
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective “organocatalysis in disguise” by the ligand sphere of chiral metal-templated complexes
Vladimir A.
Larionov
 *abc,
Ben L.
Feringa
*b and
Yuri N.
Belokon
*a
*abc,
Ben L.
Feringa
*b and
Yuri N.
Belokon
*a
aA. N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences, Vavilov Street 28, 119991 Moscow, Russian Federation. E-mail: larionov@ineos.ac.ru; yubel@ineos.ac.ru
bCentre for Systems Chemistry, Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
cPeoples’ Friendship University of Russia (RUDN University), Miklukho-Maklaya Street 6, 117198 Moscow, Russian Federation
First published on 14th July 2021
Abstract
Asymmetric catalysis holds a prominent position among the important developments in chemistry during the 20th century. This was acknowledged by the 2001 Nobel Prize in chemistry awarded to Knowles, Noyori, and Sharpless for their development of chiral metal catalysts for organic transformations. The key feature of the catalysts was the crucial role of the chiral ligand and the nature of the metal ions, which promoted the catalytic conversions of the substrates via direct coordination. Subsequently the development of asymmetric organic catalysis opened new avenues to the synthesis of enantiopure compounds, avoiding any use of metal ions. Recently, an alternative approach to asymmetric catalysis emerged that relied on the catalytic functions of the ligands themselves boosted by coordination to metal ions. In other words, in these hybrid chiral catalysts the substrates are activated not by the metal ions but by the ligands. The activation and enantioselective control occurred via well-orchestrated and custom-tailored non-covalent interactions of the substrates with the ligand sphere of chiral metal complexes. In these metal-templated catalysts, the metal served either as a template (a purely structural role), or it constituted the exclusive source of chirality (metal-centred chirality due to the spatial arrangement of achiral or chiral bi-/tridentate ligands around an octahedral metal centre), and/or it increased the Brønsted acidity of the ligands. Although the field is still in its infancy, it represents an inspiring combination of both metal and organic catalysis and holds major unexplored potential to push the frontiers of asymmetric catalysis. Here we present an overview of this emerging field discussing the principles, applications and perspectives on the catalytic use of chiral metal complexes that operate as “organocatalysts in disguise”. It has been demonstrated that these chiral metal complexes are efficient and provide high stereoselective control in asymmetric hydrogen bonding catalysis, phase-transfer catalysis, Brønsted acid/base catalysis, enamine catalysis, nucleophilic catalysis, and photocatalysis as well as bifunctional catalysis. Also, many of the catalysts have been identified as highly effective catalysts at remarkably low catalyst loadings. These hybrid systems offer many opportunities in the synthesis of chiral compounds and represent promising alternatives to metal-based and organocatalytic asymmetric transformations.
 Vladimir A. Larionov | Vladimir A. Larioniv received a MSc degree in Bioorganic Chemistry in 2011 from Baskir State University, Ufa (Russia). He obtained his PhD from A. N. Nesmeyanov Institute of Organoelement Compounds RAS (INEOS RAS), Moscow in 2014 under the supervision of Dr Victor Maleev and Prof. Yuri Belokon. Then, he carried out postdoctoral research with Prof. Eric Meggers at Philipps-Universität Marburg (Marburg, Germany) from 2015 to 2016. He visited the University of Groningen (Stratingh Institute for Chemistry, the Netherlands) in summer 2019 and worked with Nobel Prize Laureate Prof. Ben Feringa as a visiting researcher. Currently, he is a senior researcher/group leader at INEOS RAS (Moscow, Russia) and an assistant at RUDN University (Moscow, Russia). His current research is focused on asymmetric metal complex catalysis, phase-transfer catalysis, hydrogen bond donor catalysis, asymmetric amino acid synthesis (in particular, by radical chemistry) and green chemistry. |
 Ben L. Feringa | Ben L. Feringa obtained his PhD degree in 1978 at the University of Groningen in the Netherlands under the guidance of Prof. Hans Wynberg. After working as a research scientist at Shell he was appointed full professor at the University of Groningen in 1988 and named the distinguished Jacobus H. van't Hoff Professor of Molecular Sciences in 2004. He was elected foreign honorary member of the American Academy of Arts and Sciences and member of the Royal Netherlands Academy of Sciences. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, photopharmacology, self-assembly and nanosystems. |
 Yuri N. Belokon | Yuri N. Belokon was awarded an Engineer-chemist diploma from the USSR Institute of Gas and Oil Industry in 1960. He obtained his PhD from Nesmeyanov Institute of Organoelement Compounds RAS (INEOS RAS), Moscow in 1968 under the supervision of Dr V. M. Belikov. Subsequently, he stayed at INEOS RAS, and received a DSc in 1980 and made a full professor in 1990. He was head of the Laboratory of Asymmetric Catalysis at INEOS RAS from 1990 until 2009 and, at present, he is a chief scientist. His research interests include the following topics: asymmetric catalysis by chiral metal complexes and organocatalysts; asymmetric phase-transfer catalysis; asymmetric synthesis of amino acids; fixation of CO2 into cyclic carbonates; preparation of novel charge assisted hydrogen bonding frameworks and their use in organic catalysis. |
1. Introduction
Asymmetric catalysis is nowadays the most important and desirable methodology for the synthesis of chiral molecules.1–5 Noting the key role of precise control of chirality in among others drug design and natural product synthesis and the major industrial implications it came not as a surprise that the world community recognized the importance of asymmetric catalysis when in 2001 the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry to W. S. Knowles, R. Noyori, and K. B. Sharpless for their development of catalytic asymmetric hydrogenation and oxidation reactions.6 Asymmetric catalysis is widely applied in the pharmaceutical industry for the large-scale production synthesis of pharma intermediates and commercial drugs.7–9 Commonly, there are two types of chiral catalytic systems used in academia and industry: (1) chiral metal complexes10–17 and (2) organocatalysts.18–33 Nowadays, dual catalytic systems find ever-increasing applications where both chiral organo- and metal catalysts are combined to promote a target reaction.34,35The key feature of metal-catalysts is the crucial role of the metal ions which promote the catalytic conversions of the coordinated substrates. The chiral complexes based on transitional and noble metals are widely applied in asymmetric hydrogenation (Rh, Ir, Ru complexes),10,36–38 in enantioselective C–H activation (Pd, Ir, Rh, Ni, Co complexes)39–41 and cross-coupling reactions (Pd, Cu and Ni complexes),42,43 in the field of organic synthesis for the rapid construction of complex molecular architectures and for the total synthesis of natural products under mild conditions (Cu, Au, Pd and Pt complexes).44–48 Another important class of chiral metal catalysts is represented by classical Lewis acids that activate organic molecules via strong coordination binding with acidic metal centres.49–53 A small amount of a chiral metal complex (in most cases around 1 mol%) can provide a great number of enantioselective transformations leading to stereocentres at carbon, nitrogen, sulphur, silicon and other atoms.54–57
Although purely organic natural asymmetric catalysts, i.e. enzymes and chiral alkaloids,58–63 have been very well known for a long time, the elaboration of asymmetric, artificial, organic non-metal catalysis began in the early 2000s after breakthrough studies on thiourea derivative and proline catalysis.26,28,64,65 Unfortunately, most of the chiral organocatalysts were and still are less active than most metal based ones and often require long reaction times. As a consequence, their use on an industrial scale is hampered by the need for high catalyst loading in order to provide efficient catalysis and a high degree of enantioselective control.66,67 Although, recently highly efficient asymmetric organocatalysts have been developed (with requirement of <1 mol% (ppm levels) of catalyst loadings)67–70 there is still need to develop more active and alternative catalysts and related catalytic reactions. The underlying reasons for this observation can be attributed to the relatively weak non-covalent interactions of the substrate and catalyst (e.g., hydrogen bonding,71–76 ion-pairing,77–79etc.).80–83 On the other hand, non-covalent interactions play a key role in catalysis by lowering the kinetic barriers to reactions through transition state stabilization.71
Recently, another approach to asymmetric catalysis has emerged that relies on the catalytic functions of the ligands themselves boosted by the coordination to the metal ions.84–87 Several groups have independently developed a series of chiral metal complex catalysts for asymmetric transformations88–137 where the substrates are activated by the ligand sphere of metal complexes via well-orchestrated and custom-tailored non-covalent interactions (Fig. 1).80–83 In these systems the Lewis acidic metal does not activate the substrates directly but increases the catalytic properties of the ligands themselves. For instance, the Brønsted acidity of the coordinated ligands, such as NH2 groups bound to cobalt(III) ions (Fig. 1a and c), greatly increases as compared to the original non-coordinated form.91,95,98,105,109,116–118,132,136 Gladysz nicknamed these chiral systems chiral “organocatalysts in disguise”.84–87
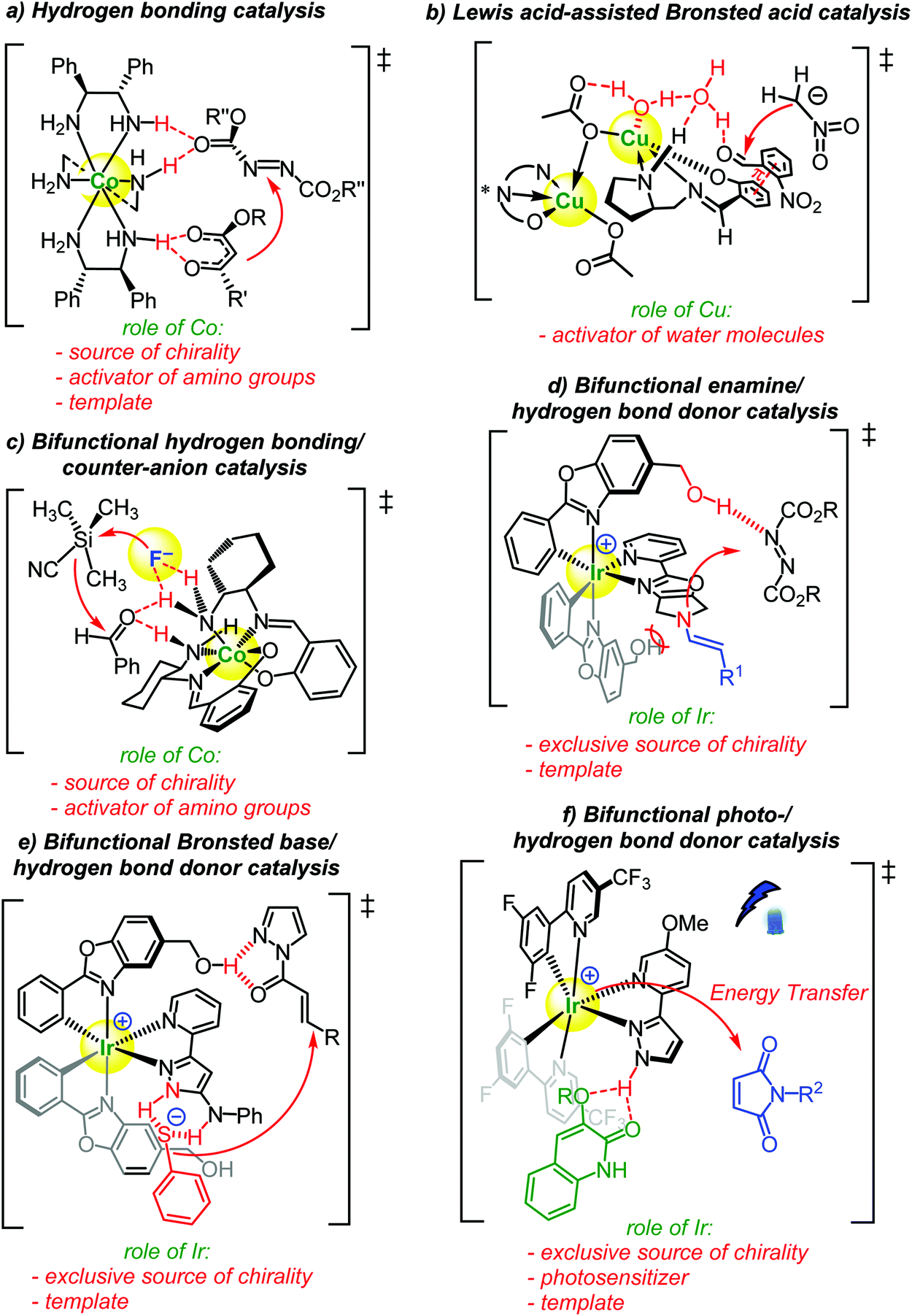 | ||
| Fig. 1 Representative examples of chiral metal-templated “organocatalysts in disguise” and the activation mode of substrates: (a) enantioselective α-amination of 1,3-dicarbonyl compounds with azodicarboxylates;117 (b) enantioselective Henry reaction;129 (c) enantioselective cyanosilylation of benzaldehyde;93 (d) enantioselective α-amination of aldehydes with azodicarboxylates;99 (e) enantioselective sulfa-Michael addition;100 (f) enantioselective intermolecular [2+2] photocycloaddition of 3-alkoxyquinolones with maleimides.130 | ||
Structurally, the metal serves either as a template (a purely structural role) (Fig. 1a and c–f), or constitutes the exclusive source of chirality (metal-centred chirality)138–149 (Fig. 1d–f) due to the spatial arrangement of achiral or chiral bi-/tridentate ligands around an octahedral metal centre. The chiral configuration is designated Λ in case when the chiral/achiral bi- or tridentate ligands form a left-handed propeller and Δ in the case when they form a right-handed propeller).138–149 In some cases, the metal ion is a Lewis acid which activates water molecules, in fact, allowing them to perform real Brønsted acid catalysts (Fig. 1b).102,129 Recently, it was also demonstrated that the metal serves as a photosensitizer activating the substrate by energy transfer (Fig. 1f).121,130
The field is a fledging one merging chiral organic catalysis and chiral inorganic coordination chemistry taking benefit from unique features of both, and holds great unexplored potential for asymmetric catalysis. It has been proven that the reported chiral metal complexes were efficient catalysts and provided high stereoselective control in asymmetric hydrogen bonding catalysis (Fig. 1),91,95–98,101,109,111,114,117,118,124,131,133 phase-transfer catalysis,95,98,105,125,126 Brønsted acid/base catalysis (Fig. 1b and e),94,100,102–104,106,107,115,122,123,126–129 enamine catalysis (Fig. 1d),99 nucleophilic catalysis,120 and photocatalysis (Fig. 1f)121,130 as well as bifunctional catalysis (Fig. 1c–f).93,116,118,121,130 Also, many of these systems have been identified as effective catalysts at remarkably low catalyst loadings, some of them even down to the parts-per-million range.111,114 Importantly, most of the catalysts presented here are robust and air stable that allows to conduct the reactions under air and in the presence of the water molecules. With this timely review of a less recognized but highly promising approach in asymmetric catalysis we aim to provide perspectives and discuss the basic principles and important developments of this new class of chiral metal catalysts with ‘non-innocent’ ligands that will operate exclusively as organocatalysts and where the activation of the substrates will take place by the ligand sphere of the metal-templated complexes.
