
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Racemases and epimerases operating through a 1,1-proton transfer mechanism: reactivity, mechanism and inhibition
Matthew D.
Lloyd
 *a,
Maksims
Yevglevskis
ab,
Amit
Nathubhai
ac,
Tony D.
James
de,
Michael D.
Threadgill
af and
Timothy J.
Woodman
a
*a,
Maksims
Yevglevskis
ab,
Amit
Nathubhai
ac,
Tony D.
James
de,
Michael D.
Threadgill
af and
Timothy J.
Woodman
a
aDrug & Target Discovery, Department of Pharmacy & Pharmacology, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: M.D.Lloyd@bath.ac.uk; Fax: +44-(0)1225-386786
bCatSci Ltd., CBTC2, Capital Business Park, Wentloog, Cardiff CF3 2PX, UK
cUniversity of Sunderland, School of Pharmacy & Pharmaceutical Sciences, Sciences Complex, Sunderland SR1 3SD, UK
dDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, People's Republic of China
fInstitute of Biological, Environmental & Rural Sciences, Aberystwyth University, Aberystwyth SY23 3BY, UK
First published on 12th April 2021
Abstract
Racemases and epimerases catalyse changes in the stereochemical configurations of chiral centres and are of interest as model enzymes and as biotechnological tools. They also occupy pivotal positions within metabolic pathways and, hence, many of them are important drug targets. This review summarises the catalytic mechanisms of PLP-dependent, enolase family and cofactor-independent racemases and epimerases operating by a deprotonation/reprotonation (1,1-proton transfer) mechanism and methods for measuring their catalytic activity. Strategies for inhibiting these enzymes are reviewed, as are specific examples of inhibitors. Rational design of inhibitors based on substrates has been extensively explored but there is considerable scope for development of transition-state mimics and covalent inhibitors and for the identification of inhibitors by high-throughput, fragment and virtual screening approaches. The increasing availability of enzyme structures obtained using X-ray crystallography will facilitate development of inhibitors by rational design and fragment screening, whilst protein models will facilitate development of transition-state mimics.
 Matthew D. Lloyd | Matthew D. Lloyd obtained his BSc (Hons) in Biological Chemistry at the University of Leicester, and a DPhil at the University of Oxford with the late Professor Sir Jack E. Baldwin FRS and Professor Chris Schofield FRS. Following postdoctoral research at Brown University and Oxford, he joined the Department of Pharmacy & Pharmacology at the University of Bath in 2002 and is a Senior Lecturer and Fellow of the Royal Society of Biology, undertaking research in chemical biology, lipid metabolism, enzymes and inhibitors as new treatments for cancer and other diseases. He is 4th Degree Black Belt in Ch’ang-Hon Taekwon-Do. |
 Maksims Yevglevskis | Maksims Yevglevskis obtained his BSc in Chemical Engineering from Riga Technical University (Latvia) in 2009 and MRes in Chemical Research from University of Bath in 2011. He then joined Matthew Lloyd's group at the University of Bath, where he obtained a PhD in 2015 working on developing assays and inhibitors of α-methylacyl-CoA racemase (AMACR). This was followed by a three-year PDRA position on the same project. He is currently working at CatSci Ltd in Cardiff and has an interest in enzymes as drug targets and biomarkers. |
 Amit Nathubhai | Amit Nathubhai obtained his BSc (hons) and MSc in Biological Chemistry from the University of Leicester and a PhD from the University of Bath with Dr Ian Eggleston. Amit performed post-doctoral studies in Medicinal Chemistry, Biochemistry and in vitro Cell Biology at the University of Bath. He is currently a Senior Lecturer at the University of Sunderland. His research interests lie at the chemistry and biology interface and emphasises development of chemical tools and drug-like molecules to dissect biological mechanisms involved in cancer, fibrosis, diabetes and obesity. He has a keen interest in fresh-water aquatics, and writing and playing music. |
 Tony D. James | Tony D. James obtained his BSc from the University of East Anglia (1986), PhD from the University of Victoria, Canada (1991), and undertook Postdoctoral Research with Seiji Shinkai (1991-95). He was a Royal Society University Research Fellow at the University of Birmingham (1995-2000) before moving to the University of Bath (2001) where he is a Professor and holds a prestigious Royal Society Wolfson Research Merit Award (2017-2022). He was awarded the Daiwa-Adrian Prize (2013), the inaugural CASE Prize (2015) and the MSMLG Czarnik Award (2018). He has >363 papers on molecular recognition and sensor design and a h-index of 72. |
 Michael D. Threadgill | Michael D. Threadgill is Emeritus Professor of Medicinal Chemistry at the University of Bath and Visiting Professor at Aberystwyth University. He obtained MA and PhD from the University of Cambridge (PhD under the supervision of Prof. Sir Alan R. Battersby FRS), PGCE from the University of Durham and DSc from the University of Bath and is a Fellow of the Royal Society of Chemistry. His research interests include developing inhibitors of the PARP superfamily of enzymes, tumour-selective delivery of drugs, identification of natural products and BNCT. He is also a keen cricketer and umpire. |
 Timothy J. Woodman | Timothy J. Woodman obtained a BSc (hons) in Chemistry from the University of Warwick (1994) and a PhD in organometallic chemistry from the same institution in 1999, studying under Dr Gerald Willey. Following a year in New Zealand as a Royal Society Fellow, he joined the research group of Professor Manfred Bochmann, initially at the University of Leeds and subsequently at the University of East Anglia. He joined the University of Bath in 2005 and is currently a Senior NMR Spectroscopist, with interests in prostate cancer and the science of chillies. Outside the University, his main interest is cricket. |
Introduction
Chirality is at the very heart of Chemical Biology. Proteins, nucleic acids, carbohydrates and many lipids are all chiral molecules, as are the overwhelming majority of their monomer precursors. In addition, many cellular metabolites also contain chiral centres. It is well-known that, for most chiral biomolecules, one particular configuration is preferred; thus proteins contain predominantly chiral amino-acids with L-configuration1,2 (S-configuration in the Cahn-Ingold-Prelog system3 except for R-cysteine and achiral glycine). Similarly, carbohydrates are or contain predominantly D-sugars, with L-ascorbic acid (vitamin C) being a well-known exception. An important consequence of the chiral nature of proteins is that, when they interact with other chiral molecules, a diastereomeric situation arises; thus, most proteins will be highly selective for a particular configuration of their interacting partners (substrate, inhibitor, allosteric effector). An important consequence of this is that different stereoisomers of chiral drugs are effectively different drugs, which will generally have different protein targets (enzyme, receptors etc.) and different pharmacokinetics.4,5 Finally, many drugs are known to undergo metabolic changes of chiral configuration in vivo,4,5e.g. ibuprofen and related ‘profens’ (reviewed in ref. 6–8) and mandelic acid.9,10 In addition the 2-(aryloxy)propanoic acid herbicides undergo changes in chiral configuration which are mediated by soil bacteria.11–13Notwithstanding the fact that most biological molecules exist overwhelmingly in one stereochemical configuration, there are many examples where minor stereoisomers play an essential role. The most well-known example of this is proteinogenic amino-acids such as alanine and glutamate, which are found in their D-configuration (R-configuration) within bacterial peptidoglycan.14–17 In most cases, these minor stereoisomers are not biosynthesised de novo but are obtained by changing the stereochemical configuration of the most abundant isomer into that of the less abundant isomer.
The enzymes which perform these changes in stereochemical configuration are known as racemases and epimerases, which have been shown to have a pivotal position in metabolism, and thus have gained significant interest as drug targets for diseases such as bacterial infections,14,18–25 Chagas disease,26–28 cancer,6,7,18,29 Alzheimer's disease and other dementias,1,2,30–32 formation of cataracts1,33 and diabetic retinopathy;34 racemase levels are also a marker of ischaemic stroke.35 Inhibition of diaminopimelate epimerase activity also potentiates cephem antibiotic activity by compromising the integrity of the bacterial cell wall.36
Low activity or concentrations of racemases/epimerases (AMACR,37 methylmalonyl-CoA epimerase38) are associated with inherited errors in metabolism and may also be associated with stroke and dementia39 and neurodegenerative diseases,40 such as Amyotrophic Lateral Sclerosis (ALS, a.k.a. motor neurone disease, Lou Gehrig's disease). Increased methylmalonic acid levels in the aging population (resulting from a decrease in methylmalonyl-CoA epimerase activity) is suggested to promote an aggressive cancer phenotype by upregulation of the SOX4 transcription factor.41 Increased levels of aspartate/glutamate racemases protect Salmonella enterica from aminoacrylate metabolic stress.42 Increased activity of the bifunctional enzyme UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in sialuria, an extremely rare genetic disorder, while knockout of the corresponding gene is lethal in mice.43,44 Mutations in this epimerase are linked to hereditary inclusion body myopathy (HIBM).44,45 In addition, O-ureidoserine racemase is involved in the biosynthesis of the antibiotic D-cycloserine,46 while a peptide epimerase is found in funnel web spider (Agelenopsis aperta) venom which interconverts two 48 amino-acid peptides differing only in the configuration at a single serine residue (Ser-46).47 Finally, racemases and epimerases are used in dynamic kinetic resolutions and other biotechnological applications.48–54
Racemases and epimerases use several different strategies to bring about changes in stereochemical configuration of their substrates, including the use of radical reactions,55–60 elimination and re-addition of nucleotides20,61 and the use of redox cofactors.18,19,62–64 An important example of a ‘epimerase’ utilising redox cofactors is decaprenylphosphoryl-β-D-ribose epimerase (DprE); however, this is not a true epimerase reaction as the oxidative and reductive reactions are catalysed by separate enzymes (DprE1 and 2, respectively) using different cofactors [flavin adenine dinucleotide (oxidised) and nicotinamide adenine dinucleotide (reduced)].65–68
By far the most common mechanism used by racemases and epimerases is the deprotonation/reprotonation14,18,20,63 (1,1-proton transfer) reaction. These enzymes fall into three classes: those which are pyridoxal 5′-phosphate (PLP)-dependent;5,50,69,70 those which use metal ions (enolase enzymes14,49,71,72); and those which are cofactor-independent (Scheme 1).6,7,18,20,63 The PLP-dependent enzymes (Scheme 1A) catalyse exchange between PLP in the internal aldimine 1 (catalytic Lys side-chain) and the external aldimine 2 (substrate α-amino group). Deprotonation69,70 of 2 results in the ylide intermediate 3 which is subsequently reprotonated from the other face to produce the external aldimine 4 with opposite configuration. In contrast, the metal-dependent enzymes, e.g. mandelate racemase, apparently perform a concerted reaction (Scheme 1B; 5–7). Solvent isotope experiments show that label is incorporated into product with little incorporation into recovered substrate,73,74 which is consistent with a concerted mechanism. However, kinetic isotope effect measurements on mandelate racemase are consistent with a stepwise reaction and a discrete deprotonated intermediate.75 Finally, most cofactor-independent racemases/epimerases utilise a concerted mechanism,5,76–78 as illustrated by glutamate racemase (Scheme 1C; 8–10). However, some cofactor-independent enzymes using substrates with acidic α-protons perform their reactions with a stepwise mechanism via a discrete enolate intermediate (e.g. α-methylacyl-CoA racemase79–81).
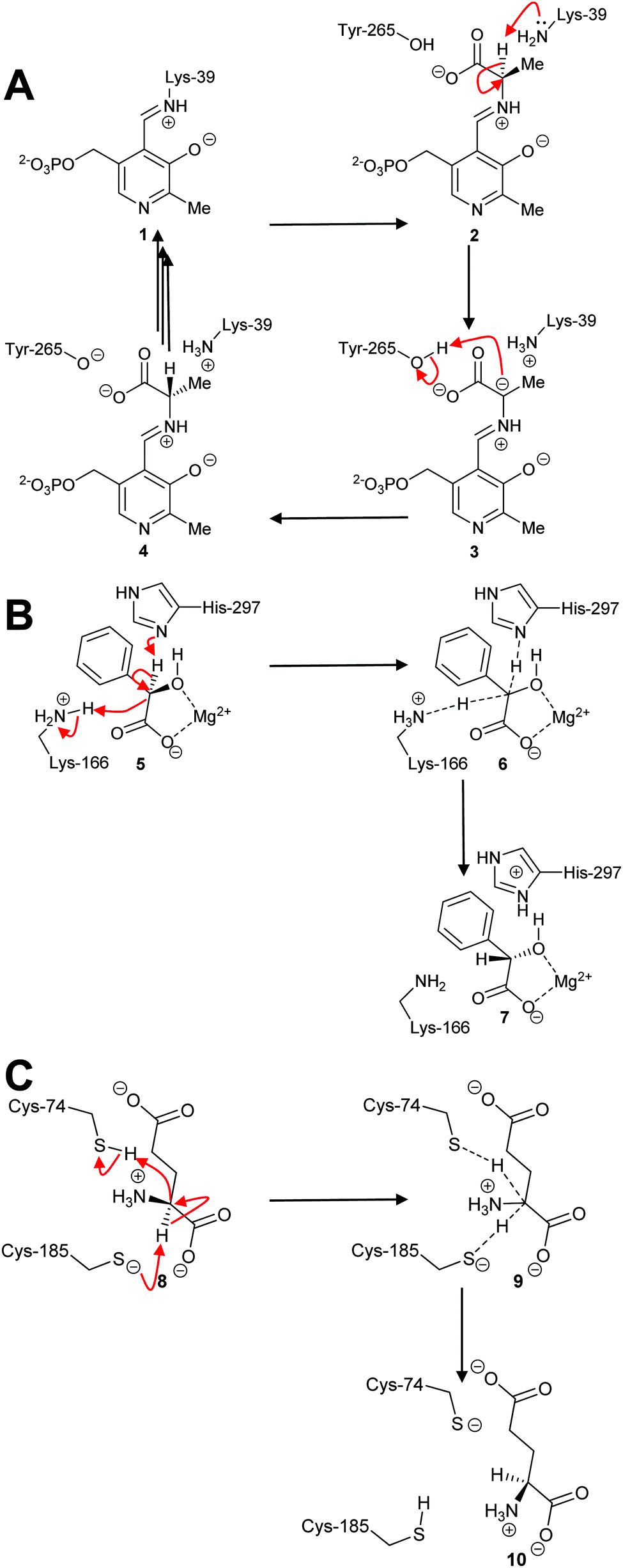 | ||
| Scheme 1 Example mechanisms of racemases and epimerases operating by a 1,1-proton transfer mechanism. (A) PLP-dependent amino acid racemases, as shown by alanine racemase;5,69 (B) Metal-dependent (enolase) enzymes, as shown by mandelate racemase;74,75 (C) Cofactor-independent racemases as shown by glutamate racemase.63,82,83 Dashed lines show bonds being broken or formed in the transition state. | ||
Enzymes which use metal ions as Lewis acids (enolase family enzymes) or are cofactor-independent are of particular interest, since they are able to perform the apparently simple 1,1-proton transfer using active site amino-acid residues and thus are model systems for understanding enzymatic reactions in general. Several of these enzymes are also important as drug targets,6,7,20–23 potential drug targets,84 or are used in biotechnological applications.48,49,51 This review will consider racemases/epimerases utilising deprotonation and deprotonation mechanisms, their reactivity and the strategies used to inhibit them.
Reactivity of racemases and epimerases
Racemisation and epimerisation reactions
On the face of it, the reaction catalysed by racemases and epimerases operating through a 1,1-proton transfer mechanism is deceptively simple, consisting of only deprotonation and deprotonation (Scheme 1). In the case of the PLP-dependent enzymes, e.g. alanine racemase, the active site is situated at the interface between two dimer subunits.69 Formation of the external aldimine between the PLP cofactor and substrate considerably enhances the acidity of the Cα–H.5,18,20,69 Stabilisation of the developing negative charge in PLP-dependent enzyme reactions requires that the broken bond is perpendicular to the PLP π-system.69,70,85The imine nitrogen between the amino-acid substrate and the PLP cofactor is thought to be protonated5 and this enhances the acidity of the Cα–H. This effect is illustrated by chemical systems which show that the pKa of zwitterionic glycine is 28.9 whilst the corresponding pKa for the zwitterionic imine between glycine and acetone is 22.86 Model studies using the glycine aldimine of pyridoxal suggest a Cα–H pKa value of 11 and 17 for when the pyridoxal aromatic hydroxy group is protonated and deprotonated, respectively.5 These studies also show that protonation of the amino-acid carboxylate further decreases the Cα–H pKa value to 6 but crystal structures suggest that this does not occur during the enzyme catalytic cycle. This is contrast to the situation in cofactor-independent racemases/epimerases, where substrate carboxylate groups are held within a hydrogen-bonding network20 or transiently protonated during the reaction.21 The pKa of the external aldimine Cα–H in the alanine racemase reaction is estimated to be 9, which is intermediate between those for the catalytic bases, Tyr-265 and Lys-39.69
The mechanism of some PLP-dependent enzymes, e.g. ornithine decarboxylase, is thought to go via a quinoid intermediate69,70 resulting from protonation of the pyridoxal nitrogen by a glutamic acid residue. The equivalent residue in alanine racemase is an arginine and the pyridoxal nitrogen is not extensively protonated69,70 (Scheme 2). Therefore, alanine racemase is thought to catalyse its reaction via a carbanion 3 not a quinoid 11 intermediate.69 Kinetic isotope effect experiments on alanine racemase are consistent with a carbanion rather than quinoid intermediate.70,87
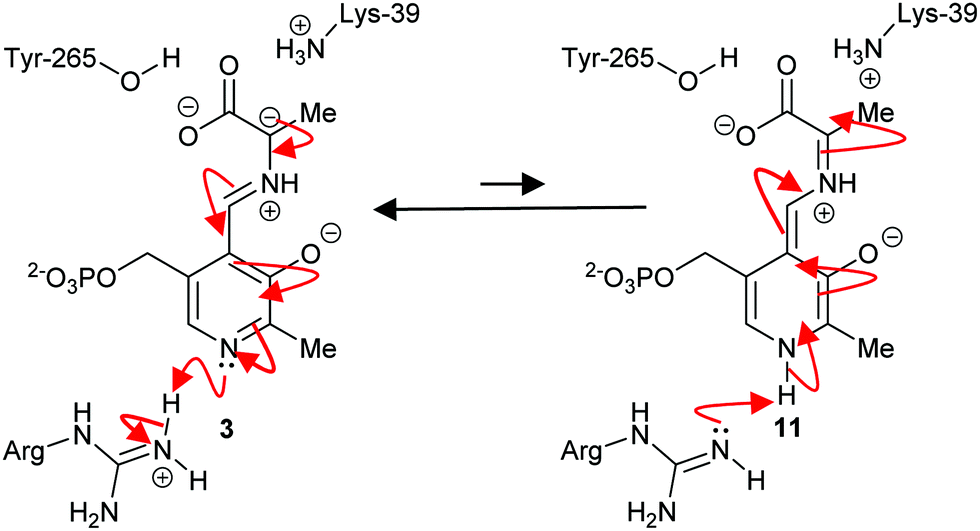 | ||
| Scheme 2 Carbanion 3 and quinoid 11 intermediates in the alanine racemase reaction.69 | ||
The situation is different for enolase-family racemases and epimerases and those which are cofactor-independent. The fundamental problem for these enzymes is how to deprotonate a substrate carbon acid (typical pKa = ∼21–2386,88,89) using active site bases with pKa values in the range 6–9 without the enhancement afforded by a PLP cofactor. Many racemase/epimerase substrates possess carboxylic acids (pKa 2–5), which are deprotonated to the negatively charged carboxylate (e.g. substrates of amino-acid racemases/epimerases20 and methylmalonyl-CoA epimerase90). Consequently, the apparent pKa of the Cα–H for these substrates will be ∼29.20,86 This effect is illustrated by chemical systems, which show that the pKa for the Cα–H of glycine in water is 28.9, while the corresponding pKa for glycine methyl ester is 21.0.86
Racemases and epimerases utilising a negatively charged substrate generally hold the carboxylate group within a hydrogen-bonding network or ion pair to disperse the negative charge.20 In some cases, the enzyme also transfers the incoming proton onto the substrate carboxylate group before it is transferred onto the Cα of the product (e.g. glutamate racemase21). Exceptions to this strategy are seen with methylmalonyl-CoA epimerase90 and mandelate racemase,91,92 where the carboxylate group is ligated to the active site Co2+ or Mg2+ ion which acts as a Lewis acid and diminishes the pKa of the Cα–H.21 Typically the carboxylate group is also held within a hydrogen-bonding network with active-site residues.5 Some racemase/epimerase substrates also contain further destabilising groups, such as ammonium groups (amino-acid racemases/epimerases),18,20 amide carbonyl groups (N-succinylamino acid racemases, dipeptide epimerases and other enolase family enzymes18,49) and OH (mandelate racemase,18,91 various sugar epimerases18). Both ammonium and OH groups are more easily deprotonated than the Cα–H. Chemical models86,93 show that the pKa of the Cα–H is diminished by 9–15 units by protonation of an adjacent amine and a number of amino-acid racemases/epimerases,20,94 including diaminopimelate epimerase and glutamate racemase, appear to protonate the amine of the substrate during the reaction. In the case of mandelate racemase18,91 and N-succinylamino-acid racemases,49 the OH or amide carbonyl groups are ligated to active-site metals such as Mg2+ (mandelate racemase18,91,92) or Co2+, Mn2+ or, occasionally, Mg2+ (N-succinylamino-acid racemases49). The rates of proton transfer for the deprotonation and reprotonation steps are generally high, with rate constants of the order of 5 × 109 to 100 × 109 M−1 s−1.86
Recent analysis18 of racemase/epimerase crystal structures, obtained in the presence of ligands, suggests that the vast majority of enzymes bind the two substrate stereoisomers using ‘mirror-image packing’, that is functional groups are held within the same position with the Cα–H on opposite sides in the different stereoisomers. In some cases, e.g. amino-acid racemases/epimerases,20 the positions of the substrate side-chain and functional groups show remarkably small differences in their positions between the stereoisomers. In other cases, e.g. AMACR/MCR6,7,18,95 which utilises substrates with large hydrophobic side-chains, the different epimers are accommodated by fixing two of the function groups (the methyl group and acyl-CoA moiety in this case) whilst the side-chain is accommodated in discrete binding sites on a hydrophobic surface at the entrance of the active site.
The active-site bases sit immediately adjacent to the Cα–H. In the vast majority of cases, the active-site bases are located on both sides of the substrate (the so called ‘two-base enzymes’), while, in a few cases (the ‘one-base’ enzymes), a single active-site base mediates catalysis.18,20,96 Many racemases/epimerases are dimers, with the active site located at the dimer interface and active-site bases contributed by both subunits;18,20,95 binding of substrate often triggers movement of the subunits from an ‘open’ to a ‘closed’ conformation, moving the active-site bases into position and desolvating the active site.20,94,97,98 In some enzymes (e.g. glutamate racemase21), this conformational change triggers a change in the conformation of the deprotonating active-site base as part of the pre-activation step which results in protonation of the substrate carboxylate group. It has also been suggested that conformational changes by ‘capping domains’, which result in the closed form of the racemase, activate the enzyme for catalysis, are important, e.g. in mandelate racemase.94 In other cases, little or no conformational changes are observed in the protein upon binding of substrate and the enzyme active site is substantially desolvated in the unbound state.20,95
Active-site bases
PLP-dependent enzymes use several different active-site bases. In alanine racemase, these are generally thought to be Tyr-265 and Lys-39 (Fig. 1).5,69,70,99 Chemical models suggest that the pKa of these active-site bases are increased to ∼21 (Lys) and ∼28 (Tyr), respectively, in the hydrophobic active site.100 This is considerably higher than the experimentally-determined Cα–H pKa value of 11101 and 9.94.87 Hence, deprotonation of the substrate is expected to be facile. In serine racemase, the corresponding active-site bases are Lys-57 and Ser-8269 and the experimentally determined external aldimine Cα–H pKa value is 9.26.87 Chemical models suggest that their active site base pKa values will be ∼21 and 33–39,100 the latter being extremely high. These pKa values will be modified by hydrogen-bonding networks within the active site, to allow deprotonation of the active-site residues and reprotonation of the carbanionic intermediate (vide supra, Schemes 1 and 2, 3).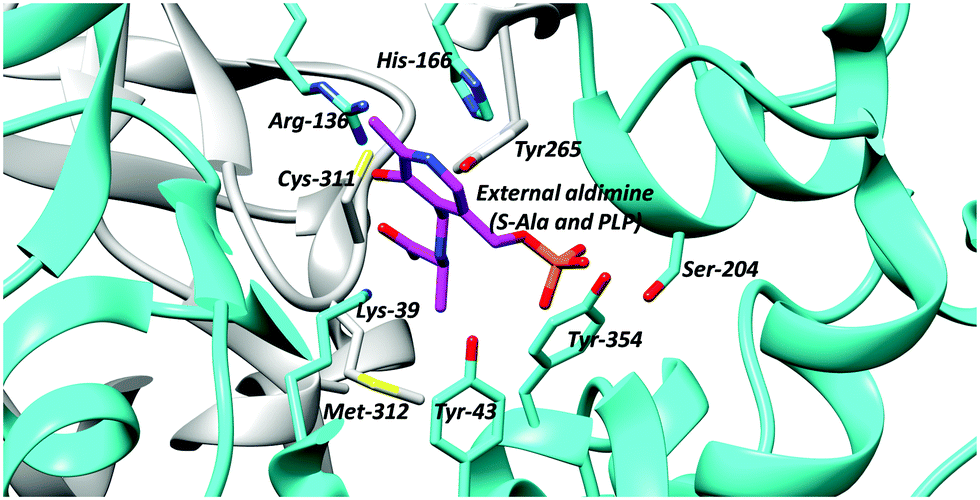 | ||
| Fig. 1 Active site residues of B. stearothermophilus alanine racemase showing the external aldimine (alanine conjugated to PLP) and active site bases Lys-39 and Tyr-265 (PDB: 1L6F).99 | ||
The N-succinylamino acid racemases and related enolase enzymes, e.g. O-succinylbenzoate synthase,102,103 utilise a pair of lysine residues as catalytic bases49,104 (Fig. 2). Chemical models100 suggest that the pKa for these lysine residues within the active site will be ∼21. The Cα–H pKa for these substrates ligated to active-site metals appears not to have been calculated, though studies on other metal-dependent enzymes (mandelate racemase)101 suggests that this will be ∼15.
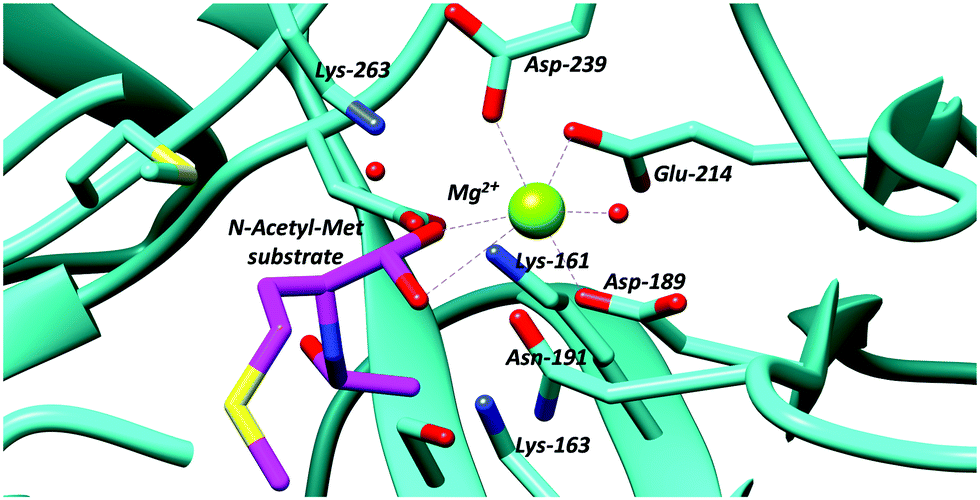 | ||
| Fig. 2 Active site residues of an N-acetyl-amino-acid racemase, showing binding of N-acetyl-methionine substrate (PDB: 4A6G).104 | ||
Several different active-site bases are used by the cofactor-independent racemases and epimerases. In most amino-acid racemases/epimerases, both active-site bases are Cys, which act as a thiolate base/thiol acid pair, catalysing deprotonation and deprotonation,18,20e.g. Cys-74 and Cys-185 in B. subtilis glutamate racemase105 (Fig. 3). Cys is favoured as an active-site base in amino-acid racemases and epimerases because it is more easily desolvated and has a lower pKa than Ser or Thr.106 Chemical models suggest that desolvation raises the pKa of the Cys residue thiol to thiolate conversion to ∼28,100 matching the expected pKa of the Cα–H of ∼29.20,86 This allows deprotonation of the Cα–H by the Cys thiolate. In contrast, the pKa values of the active-site Cys residues acting as an acid appear to be ∼6–7 to enable protonation from the opposite side. This change in pKa appears to be mediated by a dipole on the α-helices bearing the Cys thiol (at least in diaminopimelate epimerase20,107). Exceptions to this rule include aspartate/glutamate racemase from a pathogenic E. coli strain (Ecl-DER), in which one of the catalytic Cys is replaced by Thr. This enzyme catalyses irreversible conversion of S-Asp to R-Asp, which arises partly because of differences in the pKa values of the Cys and Thr side-chains and partly because of differences in the distance between the Cα–H and the catalytic bases on either side of the substrate.20,108,109 Similarly, MMP0739 aspartate/glutamate racemase from Methanococcus maripaludis possesses active-site Cys and Thr residues and is predicted to catalyse unidirectional enantiomerisation42 (the opposite catalytic base is replaced compared to the aspartate/glutamate racemase exception noted above20). The H. sapiens trans-3-hydroxy-S-proline epimerase110 also possesses an equivalent Cys-to-Thr substitution to that in MMP0739.42 However, biochemical analysis shows that this Cys-to-Thr substitution converts the latter enzyme from an epimerase into a dehydratase,110i.e. the enzyme catalyses elimination rather than racemisation/epimerisation (vide infra).
