
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron deficient borane-mediated hydride abstraction in amines: stoichiometric and catalytic processes
Shyam
Basak
a,
Laura
Winfrey
b,
Betty A.
Kustiana
a,
Rebecca L.
Melen
 *a,
Louis C.
Morrill
*a and
Alexander P.
Pulis
*b
*a,
Louis C.
Morrill
*a and
Alexander P.
Pulis
*b
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: MelenR@cardiff.ac.uk; MorrillLC@cardiff.ac.uk
bSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: a.pulis@leicester.ac.uk
First published on 3rd February 2021
Abstract
The manipulation of amino C–H bonds has garnered significant interest from the synthetic community due to its inherently high atom, step and redox economy. This Tutorial Review summarises the ability of boranes to mediate hydride abstraction from α-amino and γ-amino conjugated C–H bonds. Borane-mediated hydride abstraction results in the generation of reactive iminium hydridoborate salts that participate in a variety of stoichiometric and catalytic processes. The reactions that have utilised this unusual reactivity include those that manipulate amino scaffolds (including dehydrogenation, racemisation, isomerisation, α- and β-functionalisation, and C–N bond cleavage) and those that use amine-based reagents (transfer hydrogenation, and alkylation).
 Shyam Basak | Dr Shyam Basak obtained his PhD from Indian Institute of Technology Kharagpur under the supervision of Prof. Dipakranjan Mal in 2017. Then he joined the group of Prof. Amit Basak as a research associate in the same institute for one year. In 2018, he moved to UK to pursue postdoctoral research at Cardiff University in the lab of Dr Louis Morrill to work in the field of FLP chemistry in collaboration with Dr Rebecca Melen. Currently, he is continuing as a postdoctoral researcher in the Morrill group to work on synthetic organic electrochemistry. |
 Laura Winfrey | Laura Winfrey completed her MChem degree with a year abroad in 2019 at the University of Leicester (UK) and Kent State University (USA). Laura is currently studying for her PhD at the University of Leicester with Dr Alex Pulis where she is discovering new catalytic methods for the sustainable synthesis of amines. |
 Betty A. Kustiana | Betty A. Kustiana received a Master in Chemistry at University of Copenhagen and is currently pursuing her PhD at Cardiff University focused on the main group catalysis. |
 Rebecca L. Melen | Dr Rebecca Melen studied for her PhD degree at the University of Cambridge (UK). Following Postdoctoral studies in Toronto (Canada) and Heidelberg (Germany), she took up a position at Cardiff University (UK) in 2014 where she is now a Reader in Inorganic Chemistry. In 2018, she was awarded an EPSRC early career fellowship and she was the 2019 recipient of the RSC Harrison Meldola Memorial Prize. Her research interests include diverse aspects of main group reactivity and catalysis, including the applications of main group chemistry in organic synthesis. |
 Louis C. Morrill | Dr Louis Morrill received his PhD from the University of St Andrews in 2014 under the direction of Prof. Andrew Smith and undertook postdoctoral research at UC Berkeley with Prof. Richmond Sarpong. In June 2015, he initiated his independent research career at Cardiff University. Research in the group is focused on inventing new reactions in organic chemistry and developing sustainable catalytic methodologies for synthesis. |
 Alexander P. Pulis | Dr Alex Pulis obtained his PhD from the University of Bristol (UK) under the guidance of Prof. Varinder K. Aggarwal. In 2014, he joined Prof. Douglas Stephan at the University of Toronto (Canada) for postdoctoral studies. He then moved to the University of Manchester (UK) as a fixed term Lecturer within the group of Prof. David J. Procter. Alex began his independent career at the University of Leicester (UK) in 2018 where he explores the reactivity of main group elements and applies these finding to challenges in chemical synthesis. |
Key learning points1. Amino C(sp3)–H functionalisation processes are a powerful strategy for the synthesis of complex amines.2. Electron deficient boranes are capable of efficiently mediating hydride abstraction from a variety of amine substrates. 3. Borane-mediated hydride abstraction generates reactive intermediates that can be processed in a variety of chemical reactions. 4. Borane-mediated hydride abstraction is a versatile process that has been utilised in a variety of catalytic and stoichiometric processes, including sophisticated catalytic cycles and cascade reactions. 5. The current state of the borane-mediated hydride abstraction field and its exciting future prospects. |
1 Introduction
Hydride transfer is a fundamental reactivity principle that has found numerous applications in synthesis.1,2 One of the archetypal examples of which is during the reduction of functional groups via hydride donation from a hydridoborate (a.k.a. borohydride) anion, such as in the conversion of carbonyls to alcohols or imines to amines.2 The reverse process (hydride abstraction from a C–H bond by a borane, resulting in oxidation of the carbon atom) is far less studied and has found little utility in synthesis (Scheme 1a). However, given the great interest in C–H functionalisation strategies in synthesis due to high atom, step and redox economy, the use of borane-mediated hydride abstraction from an organic molecule is being recognised as an efficient route to generate reactive intermediates for downstream chemical processes.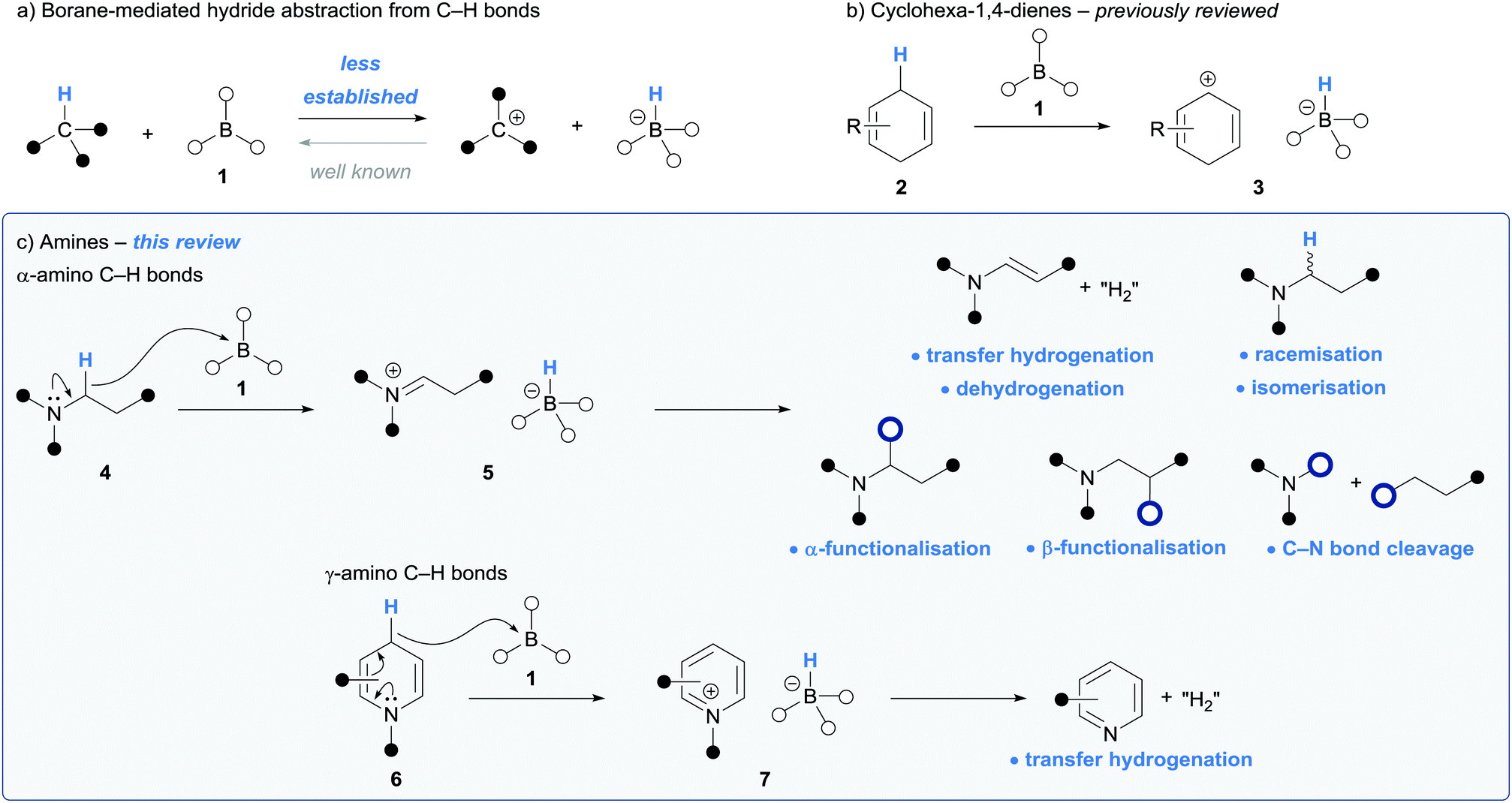 | ||
| Scheme 1 Electron deficient borane-mediated hydride abstraction from C–H bonds. | ||
There are two general classes of organic substrates that undergo hydride abstraction with an electron deficient borane (1): cyclohexa-1,4-dienes (2) and amines (4 and 6) (Scheme 1b and c). Hydride abstraction involving cyclohexa-1,4-dienes generates Wheland-type intermediates 3 and has been successfully utilised in various hydroelementation processes. This area, pioneered by Oestreich and co-workers, has been recently reviewed.3–5
Iminium ions are valuable molecules that are widely applied in synthetic applications, for example in the Mannich reaction,6 and are usually formed via the condensation of an amine and a carbonyl derivative. Amines (e.g.4 and 6) that bear α-amino and γ-amino conjugated C–H bonds respectively, undergo borane-mediated hydride abstraction to form iminium hydridoborate salts (5 and 7). This strategy is complementary to traditional approaches as the reactive iminium is formed directly from the parent amine in situ. In addition, there is great interest from the synthetic community, especially from the pharmaceutical industry,7 in efficient synthetic methods involving the C–H functionalisation of amines. Borane-mediated hydride abstraction in amines has provided an unusual but powerful method to mediate amine C–H functionalisation reactions and to activate amine-based reagents. This is reflected in the recent flurry of reports that utilise this reactivity.
This Tutorial Review will summarise processes involving borane-mediated hydride abstraction of amino C–H bonds. In both cases of α- and γ-amino conjugated hydride abstraction, the use of stoichiometric borane and then borane-catalysed processes are described. The use of tris(pentafluorophenyl)borane, B(C6F5)3, will be most frequently encountered in this context, but other boranes have also been explored more recently. We will also describe the proposed mechanistic steps and catalytic cycles that underpin these processes, focussing on the fate of both the iminium moiety and the hydridoborate counterion.
2 Borane-mediated α-amino hydride abstraction
2.1 Stoichiometric studies
This section will introduce the array of amines that undergo borane-mediated α-amino hydride abstraction with a stoichiometric quantity of borane, including zirconium and zinc coordinated N-compounds, N-aryl amines, aliphatic tertiary amines, as well as intramolecular hydride transfer. The following sections are organised based on the fate of the resultant iminium hydridoborate salt (observation only, proton transfers, nucleophilic addition to the iminium moiety, and further hydride transfer), although there will be some overlap between these topics.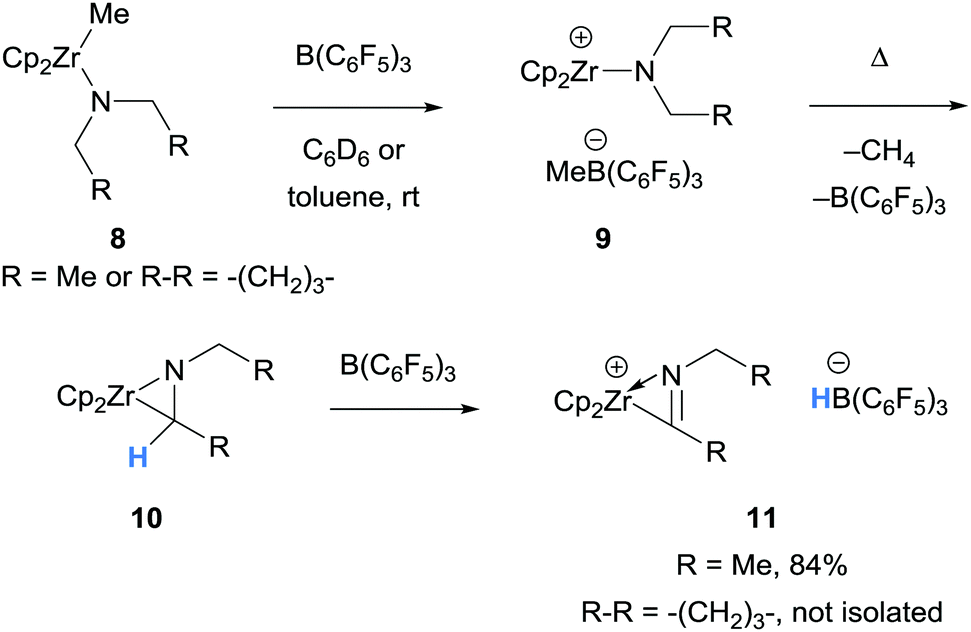 | ||
| Scheme 2 B(C6F5)3-mediated α-amino hydride abstraction in the formation of (η2-iminoacyl)zirconocene cations. Cp = cyclopentadienyl. | ||
The first example of borane-mediated α-amino hydride abstraction from simple amines was reported by Santini and co-workers in 2002 during NMR analysis of mixtures of dialkyl anilines and B(C6F5)3 (Scheme 3).9 In the case of dimethylaniline (12a), Lewis acid–base pairs 13a were observed along with iminium hydridoborate ion pair 14a, where the latter presumably forms as a result of borane-mediated hydride abstraction. On the other hand, diethyl aniline (12b) formed ammonium hydridoborate ion pair 15 and a mixture of E- and Z-iminium-borate zwitterions 17. Zwitterions 17 are formed as a result of proton transfer from iminium 14b to diethyl aniline (12b) and subsequent addition of enamine 16 to B(C6F5)3. Further examples of the formation of iminium hydridoborates and subsequent proton transfers are given in Section 2.1.2.
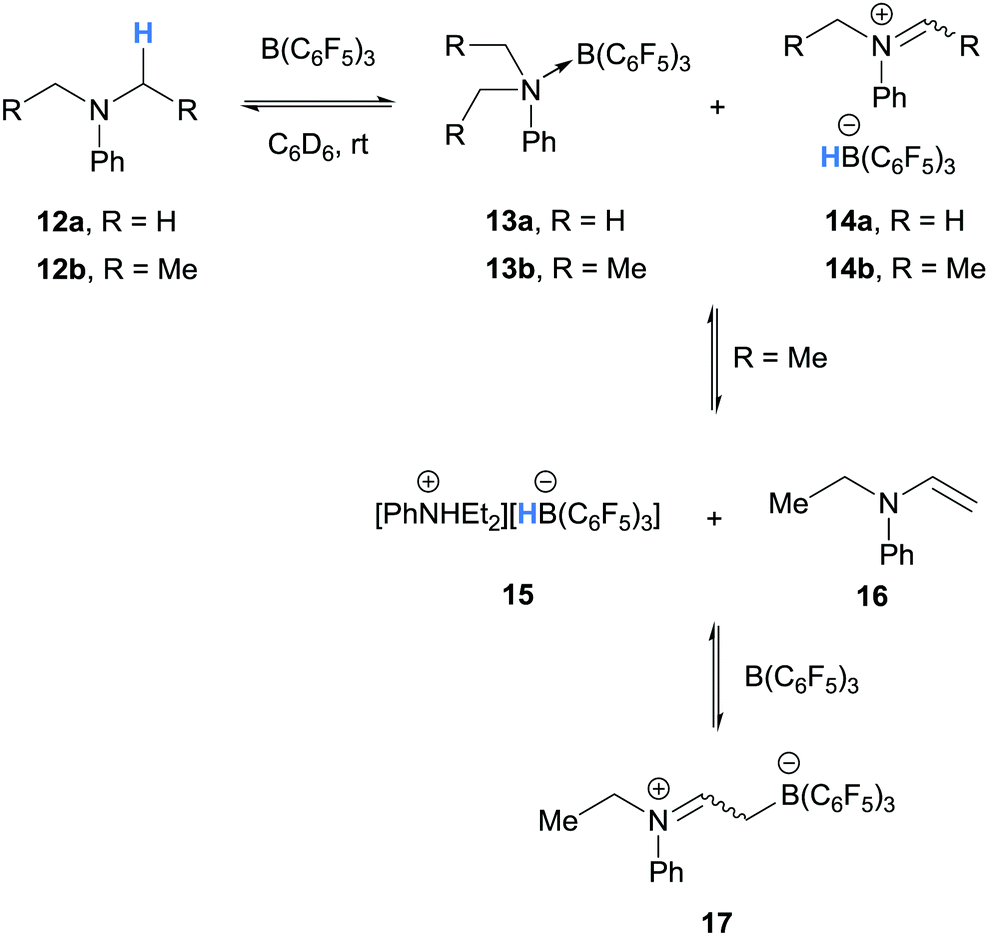 | ||
| Scheme 3 B(C6F5)3-mediated α-amino hydride abstraction from N,N-dialkylanilines. | ||
In 2006, Chen and co-workers reported that binuclear zinc enolate complexes 18 in combination with B(C6F5)3, form cationic zinc enolates 19, which were found to be highly active catalysts for acrylate polymerisation (Scheme 4).10
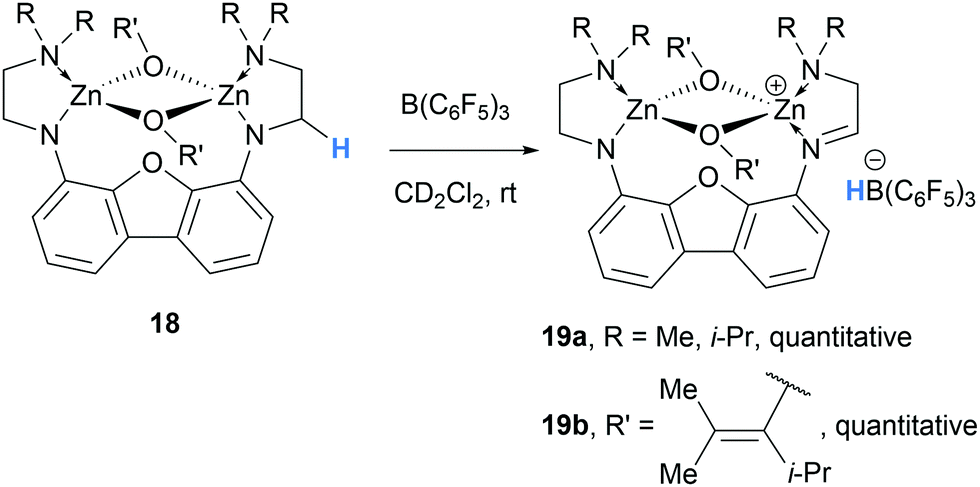 | ||
| Scheme 4 B(C6F5)3-mediated α-amino hydride abstraction in from cationic zinc enolates. | ||
In 2012, as part of a study of amine/B(C6F5)3 frustrated Lewis pairs, Erker, Stephan and co-workers further expanded the range of amines that undergo borane-mediated hydride abstraction.11 It was found that N,N-dimethylaniline (12a), used as supplied, reacted with B(C6F5)3 to produce the iminium-borohydride ion pair 14a, as in the study by Santini and co-workers.9 In contrast, hydride abstraction was not observed when N,N-dimethylaniline (12a) was carefully purified and the reaction was performed under rigorously dry conditions. The reaction of N-isopropyl aniline (20) with B(C6F5)3 resulted in hydride abstraction, where iminium-hydridoborate 21 was a minor component of an equilibrium with adduct 22 (Scheme 5a). On treatment of B(C6F5)3 with 1,4-C6H4(CH2NHt-Bu)2 (23), hydride abstraction was also observed in addition to mono- and bis-amine-borane adducts 25 and 26 (Scheme 5b). On the other hand, hydride abstraction was not observed in the reaction of benzyldimethyl amine and B(C6F5)3.
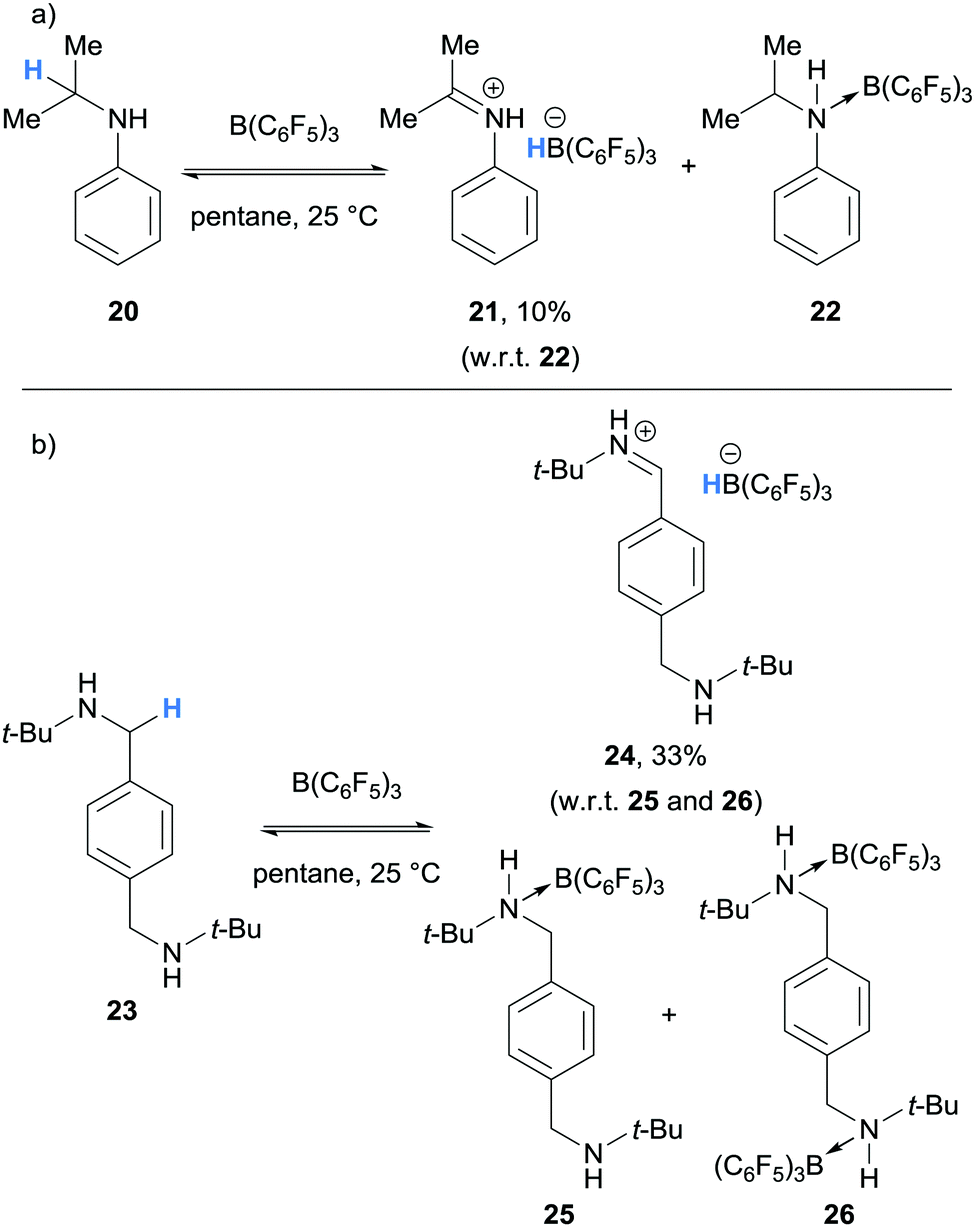 | ||
| Scheme 5 B(C6F5)3-mediated α-amino hydride abstraction from secondary amines. | ||
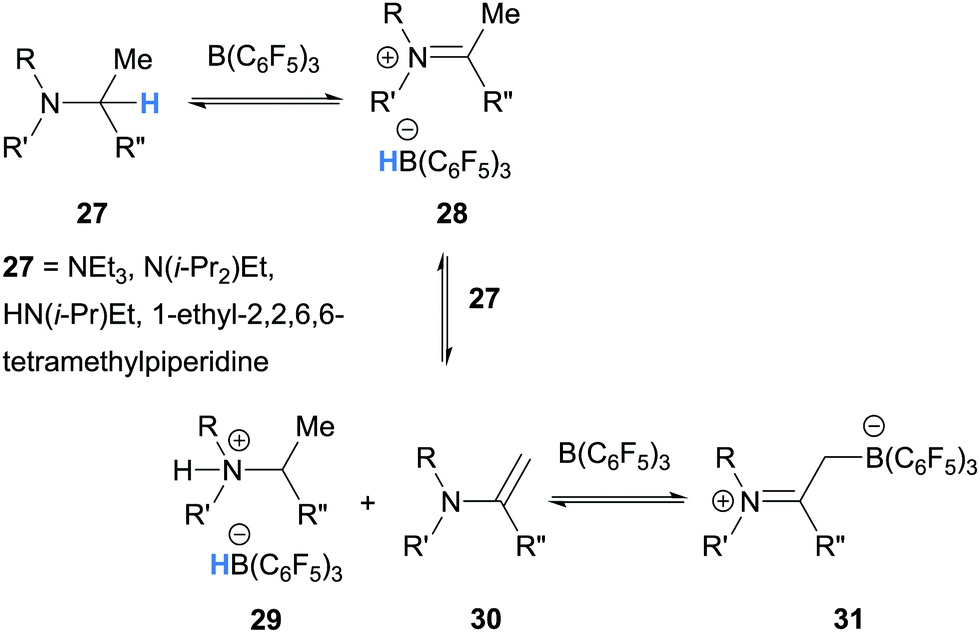 | ||
| Scheme 6 B(C6F5)3-mediated α-amino hydride abstraction from aliphatic tertiary amines. | ||
In a related process, Focante and co-workers, and Resconi and co-workers showed that the reaction of N-methylindoline (32) and B(C6F5)3 forms ammonium-hydridoborate 35 and zwitterion 36 (Scheme 7).15,16 In this case, B(C6F5)3-mediated hydride abstraction of indoline 32 and subsequent deprotonation of the iminium hydridoborate salt 33 forms a mixture of ammonium-hydridoborate 35 and indole 34. The reaction of indole 34 and B(C6F5)3 is known to form zwitterion 36.17
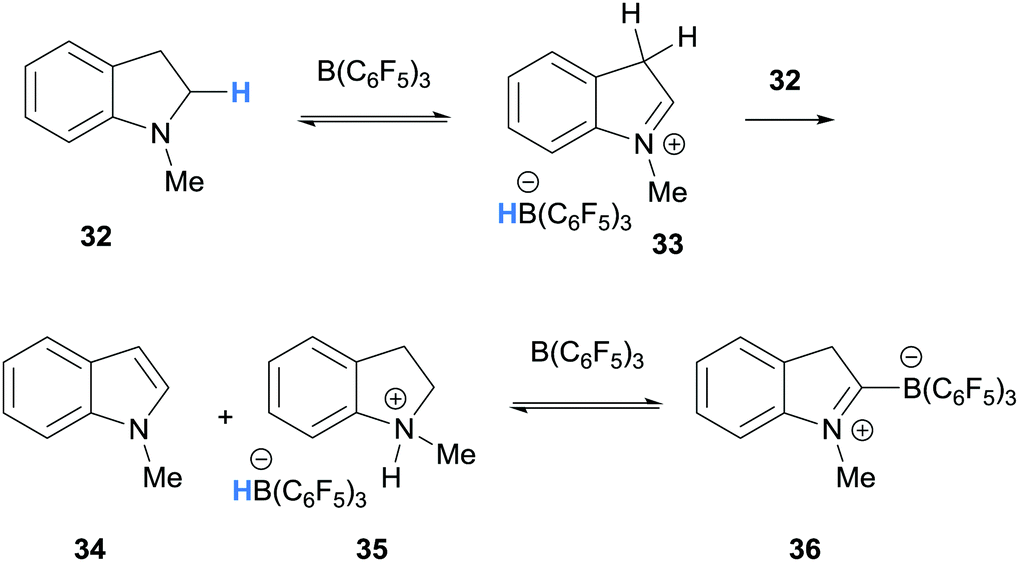 | ||
| Scheme 7 B(C6F5)3-mediated α-amino hydride abstraction from N-methylindoline. | ||
Grimme, Erker and co-workers performed a combined theoretical and experimental study on the generation and stability of α-ferrocenyl carbenium ions. Hydride abstraction was observed in the reaction of (±)-dimethylamino-[3]ferrocenophanes (±)-37a and (±)-37b with B(C6F5)3 and the ferrocene stabilised iminium hydridoborate salts (±)-38a and (±)-38b were formed (Scheme 8).18–20 The iminium moiety in 38a was also hydrolysed to form (±)-ferrocenophane ketone (±)-39.19
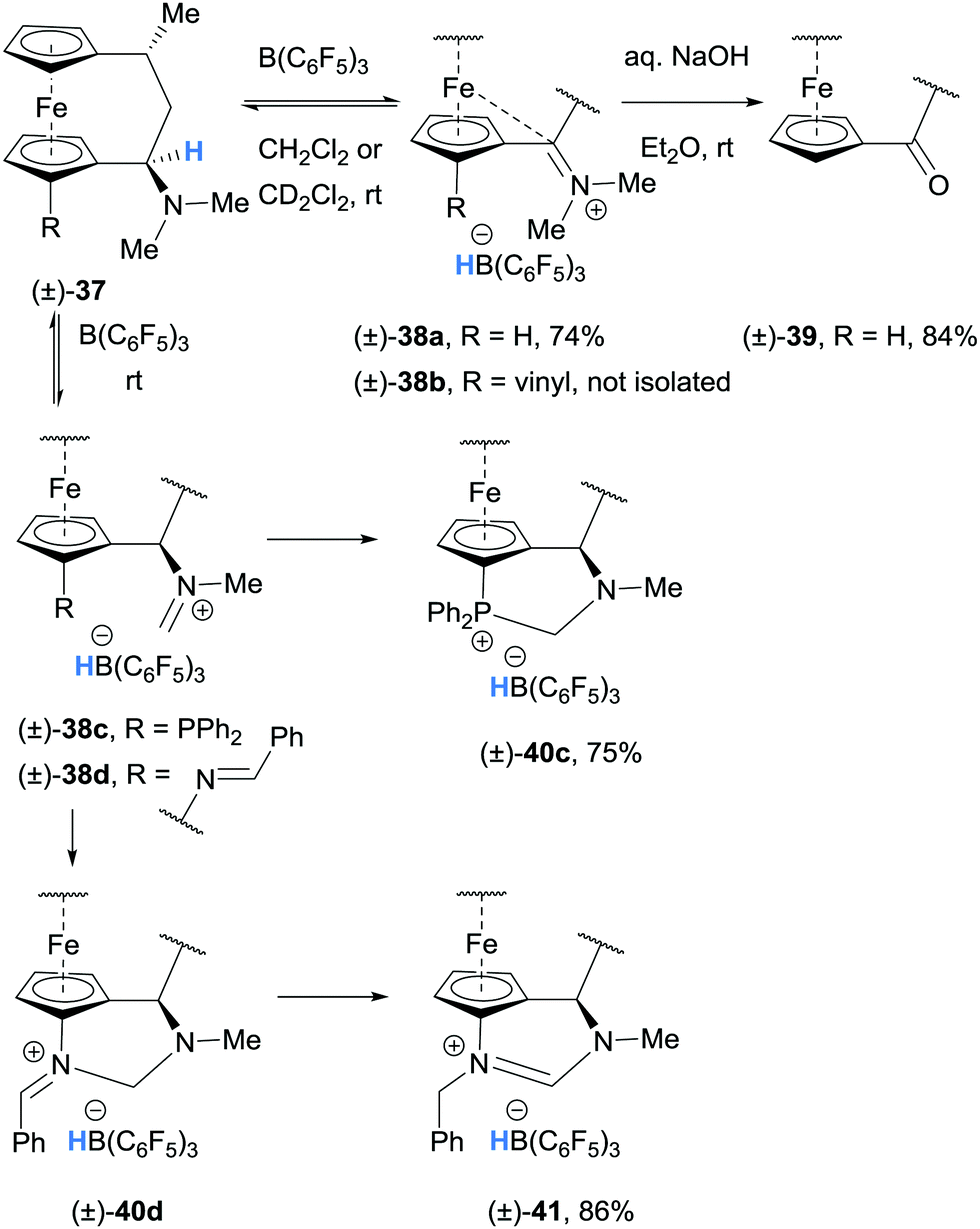 | ||
| Scheme 8 B(C6F5)3-mediated α-amino hydride abstraction from dimethylamino-[3]ferrocenophanes (±)-37 and resultant reactivity. | ||
Analogous ferrocenophanes (±)-37c and (±)-37d also underwent hydride abstraction with B(C6F5)3. In both cases, (±)-37c and (±)-37d appeared to undergo hydride abstraction at the N-CH3 substituent (rather than the C-secondary amino C–H bond as in (±)-37a and (±)-37b) resulting in formation of (±)-40c and (±)-41.21
In iminium hydridoborate (±)-38c, the pendant phosphine moiety traps the electrophilic iminium carbon to form phosphonium salt (±)-40c. Similarly, (±)-38d, that bears a pendant imine, forms (±)-41via intermediate (±)-40d (Scheme 8).
In a related process, Mercandelli, D’Alfonso, Resconi and co-workers reported the intramolecular trapping of iminium hydridoborate 43, generated via B(C6F5)3-mediated hydride abstraction in 1,8-bis(dimethylamino)naphthalene (42), with a pendant amino group (Scheme 9).22
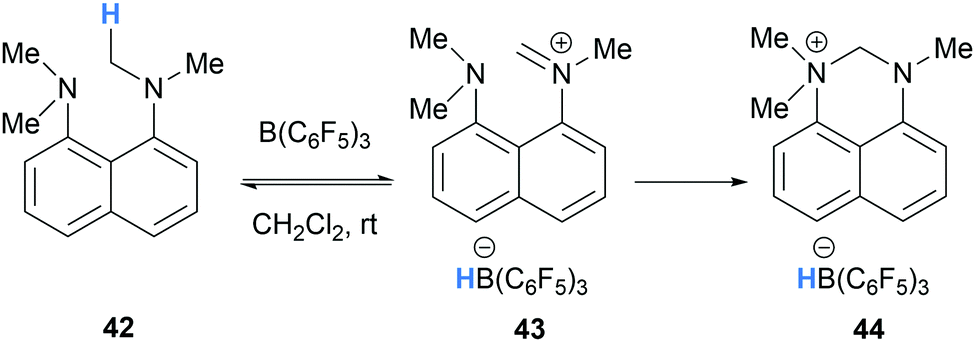 | ||
| Scheme 9 B(C6F5)3-mediated α-amino hydride abstraction from 1,8-bis(dimethylamino)naphthalene 42 and resultant intramolecular nucleophilic addition. | ||
In 2015, Chen and co-workers studied the application of a Et3N/B(C6F5)3 Lewis pair towards the polymerisation of acrylate monomers.23 Instead of polymerisation taking place, a mixture of Et3N/B(C6F5)3/methyl methacrylate (45) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) afforded methyl isobutyrate 46 in quantitative yield (Scheme 10a). NMR experiments revealed that upon mixing Et3N, B(C6F5)3 and methyl methacrylate in a 1:1:1 ratio, a mixture of zwitterion 47 and ammonium-hydridoborate salt 48 was formed. This observation was in line with previous reports of borane-mediated α-amino hydride abstraction followed by deprotonation of the iminium hydridoborate salt as described above in Section 2.1.2. The addition of one further equivalent of B(C6F5)3 presumably activates methyl methacrylate (45) for reduction by the hydridoborate counterion in 48 (Scheme 10b). Subsequent proton transfers in salt 49 results in the formation of the hydrogenated product 46.
:2:1) afforded methyl isobutyrate 46 in quantitative yield (Scheme 10a). NMR experiments revealed that upon mixing Et3N, B(C6F5)3 and methyl methacrylate in a 1:1:1 ratio, a mixture of zwitterion 47 and ammonium-hydridoborate salt 48 was formed. This observation was in line with previous reports of borane-mediated α-amino hydride abstraction followed by deprotonation of the iminium hydridoborate salt as described above in Section 2.1.2. The addition of one further equivalent of B(C6F5)3 presumably activates methyl methacrylate (45) for reduction by the hydridoborate counterion in 48 (Scheme 10b). Subsequent proton transfers in salt 49 results in the formation of the hydrogenated product 46.
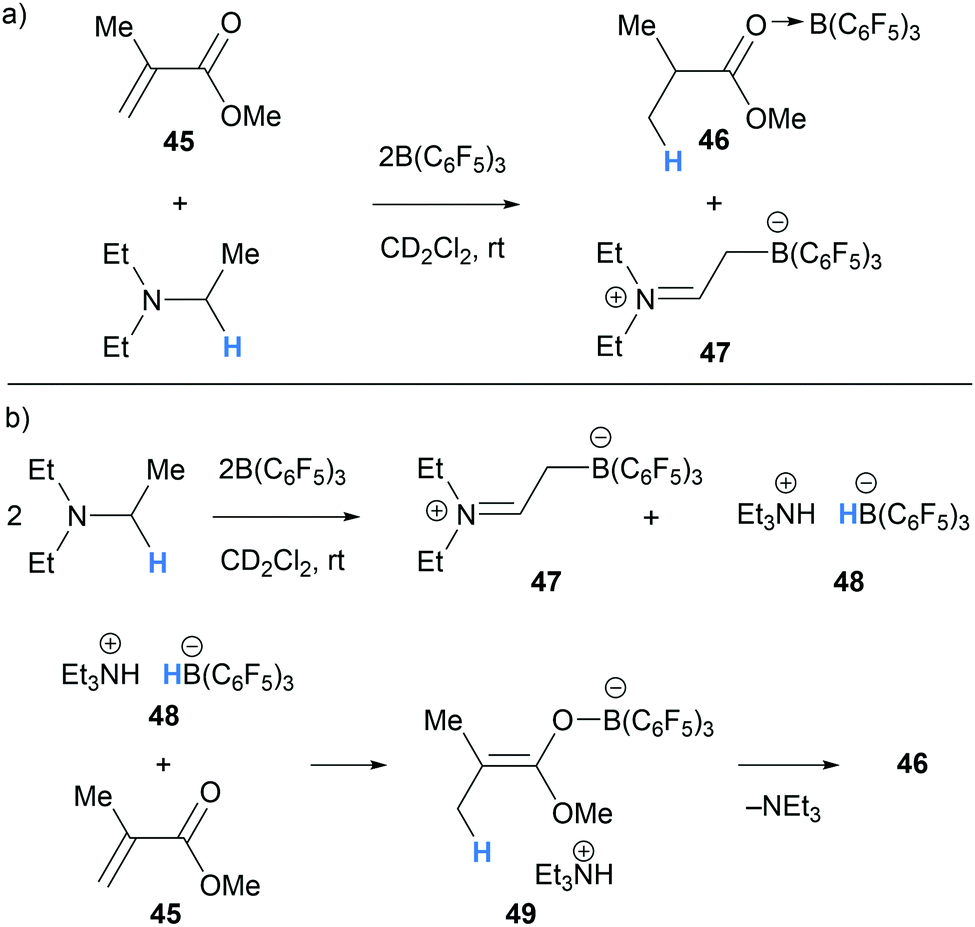 | ||
| Scheme 10 B(C6F5)3-mediated α-amino hydride abstraction utilised in transfer hydrogenation of methyl methacrylate. | ||
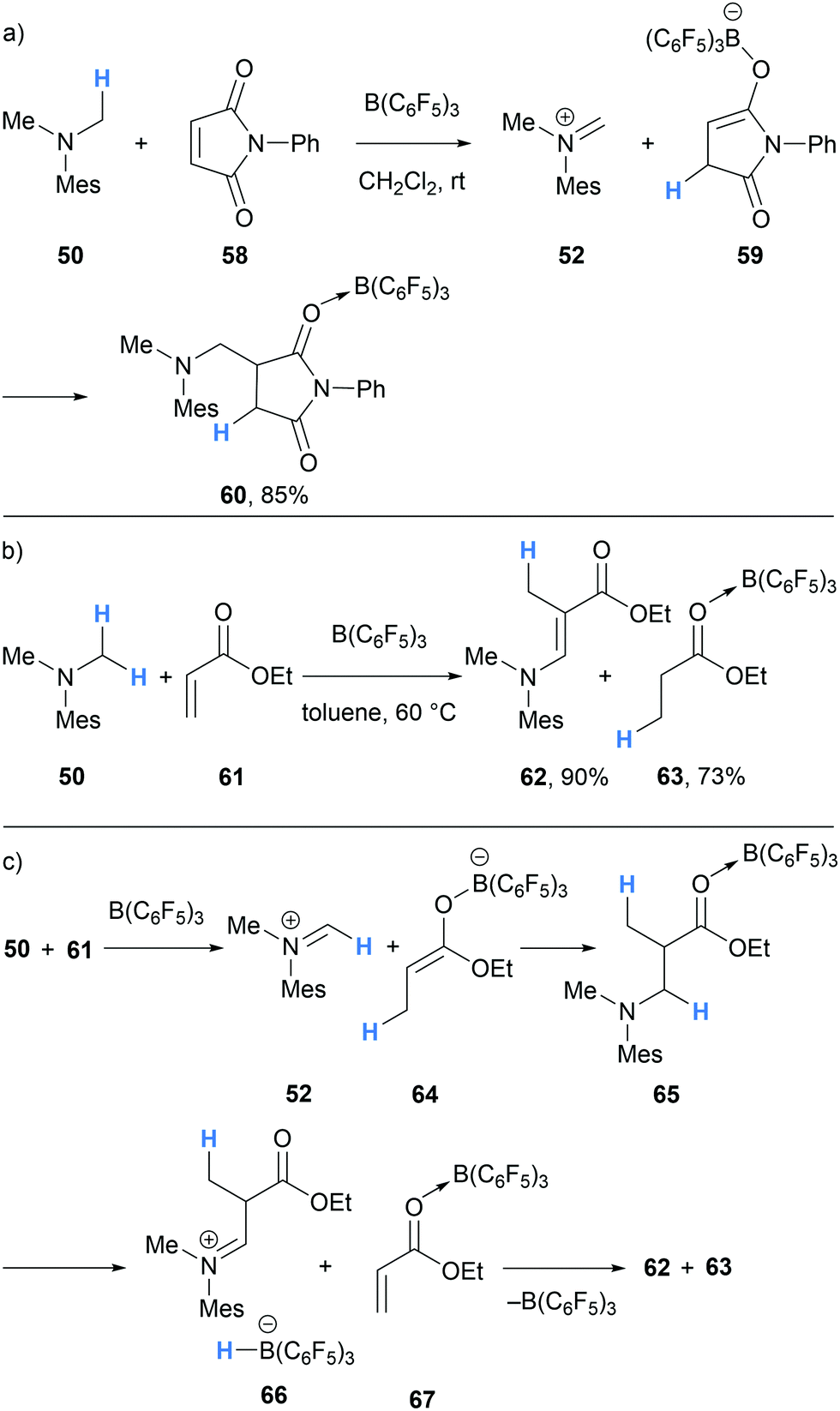
In 2017, Erker and co-workers undertook experimental and theoretical studies on the B(C6F5)3-mediated α-amino hydride abstraction from N-methyl amines in the presence of electron deficient alkynes and alkenes (Schemes 11 and 12).24 In all cases, the generated hydridoborate counterions in the iminium salts (cf.14a–14b) reduced the electron deficient alkyne or alkene to form an enolate (cf.53, 59, 64). The nucleophilic enolates reacted with the iminium electrophile in a Mannich-type process to form new C–C bonds. In the case of the reaction of N,N-dimethylmesitylamine (50), dimethylacetylenedicarboxylate (51) and B(C6F5)3, a nucleophilic allenoate 53 is formed. After C–C bond formation, the resulting product 54 isomerised to zwitterion 55 (Scheme 11a). An analogous process was reported in the reaction of 1,2,2,6,6-pentamethylpiperidine (56) and dimethylacetylenedicarboxylate (51) and B(C6F5)3 (Scheme 11b).
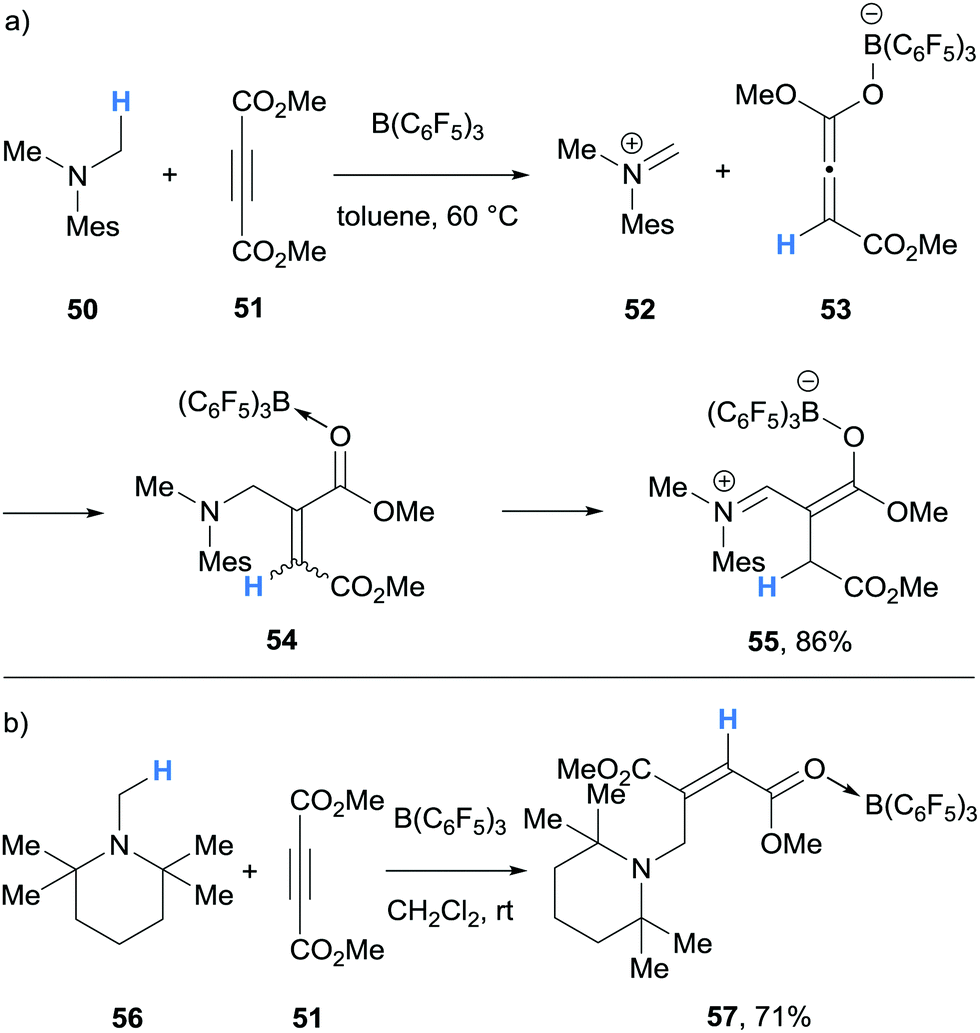 | ||
| Scheme 11 B(C6F5)3-mediated α-amino hydride abstraction and subsequent hydride transfer to electron deficient alkynes utilised in C–C bond forming reactions. Mes = 2,4,6-trimethylphenyl. | ||
 | ||
| Scheme 12 B(C6F5)3-mediated α-amino hydride abstraction and subsequent hydride transfer to electron deficient alkenes utilised in C–C bond forming reactions. | ||
Similar reactivity was also observed when α,β-unsaturated carbonyl compounds were used (Scheme 12). In the case of N-phenyl-maleimide (58), hydride abstraction and subsequent transfer generated enolate 59, which then added to the iminium carbon in 52 to form succinamide 60. The reaction of ethyl acrylate (61), amine 50 and B(C6F5)3 initially followed a similar course and formed Mannich product 65. However, 65 also underwent further B(C6F5)3-mediated hydride abstraction to form iminium hydridoborate 66, which after proton transfer, resulted in the transfer hydrogenation of ethyl acrylate to form ethyl propionate (63).
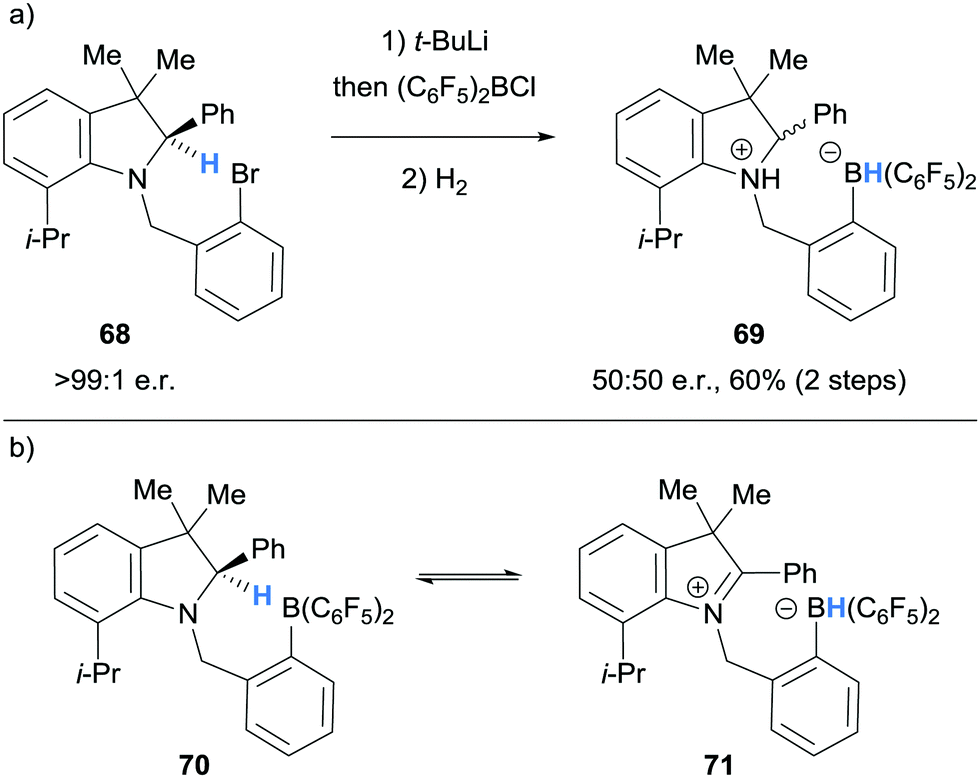
In 2011, Repo and co-workers observed racemisation during the synthesis of an ammonium hydridoborate 69 from enantioenriched amine 68 (>99:1 e.r., Scheme 13a).25 To explain this observation, it was proposed that the intermediate amino borane 70, underwent reversible intramolecular hydride abstraction from the α-amino C–H bond to the B(C6F5)2 moiety giving 71 (Scheme 13b).
 | ||
| Scheme 13 Borane-mediated intramolecular α-amino hydride abstraction and racemisation. | ||
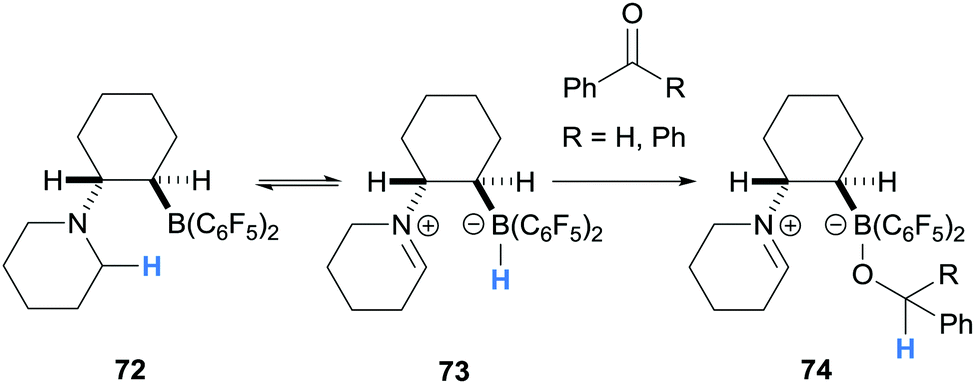
During a study of ethylene and hexylene bridged amino borane FLPs, Erker and co-workers reported the synthesis of zwitterions 74 from amino borane 72 (Scheme 14).26 In this case, intramolecular hydride abstraction from the α-amino C–H bond to the B(C6F5)2 moiety generated iminium hydridoborate 73 that reduced benzaldehyde or benzophenone yielding 74.
 | ||
| Scheme 14 Borane-mediated intramolecular α-amino hydride abstraction and subsequent carbonyl reduction. | ||
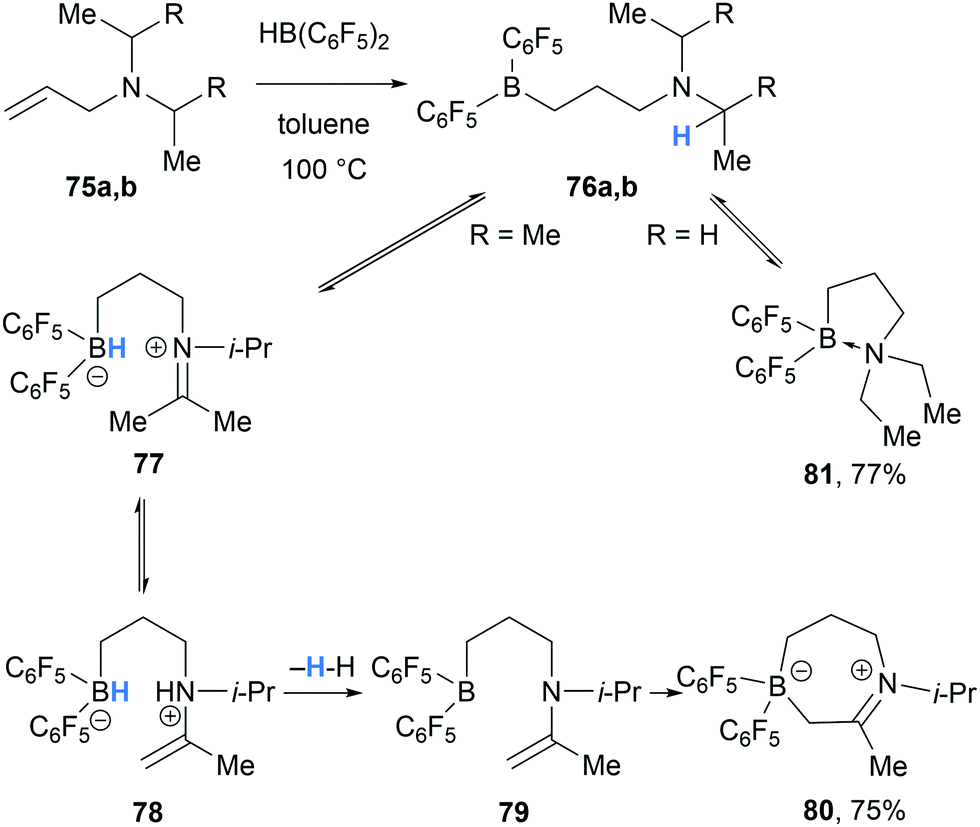
In 2012, Mitzel and co-workers reported the formation of cyclic iminium borate 80 in the reaction between HB(C6F5)2 and N,N-diisopropylallylamine (75a) (Scheme 15).27 The formation of 80 was proposed to proceed via hydroboration of 75a with HB(C6F5)2 to form amino borane 76a. Intramolecular borane-mediated hydride abstraction in 76a formed iminium hydridoborate zwitterion 77, which underwent proton transfer generating enaminium hydridoborate 78. Dehydrogenation of 78 then allowed for intramolecular nucleophilic addition of the enamine moiety to the three-coordinate borane in 79 to form the cyclic product 80. In contrast, less hindered allyl amines, such as diethylallylamine (75b), did not form the analogous cyclic product, and instead formed intramolecular Lewis adduct 81.
 | ||
| Scheme 15 Borane-mediated intramolecular α-amino hydride abstraction and subsequent dehydrogenation and cyclisation. | ||
In a related reaction, Mitzel and co-workers reported the formation of seven-membered iminium-borate 86 during the hydroboration of N,N-diallyl-N-tert-butylamine (82) with HB(C6F5)2 (Scheme 16).28 In line with previous reports, mono-hydroboration of 82 generated the amino borane 83 which undergoes intramolecular α-amino hydride abstraction to form α,β-unsaturated iminium hydridoborate 84. We propose that iminium-borate 84 then undergoes intramolecular conjugate reduction from the hydridoborate moiety to the β-position of the α,β-unsaturated iminium 84. The generated enamine 85 intramolecularly traps the electrophilic borane moiety to form the cyclic zwitterion 86.
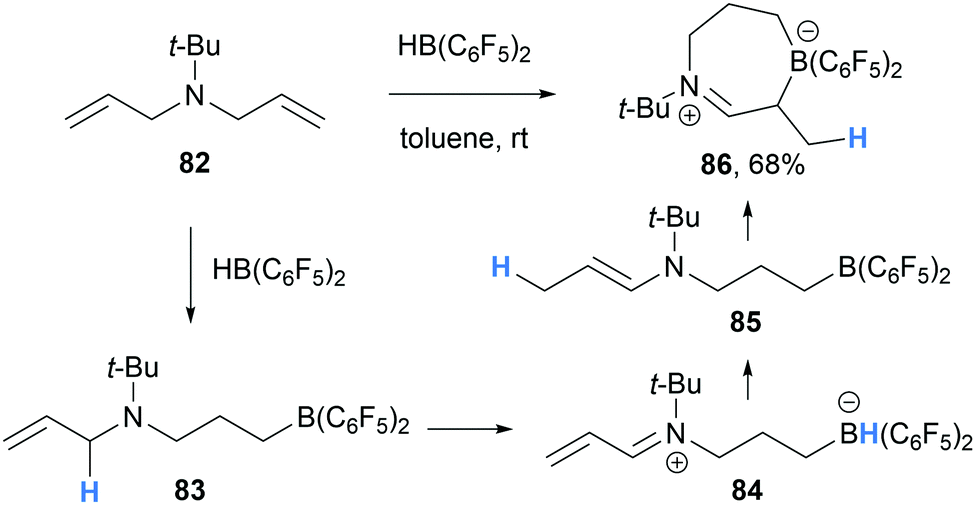 | ||
| Scheme 16 Borane-mediated intramolecular α-amino hydride abstraction and subsequent hydride transfer and cyclisation. | ||
In 2016, Erker and co-workers reported an interesting 1,2-hydride migration from an α-amino C–H bond to a borane atom (Scheme 17).29 The dimer 88 was formed after double hydroboration of N,N-diallylaniline (87) with C6F5BH2. Under dilute conditions, dimer 88 dissociates to form monomer 89 which can undergo the stereospecific 1,2-hydride migration to form cis-iminium-hydridoborate zwitterion 90. The cis-isomer 90 slowly equilibrates to a 1:1 mixture with the trans-isomer, presumably via intermolecular hydride transfer.
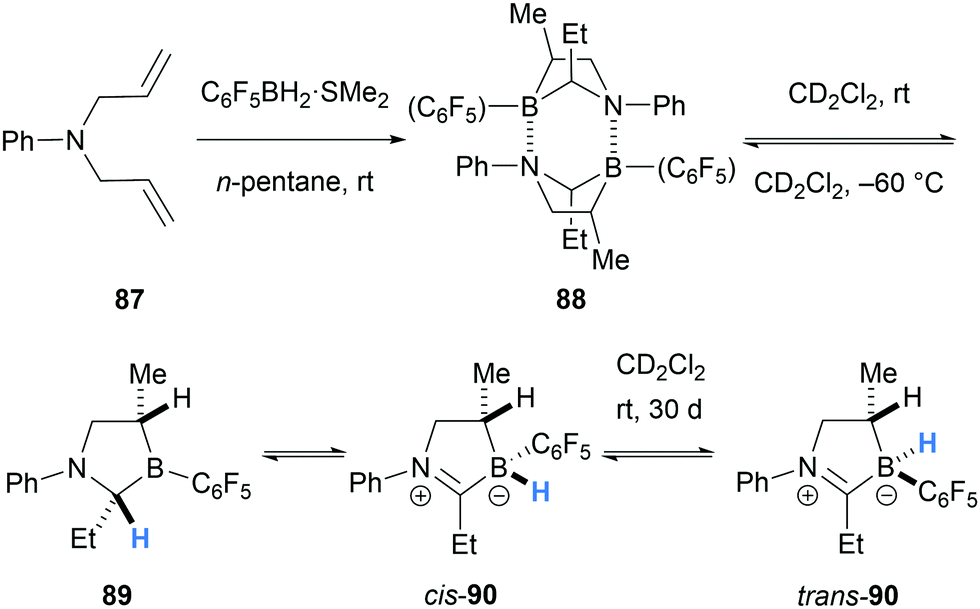 | ||
| Scheme 17 1,2-Hydride migration from an α-amino C–H bond to a borane atom. | ||
2.2 Catalytic applications
Borane-mediated α-amino hydride abstraction has been successfully utilised in a variety of catalytic manifolds by virtue of the range of processes by which the hydridoborate counterion can react in downstream processes. In this section we will discuss borane-catalysed transfer hydrogenation, dehydrogenation, racemisation, isomerisation, α- and β-functionalisation of amines, and processes that involve C–N bond cleavage. All of these transformations are made possible by the unique hydride acceptor and donation abilities of the boron Lewis acids used.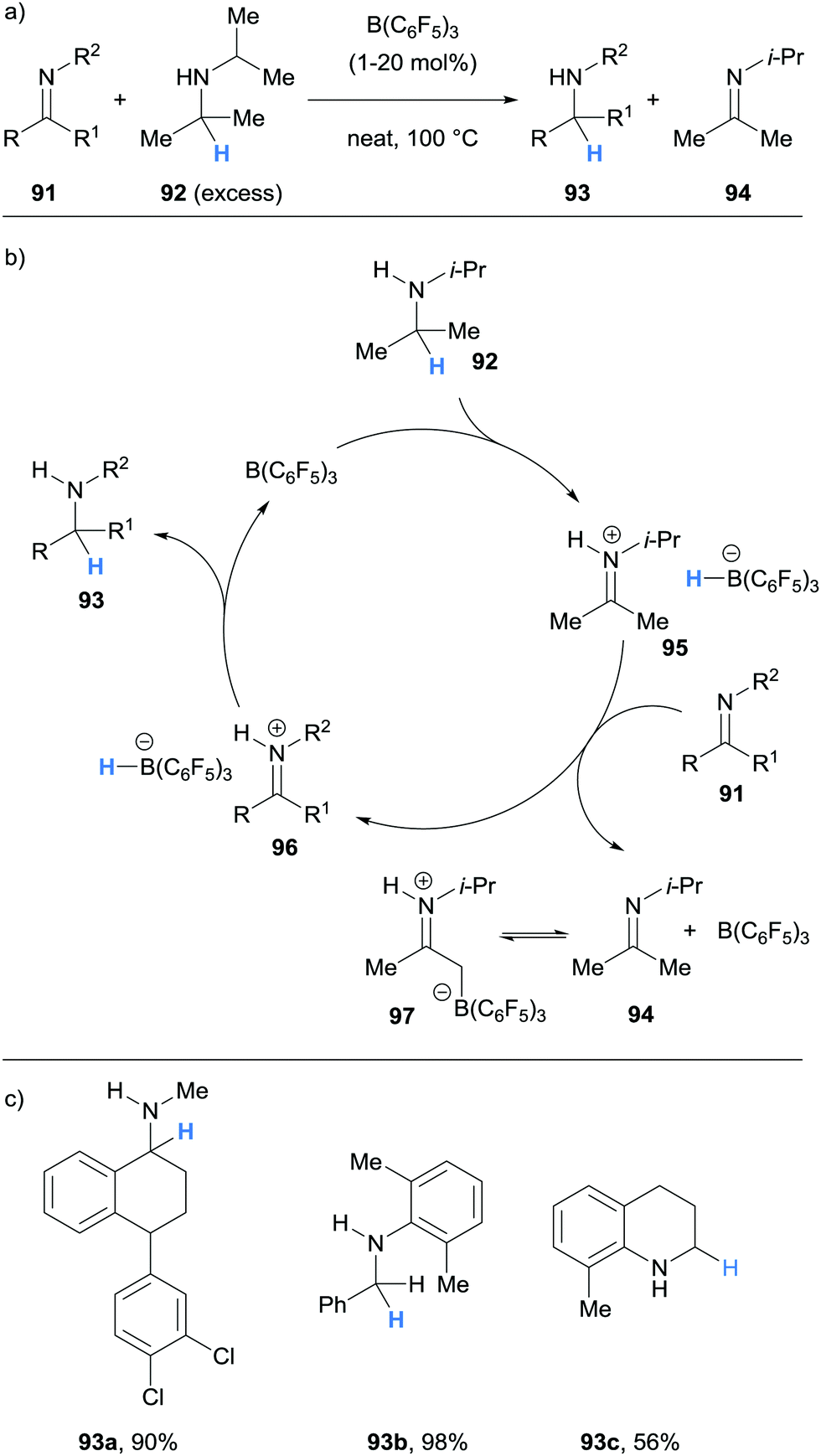 | ||
| Scheme 18 Borane-mediated hydride abstraction utilised in B(C6F5)3-catalysed transfer hydrogenation. | ||
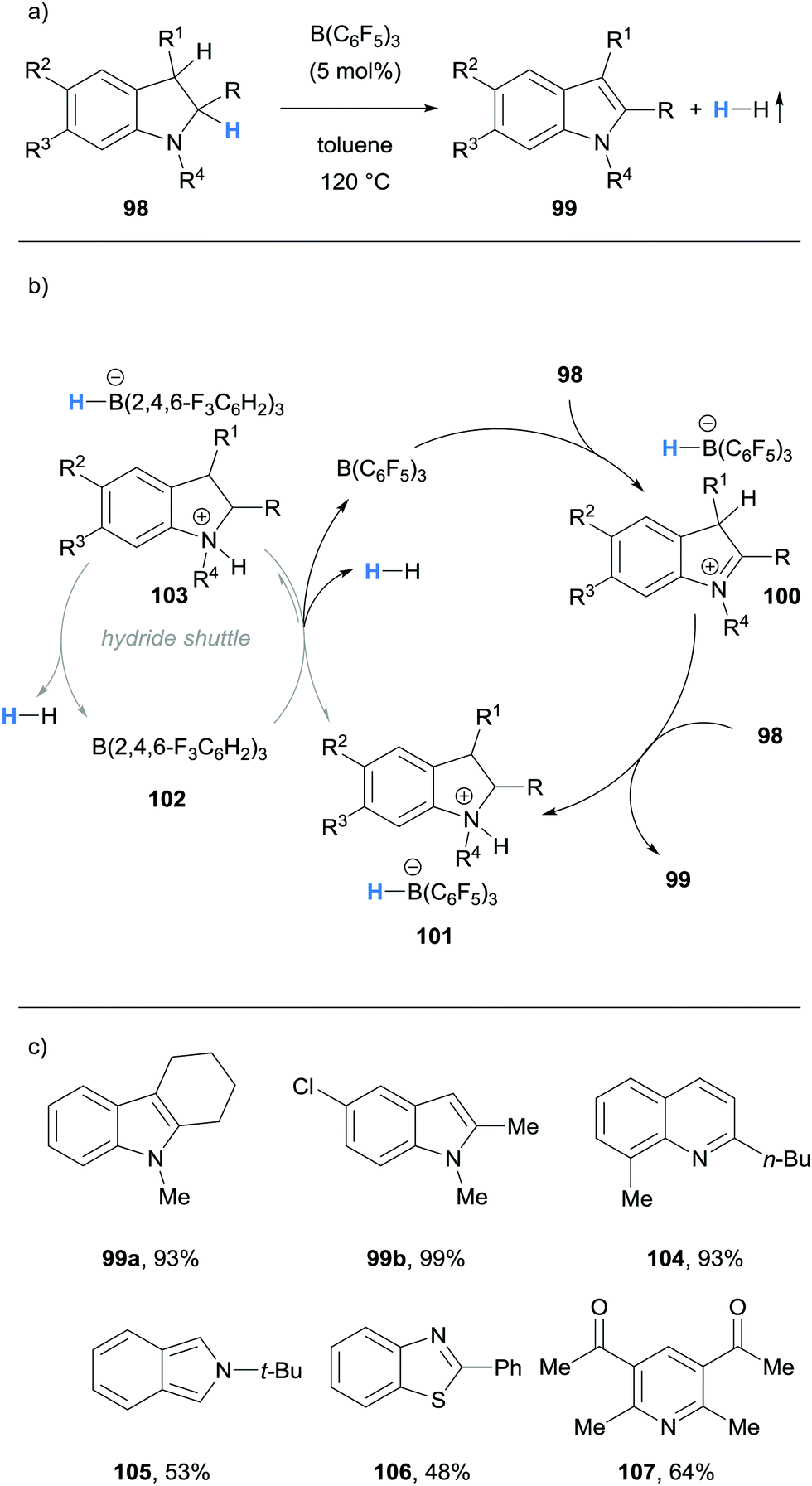 | ||
| Scheme 19 Borane-mediated hydride abstraction utilised in B(C6F5)3-catalysed acceptorless dehydrogenation. | ||
 | ||
| Scheme 20 Borane-mediated hydride abstraction utilised in B(C6F5)3-catalysed acceptorless dehydrogenation. | ||
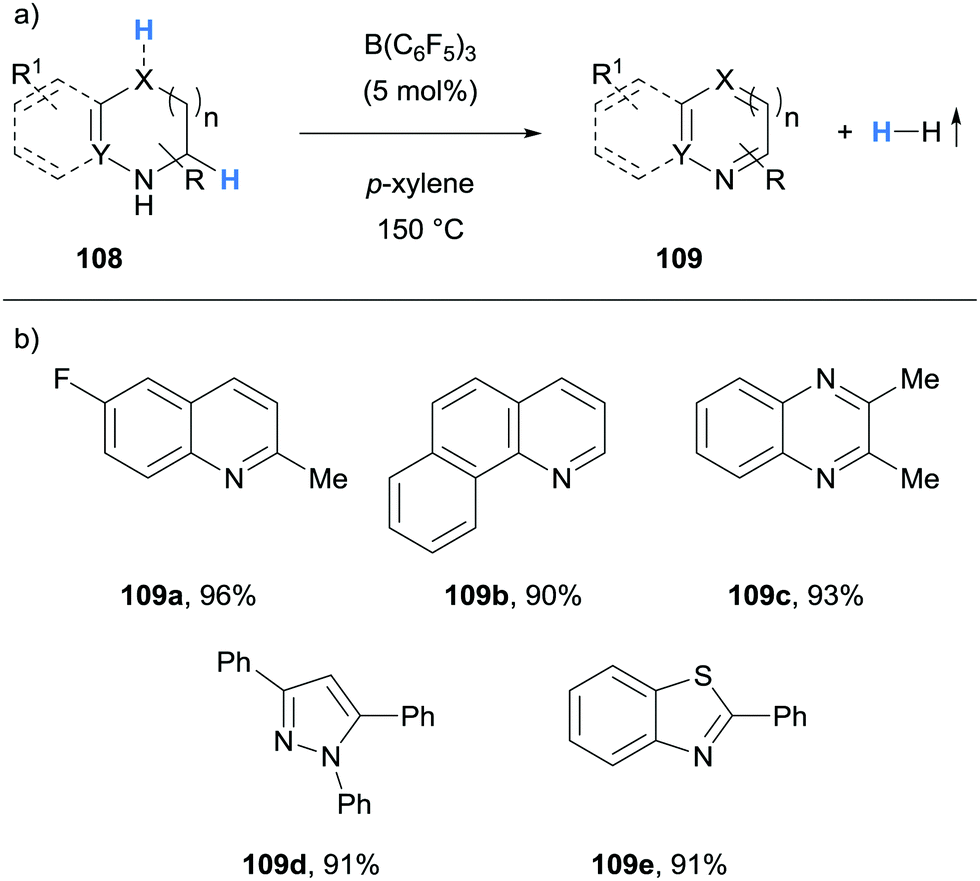
Grimme, Paradies and co-workers described the dehydrogenation of a variety of indolines 98 to the corresponding indoles 99, as well as examples of the dehydrogenation of 1,2-dihydroquinolines (cf.104), an isoindoline (cf.105), a benzothiazoline (cf.106) and dihydropyridines (cf.107) in a process mediated by hydride abstraction (Scheme 19a and c).31 Using indoles as an example, B(C6F5)3-mediated hydride abstraction occurred at the C2 position of indoline 98 to form iminium 100 (Scheme 19b). The indole product 99 was formed through subsequent deprotonation at the C3 position by a second equivalent of 98. Hydrogen gas and the B(C6F5)3 catalyst were then liberated from the ammonium hydridoborate salt 101 in what was calculated to be the rate limiting step. Interestingly, a rate enhancement was observed upon addition of the weaker Lewis acid B(2,4,6-F3C6H2)3 (102). Whilst 102 did not mediate hydride abstraction, it served as a hydride shuttle whereby the hydridoborate counterion in 103 was more basic than in 101, so that the formation of H2 was more favourable. The hydride shuttle strategy enabled the dehydrogenation of a 1,2-dihydroquinoline (cf.104), an isoindoline (cf.105), a benzothiazoline (cf.106) and dihydropyridines (cf.107).
Kanai and co-workers reported the B(C6F5)3-catalysed acceptorless dehydrogenation of N-heterocycles (Scheme 20).32 The substrate scope included the dehydrogenation of indolines, tetrahydroquinolines (cf.109a), tetrahydroquinoxalines (cf.109c), pyrazolines (cf.109d) and benzothiazolines (cf.109e). Notably, coordinating functional groups and atoms, such as thioether and methoxy groups, were tolerated.
Acknowledging the ability of B(C6F5)3 to catalyse the dehydrogenation of indolines, Zhang and co-workers made use of this in their direct C3 C–H silylation and borylation approach to substituted indoles 112 and 114 (Scheme 21).33,34 The initial role of B(C6F5)3 was to activate the silane or catecholborane to form the weak adduct 115. Adduct 115 then underwent nucleophilic attack by indoles 110, forming the iminium hydridoborate salts 116. Another equivalent of 110 deprotonated 116 to form the desired C3-functionalised product 112 or 114. Hydride transfer within 117 resulted in regeneration of B(C6F5)3 and formed indolines 113. The disproportionation reaction yields C3-functionalised indole 112 or 114 and indoline 113 at room temperature. In a bid to improve the atom economy, the temperature was increased which promoted the B(C6F5)3-mediated hydride abstraction and subsequent dehydrogenation of indoline 113 to form indole 110. The dehydrogenation presumably occurred in a related catalytic cycle to that described by Grimme and Paradies (cf.Scheme 19). The catalytic performance of B(C6F5)3 was investigated by sequentially adding batches of indole and hydrosilane or catecholborane to the same reaction mixture and analysing the yield of desired product each time. The B(C6F5)3 remained catalytically competent over 10 sequential reactions.
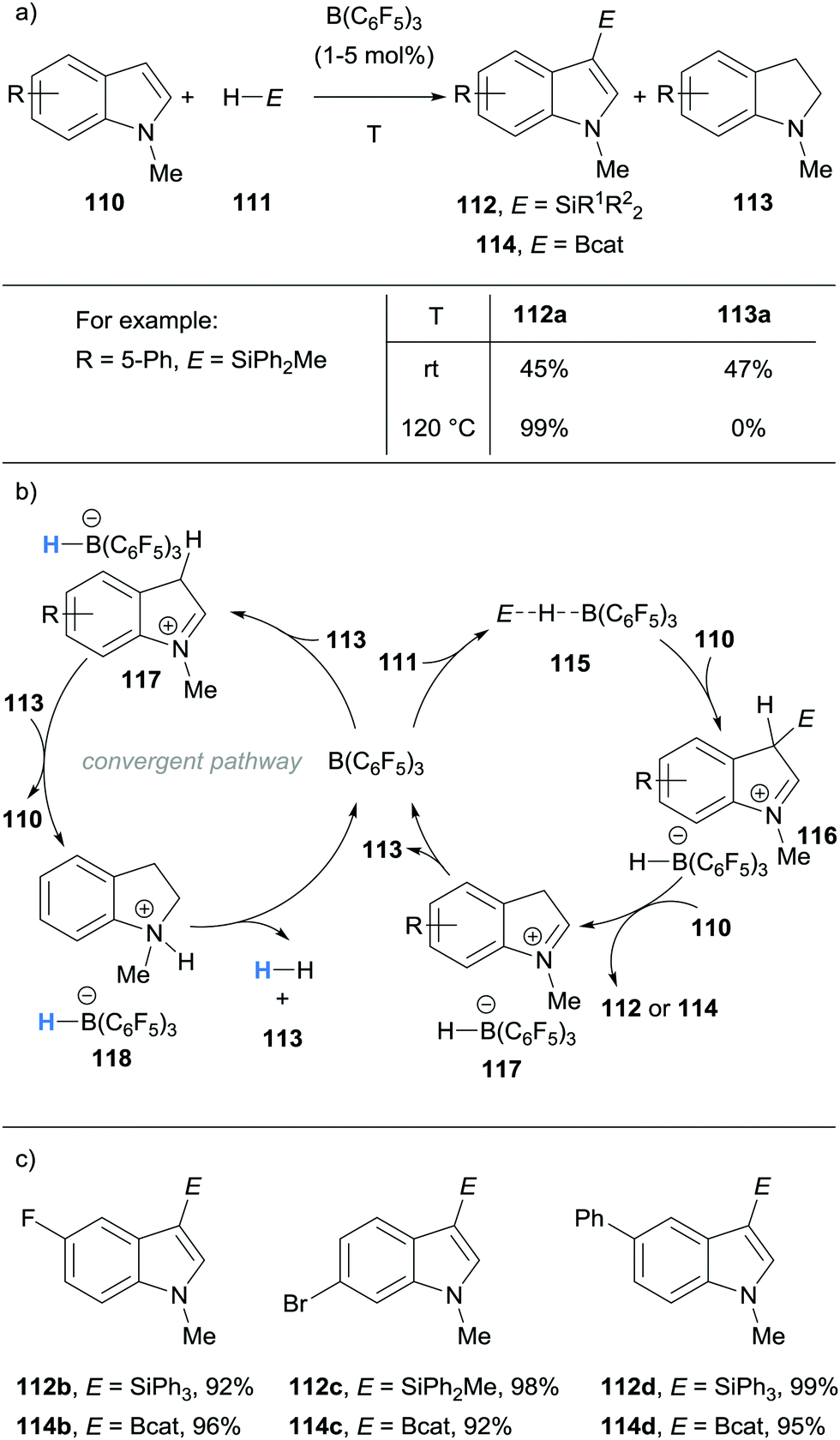 | ||
| Scheme 21 Borane-mediated hydride abstraction utilised in B(C6F5)3-catalysed convergent C3-silylation and borylation of indoles. | ||
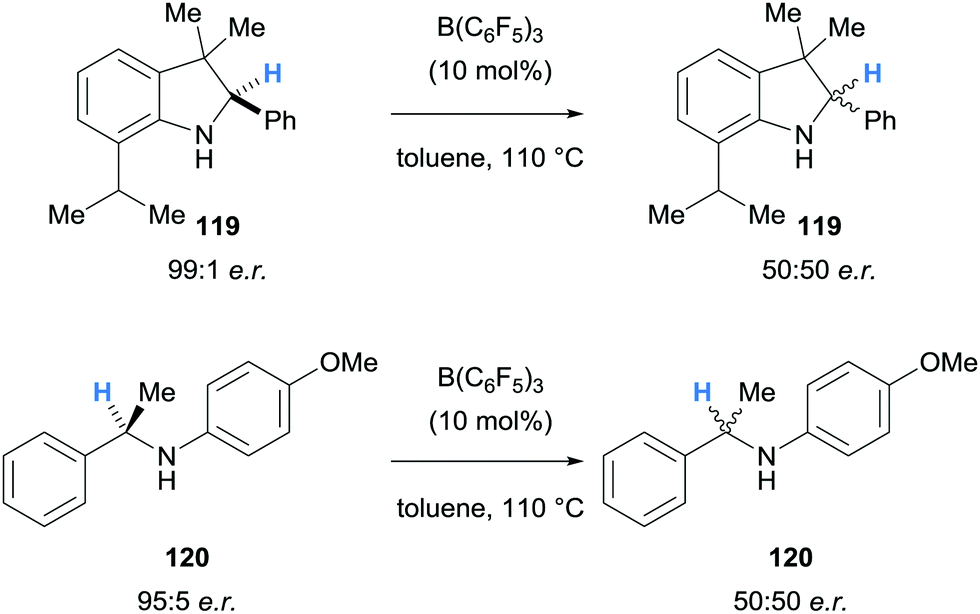 | ||
| Scheme 22 Borane-mediated hydride abstraction utilised in the racemisation of amines. | ||
In 2017, Erker and co-workers reported an example of B(C6F5)3-catalysed isomerisation of N-allyltetramethylpiperidine (121) to the corresponding enamine 122 (Scheme 23a).35 The authors proposed that isomerisation of 121 occurred via a B(C6F5)3-mediated hydride abstraction to form α,β-unsaturated iminium hydridoborate 123 (Scheme 23b). Hydride transfer from the counterion then occurred in a conjugate fashion to form enamine 122 as a mixture of cis-/trans-isomers.
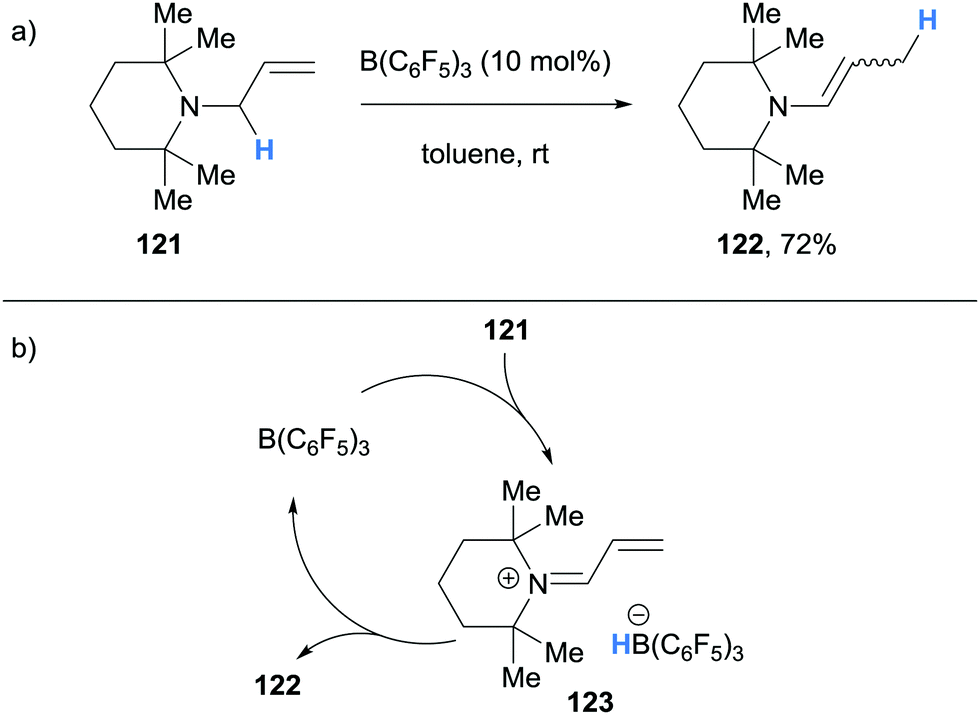 | ||
| Scheme 23 Borane-mediated hydride abstraction utilised in the isomerisation of an alkene. | ||
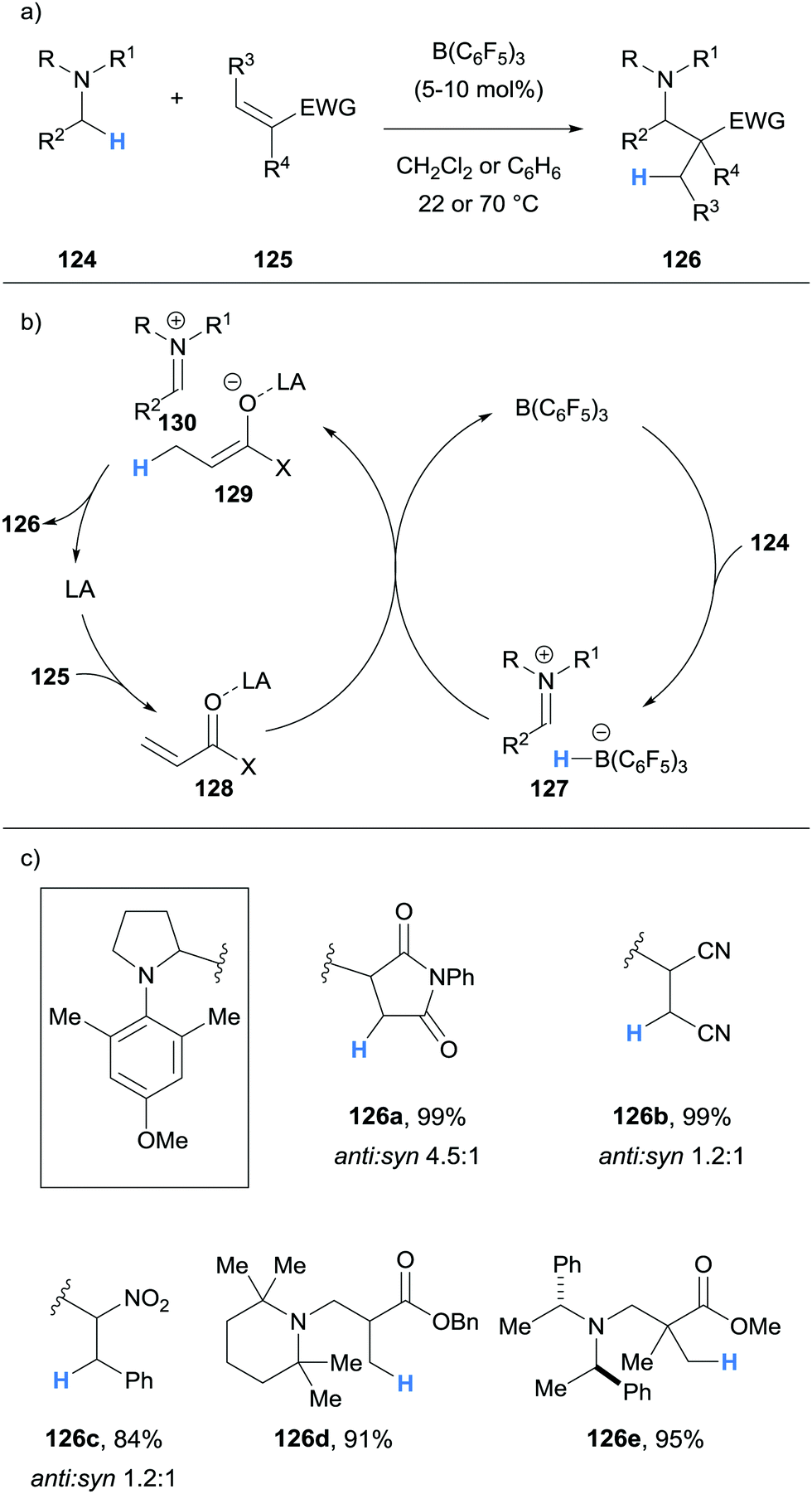 | ||
| Scheme 24 Borane-mediated α-amino hydride abstraction utilised in a redox neutral, B(C6F5)3-catalysed α-functionalisation of N-alkylamines. LA = Lewis acid. | ||
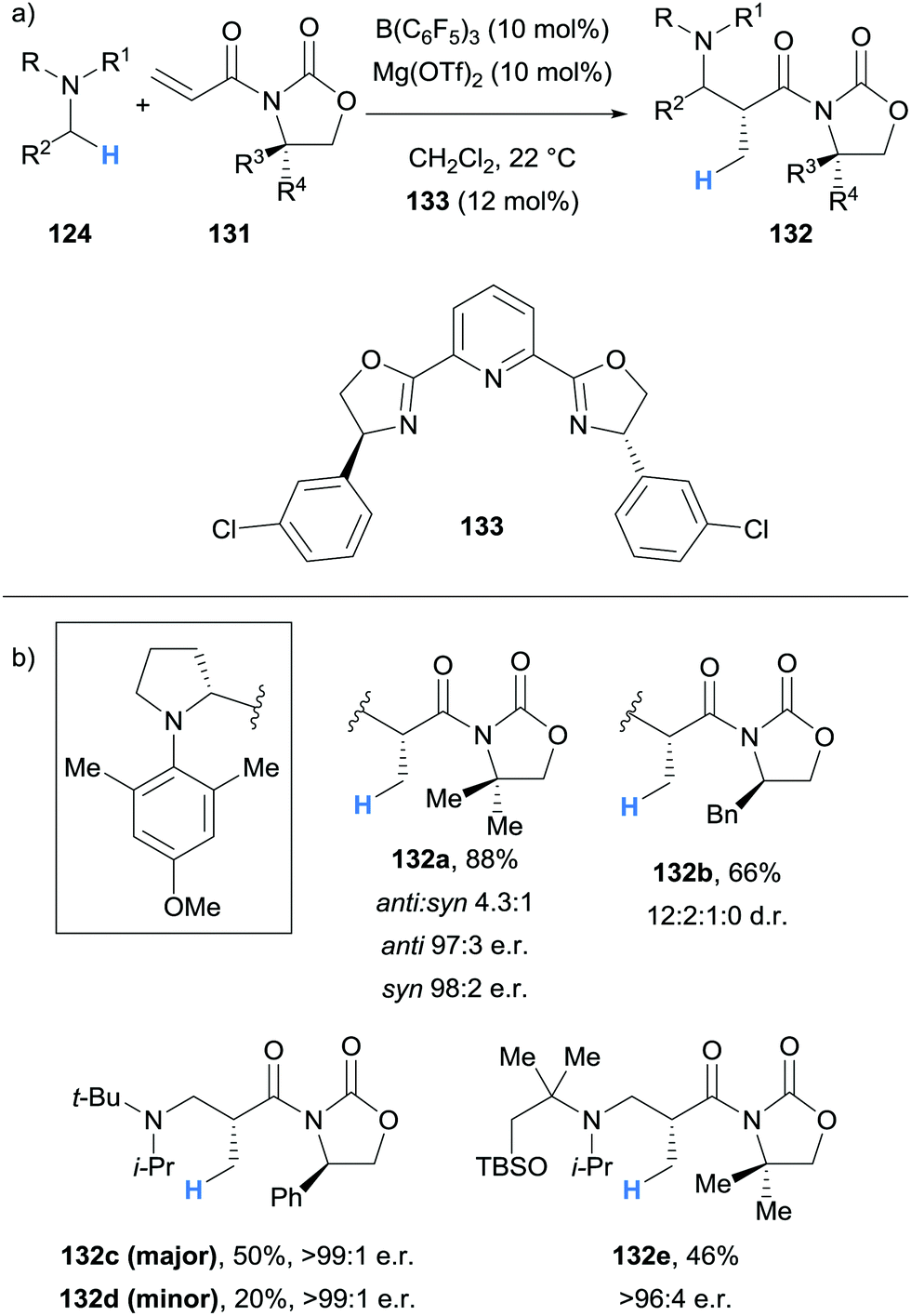 | ||
| Scheme 25 Borane-mediated hydride abstraction utilised in a redox neutral, B(C6F5)3-catalysed asymmetric α-functionalisation of N-alkylamines. | ||
Impressively, Wasa also reported an enantioselective version of the reaction, where a variety of N-aryl pyrrolidines 124 and acryloyloxazolidinones 131 were coupled to form Mannich type products 132 with good diastereoselectivity and excellent enantioselectivity (Scheme 25). In this case, the Lewis acid co-catalyst (LA) was derived from Mg(OTf)2 and chiral ligand 133. The selective activation of the acryloyloxazolidinone Michael acceptors 131 by the chiral co-catalyst was crucial to enantioselectivity, and it was suggested that the high steric hindrance of B(C6F5)3 limited its ability to coordinate to 131.
Wasa and co-workers have also reported Mannich-type reactions involving silyl enol ethers 134 and iminum ions generated via borane-mediated hydride abstraction from N-alkylamines 124 (Scheme 26a).37 After generation of the iminium hydridoborates 136, nucleophilic silyl enol ethers 134 react to form a new C–C bond and form salt 137. The hydridoborate counterion in 137 was proposed to react with the silyl moiety, thus releasing the β-amino carbonyl product 135, trimethylsilane and the B(C6F5)3 catalyst (Scheme 26b). Various amines 124, such as pyrrolidines, N,N-dialkylanilines and trialkylamines, were amenable to the process (Scheme 26c). The researchers applied this method to the synthesis of biologically important molecules such as the fluoxetine derivative 135c, a known antidepressant.
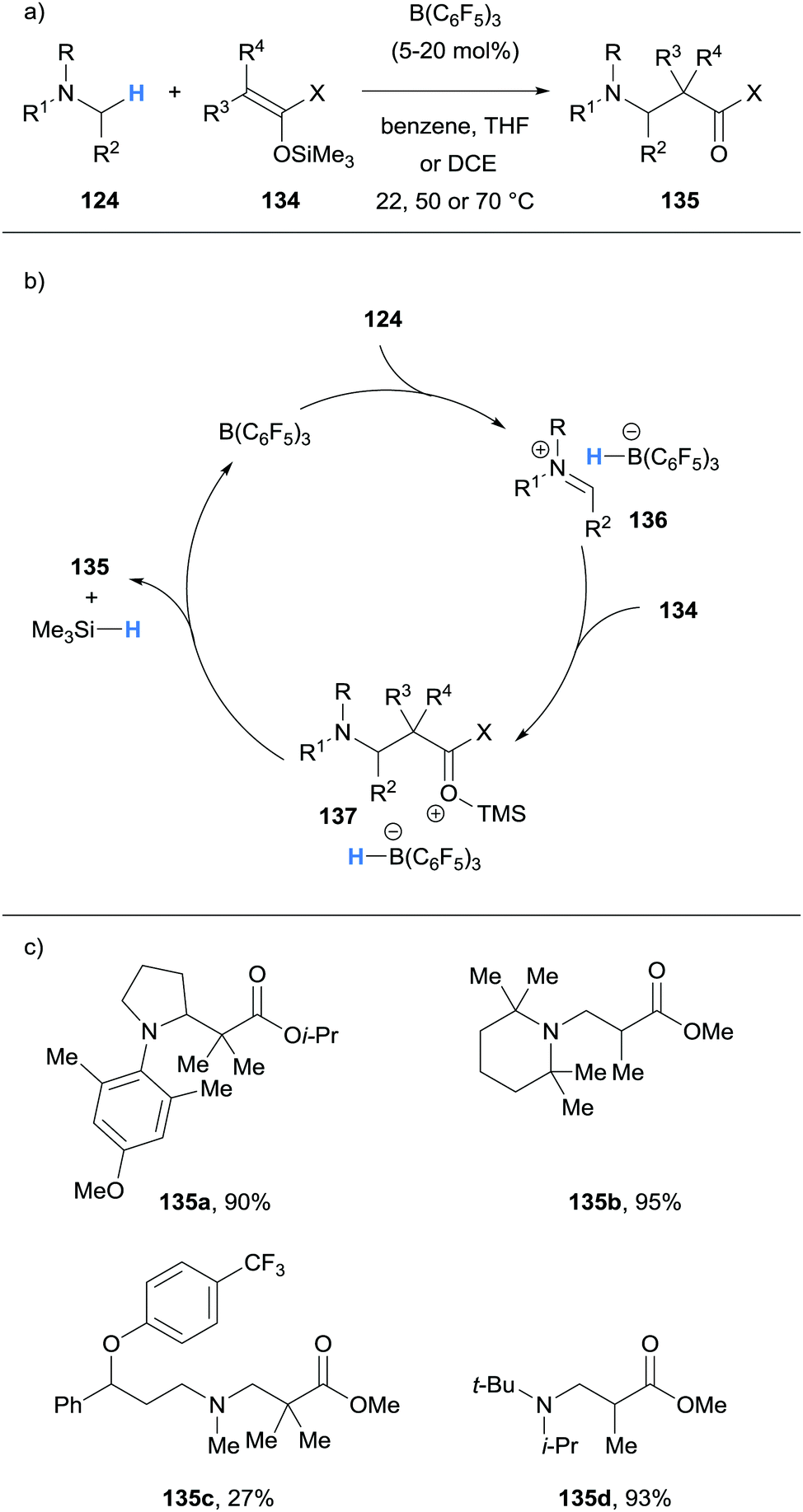 | ||
| Scheme 26 Borane-mediated hydride abstraction utilised in an α-functionalisation of N-alkylamines using silyl enol ethers. | ||
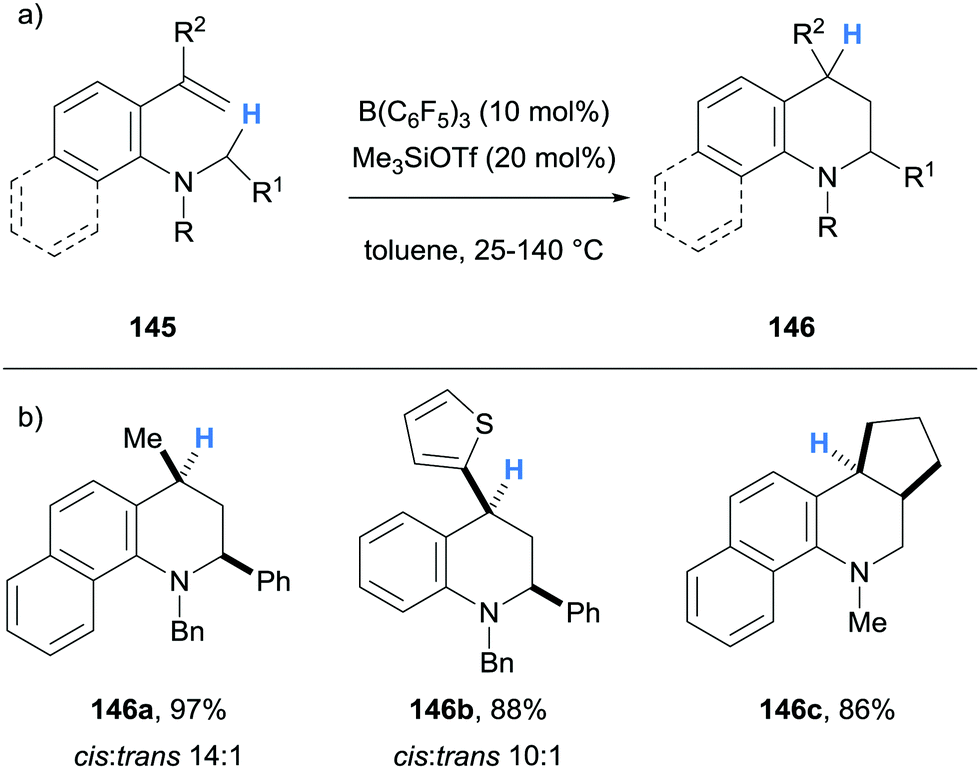
Borane-mediated hydride abstraction has also been used in the synthesis of tetrahydroquinolines by the research groups of Grimme and Paradies,38 and Wang39 (Schemes 27 and 28, respectively). In both cases, B(C6F5)3-catalysed the efficient conversion of 2-amino styrenes 138 and 145 into tetrahydroquinolines 139 and 146. In the Grimme and Paradies study, the mechanism was investigated computationally, and it was proposed that after borane-mediated hydride abstraction to form iminium hydridoborates 140, a disrotatory 6π-electrocyclisation occurred to form cationic intermediates 141. Hydride transfer from the hydridoborate anions of 141 formed the desired heterocyclic products 139 and regenerated the B(C6F5)3 catalyst (Scheme 27b). Products that bear β-amino C–H bonds, such as in 139a and 139b, were involved in an off-cycle equilibrium where further B(C6F5)3-mediated hydride abstraction formed enamines 143 and zwitterions 144 in a related process, as observed in other cases (cf.Scheme 6 above). However, NEt3 was used in the work up which converted zwitterion 144 to product 139. It was also shown that stabilisation of the benzylic carbocation in 141 was essential, as only substrates that formed tertiary carbocations were successful in the formation of tetrahydroquinolines 139. Some substrates were prone to autoxidation during the workup (cf.139b).
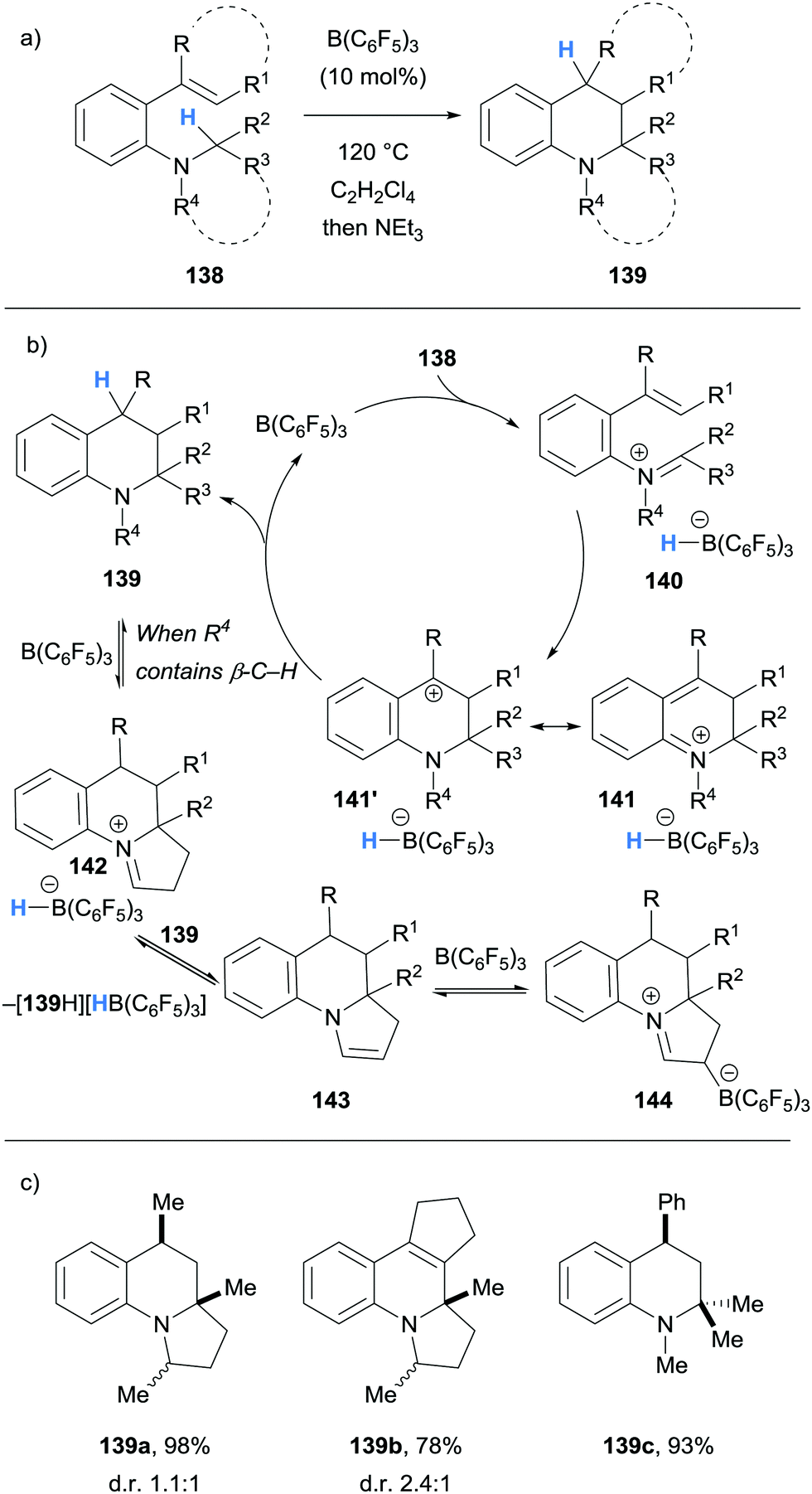 | ||
| Scheme 27 Borane-mediated hydride abstraction utilised in the B(C6F5)3-catalysed synthesis of tetrahydroquinolines. | ||
In the Wang and co-workers B(C6F5)3-catalysed synthesis of tetrahydroquinolines, Me3SiOTf (20 mol%) was used as an additive, and its role was to accept the hydride from the hydridoborate counterion (Scheme 28). The weaker Lewis acidity of Me3SiOTf facilitated subsequent donation of the hydride to the benzylic cation, which was determined to be the rate-limiting step.
 | ||
| Scheme 28 Borane-mediated hydride abstraction utilised in the B(C6F5)3-catalysed synthesis of tetrahydroquinolines. | ||
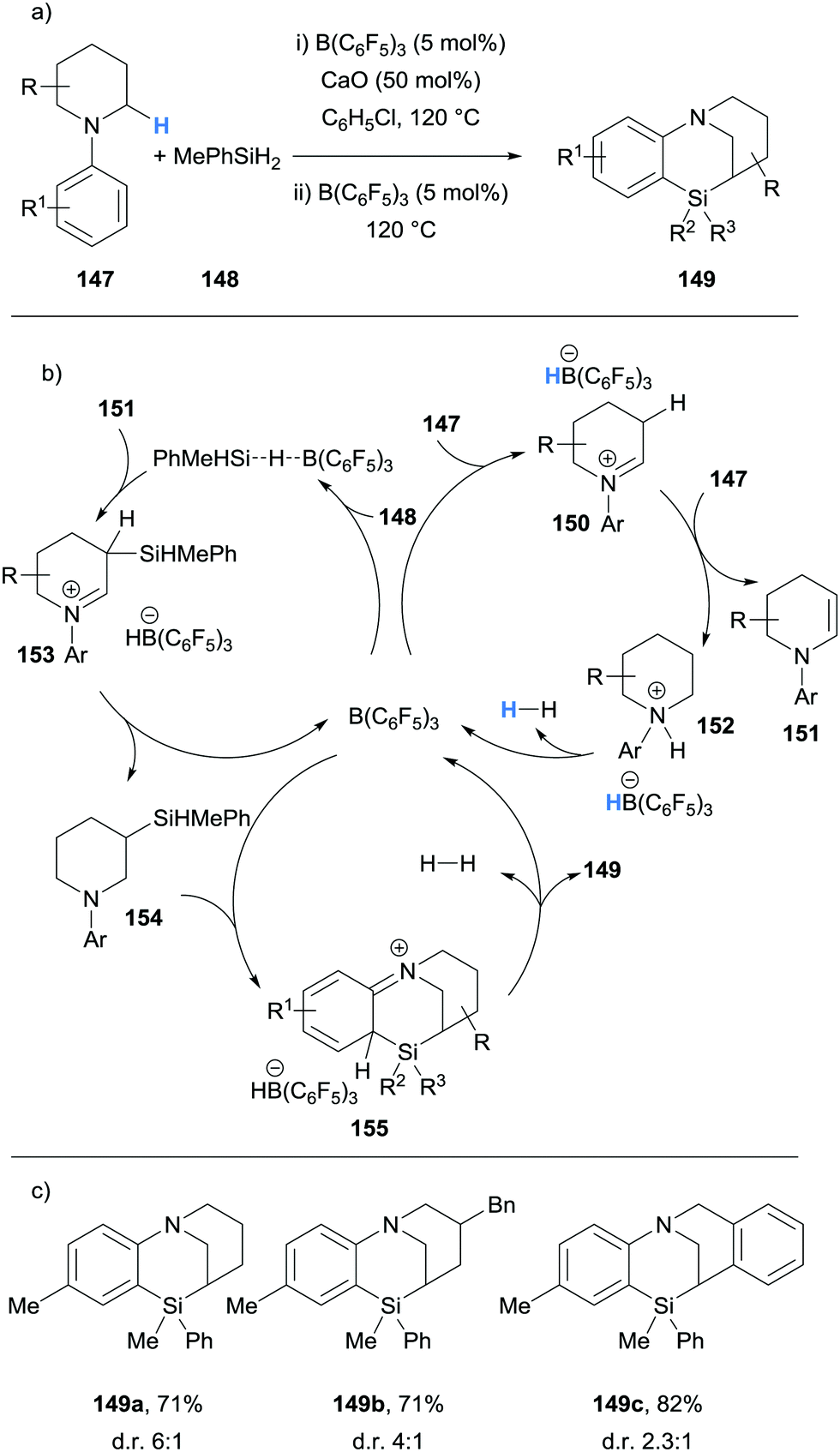
Chang and Park have described the B(C6F5)3-catalysed formation of polycyclic bridged sila-N-heterocycles 149 from N-aryl piperidines 147 and methyl phenyl silane (148) (Scheme 29).40 Experimental40 and theoretical41 studies have revealed a triple role for the B(C6F5)3 catalyst. Firstly, B(C6F5)3 mediates the hydride abstraction step that enables the formation of iminium hydridoborate 150 and subsequent formation of enamine 151 in a related process to that previously described (cf.Scheme 6). Piperidinium borohydride 152 then releases hydrogen to regenerate B(C6F5)3. The second role of B(C6F5)3 is to activate methyl phenyl silane (148) for nucleophilic addition by enamine 151. Hydride transfer from the hydridoborate counterion to the iminium moiety in 153 provides amine 154. Finally, B(C6F5)3 activates the Si–H bond for electrophilic silylation of the aromatic ring in 154, which subsequently releases hydrogen to form polycyclic bridged sila-N-heterocycles 149. The additive CaO was used and proposed to assist with the proton transfer steps of the ionic cascade mechanism.
 | ||
| Scheme 29 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed silylative cascade. | ||
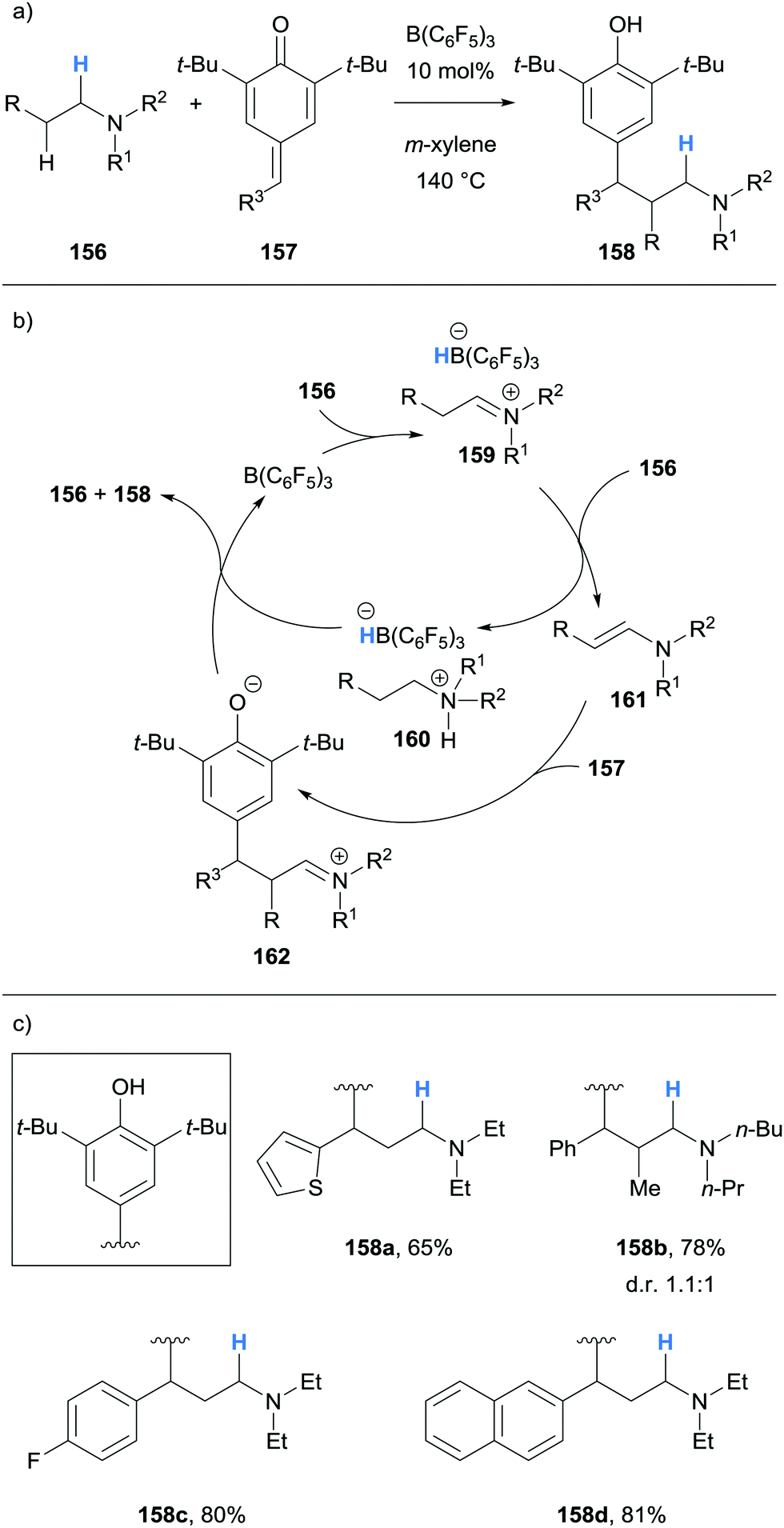
Ma and co-workers reported the β-alkylation of acyclic tertiary amines 156 with para-quinone methides 157 (Scheme 30).42 The B(C6F5)3-mediated hydride abstraction of amines 156 allowed for the formation of ammonium hydridoborate 160 and enamine 161. Conjugate addition between the nucleophilic enamine 161 and para-quinone methide 157 occurred to produce iminium 162. Protonation of 162 and hydride transfer from the ammonium hydridoborate 160 yielded β-functionalised product 158. An unsymmetrical tertiary amine was reacted where β-functionalisation occurred preferentially on ethyl instead of the n-butyl chain.
 | ||
| Scheme 30 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed β-amino alkylation. | ||
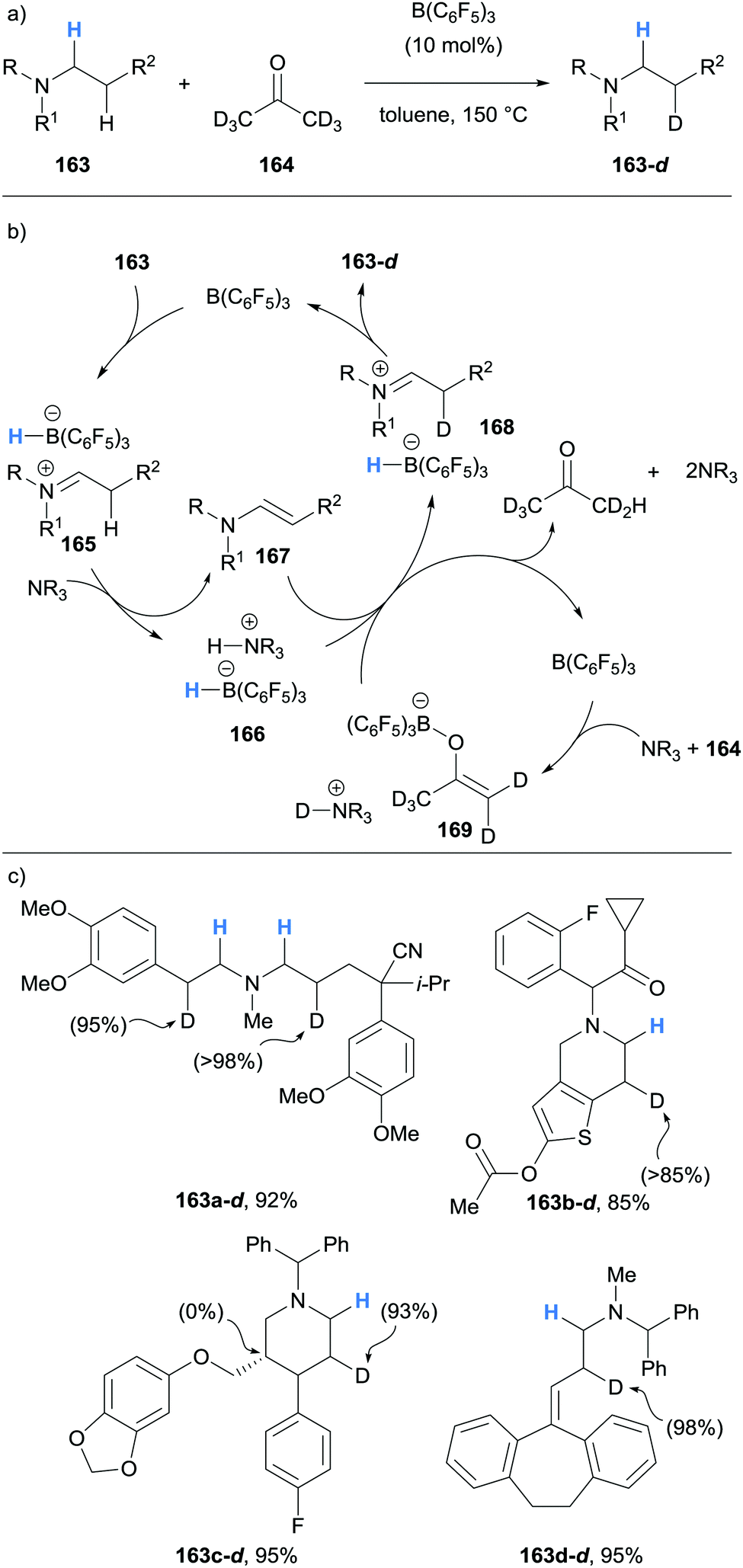
In 2019, Wasa showed that borane-mediated α-amino hydride abstraction could be used to efficiently deuterate β-amino C–H bonds of N-alkyamines 163, including pharmaceutical compounds (Scheme 31).43 As seen before, B(C6F5)3 mediated the formation of ammonium hydridoborate 166 and enamine 167. It was proposed that B(C6F5)3 also has the role of mediating FLP-like deprotonation of deuterated acetone (164) to form ammonium borate 169, which serves as the deuterium source for the deuteration of enamine 167. Impressively, pharmaceutically active compounds, with some containing Lewis acid-sensitive functional groups such as cyano, ester, amide and ketones, were successfully deuterated.
 | ||
| Scheme 31 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed β-amino deuteration. Parenthesis refer to %D incorporation. | ||
In 2020, we reported the application of borane-mediated α-amino hydride abstraction in a B(C6F5)3-catalysed C3 alkylation of indoles and oxindoles that utilised amine based alkylating agents (Scheme 32).44 The mechanism was proposed to begin with the generation of iminium hydridoborate 175 after B(C6F5)3-mediated hydride abstraction of the alkylating agent 172. The iminium 175 underwent nucleophilic attack by the indole 170 (or oxindole 171) to form a new C–C bond. Subsequent proton transfers resulted in the formation of α,β-unsaturated iminium 177, presumably via an E1cB-like elimination. Hydride transfer from the hydridoborate counterion to 177 allowed the formation of alkylated indoles 173 (and oxindoles 174).
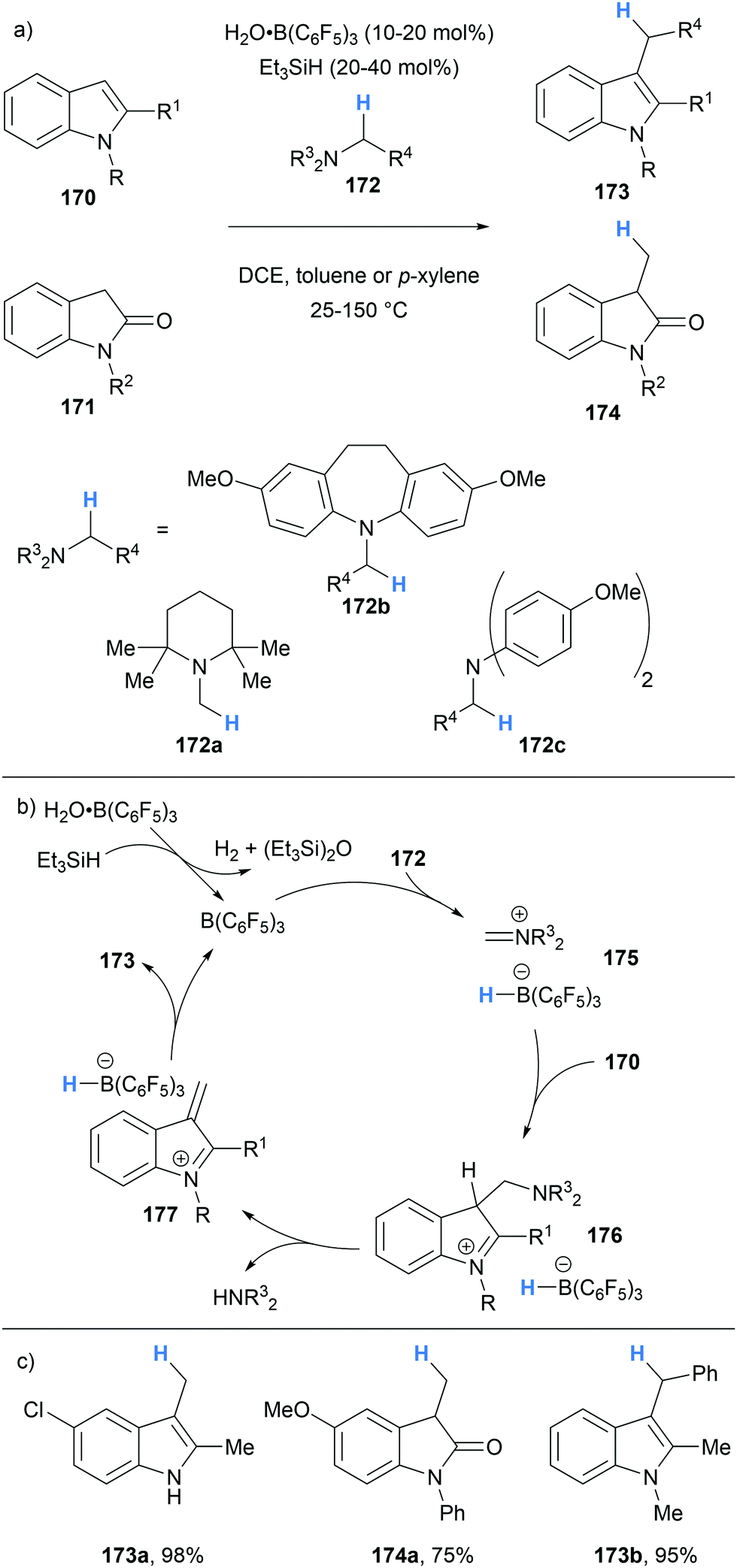 | ||
| Scheme 32 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed alkylation of indoles and oxindoles. | ||
In the same report, we also reported the use of borane-mediated hydride abstraction in a ring-opening cascade process between indoles 170 and pyrrolidines 178 to form 4-(3-indolyl)butylamines 179, a privileged motif found in several serotonergic/dopaminergic drug molecules (Scheme 33).44 The mechanism was proposed to proceed in a similar fashion to the previously described alkylation process (cf.Scheme 32), except the cyclic nature of amine 178 allowed for the amine portion to be retained in product 179 after elimination (Scheme 33). The use of Et3SiH (20 mol%) serves to free the B(C6F5)3 catalyst from its water adduct in situ. This modification allowed the authors to directly use B(C6F5)3 as received from the supplier and weigh it in air on the open bench, thus negating the use of a glove box and additional catalyst purification.
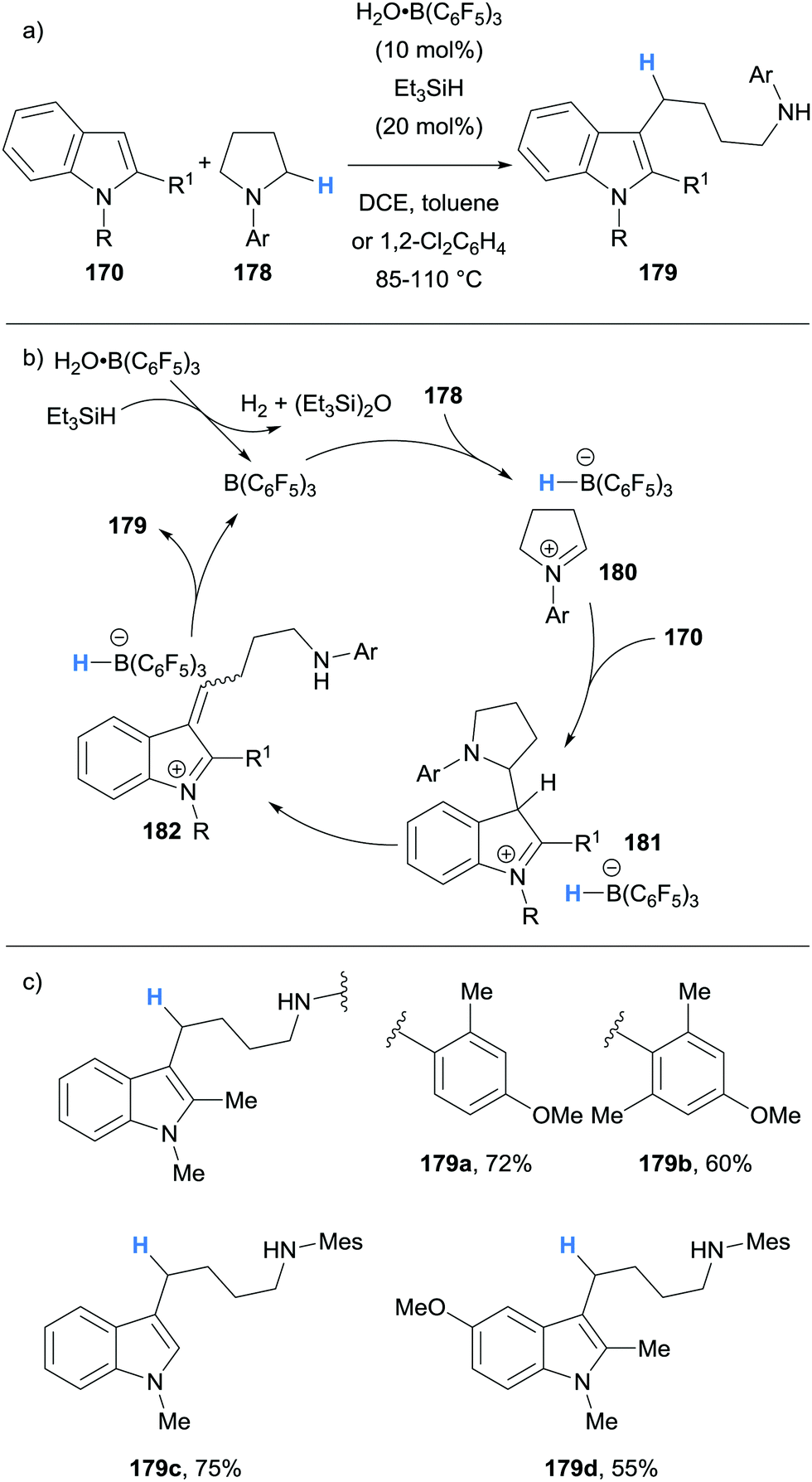 | ||
| Scheme 33 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed alkylation-ring opening cascade. | ||
Chang and co-workers described the use of borane-mediated hydride abstraction in a dehydrogenation/β-silylation/C–N bond cleavage cascade process (Scheme 34).45 Cyclic tertiary amines (183) underwent cine-silylative ring opening to form amines 185, and tertiary amines (186) underwent deconstruction to form secondary amines 187 and alkyl silanes 188. Impressively, B(C6F5)3 has multiple roles in these processes. Using the cine-silylative ring opening to illustrate the mechanism, cyclic amines 183 undergo B(C6F5)3-catalysed dehydrogenation to form enamines 190 and ammonium hydridoborate 191 in the rate limiting step. B(C6F5)3-catalysed β-silylation of enamines 190 with silanes 184 then gave silylated amine 192. Amine 192 is predisposed to form silazetidinium intermediate 193via B(C6F5)3-mediated activation of the Si–H bond. Cycloreversion cleaved the C–N bond in 193 and gave 194, which after hydride transfer from the hydridoborate counterion, gave amino alkene 195. B(C6F5)3-catalysed hydrosilylation of the alkene moiety in 195 allowed the formation of product 185. Interestingly, theoretical and experimental investigations revealed that borane-mediated hydride abstraction preferentially occurred at the more substituted carbon of the ring (cf. formation of 189) and that exo-enamine 190 formation was more favoured than the corresponding endo-enamine.
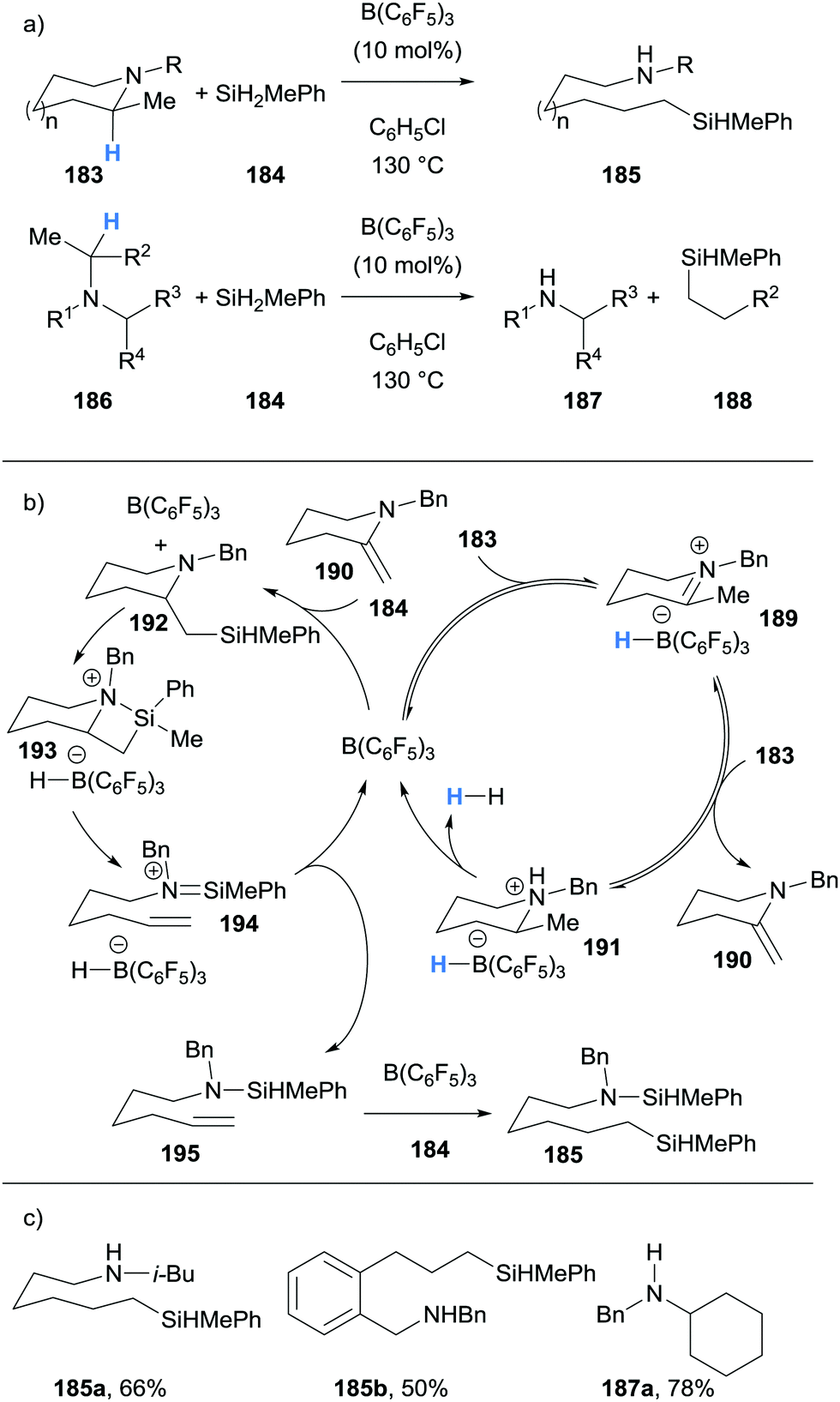 | ||
| Scheme 34 Borane-mediated hydride abstraction utilised in a B(C6F5)3-catalysed cine-silylative ring opening and tertiary amines deconstruction processes. | ||
3 Borane-mediated γ-amino hydride abstraction
In contrast to α-amino hydride donors, donation from a γ-amino conjugated C–H bond will be more familiar. 1,4-Dihydropyridines are well studied hydride donors in both a biological (e.g. NADH) and chemical (e.g. Hantzsch esters) setting.1 However, γ-hydride abstraction of conjugated amines with boranes has received relatively little attention. In this section, stoichiometric investigations of hydride abstraction from 1,4-dihydropyridines and 2-alkylideneimidazolines will be discussed. An application of this reactivity in transfer hydrogenation will also be included.3.1 Stoichiometric studies
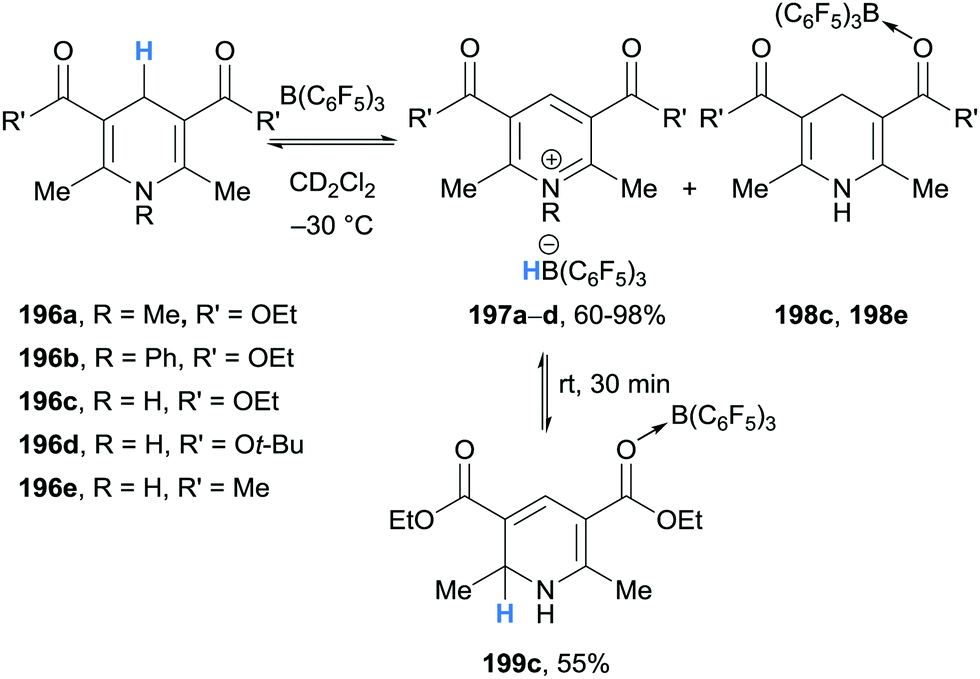 | ||
| Scheme 35 Borane-mediated γ-amino hydride abstraction from Hantzsch ester derivatives. | ||
Inspired by the naturally occurring organic hydride donor NAD(P)H, Melen and co-workers studied the B(C6F5)3-mediated hydride abstraction of N-benzyl-1,4-dihydropyridine derivatives (200a–200c).47 The reaction between N-benzyl-1,4-dihydropyridine 200a and B(C6F5)3 afforded pyridinium borohydride 201a in excellent yield via γ-amino hydride abstraction (Scheme 36a). A similar outcome was observed when carboxylic acid substituted 1,4-dihydropyridine 200b was treated with B(C6F5)3 (Scheme 36b). Interestingly, when nicotinamide derivative 200c was reacted with B(C6F5)3, a disproportionation was observed and pyridinium borate zwitterion 202c and B(C6F5)3 coordinated 1,2-dihydropyridine 203c were formed. The mechanism, supported by deuterium labelling experiments, was proposed to involve initial coordination of B(C6F5)3 to nicotinamide derivative 200c to acidify the N–H bond and allow protonation of another molecule of 200c (Scheme 36c). Borane-mediated hydride abstraction then formed a hydridoborate counterion that transfers a hydride to the iminium moiety in 205c.
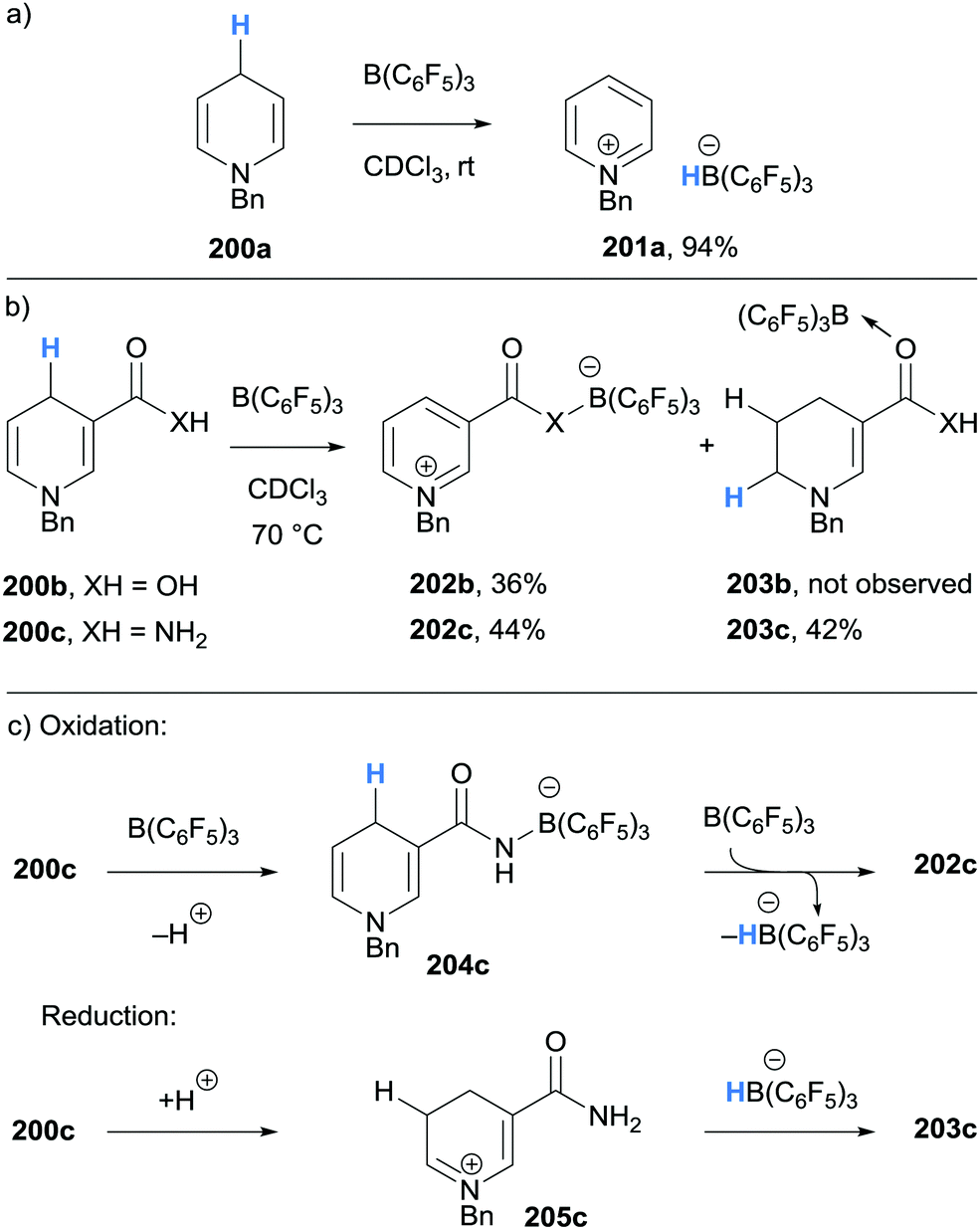 | ||
| Scheme 36 Borane-mediated γ-amino hydride abstraction from 1,4-dihydropyridine derivatives. | ||
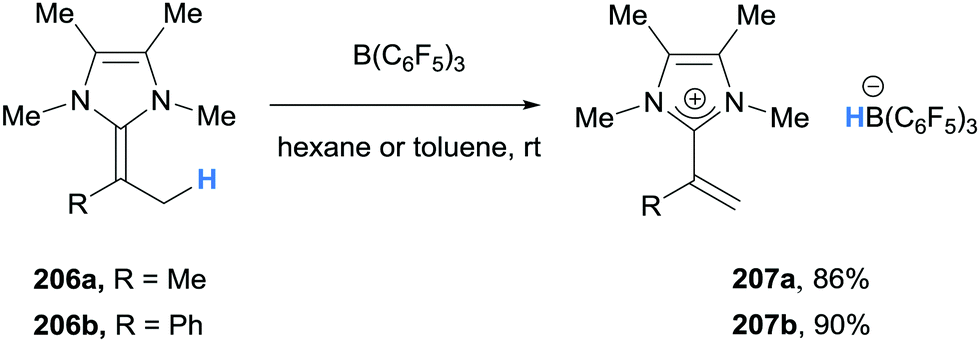 | ||
| Scheme 37 Borane-mediated γ-amino hydride abstraction from 2-alkylideneimidazolines. | ||
3.2 Catalytic applications
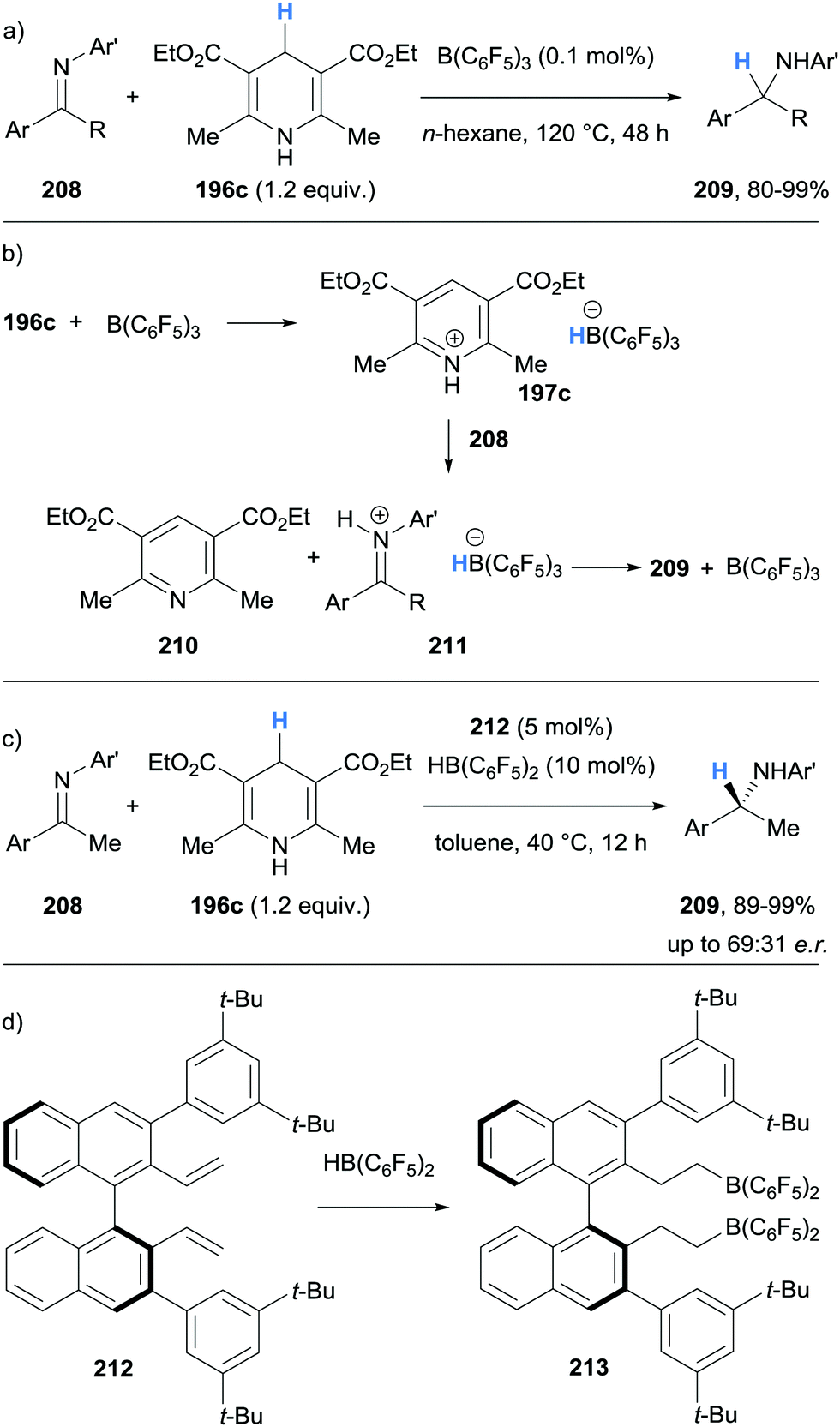 | ||
| Scheme 38 Borane-mediated γ-amino hydride abstraction from Hantzsch esters utilised in the B(C6F5)3-catalysed transfer hydrogenation of imines. | ||
4 Summary and outlook
Borane-mediated hydride abstraction has been observed in a variety of amino substrates. The most common substrate class reported are amines that bear α-amino C–H bonds where, in stoichiometric studies, iminium hydridoborates have been observed. There are a variety of chemical reactions that the iminium hydridoborate generated can undergo, which include proton transfers to form enamines, nucleophilic addition to the electrophilic iminium, and further hydride transfers.Due to the array of reactions that both the iminium and hydridoborate ions can undergo, borane-mediated hydride abstraction has been employed as a key event in many powerful borane-catalysed processes. These include novel strategies for reagent activation, such as the use of amines as H2 surrogates in transfer hydrogenation, and as alkylating agents. In addition, new methods for the manipulation of amines, including dehydrogenation, racemisation, isomerisation, α- and β-functionalisation, and C–N bond cleavage have all been reported. The application of these methods in synthesis has demonstrated the high functional group tolerance possible in processes involving borane-mediated hydride abstraction. Borane-mediated hydride abstraction involving γ-amino conjugated C–H bonds has also been reported.
Borane-mediated hydride abstraction is a relatively young field and there is no doubt that advances will continue to be made. By far the most commonly used borane has been B(C6F5)3, likely because it is commercially available, robust and well-established use in the borane catalysis arena. However, we expect that optimisation of the borane structure will be a fruitful endeavour with respect to tuning regio- and chemo-selectivity. In addition, asymmetric processes are poorly represented with respect to borane-mediated hydride abstraction and we anticipate growth in this area. Furthermore, expansion of the amine scope with respect to other classes of conjugated amines may lead to valuable methods involving remote functionalisation. Given the great interest, especially from the pharmaceutical industry,6,7 in efficient synthetic methods that manipulate C–H bonds of amines, borane-mediated hydride abstraction will continue to be exploited in novel processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. W. and A. P. P. thank the School of Chemistry, University of Leicester, for their generous support and the Royal Society for a Research Grant (No. RGS\R1\191082). S. B., B. A. K., R. L. M., and L. C. M. gratefully acknowledge the School of Chemistry, Cardiff University for generous support, the Leverhulme Trust for a Research Project Grant (No. RPG-2015-361), and the Schlumberger Foundation for a Faculty for the Future Fellowship (B. A. K.). R. L. M. would like to acknowledge the EPSRC for a research fellowship (No. EP/R026912/1).Notes and references
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS , and references therein.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC , and references therein.
- J. C. L. Walker and M. Oestreich, Synlett, 2019, 2216–2232 CAS.
- S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688–4695 RSC.
- M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 Search PubMed.
- J. F. Allochio Filho, B. C. Lemos, A. S. de Souza, S. Pinheiro and S. J. Greco, Tetrahedron, 2017, 73, 6977–7004 CrossRef CAS.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS.
- B. Temme and G. Erker, J. Organomet. Chem., 1995, 488, 177–182 CrossRef CAS.
- N. Millot, C. C. Santini, B. Fenet and J. M. Basset, Eur. J. Inorg. Chem., 2002, 3328–3335 CrossRef CAS.
- L. E. Garner, H. Zhu, M. L. Hlavinka, J. R. Hagadorn and E. Y.-X. Chen, J. Am. Chem. Soc., 2006, 128, 14822–14823 CrossRef CAS.
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367–2378 CrossRef CAS.
- A. D. Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D’Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030–5041 CrossRef.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS.
- C. Jiang, O. Blacque, T. Fox and H. Berke, Organometallics, 2011, 30, 2117–2124 CrossRef CAS.
- F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170–188 Search PubMed.
- F. Focante, PhD thesis, University of Bologna, 2005.
- F. Focante, I. Camurati, D. Nanni, R. Leardini and L. Resconi, Organometallics, 2004, 23, 5135–5141 CrossRef CAS.
- P. Liptau, M. Neumann, G. Erker, G. Kehr, R. Fröhlich and S. Grimme, Organometallics, 2004, 23, 21–25 CrossRef CAS.
- L. Tebben, M. Neumann, G. Kehr, R. Fröhlich, G. Erker, S. Losi and P. Zanello, Dalton Trans., 2006, 1715–1720 RSC.
- J.-B. Sortais, T. Voss, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2009, 7417–7418 RSC.
- K. Unverhau, G. Lübbe, B. Wibbeling, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2010, 29, 5320–5329 CrossRef CAS.
- A. D. Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D’Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030–5041 CrossRef.
- J. Chen and E. X.-Y. Chen, Molecules, 2015, 20, 9575–9590 CrossRef CAS.
- G.-Q. Chen, G. Kehr, C. G. Daniliuc, M. Bursch, S. Grimme and G. Erker, Chem. – Eur. J., 2017, 23, 4723–4729 CrossRef CAS.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS.
- S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842–1849 RSC.
- D. Winkelhaus, B. Neumann, H.-G. Stammler, R. J. F. Berger, Y. V. Vishnevskiy and N. W. Mitzel, Chem. – Eur. J., 2012, 18, 9312–9320 CrossRef CAS.
- L. A. Körte, S. Blomeyer, S. Heidemeyer, J. H. Nissen, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2016, 45, 17319–17328 RSC.
- T. Wang, L. Liu, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, Chem. – Asian J., 2016, 11, 1394–1399 CrossRef CAS.
- J. M. Farrell, Z. M. Heiden and D. W. Stephan, Organometallics, 2011, 30, 4497–4500 CrossRef CAS.
- A. F. G. Maier, S. Tussing, T. Schneider, U. Flörke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef CAS.
- M. Kojima, M. Kanai and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12224–12227 CrossRef CAS.
- Y. Han, S. Zhang, J. He and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 7399–7407 CrossRef CAS.
- S. Zhang, Y. Han, J. He and Y. Zhang, J. Org. Chem., 2018, 83, 1377–1386 CrossRef CAS.
- T. Wang, L. Wang, C. G. Daniliuc, K. Samigullin, M. Wagner, G. Kehr and G. Erker, Chem. Sci., 2017, 8, 2457–2463 RSC.
- M. Shang, J. Z. Chan, M. Cao, Y. Chang, Q. Wang, B. Cook, S. Torker and M. Wasa, J. Am. Chem. Soc., 2018, 140, 10593–10601 CrossRef CAS.
- J. Z. Chan, Y. Chang and M. Wasa, Org. Lett., 2019, 21, 984–988 CrossRef CAS.
- A. F. G. Maier, S. Tussing, H. Zhu, G. Wicker, P. Tzvetkova, U. Flörke, C. G. Daniliuc, S. Grimme and J. Paradies, Chem. – Eur. J., 2018, 24, 16287–16291 CrossRef CAS.
- J.-J. Tian, N.-N. Zeng, N. Lie, X.-S. Tu and X.-C. Wang, ACS Catal., 2019, 9, 295–300 CrossRef CAS.
- J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209–13213 CrossRef CAS.
- M. Zhou, S. Park and L. Dang, Org. Chem. Front., 2020, 7, 944–952 RSC.
- R. Li, Y. Chen, K. Jiang, F. Wang, C. Lu, J. Nie, Z. Chen, G. Yang, Y.-C. Chen, Y. Zhao and C. Ma, Chem. Commun., 2019, 55, 1217–1220 RSC.
- Y. Chang, A. Yesilcimen, M. Chao, Y. Zhang, B. Zhang, J. Z. Chan and M. Wasa, J. Am. Chem. Soc., 2019, 141, 14570–14575 CrossRef CAS.
- S. Basak, A. Alvarez-Montoya, L. Winfrey, R. L. Melen, L. C. Morrill and A. P. Pulis, ACS Catal., 2020, 10, 4835–4840 CrossRef CAS.
- J. Zhang and S. Chang, J. Am. Chem. Soc., 2020, 142, 12585–12590 CrossRef CAS.
- J. D. Webb, V. S. Laberge, S. J. Geier, D. W. Stephan and C. M. Crudden, Chem. – Eur. J., 2010, 16, 4895–4902 CrossRef CAS.
- L. C. Wilkins, N. Santi, L. Y. P. Luk and R. L. Melen, Philos. Trans. R. Soc., A, 2017, 375, 20170009 CrossRef.
- S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2301–2314 CrossRef CAS.
- Q. Wang, J. Chen, X. Feng and H. Du, Org. Biomol. Chem., 2018, 16, 1448–1451 RSC.
- Alternative mechanisms for the B(C6F5)3-catalysed transfer hydrogenation of aldehydes, p-quinone methides and fuchsones using Hantzsch ester as the hydrogen source have been proposed to occur via B(C6F5)3 activation of the reduction substrate prior to hydride transfer directly from the Hantzsch ester. See: A. Hamasaka, H. Tsuji, M. Ehara and Y. Uozumi, RSC Adv., 2019, 9, 10201–10210 RSC.
| This journal is © The Royal Society of Chemistry 2021 |