
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide-based coacervates as biomimetic protocells
Manzar
Abbas†
 ,
Wojciech P.
Lipiński†
,
Jiahua
Wang
and
Evan
Spruijt
*
,
Wojciech P.
Lipiński†
,
Jiahua
Wang
and
Evan
Spruijt
*
Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: e.spruijt@science.ru.nl
First published on 22nd February 2021
Abstract
Coacervates are condensed liquid-like droplets formed by liquid–liquid phase separation of molecules through multiple weak associative interactions. In recent years it has emerged that not only long polymers, but also short peptides are capable of forming simple and complex coacervates. The coacervate droplets they form act as compartments that sequester and concentrate a wide range of solutes, and their spontaneous formation make coacervates attractive protocell models. The main advantage of peptides as building blocks lies in the functional diversity of the amino acid residues, which allows for tailoring of the peptide's phase separation propensity, their selectivity in guest molecule uptake and the physicochemical and catalytic properties of the compartments. The aim of this tutorial review is to illustrate the recent developments in the field of peptide-based coacervates in a systematic way and to deduce the basic requirements for both simple and complex coacervation of peptides. We review a selection of peptide coacervates that illustrates the essentials of phase separation, the limitations, and the properties that make peptide coacervates biomimetic protocells. Finally, we provide some perspectives of this novel research field in the direction of active droplets, moving away from thermodynamic equilibrium.
 Manzar Abbas | Manzar Abbas obtained his PhD degree from the Institute of Process Engineering, Chinese Academy of Sciences, (CAS) in 2017. Currently, he is a Marie Curie Individual fellow at the Institute for Molecules and Materials, Radboud University, the Netherlands. He is working in the Coacervates and Soft Interfaces group. His research interests include the design of functional short peptide building blocks, self-assembly, liquid–liquid phase separation of short peptides for coacervates and biomimetic protocells, and drug delivery for phototherapy. |
 Wojciech P. Lipiński | Wojciech (Wojtek) Lipiński obtained his master degree in chemistry from Łódź University of Technology (Poland). His master internship focused on activatable cell penetrating peptides for drug delivery. He is currently working as a PhD candidate at the Radboud University Nijmegen in the group of Coacervates and Soft Interfaces, studying liquid–liquid phase separation of simple peptide derivatives and liquid-to-solid transition of model membraneless organelles. |
 Jiahua Wang | Jiahua Wang obtained his PhD degree in 2020 from East China University of Science & Technology (ECUST), China. Now he is a postdoctoral researcher in Coacervates and Soft Interfaces group at Radboud University in Nijmegen. His research interests are focused on self-assembly processes in (biological) soft matter and peptide–metal coacervates for biomimetic protocells. |
 Evan Spruijt | Evan Spruijt is a principal investigator at the Institute for Molecules and Materials, Radboud University, Nijmegen. His research is focused on phase transitions and self-organization of peptides, proteins and nucleic acids, and their role in cellular organization and the emergence of life-like systems. The current research interests in his group are mainly centred on coacervates as versatile and functional protocells. |
Key learning points(1) Liquid–liquid phase separation dictates the formation of coacervates and holds the key to control coacervate-based protocells.(2) We offer design rules and guidelines for the construction of peptide-based coacervates that mimic cellular life, based on an overview from recent literature. (3) Peptide coacervates offer promising routes to bio-inspired control over compartmentalization, sequestration and catalysis. (4) Active, dissipative systems enable further advancing peptide coacervates towards moving, growing and self-dividing protocells. (5) Insights from peptide coacervates can help to better understand phase separation in biology. |
1. Introduction
Living systems are characterized by a multitude of functionalities, including compartmentalization, metabolism, replication, and adaptation.1 How such complex, life-like behaviour could emerge from a mixture of molecules remains one of the most fundamental questions in science. Endeavours to elucidate what physical and chemical principles underlie such an emergence typically begin with an outline of the hallmarks of living systems. Of those hallmarks, compartmentalization is one of the most elemental ones: a container in which all vital reactions can take place helps to define the boundaries of a living system, it separates and protects the “self” from the outside world, and it is required to maintain concentrations and the state of essential molecules.2 Compartments likely also played an important role in bringing together the chemical building blocks of life. Without a way to contain and concentrate these scarce molecules, they could have been too diluted in primordial ponds to allow for reactions and complexation. This does not mean that other hallmarks are of secondary importance to create life-like systems; replication and adaptation are, for example, actively investigated in a diverse set of systems. Nevertheless, the focus on compartmentalization offers a tangible goal, which makes it an appealing starting point.For a long time, researchers have thus been working on designing and building increasingly plausible protocells, which are the most rudimentary compartments from which living cells could have emerged.3 Protocells should ideally be made of components that could have formed abiotically, and display or mimic characteristics commonly present in living cells. Various types of protocell models have been explored in recent years. Due to the similarity to modern cells, compartments surrounded by membranes made of lipids or fatty acids (liposomes) have been proposed as obvious candidates for protocellular compartments. Besides lipids, other types of molecules and particles have the ability to form vesicles with a form of membrane. These include amphiphilic proteins or peptides (proteinosomes), block copolymers (polymersomes) or even inorganic colloidal particles (colloidosomes). Apart from the uncertainties regarding the prebiotic pathways to synthesize some of the components, many of these protocells with a membranous shell are difficult to form spontaneously and have a limited permeability for nutrients.
An alternative type of protocell could have been formed by membraneless compartments, such as coacervates, an idea that was first proposed by Oparin in his work on the origins of life in the 1930s, following the introduction of coacervates by Bungenberg-de Jong in 1929. Coacervate droplets are formed spontaneously through the physical phenomenon of liquid–liquid phase separation, in which a liquid condensed phase that is typically rich in macromolecules, separates from an aqueous solution.4 This is a spontaneous process, equivalent to the separation of oil and water, with the main difference between phase-separated coacervate systems and oil-in-water emulsions being that water is the continuous phase both inside and outside the coacervates (see Section 2 for details). Because coacervates lack a membrane, they can easily take up and concentrate solutes from the surroundings due to different affinity of molecules towards the coexisting phases.
The spontaneous formation and the ability of both selective accumulation and dynamic exchange of cargo make coacervate droplets attractive candidates for protocells. The potentially enhanced concentration of building blocks and the chemically distinct environment inside coacervates may further accelerate reactions taking place inside, localize different reactions in the same compartment, and endow coacervates with the properties of a catalyst. However, the lack of a membrane also presents a downside: coacervates and other membraneless model protocells, such as oil droplets, are prone to coalescence and have a limited stability. In addition, coacervation typically requires long and highly charged macromolecules, such as polysaccharides, synthetic polyelectrolytes or nucleic acids, which may have been difficult to form spontaneously under prebiotic conditions. The main challenge for coacervates as protocell models is therefore to identify the simplest molecules capable of phase separating under mild conditions and to strike a balance between their tendency to fuse and their ability to sequester and exchange solutes.
In search for molecules that are capable of forming coacervate protocells, a multitude of synthetic polymers and natural polymers (proteins, polynucleotides, polysaccharides and their derivatives) has been explored.5 Recent discoveries in cell biology have demonstrated that phase separation is also a prevalent mechanism to create dynamic intracellular compartments in many cell types. In particular, proteins with low-complexity regions (LCRs) are prone to undergo phase separation.6 These findings have reinvigorated research into coacervates as protocells and led to new insights for the design of simple peptides with phase separating potential. Peptides and peptide derivatives seem to be particularly promising molecules for the construction of life-like coacervate protocells. They have a relatively simple chemical structure (peptide backbone) and a multitude of functional groups through different amino acid side chains. This means that their structure can incorporate multiple moieties responsible for intermolecular interactions leading to phase separation, but also regions with, for example, a catalytic function. At the same time a large variety of peptide building blocks could be accessed abiotically using only one type of reaction: the condensation of amino acids or peptide fragments into longer sequences.
In this review we focus on liquid–liquid phase separation of peptides, the unique properties of peptide-based coacervates and their potential role in the emergence of life-like systems. We first give a brief background of coacervates and describe the basic driving forces for coacervation. Based on recent literature reports, we then identify structural features in peptides and peptide derivatives responsible for intermolecular interactions leading to LLPS, both in artificially designed systems and in sequences of natural phase-separating proteins, from which minimal motifs for LLPS could be extracted. We divided the discussion into complex coacervate systems comprising of two peptide components or a peptide and a non-peptide component, and simple coacervate systems, which require only a single type of peptide. Most peptides that we discuss undergo liquid–liquid phase separation in solution, but some are also found to undergo liquid-to-solid transitions, or alternative self-assembly and complexation with inorganic components. This leads to reflections about possible influences of the protein-rich coacervate environment on chemical reactions. Finally, we discuss examples of active systems, in which compartmentalization is coupled to the dissipation of energy, as probably the most advanced coacervate protocell models developed to date. While a fully functional synthetic cell remains out of reach and may require more than only a coacervate compartment, active droplets with their ability to divide and self-organise may be an important step towards the development of artificial life.
2. Fundamentals of peptide phase separation
2.1 Associative, segregative and simple phase separation
Three types of liquid–liquid phase separation are commonly distinguished: segregative, associative and simple phase separation (Fig. 1a). In segregative phase separation, two soluble molecules (e.g., peptides, polymers, nucleotides) do not mix despite a favourable mixing entropy due to repulsive interactions between them. As a result, they end up in two separated phases, each enriched in one of the solutes. A classic example of segregative phase separation is that of poly(ethylene glycol) and dextran. A phase diagram is often used to summarise the occurrence of phase separation and the composition of the coexisting phases. A schematic phase diagram for segregative phase separation is shown in Fig. 1b. The phase diagram depicts the range of concentrations of the two soluble molecules (and sometimes also temperature, pH) for which the mixed state (one-phase region) and the demixed state (two-phase region) are thermodynamically stable. The boundary between these regions is called the binodal. Each point on the binodal represents a possible composition of one of the phases formed upon phase separation, and is connected to a second point corresponding to the coexisting phase via a tie line. The phase diagram is most often constructed by preparing mixtures at different relative concentrations and reporting their state (mixed/demixed) after equilibration. The approximate binodal is then drawn as the line that separates the one- and two-phase regions. A more accurate approach is to measure the concentration of both molecules in the separate phases (e.g., by absorbance, fluorescence or NMR) for different overall compositions in order to draw a series of tie lines and reconstruct the binodal by connecting these points.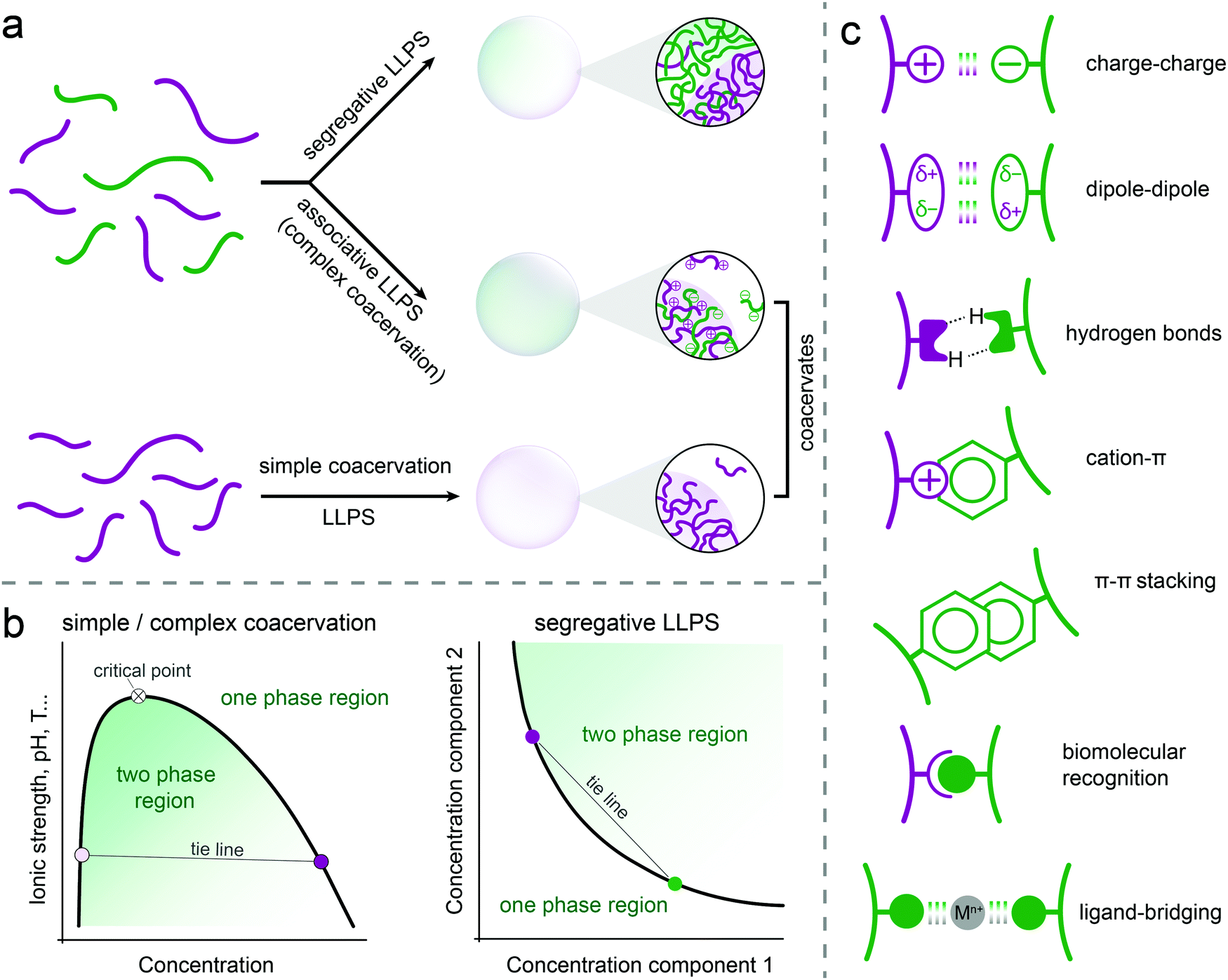 | ||
| Fig. 1 (a) Types of liquid–liquid phase separation and the formation of coacervates, (b) schematic phase diagrams of simple or complex coacervation and segregative phase separation, (c) possible interactions involved in the formation of peptide-based coacervates. | ||
In associative phase separation, two soluble molecules end up in the same phase, due to attractive interactions between them. This condensed phase is called a (complex) coacervate; it is enriched in both solutes, but still contains a significant amount of solvent (typically more than 50% by weight). The coexisting phase is depleted of both solutes, and contains mostly solvent. A classic example of associative phase separation is that of two oppositely charged polymers. A phase diagram of associative phase separation often depicts the one- and two-phase regions for a particular ratio between the two interacting molecules (e.g., equimolar, or 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge ratio), as a function of the concentration of one of them and a tuning parameter, such as temperature or salt concentration (Fig. 1b). Like for segregative phase separation, the binodal separates the two regions and tie lines connect two coexisting phases on the binodal. The tuning parameter can be used to increase or decrease the width of the two-phase region, and beyond a critical point, no phase separation occurs. Depending on the tuning parameter, this could be an upper or lower critical solution temperature (UCST/LSCT) or a critical salt concentration.
:1 charge ratio), as a function of the concentration of one of them and a tuning parameter, such as temperature or salt concentration (Fig. 1b). Like for segregative phase separation, the binodal separates the two regions and tie lines connect two coexisting phases on the binodal. The tuning parameter can be used to increase or decrease the width of the two-phase region, and beyond a critical point, no phase separation occurs. Depending on the tuning parameter, this could be an upper or lower critical solution temperature (UCST/LSCT) or a critical salt concentration.
Finally, we define simple phase separation here as the situation in which the attractive interactions are present in a single molecule, resulting in it becoming insoluble at certain solution temperature, pH and salt concentration. This phase separation also results in a condensed phase, called a simple coacervate, which has very similar properties to the complex coacervates formed by associative phase separation. Many proteins with disordered regions have been found to undergo simple phase separation, driven by a combination of π–π, cation–π, hydrogen bonding, dipole–dipole and charge interactions (Fig. 1c). From a theoretical point of view, simple coacervation is usually modelled, for example using Flory-Huggins theory, as a segregation between a polymer and a solvent. However, to highlight the analogy with associative phase separation, in which all solutes are also concentrated in a single condensed phase, we termed this simplest form of phase separation simple coacervation.
2.2 Coacervates, (micro)gels and aggregates
Coacervates are formed by associative or simple phase separation, and appear as the dense phase enriched in one or more solutes.7 Phase separation is usually induced by changes in solution temperature, pH, salt concentration or composition (e.g., addition of a second peptide), and can in theory proceed via nucleation and growth of coacervate droplets when an energy barrier must be overcome to nucleate the coacervate phase, or spinodal decomposition when any coacervate formation is energetically favourable. In many cases of protocell research involving coacervates, phase separation takes place in relatively dilute solutions by gradual increase of the concentration of, or the attractive interactions between peptides, which is likely to occur via nucleation of coacervate droplets and their growth.7This droplet state of coacervates (Fig. 2) is of interest for protocell research, but because the droplets are not stabilised, coarsening of the coacervate emulsion occurs, predominantly through coalescence and driven by a reduction of the total interfacial area. Ultimately, a macroscopically separated coacervate phase is formed, which is difficult to redisperse (Fig. 2). Coarsening can be slowed down in very dilute conditions, or completely prevented by stabilising layers.4 While relevant for coacervates as protocells, these approaches are beyond the scope of our review, as we focus primarily on the chemical requirements of peptides as building blocks of coacervate protocells.
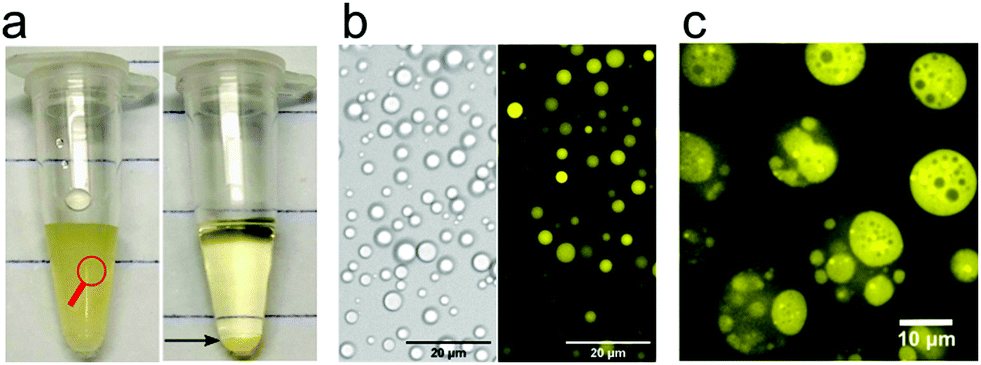 | ||
| Fig. 2 (a) Pictorial formation of coacervates of poly-L-lysine and ATP, (b) bright-field transmission and fluorescence image of coacervate droplets in the poly-L-lysine/ATP system (taken from the left panel in a) and (c) an example of a multiphase coacervate system with dual multiphase arrangement (1/2 and 2/1) containing PLys/PLys(Me)3/poly(3-sulfopropyl methacrylate). Reproduced from ref. 23 with permission from the American Chemical Society, Copyright 2020. | ||
Recent advances in phase separation of disordered proteins have enhanced the understanding of the structural and thermodynamic requirements for coacervation.8 In general, multiple weak attractive interactions, such as charge–charge, dipole–dipole, hydrogen bonds, cation–π or π–π are required (Fig. 1c).9,10 These interaction patches should be spaced by regions of higher flexibility and solubility.11 Phase separation does not occur if the line density of interacting residues is too low or if the attractive interactions are too weak to overcome the stronger repulsion and mixing entropy. On the other hand, a too high local density of interacting residues has been found to lead to kinetic trapping and the formation of solid, typically amorphous aggregates. Associative phase separation that is driven mostly by charge–charge interactions is an exception: condensation into liquid coacervates is possible even for polypeptides or nucleic acids that consist of only charged residues if the attraction forces between oppositely charged molecules are weakened by charge screening upon addition of salt.12
2.3 Molecular design rules for peptide coacervation
From the perspective of protocells, it is important to know to what extent these design principles apply to peptide-based coacervates. In the following sections, we discuss this question in detail, based on recent literature on peptide-based coacervates, but it is instructive to give some general guidelines here, which we summarized in Fig. 3.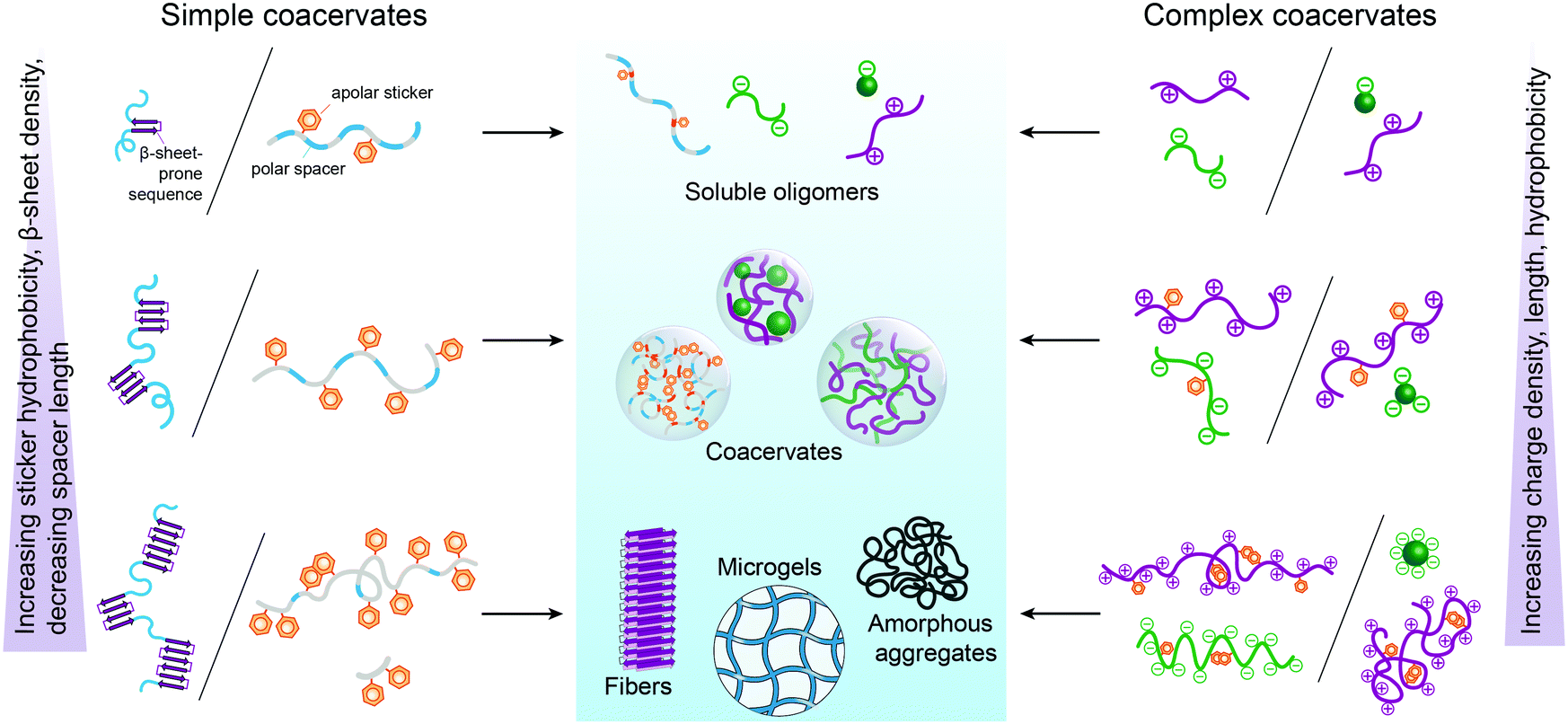 | ||
| Fig. 3 Schematically illustrated qualitative guidelines for the formation of peptide-based simple coacervates (left) and complex coacervates (right): increasing the sticker hydrophobicity, β-sheet density, charge density and total length, or decreasing the polar spacer length tends to lead initially to coacervation, but upon further increase to the formation of solid-like assemblies, including gels. For more details, see the examples in Section 3. | ||
Firstly, the length of peptides plays an important role in their phase separation propensity. Below a critical length, peptides do not phase separate at a specific temperature or salt concentration,13,14 as the interactions are not strong enough to overcome the mixing entropy. For charge-driven associative phase separation of homopeptides, the critical length is around 5–10 amino acids14 similar to the minimal length of adjacent similarly charged amino acids in peptides consisting of blocks of oppositely charged residues.15
The charge density is perhaps even more important than length. Peptide/nucleotide coacervates form more readily with nucleoside triphosphates than diphosphates, which in turn are more stable than monophosphates.16 For peptides, the critical charge density for coacervation at physiological conditions is around 0.2 per amino acid.17,18
For simple coacervation driven by hydrophobic interactions, the critical length and density appears to be even lower, as compounds with as few as two adjacent aromatic residues have been found to phase separate.19,20 In contrast to charged homopeptides in complex coacervation, a hydrophilic spacer or flanking region is essential for aromatic-rich peptides in simple coacervation to prevent kinetic trapping. However, the precise sequence requirements of peptide-based spacers remain elusive.
Finally, combinations of interaction motifs (Fig. 1c) could either strengthen or weaken each other, depending on their combination and relative positions, as illustrated by mixed cationic-hydrophobic peptides.12 Also in this case, the quantitative design rules for combining interaction motifs are, however, still incompletely understood.
3. Coacervation involving peptides
It is not uncommon for proteins or peptides to become insoluble when the pH, temperature, solvent quality or salt concentration is changed. Usually, this leads to the formation of an amorphous, solid precipitate. Similarly, protein aggregation and self-assembly typically results in solid amyloid fibrils, or nano- or micro-structures. However, recent research has shown that there are also numerous examples of peptides and proteins that separate from solution into a dense liquid phase under certain conditions. The dense liquid phase can be a simple or a complex coacervate (see Section 2), and take the form of small droplets, which have properties that make them interesting as protocells. Below we discuss different types of coacervates in which peptides are one of the major components (Table 1).| Peptides | Sequence/characteristic motifs | Other coacervate components | Ref. |
|---|---|---|---|
| a Formation of coacervates or aggregates depends on length and stereochemistry (see Fig. 3 and 5). b Multiple repeats of the motif are required to drive LLPS. c Only one example of motifs characteristic for LARKS, multiple repeats can drive reversible gelation and may be involved in LLPS. d Ctm = cystamine (2,2′-dithiobisethanamine) linker. Other linkers that result in simple coacervates with FF dipeptides are 2,2′-diaminoethyl sulphide, 2-(2-aminoethoxy)ethylamine, 2,2′-(ethylenedioxy)bis(ethylamine) and 1,4-bis 3-aminopropoxybutane. | |||
| Complex coacervates | |||
| Poly-lysine | Kn (n > 10) | ATP/ADP/RNA/Ena/Dna | 14, 16, 22 and 24 |
| Poly-arginine | Rn (n > 5) | ATP/ADP/RNA/Ena/Dna | 14 |
| RP2/RP3 | (RRASL)n (n = 2/3) | polyU/RNA | 21 and 29 |
| GFP-K72 | GFP-[GVGVP(GKGVP)9]8 | RNA | 17 |
| KVR heptapeptide | Fmoc-KVRVRVK-NH2 | ATP | 25 |
| PR20 | (PR)20 | PO43−/CO32− | 31 |
| RGG | RG/RGGb | RNA | 28 |
| GXK tripeptide | GHK/GFK | SiW11 | 33 |
| Simple coacervates | |||
| mfp-3S | GYDGYNWPYGYNGY-RYGWNKGWNGY | — | 36 |
| HBP-1 | GHGLYb | — | 9 |
| LARKS/PLD | SYSSYGQSc | — | 6 and 28 |
| FF motif | Z-FF-OH | — | 41 |
| FF motif | (FF)2-Ctmd | — | 19 |
3.1 Complex coacervates
Complex coacervates are formed as a result of non-covalent interactions between (at least) two components. One of the most classic examples of complex coacervation is phase separation in water solution of alginate (a negatively charged polysaccharide) and gelatin (a positively charged protein). However, from the point of view of protocells and the origins of life, coacervates in which the commonly used synthetic and large macromolecules are replaced by low-complexity peptides are of particular interest.Several years later, similar peptide–polynucleotide and mononucleotide coacervates were shown to be dynamically controlled by enzymatic reactions. Condensation of droplets could be driven by dephosphorylation of phosphoserine residues in an arginine-rich peptide or by phosphorylation of ADP to ATP, while the reverse reactions led to droplet dissolution.21,22 Interestingly, in the first system, composed of the peptide (RRASL)3 and poly-U, a single phosphorylation significantly changed the phase separation propensity of the peptide and resulted in coacervate dissolution. Similarly, in the ATP/poly-L-lysine system, single dephosphorylation (conversion of ATP to ADP) resulted in the dissolution of droplets. These results illustrate that the charge density is critical for the stability of coacervates: in the case of peptide phosphorylation the net line charge density was decreased from 0.4 to around 0.2 (for a single phosphorylation).
Despite the simplicity of the building blocks, coacervates formed by peptides and nucleic acids or nucleotides can give rise to a complex organisation. Recently, it was shown that mixing coacervates formed by nucleic acids (RNA or single stranded DNA) or nucleotides and different cationic polypeptides (lysine-rich elastin-like polypeptide K72 or poly-L-lysine) could result in formation of multiphase droplets.23,24 From the point of view of protocells, a container with multiple subcompartments could provide a way to separate mutually incompatible molecules or reaction pathways. Moreover, the engulfing observed in some multiphase droplet systems could provide a way in which small protocells grow and increase in complexity. Although the exact nature of intermolecular interactions that drive multiphase separation of polypeptide/nucleotide systems remains incompletely explained, it is clear that even in relatively simple mixtures of only charged peptides and nucleic acids, small differences in chemical structure between the peptides can result in significant changes in the ability to phase-separate.
Increasing the sequence complexity of peptide compounds involved in coacervation can help to understand the conjunction of multiple interaction types (Fig. 1c). The peptide derivative Fmoc-VVVRRKK-NH2 that contains in total four cationic residues and three hydrophobic residues along with the hydrophobic Fmoc group, was used by Das and co-workers.25 In their strategy, the peptide acts as a polycation and could be expected to form a complex with a nucleotide ATP as a polyanion. However, this amphiphilic peptide motif was unable to form coacervates with ATP at any concentration; instead, the strong association between the high charge density region on the peptide and the nucleotides, combined with the overall amphiphilic architecture, resulted in the formation of solid nanofibers (Fig. 4a). By scrambling the amino acid positions in the peptide motif to Fmoc-KVRVRVK-NH2, the assembly was drastically altered. The alternating charged-hydrophobic peptide no longer has an amphiphilic character, and its local charge density is decreased. As a result, this motif was found to phase separate with ATP into apparent complex coacervates (Fig. 4b). The coacervates were disassembled in the presence of ATP-ase, showing the reversibility and accessibility of the assembly.
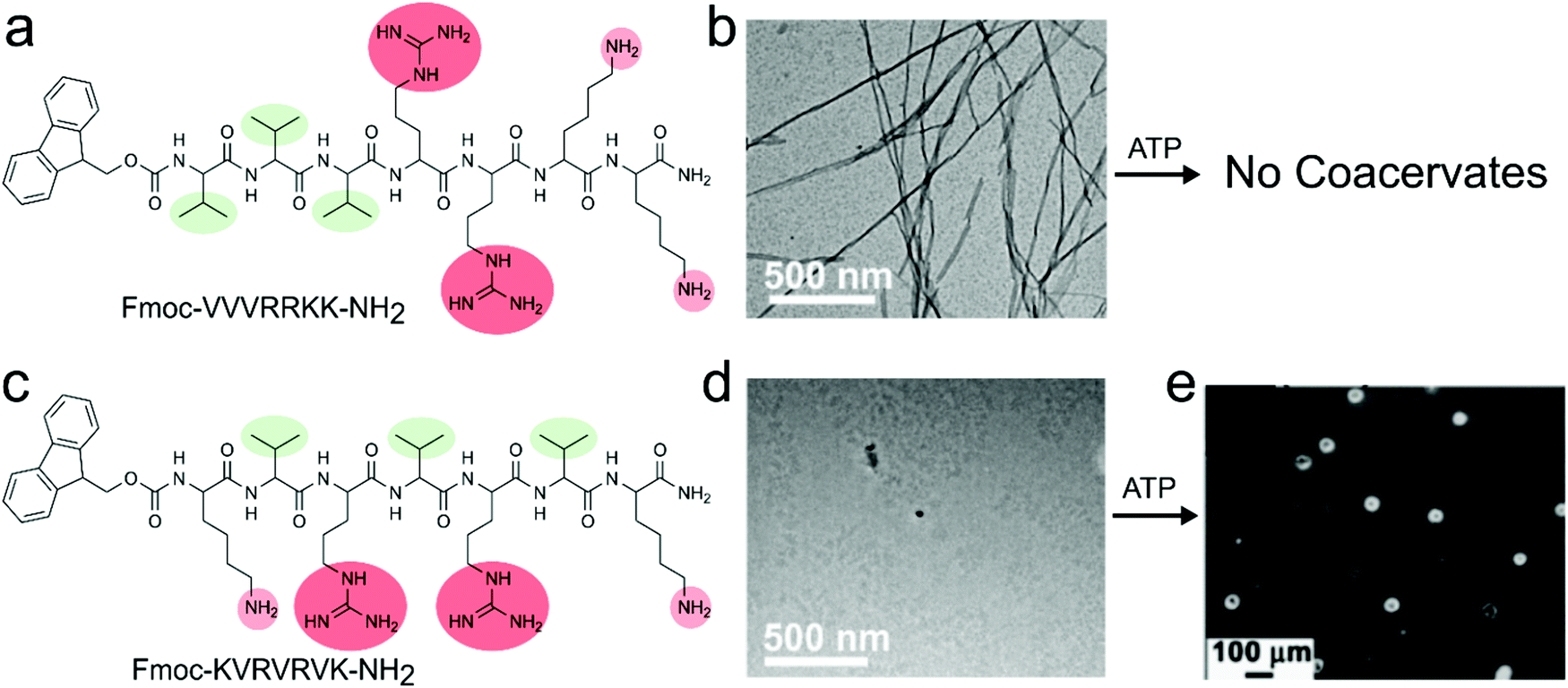 | ||
| Fig. 4 Chemical structures, assembly and complex coacervation of peptides containing both cationic and aromatic residues with ATP, (a) structure of Fmoc-VVVRRKK-NH2, (b) transmission electron microscope (TEM) image of nanofibers due to strong amphiphilic nature of compound which does not show any coacervation with ATP. (c) Structure of Fmoc-KVRVRVK-NH2, (d) TEM image of solution of monomers, (e) coacervates of Fmoc-KVRVRVK-NH2 and ATP. Reproduced from ref. 25 with permission from the Royal Society of Chemistry, Copyright 2019. | ||
These results indicate that an equal spacing of charged residues is important for complex coacervation. Moreover, they show that neighbouring non-charged residues influence the minimal peptide charge or length required for phase separation: while arginine or lysine-based homopeptides shorter than 10 amino acids do not phase separate with ATP,14 the mixed peptide Fmoc-KVRVRVK-NH2, which has only 4 charged amino acids, did phase separate, most likely due to the flanking hydrophobic residues which strengthen the charge–charge interactions. Like poly-L-lysine/ATP coacervates, these Fmoc-KVRVRVK-NH2/ATP coacervates were able to encapsulate proteins such as cytochrome c (CytC), and catalyse redox reactions in a spatiotemporally controlled manner under dissipative conditions created by ATPase. This combination of controlled assembly, localisation of catalysts and compatibility with redox chemistry makes them interesting protocell candidates.
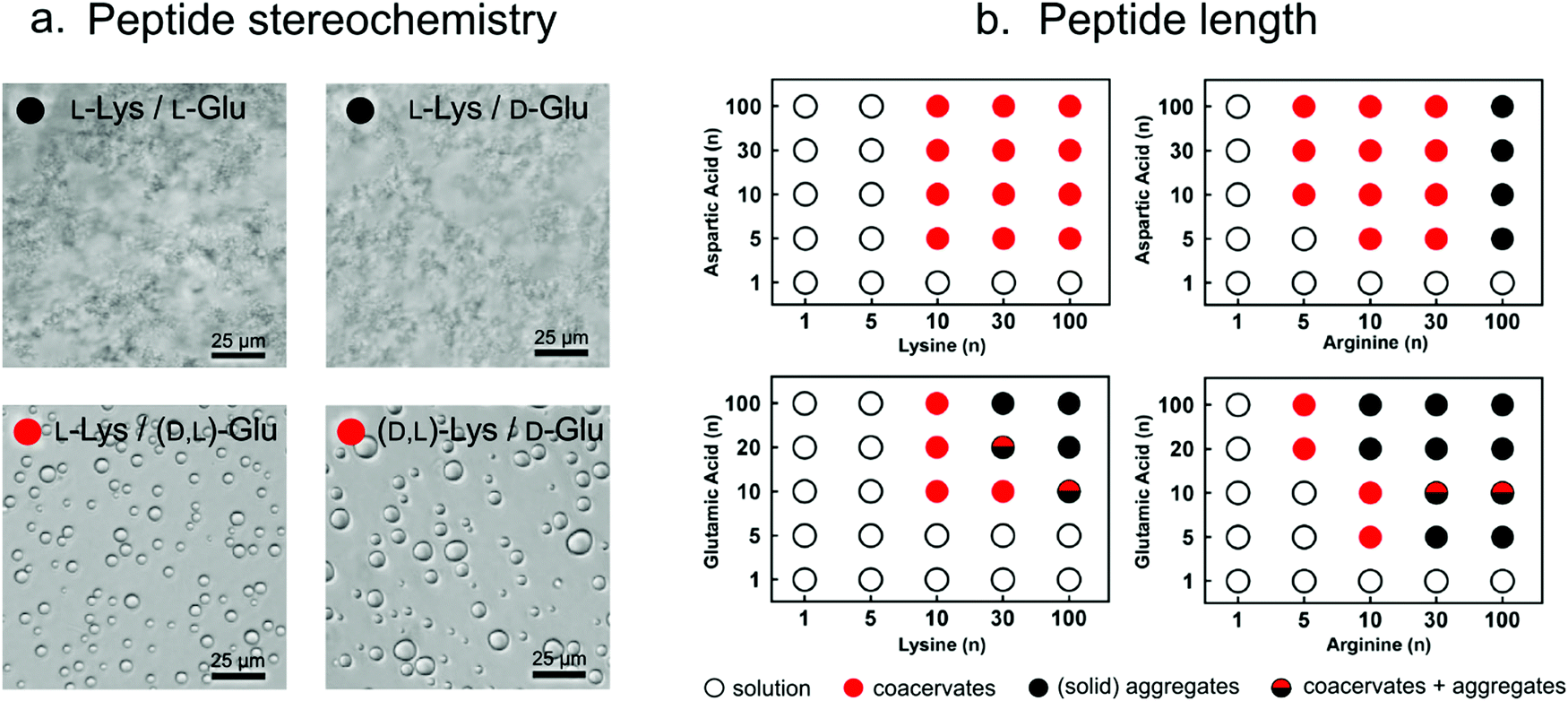 | ||
| Fig. 5 (a) Optical micrographs showing that, depending on the stereochemistry, poly-lysine and poly-glutamate can form either solid aggregates or liquid condensates. Reproduced from ref. 26 with permission from Nature publishing group, Copyright 2015. (b) Increasing peptide length results in stronger interactions and formation of liquid droplets or (upon further increase in length) solid aggregates. Reproduced from ref. 14 with permission from the Nature Publishing group, Copyright 2020. | ||
The ability to undergo liquid–liquid phase separation is not only dependent of stereochemistry, but it also on the polypeptide length, and the charge density and the chemical nature of the charged groups. Coacervate droplets can also be formed using poly-L-aspartate instead of poly-L-glutamate and poly-L-arginine instead of poly-L-lysine because of attaining the similar charge balance/interactions. Keating and co-workers found that for these combinations the minimal peptide length required for peptide–peptide coacervates is around 10 amino acids. However, in these mixtures, poly-L-arginine consistently seems to form stronger interactions than poly-L-lysine and poly-L-glutamate interacts stronger than poly-L-aspartate.10,14,27 Arginine not only shows higher potency to interact with negatively charged peptides and nucleotides via charge–charge interactions compared to lysine (due to its higher pKa), but it can also participate in the formation of multiple hydrogen bonds with, for example, phosphates, and it exhibits stronger stacking interactions with π-systems-containing molecules, due to its aromatic nature.28
Besides metal ions, small inorganic anions can also mediate complexation by bridging positively charged residues in peptides, potentially leading to phase separation. Boeynaems et al. found that arginine-rich peptides with proline-arginine repeats (PR20) can phase separate in presence of crowding agent PEG and divalent anions, such as phosphate and carbonate (Fig. 6a).31 Similarly, Yang et al. showed that large anionic counterions like sulphate, phosphate, citrate and even monoanionic nitrate with multiple binding sites can exhibit strong ion pairing between positively charged arginine moieties, which could trigger coacervation of a mussel-derived adhesive proteins ((m)fp-3F) without crowding agent (Fig. 6b).32 Typically, coacervates formed by peptides bridged by inorganic counterions with multiple binding sites (Fig. 1c) are sensitive to charge screening by monovalent salt – a high salt concentration is expected to prevent this type of phase separation. However, in other cases where coacervation is driven by intramolecular π–π interactions, addition of salt may enhance coacervation by reducing the repulsion between charged residues in the same molecule.
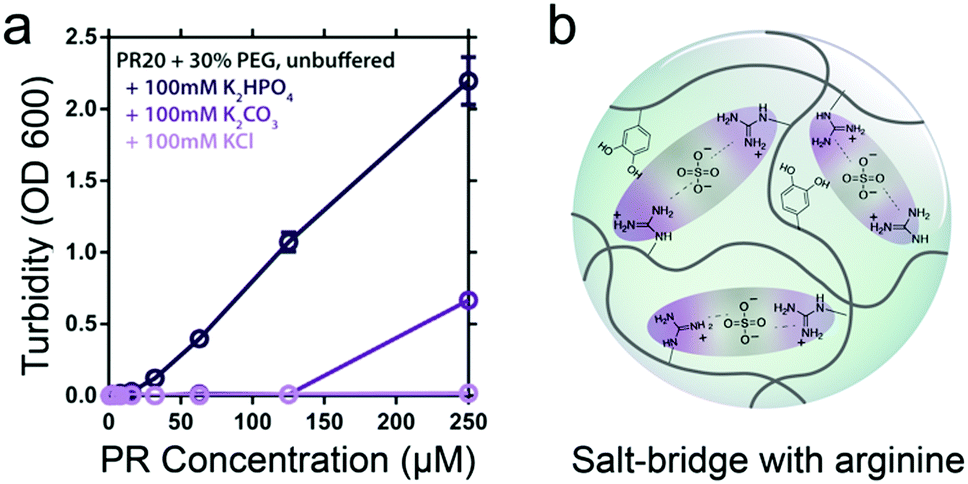 | ||
| Fig. 6 (a) PR-PEG LLPS is dependent on inorganic counterions and correlated to anionic charge. Reproduced from ref. 31 with permission from permission from the Wiley-VCH publishing group, Copyright 2017. (b) The possible interactions within coacervates of mfp-3F. | ||
Short peptides can also complex with inorganic metal clusters to form coacervates. Li et al. reported a new kind of short peptide-based coacervate by condensation of cationic tripeptides with inorganic anionic polyoxometalates (POM) (Fig. 7).33 The main driving force behind the phase separation is thought to be the attraction between protonated amine groups and the anionic POM. However, additional interactions between the peptides and POMs play an important role in phase separation. At pH 6.5, the tripeptide GVK could not form coacervates with SiW11, while GHK and GFK both could form coacervates, due to hydrogen bonding between histidine and the POM, and a more hydrophobic local environment created by phenylalanine, which could strengthen the charge–charge interactions. The presence of a histidine residue also enabled control over the physicochemical properties of the GHK-based coacervates through pH variation: at pH 4.5, the protonated histidine residue contributed to an increased charge density, leading to a lower water content and denser structure of the coacervates than at pH 6.5.
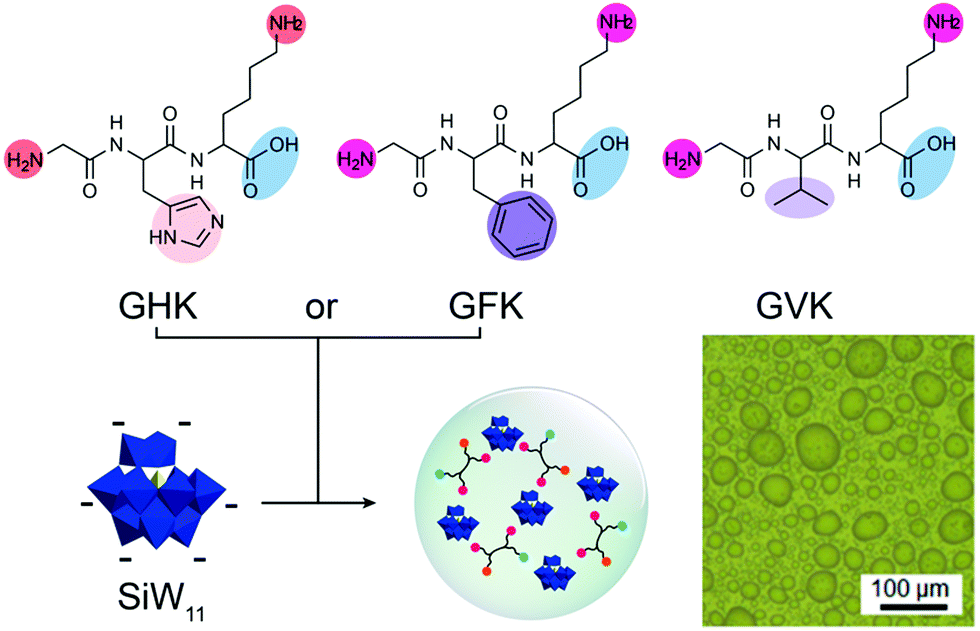 | ||
| Fig. 7 Chemical structures of tripeptides GHK, GFK, and GLK and the packing model of the peptides GHK/GFK and polyoxometalates – SiW11 coacervate and optical microscope image of coacervates. Reproduced from ref. 33 with permission from the American Chemical Society, Copyright 2019. | ||
Finally, some inorganic compounds, such as phosphate, can form polymers that interact with oppositely charged peptides in a similar manner as peptides or polynucleotides. From a protocell point of view, inorganic polyphosphate (polyP) is a particularly interesting molecule. It is one of the most ancient, compact intracellular polyanions, consisting entirely of high-energy phosphate diester linkages. It has been regarded as a chaperone that stabilises unfolding proteins, and prevents irreversible aggregation of proteins. Recently, Wang et al. showed that endogenous inorganic polyphosphate can interact with positively charged green fluorescent protein (GFP36+) and undergoes liquid–liquid phase separation near the poles of bacterial cells.34 Remarkably, RNA, which is also present in significant concentrations, did not undergo phase separation with GFP36+ under the same conditions, which was explained by the higher linear charge density of polyP. Protein/polyP phase separation is dependent on the chain length of polyP: increasing the polyP length favoured coacervate formation in vitro.
3.2 Simple coacervates
Unlike complex coacervates, simple coacervates contain a single component that is responsible for phase separation. This means that the coacervate (dense phase) is composed primarily of this single component and water. The phase separation in simple coacervate systems is also caused by weak, non-covalent intermolecular forces, which can all be present in peptides (Fig. 1c). However, contrary to complex coacervates, much less is known about the design criteria for single peptides capable of undergoing simple coacervation.From the point of view of protocells these short peptide analogues provide insight into the molecular fundamentals of peptide-based simple coacervation. In case of mfp-3S-pep, LLPS was observed to be dependent on the pH and ionic strength; a higher ionic strength induced LLPS, as salt was found to screen electrostatic repulsion between cations sufficiently to promote cation–π interactions.12 Furthermore, the LLPS range was also reduced by exchanging phenylalanine residues for 3,4-dihydroxy-L-phenylalanine (DOPA). Presence of DOPA made the peptide more hydrophilic and less prone to undergo LLPS, but it improved the adhesive properties on metal oxides, due to DOPA's ability to form hydrogen bonds. The mussel foot-derived peptides are one of the shortest non-derivatised peptides (and polymers in general) capable of simple coacervation, and therefore, they are an interesting starting point for the design of simple peptide protocells.
Gabryelczyk et al. systematically explored the HBP-1 sequence to analyse the influence of characteristic peptide motifs on LLPS. According to their study, GHGLY is the basic sequence motif that drives phase separation and 4 repeats of this sequence are the smallest number required to trigger LLPS. Histidine and tyrosine are critical for phase separation: upon increasing the pH histidine residues become deprotonated and form H-bonds with tyrosine residues, thus stabilising the condensed phase. Presumably, further increasing the pH until tyrosine is also deprotonated (around 11) would destabilise the interaction and lead to dissolution of the coacervates. They also observed that peptides containing GAGFA in the centre of all repeats instead of GHGLH did not phase separate into liquid coacervates, but formed a dense, compact hydrogel (Fig. 8).9 This change of state was explained by the enhanced hydrophobic interactions of phenylalanine amino acid in the GAGFA repeats, resulting in stronger intermolecular interactions.
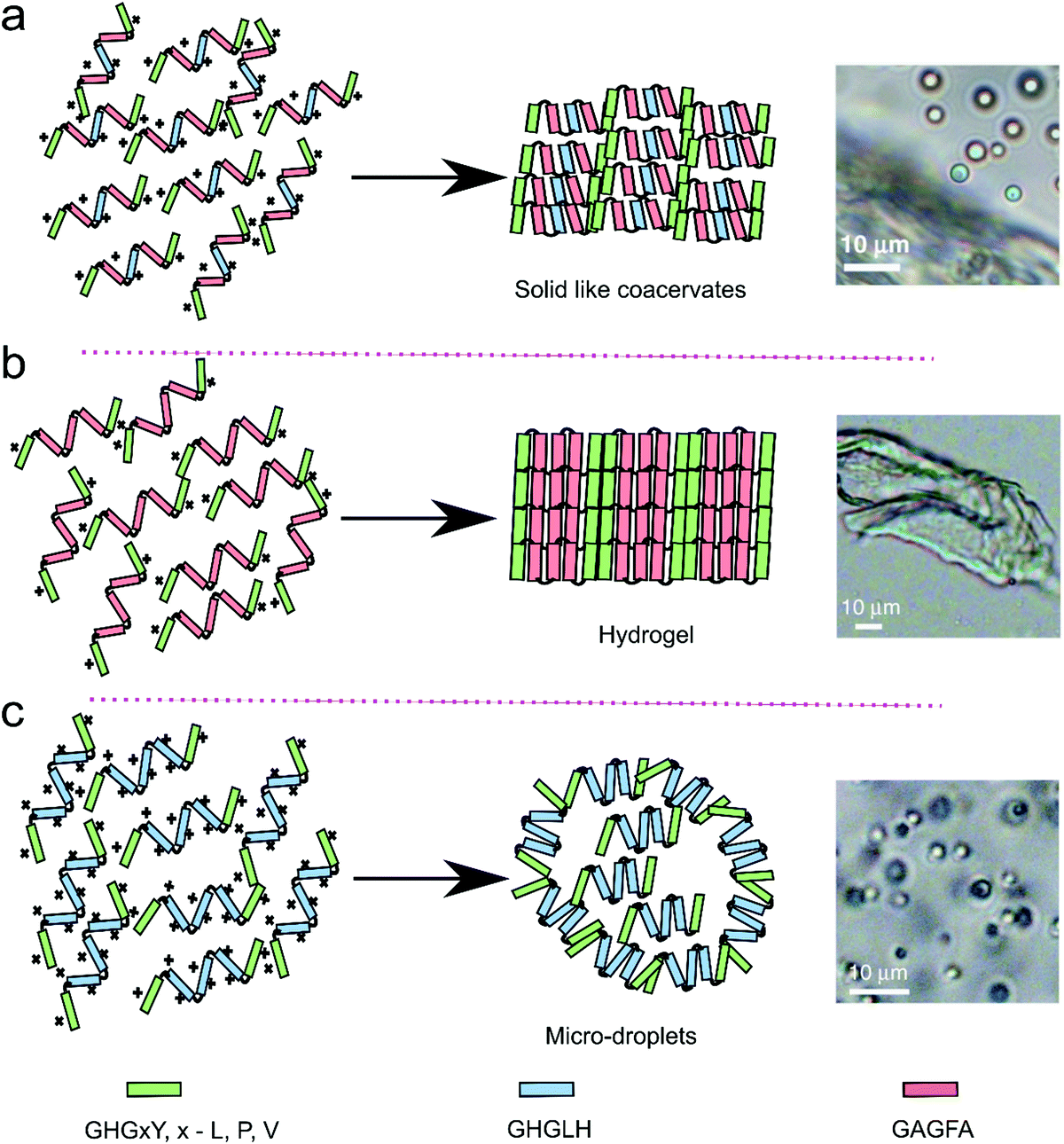 | ||
| Fig. 8 Proposed model of HBP derived peptides of three different repeats (GHGxY, x – L, P, V, GHGLH and GAGFA) for the liquid–liquid phase separation resulted into three different morphology on the basis of arrangement of the peptide repeats. Reproduced from ref. 9 with permission from the Nature Publishing group Copyright 2019. | ||
In an attempt to find shorter peptide fragments capable of phase separation, other researchers started from motifs that are well known for the self-assembling propensity. One of the best studied examples is phenylalanine dipeptide (FF) motif, which can form amyloid-like assemblies while its derivatives can also form hydrogels or nanostructures of various morphologies. One of the FF derivatives, N-carboxybenzyl protected (Z-FF), was found to form a metastable liquid phase (coacervate), which transformed in time into solid nanofibers (see Section 3.3).41 The relative stability of the coacervate droplet state could be enhanced by addition of silver ions to more than 10 hours at 10 mM Ag+. Further extending the stability of the droplet state by sequence variation or derivatisation could be interesting for protocell research.
We have recently designed short peptide derivatives that are able to undergo LLPS based on FF- and other hydrophobic dipeptides, linked together via polar spacers. This motif is based on a sticker-spacer arrangement, where the dipeptide moieties serve as stickers, and the polar linker serves as spacer. We constructed a small library of derivatives that could separate as liquid droplets or solid aggregates.19 The liquid droplets were simple coacervates, which typically formed at sub-mM concentrations of the peptide derivatives and which could retain up to 75 wt% water (solvent). Our design permitted chemical variations in both the hydrophobic sticker amino acids and the spacer, providing a new route to the formation of protocells with tuneable properties under prebiotically relevant conditions. We showed that by including a disulphide bond in the spacer, the coacervates reversibly formed dense, liquid compartments, controlled by redox chemistry. Their potential as protocells is illustrated by a variety of characteristics. They sequestered a wide range of solutes, including nucleic acids and porphyrins. They were found to facilitate melting of short RNA hairpins that were taken up by partitioning. Moreover, they acted as microreactors in which the rates of two types of addition reactions (aldol and hydrazone formation) were significantly increased by localising and concentrating the reactants and lowering the energy barrier. In short, these short peptide derivatives hold real promise as protocells due to their simple but versatile design, and offer a range of possibilities to design protocell variants with different properties.
3.3 Liquid–solid transition of peptide coacervates
Liquid–liquid phase separation of proteins into dense liquid coacervates has originally been viewed as an independent path from precipitation into solid aggregates. However, increasing evidence suggests that there is a direct link between the liquid condensed state and solid aggregate state. Studies of FUS or hnRNAP1 show that disordered regions of these proteins are responsible for both types of phase transition, and the coacervate state could be a metastable state from which amyloids or other ordered structures could nucleate.42 From the point of view of peptide-based coacervate protocells, it is important to understand if and under what conditions similar liquid-to-solid transitions could occur in systems of simpler peptides. Mann and co-workers described such a complex coacervate system of poly(diallyl-dimethylammonium chloride) (PDDA) and an Fmoc-protected D-Ala-D-Ala dipeptide.43 While at pH 8 the mixture was phase separated into two liquid phases, upon gradually decreasing the pH, the liquid coacervate droplets coarsened into fibrils, similar to recent findings with the protein FUS. Re-assembly of droplets from the obtained hydrogel could be achieved by deprotonation of Fmoc-AA peptide upon increasing the pH (Fig. 9).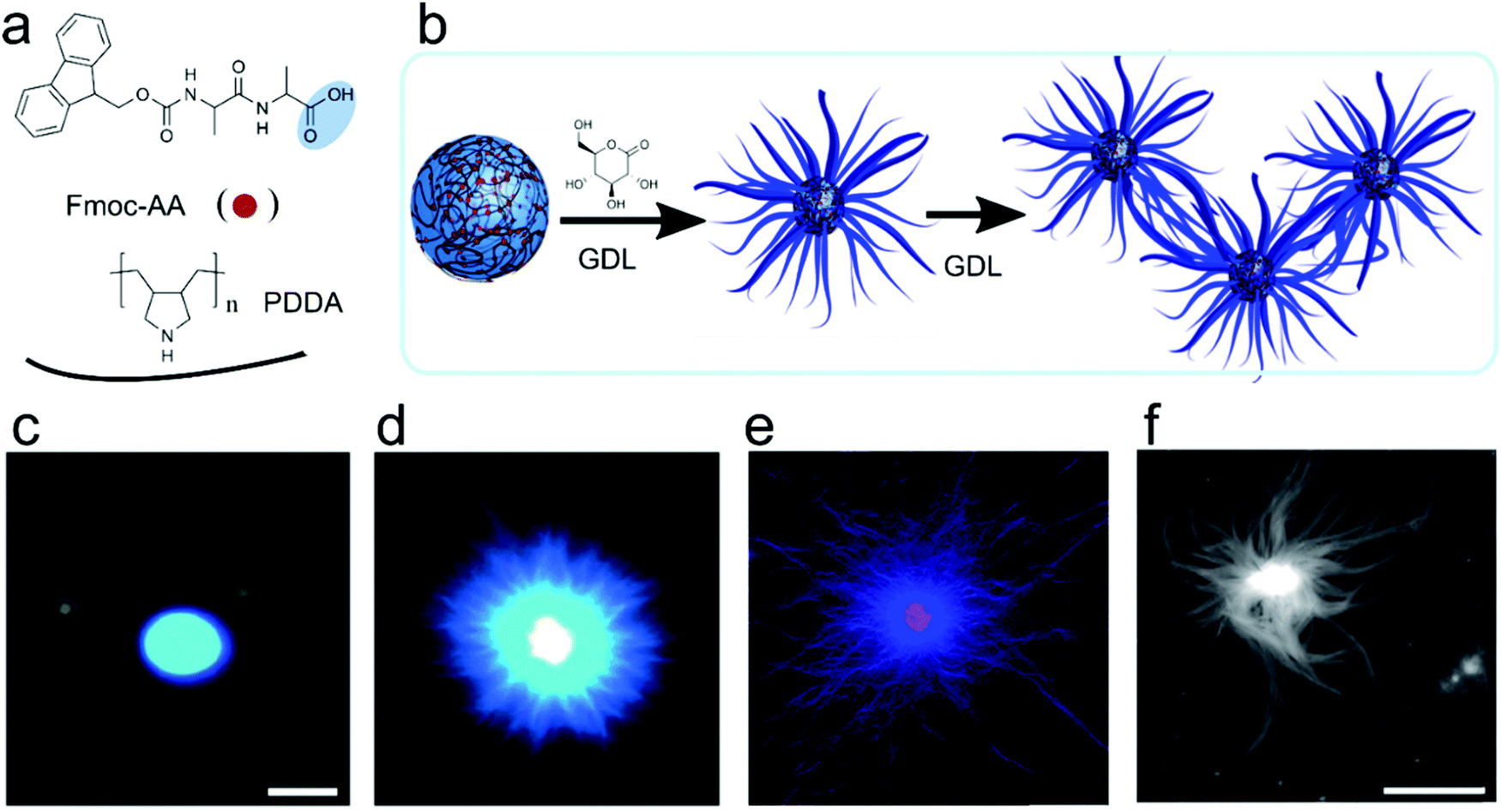 | ||
| Fig. 9 Chemical structures, schematic illustration and microscopic images for the metamorphosis of coacervates-protocells to hydrogel, (a) chemical structures of dipeptide/polymer coacervate micro droplets prepared at pH 8.5, (b) schematic illustration of reconfiguration to aster-like core–shell microstructure at pH 4.5, (c and d) PDDA/Fmoc-AA coacervates before and after addition of GDL and sequestered with Hoechst 33258 fluorescent. This change from coacervates to nanofibers induced due to slow hydrolysis of GDL in the coacervates phase, (e) PDDA/Fmoc-AA coacervates core hydrogel sequestered with 1 mol-% RITC-labelled PAH, (f) AFM images of nanofibrous gel. Reproduced from ref. 43 with permission from the Royal Society of Chemistry, Copyright 2016. | ||
A liquid–solid transition has also been observed for simple coacervates made of N-protected peptides. Droplets formed by Z-FF and some Fmoc-protected amino acids promoted nucleation of more stable fibrils (Fig. 10).41 Later, it was shown that the liquid–solid transition of Z-FF can be induced by shear, similar to silk proteins. Interestingly, the liquid–solid transition into silk-like fibrils was observed for several proteins containing low-complexity domains under shear conditions comparable to those found in cells, which suggests that cells must employ some protective strategies to prevent formation of aggregates.20
 | ||
| Fig. 10 (a) Chemical structure of short peptide and amino acid derivative, (b) microscopic image of droplets, (c) free energy landscape with the transformation of non-equilibrium to equilibrium state, Reproduced from ref. 41 with permission from the Wiley-VCH, Copyright 2019. | ||
As the liquid–solid transitions of disordered proteins are only beginning to be studied in detail, this phenomenon is still hardly looked at in peptide-based protocells. Current examples of liquid–solid transitions in peptide-based coacervates appear to rely on the stacking of aromatic protecting groups (Fmoc, carboxybenzyl) and side groups (Phe). In the case of proteins, formation of extensive β-sheets nucleated in the liquid condensed phase is believed to underlie the liquid–solid transitions observed in biology. Similar β-sheet formation was suggested to play a role in the direct formation of aggregates in mixtures of monoenantiomeric peptide complex coacervates (poly-L-lysine/poly-L-glutamate).26 It is unclear if such a structural transition can also occur in a peptide coacervate phase and what role the peptide length plays in facilitating such a transition.
4. Sequestration and catalysis
One of the most important advantages of coacervates as protocell models is their ability to sequester and concentrate a wide variety of molecules on account of the lack of a membrane barrier. By enhancing the concentrations of reactants, coacervate protocells could accelerate reactions if the corresponding rate equation remains unchanged. However, the distinct local environment of some coacervates could also change the reaction energy landscape, for instance resulting in a lowering of the energy barrier of a rate-limiting step, or in a relative stabilisation of the product compared to the reactants. The former is analogous to the action of a catalyst, and it makes coacervates potentially catalytic microreactors. There is evidence in the literature that coacervates can alter the energy landscape of, for example, hybridisation between complementary strands of DNA, and template-directed elongation of an RNA primer. In this section, we discuss specifically the ability of peptide-based coacervates to sequester guest molecules and to alter the rates of reactions between them.Stroberg and Schnell compared the confined volume of coacervate droplets to the microdroplets generated using electrospray ionisation.44 They propose that there are two types of mechanisms underlying the reaction acceleration, as outlined above: (i) increasing the local concentration of the reactants, (ii) altering the reaction environment. Increasing the concentration of reactants occurs through partitioning, largely driven by the same interactions that underlie the formation of coacervates (Fig. 1c).7 It should be mentioned that the distinction between host (i.e., peptides) and guest molecules is not always sharp, and high concentrations of guest molecules can significantly alter the properties of liquid condensates.
The main reason for accumulation of guest molecules in coacervates is their reduced internal polarity. The polarity inside coacervates is usually significantly lower than the surrounding solution; in other words, coacervates provide a more hydrophobic environment, which results in preferential accumulation of many hydrophobic organic compounds, such as Nile red, bromothymol blue or thioflavin-T dyes.19 When the concentrations of these guests reach similar levels as the local concentration of coacervate-forming peptides, the concentration effect is no longer described by a simple partitioning equilibrium but rather by an associative phase separation of peptides and guest molecules, resulting in a hybrid coacervate.
The local hydrophobic environment is not the only factor that can promote the accumulation of guest molecules inside coacervates. Charge–charge interactions between patches of positively and negatively amino acid residues and hydrogen bonding have been found to play an important role. For example, Ddx4 droplets accumulate both positively and negatively charged proteins, while neutral proteins remain excluded, and hydrogen bonding is responsible for differences in partitioning coefficients of polynucleotides poly-A and poly-N in poly-U/spermine coacervates.
Finally, large guest molecules are often sequestered into coacervates at the expense of smaller molecules that were involved in the original phase separation, even if they have the same intermolecular interactions. Polynucleotides, for example, can achieve partitioning coefficients as high as 105 in polyamine/ATP coacervates, while the local ATP concentration is about 500 times higher than the dilute phase.45 This process is primarily driven by an increase in entropy: a single polynucleotide can replace multiple small molecules, which are released from coacervate environment into the dilute phase.7 A similar effect has been observed for short peptide/peptide complex coacervates.46
The distinct local environment with a reduced polarity inside coacervate droplets not only leads to increased concentrations of certain guest molecules, it can also impact reaction energies. The “solvent” properties of coacervates might be closer to organic solvents like DMSO than to water, leading to altered energy levels of guest molecules or their products (e.g., conformation of guest polypeptides). In a very simplified view, favourable interactions between the coacervate environment and a reaction intermediate or its product would stabilise these species and result in an acceleration of a compartmentalised reaction.7 It should be mentioned here, that similar mechanism could also lead to more stable conformations of the sequestered reactants and result in reaction inhibition rather than acceleration.
One way in which the relative stability of an intermediate complex or a product can be stabilised inside coacervates is through crowding. Excluded volume interactions induced by crowding agents can favour compact or complexed states of macromolecules. It has been suggested that the high local concentration of peptides and other phase separating molecules in coacervates can have the same effect on macromolecular guests. For example, the binding between an RNA polymerase enzyme and its promotor site on a DNA plasmid was found to be 2–3 orders of magnitude stronger inside PEG-based coacervates, and the subsequent rate constant associated with transcription was six-fold higher.47 However, the chemical nature of the coacervates can play a much more important role in determining the overall reactivity than crowding alone: while transcription was possible inside poly-L-lysine/CMDex coacervates,48 overall gene expression rates were slower and the overall yield reduced, compared to PEG-based coacervates.
A combined effect of reactant concentration and an increased rate constant has recently been observed for reactions between small molecules as well. In the presence of simple coacervates of FF dipeptides linked via polar spacers, two basic condensation reactions, aldol condensation and hydrazone formation, occurred almost 50- and 12-fold faster, respectively.19 These moderately hydrophobic coacervates were found to concentrate the reactants between 2 and 5-fold, and they effected a lowering of the apparent energy barrier by up to 6 kJ mol−1. Although examples of similar rate enhancements in peptide-based coacervates remain scarce, results obtained with coacervates of synthetic polymers and nucleotides, harbouring ribozymes,49 or Ru-polyoxometalate catalysts,50 show promising results for the role of (peptide-based) coacervates as protocells with sequestration and catalytic properties.
Finally, surface catalysis, a phenomenon also observed in electrosprayed microdroplets used in the analogy by Stroberg and Schnell, could also act to enhance reactivity in coacervate droplets. Although the characterization of the surface properties of coacervates is limited, it is likely that the solution-condensate interfaces may concentrate specific molecules and aid their reaction by imposing a preferred orientation, or by their interaction with surface-adsorbed catalysts, similar to observations in microdroplets, vesicles and micelles. In summary, peptide-based coacervate hold great promise as protocellular compartments to concentrate and localise reactants, and to potentially catalyse reactions between them, but a detailed molecular understanding of the influence of the coacervate environment on reaction kinetics is still lacking, and requires a more systematic investigation of reactions in model coacervates in the future.
5. Active coacervates
To make the step from passive compartments to a plausible protocell that exhibits life-like behaviour, coacervates must move away from thermodynamic equilibrium. Living cells move, grow, divide and maintain homeostasis via a continuous turnover of chemical energy. In order to develop coacervate protocells with similar capabilities, they must be endowed with activity, and be able to use or convert chemical energy into potential mechanical work or kinetic energy. In this section we discuss how fuel-driven assembly and disassembly of coacervates can be achieved and how it could constitute a first step towards active growth and division of coacervates.Supramolecular chemistry has led the way in recent years by exploring the role of chemical reactions in out-of-equilibrium assembly. Fuel-driven molecular assemblies with spatiotemporal control behave fundamentally different from non-active in-equilibrium assemblies. They are programmable and can exhibit phenomena like motion, bistability and oscillatory responses. Similar principles of dissipative assembly can also be used in designing active coacervate-based protocells. Moreover, peptides as building blocks for coacervates offer a wide array of possibilities for the implementation of fuel-driven modifications, for example, by phosphorylation, acetylation, esterification, anhydride formation, and thiol oxidation.
Boekhoven and co-workers pioneered the use of fuel-driven chemical reaction networks to create dissipative assemblies, including compartments, with diverse morphologies. They first described a series of active fibrous and colloidal systems that were not coacervates, based on the self-assembly of Fmoc-modified acidic amino acids and tripeptides upon transformation to the anhydride.51 More recently, Donau et al. exploited the same strategy of the fuel-driven assembly to form dissipative peptide/RNA complex coacervates. They used arginine-rich peptides with a C-terminal aspartic acid, which could be converted into an anhydride by the condensing agent EDC.52 The anhydrides were found to phase separate into coacervate droplets with long polynucleotides, such as poly-U (Fig. 11). The coacervate droplets dissolved again upon hydrolysis of the anhydrides, but at higher concentrations of fuel (3x excess compared to precursor), the coacervates persisted beyond the lifetime of the anhydrides, which could indicate a side reaction or a liquid–solid transition taking place inside the coacervates. At near-stoichiometric fuel concentrations, the coacervates were dynamic and dissolved completely, through a process of vacuolisation and occasional fragmentation into satellite droplets. Interestingly, these active coacervates were found to transiently encapsulate a range of functional and folded RNA molecules, which is of importance for their potential as active protocells.
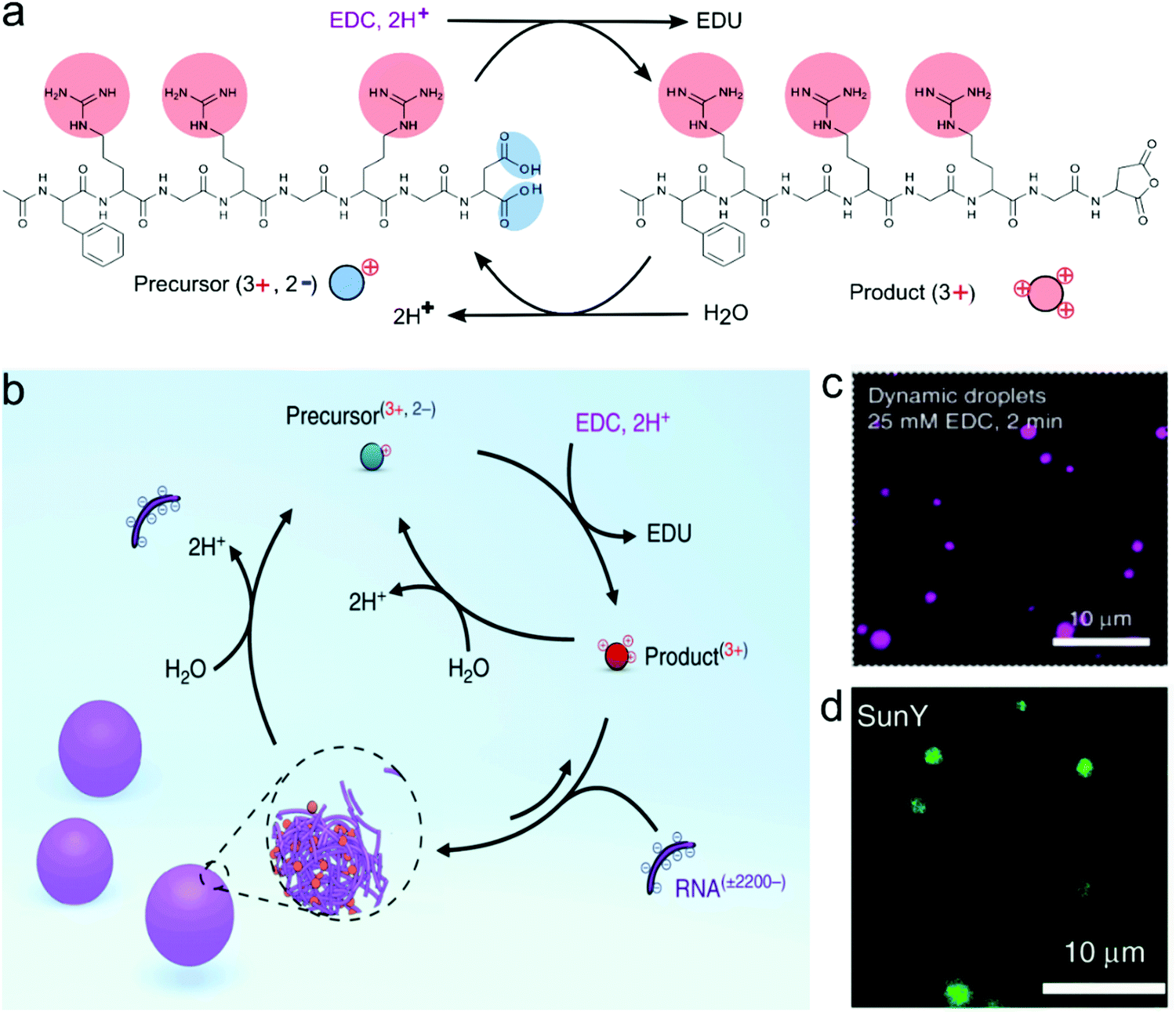 | ||
| Fig. 11 The formation of complex coacervates from a peptide and RNA driven by a chemical fuel, (a) The transformation of peptide precursors by removing the two negative charges to transient cationic anhydride as a product in the chemical reaction cycle that changes the chemical fuel EDC to waste EDU, (b) the formation of complex coacervates with RNA (poly-U). The chemical cycle controlled the influx and out flux of droplet materials. (c) Confocal images of dynamic droplets at 2 minutes after the addition of fuel. (d) The encapsulation of SunY into dynamic droplets. Reproduced from ref. 52 with permission from the Nature Publishing group Copyright 2020. | ||
Finally, Nakashima et al. developed an alternative strategy to create dissipative peptide-based complex coacervates, in which the formation and dissolution is controlled by two enzymatic reactions and two orthogonal small molecules as fuel.22 Their system is based on poly-L-lysine and either ADP or ATP as second component, which have distinctly different phase separation propensity at physiological salt concentrations. By converting ADP to ATP using pyruvate kinase fuelled by the phosphate donor phosphoenolpyruvate (PEP), coacervates were formed in a previously homogeneous solution. The lifetime of coacervates could be programmed by adding a second enzyme, hexokinase, together with glucose as penultimate phosphate acceptor. While enzymatic networks were unlikely to have been present in a protocellular setting, active coacervates controlled by enzymes can offer more control over the independent rates of condensation and dissolution, and are therefore useful as model systems to gain a better understanding of the requirements for dissipative protocells.
6. Conclusion
Compartmentalization is a fundamental property of living systems, and primitive compartments could help understand how protocells with life-like properties can emerge from mixtures of molecules. The spontaneous formation of complex or simple coacervates from peptides provides an attractive route to biomimetic protocells. Such coacervates formed by phase separation offer important advantages over other protocellular architectures, the most important of which is their ability to take up and concentrate a wide range of solutes for further reactions. In addition, it has recently emerged that coacervates can be formed from much simpler molecules (e.g., Z-FF or PRn/PO43−) than many other types of compartments, and they could thus have provided a way to concentrate building blocks and organise the assembly of a surrounding lipid shell. Instead of long, synthetic polymers, coacervates can now be made from short peptides by themselves, or combined with (oligo) nucleotides or small inorganic ions.Peptides are the ideal building blocks to create biomimetic protocells. Their diverse side group functionalities allow careful tuning of the interactions driving phase separation, the selectivity for guest molecules, and the physicochemical properties of the internal liquid phase. Moreover, they offer handles to build active systems that require or utilise the energy dissipation for functional behaviour. Even though the full range of known phase-separating peptides is still relatively modest, it is possible to deduce several core principles for phase separation that will help design the next generations of peptide-based coacervates, which have been summarized in Fig. 3.
The interactions that underlie peptide coacervation also govern the properties that make these coacervates interesting as protocells. Charge neutralisation, hydrogen bond formation and π–π stacking make the condensed interior of coacervates relatively hydrophobic and lead to the concentration of many organic guest molecules. Going a step further, the first studies have now shown that coacervates can enhance the rates of reactions of small and large molecules by either concentrating reactants, lowering the reaction barrier or stabilising intermediates or products. It can be expected that different coacervates may be tailored for different substrates and reactions. If those reactions can be driven by chemical fuel, they result in dissipative coacervate protocells, which may become capable of life-like behaviour, such as active growth, motion, and division.52 In addition, more sophisticated protocells could be made by combining coacervates with other types of compartments in a hybrid approach, or by using external (electric) fields to push coacervate-based protocells out of equilibrium, as discussed in other reviews.1–5 Overall, recent developments in peptide-based coacervates have shown that short peptides hold great promise as prebiotically plausible building blocks for (hybrid) protocells that mimic living cells.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors thank the European Research Council (ERC) for financial support under the European Union's Horizon 2020 research and innovation program under grant agreement No. 851963. M. A. gratefully acknowledges a Marie Skłodowska Curie Fellowship (project No. 839177).References
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef CAS
.
- J. P. Schrum, T. F. Zhu and J. W. Szostak, Cold Spring Harbor Perspect. Biol., 2010, 2, a002212 Search PubMed
- A. J. Dzieciol and S. Mann, Chem. Soc. Rev., 2012, 41, 79–85 RSC
- M. H. I. v. Haren, K. K. Nakashima and E. Spruijt, J. Syst. Chem., 2020, 8, 107–120 Search PubMed
- W. K. Spoelstra, S. Deshpande and C. Dekker, Curr. Opin. Biotechnol, 2018, 51, 47–56 CrossRef CAS
- M. P. Hughes, M. R. Sawaya, D. R. Boyer, L. Goldschmidt, J. A. Rodriguez, D. Cascio, L. Chong, T. Gonen and D. S. Eisenberg, Science, 2018, 359, 698–701 CrossRef CAS
- K. K. Nakashima, M. A. Vibhute and E. Spruijt, Front. Mol. Biosci., 2019, 6, 21 CrossRef CAS
- J. Wang, J. M. Choi, A. S. Holehouse, H. O. Lee, X. Zhang, M. Jahnel, S. Maharana, R. Lemaitre, A. Pozniakovsky, D. Drechsel, I. Poser, R. V. Pappu, S. Alberti and A. A. Hyman, Cell, 2018, 174, 688–699 e616 CrossRef CAS
- B. Gabryelczyk, H. Cai, X. Shi, Y. Sun, P. J. M. Swinkels, S. Salentinig, K. Pervushin and A. Miserez, Nat. Commun., 2019, 10, 5465 CrossRef
- D. Priftis, L. Leon, Z. Song, S. L. Perry, K. O. Margossian, A. Tropnikova, J. Cheng and M. Tirrell, Angew. Chem., Int. Ed., 2015, 54, 11128–11132 CrossRef CAS
- E. W. Martin, A. S. Holehouse, I. Peran, M. Farag, J. J. Incicco, A. Bremer, C. R. Grace, A. Soranno, R. V. Pappu and T. Mittag, Science, 2020, 367, 694–699 CrossRef CAS
- S. Kim, H. Y. Yoo, J. Huang, Y. Lee, S. Park, Y. Park, S. Jin, Y. M. Jung, H. Zeng, D. S. Hwang and Y. Jho, ACS Nano, 2017, 11, 6764–6772 CrossRef CAS
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, Nat. Chem., 2020, 12, 814–825 CrossRef CAS
- F. P. Cakmak, S. Choi, M. O. Meyer, P. C. Bevilacqua and C. D. Keating, Nat. Commun., 2020, 11, 5949 CrossRef CAS
- J. J. Madinya, L.-W. Chang, S. L. Perry and C. E. Sing, Mol. Syst. Des. Eng, 2020, 5, 632–644 RSC
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720–724 CrossRef CAS
- E. Te Brinke, J. Groen, A. Herrmann, H. A. Heus, G. Rivas, E. Spruijt and W. T. S. Huck, Nat. Nanotechnol., 2018, 13, 849–855 CrossRef CAS
- C. Ma, A. Malessa, A. J. Boersma, K. Liu and A. Herrmann, Adv. Mater., 2020, 32, e1905309 CrossRef
- M. Abbas, W. P. Lipiński, K. K. Nakashima, W. T. Huck and E. Spruijt, 2020 DOI:10.26434/chemrxiv.12881288.v1.
- Y. Shen, F. S. Ruggeri, D. Vigolo, A. Kamada, S. Qamar, A. Levin, C. Iserman, S. Alberti, P. S. George-Hyslop and T. P. J. Knowles, Nat. Nanotechnol., 2020, 15, 841–847 CrossRef CAS
- W. M. Aumiller, Jr. and C. D. Keating, Nat. Chem., 2016, 8, 129–137 CrossRef
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC
- T. Lu and E. Spruijt, J. Am. Chem. Soc., 2020, 142, 2905–2914 CrossRef CAS
- G. A. Mountain and C. D. Keating, Biomacromolecules, 2020, 21, 630–640 CrossRef CAS
- B. Saha, A. Chatterjee, A. Reja and D. Das, Chem. Commun., 2019, 55, 14194–14197 RSC
- S. L. Perry, L. Leon, K. Q. Hoffmann, M. J. Kade, D. Priftis, K. A. Black, D. Wong, R. A. Klein, C. F. Pierce, 3rd, K. O. Margossian, J. K. Whitmer, J. Qin, J. J. de Pablo and M. Tirrell, Nat. Commun., 2015, 6, 6052 CrossRef CAS
- B. S. Schuster, G. L. Dignon, W. S. Tang, F. M. Kelley, A. K. Ranganath, C. N. Jahnke, A. G. Simpkins, R. M. Regy, D. A. Hammer, M. C. Good and J. Mittal, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11421–11431 CrossRef CAS
- T. Kaur, M. Raju, I. Alshareedah, R. B. Davis, D. A. Potoyan and P. R. Banerjee, Nat. Commun., 2020, 12, 872 CrossRef
- P. L. Onuchic, A. N. Milin, I. Alshareedah, A. A. Deniz and P. R. Banerjee, Sci. Rep., 2019, 9, 12161 CrossRef
- J. B. Rayman, K. A. Karl and E. R. Kandel, Cell Rep., 2018, 22, 59–71 CrossRef CAS
- S. Boeynaems, E. Bogaert, D. Kovacs, A. Konijnenberg, E. Timmerman, A. Volkov, M. Guharoy, M. De Decker, T. Jaspers, V. H. Ryan, A. M. Janke, P. Baatsen, T. Vercruysse, R. M. Kolaitis, D. Daelemans, J. P. Taylor, N. Kedersha, P. Anderson, F. Impens, F. Sobott, J. Schymkowitz, F. Rousseau, N. L. Fawzi, W. Robberecht, P. Van Damme, P. Tompa and L. Van Den Bosch, Mol. Cell, 2017, 65, 1044–1055 e1045 CrossRef CAS
- B. Yang, S. Jin, Y. Park, Y. M. Jung and H. J. Cha, Small, 2018, 14, e1803377 CrossRef
- X. Li, T. Zheng, X. Liu, Z. Du, X. Xie, B. Li, L. Wu and W. Li, Langmuir, 2019, 35, 4995–5003 CrossRef CAS
- X. Wang, C. Shi, J. Mo, Y. Xu, W. Wei and J. Zhao, Angew. Chem., Int. Ed., 2020, 59, 2679–2683 CrossRef CAS
- W. Wei, Y. Tan, N. R. Martinez Rodriguez, J. Yu, J. N. Israelachvili and J. H. Waite, Acta Biomater., 2014, 10, 1663–1670 CrossRef CAS
- W. Wei, L. Petrone, Y. Tan, H. Cai, J. N. Israelachvili, A. Miserez and J. H. Waite, Adv. Funct. Mater., 2016, 26, 3496–3507 CrossRef CAS
- Y. Tan, S. Hoon, P. A. Guerette, W. Wei, A. Ghadban, C. Hao, A. Miserez and J. H. Waite, Nat. Chem. Biol., 2015, 11, 488–495 CrossRef CAS
- H. Cai, B. Gabryelczyk, M. S. S. Manimekalai, G. Gruber, S. Salentinig and A. Miserez, Soft Matter, 2017, 13, 7740–7752 RSC
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS
- J. Lu, Q. Cao, M. P. Hughes, M. R. Sawaya, D. R. Boyer, D. Cascio and D. S. Eisenberg, Nat. Commun., 2020, 11, 4090 CrossRef CAS
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef
- R. Krishna Kumar, R. L. Harniman, A. J. Patil and S. Mann, Chem. Sci., 2016, 7, 5879–5887 RSC
- W. Stroberg and S. Schnell, Biophys. J., 2018, 115, 3–8 CrossRef CAS
- E. A. Frankel, P. C. Bevilacqua and C. D. Keating, Langmuir, 2016, 32, 2041–2049 CrossRef CAS
- R. R. Poudyal, R. M. Guth-Metzler, A. J. Veenis, E. A. Frankel, C. D. Keating and P. C. Bevilacqua, Nat. Commun., 2019, 10, 490 CrossRef CAS
- E. Sokolova, E. Spruijt, M. M. Hansen, E. Dubuc, J. Groen, V. Chokkalingam, A. Piruska, H. A. Heus and W. T. Huck, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11692–11697 CrossRef CAS
- T. Y. Dora Tang, D. van Swaay, A. deMello, J. L. Ross Anderson and S. Mann, Chem. Commun., 2015, 51, 11429–11432 RSC
- B. Drobot, J. M. Iglesias-Artola, K. Le Vay, V. Mayr, M. Kar, M. Kreysing, H. Mutschler and T. D. Tang, Nat. Commun., 2018, 9, 3643 CrossRef
- P. Gobbo, L. Tian, B. Pavan Kumar, S. Turvey, M. Cattelan, A. J. Patil, M. Carraro, M. Bonchio and S. Mann, Nat. Commun., 2020, 11, 41 CrossRef CAS
- M. Tena-Solsona, B. Riess, R. K. Grotsch, F. C. Lohrer, C. Wanzke, B. Kasdorf, A. R. Bausch, P. Muller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS
- C. Donau, F. Späth, M. Sosson, B. A. K. Kriebisch, F. Schnitter, M. Tena-Solsona, H.-S. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |