
An overview from simple host–guest systems to progressively complex supramolecular assemblies
Mhejabeen
Sayed
 *ab and
Haridas
Pal
*bc
*ab and
Haridas
Pal
*bc
aRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai, 400085, India. E-mail: msayed@barc.gov.in; Fax: +91-22-25505151
bHomi Bhabha National Institute, Anushaktinagar, Mumbai, 400094, India
cAnalytical Chemistry Division, Bhabha Atomic Research Centre, Mumbai, 400085, India. E-mail: hpal@barc.gov.in
First published on 17th November 2021
Abstract
Supramolecular chemistry involving macrocyclic hosts is a highly interdisciplinary and fast-growing research field in chemistry, biochemistry, and materials science. Host–guest based supramolecular assemblies, as constructed through non-covalent interactions, are highly dynamic in nature, and can be tuned easily using their responses to various external stimuli, providing a convenient approach to achieve excellent functional materials. Macrocyclic hosts, particularly cyclodextrins, cucurbit[n]urils, and calix[n]arenes, which have unique features like possessing hydrophobic cavities of different sizes, along with hydrophilic external surfaces, which are also amenable towards easy derivatizations, are versatile cavitands or host molecules to encapsulate diverse guest molecules to form stable host–guest complexes with many unique structures and properties. Interestingly, host–guest complexes possessing amphiphilic properties can easily lead to the formation of various advanced supramolecular assemblies, like pseudorotaxanes, rotaxanes, polyrotaxanes, supramolecular polymers, micelles, vesicles, supramolecular nanostructures, and so on. Moreover, these supramolecular assemblies, with varied morphologies and responsiveness towards external stimuli, have immense potential for applications in nanotechnology, materials science, biosensors, drug delivery, analytical chemistry and biomedical sciences. In this perspective, we present a stimulating overview, discussing simple host–guest systems to complex supramolecular assemblies in a systematic manner, aiming to encourage future researchers in this fascinating area of supramolecular chemistry to develop advanced supramolecular materials with superior functionalities, for their deployment in diverse applied areas.
 Mhejabeen Sayed | Dr Mhejabeen Sayed obtained her PhD (2012) in Chemistry from the University of Mumbai, India. She joined the Chemistry Department of the School of Science, Narsee Monjee Institute of Management Studies, Mumbai, as an Assistant Professor for a brief period. Later on, she joined the Radiation and Photochemistry Division of the Bhabha Atomic Research Centre (BARC), Mumbai, India, initially as a prestigious K. S. Krishnan Research Associate, and finally joined as a Scientist, after her successful completion of a research associateship. Her research interests include supramolecular and suprabiomolecular photochemistry involving both macrocyclic hosts (cyclodextrins, calixarenes and cucurbiturils) and biomacromolecules (proteins and DNA), exploring the enhanced stabilization and solubilization of dyes/drugs and their controlled binding and release, to find their applications in drug delivery mechanisms, molecular sensors, on–off switches, catalysis, aqueous dye lasers, and so on. Her research interests also include understanding the excited-state processes, solvatochromism, and electron and proton transfer processes in biologically important dyes/drugs. |
 Haridas Pal | Prof. Haridas Pal did his PhD (1992) in Chemistry at the University of Mumbai. He did his post-doctoral studies for two years during 1994–1996 at the Institute of Molecular Sciences, Okazaki, Japan. He also visited the University of Heidelberg for six months during 2004–2005 as a visiting scientist. His major research interests include fast and ultrafast electron and proton transfer processes, solvatochromism, supramolecular chemistry, single molecule spectroscopy, and related areas. Prof. Pal received the prestigious “Homi Bhabha Science & Technology Award (2008)” from the Department of Atomic Energy and the “Bronze Medal (2011)” from the Chemical Research Society of India. For his outstanding research contributions, Prof. Pal was also elected as a fellow of the Maharashtra Academy of Sciences in 2005 and as a fellow of the National Academy of Sciences, India, in 2005. |
Introduction
Supramolecular chemistry deals with the well-organized association of smaller constituent molecular units through reversible non-covalent interactions to form complex structural assemblies that possess new properties and improved functionalities, which are entirely different from those of their individual components.1–3 The term “supramolecular chemistry” was coined by Lehn in 1978 to suggest that it is the “chemistry beyond molecules”.2 Supramolecular chemistry plays many vital roles in several biological processes, namely, selective substrate–enzyme interactions, specific DNA–protein interactions, hydrogen bond guided interactions among nucleobases in DNA and RNA, antigen–antibody interactions, and many others.2,4–6Since the mid-twentieth century, the supramolecular chemistry of synthetic macrocyclic hosts has received tremendous interest in multidisciplinary research areas in chemistry and biology. The formation of supramolecular assemblies essentially relies on the mutual recognition of the constituent molecular entities in specific manners. The first artificial host–guest complex formation was reported by Pedersen in 1967, whereby the selective recognition of alkali metal cations as guests was realized through the use of crown ether as the artificial macrocyclic host, discovered by the author.7 It should be noted that the crown ethers are indeed the first-generation macrocyclic hosts used in supramolecular host–guest chemistry.7 Since the first report by Pedersen, host–guest chemistry has become an important research area in supramolecular chemistry, providing ample opportunities in the design and construction of intriguing assemblies for various applications, especially in the analytical chemistry, sensors, nanotechnology, and biomedical domains.8–12
Contrary to conventional molecular chemistry that deals with covalent bonds, host–guest chemistry relies simply on dynamic non-covalent interactions. Thus, supramolecular systems can be modulated easily using their strong responses to various external stimuli like pH, light, salt, electrical input, temperature, enzymes, chemical agents, etc.,10–16 providing a very convenient method to achieve excellent tunability of the properties of such materials, making them very useful for applications in various areas like molecular sensors, drug delivery, nanomedicines, biosensors, catalysis, biomimetic chemistry, materials science, molecular electronics, and so on.10–12,17–21
In supramolecular host–guest complexes, the guest molecules are encapsulated either partially or entirely into the cavities of the macrocyclic cavitand or host molecules. Thus, the compatibility in the size and shape of the host cavity and the guest molecule is essential in the formation of the host–guest complex. In such systems, the mutual orientations of the host and guest units are again determined by the specific interactions that participate between the recognition sites of the macrocyclic hosts and the functional groups of the guest molecules.3,21–24 Various non-covalent interactions can collectively provide stability to the host–guest complexes and these include electrostatic, hydrophobic, hydrogen bonding, C–H–π, and van der Waals interactions. The multipoint contacts between the constituent units and collective participation of several non-covalent interactions result in the formation of supramolecular host–guest complexes with remarkable properties, which are useful for various applications.3,21–24
Though macrocyclic hosts with smaller cavity sizes usually favor the formation of simple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric complexes, hosts with larger cavity sizes can often lead to the formation of higher stoichiometric complexes. Furthermore, host–guest complexes that possess substantial amphiphilic character can also lead to the formation of functional nano-assemblies with larger complexity, namely in the form of pseudorotaxanes, rotaxanes, supramolecular polymers, micelles, vesicles, supramolecular nanotubes, and so on.21,25–29 Versatility in the derivatization of the macrocyclic hosts has additionally motivated researchers to develop new cavitand molecules with advanced functionalities and applications.22,23 Diverse classes of the host molecules discovered to date include cyclodextrins, cucurbit[n]urils, calix[n]arenes, pillarenes, crown ethers, cyclophanes, cryptands, and many others. These macrocyclic hosts have reasonably rigid hydrophobic cavities with varying dimensions and molecular recognition characteristics. Since the field of host–guest chemistry is extremely vast, in the present article we have restricted our discussion only to cyclodextrin (CD), cucurbit[n]uril (CBn), and calix[n]arene (CXn) based host molecules.
:1 stoichiometric complexes, hosts with larger cavity sizes can often lead to the formation of higher stoichiometric complexes. Furthermore, host–guest complexes that possess substantial amphiphilic character can also lead to the formation of functional nano-assemblies with larger complexity, namely in the form of pseudorotaxanes, rotaxanes, supramolecular polymers, micelles, vesicles, supramolecular nanotubes, and so on.21,25–29 Versatility in the derivatization of the macrocyclic hosts has additionally motivated researchers to develop new cavitand molecules with advanced functionalities and applications.22,23 Diverse classes of the host molecules discovered to date include cyclodextrins, cucurbit[n]urils, calix[n]arenes, pillarenes, crown ethers, cyclophanes, cryptands, and many others. These macrocyclic hosts have reasonably rigid hydrophobic cavities with varying dimensions and molecular recognition characteristics. Since the field of host–guest chemistry is extremely vast, in the present article we have restricted our discussion only to cyclodextrin (CD), cucurbit[n]uril (CBn), and calix[n]arene (CXn) based host molecules.
The CD family of the macrocyclic hosts are the classical container shaped cyclic oligosaccharides composed of D-(+)-glucopyranose units joined by α-(1–4)-glycosidic linkages, discovered by Villiers, in 1891.30 These cavitand molecules possess three-dimensional truncated cone-shaped structures, resulting in an inner hydrophobic cavity and two hydrophilic portals or rims, the narrower one being composed of the primary hydroxyl groups and the wider one being laced with secondary hydroxyl groups. Based on the number of glucopyranose units present, different CD homologs with diverse cavity sizes are available, namely αCD, βCD, and γCD, having six, seven, and eight monomer units, respectively (cf.Scheme 1). While CDs show substantial stability, biocompatibility, low toxicity, and excellent water solubility, their hydroxyl groups are also amenable towards easy functionalization.1,23,24,31–36 Accordingly, numerous CD derivatives have been synthesised with enhanced solubility and guest binding properties as compared to those of the parent CD hosts.22–24,32,33,35–38 In the literature, parent αCD and βCD hosts and their derivatives are reported to interact with organic guest molecules mostly through the formation of inclusion complexes with 1:1 stoichiometry. On the contrary, γCD and its derivatives can often accommodate two guest molecules simultaneously, as their cavities are substantially larger, leading to the formation of 1:2 host-to-guest complexes.22–24,32,33 Sometimes, other higher order complexes are also formed involving γCD and its derivatives.22,25 Using CD based hosts, various complexation studies have been reported in the literature involving different guest molecules, demonstrating simple to complicated supramolecular structures,1,22,23,25,31 some of which will be considered in this article as representative examples for discussion.
 | ||
| Scheme 1 Typical shape and chemical structure of cyclodextrin hosts. The cavity dimensions of αCD, βCD, and γCD hosts are also tabulated here for their quick comparison. | ||
In supramolecular chemistry, the CBn family of hosts have received substantial attention due to their superior recognition ability for various organic guests, particularly cationic guests. CBn hosts are highly symmetrical and quite rigid pumpkin-shaped cavitand structures constituting glycoluril units joined in a cyclic manner through methylene bridges. Depending upon the number of glycoluril units, different CBn homologs are present, namely, CB5, CB6, CB7, CB8, CB10, and CB14, having 5, 6, 7, 8, 10, and 14 glycoluril units, respectively.9,13,23,31,34,39–46 Among the CBn homologues, the CB5 to CB8 molecules (cf.Scheme 2) have been investigated quite extensively in host–guest chemistry. However, studies involving CBn derivatives are relatively rare compared to those on the parent CBn hosts, especially because of the difficulty in the derivatization of the CBn molecules.40
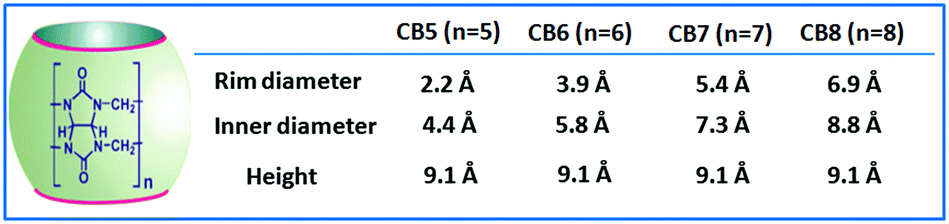 | ||
| Scheme 2 Typical shape and chemical structure of CBn hosts. The cavity dimensions of CB5, CB6, CB7, and CB8 hosts are also tabulated here for their quick comparison. | ||
The cavities of CBn hosts are very hydrophobic in nature due to their low polarity and polarizability. However, the portals of these macrocyclic cavities are reasonably polar in nature, as they are laced with highly polarisable carbonyl groups. Endowed with these exciting structural features, CBn hosts can interact quite strongly with various organic guests involving strong ion–dipole, dipole–dipole, and hydrophobic interactions in a collective manner.9,13,22,23,31,34,41,42 Accordingly, CBn hosts in general form strong inclusion complexes with many organic guests, with binding constants (Kb) typically in the order of 105 M−1, and in exceptional cases the Kb value can even become unusually high, such as in the range of 1010–1017 M−1.47,48 The water solubility of CBn hosts is in general relatively low, especially for CB6 and CB8 molecules. However, their solubility can be enhanced significantly by the addition of salt, charged guests, or using a solution with an acidic pH.31 While CB7 mostly forms 1:1 stoichiometric complexes with organic guests, its higher homolog, CB8, can often form higher-order complexes, due to the simultaneous encapsulation of two guest molecules in the significantly larger cavity of the latter host.13,25,49,50
The CXn family of cavitand molecules are considered to be third-generation macrocyclic hosts, placed after crown ethers and cyclodextrins. These host molecules have attracted considerable research interest in supramolecular chemistry, as they offer a wide range of applications in areas like biocatalysis, enzyme mimics, enzyme assays, drug delivery, pharmaceuticals, biosensing, and so on.1,6,17,19–21,51 The CXn hosts are in general quite non-volatile and have high melting points and thermal stability, making them very useful in many supramolecular applications.1,6,17,25,51 CXn molecules are formed by the cyclic oligomerization of phenolic units, connecting them through methylene bridges, resulting in cone-shaped cavitand structures. Common CXn macrocycles are CX4, CX6 and CX8, consisting of four, six, and eight phenolic units, respectively.1,17,52
An important characteristic of CXn hosts is the existence of their π-electron rich hydrophobic cavity. The CXn cavities are quite flexible and thus can readjust their shape while undergoing complexation with the guest molecules.1,19,21,25,34,52,53 The phenolic OH groups present at the CXn portals can be functionalized suitably to obtain various CXn derivatives, endowing them with the ability to encapsulate a wide variety of organic guests, metal ions, and biomolecules. Further, suitably functionalized CXn derivatives, e.g. sulfonated CXn (SCXn) hosts (cf.Scheme 3), can have drastically increased aqueous solubility and thus this enhances their prospects in various applications.15,19,21,25,53 Interestingly, SCXn hosts are capable of binding cationic, neutral, and anionic guests, cooperatively involving electrostatic (due to sulfonato groups at the wider cavity rim), hydrophobic (owing to hydrophobic host cavity), hydrogen bonding (through phenolic OH groups at the narrow cavity rim), π–π and cation–π interactions (due to the phenolic π cloud).1,15,19,21,25,53 In fact, the SCXn hosts are reported to display quite high selectivity and sensitivity towards some specific organic and biologically relevant guest molecules like drugs, amino acids and proteins, and also for monovalent and divalent metal cations.15,19,21,22,54,55
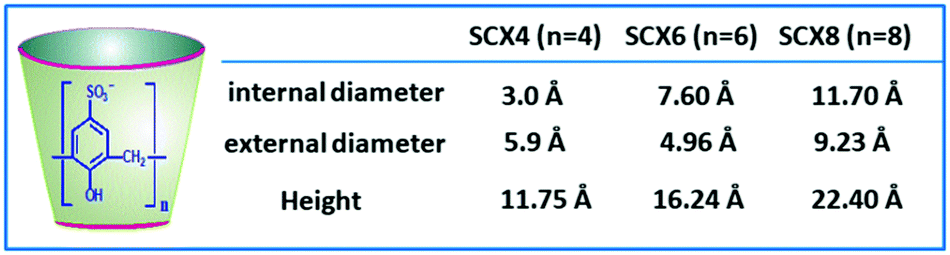 | ||
| Scheme 3 Chemical structure and typical shape of SCXn hosts. The cavity dimensions of SCX4, SCX6, and SCX8 hosts are also tabulated here for their quick comparison. | ||
The molecular recognition properties of CD, CBn, and CXn based macrocyclic hosts for diverse guest molecules have been utilized extensively to construct various host–guest complexes, which are further guided to self-organize into exotic supramolecular assemblies with exciting properties and behavior, with promising applications in various fields. To date, a variety of supramolecular assemblies have been reported, from simple host–guest complexes to pseudorotaxanes, rotaxanes, polyrotaxanes, supramolecular polymers, micelles, vesicles, one-dimensional nanofibers or nanotubes, and many other complex assemblies. In the literature, though many state-of-the-art review articles are available on supramolecular chemistry, most of them cover specific types of supramolecular systems or assemblies, and do not give an overview of the various types of supramolecular assemblies which are possible to obtain using different host–guest systems. In this perspective, thus, we intend to present an overview assimilating both simple host–guest systems and various complex supramolecular assemblies, discussing their constitutions, structural features, and applicability in a systematic manner. Our aim in this perspective is to draw the interest of researchers who are new to the area of supramolecular chemistry. We hope that this perspective will be useful for them to begin their research into supramolecular host–guest systems and will open up the way forward to explore more complex supramolecular architectures with advanced structures, functionalities, and properties for diverse applications.
Simple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 to higher order stoichiometries of host–guest complexes
:1 to higher order stoichiometries of host–guest complexes
As mentioned above, the interaction of a macrocyclic host with a guest molecule depends upon various factors, such as the dimensions of the host cavity, nature of the guest molecule, size compatibility of the host and guest molecules, their mutual orientation, and the interaction forces involved in the host–guest complex formation. In the last few decades, our group, as well as others, have extensively contributed to the field of supramolecular host–guest chemistry. In one of the studies,56 it was shown that even though both CB7 and βCD have hydrophobic cavities of quite comparable cavity dimensions, the two hosts show very contrasting complexation behaviour with the dye acridine orange (AOH+), primarily due to the differences in the characteristic molecular recognition sites present on the two host molecules, especially at their portals. AOH+ is a biologically important cationic chromophoric molecule that is extensively used as a fluorescence marker to distinguish between DNA and RNA, as well as a staining agent for cellular structures.57,58 The supramolecular host–guest interaction of AOH+ with the CB7 host results in significant modulation of the photophysical properties of the dye upon the formation of the 1:1 stoichiometric dye–host complex, which is stabilized mainly through strong ion–dipole interactions between the cationic AOH+ and the polarizable carbonyl groups present at the host portals. In contrast, AOH+ shows quite an insignificant interaction with the βCD host, presumably due to the hydrophilic nature of the AOH+ dye and the absence of any cation-receptor sites on the βCD molecule.56 Similar observations have been also found for other cationic dyes upon their interactions with cucurbituril and cyclodextrin hosts.22,59
Even though CDs do not possess any cationic receptor sites, there are several reports on the complexation of cationic guest molecules with CD based hosts. For example, 1:1 stoichiometric inclusion complex formations of cationic dyes like pyronine Y and pyronine B have been reported with βCD hosts.60 Pyronine B with N,N-diethyl substituents forms a much stronger dye–host complex with βCD than pyronine Y, which has N,N-dimethyl groups. The results are rationalized based on the relative magnitudes of the hydrophobic and specific interactions between the pyronine dyes and the electron-rich oxygen atoms present at the βCD cavity wall. In the present context, as well in the cases where the end to end lengths of the guest molecules are larger than the height of the macrocyclic hosts, two host molecules can suitably participate to encapsulate two opposite ends of the guest molecules, forming 1:2 dye-to-host stoichiometric complexes. This particular aspect has been nicely demonstrated in the case of oxazine 1 dye (OX1) upon its interaction with two cyclodextrin hosts, βCD and γCD.61 OX1 is a symmetric cationic molecule belonging to the p-benzoquinone diimine class of organic dyes. Upon its interaction with either βCD or γCD hosts, OX1 undergoes the formation of some 1:2 (dye:host) stoichiometric complexes as well as the 1:1 dye-to-host inclusion complexes, especially at the higher host concentration regions (cf.Scheme 4). The results also indicate that OX1 binds more strongly with the βCD host than γCD, which is rationalized based on the stronger hydrophobic interactions of the OX1 dye with the smaller βCD cavity as compared to those with the larger γCD cavity. Furthermore, for the OX1–γCD system, the formation of dimeric OX1–γCD complexes [(OX1)2–γCD] is also indicated, especially when higher dye concentrations are used. Due to the large cavity size of the γCD host, it can simultaneously accommodate two OX1 molecules inside its cavity, facilitated mainly by the joint participation of the π–π interactions between OX1 dyes and the hydrophobic interactions associated with the encapsulation of the dye dimer inside the γCD host. In contrast, however, the smaller βCD cavity can accommodate only one dye at a time, and thus it facilitates the disaggregation of the initially aggregated OX1 dye, if any occurs, especially at the higher dye concentrations used. Overall, the different stoichiometric complexes formed in the OX1–βCD and OX1–γCD systems are rationalized on the basis of size compatibility and the relative dimensions of the dye and the βCD and γCD host cavities.
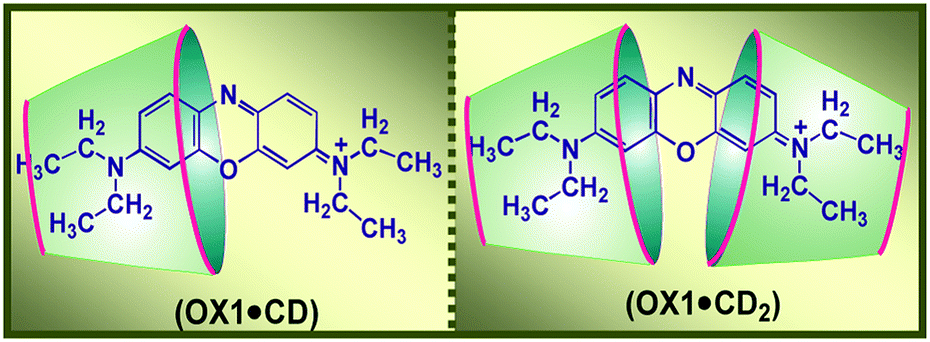 | ||
| Scheme 4 Schematic representation of the 1:1 and 1:2 dye-to-host stoichiometric complexes of OX1 with cyclodextrin hosts. The graphic has been reproduced from ref. 61 with permission from the American Chemical Society. | ||
Likewise, sanguinarine (SA) dye, a biologically active natural benzophenanthridine alkaloid, which possesses antimicrobial and anticancer activities, has also been reported to undergo both 1:1 and 1:2 dye-to-host inclusion complex formation with another host molecule, CB7, displaying a huge enhancement in the fluorescence intensity of the dye, which is attributed to the reduction in the radiationless deactivation process from the excited state of SA in its complexed form with the host molecule.62 SA in its free form is susceptible to nucleophilic attack by hydroxide anions. In this context, the authors have also reported the consequent protection of the SA dye from nucleophilic attack and photooxidation upon encapsulation by the CB7 host cavity.
Encapsulation of two or more guest molecules inside a macrocyclic host cavity to form higher order complexes is an important criterion for the formation of various extended supramolecular architectures, some of which will be discussed in forthcoming sections of this article. The formation of such higher order complexes in host–guest systems is largely determined by the chemical constitution and the cavity size of the host molecules concerned. In this respect, the neutral red (NRH+) molecule, a phenazine-based dye and a well-known fluorescence marker for biological systems, shows contrasting complexation behaviour with the higher macrocyclic homolog, CB8, as compared to the lower homolog, CB7.63 The substantial differences in the cavity dimensions of the CB7 and CB8 hosts bring out quite remarkable differences in the host–guest stoichiometry upon the interaction of NRH+ with these two hosts. Due to the smaller cavity size of CB7, NRH+ undergoes 1:1 (dye:host) stoichiometric complex formation, resulting in ∼6 fold increase in the fluorescence intensity of the dye. In contrast, due to the larger cavity size of CB8, the π-stacked dimeric NRH+ molecules are suitably accommodated within the CB8 cavity, resulting in the formation of 2:1 dye-to-host stoichiometric (NRH+)2–CB8 complexes, displaying a drastic reduction in the fluorescence intensity of the dye (cf.Scheme 5). The NRH+ dimers encapsulated inside the CB8 cavities have been further explored for the possibility of the formation of heterodimers within the host cavities through the replacement of one of the two NRH+ molecules from the 2:1 dye-to-host complexes by introducing a second guest molecule, such as tryptophan (Trp), an amino acid, into the solution. The results clearly indicate the exchange of one of the NRH+ dye molecules from the CB8 cavity for the added Trp, forming NRH+–Trp heterodimers inside the CB8 cavities. This finding was further examined in the presence of a protein, bovine serum albumin (BSA), establishing that there is a controlled release of one of the NRH+ dyes from the (NRH+)2–CB8 complexes, resulting in the formation of NRH+–CB8–BSA ternary complexes, which is quite relevant to the design of host–guest based drug delivery systems.
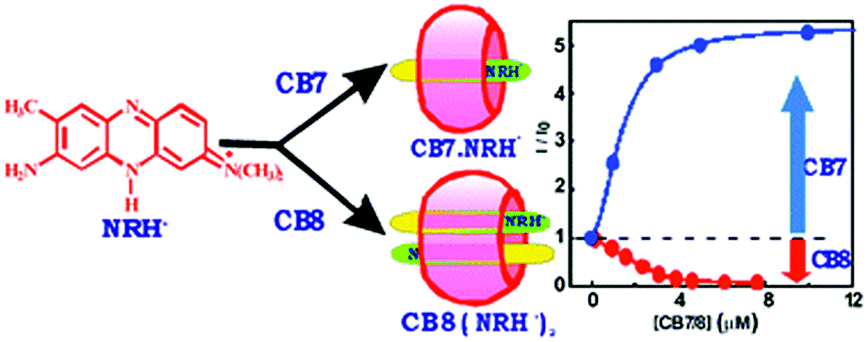 | ||
| Scheme 5 Schematic representation of the contrasting guest binding interaction of CB7 and CB8 with the NRH+ dye. The scheme has been reproduced from ref. 63 with permission from the Royal Society of Chemistry. | ||
In general, selective formations of heterodimers inside macrocyclic host cavities are difficult to achieve, though there are a few reports in the literature. Kim et al.64 reported the first example of the selective inclusion of hetero-guest pairs inside a macrocyclic host. These authors demonstrated the formation of hetero-guest pairs between the electron-deficient guest, methylviologen (MV2+), and the electron-rich guest, 2,6-dihydroxynaphthalene (HN), inside the CB8 cavity, which was mainly driven by the charge transfer (CT) interaction between the two guests, facilitated by their close contact within the host cavity (cf.Scheme 6). The authors confirmed the formation of MV2+–HN hetero-guest pairs inside the CB8 cavities through absorption, emission and NMR studies.
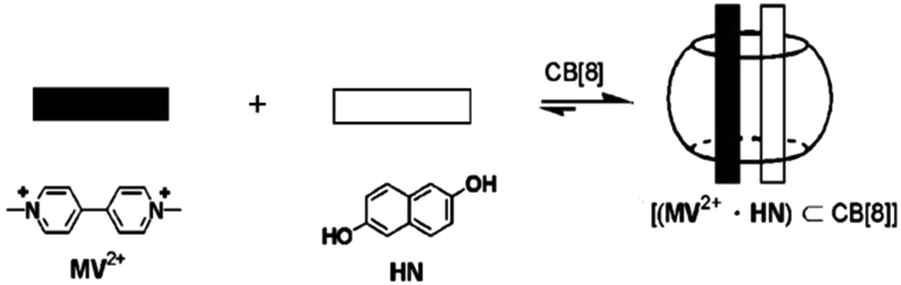 | ||
| Scheme 6 Inclusion of hetero-guest pairs inside the CB8 cavity. The scheme has been reproduced from ref. 64 with permission from Wiley-VCH Verlag GmbH. | ||
Likewise, Scherman et al.65 reported the inclusion of donor–acceptor pairs inside the CB8 cavity by screening a wide range of electron-deficient dicationic viologen guests and electron-rich aromatic guest molecules, whereby well-defined face-to-face π–π-stacking geometry of the donor and acceptor pairs formed inside the CB8 cavity, driven mainly by CT interactions. The authors observed a more stabilized CT excited state inside the CB8 cavity as compared to that in the solution environment. The authors inferred that the observed trends in the binding process for a wide range of donor–acceptor pairs inside CB8 could be better explained by considering the electrostatic interactions combined with the solvation effects. It is suggested that the heteroternary complexes formed in the present systems could be very reliable models for the in-depth study of the CT interactions.
Taking advantage of the stimulus-responsive nature of the supramolecular system, Schalley et al.66 designed pH and redox responsive heteroternary complexes, composed of CB8 as a macrocyclic host and three types of guest molecules in combination, namely, a pH-responsive phenylpyridine derivative, a redox-responsive alkyl viologen, and a neutral electron-rich naphthalene derivative. In their studied systems, both homo or hetero ternary complexes could be selectively formed in a controlled manner by applying pH and redox signals as the external stimuli. Protonated phenylpyridine guests formed a 2:1 homoternary complex with CB8, while upon deprotonation, they formed a 1:1:1 heteroternary complex along with the electron deficient alkyl viologen as the counterpart in the CB8 cavity. Reduction of the viologen resulted in the formation of viologen radical-cations, which dimerized within the CB8 cavity, forming 2:1 homoternary complexes. Thus, the authors demonstrated a unique supramolecular system illustrating an inter-conversion between heteroternary and homoternary CB8 complexes, which could be selectively controlled by using simple redox signals or pH as the stimuli, which could be explored further with logic gate implications.
It should be noted here that in host–guest interactions, the formation of externally bound exo or exclusion complexes are also possible in number of cases. Such complexation arises when the host cavity is not large enough for the guest molecule to be accommodated inside the host cavity or when there prevails a very strong non-covalent interaction between the guest molecule and anchoring functional groups present at the host portals. In this context, the interaction of Thioflavin-T (ThT), a well-known amyloid fibril marker, with two versatile macrocyclic hosts, namely CB5 and CB7, deserves to be mentioned.67 Distinct differences in the CB5 and CB7 cavity sizes bring out remarkably different complexation behaviours of the two hosts with the ThT molecule. Both CB5 and CB7 undergo 1:1 and 1:2 dye-to-host stoichiometric complexes formation with the ThT guest. Interestingly, however, with the cavity of the CB7 host being wider, it can accordingly form strong inclusion complexes with the ThT guest, while the CB5 host, which has a much narrower cavity, does not allow the guest dye to enter into the host cavity and accordingly forms exclusion complexes with the ThT guest. In both cases, the complexes formed are stabilized largely through the participation of strong ion–dipole interactions between the cationic ThT dye and the negatively charged portals of the CB host, which consist of highly polarizable carbonyl groups. In the case of the ThT–CB7 inclusion complexes, along with the above strong ion–dipole interactions, an additional hydrophobic interaction is also contributed by the CB cavity, enhancing the stability of the concerned inclusion complexes. In the reported study, the authors confirmed the proposed mechanisms by ground-state absorption, steady-state fluorescence, time-resolved fluorescence, NMR, and quantum chemical studies.
Host–guest complexation becomes more complicated but quite fascinating when there is host-induced dimerization or higher-order dye aggregate formation, especially on the external surface of the host molecules. In this context, Singh et al.68 reported a very uncommon finding of strongly emissive H aggregate formation by ThT dye, caused by the host assisted non-covalently assembled cationic dye molecules on the highly negative surface of a sulfated-β-cyclodextrin (SCD) derivative, while the free ThT in aqueous solution has extremely weak fluorescence. In SCD, the hydroxyl groups of the native βCD were functionalized with negatively charged sulfonato (SO3−) groups, and in this process the portals of the host molecules became highly negatively charged and exceedingly hydrophilic in nature, assisting the formation of H-aggregates of the dye (cf.Scheme 7). The observation in the present case was opposite to that observed with the parent βCD host, where simple host–guest inclusion complex formation prevailed, leading to the de-aggregation of dyes that happened initially to some extent for the free dye in aqueous solution. For the ThT–SCD system, the H-aggregate formation was attributed to the strong electrostatic interactions between the positively charged dye and the highly negative SCD host, arising due to the presence of multiple SO3− groups at the host portals. The emissive nature of the host assisted H-aggregates of ThT dye was attributed to the restrictions imposed on the non-radiative torsional relaxation processes in the excited state of the otherwise very flexible ThT molecules upon their strong binding to the SCD surface.
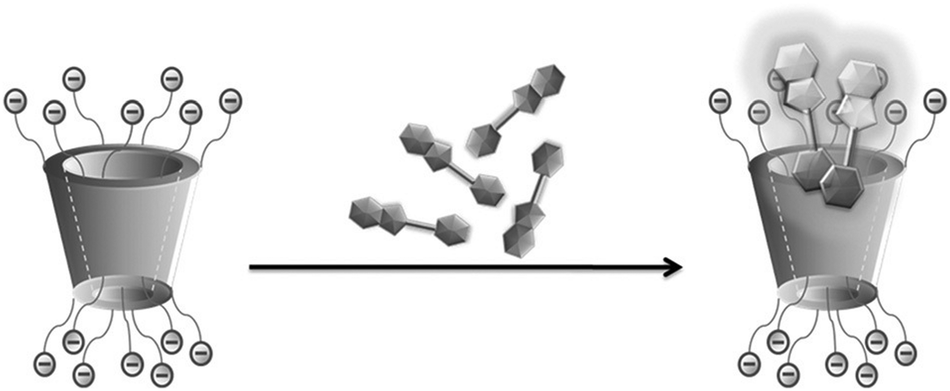 | ||
| Scheme 7 Formation of emissive aggregates of ThT on the sulfated-β-cyclodextrin (SCD) surface. The scheme has been reproduced from ref. 68 with permission from Wiley-VCH Verlag GmbH. | ||
Heyne et al.69 also reported the formation of emissive H-aggregates of the positively charged thiazole orange (TO) dye, attached non-covalently to the anionic SO3− groups present at the upper rim of the sulfated CX4 derivative (SCX4), through the formation of host–guest complexes with a 3:1 dye-to-host binding ratio. In a similar vein, our group also reported strong host assisted aggregate formation for the cationic dye AOH+ at the negatively charged portals of the SCD host, which was attributed to the strong electrostatic interactions between the cationic dye and multiple negatively charged SO3− groups present on the host portals, along with the additional π–π interactions participating between the stacked dye molecules bound on the host surface.38
Multistep complexation behaviour of organic dyes with macrocyclic hosts and their host concentration dependent sequential transformations has also been interestingly reported by our group. In one such work, the sequential changes in the mode of interaction of AOH+ dye with increasing concentrations of the sulfobutylether-β-cyclodextrin (SBEβCD) host were observed, conceptually shown in Scheme 8.37 The SBEβCD host (also known as Captisol), is a multi-anionic βCD derivative, having seven sulfobutylether groups on average attached to both the portals of the βCD scaffold that effectively extend the hydrophobic cavity height of the SBEβCD host, while the presence of the terminal SO3− groups makes the host molecules excellent cation receptors. At lower concentration of the host, AOH+ predominantly underwent dimerization at the negatively charged portal regions of the host and these complexes were stabilized mainly through strong ion–ion interactions between the cationic charge of the AOH+ dye and the negatively charged SO3− groups of the host. Interestingly, however, at higher SBEβCD concentrations, the initially formed host-bound AOH+ dimers eventually disintegrated in a competitive manner to form 1:1 monomeric AOH+–SBEβCD complexes, both as the exclusion and inclusion complexes, stabilized by the joint participation of the electrostatic interactions between the AOH+ dye and the SO3− groups of the host and the hydrophobic interactions imparted by the host cavity to the bound dye. Similarly, our group also reported the host concentration dependent changes in the host–guest complex formations upon the interaction of the AOH+ dye with water soluble anionic sulfated-CXn (SCXn) hosts, namely SCX4 and SCX6, which have highly different cavity dimensions.53 Also in these cases, at lower host concentrations, the AOH+ molecules underwent initial host assisted aggregation, stabilized by strong electrostatic interactions along with the additional contribution arising from the π–π interactions between the stacked dye molecules. At higher SCXn concentrations, however, the host-bound aggregated AOH+ molecules eventually disintegrated to form 1:1 AOH+–SCXn complexes, in both inclusion and exo fashions, stabilized by both electrostatic and hydrophobic interactions. The observed results also clearly indicated that the AOH+ dye bound more strongly with the SCX6 host as compared to the SCX4 host, indicating that the number of SO3− groups present at the SCXn portals and the size compatibility of the guest and the host cavity play dominant roles in determining the overall binding strengths for the studied AOH+–SCXn systems.
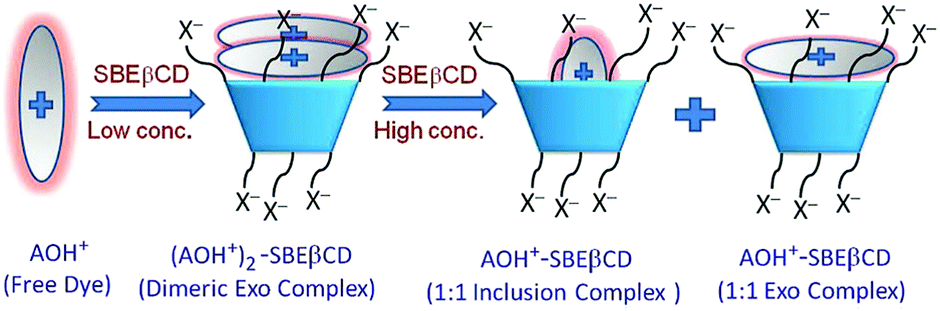 | ||
| Scheme 8 Schematic representation of the multistep binding interactions of acridine orange dye (AOH+) with changing SBEβCD host concentration. The scheme has been reproduced from ref. 37 with permission from the Royal Society of Chemistry. | ||
Even though both the CBn and SCXn hosts are known to be typical anion receptors, the above two classes of hosts can exhibit largely different complexation behaviours with guest molecules. In this context, Assaf et al. reported the contrasting complexation behaviour of CBn (CB7 and CB8) and SCXn (SCX4 and SCX5) hosts with perylene-based diimide dye (PDI), which is well-known for its self-aggregation in aqueous solution.70 Though both CBn and SCXn hosts are known to behave preferentially as cation binders, the CBn hosts reduced the self-aggregation of PDI molecules very significantly through the formation of stable dye–host inclusion complexes with a 1:2 dye-to-host binding stoichiometry, while in contrast, the SCXn hosts assisted the host-induced aggregation of the PDI molecules further, due to strong electrostatic interactions, as shown conceptually in Scheme 9.
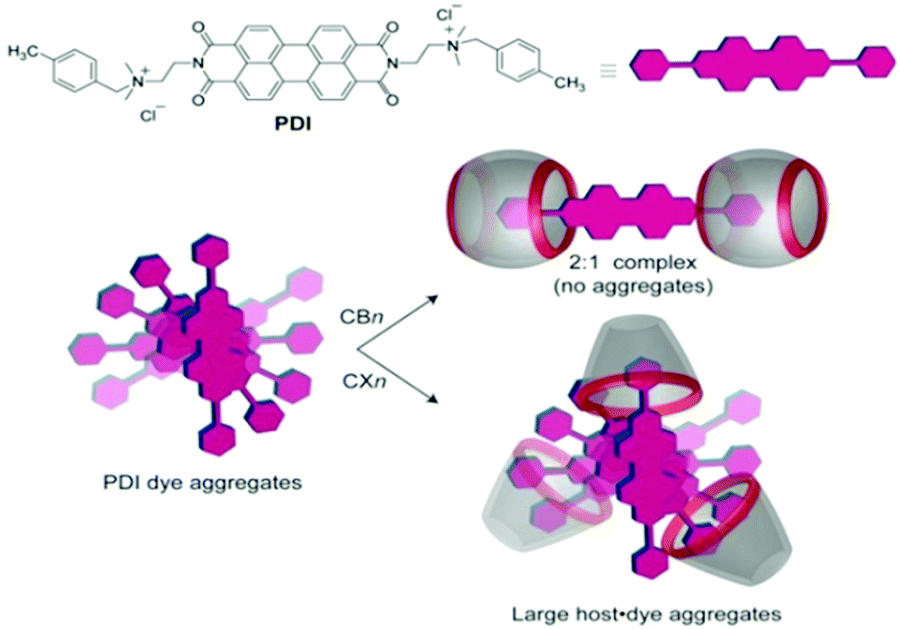 | ||
| Scheme 9 Chemical structure and symbolic representation of the PDI dye. The potential binding modes of the PDI dye with CBn and SCXn hosts are conceptually shown. The scheme has been reproduced from ref. 70 with permission from Wiley-VCH Verlag GmbH. | ||
Extensive studies have been reported in the literature on supramolecular host–guest interactions forming various types of simple to complicated host–guest complexes with various stoichiometric ratios. The inclusion of all the reported work in the present context is beyond the scope of the present article. Interested readers can, however, go through the other comprehensive compilations discussing various host–guest complexes, which are available in the literature.1,9,22,23,31,34,46
Pseudorotaxane and polypseudorotaxane systems
Mechanically interlocked molecules are of great interest to researchers due to their possibilities for constructing innovative functional materials, which are useful for the development of molecular switches, machines, sensors, logic gates, and many others.71,72 Pseudorotaxanes are examples of such interlinked systems in which long linear chain-like axle molecules are threaded through the macrocyclic hosts, which are like the wheels, driven by the non-covalent interactions between the constituent units. Unlike rotaxanes, where dumbbell shaped molecules are mechanically interlocked with macrocyclic host cavities acting as the anchoring ring, pseudorotaxanes do not possess the bulky stopper-like structures at both ends of the axle molecular units, making the assembly and dismantling of the macrocyclic host molecules through the axle molecules highly feasible.27,71–76 In the following section, we discuss some of the pseudorotaxane systems, which are considered based on their intriguing structural features and potential uses in various applied areas.Dearden et al.77 reported the formation of a pseudorotaxane system involving CB6 as the macrocyclic host and the doubly protonated 1,4-butanediamine (DAB) dication as the axle molecule. Inclusion complex formation in the present pseudorotaxane system was attributed to the charge–dipole interaction between the dicationic DAB and the electronegatively polarized carbonyl groups present at the CB6 portals.
In inclusion complex formations involving pseudorotaxane systems, steric factors and the size compatibility of the axle molecular chain and the macrocyclic host cavity play very significant roles. In this regard, Harada et al.78 reported pseudorotaxane formations involving polyethylene glycols (PEGs) of various molecular weights as the axle molecules and αCD as the anchoring macrocycle. Interestingly, the authors observed that while there was significant complex formation for the PEG molecule with the αCD host when the molecular weight of PEG was very high, e.g. 1000, PEG molecules with much lower molecular weights did not show any complex formation with the αCD host. Similar to binary pseudorotaxane systems, the formation of ternary pseudorotaxane systems is also quite possible, where two different macrocycles participate in threading a linear guest or axle molecule. In this context, Buschmann et al.79 reported the ternary complex formation of PEG with two different macrocyclic hosts, αCD and CB6, to form a ternary pseudorotaxane. The authors suggested that as both the αCD and CB6 hosts have nearly identical complexing behaviour with the hydrophobic organic molecules, due to their similar dimensions and hydrophobic cavities, the formation of ternary complexes where PEG is threaded with both αCD and CB6 hosts in a statistically distributed manner on the PEG polymer chain becomes quite possible. Similarly, Inoue and co-workers80 also established ternary pseudorotaxane formation for cationic dihexylammonium (DHA) as the axle molecule and CB6 and βCD as the macrocyclic hosts, where both the host molecules were included along the DHA chain. The unique ternary complex DHA:CB6:βCD, with a 1:1:1 stoichiometry of the components, was successfully synthesized by these authors in a stepwise manner, driven by both enthalpic and entropic forces. In the synthetic process, the 1:1 DHA–CB6 complex was formed first, due to the strong interaction between DHA and CB6. Subsequently, in the second step, the addition of βCD led to the formation of the ternary DHA:CB6:βCD (1:1:1) complex (cf.Scheme 10).
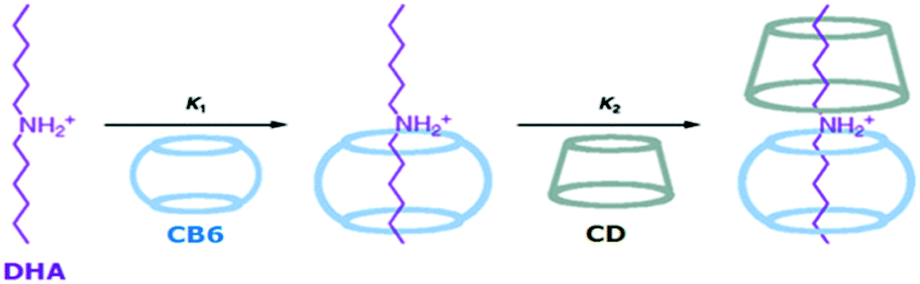 | ||
| Scheme 10 Schematic representation of the stepwise formation of the ternary CB6:βCD:DHA complex with a 1:1:1 stoichiometry. The scheme has been reproduced from ref. 80 with permission from the American Chemical Society. | ||
In another example, ternary pseudorotaxane formation was also reported using the N,N′-disubstituted methylenediimidazolium cation as the axle molecule and CB7 and CDs as the two macrocyclic hosts.81 The methylenediimidazolium cation used here had two long chain N,N′-substituents, which also had two aromatic groups at their terminal positions. For the ternary pseudorotaxane formed in the present case, the CB7 host bound the cationic diimidazolium cation, while the CD cavity encapsulated one of the aromatic residues of the two substituent groups of the axle molecule. Regarding binary and ternary pseudorotaxane formations, Vicha and co-workers82 reported more complex systems involving adamantylated-bisimidazolium salts as the guest axle molecules. In their work, four different bisimidazolium based guest molecules with one central biphenyl binding site, along with two symmetrical adamantyl binding sites at the terminal positions, were synthesized. The synthesized molecules showed pseudorotaxane formations with the βCD host, with stoichiometries of 1:1 and 1:3 for the guest to host ratios. When both βCD and CB7 hosts were used together, ternary pseudorotaxane like architectures were formed with stoichiometries of 1:1:1 or 1:2:1 for the guest to CB7 to βCD units.
It is evident that the pseudorotaxanes constructed through the incorporation of several macrocyclic moieties into the main linear chains of the guest molecules can be well defined as the polypseudorotaxanes. The length of a polypseudorotaxane system is understandably determined by the length of the linear guest molecule used. The major factors for such polypseudorotaxane formations are the specific interactions involved, as well as the size compatibility between the host ring and the encapsulated linear guest. Specific interactions between the macrocyclic hosts placed adjacent to each other in the polypseudorotaxane also play a significant role in the formation of such systems.76,83 Nostro and co-workers84 studied polypseudorotaxane formation by linear hydrophobic guest polymer chains, such as poly(propylene glycol) bis-2-aminopropyl ether (PPG-Am2) and pluronic 105 (PLU), with cyclodextrin (CD) hosts, such as βCD and γCD. It was found that the polypseudorotaxane formations in these cases were strongly dependent upon the relative sizes of the guest molecules and the host cavities of the macrocycles concerned. The authors observed that the polypsuedorotaxanes were formed significantly for PPG-Am2 involving βCD as the host and for PLU involving γCD as the host. The authors also calculated the number of threaded CD molecules per polymer chain, achieved by performing kinetic studies, and the values were found to be in the range of 15 to 19 CD molecules per polymer chain. The complexation process was found to be enthalpy driven, dominated by hydrophobic interactions, as usually happens with CD based host–guest complexes. In a noteworthy contribution, Harada et al.85 showed doubly-threaded polypseudorotaxane formations of PEG molecules with γCD hosts, in which two polymer chains were threaded across by the γ-CD hosts. Likewise, Tonelli and co-workers86 reported an interesting phenomenon of the constrained polymerization of styrene based guest molecules within the narrow channels of the γCD host. According to the authors, following the appropriate recrystallization of the CD hosts from their saturated aqueous solutions, it is possible to obtain the channel type packing of the hosts in the solid-state, where water molecules are just included within the narrow channels of the packed host molecules. Suspending such a material based on the γCD host into liquid styrene resulted in the formation of host–guest inclusion complexes where styrene molecules were included within the narrow host channels. The inclusion complexes thus obtained were then subjected to constrained polymerization, producing polystyrene molecules within the narrow γCD channels, conceptually shown in Scheme 11.
 | ||
| Scheme 11 Conceptual representation of the constrained polymerization of styrene molecules encapsulated within the narrow γCD channels. The scheme has been reproduced from ref. 86 with permission from Wiley-VCH Verlag GmbH. | ||
Kim and co-workers87 reported polypseudorotaxane formation using CB6 host threading on a polyviologen (PV) polymer that had around 10 bipyridinium units linked together by decamethylene units as the spacers (cf.Scheme 12). In the present case, the degree of CB6 threading, i.e. the number of CB6 beads per repeat unit, could be controlled between 0.1 and 1.0. The resulting CB6–PV complex was so stable that no appreciable de-threading was observed. The threaded CB6 beads were tightly confined in the middle of the decamethylene units of the PV polymer and were stabilized through hydrophobic interactions between the entrapped decamethylene unit and the CB6 cavity, along with charge–dipole interactions between the carbonyl groups of the CB6 host and the bipyridinium unit of the polymer molecule, resulting in a well-defined structure for the polypseudorotaxane system in aqueous medium. As indicated before, the stability of pseudorotaxane systems depends largely upon the size compatibility between the macrocyclic hosts and the encapsulated linear guest molecules. With this consideration, Yui and co-workers88 demonstrated biodegradable polypseudorotaxane formation by the simple mixing of aqueous solutions of poly(ε-lysine) and αCD, but they could not observe similar pseudorotaxane formation when replacing αCD by βCD and/or γCD hosts. This indicated that, due to their large cavities, βCD and γCD cannot bind the poly(ε-lysine) chains of the guest tightly enough to produce the concerned pseudorotaxane systems. In these cases, even if the complexes were formed, their stability was so low that they dissociated almost instantly and, thus, there was no appearance of the pseudorotaxane systems. In an interesting study, Harada and co-workers89 contrastingly reported the formation of polypseudorotaxane systems for the inorganic polymer poly(dimethylsiloxane) (PDMS), involving βCD and γCD hosts, but could not find similar polypseudorotaxane formation for PDMS upon using the αCD host.
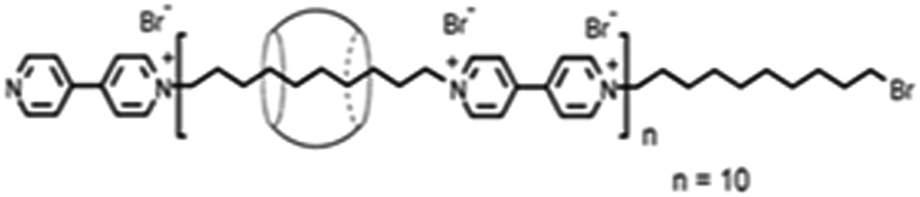 | ||
| Scheme 12 Schematic representation of the polypseudorotaxane formed in the CB6–polyviologen system. The scheme has been reproduced from ref. 87 with permission from the American Chemical Society. | ||
Very rarely observed calixarene-based polypseudorotaxanes were reported by Yamagishi and co-workers,90 using p-tert-butylcalix[8]arenes as the anchoring macrocycles and PEGs of various molecular weights as the axle molecules. In their study, the polypseudorotaxanes were formed through the polycondensation of p-tert-butylphenols with para-formaldehyde in the presence of PEG molecules, which was supported by the results obtained from FT-IR, 1H NMR, and differential scanning calorimetry (DSC) measurements. The yield and the composition of the polypseudorotaxanes formed were found to be dependent on the molecular weight of the PEGs used.
Rotaxanes and polyrotaxanes
The pseudorotaxane systems where the guest molecules (axles) particularly possess covalently attached bulky end groups, which can act as stoppers for the threaded macrocyclic hosts, are commonly known as rotaxane systems. The bulky end groups of the axle molecules are usually larger than the internal diameters of the macrocyclic hosts such that they can resist the dissociation or unthreading of the hosts from the linear guest molecules.27,73,76 In this section, we discuss some important rotaxane systems reported in the literature. The first example of a rotaxane structure based on the CB6 host was reported by Kim et al., and was synthesized using a linear guest molecule based on a spermine scaffold.91 As a guest molecule, spermine not only showed strong interactions with CB6 but it also had two amine groups at both ends, which could be substituted easily with bulky groups acting as the stoppers. In the synthesis process, the CB6 molecule was first threaded to the spermine molecule to form a pseudorotaxane. Then, bulky stopper groups like dinitrophenyl moieties were introduced at both ends of the spermine unit to form the eventual rotaxane structure (cf. Scheme 13). The strong interaction of CB6 with spermine was attributed to the hydrogen bonding interactions between the carbonyl oxygen atoms present at the CB6 portals and the hydrogen atoms of the protonated amino groups of the spermine molecule. The drastically improved solubility upon pseudorotaxane formation and the high affinity of CB6 for spermine allowed the simple, one-pot and high yield synthesis of the concerned rotaxane system.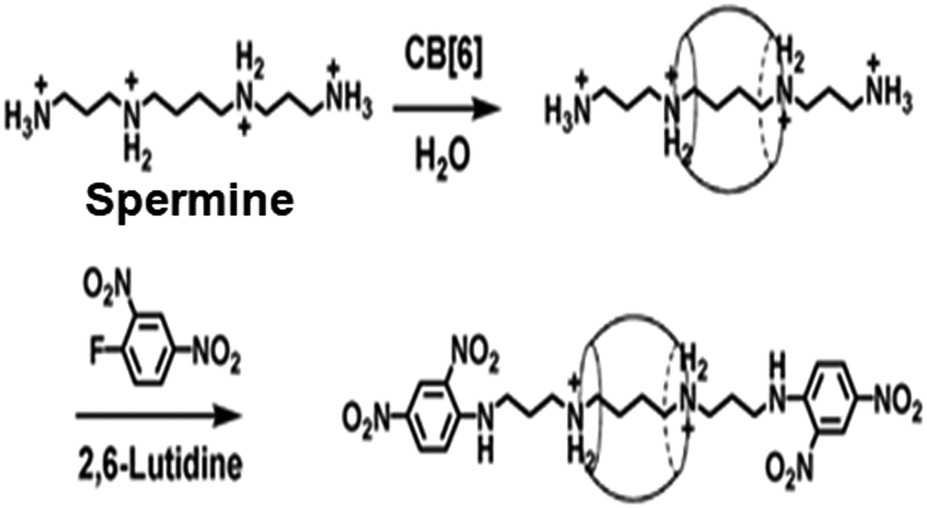 | ||
| Scheme 13 Schematic representation of CB6 threading over a spermine molecule to form a pseudorotaxane and the subsequent attaching of dinitrophenyl groups at both the ends of the spermine unit to form the final rotaxane. The scheme has been reproduced from ref. 74 with permission from the American Chemical Society. | ||
In another example, Kaifer and co-workers92 reported the formation of a CD-based rotaxane system by interacting with ω-[(ferrocenylmethyl)-dimethylammonium]alkanoic acid as the axle molecule threaded with the αCD host. In this system, the ferrocene moiety present at one end of the alkanoic acid acted as one of the stoppers, while the other end was capped through the coupling of the alkanoic acid group with a potassium 5-amino-2-naphthalene-sulfonate group via amide coupling. In the present case, two isomeric rotaxanes were formed that differ from each other in the orientation of the αCD host with respect to the end groups of the rotaxane systems, as shown in Scheme 14. Likewise, the synthesis of a two component rotaxane ([2]rotaxane) and a three component rotaxane ([3]rotaxane) was also reported by Dardeer.93 The author synthesized the [2]rotaxane system by threading βCD onto the succinic dihydrazide molecule and then trapping the end groups using chlorendic anhydride units. Similarly, the [3]rotaxane system was synthesized by threading two αCD hosts over a di-acyl molecule, namely sebacoyl chloride, and then trapping its end groups with chlorendic anhydride units. The structures of the [2]rotaxane and [3]rotaxane systems were investigated using FT-IR, 1H-NMR, 13C-NMR, 2D-NMR, and scanning electron microscopy (SEM) measurements.
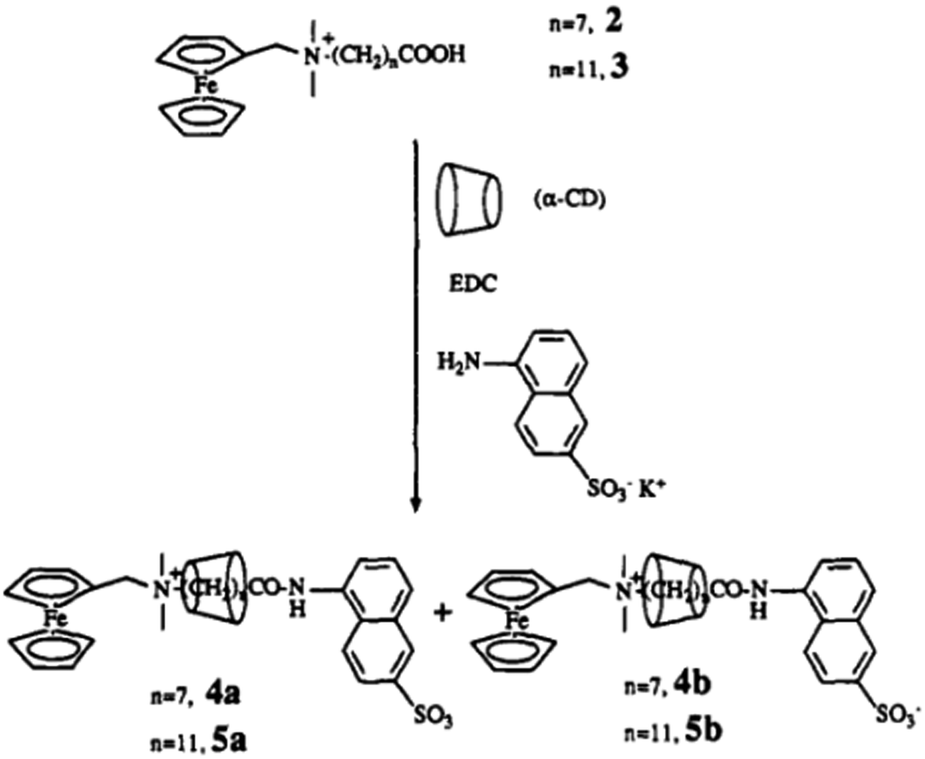 | ||
| Scheme 14 Schematic representation of a CD-based rotaxane system, synthesised by capping the free alkanoic acid end by amide coupling. The scheme has been reproduced from ref. 92 with permission from the American Chemical Society. | ||
Polyrotaxanes are in general synthesized by coupling the end groups of the polypseudorotaxanes by larger substituent groups acting as the stoppers for the threaded macrocycles.27,73–76 Based on this approach, Harada and co-workers presented a classic example of polyrotaxane formation.94 They first prepared the polyseudorotaxane using αCD as the host molecule and PEG bis(amines) as the linear guest. Subsequently, both the end amine groups of the polyseudorotaxane were capped with bulky 2,4-dinitrophenyl groups as the stoppers, forming the resultant polyrotaxane system, which the authors referred to as a molecular necklace (cf.Scheme 15).
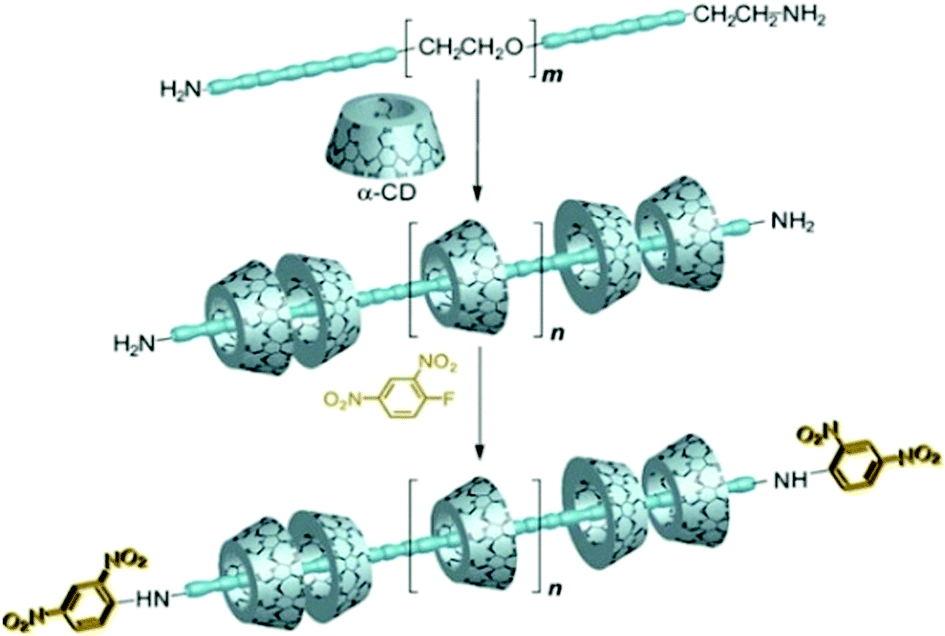 | ||
| Scheme 15 Schematic illustration of the synthesis of polyrotaxane incorporating PEG bisamines and αCD. The scheme has been reproduced from ref. 76 with permission from Wiley-VCH Verlag GmbH. | ||
Similarly, Buschmann and co-workers95 reported the synthesis of mono-, oligo-, and poly-rotaxane systems with cucurbituril hosts following interfacial condensation. The pseudorotaxane system was first prepared through complexation of the linear 1,6-hexanediarnmonium cation with the cucurbituril host. The amino groups of this system were then coupled with suitably substituted mono and diacid chlorides, namely the substituted benzoyl chloride, or the substituted benzene-dicarbonyl dichloride, producing various rotaxane systems. In the present cases, it was possible to obtain polyrotaxanes of varied lengths by controlling the amide repeating units and the number of cucurbituril beads threaded onto the overall chain. Different characterization methods like IR spectroscopy, differential thermal analysis, NMR, and elemental analyses were used to verify different structures of the rotaxanes formed. Similarly, Jia et al.96 reported CD-based polyrotaxane formations by threading βCD hosts onto bile acid diamine derivatives and coupling them with PEG-dicarbonate spacers which have tunable lengths. The resulting polypseudorotaxanes were subsequently capped at both ends with bulky monoamino-βCD molecules and, thus, this prevented the dissociation of the βCD beads from the bile acid chains (cf.Scheme 16). In the present cases, different polyrotaxane isomers could be obtained by adjusting the lengths of the PEG dicarbonate spacers. It should be mentioned that such polyrotaxane systems could be explored further for the development of new materials for uses in biology, sensing, catalysis, molecular machines, logic gates, and so on.93,94
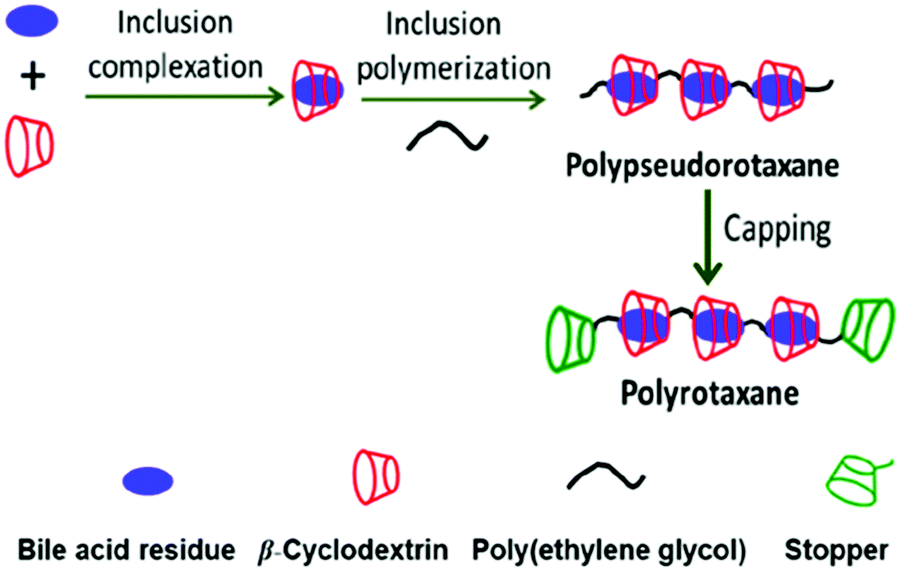 | ||
| Scheme 16 Conceptual representation of polyrotaxane formation of βCDs with PEGylated bile acid derivatives. The scheme has been reproduced from ref. 96 with permission from the American Chemical Society. | ||
Harada and co-workers reported the synthesis of a polyrotaxane based on the photoreaction of the end group of a pseudorotaxane precursor complex.97 In the present synthesis, the pseudopolyrotaxane precursor complex was first formed by threading αCD macrocycles onto a poly(propyleneglycol) (PPO) molecule that had a triphenylmethyl group at one end as a stopper and a photoreactive 2-anthryl group at the other end (cf.Scheme 17). As such, the 2-anthryl end group was small enough to act as a stopper. However, when the precursor complex was exposed to visible light, photodimerization of the 2-anthryl end group took place, resulting in the 2-anthryl photodimer as a sufficiently large end group to block the dethreading of the αCD beads from the resultant polyrotaxane.
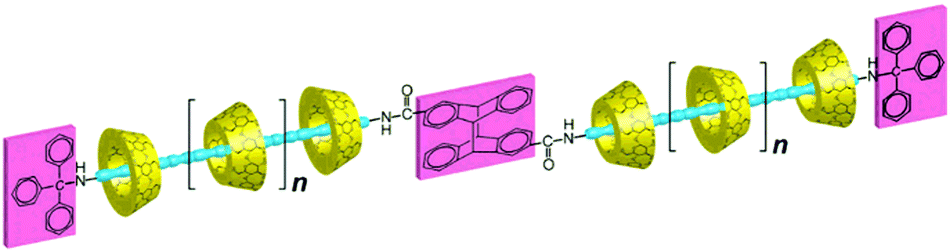 | ||
| Scheme 17 Schematic representation of synthesizing αCD based polyrotaxane via photodimerization. The scheme has been reproduced from ref. 97 with permission from the American Chemical Society. | ||
A conjugated polyrotaxane system was synthesized by Hadziioannou and coworkers by the complexation of 5,5′-dibromobithiophene molecules with αCD hosts, subsequently followed by the polymerization of these complexes through a Ni-catalyzed Yamamoto coupling reaction in DMF solvent.98 After polymerization, bulky 9-bromoanthracene molecules were finally coupled at both ends of the polypseudorotaxane to prevent the dethreading of the αCD beads, resulting in the polythiophene based polyrotaxane system. The structure of the polyrotaxane was characterized by UV–vis spectroscopy, small-angle neutron scattering (SANS), and AFM studies. It was revealed that the polyrotaxane had about seven αCD rings on its chain and the system was reasonably soluble in both water and DMSO. In another work, Geckeler et al.99 tried to polymerize the inclusion complexes of p-phenylenediamine and terephthalaldehyde with αCD to form an azomethine based polypseudorotaxane. The resulting system was then converted into polyrotaxane by capping its amino terminal directly with C60 and its aldehyde terminal through p-xylylenediamine mediated coupling with C60. The resulting C60 capped polyrotaxane was reported to be reasonably soluble in a polar aprotic solvent like DMF (cf.Scheme 18).
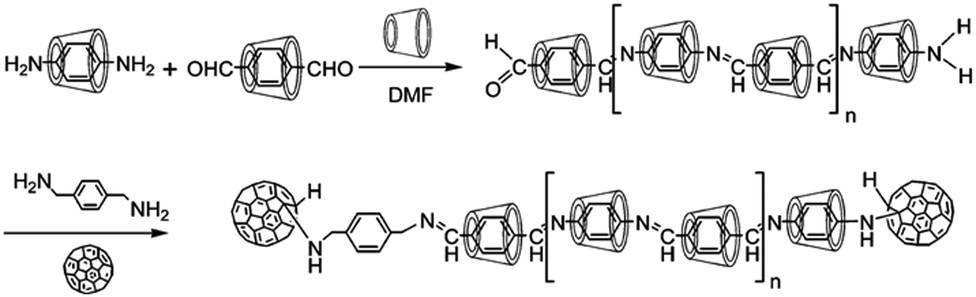 | ||
| Scheme 18 Schematic representation of the synthesis of poly(azomethine) rotaxane capped with C60 as the end groups. The scheme has been reproduced from ref. 99 with permission from the American Chemical Society. | ||
A major challenge in the synthesis of CD-based polyrotaxane systems is their low solubility in water, possibly arising due to hydrogen bonding and/or hydrophobic interactions of the CD units among the polyrotaxane molecules, leading to their aggregation. In this context, Resmerita et al. reported the synthesis of a water-soluble polyrotaxane using a CB7 and a tris-O-methylated derivative of αCD (TMαCD) as the hosts and amine-terminated poly(ethylene glycol) as the guest molecule.100 The observed results in the present cases were compared with those obtained upon using simple αCD as the host. In this study, the authors used a TMαCD host with the assumption that methyl substituents would reduce the hydrogen bonding interactions of the CD units among polyrotaxane molecules and thus increase the rotaxane solubility in water. Similarly, CB7 was used, assuming that the hydrophilic carbonyl portals of the hosts would help to increase rotaxane solubility in water and the hydrophobic cavity of the hosts would mean that the neutral guest was held more tightly as compared to in the CD hosts. In the first step of the synthesis, the CB7, TMαCD, or αCD hosts were threaded on the guest molecule, driven mainly by hydrophobic interactions, to form the corresponding pseudopolyrotaxane systems. In the next step, both the amino end groups of these polypseudorotaxanes were covalently attached with bulky bromotriphenylmethane units to form the corresponding polyrotaxane systems. The chemical structures of the so-formed polyrotaxanes were investigated using NMR, isothermal titration calorimetry (ITC), FT-IR, and quantum chemical calculations (cf.Scheme 19).
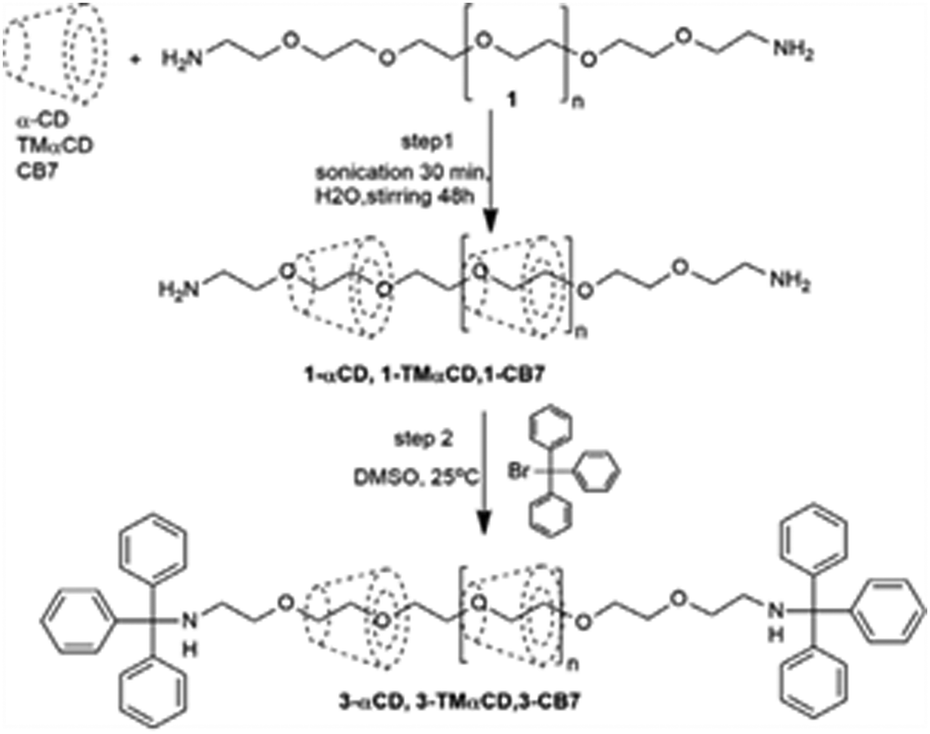 | ||
| Scheme 19 Synthetic route to the 3-αCD, 3-TMαCD and 3-CB7 polyrotaxanes. The scheme has been reproduced from ref. 100 with permission from Elsevier. | ||
Supramolecular polymers
The chemistry of supramolecular polymers combines supramolecular chemistry with polymer sciences and such intriguing systems find potential applications in various fields, such as drug delivery, shape-memory, self-repairing and self-healing, stimuli-responsive materials, and so on.25,36,40,46,101 Supramolecular polymers refer to the large molecular assemblies constructed in the form of polymeric chain structures, resulting from the union of multiple constituent units bound together through reversible non-covalent interactions. The development of macrocyclic molecule based supramolecular polymer systems depends largely on the strength of the associated host–guest interactions, which again relies on the nature of the macrocyclic hosts and the guest molecules involved.25,102 Park and co-workers designed a highly fluorescent supramolecular polymer system based on a CB8 host and a 1-methylpyridium based cyanostilbene derivative (Py+–CN–MBE) as the chromophoric guest (cf.Scheme 20).103 For this guest, its 1-methylpyridinium moiety provided very strong binding with the CB8 host. The cyanostilbene derivative used was quite non-emissive in aqueous solution, but became strongly emissive in nature upon its interaction with the CB8 host, mostly due to the formation of a linear and rigid supramolecular polymer. These authors also observed that another stilbene derivative that did not have a cyano unit actually inhibited the supramolecular polymer formation, and in the presence of CB8 there was actually a partial quenching of its fluorescence. It was proposed that such fluorescent supramolecular polymer systems could find interesting uses in chemical and biochemical sensing, as well as in bioimaging applications.103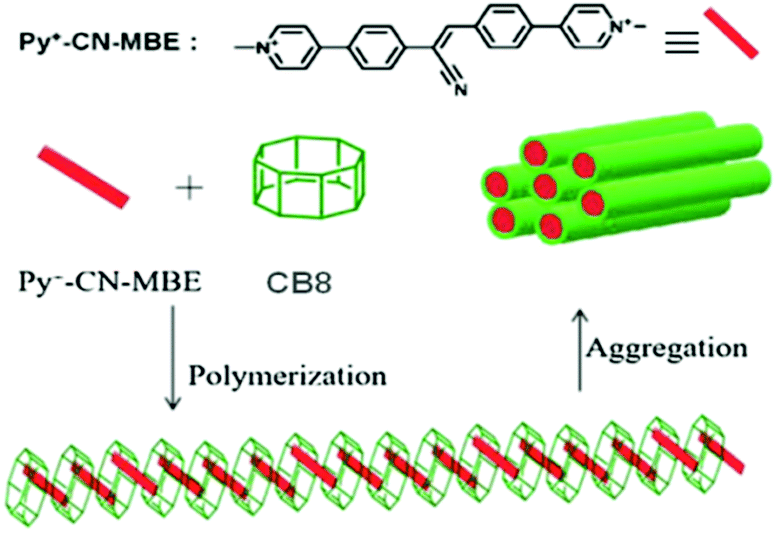 | ||
| Scheme 20 Schematic representation of the Py+–CN–MBE–CB8 based polymer formation. The scheme has been reproduced from ref. 25 with permission from Wiley-VCH Verlag GmbH. | ||
As the CB8 cavity is relatively larger, it can act as an anchor to bind the suitable end groups of two guest molecules simultaneously through their encapsulation and a relay of such interactions would eventually lead to the formation of unique supramolecular polymeric systems. In this context, CB8 based supramolecular polymerization was reported by Pang et al.104 In this case, linear supramolecular polymer formation was observed upon the direct interaction of CB8 with thiazole orange (TO) in aqueous medium and, as suggested by the authors, the process was driven by the π–π and hydrophobic interactions among the constituent units (cf.Scheme 21). Various experimental techniques like electrospray ionization mass spectrometry (ESI-MS), DLS, NMR, atomic force microscopy (AFM), and viscosimetry were employed to reveal the formation of linear supramolecular polymers in the present cases.
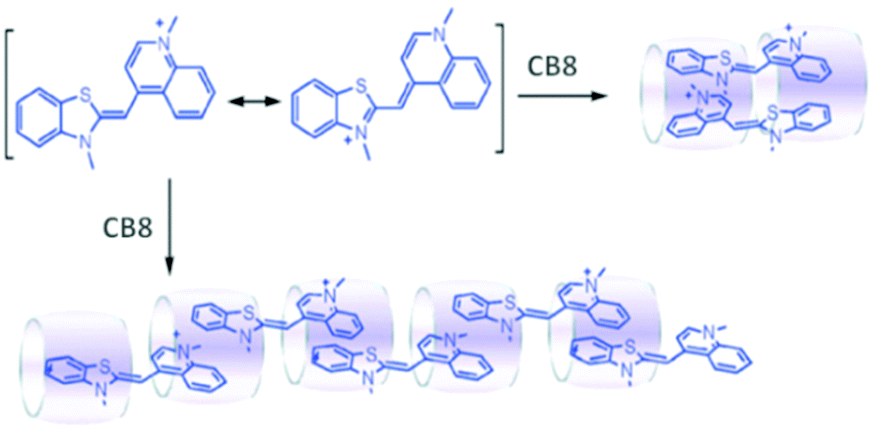 | ||
| Scheme 21 The structure of TO and schematic illustration of the TO–CB8 supramolecular polymerization driven by host-enhanced π–π interactions. The scheme has been reproduced from ref. 104 with permission from the Royal Society of Chemistry. | ||
In another example, CB8 based supramolecular polymer formation was also reported by Zhang and co-workers.105 In this study, the authors designed and synthesized a homoditopic monomer guest, i.e. 4,4′-(propane-1,3-diyl)bis[1-(anthracen-2-ylmethyl)-pyridinium] bromide (DAP), consisting of two anthracene moieties coupled through a pyridinium unit in between as the linker. When CB8 was mixed with the bifunctional anthracene derivative, it led to the formation of linear supramolecular polymer, which was driven by the π–π interactions between the encapsulated anthracene moieties of the successive guest molecules inside a CB8 host cavity. As a continuation of this work, Zhang et al.106 also reported the construction of a novel supramolecular polymer system involving both CB8 and CB7 as the macrocyclic hosts and using a unique bifunctional molecule with a p-phenylene unit in the middle and coupling two naphthalene moieties at the two ends (Naph–Phen–Naph) as the guest, facilitated and controlled by a self-sorting process. The authors showed that CB7 selectively recognized the p-phenylene unit in the middle, while CB8 underwent 2:1 (guest:host) complexation with the naphthalene units of successive guest molecules. Thus, when the Naph–Phen–Naph guest was mixed with CB7 and CB8, it resulted in the formation of the concerned linear supramolecular polymer. Interestingly, in the present system, the supramolecular polymerization process could be controlled by adjusting the CB7 repeating units. This line of research provides a general methodology to control the molecular weight and the structure of supramolecular polymers. Similarly, Thangavel et al. reported the formation of a supramolecular polymer based on the host–guest complexation of CB8 with a phenyl-substituted diazaperylenediium dication (DAP) guest.107 The structure of the resulting complex was characterized using NMR, ESI-MS, ITC, 2D NOESY (two-dimensional nuclear Overhauser enhancement spectroscopy), and DOSY (diffusion-ordered spectroscopy) measurements.
An elegant rotaxane based supramolecular polymer system was synthesized by Harada and coworkers, involving the orthogonal interactions of αCD and βCD hosts for the specially designed rotaxane assembly.108 The authors first synthesized a βCD derivative, namely, 6-aminocinnamoyl-βCD (6-aminoCiO-βCD), which then interacted with 1-adamantane carboxylic acid (AdCx), whereby the AdCx was bound to the βCD cavity and the aminocinnamoyl unit remained exposed to water. To this system, αCD was then added, whereby the phenyl moiety of the aminocinnamoyl unit was entrapped into the αCD cavity. The system was subsequently reacted with trinitrobenzene sulfonic acid (TNBS), whereby the TNBS molecule was coupled to the amino group of the aminocinnamoyl unit, forming a bulky trinitrophenyl group as a stopper to resist the escape of the αCD ring. The AdCx molecule complexed into the βCD cavity was finally removed from the system, following extraction using organic solvents, to obtain the desired rotaxane system with a free βCD cavity. The βCD cavity of this rotaxane system could then undergo complexation with the trinitrophenyl group of the other rotaxane molecule to produce the rotaxane based supramolecular polymer where αCD and βCD molecules were arranged alternatingly in a linear fashion, as shown in Scheme 22.
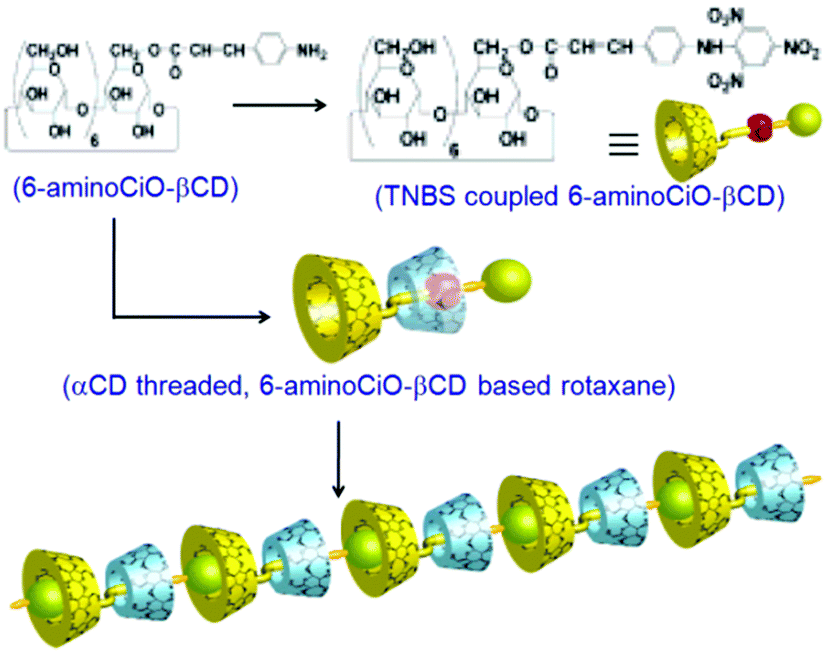 | ||
| Scheme 22 The proposed structure of a supramolecular rotaxane based polymer. The scheme has been reproduced from ref. 108 with permission from the American Chemical Society. | ||
Water-soluble sulfonatocalixarene based supramolecular polymer systems have also been reported in the literature. In this context, Liu and coworkers designed and synthesized a homoditopic bis(p-sulfonatocalix[5]arene) host molecule that bridges two calixarene moieties at their lower rims and could provide two guest binding sites.109 Supramolecular polymers with both 1D linear and 2D net-like topological structures were fabricated by employing this bis-calixarene host upon complexation with dicationic and tetracationic porphyrin guests, respectively. Interestingly, the binding stoichiometries between the bis-calixarene host and the porphyrin guests were found to be 1:5 and 2:5 with the dicationic and tetracationic porphyrins (cf.Scheme 23), respectively, such that the number of negative sulfonate groups on the calixarene moieties was equal to the number of positive charges on the porphyrin units. This thus indicates that, in the present system, the ion–ion interaction played the central role in the formation of the supramolecular polymer structures. In these supramolecular polymers, calixarenes and porphyrins acted as the effective electron donor–acceptor pairs, and thus the photoinduced electron transfer (PET) process prevailed in the present system. It was thus suggested that such supramolecular nanoarchitectures might find potential applications in areas like artificial photosynthesis, optoelectronics, and so on.107
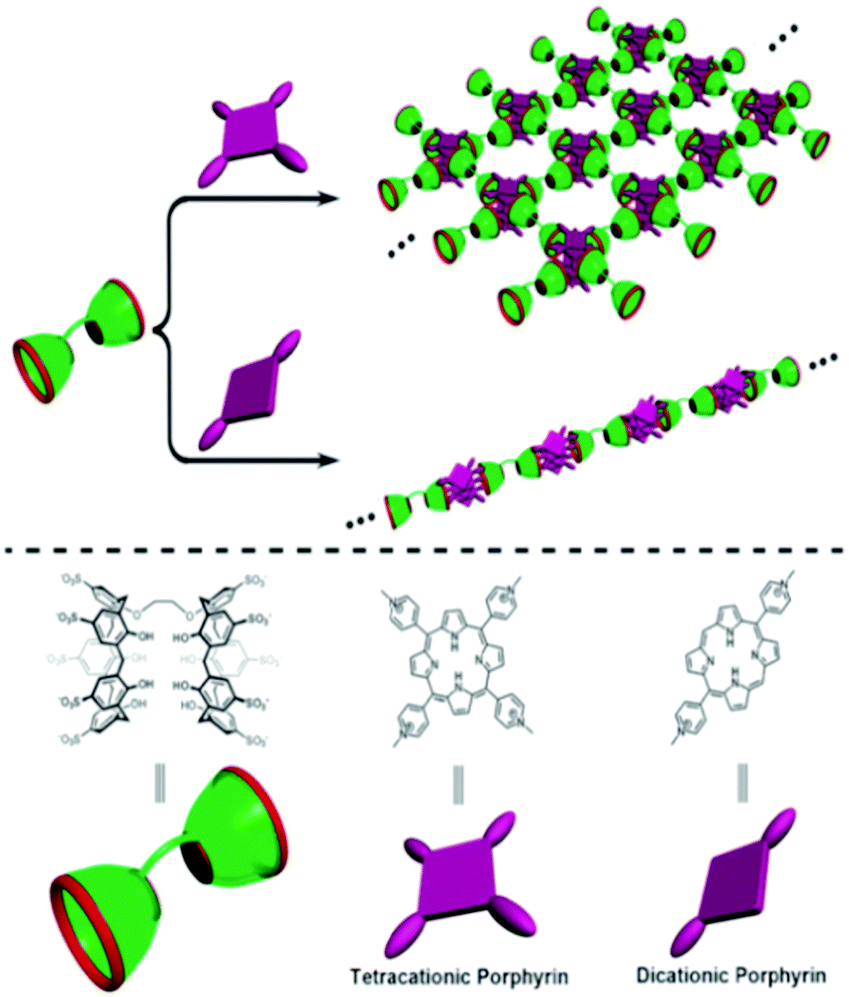 | ||
| Scheme 23 Schematic representation of the construction of a supramolecular 2D network and 1D linear polymer based on the complexation of ditopic bis(p-sulfonatocalix[5]arene) host with tetracationic and dicationic porphyrin guests, respectively. The scheme has been reproduced from ref. 102 with permission from the Royal Society of Chemistry. | ||
Liu and coworkers further reported the construction of linear supramolecular ternary polymers, taking advantage of the distinguishable binding properties of macrocyclic host molecules, such as cyclodextrins and calixarenes, involving two orthogonal host–guest interactions.110 For this purpose, the authors synthesized a heteroditopic adamantane–viologen guest and interacted it with bis-cyclodextrin and bis-calixare hosts. The adamantane moiety of the guest preferentially bound the cyclodextrin cavity, whereas the viologen motif was mainly included in the calixarene cavity. These two independent host–guest interactions that participate in an orthogonal manner effectively led to the formation of a linear supramolecular ternary polymer, as shown in Scheme 24. The authors proposed that, as both cyclodextrins and sulfonatocalixarenes are biocompatible, such polymeric systems with orthogonal interactions might be good mimics for biological processes.110
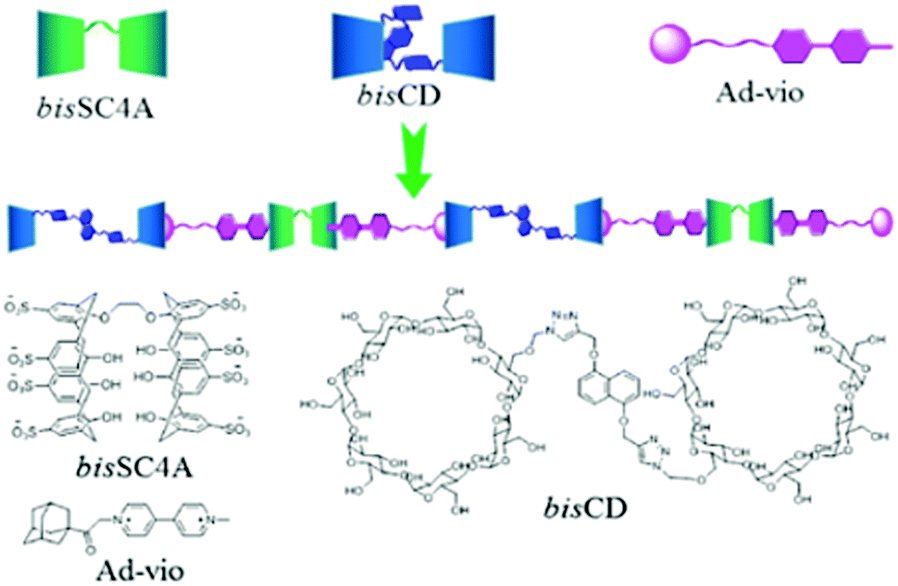 | ||
| Scheme 24 Schematic representation of bisSC4A, Ad-Vio, bisCD and the supramolecular ternary polymer [bisCD]–[Ad-Vio]–[bisSC4A]. The scheme has been reproduced from ref. 110 with permission from the Royal Society of Chemistry. | ||
Micelles from supramolecular polymers
Like conventional surfactants, supramolecular polymers are also quite amphiphilic in nature and, thus, they can also form micelles when their concentrations become higher than the critical micelle concentration (CMC). Like conventional micelles, supramolecular micellar assemblies can also be used in various applications, such as drug delivery, the purification of wastewater, as phase-transfer catalysts, and many others.17,21,29,36,50 Micelles have hydrophobic cores and hydrophilic shells, and are capable of solubilizing both hydrophobic and hydrophilic compounds inside the core or shell regions, respectively. The micellization of supramolecular polymers is dependent on the preparation conditions, interaction parameters, and ratio of the host and guest volume fractions in the polymer.17,21,29,50 Jiang et al. reported the formation of multicore and core–shell micelles from graft-like supramolecular polymers (cf.Scheme 25).111 In their study, graft-like polymers were formed, involving hydrophilic βCD derivatives as the hosts with suitable hydrophilic chains as their substituents, and adamantyl end-functionalized poly(ε-caprolactone) molecules (PCLs) as the guests. The high inclusion ability of the adamantyl group by the βCD cavity supported the formation of graft-like supramolecular polymers. These polymers formed multicore micelles under non-equilibrium conditions when their solutions in N-methyl-2-pyrrolidone (NMP) were drop-wise added into water. The sizes of these multicore micelles were found to be in the range of 180 to 220 nm. The formation of simple core–shell micelles was also observed for these polymeric systems under equilibrium conditions when the polymer solutions in NMP were dialysed against water. The diameters of these micelles were found to be around 120 nm.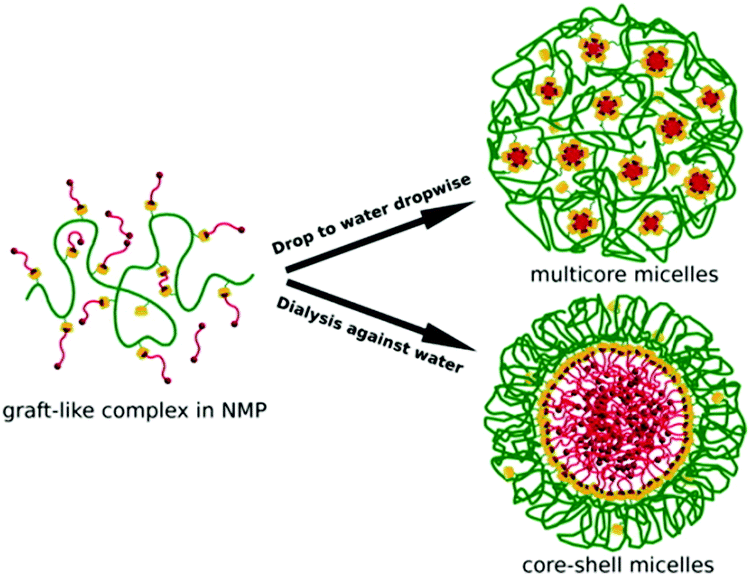 | ||
| Scheme 25 Schematic representation of the formation of multicore and core–shell micelles. The scheme has been reproduced from ref. 111 with permission from Wiley-VCH Verlag GmbH. | ||
Wang and Jiang also reported the construction of micelles using supramolecular polymers, taking advantage of the strong inclusion complex formation of βCD with adamantyl groups.112 Thus, complex formation between a hydrophobic poly(tert-butyl acrylate) polymer containing the adamantine moiety as an end group (PtBA–ADA) and a hydrophilic poly(methacrylate) polymer possessing a βCD macrocycle at the terminal (PGMA–CD) could effectively lead to the formation of micellar assemblies with diameters in the range of 100 to 200 nm, which could be adjusted by changing the βCD to ADA ratio in the solution, achieved using different mixtures of PGMA–CD and PtBA–ADA in DMF added into a large volume of water to prepare the micelles. The micelles formed in these cases had a hydrophobic core consisting of the PtBA–ADA motif and a hydrophilic shell made up mainly by PGMA–CD units. The presence of the βCD moieties on the micellar surface provided a unique opportunity to modify the surface either with hydrophobic or charged characteristics, following the encapsulation of either neutral or ionic guests into the βCD cavities. Additionally, after cross linking and core removal, these micelles could be converted into hollow spheres consisting of PGMA–CD networks, and thereby could be used for various applications, especially because large quantities of guests can be encapsulated into such cavities.
The first protein sized fluorescent supramolecular micelle systems were reported by Klymchenko et al.,113 involving the synthesis of amphiphilic calixarene derivatives possessing long alkyl chain substitutions at the phenolic OH sites (present at the lower rim) and the alkyne bearing polar quaternary ammonium substitutions at the para-positions of the constituent phenyl moieties (present at the upper rim). The synthesized calixarene derivatives could easily undergo micelle formation. In this study, the authors also cross-linked these amphiphilic host molecules with PEGylated cyanine 3 and cyanine 5 bis-azide dyes and, using these latter systems, they could construct ultrabright emissive protein-sized (∼7 nm) shell-cross-linked micellar assemblies, where covalently attached cyanine dyes decorated the corona region of the micelles, formed by the amphiphilic calixarene derivatives. The micelles thus formed showed excellent stability in aqueous and organic media. In addition, they had small hydrodynamic diameters, superior brightness, and could readily permeate into cells, and thus could be useful in biosensing and bioimaging applications.
Likewise, Zhou et al.114 designed supramolecular polymeric micelles which were constructed based on the host–guest interactions of a hydrophilic PEGylated calix[4]arene derivative with a hydrophobic photosensitizer molecule, chlorin e6 (Ce6). The inclusion complex formation of Ce6 with the calixarene derivative led to the formation of amphiphilic supramolecular polymers, which could self-assemble to form supramolecular polymeric micelles in aqueous solution, which could display intriguing stimuli-responsive properties (cf.Scheme 26). The micelles formed in the present system were about 200 nm in diameter, and were very suitable for passive drug delivery, due to their enhanced permeability and retention (EPR) effects. These properties, as well as the efficacy of the formed polymeric micelles towards photodynamic therapy (PDT), were assessed by the authors using HeLa cells as the representative cancer cell line.
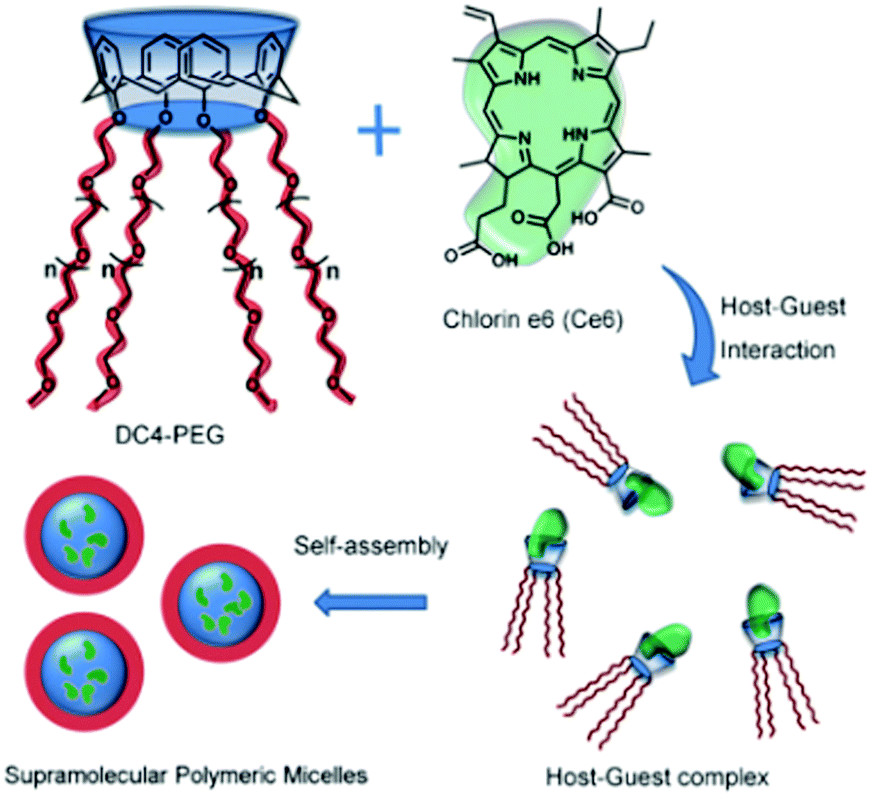 | ||
| Scheme 26 Chemical structures of the star-like calix[4]arene host, chlorin e6 guest molecule, and formation of the supramolecular polymeric micelles based on host–guest interactions. The scheme has been reproduced from ref. 114 with permission from the Royal Society of Chemistry. | ||
With regard to supramolecular polymeric micelles, García-Río et al. reported an important study involving the interaction of an amphiphilic p-sulfonatocalix[4]arene tetrabutyl ether (SC4TB) host with a trans-4-[4-(dimethylamino)styryl]-1-methylpyridinium iodide (DMSI) guest dye.115 In this study, the authors investigated the above system at concentrations both below and above the critical micellar concentration (CMC) of the supramolecular polymer. For concentrations below the CMC, the dye molecules underwent host–guest complexation with the available host molecules. Above the CMC, however, the dye molecules were seen to undergo an exchange between the recognition sites of the calixarene hosts present both in the monomeric states and in the micellar assemblies. It was also observed that above the CMC, a fraction of the guest dyes also resided in the micellar hydrophobic core. Accordingly, in this case, there was a reasonable distribution of the guest molecules between all possible microscopic localization sites of the micellar system. It was inferred that the high concentration of counter cations (e.g. Na+ ions in the studied system) at the Stern layer caused a large charge neutralization for the micellized sulfonatocalix[4]arene hosts, resulting in less efficient recognition of the cationic guest molecules at these binding sites and thus allowing their distribution at different other localization sites. As suggested by these authors, the above situation might be useful in the self-sorting process and could be employed for the efficient translocation of the guests from the host cavity to other pseudophases of the supramolecular systems.
Supramolecular micelle formations involving CBn hosts and polymer functionalized guest molecules have also been reported in the literature. Taking advantage of the ability of CB8 to encapsulate two guest molecules simultaneously, Scherman et al. reported supramolecular micelle formation involving ternary complexes of double hydrophilic block copolymer (DHBC) systems with a CB8 host.116 In this study, the authors designed one pH responsive DHBC system that consisted of a naphthalene-terminated poly(dimethylaminoethyl-methacrylate) (PDMAEMA) structure and one thermo-responsive DHBC system that comprised a methyl viologen terminated poly(N-isopropylacrylamide) (PNIPAAm) structure and followed the ternary complex formation of the DHBC systems with the CB8 host. These ternary complexes subsequently underwent self-assembly in the solution, leading to the formation of supramolecular micelles, as depicted in Scheme 27. The authors also demonstrated the usefulness of these micelles for the encapsulation and controlled release of drugs, using the chemotherapeutic drug doxorubicin as the model system. The release of the drug was found to be triggered by external stimuli like pH, temperature and the addition of competitive guests for CB8. Moreover, the combined external triggers of temperature and a competitive binder showed faster drug release than any of the stimuli alone. The authors also observed that these supramolecular micelles had faster stimuli responsive drug release than their covalent analogues, demonstrating a significant advantage of dynamic non-covalent systems over covalent systems. Furthermore, the authors also checked the applicability of the present system in human cervical carcinoma cells. A significant reduction in the viability of cells was observed upon triggering the release of the drug from the supramolecular micelles, indicating the promise of such systems in chemotherapeutic applications.
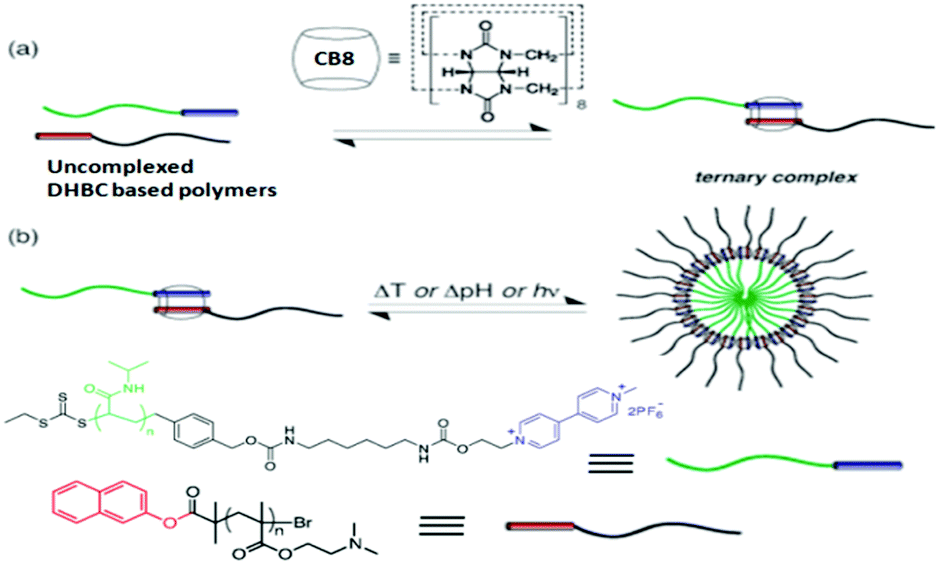 | ||
| Scheme 27 Schematic representation of (a) CB8 based ternary complex formation involving DHBC based polymers, and (b) self-assembly of the ternary complexes to form the micellar superstructure. The chemical structures of the two DHBC based polymers are also shown. The scheme has been reproduced from ref. 116 with permission from the Royal Society of Chemistry. | ||
In their studies, Scherman et al.117 have also developed supramolecular micelles that could bind and release insulin in a controlled manner. For this purpose, the authors synthesized glucose-sensitive DHBC systems, which can be held together through CB8 assisted ternary complex formation. These ternary complexes subsequently underwent self-association to produce the supramolecular micellar systems. In this study, poly(dimethylacrylamide) (PDMAAm) was used as one of the DHBC systems, along with two other DHBC systems, namely poly(acrylamidophenyl boronic acid) (PAAPBA) and poly(N-isopropylacrylamide) (PNIPAAm), used for their responses towards glucose and temperature, respectively. The sensitivity of these micelles towards drug binding and release was also investigated using insulin-loaded micelles and by following their responses to external triggers like changing temperature, glucose concentration, or the addition of a competitive guest for the CB8 cavity. A good control over the release of insulin from the supramolecular micelles was observed in their study under physiological conditions of pH ∼ 7.4 and a temperature of 37 °C. The present supramolecular approach of insulin delivery achieved by the simple use of host and guest components offers ease and versatility over the covalently bound carriers, which involve complicated synthetic procedures. Furthermore, the cytotoxicity of the present micellar system was tested using 3T3 fibroblast cells and it was observed that the present micellar systems were nontoxic, suggesting the potential of this system as the drug delivery vehicle for insulin therapies.
Vesicles from supramolecular polymers
Supramolecular vesicles can also be constructed by the self-assembly of some kind of supramolecular amphiphiles formed through the interaction of macrocyclic hosts with some guest molecules under specific conditions. Such vesicles have attracted considerable attention from researchers due to their structural simplicity, versatility, and most importantly the reversibility of their formation. These supramolecular assemblies have wide applications in various fields, such as the analysis of biological processes, light-harvesting systems, drug/gene delivery, fluorescence vesicular sensors, and so on.17,21,29,36,40,50 To date, many supramolecular vesicles based on CD, CBn, and CXn have been reported in the literature. In a study, Jung et al.118 designed and synthesised two amphiphilic βCD derivatives, namely mono[6-deoxy-6-(octadecanamido)]-βCD and mono[6-deoxy-6-(octadecenamido)]-βCD, with the aim of using these systems in the construction of supramolecular nanovesicles. It was indeed observed that these βCD derivatives self-assemble to form distinctive nanovesicles in water, which were suitably characterized using fluorescence spectroscopy, DLS, and TEM measurements. To find their practical applications, all-trans-retinol (vitamin A), a drug that is highly labile towards decomposition under light and oxygen, was loaded into these nanovesicles. It was found that, upon loading into these nanovesicles, the drug was greatly stabilized against photodegradation, demonstrating the potential of such systems in pharmaceutical applications. In a similar line, Tao et al.119 reported supramolecular vesicle formation using a linear hyperbranched supramolecular amphiphile produced through the non-covalent host–guest interaction of a hyperbranched polyglycerol modified βCD derivative (CD-g-HPG) and an adamantane-functionalized long alkyl chain (AD-C18), where strong binding of the adamantine moiety by the βCD cavity provided stability for the supramolecular amphiphile. The self-assembly of the resultant amphiphile led to the formation of supramolecular vesicles in water (cf.Scheme 28), which were demonstrated to be very stable and quite flexible, and were also amenable towards disassembling in the presence of competitive binders for the βCD cavities. | ||
| Scheme 28 Schematic representation of the formation of supramolecular vesicles involving the host–guest interactions of the amphiphilic molecules, CD-g-HPG and AD-C18. The scheme has been reproduced from ref. 119 with permission from the American Chemical Society. | ||
For p-sulfonatocalix[n]arene (SCXn, n = 4–8) hosts, because of their electron-rich cavities, negative charges at the wider portals, high aqueous solubility, and biological compatibility, these macrocycles have been realized as versatile building blocks for the construction of supramolecular amphiphile systems. Liu et al.120 reported nanoscale supramolecular binary vesicle formation, utilizing the host–guest interaction of p-sulfonatocalix[5]arene (SCX5) host with the 1-pyrenemethylaminium ion (PMA) as the guest. Simple mixing of SCX5 and PMA solutions with a charge-matching molar ratio resulted in the formation of supramolecular amphiphiles, driven mainly by enthalpic forces, which were then self-assembled to form the stimulus responsive supramolecular binary vesicles. The stimulus responsive characteristics of these supramolecular assemblies provide an added advantage over a covalent delivery system due to the reversible nature of the non-covalent interactions. Thus, the encapsulation and controlled release of the drug was checked by loading a good amount of DOX into the vesicles. Upon increasing the temperature, DOX was successfully released, accompanying the disassembly of the vesicles, demonstrating the potential of these vesicles as drug delivery models for biomedical applications. In a subsequent study, the authors also reported the formation of supramolecular polymeric vesicles through the complexation of the SCX4 host with a cationic polyammonium guest, chitosan, conceptually shown in Scheme 29.121 The formation of the supramolecular amphiphile in this case through host–guest interaction between SCX4 and chitosan was driven mainly by the dominant electrostatic interaction. The supramolecular amphiphiles thus formed were subsequently self-assembled, forming the supramolecular polymeric vesicles. The vesicles formed in this case were also found to be multi-stimuli responsive towards pH, temperature, and competitive binders, suggesting their usefulness in various applications.
 | ||
| Scheme 29 Schematic representation of the formation of supramolecular polymeric vesicles by SCX4 and chitosan. The scheme has been reproduced from ref. 121 with permission from Wiley-VCH Verlag GmbH. | ||
Kim and co-workers reported the formation of giant vesicles, driven by the CT complex formation inside CB8 cavity.122 In their study, a ternary complex was first formed with a CB8 cavity simultaneously encapsulating one methyl viologen derivative with a long (C12 and C16) aliphatic chain (MV-C12 and MV-C16), and a 2,6-dihydroxynaphthalene (DHNp) molecule as the guests. This ternary complex acted as a supramolecular amphiphile with its polar head group in the form of a CB8 moiety, and its hydrophobic tail in the form of an alkyl chain. These amphiphiles then self-assembled to form large vesicles in the solution (cf.Scheme 30), which was confirmed using NMR, mass spectroscopy, TEM, and SEM measurements. Vesicles with an average diameter of about 20 nm were formed with the ternary complexes formed by CB8 with MV-C12 and DHNp. Much larger vesicles, with diameters ranging from 400 to 950 nm, were, however, formed using the ternary complexes of CB8 with MV-C16 and DHNp. These results indicate that the size of the vesicles can be controlled by changing the alkyl chain length of the guest molecule complexing with the CB8 host.
 | ||
| Scheme 30 Schematic representation of the supramolecular vesicles formed involving ternary complexes of MV-C12 (or MV-C16), DHNp, and CB8. (a) TEM image of the vesicles formed by MV-C12, DHNp, and CB8. (b) TEM and (c) SEM images of the vesicles formed by MV-C16, DHNp, and CB8. The scheme has been reproduced from ref. 122 with permission from Wiley-VCH Verlag GmbH. | ||
Das and co-workers also reported supramolecular vesicle formation from another CB8 based ternary complex.123 In this study, a CB8 based supramolecular amphiphile was first produced by forming a CT complex within the host cavity, involving a peptide containing an azobenzene moiety as its end group and a methyl viologen derivative containing a long aliphatic chain as the two amphiphilic guest molecules. The supramolecular amphiphiles thus formed were subsequently self-assembled to form vesicles in the solution. However, it was observed that the azobenzene units underwent trans to cis isomerization upon photoirradiation, leading to the disruption of the vesicles, as the space inside the CB8 cavity was not sufficient to accommodate the cis-isomer. Additionally, the vesicles also showed reversible responses towards a competitive binder like 1-adamantylamine, leading to the rupture of the vesicles by displacing both the guest molecules from the CB8 host. Whereas when the 2,6-dihydroxynaphthalene was used as the competitive binder, it replaced the peptide unit from the CB8 cavity and is itself included in the host cavity, forming a new ternary complex. In this case, the vesicle structure remained effectively unaltered, though the peptide units were released from the initial vesicles. As the authors suggest, these stimulus-responsive vesicles may find applications in various fields, including drug delivery, sensors, molecular switches, and so on.
Supramolecular nanoparticles, nanorods, and nanosheets
The host–guest based supramolecular amphiphile scaffolds can also hierarchically self-assemble into various nanostructures, namely nanoparticles, nanorods, nanosheets, nanofibres, and others. The morphologies and functions of these nanosystems can also be tuned by carefully designing the host–guest complexes. These nanometer-sized assemblies, with varying morphologies, unique properties, and responsiveness towards different external stimuli, have shown their great potentials in various applications like drug delivery, stimuli-responsive functional materials, biosensors, etc.17,21,50 In this section, we present some of the selected work reported in this subject area.Assaf et al.124 reported the formation of contrasting self-assembled nanostructures involving the interaction of differently sized CBn homologues (n = 7 and 8) with an amphiphilic pyridinium-functionalized anthracene (AnPy) derivative. The AnPy molecule comprised a hydrophilic electron-deficient pyridinium moiety at one end and a hydrophobic electron-rich anthracene ring at the other end, with an alkyl spacer between the two units. The CB7 host, with a relatively smaller cavity, could suitably encapsulate only the pyridinium unit. In contrast, the CB8 host with a much larger cavity could easily encapsulate both pyridinium and anthracene units simultaneously, mainly driven by the associated CT interaction between the two units. The complexation of CB7 with AnPy eventually led to the formation of solid nanoparticles with diameters ranging from 400 to 1000 nm. On the other hand, complexation of CB8 with AnPy resulted in the formation of nanorods with lengths of a few micrometers and widths of about 700 nm.
In the present study, as CB7 was used as the host, the pyridinium unit of AnPy was encapsulated into the host cavity, and the anthracenyl unit of AnPy remained outside the cavity. In these cases, the anthracenyl units of different AnPy–CB7 complexes interacted among themselves through π–π stacking, resulting in the formation of nanoparticles. In contrast, when CB8 interacted with AnPy, it led to the formation of supramolecular polymeric chains through the simultaneous incorporation of anthracene and pyridinium units of different AnPy molecules into the CB8 cavities in a sequential manner. The polymeric chains thus formed could then interact with each other, leading to the formation of nanorods. The different kinds of nanostructures formed involving AnPy–CB7 and AnPy–CB8 systems are conceptually shown in Scheme 31. The resulting assemblies were seen to display stimuli responsive properties and could be assembled and disassembled by applying external stimuli like temperature and a competitive binder, demonstrating the potential of these stimulus responsive nanostructures in drug delivery applications.
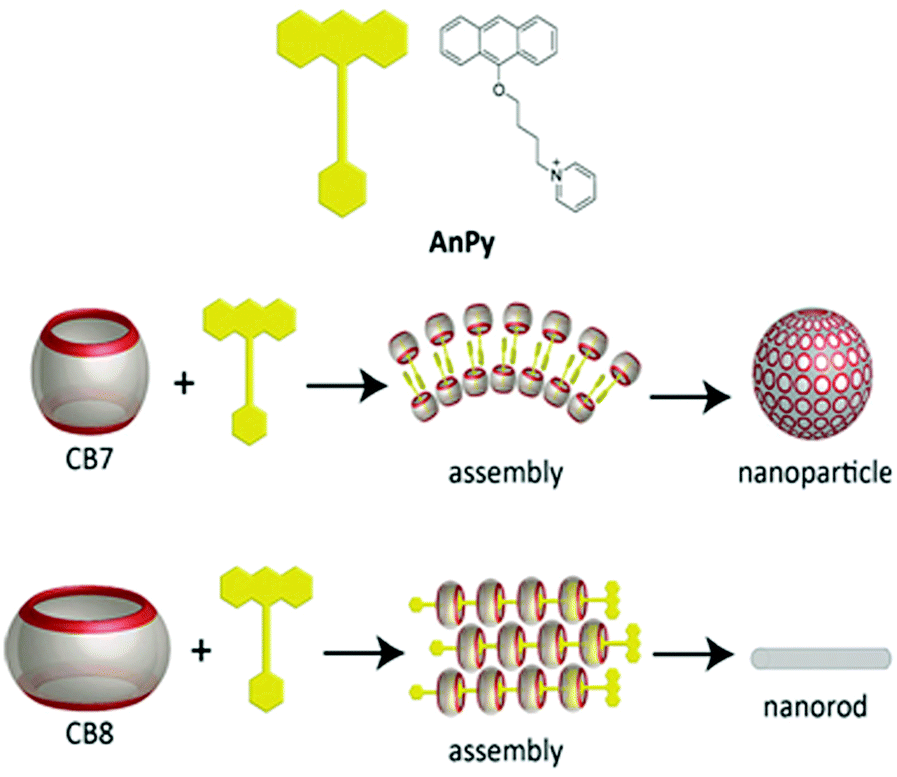 | ||
| Scheme 31 Chemical structure of the pyridinium-functionalized anthracene (AnPy) molecule, its symbolic model used and the schematic representation of the supramolecular assemblies formed by the AnPy–CB7 and AnPy–CB8 systems. The scheme has been reproduced from ref. 124 with permission from the Royal Society of Chemistry. | ||
Xing and co-workers also reported the formation of different self-assembled nanostructures in aqueous solution using the non-covalent interactions of CB7 and CB8 with the amphiphilic pyridinium-functionalized anthracene (AP) molecule.125 It was seen that the CB7 cavity mainly encapsulated the pyridinium unit, whereas the CB8 cavity encapsulated both the pyridinium and anthracene moieties simultaneously. The AP–CB7 complexes formed in the system were further self-assembled to produce spherical nanoparticles with an average diameter of about 200 nm. On the contrary, the self association of the AP–CB8 complexes led to the formation of the nanorods with widths of about 400 nm and lengths of several micrometers. Interestingly, these supramolecular assemblies were also used by the authors to construct an artificial light-harvesting system employing Eosin Y (EY) as the energy acceptor. It was seen that the fabricated light-harvesting system facilitated excitation energy transfer from the supramolecular assemblies to the EY units. The present study thus demonstrated that such supramolecular nano-assemblies have potential for uses in artificial light-harvesting systems.
Likewise, Lui et al.126 also reported the host–guest complexation mediated nanoparticle formation involving SCX4 as the macrocyclic host and the pyridinium-functionalized anthracene molecule, AnPy, as the guest molecule (cf.Scheme 32). The nanostructures formed in the present system were characterized using UV–vis, NMR, DLS, and TEM measurements. The authors showed that the AnPy–SCX4 based supramolecular assemblies were quite responsive towards temperature change and they exhibited very efficient photolysis upon irradiation with visible light in the presence of the photosensitizer, Eosin Y (EY). This approach may pave the way to construct various photoresponsive self-assembled materials and apply them in photodynamic therapy.
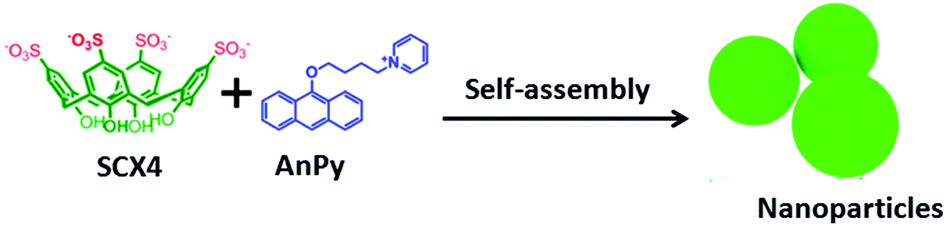 | ||
| Scheme 32 Schematic representation of the nanoparticles formed by AnPy and SCX4. The scheme has been reproduced from ref. 126 with permission from the American Chemical Society. | ||
Liu et al.127 reported the formation of supramolecular nanosheets involving strong host–guest interactions of the cationic tetranaphthalenyl-methylpyridinium conjugated porphyrin (TPOR) guest with the versatile macrocyclic host, CB7. In these cases, each naphthalenyl moiety of TPOR bound to the CB7 cavity, forming 4:1 stoichiometric (CB7)4–TPOR complexes, which acted effectively as supramolecular amphiphiles and, thus, underwent self-assembly to provide the nanostructures in the form of nanosheets (cf.Scheme 33). It was reported that the (CB7)4–TPOR based nanosheets were responsive towards a competitive guest like 1-adamantanamine hydrochloride. In addition, these nanosheets also exhibited greatly enhanced photosensitizing efficiency as compared to that of the sole TPOR molecules. The authors anticipated that such supramolecular assemblies with photosensitizing ability would find potential applications in photodynamic therapy.127
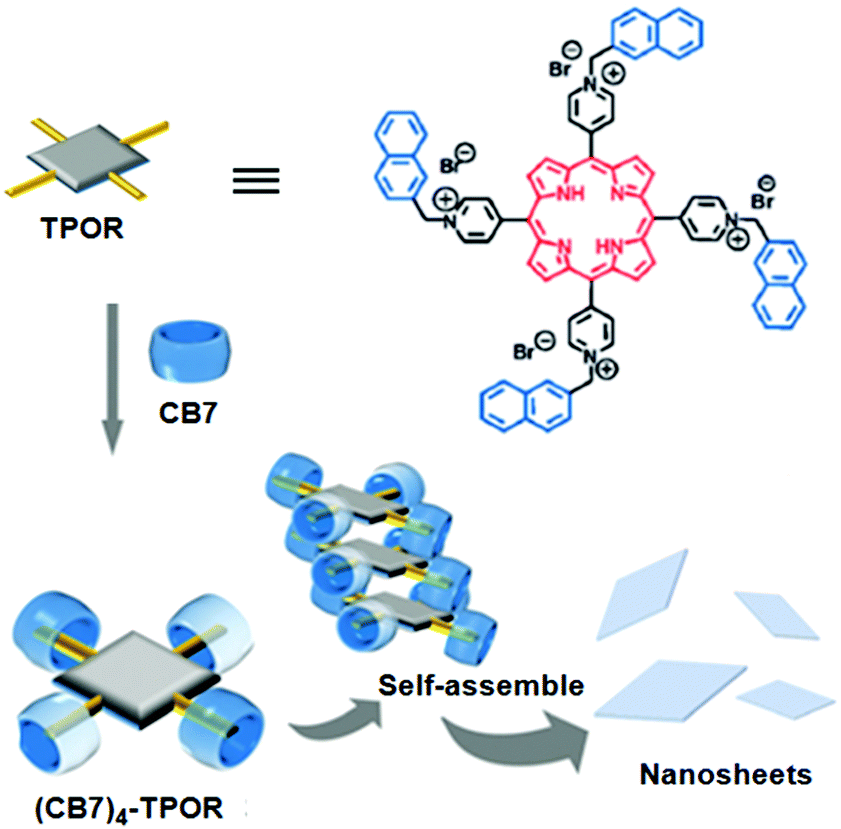 | ||
| Scheme 33 Schematic representation of the formation of nanosheets from the interaction of TPOR with CB7. The scheme has been reproduced from ref. 50 with permission from the Royal Society of Chemistry. | ||
Taking a similar perspective, Liu and co-workers128 reported the formation of rectangular nanosheets, utilising the CD based supramolecular polyrotaxane network, prepared from the Schiff base polycondensation reaction of 5,10,15,20-tetrakis(4-aminophenyl)porphyrin with the βCD encapsulated p-phthaldehyde molecule. These supramolecular polyrotaxanes then self-assembled to form highly ordered two-dimensional rectangular nanostructures, with average side lengths of about 500 nm (cf.Scheme 34). The nanosheet morphology was comprehensively characterized by NMR, FT-IR, XRD, TEM, and SEM measurements. The authors also used these nanostructures as the heterogeneous catalyst for the photooxidation of dimethylanthrancene under visible light irradiation, providing up to about 99% oxidation efficiency. This approach of preparing supramolecular polyrotaxane networks could be applied to functional materials and catalytic chemistry.
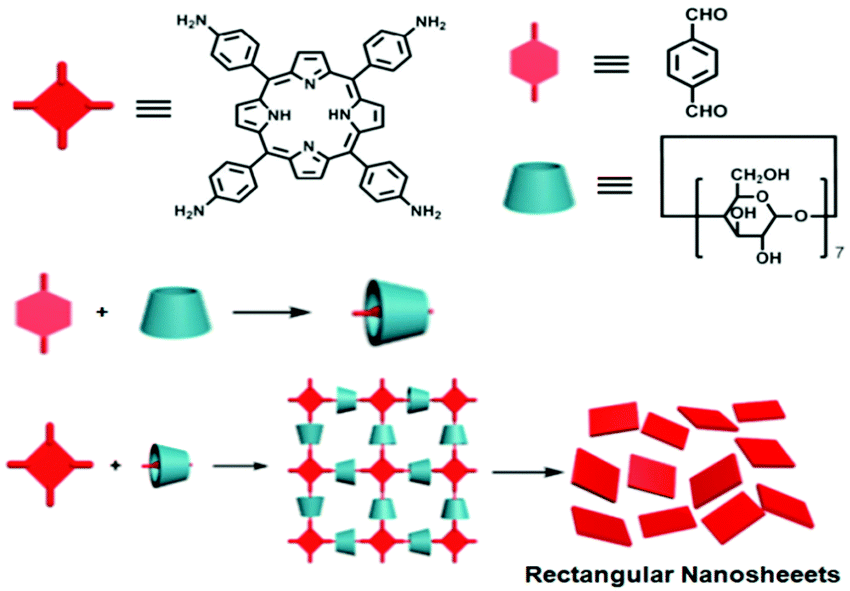 | ||
| Scheme 34 Schematic representation of the formation of rectangular nanosheets based on β-CD and porphyrins. The scheme has been reproduced from ref. 17 with permission from Wiley-VCH Verlag GmbH. | ||
Very recently, Tang and coworkers reported an excellent work where the authors constructed diversified supramolecular morphologies following the self-assembly of unique supra-amphiphiles.129 In their study, an aggregation-induced emission (AIE)-active amphiphilic scaffold was first prepared by functionalizing the 2,5-dimethoxybenzene-1,4-dicarboxaldehyde (DMA) molecule through the reaction of its two aldehyde groups with two molecules of an adamantine (Ad) derivative, namely N-(adamantan-1-yl)-8-bromooctanamide. The amphiphilic scaffold thus prepared, designated as DAdDMA, was then used to prepare a novel bola-type supra-amphiphile system, DAdDMA@2β-CD, utilizing the typical host–guest interactions of the two Ad groups of the DAdDMA molecule with the two βCD hosts. These supra-amphiphiles subsequently self-organized to provide various well-defined nano-assemblies, namely, leaf-like lamellar nanostructures, vesicles, normal nanofibers, helical nano-fibers, nano-ribbons, and toroids, as shown in Scheme 35, which were achieved by suitably adjusting the composition of the mixed solvent systems used in the study, typically the DMF–water and ethanol–water solvent mixtures, and also by changing the temperature conditions. The authors also investigated the light-harvesting properties of these nano-assemblies and found that the leaf-like lamellar nanostructures performed much better than other nano-assemblies, and could provide up to ∼94.2% energy transfer efficiency. The work is expected to stimulate many follow-up works to develop other similar supramolecular-based functional nanomaterials with more diversified topological morphologies to make them useful for many applications, ranging from biotechnology to stimuli-responsive supramolecular functional materials and designs.
 | ||
| Scheme 35 (A) The structures of DAdDMA and β-CD. (B) Fabrication of supra-amphiphile DAdDMA@2β-CD. (C) Schematic representation of formation of diversified topological morphologies from DAdDMA@2β-CD supra-amphiphiles. The scheme has been reproduced from ref. 129 with permission from Wiley-VCH Verlag GmbH. | ||
Summary and conclusions
In the current perspective, we have demonstrated the substantial progress in supramolecular host–guest chemistry from simple host–guest complexes to advanced supramolecular architectures. This perspective highlights the various supramolecular nanostructures prepared using macrocyclic hosts, such as cyclodextrins, cucurbit[n]urils, and calix[n]arenes, by utilizing their chemical constitutions and structural features and suggesting their future applications. The complex supramolecular assemblies considered here are pseudorotaxanes, rotaxanes, polyrotaxanes, supramolecular polymers, micelles, vesicles, nanoparticles, nanosheets, nanorods, etc. Though a simple host–guest system can be obtained just by the simple mixing of macrocyclic hosts and guest molecules, the design of complex supramolecular assemblies with varied morphologies and functions requires the synergetic integration of various factors like the strength of the host–guest binding, nature of the macrocyclic hosts and guest molecules used, their different interaction parameters, preparation conditions, and the ratio of the mole fractions of the guest and host used in the preparation method.In the literature, though several reviews on supramolecular chemistry are available, the assimilation of various supramolecular assemblies in one place has not been reviewed yet. Each subtopic discussed in this perspective is in itself is a central topic and, thus, discussing each subtopic in great detail is not possible. However, we have tried our best to comprehensively overview various supramolecular assemblies, ranging from simple to progressively complex supramolecular assemblies, by selecting exciting examples to showcase different supramolecular structures with unique characteristics, as discussed in this perspective. The various supramolecular assemblies discussed here are endowed with enhanced aqueous solubility, good biocompatibility, and impressive responses and amenability towards external stimuli, which can intriguingly offer enormous potential to these systems for their applications in diverse areas like biotechnology, medicines, drug delivery, sensors, functional materials, nanosciences, energy materials and many others. We hope that this perspective will be a stimulus for young researchers to carry forward their research in this fascinating area of supramolecular chemistry to create new and innovative nanoplatforms which have broad applications in the bioanalytical, biomedical, and pharmaceutical sciences. In particular, it should be mentioned that the practical applicability of supramolecular assemblies in the clinical sciences is still quite a challenging task. Thus, it is highly desirable in the subject domain discussed in the present review article for future research to be carried out into designing supramolecular assemblies with advanced properties to promote their clinical applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful to all the colleagues, collaborators, and other researchers whose findings and inferences have helped us to write this review in a comprehensive manner. The authors are also highly grateful to their home institute, the Bhabha Atomic Research Centre, Mumbai, India, for generously providing all the financial and academic support in the pursuit of the authors’ every scientific endeavor.Notes and references
- C. Jiang, Z. Song, L. Yu, S. Ye and H. He, TrAC, Trends Anal. Chem., 2020, 133, 116086 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- W. Liu and J. F. Stoddart, Chem, 2021, 7, 919–947 CAS.
- M. Sayed, B. Krishnamurthy and H. Pal, Phys. Chem. Chem. Phys., 2016, 18, 24642–24653 RSC.
- M. Sayed, B. Krishnamurthy and H. Pal, J. Mol. Liq., 2021, 336, 116146 CrossRef CAS.
- A. Sowa and J. Voskuhl, Int. J. Pharm., 2020, 586, 119595 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- A. A. Elbashir, N. F. A. Dsugi, T. O. M. Mohmed and H. Y. Aboul-Enein, Luminescence, 2014, 29, 1–7 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. Garcia, Angew. Chem., Int. Ed., 2018, 57, 2–16 CrossRef.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 1909049 CrossRef CAS.
- Y.-M. Zhang, Y.-H. Liu and Y. Liu, Adv. Mater., 2020, 32, 180615 Search PubMed.
- M. Sayed, F. Biedermann, V. D. Uzunova, K. I. Assaf, A. C. Bhasikuttan, H. Pal, W. M. Nau and J. Mohanty, Chem. – Eur. J., 2015, 21, 691–696 CrossRef CAS PubMed.
- M. Sayed and H. Pal, Phys. Chem. Chem. Phys., 2015, 17, 9519–9532 RSC.
- M. Sayed, K. Shinde, R. Shah and H. Pal, ChemistrySelect, 2016, 1, 989–999 CrossRef CAS.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan, V. D. Uzunova, W. M. Nau and H. Pal, Chem. Commun., 2008, 3681–3683 RSC.
- Z. Liu, X. Dai, Y. Sun and Y. Liu, Aggregate, 2020, 1, 31–44 CrossRef.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- M. Sayed and H. Pal, J. Mater. Chem. C, 2016, 4, 2685–2706 RSC.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 979–985 CrossRef CAS PubMed.
- Y. Chen, S. Sun, D. Lu, Y. Shi and Y. Yao, Chin. Chem. Lett., 2019, 30, 37–43 CrossRef CAS.
- S. Gürbüz, M. Idrisa and D. Tuncel, Org. Biomol. Chem., 2015, 13, 330–347 RSC.
- A. Hashidzume, H. Yamaguchi and A. Harada, Eur. J. Org. Chem., 2019, 3344–3357 CrossRef CAS.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef CAS PubMed.
- H. Zhu, L. Shangguan, B. Shi, G. Yu and F. Huang, Mater. Chem. Front., 2018, 2, 2152–2174 RSC.
- A. Villers, C. R. Fr. Acad. Sci., 1891, 112, 435–438 Search PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- M. Sayed, G. K. Gubbala and H. Pal, New J. Chem., 2019, 43, 724–736 RSC.
- M. Shaikh, Y. M. Swamy and H. Pal, J. Photochem. Photobiol., A, 2013, 258, 41–50 CrossRef CAS.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS PubMed.
- G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254–2266 RSC.
- M. Sayed, S. Jha and H. Pal, Phys. Chem. Chem. Phys., 2017, 19, 24166–24178 RSC.
- M. Sayed, S. Panjwani and H. Pal, ChemistrySelect, 2018, 3, 8131–8143 CrossRef CAS.
- M. Sayed, M. Sundararajan, J. Mohanty, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2015, 119, 3046–3057 CrossRef CAS PubMed.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- M. Sayed, S. Dutta Choudhury and H. Pal, Hybrid supramolecular assemblies of cucurbit[n]uril supported metal and other inorganic nanoparticles, in Cucurbituril-based Functional Materials, ed. D. Tuncel, RSC, 2020, pp. 95–119 Search PubMed.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, Chem. – Eur. J., 2009, 15, 12362–12370 CrossRef CAS PubMed.
- Y. Fan, R.-H. Gao, Y. Huang, B. Bian, Z. Tao and X. Xiao, Front. Chem., 2019, 7, 154 CrossRef CAS PubMed.
- C. Liu, Y. Wu, X. Han and S. Liu, New J. Chem., 2020, 44, 3185–3188 RSC.
- M. S. Mokhtar, F. O. Suliman and A. A. Elbashir, J. Inclusion Phenom. Macrocyclic Chem., 2019, 94, 31–43 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. D. Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- L. Cao, M. Sekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- A. Blanco-Gómez, P. Cortón, L. Barravecchia, I. Neira, E. Pazos, C. Peinador and M. D. García, Chem. Soc. Rev., 2020, 49, 3834–3862 RSC.
- K. M. Park, M. Y. Hur, S. K. Ghosh, D. R. Boraste, S. Kim and K. Kim, Chem. Commun., 2019, 55, 10654–10664 RSC.
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC.
- M. Shaikh, J. Mohanty, D. K. Maity, S. K. Nayak and H. Pal, J. Photochem. Photobiol., A, 2008, 195, 116–126 CrossRef CAS.
- M. Sayed, D. M. Tom and H. Pal, Phys. Chem. Chem. Phys., 2020, 22, 13306–13319 RSC.
- H. Bakirci and W. M. Nau, Adv. Funct. Mater., 2006, 16, 237–242 CrossRef CAS.
- V. Francisco, A. PiÇeiro, W. M. Nau and L. García-Río, Chem. – Eur. J., 2013, 19, 17809–17820 CrossRef CAS PubMed.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408–414 CrossRef CAS PubMed.
- S. A. Krolenko, S. Y. Adamyan, T. N. Belyaeva and T. P. Mozhenok, Cell Biol. Int., 2006, 30, 933–939 CrossRef CAS PubMed.
- F. Traganos, Z. Darzynkiewicz, T. Sharpless and M. R. Melamed, J. Histochem. Cytochem., 1977, 25, 46–56 CrossRef CAS PubMed.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132–5138 CrossRef CAS PubMed.
- B. Reija, W. Al-Soufi, M. Novo and J. V. Tato, J. Phys. Chem. B, 2005, 109, 1364–1370 CrossRef CAS PubMed.
- M. Shaikh, J. Mohanty, M. Sundararajan, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2012, 116, 12450–12459 CrossRef CAS PubMed.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061–1070 RSC.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, Phys. Chem. Chem. Phys., 2010, 12, 7050–7055 RSC.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS PubMed.
- F. Biedermann and O. A. Scherman, J. Phys. Chem. B, 2012, 116, 2842–2849 CrossRef CAS PubMed.
- S. Schoder and C. A. Schalley, Chem. Commun., 2017, 53, 9546–9549 RSC.
- S. D. Choudhury, J. Mohanty, H. P. Upadhyaya, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2009, 113, 1891–1898 CrossRef PubMed.
- N. H. Mudliar and P. K. Singh, Chem. – Eur. J., 2016, 22, 7394–7398 CrossRef CAS PubMed.
- V. Lau and B. Heyne, Chem. Commun., 2010, 46, 3595–3597 RSC.
- M. Nilam, C. Huang, S. Karmacharya, G. H. Aryal, L. Huang, W. M. Nau and K. I. Assaf, ChemistrySelect, 2020, 5, 5850–5854 CrossRef CAS.
- K. Yang, S. Chao, F. Zhang, Y. Pei and Z. Pei, Chem. Commun., 2019, 55, 13198–13210 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- F. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018 CrossRef CAS.
- K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC.
- J. Terao, S. Tsuda, Y. Tanaka, K. Okoshi, T. Fujihara, Y. Tsuji and N. Kambe, J. Am. Chem. Soc., 2009, 131, 16004–16005 CrossRef CAS PubMed.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed.
- H. Zhang, E. S. Paulsen, K. A. Walker, K. E. Krakowiak and D. V. Dearden, J. Am. Chem. Soc., 2003, 125, 9284–9285 CrossRef CAS PubMed.
- A. Harada, J. Li and M. Kamachi, Macromolecules, 1993, 26, 5698–5703 CrossRef CAS.
- H.-J. Buschmann, A. Wego, K. Jansen, E. Schollmeyer and D. Döpp, J. Inclusion Phenom. Macrocyclic Chem., 2005, 53, 183–189 CrossRef CAS.
- M. V. Rekharsky, H. Yamamura, M. Kawai, I. Osaka, R. Arakawa, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Org. Lett., 2006, 8, 815–818 CrossRef CAS PubMed.
- L. Leclercq, N. Noujeim, S. H. Sanon and A. R. Schmitzer, J. Phys. Chem. B, 2008, 112, 14176–14184 CrossRef CAS PubMed.
- P. Branna, M. Rouchal, Z. Pruckova, L. Dastychova, R. Lenobel, T. Pospíšil, K. Maláč and R. Vícha, Chem. – Eur. J., 2015, 21, 11712–11718 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Muller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- P. L. Nostro, J. R. Lopes and C. Cardelli, Langmuir, 2001, 17, 4610–4615 CrossRef.
- A. Harada, J. Li and M. Kamachi, Nature, 1994, 370, 126–128 CrossRef CAS.
- T. Uyar, M. Rusa and A. E. Tonelli, Macromol. Rapid Commun., 2004, 25, 1382–1386 CrossRef CAS.
- S. Choi, J. W. Lee, Y. H. Ko and K. Kim, Macromolecules, 2002, 35, 3526–3531 Search PubMed.
- K. M. Huh, T. Ooya, S. Sasaki and N. Yui, Macromolecules, 2001, 34, 2402–2404 CrossRef CAS.
- H. Okumura, M. Okada, Y. Kawaguchi and A. Harada, Macromolecules, 2000, 33, 4297–4298 CrossRef CAS.
- T.-A. Yamagishi, A. Kawahara, J. Kita, M. Hoshima, A. Umehara, S.-I. Ishida and Y. Nakamoto, Macromolecules, 2001, 34, 6565–6570 CrossRef CAS.
- Y.-M. Jeon, D. Whang, J. Kim and K. Kim, Chem. Lett., 1996, 503–504 CrossRef CAS.
- R. Isnin and A. E. Kaifer, J. Am. Chem. Soc., 1991, 113, 8188–8192 CrossRef CAS.
- H. M. Dardeer, J. Inclusion Phenom. Macrocyclic Chem., 2018, 91, 105–114 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- C. Meschke, H.-J. Buschmann and E. Schollmeyer, Macromol. Rapid Commun., 1998, 19, 59–63 CrossRef CAS.
- Y.-G. Jia, S. Liu, J. Wang, L. Cai, J. Jin, L. Mo, M. Gao, L. Ren and X. X. Zhu, Macromolecules, 2018, 51, 8455–8460 CrossRef CAS.
- M. Okada and A. Harada, Org. Lett., 2004, 6, 361–364 CrossRef CAS PubMed.
- M. van den Boogaard, G. Bonnet, P. van’t Hof, Y. Wang, C. Brochon, P. van Hutten, A. Lapp and G. Hadziioannou, Chem. Mater., 2004, 16, 4383–4385 CrossRef CAS.
- D. Nepal, S. Samal and K. E. Geckeler, Macromolecules, 2003, 36, 3800–3802 CrossRef CAS.
- A.-M. Resmerita, K. I. Assaf, A. I. Lazar, W. M. Nau and A. Farcas, Eur. Polym. J., 2017, 93, 323–333 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- D.-S. Guo and Y. Liu, Chem. Soc. Rev., 2012, 41, 5907–5921 RSC.
- H.-J. Kim, D. R. Whang, J. Gierschner and S. Y. Park, Angew. Chem., 2016, 128, 16147–16151 CrossRef.
- Y. Xu, M. Guo, X. Li, A. Malkovskiy, C. Wesdemiotis and Y. Pang, Chem. Commun., 2011, 47, 8883–8885 RSC.
- Y. Liu, K. Liu, Z. Wang and X. Zhang, Chem. – Eur. J., 2011, 17, 9930–9935 CrossRef CAS PubMed.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., 2014, 126, 5455–5459 CrossRef.
- A. Thangavel, M. Macias and S. Tsumaki, ACS Omega, 2020, 5, 5574–5579 CrossRef CAS PubMed.
- M. Miyauchi, T. Hoshino, H. Yamaguchi, S. Kamitori and A. Harada, J. Am. Chem. Soc., 2005, 127, 2034–2035 CrossRef CAS PubMed.
- D.-S. Guo, K. Chen, H.-Q. Zhang and Y. Liu, Chem. – Asian J., 2009, 4, 436–445 CrossRef CAS PubMed.
- H.-X. Zhao, D.-S. Guo, L.-H. Wang, H. Qian and Y. Liu, Chem. Commun., 2012, 48, 11319–11321 RSC.
- S. Ren, D. Chen and M. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4267–4278 CrossRef CAS.
- J. Wang and M. Jiang, J. Am. Chem. Soc., 2006, 128, 3703–3708 CrossRef CAS PubMed.
- I. Shulov, R. V. Rodik, Y. Arntz, A. Reisch, V. I. Kalchenko and A. S. Klymchenko, Angew. Chem., 2016, 128, 16116–16120 CrossRef.
- C. Tu, L. Zhu, P. Li, Y. Chen, Y. Su, D. Yan, X. Zhu and G. Zhou, Chem. Commun., 2011, 47, 6063–6065 RSC.
- S. Fernández-Abad, M. Pessêgo, N. Basílio and L. García-Río, Chem. – Eur. J., 2016, 22, 6466–6470 CrossRef PubMed.
- X. J. Loh, J. d. Barrio, P. P. C. Toh, T.-C. Lee, D. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed.
- X. J. Loh, M.-H. Tsai, J. del Barrio, E. A. Appel, T.-C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC.
- H. Kim, Y. Hu, D. Jeong, B.-H. Jun, E. Cho and S. Jung, Molecules, 2016, 21, 963 CrossRef PubMed.
- W. Tao, Y. Liu, B. Jiang, S. Yu, W. Huang, Y. Zhou and D. Yan, J. Am. Chem. Soc., 2012, 134, 762–764 CrossRef CAS PubMed.
- K. Wang, D.-S. Guo and Y. Liu, Chem. – Eur. J., 2010, 16, 8006–8011 CrossRef CAS PubMed.
- S. Peng, K. Wang, D.-S. Guo and Y. Liu, Soft Matter, 2015, 11, 290–296 RSC.
- Y. J. Jeon, P. K. Bharadwaj, S. Choi, J. W. Lee and K. Kim, Angew. Chem., 2002, 114, 4654–4656 CrossRef.
- J. H. Mondal, S. Ahmed, T. Ghosh and D. Das, Soft Matter, 2015, 11, 4912–4920 RSC.
- K. I. Assaf, M. A. Alnajjar and W. M. Nau, Chem. Commun., 2018, 54, 1734–1737 RSC.
- Z. Lian, M. Jiang, F. Qiao, M.-N. Chen, R.-Z. Wang, S. Zhuo and L.-B. Xing, J. Photochem. Photobiol., A, 2020, 386, 112135 CrossRef CAS.
- Y.-X. Wang, Y.-M. Zhang and Y. Liu, J. Am. Chem. Soc., 2015, 137, 4543–4549 CrossRef CAS PubMed.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285–8289 CrossRef CAS PubMed.
- W.-L. Zhou, X. Zhao, Y. Chen and Y. Liu, Org. Chem. Front., 2019, 6, 10–14 RSC.
- S. Fu, X. Su, M. Li, S. Song, L. Wang, D. Wang and B. Z. Tang, Adv. Sci., 2020, 7, 2001909 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2021 |