
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHoneycomb layered oxides: structure, energy storage, transport, topology and relevant insights
Godwill Mbiti
Kanyolo
 *a,
Titus
Masese
*bc,
Nami
Matsubara
d,
Chih-Yao
Chen
b,
Josef
Rizell
e,
Zhen-Dong
Huang
*f,
Yasmine
Sassa
e,
Martin
Månsson
d,
Hiroshi
Senoh
c and
Hajime
Matsumoto
c
*a,
Titus
Masese
*bc,
Nami
Matsubara
d,
Chih-Yao
Chen
b,
Josef
Rizell
e,
Zhen-Dong
Huang
*f,
Yasmine
Sassa
e,
Martin
Månsson
d,
Hiroshi
Senoh
c and
Hajime
Matsumoto
c
aDepartment of Engineering Science, The University of Electro-Communications, 1-5-1, Chofugaoka, Chofu, Tokyo 182-8585, Japan. E-mail: gmkanyolo@gmail.com
bAIST-Kyoto University, Chemical Energy Materials Open Innovation Laboratory (ChEMOIL), Sakyo-ku, Kyoto 606-8501, Japan
cResearch Institute of Electrochemical Energy, National Institute of Advanced Industrial Science and Technology (AIST), 1-8-31 Midorigaoka, Ikeda, Osaka 563-8577, Japan. E-mail: titus.masese@aist.go.jp
dDepartment of Applied Physics, Sustainable Materials Research & Technologies (SMaRT), KTH Royal Institute of Technology, SE-10691 Stockholm, Sweden
eDepartment of Physics, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
fKey Laboratory for Organic Electronics and Information Displays and Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications (NUPT), Nanjing, 210023, China. E-mail: iamzdhuang@njupt.edu.cn
First published on 12th February 2021
Abstract
The advent of nanotechnology has hurtled the discovery and development of nanostructured materials with stellar chemical and physical functionalities in a bid to address issues in energy, environment, telecommunications and healthcare. In this quest, a class of two-dimensional layered materials consisting of alkali or coinage metal atoms sandwiched between slabs exclusively made of transition metal and chalcogen (or pnictogen) atoms arranged in a honeycomb fashion have emerged as materials exhibiting fascinatingly rich crystal chemistry, high-voltage electrochemistry, fast cation diffusion besides playing host to varied exotic electromagnetic and topological phenomena. Currently, with a niche application in energy storage as high-voltage materials, this class of honeycomb layered oxides serves as ideal pedagogical exemplars of the innumerable capabilities of nanomaterials drawing immense interest in multiple fields ranging from materials science, solid-state chemistry, electrochemistry and condensed matter physics. In this review, we delineate the relevant chemistry and physics of honeycomb layered oxides, and discuss their functionalities for tunable electrochemistry, superfast ionic conduction, electromagnetism and topology. Moreover, we elucidate the unexplored albeit vastly promising crystal chemistry space whilst outlining effective ways to identify regions within this compositional space, particularly where interesting electromagnetic and topological properties could be lurking within the aforementioned alkali and coinage-metal honeycomb layered oxide structures. We conclude by pointing towards possible future research directions, particularly the prospective realisation of Kitaev–Heisenberg–Dzyaloshinskii–Moriya interactions with single crystals and Floquet theory in closely-related honeycomb layered oxide materials.
 Godwill Mbiti Kanyolo | Godwill Mbiti Kanyolo is a Postdoctoral Researcher in the University of Electro-Communications (Tokyo, Japan). He completed his PhD, MS and BS degrees in Fundamental Science and Engineering at the University of Electro-Communications. His current research pursuit involves theoretical and experimental studies on the interaction of the electromagnetic field with small Josephson junctions and other related condensed matter systems. He is also interested in analogue systems that test the role of geometry such as topology and gauge theory in gravitational systems, Brownian motion and quantum theory. |
 Titus Masese | Titus Nyamwaro Masese is a senior scientist at the National Institute of Advanced Industrial Science and Technology (AIST), Japan where he has developed close affiliations with AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL). He completed his postgraduate studies under the tutelage of Prof. Yoshiharu Uchimoto and his undergraduate studies under Prof. Haruyuki Inui at Kyoto University. His current area of research includes, amongst others, the syntheses and physicochemical characterisation of novel functional materials for rechargeable potassium-, magnesium- and fluoride-ion batteries. He is also currently pursuing the design of new honeycomb layered materials to test both the solid-state (electro)chemistry, ionics and physics of Kitaev quantum spin liquids. |
 Chih-Yao Chen | Chih-Yao Chen received his Master's degree in Materials Science and Engineering from National Cheng Kung University, Taiwan, and PhD degree in Energy Science from Kyoto University under the supervision of Prof. Rika Hagiwara. After working at Osaka University with Prof. Tetsuya Tsuda and Prof. Susumu Kuwabata as a postdoctoral researcher, he joined AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL) directed by Prof. Qiang Xu. His research interests focus on ionic liquids and molten salts and their application in electrochemical energy storage devices. |
 Zhen-Dong Huang | Zhen-Dong Huang received his PhD Degree (2012) in Mechanical Engineering from Hong Kong University of Science and Technology (HKUST). He worked as a program-specific researcher in the research group of Prof. Yoshiharu Uchimoto at Kyoto University. He is currently an associate professor in the Institute of Advanced Materials at Nanjing University of Posts and Telecommunications. He works on the development of high energy density nanostructured materials for various energy storage systems, such as lithium-, sodium- and potassium-ion batteries. |
 Yasmine Sassa | Yasmine Sassa is a tenure-track professor at the Chalmers University of Technology, Gothenburg, Sweden. She is the leader of the Quantum Technologies and Materials (QuTM) Laboratory, which conducts research concerning superconductivity, magnetism, skyrmions and correlated electron physics. Prof. Sassa uses state-of-the art experimental techniques, e.g. angle-resolved photoemission, resonant inelastic X-ray scattering, neutron scattering and muon spin rotation, for advanced materials characterisations of atomic, electronic, and magnetic spin structures as well as excitations. She is also an expert in thin-film growth and conducts computer modeling of electronic band structures. Prof. Sassa conducted her PhD/post doc research in Switzerland at PSI and ETHZ. |
 Martin Månsson | Martin Månsson is a tenured professor at the KTH Royal Institute of Technology, Stockholm, Sweden, where he leads the Sustainable Materials Research & Technologies (SMaRT) group. He runs a comprehensive research program concerning multifunctional materials ranging from quantum physics low-dimensional magnetism and superconductors, to novel battery and hydrogen storage materials. The experimental science is conducted at state-of-the-art large-scale research facilities using neutron scattering, muon spin rotation and synchrotron radiation techniques. Prof. Månsson is the Director of Studies for the Swedish national graduate school in neutron scattering (SwedNess). His early scientific career was conducted at PSI, ETHZ and EPFL in Switzerland. |
 Hiroshi Senoh | Hiroshi Senoh is currently a Senior Scientist at the National Institute of Advanced Industrial Science and Technology (AIST), Japan. He was born in Kyoto and raised in Kobe. Prior to joining AIST in 2004, he was involved in postdoctoral research in the field of hydrogen storage at Japan Science and Technology Agency (JST). He received his PhD in Electrochemistry from Osaka Prefecture University in 2001. His current research is focused on the electrochemical energy systems for nickel–metal hydride batteries, fuel cells, lithium-ion batteries, and next-generation rechargeable batteries. |
 Hajime Matsumoto | Hajime Matsumoto is a Chief Senior Scientist in Research Institute of Electrochemical Energy, National Institute of Advanced Industrial Science and Technology (AIST), Japan where he has been since 1996. He has also worked as an associate professor in Division of Applied Chemistry, Graduate School of Engineering, Osaka University (cross-appointment fellow) for 3 years. He is a principal investigator in a number of national projects. His main area of research is on the preparation of zero-solvents based on new perfluoroanions and their application to electrochemical devices. He was awarded the Battery Technology Prize from The Committee of Battery Technology (Electrochemical Society of Japan). |
1 Introduction
Charles Darwin famously described the honeycomb as an engineering masterpiece that is “absolutely perfect in economising labour and wax”.1 For over two millennia, scientists and philosophers alike have found a great deal of fascination in the honeycomb structures found in honeybee hives. These hexagonal prismatic wax cells built by honey bees to nest their larvae, store honey and preserve pollen are revered as a feat in precision engineering and admired for their elegance in geometry.2 The honeycomb framework offers a rich tapestry of qualities adopted in myriads of fields such as mechanical engineering, architectural design biomedical engineering, etc. (as briefly outlined in Fig. 1).2,3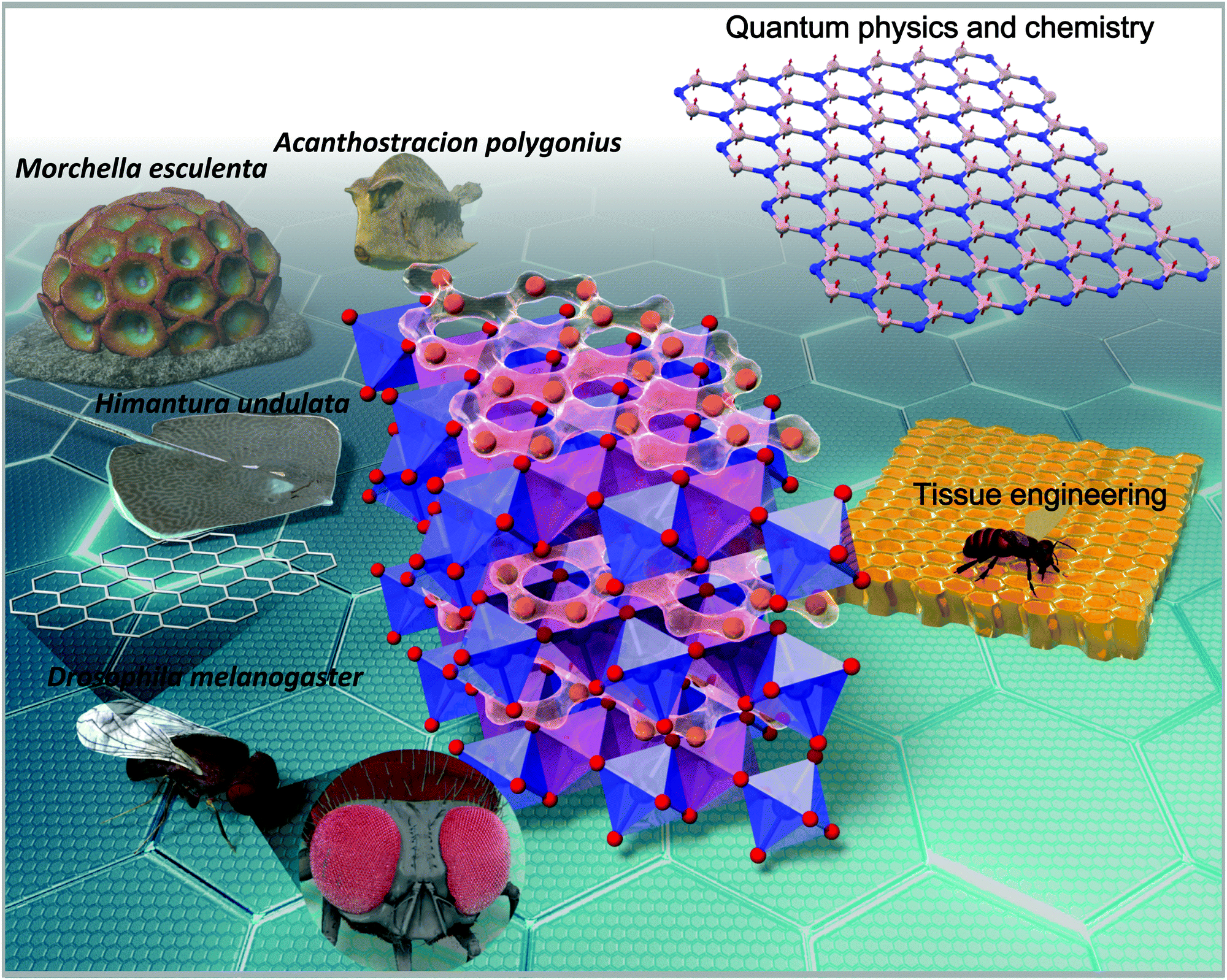 | ||
| Fig. 1 Schematic illustration of the various realisations of the honeycomb structure found not only in energy storage materials, but also as pedagogical models in condensed-matter physics, solid-state chemistry and extending to tissue engineering.2 Specific varieties of fungi (videlicet, Morchella esculenta) tend to adopt honeycomb-like structures,4 whilst insects such as the fruit flies (Drosophila melanogaster) have their wing cells in honeycomb configuration;5–7 thus endowing them with excellent rigidity. In addition, the honeycomb whip ray (Himantura undulata)8 and the honeycomb cowfish (Acanthostracion polygonius)9 have honeycomb patterns on their body that are thought to aid in their facile movement and camouflage. | ||
The discovery and isolation of graphene in 2004, not only revolutionised our understanding of two-dimensional materials but also unveiled a new platform for fabricating novel materials with customised functionalities.10,11 Two-dimensional (2D) nanostructures are crystalline systems comprising covalently bonded atom cells in planar arrangement of mesoscopic thicknesses. Due to their small size, this class of materials exhibits highly controlled and unique optical, magnetic, or catalytic properties.12–54 By substituting the constituent atoms and manipulating the atomic cell configurations, materials with remarkable physicochemical properties such as high electron mobility, unique optical and chemical functionality can be tailor-made for various technological realms such as catalysts, superconductivity, sensor applications and energy storage. Assembling such materials into vertical stacks of layered combinations allows the insertion of atoms within the stacks creating a new class of multi-layered heterostructures. A unique characteristic of these structures is that inter-layer bonds holding together the thin 2D films are significantly weaker (van der Waals bonds) than the covalent bonds within the 2D monolayers, which further augments the possibility for emergent properties exclusive to these materials.
In electrochemistry, layered frameworks composed of alkali or coinage metal atoms interposed between 2D sheets of hexagonal (honeycomb) transition metal and chalcogen (or pnictogen) oxide octahedra have found great utility as next-generation cathode materials for capacious rechargeable battery systems.55–59 The weak interlayer bonds between transition metal slabs facilitate facile mobility of intercalated atoms during the battery operation (de)insertion processes, endowing these heterostructures with ultrafast ionic diffusion rendering them exemplar high energy (and power) density cathode materials. Furthermore, the unique topological changes occurring during these electrochemical processes have been seen to induce new domains of physics entailing enigmatic optical, electromagnetic and quantum properties that promise to open new paradigms of computational techniques and theories quintessential in the field of quantum material science catapulting the discovery of materials with novel functionalities.12–53 This has created an entirely new platform of study, encompassing fields, inter alia, materials science, solid-state chemistry, electrochemistry and condensed matter physics.15,19,29,35,39,60–91
Although layered oxides encompass a broad class of materials with diverse structural frameworks and varied emergent properties, in this review, we focus on the stellar properties innate in the aforementioned class of honeycomb layered oxides comprising alkali or coinage metal atoms sandwiched between slabs consisting of transition metal oxide octahedra surrounding chalcogen or pnictogen oxide octahedra in a honeycomb configuration. To provide deeper insights, we delineate the fundamental chemistry underlying their material design along with emergent domains of physics. We further highlight their functionalities for tunable electrochemistry, superfast ionic conduction, electromagnetism and topology, as illustrated in Fig. 2. Moreover, we underpin the unexplored albeit vastly promising crystal chemistry space whilst outlining effective ways to identify regions within this compositional space, particularly where potentially interesting electromagnetic and topological properties could be lurking within the aforementioned honeycomb layered oxides. The looming challenges are also discussed with respect to the governing chemistries surrounding honeycomb layered oxides. Finally, we conclude by pointing towards possible future research directions, particularly the prospective realisation of Kitaev–Heisenberg–Dzyaloshinskii–Moriya interactions in closely-related honeycomb layered oxide materials, and their connection to Floquet theory and fabrication efforts that are not limited to single crystals.
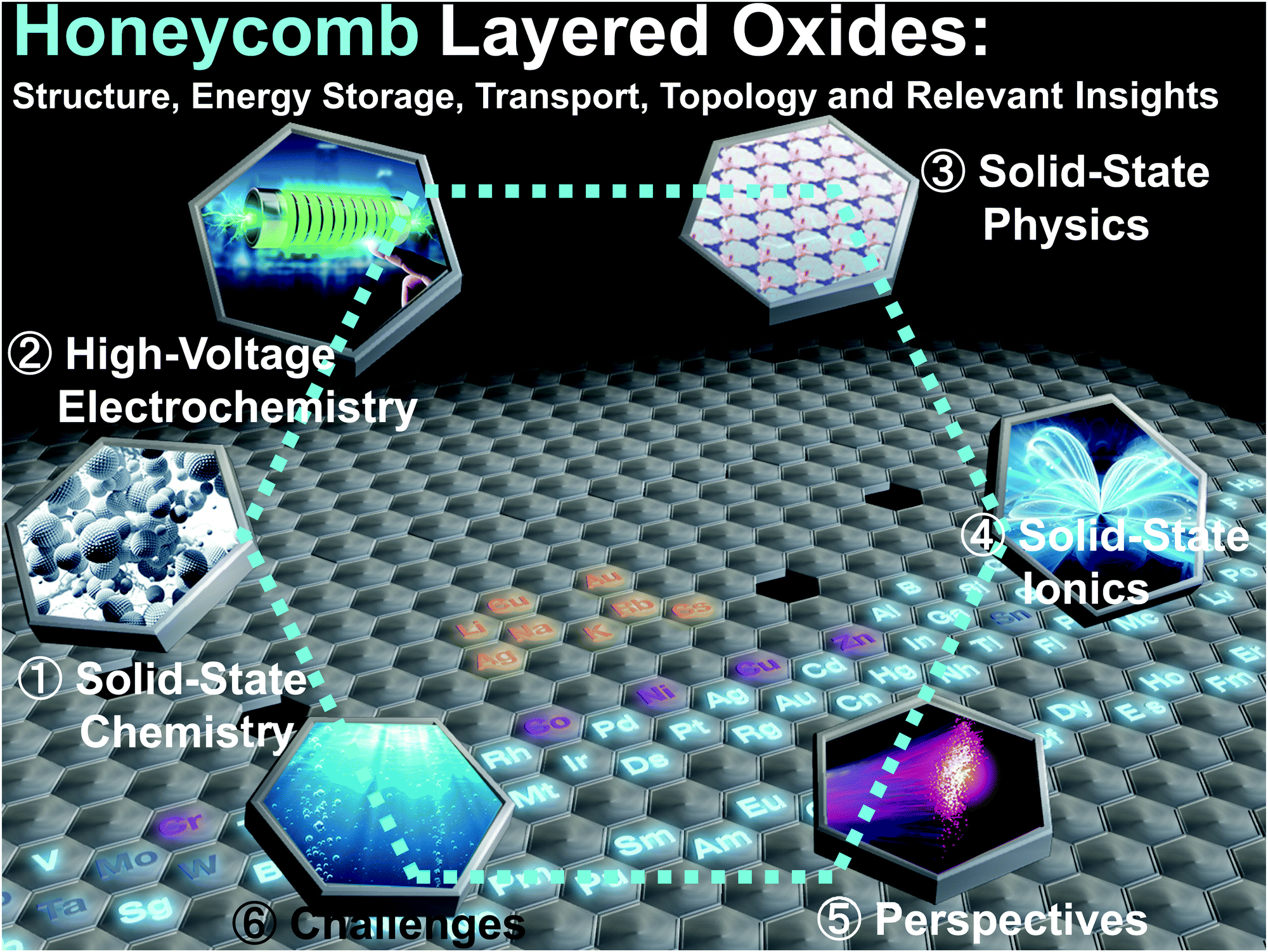 | ||
| Fig. 2 Illustration of the various sections to be covered in this review. This starts from solid-state chemistry, physics, electrochemistry to solid-state ionics. We finally adumbrate on the challenges and perspectives of these honeycomb layered oxides. | ||
2 Materials chemistry of honeycomb layered oxides
2.1 Chemical composition
The aforementioned honeycomb layered oxides generally adopt the following chemical compositions, taking into account that charge electro-neutrality is maintained and assuming ordered structures: A2+M22+D6+O62− (or equivalently as A2/3+M2/32+D1/36+O22−), A3+M22+D5+O62− (A+M2/32+D1/35+O22−), A4.5+M0.53+D6+O62− (A3/2+M1/63+D1/36+O22−), A4+M3+D5+O62− (A4/3+M1/33+D1/35+O22−), A2+D4+O32− (A4/3+D2/34+O22−), A3+A′+M3+D5+O62− (A+A′1/3+M1/33+D1/35+O22−), A4+M2+D6+O62− (A4/3+M1/32+D1/36+O22− or A8+M22+D26+O122−), amongst others (Fig. 3a).15,16,19,29,35,39,60–88,94–118,125–133 Here M denotes transition metal atoms such as Ni, Co, Mn, Fe, Cu, Zn, Cr (including Mg); D denotes Te, Sb, Bi, Nb, Ta, W, Ru, Ir, Os; A and A′ denote alkali atoms such as Li, Na, K or coinage atoms like Cu and Ag (with A ≠ A′). It is worth mentioning that the honeycomb layered framework is not limited to compositions entailing only one species of alkali atoms. Oxides compositions comprising mixed-alkali atoms such as Na3LiFeSbO6, Na2LiFeTeO6, Ag3LiRu2O6, Ag3NaFeSbO6, Ag3LiIr2O6, Ag3LiMTeO6 (M = Co, Ni), Ag3LiMSbO6 (M = Cr, Mn, Fe) and, more recently, Li3−xNaxNi2SbO6 as well as oxides with off-stoichiometric compositions such as Li3Co1.06TeO6 have been explored with the aim of merging favourable attributes from multiple species to improve various material functionalities, for instance, battery performance.89–91,136–139 Other atypical honeycomb layered oxide compositions are those encompassing alkali or mixed-alkali species atoms embedded within the transition metal slabs. Compositions such as LixMyMn1−yO2 (0 < x < 1; 0 < y < 1; M = Li, LiNi), Na3/4(Li1/4Mn3/4)O2, Na5/6(Li1/4Mn3/4)O2, Na2/3(Li1/6Mn5/6)O2, etc. have also been considered for use as high-performance battery materials,119–124,134 and can be envisaged to exhibit unique three-dimensional diffusion as they have some of their alkali atoms reside in the honeycomb layers. Although three-dimensional diffusion is covered in a later section, it is pertinent to note that this review will mainly focus on two-dimensional diffusion of cations in relation to topotactic curvature evolutions. Other classes of honeycomb layered oxide frameworks with vastly different properties and applications are also beyond the scope of this review.140–147 | ||
| Fig. 3 Combination of elements that constitute materials exhibiting the honeycomb layered structure. (a) Choice of elements for layered oxide compositions (such as A2+M22+D6+O6 (A2/3+M2/32+D1/36+O2), A3+M22+D5+O6 (A+M2/32+D1/35+O2), etc.) that can adopt honeycomb configuration of transition metal atoms. Inset shows a polyhedral view of the crystal structure of layered honeycomb oxides, with the alkali atoms (shown as brown spheres) sandwiched between honeycomb slabs (blue). (b) X-Ray diffraction (XRD) pattern of K2Ni2TeO6 (12.5% cobalt-doped) honeycomb layered oxide.92 Inset: Slab of layered oxide showing the honeycomb arrangement of nickel (Ni) atoms around non-magnetic tellurium (Te) atoms. Dashed line highlights the unit cell. (b) Adapted from ref. 92 with permission (Creative Commons licence 4.0). | ||
2.2 Preparative methods
High-temperature solid-state synthesis is often considered an expedient route to synthesise most of the above mentioned honeycomb layered oxides because their initial precursor materials usually require high temperatures to activate the diffusion of individual atoms.148 In this technique, precursors are mixed in stoichiometric amounts and pelletised to increase the contact surface area of these reactants. Finally, they are fired at high temperatures (over 700 °C) resulting in thermodynamically-stable honeycomb layered structures. The firing environment (argon, nitrogen, air, oxygen, carbon monoxide, hydrogen, etc.) needs to be adequately controlled to obtain materials with the desired oxidation states of transition metals. For example, an inert firing environment is demanded for layered oxides that contain Mn2+ and Fe2+; otherwise oxidised samples containing Mn3+ and Fe3+ are essentially formed. Nonetheless, not all high-temperature synthesis processes are as restrictive, as compositions containing Ni2+, such as A2Ni2TeO6 (A = Li, Na, K, etc.) can be synthesised under air to obtain samples that contain Ni still in the divalent state. Moreover, varying the synthesis protocols (annealing temperature and time, types of precursors, thermal ramp rate, etc.) during high-temperature synthesis not only aid in enhancing the scalability of the synthesis but also gives rise to the possibility of obtaining new polymorphs (or polytypes), as has been shown in the high-temperature synthesis of Na3Ni2BiO6, Na4NiTeO6 and Na3Ni2SbO6.57,73,82,149The topochemical ion-exchange synthesis route is also possible for honeycomb layered oxides with accessible kinetically-metastable phases.137,150–153 Despite the high binding strength amongst adjacent atoms within the honeycomb slab, the use of cations with higher charge-to-radius ratio such as Li+ in LiNO3 can drive out the Na+ atoms present in Na2Cu2TeO6 by lowering their electrostatic energy to create Li2Cu2TeO6.64 Here, the two precursors are heated together at a moderate temperature (300 °C) triggering the diffusion of Na+ and Li+. Other oxides that can be prepared via the ion-exchange route include Li3Co2SbO6, Ag3M2SbO6 (M = Ni, Co and Zn), Li2Ni2TeO6, Ag3LiGaSbO6, Ag3LiAlSbO6, Ag3Ni2BiO6 and Li3M2SbO6 (M = Fe and Mn).32,56,63,66,86 However, it is worthy to recapitulate that there exists exemplars of compounds that can be synthesised topochemically such as Ag3Co2SbO6 and Ag3LiDO6 (D = Ru, Ir) that are exceptions to the rule that ion exchange can only happen from ions with lower charge-to-radius ratio to those with higher ratios (taking into account Ag has a larger ionic radius than Li154).32,137 The syntheses of these honeycomb layered oxides are typically done at ambient pressures; however, high-pressure syntheses routes remain unexplored, a pursuit which may expand their material platforms. Equally important is the utilisation of low-temperature routes such as sol–gel and mechanochemical synthesis since they do not require a priori high-temperatures to attain thermodynamically-stable phases. Although not delved in the scope of this review, honeycomb layered oxides entailing alkaline-metal atoms as the resident cations such as SrRu2O6 and BaRu2O6 can be prepared via a low-temperature hydrothermal synthesis route.135,155,156 Finally, despite the scarce exploration of electrochemical ion-exchange157–166 of honeycomb layered oxide materials, invaluable results on the K+/Na+ ion-exchange process in Na3Ni2SbO6 has been recently reported, demonstrating this process as a promising route to pursue.167
2.3 Crystallography
To ascertain the crystal structure of honeycomb layered oxides and discern the precise location of the constituent atoms, transmission electron microscopy (TEM), neutron diffraction (ND) and X-ray diffraction (XRD) analyses can be performed on single-crystals or polycrystalline samples. Although the XRD is the most commonly used crystallography technique, it is ineffective in analysing oxides composed of lighter atoms such as Li, H, and B due to their low scattering intensity. Also, honeycomb layered oxides with elements of similar atomic number are difficult to distinguish using XRD because they diffract with similar intensity.To distinguish light elements or elements with close atomic numbers on honeycomb layered oxides, the ND is used because the neutron beam interacts directly with the nucleus hence, the ability to observe light elements. In spite of the high accuracy, the equipment remain very expensive and ND experiments require the use of very large sample amounts to obtain high-resolution data – an impediment to materials that can only be prepared on a small scale.
Although, XRD analyses accurately validate the precise crystal structure of honeycomb oxides with heavy elements such as K2Ni2TeO6 (partially doped with Co), as shown in Fig. 3b, TEM is used to obtain unequivocal information relating to the structure of materials at the atomic scale. A number of studies have reported the utilisation of TEM analyses on honeycomb layers of oxides to determine, with high-precision, the arrangement of atoms within the honeycomb lattice and the global order of atoms within the structure of materials in a honeycomb lattice.59,70,77,82,92,152,168–171 Likewise, the honeycomb lattice comprising Te surrounded by transition metals in K2Ni2TeO6 (Fig. 3b) can be seen from state-of-the-art TEM images, shown in red Fig. 4. It is worth noting that TEM analyses are expensive to conduct and may lead to damage of samples because of the strong electron beam. We also note that the sensitivity of samples to the electron beam differ even within slightly the same honeycomb layered oxide composition. For instance, Cu3Co2SbO6 is more susceptible to electron-beam damage than Cu3Ni2SbO6,70 which implies that tuning of the chemical composition of these materials can induce structural stability necessary to perform intensive TEM analyses.
 | ||
| Fig. 4 (a) High-resolution transmission electron microscopy (TEM) image of a crystallite of K2Ni2TeO6 (12.5% cobalt-doped) honeycomb layered oxide and (b) corresponding electron diffractograms taken along the [001] zone axis.92 (c) Visualisation (along the c-axis [001]) of the honeycomb configuration of Ni atoms around Te atoms (in brighter contrast) using High-Angle Annular Dark-Field Scanning TEM (HAADF-STEM). Dashed lines indicate the unit cell. (d) STEM imaging with Ni atoms (partially with Co) (in green) assuming a honeycomb fashion (as highlighted in dashed lines) and (e) STEM imaging showing Te atoms (in red) surrounded by transition metal atoms. (f) Annular Bright-Field TEM (ABF-TEM) of segments manifesting potassium atoms (in brown) assuming a honeycomb fashion and overlapped with oxygen atoms. Note that some portions of the honeycomb ordering of transition metal atoms slightly appear obfuscated, owing to sensitivity of the samples to long-time beam exposure. (a and b) Reproduced and adapted from ref. 92 under Creative Commons licence 4.0. | ||
2.4 Nomenclature
In a notation system promulgated by Hagenmuller, Delmas and co-workers,93,172 honeycomb layered oxides can also be classified according to the arrangement of their honeycomb layers (stackings) The notation comprises a letter to represent the bond coordination of A alkali, alkaline-earth or coinage metal atoms with the surrounding oxygen atoms (generally, ‘T’ for tetrahedral, ‘O’ for octahedral, or ‘P’ for prismatic) and a numeral that indicates the number of repetitive honeycomb layers (slabs) per each unit cell (mainly, ‘1’, ‘2’ or ‘3’) as shown in Table 1. For instance, Na2M2TeO6 (with M being Mg, Zn, Co or Ni) possess P2-type structures, the nomenclature arises from their repetitive two-honeycomb layers sequence in the unit cell with prismatic coordination of Na atoms with oxygen in the interlayer region.24,34,60,71,173 Structures such as O3-type stackings can be found in Na3M2SbO6 (here M = Zn, Ni, Mg or Cu) and Na3LiFeTeO6, whereas Na3Ni2SbO6 and Na3Ni2BiO6 reveal O1-type and P3-type stackings during the electrochemical extraction of alkali Na atoms.58,80–83,91,174 Note that the aforementioned oxide compositions are representative of the main stackings observed, and is by no means, an exhaustive summary. Ag- and Cu-based honeycomb layered oxides, prepared via topochemical ion-exchange, such as A3M2DO6 (A = Ag, Cu; M = Ni, Mn, Co, Zn; D = Bi, Sb) and related oxides, adopt a linear (dumbbell-like) coordination of alkali or coinage metal atoms with the adjacent two oxygen atoms with an intricate multiple stacking sequence of the honeycomb slabs.47,63,130,175 The various stacking sequences exhibited by representative honeycomb layered oxides are detailed in Fig. 5 and Table 1.| Coordination of alkali atoms with oxygen | Hagenmuller–Delmas’ notation (slab stacking sequence) | Honeycomb layered oxide compositions |
|---|---|---|
| Tetrahedral | T2 | Li2Ni2TeO6 |
| Octahedral | O1 | NaNi2BiO6−δ, Na2RuO3, Li2MO3 (M = Ni, Pt, Rh), BaRu2O6 |
| O2 | Li2MnO3, Lix(Li1/5Ni1/5Mn3/5)O2 (x < 1), Lix(Li1/4Mn3/4)O2, | |
| Na2ZrO3, Na2SnO3, Li2RuO3, SrRu2O6 | ||
| O3 | Li2Ni2TeO6, Li3Ni2BiO6, Na3Ni2BiO6, Na2Cu2TeO6, | |
| Na3M2SbO6 (M = Ni, Cu, Cr, Co, Mg, Zn), Li2Cu2TeO6 | ||
| Li3M2SbO6 (M = Ni, Cu, Co, Zn), NaRuO3, | ||
| Li2MnO3, Li(Li1/5Ni1/5Mn3/5)O2, Na7/10(Ni7/20Sn13/20)O2, | ||
| NaxNix/2Mn1−x/2O2 (1 ≤ x < 4/5), Na3LiFeSbO6, Li4MTeO6 | ||
| (M = Co, Ni, Cu, Zn), Li4MSbO6 (M = Cr, Fe, Al, Ga, Mn) | ||
| Prismatic | P2 | K2Ni2TeO6, Na2M2TeO6 (M = Mg, Zn, Co, Ni) |
| Na3/4(Li1/4Mn3/4)O2, Na2/3 (Mg7/25Mn18/25)O2, BaRu2O6, | ||
| Na5/6(Li1/4Mn3/4)O2, Na2/3(Li1/6Mn5/6)O2, | ||
| Na2/3(Mg1/4Mn3/4)O2, Na2/3(Ni1/3Mn2/3O2), Li2RuO3, | ||
| NaxNix/2Mn1−x/2O2 (2/3 ≤ x < 4/5), Na2/3Ni1/3Mn2/3O2, | ||
| P3 | Na2/3(Mg1/3Mn2/3)O2, Na2/3(Ni1/3Mn2/3)O2 | |
| Dumbbell (linear) | D | Ag3NaFeSbO6, Ag3LiMTeO6 (M = Co, Ni), |
| Ag3LiMSbO6 (M = Cr, Mn, Fe), Ag3LiM2O6 (M = Ir, Ru, Rh), | ||
| A3M2DO6 (M = Ni, Co, Mg, Zn, Mn; A = Cu, Ag; D = Bi, Sb) |
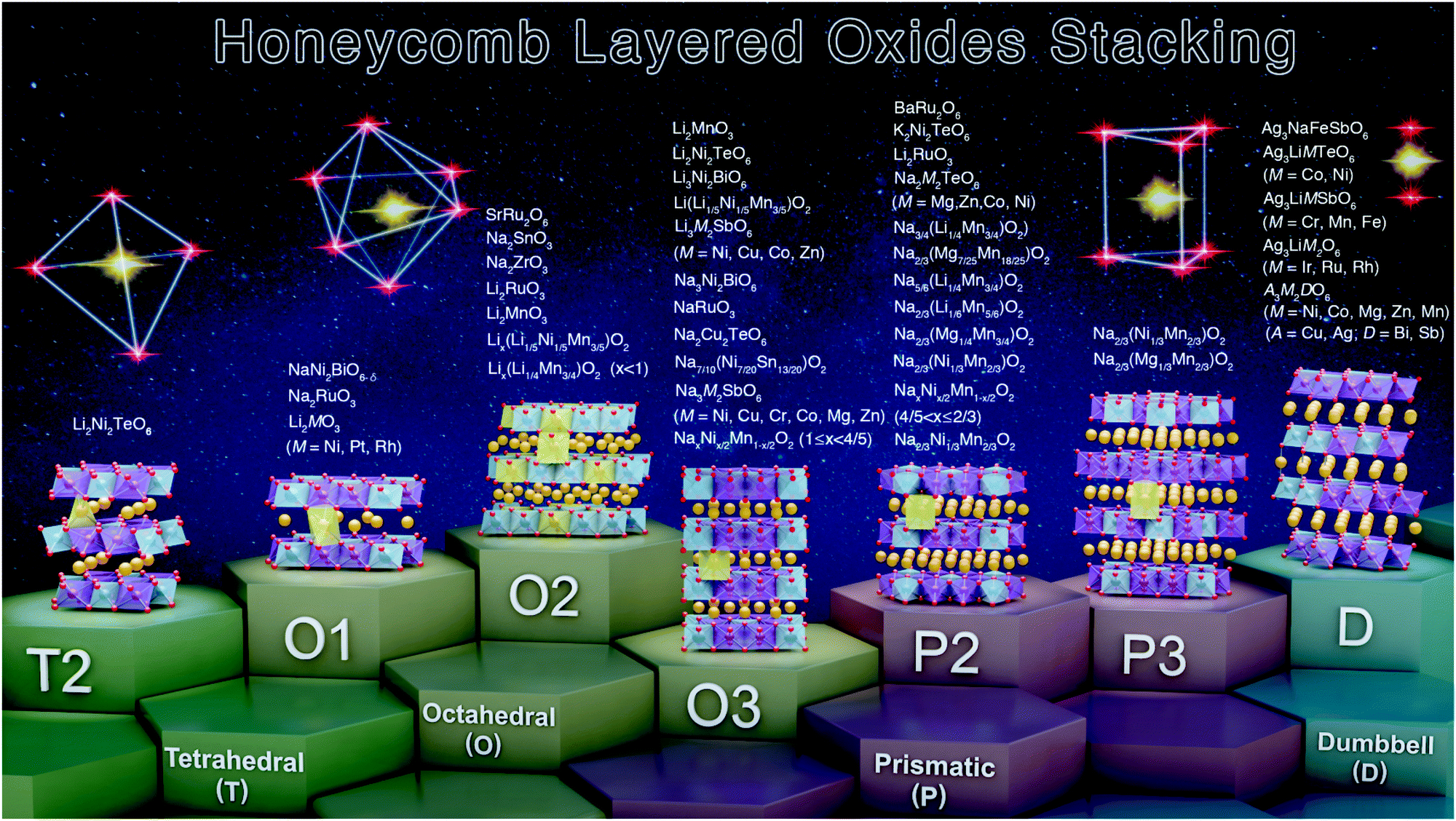 | ||
| Fig. 5 Summary of the various stacking sequences adopted by representative honeycomb layered oxides including those that entail pnictogen or chalcogen atoms in the slab layers. Note here that ‘T’, ‘O’ and ‘P’ denote the coordination of the alkali or coinage metal atoms (sandwiched between the honeycomb slabs) with the adjacent oxygen atoms of the honeycomb slab, id est, tetrahedral, octahedral and prismatic coordination, respectively. The numbers or digits (‘1’, ‘2’ and ‘3’) indicate the repetitive alkali-atom layers per unit cell, as denoted in Hagenmuller–Delmas’ notation.93 Note that the predominant stacking sequences have been shown, for ease of explanation. A more elaborate list of the stacking sequences for other honeycomb layered oxides reported thus far are provided in Table 2.15,16,19,29,35,39,60–88,94–135 | ||
2.5 Stacking sequences
The stacking sequences of the honeycomb slabs as well as the emplacement patterns of the oxygen atoms play a crucial role in the nature of emergent properties; even minuscule differences in atomistic placements could result in distinct variations of crystal frameworks with an assortment of physochemical properties.82,123,149,173,176 In general, the various manner of stackings observed in honeycomb layered oxides is contingent on the synthesis procedure, the content of alkali A atoms sandwiched between the honeycomb slabs and the nature of alkali A cations (that is, Li, Na, K and so forth).177 Different stacking sequences of the honeycomb slabs are observed in, for example, honeycomb layered oxides that comprise Na and Li atoms. Na atoms, with larger radii, tend to have a strong affinity to coordinate with six oxygen atoms; adopting octahedral (O) or prismatic (P) coordination.177 Li atoms, vide infra, have been found to possess tetrahedral (T) and octahedral coordination, as recently observed in Li2Ni2TeO6.56 Further, TEM analyses performed on oxides such as Na3Ni2BiO6, indicate assorted sequences of honeycomb ordering.82 Using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging studies, Khalifah and co-workers have broached another labeling scheme to allow the indication of the number of repetitive honeycomb layers.82 Using their notation, they illustrated that Na3Ni2BiO6 had 6 layers (6L), 9 layers (9L) and 12 layers (12L) of stacking honeycomb ordering sequence (periodicity). Such sequence of honeycomb ordering (and even stacking disorder) can be influenced by the reaction kinetics during the use of various synthesis conditions and higher orders of stacking sequences (4L, 6L, 9L, 12L, etc.) can be anticipated in honeycomb layered oxides.3 Magneto-spin models of honeycomb layered oxides
3.1 Crystal structure considerations
As aforementioned, honeycomb layered oxides mainly comprise alkali cations A+ sandwiched in a framework containing layers or slabs of M and D atoms coordinated, octahedrally, with oxygen atoms. M atoms are essentially magnetic with a valency of +2 or +3, whilst D atoms are non-magnetic and generally possess valency states (oxidation states or oxidation numbers) of +4, +5 or +6. The MO6 and DO6 octahedra assume a honeycomb configuration within the layers; DO6 octahedra being surrounded by six MO6 octahedra, as shown in Fig. 6a. Note that such an ordered honeycomb configuration of magnetic M atoms around non-magnetic D atom is contingent on their ionic radii.64,71,79,86 For instance, for honeycomb layered oxides with Te (or even Bi), as the D atoms and transition metal atoms such as Ni, M atoms, typically form ordered honeycomb configurations in oxides such as Na2Ni2TeO6, Na4NiTeO6, Na3Ni2BiO6 and, more recently, K2Ni2TeO6.24,34,57,59,60,71,82,173,174 However there is slight disorder between Te and surrounding metal atoms within the honeycomb slab as noted in Na2Zn2TeO6.178 On the other hand, in honeycomb layered oxides with Sb as the D metal atoms, such as Na3Ni2SbO6, disordered honeycomb configurations are often observed.149 This is due to the movement of Sb (D) atoms to the sites of Ni (M) atoms, which have similar ionic radii,154 a phenomenon commonly referred to as ‘cationic site mixing’. Also worthy of mention, is that the ionic radii of the sandwiched A atoms in honeycomb layered oxides has influence on the honeycomb ordering. This has been noted in Li-based honeycomb layered oxides such as Li4NiTeO6 and Li2Ni2TeO6, whereby Li atoms are located in the sites of Ni atoms.26,55,56 Hereafter, magnetism of honeycomb layered oxides with ordered honeycomb configurations of magnetic M atoms around D atoms shall be discussed, to serve as an entry point to some fundamental models of magneto-spin phenomena that have generated tremendous research interest in recent years.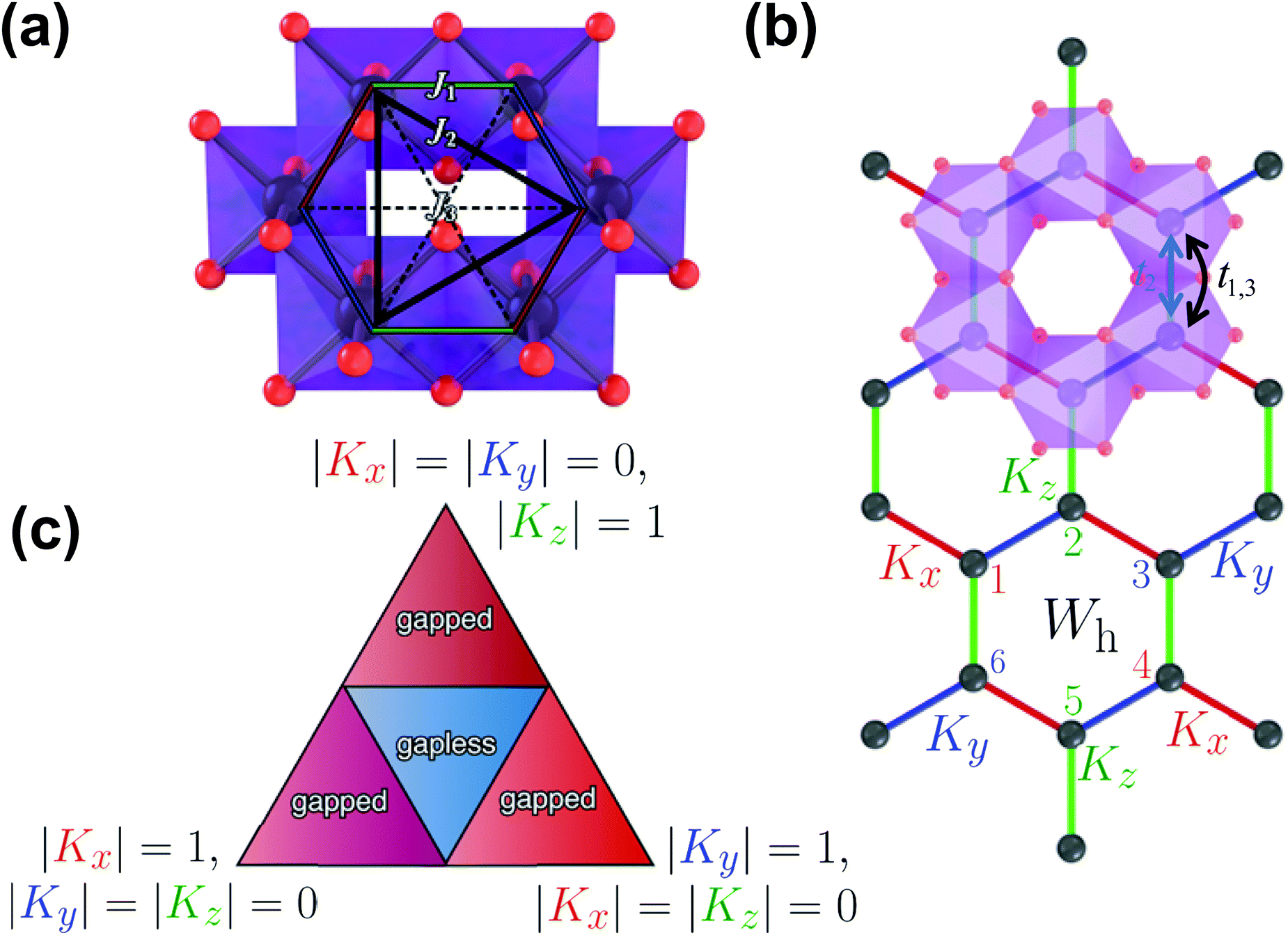 | ||
| Fig. 6 Nature of magnetic configurations adopted by honeycomb layered oxide materials. (a) Fragment of the transition metal slab showing the honeycomb configuration of transition metal atoms and the possible spin interactions with neighbouring magnetic atoms. Here Ji (where i = 1, 2 and 3) represents the magnetic exchange interactions (Kitaev, Haldane and higher-order interactions respectively) between an atom and its i-th neighbour. Transition metal atoms (in purple) are depicted surrounded by oxygen atoms (in red) in octahedral coordinations. (b) A schematic of the realisation of the Kitaev model. Kx, Ky and Kz denote the Kitaev coupling constants for the next-neighbouring transition metal bonds in the axes (depicted in red, blue and green, respectively). Wh denotes the plaquette flux operator with links labelled as ‘1’, ‘2’, ‘3’, ‘4’, ‘5’ and ‘6’. The blue (t1,3) or black (t2) double-headed arrow indicates the hopping path of an electron from one transition metal atom to an adjacent one directly or indirectly (via a shared oxygen atom) respectively along the spin-directional bonds of the honeycomb lattice. (c) The solution of the Kitaev model depicted as a phase diagram in the form of a triangle showing the spectrum constraints in the Brillouin zones as either gapped or gapless. The vertices of the triangle correspond to |Kz| = 1, |Kx| = |Ky| = 0, |Kx| = 1, |Ky| = |Kz| = 0 and |Ky| = 1, |Kx| = |Kz| = 0. | ||
The honeycomb arrangement of magnetic metal atoms (M) within the slabs of honeycomb layered oxides often leads to fascinating magnetic behaviour. This is due to the interactions generated from the spins innate in the magnetic atoms (what is commonly termed as magnetic coupling). As is explicitly shown in Fig. 6a, such interactions primarily originate from spins from the adjacent magnetic atoms (Kitaev-type interactions (denoted as J1)) within the honeycomb lattice, but they can also be influenced by spins of magnetic atoms from adjoining layers in the honeycomb configuration (id est, Haldane-type interactions (J2)). Spin interactions emanating from distant atoms may still occur and shall herein be classified as higher-order interactions (J3).28 Such magnetic interactions can be of varied fashion, spanning over short distances across the honeycomb lattice (what is termed as short-range interactions) or long distances extending to those of the adjacent honeycomb slabs (long-range interactions).
3.2 Kitaev model
In the case of J1 (type) interactions, the spin–spin interactions can either be anisotropic (Kitaev) or isotropic (Heisenberg) leading to an interaction Hamiltonian of the form, | (1a) |
![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) j = (Sxj,Syj,Szj) are the spin matrices. The sum is taken over next-neighbour interaction sites corresponding to J1 interactions. In a seminal paper, Kitaev showed that eqn (1a) with J1 = 0 is exactly soluble into a ground state of a topological superconductor in terms of a Kitaev-quantum spin liquid (K-QSL).179,180 K-QSL is a spin quantum state with long-range entanglement and short-range order of spin moments which continue to fluctuate coherently whilst still maintaining their disordered formation even at low temperatures.
j = (Sxj,Syj,Szj) are the spin matrices. The sum is taken over next-neighbour interaction sites corresponding to J1 interactions. In a seminal paper, Kitaev showed that eqn (1a) with J1 = 0 is exactly soluble into a ground state of a topological superconductor in terms of a Kitaev-quantum spin liquid (K-QSL).179,180 K-QSL is a spin quantum state with long-range entanglement and short-range order of spin moments which continue to fluctuate coherently whilst still maintaining their disordered formation even at low temperatures.
In particular, the K-QSL Hamiltonian has a conserved quantity Wh = (Sx1Sy2Sz3Sx4Sy5Sz6)h for a given (honeycomb) plaquette h, id est as shown in Fig. 6b. Each plaquette satisfies Wh2 = 1 (Wh = ±1) and the commutator [H(J1 = 0),Wh] = i∂Wh/∂t = 0 in the Heisenberg picture of quantum mechanics, which immensely simplifies calculations of the ground state properties such as spin–spin correlation functions. For instance, since Wh and H(J1 = 0) can be simultaneously diagonalised, the ground state corresponds to Wh = 1 for all plaquettes (known as the vortex-free state) whereas flipping any one plaquette to Wh = −1 corresponds to the lowest excited energy state (single vortex state) with a finite energy gap. Moreover, Wh commutes with both spin operators Sγj and Sγ′k at different sites j and k if and only if γ = γ′ and k = j ± 1 within a given plaquette. Otherwise, one can find Wh that commutes with either one of the operators but anti-commutes with the other one. Thus, using the cyclic property of the trace 〈abc〉 = 〈bca〉 = 〈cab〉 where a = SγjSγ′k, b = Wh, c = Wh, one can show that there is no long-range spin–spin correlation in the honeycomb lattice, id est 〈SγjSγ′k〉 = 〈SγjSγ′kW2h〉 = 〈WhSγjSγ′kWh〉 = 〈SγjWhSγ′kWh〉 = −〈SγjWhSγ′kWh〉 = −〈SγjSγ′kW2h〉 = −〈SγjSγ′k〉 = 0.181 This is the hallmark of a quantum spin liquid.182,183
The energy spectrum of the model is exactly soluble by mapping the spin operators in H(J = 0) to Majorana fermions operators by a Jordan–Wigner transformation.184 (Majorana fermions are uncharged propagating degrees of freedom with quantum statistics described by anti-commuting self-adjoint quantum operators that square to 1). This yields the phase diagram in the form of the triangle shown in Fig. 6c for half the Brillouin zone, where for |Kx| < |Ky| + |Kz|, |Ky| < |Kx| + |Kz| and |Kz| < |Kx| + |Ky| under the constraint |Kx| + |Ky| + |Kz| = 1, the spectrum is gapless whilst the rest of the Brillouin zone is gapped.179 The vertices of the triangle correspond to |Kz| = 1, |Kx| = |Ky| = 0, |Kx| = 1, |Ky| = |Kz| = 0 and |Ky| = 1, |Kx| = |Kz| = 0.
In the gapped phase, spin correlations decay exponentially over a length scale inversely proportional to the gap. Thus, fermionic, vortex or quasi-particle (fermion + vortex) excitations do not have long-range interactions. However, they can interact topologically as they move around each other (their worldlines in three-dimensional (3D) space satisfy specific braiding rules).179,185,186 Thus, the energy gap favours topological interactions which reveal their Abelian anyonic statistics (id est, the quantum phases acquired by particle exchange is additive). This carries major implications in topological quantum computing as it suggests the possibility of achieving the stabilisation of quantum bits (qubits), considering that any such uncharged system is extremely hard to disturb, exempli gratia by a photon, because photons do not couple to uncharged quasi-particles. Moreover, the quantum state is topologically protected by the energy gap which cuts off any other non-topological interactions. Hence, quantum computation can be carried out topologically by particle exchange exploiting the braiding rules.179,185
On the other hand, the quasi-particles in the gapless phase will have long-range interactions. Nonetheless, these can be suppressed by introducing a gap via a time-reversal symmetry breaking term  in Hamiltonian H(J1 = 0) in eqn (1a), corresponding to the interaction of the spins with an external magnetic field hγ = (hx,hy,hz). Since this term destroys the exact solubility of the Kitaev model, the model is solved perturbatively to yield a gap Δ ∼ hxhyhz/K2, where K ≡ Kx = Ky = Kz.179 Such a gapped phase admits non-Abelian quasi-particles, that are even more robust for quantum computing than the Abelian quasi-particles (id est, the quantum phase acquired by the wavefunction under the exchange of quasi-particles is non-additive).
in Hamiltonian H(J1 = 0) in eqn (1a), corresponding to the interaction of the spins with an external magnetic field hγ = (hx,hy,hz). Since this term destroys the exact solubility of the Kitaev model, the model is solved perturbatively to yield a gap Δ ∼ hxhyhz/K2, where K ≡ Kx = Ky = Kz.179 Such a gapped phase admits non-Abelian quasi-particles, that are even more robust for quantum computing than the Abelian quasi-particles (id est, the quantum phase acquired by the wavefunction under the exchange of quasi-particles is non-additive).
3.3 Kitaev–Heisenberg model
Based on the robustness of the ungapped phase (K ≡ Kx = Ky = Kz) under a finite magnetic field (![[h with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0068_20d1.gif) ≠ 0) for quantum computing, it is illustrative to consider the behaviour of this ungapped phase in eqn (1a) with a finite Heisenberg term (J1 ≡ J ≠ 0) and no magnetic field ( = 0),
≠ 0) for quantum computing, it is illustrative to consider the behaviour of this ungapped phase in eqn (1a) with a finite Heisenberg term (J1 ≡ J ≠ 0) and no magnetic field ( = 0), | (1b) |
 and J = κ
and J = κ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosφ and K = κsinφ, to yield 1) a phase diagram of the Kitaev–Heisenberg model with two distinct gapless K-QSL (disordered) phases around (φ = π/2 and φ = 3π/2 where J ≃ 0, and 2) four ordered phases given by Néel antiferromagnetic, ferromagnetic, zigzag and stripy phases, as shown in Fig. 7. Distinct phase transitions between two phases occur at peaks corresponding to −∂2E0/∂φ2, where E0 is the ground-state energy of the Kitaev–Heisenberg model, with the smallest peaks occurring at two ordered–disordered phase boundaries, signifying a relatively smoother transition compared to the typical ordered–ordered phase transitions.
cosφ and K = κsinφ, to yield 1) a phase diagram of the Kitaev–Heisenberg model with two distinct gapless K-QSL (disordered) phases around (φ = π/2 and φ = 3π/2 where J ≃ 0, and 2) four ordered phases given by Néel antiferromagnetic, ferromagnetic, zigzag and stripy phases, as shown in Fig. 7. Distinct phase transitions between two phases occur at peaks corresponding to −∂2E0/∂φ2, where E0 is the ground-state energy of the Kitaev–Heisenberg model, with the smallest peaks occurring at two ordered–disordered phase boundaries, signifying a relatively smoother transition compared to the typical ordered–ordered phase transitions.
 | ||
| Fig. 7 Various spin configurations that can be realised in honeycomb layered frameworks, described by the Kitaev–Heisenberg Hamiltonian (HKH) in eqn (1b) with coupling constants parametrised by angular variable φ, entailing magnetic atoms (transition metals that are magnetic such as Ni, Fe, Co, etc.), based on the phase diagram of the Kitaev–Heisenberg model. Phases of the Kitaev–Heisenberg model (ferromagnetic, stripy, zigzag, Néel ferromagnetic, etc.) are shown with the direction of the electron spins in the honeycomb lattice. The green arrows show the spin-up, whereas the brown arrows show spin-down alignment of the magnetic moment of the transition metal atoms. | ||
Achieving the Hamiltonian given in eqn (1b) in a condensed matter system with J = 0 is considered to be the Holy Grail of topological quantum computing.179,186 Honeycomb layered oxides consisting of alkali or coinage metal atoms sandwiched between slabs exclusively made of transition metal and chalcogen (or pnictogen) atoms, which are the prime focus of this review, typically exhibit Néel antiferromagnetic properties,15–21,23–28,31,32,34,35,37–53,60–62,91 which suggests that the Heisenberg term (J) dominates over the Kitaev term (K). Nonetheless, observing the K-QSL phase in honeycomb layered oxides based on magnetic transition atoms with 3d orbitals such as M = Co, has shown great promise due to the localised nature of the magnetic electrons which favour the charge-transfer phase188 of the Mott insulator – a prerequisite for the realisation of the Kitaev–Heisenberg model. For instance Na2Co2TeO6, has been shown to be favoured by the suppression of non-Kitaev interactions with rather moderate external magnetic fields, since external magnetic fields vastly suppress the Haldane-type and higher-order interactions.189–192 Likewise, Na3Co2SbO6 has shown a rather promising route to the realisation of K-QSL via strain or pressure control.188 Conversely, it is also possible to suppress the Kitaev-type interactions instead by designing the honeycomb lattice to be composed of alternating magnetic and non-magnetic atoms, leaving only Haldane-type interactions corresponding to additional interaction terms in the Hamiltonian;193 effectively attaining a quantum anomalous Hall insulator (or also referred to as a Chern insulator).194,195
Based on the immense interest generated by the so-called Kitaev materials,187,188,190,196–204 we shall endeavour to briefly digress into this different class of honeycomb layered oxides, namely the honeycomb iridium oxides (iridates) embodied by A2IrO3 with A = Na, Li or Cu187,197–224 and other related compounds such as RuCl3, which also hold promise to realising the Kitaev–Heisenberg Hamiltonian given in eqn (1b). These materials exist in different phases (polymorphs), often labelled with a prefix α-, β- or γ-signifying the layering of the honeycomb sublattices which facilitate the existence of stacking faults. For instance, Li2IrO3 is polymorphic with three distinct phases, α-Li2IrO3, β-Li2IrO3 and γ-Li2IrO3 which adopt the honeycomb, hyper-honeycomb and stripy honeycomb crystal structures, respectively.203,204,211,214,225–229 α-Li2IrO3 exhibits Néel antiferromagnetic behaviour below their Néel temperature (15 K), and is paramagnetic above 15 K.230 Its crystalline structure consists of IrO6 octahedra arranged in a honeycomb fashion and separated by Li+ cations, with other Li+ cations situated within the honeycomb slab. On the other hand, RuCl3 also exists in two main polymorphic phases, α-RuCl3 and β-RuCl3. However, β-RuCl3 is prepared under specific conditions at low temperatures, and irreversibly reverts to the more stable polymorphic counterpart, α-RuCl3 at approximately 500 °C. The RuCl3 layers consist of RuCl6 octahedra linked by van der Waals forces forming interlayers devoid of any cations. The discovery of high-temperature K-QSL (half-integer thermal hall conductivity) in α-RuCl3 has brought such materials into the forefront of the pursuit of the non-Abelian K-QSL231–234 and its related applications to topological quantum computing.235 In particular, the magnetic spins, originating from the Ir4+ ions or Ru3+ surrounded by the ligand O2− or Cl− ions respectively, lead to a K < 0 (ferromagnetic) Kitaev interaction via the celebrated Jackeli–Khaliullin mechanism.236
3.4 Realising the Kitaev interaction term
To have a qualitative understanding of Jackeli–Khaliullin mechanism, we briefly offer an intuitive picture of the relevant crystal structure, energy scales and interactions that are predicted to give rise to the Kitaev coupling constant K in eqn (1b). Whenever the valence electron–electron Coulomb (Hubbard) interaction strength U in a (semi-)metal is greatly larger than their ion-to-ion hopping rate t (id est, U ≫ t ≃ 0), the valence electrons tend to localise proximal to their parent ions forming a Mott insulator.237,238 On the other hand, their spins will transition from a disordered state (in this case, paramagnetic) to an ordered configuration (viz., ferromagnetic, stripy, zigzag, Néel ferromagnetic, etc.) below a transition temperature, with their magneto-spin dynamics governed to leading order by the Heisenberg term J in eqn (1b) where K ≃ 0. However, the hopping rate t is small but finite (id est in honeycomb layered materials such as iridates and α-RuCl3, U ≫ t ≠ 0), which warrants the modification of the Heisenberg model.61,188,199,201,204,212,216–224,239In iridates, the Kitaev interaction term is predicted to arise from the d orbitals of the Ir4+ ion containing 5 electrons (5d5).61,188,239 In particular, from a solid-state chemistry perspective, the 5d orbital of a free Ir4+ ion is 10-fold degenerate (dx2−y2, dz2, dxy, dyz and dxz, spin 1/2). This degeneracy is lifted by crystal field splitting, into a 4-fold degenerate eg orbital (dx2−y2 and dz2, spin 1/2) and a lower energy 6-fold degenerate t2g orbital (dxy, dyz and dxz, spin 1/2) when a Ir4+ ion is bonded with the ligand O2− ions forming the octahedral structure depicted in Fig. 6b. Since the ligand O2− ions approach the Ir4+ ions at the centre of the octahedra along the x, y and z axes, this finite energy difference Δ between the eg and t2g orbitals arises from the fact that the electrons in the dx2−y2 and dz2 orbitals have a greater Coulomb repulsion compared to the electrons in the dxy, dyz and dxz orbitals. The triplet t2g orbital experiences a further splitting into a ground jeff = 3/2 doublet state and an excited jeff = 1/2 singlet state due to spin–orbit coupling. The jeff = 1/2 state is composed of a linear combination of spin 1/2 and orbital (dxy, dyz, dxz) entangled states.
For brevity, spin–orbit coupling (SOC) is a relativistic effect where, in the inertial frame of an electron orbiting a stationary nucleus generating an electric field ![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) at a distance |
at a distance |![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) | = r with momentum
| = r with momentum ![[p with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0070_20d1.gif) , there is a finite magnetic field
, there is a finite magnetic field ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) ∝ × = || × /r, that couples to the spin of the electron, thus generating a magnetic moment proportional to · ∝ λ
∝ × = || × /r, that couples to the spin of the electron, thus generating a magnetic moment proportional to · ∝ λ![[L with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004c_20d1.gif) · = HSOC where = × is the angular momentum of the electron and λ is the spin–orbit coupling strength.201,222,223 This energy splitting can be written in terms of the total angular momentum
· = HSOC where = × is the angular momentum of the electron and λ is the spin–orbit coupling strength.201,222,223 This energy splitting can be written in terms of the total angular momentum ![[J with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004a_20d1.gif) = + as HSOC = λ(J2 − L2 − S2)/2, where we have used J2 = ( + )2 = L2 + 2· + S2. Note that the p orbital splitting (states labelled by total quantum number j) will differ from the t2g orbital splitting (states labelled by effective total quantum number jeff) by the sign of λ. Thus, taking the spin and angular momentum quantum numbers respectively as s = 1/2 and l = 1 and using 〈L2〉 = l(l + 1), 〈S2〉 = s(s + 1/2) and 〈J2〉 = j(j + 1) where j = l ± s, the energy splitting between the ground j = 1/2 singlet state and the excited j = 3/2 doublet state becomes 〈HSOC〉 = 3λ/2.240
= + as HSOC = λ(J2 − L2 − S2)/2, where we have used J2 = ( + )2 = L2 + 2· + S2. Note that the p orbital splitting (states labelled by total quantum number j) will differ from the t2g orbital splitting (states labelled by effective total quantum number jeff) by the sign of λ. Thus, taking the spin and angular momentum quantum numbers respectively as s = 1/2 and l = 1 and using 〈L2〉 = l(l + 1), 〈S2〉 = s(s + 1/2) and 〈J2〉 = j(j + 1) where j = l ± s, the energy splitting between the ground j = 1/2 singlet state and the excited j = 3/2 doublet state becomes 〈HSOC〉 = 3λ/2.240
Thus, 4 electrons (2 spin-up and 2 spin-down states) occupy the jeff = 3/2 doublet state and the remaining 1 electron (spin-up or spin-down) occupies the jeff = 1/2 singlet state, leaving a spin-down or spin-up singlet state unoccupied.224 It is this unoccupied state (hole) which has a finite U > t ≠ 0 hopping rate from one Ir4+ ion to an adjacent one. Thus, one condition sine qua non for the realisation of the Jackeli–Khaliullin mechanism is the existence of the jeff = 1/2 singlet state in a 5d transition metal ion such as Ir4+ and Ru3+, bonded with ligand ions such as O2− and Cl2− forming an octahedron.61
Since hopping occurs from the jeff = 1/2 state of a transition metal ion directly to an adjacent one (labelled as t1,3 in Fig. 6b) or indirectly via the p orbitals of a ligand ion (labelled as t2 in Fig. 6b), the second condition for the realisation of the Jackeli–Khaliullin mechanism concerns the geometric orientation of adjacent octahedra. In particular, the adjacent octahedra in honeycomb layered materials can either share a vertex (180° bond) or an edge (90° bond), which leads to either a single indirect t2 hopping path or a pair of equivalent t2 hopping paths respectively. It is the pair of t2 hopping paths in the 90° bond which results in the Jackeli–Khaliullin mechanism. This is because the Kanamori Hamiltonian224 describing the Mott insulator237,238 is perturbed by the hopping Hamiltonian with terms resulting from the pair of t2 paths, leading to destructive interference of the symmetric Heisenberg exchange terms but constructive interference of the asymmetric Kitaev exchange terms. Such a calculation results in a ferromagnetic (K < 0) Kitaev term,196,224

Typically, since spin–orbit coupling is greater for large transition metal atoms with 4d or 5d orbitals, much of the search for K-QSL has focused on their jeff = 1/2 state. Based on the Jackeli–Khaliullin mechanism, calculations are often conducted via complementary spin-wave analysis and exact diagonalisation, which indeed reveal that Kitaev coupling constant is ferromagnetic (K < 0) for A2IrO3 when (A = Li, Na).207 Unfortunately, the finite direct t1,3 hopping from the jeff = 1/2 state have been shown to restore the Heisenberg term as well as introduce another bond-independent coupling, often labelled by Γ.224 Thus, these non-Kitaev terms hinder the prospects of realising K-QSL with the conventional Jackeli–Khaliullin mechanism. Moreover, crystalline distortions of the octahedra, exempli gratia Jahn–Teller distortions introduce long-range order that smears out the K-QSL phase, hence hindering its experimental realisation.241,242
This has lent further impetus to the consideration of realisation of honeycomb layered materials beyond the Jackeli–Khaliullin mechanism, which rely not only on f orbitals but also on d orbitals.61,188,190,243 In particular, honeycomb layered oxides entailing for instance high-spin 3d7 ions such as Co2+, Fe+ and Ni3+ are considered as apposite models.61,188 Since Fe+-based honeycomb layered oxides are difficult to stabilise in the chemical compositions enumerated in Section 2, it leaves Co2+-based honeycomb layered oxides entailing pnictogen or chalcogen atoms such as A3Co2SbO6, A3Co2BiO6, A2Co2TeO6, A4CoTeO6 (where A can be Li, Na, K, Ag, Cu, Rb, etc.), amongst others as promising candidates. As for Ni3+ compounds, they can be synthesised, for instance via the (electro)chemical oxidation of Ni2+-based honeycomb layered oxides such as A2Ni2TeO6, A3Ni2SbO6, A3Ni2BiO6, A4NiTeO6 (A = Li, Na, K, Ag, Cu, etc.) and so forth. Indeed, honeycomb layered oxides such as Na2Co2TeO6, Na3Co2SbO6 and NaNi2BiO6−δ have generated traction in the search for K-QSL state.61,188
3.5 Beyond the Kitaev–Heisenberg model with higher-order interactions
For instance, the necessity to include the J2 and J3 Heisenberg couplings in the case of the Mott-insulating layered iridates A2IrO3 (A = Na, Li) into eqn (1b), thus further generalising the Kitaev–Heisenberg model has been tackled, exempli gratia, by Kimchi and You within the so-called Kitaev–Heisenberg-J2–J3 model.216 Generally, higher-order interactions are best observable when these layered oxides are cooled down to extremely low temperatures, where the thermal motion of the spins of the magnetic atoms is suppressed or negligibly small. At a unique magnetic ordering (transition) temperature, the spins align themselves in specific directions along the honeycomb configuration in various manners signifying a phase transition into new states of matter, as shown in Fig. 7. For example, a paramagnetic material transitions into antiferromagnetic when spins align in the same direction (parallel) or the opposite directions (antiparallel). Depending on the magnetic phase of matter they transition into, transition temperatures can be termed as Néel temperature or Curie temperature.244,245 Néel temperature is the transition temperature where antiferromagnetic materials become paramagnetic and vice versa. We shall focus on Néel temperature since a vast majority of the honeycomb layered oxides display antiferromagnetic transitions at low temperatures. The Néel temperatures for most honeycomb layered oxides (incorporating pnictogen or chalcogen atoms) tend to be at lower temperatures (below 40 K) as shown in Fig. 8.15–21,23–28,31,32,34,35,37–53,60–62,91 Another intriguing manifestation of anti-ferromagnetism is the manner in which the antiparallel spins align in the honeycomb configuration. The antiparallel spins may assume various conformations such as zigzag ordering or alternating stripe-like (stripy) patterns within the honeycomb slab. Zigzag spin structure has been observed in honeycomb layered oxides, such as Li3Ni2SbO6, Na3Co2SbO6, Na2Co2TeO6, amongst others.22,29,33,36,52,77 | ||
| Fig. 8 Magnetic transition temperatures of representative honeycomb layered oxide materials. (a) Various magnetic transition temperatures attained in honeycomb layered oxides that entail a change in spin configuration to antiferromagnetic states (videlicet., Néel temperature). (b) The magnetic transition temperatures of Na-based honeycomb layered oxides (that have mostly been subject of passionate research owing to their intriguing magnetism) has been highlighted for clarity to readers.15–21,23–28,31,32,34,35,37–53,60–62,91 | ||
Correspondingly, competing magnetic interactions on honeycomb lattices may induce both antiferromagnetic and ferromagnetic spin re-ordering, with the latter dominating when an external magnetic field is applied; a process referred to as spin-flop magnetism, observed in oxides such as Na3Co2SbO6 and Li3Co2SbO6.18 Moreover, depending on the distance between the spins, spiral-like or helical spin arrangements may result. Competing interactions or ‘frustrations’ may also cause the spins in a honeycomb lattice to orient haphazardly (magnetic disorder), even at low temperatures, leading to a plenitude of exotic magnetic states such as spin-glasses and spin-flop behaviour as has been noted in oxides such as Li3Co2SbO6.75,246 Complex magnetic phase diagrams as well as enigmatic interactions (Heisenberg–Kitaev interactions, Dzyaloshinskii–Moriya (DM) interactions, etc. are discussed in the last section of this review) can be envisaged in the honeycomb layered oxides.241 For instance, the Heisenberg–Kitaev model describes the magnetism in honeycomb lattice Mott insulators with strong spin–orbit coupling. An asymmetric (DM) spin interaction term in the Heisenberg–Kitaev model can be shown to lead to (anti-)vortices-like magnetic nanostructures commonly referred to as magnetic skyrmions that act as one of possible solutions describing equilibrium spin configurations in ferromagnetic/antiferromagnetic materials.247 The binding of these vortex/anti-vortex pairs over long distances in 2D constitutes a higher-order interaction that becomes finite at a certain temperature when these materials undergo a Berezinskii, Kosterlitz and Thouless (BKT) transition – an example of a topological phase transition.38,248 The possibility of these (and more) higher-order interactions demonstrates that there is room for both experimentalists and theorists in physics and chemistry to expand the pedagogical scope of honeycomb layered oxides.
4 Solid-state ion diffusion in honeycomb layered oxides
High ionic conductivity is a prerequisite for superfast ionic conductors that may serve as solid electrolytes for energy storage devices. The presence of mobile alkali atoms sandwiched in honeycomb slabs, as is present in honeycomb layered oxides, endows them with fast ionic conduction not only at high temperatures but also at room temperature. The ionic conductivity of honeycomb layered oxides reported to date, with the tellurate-based honeycomb layered oxides exhibiting the highest conductivity so far is shown in Fig. 9.15,59,90,173,249–255,257,258 Experimentally, the measurement of ionic conductivity is conducted via linear response techniques with polarised electromagnetic fields such as electrochemical impedance spectroscopy (EIS).259 EIS entails applying a low amplitude low frequency oscillating voltage (current) and measuring the current (voltage) response. The current-to-voltage ratio determines the inverse of the impedance (admittance) of the material, where the real part of the admittance Y(ω) = σ(ω) + iωε is proportional to the conductivity σ(ω) and the imaginary part is proportional to the permittivity ε of the material.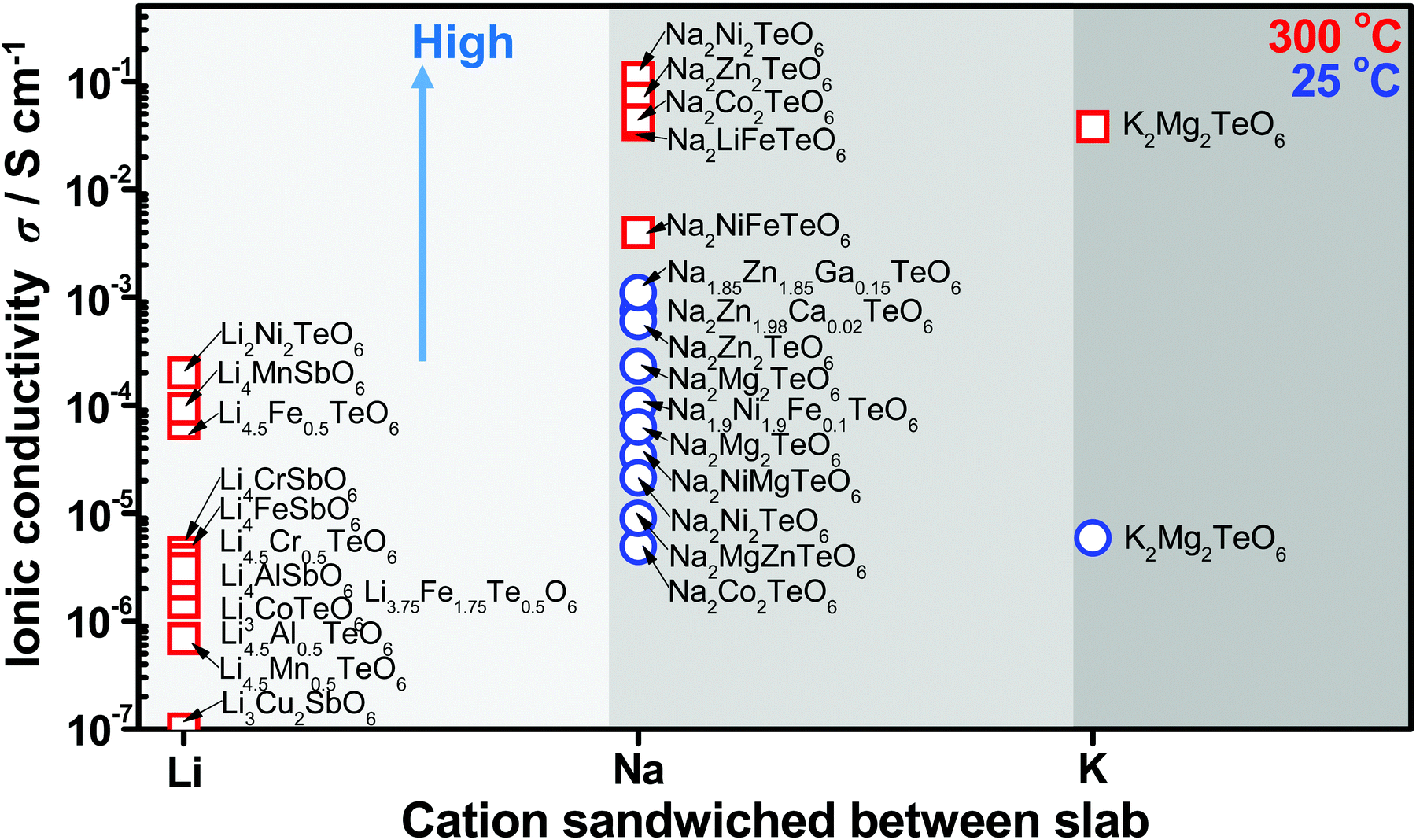 | ||
| Fig. 9 Solid-state diffusive properties of typical cations sandwiched between honeycomb slabs of various layered oxides showing values of ionic conductivity attained in honeycomb layered oxides at room temperature and also at high temperature (300 °C).59,90,173,249–255 Honeycomb layered oxides based on tellurates generally tend to show high ionic conduction, owing to the partial occupancy of alkali atoms in distinct crystallographic sites that facilitate rapid hopping diffusion mechanism. Inset shows the plot of the dependency of (normalised) conductance σ to (normalised) thermal energy kBT of the cations determined by linear response of the diffusion current to low amplitude, slowly oscillating voltage by electrochemical impedance spectroscopy (EIS). | ||
4.1 Heuristics of cationic diffusion
In order to rationalise the heuristics behind the high ionic conductivity of the honeycomb layered oxides and predict associated outcomes, it is imperative to introduce a detailed theoretical approach that incorporates the thermodynamics of the cations. In particular, the connection between the ionic conductivity of honeycomb layered oxides and other physical measurable quantities such as the diffusion of solid-state alkali cations at thermal equilibrium and very low frequencies undergoing Brownian motion satisfies the fluctuation–dissipation theorem.260 Here, we showcase this approach based on heuristic arguments that captures the diffusion aspects of ionic conductivity of the (honeycomb) layered materials.261Ionic conductivity of A cations can be heuristically modeled under a Langevin–Fick framework of equations,262,263
 | (2a) |
 | (2b) |
exp(−βEa) is the Arrhenius equation relating the diffusion coefficient to the activation energy (per mole) of diffusion (Ea), D0 is the maximal diffusion coefficient, β = 1/kBT is the inverse temperature, T is the temperature at equilibrium, kB is the Boltzmann constant, ρ(t,![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0078_20d1.gif) ) is the concentration of alkali cations,
) is the concentration of alkali cations, ![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) is their velocity vector and V(t,) is a time-dependent voltage distribution over the material. Imposing charge and momentum conservation,
is their velocity vector and V(t,) is a time-dependent voltage distribution over the material. Imposing charge and momentum conservation,  and d/dt = 0 respectively, and assuming the ionic concentration satisfies the Boltzmann distribution at thermal equilibrium, ρ(T,t,) = ρ0exp(−βqV(t,)) (where ρ0 is the ionic density at zero voltage) leads to the ionic conductivity σ = qμρ proportional to the mobility μ of the alkali cations, which satisfies the fluctuation–dissipation relation μ = βD first derived by Einstein and Smoluchowski to describe particles undergoing Brownian motion (diffusion).264–266 Based solely on eqn (2), the ionic conductivity of the alkali cations of honeycomb layered oxides, as summarised in Fig. 9 and Table 2, is related to the equilibrium temperature of the materials.
and d/dt = 0 respectively, and assuming the ionic concentration satisfies the Boltzmann distribution at thermal equilibrium, ρ(T,t,) = ρ0exp(−βqV(t,)) (where ρ0 is the ionic density at zero voltage) leads to the ionic conductivity σ = qμρ proportional to the mobility μ of the alkali cations, which satisfies the fluctuation–dissipation relation μ = βD first derived by Einstein and Smoluchowski to describe particles undergoing Brownian motion (diffusion).264–266 Based solely on eqn (2), the ionic conductivity of the alkali cations of honeycomb layered oxides, as summarised in Fig. 9 and Table 2, is related to the equilibrium temperature of the materials.
| Compound | σ 573K/S cm−1 (300 °C (573 K)) | σ 300K/S cm−1 (25 ± 3 °C (298 ± 3 K)) | E a/eV | Pellet compactness/% |
|---|---|---|---|---|
| K2Mg2TeO6 | 3.8 × 10−2 | ∼10−5 | 0.92 | ∼70 |
| Li2Ni2TeO6 | 2.0 × 10−4 | 0.80 (333–573 K) | ||
| Li3Co1.06TeO6 | 1.6 × 10−6 | |||
| Li3Cu2SbO6 | 1.0 × 10−7 | |||
| Li3.5Zn1.5BiO5.75 | 1.13 × 10−5 | 0.37 (373–573 K) | ||
| Li3.75Fe1.75Te0.5O6 | 2.21 × 10−6 | 0.60 | ||
| Li4CrSbO6 | 4.31 × 10−6 | 0.66 (300–573 K) | ||
| Li4FeSbO6 | 3.66 × 10−6 | 0.57 (300–573 K) | ||
| Li4MnSbO6 | 9.33 × 10−5 | 0.57 (300–573 K) | ||
| Li4AlSbO6 | 3.05 × 10−6 | 0.91 (300–573 K) | ||
| Li4.5Cr0.5TeO6 | 3.24 × 10−6 | 0.53 (373–573 K) | ||
| Li4.5Mn0.5TeO6 | 6.88 × 10−7 | 0.73 (373–573 K) | ||
| Li4.5Al0.5TeO6 | 1.49 × 10−6 | 0.66 (373–573 K) | ||
| Li4.5Fe0.5TeO6 | 6.76 × 10−5 | 0.70 | ||
| Na1.9Ni1.9Fe0.1TeO6 | 4.7 × 10−2 | 1 × 10−4 | 0.38 (373–623 K) | 72 |
| Na2Zn2TeO6 | (5.1–7.0) × 10−2 | 9 × 10−5 | 55–68 | |
| Na2Co2TeO6 | 4.4 × 10−2 | |||
| Na2Co2TeO6 | (3.1–4.4) × 10−2 | (3.8–4.9) × 10−6 | 0.52 (373–623 K) | 56–82 |
| Na2Mg2TeO6 | 2.3 × 10−2 | 6.3 × 10−5 | 74 | |
| Na2Mg2TeO6 | 2.3 × 10−4 | 0.341 (323–393 K) | 87.2 | |
| Na2NiFeTeO6 | ∼4.0 × 10−3 | 0.46–0.49 (343–663 K) | ||
| Na2NiFeTeO6 | 4.0 × 10−3 | |||
| Na2NiMgTeO6 | 2.13 × 10−5 | 0.59 (T < 303 K) | ||
| Na2MgZnTeO6 | 9 × 10−6 | 0.36 (T < 303 K) | ||
| Na2Zn2TeO6 | (6.29–7.54) × 10−4 | |||
| Na2Zn2TeO6 | ∼6 × 10−4 | |||
| Na2Zn2TeO6 | 5.7 × 10−4 | |||
| Na2−xZn2−xGaxTeO6 (x = 0.15) | (6.29–10.9) × 10−4 | |||
| Na2Zn2−xCaxTeO6 (x = 0–0.05) | 7.54 × 10−4 (x = 0.02) | |||
| Na2Zn2TeO6 (Ga-doped) | 8.3 × 10−4 | |||
| Na2Ni2TeO6 | (1.01–1.08) × 10−1 | (8–34) × 10−4 | 0.55 (373–623K) | 79.6–80.3 |
| Na2Ni2TeO6 | 2 × 10−6 (323 K) | ∼0.58(3) (T ≥ 383 K), 0.39 (T < 353 K) | 90 | |
| Na2LiFeTeO6 | 4 × 10−2 | 0.44–0.49 (343–663 K) | ||
In particular, the ionic conductivity computes to σ(T,t,) = qμρ(T,t,) = qD0ρ0βexp(−β{Ea + qV(t,)}). Plotting the ionic conductivity versus the normalised temperature kBT/(Ea + qV), we find that the ionic conductivity scales with the equilibrium temperature in the regime kBT/(Ea + qV) ∼ kBT/Ea, which is always satisfied in EIS measurements. For kBT/Ea < 1, raising the temperature increases the thermal motion of the cations, which in turn raises the ionic conductivity. The ionic conductivity attained in honeycomb layered oxides at room temperature (25 °C) and also at high temperature (300 °C) is shown in Fig. 9, which showcases the increase of ionic conductivity with temperature as expected.
4.2 Correlation between interslab distances and cationic diffusion
Moreover, classical motion of the alkali cations and other electromagnetic interactions along the z direction are precluded, since these materials often satisfy the condition Δz ≫ rion, where Δz is the interlayer/interslab separation distance and rion is the ionic radius of the alkali cations. This condition effectively restricts the electrodynamics in these layered materials to two dimensions (2D), and is almost always satisfied since the ionic radius of the alkali cations is correlated with interslab distance, as shown in Fig. 10a. For instance, alkali cations with large ionic radii such as K in the layered oxide K2Ni2TeO6 are restricted to the two-dimensional (2D) honeycomb diffusion channels in the ab plane (Fig. 10b), as has also been shown in Na2Ni2TeO6.267–269 Thus, the large interslab separation, together with the TeO6 octahedra acts as a barrier preventing inter-channel exchange of the alkali cations.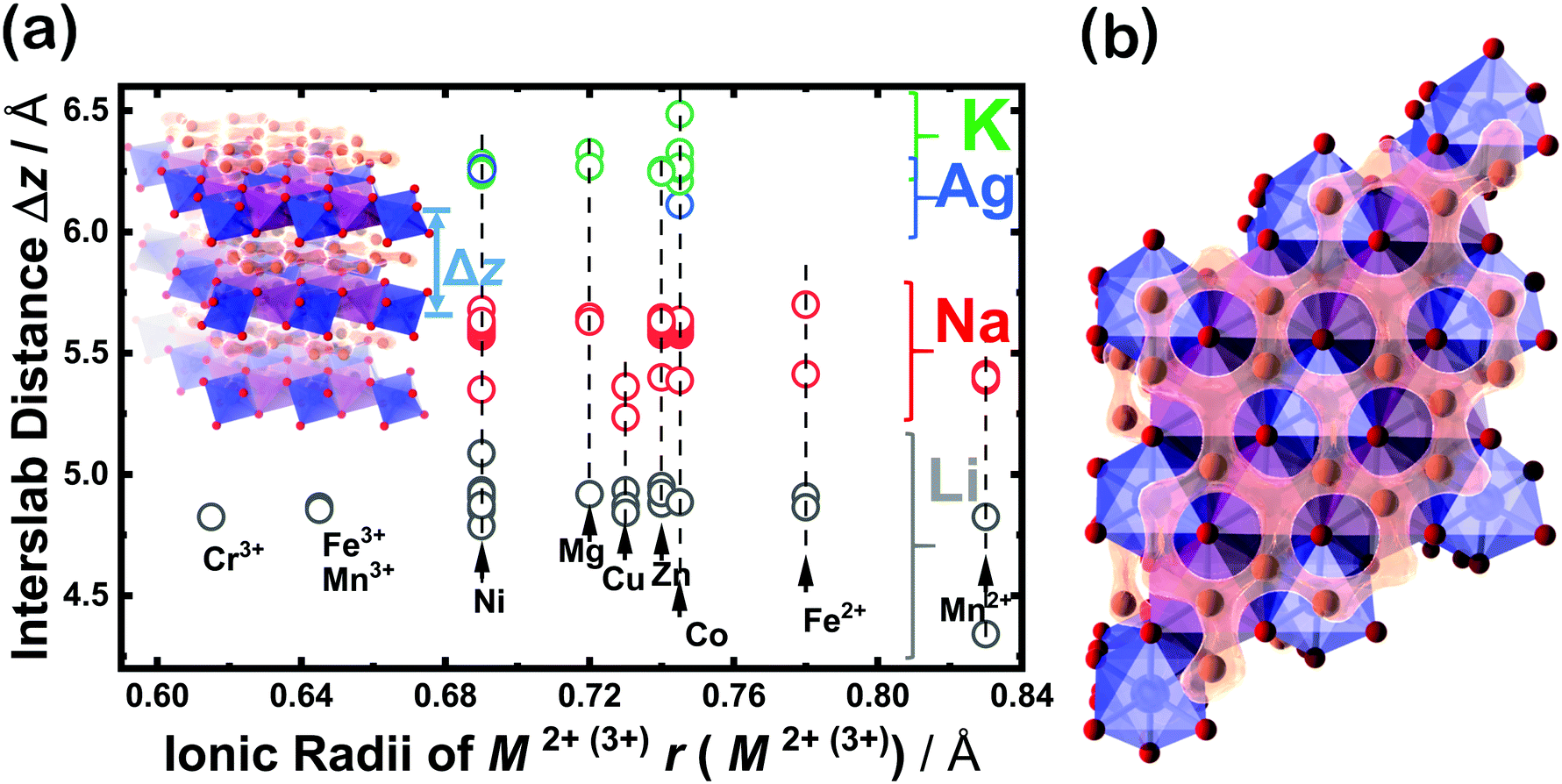 | ||
| Fig. 10 Regarding the honeycomb interslab distance and the size of sandwiched alkali or coinage metal atoms. (a) Correlation of the average interslab distance (Δz) and the size (id est, the Shannon–Prewitt ionic radius) of the sandwiched alkali ion (A) in honeycomb layered oxides adopting the following compositions: A2+M22+D6+O6 (A2/3+M2/32+D1/36+O2), A3+M22+D5+O6 (A+M2/32+D1/35+O2), A4+M3+D5+O6 (A4/3+M1/33+D1/35+O2), A4+M2+D6+O6 (A4/3+M1/32+D1/36+O2), etc. where M = Fe, Mn, Co, Ni, Cu, Zn, Mg or a combination of at least two transition or alkaline-earth metal atoms; D = Te, Sb, Bi, Nb; A = Cu, Ag, Li, Na, K.16,22,32,59,66,71,75,79,86,92,130,168,256 For clarity's sake, the error bars associated with each data point are smaller than the size of the data markers. (b) A fragment of a honeycomb layered oxide such as K2Ni2TeO6 in the ab plane, showing the two-dimensional (2D) diffusion channels of potassium ions. This figure also shows that apart from the type of alkali or coinage metal atom, where profound change in the interlayer distance can be expected, the nature of the transition metal atom M also influences the interlayer distance albeit to a smaller extent in some instances. | ||
Other factors that affect the ionic conductivity of the cations include the ionic radius of the A cations in relation to the M atoms. For instance, in the case of Li2Ni2TeO6 (where M = Ni) in comparison to Na2Ni2TeO6 and K2Ni2TeO6, Ni atoms act as impurities in the diffusion dynamics of A = Li since the interlayer separation distance in Li2Ni2TeO6 is vastly smaller compared to Na2Ni2TeO6 and K2Ni2TeO6. Electrochemically, collisions with such impurities in these honeycomb layered oxides are suppressed by the larger interslab distance in conjunction with the greater sizes of Na and K atoms relative to Li, which ensures their facile mobility within the two dimensional planes. In contrast, the smaller interslab distance and the equivalent atomic sizes of Li and Ni atoms, which lie at close proximity to the honeycomb slabs leads to the interchange of their crystallographic sites (commonly referred to as Li/Ni ‘cationic mixing’).270 Consequently, the mobility of Li is obstructed through the collisions with Ni atoms within the 2D honeycomb surface that act as impurities, a process quantum mechanically referred to as scattering. Within our heuristic approach, scattering of Li ions through collisions with Ni costs more (activation) energy than in the case of Na or K, (ELia ≫ ENaa,EKa). Thus, the Einstein–Smoluchowski relation μ = D/kBT together with Arrhenius equation D = D0exp(−Ea/kBT) leads to a smaller ionic mobility in the case of A = Li ions. This trend is indeed shown in Fig. 9 and Table 2, wherein Li-based honeycomb layered oxides, regardless of the temperature, still show inferior ionic conductivity compared to those with Na or K.
5 Electrochemistry of honeycomb layered oxides
5.1 Theoretical basis for high voltages in honeycomb layered oxides
The layered structure consisting of highly oxidisable 3d transition metal atoms in the honeycomb slabs segregated pertinently by alkali metal atoms, renders this class of oxides propitious for energy storage. In principle, classical battery electrodes rely on the oxidation (or reduction) of constituent 3d metal cations to maintain charge electro-neutrality, thus facilitating the extraction (or reinsertion) of alkali metal cations, a process referred to as ‘charge-compensation’.271 In principle, the constituent pentavalent or hexavalent d0 cations (such as Bi5+, Sb5+, Te6+, W6+ and so forth) do not participate in the charge-compensation process during battery performance. However, the highly electronegative WO66− (or TeO66−, WO66−, BiO67−, SbO67−, RuO67−, etc.) anions lower the covalency of the bonds formed between the oxygen (O) atoms and 3d transition metal (M) resulting in an increase the ionic character of M–O bonds within the honeycomb layered oxides.272 Consequently, the energy required to oxidise M cations increases, inducing a staggering increase in the voltage of related honeycomb layered oxides within the battery. This process is commonly referred to as ‘inductive effect’.273 For clarity, the electronegativity trend (based on the Pauling scale) is generally as follows: W > Ru > Te > Sb > Bi. Honeycomb layered oxides such as Li4Ni2+TeO6, and more recently, Li2Ni22+TeO6, manifest higher voltages (over 4 V) in comparison to other layered oxides or compounds containing Ni2+.26,55,56 This is rationalised by considering Te6+ as a TeO66− moiety, which being more electronegative than anions such as O2−, increases the voltage necessary to oxidise Ni2+ (or technically as redox potential) through the inductive effect (as succinctly shown in Fig. 11a).55 Voltage increase due to this inductive effect has, in particular, been noted in polyanion-based compounds when (SO4)2− are replaced either by (PO4)3− or (PO4F)4− anion moieties.274 Therefore, the inductive effect seems to be typical and represents a crucial strategy when tuning the voltages of honeycomb layered oxides. Indeed, besides Li2Ni2TeO6 and Li4NiTeO6, analogues consisting of Na (such as Na2Ni2TeO6 and Na4NiTeO6) and K alkali atoms (such as K2Ni2TeO6 and K4NiTeO6) also exhibit high voltages surpassing those of layered oxides in their respective fields.57,59,65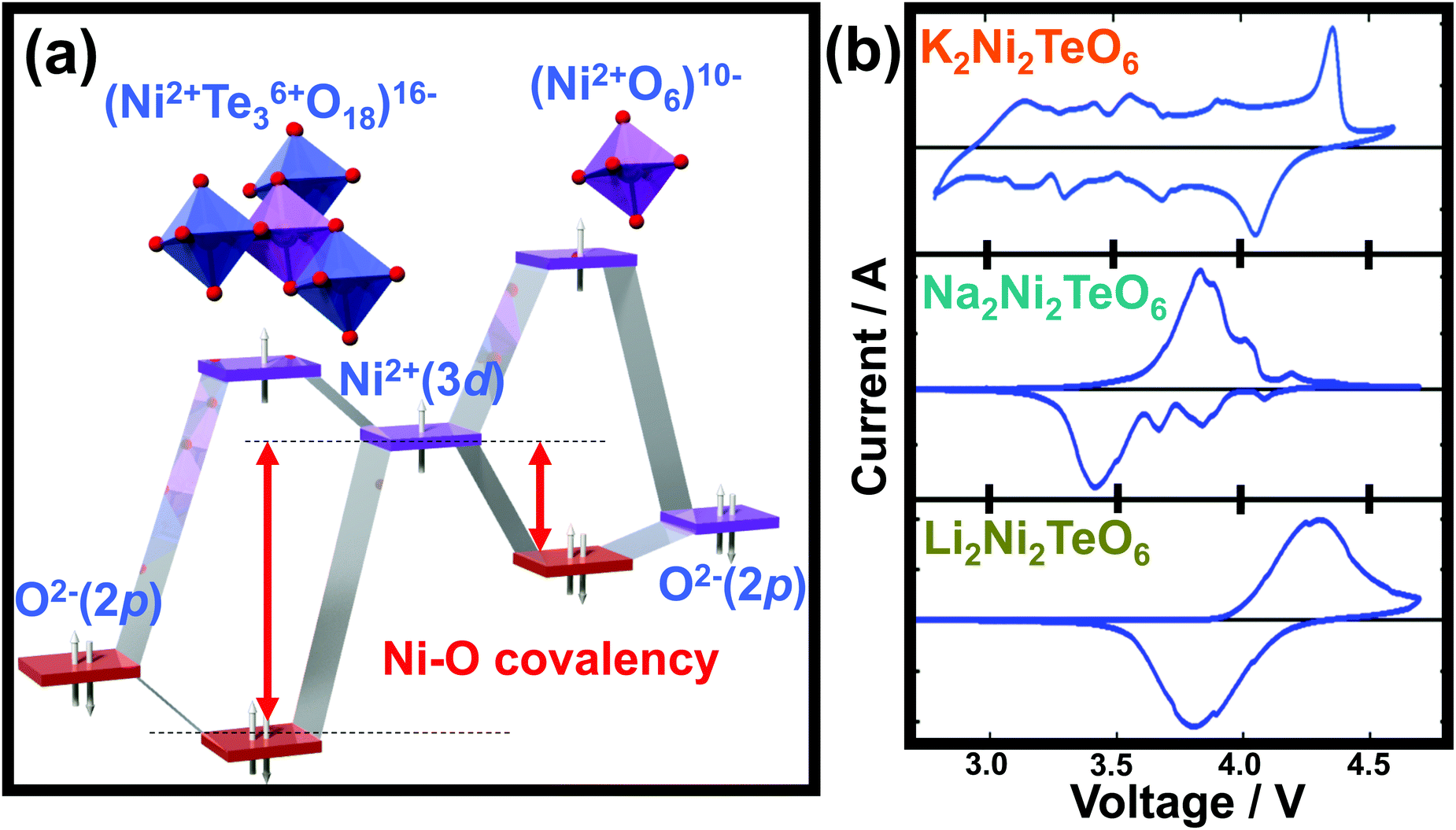 | ||
| Fig. 11 High-voltage electrochemistry of honeycomb layered oxides. (a) Molecular orbital calculations of the voltage increase arising from the ‘inductive effect’ that alters the covalency of Ni–O bonds, due to the presence of more electronegative Te atom surrounded by a honeycomb configuration of Ni atoms. (b) Voltage-response curves (technically referred to as cyclic voltammograms) of honeycomb layered compositions (A2+M22+D6+O6 (A = Li, Na, K)), showing their potential as high-voltage cathode materials for rechargeable alkali cation batteries. Technically, these cyclic voltammograms (voltage-response curves) were plotted under a scan rate of 0.1 millivolt per second. Part of the data in (b) was adapted from ref. 59 under Creative Commons licence 4.0. | ||
5.2 Electrochemical measurements
Theoretical insights regarding the high voltage innate in the aforementioned honeycomb layered oxides are validated by experimental investigations. Typical electrochemical measurements performed include: cyclic voltammetry, which assesses the voltages during charging and discharging at which the 3d transition metal redox processes occur as well as other structural changes and galvanostatic charge/discharge measurements, which principally determine pivotal battery performance metrics, inter alia, (i) the amount of alkali atoms electrochemically extracted or inserted (id est, capacity) during charging and discharging, (ii) how fast the alkali atoms can be extracted or inserted (referred to as rate performance), (iii) voltage regimes where alkali atoms are dominantly being extracted or inserted and (iv) the nature of the extraction or reinsertion process of alkali atoms, for instance, whether it occurs topotactically (referred to as a single-phase, solid-solution or monophasic behaviour) or as a multiple phase (referred to as a two-phase or biphasic behaviour). The cyclic voltammograms of A2+Ni22+Te6+O6 (A = Li, Na and K) are illustrated in Fig. 11b, depicting voltage peaks/humps around 4 V where the redox process of Ni (in this case Ni2+/Ni3+) occur during electrochemical extraction/insertion of alkali atoms. It is noteworthy that the larger the ionic radius of A is, the more pronounced the minor voltage humps are seen. This is indicative of structural changes (phase transitions) occurring, details of which shall be elaborated in a later section. Usually the voltage response curves (cyclic voltammograms) should nicely mirror each other (taking the line where the current density is set as zero to be the mirror plane in Fig. 11b). However, due to some electrochemical issues (such as inherently slow alkali-ion kinetics), the voltages at which the redox processes or structural changes occur deviate from each other as seen in Fig. 11b. This is technically referred to as ‘voltage polarisation’ or ‘voltage hysteresis’.311 Voltage polarisation can significantly be decreased by partial substitution (or doping) of the constituent 3d transition metal atoms with isovalent metals. For instance, partial doping with Zn, Mn or Mg in Na3Ni2SbO6 leads to lower voltage polarisation compared to that of the undoped Na3Ni2SbO6.296,297 Furthermore, doped oxides present higher voltages; depicting doping as another feasible route to increase the voltages of these honeycomb layered oxides.2955.3 Suitable electrolytes for high-voltage honeycomb layered oxides
Precise and adequate evaluation of the voltage responses of high-voltage cathode materials, such as the aforementioned A2Ni2TeO6, demands the utilisation of stable electrolytes that can sustain high-voltages. Conventional electrolytes consisting of organic solvents are prone to decomposition during high-voltage operations; rendering them unsuitable for the high-voltage performance innate in such honeycomb layered oxides electrodes. Ionic liquids, which comprise entirely of organic cations and (in)organic anions, are a growing class of stable and safe electrolytes exhibiting a plenitude of distinct properties; pivotal amongst them being their good electrochemical and thermal stability, low volatility and low flammability.92,299,300 These attributes assure improved safety for batteries utilising ionic liquids. Matsumoto and co-workers were amongst the first to show exemplary performance of layered oxides such as LiCoO2 with the use of ionic liquids.298–300 Honeycomb layered oxides, such as K2Ni2TeO6 and their cobalt-doped derivatives,59,92,168 have been shown to display stable performance at high-voltage operation in electrolytes comprising ionic liquids plausible candidates of which are depicted in Fig. 12. | ||
| Fig. 12 Illustration showing a selection of electrolytes (in particular ionic liquids) which guarantee the stable electrochemical performance of honeycomb layered oxides. In principle, ionic liquids consist of organic cations (pyrrolidinium, imidazolium, piperidinium, etc.) coupled with organic or inorganic anions (such as BF4−, PF6−, ClO4−, etc.) Organic cations are shown in black, whilst organic or inorganic anions are in yellow. Purity of the salts, solubility and compatibility with the honeycomb layered oxide cathode materials, amongst other factors are necessary to consider when obtaining suitable ionic liquids for high-voltage operation. Readers may further refer to the literature for more details regarding the ionic liquids.59,92,275–294 | ||
5.4 Alkali-ion kinetics and redox processes
Fast kinetics of alkali ions within an electrode during electrochemical extraction/insertion is a crucial parametric that influences the rate performance of battery performances. For instance, previous reports have attributed the good rate performance and excellent cyclability of Na3Ni2SbO6 cathode material (Fig. 13a–c) to the fast interlayer kinetics of Na+ within the highly-ordered Na3Ni2SbO6.58,301 Honeycomb layered oxides such as Na3Ni2SbO6 also exhibit preponderant rate capabilities (as shown in Fig. 13d and e) and can sustain fast Na-ion kinetics upon successive Na-ion (de)insertion (Fig. 13f).83 Pertaining to structural stability, the manner in which 3d transition metal atoms (for example Ni atoms in Na3Ni2SbO6) are arranged in a honeycomb configuration endows it not only with good thermal stability but also structural stability to endure repeatable alkali atom extraction and insertion (Na atoms in this case).169 | ||
| Fig. 13 Electrochemical performance of representative honeycomb layered oxide cathode materials. (a) Voltage-capacity plots of Na3Ni2SbO6 showing the initial (dis)charge curves under a Na-ion (de)insertion rate (current density) commensurate to 0.1C. Technically, nC rate denotes the number of hours (1/n) necessary to (de)insert alkali-ions to the full theoretical capacity (alkali ion occlusion capacity per formula unit). (b) Rate capability of Na3Ni2SbO6. (c) Capacity retention of Na3Ni2SbO6 at (de)insertion rates of 0.1C and 2C (inset). (d) Voltage-capacity plots of Na3Ni2BiO6 at a current density equivalent to 0.05C. (e) Corresponding rate performance, showing Na3Ni2BiO6 to also sustain fast Na-ion kinetics. (f) Capacity retention of Na3Ni2BiO6 at various rates. (a–c) were reproduced and adapted from ref. 58 with permission. (d–f) were reproduced and adapted from ref. 83 by permission of the Royal Society of Chemistry. | ||
Besides the high-voltage and facile alkali-ion kinetics, this class of honeycomb layered oxides can accommodate ample amounts of alkali atoms depending on the choice of both d0 (4d or 5d) cations and 3d transition metal atoms. The increase of the amount of alkali atoms accommodated within the interlayers of the honeycomb slabs implies an increase in the energy storage capacity, indicative of a high energy density. For instance, more of alkali atoms can be extracted from honeycomb layered oxide compositions such as A3+Ni22+Sb5+O6 (A = Li, Na and K) or A3+Ni22+Bi5+O6 than in A2+Ni22+Te6+O6, despite the higher molar mass of A3+Ni22+Bi5+O6 compared to A2+Ni22+Te6+O6. The voltage-capacity plots of representative honeycomb layered oxides that can be utilised as cathode materials for rechargeable alkali-ion batteries are shown in Fig. 14.55–59,83,92,149,168,169,174,295–310 Note that the capacities of these oxides have been calculated based on the manifold oxidation states of the constituent transition states that can be allowed to facilitate maximum extraction of alkali atoms from the layered structures. In principle, the theoretical capacity (Q (mA h g−1)) of a material is determined by the molar mass (M (g mol−1)) and the number of electrons (n) involved during alkali-ion extraction (charging) or insertion (discharging), in accordance with the following equation,
 | (3) |
 | ||
| Fig. 14 Voltage-capacity plots of various honeycomb layered oxides encompassing mainly pnictogen or chalcogen atoms, showing their potential as high-energy-density contenders for high-voltage alkali-ion batteries.55–59,83,92,149,168,169,174,295–310 The error bars represent the upper and lower limits of the voltages attained experimentally. Note also that the theoretical capacities have been calculated based on the change in oxidation states of transition 3d metals as charge-compensation cations. | ||
Another point of emphasis is the nature of the redox process occurring within these honeycomb layered oxides. During the charge compensation redox process, the constituent Ni cations (videlicet., Ni2+/Ni3+) are completely utilised in oxides such as A2+Ni22+Te6+O6 (A = Li, Na and K) ensuring full electrochemical extraction of the alkali atoms. However, for oxides such as A4+Ni2+Te6+O6, it is impossible to fully extract all the alkali A atoms relying on the redox process of constituent Ni atoms (Ni2+/Ni4+) alone.
5.5 Anionic redox processes
To fully tap the capacity (hence energy density) of such oxide compositions, the redox process of anions such as oxygen also have to be utilised, besides the redox process of 3d transition metal cations. Formation of ligand holes, peroxo- or superoxo-like species are expected to occur in the oxygen orbital when anionic redox processes take place, and sometimes oxygen (O2) may be liberated leading to complete structural collapse; thus affecting the cyclability/performance durability of such oxides when used as battery materials.312Anionic redox processes provide a judicious route to utilise the full capacity of electrode materials and has been a subject that has attracted humongous interest in the battery community in recent years.312–335 Apart from facilitating an increase in the redox voltage of honeycomb layered oxides, the presence of d0 cations (such as W6+, Te6+, Sb5+, etc.) also helps stabilise the anion–anion bonding that accompanies the oxygen redox chemistry. For example, the existence of highly valent W6+ (5d0) cations in Li4NiWO6 strongly stabilises the O–O bonds, thereby averting the formation of gaseous O2 following anion oxidation.69 Just like Li4NiWO6, other honeycomb layered oxides such as Li4FeSbO6 have also been found to manifest good oxygen-based redox reversibility, but it generates a large voltage hysteresis in the process.310,336 Further investigations on the oxygen-based redox reversibility are still ongoing in this field to uncover the factors underlying the large voltage hysteresis and determine ways to minimise it. What is emerging with these honeycomb layered oxides is that the presence of high-valency d0 (4d or 5d) is a crucial condition to produce not only high redox (and in some cases paradoxical) voltages, but also invoke oxygen redox chemistry aside from 3d cationic redox processes. Moreover, the possibility to expand the materials platform of these honeycomb layered compounds through partial substitution with isovalent or even aliovalent 3d transition metals, renders them as apposite model compounds to study numerous electrochemical aspects.
6 Topological phase transitions in honeycomb layered oxides
Honeycomb layered oxides are susceptible to undergoing structural changes (phase transitions) upon electrochemical alkali-ion extraction. The presence of divalent transition metals (M2+) in the honeycomb slabs plays a major role in inducing these transitions during alkali-ion reinsertion process. In principle, when alkali atoms are electrochemically extracted, the valency state (oxidation state) of the transition metal atoms residing in the honeycomb slabs increase and vice versa during the reinsertion process; earlier defined as the charge-compensation process. Voids or vacancies created during alkali atom extraction leads to enhanced electrostatic repulsion between the metal atoms residing in different slabs; leading to an increase in the interslab distance.337–3406.1 Structural changes as phase transitions
Evolution of the structural changes upon alkali-ion extraction and reinsertion can readily be discerned using X-ray diffraction (XRD) analyses. During alkali-ion extraction, (00z) Bragg diffraction peaks that reflect the honeycomb interslab planes shift towards lower diffraction angles indicating the expansion of the interslab distance/spacing. The reverse process occurs during alkali-ion reinsertion, as has been exemplified in K2Ni2TeO6 upon potassium-ion extraction and reinsertion (as shown in Fig. 15a).59 Apart from overall peak shifts observed during topotactic alkali-ion extraction and reinsertion (which technically manifests a single-phase (monophasic/solid-solution) behaviour), peak broadening or asymmetric peaks can be observed along with the disappearance of peaks and the emergence of new ones (reflecting a two-phase/biphasic behaviour). | ||
| Fig. 15 Phase transitions of honeycomb layered oxides. (a) Increase/decrease of the interslab distance (Δz) of honeycomb layered oxide K2Ni2TeO6 with charging (K+ extraction)/discharging (K+ reinsertion). (b) Crystal structural evolution of K2Ni2TeO6 upon charging and discharging, showing the occurrence of intricate phase transition mechanism. (c) Broadening and shifting of the (002) and (004) Bragg diffraction peaks that are sensitive to alkali-ion extraction/reinsertion during discharging/charging. (d) Rendition of the phase transition in these classes of layered oxides that entails complex phase transitions (mono- and bi-phasic, and amongst others), akin to a process of successively removing blocks from a complete ‘Jenga wooden blocks set’ which can account for the Devil's staircase-like voltage profiles typically observed for honeycomb layered oxides.58,59,295–297,301,307,341 (a–c) Reproduced from ref. 59 under Creative Commons licence 4.0. | ||
6.2 Stacking sequence changes as phase transitions
The phase transition behaviour of honeycomb layered cathode oxides during alkali-ion extraction (charging) and reinsertion (id est, discharging), entails intricate structural changes that affects the coordination environment of alkali atoms. For instance, electrochemical sodium (Na)-ion extraction from Na3Ni2BiO6 and Na3Ni2SbO6 during charging process leads to a sequential change in the bond coordination of Na, namely from the initial octahedral (O) coordination to prismatic (P) and finally to an octahedral (O) coordination.58,82,83,174,301 Further, the manner of stacking of repetitive honeycomb slabs per unit cell changes from 3 to 1. Thus, the phase transition of Na3Ni2BiO6 during charging process can be written in the following Hagenmuller–Delmas’ notation93,172 as previously described: O3 → P3 → O1 stacking mode. However, phase transitions can influence crucial electrochemical performance parametrics such as the rate capabilities of related oxides when used as battery materials. As such, crucial strategies have been sought to suppress the intricate phase transformation processes, for example, through doping or partial substitution of the transition metal atoms in the honeycomb slabs (exempli gratia, Na3Ni1.5M0.5BiO6 (where M = Mg, Zn, Ni, Cu)) or even the alkali atoms in for instance Na1.6Sr0.2Ni2TeO6.65,341Multiple phase transformations observed in honeycomb layered oxides during alkali-ion extraction and reinsertion have a profound effect on their electrochemical characteristics such as rate performance and nature of the voltage profiles. These intricate phase transitions lead to the appearance of staircase-like voltage profiles as is often observed in the voltage-capacity profiles of most of the reported honeycomb layered cathode oxide materials.58,59,295–297,301,307,341 Shifting of the honeycomb slabs during electrochemical alkali-ion extraction and reinsertion, or what is commonly termed as interslab gliding, has been rationalised to occur as the alkali atoms rearrange their occupying positions (alkali atom ordering). Such a complex phase transformation process can be envisioned through successively removing blocks from a complete ‘Jenga wooden blocks set’, as shown in Fig. 15b. Assuming that the ‘blocks’ are the ‘alkali atoms’, removal of these wooden blocks will lead to rearrangement of the whole Jenga set to avoid structural collapse either by sliding (gliding) or rotation (shear) of the blocks (slabs). A mechanism akin to this Jenga-like mechanism, which is further discussed below, can account for the Devil's staircase-like voltage profiles typically observed for honeycomb layered oxides.57–59,65,82,83,174,295–297,301,341
6.3 Topological order and phase transitions in honeycomb layered oxides
Phase transformation behaviour observed upon electrochemical alkali-ion extraction and reinsertion can spur enigmatic structural changes, like the aforementioned ‘Jenga-like’ transitions. A comprehensive analysis of this mechanism calls for a deeper understanding based on a more comprehensive theory. Nonetheless, we hereafter highlight an approach based on heuristics founded on geometry, topology and electromagnetic considerations.261 Readers may find it prudent to revise topics on tensor calculus, index notation, Einstein convention346 and other widely useful concepts in applied mathematics such as Gaussian curvature (Gauss–Bonnet theorem) in 2D,344,345 the Levi-Civita symbol and Chern–Simons theory.343,344,347–354 Here, we use units where Planck's constant and the speed of the massless photon in the crystal are set to unity: ℏ = c = 1.| ∂alnρ(t,x,y) = qβεabc∂bAc, | (4a) |
 | (4b) |
![[n with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006e_20d1.gif) = (0,0,1) is the normal vector to the ab plane, g is (approximately) the number of free cations, ρ ∝ exp(−βEa) is the ionic charge density with Ea the energy of the cations and
= (0,0,1) is the normal vector to the ab plane, g is (approximately) the number of free cations, ρ ∝ exp(−βEa) is the ionic charge density with Ea the energy of the cations and  , β = 1/kBT is the inverse temperature, K is the Gaussian curvature of a curved closed intrinsic surface M and ∂M is the boundary of M representing the diffusion pathways of g number of cations which form honeycomb lattice on M displayed in Fig. 16. Thus, the integral equation is simply the well-known Gauss–Bonnet theorem.344
, β = 1/kBT is the inverse temperature, K is the Gaussian curvature of a curved closed intrinsic surface M and ∂M is the boundary of M representing the diffusion pathways of g number of cations which form honeycomb lattice on M displayed in Fig. 16. Thus, the integral equation is simply the well-known Gauss–Bonnet theorem.344
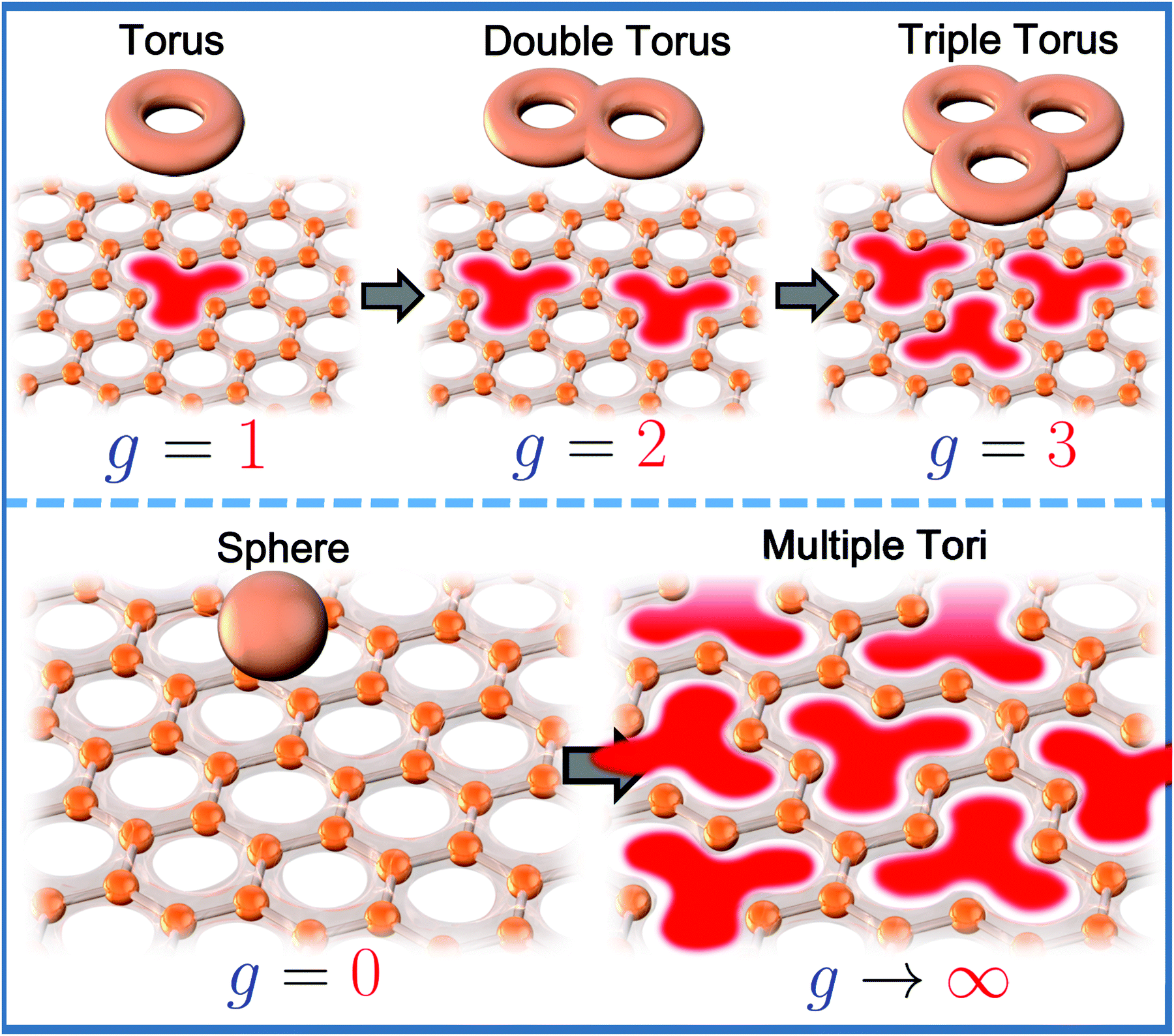 | ||
| Fig. 16 Atomic rearrangement triggered by extraction of alkali-atoms in honeycomb layered oxides such as K2Ni2TeO6, where g ∈ integer is the number of alkali-atoms extracted by applying an external voltage in the ab (x–y) plane.261 The tori denote the various geometrical objects with holes denoted as g (for genus). The tori can be mathematically mapped to the various configurations of the honeycomb lattice with ionic vacancies also denoted as g. | ||
 | ||
| Fig. 17 Topological transitions of honeycomb layered oxides.261 (a) A two-dimensional (2D) field theory relating a Chern–Simons term342,343 to the ionic concentration ρ(t,x,y) (charge density of the cations) where q is the unit charge of a single cation, β = 1/kBT is the inverse temperature and Ac = (V,Ax,Ay) is the 2D electromagnetic potential (see eqn (4a)). (b) A Gauss–Bonnet theorem344,345 relating the applied electric-field ≡ Ei = εibc∂bAc to the Euler characteristic χ = 2 − 2g and Gaussian curvature K of the honeycomb surface M, where Δz ∼ λc = 2π/m is taken to be the order of the Compton wavelength of the cations and = (0,0,1) is a vector normal to the honeycomb surface (see eqn (4b)). χ(M) is applied to estimate the transitions from the complete g = 0 honeycomb configuration to g ∈ integer quasi-stable configurations such as the three-clover atomic arrangements depicted in Fig. 16. | ||
In the special case of static equilibrium when the ionic density ρ is strictly time-independent ∂ρ/∂t = 0 and the electromagnetic vector potential is given by Ac = (V(x,y),0,0), the Chern–Simons term reduces to ∂alnρ(t,x,y) = qβεabc∂bAc → ∂ilnρ(x,y) = qβεij∂jV(x,y) which yields ρ = qμρ ≡ σ with the ansatz 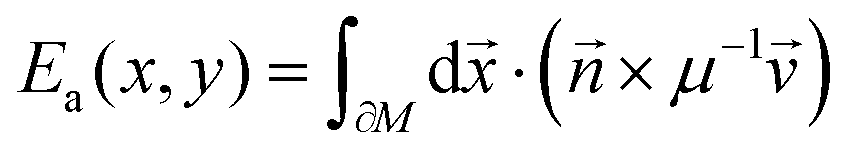 , where εij is the 2D Levi-Civita symbol and μ = D/kBT is the mobility of the cations. Hence, the energy evaluated over a closed loop Ea(∮∂) = m(g − 1) over the honeycomb surface conveniently counts the missing mass of cations within the loop, as shown in Fig. 16. Equivalently, this corresponds to the (activation) energy Ea = (g − 1)mc2 needed to render the cations mobile, where c = 1 is the speed of the massless photon in the crystal. This means that the quasi-stable configuration with g = 1 requires no activation energy to create and can be considered as a ground state of the system. However, since the other configurations are shifted by a constant energy Ea = mg from this ground state, the system contains an additional g − 1 number of stable configurations.
, where εij is the 2D Levi-Civita symbol and μ = D/kBT is the mobility of the cations. Hence, the energy evaluated over a closed loop Ea(∮∂) = m(g − 1) over the honeycomb surface conveniently counts the missing mass of cations within the loop, as shown in Fig. 16. Equivalently, this corresponds to the (activation) energy Ea = (g − 1)mc2 needed to render the cations mobile, where c = 1 is the speed of the massless photon in the crystal. This means that the quasi-stable configuration with g = 1 requires no activation energy to create and can be considered as a ground state of the system. However, since the other configurations are shifted by a constant energy Ea = mg from this ground state, the system contains an additional g − 1 number of stable configurations.
Similar to the total collapse of the pieces in Jenga at the end of the game, this process of extraction of alkali cations and subsequent restabilisation cannot continue indefinitely since eqn (4a) and (4b) remain valid only around equilibrium and the conditions of adiabatic perturbations around equilibria g values. Whence, the transformation of the complete honeycomb structure into a predominantly 3-clover configuration should induce a phase transition. One possible approach to a theoretical treatment of such transitions is to apply the Berezinskii–Kosterlitz–Thouless model248 of phase transitions to the magnetic fields (or fluxes) introduced during this dynamical Jenga phase. Another approach is to consider the phase transitions that may be triggered by sound waves in the crystal arising from rapid (non-adiabatic) extractions of the cations from the honeycomb surface. Geometrically, this entails periodically time-varying Gaussian (curvature) metric analogous to gravitational waves in the space-time geometry. When quantised, these sound waves are phonons that can mediate a weak attractive force between the positively charged fermionic cations (forming Cooper-pairs) and hence may lead to superconductivity.355,356 In contrast, an idealised approach to the dynamics of the cations has been proposed in ref. 261, where bosonic cations form a Bose–Einstein condensate357,358 below the critical temperature and their dynamics are consistent with eqn (4a) and (4b). Above the critical temperature, unpaired charged vortices appear representing diffusion channels of the cations under small curvature perturbations around g ≃ 1. Of course, further research of the physics of the Jenga mechanism including other non-adiabatic phenomena testing the validity or failures of eqn (4a) and (4b) will certainly be the focus of frontier research in the coming years.
7 High-precision measurement of diffusion and magneto-spin properties of honeycomb layered oxides
In the previous section(s), we discussed the physics and electro-chemistry of the diffusion of cations within the honeycomb layers. However, we neglected their magneto-spin interactions with the inter-layers which in turn can substantially affect their mobility and hence, their solid-state alkali-ion diffusion properties. This approximation is valid since alkali cations (exempli gratia K) are known generally to possess an inherently weak nuclear magnetic moment which barely interacts with the octahedra (exempli gratia TeO6) in the inter-layers. In particular, the diffusion dynamics of the cations is resilient to local magneto-spin interactions in the honeycomb layers since the weak magnetic fields originating from the large number of cations in the honeycomb layers tend to randomise and average out according to central limit theorem.359 This means that even though the Gaussian average (mean) of the magnetic fields vanishes, 〈Bz(t)〉 = 0, the mean-square 〈Bz(t)Bz(0)〉 ≠ 0 need not vanish. Hence, the diffusion and magneto-spin properties of the cations are encoded in the mean of the random magnetic fields in the honeycomb layers. However, measuring these properties by applying the Gaussian average over magnetic quantities is an intricate task that often proves elusive to undertake due to a scarcity of effective techniques.7.1 Considering effects of alkali-ion diffusion on muon spin-polarisation
In 2D, the Langevin equation given in eqn (2b) is replaced by, | (5a) |
∝ ![[small eta, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e8.gif) is proportional to the Langevin force, its mean-square is given by the fluctuation–dissipation theorem,260 〈Bz(t)Bz(0)〉 = 2kBTμq2f(t) ≠ 0 with f(t) a function of time. Consequently, the mobility μ (related to the diffusion coefficient by the Einstein–Smoluchowski relation μ = βD) can be determined from the mean-square of the local magnetic fields in the honeycomb layers through the (dynamic) Kubo–Toyabe (KT) function360–362 given by,
is proportional to the Langevin force, its mean-square is given by the fluctuation–dissipation theorem,260 〈Bz(t)Bz(0)〉 = 2kBTμq2f(t) ≠ 0 with f(t) a function of time. Consequently, the mobility μ (related to the diffusion coefficient by the Einstein–Smoluchowski relation μ = βD) can be determined from the mean-square of the local magnetic fields in the honeycomb layers through the (dynamic) Kubo–Toyabe (KT) function360–362 given by, | (5b) |
7.2 Rationale and methodology behind applying muon spin rotation and relaxation measurement techniques
At this juncture, it is imperative to explain the rationale for the use of muons in analysis of diffusion and magneto-spin properties of materials. Muons stand out from other members of the lepton family of elementary particles mainly owing to the following reasons:• Muons are abundant and are indeed a product of cosmic radiation (recall the Aurora Borealis and Aurora Australis). Muons can also be artificially produced using spallation sources such as ISIS Neutron and Muon Source (UK), Japan Proton Accelerator Research Complex (JPARC), TRIUMF (Canada) and Paul Scherrer Institute (PSI, Switzerland);
• The spin configuration of muons are traceable (technically, muons are 100% spin-polarised since they are produced via the decay of a (positive) pion at rest into a positron and an electron neutrino via the weak interaction, which violates parity),368–370 implying that they are easy to detect via their decay products unlike other members of the lepton elementary particles. This aspect endows μ+SR measurements with an upper edge over other resonance techniques such as nuclear magnetic resonance (NMR). In addition, muons possess a high gyromagnetic ratio (γμ = 135.5 MHz T−1) meaning that they are very sensitive to weak magnetic fields;
• Unlike electrons, muons have a finite lifetime that is appreciable; thus, μ+SR offers a unique measurement time window that complements conventional techniques such as NMR and neutron diffraction.
Detection of alkali-ion diffusion by muons first entails the embedding of muons into a sample (or muon implantation), the sample in this case being the layered oxide material. These muons are artificially produced via the bombardment of high energy protons onto a carbon (graphite) or beryllium target, as is schematically shown in Fig. 18. The muons (in this case, positive muons (anti-muons)) are then focused using a collimator to the sample where they bind with oxygen ions (O2−) to form stable μ+-O2− bonds. The implantation of muons into a honeycomb layered oxide is illustrated in Fig. 19a, where muons typically reside at the vicinity of oxygen ions at distances in the ranges of 1–1.2 Å.364 The implanted muons are initially static and are able to sense the local nuclear magnetic field in the layered oxide when it is in a paramagnetic state, a behaviour that can mathematically be expressed using a static Kubo–Toyabe (KT) function,360–362 as is also shown in Fig. 19b. When alkali-ion diffusion occurs, the local nuclear field haphazardly fluctuates and the implanted muons acquire a dynamic contribution in the KT function through the hopping rate of the cations; thus are able to sense the local field that is randomly fluctuating at an average rate. The mobility of alkali cations can be increased by temperature beyond a certain critical temperature Tc where the alkali cations become mobile, thus inducing an additional fluctuation in the local mean-square magnetic field leading to a conspicuous increase of the fluctuation (collision) rate ν0 → ν(T), where the mean-square magnetic field is given by 〈BZ(t)Bz(0)〉 = 〈Bz2(0)〉exp(−νt). Consequently, the self-diffusion coefficient  related to the diffusion coefficient, D(T) by a Boltzmann factor
related to the diffusion coefficient, D(T) by a Boltzmann factor  is accurately determined using the μ+SR measurements by considering the collisions as a Markov process359,372 over n paths of the cations in the 2D honeycomb lattice where Ni is the number of cation sites, Zi the vacancy fraction and si the length of the mean-free path between collisions.373 The correct diffusion/jump paths can then be confirmed by complementary experimental and modeling inputs deduced from, exempli gratia temperature dependent neutron/X-ray diffraction or molecular dynamics simulations.
is accurately determined using the μ+SR measurements by considering the collisions as a Markov process359,372 over n paths of the cations in the 2D honeycomb lattice where Ni is the number of cation sites, Zi the vacancy fraction and si the length of the mean-free path between collisions.373 The correct diffusion/jump paths can then be confirmed by complementary experimental and modeling inputs deduced from, exempli gratia temperature dependent neutron/X-ray diffraction or molecular dynamics simulations.
 | ||
| Fig. 18 Working principle of (anti-)muon spin relaxation (μ+SR) as a potent tool for investigating the diffusive and magnetic properties of target materials. The (anti-)muon is produced when a high energy proton beam is directed onto carbon nuclei which produce (positive) pions. The (positive) pion decays into an (anti-)muon and a muon-neutrino, which subsequently decays to a positron, an anti-muon neutrino and an electron-neutrino which escape the sample. The difference in the positron counts in the forward (F) and backward (B) detectors normalised by the total count, the asymmetry function A(t), gives the spin relaxation of the (anti-)muon in the sample. | ||
 | ||
| Fig. 19 High-precision measurement of diffusion and magneto-spin properties of honeycomb layered oxides relevant to phase transitions. (a) The anti-muon implantation into a honeycomb layered oxide framework with a stoichiometric composition of, for instance, K2Ni2TeO6. The anti-muon is expected to bind onto the oxygen ions located in the octahedral (TeO6 and NiO6) structures of the material altering the typical decay rate of the (anti-)muon. The hopping rate, ν of the diffusing potassium (K) cations along the honeycomb depends on their interaction with the anti-muons through their random nuclear magnetic fields which alters the anti-muon decay rate Δν. (b) The analysis of alkali-ion diffusion using the dynamical Kubo–Toyabe function,360–362Pz(t) which describes the relaxation of muon spin polarisation in the presence of a particular (typically Gaussian) distribution of nuclear magnetic fields of the cations in the honeycomb layered oxide material. The total asymmetry function in μ+SR experiments depends on the Kubo–Toyabe function, which depends on the decay rate of the anti-muons due to transport properties of the cations such as their hopping rate in the material. The hopping rate in turn determines the self-diffusion coefficient of the material. | ||
7.3 Muon spin rotation and relaxation diffusion coefficient measurement results
Sugiyama, Månsson and co-workers have pioneered the use of μ+SR measurements in the study of both the magneto-spin and alkali-ion diffusive properties in a wide swath of layered materials such as LiMO2 (where M = Ni and Co), LiCrO2, LiNi1/3Co1/3Mn1/3O2, Li2MnO3 and even to NaCoO2.363–367,374–410 Investigation of potassium-ion (K+) dynamics in related layered materials is particularly unwieldy, due to the innately weak nuclear magnetic moment of potassium relative to other ions such as lithium (Li) and sodium (Na). This renders it difficult to capture the dynamics of K+ in layered materials using standard techniques such as NMR spectroscopy. As discussed above, the fact that spin-polarised muons possess a strong gyromagnetic moment, makes μ+SR measurements particularly ideal for capturing the dynamics of cations such as the K+ with an extremely weak nuclear magnetic moment in materials. For clarity, the nuclear magnetic moment/gyromagnetic ratio of K (μ[39K] = 0.39 μN, 12.50 MHz T−1) is much smaller than for Li (μ[7Li] = 3.26 μN, 108.98 MHz T−1) and Na (μ[23Na] = 2.22 μN, 70.81 MHz T−1). The μ+SR asymmetry function time spectrum of honeycomb layered oxide K2Ni2TeO6 (or equivalently as K2/3Ni2/3Te1/3O2) measured below the antiferromagnetic transition temperature (26 K) as shown in Fig. 19a, is shown in Fig. 20a, where precession of the muon (spin-polarisation) occurs. It is evident that the muon precesses due to the emergence of a spontaneous internal magnetic field, resulting in a clear oscillation of the time spectrum. This is a response that is typically observed from a muon ensemble in a magnetically ordered state (in this case, antiferromagnetic Ni spin ordering in K2Ni2TeO6).371 | ||
| Fig. 20 High-precision measurement of magneto-spin and diffusion properties of honeycomb layered oxides relevant to phase transitions. (a) Presence of an antiferromagnetic spin ordering in K2Ni2TeO6 below 26 K revealed by a clear oscillation in the μ+SR time spectra. (b) The onset and evolution of K-ion diffusion revealed by an exponential increase in field fluctuation rate (= ion hopping rate) from which the activation energy (Ea) of the diffusion process can be determined.371 | ||
The dependency plot of the fluctuation rate, which is dynamically related to the hopping rate of K+ with temperature, ν(T) is shown in Fig. 20b. Between 250 K and 500 K, this fluctuation rate increases with temperature signifying the onset of diffusive motion of K+ in K2Ni2TeO6. The hopping rate nicely obeys a trend akin to Arrhenius equation ν(T) = ν0exp(−EKa/kBT) from where an activation energy commensurate to approximately EKa ≃ 120 meV is obtained.371 The diffusion coefficient can be calculated using the above hopping rate assumptions to yield a diffusion coefficient value of DK(T) = 1.2 × 10−10 cm2 s−1, which is an order of magnitude lower than that of layered materials such as LiCoO2.363,411
Caution needs to be taken when interpreting the μ+SR measurement data, as muons per se can also be mobile in inactive materials. K2Ni2TeO6 indeed shows reversible K+ extraction and insertion behaviour (thus, electrochemically active) at room temperature; thus, the onset of K+ diffusive motion that arises at T > 250 K is irrefutable. The feasibility of utilising μ+SR measurements to further unveil the intricacies of the dynamics of such cations as K+, which tend to possess low nuclear magnetic moments, will expand the pedagogical scope of cationic intercalation (insertion) and deintercalation (extraction) dynamics within honeycomb layered oxides and other layered materials.
8 Summary and future challenges for honeycomb layered oxides
This review provides an elaborate account of the exceptional chemistry and the physics that make honeycomb layered oxides a fledgling class of compounds. We explore the prospects that would result in myriads of compositions expected to be reported in the coming decades. Majority of the honeycomb layered oxides reported typically engender Li+ or Na+ as resident cations. However, the further adoption of honeycomb layered oxides with large-radii alkali ions, such as K+, Rb+ and Cs+ or even coinage metal ions such as Ag+, H+, Au+, Cu+, etc., is expected to further increase the compositional space of related compounds, unlocking manifold functionalities amongst this class of materials. In fact, preliminary theoretical computations have affirmed the feasibility of preparing honeycomb layered oxides encompassing cations such as Rb+, Cs+, Ag+, H+, Au+, Cu+, etc. to adopting, for instance, a chemical composition of A2Ni2TeO6, where A = Rb, Cs, Ag+, etc. By the same token, synthesis of honeycomb layered materials that encompass alkaline-earth metals such as A = Ba, Sr, etc. has also been proposed as another avenue of augmenting the various combinations of these materials. Indeed, studies pertaining compositions such as ARu2O6, where A = Ba, Sr, etc. have recently been reported.228 Undoubtedly, this class of materials offers an extensive platform worthy of pursuit in the coming years (Fig. 3a).To predict or interpret possible emergent features of honeycomb layered oxides, it is crucial to understand their unique structural frameworks and the local and bulk atomistic changes they undergo. A combination of crystallography techniques that include transmission electron microscopy (TEM), neutron diffraction and X-ray diffraction are expected to offer a holistic view of the arrangement of atoms within the honeycomb lattice and the global order of atoms within the honeycomb structure of the new materials. Moreover high-resolution TEM at low temperatures, as can be availed by cryogenic microscopy, is a possible route to discern the arrangement of transition metal atoms in the honeycomb lattice at low temperatures where transitions tend to occur.412 Experimental and theoretical reports on the structures of these materials has already revealed some stacking disorders with honeycomb layered species such as Na3Ni2SbO6, Na2Zn2TeO6 and Na2Ni2TeO6, associating them with emergent functionalities such as phase transitions, magnetic ground states and ionic diffusion.60,170,253,413 Although defects have been known to have both prolific and detrimental effects on honeycomb layered oxides, they still remain vastly underexplored. Nonetheless, de novo computational and experimental techniques are expected to uncover new defect physics and chemistry that will expand their uses into multiple fields.
On another front, doping offers a prospective route to availing more possibilities with a broader scope of chemical compositions that display improved electrochemical and additional magnetic properties. From an electrochemical perspective, doping with non-magnetic atoms such as magnesium or strontium generally reinforces the crystalline structure by suppressing electrochemically – driven phase transitions, whilst increasing the thermodynamic entropy of the materials. Increased entropy has added advantages that include raising the working voltage as well as facilitating multiple redox electrochemistry during battery operations as elucidated in Fig. 11 and 14. Relating to ionic conductivity, partial doping of the transition metal atoms in the honeycomb slab with aliovalent or isovalent atoms is a pertinent strategy to increase the ionic conductivity of honeycomb layered oxides. For instance, partial substitution of Zn2+ with Ga3+ in Na2Zn2TeO6 solid-state electrolyte (with a wide voltage tolerance) aids to increase the Na+-ion mobility (conductivity) due to increased formation of Na+ vacancies.249 Theoretical investigations done by Sau and co-workers have accentuated the profound effect of transition metal substitution in Na2M2TeO6 (M = Ni, Zn, Co, Cu).268,269,414 In contrast, doping with magnetic atoms, as shown in Fig. 8, reveals fascinating magnetic behaviour that places honeycomb layered oxides amongst the exotic quantum materials.
Moreover, topochemical reactions, for instance, chemical ion-exchange of Rb or silver (Ag) with potassium (K) in K2Ni2TeO6 can aid build an entire host of new oxide materials with a wide swath of physicochemical properties. Indeed, such a design strategy has proven effective in the synthesis of Ag3Ni2BiO6, for instance, via topochemical ion-exchange of Li3Ni2BiO6.66 Additionally, the introduction of alkali cations with differing ionic radii makes the tuning of the distance between the honeycomb layers (interslab/interlayer distance) possible; thus presenting avenues to tune the interlayer magnetic couplings as discussed in Fig. 10. This guarantees the feasibility to not only adjust the electrochemical properties but also to tweak the physicochemical aspects such as the magnetic dimensionality of the honeycomb lattice. This calls for further exploratory synthesis to be augmented with computational protocols in order to expedite the design of new honeycomb layered oxide compositions.
There has been significant progress in the physics entailing topological states, for which honeycomb layered oxides play a pivotal role in advancing this topical field. In this review, we have discussed the Kitaev and Haldane magnetic (spin) interactions within the honeycomb lattice that offer a path to the experimental realisation of Kitaev quantum spin liquid and a quantum anomalous Hall insulator (Chern insulator) respectively.179,180,189–192,194,195 In addition, higher-order magnetic interactions induced by the angle between the spins of the magnetic cations, introduces other interactions: mainly, the Heisenberg and asymmetric/Dzyaloshinskii–Moriya (DM) interactions.415,416 Due to the additional angular space-time dependent degree of freedom, these interactions are considered of higher order and thus very elusive to realise without the presence of, for instance, single-crystals of target honeycomb layered oxide materials. Irradiating circularly-polarised oscillating electric fields on preferably single crystals within a Floquet model (theory) is a plausible route to realising DM interactions within honeycomb layered oxide materials, as has been suggested by several authors.417–419 The primary significance of these interactions is the evaluation of magnetic skyrmions420–424 – quasi-particles that have been predicted to exist in certain magnetic condensed matter systems such in magnetic thin films either as dynamic excitations or stable/metastable configurations of spin; which shows great promise in topological quantum computing applications.193,425–427
Regarding single-crystal growth, the high thermal stability of honeycomb layered oxides, such as tellurates, bismuthates and antimonates, makes them suitable for crystallisation at high temperatures conducive for their preparation. In fact, the possibility of growing single crystals in honeycomb layered oxides (such as Na2Ni2TeO6, Na2Cu2TeO6, Na3Cu2SbO6 and Na2Co2TeO6) using high-temperature solid-state reactions has already been achieved.24,25,39 Another fascinating pursuit will be the design of thin films from honeycomb layered oxide materials, either using molecular beam epitaxy (MBE), atomic laser deposition (ALD) or pulsed laser deposition (PLD), which will aid to accurately visualise the presence of magnetic skyrmions or any emergent topological physics that covers, inter alia, superconductivity, magneto-resistance and ferro-electricity. Moreover, an extension of the μ+SR technique, namely low energy μ+SR (LEM) offers the unique possibility to tune the muon implantation depth into samples from 5–500 nm. Thus, synthesising thin film battery cells, exempli gratia from honeycomb layered oxide materials, allows one to gain unique access to depth-resolved studies of the dynamics of the cations at and across the buried interfaces, isto es the solid-state cathode/electrolyte/anode, respectively.428,429
A plethora of unprecedented amazing phenomena may also be found when honeycomb layered oxides are subjected under high-pressure (stress) conditions. This has, amongst other things, the effect of making higher-order interactions finite and thus non-negligible. In particular, exerting pressure perpendicular to the honeycomb slabs bring into play 3D interactions that may have been otherwise negligible. Experimentally, Na2Cu2TeO6 shows new bond coordination (dimerisation of Cu bonds) at high pressure, leading to a change in the magnetic properties technically referred to as magnetic phase transitions.21,98 Generally, high pressure exerted in these layered oxides can introduce defects or microstructure evolutions that may show great potential for novel functional materials. Although the global topology of honeycomb layered materials is robust against local defects, whenever these defects are related to topological invariants (exempli gratia Berry's phase),430–440 they will affect global properties of the material such as phase transitions, as exemplified in Fig. 15–17. Phase transition phenomena inherent in these classes of honeycomb layered oxide materials will certainly necessitate the use of spectroscopic techniques such as muon spin relaxation (μ+SR), as well as computational and theoretical approaches, to discern the nature of the spin interactions innate at high-pressure regimes. Moreover, resolution at the atomic-scale of related functional materials when subjected to ultra-high pressure will attract tremendous research interests in the coming years.
The heightened interest in oxide materials based on honeycomb layers is expected to spearhead the design of a new generation of materials that promise to make remarkable contributions in the fields of energy, electronic devices, catalysis, and will ultimately benefit the scientific community in a broad swath of fields in the coming decades, as can be envisaged in Fig. 21. Recent reports are also emerging on honeycomb layered oxides as photocatalysts, optical materials, superfast ionic conductors, and so forth.12–14,250–252,254,255,267,441–444 A grand challenge with most of these materials lies in their handling. Particularly for honeycomb layered oxides comprising alkali ions with large radii such as potassium and rubidium, handling demands the presence of a controlled atmosphere (videlicet, storage in argon-purged glove boxes) as they are sensitive to moisture (hygroscopic) and air. Future work should also focus on the improvement of the stability of related honeycomb layered oxides, for instance, when exposed to air; to enable handling and mass production of these materials in ambient conditions. Their instability can be contained and controlled, for example, by tuning their chemical composition. Partial substitution of the constituent transition metal atoms is a possible route, as has been noted when Na3Ni2SbO6 is partially substituted even with a minuscule amount with Mg, Mn or even Ru.295,297,445 On another front, the use of multiple transition metals in equivalent amounts has also been presented as a new route for the design of stable layered oxides (often referred to as ‘high-entropy oxides’) with unique physicochemical properties.446–448 Although this concept presents new possibilities for the design and application of honeycomb layered oxides, it is still in infancy with a lot of growth potential.
 | ||
| Fig. 21 Diversity of honeycomb structures in various realms of science and technology. The schematic highlights the future perspectives of honeycomb layered oxides that can be envisioned such as superconductivity, phase transitions, photocalysis, thermochromism, spin dynamics, photovoltaics and optics. | ||
In brief, partial substitution also induces a change in the phase transitions observed when alkali cations are electrochemically extracted, as is the case when they are used as battery materials. Hygroscopicity presents another avenue for tuning the interslab distance and editing electrochemical profiles in some materials bringing forth several advantages such as superconductive phase transitions, as has been noted in layered NaCoO2 when hydrated.449
Honeycomb layered oxides (particularly for compositions incorporating pnictogen or chalcogen atoms that have been delved in this review) can serve as high-voltage cathode materials for rechargeable batteries, as summarised in Fig. 12, exhibiting theoretically high capacities. A challenge is their safe and stable operation at high-voltage regimes; warranting the adoption of stable electrolytes that can tolerate high-voltage battery operation. Ionic liquids, which consist of organic or inorganic anions and organic cations, manifest a plenitude of desirable properties. Paramount amongst them is their low flammability, good chemical stability and excellent thermal stability.275,450 In particular, the inherently large voltage tolerance makes ionic liquids propitious when matched to high-voltage layered cathodes during battery operation. Stable performance of high-voltage layered cathode materials using piperidinium-based ionic liquids has been shown;298,300 likewise, assessment of high-voltage honeycomb layered oxides using stable electrolytes (such as ionic liquids) is a plausible route for harnessing their high electrochemical performance. A schematic list of the choice of stable electrolytes, especially ionic liquids, for honeycomb layered oxide cathode materials is furnished in Fig. 11. On another note, exotic redox chemistry can be manifested in honeycomb layered oxides. For instance, Li4FeSbO6 is currently amongst model materials to study oxygen anion redox chemistry; a topical area in battery research nowadays.69,310,312,336 Much room still exists in the search for related honeycomb layered oxides.
At this juncture it begs the question: quo vadis, honeycomb layered oxides? The rich electrochemical, magnetic, electronic, topological and catalytic properties generally innate in layered materials, indubitably present a conducive springboard to break new ground of unchartered quantum phenomena and the coexistent electronic behaviour in two-dimensional (2D) systems. It is our expectation that this will unlock unimaginable applications in the frontier fields of computing, quantum materials and internet-of-things (IoT).
Finally, the vexing question of why magnetic atoms in the slabs of these layered oxides conveniently align in a honeycomb architecture, to our knowledge, remains to be addressed; an attestation that the landscape of honeycomb layered oxide materials still remains broad and uncharted, moving forward into this new age of avant-garde innovation. An eminent mathematician has elegantly posited a solution to why bees prefer the honeycomb architecture in what now is emerging as ‘the Honeycomb conjecture’.451 Presumably, it is through a review of the materials found in nature that we can glean insights for future design in this universe of honeycomb layered oxide materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted under the auspices of the National Institute of Advanced Industrial Science Technology (AIST), Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number 19 K15685) and Japan Prize Foundation. Part of this research was also supported by the European Commission through a Marie Skłodowska-Curie Action and the Swedish Research Council – VR (Dnr. 2014-6426, 2016-06955 and 2017-05078), the Carl Tryggers Foundation for Scientific Research as well as the Swedish Foundation for Strategic Research (SSF) within the Swedish national graduate school in neutron scattering (SwedNess). The authors would also like to thank Minami Kato, Kohei Tada, Ola Kenji Forslund, Elisabetta Nocerino, Konstantinos Papadopoulos, Anton Zubayer and Keigo Kubota for fruitful discussions on this manuscript. We acknowledge that this review paper was proofread and edited through the support provided by Edfluent services. T. M. gratefully acknowledges Natsumi Ishii and Rei Ishii for the unwavering support in conducting this work.References
- C. Darwin, On the Origin of Species by Means of Natural Selection, John Murray, London, 1st edn, 1859 Search PubMed.
- Q. Zhang, X. Yang, P. Li, G. Huang, S. Feng, C. Shen, B. Han, X. Zhang, F. Jin and F. Xu, et al. , Prog. Mater. Sci., 2015, 74, 332–400 CrossRef.
- B. L. Karihaloo, K. Zhang and J. Wang, J. R. Soc., Interface, 2013, 10, 20130299 CrossRef CAS.
- P. Garca-Pascual, N. Sanjuán, R. Melis and A. Mulet, J. Food Eng., 2006, 72, 346–353 CrossRef.
- K. Sugimura and S. Ishihara, Development, 2013, 140, 4091–4101 CrossRef CAS.
- K. Ikawa and K. Sugimura, Nat. Commun., 2018, 9, 1–14 CrossRef CAS.
- M. D. de la Loza and B. Thompson, Mech. Dev., 2017, 144, 23–32 CrossRef.
- N. Yucel, A. Sakalli and A. Karahan, J. Appl. Ichthyol., 2017, 33, 530–532 CrossRef.
- J. Garca-Hernández, Coral Reefs, 2018, 37, 807 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- K. Kitagawa, T. Takayama, Y. Matsumoto, A. Kato, R. Takano, Y. Kishimoto, S. Bette, R. Dinnebier, G. Jackeli and H. Takagi, Nature, 2018, 554, 341–345 CrossRef CAS.
- R. Kadari, R. Velchuri, K. Sreenu, G. Ravi, N. R. Munirathnam and M. Vithal, Mater. Res. Express, 2016, 3, 115902 CrossRef.
- Y. Pu, Y. Liu, D. Liu, Z. Zhou, S. Ding, Z. Xia and M. Li, Int. J. Hydrogen Energy, 2018, 43, 17271–17282 CrossRef CAS.
- J. H. Roudebush, G. Sahasrabudhe, S. L. Bergman and R. J. Cava, Inorg. Chem., 2015, 54, 3203–3210 CrossRef CAS.
- V. Kumar, N. Bhardwaj, N. Tomar, V. Thakral and S. Uma, Inorg. Chem., 2012, 51, 10471–10473 CrossRef CAS.
- E. A. Zvereva, O. A. Savelieva, Y. D. Titov, M. A. Evstigneeva, V. B. Nalbandyan, C. N. Kao, J.-Y. Lin, I. A. Presniakov, A. V. Sobolev, S. A. Ibragimov, M. Abdel-Hafiez, Y. Krupskaya, C. Jähne, G. Tan, R. Klingeler, B. Büchner and A. N. Vasiliev, Dalton Trans., 2013, 42, 1550–1566 RSC.
- S. Derakhshan, H. L. Cuthbert, J. E. Greedan, B. Rahaman and T. Saha-Dasgupta, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 104403 CrossRef.
- L. Viciu, Q. Huang, E. Morosan, H. W. Zandbergen, N. I. Greenbaum, T. McQueen and R. J. Cava, J. Solid State Chem., 2007, 180, 1060–1067 CrossRef CAS.
- K. Morimoto, Y. Itoh, C. Michioka, M. Kato and K. Yoshimura, J. Magn. Magn. Mater., 2007, 310, 1254–1256 CrossRef CAS.
- S. Derakhshan, J. E. Greedan, T. Katsumata and L. M. D. Cranswick, Chem. Mater., 2008, 20, 5714–5720 CrossRef CAS.
- H.-J. Koo and M.-H. Whangbo, Inorg. Chem., 2008, 47, 128–133 CrossRef CAS.
- E. A. Zvereva, M. A. Evstigneeva, V. B. Nalbandyan, O. A. Savelieva, S. A. Ibragimov, O. S. Volkova, L. I. Medvedeva, A. N. Vasiliev, R. Klingeler and B. Buechner, Dalton Trans., 2012, 41, 572–580 RSC.
- M. Schmitt, O. Janson, S. Golbs, M. Schmidt, W. Schnelle, J. Richter and H. Rosner, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174403 CrossRef.
- R. Sankar, I. Panneer Muthuselvam, G. J. Shu, W. T. Chen, S. K. Karna, R. Jayavel and F. C. Chou, CrystEngComm, 2014, 16, 10791–10796 RSC.
- G. Xiao, Z. Xia, W. Zhang, X. Yue, S. Huang, X. Zhang, F. Yang, Y. Song, M. Wei, H. Deng and D. Jiang, Cryst. Growth Des., 2019, 19, 2658–2662 CrossRef CAS.
- E. A. Zvereva, V. B. Nalbandyan, M. A. Evstigneeva, H.-J. Koo, M.-H. Whangbo, A. V. Ushakov, B. S. Medvedev, L. I. Medvedeva, N. A. Gridina, G. E. Yalovega, A. V. Churikov, A. N. Vasiliev and B. Büchner, J. Solid State Chem., 2015, 225, 89–96 CrossRef CAS.
- Y. Itoh, J. Phys. Soc. Jpn., 2015, 84, 64714 CrossRef.
- E. A. Zvereva, M. I. Stratan, Y. A. Ovchenkov, V. B. Nalbandyan, J.-Y. Lin, E. L. Vavilova, M. F. Iakovleva, M. Abdel-Hafiez, A. V. Silhanek, X.-J. Chen, A. Stroppa, S. Picozzi, H. O. Jeschke, R. Valent and A. N. Vasiliev, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 144401 CrossRef.
- A. K. Bera, S. M. Yusuf, A. Kumar and C. Ritter, Phys. Rev. B, 2017, 95, 94424 CrossRef.
- S. K. Upadhyay, K. K. Iyer, S. Rayaprol, P. L. Paulose and E. V. Sampathkumaran, Sci. Rep., 2016, 6, 31883 CrossRef CAS.
- C. Koo, E. Zvereva, I. Shukaev, M. Richter, M. Stratan, A. Vasiliev, N. Vladimir and R. Klingeler, J. Phys. Soc. Jpn., 2016, 85, 84702 CrossRef.
- E. A. Zvereva, M. I. Stratan, A. V. Ushakov, V. B. Nalbandyan, I. L. Shukaev, A. V. Silhanek, M. Abdel-Hafiez, S. V. Streltsov and A. N. Vasiliev, Dalton Trans., 2016, 45, 7373–7384 RSC.
- A. I. Kurbakov, A. N. Korshunov, S. Y. Podchezertsev, A. L. Malyshev, M. A. Evstigneeva, F. Damay, J. Park, C. Koo, R. Klingeler, E. A. Zvereva and V. B. Nalbandyan, Phys. Rev. B, 2017, 96, 24417 CrossRef.
- S. K. Karna, Y. Zhao, R. Sankar, M. Avdeev, P. C. Tseng, C. W. Wang, G. J. Shu, K. Matan, G. Y. Guo and F. C. Chou, Phys. Rev. B, 2017, 95, 104408 CrossRef.
- E. A. Zvereva, M. I. Stratan, I. L. Shukaev, V. B. Nalbandyan and A. N. Vasil’ev, J. Exp. Theor. Phys., 2017, 124, 612–616 CrossRef CAS.
- J. Werner, W. Hergett, J. Park, C. Koo, E. A. Zvereva, A. N. Vasiliev and R. Klingeler, J. Magn. Magn. Mater., 2019, 481, 100–103 CrossRef CAS.
- P. P. Stavropoulos, D. Pereira and H.-Y. Kee, Phys. Rev. Lett., 2019, 123, 37203 CrossRef CAS.
- A. Korshunov, I. Safiulina and A. Kurbakov, Phys. Status Solidi B, 2020, 257, 1900232 CrossRef CAS.
- W. Yao and Y. Li, Phys. Rev. B, 2020, 101, 85120 CrossRef CAS.
- S. Li, V. Loganathan and A. H. Nevidomskyy, 2009, arXiv preprint, arXiv:1906.02215, https://arxiv.org/abs/1906.02215v1.
- Y. Miura, R. Hirai, Y. Kobayashi and M. Sato, J. Phys. Soc. Jpn., 2006, 75, 84707 CrossRef.
- M. Schmitt, S. Gerlach, M. Schmidt, W. Schnelle and H. Rosner, Z. Anorg. Allg. Chem., 2006, 632, 2091 CrossRef.
- Y. Miura, R. Hirai, T. Fujita, Y. Kobayashi and M. Sato, J. Magn. Magn. Mater., 2007, 310, e389–e391 CrossRef CAS.
- Y. Miura, Y. Yasui, T. Moyoshi, M. Sato and K. Kakurai, J. Phys. Soc. Jpn., 2008, 77, 104709 CrossRef.
- W. Li, S.-S. Gong, Y. Zhao and G. Su, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 184427 CrossRef.
- C. N. Kuo, T. S. Jian and C. S. Lue, J. Alloys Compd., 2012, 531, 1–4 CrossRef CAS.
- J. H. Roudebush and R. J. Cava, J. Solid State Chem., 2013, 204, 178–185 CrossRef CAS.
- B. Zhang, Y. Zhang, Z. Wang, D. Wang, P. J. Baker, F. L. Pratt and D. Zhu, Sci. Rep., 2014, 4, 6451 CrossRef CAS.
- B. Jeevanesan and P. P. Orth, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 144435 CrossRef.
- E. Lefrançois, M. Songvilay, J. Robert, G. Nataf, E. Jordan, L. Chaix, C. V. Colin, P. Lejay, A. Hadj-Azzem, R. Ballou and V. Simonet, Phys. Rev. B, 2016, 94, 214416 CrossRef.
- C. Wong, M. Avdeev and C. D. Ling, J. Solid State Chem., 2016, 243, 18–22 CrossRef CAS.
- J. Werner, W. Hergett, M. Gertig, J. Park, C. Koo and R. Klingeler, Phys. Rev. B, 2017, 95, 214414 CrossRef.
- A. Scheie, K. Ross, P. P. Stavropoulos, E. Seibel, J. A. Rodriguez-Rivera, J. A. Tang, Y. Li, H. Y. Kee, R. J. Cava and C. Broholm, Phys. Rev. B, 2019, 100, 214421 CrossRef CAS.
- Y. Mao and Y. Li, Phys. Rev. B, 2020, 102, 054414 CrossRef CAS.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, K. Guruprakash, A. Prakash, M. Doublet and J.-M. Tarascon, Chem. Commun., 2013, 49, 11376–11378 RSC.
- N. S. Grundish, I. D. Seymour, G. Henkelman and J. B. Goodenough, Chem. Mater., 2019, 31, 9379–9388 CrossRef CAS.
- Z. Yang, Y. Jiang, L. Deng, T. Wang, S. Chen and Y. Huang, J. Power Sources, 2017, 360, 319–323 CrossRef CAS.
- D. Yuan, X. Liang, L. Wu, Y. Cao, X. Ai, J. Feng and H. Yang, Adv. Mater., 2014, 26, 6301–6306 CrossRef CAS.
- T. Masese, K. Yoshii, Y. Yamaguchi, T. Okumura, Z.-D. Huang, M. Kato, K. Kubota, J. Furutani, Y. Orikasa and H. Senoh, et al. , Nat. Commun., 2018, 9, 1–12 CrossRef CAS.
- A. I. Kurbakov, A. N. Korshunov, S. Y. Podchezertsev, M. I. Stratan, G. V. Raganyan and E. A. Zvereva, J. Alloys Compd., 2020, 820, 153354 CrossRef CAS.
- Y. Motome, R. Sano, S. Jang, Y. Sugita and Y. Kato, J. Phys.: Condens. Matter, 2020, 32, 404001 CrossRef CAS.
- V. B. Nalbandyan, M. Avdeev and M. A. Evstigneeva, J. Solid State Chem., 2013, 199, 62–65 CrossRef CAS.
- N. Bhardwaj, A. Gupta and S. Uma, Dalton Trans., 2014, 43, 12050–12057 RSC.
- V. Kumar, A. Gupta and S. Uma, Dalton Trans., 2013, 42, 14992–14998 RSC.
- A. Gupta, C. Buddie Mullins and J. B. Goodenough, J. Power Sources, 2013, 243, 817–821 CrossRef CAS.
- R. Berthelot, W. Schmidt, S. Muir, J. Eilertsen, L. Etienne, A. W. Sleight and M. A. Subramanian, Inorg. Chem., 2012, 51, 5377–5385 CrossRef CAS.
- S. Laha, E. Morán, R. Sáez-Puche, M. Á. Alario-Franco, A. J. Dos santos Garcia, E. Gonzalo, A. Kuhn, F. García-Alvarado, T. Sivakumar, S. Tamilarasan, S. Natarajan and J. Gopalakrishnan, J. Solid State Chem., 2013, 203, 160–165 CrossRef CAS.
- E. McCalla, A. Abakumov, G. Rousse, M. Reynaud, M. T. Sougrati, B. Budic, A. Mahmoud, R. Dominko, G. Van Tendeloo, R. P. Hermann and J.-M. Tarascon, Chem. Mater., 2015, 27, 1699–1708 CrossRef CAS.
- Z. N. Taylor, A. J. Perez, J. A. Coca-Clemente, F. Braga, N. E. Drewett, M. J. Pitcher, W. J. Thomas, M. S. Dyer, C. Collins, M. Zanella, T. Johnson, S. Day, C. Tang, V. R. Dhanak, J. B. Claridge, L. J. Hardwick and M. J. Rosseinsky, J. Am. Chem. Soc., 2019, 141, 7333–7346 CrossRef CAS.
- J. H. Roudebush, N. H. Andersen, R. Ramlau, V. O. Garlea, R. Toft-Petersen, P. Norby, R. Schneider, J. N. Hay and R. J. Cava, Inorg. Chem., 2013, 52, 6083–6095 CrossRef CAS.
- R. Berthelot, W. Schmidt, A. W. Sleight and M. A. Subramanian, J. Solid State Chem., 2012, 196, 225–231 CrossRef CAS.
- S. Uma and A. Gupta, Mater. Res. Bull., 2016, 76, 118–123 CrossRef CAS.
- Z. He, W. Guo, M. Cui and Y. Tang, Dalton Trans., 2017, 46, 5076–5081 RSC.
- Z. He, M. Cui and C. Qiu, J. Alloys Compd., 2018, 748, 794–797 CrossRef CAS.
- M. I. Stratan, I. L. Shukaev, T. M. Vasilchikova, A. N. Vasiliev, A. N. Korshunov, A. I. Kurbakov, V. B. Nalbandyan and E. A. Zvereva, New J. Chem., 2019, 43, 13545–13553 RSC.
- J. Xu, A. Assoud, N. Soheilnia, S. Derakhshan, H. L. Cuthbert, J. E. Greedan, M. H. Whangbo and H. Kleinke, Inorg. Chem., 2005, 44, 5042–5046 CrossRef CAS.
- R. Ramlau, R. Schneider, J. H. Roudebush and R. J. Cava, Microsc. Microanal., 2014, 20, 950–951 CrossRef.
- O. A. Smirnova, V. B. Nalbandyan, A. A. Petrenko and M. Avdeev, J. Solid State Chem., 2005, 178, 1165–1170 CrossRef CAS.
- W. Schmidt, R. Berthelot, A. W. Sleight and M. A. Subramanian, J. Solid State Chem., 2013, 201, 178–185 CrossRef CAS.
- E. M. Seibel, J. H. Roudebush, H. Wu, Q. Huang, M. N. Ali, H. Ji and R. J. Cava, Inorg. Chem., 2013, 52, 13605–13611 CrossRef CAS.
- E. M. Seibel, J. H. Roudebush, M. N. Ali, K. A. Ross and R. J. Cava, Inorg. Chem., 2014, 53, 10989–10995 CrossRef CAS.
- J. Liu, L. Yin, L. Wu, J. Bai, S.-M. Bak, X. Yu, Y. Zhu, X.-Q. Yang and P. G. Khalifah, Inorg. Chem., 2016, 55, 8478–8492 CrossRef CAS.
- D. S. Bhange, G. Ali, D.-H. Kim, D. A. Anang, T. J. Shin, M.-G. Kim, Y.-M. Kang, K. Y. Chung and K.-W. Nam, J. Mater. Chem. A, 2017, 5, 1300–1310 RSC.
- D. Gyabeng, D. A. Anang and J. I. Han, J. Alloys Compd., 2017, 704, 734–741 CrossRef CAS.
- J.-Q. Yan, S. Okamoto, Y. Wu, Q. Zheng, H. D. Zhou, H. B. Cao and M. A. McGuire, Phys. Rev. Mater., 2019, 3, 74405 CrossRef CAS.
- D. K. Yadav, A. Sethi, Shalu and S. Uma, Dalton Trans., 2019, 48, 8955–8965 RSC.
- R. W. Smaha, J. H. Roudebush, J. T. Herb, E. M. Seibel, J. W. Krizan, G. M. Fox, Q. Huang, C. B. Arnold and R. J. Cava, Inorg. Chem., 2015, 54, 7985–7991 CrossRef CAS.
- A. J. Brown, Q. Xia, M. Avdeev, B. J. Kennedy and C. D. Ling, Inorg. Chem., 2019, 58, 13881–13891 CrossRef CAS.
- G. Heymann, E. Selb, M. Kogler, T. Götsch, E.-M. Köck, S. Penner, M. Tribus and O. Janka, Dalton Trans., 2017, 46, 12663–12674 RSC.
- V. B. Nalbandyan, A. A. Petrenko and M. A. Evstigneeva, Solid State Ionics, 2013, 233, 7–11 CrossRef CAS.
- W. Schmidt, R. Berthelot, L. Etienne, A. Wattiaux and M. A. Subramanian, Mater. Res. Bull., 2014, 50, 292–296 CrossRef CAS.
- K. Yoshii, T. Masese, M. Kato, K. Kubota, H. Senoh and M. Shikano, ChemElectroChem, 2019, 6, 3901–3910 CrossRef CAS.
- C. Delmas, C. Fouassier, J.-M. Réau and P. Hagenmuller, Mater. Res. Bull., 1976, 11, 1081–1086 CrossRef CAS.
- K. Navaratnarajah, K. Apostolos and C. Alexander, Sci. Rep., 2019, 9, 1–9 CrossRef.
- L. Zheng, H. Wang, M. Luo, G. Wang, Z. Wang and C. Ouyang, Solid State Ionics, 2018, 320, 210–214 CrossRef CAS.
- M. Koch-Janusz, D. Khomskii and E. Sela, Phys. Rev. Lett., 2015, 114, 247204 CrossRef.
- M. Tamaru, X. Wang, M. Okubo and A. Yamada, Electrochem. Commun., 2013, 33, 23–26 CrossRef CAS.
- D. D. Gazizova, A. V. Ushakov and S. V. Streltsov, JETP Lett., 2018, 107, 483–487 CrossRef CAS.
- K. M. Mogare, K. Friese, W. Klein and M. Jansen, Z. Anorg. Allg. Chem., 2004, 630, 547–552 CrossRef CAS.
- F. Lu, W. Zeng, H. Lin, S. Liu, X. Tian, J. Yang, J. Li and Y. Yang, Electrochim. Acta, 2019, 315, 48–57 CrossRef CAS.
- T. J. Bastow, M. Hobday, M. E. Smith and H. J. Whitfield, Solid State Nucl. Magn. Reson., 1994, 3, 49–57 CrossRef CAS.
- R. Wang, X. He, L. He, F. Wang, R. Xiao, L. Gu, H. Li and L. Chen, Adv. Energy Mater., 2013, 3, 1358–1367 CrossRef CAS.
- A. D. Robertson and P. G. Bruce, Chem. Mater., 2003, 15, 1984–1992 CrossRef CAS.
- A. Boulineau, L. Croguennec, C. Delmas and F. Weill, Chem. Mater., 2009, 21, 4216–4222 CrossRef CAS.
- Y. Zuo, B. Li, N. Jiang, W. Chu, H. Zhang, R. Zou and D. Xia, Adv. Mater., 2018, 30, 1707255 CrossRef.
- P. Strobel and B. Lambert-Andron, J. Solid State Chem., 1988, 75, 90–98 CrossRef CAS.
- J. Ma, Y.-N. Zhou, Y. Gao, X. Yu, Q. Kong, L. Gu, Z. Wang, X.-Q. Yang and L. Chen, Chem. Mater., 2014, 26, 3256–3262 CrossRef CAS.
- E. Shinova, E. Zhecheva, R. Stoyanova and G. D. Bromiley, J. Solid State Chem., 2005, 178, 1661–1669 CrossRef CAS.
- S. Okada, J.-I. Yamaki, K. Asakura, H. Ohtsuka, H. Arai, S.-I. Tobishima and Y. Sakurai, Electrochim. Acta, 1999, 45, 329–334 CrossRef CAS.
- K. Asakura, S. Okada, H. Arai, S.-I. Tobishima and Y. Sakurai, J. Power Sources, 1999, 81, 388–392 CrossRef.
- M. J. OMalley, H. Verweij and P. M. Woodward, J. Solid State Chem., 2008, 181, 1803–1809 CrossRef CAS.
- V. Hermann, S. Biswas, J. Ebad-Allah, F. Freund, A. Jesche, A. A. Tsirlin, M. Hanfland, D. Khomskii, P. Gegenwart and R. Valent, et al. , Phys. Rev. B, 2019, 100, 064105 CrossRef CAS.
- Y. Luo, C. Cao, B. Si, Y. Li, J. Bao, H. Guo, X. Yang, C. Shen, C. Feng and J. Dai, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 161121 CrossRef.
- V. Todorova and M. Jansen, Z. Anorg. Allg. Chem., 2011, 637, 37–40 CrossRef CAS.
- S. A. Kimber, I. Mazin, J. Shen, H. O. Jeschke, S. V. Streltsov, D. N. Argyriou, R. Valenti and D. I. Khomskii, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 081408 CrossRef.
- Y. Yu, P. Karayaylali, S. H. Nowak, L. Giordano, M. Gauthier, W. Hong, R. Kou, Q. Li, J. Vinson and T. Kroll, et al. , Chem. Mater., 2019, 31, 7864–7876 CrossRef CAS.
- H. Jang, W. Jin, G. Nam, Y. Yoo, J. Jeon, J. Park, M. G. Kim and J. Cho, Energy Environ. Sci., 2020, 13, 2167–2177 RSC.
- A. James and J. Goodenough, J. Solid State Chem., 1988, 74, 287–294 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Perez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS.
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S.-P. Cho, G. Yoon, M. H. Lee, S.-K. Jung and W. Yang, et al. , Nat. Mater., 2020, 19, 419–427 CrossRef CAS.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin and M. A. Pérez-Osorio, et al. , Nat. Chem., 2018, 10, 288 CrossRef CAS.
- N. Yabuuchi, R. Hara, M. Kajiyama, K. Kubota, T. Ishigaki, A. Hoshikawa and S. Komaba, Adv. Energy Mater., 2014, 4, 1301453 CrossRef.
- J. Cabana, N. A. Chernova, J. Xiao, M. Roppolo, K. A. Aldi, M. S. Whittingham and C. P. Grey, Inorg. Chem., 2013, 52, 8540–8550 CrossRef CAS.
- B. Song, E. Hu, J. Liu, Y. Zhang, X.-Q. Yang, J. Nanda, A. Huq and K. Page, J. Mater. Chem. A, 2019, 7, 1491–1498 RSC.
- H. Szillat and H. Müller-Buschbaum, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 261–264 CAS.
- C. Greaves and S. M. A. Katib, Mater. Res. Bull., 1990, 25, 1175–1182 CrossRef CAS.
- J. M. S. Skakle, M. A. Castellanos R., S. Tovar and A. R. West, J. Solid State Chem., 1997, 131, 115–120 CrossRef CAS.
- R. Nagarajan, S. Uma, M. K. Jayaraj, J. Tate and A. W. Sleight, Solid State Sci., 2002, 4, 787–792 CrossRef CAS.
- A. Gupta, V. Kumar and S. Uma, J. Chem. Sci., 2015, 127, 225–233 CrossRef CAS.
- E. A. Zvereva, M. I. Stratan, A. V. Ushakov, V. B. Nalbandyan, I. L. Shukaev, A. V. Silhanek, M. Abdel-Hafiez, S. V. Streltsov and A. N. Vasiliev, Dalton Trans., 2016, 45, 7373–7384 RSC.
- G. C. Mather, C. Dussarrat, J. Etourneau and A. R. West, J. Mater. Chem., 2000, 10, 2219–2230 RSC.
- G. C. Mather, R. I. Smith, J. M. S. Skakle, J. G. Fletcher, M. A. Castellanos, R. M. P. Gutierrez and A. R. West, J. Mater. Chem., 1995, 5, 1177–1182 RSC.
- P.-H. T. Nguyen, J. Milam-Guerrero, G. T. Tran, C. J. Bloed, A. J. Neer, A. Nguyen, T. Gredig, A. Huq, S. H. Lapidus and B. C. Melot, et al. , Inorg. Chem., 2020, 59, 7389–7397 CrossRef CAS.
- P.-F. Wang, H. Xin, T.-T. Zuo, Q. Li, X. Yang, Y.-X. Yin, X. Gao, X. Yu and Y.-G. Guo, Angew. Chem., Int. Ed., 2018, 130, 8310–8315 CrossRef.
- T. Marchandier, G. Rousse, Q. Jacquet, A. M. Abakumov, F. Fauth, C. V. Colin and J.-M. Tarascon, Chem. Mater., 2020, 32, 8471–8480 CrossRef CAS.
- C. Vallée, M. Saubanère, P. Sanz-Camacho, Y. Biecher, B. Fraisse, E. Suard, G. Rousse, D. Carlier and R. Berthelot, Inorg. Chem., 2019, 58, 11546–11552 CrossRef.
- S. Bette, T. Takayama, V. Duppel, A. Poulain, H. Takagi and R. E. Dinnebier, Dalton Trans., 2019, 48, 9250–9259 RSC.
- S. A. J. Kimber, C. D. Ling, D. J. P. Morris, A. Chemseddine, P. F. Henry and D. N. Argyriou, J. Mater. Chem., 2010, 20, 8021–8025 RSC.
- A. J. Brown, J. Liu, F. P. Marlton, M. Avdeev, B. J. Kennedy and C. D. Ling, J. Solid State Chem., 2020, 287, 121385 CrossRef CAS.
- N. Doudin, D. Kuhness, M. Blatnik, F. Netzer and S. Surnev, J. Condens. Matter Phys., 2017, 29, 234004 CrossRef CAS.
- J. Goniakowski and C. Noguera, J. Phys. Chem. C, 2020, 124, 8186–8197 CrossRef CAS.
- S. Pomp, D. Kuhness, G. Barcaro, L. Sementa, V. Mankad, A. Fortunelli, M. Sterrer, F. Netzer and S. Surnev, J. Phys. Chem. C, 2016, 120, 7629–7638 CrossRef CAS.
- L. Clark, G. Sala, D. D. Maharaj, M. B. Stone, K. S. Knight, M. T. Telling, X. Wang, X. Xu, J. Kim and Y. Li, et al. , Nat. Phys., 2019, 15, 262–268 Search PubMed.
- J. Sahoo and R. Flint, Phys. Rev. B, 2020, 101, 115103 CrossRef CAS.
- M. Rani, H. Sakurai and J. Ahmad, Phys. B, 2015, 456, 182–184 CrossRef CAS.
- S. Asai, M. Soda, K. Kasatani, T. Ono, V. O. Garlea, B. Winn and T. Masuda, Phys. Rev. B, 2017, 96, 104414 CrossRef.
- S. Streltsov, Phys. Met. Metallogr., 2018, 119, 1276–1279 CrossRef CAS.
- R. Uppuluri, A. Sen Gupta, A. S. Rosas and T. E. Mallouk, Chem. Soc. Rev., 2018, 47, 2401–2430 RSC.
- J. Ma, S.-H. Bo, L. Wu, Y. Zhu, C. P. Grey and P. G. Khalifah, Chem. Mater., 2015, 27, 2387–2399 CrossRef CAS.
- Y. Hosogi, H. Kato and A. Kudo, J. Mater. Chem., 2008, 18, 647–653 RSC.
- A. J. Martinolich and J. R. Neilson, Chem. Mater., 2017, 29, 479–489 CrossRef CAS.
- V. Todorova, A. Leineweber, L. Kienle, V. Duppel and M. Jansen, J. Solid State Chem., 2011, 184, 1112–1119 CrossRef CAS.
- C. Heubner, T. Lein, M. Schneider and A. Michaelis, J. Mater. Chem. A, 2020, 8, 16854–16883 RSC.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1046–1048 CrossRef CAS.
- T. Marchandier, Q. Jacquet, G. Rousse, B. Baptiste, A. M. Abakumov and J.-M. Tarascon, Chem. Mater., 2019, 31, 6295–6305 CrossRef CAS.
- L. Wang, J. Chu, B. Gao, J. Pan, W. Li, T. Wang, J. Feng, H. Zhang, G. Mu and F. Huang, et al. , J. Alloys Compd., 2020, 816, 152672 CrossRef CAS.
- S.-H. Bo, C. P. Grey and P. G. Khalifah, Chem. Mater., 2015, 27, 4630–4639 CrossRef CAS.
- A. R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499–500 CrossRef CAS.
- F. Capitaine, P. Gravereau and C. Delmas, Solid State Ionics, 1996, 89, 197–202 CrossRef CAS.
- Y. Orikasa, T. Masese, Y. Koyama, T. Mori, M. Hattori, K. Yamamoto, T. Okado, Z.-D. Huang, T. Minato, C. Tassel, J. Kim, Y. Kobayashi, T. Abe, H. Kageyama and Y. Uchimoto, Sci. Rep., 2014, 4, 5622 CrossRef CAS.
- J. Paulsen, C. Thomas and J. Dahn, J. Electrochem. Soc., 1999, 146, 3560–3565 CrossRef CAS.
- A. D. Robertson, A. R. Armstrong, A. J. Fowkes and P. G. Bruce, J. Mater. Chem., 2001, 11, 113–118 RSC.
- H. Gwon, S.-W. Kim, Y.-U. Park, J. Hong, G. Ceder, S. Jeon and K. Kang, Inorg. Chem., 2014, 53, 8083–8087 CrossRef CAS.
- S. Baskar, K. Sada and P. Barpanda, ECS Trans., 2017, 80, 357–364 CrossRef CAS.
- N. Naveen, W. B. Park, S. C. Han, S. P. Singh, Y. H. Jung, D. Ahn, K.-S. Sohn and M. Pyo, Chem. Mater., 2018, 30, 2049–2057 CrossRef CAS.
- J.-Y. Hwang, J. Kim, T.-Y. Yu, S.-T. Myung and Y.-K. Sun, Energy Environ. Sci., 2018, 11, 2821–2827 RSC.
- H. Kim, D.-H. Kwon, J. C. Kim, B. Ouyang, H. Kim, J. Yang and G. Ceder, Chem. Mater., 2020, 4312–4323 CrossRef.
- T. Masese, K. Yoshii, M. Kato, K. Kubota, Z.-D. Huang, H. Senoh and M. Shikano, Chem. Commun., 2019, 55, 985–988 RSC.
- P.-F. Wang, M. Weng, Y. Xiao, Z. Hu, Q. Li, M. Li, Y.-D. Wang, X. Chen, X. Yang, Y. Wen, Y.-X. Yin, X. Yu, Y. Xiao, J. Zheng, L.-J. Wan, F. Pan and Y.-G. Guo, Adv. Mater., 2019, 31, 1903483 CrossRef CAS.
- L. Xiao, Z. Ding, C. Chen, Z. Han, P. Wang, Q. Huang, P. Gao and W. Wei, iScience, 2020, 23, 100898 CrossRef CAS.
- S. Bette, T. Takayama, V. Duppel, A. Poulain, H. Takagi and R. E. Dinnebier, Dalton Trans., 2019, 48, 9250–9259 RSC.
- C. Delmas, C. Fouassier and P. Hagenmuller, Phys. B, 1980, 99, 81–85 CrossRef CAS.
- M. A. Evstigneeva, V. B. Nalbandyan, A. A. Petrenko, B. S. Medvedev and A. A. Kataev, Chem. Mater., 2011, 23, 1174–1181 CrossRef CAS.
- L. Zheng and M. N. Obrovac, J. Electrochem. Soc., 2016, 163, A2362–A2367 CrossRef CAS.
- V. Politaev and V. Nalbandyan, Solid State Sci., 2009, 11, 144–150 CrossRef CAS.
- J. Bréger, M. Jiang, N. Dupré, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466, DOI:10.1021/acs.chemrev.9b00463.
- X. Li, F. Bianchini, J. Wind, C. Pettersen, D. Wragg, P. Vajeeston and H. Fjellvåg, ACS Appl. Mater. Interfaces, 2020, 12, 28188–28198 CrossRef CAS.
- A. Kitaev, Ann. Phys., 2006, 321, 2–111 CAS.
- Y. Zhou, K. Kanoda and T.-K. Ng, Rev. Mod. Phys., 2017, 89, 025003 CrossRef.
- G. Baskaran, S. Mandal and R. Shankar, Phys. Rev. Lett., 2007, 98, 247201 CrossRef CAS.
- L. Savary and L. Balents, Rep. Prog. Phys., 2016, 80, 016502 CrossRef.
- C. Broholm, R. Cava, S. Kivelson, D. Nocera, M. Norman and T. Senthil, Science, 2020, 367, 1–9 CrossRef.
- H.-D. Chen and Z. Nussinov, J. Phys. A: Math. Theor., 2008, 41, 075001 CrossRef.
- M. Freedman, A. Kitaev, M. Larsen and Z. Wang, Bull. Amer. Math. Soc., 2003, 40, 31–38 CrossRef.
- A. Y. Kitaev, Ann. Phys., 2003, 303, 2–30 CAS.
- J. Chaloupka, G. Jackeli and G. Khaliullin, Phys. Rev. Lett., 2013, 110, 097204 CrossRef.
- H. Liu, J. Chaloupka and G. Khaliullin, 2020, arXiv preprint arXiv:2002.05441.
- R. Zhong, T. Gao, N. P. Ong and R. J. Cava, Sci. Adv., 2020, 6, 6953 CrossRef.
- H. Liu and G. Khaliullin, Phys. Rev. B, 2018, 97, 014407 CrossRef CAS.
- R. Sano, Y. Kato and Y. Motome, Phys. Rev. B, 2018, 97, 014408 CrossRef CAS.
- C. Xu, J. Feng, M. Kawamura, Y. Yamaji, Y. Nahas, S. Prokhorenko, Y. Qi, H. Xiang and L. Bellaiche, Phys. Rev. Lett., 2020, 124, 087205 CrossRef CAS.
- F. D. M. Haldane, Phys. Rev. Lett., 1988, 61, 2015–2018 CrossRef CAS.
- N. Regnault and B. A. Bernevig, Phys. Rev. X, 2011, 1, 21014 Search PubMed.
- T. Thonhauser and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 235111 CrossRef.
- S. Trebst, 2017, arXiv preprint arXiv:1701.07056.
- F. Ye, S. Chi, H. Cao, B. C. Chakoumakos, J. A. Fernandez-Baca, R. Custelcean, T. Qi, O. Korneta and G. Cao, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 180403 CrossRef.
- Y. Sizyuk, C. Price, P. Wölfle and N. B. Perkins, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 155126 CrossRef.
- E. K.-H. Lee and Y. B. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 064407 CrossRef.
- S. Choi, R. Coldea, A. Kolmogorov, T. Lancaster, I. Mazin, S. Blundell, P. Radaelli, Y. Singh, P. Gegenwart and K. Choi, et al. , Phys. Rev. Lett., 2012, 108, 127204 CrossRef CAS.
- J. G. Rau, E. K.-H. Lee and H.-Y. Kee, Annu. Rev. Condens. Matter Phys., 2016, 7, 195–221 CrossRef CAS.
- J. Knolle, G.-W. Chern, D. Kovrizhin, R. Moessner and N. Perkins, 2014, arXiv preprint arXiv:1406.3944.
- A. Glamazda, P. Lemmens, S.-H. Do, Y. Choi and K.-Y. Choi, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- Y. Singh, S. Manni, J. Reuther, T. Berlijn, R. Thomale, W. Ku, S. Trebst and P. Gegenwart, Phys. Rev. Lett., 2012, 108, 127203 CrossRef.
- J. Clancy, N. Chen, C. Kim, W. Chen, K. Plumb, B. Jeon, T. Noh and Y.-J. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195131 CrossRef.
- X. Wan, A. M. Turner, A. Vishwanath and S. Y. Savrasov, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 205101 CrossRef.
- J. Chaloupka, G. Jackeli and G. Khaliullin, Phys. Rev. Lett., 2010, 105, 027204 CrossRef.
- I. Mazin, S. Manni, K. Foyevtsova, H. O. Jeschke, P. Gegenwart and R. Valent, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 035115 CrossRef.
- S. Manni, Y. Tokiwa and P. Gegenwart, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 241102 CrossRef.
- S. H. Chun, J.-W. Kim, J. Kim, H. Zheng, C. C. Stoumpos, C. Malliakas, J. Mitchell, K. Mehlawat, Y. Singh and Y. Choi, et al. , Nat. Phys., 2015, 11, 462–466 Search PubMed.
- S. Nishimoto, V. M. Katukuri, V. Yushankhai, H. Stoll, U. K. Rößler, L. Hozoi, I. Rousochatzakis and J. Van Den Brink, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- E. C. Andrade and M. Vojta, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 205112 CrossRef.
- S. Guo, R. Zhong, W. Wang, J. Tao, D. Ni and R. J. Cava, J. Am. Chem. Soc., 2020, 142, 5389–5395 CrossRef CAS.
- T. Takayama, A. Kato, R. Dinnebier, J. Nuss, H. Kono, L. Veiga, G. Fabbris, D. Haskel and H. Takagi, Phys. Rev. Lett., 2015, 114, 077202 CrossRef CAS.
- J. Wang, J. Terzic, T. Qi, F. Ye, S. Yuan, S. Aswartham, S. Streltsov, D. Khomskii, R. K. Kaul and G. Cao, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 161110 CrossRef.
- I. Kimchi and Y.-Z. You, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 180407 CrossRef.
- J. Chaloupka and G. Khaliullin, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 024413 CrossRef.
- Y. Yamaji, Y. Nomura, M. Kurita, R. Arita and M. Imada, Phys. Rev. Lett., 2014, 113, 107201 CrossRef.
- S. Bhattacharjee, S.-S. Lee and Y. B. Kim, New J. Phys., 2012, 14, 073015 CrossRef.
- T. Suzuki, T. Yamada, Y. Yamaji and S.-I. Suga, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 184411 CrossRef.
- V. Hermann, M. Altmeyer, J. Ebad-Allah, F. Freund, A. Jesche, A. A. Tsirlin, M. Hanfland, P. Gegenwart, I. Mazin and D. Khomskii, et al. , Phys. Rev. B, 2018, 97, 020104 CrossRef CAS.
- G. Cao and P. Schlottmann, 2017, arXiv preprint arXiv:1704.06007.
- G. Cao and P. Schlottmann, Rep. Prog. Phys., 2018, 81, 042502 CrossRef.
- J. G. Rau, E. K.-H. Lee and H.-Y. Kee, Phys. Rev. Lett., 2014, 112, 077204 CrossRef.
- M. Majumder, R. Manna, G. Simutis, J. Orain, T. Dey, F. Freund, A. Jesche, R. Khasanov, P. Biswas and E. Bykova, et al. , Phys. Rev. Lett., 2018, 120, 237202 CrossRef CAS.
- V. M. Katukuri, R. Yadav, L. Hozoi, S. Nishimoto and J. Van Den Brink, Sci. Rep., 2016, 6, 29585 CrossRef CAS.
- I. Kimchi, J. G. Analytis and A. Vishwanath, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 205126 CrossRef.
- P. E. Pearce, G. Assat, A. Iadecola, F. Fauth, R. Dedryvère, A. Abakumov, G. Rousse and J.-M. Tarascon, J. Phys. Chem. C, 2020, 124, 2771–2781 CrossRef CAS.
- K. A. Modic, T. E. Smidt, I. Kimchi, N. P. Breznay, A. Biffin, S. Choi, R. D. Johnson, R. Coldea, P. Watkins-Curry and G. T. McCandless, et al. , Nat. Commun., 2014, 5, 1–6 Search PubMed.
- F. Freund, S. Williams, R. Johnson, R. Coldea, P. Gegenwart and A. Jesche, Sci. Rep., 2016, 6, 35362 CrossRef CAS.
- Y. Kasahara, T. Ohnishi, Y. Mizukami, O. Tanaka, S. Ma, K. Sugii, N. Kurita, H. Tanaka, J. Nasu and Y. Motome, et al. , Nature, 2018, 559, 227–231 CrossRef CAS.
- Y. Vinkler-Aviv and A. Rosch, Phys. Rev. X, 2018, 8, 031032 CAS.
- Y. Kasahara, K. Sugii, T. Ohnishi, M. Shimozawa, M. Yamashita, N. Kurita, H. Tanaka, J. Nasu, Y. Motome and T. Shibauchi, et al. , Phys. Rev. Lett., 2018, 120, 217205 CrossRef CAS.
- T. Yokoi, S. Ma, Y. Kasahara, S. Kasahara, T. Shibauchi, N. Kurita, H. Tanaka, J. Nasu, Y. Motome, C. Hickey, et al., 2020, arXiv preprint arXiv:2001.01899.
- D. Aasen, R. S. Mong, B. M. Hunt, D. Mandrus and J. Alicea, 2020, arXiv preprint arXiv:2002.01944.
- G. Jackeli and G. Khaliullin, Phys. Rev. Lett., 2009, 102, 017205 CrossRef CAS.
- A. Georges, G. Kotliar, W. Krauth and M. J. Rozenberg, Rev. Mod. Phys., 1996, 68, 13 CrossRef CAS.
- M. Imada, A. Fujimori and Y. Tokura, Rev. Mod. Phys., 1998, 70, 1039 CrossRef CAS.
- S. M. Winter, A. A. Tsirlin, M. Daghofer, J. van den Brink, Y. Singh, P. Gegenwart and R. Valenti, J. Condens. Matter Phys., 2017, 29, 493002 CrossRef.
- R. G. Burns and R. G. Burns, Mineralogical applications of crystal field theory, Cambridge University Press, 1993, vol. 5 Search PubMed.
- S. M. Winter, Y. Li, H. O. Jeschke and R. Valent, Phys. Rev. B, 2016, 93, 214431 CrossRef.
- H. Jiang, R. I. Gomez-Abal, P. Rinke and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045108 CrossRef.
- S.-H. Jang, R. Sano, Y. Kato and Y. Motome, Phys. Rev. B, 2019, 99, 241106 CrossRef CAS.
- N. A. Spaldin, Magnetic materials: fundamentals and device applications, Cambridge University Press, Cambridge, 2006, pp. 89–106 Search PubMed.
- C. Kittel, Introduction to Solid State Physics, John Wiley & Sons, New York, 8th edn, 2005 Search PubMed.
- M. Bieringer, J. E. Greedan and G. M. Luke, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 6521–6529 CrossRef CAS.
- U. K. Rößler, A. N. Bogdanov and C. Pfleiderer, Nature, 2006, 442, 797–801 CrossRef.
- J. M. Kosterlitz and D. J. Thouless, J. Phys. C: Solid State Phys., 1973, 6, 1181–1203 CrossRef CAS.
- Y. Li, Z. Deng, J. Peng, E. Chen, Y. Yu, X. Li, J. Luo, Y. Huang, J. Zhu, C. Fang, Q. Li, J. Han and Y. Huang, Chem. – Eur. J., 2018, 24, 1057–1061 CrossRef CAS.
- Y. Li, Z. Deng, J. Peng, J. Gu, E. Chen, Y. Yu, J. Wu, X. Li, J. Luo, Y. Huang, Y. Xu, Z. Gao, C. Fang, J. Zhu, Q. Li, J. Han and Y. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 15760–15766 CrossRef CAS.
- J.-F. Wu, Q. Wang and X. Guo, J. Power Sources, 2018, 402, 513–518 CrossRef CAS.
- Z. Deng, J. Gu, Y. Li, S. Li, J. Peng, X. Li, J. Luo, Y. Huang, C. Fang, Q. Li, J. Han, Y. Huang and Y. Zhao, Electrochim. Acta, 2019, 298, 121–126 CrossRef CAS.
- F. Bianchini, H. Fjellvåg and P. Vajeeston, J. Phys. Chem. C, 2019, 123, 4654–4663 CrossRef CAS.
- J.-F. Wu, Z.-Y. Yu, Q. Wang and X. Guo, Energy Storage Mater., 2020, 24, 467–471 CrossRef.
- M. Dubey, A. Kumar, S. Murugavel, G. V. Prakash, D. A. Jose and C. R. Mariappan, Ceram. Int., 2020, 46, 663–671 CrossRef CAS.
- C.-Y. Chen, J. Rizell, G. M. Kanyolo, T. Masese, Y. Sassa, M. Månsson, K. Kubota, K. Matsumoto, R. Hagiwara and Q. Xu, Chem. Commun., 2020, 56, 9272–9275 RSC.
- K. Sau and P. P. Kumar, J. Phys. Chem. C, 2015, 119, 18030–18037 CrossRef CAS.
- K. Sau, Ionics, 2016, 22, 2379–2385 CrossRef CAS.
- A. Lasia, Modern Aspects of Electrochemistry, Springer, Boston, 1999, pp. 143–248 Search PubMed.
- J. Weber, Phys. Rev., 1956, 101, 1620–1626 CrossRef.
- G. M. Kanyolo and T. Masese, Sci. Rep., 2020, 10, 13284 CrossRef CAS.
- A. Fick, J. Membr. Sci., 1995, 100, 33–38 CrossRef CAS.
- P. Langevin, C. R. Acad. Sci., 1908, 146, 530–533 CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- M. Von Smoluchowski, Ann. Phys., 1906, 326, 756–780 CrossRef.
- R. P. Feynman, R. B. Leighton and M. Sands, The Feynman Lectures on Physics, Addison-Wesley, 2nd edn, 2005, ch. 41, vol. 1 Search PubMed.
- A. K. Bera and S. M. Yusuf, J. Phys. Chem. C, 2020, 124, 4421–4429 CrossRef CAS.
- K. Sau, AIP Conf. Proc., 2016, 020119 CrossRef.
- K. Sau and P. P. Kumar, J. Phys. Chem. C, 2015, 119, 1651–1658 CrossRef CAS.
- Y.-M. Choi, S.-I. Pyun and S.-I. Moon, Solid State Ionics, 1996, 89, 43–52 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- A. K. Padhi, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS.
- K. Matsumoto, J. Hwang, S. Kaushik, C.-Y. Chen and R. Hagiwara, Energy Environ. Sci., 2019, 12, 3247–3287 RSC.
- T. Yamamoto, K. Matsumoto, R. Hagiwara and T. Nohira, J. Phys. Chem. C, 2017, 121, 18450–18458 CrossRef CAS.
- J. Hwang, K. Matsumoto and R. Hagiwara, Adv. Sustainable Syst., 2018, 2, 1700171 CrossRef.
- K. Kubota, T. Nohira and R. Hagiwara, Electrochim. Acta, 2012, 66, 320–324 CrossRef CAS.
- C.-Y. Chen, T. Kiko, T. Hosokawa, K. Matsumoto, T. Nohira and R. Hagiwara, J. Power Sources, 2016, 332, 51–59 CrossRef CAS.
- K. Matsumoto, T. Hosokawa, T. Nohira, R. Hagiwara, A. Fukunaga, K. Numata, E. Itani, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2014, 265, 36–39 CrossRef CAS.
- K. Kubota, T. Nohira, T. Goto and R. Hagiwara, Electrochem. Commun., 2008, 10, 1886–1888 CrossRef CAS.
- A. Fukunaga, T. Nohira, Y. Kozawa, R. Hagiwara, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2012, 209, 52–56 CrossRef CAS.
- C. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 238, 296–300 CrossRef CAS.
- R. Hagiwara and Y. Ito, J. Fluorine Chem., 2000, 105, 221–227 CrossRef CAS.
- D. Monti, E. Jónsson, M. R. Palacn and P. Johansson, J. Power Sources, 2014, 245, 630–636 CrossRef CAS.
- N. Wongittharom, C.-H. Wang, Y.-C. Wang, C.-H. Yang and J.-K. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 17564–17570 CrossRef CAS.
- C.-H. Wang, Y.-W. Yeh, N. Wongittharom, Y.-C. Wang, C.-J. Tseng, S.-W. Lee, W.-S. Chang and J.-K. Chang, J. Power Sources, 2015, 274, 1016–1023 CrossRef CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira, C. Ding, T. Yamamoto and R. Hagiwara, Electrochim. Acta, 2014, 133, 583–588 CrossRef CAS.
- A. Fukunaga, T. Nohira, R. Hagiwara, K. Numata, E. Itani, S. Sakai and K. Nitta, J. Appl. Electrochem., 2016, 46, 487–496 CrossRef CAS.
- S. A. M. Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef.
- C.-Y. Chen, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 237, 52–57 CrossRef CAS.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS.
- H. Ohno, Electrochemical aspects of ionic liquids, John Wiley & Sons, 2005 Search PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- Y. Kee, N. Dimov, A. Staykov and S. Okada, Mater. Lett., 2016, 183, 187–190 CrossRef CAS.
- F. Aguesse, J.-M. Lopez del Amo, L. Otaegui, E. Goikolea, T. Rojo and G. Singh, J. Power Sources, 2016, 336, 186–195 CrossRef CAS.
- Y. Kee, B. Put, N. Dimov, A. Staykov and S. Okada, Batteries Supercaps, 2020, 3, 390–391 CrossRef CAS.
- H. Matsumoto, H. Sakaebe and K. Tatsumi, J. Power Sources, 2005, 146, 45–50 CrossRef CAS.
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 160, 1308–1313 CrossRef CAS.
- H. Sakaebe and H. Matsumoto, Electrochem. Commun., 2003, 5, 594–598 CrossRef CAS.
- P.-F. Wang, H.-R. Yao, Y. You, Y.-G. Sun, Y.-X. Yin and Y.-G. Guo, Nano Res., 2018, 11, 3258–3271 CrossRef CAS.
- J. Bao, D. Wu, Q. Tang, Z. Ma and Z. Zhou, Phys. Chem. Chem. Phys., 2014, 16, 16145–16149 RSC.
- S. Kim, M. Aykol, V. I. Hegde, Z. Lu, S. Kirklin, J. R. Croy, M. M. Thackeray and C. Wolverton, Energy Environ. Sci., 2017, 10, 2201–2211 RSC.
- B. Huang, H. Wang, S. Zhang and C. Deng, Electrochim. Acta, 2020, 334, 135644 CrossRef CAS.
- C. Wang, G. Zhang, W. Qu, H. Wang, S. Zhang and C. Deng, J. Mater. Chem. A, 2019, 7, 1797–1809 RSC.
- E. Paharik, K. Rudman and G. Koenig, ECS Trans., 2017, 80, 259–266 CrossRef CAS.
- H. Dai, C. Yang, X. Ou, X. Liang, H. Xue, W. Wang and G. Xu, Electrochim. Acta, 2017, 257, 146–154 CrossRef CAS.
- J. Han, Y. Niu, Y. Zhang, J. Jiang, S.-J. Bao and M. Xu, J. Solid State Electrochem., 2016, 20, 2331–2335 CrossRef CAS.
- C. Zhao, Y. Lu, L. Chen and Y.-S. Hu, Nano Res., 2019, 12, 2018–2030 CrossRef CAS.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák and J.-M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS.
- M. Stern and A. L. Geaby, J. Electrochem. Soc., 1957, 104, 56 CrossRef CAS.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J.-M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS.
- S. Laha, S. Natarajan, J. Gopalakrishnan, E. Morán, R. Sáez-Puche, M. Alario-Franco, A. Dos Santos-Garcia, J. Pérez-Flores, A. Kuhn and F. Garca-Alvarado, Phys. Chem. Chem. Phys., 2015, 17, 3749–3760 RSC.
- J. Hong, W. E. Gent, P. Xiao, K. Lim, D.-H. Seo, J. Wu, P. M. Csernica, C. J. Takacs, D. Nordlund, C.-J. Sun, K. H. Stone, D. Passarello, W. Yang, D. Prendergast, G. Ceder, M. F. Toney and W. C. Chueh, Nat. Mater., 2019, 18, 256–265 CrossRef CAS.
- M. B. Yahia, J. Vergnet, M. Saubanère and M.-L. Doublet, Nat. Mater., 2019, 18, 496–502 CrossRef.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- B. Li and D. Xia, Adv. Mater., 2017, 29, 1701054 CrossRef.
- H. Xu, S. Guo and H. Zhou, J. Mater. Chem. A, 2019, 7, 23662–23678 RSC.
- M. Li, T. Liu, X. Bi, Z. Chen, K. Amine, C. Zhong and J. Lu, Chem. Soc. Rev., 2020, 49, 1688–1705 RSC.
- D. Foix, M. Sathiya, E. McCalla, J.-M. Tarascon and D. Gonbeau, J. Phys. Chem. C, 2016, 120, 862–874 CrossRef CAS.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi and A. Nakao, et al. , Nat. Commun., 2016, 7, 1–10 Search PubMed.
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C.-J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák and G. Van Tendeloo, et al. , Science, 2015, 350, 1516–1521 CrossRef CAS.
- M. Oishi, K. Yamanaka, I. Watanabe, K. Shimoda, T. Matsunaga, H. Arai, Y. Ukyo, Y. Uchimoto, Z. Ogumi and T. Ohta, J. Mater. Chem. A, 2016, 4, 9293–9302 RSC.
- Y. Xie, M. Saubanère and M.-L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- A. Watanabe, K. Yamamoto, T. Uchiyama, T. Matsunaga, A. Hayashi, K. Maeda, H. Kageyama and Y. Uchimoto, ACS Appl. Energy Mater., 2020, 3, 4162–4167 CrossRef CAS.
- K. Yamamoto, Y. Zhou, N. Yabuuchi, K. Nakanishi, T. Yoshinari, T. Kobayashi, Y. Kobayashi, R. Yamamoto, A. Watanabe and Y. Orikasa, et al. , Chem. Mater., 2020, 32, 139–147 CrossRef CAS.
- T. Masese, C. Tassel, Y. Orikasa, Y. Koyama, H. Arai, N. Hayashi, J. Kim, T. Mori, K. Yamamoto and Y. Kobayashi, et al. , J. Phys. Chem. C, 2015, 119, 10206–10211 CrossRef CAS.
- M. Oishi, C. Yogi, I. Watanabe, T. Ohta, Y. Orikasa, Y. Uchimoto and Z. Ogumi, J. Power Sources, 2015, 276, 89–94 CrossRef CAS.
- M. Oishi, T. Fujimoto, Y. Takanashi, Y. Orikasa, A. Kawamura, T. Ina, H. Yamashige, D. Takamatsu, K. Sato and H. Murayama, et al. , J. Power Sources, 2013, 222, 45–51 CrossRef CAS.
- T. Masese, K. Yoshii, K. Tada, M. Kato, S. Uchida, K. Kubota, T. Ina, T. Okumura, Z.-D. Huang and J. Furutani, et al. , Energy Technol., 2020, 2000039 CrossRef CAS.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- B. M. De Boisse, G. Liu, J. Ma, S.-I. Nishimura, S.-C. Chung, H. Kiuchi, Y. Harada, J. Kikkawa, Y. Kobayashi and M. Okubo, et al. , Nat. Commun., 2016, 7, 1–9 Search PubMed.
- P. Rozier, M. Sathiya, A.-R. Paulraj, D. Foix, T. Desaunay, P.-L. Taberna, P. Simon and J.-M. Tarascon, Electrochem. Commun., 2015, 53, 29–32 CrossRef CAS.
- M. Assadi, M. Okubo, A. Yamada and Y. Tateyama, J. Mater. Chem. A, 2018, 6, 3747–3753 RSC.
- M. Jia, H. Wang, H. Wang, Y. Chen, C. Guo and L. Gan, J. Solid State Chem., 2017, 254, 132–137 CrossRef CAS.
- I. Saadoune and A. Maazaz, J. Solid State Chem., 1996, 122, 111 CrossRef CAS.
- L. Croguennec, C. Pouillerie and C. Delmas, J. Electrochem. Soc., 2000, 147, 1314 CrossRef CAS.
- Y. Hironaka, K. Kubota and S. Komaba, Chem. Commun., 2017, 53, 3693–3696 RSC.
- P.-F. Wang, H.-R. Yao, X.-Y. Liu, Y.-X. Yin, J.-N. Zhang, Y. Wen, X. Yu, L. Gu and Y.-G. Guo, Sci. Adv., 2018, 4, eaar6018 CrossRef.
- P.-F. Wang, Y.-J. Guo, H. Duan, T.-T. Zuo, E. Hu, K. Attenkofer, H. Li, X. S. Zhao, Y.-X. Yin, X. Yu and Y.-G. Guo, ACS Energy Lett., 2017, 2, 2715–2722 CrossRef CAS.
- A. Zee, Quantum field theory in a nutshell, Princeton University Press, 2010, vol. 7 Search PubMed.
- R. Jackiw and E. J. Weinberg, Phys. Rev. Lett., 1990, 64, 2234 CrossRef.
- C. B. Allendoerfer and A. Weil, Trans. Amer. Math. Soc., 1943, 53, 101–129 CrossRef.
- B. Dubrovin, A. Fomenko and S. Novikov, Modern geometry methods—and applications: Part II: The geometry and topology of manifolds, Springer Science & Business Media, 2012, vol. 104 Search PubMed.
- A. Einstein, et al. , Ann. Phys., 1916, 49, 769–822 CrossRef CAS.
- G. V. Dunne, Topological aspects of low dimensional systems, Springer, Berlin Heidelberg, 2007, pp. 177–263 Search PubMed.
- M. Marino, Rev. Mod. Phys., 2005, 77, 675 CrossRef CAS.
- G. V. Dunne, R. Jackiw and C. A. Trugenberger, Phys. Rev. D: Part. Fields, 1990, 41, 661 CrossRef.
- E. Witten, Chern-Simons Gauge Theory: 20 Years After, American Mathematical Society/International Press, 2011, vol. 50, p. 347 Search PubMed.
- S. Alexander and N. Yunes, Phys. Rep., 2009, 480, 1–55 CrossRef CAS.
- S. Axelrod and I. M. Singer, 1993, arXiv, preprint, arXiv:hep-th/9304087,https://arxiv.org/abs/hep-th/9304087v1.
- M. V. Berry, Proc. R. Soc. London, Ser. A, 1984, 392, 45 Search PubMed.
- J. H. Schwarz, J. High Energy Phys., 2005, 2004, 078 CrossRef.
- L. N. Cooper, Phys. Rev., 1956, 104, 1189–1190 CrossRef CAS.
- J. Bardeen, Science, 1973, 181, 1209–1214 CrossRef CAS.
- E. P. Gross, J. Math. Phys., 1963, 4, 195 CrossRef CAS.
- L. P. Pitaevskii, J. Exp. Theor. Phys., 1961, 13, 451 Search PubMed.
- P. Billingsley, Probability and Measure, John Wiley & Sons, 3rd edn, 1995 Search PubMed.
- R. Kubo and T. Toyabe, Ampere (Ljubljana, 1966) ed R Blinc, Amsterdam, North-Holland, 1967, vol. 810 Search PubMed.
- R. Hayano, Y. Uemura, J. Imazato, N. Nishida, T. Yamazaki and R. Kubo, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 850 CrossRef CAS.
- T. Yamazaki, Hyperfine Interact., 1979, 6, 115–125 CrossRef CAS.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Phys. Rev. Lett., 2009, 103, 147601 CrossRef.
- M. Månsson and J. Sugiyama, Phys. Scr., 2013, 88, 68509 CrossRef.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, O. Ofer, M. Månsson, J. H. Brewer, E. J. Ansaldo, K. H. Chow, Y. Ikedo, Y. Miyake, K. Ohishi, I. Watanabe, G. Kobayashi and R. Kanno, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 54430 CrossRef.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, Y. Ikedo, Y. Miyake, O. Ofer, M. Månsson, E. J. Ansaldo, K. H. Chow, G. Kobayashi and R. Kanno, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 54111 CrossRef.
- J. Sugiyama, K. Mukai, H. Nozaki, M. Harada, K. Kamazawa, Y. Ikedo, M. Månsson, O. Ofer, E. J. Ansaldo, J. H. Brewer, K. H. Chow, I. Watanabe, Y. Miyake and T. Ohzuku, Phys. Procedia, 2012, 30, 105–108 CrossRef CAS.
- T.-D. Lee and C.-N. Yang, Phys. Rev., 1956, 104, 254 CrossRef CAS.
- S. Blundell, Contemp. Phys., 1999, 40, 175–192 CrossRef CAS.
- A. Yaouanc and P. D. De Reotier, Muon spin rotation, relaxation, and resonance: applications to condensed matter, Oxford University Press, 2011, vol. 147 Search PubMed.
- N. Matsubara, E. Nocerino, O. K. Forslund, A. Zubayer, P. Gratrex, D. Andreica, J. Sugiyama, Z. Guguchia, S. Cottrell, A. Kalaboukhov, Y. Sassa, T. Masese and M. Månsson, 2020, arXiv, preprint, arXiv:2003.05805, https://arxiv.org/abs/2003.05805v1.
- E. Behrends, Introduction to Markov chains, Springer, 2000, vol. 228 Search PubMed.
- R. J. Borg and G. J. Dienes, An introduction to solid state diffusion, Elsevier, 2012 Search PubMed.
- J. Sugiyama, Y. Ikedo, O. Ofer, M. Månsson, E. J. Ansaldo, J. H. Brewer, K. H. Chow, H. Sakurai and E. Takayama-Muromachi, J. Phys. Soc. Jpn., 2009, 78, 84715 CrossRef.
- J. Sugiyama, M. Månsson, Y. Ikedo, T. Goko, K. Mukai, D. Andreica, A. Amato, K. Ariyoshi and T. Ohzuku, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 184411 CrossRef.
- J. Sugiyama, Y. Ikedo, M. Månsson, J. H. Brewer, S. L. Stubbs, E. J. Ansaldo, K. H. Chow, J. S. Lord, H. Ohta, C. Michioka and K. Yoshimura, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 214505 CrossRef.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, J. Phys.: Conf. Ser., 2010, 225, 12052 CrossRef.
- J. Sugiyama, Y. Ikedo, K. Mukai, H. Nozaki, M. Månsson, O. Ofer, M. Harada, K. Kamazawa, Y. Miyake, J. H. Brewer, E. J. Ansaldo, K. H. Chow, I. Watanabe and T. Ohzuku, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 224412 CrossRef.
- J. Sugiyama, H. Nozaki, M. Månsson, K. Prša, D. Andreica, A. Amato, M. Isobe and Y. Ueda, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 214407 CrossRef.
- J. Sugiyama, K. Mukai, H. Nozaki, M. Harada, M. Månsson, K. Kamazawa, D. Andreica, A. Amato and A. D. Hillier, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 24409 CrossRef.
- J. Sugiyama, H. Nozaki, K. Miwa, H. Yoshida, M. Isobe, K. Prša, A. Amato, D. Andreica and M. Månsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 184417 CrossRef.
- J. Sugiyama, H. Nozaki, K. Mukai, M. Harada, M. Månsson and A. Hillier, Solid State Ionics, 2014, 262, 901–903 CrossRef CAS.
- J. Sugiyama, H. Nozaki, M. Harada, Y. Higuchi, H. Sakurai, E. J. Ansaldo, J. H. Brewer, L. Keller, V. Pomjakushin and M. Månsson, Phys. Procedia, 2015, 75, 868–875 CrossRef CAS.
- J. Sugiyama, I. Umegaki, D. Andreica, C. Baines, A. Amato, M. Guignard, C. Delmase and M. Månsson, RSC Adv., 2015, 5, 18531–18537 RSC.
- J. Sugiyama, H. Nozaki, I. Umegaki, K. Mukai, K. Miwa, S. Shiraki, T. Hitosugi, A. Suter, T. Prokscha, Z. Salman, J. S. Lord and M. Månsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 14417 CrossRef.
- J. Sugiyama, H. Nozaki, I. Umegaki, K. Mukai, S. P. Cottrell, S. Shiraki, T. Hitosugi, Y. Sassa, A. Suter, Z. Salman, T. Prokscha and M. Månsson, JPS Conf. Proc., 2018, 21, 011015 Search PubMed.
- J. Sugiyama, H. Nozaki, I. Umegaki, Y. Higuchi, S. P. Cottrell, S. Hori, R. Kanno and M. Månsson, JPS Conf. Proc., 2018, 21, 011016 Search PubMed.
- J. Sugiyama, D. Andreica, O. K. Forslund, E. Nocerino, N. Matsubara, Y. Sassa, Z. Guguchia, R. Khasanov, F. L. Pratt, H. Nakamura and M. Månsson, Phys. Rev. B, 2020, 101, 174403 CrossRef CAS.
- Y. Ikedo, J. Sugiyama, O. Ofer, M. Månsson, H. Sakurai, E. Takayama-Muromachi, E. J. Ansaldo, J. H. Brewer and K. H. Chow, J. Phys.: Conf. Ser., 2010, 225, 12017 CrossRef.
- K. Mukai, J. Sugiyama, Y. Ikedo, H. Nozaki, K. Kamazawa, D. Andreica, A. Amato, M. Månsson, J. H. Brewer, E. J. Ansaldo and K. H. Chow, J. Phys. Chem. C, 2010, 114, 11320–11327 CrossRef CAS.
- H. Ohta, M. Månsson, Y. Ikedo, J. Sugiyama, C. Michioka, K. Yoshimura, J. H. Brewer, E. J. Ansaldo, S. L. Stubbs, K. H. Chow and J. S. Lord, Phys. C, 2010, 470, S755–S757 CrossRef CAS.
- I. Umegaki, Y. Higuchi, H. Nozaki, Y. Kondo, H. Oka, Y. Makimura, O. K. Forslund, M. Månsson, S. P. Cottrell and J. Sugiyama, JPS Conf. Proc., 2019, 25, 011009 Search PubMed.
- O. K. Forslund, D. Andreica, Y. Sassa, H. Nozaki, I. Umegaki, E. Nocerino, V. Jonsson, O. Tjernberg, Z. Guguchia, Z. Shermadini, R. Khasanov, M. Isobe, H. Takagi, Y. Ueda, J. Sugiyama and M. Månsson, Sci. Rep., 2019, 9, 1141 CrossRef.
- Y. Sassa, S. Wang, J. Sugiyama, A. Amato, H. M. Rønnow, C. Rüegg and M. Månsson, JPS Conf. Proc., 2018, 21, 011010 Search PubMed.
- O. K. Forslund, D. Andreica, Y. Sassa, H. Nozaki, I. Umegaki, V. Jonsson, Z. Guguchia, Z. Shermadini, R. Khasanov, M. Isobe, H. Takagi, Y. Ueda, M. Månsson and J. Sugiyama, JPS Conf. Proc., 2018, 21, 011006 Search PubMed.
- H. Nozaki, H. Sakurai, I. Umegaki, E. J. Ansaldo, G. D. Morris, B. Hitti, D. J. Arseneau, D. Andreica, A. Amato, M. Månsson and J. Sugiyama, JPS Conf. Proc., 2018, 21, 011005 Search PubMed.
- I. Umegaki, H. Nozaki, M. Harada, M. Månsson, H. Sakurai, I. Kawasaki, I. Watanabe and J. Sugiyama, JPS Conf. Proc., 2018, 21, 011018 Search PubMed.
- M. Månsson, O. K. Forslund, H. Nozaki, I. Umegaki, S. Shiraki, T. Hitosugi, T. Prokscha, Z. Salman, A. Suter, Y. Sassa and J. Sugiyama, JPS Conf. Proc., 2018, 21, 011025 Search PubMed.
- O. K. Forslund and M. Månsson, Muons as an Optimal Probe for Future All Solid-State Energy Devices: A Brief Introduction and Review, Montreal, 2017, pp. 84–101 Search PubMed.
- H. Nozaki, H. Sakurai, O. Ofer, E. J. Ansaldo, J. H. Brewer, K. H. Chow, V. Pomjakushin, L. Keller, K. Prša, K. Miwa, M. Månsson and J. Sugiyama, Phys. B, 2018, 551, 137–141 CrossRef CAS.
- I. Umegaki, S. Kawauchi, H. Sawada, H. Nozaki, Y. Higuchi, K. Miwa, Y. Kondo, M. Månsson, M. Telling, F. C. Coomer, S. P. Cottrell, T. Sasaki, T. Kobayashi and J. Sugiyama, Phys. Chem. Chem. Phys., 2017, 19, 19058–19066 RSC.
- K. Mukai, Y. Ikedo, K. Kamazawa, J. H. Brewer, E. J. Ansaldo, K. H. Chow, M. Månsson and J. Sugiyama, RSC Adv., 2013, 3, 11634–11639 RSC.
- K. Mukai, D. Andreica, Y. Ikedo, H. Nozaki, M. Månsson, A. Amato and J. Sugiyama, J. Appl. Phys., 2013, 113, 53904 CrossRef.
- H. Nozaki, S. Ohta, M. Harada, M. Månsson, D. Sheptyakov, V. Pomjakushin, I. Watanabe, Y. Ikedo, Y. Miyake and J. Sugiyama, JPS Conf. Proc., 2014, 2, 010303 Search PubMed.
- M. Månsson, H. Nozaki, J. M. Wikberg, K. Prša, Y. Sassa, M. Dahbi, K. Kamazawa, K. Sedlak, I. Watanabe and J. Sugiyama, J. Phys.: Conf. Ser., 2014, 551, 12037 CrossRef.
- Y. Ikedo, J. Sugiyama, K. Mukai, M. Månsson, T. Goko, D. Andreica and A. Amato, J. Phys.: Conf. Ser., 2010, 225, 12016 CrossRef.
- O. Ofer, Y. Ikedo, T. Goko, M. Månsson, J. Sugiyama, E. J. Ansaldo, J. H. Brewer, K. H. Chow and H. Sakurai, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 94410 CrossRef.
- K. H. Chow, M. Månsson, Y. Ikedo, J. Sugiyama, O. Ofer, E. J. Ansaldo, J. H. Brewer, M. Isobe, H. Gotou, T. Yagi, Y. Ueda and C. Baines, Phys. Procedia, 2012, 30, 117–120 CrossRef CAS.
- O. Ofer, J. Sugiyama, J. H. Brewer, M. Månsson, K. Prša, E. J. Ansaldo, G. Kobayashi and R. Kanno, Phys. Procedia, 2012, 30, 160–163 CrossRef CAS.
- J. M. Wikberg, M. Månsson, M. Dahbi, K. Kamazawa and J. Sugiyama, Phys. Procedia, 2012, 30, 202–205 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- J. J. De Yoreo and N. A. J. M. Sommerdijk, Nat. Rev. Mater., 2016, 1, 16035 CrossRef CAS.
- X. Li, F. Bianchini, J. Wind, P. Vajeeston, D. Wragg and H. Fjellvåg, J. Electrochem. Soc., 2019, 166, A3830–A3837 CrossRef CAS.
- K. Sau, AIP Conf. Proc., 2016, 020119 CrossRef.
- T. Moriya, Phys. Rev., 1960, 120, 91–98 CrossRef CAS.
- I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241–255 CrossRef CAS.
- S. A. Owerre, J. Phys.: Condens. Matter, 2016, 28, 386001 CrossRef CAS.
- S. A. Owerre, J. Phys. Commun., 2017, 1, 21002 CrossRef.
- S. A. Owerre, Sci. Rep., 2019, 9, 7197 CrossRef CAS.
- C. Felser, Angew. Chem., Int. Ed., 2013, 52, 1631–1634 CrossRef CAS.
- G. Finocchio, F. Büttner, R. Tomasello, M. Carpentieri and M. Kläui, J. Phys. D: Appl. Phys., 2016, 49, 423001 CrossRef.
- A. Fert, N. Reyren and V. Cros, Nat. Rev. Mater., 2017, 2, 1–15 Search PubMed.
- N. Nagaosa and Y. Tokura, Nat. Nanotechnol., 2013, 8, 899 CrossRef CAS.
- A. Fert, V. Cros and J. Sampaio, Nat. Nanotechnol., 2013, 8, 152–156 CrossRef CAS.
- C. Nayak, S. H. Simon, A. Stern, M. Freedman and S. Das Sarma, Rev. Mod. Phys., 2008, 80, 1083–1159 CrossRef CAS.
- K. Plekhanov, G. Roux and K. Le Hur, Phys. Rev. B, 2017, 95, 45102 CrossRef.
- A. R. Wright, Sci. Rep., 2013, 3, 2736 CrossRef.
- E. Morenzoni, T. Prokscha, A. Suter, H. Luetkens and R. Khasanov, J. Condens. Matter Phys., 2004, 16, S4583 CrossRef CAS.
- J. Sugiyama, H. Nozaki, I. Umegaki, K. Mukai, K. Miwa, S. Shiraki, T. Hitosugi, A. Suter, T. Prokscha, Z. Salman, J. S. Lords and M. Månsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 014417 CrossRef.
- E. Cohen, H. Larocque, F. Bouchard, F. Nejadsattari, Y. Gefen and E. Karimi, Nat. Rev. Phys., 2019, 1, 437–449 CrossRef.
- G. M. Kanyolo, 2019, arXiv, preprint, arXiv:1909.00778, https://arxiv.org/abs/1909.00778v2.
- Y. Liu, Y. Xu and W. Duan, Natl. Sci. Rev., 2018, 5, 314–316 CrossRef CAS.
- P. Ao and D. J. Thouless, Phys. Rev. Lett., 1993, 70, 2158 CrossRef.
- D. Loss, P. Goldbart and A. Balatsky, Phys. Rev. Lett., 1990, 65, 1655 CrossRef.
- A. Tomita and R. Y. Chiao, Phys. Rev. Lett., 1986, 57, 937 CrossRef CAS.
- J. Zak, Phys. Rev. Lett., 1989, 62, 2747 CrossRef.
- J. Samuel and R. Bhandari, Phys. Rev. Lett., 1988, 60, 2339 CrossRef.
- M.-C. Chang and Q. Niu, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 7010 CrossRef CAS.
- A. Aronov and Y. B. Lyanda-Geller, Phys. Rev. Lett., 1993, 70, 343 CrossRef.
- D. Xiao, M.-C. Chang and Q. Niu, Rev. Mod. Phys., 2010, 82, 1959 CrossRef CAS.
- A. K. Bera, N. Shah and S. M. Yusuf, AIP Conf. Proc., 2019, 2115, 30568 CrossRef.
- C. Pei, C. Hou, Y. Li, G. Yao, Z. Ren, P. Liu and H. Zhang, J. Alloys Compd., 2019, 792, 46–49 CrossRef CAS.
- H. Li, P. Zhang, X. Chen, B. Tang, S. Yu, J. Lu and S. Zhang, J. Alloys Compd., 2020, 155043 CrossRef CAS.
- X. Shi, H. Zhang, D. Zhang, C. Liu, L. Shi and X. Wang, Ceram. Int., 2020, 46, 16038–16046 CrossRef CAS.
- L. Xiao, Z. Din, Q. Huang, C. Chen, Y. Feng, C. Liang, P. Gao and W. Wei, Acta Mater., 2020, 199, 34–41, DOI:10.2139/ssrn.3581372.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya and H. Hahn, et al. , Nat. Commun., 2018, 9, 1–9 CrossRef.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y.-S. Hu, Angew. Chem., Int. Ed., 2020, 59, 264–269 CrossRef CAS.
- K. Takada, H. Sakurai, E. Takayama-Muromachi, F. Izumi, R. A. Dilanian and T. Sasaki, Nature, 2003, 422, 53–55 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- T. C. Hales, Discrete Comput. Geom., 2001, 25, 1–22 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |