
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Quantum tunneling dynamical behaviour on weakly bound complexes: the case of a CO2–N2 dimer
Miguel
Lara-Moreno
a,
Thierry
Stoecklin
a,
Philippe
Halvick
*a and
Majdi
Hochlaf
b
aUniversité de Bordeaux, ISM, CNRS UMR 5255, 33405, Talence, France. E-mail: philippe.halvick@u-bordeaux.fr
bUniversité Gustave Eiffel, COSYS/LISIS, 5 Bd Descartes, 77454 Champs-sur-Marne, France
First published on 23rd April 2021
Abstract
Correction for ‘Quantum tunneling dynamical behaviour on weakly bound complexes: the case of a CO2–N2 dimer’ by Miguel Lara-Moreno et al., Phys. Chem. Chem. Phys., 2019, 21, 3550–3557, DOI: 10.1039/c8cp04465a.
About one year after the publication of our original article1 reporting the first calculations of the rovibrational bound states of the van der Waals system CO2–N2, the lowest intermolecular bending frequency v2 was measured by Barclay et al.2 They found a large discrepancy between the calculated and measured frequencies, the calculated frequency being more than two times larger than the measured one. This discrepancy motivated Quintas-Sánchez et al.3 to perform new calculations, using a new accurate potential energy surface (PES). Conversely to the original article, their calculation of the lowest intermolecular bending frequency is in excellent agreement with the experimental one. This raised questions on the accuracy of our calculations. The PES we have used (PES-1) is expected to be slightly less accurate than the one (PES-2) used by Quintas-Sánchez et al. Both PESs have been generated by the explicitly-correlated coupled-cluster theory (CCSD(T)-F12), with a quadruple-zeta basis set for PES-1 and with an extrapolation based on triple- and quadruple-zeta basis sets for PES-2. The two PESs are quite similar. The comparison of the energies of the stationary points shows differences of a few cm−1. Since it was difficult to attribute the large error on frequency to inaccuracies of PES-1, we turned our attention to the method used for the rovibrational states calculations. Quintas-Sánchez et al.'s and our methods are based on a Hamiltonian including all the allowed motions of two rigid rotors. Even if the technical details of the numerical methods used to solve the Hamiltonian equation are different, both methods are expected to produce accurate results as long as the rovibrational basis set used to represent the Hamiltonian is large enough. Therefore we could not identify a reasonable cause for the large error on the intermolecular bending frequency, leading us to consider the possibility of an error in the solving process of the Hamiltonian equation. After a meticulous screening of the source code, we found an error which is triggered when both monomers have a center of symmetry. This programming error suppresses part of the angular contributions of the PES. After rectifying this error, we have computed a new set of energy levels and wavefunctions, using exactly the same method and the same parameters as defined previously in the original article. Nevertheless, a supplementary check of the convergence with respect to the angular basis set has been done. We use 15 rotational functions for CO2 and 8 for N2. Since we build a Hamiltonian matrix for each irrep of the G8 group, then only symmetric or antisymmetric angular functions are necessary in each Hamiltonian matrix representation. Thus the maximum values of the angular quantum numbers are 29 and 15 for CO2 and N2 respectively. However, Quintas-Sánchez et al. have used the maximum value of 37 in their calculations, suggesting that our angular basis set was too small. A new calculation with 19 angular functions both for CO2 and N2 has shown that the lowest rovibrational energies are changed by less than ∼0.01 cm−1.
We report in Table 1 the new values of the energies which replace the values previously published in the original article. The energies of the ground state and the levels involving only excitation of the v1 and v3 modes are lowered by 2 or 3 cm−1. The excited levels of the v4 mode are lowered by ∼6 cm−1 or more, and it is for the v2 mode that the largest decrease is observed, about 24 cm−1 per quantum. A high density of rovibrational levels is noticed. The number of levels for which we have been able to assign quantum numbers is much smaller than in the original work. Indeed, since there are now more coupling terms arising from PES-1, the nodal structures of highly excited states are much more intricate and therefore the contour plots of the wavefunctions allow us to assign quantum numbers only for a limited set of levels. The four van der Waals fundamental frequencies can be obtained from the rovibrational energies in Table 1. They are listed in Table 2, along with the calculated frequencies of Quintas-Sánchez et al. and the measured frequency.2 Our new calculated frequencies are in good agreement with those of Quintas-Sánchez et al., except for the v2 frequency where a significant difference of ∼4 cm−1 is observed. In order to check this discrepancy, we have recalculated the rovibrational energies for PES-1 with the WAVR4 code4 and a convergence threshold of 0.1 cm−1. As shown in Table 2, there is an excellent agreement between the results from our code and those of WAVR4. The difference found between our v2 frequency and the one of Quintas-Sánchez et al. can then be attributed unequivocally to inaccuracies of PES-1.
| v 1 | v 2 | v 3 | v 4 | ε | Energy | Γ i | |ΔE| |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | + | −227.389 |