2. Asymmetric hydrogen bonding catalysis with chiral-at-metal complexes
2.1 ‘Werner-type’ chiral cobalt(III) complexes as hydrogen bond donor catalysts
In 1911, Alfred Werner reported the preparation of enantiopure tris(ethylenediamine) complexes Λ/Δ-[Co(en)3]X3 with X− = Br−, Cl−, or NO3−.150–154 Since 2008, Gladysz and co-workers explored ‘Werner-type’ chiral octahedral cobalt(III) complexes as chiral hydrogen bond donor catalysts in asymmetric reactions (Fig. 2).91,109,116,117,124,132,133,136 | ||
| Fig. 2 ‘Werner-type’ chiral octahedral cobalt(III) complexes as asymmetric hydrogen bond donor catalysts.91,109,116,117,124,132,133,136 | ||
It was proven by single crystal X-ray crystallographic analysis that the counter anions formed multidentate hydrogen bonds with the amino groups of the cobalt(III) complexes.155,156 Evidently, the Brønsted acidity of the NH2 groups was enhanced significantly. Unfortunately, the complexes were very poorly soluble in organic solvents where most organic reactions are routinely conducted. To solve this problem, simple halogen counter-anions were substituted by hydrophobic tetrakis[(3,5-trifluoromethyl)phenyl]borate anions (BArf−) under phase-transfer conditions to generate Δ-1.91 The Gladysz group demonstrated that Δ-1 catalysed the Michael addition of dimethyl malonate to cyclopentenone, giving the corresponding addition product in a 78% yield and with moderate enantioselectivity up to 33% ee (Scheme 1).91 The authors postulated that activation of the substrate by hydrogen bond donation from the chiral complex cation was a driving force for the conversion and governing stereocontrol. Subsequently, effects of the metal and charge upon enantioselectivities were analyzed.133 It was demonstrated that a number of properties appeared to correlate to the NH Brønsted acidity order ([Pt(en)3]4+ > [Cr(en)3]3+ > [Co(en)3]3+ > [Rh(en)3]3+ > [Ir(en)3]3+.133
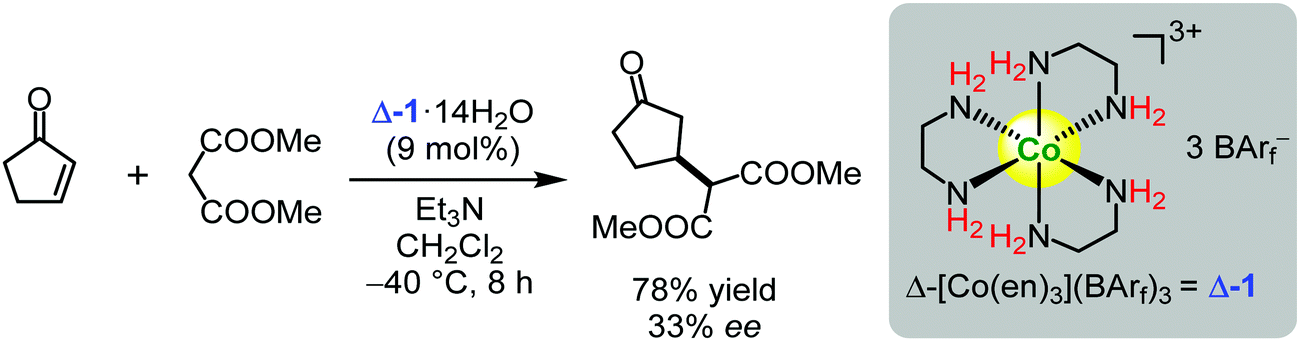 | ||
| Scheme 1 Enantioselective Michael addition of dimethyl malonate to cyclopentenone catalysed by the chiral ‘Werner-type’ cobalt(III) complex Δ-1 (Gladysz, 2008).91 | ||
In 2015, Gladysz and co-workers introduced a modified chiral ‘Werner type’ complex Λ(S,S)-2a containing a chiral (S,S)-1,2-diphenylethylenediamine ligand instead of achiral tris(ethylenediamine) ligands (Scheme 2).109 The complex was a highly efficient catalyst in terms of yield and enantioselectivity for the asymmetric addition of dimethyl malonates to nitroalkenes giving the corresponding Michael products in up to 98% yield and with up to 98% ee (Scheme 2).109
 | ||
| Scheme 2 Enantioselective Michael addition of dimethyl malonates to nitroalkenes catalysed by the chiral cobalt(III) complex Λ(S,S)-2a (Gladysz, 2015).109 | ||
The authors expanded the application of the (S,S)-1,2-diphenylethylenediamine-based cobalt(III) complex Λ(S,S)-2a to catalyse other important asymmetric transformations. Accordingly, the catalyst, Δ(S,S)-2b with the inverted configuration at the metal centre (Λ → Δ) and containing two B(C6F5)4− anions and one chloride anion, was applied in the Michael addition of various 1,3-dicarbonyl compounds to di-tert-butyl azodicarboxylate i.e. an important amination to provide e.g. amino acid precursors.117 The products were obtained with very high enantioselectivities (up to >99% ee) and excellent yields (up to 99%) (Scheme 3).117
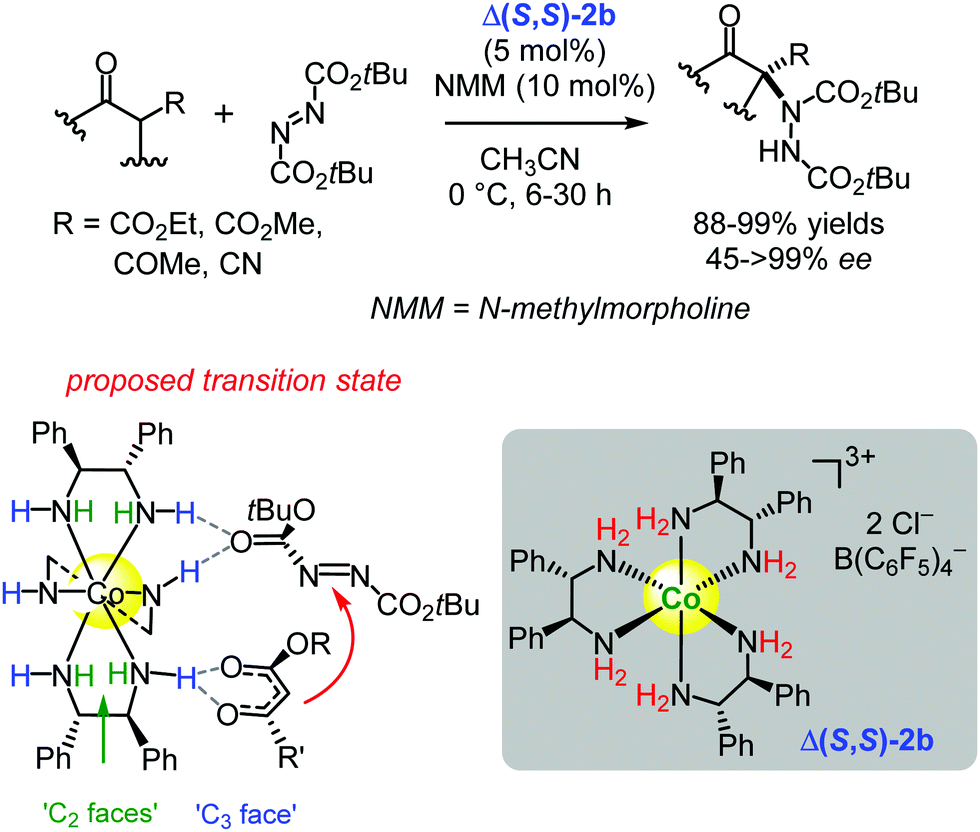 | ||
| Scheme 3 Enantioselective α-amination of 1,3-dicarbonyl compounds catalysed by the chiral cobalt(III) complex Δ(S,S)-2b (Gladysz, 2016).117 The possible transition state shows the proposed hydrogen-bonding interactions between the catalyst and the substrates (Adapted with permission from ref. 117. Copyright © 2016 American Chemical Society). | ||
The authors proposed that the substrate activation may take place at the sterically less congested ‘C3-faces’ of Δ(S,S)-2bvia their three roughly syn-periplanar-oriented NH bonds (Scheme 3). However, as they indicated there is yet not enough evidence to pinpoint the precise mode of substrate activation, and the activation may alternatively occur through NH interactions at the more congested ‘C2-faces’ or through ‘mixed’ NH interactions at both the ‘C3-faces’ and the ‘C2-faces’ (Scheme 3).117
The chiral complexes Δ(S,S)-2c and Λ(S,S)-2d were also applied as catalysts for asymmetric additions of stabilized carbanions to in situ generated imines formed from α-amino sulfones (Schemes 4 and 5).124 Complex Λ(S,S)-2d differs from Δ(S,S)-2c with the inverted configuration at the metal centre, and it features one B(C6F5)4− counter-anion instead of one BArf− counter-anion. Complex Δ(S,S)-2c was an efficient catalyst for the asymmetric addition of malonates to in situ generated Boc- and Cbz-protected imines, giving the desired products with yields of 90–97% and enantioselectivities ranging from good to excellent (79–99% ee) (Scheme 4).124
 | ||
| Scheme 4 Enantioselective additions of malonates to in situ generated imines catalysed by the chiral cobalt(III) complex Δ(S,S)-2c (Gladysz, 2017).124 | ||
 | ||
| Scheme 5 Enantioselective additions of nitroalkanes to in situ generated imines catalysed by the chiral cobalt(III) complex Λ(S,S)-2d (Gladysz, 2017).124 | ||
In turn, Λ(S,S)-2d efficiently catalysed the asymmetric addition of nitroalkanes to in situ generated Boc-protected aryl imines. The corresponding products were obtained in yields up to 93% and enantioselectivities up to 91% ee (Scheme 5).124
Quite recently, the catalyst Δ(S,S)-2c was applied for the enantioselective generation of quaternary carbon stereocenters by the addition of substituted cyanoacetate esters to acetylenic esters (Scheme 6).136 The desired products were obtained in high yields with high Z-selectivity and enantioselectivity up to 99% ee.136
 | ||
| Scheme 6 Enantioselective additions of substituted cyanoacetate esters to acetylenic esters catalysed by the chiral cobalt(III) complex Δ(S,S)-2c (Gladysz, 2020).136 | ||
Recently, the Gladysz group also demonstrated that the chiral cobalt(III) complexes based on sepulchrate and sarcophagine157,158 (for example, Λ-3) could act as enantioselective hydrogen bond donor catalysts in Michael type C–C and C–N bond forming reactions (Scheme 7).131 Although the enantioselectivities of the reported reactions were rather low (30–57% ee), the authors proposed future directions for improvement of the catalyst's efficiencies.131
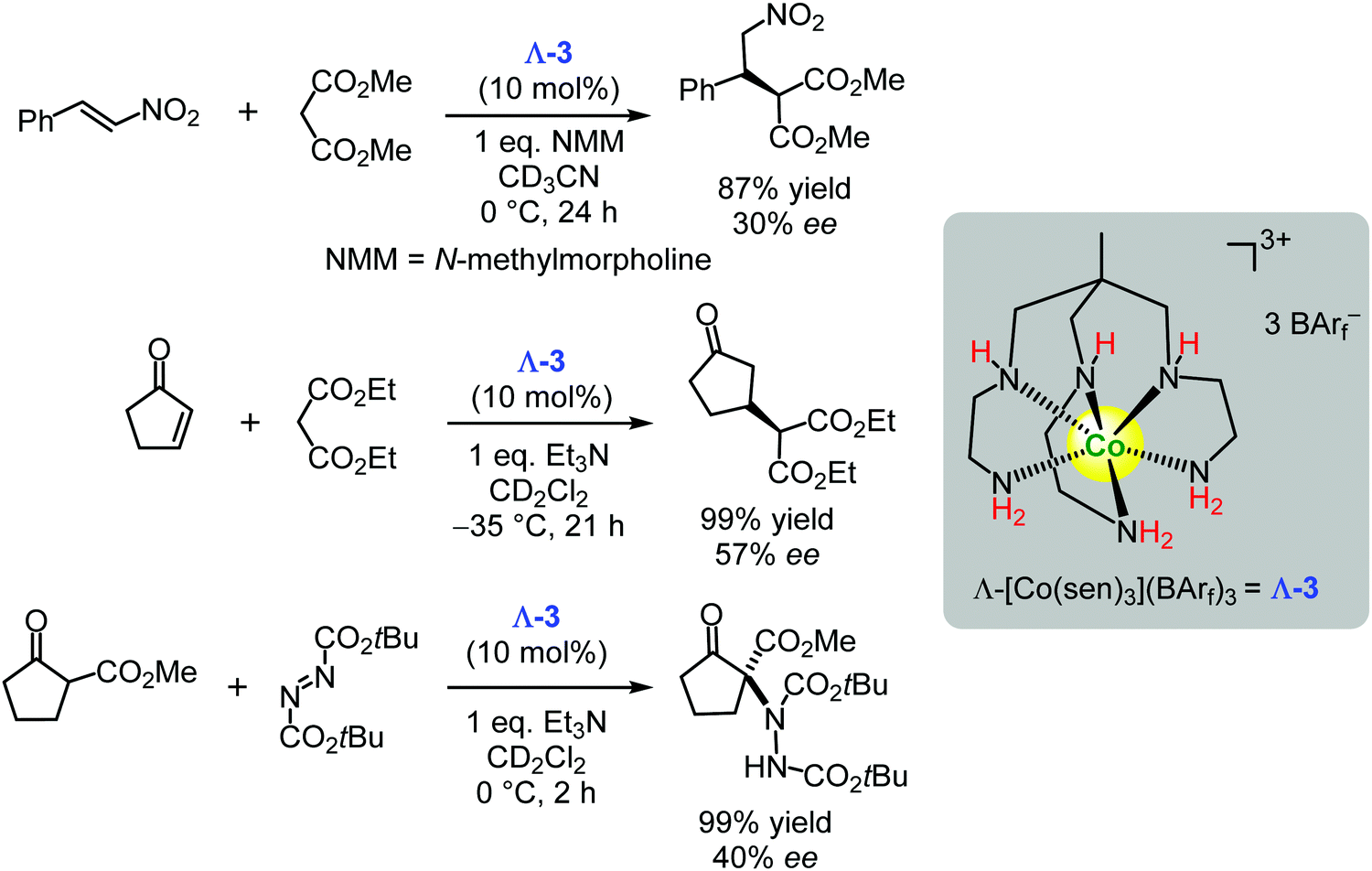 | ||
| Scheme 7 Enantioselective Michael type additions catalysed by the chiral cobalt(III) complex Λ-3 (Gladysz, 2019).131 | ||
Generally, in the case of ‘Werner-type’ cobalt(III) complexes of Δ-1 type, the metal serves as a template, in some cases the exclusive source of chirality, and as an activator of the Brønsted acidity of amino groups, in fact, making them efficient hydrogen bond donors. It also becomes evident from these studies that control of reactivity and stereoselectivity can be achieved through choice of metal ions, ligands and counterions holding promise for fine tune for a wide range of chiral transformations.
2.2 Chiral-at-metal iridium(III) complexes as dual hydrogen bond donor/acceptor catalysts
In 2013, Meggers and Gong introduced a novel class of chiral-only-at-metal148 octahedral bis-cyclometalated iridium(III) complexes as highly efficient asymmetric hydrogen bond donor/acceptor catalysts for enantioselective transformations (Fig. 3).96 Since then, these catalysts have been widely employed in the enantioselective conjugate reduction of β,β-disubstituted nitroalkenes with Hantzsch esters and Friedel–Crafts alkylation of indoles with β,β-disubstituted nitroalkenes (Schemes 8–11).96,97,108,111–114 | ||
| Fig. 3 Chiral-at-metal octahedral bis-cyclometalated iridium(III) complexes as asymmetric hydrogen bond donor/acceptor catalysts.96,97,108,111–114 | ||
 | ||
| Scheme 8 Enantioselective reduction of β,β-disubstituted nitroalkenes with a Hantzsch ester catalysed by the chiral iridium(III) complex Λ-4 (Gong and Meggers, 2013).96 The 3D model shows the proposed hydrogen-bonding interactions between the catalyst and the substrates (Reprinted with permission from ref. 96. Copyright © 2013 American Chemical Society). | ||
 | ||
| Scheme 9 Enantioselective reduction of β,β-disubstituted nitroalkenes with a Hantzsch ester catalysed by the chiral C2-symmetric iridium(III) complex Λ-7 (Gong and Meggers, 2016).111 | ||
 | ||
| Scheme 10 Enantioselective Friedel–Crafts alkylation of indoles with nitroalkenes catalysed by the chiral iridium(III) complexes Λ-5–6 (Gong and Meggers, 2013-2016).97,108,112,113 The 3D models show the proposed hydrogen-bonding interactions between the catalyst and the substrates (Reprinted with permissions from ref. 97 and 112. Copyright © 2013, 2016 Wiley-VCH Verlag GmbH & Co. KGaA; reproduced from ref. 113 with permission from the Royal Society of Chemistry, copyright © 2016). | ||
 | ||
| Scheme 11 Enantioselective Friedel–Crafts alkylation of indoles with α-substituted β-nitroacrylates catalysed by the chiral iridium(III) complex Λ(R,R)-8 (Gong and Meggers, 2016).114 The 3D model shows the proposed hydrogen-bonding interactions between the catalyst and the substrates (Reprinted with permission from ref. 114. Copyright © 2016 American Chemical Society). | ||
They demonstrated that the iridium(III) complex Λ-4 efficiently catalysed the enantioselective reduction of β,β-disubstituted nitroalkenes with a Hantzsch ester as the reducing agent at remarkably low catalyst loadings (up to 0.1 mol%) (Scheme 8).96 The corresponding reduced (R)-nitroalkane products were obtained in high yields with excellent enantioselectivities (93–99% ee) (Scheme 8).96
Based on the experimental results, the authors proposed a model for the activation of the substrates via hydrogen-bonding interactions between the catalyst and the substrates (Scheme 8). They assumed that the amidopyrazole moiety was responsible for activating the nitroalkene by double hydrogen bonding (similar to chiral thioureas159–162), which increased the electrophilicity of the nitroalkene, while one of the two cyclometalated phenylbenzoxazole ligands placed an OH group in the proper position to increase the hydride donor propensity of the Hantzsch ester through the formation of a hydrogen bond between the NH group of the Hantzsch ester and the lone pair of the OH group (Scheme 8).96
Subsequently a second-generation metal-templated hydrogen bonding catalyst Λ-7 was developed, which was C2-symmetrical and therefore contained two catalytic sites per molecule of Λ-7. The complex efficiently catalysed the enantioselective reduction of β,β-disubstituted nitroalkenes with a Hantzsch ester (Scheme 9).111
The authors reported impressive results using only 0.004 mol% of complex Λ-7 (40 ppm). The corresponding chiral (R)-nitroalkanes were obtained in high yields and excellent enantioselectivities (91–98% ee) with turnover numbers (TON) as high as 20250.111
Based on X-ray crystallography, NMR spectroscopy, computational investigations, and kinetic experiments it was shown that the asymmetric catalysis was merely driven by hydrogen-bonding and van der Waals interactions. Furthermore, numerous experiments revealed that there was no direct involvement of the metal ion in the transition state of the reactions and confirmed that the metal exerted a purely structural role and thus indirectly served as the source of chirality.111
After these pioneering reports, Meggers and co-workers applied the catalytic system to enantioselective Friedel–Crafts alkylation type reactions (Scheme 10).97,108,112–114 For instance a modified version of the octahedral chiral-only-at-metal iridium(III) complex Λ-5 with the stronger H-bond acceptor N,N-diethylcarboxamide efficiently catalysed the asymmetric Friedel–Crafts alkylation of indoles with the α-substituted-β-nitroacrylates as electrophiles (Scheme 10a).97 The “conjugate-addition products” were obtained in yields in the range of 72–97% and enantioselectivities from good to excellent (76–98% ee).97
The same chiral-at-metal iridium(III) complex Λ-5 was shown to be an excellent catalyst for the asymmetric Friedel–Crafts β-alkylation of 2,5-disubstituted pyrroles with the α-substituted-β-nitroacrylates.113 The coupling gave valuable chiral building blocks containing all-carbon quaternary stereocentres with yields in the range of 75–98% and high enantioselectivities (86 → 99% ee) (Scheme 10b).113 It was proposed that the stereocontrol in the challenging construction of the quaternary stereocentres was due to the formation of three hydrogen bonds by the catalyst with the substrates. One of those fixed the nucleophile, and the other two properly oriented the nitro-group of the electrophile as illustrated in Scheme 10a and b.97,113
Expanding the substrate class to 2-nitro-3-arylacrylates (Scheme 10c),108 a metal-templated iridium(III) complex Λ-6a, in which the benzoxazole ligands of Λ-5 were replaced by benzothiazoles, was shown to effectively catalyses the asymmetric Friedel–Crafts alkylation of indoles with α-nitroacrylates giving the corresponding products in up to 97% yield and up to 98% ee, albeit the diastereoselectivity was low (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30).108
:30).108
The same authors reported the application of a new member of the iridium(III) family designated Λ-6b. The modification consisted of the introduction of 2,6-dimethyl phenyl substituents at the metalated phenyl moiety. An asymmetric Friedel–Crafts alkylation of indoles with (Z)-1-bromo-1-nitrostyrenes (Scheme 10d) provided the desired products with 78–89% yields and with enantioselectivities up to 96%; although, the diastereosectivity control was low (up to 74:26% dr).112 Notably, the bromo substituent played an important role in delivering high enantioselectivity, in contrast to unsubstituted nitrostyrene.112
A next-generation catalyst Λ(R,R)-8 for the asymmetric Friedel–Crafts alkylation of indoles with α-substituted β-nitroacrylates was introduced by modifying the core of the benzothiazole ligand and applying the C2-symmetrical bis-pyrazole ligand (Scheme 11).114 The introduction of the 8-membered prolinol ether containing lactam moiety into the catalyst Λ(R,R)-8 allowed them to fix the carboxamide in a well-defined conformation, and this led to the improvement in the catalytic performance of the iridium(III) complex. In most cases, the desired products were obtained in excellent yields with high enantioselectivities in the presence of only 0.1–0.2 mol% of the catalyst Λ(R,R)-8.114
Scheme 11 illustrates the activation mode of both substrates via hydrogen bonding interactions which bring the indole nucleophile and the nitro olefin into a perfect position for the proposed ternary transition state, leading to a high catalytic activity and enantioselective control exerted by Λ(R,R)-8.
Essentially all of the chiral-at-metal catalysts elaborated by the Meggers group contain hydrogen-bonding donor/acceptor motifs enabling bifunctional ligand-sphere-mediated “organocatalysis”.96,97,108,111–114 In these complexes the metal ion mainly serves as a template to construct well-defined chiral octahedral structure to provide high stereocontrol in asymmetric transformations. It was clearly demonstrated that the construction of a proper chiral environment by modifying a ligand sphere provides high enantioselectivity in a presence of low catalyst loadings (sometimes even in ppm amounts). This finding will help in future to develop new efficient catalytic systems.
3. Asymmetric phase-transfer catalysis facilitated by cationic chiral cobalt(III) complexes via hydrogen bonding with the substrates
In 2012, we introduced a new class of chiral catalysts based on chiral positively charged stereochemically inert octahedral cobalt(III) complexes of the Λ(R,R)-9 type.93 These complexes were easily prepared from the Schiff bases of chiral 1,2-diaminocyclohexane hydrochloride and salicylaldehydes and the subsequent reaction of the resulting Schiff base ligand with cobalt(III) salt.95 X-ray single crystal analysis of the Λ(R,R)-9a showed that the complex cation formed a set of hydrogen bonds with the halide counter-anion (Fig. 4).95 | ||
| Fig. 4 X-ray structure of Λ(R,R)-9a with chlorine counter-anion. Hydrogen atoms at carbon atoms and tert-butyl groups are omitted for clarity (CCDC 927098).95 | ||
The complex was highly soluble in CH2Cl2 and the counter-anion could be readily exchanged for other anions under phase-transfer conditions, simply by washing a CH2Cl2 solution of the complex several times with aqueous solutions of the desired sodium or potassium halides.95 To test the catalytic potency of the novel family of complexes, the asymmetric phase-transfer alkylation of O’Donnell's substrate,24,163 leading to the asymmetric synthesis of valuable chiral α-amino acids, was chosen. The reaction is routinely used as a benchmark reaction to test any new phase-transfer (PT) catalysts.163–169 The chiral cobalt(III) complex Λ(R,R)-9a efficiently catalysed the alkylation reaction with a variety of alkyl halides furnishing the amino acid derivatives with 86–92% ee (Scheme 12).95 It was proposed that the reactions proceeded via the phase-transfer mechanism164 in which the key steps were the formation of chiral contact-ion pairs formed between the catalyst cation and the emerging enolate of O’Donnell's substrate. The enolate ion was then transferred into the organic phase where the stereoselective alkylation took place.
 | ||
| Scheme 12 Enantioselective alkylation and Michael addition of O’Donnell's substrate catalysed by the chiral cobalt(III) complexes Λ(R,R)-9 (Belokon, 2013–2014).95,98 | ||
Another closely related PTC reaction catalysed by the chiral cobalt(III) complexes Λ(R,R)-9a,b was the Michael addition of O’Donnell's substrate to a variety of EWG-activated olefins.98 The scope of the reaction with respect to various Michael acceptors was broad providing the adducts in yields up to 97%, diastereoselectivities up to >99:1 and enantioselectivities up to 96% ee (Scheme 12).98
Evidently, it was the attack of an electrophile on the less shielded face of the enolate/catalyst associate that determined the eventual configuration of the product. In order to rationalize the stereoselectivity of the alkylation, DFT calculations of the anticipated ion pairs were performed which revealed that it was the Z-enolate of O’Donnell's substrate which formed the most stable ion pair with the Δ(S,S)-9a cation (Fig. 5).98 The formation of hydrogen bonds between the NH groups and the carbonyl oxygen of the Z-enolate secured the most stable orientation of the enolate relative to the complex moiety. As a result, the si-side was shielded by the complex and the re-side opened to electrophilic attack. Importantly, the calculations were fully supported by the experiments, because Δ(S,S)-9a produced amino acids of R-configuration (Fig. 5).98
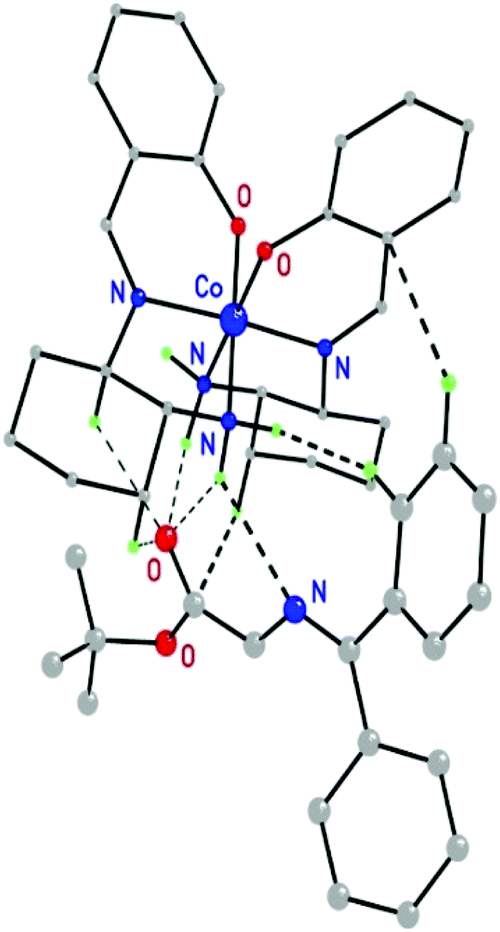 | ||
| Fig. 5 Energetically favoured optimized structure of the ion pairs formed between the Z-configured enolate of O’Donnell's substrate and the cobalt(III) complex cation of the Λ(R,R)-9.98 Only hydrogen atoms involved in interactions between the participating species are shown (highlighted in green) (Reprinted with permission from ref. 98. Copyright © 2014 Wiley-VCH Verlag GmbH & Co. KGaA). | ||
It was also demonstrated that the chiral cobalt(III) complex Λ(R,R)-9a catalysed the asymmetric epoxidation of chalcones with hydrogen peroxide under PTC conditions (Scheme 13).105 The experiments were conducted in different solvents, but methyl tert-butyl ether to which some amount of tBuOK was added provided the best results (47–99% yields and up to 55% ee).105
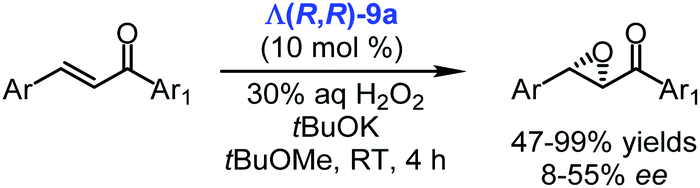 | ||
| Scheme 13 Enantioselective epoxidation of chalcones catalysed by the chiral cobalt(III) catalyst Λ(R,R)-9a (Belokon, 2015).105 | ||
Recently, a new class of amino acid precursors was introduced, much easier to prepare than O’Donnell's substrate, based on Schiff bases of different glycine esters and 2-hydroxybenzophenone.125 The chiral cobalt(III) complex Λ(R,R)-10 was applied to the asymmetric catalytic alkylation giving the desired (S)-phenyl alanine derivative with moderate yield (69%) and enantioselectivity (70% ee) (Scheme 14).125
 | ||
| Scheme 14 Enantioselective alkylation of the Schiff base of glycine ester and 2-hydroxybenzophenone with benzyl bromide catalysed by the chiral cobalt(III) complex Λ(R,R)-10 (Belokon, 2018).125 | ||
As in case of the ‘Werner type’ chiral cobalt(III) complexes applied as catalysts by Gladysz group, the metal ion in the complexes Λ(R,R)-9-10 serves as a template and as a source of chirality and, in addition, activates the coordinated NH2 groups, making them efficient hydrogen bond donors. An important, future perspective of this approach is that by modifying the structure of the ligand and changing the metal source it will be possible to improve the catalytic activity of these complexes and to create a multitask catalytic system for a broad range of asymmetric transformations.
4. Asymmetric Brønsted acid catalysis with chiral metal complexes
4.1 Protonated chiral anionic cobalt(III) complexes as Brønsted acid catalysts
In this subsection we will discuss chiral anionic octahedral cobalt(III) complexes which in contrast to previous ones are based on chiral amino acids and have a negative charge (Fig. 6). As a result, these type of cobalt(III) complexes have different properties and, respectively, catalyse completely different type of asymmetric reactions. | ||
| Fig. 6 Chiral octahedral anionic and coordinatively-inert cobalt(III) complexes based on the Schiff bases of α-amino acids and salicylaldehydes.88–90,92,94 | ||
In 1977, some of us introduced a new family of chiral octahedral anionic coordinatively-inert cobalt(III) complexes based on the Schiff bases of α-amino acids and salicylaldehydes (Fig. 6).170 A stereogenic cobalt(III) centre is coordinated by two identical chiral tridentate salicylimine ligands, and the complexes are easily available via a one-pot protocol as a mixture of the two diastereomeric complexes Λ(S,S) and Δ(S,S).170 The resulting diastereomeric complexes are readily purified and separated via flash chromatography on alumina, and the introduction of a counter-cation of choice (including proton H+) is achieved via ion-exchange chromatography as the final synthetic step.88–90,92,94 The chiral cobalt(III) complexes were introduced for the first time by Belokon et al. in 2004 as asymmetric catalysts for the enantioselective cyanosilylation of benzaldehyde.88 It should be noted that the chiral anionic cobalt(III) catalysts operated in solution as a mixture of assemblies. This was confirmed by the dependence of the product ee on the catalyst concentration and DOSY experiments.90
Following this pioneering work, Maleev and co-workers demonstrated that the octahedral diastereoisomerically pure cobalt(III) complex Δ(S,S)-11a could be converted into its protonated form with a cation exchange resin. The acidic form of the complex served as a chiral Brønsted acid catalyst in the asymmetric aza-Diels–Alder reaction between substituted imines and cyclohexenone, which gave the corresponding cycloaddition products in 16–77% ee (for the endo diastereomer) with endo/exo ratios of up to 4:1 (Scheme 15).94 The authors assumed that the imines were activated via protonation of the imino nitrogen and that the ion pair of the iminium cation and the chiral complex counter-anion were responsible for the asymmetric induction in the subsequent cycloaddition step.77,171 Furthermore, the enone is probably activated by the anionic cobalt complex through a deprotonation step. Interestingly, the Λ(S,S)-diastereomer provided the cycloaddition product in almost racemic form (6% ee).94
 | ||
| Scheme 15 Enantioselective aza-Diels–Alder reaction catalysed by the chiral cobalt(III) complex Δ(S,S)-11a (Maleev, 2013).94 | ||
Subsequently, the Yu group employed the another representative of the family of the complexes Λ(S,S)-11b derived from (S)-tert-leucine and 3,5-di-tert-butylsalicylaldehyde as a catalyst for the asymmetric synthesis of 4-aminofuranobenzopyrans, starting from salicylaldimines and 2,3-dihydrofuran in 31–95% yields, with cis/trans ratios of up to >20:1, and 50–92% ee (Scheme 16).123 Similar to the reaction presented in Scheme 14, the authors assumed that the imine was protonated by the Brønsted acidic cobalt(III) complex to form a chiral-complex-associated iminium ion. As a result, via protonation (involving the more electrophilic imine), a stereoselective Mannich reaction occurs with 2,3-dihydrofuran (Scheme 16), which is followed by an intramolecular ketalization to afford the desired 4-aminofuranobenzopyrans.123
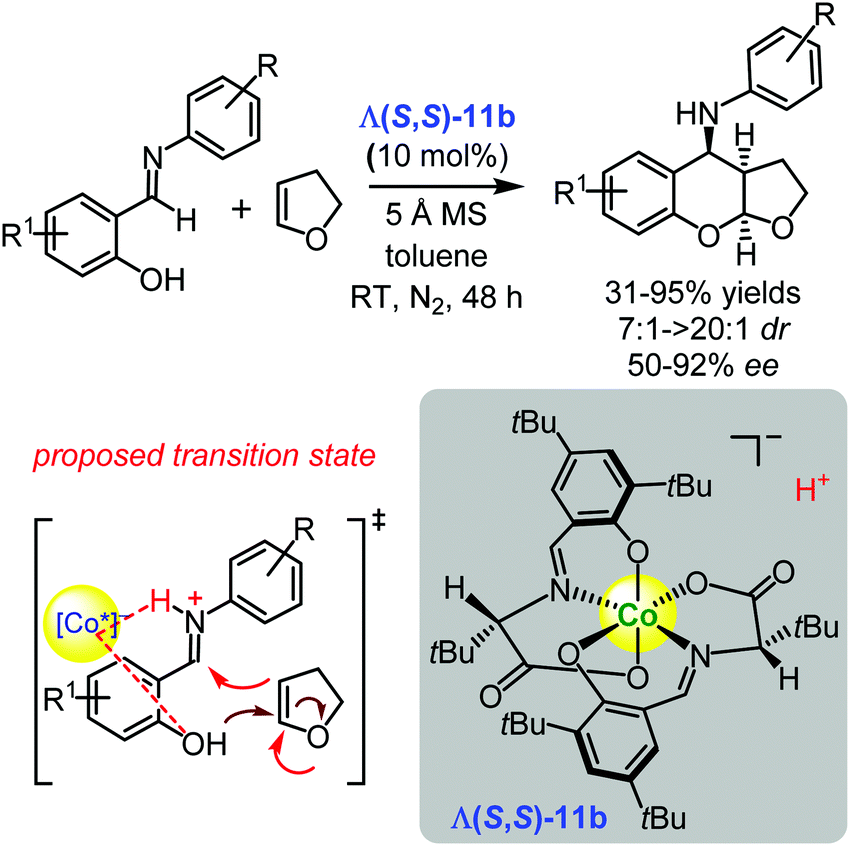 | ||
| Scheme 16 Enantioselective Povarov reaction catalysed by the chiral cobalt(III) complex Λ(S,S)-11b (Yu, 2017).123 | ||
4.2 Chiral iridium(III) and ruthenium(II) complexes with a coordinated water molecule and hydroxyl group as Brønsted acid catalysts
In 2014, Carmona and co-workers published seminal results demonstrating that water molecules coordinated to iridium(III) and ruthenium(II) ion centres became acidic enough to convert the complexes into Brønsted acids which, in turn, catalysed the activation of electrophiles in asymmetric transformations.102–104 In other words, the chiral iridium(III) complex (SIr,SC)-12 became a chiral Lewis acid-assisted Brønsted acid49–51 which promoted the enantioselective Friedel–Crafts hydroxyalkylation of indoles with 3,3,3-trifluoropyruvates furnishing the indole derivatives in 92–97% yields and up to 86% ee (Scheme 17).102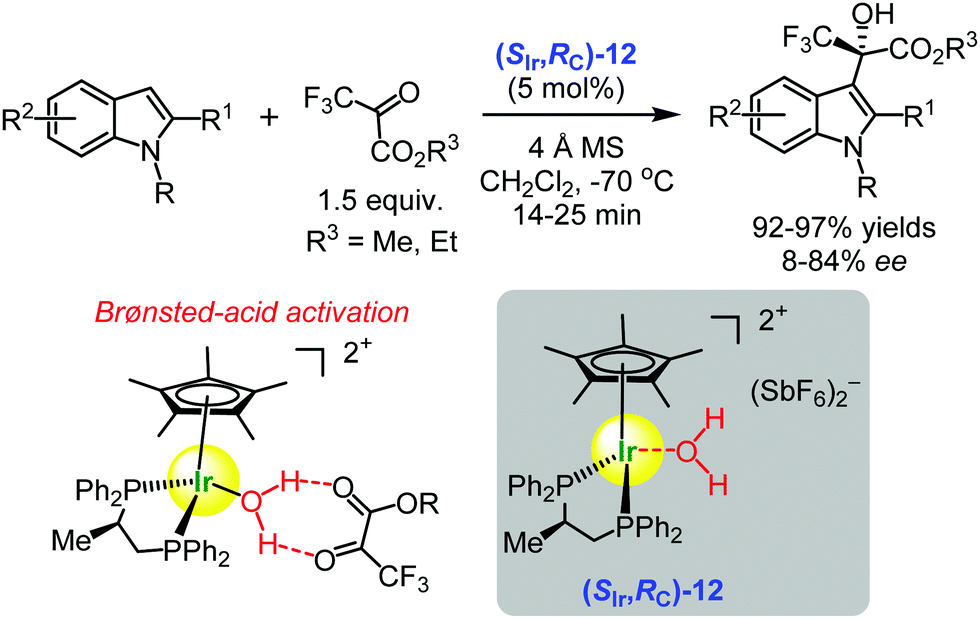 | ||
| Scheme 17 Enantioselective Friedel–Crafts hydroxyalkylation of indoles with 3,3,3-trifluoropyruvates catalysed by the chiral iridium(III) complex (SIr,SC)-12 (Carmona and Maseras, 2014).102 The proposed Brønsted-acid activation mode is illustrated. | ||
On the basis of DFT calculations, the authors assumed that the hydrogen bonding interactions of the protons of water molecule with the carbonyl pyruvate group was sufficient to activate 3,3,3-trifluoropyruvate for the nucleophilic attack by the indole molecule. It was proposed that the catalytic role of the metal complex was in the modulation of the Brønsted-acid/base properties of the coordinated water. The acidity of water was enough to protonate the pyruvate–indole pair, and the resulting basic hydroxyl group deprotonated the ensuing intermediate. Most probably, the two electron-withdrawing groups (CF3 and CO2Et) precluded direct pyruvate carbonyl coordination to the metal.102
Based on this design Carmona and co-workers demonstrated that the chiral ruthenium(II) complex (SC1,RC2)-13 derived from (1S,2R)-1-(diphenylphosphanyl)-3-methoxy-1-phenylpropan-2-ol ((SC1,RC2)-phosphane-hydroxyl ligand) catalysed asymmetric Diels–Alder and Friedel–Crafts reactions (Scheme 18).103 In general, good conversions were achieved in the case of the Diels–Alder reaction of cyclopentadiene and trans-β-nitrostyrene or methacrolein. However, low enantioselectivities for both reactions were observed (only up to 12%).103 In contrast, the Friedel–Crafts reactions gave the products in high yields (up to 99%) and with enantioselectivities up to 81% ee (Scheme 18).103 The large difference in the enantioselectivities of both reactions might be attributed to the significant involvement of the background condensation in the Diels–Alder case leading to racemic products.
 | ||
| Scheme 18 Enantioselective Diels–Alder reaction between cyclopentadiene and trans-β-nitrostyrene or methacrolein and Friedel–Crafts reaction between indole and methyl 3,3,3-trifluoropyruvate or trans-β-nitrostyrene catalysed by the chiral ruthenium(II) complex (SC1,RC2)-13 (Carmona, Lamata, Rodríguez, 2014).103 | ||
Based on calculations and crystallographic as well as spectroscopic data, the authors concluded that the chiral ruthenium(II) complex (SC1,RC2)-13 was an active metal-templated Brønsted acid catalyst for both the Diels–Alder and Friedel–Crafts reactions in which the asymmetric activation of carbonyl- and nitro-containing substrates occurred through hydrogen bonding with the ligand hydroxyl group coordinated to metal.103
In a follow up study the application of the chiral half-sandwich rhodium(III) (RP)-14 and iridium(III) (RP)-15 complexes based on hydroxymethylpyridine and (R)-phosphoramidite ligands as Brønsted acid catalysts for the enantioselective Friedel–Crafts addition of indoles to 3,3,3-trifluoropyruvate was reported (Scheme 19).104 The indole derivatives were obtained with enantioselectivities in a range of 42–82% ee.104
 | ||
| Scheme 19 Enantioselective Friedel–Crafts hydroxyalkylation of indoles with 3,3,3-trifluoropyruvates catalysed by chiral rhodium(III) (RP)-14 or iridium(III) complexes (RP)-15 (Carmona and Lamata, 2014).104 | ||
In conclusion, the studies of the Carmona group demonstrated that the metal moiety acted as a Lewis acid, enhancing the acidity of the protons of water molecule or hydroxyl groups, and as a chiral fragment governing the stereochemistry of the process. These data supported the previously assumed concept of Lewis acid-assisted Brønsted acid catalysis that was formulated by Yamomoto and co-workers.49–51
4.3 Water coordinated chiral copper(II) complex as a Brønsted acid catalyst
Recently, some of us introduced a new chiral copper(II) catalyst (S)-16 for the enantioselective Henry reaction (Scheme 20).129 The catalyst was prepared from Cu(OAc)2 and a Schiff base derived from (S)-2-(aminomethyl)pyrrolidine and 3,5-di-tert-butylsalicylaldehyde. The condensation of nitromethane and o-nitrobenzaldehyde in the presence of 10 mol% water and the copper complex (S)-16 furnished the nitro alcohol in a 78% yield and 77% ee.129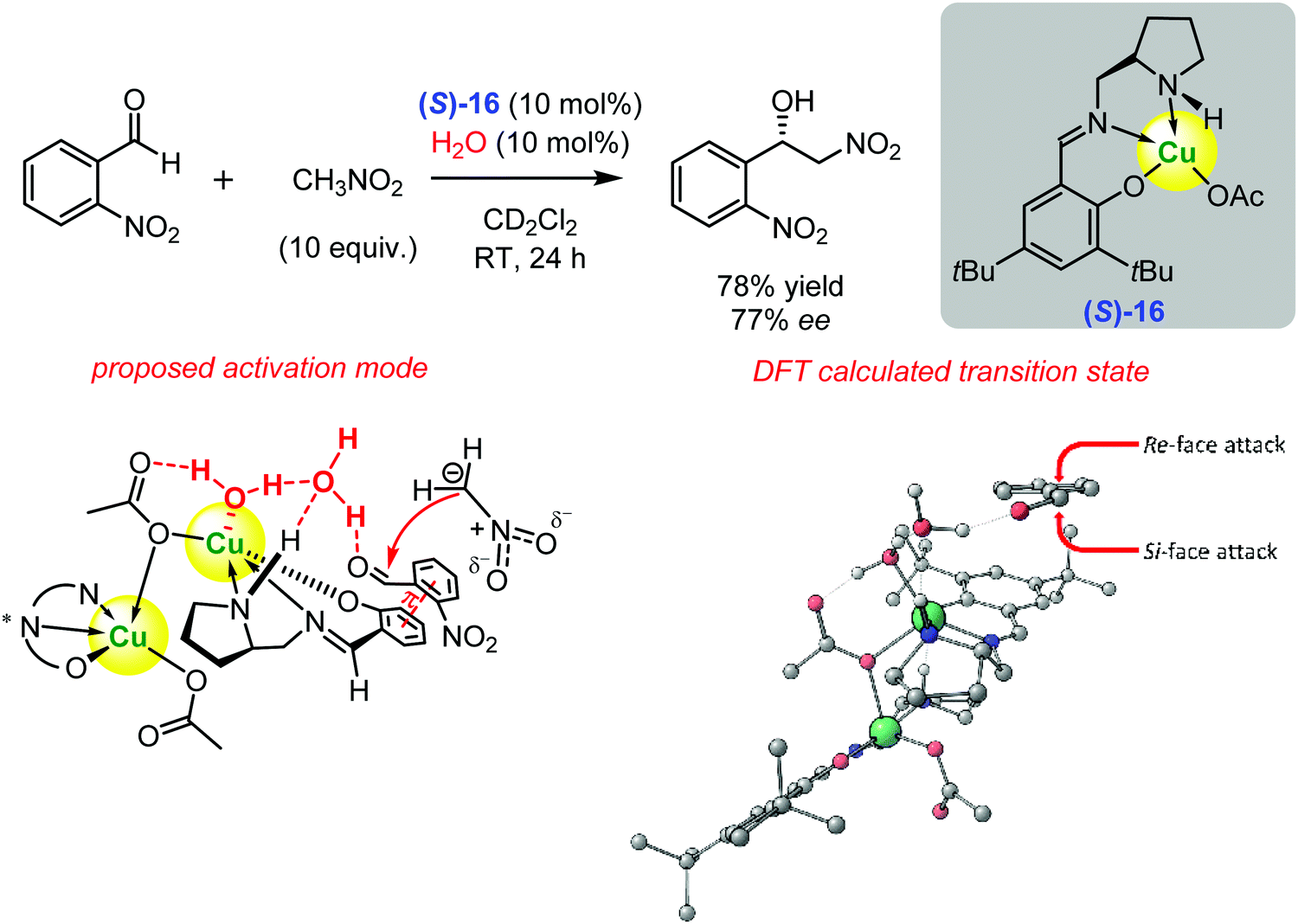 | ||
| Scheme 20 Enantioselective Henry reaction catalysed by the chiral copper(II) complex (S)-16 (Larionov and Belokon, 2019).129 The proposed activation mode and the DFT calculated transition state are illustrated (Reprinted with permission from ref. 129. Copyright © 2019 American Chemical Society). | ||
Based on the combined set of experimental data, spectroscopic data, and quantum-chemical calculations, it was proven that the copper(II) ion was not involved in the direct Lewis acid type activation of the aldehyde carbonyl group. Instead, the Lewis acid centre coordinated with water molecules, making them highly efficient Brønsted acid catalysts (Scheme 20). In addition, the coordinated water formed hydrogen bonds with the nitromethane, increasing its CH acidity.129
The studies discussed in this subsection clearly show a great potential of the chiral metal-templated Brønsted acids for various asymmetric transformations. For instance, the protonated cobalt(III) complexes operating as chiral Brønsted acids nicely compliment chiral phosphoric acids and can be applied in the future to solve problems arising in case of the use of chiral phosphoric acids. On the other hand, the efficiency of the previously studied metal catalysts containing some traces of water were underestimated as the water content of the reaction was not taken into account and assessed. In this context, it was clearly demonstrated by the Carmona and our groups that water molecule coordinated to Lewis acid center become a Brønsted acid catalyst and efficiently drive the asymmetric transformations with high stereocontrol.
5. Asymmetric ion-pairing catalysis with chiral cobalt(III) complexes under phase-transfer conditions
The Gong and Yu groups extended the application of the Δ(S,S)-11a type cobalt(III) complexes as anionic phase-transfer catalysts in various asymmetric transformations, where enantiocontrol provided via ion-pair formation between the substrates and catalyst.110,122,126–128 For example, they demonstrated that the diastereomeric cobalt(III) complexes Λ(S,S)-11c and Δ(S,S)-11c, which were based on the Schiff base of (S)-valine and 3,5-di-tert-butylsalicylaldehyde, efficiently catalysed the enantioselective bromoaminocyclization of γ-amino alkenes with N-bromosuccinimide as the Br+ source (Scheme 21). This resulted in the corresponding 2-substituted pyrrolidines (both enantiomers) with ee values of 53–98% and with 47–99% yields.122 Interestingly, these results stand in contrast to the aforementioned examples where the diastereomers (Λ vs. Δ) were unable to provide any significant enantioinduction and/or conversion.88,94,110 | ||
| Scheme 21 Enantioselective bromoaminocyclization of γ-amino alkenes catalysed by the diastereomeric chiral cobalt(III) complexes Δ(S,S)-11c and Λ(S,S)-11c (Yu and Gong, 2017)122 (Adapted with permission from ref. 86. Copyright © 2018 Elsevier). | ||
More recently Yu and co-workers applied the complex Λ(S,S)-11c as a catalyst for the asymmetric synthesis of 3-bromohexahydropyrrolo[2,3-b]indoles (Scheme 22).126 Accordingly, the bifunctional complex Λ(S,S)-11c efficiently catalysed the enantioselective bromocyclization of protected tryptamines with readily available N-bromosuccinimide (NBS) under air. This afforded valuable key building blocks for cyclotryptamine alkaloids with 18–87% enantioselectivities and with 42–99% yields (Scheme 22).126
 | ||
| Scheme 22 Enantioselective bromocyclization of tryptamines catalysed by the chiral cobalt(III) complex Λ(S,S)-11c (Yu, 2018).126 The proposed transition state is illustrated. | ||
A proposal for the transition state of this transformation involves the Brønsted acidic cobalt(III) complex exchanging its proton for a bromide cation, with NBS forming the bromide complex in situ (Scheme 22). Then, the obtained complex, serving as a chiral brominating reagent, generates the chiral bromonium ion-pair with the tryptamine, which is stabilized by the hydrogen-bonding interaction and controls the stereoselectivity in the subsequent bromocyclization step (Scheme 22).126
It was also demonstrated that the complex Λ(S,S)-11c catalysed the enantioselective intermolecular iodoacetalization of vinyl ethers (such as 3,4-dihydro-2H-pyran, 2,3-dihydrofuran, ethyl vinyl ether) with alcohols and N-iodosuccinimide (NIS), furnishing the 3-iodoacetals in 61–99% yields and with modest enantioselectivities (10–66% ee) (Scheme 23).127 Next, the authors expanded the method by applying it to the synthesis of valuable 2-deoxy-2-iodoglycosides, including monosaccharides and disaccharides, and 2-deoxy-2-iodoglycosyl carboxylates via the diastereoselective iodoglycosylation or iodocarboxylation of glycals with a variety of alcohols/carboxylic acids and NIS (up to 9:1 dr) (Scheme 23).128 As in previous work,126 it was proposed that the exchange reaction with NIS, and then the chiral iodonium ion-pair formation, was responsible for the asymmetric induction in the subsequent iodoacetalization or iodoglycosylation steps.
 | ||
| Scheme 23 Enantioselective intermolecular iodoacetalization of enol ethers and diastereoselective iodoglycosylation and iodocarboxylation of glycals with alcohols or carboxylic acids catalysed by the chiral cobalt(III) complex Λ(S,S)-11c (Yu, 2018–2019).127,128 | ||
The examples discussed in this subsection clearly show a great potential of the chiral cobalt-templated complexes as efficient anionic phase-transfer catalysts for the asymmetric bromocyclization and iodination transformations. Notably, the addition of molecular sieves (MS) in the presented reactions slightly improved the reaction enantioselectivity although no reasonable explanation of the phenomena was put forward.122,123,126–128
6. Asymmetric nucleophilic catalysis by chiral-at-metal iridium(III) complex
In 2017, Meggers and co-workers introduced a new type of nucleophilic catalysts based on the chiral-only-at-metal iridium(III) complex Λ-17.120 The catalyst promoted the asymmetric Steglich rearrangement of O-acylated azlactones and furnished the desired valuable products in 68–99% yields and up to 96% ee (Scheme 24a).120 The related enantioselective Black rearrangement of O-acylated benzofuranones gave the target product with excellent yields (up to 99%) and 30–94% ee (Scheme 24b).120 In addition, the Λ-17 was an efficient catalyst for the enantioselective reaction between aryl alkyl ketenes and 2-cyanopyrrole to provide the corresponding α-chiral N-acyl pyrroles with excellent yields (96–99%) and enantioselectivity (up to 95% ee) (Scheme 24c).120 | ||
| Scheme 24 Enantioselective Steglich rearrangement of O-acylated azlactones, Black rearrangement of O-acylated benzofuranones, and the asymmetric reaction between aryl alkyl ketenes and 2-cyanopyrrole catalysed by the chiral iridium(III) complex Λ-17 (Meggers, 2017).120 | ||
The combined experimental and spectroscopic data, including single crystal X-ray analysis of the catalysis intermediate analogue (Fig. 7), indicate that the imidazoquinoline ligand of the chiral-only-at-metal iridium(III) complex Λ-17 was the nucleophilic part of the successful catalyst. Moreover, a chiral recognition model for the transition state was proposed based on both X-ray analysis data and quantum-chemical calculations.120
 | ||
| Fig. 7 Three-dimensional structure of the catalyst intermediate analogue for the Black rearrangement (CCDC 1536879).120 | ||
To summarise, the metal ion fulfils here a purely structural role and constitutes the exclusive source of metal-centred chirality by forming a well-defined molecular pocket to achieve excellent stereocontrol, while the actual catalysis is mediated through the carefully arranged and rigidly fixed nucleophilic imidazoquinoline ligand. This result show how it is possible to create a multitask catalytic system for a set of asymmetric reactions by the ligand design. For instance, a well-organized chiral environment provided high enantioselectivities.
7. Asymmetric bifunctional Brønsted base/hydrogen bond donor catalysis by chiral metal complexes
7.1 Chiral-only-at-metal iridium(III) complexes as bifunctional Brønsted base/hydrogen bond donor catalysts
In 2014, Meggers and Gong introduced a novel class of the chiral-only-at-metal octahedral bis-cyclometalated iridium(III) complexes Λ-18a–c, featuring a basic 3-(N-phenylamino)-5-(pyridin-2-yl)pyrazolato bidentate ligand (Scheme 25).100 The complex Λ-18a efficiently catalysed the enantioselective sulfa-Michael addition of N-pyrazolylcrotonates with aryl thiols at remarkably low catalyst loadings (up to 0.02 mol%) resulting in the corresponding products in high yields (85–98%) and excellent enantioselectivities (91–97% ee) (Scheme 25a).100 The efficiency of the catalyst was attributed to dual mode activation based on the ability of the ligand to serve as both an asymmetric chiral Brønsted base and a hydrogen bonding entity. Importantly, the hexyl groups were introduced into the benzoxazole ligand to improve the solubility of the Λ-18a–c in aprotic solvents. | ||
| Scheme 25 Enantioselective sulfa-Michael and aza-Henry reactions catalysed by the chiral iridium(III) complexes Λ-18a–c (Gong and Meggers, 2014–2016).100,106,107,115 | ||
Next, the catalyst structure was modified by the introduction of 3,5-di-tert-butyl phenyl groups into the benzoxazole ligand at the metalated phenyl moiety. The resulting complex Λ-18b was employed as a catalyst for the aza-Henry (nitro-Mannich) reaction (Scheme 25b),100 between phenylnitromethane and N-Boc-Schiff bases of aldehydes affording the (1R,2S)-nitro-N-Boc-amines with excellent yields and high enantioselectivity (90–97% ee) and diastereocontrol (>20:1 dr in most cases).
For the sulfa-Michael addition, the authors proposed a transition state model including the deprotonation of the acidic thiol by the basic aminopyrazole ligand leading to the formation of the ion pair of the thiolate ion and the cationic iridium complex stabilized by a double hydrogen bonding (Fig. 8). Simultaneously, the electrophilic acceptor, α,β-unsaturated N-acyl pyrazole, was activated via a hydrogen bonding with the hydroxyl group of the benzoxazole moiety. Thereby, both substrates were activated and juxtapositioned to allow the Michael addition to proceed with high-rate acceleration and high asymmetric induction (Fig. 8).100
 | ||
| Fig. 8 The proposed Brønsted-base/hydrogen bond activation mode in a sulfa-Michael addition reaction is illustrated.100 | ||
Similarly, the efficient control of the aza-Henry reaction was attributed to the bifunctional mode of action in which the iridium complex Λ-18b simultaneously deprotonated the nitro substrate and activated the N-Boc-Schiff base through hydrogen bonds properly arranging the substrates for the stereoselective coupling.100
The application of the iridium(III) complex Λ-18a was expanded to the enantioselective sulfa-Michael addition of aromatic thiols to α,β-unsaturated γ-oxoesters bearing an imidazole substituent in the δ position (Scheme 25c).106 The chiral products were obtained in high yields (up to 99%) and up to 97% ee.106
The same iridium(III) complex Λ-18a was also proven to be an efficient chiral catalyst for the asymmetric aza-Henry reaction between isatin-derived ketimines and aryl nitromethanes affording the corresponding oxindoles with a quaternary stereocenter at the 3-position with excellent yields and enantioselectivities up to 98% ee (Scheme 25d).107
Moreover, Meggers and co-workers could expand the scope of the substrates to less acidic alkyl thiols in Michael additions by modifying a metal-templated Brønsted base catalyst via tuning its basicity (Scheme 25e).115 The basicity of the iridium-coordinated pyrazolato ligand was increased from the approximate pKa = 16 for the conjugate acid in MeCN to pKa = 19 by introducing a pyrrolidine moiety at the 5-position of the pyridyl fragment. The increase in basic properties of the generated Λ-18c was sufficient to deprotonate aliphatic thiols (nBuSH: pKa = 17.0 in DMSO).115 Expectedly, the enantioselective sulfa-Michael addition of alkyl thiols to α,β-unsaturated N-acylpyrazoles furnished the desired products with high yields (71–99%) and enantioselectivities (86–98% ee) (Scheme 25e).115
7.2 Chiral ruthenium(II) complex as a bifunctional Brønsted base/hydrogen bond donor catalyst
In 2014, Gladysz and co-workers developed a metal-templated catalyst (SRuRCRC)-19 based on a ruthenium(II) complex featuring a chiral 2-guanidinobenzimidazole ligand for the enantioselective Michael addition of malonates to nitroalkenes (Scheme 26).101 Interestingly, the chiral ligand itself gave the desired products with average conversions (up to 70%) and modest enantioselectivity (only 68% ee) within an unacceptably long reaction time of 5 days.101 In contrast, the metal coordinated complex (SRuRCRC)-19 efficiently catalysed the reaction with yields up to >99% and high enantiocontrol (83 → 99% ee) in only during 1–2 days (Scheme 25).101 | ||
| Scheme 26 Enantioselective additions of malonate esters to nitroalkenes catalysed by the chiral ruthenium(II) complex (SRuRCRC)-19 (Gladysz, 2014).101 Proposed models for the addition of malonate esters to trans-β-nitrostyrene catalysed by the bifunctional ruthenium catalyst is illustrated. | ||
The authors propose that the ruthenium(II) complex (SRuRCRC)-19 operated in the same way as the Takemoto's catalyst159–162 and was responsible for both the activation of nitrostyrene via the formation of a set of hydrogen bonds and the deprotonation of malonate by the basic tertiary amine moiety (Scheme 26). Furthermore, the enantioselection was rationalized by computational studies.134 Notably, the chiral ruthenium(II) complex (SRuRCRC)-19 and the chiral ligand itself furnished the enantiomer with the same configuration, indicating that the ligand-based carbon stereocentre, and not the ruthenium centre, controls the configuration of the new carbon stereocentre in the product.
7.3 ‘Werner-type’ chiral-at-metal cobalt(III) complex as a bifunctional Brønsted base/hydrogen bond donor catalyst
Taking a bifunctional catalyst approach, Gladysz and co-workers improved the performance of Δ-[Co(en)3](BArf)3 = Δ-1 (see Fig. 2, only 33% ee of the adduct) in the Michael addition reaction. They modified the ligand framework by introducing a 3-(dimethylamino)propyl residue with S-configuration into one of the three ethylenediamine moieties producing the Λ(S)-20 (Scheme 27).116 One advantage of this catalyst was its intrinsic basicity which abolished any need for an external base. The complex Λ(S)-20 catalysed the Michael addition of malonates to nitroalkenes giving the corresponding addition products with excellent yields (up to 98%) and stereoselectivity (up to 99% ee) (Scheme 27).116 The ‘correct’ length of the linker between the tertiary amine and the modified ethylenediamine ligand was shown to be crucial for achieving a high enantioinduction, which demonstrated that the tertiary amine's mechanistic task must lie well beyond that of a simple Brønsted base.116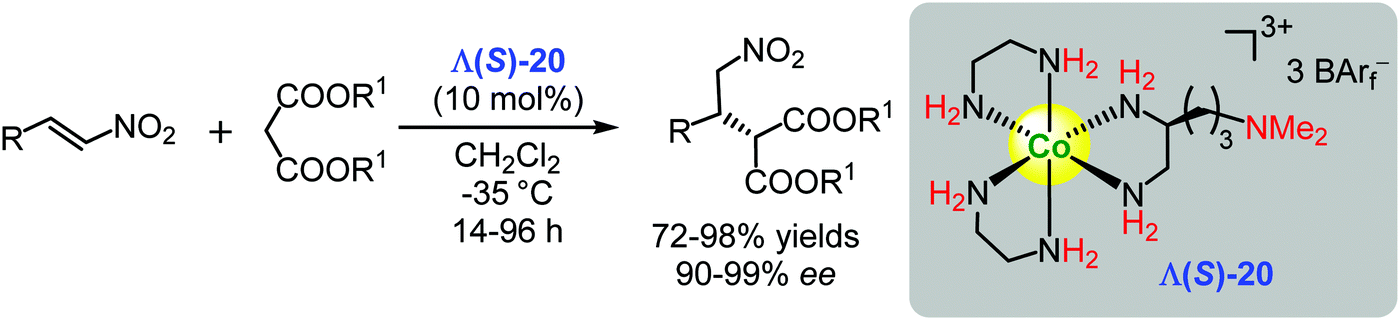 | ||
| Scheme 27 Enantioselective conjugate addition of malonates to nitroalkenes catalysed by the bifunctional chiral Co(III) complex Λ(S)-20 (Gladysz, 2016).116 | ||
To conclude, the chiral metal-templated Brønsted bases present a promising class of catalysts where the metal ion serves as a template and, in some cases an exclusive source of chirality. The described concept allows to construct catalytic systems in which several active catalytic centres are combined close to each other in a well-defined chiral manner that provides high stereocontrol (sometimes at remarkably low catalyst loadings).100 In contrast, this approach is difficult to realize in case of classical organocatalysts that explain why the low enantioselectivities are observed in some challenging asymmetric transformations.
8. Asymmetric bifunctional enamine/hydrogen bond donor catalysis by chiral-at-metal iridium(III) complex
In contrast a bifunctional chiral-only-at-metal iridium(III) catalyst Λ-21, introduced by Meggers does not rely on the previously reported double hydrogen-bonding donor motif (Scheme 28).99 The complex Λ-21 was found to be an efficient catalyst for the asymmetric α-amination of enolizable aldehydes with azodicarboxylates giving the oxazolidinone products in up to 96% yield and with up to 97% ee (Scheme 28).99 | ||
| Scheme 28 Enantioselective α-amination of aldehydes with azodicarboxylates catalysed by the chiral iridium(III) complex Λ-21 (Meggers, 2014).99 | ||
As depicted in Scheme 28, the iridium(III) complex Λ-21 was supposed to operate as a bifunctional cooperative enamine/hydrogen-bonding catalyst. It was proposed that the cyclic secondary amine moiety of the complex Λ-21 activated the enolizable aldehydes by an intermediate enamine formation, whereas the hydroxymethyl group activated azodicarboxylates by acting as a hydrogen-bond donor which triggered the conjugate addition reaction with the enamine. Next, the resulting iminium species hydrolysed, which gave chiral α-hydrazino aldehydes as primary products.99 As these products were prone to racemization, they were usually reduced in situ to the corresponding alcohols and then cyclized to the depicted oxazolidinones. It was shown that, in contrast to classical proline catalysis, a well-organized structure of the iridium-templated complex bearing pyrrolidine moiety in combination with the hydrogen bond donor fragment allow to reach a high enantioselectivity at low catalyst loading.
9. Asymmetric bifunctional hydrogen bond/counter-anion catalysis by chiral cationic cobalt(III) complexes
A proof of principle was demonstrated by some of us that the chiral cobalt(III) complexes Λ(R,R)-9c–e could serve as bifunctional catalysts for the asymmetric cyanosilylation of benzaldehyde with trimethylsilyl cyanide (Scheme 29).93 The corresponding cyanosilylation product was obtained in up to 99% yield but only with rather low enantioselectivity (up to 27% ee).93 Although this result was less encouraging in terms of enantioselectivity, an interesting aspect of the reaction was observed: the variation of the halide counter-anion did not have a significant effect on the enantioselectivity, but surprisingly influenced the yield and the reaction rate. Kinetic experiments revealed that the complex Λ(R,R)-9d with a fluoride counter-anion was in fact several orders of magnitude more active than the chloride featuring complex Λ(R,R)-9c.93 An additional nucleophilic activation of a silicon atom by the fluoride anion is a well-established phenomenon.172 It was proposed that the amino groups of the cobalt(III) complex activates and aligns benzaldehyde via a hydrogen-bonding, while the fluoride anion is also fixed via a hydrogen-bonding interaction. The approach of trimethylsilyl cyanide to the spatially constrained fluoride anion triggers a nucleophilic attack of the fluoride anion on the silicon centre of trimethylsilyl cyanide, so that the cyanide anion attacked the activated benzaldehyde, which finally led to the formation of the cyanosilylation product (Scheme 29).93 | ||
| Scheme 29 Enantioselective cyanosilylation of benzaldehyde catalysed by the chiral cobalt(III) complexes Λ(R,R)-9c–e (Belokon, 2012).93 A plausible structure of a transition state for the cyanosilylation of benzaldehyde is illustrated (Adapted with permission from ref. 86. Copyright © 2018 Elsevier). | ||
Next it was demonstrated that the cobalt(III) complex Δ(S,S)-9f with an iodide counter-anion can be a suitable catalyst for the kinetic resolution of a racemic chalcone epoxide with CO2 providing the cyclic carbonate with 72% conversion, and the remaining epoxide was partially enriched (50% ee) (Scheme 30).105,118
 | ||
| Scheme 30 Kinetic resolution of chalcone epoxide with CO2 catalysed by the chiral cobalt(III) complex Δ(S,S)-9f (Belokon, 2015–2016).105,118 | ||
A plausible mechanism for the cycloaddition reaction is depicted in Scheme 31. At first, chalcone epoxide gets activated and aligned via a hydrogen-bonding by the cobalt(III) complex Δ(S,S)-9f (Scheme 31). The associated iodide anion subsequently opens the epoxide ring, and CO2, which is also probably activated and aligned in close proximity via a hydrogen-bonding, is then attacked by the nucleophilic oxyanion. Then, the five-membered cyclic carbonate is released along with the intramolecular cyclization.118
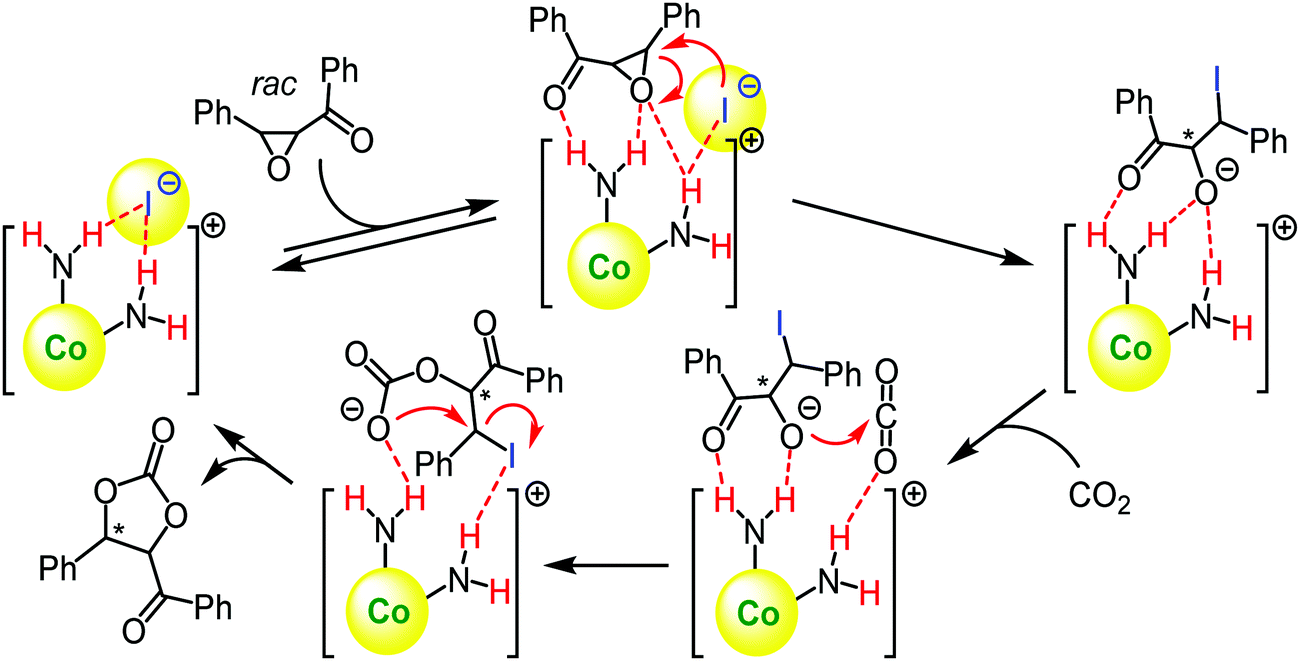 | ||
| Scheme 31 Plausible schematic mechanism for the kinetic resolution of chalcone epoxide via ring opening with CO2 catalysed by the chiral cobalt(III) complex Δ(S,S)-9f (Adapted with permission from ref. 86. Copyright © 2018 Elsevier).118 | ||
The low enantioselectivities observed probably can be improved by further ligand modification and/or metal ion exchange. It should be emphasized, the kinetic resolution of epoxides with CO2 is very challenging process and there are only few examples in literature where high s-factors were achieved.173–177
10. Asymmetric bifunctional photoactivated/hydrogen bonding catalysis by chiral-at-metal iridium(III) complexes
Merging chiral-at-metal catalysis with photochemistry Baik and Yoon presented the chiral-only-at-metal iridium(III) catalyst Λ-22a, featuring a pyridylpyrazole hydrogen-bonding ligand,121 which is based relies on the previously reported Meggers’ catalytic system96 (Scheme 32). Complex Λ-22a was found to be an efficient bifunctional catalyst for the enantioselective intramolecular [2+2]-photocycloaddition of quinolones, giving the cyclic products in up to 98% yield and with up to 91% ee (Scheme 32).121 | ||
| Scheme 32 Enantioselective intramolecular [2+2] photocycloaddition of quinolones catalysed by the chiral iridium(III) complex Λ-22a (Baik and Yoon, 2017).121 | ||
The present enantiopure iridium complex functionalized with a hydrogen-bonding domain served as a highly enantioselective triplet sensitizer. Here, the excited state of the iridium(III) complex was the triplet energy donor, and the well-orchestrated quinolone substrate was the acceptor. Finally, the high enantiocontrol was provided by the hydrogen bond formation and π–π interactions of the substrate with the ligand sphere of the catalyst.121
A slightly modified octahedral chiral-only-at-metal iridium(III) complex Λ-22b, was subsequently used, in which methoxy groups were introduced in a pyridylpyrazole moiety, showing remarkable performance as a bifunctional photo-/hydrogen bond donor catalyst in the asymmetric intermolecular [2+2]-photocycloaddition of 3-alkoxyquinolones with maleimides to afford the valuable products with excellent yields and high enantioselectivities (up to 99% ee) and diastereocontrol up to 9:1 dr (Scheme 33).130
 | ||
| Scheme 33 Enantioselective intermolecular [2+2] photocycloaddition of 3-alkoxyquinolones with maleimides catalysed by the chiral iridium(III) complex Λ-22b (Baik and Yoon, 2019).130 | ||
In comparison with their previous results,121 the authors conclude that the energy transfer to maleimide proceeds from the quinolone substrate associated with the pyrazole moiety of the iridium complex. The resulting triplet-state maleimide reacts with the quinolone aligned via a hydrogen-bonding within the well-defined chiral environment provided by the iridium stereocentre, affording the cycloadduct with high ee.130
Generally, in these chiral-only-at-metal iridium(III) complexes, the metal serves as a template, the exclusive source of chirality providing a well-organized chiral ambience, and as a photosensitizer to generate the triplet-state of the substrate. The main advantage here, in contrast to Meggers’ catalysts, that a proper ligand design allowed to construct a suitable catalyst for the asymmetric photoinitiated reactions. Currently, photoredox catalysis is one of the most rapidly developing areas of chemical research and a number of catalytic systems are exists for asymmetric transformations today.178–183 However, in many cases, the combination of chiral catalyst with photosensitizer are required for the successful reaction.184–186 Although several chiral pure one-component organocatalysts were recently elaborated187 there is still a great demand in efficient multitask chiral catalytic systems for the photoredox chemistry. In this context, the chiral iridium(III) complexes developed by Yoon are promising one-component catalysts which are not require an external photosensitizer and, at same time are powerful stereoinductors. This perspective approach for construction of chiral photocatalysts will give new opportunities in future of photoredox chemistry and allow to use them in challenging transformations.
11. Chiral ferrocene-based organocatalysts
In this review, we cannot ignore the chiral ferrocene-based complexes that are broadly used as organocatalysts in numerous asymmetric reactions.188–199 These catalysts have been thoroughly summarized in review articles188–191 and selected examples of them are depicted in Fig. 9. Since the pioneering works of Fu and co-workers on ferrocene-based planar-chiral derivatives,189,190,192–197 several ferrocene-based nucleophilic organocatalysts have been developed with versatile applications in enantioselective catalysis.191 The majority of these complexes are used as efficient nucleophilic catalysts for the asymmetric transformation of ketenes into chiral valuable building blocks.189–199 Here, the main role of an iron(II) ion is that it serves as a template providing planar chirality. As this field was extensively reviewed and summarized earlier,188–191 we do not discuss the reported studies and approaches in detail in the current manuscript. | ||
| Fig. 9 The examples of the chiral ferrocene-based organocatalysts used for asymmetric reactions.188–199 | ||
12. Summary and outlook
In this review article, we have focused on chiral metal-templated complexes that serve as chiral catalysts in various asymmetric transformations. In contrast to classical metal complex catalysts, this class of promoters holds a great unexplored potential for the asymmetric catalysis and the advances using these approaches have so far only partly been reviewed. However, they represent – from a practical point of view – a favourable and capable class of chiral catalysts, where the substrates are activated by the ligand sphere of metal-templated complexes via well-orchestrated and custom-tailored non-covalent or ion-paired interactions. The most fascinating chiral metal catalysts contain hydrogen-bonding donor/acceptor motifs, nucleophilic and amine moieties enabling ligand-sphere-mediated “organocatalysis”. Such chiral metal complex systems can be called chiral “organocatalysts in disguise” due to the metal centre being only a spectator in the direct activation of the substrates. In contrast to classical organocatalysts, the described methods allow to construct chiral metal-templated catalytic systems with a well-defined chiral environment in which several active catalytic centres are properly combined close to each other in space that provides a high sterecontrol (sometimes at remarkably low catalyst loadings) in asymmetric transformations. ln addition, by the variation of metal ion it is possible to regulate the acidity of the ligands, which in turn lead to the increase of activity of the whole catalytic system. This feature will help to solve one of the main challenges in organocatalysis – an acid–base factor because further increase of the acidity of the Brønsted acid may impair its catalytic performance.75 Moreover, in contrast to classical organocatalysts, having the metal ion in the system will provide great opportunities to use these types of complexes in enantioselective (photo-)redox reactions where the electrons will be transferred or accepted via the ligand sphere.Essentially, the metal serves the following roles:
1. It is a template (a structural role as a ‘glue’ to spatially arrange the participating organocatalytic ligands) and the exclusive source of chirality (metal-centred chirality, namely a chiral propeller-shaped arrangement of bidentate or tridentate ligands around an octahedral metal centre). The octahedral arrangement of the organic ligands allows one to custom-tailor non-covalent interactions with the participating substrates in a three-dimensional manner, which would be challenging to realize and to optimize with purely organic catalyst scaffolds.
2. It is an activator of the ligand amino groups, allowing tuning of hydrogen bond properties making them efficient hydrogen bond donors.
3. It can act as a photosensitizer in combination with hydrogen-bond donor ability.
It has been proven that the reported chiral metal complexes were highly efficient in providing excellent stereocontrol in asymmetric hydrogen bonding catalysis, phase-transfer catalysis, Brønsted acid/base catalysis, enamine catalysis, nucleophilic catalysis, and photocatalysis as well as bifunctional catalysis. Also, in contrast to classical organocatalysts, many of them have been identified as being highly effective at remarkably low catalyst loadings. It should be noted that most of the presented catalysts are robust and air stable, allowing to conduct the reactions under air and moisture conditions. Furthermore, the chiral metal-templated complexes are already used not only in catalysis but also as chirality sensing agents200–203 and in biochemistry.204–207 There are outstanding future perspectives for this in the field of asymmetric catalysis in that control of reactivity and stereoselectivity can be achieved through choice of metal ions, ligands and counter-ions, holding great promise for fine tuning for a wide range of chiral transformations. We believe that the various approaches discussed here will have a fresh impact on the development of new chiral metal complex catalysts with ‘non-innocent’ ligands that can operate exclusively as organocatalysts, and where the activation of the substrates will take place in the ligand sphere of the metal-templated complex and the enantioselectivity control will be provided by the chiral environment of the metal complex. Further bridging the worlds of metal coordination chemistry, ligand design and organic catalysis is promising a bright future for “organocatalysis in disguise”.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Russian Science Foundation (RSF grant No. 20-13-00155). B.L.F acknowledges financial support by the Ministry of Education, Culture and Science (Gravitation Program no. 024.001.035). The publication has been supported by the RUDN University Strategic Academic Leadership Program (the access to electronic databases). The access to literature resources were performed with support from the Ministry of Science and Higher Education of the Russian Federation.Notes and references
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis I-III, Springer-Verlag, Berlin Heidelberg, Berlin, 2001 Search PubMed.
- K. Mikami and M. Lautens, New Frontiers in Asymmetric Catalysis, Wiley, Hoboken, 1st edn, 2007 Search PubMed.
- P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, 2009 Search PubMed.
- I. Ojima, Catalytic Asymmetric Synthesis, 3rd ed., Wiley, Hoboken, 2010 Search PubMed.
- http://www.nobelprize.org/prizes/chemistry/2001/summary/ .
- J. M. Hawkins and T. J. N. Watson, Angew. Chem., Int. Ed., 2004, 43, 3224–3228 CrossRef CAS PubMed.
- N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291–1299 CrossRef CAS.
- H.-U. Blaser and H.-J. Federsel, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, Wiley-VCH, Weinheim, 2nd edn, 2010 Search PubMed.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS PubMed.
- G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS.
- B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS.
- T. Hayashi, Acc. Chem. Res., 2000, 33, 354–362 CrossRef CAS.
- M. J. Burk, Acc. Chem. Res., 2000, 33, 363–372 CrossRef CAS PubMed.
- W. S. Knowles and R. Noyori, Acc. Chem. Res., 2007, 40, 1238–1239 CrossRef CAS PubMed.
- A. Berkessel and H. Groger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- B. List, Asymmetric Organocatalysis in Topics in Current Chemistry, Springer-Verlag Berlin Heidelberg, Berlin, 2009 Search PubMed.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS.
- B. List, Adv. Synth. Catal., 2004, 346, 1021 CrossRef CAS.
- J. Seayada and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS.
- M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef PubMed.
- D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS.
- G. C. Fu, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS.
- B. List, Acc. Chem. Res., 2004, 37, 548–557 CrossRef CAS.
- S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570–579 CrossRef CAS PubMed.
- W. Notz, F. Tanaka and C. F. Barbas, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS.
- S. Hoffmann, A. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS.
- D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- P. Etayoa and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- S. Motevalli, Y. Sokeirik and A. Ghanem, Eur. J. Org. Chem., 2016, 1459–1475 CrossRef CAS.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS.
- S. Shi, K. S. Nawaz, M. K. Zaman and Z. Sun, Catalysts, 2018, 8, 90 CrossRef.
- I. D. G. Watson and F. D. Toste, Chem. Sci., 2012, 3, 2899–2919 RSC.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS.
- C. I. Stathakis, P. L. Gkizis and A. L. Zografos, Nat. Prod. Rep., 2016, 33, 1093–1117 RSC.
- Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256–2275 RSC.
- J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465–479 CrossRef.
- H. Yamamoto, Lewis Acid Reagents: A Practical Approach, Oxford University Press, Oxford, 1999 Search PubMed.
- H. Yamamoto, Lewis Acids in Organic Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS.
- M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- Y. Minko and I. Marek, Chem. Commun., 2014, 50, 12597–12611 RSC.
- J.-S. Yu, H.-M. Huang, P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, ACS Catal., 2016, 6, 5319–5344 CrossRef CAS.
- Y.-M. Cui, Y. Lin and L.-W. Xu, Coord. Chem. Rev., 2017, 330, 37–52 CrossRef CAS.
- J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS.
- H. Wynberg, Top. Stereochem., 1986, 16, 87–129 CAS.
- H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 16, 4057–4060 CrossRef.
- S. Colonna, H. Hiemstra and H. Wynberg, J. Chem. Soc., Chem. Commun., 1978, 238–239 RSC.
- G. Snatzke, H. Wynberg, B. Feringa, B. G. Marsman, B. Greydanus and H. Pluim, J. Org. Chem., 1980, 45, 4094–4096 CrossRef CAS.
- S. Colonna, A. Re and H. Wynberg, J. Chem. Soc., Perkin Trans. 1, 1981, 547–552 RSC.
- J. Gawronski, K. Gawronska and H. Wynberg, J. Chem. Soc., Chem. Commun., 1981, 307–308 RSC.
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS.
- W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386–7387 CrossRef CAS.
- Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 CrossRef CAS.
- F. Giacalone, M. Gruttadauria, P. Agrigento and R. Notoa, Chem. Soc. Rev., 2012, 41, 2406–2447 RSC.
- S. Y. Park, J.-W. Lee and C. E. Song, Nat. Commun., 2015, 6, 7512 CrossRef.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200–13203 CrossRef CAS.
- H. Y. Bae, D. Höfler, P. S. J. Kaib, P. Kasaplar, C. K. De, A. Döhring, S. Lee, K. Kaupmees, I. Leito and B. List, Nat. Chem., 2018, 10, 888–894 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299–4306 RSC.
- X. Yu and W. Wang, Chem. – Asian J., 2008, 3, 516–532 CrossRef CAS PubMed.
- T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 2633–2646 CrossRef CAS.
- M. C. Gimeno and R. P. Herrera, Eur. J. Org. Chem., 2020, 1057–1068 CrossRef CAS.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS.
- M. R. Vitale, S. Oudeyer, V. Levacher and J.-F. Brière, Radical and Ion-pairing Strategies in Asymmetric Organocatalysis, ISTE Press – Elsevier, 2016 Search PubMed.
- F. Legros, S. Oudeyer and V. Levacher, Chem. Rec., 2017, 17, 429–440 CrossRef CAS.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef CAS PubMed.
- K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- L. Gong, L.-A. Chen and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10868–10874 CrossRef CAS.
- S. K. Ghosh, A. Ehnbom, K. G. Lewis and J. A. Gladysz, Coord. Chem. Rev., 2017, 350, 30–48 CrossRef CAS.
- T. Cruchter and V. A. Larionov, Coord. Chem. Rev., 2018, 376, 95–113 CrossRef CAS.
- A. R. Wegener, C. Q. Kabes and J. A. Gladysz, Acc. Chem. Res., 2020, 53, 2299–2313 CrossRef CAS PubMed.
- Y. N. Belokon, A. G. Bulychev, V. I. Maleev, M. North, I. L. Malfanov and N. S. Ikonnikov, Mendeleev Commun., 2004, 14, 249–250 CrossRef.
- Y. N. Belokon, V. I. Maleev, I. L. Mal’fanov, T. F. Savel’eva, N. S. Ikonnikov, A. G. Bulychev, D. L. Usanov, D. A. Kataev and M. North, Russ. Chem. Bull., 2006, 115, 821–827 CrossRef.
- Y. N. Belokon, V. I. Maleev, D. A. Kataev, I. L. Mal’fanov, A. G. Bulychev, M. A. Moskalenko, T. F. Savel’eva, T. V. Skrupskaya, K. A. Lyssenko, I. A. Godovikov and M. North, Tetrahedron: Asymmetry, 2008, 19, 822–831 CrossRef CAS.
- C. Ganzmann and J. A. Gladysz, Chem. – Eur. J., 2008, 14, 5397–5400 CrossRef CAS PubMed.
- Y. N. Belokon’, V. I. Maleev, D. A. Kataev, T. F. Savel’eva, T. V. Skrupskaya, Y. V. Nelyubina and M. North, Tetrahedron: Asymmetry, 2009, 20, 1746–1752 CrossRef.
- Y. N. Belokon, V. A. Larionov, A. F. Mkrtchyan, V. N. Khrustalev, A. Nijland, A. S. Saghiyan, I. A. Godovikov, A. S. Peregudov, K. K. Babievsky, N. S. Ikonnikov and V. I. Maleev, Russ. Chem. Bull., 2012, 61, 2252–2260 CrossRef CAS.
- V. I. Maleev, T. V. Skrupskaya, L. V. Yashkina, A. F. Mkrtchyan, A. S. Saghyan, M. M. Il’in and D. A. Chusov, Tetrahedron: Asymmetry, 2013, 24, 178–183 CrossRef CAS.
- Y. N. Belokon, V. I. Maleev, M. North, V. A. Larionov, T. F. Savel’yeva, A. Nijland and Y. V. Nelyubina, ACS Catal., 2013, 3, 1951–1955 CrossRef CAS.