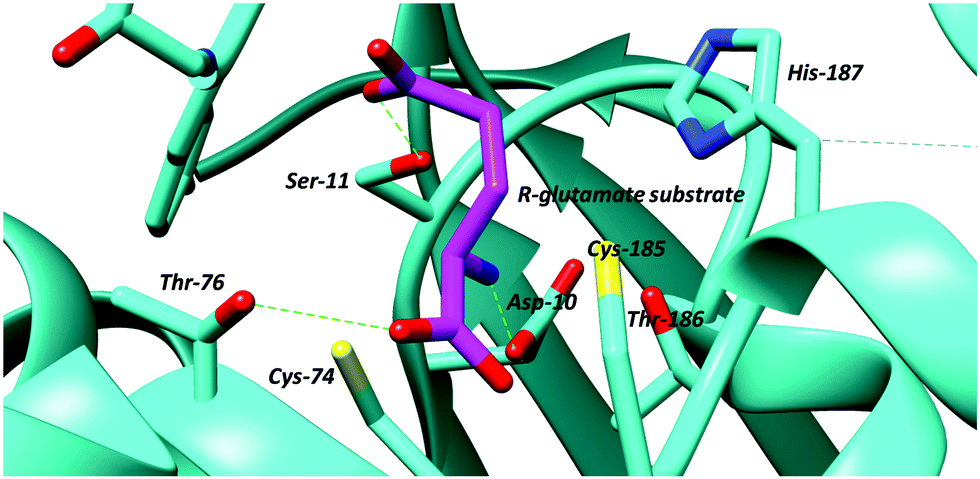 | ||
| Fig. 3 Active site of glutamate racemase from B. subtilis showing bound R-glutamate substrate and active site bases, the cysteine residues, Cys-74 and Cys-185 (PDB: 1ZUW).105 Hydrogen bonds are shown as green dashed lines. | ||
Other racemases and epimerases use a variety of active-site bases, including Cys/Cys (allantoin racemase71), His/Lys (mandelate racemase71), Glu/Glu or Asp/Asp (several different epimerases acting on sugar substrates71), Glu/Glu (methylmalonyl-CoA epimerase90), Tyr/Glu (heparin sulfate D-glucuronosyl C-5 epimerase71), Glu/His or Tyr/His (various sugar mutarotatases71), Lys/Lys (various N-succinylamino-acid racemases and enolase family racemases18,49), an Asp/His pair and Tyr (dTDP-diphosphate-4-keto-6-deoxyglucose 3,5-epimerase a.k.a. RmlC),111 and a Glu/His pair and Asp (AMACR and MCR71,81,95,112). N-Acetylmannosamine-6-phosphate 2-epimerase appears to be an exception to this rule, as only one active site base/acid (Lys) has been identified.18,96
The active sites of these other racemases and epimerases also exclude bulk solvent.95 Chemical models100 again suggest that the pKa of these active-site bases are correspondingly increased to ∼29 (His), ∼21 (Lys), ∼22 (Asp and Glu), and ∼28 (Tyr), again matching approximately the expected pKa values of the substrate Cα–H. Each of these bases participates in a hydrogen-bonding network with other active site residues and, in some cases, active-site ordered waters.
An often-overlooked consideration in the catalytic mechanism is the hydrogen bonding between the electron-deficient Cα–H (which are activated by adjacent carbonyl groups) and active-site bases. This is of relevance for all proteins, since all protein amino-acid residues are capable of forming such bonds.113 These hydrogen bonds tend to be moderately weak (8 to 10.6 kJ mol−1 when bonding to water compared to 18.9 kJ mol−1 for an ‘typical’ intra-molecular bond114). In addition, amino-acids and other racemase/epimerase substrates will also be able to form such bonds. The case of the Cα–H/His/Glu hydrogen bond in AMACR/MCR is particularly interesting in this regard (Fig. 4), as the hydrogen bond resembles that in the catalytic triad of chymotrypsin and related hydrolytic enzymes which has been studied in detail.115
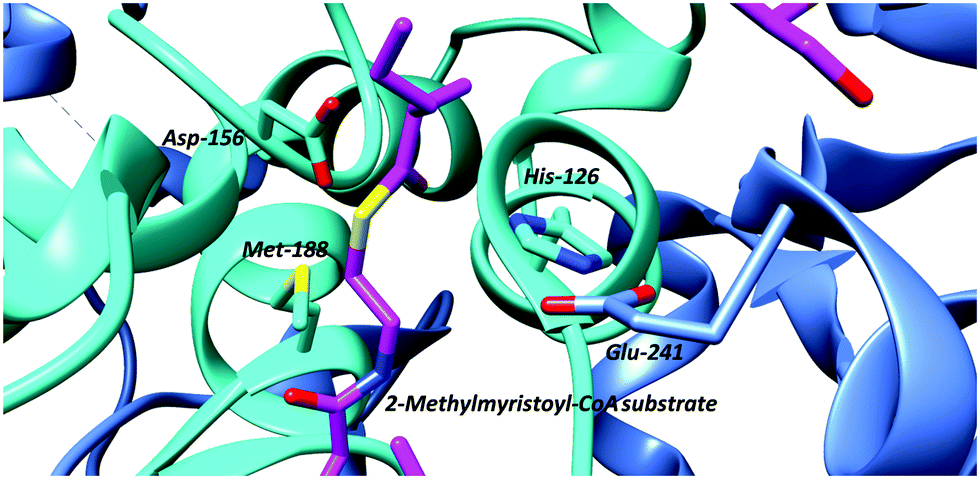 | ||
| Fig. 4 Active-site arrangement of α-methylacyl-CoA racemase (MCR) from M. tuberculosis showing binding of binding of 2-methyltetradecanoyl-CoA substrate (PDB: 2GCI).95 Active site bases include Asp-156 and the His-126/Glu-241 pair, with Glu-241 contributed by the second monomer subunit. The His-126/Glu-241 pair removes the α-proton of the S-2-methylacyl-CoA substrate whilst Asp-156 protonates the enolate intermediate.6,7,81,95 The roles of these residues are reversed for the R-2-methylacyl-CoA substrate. Met-188 stabilises formation of the enolate intermediate. | ||
Concerted versus stepwise reactions
The PLP-dependent enzymes have been extensively studied and a series of mechanistic and computational studies show the presence of a carbanionic intermediate (vide supra, Schemes 1 and 2, 3),5,50,69,70,101 indicating a step-wise reaction. Kinetic isotope effect studies on alanine racemase are also consistent with a carbanionic intermediate.87 Alanine racemase catalyses Cα–H exchange but the stereochemical course of this reaction was not determined,116 although non-stereoselective incorporation of label into substrate is expected because of the stability of the carbanionic intermediate.Studies investigating isotopic incorporation from solvent into substrates have been particularly informative about the concertedness of mechanism in other enzymes. For the majority of enolase family and cofactor-independent racemases and epimerases, isotopic incorporation is observed into the product but very little incorporation is observed into the substrate, e.g. glutamate racemase,78 proline racemase,77 mandelate racemase,74 2-methylmalonyl-CoA epimerase73 and a racemase mediating post-translational modification of peptides.117 This is consistent with a concerted reaction. Monitoring the progress of the reaction by these enzymes in isotopically labelled solvent using circular dichroism typically results in an over-shoot of the equilibrium position, e.g. as has been observed for mandelate racemase.74 This results from isotopic incorporation into product only with a significant kinetic deuterium isotope effect affecting the reverse reaction. These results further support a mechanism in which two-base enzymes catalyse a microscopic enantiomerisation reaction, with asynchronously concerted deprotonation and reprotonation.76 Such a mechanism minimises the formation of a highly unstable doubly deprotonated intermediate and hence partly overcomes the effect of destabilising groups adjacent to the Cα–H (i.e. the carboxylate).
In contrast to the above is the observation that incubation of substrates with AMACR in 2H2O results in a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 incorporation of deuterium into substrate and product. This has been interpreted as formation of a discrete deprotonated intermediate followed by deuteration from either side.79,80 Analysis of the crystal structure of the M. tuberculosis homologue, MCR, shows catalytic residues on both sides of the substrate (the His-126/Glu-241 pair and Asp-156; Fig. 4) and are consistent with the formation of an enolate intermediate.81,95,112 Thus, AMACR and MCR fundamentally differ in their mechanisms from most other cofactor-independent racemases and epimerases, in that they catalyse microscopic racemisation rather than epimerisation.8,79,80 Incorporation of deuterium from solvent is also catalysed by hydantoin racemase via an enolate intermediate118 and is expected to be non-stereoselective but this has not yet been verified.
:1 incorporation of deuterium into substrate and product. This has been interpreted as formation of a discrete deprotonated intermediate followed by deuteration from either side.79,80 Analysis of the crystal structure of the M. tuberculosis homologue, MCR, shows catalytic residues on both sides of the substrate (the His-126/Glu-241 pair and Asp-156; Fig. 4) and are consistent with the formation of an enolate intermediate.81,95,112 Thus, AMACR and MCR fundamentally differ in their mechanisms from most other cofactor-independent racemases and epimerases, in that they catalyse microscopic racemisation rather than epimerisation.8,79,80 Incorporation of deuterium from solvent is also catalysed by hydantoin racemase via an enolate intermediate118 and is expected to be non-stereoselective but this has not yet been verified.
The above results can be rationalised based on the pKa values for the deprotonation of the substrate. The pKa of Cα–H for a thioester is 21,86,88 while the pKa values for Cα–H for amino-acid zwitterions is 29,20,86 for simple carboxylates is 33 and for simple amides 28.4.86 Therefore, concerted reactions occur with substrates containing relatively unactivated Cα–H (high pKa values), with consequent asymmetrical isotopic incorporation. This explains the behaviour of peptide epimerases,117 which are observed to undergo concerted reactions. These peptide substrates have pKa values of ∼26–31 for Cα–H, although these values are dependent on both N- and C-substituents and the protonation status of amine groups.86 This model also allows prediction of enzymatic behaviour for uncharacterised racemases/epimerases, e.g. hydantoin racemase,118 based on pKa values for Cα–H. The proposed model also casts doubt on the use of isotopic labelling studies to differentiate between ‘two-base’ and ‘one-base’ enzymes (reviewed in ref. 92). It has previously been proposed that near-symmetrical isotopic incorporation into substrate and product is indicative of ‘internal return’, i.e. a ‘one-base’ mechanism. The results on AMACR79,80 (reviewed in ref. 6 and 7) show that this behaviour is also observed with ‘two-base’ enzymes with activated Cα–H, as it is known that AMACR/MCR possesses appropriate active-site bases on both sides of the substrate.
Elimination reactions
Several racemases/epimerases catalyse elimination reactions, in addition to racemisation/epimerisation. With the exception of the ‘mutant’ H. sapiens trans-3-hydroxy-S-proline epimerase containing a Cys-to-Thr substitution noted above110 giving rise to dehydratase activity, and the Labrenzia aggregata cis-3-hydroxy-S-proline racemase/dehydratase (IAM 12614)119 (vide infra), all of the known elimination reactions take place with unnatural substrates. The vast majority of these unnatural substrates are halogen derivatives,20,47,63,82,120–126 with only a few exceptions.63,119,124,127,128 The deprotonation step in the elimination reaction is highly similar to that described for racemisation/epimerisation (vide supra).Several PLP-dependent racemases catalyse elimination reactions.129–133 The classic example is alanine racemase (Scheme 3) which β-eliminates halogens from 3-fluoroalanine 12 and 3-chloroalanine 13.132O-Carbamoyl-R-serine 14R and O-acetyl-R-serine 15R act as irreversible inhibitors whilst O-carbamoyl-S-serine 14S and O-acetyl-S-serine 15S are reversible competitive inhibitors.132 3-Fluoroalanine 12 is a potent inactivator of alanine racemase. 3-Chloroalanine 13 and O-carbamoyl-S-serine 14 and O-acetyl-S-serine 15 also act as substrates. These substrates result in the formation of 2-aminoacrylate 16, which tautomerises to pyruvate 17 with a partition coefficient of between 790 and 920 to 1 (catalytic conversion/inactivation).
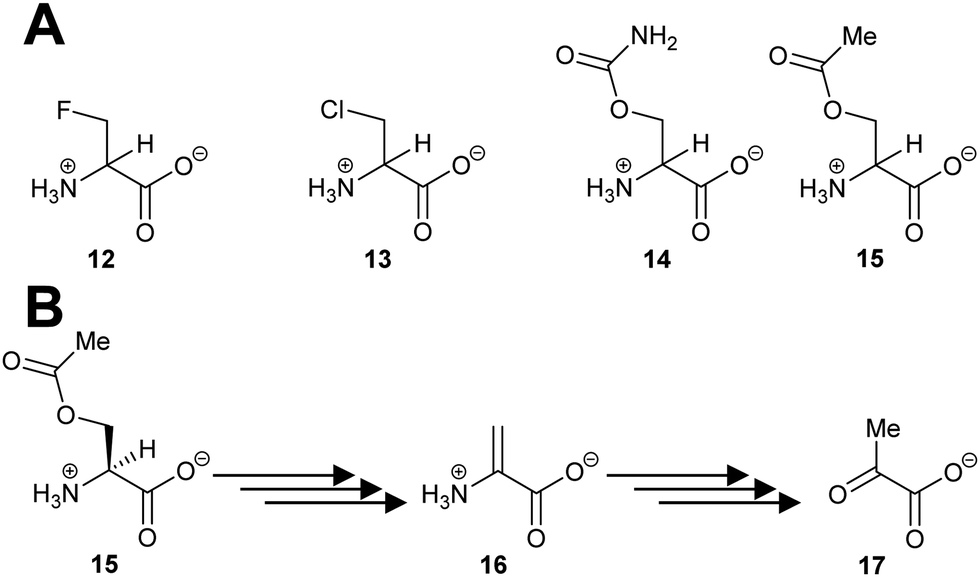 | ||
| Scheme 3 (A) Structures of eliminating inhibitors and substrates of E. coli alanine racemase; (B) conversion of O-acetyl-S-serine 15S to pyruvate 17 by alanine racemase.132 | ||
There have also been several studies on the elimination reaction catalysed by H. sapiens serine racemase.129–131,133 The wild-type enzyme has a ca. 4-fold preference for β-elimination over racemisation of S-serine.129,131 Other substrates can also undergo β-elimination, including S-serine-O-sulfate and S-threo-hydroxyaspartate.131 The enzyme is allosterically activated by divalent metal ions (with Mn2+ being the strongest) and ATP,129,133 and activity is potentiated by halide anions.130 The elimination reaction catalysed by serine racemase is thought to control levels of R-serine in neurons133 and, hence, modulate the activity of NMDA receptors;129,131,133 over-activation of the NMDA receptor has been shown to result in neuronal cell death.133 This is, however, at the expense of producing highly electrophilic 2-aminoacrylate 16.129,131,133
Enolase family enzymes, such as P. putida mandelate racemase126 and L. aggregata cis-3-hydroxy-S-proline racemase/dehydratase (IAM 12614),119 are also able to catalyse elimination reactions. Mandelate racemase was able to catalyse elimination of chlorine from 3-chlorolactate 18 to give pyruvate 19 (Scheme 4A).126 The mechanistic details of the reaction was not determined but it is assumed to occur by E2 anti-elimination to give the enol 20 followed by tautomerisation.126 However, the possibility of a E1cb-type mechanism via an enediolate type intermediate cannot be discounted. The elimination of chlorine from 3-chlorolactate 18 by mandelate racemase is reminiscent of the elimination of HCl from 3-chloroalanine 13 by glutamate racemase, which also gives pyruvate 17 as a product (vide infra, Scheme 12).134 This result contrasts with the earlier observation on P. putida mandelate racemase with 3,3,3-trifluorolactate 21, which undergoes racemisation. β-Elimination to give 22 is not observed (Scheme 4B).91
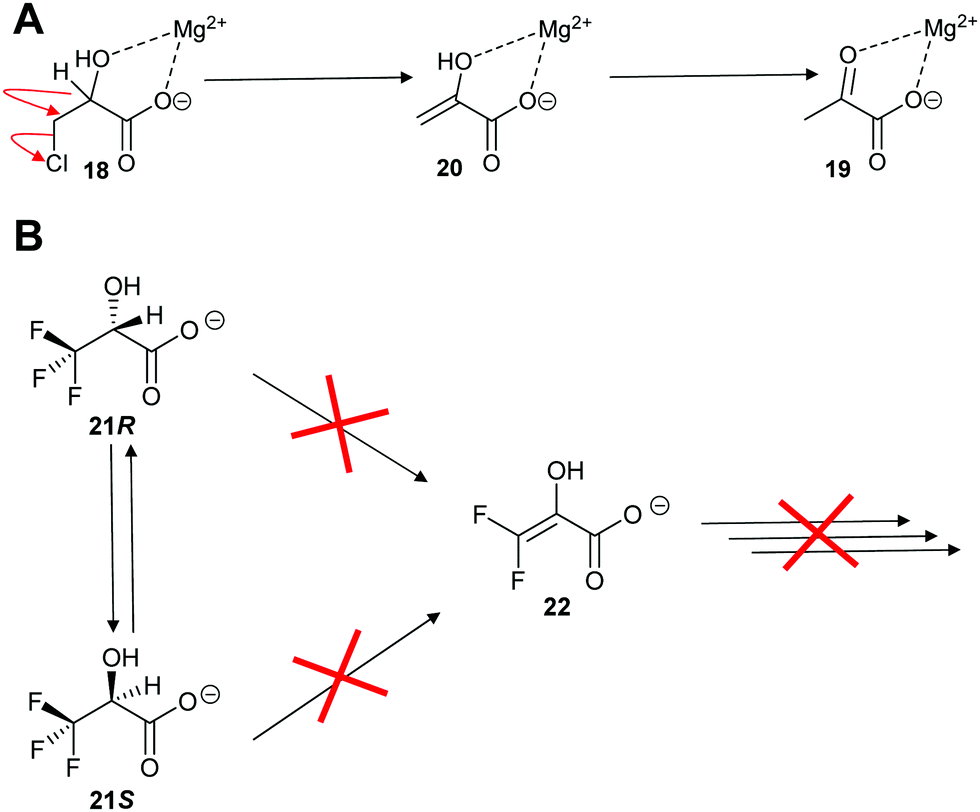 | ||
| Scheme 4 Reaction of halogen substrates with P. putida mandelate racemase.91,126 (A) Elimination of chlorine from 3-chlorolactate 18;126 (B) expected elimination of 3,3,3-trifluorolactate 21 to give 22.91 | ||
L. aggregata cis-3-hydroxy-S-proline racemase/dehydratase (IAM 12614)119 catalyses both racemisation and β-elimination reactions with its substrate 23, in a 3 to 2 ratio (Scheme 5). The β-elimination reaction is proposed to go via an enediolate intermediate 24, although it may be a more concerted E2-like reaction. The cis substrate allows for anti-elimination of the hydroxy group to give the enamine product 26, which subsequently tautomerises to Δ-pyrroline-2-carboxylate 27 (Scheme 5). Alternatively, epimerisation to give 25 can occur. It is notable that 27 is a known inhibitor of T. cruzi proline racemase.135
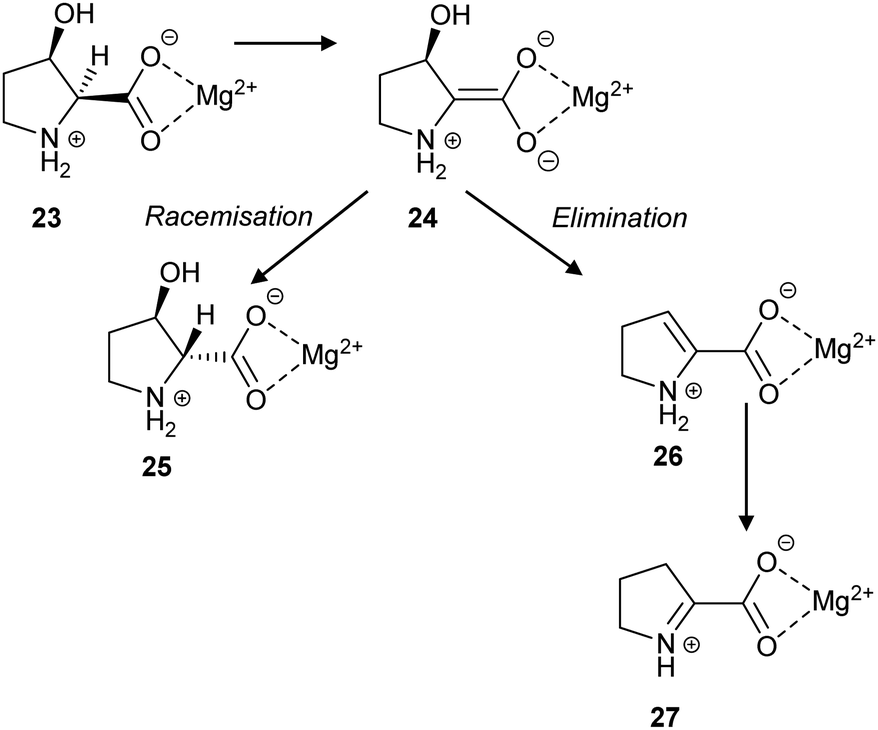 | ||
| Scheme 5 The racemisation and elimination reactions catalysed by cis-3-hydroxy-S-proline racemase/dehydratase.119 | ||
The cofactor-independent enzymes diaminopimelate epimerase124 and glutamate racemase128 are able to eliminate N-hydroxy substrates. In the case of glutamate racemase, deprotonation of substrate 28 results in elimination of hydroxide or water with formation of imine 29, which is hydrolysed to 2-oxoglutarate 30 (Scheme 6).
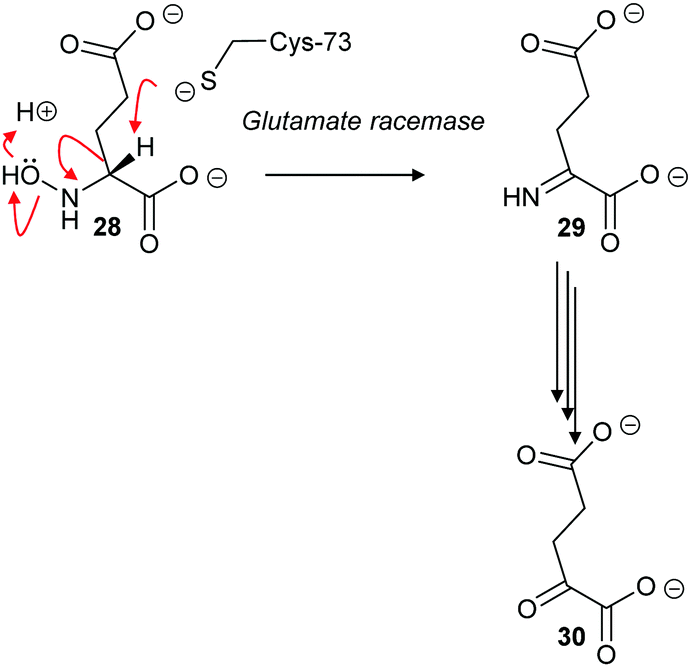 | ||
| Scheme 6 Elimination of N-hydroxy-R-glutamate 28 by an E2 mechanism followed by hydrolysis of imine 29 to form 2-oxoglutarate 30.128 | ||
With aliphatic substrates containing β-fluorine or β-chlorine, the presence of the halogen increases the acidity of the Cα–H,120,136 and, hence, these elimination substrates tend to be converted with somewhat higher efficiency than their racemisation/epimerisation equivalents.120 With diaminopimelate epimerase,121 only stereoisomers allowing an antiperiplanar conformation between the Cα–H and the fluorine underwent elimination, with substrates not allowing an antiperiplanar conformation undergoing epimerisation instead. Similarly, mutant glutamate racemases (in which the active-site Cys bases were mutated to Ser) eliminated either 2R,3R- or 2S,3S-3-chloroglutamate stereoisomers 31R and 31S with anti-elimination (Scheme 7), depending on which active site Cys residue was still present.82 The resulting enamine 32 tautomerises to imine 29, which is hydrolysed to 2-oxoglutarate 30. These results are consistent with a substantially concerted (E2) mechanism.137
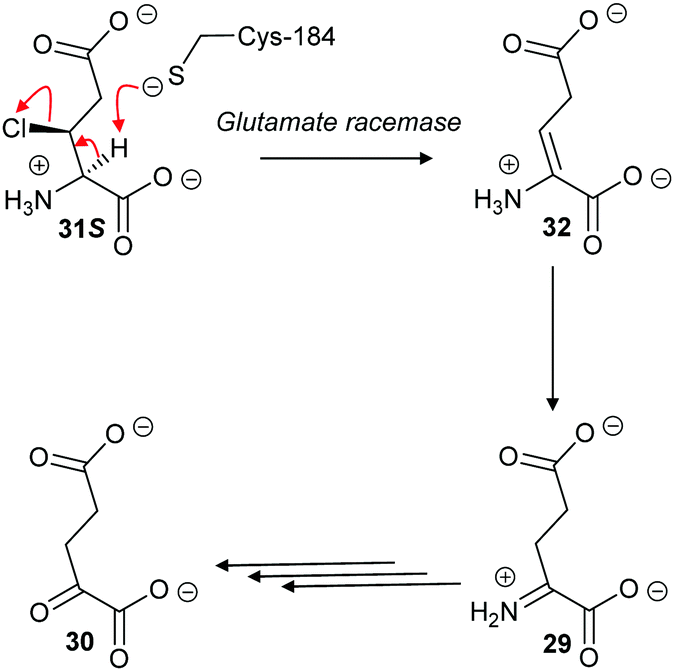 | ||
| Scheme 7 β-Elimination of 2S,3S-3-chloroglutamate 31S by Lactobacillus glutamate racemase to give enamine 32. Tautomerisation to imine 29 followed by hydrolysis gives the resulting 2-oxoglutarate 30.82 | ||
The above results contrast with those observed with AMACR, in which epimeric substrates 33 and 34 were eliminated to the same product 35,120 consistent with an E1cb mechanism through the enolate intermediate 36 (Scheme 8).137 These results are inconsistent with an E2-elimination because the substrate requires a conformation in which the α-H and the fluorine are anti-; epimer 33 can adopt such a conformation but epimer 34 cannot. Interestingly, compounds closely related to 33 and 34 were synthesised136 and tested as inhibitors of native rat AMACR and no elimination of fluoride was observed.136 These inhibitors136 had the same configuration as 34 (and its epimer with opposite C2 and C3 configurations) but, in view of the subsequent report,120 this is a surprising observation.
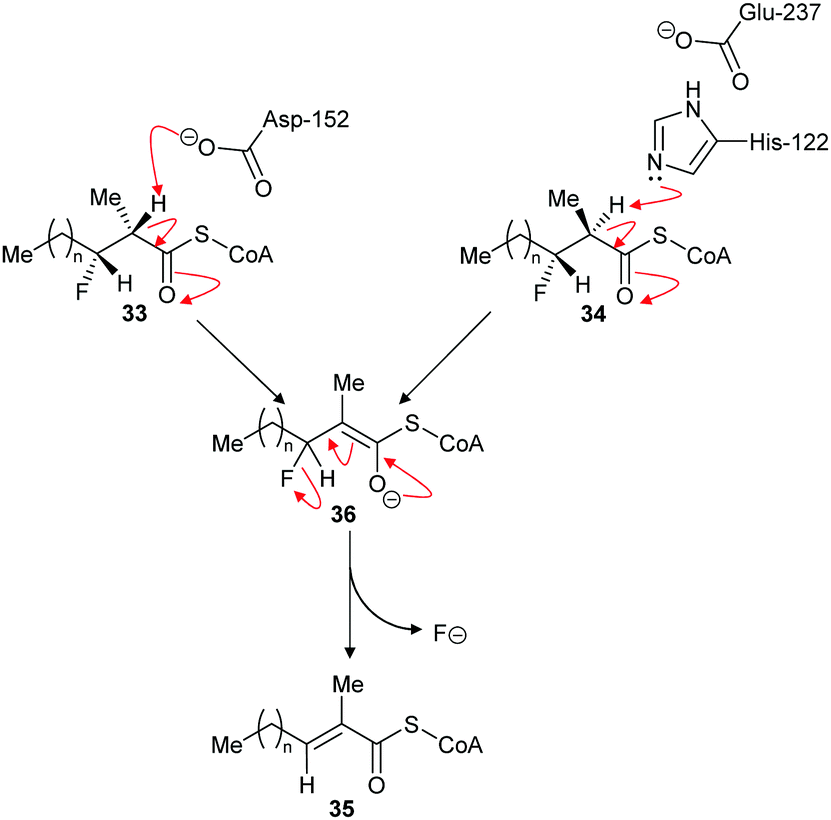 | ||
| Scheme 8 Elimination of fluoride from substrates by α-methylacyl-CoA racemase.120 For substrates 33 and 34, n = 6. For inhibitors (34 and its epimer with opposite C2 and C3 configuration) tested on native rat enzyme reported not to eliminate,136n = 12. Enzyme catalytic residue numbers are those for human AMACR.6,7 | ||
However, this is not the only example where an expected elimination reaction did not take place. Nagar et al. investigated trifluorolactate (2-hydroxy-3,3,3-trifluoropropanoate) 9R and 9S as substrates for mandelate racemase (vide supra, Scheme 4).91 Kinetic analysis showed that Km values for trifluorolactate 9 were unexpectedly similar to the natural substrate, mandelate (1.2–1.74 mM and 1.0–1.2 mM, respectively) and lower than the predicted Km value of ∼10 mM. In contrast, kcat values were reduced by ∼318-fold, with kcat/Km reduced by ∼430-fold. 19F NMR analysis showed that no fluoride was eliminated during the reaction and, hence, 10 was not formed.91 The lack of a β-elimination reaction is unexpected, because the trifluorolactate 9 is able to take up the required anti-conformation for an E2 reaction when in a staggered conformation. Clearly, the mandelate racemase-catalysed racemisation of trifluorolactate is faster than the elimination reaction. The reasons for this are unclear but it could be related to the presence of multiple fluorine atoms within the substrate.138 If loss of fluoride is asynchronous with abstraction of the Cα–H, this will result in generation of a positive charge on the β-carbon. The presence of two additional fluorine atoms will destabilise formation of this transition state. However, it is notable that E. coli dipeptide epimerase (YcjG) eliminates fluoride from S-alanyl-R,S-difluoroalanine in preference to epimerisation,125 suggesting that other factors are also at play such as the extent of δ+ charge stabilisation in the transition state.
Methods for determining racemase and epimerase activity
Racemases and epimerases are simple enzymes, in the sense that they only have one substrate and product (a uni-uni reaction), which is a characteristic they share with other isomerases. A consequence of racemases and epimerases accepting both stereochemical configurations of their substrates is that their reaction will, in most cases, be readily reversible and kcat/Km values are likely to be similar for the reactions in both directions (which is required by the Haldane relationship79,80,139 for an equilibrium constant of ∼1). Hence the rate for the reverse reaction when determining initial rates is likely to be significant and this must be corrected for in any kinetic study, such as the determination of inhibitor potency.Several different assays exist for measuring racemase/epimerase activity (Table 1). One approach is to measure rates at very early time points where the reverse reaction will have less impact. Typically, the enzyme reaction is followed using techniques such as optical rotation46,140–145 or circular dichroism,23,91,107,146–150 allowing a time-course to be determined. These assays are ideal, in that they allow much more accurate determination of initial rates,151 although correction for the reverse reaction should still be performed. Indeed, circular dichroism is by far the most common method of detecting enzymatic activity (Table 1), although it is noted that these are kinetic studies designed to measure Km, kcat and kcat/Km (vide infra).
| Enzyme | Type | Substrate | Reaction | Assay type | K m (μM) | k cat (s−1) | k cat/Kma (M−1 s−1) |
|---|---|---|---|---|---|---|---|
|
a
k
cat/Km values are calculated from the reported the kcat and Km values given in the paper.
b
k
cat values are calculated from reported Vmax values in μmol min−1 mg−1 and reported molecular weights of 42500177 and 41200 Da,178 respectively. Abbreviations used: AMACR, α-methylacyl-CoA racemase; CoA, coenzyme A; HPLC, high-performance liquid chromatography; PLP, pyridoxal 5′-phosphate; UPLC, Ultra Performance Liquid Chromatography. |
|||||||
| Baccilus psychrosccharolyticus alanine racemase177 | PLP-dependent | S-Alanine | Racemisation | Polarimetry | 17900 |
715b | 39944 |
| Baccilus psychrosccharolyticus alanine racemase177 | PLP-dependent | R-Alanine | Racemisation | Polarimetry | 12200 |
1417b | 116147 |
| Corbicula japonica Alanine racemase178 | PLP-dependent | S-Alanine | Racemisation | Coupled enzyme (D-amino acid oxidase) | 22600 |
430b | 19026 |
| Corbicula japonica Alanine racemase178 | PLP-dependent | R-Alanine | Racemisation | Coupled enzyme (NAD+-dependent) | 9200 | 196b | 21304 |
| Tolypocladium inflatum alanine racemase179 | PLP-dependent | S-Alanine | Racemisation | Coupled enzyme (D-amino acid oxidase) | 7000 | 3.8 | 543 |
| Tolypocladium inflatum alanine racemase179 | PLP-dependent | R-Alanine | Racemisation | Coupled enzyme (NAD+-dependent) | 2700 | 1.51 | 559 |
| E. coli alanine racemase134 | PLP-dependent | S-Alanine | Racemisation | Coupled enzyme (NAD+-dependent) | 340 ± 10 | 170 ± 2 | 500000 |
| B. subtilis alanine racemase134 | PLP-dependent | S-Alanine | Racemisation | Coupled enzyme (NAD+-dependent) | 5900 ± 900 | 1190 ± 70 | 201695 |
| M. tuberculosis alanine racemase134 | PLP-dependent | S-Alanine | Racemisation | Coupled enzyme (NAD+-dependent) | 3700 ± 600 | 37 ± 2 | 10000 |
| L. otakiensis isoleucine 2-epimerase180 | PLP-dependent | 2S-Isoleucine | Epimerisation | UPLC | 5000 ± 80 | 502 ± 16.2 | 100400 |
| L. otakiensis isoleucine 2-epimerase180 | PLP-dependent | 2R-Allo-Isoleucine | Epimerisation | UPLC | 13200 ± 644 |
939 ± 26.8 | 71136 |
| H. sapiens serine racemase131 | PLP-dependent | S-Serine | Racemisation | Coupled enzyme (D-amino acid oxidase) | 7800 ± 700 | 0.205 ± 0.007 | 26.3 |
| H. sapiens serine racemase131 | PLP-dependent | S-Serine | Elimination | Coupled enzyme (NADH-dependent) | 10000 ± 600 |
0.97 ± 0.028 | 97.0 |
| H. sapiens serine racemase131 | PLP-dependent | S-Serine-O-sulfate | Elimination | Coupled enzyme (NADH-dependent) | 1200 ± 100 | 12.06 ± 0.16 | 10050 |
| H. sapiens serine racemase131 | PLP-dependent | S-Threo-3-hydroxyaspartate | Elimination | Coupled enzyme (NADH-dependent) | 2500 ± 300 | 23.33 ± 0.266 | 9332 |
| P. putida mandelate racemase91 | Enolase | S-Mandelate | Racemisation | Circular dichroism | 1000 ± 100 | 637 ± 31 | 637000 |
| P. putida mandelate racemase91 | Enolase | R-Mandelate | Racemisation | Circular dichroism | 1200 ± 200 | 792 ± 19 | 660000 |
| P. putida mandelate racemase91 | Enolase | S-Trifluorolactate | Racemisation | Circular dichroism | 1740 ± 80 | 2.5 ± 0.3 | 1437 |
| P. putida mandelate racemase91 | Enolase | R-Trifluorolactate | Racemisation | Circular dichroism | 1200 ± 200 | 2.0 ± 0.2 | 1667 |
| Amycolatopsis sp. Ts-1- 60 N-acyl amino acid racemase104 | Enolase | N-Acetyl-S-Methionine | Racemisation | HPLC | 18000 |
20 | 1111 |
| Amycolatopsis sp. Ts-1- 60 N-acyl amino acid racemase104 | Enolase | N-Acetyl-R-Methionine | Racemisation | HPLC | 40000 |
14 | 350 |
| N-acylamino acid racemase181 | Enolase | N-Acetyl-R-methionine | Racemisation | HPLC | 11470 ± 1360 |
0.809 ± 0.027 | 70.6 |
| N-acylamino acid racemase181 | Enolase | N-Acetyl-R-methionine | Racemisation | Coupled enzyme (acylase, D-amino acid oxidase), colorimetric | 23410 ± 2120 |
2.55 ± 0.09 | 108.9 |
| LvNSAR/OSBS182 | Enolase | N-Succinyl-R-phenylglycine | Racemisation | Polarimetry | 2700 ± 540 | 2.2 ± 0.2 | 815 |
| RcNSAR/OSBS182 | Enolase | N-Succinyl-R-phenylglycine | Racemisation | Polarimetry | 1800 ± 230 | 15 ± 4 | 8333 |
| AmedNSAR182 | Enolase | N-Succinyl-R-phenylglycine | Racemisation | Polarimetry | 2800 ± 550 | 74 ± 7 | 26429 |
| ExiOSBS182,183 | Enolase | N-Succinyl-S-phenylglycine | Racemisation | Polarimetry | 1700 ± 500 | 0.07 ± 0.006 | 41.2 |
| GkNSAR/OSBS182,184 | Enolase | N-Succinyl-S-phenylalanine | Racemisation | Polarimetry | 800 ± 200 | 19 ± 1 | 23750 |
| AmyNSAR182,185 | Enolase | N-Succinyl-S-phenylglycine | Racemisation | Polarimetry | 1000 ± 10 | 42 ± 2 | 42000 |
| DrNSAR182,186 | Enolase | N-Succinyl-S-phenylglycine | Racemisation | Polarimetry | 1400 ± 200 | 520 ± 30 | 371429 |
| M. aeruginosa Aspartate racemase (McyF)187 | Cofactor-independent | S-Aspartate | Racemisation | Coupled enzyme (D-amino acid oxidase) | 22900 ± 2100 |
42.5 ± 1.3 | 1856 |
| M. tuberculosis diaminopimelate epimerase167 | Cofactor-independent | S,S-Diaminopimelate | α-3H for α-1H exchange | Isotopic wash-out from 3H-labelled substrate | 166 | 0.1465 | 882.5 |
| B. subtilis glutamate racemase166 | Cofactor-independent | S-Glutamate | Racemisation | Circular dichroism | 14000 ± 1000 |
42 ± 2 | 3000 |
| B. subtilis glutamate racemase166 | Cofactor-independent | R-Glutamate | Racemisation | Circular dichroism | 1240 ± 80 | 4.72 ± 0.09 | 3806 |
| F. nucleatum glutamate racemase166 | Cofactor-independent | S-Glutamate | Racemisation | Circular dichroism | 1040 ± 70 | 17.4 ± 0.8 | 16730 |
| F. nucleatum glutamate racemase166 | Cofactor-independent | R-Glutamate | Racemisation | Circular dichroism | 1700 ± 100 | 26 ± 1 | 15294 |
| C. sticklandii proline racemase188 | Cofactor-independent | S-Proline | Racemisation | Circular dichroism | 5700 ± 500 | 97 ± 5 | 17018 |
| H. sapiens UDP-N-acetylglucosamine 2-epimerase44 | Cofactor-independent | UDP-N-acetylglucosamine | Epimerisation | Coupled enzyme (NADH) | 33.1 ± 4.2 | 11.8 ± 2.0 | 356495 |
| C. sticklandii proline racemase188 | Cofactor-independent | R-Proline | Racemisation | Circular dichroism | 3900 ± 400 | 51 ± 1 | 13077 |
| B. subtilis RacX168 | Cofactor-independent | S-Lysine | Racemisation | HPLC | 27900 ± 2670 |
0.0013 ± 0.000083 | 0.047 |
| Streptomyces O-ureidoserine racemase46 | Cofactor-independent | S-O-Ureidoserine | Racemisation | Circular dichroism | 12000 |
475 | 39583 |
| Streptomyces O-ureidoserine racemase46 | Cofactor-independent | R-O-Ureidoserine | Racemisation | Circular dichroism | 32000 |
1450 | 45312 |
| E. coli YgeA168 | Cofactor-independent | S-Homoserine | Racemisation | HPLC | 171000 ± 21100 |
0.130 ± 0.001 | 0.76 |
| E. coli YgeA168 | Cofactor-independent | R-Homoserine | Racemisation | HPLC | 25100 ± 4170 |
0.019 ± 0.0011 | 0.76 |
| Agelenopsis aperta peptide epimerase117 | Cofactor-independent | N-Acetyl-Gly-Leu-S-Ser-Phe-Ala | Racemisation | HPLC | 8000 ± 1400 | 0.076 ± 0.005 | 9.5 |
| Agelenopsis aperta peptide epimerase117 | Cofactor-independent | N-Acetyl-Gly-Leu-R-Ser-Phe-Ala | Racemisation | HPLC | 1100 ± 300 | 0.0058 ± 0.0005 | 5.3 |
| M. tuberculosis AMACR146 | Cofactor-independent | S-Ibuprofenoyl-CoA | Racemisation | Circular dichroism | 86 ± 6 | 450 ± 14 | 5232558 |
| M. tuberculosis AMACR146 | Cofactor-independent | R-Ibuprofenoyl-CoA | Racemisation | Circular dichroism | 48 ± 5 | 291 ± 30 | 6062500 |
| H. sapiens AMACR152 | Cofactor-independent | Pristanoyl-CoA | α-3H for α-1H exchange | Isotopic wash-out from 3H-labelled substrate | 85.6 ± 17.1 | 0.08855 ± 0.006 | 1034 |
| H. sapiens AMACR127 | Cofactor-independent | 2R,S-3-(2,4-Dinitrophenoxy)-2-methylpropanoyl-CoA | Elimination | Direct colorimetric | 56 ± 5.9 | 0.088 | 1571 |
A second alternative when analysing substrates undergoing racemisation or epimerisation is to use HPLC of a diastereoisomeric substrate/product mixture at a fixed time point,37,136 although the differences in energies between diastereoisomers means that these substrates may behave differently from natural enantiomeric substrates. Alternatively, a product containing one chiral centre can be derivatised using a chiral reagent and analysed by HPLC, GC or NMR.8,79,80 The latter approach is time-consuming, as several time-points for each reaction should be analysed and can be technically challenging, especially when working with the low amounts of product typically obtained from enzymatic reactions. Chiral HPLC is an option for separating enantiomeric substrates, although there appear to be no examples of this having been used.
A second approach is to measure exchange of the α-proton with isotopically labelled substrates82,95,152–154 or solvent,8,73,78–80,107 measuring reaction extent by scintillation counting, mass spectrometry or NMR. Such approaches will introduce significant kinetic isotope effects155–158 and deprotonation and reprotonation rates will be markedly different from each other although the extent of this will depend on levels of conversion of substrate and whether the transition states are early or late.107 Consequently, careful design of experiments is needed, especially where precise rate measurements are required. These approaches are often used in mechanistic studies where isotopic distribution in substrate and product is measured (vide supra).
A third approach is to make the enzymatic reaction irreversible. This can be achieved using an irreversible coupled enzyme to remove the reaction product.107,159,160 There are a number of examples of the use of coupling enzymes in kinetic studies determining Km and kcat values (Table 1). The most common coupling enzymes used are D-amino acid oxidase and NAD-dependent oxidoreductases. Coupled enzyme assays are the second most common method of assessing enzymatic activity. Similarly, an unnatural substrate which undergoes an irreversible elimination reaction82,83,120,127,161 can also be used. Typical examples of eliminated groups include water (from amino-acid hydroxamate derivatives83,162), bromide,122,123 chloride47,82,124 and fluoride120,121,124,125,161 as described above. The products from these elimination reactions often need to be assayed using coupling enzymes83,121 or low-throughput spectroscopic techniques such as NMR.120,121,161 Attempts to use fluoride sensors to measure enzymatic activity with substrates eliminating fluorine has met with limited success.161 A notable example of this approach is the elimination reaction of an unnatural acyl-CoA substrate 37 by AMACR to give 2,4-dinitrophenoxide 38 and acyl-CoA 39 (Scheme 9);127 this assay was used in a high-throughput screening campaign of 20387 compounds which identified novel pyrazoloquinolines and pyrazolopyrimidines as inhibitors163 and also in the first extensive inhibitor structure–activity relationship studies on any racemase/epimerase (vide infra).164,165
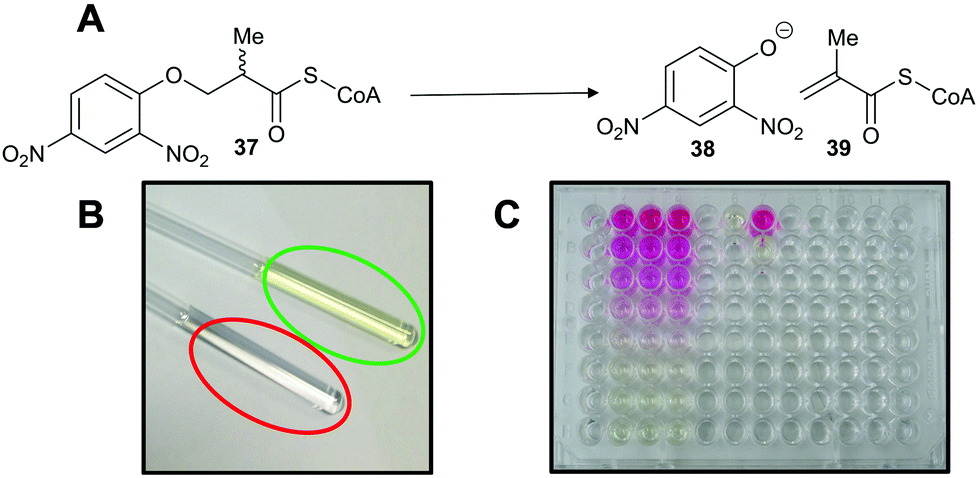 | ||
| Scheme 9 Colorimetric assay for α-methylacyl-CoA racemase (AMACR) based on elimination of 2,4-dinitrophenolate 38 from the acyl-CoA substrate 37.127 (A) Reaction catalysed by AMACR; (B) assay samples showing reaction with heat-inactivated enzyme (red circle) and active enzyme (green circle) showing absorbance at 354 nm;127 (C) measurement of dose–response curve for Rose Bengal (a known inhibitor of AMACR127,152) using the colorimetric assay. Schemes 9B and C have been reproduced from Yevglevskis et al., 2017127 with permission from the Royal Society of Chemistry. | ||
Catalytic efficiency of racemases and epimerases
Kinetic parameters for racemases/epimerases can vary quite widely (Table 1). Km values for amino-acid racemases tend to be in the low mM range, although there are several examples where much higher Km values have been measured. Typical examples include Fusobacterium nucleatum and B. subtilis glutamate racemase, which have Km values of 1.04 and 1.07 mM and 14 and 1.24 mM, respectively.166 In contrast, O-ureidoserine racemase which has reported Km values46 of 12 and 32 mM for S- and R-O-ureidoserine, respectively. In contrast, the Km for S,S-diaminopimelate (2,6-diaminoheptanedioic acid) for M. tuberculosis diaminopimelate epimerase is only 166 μM,167 significantly lower than the Km values for other amino-acid racemases/epimerases. The relatively high Km values for most amino-acid racemases/epimerases are undoubtedly a consequence of these enzymes converting small and relatively unfunctionalised substrates. The same trend is observed for mandelate racemase, which has Km values of 1.0 and 1.2 mM for S- and R-mandelate (2-hydroxyphenylacetate), respectively.91 Racemases/epimerases with larger substrates tend to have lower Km values, as there is more opportunity for binding interactions. For example, human AMACR has a Km value of ∼86 μM for pristanoyl-CoA,152 while Km values for S- and R-ibuprofenoyl-CoA are 86 and 48 μM for the M. tuberculosis homologue.146 These lower Km values are generally accompanied by lower kcat values (Table 1).Catalytic efficiency is quantified using kcat/Km values (Table 1). Again, these can vary quite widely but many racemases/epimerases have relatively modest efficiencies. For example, O-ureidoserine racemase is quite efficient, with reported kcat/Km values46 of 39583 and 45312 M−1 s−1. Similarly, kcat/Km is reported to be 16730 and 15294 M−1 s−1 for F. nucleatum glutamate racemase for S- and R-Glu, while the corresponding values are 3000 and 3806 M−1 s−1 for the B. subtilis enzyme.166 In contrast, M. tuberculosis diaminopimelate epimerase167 has a very modest kcat/Km of 883 M−1 s−1. On the other hand, RacX168 has extremely low kcat/Km values of 2.86 and 3.23 M−1 s−1 for S- and R-Lys, while YgeA168 has kcat/Km values of 45.8 and 45.8 M−1 s−1 for S- and R-His.
k cat/Km values for other racemases and epimerases tend to be higher and this is often related to the lower Km values observed for these larger substrates. For example, mandelate racemase (6.2 and 6.5 × 105 M−1 s−1 for S- and R-mandelate91) and the M. tuberculosis homologue of AMACR (5.23 × 106 and 6.0 × 106 M−1 s−1 for S- and R-ibuprofenoyl-CoA,146 respectively). Finally, N-succinylamino acid racemases and N-acetylamino acid racemases exhibit highly variable kcat/Km values (Table 1).
It is noteworthy that even the most efficient racemases/epimerases have kcat/Km values well below the theoretical diffusion-controlled maximum of ∼1 × 109 M−1 s−1.169 As proton-transfer reactions are extremely fast (between 5 × 109 and 1 × 1011 M−1 s−1),86 rates may be limited by binding of substrate, release of product or conformational changes in the protein. A survey of kcat/Km values for various enzymes169 shows that, for most enzymes, they are around 105 to 109 M−1 s−1, with the most efficient enzyme (superoxide dismutase) having a kcat/Km of 7 × 109 M−1 s−1. Moreover, kcat/Km values for most enzyme-catalysed reactions appear to be diffusion-limited.169 There have been few detailed kinetic studies on racemases/epimerases but studies on mandelate racemase using mandelate as a substrate show that both kcat and kcat/Km are affected by increasing the viscosity of the solvent.170,171 This indicates that both binding of substrate and release of product are partly rate-limiting, although the effects on kcat are more extensive than those on kcat/Km indicating that that release of product is more sensitive to solvent viscosity than binding of substrate.172 In contrast, poorer substrates of mandelate racemase91 or less active mutants of the enzyme173 tend to be unaffected by increasing solvent viscosity, suggesting that rates are limited by the chemical reaction or other processes, e.g. conformational changes in the protein. Although the kcat/Km values for mandelate racemisation is relatively modest (6.2 and 6.5 × 105 M−1 s−1)91 compared to these other enzymes, it should be noted that racemisation of mandelate is a ‘difficult’ reaction as judged by the estimated half-life for the spontaneous uncatalysed reaction of 9.8 × 104 year.169 Thus, mandelate racemase is providing a considerable enhancement (an effective molarity of ∼4.87 × 106 M). It is unclear whether racemases/epimerases with lower kcat/Km values are limited by diffusion, chemical reactivity or other processes, or whether these low efficiencies result from a low amount of active enzyme within the enzyme preparation.
Drug design strategies for inhibiting racemases and epimerases
As noted above, many racemases and epimerases are drug targets for various diseases. The following is a survey of different strategies for the development of inhibitors.Substrate/product analogues
Exploiting the differences in side-chain conformation of different racemase/epimerase substrate stereochemical isomers can be a particularly fruitful strategy for the development of inhibitors. A significant advantage of these inhibitors is that they are achiral when identical sidechains are used. The substrate/product analogue approach works particularly well for racemases/epimerases possessing discrete side-chain-binding pockets for the different stereoisomers, e.g. mandelate racemase139 and M. tuberculosis α-methylacyl-CoA racemase (MCR).149 It can also work for enzymes with more subtle changes in side-chain conformation, e.g. aspartate racemase174 and glutamate racemase,175 although the potency of inhibition tends to be more modest. In many respects, substrate/product analogues are the equivalent of bisubstrate inhibitors of other enzymes,176 which often give rise to potent inhibition.Several substrate/product analogues have been reported as inhibitors of amino-acid racemases (Fig. 5). For example, citrate 40 was shown by X-ray crystallography to bind as a substrate/product analogue to aspartate racemase.174 Citrate 40 behaves as a competitive inhibitor, although the potency was very low (Ki = 7.4 mM vs. Km = 0.74 mM for L-aspartate). Pal et al. designed cyclic inhibitors of glutamate racemase, in which the ring mimicked the side-chain positions for the different stereoisomers of glutamate, including compound 41.175 This proved to be a partial non-competitive inhibitor, although potency was modest (Ki = 3.1 mM vs. Km = 1.41 mM for substrate S-glutamate).175 In contrast, substrate/product analogues were poor inhibitors of serine racemase (e.g.42, mixed competitive inhibition; Ki = 167 mM and Ki’ = 661 mM vs. Km = 19 mM)188 and proline racemase (e.g.43, non-competitive inhibition; Ki = 111 mM vs. Km = 5.7 mM).188 Proline racemase is known to have an extremely confined active site in the ‘closed form’ of the enzyme,26,27 which binds substrates and inhibitors.
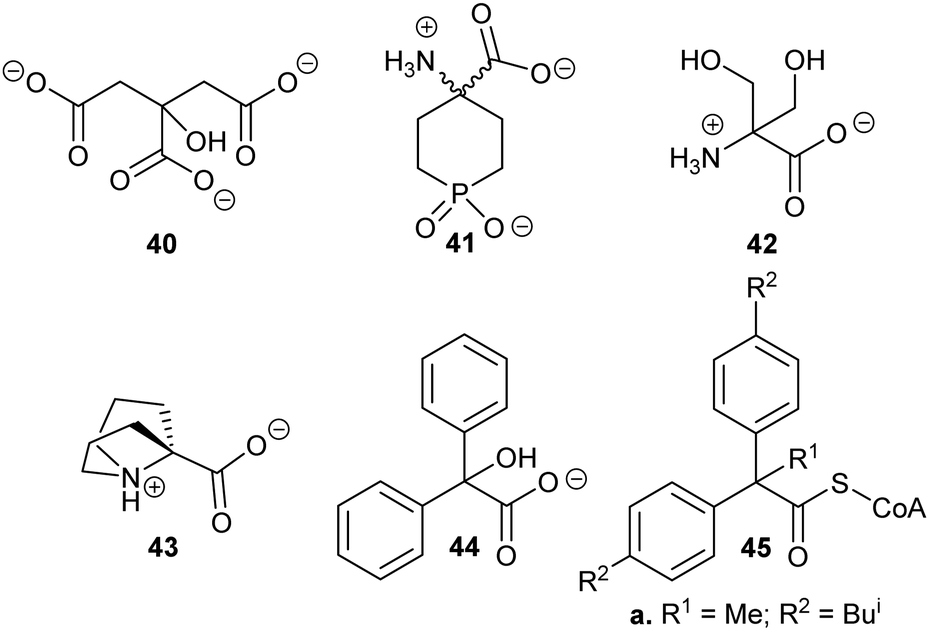 | ||
| Fig. 5 Structures of representative substrate–product analogues which are inhibitors of aspartate racemase (40),174 glutamate racemase (41),175 serine racemase (42),188 proline racemase (43),188 mandelate racemase (44)139 and M. tuberculosis α-methylacyl-CoA racemase (MCR) (45).149 | ||
There have only been two substrate/product analogue studies on non-amino-acid racemases. Mandelate racemase substrate/product analogues139 bind with similar affinity to the substrate [e.g. benzilate (2,2-diphenyl-2-hydroxyacetate) 44, Ki = 0.67 mM vs. Km = 0.70 and 0.54 mM for R- and S-mandelate, respectively139]. Similarly, a substrate–product analogue of ibuprofenoyl-CoA (Fig. 5, 45a) was a competitive inhibitor of the M. tuberculosis homologue of AMACR (MCR) and showed about a 6-fold increase in binding affinity (Ki = 16.9 μM vs. Km = 106 μM) compared to ibuprofenoyl-CoA, undoubtedly due to the side-chain of the inhibitor binding to both the R- and S- subsites.149
Enhancing acidity of the α-proton and alternative substrates
A number of racemases/epimerases have alternative substrates which undergo changes in stereochemical configuration8,91,127,164,189 or elimination.63,82,120–124,128,161,164 Efficiency of inhibition is dependent on concentrations of inhibitor and their catalytic efficiency as substrates (kcat/Km). Alternative substrates are usually competitive inhibitors (for example see127), which means that inhibition can be overcome by high concentrations of the substrate whose conversion is being inhibited.Efficient inhibition can be achieved by increasing the acidity of the Cα–H, e.g. by use of trifluoromethyl group (Fig. 6, 47 and 48).136,190 The trifluoromethyl group lowers the energy of the enolate intermediate81 in the AMACR reaction;136 intermediates generally bind tightly to enzymes and more closely resemble the transition states of the reaction.94 The presence of a sulfur atom immediately adjacent to the substrate Cα–H is also an effective strategy for increasing acidity (vide infra, Fig. 29).165
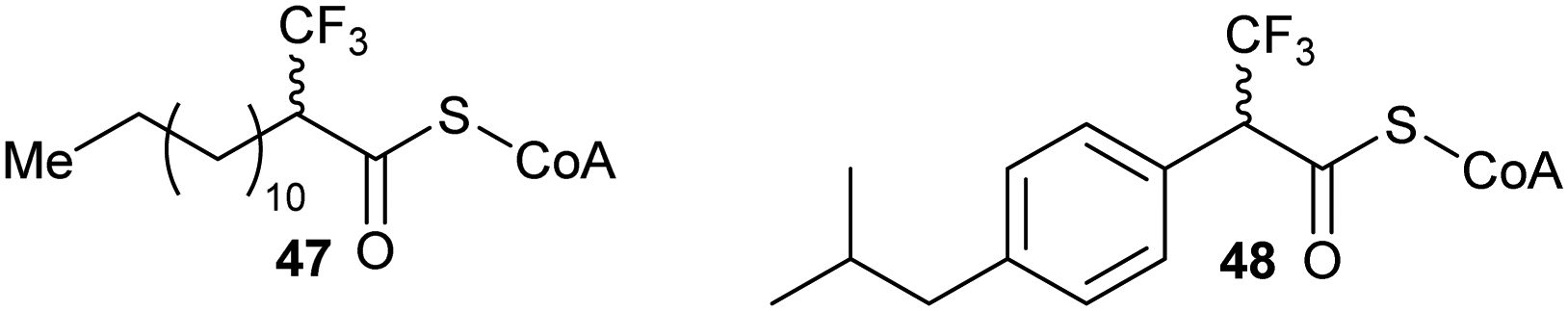 | ||
| Fig. 6 Representative inhibitors with increased Cα–H acidity.136,165,190 | ||
Preventing the removal of the α-proton
These types of inhibitors fall into two types: those in which the Cα–H has been replaced by an alternative group and those with neighbouring groups which decrease the acidity of the Cα–H. A number of different groups have been used to replace the Cα–H (in addition to the substrate/product analogues with a second side-chain noted above), including fluorine atoms, e.g.49,191 and methylene groups, e.g.50192 (Fig. 7).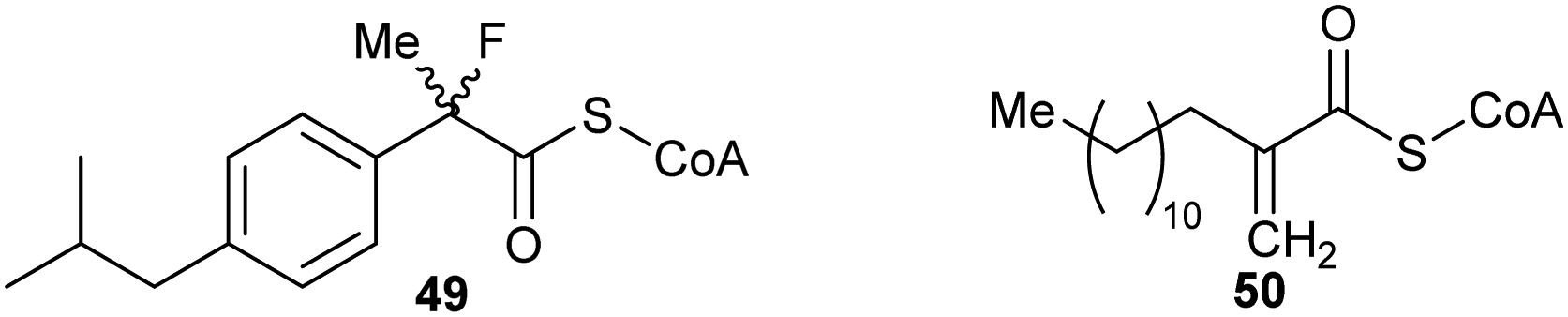 | ||
| Fig. 7 Representative inhibitors in which the Cα–H is replaced.191,192 | ||
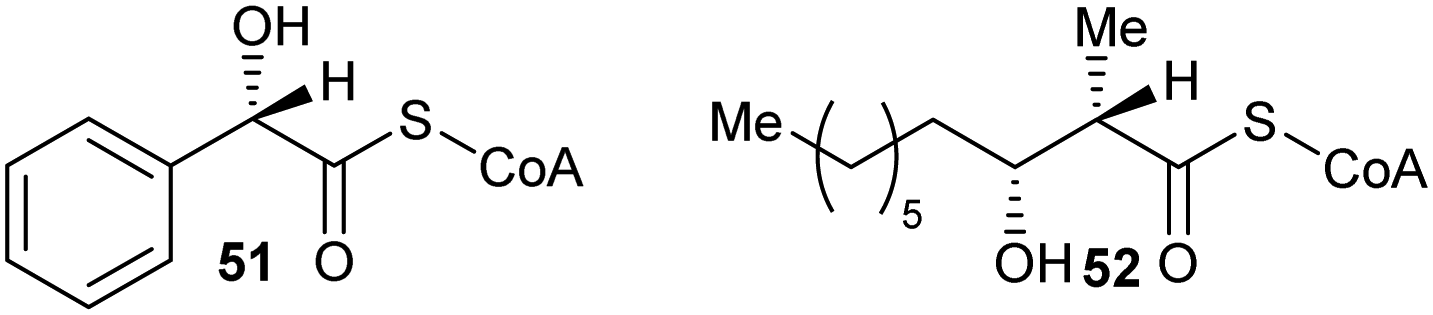
Inhibitors can also have substituents adjacent to the Cα–H, which raise the energy of the deprotonated intermediate, such as hydroxy groups as exemplified by 519,164 and 52164 (Fig. 8). Exchange of the Cα–H was shown not to occur by incorporation studies in 2H2O and 1H NMR analyses when 51 and 52 were tested as substrates for AMACR.9,164 In all cases, these approaches tend to give rise to moderate inhibitors, as judged by the ratio of IC50/Km or Ki/Km values.
 | ||
| Fig. 8 Representative inhibitors in which the acidity of Cα–H is decreased.164,191 | ||
Transition-state and intermediate analogues
Transition-state analogues are widely recognised as potent drugs.135,193,194 This approach has been relatively under-used as a strategy for inhibition of racemases and epimerases, although the few examples show that highly potent inhibitors can be obtained.An early example is proline racemase, which is inhibited by pyrrole-2-carboxylate 53 and Δ-pyrroline-2-carboxylate 27 (reviewed in ref. 135) (Fig. 9A). Relatively high concentrations of these compounds are required for inhibition in vitro of the enzyme (about 10 × that of substrate) and they should be considered as inhibitors in which the Cα–H is replaced (vide supra). X-ray crystallographic analysis showed that pyrrole-2-carboxylate binds within the T. cruzi active site (Fig. 9B) between the catalytic bases Cys-130 and Cys-300.195 However, despite its relatively low potency, pyrrole-2-carboxylate 53 reduced invasion of T. cruzi in infected mammalian cell models and also reduced differentiation of the parasite from the amastigote form into trypomastigotes.28 A number of more water-soluble analogues (e.g.54 and 55) were tested for their ability to inhibit the enzyme but these proved to have lower potency.26 Compounds 53, 54 and 55 had similar lipophilicity (calculated logP values of −2.41, −2.33 and −2.20, respectively) and the loss of inhibitory activity is likely to be related to the difficulties of accommodating the bulky halogen in the highly restricted active site. The halogen atom in 54 and 55 is also likely to force the carboxylate group out of plane, and this is expected to have a significant impact on binding affinity.
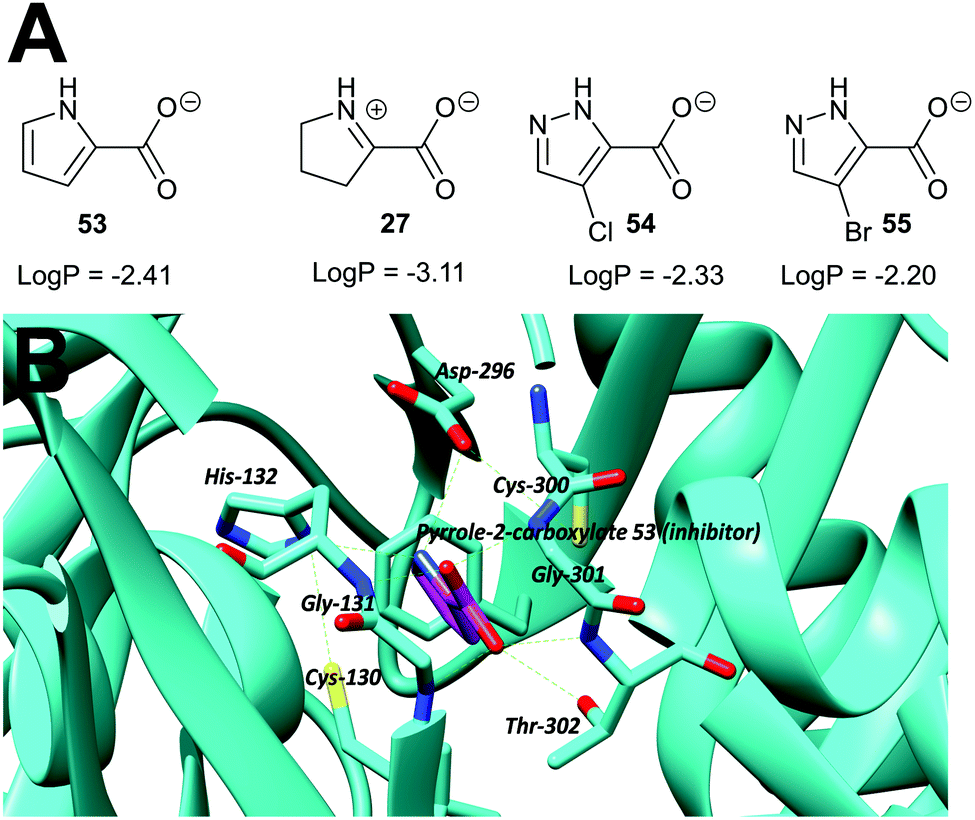 | ||
| Fig. 9 (A) Structures of proline racemase transition-state analogues.135 LogP values were calculated using: https://www.molinspiration.com/cgi-bin/properties. LogP, log10 (ratio of concentrations of drug in octan-1-ol and water at equilibrium); (B) X-ray crystal structure of pyrrole-2-carboxylate 53 bound within the active site of proline racemase from T. cruzi,195 showing the catalytic bases Cys-130 and Cys-300. Hydrogen bonds are shown as green dashed lines. | ||
A more recent example of the use of transition-state analogues are the carbamate inhibitors of α-methylacyl-CoA racemase (Fig. 10),191 which mimic the transition state (or enolate intermediate), giving rise to highly potent inhibition.127,164,191 Although the carbamate inhibitor 56 is by far the most potent AMACR inhibitor reported to date, it has limited utility because acyl-CoAs violate Lipinski guidelines and inhibitors are delivered to cells as the carboxylic acid pro-drug. Unfortunately, the acid pro-drug in this case would be a carbamate 57 which may readily decompose164 especially under acidic conditions or in the presence of cellular nucleophiles.
 | ||
| Fig. 10 Structure of the enolate intermediate analogue as an inhibitor of AMACR191 and the unstable carbamate pro-drug. | ||
There are several other examples of using analogues of the deprotonated intermediate as inhibitors. For example, the conversion of mandelate by mandelate racemase is proposed to go through an aci-carboxylate intermediate (Fig. 11, 58).196 Several mandelate racemase inhibitors of this type have been reported, including the phosphonate inhibitors196,197 such as the highly potent inhibitor 59 (Ki = 4.7 μM vs. Km of 1.0 and 1.2 mM for R- and S-mandelate, respectively). The phosphonate group in 59 possesses two negatively charged oxygen atoms, and hence resembles the aci-carboxylate intermediate 58. Similarly, cupferron 60 and N-hydroxyformanilide 61 also act as analogues of the deprotonated intermediate 58 because they have an extended planar system of sp2-hybridised atoms, whilst benzohydroxamate 62 is a hydroxamate. Inhibitors 59–62 (Fig. 11) strongly ligate to the metal in the active site of mandelate racemase (Ki values of 2.7, 2.8 and 9.3 μM, respectively).198
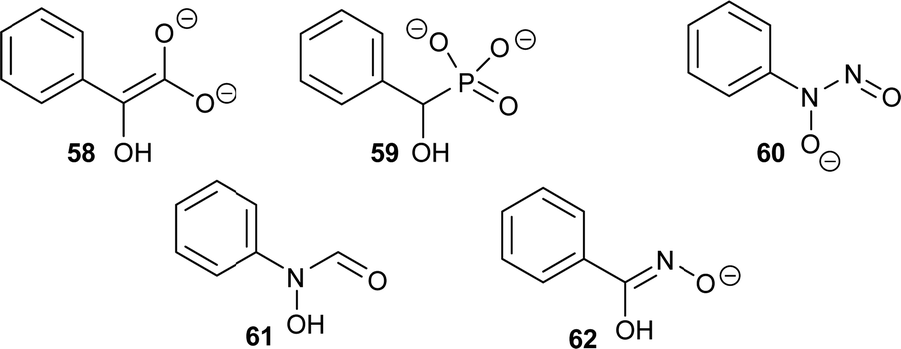 | ||
| Fig. 11 Inhibitors of mandelate racemase (59–62)196–198 resembling the aci-carboxylate deprotonated intermediate 58.196 | ||
Allosteric inhibition
Allosteric inhibition arises from inhibitors binding somewhere other than at the enzyme active site. The uncompetitive type of inhibition observed through enzyme kinetics arises from binding of the inhibitor to the enzyme-substrate complex with (almost) no binding to unoccupied enzyme199 and, hence, must arise from binding at an allosteric site.Glutamate racemase is the only racemase/epimerase for which confirmed allosteric inhibitors have been reported. Lundqvist et al. identified an uncompetitive inhibitor 63 during a high-throughput screening campaign on the H. pylori enzyme (Fig. 12).200 The inhibitor-binding site is remote from the active site.21,200 A second cryptic inhibitor-binding site was subsequently identified in the B. anthracis enzyme by virtual screening, which led to the identification of pyridine-2,6-dicarboxylate (dipicolinate) 64 as an inhibitor (Fig. 12), with Ki = 1.9 mM.25 Further studies on 37 showed that inhibitor binding resulted in the active-site Cys185 adopting a conformation in which the SH group points away from glutamate Cα–H.21 It is also noted that some uncompetitive inhibitors of α-methylacyl-CoA racemase were recently identified (vide infra, Fig. 15),163 implying that they bind to an allosteric site, although the exact binding site has not been confirmed.
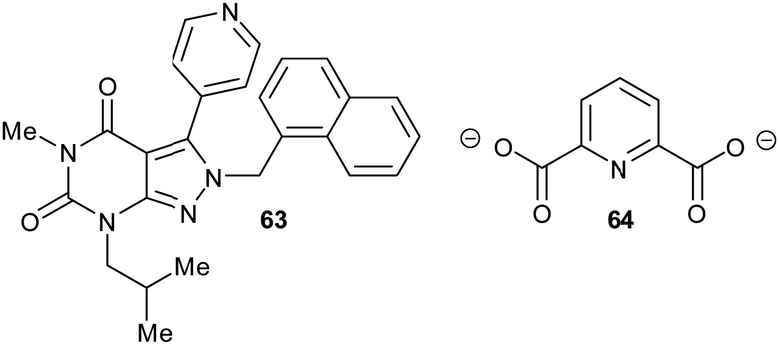 | ||
| Fig. 12 Structures of the allosteric inhibitors 63 (H. pylori glutamate racemase)200 and pyridine-2,6-dicarboxylate 64 (B. anthracis glutamate racemase).25 The ionisation state of 64 which is shown is that used in the virtual screen. | ||
Covalent inhibition
Inhibitors which form a covalent bond to their targets are enjoying a resurgence because of their potential for long-lasting effects and strong affinity for the target, amongst other benefits.201–207 Indeed, around 30% of all approved clinical drugs acting on enzymes are covalent inhibitors.202,207 Covalent inhibitors can cause either reversible or irreversible inhibition of their target.204–208 There is a perception that covalent inhibitors are non-selective and hence are less useful. However, studies have shown that high selectivity for the target enzyme can be achieved.203–210 Modification of the substituents around the electrophile can also further enhance selectivity,210–212 especially for electrophiles modifying cysteine residues212–215 (which are the catalytic bases in many cofactor-independent amino-acid racemases and epimerases5,18,20,22). Electrophilic properties can be predicted using the ‘electrophilicity index’.209Covalent inhibition of racemases and epimerases has been previously investigated. Both diaminopimelate epimerase20,124 and α-methylacyl-CoA racemase127,152 have been shown to be inhibited by non-specific protein-modification agents. In each case, these are cysteine-reactive compounds such as iodoacetamide, ebselen (2-phenyl-1,2-benzoisoselenazol-3(2H)-one) and ebselen oxide. It is also noted that mandelate racemase undergoes covalent inhibition by 3-hydroxypyruvate because of formation of an imine between the inhibitor and Lys-166, one of the active-site bases.216 There have also been several attempts to design irreversible inhibitors rationally, most notably the aziridine inhibitors of diaminopimelate epimerase.20,124,217,218 A recent example of rational covalent inhibitor design is seen with O-ureidoserine racemase (which interconverts S- and R- O-ureidoserine 65), which is irreversibly inhibited by oxiranes R- and S-66 to give covalent adducts (Scheme 10).46
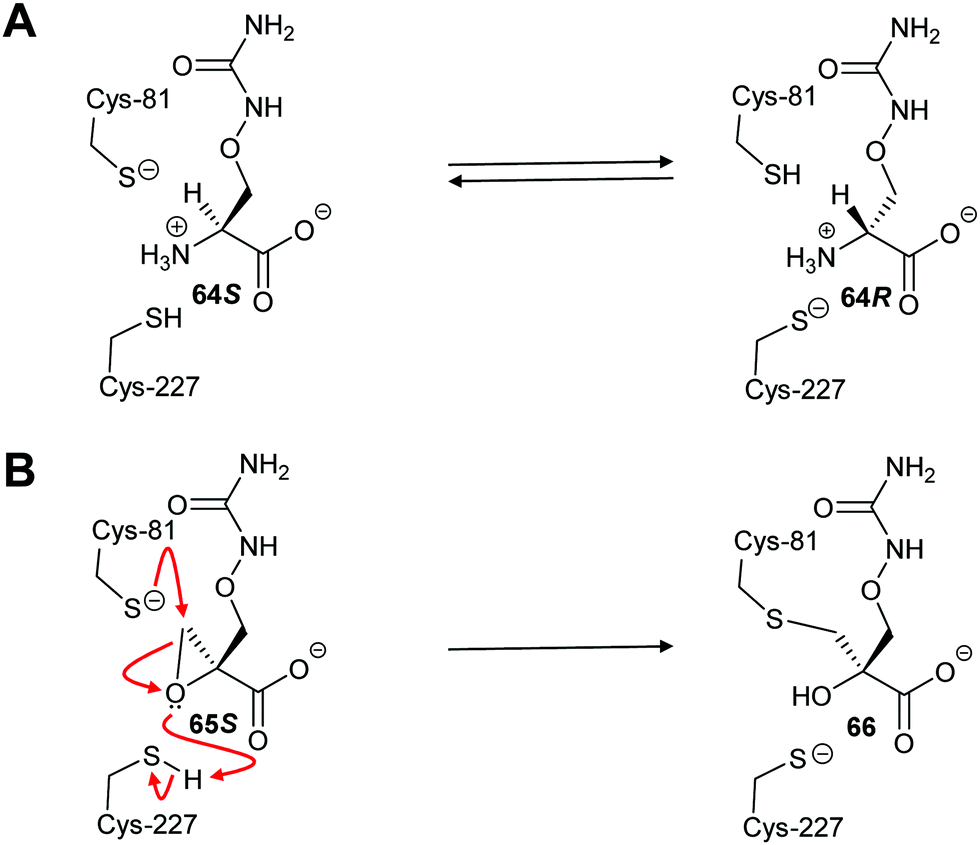 | ||
| Scheme 10 (A) Interconversion of O-ureidoserine substrate isomers 64S and 64R by O-ureidoserine racemase. (B) Inactivation of O-ureidoserine racemase by epoxide 65S to give covalent adduct 66. The roles of the Cys residues are reversed for the enantiomeric epoxide 65R.46 | ||
An irreversible inhibitor of B. subtilis glutamate racemase was also identified by virtual screening (vide infra),23,24 and was proposed to bind close to the catalytic cysteine residues.22 The inhibitor is proposed to modify irreversibly one of these thiols by conjugate addition (a.k.a. Michael addition).202,205,206 The hit compound 67a and several analogues 67b–67d (Scheme 11A) were subsequently shown to be irreversible inhibitors.22 Compounds 67a and 67c proved to be non-saturating inhibitors. In contrast, 67b and 67d displayed saturating inhibition, consistent with modification of active site residues. Further experiments showed that inhibition was reversible, consistent with a reversible conjugate addition via enolate 68a to give the product 69a (Scheme 11B). Mass spectrometric analysis of wild-type and C74A mutant glutamate racemase following incubation with 67a confirmed modification of Cys-74, one of the active-site bases. Compound 67a was unreactive with 2-mercaptoethanol under the assay conditions,22 showing that conjugate addition to thiols only occurred in the presence of the high nucleophilic Cys-74 in the enzyme active site. The rhodanine warhead (Scheme 11A) is recognised as a common motif found in pan-assay interference compounds (PAINs), which give rise to false positive or intractable leads in high-throughput screening campaigns.219 These rhodanine glutamate racemase inhibitors showed activity against various bacterial strains, including various methicillin-resistant S. aureus strains.22
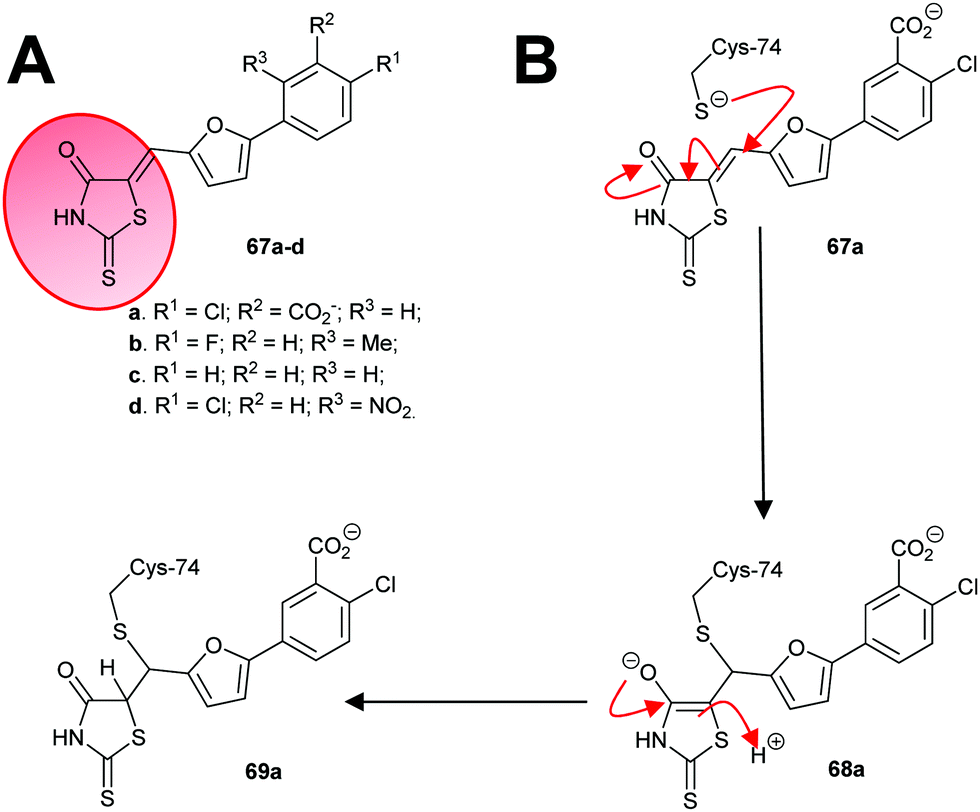 | ||
| Scheme 11 (A) Structures of irreversible inhibitors of B. subtilis glutamate racemase.22 The rhodanine motif is highlighted in red; (B) inactivation of glutamate racemase by 68a by 1,4-conjugate addition. | ||
In a second study, 3-chloroalanine 70 (β-chloroalanine; a poor inhibitor of PLP-dependent alanine racemase134) was shown to irreversibly inactivate glutamate racemase from M. tuberculosis.134 Non-saturating kinetics where observed for the S-isomer with a second-order rate constant of 2.7 M−1 s−1. Mass spectrometric analysis of peptides showed that 3-chloro-S-alanine (70S) reacted at Cys-185, while 3-chloro-R-alanine (70R) reacted at Cys-74. In the glutamate racemase reaction, R-glutamate is deprotonated by Cys-74 whilst S-glutamate is deprotonated by Cys-185 during enantiomerisation, i.e. the active-site Cys acting as an acid is derivatised by 3-chloro-alanine 70.
The authors proposed134 that the adduct was a pyruvate derivative, based on the observation that pyruvate 71 was generated upon treatment of the enzyme with 3-chloro-alanine 70 but their proposed mechanism is very unlikely. Two more likely mechanisms can be envisaged (Scheme 12, pathways A and B) based on the observed increase in mass of ∼87 Da. In pathway A, removal of the Cα–H of 70S by Cys-74 results in elimination of HCl, yielding the aminoacrylate complex 72. This is followed by conjugate addition of Cys-185 to give 73. However, complex 72 is achiral and non-specific derivatisation of the active site Cys residues might be expected if 72 resulted in alkylation. Pathway B, via the aziridine intermediate 74, preserves the chirality of reaction and gives rise to the same adduct 73. However, the active site Cys residues are relatively distant from the α-amino group, making pathway B less likely. Digestion of the derivatised enzyme and mass spectrometric analysis shows the presence of nitrogen within the enzyme-inhibitor adduct, discounting the possibility that the adduct is a pyruvate derivative (Scheme 12, pathway C). The observed pyruvate 17 generated in the reaction arises from tautomerisation of aminoacrylate 72 to the imine followed by hydrolysis, i.e.70 behaves as a substrate as well as an inhibitor.220
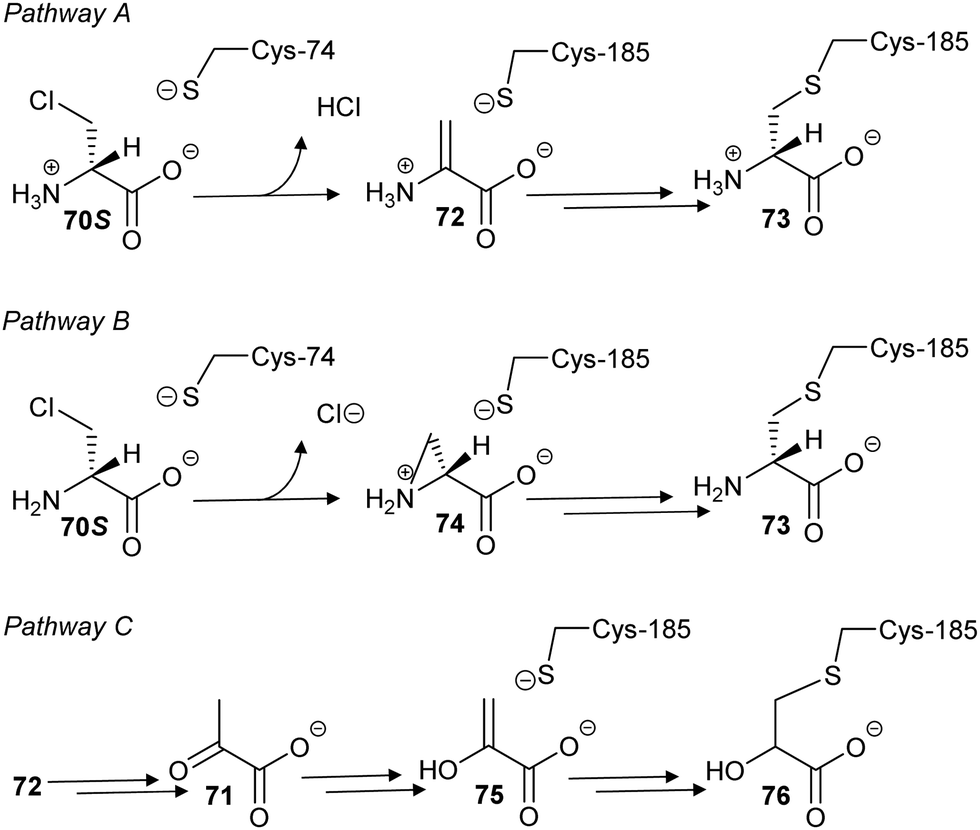 | ||
| Scheme 12 Proposed mechanisms of glutamate racemase inactivation by 3-chloro-S-alanine 70S (β-chloro-S-alanine). | ||
Similarly, two covalent inhibitors (77 and 78) of T. cruzi proline racemase were identified by virtual screening.26 The inhibitors were proposed to modify the active-site cysteine residues26 by conjugate addition202,205,206 and this was subsequently confirmed by X-ray crystallography.27 The active compounds (Scheme 13A) each have a double bond in conjugation with a carboxylate and a ketone26,27 and X-ray crystallography showed that conjugate addition occurred towards the ketone.27 This is unsurprising as ketone carbons are more δ+ than carboxylic acids/carboxylates and hence conjugate addition is expected to occur towards the ketone. The most active compound of those subsequently investigated (NG-P27, 79)27 was active against T. cruzi in infected mammalian cells. It is also notable that one of the original compounds26 (5-bromo-4-oxopent-2-enoate 78) is divalent and reacts with both active-site cysteine residues, cross-linking the enzyme to give adduct 80 (Scheme 13B).27
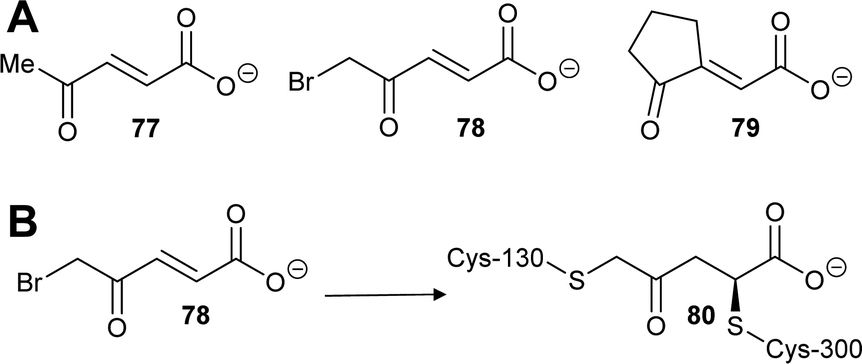 | ||
| Scheme 13 (A) Structures of highly active covalent inhibitors of proline racemase;26,27 (B) Reaction of 78 with catalytic cysteine residues in proline racemase by conjugate addition and SN2 reaction to give a cross-linked adduct 80.27 | ||
Virtual screening and structure-based fragment screening
Virtual screening of drug targets with a compound library is a well-established method in drug discovery.221–225 These approaches can utilise artificial intelligence to optimise the process223 or negative design222 to remove compounds which are poor prospects.There are only a few examples of virtual screening being used for identification of inhibitors of racemases/epimerases and all have been for amino-acid racemases. For example, Skariyachan et al. conducted a screen of a virtual natural products library against diaminopimelate epimerase, amongst several other targets, identifying limonin 81 as a hit (Fig. 13).226 Limonin 81 and several other hits showed dose-dependent activity against a clinical strain of multi-drug resistant Acinetobacter baumannii.
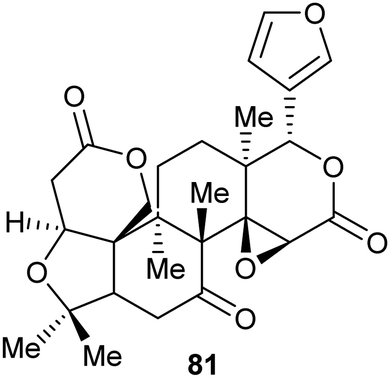 | ||
| Fig. 13 Structure of limonin 81.226 | ||
Studies on B. subtilis glutamate racemase used ab initio quantum mechanics/molecular mechanics to probe transition states in the reaction.23 A strong correlation between computational and experimentally determined binding of known inhibitors was observed. The same study23 used a enzymatic transition state conformation in a virtual screen of over one million compounds followed by experimental testing. Although no tight-binding inhibitors were identified, several common motifs for competitive inhibitors were identified. A subsequent virtual screening study on the same enzyme24 identified several competitive inhibitors such as 64 and 82, with the two most potent inhibitors, 83 and 84, having Ki values of 59 and 42 μM (Fig. 14).
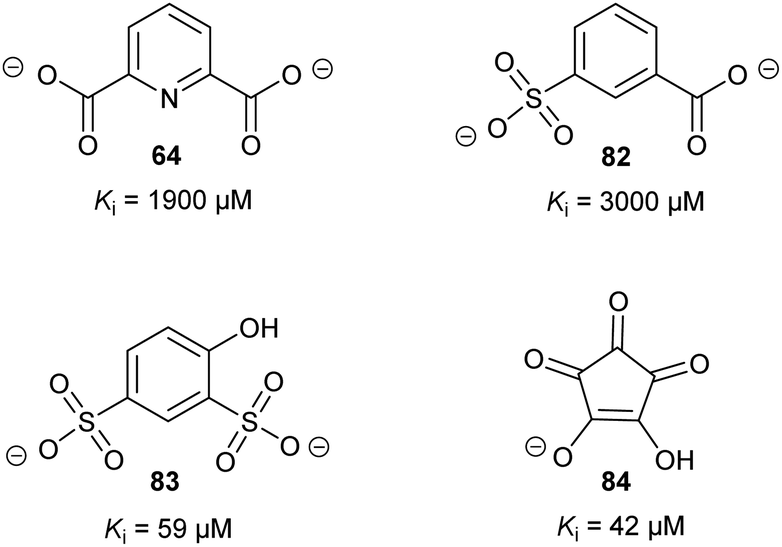 | ||
| Fig. 14 Representative inhibitors of B. subtilis glutamate racemase identified using virtual high-throughput screening.23,24 The inhibitor ionisation state shown are those that were used in the virtual screen. | ||
 | ||
| Fig. 15 Representative structures of pyrazoloquinoline and pyrazolopyrimidine AMACR inhibitors.16385a, R1 = R2 = H, uncompetitive inhibition, Ki = 4.8 ± 0.7 μM; 85b, R1 = R2 = F, mixed competitive inhibition, Ki = 2.4 ± 0.9 μM; 86, uncompetitive inhibition, Ki = 4.6 ± 0.4 μM. | ||
Virtual screening has also been used against proline racemase from T. cruzi, the causative agent of Chagas disease (vide supra).26 Proline racemase without a ligand exists in an ‘open’ conformation. Upon binding of an inhibitor, a ‘closed’ conformation is adopted, such that a very restricted active site is produced, which prevents design of inhibitors using standard approaches. The virtual screening study generated forty-nine intermediate conformations en route from the ‘open’ to ‘closed’ conformations. Four of these conformations were used in virtual screens of 31000 compounds. These screens led to the identification of covalent inhibitors (vide supra, Scheme 13), which showed dose-dependent activity against T. cruzi in infected mammalian cells.26,27
High-throughput screening and related approaches
High-throughput screening is an under-utilised approach to discovering inhibitors of racemases and epimerases. High-throughput screening offers a number of advantages, including the possibility of discovering inhibitors which are not competitive (which is the mode of inhibition often observed for active-site-directed inhibitors).227Several different in vitro assays have been used in discovery campaigns. Release of tritium (3H+) from a radiolabelled substrate into solvent was used in a screen of ∼5000 compounds against human α-methylacyl-CoA racemase (AMACR).152 Crucially, the assay requires several steps, including chromatographic separation of residual (acyl-CoA) substrate from tritiated water product. Therefore, this assay is not ideally suited for high-throughput or fragment-screening campaigns. This study152 identified a number of non-specific protein-modifying and degrading agents, such as ebselen, ebselen oxide and Rose Bengal.127,152
A subsequent high-throughput screen on human AMACR163 made use of an eliminating substrate 37 (vide supra, Scheme 9) producing 2,4-dinitrophenoxide 38.127 Conveniently, this allowed identification of inhibitors based on absorbance changes over the time course in a continuous assay. The screen identified a series of mixed competitive and uncompetitive pyrazoloquinolines, e.g.85, and pyrazolopyrimidines, e.g.86 (Fig. 15). The use of a chromogenic substrate127 allows real-time monitoring of the enzymatic reaction but substrates of this type can only be used with a few racemases/epimerases.
Two studies have made use of high-throughput screens with coupling enzymes. The first study, by Lundqvist et al., conducted a high-throughput screen of 385861 compounds against H. pylori glutamate racemase.200 No details of the actual screen are given but the authors used two different assays for assessing identified inhibitor activity: conversion of S- to R-glutamate was coupled to UDP-N-acetyl-muramic acid-alanine: R-glutamate ligase (MurD) and purine nucleoside phosphorylase with monitoring of the reaction at 360 nm. In the second assay, conversion of R- to S-glutamate was coupled to S-glutamate dehydrogenase with spectrophotometric monitoring of conversion of NAD+ to NADH. These screens led to the identification of an uncompetitive inhibitor (vide supraFig. 12, 63), which was subsequently shown to exert its effect by changing the conformation of the catalytic base, Cys-185, such that it points away from the glutamate substrate Cα–H.
In a second example228 of a coupled assay, dTDP-6-deoxy-D-xylo-4-hexopyranosid-4-ulose 3,5-epimerase (RmlC) was coupled to the subsequent enzyme in the biosynthetic pathway, which is NADP+-dependent, with activity being followed by decreasing fluorescence of NADPH at 460 nm. This led to the identification of a series of inhibitors, including some with potency in the nM range e.g.87 (Fig. 16).228 Use of coupling enzymes, such as in these examples, is a standard approach in high-throughput and fragment-screening campaigns227 (vide infra), although it is always necessary to check if the hits are inhibiting the desired target or the coupling enzyme.
 | ||
| Fig. 16 Structure of the most potent hit 87 identified by high-throughput screening inhibiting RmlC. 61 is a fully reversible, competitive inhibitor with IC50 = 200 nM.228 | ||
Racemases used in biotechnological applications have been assayed in several different ways, typically using oxidase enzymes of various types. These assays could be adapted for high-throughput screening for inhibitors. For example, mutant mandelate racemases were assayed using mandelate dehydrogenase, which uses NAD+. Conveniently, the ketoacid product can be assayed using 2,4-dinitrophenylhydrazine (2,4-DNPH) at alkaline pH with the final 2,4-dinitrophenylhydrazone product absorbing at 450 nm.54 Ketoacids are also be produced by the action of amino-acid racemases and epimerases on amino-acid hydroxamate and other eliminating substrates82,128 (vide supra, Schemes 3, 4, 6 and 7). Alternatively, the NAD(P)H product from dehydrogenases can be assayed using diaphorases23,125 or by direct monitoring of absorbance or fluorescence.
Similarly, hydrogen peroxide is produced by several oxidative enzymes, including D-amino-acid oxidase, which can be conveniently assayed using horseradish peroxidase.174,229 Notably, an assay based on D-amino-acid oxidase/horseradish peroxidase was used to evaluate rationally designed inhibitors of proline racemase26 and in the high-throughput screening of alanine racemase.230 Similarly, a continuous assay for N-acetylamino-acid racemases was developed using the R- substrate 88R (Scheme 14).181 A stereoselective deacetylase was used to convert the S- product 88S to the corresponding S-amino acid 89S and acetate 90. L-Amino acid oxidase was used to produce H2O2 from 89S, which was quantified by the horseradish peroxidase-catalysed oxidation of dianisidine 90 to give the coloured oxidation product 91.
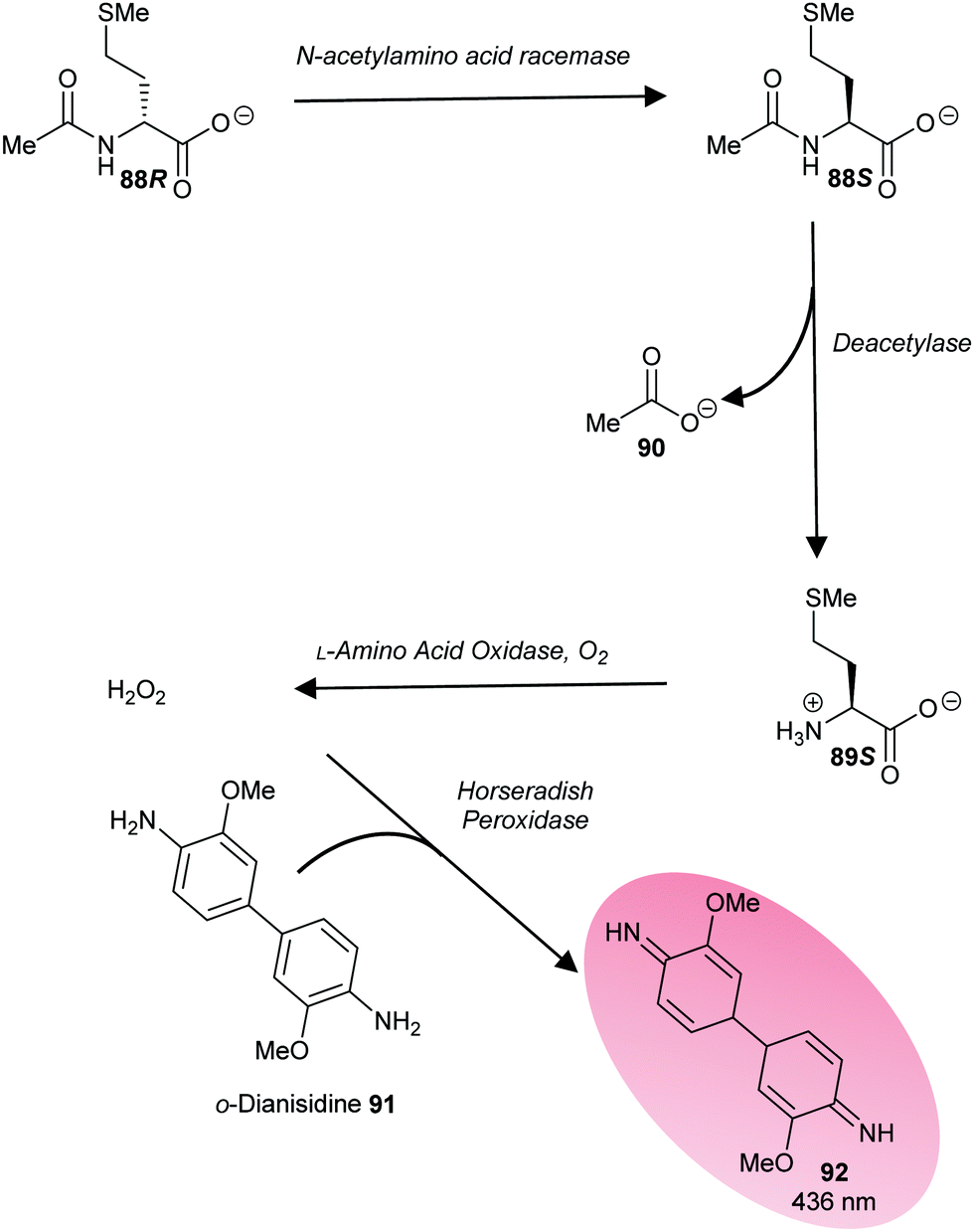 | ||
| Scheme 14 Continuous assay of N-acetyl-amino acid racemases using a three-enzyme coupling system to give a coloured product 92 absorbing at 436 nm.181 | ||
In additional, several racemases/epimerases eliminate HF from fluorine-containing substrates (vide supra, Scheme 8),120,121 which could potentially be assayed using fluoride sensors, although this can be challenging in aqueous systems.161 Finally, microscale, medium-throughput polarimetric assays offer the possibility of direct observation of the change in chirality142 during screening.
Inhibitors of racemases and epimerases
Racemases and epimerases play pivotal roles in metabolism and are excellent drug targets. The following is a survey of recent advances in drug development, focussing on recently reported small-molecule inhibitors. Inhibition of many amino-acid racemases/epimerases has been the subject of a recent excellent review20 and readers are referred to this and the above sections for details of studies on inhibition of diaminopimelate racemase,20,124,217,218,226 proline racemase,26,27,135,188 hydroxyproline epimerase,20 aspartate racemase,20 serine racemase,188 isoleucine epimerase20 and O-ureidoserine racemase.46 Studies on inhibition of mandelate racemase91,139,196–198,216 are detailed in the section on inhibition strategies above. There have been no reported studies on inhibition of EcL-DER, RacX, YgeA, McyF, YcjG or N-acetylmannoseamine-6-phosphate 2-epimerase or N-acetylamino-acid or N-succinylamino-acid racemases since 2015.PLP-dependent racemases
An important inhibitor of alanine racemase is D-cycloserine, a natural product used in the treatment of drug resistant tuberculosis.233,234 It is notable that the biosynthetic pathway for D-cycloserine contains a reaction catalysed by a racemase, O-ureidoserine racemase46 (vide supra, Scheme 10A). D-Cycloserine 93 is a relatively non-specific antibiotic and targets M. tuberculosis alanine racemase and D-Ala-D-Ala ligase.233,234 Inhibition of alanine racemase is proposed to occur by formation of the external aldimine 94 followed by reversible rearrangement to the ketimine 95 and isoxazole 96 (Scheme 15).233 Stereoselective isotope exchange with solvent is observed when the reaction is carried out in 2H2O, with D-cycloserine 93 incorporating deuterium at the α-position without a change in stereochemical configuration. Deuteration appears to arise by exchange of the α-proton. Incubations of alanine racemase with D-cycloserine 93 also result in the formation of isoxazole 97.233 The authors propose a complex rearrangement of the keto tautomer of 96 but this seems unlikely. A simpler explanation is that alanine racemase catalyses hydrolysis of aldimine 94 using a hydrogen-bonded water molecule, to form the linear aldimine 98 directly. This could undergo imine exchange to form 99 but a more likely scenario is that 97 is released from 98 which uses its hydroxylamine to form the aldimine complex 99 directly (the pKa for the conjugate acid of the α-NH2 and γ-O-NH2 groups are 9.14 and 3.16,235 respectively, and, hence, the γ-O-NH2 group will be uncharged at neutral pH). Thus, D-cycloserine 93 is both a substrate (undergoing α-proton exchange and hydrolysis) and an irreversible inhibitor of alanine racemase, consistent with the observation that M. tuberculosis alanine racemase is not fully inhibited even by high concentrations of 93.
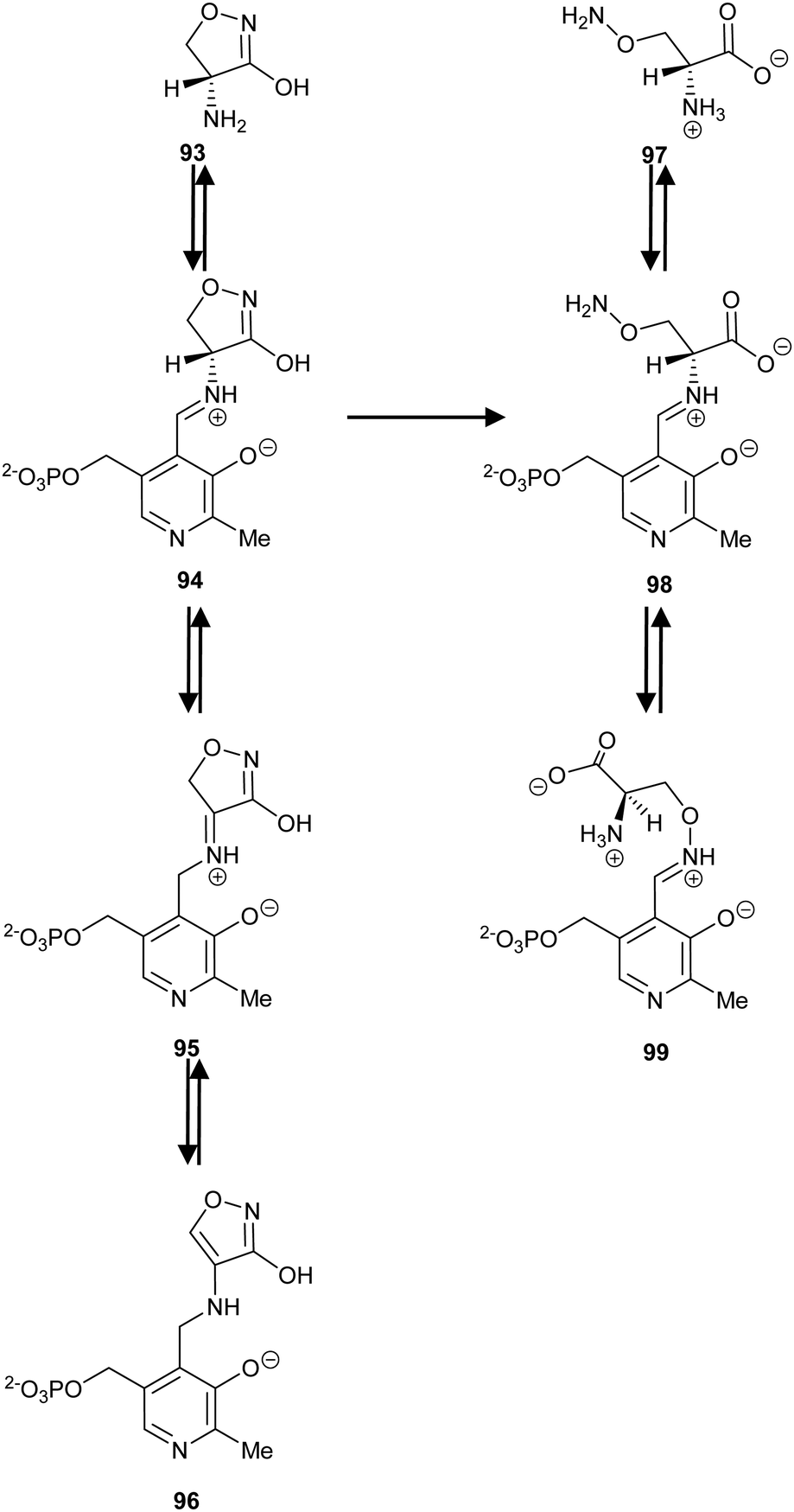 | ||
| Scheme 15 Inhibition of alanine racemase by D-cycloserine 93. Note that 96 was proposed to be directly converted into 99233 but it is more likely that 94 undergoes hydrolysis to form 98, which releases 97. This directly forms two different external aldimines, 98 or 99, respectively (see text for details). | ||
A series of tetrazole-peptide derivatives were also designed and synthesised as inhibitors of alanine racemase in bacterial cells (Fig. 17).236 The tetrazole group is a well-established bioisostere of the carboxylate group,236 and hence 5-(1-aminoethyl)tetrazole 100 should behave as an analogue of alanine. S- and R-5-(1-aminoethyl)tetrazole 100 (AET) were inactive when tested against a series of Gram-negative and -positive bacteria236 but this is unsurprising as it is known that alanine is imported into bacterial cells as an oligopeptide. Indeed, fosfalin 101 is delivered to bacterial cells as a “dipeptide” alafosfalin 102.231,236
 | ||
| Fig. 17 Structures of 5-(1-aminoethyl)tetrazole 100, fosfalin 101, alafosfalin 102, and dipeptide analogues 103a-d and tripeptide analogue 104.236 | ||
Therefore, a series of di- and tripeptide derivatives were synthesised and tested.236 The SS-Ala-Ala analogue 103a was active against several Gram-negative species, whilst the SS-norvalene-AET 103b, SS-Leu-AET 103c and SS-Phe-AET 103d analogues were active against several Gram-positive species. The SSS-Ala-Ala-AET analogue 104 showed similar activity to 103a. N-Succinyl derivatives of these peptide analogues were largely inactive. It was not determined if S- and R-5-(1-aminoethyl)tetrazole 100 or any of the peptide analogues were inhibitors of alanine racemase and hence the mechanism of antibacterial activity has not been confirmed.
High-throughput screening of small-molecule and fungal extract libraries against Aeromonas hydrophila alanine racemase has also been performed. This screen identified several previously unknown inhibitors (Fig. 18) of moderate potency (IC50 = 6.6 to 18.5 μM), including homogentisic acid 105 and hydroxyquinone 106.230D-Cycloserine 93 (the control inhibitor) had an IC50 of 5.4 μM under the conditions used in this screen. Kinetic analysis showed that homogentisic acid 105 was a competitive inhibitor (Ki = 51.7 μM) whilst hydroxyquinone 106 was a non-competitive inhibitor (Ki = 212 μM). These two compounds showed antibacterial activity against A. hydrophila, a Gram-negative anaerobic pathogen. Anabellamide 107 was a potent inhibitor of alanine racemase in vitro (IC50 = 6.6 μM) but was inactive in cellular assays (Fig. 18).
 | ||
| Fig. 18 Structures of alanine racemase inhibitors identified by high-throughput screening.230 Abbreviation used: Ph, phenyl. | ||
Subsequently, a study investigating inhibition of Lactobacillus buchneri isoleucine 2-epimerase by substrate/product analogues was reported.189 Two groups of inhibitors (Fig. 19) were investigated, based on the structure of the substrate S-isoleucine 108. These inhibitors fell into two classes: those with a single modified sidechain (109–111); and those with dual sidechains (112), which are similar to the substrate–product analogues that inhibit other racemases and epimerases.139,149,150,174,188,197,198 The 2R- and 2S- enantiomers of 109 were substrates of the enzyme, although these are converted with an efficiency of only ∼50% to 80% of that of the natural substrates (as judged by kcat/Km values180,189) and, hence, would not be very effective competitive inhibitors. On the other hand, 110 which possesses an additional methyl group on the sidechain was not a substrate but instead behaved as a pure competitive inhibitor (Ki values of 1.5 and 2.9 mM for the 2R and 2S enantiomers, respectively). Compound 111 which has a cyclic sidechain was also an alternative substrate and was converted with very similar efficiencies to 109 (as judged by kcat/Km values). The synthesised compounds with dual sidechains (112, n = 1, 2 and 3) were rather poor inhibitors, although potency increased with increasing size and hydrophobicity of the sidechain (Ki = 144, 19 and 11 mM, respectively).
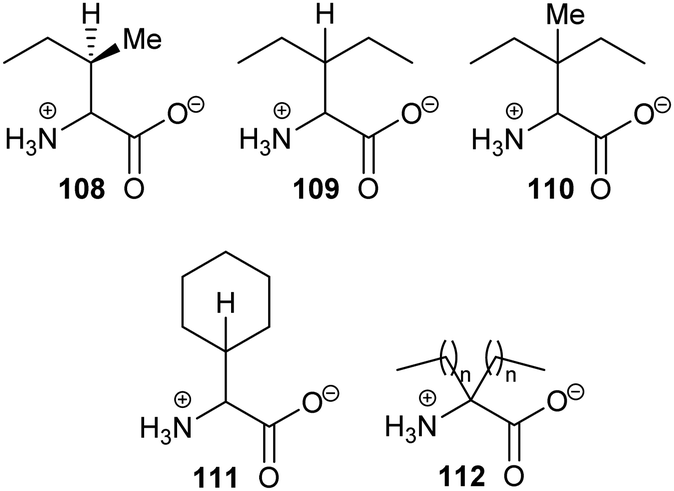 | ||
| Fig. 19 structures of isoleucine 108 and inhibitors of isoleucine 2-epimerase.189 For 112, n = 1, 2 and 3. | ||
A large number of studies have appeared on the biological and pathological role of serine racemase in the last five years35,69,129,241–244 but only one study has reported synthesis and evaluation of inhibitors.239 This study239 elaborated a potent hit (Fig. 20, 113) identified in a previous virtual screen.245 Of the synthesised compounds, five showed potent inhibition of serine racemase in vitro. Two of these compounds were similar in structure to the original hit and IC50 values were determined for the three other compounds (140, 270 and 280 μM for 114, 115 and 116, respectively). This compares to an IC50 value of 770 μM for malonate, a standard inhibitor. Further studies showed 114 reduced NDMA receptor activation by ∼1.4-fold, consistent with engagement of the target in vivo.
 | ||
| Fig. 20 Structures of inhibitors of serine racemase.239,245 | ||
Cofactor-independent racemases and epimerases
Malapati et al. have reported a series of medium-throughput screening studies on M. tuberculosis glutamate racemase using thermal-shift assays (Fig. 21).246–248 Structure–activity relationship (SAR) studies led to inhibitors 117–119 with low μM IC50 values. Non-competitive inhibition was assigned based on the observed changes within the thermal shift assay, although this was not confirmed by enzyme activity assays. Docking studies suggested that these compounds bound to an allosteric binding site.
 | ||
| Fig. 21 Structures of leads reported by Malapati et al.246–248 | ||
In addition, Duvall et al. reported phenotypic screening of a diversity-orientated synthetic collection (∼100000 compounds) against Clostridium difficile and other bacterial strains under anaerobic conditions.249 One of the hits (BRD0761, Fig. 22, 120) showed minimum inhibitory concentrations of 0.06–0.25 μg mL−1 (0.13–0.55 μM) against various C. difficile strains, with much higher MIC values against other anaerobes, while its epimer BRD3141 121 was also active (Fig. 22). BRD0761 inhibited uptake of [14C]-N-acetylglucosamine into bacteria in a dose-dependent manner, suggesting that it targeted bacterial cell wall biosynthesis. The target was identified from resistance mutants as glutamate racemase and a binding model was produced based on the X-ray crystal structure of H. pylori enzyme. Dosing of mice with 120 protected them from C. difficile infection.
 | ||
| Fig. 22 Structures of BRD0761 120 and BRD3141 121.249 | ||
A series of N-acetylglucosamines and N-acetylmannosamines, some with modified UDP moieties, have been previously developed as inhibitors but had modest potency (reviewed in ref. 253). Nieto-Garcia et al., reported a series of inhibitors in which the C6 hydroxy group was replaced with sulfur or selenium (Fig. 23).253 The diselenide inhibitor 122 proved to be highly potent (IC50 = 8.5 μM) compared to the other inhibitors (IC50 values of 1.9 to >10 mM). The dimeric monoselenide inhibitor 123 was much less potent (IC50 = 3.0 mM). The corresponding disulfide analogue 124 was also much less active than 122 (IC50 = 4.2 mM), showing the importance of the diselenide unit for potent inhibition. The much higher potency of 122 compared to the other inhibitors could be due to bond length or flexibility of the linker.253 Small-molecule diselenide bonds have been reported as having a bond length of 2.29 Å,254 while disulfide bonds (in proteins) have a corresponding bond length of 2.05 Å.255 It has also been suggested that van der Waals interactions and hydrogen bonding potential may also be important in determining inhibitory potency.253 Diselenide 122 was a competitive inhibitor with a Ki value of 15.7 μM.253
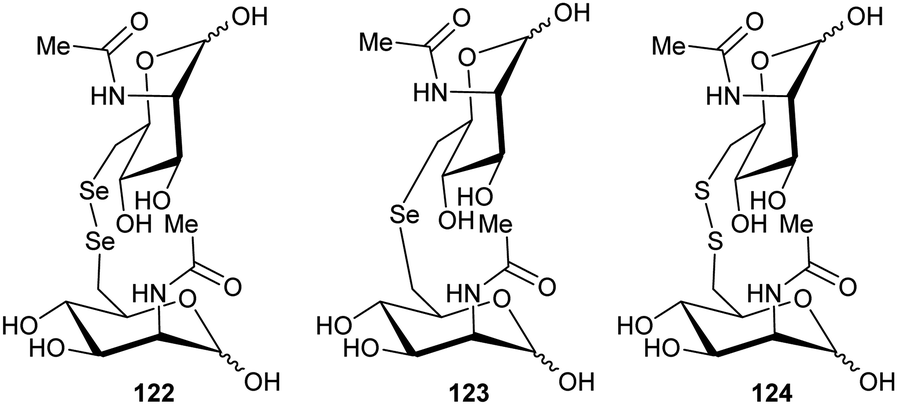 | ||
| Fig. 23 Structures of (di)selenide and disulfide inhibitors of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase.253 | ||
Hinderlich et al. reported a high-throughput screening campaign using a library of 41536 compounds and a luciferase assay to measure ATP depletion.252 The N-acetylmannosamine substrate was used at 33 μM, close to its Km value, with an average Z′ value (a measure of the ability of the assay to discriminate between a hit and random noise227,256) of 0.78. Compounds were screened at 13 μM, yielding 252 hits of which 174 were analysed using dose–response curves, yielding 46 inhibitors with IC50 < 33 μM. Further analysis and counter-screening against yeast hexokinase yielded several leads 125–128 (Fig. 24). The IC50 values did not significantly change with changing concentrations of ATP, suggesting that they were non-competitive. Modelling studies suggested that 125, 126 and 128 bound in the N-acetylmannosamine-binding site in the closed form of the enzyme, although 127 was larger than the available site, suggesting it might be bound to the open conformation.252
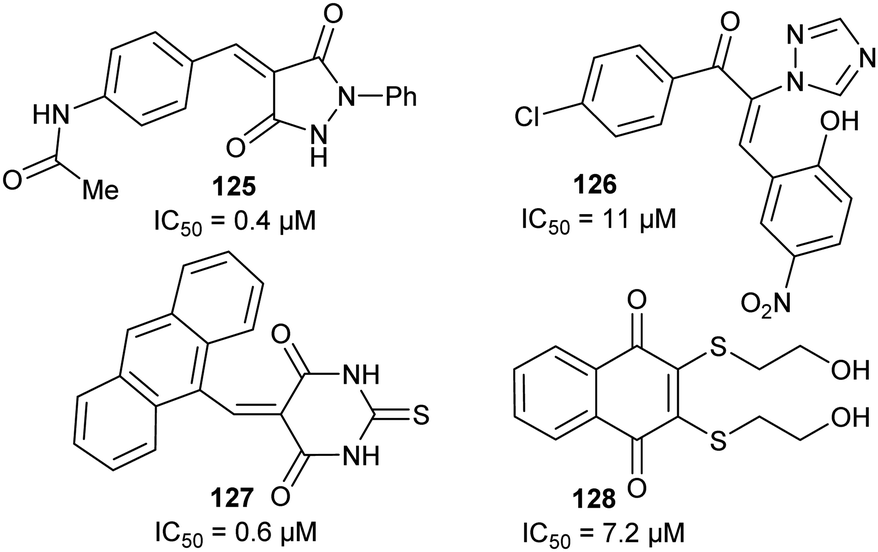 | ||
| Fig. 24 Structures of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase inhibitors.252 | ||
UDP-N-acetylglucosamine epimerase/N-acetylmannosamine kinase is also one of only two racemases or epimerases to be subjected to a fragment-screening campaign.257 A library of 281 fluorinated fragments were screened at 50 μM using 19F NMR and binding of inhibitor was confirmed by competition with N-acetylmannosamine and ATP, yielding 23 hits. Of these, compound 129 was also shown to inhibit in a coupled enzyme assay and so was chosen for development. Analogues of 129 were screened, leading to identification of 130 which was optimised to 131 (Fig. 25). Modelling of the binding of the inhibitor suggested that 131 bound to the active-site Mg2+ used in the kinase reaction, near the catalytic site. However, these compounds were not developed into more potent leads.
 | ||
| Fig. 25 Structure of hit 129 and derived inhibitors 130 and 131.257 | ||
Several inhibitors of RmlC have been previously characterised (including the high-throughput screening inhibitors noted above;228vide supra, Fig. 16), although many have limited aqueous solubility.259 van der Beek et al. conducted a fragment-screening campaign with a commercial library using bio-layer interferometry.259 A library of ∼1000 fragments was screened at 200 μM with twelve hits. Of these, seven compounds showed dose-dependent enzyme inhibition and inhibited bacterial growth. Three hits (Fig. 26, 132–134) with diverse structures inhibited both RmlB (the preceding enzyme in the biosynthetic pathway) and RmlC.
 | ||
| Fig. 26 Structures of fragment screening hits active against RmlC.259 | ||
Sasikala et al. also conducted a virtual screen of RmlC and identified several potential inhibitors of the Vibrio vulnificus enzyme (also known as WbpP).261 However, none of these compounds were confirmed as hits in biochemical or biophysical screens.
AMACR has been the subject of several previous inhibitor studies as well as structural studies on the M. tuberculosis homologue (MCR), with literature up to the end of 2012 having been previously reviewed.6,7 Following on from previous reports,136 Carnell et al. reported a series of acyl-CoA inhibitors with modified cores.191 The reported several new inhibitors (Fig. 27) including (±)-α-fluoroibuprofenoyl-CoA 49 (in which the Cα–H was replaced by fluorine), a chloro derivative 135, and N-dodecanoyl-R-alanyl-CoA 136. Inhibitor 49 replaces the Cα–H with a Cα-F, effectively removing the α-proton. Substitution of hydrogen with fluorine is commonly used in drug design because of the similar atomic radii (1.10 vs. 1.35 Å),267 bond lengths to carbon (1.08 to 1.11 vs. 1.26 to 1.41 Å)267 and the high C-F bond energies (typically >456 kJ mol−1).138,268 Inhibitors 135 and 136 are expected to form the enolate intermediate more easily,191 although this was not actually proven. Model studies suggest the Cα–H pKa for 136 should be ∼14.5–16.586,191 compared to a Cα–H pKa of ∼21 for standard acyl-CoA esters.86,88 Inhibitors 49 and 135 had IC50 values of 324 and 570 nM.191N-Dodecanoyl-R-alanyl-CoA 136 was less potent, with an IC50 value of 2300 nM, which is probably be due to lower stabilisation of the negatively charged intermediate.191 The known inhibitor (±)-2-trifluoromethyltetradecanoyl-CoA13647 had an IC50 of 156 nM.191 Significantly, two potent carbamate inhibitors 56 and 137 as analogues of the intermediate enolate were reported (IC50 = 98 and 1000 nM). Later studies127,164 showed that the carbamate inhibitor 56 was highly potent compared to other inhibitors (IC50 = ∼0.4 nM using the colorimetric assay127).
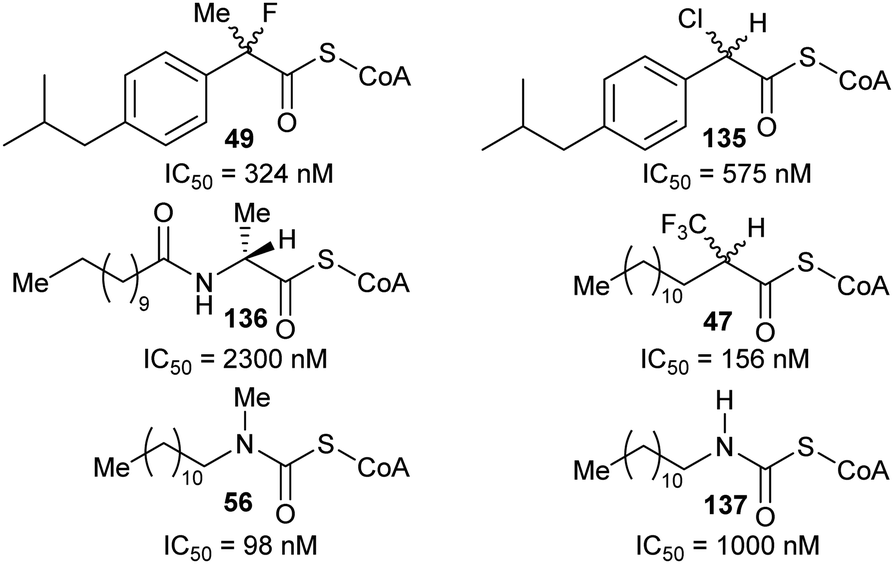 | ||
| Fig. 27 Structures of rationally designed AMACR inhibitors reported by Carnell et al.191 | ||
Also following on from the Carnell et al. study in 2007,136 Festuccia et al.190 reported the synthesis and testing of trifluoroibuprofenoyl-CoA (Fig. 28, 48). Limited kinetic analysis suggested non-competitive inhibition by this compound with a Ki = 1.7 μM. This result is notable because non-competitive inhibition of enzymes is rather rare (reviewed in ref. 227) and this is the only example of a non-competitive inhibitor reported for AMACR (and one of only a few for racemases/epimerases in general). Non-competitive inhibition is inconsistent with the inhibitor acting as an alternative substrate but instead arises through allosteric inhibition, stabilisation of an inactive conformation or covalent modification of the target.227 The basis for inhibition of AMACR by trifluoroibuprofenoyl-CoA is unclear, although elimination of fluoride is not reported. Treatment of cultured androgen-dependent and -independent prostate cancer cells with the pro-drug trifluoroibuprofen (Fig. 28, 138) resulted in arrest at G2/M in the cell cycle and a host of other changes, including induction of apoptosis.190 Tumour growth in androgen-dependent and -independent prostate cancer xenograft mouse models was also significantly reduced by treatment with this agent.190
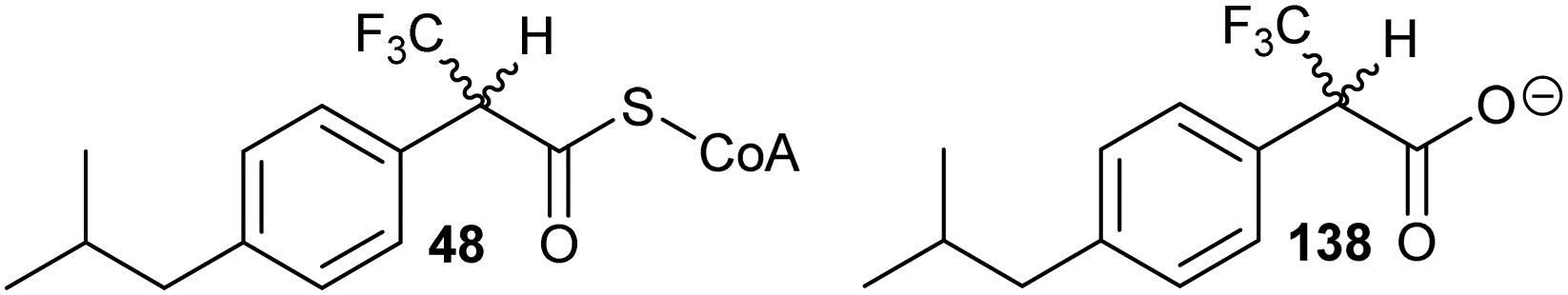 | ||
| Fig. 28 Structures of trifluoroibuprofenoyl-CoA 48 and the trifluoroibuprofen pro-drug 138.190 | ||
The advent of the AMACR colorimetric assay (vide supra, Scheme 9)127 has enabled much more thorough testing of inhibitors than had been previously possible, including determination of IC50 and Ki values and of reversibility of inhibition. This also enabled the first structure–activity relationship studies to be conducted. The first studies127,164 looked at a series of known AMACR inhibitors and substrates. A second study165 looked at a focussed series of 2-(arylthio)propanoyl-CoA inhibitors; the presence of the side-chain sulfur atom resulted in increased acidity of the Cα–H (previous studies on straight-chain acyl-CoAs and their 3-thia analogues showed that the presence of the sulfur reduces the pKa of the Cα–H to ∼15–16.5,269,270 compared to ∼21 for the corresponding acyl-CoA86,88). Many of these 2-(arylthio)propanoyl-CoA inhibitors were equipotent to fenoprofenoyl-CoA but optimisation of the inhibitor side-chain resulted in increased potency, e.g.139, IC50 = 22.3 nM.165 A 2-(arylsulfonyl)propanoyl-CoA inhibitor 140 was also synthesised in the hope that the presence of the sulfonyl group would further increase Cα–H acidity but this proved to be a poor inhibitor (Fig. 29).165
 | ||
| Fig. 29 Structure of the most potent 2-(arylthio)propanoyl-CoA inhibitor 139 and the poorly active 2-(arylsulfonyl)propanoyl-CoA 140 of human AMACR.165 | ||
Plotting pIC50 values for all inhibitors127,164,165 characterised by the AMACR colorimetric assay127 against calculated logP values (Fig. 30) showed that inhibitor potency was positively correlated with logP. The 2-(arylsulfonyl)propanoyl-CoA inhibitor 140 was highly hydrophilic,165 suggesting that this was the reason for its unexpected low potency. Although the 2-(arylthio)propanoyl-CoA inhibitors, such as 139, were highly potent in enzyme assays in vitro (IC50 = 22–520 nM), the carboxylic acid pro-drugs did not show any appreciable inhibition of androgen-dependent or -independent prostate cancer cells,165 possibly due to oxidation of the inhibitor pro-drug sulfur to the sulfoxide or sulfone.
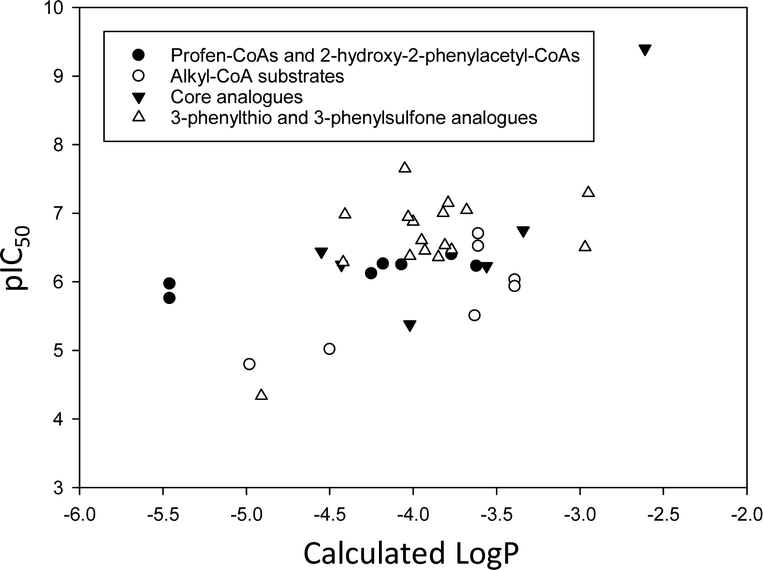 | ||
| Fig. 30 Potency of acyl-CoA inhibitors of AMACR, as measured by pIC50 as a function of calculated logP values. Inhibitors are as follows (with compound numbers from the original papers in parentheses): Ibuprofenoyl-CoA and analogues (5–11);164 straight-chain acyl-CoAs and other substrates (12–14 and 18–21);164 inhibitors with modified acyl-CoA cores (4, 22–26);164 and 2-arylthiapropanoyl-CoAs and 2-arylsulfonylpropanoyl-CoA (7a–7n, 10b).165 LogP values were calculated using: https://www.molinspiration.com/cgi-bin/properties. LogP, log10 (ratio of concentrations of drug in octan-1-ol and water at equilibrium); pIC50, −log10IC50. | ||
Since the last review,7 two studies featuring rational design of inhibitors for the M. tuberculosis AMACR homologue, MCR, have been published. The first study149 describes the synthesis and testing of several substrate/product acyl-CoA inhibitors (vide supra), in which the α-proton is replaced by a second side-chain in the inhibitor. The presence of the second sidechain increases potency of inhibition by ∼6-fold, although the measured absolute potency is relatively modest (e.g. 16.9 cf. 106 μM for 45avs. ibuprofenoyl-CoA). One of these inhibitors (Fig. 31A, 45b) has a α-proton in place of the α-methyl group and, as predicted, this does not undergo enzyme-catalysed exchange with solvent consistent with the α-proton being located in the methyl-binding site of the enzyme. The study is also notable in that several carboxylic acid precursors are also inhibitors, albeit with IC50 values in the mM range.149 Similar to the above AMACR inhibitors (vide supra, Fig. 30), potency of inhibition of MCR is also related to calculated logP values (Fig. 31).
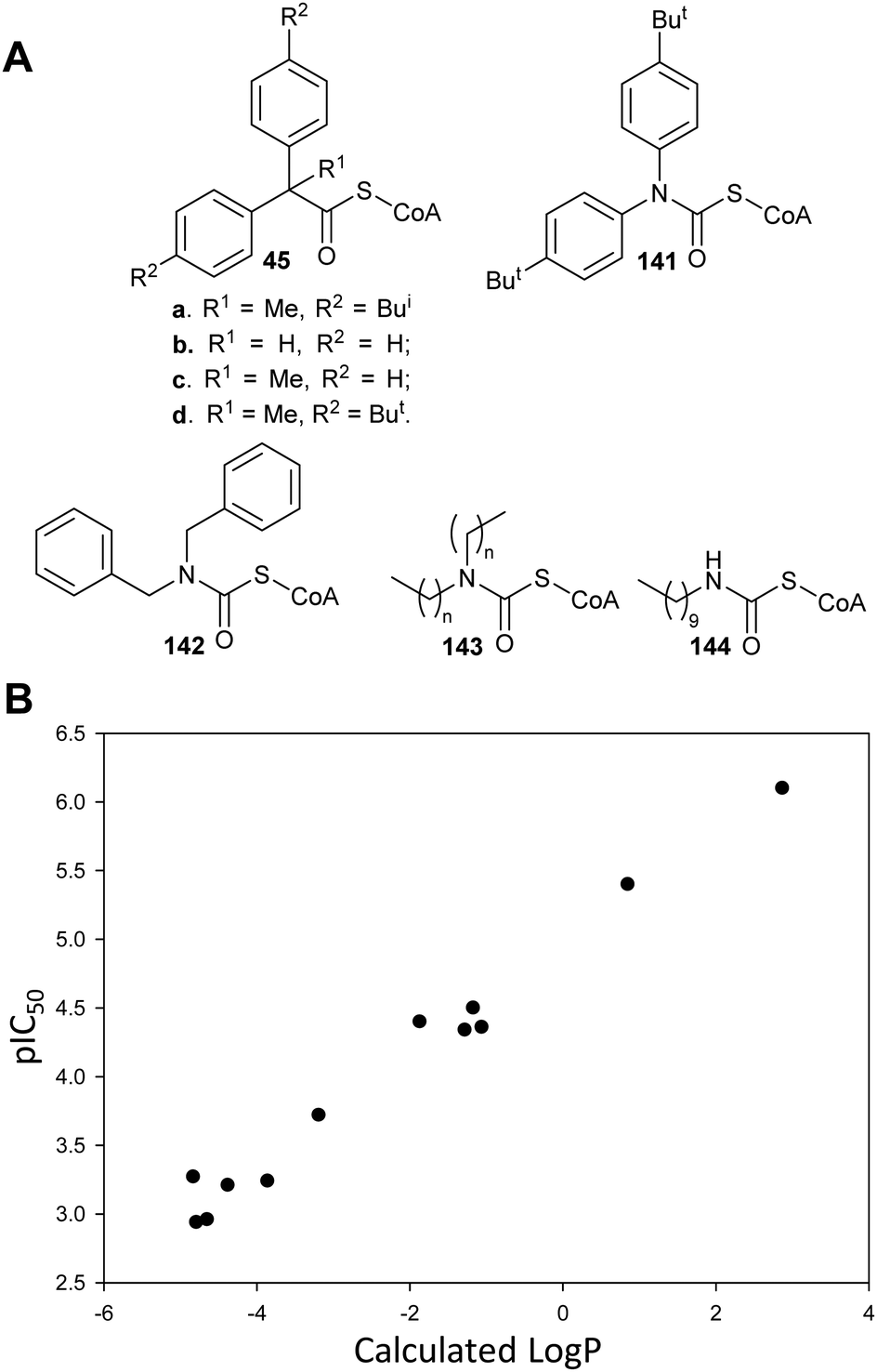 | ||
| Fig. 31 (A) Structures of substrate–product analogues149,150 inhibiting the M. tuberculosis homologue of AMACR (MCR). For 143, n = 3, 5, 7, 9 or 11; (B) acyl-CoA inhibitor potency as measured by pIC50 as a function of calculated LogP values. LogP values were calculated using: https://www.molinspiration.com/cgi-bin/properties. LogP, log10 (ratio of concentrations of drug in octan-1-ol and water at equilibrium); pIC50, −log10IC50. | ||
Following from the observation that carbamate analogues are highly potent AMACR inhibitors,127,164,191 Pal et al.150 synthesised and tested carbamate analogues 141–144 of their substrate/product inhibitors against MCR (Fig. 31A).149 Inhibition is reported to be competitive, although the Lineweaver–Burk plots for some analogues, e.g.143 (n = 5), suggested mixed competitive inhibition. Surprisingly, several of these analogues show irreversible inhibition, in marked contrast to the carbamate AMACR inhibitors which are fully reversible.127,164 Inhibitors with long alkyl chains (n = 9 and 11) show saturating loss of activity with maximum kinact values of ∼0.4 min−1 consistent with being active-site directed. Analogues with less lipophilic side-chains (143, n = 3) or a single side-chain (144) showed a non-saturating loss of enzymatic activity with a rate constant of 0.016–0.04 min−1.150 Inhibition was not reversed upon dialysis but no protein modification was observed by mass spectrometry. This observation is consistent with either non-covalent slow-binding inhibition, resulting in a long-lived enzyme-inhibitor complex, or irreversible inhibition resulting in a covalent modification of the protein, which is labile under mass spectrometric conditions.
Identification of AMACR inhibitors by high-throughput screening has also been reported.163 Unlike the previous study,152 the identified inhibitors were not non-specific protein modification agents.163 A number of pyrazoloquinolines and pyrazolopyrimidines were identified (vide supra, Fig. 15, 85a, 85b and 86), and some structure–activity relationships were observed.163 The identified inhibitors displayed either mixed competitive or uncompetitive inhibition. The latter is a rare type of inhibition and arises from binding of inhibitor to the enzyme-substrate complex.
Conclusions
Racemases and epimerases occupy a unique position in metabolism, in that they are the only major class of enzymes which can use substrates with both configurations at a chiral centre. Because of this, many racemases and epimerases are excellent drug targets and several have been extensively investigated as such, e.g. glutamate racemase.20–25,134,175,200,249 Use of inhibitors with the same configuration as the less abundant substrate (often D- or R-enantiomer) potentially offers additional benefits in that these isomers may be less prone to off-target binding and may have reduced drug metabolism, with consequent reductions in toxicities and longer durations of action.However, efforts to develop drugs targeted against racemases and epimerases have been largely limited to rational design campaigns, with the few notable exceptions detailed above. Development of inhibitors which are alternative substrates has met with limited success, in part because several of the effective inhibitors are rapidly depleted in vivo whilst the effects of less effective inhibitors are readily overcome by the physiological substrate. Moreover, these inhibitors are necessarily chiral and there had been a move away from chiral drugs towards drugs with fewer sp3 carbons.227 This is despite a growing realisation that the attrition rate is higher for ‘flatter’ drugs271,272 and that licenced drugs have a higher average proportion of sp3 centres than molecules published in The Journal of Medicinal Chemistry.273 Consequently there has been a more recent move towards structures with a higher proportion of sp3 and chiral centres.274 Similarly, racemase/epimerase inhibitors in which the Cα–H is replaced or where deprotonation is made more difficult tend not to be highly potent. The use of substrate–product analogues as inhibitors has also met with variable success. This strategy tends to work relatively well for racemases and epimerases in which large changes in substrate side-chain position occur during the reaction. Enzymes catalysing reactions resulting in limited changes in side-chain position and/or with small or sterically hindered active sites tend to be poorly inhibited by this type of compound.
There are relatively few developed inhibitors which are analogues of the transition state/deprotonated intermediate, perhaps because the early inhibitors developed against proline racemase were not highly effective and one of these inhibitors was an unstable imine.135 Some covalent inhibitors have been developed by rational design or identified by screening techniques.20,22–24,26,27,46,124,134,217,218 There has been renewed interest in the development of covalent inhibitors in recent years, prompted by a number of covalent drugs coming into clinical use.201,205,207,208 Covalent drugs acting on racemases and epimerases have all been directed against enzymes using active-site cysteine thiols20,22–24,26,27,46,124,134,217,218 (amino-acid racemases and epimerases). Covalently reacting drugs containing electrophiles reacting with other active-site bases205,208 have been under-explored. Both transition-state analogues135,193,194 and covalent inhibitors201,204–206,208 offer the potential for high potency and long duration of action and are potentially fertile ground for the future development of inhibitors.
Screening approaches227 have also been under-used to identify novel inhibitors. There are only five high-throughput screening campaigns in the literature152,163,200,228,252 and only two fragment-screening campaigns.257,259 Almost all racemases and epimerases catalyse reversible reactions and this places restriction on these assays but these can be overcome by using an elimination substrate or irreversible coupling enzyme (see section on enzyme assays for examples). The use of coupling enzymes also enables assays based on fluorescence or absorbance to be used, which are readily adaptable to high-throughput screening formats.227 Direct assaying of racemase or epimerase activity may also be possible using fluorescence anisotropy to monitor ligand binding.227
Fragment screening using assays of enzyme activity227,275–279 or biophysical techniques275–280 (particularly X-ray crystallography274–277,279–281) hold significant promise, although the different screening techniques have advantages and disadvantages280 and different tendencies towards false positive and negative results.279 There is also a balance to be struck between fragment complexity and affinity to maximise chances of success.279 Screening of fragment libraries for direct identification of inhibitors is particularly appealing for enzymes with small, enclosed active sites, e.g. proline racemase,26,27 as the amino-acid substrates are small fragments themselves (Mw = 89–204 Da). There have been a number of studies on the screening of small fragments (which will generally have low affinity279), including one using virtual screening initially to triage compounds which resulted in a 40% hit rate for a very small fragment library (fifteen compounds).282 Similarly, fragment-based screening holds promise for development of inhibitors of enzymes with larger active sites,227,275–280 although there are challenges associated with identification of different fragments which bind simultaneously and also in the elaboration of fragment hits into leads.274,283
It is important that inhibitors produced by rational design, identified by screening and other approaches are fully characterised to determine if covalent modification of the target is occurring. There are examples of rationally designed racemase inhibitors intended to be reversible which appear to exert their effects by covalent modification of the racemase target.150 Several inhibitors identified by screening approaches239,252,259 could also potentially inhibit their targets by covalent modification. Unselective modification of off-target proteins or other biological molecules could give rise to significant toxicities.201–205,207–209,215 Therefore, it is important to balance this potential draw-back with the advantages of covalent inhibition.
Abbreviations used
| AET | 5-(1-Aminoethyl)tetrazole |
| AMACR | Human α-methylacyl-CoA racemase (a.k.a. P504S) spliced variant 1A |
| DprE | Decaprenylphosphoryl-β-D-ribose epimerase |
| EcL-DER | E. coli L-aspartate/L-glutamate racemase |
| IAM 12614 | L. aggregata cis-3-hydroxy-S-proline racemase/dehydratase |
| MCR | α-Methylacyl-CoA racemase from M. tuberculosis |
| McyF | Microcystis aeruginosa aspartate racemase |
| MMP0739 | Aspartate/glutamate racemase from Methanococcus maripaludis |
| Ph | phenyl |
| pIC50 | −Log10(IC50) |
| PLP | Pyridoxal 5′-phosphate |
| RacX | B. subtilis arginine, lysine and ornithine racemase |
| RmlC | Deoxythymidine diphosphate-4-keto-6-deoxyglucose 3,5-epimerase |
| YgeA | E. coli homoserine racemase |
| YcjG | E. coli alanyl dipeptide epimerase. Standard one- and three-letter amino-acid codes are used. |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors have been funded by Prostate Cancer UK (grants S10-03 and PG14-009), LifeArc, Biochemical Society Summer Vacation Studentships, the Cancer Research @ Bath network and a Bath-Shandong Undergraduate Exchange Studentship. TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01). Diagrams in Schemes 9B and C have been reproduced from Yevglevskis et al., 2017127 with permission from the Royal Society of Chemistry.Notes and references
- N. Fujii, T. Takata, N. Fujii, K. Aki and H. Sakaue, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 840–847 CrossRef CAS PubMed.
- J. Sasabe and M. Suzuki, Keio J. Med., 2019, 68, 1–16 CrossRef CAS PubMed.
- R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl., 1966, 5, 385–415 CrossRef CAS.
- A. Ballard, S. Narduolo, H. O. Ahmad, D. A. Cosgrove, A. G. Leach and N. J. Buurma, Expert Opin. Drug Discovery, 2019, 14, 527–539 CrossRef CAS PubMed.
- A. Ballard, S. Narduolo, H. O. Ahmed, N. I. Keymer, N. Asaad, D. A. Cosgrove, N. J. Buurma and A. G. Leach, Chem. – Eur. J., 2020, 26, 3661–3687 CrossRef CAS PubMed.
- M. D. Lloyd, D. J. Darley, A. S. Wierzbicki and M. D. Threadgill, FEBS J., 2008, 275, 1089–1102 CrossRef CAS PubMed.
- M. D. Lloyd, M. Yevglevskis, G. L. Lee, P. J. Wood, M. D. Threadgill and T. J. Woodman, Prog. Lipid Res., 2013, 52, 220–230 CrossRef CAS PubMed.
- T. J. Woodman, P. J. Wood, A. S. Thompson, T. J. Hutchings, G. R. Steel, P. Jiao, M. D. Threadgill and M. D. Lloyd, Chem. Commun., 2011, 47, 7332–7334 RSC.
- M. Yevglevskis, C. R. Bowskill, C. C. Y. Chan, J. H.-J. Heng, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Org. Biomol. Chem., 2014, 12, 6737–6744 RSC.
- L.-B. Gao, J.-Z. Wang, T.-W. Yao and S. Zeng, Chirality, 2012, 24, 86–95 CrossRef CAS PubMed.
- T. Poiger, M. D. Müller, H.-R. Buser and I. J. Buerge, J. Agric. Food Chem., 2015, 63, 2583–2590 CrossRef CAS PubMed.
- H.-R. Buser and M. D. Müller, Environ. Sci. Technol., 1997, 31, 1960–1967 CrossRef CAS.
- M. D. Müller and H.-R. Buser, Environ. Sci. Technol., 1997, 31, 1953–1959 CrossRef.
- Y. Ogasawara and T. Dairi, Front. Microbiol., 2018, 9, 156 CrossRef PubMed.
- S. A. Cochrane and C. T. Lohans, Eur. J. Med. Chem., 2020, 194, 112262 CrossRef CAS PubMed.
- T. Do, J. E. Page and S. Walker, J. Biol. Chem., 2020, 295, 3347–3361 CrossRef CAS PubMed.
- A. J. F. Egan, J. Errington and W. Vollmer, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS PubMed.
- S. L. Bearne, Chem. – Eur. J., 2020, 26, 10367–10390 CrossRef CAS PubMed.
- R. V. Chikhale, M. A. Barmade, P. R. Murumkar and M. R. Yadav, J. Med. Chem., 2018, 61, 8563–8593 CrossRef CAS PubMed.
- C. Fischer, Y. C. Ahn and J. C. Vederas, Nat. Prod. Rep., 2019, 36, 1687–1705 RSC.
- K. R. Witkin, N. R. Vance, C. Caldwell, Q. Li, L. Yu and M. A. Spies, ChemMedChem, 2020, 15, 376–384 CrossRef CAS PubMed.
- N. R. Vance, K. R. Witkin, P. W. Rooney, Y. Li, M. Pope and M. A. Spies, ChemMedChem, 2018, 13, 2514–2521 CrossRef CAS PubMed.
- M. A. Spies, J. G. Reese, D. Dodd, K. L. Pankow, S. R. Blanke and J. Baudry, J. Am. Chem. Soc., 2009, 131, 5274–5284 CrossRef CAS PubMed.
- K. L. Whalen, K. L. Pankow, S. R. Blanke and M. A. Spies, ACS Med. Chem. Lett., 2010, 1, 9–13 CrossRef CAS PubMed.
- K. L. Whalen, K. B. Tussey, S. R. Blanke and M. A. Spies, J. Phys. Chem. B, 2011, 115, 3416–3424 CrossRef CAS PubMed.
- A. Berneman, L. Montout, S. Goyard, N. Chamond, A. Cosson, S. d'Archivio, N. Gouault, P. Uriac, A. Blondel and P. Minoprio, PLoS One, 2013, 8, e60955 CrossRef CAS PubMed.
- P. de Aguiar Amaral, D. Autheman, G. Dias de Melo, N. Gouault, J.-F. Cupif, S. Goyard, P. Dutra, N. Coatnoan, A. Cosson, D. Monet, F. Saul, A. Haouz, P. Uriac, A. Blondel and P. Minoprio, PLoS Neglected Trop. Dis., 2018, 12, e0006853 CrossRef CAS PubMed.
- L. Coutinho, M. Alves Ferreira, A. Cosson, M. Meuser Batista, D. da Gama Jaén Batista, P. Minoprio, W. M. Degrave, A. Berneman and M. de Nazaré Correia Soeiro, Mem. Inst. Oswaldo Cruz, 2009, 104, 1055–1062 CrossRef PubMed.
- S. Zha, S. Ferdinandusse, S. Denis, R. J. Wanders, C. M. Ewing, J. Luo, A. M. De Marzo and W. B. Isaacs, Cancer Res., 2003, 63, 7365–7376 CAS.
- R. Inoue, K. Hashimoto, T. Harai and H. Mori, J. Neurosci., 2008, 28, 14486–14491 CrossRef CAS PubMed.
- C.-H. Chang, C.-H. Lin and H.-Y. Lane, Int. J. Mol. Sci., 2020, 21, 2676 CrossRef CAS PubMed.
- W. J. Griffiths and Y.-Q. Wang, Prostaglandins Other Lipid Mediators, 2020, 147, 106381 CrossRef CAS PubMed.
- M. Y. S. Hooi and R. J. W. Truscott, Age, 2011, 33, 131–141 CrossRef CAS PubMed.
- H. Jiang, J. Fang, B. Wu, G. Yin, L. Sun, J. Qu, S. W. Barger and S. Wu, J. Neuroinflammation, 2011, 8, 119 CrossRef CAS PubMed.
- A. Steliga, P. Kowiański, E. Czuba, M. Waśkow, J. Moryś and G. Lietzau, Transl. Stroke Res., 2019, 11, 553–579 CrossRef PubMed.
- K. R. Baker, H. H. Sigurðardóttir, B. Jana and L. Guardabassi, Antimicrob. Agents Chemother., 2017, 61, e01773–e01716 CrossRef CAS PubMed.
- S. Ferdinandusse, S. Denis, P. T. Clayton, A. Graham, J. E. Rees, J. T. Allen, B. N. McLean, A. Y. Brown, P. Vreken, H. R. Waterham and R. J. A. Wanders, Nat. Genet., 2000, 24, 188–191 CrossRef CAS PubMed.
- C. M. Dobson, A. Gradinger, N. Longo, X. Wu, D. Leclerc, J. Lerner-Ellis, M. Lemieux, C. Belair, D. Watkins, D. S. Rosenblatt and R. A. Gravel, Mol. Genet. Metab., 2006, 88, 327–333 CrossRef CAS PubMed.
- M. Andréasson, R. H. Zetterström, U. von Döbeln, A. Wedell and P. Svenningsson, Int. J. Mol. Sci., 2019, 20, 2631 CrossRef PubMed.
- N. F. Liachko, A. D. Saxton, P. J. McMillan, T. J. Strovas, C. D. Keene, T. D. Bird and B. C. Kraemer, PLoS Genet., 2019, 15, e1008526 CrossRef CAS PubMed.
- A. P. Gomes, D. Ilter, V. Low, J. E. Endress, J. Fernandez-Garcia, A. Rosenzweig, T. Schild, D. Broekaert, A. Ahmed, M. Planque, I. Elia, J. Han, C. Kinzig, E. Mullarky, A. P. Mutvei, J. Asara, R. de Cabo, L. C. Cantley, N. Dephoure, S. M. Fendt and J. Blenis, Nature, 2020, 585, 283–287 CrossRef CAS PubMed.
- K. M. Hodge-Hanson, A. Zoino and D. M. Downs, J. Bacteriol., 2018, 200, e00751–e00717 CrossRef CAS PubMed.
- D. Kreuzmann, R. Horstkorte, G. Kohla, C. Kannicht, D. Bennmann, A. Thate and K. Bork, ChemBioChem, 2017, 18, 1188–1193 CrossRef CAS PubMed.
- S.-C. Chen, C.-H. Huang, S.-J. Lai, C. S. Yang, T.-H. Hsiao, C.-H. Lin, P.-K. Fu, T. P. Ko and Y. Chen, Sci. Rep., 2016, 6, 23274 CrossRef CAS PubMed.
- R. Singh and R. Arya, Mol. Neurobiol., 2016, 53, 3088–3101 CrossRef CAS PubMed.
- Y.-C. Ahn, C. Fischer, M. J. van Belkum and J. C. Vederas, Org. Biomol. Chem., 2018, 16, 1126–1133 RSC.
- A. S. Murkin and M. E. Tanner, J. Org. Chem., 2002, 67, 8389–8394 CrossRef CAS PubMed.
- M. M. Musa, Chirality, 2020, 32, 147–157 CrossRef CAS PubMed.
- S. Martínez-Rodríguez, P. Soriano-Maldonado and J. A. Gavira, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140377 CrossRef PubMed.
- J. F. Rocha, A. F. Pina, S. F. Sousa and N. M. F. S. A. Cerqueira, Catal. Sci. Technol., 2019, 9, 4864–4876 RSC.
- Y. Zhang, Y. Zhang and O. Ramström, Catal. Rev.: Sci. Eng., 2020, 62, 66–95 CrossRef CAS PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, D. S. Froese, E. Krysztofinska, W. W. Yue and C. J. Schofield, ChemBioChem, 2016, 17, 471–473 CrossRef CAS PubMed.
- A. Tøndervik, G. Klinkenberg, F. L. Aachmann, B. I. G. Svanem, H. Ertesvåg, T. E. Ellingsen, S. Valla, G. Skjåk-Braek and H. Sletta, Biomacromolecules, 2013, 14, 2657–2666 Search PubMed.
- C. Yang, L. Ye, J. Gu, X. Yang, A. Li and H.-W. Yu, Appl. Microbiol. Biotechnol., 2017, 101, 1063–1072 CrossRef CAS PubMed.
- R. Ushimaru, Z. Chen, H. Zhao, P.-H. Fan and H.-W. Liu, Angew. Chem., Int. Ed., 2020, 59, 3558–3562 CrossRef CAS PubMed.
- A. Benjdia, A. Guillot, P. Ruffié, J. Leprince and O. Berteau, Nat. Chem., 2017, 9, 698–707 CrossRef CAS PubMed.
- S. W. Fuchs, G. Lackner, B. I. Morinaka, Y. Morishita, T. Asai, S. Riniker and J. Piel, Angew. Chem., Int. Ed., 2016, 55, 12330–12333 CrossRef CAS PubMed.
- F. Kudo, S. Hoshi, T. Kawashima, T. Kamachi and T. Eguchi, J. Am. Chem. Soc., 2014, 136, 13909–13915 CrossRef CAS PubMed.
- B. I. Morinaka, A. L. Vagstad and J. Piel, in Marine Enzymes and Specialized Metabolism, Pt A, ed. B. S. Moore, 2018, vol. 604, pp. 237–257 Search PubMed.
- A. L. Vagstad, T. Kuranaga, S. Püntener, V. R. Pattabiraman, J. W. Bode and J. Piel, Angew. Chem., Int. Ed., 2019, 58, 2246–2250 CrossRef CAS PubMed.
- S. T. M. Allard, M. F. Giraud and J. H. Naismith, Cell. Mol. Life Sci., 2001, 58, 1650–1665 CrossRef CAS PubMed.
- J. A. Rankin, R. C. Mauban, M. Fellner, B. Desguin, J. McCracken, J. Hu, S. A. Varganov and R. P. Hausinger, Biochemistry, 2018, 57, 3244–3251 CrossRef CAS PubMed.
- M. E. Tanner, Acc. Chem. Res., 2002, 35, 237–246 CrossRef CAS PubMed.
- M. Alfano and C. Cavazza, Protein Sci., 2020, 29, 1071–1089 CrossRef CAS PubMed.
- M. Brecik, I. Centarova, R. Mukherjee, G. S. Kolly, S. Huszar, A. Bobovska, E. Kilacskova, V. Mokosova, Z. Svetlikova, M. Sarkan, J. Neres, J. Kordulakova, S. T. Cole and K. Mikusova, ACS Chem. Biol., 2015, 10, 1631–1636 CrossRef CAS PubMed.
- G. Degiacomi, J. M. Belardinelli, M. R. Pasca, E. De Rossi, G. Riccardi and L. R. Chiarelli, Appl. Sci., 2020, 10, 623 CrossRef CAS.
- J. Gawad and C. Bonde, Chem. Cent. J., 2018, 12, 72 CrossRef CAS PubMed.
- G. Riccardi, M. R. Pasca, L. R. Chiarelli, G. Manina, A. Mattevi and C. Binda, Appl. Microbiol. Biotechnol., 2013, 97, 8841–8848 CrossRef CAS PubMed.
- J. Liang, Q. Han, Y. Tan, H. Z. Ding and J. Y. Li, Front. Mol. Biosci., 2019, 6, 4 CrossRef CAS PubMed.
- M. D. Toney, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1407–1418 CrossRef CAS PubMed.
- S. L. Bearne, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 619–630 CrossRef CAS PubMed.
- M. E. Glasner, D. P. Truong and B. C. Morse, FEBS J., 2020, 287, 1323–1342 CrossRef CAS PubMed.
- J. Q. Fuller and P. F. Leadlay, Biochem. J., 1983, 213, 643–650 CrossRef CAS PubMed.
- V. M. Powers, C. W. Koo, G. L. Kenyon, J. A. Gerlt and J. W. Kozarich, Biochemistry, 1991, 30, 9255–9263 CrossRef CAS PubMed.
- B. Mitra, A. T. Kallarakal, J. W. Kozarich, J. A. Gerlt, J. G. Clifton, G. A. Petsko and G. L. Kenyon, Biochemistry, 1995, 34, 2777–2787 CrossRef CAS PubMed.
- A. Rubinstein and D. T. Major, J. Am. Chem. Soc., 2009, 131, 8513–8521 CrossRef CAS PubMed.
- G. J. Cardinal and R. H. Abeles, Biochemistry, 1968, 7, 3970–3978 CrossRef PubMed.
- K. A. Gallo, M. E. Tanner and J. R. Knowles, Biochemistry, 1993, 32, 3991–3997 CrossRef CAS PubMed.
- D. J. Darley, D. S. Butler, S. J. Prideaux, T. W. Thornton, A. D. Wilson, T. J. Woodman, M. D. Threadgill and M. D. Lloyd, Org. Biomol. Chem., 2009, 7, 543–552 RSC.
- D. J. Darley, D. S. Butler, S. J. Prideaux, T. W. Thornton, A. D. Wilson, T. J. Woodman, M. D. Threadgill and M. D. Lloyd, Org. Biomol. Chem., 2009, 7, 5272 RSC.
- S. Sharma, P. Bhaumik, W. Schmitz, R. Venkatesan, J. K. Hiltunen, E. Conzelmann, A. H. Juffer and R. K. Wierenga, J. Phys. Chem. B, 2012, 116, 3619–3629 CrossRef CAS PubMed.
- M. E. Tanner, K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3998–4006 CrossRef CAS PubMed.
- S. Glavas and M. E. Tanner, Biochemistry, 1999, 38, 4106–4113 CrossRef CAS PubMed.
- R. J. Stern, T.-Y. Lee, T.-J. Lee, W. Yan, M. S. Scherman, V. D. Vissa, S.-K. Kim, B. L. Wanner and M. R. McNeil, Microbiology, 1999, 145, 663–671 CrossRef CAS PubMed.
- H. C. Dunathan, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 712–716 CrossRef CAS PubMed.
- T. L. Amyes and J. P. Richard, Synlett, 2017, 1407–1421 CAS.
- M. A. Spies and M. D. Toney, Biochemistry, 2003, 42, 5099–5107 CrossRef CAS PubMed.
- J. P. Richard and T. L. Amyes, Curr. Opin. Chem. Biol., 2001, 5, 626–633 CrossRef CAS PubMed.
- G. Williams, E. P. Maziarz, T. L. Amyes, T. D. Wood and J. P. Richard, Biochemistry, 2003, 42, 8354–8361 CrossRef CAS PubMed.
- A. A. McCarthy, H. M. Baker, S. C. Shewry, M. L. Patchett and E. N. Baker, Structure, 2001, 9, 637–646 CrossRef CAS.
- M. Nagar, A. Narmandakh, Y. Khalak and S. L. Bearne, Biochemistry, 2011, 50, 8846–8852 CrossRef CAS PubMed.
- J. A. Gerlt, G. L. Kenyon, J. W. Kozarich, D. J. Neidhart, G. A. Petsko and V. M. Powers, Curr. Opin. Struct. Biol., 1992, 2, 736–742 CrossRef CAS.
- J. Crugeiras, A. Rios, E. Riveiros, T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 2008, 130, 2041–2050 CrossRef CAS PubMed.
- T. L. Amyes and J. P. Richard, Biochemistry, 2013, 52, 2021–2035 CrossRef CAS PubMed.
- P. Bhaumik, W. Schmitz, A. Hassinen, J. K. Hiltunen, E. Conzelmann and R. K. Wierenga, J. Mol. Biol., 2007, 367, 1145–1161 CrossRef CAS PubMed.
- M. C. Pélissier, C. Sebban-Kreuzer, F. Guerlesquin, J. A. Brannigan, Y. Bourne and F. Vincent, J. Biol. Chem., 2014, 289, 35215–35224 CrossRef PubMed.
- J. P. Richard, T. L. Amyes, B. Goryanova and X. Zhai, Curr. Opin. Chem. Biol., 2014, 21, 1–10 CrossRef CAS PubMed.
- H.-Y. Sagong and K.-J. Kim, Sci. Rep., 2017, 7, 42318 CrossRef CAS.
- A. Watanabe, T. Yoshimura, B. Mikami, H. Hayashi, H. Kagamiyama and N. Esaki, J. Biol. Chem., 2002, 277, 19166–19172 CrossRef CAS PubMed.
- F. Ding, J. M. Smith and H. Wang, J. Org. Chem., 2009, 74, 2679–2691 CrossRef CAS PubMed.
- M. D. Toney, Front. Bioengin. Biotechnol., 2019, 7, 25 CrossRef PubMed.
- E. A. T. Ringia, J. B. Garrett, J. B. Thoden, H. M. Holden, I. Rayment and J. A. Gerlt, Biochemistry, 2004, 43, 224–229 CrossRef PubMed.
- J. B. Thoden, E. A. T. Ringia, J. B. Garrett, J. A. Gerlt, H. M. Holden and I. Rayment, Biochemistry, 2004, 43, 5716–5727 CrossRef CAS PubMed.
- S. Baxter, S. Royer, G. Grogan, F. Brown, K. E. Holt-Tiffin, I. N. Taylor, I. G. Fotheringham and D. J. Campopiano, J. Am. Chem. Soc., 2012, 134, 19310–19313 CrossRef CAS.
- S. N. Ruzheinikov, M. A. Taal, S. E. Sedelnikova, P. J. Baker and D. W. Rice, Structure, 2005, 13, 1707–1713 CrossRef CAS PubMed.
- J. P. Richard and T. L. Amyes, Bioorg. Chem., 2004, 32, 354–366 CrossRef CAS PubMed.
- C. W. Koo and J. S. Blanchard, Biochemistry, 1999, 38, 4416–4422 CrossRef CAS PubMed.
- J.-W. Ahn, J. H. Chang and K.-J. Kim, FEBS Lett., 2015, 589, 3842–3847 CrossRef CAS PubMed.
- X. Liu, F. Gao, Y. Ma, S. Liu, Y. Cui, Z. Yuan and X. Kang, FEBS Lett., 2016, 590, 1262–1269 CrossRef CAS PubMed.
- W. F. Visser, N. M. Verhoeven-Duif and T. J. de Koning, J. Biol. Chem., 2012, 287, 21654–21662 CrossRef CAS PubMed.
- D. K. Dhaked, M. B. Divya and L. Guruprasad, Prog. Biophys. Mol. Biol., 2019, 145, 52–64 CrossRef CAS PubMed.
- X. Li, Q.-C. Zheng and H.-X. Zhang, Int. J. Quantum Chem., 2012, 112, 619–624 CrossRef CAS.
- S. Scheiner, Struct. Chem., 2019, 30, 1119–1128 CrossRef CAS.
- S. Scheiner, T. Kar and Y. L. Gu, J. Biol. Chem., 2001, 276, 9832–9837 CrossRef CAS.
- S. Scheiner, Isr. J. Chem., 2009, 49, 139–147 CrossRef CAS.
- U. M. Babu and R. B. Johnston, Biochem. Biophys. Res. Commun., 1974, 58, 460–466 CrossRef CAS PubMed.
- S. D. Heck, W. S. Faraci, P. R. Kelbaugh, N. A. Saccomano, P. F. Thadeio and R. A. Volkmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4036–4039 CrossRef CAS PubMed.
- A. Wiese, M. Pietzsch, C. Syldatk, R. Mattes and J. Altenbuchner, J. Biotechnol., 2000, 80, 217–230 CrossRef CAS.
- X. S. Zhang, R. Kumar, M. W. Vetting, S. W. Zhao, M. P. Jacobson, S. C. Almo and J. A. Gerlt, J. Am. Chem. Soc., 2015, 137, 1388–1391 CrossRef CAS PubMed.
- M. Yevglevskis, G. L. Lee, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Chem. Commun., 2014, 50, 14164–14166 RSC.
- M. H. Gelb, Y. Lin, M. A. Pickard, Y. Song and J. C. Vederas, J. Am. Chem. Soc., 1990, 112, 4932–4942 CrossRef CAS.
- D. T. Lin, V. M. Powers, L. J. Reynolds, C. P. Whitman, J. W. Kozarich and G. L. Kenyon, J. Am. Chem. Soc., 1988, 110, 323–324 CrossRef CAS.
- J. A. Landro, A. T. Kallarakal, S. C. Ransom, J. A. Gerlt, J. W. Kozarich, D. J. Neidhart and G. L. Kenyon, Biochemistry, 1991, 30, 9274–9281 CrossRef CAS PubMed.
- R. J. Cox, A. Sutherland and J. C. Vederas, Bioorg. Med. Chem., 2000, 8, 843–871 CrossRef CAS.
- H. Zhou, D. M. Z. Schmidt, J. A. Gerlt and W. A. van der Donk, ChemBioChem, 2003, 4, 1206–1215 CrossRef CAS PubMed.
- K. Oki, F. S. Lee and S. L. Mayo, Protein Eng., Des. Sel., 2019, 32, 261–270 CrossRef CAS PubMed.
- M. Yevglevskis, G. L. Lee, A. Nathubhai, Y. D. Petrova, T. D. James, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Chem. Commun., 2017, 53, 5087–5090 RSC.
- S. Glavas and M. E. Tanner, Bioorg. Med. Chem. Lett., 1997, 7, 2265–2270 CrossRef CAS.
- D. L. Graham, M. L. Beio, D. L. Nelson and D. B. Berkowitz, Front. Mol. Biosci., 2019, 6, 8 CrossRef CAS PubMed.
- M. Marchetti, S. Bruno, B. Campanini, S. Bettati, A. Peracchi and A. Mozzarelli, Amino Acids, 2015, 47, 163–173 CrossRef CAS PubMed.
- D. L. Nelson, G. A. Applegate, M. L. Beio, D. L. Graham and D. B. Berkowitz, J. Biol. Chem., 2017, 292, 13986–14002 CrossRef CAS.
- E. Wang and C. Walsh, Biochemistry, 1978, 17, 1313–1321 CrossRef CAS PubMed.
- H. Wolosker, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1558–1566 CrossRef CAS PubMed.
- G. A. Prosser, A. Rodenburg, H. Khoury, C. de Chiara, S. Howell, A. P. Snijders and L. P. S. de Carvalho, Antimicrob. Agents Chemother., 2016, 60, 6091–6099 CrossRef CAS.
