
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptides as a platform for targeted therapeutics for cancer: peptide–drug conjugates (PDCs)
Bethany M.
Cooper
 a,
Jessica
Iegre
a,
Daniel H.
O' Donovan
b,
Maria
Ölwegård Halvarsson
c and
David R.
Spring
*a
a,
Jessica
Iegre
a,
Daniel H.
O' Donovan
b,
Maria
Ölwegård Halvarsson
c and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bOncology R&D, AstraZeneca, Cambridge, CB4 0WG, UK
cMedicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
First published on 21st December 2020
Abstract
Peptides can offer the versatility needed for a successful oncology drug discovery approach. Peptide–drug conjugates (PDCs) are an emerging targeted therapeutic that present increased tumour penetration and selectivity. Despite these advantages, there are still limitations for the use of peptides as therapeutics exemplified through their slow progression to get into the clinic and limited oral bioavailability. New approaches to address these problems have been studied and successfully implemented to enhance the stability of peptides and their constructs. There is great promise for the future of PDCs with two molecules already on the market and many variations currently undergoing clinical trials, such as bicycle-toxin conjugates and peptide–dendrimer conjugates. This review summarises the entire process needed for the design and successful development of an oncology PDC including chemical and nanomaterial strategies to enhance peptide stability within circulation, the function of each component of a PDC construct, and current examples in clinical trials.
 Bethany M. Cooper | Bethany M. Cooper received her MChem degree from the University of Leeds in 2018, having completed her 3rd year of study at Lubrizol Ltd, Hazelwood. On return to the University of Leeds her final year was spent under the supervision of Professor Steve Marsden. In 2019, she started her PhD studies at the University of Cambridge under the supervision of Professor David Spring and industrial supervisor Dr Maria Ölwegård-Halvarsson, where her research has focused on peptide stapling methodologies for use within therapeutics. |
 Jessica Iegre | Jessica Iegre was born in Italy and obtained her MSci in Medicinal Chemistry and Pharmaceutical Technology at the University of Pisa, Italy in 2013. The same year, she joined the AstraZeneca IMED Graduate programme in Göteborg, Sweden where she spent 2 years working across three different departments: medicinal chemistry, DMPK and computational chemistry. In 2015 she joined the Spring group at the University of Cambridge, and she obtained her PhD in chemical biology in 2019. Jessica is currently a Postdoctoral Research Associate in the group, and she is developing novel stapled peptides to inhibit medicinally relevant protein–protein interactions. |
 Daniel H. O'Donovan | Daniel Hillebrand O' Donovan is currently Associate Principal Scientist in Early Oncology R&D at AstraZeneca, Cambridge, UK. Following PhD studies in medicinal chemistry at Trinity College Dublin, he moved to Germany for postdoctoral studies at the Max Planck Institute for Kohlenforschung followed by a Marie Curie fellowship at the University of Oxford, UK. Since joining AstraZeneca in 2016, he has worked in a variety of research areas including antihormonal therapies for breast cancer, epigenetics, immuno-oncology and targeted protein degradation. |
 Maria Ölwegård Halvarsson | Dr Maria Ölwegård-Halvarsson is a Senior Research Scientist in the New Modalities group, Department of Medicinal Chemistry, AstraZeneca, Sweden. In her current role she develops linker chemistry and synthesizes drug conjugates within the targeting delivery platform. She obtained her PhD in organic chemistry from Gothenburg University in 1990 and then spent four years at the Department of Medicinal Biochemistry, Gothenburg University. She joined AstraZeneca in 1995, working three years in the isotope labelling group and has a 25 year commitment to the company, synthesizing drug molecules within the cardiovascular and metabolic disease areas in lead optimization and lead generation phase. |
 David R. Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
Key learning points1. A variety of methods to address peptide chemical and enzymatic stability can be implemented.2. The design of a PDC requires understanding of the mechanism of action intended and hence environmental stimuli (pH, GSH and enzymes). 3. The function and rationale behind the design of each component of a PDC. 4. The stability of the PDC construct can be improved through the use of nanomaterials. 5. Bicycle-toxin conjugates (BTCs) and peptide-dendrimer conjugates are emerging constructs that fall under the umbrella term PDCs. |
Introduction
Traditionally, research within drug discovery falls into two groups: small molecules (<500 Da) and biologics (>5000 Da).1 Peptides are placed within the molecular weight range that is typically under-represented in the pharmaceutical company pipelines. A peptide is defined by the FDA as a polymer composed of less than 40 amino acids (500–5000 Da).2 Over recent years, the research community is acknowledging the many advantages that peptides bring over small molecules and biologics. These include simpler design, ability to interact with underexplored targets, cheaper synthesis, decreased immunogenicity and enhanced tissue penetration.1 To date in the U.S., Europe and Japan markets, there are more than 100 peptide drugs used to treat a range of diseases.2 Financially, the peptide market is lucrative as it is estimated to be worth £11–16 billion annually by 2019.2 However, there is still a significant challenge for the pharmaceutical industry to get peptides to market with many adopting greener peptide synthesis techniques at increased costs than traditional approaches.Peptides can offer a multifunctional approach – in addition to being biologically active, they are excellent at transporting cargos to the desired targets. Their use within targeted therapeutics is an exciting area of research with great promise in the future with particular focus in, but not limited to, oncology. Witnessing the current success and investment into many antibody–drug conjugates (ADCs), the equivalent peptide–drug conjugates (PDCs) show promise for the future of the use of peptides within this setting. This review will highlight the excellence and limitations of peptides, their use in PDCs for advancing targeted cancer therapeutics and will consider how the specific tumour microenvironment can aid the design of a PDC. In addition, the review provides an examination of how the stability of PDCs in circulation can be enhanced through both chemical modifications and material science – a topic rarely discussed but extremely valuable to the successful development of new peptide drugs.
ADME & PK considerations for peptides
When considering therapeutic agents, it is crucial to analyse pharmacokinetic (PK) properties such as absorption, distribution, metabolism and excretion (ADME) as well as pharmacodynamic (PD) properties. Traditionally, PK properties of peptides tend to differ substantially from those of small drug molecules.3 A significant limitation of peptides is their limited or non-existent oral bioavailability leading to administration through intravenous (IV) injection. One of the most convenient methods of administration is oral as it allows the patient to take the medication independently, unlike IV that often requires a clinical setting.A small molecule is defined as ‘drug-like’ if it satisfies the criteria of Lipinski's Rule of 5 (Ro5). These rules focus on several fundamental factors including the molecular weight (<500 Da), ≤5 H-bond donors, ≤10 H-bond acceptors and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (<5): molecules that satisfy the Ro5 are likely to be orally bioavailable.4 Peptides do not fit into the Ro5 criteria due to their relatively large size (500–5000 Da) in comparison to small molecules (typically <500 Da). Despite peptides not satisfying the Ro5 criteria, the rule does not indicate that a peptide cannot become a drug as proven through many peptides on the market and in clinical trials.4
P (<5): molecules that satisfy the Ro5 are likely to be orally bioavailable.4 Peptides do not fit into the Ro5 criteria due to their relatively large size (500–5000 Da) in comparison to small molecules (typically <500 Da). Despite peptides not satisfying the Ro5 criteria, the rule does not indicate that a peptide cannot become a drug as proven through many peptides on the market and in clinical trials.4
Santos et al. analysed peptides approved by the FDA between 2012–2016 to allow comparison to the Ro5.4 They determined that the most orally available peptides were indeed up to a MW of 1200 Da and displayed a logP within the range of 5–8. Close interpretation of the results found that orally available peptides had 5 times more H-bond donors and acceptors than what was considered as acceptable by Lipinski's Ro5 for small molecules.4
Oral administration is challenging for both biologics and peptides and in many cases, it may not be feasible. The whole journey of oral administration through the gastrointestinal tract is problematic, starting with the enzymes amylase and lipase found within saliva that break down the peptides into smaller molecules (Fig. 1).5 On arrival into the stomach, the peptide is subjected to harsh acidic conditions and proteolysis by cathepsin and pepsin. Even if the peptide successfully remains intact to this point, the lumen of the small intestine experiences a pH change and has a vast number of proteolyzing enzymes including trypsin, chymotrypsin and carboxypeptidase.5
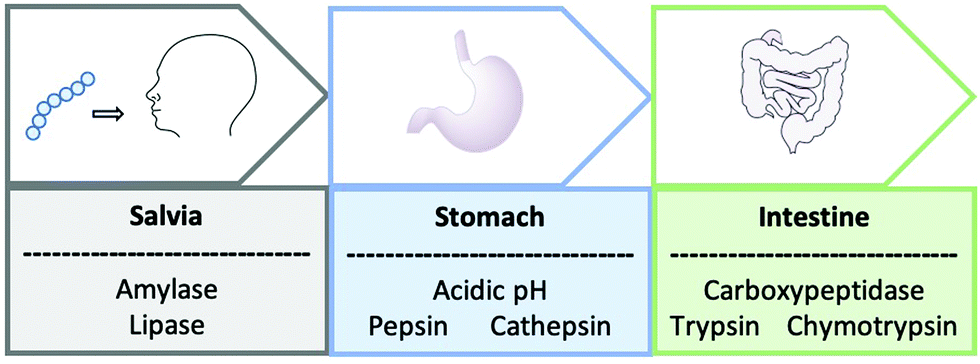 | ||
| Fig. 1 A schematic to represent the journey of an orally administrated peptide through the gastrointestinal tract. | ||
Compared to biologics, peptides have a much shorter circulatory half-life (days vs. weeks) resulting in the need for sub-optimal frequent drug administrations. The impact of this is witnessed through the advancement of ADCs and a slower progression of PDCs. The lifetime of a hydrophilic peptide in circulation is determined by many soluble enzymes in the blood and at membranes. Exopeptidases are a class of enzymes that can be split into two subgroups: amino- and carboxypeptidases and target the N- and C-terminal, respectively.6 It is these enzymes that are responsible for the chemical instability and the breakdown of peptides in the bloodstream.
Short half-lives are also experienced due to rapid renal clearance, resulting in many hampered peptide in vivo studies and ultimately the pursuit of the peptide as a drug. Found within the kidney are glomeruli pores that have a size of ∼8 nm; circulating peptides that are less than 25 kDa filter through the glomeruli, and are not reabsorbed through the renal tubule.3 Considering such limitations, it is not surprising that many oral peptide drug candidates have entered clinical trials, but with limited success and overall are restricted to the area of endocrine disorders. Formulations are helpful in this setting and have been successfully used in the development of Semaglutide, the most recently FDA approved orally available peptide drug used to treat Type 2 diabetes (September 2019). Sodium N-[8-(2-hydroxybenzoyl)amino caprylate] (SNAC) is used with Semaglutide to form a co-formulation.7 SNAC works as a buffering agent within the stomach, which in turn diminishes the activity of proteolyzing enzymes including pepsin where the maximum activity is experienced at a pH within the range of 2–4.7 Despite Semaglutide's approval, the delivery of peptides orally still has a long way to develop before we see more in the clinic.
Several ways can be used to try and improve the ADME properties of peptides such as the improvement of the cell permeability, enhancement of chemical and proteolysis stability and reduction of renal clearance overall resulting in the extension of the circulatory half-life. The extended half-life is beneficial both economically and for the patients’ compliance. The next part of the review will focus on the ways of modifying a peptide to achieve such improvements.
Improving enzymatic and chemical stability of peptides by chemical modifications
Cyclisation
Cyclisation techniques have been used widely in the peptide field and achieved in several ways from cyclising head to tail, head/tail to side chain or side chain to side chain.A type of side chain to side chain cyclisation is called stapling, a technique that enables the peptide to be locked into a desired conformation. Peptide stapling is used commonly to enhance a peptide's secondary structure such as α-helices and β-turns, which can improve the binding affinity to the target and enhance ADME properties.
There are two subgroups of peptide stapling (PS): one-component (1C) and two-component (2C). In one-component peptide stapling (1C-PS) there is an intramolecular linkage between often unnatural amino acid side chains and can allow cyclisation depending on the secondary structure.
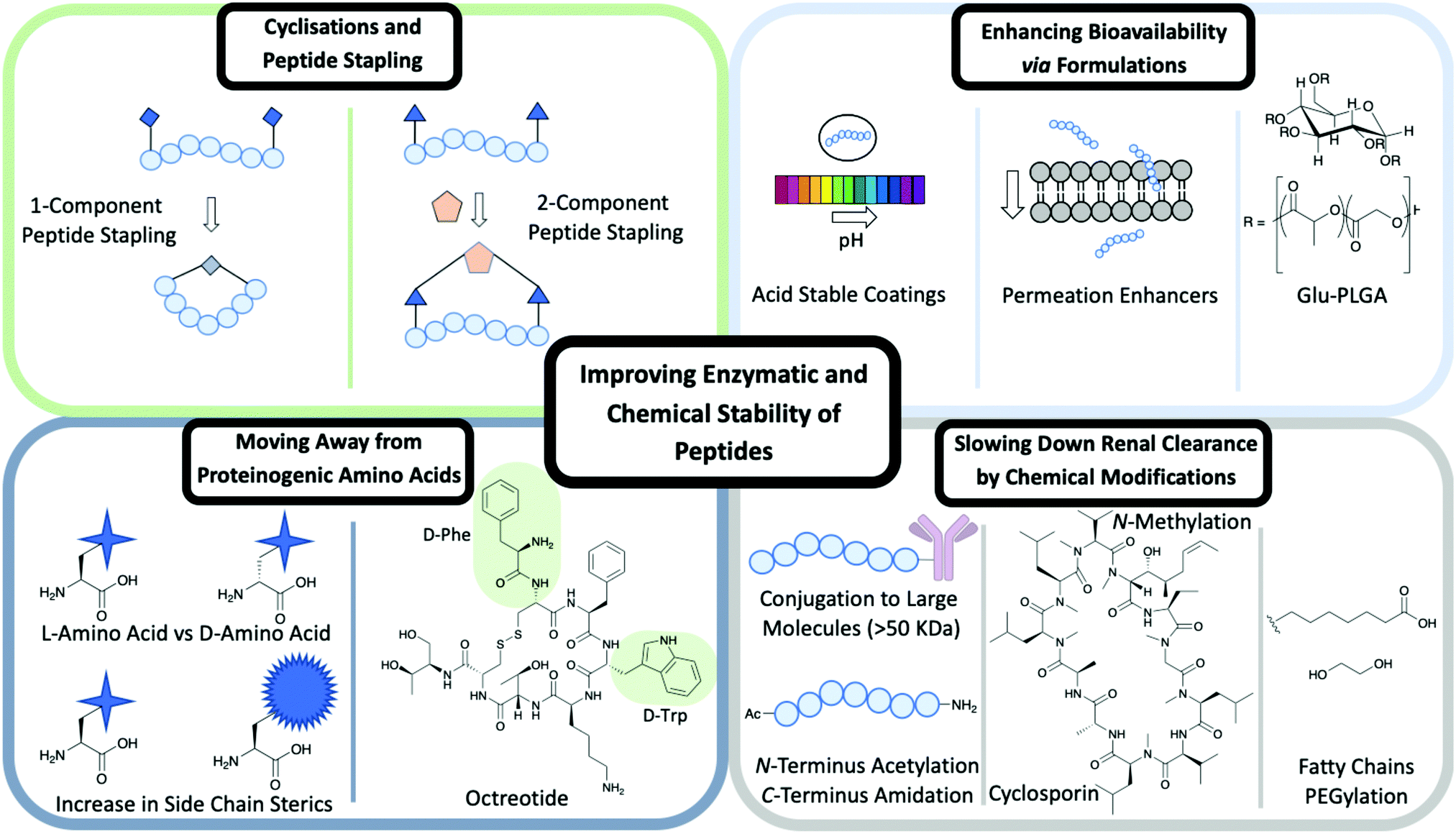 | ||
| Fig. 2 Different methods used to improve the enzymatic and chemical stability of peptides including: cyclisations and peptide stapling, formulations, moving away from proteinogenic amino acids and chemical modifications. | ||
One of the first examples of 1C-PS was by Blackwell and Grubbs through the use of ring-closing metathesis (RCM) of O-allyl serine residues.8 1C-PS is not without its limitations. For example, modifications would result in the entire peptide being resynthesized, which could prove costly in both time and materials. However, 1C-PS has proved successful in many cases exemplified by the first stapled peptide ALRN-5281 to reach clinical trials and complete Phase I.9 ALRN-5281 is used to treat adult growth hormone deficiency and was cyclised through the use of 1C-PS via RCM.9
Peptide stapling more recently has moved the focus away from 1C-PS to two-component peptide stapling (2C-PS) (Fig. 2). The use of 2C-PS offers many advantages over 1C-PS and allows synthetic versatility through the use of linear peptides and separate staples. 2C-PS allows modifications to be made at a late stage if needed to the peptide or the staple, which is highly beneficial for an optimization campaign. 2C-PS has been widely applied to bicycles therapeutics developed by Christian Heinis and Sir Greg Winter.10 The overall concept of bicycles is the cross-linking of three cysteine residues to a tri-functionalised linker to form a bicycle construct. More details on the application of bicycles for use within drug conjugates are detailed later in the review.
Moving away from proteinogenic amino acids
Side chains of amino acids offer another excellent source for modification with many papers being published recently on direct amino acid modification. Increasing the steric bulk of the side chains results in increased stability, as enzyme recognition is disrupted.6One way to increase the overall stability of the full peptide is to swap L-amino acids to D-amino acids. The D-amino acid sequence has a decreased substrate recognition and binding affinity for proteolytic enzymes.3 An example of increasing the half-life of a biologically active peptide is modifying Somatostatin to Octreotide used to treat gastrointestinal tumours.3,6 Octreotide's amino acid sequence incorporates two D-amino acids, whereas Somatostatin is only composed of L-amino acids (Fig. 2). The resulting half-life increases from a few minutes for Somatostatin to 1.5 hours for Octreotide hence enhancing favourable PK properties.11 Despite this example highlighting the benefits of L- to D-amino acid exchange, there are a few cases in which D-amino acid-containing peptides show a reduced half-life in comparison to the L-analogue.3 It is important to consider the effects that such modifications could have on the overall secondary structure of the peptide and on any intra- or intermolecular interactions.3 An alternative is the use of D-peptides. These peptides are mirror images of the L-amino acid-containing counterpart and are composed fully of D-amino acids. The Kay group have recently developed D-peptides for use as HIV entry inhibitors.12
Slowing down renal clearance by chemical modifications
The overall net charge for the peptide sequence is an important consideration for renal clearance. Peptides that acquire a net negative charge tend to exhibit a longer half-life in comparison to those with a net positive charge.13 The presence of anionic carbohydrate moieties found within the kidney's glomerular membrane limit the filtration of anionically charged species into the urine.13Another approach is the conjugation of peptides with larger molecules (>50 kDa) to increase lipophilicity and their binding to albumin, thereby improving the PK and PD properties of peptides.13 It is the enhanced steric that prevent the conjugate from being filtered out through the kidneys and allow a longer circulation period. Modifications at the N- and C-terminus help slow down renal clearance. Typically the breakdown by exopeptidases of a peptide sequence occurs at the N- or C-terminus.6
There are ways this can be prevented by modifying the terminus to increase proteolysis resistance. An amide bond between the C- and N-terminus achieved through a cyclisation reaction has been shown to prevent enzyme degradation.6 If cyclisation is not the preferred structure for binding, then N-acetylation and C-amidation can be an alternative to enhance resistance to proteolysis. These modifications have proven successful in a range of studies on Somatostatin, where the half-life of the modified molecule was extended when compared to the unmodified peptide.3
N-Methylation of amide bonds is another modification that enhances metabolic resistance. N-Methylation increases the steric hindrance and allows tuning of the peptide conformation.14 Cyclosporin is an example of a naturally occurring peptide used as an immunosuppressive drug (Fig. 2). Cyclosporin is a hepta-N-methylated cyclic undecapeptide, with an oral bioavailability of 29%.14
The use of polyethylene glycol (PEG) has been vastly explored in slowing down renal clearance and by increasing the binding to plasma proteins such as albumins. PEG is an ideal candidate for modification: it is cheap, biocompatible, hydrophilic, non-toxic and non-immunogenic.6 The promise of PEGylation for the modification of peptides is highlighted by a number of examples, which are discussed herein.
RGD is a homing tripeptide (see later), whose sequence is able to allow the HM-3 peptide to selectively bind to specific target sites that display high levels of integrins within tumours.13,15 The original HM-3 peptide had limited effect as a consequence of its short half-life and required twice a day administration. There was a need to enhance the peptide's half-life to reduce the number of administrations needed. Methoxy-poly(ethylene glycol)-aldehyde (mPEG-ald) was the PEG linker of choice for attachment at the N-terminus. Upon this modification, there was a 5.86 fold increase in half-life in male rat studies when compared to the unmodified HM-3 peptide.15
Another PEGylated peptide is PEG-adrenomedullin (PEG-ADM) by Bayer used in patients suffering from Acute Respiratory Distress Syndrome (ARDS) associated with lung failure.16 The peptide was enrolled into Phase 2 clinical trials in August 2020 with the predicted end date of early 2023.16
PEG is not the only molecule used in conjugation to the peptide approaches to slow down renal clearance. Other widely used examples include polysialic acids (PSA), a homopolymer, and hydroxyethyl starch (HES), a branched amylopectin.13
The addition of fatty chains has been an effective method as an addition to peptides to increase the half-life. Glucagon-like peptide (GLP-1) receptor agonists have been used to control blood sugar levels in patients with Type 2 diabetes.17 One of the earliest example is Exenatide, an analogue of a nonhuman peptide that in 2005 had twice-daily administration and an IV half-life of 30 minutes. In 2009 Liraglutide, a near analogue of human GLP-1, had a fatty acid chain with a spacer joined to the main peptide backbone for binding to albumin (Fig. 3). Liraglutide represented a significant improvement of GLP-1 with an extended IV half-life of 8–10 hours and once-daily administration.17 Semaglutide is a GLP-1 agonist, with a γGlu-2xOEG linker to a C18 fatty chain (Fig. 3).17 The determined IV half-life was 46.1 hours through studies involving mini pigs, enabling once-weekly doses – a vast improvement to the early GLP-1 agonists.17
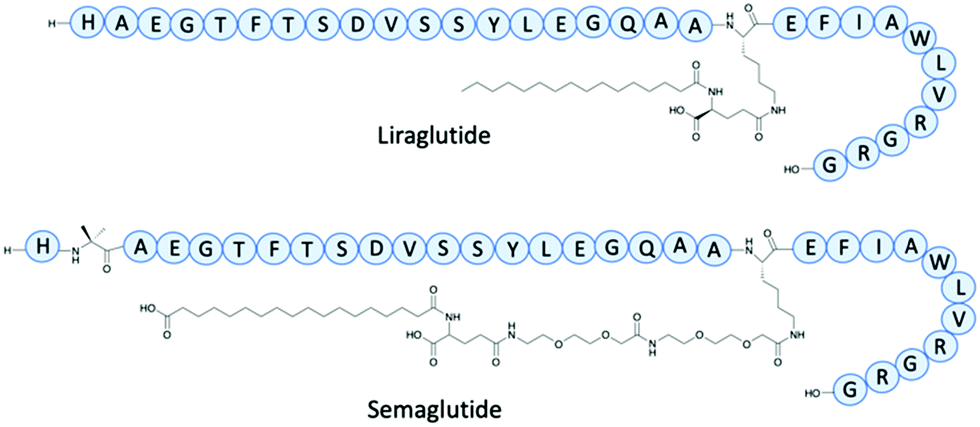 | ||
| Fig. 3 The structures of Liraglutide and Semaglutide. | ||
Enhancing bioavailability via formulations
Several methods have been used previously in the literature to enhance the oral bioavailability of peptide therapeutics via formulations.5 These can include permeation enhancers and acid-stable coatings (Fig. 2).5 Permeation enhancers are able to transport the peptide through epithelial cells – an alternative route is available through intercellular junction and adhesion protein interference resulting in a paracellular route.5 The use of acid-stable coatings to improve the oral availability of a drug is a widely used approach. These coatings are pH active, where at low pH in the stomach the coating remains intact; as the peptide moves to the intestine the pH rises and the coating breaks down to release the contents. The introduction of citric acid can be used to help neutralise the optimum basic pH conditions for a range of gastrointestinal peptidases, hence slowing down degradation caused by peptidases.5Formulations can be used to enhance the bioavailability of IV administrated peptides. The FDA approved Sandostatin LAR is an excellent example of this in which Octreotide is encapsulated in a glucose-poly(lactide-co-glycolide) (Glu-PLGA) star-shaped polymer (Fig. 2).18
Improving the overall chemical and enzymatic stability of a peptide using the techniques discussed is beneficial for the discovery of new therapeutics including targeted therapies exemplified by peptide–drug conjugates. These concepts will be discussed in the following section.
Cancer therapy and targeted drug delivery
Cancer is among the leading contributors to human mortality and disease, with nearly 50% of people being diagnosed with cancer in their lifetime.19 In 2000 and 2010, two landmark publications defined the “Hallmarks of Cancer”, characterising all cancers with eight common traits: cancer cells stimulate their own growth, lack sensitivity to anti-growth signals, evade programmed cell death (apoptosis), can divide indefinitely, can sustain blood vessel formation (angiogenesis), invade other tissues and metastasise, deregulate metabolism and evade the immune system.20,21 Our growing understanding of cancer cell biology has enabled significant advances in treating this disease.Subject to the stage and tumour type, patients are treated with one or a combination of the following options: surgery, radiotherapy or pharmacotherapy.
Traditional pharmacotherapy (chemotherapy) is characterised by cytotoxic drug regimens which target rapidly dividing cells through inhibiting mitosis and is associated with serious side-effects such as bone marrow and gastrointestinal toxicity. Even if the tumour is successfully eradicated, lasting damage may continue to affect healthy tissues and residual cancer cells may result in relapse of the disease.
Fortunately, newer molecular therapies can improve patient outcomes and reduce toxicity by targeting the unique characteristics of tumour cells including the cell morphology (leaky cell membranes), lower pH, increased glutathione (GSH) and enzyme presence (Fig. 4).22
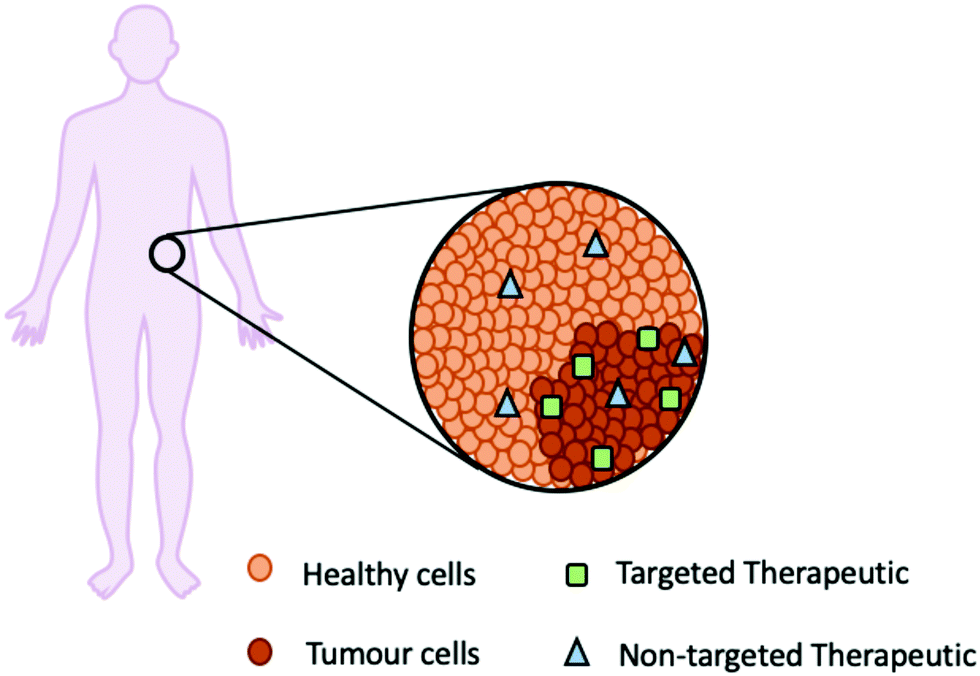 | ||
| Fig. 4 A schematic representation of how a targeted therapeutic (green square) is specific for the tumour cells in comparison to a non-targeted therapeutic (blue triangle). | ||
Antihormonal therapy is often effective for tumours whose growth is driven by endocrine signalling, such as breast and prostate cancers. Drugs which selectively inhibit an oncogenic mutant protein while sparing the unchanged (wild-type) protein are another successful approach, such as Gleevec (imatinib), a tyrosine kinase inhibitor which has revolutionised the treatment of Chronic Myelogenous Leukemia (CML).23 By targeting cancer's ability to evade the immune response, checkpoint inhibitor antibodies such as Keytruda (pembrolizumab) and Imfinzi (durvalumab) can rekindle the immune system's ability to recognise and eliminate tumours, providing new treatment options for recalcitrant tumours such as non-small lung cell cancer (NSCLC) and Hodgkin's lymphoma. Following treatment with all of these drugs, cancers may further mutate to develop resistance to targeted therapies; these resistance mutations provide challenges but also new opportunities for drug discovery.24
Antibody–drug conjugates (ADCs) are another promising line of targeted therapy composed of an antibody which binds to a protein highly expressed in tumour cells (a tumour antigen), connected via a linker to a cytotoxic small molecule payload. In principle, this design enables the targeted delivery of a drug to the tumour, killing cancer cells while sparing healthy tissue and providing a wider therapeutic margin than traditional chemotherapy. In 2000, Mylotarg (gemtuzumab ozogamicin) became the first ADC approved by the FDA, combining an antibody to the CD33 antigen expressed in leukaemia cells with a cytotoxic payload derived from a calicheamicin natural product. Over the past 20 years, there have been significant advances in this field exemplified by 5 ADCs approved between 2008 to 2018 and 3 ADCs approved by the FDA in 2019 for various cancers, including AstraZeneca and Daiichi's Enhertu (trastuzumab deruxtecan) for the treatment of HER2 metastatic breast cancer, Roche's Polivy (polatuzumab vedotin) for large B-cell lymphoma, and Astella and Seattle Genetics’ Padcev (enfortumab vedotin-ejfv) for urothelial bladder cancer.25 The most recent ADC being approved in August 2020 was GSK's Blenrep (belantamab mafodotin-blmf) for relapsed or refractory multiple myeloma, bring the total to 9 FDA approved ADCs as of September 2020.26
Despite continuing progress for ADCs, these therapies present several drawbacks. As cytotoxic drugs, ADCs can incur significant toxicity; in the case of Mylotarg, the FDA issued a black box warning for potentially life-threatening side effects in patients who did not receive stem cell transplantation.27 ADCs can also generate dangerous immune reactions, yielding toxicity and halting further therapy.28 Furthermore, the complicated structure of ADCs results in high production costs, in some cases restricting access to these drugs as their cost-effectiveness may be called into question by health insurers. Owing to their high molecular weight and protein-like physicochemical properties, ADCs also suffer from restricted distribution which can prevent these therapies from penetrating some types of solid tumours and limiting their efficacy.
To overcome these issues, many research groups have explored alternative approaches such as peptide-based drugs, protein–protein interaction inhibitors (PPIs) and drug delivery transporters. This review will focus on the latter and describe how peptides can be used within peptide–drug conjugates (PDCs) for the targeted treatment of cancer (Fig. 5).
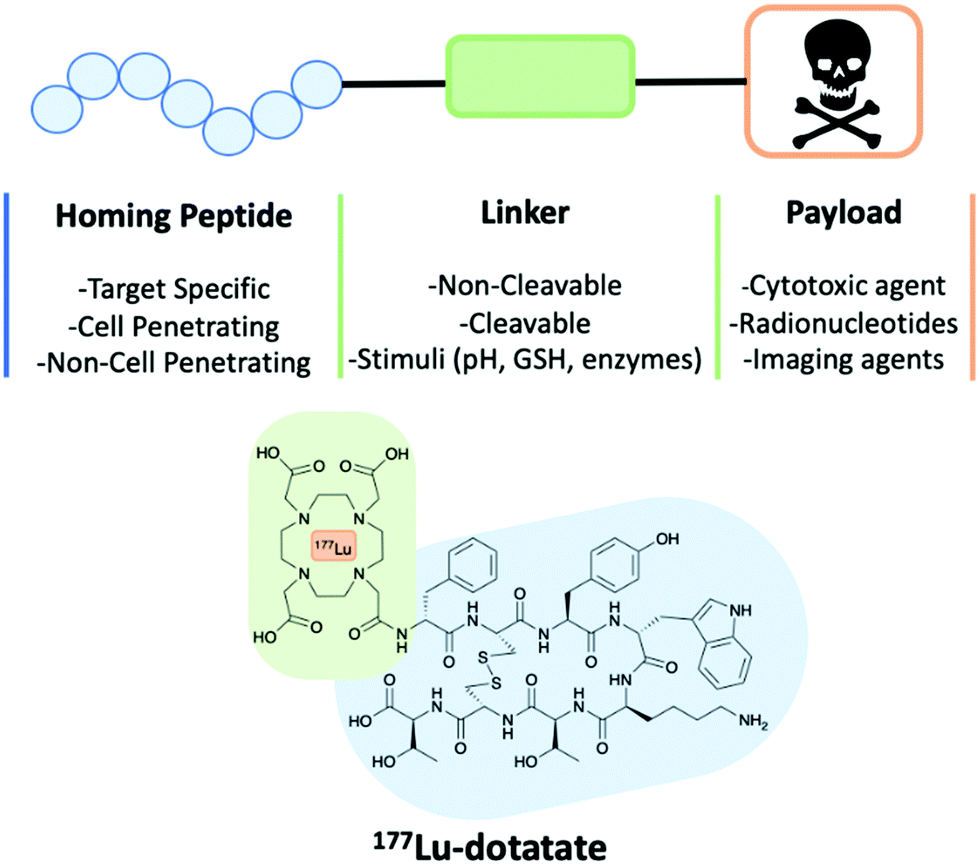 | ||
| Fig. 5 A schematic of a peptide–drug conjugate construct consisting of a homing peptide, linker and payload. The structure of 177Lu-dotatate an FDA approved peptide–drug conjugate. | ||
Peptide–drug conjugates (PDCs)
Peptide–drug conjugates are a class of targeted therapeutics, with a similar construct to that of ADCs and only differing through the homing device. A PDC is composed of three vital components: a homing peptide, a linker and a cytotoxic payload (Fig. 5). All three work in synergy to deliver cytotoxins through targeting the selected receptor of a tumour cell. As discussed earlier, the ADC market has been fast-paced, but unfortunately, this is not the same case for PDCs. A contributing factor is that methodologies that overcome the intrinsic poor PK properties of the peptides have only recently emerged, and pharmaceutical companies have started expanding their pipelines to accommodate peptide therapeutics only in the last decade.There is currently only one therapeutic PDC on the market, 177Lu-dotatate, but many more are in various phases of the pipeline. 177Lu-dotatate is used to treat gastroenteropancreatic neuroendocrine tumours (GEP-NETs) and was the first FDA approved PDC (Fig. 5).29 Somatostatin is the homing peptide, which is conjugated to a cytotoxic radiotherapeutic agent 177Lu through an amide linker. 177Lu-dotatate is IV administrated once every 8 weeks usually for a total of four cycles of treatments.29
Earlier this year, Theratechnologies Inc. released a statement about two advanced PDCs in their pipeline: TH1902 and TH1904.30 TH1902 is a PDC with a Docetaxel payload and used to treat triple-negative breast cancer as well as ovarian cancer.
TH1904 uses a doxorubicin payload and is used to treat ovarian cancer. Both target Sortilin 1 (SORT1) receptors, that are overexpressed in numerous cancers including triple-negative breast cancer, ovarian, lung, colorectal, skin and pancreatic.30
In vivo studies identified good accumulation of the PDCs in ovarian cancer with limited off-target delivery in healthy cells.30
These PDCs are examples of therapeutics, however, PDCs can be used as successful diagnostic tools by employing the use of radionucleotides.
On constructing a PDC, it is key to understand the role of each component to fully appreciate the design. Considerations include the mechanism of action and how alternative approaches can improve the current limitations of this emerging modality.
Homing peptide
A homing peptide is a selected peptide that is chosen for its specific targeting capabilities of protein receptors found overexpressed at tumour tissues. In the case of PDCs, the homing peptide will direct the whole PDC construct to the targeted cell and limit off-target delivery. These homing peptides often have precedent in the literature for having a strong binding affinity to the target site within the nanomole magnitude. Several techniques can be used to determine their binding affinity including surface plasmon resonance (SPR), biolayer interferometry (BLI) and isothermal titration calorimetry (ITC).The secondary structure of the homing peptide has a pronounced effect on its binding affinity. Therefore, structural information is important when seeking to increase the binding affinity of the homing peptide through stabilisation of the secondary structure. The most common examples of peptide secondary structure include α-helix, β-sheet and random coil. On the attachment of the linker to allow conjugation to the cytotoxin, the secondary structure of the homing peptide must be retained, and the linker should not disrupt binding. There is a vast range of homing peptides for many targets detailed in a recent review by Vrettos et al. and summarised in Table 1.19
| Homing peptide | Receptor |
|---|---|
| RGD (tripeptide – arginine, glycine, aspartic acid) | Integrins (α5β1, α8β1 and αIIbβ3) |
| GnRH (gonadotropin-releasing hormone) | GnRH-R (receptor version of the hormone) |
| SST (somatostatin) | SSTR1-5 (somatostatin receptor) |
| EGF (epidermal growth factor) | EGFR: HER1, HER2, HER3, HER4 |
| Angiopep-2 | LRP-1 (low-density lipoprotein receptor-related protein-1) |
Most homing peptides reported to date are linear. Even though they show good binding, there are several drawbacks including degradation by enzymes at the termini, chemical instability and fast renal-clearance. A way to overcome these limitations is through the use of cyclisation or peptide stapling of the linear peptide as described earlier.
A study by Lu and co-workers demonstrated how a stapled RGD peptide was used as a homing peptide that targets αvβ3 integrin (Fig. 6).31 The peptide can be used to modify a nanoparticle to effectively deliver a drug to target glioblastoma multiforme (GBM), an aggressive CNS tumour with poor prognosis.31 The RGD tripeptide has precedent for targeting αvβ3 integrins expressed on glioma cells and overcoming the blood brain tumour barrier (BBTB). During the early stages of glioma, therapeutics can be hindered as the blood brain barrier (BBB) still remains intact. Therefore, a suitable vehicle to overcome such barrier is advantageous. The stapled RGD peptide was modified with two unnatural amino acids and cyclised through an RCM reaction. SPR determined the binding affinity of the stapled RGD peptide with αvβ3 and the KD was found to be 17.8 μM.31 Results indicated the stapled peptide proved successful in targeting the overexpressed αvβ3 integrins in the tumour environment. It was also determined that the stapled peptide allowed the vital delivery of the cytotoxin across the BBB to allow efficient targeting GBM.31
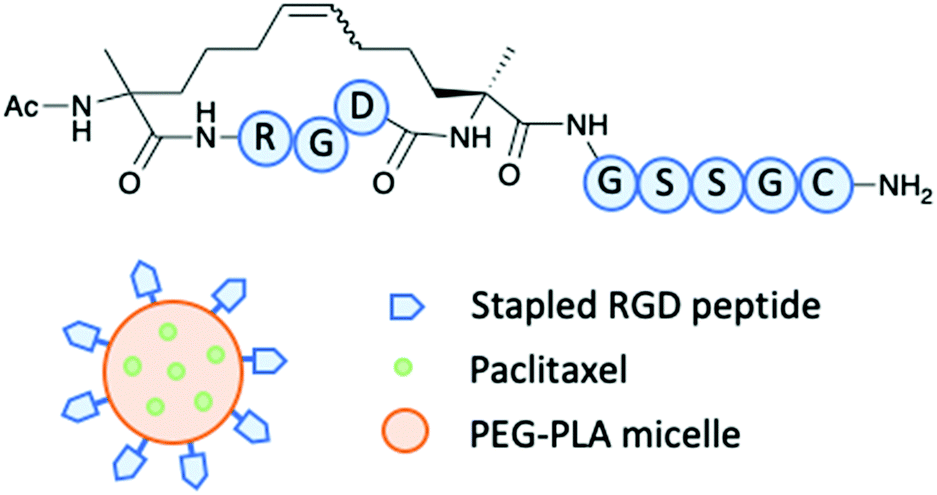 | ||
| Fig. 6 Lu and co-workers stapled RGD peptide to modify a Paclitaxel loaded PEG–PLA micelle.31 | ||
Tripodi et al. recently established how NGR can form a conjugate with cytotoxin Daunorubicin.32 NGR is a homing tripeptide and is able to distinguish CD13 receptors found on tumour cells. Daunorubicin was conjugated to the cyclic NGR peptide through an oxime-linkage and a cleavable tetrapeptide GFLG linker. Cathepsin B, found overexpressed within many tumour environments, cleaved the GFLG linker to release the Daunorubicin payload. Two conjugates were tested featuring the cyclic peptides KNGRG and NleNGRG.32 The cell lines tested were Kaposi's Sarcoma (KS) that are CD13(+) and HT-29 human colorectal adenocarcinoma cells that are CD13(−), the control being MRC-5 fibroblasts. It was concluded that the conjugate with Nle instead of Lys was the more efficient conjugate for uptake even at low concentrations. The mice could tolerate both conjugates well, with no significant changes in body weight observed. Liver toxicity is an important control for drug metabolism and both the conjugates were found to be selective without toxic side effects unlike free Daunorubicin.32
Some homing peptides can also act as cell penetrating peptides (CPPs). A CPP often displays several characteristic features to aid transport across cell membranes: hydrophobicity, amphipathicity and net positive charge.33 If a cell penetrating homing peptide is attached, through the use of a linker, to a cargo including therapeutic peptides, small molecule cytotoxins and proteins – the homing peptide not only directs the cargo to the desired tissue but allows for its internalisation. Cationic CPPs present limitations such as their inability to selectively home to the target resulting in unspecific cell uptake. For this reason, PDC featuring anionic CPPs are often used to aid specificity for tumour cells.34 The homing ability of cell penetrating homing peptide can also be enhanced by incorporating a stimuli-responsive linker as described in the following section.
Linker
Various linker technologies have been used depending on the desired PDC mechanism of action. The linker must demonstrate stability within the circulation in order to prevent the premature and unspecific release of the drug. Pre-release is a cause for concern for PDCs as the build-up of cytotoxins can lead to dangerous or even fatal side effects. It is also important to consider the fate of the linker when it is metabolised within the body, and hence the resulting toxicity of the products formed.As with ADCs, the linker used for PDCs can be either cleavable or non-cleavable. The cleavage of a cleavable linker can be enzyme or chemically mediated. Whereas, a non-cleavable linker is not triggered by environmental stimuli such a change in pH, tripeptide glutathione (GSH) or enzymes found within the tumour microenvironment.35
Cleavable linkers
There are several chemical motifs under the umbrella of chemically cleavable linkers including acid cleavable, reducible disulfides and linkers which are cleaved by exogenous stimuli. Often endogenous stimuli are easier to control and present several advantages: the patient's biology should not affect linker cleavage rates and there is still cleavage when the endogenous trigger concentrations are low. A detailed review of all aspects of cleavable linkers was published in 2019 by Bargh et al.35A chemically cleavable linker that directly relies on the lysosomal pH for efficient payload release is the acid-cleavable N-acyl hydrazine linker (Fig. 7). The linker is cleaved at low pH in lysosomes (pH 4.5–5.0), whereupon cleavage a ketone adduct with the cytotoxic payload attached and a hydrazide is formed.35 An FDA approved ADC by Pfizer, gemtuzumb ozogamicin (Mylotarg) successfully used this linker technology.35 The stability of the hydrazone at various pHs (4.5 to 7.4) both in vitro and in vivo in mice was found to be good. However, despite the successful usage of this linker technology, the field has moved away from acid cleavable linkers and there is increasing interest towards enzyme cleavable linkers.35
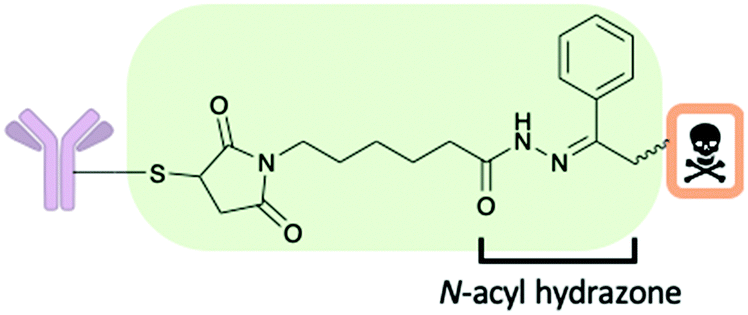 | ||
| Fig. 7 An antibody–drug conjugate construct with a cleavable pH sensitive N-acyl hydrazine linker. | ||
Enzyme cleavable linkers are popular choices for both ADCs and PDCs, with selective release of the drug at the target site and limited, if any, pre-release within the circulation.38 A widely used group of enzyme cleavable linkers are the dipeptides: Val-Ala or Val-Cit (Fig. 8). In which both dipeptides show good stability within the human circulation. Cleavage of the dipeptides occurs only in the presence of cathepsin or carboxylesterase 1 (CES1c) in mouse plasma.35 The cleavage subsequently triggers a release mechanism of the drug through a para-aminobenzyl carbamate (PABC) (Fig. 8).38 However, the instability of such linkers within mouse plasma causes problems in pre-clinical in vivo studies due to prelease as a result of hydrolysis. Therefore, the efficiency of in vivo studies are compromised.38
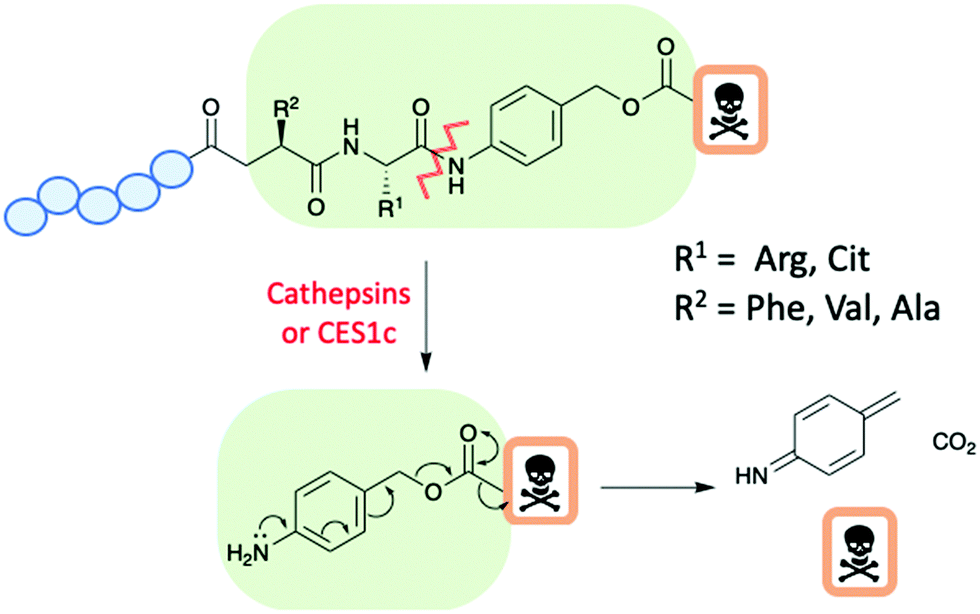 | ||
| Fig. 8 A schematic of a peptide–drug conjugate with an enzyme cleavable dipeptide linker and para-aminobenzyl carbamate spacer. | ||
A recent advancement within the cleavable linker field is the development of an arylsulfate linker cleaved by lysosomal sulfatases (Fig. 9).38 The arylsulfate linker would perform a 1,6-elimination upon cleavage to release the unmodified drug. Benefits of this linker include stability in human serum and mouse plasma, hydrophilic and selective payload release at target. In this study, the synthesised ADC with the arylsulfate linker and monomethyl auristatin E (MMAE) payload was potent and selective for human epidermal growth factor receptor 2 (HER2) positive cell lines.38
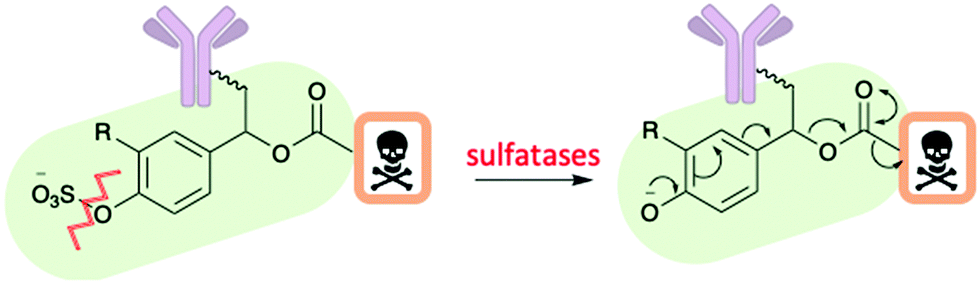 | ||
| Fig. 9 The cleavage mechanism for an arylsulfate linker by Bargh et al.38 | ||
Enhertu is an ADC approved by the FDA in December 2019, for the treatment of HER2-positive breast cancer. It is composed of a humanized anti-HER2 IgG1 monoclonal antibody (mAb), a maleimide tetrapeptide linker which conjugates the mAb to the cytotoxic topoisomerase inhibitor Dxd (exatecan derivative).39 The tetrapeptide linker is cleaved by enzymes within the lysosome to release the cytotoxic payload.
Non-cleavable linkers
Alternative to cleavable linkers are non-cleavable linkers. These linkers are not activated through external stimuli such as a chemically induced one.35 A non-cleavable linkers mechanism of action starts with the peptide/mAb being metabolised to leave the payload-linker construct, which can go on to escape out of the endosome/lysosome to kill the cell.35 Although cleavable linkers are preferred to non-cleavable for the development of targeted therapeutics, they present increased stability in circulation. The most recent FDA approved ADC, Blenrep, uses a maleimidocaproyl protease-resistant non-cleavable linker, to conjugate the anti-B cell maturation antigen (BCMA) to the microtubule inhibitor monomethyl auristatin F (MMAF).26 The choice of cleavable or non-cleavable linkers depends on the overall needs for the design and mode of action of the targeted therapeutic.Payloads
There are a range of cytotoxic drugs available for cancer treatment, but often with each drug comes a range of limitations including poor PK properties. However, the most limiting aspect is the unselective manner of the drug to target cancer cells, causing harm to healthy cells and resulting in severe side effects. Targeted therapeutics offer a way to repurpose drugs which have already been FDA approved at a significant cost. The use of a peptide allows for specific targeted therapy, consequently resulting in the enhancement of several properties for example the therapeutic window. As a result of the cytotoxin being attached to a peptide, the dose of the cytotoxin will be reduced as a greater proportion is reaching the target – typically an increased dose is needed to compensate for the off-site delivery. There are several criteria that determine which cytotoxic payload to use in PDCs including stability within circulation, the demonstration of high potency, its release through the cleavage of the linker, and there must be a viable attachment point for the connectivity to the linker.37 The cytotoxic payloads chosen usually have a low IC50, typically within the nanomolar range.19 Examples of payloads used within PDCs include Doxorubicin, Taxol, Daunorubicin, Gemzar and Mertansine (Fig. 10).19 However, the cytotoxic payloads can include radionucleotides in the form of the FDA approved 177Lu-dotatate (Fig. 5).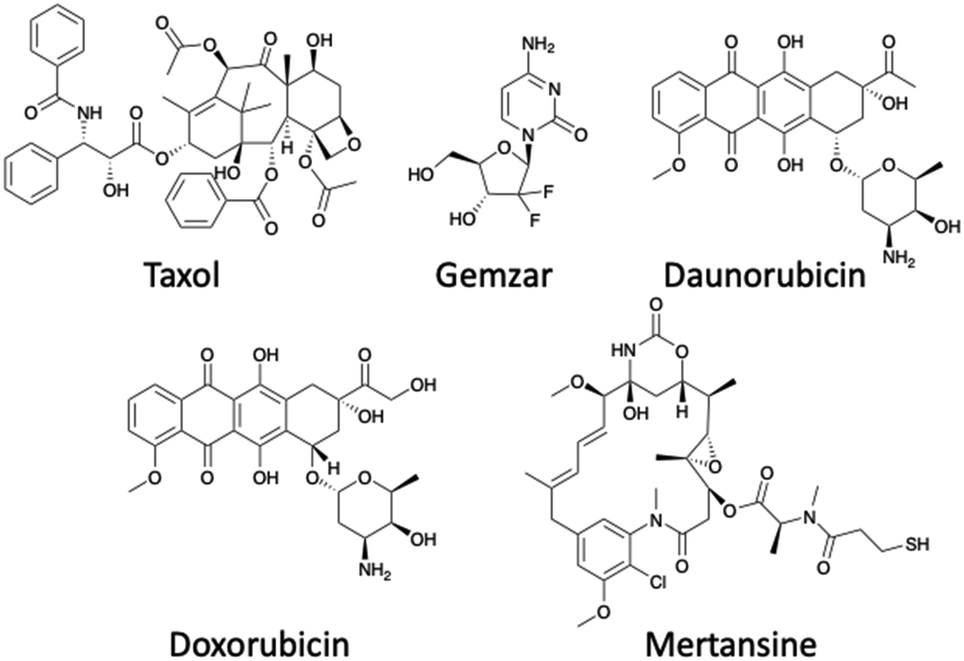 | ||
| Fig. 10 Structures of payloads used within the peptide–drug conjugate field. | ||
PDCs are not limited to only the use as therapeutics but are widely used as imaging agents exemplified by the first FDA approved radionucleotide-containing PDC, 111In-DTPA-Octreotide (Octreoscan) used for treatment against neuroendocrine tumours. However, studies have showed that Octreoscan has limited effect on tumour regression, and as a result, it is mainly used for diagnostic purposes. In contrast, the 177Lu-dotatate showed an increased period for progression-free survival and has greater potential as a therapeutic.40
Radionucleotides allow imaging of the tumour through various scanning techniques to determine the precise location of the tumour.40 The use of conjugates offers a significant advantage as the homing peptide binds selectively to the receptors on the target with very little off-site targeting. PET images are obtained when the conjugate is labelled with a positron-emitting radioisotope, for example, gallium-68 (68Ga), copper-64 (64Cu) and fluorine-18 (18F). Single-photon emission computed tomography (SPECT) imaging can also be employed for tumours when radioisotopes that are gamma-emitting are used, for example, iodine-123 (123I) and technetium-99m (99mTc).40 In order to incorporate the imaging agent into the peptide, bi-functional chelating agents are often used: 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and diethylenetriaminepentaacetic acid (DTPA) are examples of the most frequently used within this context.40
A PDC is a versatile approach that offers a great opportunity for the delivery of payloads and imaging agents to identify the tumour location or to determine tumour progression.
How a PDC targets the desired tissue
The mechanism of action for a PDC varies depending on several factors including the linker and homing peptide. As detailed earlier, a cleavable linker can be cleaved in the presence of stimuli at a specific pH or enzymes. The location of the stimuli will determine the mechanism of action. One scenario is where a PDC will follow a similar process to that of an ADC, first internalisation and then intracellular cleavage to release the cytotoxin. An alternative scenario occurs when cleavage occurs outside the cell followed by internalisation of the cytotoxin. Homing peptides also play a vital role in the mechanism of action as they can be cell penetrating peptides or non-cell penetrating peptides. Traditionally, a non-cell penetrating homing peptide binds with the target receptor overexpressed on tumour cells triggering receptor-mediated endocytosis and internalisation. Within the early sorting endosome, the PDC becomes unbound from the receptor moving into the late endosome, and the receptor is recycled back to the surface. The PDC finally enters the lysosome, where the reduced pH or the presence of specific enzymes cleave the PDC and releases the cytotoxin (Fig. 11A).41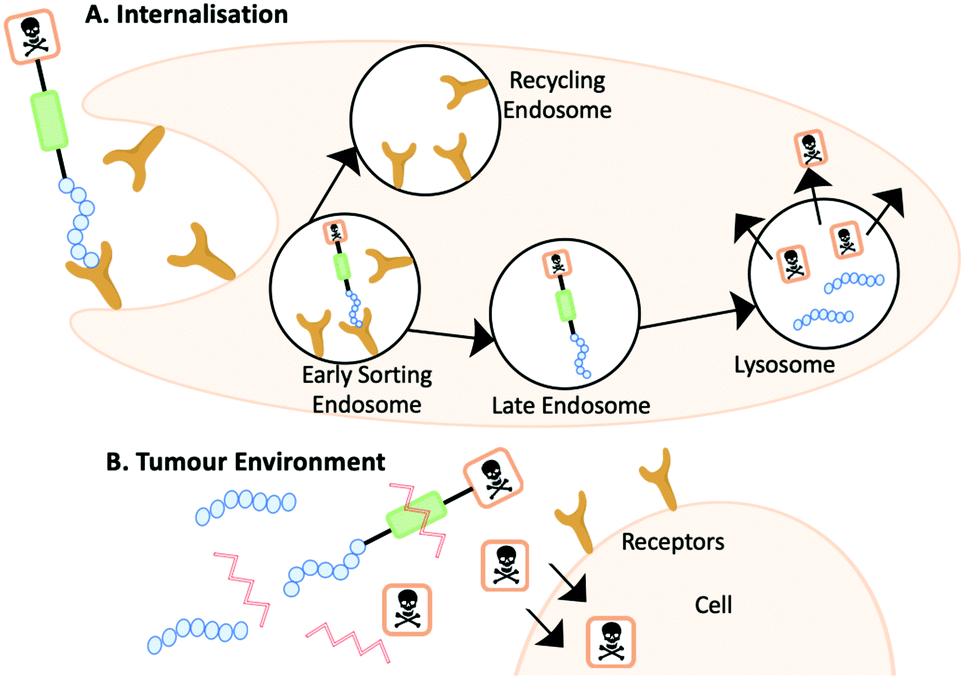 | ||
| Fig. 11 (A) The internalisation mechanism of action where the PDC is cleaved within the lysosome. (B) The PDC is cleaved outside of the cell and the cytotoxin diffuses into the cell. | ||
Alternative mechanism of actions have been reported including a PDC to target metastatic breast cancer with doxorubicin by You et al. (Fig. 11B).42 The PDC undergoes a different pathway upon entry into the tumour environment as the PDC is cleaved by MMP-2 prior to internalising. Doxorubicin diffuses the membrane of the tumour cells and can exert its bioactivity.42 On designing a PDC it is important to consider the target and the stimuli present to ensure that the proposed mechanism of action is fulfilled.
Stability of PDCs
One of the main drawbacks of PDCs is, similar to peptides, their poor circulation stability and fast renal clearance. A PDC must be stable within circulation to prevent the pre-release of the cytotoxic payload and lead to systemic exposure. The use of a range of nanoparticles has been studied to enhance the inherently poor stability of PDCs, examples of which will be discussed herein.A way to overcome the poor stability in circulation which bridges multiple disciplines is the conjugation of the PDC to gold nanoparticles (AuNPs). Due to their desirable physio-chemical, safety properties, relative ease of synthesis and longer circulation half-life, AuNPs are an excellent addition to a PDC to increase the overall stability.43 Kalimuthu et al. reported that PDCs developed to treat A20 murine lymphoma cells displayed a >90 times increase in the PDC half-life when conjugated to PEG-coated AuNPs to produce a selective PEG-AuNP-PDC.43
A recent dual-functional approach by Zhang and co-workers demonstrates the use of nanoparticles to increase the stability of a prodrug PDC.44 The rationale behind the design was based around photothermal therapy (PTT), a non-invasive form of antitumour therapy that involves the use of near-infrared (NIR) light (Fig. 12). The design used a hollow Cu sulphide nanoparticle (HCuS) to encapsulate the PDC (cRGD-SMCC-DM1). The PDC comprises of three components: the integrin RGD homing peptide, the SMCC non-cleavable thioether linker and the DM1 cytotoxic payload. HCuS nanoparticles present a fluorescently labelled amphiphilic copolymer fPEDC on the surface, termed P@HCuS. fPEDC copolymers promote several properties: they act as a chromophore for detection, aid stabilisation and are pH/redox-sensitive.44 Studies on a triple-negative breast cancer cell line, MDA-MB-231, reported that upon laser treatment, the PDC/P@HCuS nanoparticles resulted in ∼12.1% early apoptosis and >70% late apoptosis, higher than P@HCuS alone.44 Maximum distribution was observed after 24 hours from the injection of the PDC/P@HCuS complex in MDA-MB-231 tumour bearing mice. Laser irradiation localised the complex both intra- and extracellularly. The fPEDC is redox/pH-sensitive and hence the linker was cleaved inside the tumour cells. The extracellular PDCs, through a process of receptor-mediated endocytosis, entered the cells and enzymatically degraded in the lysosome where DM1 was released. The nanosystem construct PDC/P@HCuS had excellent circulation times with up to 3 weeks of anti-tumour efficacy from initial NIR laser irradiation.44
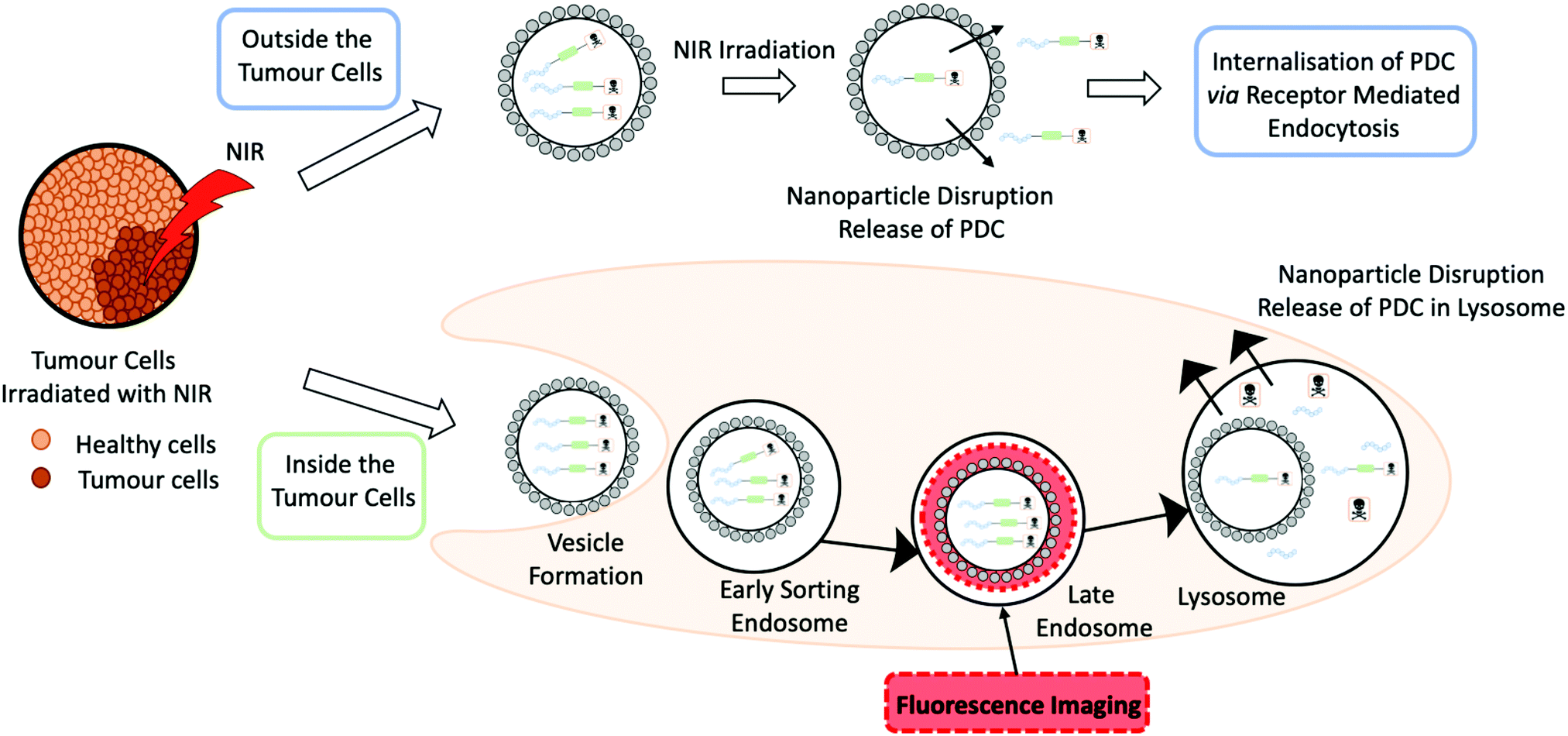 | ||
| Fig. 12 A schematic to represent a photothermal therapy (PTT) both inside and outside the tumour cells by Zhang and co-workers. In the schematic the grey circle represents a Cu sulphide nanoparticles, loaded with a peptide–drug conjugate that is irradiated with near infrared (NIR) light.44 | ||
Enhancing the PK properties of the PDC through nanomaterials is an advancing area of research and offers excellent potential to get PDCs to clinical trials.
Administration routes for PDCs
Like ADCs, PDCs are not administrated orally and need to be given through IV injection to the patient. Further development is needed within this area, as oral administration is extremely advantageous for the prospects of therapies and the most accessible way to receive treatment. The limitations of orally administrating peptides are described earlier in the review, along with methods which enhance their chemical and enzymatic stability. The following section will focus on new nanomaterial methods to deliver peptides and proteins in a controlled manner.Whitehead and co-workers have recently released a novel way to deliver a peptide and proteins through oral administration.45 They take advantage of small, negatively charged particles which can enhance the permeability of the protein. It was determined that silica particles of 50 nm were the most effective in the delivery of insulin orally. The carrying system was gel capsules loaded with insulin that were mouse-specific and coated with a pH-responsive polymer, named Eudragit L100-55. The polymer was specifically designed to only allow drug release in the range of pH 5.0–5.5 experienced in the small intestine of the mouse. Both the silica nanoparticles and the modified insulin were orally administrated to the mouse, and blood glucose levels were tested every hour to determine the glucose-lowering effect. The mice treated with 200 mg kg−1 of silica nanoparticles displayed hypoglycaemia for 10 hours from administration.45 The proposed hypothesis for these observations states that the silica nanoparticles bind with endothelial cells and open the tight junctions to allow the peptide through. This process is reversible and further investigation into inflammation has confirmed there is no damage to the endothelial cells. Although this is only one example of this kind, the use of silica nanoparticles appears to be a promising approach to increase oral bioavailability safely. Further investigation is needed to closely investigate effects in non-human primates to test the feasibility for clinical trials.45 Approaches to orally administered peptides could, in the future, be an exciting new strategy to bring PDC candidates to clinical trials.
Latest cancer targeting PDCs in development and clinical trials
PDCs is an umbrella term for many different conjugates using different types of peptides. For example, bicycle-toxin conjugates and peptide-dendrimer conjugates both have shown promise as drug delivery systems.A bicycle peptide is typically between 9–20 amino acids long and has 3 cysteine residues within the sequence.10 These cysteine residues react with a small molecule linker to constrain the peptide in a rigid conformation (Fig. 13).46 The bicycles can be used as transporters for drug molecules through bicycle-toxin conjugates (BTCs). These conjugates offer several advantages over ADCs which include deeper tumour penetration, rapid extravasation and slower renal clearance.46 The drug is attached to the bicycle peptide ensuring the conformation is not hampered.
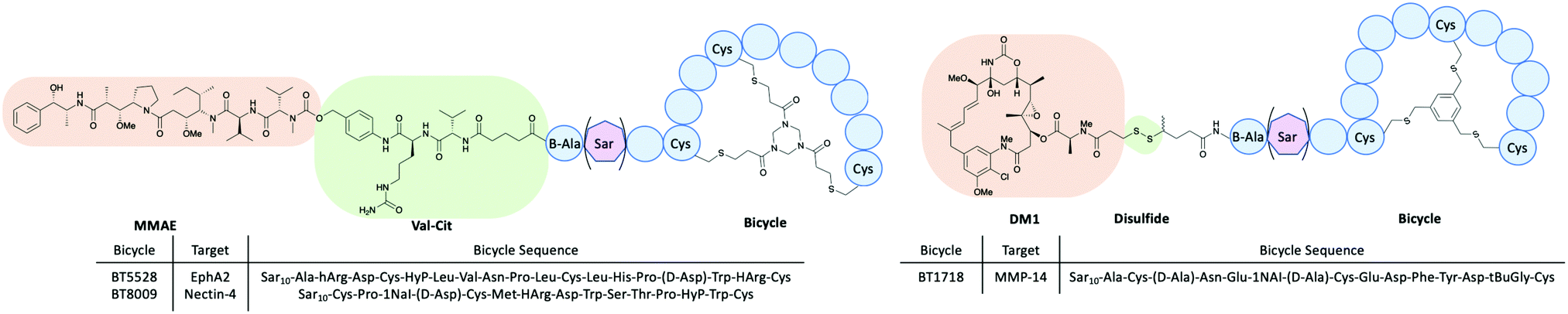 | ||
| Fig. 13 The structures of bicycle-toxin conjugates BT5528, BT8009 and BT1718 developed by Bicycle Therapeutics.46,47 | ||
There are currently several BTCs in clinical trials from Bicycle Therapeutics including BT1718, BT5528 and BT8009 all targeting specific tumours (Fig. 13).47 BT1718 is a BTC from Bicycle Therapeutics currently in Phase I/IIa sponsored by Cancer Research UK. The target for this BTC is membrane type 1 matrix metalloproteinase (MMP-14), which is overexpressed in several tumours for example ovarian, lung, breast, bladder and endometrial. BT1718 is composed of the specific targeting bicycle conjugated through a cleavable disulfide bond to the MD1 cytotoxin. The Phase I dosage helped stabilise the tumour in 54% of the candidates and seemed well tolerated, allowing the progression to Phase II.47
BT5528 is a BTC in Phase I/II trial targeting Ephrin type-A receptor 2 (EphA2) which is overexpressed in a number of tumours and a member of the tyrosine kinases family. This family of receptors is responsible for many important roles including cell migration, adhesion, proliferation and differentiation. BT5528 is composed of the targeting bicycle conjugated to an MMAE payload through an enzyme cleavable Val-Cit linker. The dose administrated in Phase I/II clinical trials seems to be well-tolerated.46,47
BT8009 is another BTC in clinical trials targeting Nectin-4 which is overexpressed in a number of cancers including lung, breast, bladder and pancreatic. This BTC has the targeting bicycle and similar architecture to BT5528 with an MMAE payload conjugated together through a Val-Cit cleavable linker. BT8009 has displayed promising results in clinical trials and demonstrated to be selective in pre-clinical models. Toxicity studies highlighted levels of toxicity which are correlated to the MMAE payload and limited off-target toxicity.47
Dendrimers are characterised by their highly branched architecture which is composed of several layers including the core, generations and outer shell where functional groups are exposed (Fig. 14). Dendrimers have many applications in a host of scientific fields: pharmaceutical, nanotechnology and biomedical to name a few.48 Specifically, peptide dendrimers will be the focus of this section. This class of dendrimer differs from linear peptides and polymers. Several key properties are unique to dendrimers including thermal performance, viscosity and use for encapsulation. Peptide dendrimers can be subcategorised into two groups: covalent (Type I and II) and noncovalent (Type III). Covalent peptide dendrimers are built on natural or unnatural amino acids; in contrast, noncovalent peptide dendrimers are not attached to multiple frameworks through the periphery.48
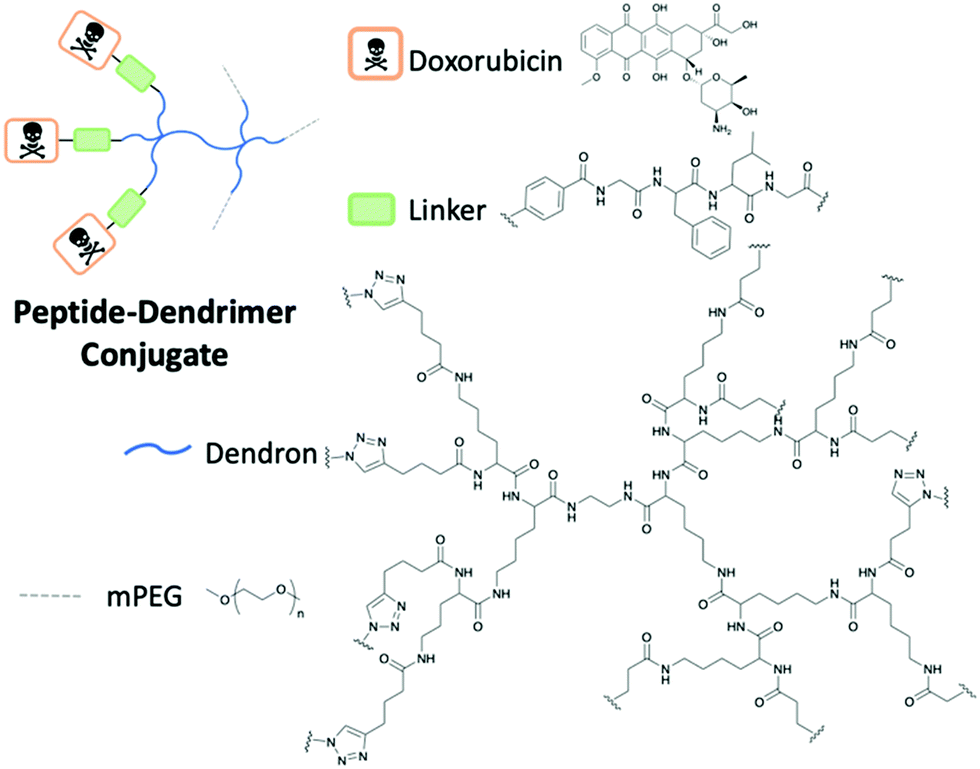 | ||
| Fig. 14 The structure of the peptide-dendrimer conjugate by Gu and co-workers to deliver Doxorubicin.49 | ||
Peptide dendrimers have been studied as useful drug delivery mechanisms; the attachment of the drug can occur either through a covalent bond or through noncovalent encapsulation of the drug. The encapsulation of the drug occurs in dendrimer voids through hydrogen bonds, electrostatic or hydrophobic. The use of peptide dendrimers is seen as beneficial due to their tuneable amino acid features as well as good biocompatibility.48
A study by Gu and co-workers highlighted the success of peptide dendrimer drug conjugates to selectively deliver Doxorubicin (DOX) to the target cancer site.49 Polyethylene glycol (PEG) has been shown to be biocompatible in the body as it exhibits good solubility, limited toxicity and immunogenicity, flexible and low protein absorption. The incorporation of PEG in the dendrimer enhanced the PK properties of the dendrimer conjugate through the extension of the half-life. The designed peptide PEG dendrimer DOX conjugate uses the tetrapeptide GFLG cleavable linker to conjugate peptide dendrimer to the DOX through click chemistry (Fig. 14). The overexpression of cathepsin B cleaved the GFLG linker to release the drug after endocytosis.49 The results of the study determined that in mice with 4T1 murine breast cancer, the use of the dendrimer nanoparticle with DOX was significantly more efficient in reducing the tumour size, in comparison to free DOX. The weight of the mice was stable throughout the study, concluding that the nanoparticle was well tolerated in vivo.49
A recent example (September 2019) from Oliveria and co-workers demonstrated how peptide dendrimers have been effectively used to treat colorectal cancer with controlled gemcitabine (GEM) drug delivery.50 Carboxymethylchitosan/poly(amidoamine) (CMCht/PAMAM) dendrimer nanoparticles were developed into target-specific drug delivery vehicles through the conjugation of the YIGSR peptide to gemcitabine. The 5-mer peptide has shown to bind selectivity to the laminin receptor (LR) found in many carcinomas. The YIGSR conjugated peptide dendrimer showed selectivity with internalisation for HCT-116 cancer cells that have a high expression of the receptor LR. The GEM release occurred intracellularly and after 72 hours there was 31.15% cell death.50 Future work for this dendrimer conjugate includes testing on other colorectal cancer cell lines.50
Conclusions
Despite peptides being currently under-represented in clinical trials in comparison to small molecules and biologics, they provide exquisite versatility that can aid the design of targeted therapeutics. A peptide can offer many properties a biologic is unable to, including enhanced tumour penetration, decreased immunogenicity and cheaper synthesis. A drawback of peptides is their fast-renal clearance, as a consequence of their relatively small size (<5000 Da). This issue has been addressed through various methods such as chemical modifications and physical techniques (cyclisation, peptide stapling, formulations). These established methods have proven to slow down the renal clearance, as a result, they are highly beneficial for clinical studies. Peptide–drug conjugates (PDCs) are an advancing area of research with great promise for the future, as witnessed by the two PDCs on the market. Furthermore, PDCs with the correct design and target could make an impact on the targeted therapeutic market by delivering safer medicines. There are already PDCs in the form of BTCs in clinical trials to treat a range of cancers. The combination of material science through the use of nanoparticles and PDCs is an attractive strategy that could solve the stability problems experienced with peptides. This review highlights the potential of peptides as a diverse tool for targeted drug delivery. Despite recent advances, there remains a number of critical challenges that hamper the potential of peptides as drugs. In particular, there is a need for greener peptide synthesis and technologies to overcome the limited shelf life of a peptide.In conclusion, recent advances in both chemistry and biology have expanded our understanding of the benefits and limitations of peptides setting up the foundations for significant advancements in the PDC as targeted therapy.
Conflicts of interest
There are no conflicts to declare.References
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136 CrossRef CAS PubMed.
- Impact Story: Developing the Tools to Evaluate Complex Drug Products: Peptides, https://www.fda.gov/drugs/regulatory-science-action/impact-story-developing-tools-evaluate-complex-drug-products-peptides, accessed 28 August 2020.
- L. Di, AAPS J., 2014, 17, 134–143 CrossRef PubMed.
- G. B. Santos, A. Ganesan and F. S. Emery, ChemMedChem, 2016, 11, 2245–2251 CrossRef CAS PubMed.
- D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS PubMed.
- M. Werle and A. Bernkop-Schnürch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- J. D. Bucheit, L. G. Pamulapati, N. Carter, K. Malloy, D. L. Dixon and E. M. Sisson, Diabetes Technol. Ther., 2020, 22, 10–18 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS PubMed.
- ClinicalTrials.gov, Phase 1 Safety Study of ALRN-5281 in Healthy Subjects, https://clinicaltrials.gov/ct2/show/NCT01775358, accessed 5 January 2020.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502 CrossRef CAS PubMed.
- A. G. Harris, Gut, 1994, 35, S1–S4 CrossRef CAS PubMed.
- Y. Nishimura, J. Nicholas Francis, O. K. Donau, E. Jesteadt, R. Sadjadpour, A. R. Smith, M. S. Seaman, B. D. Welch, M. A. Martin and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22436–22442 CrossRef CAS PubMed.
- H. Wu and J. Huang, Protein Pept. Lett., 2018, 25, 514–521 CrossRef CAS PubMed.
- A. F. B. Räder, F. Reichart, M. Weinmüller and H. Kessler, Bioorg. Med. Chem., 2018, 26, 2766–2773 CrossRef PubMed.
- K. Zhou, X. Zheng, H. M. Xu, J. Zhang, Y. Chen, T. Xi and T. Feng, Bioconjugate Chem., 2009, 20, 932–936 CrossRef CAS PubMed.
- ClinicalTrials.gov, This Study Collects Information on the Safety of Inhaled Pegylated Adrenomedullin (PEG-ADM), How the Drug is Tolerated and How it Affects Patients Suffering From a Type of Lung Failure That Cause Fluid to Build up in the Lungs Making Breathing Difficult, https://clinicaltrials.gov/ct2/show/NCT04417036, accessed 20 July 2020.
- J. Lau, P. Bloch, L. Schäffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- J. Hadar, S. Skidmore, J. Garner, H. Park, K. Park, Y. Wang, B. Qin and X. Jiang, J. Controlled Release, 2019, 304, 75–89 CrossRef CAS PubMed.
- E. I. Vrettos, G. Mező and A. G. Tzakos, Beilstein J. Org. Chem., 2018, 14, 930–954 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- Q. He, J. Chen, J. Yan, S. Cai, H. Xiong, Y. Liu, D. Peng, Z. Liu and M. Mo, Asian J. Pharm. Sci., 2019, 15, 19–44 Search PubMed.
- C. Gambacorti-Passerini, L. Antolini, F. X. Mahon, F. Guilhot, M. Deininger, C. Fava, A. Nagler, C. M. Della Casa, E. Morra, E. Abruzzese, A. D’Emilio, F. Stagno, P. Le Coutre, R. Hurtado-Monroy, V. Santini, B. Martino, F. Pane, A. Piccin, P. Giraldo, S. Assouline, M. A. Durosinmi, O. Leeksma, E. M. Pogliani, M. Puttini, E. Jang, J. Reiffers, M. G. Valsecchi and D. W. Kim, J. Natl. Cancer Inst., 2011, 103, 553–561 CrossRef CAS PubMed.
- R. A. Ward, S. Fawell, N. Floc’h, V. Flemington, D. McKerrecher and P. D. Smith, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00383.
- A. Keown, With 3 Approvals in 2019, Companies See Great Future for ADC Therapies, https://www.biospace.com/article/with-3-approvals-in-2019-companies-see-great-future-for-adc-therapies/, accessed 12 August 2020.
- P. Hofland, GSK's Belantamab Mafodotin Approved as Monotherapy for Adult Patients with R/R Multiple Myeloma, https://www.oncozine.com/gsks-belantamab-mafodotin-approved-as-monotherapy-for-adult-patients-with-r-r-multiple-myeloma/, accessed 25 August 2020.
- F. J. Giles, H. M. Kantarjian, S. M. Kornblau, D. A. Thomas, G. Garcia-Manero, T. A. Waddelow, C. L. David, A. T. Phan, D. E. Colburn, A. Rashid and E. H. Estey, Cancer, 2001, 92, 406–413 CrossRef CAS PubMed.
- M. B. Hock, K. E. Thudium, M. Carrasco-Triguero and N. F. Schwabe, AAPS J., 2015, 17, 35–43 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, FDA approves lutetium Lu 177 dotatate for treatment of GEP-NETS, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lutetium-lu-177-dotatate-treatment-gep-nets, accessed 12 January 2020.
- Theratechnologies Inc., New Data Show Theratechnologies’ SORT1+ Technology Is Effective in Many Treatment-Resistant Cancers, https://www.theratech.com/wp-content/uploads/2020/06/Press-release-AACR-June-22-EN-FINAL.pdf, accessed 11 September 2020.
- H. Ruan, X. Chen, C. Xie, B. Li, M. Ying, Y. Liu, M. Zhang, X. Zhang, C. Zhan, W. Lu and W. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 17745–17756 CrossRef CAS PubMed.
- A. A. P. Tripodi, I. Ranđelović, B. Biri-Kovács, B. Szeder, G. Mező and J. Tóvári, Pathol. Oncol. Res., 2019, 26, 1879–1892 CrossRef PubMed.
- I. Vhora, S. Patil, P. Bhatt and A. Misra, Advances in Protein Chemistry and Structural Biology, Academic Press Inc., 2015, vol. 98, pp. 1–55 Search PubMed.
- N. Q. Shi, W. Gao, B. Xiang and X. R. Qi, Int. J. Nanomed., 2012, 7, 1613–1621 CAS.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- D. Rotin, B. Robinson and I. F. Tannock, Cancer Res., 1986, 46, 2821–2826 CAS.
- M. Poreba, FEBS J., 2020, 287(10), 1936–1969 CrossRef CAS PubMed.
- J. D. Bargh, S. J. Walsh, A. Isidro-Llobet, S. Omarjee, J. S. Carroll and D. R. Spring, Chem. Sci., 2020, 11, 2375–2380 RSC.
- U.S. Food and Drug Administration, Drug Trials Snapshot: ENHERTU, https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshot-enhertu, accessed 28 August 2020.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Front. Chem., 2020, 8, 571 CrossRef CAS PubMed.
- S. Majumdar and T. J. Siahaan, Med. Res. Rev., 2012, 32, 637–658 CrossRef CAS PubMed.
- Y. You, Z. Xu and Y. Chen, Drug Delivery, 2018, 25, 448–460 CrossRef CAS PubMed.
- K. Kalimuthu, B. C. Lubin, A. Bazylevich, G. Gellerman, O. Shpilberg, G. Luboshits and M. A. Firer, J. Nanobiotechnol., 2018, 16, 34 CrossRef PubMed.
- Y. Sun, Y. Liang, W. Dai, B. He, H. Zhang, X. Wang, J. Wang, S. Huang and Q. Zhang, Nano Lett., 2019, 19, 3229–3237 CrossRef CAS PubMed.
- N. G. Lamson, A. Berger, K. C. Fein and K. A. Whitehead, Nat. Biomed. Eng., 2020, 4, 84–96 CrossRef CAS PubMed.
- G. E. Mudd, A. Brown, L. Chen, K. van Rietschoten, S. Watcham, D. P. Teufel, S. Pavan, R. Lani, P. Huxley and G. S. Bennett, J. Med. Chem., 2020, 63, 4107–4116 CrossRef CAS PubMed.
- B. Therapeutics, Programs, https://www.bicycletherapeutics.com/programs/#bicycle-conjugates, accessed 14 May 2020.
- R. Sapra, R. P. Verma, G. P. Maurya, S. Dhawan, J. Babu and V. Haridas, Chem. Rev., 2019, 119, 11391–11441 CrossRef PubMed.
- N. Li, N. Li, Q. Yi, K. Luo, C. Guo, D. Pan and Z. Gu, Biomaterials, 2014, 35, 9529–9545 CrossRef CAS PubMed.
- M. R. Carvalho, C. R. Carvalho, F. R. Maia, D. Caballero, S. C. Kundu, R. L. Reis and J. M. Oliveira, Adv. Ther., 2019, 2, 1900132 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |