
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective modification strategies in antibody–drug conjugates
Stephen J.
Walsh†
 ab,
Jonathan D.
Bargh†
a,
Friederike M.
Dannheim†
a,
Abigail R.
Hanby†
a,
Hikaru
Seki†
a,
Andrew J.
Counsell†
a,
Xiaoxu
Ou†
a,
Elaine
Fowler†
a,
Nicola
Ashman†
a,
Yuri
Takada†
a,
Albert
Isidro-Llobet
c,
Jeremy S.
Parker
d,
Jason S.
Carroll
b and
David R.
Spring
*a
ab,
Jonathan D.
Bargh†
a,
Friederike M.
Dannheim†
a,
Abigail R.
Hanby†
a,
Hikaru
Seki†
a,
Andrew J.
Counsell†
a,
Xiaoxu
Ou†
a,
Elaine
Fowler†
a,
Nicola
Ashman†
a,
Yuri
Takada†
a,
Albert
Isidro-Llobet
c,
Jeremy S.
Parker
d,
Jason S.
Carroll
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bCancer Research UK Cambridge Institute, University of Cambridge, Robinson Way, Cambridge, CB2 0RE, UK
cGSK, Gunnels Wood Road, Stevenage, SG1 2NY, UK
dEarly Chemical Development, Pharmaceutical Development, R&D, AstraZeneca, Macclesfield, UK
First published on 8th December 2020
Abstract
Antibody–drug conjugates (ADCs) harness the highly specific targeting capabilities of an antibody to deliver a cytotoxic payload to specific cell types. They have garnered widespread interest in drug discovery, particularly in oncology, as discrimination between healthy and malignant tissues or cells can be achieved. Nine ADCs have received approval from the US Food and Drug Administration and more than 80 others are currently undergoing clinical investigations for a range of solid tumours and haematological malignancies. Extensive research over the past decade has highlighted the critical nature of the linkage strategy adopted to attach the payload to the antibody. Whilst early generation ADCs were primarily synthesised as heterogeneous mixtures, these were found to have sub-optimal pharmacokinetics, stability, tolerability and/or efficacy. Efforts have now shifted towards generating homogeneous constructs with precise drug loading and predetermined, controlled sites of attachment. Homogeneous ADCs have repeatedly demonstrated superior overall pharmacological profiles compared to their heterogeneous counterparts. A wide range of methods have been developed in the pursuit of homogeneity, comprising chemical or enzymatic methods or a combination thereof to afford precise modification of specific amino acid or sugar residues. In this review, we discuss advances in chemical and enzymatic methods for site-specific antibody modification that result in the generation of homogeneous ADCs.

| David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. Jason Carroll is Professor of Molecular Oncology at the University of Cambridge and a Senior Group Leader at the Cancer Research UK Cambridge Institute. Jeremy Parker is Senior Director and Head of Early Chemical Development at AstraZeneca in Macclesfield. Albert Isidro-Llobet is an Investigator and Associate Fellow at GSK in Stevenage. Stephen Walsh, Jonathan Bargh, Friederike Dannheim, Abigail Hanby, Hikaru Seki, Andrew Counsell, Xiaoxu Ou, Elaine Fowler, Nicola Ashman and Yuri Takada are members of the Spring lab in the University of Cambridge Department of Chemistry. Stephen Walsh is also a postdoctoral research associate in the Carroll lab in the Cancer Research UK Cambridge Institute. Photos are in order of the author list (left to right, top to bottom). |
1 Introduction
Antibody–drug conjugates (ADCs) are a class of targeted therapeutics, typically developed for the treatment of cancer. By harnessing the cell selectivity of monoclonal antibodies (mAbs) and the cytotoxicity of small molecule toxins, malignant cells can be selectively destroyed whilst sparing healthy tissue.1,2 Critical to the success of this strategy is a covalent linker between the two therapeutic components, which facilitates the ADC's mechanism of action.3 This marriage of macromolecular biology and small molecule chemistry is at the heart of the clinical success of ADCs. Indeed, the field has enjoyed significant clinical and commercial success in recent years, with nine ADCs receiving approval from the US Food and Drug Administration and >80 others in clinical development (Fig. 1).4–13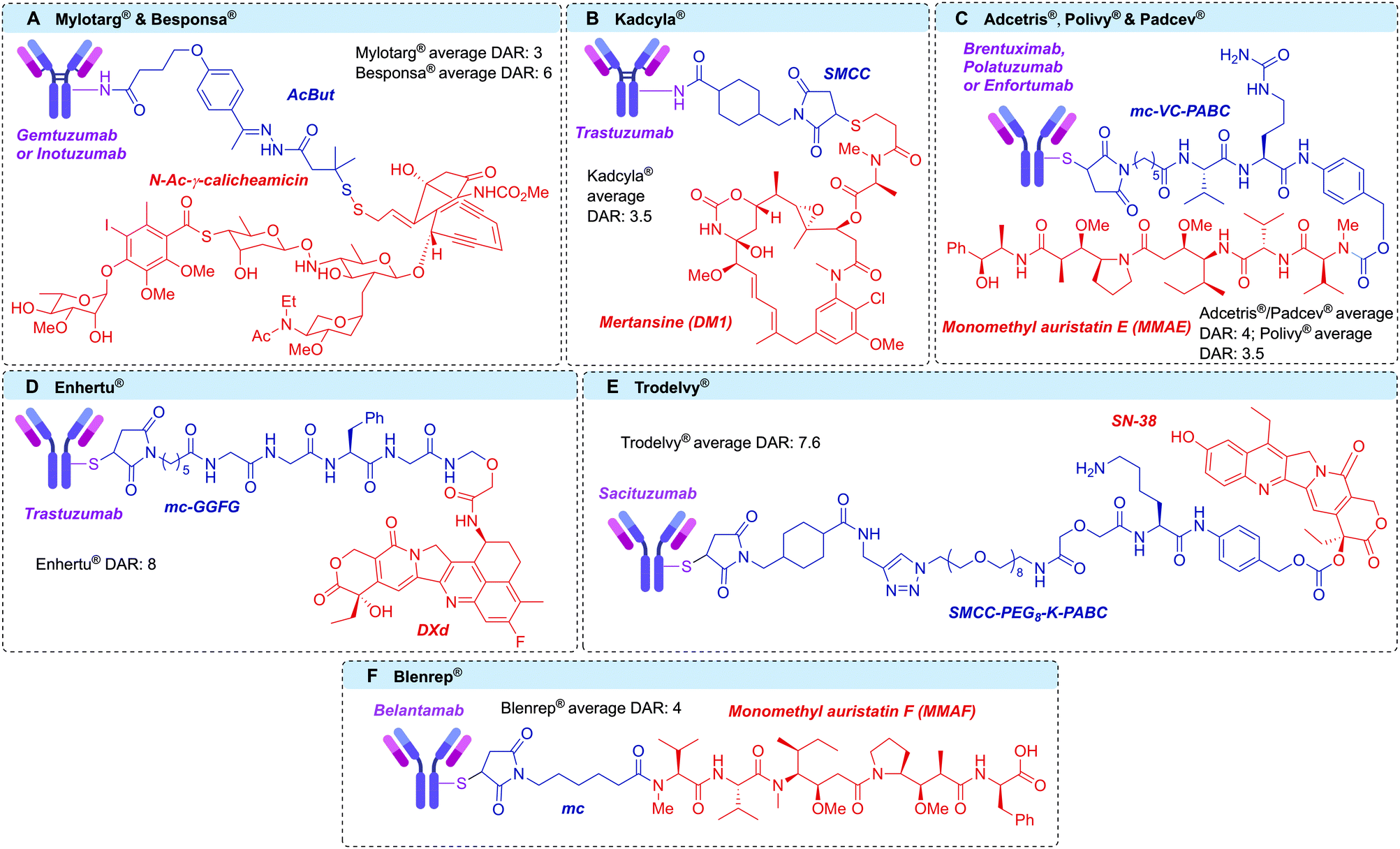 | ||
| Fig. 1 Structures of the clinically-approved ADCs, with linkers in blue and payloads in red. (A) Gemtuzumab ozogamicin (Mylotarg®) and inotuzumab ozogamicin (Besponsa®); (B) trastuzumab emtansine (Kadcyla®); (C) brentuximab vedotin (Adcetris®); polatuzumab vedotin (Polivy®) and enfortumab vedotin (Padcev®); (D) trastuzumab deruxtecan (Enhertu®); (E) sacituzumab govitecan (Trodelvy®); (F) belantamab mafodotin (Blenrep®). AcBut = 4-(4-acetylphenoxy)butanoic acid, SMCC = succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate, MC = maleimidocaproyl, PABC = p-aminobenzyloxycarbonyl. | ||
The antibody portion of the ADC targets and binds surface receptors that are expressed at discernibly higher levels on cancer cells compared to healthy tissue, thereby allowing selective targeting.1 Once bound, the ADC–antigen complex is usually internalised into the malignant cell and trafficked through the endosomes and lysosomes. At this point the drug can be liberated from the antibody, thus enabling its cytotoxic function. In the case of a ‘non-cleavable’ linker the release of the cytotoxic metabolite occurs by lysosomal degradation of the antibody into its constituent amino acids, releasing the payload with the linker and amino acid appendage.14–16 More commonly, a ‘cleavable’ linker is employed, in which a chemical- (e.g. low pH17 or glutathione18,19) or enzyme- (e.g. protease,20 phosphatase,21,22 glycosidase23,24 or sulfatase25) sensitive trigger is incorporated.26 Cleavable linker technology therefore enables the selective release of an unmodified payload at the target cell. Whilst an important ADC component, cleavable linker technologies have been discussed elsewhere and will not be the focus of this review, which focuses on linker-antibody attachment chemistry.26
Currently, all ADCs in clinical and preclinical development incorporate antibodies of the immunoglobulin G (IgG) isotype. IgGs can be divided into four subclasses: IgG1, IgG2, IgG3 and IgG4. The four subclasses have approximately 90% sequence homology, but vary in serum stability, number of interchain disulfide bonds, and their ability to activate the immune system via antibody-dependent cellular cytotoxicity (ADCC) or the complement pathway (Fig. 2).27 Traditionally, IgG1 has been utilised the most in ADC development, due to its favourable balance of long serum half-life and moderate to strong immune activation. However, IgG4 has also been employed in cases where less immune activation is desirable.28 Both IgG1 and IgG4 contain a total of 16 disulfide bonds per antibody. Of these, 12 are intrachain bonds and 4 are interchain bonds. While interchain bonds are highly solvent exposed and may easily be reduced and/or modified by chemical methods, the intrachain bonds are buried within the globular fold of the protein and are therefore unreactive to chemical modification unless harsh denaturing conditions are applied.29 In contrast to IgG1, native IgG4 molecules can undergo dynamic Fab arm exchange which may reduce their efficacy in vivo and lead to undesired off-target effects. However, this can be prevented through a S228P mutation in the hinge region of the heavy chain, as in the case of clinically approved ADCs Mylotarg® and Besponsa®.30,31 Currently, all other approved ADCs utilise IgG1 antibodies.
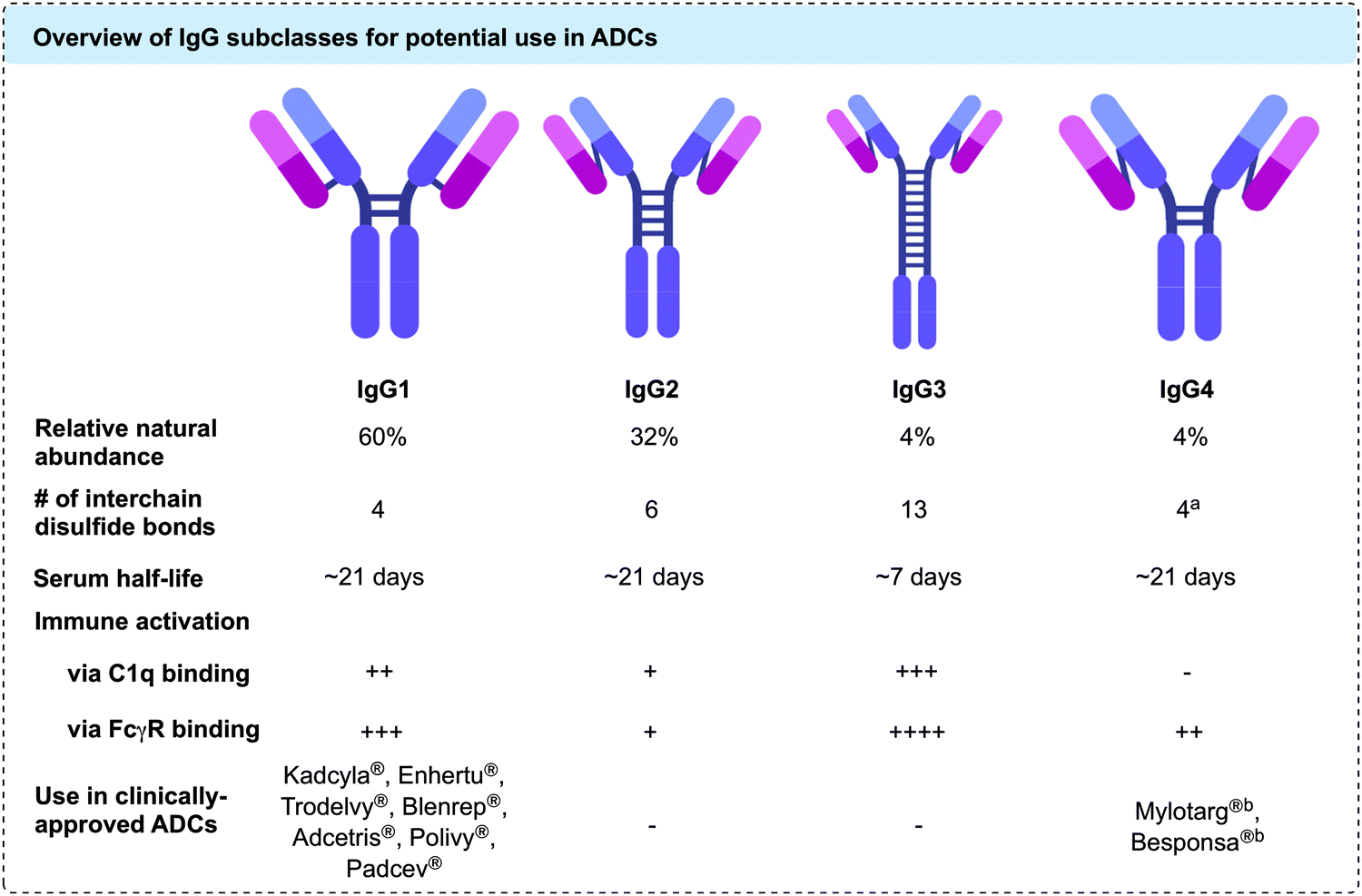 | ||
Fig. 2 Overview of IgG subclasses for potential use in ADCs; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Hinge region disulfides are labile, enabling spontaneous Fab arm exchange with other IgG4 antibodies in vivobFab arm exchange is prevented through S228P mutation in the hinge region. Hinge region disulfides are labile, enabling spontaneous Fab arm exchange with other IgG4 antibodies in vivobFab arm exchange is prevented through S228P mutation in the hinge region. | ||
1.1 ADC requirements
ADC research has progressed significantly over the past 30 years.32 A wealth of knowledge now exists on the specific requirements for the three individual ADC components; the antibody, the cytotoxic drug and the linker. Whilst the natures of the target antigen, antibody,1,28,33 linker-drug attachment16,26 and payload34–36 are all crucial to the pharmacology of an ADC, this review will focus on the developments in conjugation technology.Early ADC research focused on the properties of the eponymous antibody and drug components, with little emphasis on the linker.32,37 However, extensive research has revealed the importance of bioconjugation and the resulting linker-antibody attachments. To maximise the ADC's anti-tumour efficacy and safety, a number of key linker-antibody attachment attributes have been identified: (1) the attachment motif must be highly stable in circulation to avoid premature drug release, which can lower ADC efficacy and cause toxicity in healthy tissue;38,39 (2) the number of linker-payloads per antibody should be optimised for potency without compromising safety;40 (3) the location of attachment on the antibody should not interfere with the antibody's function; and (4) the conjugation reaction should efficiently and selectively facilitate modification of the antibody in a controlled and consistent manner.41
1.2 Drug-to-antibody ratio and conjugation site
The stoichiometry of the linker-payloads on the antibody is referred to as the drug-to-antibody ratio (DAR). Given the limited number of ADCs that each target cell can internalise, it is desirable to maximise the DAR to increase potency (Fig. 3A).42 However, the cytotoxins used in ADC research to achieve the desired potency tend to be large, lipophilic species. As such, increasing the DAR extensively can cause an increase in protein aggregation and an associated increase in ADC clearance, in turn returning diminished efficacy and safety.40,43 A fine balance must be achieved to obtain the desired activity without eradicating the pharmacokinetic properties of the antibody. The optimal DAR is highly dependent on the nature of the linker and payload, but for most commonly used linker-payloads it is ca. 2–4.40 However, in a handful of cases a DAR as high as 8 has safely been achieved through the use of hydrophilic linker-payloads, as exemplified with the clinically approved Enhertu® and Trodelvy®.12,42,44–46 It is also important to note that although an ADC synthesised via heterogeneous conjugation methods may have an average DAR of 2–4, there will be a distribution within this where some antibody molecules will be loaded with significantly higher or lower numbers of payloads relative to the reported average DAR.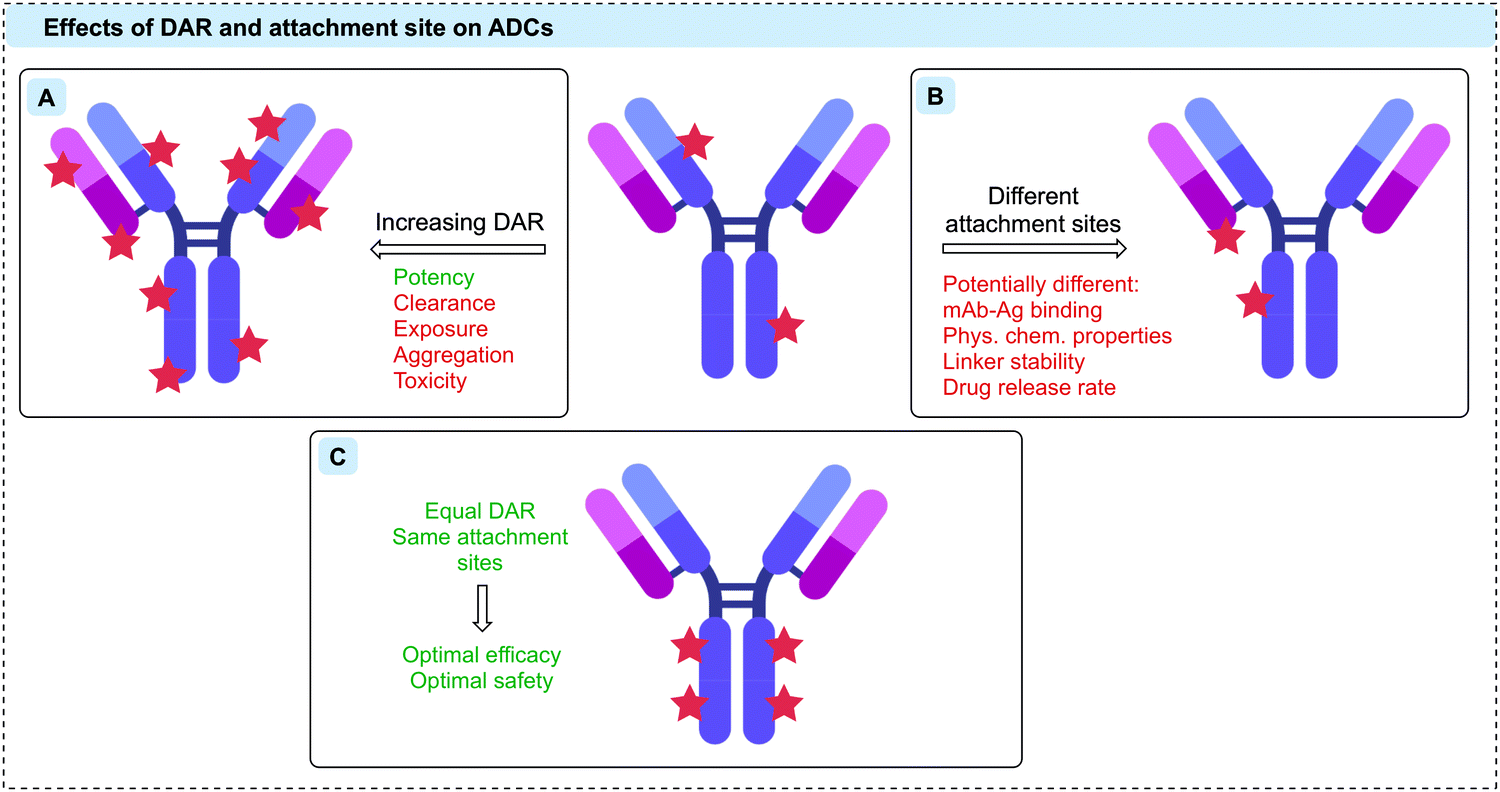 | ||
| Fig. 3 The therapeutic effects of (A) increasing DAR, (B) different attachment sites, and (C) optimal DAR and conjugation sites on ADCs. | ||
As well as the drug loading, the attachment site of the linker-payload to the antibody is also an essential consideration (Fig. 3B).38,39 It is critical that the attachment should be distal to the antigen-binding region, leaving antibody binding and internalisation unaffected. Furthermore, the attachment site can also have a dramatic effect on linker stability, which determines the rate of drug release both in circulation and at the tumour site.38,39
Many of the issues with early generation ADCs were attributed to the conjugation strategies that were employed, which led to heterogeneous and often unstable bioconjugates. To achieve optimal efficacy and safety, it is now widely accepted that ADCs with homogeneous DAR and attachment sites can generate superior therapeutics (Fig. 3C).47 Given the huge number of reactive residues in an IgG, ADCs thus represent one of the most challenging applications of protein bioconjugation.48 Advances in site-selective protein modification have enabled the development of a new generation of ADCs that fulfil these homogeneity requirements. Site-selective modification can be defined as chemo- and regio-selective protein modification and will be referred to as such hereafter.
In this review, we will discuss strategies for the construction of homogeneous ADCs including the most recent advances in the field. First, both chemical and enzymatic methods that facilitate amino acid modification will be discussed. This will be followed by a discussion of the developments made in the modification of the carbohydrate moiety of antibodies.
2 Amino acid modification – chemical methods
2.1 Stochastic conjugation with naturally occurring amino acids
Proteins can be considered meta-stable molecules. Thus, bioconjugation reactions between proteins and small molecules must meet a strict set of requirements to maintain protein structure and function. It is imperative that these reactions proceed in aqueous buffer under mild conditions (temperatures ca. 37 °C, pH 5–9, <15% organic co-solvent, low (μM) protein concentration, <500 equivalents reagent).49–53 While extensive work over recent decades has resulted in the generation of a toolbox of bioorthogonal reactions (reactions that can occur in living systems without affecting that system), such as the copper-catalysed azide–alkyne cycloaddition (CuAAC), the strain-promoted azide–alkyne cycloaddition (SPAAC) and the inverse electron demand Diels–Alder (iEDDA),54–56 the natural reactivity of canonical amino acids such as lysine or cysteine has classically been exploited for the creation of protein conjugates. Indeed, all nine of the currently approved ADCs are synthesised via modification of either of these amino acids. Furthermore, other applications requiring modified antibodies, such as antibody–enzyme conjugates (for antibody-directed enzyme prodrug therapy [ADEPT]57,58 or enzyme-linked immunosorbent assay [ELISA]),59 as well as antibody–radioisotope60 conjugates have generally employed stochastic lysine/cysteine modification techniques.Lysine residues offer a facile method for bioconjugation due to their high natural abundance, surface accessibility and the nucleophilicity of the ε-amino side chain. IgG1 antibodies contain approximately 85 lysine residues, of which more than 40 are typically modifiable.61 Although the average DAR can be guided by reagent stoichiometry and reaction conditions, control of the conjugation site is essentially impossible and millions of different species can be generated in every synthetic batch (Fig. 4A).61 Moreover, lysine residues decorate the entire surface of an antibody; therefore, their modification can impede antigen recognition, thus limiting the efficacy. Despite these shortcomings, Mylotarg®, Kadcyla® and Besponsa® all employ lysine bioconjugation and are therefore administered as a heterogeneous mixture of products. Remarkably, in the case of Mylotarg®, 50% of the mAbs are unconjugated (DAR = 0), with the remaining species averaging DAR = 6, affording an overall average DAR ∼ 3.62
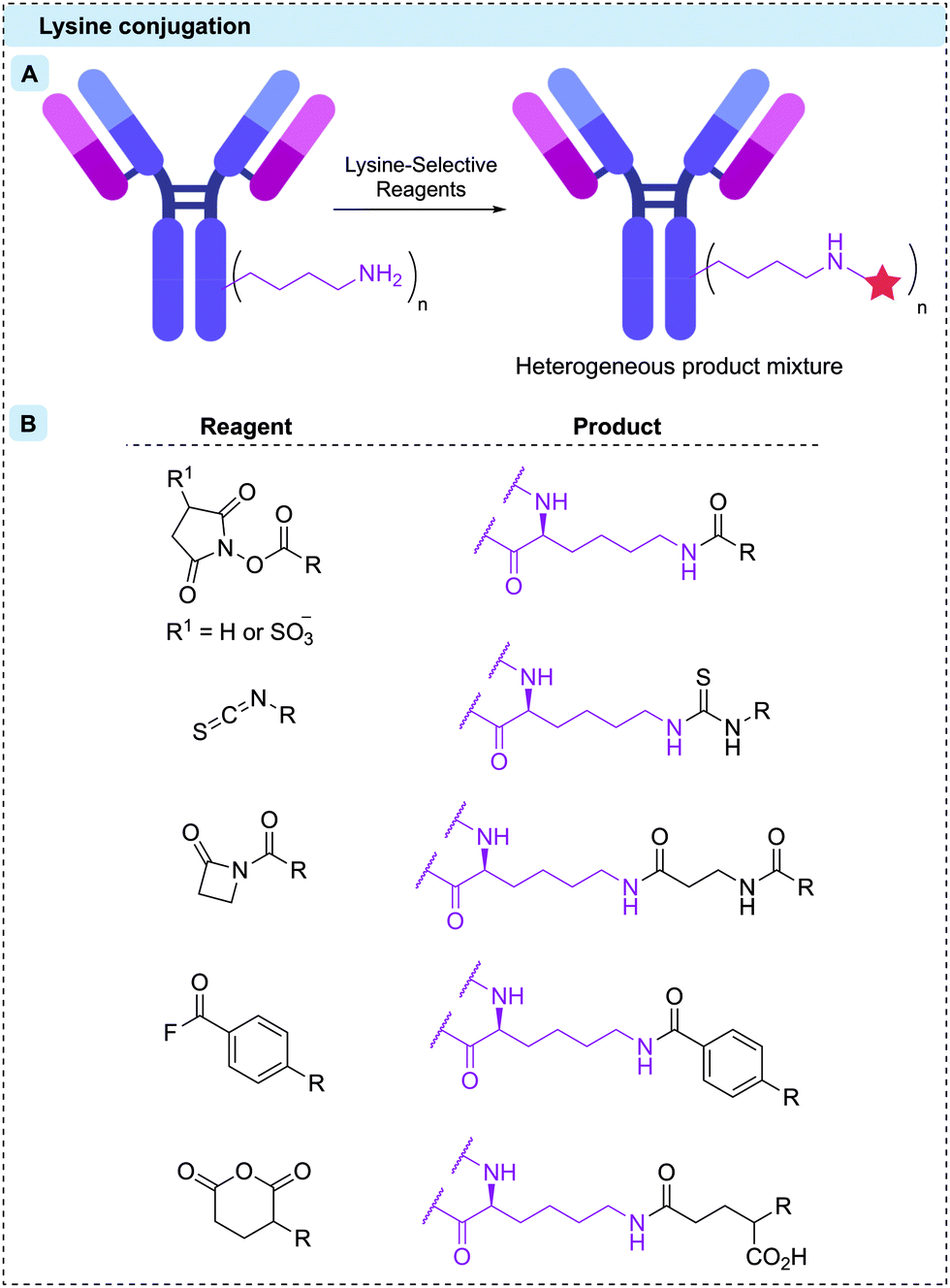 | ||
| Fig. 4 (A) Stochastic reaction with surface-exposed lysine residues results in a heterogeneous product; (B) structures of lysine-selective reagents and their products upon conjugation. | ||
A range of lysine-selective reagents have been developed (Fig. 4B). N-Hydroxysuccinimide (NHS) esters (and their more soluble 3-sulfonated analogues) are by far the most commonly used, due to their rapid lysine reactivity and the stability of the amide product. However, other amino acids such as cysteine and tyrosine, can also be modified during NHS ester reactions, forming less stable linkages.63–66 These less stable linkages can release the linker-payload prematurely, potentially causing toxicity and lowering efficacy.63 Other lysine-selective reagents that have successfully been used to construct ADCs include isothiocyanates,67 β-lactams,68 acyl fluorides,69 and mixed anhydrides70 (Fig. 4B).
Cysteine residues offer a particularly attractive target for protein bioconjugation due to their low natural abundance and the exceptionally high nucleophilicity of the deprotonated thiolate side chain. For IgG1 modification, naturally occurring cysteine residues can be unmasked by reduction of the four interchain disulfide bonds, revealing up to eight reactive thiol residues.71 Subsequent reaction with soft electrophiles affords selective bioconjugation at the eight different sites (Fig. 5A). In some cases, including the approved ADCs Enhertu® and Trodelvy®, a resulting DAR 8 conjugate has been achieved with high homogeneity, efficacy and safety. However, a DAR of 8 is not suitable for many linker-payloads.72 Creation of ADCs with an average DAR of 2–4 therefore requires partial disulfide reduction/reoxidation and controlled linker-payload stoichiometry.71,73 The resulting ADCs are inescapably heterogeneous, although with less variability than is seen with stochastic lysine conjugation.
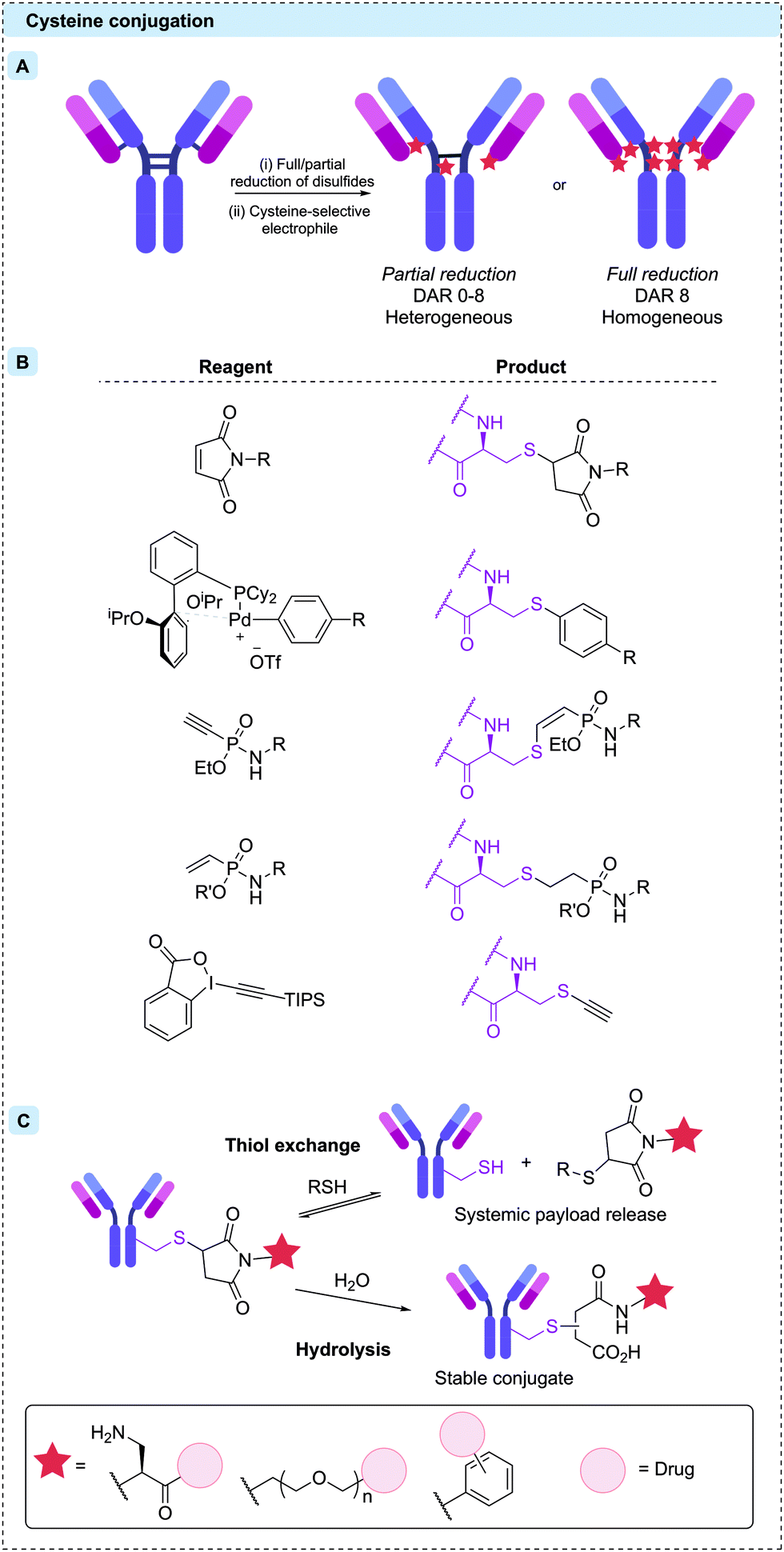 | ||
| Fig. 5 (A) Reduction of interchain disulfides reveals thiol residues, reactive towards soft electrophilic reagents. Partial reduction results in heterogeneous product distributions, but full reduction and complete reaction of all eight reactive cysteines results in DAR 8 conjugates. (B) Structures of cysteine-selective reagents and their products upon conjugation. (C) The post-conjugation reactions of thiosuccinimide linkages (top). The retro-Michael addition and subsequent maleimide–thiol reaction, resulting in overall thiol-exchange (bottom). Hydrolysis of the succinimide moiety creates a stable chemical linkage. The structures of these self-hydrolysing maleimides are shown in the solid box. | ||
Cysteine modification occurs most commonly by 1,4-conjugate addition to N-substituted maleimides. Maleimides are particularly attractive reagents due to their synthetic accessibility and rapid reaction rates with cysteine under mild conditions. Indeed, Adcetris®, Polivy®, Padcev®, Enhertu®, Trodelvy® and Blenrep® are all synthesised via maleimide modification of cysteines, as are the majority of ADCs currently in clinical trials. However, the resulting thiosuccinimide conjugates are inherently unstable, due to their propensity towards retro-Michael addition (Fig. 5C).74 In circulation, the prematurely released maleimide-payload can then react with plasma thiols or diffuse into nearby cells, causing a reduction in efficacy and/or safety.38,75 This instability can be mitigated by forcing post-conjugation hydrolysis of the thiosuccinimide, creating a stable chemical linkage. Accordingly, a number of “self-hydrolysing” maleimides have now been developed, with ring-opening catalysed by adjacent functional groups such as primary amine, polyethylene glycol (PEG) and N-aryl amongst the most promising.76–79 Other reagents including α-halocarbonyls,80 palladium oxidative-addition complexes,81 ethynylphosphonamidates,82,83 vinylphosphonites84 and ethynylbenziodoxolones85 have also been used to synthesise ADCs with stable thioether bonds via modification of reduced interchain disulfides (Fig. 5B).
2.2 Engineered cysteines
Genetic modification of the number of accessible cysteine residues on an antibody surface has emerged as a popular method to achieve the desired site-selective and homogeneous modification. The earliest example using engineered antibodies decreased the number of interchain disulfides by replacing one of the cysteine residues with a different amino acid, resulting in fewer reactive cysteine residues. Mutants of the anti-CD30 IgG1 antibody cAC10, were generated by replacing select cysteine residues with serines.86 Five mutants with different cysteine positions were modified with maleimidocaproyl-valine-citrulline-para-aminobenzoyl-monomethyl auristatin E (mc-Val-Cit-PABC-MMAE) to generate homogeneous ADCs with a DAR of either 2 or 4 (Fig. 6). Characterisation of these conjugates showed that mutagenesis did not impede antigen binding or in vitro cytotoxicity. However, in vivo mouse xenograft models revealed these homogeneous ADCs had similar efficacy and therapeutic indices compared to analogous heterogeneous ADCs with similar average DARs. The authors concluded that the benefits from improved homogeneity may have been offset by the removal of interchain disulfide bonds.87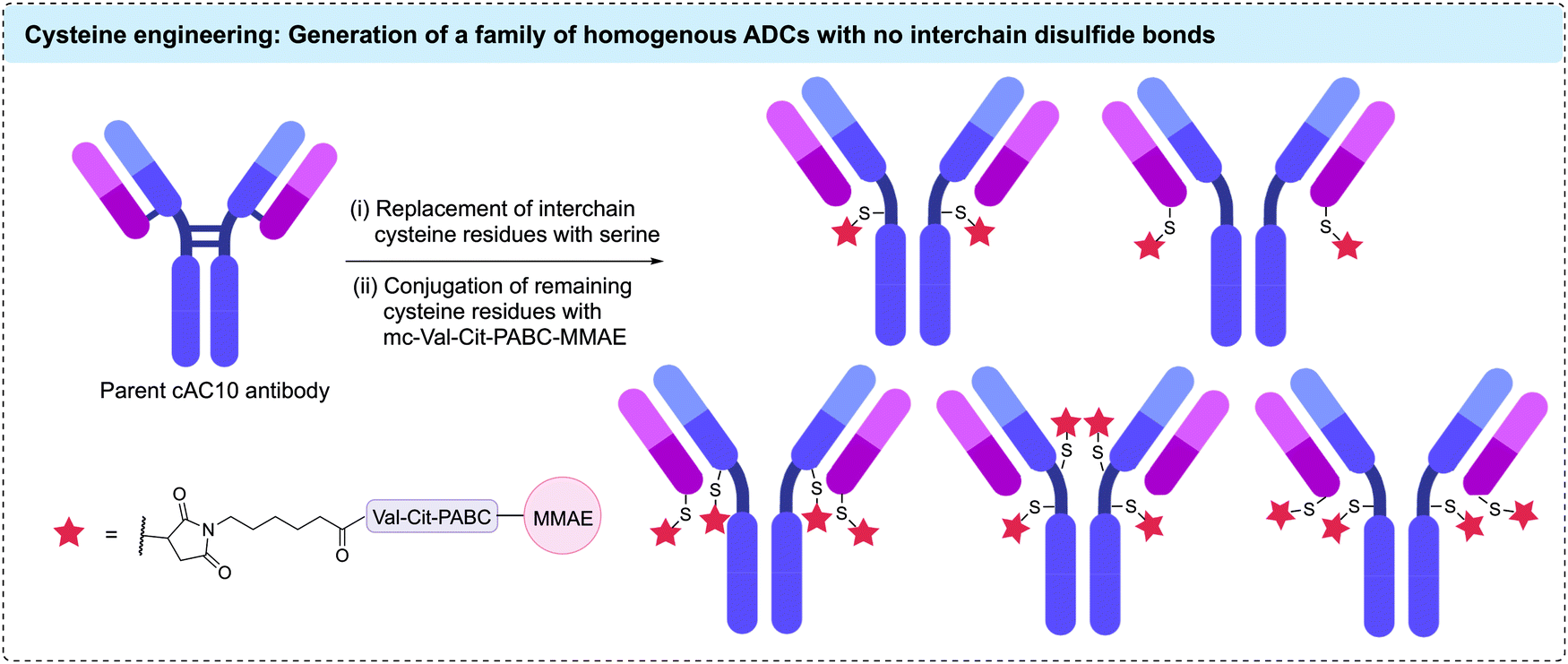 | ||
| Fig. 6 Replacement of interchain cysteine residues with serine residues enables the generation of homogenous ADCs with DARs of 2 or 4 via cysteine conjugation. | ||
A significant advance was made by the development of antibodies with additional engineered cysteine residues. Introduction of non-native cysteine residues that are not involved in structural disulfide bonding can facilitate functionalisation with cytotoxic payloads. This approach offers a number of advantages: (1) modification of these unpaired cysteines with payloads will give homogeneous ADCs with a defined attachment site and drug stoichiometry; and (2) all native immunoglobulin disulfide bonds will be retained, potentially improving the stability and endogenous biology of the antibody. Identification of potential mutation sites is typically achieved using computational modelling, screening of model systems, or high-throughput scanning.88–90 The mutated antibodies are then extensively characterised for stability, binding, aggregation, clearance and cytotoxicity.
Seminal work by Junutula et al. first introduced cysteine-engineered antibodies for biotherapeutic development, termed THIOMAB™.91 In this study, the engineered cysteine was installed on an anti-MUC16 antibody by mutation of heavy chain alanine 114 (HC-A114). The authors found that expression of the mutated antibodies in Chinese hamster ovary (CHO) cells generated the THIOMAB with the engineered cysteine residues capped as disulfides with cysteine or glutathione. Therefore, a procedure of partial reduction (using tris(2carboxyethyl)phosphine [TCEP] or dithiothreitol [DTT]), purification, and re-oxidation of the interchain disulfide bonds (using CuSO4 or dehydro-ascorbic acid) was required to reveal the reactive thiols. This partially reduced antibody was then treated with mc-Val-Cit-PABC-MMAE, producing a highly homogeneous ADC with an average DAR of 1.6 (Fig. 7A). This ADC was then compared with an analogous heterogeneous anti-MUC16 ADC (average DAR of 3.1) synthesised via modification of native interchain disulfides. An in vivo rat xenograft model revealed that the homogeneous ADC was at least as efficacious as the heterogeneous ADC despite its lower drug loading. Evaluation of the safety profile of both ADCs in rats and monkeys indicated that the heterogeneous ADC caused significant adverse effects. In contrast, the homogeneous ADC derived from the engineered antibody displayed no significant adverse effects, with all parameters essentially identical to vehicle-treated animals. Furthermore, the clearance rate for the homogeneous ADC was markedly slower than that of the heterogeneous ADC. Following these promising results, optimisation of the conjugation reaction yielded an ADC with a DAR of 2.
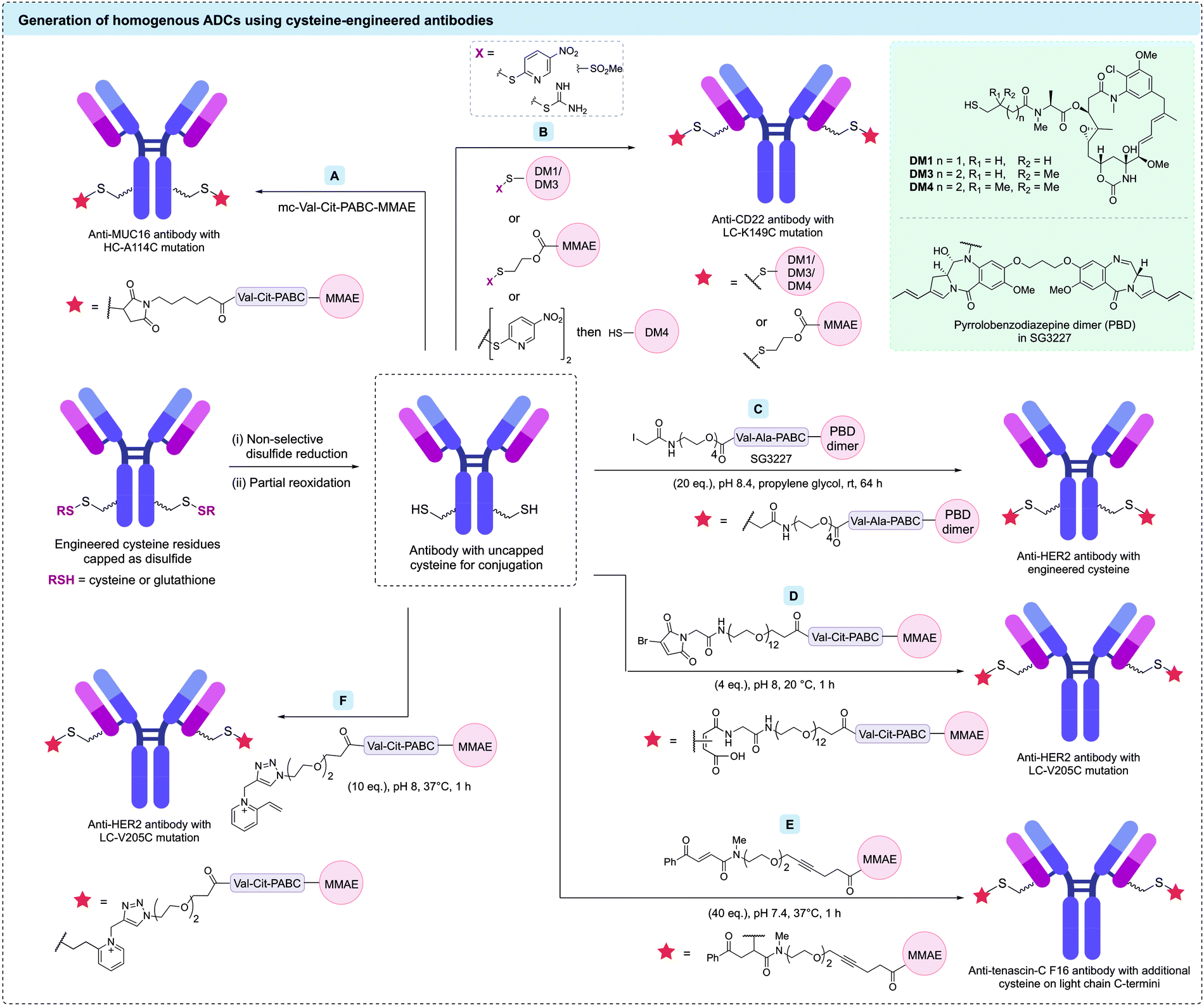 | ||
| Fig. 7 Strategies to functionalise cysteine-engineered antibodies to generate homogeneous ADCs. After revealing uncapped cysteine residues via reduction and re-oxidation steps, these cysteine-containing antibodies have been modified using (A) maleimides; (B) direct disulfide formation; (C) iodoacetamides; (D) bromomaleimides; (E) carbonylacrylic reagents; (F) N-alkyl vinylpyridine salts. | ||
Many cysteine-engineered antibodies require the initial two-step reduction–reoxidation process reported by Junutula et al. to remove the capping disulfide prior to conjugation. However, one report by Shinmi et al. described the expression of a trastuzumab variant (LC-Q124C) that was isolated with no capping moiety, due to the sterically hindered location of the cysteine.92 Modification of this residue with mc-Val-Cit-PABC-MMAE produced a homogeneous ADC with a DAR of 2.
In a related study by Sussman et al., several anti-CD70 (h1F6) antibodies were engineered to contain cysteine mutations (heavy chain modifications of S239C, E269C, K326C or A327C), facilitating site-selective modification with a non-cleavable mc-monomethyl auristatin F (MMAF) linker-payload.93 These ADCs showed significantly different anti-tumour efficacies in vivo, despite similar in vitro potencies. To provide a rationale for this disparity, linker stability and thiosuccinimide ring opening were investigated. The authors found that ADCs with the slowest rate of thiosuccinimide ring opening also had the most stable linkage and in vivo potency. This trend is opposite to that reported by Shen et al.75 and others,76 who found that increased thiosuccinimide ring opening resulted in improved linkage stability and efficacy. Hydrophobic interaction chromatography (HIC) of the ADCs suggested that the conjugation site had a dramatic effect on the hydrophobicity of the conjugate. This in turn affected the stability of the thiosuccinimide linkage with the most hydrophobic ADCs demonstrating the slowest rate of hydrolysis and highest stability. The contrasting observations from these studies demonstrate the importance of optimising the site of cysteine engineering for each antibody to ensure sufficient stability.
In addition to traditional cysteine engineering methods via amino acid mutation, Dimasi et al. used cysteine insertion on an anti-EphA2 antibody.94 Six cysteine-engineered antibodies comprising additional cysteines inserted before and after positions HC-S239, HC-A114, and LC-V205, were produced and evaluated for ADC development. Modification of the inserted cysteine with a maleimide-pyrrolobenzodiazepine (PBD) dimer linker-payload generated a series of homogeneous DAR 2 ADCs. It was found that the ADC synthesised from the antibody containing a cysteine insertion after HC-S239 displayed the most favourable characteristics, with high in vivo plasma stability and dose dependant in vitro cytotoxicity observed. Interestingly, although this ADC maintained binding with its target antigen and the neonatal Fc receptor (FcRn), it displayed significantly reduced binding with Fcγ receptors (FcγRs). Reduced binding with FcγRs may be beneficial, as emerging studies suggests that this binding mode can lead to non-specific uptake of ADCs.95
Pillow et al. and Sadowsky et al. have also shown that drug attachment can be achieved through formation of a mixed disulfide.19,104 Mixed disulfide bonds were formed by treating the antibody with an drug molecule bearing an activated thiol group, or alternatively by first activating the antibody's cysteine residues with 2,2′-dithiobis(5-nitropyridine), followed by reaction with a thiol-bearing drug molecule (Fig. 7B). Plasma stable constructs were generated using these methods on an anti-CD22 antibody with a cysteine mutation (LC-K149C). Drug release in disulfide-linked ADCs occurs by protein catabolism in the target cell to give a cysteine–drug conjugate, followed by disulfide reduction in the cytosol to release the free payload. The disulfide linkage approach could also be applied to amine-functionalised payloads (e.g. MMAE), using immolative 2-mercaptoethyl carboxy groups. In a related study, Vollmar et al. investigated the basis of the site-dependent stability of these disulfide bonds by comparing five trastuzumab mutants: HC-A118C, HC-A140C, LC-S121C, LC-K149C, and LC-V205C.105 The authors found cysteine pKa to have a greater influence on disulfide stability compared to steric effects – cysteine residues with the highest pKa resulted in the most stable disulfide linkages.
Although the majority of cysteine engineered ADCs have a DAR of 2, a small number of reports have aimed to utilise engineered cysteines to generate higher DAR ADCs. For example, Pillow et al. generated a DAR 6 antibody-PROTAC by conjugating a chimeric BRD4 degrader (GNE-987) to engineered LC-K149C, HC-L174C, and HC-Y373C cysteine residues of an anti-CLL1 antibody via a methanethiosulfonyl (MTS) disulfide linkage.106 In EOL-1 and HL-60 mouse xenografts, the anti-CLL1 ADC was shown to cause complete tumour regression after a single IV dose at either 5 or 10 mg kg−1. Separately, Neumann et al. generated DAR 10 ADCs via modification of the 8 interchain disulfide cysteines plus two additional engineered cysteines.107 An anti-CD1232 antibody with two additional cysteine residues (HC-S239C) was conjugated to maleimide-modified nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. In HNT-34 AML xenograft, the DAR 10 ADC induced rapid tumor regression after single administration of 10 mg kg−1 and sustained the tumor regression after subsequent doses. Linker technologies have been developed to allow DAR 4 ADCs to be generated through modification of two cysteine residues. For example, Kumar et al. have combined a cysteine-engineered anti-HER2 antibody with a bis-functionalised maleimide reagent to generate DAR 4 ADCs with two different drug payloads (Fig. 8).108 Cysteine modification with a maleimide reagent containing both ketone and alkyne reactive groups was followed by oxime ligation with an aminooxy-Val-Cit-PABC-MMAE payload and CuAAC with azido-Val-Ala-PABC-PBD to generate a homogeneous ADC with two MMAE and two PBD payloads. Although in vitro studies showed that the more potent PBD cytotoxin dominated the cytotoxic properties of the ADC, this study demonstrated that varied functional moieties can be installed on cysteine-engineered antibodies.
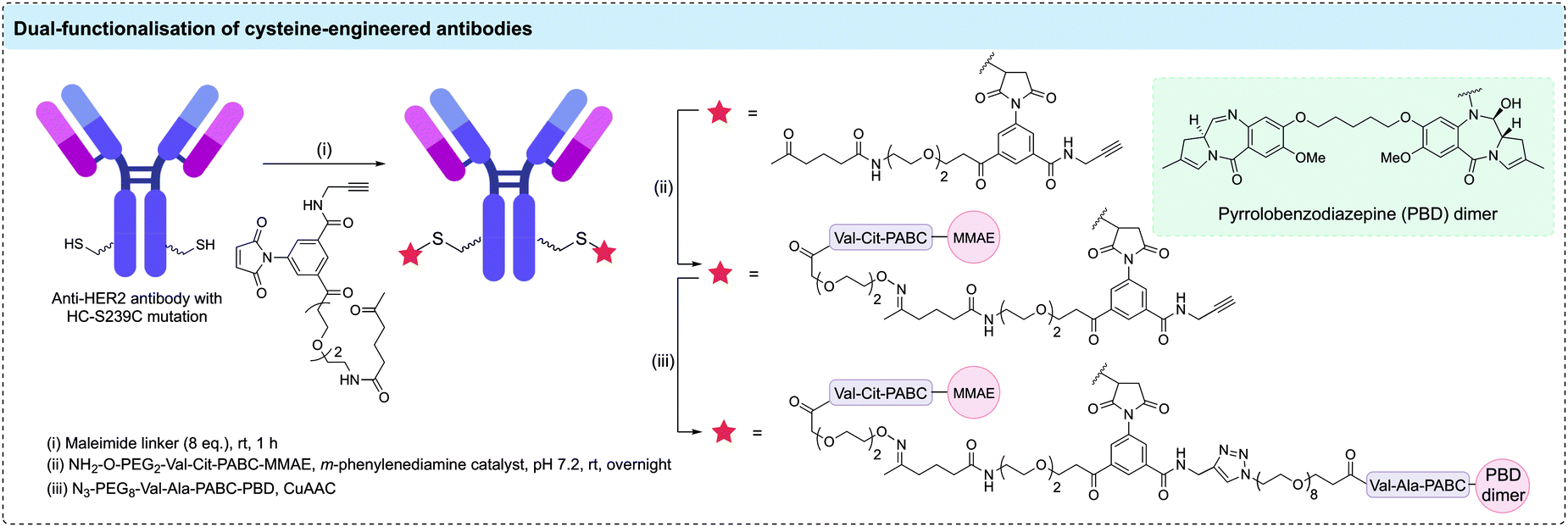 | ||
| Fig. 8 Dual functionalisation of an anti-HER2 antibody containing two additional cysteine residues. A maleimide reagent containing orthogonal alkyne and ketone handles enabled conjugation of MMAE and PBD dimer payloads. | ||
Although ADCs are usually developed for the treatment of cancer, cysteine-engineered ADCs have been developed as antibiotics. To target intracellular methicillin-resistant S. aureus bacteria, Lehar et al. designed antibody–antibiotic conjugates that are activated specifically inside mammalian cells.115 A human IgG1 which targets wall teichoic acids of S. aureus was first engineered with a LC-V205C mutation and subsequently modified with a rifamycin derivative (an antibacterial that targets bacterial RNA polymerase) via a maleimide-Val-Cit cleavable linker. Compared to vancomycin treatment, these antibody–antibiotic conjugates were superior in treating MRSA in vitro and in vivo. A Phase I clinical trial demonstrated favourable safety and pharmacokinetic profile of the anti-S. aureus antibody–antibiotic conjugate in healthy volunteers (ClinicalTrials.gov Identifier: NCT02596399),116 and further clinical trials are currently underway to treat patients with S. aureus infections (ClinicalTrials.gov Identifier: NCT03162250).
As demonstrated in this section, the use of cysteine-engineered antibodies provides reactive handles to reliably synthesise site-specifically modified, homogenous ADCs. Despite the requirement to optimise the most suitable position for cysteine engineering, this method has been used to generate numerous ADCs comprising various antibody-linker-payload combinations against a range of malignancies. Further development of cysteine-engineered antibodies and their progression to clinical trials is expected.
2.3 Disulfide rebridging
Disulfide rebridging involves the reduction of the four interchain disulfide bonds in an IgG1 antibody followed by reaction with a cysteine-selective cross-linking reagent. The bis-reactive reagent enables the reconnection of the polypeptide chains while simultaneously installing drug molecules or bioorthogonal functionalities amenable to further modification. By covalently reconnecting the cysteine residues, the stabilising effect of the disulfide bonds is maintained and a controlled loading of one linker molecule per disulfide can be achieved. Depending on the number of drug molecules attached to each linker, a DAR of 4, 8 or 16 has been attained in this way.117 Since the conjugation utilises native cysteine residues in the antibody hinge region, no alteration of the genetic code or the glycosylation pattern is required. The three most established disulfide rebridging technologies are bissulfone reagents, next-generation maleimides (NGMs) and pyridazinediones (PDs); however, in recent years numerous other methods have emerged, including the use of arylene dipropiolonitrile (ADPN), divinylpyrimidine (DVP), dibromomethyl heterocycles (C-Lock™), dichloroacetone or platinum(II) complexes. Here, we will give an overview of the applications and benefits of each strategy and discuss the overall utility of disulfide rebridging in ADC development.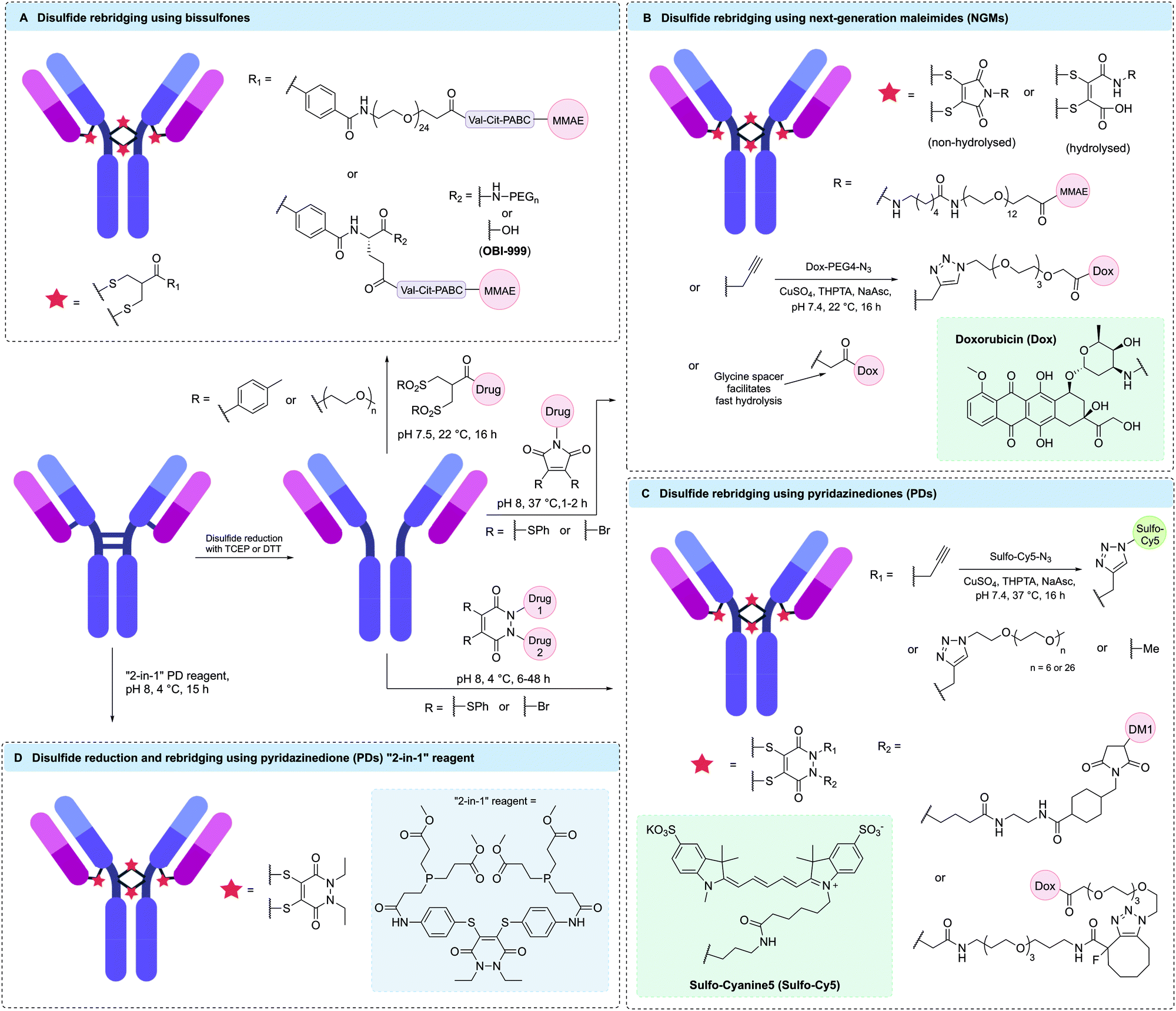 | ||
| Fig. 9 Selected examples of homogenous ADCs generated via disulfide rebridging using (A) bissulfones, (B) next-generation maleimides (NGMs), (C) pyridazinediones (PDs) or (D) “2-in-1” PD reagents. | ||
It was hypothesised that premature degradation of the cleavable Val-Cit unit in circulation was lowering the efficacy of bissulfone-reacted ADCs. The mouse plasma stability of the Val-Cit motif is known to be affected by linker structure (including PEGylation pattern) and attachment point.39,122 Therefore, Pabst et al. investigated the effects of PEGylation on the potency and stability of bissulfone-conjugated ADCs containing a Val-Cit-PABC cleavage mechanism and found that ADCs with branched or non-PEGylated linkers were significantly more stable in vivo in mice than bissulfone ADCs with linear PEGylated linkers.123 Furthermore, the branched linker ADC demonstrated superior stability compared to the approved heterogeneous anti-CD30 ADC Adcetris® and showed excellent in vivo efficacy with complete tumour regression observed after treatment with a single dose at 1 mg kg−1 in a CD30-positive mouse xenograft (Fig. 9A). In a different study, it was found that cyclisation of the branched PEG chains could improve the efficacy of bissulfone ADCs even further and allow for complete tumour regression in the same mouse model at a dosage of 0.8 mg kg−1.124
Abzena is currently licensing bissulfone linker technology to various pharmaceutical companies under the tradename ThioBridge™. This arrangement has led to the development of multiple drugs in preclinical and clinical trials. For example, OBI-999 is an ADC developed by OBI Pharma composed of an MMAE payload attached to a humanised anti-Globo H antibody via a bissulfone linker (Fig. 9A).36,125 The ADC has recently received FDA orphan drug designation for the treatment of pancreatic and gastric cancer based on promising preclinical data. OBI is currently recruiting patients with locally advanced or metastatic solid tumours, including gastric, pancreatic, oesophageal and colorectal cancers, for a Phase I/II study (ClinicalTrials.gov Identifier: NCT04084366). Another bissulfone-based ADC, HTI-1511, was developed by Halozyme and contains an MMAE payload conjugated to an EGFR-targeting antibody. This ADC has demonstrated promising tumour growth inhibition or regression in patient-derived xenograft models in mice, including models with KRAS/BRAF mutations typically associated with resistance to EGFR-targeted therapy and poor outcome.36,126 Furthermore, HTI-1511 has been shown to be well tolerated in primate models.
A detailed procedure for the use of bissulfone reagents was recently published by Bird et al., potentially aiding its more widespread use.127 Typical reaction conditions are very mild and involve incubation of reduced antibody with 5–6 equivalents of bissulfone in pH 7.5 buffer at 22 °C for 16 hours. Conversion to the desired DAR 4 species is typically in the range of 75–85% and further purification by HIC is often performed to attain fully homogenous ADCs with a DAR 4 content of >95%.123 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) data of OBI-999 suggests that the ADC exists as a mixture of two isomeric species; one being the natively rebridged species in which all polypeptide chains are rebridged in an interchain fashion and the other being a “half-antibody” species which has lost the covalent linkage between the antibody heavy chains due to the formation of non-native intrachain bridges between the cysteines in hinge region of the heavy chains. Whether this heterogeneity has any impact on ADC performance is unclear (vide infra).
Like traditional maleimides, NGMs are unstable in the presence of free thiols unless hydrolysed to the maleamic acid form.103 Nunes et al. showed that complete hydrolysis of an NGM-containing antibody-conjugate could be achieved by incubation in pH 8.4 buffer for 72 hours and the resulting hydrolysed conjugate was fully stable in human plasma for 7 days.130 This hydrolysis protocol was used in the synthesis of a DAR 4 ADC via reaction of trastuzumab with a dithiophenolmaleimide linker containing a non-cleavable PEG12 spacer and an MMAE payload (Fig. 9B). The ADC was more efficacious than unmodified trastuzumab in vivo, with three doses at 20 mg kg−1 affording complete tumour regression in mouse models.131
Building on this work, the in vivo stability and efficacy of an NGM ADC was directly compared to a heterogeneous ADC with an average DAR of 4 synthesised via maleimide modification of interchain disulfides.132 A non-cleavable dibromomaleimide linker was employed to connect MMAF to trastuzumab or the anti-CD98 antibody IGNX, generating homogenous DAR 4 ADCs. In both cases, the NGM ADCs had remarkably increased circulation half-life in mice (184 h vs. 130 h) and achieved improved tumour regression compared to their heterogeneous counterparts.
Despite these promising results, the requirement for 72 hour-hydrolysis to ensure stability was still considered a major drawback of NGMs. This process was accelerated by Morais et al. by increasing electron deficiency and steric bulk around the maleimide motif.133 More specifically, it was found that the incorporation of a glycine-derived motif adjacent to the maleimide ring reduced the hydrolysis time from 72 hours to just 1 hour (Fig. 9B).
Most reported NGM ADCs utilise non-cleavable linkers; however, their usage with cathepsin-cleavable linkers has also been reported. Bryden et al. showed that Val-Ala and Val-Cit dipeptides could be incorporated into NGM linkers.134 In the first instance, the hydrophobic nature of the Val-Ala motif was reported to cause a significant reduction in reactivity and only the Val-Cit-NGM linker was able to yield >50% of the desired DAR 4 ADC. However, later incorporation of PEG chains off-set the hydrophobicity issue and allowed for the generation of viable ADCs with either dipeptide motif.
Recently, significant effort has been invested into exploring the effects of different substitution patterns on NGM stability and homogeneity. For example, Forte et al. have reported that diiodomaleimides might be superior to dibromomaleimides in terms of reagent stability.135 Furthermore, Feuillâtre et al. reported the use of hybrid thiobromomaleimides (TBMs) which were shown to produce homogenous ADCs with a marginally narrower DAR distribution compared to dibromomaleimide and dithiophenolmaleimide reagents.136
NGMs have repeatedly demonstrated the ability to generate ADCs with high levels of homogeneity. Optimisation of the ring hydrolysis has significantly improved the utility of this method and allowed for the synthesis of ADCs with excellent plasma stability. In addition to being compatible with a large range of traditional payloads such as MMAE and doxorubicin, NGM linkers have recently been shown to allow for the generation of ADCs with PROTAC payloads, showcasing their broad applicability.137 The NGM linker platform is currently marketed by ThioLogics.
PD reagents can be modified for the generation of dual-functional conjugates, as each of the two ring nitrogens are easily decorated with orthogonal click handles. In 2015, Maruani et al. utilised this capability by modifying trastuzumab with a dibromopyridazinedione linker containing both a terminal alkyne and a strained alkyne.139 These handles were subsequently functionalised with a doxorubicin payload and a fluorophore via orthogonal CuAAC or SPAAC reactions to yield a DAR 4 ADC containing four cytotoxic payloads and four fluorophores (Fig. 9C). Despite the double modification, the ADC maintained high affinity for its target antigen and was completely selective for antigen-positive cell lines in vitro.
The ability to add a second functional moiety to an antibody using PD reagents has also been exploited to mask the hydrophobicity of the attached payload.140 PD reagents functionalised with a DM1 payload and either a PEG6 or PEG26 chain were shown to react efficiently with reduced trastuzumab to generate DAR 4 ADCs of similar homogeneity (Fig. 9C). In vivo, both ADCs showed comparable activity to T-DM1 and lead to complete tumour regression after administration of 2 doses at 10 mg kg−1 in a mouse xenograft model.
Whilst regular PD linkers react with one disulfide each to generate DAR 4 ADCs, Lee et al. exploited the second functionalisation vector of the PD scaffold to connect two reagents, thus creating a single linker that could react with four cysteine residues.141 This modality was amenable to attachment of a single alkyne handle, which enabled the synthesis of antibody conjugates with a controlled loading of 2 payload molecules. The concept was validated by the generation of a homogenous DAR 2 doxorubicin ADC.
In another approach, incorporation of a dendritic spacer into a dibromopyridazinedione reagent enabled the synthesis of a homogenous trastuzumab ADC with a DAR of 16 via attachment of four porphyrin-based photosensitiser payloads to each PD-dendrimer.142 This ADC showed excellent cytotoxicity in antigen-positive cell lines when exposed to light but exerted no effect in the dark, thus demonstrating the compatibility of PD linkers with photosensitiser payloads.
Post-conjugation functionalisation of PD linkers has mainly been achieved via CuAAC or SPAAC reactions; however, other types of click chemistry may also be used. For example, Marquard et al. recently reported the synthesis of a dibromopyridazinedione reagent with a trans-cyclooctene (TCO) functionalisation handle. This PD-TCO reagent was successfully conjugated to three therapeutically relevant IgG1 antibodies (trastuzumab, cetuximab and rituximab) and were further functionalised with tetrazine-containing fluorophores.143 All three conjugates were highly homogenous and could be recovered in high yields. Notably even rituximab, which is known to exhibit >90% protein loss after TCO modification via traditional NHS-ester chemistry, could be recovered in 83% yield after PD-TCO modification. This showcased a clear advantage of disulfide rebridging chemistry over stochastic lysine modification in terms of protein recovery when dealing with aggregation-prone antibodies.
PD reagents are currently considered to be among the best disulfide rebridging agents in terms of ADC homogeneity and have resulted in >90% conversion to the desired DAR 4 conjugate in most of the studies discussed above. However, variable patterns of cysteine connectivity have been reported with half-antibody content ranging from 5% to 95% depending on reagent and reaction conditions.139,140 Although the effect of variable cysteine connectivity on ADC performance remains unknown, methods for reducing half-antibody formation with PD linkers have been explored. For example, Lee et al. found that half-antibody formation could be lowered by the use of a “2-in-1” reagent. The reagent was designed to effect both disulfide reduction and rebridging, thus lowering the residence time of the reduced cysteine residues and their potential for scrambling.144 The dithioaryl(TCEP)pyridazinedione reagent was used for the modification of trastuzumab and led to reduced half-antibody formation in comparison to a two-step reduction-rebridging protocol using TCEP and a regular dibromo- or dithiopyridazinedione linker (Fig. 9D). However, the reagent was not suitable for long-term storage due to poor stability. For this reason, Bahou et al. sought to optimise the PD bioconjugation protocol as an alternative way of maximising full antibody formation without the need for bespoke “2-in-1” reagents.145 It was thus found that the application of an in situ protocol, in which the addition of the PD linker precedes addition of the reducing agent, in combination with a decrease in reaction temperature from 37 °C to 4 °C, led to a significant reduction in half-antibody formation and an improvement in overall ADC homogeneity.
PDs, like NGMs, are among the best disulfide rebridging reagents in terms of reaction kinetics and product homogeneity. Furthermore, the “plug-and-play” aspect allows for facile functionalisation with multiple different payloads via orthogonal click handles is appealing for the synthesis of dual-functional ADCs. Like NGMs, PDs are currently marketed by ThioLogics.
C-Lock™ is a proprietary technology developed by Concortis Biotherapeutics encompassing the use of dibromomethyl heterocycles such as dibromomethylquinoxaline as disulfide rebridging linkers (Fig. 10A). In 2013, Concortis was acquired by Sorrento Therapeutics which used C-Lock™ technology to develop STI-6129, an ADC comprising a duostatin payload and a CD38-targeting antibody, for the treatment of haematological malignancies. STI-6129 showed promising activity in preclinical studies and has recently entered Phase I clinical trials for the treatment of relapsed or refractory systemic AL amyloidosis (ClinicalTrials.gov Identifier: NCT04316442). C-Lock™ technology has also been applied by Zova Biotherapeutics who in 2019 published the development of ZV0508 – an ADC composed of a duostatin payload linked to an antibody targeting 5T4 oncofetal glycoprotein via a dibromomethylquinoxaline linker.146 HIC analysis of this ADC showed excellent conversion (>90%) to the DAR 4 ADC after incubation with just 5 molar equivalents of linker. However, as with other ADCs derived from disulfide rebridging, analysis by CE-SDS showed that Zova's C-Lock™ ADC was comprised of a mixture of full and half-antibody species. Nonetheless, ZV0508 displayed excellent tolerability and potency in preclinical in vivo investigations where it outperformed an analogous ADC generated using maleimide conjugation. Zova Biotherapeutics has since filed patent applications for several anti-HER2 and anti-5T4 ADCs containing C-Lock™ linkers.147,148
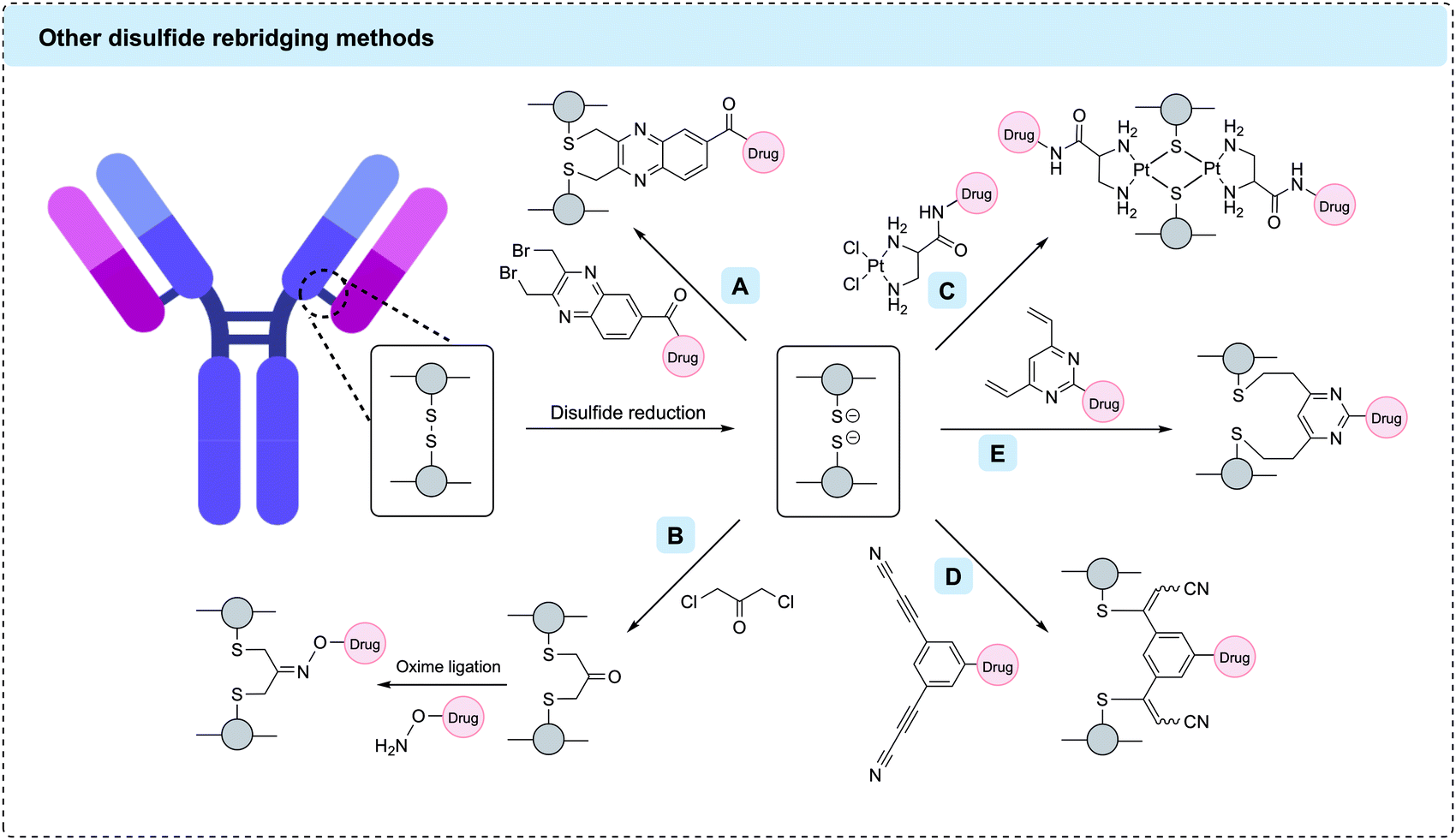 | ||
| Fig. 10 Other disulfide rebridging methods for the generation of homogenous ADCs. (A) C-Lock™ reagents such as dibromomethylquinoxaline; (B) dichloroacetone conjugation followed by oxime ligation; (C) Pt(II)-based reagents; (D) arylene dipropiolonitrile (ADPN); (E) divinylpyrimidine (DVP). | ||
Concurrently, Novartis has reported the use of 1,3-dihaloacetone reagents such as 1,3-dichloroacetone or 1,3-dibromoacetone for the rebridging of antibody disulfides. In the resulting conjugates, each pair of cysteines is connected through a three-carbon tether containing a reactive ketone. This ketone can then be further reacted with a hydroxylamine-modified payload via oxime ligation (Fig. 10B). In one application, this approach was used for the construction of an anti-HER2 ADC with a DAR of 3.8.149 In a different application, the introduced ketones were linked together by a linker-payload containing two hydroxylamine groups, thus enabling the generation of a MMAF-containing anti-HER2 ADC with a reported DAR of 1.8.150 Both ADCs were shown to be highly homogenous by mass spectrometry and SDS-PAGE with approximately 90% conversion to the desired product and minimal half-antibody formation.
Invictus oncology has developed platinum(II) reagents for disulfide rebridging (Fig. 10C).151 A Pt(II)-based linker-payload was generated by tethering the topoisomerase inhibitor camptothecin to a bivalent amine ligand which was subsequently complexed with Pt(II) chloride. This linker-payload reacted with reduced disulfides in trastuzumab, rituximab or cetuximab to generate ADCs with a DAR of 8. The ADCs showed increased stability and homogeneity versus analogous maleimide conjugates, although a significant amount of half-antibody formation was observed. The biological activity of the cetuximab ADC was validated in vitro and in vivo. The ADC was only marginally more active than unmodified cetuximab; however, this may be due to the choice of payload or release mechanism rather than the linker.
In addition to these industrially developed methods, multiple academic labs have partaken in the expansion of disulfide rebridging methods. For instance, Koniev et al. published a reduction-rebridging strategy for the generation of ADCs using arylene dipropiolonitrile (ADPN) linkers in 2018 (Fig. 10D).152 This work was inspired by previous findings that arylene monopropiolonitrile linkers formed very stable linkages with cysteine residues.153 To translate this into a rebridging strategy, three regioisomers of ADPN were compared. It was found that meta-ADPNs gave superior conversion over ortho- or para-ADPNs, albeit yielding a mixture of full and half-antibody species. The meta-ADPN linker was subsequently utilised to generate a trastuzumab ADC containing MMAE and a β-galactosidase-cleavable linker. Native mass spectrometry revealed ∼50% conversion to the desired DAR 4 species, with significant amounts of DAR 3 and DAR 5 species present. The ADC showed comparable cytotoxicity to T-DM1 when evaluated in vitro.
In 2019, Walsh et al. reported the use of divinylpyrimidine (DVP) reagents as disulfide bridging reagents for the generation of ADCs.154 Like meta-ADPNs, DVPs enable rebridging via two consecutive Michael-addition reactions; however, rather than installing a five-carbon bridge, DVPs insert a flexible seven-carbon bridge between the two cysteine residues (Fig. 10E). This approach was used for the synthesis of several trastuzumab-based DAR 4 ADCs containing cathepsin-cleavable, sulfatase-cleavable, or non-cleavable spacers and an MMAE, hemiasterlin or doxorubicin payload.25,154,155 In all cases >90% conversion to the desired DAR 4 species was observed, which existed as mixtures of full and half-antibody formats. The conjugates displayed complete stability in human plasma over 14 days and were highly potent and selective in vitro. Recently, the scope of the methodology was further expanded by the development of a dual-functional DVP linker.156 This linker enabled efficient dual functionalisation of trastuzumab with MMAE and a fluorophore without causing any negative effects on the activity of the antibody or either payload. Divinyltriazine reagents have also been shown to generate antibody conjugates with a payload loading of 4, with the rebridging proceeding efficiently using near-stoichiometric quantities of reagent.157
Many of the current rebridging methods suffer from the formation of half-antibody species which originate from non-native intrachain rebridging of the hinge region disulfides (vide supra). Such half-antibody species lack the native covalent link between the two heavy chains but remain held together by strong non-covalent interactions. The impact of these species on the physicochemical properties and binding of ADCs depends on the particular system. In 2015, Lyon et al. compared the plasma clearance rate of unmodified disulfide-containing antibodies and maleimide-modified antibodies in which all interchain disulfides had been reduced. No significant difference was observed, indicating that interchain disulfide bonds are not essential to antibody stability and their absence does not negatively affect clearance.42 However, a more recent investigation by Bahou et al. produced contrasting results.158 Dibromo- and dichloropyridazinedione linkers were used to generate antibody-conjugates with varying homogeneity ranging from 10–50% half-antibody content. The thermal stability, aggregation potential and antigen-binding affinity of these conjugates were compared and the conjugate with the higher half-antibody content was shown to perform marginally worse than its more homogenous counterpart in many of the assays. No comparison between the performance of analogous half-antibody and full-antibody ADCs was undertaken. Therefore, it appears that more research into the nature and importance of half-antibody formation is warranted.
2.4 Non-canonical amino acids
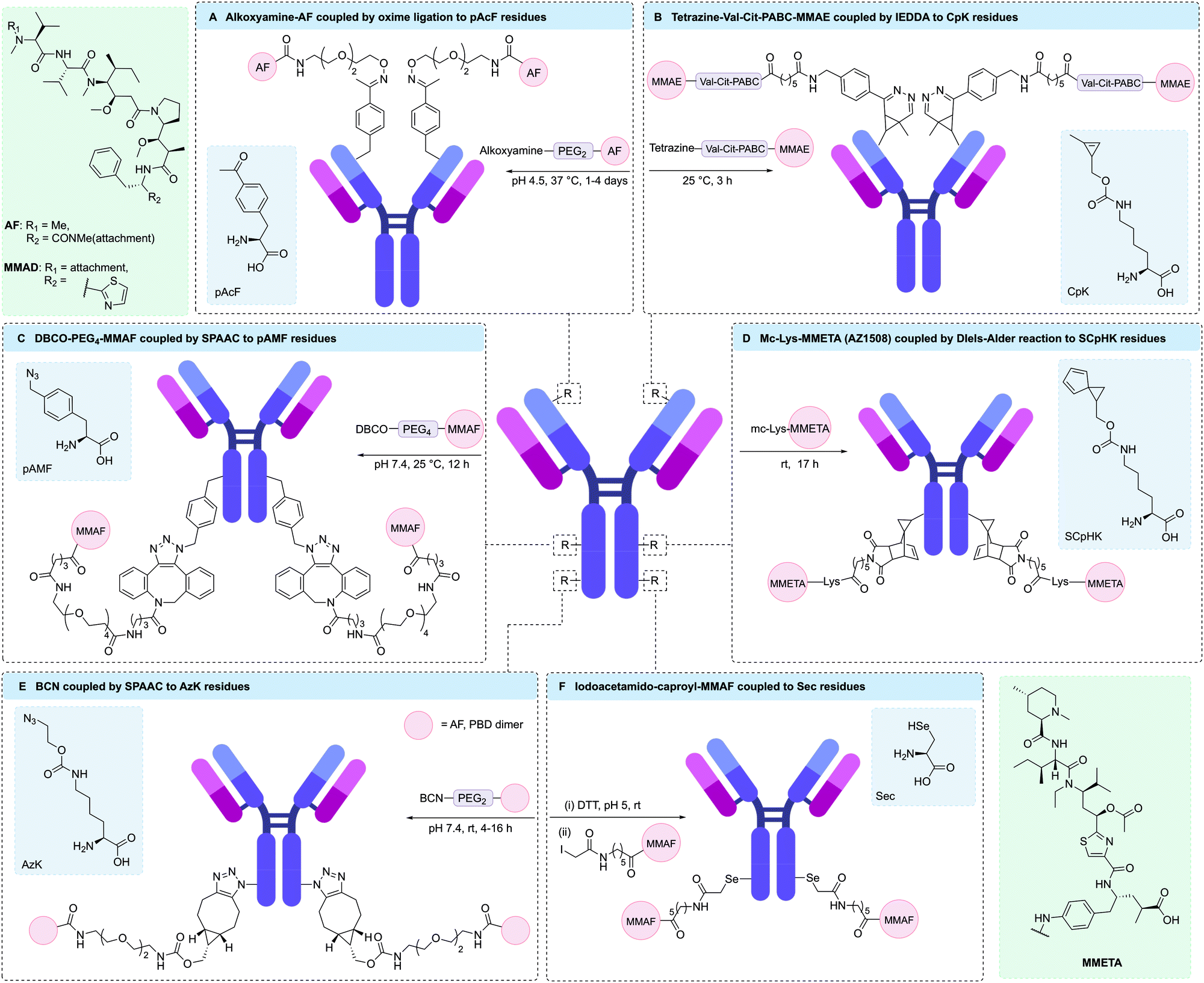 | ||
| Fig. 11 Unnatural amino acids and site-specific bioconjugation. (A) pAcF has a ketone side chain which can participate in oxime ligation reactions; (B) CpK has a cyclopropene side chain which can participate in IEDDA reactions; (C) pAMF has a an azide side chain which can undergo click reactions; (D) SCpHK has a spiro[2.4]hepta-4,6-diene side chain which can participate in Diels–Alder reactions; (E) AzK has a an azide side chain which can undergo click reactions; (F) Sec has a selenol side chain which can undergo reduction and subsequent nucleophilic substitution reactions. | ||
Antibodies engineered to incorporate pAcF have also been used to selectively target immune cells through the CD11a antigen. Using an anti-CD11a antibody conjugated to a liver X receptor agonist, Lim et al. were able to target macrophages to reverse cholesterol transport and limit inflammation, whilst preventing undesirable lipogenic effects in hepatocytes.180 To achieve this, pAcF was site-specifically incorporated into a single site on both heavy chains of an anti-CD11a antibody. Next, an aminooxy-functionalised Liver X receptor agonist was conjugated to the engineered antibody through an oxime linkage, affording the desired DAR 2 ADC. Yu et al. were also able to use an anti-CD11a antibody engineered to incorporate pAcF in both heavy chains to selectively deliver a phosphodiesterase 4 inhibitor for the treatment of inflammatory conditions.181 Modification of the engineered antibody with an analogue of GSK256066, a known phosphodiesterase 4 inhibitor, via an oxime linkage proceeded efficiently to give an ADC with a DAR of 2. The resulting ADC was shown to rapidly internalise into immune cells and suppress lipopolysaccharide-induced TNFα secretion in monocytes, as well as significantly reducing inflammatory cytokine production.
To aid the synthesis of more homogenous ADCs, Sato and co-workers have developed a novel platform for cell-free expression of ncAA-containing antibodies with faster reaction kinetics for conjugation via SPAAC.185 This involved the engineering of an aaRS/RNA pair to enable incorporation of either pAzF or para-azidomethylphenylalanine (pAMF) into aglycosylated trastuzumab. It was hypothesised that pAMF would enable more rapid SPAAC conjugation compared to pAzF, due to the azido group being further from the electron withdrawing phenyl ring, hence generating ADCs with more precise drug-loading. A number of pAMF containing antibodies have since been produced via this CFPS expression system, with the resulting engineered antibodies being successfully conjugated to dibenzylcyclooctyne (DBCO)-MMAF, DBCO-maytansinoid or DBCO-Val-Cit-hemiasterlin payloads to give highly potent ADCs (Fig. 11C).174,185–187 Ahn et al. were also able to use this technology to introduce two DBCO-functionalised bifunctional chelators to trastuzumab, thus enabling the incorporation of radioisotopes for positron emission tomography.188 A modified CFPS system enabled incorporation of 2, 4, 6, or 8 pAMF residues into an anti-HER2 antibody, which could then undergo conjugation to DBCO-maytansine to yield ADCs with DAR values of 1.77, 3.83, 5.82 and 7.43, respectively.174 These ADCs were tested against several HER2-expressing cell-lines (SKBR3, BT474, MDA-MB-453, and JIMT1), showing a general trend of higher potency with increasing DAR.
In addition to alanine-based ncAAs, several lysine analogues, such as AzK, have also been site-specifically incorporated into antibodies. For example, Zhou and co-workers used AzK-containing rituximab to generate a dodecane tetraacetic acid-rituximab conjugate via SPAAC.189 Subsequent radiolabelling of the chelate-modified antibodies with 64Cu resulted in a homogeneous radioconjugate with two chelates per antibody. Marelli and co-workers were also able to express AzK in antibodies, enabling the subsequent generation of ADCs with AF, PBD dimer, or tubulysin payloads (Fig. 11E).167,175 In one example, an anti-HER2 antibody expressing AzK on each heavy chain was conjugated to DBCO-tubulysin to give an ADC with a DAR ∼ 2. In vitro assays demonstrated the potent and selective cytotoxicity of the resulting ADC.167
In eukaryotes, Sec is incorporated into polypeptides in response to the UGA stop codon when a Sec incorporation sequence (SECIS) is present in the 3′ untranslated region of the mRNA.193,194 First-generation selenomabs were engineered to incorporate one or two C-terminal Sec residues by inserting the UGA codon and SECIS at the 3′ end of its encoding gene.193,195 The resulting recombinant antibodies were shown to fully retain their antigen binding capabilities. However, competition between Sec incorporation and termination at the UGA codon, led to low Sec incorporation efficiency.195 Indeed, inefficient Sec incorporation via the UGA stop codon and SECIS element remains a challenge that requires further optimisation.
Rader and co-workers have exploited the high reactivity of Sec under mildly acidic conditions for selective modification of a trastuzumab-based selenomab via reaction with iodoacetamide-modified MMAF.196 Initial studies positioned the Sec residue at the C-terminus of the antibody. However, competition between Sec insertion and termination at the UGA codon resulted in a mixture of IgG-stop and IgG-Sec-MMAF proteins, thus yielding a selenomab-drug conjugate with an average DAR of 0.6. Alternatively, by positioning Sec residues in the CH3 domains of trastuzumab at HC-396 the incorporation of two Sec residues was achieved. In addition, higher conjugation efficiency of the resulting selenomab with iodoacetamide-modified MMAF was observed due to the higher solvent accessibility of the Sec residues (Fig. 11F). Indeed, a DAR 2 iodoacetamide-based selenomab-drug conjugate was produced, which showed excellent stability, selectivity, and potency in both in vitro and in vivo mouse models.
 | ||
| Fig. 12 Dual functional ADCs generated via GCE. (A) Microtubulin inhibitor AF and fluorescent dye AlexaFluor 488 were conjugated to a HER2-targeting antibody via site-specific conjugation at the engineered pAcF and AzK residues, respectively; (B) DNA crosslinking agent PNU-159682 and tubulin polymerisation inhibitor MMAF were conjugated to a HER2-targeting thio-selenomab via site-specific conjugation at the engineered Sec and Cys residues, respectively. | ||
More recently, thio-selenomabs have also facilitated the generation of dual functional antibodies through genetic incorporation of an ncAA.200,201 These antibodies contain both engineered Sec and cysteine residues, enabling bioorthogonal conjugation of two distinct payloads. Indeed, this dual conjugation method was used by Nilchan et al. to generate an anti-HER2 ADC that combined both the tubulin-targeting payload MMAF and the DNA-damaging payload PNU-159682 (Fig. 12B).200 This required engineering of trastuzumab to contain Sec (HC-S396) and additional cysteine residues (HC-A114C) in both heavy chains. Subsequent site-selective dual modification was achieved via reaction of Sec with iodoacetamide-functionalised PNU-159682, followed by reaction of cysteine residues with methylsulfone phenyloxadiazole-MMAF to produce a serum stable bioconjugate. Treatment of human breast cancer cells with the resulting ADC suggested distinct mechanisms of action for each payload upon analysis of cell cycle arrest. However, the ADC combining PNU-159682 and MMAF did not demonstrate improved potency compared to an ADC functionalised with PNU-159682 alone.
Two further ADCs currently in Phase I clinical trials, STRO-001 and STRO-002, have been developed by Sutro.186,187 These ADCs were generated using a cell-free platform, XpressCF+™, which has an engineered RF1 mutant to facilitate efficient incorporation of pAMF at positions designated by a UGA codon.174 In STRO-001, pAMF was incorporated into each heavy chain of an anti-CD74 antibody by replacing the codon corresponding to HC-F404 with an amber codon.187 Subsequent SPAAC between each pAMF and a DBCO-functionalised maytansinoid payload conjugated via a non-cleavable linker gave STRO-001; an ADC with a DAR of 2. Preliminary trial data has shown that STRO-001 is generally well tolerated and has encouraging anti-tumour activity in a group of patients with pre-treated diffuse large B-cell lymphoma (ClinicalTrials.gov Identifier: NCT03424603).169 In contrast, STRO-002 is a DAR 4 ADC composed of an anti-folate receptor alpha human IgG1 antibody conjugated to a cleavable drug-linker (SC239) containing the tubulin-targeting 3-aminophenyl hemiasterlin payload SC209.186 To generate this ADC, four pAMF residues were incorporated into the antibody at two defined sites on each heavy chain. These sites were then conjugated to SC239 via a cleavable Val-Cit-PABC linker functionalised with DBCO. Studies in patients with solid tumours have shown that this ADC is also well-tolerated, with mostly mild adverse effects, and the clinical benefits are promising (ClinicalTrials.gov Identifier: NCT03748186).170 Indeed, based on a trial of 20 ovarian cancer patients STRO-002 dose levels of 2.9 mg kg−1 or higher led to one partial response and 14 patients with stable disease in initial post-baseline scans. Furthermore, 13 patients had a ≥50% reduction or normalisation of cancer biomarkers.
In addition to these aforementioned ADCs in clinical trials, several more ADCs containing ncAAs are also progressing towards Phase I trials. Whilst production challenges still exist, recent efforts have successfully increased both protein expression and reaction kinetics. Thus, the applicability of this approach for the generation of clinically useful ADCs is rapidly increasing.
2.5 C-/N-terminal modifications
Modification of either the N- or C-terminus of an antibody has proven to be a viable strategy for generating homogeneous ADCs. Antibody modification at these positions enables simple and site-specific introduction of bioorthogonal motifs or affinity tags that facilitate further functionalisation. Importantly, the C-terminal positions are generally distal from critical antigen binding regions, which typically leads to complete retention of antibody binding specificity upon modification. Accordingly, C-terminal modifications have broad applicability across antibodies and the wider protein modification field. N-Terminal modifications have also been prolifically used for site-specific protein conjugation.205 However, this approach is more challenging to apply to antibodies due to the close proximity to the vital receptor binding region. Hence, modifications at the N-terminus must be carefully monitored to ensure that conjugation does not hinder antigen binding affinity. The terminal α-amine group has a pKa of 6–8, making it more nucleophilic than lysine residues under milder conditions.205 Hence, this terminal position can be selectively targeted in the presence of other aliphatic amines at low or neutral pH. Furthermore, the microenvironment at the terminal positions often differs to that of other regions within the protein structure, a factor that has been harnessed to enhance selectivity for conjugation.Bioorthogonal functionalities suitable for site-selective conjugation can be introduced to the terminal positions of an antibody using synthetic strategies. For example, several chemical methods detail the introduction of reactive aldehyde handles onto the antibody termini, which can be further modified to enable the generation of homogeneous ADCs.
N-Terminal transamination is one strategy used to synthesise ADCs in a site-selective manner via conversion of the amino N-terminus to an aldehyde functional group. N-Methylpyridinium-4-carboxaldehyde benzenesulfonate salt (Rapoport's salt) was found to transaminate glutamate-terminal proteins,206 installing an aldehyde or ketone functional handle suitable for conjugation through oxime or hydrazone linkages (Fig. 13A). Trastuzumab was used to examine the efficiency of this conjugation approach in an antibody context. Initially, unaltered trastuzumab containing an EVQ motif at the heavy chain N-terminus was treated with Rapoport's salt, yielding 67% of the conjugation product. No modification of the light chain terminal, which contained a DIQ sequence, was observed. Next, modified antibody constructs were prepared whereby the N-termini of both the heavy and light chains were modified to append an EES peptide motif, which was discovered to have enhanced and more selective reactivity with Rapoport's salt. Conjugation of this modified antibody via oxime ligation gave 56% and 68% yield on the light and heavy chains, respectively. Using this technique, AlexaFluor 488 antibody conjugates were prepared and analysis demonstrated that N-terminal modification did not hinder antigen binding, validating this approach for ADC preparation.
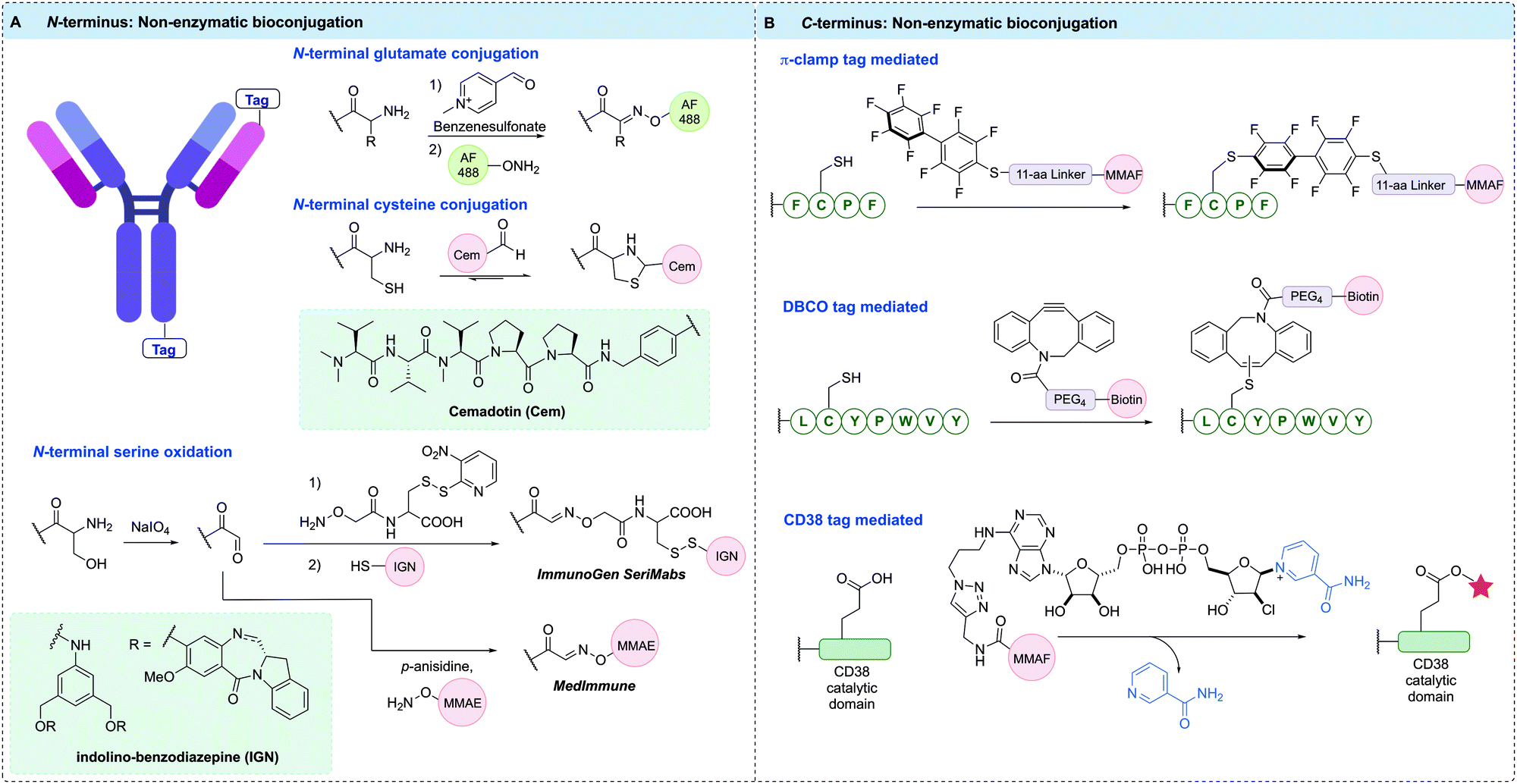 | ||
| Fig. 13 Non-enzymatic bioconjugation methods used to construct homogeneous ADCs at the: (A) N-terminal position, where terminal glutamate and cysteine residues can be selectively modified with aldehyde functionalised payloads, and Ser residues introduced can be oxidised to aldehydes, which can be trapped using alkoxyamine functionalised payloads to form oxime linked conjugates; (B) π-clamp peptide sequence selectively reacts with perfluoroaromatic probe, DBCO tagged antibody selectively reacts with DBCO reagents via the thiol–yne reaction, CD38 tag reacts with covalent inhibitor-tagged payload, forming a stable arabinosyl ester. | ||
The aldehyde moiety has also been exploited for the modification of cysteine residues appended to antibody N-termini (Fig. 13A).207 This technique generated a homogeneous thiazolidine linked ADC through reaction of the 1,2-aminothiol functionality found on the anti-fibronectin F8 antibody N-terminus cysteine residues with an aldehyde-containing drug. The drug used in this proof-of-concept study was cemadotin, an analogue of dolastatin-15. The aldehyde-containing derivative was found to have comparable cytotoxicity to the parent compound, validating its use in ADC development. The resultant ADC enabled slow in vitro release of the toxic payload through hydrolytic cleavage of the thiazolidine linkage. The antibody specifically targets cancer cells and upon internalisation, releases the free aldehyde-containing drug in a traceless fashion. An interesting application of this technology is in the masking of aldehyde functional groups present in drugs. Aldehyde functionalities generally suffer from oxidation, potential for epimerisation or reactivity with various biomolecules; therefore, antibody conjugation is a useful strategy to mitigate their off-target effects.
Another important strategy for homogeneous ADC generation involves the selective oxidation of a serine residue genetically appended to the antibody N-terminus (Fig. 13A). Such a serine-containing antibody can then undergo sodium periodate-mediated oxidation, generating an aldehyde handle. The aldehyde can subsequently react with alkoxyamine functionalised payloads under mild conditions to form oxime linked conjugates. This technology was reported by both ImmunoGen and MedImmune in 2015. ImmunoGen designated this serine modification strategy as SeriMabs and following the serine oxidation, condensed the resulting aldehyde with a dithiopyridine-containing heterobifunctional linker, generating a stable oxime bond.208 The dithiopyridine groups could then be reacted with thiol-containing payloads, forming a disulfide linked DAR 2 ADC in >90% yield. As such, the SeriMab technology was utilised to conjugate a mono-imine containing indolino-benzodiazepine (IGN) DNA-alkylating payload connected to the N-terminal aldehyde via oxime ligation.209 The generated ADC exhibited comparable antigen binding affinity to the unconjugated antibody, and was highly potent in vitro and in vivo. Importantly, the oxime linkage was determined to be stable in vivo for 3 days, with an approximate payload release t1/2 of >10 days. An MMAE-conjugated ADC with a DAR of 2 was also prepared by MedImmune using the same serine oxidation strategy applied to an anti-EphA2 antibody.210 This ADC exhibited good hydrolytic and serum stability and had high potency in vitro against PC-3 cell lines whilst also displaying good in vivo efficacy. Additionally, a DAR 4 ADC was generated by modifying terminal serine residues on both the light and heavy chains.
Site-specific bioconjugation can also be achieved by modifying the microenvironment of an amino acid, which can activate a specific amino acid residue in the presence of other reactive species (Fig. 13B). One recent example of this approach installed a so-called π-clamp peptide sequence (FCPF) to direct site-selective modification of the peptide cysteine with perfluoroaromatic reagents in the presence of competing cysteine residues.211 The π-clamp sequence was initially appended to the C-termini of trastuzumab heavy chains. Selective cysteine conjugation was achieved by introducing an MMAF-conjugated perfluoroaromatic probe, which rapidly reacted with the π-clamp cysteine residue via a SNAr reaction under mild, reducing conditions. Notably, none of the native interchain cysteines displayed any reactivity with this reagent under reducing conditions. The resultant ADC retained HER2 affinity and exhibited high in vitro potency and selectivity. The conjugation technology was also applied to the selective modification of a C225 antibody appended with the π-clamp, indicating the generality of this strategy.
A further example of microenvironment modification harnessed the thiol–yne reaction to achieve rapid and site-selective conjugation of DBCO reagents to a seven-residue peptide tag (LCYPWVY) introduced at the protein C-terminus (Fig. 13B).212 The cysteine-containing peptide sequence (DBCO-tag) acts as an affinity tag to enable highly regioselective cysteine modification. A DBCO-tagged antibody was generated by genetically fusing the DBCO-tag to the heavy chain C-terminus of trastuzumab. The modified antibody was then treated with biotin-conjugated DBCO under reducing conditions, generating 90% mono-labelled antibody, and importantly, leaving the eight native cysteines intact. The antigen binding affinity of the resultant antibody was unaffected, and the thiol enol ether linkage was highly stable to glutathione under physiological conditions for over four days. This method is highly versatile due to the multitude of commercially available DBCO reagents, which may enable rapid and efficient synthesis of ADCs.
CD38 is an adenosine diphosphate (ADP)-ribosyl cyclase that catalyses the production of ADP-ribose and cyclic ADP-ribose from nicotinamide adenine dinucleotide (NAD+). Covalent inhibitors of CD38 have been developed that react with catalytic E226 to form a stable arabinosyl ester. Dai et al. exploited these developments via fusion of the catalytic domain of CD38 to the heavy chain C-terminus or light chain N-terminus of trastuzumab.213 Subsequent reaction of the fusion antibody with an MMAF-modified covalent inhibitor of CD38 generated a pair of DAR 2 ADCs, as evidenced by MS (Fig. 13B). The ADCs retained antigen affinity and selectivity, demonstrated high stability in mouse plasma, and were highly potent in vitro and in vivo.
Several different chemical methods for termini modification have been utilised for antibody modification. Importantly, modification at either the N- or C-termini of both heavy and light chains appears possible without detriment to antibody binding of target receptors or Fc receptors.
2.6 Other chemical methods
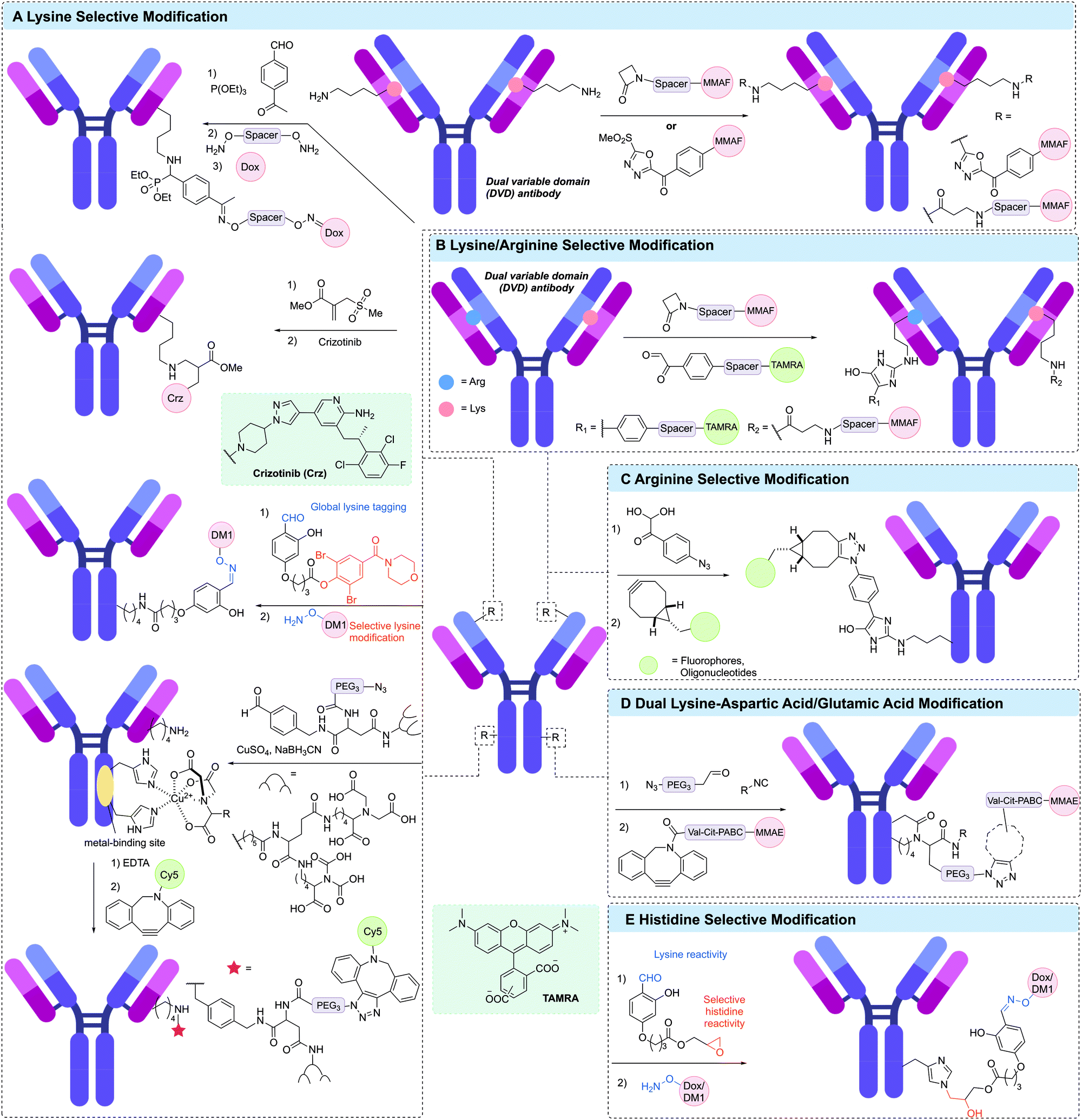 | ||
| Fig. 14 Other residue-selective modification for generating ADCs. (A) Lysine selecive modification via a phospha-Mannich reaction (left top), using sulfonyl acrylate reagents (left second), using linchpin directed modification reagent (left third), of catalytic lysine in variable domain using β-lactam derivative and methylsulfone phenyloxadiazole (MS-PODA) derivative (right top), and using metal-binding site-directed modification (bottom left); (B) lysine/arginine selective modification using β-lactam and phenylglyoxal derivatives; (C) arginine selective modification usig 4-azidophenyl glyoxal (APG) reagent; (D) dual lysine-aspartic acid/glutamic acid modification using aldehyde and isocyanide-modified compounds; (E) histidine selective modification using linchpin directed modification. | ||
A unique approach to selective antibody modification has involved the introduction of the variable domain from the aldolase antibody 38C2 (or h38C2 for the human variant) into the antibody scaffold to generate a dual variable domain (DVD) antibody. The variable domain of 38C2 contains a catalytic lysine with low pKa (∼6) in its enzyme active site. In one use of this DVD format, Rader and co-workers generated a DVD based on trastuzumab and the variable domain of h38C2, which was site-selectively modified via reaction with 1,3-diketone reagents or a β-lactam derivative of MMAF in >95% conversion (Fig. 14A).216–218 This anti-HER2 DAR 2 ADC demonstrated exquisite in vitro and in vivo activity and selectivity. Based on these results, anti-CD138 and anti-CD79b ADCs were also generated from DVD antibodies. Although these antibodies are ∼50 kDa larger than typical IgGs, the authors noted that they retain similar pharmacokinetic properties. The reactive lysine in the catalytic pocket was also amenable to selective modification with methylsulfone phenyloxadiazole (MS-PODA) modified payloads (Fig. 14A).219,220
Mortensen et al. exploited the metal-binding ability of proteins to site-selectively modify lysine residues in a range of therapeutic antibodies.221,222 Treatment of the antibody with a metal (CuSO4) and a small molecule metal coordination ligand, which was further functionalised with an aldehyde enabled modification of specific lysines in the vicinity of the metal binding site via reductive amination of the aldehyde (Fig. 14A). Incorporation of an azide in the metal chelator enabled further modification via SPAAC with a DBCO-Cy5 payload. Removal of the Cu2+ was also achieved by treatment with EDTA. This strategy was used to modify trastuzumab, rituximab and cetuximab with modification of trastuzumab shown to occur primarily at LC-K190 and HC-K136.
Among the numerous Fc-binding domains (FcBDs), the ZZ-domain, a modified dimer of the IgG binding site of protein A from Staphylococcus aureus,228 is widely used. Mazor et al. generated an IgG-binding peptide-toxin fusion, ZZ-PE38 by genetically fusing the ZZ-domain to a truncated Pseudomonas exotoxin A (PE38).229 This strategy was used to generate non-covalent antibody–toxin conjugates targeting HER2, CD24 and EGFR, with each demonstrating efficient tumour regression in vivo.230–232
Instead of the FcBD protein, Park et al. have reported a peptide-directed photo-crosslinking (PEDIP) approach to covalently modify the Fc region of an antibody.233 A 13 residue peptide (called Fc-III) was identified as binding the Fc region of IgG antibodies. Incorporation of para-benzoyl-phenylalanine (BPA) into the Fc-III peptide enabled covalent modification of HC-M252 in trastuzumab upon irradiation with ultraviolet light (Fig. 15A). Further modification of Fc-III with another truncated analogue of Pseudomonas exotoxin A (PE24) enabled generation of a trastuzumab-PE24 conjugate with 1 toxin per antibody. Similarly, Vance et al. utilised the same BPA-containing Fc-III peptide to install a protected thiol site-selectively on trastuzumab. Subsequent thiol deprotection enabled further modification with maleimide-Val-Cit-PABC-MMAE to generate a DAR 1.9 ADC. The authors also demonstrated the high stability of the conjugate in human plasma and potent in vitro cytotoxicity.234
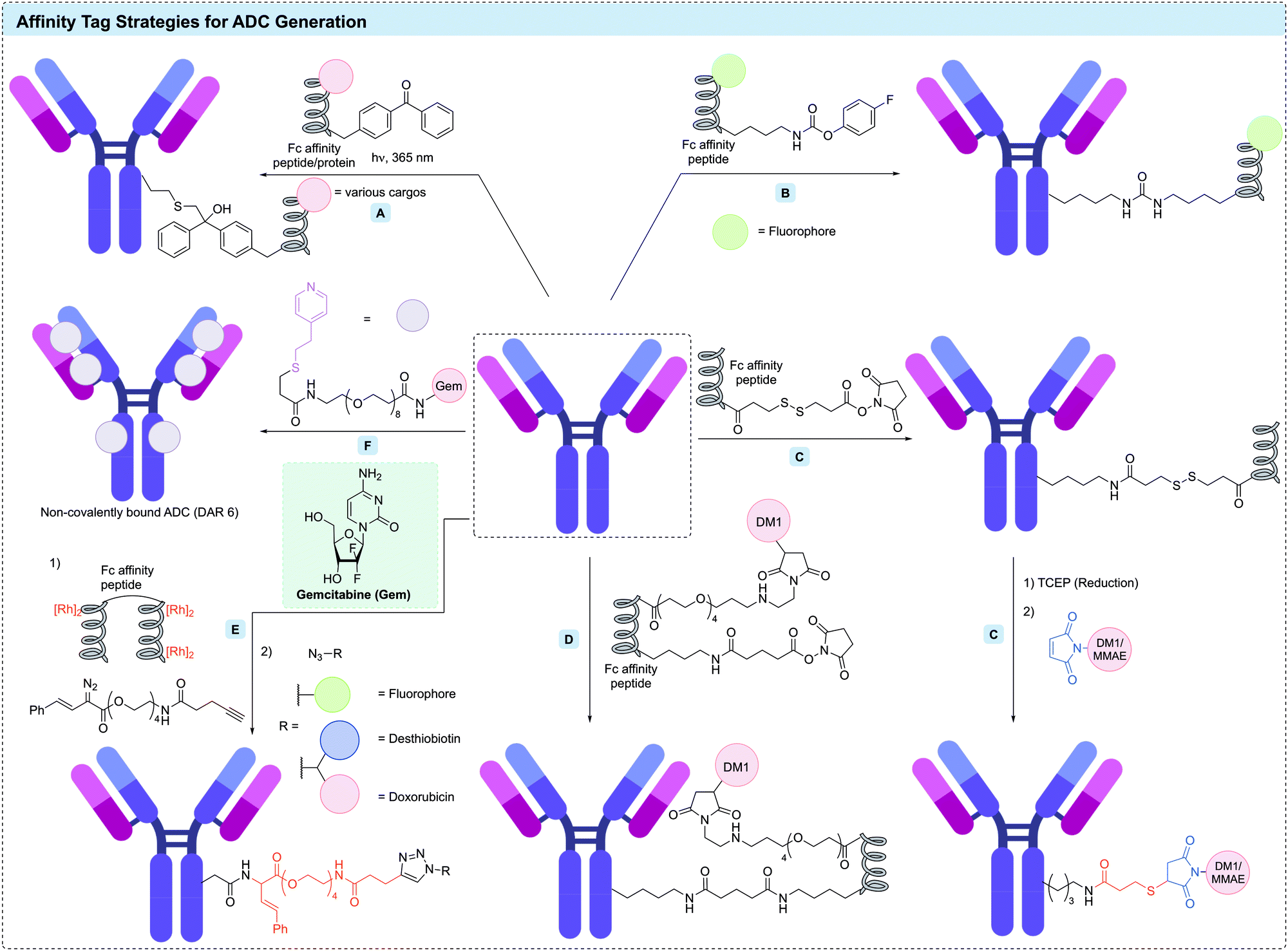 | ||
| Fig. 15 Affinity tag strategy for ADC generation. (A) Photoaffinity labeling strategy using Fc affinity peptide/protein; (B) affinity peptide in combination with the activated ester method to modify lysine resdues; (C) traceless affinity peptide labeling using the cleavable linker in combination with activated ester method (AJICP); (D) the use of an Fc-affinity peptide with an appended N-hydroxysuccinimide ester (NHS); (E) traceless affinity peptide labeling using the metallopeptide method; (F) self-assembling strategy via interaction of the antibody with a mercaptoethylpyridine (MEP) derivative. | ||
Kishimoto et al. have reported the use of an Fc-directed peptide (17 residues, similar to Fc-III) with an appended NHS ester to enable selective modification of Fc lysines (Fig. 15D).235 This conjugation reaction completed within 15 minutes under mild conditions and produced the desired conjugate in high yield. This method enabled site-specific modification of HC-K248 in the Fc region of trastuzumab. In one example, further functionalisation of the affinity peptide with DM1 yielded ADCs with a DAR of 1 or 2. The DAR 1 trastuzumab-peptide conjugate maintained its affinity for Fc receptors (FcRn, FcγRI, and FcγRIIIa); however, this affinity was completely lost in the DAR 2 ADC. It was hypothesised that this loss of FcRn binding was caused by overlap between the binding site of the Fc-III-derived peptide and FcRn.
Yu et al. have described the use of the B domain of protein A to direct site-selective modification of an Fc lysine in trastuzumab.236 A library of modified B domains containing ncAAs with lysine-reactive side chains (e.g. acrylamide, isothiocyanate or carbamate) were reacted with trastuzumab. The carbamate-containing peptide was shown to cross-link efficiently with the antibody heavy chain K337, forming a stable urea linkage (Fig. 15B). This approach was used to obtain a fluorescein-labelled antibody with a fluorophore loading of 2.
Gupta et al. reported the novel type of non-covalent ADCs that self-assemble via interaction of the antibody with a small molecule affinity ligand (Fig. 15F).245 4-Mercaptoethylpyridine (MEP), which is used in antibody purification because of its ability to selectively bind distinct regions within both the Fab and Fc fragments was used to facilitate this non-covalent conjugation.246,247 This method yielded homogeneous DAR 6 ADCs via reaction of trastuzumab or cetuximab with MEP-modified gemcitabine. The modification reaction proceeded rapidly (<8 min) by binding at two conserved binding sites on each Fab and two in the Fc region. Furthermore, the structure, binding specificity, and affinity of the antibody were retained after modification. Remarkably, the conjugates were highly stable in plasma and showed excellent anti-tumour efficacy in mice with non-small cell lung cancer xenografts.
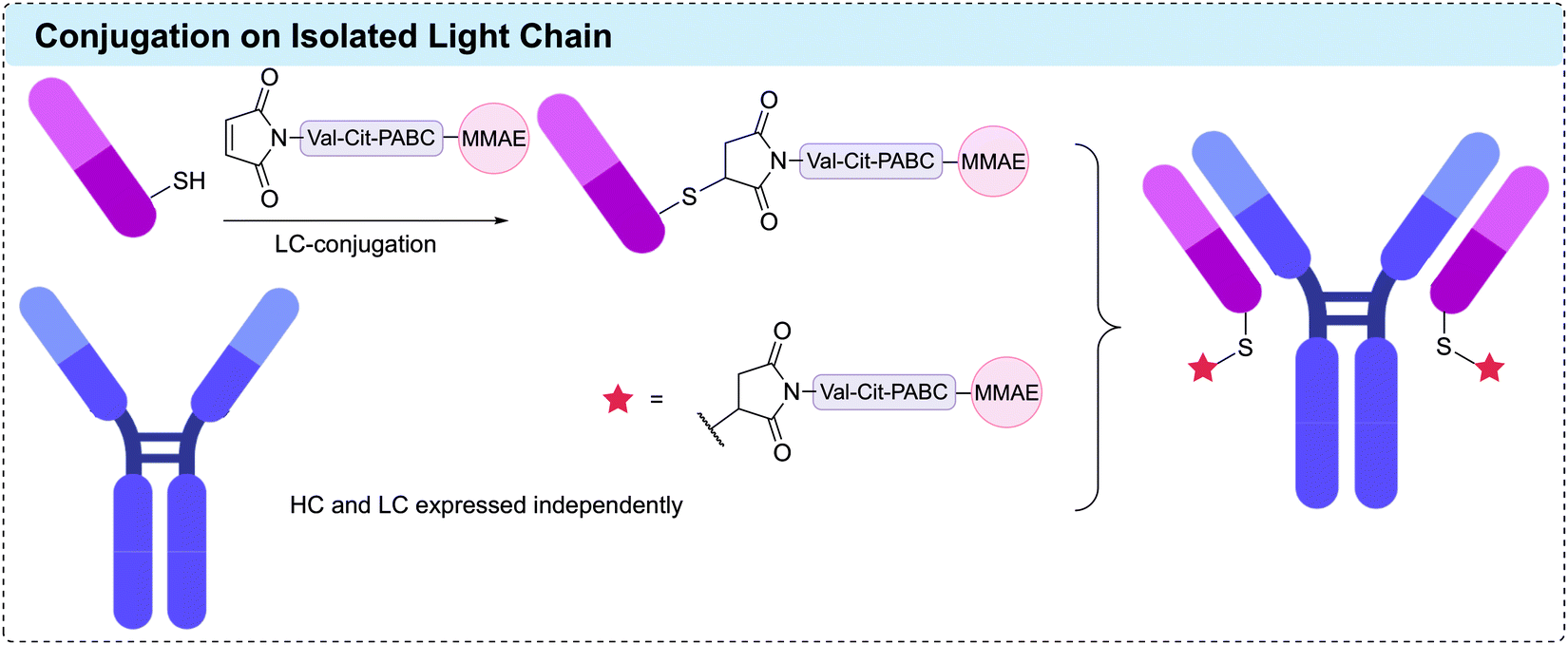 | ||
| Fig. 16 Strategy to obtain DAR 2 homogeneous ADCs via assembly from independently produced heavy chain and MMAE-conjugated light chain. Each chain was independently produced and purified, and the LC modified via reaction with mc-Val-Cit-PABC-MMAE. | ||
3 Amino acid modification – enzymatic methods
Enzymes have been frequently used to achieve site-selective antibody modification due to their high specificity and mild reaction conditions. Enzymes can either directly attach a payload to a specific amino acid sequence or introduce a reactive functionality on the antibody that can be further functionalised with the desired payload. Both approaches have been successfully utilised to generate homogenous ADCs.3.1 Transglutaminase
Amongst the enzymatic methods available for attachment of a payload to an antibody, microbial transglutaminases (mTG) have proven themselves particularly useful.249 Protein-glutamine γ-glutamyltransferases (transglutaminases) are a class of enzyme that catalyse the acyl transfer reaction between the γ-carboxyamide group of glutamine residues and 6-amino-groups of lysine residues, to form intra- and intermolecular N6-(5-glutamyl)-lysine isopeptide crosslinks with subsequent release of ammonia.250 The unique reactivity of transglutaminases has been exploited as a diverse tool for the post-translational modification of proteins. The transglutaminase from actinobacterium Streptomyces mobaraensis was first isolated by Ando et al. in 1989.251,252In the context of IgG modification, a number of distinct approaches to effecting specific modification have emerged (Fig. 17). Native or engineered residues can be targeted; however, large proteins such as human IgG contain a high number of surface-exposed Gln and Lys residues, presenting a potential challenge to control the site-selectivity and loading of a proposed transamidation.253 In many successful approaches, the antibody adopts the role as the acyl-donor substrate, given that amino acids neighbouring Gln are known to exert a greater influence on mTG specificity.251
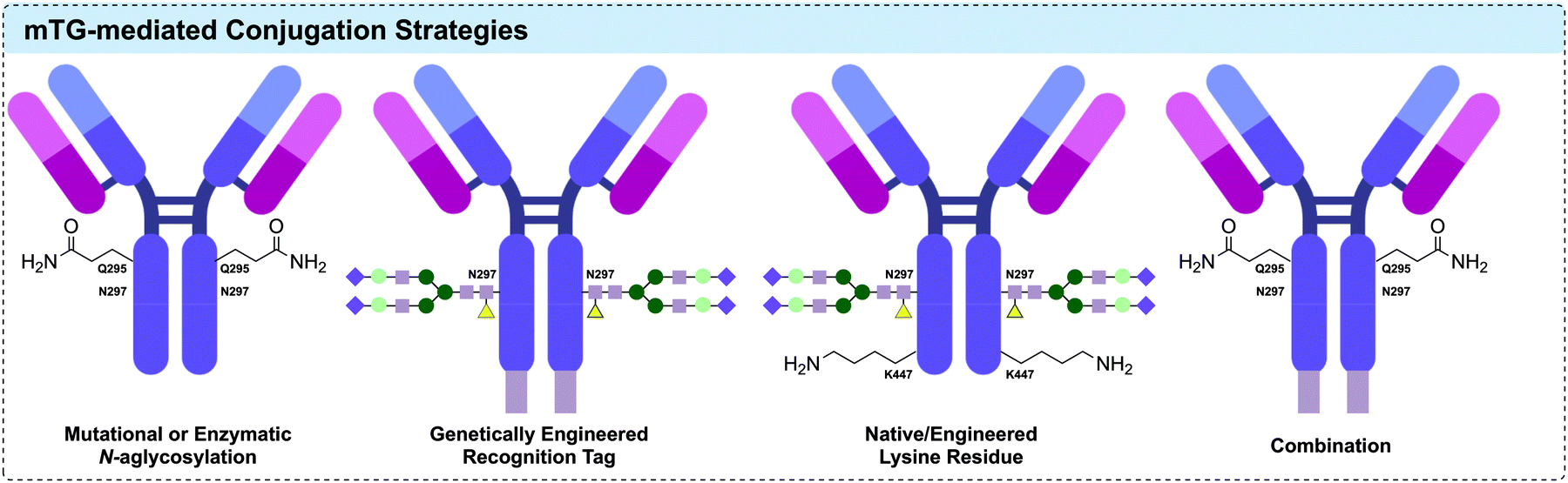 | ||
| Fig. 17 Approaches to mTG-mediated site-specific conjugation to IgG1 for the generation of ADCs (L to R): exposure of native Q295 via enzymatic or mutational removal of N-linked heavy chain glycans; genetic incorporation and expression of mTG specific recognition motif; conjugation via native/engineered lysine residue; combination of multiple approaches allowing for dual functionalisation. | ||
The first systematic investigation of TG-mediated conjugation to native antibodies was reported in 2008 by Schibli and co-workers.256 mTG (from S. mobaraensis), and recombinant His-tagged human transglutaminase 2 (TG2) were comparatively investigated for their ability to catalyse attachment of a Cy3 fluorescent probe to three types of IgG. A wide screen of conditions was undertaken, encompassing pH, enzyme/substrate concentrations, and the side chain used for attachment (lysine or glutamine). In-gel fluorescence following SDS-PAGE revealed an almost exclusive labelling of the heavy chain. Reflecting the increased number of exposed lysine residues and the known specificity of TGs towards glutamine-containing sequences, conjugation with glutamine-modified substrates was found to give a 6-fold increase in labelling compared to that of lysine-modified payloads. Modification of native lysine residues could be achieved however, with antibody-to-fluorophore ratios (FAR) ranging between 0.3 and 0.5. Crucially, the authors observed that conjugation with lysine-substrates was far superior when attempted on an aglycosylated antibody, producing a FAR of 1.0. It was suggested that the single-site mutation (N297Q), which either provides an additional glutamine coupling site or induces structural changes in the IgG to increase accessibility of other glutamine residues. More light was shed on the surprising influence of aglycosylation towards lysine-substrate conjugation in a follow-up study, in which it was demonstrated that mTG-mediated functionalisation yielded immunoconjugates with defined stoichiometry of loading at site-specific positions in the Fc region (Fig. 18B).257 With the aglycosylated anti-L1-CAM antibody chCE7 (bearing the N297Q mutation), controlled attachment of four labels per antibody was achieved. Tryptic digestion followed by MALDI-TOF MS analysis unambiguously assigned Q295 and N297Q as the sites of conjugation. Prior work had suggested that removal of the Fc glycans results in greater mobility of the C/E (Q295-T299) loop near the proposed site of modification,258–260 and so N-glycosidase F (PNGase F) was applied to enzymatically deglycosylate native chCE7 and rituximab (Fig. 18A). Specific attachment to the now-accessible Q295 residue was achieved via mTG-mediated conjugation following the deglycosylation, was again confirmed by MS. This approach was similarly adopted to functionalise aglycosylated chCE7 with a DOTA metal chelator.261
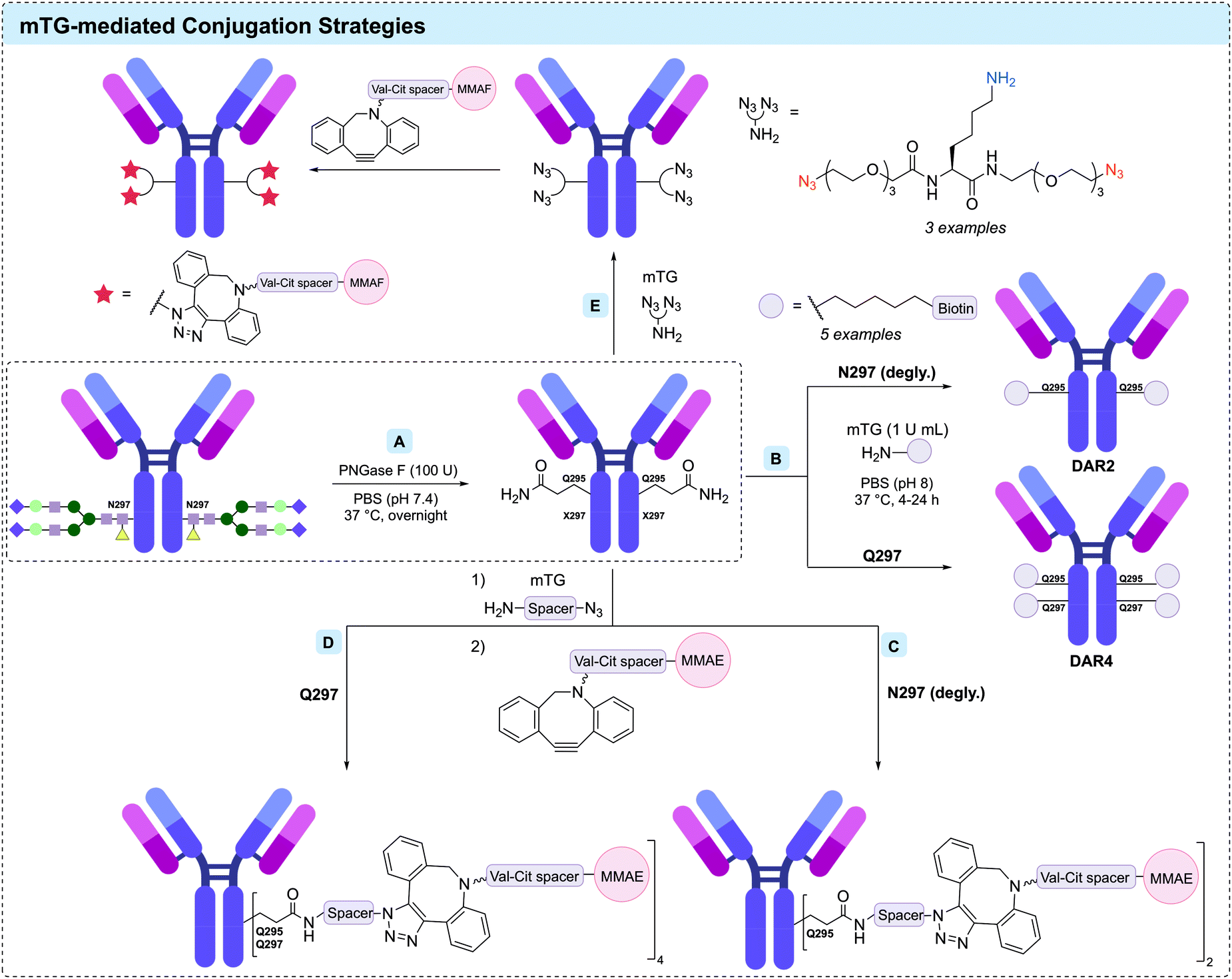 | ||
| Fig. 18 mTG-mediated conjugation strategies via heavy chain Q295: (A) deglycosylation of N297 with PNGase F, enabling accessibility of Q295; (B) conjugation of amine payloads yields final products with controlled substrate:antibody ratio of either 2 (N297) or 4 (where N297Q mutation provides additional accessible glutamines); (C and D) two-step chemo-enzymatic method for site-specific functionalisation enables assembly of DAR2 or DAR4 ADCs depending upon residue 297; and (E) branched azide-linkers (example shown) facilitate installation of two payloads per antibody attachment site via chemo-enzymatic protocol. | ||
The deglycosylation approach was augmented by Dennler et al. in 2014, with their development of a two-step chemoenzymatic protocol for the controlled assembly of ADCs comprising MMAE and MMAF payloads (Fig. 18C).262,263 The anti-HER2 antibody trastuzumab was first deglycosylated with PNGase F, followed by mTG-mediated enzymatic ligation of a small molecular weight amine substrate (40-fold molar equivalents per conjugation site) overnight in PBS at 37 °C. This substrate contained a reactive vector to facilitate post-conjugation payload functionalisation, either a sulfhydryl group for subsequent conjugation with a maleimide, or an azide allowing for SPAAC. A DAR of 2.0 was achieved via the SPAAC route, and 1.8 for the thiol-maleimide. In comparison, direct enzymatic ligation of the spacer-auristatin derivatives proved inferior for a multitude of reasons. Apart from eliciting DARs between 1.0 and 1.6, extensive optimisation of each substrate was deemed necessary, and the hydrophobicity of the payloads likely to necessitate addition of organic co-solvent, which lowers mTG activity. Furthermore, the increased reactivity and selectivity of the initially attached small substrate allowed the authors to use near-stoichiometric quantities of the toxins (2.5 equivalents), exemplifying the potential cost benefit to this approach.
Lhospice et al. utilised this chemoenzymatic approach to produce novel ADCs from an aglycosylated variant of anti-CD30 antibody cAC10, containing the N297Q mutation (Fig. 18D).47 The antibodies were assembled either through direct conjugation of the amine-Val-Cit-MMAE, or an amino-azide reagent followed by SPAAC to attach the payload. The latter, two-step approach required only 10 molar equivalents of the azide reagent, and 5 molar equivalents of the MMAE-payload—compared to the 80 molar equivalents required for direct enzymatic conjugation of the amine-Val-Cit-MMAE payload. LC-MS analyses confirmed the two-step chemoenzymatic approach to elicit products with higher homogeneity (DAR 4.0) than that of the direct ligation (DAR 3.6). The conjugates were found to exhibit in vitro EC50 levels comparable to that of Adcetris®. All four ADCs were stable in cynomolgus monkey, human, and rat plasma for at least 1 week. In vivo pharmacokinetic studies in rat were consistent with this result, with no change in DAR observed after 2 weeks. Furthermore, 125I radiolabelling revealed superior tumour uptake, and slower blood clearance, in mice compared to those of Adcetris®. Crucially, in vivo studies estimated the maximum tolerated dose of the homogeneous ADCs in Wistar rats to be greater than 3 times that of Adcetris®.
