
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtroposelective transformation of axially chiral (hetero)biaryls. From desymmetrization to modern resolution strategies
José A.
Carmona†
 a,
Carlos
Rodríguez-Franco†
a,
Rosario
Fernández
*b,
Valentín
Hornillos
*a and
José M.
Lassaletta
*a
a,
Carlos
Rodríguez-Franco†
a,
Rosario
Fernández
*b,
Valentín
Hornillos
*a and
José M.
Lassaletta
*a
aInstituto de Investigaciones Químicas (CSIC-US) and Centro de Innovación en Química Avanzada (ORFEO-CINQA), C/Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: jmlassa@iiq.csic.es
bDepartamento de Química Orgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Sevilla, C/Prof. García González, 1, 41012 Sevilla, Spain
First published on 25th January 2021
Abstract
This tutorial review provides a systematic overview of the available methodologies for the atroposelective transformation of (heterobiaryl)biaryl precursors toward the synthesis of enantiomerically enriched products and the conceptual aspects associated to each type of transformation. Depending on the presence or absence of symmetry in the starting material and the participation of racemization or dynamization events along the process, several strategies have been developed, including desymmetrization, classical kinetic resolution (KR), dynamic kinetic resolution (DKR) and dynamic kinetic asymmetric transformation (DYKAT). Seminal contributions and a handful of selected examples are discussed to illustrate the potential of these synthetic tools.
 José A. Carmona | José Alberto Carmona studied chemistry at the University of Seville and received his PhD in 2019 under the supervision of Prof. José M. Lassaletta, Prof. Rosario Fernández and Dr Valentín Hornillos. His current research interests include the development of novel synthetic methodologies in the field of asymmetric catalysis, with emphasis on metal catalysis and organocatalysis. |
 Carlos Rodríguez-Franco | Carlos Rodríguez Franco was born in Seville (Spain) in 1995. He studied chemistry at University of Seville and commenced PhD studies in the group of Asymmetric Catalysis at the Instituto de Investigaciones Químicas (CSIC) in 2019, exploring new methodologies for the atroposelective synthesis of axially chiral biaryls involving dynamization processes. |
 Rosario Fernández | Rosario Fernández studied chemistry at the University of Sevilla and received both her BS degree (1980) and her PhD degree (1985) under the supervision of Prof. Antonio Gómez Sánchez. She was a NATO postdoctoral fellow at the University of Paris-Sud (Orsay, France) in the laboratory of Prof. Serge David from 1986 to 1987. In 1987 she returned to the University of Sevilla, where she was promoted to Associate Professor. In 2008 she became a Full Prof. at the same University. Her current research interests include asymmetric synthesis and enantioselective catalysis, in both aspects, asymmetric metal catalysis and organocatalysis. |
 Valentín Hornillos | Valentín Hornillos (Toledo, 1978) obtained his PhD from the University Complutense de Madrid in 2009, working on the synthesis of fluorescent lipid drugs. In 2007, he also spent four months in the laboratories of Novartis in Vienna. From 2010 until 2015, he was a postdoctoral fellow in the group of Professor Feringa at the University of Groningen and in 2015 he moved to the IIQ, CSIC in Seville as a Talent Hub Fellow. Since 2019, he has been Ramón y Cajal researcher at the University of Seville. His research interests include the development of new transition metal-catalyzed enantioselective processes. |
 José M. Lassaletta | José María Lassaletta received his PhD in 1990 at the University of Seville. He then performed postdoctoral stages in the ‘Instituto de la Grasa y sus Derivados’ (CSIC) and the University of Konstanz, (Germany) with Prof. R. R. Schmidt. In 1995 he moved to the ‘Instituto de Investigaciones Químicas’ (CSIC, Seville), where he promoted to Tenured Scientist (1996), Research Scientist (2005) and Research Professor (2009). He has been recognized with the ‘Felix Serratosa’ Lecture (2011) and the ‘Ignacio Ribas’ Medal from the Royal Society of Chemistry (2017). His current interests focus on asymmetric homogeneous catalysis and development of synthetic methodologies. |
Key learning points1. Desymmetrization can be applied to achiral (hetero)biaryls featuring or lacking restricted rotation around the pro-stereogenic axis.2. Classical kinetic resolution still remains as one of the most useful tools for the synthesis of BINOL, BINAM or NOBIN derivatives. 3. In dynamic kinetic resolutions, the racemization of (hetero)biaryls is facilitated by covalent bridges or intramolecular bonding interactions. 4. Dynamic kinetic asymmetric cross-coupling of heterobiaryl electrophiles is a powerful tool for the synthesis of axially chiral derivatives. |
1. Introduction
Axial chirality in biaryl compounds originates when the rotational barrier (or atropisomerization barrier) around the aryl–aryl bond is high enough to avoid the interconversion of the resulting enantiomers (atropisomers) at room temperature. The configurational stability of the stereogenic axis critically depends upon the number and size of the groups at the ortho positions around the axis (2,2′ and 6,6′ in typical 6-membered biaryls). Axially chiral biaryls are widely used as the key structural motif of chiral ligands which have found a plethora of applications in asymmetric catalysis.1 Well established examples illustrated in Fig. 1 include C2-symmetric binaphthyls such as the ‘privileged structures’ BINOL and BINAP, as well as BINAM derivatives or monodentate ligands such as BINEPINEs. Additionally, non-C2-symmetric derivatives such as MOP, MAP or heterobiaryl derivative QUINAP have also found countless applications in this field. Finally, selected representatives of axially chiral compounds with relevant applications in asymmetric organocatalysts are also shown, including phase transfer catalysts (PTCs), chiral phosphoric acids (CPAs) and bifunctional thiourea H-bond donors.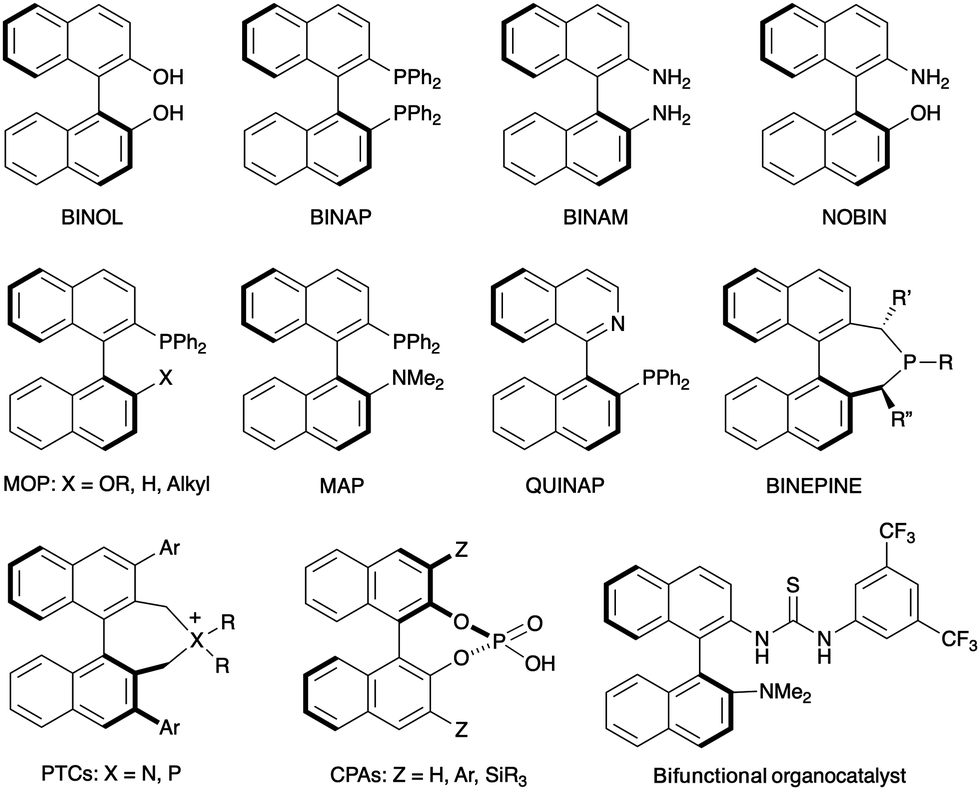 | ||
| Fig. 1 Axially chiral (hetero)biaryls used as ligands or catalysts in asymmetric catalysis. Selected examples. | ||
Axially chiral biaryls are also key structural motifs in a number of natural products and bioactive compounds featuring antibiotic, antiviral or anticancer activities.1,2 The selected examples collected in Fig. 2 include well known vancomycin, (a marketed glycopeptidic antibiotic prescribed to treat a number of bacterial infections), Michellamine B (an alkaloid which has been found to inhibit HIV viral replication), gossypol, (a terpenoid aldehyde featuring contraceptive and antimalarial properties), steganacin (an antileukemic lignan lactone which inhibits tubulin formation) and Marinopyrrole A (a marine natural product exhibiting significant antibiotic and anticancer activities).
 | ||
| Fig. 2 Examples of natural products featuring axial chirality. | ||
Owing to the importance of axially chiral biaryls, development of new methods providing access to new structures is a hot topic that occupies the interest of many research groups in asymmetric catalysis. Essentially, there are five different approaches for the catalytic atroposelective synthesis of axially chiral biaryl derivatives (Scheme 1). The most straightforward method consists of the direct formation of the axis by a C–C bond forming asymmetric cross-coupling, a challenging reaction that requires highly efficient catalysts providing the required stereocontrol in combination with a high catalytic activity for the coupling of hindered ortho-substituted substrates.3 A second approach is based on oxidative coupling reactions, a methodology that has a narrower scope of substrates but does not require pre-functionalization of the starting materials.4 A third alternative consist of central-to-axial chirality transfer processes, in which one or more stereogenic centers are destroyed with simultaneous installation of axial chirality.5 Alternatively, methodologies based on the construction of aromatic rings by cycloadditions or cyclizations, using either metal catalysis or organocatalysis are also versatile alternatives for the asymmetric synthesis of (hetero)biaryls.6 Finally, these compounds can be accessed from pre-existing, readily available (hetero)biaryl precursors. A variety of conceptually diverse methodologies are available for such transformation, which can be rationally classified according to the characteristics of the starting material and the nature of the process (Scheme 2). Thus, four possible approaches can be considered, depending on the following questions: (i) is the starting biaryl chiral or achiral? (ii) is it configurationally stable under the reaction conditions? and (iii) is there a racemization process involved in the reaction? Using the above classification as a guide, this tutorial review aims to provide an overview of modern concepts and strategies that have been recently developed for the atroposelective transformation of pre-existing axially chiral biaryls, limiting the scope to approaches based exclusively on enantioselective catalysis.
 | ||
| Scheme 1 Main approaches for the catalytic enantioselective synthesis of axially chiral (hetero)biaryls. | ||
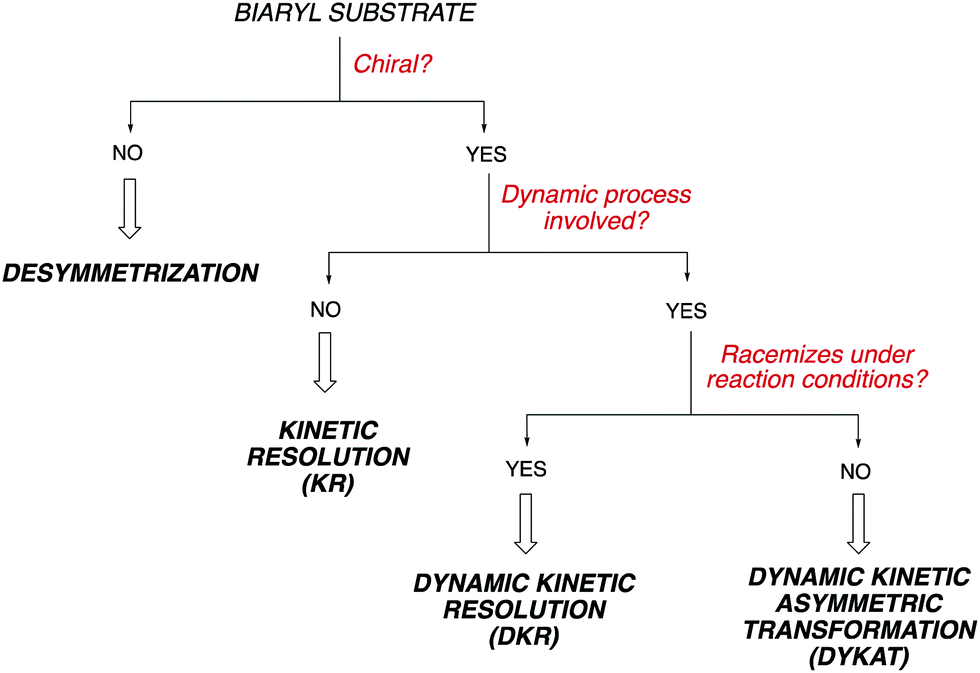 | ||
| Scheme 2 Classification of atroposelective transformation of (hetero)biaryl scaffolds. | ||
2. Desymmetrization
Two types of desymmetrization reactions can be considered from biaryl substrates, depending on whether the starting material features a restricted rotation around a prochiral axis (Section 2.1), or, on the contrary, there is a fast interconversion of conformational enantiomers (Sections 2.2 and 2.3)2.1. Desymmetrization of substrates with restricted rotation of the prochiral axis
Biaryl substrates with three or more ortho substituents are in general characterized by a restricted rotation around the aryl–aryl bond. When any of the two aryl groups is symmetrically substituted, a perpendicular plane of symmetry appears, which must be eliminated in order to generate axial chirality. In practice, there are two different approaches to break the symmetry of these type of substrates (Scheme 3): | ||
| Scheme 3 Desymmetrization routes for the asymmetric synthesis of (hetero)biaryls. | ||
(a) Functional group transformation of enantiotopic substituents (typically at ortho positions). Such transformations (e.g., A into B) break the symmetry of the molecule and hence, results in the generation of a stereogenic axis. A seminal contribution by Hayashi and co-workers7 is based on this strategy: starting from achiral biaryl bistriflates, an enantioselective Kumada coupling was developed by using PdCl2[(S)-Phephos] [Phephos = 2-(dimethylamino)-1-(diphenylphosphino)-3-phenylpropane] as the catalyst (Scheme 4A). More recently, Cheong, Smith and co-workers8 have exploited chiral bases BTM and HyperBTM as acyl transfer catalysts for the acylative desymmetrization of biaryl diols (Scheme 4B). Remarkably, a divergent stereoselectivity was observed depending on the substitution pattern of the substrate and the catalyst used.
 | ||
| Scheme 4 Catalytic biaryl desymmetrization. (A) Ni-Catalysed Kumada coupling. (B) Organocatalytic acylation. (C) Organocatalytic electrophilic bromination. | ||
(b) Introduction of additional substituents Z in the symmetrically substituted (hetero)aromatic ring responsible for the existence of the mirror plane. In this way, the symmetry is also lost and a configurationally stable stereogenic axis is generated. In a representative example of this strategy, the group of Akiyama developed an enantioselective synthesis of biaryls through asymmetric electrophilic bromination of dihydroxybiphenyls. Using a chiral phosphoric acid (CPA) as the catalyst and N-bromophthalimide (NBP) as the brominating reagent, excellent enantioselectivities of the monobrominated product were achieved (Scheme 4C).9 The CPA orchestrates the formation of a highly organized hydrogen-bonding network with the substrate and the brominating agent, enabling the reaction to occur in a high enantioselective manner.
Interestingly, the examples shown above share a common feature: in all cases, good enantioselectivities were achieved in the initial desymmetrization event. However, a second step leading to achiral products takes place with a significant kinetic resolution. In all cases the minor enantiomer reacts faster, providing further enantioenrichment of the product, as illustrated in Scheme 5.
 | ||
| Scheme 5 Tandem desymmetrization/kinetic resolution. | ||
2.2. Desymmetrization of configurationally labile biaryl substrates via C–H activation/functionalization‡
Additional methodologies for atroposelective desymmetrization have been developed from biaryl precursors that do not feature symmetric substitution patterns in any of the aryl units but, not showing restricted rotations around the pro-stereogenic axis, are a rapidly interconverting mixture of conformational enantiomers, and therefore achiral. As a matter of fact, the configurational stability of biaryl atropisomers critically depends on the number and size of substituents at the ortho positions. Therefore, the introduction of additional ortho substituents in this type of substrates is expected to increase the rotation barrier significantly and might ultimately result in the generation of axial chirality in the products.Directed metal-catalysed C–H activation/functionalization is a well-established methodology which provides diverse possibilities within this strategy. A relevant example is the atroposelective Rh-catalysed oxidative coupling of 1-phenylbenzo[h]isoquinolines with styrenes or electron poor alkenes. This transformation, developed by You and co-workers,10 proceeds with high levels of enantioselectivity when chiral spirocyclic cyclopentadienyl (Cp) ligands are used (Scheme 6A). Rhodium catalysis has also been used by the same group to perform a highly efficient arylation of heterobiaryls,11 using in this case a TADDOL-derived monodentate phosphonite as the optimal ligand to achieve excellent enantioselectivities (Scheme 6B). Other transition metals have also been used in atroposelective desymmetrizations. For instance, Pd(OAc)2 has been used as the catalyst in the atroposelective oxidative alkenylation of biaryl aldehydes. This innovative strategy developed by Shi and co-workers12 is based on the generation of a transient chiral auxiliary by in situ condensation of the substrate with sub-stoichiometric amount of free amino acids, reaching excellent enantioselectivities in their reaction with activated alkenes (Scheme 6C). The same group also used Pd(OAc)2 in combination with chiral spiro phosphoric acids for the efficient, highly enantioselective catalytic desymmetrization of two different types of substrates via oxidative C–H alkenylation: First, the heterocyclic N atom of 8-arylquinolines13 was used as the directing group using STRIP as an optimal ligand and AgOAc as the oxidant (Scheme 6D). Second, a similar spirocyclic phosphoric acid (SPA) in combination with Ag2CO3 as the terminal oxidant was applied to the desymmetrization of biaryl amines (Scheme 6E). Remarkably, the olefination can be performed using the free amine as a directing group.14
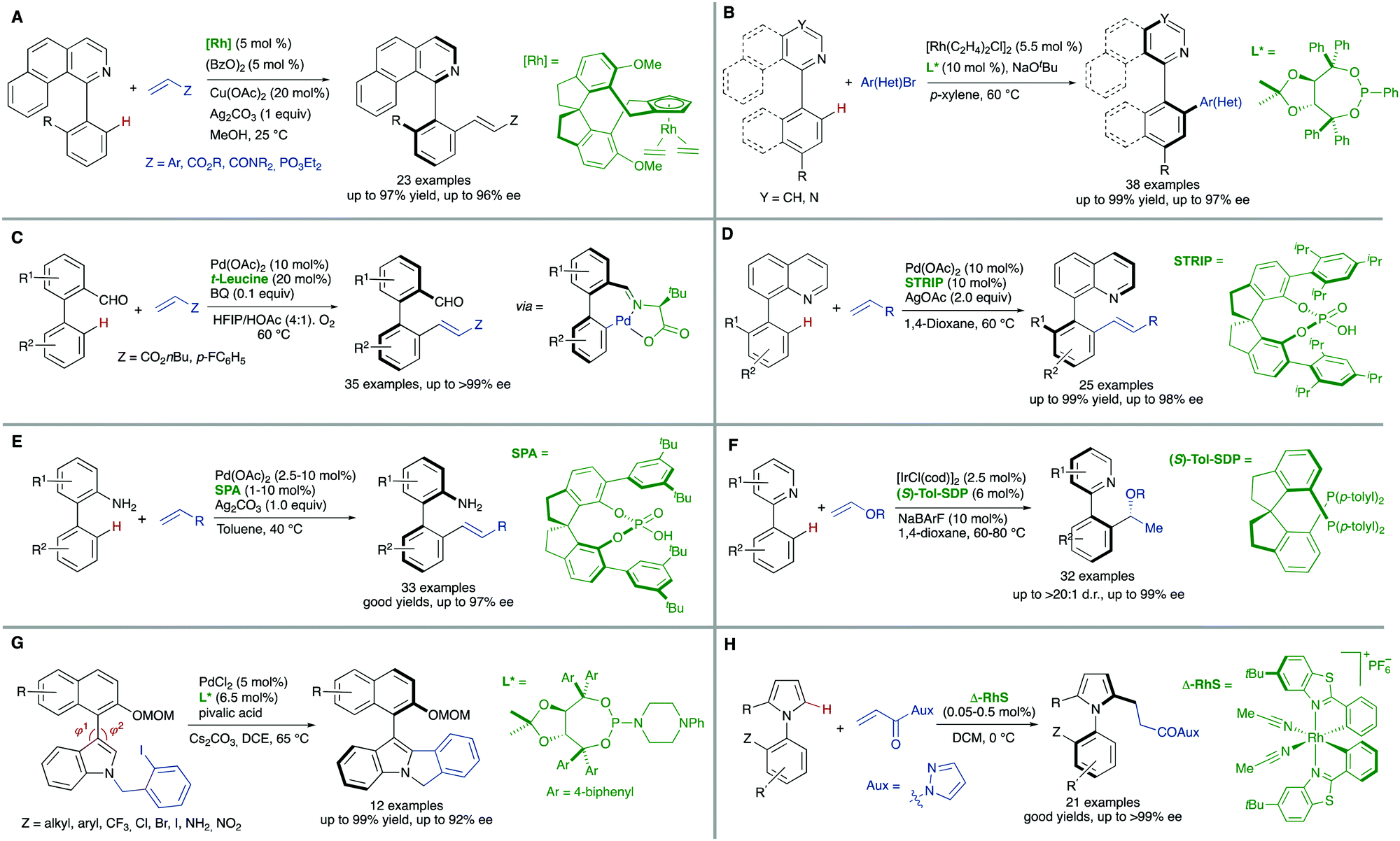 | ||
| Scheme 6 Desymmetrization via metal-catalysed C–H activation/functionalization: selected examples. (A) Directed Rh-catalysed oxidative olefination. (B) Rhodium catalysed arylation (C–E): directed Pd-catalysed oxidative alkenylation. (F) Iridium-alkylation via hydroarylation. (G) Pd-Catalysed arylation. (H) Rh-Catalysed alkylation via conjugate addition. | ||
Iridium is particularly efficient in this context and has been used in the hydroarylation of vinyl ethers and strained alkenes with heterobiaryls (Scheme 6F). Using [IrI/Tol-SDP] complexes (SDP = spiro diphosphine), simultaneous generation of central and axial chirality was achieved with high regio-, diastereo- and enantioselectivity.15 Deuterium labelling and computational evidences suggest that a modified Chalk–Harrod-type mechanism operates, and that the migratory insertion into the Ir–CAryl bond is the selectivity-determining step.
The successful implementation of desymmetrization strategies based on C–H activation/functionalization usually requires that at least two of the ortho positions remain unsubstituted in the starting material to make the stereogenic axis labile. However, the presence of a smaller five-membered ring provokes a widening of the angles φ1 and φ2 involved in the configurational stability of the product (Scheme 6G). This circumstance was exploited by Gu and co-workers in the Pd-catalysed intramolecular arylation of tri-ortho substituted 3-naphthyl N-(2-iodobenzyl)indoles, which proceeds with good enantioselectivities using TADDOL-derived phosphoramidites.16 The participation of directing groups is not an essential requisite to perform atroposelective transformation of biaryls using C–H activation. For instance, Houk, Meggers and co-workers17 used a chiral-at-the-metal, cationic Rh complex in the atroposelective alkylation of a wide scope of N-aryl pyrroles by conjugate addition to N-acryloyl-1H-pyrazole, reaching excellent enantioselectivities for a wide scope of substrates (Scheme 6H).
In addition to these metal-catalysed transformations, the organocatalytic asymmetric bromination of biaryls developed by the group of Miller is also a seminal example of this strategy (Scheme 7).18 In this approach, a tripeptide catalyst was used to perform an atroposelective electrophilic aromatic substitution using NBS as the bromination reagent. Starting from 3′-hydroxy biphenyl carboxylic acids, tribromination of the electron-rich phenol unit proceeds with good yields and >90% ee in most cases. A docking model based on multiple H-bond interactions explains the observed stereoselectivity.
 | ||
| Scheme 7 Organocatalytic bromination by Miller and co-workers. | ||
2.3. Desymmetrization of biaryls via ring-opening reactions‡
A third type of desymmetrization consists of the atroposelective ring opening of five-membered bridged biaryl structures. In these compounds, the angles φ1 and φ2 involved in the stereo-chemical stability of the biaryl axis are forced to be wider than the standard 120°, facilitating a rapid isomerization equilibrium between the two atropisomeric conformers (Scheme 8). In ortho/ortho′ substituted derivatives (R or R′ ≠ H) the ring opening reactions are facilitated by a significant release of torsional strain, resulting in milder reaction conditions. Hayashi and co-workers reported in 2002 the first example of this strategy, namely the Ni-catalysed C–S bond cleavage of dinaphtho[2,1-b:1′,2′-d]thiophene via asymmetric cross-coupling with different Grignard reagents (Scheme 9).19 This methodology afforded biaryl thiols with moderate-to-good yields, and high enantioselectivities were achieved by using a chiral phosphino-oxazoline ligand. Continuing with these studies, the same group also developed a Pd-catalysed carbonylation of a dinaphthaleneiodonium salt.20 The C–I bond cleavage/ring-opening reaction afforded methyl 2′-iodo-[1,1′-binaphthalene]-2-carboxylate although, unfortunately, the enantioselective version using (R)-BINAP as the ligand afforded poor results in terms of reactivity and enantioselectivity (Scheme 10A). It was not until recently, in 2018, that Gu and co-workers reported a highly atroposelective ring opening reaction of five-membered-bridged diaryliodonium salts.21 Using preformed cationic Cu(PyBOX)2 [PyBOX = bis(oxazolinyl)pyridine] complexes as catalysts, these salts were efficiently coupled with amines to afford axially chiral biaryl amino iodides with excellent yields and enantioselectivities in most cases (Scheme 10B). It was found that two substituents in the ortho positions of the biaryl iodonium salt were necessary for the reaction to occur efficiently. The loss of planarity induced by steric hindrance decreases the bond dissociation energy (BDE) of the C–I bonds in the cyclic iodonium salt, thus allowing the reaction to be carried out under mild conditions, as required to preserve the C–I bond of the products unreacted. Non-symmetrical cyclic diaryliodonium salts are also suitable substrates, and good regioselectivities were achieved by introducing an extra group adjacent to the C–I bond position. More recently, an extension of the method to enable reactions with protected O-alkylhydroxylamines, including sulfonamides and carbamates, has been also developed (Scheme 10C). In this case, a different CuI/BOX (BOX = bisoxazoline) complex was used as the catalyst.22 Aiming to introduce new functionalities into this system, the same group reported a Cu-catalysed asymmetric thiolative ring opening reaction for the synthesis of atropisomeric 2-thio-2′-iodo-1,1′-biphenyl derivatives.23 In this case, the use of potassium thiocarboxylates as nucleophiles and Cu(CH3CN)4PF6/PhBOX as the optimal catalytic system delivered the desired thioesters satisfactorily in terms of reactivity and enantioselectivity (Scheme 10D). These diaryliodoniums were also used in the synthesis of lactone-bridged biaryl atropisomers.24 The method consisted of a Cu-catalysed asymmetric acyloxylation, followed by a Pd-catalysed diastereoselective Heck cyclization, to deliver bridged biaryl atropisomers with both axial and central chirality (Scheme 10E). The first step, a reaction between the diaryliodonium and an α,β-unsaturated carboxylic acid was achieved with excellent enantioselectivities using Cu(OTf)2/PhBOX as the catalytic system. It is worth noting that, unlike the reaction with thioacetic acids, carboxylic acids could be directly used, instead of employing their corresponding sodium or potassium salts. The obtained biaryl α,β-unsaturated esters were then subjected to a Pd-catalysed Heck cyclization to afford the final products without erosion of the enantiomeric purity and with perfect diastereoselectivity. A significant extension of the scope for the Cu-catalysed acyloxylation of diaryliodonium salts was later reported by Zhang and co-workers.25 Using similar catalyst and conditions, axially chiral iodo carboxylates were isolated with excellent yields and enantioselectivities. Remarkably, the method was applied to the synthesis of five prescribed drugs. Aiming to introduce a phosphine functionality into the biaryl atropisomeric skeletons, Gu and co-workers reported also a Cu-catalysed ring-opening/oxidative phosphorylation reaction of cyclic diaryliodonium salts and diarylphosphine oxides (Scheme 10F).26 During preliminary experiments designed to obtain axially chiral phosphine oxides, an unexpected overoxidized phosphinate was also observed. The addition of TEMPO resulted in higher chemoselectivity toward the later, allowing for the preparation of a large variety of phosphinates in very good overall yields and excellent enantioselectivities. 18O-labelling experiments confirmed that the oxidation assisted by TEMPO takes place before the C–O bond forming step. A phosphine oxide transfer reaction over the synthesized phosphinates was also developed by reaction with tBuLi or tBuMgCl·LiCl, to achieve 2′-hydroxyl biaryl phosphine oxides with moderate-to-good yields and absolute retention of the configuration. | ||
| Scheme 8 Desymmetrization of biaryls featuring five-membered bridges. | ||
 | ||
| Scheme 9 Desymmetrization of dinaphtho[2,1-b:1′,2′-d]thiophene via Ni-catalysed coupling with Grignard reagents. | ||
 | ||
| Scheme 10 Desymmetrization of cyclic biaryl iodonium salts. (A) Pd-Catalysed carbonylation. (B) Cu-Catalysed amination. (C) Cu-Catalysed hydroxyamination. (D) Cu-Catalysed thiolation. (E) Cu-Catalysed acyloxylation. (F) Cu-Catalysed oxidative phosphorylation. | ||
In 2019, Gu and co-workers also reported a more challenging ring-opening reaction of 9H-fluoren-9-ols via Pd-catalysed C–C bond cleavage and coupling with aryl bromides (Scheme 11).27 As in previous cases, ortho,ortho′-disubstitution in the five-membered bridged biaryl scaffold increases the torsional strain and reduces the inherently high bond dissociation energy (BDE). Forty-four heterobiaryl ketones were obtained with very good overall reactivities and enantioselectivities using chloro(2′-amino-1,1′-biphenyl-2-yl)palladium(II) dimer and a chiral TADDOL-based phosphoramidite ligand.
 | ||
| Scheme 11 Desymmetrization of 9H-fluoren-9-ols via C–C bond activation. | ||
3. Kinetic resolutions (KRs)
Alternative solutions are required for those starting materials lacking symmetry, and therefore chiral. In cases where there are no dynamic processes involved, a classical kinetic resolution (KR) remains as the only available strategy. In order to achieve a successful KR, different rates for the reaction of the two atropisomers of the substrate are required. Ideally, one of them is fully transformed into the desired product, while the other remains unreacted (Scheme 12). Although there is a limitation regarding the maximum 50% yield, kinetic resolution is still one of the most common methods for the synthesis of enantioenriched biaryls and has been extensively applied to the synthesis of BINOL, BINAM and NOBIN, among other biaryl derivatives. A seminal contribution by Tokunaga, Tsuji and co-workers28 showed in 2005 that this approach can be efficiently applied to the Pd-catalysed methanolysis of vinyl ethers of axially chiral 2,2′-dihydroxy-1,1′-biaryls. Using Pd(OAc)2 and a (R,R)-1,2-cyclohexanediamine ligand as the catalytic system, a variety of BINOL derivatives were obtained with high selectivity and generality (Scheme 13A). Later in 2013, Maruoka and co-workers reported on the kinetic resolution of axially chiral 2-(N-sulfonylamino)-1,1′-biaryls by phase-transfer-catalysed allylation.29 Using binaphthyl-derived chiral quaternary ammonium salts, the allylation proceeds with excellent selectivities (S factor up to 43) (Scheme 13B). Sibi and co-workers developed conceptually novel fluxionally chiral DMAP derivatives that were successfully applied as acylation organocatalysts for the kinetic resolution of a series of BINOL derivatives.30 Using isobutyric anhydride as the reagent, selectivity factors of up to 51 were achieved (Scheme 13C). A different organocatalytic kinetic resolution of BINOL and NOBIN derivatives via acylation was developed in the same year by Zhao and co-workers.31 Their approach makes use of in situ generation of N-heterocyclic carbenes for the activation of functionalized aldehydes via acyl azolium intermediates. Using an aminoindanol derived triazolium precatalyst and α-benzoyloxy isovaleraldehyde as the reagent, excellent levels of enantioselectivity of the unreacted starting materials were achieved (Scheme 13D). Very recently, Yang and co-workers reported a versatile, scalable protecting group-free kinetic resolution of unprotected BINAM and NOBIN derivatives.32 The method makes use of an organocatalytic N-amination with azodicarboxylates reaching impressive selectivities (s factor up to 420) in the formation of the corresponding triazanes when a hindered CPA (TRIP) is used as the catalyst (Scheme 13E). A kinetic resolution approach has also been developed by You and co-workers to afford appealing functionalized heterobiaryls.33 In this case, the resolution is achieved by a Pd(II)-catalysed C–H iodination of 1-(1)-naphthylisoquinoline N-oxides (Scheme 13F). Using the oxygen atom of the substrate as the directing group for the initial C–H activation step and a N-protected phenylalanine ligand, iodination with N-iodosuccinimide takes place efficiently and with high levels of selectivity (S factor up to 27). The group of Zhou has also performed a kinetic resolution of heterobiaryls, using in this case organocatalytic asymmetric transfer hydrogenation of 5- or 8-aryl substituted quinolines with a Hantzsch ester as the hydrogen source and a chiral phosphoric acid (CPA) as the catalyst (Scheme 13G).34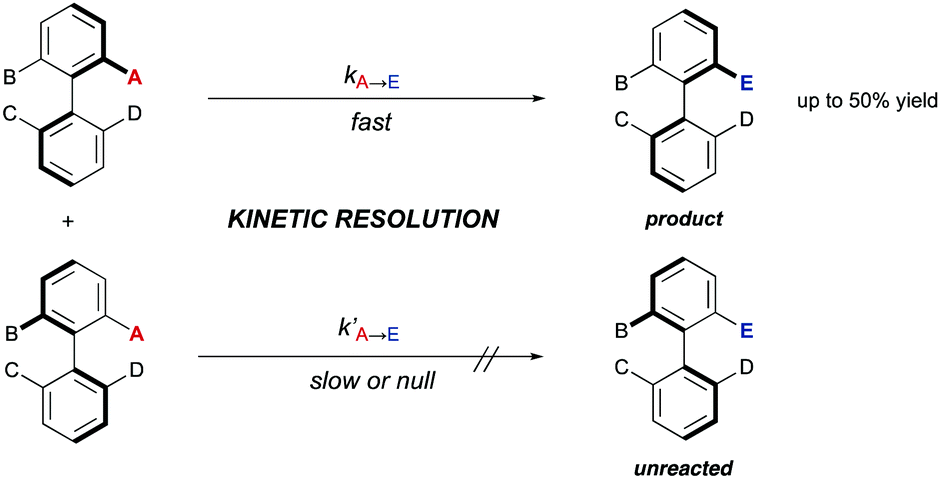 | ||
| Scheme 12 Classical kinetic resolution of biaryls. | ||
 | ||
| Scheme 13 Atroposelective synthesis of (hetero)biaryl via kinetic resolution: Selected examples. (A) Pd-Catalysed methanolysis of vinyl ethers. (B) Phase-transfer-catalysed N-allylation. (C and D) Organocatalytic O-acylation. (E) Organocatalytic N-amination. (F) Pd-Catalysed C–H iodination. (G) Organocatalytic transfer hydrogenation. | ||
While the above examples clearly demonstrate the practical value of the classical kinetic resolution in the synthesis of axially chiral (hetero)biaryls, the inherent limitation of a maximum 50% yield has stimulated intensive investigations to solve this problem by designing dynamic processes that can be classified into two different groups depending on the nature of the dynamization: dynamic kinetic resolution (DKR), or dynamic kinetic asymmetric transformation (DYKAT).
4. Dynamic kinetic resolutions of preformed chiral biaryls (DKRs)
A DKR scenario situation appears when the racemic biaryl substrate is able to racemize in the reaction medium, ensuring the constant transformation of the less reactive atropisomer into the more reactive one, and then to the desired enantioenriched product after a catalytic transformation. This interconversion process enables the transformation of both atropisomers into a single stereoisomeric product in a quantitative theoretical yield. For an efficient DKR to occur, the racemization rate (krac) needs to be equal or higher than the reaction rate for the more reactive atropisomer (k in Scheme 14). In a dynamic kinetic resolution, the racemization process does not necessarily involve a chiral catalyst. In general terms, the DKR will provide higher enantiomeric excesses than the classic KR because the continuous racemization on the substrate will prevent the build-up of the opposite atropisomer from the starting material that affects negatively the KR.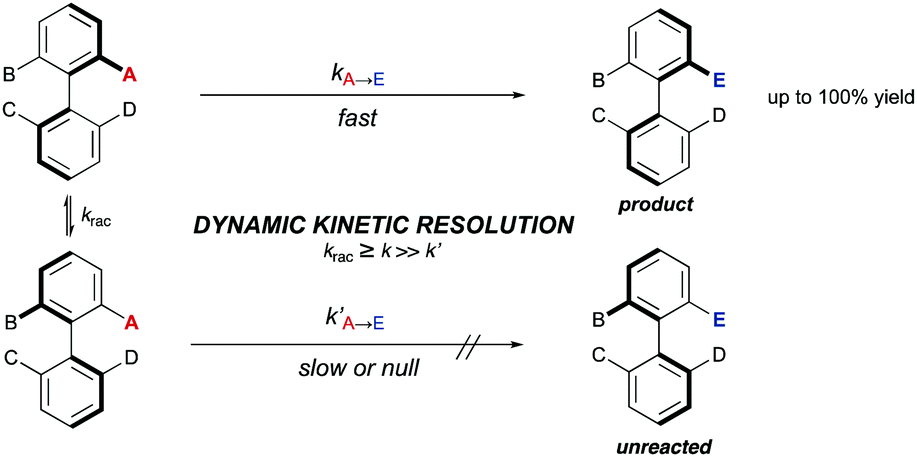 | ||
| Scheme 14 Dynamic kinetic resolution of biaryls. | ||
4.1 Bringmann's biaryl ‘lactone concept’ and related ring-opening reactions‡
The ‘lactone concept’ was first introduced in 1992 by Gerhard Bringmann in a report on the atropo-enantioselective lactone ring opening with a chiral hydride transfer reagent derived from a borane species (Scheme 15).35 This approach is based on the formation of biaryls featuring a lactone bridge which was shown to reduce substantially the racemization barrier between the atropisomers, being ultimately responsible for the configurational instability of the biaryl axis. Then, an atroposelective transformation is performed to cleave the lactone bridge, leading to the corresponding axially chiral biaryl compounds, which, lacking the bridge, are configurationally stable (Scheme 15). The asymmetric ring-opening of configurationally unstable biaryl lactones was later extended to different reactions beyond the lactone reduction, including atropo-enantio- and atropo-diastereoselective cleavage with N- and O-nucleophiles, and the methodology constitutes a very powerful tool that has been widely applied in the total synthesis of natural products containing a stereogenic axis.36 This strategy, however, showed initially an important limitation regarding the need for stoichiometric amounts of the chiral nucleophile responsible for the lactone bridge cleavage. The first example of a catalytic atroposelective reduction of biaryl lactones was reported by Bringmann and co-workers in 2002. Using 10 mol% of the Corey–Bakshi–Shibata (CBS) reagent as the catalyst and borane as the terminal reducing agent, the corresponding axially chiral diol was obtained in high yield and an 88% ee, slightly lower than that for the stoichiometric reaction (Scheme 16A).37 This achievement represents an important progress to the ‘lactone strategy’ and captured the attention of other authors aiming to expand the synthetic utility of this system. In 2008, Yamada and co-workers developed an improved catalytic atropo-enantioselective reduction of biaryl lactones for the synthesis of axially chiral biaryls (Scheme 16B).38 This contribution expanded the scope of biaryl structures that was limited to only two examples in the original Bringmann's report. In this case, sodium borohydride was employed to generate a chiral Co–H complex responsible for the reduction of the carbonyl functionality. HPLC analysis of the starting biaryl lactone at different temperatures helped the authors to determine that, at 30 °C, the atropisomerization of the substrate was taking place fast enough to enable an efficient dynamic kinetic resolution. A major breakthrough in the field was reported in 2016 by the group of Wang: using a chiral bifunctional quinine-based thiourea organocatalysts, the atroposelective transesterification of Bringmann's lactones proceeds with enantiomeric excesses up to 99%. In this report, the authors highlighted the effect of a synergistic activation mode of the thiourea as H-bond donor to the carbonyl, and the sp3 quinine nitrogen as H-bond acceptor to activate the alcohol, crucial to achieve high selectivities (Scheme 16C).39 More recently, in 2019, a [Cu]–H-catalysed atroposelective reduction of Bringmann's lactones was developed by Zhang and co–workers using Cu(OAc)2/(R)-DTBM-SEGPHOS as the catalytic system in combination with NaOtBu as the base and PMHS as reductant. Biaryl diols were obtained with very good overall yields and enantioselectivities, and different functional groups, including halides, cyano and nitro, were well tolerated, allowing for further modifications (Scheme 16D).40 Interestingly, asymmetric [Cu]–H-catalysis represents an uncommon method for the reduction of esters. | ||
| Scheme 15 The ‘lactone concept’ and representative diastereoselective transformations. | ||
 | ||
| Scheme 16 Catalytic asymmetric ring-opening of biaryl lactones. (A) Reduction by borane. (B) Co-Catalysed reduction by NaBH4. (C) Organocatalytic transesterification. (D) Cu-Catalysed reduction by silane. | ||
A related strategy was developed by Akiyama and co-workers41 for the dynamic kinetic resolution of biaryl lactols or hydroxy aldehydes via asymmetric transfer hydrogenation of the intermediate biaryl hemiaminals formed in the reaction with aromatic amines (Scheme 17A). Using chiral phosphoric acid (CPA) catalysts and a Hantzsch ester as the hydrogen source, enantiodivergent reactions were developed, the absolute configuration depending on the structure of the reacting aniline: ortho-hydroxyanilines favour the formation of the R isomer, while the use of the meta-hydroxyanilines inverts the atroposelectivity to furnish the S isomer of the biaryl. In this way, a variety of axially chiral amino alcohols can be obtained in high yield and good enantioselectivities. As in the lactone cases discussed above, the formation of cyclic intermediates, facilitated in this case by ring closing-ring opening events, is the key for the dynamization of the stereogenic axis. Inspired by these results, Wang and co-workers later developed a related redox-neutral dynamic kinetic resolution of biaryl diols (Scheme 17B).42 The strategy is based on the use of Cp*IrHTsDPEN (Cp* = cyclopentadienyl, DPEN = diphenylethylenediamine) complex as a hydrogen borrowing catalyst. After oxidation of the starting material by dehydrogenation, the resulting aldehyde reacts with anilines in the presence of an achiral Brønsted acid (F5C6CO2H). Ensuing hydrogenation of the configurationally labile products affords the corresponding amino alcohols in good yields and good to excellent enantioselectivities. Additionally, the same authors reported in parallel a Ru-catalysed atroposelective reductive amination via dynamic kinetic resolution (Scheme 17C).43 This methodology expanded the scope to both primary and secondary alkyl amines leading to a variety of amino alcohols in good to excellent yields and enantioselectivities.
 | ||
| Scheme 17 Atroposelective approaches for the synthesis of axially chiral biaryl amino alcohols. (A) Reductive amination via organocatalytic transfer hydrogenation. (B) Ir-Catalysed redox-neutral amination. (C) Ru-Catalysed reductive amination. | ||
4.2 DKR enabled by noncovalent interactions in cyclic transition states or intermediates‡
In the precedent section, the required racemization for a dynamic kinetic resolution to occur is facilitated by the presence or formation of a covalently bridge, which is then cleaved in a subsequent stereochemistry-determining step. Alternatively, the labilization of the stereogenic axis can also be facilitated by formation of six-membered ring transition states or intermediates. In 2016, Clayden and co-workers developed a biocatalytic reduction for the atropo-enantioselective synthesis of axially chiral heterobiaryl N-oxides via dynamic kinetic resolution (Scheme 18).44 Particularly, a ketoreductase (KRED 130) was used for the reduction of a configurationally unstable heterobiaryl ketone under mild conditions, while glucose is oxidized to gluconic acid. In this system, the dynamization process for the atropisomerization of the substrate is enabled by an intramolecular interaction between the N-oxide oxygen atom and the aldehyde carbonyl atom, in a six-membered transition state that was supported by molecular modelling, closely resembling the mechanism for atropisomerization at Bringmann's lactones. In contrast to the latter, however, there is no covalent bond formation in this case, but a transition state TS with a bonding interaction. It is also worth mentioning that the authors determined that the reaction with the aldehyde derived from 1-(1-naphthyl)isoquinoline N-oxide was not equally efficient due to the slow racemization rate of this hindered tetra-ortho-substituted system, providing a simple kinetic resolution instead. | ||
| Scheme 18 Dynamic kinetic resolution via biocatalytic reduction by Clayden and co-workers. | ||
Hornillos, Lassaletta and co-workers have recently developed a related asymmetric Zn-catalysed hydrosilylation of configurationally labile heterobiaryl ketones.45 In this case, the atropisomerization of the substrates takes place through a 5-membered cyclic transition state, in which a Lewis acid–base interaction between the isoquinoline nitrogen and the carbonyl group promotes the dynamization of the system. Several studies, including HPLC analysis and DFT calculations, supported a labilization mechanism through formation of a quasi-zwitterionic transition state featuring the partial formation of a C–N bond. Although the calculated racemization barrier is relatively high (22.1 kcal mol−1), the Lewis acidity of the aldehyde carbonyl group should be significantly enhanced by coordination with the acidic Zn complex, further increasing the efficiency of the bonding interaction and reducing the racemization barrier. Secondary alcohols bearing both axial and central chirality were obtained via diastereo- and highly enantioselective DKR (Scheme 19).
 | ||
| Scheme 19 Dynamic kinetic resolution of heterobiaryl ketones via Zn-catalysed hydrosilylation. | ||
A different strategy for the labilization of the stereogenic axis in BINOL derivatives was developed by Akai and co-workers,46 who discovered that a RuII catalyst efficiently racemises BINOL derivatives under mild conditions compatible with lipases, as required for a biocatalytic acylation (Scheme 20). In particular, using η5-Ph5CpRu(CO)2Cl as the racemization catalyst and immobilized Pseudomonas sp. lipoprotein lipase LIP301, a variety of axially chiral C1- and C2-symmetric diols were obtained with excellent yields and enantioselectivities after hydrolysis of the monoacetylated products. Experimental evidence suggests that the racemization takes place by oxidation of the deprotonated substrate/RuII complex by molecular oxygen, followed by a single electron transfer (SET) process in the resulting RuIII binaphtholate complex. In this way, a radical intermediate bearing a sp3 carbon at C1 is formed, thereby enabling the free rotation around the C–C bond before regenerating the stereogenic axis in the racemate.
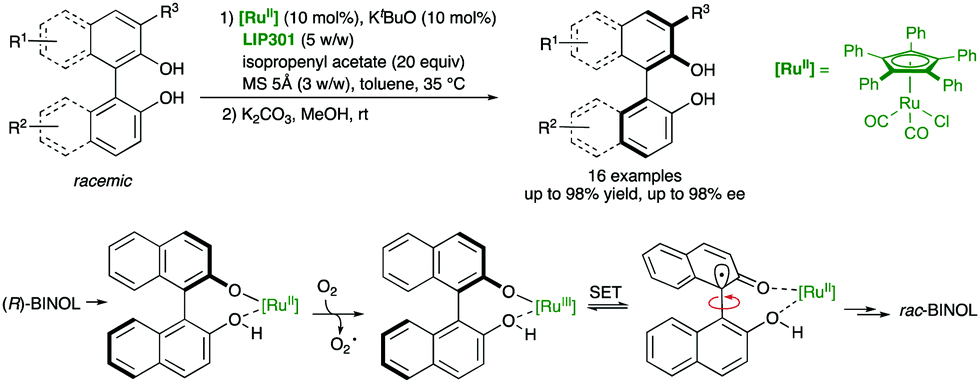 | ||
| Scheme 20 Dynamic kinetic resolution of BINOL derivatives via biocatalytic acylation and Ru-catalysed racemization mechanism. | ||
4.3 DKR via protonation–deprotonation of enolates
Interconversion of enantiomeric enolates through a protonation–deprotonation sequence has also been reported as a versatile approach for the dynamic atroposelective synthesis of axially chiral biaryls. In this context, Smith and co-workers reported in 2017 an enantioselective organocatalytic cation-directed O-alkylation of racemic 1-aryl-2-tetralones. After oxidation, the process provides easy access to non-C2-symmetric BINOL derivatives in good overall yields and up to 96% ee.47 This strategy is based on the atroposelective formation of enol ethers from the corresponding ketones, using a chiral quinidine-derived ammonium salt as a phase transfer catalyst and benzyl iodide as the alkylating agent. Interestingly, C-alkylation was outcasted due to steric hindrance. These enantioenriched enol ethers were then subjected to dehydrogenative aromatization using DDQ as the oxidant, without compromising the enantiomeric purity of the products (Scheme 21). Preliminary studies allowed a plausible mechanism to be proposed in which a fast interconversion of enantiomeric enolates takes place through protonation–deprotonation equilibrium. The chiral ammonium counterion differentiates between the rapidly equilibrating atropisomeric enolates, leading to the obtention of enantioenriched enol ethers via DKR. The study of the configurational stability of the enol ether derived from the model substrate showed a calculated rotational barrier of 32 kcal mol−1. Subsequent oxidation with DDQ at room temperature consequently proceeds with any loss of enantiomeric excess. | ||
| Scheme 21 Dynamic kinetic resolution of axially chiral enolates via alkylation and racemization mechanism. | ||
5. Dynamic kinetic asymmetric transformations (DYKAT)
In some cases, there are no mechanisms to promote the racemization of biaryl substrates that are configurationally stable by virtue of a high rotational barrier, while the reaction conditions do not alter this situation. In a dynamic kinetic asymmetric transformation (DYKAT), however, the dynamization event is mediated by a chiral catalyst, involving the formation of either diastereomeric intermediates in which the configurational stability is compromised (type I, Scheme 22A), or a common chiral intermediate en route to the desired products (type II, Scheme 22B). | ||
| Scheme 22 Dynamic kinetic asymmetric transformation. | ||
To the best of our knowledge, all the reported examples of the application of a DYKAT strategy deal with the resolution of heterobiaryl structures. In this context, our research group reported in 2013 a Pd-catalysed asymmetric arylation of heterobiaryl triflates with arylboroxine (Scheme 23A), reaching good yields and high enantioselectivities using a TADDOL-derived phosphoramidite as the optimal ligand.48 The strategy relies on two key assumptions: (I) The N atom of the heterocycle is a better ligand than triflate and, upon oxidative addition, it incorporates into the coordination sphere of the PdII center to form cationic cyclic intermediates IOA (Scheme 23B). (II) The geometry of the five-membered palladacycle results in a widening of the angles φ1 and φ2 involved in the stabilization of the stereogenic axis, facilitating a fast interconversion of diastereomeric structures IOA and ent-IOA and, hence, making possible the envisaged dynamic kinetic cross-coupling reaction. The starting hypotheses were supported by single crystal X-ray analysis of an independently synthesized oxidative addition intermediate (Scheme 23C), showing indeed the anticipated cyclic, cationic structure and the widening of the mentioned φ1 and φ2 angles.
 | ||
| Scheme 23 Dynamic kinetic asymmetric Suzuki–Miyaura cross-coupling and dynamization mechanism. | ||
In parallel, Virgil, Stoltz and co-workers reported on the Pd-catalysed coupling of 1-(isoquinolin-1-yl)naphthalen-2-yl triflate with diphenylphosphine for the synthesis of QUINAP (Scheme 24A).49 Unlike the analogous bromide, it was found that the use of triflate as the coupling partner enables a dynamic process involving isomerization of an arylpalladium intermediate. As in the precedent case, this isomerization is proposed to take place between diastereomeric oxidative addition intermediates. In this case, however, the atropisomerization was suggested to proceed via a transition structure stabilized by an agostic interaction between the isoquinoline peri-hydrogen and the cationic Pd center. Some years later, our group reported a more comprehensive study of the Pd-catalysed C–P coupling reaction between heterobiaryl sulfonates and trialkylsilylphosphines. Using a similar JOSIPHOS-type bidentate phosphine, the methodology was used for the synthesis of several families of enantiomerically enriched heterobiaryl phosphines including QUINAP, PINAP, and QUINAZOLINAP analogues (Scheme 24B), accomplished with good yields and enantioselectivities.50 Although the previously mentioned crystallographic data supports a dynamization via cationic oxidative addition intermediates, DFT calculations were performed to further clarify which of the proposed mechanisms is favored. Using trimethylphosphine as a model ligand, it was found out that there exists a much higher stabilization in intermediate IOA, with isoquinolyl nitrogen coordinated to Pd, compared to intermediate IOA′ stabilized by an agostic interaction (ΔΔG‡ = +9.3 kcal mol) (Scheme 24C). This large energy difference is enough to completely discard the participation of IOA′ in the mechanism. Additionally, the calculated racemization barrier for IOA (ΔG‡ = 18.7 kcal mol−1) and the racemization rate (krac = 0.57 s−1) under the reaction conditions are in full agreement with the experimental results.
 | ||
| Scheme 24 Dynamic kinetic C–P coupling from heterobiaryl sulfonates. | ||
This DYKAT strategy could be extended to the synthesis of different functionalized heterobiaryls. The atroposelective dynamic kinetic asymmetric Buchwald–Hartwig amination of heterobiaryls is particularly challenging for the limited configurational stability of the products and the need to prevent hydrolytic cleavage of the heterobiaryl sulfonates used as the starting material in the precedent examples. In this case, however, heterobiaryl bromides are suitable substrates and the coupling with primary alkyl- and arylamines can be accomplished under mild conditions to afford the desired products in excellent yields and enantioselectivities when a combination of Pd(dba)2 and (S)-QUINAP is used as the catalyst system and NaOtBu as the base.51 Experimental evidences, including X-ray diffraction analysis of key intermediates, support the mechanism shown in Scheme 25A, in which the base plays a dual role, behaving also as an efficient bromide scavenger thanks to the low solubility of NaBr in toluene.
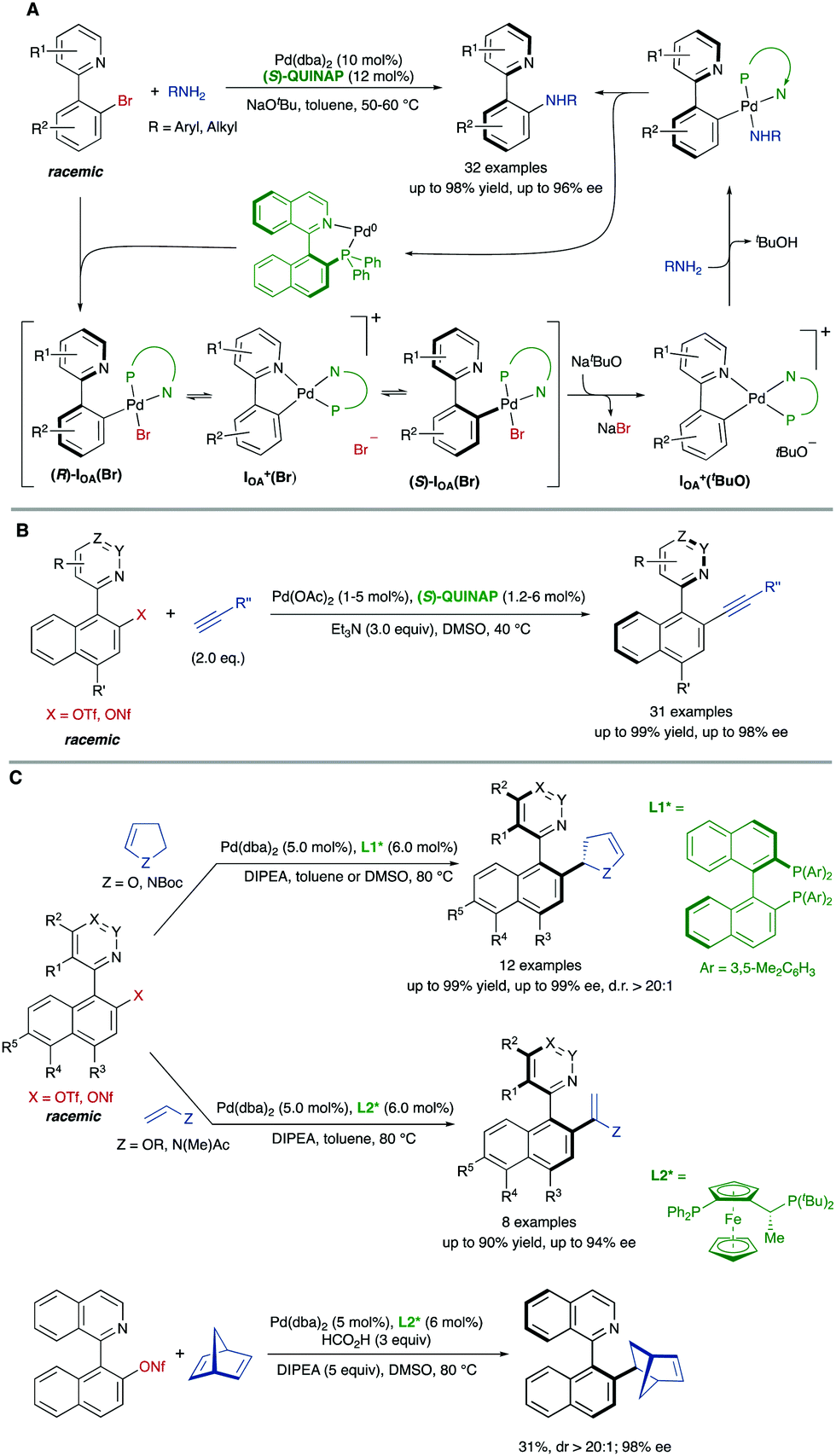 | ||
| Scheme 25 Dynamic kinetic asymmetric Pd-catalysed cross-coupling of heterobiaryl electrophiles via cationic, cyclic oxidative addition intermediates. | ||
(S)-QUINAP was also the ligand that provided the best results on the dynamic kinetic Cu-free atroposelective Sonogashira alkynylation of configurationally stable heterobiaryl sulfonates,52 enabling the synthesis of axially chiral heterobiaryl alkynes with a broad reaction scope (Scheme 25B). Aromatic and aliphatic terminal alkynes are well tolerated as coupling partners, and the catalyst loading could be decreased to 1 mol% when the reaction was performed at 2 mmol or larger scale.
Finally, our group developed also a dynamic kinetic asymmetric Pd-catalysed Heck reaction of heterobiaryl sulfonates with electron-rich olefins.53 Using (R)-DM-BINAP as the ligand, the coupling with 2,3-dihydrofuran or N-Boc protected 2,3-dihydropyrrole afforded the desired products with a simultaneous control of axial and central chirality (Scheme 25C). Different biaryl scaffolds were tested with excellent enantioselectivities and perfect diastereoselectivities. The reaction was also carried out with acyclic enol ethers and enamides to afford axially chiral heterobiaryl alkenes in high yields and enantioselectivities in the presence of a JOSIPHOS-type ligand. Preliminary studies using formic acid as the hydride source demonstrated that this Heck reaction could be performed with strained alkenes such as norbornadiene, achieving the reduced Heck product with a promising 98% ee and d.r. > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, although a lower reactivity was observed. DFT calculations showed that β-hydride elimination is the stereocontrolling step, where a large energy difference between the isomeric transition states accounts for the observed level of enantioselectivity and the observe absolute and relative configuration.
:1, although a lower reactivity was observed. DFT calculations showed that β-hydride elimination is the stereocontrolling step, where a large energy difference between the isomeric transition states accounts for the observed level of enantioselectivity and the observe absolute and relative configuration.
Conclusions and outlook
In recent years, intensive research activity focused on the development of methodologies for the synthesis of axially chiral biaryls and heterobiaryls. This phenomenon is in turn a direct consequence of the great perspectives for these compounds in diverse fields, including drug discovery, functional materials, and asymmetric catalysis, where they are key players as ligands or organocatalysts, being also particularly well suited for applications in emerging areas such as molecular motors design.54 Thus, many original strategies have been developed to fill significant gaps and expand the chemical space of this class of compounds. In many cases, transformation of readily available achiral or racemic (hetero)biaryl precursors appears as the most convenient approach in terms of efficiency and step economy, while complementing the scope of alternative approaches based of cross-coupling, chiral-to-axial chirality transfer or de novo ring construction.In this context, a few methodologies including kinetic resolutions or the diastereoselective version of the lactone ring opening have been developed time ago, but it has been only recently that a number of innovative strategies have appeared, improving significantly the efficiency for the preparation of established families of axially chiral derivatives and offering solutions for the synthesis of previously inaccessible families of (hetero)biaryls.
From a conceptual viewpoint, readers will find out that most of the desymmetrization and (dynamic) resolution strategies outlined along this review are distinct to those known for chiral compounds based on central chirality. Thus, while the racemization of stereogenic centres requires bond breaking/bond forming events, the generation of a stereogenic axis requires specifically designed approaches, as discussed along this review. Many of the most recent contributions highlighted herein are creative and conceptually novel strategies that are likely to stimulate further developments, ultimately enriching the prospect of this exciting group of compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish Ministerio de Ciencia e Innovación (Grants PID2019-106358GB-C21, PID2019-106358GB-C22, contract RYC-2017-22294 for V. H., fellowship FPU18/02677 for C. R.-F.), European funding (ERDF) and Junta de Andalucía (Grants P18-FR-3531, P18-FR-644, US-1262867, US-1260906, and fellowship for J. A. C.).Notes and references
- For a recent monography see: Atropisomerism and Axial Chirality, ed. J. M. Lassaletta, World Scientific, London, 2019 Search PubMed.
- J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131–190 CrossRef CAS.
- C. Zheng and S.-L. You, Asymmetric Oxidative Biaryl Coupling Reactions, RSC Catalysis Ser., 2015, ch. 3, vol. 25, pp. 92–125 Search PubMed.
- H. Yang, J. Chen and L. Zhou, Chem. – Asian J., 2020, 15, 2939–2951 CrossRef CAS.
- A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC.
- T. Hayashi, S. Niizuma, T. Kamikawa, N. Suzuki and Y. Uozumi, J. Am. Chem. Soc., 1995, 117, 9101–9102 CrossRef CAS.
- E. S. Munday, M. A. Grove, T. Feoktistova, A. C. Brueckner, D. M. Walden, C. M. Young, A. M. Z. Slawin, A. D. Campbell, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 7897–7905 CrossRef CAS.
- K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964–3970 CrossRef CAS.
- J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS.
- Q. Wang, Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2019, 141, 9504–9510 CrossRef CAS.
- Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS.
- J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, L. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708–6712 CrossRef CAS.
- B.-B. Zhan, L. Wang, J. Luo, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 3568–3572 CrossRef CAS.
- A. Romero-Arenas, V. Hornillos, J. Iglesias-Sigüenza, R. Fernández, J. López-Serrano, A. Ros and J. M. Lassaletta, J. Am. Chem. Soc., 2020, 142, 2628–2639 CrossRef CAS.
- C. He, M. Hou, Z. Zhu and Z. Gu, ACS Catal., 2017, 7, 5316–5320 CrossRef CAS.
- C.-X. Ye, S. Chen, F. Han, X. Xie, S. Ivlev, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 13552–13556 CrossRef CAS.
- J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251–1255 CrossRef CAS.
- T. Shimada, Y.-H. Cho and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 13396–13397 CrossRef CAS.
- A. Kina, H. Miki, Y.-H. Cho and T. Hayashi, Adv. Synth. Catal., 2004, 346, 1728–1732 CrossRef CAS.
- K. Zhao, L. Duan, S. Xu, J. Jiang, Y. Fu and Z. Gu, Chem, 2018, 4, 599–612 CAS.
- Q. Li, M. Zhang, S. Zhan and Z. Gu, Org. Lett., 2019, 21, 6374–6377 CrossRef CAS.
- M. Hou, R. Deng and Z. Gu, Org. Lett., 2018, 20, 5779–5783 CrossRef CAS.
- X. Xue and Z. Gu, Org. Lett., 2019, 21, 3942–3945 CrossRef CAS.
- K. Zhu, K. Xu, Q. Fang, Y. Wang, B. Tang and F. Zhang, ACS Catal., 2019, 9, 4951–4957 CrossRef CAS.
- L. Duan, K. Zhao, Z. Wang, F.-L. Zhang and Z. Gu, ACS Catal., 2019, 9, 9582–9858 Search PubMed.
- R. Deng, J. Xi, Q. Li and Z. Gu, Chem, 2019, 5, 1–13 Search PubMed.
- H. Aoyama, M. Tokunaga, J. Kiyosu, T. Iwasawa, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2005, 127, 10474–10475 CrossRef CAS.
- S. Shirakawa, X. Wu and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 14200–14203 CrossRef CAS.
- G. Ma, Y. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014, 53, 11818–11821 CrossRef CAS.
- S. Lu, S. B. Poh and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 11041–11045 CrossRef CAS.
- W. Liu, Q. Jiang and X. Yang, Angew. Chem., Int. Ed., 2020, 59, 23598–23602 CrossRef CAS.
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741–2745 CrossRef CAS.
- J. Wang, M.-W. Chen, Y. Ji, S.-B. Hu and Y.-G. Zhou, J. Am. Chem. Soc., 2016, 138, 10413–10416 CrossRef CAS.
- G. Bringmann and T. Hartung, Angew. Chem., Int. Ed. Engl., 1992, 31, 761–762 CrossRef.
- G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS.
- G. Bringmann, M. Breuning, P. Henschel and J. Hinrichs, Org. Synth., 2002, 79, 72 CrossRef CAS.
- T. Ashizawa, S. Tanaka and T. Yamada, Org. Lett., 2008, 10, 25212524 CrossRef.
- C. Yu, H. Huang, X. Li, Y. Zhang and W. Wang, J. Am. Chem. Soc., 2016, 138, 69566959 Search PubMed.
- L. Hu, G.-Q. Chen, B.-J. Lin, Q.-W. Zhang, Q. Yin and X. Zhang, Org. Lett., 2019, 21, 5575–5580 CrossRef CAS.
- K. Mori, T. Itakura and T. Akiyama, Angew. Chem., Int. Ed., 2016, 55, 11642–11646 CrossRef CAS.
- J. Zhang and J. Wang, Angew. Chem., Int. Ed., 2018, 130, 474–478 CrossRef.
- D. Guo, J. Zhang, B. Zhang and J. Wang, Org. Lett., 2018, 20, 6284–6288 CrossRef CAS.
- S. Staniland, R. W. Adams, J. J. W. McDouall, I. Maffucci, A. Contini, D. M. Grainger, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 10755–10759 CrossRef CAS.
- V. Hornillos, J. A. Carmona, A. Ros, J. Iglesias-Sigüenza, J. López-Serrano, R. Fernández and J. M. Lassaletta, Angew. Chem., Int. Ed., 2018, 57, 3777–3781 CrossRef CAS.
- G. A. I. Moustafa, Y. Oki and S. Akai, Angew. Chem., Int. Ed., 2018, 57, 10278–10282 CrossRef CAS.
- J. D. Joliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558–562 CrossRef.
- A. Ros, B. Estepa, P. Ramírez-López, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2013, 135, 15730–15733 CrossRef CAS.
- V. Bhat, S. Wang, B. M. Stoltz and S. C. Virgil, J. Am. Chem. Soc., 2013, 135, 16829–16832 CrossRef CAS.
- P. Ramírez-López, A. Ros, B. Estepa, R. Fernández, B. Fiser, E. Gómez-Bengoa and J. M. Lassaletta, ACS Catal., 2016, 6, 3955–3964 CrossRef.
- P. Ramírez-López, A. Ros, A. Romero-Arenas, J. Iglesias-Sigüenza, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2016, 138, 12053–12056 CrossRef.
- V. Hornillos, A. Ros, P. Ramírez-López, J. Iglesias-Sigüenza, R. Fernández and J. M. Lassaletta, Chem. Commun., 2016, 52, 14121–14124 RSC.
- J. A. Carmona, V. Hornillos, P. Ramírez-López, A. Ros, J. Iglesias-Sigüenza, E. Gómez-Bengoa, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2018, 140, 11067–11075 CrossRef CAS.
- B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, Nat. Chem., 2016, 8, 860–866 CrossRef CAS.
Footnotes |
| † These authors contribute equally. |
| ‡ A characteristic aspect of axially chiral compounds is that, in most cases, the racemization process does not involve bond breaking/bond forming events. Thus, fluxional biaryls can be classified as achiral or racemic depending on the energy required for the atropisomers to interconvert at a given temperature. Considering that the barriers for the atropisomerization are not always known, the difference between desymmetrization of fluxional biaryls as discussed in Sections 2.2 and 2.3 and dynamic kinetic resolution as discussed in Sections 4.1 and 4.2 is not always clearly defined and might be controversial. In fact, some of the transformations that have been classified herein as desymmetrizations were described as DKR in the original publications. In this review, biaryls that feature estimated interconversion barriers under ca. 15 kcal mol−1 have been considered achiral, although in some cases there are no precise data available. |
| This journal is © The Royal Society of Chemistry 2021 |