
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the effect of metal loading on the reducibility and redox chemistry of ceria supported Pd catalysts†
Adam H.
Clark‡
 a,
Huw R.
Marchbank§
a,
David
Thompsett
b,
Janet M.
Fisher
b,
Alessandro
Longo
cd,
Kevin A.
Beyer
e,
Timothy I.
Hyde
b and
Gopinathan
Sankar
*a
a,
Huw R.
Marchbank§
a,
David
Thompsett
b,
Janet M.
Fisher
b,
Alessandro
Longo
cd,
Kevin A.
Beyer
e,
Timothy I.
Hyde
b and
Gopinathan
Sankar
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: g.sankar@ucl.ac.uk
bJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading RG4 9NH, UK
cI20, ESRF-The European Synchrotron, CS40220, 38043 Grenoble, Cedex 9, France
dCNR-ISMN, Consiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati, Via Ugo La Malfa 153, 90146 Palermo, Italy
eX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, USA
First published on 16th December 2021
Abstract
The effect of Pd loading on the redox characteristics of a ceria support was examined using in situ Pd K-edge XAS, Ce L3-edge XAS and in situ X-ray diffraction techniques. Analysis of the data obtained from these techniques indicates that the onset temperature for the partial reduction of Ce(IV) to Ce(III), by exposure to H2, varies inversely with the loading of Pd. Whilst the onset and completion temperatures of the reduction of Ce(IV) to Ce(III) are different, both samples yield the same maximal fraction of Ce(III) formation independent of Pd loading. Furthermore, the partial reduction of Ce is found to be concurrent with the reduction of PdO and demonstrated that the presence of metallic Pd is necessary for the reduction of the CeO2 support. Upon passivation by room temperature oxidation, a full oxidation of the reduced ceria support was observed. However, only a mild surface oxidation of Pd was identified. The mild passivation of the Pd is found to lead to a highly reactive sample upon a second reduction by H2. The onset of the reduction of Pd and Ce has been demonstrated to be independent of the Pd loading after a mild passivation with both samples exhibiting near room temperature reduction in the presence of H2.
Introduction
Ceria (CeO2) has extensive uses in the field of catalysis1 as well as other important industrial applications.2–5 Platinum group metals (PGMs)6 and transition metals7 can be loaded onto a ceria support either separately, or in conjunction with other metals e.g. bimetallic systems. Once dispersed, the metals appear as either single particles, or atomic clusters.8–10 The catalytic properties of the material change as a function of the particle or cluster size. As the particle volume decreases, the surface area increases, resulting in an increase in their efficiency.11 An added benefit of a reduced volume is reduced cost.12 Additionally, the combination of PGMs and their support can improve the oxygen storage capacity (OSC) and redox operation.13,14 The OSC of the ceria combined with the catalytic properties of the noble metal nanoparticles makes for a highly useful material.15 It has been suggested that the support enhances the activity of noble metal catalysis due to strong metal support interactions (SMSIs).16,17 There are several ways by which the SMSI effect can be explained e.g. formation of PGM–O–Ce bonds,18 alloy formation,19 diffusion of the PGM into the support and/or encapsulation of the PGM by the support,20 and partial reduction of ceria by the PGM creating bridging hydroxyl moieties.21 In general, the reduction of an oxide support changes the chemical properties of the noble metals dispersed on its surface. In addition to being a reversible process,22 the reducibility of the support is improved. H2 dissociates into atomic hydrogen by the noble metal while nearby oxygen atoms located on the support surface are removed. The localization of oxygen vacancy electrons results in the reduction of Ce(IV) to Ce(III).23–25 Charge transfer can occur via electronic interactions between the respective components in the system.26–29Chemical interactions, such as redox reactions, complicate these interactions and often influence the catalytic performance and reducibility of the loaded metal.30–32 Among the various PGMs, the use of Pd on ceria has been well documented such as in automotive exhaust catalysis,33 steam reforming,34 methanol synthesis,35 abatement of methane,36 C–H bond activation37 and in various catalytic oxidation processes.38–41 In order to understand the reduction properties of palladium ions and their influence on the reactivity of a ceria support, it is necessary to study both the short and long range structures of the system and to obtain element specific information probing the supported Pd metal ions. This is accomplished by combining suitable, complimentary structural methods. This includes X-ray absorption spectroscopy (XAS), which comprises X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), an element specific technique and useful to determine short-range geometric and electronic structural information. On the other hand, X-ray diffraction (XRD) is a powerful technique in determining the long-range structural order, respectively, present in a given system, but is less informative on dispersed metal ions and nano-crystalline metal particles which are present in small concentrations. Therefore, it is ideal to use a combination of these methods to probe the structure of a catalytic system to determine how the metal ions and supports dynamically interact in catalysis. Here we report the use of in situ Pd K-edge, Ce L3-edge, and XRD techniques to determine the effect of the concentration of palladium on the reactivity of the Pd/CeO2 system.
Experimental
The high-surface area ceria support was obtained from Rhodia-Solvay. The surface area of the support material was measured using BET analysis of 130 m2 g−1. The supported Pd catalysts were made by an incipient wetness impregnation method. The appropriate amounts of Pd nitrate solution (Johnson Matthey) were used to load palladium onto the ceria support. Materials were dried at 105 °C and calcined at 500 °C and these as-prepared catalysts were used for further in situ and ex situ characterization studies. XRD performed on the as-received samples is shown in Fig. S1 in the ESI† demonstrating phase purity and an average ceria crystallite size of approximately 5 nm and is reported in Table S1 (ESI†) and has previously been reported.42 ICP analysis quantifying the Pd content of the 1 wt% and 5 wt% Pd samples and the impurity content of the high surface area (HSA) ceria support is reported in Tables S2 and S3 (ESI†) respectively.In situ XAS data of the 1 and 5 wt% Pd supported on ceria were acquired at the Ce L3-edge (5723 eV) and Pd K-edge (24350 eV). XAS spectra were collected in step scan acquisition mode using transmission geometry at the BM26A beamline,37 ESRF, equipped with a Si(111) double crystal monochromator. Measurements of all the samples were carried out in a transmission mode using ionization chambers. In a typical experiment on the Ce L3-edge, pellets were made from 4 mg of the ceria samples mixed with 40 mg of fumed silica. On the Pd K-edge typically 100 mg of the ceria samples were used. The samples were purged under N2 prior to exposing the catalyst in a flow of 5% H2/N2. Data were obtained at room temperature followed by measurements at various temperatures during the temperature ramp from room temperature to 450 °C at 5 °C min−1. The samples were then cooled in 5% H2/N2 to room temperature whilst data were collected at various temperatures during this process. Between the first and second cycles of H2 treatment the samples were exposed to synthetic air at room temperature for 30 minutes. The second reduction cycle was only performed in part, heating to 100 °C in 5% H2/N2 at 5 °C min−1 on the Pd K-edge, as our aim was to monitor the reduction at the initial stages of the reaction. Data processing and analysis were performed using Athena.43 Linear combination fitting (LCF) analysis of the Ce L3-edge XANES was performed using a freshly calcined sample consisting of the bare ceria support and cerium nitrate as reference materials representing Ce(IV) and Ce(III), respectively. Similarly, LCF analysis on the Pd K-edge was performed using a Pd metal foil and PdO as reference materials.
X-ray diffraction patterns were obtained from the 11-ID-B beamline at the APS, using a 2D detector. The wavelength used was λ = 0.1430 Å with an obtainable Q range of 0.55 and 27.93 Å−1. The samples were prepared using a sieve fraction between 100 and 150 μm to aid in gas flow through the sample. These were loaded into a 0.9 mm (internal diameter) fused silica capillary with a sample bed length of approximately 1 cm, and quartz wool plugs were used on either side of the sample to inhibit the movement of the sample with gas flow. Metal furnace heating elements mounted above and below the capillary were used to control the temperature.44 All the samples were measured under a 3.5% H2/He atmosphere during continuous heating and cooling at a rate of 10 °C min−1 between 30 °C and 450 °C. The samples were also held at 450 °C for 10 minutes prior to cooling. Between the first and second cycles of H2 treatment the samples were exposed to 5% O2/He at room temperature. XRD patterns were azimuthally integrated using FIT2D.45 The XRD patterns were then refined using the GSAS software46 with a batch Rietveld refinement method between 1.8 and 22° 2Θ.
Results and discussion
First, we discuss the results of temperature programmed reduction experiments and then the XAS analysis followed by XRD. In Fig. 1 the TPR profiles are reported as normalized by the integral intensity for 1 wt% Pd/CeO2, 5 wt%Pd/CeO2 and the high surface area CeO2 support to highlight the temperature at which H2 consumption occurs. The 5 wt% Pd/CeO2 sample exhibits a maximum of H2 uptake at approximately 65 °C, while the 1 wt% Pd/CeO2 sample is shifted to approximately 155 °C, and HSA CeO2 begins to show H2 uptake only above 300 °C.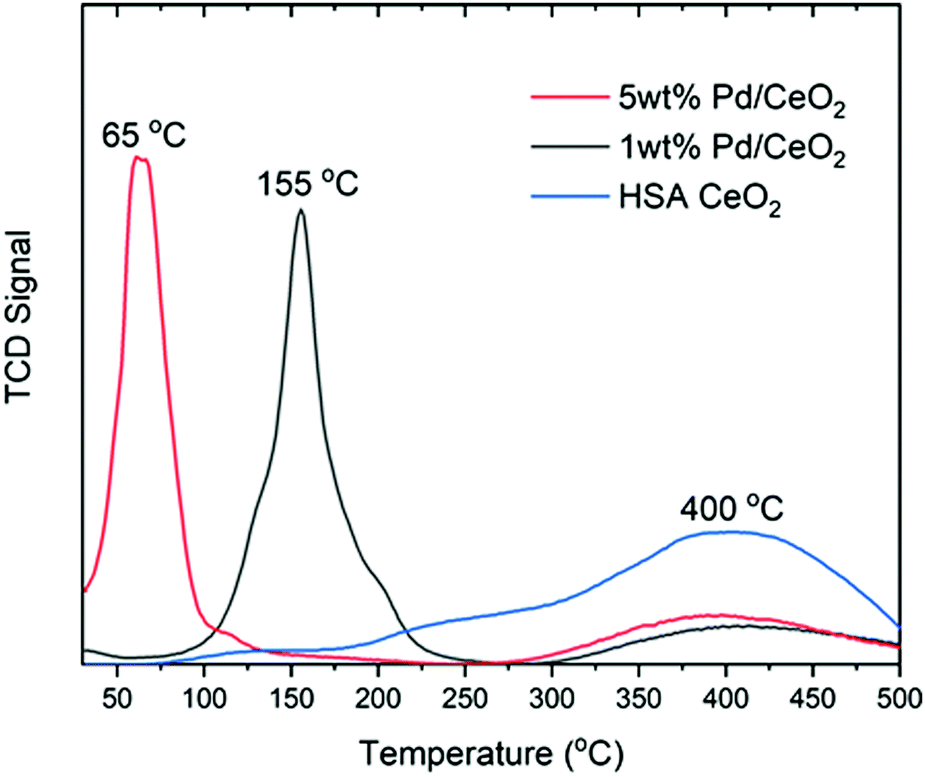 | ||
| Fig. 1 TPR results from the Pd loaded ceria samples. TPR traces giving the normalized integral intensity TCD signal for 1 wto Pd/CeO2, 5 wt% Pd/CeO2 and the CeO2 support shown in black, red and blue respectively for the first cycle of reduction. | ||
From these measurements it is clearly evidenced that the higher the content of Pd present in the sample the maximum in the H2 consumption appears at lower temperature. However, while TPR measurements give a strong indication that the content of Pd strongly influences the temperature of reduction, the method is insensitive to the individual components of the sample and cannot elucidate whether there is a synergistic effect between the reduction of Pd(II) to the metallic state and the formation of oxygen vacancies and reduction to Ce(III) in the CeO2 support.
Pd K-edge XAS
To address the question of the role of Pd in promoting the reduction of CeO2 we have employed XAS at both the Pd K-edge and Ce L3-edge to provide detailed insight into the electronic structure changes of Pd and Ce ions along with XRD to monitor the geometric structural evolution. In Fig. 2 the Pd K-edge XANES spectra of 1 and 5 wt% Pd/CeO2 catalysts recorded during reaction with hydrogen and heating to 450 °C, followed by cooling in a H2/N2 atmosphere and subsequently room temperature exposure to synthetic air, finally followed by reduction up to ca. 100 °C (see Fig. 2A and B) are shown. To analyse the Pd K-edge data multivariate curve resolution (MCR) was performed using an alternative least squares approach.47,48 While typically analysis is performed using a linear combination fitting (LCF) method, here the nano-crystalline nature of the supported Pd leads to significant dampening of the oscillation in the post-edge region which results is a significant misfit between experimental and fitted data. MCR combats this by being able to computationally separate the pure spectral components present in a dataset consisting of multiple spectra obtained during an in situ experiment. Using MCR analysis it was possible to extract the significant components which, when compared to standard reference materials, can be readily identified as oxidic Pd(II) and nano-crystalline metallic Pd(0) species, see Fig. S2 (ESI†).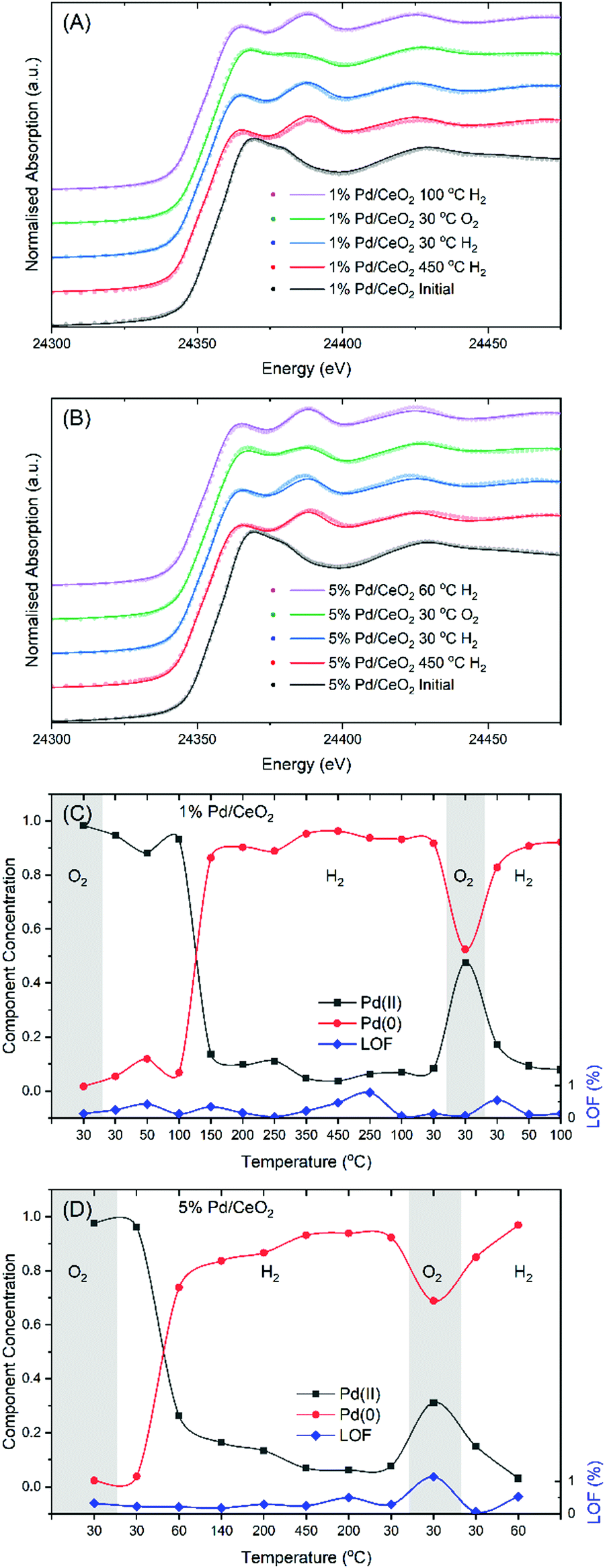 | ||
| Fig. 2 Example of stacked (offset for clarity) Pd K-edge data showing the MCR analysis of the 1 wt% Pd/CeO2 (A) and 5 wt% Pd/CeO2 (B) samples after the initial reduction in 5% H2/N2 at 450 °C, cooled to 30 °C in 5% H2/N2, after passivation with synthetic air and after the second reduction in 5% H2/N2 spectra in black, red, blue, green and purple respectively. The raw data are shown as points whereas the MCR fitting is given as solid lines. The results of MCR analysis giving the proportion of oxidic (black), metallic (red) and the lack of fit % (blue) for the 1 wt% Pd/CeO2 (C) and 5 wt% Pd/CeO2 (D) samples. | ||
The XANES data of both the 1 wt% Pd/CeO2 and 5wt% Pd/CeO2 samples in their respective initial states clearly resemble that of PdO thus confirming the presence of Pd(II) in the as-prepared catalysts (black curves in Fig. 2A and B). Upon reduction by heating in 5% H2/N2, the XANES data of both 1 and 5wt% samples are similar to those of the Pd metal, both in the edge position and on the top of the edge and are interpreted as the formation of nano-crystalline Pd particles on the surface of the CeO2 support (red curve). While at 450 °C, both samples are shown to be metallic, the reduction onset temperatures are notably different. The 1 wt% Pd/CeO2 sample requires a temperature more than 100 °C before the Pd is reduced to a metallic state, see Fig. 2C. However, the majority of the 5wt% Pd/CeO2 sample is reduced by 60 °C, Fig. 2D. These findings demonstrate that the reducibility of Pd is likely hindered by a greater dispersion of Pd on the CeO2 surface.
To investigate this point further, EXAFS analysis was performed on the data collected after reduction and cooling to 30 °C, while remaining in the H2 atmosphere. Curve fitting of the EXAFS can be used to estimate the Pd particle size through the determination of the average Pd–Pd coordination number. The results of the EXAFS fitting using Artemis49 are given in Table 1. By using the empirically derived Hill equation,50 coordination numbers of 5.9 and 7.8 equate to approximately 0.4 nm and 0.7 nm spherical particle size for the 1 wt% Pd and 5 wt% Pd samples respectively. The EXAFS analysis confirms the well dispersed nature of Pd in these supported samples even after reduction in hydrogen with a smaller Pd particle size obtained for 1 wt% Pd/CeO2. Figures demonstrating the quality of fitting to the R- and k-space are given in the ESI,† Fig. S3.
| Sample | Scattering path | R (Å) | σ 2 (Å2) | C.N. |
|---|---|---|---|---|
| 1 wt% Pd/CeO2 | Pd–Pd | 2.78 ± 0.02 | 0.008 ± 0.003 | 5.9 ± 0.6 |
| 5 wt% Pd/CeO2 | Pd–Pd | 2.79 ± 0.02 | 0.009 ± 0.002 | 7.8 ± 0.8 |
After the samples were cooled to room temperature, passivation in synthetic air was performed which results in a partial oxidation of Pd(0) to Pd(II). The degree of oxidation is shown to depend on the Pd content and may be related to the metallic Pd cluster size as the 1 wt% Pd/CeO2 sample oxidises to a greater degree than the 5 wt% Pd/CeO2 sample. The white line intensity in Fig. 1A and B demonstrated that the two samples differ with an increased intensity observable for the 1 wt% Pd/CeO2 sample. However, neither sample can be observed to undergo a full oxidation with respect to Pd ions, within the 30 minute exposure to synthetic air at 30 °C. From the MCR analysis reported in Fig. 1C and D it is revealed that 1 wt% Pd/CeO2 converts to approximately 45% Pd(II), while for 5 wt% Pd/CeO2 the degree of oxidation is only 30%. A direct overlay of the different spectra is given in supplementary Fig. S4 (ESI†) for clarity. A possible explanation for this result is that the smaller Pd particles found on 1 wt% Pd/CeO2 have a greater surface area to volume ratio resulting in more surface atoms being exposed and readily oxidized. A second reduction step was then performed by heating in 5% H2/N2 with both samples showing a rapid reduction back to metallic Pd at temperatures below 100 °C, as seen in the final state (purple curve) in Fig. 2A and B.
Ce L3-edge XAS
Whilst XAS experiments performed at the Pd K-edge give access to the oxidation state and local coordination geometry of the Pd in the CeO2 supported Pd samples, analysis of the Ce L3-edge provides information on the oxidation state Ce in the CeO2 support. In Fig. 3A and B we show the Ce L3-edge XANES measured in the initial state, reduced at 450 °C and after cooling in 5% H2/N2 at 30 °C. In Fig. 3C and D we show the results of LCF analysis of the Ce L3-edge data giving the fractions of Ce(III) during the in situ reduction experiment; the example fitting is shown in Fig. S5 (ESI†). | ||
| Fig. 3 Example Ce L3-edge data from the 1 wt% Pd/CeO2 (A), and 5 wt% Pd/CeO2 (B) samples are shown for the initial as received material, after reduction at 450 °C, after cooling to 30 °C as black, red and blue curves respectively. The results of the linear combination fitting, represented by the fraction of Ce(III), performed using a cerium nitrate hexahydrate standard for Ce(III) and a fully oxidized high surface area ceria support material for Ce(IV) are given for 1 wt% Pd/CeO2 (C) and 5 wt% Pd/CeO2 (D). | ||
It appears that, whilst both samples show partial reduction of Ce(IV) to Ce(III), the reduction onset temperature is dependent on the Pd content in the sample. The 5 wt% Pd/CeO2 sample shows that the maximal extent of reduction, ∼15% Ce(III), is achieved below 150 °C. However, the 1wt% Pd/CeO2 sample has an onset of reduction occurring above 150 °C with the maximum Ce(III) content being achieved only at approximately 350 °C. In our previous work,51 we were able to identify that in pure ceria the reduction process has an onset temperature >300 °C and is temperature reversible implying a mechanism without the formation of oxygen vacancies. Based on the degree of reduction observed from the Ce L3-edge XAS, approximately 15 mol% Ce3+ ions, the stoichiometry of the reduced ceria is estimated as CeO1.85 and is broadly in the range expected for this class of materials as reported elsewhere.1
When considering the CeO2 supported Pd samples, the reduction process is clearly demonstrated to be promoted by Pd to lower temperature and is temperature irreversible due to the consequence of oxygen vacancy formation. These results demonstrate that the reduction to Ce(III) is promoted by Pd and directly agrees with the Pd K-edge analysis and suggests that after reduction of the initial Pd(II), the extraction of oxygen from the ceria lattice occurs. This suggests that either reverse oxygen spill-over to the Pd ceria interface or the spill-over of hydrogen from the Pd surface drives the reduction of the ceria support. Furthermore, when compared to temperature programmed reduction experiments, shown in Fig. 1, only a single event for H2 uptake is observed. This suggests that as soon as PdO is reduced the reduction of ceria proceeds.
In situ XRD
To further support the above presented results, experiments were conducted with X-ray diffraction. Here XRD has been analysed to determine the lattice parameter of the CeO2 fluorite phase and where possible the evolution of the metallic Pd phase during the in situ reduction experiments. In Fig. 4A we show the refined CeO2 lattice parameter from Rietveld analysis for the 1 wt% Pd/CeO2 and 5 wt% Pd/CeO2 samples in blue and black respectively. The temperature is denoted in the red curve and related to the right Y axis. Typical best fits for the Rietveld analysis are given in Fig. S6 and S7 (ESI†) for the 1 wt% Pd/CeO2 and 5 wt% Pd/CeO2 samples respectively showing a very strong agreement between the data and the fluorite CeO2 model at all stages of the reduction experiments. | ||
| Fig. 4 Rietveld analysis of the fluorite CeO2 phase giving the lattice parameters of 1 wt% Pd/CeO2 and 5 wt% Pd/CeO2 in blue and black respectively. (A) The first reduction cycle with lattice parameters on the left Y axis and temperature on the right Y axis. (B) The second reduction cycle. Note that the time in the x-axis in (B) is a continuation of the experiment reported in (A). Figures (C) and (D) give the instantaneous lattice expansion derived from the heating portion of first and second reduction cycles respectively. | ||
It is clear from Fig. 4A that the onset of an increase in the lattice parameter takes place above room temperature, after admitting 3.5% H2/N2 into the capillary. While both samples exhibit a strong lattice expansion upon heating in H2, the slight horizontal offset during the first reduction cycle, Fig. 4A, between the 1 wt% Pd/CeO2 and 5 wt% Pd/CeO2 samples evidences a difference in temperature at which the lattice expansion occurs. After the initial rapid expansion of the fluorite CeO2 lattice, an approximately linear expansion can be seen with continuous heating up to 450 °C. The initial rapid increase in the lattice parameter is likely to be associated with the formation of Ce(III) ions, as indicated in the Ce L3-edge XANES analysis. The formation of Ce(III) leads to an expansion of the ceria lattice due to their larger ionic radii as compared to the Ce(IV) ions.52 However, secondary to the expansion due to the formation of Ce(III) ions within the fluorite lattice, the linear thermal expansion also occurs. For ceria the linear thermal expansion coefficient is reported to be approximately 10 × 10−6 K−1. Upon cooling in a hydrogen atmosphere, the respective lattice parameters are observed to decrease linearly which is suggested to be due to the thermal contraction. On reaching room temperature, the respective lattice parameter values remain higher than the initial starting values suggesting an irreversible structure change, consistent with the Ce L3-edge XAS analysis findings.
By considering the instantaneous expansion coefficient it is possible to deconvolute the two competing expansion effects and is described by:

However, when considering the second reduction cycle performed after mild passivation through room temperature reaction with air, shown in Fig. 4B, it is worth noting that the lattice parameter of CeO2 returns to its starting value as soon as air was introduced after the first cycle of reduction, suggesting a complete reoxidation of reduced Ce(III) at this stage. Upon re-introducing 3.5% H2/N2 after the reoxidation, both 1% and 5% samples exhibit a near room temperature partial reduction of Ce(IV) to Ce(III). The respective lattice parameters rapidly increase to almost the same value observed for their first cycle, at a temperature well below 100 °C. Subsequent increases in temperature linearly increase the lattice parameter similar to that observed for the first cycle of reduction. The instantaneous expansion coefficient clearly shows that an inflection (see Fig. 4D) in the ceria lattice parameter is observed at approximately 50 °C for both the 1 wt% and 5 wt% loadings of Pd. When considering the instantaneous lattice expansion coefficient this process can be observed to occur rapidly resulting in an instantaneous expansion coefficient maximum of approximately 250 × 10−6 K−1. This confirms the previously discussed results from the Pd K-edge XAS measurements, and the mild passivation of the Pd by room temperature oxidation can be readily reduced and thus can promote the reduction of ceria.
After close inspection of the XRD patterns for the 5 wt% Pd/CeO2 sample a very weak reflection associated with the (111) reflection of metallic Pd can be seen at 2θ of approximately 3.65 °. The formation of the metallic Pd phase is evidenced in the colourmap given in Fig. 5A showing a slight increase in intensity (on a logarithmic scale) with a change from dark blue to light blue colour. To extract information on the lattice parameter of the metallic Pd from the (111) reflection a custom peak fitting tool was written in python. At the start of the experiment the Pd was noted as being oxidic Pd(II) from XAS measurements and therefore no metallic Pd is expected to be present allowing for the baseline correction to be achieved through subtraction of the first dataset corresponding to the initial starting material. Fig. S8 (ESI†) gives the surface colour map of the baseline corrected data which were used for further analysis. Examples of the quality of the peak fitting are given in Fig. S9 (ESI†). Attempts to also perform the same analysis on 1 wt% Pd/CeO2 were made; however, no peak related to the Pd(111) reflection could be clearly detected as a consequence of the lower Pd content and expected lower crystallinity. The colour map for the 1 wt% Pd/CeO2 sample is given in supplementary Fig. S10 (ESI†).
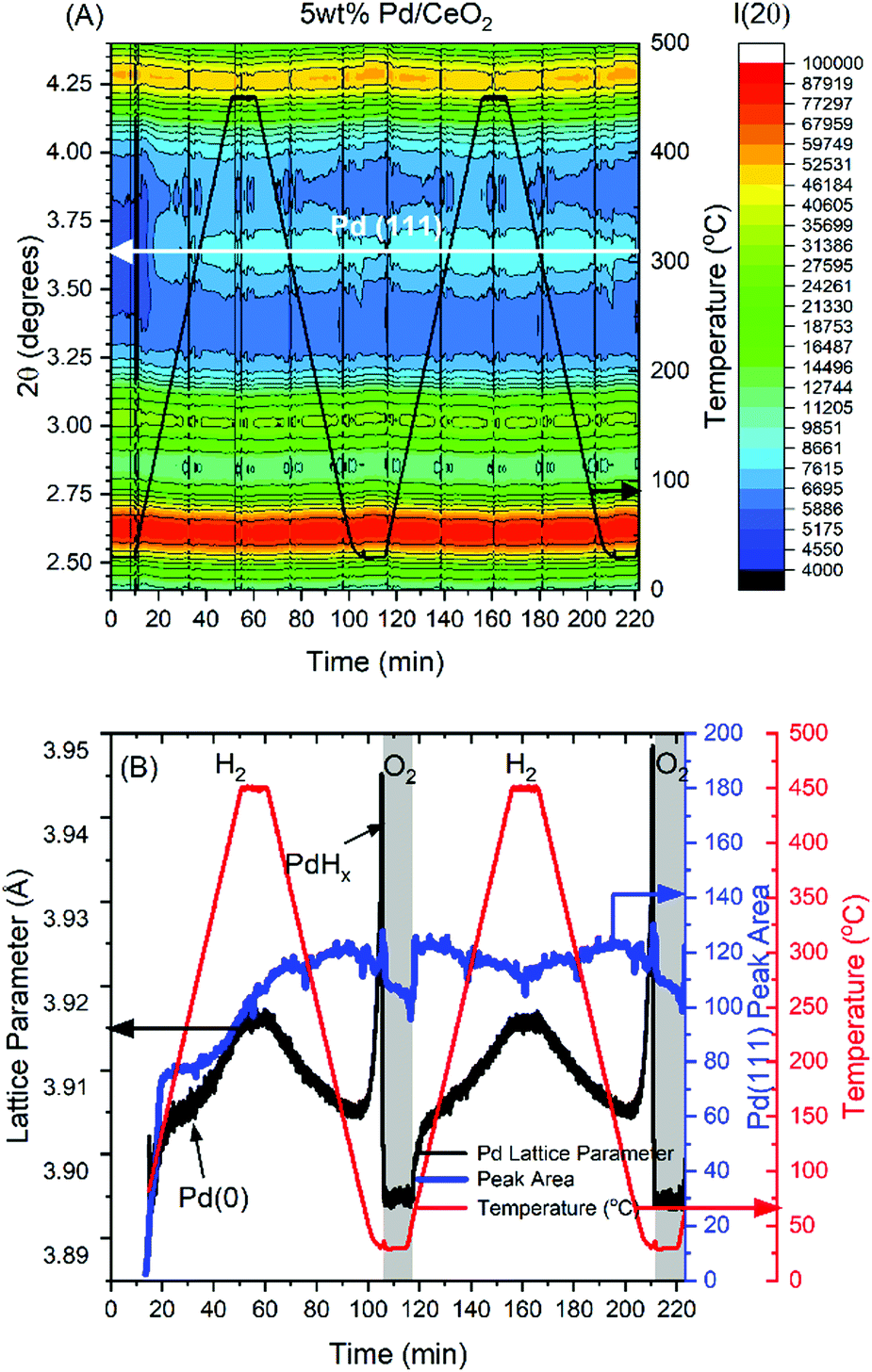 | ||
| Fig. 5 (A) Surface colourmap illustrating selected XRD data collected on the 5% Pd/CeO2 sample around the Pd(111) reflection region. Temperature is indicated with the black trace associated with the right Y axis. (B) The results of the Gaussian peak fitting to the Pd(111) reflection yielding the Pd lattice parameter and peak area in black and blue respectively. The temperature is given as the red trace associated with the second left Y axis. | ||
The baseline corrected data for the 5 wt% Pd/CeO2 sample were used for sequential fitting with a single Gaussian peak to model the position and area of the peak giving insight directly into the Pd lattice parameter evolution and relative crystallinity throughout. The results of the Gaussian peak fitting are shown in Fig. 5B for the first and second reduction cycles. On first inspection the formation of crystalline Pd0 was found to occur above approximately 100 °C with a rapid increase in the peak area occurring during the initial phase of heating. Given that XRD analysis is only sensitive to the long range structure, the initial reduction to a disordered Pd may be missed and could explain the temperature inconsistency between XAS which showed almost full Pd0 at approximately 60 °C and the XRD analysis which reveals crystalline Pd0 formation at around 100 °C.
With further heating of the sample, the Pd cubic lattice parameter is observed to increase with a maximum of 3.918 Å, in line with the expected lattice parameter taking into account thermal expansion. Concurrent to this, the relative crystallinity, observed from the peak area shown in Fig. 5B in blue, is observed to increase suggesting a partial sintering of the metallic Pd phase during heating. During cooling the lattice parameter is noted to decrease until around 50 °C before a rapid and substantial increase during further cooling to room temperature. This expansion is due to the formation of a PdHx phase known to occur when Pd is treated under a hydrogen containing atmosphere at low temperature. Upon exposure to oxygen at room temperature the PdHx phase is removed and the Pd lattice parameter relaxes to 3.896 Å. In the Pd K-edge XAS spectra shown in Fig. 2 the exposure to oxygen leads to a partial oxidation to Pd(II) and from the XRD analysis it is found that this process is concurrent with only a very minor decrease in the Pd(111) reflection peak area. This result evidences that the passivation is only a surface oxidation process with the core remaining metallic. During the second reduction cycle, the lost intensity of the Pd(111) reflection is rapidly recovered suggesting that the surface PdO species is reduced as soon as hydrogen is reintroduced into the sample, in line with the results obtained from the Pd K-edge XAS.
Conclusions
In summary, combined in situ multi-edge XAS and X-ray scattering studies enabled us to determine the effect of palladium loading on a high-surface area support on the reactivity of both Pd(II) and Ce(IV) ions. The results clearly indicate that there is a promotion of the partial reduction of ceria due to the presence of Pd. The temperature at which the promoted reduction temperature of the ceria is also noted to inversely depend on the wt% loading of the Pd onto the ceria support; a higher loading of Pd leads to a lower reduction temperature of the Pd and the ceria support. This finding could be attributed to the strong interaction of metal ions with the support preventing the reduction of the initial PdO to metallic Pd. The promotion of the CeO2 reduction is interpreted as the extraction of oxygen from the ceria lattice and is suggested to be due to a hydrogen spill over mechanism or reverse oxygen spill over mechanism.More importantly, the findings obtained from the Pd K-edge XAS and XRD studies clearly demonstrate that passivation in air at room temperature only partially oxidises the supported Pd while a complete reoxidation of the reduced ceria (CeO1.85) is observed. Based on this ceria lattice parameter change, we conclude that the restoration of the full stoichiometric fluorite phase CeO2 takes place. Furthermore, the second reduction cycle, observed by XRD, demonstrates that the reduction of ceria, observed indirectly by analysis of the lattice parameter, is promoted to near room temperature irrespective of the Pd wt% loading. The Pd K-edge data corroborate this result showing that the Pd also undergoes near room temperature reduction by hydrogen following the mild passivation irrespective of the Pd wt% loading. These results indicate that with an initial pre-treatment cycle and subsequent passivation, the promoted reduction of ceria can be reduced to near room temperature without the requirement of high Pd wt% loadings and may have a direct impact on the relevant fields of catalysis where these classes of materials are currently utilised.
Author contributions
AHC and HRM performed the experiments, analysis, interpretation and manuscript drafting. DT and JF provided the samples used in the study. AL and KB optimised the beamlines for the experiments performed at ESRF and APS respectively and provided user support throughout. TIH and GS contributed to the drafting of the manuscript, interpretation of results and supervision of both AHC and HRM.Conflicts of interest
The authors do not have any conflict of interest to declare.Acknowledgements
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank EPSRC and Johnson Matthey Plc for Case Awards (HRM and AHC). We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities.Notes and references
- A. Trovarelli, Catalysis by Ceria and Related Materials, Imperial College Press, London, 2002, vol. 2 Search PubMed.
- R. M. Heck and R. J. Farrauto, Appl. Catal., A, 2001, 221, 443–457 CrossRef CAS.
- R. Craciun, B. Shereck and R. J. Gorte, Catal. Lett., 1998, 51, 149–153 CrossRef CAS.
- P. Pantu, K. Kim and G. R. Gavalas, Appl. Catal., A, 2000, 193, 203–214 CrossRef CAS.
- Y. I. Matatov-Meytal and M. Sheintuch, Ind. Eng. Chem. Res., 1998, 37, 309–326 CrossRef CAS.
- H. Renner, G. Schlamp, I. Kleinwächter, E. Drost, H. M. Lüschow, P. Tews, P. Panster, M. Diehl, J. Lang, T. Kreuzer, A. Knödler, K. A. Starz, K. Dermann, J. Rothaut, R. Drieselmann, C. Peter and R. Schiele, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2001 Search PubMed.
- D. Astruc, Nanoparticles and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 1–48 Search PubMed.
- A. Neitzel, A. Figueroba, Y. Lykhach, T. Skála, M. Vorokhta, N. Tsud, S. Mehl, K. Ševčíková, K. C. Prince, K. M. Neyman, V. Matolín and J. Libuda, J. Phys. Chem. C, 2016, 120, 9852–9862 CrossRef CAS.
- N. Acerbi, S. Golunski, S. C. Tsang, H. Daly, C. Hardacre, R. Smith and P. Collier, J. Phys. Chem. C, 2012, 116, 13569–13583 CrossRef CAS.
- J. Zhou, A. P. Baddorf, D. R. Mullins and S. H. Overbury, J. Phys. Chem. C, 2008, 112, 9336–9345 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- J. A. Rabó, Catal. Today, 1994, 22, 201–233 CrossRef.
- L. F. Liotta, A. Longo, A. Macaluso, A. Martorana, G. Pantaleo, A. M. Venezia and G. Deganello, Appl. Catal., B, 2004, 48, 133–149 CrossRef CAS.
- L. F. Liotta, A. Longo, G. Pantaleo, G. Di Carlo, A. Martorana, S. Cimino, G. Russo and G. Deganello, Appl. Catal., B, 2009, 90, 470–477 CrossRef CAS.
- H. Asakura, S. Hosokawa, K. Beppu, K. Tamai, J. Ohyama, T. Shishido, K. Kato, K. Teramura and T. Tanaka, Catal. Sci. Technol., 2021, 11, 6182–6190 RSC.
- J. Graciani, A. B. Vidal, J. A. Rodriguez and J. F. Sanz, J. Phys. Chem. C, 2014, 118, 26931–26938 CrossRef CAS.
- M. Ahmadi, H. Mistry and B. Roldan Cuenya, J. Phys. Chem. Lett., 2016, 7, 3519–3533 CrossRef CAS PubMed.
- H. Hirata, K. Kishita, Y. Nagai, K. Dohmae, H. Shinjoh and S. Matsumoto, Catal. Today, 2011, 164, 467–473 CrossRef CAS.
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. Larese, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 1999, 50, 175–206 CrossRef CAS.
- S. Suhonen, M. Valden, M. Hietikko, R. Laitinen, A. Savimäki and M. Härkönen, Appl. Catal., A, 2001, 218, 151–160 CrossRef CAS.
- G. Jacobs, U. M. Graham, E. Chenu, P. M. Patterson, A. Dozier and B. H. Davis, J. Catal., 2005, 229, 499–512 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- O. V. Safonova, A. A. Guda, C. Paun, N. Smolentsev, P. M. Abdala, G. Smolentsev, M. Nachtegaal, J. Szlachetko, M. A. Soldatov, A. V. Soldatov and J. A. Van Bokhoven, J. Phys. Chem. C, 2014, 118, 1974–1982 CrossRef CAS.
- C. Loschen, S. T. Bromley, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2007, 111, 10142–10145 CrossRef CAS.
- T. Baidya, A. Gayen, M. S. Hegde, N. Ravishankar and L. Dupont, J. Phys. Chem. B, 2006, 110, 5262–5272 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
- D. O. Scanlon, B. J. Morgan and G. W. Watson, Phys. Chem. Chem. Phys., 2011, 13, 4279–4284 RSC.
- S. Golunski and A. Walker, Chem. Commun., 2000, 1593–1594 RSC.
- G. Sankar, C. N. R. Rao and T. Rayment, J. Mater. Chem., 1991, 1, 299–300 RSC.
- C. M. Y. Yeung and S. C. Tsang, J. Phys. Chem. C, 2009, 113, 6074–6087 CrossRef CAS.
- J. Lin, L. Yang, T. Wang and R. Zhou, Phys. Chem. Chem. Phys., 2017, 19, 7844–7852 RSC.
- R. Craciun, W. Daniell and H. Knözinger, Appl. Catal., A, 2002, 230, 153–168 CrossRef CAS.
- N. Tsubaki and K. Fujimoto, Top. Catal., 2003, 22, 325–335 CrossRef CAS.
- J. Ma, Y. Lou, Y. Cai, Z. Zhao, L. Wang, W. Zhan, Y. Guo and Y. Guo, Catal. Sci. Technol., 2018, 8, 2567–2577 RSC.
- L. M. Misch, J. A. Kurzman, A. R. Derk, Y. Il Kim, R. Seshadri, H. Metiu, E. W. McFarland and G. D. Stucky, Chem. Mater., 2011, 23, 5432–5439 CrossRef CAS.
- M. Danielis, S. Colussi, C. De Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Catal. Sci. Technol., 2019, 9, 4232–4238 RSC.
- E. M. Slavinskaya, T. Y. Kardash, O. A. Stonkus, R. V. Gulyaev, I. N. Lapin, V. A. Svetlichnyi and A. I. Boronin, Catal. Sci. Technol., 2016, 6, 6650–6666 RSC.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- S. Roy and M. S. Hegde, Catal. Commun., 2008, 9, 811–815 CrossRef CAS.
- H. R. Marchbank, A. H. Clark, T. I. Hyde, H. Y. Playford, M. G. Tucker, D. Thompsett, J. M. Fisher, K. W. Chapman, K. A. Beyer, M. Monte, A. Longo and G. Sankar, ChemPhysChem, 2016, 17, 3494–3503 CrossRef CAS PubMed.
- D. S. Mannel, M. S. Ahmed, T. W. Root and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 1690–1698 CrossRef CAS PubMed.
- P. J. Chupas, K. W. Chapman, C. Kurtz, J. C. Hanson, P. L. Lee and C. P. Grey, J. Appl. Crystallogr., 2008, 41, 822–824 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Häusermann, High Press. Res., 1996, 14, 235–248 CrossRef.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. Voronov, A. Urakawa, W. van Beek, N. E. Tsakoumis, H. Emerich and M. Rønning, Anal. Chim. Acta, 2014, 840, 20–27 CrossRef CAS PubMed.
- A. De Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964–4976 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- A. H. Clark, K. A. Beyer, S. Hayama, T. I. Hyde and G. Sankar, Chem. Mater., 2019, 31, 7744–7751 CrossRef CAS.
- R. K. Hailstone, A. G. DiFrancesco, J. G. Leong, T. D. Allston and K. J. Reed, J. Phys. Chem. C, 2009, 113, 15155–15159 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04654c |
| ‡ Current address: Paul Scherrer Institut, Villigen, CH-5232 Switzerland. |
| § Current Address: Johnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading RG4 9NH, UK. |
| This journal is © the Owner Societies 2022 |