
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Classic highlights in porphyrin and porphyrinoid total synthesis and biosynthesis
Mathias O.
Senge
 *a,
Natalia N.
Sergeeva
*b and
Karl J.
Hale
*c
*a,
Natalia N.
Sergeeva
*b and
Karl J.
Hale
*c
aSchool of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, The University of Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: sengem@tcd.ie; Web: http://www.twitter.com/mathiassenge
bSchool of Chemistry, University of Leeds, LS2 9JT Leeds, UK. E-mail: n.sergeeva@leeds.ac.uk
cThe School of Chemistry & Chemical Engineering, The Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, Northern Ireland, UK. E-mail: k.j.hale@qub.ac.uk
First published on 24th February 2021
Abstract
Porphyrins feature prominently in nature, be it as enzymatic cofactors, electron and exciton shuffles, as photoactive dyes, or as signaling substances. Their involvement in the generation, storage and use of oxygen is pivotal to life, while their photochemical properties are central to the biochemical functioning of plants. When complexed to metals, porphyrins can engage in a multitude of contemporary applications ranging from solar energy generation to serving as catalysts for important chemical reactions. They are also able to function as useful theranostic agents, and as novel materials for a wide range of applications. As such, they are widely considered to be highly valuable molecules, and it almost goes without saying that synthetic organic chemistry has dramatically underpinned all the key advances made, by providing reliable access to them. In fact, strategies for the synthesis of functionalized porphyrins have now reached a state of refinement where pretty well any desired porphyrin can successfully be synthesized with the approaches that are available, including a cornucopia of related macrocycle-modified porphyrinoids. In this review, we are going to illustrate the development of this exciting field by discussing a number of classic syntheses of porphyrins. Our coverage will encompass the natural protoporphyrins and chlorophylls, while also covering general strategies for the synthesis of unsymmetrical porphyrins and chlorins. Various industrial syntheses of porphyrins will also be discussed, as will other routes of great practical importance, and avenues to key porphyrinoids with modified macrocycles. A range of selected examples of contemporary functionalization reactions will be highlighted. The various key syntheses will be described and analyzed from a traditional mechanistic organic chemistry perspective to help student readers, and those who are new to this area. The aim will be to allow readers to mechanistically appreciate and understand how many of these fascinating ring-systems are built and further functionalized.
 Mathias O. Senge | Mathias O. Senge, received his PhD from the Philipps Universität Marburg in 1989. After a postdoctoral fellowship with Prof. Kevin M. Smith at UC Davis, habilitation in Organic Chemistry in 1996 at the Freie Universität Berlin, Heisenberg fellowship at FU Berlin and UC Davis, he became Professor of Organic Chemistry at the Universität Potsdam. Since 2005 he holds the Chair of Organic Chemistry at Trinity College Dublin. Mathias is recipient of fellowships from the Studienstiftung des Deutschen Volkes, the Deutsche Forschungsgemeinschaft, and a Science Foundation Ireland Research Professorship. Currently a Hans Fischer Senior Fellow at the Institute of Advanced Study at the Technical University Munich his interests include synthetic organic chemistry, hydrocarbon scaffolds, tetrapyrroles, photonics, and history of science. |
 Natalia N. Sergeeva | Natalia N. Sergeeva was born in St. Petersburg, Russia. She graduated as Diploma-Chemist from Saint-Petersburg State University. She completed her PhD thesis in organic synthesis with Prof. Klaus Burger at the Universität Leipzig, Germany. She undertook a postdoctoral appointment at the University of Potsdam and a research fellowship at Trinity College Dublin. She joined Leeds as a Lecturer in 2013. Her main interests are applied photochemistry, organic and hybrid materials for use in (photo)catalysis, dyes, optical filters for solar protection, and photochemical processes in materials and biology. |
 Karl J. Hale | Karl J. Hale received his PhD in Synthetic Carbohydrate Chemistry from King's College London in 1985. After 4 years of postdoctoral research in Professor Amos B. Smith III's group at Penn, Karl worked briefly at F. Hoffmann-La Roche, USA. From 1990–2007, he served on the Faculty of University College London, where he was Professor of Chemistry. Since 2007 he has been Professor of Organic and Medicinal Chemistry at Queen's University Belfast, where he has extensive research interests in complex natural product total synthesis. His achievements in this field have been recognized with numerous honors that include the 2007 Liebig Lectureship of the German Chemical Society and the 2011 RSC Bader Award. |
1. Introduction
Porphyrins are ubiquitous macrocyclic pigments and cofactors in Nature that are frequently referred to as the “pigments of life”.1 The term porphyrin actually derives from the Greek word for “purple” (porphyra) and it was first ascribed to these compounds on account of their often vivid purple and red colors. Porphyrins constitute a unique structural class of compounds that play many pivotal roles in Nature ranging from oxygen transport and electron transfer, to methanogenesis and photosynthesis. They can also function as novel oxidation catalysts. In medicine, they frequently serve as drugs for photodynamic therapy, and their involvement in the onset of diseases such as malaria, porphyrias, and various neuropsychiatric disorders is now well documented. Additionally, they have emerged as crucial regulatory effectors for many biochemical processes. For example, in plant physiology, they function as reaction center and accessory pigments in photosynthesis, while the essential role of heme 1 (Fig. 1) as a cofactor in biochemical catalysis, electron transport and as a respiratory pigment, needs no further elaboration. Industrially, they are some of the most important fine chemicals in existence, being employed in an ever-expanding array of applications that include pigments and oxidation catalysts, cancer therapeutics, artificial photosynthesis mediators, sensors, non-linear optical devices, theranostic agents and nanomaterials.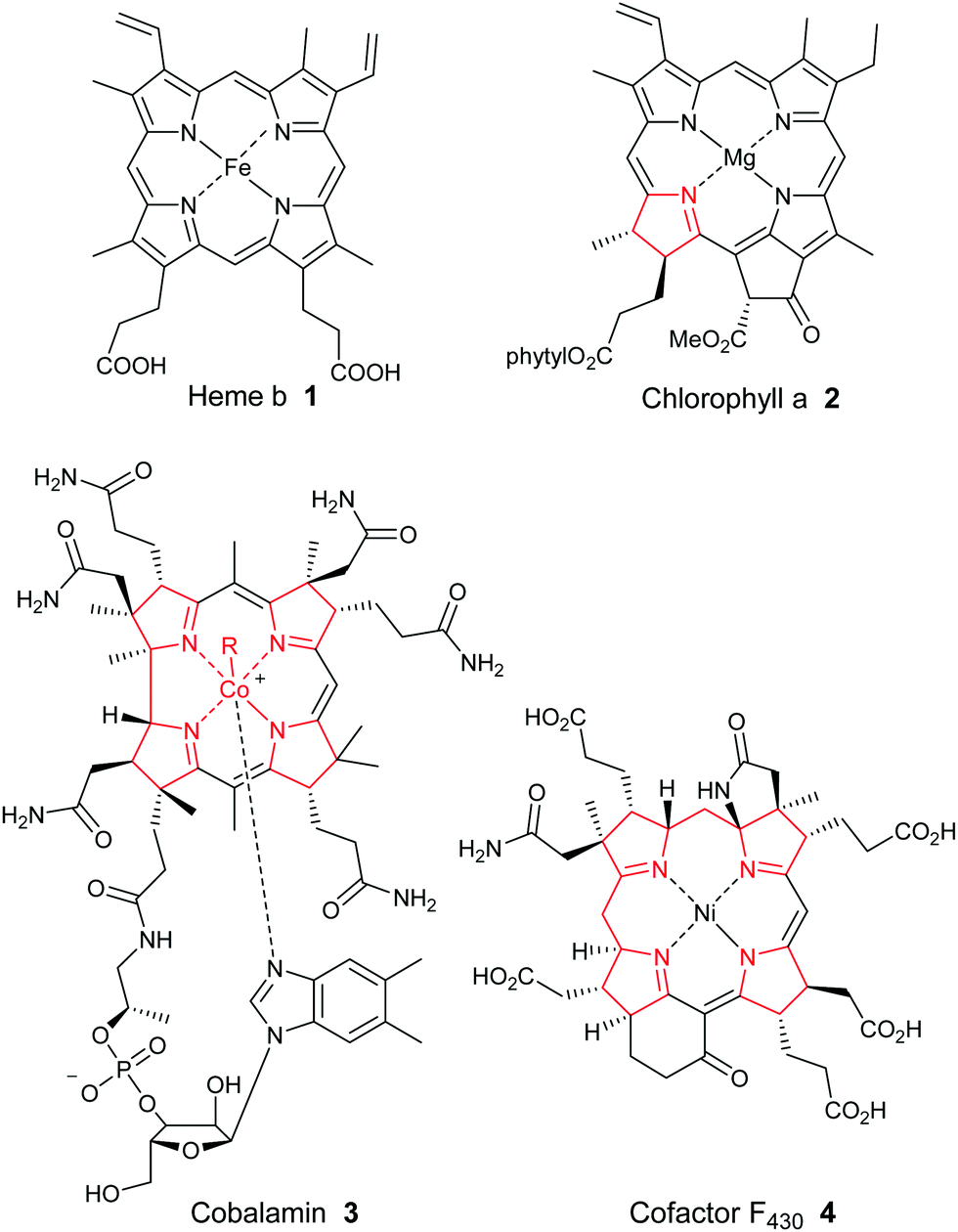 | ||
| Fig. 1 Selected natural cyclic tetrapyrroles. | ||
Because of their biological relevance, heme b 1, chlorophyll a 2, the highly-reduced corrinoids (cobalamin 3 and vitamin B12) and the corphins (e.g., cofactor F4304) (Fig. 1) have each been the target of numerous synthetic studies over the years, some dating back well over a century.2,3 The importance of these studies is illustrated by the fact that several Nobel prizes have been awarded wholly, or in part, for organic synthesis studies in this area. Examples have included the 1915 prize to Willstätter for his work on establishing the constitution of the chlorophylls, the 1930 award to Hans Fischer for his synthesis of haemin, and the 1965 Nobel prize that was awarded to R. B. Woodward “for his outstanding achievements in the art of organic synthesis”, which almost certainly took into account his masterly total synthesis of chlorophyll a. Woodward later crowned that remarkable achievement with a spectacular formal total synthesis of vitamin B12 with Eschenmoser in 1972, and a later full total synthesis in 1976. To this day, cobyric acid and vitamin B12 represent the most complex porphyrins ever synthesized (see Scheme 7 and Section 2.4 for a summary of their synthetic efforts).
Porphyrins are [18π]-electron heteroaromatic compounds in which the aromatic character of the underlying tetrapyrrole moiety, and the reactivity of the functional groups present in their side chains, governs their rich chemistry. Three different classes of 24-atom macrocyclic tetrapyrrole are found in Nature, and each is differentiated by their respective degree of oxidation. The various classes of porphyrin are: the porphyrins themselves (5, e.g., hemes), the chlorins (6, e.g., chlorophylls and bacteriochlorophylls c–e), and lastly, the (iso)bacteriochlorins (7a and 7b, e.g., bacteriochlorophylls a and b or sirohydrochlorin, respectively) (Fig. 2). The chemistry of porphyrins is governed by five different positions: the core nitrogen atoms, the central metal ions (if present), the α- (Ca = 1, 4, 6, 9, 11, 14, 16, 19) and β-pyrrole (Cb = 2, 3, 7, 8, 12, 13, 17, 18) positions, and the connecting Cm-positions (often called meso in the older literature = 5, 10, 15, 20) (see formula 5). Each has the capacity to coordinate many different types of metal ions within the macrocyclic inner core of nitrogen atoms. This, along with their conformational flexibility,4 their rich electro- and photo-chemistry, the variability of their side chains, and their ability to interact with apoproteins,5 accounts for their unique role in Nature and their widespread utility in the applied sciences.6 Even more reduced species are based on the framework exemplified by F430 4 and the 23-atom corrin system 8. This is complemented by numerous synthetic porphyrinoids with altered (expanded, contracted, isomeric, ring- or atom-modified) macrocycles.7
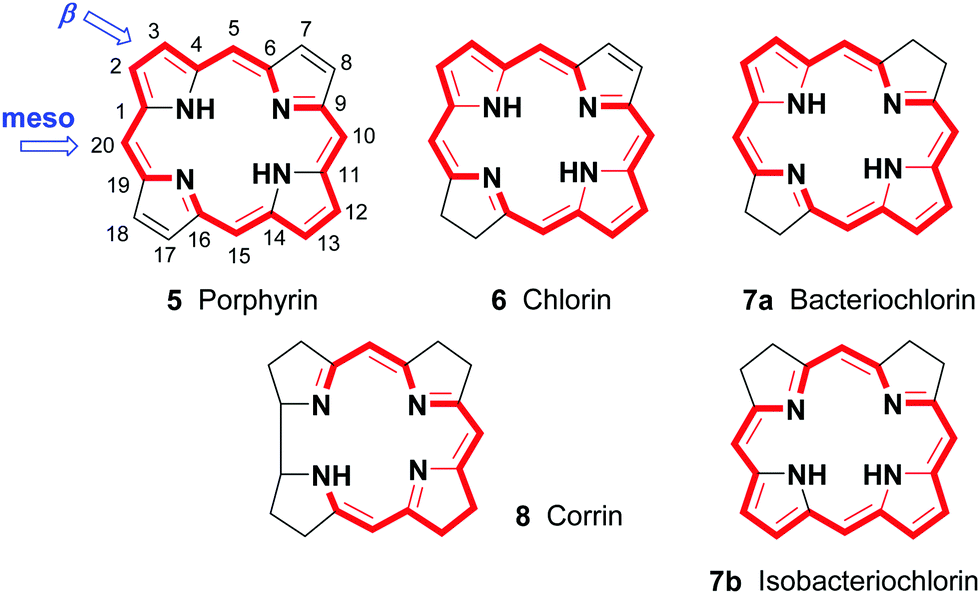 | ||
| Fig. 2 Fundamental tetrapyrrole skeletons in Nature. | ||
With respect to their chemistry, synthesis and reactivity, much valuable information on porphyrins can still be found in many of the older books. Willstätter's and Fischer's classic texts2,8 were followed, many years later, by a much more modern-day monograph on porphyrins in 1975 by Smith.9 Thereafter, a six volume series appeared from Dolphin in 1978,10 and this was followed by Scheer's authoritative book on chlorophylls,11 and most recently, by the multi-volume series, edited by Kadish, Smith and Guilard.6 The latter now comprises over 50 volumes and indicates that a comprehensive review of the porphyrin literature is no longer possible. Currently, approximately 2500 papers are published annually on porphyrins. Thus, in this brief review, we can only touch on a few of the many synthetic highlights in this area, and our primary aim has been to use examples of past, as well as current syntheses, to illustrate how important concepts and design strategies have developed in porphyrin chemistry.
Retrosynthetically, a range of different approaches can be envisaged for the synthesis of porphyrins (Scheme 1). In the end, however, the precise synthetic strategy chosen is often determined by whether the appropriate pyrrole precursors are readily available. Thus, the simplest way of disassembling a porphyrin is to split the parent structure into its four constituent pyrrole units and to couple these with appropriate aldehydes under acid-catalyzed conditions; typically, this is then followed by an oxidation of the non-aromatic porphyrinogen intermediate to the target porphyrin (path A).12 Obviously, this mixed condensation path can sometimes be attended by the formation of unsymmetrical porphyrin product mixtures. Alternative approaches therefore use a variety of 2-pyrrole component precursors 11 to effect similar condensations (path D), or they rely on the total synthesis of open-chain bilanes 9 (path B) followed by ring-closure reactions.
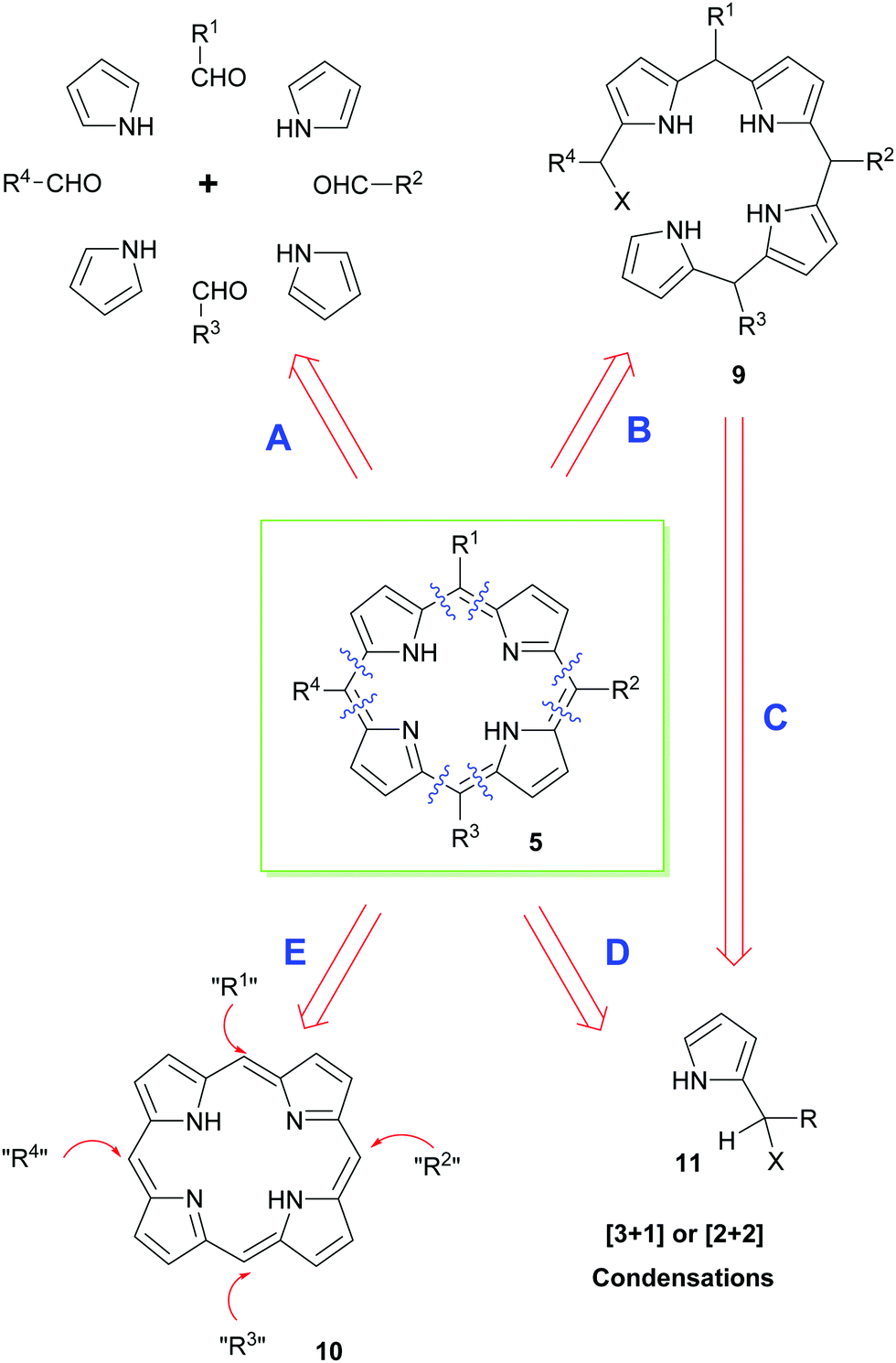 | ||
| Scheme 1 Some of the approaches used to build up porphyrins. | ||
Until very recently, most acid-catalyzed condensation methods were accompanied by competitive scrambling of the pyrrole units during the key porphyrin ring-assembly step. Typically, such rearrangements proceed by acid-mediated cleavage and recondensation of the pyrrole units from the nascent oligopyrrole chain as the porphyrin ring-assembly occurs.13 As will be seen below, this situation is often less problematical with β-substituted porphyrins, e.g., naturally-occurring heme and chlorophyll derivatives. Nonetheless, because of the aforementioned difficulties, significant effort has been expended on the development of alternative syntheses that exploit functional group interconversions (FGIs) and novel coupling reactions to introduce the requisite side chains (i.e. that functionalize unsubstituted porphyrin 5 according to path E in Scheme 1). In the coming sections, we will outline some of the main approaches used for securing meso- or β-substituted porphyrins.
2. Classic syntheses of natural porphyrins
Due to their biological significance, haemin and chlorophyll a (2) became primary synthetic targets of the early porphyrin chemists until syntheses were successfully accomplished of the former in 192814 and the latter in 1960,15 respectively. Despite the importance of these ventures, and the significant effort that was put into building related structures, only limited advances were made thereafter with regard to improving the original chemistry developed for the preparation of these molecules. Even today, these two monumental synthetic achievements stand out in terms of their planning, logic, and efficiency; which is not something that one can often say for many syntheses accomplished in the early-to-middle part of the 20th Century.16,17 This fact notwithstanding, it is still often easier to isolate the relevant compounds from their natural sources rather than synthesize them de novo.2.1 The type-isomer problem
All naturally-occurring porphyrins are unsymmetrically β-substituted macrocycles. Formally, natural porphyrins are derived by tetramerization of porphobilinogen 16, a pyrrole with one acetic acid and one propionic acid β-substituent. Tetracyclization of a pyrrole with two different β-residues under standard condensation conditions yields four different porphyrins. Historically, these are known as ‘type-isomers’ (illustrated for the etioporphyrins 12–15 in Fig. 3). Biosynthetically the fourth pyrrole unit (D) is inserted in reverse fashion and all biological porphyrins are descendants from the type III uroporphyrinogen isomer 17. During subsequent biosynthetic steps some of these side chains are converted to methyl and vinyl groups, and the macrocycle of heme b 1 has three different types of β-substituents (methyl, vinyl, propionic acid) and thus is based on the protoporphyrin IX type isomer 18.18 Solving the problem of how to generate only one of these isomers lies at the heart of any natural porphyrin synthesis and the same issue arises when targeting non-natural porphyrins with different meso- and/or β-substituents.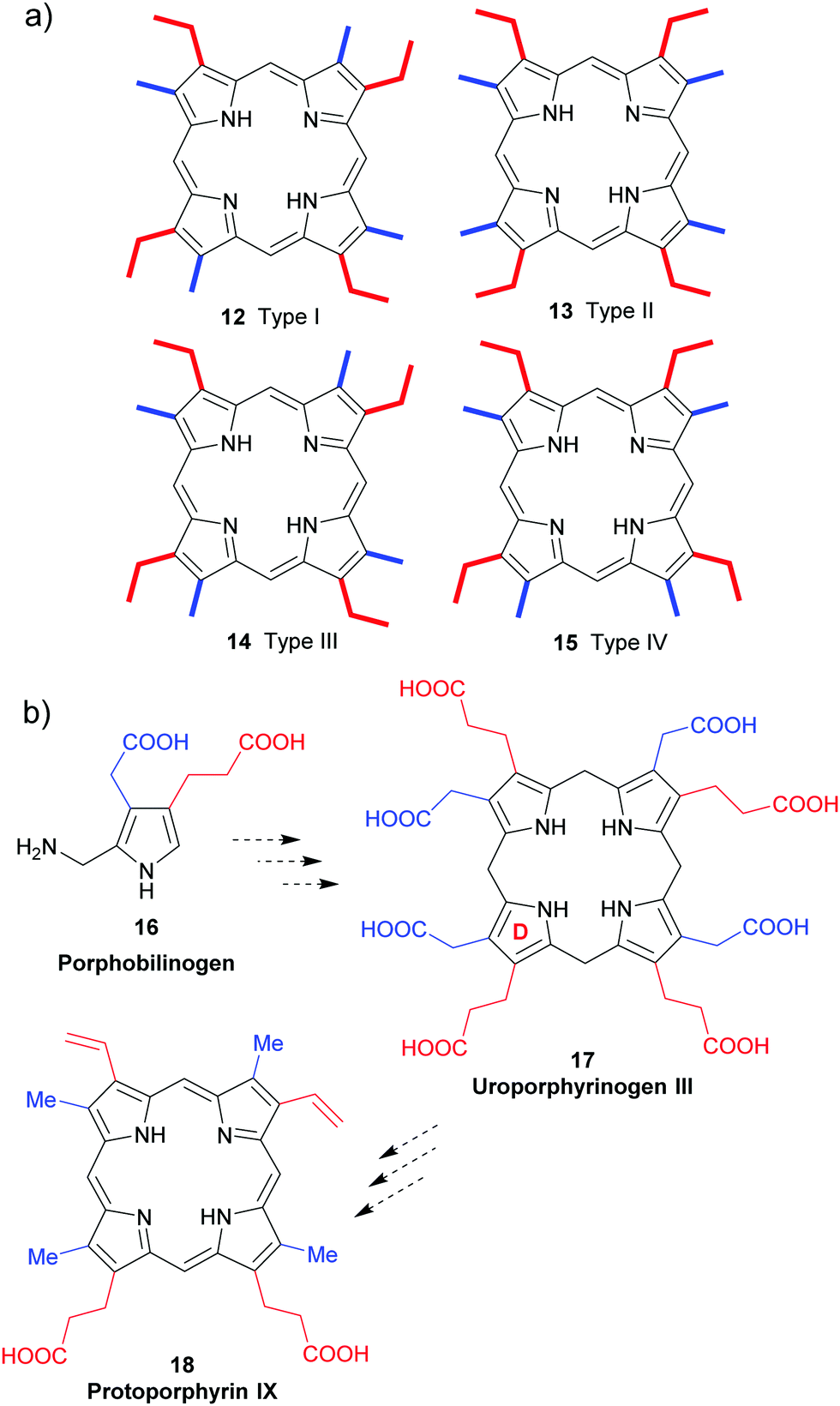 | ||
| Fig. 3 The type isomer ‘problem’ in porphyrin synthesis. (a) Etioporphyrin type isomers. (b) Snapshot of protoporphyrin IX biosynthesis. | ||
2.2 Total synthesis of haemin (Fischer)
As just outlined, the main problem that has to be confronted in the synthesis of haemin (the iron(III) analogue of heme b 1) and chlorophyll a stems from the unsymmetrical way in which the pyrrole ring-D unit is incorporated into the tetrapyrrole array. While rings-A, -B, and -C have their β-substituents derived from the same 3-acetic acid and 4-propionic acid residues, ring-D is functionalized with the acetic acid residue in the 4-position, and the propionic acid group in position-3. This is achieved in vivo through inversion of one of the pyrrole rings.How Fischer decided to address the synthesis of haemin [chloro(protoporphyrinato IX)iron(III) 19] (Scheme 2) was simply to split the tetracyclic pyrrole at its midriff, into two new dipyrromethene halves: the northern sector 23 and the southern sector 24.19 These, in turn, were to be derived from the respective pyrrole subunits (25 + 26 and 2 × 27) with ring-closure to 22 ultimately being achieved by a tandem [2 + 2]-condensation/cyclization in which the enamino vinyl group of 23 would serve as the initial attacking nucleophile on 24.
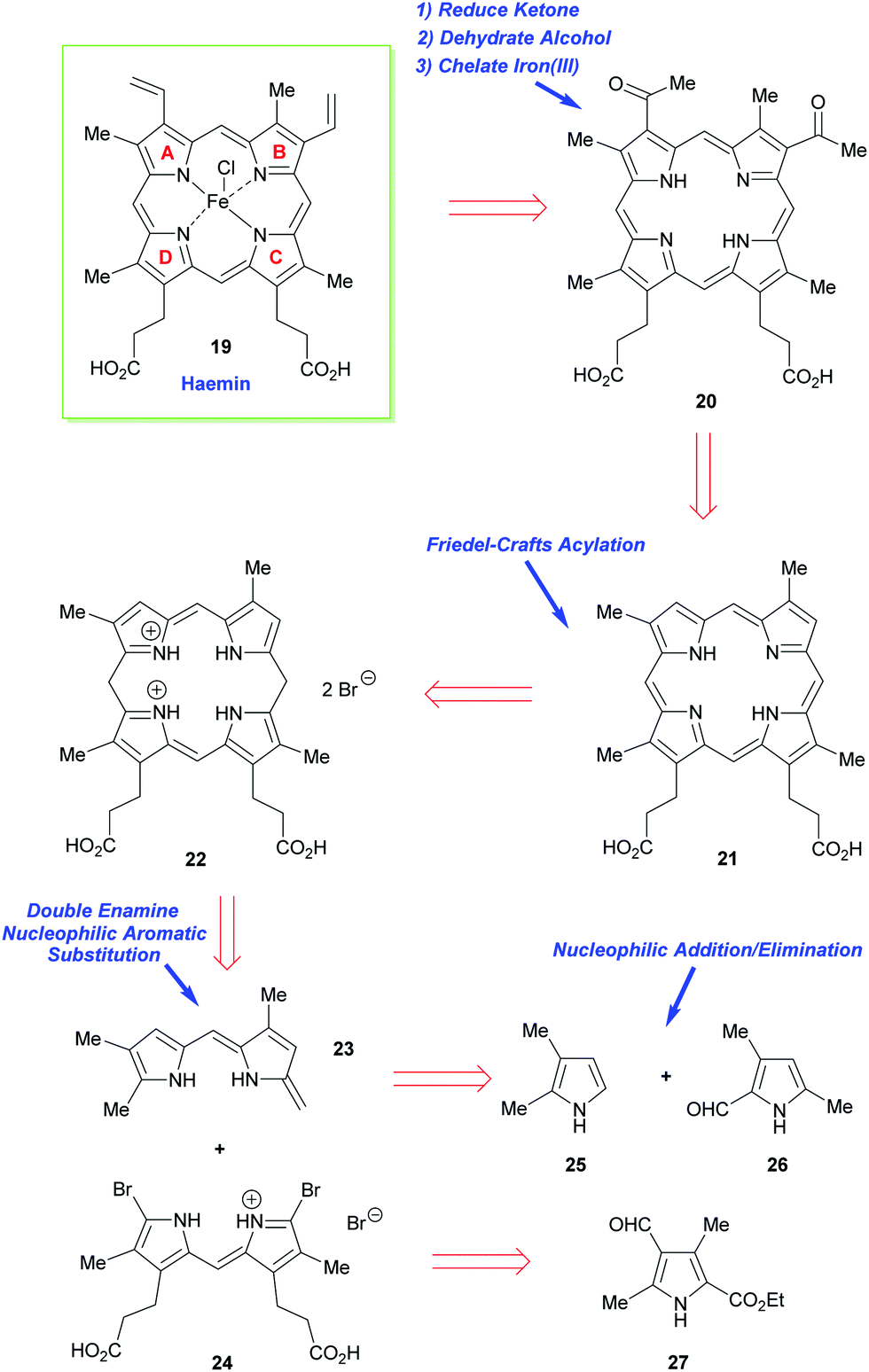 | ||
| Scheme 2 Fischer's retrosynthetic analysis of haemin. | ||
The forward synthesis that eventually led to haemin is summarized in Scheme 3. The all-important attachment of the vinyl groups to the haemin skeleton (a long-standing problem in porphyrin chemistry)20 was a highly challenging task for Fischer, since he only had harsh reaction conditions and reagents at his disposal. Likewise, the synthesis was less logically planned than it appears in Scheme 2 but governed by an ‘intuitive feel’ of what reactions might prove useful. Even so, he did manage to accomplish the synthesis successfully, by initially targeting the deuteroporphyrin 21 in his route,19b and performing a Friedel–Crafts C-acetylation mediated by Fe(III) and AlCl3, followed by a Meerwein–Ponndorf–Verley type-reduction with potassium ethoxide. This was followed by an acid-mediated alcohol elimination and an Fe(III) complexation to complete this pioneering achievement (note, while the natural heme contains Fe(II), haemin is an Fe(III) species).
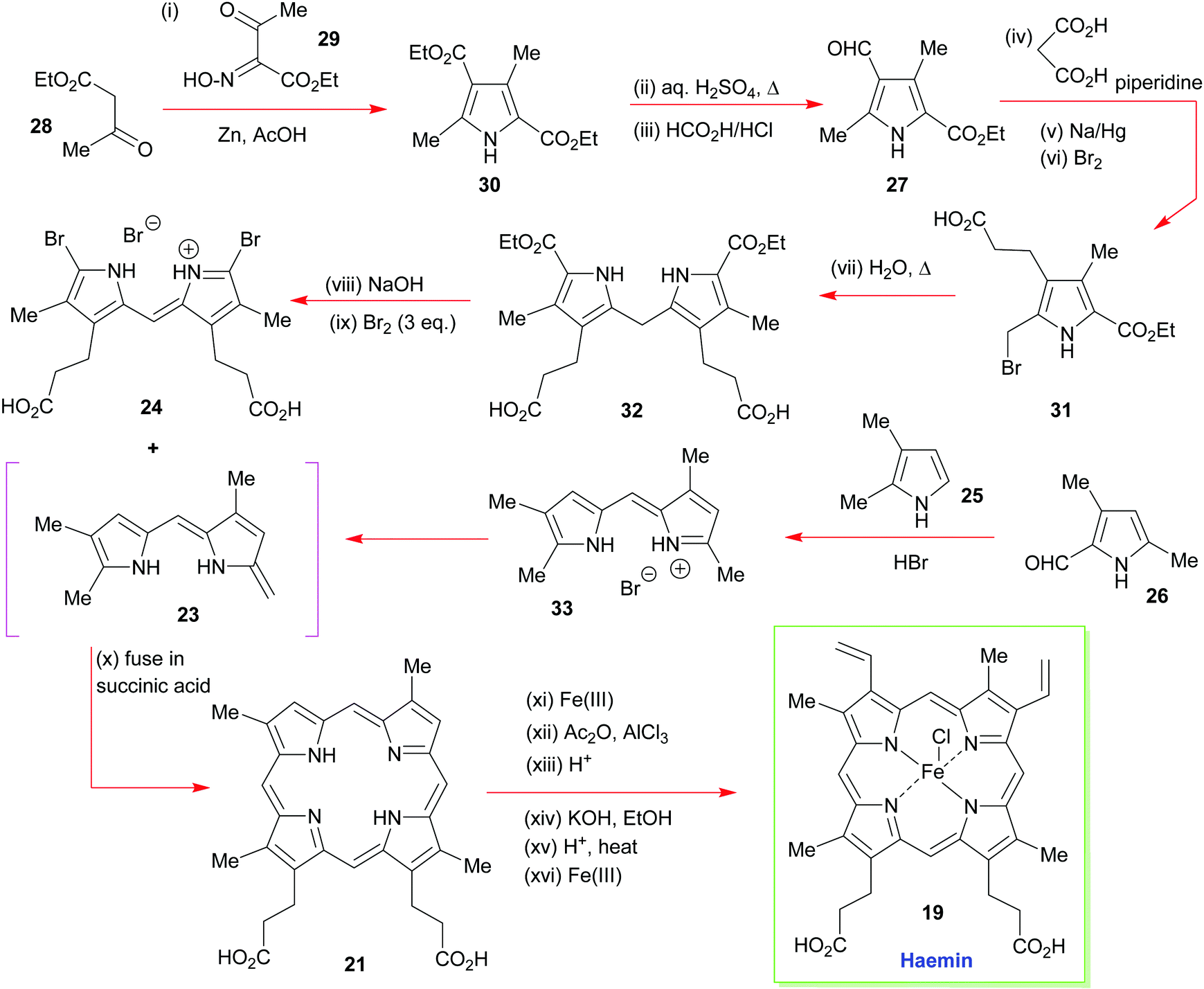 | ||
| Scheme 3 A summary of Fischer and Zeile's synthesis of haemin.19a | ||
2.3 Total synthesis of chlorophyll a (Woodward)
Thirty years later, the great R. B. Woodward tackled the synthesis of chlorophyll a (2) by making its chlorin e6 trimethyl ester precursor 34 his primary target.15,16 While conceptually the main problem in both syntheses revolved around construction of an unsymmetrical arrangement of the ABCD-(tetra)pyrrole, for chlorophyll a this presented many added challenges. On the one hand it required a synthesis of a dihydroporphyrin (i.e., a chlorin), while on the other, it demanded a synthesis of an isocyclic pentanone ring-E fragment, which was anything but a trivial task. The multiple arrays of remote asymmetric centers within chlorophyll a also presented a very severe synthetic challenge, and they must have seemed even more foreboding at the time Woodward addressed the problem.While it was well known that chlorophyll a (in the form of the respective pheophorbide) could be prepared via a Dieckmann cyclization from 34,21,22 this precursor did carry two aza-enolizable positions, but fortunately the pathway leading to the desired cyclopentenone was the one favored.
Woodward's plan for the synthetic assembly of 34 (Scheme 4) was quite distinct from Fischer's earlier synthesis of haemin. Woodward cut the tetrapyrrole ring system of 34 into the “Western” and “Eastern” halves 41 and 42 and he envisioned moving forward via the tethering strategy shown. Because pyrrole aldehydes were well known to have low electrophilicity (due to their conjugative enolization with the pyrrole N-atom), and difficulties had already been encountered during an earlier attempted intermolecular union of structurally-related “Eastern” and “Western” fragments, Woodward opted to establish the C(4)–C(5) bond between the intermediates 41 and 42 in, a then, quite revolutionary way. In this regard, he would initially brace the two units together (as in 40) and then rely upon the higher nucleophilicity of the pyrrole α-position to sequentially connect the C(4) and C(5) carbons together, and the C(15) and C(16)-carbons would be conjoined subsequently (in the manner shown in 39). This would, of course, give rise to a nonasubstituted porphyrin 38 after further functional group manipulation. Other critical elements in Woodward's synthetic plan were: (i) his recognition that thioaldehydes are more electrophilic than normal aldehydes, due to the greater dipolar character of thiocarbonyls, and (ii) the understanding that the newly installed C(15)-substituent would set up a sterically-strained ring system that would greatly impact the reactivity of the flexible porphyrin to allow a porphyrin 38 → purpurin 37 conversion. However, Woodward's approach would require a late-stage regiochemical correction of the mode of annulation in ring-D. This would entail the purpurin 37 being photo-oxidatively transformed into 36 and its α-keto ester being jettisoned en route to 35, which would potentially be elaborated into 34.
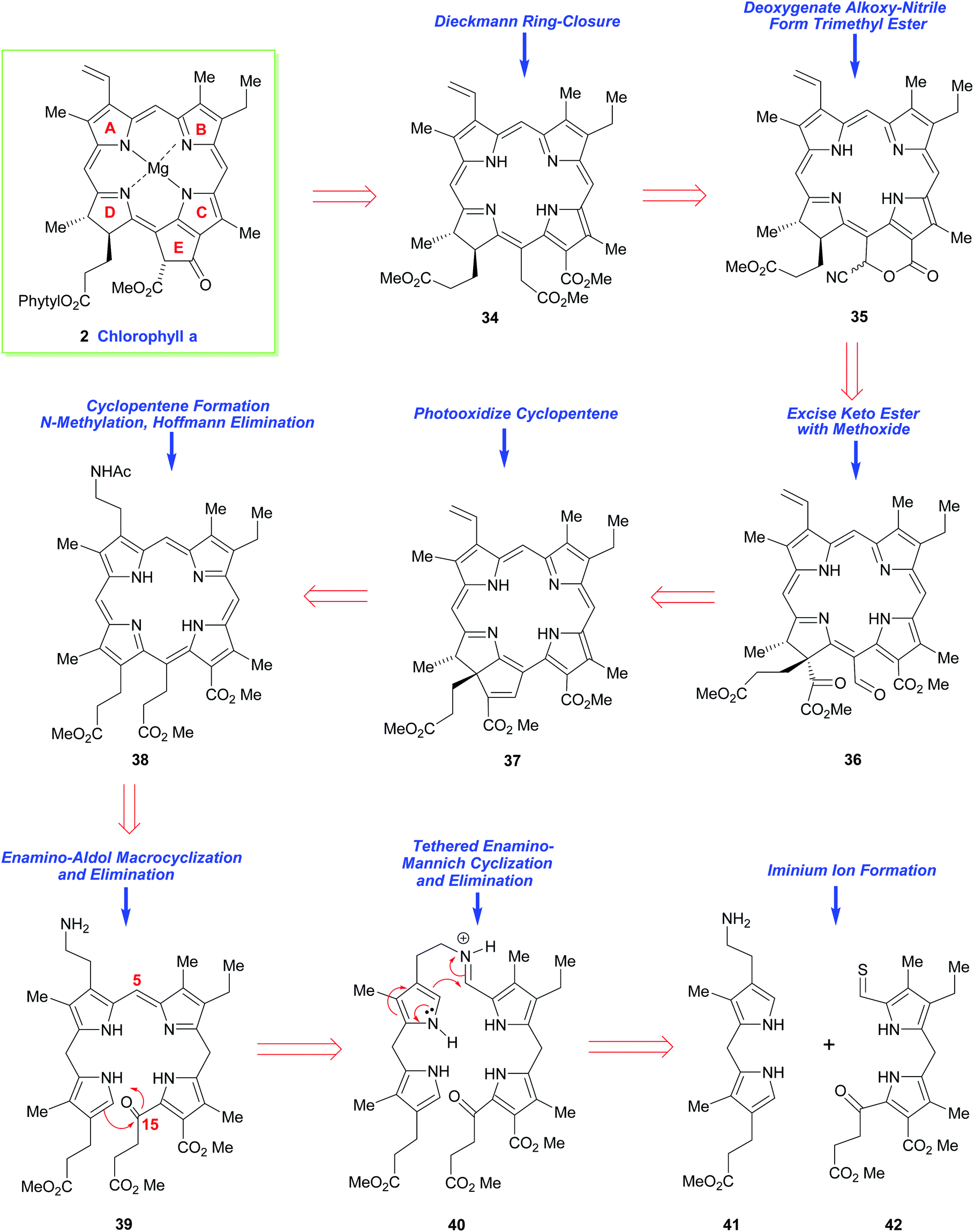 | ||
| Scheme 4 Woodward's retrosynthetic analysis of chlorophyll a. | ||
Woodward commenced his synthesis (Schemes 5 and 6) from readily available tetrasubstituted pyrrole 43 (Scheme 5) (available from Knorr's pyrrole23) where the dicyanoalkene moiety would serve as a base-removable protecting group for a, soon to be unmasked, pyrrole aldehyde via a malononitrile anion elimination. Initially, Woodward regioselectively chlorinated 43 with sulfuryl chloride in acetic acid to obtain 44 and this was then subjected to a Friedel–Crafts-type alkylation with the pyrrole 45; the end result was 46 which itself was subjected to a further round of electrophilic aromatic substitution, on this occasion, with the acid chloride 47 in the presence of anhydrous zinc chloride. Treatment of the product 48 with dilute sodium hydroxide nicely unveiled the pyrrolo-aldehyde unit that was needed for formation of the iminium hydrobromide 49, but this overall sequence did require a reinstatement of the methyl ester groups along the way. A major discovery by Woodward's team was that if the iminium ion 49 was exposed to hydrogen sulfide under basic conditions, it could be smoothly converted into the thioaldehyde 42 without perturbation of the pendant ketone.
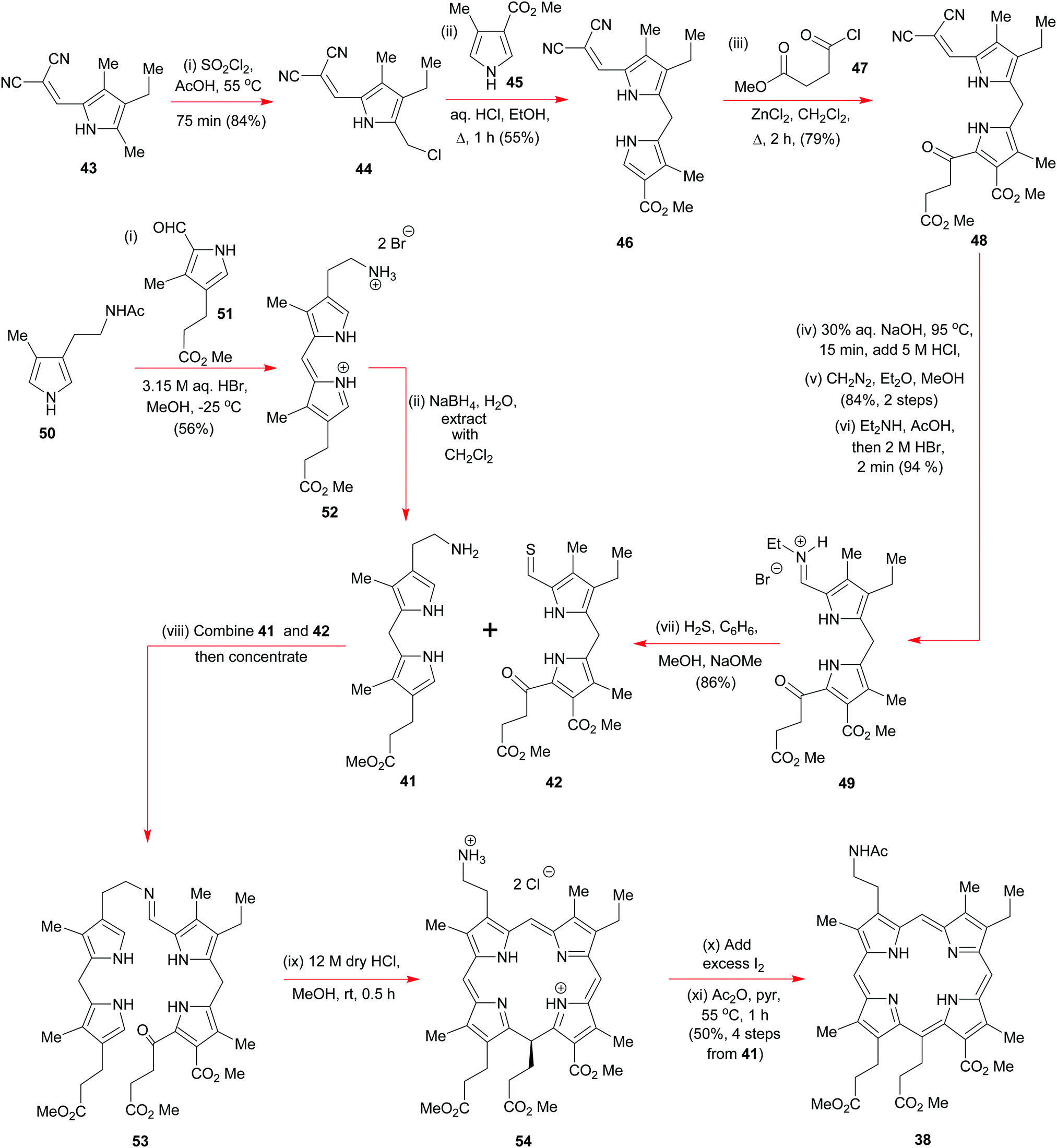 | ||
| Scheme 5 The early stages of Woodward's total synthesis of chlorophyll a. | ||
 | ||
| Scheme 6 Woodward's completion of the total synthesis of chlorophyll a. | ||
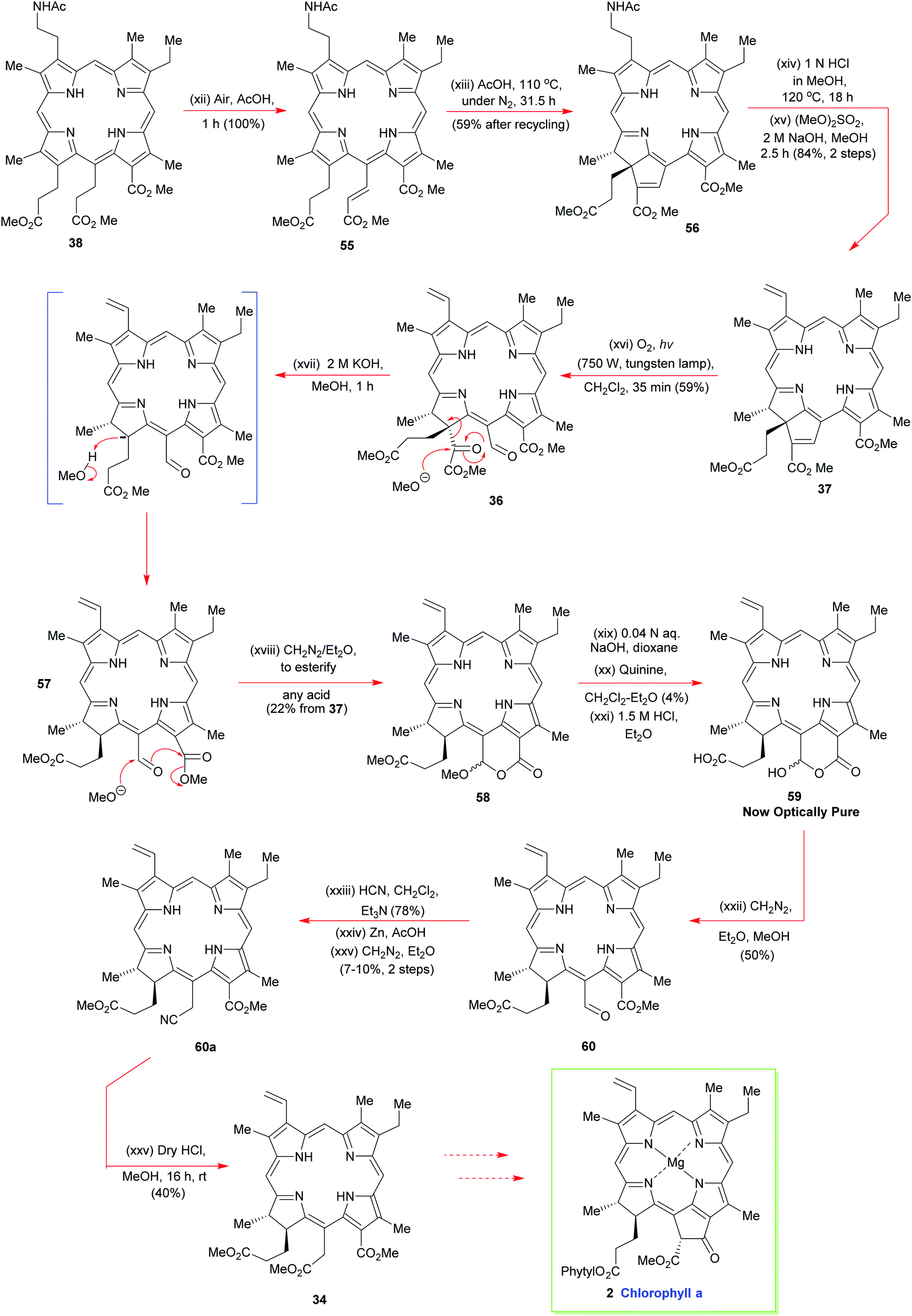
Discovery of the latter transformation was an important breakthrough, for it now paved the way for the key intermolecular Schiff-base tethering-operation, and for the subsequent intramolecular electrophilic aromatic substitution that was needed to establish the C(5)–C(6) bond. Formation of the imine 53 from 41 and 42 required no catalysis at all, but before the key C–C-bonding steps could be effected, 53 had to be exposed to concentrated anhydrous HCl in methanol. Initially this led to 54via40 but upon iodine oxidation and N-acetylation, the more stable porphyrin 38 was obtained in 50% overall yield for the four steps from 41 and 42. In his 1961 Pure Appl. Chem. article,15b Woodward drew special attention to the fact that well over 50 g of 38 was prepared by his group, attesting to the reliability of the chemistry in skillful hands, and its ability to be scaled up.
A truly remarkable reaction sequence now followed. It was observed that 38 spontaneously oxidized to 55 in hot acetic acid in the presence of air (Scheme 6). Further prolonged heating of 55 in acetic acid at 110 °C then set up an equilibrium between 55 and 56 from which the ring-annulated 56 could be isolated in good yield after multiple recycling schedules; this cyclization beautifully installed the two stereocenters of ring-D. Hot aqueous HCl in methanol then detached the N-acetyl grouping from 56 to allow exhaustive N-methylation to proceed on the primary amine, using base and dimethylsulfate. It is noteworthy that the pyrrole N-atoms were left completely untouched by these reaction conditions and, significantly, in situ Hoffmann elimination of the quaternary ammonium salt proceeded thereafter with great facility.
Woodward now implemented his key photo-oxidation step on 37 to fracture its cyclopentene ring system and nicely expose the all-important meso side chain that was needed for further synthetic progress towards the target (Scheme 6). The α-keto ester unit was then excised from 36, in the manner shown, using potassium methoxide. The final resting place for this methanolysis sequence was lactone 58. It was then hydrolyzed with dilute aqueous sodium hydroxide to give the lactone hemiacetal 59, which was then subjected to an optical resolution with quinine. The elaboration of optically pure 59 now involved esterification and installation of the C(15)-acetic acid sidechain to yield 34via60a. Overall, the synthesis of 34 required 46 individual steps15 and it remains an unsurpassed highlight in tetrapyrrole chemistry, its only real weak point being its optical resolution step. However, this aside, if the synthesis was racemic, it would otherwise be remarkably efficient, and still hold its own with many modern-day porphyrin syntheses, which makes Woodward's superb achievement even more remarkable.
2.4 Total synthesis of vitamin B12 (Woodward, Eschenmoser)
The total synthesis of vitamin B1224 was probably the most complex and longest synthetic endeavor ever undertaken in preparative porphyrin chemistry. This 60-step synthesis was completed in 1973 by the combined teams of Woodward and Eschenmoser.25 Preceding decades of work had seen contributions from many eminent chemists. This included the early work on vitamin B12 degradation by Alexander R. Todd and Alan W. Johnson, the landmark structure elucidation of cobyrinic acid by Dorothy Crowfoot-Hodgkin in 1956, and identification of the adenosyl ligand by Horace Barker which all helped to firmly establish its structure.26Looking back over the vitamin B12 total synthesis venture, it involved a veritable Who's–Who of top chemists; with contributions from Jean-Marie Lehn, Ian Fleming, John Carnduff, Subramania Ranganathan, Roald Hoffmann, Heinz Gschwend, Ivan Ernest, Ernest Hamanaka, Yoshito Kishi, Alexander Wick, Kevin M. Smith, Edward D. Brown, Kenneth Richardson, D. John Faulkner, Elmar Konz, Romeo Paioni, Helmut Hamberger, Stanley Roberts, Volker Jager, Pat Confalone, Kaspar Burri, Walter Fuhrer, Hans Maag, Dennis Keith, and Mark Wuonola, to name only a few.27,28 Overall, this momentous task took more than 16 years to complete, involved over 100 chemists, conquering 80 synthetic steps, and it provided crucial experimental support for important concepts such as the Woodward–Hoffmann rules.28 It will always remain a landmark in the annals of chemical synthesis.29 Taking into account the chemical and analytical limitations of the time (NMR resolution, sample requirements for standard analytical techniques), it is one of the most challenging and arduous organic syntheses ever undertaken.
To describe in detail the total synthesis of vitamin B12 alone would require as many pages as the present review, and this mammoth task has already been undertaken with care by Nicolaou and coworkers in a series of masterly reviews,29 as well as by Eschenmoser himself in 2015.25h Given this, we have thus confined ourselves here to providing a detailed summary of the original Woodward–Eschenmoser joint synthetic plan (see Scheme 7, strategy 1) and we have then gone on to show the later stages of Eschenmoser's independent second route (strategy 2) which intersected with the same fundamental macrocycle 63 and other intermediates of the first generation Woodward–Eschenmoser route, but which developed much improved, higher yielding, reactions for effecting the final 8-step end game needed to secure cobyric acid (61), having learnt much from the original first-generation pathway that had been developed jointly with Woodward.
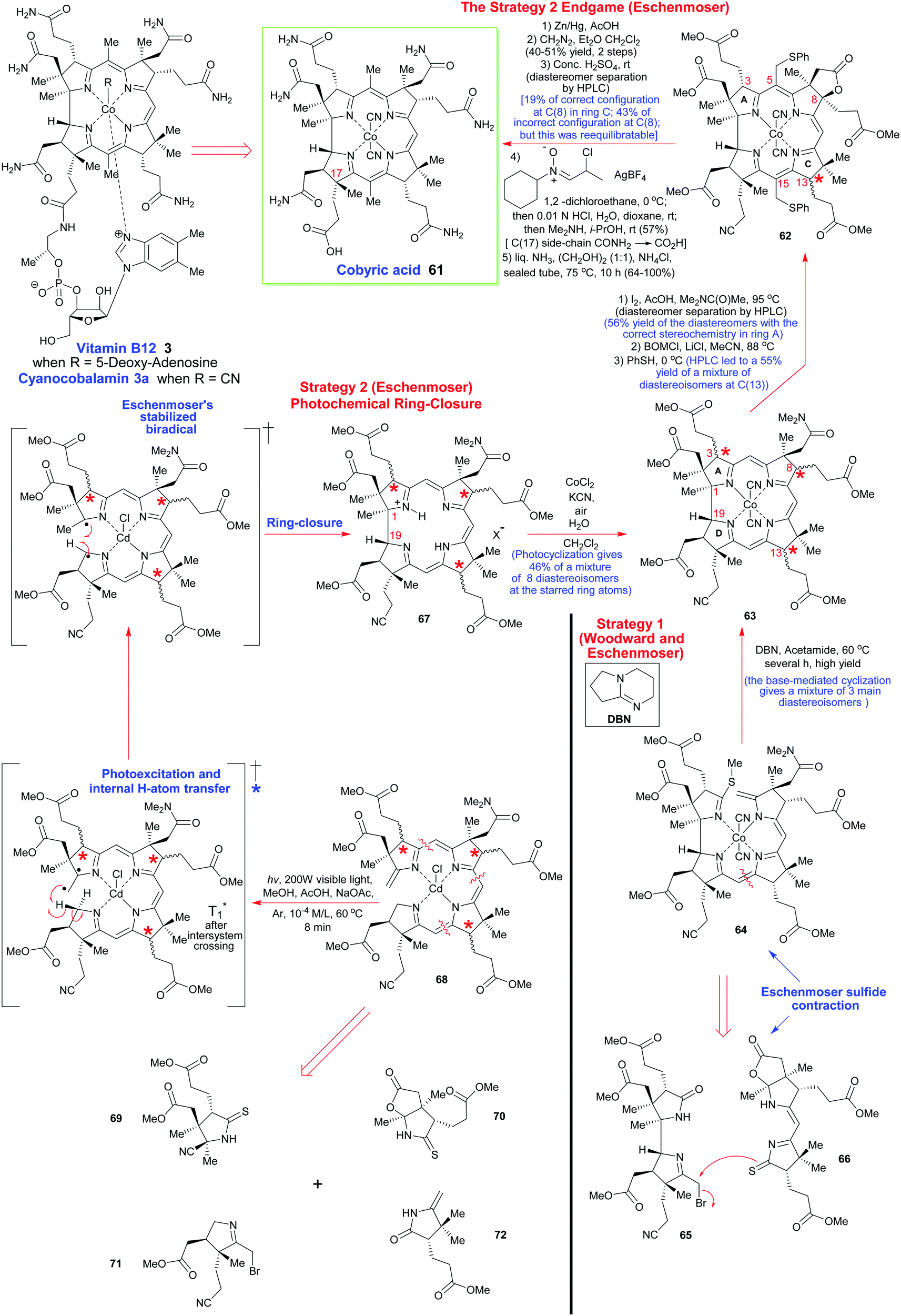 | ||
| Scheme 7 A summary of the key steps involved in Woodward's and Eschenmoser's joint synthesis of cobyric acid (strategy 1) and Eschenmoser's independent total synthesis (strategy 2) which came later. | ||
Now given that the transformations for appending the adenosyl ligand had already been described by Friedrich et al.,30 both synthetic ventures initially stopped at this point, with completion of a total synthesis of cobyric acid 61.25j However, it is now widely known that Woodward and Wuonola repeated the latter chemistry to complete a full total synthesis on March 17, 1976.28
How the two synthetic strategies 1 and 2 differed from one another was in the synthetic end games that they employed, and in the way they both went about building up the key cyclization precursors 64 and 68, as well as how they effected their respective macrocyclic ring-closures. In the original collaborative strategy 1, Woodward and Eschenmoser favored a [2 + 2]-type union for crafting 64, while in Eschenmoser's later stand-alone pathway, a tetramerization approach was favored for the assembly of 68.
Strategically, and taking into account both the successes and the weaknesses of the original ‘end game’ for securing vitamin B12, Eschenmoser's second-generation pathway once more utilized the same C(8)-lactone that had previously featured in Woodward and Eschenmoser's first-generation route to cobyric acid 61. This was once more selected to allow the requisite C(5)- and C(10)-C-methylations to be successfully achieved.
However, on this second pass through, the Eschenmoser team were now fully aware of the tremendous stereochemical lability of the C(3), the C(8) and the C(13) stereocenters in their intermediates post-macrocyclization, and they thus approached the second-generation synthesis with a different mind-set. As Eschenmoser and Winter succinctly put it: “Three of the centers of chirality [marked by stars in Scheme 7] proved to be configurationally labile in the intermediates of the chromophore synthesis.” Recognition of this fact, and a subsequent judicious use of preparative HPLC at various key points in the final reaction sequence did, of course, allow good synthetic progress to eventually be made, and a new improved, second-generation, route was thus crafted (see Scheme 7).
In a new second strategy, the initial aim was to re-synthesize the Woodward–Eschenmoser intermediate 63 by a new photochemical cyclization reaction that involved the seco-corrin 68, which itself existed as a mixture of diastereoisomers. The key macrocyclization produced a mixture of 8 different stereoisomers in total (which was a much greater number than in strategy 1), but this highly intimidating number of isomers could quickly be reduced to just 4 through the judicious use of a novel iodinative lactonization tactic involving I2, AcOH, and Me2NC(O)Me at 95 °C which stereospecifically annulated a cis-fused γ-butyrolactone ring onto ring B, to thus impose stereochemical specificity at C(8). Despite 4 stereoisomers now being formed, still, this represented a considerable improvement in the overall complexity of the ensuing mixture, for after fractionation of these stereoisomers by preparative HPLC, a collection of stereoisomers of structure 62 could thereafter be obtained in 56% overall yield, which now had the correct configuration within ring A.
This partial purification thus set up an exceedingly clever double enamine-benzyloxymethylation reaction for the regioselective C-alkylation of C(5) and C(15). This process proceeded with total regiocontrol, due to the C(10) carbon atom being strongly sterically shielded by the C-ring geminal dimethyl groups at C(12). As with strategy 1, this approach was again allied with a conceptually similar thiophenylation/reductive desulfurization/lactone C(8)-deoxygenation tactic, except now, the key multi-step triple reduction was performed on the 62 diastereoisomeric mixture with Zn/Al amalgam in AcOH. The latter reagent implemented the desired transformation quite effortlessly and in greatly improved yield (40–51%), which is really quite impressive given the enormity of the three reactions that were concurrently being effected, on multiple diastereoisomers! The greatly increased efficiency of this new triple reduction protocol contrasted sharply with Woodward and Eschenmoser's original strategy 1, where the reduction sequence used RANEY® Ni for this purpose, and only delivered a quite low yield of the desired C(7)-carboxylic acid and, thereafter, the requisite methyl esters following diazomethane treatment. However, even this new Zn/AcOH reduction procedure still did not permit fractionation of the C(8)-deoxygenated diastereomeric mixture of methyl esters that was so obtained.
To effect the desired separation, it proved necessary to advance forward by one step, and convert the various nitrile-containing side-chains present at C(17) in the different diastereoisomers to primary amides by acid hydrolysis with concentrated sulfuric acid. This method for amide generation from the individual nitrile-containing components had previously been used by Woodward and Eschenmoser in their first generation strategy to cobyric acid (61), and was reported to give rise to a 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28 (approx. 2.6:1) mixture of C(8)-epimers enriched in the undesired neo configuration needed for the acquisition of 61. Significantly, this acid hydrolysis step was accompanied by total epimerization of the C(13)-stereocenter to the natural stereochemistry found in cobyric acid (61) itself!
:28 (approx. 2.6:1) mixture of C(8)-epimers enriched in the undesired neo configuration needed for the acquisition of 61. Significantly, this acid hydrolysis step was accompanied by total epimerization of the C(13)-stereocenter to the natural stereochemistry found in cobyric acid (61) itself!
So now, really quite amazingly, the Eschenmoser team had successfully managed to pivot their synthesis away from being a highly complex mixture of eight different stereoisomers to one that consisted of just two C(8)-diastereoisomers, which could now be easily separated by preparative HPLC. Unfortunately though, another highly troublesome obstacle lay ahead. It transpired that the diastereoisomer with the desired correct side-chain stereochemistry at C(8) was actually the minor component of these two separated products, and it could only be isolated in a rather low 19% yield. This contrasted with the undesired C(8)-epimer, which was isolated in a far more substantial 43% yield.
Nonetheless, the Eschenmoser team was still not stopped, for the undesired C(8)-epimer (which otherwise had correct stereochemistry in rings A, C, and D) could be further re-equilibrated to the aforementioned 2.6:1 mixture of C(8)-epimers by a further treatment with conc. H2SO4. This did, of course, mean that following multiple cycles of acid re-equilibration and HPLC separation, most of the synthetic material at hand could eventually be advanced forward to cobyric acid 61 productively.
So, although this was not one of the most convenient of situations that the Eschenmoser team had to contend with, it was certainly not an insurmountable barrier to progress, and it is a great testament to their tremendous experimental skill and resolve that they did not let this difficulty overwhelm them. In what was now yet another masterstroke of great genius, the Eschenmoser team next identified conditions that could mildly and successfully hydrolyze the newly created amide side-chain at C(17) to the corresponding acid without causing concomitant ester hydrolysis. For this, a refined three step sequence was devised that involved: (1) treatment of the amide with N-cyclohexyl-α-chloro-propionaldonitrone and AgBF4 in 1,2-dichlorethane at 0 °C; (2) hydrolysis of the resulting mixed iminium ion/nitrone intermediate with 0.01 N aq. HCl, and (3) chemoselective aminolysis of the resultant aldehydo-ester with dimethylamine, which kept all of the pendant ester functionality intact. Given the significant complexity of these three transformations, we have presented a mechanistic rationale for this outstanding sequence in footnote 25i of this manuscript, to allow readers to see what was exactly done here, and how this achieved the C(17)-side-chain –CN–CO2H conversion.25i
The final step in the synthetic acquisition of cobyric acid (61) is the same as in strategy 1. It involved a high temperature, oxygen-free, ammonolysis of the methyl ester groups with liquid ammonia in ethylene glycol at 75 °C, which had to be conducted in the presence of NH4Cl. If these carefully defined conditions were not strictly adhered to, and the NH4Cl was omitted, then calamity ensued, with C(8)-lactamization proceeding detrimentally to give a compound known as dehydrocobyric acid! However, if the NH4Cl was present, then the desired cobyric acid (61) was produced in 64–100% yield! Woodward attributed this original successful outcome to the NH4Cl preventing the formation of CONH− groupings and it also protonating the ester carbonyls, thereby enhancing their electrophilicity, although he joked about many organic chemists probably not necessarily accepting such a mechanistic proposition!
As always in published total syntheses, one must always be careful when belatedly analyzing routes and considering their apparent retrosynthetic planning. While very often these might appear to be perfectly logical to the contemporary reader, many years later with the benefit of hindsight and advances in knowledge, the situation at the time is often much less logical, less planned, and often characterized by a going back and forth between individual reactions.
In the ‘A–B’- and the ‘A–D’-approaches shown here there was a reliance on cyclization reactions of preformed ‘linear’ tetrapyrroles. In the former, the thioether 64 was prepared from a bispyrrole 65 and dipyrromethene 66 precursor with the fusion of the Eastern and Western halves ultimately being achieved using Eschenmoser's excellent sulfur-contraction method, which helped forge the key C/D-ring connection.31 Linking the A- and B-rings was then achieved by base-catalyzed (DBN) cyclization of the cobalt complex. Significantly, Woodward indicated that this ring-closure method only afforded three diastereoisomers of undefined configuration, but as we have seen, such mixtures do not in any way disbar further synthetic progression or demand diastereomer separation.
Contrastingly, Eschenmoser's ‘A–D’ route employed the seco-corrin 68 as its key intermediate. It was built up from the four pyrrole units 69–72. Quite spectacularly, 68 underwent Cd(II)-templated ring-closure and subsequent cobalt complexation in 46% yield when photoirradiated with high-energy visible light under the 60 °C conditions shown in Scheme 7; a process that afforded a mixture of 8 stereoisomers in which the key C(1)/C(19)-bond was formed with 95% stereoselectivity. It will be noted that analogous Co(III)-complexed seco-corrins fail to undergo this photocyclization due to low-lying metal d-state quenching of the photoexcited complexes. Mechanistically, this photochemical ring-closure reaction is really quite fascinating and most chemists will have their own view of how it is probably proceeding. Given that this is meant to be an instructional teaching review, we will discuss potential mechanisms in more detail now.
Because structurally analogous Cd-seco-corrin complexes showed no fluorescence following photoexcitation, this naturally led Eschenmoser to propose that this must be due to them losing their initial excitation energy by non-radiative decay, which implied that the initially formed, spin-paired, π–π* singlet excited state of these photoexcited seco-corrin complexes was undergoing fast intersystem crossing (ISC). Such ISCs typically occur over a timescale that is anywhere between 10−6 to 10−11 s−1, and they are also always accompanied by electron spin inversion which gives the triplet excited complexes, having one unpaired electron residing in each of the π and π* orbitals. The Eschenmoser team thus suggested that it was most likely the triplet excited state of 68 that was involved in the key photocyclization reaction, and that it was possibly proceeding via a photochemically-allowed antarafacial 1,16 H-atom shift, followed by a thermally-allowed (and induced) antarafacial 1,15π–σ macrocyclic electrocyclization in the ground state. As such, and with respect to the sigmatropic shift specifically, it was pointed out that this was formally a conjugated 15-center-16-π-electron system that was undergoing this internal rearrangement, and as such, it was fully in accord with the photochemical Woodward–Hoffmann (WH) rules. Eschenmoser's only other statement on mechanism concerned the intermediary biradical that was being invoked following the internal 1,16 H-atom sigmatropic shift. Specifically, it was stated that whether it ever attained “the status of an intermediate remains unknown”.32
While the above analysis has presented the Eschenmoser team's view of what is happening in the conversion of 68 into 67, one can envisage alternative pathways by which the key macrocyclizing C(1)/C(19)-triplet biradical could potentially be arising. Thus, because the initially formed triplet excited state of 68 would almost certainly encounter significant steric and torsional strain during its attempts to attain the requisite transition state needed for concerted antarafacial sigmatropic H-atom rearrangement, it is highly likely that the aforementioned H-atom transfer occurred in a stepwise manner, proceeding by the pathway depicted in Scheme 7.
In this regard, if one assumes that such a mechanistically supported excited triplet biradical is indeed being generated from 68 (see Scheme 7), and it has a quite long lifetime in solution (as T1 excited states often do), then one can easily imagine that the exocyclic non-stabilized C(1)-primary ring-A –CH2˙ radical of this biradical would comfortably be able to rotate its central C–C bond to position itself close to the desired C(19) H-atom to bring about its abstraction. Such unrestricted bond rotations typically occur over picosecond timeframes, which would make such a proposal highly reasonable. Moreover, because a similar stabilized iminylmethyl-type radical would now concurrently be generated at C(1), one could further envision such a new C(1)/C(19) biradical engaging in a rapid macrocyclization, with a rate constant roughly of the order of 109 s−1, due to it being solvent caged, and solvent-caged radical pairs typically reacting very rapidly with one another (see Scheme 7).
An intramolecular radical–radical coupling of this type would also probably require far less atomic distortion to occur to achieve the requisite orbital alignment necessary for bonding, particularly when compared with its antarafacial electrocyclizative alternative. For this very reason, one of us in particular (KJH), strongly favors this alternate mechanistic hypothesis.
While conceptually, the latter H-atom abstraction/tandem biradical ring-closure proposal is far easier to follow and see than its Woodward–Hoffmann mechanistic alternative, both hypotheses very clearly intersect at the same Eschenmoser C(1)/C(19)-biradical intermediate, which can potentially be invoked to undergo ring-closure by either of the suggested pathways; either electrocyclization or solvent caged radical–radical coupling. Moreover, Eschenmoser has not rejected the idea of such a C(1)/C(19)-biradical intermediate being reasonably long-lived. Far from it! In fact, it was he who first invoked such a biradical existing, and it has featured repeatedly in many of his B12 synthesis papers.
Both paths also arrive at the same desired end-point, namely the formation of 67 as mixture of 8 stereoisomers where C(1)/C(19) bond is constructed with 95% stereoselectivity. The fact also that the A/D bond forming process is not completely stereoselective further suggests that an alternative non-Woodward–Hoffmann process might very well be competitively or even exclusively operating.
In this aspect, this issue of A/D bond stereochemistry was discussed most uncompromisingly and without any reference to the WH concepts of orbital symmetry in the earlier 1973 report of Woodward on his vitamin B12 synthesis with Eschenmoser,25c where he described the independent photochemical cyclization work that was being conducted in the Eschenmoser laboratory as follows: “Now at the time this trace was produced (referring to the HPLC trace of the photochemical cyclization of compound 68 in Scheme 7), the photocyclization of an intermediate containing no bond between rings A and D was carried out on a zinc derivative; the formation of the A/D bond was non-stereospecific, and led to a complicated mixture of substances separated by liquid–liquid chromatography. Five of them were the same as the five produced in the oxidative cyclization; then there were several more which represent substances differing in stereochemistry at the A/D juncture. So the photochemical method in its early stages was non-stereospecific but it has now been found that using a corresponding cadmium derivative in the photoreaction, a stereospecific cyclization takes place almost exclusively to give materials of the natural series”. So, very clearly, there must be other mechanistic possibilities, and Woodward implicitly acknowledged this absolute fact with the statement he made, which makes no attempt to rigidly tie the Eschenmoser results to his own WH theory.
Consequentially, the only items up for further mechanistic discussion are whether it is actually a formal primary radical H-atom abstraction that is generating the key ring-closing Eschenmoser biradical, or whether it is a genuine 1,16 H-atom sigmatropic shift that is creating it. Also, it is not at all clear whether the actual macrocyclizative ring-closure is a formal electrocyclization or whether it is just a biradical cage combination? Both pathways can potentially be followed, with the different triplet forming Zn- and Cd-seco-corrin complexes and their metals have differing abilities to constrain the respective seco-corrin C(1)/C(19) triplet biradical to react in a highly stereoselective manner. Clearly, the remarkable mechanistic course of these metal-templated photocyclizations is one that holds great fascination, and by discussing these new mechanistic possibilities, at least some alternative ways of thinking about how these reactions are proceeding has been provided; thought that might allow this exquisite photocyclization reaction to be further refined by future generations of chemists.
While for some readers of this article, such deep mechanistic analysis might seem somewhat pernickety, to connoisseurs of mechanism, such discussion will no doubt be ambrosia and, hopefully, the present account will stimulate further model work aimed at testing these two mechanistic hypotheses. In this regard, and whatever mechanistic pathways are actually being followed, one thing is certain: that the photochemical conversion of 68 into 67 and thereafter 63 is a reaction of great synthetic significance for corrin ring-construction.
Now before we depart fully from the vitamin B12 story, and these monumental synthetic achievements of Woodward and Eschenmoser, which are clearly a tour de force, we would like to draw attention to the syntheses of all of the various pyrrolic building blocks, which themselves involve much conceptually elegant synthetic planning. Indeed, the respective syntheses of each of these different sub-units contain many highlights. We therefore guide the interested reader to a very careful study of these landmark papers.
Also, before leaving our discussion of the vitamin B12 synthesis, we do think it important that we comment further on the diastereomeric mixture issues that Woodward and Eschenmoser faced in their collective synthetic conquests of this molecule. We have been careful not to gloss over the stereochemical control problems that had to be confronted during the later stages of their respective two total syntheses, since similar difficulties will again be very likely faced in any future total synthesis of the vitamin B12 molecule, or other structurally-related porphyrins such as cofactor F430 (4). It is imperative therefore that readers fully appreciate the problems that are likely to arise, so that due synthetic planning can be made.
Indeed, it is probably fair to say that the issue of stereocenter fragility was one of the really key aspects of the vitamin B12 synthetic venture that Woodward and Eschenmoser both went to great pains to point out in all of their various publications.25 This was done not just to alert the community to the very existence of this problem, but also to teach us how to overcome it.
Indeed, it is a true testament to the high scientific integrity of the Woodward and Eschenmoser teams that they never ever shied away from highlighting the dramatic C(3), C(8) and C(13) stereocenter lability issues that they constantly had to battle against in their advanced macrocyclized products, and the fact that they were able to recognize that the loss of pre-ordained stereochemistry was occurring in their systems, with the limited spectroscopic tools that they then had available to them at the time, is truly remarkable.
In fact, so profound were the mixture problems that they were repeatedly confronting, that Woodward even went so far as to provide us with a number of typical HPLC traces of the various reaction mixtures that the Harvard and Zurich teams had obtained from the various different macrocyclizations that they had performed, just to make sure that we were fully aware of how severe these issues actually were. Eschenmoser's 1977 and 2015 reports also went to great pains to point out the stereochemical control issues.25d,h By being so unambiguous in their discussion of this matter, we were thus all able to see what was actually involved in a successful synthesis of vitamin B12.
Therefore to gloss over the issue of stereochemical scrambling of the aza-enolizable stereocenters in Woodward and Eschenmoser's Co-complexed ring imines, is only to diminish the achievements of these two great chemistry giants, for it seemingly relegates the stereochemical fragility issue to triviality status, and it denies the scale of the massive problems that were actually faced in many of the subsequent steps that had to be effected post-macrocyclization.
Indeed when Woodward and Eschenmoser first developed their ideas about using metal ion-templating to facilitate their macrocyclization reactions, they themselves probably never suspected for one moment that their Co- and Cd-complexed macrocyclic imine intermediates would ever experience such profound and dramatic weakening of their C–H bonds at C(3), C(8) and the C(13)- in the resulting ring-closed corrins. It is vital therefore that these stereochemical lability issues are properly recognized and understood here for what they are for all newcomers to the field.
Moreover, the fact that Eschenmoser was able to take such a highly complex mixture of eight different stereoisomeric macrocycles in his second-generation B12 synthesis, and then to so dramatically reduce it to just two C(8)-corrin epimers, is really quite remarkable, particularly when one considers that his team was also able to satisfactorily separate their desired C(8)-epimer by chromatography, and they were able to re-equilibrate and recycle their other undesired epimer by a simple acid treatment and further HPLC.
In our opinion, even today, almost 50 years on, we doubt that anyone would likely better Eschenmoser's second-generation synthetic end game to vitamin B12, most especially when there is this overwhelming systemic preference for producing the C(8)-epi (neo) stereochemistry in late-stage precursors that are subjected to acid treatment. So, with this final departing epilogue, we leave the beguiling Woodward and Eschenmoser vitamin B12 total synthesis story for colleagues to derive further inspiration from and ponder over, because of its extremely clever planning. In this regard, it will always remain an enduring crowned masterpiece of synthetic organic chemistry excellence in our field.
2.5 Nature's way – biosynthesis
A related corphin molecule is cofactor F430 (4).33 Involved in the methanogenesis of archaebacteria its structure is as complex as that of vitamin B12 but it has yet to find the attention of synthetic chemists, possibly due a lack of funding for such endeavors by global funding agencies. On the other hand, our colleagues from biochemistry have been funded generously and most enzymes involved in cofactor F430's biosynthetic pathway have been elucidated.18b,34 Hence, this presents an opportunity to outline ‘Nature's way’ of synthesizing such structures, i.e. the biosynthesis of tetrapyrroles.As is the case with the total synthesis of natural porphyrins, their biosynthesis is complex, and often involves many steps. The complexity of these pathways increases even further if one considers the various classes of natural porphyrin and one also takes into account aerobic versus anaerobic pathways, and the many different phylae and salvage pathways (Fig. 4). This topic is almost as broad as that of the total synthesis and has been covered in detail in many comprehensive reviews.1,11,18,35 Historically, porphyrin chemists made crucial contributions to elucidating individual steps. For example, it is easy to recall the significant contributions made by Sir Alan R. Battersby1,36 and A. Ian Scott.37 Today, however, most of the progress is being driven by molecular biology methods. Hence, only a brief account will be given in the following section of this review.
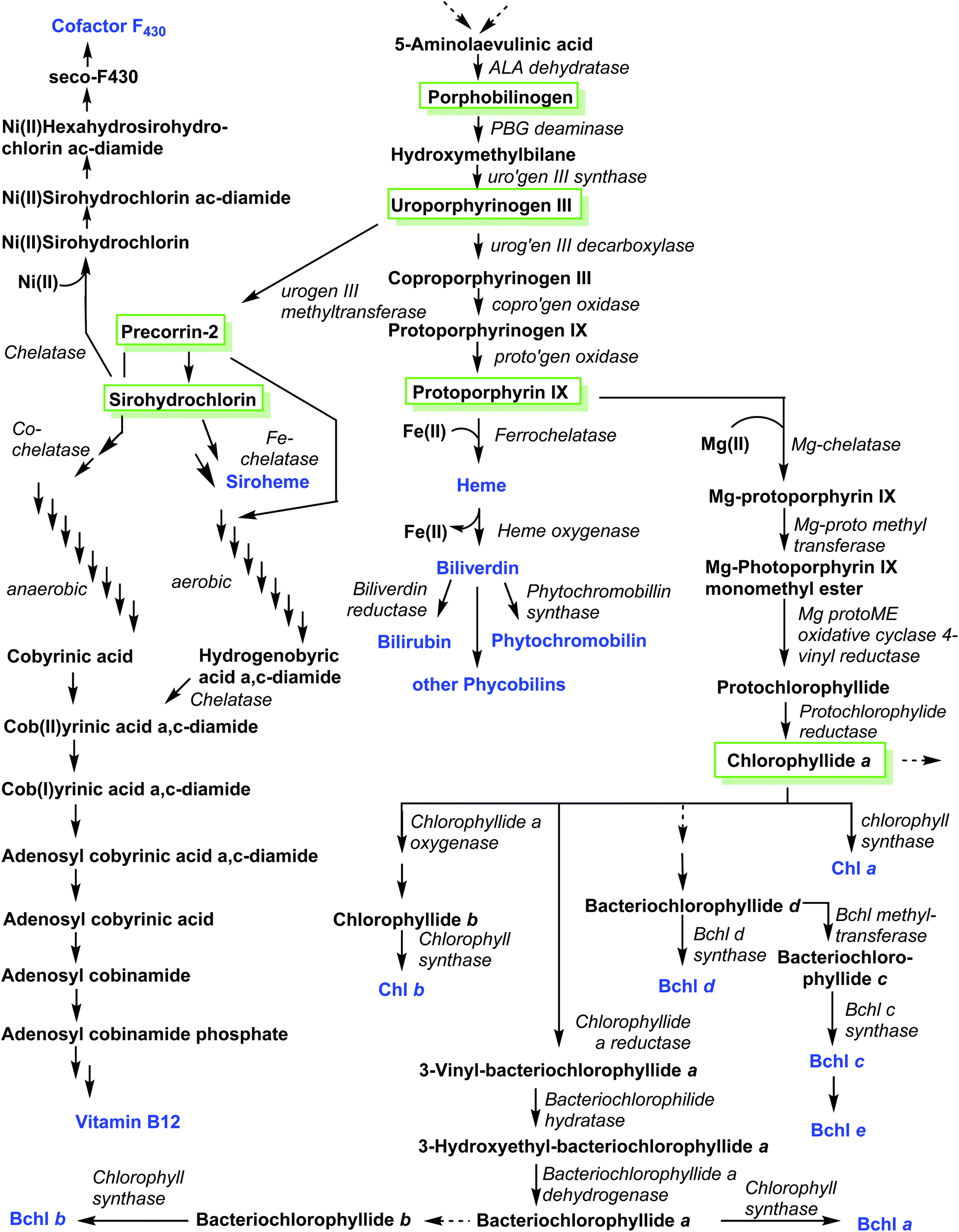 | ||
| Fig. 4 Overview of tetrapyrrole biosynthesis. | ||
If we take the number of synthetic steps and enzymes involved, then Nature has as much to do as Fischer, Woodward, Eschenmoser and coworkers in synthesizing the pigments of life, but perhaps with still a bit more selectivity.25h,25i One only needs to contrast the structure of uroporphyrinogen III 17 with that of cobyric acid 61 or F430 4 to appreciate the high degree of chemical complexity which can be added to this cyclic tetrapyrrolic precursor in the later biosynthetic steps. Scheme 8 summarizes the overall key steps in the biosynthesis of porphyrins.
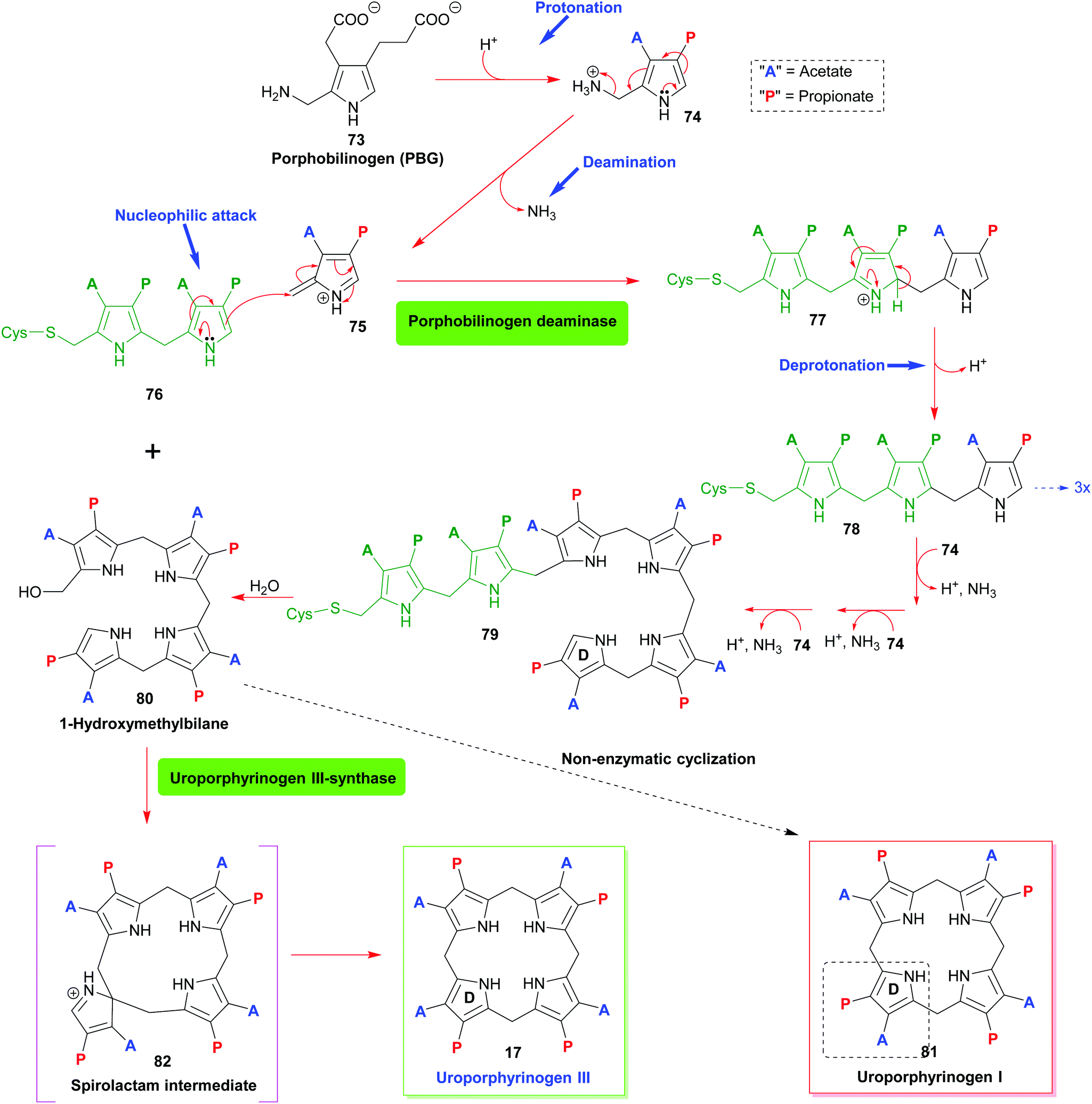 | ||
| Scheme 8 Biosynthetic construction of the porphyrinogen: enzymatic conversion of porphobilinogen into 1-hydroxymethylbilane by porphobilinogen deaminase and generation of uroporphyrinogen III through action of uroporphyrinogen III-synthase. | ||
The first pyrrole intermediate is porphobilinogen (PBG) 73, four fragments of which are combined to form the tetrapyrrole. And here we already encounter two specific and unique aspects of porphyrin biosynthesis. The tetrapyrrolic macrocycle is not the result of a simple tetramerization reaction, as illustrated in Scheme 1. The biosynthesis employs approach B, shown in Scheme 1, i.e. the synthesis and then cyclization of a bilane.
However, this natural 1-hydroxymethylbilane 80, is actually derived from a hexapyrrane 79 (Scheme 8). Porphobilinogen deaminase,38 the enzyme catalyzing the construction of the bilane contains a dipyrromethane cofactor 76 which acts as an anchor onto which four additional pyrroles are added sequentially through protonation, deamination, nucleophilic attack, and deprotonation sequences.
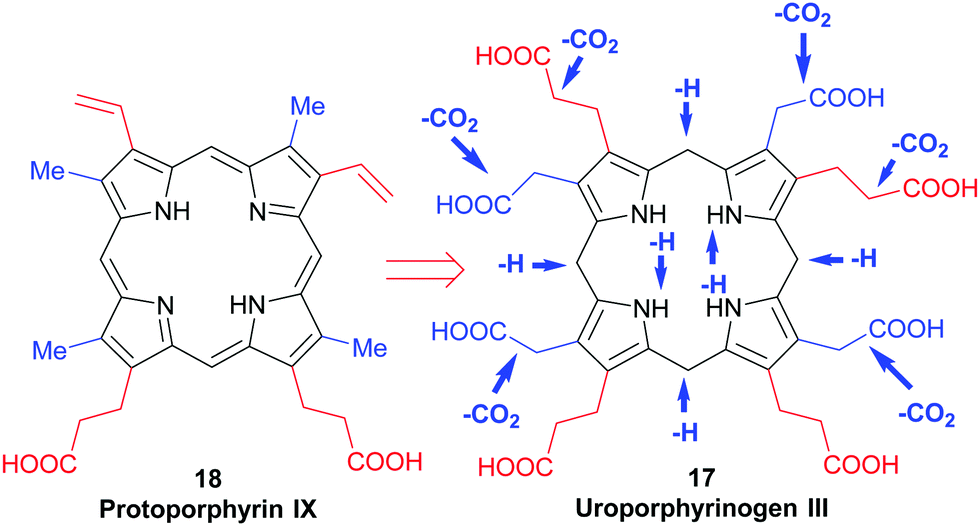
The hydrolytically released bilane 80 now contains all four required pyrrole units. Recalling the type isomer problem outlined in Fig. 3, a closer inspection shows that the positioning of the acid side chains in ring D is not the one found in the end products. The key role of enzymatic catalysis is evidenced by simple chemical (aka acid-catalyzed) ring-closure experiments which yield uroporphyrinogen I 81. Hence, the natural catalyst, the enzyme uroporphyrinogen III-synthase facilitates an intricate reaction mechanism. Ring-closure of 80 is achieved in such a way as to generate an intermediary spirolactam 82, which is then converted into uroporphyrinogen III 17, which now has the ‘correct’ regiochemistry of the acetic and propionic acid side chains. In effect, pyrrole ring D is inverted during these biosynthetic steps.
Elucidation of the mechanism was aided significantly by the elaborate synthetic efforts by Battersby, whose group over many decades developed intricate syntheses of related spiro compounds and used them as inhibitors of the natural system.39 The relevance of enzymatic control of tetrapyrrole biosynthesis is also indicated by the fact that impairment thereof gave rise to porphyrias, a class of mostly genetic diseases where errors in the biosynthesis result in an accumulation of non-natural porphyrins with attendant clinical symptoms.40
Uroporphyrinogen III (17) is the only biosynthetic intermediate common to all natural tetrapyrroles. The following step to generate the aromatic system of protoporphyrin IX involves first the decarboxylation of all four acetic acid side chains in the nonaromatic porphyrinogen 17 to form the tetramethyl coproporphyrinogen derivative (Scheme 9). A second decarboxylation event then occurs, of the two propionic acid groups at the 3- and 8-positions; this action stations vinyl groups at these positions and leads to formation of protoporphyrinogen (Scheme 9). Finally, stepwise oxidation and removal of six hydrogen atoms gives the aromatic protoporphyrin IX 18. The latter is the common precursor for the natural porphyrins, their degradation products and photo- and phytobilins.
 | ||
| Scheme 9 Steps needed to generate the conjugated aromatic system of natural porphyrin-type tetrapyrroles. | ||
After insertion of Mg(II), photosynthetic organisms use 18 also for the biosynthesis of the (bacterio)chlorophylls. Additional side chain manipulations can also occur. For example, formation of the isocyclic pentanone ring by cyclization of the C13-propionic acid group onto the C15 meso position through protochlorophyllide oxidoreductases. Here, the key step is the reduction of the C17, C18-double bond to yield the chlorin (dihydroporphyrin) macrocycle of the chlorophylls, for which both light-dependent and dark pathways are known to operate.41
A comparison of the macrocycles of vitamin B12 and F430 with the hemes and chlorophylls underlines the difference in biosynthetic strategies needed. For the latter, fully conjugated 18π aromatic macrocycles with 20 carbon atoms are needed. The former has highly reduced pyrroline rings and no fully conjugated π-electron ring systems. The key anaerobic intermediate for these highly reduced porphyrinoids is sirohydrochlorin 83, formally an isobacteriochlorin (2,3,7,8-tetrahydroporphyrin). This is derived from 17via two sequential deprotonation/methylation sequences involving a meso methylene unit and the neighboring β-position of a pyrrole ring, which yields precorrin-2 (via precorrin-1), which is then oxidized to 83.42
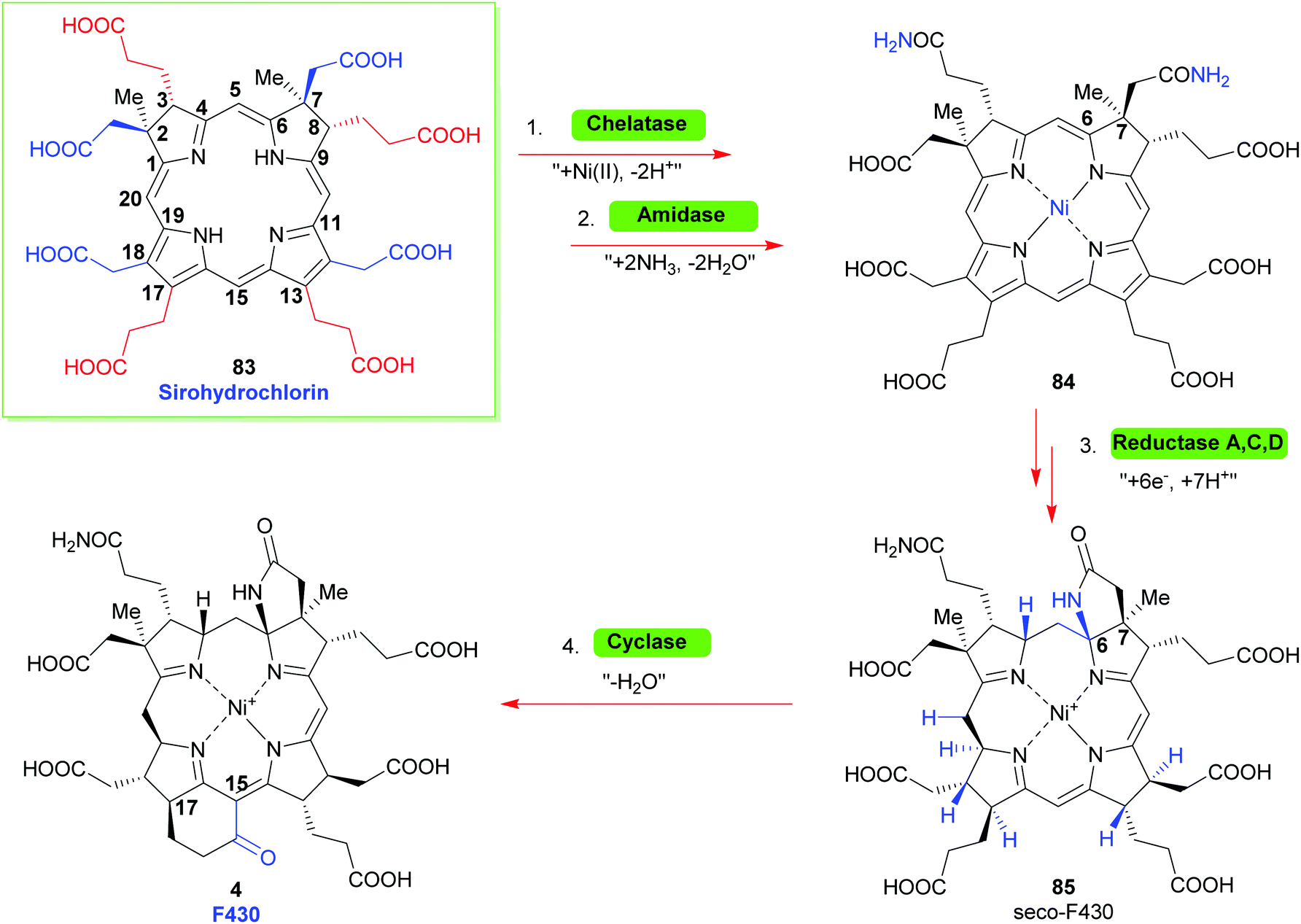
Looking towards the terminal steps involved in the biosynthesis of cofactor F430 (Scheme 10)34 these involve metalation with Ni(II), amidation of the residues at C3 and C7 to give 84, reduction of various Ca, Cb and Cm positions and cyclization of a C6 amide onto the C6 position yield the lactam seco-F430 85. This is then followed by formation of cyclic ketone between C15/C17 to give F430.
 | ||
| Scheme 10 Overview of the biosynthetic transformations from sirohydrochlorin to F430. | ||
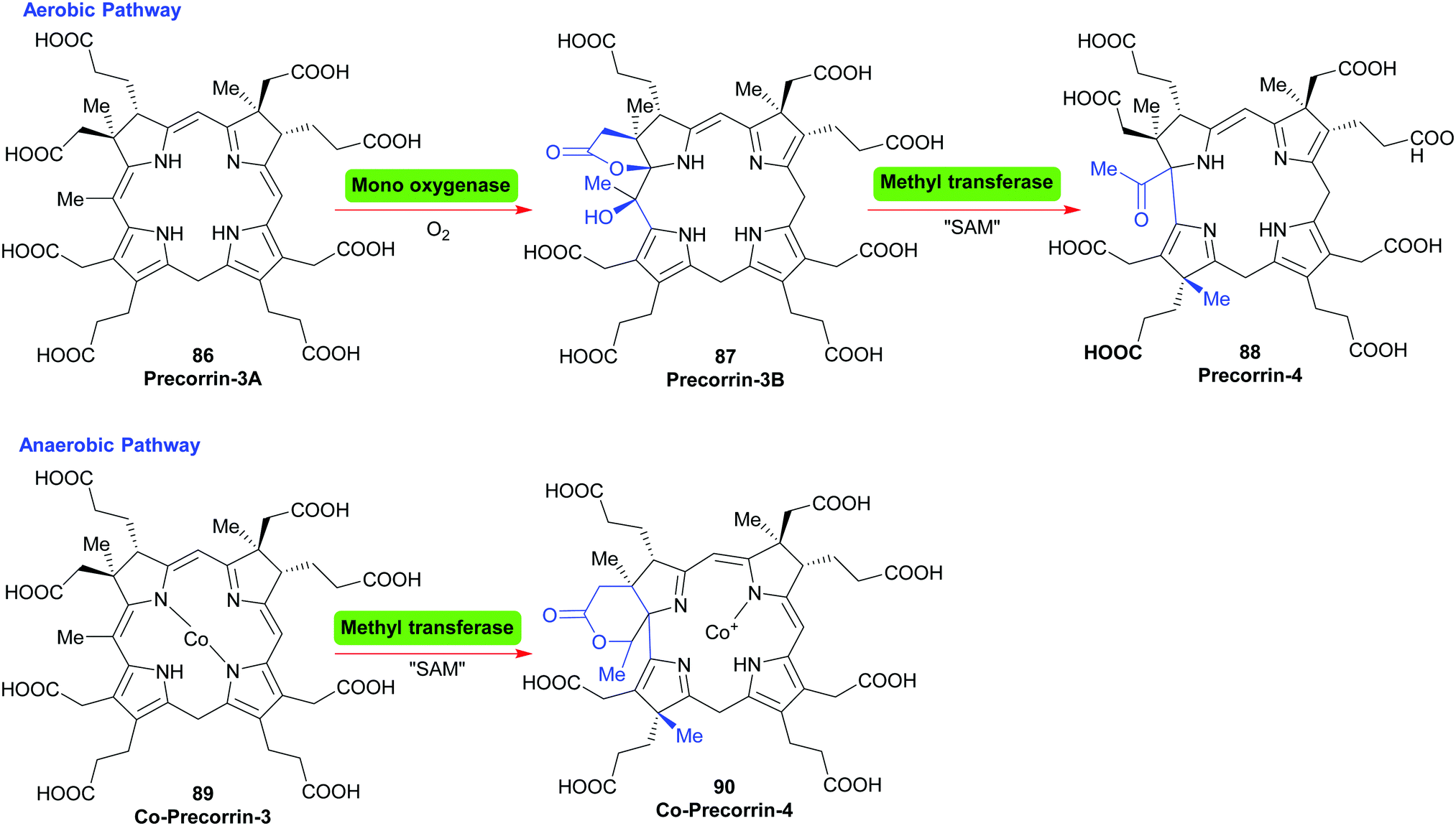
For the cobalamins the situation is even more complicated with about 30 biosynthetic steps and the existence of both an aerobic and an anaerobic pathway.43 The main differences lie in the requirement for molecular O2 and in when the cobalt insertion takes place; either ‘late’ in the aerobic pathway, or ‘early’, following formation of 17 in the anaerobic one.43c For an organic chemist, the intriguing question is how the macrocycle is contracted, i.e. the C20- → C19-conversion?
In the aerobic pathway, this key step involves the formation of a hydroxylated γ-lactone (precorrin-3B 87) from precorrin-3A 86. The contraction then involves the lactone taking part in a masked pinacol rearrangement, which pushes the methylated fragment out of the ring system to give precorrin-4 88. Subsequently, this two-carbon fragment is cleaved off as acetic acid, and is followed by other steps and a late cobalt insertion (Scheme 11).43a In the anerobic pathway, Co-precorrin-3 89 is methylated at C17 and undergoes ring contraction with formation of a δ-lactone 90. The exact details and sequence of the events that follow are not clear. Probably initially methylation occurs at C12; this is then likely followed by reduction at C10, cleavage of acetaldehyde and then further methylation/reduction steps to give cobyrinic acid.
 | ||
| Scheme 11 The ring contraction step in the biosynthesis of cobalamins. SAM = S-adenosyl methionine; Nature's methylating agent. | ||
3. Modern day strategies to various porphyrin workhorses
3.1 Symmetric meso-tetrasubstituted porphyrins (Lindsey)
Due to their symmetry, the synthesis of S4-symmetric porphyrins is reasonably easy and, for meso-substituted porphyrins, can be readily achieved by the simple two-step, one-pot, reaction shown in Scheme 12. Thus, meso-aryl and meso-alkyl porphyrins 9244 are some of the most widely used “workhorses” in porphyrin chemistry. Ignoring the phthalocyanines for now (see Section 3.4.1), 5,10,15,20-tetraphenylporphyrin 92 (R = Ph) is probably the most widely studied porphyrin in industry and academia. Its synthesis involves the simple mixing of pyrrole with benzaldehyde under acidic conditions, followed by oxidation of the intermediary porphyrinogen 91 (R = Ph) to the aromatic porphyrin. Many different conditions44 have been studied and developed for this synthesis, including the older Rothemund and Adler–Longo procedures.45 In our experience, however, the Lindsey conditions46,47 for accessing this and other meso-aryl44 and -alkyl48 porphyrins 92 remain the best and most general currently available. That approach employs either BF3·Et2O or trifluoroacetic acid as the acid-promoter for the porphyrinogen synthesis step and uses either chloranil or DDQ as the oxidant for converting the latter into the porphyrin.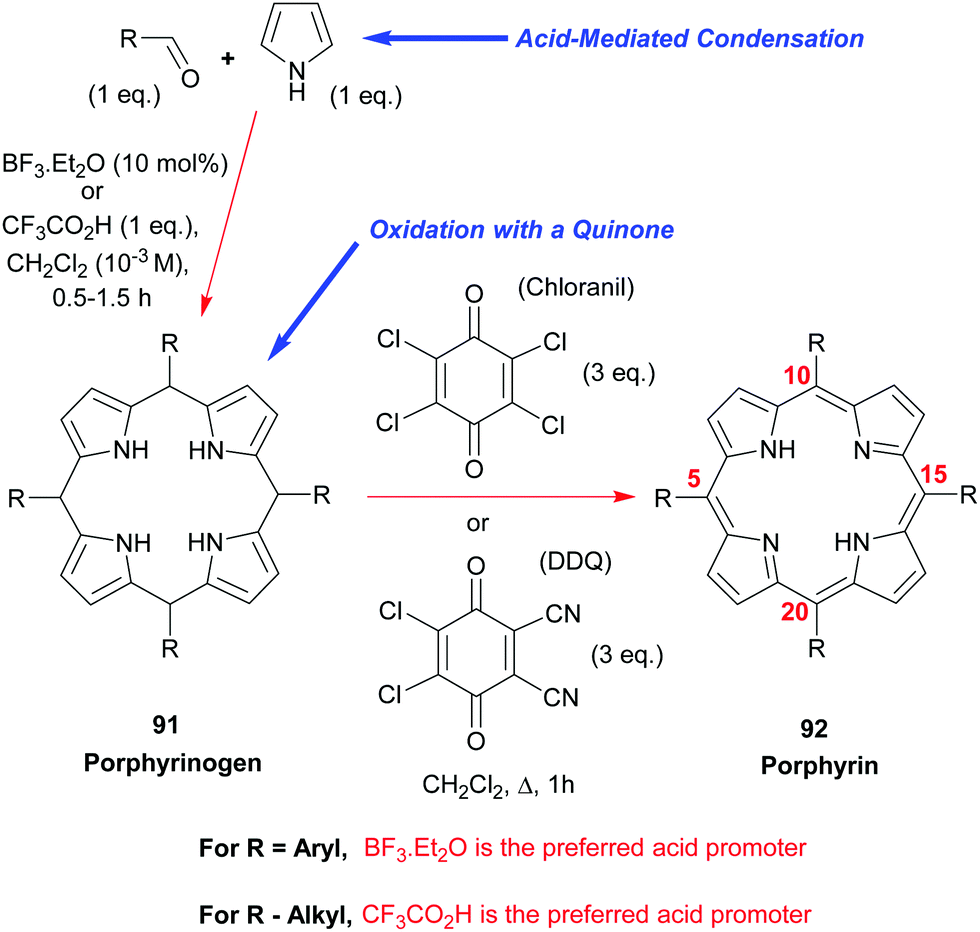 | ||
| Scheme 12 Lindsey's general method for the synthesis of meso-tetrasubstituted porphyrins. | ||
In the case of the BF3·Et2O-catalyzed reactions, it is often advantageous (in terms of final product yield) to include a water-scavenger such as triethyl orthoacetate in the reaction mixture, but this additive confers no benefit at all when the porphyrinogen assembly step is conducted with trifluoroacetic acid. Via the Lindsey protocol, yields as high as 50–55% could be realized in the synthesis of 5,10,15,20-tetraphenylporphyrin 92 (R = Ph) using catalytic BF3·Et2O and triethyl orthoacetate as the condensing system, and chloranil as the oxidant. By way of contrast, when DDQ was used as the oxidant the yields were typically lower and usually sat at the 45–50% level. However, the latter has the advantage of circumventing the formation of the respective chlorin as a side-product.
While this method does provide a very wide range of meso-substituted porphyrins from non-hindered aldehydes that have good solubility in CH2Cl2, it does not work well with sterically hindered aldehydes such as mesitaldehyde, 9-anthraldehyde, 2-norbornene-carboxaldehyde, and pivaldehyde, nor does it perform to high specifications with alkyl or aryl α,β-unsaturated enals (e.g., cinnamaldehyde, phenylpropargaldehyde, or (E)-2-octenal). Aldehydes with low solubility in CH2Cl2, such as paraformaldehyde, are also problematic.
3.2 Symmetric octa-β-substituted porphyrins
The next most popular porphyrin in industrial and academic use is 2,3,7,8,12,13,17,18-octaethylporphyrin 102 (Scheme 13). It provides a perfect example of the synthesis of a β-alkyl substituted porphyrin.49,50 Pioneered by Inhoffen in 1966,49 the synthesis is best commenced with the construction of an appropriate α-substituted 3,4-diethylpyrrole (e.g., 99, Scheme 13).51 This is then subjected to an acid-promoted cyclization to yield the porphyrinogen which is subsequently oxidized to the porphyrin 102. Often just air is sufficient to mediate this step. The aforementioned pathway illustrates the main strategic difference between the syntheses used for securing symmetric meso-substituted porphyrins and those for porphyrins of the β-octaalkyl series. While in the former, the carbon atom for the meso-position is provided by the aldehyde, in the latter, it is provided by a pyrrole with a hydroxyalkyl substituent at the α-position. As can be seen from Scheme 13, conditions like those used for the synthesis of the meso-tetrasubstituted porphyrins can be used with α-free pyrroles (for example, after decarboxylation of 99), and this approach is now exploited routinely for the synthesis of dodecasubstituted and other porphyrins.4,52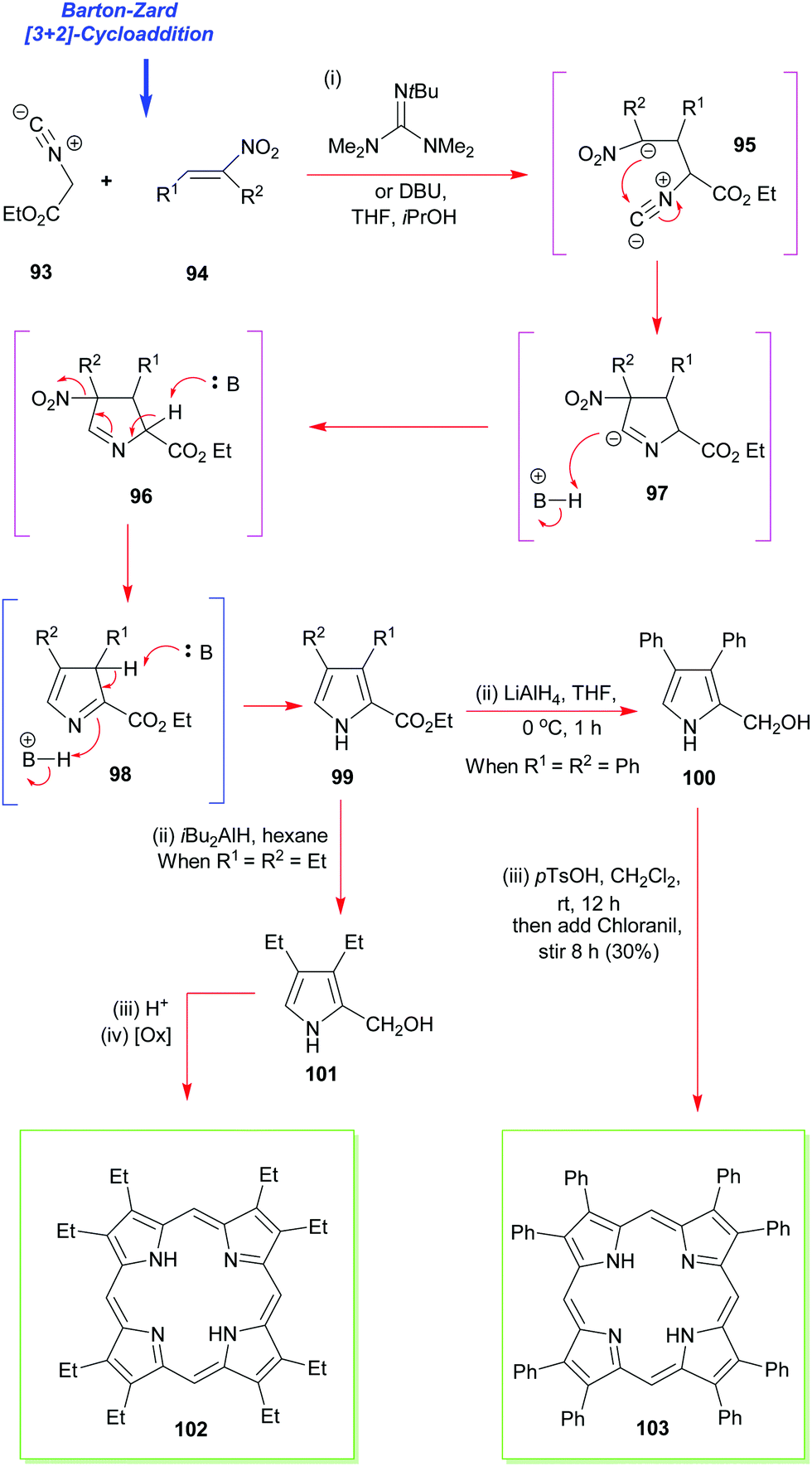 | ||
| Scheme 13 The Barton–Zard pyrrole synthesis in the synthesis of 2,3,7,8,12,13,17,18-octaethyl- and octaphenylporphyrins 102 and 103. | ||
While there are many ways to construct 2,3,4-substituted pyrroles such as 99, including functionalization of a 3,4-dialkylpyrrole with formaldehyde, or Mannich reaction of the same with dimethylamine and formaldehyde, the most significant advance in the area has been the Barton–Zard pyrrole synthesis.53 It relies on the [3+2] cycloaddition of ethyl isocyanoacetate 93 to a nitroalkene 94 to yield an ethyl pyrrole carboxylate (e.g., 99) which is trisubstituted. This can then be reduced to the pyrrole-2-carbinol 100 and the latter tetramerized to the porphyrin. The method is applicable to the synthesis of almost any desired 3,4-disubstituted pyrrole,54 and it has now benefitted numerous porphyrin total syntheses ever since it was first invented by Barton and Zard. These have included the total synthesis of 2,3,7,8,12,13,17,18-octaphenyl-porphyrin 103 as well as 2,3,7,8,12,13,17,18-octaethyl-porphyrin 102 (via101).
Additional developments, from Ono's group, have included the reaction of β-nitrostyrenes 104 with aqueous TiCl3 in 1,4-dioxane which has allowed the facile synthesis of 3,4-diarylpyrroles 105 and the construction of β-aryl- and β-heteroaryl-porphyrins (e.g., 106, Scheme 14).55 The latter is also a typical example of how to access 2,3,5,7,8,10,12,13,15,17,18,20-dodecasubstituted porphyrins, which are of interest due to their highly non-planar macrocyclic structures which have altered reactivity and physicochemical properties.4,52,56
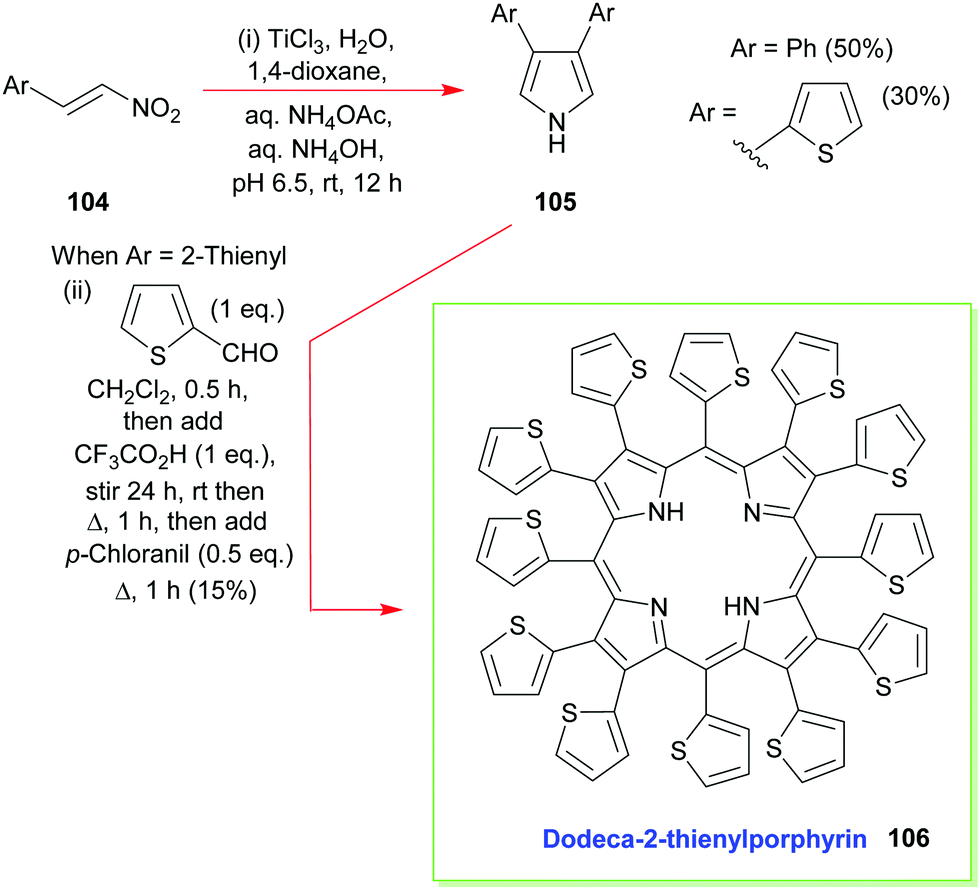 | ||
| Scheme 14 Ono's Ti(III)-mediated synthesis of 3,4-diaryl- and 3,4-diheteroaryl-pyrroles and the subsequent use of these intermediates in the synthesis of dodeca-2-thienylporphyrin 106. | ||
The nonplanarity arise from peri-interactions of the β- and meso-substituents (steric hindrance). As a result, such porphyrins often exhibit highly distorted macrocycles, are highly basic, and have many unique features. These range from very fast metalation rates, stabilization of metals in high oxidation states, organocatalytic ability,56b and addition reactions at the Cm positions, to very different photophysical properties.4,56 The conformational flexibility of the porphyrin system evidenced by these ‘highly substituted’ porphyrins also serves – in part – as an explanation for the functional variability of the natural porphyrins.5
Ono has also reported on the synthesis and use of various 4,7-dihydro-4,7-ethano-2H-isoindoles in the creation of soluble precursors of tetrabenzoporphyrins (Scheme 15).57 One such intermediate 111 was prepared by a multistep sequence that commenced with a Diels–Alder reaction between 1,3-cyclohexadiene 107 and β-phenylsulfonylnitroethylene 108.55 This was followed by an in situ E1cb elimination of the phenylsulfone moiety from 109 and a Barton–Zard pyrrole ring-construction with ethyl isocyanoacetate 93 to secure 111 which was reduced. Porphyrin construction then proceeded from 112via the p-TsOH-induced assembly of the porphyrinogen followed by oxidation with p-chloranil. Together these steps delivered the highly soluble porphyrin 113 which was then converted through to the sparingly soluble tetrabenzoporphyrin by the retro-Diels–Alder reaction which liberated the target 114 plus ethylene.57
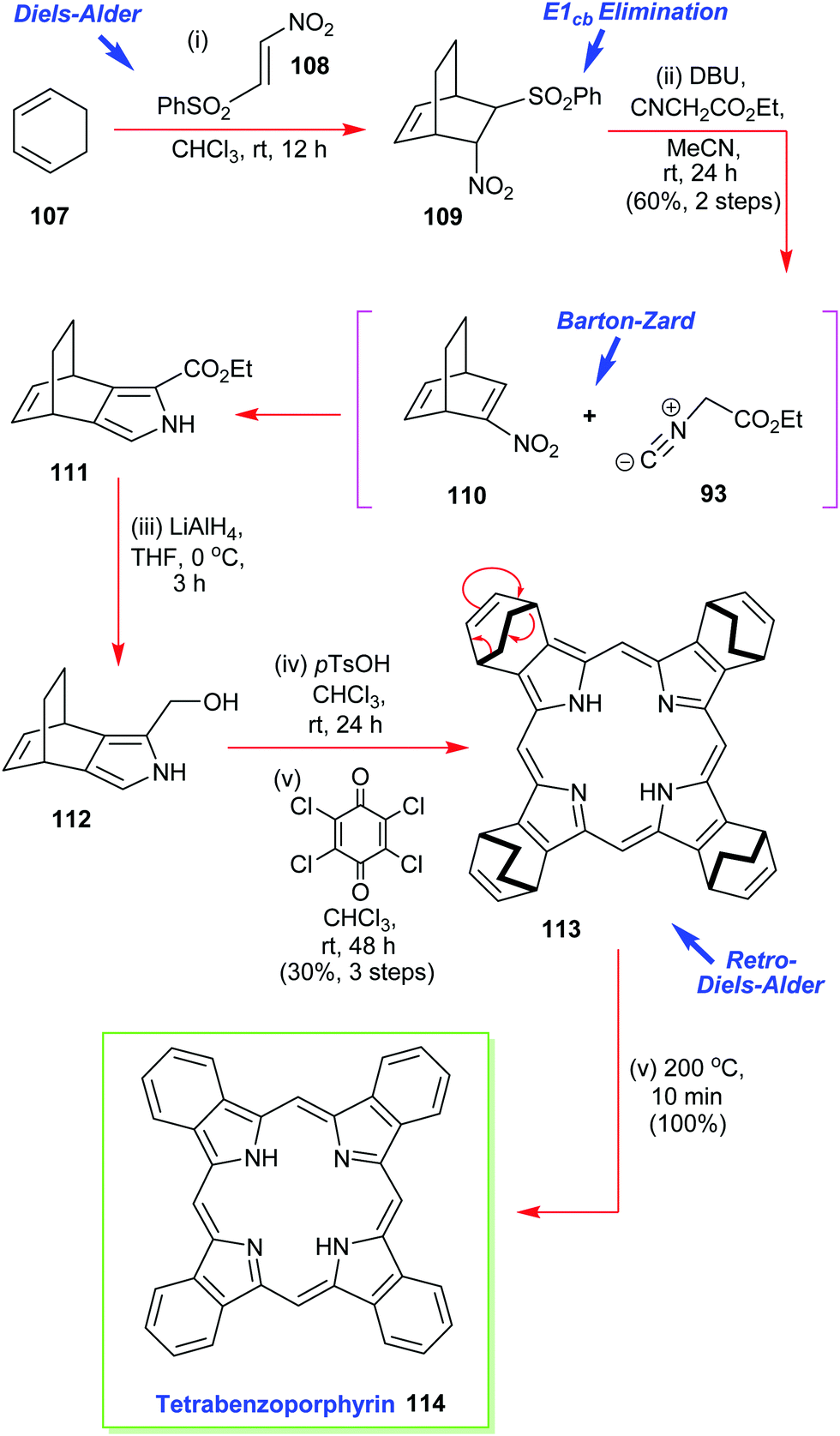 | ||
| Scheme 15 Ono's Barton–Zard masked-isoindole route to the sparingly soluble tetrabenzoporphyrin 114. | ||
It must be pointed out that tetrabenzoporphyrin 114 was previously considered to be a very difficult target molecule to prepare and subsequently manipulate, in pure form, due to its extremely high insolubility in many common solvents. Ono's isoindole route to this type of molecule thus represents an elegant and major advance in the area.
To further exemplify the generality and power of the new masked isoindole protocol for benzoporphyrin synthesis, Ono deesterified 111 and conducted an in situ high-temperature decarboxylation to access the 3,4-disubstituted pyrrole 115 which was then submitted to the standard Lindsey porphyrin assembly conditions with 2-thiophene-carboxaldehyde to yield 116 (Scheme 16).57 The latter molecule was duly elaborated into the 5,10,15,20-tetrakis(2-thienyl)-tetrabenzoporphyrin 117 using the same retro-Diels–Alder/ethylene extrusion tactic.57
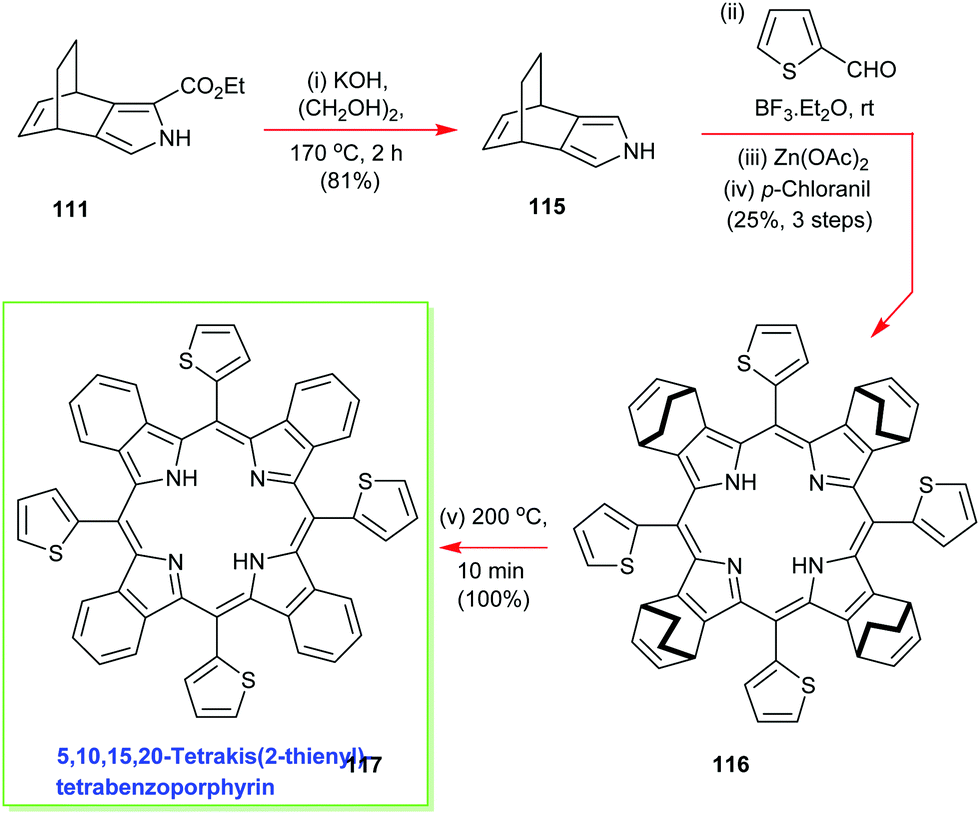 | ||
| Scheme 16 Ono's masked-isoindole route to 5,10,15,20-tetrakis(2-thienyl)tetrabenzoporphyrin 117. | ||
In the same paper, Ono also reports that is possible to prepare mixed β-octasubstituted porphyrins with a lone isoindole component (Scheme 17).57 Yet again, his retro-Diels–Alder ethylene extrusion tactic featured at the final step of the synthesis of porphyrin 126. This is also an example of how to construct porphyrins through a [3+1] approach, which we will discuss in more detail below (see Section 4.3). Other notable features of this synthesis were its exemplification of the decarboxylative nucleophilic addition of in situ-generated pyrrole 2-carboxylic acids to pyrrole 2,5-dialdehydes for porphyrinogen assembly. The mechanistic workings of this step are detailed in Scheme 17.
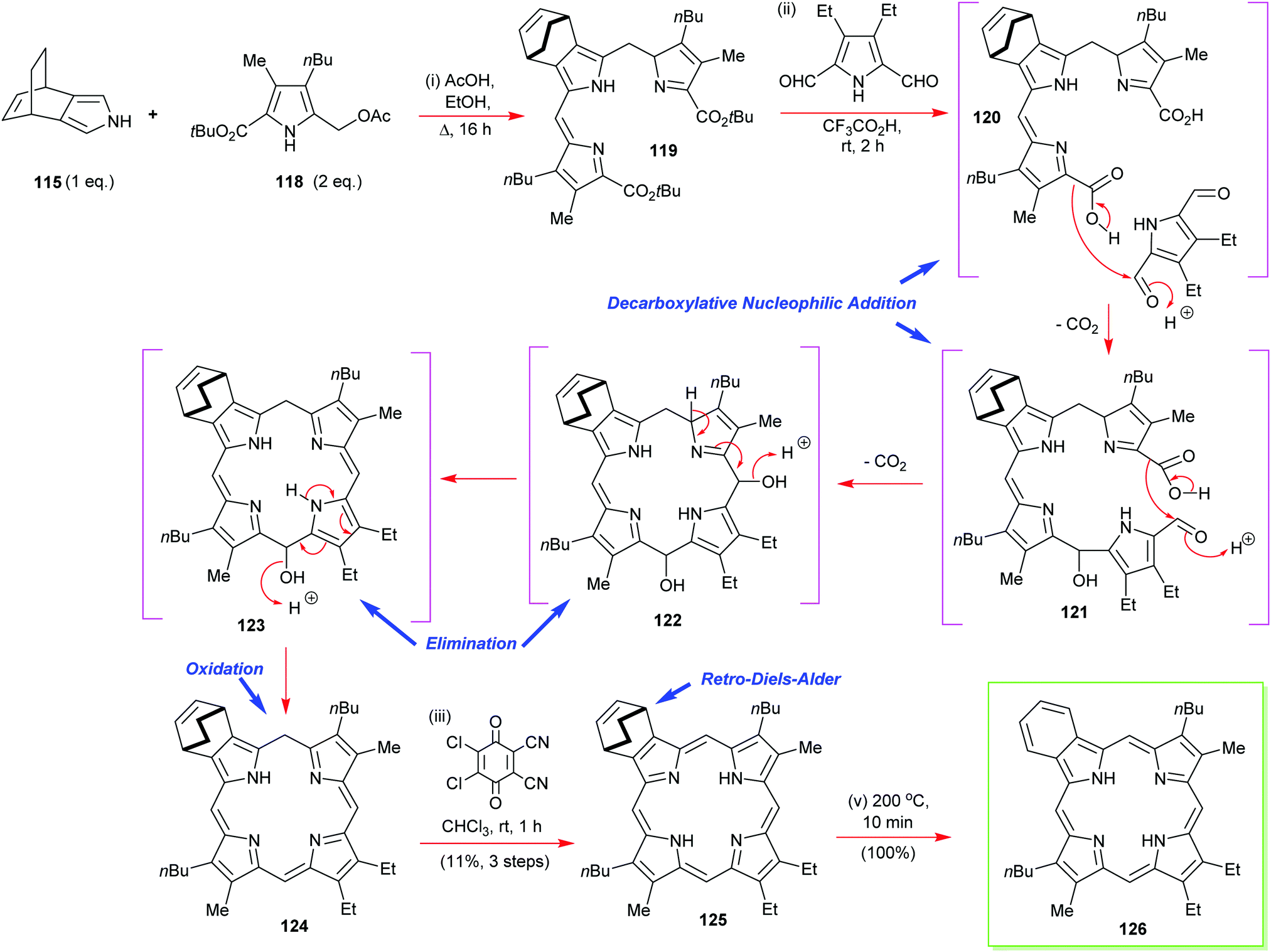 | ||
| Scheme 17 Ono's synthesis of a monobenzoporphyrin 126. | ||
3.3 The ‘parent’ of all – porphyrin
Structurally the simplest of all porphyrins, namely, porphyrin (5) itself (sometimes still called ‘porphine’), is one of the most difficult to work with.58 Like tetrabenzoporphyrin 114 it easily forms π-aggregates and is insoluble in many solvents. First prepared by Fischer and Gleim in <0.1% yield59 it took many decades until acid-catalyzed condensation reactions of 2-hydroxymethylpyrrole reached 15% using a water/4-methyl-2-pentanone solvent system that yielded 30 mg of product.60More practical in terms of simplicity, concentration, purification, and gram-scale synthesis is an approach from Lindsey's laboratory.61 The reaction of 1-formyldipyrromethane 127 in the presence of air gave the much more soluble (porphyrinato)magnesium(II) 128 in good yields of 30–40% (Scheme 18). The magnesium complex obtained from this template [2+2] condensation can easily be transformed into the free base using TFA and DCM.
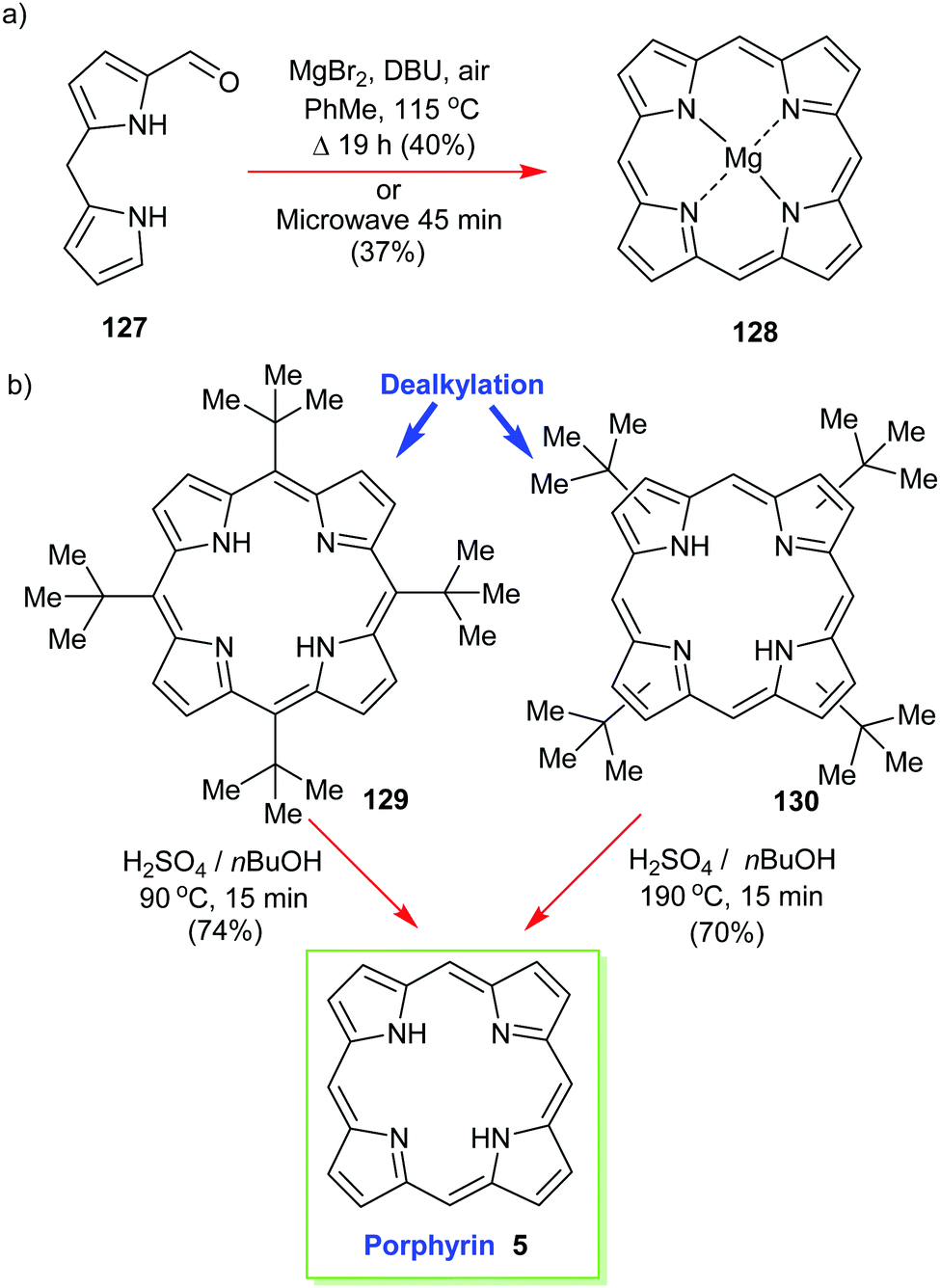 | ||
| Scheme 18 Syntheses of porphyrin 5. (a) Lindsey's template approach, (b) Neya and Funasaki's dealkylation method. | ||
Another ‘textbook’ approach is to first prepare a soluble functionalized porphyrin as a precursor and to then use an acid-catalyzed dealkylation to ‘deprotect’ the unsubstituted porphyrin 5. Below, in Scheme 18, we see the nonplanar 5,10,15,20-tetra(tert-butyl)porphyrin 129 (or 130) being used in this way.62 Pioneered by Neya and Funasaki,63 this method gives high yields and liberates the target in a solution where it can be used for further reactions.62b
3.4 Industrial scale porphyrinoid dyes
Only very few porphyrin derivatives have been prepared on an industrial scale viz. on a scale of more than a few kgs. Here we highlight two examples: the synthetic phthalocyanines, for which no natural counterparts exist; and the ‘chlorophyllins’ (see Section 3.4.2), which are derived from natural chlorophylls. The commercial use of porphyrins will no doubt increase over the coming years, with many applications already, most notably in the materials and biomedical areas, which utilize synthetic porphyrins, albeit still on a small (<kg) scale.64,65 Potentially, the promise and increasing utility of porphyrins in photovoltaics will add another large scale application.66000 tons annually. Phthalocyanines (Pc) have conjugated planar heteroaromatic systems that contain 18π-electrons and conform to the Hückel rule. Their pyrrole rings concurrently contribute to the aromatic character and result in highly conjugated planar systems of excellent stability. For example, they can be thermally sublimed at temperatures above 500 °C without risk of decomposition and they exhibit high light stability (lightfastness). These properties are highly desirable in many technical applications and make them attractive industrial colorants.67 Originally, phthalocyanines were produced as pigments that were generally insoluble in the application medium. Since then, however, technical processes including sulfonation and chloromethylation have been developed to address these solubility limitations and now they can also be manufactured in dye forms.
The accidental discovery of the phthalocyanines dates back more than a hundred years to 1907 when it was first encountered as a blue impurity formed in the reaction of phthalimide with acetic anhydride. It was subsequently met again en route to 1,2-dicyanobenzene(phthalonitrile) in the reaction of 1,2-dibromobenzene with copper(I) cyanide in boiling quinoline. Its first accidental industrial preparation was in 1928 at a manufacturing company known as Scottish Dyes in Grangemouth, Scotland. The formal chemical structure of phthalocyanines was resolved in 1933 by Linstead.68
Typically, phthalocyanines are produced as transition metal complexes using a templated synthesis. This is due to a perfect fit between the size of a metal ion and inner space within the macrocycle. Of great industrial relevance are the complexes of transition metals such as Cu, Co, Ni. For example, the copper phthalocyanines known as phthalocyanine green and phthalocyanine blue are among the most important industrial pigments. While the manufacture of phthalocyanines is mainly dedicated to customers in the coloration and printing industries, non-colorative applications have also been extensively explored.
Phthalocyanines 131 can be prepared through several routes,67,69 with the most popular synthetic pathways being illustrated in Scheme 19. However, only two organic precursors, phthalic anhydride 134 and phthalonitrile 132 have gained industrial importance due to the costs of the starting materials, the economy of the technical processes, and the yields of the final pigments. Both precursors are commonly synthesized via the oxidation of the petrochemical, o-xylene 137, in the presence of a catalyst (Scheme 20).
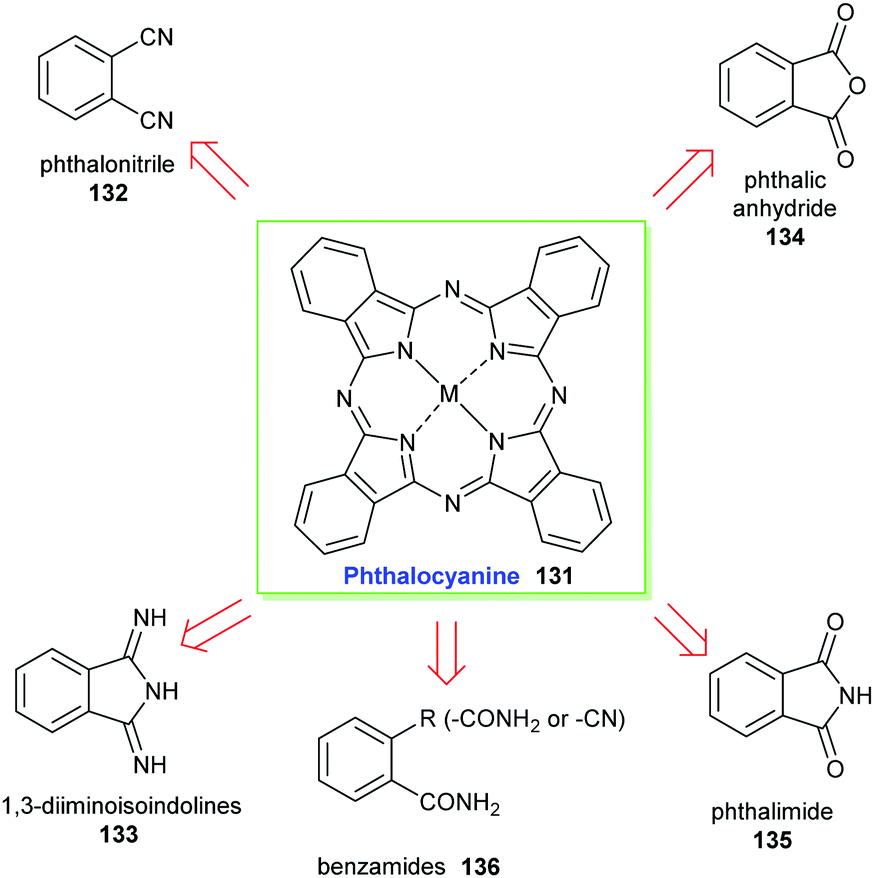 | ||
| Scheme 19 Retrosynthetic analysis for phthalocyanine 131. | ||
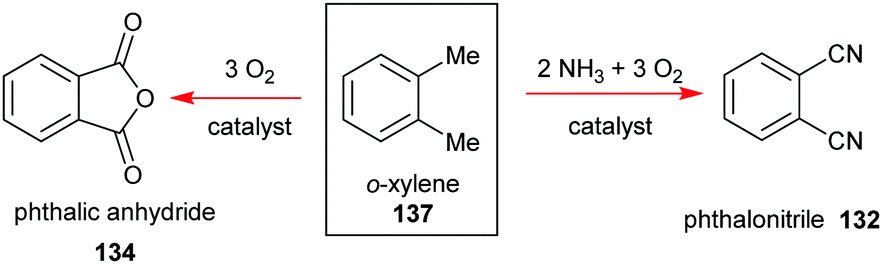 | ||
| Scheme 20 Syntheses phthalic anhydride and phthalonitrile as precursors for the industrial synthesis of phthalocyanines. | ||
The phthalic anhydride/urea process developed in England and Germany is the most significant, while the use of phthalonitrile is only second in pigment manufacture. Both pathways bear significant mechanistic similarities in chemical steps, e.g., coordination to a metal ion before tetramerization, followed by macrocyclic ring-closure and two-electron reduction. Phthalocyanines can be produced by batch or solvent methods; typically, a high boiling point solvent is utilized in the latter. Both preparation methods have their pros and cons. Generally, the phthalic anhydride/urea approach leads to non-halogenated phthalocyanines. However, partially chlorinated or brominated pigments can be produced, when the corresponding mono-substituted phthaloanhydride is used. Similarly, sulfonic groups can be introduced into the macrocycle. In contrast, the phthalonitrile method is mainly used to manufacture ring-chlorinated species, where the degree of halogenation can be controlled by the choice of a metal catalyst. Thus, the use of Cu(I)chloride produces “semi-chloro” phthalocyanine (CuPc–Cl0.5) and Cu(II)chloride affords (CuPc–Cl).
Formally, the phthalic anhydride/urea process can be illustrated using the reaction pathway given in Scheme 21. A common preparation method includes heating urea, phthalic anhydride, and a copper salt (e.g., CuCl, CuCl2, CuCO3, or CuSO4) in the presence of a molybdenum catalyst such as ammonium molybdate or molybdenum trioxide. Firstly, phthalic anhydride 134 reacts with ammonia that is liberated from the decomposition of urea to form phthalimide 135 and subsequently, mono-iminophthalimide 138. Together, they produce diiminophthalimide (1-amino-3-iminoisoindoline 139); the sequential process essentially being promoted by a molybdenum catalyst. The next step is a self-condensation of diiminophthalimide to form a polyisoindolenine 140 followed by complexation with copper ions. The ring-closure step is assisted by the release of ammonia and a successive reduction to yield (phthalocyaninato)copper(II) (CuPc, 141).
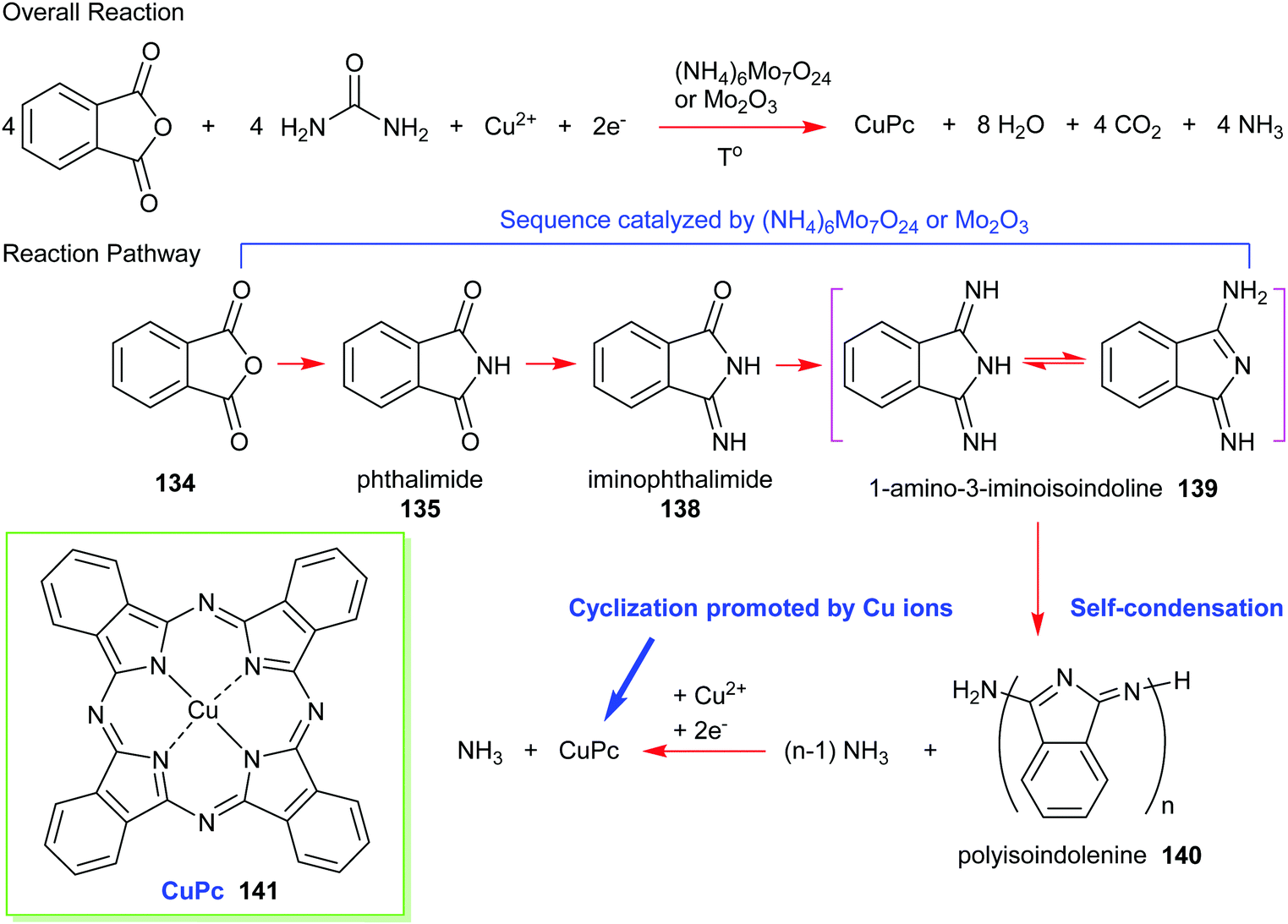 | ||
| Scheme 21 Synthetic pathway to CuPc 141 starting from phthalic anhydride and urea. | ||
A synthesis of phthalocyanines using phthalonitrile had been developed shortly before the onset of the World War 2.68b,70 In this approach, phthalonitrile 132 was heated in the presence of copper bronze or a copper salt at elevated temperatures with or without a solvent. Subsequent studies elucidated the mechanism of this process including the role of copper and its oxidation states.71 A modern variant of the synthesis is illustrated in Scheme 22.
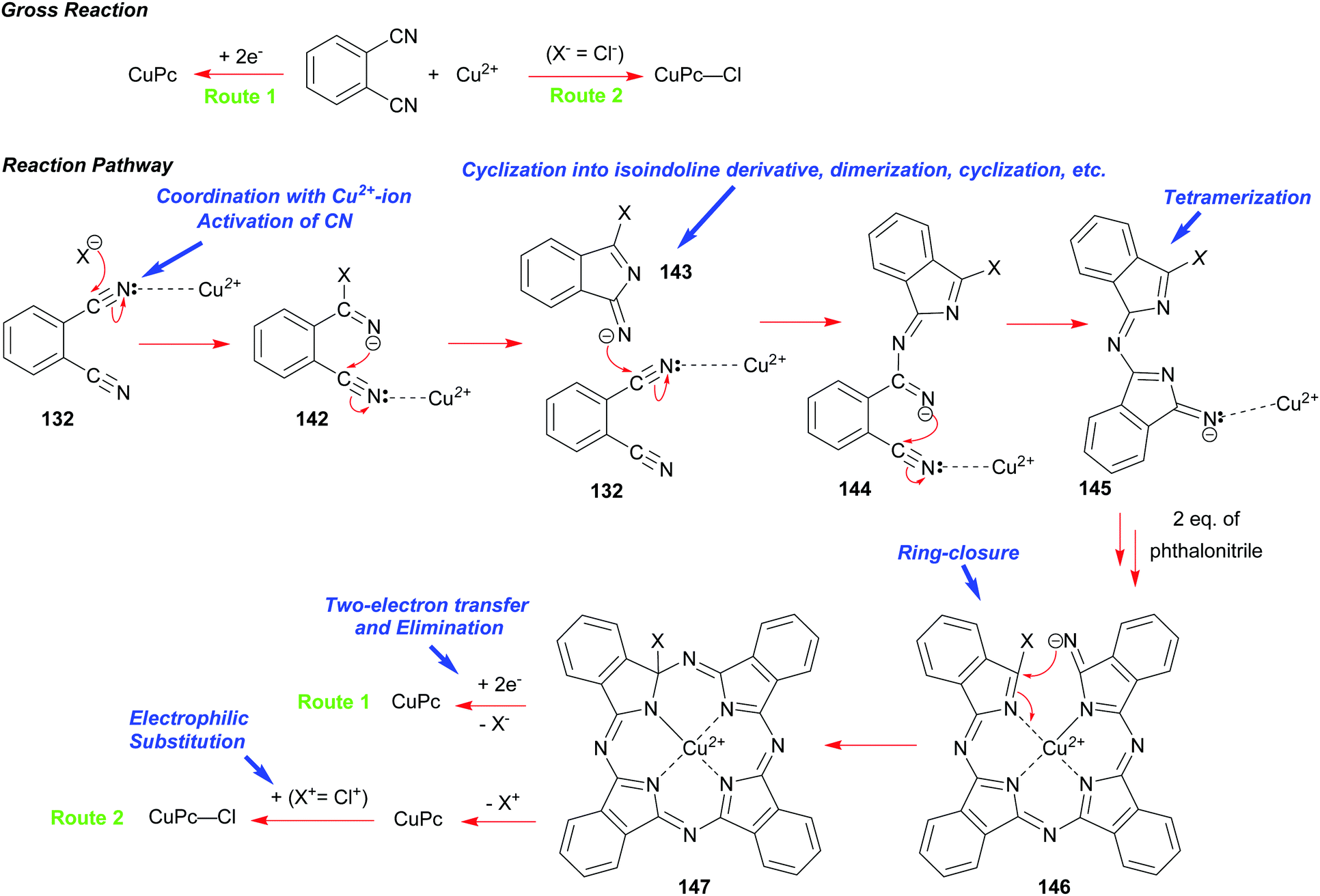 | ||
| Scheme 22 Synthetic pathways to CuPc 141 and CuPc–Cl starting from phthalonitrile. | ||
The initial step involves the coordination of one of the CN-groups of phthalonitrile 132 with a copper ion followed by a nucleophilic attack on the activated CN group usually by a copper counter anion (X−) such as chloride to give 142. Subsequently, this event promotes a cyclization into an isoindoline derivative 143, which acts as a nucleophile in a successive attack on the activated CN-group of another phthalonitrile 132. This sequence of transformations repeats, eventually leading to the formation of a tetrameric intermediate 146. A subsequent ring-closure facilitated by Cu-ion takes place to form the macrocycle 147. When copper metal is used as the reactant (Route 1), two-electron transfer from Cu(0) is required to allow the elimination of X− to yield the CuPc macrocycle. Thus, Cu(0) is oxidized into Cu(II) that can participate further in the reaction. However, elimination of (X+) is proposed, when a Cu(II) salt is used instead of Cu(0) to yield CuPc (Route 2). Importantly, when Cu(II)chloride is used as the reagent, a further electrophilic attack by Cl+ on CuPc takes place leading to (CuPc–Cl).
Chlorophylls are relatively unstable; hence, most of these materials contain chemically altered derivatives of chlorophyll a/b (2/148) generically called ‘chlorophyllin’ which was first described by Willstätter.2,73 Prepared by saponification of the phytyl side chain with sodium hydroxide and exchange of the central magnesium atom for copper or other metal ions it is an inhomogenous, water-soluble and photostable material. As outlined in Scheme 23 the harsh reaction conditions yield a mixture of compounds (pheophorbides, phytochlorins and rhodochlorins, e.g., 150) that rarely contains a single compound as inferred by trade names such as ‘copper-chlorophyllin’ (149) or ‘zinc-chlorophyllin’.
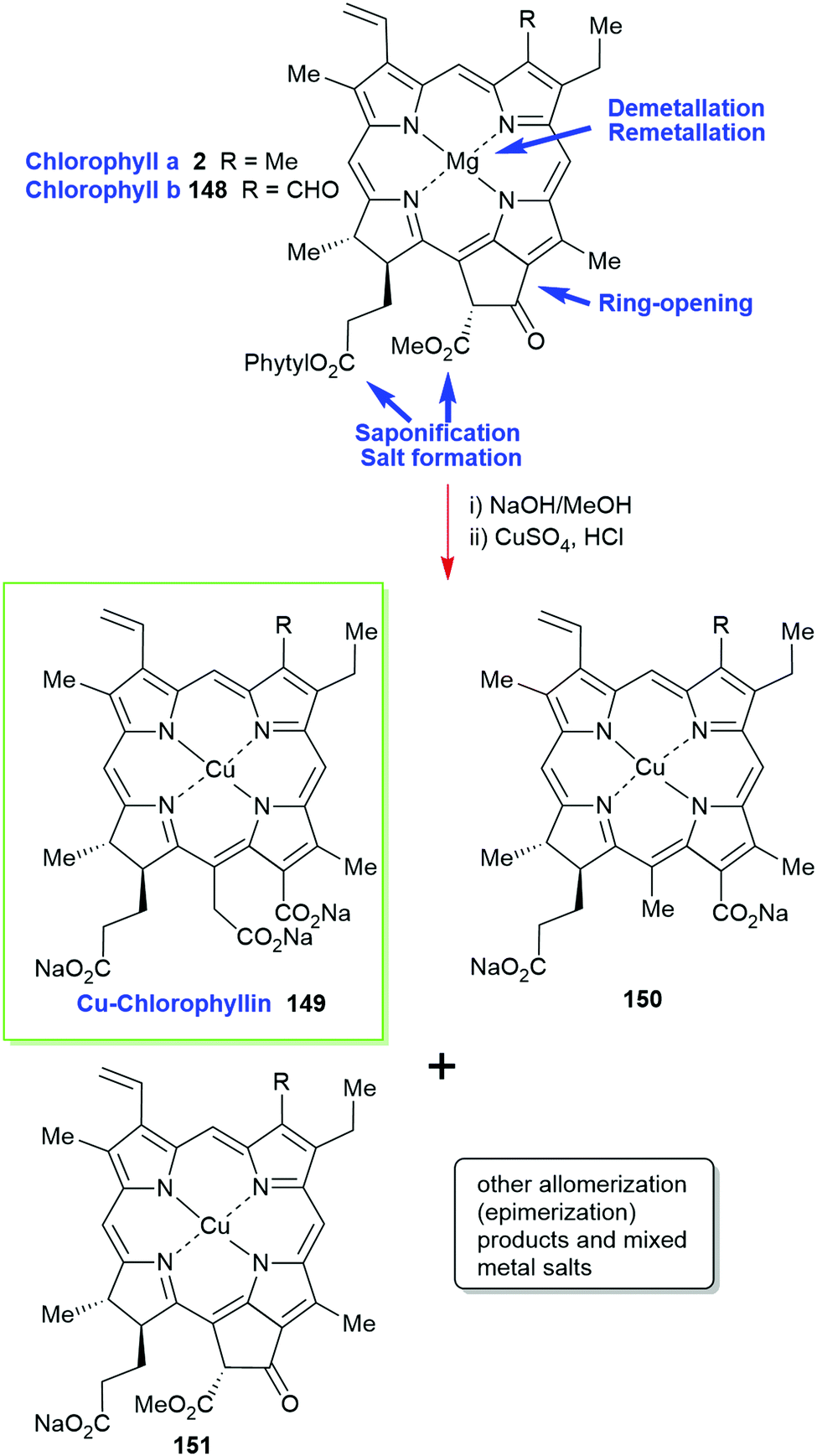 | ||
| Scheme 23 Preparation of’chlorophyllin’. | ||
4. Alternative strategies to β-substituted porphyrins
4.1 Monopyrrole tetramerization (Smith)
Going back to β-octasubstituted porphyrins, the following section illustrates approaches for the preparation of systems with different β-substituents/pyrrole units. Porphyrins, as ‘tetra’pyrroles, require four pyrrole building blocks and thus, in principle, a [4 × 1] approach can be considered. Obviously, symmetry considerations limit the synthetic possibilities, e.g., “monopyrrole tetramerizations can only yield a structurally unique product if the 3- and 4-substituents in the monopyrrole are identical”.50 Tetramerization is achieved as a ‘linear’ tetrapyrrole adopts a helical conformation, especially with bulky 3,4-substituents, bringing the two ends close together, facilitating cyclization over polymerization.Using pyrroles with two different β-substituents typically results in mixtures as the acidic reaction conditions promote ‘scrambling’,47i.e. cleavage of the growing pyrrole chain and recondensation. If acidic conditions can be circumvented, e.g., by using pyrrole 152 with a leaving group which can be cleaved under neutral conditions, then the preparation of the type I porphyrins (such as 12) is possible in some cases, notably for coproporphyrin I tetramethyl ester 153 (Scheme 24).74 A similar approach using two pyrroles 154 and 155 gave the D2h symmetric porphyrin 156.75
 | ||
| Scheme 24 Examples of pyrrole tetramerization reactions from Smith's group. | ||
4.2 [2+2] Condensation reactions (MacDonald)
Most modern porphyrin syntheses exploit symmetric or unsymmetric dipyrroles as condensation precursors, and the majority of these are distinguished by the type of dipyrrolic intermediate they employ (Fig. 5). For example, Fischer's classic haemin synthesis (and others) relied on the use of dipyrromethenes 157.8 The latter are typically derived from dipyrromethanes 158 by oxidation and, due to their stability (and the acid-sensitivity of dipyrromethenes 157), such intermediates have featured prominently in the porphyrin syntheses published in the early part of the last century.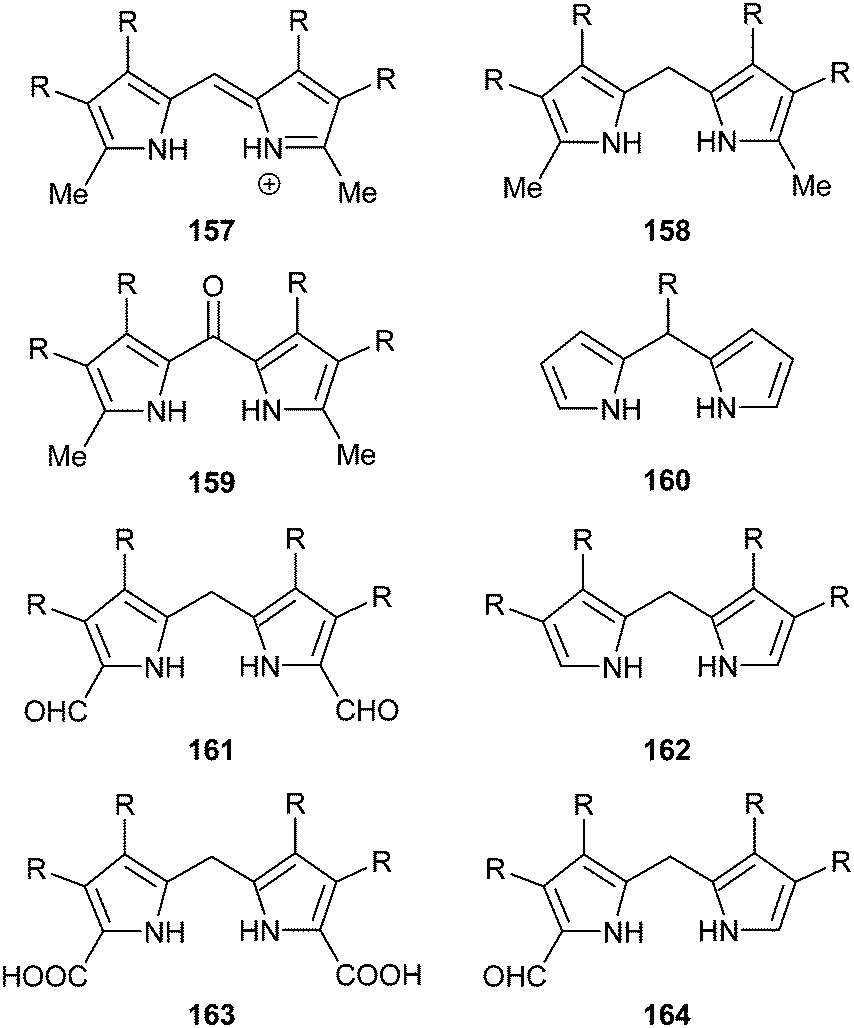 | ||
| Fig. 5 Some generalized types of dipyrrolic intermediates often used in [2+2]-porphyrin assembly. | ||
Unsymmetrical systems require the use of protecting group chemistry to differentiate the substituents on the two pyrrole units while symmetrical systems are relatively easily prepared by self-condensation methods. A very detailed analysis of the various strategies and intermediates is given in a review by Kevin Smith which serves as probably the best overview presently available.76 He, and others, have pioneered many of the new advances in porphyrin synthesis over the past 50 years, and their strategies have used dipyrromethanes 158,77 dipyrroketones 159,8 and meso-substituted dipyrromethanes 160.78 All of these intermediates have also featured in the various [2+2]-condensation methods developed in the past. The most significant development in this regard has been the so-called MacDonald [2+2]-cyclocondensation.79,80
The MacDonald cyclocondensation relies on the acid-catalyzed union of a 1,9-diformyldipyrromethane 161 with a 1,9-di-unsubstituted dipyrromethane 162 or a 1,9-dicarboxylic acid 163. The method works extremely well and can give porphyrins in yields as high as 50–60%, as evidenced by MacDonald's synthesis of the uroporphyrin II octamethyl ester (167) (Scheme 25)79 which was accomplished by exposing an acetic acid solution of dialdehyde 165 and bis-pyrrole 166 to catalytic 56% hydroiodic acid in acetic acid with the continuous bubbling of air through the reaction mixture for 24 h and the rigorous exclusion of light. As can be seen from this example, it is a very simple protocol to effect, and it generally gives very good results. However, like Fischer's approach, it is limited by its symmetry restrictions, with one of the two dipyrromethane coupling partners needing to be symmetrical about the meso position for good results to be obtained. A convenient application of this technology is the self-condensation of 1-formyl-9-unsubstituted dipyrromethane 164 for the preparation of centrosymmetric porphyrins.
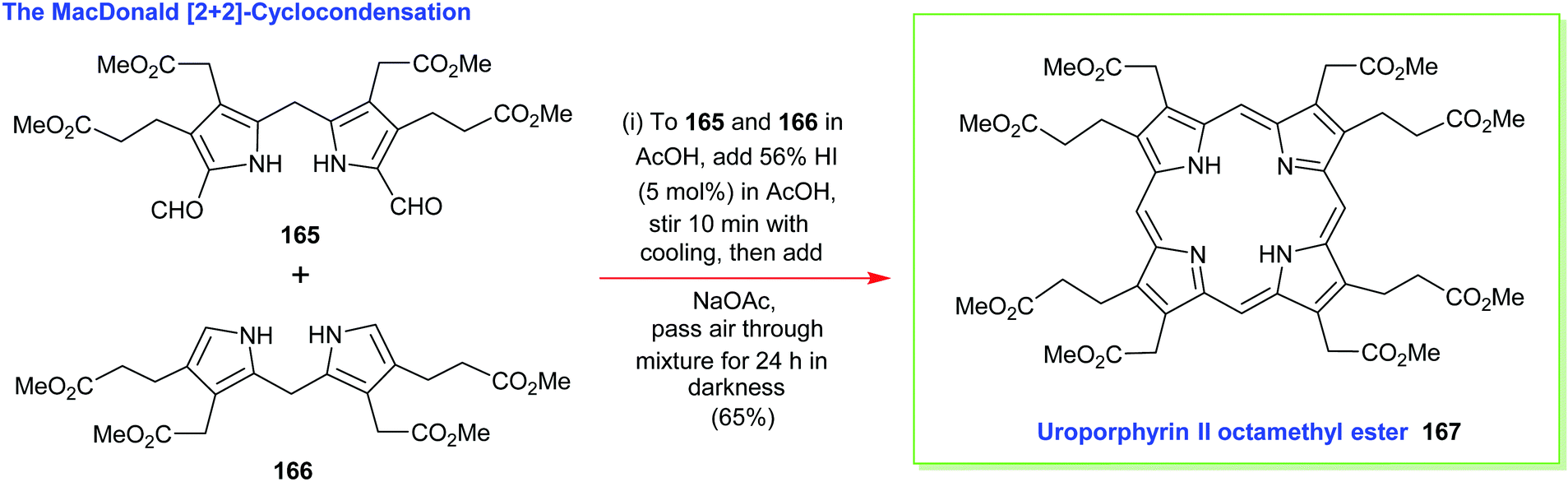 | ||
| Scheme 25 MacDonald's [2+2]-cyclocondensation route to uroporphyrin II octamethyl ester 167. | ||
4.3 [3+1] Condensation reactions
Another approach to the synthesis of porphyrins is a [3+1]-strategy.81 This requires the synthesis of a tripyrrolic precursor, typically a tripyrrane82 which is then condensed with a monopyrrole unit. In many cases, the latter carries 5,10-meso-substituents on the central pyrrole. Significant problems can sometimes be the acid-sensitivity of the tripyrrane and the possibility of pyrrole-scrambling. However, this was not an issue in the example shown in Scheme 26 from Lash which details another approach to tetrahydromono-benzoporphyrins (173).83 Another example of a [3+1]-cyclocondensation has previously been seen in Scheme 17. Such approaches can thus be useful, and they have found widespread application in the preparation of different pyrrole-modified porphyrins.84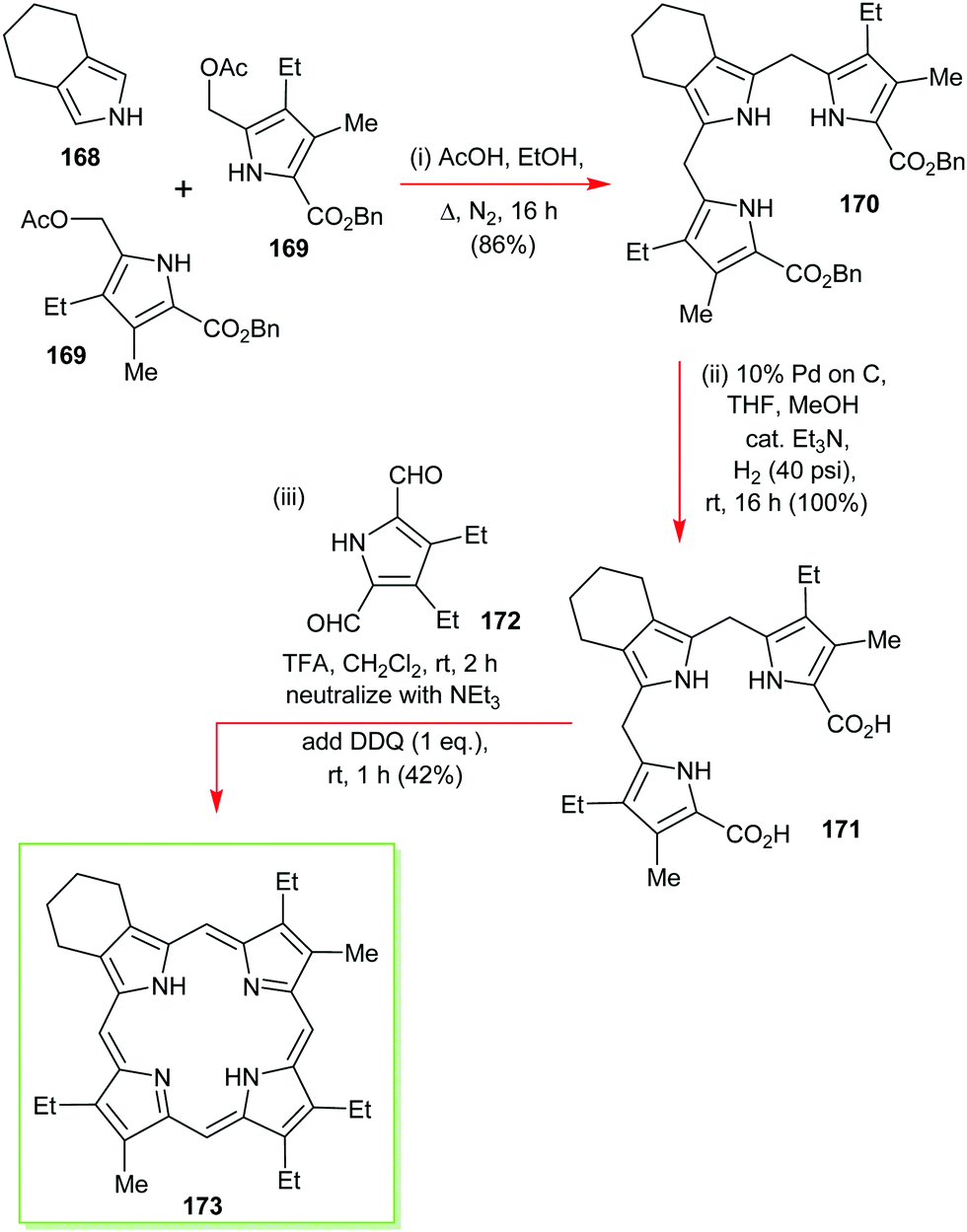 | ||
| Scheme 26 Lash's synthesis of a mixed monotetrahydrobenzoporphyrin 173. | ||
4.4 Cyclization strategies
Naturally, the synthesis of a completely unsymmetrical β-substituted porphyrin requires the use of an appropriately substituted open-chain tetrapyrrole precursor. Like the dipyrrolic components for [2+2]-condensations, various precursor molecules exist and all of them have been used, to some extent, in porphyrin syntheses over the years.76 Examples include the bilanes 174, the a-and b-bilenes 175 and 176, the a,c-biladienes 177 and 178, and a,b,c-bilatriene 179 (Fig. 6).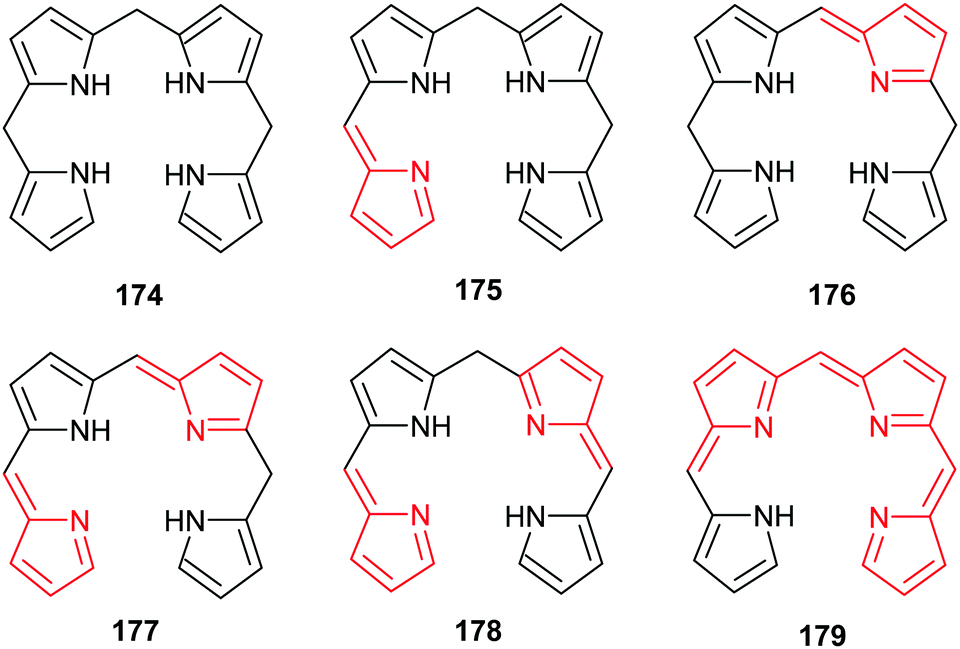 | ||
| Fig. 6 Some generalized types of tetrapyrrole intermediates often used in porphyrin assembly. | ||
The oldest example of such an approach dates back more than sixty years and involved the attempted synthesis of etioporphyrin II (13) (Scheme 27).85 In this, the reaction of the 1-unsubstituted dipyrromethane 182 with formic acid/HBr (5:1) was believed to give a bilane 183 after catalytic reduction. Hydrolysis of its esters then afforded the diacid 184 which underwent cyclization in concentrated formic acid to yield etioporphyrin II (13) of 70% purity. Mechanistically, the formation of 183 is thought to involve a formylation of the unsubstituted pyrrole ring of 182 with HCO2H to give a dipyrrole aldehyde which then condenses with another unsubstituted molecule of 182 to provide the bilane alcohol which is brominated and/or eliminated to the bilene before being catalytically reduced. Yet again, formic acid also monoformylates 184 with loss of water and CO2, and a decarboxylative nucleophilic addition also proceeds (with further loss of water and CO2) to give 13. While today we are aware of the acid-lability of bilenes and bilanes, this was a totally new territory in 1952, and this experiment must have produced a mixture of several etioporphyrin isomers and not just the etioporphyrin II that is shown. Subsequent efforts focused initially on a-bilenes, which were found to give mixtures of porphyrins,86 and then on the more commonly used b-bilenes.87
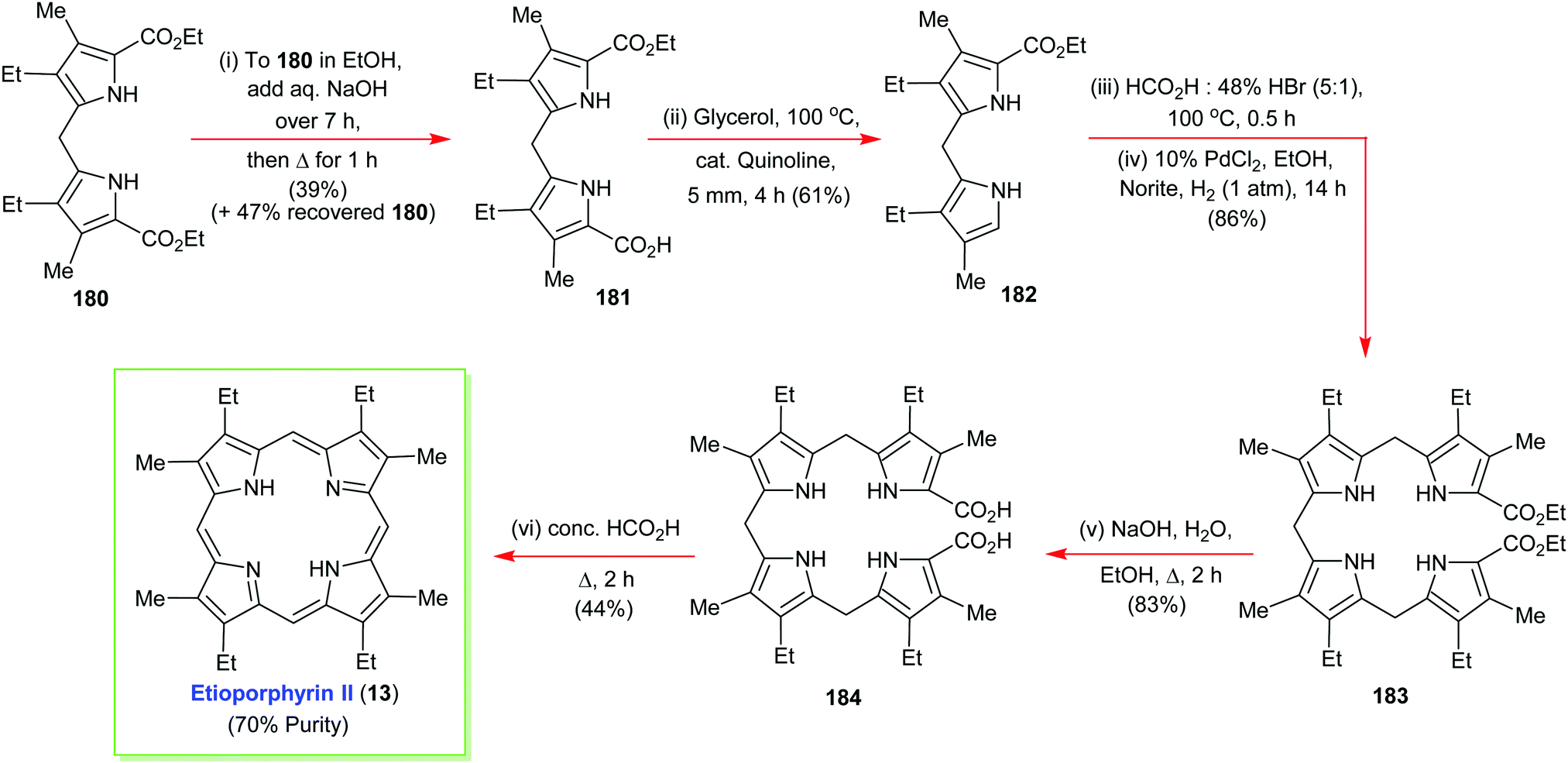 | ||
| Scheme 27 The Corwin–Coolidge bilane pathway to etioporphyrin II 13 of 70% purity. | ||
a,c-Biladienes have turned out to be some of the most versatile building blocks in synthetic porphyrin chemistry, exhibiting rich cyclization chemistry.88 In practical terms, two cyclization methods have stood the test of time. The older method of Alan W. Johnson involves the use of metal(II) salts, typically copper(II), as templates for cyclization.89–91 This method often works well with symmetric 1,19-dimethyl-a,c-biladienes 185 (Scheme 28) but has the disadvantage that it always yields a metal-complexed porphyrin 186, and removal of the central metal typically requires rather forcing reaction conditions; i.e. treatment with sulfuric or trifluoroacetic acids. The originally formulated Johnson-Kay methodology also typically mandated high temperatures to obtain a successful outcome and, while often reliable, the yields can, on occasion, be rather variable (20–75%). A room temperature cyclization method has been devised by Smith and Minnetian91 that operates in the presence of other metal oxidants, but even this does not overcome the overall requirement for an acid-induced demetallation step.
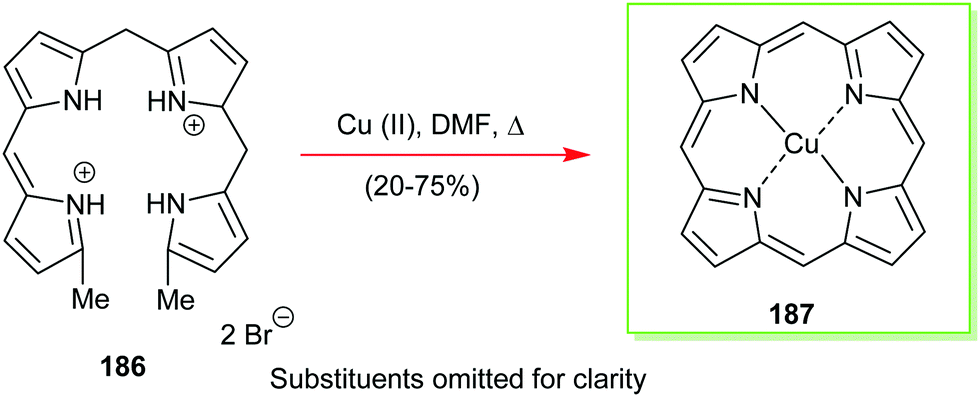 | ||
| Scheme 28 The Johnson-Kay copper-templated pathway to porphyrins. | ||
A significant development in the area, therefore, was Smith's discovery that a,c-biladiene salts 187 can undergo an electrochemical cyclization to porphyrins 188 (Scheme 29).92 Mechanistically, this reaction is intriguing, and Smith's probing has led to the pathway presented in Scheme 30.
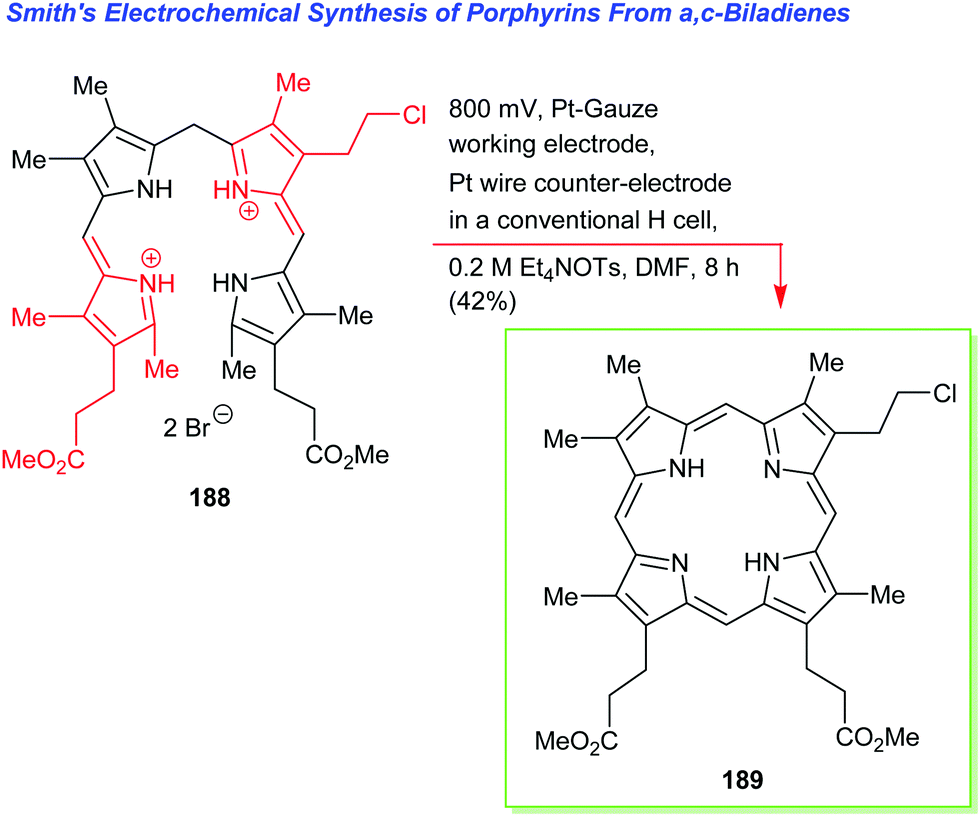 | ||
| Scheme 29 Kevin Smith's electrochemical synthesis of porphyrins from a,c-biladienes. | ||
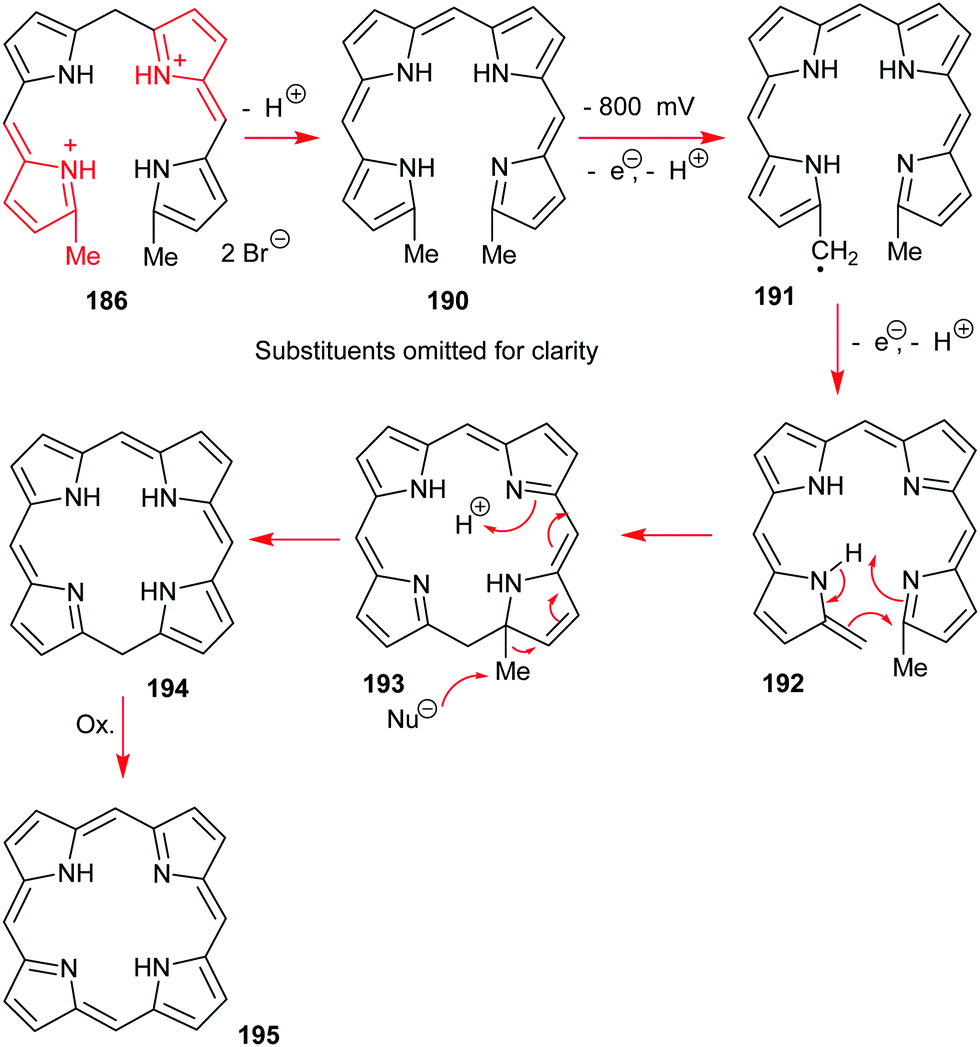 | ||
| Scheme 30 The mechanism of Smith's electrochemical synthesis of porphyrins from a,c-biladienes. | ||
When allied with recent developments in a,c-biladiene construction (Scheme 31), the electrochemical cyclization method of Smith now circumvents many of the symmetry limitations associated with earlier approaches to porphyrins.89–91 As outlined in Scheme 31, the key steps that are effected, these days, are sequential “counterclockwise” (left pathway)89c or “clockwise” (right pathway)90 routes, which utilize different carboxylic esters. For example, catalytic hydrogenation of 196 affords the monocarboxylic acid dipyrromethane 197 which can then be condensed under mild conditions with the 2-formyl-5-methylpyrrole 199 to give the tripyrrane salt 201. Cleavage of the t-butyl ester with TFA followed by condensation with another 2-formylpyrrole 200 then affords the 1,19-dimethyl-a,c-biladiene 203, which can then be cyclized using either Cu(II), Cr(III) or electrochemical methods to the porphyrin 204. Similar strategies can be employed in the complementary clockwise route which involves the dipyrromethane 198 being condensed with the pyrrole 200 to give 202 followed by standard conversion to 203 and thence 204.
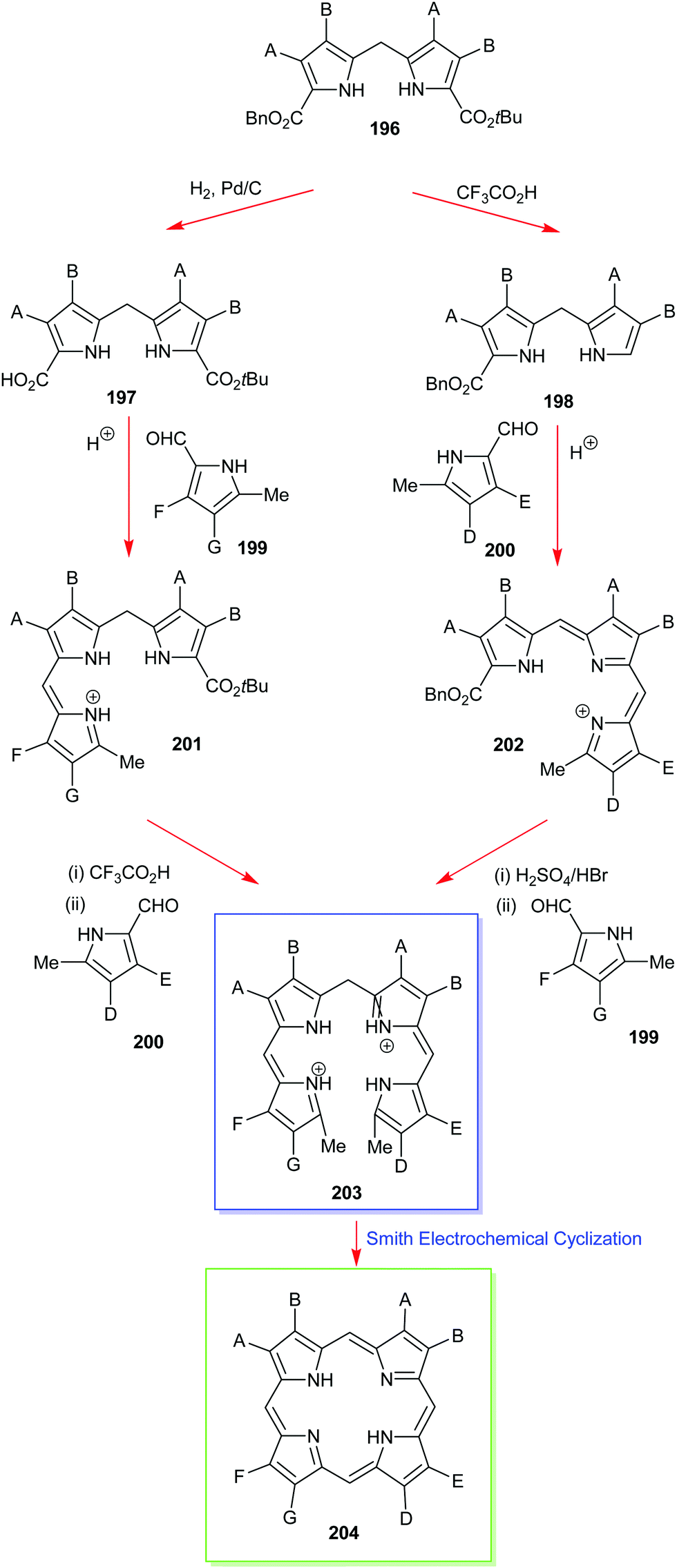 | ||
| Scheme 31 Some developments in the construction of a,c-biladienes that can be used in porphyrin ring construction. | ||
5. Strategies to unsymmetric meso-substituted porphyrins
5.1 Target systems
Entirely analogous to the situation with β-substituted porphyrins, the main synthetic problems for meso-substituted porphyrins arise from symmetry considerations. While a veritable cornucopia of symmetric 5,10,15,20-tetrasubstituted porphyrins has been prepared through simple pyrrole tetramerization (see Section 4.1), progress in the synthesis of the less symmetric derivatives of the porphyrin “alphabet soup” (Fig. 7)47,64 has somewhat lagged behind. For a comprehensive overview of the various condensation methods and the historical development of this subject see the excellent review by Lindsey.44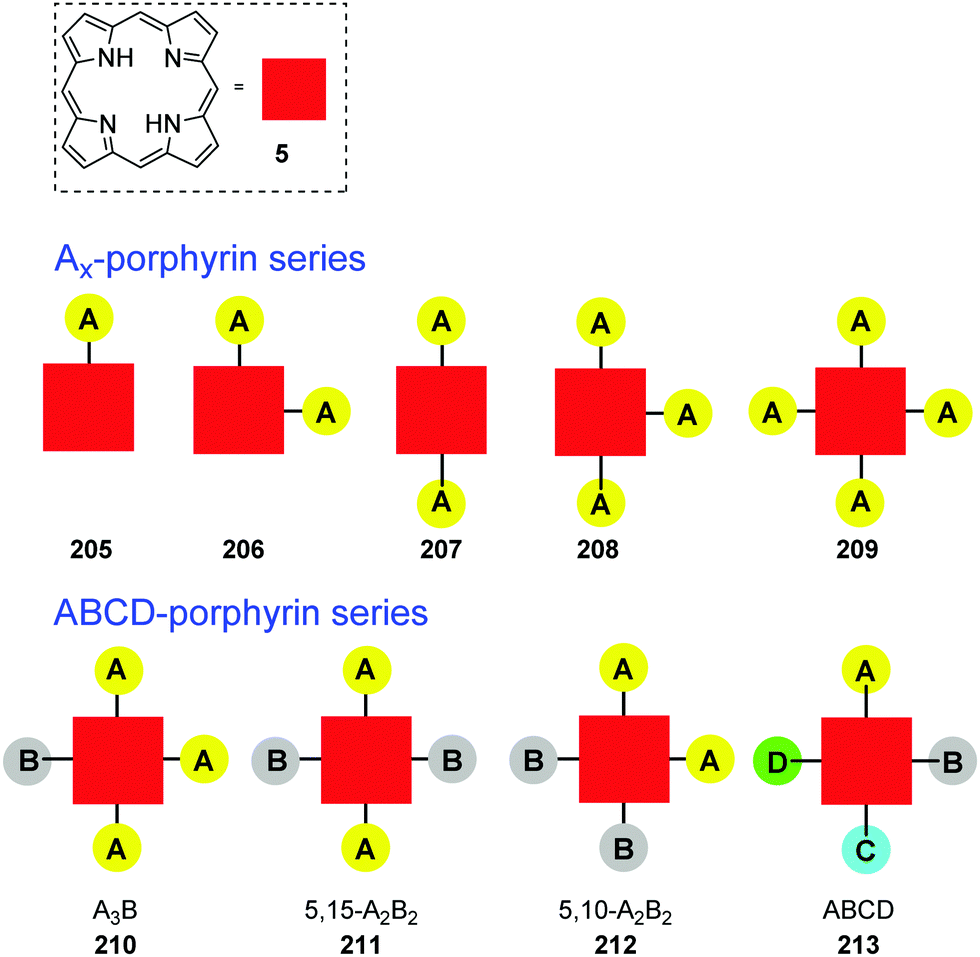 | ||
| Fig. 7 Selected examples of different meso-substituted porphyrins. | ||
With regard to fundamental condensation methods, historically, the main advances were the Rothemund method (condensation in a sealed vessel),45a the Adler and Longo method (acid catalysis),45b,93 Volz's use of pyrrole carbinols,94 and finally, the two-step, one-flask Lindsey method, which typically uses BF3·Et2O as an acid catalyst and chloranil as an oxidant.46
Porphyrins with lower symmetry than S4 have been studied for about 40 years. As might be expected, initial work focused on 5,15-disubstituted porphyrins (also often called “trans porphyrins”), e.g., 207 and related derivatives with β-substituents.95 Their synthesis is relatively straightforward and only requires the reaction of an aldehyde with either an unsubstituted or 2-substituted dipyrromethane.96 Later work focused on accessing 5,15-disubstituted porphyrins with two different substituents (AB) through use of two different dipyrromethanes which could not self-condense. Examples of this are reactions of a 1,9-diformyldipyrromethane with a 1,9-unsubstituted dipyrromethane or mixed condensations.97 Similarly, access to 5,10-disubstituted (206,“cis”-A2) porphyrins was gained late and involved either a [2+2] condensation of a dipyrromethane dicarbinol with dipyrromethane98 or a [3+1] condensation of tripyrrane with pyrrole and aldehydes.62b,99
The entry into the less symmetric members of the “porphyrin alphabet soup”,64e.g., 210–213, initially involved so-called mixed condensations, where different aldehydes were used and which required, an often tedious, chromatographic separation of the products. Until the last decade attempts to prepare such compounds via total synthesis were rare. A typical example of an early synthesis of an ABCD porphyrin from Smith's laboratory (216) is shown in Scheme 32.
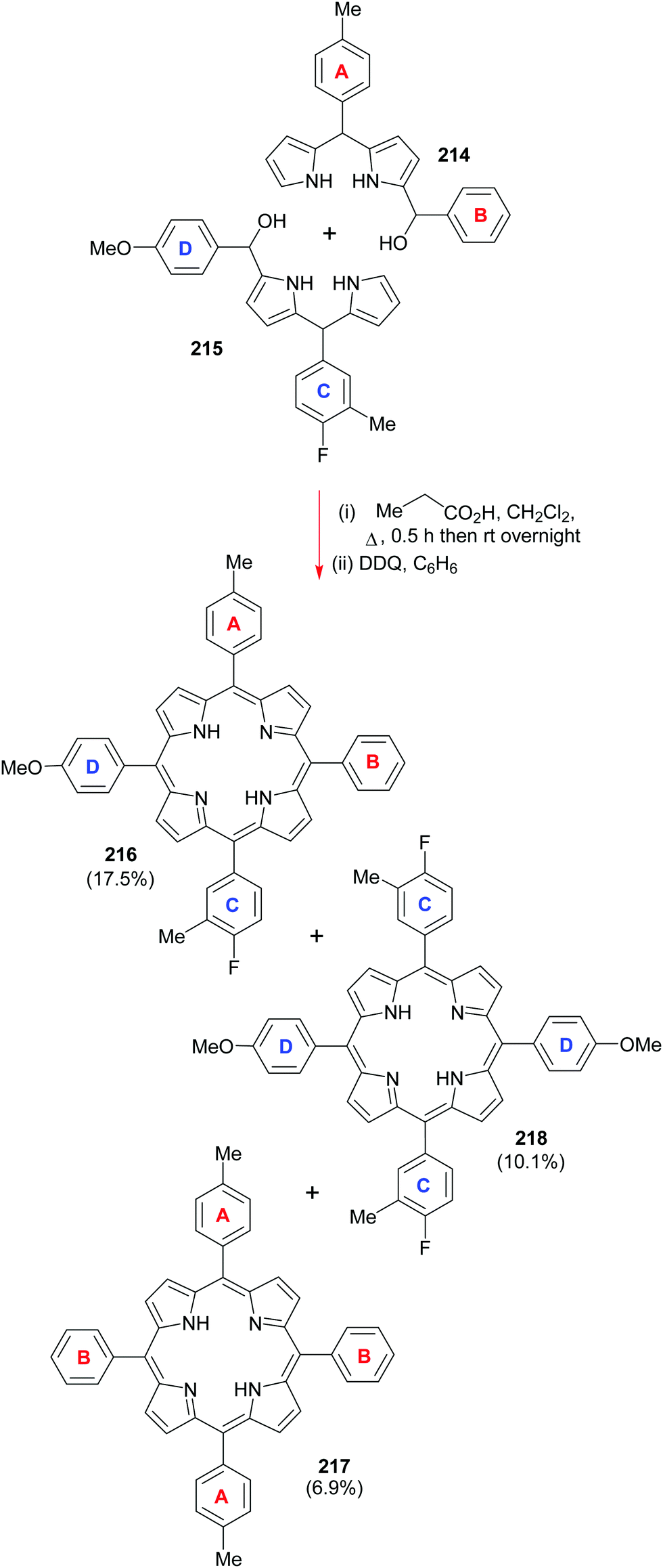 | ||
| Scheme 32 Smith's [2+2]-fragment cross-condensation route to the meso-substituted porphyrin 216 where all four rings are differentially substituted (ABCD). | ||
In this method, the stepwise synthesis of a dipyrromethane is conducted. It is then C-acylated and reduced to the carbinol. These can then either be self-condensed or used for a mixed condensation as was done in Scheme 32.100 However, these methods were only applied to meso-arylporphyrins. Similar approaches, [3+1]-condensations and reactions of bilanes with aldehydes have likewise been used for the synthesis of the so-called A2B2 porphyrins, notably of the type 212.101,102 Several approaches have also been developed for members of the Ax or ABCD series. In most cases, however, these have been targeted at specific types of porphyrin, such as meso-formyl derivatives103 and a survey of the developments in the last decade has been provided.104
5.2 A general approach to ABCD-type porphyrins (Lindsey)
The first, nearly general, method for the synthesis of meso-tetrasubstituted porphyrins was developed by Lindsey, who focused on relatively short and large-scale total syntheses. For example, his group prepared 5,10-disubstituted zinc(II)porphyrins through the condensation of 1,9-bis(N,N-dimethylaminomethyl)dipyrromethane and dipyrromethane in ethanol at reflux, in the presence of zinc acetate, followed by oxidation with DDQ.105 This synthesis was compatible with a variety of substituents (e.g., alkyl, aryl, ester, acetal) and gave 5,15-AB-, A2- and A-porphyrins. A different route utilized the 1,9-diformylation of a dipyrromethane, the conversion of the diformyldipyrromethane to the bis(imino) derivative and the condensation of this compound with a dipyrromethane to prepare 5,15-AB porphyrins.106 Some limitations still exist; the possible substituents included aryl/alkyl, aryl/aryl, but not alkyl/alkyl combinations; for A-porphyrins the substituent also had to be aryl.Scheme 33 outlines the general approach taken by Lindsey for creating meso-ABCD-porphyrins 222. Here the directed synthesis of a 1-brominated-19-acylbilane 221 was performed via Sc(OTf)3 or Yb(OTf)3-catalyzed condensation of a 1-acyldipyrromethane 220 with a 9-protected dipyrromethane-1-carbinol 219 (derived from a 9-protected 1-acyldipyrromethane).107 A bromo residue was found to be the best leaving group for the final cyclization. In the case of the Sc(III)-promoted reactions, the use of 2,6-di-tert-butylpyridine proved necessary, but the latter required chromatographic removal. This was not the case for Yb(OTf)3, which could also be used very successfully in much smaller quantities. A one-pot DBU-assisted, transformation of the 1-protected 19-acylbilane under metal-templating conditions was also developed that gave the corresponding metalloporphyrins 222 in good yield. Indeed, via this approach, the zinc and magnesium porphyrins 223 and 224 were synthesized in 50% and 69% yield, respectively.
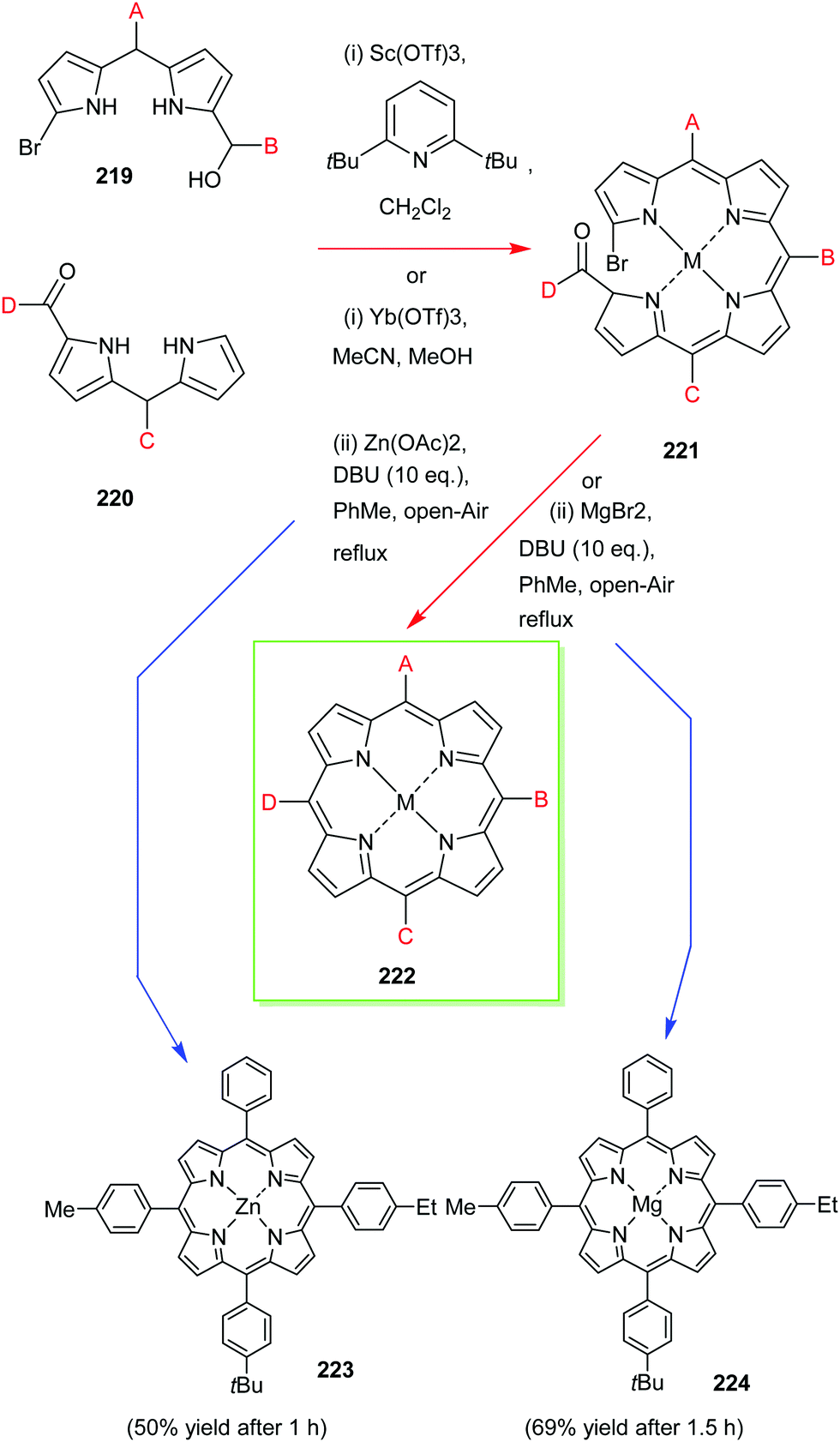 | ||
| Scheme 33 Lindsey's strategy for securing unsymmetric meso-tetrasubstituted porphyrins. | ||
6. Strategies for the synthesis of chlorins
Chlorins are 18π-electron aromatic tetrapyrroles in which the C(2)–C(3)-bond of ring A is fully saturated. Total, as opposed to partial, syntheses of chlorins have been rare since Woodward and Eschenmoser's synthesis of vitamin B12 and Woodward's earlier efforts on chlorophyll a. Even so, there have some notable synthetic accomplishments in this area in recent times, including Battersby's racemic synthesis of bonellin dimethyl ester, Battersby's powerful asymmetric route to heme d1 methyl ester, Kishi's stunning synthesis of tolyporphin A O,O-diacetate, and Jacobi's multiple synthetic strategies to various unsymmetrical chlorins. Together, these are all major highlights in the chlorin total synthesis area, and so now we will discuss each of these pieces of work in detail.6.1 Total synthesis of (±)-bonellin dimethyl ester (Battersby)
One especially noteworthy chlorin synthesis of the modern era is that of (±)-bonellin dimethyl ester 236 by Battersby and coworkers at Cambridge108 which exemplified a powerful new photochemical macrocyclization strategy that had been developed by the Battersby group some years earlier.16b,108–110 Bonellin dimethyl ester is a sex-differentiation compound of the echiura worm.111 Battersby's pathway (Scheme 34) was formulated around a condensation of the western hemisphere 233 with the eastern hemisphere 234 to obtain 235 which was subsequently photocyclized.108 The latter reaction took place over 7 days to produce a 20–36% yield of product. However, if the yield was based upon recovered starting material, it typically amounted to 95–97%. As with the Eschenmoser vitamin B12 photocyclization, the Battersby photocyclization is mechanistically intriguing and potentially involves a delocalized triplet biradical undergoing a conrotatory, antarafacial, 18π-electrocyclization, via a helical conformation which promotes satisfactory orbital overlap. However, equally, one can also envision macrocyclization proceeding via simple biradical cage combination, which one would expect to be very fast. In either case, the initially formed cycloadduct undergoes subsequent tautomerization and E1cb elimination to afford the desired macrocycle 236.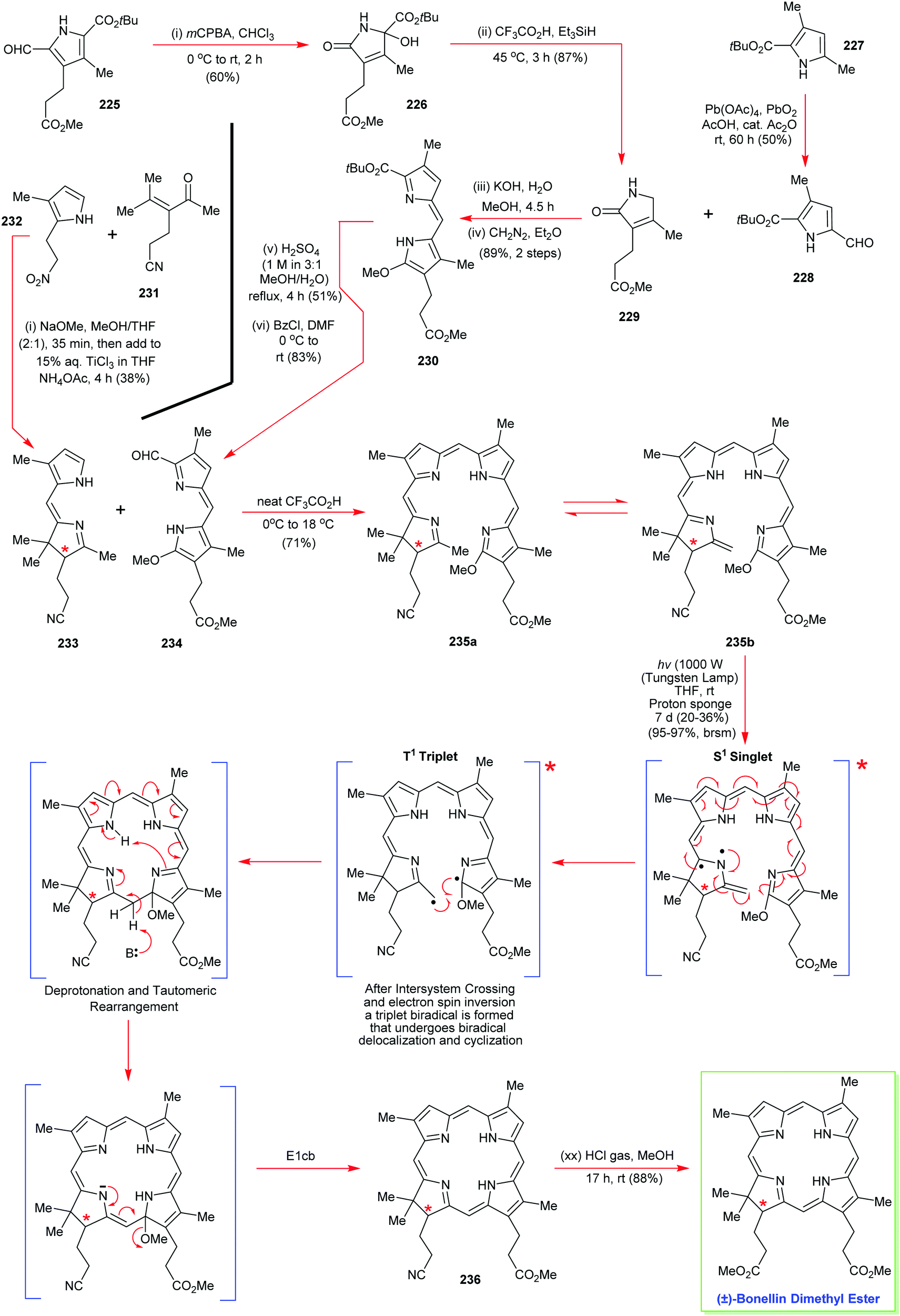 | ||
| Scheme 34 Battersby's total synthesis of racemic bonellin dimethyl ester. Two mechanistic possibilities exist for the key photocyclization (see main text for more discussion). | ||
The final step of Battersby's synthesis was the conversion of the product nitrile into the bonellin methyl ester with gaseous HCl in MeOH.
6.2 Total synthesis of (2R,7R)-heme d1 methyl ester (Battersby)
Another landmark chlorin total synthesis of the modern era is Battersby's enantioselective pathway to the heme d1 methyl ester 237.112 Heme d1 is one of the two heme units found within the bacterial enzyme cytochrome cd1.112 Although its gross chemical structure was elucidated in the mid-1980s, as a result of Chang and Wu's synthesis of a mixture of all the possible racemic diastereoisomers of this structure,113 questions still remained concerning the absolute stereochemistry of the C(2)- and C(7)-positions in rings-A and -B. Based upon a biosynthetic hunch that heme d1 and sirohydrochlorin were structurally related, and that the absolute stereochemistry of both molecules was the same, Battersby and his team attempted the total synthesis of the (2R,7R)-stereoisomer, and happily, subsequent correlations revealed that their hypothesis was in fact correct. We will therefore discuss this elegant second-generation route to the heme d1 methyl ester since it reports many noteworthy reactions of general significance in the porphyrin field.Battersby's plan for securement of the heme d1 methyl ester (Scheme 35) centered on implementation of a regioselective dihydroxylation reaction on the 3,8-dioxoisobacteriochlorin 240a with OsO4, which would mirror an earlier chemical precedent set by Chang and Wu in their earlier mixture synthesis.113 The Battersby team would thereafter effect an acid-mediated dehydration reaction on the diol 239 to obtain 238 which, in turn, would be subjected to an acid-induced enolization and an alkene migration step to furnish the methyl ester of the natural product. Compound 240a and its tautomer 240b were themselves considered derivable from 241 by allylic oxidation and 241 was, in turn, deemed accessible from an application of Battersby's photochemical 18π-antarafacial macrocyclization reaction on 242. The remaining retrosynthetic steps split 242 into 243 and 244, and 244 was itself considered derivable through a site-selective Wittig union between 245 and 246, while 243 was thought accessible from a Grignard union between thiopyridyl ester 248 and the pyrrole anion 247.
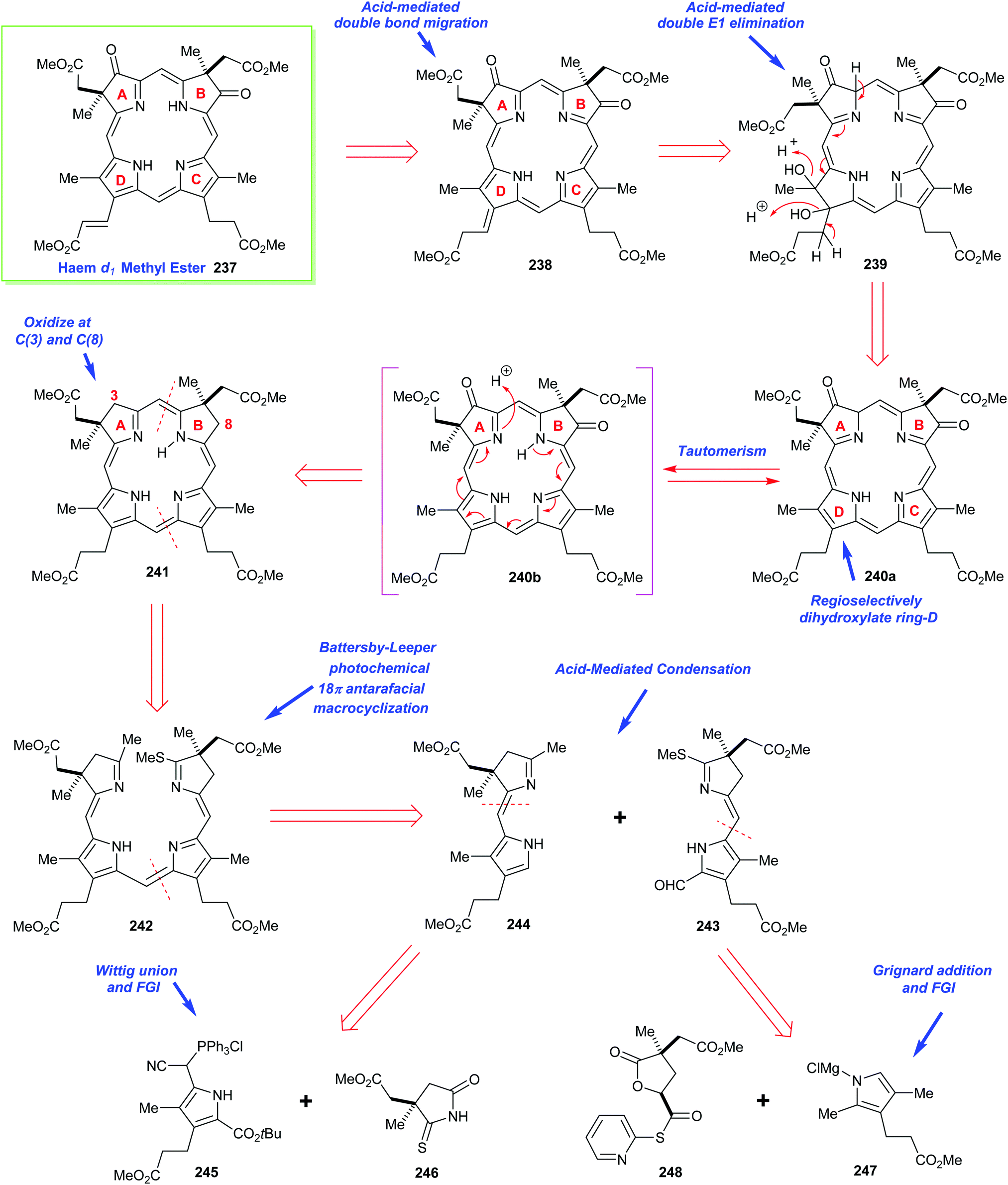 | ||
| Scheme 35 Battersby's retrosynthetic analysis of the heme d1 methyl ester. | ||
Battersby initially devised the route detailed in Scheme 36 for the accession of 244. It set off from diethyl (R)-malate 249 and used a Seebach double alkylation reaction to set the quaternary asymmetric center at C(2) with a high level of stereocontrol (dr 96:4) in favor of the desired stereoisomer 250. Although the free radical reduction of the xanthate derived from 250 was indeed viable, for larger scale work, the zinc-mediated reduction of chloride 251 proved far more preferable. Of course, this necessitated a potentially problematical chloride displacement reaction being effected on a neopentylic carbon. Nevertheless, by using triphenylphosphine, excess carbon tetrachloride and acetonitrile, this reaction was accomplishable, the desired chloride 251 being obtained in 74% yield. The zinc reduction step also proceeded well to give, after ester hydrolysis, the diacid 252.
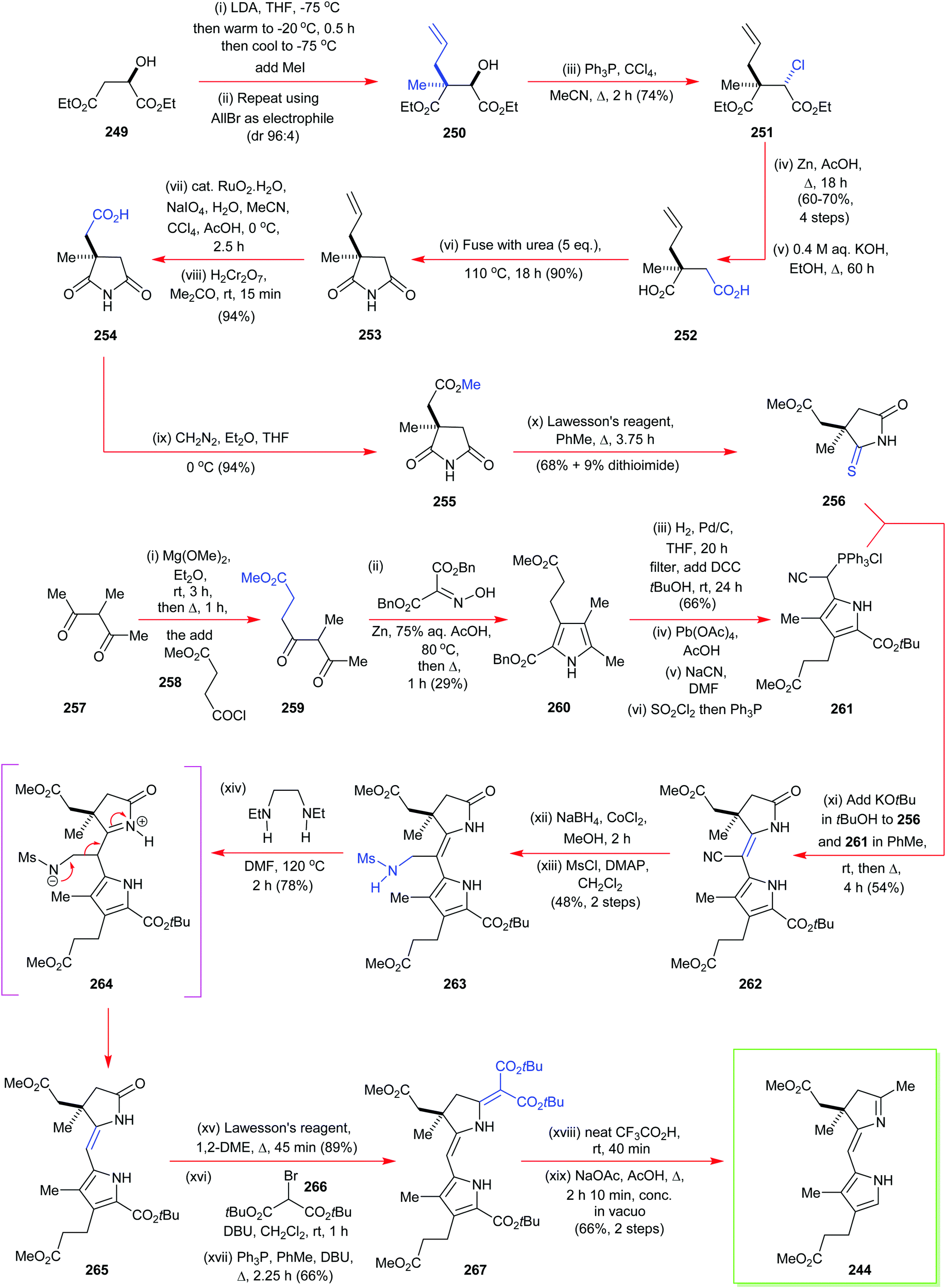 | ||
| Scheme 36 Battersby's first-generation synthesis of the Western hemisphere 244 of the heme d1 methyl ester. | ||
Formation of the succinimide 253 was now the order of the day, and it was accomplished by performing a high temperature fusion reaction with solid urea at 110 °C for 18 h. Oxidative cleavage of the alkene in 253 was next attempted. Rather surprisingly, catalytic RuO4 oxidation did not accomplish the conversion of 253 directly to the acid 254, the latter event requiring a further exposure of the initially produced acid/aldehyde mixture to Jones’ reagent. Diazomethane treatment thereafter fashioned ester 255 and significantly, the more electron-rich (but more sterically hindered) carbonyl was regioselectively thionated. Although a small amount of the undesired dithioimide was formed, happily, the bulk of the reaction mixture was enriched in 256.
Battersby now reacted 256 with the pyrrole phosphonium chloride 261 which itself had been prepared from the 1,3-diketone 257 by a six-step sequence. It was generally found that the use of potassium tert-butoxide gave the best results in this key Wittig union, and naturally the thioamide reacted preferentially, due the greater electrophilicity of a thioamide group, brought about by the significant dipolar character of a thiocarbonyl. Whilst the stabilized Wittig process worked reasonably well, it did carry with it the burden of having to excise the surplus cyano functionality from 262. In a very cunning synthetic maneuver, Battersby chemoselectively reduced the nitrile to the primary amine with sodium borohydride that had been doped with cobalt(II) chloride. Remarkably, this reductant succeeded beautifully in this task, which is notable given the potentially conflicting presence of the three ester groups and the lactam carbonyl. Success in this step permitted an N-mesylation to be performed to obtain 263 and the use of a retro-Mannich reaction to excise the surplus carbon; the latter involved the anion 264.
In order to transform the lactam carbonyl into the pyrrolidine imine 244, Battersby now converted 265 into a thiolactam and alkylated the latter on its S-atom with the bromomalonate 266. An Eschenmoser-style S-extrusion reaction was then effected on this intermediate with triphenylphosphine and DBU in toluene at reflux to obtain 267. Neat trifluoroacetic acid then deprotected the three tert-butyl esters from 267 to unmask a malonic acid, which was exhaustively decarboxylated by heating with sodium acetate in acetic acid at reflux. The result was 244, formed in 66% yield for these last two steps.
Following these opening moves, Battersby improved his synthetic route to 265 and thence 244 (Scheme 37). In his new pathway to 265 and 244, the chiral butenolide 268 was utilized as the starting material and proceedings were commenced with a higher order allylcuprate addition to obtain 269. Although this addition proceeded with good stereocontrol, due to the bulky –CH2OTr group hindering an underside attack on 268, it did only proceed in 25% yield. Fortunately, this low yield was encountered at the very beginning of the synthesis, which meant that suitable scale-up could allow decent quantities of the desired product to be advanced forward. The next reactions attempted were acidic removal of the O-trityl ether from 269 and Jones oxidation to the acid followed by O-benzylation. Benzyl ester 270 then had its alkene linkage oxidatively degraded. Use of ruthenium tetraoxide and sodium periodate furnished the desired acid alongside a substantial quantity of aldehyde. To gain a complete conversion to the carboxylic acid, a Jones oxidation was performed on this mixture. Diazomethane then procured methyl ester 271. Hydrogenation subsequently unmasked the acid needed for conversion into the thiopyridyl ester which was reacted with the pyrrole anion 272. The pyrrole unit then had its exo-methyl group adjacent to the ring-N atom chlorinated, and this activated chloride was hydrolyzed, and the resulting alcohol converted to the tert-butyl ester by oxidation and esterification. The ketone within 274 was then reduced with NaBH4, the alcohol O-acetylated, and this O-acetate thermally eliminated by heating at 200 °C. The ene-lactone 275 was then subjected to an ammonolysis-lactamization reaction to secure 265 but this process did not go all the way to completion, it co-produced the lactone 276 as the major product. Fortunately, this intermediate could be converted through to 265 in a further two steps by methanolysis and acid-induced elimination of the intermediary lactam hemiaminal.
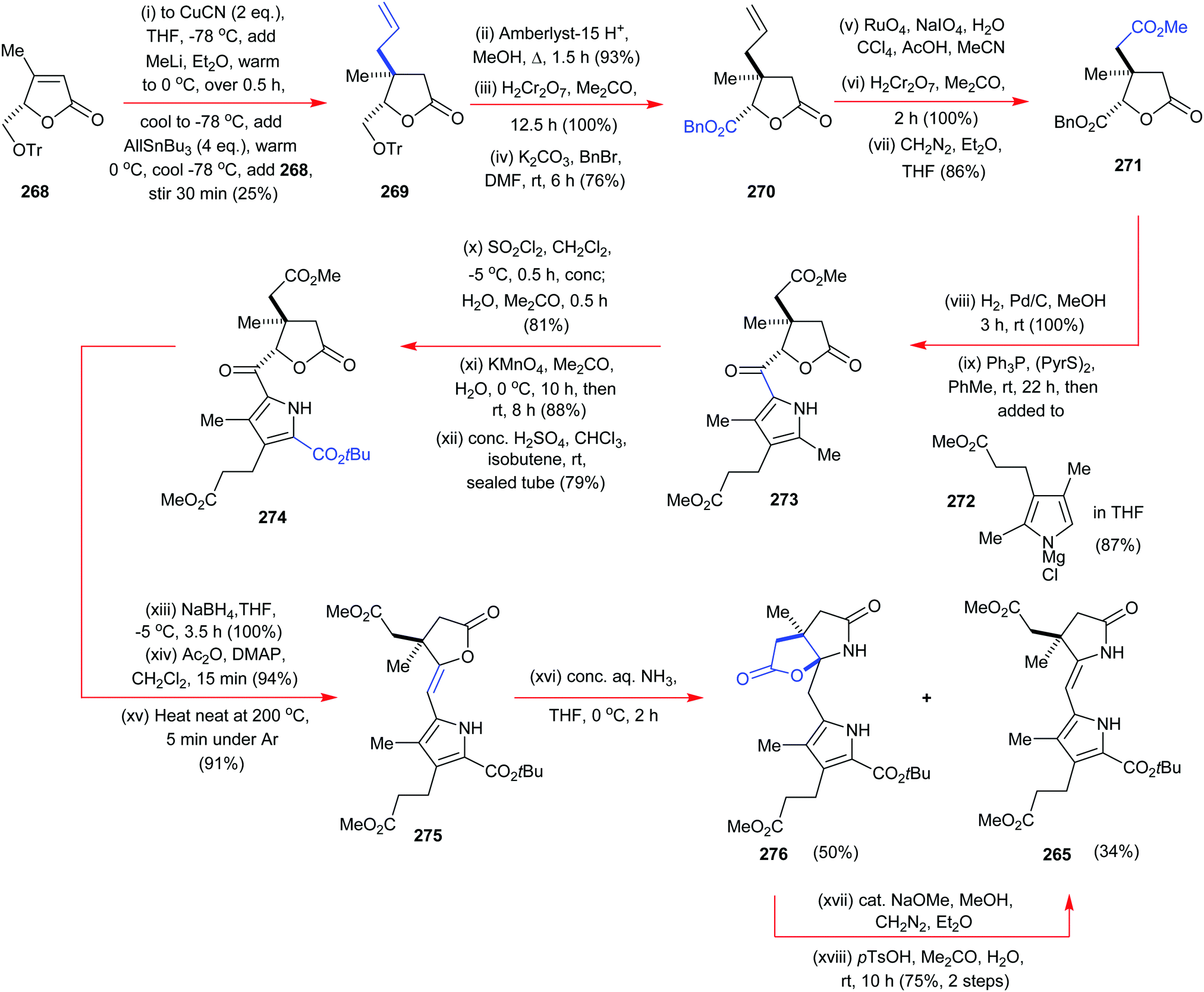 | ||
| Scheme 37 Battersby's second-generation synthesis of the Western hemisphere advanced fragment 265 for the heme d1 methyl ester. | ||
The partner fragment for lactam 244 was the aldehyde 243 and its synthesis is presented in Scheme 38. In this pathway, a Koga-type alkylation on a C(5)-O-etherified butyrolactone was initially allied with an O-deprotection/alcohol oxidation sequence to secure the acid 277. This acid was then O-benzylated prior to having its terminal alkene oxidatively degraded with catalytically generated RuO4, which initially dihydroxylated the alkene prior to effecting 1,2-diol cleavage and aldehyde oxidation. Because the latter oxidation never usually went all of the way through to completion, but instead unexpectedly furnished a mixture of aldehyde and carboxylic acid, a chromic acid oxidation was enlisted to fully convert this mixture entirely to the carboxylic acid, which was esterified with diazomethane before the O-benzyl ester was hydrogenolyzed to provide the desired carboxylic acid 278. The latter was then converted into the thiopyridyl ester under classical Mukaiyama conditions, to allow a subsequent nucleophilic addition of the pyrrolomagnesium anion 272 to proceed followed by elimination, in what was formally an electrophilic aromatic substitution reaction that occurred in 94% yield.
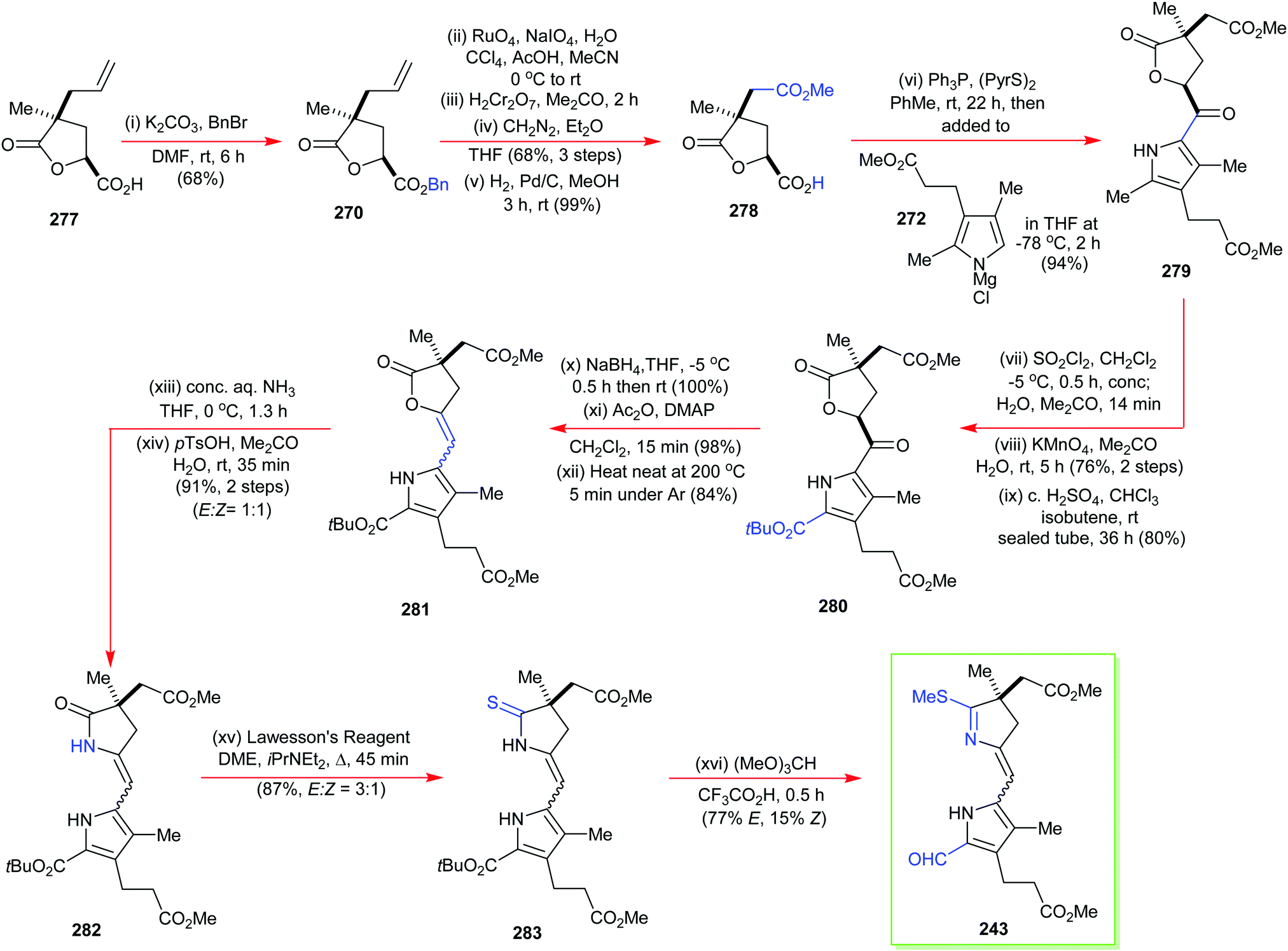 | ||
| Scheme 38 Battersby's pathway the Eastern hemisphere fragment 243 for the heme d1 methyl ester. | ||
A remarkable sequence of reactions now followed. Specifically, a site-selective dichlorination of the C(2)-Me in the pyrrole was achieved by low temperature treatment of 279 with 2 equiv. of sulfuryl chloride. This created a geminal dichloromethyl at that position with high regiocontrol, presumably due to in situ N-chlorosulfation of the pyrrole N-atom, which inductively acidified the neighboring H-atoms of the nearby C(2)-Me to a much greater degree than those within the C(4)-Me, to allow sequential enolization and chlorination to occur twice at the C(2)-Me with high selectivity, and without geminal dichlorination of the keto group. The dichloromethyl grouping was then immediately hydrolyzed to an aldehyde to allow for subsequent aldehyde oxidation to the corresponding acid with aqueous KMnO4 in acetone under neutral conditions. The combined sequence proceeded in a noteworthy 76% yield for the two steps. Isobutene treatment under acidic conditions thereafter furnished the desired t-butyl ester 280 in 80% yield in one of the outstanding sequences of the synthesis.
The Battersby team next addressed the reductive removal of the ketone functionality from 280. This was done by NaBH4 treatment to give a pair of alcohols which were O-acetylated and thermolytically eliminated.
Yet more elegant transformations now followed, including a regioselective ammonolysis of the hindered lactone in 281 without perturbation of the two pendant methyl ester groups. This liberated an amide-hemiaminal mixture that underwent p-TsOH-mediated eliminative dehydration to give 282. A high yielding (87%) thionation of the more nucleophilic lactam carbonyl in 282 next afforded the thionoamide 283 as a 3:1 E:Z mixture, and this level of selectivity was further improved after formylation. The latter was achieved, with concurrent thiomethylation, by sequential treatment with TFA, addition of (MeO)3CH, and subsequent addition of H2O. Although Battersby et al. never gave their thoughts on what was actually happening in this multi-step one-pot sequence, presumably, it commences with the loss of isobutene to give the corresponding pyrrole 2-carboxylic acid. Subsequent reaction of the acid with the trimethylorthoformate then possibly yielded a dimethyl acetal ester which spontaneously decarboxylated with internal alkylation. Alternatively, the pyrrole might itself be staging an attack on the in situ-generated (MeO)(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+–Me ion, with subsequent decarboxylation subsequently restoring pyrrole ring aromaticity. Concomitantly, the thionoamide S-atom must itself be engaging in a nucleophilic attack on the (MeO)(H)CO+–Me ion that is being generated from the (MeO)3CH under the initially anhydrous conditions. This would install the methylthio-imine functionality that is positioned within 243. In turn, the aldehyde group would be liberated from the newly installed acetal by simple aqueous acid hydrolysis which, quite remarkably, leaves the thiomethyl-imine undisturbed.
O+–Me ion, with subsequent decarboxylation subsequently restoring pyrrole ring aromaticity. Concomitantly, the thionoamide S-atom must itself be engaging in a nucleophilic attack on the (MeO)(H)CO+–Me ion that is being generated from the (MeO)3CH under the initially anhydrous conditions. This would install the methylthio-imine functionality that is positioned within 243. In turn, the aldehyde group would be liberated from the newly installed acetal by simple aqueous acid hydrolysis which, quite remarkably, leaves the thiomethyl-imine undisturbed.
Battersby united pyrrole 244 with the aldehyde 243via a trifluoroacetic acid mediated condensation reaction that gave 242 as the initial intermediate. The latter was then irradiated for 90 h with UV light from a high-power tungsten lamp that had been passed through a potassium dichromate solution filter. These conditions induced macrocyclization to give 241 in a quite respectable 53% yield and, significantly, more than 100 mg was successfully prepared by this process.
The time had now come to perform the challenging chemoselective oxidation of 241 to obtain 240a/240b. In the end, selenium dioxide accomplished the desired transformation in 42% yield when 1,4-dioxane was used as the reaction solvent, and the reactants were heated at reflux for 2 h. The next phase of the synthesis was dihydroxylation of rings C and D in 240a/240b with osmium tetraoxide, which furnished 239 and 284 as a mixture but in very low overall conversion. Fortunately, the starting diketone 240a/240b could be readily recovered and recycled. Finally, in order to obtain the heme d1 methyl ester, the unseparated mixture of 239 and 284 was heated with conc. hydrochloric acid in benzene at reflux for 3.5 h. The desired target 237 was isolated in 13% yield based upon unrecovered 240a/240b and it was formed alongside 4% of 285 calculated similarly (Scheme 39).
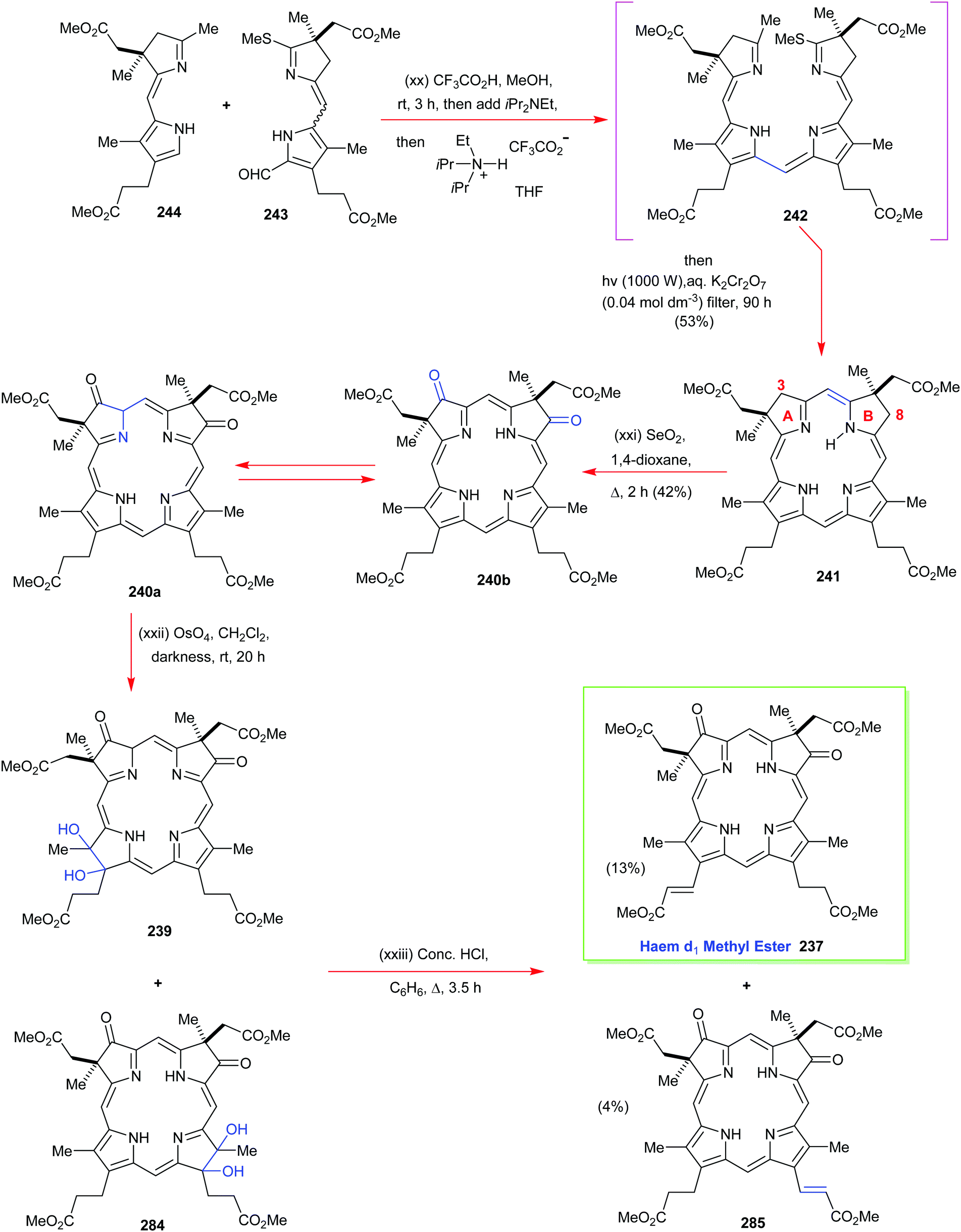 | ||
| Scheme 39 Battersby's completion of the total synthesis of the heme d1 methyl ester 237 from 243 and 244. | ||
Although this synthesis did ultimately allow the absolute stereochemistry of heme d1 to be firmly established, and it also showed off Battersby's photomacrocyclization chemistry for building up complex porphyrin ring-systems, it did falter in its very final stages, with its very low yielding dihydroxylation step which struggled to match the task at hand. Despite this, the dihydroxylation method and related reactions continue to remain a widely used protocol for the transformation of porphyrins into chlorins and has given rise to a whole range of novel porphyrins based on Brückner's ‘breaking and mending’ strategy.114 Notwithstanding the dihydroxylation problems, the Battersby synthesis did still have many highlights, including the remarkable diketone installation step to obtain 240a/b, which used selenium dioxide as the oxidant. The synthesis thus confirmed that it was indeed possible to install diketone functionality of this sort into chlorins by imine-type allylic oxidation, and undoubtedly this outcome spurred Kishi, Minehan and Wang's subsequent synthetic efforts on tolyporphin A O,O-diacetate. Although the latter synthesis did ultimately install the diketo functionality of this molecule with a different reagent system, it did still perform an oxidation of this type. Kishi's synthesis of the corrected structure (+)-tolyporphin A diacetate will now be discussed.
6.3 Total synthesis and structure confirmation of (+)-tolyporphin A diacetate (Kishi)
In the early 1990s, Moore and coworkers isolated a structurally unprecedented dimeric C-glycoside porphyrin, which they named (+)-tolyporphin A, from the lipophilic extract of the cyanophyte microalga Tolypothrix nodosa.115 Apart from its unusual structure (286, Fig. 8), the one property that made this compound of special interest was its ability to reverse multidrug resistance in a vinblastine-resistant human ovarian carcinoma cell line. Together, these combined facets made the originally proposed structure for (+)-tolyporphin A diacetate a highly prized target. Unfortunately, however, the initially assigned structure for (+)-tolyporphin A was later proven to be incorrect by Minehan and Kishi116 when they synthesized the proposed compound, only to find that its O,O-diacetate derivative did not match up with that derived from the natural product!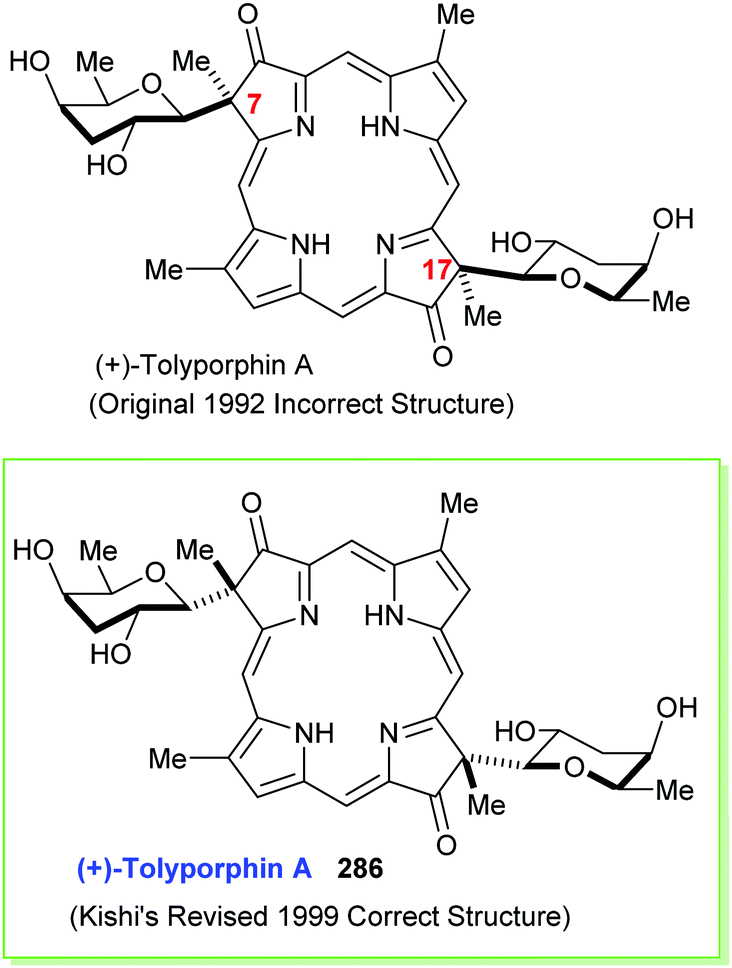 | ||
| Fig. 8 Structure of tolyporphin A. | ||
These events, and further structural investigations by Kishi and Moore,117 subsequently led to the proposal of a new structure for tolyporphin A in which the quaternary stereocenters at C(7) and C(17) were now inverted. However, to confirm this structural assignment, yet another chemical synthesis was required. Wang and Kishi118 duly took up this synthetic baton in the late 1990s, successfully completing the task in mid-1999, to thereby confirm the new stereochemical assignment. Herein, we now discuss this synthesis in detail, since it is the epitome of elegance and excellent design as far as modern-day porphyrin total synthesis is concerned.
One of the major issues that must be contended with in any synthetic plan for the tolyporphin A O,O-diacetate is when to actually create its two β-linked C-glycosidic bonds. Although, in principle, one can retrosynthetically disconnect both sugar motifs immediately, and perform a late-stage enolate C-glycosylation to forge the link between the sugar and the porphyrin ring, the prospects for simultaneously controlling the quaternary carbon stereocenter and the β-glycosidic bond stereochemistry, whilst concurrently preserving all of the other delicate functionality present within the dioxoporphyrin ring, all appear to be far too risky to leave to the final stages of any synthesis. Kishi thus elected to attach the two sugar units quite early on, in his forward-advancing route, initially securing the stereochemistry of the newly induced stereocenters, and then building up the delicate porphyrin ring system in distinct stages.
Specifically, his plan was to use an Eschenmoser thiolactam S-alkylation/episulfide ring-contraction119 (in modified form) to fashion the fragments 294, 295 and 296 (Scheme 40). An eneamide O-methylation reaction would then establish 293, ready for prosecution of a Lewis acid-assisted internal nucleophilic addition to create the carbocation 292 which would then be expected to fragment into 291. A base-induced retro-aldol type cleavage would thereafter convert 290 into 289 and this would be oxidatively demetallated. The sugar protecting groups would then be exchanged, and a Battersby-style heme d1 methyl ester oxidation performed on the C(8)- and C(18)-methylenes to install the final two keto-groups present in the target.
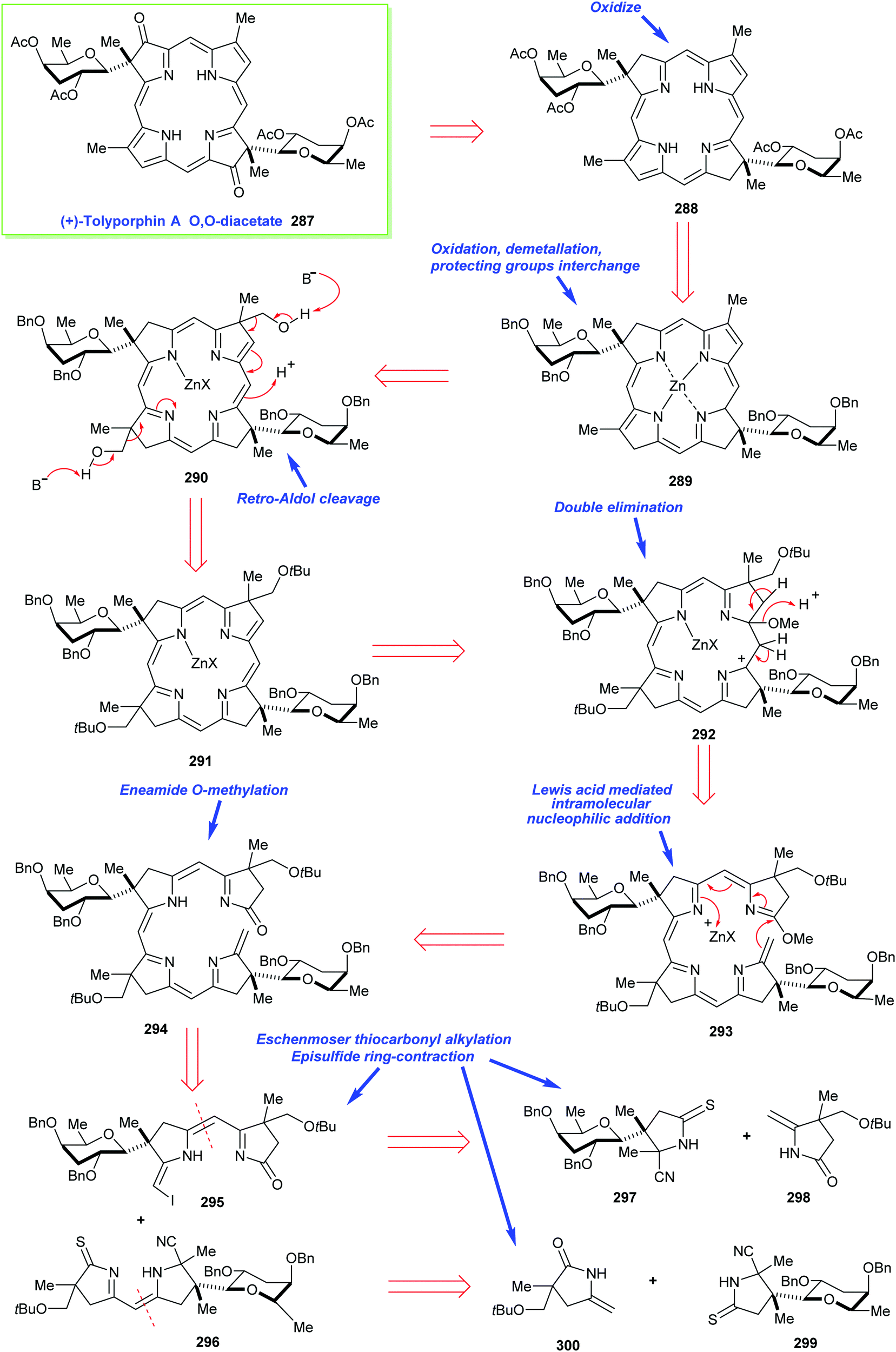 | ||
| Scheme 40 Kishi's retrosynthetic analysis of his corrected structure for (+)-tolyporphin A O,O-diacetate 287. | ||
With this overall planning in mind, Kishi and coworkers embarked on their synthesis of the C-glycosylated vinyl iodide 295 from protected 3,6-dideoxy-D-galactoside 301; their route is shown in Scheme 41. It commenced with the selective acetolysis of 301 to obtain glycosyl acetate 302. A trimethylsilyl triflate-mediated addition of the silyl ketene acetal 303 to the glycosyl cation derived from 302 next yielded a 3:1 mixture of β:α epimers with 10:1 selectivity in favor of the desired epimer 304 at C(7). Following O-desilylation of this product mixture with TBAF, the desired product was chromatographically separated in 60% overall yield from 301. After methyllithium addition to the lactone, compound 305 was isolated which, after in situ lactol ring-opening to the 1,2-diol and oxidative cleavage with Pb(OAc)4, produced an aldehydo–ketone which underwent Pinnick oxidation to the acid 306 in good yield. Mixed anhydride formation and ammonolysis subsequently afforded an amide, which smoothly dehydrated when heated with mild base at 180 °C under Dean–Stark conditions to give 307. This imino-lactam was then condensed with cyanide and the product thionated with Lawesson's reagent to secure the desired thioamide-nitrile 297, and significantly the nitrile group survived this thionation procedure.
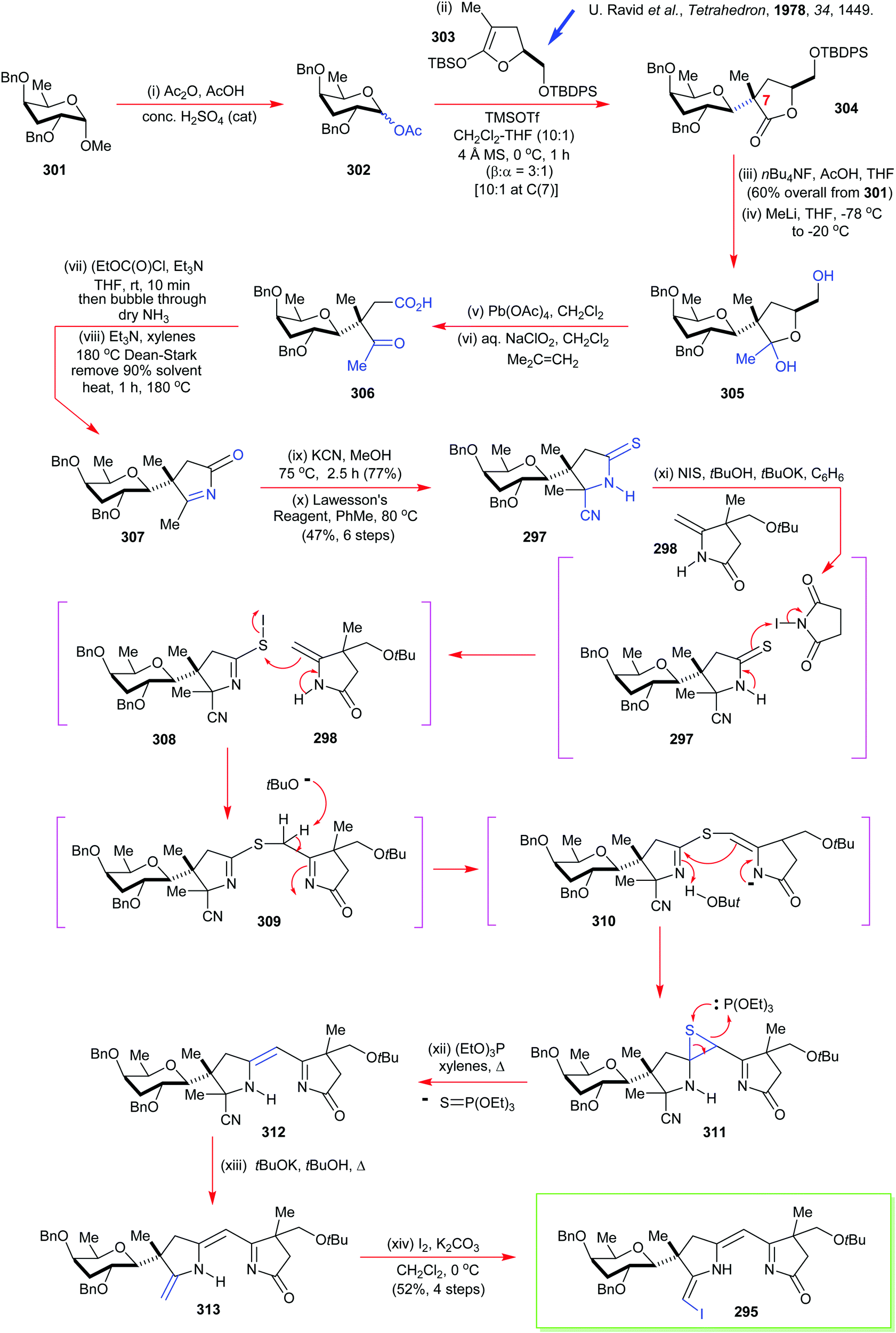 | ||
| Scheme 41 Kishi's route to vinyl iodide 295. | ||
A novel variant of the Eschenmoser episulfide ring contraction119 was now used to join 297 to 298 to obtain 311 and thence 312. Mechanistically, this highly complex reaction sequence possibly involves the thioamide 297 initially attacking the N-iodosuccinimide to obtain 308 and its iodo substituent then being displaced from the sulfur by 298. Although this was the key fragment-unifying step, it will be immediately noted that in 309 only a thioether linkage connects the original two reaction partners together. For C–C bond union to occur, a base had to be added to bring about the deprotonation of 309 to create the enamino anion 310. Once this happened, an excellent nucleophile was now sited in close proximity to a very good electrophile. Accordingly, the formation of episulfide 311 could now occur and this intermediate, once heated with triethyl phosphite subsequently underwent the key Eschenmoser C–C-bond forming ring-contraction with extrusion of (EtO)3PS. The end result of this rather enchanting sequence was, of course, 312 which, after elimination, gave the enamine intermediate 313 which could be iodinatively eliminated to procure 291 in a most impressive 52% overall yield from 297.
The next phase of the synthesis was the construction of 321 by a 2-step Eschenmoser union between 297 and 300 (Scheme 42) followed by a thionation on 314. A second variant of the Eschenmoser process was now devised for building up the open chain tetrapyrrole 319 from 295 and 296. Mechanistically, the sequence can be envisaged as successively proceeding via the intermediates 315, 316, and 317 with the Ni-complexed episulfide 317a undergoing the key Eschenmoser desulfurative olefination to give 318, en route to 319. The latter intermediate was then demetallated and eliminatively decyanated to provide 320. Ionic macrocyclization was next accomplished efficiently by O-methylation of the enamide 320 and Lewis acid-assisted nucleophilic addition and elimination mediated by ZnCl2 in C6H6/MeOH which gave 321 in 48% overall yield over 6 steps.
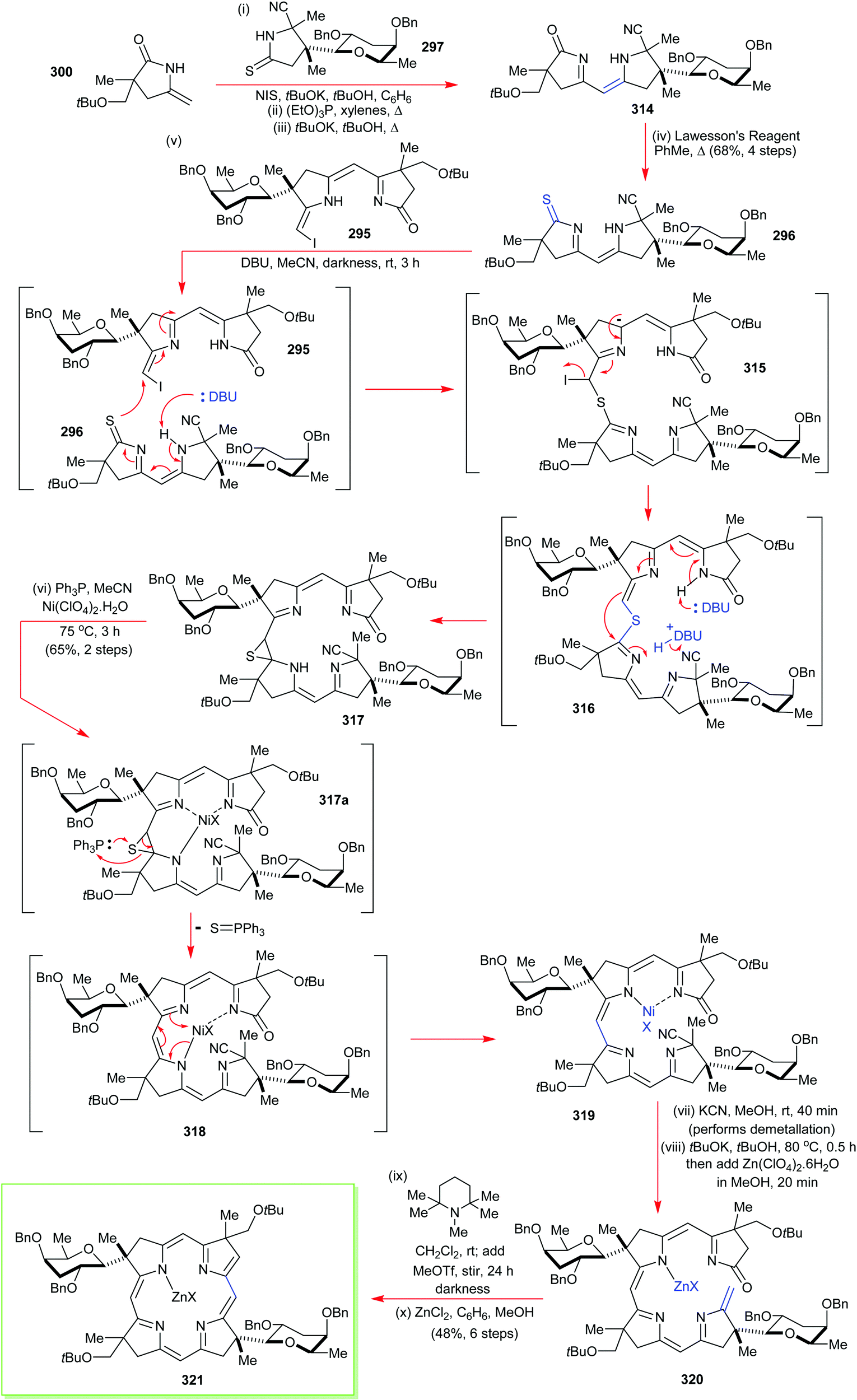 | ||
| Scheme 42 Kishi's pathway to 321. | ||
The final stages of Kishi's synthesis (Scheme 43) involved expulsion of the –CH2OtBu units from 321 by trifluoroacetic acid mediated O-dealkylation and base-induced retro-aldol cleavage with KOtBu. Together these two steps secured the zinc bacteriochlorin 322 whose benzyl ethers were detached, followed by O-acetylation of tetraol 323. Considerable effort went into accomplishing this step before it was eventually discovered that chromium trioxide and 3,5-dimethylpyrazole could accomplish this goal very nicely. Although the 30% yield attained in this step might not seem that high, when one considers the extreme chemical sensitivity of this system, such a yield is not so disappointing. Importantly, however, it did allow the structure of the natural product to be determined at the two quaternary centres and it confirmed that their stereochemistry was opposite to that originally assigned by Moore. In the modern era, this total synthesis of 287 probably represents the state of the art; certainly in terms of a complex chlorin natural product target that has been successfully synthesized to high standards.
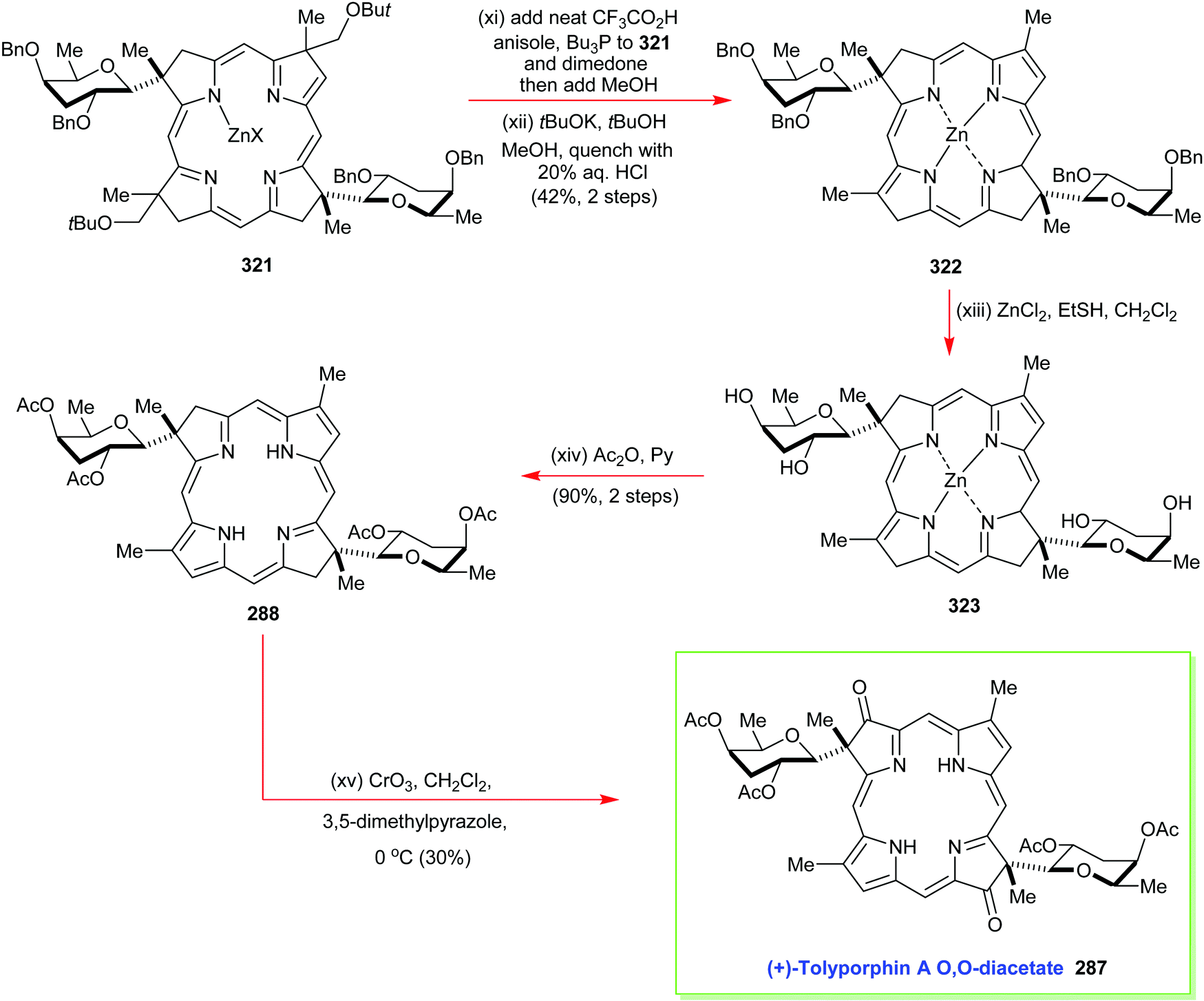 | ||
| Scheme 43 Kishi's end game for (+)-tolyporphin A O,O-diacetate. | ||
6.4 Synthetic strategies to novel chlorin structures (Jacobi)
Jacobi and coworkers have formulated a number of very effective non-traditional synthetic approaches to various dipyrrole structures of value for non-symmetric chlorin synthesis. One of these routes120 to fragments of general structure 332, where the A-, B-, and C-substituents are either alkyl or aryl, is exemplified in Scheme 44. In this, a functionalized pyrrole iodide 324 is reacted with a Pd(0)-catalyst to give a pyrrole-Pd(II) species that can then go on to participate in a heteroatom-directed carbopalladation reaction with the acetylenic acid 325 in which an acyloxypalladium(II) intermediate 327 is believed to be generated that undergoes reductive elimination to give the lactone 328. Jacobi has processed such structures in a number of ways, one of which involves reacting the lactone with liquid NH3 in THF at −33 °C and inducing lactamization of the product amido-ketone with commercially available montmorillonite K-10 clay. The product lactams 329 are then thionated, S-alkylated with iodomethane and subjected to Negishi cross-coupling to secure molecules of general structure 332.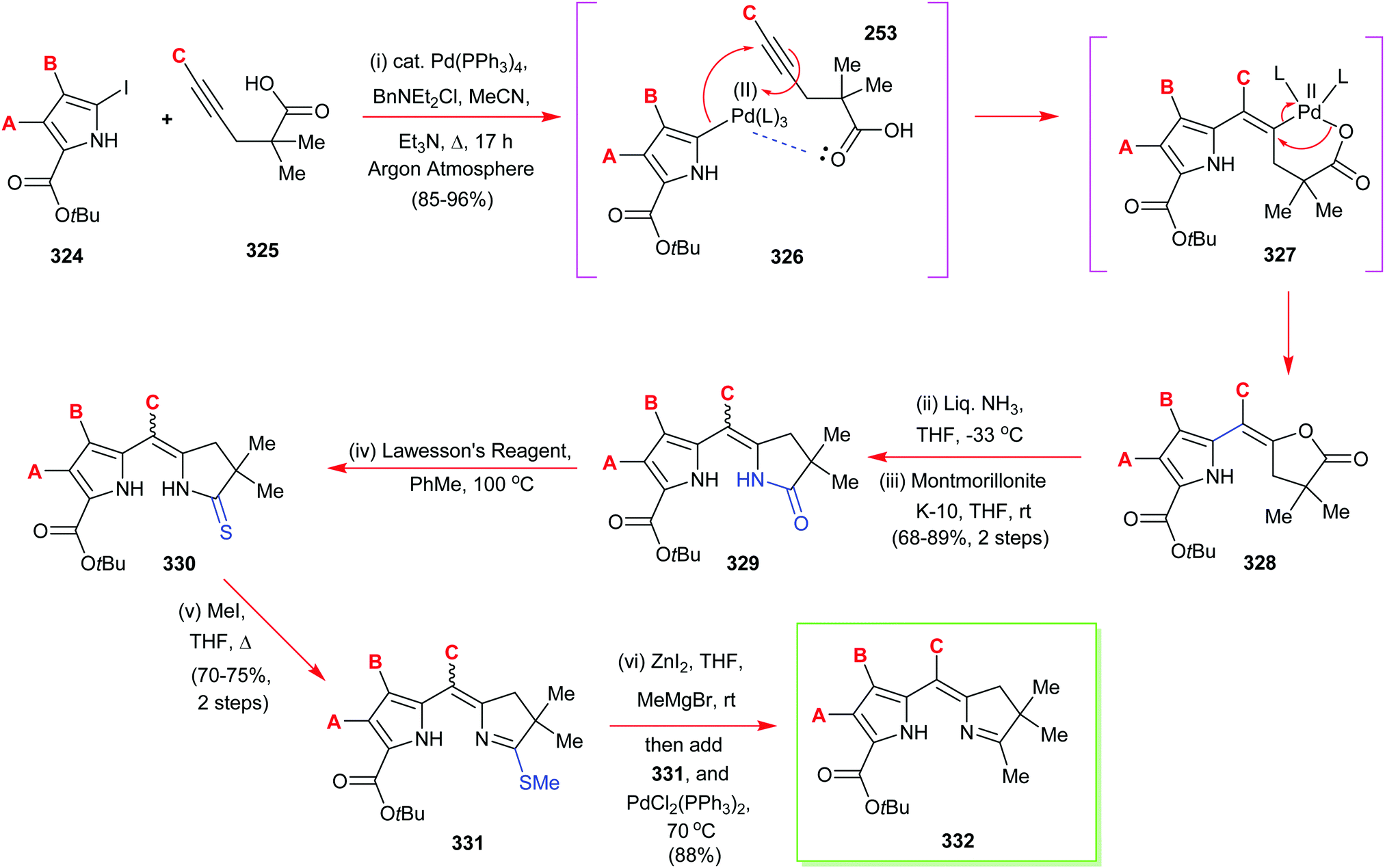 | ||
| Scheme 44 Jacobi's general route to multifunctionalized dipyrrole fragments of general structure 332. | ||
Besides this pathway to molecules of the 332 class, Jacobi has proposed several other routes to compounds of this type, one of which also proceeds from the lactone 328 and involves a Tebbe-type olefination with a titanium alkylidene (Scheme 45).120 The intermediary enol ethers 333 are then hydrolyzed with aqueous acid to the respective diketone intermediates 334 which are then subjected to amination and cyclization with Montmorillonite K-10 clay.
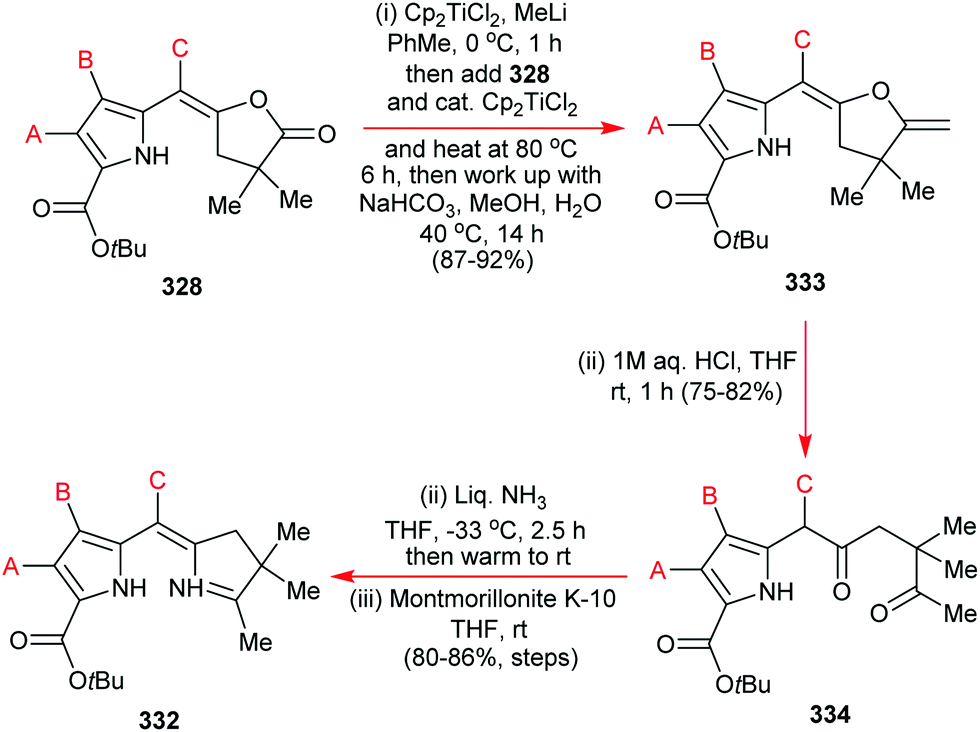 | ||
| Scheme 45 Jacobi's second general route to multifunctionalized dipyrrole fragments of structure 332. | ||
To obtain reduced dipyrroles such as 337, which lack a meso-substituent, Jacobi has recommended the conditions shown in Scheme 46 for performing the amination step.120 Apparently diketone intermediates such as 336 are highly susceptible to undergoing internal aldol cyclization when attempts are made to ring-close them with the majority of ammonia sources. However, when ammonium acetate is used for amination in DMF at 0 °C, this complication can be circumvented, with even better results typically being obtained when ammonium propionate (NH4+EtCO2−) is substituted for NH4OAc in the amination step, the yields typically being raised by a further 20%.
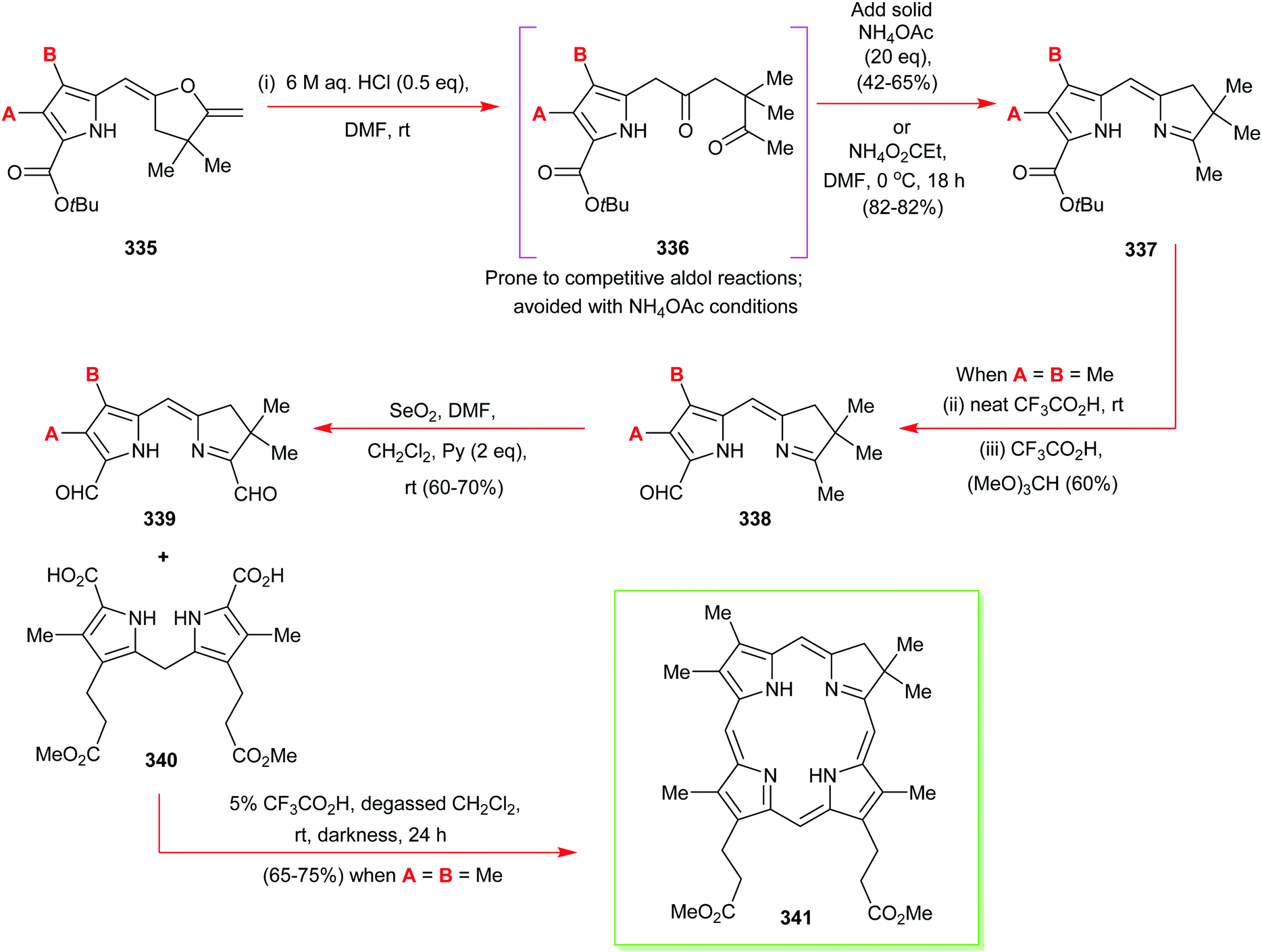 | ||
| Scheme 46 Jacobi's route to dipyrrole fragments of general structure 341 lacking a meso-substituent and an example of their use for non-symmetrical chlorin formation. | ||
With these various partially reduced dipyrroles in hand, Jacobi set about examining how they could be used for the formation of unsymmetrical chlorins.121 In the case of 337 (Scheme 46), such structures were carried forward to 338 by decarboxylative formylation (see Scheme 38 for a related transformation), and the pyrromethane unit was oxidized with selenium dioxide. Dialdehyde 339 (where A = B = Me) was then doubly decarboxylatively condensed with diacid 340 to obtain 341 in very respectable yield (65–75%). The Jacobi methodology has thus taken the synthesis of unsymmetrical chlorins such as 342 to new heights of efficiency and sophistication, using conditions analogous to other modern-day porphyrin syntheses for the key condensation and cyclization steps. In yet another powerful development from Jacobi's laboratory, mixed dipyrroles such as 343 have also been used for the highly regiocontrolled assembly of meso-substituted chlorins such as 344 (Scheme 47). In this approach, the acid component of the 343 structure participates in an acid-promoted site-selective decarboxylative aldol condensation with the highly electrophilic α-imino-aldehyde unit of 342, which is preferentially activated by protonation of the imine. Following this union, elimination of water subsequently ensues to give an uncyclized tetrapyrrole in which there is now a pyrrole t-butyl ester internally tethered to a pyrrole aldehyde. Increasing the concentration of trifluoroacetic acid to 25%, and maintaining darkness and an oxygen-free atmosphere, then induces a cleavage of the tert-butyl ester to give the terminal pyrrole carboxylic acid, which decarboxylates, prior to undergoing an internal aldol condensation with accompanying elimination of H2O to produce 344 in 87% yield. Significantly, all the pyrrole ring systems in chlorin 344 are in different environments and are non-equivalent. Clearly this extremely valuable work from Jacobi's lab has greatly improved synthetic access to chlorin structures and represents a major landmark in this important area.
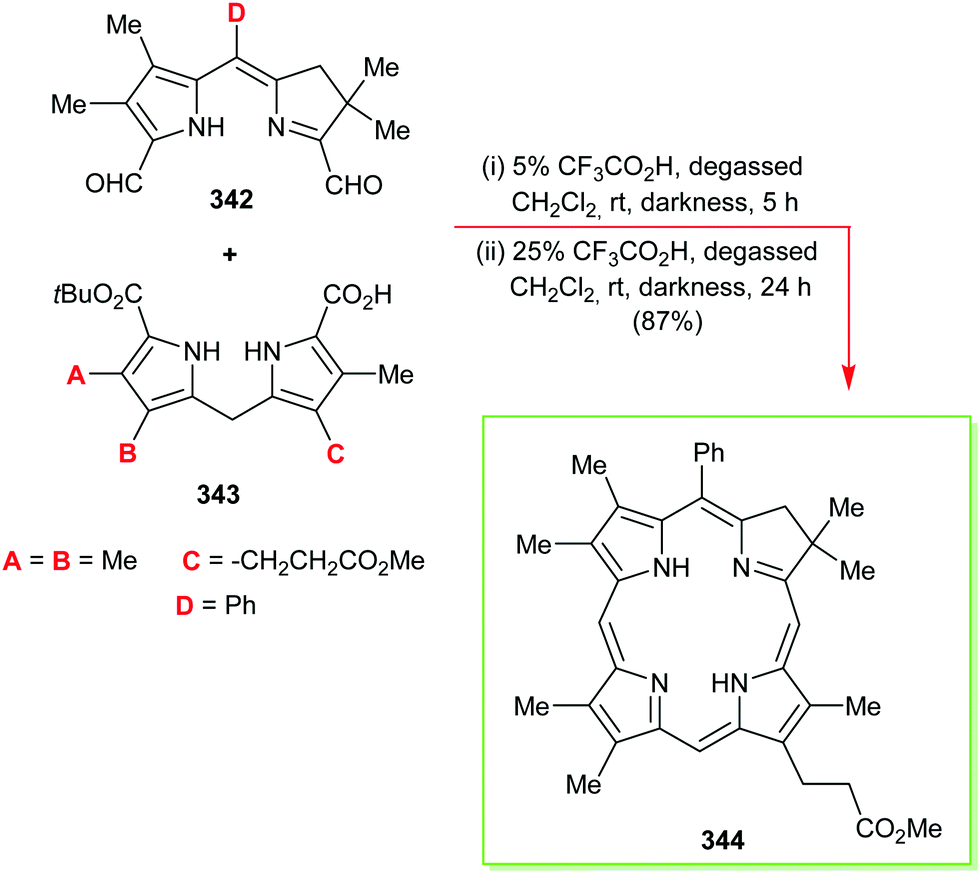 | ||
| Scheme 47 Jacobi's use of dipyrrole acids of general structure 344 to create unsymmetrical chlorins with a positionally guaranteed meso-substituent. | ||
Notably, the characteristic geminal dimethyl system found in reduced chlorin ring systems of this sort has been widely adopted as a stabilizing building block in chlorin ring construction for quite some time (since it prevents re-oxidation to the porphyrin).122 In recent times, this feature has mostly been applied in the construction of meso-substituted systems and the so-called sparsely substituted chlorins by Lindsey's group.123,124Scheme 48 illustrates the three main approaches taken by Montforts, Jacobi and Lindsey to gem-dimethyl chlorins. Given these earlier precedents and latest methodologies, we expect to see additional synthetic work in this direction over the coming years simply because of the elegant simplicity of these methods. This is aptly shown in two major reviews by Lindsey, which illustrate the contemporary synthetic chemistry of such chlorins in outstanding detail.124,125
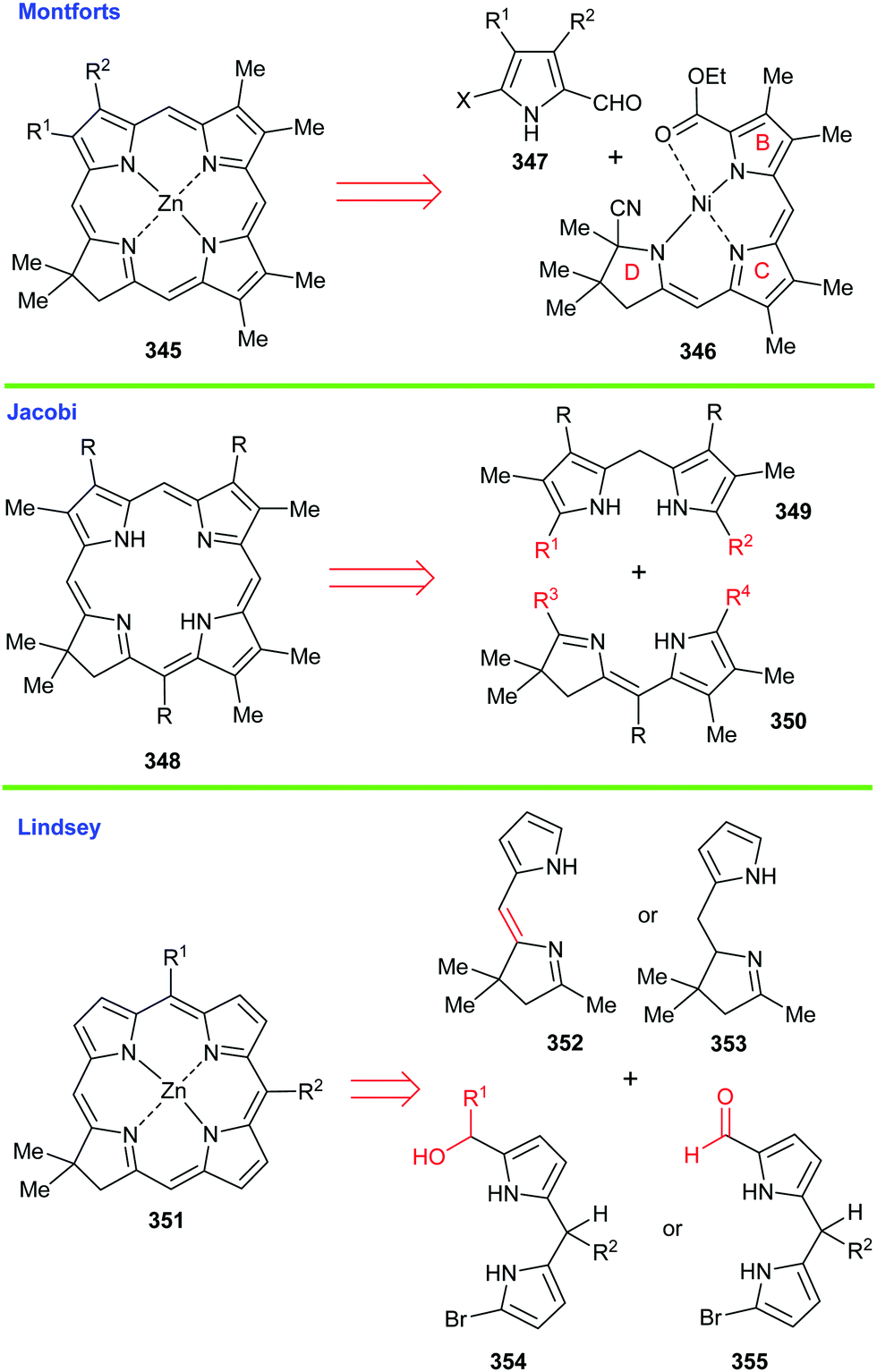 | ||
| Scheme 48 Synthetic strategies for gem-dimethylchlorins by Montforts, Jacobi, and Lindsey. | ||
7. Syntheses that rely upon functionalization reactions and functional group interconversion
Despite the many advances that have been made since Willstätter's early investigations,2 almost all practical porphyrin syntheses, and most especially those aimed at delivering material for biological studies or other applications, are partial syntheses as opposed to total syntheses. Chlorophyll derivatives are routinely prepared from phytochlorin derivatives derived from plant-extracted chlorophyll72 and the same is true for heme derivatives, where haemin and deuteroporphyrin are still typical starting materials.126 As already outlined, thousands of porphyrins have been prepared starting from 2,3,7,8,12,13,17,18-octaethylporphyrin or 5,10,15,20-tetraarylporphyrins. Thus, most porphyrins are, these days, prepared through functional group interconversions (FGI) from other more readily available porphyrins. The synthetic organic chemistry literature on these reactions easily exceeds 10000 papers since WW II.1,64,104 Clearly, such a high volume of work cannot possibly be covered here, and so the following section will thus only give a few indicative examples of such approaches.
In simple terms, the β-positions can undergo [2+1], [2+3] and [2+4] cycloadditions127 (see Scheme 49 for an example of a Diels–Alder reaction of the porphyrin-‘ene’ 358 with the diene 357 to give 359),127a and SR, AE, AR, and AN reactions, some of which have been used to convert porphyrins into the respective chlorins. The latter is illustrated in Scheme 50 with the classic epoxidation reaction of 2,3,7,8,12,13,17,18-octaethylporphyrin 102 yielding first 360, then the dihydroxychlorin 361 and finally the oxochlorin 362 after an in situ pinacol-pinacolone rearrangement.8,114,128
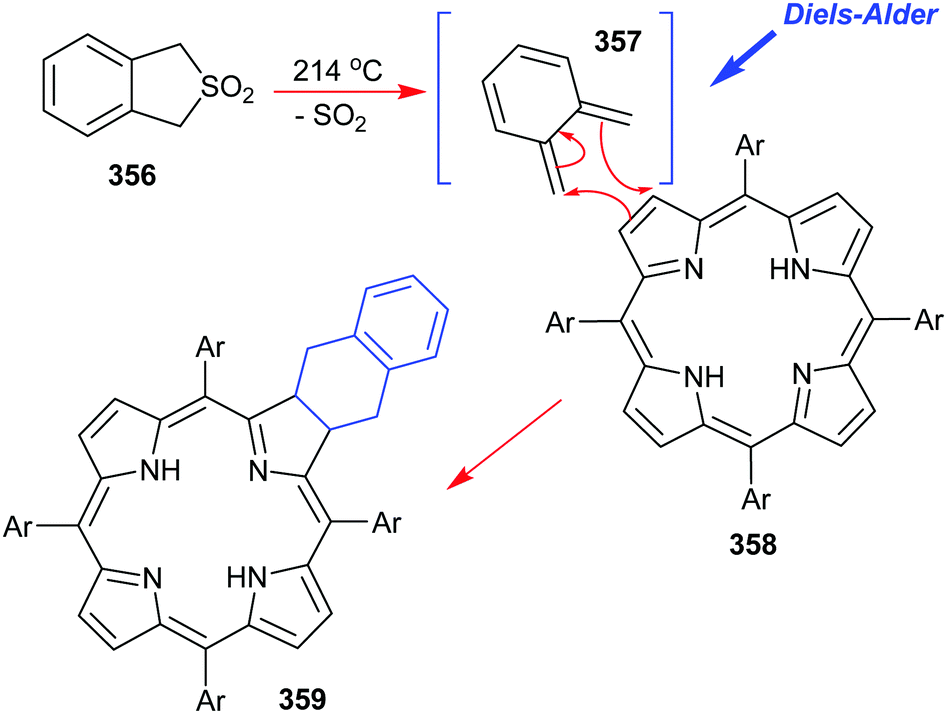 | ||
| Scheme 49 Exemplary cycloaddition reaction involving porphyrins. Cavaleiro and coworkers’ use of meso-tetraarylporphyrins 358 as dienophile in a Diels–Alder reaction with ortho-benzoquinodimethane 357 to give chlorin 359 (plus other products). | ||
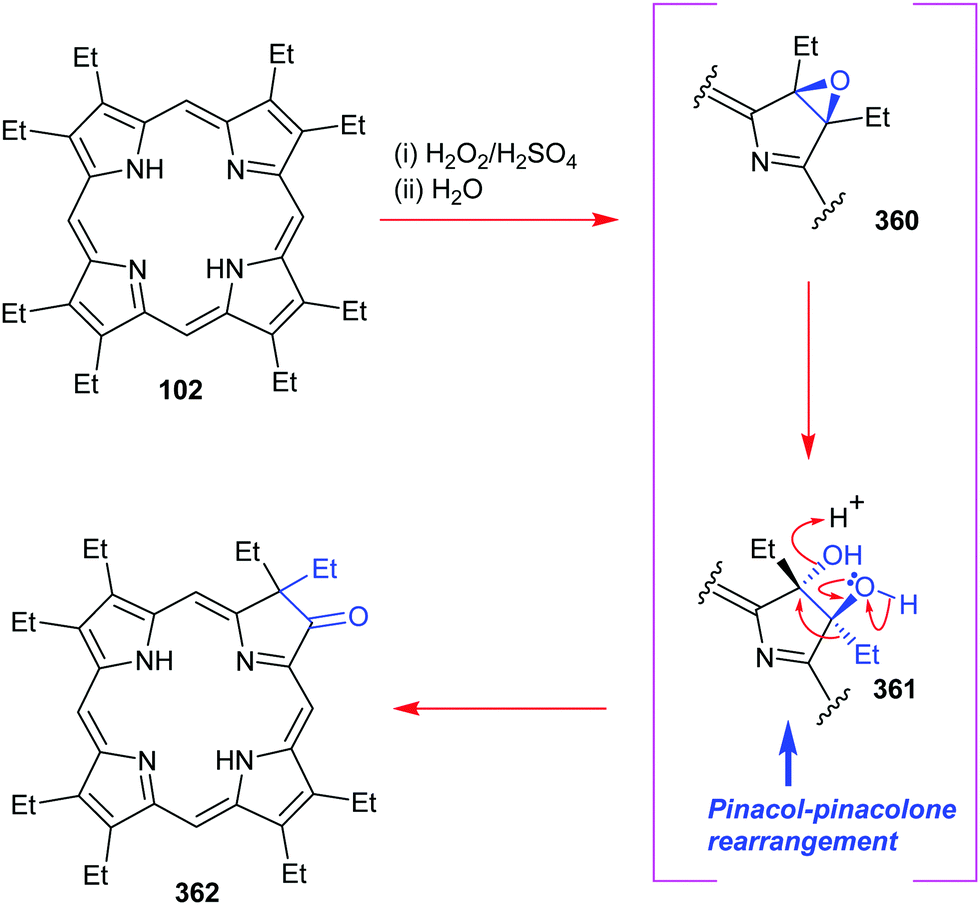 | ||
| Scheme 50 Epoxidation and rearrangement of 2,3,7,8,12,13,17,18-octaethylporphyrin 102 to yield oxochlorin 362. The reaction also yields regioisomeric dioxo(iso)bacteriochlorins. | ||
For the meso-positions a range of nitration, halogenation and formylation reactions have been developed. Most suitable for synthetic purposes are Pd-catalyzed coupling reactions, most notably the Suzuki and Sonogashira variants which have opened new vistas in porphyrin chemistry.64,129,130 When allied with even newer technologies such as ring-closing metathesis, such chemistry can take on even greater significance, since it opens up the possibility of controlled formation of many unusual ring-annulated porphyrins.130
One example of the application of such combined technologies is provided by Smith's very elegant pathway to the dibenzoporphyrin 367 (Scheme 51)131 which nicely confederates the Crossley bromination132 with a Pd(0)-catalyzed Suzuki cross-coupling and a Grubbs RCM reaction to give, after DDQ oxidation, a dibenzoporphyrin that is meso-tetrasubstituted. Clearly, other possibilities for forming ring-fused porphyrin polycycles exist due to the creative adaption and licensing of these protocols. Obviously, it is impossible here to give even a selective overview of what could be done in this regard. Nevertheless, it will be appreciated that many synthetic possibilities exist.
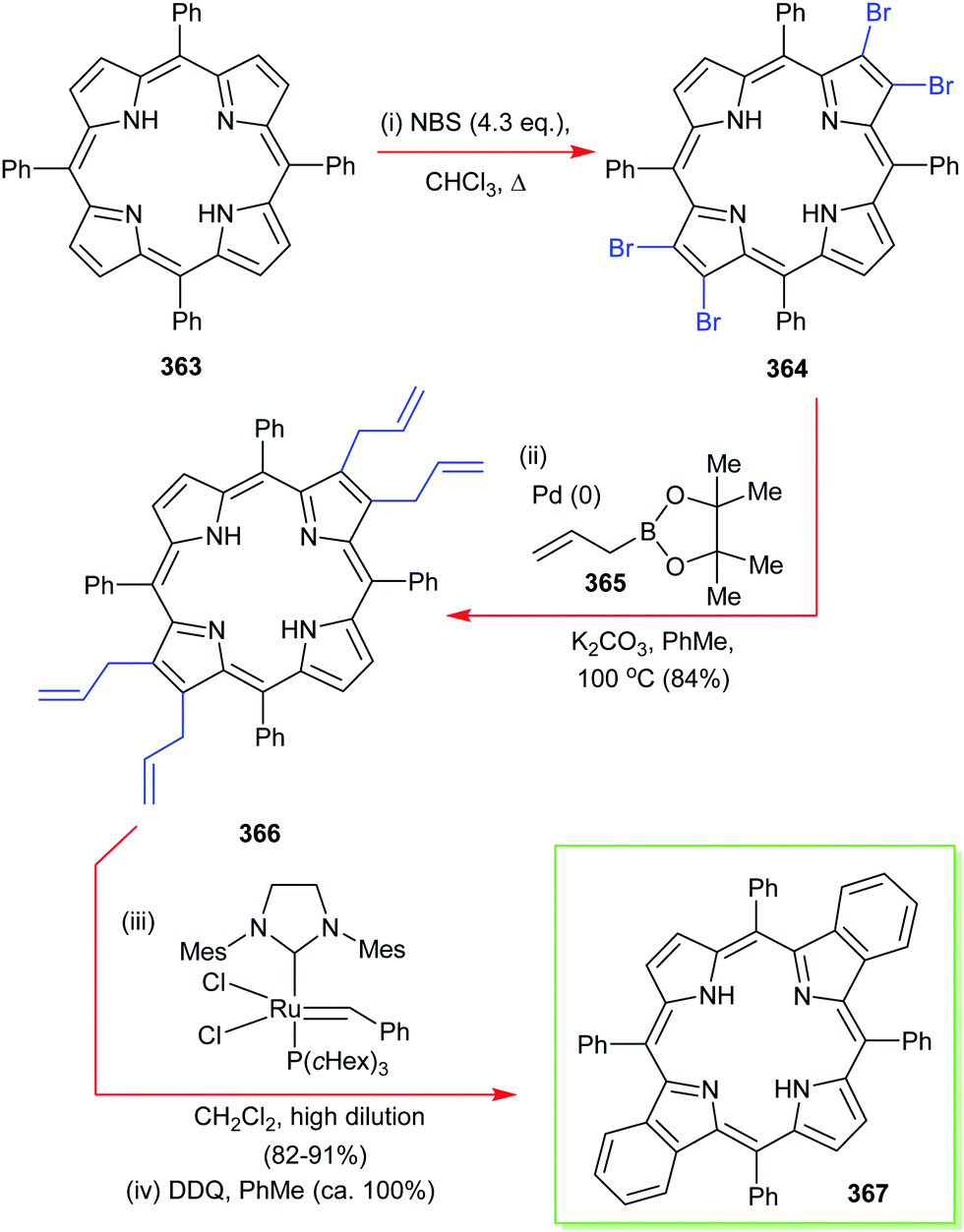 | ||
| Scheme 51 Smith's combined use of the Crossley bromination, Suzuki coupling, RCM and oxidation to secure dibenzoporphyrins. | ||
From a purely personal view, three areas stand out. First, there is the broad range of meso and β-functionalization reactions based on Heck, Suzuki, and Sonogashira couplings and newer developments such as Buchwald–Hartwig reactions.130 Together with older methods, such as the Vilsmeier formylation and C-nitration for example, this, in principle, allows the generation of any desired meso-substituted porphyrin provided a sequential application of the individual reactions is possible. If applied to unsubstituted porphyrin 5 in a rational manner and sequence, another entry into the ABCD porphyrins 213 would be achievable.
Many of these reactions require haloporphyrins as precursors and it is difficult to introduce only one bromine atom or a second in a regioselective manner into the porphyrin system. However, the situation becomes easier if one or more of the required meso-substituents are already present. Thus, most applications of the Pd-catalyzed reactions use A2205/206 or A3208 porphyrins as starting materials. Scheme 52 gives classic examples of the emerging use of transition metal-catalysis in porphyrin functionalization in the early 1990s.133 The heights to which such strategies can be taken are illustrated by the combination of transition metal catalysis with supramolecular chemistry in Anderson's Vernier-templated synthesis of a porphyrin 12mer 375 from the tetramer 374.134
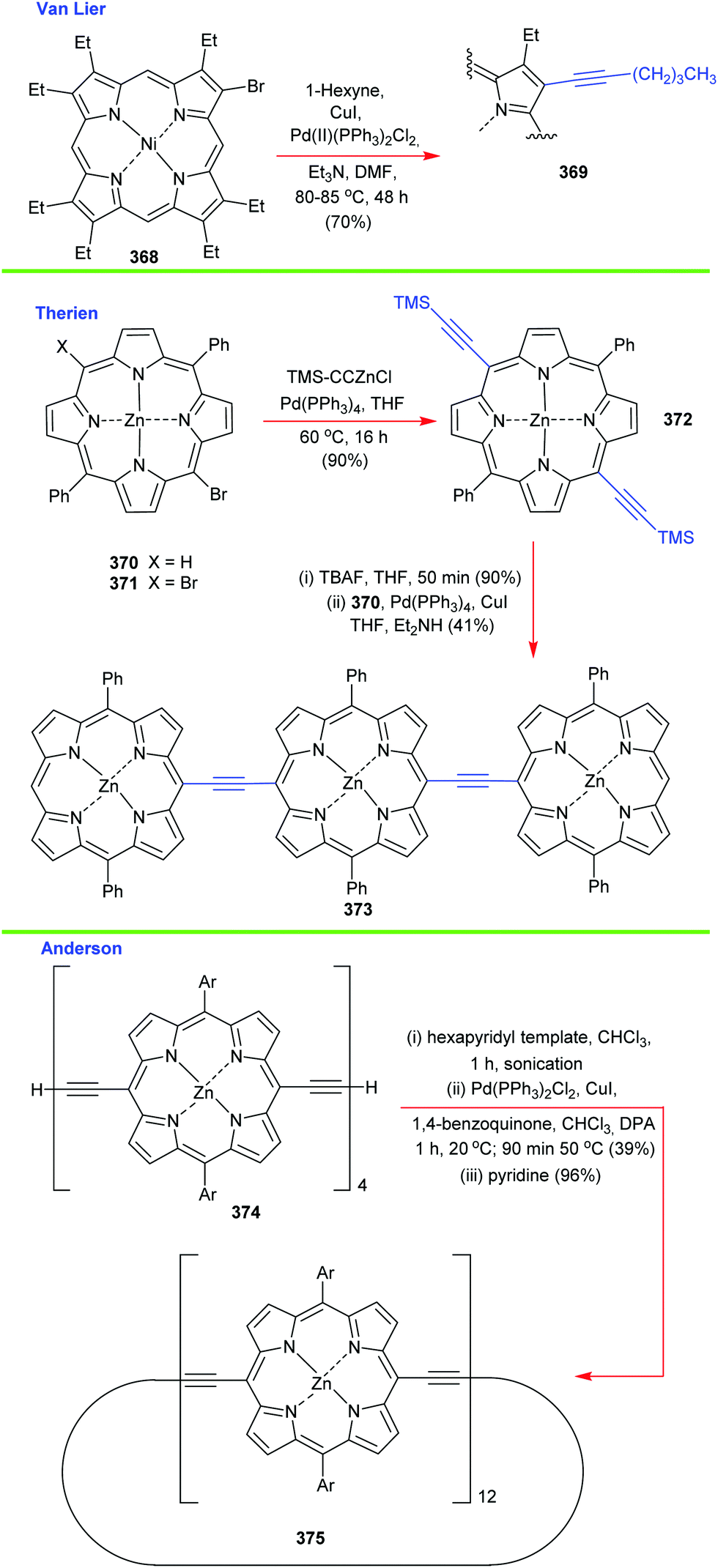 | ||
| Scheme 52 Examples of alkynyl-porphyrins pioneered by van Lier, Therien and Anderson. | ||
Secondly, these methods are complemented by the direct nucleophilic substitution of meso-positions with organolithium reagents developed by Kalisch and Senge (Scheme 53).135 For metalloporphyrins 376, the key intermediate is the Meisenheimer-complex 379 initially formed by attack of the nucleophile.136a Depending on the reaction conditions and work-up, this method gives access to a broad range of products.136,137 Reaction at low temperature and quenching with water and oxidation gives the meso-substituted porphyrin 381135,136b (or with excess RLi, the 5,10-disubstituted porphyrin 385)99 while addition of an organic electrophile prior to hydrolysis gives the unsymmetrically disubstituted porphyrin 382.136c The same reaction using higher temperatures during the nucleophilic attack yields 5,15-porphodimethenes (calixphyrins) 380 which are resistant to common oxidants.136d Omitting the hydrolysis step and immediate addition of an oxidant to a solution of 379 yields radicals which dimerize to the meso–meso-linked bisporphyrin 383.136e Using bulky organolithium reagents or addition of Pd-catalysts shifts the reaction to the sterically less hindered β-positions and yields chlorins 377,136f,137 and phlorins,137 or 5,10-porphodimethenes 378.136g
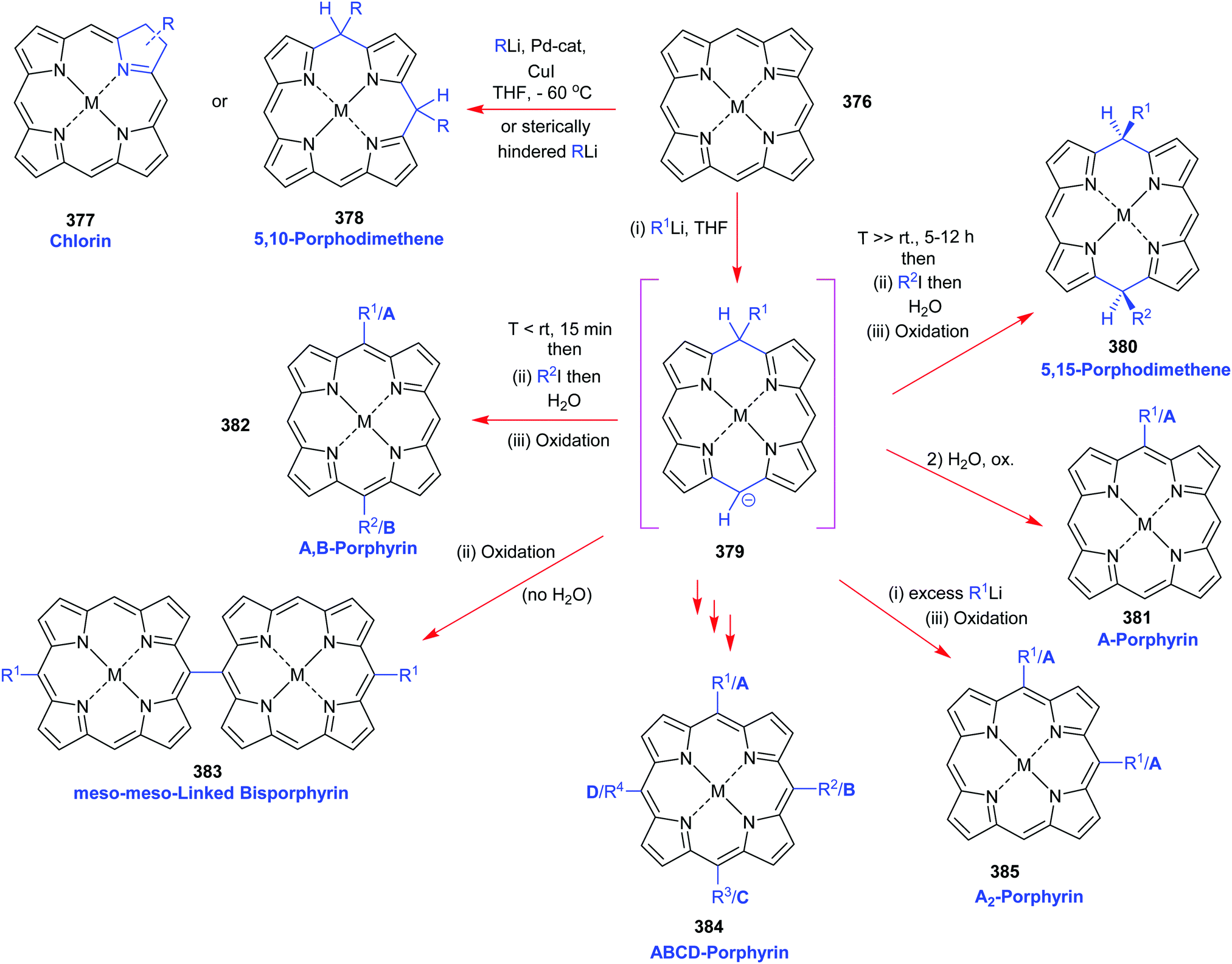 | ||
| Scheme 53 General scheme of the reaction of metalloporphyrins with organolithium reagents. | ||
These SNAr reactions can be carried out sequentially four times at all four meso-positions (387 to 391) (Scheme 54) and, in principle, can allow the generation of ABCD porphyrins.135,138 They also allow the synthesis of 2,3,5,7,8,10,12,13,15,17,18,20-dodecaalkylporphyrins such as 391,138c which are inaccessible via condensation methods due to the formation of non-oxidizable porphodimethenes such as 392.
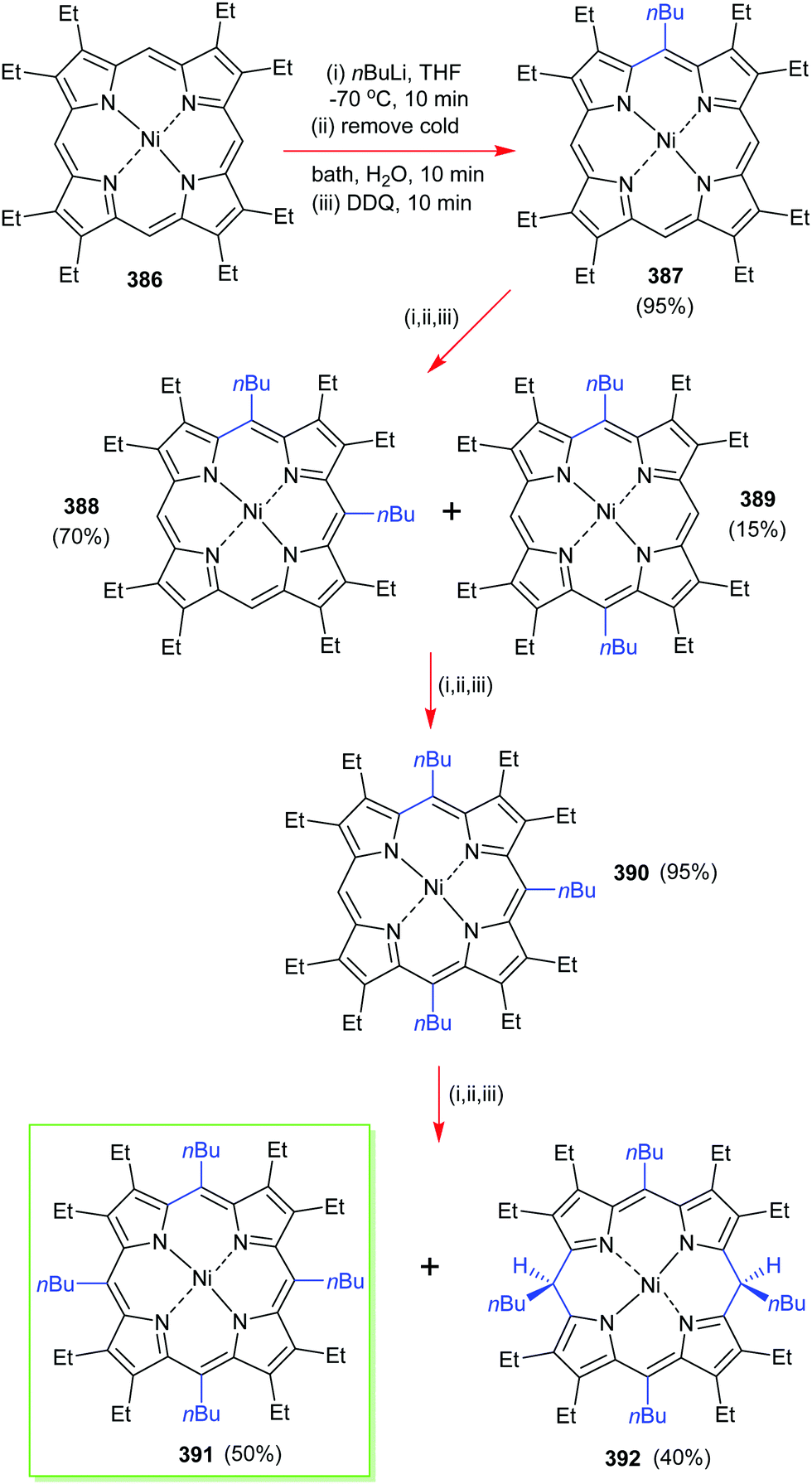 | ||
| Scheme 54 Synthesis of (5,10,15,20-tetra(n-butyl)-2,3,7,8,12,13,17,18-octaethylporphyrinato)nickel(II) via sequential SNAr reaction with nBuLi. | ||
Thirdly, Neya and Funasaki's63 acid-catalyzed dealkylation of 5,10,15,20-tetra-tert-butylporphyrin 129 gives a convenient method for preparing unsubstituted porphyrin 5 as a starting point for FGI reactions (see Scheme 18). Complemented by condensation methods,61 this was used by us for an entry into the A 205 and 5,10-A2206 porphyrin systems.62b Starting with these and using the various meso-substitution reactions at hand, ABCD porphyrins of all types can now be prepared in a few steps.44,47,64,138a,b
To illustrate the practical and contemporary use of such strategies let us consider the synthesis of compound “YD2-o-C8” (398). This and related compounds gained fame in Grätzel's efforts to develop porphyrin-sensitized solar cells with record power conversion efficiencies of 12% in 2011.139Scheme 55 outlines the original synthesis, which involves bromination and metalation of 393 to 394, Sonogashira alkynylation to 395, bromination to 396, Buchwald–Hartwig amination (397) and deprotection with TBAF; followed by a final Pd2(dba)3-catalyzed Sonogashira reaction to obtain 398. Porphyrins of such practical relevance are also some of the few for which alternative and optimized synthetic sequences have been investigated.140
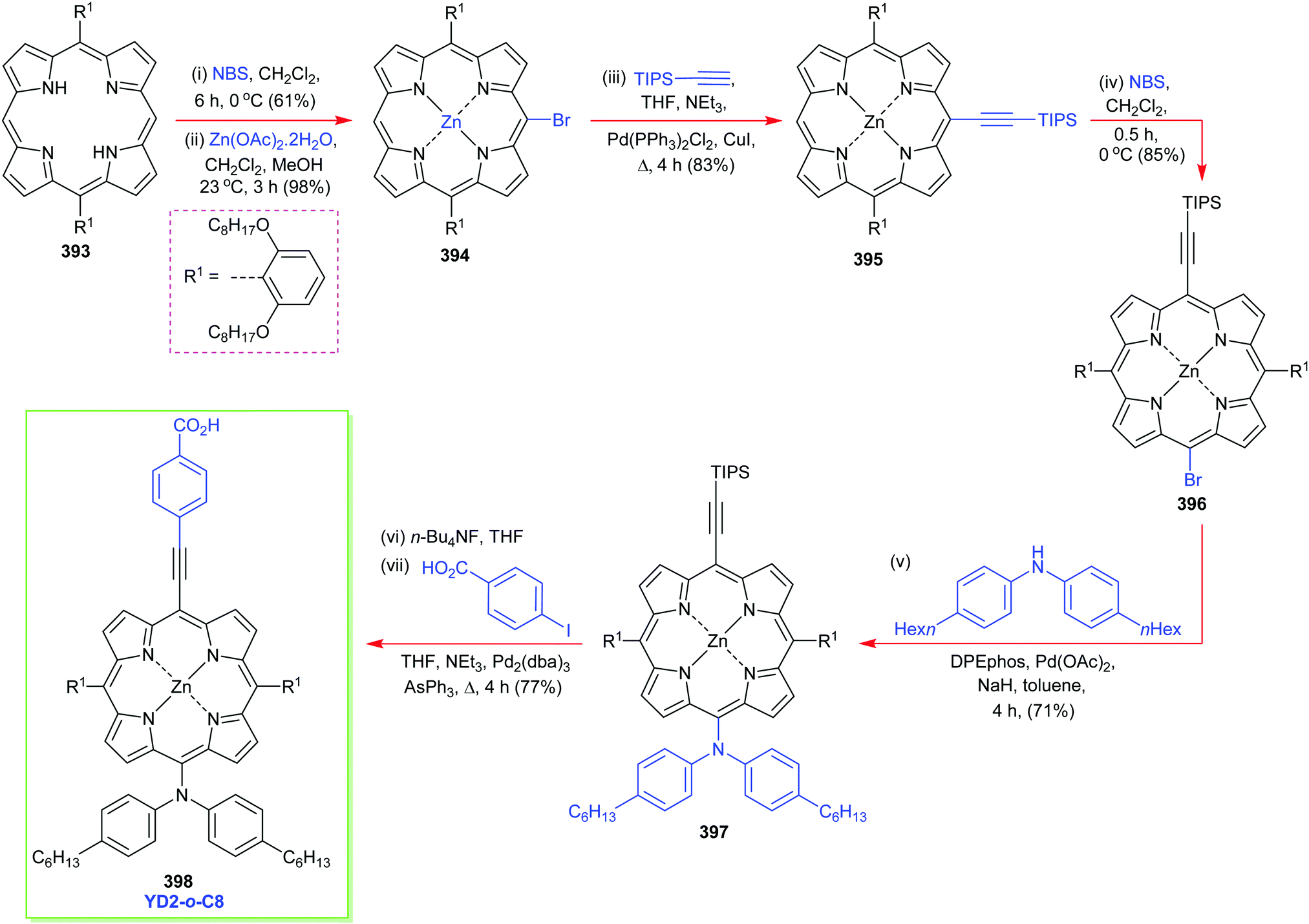 | ||
| Scheme 55 Syntheses of donor–acceptor compounds for dye-sensitized solar cells. | ||
All this leaves out the perhaps even more voluminous body of work on porphyrin arrays, materials, drug candidates, donor–acceptor complexes, expanded and contracted porphyrinoids (see below) and more. Often involving standard macrocycle substitution and FGI reactions, this area of research includes many notable achievements, e.g., radical coupling and oxidative fusing reactions and more, as exemplified and embodied by Osuka's manifold contributions to the field.141
To give only one example of this fascinating area of porphyrin synthesis, Scheme 56 illustrates the synthesis of a directly fused tetrameric porphyrin sheet by Kim, Kobayashi, Osuka and coworkers.142 This was prepared by first generating the singly meso–meso-fused tetramer 401 through a stepwise coupling sequence of 399. Final oxidative coupling of 400 with Sc(OTf)3 then gave the tetramer 401. A similarly challenging synthesis was Anderson's synthesis of a tetraanthracene fused porphyrin.143 This required first the in situ generation of the pyrrole-carbinol 403 from 402 for the synthesis of the respective free base tetraanthracenylporphyrin. After metalation to obtain 404 this could be fused to the target compound 405.
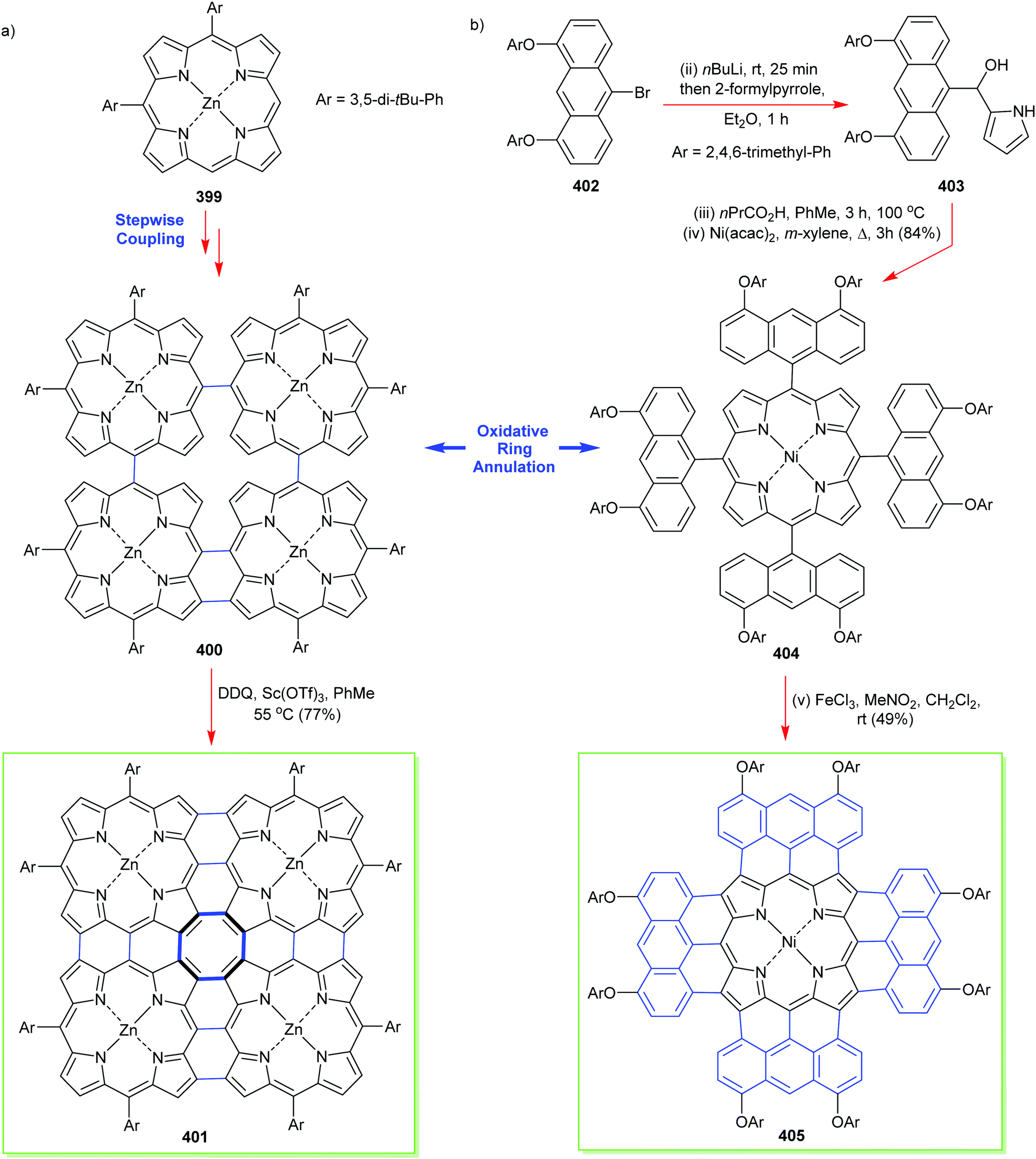 | ||
| Scheme 56 Oxidative fusing reaction of porphyrins. (a) Osuka's synthesis of a tetrameric porphyrin sheet and (b) Anderson's synthesis of a porphyrin fused to four anthracene units. | ||
Another intriguing development in porphyrin chemistry has been the advent of on-surface synthesis spearheaded by colleagues from physics working on interfacial chemistry. This typically involves depositing the starting material on metal surfaces at interfaces, pre-organizing them, e.g., using principles from supramolecular chemistry, followed by annealing reactions.144 A typical example related to the systems shown in Scheme 56 is the preparation of a β–β,meso–meso,β–β-triply fused bisporphyrin by Auwärter and colleagues (Scheme 57). Deposition of unsubstituted porphyrin (5) under ultrahigh-vacuum conditions and annealing (heating) at 720 K gave the dimer 406via temperature-induced covalent dehydrogenative coupling.145
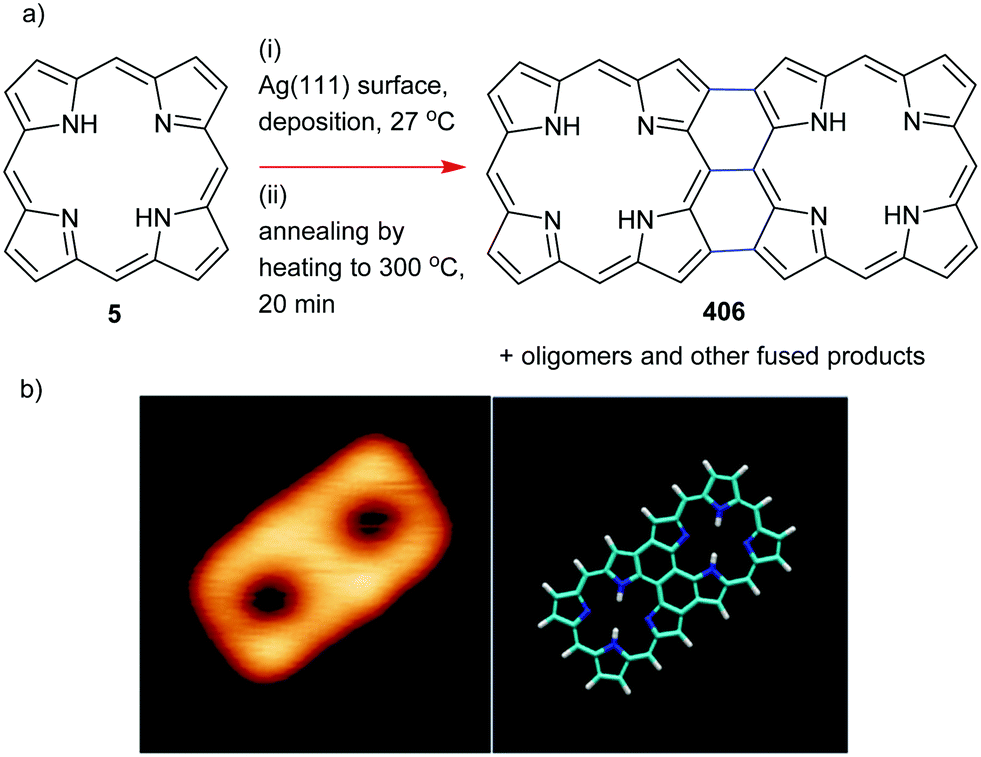 | ||
| Scheme 57 On-surface synthesis of a triply-linked bisporphyrin. (a) Synthetic scheme. (b) STM photograph and model of 406;145 courtesy of W. Auwärter, TU Munich. | ||
8. The new breed
8.1 The family of porphyrinoids
So, is this it? As much complexity as the few hundreds of natural porphyrins provide, this is just the tip of the iceberg. There are many more members of the porphyrin family than just 18π-electron systems such as 5, with 20 carbon and 4 nitrogen atoms in a 24-atom macrocycle. The family of synthetic porphyrinoid macrocycle types is ever expanding, some of which, at a first glance, appear to have only a passing resemblance to the natural systems. Fig. 9 illustrates the main classes of porphyrinoids. Clearly, we could go on with ‘Classics of porphyrinoid chemistry’ for a few hundred pages.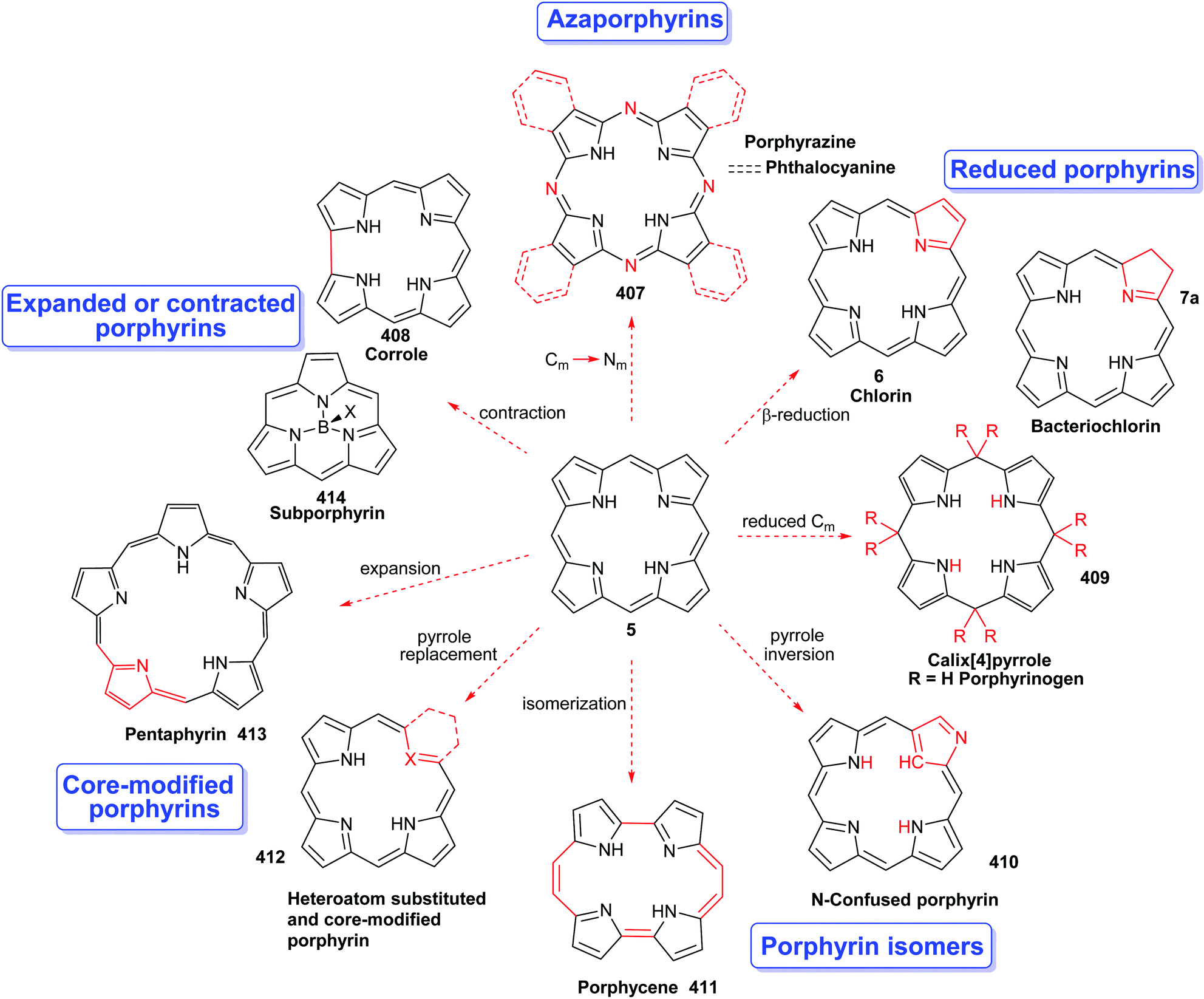 | ||
| Fig. 9 Illustration of the main members of the porphyrin family and their relationship with the parent compound. | ||
Some of these are easily related to each other, e.g., the chlorins 6 as reduction products of porphyrins, or the porphyrinogens 409, which we have encountered as intermediates in the (bio)synthesis of porphyrins. Likewise, the phthalocyanines can be considered a subclass of porphyrazines (407), i.e. are 5,10,15,20-tetraazaporphyrins. These, and the other structures shown are conceptionally related to porphyrins via exchange, deletion or addition of individual atoms or whole fragments of the cyclic tetrapyrrole system. For example, corrole 408, like the corrin systems discussed above, lacks one Cm bridge and can be viewed as a ‘nor-porphyrin’; while it is also possible to convert a corrole to a porphyrin.146 Other systems can be envisaged by removal (408), inversion (410) or addition (413) of pyrrole units; exchange of one or more pyrrole units for carba- or heterocycles (412); or a ‘reshuffling’ of the bridging Cm units (411).
One way to make sense of this bewildering complexity is to expand our viewpoint and look at porphyrinoids as conjugated cyclic compounds. This goes back to Emanuel Vogel's annulene concept, which provides a framework for comparison and stands at the beginning of the era of macrocycle modified porphyrins (Fig. 10).147 Having worked with bridged [14]annulenes such as 415 (where 14 is the number of π-electrons), he realized the relationship between syn-1,6:8,13-bisimino[14]annulene 416 and porphyrin 5. The latter can be considered as a doubly NH-bridged diaza[18]annulene (highlighted in formula 417), and is thus related to the parent [18]annulene 418. In this system, benzene is simply [6]annulene.
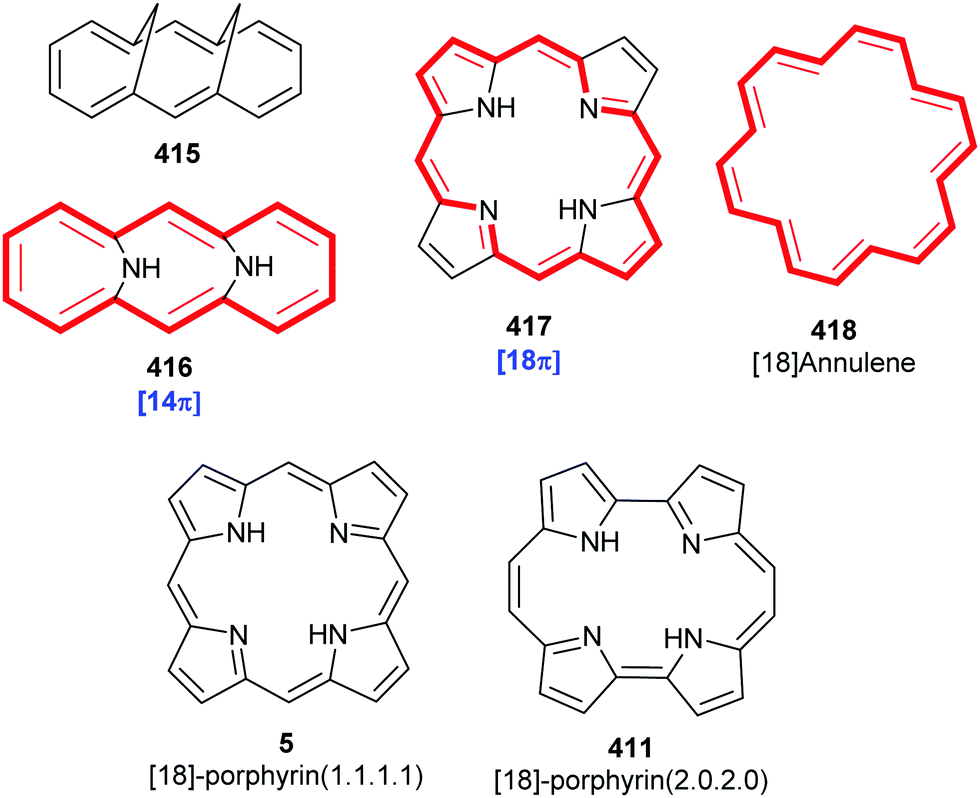 | ||
| Fig. 10 Emanuel Vogel's annulene concept applied to porphyrins. | ||
The interrelationship between the various systems shown in Fig. 9 can then be considered purely based on alterations made to the core annulene system, be it expansion, contraction, transposition, or substitution of individual atoms therein. Thus, porphyrin 5 is now viewed as a [18]porphyrin(1.1.1.1) system, where the numbers in brackets (a.b.c.d.) indicate the number of carbon atoms in the meso positions. The isomeric porphycene 411, which also has an 18 π-electron system but a different arrangement of the skeleton atoms then is [18]porphyrin(2.0.2.0) and so forth. Taking this one step further, Vogel's concept also illustrates how ‘porphyrins’ can have less or more than 18 π-electrons, and why antiaromatic or Möbius-aromatic cyclic oligopyrroles are often discussed in the context of ‘porphyrinoid chemistry’.148
So, whilst aromatic [18]porphyrins are just the beginning, they are at the heart of a ‘superfamily’ of porphyrinoid macrocycles. Each subclass has now been described in hundreds of publications and overall the field of atypical porphyrinoids encompasses several thousand papers. Many of the new systems possess a rich organometallic chemistry, conformational flexibility and can undergo skeletal rearrangement reactions.149 Staying in the overall context of this contribution we will highlight this area by only a handful of classic examples.
8.2 Porphyrin isomers
The first textbook case for the annulene concept was the synthesis of constitutional isomers of porphyrin, six of which can be envisaged with a 4N core. Indeed, Vogel's group accomplished the synthesis of porphycene 411 in 1986 (Scheme 58).150 In this isomer of porphyrin, two of the meso carbons are transposed to give a [18]-porphyrin(2.0.2.0) system. This indigo-blue compound is strongly fluorescent and has physicochemical properties very similar to porphyrin. | ||
| Scheme 58 Vogel's synthesis of porphycene. | ||
The synthesis involved preparation of 2,2′-bipyrrole 420via dehydrogenation of 419. Vilsmeier–Haack formylation to 421 then allowed a McMurry-type coupling with low-valent titanium. The reaction is presumed to pass through intermediate 422, which was readily oxidized by air. Despite the low yield, this synthesis changed the porphyrin field forever and gave birth to much of what followed later in ‘porphyrinoid chemistry’. Subsequent works significantly improved yields and provided alternate pathways to substituted porphycenes.151
Another breakthrough occurred only eight years later when the groups of Furuta and Latos-Grażyński independently reported on N-confused porphyrin 410; technically an 2-aza-21-carbaporphyrin with an inverted pyrrole ring.152 Pyrrole condensation reactions are notoriously complex and when the acid-catalyzed condensation45 of pyrrole with benzaldehyde or p-tolylaldehyde was investigated, compounds of general structure 423 were obtained in yields of 4–7% (Scheme 59) with the standard porphyrin still being the main product.153,154
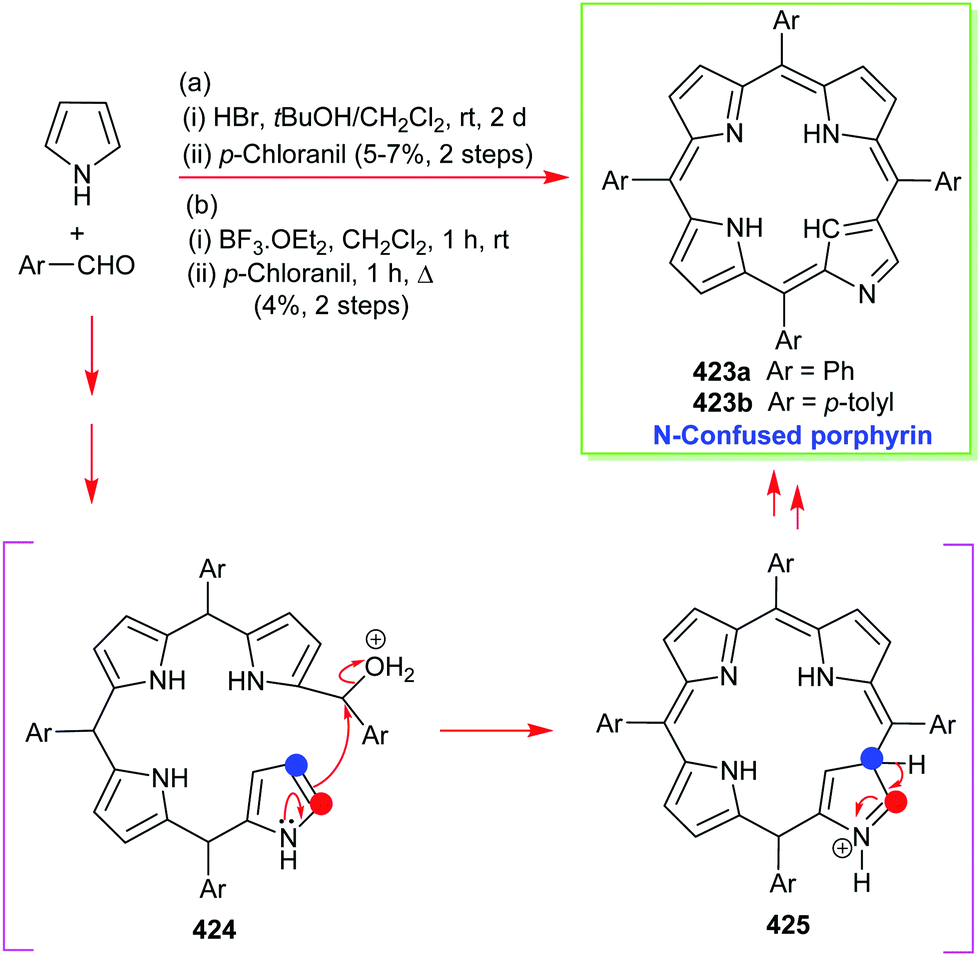 | ||
| Scheme 59 The syntheses of N-confused porphyrins by (a) Furuta and (b) Latos-Grażyński. A depictive SN2 type reaction mechanism has been drawn here for clarity. However SN1 type ring-closure mechanisms can equally well be envisaged. Please note that the tetraarylporphyrin product dominates in these reactions as the primary reaction product (see text). However, the N-confused porphyrins are also formed in 4–7% yield as by-products. | ||
Mechanistically, the key to the formation of these products lies in how the intermediary porphyrinogen12 is constructed. Pyrrole/aldehyde condensations are basically oligomerization reactions. Once the growing chain reaches a tetrameric state, it is believed to adopt a helical conformation and an intramolecular condensation step occurs.50 The latter typically involves 424 having its the α-pyrrole carbon forming the ring-closing bond to ultimately yield the standard tetraarylporphyrin. However, a rotation of a terminal pyrrole ring in 424 can beautifully position the β-carbon for an attack on the protonated carbinol. Oxidation then completes the final sequence to 423 (Scheme 59).
Again, the subsequent development of this field has been rapid with improved syntheses; related (multiply) confused porphyrins having been synthesized and notably, Latos-Grażyński developing a transformative organometallic chemistry concept originating with these compounds.149a,c
8.3 Contract or expand?
The aforementioned examples of porphyrin isomers have shown that it is possible to reshuffle the macrocyclic atoms. And Nature has shown that we can contract the porphyrin macrocycle by excising one meso-bridge to corrins. Staying with the latter for a moment, the corrinoids are non-aromatic, which raises the question of whether aromatic compounds related to porphyrins, but lacking the meso-bridge, are possible? Earlier studies had looked at corroles 408 in their relationship to corrin 8 and indicated this, but another chapter in porphyrin chemistry was opened in 1999, when the groups of Gross in Israel and Paolesse in Italy reported on the synthesis of meso-aryl-corroles (Scheme 60).155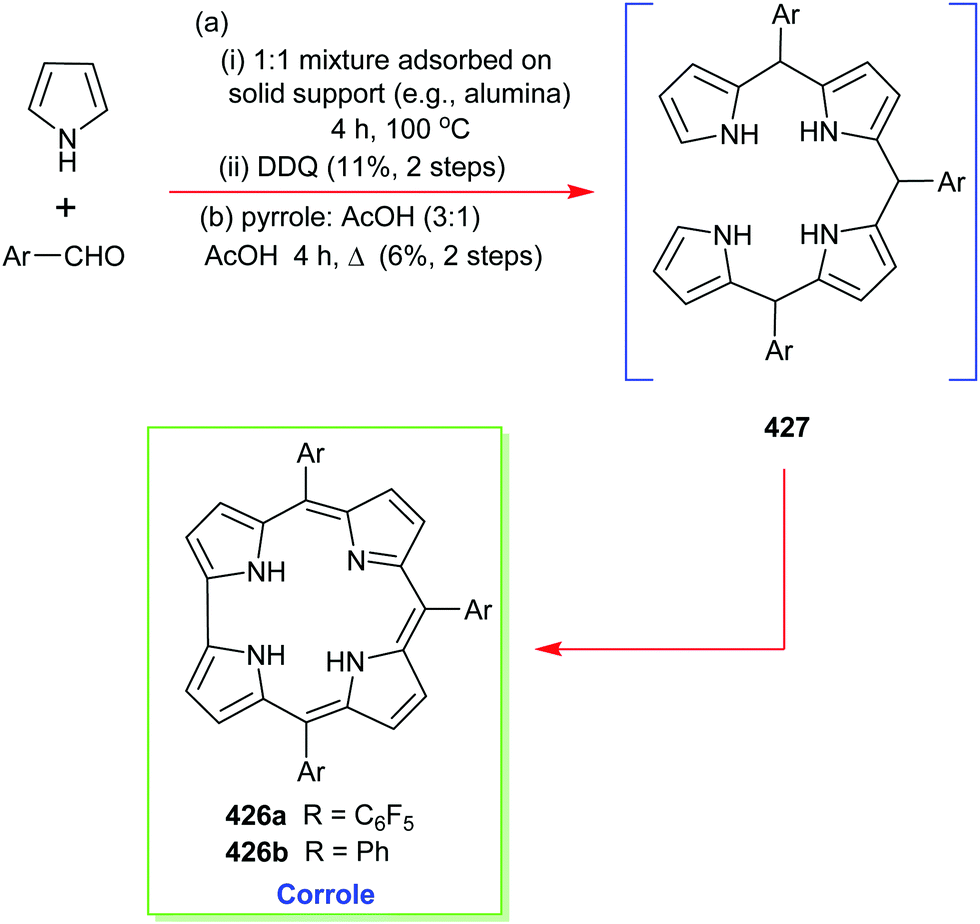 | ||
| Scheme 60 The synthesis of 5,10,15-triarylcorroles by (a) Gross and (b) Paolesse. | ||
The approach is similar to that just seen in Scheme 59. Pyrrole and the aldehyde are mixed and heated under acidic conditions. Presumably, the key intermediate is 427, which is then oxidatively fused. Thus, the difference between normal porphyrin synthesis and corrole synthesis lies in trapping the bilane 427 prior to its reaction with the fourth aldehyde. This field has also not stood still. Improvements in synthetic methods, most notably by Gryko, have resulted in a rich application chemistry in catalysis and biomedicine, and because of the unusual coordination chemistry (3 × NH), different from that of porphyrins (2 × NH), there has been and continues to be elicited, widespread interest.156
Going to the other extreme, can we also expand porphyrins? Inserting just one additional carbon atom into a meso-bridge yields a homoporphyrin. Clearly, this will not be aromatic and its physicochemical properties are more akin to those of an a,b,c-bilatriene 179. This was shown in Callot's first synthesis of a carba-homoporphyrin 429 (Scheme 61).157 Here, an exo/endo mixture of 429 was obtained upon treating the N-substituted porphyrin 428 with Ni(II)(acac)2. The mechanism of this rearrangement reaction was identified by careful isolation of two key intermediates through a judicious modification of the reaction conditions. In the envisaged pathway, the nickel salt 430 is initially formed, whose α-H-atoms in the N-ester component are now rendered acidic by the Ni(II)-coordination event, analogously to that seen in the vitamin B12 synthesis. As a result, they can easily be deprotonated by the action of a very mild base, such as Et3N, to yield an ester enolate 430aa that undergoes internal Michael-ype addition to give the aziridine 431. The latter then undergoes a thermal rearrangement to the homoporphyrin 429, possibly by 432 engaging in a cyclopropane ring-opening via the mechanism shown.
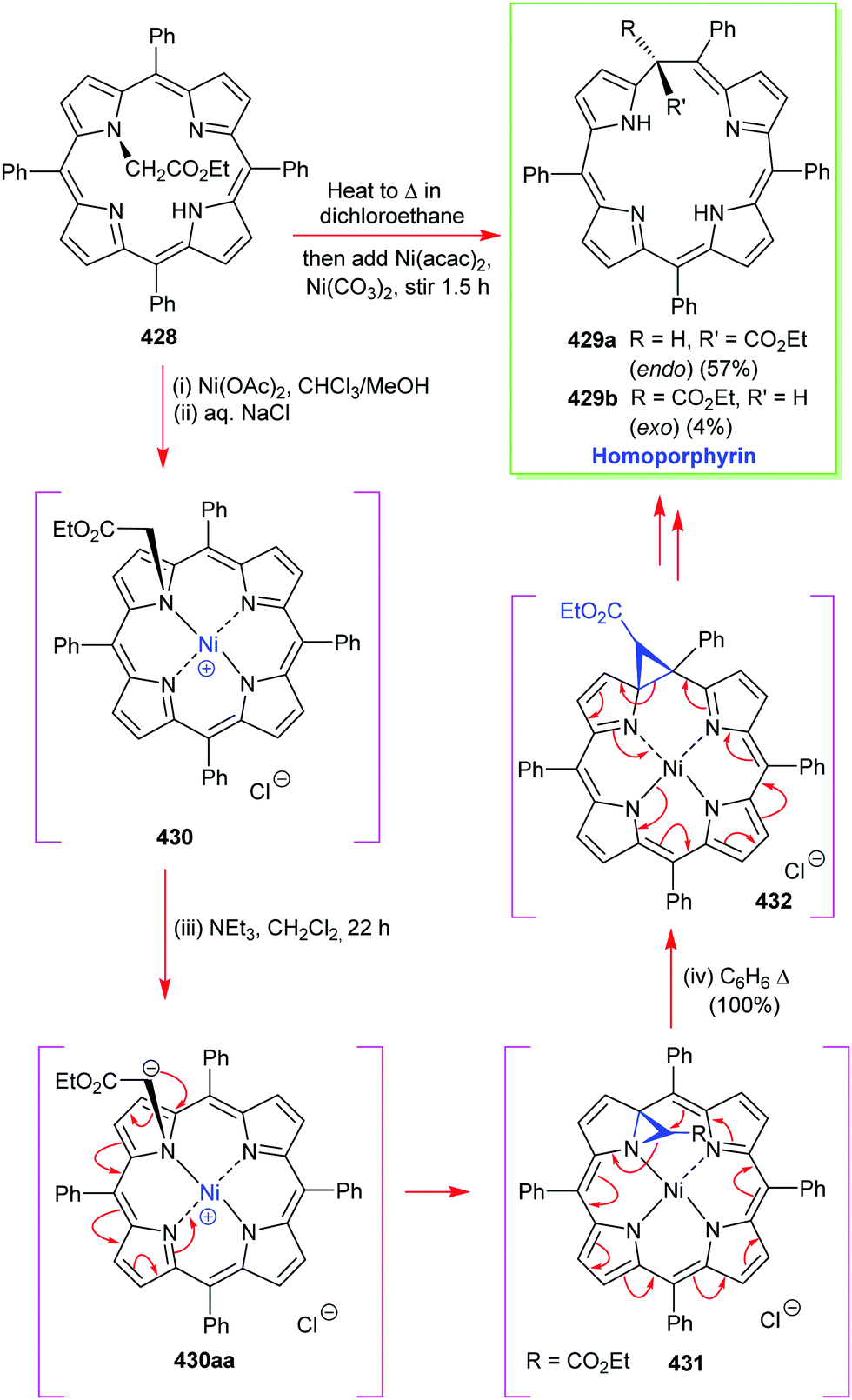 | ||
| Scheme 61 Callot's synthesis of a homoporphyrin. | ||
In order to make expanded aromatic porphyrins one can go back to Vogel's annulene concept. In the late 1990s, Burchard Franck, who also made many seminal contributions to the chemistry of porphyrinoids, surmised that [30]annulenes 433 could be prepared. While such compounds cannot be prepared based on benzene units, he realized that cyclopolyenes might be stabilized by the incorporation of pyrrole units. With previous experience from the synthesis of an aromatic tetravinylogous [26]-porphyrin158 his team targeted 438 (Scheme 62).159
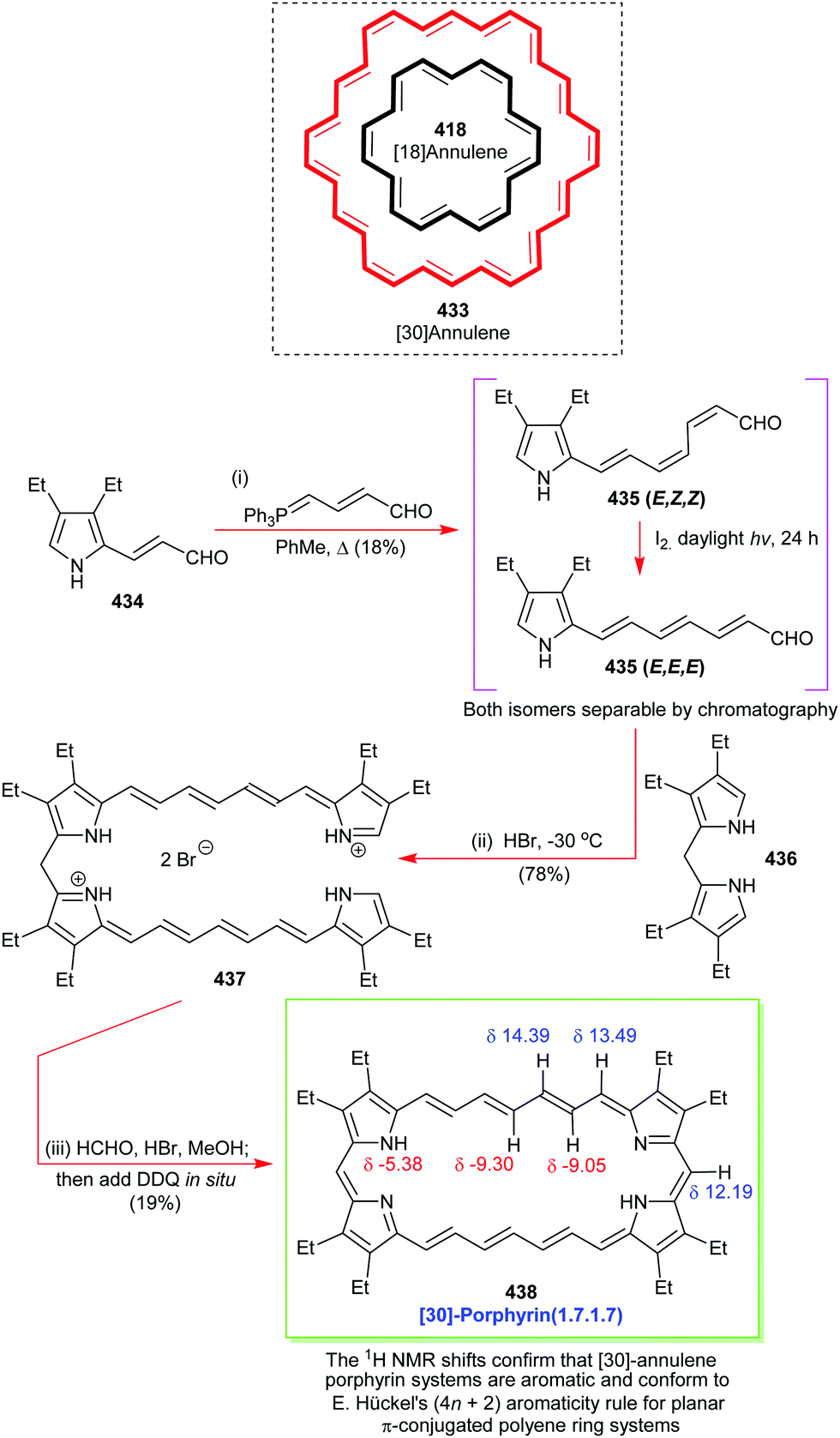 | ||
| Scheme 62 Franck and Eickmeier's synthesis of a hexavinylogous 30π-electron porphyrin annulene. | ||
Its synthesis commenced with pyrrolylacrolein 434 which was chain extended to 435via a Wittig reaction. The aldehyde was then reacted with the dipyrromethane 436 to yield the hexavinylogous biladiene bishydrobromide (formally, a bilaoctaene) 437 in good yield. Cyclocondensation with formaldehyde then proceeded in HBr/MeOH and was followed by in situ oxidation with DDQ to give the target pyrrolyl [30]annulene which showed the anticipated extensive ring current and anisotropy typical of a planar π-conjugated (4n + 2) aromatic ring system.
Having extended the carbon bridges, can one also add pyrrole units, for example, as shown for the pentaphyrin 413? Here, we go back to back to an older study from Woodward, of chlorophyll and vitamin B12 fame. During work on the latter project, his group serendipitously discovered a new dye with a brilliant blue color and named it ‘sapphyrin’.160 Their key observation was that reaction of the open-chain tetrapyrrole 439 with formic acid and HBr, followed by oxidation with iodine did not give the expect corrole. Rather it yielded 440 as a by-product (Scheme 63). Mechanistically, this was surmised to involve condensation of the two terminal pyrroles in 439 with formic acid, α-protonation and cleavage to the pentapyrrolic system, followed by cyclization.161 This was then followed by more logical targeted approaches, e.g., [4+1] cyclizations as in 439 + 441 or [3+2] approaches. As shown in formula 442 sapphyrin is a 22 π-electron compound and this ground-breaking discovery elicited a whole range of related compounds and wide-ranging studies, notably by Sessler.160,162
 | ||
| Scheme 63 Woodward's discovery of sapphyrin. | ||
Many of the applications of expanded porphyrins relate to their facile protonation and use as sensors and receptors. Like nonplanar porphyrins56 protonation facilitates conformational changes and anion binding.160 Core-protonated porphyrinoids thus have a rich coordination chemistry, which is totally of that of ‘normal’ porphyrins which are mostly used as metal coordinating ligands.
8.4 The simplest of all
Sessler's group in Austin was also the one to transform porphyrinoid chemistry by focussing on the calix[4]pyrroles 409. In the porphyrin chemist's parlance, porphyrinogens, and their more oxidized variants, such as porphomethenes, porphodimethenes, and porphotrimethenes (aka calixphyrins) have found wide interest as sensing agents and molecular scaffolds.7,162,163 The more conventional nomenclature as ‘calix’ systems relates to their analogy to the calix[n]arenes. The calix[4]pyrroles are hexahydroporphyrins and the presence of the sp3-hybridized bridging carbons allows for flexibility in the orientation of the pyrrole units; hence, their utility for interaction with other molecules. They, are, however, non-aromatic.For their first synthesis, we must go back to almost the beginning of modern organic chemistry, to Adolf von Baeyer. In 1886 he reported the reaction of acetone with pyrrole in the presence of HCl to yield a white powder which he named “Acetonpyrrol”.164 The relationship of this calix[4]pyrrole 443 to our porphyrin story should be clear. The presence of the gem-dimethyl Cm units and absence of any meso-hydrogen atoms prevented oxidation to the porphyrin. Every modern, or future, development has a beginning in the past and so it is perhaps fitting that we close our walk through synthetic porphyrin chemistry, with probably the simplest and oldest of all rational, ‘total’ syntheses of a member of the porphyrin family (Scheme 64).
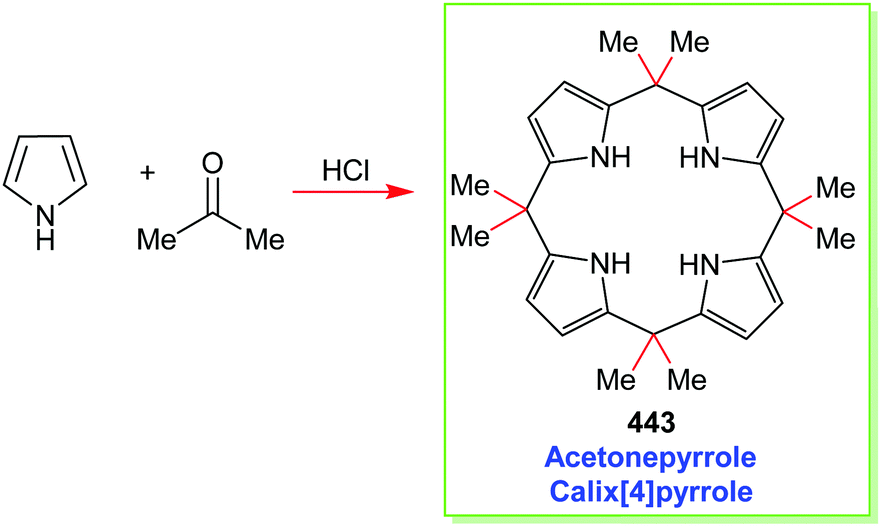 | ||
| Scheme 64 Baeyer's synthesis of acetonepyrrole. | ||
9. Closing statement
In closing, we hope that you have enjoyed this tutorial-type journey on which we have taken you, through present and past synthetic porphyrin chemistry. Our aim has been to give the non-specialist, but still expert, synthetic organic chemist, a starting point from where to commence their own synthetic endeavors in the porphyrin field, but we have consciously aimed at making our review readily accessible to postgraduate students. Throughout the article, we have attempted to summarize the main advances that have occurred in recent years in the technologies available for the synthetic construction of complex porphyrins, and we have tried to place these developments in historical context, since much of the early chemistry used for building up such structures continues to be of relevance today. The timespan of coverage has been quite long, simply because this field has evolved at a much slower pace, and major synthetic advances have been much harder to come by than in other disciplines where many more groups have operated. However, as you will have seen, there remain many synthetic challenges and problems still to be satisfactorily addressed in this extremely difficult field which, as Battersby's and Kishi's total synthesis efforts indicate, continues to attract the very brightest and most able organic chemists of the contemporary modern era. Considering the many possibilities already realized, and others yet to be achieved, in macrocycle-modified porphyrinoids,165 and the wealth of functionalization reactions developed in the past three decades alone, porphyrins and related family members are clearly a vibrant and very active area of chemistry. We look forward to further synthetic progress being made over the coming decades, as other eminent organic chemists recognize the issues that remain and are drawn to make powerful contributions to the field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 764837 and through a grant from Science Foundation Ireland (IvP 13/IA/1894). We are indebted to Prof. Willi Auwärter (Technische Universität München) for providing the photographs in Scheme 57.Notes and references
- (a) A. R. Battersby, Nat. Prod. Rep., 2000, 17, 507–526 RSC; (b) L. Milgrom, The Colors of Life, Oxford University Press, Oxford, 1997 Search PubMed.
- R. Willstätter and A. Stoll, Untersuchungen über Chlorophyll. Methoden und Ergebnisse, J. Springer, Berlin, Germany, 1913 Search PubMed.
- A masterly historical account of some of the early work is given D. A. Lightner, Bilirubin: Jekyll and Hyde Pigment of Life, Springer, Wien, 2013 Search PubMed.
- M. O. Senge, Chem. Commun., 2006, 243–256 RSC.
- M. O. Senge, S. A. MacGowan and J. M. O’Brien, Chem. Commun., 2015, 51, 17031–17063 RSC.
- (a) K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, 2000, p. 2003 Search PubMed; (b) K. M. Kadish, K. M. Smith and R. Guilard, Handbook of Porphyrin Science, World Scientific, Singapore, 2010 Search PubMed.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted & Isomeric Porphyrins, Pergamon, Oxford, 1997 Search PubMed.
- H. Fischer and H. Orth, Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1937 Search PubMed.
- K. M. Smith, Porphyrins and Metalloporphyrins, Elsevier, Amsterdam, 1975 Search PubMed.
- D. Dolphin, The Porphyrins, Academic Press, New York, 1978 Search PubMed.
- H. Scheer, Chlorophylls, CRC Press, Boca Raton, 1991 Search PubMed.
- D. Dolphin, J. Heterocycl. Chem., 1970, 7, 275–283 CrossRef CAS.
- G. R. Geier III, B. J. Littler and J. S. Lindsey, J. Chem. Soc., Perkin Trans. 2, 2001, 701–711 RSC.
- A. Treibs, Das Leben und Wirken von Hans Fischer, Hans Fischer-Gesellschaft, München, 1971 Search PubMed.
- (a) R. B. Woodward, W. A. Ayer, J. M. Beaton, F. Bickelhaupt, R. Bonnett, P. Buchschacher, G. L. Closs, H. Dutler, J. Hannah, F. P. Hauck, S. Itô, A. Langemann, E. Le Goff, W. Leimgruber, W. Lwowski, J. Sauer, Z. Valenta and H. Volz, J. Am. Chem. Soc., 1960, 82, 3800–3802 CrossRef CAS; (b) R. B. Woodward, Pure Appl. Chem., 1961, 2, 383–404 CAS.
- (a) R. B. Woodward, W. A. Ayer, J. M. Beaton, F. Bickelhaupt, R. Bonnett, P. Buchschacher, G. L. Closs, H. Dutler, J. Hannah, F. P. Hauck, S. Itô, A. Langemann, E. Le Goff, W. Leimgruber, W. Lwowski, J. Sauer, Z. Valenta and H. Volz, Tetrahedron, 1990, 46, 7599–7659 CrossRef CAS; (b) For a further analysis of Woodward's chlorophyll total synthesis and a discussion of other recent chlorin total syntheses including bonellin methyl ester and tolyporphin A, see: Y. Liu, S. Zhang and J. S. Lindsey, Nat. Prod. Rep., 2018, 35, 879–901 RSC.
- K. M. Smith, New J. Chem., 2016, 40, 5644–5649 RSC.
- (a) M. J. Warren and A. G. Smith, Tetrapyrroles: Birth, Life and Death, Springer-Verlag, New York, 2009 CrossRef; (b) D. A. Bryant, C. N. Hunter and M. J. Warren, J. Biol. Chem., 2020, 295, 6888–6925 CrossRef CAS.
- (a) H. Fischer and K. Zeile, Liebigs Ann. Chem., 1929, 468, 98–116 CrossRef CAS; (b) H. Fischer and A. Kirstahler, Liebigs Ann. Chem., 1928, 466, 178–188 CrossRef CAS.
- G. W. Kenner, S. W. McCombie and K. M. Smith, Liebigs Ann. Chem., 1973, 1329–1338 CrossRef CAS.
- H. Fischer and W. Lautsch, Liebigs Ann. Chem., 1937, 528, 265–275 CrossRef CAS.
- K. M. Smith and W. M. Lewis, Tetrahedron, 1981, 37, 399–403 CrossRef.
- L. Knorr, Liebigs Ann. Chem., 1886, 236, 290–332 CrossRef.
- (a) K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346–4363 RSC; (b) J. R. Roth, J. G. Lawrence and T. A. Bobik, Annu. Rev. Microbiol., 1996, 50, 137–181 CrossRef CAS.
-
(a) R. B. Woodward, Pure Appl. Chem., 1968, 17, 519–547 CAS;
(b) R. B. Woodward, Pure Appl. Chem., 1971, 25, 283–304 CAS;
(c) R. B. Woodward, Pure Appl. Chem., 1973, 33, 145–178 CAS;
(d) A. Eschenmoser and C. E. Wintner, Science, 1977, 196, 1410–1420 CrossRef CAS;
(e) A. Eschenmoser, Q. Rev., Chem. Soc., 1970, 24, 366–415 RSC;
(f) A. Eschenmoser, Pure Appl. Chem., 1963, 7, 297–316 CAS;
(g) A. Eschenmoser, Naturwissenschaften, 1974, 61, 513–525 CrossRef CAS;
(h) A. Eschenmoser, Helv. Chim. Acta, 2015, 98, 1483–1600 CrossRef CAS . This excellent, first-hand, account of the vitamin B12 synthesis by Albert Eschenmoser discusses the Zurich photocyclization route to B12 (strategy 2) in great detail. It stands out because of the many unique insights it provides into the key mechanistic concepts that ultimately led to the ETH team testing their novel macrocyclization approach;
(i) A possible mechanistic rationale of this remarkable C(17)-side-chain –CN to –CO2H conversion initiated with N-cyclohexyl-α-chloro-propionaldonitrone and AgBF4 invokes in situ generation of the alkenyl nitrone followed by a Michael addition of the amide and dual hydrolysis of the intermediary nitrone/iminium ion. The resulting aldehydo-ester is then thought to be cleaved by the dimethylamine via the mechanism shown:
; (j) For Eschenmoser's later much simpler, and totally regioselective, chemical conversion of the heptakis(cyanomethyl)-cobyrinate into vitamin B12, see reference 25h above and the following publication: A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1988, 27, 5–39 CrossRef :
This has to be one of the most remarkable and exquisitely regioselective chemical reactions ever performed and because this is a tutorial style review, we could not possibly fail to mention it. - (a) D. C. Hodgkin, J. Kampfer, M. Mackay, J. Pickworth, K. N. Trueblood and J. G. White, Nature, 1956, 178, 64–66 CrossRef CAS; (b) H. A. Barker, R. D. Smyth, H. Weissbach, J. I. Toohey, J. N. Ladd and B. E. Volcani, J. Biol. Chem., 1960, 235, 480–488 CrossRef CAS.
- For an excellent account of the vitamin B12 synthesis, containing many personal accounts and reminiscences of the various postdoctoral fellows involved, and plenty of unique historical photographs and perspectives, see: G. W. Craig, J. Porphyrins Phthalocyanines, 2016, 20, 1–20 CrossRef CAS.
- J. I. Seeman, Angew. Chem., Int. Ed., 2017, 56, 10228–10245 CrossRef CAS.
- (a) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS; (b) K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis: Targets, Strategies, Methods, Wiley-VCH, Weinheim, 1996 Search PubMed.
- W. Friedrich, G. Gross, K. Bernhauser and P. Zeller, Helv. Chim. Acta, 1960, 43, 704–712 CrossRef CAS.
- Y. Yamada, D. Miljkovic, P. Wehrli, B. Golding, P. Löliger, R. Keese, K. Müller and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1969, 8, 343–348 CrossRef CAS.
- R. Hoffmann and R. B. Woodward, Acc. Chem. Res., 1968, 1, 17–22 CrossRef CAS.
- (a) U. Ermler, W. Grabarse, S. Shima, M. Goubeaud and R. K. Thauer, Science, 1997, 278, 1457–1462 CrossRef CAS; (b) S. Scheller, M. Goenrich, R. Boecher, R. K. Thauer and B. Jaun, Nature, 2010, 465, 606–608 CrossRef CAS; (c) S. Mayr, C. Latkoczy, M. Krüger, D. Günther, S. Shima, R. K. Thauer, F. Widdel and B. Jaun, J. Am. Chem. Soc., 2008, 130, 10758–10767 CrossRef CAS.
- (a) K. Zheng, P. D. Ngo, V. L. Owens, X.-P. Yang and S. O. Mansoorabadi, Science, 2016, 354, 339–342 CrossRef CAS; (b) S. J. Moore, S. T. Sowa, C. Schuchardt, E. Deery, A. D. Lawrence, J. V. Ramos, S. Billig, C. Birkemeyer, P. T. Chivers, M. J. Howard, S. E. J. Rigby, G. Layer and M. J. Warren, Nature, 2017, 543, 78–82 CrossRef CAS.
- (a) Ciba Foundation, The Biosynthesis of the Tetrapyrrole Pigments, Ciba Foundation Symposium 180, John Wiley and Sons, 1994 Search PubMed; (b) R. S. Ajioka, J. D. Phillips and J. P. Kushner, Biochim. Biophys. Acta, 2006, 1763, 723–736 CrossRef CAS; (c) M. O. Senge, A. A. Ryan, K. A. Letchford, S. A. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef; (d) P. Brzezowski, A. S. Richter and B. Grimm, Biochim. Biophys. Acta, 2015, 1847, 968–985 CrossRef CAS.
- (a) A. R. Battersby, Bioorg. Med. Chem., 1996, 4, 937–964 CrossRef CAS; (b) F. Blanche, B. Cameron, J. Crouzet, L. Debussche, D. Thibaut, M. Vuilhorgne, F. J. Leeper and A. R. Battersby, Angew. Chem., Int. Ed. Engl., 1995, 34, 383–411 CrossRef CAS.
- (a) C. A. Roessner and A. I. Scott, J. Bacteriol., 2006, 188, 7331–7334 CrossRef CAS; (b) A. I. Scott, Heterocycles, 1998, 47, 1051–1066 CrossRef CAS.
- (a) G. V. Louie, P. D. Brownlie, R. Lambert, J. B. Cooper, T. L. Blundell, S. P. Wood, M. J. Warren, S. C. Woodcock and P. M. Jordan, Nature, 1992, 359, 33–39 CrossRef CAS; (b) M. A. A. Mathews, H. L. Schubert, F. G. Whitby, K. J. Alexander, K. Schadick, H. A. Bergonia, J. D. Phillips and C. P. Hill, EMBO J., 2001, 20, 5832–5839 CrossRef CAS.
- (a) W. M. Stark, C. J. Hawker, G. J. Hart, A. Philippides, P. M. Petersen, J. D. Lewis, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1993, 2875–2892 RSC; (b) W. M. Stark, M. G. Baker, F. J. Leeper, P. R. Raithby and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1988, 1187–1201 RSC.
- H. Puy, L. Gouya and J.-C. Deybach, Lancet, 2010, 375, 924–937 CrossRef CAS.
- D. J. Heyes and C. N. Hunter, Trends Biochem. Sci., 2005, 30, 642–649 CrossRef CAS.
- S. Storbeck, S. Saha, J. Krausze, B. U. Klink, D. W. Heinz and G. Layer, J. Biol. Chem., 2011, 286, 26754–26767 CrossRef CAS.
- (a) S. J. Moore and M. J. Warren, Biochem. Soc. Trans., 2012, 40, 581–586 CrossRef CAS; (b) S. Gu, O. Sushko, E. Deery, M. J. Warren and R. W. Pickersgill, Sci. Rep., 2015, 5, 16943 CrossRef CAS; (c) M. J. Warren, E. Rauz, H. L. Schubert and J. C. Escalante-Semerena, Nat. Prod. Rep., 2002, 19, 390–412 RSC; (d) A. I. Scott, N. J. Stolowich, J. Wang, O. Gawatz, E. Fridrich and G. Müller, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14316–14319 CrossRef CAS.
- J. S. Lindsey, Synthesis of meso-Substituted Porphyrins, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2000, vol. 1, pp. 45–118 Search PubMed.
- (a) P. Rothemund, J. Am. Chem. Soc., 1935, 57, 2010–2011 CrossRef CAS; (b) A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS.
- M. O. Senge, I. Bischoff, N. Y. Nelson and K. M. Smith, J. Porphyrins Phthalocyanines, 1999, 3, 99–116 CrossRef CAS.
- H. H. Inhoffen, J.-H. Fuhrhop, H. Voigt and H. Brockmann Jr., Liebigs Ann. Chem., 1966, 695, 133–143 CrossRef CAS.
- M. G. H. Vicente and K. M. Smith, Curr. Org. Synth., 2014, 11, 3–28 CrossRef CAS.
- J. L. Sessler, A. Mozaffari and M. R. Johnson, Org. Synth., 1992, 70, 68–78 CrossRef CAS.
- B. Evans, K. M. Smith and J.-H. Fuhrhop, Tetrahedron Lett., 1977, 443–446 CrossRef CAS.
- D. H. R. Barton and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1985, 1098–1100 RSC.
- D. H. R. Barton, J. Kervagoret and S. Z. Zard, Tetrahedron, 1990, 46, 7587–7598 CrossRef CAS.
- N. Ono, H. Miyagawa, T. Ueda, T. Ogawa and H. Tani, J. Chem. Soc., Perkin Trans. 1, 1998, 1595–1602 RSC.
- (a) M. Kielmann and M. O. Senge, Angew. Chem., Int. Ed., 2019, 58, 418–441 CrossRef CAS; (b) M. Roucan, M. Kielmann, S. J. Connon, S. S. R. Bernhard and M. O. Senge, Chem. Commun., 2018, 54, 26–29 RSC.
- S. Ito, T. Murashima, H. Uno and N. Ono, Chem. Commun., 1998, 1661–1662 RSC.
- M. O. Senge and M. Davis, J. Porphyrins Phthalocyanines, 2010, 14, 557–567 CrossRef CAS.
- H. Fischer and W. Gleim, Liebigs Ann., 1936, 521, 157–160 CrossRef.
- P. E. Ellis Jr. and W. A. Langdale, J. Porphyrins Phthalocyanines, 1997, 1, 305–307 CrossRef.
- D. K. Dogutan, M. Ptaszek and J. S. Lindsey, J. Org. Chem., 2007, 72, 5008–5011 CrossRef CAS.
- (a) T. Ema, M. O. Senge, N. Y. Nelson, H. Ogoshi and K. M. Smith, Angew. Chem., Int. Ed. Engl., 1994, 33, 1879–1881 CrossRef; (b) C. Ryppa, M. O. Senge, S. S. Hatscher, E. Kleinpeter, P. Wacker, U. Schilde and A. Wiehe, Chem. – Eur. J., 2005, 11, 3427–3442 CrossRef CAS.
- (a) S. Neya and N. Funasaki, Tetrahedron Lett., 2002, 43, 1057–1058 CrossRef CAS; (b) S. Neya, J. S. Quan, T. Hoshino, M. Hata and N. Funasaki, Tetrahedron Lett., 2004, 45, 8629–8630 CrossRef CAS.
- M. O. Senge, Chem. Commun., 2011, 47, 1943–1960 RSC.
- B. Habermeyer and R. Guilard, Photochem. Photobiol. Sci., 2018, 17, 1675–1690 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS.
- G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000–2015 RSC.
- (a) A. Braun and J. Tscherniak, Ber. Dtsch. Chem. Ges., 1907, 40, 2709–2714 CrossRef CAS; (b) H. de Diesbach and E. von der Weid, Helv. Chim. Acta, 1927, 10, 886–888 CrossRef CAS; (c) R. P. Linstead, Rep. Br. Assoc. Adv. Sci., 1933, 465–466 Search PubMed.
- (a) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS; (b) D. Wohlre, in Phthalocyanine: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH Publications, New York, 1989 Search PubMed; (c) J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281–321 CrossRef CAS; (d) P. Erk and H. Hengelsberg, Phthalocyanine Dyes and Pigments, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2003, vol. 13, pp. 105–149 Search PubMed.
- (a) R. P. Linstead and A. R. Lowe, J. Chem. Soc., 1934, 1022–1027 RSC; (b) C. E. Dent and R. P. Linstead, J. Chem. Soc., 1934, 1027–1031 RSC.
- (a) V. F. Borodkin, Zh. Prikl. Khim., 1958, 31, 813–816 CAS; (b) F. Baumann, B. Bienert, G. Rösch, H. Vollman and W. Wolf, Angew. Chem., Int. Ed. Engl., 1956, 68, 133–150 CrossRef CAS; (c) R. M. Christie and D. D. Deans, J. Chem. Soc., Perkin Trans. 2, 1989, 193–198 RSC.
- A. A. Ryan and M. O. Senge, Photochem. Photobiol. Sci., 2015, 14, 638–660 CrossRef CAS.
- R. Willstätter and W. Mieg, Liebigs Ann. Chem., 1906, 350, 1–47 CrossRef.
- L. T. Nguyen and K. M. Smith, Tetrahedron Lett., 1996, 37, 7177–7180 CrossRef CAS.
- L. T. Nguyen, M. O. Senge and K. M. Smith, J. Org. Chem., 1996, 61, 998–1003 CrossRef CAS.
- K. M. Smith, Strategies for the Synthesis of Octaalkylporphyrin Systems, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2000, vol. 1, pp. 1–43 Search PubMed.
- J. A. S. Cavaleiro, A. M. d’A. Rocha Gonsalves, G. W. Kenner and K. M. Smith, J. Chem. Soc., Perkin Trans. 1, 1973, 2471–2478 RSC.
- C. H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427–11440 CrossRef CAS.
- G. P. Arsenault, E. Bullock and S. F. MacDonald, J. Am. Chem. Soc., 1960, 82, 4384–4389 CrossRef CAS.
- T. D. Lash, J. Porphyrins Phthalocyanines, 2016, 20, 855–888 CrossRef CAS.
- (a) A. M. d'A. Rocha Gonsalves, G. W. Kenner and K. M. Smith, Tetrahedron Lett., 1972, 13, 2203–2206 CrossRef; (b) A. Boudif and M. Momenteau, J. Chem. Soc., Chem. Commun., 1994, 2069–2070 RSC.
- J. L. Sessler, M. R. Johnson and V. Lynch, J. Org. Chem., 1987, 52, 4394–4397 CrossRef CAS.
- T. D. Lash, J. Porphyrins Phthalocyanines, 1997, 1, 29–44 CrossRef CAS.
- T. D. Lash, Eur. J. Org. Chem., 2007, 5461–5481 CrossRef CAS.
- A. H. Corwin and E. C. Coolidge, J. Am. Chem. Soc., 1952, 74, 5196–5197 CrossRef CAS.
- J. Ellis, A. H. Jackson, A. C. Jain and G. W. Kenner, J. Chem. Soc., 1964, 1935–1949 RSC.
- P. S. Clezy, Aust. J. Chem., 1991, 44, 1163–1193 CrossRef CAS.
- K. M. Smith, Cyclizations of a,c-Biladiene Salts to Give Porphyrins and Their Derivatives, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2000, vol. 1, pp. 119–148 Search PubMed.
- (a) A. W. Johnson and I. T. Kay, J. Chem. Soc., 1961, 2418–2423 RSC; (b) P. S. Clezy and A. Liepa, Aust. J. Chem., 1971, 24, 1027–1040 CrossRef CAS; (c) J. A. P. B. de Almeida, G. W. Kenner, J. Rimmer and K. M. Smith, Tetrahedron, 1976, 32, 1793–1799 CrossRef.
- K. M. Smith and G. W. Craig, J. Org. Chem., 1983, 48, 4302–4306 CrossRef CAS.
- K. M. Smith and O. M. Minnetian, J. Chem. Soc., Perkin Trans. 1, 1986, 277–280 RSC.
- D. Jeyakumar, K. M. Snow and K. M. Smith, J. Am. Chem. Soc., 1988, 110, 8562–8564 CrossRef CAS.
- A. D. Adler, F. R. Longo and W. Shergalis, J. Am. Chem. Soc., 1964, 86, 3145–3149 CrossRef CAS.
- H. Volz, M. Hassler and H. Schäffer, Z. Naturforsch., 1986, 41b, 1265–1272 CAS.
- M. J. Gunter and L. N. Mander, J. Org. Chem., 1981, 46, 4792–4795 CrossRef CAS.
- C. Brückner, J. J. Posakony, C. K. Johnson, R. W. Boyle, B. R. James and D. Dolphin, J. Porphyrins Phthalocyanines, 1998, 2, 455–465 CrossRef.
- A. Wiehe, Y. M. Shaker, J. C. Brandt, S. Mebs and M. O. Senge, Tetrahedron, 2005, 61, 5535–5564 CrossRef CAS.
- R. P. Briñas and C. Brückner, Tetrahedron, 2002, 58, 4375–4381 CrossRef.
- S. Hatscher and M. O. Senge, Tetrahedron Lett., 2003, 44, 157–160 CrossRef CAS.
- D. M. Wallace, S. H. Leung, M. O. Senge and K. M. Smith, J. Org. Chem., 1993, 58, 7245–7257 CrossRef CAS.
- M. Barbero, S. Cadamuro, I. Degani, R. Fochi, A. Gatti and V. Regondi, J. Org. Chem., 1988, 53, 2245–2250 CrossRef CAS.
- C.-H. Lee, F. Li, K. Iwamoto, J. Dadok, A. A. Bothner-By and J. S. Lindsey, Tetrahedron, 1995, 51, 11645–11672 CrossRef CAS.
- (a) M. O. Senge, S. S. Hatscher, A. Wiehe, K. Dahms and A. Kelling, J. Am. Chem. Soc., 2004, 126, 13634–13635 CrossRef CAS; (b) A. Balakumar, K. Muthukumaran and J. S. Lindsey, J. Org. Chem., 2004, 69, 5112–5115 CrossRef CAS.
- (a) M. O. Senge and J. Richter, J. Porphyrins Phthalocyanines, 2004, 8, 934–953 CrossRef CAS; (b) S. Horn, K. Dahms and M. O. Senge, J. Porphyrins Phthalocyanines, 2008, 12, 1053–1077 CrossRef CAS.
- D. Fan, M. Taniguchi, Z. Yao, S. Dhanalekshmi and J. S. Lindsey, Tetrahedron, 2005, 61, 10291–10302 CrossRef CAS.
- M. Taniguchi, A. Balakumar, D. Fan, B. E. McDowell and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2005, 9, 554–574 CrossRef CAS.
- D. K. Dogutan, S. H. H. Zaidi, P. Thamyongkit and J. S. Lindsey, J. Org. Chem., 2007, 72, 7701–7714 CrossRef CAS.
- (a) C. J. Dutton, C. J. R. Fookes and A. R. Battersby, J. Chem. Soc., Chem. Commun., 1983, 1237–1238 RSC; (b) A. R. Battersby, C. J. Dutton and C. J. R. Fookes, J. Chem. Soc., Perkin Trans. 1, 1988, 1569–1576 RSC.
- (a) P. J. Harrison, Z.-C. Sheng, C. J. R. Fookes and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1987, 1667–1678 RSC; (b) A. R. Battersby, C. J. Dutton, C. J. R. Fookes and S. P. D. Turner, J. Chem. Soc., Perkin Trans. 1, 1988, 1557–1567 RSC.
- (a) For an alternative synthesis of (±)-bonellin dimethyl ester, see: F.-P. Montforts and U. M. Schwartz, Liebigs Ann. Chem., 1991, 709–725 CrossRef CAS; (b) D. Kusch, E. Töllner, A. Lincke and F.-P. Montforts, Angew. Chem., Int. Ed. Engl., 1995, 34, 784–787 CrossRef.
- (a) F. Baltzer, Pubbl. Stn. Zool. Napoli, 1925, 6, 223–287 Search PubMed; (b) F. B. D. De Silva, Varkbl. Biol., 1934, 15, 221 Search PubMed.
- (a) J. Micklefield, M. Beckmann, R. L. Mackman, M. H. Block, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1997, 2123–2138 RSC; (b) R. L. Mackman, J. Micklefiedl, M. H. Block, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1997, 2111–2122 RSC; (c) C. J. Aucken, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1997, 2099–2110 RSC.
- (a) W. Wu and C. K. Chang, J. Am. Chem. Soc., 1987, 109, 3149–3150 CrossRef CAS; (b) C. K. Chang and W. Wu, J. Biol. Chem., 1986, 261, 8593–8596 CrossRef CAS.
- C. Brückner, Acc. Chem. Res., 2016, 49, 1080–1092 CrossRef.
- M. R. Prinsep, F. R. Caplan, R. E. Moore, G. M. L. Patterson and C. D. Smith, J. Am. Chem. Soc., 1992, 114, 385–387 CrossRef CAS.
- T. G. Minehan and Y. Kishi, Angew. Chem., Int. Ed., 1999, 38, 923–925 CrossRef CAS.
- T. G. Minehan, L. Cook-Blumberg, Y. Kishi, M. R. Prinsep and R. E. Moore, Angew. Chem., Int. Ed., 1999, 38, 926–928 CrossRef CAS.
- W. Wang and Y. Kishi, Org. Lett., 1999, 1, 1129–1132 CrossRef CAS.
- (a) A. Eschenmoser, Q. Rev., Chem. Soc., 1970, 24, 366–415 RSC; (b) M. Roth, E. Dubs, E. Götschi and A. Eschenmoser, Helv. Chim. Acta, 1971, 54, 710–734 CrossRef CAS; (c) E. Götschi, W. Hunkeler, H.-J. Wild, P. Schneider, W. Fuhrer, J. Gleason and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1973, 12, 910–912 CrossRef.
- (a) W. G. O’Neal, W. P. Roberts, I. Ghosh and P. A. Jacobi, J. Org. Chem., 2005, 70, 7243–7251 CrossRef; (b) W. G. O’Neal, W. P. Roberts, I. Ghosh, H. Wang and P. A. Jacobi, J. Org. Chem., 2006, 71, 3472–3480 CrossRef.
- W. G. O’Neal and P. A. Jacobi, J. Am. Chem. Soc., 2008, 130, 1102–1108 CrossRef.
- (a) F.-P. Montforts, B. Gerlach and F. Hoper, Chem. Rev., 1994, 94, 327–347 CrossRef CAS; (b) F. P. Montforts and U. M. Schwartz, Liebigs Ann. Chem., 1985, 1228–1253 CrossRef CAS.
- C. Muthiah, M. Ptaszek, T. M. Nguyen, K. M. Flack and J. S. Lindsey, J. Org. Chem., 2007, 72, 7736–7749 CrossRef CAS.
- For a first-rate, comprehensive, account of this topic, see: J. S. Lindsey, Chem. Rev., 2015, 115, 6534–6620 CrossRef CAS.
- M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344–535 CrossRef CAS.
- (a) K. M. Smith, Acc. Chem. Res., 1979, 12, 374–381 CrossRef CAS; (b) V. Yu Pavlov, Russ. J. Org. Chem., 2007, 43, 1–34 CrossRef; (c) J. M. O’Brien, E. Sitte, K. J. Flanagan, H. Kühner, L. J. Hallen, D. Gibbons and M. O. Senge, J. Org. Chem., 2019, 84, 6158–6173 CrossRef; (d) E. Sitte and M. O. Senge, Eur. J. Org. Chem., 2020, 3171–3191 CrossRef CAS.
- (a) A. C. Tomé, P. S. S. Lacerda, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Chem. Commun., 1997, 1199–1200 RSC; (b) A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 408–414 CrossRef.
- (a) R. Bonnett, D. Dolphin, A. W. Johnson, D. Oldfield and G. F. Stephenson, Proc. Chem. Soc., 1964, 371–372 CAS; (b) H. H. Inhoffen and W. Nolte, Tetrahedron Lett., 1967, 23, 2185–2187 CrossRef.
- (a) M. G. H. Vicente, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2000, vol. 1, pp. 149–199 Search PubMed; (b) L. Jaquinod, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, USA, 2000, vol. 1, pp. 201–237 Search PubMed.
- (a) N. N. Sergeeva, M. O. Senge and A. Ryan, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 3, pp. 325–366 Search PubMed; (b) S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS.
- L. Jiao, E. Hao, F. R. Fronczek, M. G. H. Vicente and K. M. Smith, Chem. Commun., 2006, 3900–3902 RSC.
- (a) M. J. Crossley, P. L. Burn, S. S. Chew, F. B. Cuttance and I. A. Newsom, J. Chem. Soc., Chem. Commun., 1991, 1564–1566 RSC; (b) M. J. Crossley, P. L. Burn, S. J. Langford, S. M. Pyke and A. G. Stark, J. Chem. Soc., Chem. Commun., 1991, 1567–1568 RSC.
- (a) H. Ali and J. E. van Lier, Tetrahedron, 1994, 50, 11933–11944 CrossRef CAS; (b) V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105–1111 CrossRef CAS; (c) H. L. Andersson, Chem. Commun., 1999, 2323–2330 RSC.
- (a) M. C. O’Sullivan, J. F. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O’Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef; (b) V. Kondratuk, J. K. Sprafke, M. C. O’Sullivan, L. M. A. Perdigao, A. Saywell, M. Malfois, J. N. O’Shea, P. H. Beton, A. L. Thompson and H. L. Anderson, Chem. – Eur. J., 2014, 20, 12826–12834 CrossRef.
- (a) W. W. Kalisch and M. O. Senge, Angew. Chem., Int. Ed., 1998, 37, 1107–1109 CrossRef CAS; (b) M. O. Senge, Acc. Chem. Res., 2005, 38, 733–743 CrossRef CAS.
- (a) X. Feng, I. Bischoff and M. O. Senge, J. Org. Chem., 2001, 66, 8693–8700 CrossRef CAS; (b) A. Wiehe, C. Ryppa and M. O. Senge, Org. Lett., 2002, 4, 3807–3809 CrossRef CAS; (c) X. Feng and M. O. Senge, Tetrahedron, 2000, 56, 587–590 CrossRef CAS; (d) I. Bischoff, X. Feng and M. O. Senge, Tetrahedron, 2001, 57, 5573–5583 CrossRef CAS; (e) M. O. Senge and X. Feng, J. Chem. Soc., Perkin Trans. 1, 2000, 3615–3621 RSC; (f) M. O. Senge, W. W. Kalisch and I. Bischoff, Chem. – Eur. J., 2000, 6, 2721–2738 CrossRef CAS; (g) N. N. Sergeeva, Y. M. Shaker, E. M. Finnigan, T. McCabe and M. O. Senge, Tetrahedron, 2007, 63, 12454–12464 CrossRef CAS.
- B. Krattinger and H. J. Callot, Eur. J. Org. Chem., 1999, 1857–1867 CrossRef CAS.
- (a) M. O. Senge and I. Bischoff, Eur. J. Org. Chem., 2001, 1735–1751 CrossRef CAS; (b) M. O. Senge, Y. M. Shaker, M. Pintea, C. Ryppa, S. S. Hatscher, A. Ryan and Y. Sergeeva, Eur. J. Org. Chem., 2010, 237–258 CrossRef CAS; (c) M. O. Senge, M. W. Renner, W. W. Kalisch and J. Fajer, J. Chem. Soc., Dalton Trans., 2000, 381–385 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- (a) K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem. – Eur. J., 2013, 19, 17075–17081 CrossRef CAS; (b) A. Meindl, S. Plunkett, A. A. Ryan, K. J. Flanagan, S. Callaghan and M. O. Senge, Eur. J. Org. Chem., 2017, 3565–3583 CrossRef CAS.
- (a) H. Shinokubo and A. Osuka, Chem. Commun., 2009, 1011–1021 RSC; (b) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- Y. Nakammura, N. Aratani, H. Shinokubo, A. Takagi, T. Kawai, T. Matsumoto, Z. S. Yoon, D. Y. Kim, T. K. Ahn, D. Kim, A. Muranaka, N. Kobayashi and A. Osuka, J. Am. Chem. Soc., 2006, 128, 4119–4127 CrossRef.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef.
- A. Wiengarten, K. Seufert, W. Auwärter, D. Ecija, K. Diller, F. Allegretti, F. Bischoff, S. Fischer, D. A. Duncan, A. C. Papageorgiou, F. Klappenberger, R. G. Acres, T. H. Ngo and J. V. Barth, J. Am. Chem. Soc., 2014, 136, 9346–9354 CrossRef CAS.
- C. P. Gros, J.-M. Barbe, E. Espinosa and R. Guilard, Angew. Chem., Int. Ed., 2006, 45, 5642–5645 CrossRef CAS.
- (a) E. Vogel, Pure Appl. Chem., 1993, 65, 143–152 CAS; (b) E. Vogel, Angew. Chem., Int. Ed., 2011, 50, 4278–4287 CrossRef CAS.
- (a) T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS; (b) M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef.
- (a) P. J. Chmielewski and L. Latos-Grażyński, Coord. Chem. Rev., 2005, 249, 2510–2533 CrossRef CAS; (b) B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS; (c) B. Szyszko and L. Latos-Grażyński, Chem. Soc. Rev., 2015, 44, 3588–3616 RSC; (d) M. O. Senge and N. N. Sergeeva, Angew. Chem., Int. Ed., 2006, 45, 7492–7495 CrossRef CAS.
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1986, 25, 257–259 CrossRef.
- D. Sánchez-García and J. L. Sessler, Chem. Soc. Rev., 2008, 37, 215–232 RSC.
- (a) H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS; (b) P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- Historically, the existence of such compounds was proposed by Linus Pauling and putatively observed by Melvin Calvin in the 1940s; M. O. Senge, Angew. Chem., Int. Ed., 2011, 50, 4272–4277 CrossRef CAS.
- A. Srinivasan and H. Furuta, Acc. Chem. Res., 2005, 38, 10–20 CrossRef CAS.
- (a) Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS; (b) R. Paolesse, L. Jaquinod, D. J. Nurco, S. Mini, F. Sagone, T. Boschi and K. M. Smith, Chem. Commun., 1999, 1307–1308 RSC.
- (a) I. Aviv-Harel and Z. Gross, Chem. – Eur. J., 2009, 15, 8382–8394 CrossRef CAS; (b) D. T. Gryko, Eur. J. Org. Chem., 2002, 1735–1743 CrossRef CAS; (c) I. Aviv-Hare and Z. Gross, Coord. Chem. Rev., 2011, 255, 717–736 CrossRef.
- (a) H. J. Callot and T. Tschamber, Tetrahedron Lett., 1974, 15, 3155–3158 CrossRef; (b) H. J. Callot and T. Tschamber, J. Am. Chem. Soc., 1975, 97, 6175–6178 CrossRef CAS.
- T. Wessel, B. Franck, M. Möller, U. Rodewald and M. Läge, Angew. Chem., Int. Ed. Engl., 1993, 32, 1148–1151 CrossRef.
- C. Eickmeier and B. Franck, Angew. Chem., Int. Ed. Engl., 1997, 36, 2213–2215 CrossRef CAS.
- J. L. Sessler and J. M. Davis, Acc. Chem. Res., 2001, 34, 989–997 CrossRef CAS.
- V. J. Bauer, D. L. J. Clive, D. Dolphin, J. B. Paine III, F. L. Harris, M. M. King, J. Loder, S. W. C. Wang and R. B. Woodward, J. Am. Chem. Soc., 1983, 105, 6429–6436 CrossRef CAS.
- J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef CAS.
- (a) P. A. Gale, J. L. Sessler and V. Král, Chem. Commun., 1998, 1–8 RSC; (b) P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184–2185 CrossRef.
- For a recent overview on this area see a special edition in Chem. Rev.: J. L. Sessler, Z. Gross and H. Furuta, Chem. Rev., 2017, 117, 2201–2202 CrossRef CAS.
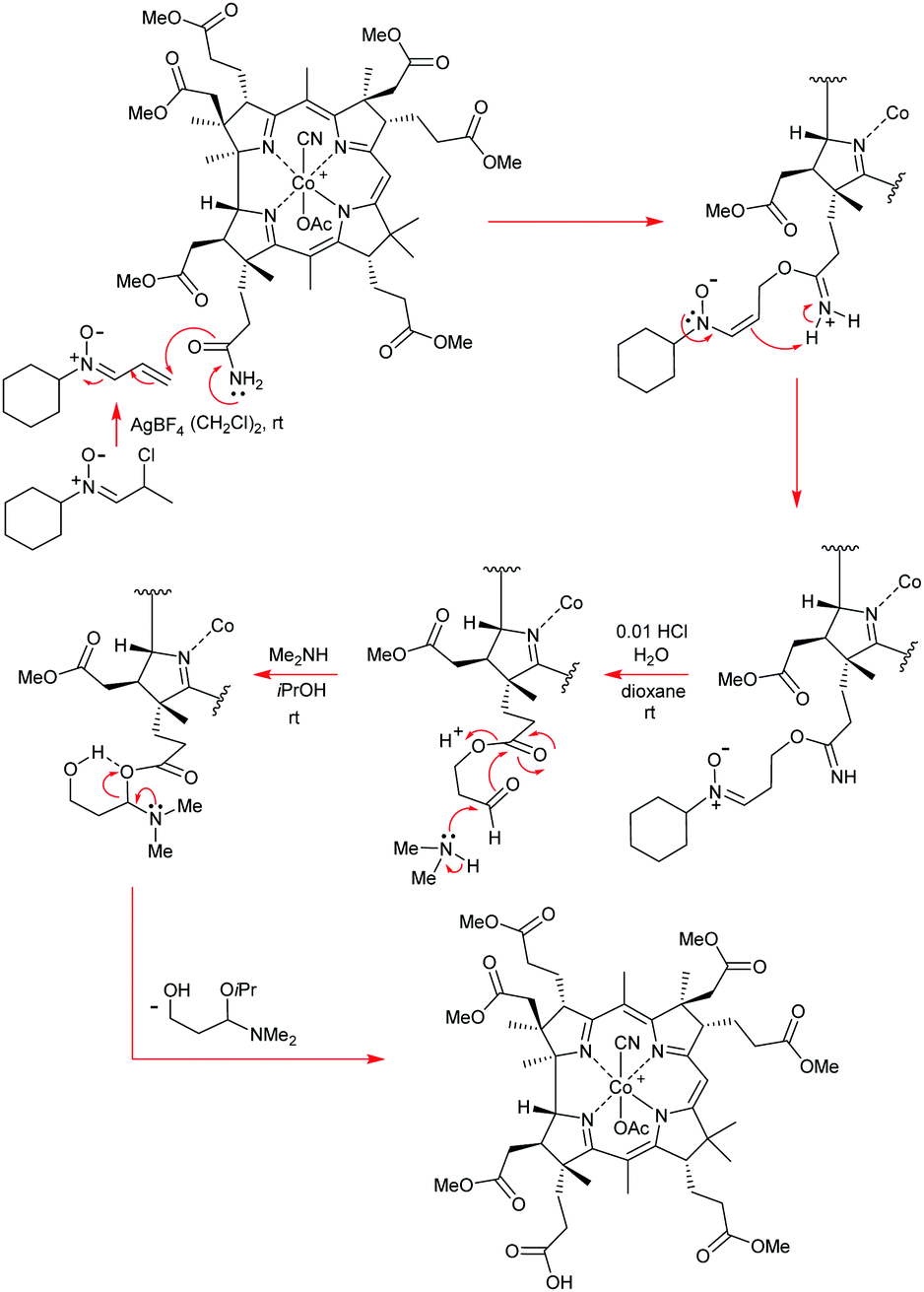
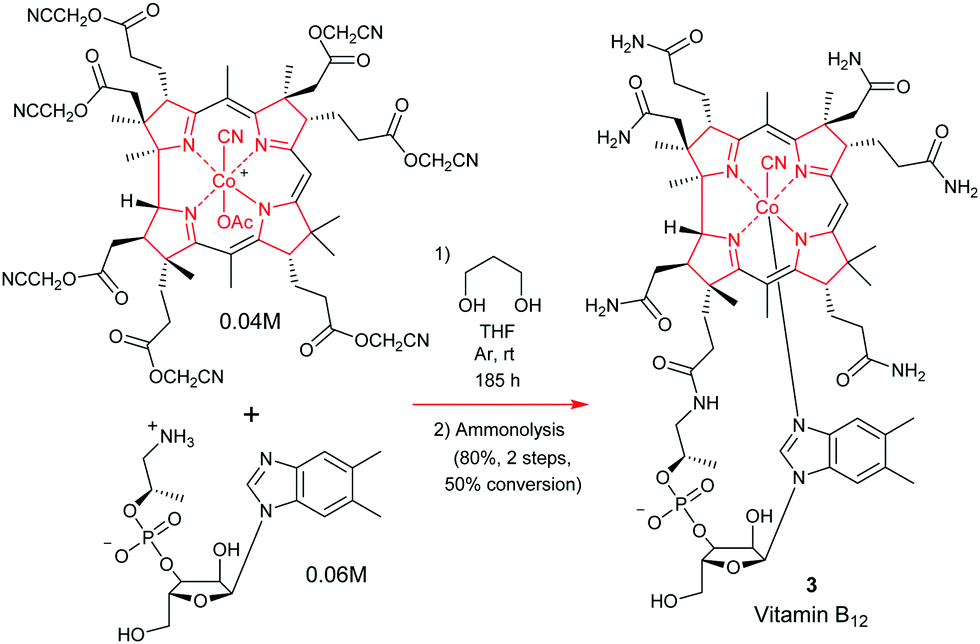
| This journal is © The Royal Society of Chemistry 2021 |