
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical switching of positronium triplet quenching†
Philipp
Brunner
 *,
Eva-Maria
Steyskal
and
Roland
Würschum
*,
Eva-Maria
Steyskal
and
Roland
Würschum
Institute of Materials Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria. E-mail: philipp.brunner@tugraz.at
First published on 4th November 2021
Abstract
Switching of positronium triplet quenching could successfully be demonstrated by electrochemical means in an aqueous K3[Fe(CN)6] electrolyte. For this purpose a suitable cell was designed to combine positron annihilation with electrochemical measurements. Highly reversible substantial variations of the mean positron lifetime τm could be observed upon electrochemical switching between the oxidation states Fe(CN)63− and Fe(CN)64−, arising from oxidation of positronium by Fe(CN)63−. Dynamic in situ measurements in dependence of potential exhibit a hysteresis like behavior of τm which perfectly correlates with the shift between the reduction and oxidation peaks simultaneously monitored by cyclic voltametry.
1 Introduction
In situ measurement techniques allow direct monitoring of chemical and physical processes taking place on a microscopic scale in the sample of interest. So far, methods like X-ray and neutron diffraction, scanning probe and electron microscopy, optical techniques like Raman spectroscopy, or magnetometry are used especially in electrochemistry to investigate in situ, e.g., energy storage materials.1,2 Recently, our group introduced positron annihilation as in situ tool and demonstrated first operando positron lifetime measurements under full electrochemical control to study charging-induced defects in battery electrodes.3 In this work we further expand the application of positron annihilation and monitor for the first time in situ the switching of triplet quenching in aqueous electrolytes by electrochemical means.Positron and positronium chemistry has a wide area of application in analytical, organic, inorganic and physical chemistry,4,5 where, e.g., different kinds of solutions, polymers like polyethylene or porous materials such as zeolites are investigated.6–9 Positronium (Ps), a highly sensitive chemical probe, is a hydrogen-like bound state of an electron (e−) and a positron (e+), which either forms in the singlet state (para-Ps), with opposite spin direction of e− and e+, or in the triplet state (ortho-Ps) with spin-parallel configuration. para-Ps undergoes rapid annihilation in two γ-quanta with a short lifetime of 125 ps in vacuum, whereas ortho-Ps due to annihilation in three γ-quanta exhibits a much longer lifetime of about 140 ns. In condensed matter, e+ of ortho-Ps undergoes so-called pick-off annihilation with an electron of the surrounding molecules having opposite spin direction, which results in a two γ-annihilation with drastically reduced lifetime in the range 1 to 5 ns compared to the lifetime of ortho-Ps in vacuum. Furthermore, Ps is able to react with solved ions in aqueous solutions, e.g., via Ps oxidation, bound-state formation, or spin conversion, which also results in a reduction of the ortho-Ps lifetime. Beside these so called quenching mechanisms, some solutes also inhibit the Ps formation, which causes a decrease in the measured ortho-Ps intensity.4,5
Up to now, only ex situ positron annihilation studies have been performed in aqueous solutions. One particularly interesting species is the Fe(CN)63−/4− redox couple,10 used for a proof of concept in this work. Fe(CN)63− ions effectively oxidize Ps and, therefore, rapidly decrease the ortho-Ps lifetime, whereas in the reduced Fe(CN)64− state, a negligible quenching reaction and, therefore, a significantly longer Ps lifetime compared to Fe(CN)63− occurs.11–14 The present work aims at in situ control and monitoring of Ps quenching by electrochemically switching between these two oxidation states (Fe(CN)63− ↔ Fe(CN)64−) and simultaneously measuring the e+ lifetime.
2 Experimental
2.1 Electrochemical Cell
To combine electrochemical measurements with positron lifetime spectroscopy, a suitable cell set-up was designed (see Fig. 1). As shown in figure part a, the cell consists of two identically constructed sub-cells (upper and lower part), made mainly of polyetheretherketone (PEEK). Between these two sub-cells the e+ source is placed (for more details see Section 2.2). The whole cell set-up is mounted between the detectors of the positron lifetime spectrometer. The counter electrode (CE) is connected via a salt bridge to the sub-cell. On the CE side 0.1 mol L−1 KCl was used as electrolyte.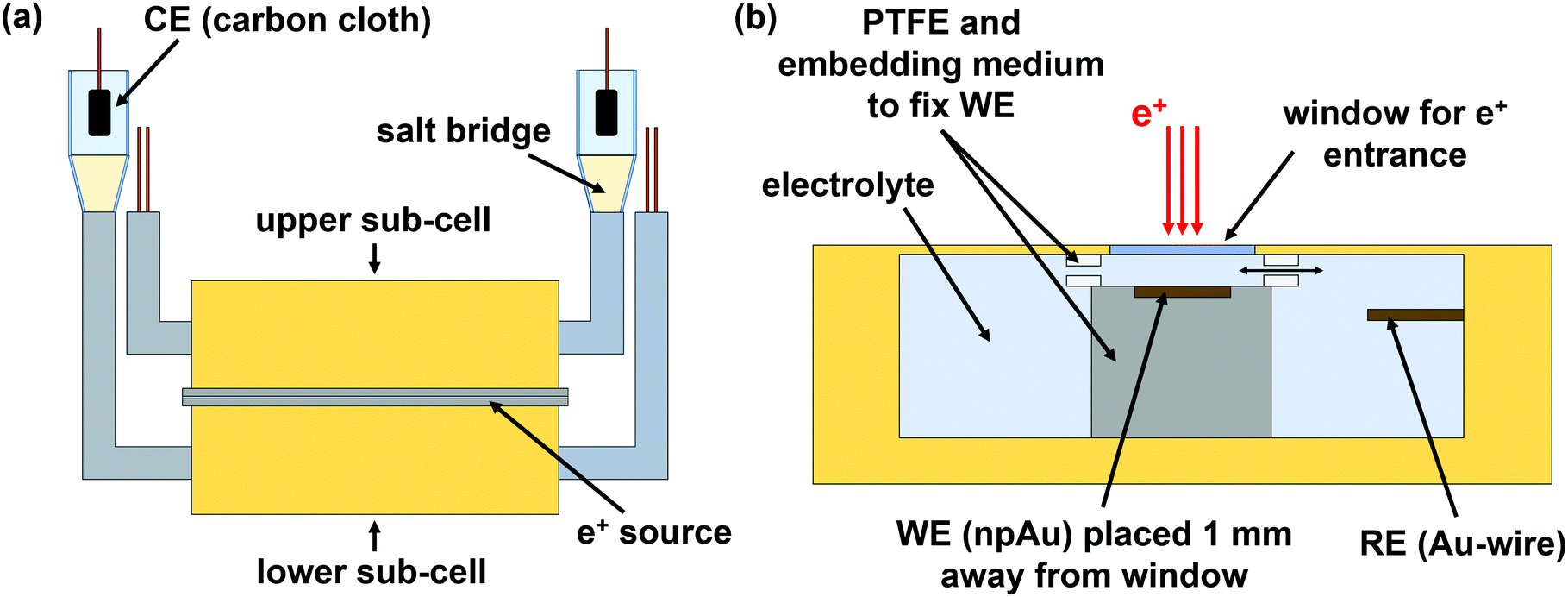 | ||
| Fig. 1 Cell designed to combine electrochemical with positron lifetime measurements. (a) The cell consists of two identically constructed sub-cells (upper and lower part). The counter electrode (CE) is connected via a salt bridge to the cell. Between the two sub-cells, the e+ source is placed. (b) Assembling inside the sub-cell showing the window for e+ entrance, the working electrode (WE), and the gold reference electrode (RE). e+ annihilation occurs in the 1 mm thick electrolyte layer above the WE. | ||
The assembling inside the sub-cell is depicted in Fig. 1b. The window for e+ entrance consists of a 15 μm thick hostaphan foil. The working electrode (WE), fixed between two PTFE pieces, is placed 1 mm away from the e+ entrance window in order to ensure that e+ annihilate quantitatively in the electrolyte. For electrolyte exchange, the PTFE pieces contain gaps. The reference electrode (RE) is placed next to the WE.
An AuAg (25at%75at%) alloy was produced by arc melting pieces of silver wire (0.5 mm diameter, 99.9%, chemPUR) and gold granules (99.99%, chemPUR) in corresponding mass ratio. A Mini Arc Melter MAM 1 from the Edmund Bühler GmbH was used as arc melter.
As reference electrodes (RE) gold wires, as counter electrodes (CE) carbon cloths connected to gold wires were used. All given potentials refer to the gold RE.