- G. B. Evans, V. L. Schramm and P. C. Tyler, MedChemComm, 2018, 9, 1983–1993 RSC.
- A. J. Carnell, I. Hale, S. Denis, R. J. A. Wanders, W. B. Isaacs, B. A. Wilson and S. Ferdinandusse, J. Med. Chem., 2007, 50, 2700–2707 CrossRef CAS PubMed.
- J. R. Mohrig, Acc. Chem. Res., 2013, 46, 1407–1416 CrossRef CAS PubMed.
- Y. Pan, ACS Med. Chem. Lett., 2019, 10, 1016–1019 CrossRef CAS PubMed.
- F. Siddiqi, J. R. Bourque, H. Jiang, M. Gardner, M. St Maurice, C. Blouin and S. L. Bearne, Biochemistry, 2005, 44, 9013–9021 CrossRef CAS PubMed.
- S.-Y. Choi, N. Esaki, T. Yoshimura and K. Soda, J. Biochem., 1992, 112, 139–142 CrossRef CAS PubMed.
- S. Sawada, Y. Tanaka, S. Hayashi, M. Ryu, T. Hasegawa, Y. Yamamoto, N. Esaki, K. Soda and S. Takahashi, Biosci., Biotechnol., Biochem., 1994, 58, 807–811 CrossRef CAS.
- D. L. Schonfeld and U. T. Bornscheuer, Anal. Chem., 2004, 76, 1184–1188 CrossRef PubMed.
- H. Stecher, A. Hermetter and K. Faber, Biotechnol. Tech., 1998, 12, 257–261 CrossRef CAS.
- T. L. Thaler, P. R. Gibbs, R. P. Trebino and A. S. Bommarius, Astrobiology, 2006, 6, 901–910 CrossRef CAS PubMed.
- T. Yamauchi, S.-Y. Choi, H. Okada, M. Yohda, H. Kumagai, N. Esaki and K. Soda, J. Biol. Chem., 1992, 267, 18361–18364 CrossRef CAS.
- D. Ouazia and S. L. Bearne, Anal. Biochem., 2010, 398, 45–51 CrossRef CAS PubMed.
- X. Z. Wang, C. C. Chen, T. Shen and J. Y. Zhang, PeerJ, 2019, 7, 8300 CrossRef PubMed.
- K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3981–3990 CrossRef CAS PubMed.
- M. Pal, M. Khanal, R. Marko, S. Thirumalairajan and S. L. Bearne, Chem. Commun., 2016, 52, 2740–2743 RSC.
- M. Pal, N. M. Easton, H. Yaphe and S. L. Bearne, Bioorg. Chem., 2018, 77, 640–650 CrossRef CAS PubMed.
- M. D. Olp, K. S. Kalous and B. C. Smith, BMC Bioinf., 2020, 21, 186 CrossRef CAS PubMed.
- B. A. P. Wilson, H. Wang, B. A. Nacev, R. C. Mease, J. O. Liu, M. G. Pomper and W. B. Isaacs, Mol. Cancer Ther., 2011, 10, 825–838 CrossRef CAS PubMed.
- C. Kumar-Sinha, R. B. Shah, B. Laxman, S. A. Tomlins, J. Harwood, W. Schmitz, E. Conzelmann, M. G. Sanda, J. T. Wei, M. A. Rubin and A. M. Chinnaiyan, Am. J. Pathol., 2004, 164, 787–793 CrossRef CAS.
- P. F. Leadlay and J. Q. Fuller, Biochem. J., 1983, 213, 635–642 CrossRef CAS PubMed.
- T. L. Amyes and J. P. Richard, in Measurement and Analysis of Kinetic Isotope Effects, ed. M. E. Harris and V. E. Anderson, 2017, vol. 596, pp. 163–177 Search PubMed.
- P. L. Fernandez and A. S. Murkin, Molecules, 2020, 25, 1933 CrossRef CAS PubMed.
- C. I. F. Watt, J. Phys. Org. Chem., 2010, 23, 561–571 CrossRef CAS.
- K. Y. Wong, Y. Q. Xu and L. Xu, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 1782–1794 CrossRef CAS PubMed.
- M. Katane, K. Nakayama, T. Kawata, Y. Yokoyama, Y. Matsui, Y. Kaneko, S. Matsuda, Y. Saitoh, T. Miyamoto, M. Sekine and H. Homma, J. Pharm. Biomed. Anal., 2015, 116, 109–115 CrossRef CAS PubMed.
- P. P. VanVeldhoven, K. Croes, M. Casteels and G. P. Mannaerts, Biochim. Biophys. Acta, 1997, 1347, 62–68 CrossRef CAS.
- M. Yevglevskis, G. L. Lee, J. Sun, S. Zhou, X. Sun, G. Kociok-Köhn, T. D. James, T. J. Woodman and M. D. Lloyd, Org. Biomol. Chem., 2016, 14, 612–622 RSC.
- L. K. P. Lam, L. D. Arnold, T. H. Kalantar, J. G. Kelland, P. M. Lanebell, M. M. Palcic, M. A. Pickard and J. C. Vederas, J. Biol. Chem., 1988, 263, 11814–11819 CrossRef CAS.
- Y. D. Petrova, K. Wadda, A. Nathubhai, M. Yevglevskis, P. J. Mitchell, T. D. James, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Bioorg. Chem., 2019, 92, 103264 CrossRef CAS PubMed.
- M. Yevglevskis, G. L. Lee, A. Nathubhai, Y. D. Petrova, T. D. James, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Bioorg. Chem., 2018, 79, 145–154 CrossRef CAS PubMed.
- M. Yevglevskis, A. Nathubhai, K. Wadda, G. L. Lee, S. Al-Rawi, T. Jiao, P. J. Mitchell, T. D. James, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Bioorg. Chem., 2019, 92, 103263 CrossRef CAS PubMed.
- J. Mackie, H. Kumar and S. L. Bearne, FEBS Lett., 2018, 592, 3399–3413 CrossRef CAS PubMed.
- V. Usha, L. G. Dover, D. L. Roper and G. S. Besra, FEMS Microbiol. Lett., 2008, 280, 57–63 CrossRef CAS PubMed.
- T. Miyamoto, M. Katane, Y. Saitoh, M. Sekine and H. Homma, Amino Acids, 2017, 49, 1885–1894 CrossRef CAS PubMed.
- M. G. Snider, B. S. Temple and R. Wolfenden, J. Phys. Org. Chem., 2004, 17, 586–591 CrossRef CAS.
- M. St Maurice and S. L. Bearne, Biochemistry, 2002, 41, 4048–4058 CrossRef PubMed.
- M. L. Harty, A. N. Sharma and S. L. Bearne, Metallomics, 2019, 11, 707–723 CrossRef CAS PubMed.
- G. Gadda and P. Sobrado, Biochemistry, 2018, 57, 3445–3453 CrossRef CAS PubMed.
- M. Nagar, H. Kumar and S. L. Bearne, Protein Eng., Des. Sel., 2018, 31, 135–145 CrossRef CAS PubMed.
- A. Ohtaki, Y. Nakano, R. Iizuka, T. Arakawa, K. Yamada, M. Odaka and M. Yohda, Proteins: Struct., Funct., Bioinf., 2008, 70, 1167–1174 CrossRef CAS PubMed.
- M. Pal and S. L. Bearne, Bioorg. Med. Chem. Lett., 2014, 24, 1432–1436 CrossRef CAS PubMed.
- P. B. Le Calvez, C. J. Scott and M. E. Migaud, J. Enzym. Inhib. Med. Chem., 2009, 24, 1291–1318 CrossRef CAS PubMed.
- Y. Okubo, K. Yokoigawa, N. Esaki, K. Soda and H. Kawai, Biochem. Biophys. Res. Commun., 1999, 256, 333–340 CrossRef CAS PubMed.
- T. Nomura, I. Yamamoto, F. Morishita, Y. Furukawa and O. Matsushima, J. Exp. Zool., 2001, 289, 1–9 CrossRef CAS.
- M. L. di Salvo, R. Florio, A. Paiardini, M. Vivoli, S. D'Aguanno and R. Contestabile, Archives Biochem. Biophys., 2013, 529, 55–65 CrossRef CAS PubMed.
- Y. Mutaguchi, T. Ohmori, T. Wakamatsu, K. Doi and T. Ohshima, J. Bacteriol., 2013, 195, 5207–5215 CrossRef CAS PubMed.
- G. Sánchez-Carrón, T. Fleming, K. E. Holt-Tiffin and D. J. Campopiano, Anal. Chem., 2015, 87, 3923–3928 CrossRef PubMed.
- D. Odokonyero, A. W. McMillan, U. A. Ramagopal, R. Toro, D. P. Truong, M. Z. Zhu, M. S. Lopez, B. Somiari, M. Herman, A. Aziz, J. B. Bonanno, K. G. Hull, S. K. Burley, D. Romo, S. C. Almo and M. E. Glasner, Biochemistry, 2018, 57, 3676–3689 CrossRef CAS PubMed.
- A. M. Brizendine, D. Odokonyero, A. W. McMillan, M. Z. Zhu, K. Hull, D. Romo and M. E. Glasner, Biochem. Biophys. Res. Commun., 2014, 450, 679–684 CrossRef CAS PubMed.
- A. Sakai, D. F. Xiang, C. F. Xu, L. Song, W. S. Yew, F. M. Raushel and J. A. Gerlt, Biochemistry, 2006, 45, 4455–4462 CrossRef CAS PubMed.
- A. W. McMillan, M. S. Lopez, M. Z. Zhu, B. C. Morse, I. C. Yeo, J. Amos, K. Hull, D. Romo and M. E. Glasner, Biochemistry, 2014, 53, 4434–4444 CrossRef CAS PubMed.
- D. Odokonyero, A. Sakai, Y. Patskovsky, V. N. Malashkevich, A. A. Fedorov, J. B. Bonanno, E. V. Fedorov, R. Toro, R. Agarwal, C. X. Wang, N. D. S. Ozerova, W. S. Yew, J. M. Sauder, S. Swaminathan, S. K. Burley, S. C. Almo and M. E. Glasner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8535–8540 CrossRef CAS PubMed.
- D. D. Cao, C. P. Zhang, K. Zhou, Y. L. Jiang, X. F. Tan, J. Xie, Y. M. Ren, Y. X. Chen, C. Z. Zhou and W. T. Hou, Biochem. Biophys. Res. Commun., 2019, 514, 1108–1114 CrossRef CAS PubMed.
- M. Harty, M. Nagar, L. Atkinson, C. M. LeGay, D. J. Derksen and S. L. Bearne, Bioorg. Med. Chem. Lett., 2014, 24, 390–393 CrossRef CAS PubMed.
- N. T. Sorbara, J. W. M. MacMillan, G. D. McCluskey and S. L. Bearne, Org. Biomol. Chem., 2019, 17, 8618–8627 RSC.
- C. Festuccia, G. L. Gravina, A. Mancini, P. Muzi, E. Di Cesare, R. Kirk, M. Smith, S. Hughes, R. Gibson, L.-Y. Lian, E. Ricevuto and A. J. Carnell, Anti-Cancer Agents Med. Chem., 2014, 14, 1031–1041 CrossRef PubMed.
- A. J. Carnell, R. Kirk, M. Smith, S. McKenna, L.-Y. Lian and R. Gibson, ChemMedChem, 2013, 8, 1643–1647 CAS.
- A. Morgenroth, E. A. Urusova, C. Dinger, E. Al-Momani, T. Kull, G. Glatting, H. Frauendorf, O. Jahn, F. M. Mottaghy, S. N. Reske and B. D. Zlatopolskiy, Chem. – Eur. J., 2011, 17, 10144–10150 CrossRef CAS PubMed.
- V. L. Schramm, Acc. Chem. Res., 2015, 48, 1032–1039 CrossRef CAS PubMed.
- V. L. Schramm, Chem. Rev., 2018, 118, 11194–11258 CrossRef CAS PubMed.
- A. Buschiazzo, M. Goytia, F. Schaeffer, W. Degrave, W. Shepard, C. Gregoire, N. Chamond, A. Cosson, A. Berneman, N. Coatnoan, P. M. Alzari and P. Minoprio, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1705–1710 CrossRef CAS PubMed.
- M. Nagar and S. L. Bearne, Biochemistry, 2015, 54, 6743–6752 CrossRef CAS PubMed.
- M. Nagar, A. D. Lietzan, M. St Maurice and S. L. Bearne, Biochemistry, 2014, 53, 1169–1178 CrossRef CAS PubMed.
- A. D. Lietzan, M. Nagar, E. A. Pellmann, J. R. Bourque, S. L. Bearne and M. St Maurice, Biochemistry, 2012, 51, 1160–1170 CrossRef CAS PubMed.
- S. M. Buker, P. A. Boriack-Sjodin and R. A. Copeland, SLAS Discovery, 2019, 24, 515–522 CAS.
- T. Lundqvist, S. L. Fisher, G. Kern, R. H. A. Folmer, Y. F. Xue, D. T. Newton, T. A. Keating, R. A. Alm and B. L. M. de Jonge, Nature, 2007, 447, 817–822 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- M. H. Johansson, Mini-Rev. Med. Chem., 2012, 12, 1330–1344 CAS.
- A. K. Ghosh, I. Samanta, A. Mondal and W. R. Liu, ChemMedChem, 2019, 14, 889–906 CrossRef CAS PubMed.
- A. Tuley and W. Fast, Biochemistry, 2018, 57, 3326–3337 CrossRef CAS PubMed.
- S. Ray and A. S. Murkin, Biochemistry, 2019, 58, 5234–5244 CrossRef CAS PubMed.
- S. E. Dalton and S. Campos, ChemBioChem, 2020, 21, 1080–1100 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- M. R. Hermann, A. Pautsch, M. A. Grundl, A. Weber and C. S. Tautermann, J. Comput.-Aided Mol. Des., 2021, 35, 531–539 CrossRef CAS PubMed.
- T. E. Speltz and R. E. Moellering, Nat. Chem., 2020, 12, 885–887 CrossRef CAS PubMed.
- C. Jöst, C. Nitsche, T. Scholz, L. Roux and C. D. Klein, J. Med. Chem., 2014, 57, 7590–7599 CrossRef.
- H. Johansson, Y. C. I. Tsai, K. Fantom, C. W. Chung, S. Kumper, L. Martino, D. A. Thomas, H. C. Eberl, M. Muelbaier, D. House and K. Rittinger, J. Am. Chem. Soc., 2019, 141, 2703–2712 CrossRef CAS PubMed.
- D. McShan, S. Kathman, B. Lowe, Z. Y. Xu, J. Zhan, A. Statsyuk and I. V. Ogungbe, Bioorg. Med. Chem. Lett., 2015, 25, 4509–4512 CrossRef CAS PubMed.
- S. G. Kathman, I. Span, A. T. Smith, Z. Y. Xu, J. Zhan, A. C. Rosenzweig and A. V. Statsyuk, J. Am. Chem. Soc., 2015, 137, 12442–12445 CrossRef CAS PubMed.
- S. G. Kathman, Z. Y. Xu and A. V. Statsyuk, J. Med. Chem., 2014, 57, 4969–4974 CrossRef CAS PubMed.
- M. Nagar, B. N. Wyatt, M. St Maurice and S. L. Bearne, Biochemistry, 2015, 54, 2747–2757 CrossRef CAS.
- B. Pillai, M. M. Cherney, C. M. Diaper, A. Sutherland, J. S. Blanchard, J. C. Vederas and M. N. G. James, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8668–8673 CrossRef CAS PubMed.
- B. Pillai, V. A. Moorthie, M. J. van Belkum, S. L. Marcus, M. M. Cherney, C. M. Diaper, J. C. Vederas and M. N. G. James, J. Mol. Biol., 2009, 385, 580–594 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS.
- B. S. Axelsson, H. G. Floss, S. Lee, A. Saeed, P. A. Spencer and D. W. Young, J. Chem. Soc., Perkin Trans. 1, 1994, 2137–2142 RSC.
- X. C. Yan, J. M. Sanders, Y.-D. Gao, M. Tudor, A. M. Haidle, D. J. Klein, A. Converso, C. A. Lesburg, Y. Zang and H. B. Wood, J. Chem. Inform. Model., 2020, 60, 4144–4152 CrossRef CAS PubMed.
- Z.-Y. Yang, J.-H. He, A.-P. Lu, T.-J. Hou and D.-S. Cao, J. Med. Chem., 2020, 63, 4411–4429 CrossRef CAS PubMed.
- X. Yang, Y. Wang, R. Byrne, G. Schneider and S. Yang, Chem. Rev., 2019, 119, 10520–10594 CrossRef CAS.
- A. Kumar and K. Y. J. Zhang, Methods, 2015, 71, 26–37 CrossRef CAS PubMed.
- N. Weill, E. Therrien, V. Campagna-Slater and N. Moitessier, Curr. Pharm. Des., 2014, 20, 3338–3359 CrossRef CAS PubMed.
- S. Skariyachan, M. Manjunath and N. Bachappanavar, J. Biomol. Struct. Dyn., 2019, 37, 1146–1169 CrossRef CAS.
- M. D. Lloyd, J. Med. Chem., 2020, 63, 10742–10772 CrossRef PubMed.
- S. Sivendran, V. Jones, D. Sun, Y. Wang, A. E. Grzegorzewicz, M. S. Scherman, A. D. Napper, J. A. McCammon, R. E. Lee, S. L. Diamond and M. McNeil, Bioorg. Med. Chem., 2010, 18, 896–908 CrossRef CAS PubMed.
- S. C. Willies, J. L. White and N. J. Turner, Tetrahedron, 2012, 68, 7564–7567 CrossRef CAS.
- Y. P. Wang, C. Yang, W. Xue, T. Zhang, X. P. Liu, J. S. Ju, B. H. Zhao and D. Liu, BMC Microbiol., 2017, 17, 122 CrossRef.
- M. A. Azam and U. Jayaram, J. Enzym. Inhib. Med. Chem., 2016, 31, 517–526 CrossRef CAS.
- M. Azam and U. Jayaram, J. Enzym. Inhib. Med. Chem., 2018, 33, 726 CrossRef.
- C. de Chiara, M. Homsak, G. A. Prosser, H. L. Douglas, A. Garza-Garcia, G. Kelly, A. G. Purkiss, E. W. Tate and L. P. S. de Carvalho, Nat. Chem. Biol., 2020, 16, 686–694 CrossRef CAS PubMed.
- W. L. Hong, L. F. Chen and J. P. Xie, Expert Opin. Ther. Targets, 2014, 18, 691–701 CrossRef CAS PubMed.
- A. Boyar and R. E. Marsh, J. Am. Chem. Soc., 1982, 104, 1995–1998 CrossRef CAS.
- L. A. Kondacs, S. Orenga, R. J. Anderson, E. C. L. Marrs, J. D. Perry and M. Gray, Molecules, 2020, 25, 1315 CrossRef CAS PubMed.
- H. E. Hoffman, J. Jiraskova, M. Ingr, M. Zvelebil and J. Konvalinka, Protein Expression Purif., 2009, 63, 62–67 CrossRef CAS PubMed.
- J. Jirásková-Vaníčková, R. Ettrich, B. Vorlová, H. E. Hoffman, M. Lepšík, P. Jansa and J. Konvalinka, Curr. Drug Targets, 2011, 12, 1037–1055 CrossRef PubMed.
- H. Mori, R. Wada, S. Takahara, Y. Horino, H. Izumi, T. Ishimoto, T. Yoshida, M. Mizuguchi, T. Obita, H. Gouda, S. Hirono and N. Toyooka, Bioorg. Med. Chem., 2017, 25, 3736–3745 CrossRef CAS.
- M. A. B. MacKay, M. Kravtsenyuk, R. Thomas, N. D. Mitchell, S. M. Dursun and G. B. Baker, Front. Psychiatry, 2019, 10, 25 CrossRef.
- M. J. Wu, Y. M. Liu, H. Zhang, M. L. Lian, J. Chen, H. Y. Jiang, Y. Xu, G. Shan and S. Z. Wu, Exp. Eye Res., 2019, 182, 93–100 CrossRef CAS.
- F. Marchesani, S. Bruno, G. Paredi, S. Raboni, B. Campanini and A. Mozzarelli, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 813–821 CrossRef CAS PubMed.
- S. Beltran-Castillo, J. J. Trivino, J. Eugenin and R. von Bernhardi, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140447 CrossRef CAS.
- A. V. Canosa, S. Faggiano, M. Marchetti, S. Armao, S. Bettati, S. Bruno, R. Percudani, B. Campanini and A. Mozzarelli, Sci. Rep., 2018, 8, 9016 CrossRef.
- H. Mori, R. Wada, J. Li, T. Ishimoto, M. Mizuguchi, T. Obita, H. Gouda, S. Hirono and N. Toyooka, Bioorg. Med. Chem. Lett., 2014, 24, 3732–3735 CrossRef CAS.
- P. Malapati, V. S. Krishna and S. Dharmarajan, Int. J. Mycobact., 2018, 7, 76–83 CrossRef CAS PubMed.
- P. Malapati, V. S. Krishna, R. Nallangi, N. Meda, R. R. Srilakshmi and D. Sriram, Bioorg. Med. Chem., 2018, 26, 177–190 CrossRef CAS PubMed.
- P. Malapati, V. S. Krishna, R. Nallangi, R. R. Srilakshmi and D. Sriram, Eur. J. Med. Chem., 2018, 145, 23–34 CrossRef CAS PubMed.
- J. R. Duvall, L. Bedard, A. M. Naylor-Olsen, A. L. Manson, J. A. Bittker, W. Sun, M. E. Fitzgerald, Z. He, M. D. Lee, J.-C. Marie, G. Muncipinto, D. Rush, D. Xu, H. Xu, M. Zhang, A. M. Earl, M. A. Palmer, M. A. Foley, J. P. Vacca and C. A. Scherer, ACS Infect. Dis., 2017, 3, 349–359 CrossRef CAS.
- E. C. de Azevedo and A. S. Nascimento, J. Struct. Biol., 2019, 207, 158–168 CrossRef CAS.
- C. W. Tan, C. H. H. Hor, S. S. Kwek, H. K. Tee, I. C. Sam, E. L. K. Goh, E. E. Ooi, Y. F. Chan and L. F. Wang, Emerging Microbes Infect., 2019, 8, 426–437 CrossRef CAS.
- S. Hinderlich, M. Neuenschwander, P. R. Wratil, A. Oder, M. Lisurek, L. D. Nguyen, J. P. von Kries and C. P. R. Hackenberger, ChemBioChem, 2017, 18, 1279–1285 CrossRef CAS PubMed.
- O. Nieto-Garcia, P. R. Wratil, L. D. Nguyen, V. Böhrsch, S. Hinderlich, W. Reutter and C. P. R. Hackenberger, Chem. Sci., 2016, 7, 3928–3933 RSC.
- R. E. Marsh, Acta Crystallogr., 1952, 5, 458–462 CrossRef CAS.
- M. A. Sun, Y. J. Wang, Q. Zhang, Y. J. Xia, W. Ge and D. J. Guo, BMC Genomics, 2017, 18, 279 CrossRef PubMed.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- J. Aretz, P. R. Wratil, E. C. Wamhoff, H. G. Nguyen, W. Reutter and C. Rademacher, Can. J. Chem., 2016, 94, 920–926 CrossRef CAS.
- S. L. van der Beek, Y. Le Breton, A. T. Ferenbach, R. N. Chapman, D. M. F. van Aalten, I. Navratilova, G. J. Boons, K. S. McIver, N. M. van Sorge and H. C. Dorfmueller, Mol. Microbiol., 2015, 98, 946–962 CrossRef CAS.
- S. L. van der Beek, A. Zorzoli, E. Çanak, R. N. Chapman, K. Lucas, B. H. Meyer, D. Evangelopoulos, L. P. S. de Carvalho, G.-J. Boons, H. C. Dorfmueller and N. M. van Sorge, Mol. Microbiol., 2019, 111, 951–964 CrossRef CAS PubMed.
- P. A. Mann, A. Müller, K. A. Wolff, T. Fischmann, H. Wang, P. Reed, Y. Hou, W. J. Li, C. E. Müller, J. Y. Xiao, N. Murgolo, X. W. Sher, T. Mayhood, P. R. Sheth, A. Mirza, M. Labroli, L. Xiao, M. McCoy, C. J. Gill, M. G. Pinho, T. Schneider and T. Roemer, PLoS Pathog., 2016, 12, e1005585 CrossRef PubMed.
- D. Sasikala, J. Jeyakanthan and P. Srinivasan, J. Recept. Signal Transduction, 2016, 36, 515–530 CrossRef CAS PubMed.
- M. de Nóbrega, H. L. Cilião, M. F. de Souza, M. R. de Souza, J. M. Serpeloni, P. E. Fuganti and I. M. de Syllos Cólus, Genet. Mol. Biol., 2020, 43, e20180329 CrossRef PubMed.
- H. Xie, L. Nie, M. Zhang, Z. Su, X. Chen, M. Xu, J. Gong, N. Chen and Q. Zhou, Exp. Ther. Med., 2020, 19, 1806–1816 CAS.
- Z. Zhang, M. Garzotto, E. W. Davis, M. Mori, W. A. Stoller, P. E. Farris, C. P. Wong, L. M. Beaver, G. V. Thomas, D. E. Williams, R. H. Dashwood, D. A. Hendrix, E. Ho and J. Shannon, Nutr. Cancer, 2020, 72, 74–87 CrossRef CAS.
- H. Lee, M. Kim, S. H. Kim, Q. Tran, G. Kong, C. Kim, S. H. Kwon, J. Park, J. B. Park, S. Park and J. Park, Front. Oncol., 2020, 10, 550673 CrossRef PubMed.
- K. Todorova, S. Hayrabedyan, T. Shamov, M. Karaivanov, A. Kuzmanov, S. Kyurkchiev and I. Kehayov, C. R. Acad. Bulg. Sci., 2007, 60, 1123–1126 CAS.
- J. Mann, Chem. Soc. Rev., 1987, 16, 381–436 RSC.
- B. M. Johnson, Y. Z. Shu, X. L. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS.
- S. M. Lau, R. K. Brantley and C. Thorpe, Biochemistry, 1988, 27, 5089–5095 CrossRef CAS PubMed.
- P. Vock, S. Engst, M. Eder and S. Ghisla, Biochemistry, 1998, 37, 1848–1860 CrossRef CAS.
- R. Zhang, P. J. McIntyre, P. M. Collins, D. J. Foley, C. Arter, F. von Delft, R. Bayliss, S. Warriner and A. Nelson, Chem. – Eur. J., 2019, 25, 6831–6839 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405–6416 CrossRef CAS PubMed.
- F. Giordanetto, C. Jin, L. Willmore, M. Feher and D. E. Shaw, J. Med. Chem., 2019, 62, 3381–3394 CrossRef CAS PubMed.
- D. A. Erlanson, I. J. P. de Esch, W. Jahnke, C. N. Johnson and P. N. Mortenson, J. Med. Chem., 2020, 63, 4430–4444 CrossRef CAS PubMed.
- C. N. Johnson, D. A. Erlanson, W. Jahnke, P. N. Mortenson and D. C. Rees, J. Med. Chem., 2018, 61, 1774–1784 CrossRef CAS PubMed.
- C. N. Johnson, D. A. Erlanson, C. W. Murray and D. C. Rees, J. Med. Chem., 2017, 60, 89–99 CrossRef CAS.
- P. N. Mortenson, D. A. Erlanson, I. J. P. de Esch, W. Jahnke and C. N. Johnson, J. Med. Chem., 2019, 62, 3857–3872 CrossRef CAS PubMed.
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, J. Med. Chem., 2016, 59, 8189–8206 Search PubMed.
- P. Kirsch, A. M. Hartman, A. K. H. Hirsch and M. Empting, Molecules, 2019, 24, 4309 CrossRef CAS PubMed.
- M. R. Bentley, O. V. Ilyichova, G. Wang, M. L. Williams, G. Sharma, W. S. Alwan, R. L. Whitehouse, B. Mohanty, P. J. Scammells, B. Heras, J. L. Martin, M. Totsika, B. Capuano, B. C. Doak and M. J. Scanlon, J. Med. Chem., 2020, 63, 6863–6875 CrossRef CAS PubMed.
- S. W. Draxler, M. Bauer, C. Eickmeier, S. Nadal, H. Nar, D. Rangel Rojas, D. Seeliger, M. Zeeb and D. Fiegen, J. Med. Chem., 2020, 63, 5856–5864 CrossRef CAS.
- A. Bancet, C. Raingeval, T. Lomberget, M. Le Borgne, J.-F. Guichou and I. Krimm, J. Med. Chem., 2020, 63, 11420–11435 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |