
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantum chemical assessment of the molecular area corresponding to the onset of the LE–LC phase transition for amphiphilic 2D monolayers at the air/water interface
Elena S.
Kartashynska
 ab and
Dieter
Vollhardt
*c
ab and
Dieter
Vollhardt
*c
aL.M. Litvinenko Institute of Physical Organic and Coal Chemistry, 70 R. Luxemburg Str., Donetsk 83114, Ukraine
bDonetsk National Technical University, 58 Artema Str., Donetsk 83000, Ukraine
cMax Planck Institute of Polymer Research, Mainz D-55128, Germany. E-mail: vollhardtd@mpip-mainz.mpg.de
First published on 26th October 2021
Abstract
An approach for the assessment of the area per surfactant molecule in a monolayer at the onset of the LE–LC phase transition (Ac) is proposed based on the quantum chemical approach and a thermodynamic model for amphiphilic monolayers, which takes into account the nonideality of the mixing entropy. The values of the Gibbs’ clusterization energy for small surfactant associates, as well as the geometric parameters of the monolayer unit cells, were used, previously calculated using the semiempirical PM3 method for eight classes of amphiphilic compounds: saturated and ethoxylated alcohols, saturated and unsaturated cis-carboxylic acids, α-hydroxylic and α-aminoacids, N-acyl-substituted alanine and dialkyl-substituted melamine. The obtained Ac values are in satisfactory agreement with the available experimental data. This allows using the proposed approach for prognostic purposes in the cases where there are no corresponding π–A isotherms for necessary surfactants, but there are calculated thermodynamic and structural parameters of its clusterization.
Introduction
Interest in studying the nonionic surfactant behavior at different interfaces is dictated by the variety of the properties of their films and the possibility of their changing. A wide range of experimental techniques and instrumental capabilities made it possible to reveal the regularities of 2D aggregate formation of amphiphilic compounds on different surfaces depending on various external conditions (temperature, pH, and the presence of organic and inorganic ions). Attention to the properties of Langmuir monolayers is also due to attempts to represent the structure of biological membranes through a combination of individual monolayers, which shows Langmuir monolayers as interesting objects for research in the framework of membrane biophysics and biosensors. The possibility of transferring surfactant monolayers to numerous solid surfaces with the multilayer formation made it possible to design optical, anticorrosive, and antifriction coatings with predetermined properties.A large number of studies have been devoted to describing the aggregation process of surfactant molecules into a monolayer.1 Even at the first stage of the study of monolayers it was shown that molecules are characterized by three possible aggregate states: gaseous, liquid, and solid. Later, four types of films were identified and characterized in detail: gaseous, gaseous-expanded, liquid-expanded, and condensed.2 Over time the classification has enlarged due to the detailed description of individual states of the films.3,4 The existence of different monolayer phases was concluded from the surface pressure–molecular area (π–A) isotherms. The π–A isotherms show a sharp break, when a main phase transition from a fluid (gaseous (G), liquid-expanded (LE)) to the condensed (liquid-condensed (LC), solid (S)) phases takes place. These parts of the π–A isotherms represent the transition region to the condensed phase and are decisive for the features of the condensed monolayer phases.
The fluid/condensed transitions were considered thermodynamically in the framework of a quasi-chemical approach. The quasi-chemical monomer/aggregate equilibrium model based on a combination of the generalized Volmer's equation and the equation of state for the case of monomers and large aggregates (area A < Ac) was developed and frequently used to describe the π–A isotherms of various amphiphilic monolayers.5,6 In any case, the amphiphilic nature of surfactants determines the complexity of the behavior of their monolayers at the water surface. The formation of one or another monolayer type depends on the competition of the CH⋯HC interactions between hydrocarbon chains and the polar interaction of head groups (hydrogen bonding network, chiral interaction, etc.).
For almost two decades ago we proposed and successfully tested a quantum chemical model for assessment of the thermodynamic parameters of surfactant clusterization. Within the framework of this model more than ten classes of nonionic surfactants were considered, for which the following were determined: threshold chain lengths, which allow the formation of condensed monolayers, the “temperature effect” of clusterization, the geometric parameters of the unit cells of monolayers, the peculiarities of the dendritic film formation, and the effect of the clusterization process on the pKa value.7–10 The calculated values of the thermodynamic parameters of clusterization based on the quantum chemical approach are in qualitative agreement with the results of a thermodynamic theory, based on the use of the Volmer-type equation for the phase transition gaseous monolayer – liquid-expanded monolayer.5 Taking into account the nonideality of the mixing entropy for monomers and clusters in the fluid-condensed transition region makes it possible to describe this region of the π–A isotherm.6 Calculation of the Gibbs’ energy of clusterization per monomer in this theory is associated with parameters such as Ac, that is the molecular area which corresponds to the onset of the phase transition on the experimentally obtained π–A isotherm, and ω(Cl) – the area per monomer in a cluster, the value of which is set based on the best agreement with the given isotherm. In this regard the question arises whether it is possible to solve the inverse problem: to estimate the area per monomer molecule at the onset of the phase transition in the monolayer using the available calculated values of the Gibbs’ clusterization energy (ΔGCl/m) per surfactant molecule. It is the purpose of the present work to estimate Ac on the basis of the values ΔGCl/m and ω(Cl) assessed within the semiempirical quantum chemical method PM3 using the thermodynamic model additionally taking into account the nonideality of the mixing entropy.11,12
Theoretical models
There are two theories used in this paper for the calculation of Ac and its comparison with the existing experimental data. The equation of state for the surfactant monolayer is represented as the generalized Volmer's equation that describes the multicomponent insoluble monolayer.13 It is valid for the bimodal distribution of aggregates (large clusters and monomers) and accounts for nonideal entropy of mixing of molecules with different sizes on the basis of the Flory–Huggins theory.14,15 The formula for it was obtained from ref. 16, so here we will not discuss this point in detail. This equation was used for fitting the experimental π–A isotherms and finding several parameters like ω(Cl) (the area per monomer in a cluster), Ac (molecular area corresponding to the onset of the phase transition) and m (the number of monomers in the aggregate during the phase transition) for the considered surfactant monolayers. The further calculation of the thermodynamic parameters of clusterization within the described thermodynamic theory is based on the experimentally obtained temperature dependences of ω(Cl) and Ac. The theoretical calculations and justifications for this are described in detail in ref. 11 using the quasi-chemical model of 2D aggregation.12 According to the quasi-chemical model the corresponding equilibrium constant Km = exp(mμ01 − μ0m)/RT is proportional to the area per surfactant molecule Ac during aggregation. Here, μm and μ1 are the chemical potentials of the cluster and monomer, m is the number of monomers in the cluster, and μm = mμ1. In this case, the chemical potentials of the monomers in the cluster (μ1(m)) and outside it (μ1) are equal. Taking into account that this equality leads to the constancy of the surface concentration of free monomers in the region of the phase transition Γ1 = Γc = constant and assuming with a certain degree of error that the areas per monomer in the cluster and outside it are also equal, we can obtain an expression for Ac in the 2D aggregation process: | (1) |
ΔGCl/m = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(ω(Cl)/Ac) ln(ω(Cl)/Ac) | (2) |
The quantum chemical model for the calculation of the thermodynamic parameters of surfactant clusterization is based on the following key points obtained on the basis of an analysis of numerous experimental and theoretical data on surfactant film formation (cf. ref. 7 for example):
– the fundamental contribution to the clusterization process is made by intermolecular CH⋯HC-interactions between the methylene groups of the hydrocarbon chains of interacting molecules;
– taking into account CH⋯HC interactions realizing only between two alkyl groups of molecular chains located opposite to each other due to the rapid decrease in the energy of these interactions (inversely proportional to the sixth power of the distance);
– intermolecular CH⋯HC-interactions are pairwise additive;
– the thermodynamic parameters of clusterization of the studied surfactants are calculated using the supramolecular approximation as the difference between the thermodynamic parameters of aggregate formation and the corresponding number of thermodynamic parameters of the monomers comprised in the cluster (for example, ΔHClT,m = ΔH0T − m·ΔH0T,mon, ΔSClT,m = S0T − m·S0T,mon, and ΔGClT,m = ΔHClT,m − T·ΔSClT,m, where ΔH0T and S0T are the enthalpy and entropy of the corresponding clusters at temperature T, ΔH0T,mon and S0T,mon are the enthalpy and entropy of the corresponding monomers at the same temperature T, and m is the number of monomers in the cluster.);
– an additive scheme is constructed on the basis of the direct calculation data for the thermodynamic parameters of formation and clusterization of small surfactant clusters; it represents the values of the required parameters as the total contributions of CH⋯HC interactions realized in the cluster and the interactions between the hydrophilic parts of surfactant molecules;
– the interface is taken into account implicitly through its orienting and stretching action.
According to our model, the expression for the clusterization Gibbs’ energy per monomer molecule of the clusters of the substituted alkanes has a linear form (see, for instance ref. 7):
| ΔGCl/m = Vi + Ui·Ka/m, | (3) |
Thus, having the values ΔGCl/m obtained as a result of quantum chemical calculations, and using eqn (2) of the thermodynamic model described above, it is possible to estimate the area per surfactant molecule during the LE–LC phase transition.
The calculated values of ΔGCl/m for the surfactant associates were estimated in the Mopac2000 software package within the framework of the PM3 quantum chemical semiempirical method. As shown in a number of our studies, it allows predicting with a sufficient degree of accuracy the threshold length of the surfactant alkyl chain at which the formation of condensed monolayers is possible, as well as estimating the geometric parameters of the unit cell of the regarded films. This enables using this method in the present work as well.
Results and discussion
Structures of surfactant associates
In the present work we consider eight classes of nonionic surfactants: saturated and ethoxylated alcohols, saturated and cis-unsaturated carboxylic acids, α-hydroxylic and α-amino acids, N-acyl-substituted alanine, and dialkyl-substituted melamine. Their structural and thermodynamic parameters of clusterization were estimated within the framework of the PM3 method. For individual representatives of these classes of compounds there are experimental π–A isotherms obtained at different temperatures, which make it possible to evaluate the thermodynamics of aggregation according to the thermodynamic model.5,6 Almost all of the surfactant classes under consideration have an oblique unit cell of the condensed monolayers with the exception of surfactants with a minimum size of hydrophilic groups: ethoxylated alcohols with a hexagonal structure, as well as saturated alcohols and carboxylic acids, for which the formation of monolayers with hexagonal and oblique elementary cells is practically isoenergetic.17,18As shown in previous studies,18–20 mostly trimers are the basic units for clusterization of surfactants with a hexagonal unit cell of the monolayer. While during the formation of an oblique monolayer the LE–LC phase transition occurs on the basis of surfactant dimers.21 This is schematically illustrated in Fig. 1 and 2 by the example of aliphatic alcohols. Here, the corresponding unit cells are highlighted in green, the arrows show the conditional direction from the carbon atom to one of the hydrogen atoms in the CH2 fragment for clarity of the differences in the method of molecular packing in a monolayer. The area per monomer molecule in the film can be calculated using the distances between the surfactant molecules in the unit cell of a monolayer using the formula as it was done by Ivanova and co-workers in ref. 22:
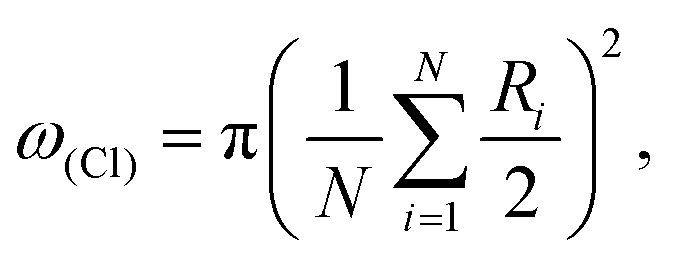 | (4) |
 | ||
| Fig. 1 Scheme of the formation of hexagonal films of aliphatic alcohols. | ||
 | ||
| Fig. 2 Scheme of the formation of oblique films of aliphatic alcohols. | ||
The calculated values of ω(Cl) for the considered surfactants are given in Table 1. It is to be noted that the experimental values of ω(Cl) for comparison with the calculated ones were obtained on the basis of the available data from grazing incidence X-ray diffraction (GIXD) studies of amphiphilic monolayers. The geometrical parameters of the unit cells of monolayers, obtained as result of X-ray structural analysis, make it possible to calculate the value of ω(Cl) using eqn (4).
| Chain length | A c, Å2 | ω (Cl), Å2 | m | |||
|---|---|---|---|---|---|---|
| Calc. | Exptl | Calc. | Exptl | Calc. | Exptl | |
| a The structure of the unit cell of the monolayer is hexagonal. b The structure of the unit cell of the monolayer is oblique; *, **, and *** monolayers are built on the basis of 1, 3 and 5 conformers, respectively. | ||||||
| Saturated alcohols | ||||||
| C14 | 31.3 (25 °C)b | 40.2 (25 °C) | 14.60 (hex.) | 17.1 (hex.)29,30 | 3 | 3.16 (20 °C) |
| 31.8 (40 °C)a | 35.6 (30 °C) | 2 | ||||
| C16 | 47.4 (25 °C)b | 33.0 (40 °C) | 19.13 (obl.) | 2 | 2.75 (25 °C)19 | |
| 28.6 (40 °C)b | 29.5 (45 °C) | |||||
| Ethoxylated alcohols (CnE1) | ||||||
| C14 | 38.5 (25 °C) | 54.3 (5 °C) | 14.60 (hex.) | — (hex.) | 3 | 331 |
| Carboxylic acids | ||||||
| C14 | 26.6 (25 °C)b | 30.0 (20 °C) | 14.60 (hex.) | 19.8–20.232 | 3 | 2–334 |
| 22.8 (25 °C)b | 19.13(obl.) | 20.0 (hex.)33 | ||||
| C20 | 27.6 (25 °C)a | 2 | ||||
| cis-Unsaturated carboxylic acids | ||||||
| C19 | 29.9 (25 °C) | 30.0 (25 °C) for C22 | 23.23 (obl.) | 23.4 (hex.)35 | 2 | 236 |
| C20 | 26.0 (25 °C) | 25.0 (25 °C) for C24 | ||||
| α-Hydroxylic acids | ||||||
| C16 | 36.2 (25 °C)** | 36.5 (30 °C) | 26.11* | 19.5–21.4 (obl.) for enantiomeric compounds with 9.11.12-positions of OH group37,38 | 2 | 1.625 (30 °C) |
| 31.3 (35 °C) | 1.48 (35 °C) | |||||
| 28.7 (45 °C) | 1.38 (45 °C)21 | |||||
| C18 | 29.4 (40 °C)* | 38.0 (36 °C) | 25.36** (obl.) | 20.8 (hex.) for racemic ones with 2-position39 | 2.0 (36 °C) | |
| 34.0 (41 °C) | 1.76 (41 °C) | |||||
| 31.2 (44 °C) | 1.52 (44 °C)21 | |||||
| α-Amino acids | ||||||
| C13 | 32.7 (5 °C)** | — | 28.57* | 30.1 (obl.)40 | 2 | — |
| 38.3 (5 °C)*** | 29.05** | |||||
| C15 | 29.6 (25 °C)* | 31.09*** (obl.) | ||||
| 31.2 (25 °C)** | ||||||
| 35.3 (25 °C)*** | ||||||
| N-Acyl-substituted alanine | ||||||
| C16 | 34.9 (20 °C) | 52.0 (20 °C) | 30.63 (obl.) | 31.2 (obl.)41 | 2 | 1.9–2.2 |
| C18 | 37.2 (20 °C) | 40.0 (26 °C) for C14 | (20–32 °C)42 | |||
| 32.6 (25 °C) | ||||||
| Dialkyl-substituted melamine | ||||||
| C10 | 55.1 (25 °C) | 49.8 (25 °C) | 51.24 (obl.) | 20.6 (obl.)43 | 1 | 0.91(25 °C)44–46 |
The calculated values on the basis of the PM3 method and the experimental values of the area per surfactant molecule in the cluster are in fairly good agreement with each other. The available discrepancies are in average about 2.5 Å2, which corresponds to no more than 15%, in particular for alcohols. It is to be noted that for the hexagonal packing of monolayers of alcohols and carboxylic acids the calculated value of ω(Cl) turned out to be lower than the cross-sectional area of the hydrocarbon chain (∼16.8 Å2), which can be explained by some overestimation of the energy of CH⋯HC interactions and, accordingly, the underestimation of the length of this bond in the PM3 method by an average of 0.5 Å in comparison with other methods.23–28
It should be noted that in the case of hydroxylic acids the available experimental data concern only compounds with an OH group in the 9, 11, and 12 positions, while for the compounds with an alcohol group in the second position there are no such data. From ref. 37 and 38 it should be noted that in the case of the α-position of the hydroxyl group in the carboxylic acid molecule both functional groups – carboxylic and hydroxylic – behave as one hydrophilic part of the molecule, which determines some structural features of the resulting monolayers. In this case it is possible (with some error) to focus on the structural parameters of α-amino acid monolayers, since the hydrophilic part of α-hydroxylic acids is only 3% smaller in area than that of α-amino acids. Then it can be assumed that ω(Cl) for α-hydroxylic acids will be practically the same as for α-amino acids.
The second point that requires discussion is disubstituted melamine. The available data of X-ray diffraction analysis show that the in-plane molecule area Axy for them is in the order of 19–20 Å2,43 which practically coincides with the cross-sectional area of one hydrocarbon chain of 20.62 Å2. Then, for the entire compact dialkylmelamine molecule this value should be at least doubled without taking into account the length of the intramolecular CH⋯HC bonds between the alkyl chains of the substituted melamine. Taking into account these intramolecular bonds the area per molecule of dialkyl-substituted melamine increases by about half the cross-sectional area of one hydrocarbon chain and will be about 50 Å2. The same conclusion follows from the geometric parameters of the unit cell: a = 4.4 Å and b = 5.0 Å, and the angle between them is θ = 116.4°.43 Such parameters are realized only if we consider the dialkyl-substituted melamine dimer, which has four hydrocarbon chains arranged in a parallelogram in cross-section. The value of ω(Cl) for this class of surfactants was calculated by us on the basis of dialkylmelamine tetramers optimized in the PM3 method.46
It can be seen that with an increase in the volume of the hydrophilic part of the surfactant, the value of ω(Cl) increases. Moreover, in the case that the film formation with oblique and hexagonal unit cells is possible for a surfactant, the smallest area per molecule falls on the latter structure. The obtained values of ω(Cl) also agree with the regularities of the geometric parameters of unit cells revealed in our previous work.47 It was shown that an increase in the linear dimensions of the hydrophilic parts of amphiphilic molecules leads to a more tilted orientation of molecules in the condensed monolayer due to the steric hindrances in the orientation of voluminous functional groups opposite to each other.
Thermodynamic parameters of surfactant associates
In our work devoted to the study of the thermodynamics of surfactant clusterization,7 enthalpy, entropy, and Gibbs’ energy of amphiphilic clusterization were estimated using the additive scheme. For this purpose, the formation thermodynamic parameters of the monomers and also the small associates (as a rule, dimers, trimers, tetramers, and hexamers) were calculated using the supermolecule approximation. After that the required thermodynamic clusterization parameter was calculated as the difference between the corresponding formation values for the associates and m molecules of monomers. It should be noted that the thermodynamic model11,12 makes it possible to estimate the value of aggregation number m, i.e. we can say on the basis of which the LE–LC phase transition of small associates occurs. Analysis of the available experimental data on the considered surfactant classes19,21,31,34,36,42,44–46 have shown that for monolayers with a hexagonal unit cell m ≈ 3, and for films with oblique one – m ≈ 2, with the exception for disubstituted melamine, for which this phase transition realizes on the basis of monomers. This is quite understandable, since the dialkyl-substituted melamine molecule itself has two hydrocarbon chains, participating in the formation of intramolecular CH⋯HC interactions. Thus, according to the clusterization thermodynamics, the dialkyl-substituted melamine monomer acts as a dimer of other surfactant classes with one hydrocarbon chain.With a temperature increase, a larger number of small aggregates dissociate. In the case of surfactants with aggregation number m > 2–3, the temperature increase leads to a decrease of m and prevailing number of dimers instead of trimers participating in the phase transition. In this regard, here we use the values of the Gibbs’ clusterization energy of dimers and trimers to calculate Ac according to eqn (2). In this case this equation involves the ΔGCl values per monomer of the corresponding associate. The values ΔGCl/m calculated using the PM3 method and estimated from the experimental π–A isotherms for small surfactant clusters are given in Table 2. As already mentioned, the PM3 method overestimates the energy of intermolecular CH⋯HC interactions.48 This leads to the fact that the clusterization Gibbs’ energy per monomer of associates and films of amphiphilic compounds is almost two times higher than the corresponding value estimated on the basis of experimental data using the theoretical model, described in ref. 5. In addition, in the work devoted to the assessment of the pKa value of carboxylic acid monolayers,10 it was also shown that the calculated values ΔGCl/m obtained by the PM3 method should be halved to obtain adequate pKa values consistent with the available experiment. In this regard for Ac calculation the ΔGCl/m values estimated on the basis of quantum chemical calculations were halved and are listed in Table 2 in the “calculation with correction” column.
| Cluster type (number of carbon atoms in the chain) | ΔGCl/m, kJ mol−1 | ||
|---|---|---|---|
| Calculation without correction | Calculation with correction | Experiment | |
| Saturated alcohols | |||
| Dimer, s: | C13: −0.93 (25 °C)17 | ||
| C14 | −2.44 (25 °C) | −1.22 (25 °C) | |
| C16 | −4.50 (25 °C); | −2.25 (25 °C); | |
| −2.10 (40 °C) | −1.05 (40 °C) | ||
| Trimer 2 (C14) | −4.05 (40 °C) | −2.02 (40 °C) | |
| Ethoxylated alcohols | |||
| Trimer 1 (C14) | −4.43 (5 °C) | −2.21 (5 °C) | — |
| Carboxylic acids | |||
| Trimer 1 (C14) | −2.98 (25 °C) | −1.49 (25 °C) | C13: −0.7 |
| Trimer 2 (C14) | −2.22 (25 °C) | −1.11 (25 °C) | C14: −1.2 |
| Dimer (C14) | −1.49 (25 °C) | −0.74 (25 °C) | C15: −1.7 |
| C16: −2.217 | |||
| cis-Unsaturated carboxylic acids | |||
| Dimer, p: | C22: −(0.75–1.5) | ||
| C19 | −1.25 (25 °C) | −0.62 (25 °C) | (4–12 °C) |
| C20 | −0.55 (25 °C) | −0.27 (25 °C) | C24: −(1.1–2.0) |
| Dimer, s (C20) | −1.25 (25 °C) | −0.62 (25 °C) | (20–27 °C)36 |
| α-Hydroxylic acids | |||
| Dimer 3, s (C16) | −1.77 (25 °C) | −0.88 (25 °C) | C16: −(0.53–0.76) |
| (30–40 °C) | |||
| Dimer 1, p (C18) | −0.62 (40 °C) | −0.31 (40 °C) | C18: −(0.58–0.76) |
| (36–44 °C)21 | |||
| α-Amino acids | |||
| Dimer 1, p (C15) | −0.17 (25 °C) | −0.08 (25 °C) | — |
| Dimer 3, p | |||
| C13 | −0.55 (5 °C) | −0.27 (5 °C) | |
| C15 | −0.36 (25 °C) | −0.18 (25 °C) | |
| Dimer 5, p | |||
| C13 | −0.97 (5 °C) | −0.48 (5 °C) | |
| C15 | −0.63 (25 °C) | −0.31 (25 °C) | |
| N-Acyl-substituted alanine | |||
| Dimer, p (C16) | −0.63 (20 °C) | −0.31 (20 °C) | C14: −(0.59–0.99) |
| Dimer, s (C18) | −0.95 (20 °C) | −0.47 (20 °C) | (20–32 °C)42 |
| −0.31 (25 °C) | −0.15 (25 °C) | ||
| Dialkyl-substituted melamine | |||
| Monolayer (C10) | −0.36 (25 °C) | −0.18 (25 °C) | −5.2 for C1146 |
It should also be noted that for the classes of amphiphilic compounds presented here, we previously considered several stable conformations of monomers, on the basis of which associates were subsequently constructed and their thermodynamic parameters were estimated.17–21,34,42,46 They, in turn, were used in the additive scheme to assess the thermodynamics of the formation of the corresponding 2D films. Here we present the thermodynamic and structural parameters of the formation of small clusters for the surfactant conformers most energetically favorable for each class. They served as structural units of the corresponding most energetically favorable monolayers, the clusterization thresholds of which are consistent with the available experimental data for the compounds under consideration. The only exceptions are α-hydroxylic and α-amino-substituted carboxylic acids, for which two (conformers 1 and 3) and three conformations (conformers 1, 3, and 5), respectively, were taken into account. Among them, the most energetically favorable films are monolayers based on the third and first monomers of the substituted acids, respectively. It should be also mentioned that for dialkyl-substituted melamine the given value ΔGCl/m refers to the formation of a 2D monolayer (not a small cluster), since the experiment has shown that the aggregation number for this class of surfactants m is equal to 0.91.46 It shows that the LE–LC phase transition occurs on the basis on monomers. The fact that the value of m is slightly smaller than unity indicates a small amount of monomers during the phase transition, in which alkyl chains are isolated, i.e., there are no intramolecular CH⋯HC interactions in the monomer.
The calculated values ΔGCl/m with correction were compared with the corresponding values estimated on the basis of the experimentally obtained π–A isotherms. It can be seen from Table 2 that for almost all regarded classes of amphiphilic compounds, the corrected values ΔGCl/m are in good agreement with the available experimental values with the exception of dialkyl-substituted melamines. The experiment was carried out considering a compound with eleven carbon atoms in an alkyl chain.
Analysis of Ac values for surfactant monolayers
The obtained calculated values of Ac are summarized in the second column of Table 1. As can be seen, the calculated data agree with the experimental data. For aliphatic alcohols the best agreement of the data is characteristic for the case when the film is formed on the basis of dimers with the oblique unit cell structure. However, if we extrapolate the Ac data for tetradecanol at a temperature of 40 °C, then we can determine the error in the Ac estimation during the formation of the hexagonal structure of a 2D film. It will be only 7% worse than for the above-described oblique monolayer structure. Among the considered amphiphilic compounds, the largest error in the assessment of Ac is 30% for ethoxylated alcohols (CnE1) and N-acyl derivatives of alanine – 30%. This can be apparently explained by the bulky hydrophilic part of the N-acyl-alanine molecule and the flexibility of CnE1 molecules, which leads to a large number of possible conformations realizing in a monolayer and having different surface coverage areas, which are extremely difficult to take into account during quantum chemical modelling. Among the regarded amphiphilic compounds there are no experimental data for comparison only with α-amino acids. However, as mentioned earlier, their hydrophilic part occupies a slightly larger area than that of α-hydroxylic acids, and both surfactant classes have the same oblique structure of monolayers. In this regard, it can be assumed that the Ac values for α-substituted acids will be practically the same, which can be seen from Table 1.It should be noted that experimental data on Ac are available only for several representatives of the homologous series of the surfactant classes. Therefore, the obtained calculated data have prognostic character. However, they reflect adequately the experimental trend for the LE–LC transition that with an increase in temperature the area per surfactant molecule of a fixed alkyl chain length decreases, and vice versa, with an increase in the surfactant chain length at a fixed temperature, the value of Ac increases. This is consistent with the “temperature effect” of clusterization, registered experimentally49–51 and described on the basis of quantum chemical calculations.52 This is due to the fact that lengthening the hydrocarbon chain of the surfactant by two CH2-fragments is equivalent to a decrease in the temperature of the clusterization process by 10–20°.53 It is known that an increase in temperature leads to a more intense thermal motion of molecules. Therefore, for the phase transition to occur in Langmuir monolayers greater compression is required and, accordingly, a smaller area of the surfactant molecules for their subsequent association. This in turn is due to the implementation of CH⋯HC interactions between the alkyl substituents of the hydrophobic parts of the surfactant and vice versa, a larger number of CH⋯HC interactions are realized between longer-chain amphiphilic molecules. They provide a negative contribution to the clusterization Gibbs’ energy, which ensures monolayer formation at a higher temperature and, correspondingly, at a higher value of area per molecule at the onset of the LE–LC phase transition.
Conclusions
In the present work an approach is proposed to estimate the Ac area per surfactant molecule at the beginning of the liquid-expanded–liquid-condensed (LE/LC) monolayer phase transition. This approach is based on the use of two schemes: a quantum chemical model for determining the thermodynamic and structural parameters of surfactant clusterization and a thermodynamic model for the behavior of amphiphilic monolayers by taking into account the nonideality of the mixing entropy. The clusterization Gibbs’ energy of small surfactant clusters, as well as the structural parameters of the monolayer unit cells calculated in the quantum chemical semiempirical PM3 method, was used to estimate Ac within the thermodynamic model. The calculation showed that(1) the assessed values of the area per monomer in a cluster ω(Cl) and Ac are in satisfactory agreement with the available experimental data of GIXD studies and π–A isotherms, and the root-mean-square error of the Ac estimated for eight classes of surfactants under consideration was 18%,
(2) with an increase in the temperature, the Ac for a surfactant with a given alkyl chain length decreases, and, conversely, with an increase in the surfactant chain length at a fixed temperature, the value of Ac increases.
The proposed approach has a predictive value, since it allows one to estimate the value of Ac for amphiphilic compounds for which there are no experimental dependences of surface pressure on the area per molecule using the calculated thermodynamic and structural parameters of clusterization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. V. acknowledges the versatile assistance by Prof. Hans-Jürgen Butt (director of the Max Planck Institute of Polymer Research). The authors acknowledge the contribution of Dr V. B. Fainerman (SINTERFACE Technologies) to the theoretical processing of experimental data. Open Access funding provided by the Max Planck Society.References
- N. K. Adam, The Physics and Chemistry of Surfaces, Clarendon Press, Oxford, 1938, p. 396 Search PubMed.
- G. L. Gaines, Jr., Insoluble Monolayers at Liquid - Gas Interfaces, Interscience, New-York, 1966, p. 386 Search PubMed.
- W. D. Harkins, The Physical Chemistry of Surface Films, Reinhold Pbl., New-York, 1952, p. 413 Search PubMed.
- D. Dervichian, The Chemistry of Fats and Other Lipids, Intr. Press, London, 1954, vol. 2, p. 193 Search PubMed.
- V. B. Fainerman and D. Vollhardt, J. Phys. Chem. B, 1999, 103, 145–150 CrossRef CAS.
- V. B. Fainerman and D. Vollhardt, J. Phys. Chem. B, 2008, 112, 1477–1481 CrossRef CAS PubMed.
- Yu. B. Vysotsky, E. S. Kartashynska, E. A. Belyaeva, V. B. Fainerman, D. Vollhardt and R. Miller, in Computational Methods for Complex Liquid-Fluid Interfaces, ed. M. Karbaschi, R. Miller and M. T. Rahni, CRC Press, 2016, pp. 199–249 Search PubMed.
- Yu. B. Vysotsky, E. S. Fomina, V. B. Fainerman, D. Vollhardt and R. Miller, Phys. Chem. Chem. Phys., 2013, 15, 11623–11628 RSC.
- Yu. B. Vysotsky, E. S. Kartashynska, E. A. Belyaeva, D. Vollhardt, V. B. Fainerman and R. Miller, J. Phys. Chem. C, 2015, 119, 18404–18413 CrossRef CAS.
- Yu. B. Vysotsky, E. S. Kartashynska, D. Vollhardt and V. B. Fainerman, J. Phys. Chem. C, 2020, 124, 13809–13818 CrossRef.
- D. Vollhardt, V. B. Fainerman and S. Siegel, J. Phys. Chem. B, 2000, 104, 4115–4121 CrossRef CAS.
- V. B. Fainerman, D. Vollhardt and V. Melzer, J. Phys. Chem., 1996, 100, 15478–15482 CrossRef CAS.
- V. B. Fainerman and D. Vollhardt, J. Phys. Chem. B, 1999, 103, 145–150 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1941, 9, 660–661 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS.
- V. B. Fainerman, E. H. Lucassen-Reynders and R. Miller, Adv. Colloid Interface Sci., 2003, 106, 237–259 CrossRef CAS PubMed.
- Yu. B. Vysotsky, V. S. Bryantsev, V. B. Fainerman and D. Vollhardt, J. Phys. Chem. B, 2002, 106, 11285–11294 CrossRef CAS.
- E. S. Fomina, Yu. B. Vysotsky, E. A. Belyaeva, D. Vollhardt, V. B. Fainerman and R. Miller, J. Phys. Chem. C, 2014, 118, 4122–4130 CrossRef CAS.
- E. S. Kartashynska, Y. B. Vysotsky, E. A. Belyaeva, V. B. Fainerman, D. Vollhardt and R. Miller, Phys. Chem. Chem. Phys., 2014, 16, 25129–25142 RSC.
- Yu. B. Vysotsky, E. A. Belyaeva, E. S. Fomina, D. Vollhardt, V. B. Fainerman and R. Miller, Phys. Chem. Chem. Phys., 2014, 16, 3187–3199 RSC.
- E. S. Fomina, Yu. B. Vysotsky, D. Vollhardt, V. B. Fainerman and R. Miller, Soft Matter, 2013, 9, 7601–7616 RSC.
- A. Ivanova, A. Tadjer, N. Tyutyukov and B. Radoev, J. Phys. Chem. A, 2005, 109, 1692–1702 CrossRef CAS PubMed.
- N. K. V. Monteiro and C. L. Firme, J. Phys. Chem. A, 2014, 118, 1730–1740 CrossRef CAS PubMed.
- A. Goursot, T. Mineva, R. Kevorkyantz and D. Talbi, J. Chem. Theory Comput., 2007, 3, 755–763 CrossRef CAS PubMed.
- J. Echeverrıa, G. Aullon, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- M. Dubecky, P. Jurecka, R. Derrian, P. Hobza, M. Otyepka and L. Mitas, J. Chem. Theory Comput., 2013, 9, 4287–4292 CrossRef CAS PubMed.
- R. S. Paton and J. M. Goodman, J. Chem. Inf. Model., 2009, 49, 944–955 CrossRef CAS PubMed.
- D. J. Wolstenholme and T. S. Cameron, J. Phys. Chem. A, 2006, 110, 8970–8978 CrossRef CAS PubMed.
- D. Vollhardt and V. B. Fainerman, Adv. Colloid Interface Sci., 2010, 154, 1–19 CrossRef CAS PubMed.
- J. Majewski, R. Popovitz-Biro, W. G. Bouwman, K. Kjaer, J. Als-Nielsen, M. Lahav and L. Leiserowitz, Chem. – Eur. J., 1995, 1, 304–311 CrossRef CAS.
- S. I. Karakashev, A. V. Nguyen and J. D. Miller, Adv. Polym. Sci., 2008, 218, 25–55 CrossRef CAS.
- K. Kjaer, J. Als-Nielsen, C. A. Helm, P. Tippman-Krayer and H. Möhwald, J. Phys. Chem., 1989, 93, 3200–3206 CrossRef CAS.
- I. R. Peterson, G. Brezesinski, B. Struth and E. Scalas, J. Phys. Chem. B, 1998, 102, 9437–9442 CrossRef CAS.
- Yu. B. Vysotsky, D. V. Muratov, F. Boldyreva, V. B. Fainerman, D. Vollhardt and R. Miller, J. Phys. Chem. B, 2006, 110, 4717–4730 CrossRef CAS PubMed.
- D. Vollhardt, J. Phys. Chem. C, 2007, 111, 6805–6812 CrossRef CAS.
- Yu. B. Vysotsky, E. A. Belyaeva, D. Vollhardt, E. V. Aksenenko and R. Miller, J. Phys. Chem. B, 2009, 113, 4347–4359 CrossRef CAS PubMed.
- D. Vollhardt, S. Siegel and D. A. Cadenhead, Langmuir, 2004, 20, 7670–7677 CrossRef CAS PubMed.
- D. Vollhardt, S. Siegel and D. A. Cadenhead, J. Phys. Chem. B, 2004, 108, 17448–17456 CrossRef CAS.
- G. Weidemann, G. Brezesinski, D. Vollhardt and H. Möhwald, Langmuir, 1998, 14, 6485–6492 CrossRef CAS.
- I. Weissbuch, M. Berfeld, W. Bouwman, K. Kjaer, J. Als-Nielsen, M. Lahav and L. Leiserowitz, J. Am. Chem. Soc., 1997, 119, 933–942 CrossRef CAS.
- N. Nandi and D. Vollhardt, Chem. Rev., 2003, 103, 4033–4075 CrossRef CAS PubMed.
- E. S. Kartashynska, Yu. B. Vysotsky, V. B. Fainerman, D. Vollhardt and R. Miller, Colloids Surf., A, 2018, 546, 346–359 CrossRef CAS.
- D. Vollhardt, V. B. Fainerman and F. Liu, J. Phys. Chem. B, 2005, 109, 11706–11711 CrossRef CAS PubMed.
- D. Vollhardt, F. Liu, R. Rudert and W. He, J. Phys. Chem. B, 2005, 109, 10849–10857 CrossRef CAS PubMed.
- N. M. Kovalchuk, D. Vollhardt, V. B. Fainerman and E. V. Aksenenko, J. Phys. Chem. B, 2007, 111, 8283–8289 CrossRef CAS PubMed.
- Yu. B. Vysotsky, A. A. Shved, E. A. Belyaeva, E. V. Aksenenko, V. B. Fainerman, D. Vollhardt and R. Miller, J. Phys. Chem. B, 2009, 113, 13235–13248 CrossRef CAS PubMed.
- Yu. B. Vysotsky, E. S. Kartashynska, E. A. Belyaeva, D. Vollhardt, V. B. Fainerman and R. Miller, J. Phys. Chem. C, 2015, 119, 5523–5533 CrossRef CAS.
- G. I. Csonka, J. C. Angyan and G. I. Csonka, THEOCHEM, 1997, 393, 31–38 CrossRef CAS.
- M. K. Durbin, M. C. Shih, A. Malik, P. Zschack and P. Dutta, Colloids Surf., A, 1995, 102, 173–179 CrossRef CAS.
- N. Islam and T. Kato, J. Colloid Interface Sci., 2006, 294, 288–294 CrossRef PubMed.
- K. S. Birdi, Self-assembly monolayer structures of lipids and macromolecules at interfaces, Springer, New-York, 1999, p. 387 Search PubMed.
- Yu. B. Vysotsky, E. S. Fomina, E. A. Belyaeva, D. Vollhardt, V. B. Fainerman and R. Miller, J. Phys. Chem. B, 2012, 116, 8996–9006 CrossRef CAS PubMed.
- A. M. Bibo and I. R. Peterson, Adv. Mater., 1990, 2, 309–311 CrossRef CAS.
| This journal is © the Owner Societies 2021 |