2.2 Measurement devices
The e+ lifetime measurements were performed with an analogous fast–fast positron lifetime spectrometer with a time resolution of 165 ps. For a more detailed description of the measurement set-up see, e.g., Keeble et al.18 The e+ source consists of 22NaCl encapsulated between 5 μm thick Al foils. For protecting the e+ source, it was covered on both sides by a 15 μm thick Hostaphan foil and clamped between two Al frames. Spectra were recorded for 30 min and subsequently summed up. The total amount of counts of the final spectrum depend on the type of measurement done (for more details see Section 2.3). The recorded spectra were analyzed via a three components fit after source and Hostaphan foil correction by means of the program PALSFIT.19 A silicon reference sample was used to determine the contribution of the source and the Hostaphan foil consisting of two components of 465 ps and 1700 ps with relative intensities of 10% and 3%. For this purpose in the common sample–source–sample set-up two Hostaphan foils were placed between the source (22NaCl–Al foil arrangement) and the sample. A detailed description of the fitting models used by PALSFIT, including the determination of numerical uncertainties, is given by Kirkegaard et al.20 Due to the correlations of the lifetime components τi and their relative intensities Ii, the covariance matrix is used to estimate the numerical uncertainty of the mean e+ lifetime τm (see eqn (97) in ref. 20).For the electrochemical measurements an AUTOLAB potentiostat (AUT50611, METROHM) and the NOVA 1.11 software were used.
2.3 Measurement procedures
Table 1 lists the electrolytes used for the different types of measurements. An alkaline environment was chosen to avoid the formation of prussian blue.21,22| Beaker | Static switch | Dynamic | Ex situ | |
|---|---|---|---|---|
| 1 mmol L−1 KOH | X | X | X | |
| 30 mmol L−1 K3[Fe(CN)6] + 1 mmol L−1 KOH | X | X | X | X |
| 50 mmol L−1 K3[Fe(CN)6] + 1 mmol L−1 KOH | X | X | X | |
| 30 mmol L−1 K4[Fe(CN)6] + 1 mmol L−1 KOH | X | |||
| 50 mmol L−1 K4[Fe(CN)6] + 1 mmol L−1 KOH | X |
The static in situ studies were performed with applied voltages of 150 mV and −350 mV measuring for 9.5 h in the case of K3[Fe(CN)6] with 1 mmol L−1 KOH and for 8 h in the case of 1 mmol L−1 KOH reference. The potential was varied with a scan rate of 2 mV s−1 upon switching in order to prevent potential jumps. A cutout of the measurement procedure is given in the ESI.† During the entire measurement sequence, e+ lifetime spectra were recorded and the cell current was monitored. The first two e+ lifetime subspectra at each potential were excluded. Spectra taken for K3[Fe(CN)6] contain at least 4.0 × 105 counts, for 1 mmol L−1 KOH reference at least 2.8 × 105 counts.
For the dynamic in situ measurement, a linear potential sweep with 2 mV s−1 was performed from the OCP value to 150 mV. Thereafter a CV with one cycle between −350 and 150 mV and a scan rate of 5.6 μV s−1 was conducted and simultaneously e+ lifetime spectra were recorded. The significantly slower scan rate for this type of measurement, as compared to, e.g., the beaker measurement, is required in order to obtain a sufficient amount of counts per recorded spectra. Five 30 min spectra were summed up to one total spectrum with about 1.0 × 105 counts. Each of these total spectra corresponds to one τm data point in Fig. 3. Each τm data point is assigned to the potential value corresponding to the middle of the linear voltage sweep over the 2.5 h measuring time.
