
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the chemistry of ketyl radicals†
Áron
Péter
 ,
Soumitra
Agasti
,
Oliver
Knowles
,
Emma
Pye
and
David J.
Procter
*
,
Soumitra
Agasti
,
Oliver
Knowles
,
Emma
Pye
and
David J.
Procter
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, UK. E-mail: david.j.procter@manchester.ac.uk
First published on 23rd March 2021
Abstract
Ketyl radicals are valuable reactive intermediates for synthesis and are used extensively to construct complex, functionalized products from carbonyl substrates. Single electron transfer (SET) reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of aldehydes and ketones is the classical approach for the formation of ketyl radicals and metal reductants are the archetypal reagents employed. The past decade has, however, witnessed significant advances in the generation and harnessing of ketyl radicals. This tutorial review highlights recent, exciting developments in the chemistry of ketyl radicals by comparing the varied contemporary – for example, using photoredox catalysts – and more classical approaches for the generation and use of ketyl radicals. The review will focus on different strategies for ketyl radical generation, their creative use in new synthetic protocols, strategies for the control of enantioselectivity, and detailed mechanisms where appropriate.
O bond of aldehydes and ketones is the classical approach for the formation of ketyl radicals and metal reductants are the archetypal reagents employed. The past decade has, however, witnessed significant advances in the generation and harnessing of ketyl radicals. This tutorial review highlights recent, exciting developments in the chemistry of ketyl radicals by comparing the varied contemporary – for example, using photoredox catalysts – and more classical approaches for the generation and use of ketyl radicals. The review will focus on different strategies for ketyl radical generation, their creative use in new synthetic protocols, strategies for the control of enantioselectivity, and detailed mechanisms where appropriate.
 Áron Péter | Áron Péter received his BSc degree from the Eötvös Loránd University (Hungary) in 2018. He completed his thesis work in the Servier Research Institute focusing on the synthesis of biologically active scaffolds. He then moved to the University of Manchester to conduct his MSc studies under the guidance of Professor David J. Procter on the use of SmI2 in total synthesis. Following this, he received a prestigious Dean's Award and stayed in the Procter group for his PhD studies. His current research is focused on the development of new processes mediated by SmI2. |
 Soumitra Agasti | Soumitra Agasti was born in West Bengal, India. He received his Master's degree (Organic Chemistry) from the Indian Institute of Technology (IIT) Bombay in 2012 and subsequently carried out PhD studies with Professor Debabrata Maiti. He joined the group of Professor David J. Procter as an EPSRC postdoctoral fellow in 2018 and is currently a Marie Sklodowska-Curie Fellow developing SmI2-catalyzed coupling reactions. |
 Oliver Knowles | Oliver J. Knowles received his MChem degree from Newcastle University in 2019, carrying out a final year research project within the Centre for Cancer on the synthesis of novel cell cycle protein inhibitors. In 2019 he joined the group of Professor David J. Procter for his PhD studies, funded by the BBSRC DTP. His research interests include the use of bio-inspired catalysts to facilitate novel radical chemistry. |
 Emma Pye | Emma Pye grew up in Sheffield where she completed her MChem degree at the University of Sheffield, carrying out a final year research project on the synthesis of pyridine boronic acid derivatives. During her undergraduate studies she spent a year in industry as a process chemist at GSK. In 2020, she joined the group of Professor David J. Procter to carry out her PhD studies, funded by the EPSRC iCAT CDT. Her research interests include the integration of radical cyclization cascades with biocatalysis. |
 David J. Procter | David J. Procter was born in Leyland in Lancashire, England. He obtained his BSc in Chemistry from the University of Leeds in 1992 and his PhD in 1995 working with Professor Christopher Rayner on organosulfur chemistry. He then spent two years as a Postdoctoral Research Associate with Professor Robert Holton at Florida State University in Tallahassee, USA working on the synthesis of Taxol. In late 1997 he took up a Lectureship at the University of Glasgow in Scotland. In 2004, he moved to the University of Manchester. David was promoted to Professor in 2008. |
Key learning points• The strategies available for ketyl radical generation.• The structure and reactivity of ketyl radicals. • How ketyl radicals can be generated under catalytic conditions. • How the enantioselectivity of reactions involving ketyl radicals can be controlled. • How ketyl radicals can be harnessed in target synthesis. |
1. Introduction
Ketyl radicals, and ketyl radical anions, are highly valuable, functionalized, reactive intermediates. Since their discovery in 1891, they have played a significant role in synthesis (Fig. 1).1 For example, ketyl radicals are readily generated from carbonyl compounds and thus play a pivotal role in strategies for carbonyl umpolung reactivity. Ketyl radicals and ketyl radical anions are one of the most well-studied reactive intermediates and are commonly exploited in powerful methods for C–C bond formation. A wide range of often complex molecular architectures is accessible by harnessing the generation and reaction of ketyl radicals in radical approaches that complement more usual ionic protocols. Recent years have seen the development of methodologies that generate and exploit ketyl radicals in innovative new ways. For example, single electron transfer (SET), proton-coupled electron transfer (PCET), hydrogen atom abstraction (HAT), and halogen atom abstraction (XAT) have all been used to access ketyl radicals (Fig. 1).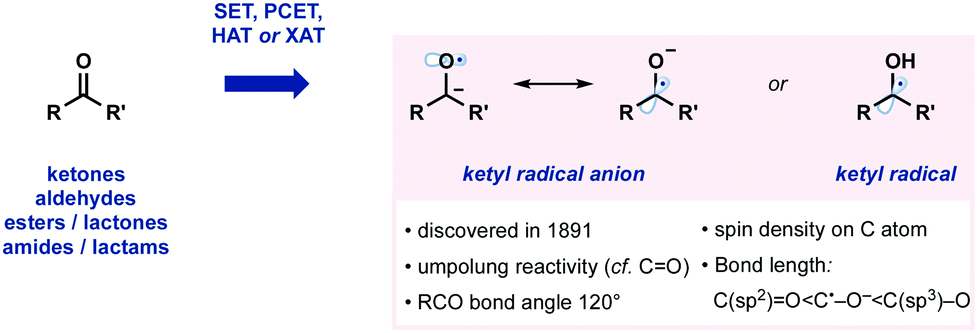 | ||
| Fig. 1 Some properties of ketyl radicals and their anions. | ||
The structural properties of ketyl radicals, and their anions, closely resemble carbonyl compounds in terms of bond angle. In contrast, in ketyl radical anions, the C˙–O− bond is longer than a C(sp2)O, but shorter than a C(sp3)–O bond. DFT and other studies have shown that the spin density mainly resides on the carbon atom of ketyl radical anions.1
Covering the literature of the past 10 years, this tutorial review highlights exciting developments in the chemistry of ketyl radicals by comparing the varied contemporary – for example, using photoredox catalysts – and more classical approaches for the generation and use of ketyl radicals. The review will focus on different strategies for ketyl radical generation, their creative use in new synthetic protocols, strategies for the control of enantioselectivity, and the proposed mechanisms of reactions involving the reactive intermediates. The overview will be an invaluable introduction to the field and a useful resource for synthetic chemists wishing to rationally design new synthetic processes that exploit the unique chemistry of ketyl radicals.
2. Overview of ketyl radical generation
The aldehyde and ketone functional groups are fundamentally important handles for synthesis. In general, polarization of the carbon–oxygen double bond (CO) results in an electrophilic centre at the carbon atom. Therefore, traditionally, these species were functionalised by strong nucleophiles, such as RMgX or RLi. The formation of ketyl radicals, or ketyl radical anions, possessing a nucleophilic carbon radical offers a means of inverting the reactivity of the starting carbonyl compound and using them in alternative C–C bond forming reactions with non-nucleophilic partners.
The formation of ketyl radicals by SET reduction does, however, suffer from a major limitation; the high reduction potential of aldehydes (Ep/2 (benzaldehyde/benzaldehyde˙−) = −1.93 V vs. SCE in MeCN) and ketones (Ep/2 (acetophenone/acetophenone˙−) = −2.11 V vs. SCE in MeCN). To overcome the high barriers to reduction, strong single electron reducing agents based on K,2 Zn,3 Mn,4 Ti,5 and Sn6 have been used. Arguably, samarium diiodide (SmI2), remains the reagent of choice for ketyl radical generation from aldehydes and ketones due to its selectivity, versatility, commercial availability, and ease of use.
With the advent of photoredox catalysis, new photochemical approaches for the generation of ketyl radicals from activated carbonyl compounds have emerged. Following excitation, SET from highly reducing photocatalysts to aldehydes and ketones forms ketyl radicals or ketyl radical anions. Attractively, ketyl radicals can be accessed without recourse to the use of radical initiators or stoichiometric metallic reductants. The resultant ketyl radicals can be used in various C–C bond forming reactions, including important ketyl–olefin couplings.
Homocoupling of ketyl radicals can lead to ‘pinacol’ by-products. However, this unwanted pathway can be suppressed by using a reactive, polarity-matched coupling partner, employing an excess of the coupling partner, or by maintaining a low concentration of ketyl radical anion. Intramolecular processes are less-likely to suffer from unwanted pinacol coupling.7
The utility of ketyl radicals is perhaps best illustrated by their privileged status in natural product total synthesis; this status arises from the high chemo-, regio-, and stereoselectivity of their reactions and the functionalized products that result. An ever-growing number of total syntheses of challenging targets have been unlocked by the chemistry of ketyl radicals. Furthermore, ketyl radicals have recently been exploited in enzymatic reactions8 and in polymerisations9 that deliver high value products.
3. Generation of ketyl radicals using metals, pseudo-metals and organocatalysts
Strong single-electron reductants, such as K,2 Zn,3 Mn,4 Ti5 and Sn,6 as well as electrochemical conditions, have been widely employed for the generation of ketyl radicals. The requirement for stoichiometric amounts of reductant can undermine the synthetic utility of these transformations. However, the use of SmI2 for ketyl radical formation has remained popular due to its versatility—a variety of carbonyl compounds can be converted to the corresponding ketyl radicals using the reagent – tuneability, commercial availability, and ability to control the stereochemical course of subsequent radical couplings by coordination to Lewis basic sites in substrates.10–12In a classical approach, Cheng and co-workers reported the coupling of aryl/heteroaryl ketones and aldehydes 1 with a range of electron deficient alkenes 2, including acrylates, acrylamides, acrylonitriles, and vinyl sulfones, to furnish γ-hydroxyl butyric acid derivatives 3 (Scheme 1).13 Importantly, the reaction does not require strong single-electron reducing agents, such as alkali metals, and instead uses inexpensive Zn metal. The process is compatible with a variety of substituted aryl and heteroaryl groups and provides straightforward access to simple γ-hydroxyl butyric acid derivatives. Interestingly, the Zn/H2O reducing system was used at room temperature under an atmosphere of NH3; Zn metal or a combination of Zn metal powder with other amines failed to deliver the desired coupling products.
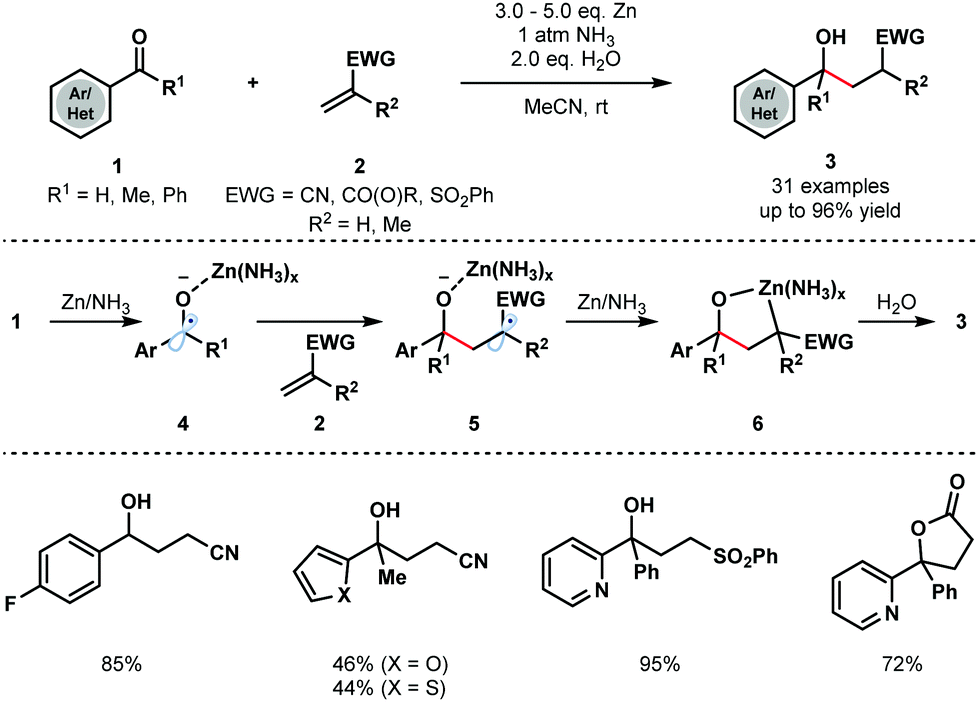 | ||
| Scheme 1 Zinc–NH3-mediated ketyl olefin couplings (Cheng, 2013). | ||
Control experiments revealed that ammonia does not activate zinc but is necessary to form a more reducing Zn/NH3 complex. SET reduction of 1 furnishes zinc ketyl radicals 4 that add to the alkene substrates 2 to give radicals 5. Further reduction is thought to then afford five-membered oxametallacycles 6 that undergo protonation to give the reductive coupling products 3.
Pinacol coupling typically involves reduction of a ketone or aldehyde precursor using a stoichiometric SET reductant, followed by homocoupling of the resultant ketyl radical anion. Reversibility of the pinacol coupling process has not previously been utilized in synthesis to generate ketyl radicals. For the first time, in 2016, Studer and co-workers exploited the reversibility of the pinacol coupling in a transition-metal-free C–C bond forming process involving aryl iodides 8 and ketyl radicals 11 (Scheme 2).7 The method was compatible with symmetrical and unsymmetrical pinacols and tolerated a range of aryl iodides bearing ortho heteroatom substituents. Upon deprotonation with NaH, readily accessible pinacols 7 were covered to Na-pinacolates 10 that are in equilibrium with sodium–ketyl radicals 11. Addition of the ketyl radicals 11 to arenes 8, facilitated by coordination of the ortho heteroatom substituents on the aryl iodide to the sodium cation, generates cyclohexadienyl radicals 12. Fragmentation of 12 releases iodine radical and generates sodium alkoxides 13, which upon protonation furnish the tertiary alcohol products 9 in good yield. The iodide radical is reduced by the ketyl radicals 11 to form ketones 14 and sodium iodide. The major byproducts, alcohols 15, are thought to arise from the reduction of ketones 14 by the NaH/NaI couple.
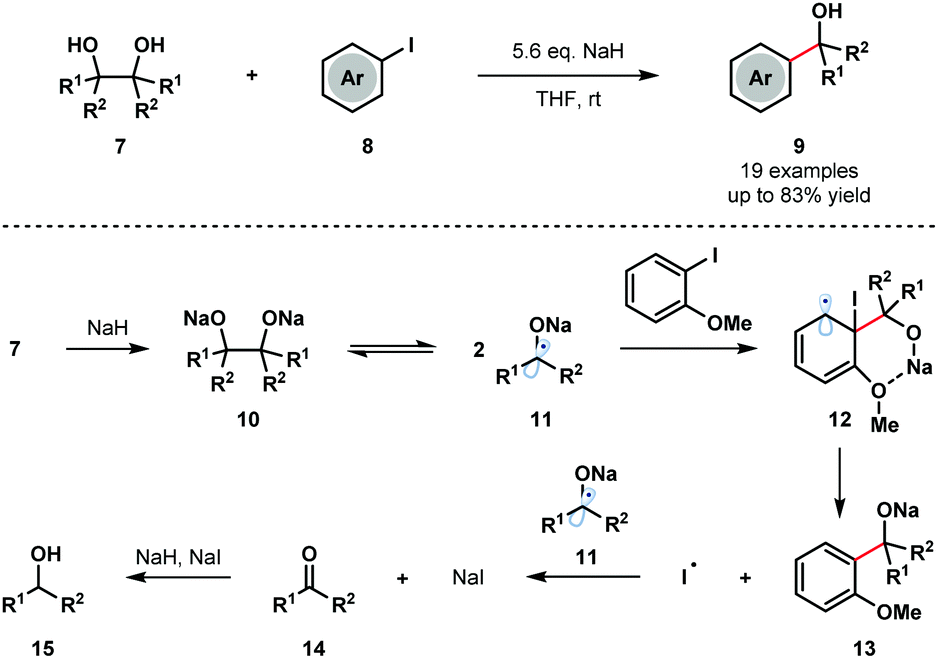 | ||
| Scheme 2 Cross-coupling of sodium ketyl radicals and aryl iodides to give tertiary benzylic alcohols (Studer, 2016). | ||
In 2012, Streuff and co-workers reported the enantioselective reductive cyclization of ketonitriles 16 using the enantiopure Ti(IV)-precatalyst 18 (Scheme 3).14 Enantioselective Ti(III)-mediated radical processes remain rare and the use of a catalytic amount of chiral transition metal catalyst alongside an inexpensive terminal reductant is attractive. The reaction was shown to tolerate aliphatic ketones, aryl ketones bearing electron-donating and electron-withdrawing substituents, and heteroaryl ketones.
 | ||
| Scheme 3 Ketyl radical intermediates in a Ti(III)-catalyzed enantioselective cyclization of ketonitriles (Streuff, 2012). | ||
In situ formation of the low-valent enantiopure Ti(III) catalyst is followed by its coordination to the carbonyl group of substrates 16 and subsequent SET reduction to provide Ti(IV)–ketyl radicals 19. Enantioselective radical 5-exo-dig cyclization involving the nitrile group and one enantiopic face of the ketyl radicals furnishes N-centred radicals 20. These radicals are quickly reduced by the Ti(III) catalyst, and the resultant anions protonated, to give the titanium(IV) alkoxide intermediates 21 which are subsequently quenched by TMSCl, liberating the α-oxygenated cyclic imines 22. Finally, hydrolysis during workup furnishes ketone products 17. The Ti(III) catalyst is regenerated by the super-stoichiometric co-reductant, zinc dust. Transition state 23 was proposed to explain the origin of enantioselectivity in the reaction.
In 2018, Li and co-workers reported an intriguing route to ketyl radical derivatives that involves the SET reduction of ketones/aldehydes by a pyridine–boryl radical under thermal conditions (Scheme 4).15 This metal-free reductive coupling of aldehydes and ketones 24 with alkenes 25 boasts a broad substrate scope and good functional group compatibility. This method provides a new, metal-free approach for the generation of ketyl radical intermediates for exploitation in C–C bond formation.
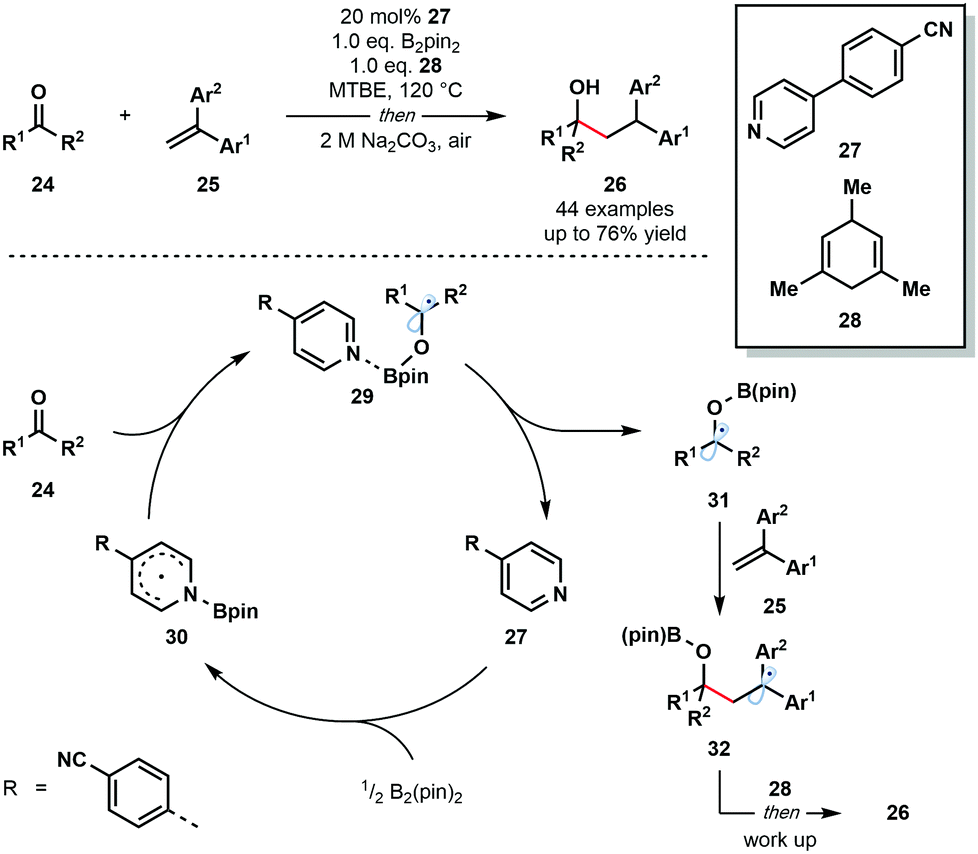 | ||
| Scheme 4 Carbonyl–alkene coupling featuring pyridine–boryl radicals (Li, 2018). | ||
The mechanism is thought to begin by the activation of the B–B bond of B2pin2 by a pyridine-derived organocatalyst 27 and formation of pyridine–boryl radical intermediate 30. The addition of this radical to carbonyl substrates 24 generates pyridine complexed boron-containing ketyl radicals 29. An alternative SET event between 30 and 24 was excluded based on DFT calculations. Dissociation of the pyridine catalyst allows ketyl radicals 31 to react with radical traps, such as 25, to yield diaryl-stabilized radical intermediates 32. Hydrogen atom abstraction from the sacrificial donor 28, followed by work up, affords the final reductive coupling products 26.
The lanthanide reductant samarium diiodide (SmI2) is a versatile and selective reagent for the generation of ketyl radical anions; coordination of Sm(III) to the radicals often allows the stereochemistry of subsequent C–C bond-forming processes to be controlled as the metal coordinates to Lewis basic sites in the coupling partners. The use of water as an additive in reactions of SmI2 in THF allows the generation of previously inaccessible ketyl radicals, for example, those derived from lactones and other aliphatic carboxylic acid derivatives. This is due to the increased reduction potential of the SmI2–H2O system (Ered1/2 (Sm(III)/Sm(II)) = −1.3 V vs. SCE in THF/DME), compared to SmI2 alone (Ered1/2 (Sm(III)/Sm(II)) = −0.89 V vs. SCE in THF), and the H2O coordinated to Sm providing a proximal proton source for the quenching of reactive intermediates. It has also been proposed that PCET is involved in reactions of SmI2–H2O. Recent studies have shown that this reagent system can also be used to generate unusual ketyl radical anions by SET reduction of the carbonyl groups in esters and amides.10–12 Inorganic additives (e.g. LiCl, LiBr, NiCl2, and FeCl3) and organic additives (e.g. HMPA, TPPA, and other Lewis basic ligands) are commonly used in conjunction with proton sources (MeOH, t-BuOH, H2O) to fine-tune the selectivity and reactivity of SmI2. Although the reagent has been used in catalytic amounts in a handful of studies, the requirement for a super-stoichiometric terminal reductant has limited widespread uptake of these catalytic methods.16 Against this backdrop, Procter and co-workers have recently reported efficient SmI2-catalyzed cyclizations that involve the generation of ketyl radical anions by SET reduction of ketone carbonyl groups by SmI2 and that proceed under mild conditions and in the absence of stoichiometric co-reductants.17
In 2012, Procter and co-workers reported the reductive cyclization of alkenyl-, alkynyl-, and allenyllactones to form oxygenated cycloheptanes using the SmI2–H2O system; allenyllactone substrates selectively produced cycloheptanes bearing three new stereocentres (Scheme 5).18 A variety of substituents on the lactone ring, and on the allene, were tolerated in these processes and products were obtained in good yield and with excellent diastereocontrol. The proposed mechanism for the reductive cyclization of alkenyllactones and allenyllactones proceeds via coordination of SmI2 to 33, followed by SET reduction of the lactone carbonyl, generating Sm(III) ketyl radicals 35. 5-Exo-trig cyclization and subsequent reduction by another equivalent of SmI2 affords hemiketal intermediates 34, which are in equilibrium with their ring-opened forms 36. Further SET reduction of these intermediates gives diols 37 upon protonation. The reductive cyclization of allenyllactones 38 is postulated to proceed in a similar fashion, to give, in this case, cycloheptanone intermediates that undergo conjugate reduction to give hemiketals 39 or complex cycloheptan-1,4-diols 40.18
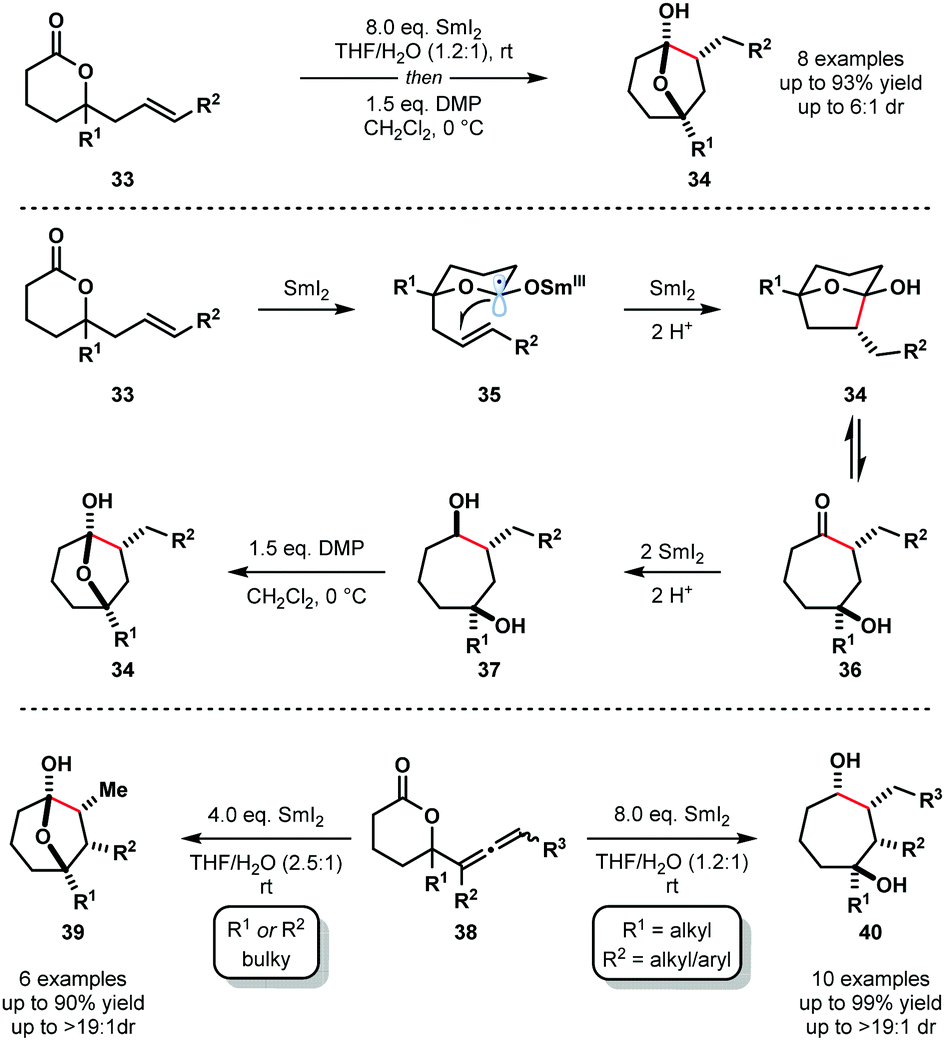 | ||
| Scheme 5 Ketyl radicals in lactone–alkene couplings using SmI2–H2O (Procter, 2012). | ||
Furthermore, selective cascade processes of alkenyl and allenyllactones can be used to generate complex bicyclic architectures 42 and 43 (Scheme 6). In these lactone cyclization cascades, the second Sm(III) ketyl radical intermediates encountered along the mechanistic pathway – i.e. those generated from ketone intermediates like 36 – are intercepted by tethered alkenes, furnishing bicyclic tertiary alcohols 42 or 43 upon work up.18
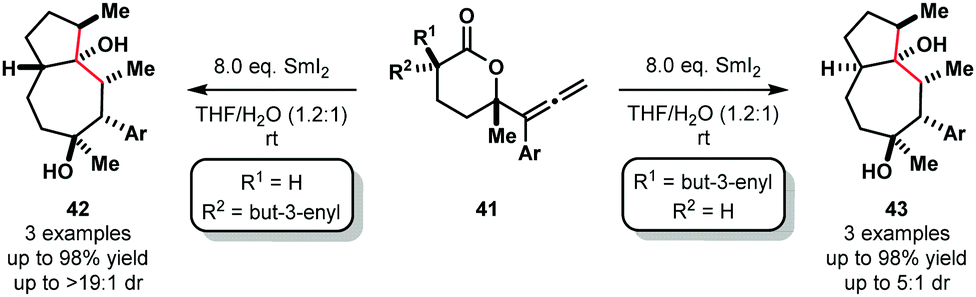 | ||
| Scheme 6 Ketyl radicals in lactone cascade cyclizations to give fused bicycles using SmI2–H2O (Procter, 2012). | ||
The synthetic potential of enantioselective radical cyclization cascades remains largely unexplored. In particular, the use of chiral ligands to exert enantiocontrol in SmI2-mediated reactions, including cascade reactions, is a long-standing challenge. In 2017, Procter and co-workers reported the SmI2-mediated, enantioselective desymmetrizing ketyl–olefin radical cyclizations and cyclization cascades of dienyl β-ketoesters (Scheme 7).19 Readily available, recyclable, enantiopure aminodiol ligand L1 facilitated the enantioselective cyclization of substrates 44 and 45 and the efficient formation of 46 and 47. This is the first example of a highly enantioselective SmI2-mediated ketyl–olefin cyclization. MeOH was used as a sacrificial proton source to preserve the integrity of the chiral aminodiol ligand L1. Furthermore, MeOH was found to increase the level of enantiocontrol observed and likely coordinates to the samarium metal centre. Substrates containing up to five new stereocentres were isolated in good yield and with high enantiopurity. The proposed mechanism begins by coordination of the two-point-binding substrates, 44 or 45, to SmI2 bearing chiral ligand L1. Next, SET from Sm(II) to the ketone moiety affords Sm(III) ketyl radicals 48, which undergo enantiodetermining cyclization to give radicals 49. Trapping of radical intermediates 49 by the proximal tethered alkene yields intermediates 50 that undergo further SET reduction and protonation to give 46.
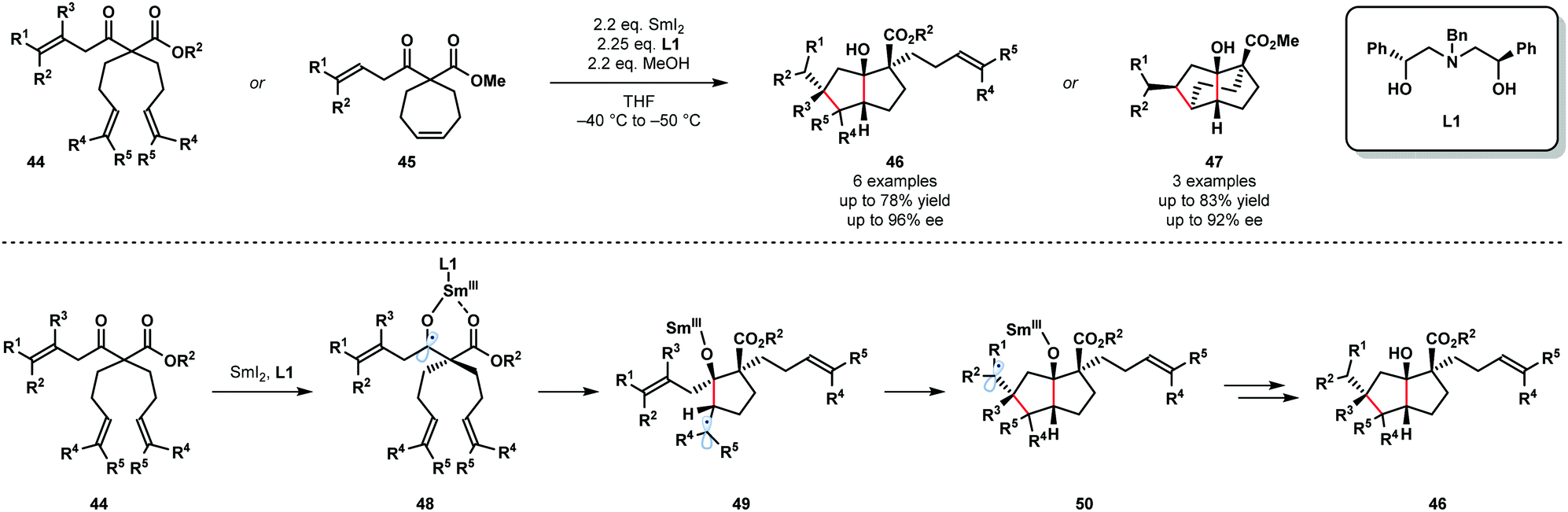 | ||
| Scheme 7 Enantioselective cyclization cascades of Sm(III) ketyl radicals (Procter, 2017). | ||
N-Heterocyclic carbenes (NHCs) are also known to facilitate radical reactions that involve ketyl radicals. In particular, deprotonated Breslow intermediates, generated from aldehydes using NHCs, are useful SET reductants. In 2019, the Ohmiya group reported a decarboxylative coupling of aryl aldehydes 51 and redox-active esters 52 to produce sterically congested aryl alkyl ketones 53 (Scheme 8).20 This methodology allows for the use of widely-available carboxylic acids as coupling partners in radical–radical couplings involving ketyl radicals. The selectivity for products of cross-coupling, rather than homo-coupling, is striking. The process tolerates a wide range of functional groups in both partners, however, only tertiary and secondary carboxylic acid partners were used and the scope with aliphatic aldehydes was not explored. The reaction is thought to begin by deprotonation of N1 to yield the active catalytic NHC species 54. The neutral Breslow intermediates 55 are formed by addition of 54 to aldehydes 51. Deprotonation then yields the reducing intermediates 56 (Ered1/2 (57/56) = −0.96 V vs. SCE in MeCN) and SET reduction of 52 provides alkyl radicals 58 and ketyl radicals 57. Redox-active esters 52 (Ered1/2 (52/52˙−) ≤ −1.28 V vs. SCE in MeCN) are likely activated by Cs+ prior to SET reduction. Finally, radical–radical coupling between 57 and 58, followed by the elimination of 54, affords congested ketones 53 and closes the catalytic cycle.
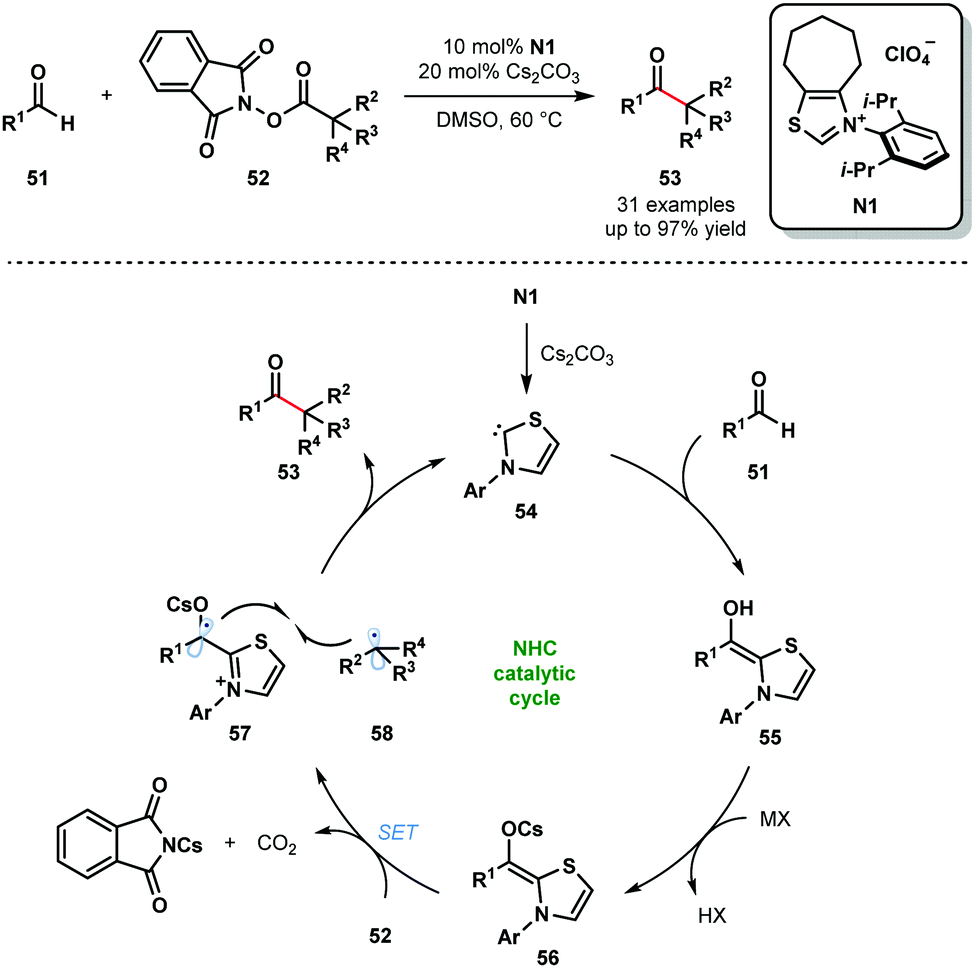 | ||
| Scheme 8 Ketyl radicals in NHC-catalysed decarboxylative coupling of activated carboxylic acids and aldehydes (Ohmiya, 2019). | ||
4. Light induced ketyl radical generation
Visible light-mediated photoredox catalysis is now established as a powerful tool to access ketyl radicals,21–26 thus complementing traditional approaches to the reactive intermediates. Upon visible light excitation, photocatalysts can engage in SET processes that generate radicals, thus avoiding the use of classical radical initiators or stoichiometric metal reagents.In 1978, Kellogg et al. reported the use of RuII(bpy)3Cl2 as a catalyst for the SET reduction and fragmentation of aryl carbonyl compounds bearing α-sulfonium groups.27 The process likely proceeds through the generation of ketyl radicals. Since then, visible light-mediated photoredox catalysis has become widely recognized as an efficient strategy for the production of ketyl radicals from aldehydes and ketones under mild conditions. The resulting ketyl radicals have been exploited in a diverse array of C–C bond forming reactions that deliver high-value, and often complex, molecular architectures. This section of the tutorial review highlights recent, significant advancements made in the chemistry of photocatalytically generated ketyl radicals.
Most mechanisms in this section are depicted as closed photocatalytic cycles, as suggested by the original authors. However, it is important to note that, in the absence of quantum yield measurements, for example, radical chain processes can not be ruled out.28
4.1 Intramolecular transformations
In 2013, Knowles and co-workers reported a catalytic intramolecular ketyl–olefin coupling reaction that is facilitated by proton coupled electron transfer (PCET) (Scheme 9).29 This seminal study constitutes the first synthetically useful example of ketyl radical generation using visible-light. The cyclisation was achieved using catalytic Ru(bpy)3(BArF)2 and diphenyl phosphoric acid, and the terminal reductant, 2-phenyl-dihydrobenzothiazoline (BT), in the presence of visible light irradiation, and was used to deliver substituted cyclopentanes, tetrahydrofurans and pyrrolidines. The scope focuses on aryl alkyl ketones and is limited to activated alkenes. Visible light-mediated photoexcitation of the ground state photocatalyst, RuII(bpy)32+, followed by reductive quenching of the excited photocatalyst, RuII(bpy)32+*, by BT forms the SET reductant RuI(bpy)3+. Concurrently, the Brønsted acid co-catalyst activates the ketone functionality in 59 by a hydrogen bonding interaction. Subsequent PCET delivers ketyl radical intermediates 61. The resultant ketyl radicals 61 undergo facile intramolecular radical addition onto the double bond forming a stabilised radical species 62. Hydrogen atom transfer from BT to 62 delivers products 60 and radical 63. RuII(bpy)32+ is reduced by radical 63 to regenerate RuI(bpy)3+ and carbocationic intermediate 64. Finally, deprotonation of 64 by the phosphate anion regenerates the Brønsted acid co-catalyst and closes the catalytic cycle.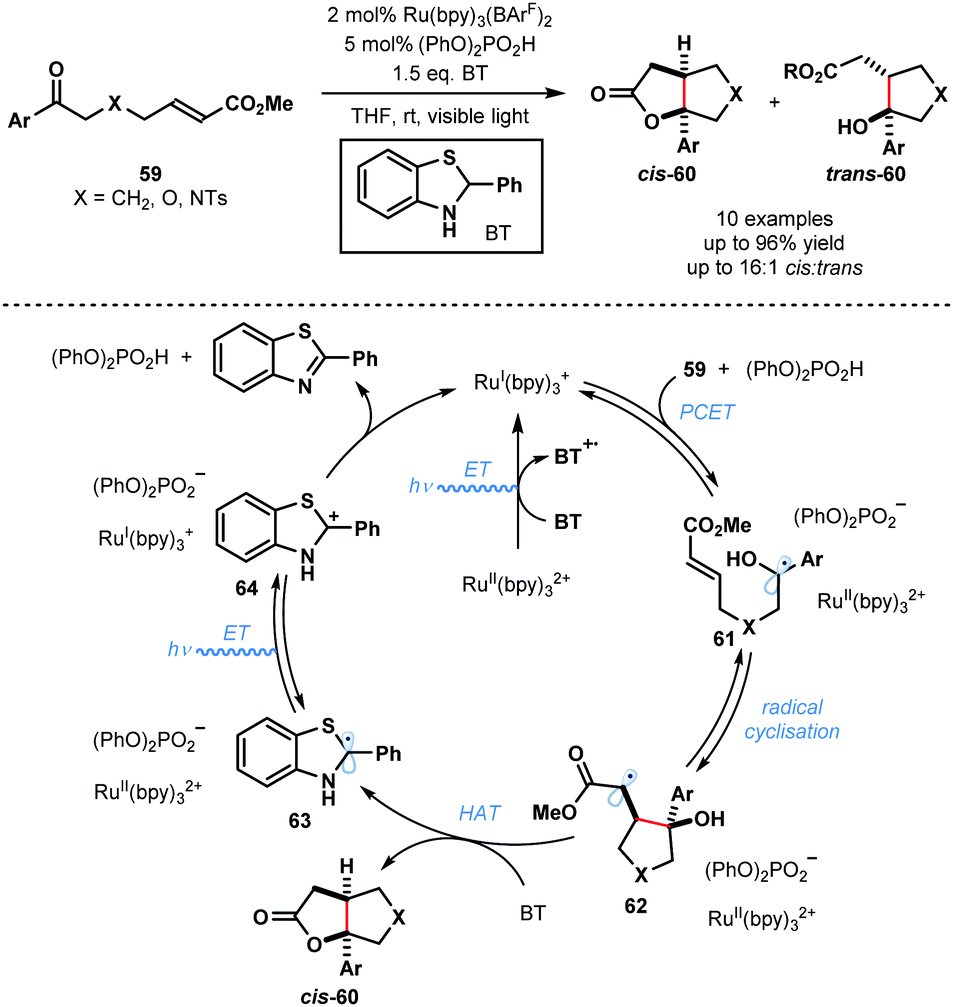 | ||
| Scheme 9 Proton-coupled electron transfer in photocatalytic ketyl–olefin cyclisations (Knowles, 2013). | ||
In the same year, Knowles and co-workers reported the use of PCET for ketyl radical generation in enantioselective catalysis (Scheme 10).30 The intramolecular reductive coupling of ketones and hydrazones, involving ketyl radicals 67, used a chiral phosphoric acid (CPA) catalyst to control the enantioselectivity of the reaction. A range of syn-1,2-hydrazo alcohols is accessible with excellent levels of diastereo- and enantiocontrol using catalyst CPA 1 in combination with the photoredox catalyst IrIII(ppy)2(dtbpy)PF6. This is the first example of an enantioselective, photoredox aza-pinacol coupling. Building on their earlier work (Scheme 9), the reaction is thought to be initiated by the off-cycle excitation of the photocatalyst to give [IrIII]* which is then converted to [IrII] by the sacrificial reductant, HE (Hantzsch ester). Concerted PCET from [IrII] to the complex of ketones 65 and CPA 1 leads to the formation of phosphate bound ketyl radical intermediates 67. Enantioselective radical cyclization, followed by HAT from HE affords the enantioenriched, cyclic products 66 and HE˙. Excitation of [IrIII] and oxidation of HE˙ by the resulting [IrIII]* produces pyH+ and regenerates the catalytically active [IrII]. Finally, the conjugate anion of CPA 1 deprotonates pyH+ and returns the CPA 1 catalyst.
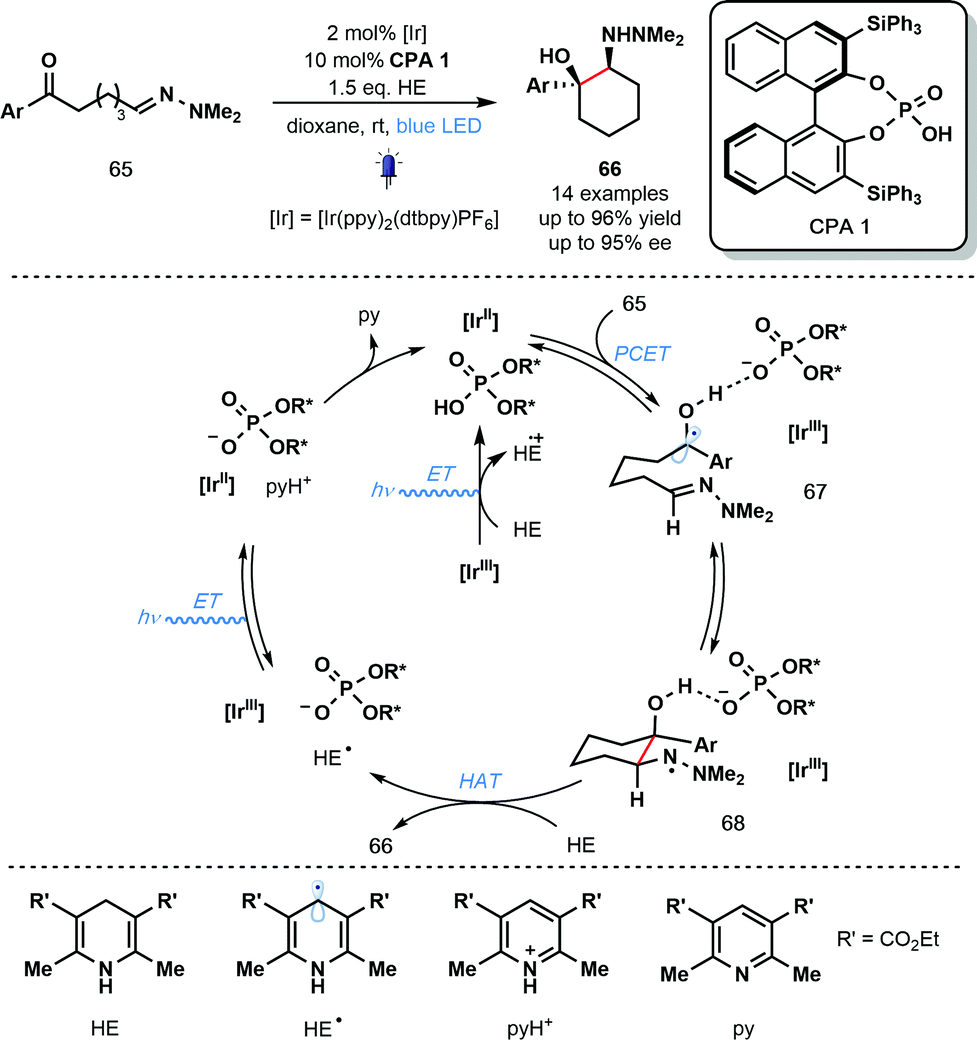 | ||
| Scheme 10 Photocatalytic enantioselective aza-pinacol cyclization employing a chiral phosphoric acid and exploiting PCET (Knowles, 2013). | ||
In 2019, Hyster and co-workers reported the use of the flavin-dependent ene-reductase, MorB (Morphinone Reductase), in conjunction with Ru(bpy)3Cl2 and visible light to achieve the enantioselective reduction of aryl ketones via enzyme-bound ketyl radical anion intermediates (Scheme 11).8 Mechanistic studies indicated that binding of the carbonyl group by the active site of MorB lowers the reduction potential of the ketone moiety and thus facilitates its reduction by RuI(bpy)3+. HAT from FMNhq then quenches the resulting ketyl radical anion. The photocatalytic cycle is closed by an electron transfer process between FMNsq and RuII(bpy)32+*. The novel marriage of photo- and bio-catalysis afforded enantioenriched alcohols 70 in good to excellent yield with moderate enantioselectivity. Despite modest scope and selectivity, this exciting approach paves the way for future marriages of photo- and bio-catalysis featuring ketyl radicals.
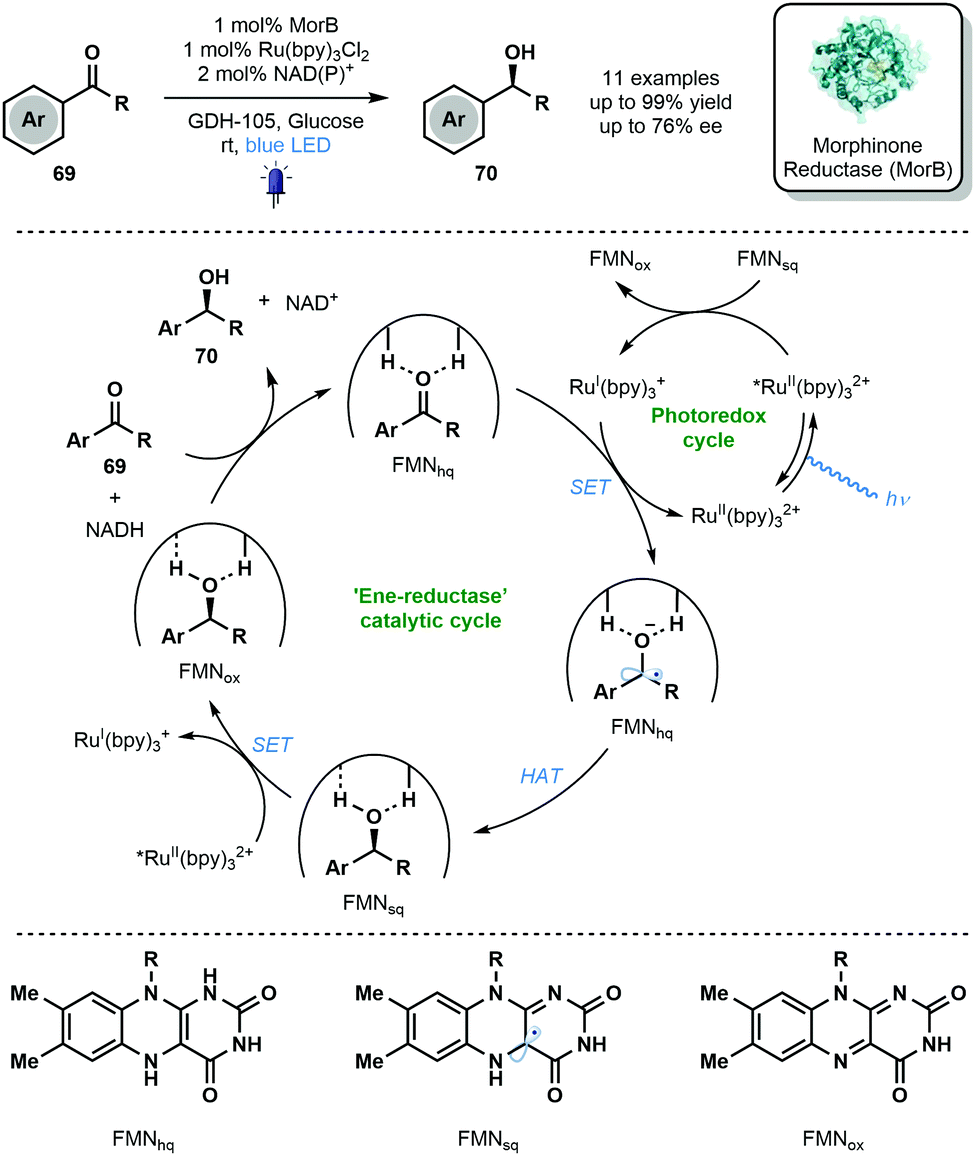 | ||
| Scheme 11 Enantioselective reduction of aromatic ketones through the integration of enzymatic and photoredox catalysis (Hyster, 2019). | ||
In 2020, Ye and co-workers reported a photocatalytic ketyl–ynamide coupling that triggers a radical Smiles rearrangement and allows efficient access to a range of 2,3-substituted indoles (72) and 3,4-substituted isoquinolines (73) (Scheme 12).31 This is the first radical Smiles rearrangement featuring ynamides. The process tolerates a range of important substituents, including, halide, cyano, methoxy, and trifluoromethyl. The postulated mechanism begins with reduction of 71 by [IrII] and formation of ketyl radicals 74. Regioselective ketyl–ynamide coupling generates alkenyl radicals 75 and triggers a Smiles rearrangement to give 76. Subsequent steps, featuring a number of SET events, yield products 72 or 73via intermediates 77–80.
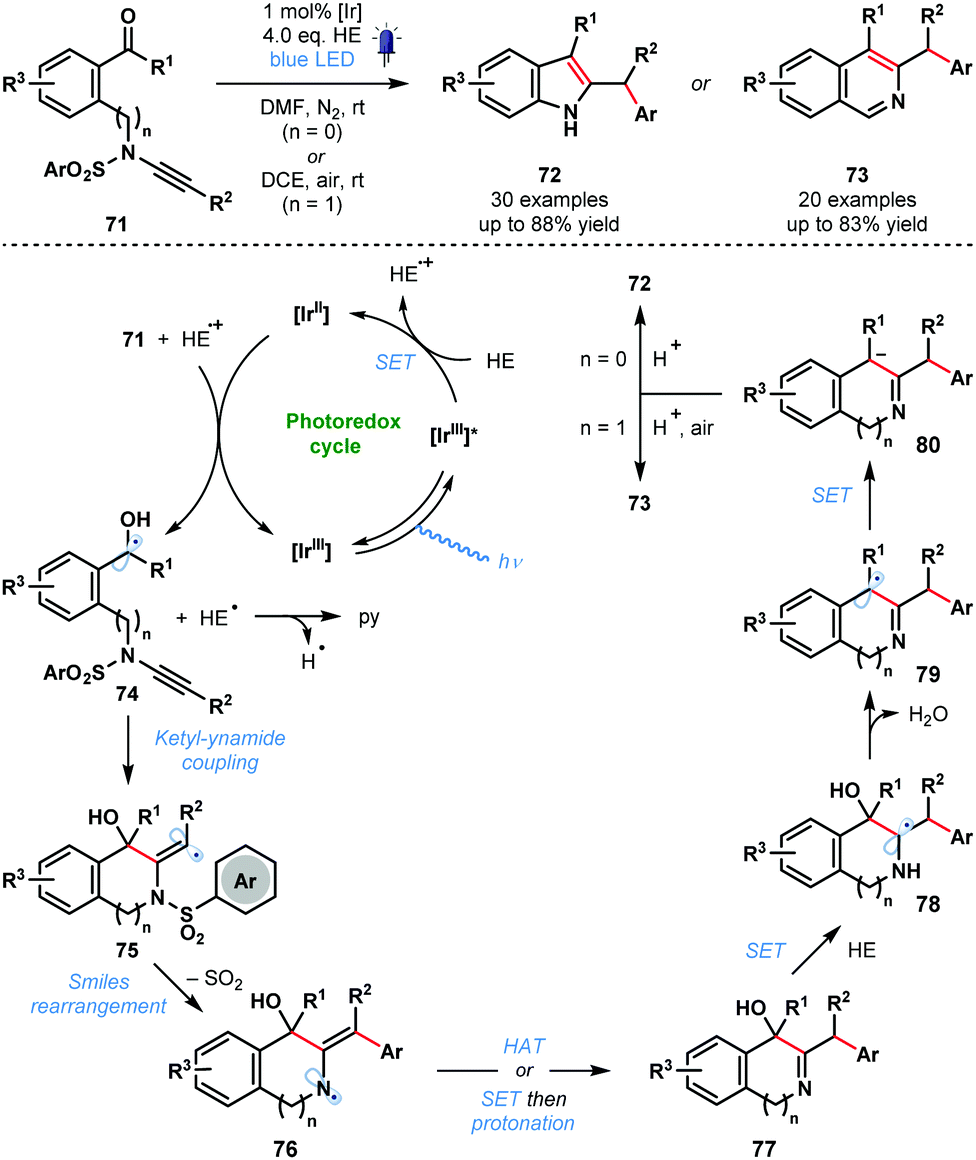 | ||
| Scheme 12 Photocatalytic synthesis of substituted indoles and isoquinolines by ketyl–ynamide coupling and radical Smiles rearrangement (Ye, 2020). | ||
4.2 Intermolecular couplings
An early dual photocatalytic and organocatalytic process was developed by MacMillan and co-workers for the direct β-functionalisation of cyclic ketones using aryl ketones (Scheme 13).32 A range of substituted aryl ketones 82 served as coupling partners with cyclic ketones 81, in the presence of 1 mol% IrIII(ppy)3 and 20 mol% azepane, to afford γ-hydroxyketones 83 in high yield. Importantly, this method complements traditional ketone functionalisation at the carbonyl carbon and at the α-position. Furthermore, selective radical–radical cross-coupling is observed and issues of homo-coupling are avoided. Oxidative quenching of the excited photocatalyst IrIII(ppy)3* by the acid-activated aryl ketones 82 generates ketyl radical anions 84 and IrIV(ppy)3+. Enamines 85, formed by condensation of azepane with ketones 81, reduce IrIV(ppy)3+ and close the photocatalytic cycle. Deprotonation of 86 generates 87 and radical–radical coupling between ketyl radical anion 84 and 87 forms intermediate 88. Subsequent hydrolysis of 88 liberates the β-functionalised cyclic ketone products 83 and regenerates azepane.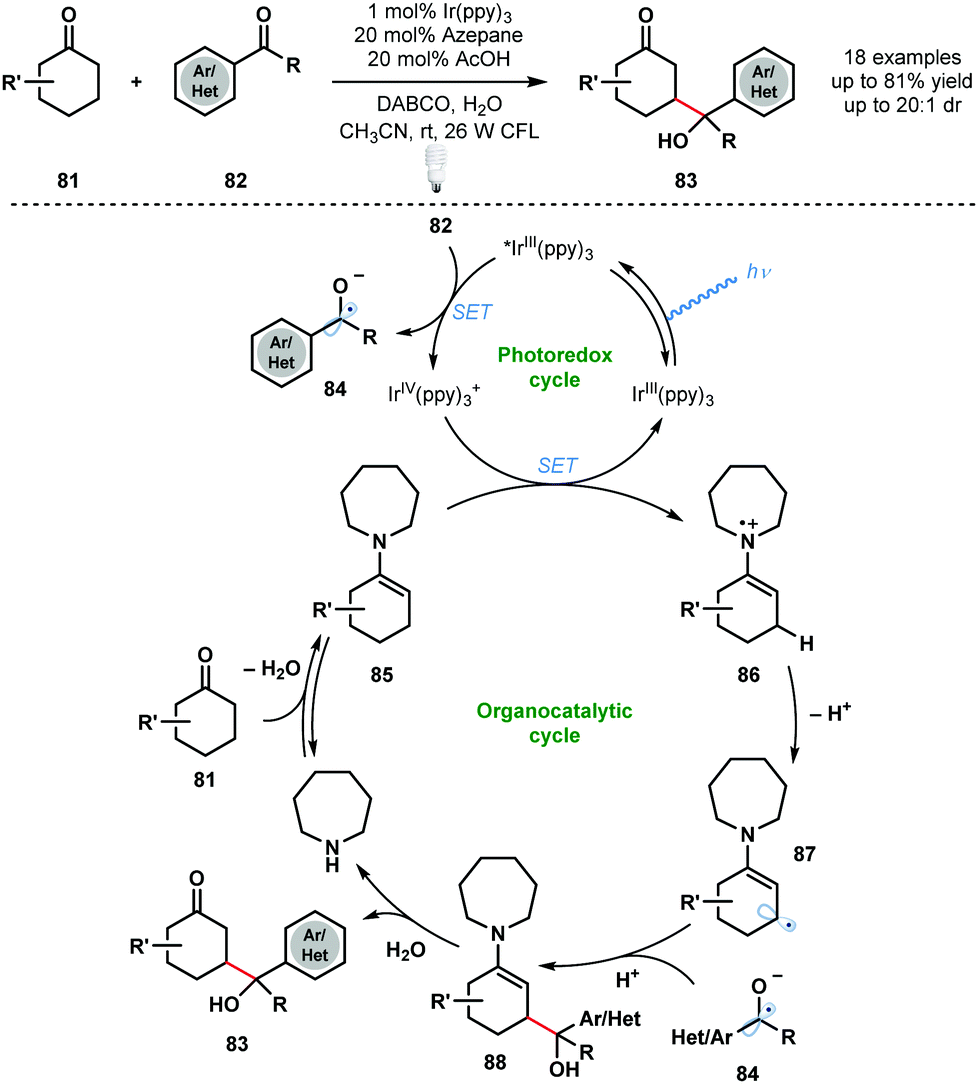 | ||
| Scheme 13 Dual photocatalytic and organocatalytic, direct β-functionalization of cyclic ketones (MacMillan, 2013). | ||
In 2014, Yoon and co-workers reported the intermolecular asymmetric [2+2] photocycloaddition of α,β-unsaturated ketones, 89 and 90, exploiting a dual catalyst system involving a photocatalyst and a chiral Lewis acid catalyst (Scheme 14).33 The major challenge involved in the development of an enantioselective [2+2] photocycloaddition reaction is the facile uncatalyzed, racemic background reaction mediated by light. By employing a photocatalyst, RuII(bpy)3Cl2, that adsorbs at wavelengths where the substrates are not activated, and by using a Lewis acid to activate the enones towards SET and stabilize the resulting radical species, the racemic background reaction was overcome. Thus, using a Lewis acid, such as Eu(OTf)3, bearing a chiral ligand, L2 or L3, promoted the enantioselective [2+2] reaction. The reaction mechanism starts with the photoexcitation of RuII(bpy)32+ to RuII(bpy)32+*. Reductive quenching of the excited state photocatalyst by i-Pr2NEt affords i-Pr2Net˙+ and RuI(bpy)3+. RuI(bpy)3+ then reduces the Lewis acid activated enone (92) by SET and produces delocalised ketyl radical anion (93). Subsequently, enone radical anion 93 undergoes enantioselective [2+2] cycloaddition with 90 to produce ketyl radical intermediate 94. Ketyl radical 94 is converted to products 91 or 91′ either by reduction of i-Pr2Net˙+ or by reduction of another molecule of 92 in a chain propagation step.28
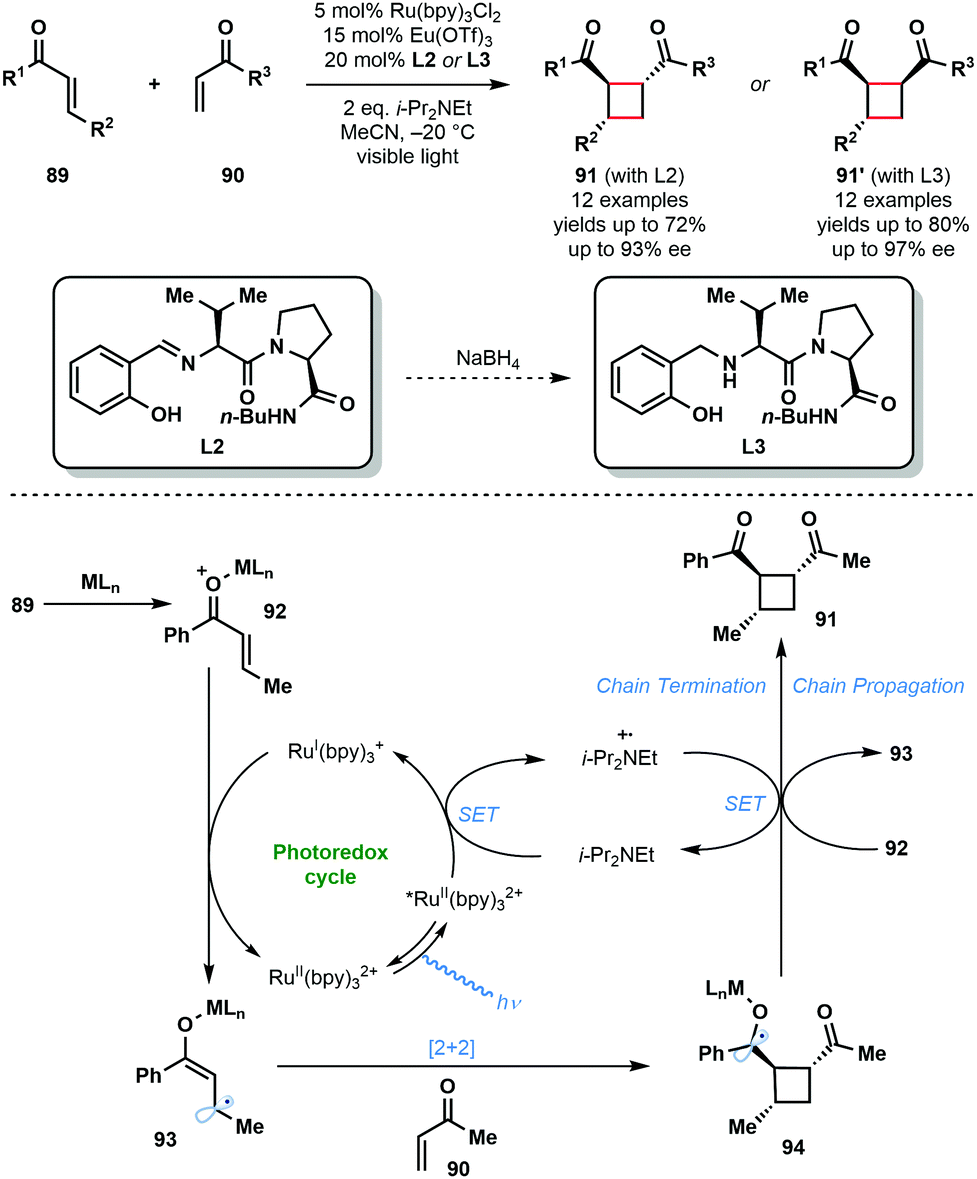 | ||
| Scheme 14 Enantioselective [2+2] photocycloaddition involving α,β-unsaturated ketones and using a transition–metal photocatalyst and Lewis acid co-catalyst. (Yoon, 2014). | ||
In 2015, Rueping and co-workers utilised photocatalytically generated ketyl radical anions, in conjunction with carbonyl activation by an amine radical cation, in the homo-coupling of aldehydes and ketones (Scheme 15).34 The mild method provides simple and convenient catalytic access to symmetrical pinacols 96 and diamines with good functional group tolerance. A variety of vicinal diols was synthesized in moderate to excellent yield using the approach. The proposed mechanism begins with quenching of the excited photocatalyst [IrIII]* by NBu3, the sacrificial reductant. Amine radical cation 97 is then thought to activate the weakly basic carbonyl groups of ketones 95 through formation of complexes 99 or 100. Ketyl radical anion 101 is formed after SET reduction by [IrII] and dimerization followed by protonation affords the vicinal diol products 96. The proposal of a dual role for NBu3 in the transformation – sacrificial electron donor and substrate activator – is noteworthy.
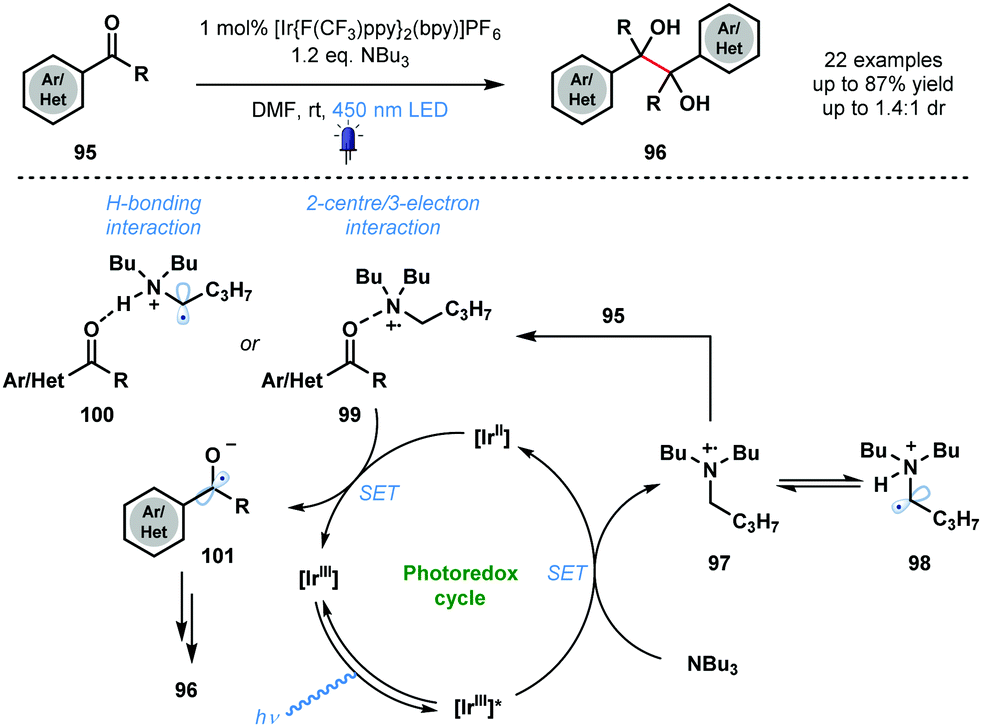 | ||
| Scheme 15 Photocatalytic homo-coupling of aldehydes and ketones (Rueping, 2015). | ||
In 2016, in a related process, Chen and co-workers reported a visible light-mediated ‘polarity-reversed’ allylation reaction of aldehydes and ketones that involves ketyl radicals (Scheme 16).35 These are the first visible-light induced, general allylation reactions of ketyl radicals. A Hantzsch ester is thought to act as both an electron/proton donor and as the activator of the carbonyl group in this transformation. After reductive quenching of the excited photocatalyst, HE˙+ activates the aldehyde/ketone partner 102 by a hydrogen bonding interaction prior to PCET and formation of the ketyl radical intermediates 106. Intermolecular radical addition of 106 to allyl sulfones 103, and radical elimination of the phenylsulfonyl group, yields adducts 104. A quantum yield (ϕ) of 0.8 was calculated for the reaction and a closed catalytic cycle was therefore proposed.
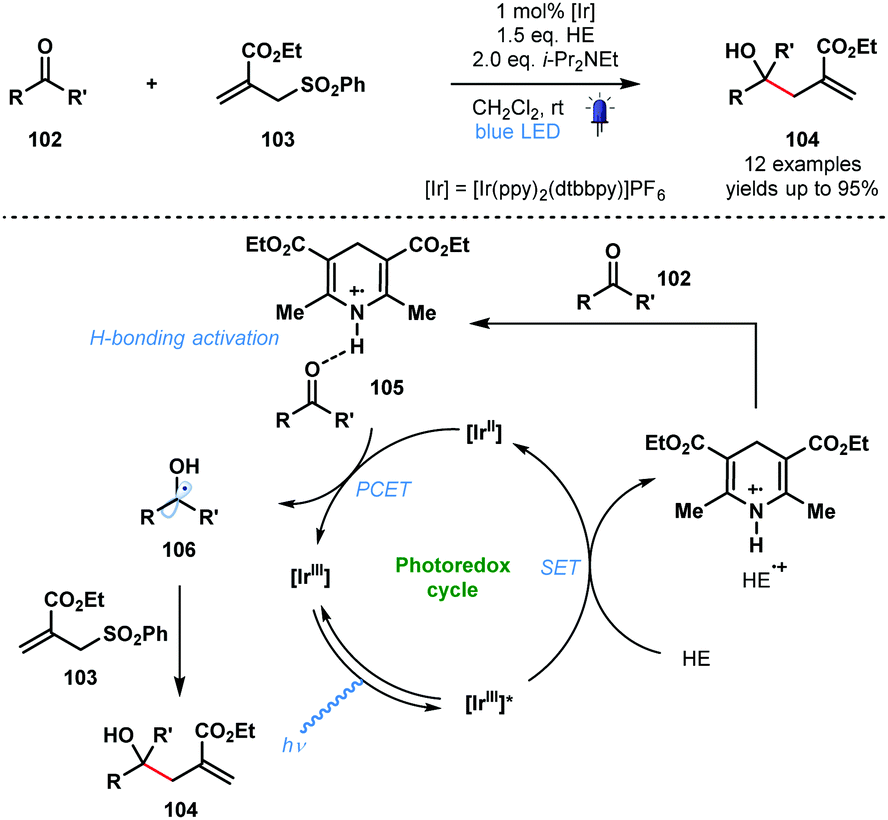 | ||
| Scheme 16 Photocatalytic allylation of aldehydes and ketones involving ketyl radicals (Chen, 2016). | ||
Building on the work of Knowles, Ngai and co-workers reported intermolecular photocatalytic ketyl–olefin couplings that employ a Lewis acid and a protic acid, to activate each of the coupling partners. The synergistic Lewis acid catalysis and photocatalysis allows the reductive coupling of alkenylpyridines with carbonyl and iminyl derivatives (Scheme 17).36 A wide range of complex substrates, including amino acid derivatives and natural products, could be converted to secondary alcohol adducts in moderate to good yield using RuII(bpy)3(PF6)2 as the photocatalyst and La(OTf)3 as the Lewis acid co-catalyst. Hantzsch ester (HE) was utilised as a sacrificial electron and hydrogen atom donor. While a useful way to couple aryl aldehydes and alkenes, the method requires alkenyl pyridine partners with a Lewis basic site for activation. The proposed mechanism begins with visible light irradiation of the ground-state photocatalyst, RuII(bpy)32+ to afford RuII(bpy)32+*. The excited photocatalyst was reductively quenched by the Hantzsch ester radical (HE˙) to form RuI(bpy)3+ and the corresponding pyridinium salt (pyH+). Aldehyde substrates 107, activated as their hydrogen bond adducts 110 by the in situ generated pyH+, undergo a PCET process involving RuI(bpy)3+ to give ketyl radicals 111. Subsequent regioselective addition to Lewis acid-activated vinyl pyridines 112 forms stabilised radicals 113. Hydrogen atom abstraction (HAT) from Hantzsch ester (HE) affords 114 and HE˙. Finally, the Lewis acid co-catalyst releases products 109 and activates a new molecule of substrate.
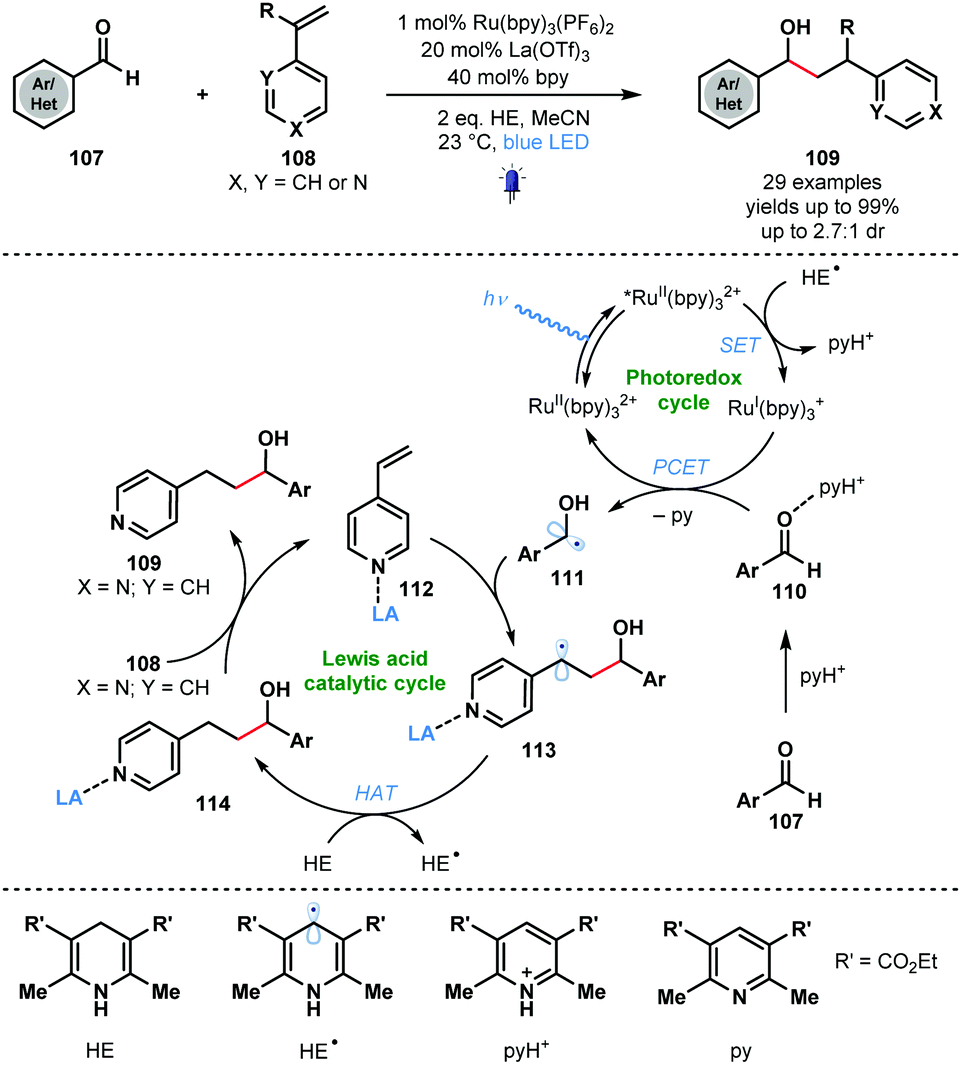 | ||
| Scheme 17 Synergistic Lewis acid/photoredox catalysis in intermolecular ketyl–olefin couplings involving aldehydes and alkenylpyridines (Ngai, 2017). | ||
Other Lewis acids can be used to activate carbonyls and aid the formation of ketyl radicals. In 2018, Huang and co-workers reported a convergent asymmetric synthesis of vicinal amino alcohols that exploits a dual Lewis acid and photocatalytic approach (Scheme 18).37 Analogous couplings of aldehydes/ketones and nitrones have been achieved using stoichiometric SmI2, however, these exploit chiral auxiliaries. A range of functionality was tolerated in the aldehyde partner and nitrone partners bearing linear, branched and cyclic alkyl motifs were successfully employed. Again, the photocatalytic cycle begins with the formation RuII(bpy)32+* by photoexcitation of RuII(bpy)32+ followed by reductive quenching of the excited state of the photocatalyst by the sacrificial reductant, N,N,N′,N′-tetraethylethylenediamine (TEEDA), producing the corresponding N-centred radical cation. Simultaneously, nitrone 115 and aldehyde 116 are complexed by a scandium Lewis acid derived from enantiopure ligand L4. Reduction of the activated aldehyde in 118 by RuI(bpy)3+ delivers the complexed ketyl radical intermediate 119 and RuII(bpy)32+. This intermediate undergoes enantioselective radical addition to the bound nitrone to form the N-centred radical cationic intermediate 120. Subsequent HAT from TEEDA˙+ affords 121 which upon protonation delivers the enantioenriched vicinal hydroxyamino alcohol product 117. The catalytic cycle is closed by the chiral Lewis acid engaging another molecule of both 115 and 116.
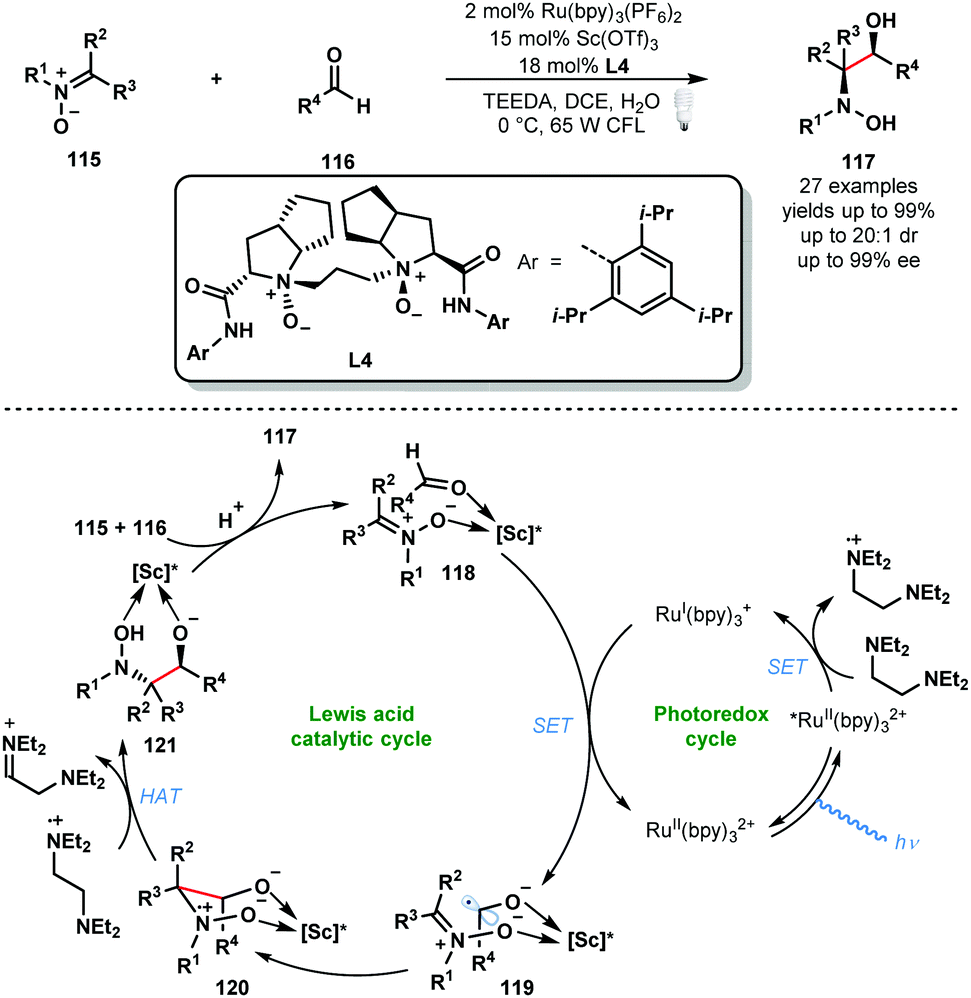 | ||
| Scheme 18 Enantioselective photocatalytic cross-coupling of nitrones and aldehydes (Huang, 2018). | ||
In the same year, Wang and co-workers developed a complementary approach using RuII(bpy)32+ to access vicinal amino alcohols and diamines that involves the photocatalytic radical cross-coupling of aldehydes, ketones, and imines with N-arylamines (Scheme 19).38 The process exhibits good functional group tolerance, with ester, nitrile, halide, and heteroaryl substituents being compatible, however, low diastereocontrol was observed. As seen before, the carbonyl substrates must first be activated by the in situ generated conjugate acid of DABCO, to give complex 125. PCET then delivers the DABCO-bound ketyl radical complex 126. The photocatalytic cycle is closed by SET oxidation of the N-arylamine substrates 123 and regeneration of the ground state catalyst. Proton transfer from 127 to complex 126 yields the carbon-centred radicals 128 and 129, as well as protonated DABCO, which undergo radical–radical coupling to afford the desired 1,2-amino alcohol products 124. Based on ‘light on/light off’ experiments, the authors excluded a radical chain mechanism, however, such experiments are not conclusive.28
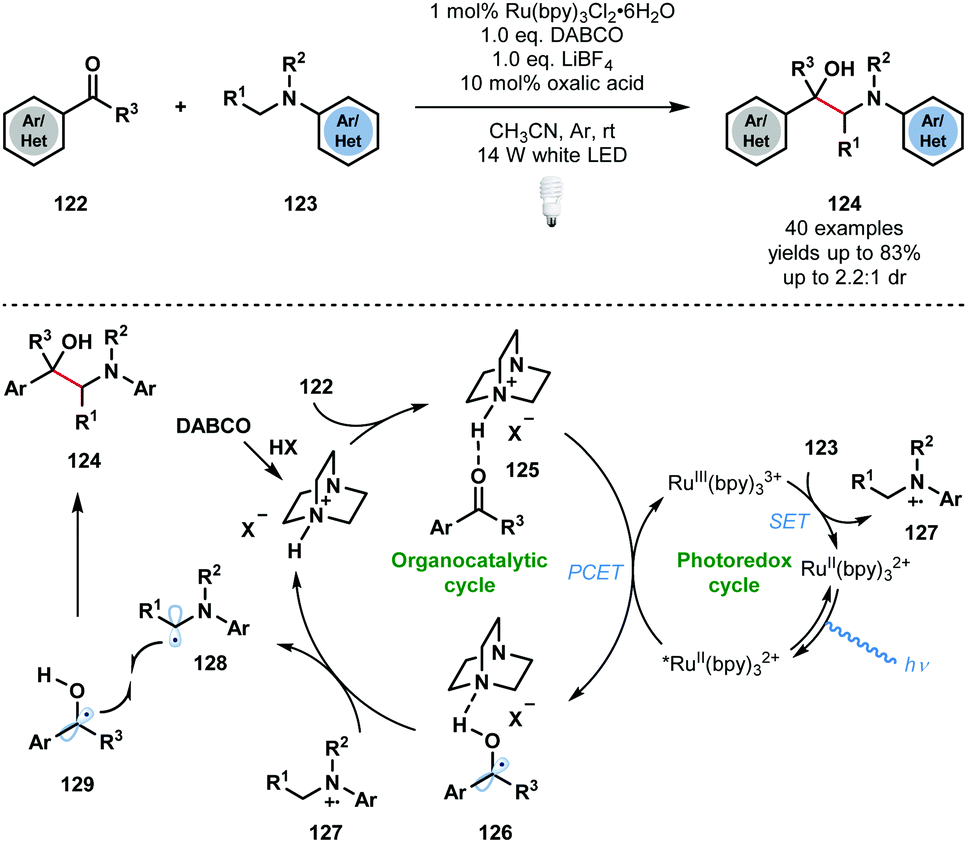 | ||
| Scheme 19 Photocatalytic cross-coupling of N-aryl amines with aldehydes and ketones, enabled by PCET (Wang, 2018). | ||
In 2018, Nagib and co-workers reported a redox-neutral strategy that delivers α-acetoxy vinyl iodides and involves ketyl radical derivatives (Scheme 20).39 The method is simple, uses commercial coupling partners, and yields synthetically useful vinyl iodides with high Z-selectivity. Interestingly, even formaldehyde can be used to generate the corresponding α-acetoxy vinyl iodide after coupling with TMS-acetylene. While steric factors prevented this method being extended to simple ketones, trifluoroacetone was used successfully. Furthermore, electron-rich alkenes, in place of alkynes, also underwent successful coupling. Most photocatalysed methods that deliver ketyl radicals use a sacrificial terminal reductant, such as an amine. In this redox-neutral approach, the carbonyl reduction step is replaced by a halogen atom abstraction step that forms ketyl radical derivatives. To close the catalytic cycle, the halogen atom is returned and product is formed. Acetyl iodide, in the presence of a zinc Lewis acid catalyst, activates aldehydes 130 as adducts 133, and visible light-mediated homolytic cleavage of Mn2(CO)10 generates a 17-electron complex, Mn(CO)5. This species readily accepts I˙ from the weak C–I bond of 133. The resulting ketyl radical derivatives 134 add to the terminal alkyne partners 131 to form vinyl radical intermediates 135. These intermediates are quenched by MnI(CO)5 to regenerate the active Mn(CO)5. Importantly, this atom transfer radical addition (ATRA) mechanism precludes the formation of reduced product 136. Finally, a Mn-catalysed isomerization via vinyl radical intermediates ensures that high Z-selectivity is seen in the formation of products 132. An alternative, radical chain mechanism was deemed unlikely based on chain initiation and cross-over experiments.
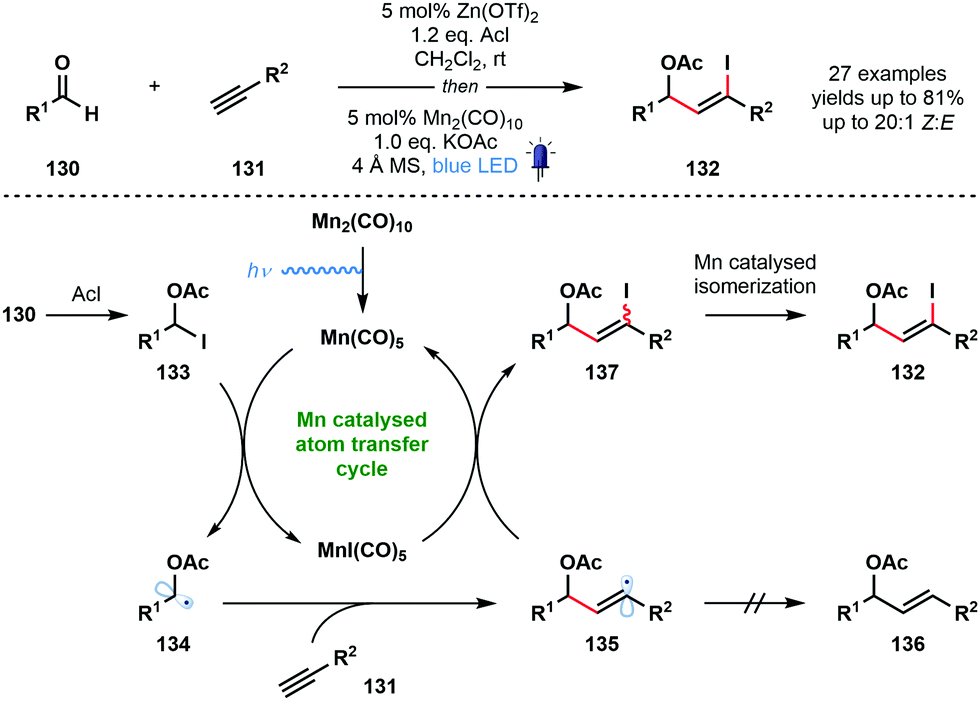 | ||
| Scheme 20 A redox neutral pathway for the generation of ketyl radical derivatives and their coupling with alkynes (Nagib, 2018). | ||
Also in 2018, Meggers and co-workers employed a chiral-at-metal photocatalyst (Δ-RhS) in a photocatalytic enantioselective [3+2] photocycloaddition involving cyclopropyl ketones and alkenes or alkynes (Scheme 21).40 Enantioenriched cyclopentane- and cyclopentene-derivatives were synthesized in excellent yield with excellent levels of enantio- and diastereocontrol. Various unsaturated partners were successful ranging from simple acrylates to highly functionalized drug derivatives. Two-point coordination of the cyclopropyl ketone substrates 138 affords complexes 143 which are excited by visible light to give 144. This strongly oxidising species undergoes SET reduction by DIPEA to give the ketyl radical intermediates 145 which subsequently fragment to form Rh-bound enolate radicals 146. Intermolecular radical addition to the alkene or alkyne partners, 139 or 140, respectively, is followed by an enantiodetermining radical cyclisation event – controlled by the helical chirality of the Rh complex – that yields highly-reducing ketyl radical complexes 147. Back electron donation to DIPEA˙+ and ligand exchange regenerates complexes 143 and liberates products 141 or 142. Alternatively, quantum yield measurements supported a radical chain process in which ketyl radicals 147 can directly reduce excited complexes 144.
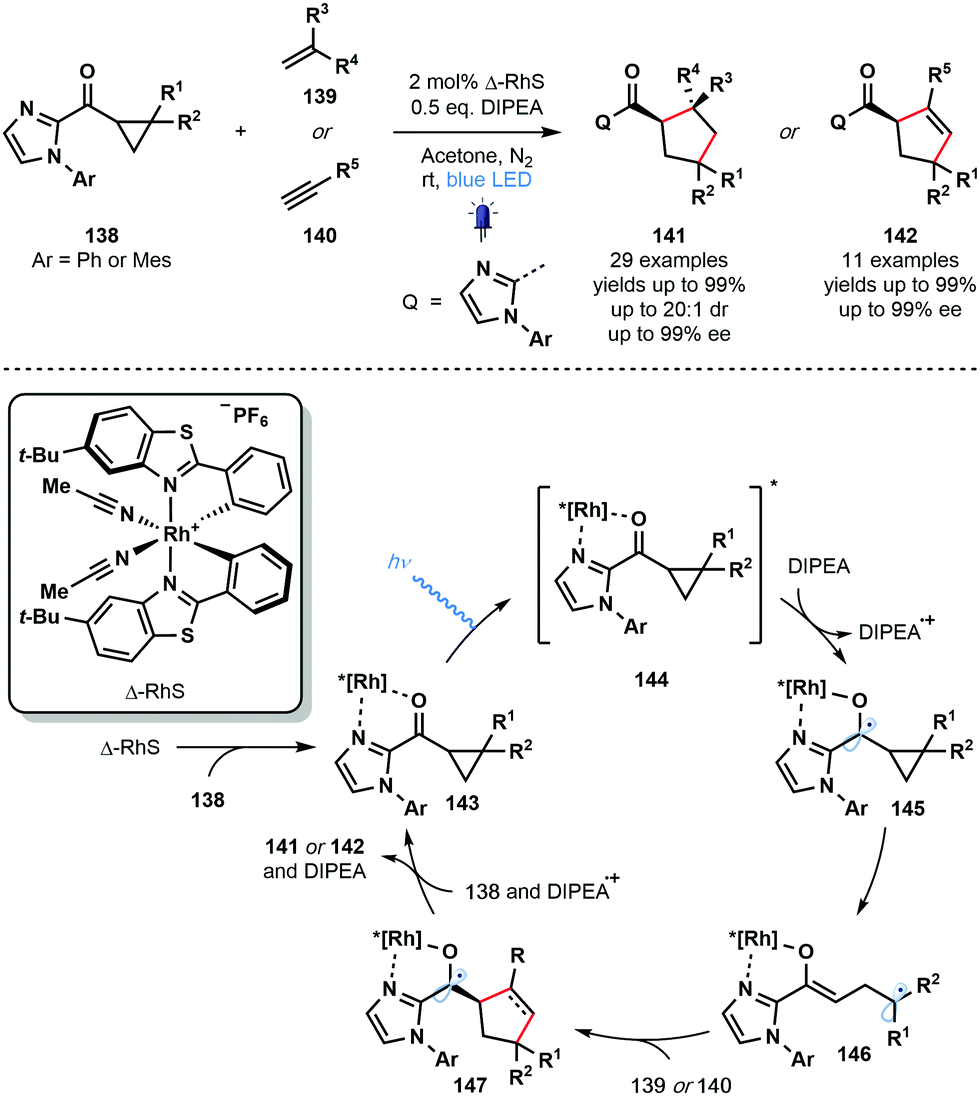 | ||
| Scheme 21 Enantioselective, Rh-catalysed, visible light-mediated [3+2] cycloaddition of cyclopropyl ketones and alkenes or alkynes (Meggers, 2018). | ||
Organic photocatalysts can also be used to generate ketyl radicals from activated ketones. In 2018, the Jiang group reported a redox-neutral approach to valuable, enantioenriched 1,2-amino alcohols 150 from N-aryl glycines 148 and vicinal diketones 149 using photocatalyst DPZ in conjunction with enantiopure CPA2 which serves as a dual H-bond donor and acceptor catalyst (Scheme 22).41 This radical–radical coupling shows good functional group tolerance and can be extended to isatin partners. The high yields and high enantiocontrol observed renders this a practical enantioselective approach to vicinal aminoalcohols.
 | ||
| Scheme 22 Enantioselective, photocatalytic radical-coupling of activated ketones and N-aryl glycines (Jiang, 2018). | ||
Glycine derivatives 148 are readily oxidised by the excited dicyanopyrazine-derived photocatalyst DPZ to afford α-amino radicals 152 after deprotonation and decarboxylation. Activated ketones 149 are then reduced to close the photocatalytic cycle yielding ketyl radical intermediates 153. Double H-bonding interactions are thought to facilitate capture of 152 and 153 by CPA2 and enantioselective radical–radical coupling gives the desired products 150 in good yield and with excellent enantiocontrol.
Violet light has also been used to generate ketyl radicals from aldehydes and ketones. In 2018, König reported a photocatalytic synthesis of homoallylic and homobenzylic alcohols from readily available allyl or benzyl bromides and aromatic aldehydes or ketones that is reminiscent of a Barbier-type coupling (Scheme 23).42 Yields were moderate in many cases, possibly due to competing pinacol formation; products of homocoupling were observed in some cases. However, this process does allow the use of stoichiometric metal reagents, typically associated with Barbier-type processes, to be avoided. The reductive radical–radical cross-coupling reaction uses phenoxazine-derived organic photocatalyst PC and DIPEA as a sacrificial electron donor and proton source. Mechanistic investigations suggested that the reduction potential of carbonyl partners 154 were significantly reduced by simultaneous activation by DIPEA and LiBF4. The excited photocatalyst PC* reduces carbonyl species 154 to deliver the ketyl radical species 157. DIPEA closes the photoredox cycle and is thought to act as a SET reductant during the formation of stabilised radicals 158 from, for example, allyl bromides. Radical–radical coupling between 157 and 158 delivers the alcohol products 156 in good yield.
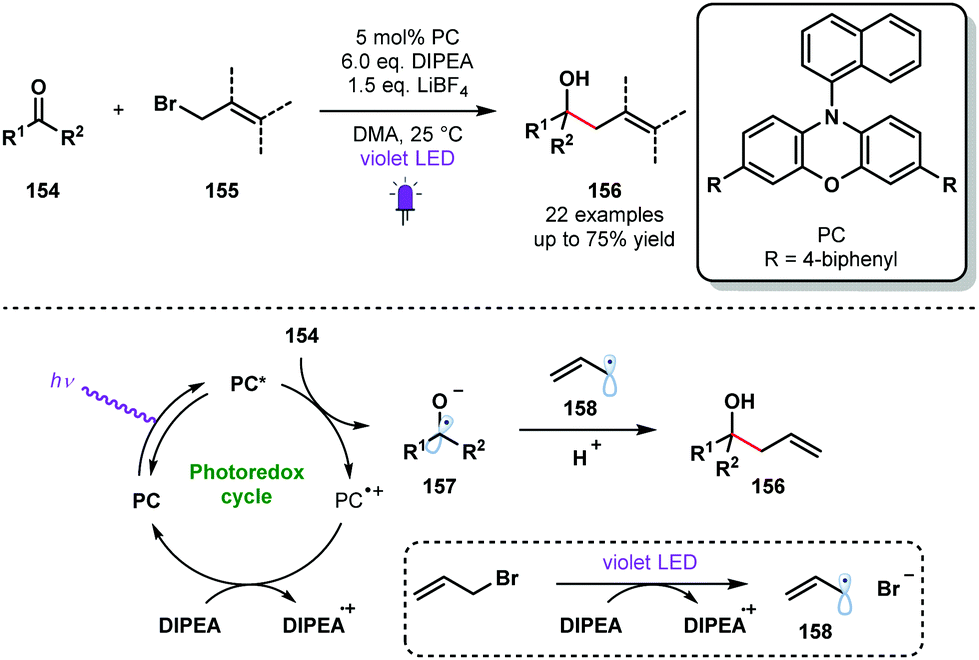 | ||
| Scheme 23 Violet light-mediated allylation and benzylation of aldehydes and ketones (König, 2018). | ||
In 2014, Studer and Curran introduced the term ‘electron catalysis’ to describe SET chain and relay processes.43 Four years later, Studer and co-workers exploited such a process in an efficient method for the α-perfluoroalkylation/β-alkenylation of unactivated alkenes that is driven by the formation of ketyl radical anions (Scheme 24).44
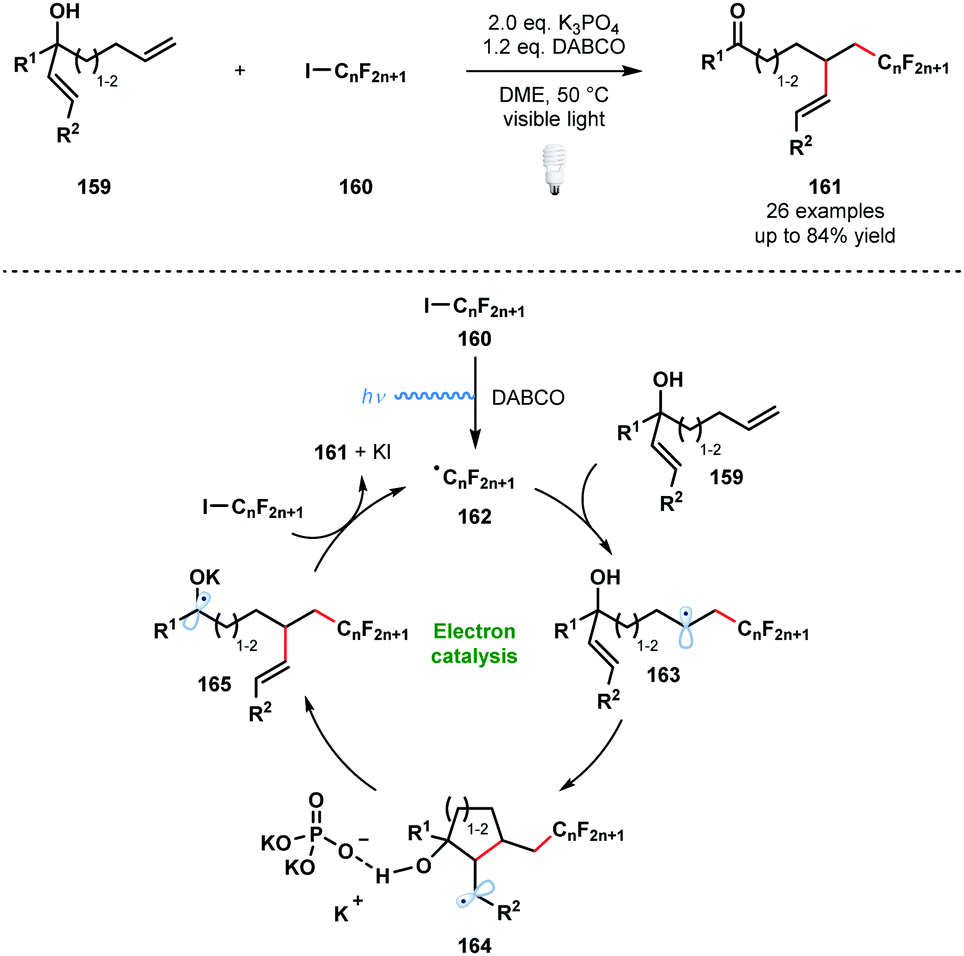 | ||
| Scheme 24 Visible light-initiated 1,2-functionalisation of alkenes by radical perfluoroalkylation/alkenyl migration (Studer, 2018). | ||
Visible light irradiation of the halogen-bond complex between DABCO and 160 affords the perfluoroalkyl radicals 162 and initiates the process. Radicals 162 add to the more accessible terminal alkene to give 163. Radical 5-exo-trig or 6-exo-trig cyclisation leads to intermediates 164 bound to an orthophosphate anion by hydrogen bonding. This interaction is thought to be key in activating the β-C–C bond towards homolytic cleavage. Concurrent deprotonation and β-C–C bond cleavage affords ketyl radical 165 which reduces 160 to sustain the chain process and yield the 1,2-functionalised alkenes 161. An analogous α-perfluoroalkyl-β-alkynylation of alkenes was reported earlier by the same team.45
In 2020, Studer and co-workers applied their approach to the visible light-initiated allylation of radicals using homoallylic alcohols (Scheme 25).46 In this variant, perfluoroalkyl radicals and simple alkyl radicals – formed from pyridinium salts 168 – were generated and engaged in radical additions to homoallylic alcohols 166 to yield allylated products 169 after base-promoted homolytic cleavage of the Cα–Cβ bond. This SET chain process is sustained by the reduction of 167 or 168 by the resultant ketyl radicals 175 or 176.
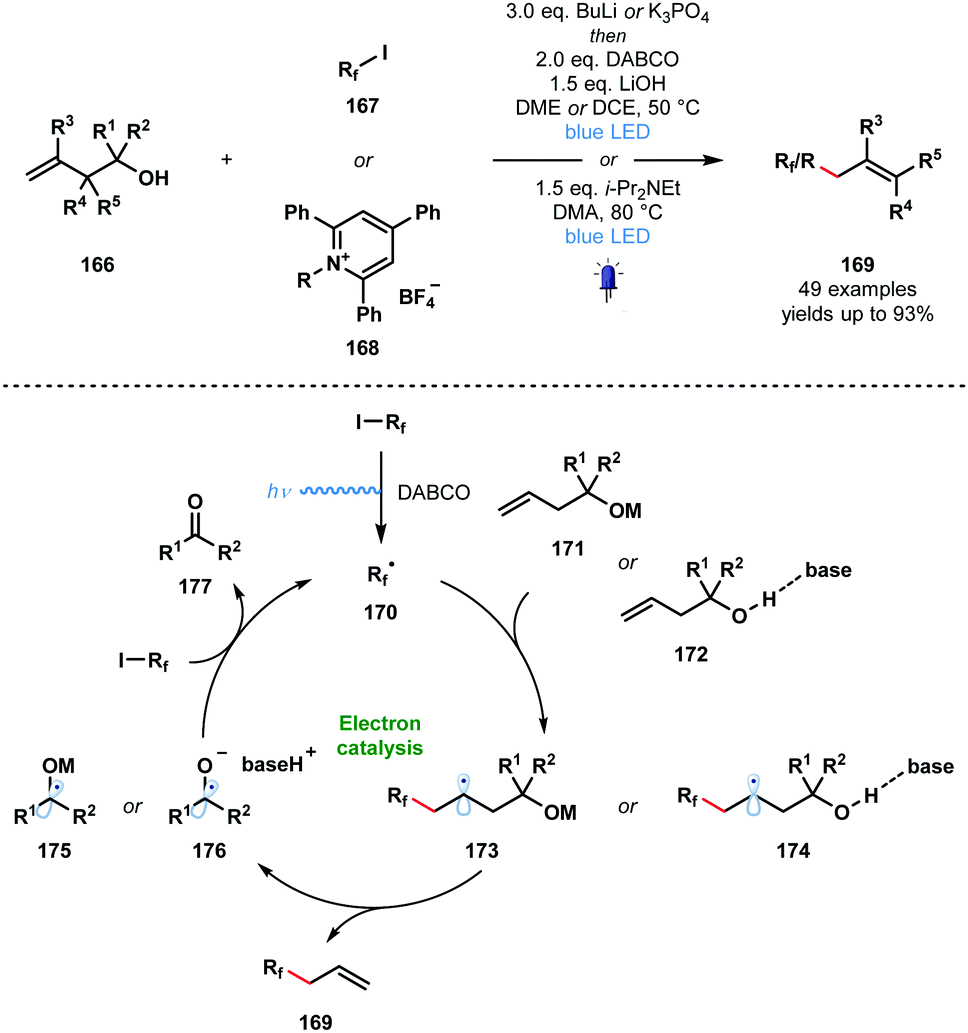 | ||
| Scheme 25 Visible light-initiated radical allylation using homoallylic alcohols and base (Studer, 2020). | ||
In 2020, the Huang group described a photocatalytic Minisci-type reaction that exploits simple aldehydes as alkylating agents (Scheme 26).47 In particular, under the mild reaction conditions, products of the benzylation of heteroarenes can be obtained; an elusive process in Minisci chemistry. The reaction tolerates a wide range of functionalities, however, the two simple pyridine substrates tested – 2-Ph-pyridine and 2-Bn-pyridine – gave low yields of the corresponding benzylated products. The excited photocatalyst [IrIII]* is thought to be reductively quenched by bromide to afford Br˙ which abstracts a hydrogen atom from Et3SiH to form silyl-centred radical Et3Si˙. Coupling of this silyl radical to aldehydes 179 afforded ketyl radical derivatives 181 that undergo subsequent radical addition to protonated pyridine derivatives 178 to yield radical cations 182. Deprotonation and loss of Et3SiOH delivers carbon-centred radicals 184. Finally, SET from [IrII] followed by protonation liberates the alkylated products 180.
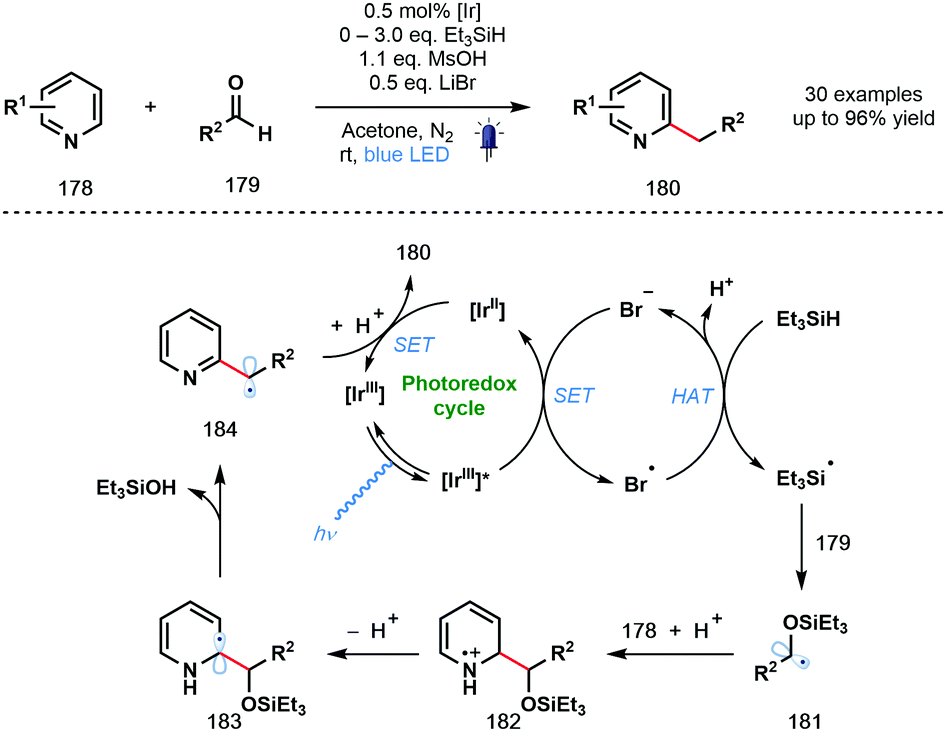 | ||
| Scheme 26 Photocatalytic Minisci-type reactions using ketyl-radical derivatives generated from aldehydes (Huang, 2020). | ||
5. Electrochemically generated ketyl radicals
Baran et al. have recently developed a user-friendly, electrochemical, intermolecular ketyl–olefin coupling using an undivided cell setup (Scheme 27).48 Previous electrochemical methods reported by Shono and co-workers mainly focused on intramolecular processes.48 Furthermore, past reports were limited by the challenges of working with a divided cell setup under an inert atmosphere and the requirement for a significant excess of the ketone partner. The new protocol allows for the use of simple alkenes 186 and unactivated ketones 185, and thus complements known protocols using SmI2, and other single-electron reducing agents, that require activated alkene partners in an intermolecular setting. Under the optimised reaction conditions, an inexpensive sacrificial anode (Zn) was used in combination with only two equivalents of the ketone partner. Conveniently, the reaction uses an undivided cell, can be run under air, no precautions to exlude moisture are necessary, and the process has been shown to tolerate a wide range of functional groups. While ketones bearing α-heteroatom substituents proved to be ineffective partners, their fragmentation provided useful support for the intermediacy of a ketyl radical. Importantly, the strong adsorption of 188 to the Sn cathode changes its electronic properties and reactivity and thus facilitates its addition to unactivated alkene coupling partners 186. | ||
| Scheme 27 Electrochemical ketyl–olefin coupling using unactivated ketones and unactivated alkenes (Baran, 2020). | ||
6. Ketyl radicals in natural product synthesis
The chemistry of ketyl radicals, accessed in various ways, continues to play a key role in total synthesis. The reagent's versatility means that SmI2 is often the reagent of choice in many studies.49 For example, Kwon's 2016 synthesis of (−)-actinophyllic acid (195) compares the suitability of various SET reductants for the pivotal step in the synthesis; a ketone–ester pinacol coupling (Scheme 28).50 Several reagents, including those based on Ti(III), Li, and Na, failed to convert 193 to the desired product 194. However, SmI2, in conjunction with 10 eq. of t-BuOH, delivered the caged scaffold of 195 in nearly quantitative yield. Interestingly, the amount of proton source proved crucial; the use of >10 eq. of t-BuOH reduced the reaction rate and <0 eq. of t-BuOH favoured the formation of rearranged product 197, presumably via intermediate 196 which then underwent a retro-aldol/aldol cascade.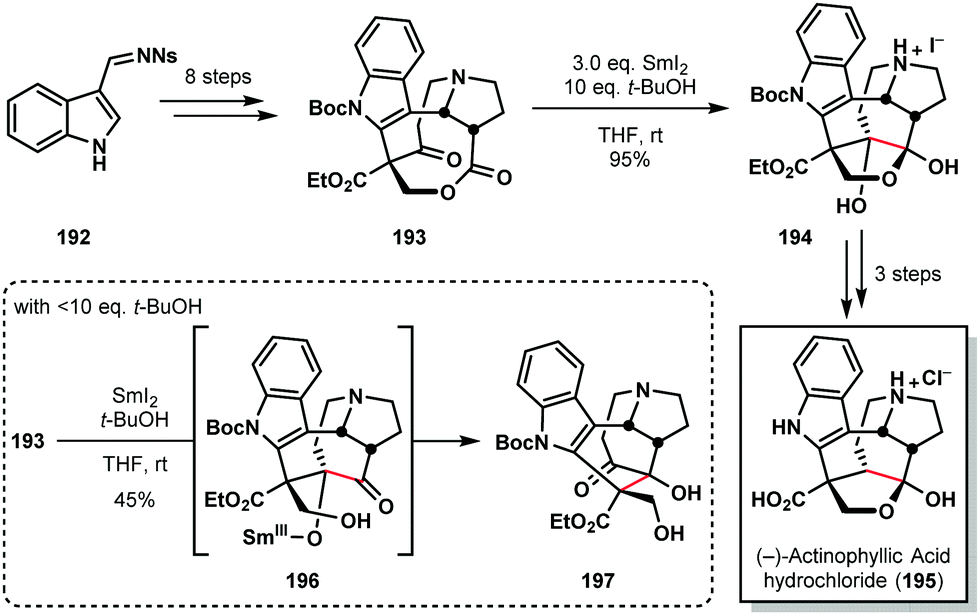 | ||
| Scheme 28 A ketyl radical in the pivotal ketone–ester pinacol coupling en route to (−)-actinophyllic acid (Kwon, 2016). | ||
The 2016 enantioselective total synthesis of (+)-steenkrotin (205) A by Ding et al. provides a striking illustration of the differences in behaviour of tin and samarium ketyl radicals (Scheme 29).51 The synthetic plan relied on a ketyl–olefin cyclisation of aldehyde 198 to build the tetracyclic core of the natural product 199. Rather than delivering 199, treatment of 198 with n-Bu3SnH in refluxing benzene gave undesired, rearrangement product 200 in 63% yield. Formation of 200 involves conversion of the aldehyde to the tin ketyl radical followed by intramolecular addition to the enone in an undesired 5-exo-trig fashion to form radical 201. A Beckwith–Dowd ring expansion, via cyclopropane 202, gives radical 203 which is quenched by n-Bu3SnH. The desired 6-endo-trig cyclization was achieved in a highly stereoselective manner by treatment of aldehyde 198 with SmI2 at room temperature. The oxophilic SmIII ion of the ketyl radical coordinates to the enone moiety, controlling the regioselectivity of the cyclization by lowering the energy of the π* orbital of the olefin (204).
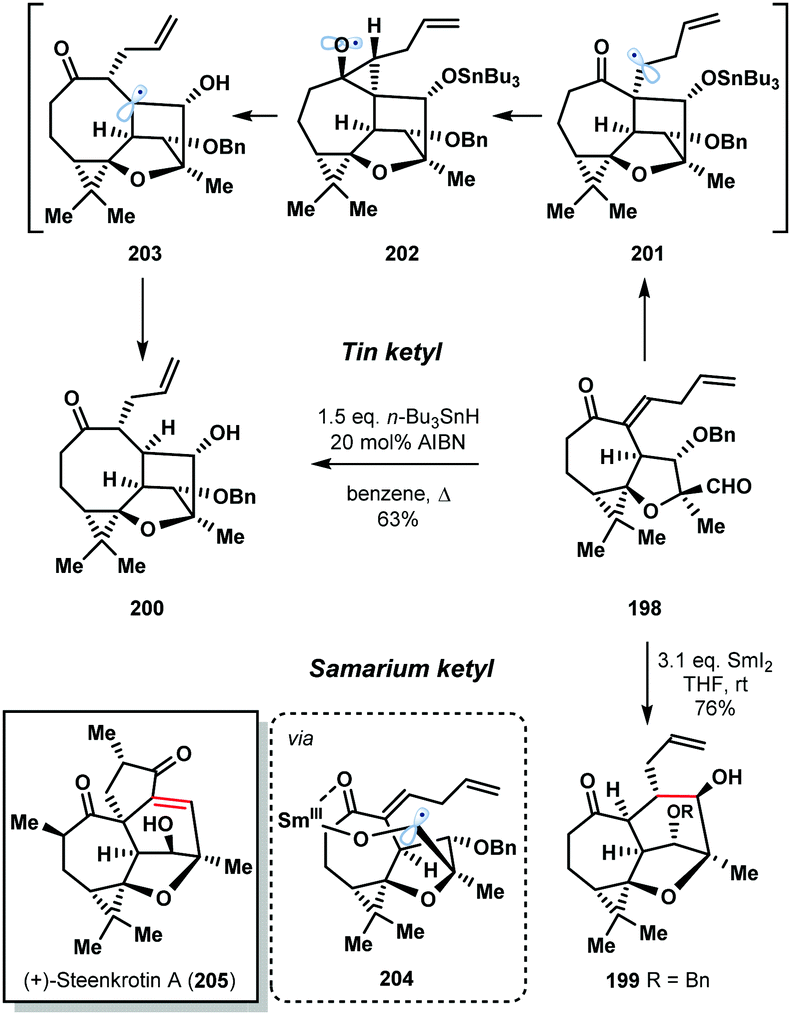 | ||
| Scheme 29 Contrasting the reactivity of tin and samarium ketyl radicals in the total synthesis of (+)-steenkrotin A (Ding, 2016). | ||
In the context of total synthesis, the most impressive applications of SmI2 for ketyl radical generation result in cascade processes that assemble complex architectures with exquisite control.49
For example, Procter's total synthesis of (+)-pleuromutilin (208) features a SmI2-mediated radical cascade cyclisation that forges the five- and eight-membered rings of the target and sets four contiguous stereocentres with high diastereocontrol (Scheme 30).52 The selective conversion of dialdehyde 206 to tricycle 207, using SmI2 in the presence of t-BuOH, proceeded in 88% yield on gram scale. This cascade reaction again highlights the high control often seen in SmI2-mediated ketyl radical reactions that arises from coordination of samarium to Lewis basic sites in the substrate; chemoselective formation of ketyl radical 209, a chelation-controlled anti-5-exo-trig cyclization (delivering 210), and a diastereoselective aldol cyclization of Sm(III)-enolate 211 furnishes 207.
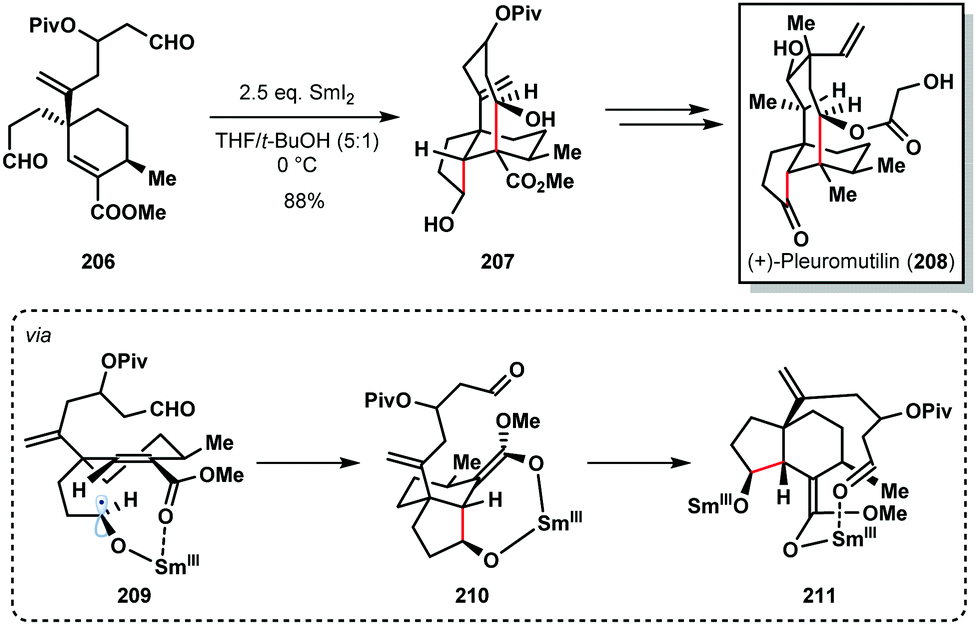 | ||
| Scheme 30 SmI2-mediated ketyl radical formation in a cyclization cascade en route to (+)-pleuromutilin (Procter, 2013). | ||
In 2018, Reisman and co-workers also achieved the synthesis of (+)-pleuromutilin (208) by using the SmI2–H2O reagent system to construct the eight-membered ring of the natural product with high diastereocontrol (Scheme 31).53 Interestingly, treatment of aldehyde 212 with SmI2, followed by work-up under air, gave undesired carboxylic acid 214 in 41% yield; exposure of Sm(III)-enolate intermediate 215 to oxygen results in the formation of an α-peroxyketone that undergoes oxidative ring scission. However, under rigorously anaerobic conditions, trapping of enolate 215 with TMSCl, followed by aqueous work up, provided the desired tricycle 213 in 93% yield with near perfect diastereocontrol. In the absence of water, the ketyl–olefin cyclization resulted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers at C14.
:1 mixture of diastereomers at C14.
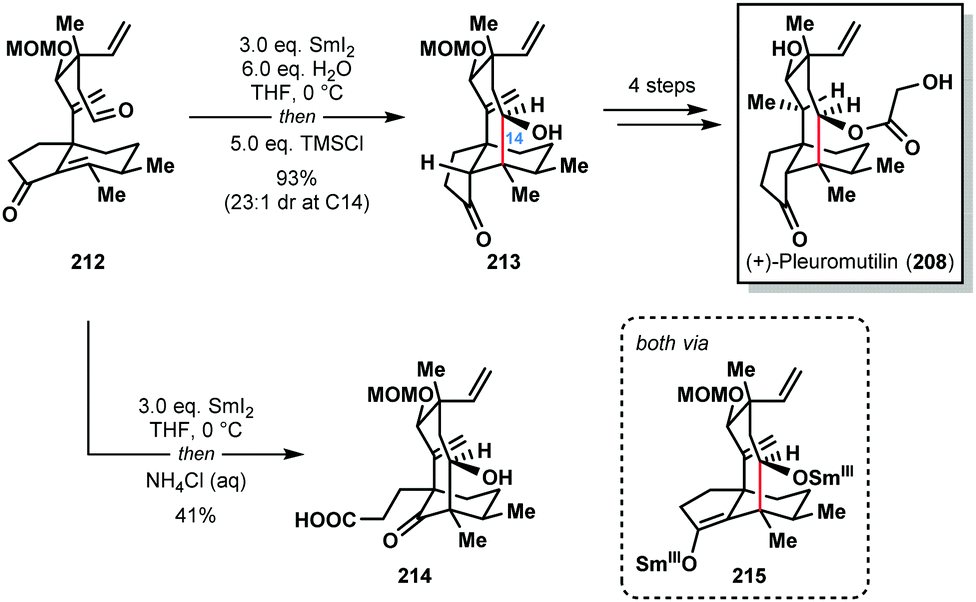 | ||
| Scheme 31 A SmI2-mediated ketyl–olefin cyclization in the total synthesis of (+)-pleuromutilin (Reisman, 2018). | ||
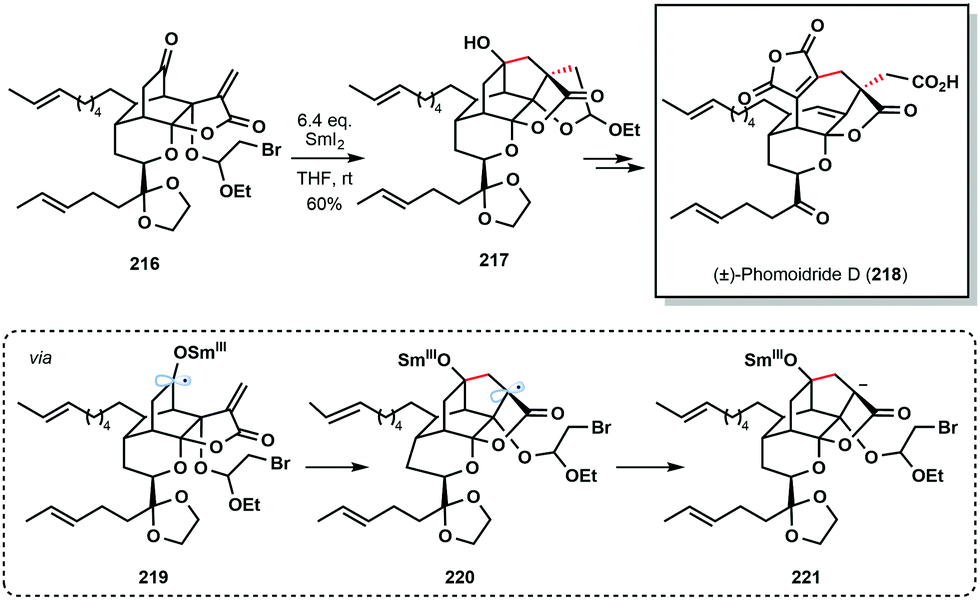
Another prime example of a cascade reaction employing a ketyl radical generated by SmI2 comes from the 2018 total synthesis of (±)-phomoidride D (218) by Wood and co-workers (Scheme 32).54 Treatment of ketone 216 with SmI2 triggered a 5-endo-trig/5-exo-tet cyclization sequence; 5-endo-trig cyclization of ketyl radical 219, followed by reduction, and an unusual intramolecular alkylation of an organosamarium (221), delivered advanced intermediate 217.
 | ||
| Scheme 32 SmI2-mediated ketyl radical formation in a cyclization cascade approach to (±)-phomoidride D (Wood, 2018). | ||
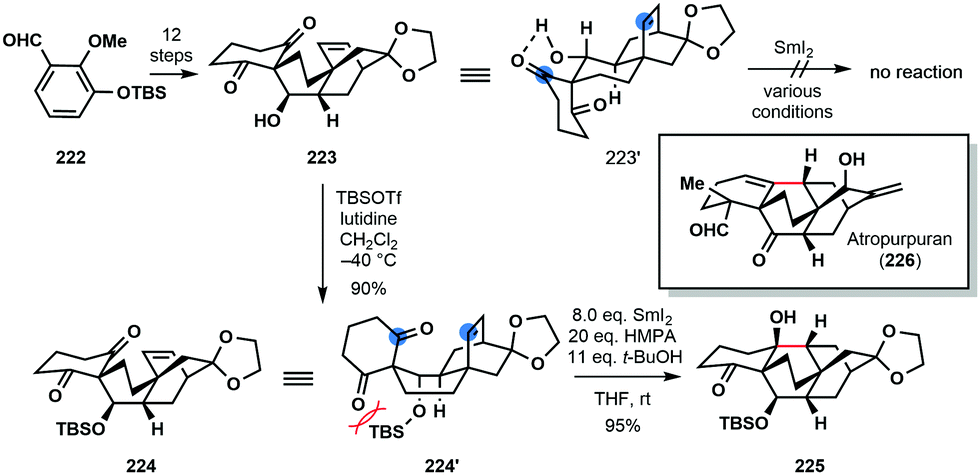
In the 2016 total synthesis of (±)-atropurpuran (226) by Qin and co-workers, an understanding of conformation proved key in the development of a SmI2-mediated ketyl–olefin cyclization (Scheme 33).55 Upon exposure to SmI2 under a variety of conditions, the proposed radical cyclization of secondary alcohol 223 proved unsuccessful and only starting material was returned. It was postulated that 223 adopts chair conformation 223′, featuring an intramolecular hydrogen bond between the hydroxyl and a ketone carbonyl; thus, the ketyl radical, formed upon ketone reduction, and the alkene partner are not sufficiently close for coupling. Protection of 223 as a bulky TBS ether breaks up the hydrogen bond interaction and switches the conformation of 224 to that of a boat (224′). Treatment of TBS-protected alcohol 224 with SmI2 in the presence of HMPA resulted in smooth cyclization to deliver the core of the natural product 225 in 95% yield.
 | ||
| Scheme 33 Control of conformation in a SmI2-mediated ketyl–olefin cyclization en route to (±)-atropurpuran (Qin, 2016). | ||
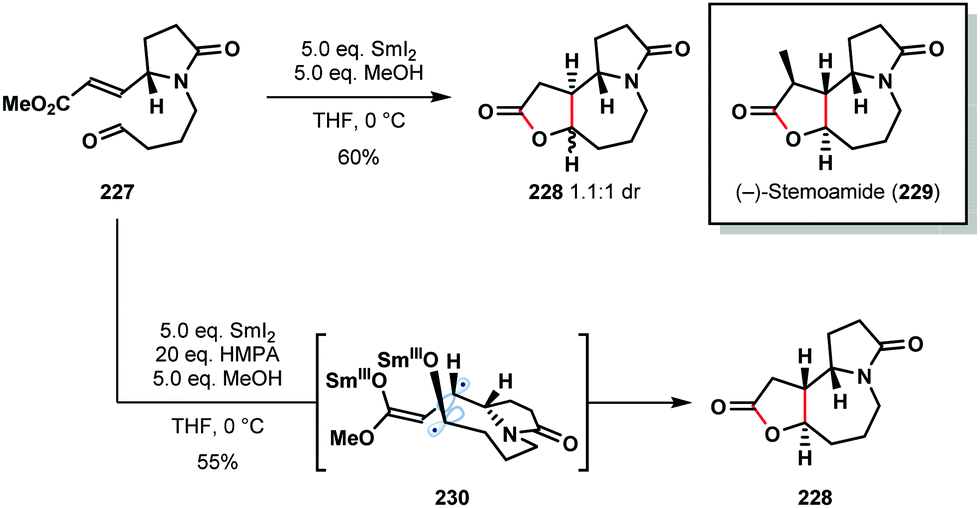
It is well-known that additives, such as HMPA and proton sources, play a crucial role in SmI2-mediated reactions. For example, HMPA can not only promote couplings but can also alter the stereoselectivity of C–C bond formation. Honda's 2011 total synthesis of (−)-stemoamide (229) provides an illustration of the dramatic impact HMPA can have on SmI2-mediated ketyl–olefin couplings (Scheme 34).56 In the absence of HMPA, aldehyde 227 underwent cyclization to give 228 as a 1.1:1 diastereoisomeric mixture in 60% yield. By employing HMPA in conjunction with SmI2, 228 was obtained as a single diastereomer in 55% yield. Interestingly, the selective coupling of diradical intermediate 230 was proposed; HMPA is known to significantly increase the reduction potential of SmI2 and may allow access to diradical 230. It is also likely that HMPA breaks up chelated transition states that lead to the alternative diastereoisomer. Finally, the coupling of Z-227 gave a similar reaction outcome; this supports an ‘alkene first’ mechanism in which the alkene geometry is lost prior to coupling and thus has little effect on the product distribution.
 | ||
| Scheme 34 The effect of HMPA on a SmI2-mediated ‘ketyl–olefin’ cyclization in the total synthesis of (−)-stemoamide (Honda, 2011). | ||
7. Conclusions
Ketyl radicals are highly versatile reactive intermediates, well-known for their synthetic utility in C–C bond forming reactions and their ability to forge complex molecular architectures. Ketyl radicals are still commonly generated using stoichiometric amounts of SET reducing agents – such as K, Zn, Ti and, most importantly, SmI2 – and these processes continue to underpin the pivotal steps in many high profile total syntheses. However, the advent of photoredox catalysis and the growth of transition–metal catalyzed radical processes, in particular, has led to a renaissance in the chemistry of ketyl radicals. For example, striking advances in the field exploit visible light-mediated photocatalytic SET, PCET, HAT, and halogen atom transfer processes that deliver ketyl radicals for C–C bond formation. Finally, recent innovative reports have described how the enantioselectivity of reactions involving ketyl radicals can be controlled. We look forward to further advances in the generation and selective harnessing of ketyl radicals, particular in the context of sustainable catalysis, and increased application of the versatile reactive intermediates to synthetic challenges in both academic and industrial settings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Manchester (Dean's Award to Á. P.), the European Union Horizon 2020 (Fellowship to S. A.; Marie Sklodowska-Curie grant 891623-SmART), BBSRC (DTP Studentship to O. K.), EPSRC (iCAT CDT Studentship to E. P.; EP/S023755/1).Notes and references
- Y. Kratish, D. Pinchuk, A. Kaushansky, V. Molev, B. Tumanskii, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2019, 58, 18849–18853 CrossRef CAS PubMed and references therein.
- G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751–9757 CrossRef CAS PubMed.
- Y. Yamamoto, R. Hattori, T. Miwa, Y.-I. Nakagai, T. Kubota, C. Yamamoto, Y. Okamoto and K. Itoh, J. Org. Chem., 2001, 66, 3865–3870 CrossRef CAS.
- A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 2533–2534 CrossRef.
- P. Bichovski, T. M. Haas, D. Kratzert and J. Streuff, Chem. – Eur. J., 2015, 21, 2339–2342 CrossRef CAS.
- L. P. T. Hong, C. Chak and C. D. Donner, Org. Biomol. Chem., 2013, 11, 6186–6194 RSC.
- X. Tang and A. Studer, Org. Lett., 2016, 18, 4448–4450 CrossRef CAS.
- B. A. Sandoval, S. I. Kurtoic, M. M. Chung, K. F. Biegasiewicz and T. K. Hyster, Angew. Chem., Int. Ed., 2019, 58, 8714–8718 CrossRef CAS.
- E. Dolci, V. Froidevaux, C. Joly-Duhamel, R. Auvergne, B. Boutevin and S. Caillol, Polym. Rev., 2016, 56, 512–556 CrossRef CAS.
- Á. Péter and D. J. Procter, CHIMIA, 2020, 74, 18–22 CrossRef and references therein.
- X. Just-Baringo and D. J. Procter, Acc. Chem. Res., 2015, 48, 1263–1275 CrossRef CAS.
- S. Shi and M. Szostak, Org. Lett., 2015, 17, 5144–5147 CrossRef CAS PubMed.
- C.-H. Yeh, R. P. Korivi and C.-H. Cheng, Adv. Synth. Catal., 2013, 355, 1338–1344 CrossRef CAS.
- J. Streuff, M. Feurer, P. Bichovski, G. Frey and U. Gellrich, Angew. Chem., Int. Ed., 2012, 51, 8661–8664 CrossRef CAS PubMed.
- J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664–3671 RSC.
- E. J. Corey and G. Z. Zheng, Tetrahedron Lett., 1997, 38, 2045–2048 CrossRef CAS.
- H.-M. Huang, J. J. W. McDouall and D. J. Procter, Nat. Catal., 2019, 2, 211–218 CrossRef CAS.
- D. Parmar, H. Matsubara, K. Price, M. Spain and D. J. Procter, J. Am. Chem. Soc., 2012, 134, 12751–12757 CrossRef CAS.
- N. Kern, M. P. Plesniak, J. J. W. McDouall and D. J. Procter, Nat. Chem., 2017, 9, 1198–1204 CrossRef CAS PubMed.
- T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed.
- M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Chem. Rev., 2017, 1, 0052 CrossRef CAS.
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS.
- L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS.
- Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C. Zhu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 3636–3644 CrossRef CAS.
- F. R. Petronijević, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323–18326 CrossRef.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS.
- L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS.
- K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed.
- C.-X. Ye, Y. Y. Melcamu, H.-H. Li, J.-T. Cheng, T.-T. Zhang, Y.-P. Ruan, X. Zheng, X. Lu and P.-Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef.
- Q. Xia, H. Tian, J. Dong, Y. Qu, L. Li, H. Song, Y. Liu and Q. Wang, Chem. – Eur. J., 2018, 24, 9269–9273 CrossRef CAS.
- L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225–229 CrossRef CAS.
- X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454–5458 CrossRef CAS.
- Y. Liu, X. Liu, J. Li, X. Zhao, B. Qiao and Z. Jiang, Chem. Sci., 2018, 9, 8094–8098 RSC.
- A. L. Berger, K. Donabauer and B. König, Chem. Sci., 2018, 9, 7230–7235 RSC.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS.
- X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888–6892 RSC.
- M. Lübbesmeyer, E. G. Mackay, M. A. R. Raycroft, J. Elfert, D. A. Pratt and A. Studer, J. Am. Chem. Soc., 2020, 142, 2609–2616 CrossRef.
- Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- P. Hu, B. K. Peters, C. A. Malapit, J. C. Vantourout, P. Wang, J. Li, L. Mele, P.-G. Echeverria, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 20979–20986 CrossRef CAS and references therein.
- M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS.
- L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298–3301 CrossRef CAS.
- S. Pan, B. Gao, J. Hu, J. Xuan, H. Xie and H. Ding, Chem. – Eur. J., 2016, 22, 959–970 CrossRef CAS.
- N. J. Fazakerley, M. D. Helm and D. J. Procter, Chem. – Eur. J., 2013, 19, 6718–6723 CrossRef CAS.
- E. P. Farney, S. S. Feng, F. Schäfers and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 1267–1270 CrossRef CAS.
- J. C. Leung, A. A. Bedermann, J. T. Njardarson, D. A. Spiegel, G. K. Murphy, N. Hama, B. M. Twenter, P. Dong, T. Shirahata, I. M. McDonald, M. Inoue, N. Taniguchi, T. C. McMahon, C. M. Schneider, N. Tao, B. M. Stoltz and J. L. Wood, Angew. Chem., Int. Ed., 2018, 57, 1991–1994 CrossRef CAS PubMed.
- J. Gong, H. Chen, X.-Y. Liu, Z.-X. Wang, W. Nie and Y. Qin, Nat. Commun., 2016, 7, 12183 CrossRef CAS.
- T. Honda, T. Matsukawa and K. Takahashi, Org. Biomol. Chem., 2011, 9, 673–675 RSC.
Footnote |
| † Dedicated to Prof. Ilhyong Ryu on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2021 |