|
<0.001 |

|
|||||||
| 0 | 1 | 0 | 0 | + | −202.343 |

|
<0.001 |

|
|||||||
| 1 | 0 | 0 | 0 | − | −195.032 |
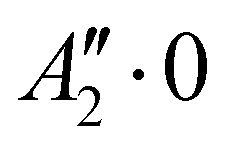
|
0.006 |
| −195.026 |
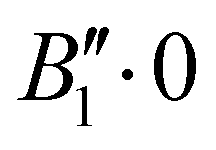
|
||||||
| 0 | 2 | 0 | 0 | + | −186.835 |

|
<0.001 |
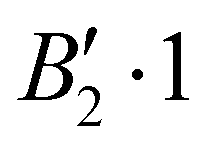
|
|||||||
| 0 | 0 | 1 | 0 | + | −180.619 |
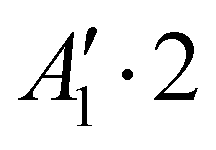
|
0.005 |
| −180.614 |

|
||||||
| 1 | 1 | 0 | 0 | − | −170.900 |
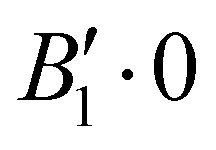
|
0.003 |
| −170.897 |
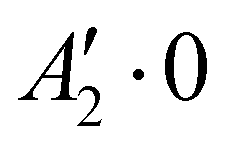
|
||||||
| 0 | 3 | 0 | 0 | + | −170.018 |

|
0.004 |
| −170.015 |
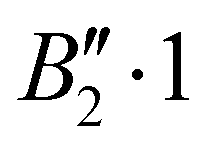
|
||||||
| 0 | 0 | 0 | 1 | + | −167.701 |

|
<0.001 |

|
|||||||
| 2 | 0 | 0 | 0 | + | −165.583 |
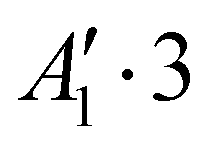
|
0.132 |
| −165.451 |
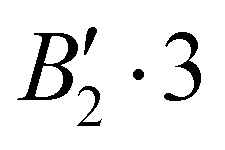
|
||||||
| 0 | 1 | 1 | 0 | + | −158.474 |

|
0.003 |
| −158.472 |

|
||||||
| 1 | 2 | 0 | 0 | − | −155.267 |

|
0.019 |
| −155.249 |

|
||||||
| 1 | 0 | 1 | 0 | − | −152.142 |

|
0.268 |
| −151.874 |

|
||||||
| 0 | 2 | 1 | 0 | + | −151.558 |

|
0.008 |
| −151.550 |
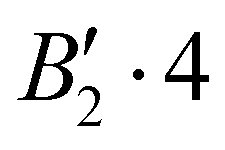
|
||||||
| 0 | 0 | 0 | 2 | + | −147.632 |

|
0.012 |
| −147.620 |

|
||||||
| 2 | 1 | 0 | 0 | + | −142.852 |

|
0.102 |
| −142.751 |
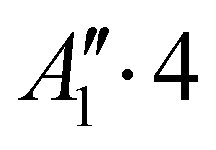
|
||||||
| 1 | 0 | 0 | 1 | − | −140.883 |

|
0.090 |
| −140.792 |

|
||||||
| 0 | 0 | 2 | 0 | + | −140.203 |

|
0.013 |
| −140.191 |
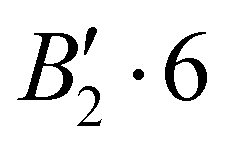
|
||||||
| 3 | 0 | 0 | 0 | − | −139.665 |

|
1.417 |
| −138.248 |

|
In Tables 2 and 3 we report the corrected values of the rotational constants and selected rotational transitions, respectively. As can be seen, the corrected calculated values are closer to the measured values than the previous calculated values. However, the agreement with the measured values is not as good as the one obtained by Quintas-Sánchez et al. This confirms the higher accuracy of PES-2 developed by the latter authors.
 . Relative errors with respect to experimental data are given in percent inside parentheses
. Relative errors with respect to experimental data are given in percent inside parentheses
|
|
Exp.5 | Corrected | Original1 | Quintas-Sánchez et al.3 |
|---|---|---|---|---|
| 101–000 | — | 3808.703 | 3880.484 | 3805.214 |
| 202–101 | 7608.377 | 7648.669 (0.53) | 7527.429 (1.06) | 7602.542 (0.08) |
| 303–202 | 11388.436 | 11461.285 (0.64) | 11277.417 (0.98) | 11384.128 (0.04) |
| 404–303 | 15148.195 | 15294.977 (0.97) | 15003.717 (0.95) | 15142.260 (0.04) |
| 505–404 | 18877.125 | 18965.259 (0.47) | 18698.230 (0.95) | 18869.660 (0.04) |

Rovibrational states localised in the region of the secondary minimum were identified and discussed in the original article. In the present correction, we re-investigate this issue. Selected plots of the rovibrational wavefunctions are presented in Fig. 1. The ground state  and the first excited state
and the first excited state 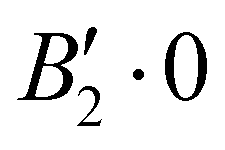 are quasi-degenerate and arise from the symmetric and antisymmetric combinations of the wavefunctions localized in the two identical potential wells connected by the exchange of the N atoms. In contrast to what we found previously,1 the state
are quasi-degenerate and arise from the symmetric and antisymmetric combinations of the wavefunctions localized in the two identical potential wells connected by the exchange of the N atoms. In contrast to what we found previously,1 the state  is now free of any nodal plane and the state
is now free of any nodal plane and the state  has a single nodal plane.
has a single nodal plane.
 | ||
| Fig. 1 2D contour plots of the wavefunctions for selected states. For each point (θ1, θ2), the wavefunction has been calculated by taking ϕ = 90° and the value of R which maximizes the probability density. | ||
A second difference with the original work is related to the localization of some wavefunctions in the region of the secondary minimum. PES-1 has a very shallow secondary minimum, located at θ1 = 0° or 180° and θ2 = 90°. This minimum does not exist in PES-2; instead there is a saddle point. Since the secondary minimum is very flat, with a well depth of only 0.78 cm−1, it is not surprising that a change of basis set is sufficient to transform it into a saddle point. As PES-2 was developed on the basis of more accurate ab initio data than PES-1, we can assume that this stationary point is a saddle point and that the secondary minimum is an artifact of the ab initio model used for PES-1. Nevertheless, a systematic ab initio investigation with a series of growing basis sets could be necessary to determine with confidence the properties of this stationary point. Fig. 1 shows several examples of wavefunctions which have a significant probability density in the region of the secondary minimum, but in contrast with the wavefunctions presented in the original article, they have now also a significant probability density in the region of the global minimum. Fig. 1 replaces Fig. 5 of the original article. The other conclusions and findings of the original paper remain unchanged.
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
References
- M. Lara-Moreno, T. Stoecklin, P. Halvick and M. Hochlaf, Quantum tunneling dynamical behaviour on weakly bound complexes: the case of a CO2–N2 dimer, Phys. Chem. Chem. Phys., 2019, 21, 3550–3557 RSC.
- A. J. Barclay, A. R. W. McKellar and N. Moazzen-Ahmadi, Spectra of CO2–N2 dimer in the 4.2 μm region: Symmetry breaking of the intramolecular CO2 bend, the intermolecular bend, and higher K-values for the fundamental, J. Chem. Phys., 2020, 153(1), 014303 CrossRef CAS PubMed.
- E. Quintas-Sánchez, R. Dawes, X.-G. Wang and T. Carrington, Computational study of the rovibrational spectrum of CO2–N2, Phys. Chem. Chem. Phys., 2020, 22(39), 22674–22683 RSC.
- I. N. Kozin, M. M. Law, J. Tennyson and J. M. Hutson, New vibration–rotation code for tetraatomic molecules exhibiting wide-amplitude motion: WAVR4, Comput. Phys. Commun., 2004, 163(2), 117–131 CrossRef CAS.
- D. J. Frohman, E. S. Contreras, R. S. Firestone, S. E. Novick and W. Klemperer, Microwave spectra, structure, and dynamics of the weakly bound complex, N2–CO2, J. Chem. Phys., 2010, 133(24), 244303 CrossRef PubMed.
| This journal is © the Owner Societies 2021 |