3 Results and discussion
The electrochemical behavior of the Fe(CN)63−/4− redox couple is shown in Fig. 2. Cyclic voltammograms (CVs) of 30 mmol L−1 K3[Fe(CN)6] and 1 mmol L−1 KOH (blue), and of 50 mmol L−1 K3[Fe(CN)6] and 1 mmol L−1 KOH (red) were recorded in a beaker. The shapes of the measured CVs correspond well to literature.10,24 Upon applying negative (cathodic) potentials and passing the reduction peak between −250 and −300 mV, Fe(CN)63− reduces to Fe(CN)64−. Changing the scan direction and applying positive (anodic) potentials leads to the reverse reaction, i.e., the oxidation of Fe(CN)64− to Fe(CN)63− upon passing the oxidation peak between −75 and −50 mV. In the CV of the 1 mmol L−1 KOH reference electrolyte (gray) no reduction and oxidation peaks are visible in contrast to the CVs of 30 mmol L−1 and 50 mmol L−1 K3[Fe(CN)6], i.e., there are no detectable changes in the electrolyte or on the electrode surface in the applied potential range associated with KOH. | ||
| Fig. 2 Cyclic voltammograms (CVs) of 1 mmol L−1 KOH (gray), of 30 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH (blue), and of 50 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH (red) measured between −400 and 200 mV with a scan rate of 5 mV s−1. The arrows indicate the scan direction. Note that these CVs were conducted in a beaker, where the npAu electrode served as working and a gold wire as reference electrode; the carbon cloth, which was used as counter electrode, was connected via the salt bridge to the beaker. On the counter electrode side 0.1 mol L−1 KCl was used as electrolyte. | ||
As a first step, ex situ measurements (without applied potentials) of the Fe(CN)63−/4− redox couple and the 1 mmol L−1 KOH reference solution are made with the cell set-up used for in situ measurements (set-up described in Fig. 1). In Table 2 the fitted mean e+ lifetimes τm and the e+ lifetime components τi with relative intensities Ii are shown. The ortho-Ps lifetimes τ3 for 30 and 50 mmol L−1 K4[Fe(CN)6] ranging from 1810 to 1830 ps correspond very well to the values of about 1800 ps reported in literature.11 In contrast to Fe(CN)64−, Fe(CN)63− strongly oxidizes Ps and, therefore, drastically reduces τ3.11–14 The intensities for the τ3 components are fixed in the fitting procedure, since according to Duplatre et al.11 a similar Ps inhibition behavior can be expected for both oxidation states. A fit with fixed I3 gives τ3 components with 996 and 789 ps for 30 and 50 mmol L−1 K3[Fe(CN)6] respectively, in good accordance with 865 and 645 ps given in literature.11 For the sake of completeness and for reasons of traceability the fitting results for K3[Fe(CN)6] with unfixed I3 components are given in Table 2. Without fixation, smaller I3 and longer τ3 values were determined for K3[Fe(CN)6], which may arise from analysis uncertainties due to strong numerical correlations between I3 and τ3.
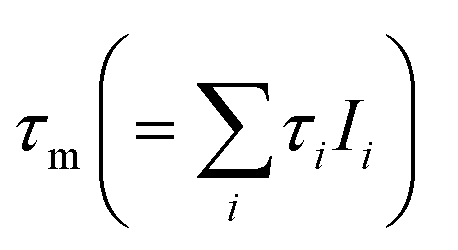 and e+ lifetime components τi with relative intensities Ii (i = 1, 2, 3); values in brackets: numerical uncertanties; fix: component fixed
and e+ lifetime components τi with relative intensities Ii (i = 1, 2, 3); values in brackets: numerical uncertanties; fix: component fixed
| τ 1 (ps) | I 1 (%) | τ 2 (ps) | I 2 (%) | τ 3 (ps) | I 3 (%) | τ m (ps) | |
|---|---|---|---|---|---|---|---|
| 1 mmol L−1 KOH | 213 (6) | 53.8 (4.5) | 416 (24) | 31.7 (4.2) | 1862 (36) | 14.5 (0.4) | 516 (3) |
| 30 mmol L−1 K4[Fe(CN)6] | 210 (10) | 48.4 (7.8) | 365 (23) | 39.3 (7.6) | 1811 (37) | 12.3 (0.3) | 468 (3) |
| 30 mmol L−1 K3[Fe(CN)6] | 217 (7) | 50.3 (4.8) | 442 (28) | 41.8 (3.7) | 1187 (80) | 7.9 (1.3) | 388 (2) |
| 30 mmol L−1 K3[Fe(CN)6] | 200 (8) | 37.6 (4.2) | 373 (10) | 50.1 (4.2) | 996 (10) | 12.3 (fix) | 385 (1) |
| 50 mmol L−1 K4[Fe(CN)6] | 216 (8) | 54.5 (6.1) | 392 (25) | 33.7 (5.8) | 1832 (40) | 11.8 (0.3) | 466 (3) |
| 50 mmol L−1 K3[Fe(CN)6] | 226 (9) | 49.5 (6.1) | 437 (30) | 46.3 (4.8) | 1106 (160) | 4.2 (1.7) | 360 (1) |
| 50 mmol L−1 K3[Fe(CN)6] | 200 (13) | 30.4 (6.0) | 353 (11) | 57.8 (6.0) | 789 (11) | 11.8 (fix) | 358 (1) |
The observed τ3-variation is considered to arise exclusively from Fe(CN)63− and Fe(CN)64−, since both OH− ions at low concentrations and K+ counterions show negligible quenching and inhibition properties.25,26. The fitted τ1 and τ2 components for 1 mmol L−1 KOH, K3[Fe(CN)6] and K4[Fe(CN)6], as well as the τ3 component for 1 mmol L−1 KOH correspond well to values found in literature for pure water.8,27
The mean e+ lifetimes 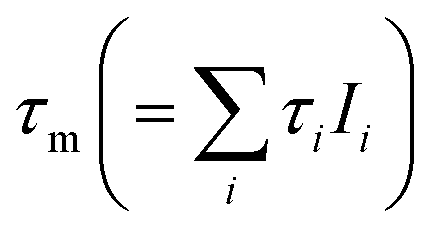 also shown in Table 2 reflect the variation of the Ps component τ3. The reduction in τm for K3[Fe(CN)6] (388 and 360 ps) compared to K4[Fe(CN)6] (468 and 466 ps) is due to Ps oxidation. The reduction in τm for K4[Fe(CN)6] (468 and 466 ps) compared to 1 mmol L−1 KOH (516 ps) is a result of Ps inhibition.11 For the in situ measurements, presented next, only the mean e+ lifetimes are quoted, since less counts could be recorded per spectrum, but τm can still be determined with high reliability even for reduced statistical accuracy.
also shown in Table 2 reflect the variation of the Ps component τ3. The reduction in τm for K3[Fe(CN)6] (388 and 360 ps) compared to K4[Fe(CN)6] (468 and 466 ps) is due to Ps oxidation. The reduction in τm for K4[Fe(CN)6] (468 and 466 ps) compared to 1 mmol L−1 KOH (516 ps) is a result of Ps inhibition.11 For the in situ measurements, presented next, only the mean e+ lifetimes are quoted, since less counts could be recorded per spectrum, but τm can still be determined with high reliability even for reduced statistical accuracy.
At first, static in situ measurements at defined potentials were performed with the electrolyte being either in the Fe(CN)63− (150 mV) or the Fe(CN)64− state (−350 mV). Fig. 3 shows the mean e+ lifetimes τm upon voltage switching between 150 mV and −350 mV for K3[Fe(CN)6] (30 mmol L−1: blue, circle; 50 mmol L−1: red, square) with 1 mmol L−1 KOH and for the pure 1 mmol L−1 KOH reference (gray, diamont). Highly reversible changes in τm of about 70 ps for 30 mmol L−1 and 90 ps for 50 mmol L−1 are observed by switching the oxidation state from Fe(CN)63− to Fe(CN)64− and vice versa. For the 1 mmol L−1 KOH reference, only a minor variation of τm within the scope of the error margin occurs upon voltage switching. This clearly demonstrates that the observed switching of τm exclusively results from the Fe(CN)63−/4− redox couple, i.e., from the switching of Ps triplet quenching which represents the central result of the present work.
 | ||
| Fig. 3 Mean e+ lifetimes τm of 30 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH (blue, circle) and 50 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH (red, square) measured in situ at potentials of 150 mV and −350 mV. According to Fig. 2, 150 mV corresponds to the Fe(CN)63−, −350 mV to the already reduced Fe(CN)64−. For comparison, τm-values of ex situ measurements (without applied potential) of K3[Fe(CN)6] and K4[Fe(CN)6] with 1 mmol L−1 KOH are shown (30 mmol L−1: blue, dotted line; 50 mmol L−1: red, dash–dotted line). In addition, τm-values are plotted for the 1 mmol L−1 KOH reference electrolyte measured in situ (gray, diamond) and measured ex situ (gray, dashed line). Unless shown, error bars of the data points are smaller than the marker size. | ||
The τm values of the in situ and ex situ measurements for both oxidation states match pretty well. The slightly reduced total variation of τm may be due to a slightly incomplete oxidation and reduction since the static positron lifetime measurements were recorded over a time span of several hours, starting upon voltage switching to achieve sufficiently high statistical accuracy. In fact, omitting the initial part of the measurements yield higher τm-variations, however, on expense of statistical accuracy. The final reduction (last measurement at 150 mV) yields clearly longer τm-values of 398 ps for 30 mmol L−1 and 382 ps for 50 mmol L−1 Fe(CN)63−, as compared to 388 and 367 ps for the initial measurement at 150 mV. This deviation may arise from electrolyte degradation, as indicated by a slight turbidity of the electrolyte, perhaps due to a formation of Fe3+/Fe2+-cyanide colloids or of a small fraction of prussian blue dispersed in the electrolyte.10,21,22
Next, dynamic in situ measurements are presented (see Fig. 4). Here, the mean e+ lifetime τm is measured for 30 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH in dependence of a slowly changing potential U of 5.6 μV s−1 between 150 and −350 mV, simultaneously monitoring the current I as plotted as CV. The dynamic CV in Fig. 4 slightly differs from the CV in Fig. 2 due to the parallel connection of the two sub-cells and the significantly slower scan rate. The mean e+ lifetimes in both oxidation states perfectly fit with the ex situ measurements (black, dashed lines) indicating that the entire electrolyte undergoes reduction and oxidation upon cycling. The τm − U variation exhibits a remarkable hysteresis like behavior which exactly resembles the shift between the reduction and oxidation peak according to the CV. Starting in the Fe(CN)63− state, upon reduction to Fe(CN)64− at around −90 mV, τm strongly increases. Reversing the scan direction, upon oxidation to Fe(CN)63− at around 50 mV, τm strongly decreases to the starting value. The dynamic scan impressively shows that also the region of mixed oxidation state is accessible to positron annihilation yielding τm-values in between those of the fully oxidized or reduced state.
 | ||
| Fig. 4 Dynamic in situ measurement of the mean e+ lifetime τm in 30 mmol L−1 K3[Fe(CN)6] with 1 mmol L−1 KOH upon slowly changing potential U with a rate of 5.6 μV s−1 between 150 and −350 mV. The simultaneously measured current I is plotted in a cyclic voltammogram (CV). The arrows indicate the scan direction. The ex situ measurements (without applied voltage) of 30 mmol L−1 K3[Fe(CN)6] and K4[Fe(CN)6], each with 1 mmol L−1 KOH, are plotted as black, dashed lines, for comparison. | ||
4 Conclusion
In conclusion, electrochemically controlled positronium triplet quenching in an aqueous electrolyte (K3[Fe(CN)6]) was for the first time successfully in situ monitored. Switching between two different potentials, corresponding to Fe(CN)63− and Fe(CN)64− oxidation states, show highly reversible changes in the τm-value, which could be associated with Ps oxidation by Fe(CN)63−. Dynamic measurements allow to exactly correlate Ps annihilation charactersitics with cyclic voltametry. As an outlook, the high sensitivity of the e+ lifetime towards already small changes in the oxidation state of the molecules in the electrolyte can be used, e.g., to study the charging/discharging behavior in an iron-based aqueous redox flow battery (IBA-RFB). In IBA-RFBs the Fe2+/Fe3+ redox couple is used as the catholyte.28 Fe2+ and Fe3+ are known to affect Ps formation and annihilation quite differently,11,13 making this application relevant redox couple highly suitable to study the charging–discharging behavior of the catholyte in IBA-RFBs by in situ positron annihilation. A further field of application would be to change in situ the concentration depending Ps quenching (spin conversion or oxidation) and inhibition behavior of, e.g., Cu2+ ions,29,30 by electrochemical means.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to Wolfgang Sprengel (Institute of Materials Physics, TU Graz) for technical support and for valuable comments. Furthermore the authors thank Gregor Klinser (former member of the Institute of Materials Physics, TU Graz) for the support during the preparation of the in situ e+ measurements. The work was performed in the framework of the interuniversity cooperation of TU Graz and Uni Graz on natural sciences (NAWI Graz).Notes and references
- P. Harks, F. Mulder and P. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS.
- S. Topolovec, H. Krenn and R. Würschum, Rev. Sci. Instrum., 2015, 86, 063903 CrossRef PubMed.
- G. Klinser, H. Kren, S. Koller and R. Würschum, Appl. Phys. Lett., 2019, 114, 013905 CrossRef.
- O. E. Mogensen, Positron annihilation in chemistry, Springer, 1995 Search PubMed.
- J. Y. Jean, P. E. Mallon and D. M. Schrader, Principles and applications of positron and positronium chemistry, World Scientific, 2003 Search PubMed.
- J. Serna, J. C. Abbe and G. Duplătre, Phys. Status Solidi A, 1989, 115, 389–397 CrossRef.
- W. Magalhaes, J. C. Abbé and G. Duplâtre, Chem. Phys., 1989, 136, 141–149 CrossRef CAS.
- G. Duplatre, A. Haessler and J. Abbe, J. Phys. Chem., 1985, 89, 1756–1760 CrossRef CAS.
- Z. Kajcsos, L. Liszkay, G. Duplâtre, L. Lohonyai, L. Varga, K. Lázár, G. Pál-Borbély, H. Beyer, P. Caullet and J. Patarin, et al. , Radiat. Phys. Chem., 2003, 68, 363–368 CrossRef CAS.
- N. Frenzel, J. Hartley and G. Frisch, Phys. Chem. Chem. Phys., 2017, 19, 28841–28852 RSC.
- G. Duplatre, J. Talamoni, J. C. Abbe and A. Haessler, Radiat. Phys. Chem., 1984, 23, 531–536 CrossRef CAS.
- A. L. F. Lazzarini and E. Lazzarini, Int. J. Radiat. Appl. Instrum., Part C. Radiat. Phys. Chem., 1990, 36, 377–381 CAS.
- S. Tao, Appl. Phys., 1976, 10, 67–79 CAS.
- A. L. Nichols, R. E. Wild, L. J. Bartal and H. J. Ache, Appl. Phys., 1974, 4, 37–40 CAS.
- E.-M. Steyskal, M. Seidl, M. Graf and R. Würschum, Phys. Chem. Chem. Phys., 2017, 19, 29880–29885 RSC.
- C. Lakshmanan, R. Viswanath, S. Polaki and R. Rajaraman, AIP Conf. Proc., 2015, 140033 CrossRef.
- S. Cattarin, D. Kramer, A. Lui and M. M. Musiani, J. Phys. Chem., 2007, 111, 12643–12649 CrossRef CAS PubMed.
- D. J. Keeble, U. Brossmann, W. Puff and R. Würschum, Charact. Mater., 2002, 1–28 Search PubMed.
- J. V. Olsen, P. Kirkegaard, N. J. Pedersen and M. Eldrup, Phys. Status Solidi C, 2007, 4, 4004–4006 CrossRef CAS.
- P. Kirkegaard, J. V. Olsen and M. Eldrup, PALSfit3: A software package for analysing positron lifetime spectra, Technical University of Denmark, 2017 Search PubMed.
- A. Abbaspour and M. A. Kamyabi, J. Electroanal. Chem., 2005, 584, 117–123 CrossRef CAS.
- R. Yang, Z. Qian and J. Deng, J. Electrochem. Soc., 1998, 145, 2231 CrossRef CAS.
- R. G. Compton and C. E. Banks, Understanding voltammetry, World Scientific, 2018 Search PubMed.
- Y. Iwasaki, T. Horiuchi, M. Morita and O. Niwa, Surf. Sci., 1999, 427, 195–198 CrossRef.
- C. Beling and F. Smith, Radiat. Phys. Chem., 1984, 23, 571–577 CrossRef CAS.
- G. Duplatre, J. C. Abbé, J. Talamoni, J. C. Machado and A. Haessler, Chem. Phys., 1981, 57, 175–183 CrossRef CAS.
- P. Stepanov, F. Selim, S. Stepanov, A. Bokov, O. Ilyukhina, G. Duplâtre and V. Byakov, Phys. Chem. Chem. Phys., 2020, 22, 5123–5131 RSC.
- H. Zhang and C. Sun, J. Power Sources, 2021, 493, 229445 CrossRef CAS.
- P. De Natale, A. Dupasquier, A. Fantola-Lazzarini, E. Lazzarini and A. Rolando, Il Nuovo Cimento D, 1993, 15, 1215–1228 CrossRef.
- B. Ganguly, V. Subrahmanyam and P. Sarkar, J. Radioanal. Nucl. Chem., 1994, 181, 201–209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03828a |
| This journal is © the Owner Societies 2021 |