
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The porphyrin center as a regulator for metal–ligand covalency and π hybridization in the entire molecule†
Robby
Büchner
 *a,
Mattis
Fondell
b,
Robert
Haverkamp
b,
Annette
Pietzsch
b,
Vinícius
Vaz da Cruz
*b and
Alexander
Föhlisch
ab
*a,
Mattis
Fondell
b,
Robert
Haverkamp
b,
Annette
Pietzsch
b,
Vinícius
Vaz da Cruz
*b and
Alexander
Föhlisch
ab
aInstitute of Physics and Astronomy, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: rbuechner@uni-potsdam.de; Tel: +49 30 806213967
bInstitute for Methods and Instrumentation for Synchrotron Radiation Research, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: vinicius.vaz_da_cruz@helmholtz-berlin.de; Tel: +49 30 806214987
First published on 29th October 2021
Abstract
The central moiety of porphyrins is shown to control the charge state of the inner complex and links it by covalent interaction to the peripheral substituents. This link, which enables the versatile functions of porphyrins, is not picked up in the established, reduced four orbital picture [Gouterman, J. Mol. Spectrosc., 1961, 6, 138]. X-ray absorption spectroscopy at the N K-edge with density functional theory approaches gives access to the full electronic structure, in particular the π* manifold beyond the Gouterman orbitals. Systematic variation of the central moiety highlights two linked, governing trends: The ionicity of the porphyrin center increases from the aminic N–H to N–Cu to N–Zn to N–Mg to the iminic N:. At the same time covalency with peripheral substituents increases and compensates the buildup of high charge density at the coordinated nitrogen sites.
1 Introduction
While the class of porphyrins and derivatives is of versatile use and of significant importance both in nature and technology, each function is fulfilled by a porphyrin with particular constitution: protoporphyrin IX, for instance, contains a center of two hydrogen atoms, which are replaced by magnesium in plants to form chlorophyll or iron in vertebrates to form haeme. Upon iron deficiency zinc protoporphyrin is synthesized by the body, instead, leading to anemia.1 If the human porphyrin metabolism is substantially impaired, protoporphyrin IX accumulates in the body causing photoallergy and hepatic disease.2 Analogously to the necessity for an iron center in heame, the magnesium center of chlorophyll is essential for light-harvesting in plants. For the technical imitation of photosynthesis, however, copper3 or zinc4 are used as central atoms, since isolated chlorophyll easily disintegrates.5 In contrast, the high stability of copper chlorophyllins allows their application in the food industry,6 catalysis,7 and medicine.8 The use of chlorophyllins is also supported by their high aqueous solubility. The ability of porphyrins and derivatives to dissolve in polar solvents, bind to synthetic surfaces or proteins in a specific geometric arrangement are governed by the peripheral substituents which differ for each before mentioned application in addition to the central moiety.Considering the vast variety of porphyrins, it is remarkable that all of them show similar optical properties (see UV/VIS spectra in the ESI†). The strong UV (Soret or B band) and weaker visible (Q) bands of all porphyrins have been explained by π–π* transitions from the two highest occupied molecular orbitals (HOMO, HOMO−1 – see Fig. 1a) to the two lowest unoccupied molecular orbitals (LUMO, LUMO+1) by Gouterman in Gouterman 1961.9 This four orbital model explains the intensity differences and the splitting of the visible bands in H2TPP by assuming the HOMO(+1) to be accidentally degenerate. This condition can be weakened by mixing of metal pz atomic orbitals with the HOMO or electron donating/accepting side chains. However, optical spectroscopy of porphyrins is rather insensitive to electronic structure changes upon peripheral substitution or exchange of the central moiety. Since past experimental electronic structure studies were focused on the Gouterman orbitals, theoretical approaches lack precision on the π manifold10 and the description of differences between the porphyrin species.
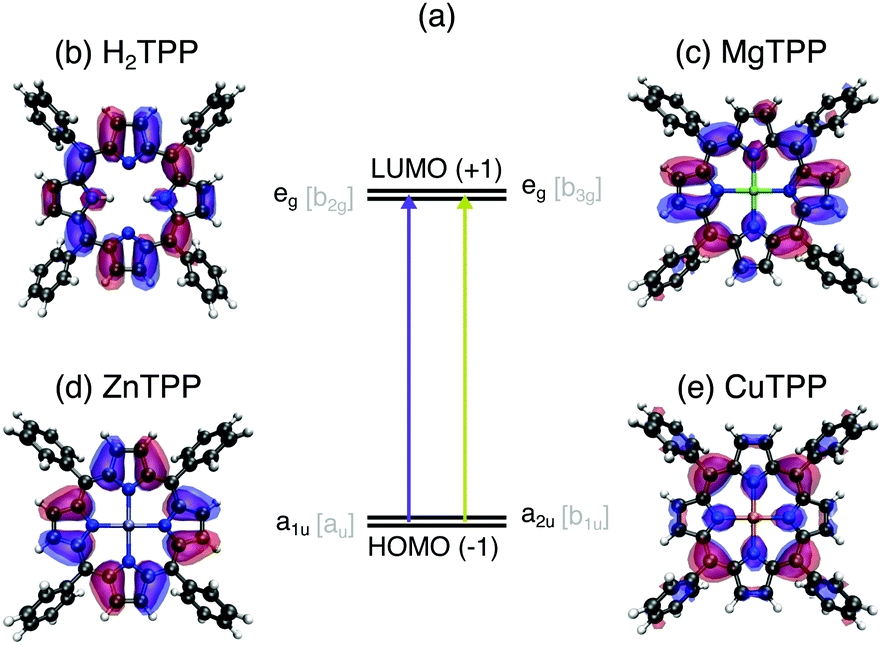 | ||
| Fig. 1 Frontier orbitals of the series of tetraphenylporphyrin (TPP) cores in relation to optical properties: (a) optical transitions between the near-degenerate LUMO/LUMO+1 (b and c) and HOMO−1/HOMO (d and e) orbitals of all porphyrins give rise to the common UV/VIS absorption bands – the foundation for the Gouterman model. Illustrations of these Gouterman orbitals (named after their symmetry in the D4h [D2h] point group) in the TPPs: (b) eg [b2g] of H2TPP, (c) eg [b3g] of MgTTP, (d) a1u [au] of ZnTTP, (e) a2u [b1u] of CuTTP. | ||
In this work, we investigate the influence of a 2H+ (Fig. 1b), Mg2+ (Fig. 1c), Zn2+ (Fig. 1d), and Cu2+ center (Fig. 1e) on the electronic structure of tetraphenylporphyrins (TPPs) and their carboxylated derivatives (TCPPs) in comparison to the parent, unsubstituted porphyrins (Ps). For this purpose, we exploit the element selectivity of Near-Edge X-ray Absorption Spectroscopy (NEXAFS) at the nitrogen atoms, which link the porphyrin macrocycle to the central moiety. To uncover the nature of the porphyrin transition beyond the Gouterman orbitals, density functional theory calculations are carried out for a range of functionals both with the transition potential (TP-DFT) and time dependent approach (TD-DFT). Thereby we establish the porphyrin center as a regulator for the hybridization in the entire molecule controlling both the ionicity of the metal–ligand bond and the π covalency of the macrocycle with peripheral substituents.
2 Methods
2.1 Experimental details
For the experimental investigation 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrins (TCPPs) have been selected, since the carboxyl groups in the para position of each phenyl substituent drastically increase the solubility of the per se mostly nonpolar porphyrins in aqueous solution. These TPP derivatives were synthesized by Por-Lab (Scharbeutz, Germany) with a minimum purity of 95%. To ensure a maximum concentration of the investigated solutions, the powder samples were dissolved in a 0.01 N NaOH solution. After one hour of ultrasonic treatment, they were filtered through a 25–50 μm filter. The final concentrations were determined by UV/VIS absorption measurements (Shimadzu UV-2700) yielding approximately 3.0 mM for H2TCPP, 3.6 mM for MgTCPP, 4.6 mM for ZnTCPP, and 2.9 mM for CuTPP.In order to directly access the absorbance of soft X-rays by transmission measurements, the thickness of the sample needs to be in the order of a few μm. Such a condition is provided by the nmTransmission NEXAFS endstation,11 where the liquid is pushed into the vacuum chamber through two 30 μm sized nozzles. Upon collision, the two jets form a liquid sheet, whose thickness can be adjusted to suit the penetration depth of soft X-rays. In the present study, a flow rate of 1.6 ml min−1 was used resulting in thicknesses of approximately 4 μm. The continuous replenishment of the solutions also prevents radiation induced sample damage.
The experiment was prepared at beamline UE49-SGM12 (Bessy II, Helmholtz-Zentrum Berlin) with the EDAX endstation13 and finally conducted at the neighboring beamline UE52-SGM.14 The spectra were on average acquired for 22 s for each 0.05 eV step. The bandwidth of the incoming X-rays was 0.13 eV.
The resulting spectra were calibrated by the NEXAFS signature of co-dissolved N2 at 400.84 eV.15,16 The solvent background including the N2 features has been measured separately, fitted and subtracted from the sample spectra yielding the pure NEXAFS of aqueous carboxylated TPPs.
2.2 Computational details
Both 5,10,15,20-tetraphenylporphyrins (TPPs, as shown in Fig. 1) and the parent Ps, in which four hydrogen atoms replace the phenyl groups, were investigated by electronic structure calculations to distinguish the effects of substitution and different central atoms.All geometry optimizations and TD-DFT calculations were carried out with the Orca package.17 Molecular geometries were optimized with the PBE018 functional and def2-TZVP(-f)19 basis set without symmetry constraints to account for the tilt of the phenyl substituents (see Section 3.3). The RIJCOSX approximation20 was used with the def2/J21 auxiliary basis set. Becke–Johnson damping22,23 was used for dispersion correction. For the TD-DFT X-ray absorption spectrum calculations the def2-TZVP basis was used for all atoms except for the central metal atom and the nitrogen atoms, on which the diffuse def2-TZVPD basis set was adopted. In the calculations, the near-degenerate core-orbitals were localized and the multiplicity of the states involved has been restricted to doublet–doublet (CuTPP) and singlet–singlet excitations (H2TPP, MgTPP, ZnTPP). For the simulation of the aqueous environment (resulting in the spectra shown in Fig. 4a) the conductor-like polarizable continuum model24 (CPCM) has been used both in the geometry optimization and TD-DFT calculations.
The choice of the density functional kernel for porphyrins poses a difficult task, specially concerning the X-ray absorption spectrum simulations. Although, the lowest π* resonance is typically well described by global hybrids25 and consequently all functionals tested here were able to correctly describe the transition from the N 1s orbital to the eg Gouterman orbitals. For large conjugated systems, such as porphyrins, additional π* resonances occur below the ionization threshold, which might not be properly described by a functional with large exact exchange fraction. On the one hand, the description of higher lying π* orbitals is worse for global hybrids with a high fraction of exact exchange (see DFT benchmark in the ESI†). On the other hand, the description of metal–ligand charge transfer states requires hybrid functionals, including large fractions of Hartree–Fock exchange (HFX).10,26,27 Since no clear single compromise could be reached we have used the BLYP28,29 functional for analyzing the transitions of π character, while the transitions affected by metal–ligand interactions were studied by the BHandHLYP30 functional.
To assess the role of core–hole relaxation, additional spectral calculations based on the transition potential (TP-DFT) method were carried out. The PSIXAS31 plugin for PSI432 was used and the same basis sets used for the TD-DFT calculations described above to enable comparability. The excited state was calculated in the presence of a half core hole.
The resulting transition energies were shifted according to the first experimental resonance of MgTCPP (−1.96 eV for TP-DFT BLYP, +20.29 eV for TD-DFT BLYP, +1.26 eV for TD-DFT BHandHLYP) or the respective resonance for which interspecies shifts are plotted (−0.21 eV for the second resonance in Fig. 4b). The final spectra were obtained by convolution with a Voigt profile of 0.13 eV33 (Lorentzian FWHM) and 0.20 eV (Gaussian FWHM) broadening.
Isosurface plots are displayed for the virtual Kohn–Sham orbitals obtained by ground state calculation of CuTPP with an isovalue of 0.02. The orbitals for the remaining systems are visually analogous.
3 Results and discussion
In Fig. 2(a) the N K-edge NEXAFS of carboxylated TPPs with 2H+, Mg2+, Zn2+, and Cu2+ as central moiety are shown. It can be seen that three resonances appear for each nitrogen atom in a specific chemical environment. The decomposition of the H2TPP spectrum in transitions of the aminic (N–H, solid lines) and iminic nitrogen atoms (N:, dashed lines) was firstly proposed by Narioka et al.34 and is confirmed by our calculations. The interpretation of the experimental features, which will be discussed below, is summarized in Fig. 2(b). In short, the 1eg (blue), b2u (pink), and 3eg (dark green) resonances are observed for each sample and nitrogen site. To enable comparability to past studies with symmetry restriction or different substituents, (instead of N(1s) → HOMO, N(1s) →…) the transitions are classified by the symmetry of the vacant orbital in the idealized D4h point group of the porphyrin macrocycle and numbered by the respective occurrence above the Fermi level. The nomenclature for the Gouterman orbitals in the reduced idealized symmetry of H2TPP (D2h) is given in Fig. 1. | ||
| Fig. 2 (a) Experimental N K-edge NEXAFS of carboxylated TPPs. The interpretation of the main resonances (see ESI† for spectral decomposition) is indicated by the color of the dashed (excitation from an iminic nitrogen atom) and solid vertical lines (other nitrogen environments): 1eg (blue), b1g (orange), b2u (pink), 3eg (dark green), a2u (red), shape resonance (gray). (b) Schematic representation of the transitions with equivalent line styles. The orbitals which are involved in the NEXAFS of all investigated species are illustrated in opaque colors. | ||
It should be noted, that the electronic transitions observed in the X-ray absorption spectrum at typical light element K-edges are dressed by sizable Franck–Condon progressions, which combined with the electronic transition dipole moment determine the intensity ratios.35–37 The role of vibrational excitations is also evidenced by the large width of the observed near-edge resonances.
The detailed assignment of the experimental features is based on the DFT calculations being shown in Fig. 3. As explained above, the BLYP functional is expected to yield reasonable results for the delocalized π* states of the electron-rich porphyrin macrocycle. The comparison of TD-DFT and TP-DFT calculations allows to assess the role of both configuration interactions (CI) and relaxation effects. For investigating the metal–ligand interactions and substituent effects in aqueous solution we refer to Fig. 4(a), where the TD-DFT BHandHLYP spectrum simulations are shown for all investigated TPPs and their phenyl-free P analogs.
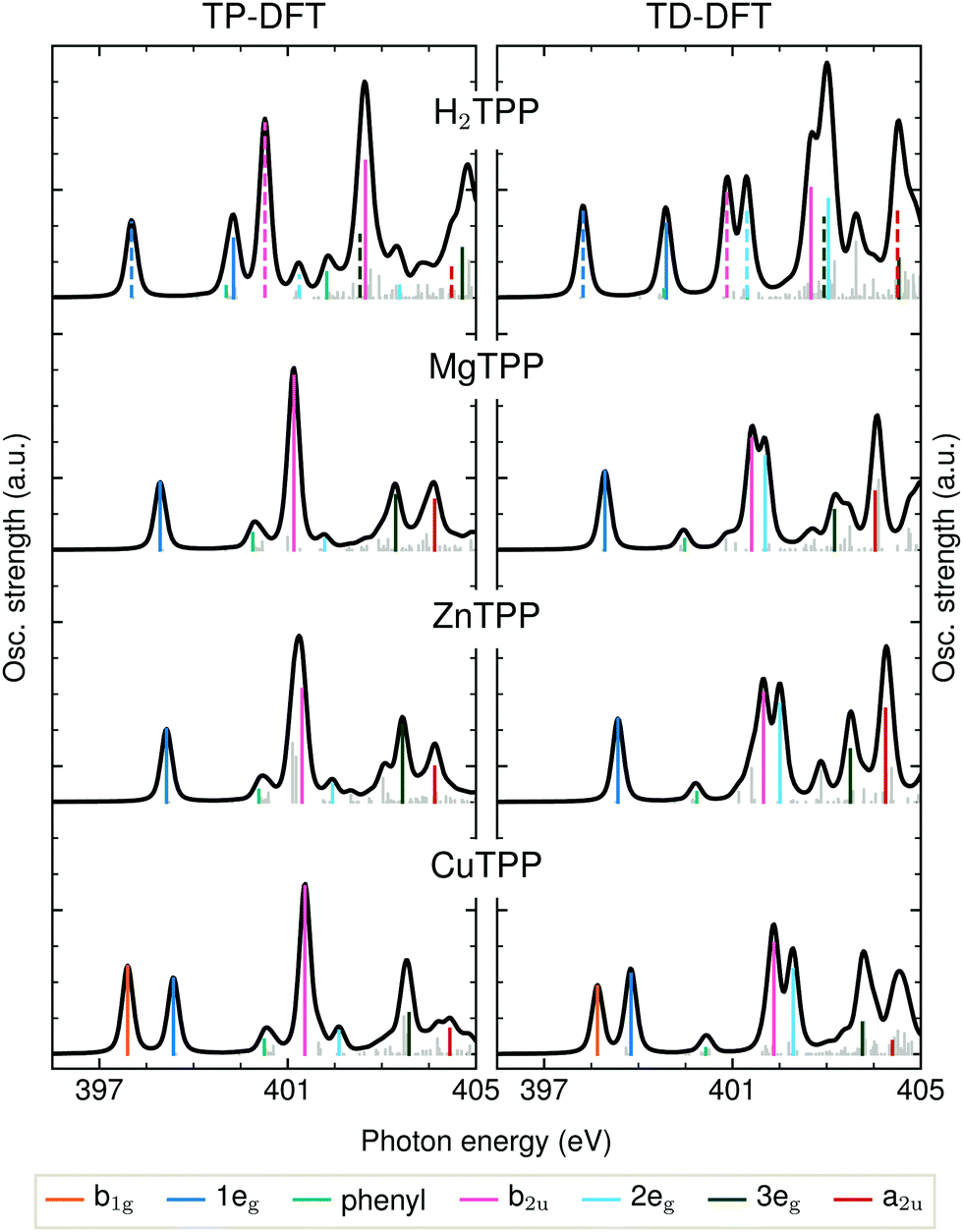 | ||
| Fig. 3 Transition potential and time dependent DFT calculations of the TPP N K-edge with the BLYP functional. Dashed transition result from the excitation of a core electron of an iminic nitrogen atom. | ||
 | ||
| Fig. 4 Blue-shift of the 1eg and b2u transition energies from iminic nitrogen (N:) in H2TPP, MgTPP, ZnTPP, CuTPP to aminic nitrogen (N–H) in H2TPP: (a) TD-DFT BHandHLYP spectral simulations with implicit solvation for the TPP series (black) and their phenyl-free P equivalents (gray). (b) Comparison of the calculated, experimental and negative Kohn–Sham orbital energy shifts. | ||
3.1 Spectral interpretation
The first peak in all experimental spectra (397.9–400.1 eV, blue in Fig. 2a) is the 1eg(π*) resonance. The vacant orbitals of the underlying transition are the Gouterman LUMO and LUMO+1, which also participate in the UV/VIS transitions (see Fig. 1). There is general consensus in literature about the assignment of this feature,34,38–43 which is not the case for the subsequent resonances.The narrow 1eg shoulder (399.5 eV, orange), that is only observed for a Cu2+ center, results from the half occupied Cu 3dx2−y2 orbital which mixes with the porphyrin σ-type b1g orbital in D4h symmetry. Early solid state measurements of copper porphyrins39,40,44 have not been able to resolve this feature. Mangione et al.43 assigned the b1g resonance to an 1eg shoulder to lower energies – similar to the one which we observe for all samples around 398 eV. Since radiation damage can be excluded for the setup used (see section 2.1) and this shoulder is also visible for the closed-shell ZnTCPP we attribute this feature to thermal geometric distortions. For CuTCPP we interpret the 399.5 eV shoulder on the opposite side of the 1eg peak as σ* resonance. This relative position is in agreement with linear dichroism measurements at the N K-edge of copper phthalocyanine,45 explains the existence of a similar shoulder in iron porphyrin in solution,46 and is theoretically backed up by our spectrum calculations and the ones by Schmidt et al.47 for another transition metal TPP.47 For CuTPP, the transition appears in both DFT BLYP calculations (see Fig. 3) below the 1eg resonance similar to the calculations done by Mangione et al.43 The energetic position above the 1eg resonance, being proposed in this work, is based on the benchmarked functionals with a large fraction of HFX as BHandHLYP (see Fig. 4a). This behavior is expected, as hybrid functionals describe charge transfer states more precisely than pure functionals.10
The second intense peak in the experimental spectra (400.1–402.0 eV, pink) has been interpreted as b1g47 or substituent feature46,48 in the past. The strong intensity of this peak in all carboxylated metal TPP spectra and the 1eg shoulder, which is only observed for CuTCPP, speak against the former assignment. A substituent transition – in our case phenyl(π*) – is indeed observed in this energy range for TD-DFT based calculations with low or no HFX (cyan in Fig. 3). However, the comparison with experimental octaethylporphyrin40 and especially metal P NEXAFS spectra41,44,49 (without any outer substituent) make clear that this peak belongs to the π* signature of the porphyrin macrocycle. The second porphyrin peak (P b2u in Fig. 4a) results from the b2u(π*) transition. This feature is prominent in all calculations shown. When the TP-DFT and TD-DFT calculations are compared, it can be noted that the b2u resonance is lowered in energy yielding a better agreement with the experimental positions for TP-DFT. At the same time, the 2eg transition is quenched following the general pattern of the core–hole effect to concentrate the excitation intensity on the energetically lower transitions.50
The quenching of the 2eg peak by core–hole relaxation is in agreement with results found in literature.34,44 Nevertheless, the partial density of unoccupied states has been used to explain the next experimental feature around 401.7 eV (dark green).38,39,44 As the 3eg feature has a strong intensity for all calculations with low HFX (see Fig. 3 and ESI†), we assign the third prominent feature to the 3eg(π*) transition.
In agreement with previous calculations,34 the experimental shoulder around the ionization potential of the metal (M) porphyrins (402.9–403.4 eV, red) can be interpreted as a2u transition. This molecular orbital is formed by hybridization of the porphyrin a2u(π*) with M pz orbitals.
Above 405 eV, shape resonances can be identified in all experimental spectra (gray). Noticeable is the small blue-shift from MgTCPP to ZnTCPP to CuTCPP indicating a steady contraction of the M–N bond.51 This interpretation agrees with the calculated [and experimentally determined52] TPP bond length of 2.06 Å for Mg–N, 2.04 Å [2.04 Å] for Zn–N, 2.01 Å [1.98 Å] for Cu–N.
3.2 Covalency of the central metal complex
As shown in Fig. 4(b), the experimental 1eg resonance energy increases from N: in H2TCPP (397.9 eV) to MgTCPP (398.3 eV) to ZnTCPP (398.5 eV) to CuTCPP (398.7 eV) to N–H in H2TCPP (400.1 eV), which is well reproduced by TD-DFT BHandHLYP calculations for the TPP core with implicit solvation. This observation might be surprising, as the 1eg Gouterman orbital is known to be little influenced by the choice of the central moiety.10,53 Also differences in the electron–hole interaction between the TPPs and derivatives are expected to be marginal, as the large conjugate system leads to an equally low contribution of the highly delocalized π* orbital at the excited nitrogen atom for all investigated species.50As pointed out by García-Lastra et al.,40 the energy of the 1eg near-edge resonances mainly depends on the electron density at the nitrogen atoms. This is in line with our calculations as far as the negative Kohn–Sham orbital energies (with 50% HFX) are comparable with ionization potentials. Since a higher electron density at the nitrogen site leads to better screening of the core–hole and thereby lowers the 1eg excitation energy, the negative charge at the nitrogen atoms must decrease in the order: N: in H2TPP, MgTPP, ZnTPP, CuTPP, N–H in H2TPP (and equally for the TCPPs). Accordingly, the covalency of the N–X (X = Mg, Zn, Cu, H) bond increases. This finding is also in line with the more contracted N–X bond in CuTPP compared to ZnTPP and ZnTPP compared to MgTPP (as discussed above).
The Löwdin orbital populations of the BHandHLYP calculations with implicit solvation reveal an evolution of the complex covalent bond along the series: The N–Mg interaction is mainly characterized by the overlap of magnesium and nitrogen s and px/py orbitals (in the porphyrin plane). For the other metal porphyrins, an increased hybridization with metal dx2−y2 and dz2 atomic orbitals is noticeable. From zinc to copper, π bonding to metal dxz/dyz orbitals gains in importance. That is in line with the established strong mixing of unoccupied metal d and porphyrin π orbitals for open shell transition metals.53 In the NEXAFS spectra, the high covalency of the copper complex becomes apparent by the b1g ligand-to-metal charge transfer state, which is missing for the other porphyrins.
Hence, the systematic shift of the 1eg X-ray absorption resonance is a measure of the charge transfer from the porphyrin macrocycle to the metal center. MgTCPP is the most ionic species with an electron density at the nitrogen sites close to the iminic nitrogen atoms in H2TCPP. By orbital re-hybridization the ionicity is reduced for ZnTCPP. The strongest metal–porphyrin interactions are observed in CuTCPP where charge delocalization over both the porphyrin macrocycle and the central complex causes a decrease of the nitrogen charge density, which is lower only for N–H in H2TCPP.
3.3 Covalency to peripheral substituents
As seen in Fig. 4, the central metal atom within the porphyrin ring not only affects the 1eg resonance position but also the energy of the b2u feature. Especially from the comparison of MgTCPP and ZnTCPP (see Fig. 4b) it becomes clear that the interspecies shift of the b2u resonance exceeds the chemical shift of the N 1s core level discussed above.A shift of the b2u resonance has been experimentally established upon substitution of NiP forming NiTPP.41 The observed shift is well reproduced by our TD-DFT calculations with BHandHLYP (see ESI†). Analogously, the calculated spectra of P and TPP are compared for the iminic, magnesium, zinc, copper, and aminic center in Fig. 4(a). It can be seen that the enhanced experimental red-shift of MgTPP is reflected in a larger calculated MgP → MgTPP shift compared to CuP → CuTPP‡.
The experimental evidence for the red-shift upon P substitution with phenyl groups is mirrored in the calculations by CI mixing of the b2u transition with those to covalent porphyrin–phenyl orbitals (see phenyl orbital plot in Fig. 2b). In other words, the rehybridization and mixing of porphyrin and phenyl orbitals and their contribution in the CI expansion continuously decreases from MgTPP to ZnTPP to CuTPP.
The influence of peripheral substituents on the porphyrin electronic structure has rarely been discussed42,43,53 or explicitly excluded40 in literature. For TPPs this simplification has been justified by the assumption that the phenyl groups are oriented perpendicular (90°) to the porphyrin macrocycle.34 However, in practice there is a competition between steric repulsion and π overlap.54 Together with geometric distortions induced by the environment this competition leads to phenyl tilts down to 20° on surfaces.44 In our calculations, the phenyl tilt amounts to approximately 66°, which is in agreement with X-ray crystallographic data.55 In accordance with the assumed increase of phenyl-macrocycle π overlap the calculated phenyl tilt slightly decreases from CuTPP to ZnTPP to MgTPP (see ESI†).
The rise in intensity from CuTCPP to ZnTCPP to MgTCPP around 400 eV (see Fig. 2) can be interpreted as an indicator for increasing covalency leading to the formation of a N(1s) → phenyl(π*) feature in this energy region. This effect would be governed by mixing of the N 2pz orbital with the phenyl π* one. However, due to strong vibrational broadening of the intense π* resonances the identification of the phenyl/substituent peak is problematic, nevertheless, its indirect presence can be seen in the shift of the b2u resonance.
Experimental evidence for a change in absorbance around 400 eV in connection to a variation of the substituents is provided by Mangione et al.43 In their calculations the authors find an increased mixing between porphyrin and substituent orbitals upon fluorine decoration of the phenyl groups in CuTPP opposed to H2TPP.
In our series the macrocycle-substituent covalency is increased for a more ionic porphyrin center. That way, the lack of charge transfer with the metal for ZnTPP/ZnTCPP and especially MgTPP/MgTCPP is compensated by an increased charge delocalization onto the substituents. This observation matches with the behaviour of haem, that reduction of the system leads to a global increase of the porphyrin charge density including non-conjugate substituents.56
4 Conclusions
By the experimental determination and theoretical description of the N K-edge NEXAFS of a series of porphyrins, we have been able to clarify the interpretation of higher π* resonances beyond those of the well-known Gouterman orbitals. We find that even though the electron–hole interaction is of minor importance for relative excitation energies (due to the highly delocalized π system), relaxation effects are crucial for the estimation of excitation intensities. The TP-DFT method is therefore well suited for the description of porphyrin core-excited states. Charge transfer states, however, are only correctly described by hybrid functionals with high HFX, which currently yield theoretically sound results only for TD-DFT.The experimental and theoretical analysis of systematic interspecies shifts uncovered two trends: The ionicity of the porphyrin center increases from N–H to N–Cu to N–Zn to N–Mg to N:. At the same time the covalency with peripheral substituents increases to compensate for the high charge density at the nitrogen sites.
These findings directly relate to the different functions of metal porphyrins and derivatives. While the ionic character of Mg2+macrocycle2− is essential for the charge transfer from negatively charged chlorophyll radicals during photosynthesis, it also gives rise to the low stability of the isolated molecule.5 The substitution of magnesium by copper increases the stability of the complex enabling the human utilization beyond its photochemical properties. In the context of technical light-harvesting the observed metal controlled π hybridization of porphyrin and phenyl orbitals are of great interest. Cordones et al.48 raised the importance of electron localization in the excited state for the efficiency of dye-sensitized solar cells. Zinc porphyrins, which can – based on our results – be seen as a tradeoff between stability and low mixing of metal and porphyrin states, serve as excellent electron acceptors.48
Taken together, the investigation of TPP/TCPP orbitals beyond the Gouterman picture revealed the porphyrin center as a regulator for covalency in the entire molecule. In MgTPP/MgTCPP the central N–Mg bond is most ionic so that π hybridization with peripheral substituents is enhanced in compensation. CuTPP/CuTCPP is a counter example with a rather covalent metal–ligand interaction and low π hybridization to the phenyl substituents. Thereby we uncovered the connection between porphyrin constitution and resulting chemical properties which enables the versatile use of porphyrins and derivatives in general.
Author contributions
R. B.: data curation, investigation, project administration, visualization, writing – original draft; M. F.: resources, investigation; R. H.: investigation; A. P.: supervision; V. V. C.: formal analysis, investigation, supervision; A. F.: conceptualization, funding acquisition, supervision; all: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Mathias Senge for sharing his expertise on the chemistry of porphyrins. A. F. and R. B. acknowledge funding from the ERC-ADG-2014 – Advanced Investigator Grant No. 669531 EDAX under the Horizon 2020 EU Framework Program for Research and Innovation. The authors thank the Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime.References
- A. A. Lamola and T. Yamane, Science, 1974, 186, 936–938 CrossRef CAS PubMed.
- D. Todd, Br. J. Dermatol., 1994, 131, 751–766 CrossRef CAS PubMed.
- G. Calogero, G. Di Marco, S. Caramori, S. Cazzanti, R. Argazzi and C. A. Bignozzi, Energy Environ. Sci., 2009, 2, 1162–1172 RSC.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- W. Kaim, B. Schwederski and A. Klein, Bioinorganic Chemistry – Inorganic Elements in the Chemistry of Life: An Introduction and Guide, Wiley, Chichester, 2013 Search PubMed.
- A. A. Ryan and M. O. Senge, Photochem. Photobiol. Sci., 2015, 14, 638–660 CrossRef CAS PubMed.
- B. Gajewska, S. Raccio, K. J. Rodriguez and N. Bruns, Polym. Chem., 2019, 10, 125–135 RSC.
- R. Dashwood, Int. J. Oncol., 1997, 10, 721–727 CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- J. R. de Souza, M. M. F. de Moraes, Y. A. Aoto and P. Homem-de Mello, Phys. Chem. Chem. Phys., 2020, 22, 23886–23898 RSC.
- M. Fondell, S. Eckert, R. M. Jay, C. Weniger, W. Quevedo, J. Niskanen, B. Kennedy, F. Sorgenfrei, D. Schick, E. Giangrisostomi, R. Ovsyannikov, K. Adamczyk, N. Huse, P. Wernet, R. Mitzner and A. Föhlisch, Struct. Dyn., 2017, 4, 054902 CrossRef PubMed.
- A. Pietzsch and S. Eisebitt, J. Large-Scale Res. Facil., 2016, 2, A54 CrossRef.
- K. Kunnus, I. Rajkovic, S. Schreck, W. Quevedo, S. Eckert, M. Beye, E. Suljoti, C. Weniger, C. Kalus, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, K. J. Gaffney, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, S. Techert, P. Wernet and A. Föhlisch, Rev. Sci. Instrum., 2012, 83, 123109 CrossRef PubMed.
- P. S. Miedema, W. Quevedo and M. Fondell, J. Large-Scale Res. Facil., 2016, 2, A27 Search PubMed.
- R. Flesch, A. A. Pavlychev, J. J. Neville, J. Blumberg, M. Kuhlmann, W. Tappe, F. Senf, O. Schwarzkopf, A. P. Hitchcock and E. Rühl, Phys. Rev. Lett., 2001, 86, 3767–3770 CrossRef CAS PubMed.
- A. W. Gillespie, F. L. Walley, R. E. Farrell, T. Z. Regier and R. I. R. Blyth, J. Synchrotron Radiat., 2008, 15, 532–534 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- T. Fransson, I. E. Brumboiu, M. L. Vidal, P. Norman, S. Coriani and A. Dreuw, J. Chem. Theory Comput., 2021, 17, 1618–1637 CrossRef CAS PubMed.
- Z.-L. Cai, M. J. Crossley, J. R. Reimers, R. Kobayashi and R. D. Amos, J. Phys. Chem. B, 2006, 110, 15624–15632 CrossRef CAS PubMed.
- I. E. Brumboiu, G. Prokopiou, L. Kronik and B. Brena, J. Chem. Phys., 2017, 147, 044301 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Ehlert and T. Klamroth, J. Comput. Chem., 2020, 41, 1781–1789 CrossRef CAS PubMed.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill and T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CAS.
- C. Nicolas and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 267–272 CrossRef CAS.
- S. Narioka, H. Ishii, Y. Ouchi, T. Yokoyama, T. Ohta and K. Seki, J. Phys. Chem., 1995, 99, 1332–1337 CrossRef CAS.
- F. K. Gel'Mukhanov, L. Mazalov and A. Kondratenko, Chem. Phys. Lett., 1977, 46, 133–137 CrossRef.
- J. Stöhr, NEXAFS Spectroscopy, Springer, San Jose, 1992 Search PubMed.
- V. Vaz da Cruz, N. Ignatova, R. C. Couto, D. A. Fedotov, D. R. Rehn, V. Savchenko, P. Norman, H. Ågren, S. Polyutov, J. Niskanen, S. Eckert, R. M. Jay, M. Fondell, T. Schmitt, A. Pietzsch, A. Föhlisch, F. Gel'mukhanov, M. Odelius and V. Kimberg, J. Chem. Phys., 2019, 150, 234301 CrossRef PubMed.
- T. Okajima, Y. Yamamoto, Y. Ouchi and K. Seki, J. Electron Spectrosc. Relat. Phenom., 2001, 114-116, 849–854 CrossRef CAS.
- I. Reid, Y. Zhang, A. Demasi, A. Blueser, L. Piper, J. E. Downes, A. Matsuura, G. Hughes and K. E. Smith, Appl. Surf. Sci., 2009, 256, 720–725 CrossRef CAS.
- J. M. García-Lastra, P. L. Cook, F. J. Himpsel and A. Rubio, J. Chem. Phys., 2010, 133, 151103 CrossRef PubMed.
- G. I. Svirskiy, A. V. Generalov, A. Y. Klyushin, K. A. Simonov, S. A. Krasnikov, N. A. Vinogradov, A. L. Trigub, Y. V. Zubavichus, A. B. Preobrazhenski and A. S. Vinogradov, Phys. Solid State, 2018, 60, 581–591 CrossRef CAS.
- M. V. Nardi, R. Verucchi, L. Pasquali, A. Giglia, G. Fronzoni, M. Sambi, G. Mangione and M. Casarin, Phys. Chem. Chem. Phys., 2015, 17, 2001–2011 RSC.
- G. Mangione, M. Sambi, S. Carlotto, A. Vittadini, G. Ligorio, M. Timpel, L. Pasquali, A. Giglia, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 24890–24904 RSC.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- R. Golnak, J. Xiao, K. Atak, J. S. Stevens, A. Gainar, S. L. M. Schroeder and E. F. Aziz, Phys. Chem. Chem. Phys., 2015, 17, 29000–29006 RSC.
- N. Schmidt, R. Fink and W. Hieringer, J. Chem. Phys., 2010, 133, 054703 CrossRef PubMed.
- A. A. Cordones, C. D. Pemmaraju, J. H. Lee, I. Zegkinoglou, M.-E. Ragoussi, F. J. Himpsel, G. de la Torre and R. W. Schoenlein, J. Phys. Chem. Lett., 2021, 12, 1182–1188 CrossRef CAS PubMed.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, A. Wiengarten, S. Joshi, K. Seufert, D. Écija, W. Auwärter and J. V. Barth, J. Chem. Phys., 2013, 138, 154710 CrossRef CAS PubMed.
- H. Oji, R. Mitsumoto, E. Ito, H. Ishii, Y. Ouchi, K. Seki, T. Yokoyama, T. Ohta and N. Kosugi, J. Chem. Phys., 1998, 109, 10409–10418 CrossRef CAS.
- F. W. Lytle, R. B. Greegor and A. J. Panson, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 1550–1562 CrossRef CAS PubMed.
- E. B. Fleischer, C. K. Miller and L. E. Webb, J. Am. Chem. Soc., 1964, 86, 2342–2347 CrossRef CAS.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2002, 117, 205–219 CrossRef CAS.
- T. A. Jung, R. R. Schlittler and J. K. Gimzewski, Nature, 1997, 386, 696–698 CrossRef CAS.
- S. J. Silvers and A. Tulinsky, J. Am. Chem. Soc., 1967, 89, 3331–3337 CrossRef CAS PubMed.
- M. P. Johansson, M. R. Blomberg, D. Sundholm and M. Wikström, Biochim. Biophys. Acta, Gen. Subj., 2002, 1553, 183–187 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: UV/VIS spectra, NEXAFS decomposition, Molecular geometries, and DFT benchmark. See DOI: 10.1039/d1cp03944j |
| ‡ The b2u feature of ZnP cannot be unambiguously assigned in the TD-DFT BHandHLYP calculation, but seems to be lowered in energy due to strong CI with transitions to mixed zinc atomic and porphyrin π* orbitals. |
| This journal is © the Owner Societies 2021 |