In the wake of failures in efficacy studies of ADCs comprising payloads of extremely high potency (e.g. auristatins, maytansines), there has been renewed consideration for the use of less potent cytotoxins.264 Newer linker formats that facilitate higher drug loadings of moderately active chemotherapeutic agents have therefore been developed, to maintain efficacy without the associated toxicity. In 2017, Anami et al. reported a series of branched azide linkers that were suitable as acyl-acceptor substrates for mTG-mediated conjugation.265 Each linker contained two azide groups (Fig. 18E), which was attached via mTG-mediated conjugation to an N297A mutant anti-HER2 antibody. This facilitated attachment of a Val-Cit-MMAF unit via SPAAC, yielding an average DAR of 3.9. The same protocol was undertaken with an N297Q mutant anti-HER2 antibody, producing an average DAR of 7.4. The ADCs exhibited poor stability in mouse plasma, due to the known susceptibility of the Val-Cit motif to extracellular carboxylesterase.122 The authors sought to remedy this in further investigations, where they developed variants of the Val-Cit motif, which were incorporated through their branched azide linker.266 In this study, mTG-synthesised ADCs containing Glu-Val-Cit or Asp-Val-Cit demonstrated superior stability, retained cathepsin activity and efficacy in a series of in vivo investigations.
mTG-conjugation to mutationally or enzymatically aglycosylated antibodies has been utilised in several broader investigations and applications, including dual modification.267 A one pot dual functionalisation, pairing mTG-mediated conjugation with lipoate-acid ligase A-mediated ligation with genetic incorporation of a peptide tag into either the heavy or light chain of trastuzumab has been reported.268,269 These two conjugation methods were used to install linkers containing orthogonal functional vectors (azide, and strained alkene), allowing for ‘click’-installation of FRET-paired fluorophores. A variety of constructs were assembled, with or without orthogonal cleavable spacers, allowing for removal of the payload from a desired position with either MMP2 or cathepsin B. Additionally, Puthenveetil et al. developed a methodology for solid-phase, site-specific dual conjugation of linker-payloads to antibodies.270 The procedure comprised binding of deglycosylated mutant trastuzumab containing engineered cysteine residues to protein A agarose beads, allowing for solid-phase mTG-mediated conjugation of an amino-BCN linker. The capped mutant cysteines were then exposed under reducing conditions, allowing for maleimide conjugation. Finally, an azide-bearing payload underwent a SPAAC with the BCN motif to generate the dual functional ADCs. Non-glycosylated approaches to antibody modification are not limited to toxin attachment. Other reports have detailed the attachment of azide polymers,271 photosensitiser motifs272 or virus nanoparticles.273
Recent research offers some reservations concerning the selectivity of mTG for the conserved PWEEQYNST sequence (containing Q295) in the Fc region.274 Trastuzumab was deglycosylated with PNGase F, and then a small azidoamine was attached with mTG. LC-MS and SDS-PAGE analysis suggested complete conversion with no evidence of light-chain modification. Tryptic digestion followed by tandem MS of the conjugate confirmed modification at the expected position. However, under more forcing conditions, triply modified species were also detected. The third site was unambiguously identified as Q3H (trastuzumab labelling), close to the N-terminal CDR1 epitope-binding region. The revelation is potentially problematic, highlighting the need for continued development of analytical methods for assessing homogeneity, and the possible scope for enhancing specificity through advances in areas such as engineered transglutaminases.
All of the methods described above require the absence of the Fc N-glycans in order to facilitate access to the conserved Q295 site. IgG glycosylation influences the stability and conformation of the Fc region and is critical for antibody recognition by Fc receptors.275 The consequence of glycosylation upon antibody pharmacokinetics in humans is unclear, with investigations on aglycosylated antibodies revealing variable stability, aggregation propensity, and half-life.276,277 Advances in engineered mTGs may preclude this dilemma, a recent example of which was reported by Dickgiesser et al.278 Utilising a semi-rational combinatorial approach based upon sequence alignments with related TGs, the authors generated a library of mTG mutants was generated, which facilitated efficient and site-specific conjugation to native glycosylated antibodies at Q295. Remarkably, ADCs with DAR ranging between 1.9 and 2.1 were achieved with several of the mutant mTGs despite the presence of the N-glycans. Tandem MS confirmed Q295 as the conjugation site, with only a minor by-product of DAR 3 (the attachment site of which was not identified). This emerging approach promises to simply expand the Q295 strategy, without detriment to the pharmacokinetic properties of the produced conjugates.
Farias and co-workers developed a methodology for the characterisation of mTG-mediated amino-PEG6 propionyl MMAD (AmPEG6-MMAD) payload attachment.279 The authors attached the AmPEG6-MMAD payload to engineered glutamine tags (LLQGA) installed at various positions of an anti-M1S1 antibody C16 (Fig. 19A). The conjugates were investigated with a combination of native MS, peptide mapping and in-source fragmentation analysis. A small amount of off-target conjugation was observed at Q295, which was attributed to the small amount of aglycosylated antibody typically produced when expressed in CHO or HEK293 systems. This off-target conjugation was mitigated by utilising a Q295N mutant antibody, resulting in approximately 99.8% site-specificity at the installed tag. The C-termini LLQGA approach is a commonly adopted mTG-ligation strategy and has seen use in the assembly of related biologics, such as antibody–oligonucleotide conjugates.280
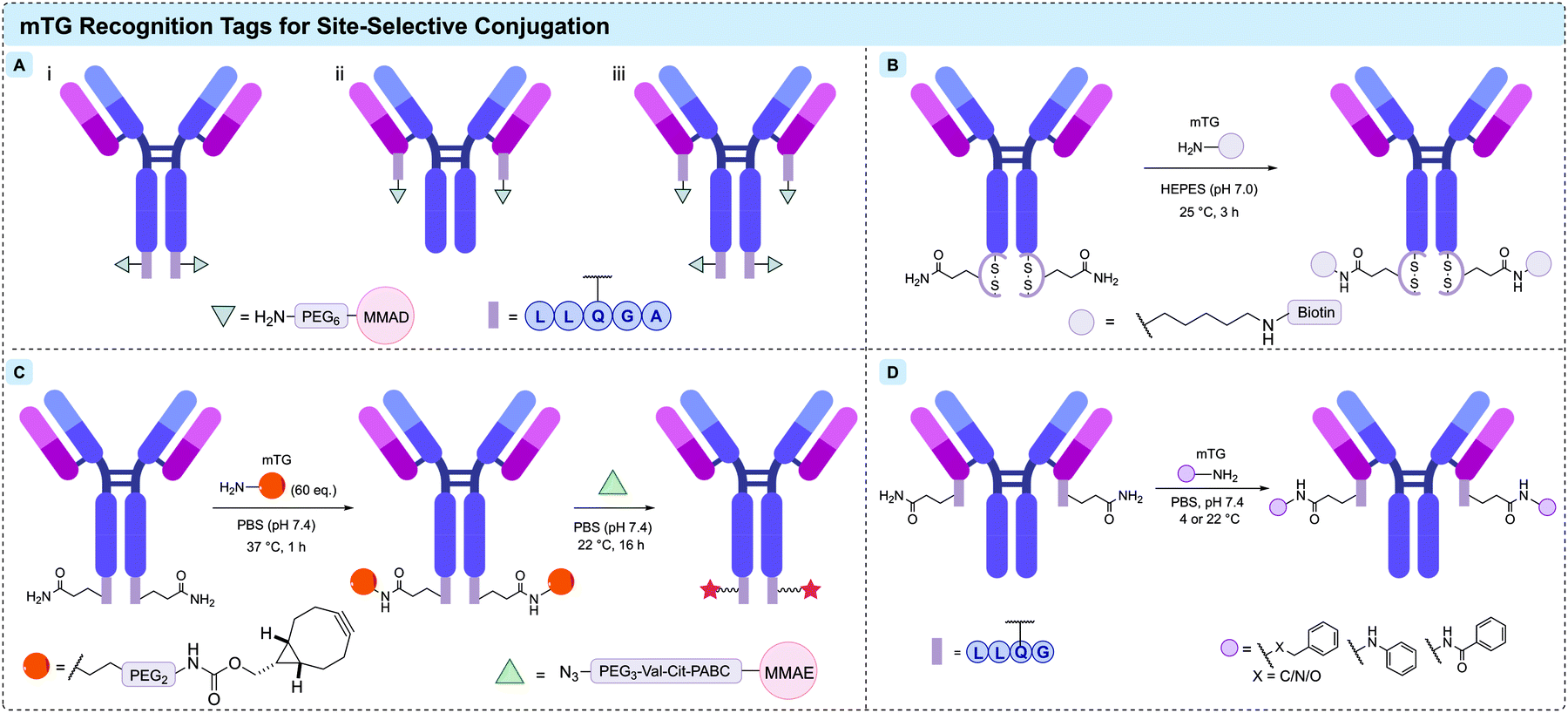 | ||
| Fig. 19 Approachs to mTG-mediated conjugation via recognition tags: (A) incorporation of AmPEG6-MMAD linker-payloads with LLQGA tag at antibody heavy chain C-termini (i), light chain C-termini (ii), and light and heavy chain C-termini (iii); (B) conformationally strained DAIP-derived sequence facilitates efficient mTG-conjugation; (C) two-step chemo-enzymatic attachment via SPIp derived recognition motif; (D) recent expansion in scope of substrates amenable to mTG-mediated conjugation. | ||
Strop et al. utilised the engineered recognition motif strategy, in combination with N297Q aglycosylation, to investigate whether the pharmacokinetic limitations of high DAR ADCs could be overcome through control over the site of conjugation.281 ADCs comprising the anti-M1S1 antibody C16 and a non-cleavable MMAD payload, were assembled with DARs of 2, 4, 6 and 8. In vitro toxicity assays against BxPC3 (high M1S1 expression) and Colo205 (moderate M1S1 expression) cell lines were conducted, comparing the response to that of similarly loaded ADCs assembled via conventional maleimide–cysteine conjugation. Expectedly, the higher DAR ADCs were the most potent. All ADCs exhibited pM toxicity against BxPC3, however those of DAR 6 and 8 maintained this level of potency against Colo205. In vivo studies revealed that the mTG-assembled DAR 8 conjugate outperformed its conventionally assembled counterpart, inducing long-term tumour growth inhibition. Schneider et al. recently reported the development of ‘dextramabs’, a novel linker platform for ADCs comprising a hydrophilic polysaccharide scaffold of desired length, allowing for the assembly of high DAR ADCs.282 The polysaccharide scaffold contains repeating azide-bearing glucose units, allowing for SPAAC functionalisation following mTG-mediated conjugation to the antibody. Functionalisation with DBCO-PEG3-Val-Cit-PABC-MMAE payloads generated ADCs of 2, 4, 8 and 11. Sub-nanomolar inhibitory activity was observed for DAR 8 and 11 ADCs (IC50 = 0.1 nM). No significant change in binding was observed, and thermal shift assays suggested no loss in stability compared to trastuzumab.
A focused study on the influence of conjugation site on the stability of Val-Cit-PABC linkers was undertaken by Dorywalska et al. in 2015.39 A range of anti-M1S1 C16 conjugates bearing aminocaproyl-Val-Cit-PABC-Aur0101 (an auristatin payload) linker-payloads at a variety of engineered Q-tags were synthesised and incubated in mouse, rat, cynomolgus monkey, and human plasmas, followed by purification and analysis via HIC and MS. The ADCs remained stable independent of conjugation site, and blood type—with the exception of mouse blood, in which stability varied widely depending upon the site of attachment. Cathepsin B activity for the Val-Cit cleavage was also found to depend upon the site of conjugation. A positive correlation between linker stability (in mouse plasma) and anti-BxPC3 IC50 values was observed in in vitro cytotoxicity studies. Further systematic characterisation was later reported.283
Wong et al. have also reported the development of epidermal growth factor receptor (EGFR) targeting ADCs.284 A low-affinity anti-EGFR ADC (RN765C) was produced by insertion of a glutamine tag (GGLLQGPP) at the C-terminus of the light-chain of the antibody, followed by mTG-mediated conjugation with the linker-payload AcLys-Val-Cit-PABC-PF-06380101. Conjugation to the antibody was undertaken with 10-fold molar excess of the linker-payload at 37 °C for 24 hours, resulting in a DAR of 1.93–2.0 following purification. This ADC exhibited sub-nanomolar in vitro EC50 across a range of cell lines—yet was less potent towards normal human keratinocytes, which also express EGFR albeit at lower levels. RN765C was highly efficacious in vivo, with sustained solid tumour regression achieved with a single dose of 1.5 mg kg−1. The same linker-payload was used by Strop et al. in designing a novel Trop-2-targeting ADC, RN927C.285 Accordingly, under similar conditions to RN765C, the AcLys-Val-Cit-PABC-PF-06380101 linker-payload was attached via mTG-mediated conjugation to an mTG-tag (LLQGA) fused to the heavy chain C-terminus. A single HIC purification step resulted in a homogeneous conjugate with a measured DAR of 2.0. Cell line and patient-derived xenograft in vivo mouse models exhibited sustained regression with a single dose of RN927C at 0.75–3 mg kg−1. A Phase I dose-escalation study was undertaken with RN927C on thirty-one patients with advanced or metastatic solid tumours (ClinicalTrials.gov Identifier: NCT02122146). Disappointingly, no partial or complete responses were observed, and the study was terminated early due to excess toxicity.286
Researchers from Pfizer utilised mTG-mediated conjugation to construct ADCs to investigate the dependency between intracellular trafficking and non-cleavable ADC-mediated cell killing.15 The investigated conjugates comprised anti-Trop-2 or anti-APLP2 (known to travel directly to lysosomes following endocytosis) IgGs. DAR 2 ADCs were constructed via mTG-mediated attachment to GGLLQGPP glutamine tags fused to the light chain C-termini, and DAR 4 ADCs via Q295/Q297 on N297Q mutant IgGs. Am-PEG6-MMAD was used as the linker-payload, conjugated under standard conditions. In growth inhibition studies against SKOV3 cells, APLP2-ADC (0.11 nM) exhibited a significantly lower EC50 compared to Trop-2-ADC (5.95 nM), despite SKOV3 Trop-2 expression being approximately double that of APLP2. The superior efficacy of APLP2-ADC over Trop-2-ADC was maintained in in vivo xenograft models.
The demonstrated stability and homogeneity of mTG-mediated conjugation techniques have prompted their application in ADC designs where safety is especially critical. In 2019, Ratnayake et al. (Pfizer) developed a series of HER2-targeting ADCs bearing depsipeptides, an extremely potent class of cytotoxins, at different attachment sites.287 The lead conjugate PF-06888667, comprising the depsipeptide SW-163D conjugated via a cleavable AcLys-Val-Cit-PABC-DMAE (dimethylethylenediamine) linker, exhibited subnanomolar in vitro cytotoxicity against N87 and MDA-MB-361-DYT2 cell lines. Complete tumour regression was observed in an N87 xenograft mouse model at a dosage ten-fold lower than that of the approved T-DM1. The study highlights the importance of rational design in payload site and attachment method.
There is a growing body of work on the optimisation of the sequence and location of recognition tags to improve conjugation efficiency and specificity. In 2015 Siegmund et al. reported the development of a rationally designed glutamine tag (GECTYFQAYGCTE), informed from a sequence analysis and crystal structure of dispase autolysis inducing protein (DAIP), a natural substrate for bacterial transglutaminase.288 Flanking cysteine residues were included to mimic the loop in DAIP by rigidifying the β-turn following disulfide formation (Fig. 19B). Kinetic studies of the mTG-mediated biotinylation of this oligopeptide resulted in significantly faster conjugation in comparison to an analogous sequence containing two asparagine residues instead of the two cysteine residues. Both tags were incorporated to the C-termini of the heavy chains of anti-EGFR antibody cetuximab. Both sequences were found by Western blot and MALDI TOF-MS analysis to mediate site-specific conjugation. Microscopy and flow cytometry demonstrated that the conjugation did not impair EGFR binding.
A novel recognition motif derived from a native substrate from the host of the S. mobaraensis bacterial transglutaminase, Streptomyces papain inhibitor (SPIp), was recently reported (Fig. 19C).289 An analysis of the substrate preference of mTG within a series of synthetic peptides derived from sequences within the papain inhibitor, as well as DAIP, was conducted. Anti-HER2 ADCs were assembled via recognition tags fused to the heavy chain C-termini, attachment with amino-PEG2-BCN linkers, followed by SPAAC with the azido-PEG3-Val-Cit-PABC-MMAE payload. For the ADC assembled from the lead sequence, a DAR of 1.81 was determined by HIC, and an IC50 of 151 pM against HER2+ SK-BR-3 cells. In contrast, a comparable ADC assembled via a simpler LLQG tag yielded a DAR of 0.48 and exhibited an IC50 of 1.9 nM. Yamazoe et al. have recently reported a high-throughput protocol for assisting the determination of optimal locations for drug conjugation.290 mTG-conjugation was utilised to generate a library of ADCs from a pool of mutated antibodies, which were analysed and validated via MS. The screening allowed for several new tags and sites to be identified and may prove to be an important tool for future development and optimisation of homogeneous ADCs.
3.2 Enzymatic C-/N-terminal modification
Several enzymatic protocols have been developed over the past decade to facilitate selective modification of the N- or C-termini of the antibody chains, often by genetic fusion of an enzyme-specific recognition tag. One such protocol involves the use of formylglycine-generating enzyme (FGE), which oxidises cysteine residues to a formylglycine unit in a CXPXR consensus sequence (X = any amino acid except proline).304 Aldehyde modified antibodies generated using this SMARTag® technology have been conjugated with hydrazine- or hydroxylamine-functionalised payloads (Fig. 20A). The hydrazine-iso-Pictet–Spengler (HIPS) ligation achieves rapid bioconjugation under mild conditions, forming hydrolytically stable protein conjugates.305 An in-depth study on the HIPS ligation revealed that C-terminus tagging resulted in improved pharmacokinetics and in vivo efficacy compared to alternative conjugation positions.306 A CD22-targeting ADC, TRPH-222 (CAT-02-106), generated using the SMARTag® HIPS bioconjugation technology, has recently entered Phase I clinical trials for the treatment of lymphoma with an expected completion date of 2022 (NCT03682796). TRPH-222 comprises a CD22-targeting antibody, which contains a formylglycine residue on each heavy chain, conjugated to a maytansinoid payload via a non-cleavable linker. TRPH-222 has a DAR of 2, and exhibited promising efficacy and safety properties during pre-clinical evaluation.307,308 Interim results from the Phase I clinical trial have recently indicated that TRPH-222 was well-tolerated in 19 non-Hodgkin's lymphoma patients with manageable side effects.309 Additionally, this ADC has shown early signs of efficacy at doses of 0.6 to 5.6 mg kg−1 administered every three weeks, including five complete responses. Additionally, an anti-HER2 DAR 2 ADC generated via FGE-mediated aldehyde insertion at the C-terminus fusion CXPXR tag followed by HIPS conjugation of a DM1 payload, has shown promising preclinical activity and tolerability compared to the approved T-DM1.310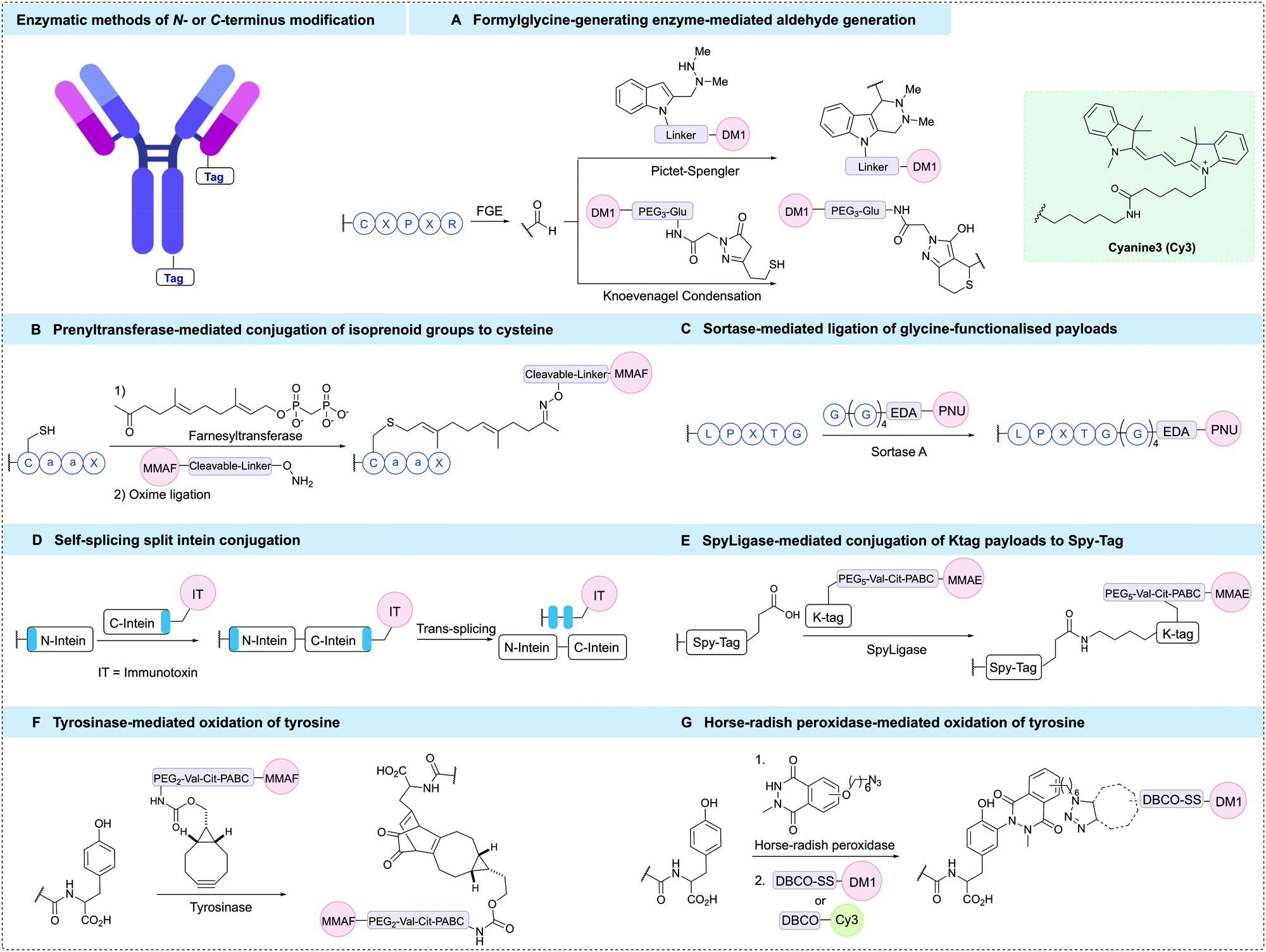 | ||
| Fig. 20 Enzymatic bioconjugations methods for C-terminal modification; (A) formylglycine-generating enzyme (FGE) oxidises a specific Cys residue to an aldehyde suitable for Pictet–Spengler or Knoevenagel conjugation; (B) prenyltransferase-mediated conjugation of functionalised isoprenoid groups to cysteine in CaaX and subsequent oxime ligation reaction; (C) sortase-mediated antibody conjugation (SMAC) technology; (D) self-splicing split inteins form a new amide bond between extein units (blue) whilst excising to form intein protein; (E) SpyLigase-mediated conjugation of a SpyTag and KTag sequence; (F) tyrosinase-mediated strategy for the oxidation of tyrosine and subsequent strain-promoted cycloaddition; (G) Horse-radish peroxidase tyrosine oxidation strategy. | ||
A recently reported alternative strategy for formylglycine conjugation developed novel N-pyrrolyl alanine Pictet–Spengler reagents that exchanged the indole heterocycle for a pyrrole ring.311 This approach enabled rapid antibody bioconjugation with an easily synthesised reagent. Additionally, formylglycine-tagged antibodies generated by FGE have also been exploited using pyrazolone reagents, whereby ligation occurs via tandem Knoevenagel condensation-trapping reactions (Fig. 20A).312,313
Further enzymatic conjugation technologies include protein prenyltransferases, a family of enzymes that can append isoprenoid groups to a cysteine residue within a CaaX recognition tag (where a is an aliphatic amino acid, and X is an amino acid that determines specificity for a particular isoprenoid transferase). These enzymes are typically responsible for post-translational modifications and have been successfully applied to the single and dual modification of proteins, achieving high conversions and selectivity.314–316 Additionally, the technology has been used for the generation of homogeneous ADCs (Fig. 20B). Initial work involved the modification of an antibody C-terminus CaaX tag with an isoprenyl derivative containing a reactive group (e.g. ketone, azide) that was subsequently modified using bioorthogonal chemistry, including oxime formation or azide–alkyne cycloadditions.317 This technology is known as ConjuALL™ and was developed by LegoChem Biosciences. Recently, the prenylation conjugation platform has been utilised to generate a HER2 targeting ADC, LCB14-0110. This ADC was synthesised through prenyltransferase-mediated conjugation of a ketone-functionalised isoprenoid to trastuzumab with a C-terminus appended CaaX motif, which was then subjected to an oxime ligation with a β-glucuronidase-cleavable MMAF payload.318 The ADC exhibited high stability during preclinical evaluation with a promising PK profile, and is currently in Phase I clinical trials in China.
Sortase-mediated antibody conjugation (SMAC) technology™ is an additional enzymatic ligation approach developed by NBE-therapeutics. SMAC-technology™ uses S. aureus sortase A, which is a transpeptidase that cleaves the amide bond between threonine and glycine residues in the LPXTG (X = any amino acid) pentapeptide motif, and subsequently catalyses the attachment of glycine-functionalised payloads to the newly generated C-terminus (Fig. 20C). The sortase recognition motif and a Strep II tag, which was used to aid removal of unreacted antibody, were fused to the light and heavy chain C-terminus of different antibodies, including brentuximab and trastuzumab. Sortase-mediated conjugation was then used to attach a series of penta-glycine tagged payloads containing maytansine or MMAE to generate homogeneous ADCs analogous to Kadcyla® and Adcetris®.319 The sortase conjugated analogues were found to exhibit similar in vitro potency to the clinically approved ADCs in cell viability assays. Additionally, conjugation of the payload to the antibody was shown to have no adverse effect on the antigen binding. Reaction efficiencies exceeding 80% were typically achieved with various payloads and antibodies, indicating the generality of this approach. Next, the in vivo potency of the trastuzumab derived ADC was studied in a HER2-positive SKOV3 cell line xenograft mouse model. Once more, the sortase conjugated ADC exhibited comparable potency to Kadcyla®. In another example, sortase-mediated conjugation was used to generate an anthracycline based ADC targeting HER2. This ADC demonstrated high serum stability and efficacy in a HER2-positive in vivo model.320 Typically, the conjugation efficiency was ca. 85%, corresponding to a DAR of ∼3.4, which could be increased to 4.0 after StrepTactin affinity chromatography purification. However, the conjugation efficiency was highly dependent on the linker-payload composition. When the extremely toxic nemorubicin metabolite PNU-159682 was used as the payload, over 95% conjugation efficiency to the anti-HER2 antibody was achieved without enrichment (DAR ca. 4.0). The non-cleavable ADCs generated were shown to be highly potent, effective on cells that expressed only moderate target antigen levels and had high in vitro and in vivo serum stability. Interestingly, incorporation of a cleavable linker component had little effect on the cytotoxicity of these ADCs, implying that the mechanism of action of the payload was unaffected by the linker in the non-cleavable variants. Additional mechanistic in vivo studies on the PNU-based ADC found that some breast cancer resistance mechanisms could be overcome using these novel ADCs.321 With promising preclinical data, NBE-therapeutics expects the first SMAC-derived ADC, NBE-002, to reach Phase I clinical trials in mid to late 2020.
Sortase-mediated conjugation has also been applied to dual-labelling strategies in combination with butelase 1, an enzyme that ligates a specific NHV amino acid motif to nucleophilic payloads. These two enzymes have recently been harnessed in an orthogonal fashion for the one-pot site selective generation of dual-labelled ADCs.322 Additional methods that combine sortase A with the π-clamp conjugation technology have also been developed to achieve an efficient antibody dual labelling technology.323
Self-splicing split inteins can be used for the generation of homogeneous antibody immunotoxin conjugates (Fig. 20D). Inteins are proteins flanked by exteins that self-excise to assemble proteins after mixing the N- and C-terminal intein components. A new amide bond is generated between the extein units, which can be used for conjugation of a payload to an antibody. Pirzer et al. applied the split inteins strategy to antibody modification by appending a Gly-Ser linker connected to the 11 amino acid intein sequence to the heavy chain C-termini of trastuzumab and an EGFR-targeting antibody.324 The immunotoxins used in this study were a truncated variant of Pseudomonas Exotoxin A, and gelonin. These immunotoxins were appended with a 143 amino acid C-terminal intein sequence connected to a maltose binding protein, which was designed to improve solubility and expression reproducibility. The tagged antibody and toxin were then combined under reducing conditions and protein trans-splicing occurred with an overall coupling efficiency of 50–70%. Unconjugated antibody was removed through immobilised metal affinity chromatography, yielding highly toxic final constructs with a toxin-antibody ratio of ca. 1.3. Importantly, immunotoxin conjugation to the antibody did not hinder antigen binding for either antibody. Next, in vitro cell viability assays demonstrated high levels of both selectivity and cytotoxicity for the ADCs. For the Exotoxin A derived ADCs, IC50 values ranged between 0.8–3.9 pM, approximately 10-fold more potent compared to the gelonin ADCs (IC50 values of 12–68 pM). The difference in activity was proposed to be a result of more efficient transport of the Exotoxin A payload from endosomes to the cytosol, observed by confocal microscopy. This split intein technology is a valuable addition to the conjugation toolbox, and future optimisation of linker lengths and composition has been proposed to improve the conjugation efficiencies.
SpyLigase has also been used to generate homogeneous ADCs (Fig. 20E).325 This enzyme catalyses formation of an isopeptide bond between SpyTag and KTag recognition peptides. In a recent report, an MMAE payload was connected to the KTag 10 amino acid sequence, and the SpyTag 13 amino acid sequence was fused to the C-terminus of the anti-EGFR antibody cetuximab. In the presence of SpyLigase, combination of the antibody and the KTag payload generated homogeneous ADCs under mild conditions with ca. 80% conjugation efficiency and a DAR of ca. 1.7. Following purification by protein A chromatography, the in vitro cytotoxicity of the resulting ADC was analysed. High potency was observed against EGFR-positive breast cancer cell lines for both non-cleavable and cleavable ADCs, with IC50 values of 0.2 and 0.1 nM respectively.
Tyrosine-specific bioconjugation has recently shown great promise for controlled protein conjugation.326 Factors including its relatively low abundance, hydrophobicity, and π–π stacking generally limits the surface exposure of tyrosine residues, which leads to low accessibility for protein conjugation. Tyrosinase is an enzyme that efficiently oxidises exposed tyrosine residues to 1,2-quinones, which can react with various functionalities to conjugate payloads (Fig. 20F). One example utilised a BCN-tagged MMAF linker-drug for strain promoted, oxidation-controlled quinone–alkyne cycloadditions (SPOCQ).326 A tyrosine residue connected to a tetra-glycine linker (G4Y) was genetically fused to the light chain C-terminus of trastuzumab and an anti-influenza AT1002 antibody. The antibodies were then oxidised with tyrosinase and treated with the BCN-tagged cleavable MMAF payload to rapidly form ADCs with reasonable conjugation efficiency under mild conditions. Overall, this approach was somewhat limited by the non-specific reaction of nucleophilic amino acid residues proximal to the 1,2-quinone, reducing the conjugation efficiency.
3.3 Other enzymatic methods
In another approach to tyrosine modification, Sato et al. have reported selective modification of surface-exposed trastuzumab tyrosines (Fig. 20G).327 In this protocol, horseradish peroxidase (HRP) catalysed single electron transfer modification of tyrosine phenol with N-methylated luminol derivatives containing a pendant azide. Modification was found to occur on tyrosines in the complementarity determining region (HC-Y53). The installed azide was then treated with DBCO-functionalised payloads, including Cy3 or Cy5 fluorophores or toxin DM1, which generated a DAR 2 ADC. The resulting conjugates were used for in vivo imaging or therapeutic evaluation.Similar to histidine, serine modification often proves difficult due to competition from other more nucleophilic residues. However, Grünewald et al. have reported an efficient enzymatic strategy using phosphopantetheinyl transferases (PPTases), which recognise serine residues in specific amino acid sequences and can transfer payloads from coenzyme A (CoA)-modified substrates.328 PPTase recognition sequences (ca. 11 amino acids) were inserted into the CH3 domain of trastuzumab. Treatment of the serine-tagged antibody with an MMAF-coenzyme A reagent generated a homogeneous DAR 2 ADC, which exhibited cytotoxicity in vitro against HER2-positive cell lines and significant tumour regression in vivo in a HER2-positive xenograft model. Further developments by Grünewald et al. applied a two-step strategy, in which CoA analogues containing bioorthogonal functional groups were first introduced using a shorter 6 amino acid PPTase recognition sequence, prior to chemical payload conjugation.329
4 Glycan modification
Within the CH2 domain of all IgG antibodies, there is a conserved site for glycosylation at HC-N297. The complex biantennary N-glycan exists as a mixture of glycoforms, for example, serum IgG purified from a healthy individual may contain up to 33 different major and minor glycoforms.330,331 Each glycoform contains a core heptasaccharide alongside additional monosaccharides. The majority of therapeutic mAbs produced in CHO or HEK293 cell lines contain a mixture of G0F, G1F and G2F glycoforms where G = galactose, F = fucose, and numbers (0–2) indicate the number of terminal galactose moieties (Fig. 21).332 Conjugation of ADC payloads to the N-glycan is attractive as: (1) it is spatially distant from the complementarity determining regions (CDRs) through which antibody-antigen recognition occurs; (2) the glycosylation pattern is well conserved across antibody types, which simplifies widespread modification of antibodies; and 3) the carbohydrate is chemically distinct from the polypeptide backbone of the antibody, enabling site-specific conjugation. | ||
| Fig. 21 Glycosylation pattern of IgG1 antibodies (top) and oxidation strategies for antibody glycan modification (bottom). (A) Oxidation of sialic acid residues followed by reaction with aminooxy, ThioPz or ABAO payloads. (B) Oxidation of fucose residues followed by hydrazone formation. | ||
4.1 Antibody glycoengineering via oxidation
Since the 1960s, glycans of glycoproteins have been utilised as a site for conjugation for a variety of labelling applications. Early methods relied on the oxidation of vicinal cis diols present at the glycan terminus.333 In 1984, O’Shannessy et al. described a new method for antibody biotinylation via sodium periodate (NaIO4)-mediated oxidation of terminal monosaccharide cis diols to aldehydes.334 Reaction with biotin-hydrazide then produced a biotin labelled antibody containing a hydrazone linkage. In a similar approach, Chua et al. reported the use of both periodate and galactose oxidase to produce reactive aldehydes on terminal galactose residues, which similarly utilised hydrazide-functionalised payloads to afford antibody modification.335This strategy was first applied to ADC production in 1989 by Laguzza et al. who modified a series of antibodies via reaction of hydrazide-modified vinca alkaloid cytotoxins with periodate-generated aldehydes on the antibody glycan. Using this method, ADCs were obtained in high yield with DARs ranging from 4–6.336 Similarly, in 1993, Hinman et al. prepared hydrazide-functionalised calicheamicin derivatives, which were conjugated to the oxidised glycans of an anti-MUC1 antibody. The produced ADCs retained target antigen binding and were selectively cytotoxic to antigen positive cells in vitro and efficacious in an in vivo mouse model.337 Finally, in 1999, Stan et al. produced ADCs by a procedure involving antibody desialylation (using neuraminidase) followed by galactose oxidation (using galactose oxidase).338 Conjugation of doxorubicin was subsequently achieved via reaction of its daunosamine motif with the oxidised galactose units to produce ADCs with a DAR of 3.7. This ADC was almost four times more efficacious than an analogous lysine-conjugated ADC with an average DAR of 7.8 in an in vivo xenograft mouse model. It was hypothesised that the superior homogeneity of the site-specific galactose conjugated ADC generated an enhanced pharmacokinetic profile, thus explaining these observations.
These approaches effectively demonstrated that the N-glycan offers an alternative modification site to amino acids. However, early methods did not account for the innate heterogeneity of antibody glycans, which exist as a mixture of glycoforms. Therefore, the direct oxidation of antibodies with varying levels of oxidisable glycan fucose, galactose and sialic acid residues still resulted in a heterogeneous mixture of conjugates. To overcome this, Neri and co-workers developed a cell line that enabled the production of an anti-fibronectin F8 antibody consisting solely of the G0F glycoform.339 Periodate oxidation of the antibody fucose residues produced in this cell line then provided excellent conversion to the desired product (>95% conversion) with a single aldehyde on each heavy chain (Fig. 21B). Treatment of the aldehyde-containing antibody with a hydrazine-modified auristatin cytotoxin, produced a homogeneous ADC with a DAR of 2.
A drawback of this fucose oxidation method is the high concentrations of NaIO4 (10 mM) required. Such high concentrations are known to cause unwanted oxidation of other amino acid side chains of the antibody. Particularly, oxidation of the methionine residues (HC-M252 and HC-M428) in the neonatal Fc receptor (FcRN) binding site can decrease FcRn binding and thus reduces the serum half-life of the antibody and the overall efficacy of the therapeutic.340,341 To avoid such high oxidant concentrations, Zhou et al. have developed a method to introduce periodate-sensitive sialic acid residues into the antibody N-glycan.342 The N-glycan was first enzymatically remodelled in vitro using β-1,4-galactosyltransferase (β-1,4-Gal-T1) and α-2,6-sialyltransferase (Sia T) to transfer galactose and sialic residues onto the native glycans (Fig. 21A). This produced near homogeneous monosialylated glycans. The two installed terminal sialic acids could then be oxidised with just 1 mM NaIO4 and conjugated to aminooxy functionalised drugs to produce homogeneous ADCs with oxime linkages. This strategy was shown to be effective on three antibodies; in one such example, an anti-HER2 antibody was modified with two different cytotoxic payloads, producing ADCs with DARs of 1.6 and 3.9. These ADCs demonstrated good in vitro activity and specificity toward HER2 positive cells. Despite the significant decrease in NaIO4 required, ∼30% of HC-M252 and ∼10% of HC-M428 residues were oxidised during this reaction, resulting in a 10% reduction in FcRn binding compared to the trastuzumab control. However, this small reduction of FcRn binding only had a marginal effect on serum half-life in vivo. In addition to the use of 10-fold lower periodate concentrations, this method also produces stable oxime linkages, superior to hydrazone linkages that have displayed liability in circulation and associated off-target payload release.336
A drawback of the oxidative sialate method is the low drug loading capability compared to other amino acid conjugation methods: only one sialic acid is introduced per heavy chain, despite the presence of two galactose acceptors, and the oxime ligation reaction at these sites has garnered capricious conjugation efficiencies, with variable DAR ADCs produced. To fully exploit the utility of this method, optimisation of the conjugation reaction to improve the homogeneity of the synthesised ADCs is desirable. To this end, Huang et al. have recently reported the development of 2-aminobenzamidoxime (ABAO) and mercaptoethylpyrazolone (ThioPz) reagents to expand the structural diversity of linkers used in glycosite-specific ADCs (Fig. 21).343 Both reagents were shown to react efficiently, rapidly and selectively with N-glycan aldehydes produced by chemoenzymatic glycan remodelling, as previously described.342 The resulting ADCs displayed high levels of homogeneity and selective in vitro cytotoxicity. Importantly, the ADCs were also highly stable under physiological conditions, in contrast to hydrazone linked ADCs often generated by glycan conjugation. Furthermore, a ThioPz-derived antibody conjugate displayed a plasma half-life that was superior to that of an analogous oxime conjugate. Therefore, these reagents may enable further optimisation of the overall pharmacological properties of glycosite-specific ADCs and enable the wider application of glycan conjugation in the future.
4.2 Endoglycosidase for glycan remodelling
Endoglycosidases are enzymes that catalyse the hydrolytic cleavage of polysaccharide chains between non-terminal sugar residues.344 Their complementary glycosynthases, typically generated from site-specific mutations at the active sites, can catalyse transglycosylation, whereby upon hydrolysis, a sugar moiety is attached to the new terminal residue. Several endoglycosidases have been discovered that exhibit specific activity to trim and functionalise the core structure of the glycan chains on antibodies. For example, both Endo-A from Arthrobactor protophormiae and Endo-H from Streptomyces plicatus are specific for high-mannose type N-glycans and Endo-D from Streptococcus pneumoniae targets the chitobiose core of N-glycans.345–348 However, the use of wild-type endoglycosidase for the modification of antibody glycans is limited as only native N-glycan donors are tolerated and both the products and substrates can be hydrolysed via a substrate-assisted mechanism resulting in low transglycosylation yields (5–20%) (Fig. 22).332,348 To overcome these limitations, several studies have explored the use of sugar oxazolines, (intermediate mimicking substrates), as the activated donor substrates for the modification of glycoproteins.348 This strategy generally involves two steps; first, N-glycan chains are trimmed by a native endoglycosidase, which is subsequently followed by transglycosylation with an engineered endoglycosidase to attach a new, non-native sugar moiety.332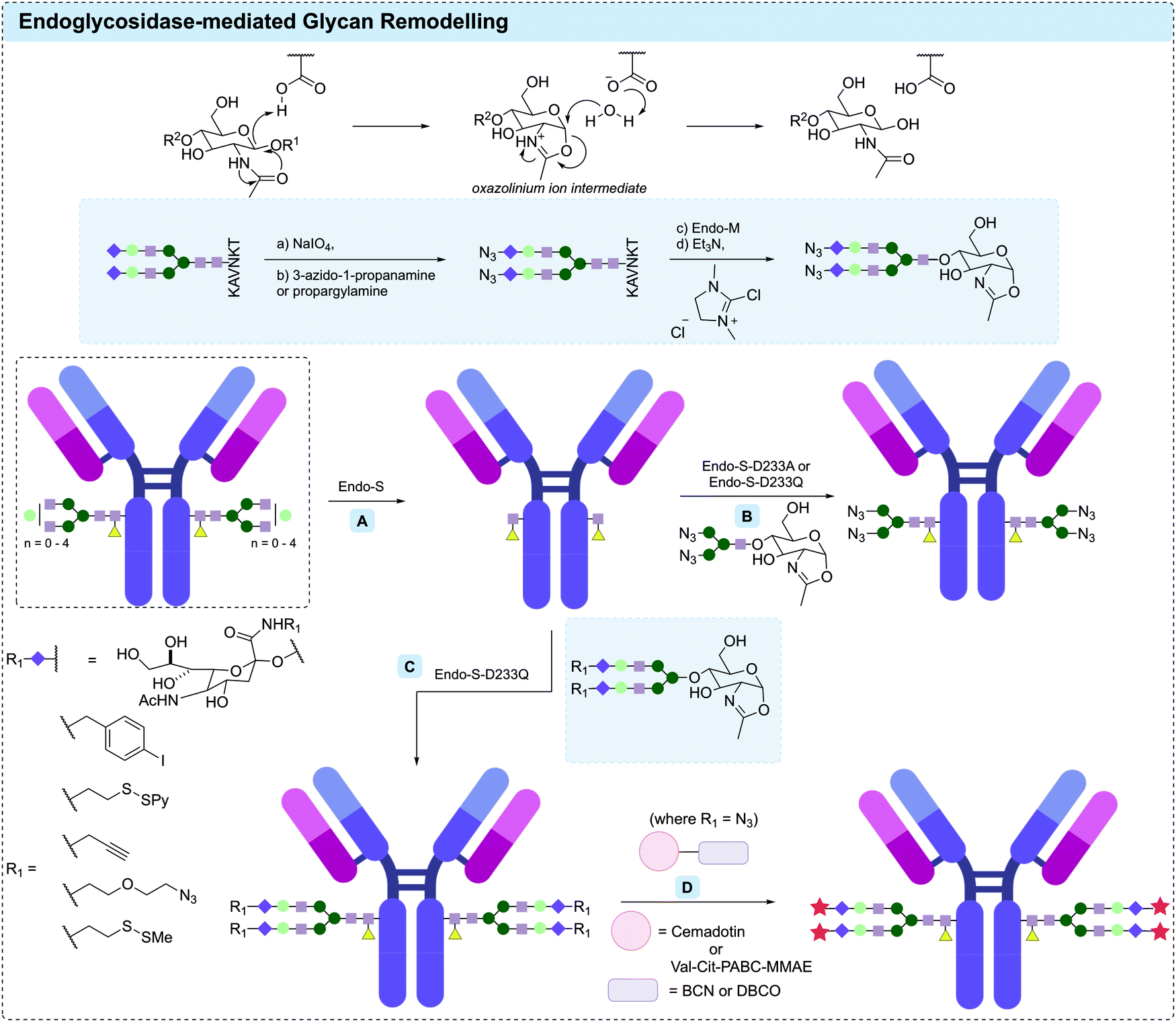 | ||
| Fig. 22 Wild-type endoglycosidase can catalyse the hydrolysis of antibody glycans via a substrate-assisted mechanism. Endoglycosidases and their glycosynthases-mediated glycan remodelling strategy: (A) deglycosylating antibody with Endo-S to unveil the innermost GlcNAc; (B) transglycosylation of core-GlcNAc of intact antibody with N3-Man3-GlcNAc under the catalysis of either Endo-S-D233A or Endo-S-D233Q; (C) Endo-S-D233Q-catalysed transglycosylation of core-GlcNAc of intact antibody with biantennary sugar oxazolines bearing reactive handles; (D) subsequent SPAAC reaction with cytotoxic payloads to generate ADCs with controlled DAR ratio. | ||
In 2012, Goodfellow et al. reported that Endo-S, a bacterial endoglycosidase from Streptococcus pyogenes, performs efficient remodelling of complex biantennary antibody N-glycan chains.349 Later that year, Wang and co-workers identified D233 in Endo-S as a key residue in promoting the formation of oxazolinium ion intermediate, which thus contributes to the undesired hydrolysis of polysaccharide substrates.350 The mutation of D233 to alanine or glutamine prevents the catalytic hydrolysis of polysaccharides while maintaining the glycosylation efficiency with sugar oxazoline donors. Thus, two resulting glycosynthase mutants, Endo-S-D233A and Endo-S-D233Q, were generated. Indeed, both enzymes showed remarkable efficiency in transglycosylating the core-GlcNAcs of intact antibodies from glycan oxazolines with high yield (Fig. 22). To demonstrate the utility of this strategy, rituximab was deglycosylated with wild-type Endo-S and transglycosylated by reaction with azide-oxazoline N3-Man3-GlcNAc under Endo-S-D223Q catalysis to generate a homogeneous antibody with four bioorthogonally reactive azide handles.
The homogeneous glycoforms generated from evolved Endo-S catalysed glycan-trimming or transglycosylation has been widely used to study the effect of glycan modification on the binding affinity of antibody towards Fcγ receptors, probing other biological activity, and for the generation of ADCs.351–354 For example, Davis and co-workers chemically functionalised the terminal sialic acid residues of biantennary sugar oxazolines with various reactive handles, including alkyne, azide, disulfide and phenyliodide moieties, and incorporated these oxazoline derivatives onto trastuzumab using the Endo-S-D233Q mutant (Fig. 22).353 The remodelled trastuzumab bearing azide handles were conjugated with cemadotin via a SPAAC reaction to generate heterogeneous ADCs with DARs of 2, 3 and 4 (Fig. 22). Compared to unfunctionalised trastuzumab, the DAR 3 ADC displayed enhanced in vitro cytotoxicity against HER2-positive SK-BR-3 cells with an EC50 of ∼800 pM.
Several studies have used directed evolution to optimise endoglycosidase transglycosidation of antibodies.355–361 Huang and co-workers have recently described the use of two of these enzymes (Endo-M and Endo-S-D233Q) to facilitate the efficient attachment of azide-containing sialic acid residues (Fig. 23).362 Subsequent CuAAC was used to attach cleavable and non-cleavable MMAE payloads, generating homogeneous ADCs with a DAR of 3.8. In vitro evaluation of these ADCs revealed that the ADC bearing a cleavable Val-Cit motif more efficiently inhibited the growth of HER2-positive SK-BR-3 cells with an EC50 of 0.09 μg mL−1, compared to the commercial trastuzumab (EC50 of 0.4 μg mL−1).
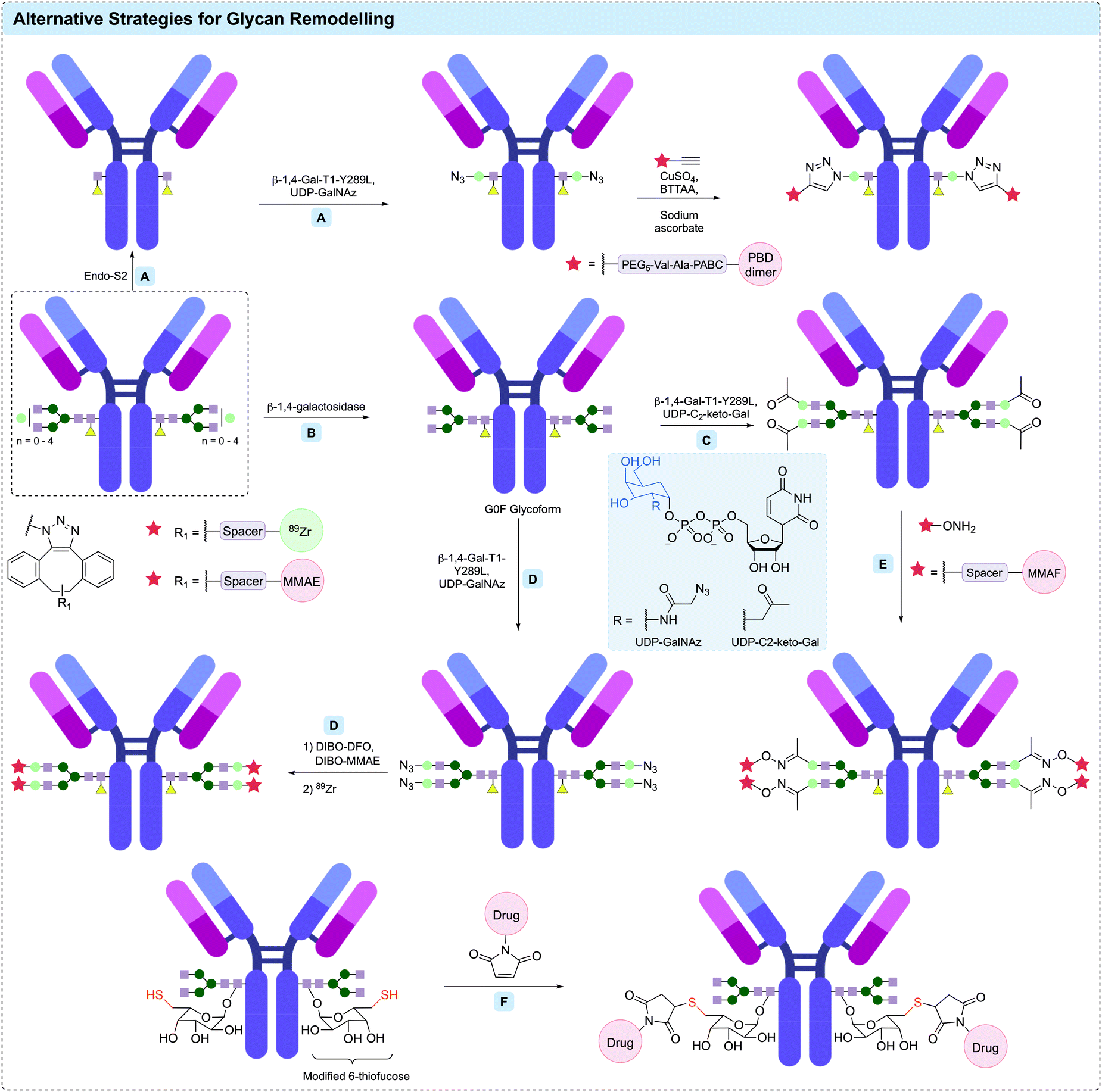 | ||
| Fig. 23 β-1,4-Galactosyltransferases and fucosyltransferase for glycan remodelling: (A) β-1,4-Gal-T1-Y289L-catalysed transglycosylation of core-GlcNAc of intact antibody followed by CuAAC reaction to PBD dimer led to the generation of a prostate-cancer-cell-targeting ADC; (B) β-1,4-galactosidase-catalysed trimming of heterogeneous antibody to give G0F glycoform; (C) incorporation of C2-keto-Gal on G0F glycoform under the catalysis of β-1,4-Gal-T1-Y289L; (D) incorporation of GalNAz on G0F glycoform under the catalysis of β-1,4-Gal-T1-Y289L followed by CuAAC reaction to generate a 89Zr-MMAE dual labelled ADC; (E) oxime ligation with MMAF payload to generate an ADC with a DAR of 4; (F) Michael addition of maleimide–drug molecule to modified 6-thiofucose residues of antibody glycans to generate an ADC with improved homogeneity. | ||
Overall, the development of glycan trimming techniques using endoglycosidases and their corresponding glycosynthases has proved to be an efficient method for providing access to homogeneous antibody glycoforms, which can then be functionalised using other glycan modification techniques (i.e. oxidation to terminal aldehydes) to allow bioconjugation of payloads and ultimately facilitate synthesis of homogeneous ADCs.
4.3 β-1,4-Galactosyltransferases
Another key glycan remodelling method that has emerged in the past two decades is the use of evolved β-1,4-galactosyltransferase (β-1,4-Gal-T1) mutants, which have the ability to incorporate modified N-acetylgalactosamine (GalNAc) moieties to terminal N-acetylglucosamine (GlcNAc) residues on the antibody glycan. β-1,4-Gal-T1 is a type of glycosyltransferase that transfers galactose (Gal) from its uridine diphosphate (UDP)-precursor to GlcNAc residues at the non-reducing end of glycoprotein carbohydrates.363,364 A single point mutation (β-1,4-Gal-T1-Y289L) was shown to significantly increase the efficiency of GalNAc transfer whilst retaining high GlcNAc transfer activity (Fig. 23).363In 2009, Qasba and co-workers described the first site-specific antibody conjugation via β-1,4-Gal-T1-Y289L catalysis using C2-keto-Gal and N-azidoacetylgalactosamine (GalNAz) as donor substrates.365 First, four therapeutic antibodies were trimmed by β-1,4-galactosidase to remove any terminal galactose residues. The resulting homogeneous G0 glycoforms were then galactosylated with either C2-keto-Gal and GalNAz in excellent efficiency via β-1,4-Gal-T1-Y289L catalysis, followed by functionalisation of the azide or ketone handles. For example, a fluorophore-antibody conjugate was generated by oxime ligation of C2-keto galactosylated trastuzumab with AlexaFluor 488 C5-aminooxyacetamide. Pleasingly, both ELISA and fluorescence activated cell sorting (FACS) analysis indicated that the antigen-binding function of the antibodies remained unaltered after glycan remodelling and functionalisation.
The first ADC generated using the β-1,4-Gal-T1-Y289L remodelling strategy was reported by Zhu et al. in 2014.366 The G2F glycoform of the HER2 targeting antibody, m860 was modified using β-1,4-Gal-T1-Y289L to incorporate terminal C2-keto-Gal residues. Conjugation of an aminooxy MMAF derivative via oxime ligation generated an anti-HER2 ADC with a DAR of 4. This ADC maintained comparable binding affinity towards FcγRIIIa and FcγRI receptors to that of the native antibody, and exhibited potent in vitro cell-killing activity towards HER2-positive JIMT-1 breast cancer cells, which are trastuzumab-resistant.
A similar strategy has also been applied to the modification of an anti-EphA2 antibody.367 First, the glycan of the anti-EphA2 antibody was almost completely removed using the Endo-S2 glycosidase, which cut after the first GlcNAc on the antibody. GalNAz residues were then installed via β-1,4-Gal-T1-Y289L catalysis, followed by CuAAC reaction to conjugate the PBD dimer payload, SG3364. This generated a DAR 2 ADC, which displayed enhanced in vitro cell-killing activity against prostate cancer cells and successfully suppressed tumour growth over 42 days in an in vivo mouse xenograft model.
This three-step strategy (use of Endo-S glycan trimming, GalNAz addition via β-1,4-Gal-T1-Y289L followed by CuAAC-mediated payload attachment), termed GlycoConnectTM, has also been applied to the generation of superior anti-HER2 ADCs with enhanced activity compared to T-DM1.368 In this study, doxorubicin, MMAF, maytansine and duocarmycin SA were all conjugated via click-reactions with the azido-modified trastuzumab variants, to generate a library of ADCs with a defined DAR of 2. ADCs bearing either cleavable or non-cleavable linkers all displayed remarkable cell-killing activity against a HER2-positive cell line (SK-BR-3), while having little effect against a HER2-negative cell line (MDA-MB-231). In a subsequent mouse PDX model, both cleavable and non-cleavable trastuzumab–MMAF conjugates resulted in complete tumour regression after a single administration at 9 mg kg−1. In contrast, at the same concentration, the approved anti-HER2 ADC Kadcyla® was significantly less efficacious.
The deglycosylation-remodelling strategy has also been widely applied to generate radioimmunoconjugates. For example, in 2013, Zeglis et al. applied the chemoenzymatic strategy for site-selective radiolabeling of antibody glycans through a modular four-step approach.369 After deglycosylating the prostate-specific membrane antigen (PSMA)-targeting antibody J591 with β-1,4-galactosidase, a GalNAz residue was incorporated on the glycan chains of J591 by β-1,4-Gal-T1-Y289L catalysis. A chelator-modified DIBO was then introduced to the glycan via a SPAAC reaction at room temperature, followed by radiolabeling with 89Zr. This formed a radioimmunoconjugate with an average chelator-to-antibody ratio of 2.8. In their following work, the deglycosylation-remodelling strategy was optimised into a one-pot reaction in which GalNAz was incorporated into the Fc-glycan of a colorectal cancer-targeting antibody huA33, followed by modification with AlexaFluor 680 and radiolabelling with 89Zr to generate a dual functionalised radioimmunoconjugate with 3.4 89Zr/mAb and 1.6 AlexaFluor 680/mAb.370
Recently, Zeglis and co-workers have applied this one-pot deglycosylation-remodeling strategy to generate a HER2-targeting ADC for use in PET imaging (Fig. 23). First, a GalNAz moiety was installed on native trastuzumab via β-1,4-galactosidase deglycosylation and subsequent β-1,4-Gal-T1-Y289L glycosidation.371 Subsequent SPAAC reaction with a 1:1 mixture of MMAE-DIBO and chelator-DIBO, followed by 89Zr radiolabeling lead to the formation of an ADC with an average DAR of 1.7 and 2.1 89Zr/mAb. This dual functionalised ADC was then evaluated in an in vivo study in athymic nude mice bearing subcutaneous HER2-expressing BT474 xenografts. Treatment at 10 mg kg−1 resulted in a 90% reduction of tumour volume over 20 days, while the administration of 10 mg kg−1 native trastuzumab did not result in significant tumour reduction (<30%).
4.4 Other glycan remodelling techniques
Finally, site-specific glycan modification has also been achieved by hijacking the pathway that attaches fucose residues to antibody carbohydrates.372 By introducing modified fucose-substrates into CHO cell lines, these unnatural fucoses can be incorporated in antibody glycans in place of fucose by fucosyltransferases. For example, 6-thiofucose peracetate, which contains a reactive thiol handle for site-specific glycan conjugation has been incorporated into a range of antibodies with 60–70% efficiency. A reduction and re-oxidation strategy allowed specific attachment of drug payloads to the glycan 6-thiofucose, producing ADCs with improved homogeneity compared to those produced by conjugation through hinge region disulfides (Fig. 23). Additionally, the conjugates had improved resistance toward retro-Michael addition reactions, resulting in improved stability compared to cysteine thiol-maleimide ADCs. Although further investigation of these ADCs in vivo is warranted, this method also avoids unwanted methionine oxidation encountered by glycan oxidation strategies, and the ADCs produced demonstrated superior cytotoxicity compared to their heterogeneous counterparts.Glycan engineering encompasses a broad range of techniques to provide access to homogeneous ADCs, with specific conjugation at a site that does not negatively impact antibody-antigen binding. Additionally, glycoengineering approaches avoid the need to engineer the amino acid sequence. However, they do require specific reagents, enzymes and enzyme mutants and are limited in drug loading capabilities. Furthermore, evaluation of the effects of glycan modification on Fc–Fc receptor interactions on the overall pharmacology of these ADCs will be important in further development.
5 Conclusions and outlook
From first generation ADCs that were primarily synthesised via stochastic lysine or cysteine modification, it is clear that significant advances have been made in site-selective antibody modification enabling the widespread synthesis of homogeneous ADCs. Each of these new methodologies has their own advantages and disadvantages (Table 1). Indeed, there are several instances of contrasting biological results obtained for the same (or highly similar) synthetic strategies with differing antibodies or payloads.| Technology | Benefits | Limitations |
|---|---|---|
| Engineered cysteines | • Homogeneity | • Genetic engineering required |
| • Tuneable reactivity/stability through site of modification alteration | • Typically limited to DAR 2 | |
| Disulfide rebridging | • Homogeneity | • Intrachain misbridging |
| • Native amino acid sequence and glycosylation | • Typically limited to DAR 4 | |
| Non-canonical amino acids | • Homogeneity | • Genetic engineering required |
| • Tuneable reactivity/stability through site of modification alteration | • Lower antibody expression yields often observed | |
| Terminus modification | • Homogeneity | • Genetic engineering required |
| • DAR alteration possible | • Modification may affect binding interactions | |
| Transglutaminase | • Homogeneity | • Often requires aglycosylated/deglycosylated antibodies or genetic engineering |
| • DAR alteration possible | ||
| Other enzymatic methods | • Homogeneity | • Typically require genetic engineering to install recognition sequence |
| • DAR alteration possible | ||
| Glycan modification | • Homogeneity | • Glycosylation profile is important in immune recognition |
| • No alteration of amino acid sequence | ||
In addition to the conjugation methodologies, it is paramount that suitable analytical techniques are available to study the reactions and the resulting ADC product. These techniques will also be required to study the in vivo metabolism or biotransformation of these ADCs. Combining techniques such as native mass spectrometry and chromatographic analysis (e.g. HIC or HPLC) will be useful in this regard.373 It is clear that the technological capability is already in place to achieve this. However, widespread dissemination of the precise methods and set-up parameters is required to further expand their utility.
As we move into an era where a larger number of ADCs synthesised using site-selective modification methods are undergoing clinical evaluation, a significant increase in data regarding the clinical effects of site-selectivity will be obtained. Utilisation of artificial intelligence and machine learning by the ADC community may help accelerate development procedures by highlighting the most beneficial conjugation method for a particular disease-target-antibody–drug-linker combination. However, thus far, these have proven inherently difficult to predict and it is unclear if machine learning technologies will be able to do so in the near future.
Ultimately, widespread further use of many of the methods described here will rely on the clinical success of themselves or analogous counterparts. It is possible that a single method will emerge as the most suitable for widespread ADC development. However, it is equally possible that this will not occur or that a single method will become prevalent for specific payloads, antibodies or indications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. B. acknowledges an iCASE studentship from GlaxoSmithKline and the EPSRC. F. M. D. acknowledges the BBSRC and AstraZeneca for funding. A. H. acknowledges the EPSRC and Cambridge Trusts for funding. H. S. A. J. C. and X. O. acknowledges Trinity College Cambridge for a Geoffrey Moorehouse Gibson Scholarship, a Krishnan-Ang Studentship and a Trinity Henry-Barlow Scholarship, respectively. Y. T. acknowledges financial support from The Uehara Memorial Foundation and the JSPS for a Research Fellowship for Young Scientists. The Spring lab acknowledges general lab support from the EPSRC, BBSRC, MRC and Royal Society.References
- P. J. Kennedy, C. Oliveira, P. L. Granja and B. Sarmento, Pharmacol. Ther., 2017, 177, 129–145 CrossRef CAS.
- J. R. Adair, P. W. Howard, J. A. Hartley, D. G. Williams and K. A. Chester, Expert Opin. Biol. Ther., 2012, 12, 1191–1206 CrossRef CAS.
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS.
- L. Gauzy-Lazo, I. Sassoon and M.-P. P. Brun, SLAS Discovery, 2020, 1–26 Search PubMed.
- M. Nejadmoghaddam, A. Minai-tehrani and R. Ghahremanzadeh, Avicenna J. Med. Biotechnol., 2019, 11, 3–23 Search PubMed.
- P. F. Bross, J. Beitz, G. Chen, X. H. Chen, E. Duffy, L. Kieffer, S. Roy, R. Sridhara, A. Rahman, G. Williams and R. Pazdur, Clin. Cancer Res., 2001, 7, 1490–1496 CAS.
- Y. N. Lamb, Drugs, 2017, 77, 1603–1610 CrossRef CAS.
- A. Ballantyne and S. Dhillon, Drugs, 2013, 73, 755–765 CrossRef CAS.
- N. C. Richardson, Y. L. Kasamon, H. Chen, R. A. de Claro, J. Ye, G. M. Blumenthal, A. T. Farrell and R. Pazdur, Oncologist, 2019, 24, e180–e187 CrossRef CAS.
- E. D. Deeks, Drugs, 2019, 79, 1467–1475 CrossRef.
- K. S. Hanna, Drugs, 2020, 80, 1–7 CrossRef.
- S. J. Keam, Drugs, 2020, 80, 501–508 CrossRef CAS.
- Y. Y. Syed, Drugs, 2020, 80, 1019–1025 CrossRef CAS.
- H. K. Erickson, W. C. Widdison, M. F. Mayo, K. Whiteman, C. Audette, S. D. Wilhelm and R. Singh, Bioconjugate Chem., 2010, 21, 84–92 CrossRef CAS.
- R. M. DeVay, K. Delaria, G. Zhu, C. Holz, D. Foletti, J. Sutton, G. Bolton, R. G. Dushin, C. Bee, J. Pons, A. Rajpal, H. Liang, D. Shelton, S.-H. Liu and P. Strop, Bioconjugate Chem., 2017, 28, 1102–1114 CrossRef CAS.
- B. Nolting, Methods Mol. Biol., 2013, 1045, 71–100 CrossRef.
- P. R. Hamann, L. M. Hinman, I. Hollander, C. F. Beyer, D. Lindh, R. Holcomb, W. Hallett, H. R. Tsou, J. Upeslacis, D. Shochat, A. Mountain, D. A. Flowers and I. Bernstein, Bioconjugate Chem., 2002, 13, 47–58 CrossRef CAS.
- G. D. Lewis Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons, E. Mai, W. A. Blättler, J. M. Lambert, R. V. J. Chari, R. J. Lutz, W. L. T. Wong, F. S. Jacobson, H. Koeppen, R. H. Schwall, S. R. Kenkare-Mitra, S. D. Spencer and M. X. Sliwkowski, Cancer Res., 2008, 68, 9280–9290 CrossRef CAS.
- T. H. Pillow, J. D. Sadowsky, D. Zhang, S. F. Yu, G. Del Rosario, K. Xu, J. He, S. Bhakta, R. Ohri, K. R. Kozak, E. Ha, J. R. Junutula and J. A. Flygare, Chem. Sci., 2016, 8, 366–370 RSC.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS.
- J. C. Kern, D. Dooney, R. Zhang, L. Liang, P. E. Brandish, M. Cheng, G. Feng, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, Y. Sun and R. M. Garbaccio, Bioconjugate Chem., 2016, 27, 2081–2088 CrossRef CAS.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. C. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS.
- S. C. Jeffrey, J. B. Andreyka, S. X. Bernhardt, K. M. Kissler, T. Kline, J. S. Lenox, R. F. Moser, M. T. Nguyen, N. M. Okeley, I. J. Stone, X. Zhang and P. D. Senter, Bioconjugate Chem., 2006, 17, 831–840 CrossRef CAS.
- S. Kolodych, C. Michel, S. Delacroix, O. Koniev, A. Ehkirch, J. Eberova, S. Cianférani, B. Renoux, W. Krezel, P. Poinot, C. D. Muller, S. Papot and A. Wagner, Eur. J. Med. Chem., 2017, 142, 376–382 CrossRef CAS.
- J. D. Bargh, S. J. Walsh, A. Isidro-Llobet, S. Omarjee, J. S. Carroll and D. R. Spring, Chem. Sci., 2020, 11, 2375–2380 RSC.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- G. Vidarsson, G. Dekkers and T. Rispens, Front. Immunol., 2014, 5, 520 Search PubMed.
- R. M. Hoffmann, B. G. T. Coumbe, D. H. Josephs, S. Mele, K. M. Ilieva, A. Cheung, A. N. Tutt, J. F. Spicer, D. E. Thurston, S. Crescioli and S. N. Karagiannis, Oncoimmunology, 2018, 7, e1395127 CrossRef.
- H. Liu and K. May, mAbs, 2012, 4, 17–23 CrossRef.
- P. Herbener, K. Schönfeld, M. König, M. Germer, J. M. Przyborski, K. Bernöster and J. Schüttrumpf, PLoS One, 2018, 13, e0195823 CrossRef.
- A. F. Labrijn, A. O. Buijsse, E. T. J. Van Den Bremer, A. Y. W. Verwilligen, W. K. Bleeker, S. J. Thorpe, J. Killestein, C. H. Polman, R. C. Aalberse, J. Schuurman, J. G. J. Van De Winkel and P. W. H. I. Parren, Nat. Biotechnol., 2009, 27, 767–771 CrossRef CAS.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS.
- K. T. Xenaki, S. Oliveira and P. M. P. van Bergen en Henegouwen, Front. Immunol., 2017, 8, 1287 CrossRef.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS.
- S. Yaghoubi, M. H. Karimi, M. Lotfinia, T. Gharibi, M. Mahi-Birjand, E. Kavi, F. Hosseini, K. Sineh Sepehr, M. Khatami, N. Bagheri and M. Abdollahpour-Alitappeh, J. Cell. Physiol., 2020, 235, 31–64 CrossRef CAS.
- D. J. Newman, Mar. Drugs, 2019, 17, 324 CrossRef CAS.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, G. F. Filzen, C. J. O’Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. Galindo Casas, K. Delaria, V. Lui, K. Poulsen, C. Loo, S. Krimm, G. Bolton, L. Moine, R. Dushin, T.-T. Tran, S.-H. Liu, M. Rickert, D. Foletti, D. L. Shelton, J. Pons and A. Rajpal, Bioconjugate Chem., 2015, 26, 650–659 CrossRef CAS.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS.
- A. Bardia, I. A. Mayer, L. T. Vahdat, S. M. Tolaney, S. J. Isakoff, J. R. Diamond, J. O’Shaughnessy, R. L. Moroose, A. D. Santin, V. G. Abramson, N. C. Shah, H. S. Rugo, D. M. Goldenberg, A. M. Sweidan, R. Iannone, S. Washkowitz, R. M. Sharkey, W. A. Wegener and K. Kalinsky, N. Engl. J. Med., 2019, 380, 741–751 CrossRef CAS.
- T. Satomaa, H. Pynnönen, A. Vilkman, T. Kotiranta, V. Pitkänen, A. Heiskanen, B. Herpers, L. Price, J. Helin, J. Saarinen, T. Satomaa, H. Pynnönen, A. Vilkman, T. Kotiranta, V. Pitkänen, A. Heiskanen, B. Herpers, L. S. Price, J. Helin and J. Saarinen, Antibodies, 2018, 7, 15 CrossRef CAS.
- W. Viricel, G. Fournet, S. Beaumel, E. Perrial, S. Papot, C. Dumontet and B. Joseph, Chem. Sci., 2019, 10, 4048–4053 RSC.
- F. Lhospice, D. Brégeon, C. Belmant, P. Dennler, A. Chiotellis, E. Fischer, L. Gauthier, A. Boëdec, H. Rispaud, S. Savard-Chambard, A. Represa, N. Schneider, C. Paturel, M. Sapet, C. Delcambre, S. Ingoure, N. Viaud, C. Bonnafous, R. Schibli and F. Romagné, Mol. Pharm., 2015, 12, 1863–1871 CrossRef CAS.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Elsevier Inc., 3rd edn, 2013 Search PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- F. Searle, C. Bier, R. G. Buckley, S. Newman, R. B. Pedley, K. D. Bagshawe, R. G. Melton, S. M. Alwan and R. F. Sherwood, Br. J. Cancer, 1986, 53, 377–384 CrossRef CAS.
- R. G. Melton, Adv. Drug Delivery Rev., 1996, 22, 289–301 CrossRef CAS.
- S. E. Winston, S. A. Fuller, M. J. Evelegh and J. G. R. Hurrell, Curr. Protoc. Mol. Biol., 2000, 50, 11.1.1–11.1.7 Search PubMed.
- T. E. Witzig, Cancer Chemother. Pharmacol., 2001, 48, S91–S95 CrossRef.
- L. Wang, G. Amphlett, W. A. Blättler, J. M. Lambert and W. Zhang, Protein Sci., 2005, 14, 2436–2446 CrossRef CAS.
- A. Wakankar, Y. Chen, Y. Gokarn and F. S. Jacobson, mAbs, 2011, 3, 164–175 CrossRef.
- H.-W. Chih, B. Gikanga, Y. Yang and B. Zhang, J. Pharm. Sci., 2011, 100, 2518–2525 CrossRef CAS.
- S. Kalkhof and A. Sinz, Anal. Bioanal. Chem., 2008, 392, 305–312 CrossRef CAS.
- S. Mädler, C. Bich, D. Touboul and R. Zenobi, J. Mass Spectrom., 2009, 44, 694–706 CrossRef.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495 RSC.
- D. S. Wilbur, M. K. Chyan, H. Nakamae, Y. Chen, D. K. Hamlin, E. B. Santos, B. T. Kornblit and B. M. Sandmaier, Bioconjugate Chem., 2012, 23, 409–420 CrossRef CAS.
- M. Hayakawa, N. Toda, N. Carrillo, N. J. Thornburg, J. E. Crowe and C. F. Barbas, ChemBioChem, 2012, 13, 2191–2195 CrossRef CAS.
- I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjugate Chem., 2017, 28, 1452–1457 CrossRef CAS.
- M. Moreau, O. Raguin, J. M. Vrigneaud, B. Collin, C. Bernhard, X. Tizon, F. Boschetti, O. Duchamp, F. Brunotte and F. Denat, Bioconjugate Chem., 2012, 23, 1181–1188 CrossRef CAS.
- M. M. C. Sun, K. S. Beam, C. G. Cerveny, K. J. Hamblett, R. S. Blackmore, M. Y. Torgov, F. G. M. Handley, N. C. Ihle, P. D. Senter and S. C. Alley, Bioconjugate Chem., 2005, 16, 1282–1290 CrossRef CAS.
- P. L. Ross and J. L. Wolfe, J. Pharm. Sci., 2016, 105, 391–397 CrossRef CAS.
- H. Liu, C. Chumsae, G. Gaza-Bulseco, K. Hurkmans and C. H. Radziejewski, Anal. Chem., 2010, 82, 5219–5226 CrossRef CAS.
- P. A. Szijj, C. Bahou and V. Chudasama, Drug Discovery Today Technol., 2018, 30, 27–34 CrossRef.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS.
- L. N. Tumey, M. Charati, T. He, E. Sousa, D. Ma, X. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Bioconjugate Chem., 2014, 25, 1871–1880 CrossRef CAS.
- R. J. Christie, R. Fleming, B. Bezabeh, R. Woods, S. Mao, J. Harper, A. Joseph, Q. Wang, Z. Q. Xu, H. Wu, C. Gao and N. Dimasi, J. Controlled Release, 2015, 220, 660–670 CrossRef CAS.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS.
- M.-A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS.
- M. Kasper, A. Stengl, P. Ochtrop, M. Gerlach, T. Stoschek, D. Schumacher, J. Helma, M. Penkert, E. Krause, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11631–11636 CrossRef CAS.
- M. A. Kasper, M. Glanz, A. Oder, P. Schmieder, J. P. Von Kries and C. P. R. Hackenberger, Chem. Sci., 2019, 10, 6322–6329 RSC.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS.
- C. F. McDonagh, E. Turcott, L. Westendorf, J. B. Webster, S. C. Alley, K. Kim, J. Andreyka, I. Stone, K. J. Hamblett, J. A. Francisco and P. Carter, Protein Eng., Des. Sel., 2006, 19, 299–307 CrossRef CAS.
- S. D. Gillies and J. S. Wesolowski, Hum. Antibodies, 1990, 1, 47–54 CAS.
- L. N. Tumey, F. Li, B. Rago, X. Han, F. Loganzo, S. Musto, E. I. Graziani, S. Puthenveetil, J. Casavant, K. Marquette, T. Clark, J. Bikker, E. M. Bennett, F. Barletta, N. Piche-Nicholas, A. Tam, C. J. O’Donnell, H. P. Gerber and L. Tchistiakova, AAPS J., 2017, 19, 1123–1135 CrossRef CAS.
- J. R. Junutula, S. Bhakta, H. Raab, K. E. Ervin, C. Eigenbrot, R. Vandlen, R. H. Scheller and H. B. Lowman, J. Immunol. Methods, 2008, 332, 41–52 CrossRef CAS.
- R. Ohri, S. Bhakta, A. Fourie-O’Donohue, J. Dela Cruz-Chuh, S. P. Tsai, R. Cook, B. Wei, C. Ng, A. W. Wong, A. B. Bos, F. Farahi, J. Bhakta, T. H. Pillow, H. Raab, R. Vandlen, P. Polakis, Y. Liu, H. Erickson, J. R. Junutula and K. R. Kozak, Bioconjugate Chem., 2018, 29, 473–485 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS.
- D. Shinmi, E. Taguchi, J. Iwano, T. Yamaguchi, K. Masuda, J. Enokizono and Y. Shiraishi, Bioconjugate Chem., 2016, 27, 1324–1331 CrossRef CAS.
- D. Sussman, L. Westendorf, D. W. Meyer, C. I. Leiske, M. Anderson, N. M. Okeley, S. C. Alley, R. Lyon, R. J. Sanderson, P. J. Carter and D. R. Benjamin, Protein Eng., Des. Sel., 2018, 31, 47–54 CrossRef CAS.
- N. Dimasi, R. Fleming, H. Zhong, B. Bezabeh, K. Kinneer, R. J. Christie, C. Fazenbaker, H. Wu and C. Gao, Mol. Pharm., 2017, 14, 1501–1516 CrossRef CAS.
- H. Uppal, E. Doudement, K. Mahapatra, W. C. Darbonne, D. Bumbaca, B. Q. Shen, X. Du, O. Saad, K. Bowles, S. Olsen, G. D. L. Phillips, D. Hartley, M. X. Sliwkowski, S. Girish, D. Dambach and V. Ramakrishnan, Clin. Cancer Res., 2015, 21, 123–133 CrossRef CAS.
- G. C. Kemp, A. C. Tiberghien, N. V. Patel, F. D’Hooge, S. M. Nilapwar, L. R. Adams, S. Corbett, D. G. Williams, J. A. Hartley and P. W. Howard, Bioorganic Med. Chem. Lett., 2017, 27, 1154–1158 CrossRef CAS.
- J. P. M. Nunes, V. Vassileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Saéz, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. De Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior, G. J. L. Bernardes and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869–10880 CrossRef CAS.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef CAS.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS.
- J. D. Sadowsky, T. H. Pillow, J. Chen, F. Fan, C. He, Y. Wang, G. Yan, H. Yao, Z. Xu, S. Martin, D. Zhang, P. Chu, J. Dela Cruz-Chuh, A. O’Donohue, G. Li, G. Del Rosario, J. He, L. Liu, C. Ng, D. Su, G. D. Lewis Phillips, K. R. Kozak, S. F. Yu, K. Xu, D. Leipold and J. Wai, Bioconjugate Chem., 2017, 28, 2086–2098 CrossRef CAS.
- B. S. Vollmar, B. Wei, R. Ohri, J. Zhou, J. He, S. F. Yu, D. Leipold, E. Cosino, S. Yee, A. Fourie-O’Donohue, G. Li, G. L. Phillips, K. R. Kozak, A. Kamath, K. Xu, G. Lee, G. A. Lazar and H. K. Erickson, Bioconjugate Chem., 2017, 28, 2538–2548 CrossRef CAS.
- T. H. Pillow, P. Adhikari, R. A. Blake, J. Chen, G. Del Rosario, G. Deshmukh, I. Figueroa, K. E. Gascoigne, A. V. Kamath, S. Kaufman, T. Kleinheinz, K. R. Kozak, B. Latifi, D. D. Leipold, C. Sing Li, R. Li, M. M. Mulvihill, A. O’Donohue, R. K. Rowntree, J. D. Sadowsky, J. Wai, X. Wang, C. Wu, Z. Xu, H. Yao, S. F. Yu, D. Zhang, R. Zang, H. Zhang, H. Zhou, X. Zhu and P. S. Dragovich, ChemMedChem, 2020, 15, 17–25 CrossRef CAS.
- C. S. Neumann, K. C. Olivas, M. E. Anderson, J. H. Cochran, S. Jin, F. Li, L. V. Loftus, D. W. Meyer, J. Neale, J. C. Nix, P. G. Pittman, J. K. Simmons, M. L. Ulrich, A. B. Waight, A. Wong, M. C. Zaval, W. Zeng, R. P. Lyon and P. D. Senter, Mol. Cancer Ther., 2018, 17, 2633–2642 CrossRef CAS.
- A. Kumar, K. Kinneer, L. Masterson, E. Ezeadi, P. Howard, H. Wu, C. Gao and N. Dimasi, Bioorganic Med. Chem. Lett., 2018, 28, 3617–3621 CrossRef CAS.
- J. Harper, C. Lloyd, N. Dimasi, D. Toader, R. Marwood, L. Lewis, D. Bannister, J. Jovanovic, R. Fleming, F. D’Hooge, S. Mao, A. M. Marrero, M. Korade, P. Strout, L. Xu, C. Chen, L. Wetzel, S. Breen, L. Van Vlerken-Ysla, S. Jalla, M. Rebelatto, H. Zhong, E. M. Hurt, M. J. Hinrichs, K. Huang, P. W. Howard, D. A. Tice, R. E. Hollingsworth, R. Herbst and A. Kamal, Mol. Cancer Ther., 2017, 16, 1576–1587 CrossRef CAS.
- F. Li, M. K. Sutherland, C. Yu, R. B. Walter, L. Westendorf, J. Valliere-Douglass, L. Pan, A. Cronkite, D. Sussman, K. Klussman, M. Ulrich, M. E. Anderson, I. J. Stone, W. Zeng, M. Jonas, T. S. Lewis, M. Goswami, S. A. Wang, P. D. Senter, C. L. Law, E. J. Feldman and D. R. Benjamin, Mol. Cancer Ther., 2018, 17, 554–564 CrossRef CAS.
- J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, G. D. L. Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS.
- S. Adams, A. Wilhelm, L. Harvey, C. Bai, N. Yoder, Y. Kovtun, T. Chittenden and J. Pinkas, Blood, 2016, 128, 2832 CrossRef.
- F. Zammarchi, S. Corbett, L. Adams, M. Mellinas-Gomez, P. Tyrer, S. Dissanayake, S. Sims, K. Havenith, S. Chivers, D. G. Willimas, P. W. Howard, J. A. Hartley and P. van Berkel, Blood, 2016, 128, 4176 CrossRef.
- W. Tang, X. Huang, Z. Ou, H. Yan, J. Gan, Q. Dong, B. Tan, Y. Yang, Y. Guo, S. Li, B. Thomas and J.-C. Yu, Cancer Res., 2019, 79, P6-20-16 Search PubMed.
- S. M. Lehar, T. Pillow, M. Xu, L. Staben, K. K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. L. Hazenbos, J. Hiroshi Morisaki, J. Kim, S. Park, M. Darwish, B. C. Lee, H. Hernandez, K. M. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. P. Lyssikatos, M. Strandh, K. Koefoed, P. S. Andersen, J. A. Flygare, M. Wah Tan, E. J. Brown and S. Mariathasan, Nature, 2015, 527, 323–328 CrossRef CAS.
- M. Peck, M. E. Rothenberg, R. Deng, N. Lewin-Koh, G. She, A. V. Kamath, M. Carrasco-Triguero, O. Saad, A. Castro, L. Teufel, D. S. Dickerson, M. Leonardelli and J. A. Tavel, Antimicrob. Agents Chemother., 2019, 63, e02588 CrossRef CAS.
- N. Forte, V. Chudasama and J. R. Baker, Drug Discovery Today Technol., 2018, 30, 11–20 CrossRef.
- F. A. Liberatore, R. D. Comeau, J. M. McKearin, D. A. Pearson, B. Q. Belonga, S. J. Brocchini, J. Kath, T. Phillips, K. Oswell and R. G. Lawton, Bioconjugate Chem., 1990, 1, 36–50 CrossRef CAS.
- R. B. del Rosario, R. L. Wahl, S. J. Brocchini, R. G. Lawton and R. H. Smith, Bioconjugate Chem., 1990, 1, 51–59 CrossRef CAS.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS.
- P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J. W. Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharm., 2015, 12, 1872–1879 CrossRef CAS.
- M. Dorywalska, R. Dushin, L. Moine, S. E. Farias, D. Zhou, T. Navaratnam, V. Lui, A. Hasa-Moreno, M. G. Casas, T. T. Tran, K. Delaria, S. H. Liu, D. Foletti, C. J. O’Donnell, J. Pons, D. L. Shelton, A. Rajpal and P. Strop, Mol. Cancer Ther., 2016, 15, 958–970 CrossRef CAS.
- M. Pabst, W. McDowell, A. Manin, A. Kyle, N. Camper, E. De Juan, V. Parekh, F. Rudge, H. Makwana, T. Kantner, H. Parekh, A. Michelet, X. B. Sheng, G. Popa, C. Tucker, F. Khayrzad, D. Pollard, K. Kozakowska, R. Resende, A. Jenkins, F. Simoes, D. Morris, P. Williams, G. Badescu, M. P. Baker, M. Bird, M. Frigerio and A. Godwin, J. Controlled Release, 2017, 253, 160–164 CrossRef CAS.
- A. Godwin, A. Kyle and N. Evans, Conjugates and conjugating reagents comprising a linker that includes at least two (–CH2–CH2–0–) units in a ring, WO2017/178828, 2017 Search PubMed.
- M. N. Chang, J.-S. Lai, W.-F. Li, I.-J. Chen, Y.-C. Tsai and K.-C. Chen, Conjugated biological molecules, pharmaceutical compositions and methods, US2018/0193481, 2018 Search PubMed.
- L. Huang, B. Veneziale, M. Frigerio, G. Badescu, X. Li, Q. Zhao, J. Bahn, J. Souratha, R. Osgood, C. Zhao, K. Phan, J. Cowell, S. Rosengren, J. Parise, M. Pabst, M. Bird, W. McDowell, G. Wei, C. Thompson, A. Godwin, M. Shepard and C. Thanos, Cancer Res., 2016, 76, 1217 Search PubMed.
- M. Bird, J. Nunes and M. Frigerio, Methods in Molecular Biology, Humana Press Inc., 2020, vol. 2078, pp. 113–129 Search PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J. W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T. S. Migone, E. H. Van Der Horst, R. L. Halcomb and D. Y. Jackson, Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- F. Bryden, C. Martin, S. Letast, E. Lles, I. Viéitez-Villemin, A. Rousseau, C. Colas, M. Brachet-Botineau, E. Allard-Vannier, C. Larbouret, M. C. Viaud-Massuard and N. Joubert, Org. Biomol. Chem., 2018, 16, 1882–1889 RSC.
- N. Forte, M. Livanos, E. Miranda, M. Morais, X. Yang, V. S. Rajkumar, K. A. Chester, V. Chudasama and J. R. Baker, Bioconjugate Chem., 2018, 29, 486–492 CrossRef CAS.
- O. Feuillâtre, C. Gély, S. Huvelle, C. B. Baltus, L. Juen, N. Joubert, A. Desgranges, M. C. Viaud-Massuard and C. Martin, ACS Omega, 2020, 5, 1557–1565 CrossRef.
- M. Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306–1312 CrossRef CAS.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS.
- S. Shao, M.-H. Tsai, J. Lu, T. Yu, J. Jin, D. Xiao, H. Jiang, M. Han, M. Wang and J. Wang, Bioorg. Med. Chem. Lett., 2018, 28, 1363–1370 CrossRef CAS.
- M. T. W. Lee, A. Maruani, D. A. Richards, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2017, 8, 2056–2060 RSC.
- F. Bryden, J. M. M. Rodrigues, H. Savoie, V. Chudasama, A. Beeby, M. H. Y. Cheng, R. W. Boyle and A. Maruani, Bioconjugate Chem., 2017, 29, 176–181 CrossRef.
- A. N. Marquard, J. C. T. Carlson and R. Weissleder, Bioconjugate Chem., 2020, 31, 1616–1623 CrossRef CAS.
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016, 7, 799–802 RSC.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- B. Shi, M. Wu, Z. Li, Z. Xie, X. Wei, J. Fan, Y. Xu, D. Ding, S. H. Akash, S. Chen and S. Cao, Cancer Med., 2019, 8, 1793–1805 CrossRef CAS.
- Z. Miao, T. Zhu, K. B. Alisher, G. Chen, Z. LI and S. Cao, Anti-HER2 antibody–drug conjugate and applications thereof, US2019/0091345, 2019 Search PubMed.
- Z. Miao, T. Zhu, A. B. Khasanov, S. Cao, Z. Li and M. Wu, Anti-5T4 antibody–drug conjugate and use thereof, US2019/0374651, 2019 Search PubMed.
- Q.-Y. Hu and H. Imase, Methods for making conjugates from disulfide-containing proteins, WO2014/083505, 2014 Search PubMed.
- Q.-Y. Hu and M. Allan, Method for making conjugates from disulfide-containing proteins, US2019/0381126, 2019 Search PubMed.
- N. Gupta, J. Kancharla, S. Kaushik, A. Ansari, S. Hossain, R. Goyal, M. Pandey, J. Sivaccumar, S. Hussain, A. Sarkar, A. Sengupta, S. K. Mandal, M. Roy and S. Sengupta, Chem. Sci., 2017, 8, 2387–2395 RSC.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Med. Chem. Commun., 2018, 9, 827–830 RSC.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J. Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 197–200 CrossRef CAS.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- J. Charoenpattarapreeda, S. J. Walsh, J. S. Carroll and D. R. Spring, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010090.
- S. J. Walsh, J. Iegre, H. Seki, J. D. Bargh, H. F. Sore, J. S. Parker, J. S. Carroll and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4224–4230 RSC.
- A. J. Counsell, S. J. Walsh, N. S. Robertson, H. Sore and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4739–4743 RSC.
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS.
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS.
- S. M. Hancock, R. Uprety, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 14819–14824 CrossRef CAS.
- P. R. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. H. Geierstanger and P. G. Schultz, Angew. Chem., Int. Ed., 2009, 48, 4052–4055 CrossRef CAS.
- O. Vargas-Rodriguez, A. Sevostyanova, D. Söll and A. Crnković, Curr. Opin. Chem. Biol., 2018, 46, 115–122 CrossRef CAS.
- H. Neumann, A. L. Slusarczyk and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 2142–2144 CrossRef CAS.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS.
- G. Roy, J. Reier, A. Garcia, T. Martin, M. Rice, J. Wang, M. Prophet, R. Christie, W. Dall’Acqua, S. Ahuja, M. A. Bowen and M. Marelli, mAbs, 2020, 12, 1684749 CrossRef.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Tran, Y. Sun, T. Phuong, R. Barnett, B. Hehli, F. Song, M. J. DeGuzman, S. Ensari, J. K. Pinkstaff, L. M. Sullivan, S. L. Biroc, H. Cho, P. G. Schultz, J. DiJoseph, M. Dougher, D. Ma, R. Dushin, M. Leal, L. Tchistiakova, E. Feyfant, H.-P. Gerber and P. Sapra, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1766–1771 CrossRef CAS.
- A. Molina, N. Shah, A. Y. Krishnan, N. D. Shah, J. M. Burke, J. M. Melear, A. I. Spira, L. L. Popplewell, C. B. Andreadis, S. Chhabra, J. P. Sharman, J. L. Kaufman, J. B. Cohen, R. Niesvizky, T. G. Martin, C. DiLea, J. Kuriakose, S. L. Matheny and J. P. Leonard, Hematol. Oncol., 2019, 37, 324–325 CrossRef.
- R. W. Naumann, D. Uyar, J. W. Moroney, F. S. Braiteh, R. J. Schilder, J. P. Diaz, E. Hamilton, L. P. Martin, D. M. O. Malley, R. T. Penson, C. Dilea, M. Palumbo, V. Dealmeida, L. P. Martin, D. M. O. Malley, R. T. Penson, C. Dilea, M. Palumbo, V. Dealmeida, S. Matheny and A. Molina, Am. Assoc. Cancer Res. Virtual Annu. Meet., 2020 Search PubMed.
- G. Yin, E. D. Garces, J. Yang, J. Zhang, C. Tran, A. R. Steiner, C. Roos, S. Bajad, S. Hudak, K. Penta, J. Zawada, S. Pollitt and C. J. Murray, mAbs, 2012, 4, 217–225 CrossRef.
- S. T. Jung, T. H. Kang, W. Kelton and G. Georgiou, Curr. Opin. Biotechnol., 2011, 22, 858–867 CrossRef CAS.
- M. Pott, M. J. Schmidt and D. Summerer, ACS Chem. Biol., 2014, 9, 2815–2822 CrossRef CAS.
- G. Yin, H. T. Stephenson, J. Yang, X. Li, S. M. Armstrong, T. H. Heibeck, C. Tran, M. R. Masikat, S. Zhou, R. L. Stafford, A. Y. Yam, J. Lee, A. R. Steiner, A. Gill, K. Penta, S. Pollitt, R. Baliga, C. J. Murray, C. D. Thanos, L. M. McEvoy, A. K. Sato and T. J. Hallam, Sci. Rep., 2017, 7, 3026 CrossRef.
- M. P. VanBrunt, K. Shanebeck, Z. Caldwell, J. Johnson, P. Thompson, T. Martin, H. Dong, G. Li, H. Xu, F. D’Hooge, L. Masterson, P. Bariola, A. Tiberghien, E. Ezeadi, D. G. Williams, J. A. Hartley, P. W. Howard, K. H. Grabstein, M. A. Bowen and M. Marelli, Bioconjugate Chem., 2015, 26, 2249–2260 CrossRef CAS.
- B. Oller-Salvia, G. Kym and J. W. Chin, Angew. Chem., Int. Ed., 2018, 57, 2831–2834 CrossRef CAS.
- A. H. Amant, F. Huang, J. Lin, D. Lemen, C. Chakiath, S. Mao, C. Fazenbaker, H. Zhong, J. Harper, W. Xu, N. Patel, L. Adams, B. Vijayakrishnan, P. W. Howard, M. Marelli, H. Wu, C. Gao, J. Read de Alaniz and R. J. Christie, Bioconjugate Chem., 2019, 30, 2340–2348 CrossRef.
- S. A. Kularatne, V. Deshmukh, J. Ma, V. Tardif, R. K. V. Lim, H. M. Pugh, Y. Sun, A. Manibusan, A. J. Sellers, R. S. Barnett, S. Srinagesh, J. S. Forsyth, W. Hassenpflug, F. Tian, T. Javahishvili, B. Felding-Habermann, B. R. Lawson, S. A. Kazane and P. G. Schultz, Angew. Chem., Int. Ed., 2014, 53, 11863–11867 CrossRef CAS.
- D. Jackson, J. Atkinson, C. I. Guevara, C. Zhang, V. Kery, S.-J. Moon, C. Virata, P. Yang, C. Lowe, J. Pinkstaff, H. Cho, N. Knudsen, A. Manibusan, F. Tian, Y. Sun, Y. Lu, A. Sellers, X.-C. Jia, I. Joseph, B. Anand, K. Morrison, D. S. Pereira and D. Stover, PLoS One, 2014, 9, e83865 CrossRef.
- R. K. V. Lim, S. Yu, B. Cheng, S. Li, N.-J. Kim, Y. Cao, V. Chi, J. Y. Kim, A. K. Chatterjee, P. G. Schultz, M. S. Tremblay and S. A. Kazane, Bioconjugate Chem., 2015, 26, 2216–2222 CrossRef CAS.
- S. Yu, A. D. Pearson, R. K. V. Lim, D. T. Rodgers, S. Li, H. B. Parker, M. Weglarz, E. N. Hampton, M. J. Bollong, J. Shen, C. Zambaldo, D. Wang, A. K. Woods, T. M. Wright, P. G. Schultz, S. A. Kazane, T. S. Young and M. S. Tremblay, Mol. Ther., 2016, 24, 2078–2089 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- P. E. Brandish, A. Palmieri, S. Antonenko, M. Beaumont, L. Benso, M. Cancilla, M. Cheng, L. Fayadat-Dilman, G. Feng, I. Figueroa, J. Firdos, R. Garbaccio, L. Garvin-Queen, D. Gately, P. Geda, C. Haines, S. Hseih, D. Hodges, J. Kern, N. Knudsen, K. Kwasnjuk, L. Liang, H. Ma, A. Manibusan, P. L. Miller, L. Y. Moy, Y. Qu, S. Shah, J. S. Shin, P. Stivers, Y. Sun, D. Tomazela, H. C. Woo, D. Zaller, S. Zhang, Y. Zhang and M. Zielstorff, Bioconjugate Chem., 2018, 29, 2357–2369 CrossRef CAS.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS.
- X. Li, C. Abrahams, S. Zhou, S. Krimm, R. Henningsen, H. Stephenson, J. Hanson, M. R. Masikat, K. Bajjuri, T. Heibeck, C. Tran, G. Yin, J. Zawada, G. Sarma, J. Chen, M. Bruhns, W. Solis, A. Steiner, A. Galan, T. Kline, R. Stafford, A. Yam, V. I. De Almeida, M. Lupher and T. Hallam, Cancer Res., 2018, 78, 1782 Search PubMed.
- C. L. Abrahams, X. Li, M. Embry, A. Yu, S. Krimm, S. Krueger, N. Y. Greenland, K. W. Wen, C. Jones, V. DeAlmeida, W. A. Solis, S. Matheny, T. Kline, A. Y. Yam, R. Stafford, A. P. Wiita, T. Hallam, M. Lupher and A. Molina, Oncotarget, 2018, 9, 37700–37714 CrossRef.
- S. H. Ahn, B. A. Vaughn, W. A. Solis, M. L. Lupher, T. J. Hallam and E. Boros, Bioconjugate Chem., 2020, 31, 1177–1187 CrossRef CAS.
- Y. Wu, H. Zhu, B. Zhang, F. Liu, J. Chen, Y. Wang, Y. Wang, Z. Zhang, L. Wu, L. Si, H. Xu, T. Yao, S. Xiao, Q. Xia, L. Zhang, Z. Yang and D. Zhou, Bioconjugate Chem., 2016, 27, 2460–2468 CrossRef CAS.
- C. Koehler, P. F. Sauter, M. Wawryszyn, G. E. Girona, K. Gupta, J. J. M. Landry, M. H.-Y. Fritz, K. Radic, J.-E. Hoffmann, Z. A. Chen, J. Zou, P. S. Tan, B. Galik, S. Junttila, P. Stolt-Bergner, G. Pruneri, A. Gyenesei, C. Schultz, M. B. Biskup, H. Besir, V. Benes, J. Rappsilber, M. Jechlinger, J. O. Korbel, I. Berger, S. Braese and E. A. Lemke, Nat. Methods, 2016, 13, 997–1000 CrossRef CAS.
- A. H. St. Amant, F. Huang, J. Lin, K. Rickert, V. Oganesyan, D. Lemen, S. Mao, J. Harper, M. Marelli, H. Wu, C. Gao, J. Read de Alaniz and R. J. Christie, Angew. Chem., Int. Ed., 2019, 58, 8489–8493 CrossRef CAS.
- T. Hofer, L. R. Skeffington, C. M. Chapman and C. Rader, Biochemistry, 2009, 48, 12047–12057 CrossRef CAS.
- T. Hofer, J. D. Thomas, T. R. Burke and C. Rader, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12451–12456 CrossRef CAS.
- D. L. Hatfield and V. N. Gladyshev, Mol. Cell. Biol., 2002, 22, 3565–3576 CrossRef CAS.
- X. Li, J. Yang and C. Rader, Methods, 2014, 65, 133–138 CrossRef CAS.
- X. Li, C. G. Nelson, R. R. Nair, L. Hazlehurst, T. Moroni, P. Martinez-Acedo, A. R. Nanna, D. Hymel, T. R. Burke and C. Rader, Cell Chem. Biol., 2017, 24, 433–442 CrossRef CAS.
- Y. Zheng, P. S. Addy, R. Mukherjee and A. Chatterjee, Chem. Sci., 2017, 8, 7211–7217 RSC.
- D. L. Dunkelmann, J. C. W. Willis, A. T. Beattie and J. W. Chin, Nat. Chem., 2020, 12, 535–544 CrossRef CAS.
- H. Xiao, A. Chatterjee, S. Choi, K. M. Bajjuri, S. C. Sinha and P. G. Schultz, Angew. Chem., Int. Ed., 2013, 52, 14080–14083 CrossRef CAS.
- N. Nilchan, X. Li, L. Pedzisa, A. R. Nanna, W. R. Roush and C. Rader, Antib. Ther., 2019, 2, 71–78 CAS.
- X. Li, J. T. Patterson, M. Sarkar, L. Pedzisa, T. Kodadek, W. R. Roush and C. Rader, Bioconjugate Chem., 2015, 26, 2243–2248 CrossRef CAS.
- M. Barok, V. Le Joncour, A. Martins, J. Isola, M. Salmikangas, P. Laakkonen and H. Joensuu, Cancer Lett., 2020, 473, 156–163 CrossRef CAS.
- J. T. Snyder, M.-C. Malinao, J. Dugal-Tessier, J. E. Atkinson, B. S. Anand, A. Okada and B. A. Mendelsohn, Mol. Pharm., 2018, 15, 2384–2390 CrossRef CAS.
- N. Rudra-Ganguly, P. M. Challita-Eid, C. Lowe, M. Mattie, S.-J. Moon, B. A. Mendelsohn, M. Leavitt, C. Virata, A. Verlinsky, L. Capo, M. S. Chang, D. L. Russell, B. Randhawa, G. Liu, R. Hubert, M. Brodey, H. Aviña, C. Zhang, J. D. Abad, B. Anand, S. Karki, Z. An, R. Luethy, F. Doñate, D. S. Pereira, K. Morrison, I. B. J. Joseph and D. R. Stover, Cancer Res., 2016, 76, 574 Search PubMed.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS.
- L. Harris, D. Tavares, L. Rui, E. Maloney, A. Wilhelm, J. Costoplus, K. Archer, M. Bogalhas, L. Harvey, R. Wu, X. Chen, X. Xu, S. Connaughton, L. Wang, K. Whiteman, O. Ab, E. Hong, W. Widdison, M. Shizuka, M. Miller, J. Pinkas, T. Keating, R. Chari and N. Fishkin, Cancer Res., 2015, 75, 647 Search PubMed.
- D. Vitharana, A. Wilhelm, L. Harris, K. Archer, M. Shizuka, E. Maloney, O. Ab, R. Laleau, X. Sun, J. Pinkas, M. Miller, R. Chari, T. Keating and N. Fishkin, Cancer Res., 2016, 76, 2965 Search PubMed.
- P. Thompson, B. Bezabeh, R. Fleming, M. Pruitt, S. Mao, P. Strout, C. Chen, S. Cho, H. Zhong, H. Wu, C. Gao and N. Dimasi, Bioconjugate Chem., 2015, 26, 2085–2096 CrossRef CAS.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS.
- C. Zhang, P. Dai, A. A. Vinogradov, Z. P. Gates and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 6459–6463 CrossRef CAS.
- Z. Dai, X. N. Zhang, F. Nasertorabi, Q. Cheng, J. Li, B. B. Katz, G. Smbatyan, H. Pei, S. G. Louie, H. J. Lenz, R. C. Stevens, Y. Zhang, Y. Zhang, Y. Zhang and Y. Zhang, Sci. Adv., 2020, 6, eaba6752 CrossRef CAS.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- C. Rader, S. C. Sinha, M. Popkov, R. A. Lerner and C. F. Barbas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5396–5400 CrossRef CAS.
- A. R. Nanna, X. Li, E. Walseng, L. Pedzisa, R. S. Goydel, D. Hymel, T. R. Burke, W. R. Roush and C. Rader, Nat. Commun., 2017, 8, 1112 CrossRef.
- A. R. Nanna, A. V. Kel’in, C. Theile, J. M. Pierson, Z. X. Voo, A. Garg, J. K. Nair, M. A. Maier, K. Fitzgerald and C. Rader, Nucleic Acids Res., 2020, 48, 5281–5293 CrossRef.
- D. Hwang, K. Tsuji, H. Park, T. R. Burke and C. Rader, Bioconjugate Chem., 2019, 30, 2889–2896 CrossRef CAS.
- D. Hwang and C. Rader, Biomolecules, 2020, 10, 764 CrossRef CAS.
- M. R. Mortensen, M. B. Skovsgaard, A. H. Okholm, C. Scavenius, D. M. Dupont, C. B. Rosen, J. J. Enghild, J. Kjems and K. V. Gothelf, Bioconjugate Chem., 2018, 29, 3016–3025 CrossRef CAS.
- M. B. Skovsgaard, T. E. Jeppesen, M. R. Mortensen, C. H. Nielsen, J. Madsen, A. Kjaer and K. V. Gothelf, Bioconjugate Chem., 2019, 30, 881–887 CrossRef CAS.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS.
- S. R. Adusumalli, D. G. Rawale, K. Thakur, L. Purushottam, N. C. Reddy, N. Kalra, S. Shukla and V. Rai, Angew. Chem., Int. Ed., 2020, 59, 10332–10336 CrossRef CAS.
- I. Dovgan, S. Erb, S. Hessmann, S. Ursuegui, C. Michel, C. Muller, G. Chaubet, S. Cianférani and A. Wagner, Org. Biomol. Chem., 2018, 16, 1305–1311 RSC.
- D. Hwang, N. Nilchan, A. R. Nanna, X. Li, M. D. Cameron, W. R. Roush, H. J. Park and C. Rader, Cell Chem. Biol., 2019, 26, 1229 CrossRef CAS.
- C. Sornay, S. Hessmann, S. Erb, I. Dovgan, A. Ehkirch, T. Botzanowski, S. Cianférani, A. Wagner and G. Chaubet, Chem. – Eur. J., 2020, 26, 13797–13805, DOI:10.1002/chem.202002432.
- B. Nilsson, T. Moks, B. Jansson, L. Abrahmsén, A. Elmblad, E. Holmgren, C. Henrichson, T. A. Jones and M. Uhlén, Protein Eng., 1987, 1, 107–113 CrossRef CAS.
- Y. Mazor, I. Barnea, I. Keydar and I. Benhar, J. Immunol. Methods, 2007, 321, 41–59 CrossRef CAS.
- Y. Mazor, R. Noy, W. S. Wels and I. Benhar, Cancer Lett., 2007, 257, 124–135 CrossRef CAS.
- S. Shapira, A. Shapira, A. Starr, D. Kazanov, S. Kraus, I. Benhar and N. Arber, Gastroenterology, 2011, 140, 935–946 CrossRef CAS.
- I. Barnea, R. Ben-Yosef, V. Karaush, I. Benhar and A. Vexler, Head Neck, 2013, 35, 1171–1177 CrossRef.
- J. Park, Y. Lee, B. J. Ko and T. H. Yoo, Bioconjugate Chem., 2018, 29, 3240–3244 CrossRef CAS.
- N. Vance, N. Zacharias, M. Ultsch, G. Li, A. Fourie, P. Liu, J. LaFrance-Vanasse, J. A. Ernst, W. Sandoval, K. R. Kozak, G. Phillips, W. Wang and J. Sadowsky, Bioconjugate Chem., 2019, 30, 148–160 CrossRef CAS.
- S. Kishimoto, Y. Nakashimada, R. Yokota, T. Hatanaka, M. Adachi and Y. Ito, Bioconjugate Chem., 2019, 30, 698–702 CrossRef CAS.
- C. Yu, J. Tang, A. Loredo, Y. Chen, S. Y. Jung, A. Jain, A. Gordon and H. Xiao, Bioconjugate Chem., 2018, 29, 3522–3526 CrossRef CAS.
- J. Ohata and Z. T. Ball, J. Am. Chem. Soc., 2017, 139, 12617–12622 CrossRef CAS.
- B. V. Popp and Z. T. Ball, Chem. Sci., 2011, 2, 690–695 RSC.
- K. Yamada, N. Shikida, K. Shimbo, Y. Ito, Z. Khedri, Y. Matsuda and B. A. Mendelsohn, Angew. Chem., Int. Ed., 2019, 58, 5592–5597 CrossRef CAS.
- Y. Matsuda, V. Robles, M. C. Malinao, J. Song and B. A. Mendelsohn, Anal. Chem., 2019, 91, 12724–12732 CrossRef CAS.
- Y. Matsuda, C. Clancy, Z. Tawfiq, V. Robles and B. A. Mendelsohn, ACS Omega, 2019, 4, 20564–20570 CrossRef CAS.
- Y. Matsuda, K. Yamada, T. Okuzumi and B. A. Mendelsohn, Org. Process Res. Dev., 2019, 23, 2647–2654 CrossRef CAS.
- Y. Matsuda, M. Leung, T. Okuzumi and B. Mendelsohn, Antibodies, 2020, 9, 16 CrossRef CAS.
- Y. Matsuda, M. C. Malinao, V. Robles, J. Song, K. Yamada and B. A. Mendelsohn, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1140, 121981 CrossRef CAS.
- N. Gupta, A. Ansari, G. V. Dhoke, M. Chilamari, J. Sivaccumar, S. Kumari, S. Chatterjee, R. Goyal, P. K. Dutta, M. Samarla, M. Mukherjee, A. Sarkar, S. K. Mandal, V. Rai, G. Biswas, A. Sengupta, S. Roy, M. Roy and S. Sengupta, Nat. Biomed. Eng., 2019, 3, 917–929 CrossRef CAS.
- L. N. Lund, P. E. Gustavsson, R. Michael, J. Lindgren, L. Nørskov-Lauritsen, M. Lund, G. Houen, A. Staby and P. M. Hilaire, J. Chromatogr. A, 2012, 1225, 158–167 CrossRef CAS.
- D. Q. Lin, H. F. Tong, H. Y. Wang and S. J. Yao, J. Phys. Chem. B, 2012, 116, 1393–1400 CrossRef CAS.
- M. Farràs, J. Miret, M. Camps, R. Román, Ó. Martínez, X. Pujol, S. Erb, A. Ehkirch, S. Cianferani, A. Casablancas and J. J. Cairó, mAbs, 2020, 12, 1702262 CrossRef.
- H. Schneider, L. Deweid, O. Avrutina and H. Kolmar, Anal. Biochem., 2020, 595, 113615 CrossRef CAS.
- M. Griffin, R. Casadio and C. M. Bergamini, Biochem. J., 2002, 368, 377–396 CrossRef CAS.
- H. Ando, M. Adachi, K. Umeda, A. Matsuura, M. Nonaka, R. Uchio, H. Tanaka and M. Motoki, Agric. Biol. Chem., 1989, 53, 2613–2617 CAS.
- H. S. Mostafa, Biocatal. Biotransformation, 2020, 38, 161–177 CrossRef CAS.
- P. Strop, Bioconjugate Chem., 2014, 25, 855–862 CrossRef CAS.
- A. Josten, L. Haalck, F. Spener and M. Meusel, J. Immunol. Methods, 2000, 240, 47–54 CrossRef CAS.
- A. Josten, M. Meusel and F. Spener, Anal. Biochem., 1998, 258, 202–208 CrossRef CAS.
- T. L. Mindt, V. Jungi, S. Wyss, A. Friedli, G. Pla, I. Novak-Hofer, J. Grünberg and R. Schibli, Bioconjugate Chem., 2008, 19, 271–278 CrossRef CAS.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS.
- S. Matsumiya, Y. Yamaguchi, J. ichi Saito, M. Nagano, H. Sasakawa, S. Otaki, M. Satoh, K. Shitara and K. Kato, J. Mol. Biol., 2007, 368, 767–779 CrossRef CAS.
- M. J. Feige, S. Nath, S. R. Catharino, D. Weinfurtner, S. Steinbacher and J. Buchner, J. Mol. Biol., 2009, 391, 599–608 CrossRef CAS.
- S. Krapp, Y. Mimura, R. Jefferis, R. Huber and P. Sondermann, J. Mol. Biol., 2003, 325, 979–989 CrossRef CAS.
- J. Grünberg, S. Jeger, D. Sarko, P. Dennler, K. Zimmermann, W. Mier and R. Schibli, PLoS One, 2013, 8, e60350 CrossRef.
- P. Dennler, A. Chiotellis, E. Fischer, D. Brégeon, C. Belmant, L. Gauthier, F. Lhospice, F. Romagne and R. Schibli, Bioconjugate Chem., 2014, 25, 569–578 CrossRef CAS.
- P. Dennler, R. Schibli and E. Fischer, Methods Mol. Biol., 2013, 1045, 205–215 CrossRef.
- N. Bodyak and A. V. Yurkovetskiy, Innovations for Next-Generation Antibody-Drug Conjugates, 2018, pp. 215–240 Search PubMed.
- Y. Anami, W. Xiong, X. Gui, M. Deng, C. C. Zhang, N. Zhang, Z. An and K. Tsuchikama, Org. Biomol. Chem., 2017, 15, 5635–5642 RSC.
- Y. Anami, C. M. Yamazaki, W. Xiong, X. Gui, N. Zhang, Z. An and K. Tsuchikama, Nat. Commun., 2018, 9, 2512 CrossRef.
- M. J. Costa, J. Kudaravalli, J. T. Ma, W. H. Ho, K. Delaria, C. Holz, A. Stauffer, A. G. Chunyk, Q. Zong, E. Blasi, B. Buetow, T. T. Tran, K. Lindquist, M. Dorywalska, A. Rajpal, D. L. Shelton, P. Strop and S. H. Liu, Sci. Rep., 2019, 9, 2443 CrossRef.
- D. N. Thornlow, E. C. Cox, J. A. Walker, M. Sorkin, J. B. Plesset, M. P. Delisa and C. A. Alabi, Bioconjugate Chem., 2019, 30, 1702–1710 CrossRef CAS.
- J. A. Walker, J. J. Bohn, F. Ledesma, M. R. Sorkin, S. R. Kabaria, D. N. Thornlow and C. A. Alabi, Bioconjugate Chem., 2019, 30, 2452–2457 CrossRef CAS.
- S. Puthenveetil, S. Musto, F. Loganzo, L. N. Tumey, C. J. O’Donnell and E. Graziani, Bioconjugate Chem., 2016, 27, 1030–1039 CrossRef CAS.
- S. Beck, J. Schultze, H. J. Räder, R. Holm, M. Schinnerer, M. Barz, K. Koynov and R. Zentel, Polymers, 2018, 10, 141 CrossRef.
- A. Sadiki, E. M. Kercher, H. Lu, R. T. Lang, B. Q. Spring and Z. S. Zhou, Photochem. Photobiol., 2020, 96, 596–603 CrossRef CAS.
- J. Park, P. L. Chariou and N. F. Steinmetz, Bioconjugate Chem., 2020, 31, 1408–1416 CrossRef CAS.
- C. Marculescu, A. Lakshminarayanan, J. Gault, J. C. Knight, L. K. Folkes, T. Spink, C. V. Robinson, K. Vallis, B. G. Davis and B. Cornelissen, Chem. Commun., 2019, 55, 11342–11345 RSC.
- E. B. Irvine and G. Alter, Glycobiology, 2020, 30, 241–253 CrossRef.
- D. Reusch and M. L. Tejada, Glycobiology, 2015, 25, 1325–1334 CrossRef CAS.
- K. Zheng, C. Bantog and R. Bayer, mAbs, 2011, 3, 568–576 CrossRef.
- S. Dickgiesser, M. Rieker, D. Mueller-Pompalla, C. Schröter, J. Tonillo, S. Warszawski, S. Raab-Westphal, S. Kühn, T. Knehans, D. Könning, J. Dotterweich, U. A. K. Betz, J. Anderl, S. Hecht and N. Rasche, Bioconjugate Chem., 2020, 31, 1070–1076 CrossRef CAS.
- S. E. Farias, P. Strop, K. Delaria, M. Galindo Casas, M. Dorywalska, D. L. Shelton, J. Pons and A. Rajpal, Bioconjugate Chem., 2014, 25, 240–250 CrossRef CAS.
- I. J. Huggins, C. A. Medina, A. D. Springer, A. Van Den Berg, S. Jadhav, X. Cui and S. F. Dowdy, Molecules, 2019, 24, 3287 CrossRef CAS.
- P. Strop, K. Delaria, D. Foletti, J. M. Witt, A. Hasa-Moreno, K. Poulsen, M. G. Casas, M. Dorywalska, S. Farias, A. Pios, V. Lui, R. Dushin, D. Zhou, T. Navaratnam, T.-T. Tran, J. Sutton, K. C. Lindquist, B. Han, S.-H. Liu, D. L. Shelton, J. Pons and A. Rajpal, Nat. Biotechnol., 2015, 33, 694–696 CrossRef CAS.
- H. Schneider, L. Deweid, T. Pirzer, D. Yanakieva, S. Englert, B. Becker, O. Avrutina and H. Kolmar, ChemistryOpen, 2019, 8, 354–357 CrossRef CAS.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. Galindo Casas, K. Delaria, V. Lui, K. Poulsen, J. Sutton, G. Bolton, D. Zhou, L. Moine, R. Dushin, T.-T. Tran, S.-H. Liu, M. Rickert, D. Foletti, D. L. Shelton, J. Pons and A. Rajpal, PLoS One, 2015, 10, e0132282 CrossRef.
- O. K. Wong, T. T. Tran, W. H. Ho, M. G. Casas, M. Au, M. Bateman, K. C. Lindquist, A. Rajpal, D. L. Shelton, P. Strop and S. H. Liu, Oncotarget, 2018, 9, 33446–33458 CrossRef.
- P. Strop, T.-T. Tran, M. Dorywalska, K. Delaria, R. Dushin, O. K. Wong, W.-H. Ho, D. Zhou, A. Wu, E. Kraynov, L. Aschenbrenner, B. Han, C. J. O’Donnell, J. Pons, A. Rajpal, D. L. Shelton and S.-H. Liu, Mol. Cancer Ther., 2016, 15, 2698–2708 CrossRef CAS.
- G. T. King, K. D. Eaton, B. R. Beagle, C. J. Zopf, G. Y. Wong, H. I. Krupka, S. Y. Hua, W. A. Messersmith and A. B. El-Khoueiry, Invest. New Drugs, 2018, 36, 836–847 CrossRef CAS.
- A. S. Ratnayake, L. P. Chang, L. N. Tumey, F. Loganzo, J. A. Chemler, M. Wagenaar, S. Musto, F. Li, J. E. Janso, T. E. Ballard, B. Rago, G. L. Steele, W. Ding, X. Feng, C. Hosselet, V. Buklan, J. Lucas, F. E. Koehn, C. J. O’Donnell and E. I. Graziani, Bioconjugate Chem., 2019, 30, 200–209 CrossRef CAS.
- V. Siegmund, S. Schmelz, S. Dickgiesser, J. Beck, A. Ebenig, H. Fittler, H. Frauendorf, B. Piater, U. A. K. Betz, O. Avrutina, A. Scrima, H. L. Fuchsbauer and H. Kolmar, Angew. Chem., Int. Ed., 2015, 54, 13420–13424 CrossRef CAS.
- A. Ebenig, N. E. Juettner, L. Deweid, O. Avrutina, H. Fuchsbauer and H. Kolmar, ChemBioChem, 2019, 20, 2411–2419 CrossRef CAS.
- S. Yamazoe, J. M. Hogan, S. M. West, X. A. Deng, S. Kotapati, X. Shao, P. Holder, V. Lamba, M. Huber, C. Qiang, S. Gangwar, C. Rao, G. Dollinger, A. Rajpal and P. Strop, Bioconjugate Chem., 2020, 31, 1199–1208 CrossRef CAS.
- H. Abe, M. Goto and N. Kamiya, Chem. Commun., 2010, 46, 7160 RSC.
- P. R. Spycher, C. A. Amann, J. E. Wehrmüller, D. R. Hurwitz, O. Kreis, D. Messmer, A. Ritler, A. Küchler, A. Blanc, M. Béhé, P. Walde and R. Schibli, ChemBioChem, 2017, 18, 1923–1927 CrossRef CAS.
- J. L. Spidel, B. Vaessen, E. F. Albone, X. Cheng, A. Verdi and J. B. Kline, Bioconjugate Chem., 2017, 28, 2471–2484 CrossRef CAS.
- J. L. Spidel and E. F. Albone, Methods in Molecular Biology, Humana Press Inc., 2019, vol. 2033, pp. 53–65 Search PubMed.
- T. I. Chio, B. R. Demestichas, B. M. Brems, S. L. Bane and L. N. Tumey, Angew. Chem., Int. Ed., 2020, 59, 13814–13820 CrossRef CAS.
- S. R. Benjamin, C. P. Jackson, S. Fang, D. P. Carlson, Z. Guo and L. N. Tumey, Mol. Pharm., 2019, 16, 2795–2807 CrossRef CAS.
- K. Yokoyama, H. Utsumi, T. Nakamura, D. Ogaya, N. Shimba, E. Suzuki and S. Taguchi, Appl. Microbiol. Biotechnol., 2010, 87, 2087–2096 CrossRef CAS.
- L. Deweid, L. Neureiter, S. Englert, H. Schneider, J. Deweid, D. Yanakieva, J. Sturm, S. Bitsch, A. Christmann, O. Avrutina, H. Fuchsbauer and H. Kolmar, Chem. – Eur. J., 2018, 24, 15195–15200 CrossRef CAS.
- K. Buettner, T. C. Hertel and M. Pietzsch, Amino Acids, 2012, 42, 987–996 CrossRef CAS.
- K. Ohtake, T. Mukai, F. Iraha, M. Takahashi, K. I. Haruna, M. Date, K. Yokoyama and K. Sakamoto, ACS Synth. Biol., 2018, 7, 2170–2176 CrossRef CAS.
- B. Böhme, B. Moritz, J. Wendler, T. C. Hertel, C. Ihling, W. Brandt and M. Pietzsch, Amino Acids, 2020, 52, 313–326 CrossRef.
- W. Steffen, F. C. Ko, J. Patel, V. Lyamichev, T. J. Albert, J. Benz, M. G. Rudolph, F. Bergmann, T. Streidl, P. Kratzsch, M. Boenitz-Dulat, T. Oelschlaegel and M. Schraeml, J. Biol. Chem., 2017, 292, 15622–15635 CrossRef CAS.
- T. Kashiwagi, K. ichi Yokoyama, K. Ishikawa, K. Ono, D. Ejima, H. Matsui and E. ichiro Suzuki, J. Biol. Chem., 2002, 277, 44252–44260 CrossRef CAS.
- D. York, J. Baker, P. G. Holder, L. C. Jones, P. M. Drake, R. M. Barfield, G. T. Bleck and D. Rabuka, BMC Biotechnol., 2016, 16, 23 CrossRef.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS.
- P. M. Drake, A. E. Albers, J. Baker, S. Banas, R. M. Barfield, A. S. Bhat, G. W. de Hart, A. W. Garofalo, P. Holder, L. C. Jones, R. Kudirka, J. McFarland, W. Zmolek and D. Rabuka, Bioconjugate Chem., 2014, 25, 1331–1341 CrossRef CAS.
- P. M. Drake, A. Carlson, J. M. McFarland, S. Bañas, R. M. Barfield, W. Zmolek, Y. C. Kim, B. C. B. Huang, R. Kudirka and D. Rabuka, Mol. Cancer Ther., 2018, 17, 161–168 CrossRef CAS.
- A. P. Maclaren, N. Levin and H. Lowman, Experimental and Molecular Therapeutics, American Association for Cancer Research, 2018, vol. 78, pp. 835–835 Search PubMed.
- Triphase Accelerator and Catalent Announce Interim Results of a Dose Escalation Phase 1 Clinical Trial of TRPH-222 in Patients with Non-Hodgkin's Lymphoma, https://www.prweb.com/releases/triphase_accelerator_and_catalent_announce_interim_results_of_a_dose_escalation_phase_1_clinical_trial_of_trph_222_in_patients_with_non_hodgkins_lymphoma/prweb17129205.htm, accessed 16 July 2020.
- R. Barfield, Y. C. Kim, S. Chuprakov, F. Zhang, M. Bauzon, A. Ogunkoya, D. Yeo, C. Hickle, M. Pegram, D. Rabuka and P. Drake, Mol. Cancer Ther., 2020, 19, 1866–1874, DOI:10.1101/2020.03.13.991448.
- S. Pomplun, M. Y. H. Mohamed, T. Oelschlaegel, C. Wellner and F. Bergmann, Angew. Chem., Int. Ed., 2019, 58, 3542–3547 CrossRef CAS.
- R. Kudirka, R. M. Barfield, J. McFarland, A. E. Albers, G. W. de Hart, P. M. Drake, P. G. Holder, S. Banas, L. C. Jones, A. W. Garofalo and D. Rabuka, Chem. Biol., 2015, 22, 293–298 CrossRef CAS.
- R. A. Kudirka, R. M. Barfield, J. M. McFarland, P. M. Drake, A. Carlson, S. Bañas, W. Zmolek, A. W. Garofalo and D. Rabuka, ACS Med. Chem. Lett., 2016, 7, 994–998 CrossRef CAS.
- M. Rashidian, J. M. Song, R. E. Pricer and M. D. Distefano, J. Am. Chem. Soc., 2012, 134, 8455–8467 CrossRef CAS.
- Y. Zhang, M. J. Blanden, C. Sudheer, S. A. Gangopadhyay, M. Rashidian, J. L. Hougland and M. D. Distefano, Bioconjugate Chem., 2015, 26, 2542–2553 CrossRef CAS.
- J. Lee, H.-J. Choi, M. Yun, Y. Kang, J.-E. Jung, Y. Ryu, T. Y. Kim, Y. Cha, H.-S. Cho, J.-J. Min, C.-W. Chung and H.-S. Kim, Angew. Chem., Int. Ed., 2015, 54, 12020–12024 CrossRef CAS.
- Y. Kim, T. Park, S. Woo, H. Lee, S. Kim, J. Cho, D. Jung, Y. Kim, H. Kwon, K. Oh, Y. Chung and Y. Park, Antibody-active agent conjugates and methods of use, US20150105541A1, USA, 2015 Search PubMed.
- B. ill Lee, M.-H. Park, J.-J. Byeon, S.-H. Shin, J. Choi, Y. Park, Y.-H. Park, J. Chae and Y. G. Shin, Molecules, 2020, 25, 1515 CrossRef.
- R. R. Beerli, T. Hell, A. S. Merkel and U. Grawunder, PLoS One, 2015, 10, e0131177 CrossRef.
- N. Stefan, R. Gébleux, L. Waldmeier, T. Hell, M. Escher, F. I. Wolter, U. Grawunder and R. R. Beerli, Mol. Cancer Ther., 2017, 16, 879–892 CrossRef CAS.
- L. D’Amico, U. Menzel, M. Prummer, P. Müller, M. Buchi, A. Kashyap, U. Haessler, A. Yermanos, R. Gébleux, M. Briendl, T. Hell, F. I. Wolter, R. R. Beerli, I. Truxova, Š. Radek, T. Vlajnic, U. Grawunder, S. Reddy and A. Zippelius, J. Immunother. Cancer, 2019, 7, 16 CrossRef.
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, Bioconjugate Chem., 2018, 29, 3245–3249 CrossRef CAS.
- M. D. Lee, W. Y. Tong, T. Nebl, L. A. Pearce, T. M. Pham, A. Golbaz-Hagh, S. Puttick, S. Rose, T. E. Adams and C. C. Williams, Bioconjugate Chem., 2019, 30, 2539–2543 CrossRef CAS.
- T. Pirzer, K.-S. Becher, M. Rieker, T. Meckel, H. D. Mootz and H. Kolmar, ACS Chem. Biol., 2018, 13, 2058–2066 CrossRef CAS.
- V. Siegmund, B. Piater, B. Zakeri, T. Eichhorn, F. Fischer, C. Deutsch, S. Becker, L. Toleikis, B. Hock, U. A. K. Betz and H. Kolmar, Sci. Rep., 2016, 6, 39291 CrossRef CAS.
- J. J. Bruins, A. H. Westphal, B. Albada, K. Wagner, L. Bartels, H. Spits, W. J. H. van Berkel and F. L. van Delft, Bioconjugate Chem., 2017, 28, 1189–1193 CrossRef CAS.
- S. Sato, M. Matsumura, T. Kadonosono, S. Abe, T. Ueno, H. Ueda and H. Nakamura, Bioconjugate Chem., 2020, 31, 1417–1424 CrossRef CAS.
- J. Grünewald, H. E. Klock, S. E. Cellitti, B. Bursulaya, D. McMullan, D. H. Jones, H. P. Chiu, X. Wang, P. Patterson, H. Zhou, J. Vance, E. Nigoghossian, H. Tong, D. Daniel, W. Mallet, W. Ou, T. Uno, A. Brock, S. A. Lesley and B. H. Geierstanger, Bioconjugate Chem., 2015, 26, 2554–2562 CrossRef.
- J. Grünewald, Y. Jin, J. Vance, J. Read, X. Wang, Y. Wan, H. Zhou, W. Ou, H. E. Klock, E. C. Peters, T. Uno, A. Brock and B. H. Geierstanger, Bioconjugate Chem., 2017, 28, 1906–1915 CrossRef.
- M. Pučić, A. Knežević, J. Vidič, B. Adamczyk, M. Novokmet, O. Polašek, O. Gornik, S. Šupraha-Goreta, M. R. Wormald, I. Redžic, H. Campbell, A. Wright, N. D. Hastie, J. F. Wilson, I. Rudan, M. Wuhrer, P. M. Rudd, D. Josić and G. Lauc, Mol. Cell. Proteomics, 2011, 10, 1–15 CrossRef.
- M. Wuhrer, J. C. Stam, F. E. Van De Geijn, C. A. M. Koeleman, C. T. Verrips, R. J. E. M. Dolhain, C. H. Hokke and A. M. Deelder, Proteomics, 2007, 7, 4070–4081 CrossRef CAS.
- L.-X. Wang, X. Tong, C. Li, J. P. Giddens and T. Li, Annu. Rev. Biochem., 2019, 88, 433–459 CrossRef CAS.
- A. Morell, G. Ashwell, A. Irvine, I. Sternlieb and I. H. Cheinberg, J. Biol. Chem., 1968, 243, 155–159 CAS.
- D. J. O’Shannessy, M. J. Dobersen and R. H. Quarles, Immunol. Lett., 1984, 8, 273–277 CrossRef.
- M.-M. Chua, S.-T. Fan and F. Karush, BBA, Biochim. Biophys. Acta, Gen. Subj., 1984, 800, 291–300 CrossRef CAS.
- B. C. Laguzza, J. J. Starling, A. L. Baker, T. F. Bumol, J. R. F. Corvalan, C. L. Nichols, S. L. Briggs, G. J. Cullinan and D. A. Johnson, J. Med. Chem., 1989, 32, 548–555 CrossRef CAS.
- L. M. Hinman, P. R. Hamann, R. Wallace, A. T. Menendez, F. E. Durr and J. Upeslacis, Cancer Res., 1993, 53, 3336–3342 CAS.
- A. C. Stan, D. L. Radu, S. Casares, C. A. Bona and T. D. Brumeanu, Cancer Res., 1999, 59, 115–121 CAS.
- K. Zuberbühler, G. Casi, G. J. L. Bernardes and D. Neri, Chem. Commun., 2012, 48, 7100–7102 RSC.
- C. Vaccaro, J. Zhou, R. J. Ober and E. S. Ward, Nat. Biotechnol., 2005, 23, 1283–1288 CrossRef CAS.
- W. Wang, J. Vlasak, Y. Li, P. Pristatsky, Y. Fang, T. Pittman, J. Roman, Y. Wang, T. Prueksaritanont and R. Ionescu, Mol. Immunol., 2011, 48, 860–866 CrossRef CAS.
- Q. Zhou, J. E. Stefano, C. Manning, J. Kyazike, B. Chen, D. A. Gianolio, A. Park, M. Busch, J. Bird, X. Zheng, H. Simonds-Mannes, J. Kim, R. C. Gregory, R. J. Miller, W. H. Brondyk, P. K. Dhal and C. Q. Pan, Bioconjugate Chem., 2014, 25, 510–520 CrossRef CAS.
- Faridoon, W. Shi, K. Qin, Y. Tang, M. Li, D. Guan, X. Tian, B. Jiang, J. Dong, F. Tang and W. Huang, Org. Chem. Front., 2019, 6, 3144–3149 RSC.
- J. Garbe and M. Collin, J. Innate Immun., 2012, 4, 121–131 CrossRef CAS.
- P. W. Robbins, R. B. Trimble, D. F. Wirth, C. Hering, F. Maley, G. F. Maley, R. Das, B. W. Gibson, N. Royal and K. Biemann, J. Biol. Chem., 1984, 259, 7577–7583 CAS.
- N. Takahashi, Biochem. Biophys. Res. Commun., 1977, 76, 1194–1201 CrossRef CAS.
- J. Sjögren, R. Lood and A. Nägeli, Glycobiology, 2019, 30, 254–267 CrossRef.
- Y. Wei, C. Li, W. Huang, B. Li, S. Strome and L. X. Wang, Biochemistry, 2008, 47, 10294–10304 CrossRef CAS.
- J. J. Goodfellow, K. Baruah, K. Yamamoto, C. Bonomelli, B. Krishna, D. J. Harvey, M. Crispin, C. N. Scanlan and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 8030–8033 CrossRef CAS.
- W. Huang, J. Giddens, S. Q. Fan, C. Toonstra and L. X. Wang, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef CAS.
- M. Kurogochi, M. Mori, K. Osumi, M. Tojino, S. Sugawara, S. Takashima, Y. Hirose, W. Tsukimura, M. Mizuno, J. Amano, A. Matsuda, M. Tomita, A. Takayanagi, S.-I. Shoda and T. Shirai, PLoS One, 2015, 10, e0132848 CrossRef.
- C. W. Lin, M. H. Tsai, S. T. Li, T. I. Tsai, K. C. Chu, Y. C. Liu, M. Y. Lai, C. Y. Wu, Y. C. Tseng, S. S. Shivatare, C. H. Wang, P. Chao, S. Y. Wang, H. W. Shih, Y. F. Zeng, T. H. You, J. Y. Liao, Y. C. Tu, Y. S. Lin, H. Y. Chuang, C. L. Chen, C. S. Tsai, C. C. Huang, N. H. Lin, C. Ma, C. Y. Wu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10611–10616 CrossRef CAS.
- T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361–2367 CrossRef CAS.
- S. Manabe, Y. Yamaguchi, K. Matsumoto, H. Fuchigami, T. Kawase, K. Hirose, A. Mitani, W. Sumiyoshi, T. Kinoshita, J. Abe, M. Yasunaga, Y. Matsumura and Y. Ito, Bioconjugate Chem., 2019, 30, 1343–1355 CrossRef CAS.
- J. Sjögren, W. B. Struwe, E. F. J. Cosgrave, P. M. Rudd, M. Stervander, M. Allhorn, A. Hollands, V. Nizet and M. Collin, Biochem. J., 2013, 455, 107–118 CrossRef.
- T. Li, X. Tong, Q. Yang, J. P. Giddens and L. X. Wang, J. Biol. Chem., 2016, 291, 16508–16518 CrossRef CAS.
- S. S. Shivatare, L. Y. Huang, Y. F. Zeng, J. Y. Liao, T. H. You, S. Y. Wang, T. Cheng, C. W. Chiu, P. Chao, L. T. Chen, T. I. Tsai, C. C. Huang, C. Y. Wu, N. H. Lin and C. H. Wong, Chem. Commun., 2018, 54, 6161–6164 RSC.
- J. P. Giddens, J. V. Lomino, M. N. Amin and L. X. Wang, J. Biol. Chem., 2016, 291, 9356–9370 CrossRef CAS.
- C. P. Liu, T. I. Tsai, T. Cheng, V. S. Shivatare, C. Y. Wu, C. Y. Wu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 720–725 CrossRef CAS.
- S. Manabe, Y. Yamaguchi, J. Abe, K. Matsumoto and Y. Ito, R. Soc. Open Sci., 2018, 5, 171521 CrossRef.
- M. Iwamoto, Y. Sekiguchi, K. Nakamura, Y. Kawaguchi, T. Honda and J. Hasegawa, PLoS One, 2018, 13, e0193534 CrossRef.
- F. Tang, Y. Yang, Y. Tang, S. Tang, L. Yang, B. Sun, B. Jiang, J. Dong, H. Liu, M. Huang, M. Y. Geng and W. Huang, Org. Biomol. Chem., 2016, 14, 9501–9518 RSC.
- B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2002, 277, 20833–20839 CrossRef CAS.
- B. Ramakrishnan, P. S. Shah and P. K. Qasba, J. Biol. Chem., 2001, 276, 37665–37671 CrossRef CAS.
- E. Boeggeman, B. Ramakrishnan, M. Pasek, M. Manzoni, A. Puri, K. H. Loomis, T. J. Waybright and P. K. Qasba, Bioconjugate Chem., 2009, 20, 1228–1236 CrossRef CAS.
- Z. Zhu, B. Ramakrishnan, J. Li, Y. Wang, Y. Feng, P. Prabakaran, S. Colantonio, M. A. Dyba, P. K. Qasba and D. S. Dimitrov, mAbs, 2014, 6, 1190–1200 CrossRef.
- P. Thompson, E. Ezeadi, I. Hutchinson, R. Fleming, B. Bezabeh, J. Lin, S. Mao, C. Chen, L. Masterson, H. Zhong, D. Toader, P. Howard, H. Wu, C. Gao and N. Dimasi, ACS Med. Chem. Lett., 2016, 7, 1005–1008 CrossRef CAS.
- R. van Geel, M. A. Wijdeven, R. Heesbeen, J. M. M. Verkade, A. A. Wasiel, S. S. van Berkel and F. L. van Delft, Bioconjugate Chem., 2015, 26, 2233–2242 CrossRef CAS.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 CrossRef CAS.
- B. M. Zeglis, C. B. Davis, D. Abdel-Atti, S. D. Carlin, A. Chen, R. Aggeler, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2014, 25, 2123–2128 CrossRef CAS.
- P. Adumeau, D. Vivier, S. K. Sharma, J. Wang, T. Zhang, A. Chen, B. J. Agnew and B. M. Zeglis, Mol. Pharm., 2018, 15, 892–898 CrossRef CAS.
- N. M. Okeley, B. E. Toki, X. Zhang, S. C. Je, P. J. Burke, S. C. Alley and P. D. Senter, Bioconjugate Chem., 2013, 24, 1650–1655 CrossRef CAS.
- R. Neupane and J. Bergquist, Eur. J. Mass Spectrom., 2017, 23, 417–426 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |