, T. Haven and L. Ackermann, Org. Lett., 2017, 19, 3795–3798 CrossRef CAS PubMed.
- L. Ilies, S. Ichikawa, S. Asako, T. Matsubara and E. Nakamura, Adv. Synth. Catal., 2015, 357, 2175–2179 CrossRef CAS.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 7660–7663 CrossRef CAS PubMed.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132–10135 CrossRef CAS PubMed.
- W. Xu and N. Yoshikai, ChemSusChem, 2019, 12, 3049–3053 CrossRef CAS PubMed.
- Y. Aihara, J. Wuelbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438–446 CrossRef CAS.
- T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162–3165 CrossRef CAS PubMed.
- D. Liu, L. Yu, Y. Yu, Z. Xia, Z. Song, L. Liao, Z. Tan and X. Chen, Eur. J. Org. Chem., 2019, 6930–6934 CrossRef CAS.
- W. Liu, G. Cera, J. C. A. Oliveira, Z. Shen and L. Ackermann, Chem. – Eur. J., 2017, 23, 11524–11528 CrossRef CAS PubMed.
- T. Sato, T. Yoshida, H. H. Al Mamari, L. Ilies and E. Nakamura, Org. Lett., 2017, 19, 5458–5461 CrossRef CAS PubMed.
- (a) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS; (b) N. Della Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (c) Q. Gao, Y. Shang, F. Song, J. Ye, Z.-S. Liu, L. Li, H.-G. Cheng and Q. Zhou, J. Am. Chem. Soc., 2019, 141, 15986–15993 CrossRef CAS PubMed.
- B. Mariampillai, J. Alliot, M. Li and M. Lautens, J. Am. Chem. Soc., 2007, 129, 15372–15379 CrossRef CAS PubMed.
- J. E. Wilson, Tetrahedron Lett., 2016, 57, 5053–5056 CrossRef CAS.
- Z. Dong, G. Lu, J. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2018, 140, 8551–8562 CrossRef CAS PubMed.
- X. Sui, T. A. Grigolo, C. J. O’Connor and J. M. Smith, Org. Lett., 2019, 21, 9251–9255 CrossRef CAS PubMed.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed.
- T. Yang, C. Kong, S. Yang, Z. Yang, S. Yang and M. Ehara, Chem. Sci., 2020, 11, 113–125 RSC.
- (a) G. Cheng, P. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 8183–8186 CrossRef CAS PubMed; (b) J. Liu, Q. Ding, W. Fang, W. Wu, Y. Zhang and Y. Peng, J. Org. Chem., 2018, 83, 13211–13216 CrossRef CAS PubMed.
- J. Wang, Z. Dong, C. Yang and G. Dong, Nat. Chem., 2019, 11, 1106–1112 CrossRef CAS PubMed.
- Z. Wu, N. Fatuzzo and G. Dong, J. Am. Chem. Soc., 2020, 142, 2715–2720 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- (a) W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803–1822 RSC; (b) M. A. J. Duncton, Med. Chem. Commun., 2011, 2, 1135–1161 RSC; (c) A. C. Sun, R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Synthesis, 2019, 1063–1072 CAS; (d) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed; (e) F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79–96 CrossRef CAS; (f) D. C. Harrowven and B. J. Sutton, Prog. Heterocycl. Chem., 2005, 16, 27–53 Search PubMed; (g) F. Minisci, E. Vismara and F. Fontana, J. Org. Chem., 1989, 54, 5224–5227 CrossRef CAS; (h) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489–519 CrossRef CAS.
- (a) M. Levy and M. Szwarc, J. Am. Chem. Soc., 1955, 77, 1949–1955 CrossRef CAS; (b) B.-M. Bertilsson, B. Gustafsson, I. Kühn, K. Torssell and A. Shimizu, Acta Chem. Scand., 1970, 24, 3590–3598 CrossRef CAS; (c) F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 4411–4416 CrossRef CAS; (d) A. Sugimori and T. Yamada, Bull. Chem. Soc. Jpn., 1986, 59, 3911–3915 CrossRef CAS.
- R. D. Bach, P. Y. Ayala and H. B. Schlegel, J. Am. Chem. Soc., 1996, 118, 12758–12765 CrossRef CAS.
- (a) F. Minisci, Synthesis, 1973, 1–24 CAS; (b) A. Citterio, F. Minisci, O. Porta and G. Sesana, J. Am. Chem. Soc., 1977, 99, 7960–7968 CrossRef CAS; (c) F. Minisci and O. Porta, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, 1974, vol. 16, pp. 123–180 Search PubMed.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- G. Li, S. Yang, B. Lv, Q. Han, X. Ma, K. Sun, Z. Wang, F. Zhao, Y. Lv and H. Wu, Org. Biomol. Chem., 2015, 13, 11184–11188 RSC.
- P.-Z. Zhang, J.-A. Li, L. Zhang, A. Shoberu, J.-P. Zou and W. Zhang, Green Chem., 2017, 19, 919–923 RSC.
- S. Jin, H. Yao, S. Lin, X. You, Y. Yang and Z. Yan, Org. Biomol. Chem., 2020, 18, 205–210 RSC.
- H. Zhuang, R. Zeng and J. Zou, Chin. J. Chem., 2016, 34, 368–372 CrossRef CAS.
- X. Rong, L. Jin, Y. Gu, G. Liang and Q. Xia, Asian J. Org. Chem., 2020, 9, 185–188 CrossRef CAS.
- N. Zhu, J. Zhao and H. Bao, Chem. Sci., 2017, 8, 2081–2085 RSC.
- C. Sen and S. C. Ghosh, Adv. Synth. Catal., 2018, 360, 905–910 CrossRef CAS.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS.
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219–2232 CrossRef CAS.
- W. Liu, X. Yang, Z.-Z. Zhou and C.-J. Li, Chem, 2017, 2, 688–702 CAS.
- T. McCallum, S. P. Pitre, M. Morin, J. C. Scaiano and L. Barriault, Chem. Sci., 2017, 8, 7412–7418 RSC.
- M. Zidan, A. O. Morris, T. McCallum and L. Barriault, Eur. J. Org. Chem., 2020, 1453–1458 CrossRef CAS.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem. – Eur. J., 2017, 23, 2537–2541 CrossRef CAS.
- J. Genovino, Y. Lian, Y. Zhang, T. O. Hope, A. Juneau, Y. Gagné, G. Ingle and M. Frenette, Org. Lett., 2018, 20, 3229–3232 CrossRef CAS PubMed.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. G. M. Dhar, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed.
- W. Xue, Y. Su, K.-H. Wang, R. Zhang, Y. Feng, L. Cao, D. Huang and Y. Hu, Org. Biomol. Chem., 2019, 17, 6654–6661 RSC.
- X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed.
- M. K. Eberhardt and R. Colina, J. Org. Chem., 1988, 53, 1071–1074 CrossRef CAS.
- K. Kawai, Y.-S. Li, M.-F. Song and H. Kasai, Bioorg. Med. Chem. Lett., 2010, 20, 260–265 CrossRef CAS PubMed.
- R. Caporaso, S. Manna, S. Zinken, A. R. Kochnev, E. R. Lukyanenko, A. V. Kurkin and A. P. Antonchick, Chem. Commun., 2016, 52, 12486–12489 RSC.
- R. A. Garza-Sanchez, T. Patra, A. Tlahuext-Aca, F. Strieth-Kalthoff and F. Glorius, Chem. – Eur. J., 2018, 24, 10064–10068 CrossRef CAS PubMed.
- S. Jiang, Z. Yang, Z. Guo, Y. Li, L. Chen, Z. Zhu and X. Chen, Org. Biomol. Chem., 2019, 17, 7416–7424 RSC.
- J. Gui, Q. Zhou, C. M. Pan, Y. Yabe, A. C. Burns, M. R. Collins, M. A. Ornelas, Y. Ishihara and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4853–4856 CrossRef CAS.
- Q. Zhang, W. A. Van Der Donk and W. Liu, Acc. Chem. Res., 2012, 45, 555–564 CrossRef CAS PubMed.
- (a) Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS; (b) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed; (c) Y. Fujiwara, J. A. Dixon, F. O’hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- L. Wang, J. Zhao, Y. Sun, H.-Y. Zhang and Y. Zhang, Eur. J. Org. Chem., 2019, 6935–6944 CrossRef CAS.
- Q. Huang and S. Z. Zard, Org. Lett., 2018, 20, 1413–1416 CrossRef CAS PubMed.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (b) H. Huang, K. Jia and Y. Chen, Angew. Chem., 2015, 127, 1901–1904 CrossRef; (c) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS; (d) T. Koike and M. Akita, Org. Biomol. Chem., 2016, 14, 6886–6890 RSC.
- G. X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- V. S. Kudale and J.-J. Wang, Green Chem., 2020, 22, 3506–3511 RSC.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- K. C. Nicolaou, A. E. Koumbis, S. A. Snyder and K. B. Simonsen, Angew. Chem., Int. Ed., 2000, 39, 2529–2533 CrossRef CAS.
- H. Andersson, F. Almqvist and R. Olsson, Org. Lett., 2007, 9, 1335–1337 CrossRef CAS PubMed.
- L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058–1068 RSC.
- F. Zhang and X.-F. Duan, Org. Lett., 2011, 13, 6102–6105 CrossRef CAS PubMed.
- O. V. Larionov, D. Stephens, A. Mfuh and G. Chavez, Org. Lett., 2014, 16, 864–867 CrossRef CAS PubMed.
- W. Jo, J. Kim, S. Choi and S. H. Cho, Angew. Chem., Int. Ed., 2016, 55, 9690–9694 CrossRef CAS.
- L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020–18021 CrossRef CAS.
- S. Han, P. Chakrasali, J. Park, H. Oh, S. Kim, K. Kim, A. K. Pandey, S. H. Han, S. B. Han and I. S. Kim, Angew. Chem., Int. Ed., 2018, 57, 12737–12740 CrossRef CAS.
- P. Ghosh, N. Y. Kwon, S. Han, S. Kim, S. H. Han, N. K. Mishra, Y. H. Jung, S. J. Chung and I. S. Kim, Org. Lett., 2019, 21, 6488–6493 CrossRef CAS.
- J. L. Jeffrey and R. Sarpong, Org. Lett., 2012, 14, 5400–5403 CrossRef CAS PubMed.
- W. Jo, S.-y. Baek, C. Hwang, J. Heo, M.-H. Baik and S. H. Cho, J. Am. Chem. Soc., 2020, 142, 13235–13245 CrossRef CAS PubMed.
- P. Ghosh, N. Y. Kwon, S. Kim, S. Han, S. H. Lee, W. An, N. K. Mishra, S. B. Han and I. S. Kim, Angew. Chem., Int. Ed., 2021, 60, 191–196 CrossRef CAS PubMed.
- W. An, S. B. Choi, N. Kim, N. Y. Kwon, P. Ghosh, S. H. Han, N. K. Mishra, S. Han, S. Hong and I. S. Kim, Org. Lett., 2020, 22, 9004–9009 CrossRef CAS.
- J. G. Rodríguez and A. Urrutia, J. Heterocycl. Chem., 1999, 36, 129–135 CrossRef.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS.
- L. Pouységu, M. Marguerit, J. Gagnepain, G. Lyvinec, A. J. Eatherton and S. Quideau, Org. Lett., 2008, 10, 5211–5214 CrossRef PubMed.
- (a) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514–6515 CrossRef CAS PubMed; (b) R. Ding and B. Yu, Asian J. Org. Chem., 2018, 7, 2427–2430 CrossRef CAS; (c) S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070–1082 RSC.
- W. Rauf and J. M. Brown, Angew. Chem., Int. Ed., 2008, 47, 4228–4230 CrossRef CAS.
- (a) M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806–13809 CrossRef CAS PubMed; (b) R. D. Dolewski, M. C. Hilton and A. McNally, Synlett, 2018, 08–14 CAS; (c) R. G. Anderson, B. M. Jett and A. McNally, Angew. Chem., Int. Ed., 2018, 57, 12514–12518 CrossRef CAS PubMed.
- X. Zhang and A. McNally, ACS Catal., 2019, 9, 4862–4866 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS; (b) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS.
- (a) Z.-T. He, H. Li, A. M. Haydl, G. T. Whiteker and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 17197–17202 CrossRef CAS; (b) A. M. Haydl and J. F. Hartwig, Org. Lett., 2019, 21, 1337–1341 CrossRef CAS PubMed.
- F. Serpier, F. Pan, W. S. Ham, J. Jacq, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 10697–10701 CrossRef CAS.
- (a) P. W. Ayers and M. Levy, Theor. Chem. Acc., 2000, 103, 353–360 Search PubMed; (b) R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC; (c) B. Cox, V. Zdorichenko, P. B. Cox, K. I. Booker-Milburn, R. Paumier, L. D. Elliott, M. Robertson-Ralph and G. Bloomfield, ACS Med. Chem. Lett., 2020, 11, 1185–1190 CrossRef CAS PubMed.
- E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2006, 128, 8350–8357 CrossRef CAS PubMed.
- S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed.
- D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190–7191 CrossRef CAS PubMed.
- S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS PubMed.
- K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906–3911 RSC.
- K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS PubMed.
- B. Wang, X. Wu, R. Jiao, S.-Y. Zhang, W. A. Nack, G. He and G. Chen, Org. Chem. Front., 2015, 2, 1318–1321 RSC.
- R. Kumar, R. Sharma, R. Kumar and U. Sharma, Org. Lett., 2020, 22, 305–309 CrossRef CAS.
- G. Sun, X. Zou, J. Wang and W. Yang, Org. Chem. Front., 2020, 7, 666–671 RSC.
- K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly and M. C. White, Nature, 2020, 580, 621–627 CrossRef CAS PubMed.
- (a) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed; (b) B. Maity, C. Zhu, H. Yue, L. Huang, M. Harb, Y. Minenkov, M. Rueping and L. Cavallo, J. Am. Chem. Soc., 2020, 142, 16942–16952 CrossRef CAS PubMed.
- P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- (a) E. Boess, M. Van Hoof, S. L. Birdsall and M. Klussmann, J. Org. Chem., 2020, 85, 1972–1980 CrossRef CAS; (b) J. P. Barham, M. P. John and J. A. Murphy, Beilstein J. Org. Chem., 2014, 10, 2981–2988 CrossRef PubMed.
- Z. Cheng, Z. Yu, S. Yang, H. C. Shen, W. Zhao and S. Zhong, J. Org. Chem., 2017, 82, 13678–13685 CrossRef CAS.
- J. M. Gil-Negrete, J. Pérez Sestelo and L. A. Sarandeses, J. Org. Chem., 2019, 84, 9778–9785 CrossRef CAS PubMed.
- A. Paul and D. Seidel, J. Am. Chem. Soc., 2019, 141, 8778–8782 CrossRef CAS PubMed.
- W. Chen, A. Paul, K. A. Abboud and D. Seidel, Nat. Chem., 2020, 12, 545–550 CrossRef CAS PubMed.
- D. Vasu, A. L. Fuentes de Arriba, J. A. Leitch, A. de Gombert and D. J. Dixon, Chem. Sci., 2019, 10, 3401–3407 RSC.
- J. P. Klinman, J. Biol. Chem., 1996, 271, 27189–27192 CrossRef CAS.
- J. Zhao, T. Nanjo, E. C. de Lucca, Jr. and M. C. White, Nat. Chem., 2019, 11, 213–221 CrossRef CAS.
- J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski and M. C. White, J. Am. Chem. Soc., 2015, 137, 14590–14593 CrossRef CAS.
- L. Qin, M. Sharique and U. K. Tambar, J. Am. Chem. Soc., 2019, 141, 17305–17313 CrossRef CAS.
- M. Liu, Z. Qiu, L. Tan, R. T. Rashid, S. Chu, Y. Cen, Z. Luo, R. Z. Khaliullin, Z. Mi and C.-J. Li, ACS Catal., 2020, 10, 6248–6253 CrossRef CAS.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS.
- M. Liu, Y. Wang, X. Kong, R. T. Rashid, S. Chu, C.-C. Li, Z. Hearne, H. Guo, Z. Mi and C.-J. Li, Chem, 2019, 5, 858–867 CAS.
- P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nat. Chem., 2017, 9, 140–144 CrossRef CAS.
- (a) P. Beak, S. T. Kerrick, S. Wu and J. Chu, J. Am. Chem. Soc., 1994, 116, 3231–3239 CrossRef CAS; (b) D. Stead, G. Carbone, P. O’Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260–7261 CrossRef CAS PubMed; (c) J. D. Firth, P. O’Brien and L. Ferris, J. Am. Chem. Soc., 2016, 138, 651–659 CrossRef CAS PubMed.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581–585 CrossRef CAS PubMed.
- C. Liao and F. P. Seebeck, Angew. Chem., Int. Ed., 2020, 59, 7184–7187 CrossRef CAS.
Footnotes |
| † First joint authors. |
| ‡ Second joint authors. |
| This journal is © The Royal Society of Chemistry 2021 |