- L.-A. Chen, W. Xu, B. Huang, J. Ma, L. Wang, J. Xi, K. Harms, L. Gong and E. Meggers, J. Am. Chem. Soc., 2013, 135, 10598–10601 CrossRef CAS PubMed.
- L.-A. Chen, X. Tang, J. Xi, W. Xu, L. Gong and E. Meggers, Angew. Chem., Int. Ed., 2013, 52, 14021–14025 CrossRef CAS.
- V. I. Maleev, M. North, V. A. Larionov, I. V. Fedyanin, T. F. Savel’yeva, M. A. Moscalenko, A. F. Smolyakov and Y. N. Belokon, Adv. Synth. Catal., 2014, 356, 1803–1810 CrossRef.
- H. Huo, C. Fu, C. Wang, K. Harms and E. Meggers, Chem. Commun., 2014, 50, 10409–10411 RSC.
- J. Ma, X. Ding, Y. Hu, Y. Huang, L. Gong and E. Meggers, Nat. Commun., 2014, 5, 5531 CrossRef.
- T. Mukherjee, C. Ganzmann, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2014, 33, 6723–6737 CrossRef CAS.
- D. Carmona, M. P. Lamata, A. Sánchez, F. Viguri, R. Rodríguez, L. A. Oro, C. Liu, S. Díez-González and F. Maseras, Dalton Trans., 2014, 43, 11260–11268 RSC.
- D. Carmona, M. P. Lamata, P. Pardo, R. Rodríguez, F. J. Lahoz, P. García-Orduña, I. Alkorta, J. Elguero and L. A. Oro, Organometallics, 2014, 33, 616–619 CrossRef CAS.
- D. Carmona, P. Lamata, A. Sánchez, P. Pardo, R. Rodríguez, P. Ramírez, F. J. Lahoz, P. García-Orduña and L. A. Oro, Organometallics, 2014, 33, 4016–4026 CrossRef CAS.
- V. A. Larionov, E. P. Markelova, A. F. Smol’yakov, T. F. Savel’yeva, V. I. Maleev and Y. N. Belokon, RSC Adv., 2015, 5, 72764–72771 RSC.
- X. Ding, H. Lin, L. Gong and E. Meggers, Asian J. Org. Chem., 2015, 4, 434–437 CrossRef CAS.
- Y. Hu, Z. Zhou, L. Gong and E. Meggers, Org. Chem. Front., 2015, 2, 968–972 RSC.
- J. Liu, L. Gong and E. Meggers, Tetrahedron Lett., 2015, 56, 4653–4656 CrossRef CAS.
- K. G. Lewis, S. K. Ghosh, N. Bhuvanesh and J. A. Gladysz, ACS Cent. Sci., 2015, 1, 50–56 CrossRef CAS.
- J. Yu, H.-J. Jiang, Y. Zhou, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2015, 54, 11209–11213 CrossRef CAS PubMed.
- W. Xu, M. Arieno, H. Löw, K. Huang, X. Xie, T. Cruchter, Q. Ma, J. Xi, B. Huang, O. Wiest, L. Gong and E. Meggers, J. Am. Chem. Soc., 2016, 138, 8774–8780 CrossRef CAS PubMed.
- K. Huang, Q. Ma, X. Shen, L. Gong and E. Meggers, Asian J. Org. Chem., 2016, 5, 1198–1203 CrossRef CAS.
- Q. Ma, L. Gong and E. Meggers, Org. Chem. Front., 2016, 3, 1319–1325 RSC.
- W. Xu, X. Shen, Q. Ma, L. Gong and E. Meggers, ACS Catal., 2016, 6, 7641–7646 CrossRef CAS.
- X. Ding, C. Tian, Y. Hu, L. Gong and E. Meggers, Eur. J. Org. Chem., 2016, 887–890 CrossRef CAS.
- S. K. Ghosh, C. Ganzmann, N. Bhuvanesh and J. A. Gladysz, Angew. Chem., Int. Ed., 2016, 55, 4356–4360 CrossRef CAS.
- A. Kumar, S. K. Ghosh and J. A. Gladysz, Org. Lett., 2016, 18, 760–763 CrossRef CAS.
- Y. A. Rulev, V. A. Larionov, A. V. Lokutova, M. A. Moskalenko, O. L. Lependina, V. I. Maleev, M. North and Y. N. Belokon, ChemSusChem, 2016, 9, 216–222 CrossRef CAS PubMed.
- V. A. Larionov, S. M. Peregudova, V. I. Maleev and Y. N. Belokon, Russ. Chem. Bull., 2016, 65, 685–688 CrossRef CAS.
- T. Cruchter, M. G. Medvedev, X. Shen, T. Mietke, K. Harms, M. Marsch and E. Meggers, ACS Catal., 2017, 7, 5151–5162 CrossRef CAS.
- K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186–17192 CrossRef CAS PubMed.
- H.-J. Jiang, K. Liu, J. Yu, L. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 11931–11935 CrossRef CAS.
- H.-J. Jiang, K. Liu, J. Wang, N. Li and J. Yu, Org. Biomol. Chem., 2017, 15, 9077–9080 RSC.
- H. Joshi, S. K. Ghosh and J. A. Gladysz, Synthesis, 2017, 3905–3915 CAS.
- Z. T. Gugkaeva, V. A. Larionov, M. A. Moskalenko, V. N. Khrustalev, Y. V. Nelyubina, A. S. Peregudov, A. T. Tsaloev, V. I. Maleev and Y. N. Belokon, Synthesis, 2018, 607–616 CAS.
- K. Liu, H.-J. Jiang, N. Li, H. Li, J. Wang, Z.-Z. Zhang and J. Yu, J. Org. Chem., 2018, 83, 6815–6823 CrossRef CAS.
- N. Li, H. Yu, R. Wang, J. Shen, W.-Q. Wu, K. Liu, T.-T. Sun, Z.-Z. Zhang, C.-Z. Yao and J. Yu, Tetrahedron Lett., 2018, 59, 3605–3608 CrossRef CAS.
- R. Wang, W.-Q. Wu, N. Li, J. Shen, K. Liu and J. Yu, Synlett, 2019, 1077–1084 CAS.
- V. A. Larionov, L. V. Yashkina, M. G. Medvedev, A. F. Smol’yakov, A. S. Peregudov, A. A. Pavlov, D. B. Eremin, T. F. Savel’yeva, V. I. Maleev and Y. N. Belokon, Inorg. Chem., 2019, 58, 11051–11065 CrossRef CAS PubMed.
- J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141(34), 13625–13634 CrossRef CAS.
- W. J. Maximuck and J. A. Gladysz, Mol. Catal., 2019, 473, 110360 CrossRef CAS.
- C. Q. Kabes, W. J. Maximuck, S. K. Ghosh, A. Kumar, N. Bhuvanesh and J. A. Gladysz, ACS Catal., 2020, 10, 3249–3263 CrossRef CAS.
- W. J. Maximuck, C. Ganzmann, S. Alvi, K. R. Hooda and J. A. Gladysz, Dalton Trans., 2020, 49, 3680–3691 RSC.
- T. Wititsuwannakul, T. Mukherjee, M. B. Hall and J. A. Gladysz, Organometallics, 2020, 39, 1149–1162 CrossRef CAS.
- T. Mukherjee, S. K. Ghosh, T. Wititsuwannakul, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2020, 39, 1163–1175 CrossRef CAS.
- Q. H. Luu and J. A. Gladysz, Chem. – Eur. J., 2020, 26, 10230–10239 CrossRef CAS PubMed.
- T. F. Savel’yeva, O. V. Khromova, V. A. Larionov, A. F. Smol’yakov, I. V. Fedyanin, Y. N. Belokon and V. I. Maleev, Catalysts, 2021, 11, 152 CrossRef.
- J.-L. Pierre, Coord. Chem. Rev., 1998, 178–180, 1183–1192 CrossRef CAS.
- U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302–322 CrossRef PubMed.
- H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1194–1208 CrossRef CAS.
- P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125–143 CrossRef CAS.
- C. Ganter, Chem. Soc. Rev., 2003, 32, 130–138 RSC.
- H. Amouri and M. Gruselle, Chirality in Transition Metal Chemistry. Molecules, Supramolecular Assemblies and Materials, Wiley, Chichester, 2008 Search PubMed.
- E. Meggers, Eur. J. Inorg. Chem., 2011, 2911–2926 CrossRef CAS.
- E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637–1651 RSC.
- Z.-Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal. Sci. Technol., 2015, 5, 3441–3451 RSC.
- A. Ehnbom, S. K. Ghosh, K. G. Lewis and J. A. Gladysz, Chem. Soc. Rev., 2016, 45, 6799–6811 RSC.
- L. Zhang and E. Meggers, Chem. – Asian J., 2017, 12, 2335–2342 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50(2), 320–330 CrossRef CAS PubMed.
- A. Werner, Chem. Ber., 1911, 44, 1887–1898 CrossRef CAS.
- A. Werner, Chem. Ber., 1911, 44, 2445–2455 CrossRef.
- A. Werner, Chem. Ber., 1911, 44, 3272–3278 CrossRef.
- A. Werner, Chem. Ber., 1911, 44, 3279–3284 CrossRef.
- A. Werner, Chem. Ber., 1912, 45, 121–130 CrossRef CAS.
- T. Mizuta, K. Toshitani, K. Miyoshi and H. Yoneda, Inorg. Chem., 1990, 29, 3020–3026 CrossRef CAS.
- T. Mizuta, K. Sasaki, H. Yamane, K. Miyoshi and H. Yoneda, Bull. Chem. Soc. Jpn., 1998, 71, 1055–1064 CrossRef CAS.
- S. Ribeiro, L. Cunha-Silva, S. S. Balula and S. Gago, New J. Chem., 2014, 38, 2500–2507 RSC.
- L. R. Gahana and J. M. Harrowfield, Polyhedron, 2015, 94, 1–51 CrossRef.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576–6579 CrossRef CAS PubMed.
- M. P. Sibi and K. Itoh, J. Am. Chem. Soc., 2007, 129, 8064–8065 CrossRef CAS PubMed.
- R. P. Herrera, D. Monge, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, Org. Lett., 2007, 9, 3303–3306 CrossRef CAS PubMed.
- M. J. O'Donnell, Aldrichimica Acta, 2001, 34, 3–15 Search PubMed.
- B. Lygo and B. I. Andrews, Acc. Chem. Res., 2004, 37, 518–526 CrossRef CAS PubMed.
- T. Ooi and K. Maruoka, Acc. Chem. Res., 2004, 37, 526–533 CrossRef CAS PubMed.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656–5682 CrossRef CAS PubMed.
- K. Maruoka, Chem. Rec., 2010, 10, 254–259 CrossRef CAS.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS.
- M. J. O'Donnell, Tetrahedron, 2019, 75, 3667–3696 CrossRef.
- Y. N. Belokon’, V. M. Belikov, S. V. Vitt, T. F. Savel'eva, V. M. Burbelo, V. I. Bakhmutov, G. G. Aleksandrov and Y. T. Struchkov, Tetrahedron, 1977, 33, 2551–2564 CrossRef.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS.
- C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371–1448 CrossRef CAS.
- X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS PubMed.
- W.-M. Ren, G.-P. Wu, F. Lin, J.-Y. Jiang, C. Liu, Y. Luo and X.-B. Lu, Chem. Sci., 2012, 3, 2094–2102 RSC.
- J. Qin, V. A. Larionov, K. Harms and E. Meggers, ChemSusChem, 2019, 12, 320–325 CrossRef CAS PubMed.
- N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- X. Wu, J. A. Castro-Osma and M. North, Symmetry, 2016, 8, 4 CrossRef.
- E. Meggers, Chem. Commun., 2015, 51, 3290–3301 RSC.
- C. Wang and Z. Lu, Org. Chem. Front., 2015, 2, 179–190 RSC.
- R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS.
- Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278–290 RSC.
- C. Jiang, W. Chen, W.-H. Zheng and H. Lu, Org. Biomol. Chem., 2019, 17, 8673–8689 RSC.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- H. Huo and E. Meggers, Chimia, 2016, 70, 186–191 CrossRef CAS PubMed.
- Y. Yin, X. Zhao, B. Qiao and Z. Jiang, Org. Chem. Front., 2020, 7, 1283–1296 RSC.
- F. Burg and T. Bach, J. Org. Chem., 2019, 84, 8815–8836 CrossRef CAS.
- L.-X. Dai and X.-L. Hou, Chiral ferrocenes in asymmetric catalysis: synthesis and applications, Wiley-VCH, Weinheim, 2010 Search PubMed.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS PubMed.
- J.-C. Zhu, D.-X. Cui, Y.-D. Li, R. Jiang, W.-P. Chen and P.-A. Wang, ChemCatChem, 2018, 10, 907–919 CrossRef CAS.
- J. C. Ruble and G. C. Fu, J. Org. Chem., 1996, 61, 7230–7231 CrossRef CAS PubMed.
- J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532–11533 CrossRef CAS.
- B. L. Hodous, J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1999, 121, 2637–2638 CrossRef CAS.
- B. L. Hodous and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 1578–1579 CrossRef CAS PubMed.
- X. Dai, T. Nakai, J. A. C. Romero and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 4367–4369 CrossRef CAS PubMed.
- J. M. Berlin and G. C. Fu, Angew. Chem., Int. Ed., 2008, 47, 7048–7050 CrossRef CAS PubMed.
- A. A. Ibrahim, P.-H. Wei, G. D. Harzmann and N. J. Kerrigan, J. Org. Chem., 2010, 75, 7901–7904 CrossRef CAS PubMed.
- S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149–15153 CrossRef CAS PubMed.
- Q. H. Luu, K. G. Lewis, A. Banerjee, N. Bhuvanesh and J. A. Gladysz, Chem. Sci., 2018, 9, 5087–5099 RSC.
- S. Jang and H. Kim, Org. Lett., 2020, 22, 4185–4189 CrossRef CAS.
- M. Alimohammadi, A. Hasaninejad, Q. H. Luu and J. A. Gladysz, J. Org. Chem., 2020, 85, 11250–11257 CrossRef CAS.
- S. Jang and H. Kim, Asian J. Org. Chem., 2021, 10, 886–890 CrossRef CAS.
- S. K. Gupta, P. B. Hitchcock, Y. S. Kushwah and G. S. Argal, Inorg. Chim. Acta, 2007, 360, 2145–2152 CrossRef CAS.
- J. Zhang, F. Pan, H. Cheng and W. Du, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2010, 40, 211–215 CrossRef CAS.
- S. Jone Kirubavathy, R. Velmurugan, R. Karvembu, N. S. P. Bhuvanesh, K. Parameswari and S. Chitra, Russ. J. Coord. Chem., 2015, 41, 345–352 CrossRef.
- L. Xue, D. Deng and Q. Wang, Russ. J. Coord. Chem., 2017, 43, 181–188 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |