
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCritical role of H-aggregation for high-efficiency photoinduced charge generation in pristine pentamethine cyanine salts†
George C.
Fish
 a,
Juan Manuel
Moreno-Naranjo‡
a,
Andreas
Billion
b,
Daniel
Kratzert
b,
Erwin
Hack
c,
Ingo
Krossing
b,
Frank
Nüesch
de and
Jacques-E.
Moser
*a
a,
Juan Manuel
Moreno-Naranjo‡
a,
Andreas
Billion
b,
Daniel
Kratzert
b,
Erwin
Hack
c,
Ingo
Krossing
b,
Frank
Nüesch
de and
Jacques-E.
Moser
*a
aPhotochemical Dynamics Group, Institute of Chemical Sciences and Engineering, École polytechnique fédérale de Lausanne, Station 6, 1015 Lausanne, Switzerland. E-mail: je.moser@epfl.ch
bInstitut für Anorganische und Analytische Chemie, and Freiburger Materialforschungszentrum, Universität Freiburg, Albertstrasse 21, 79104 Freiburg, Germany
cLaboratory for Transport at Nanoscale Interfaces, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, 8600 Dübendorf, Switzerland
dLaboratory for Functional Polymers, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, 8600 Dübendorf, Switzerland
eInstitute of Materials, School of Engineering, École polytechnique fédérale de Lausanne, Station 12, 1015 Lausanne, Switzerland
First published on 8th October 2021
Abstract
The mechanism of photoinduced symmetry-breaking charge separation in solid cyanine salts at the base of organic photovoltaic and optoelectronic devices is still debated. Here, we employ femtosecond transient absorption spectroscopy (TAS) to monitor the charge transfer processes occurring in thin films of pristine pentamethine cyanine (Cy5). Oxidized dye species are observed in Cy5-hexafluorophosphate salts upon photoexcitation, resulting from electron transfer from monomer excited states to H-aggregates. The charge separation proceeds with a quantum yield of 86%, providing the first direct proof of high efficiency intrinsic charge generation in organic salt semiconductors. The impact of the size of weakly coordinating anions on charge separation and transport is studied using TAS alongside electroabsorption spectroscopy and time-of-flight techniques. The degree of H-aggregation decreases with increasing anion size, resulting in reduced charge transfer. However, there is little change in carrier mobility, as despite the interchromophore distance increasing, the decrease in energetic disorder helps to alleviate the trapping of charges by H-aggregates.
Introduction
Cyanine dyes, and their derivatives, have a long history in the field of small molecule-based organic photovoltaics (OPV).1 The beneficial properties of cyanine dyes, such as their high extinction coefficients (typically exceeding 105 L mol−1 cm−1)2 mean that they have traditionally been used in planar architecture devices, generally in combination with fullerene acceptor materials, with device efficiencies of up to 3.7% being achieved.3 Their aforementioned high extinction coefficients mean that thin active layers, of around 20 nm thickness, can be employed allowing them to circumvent issues that typically thwart the performance of planar architecture devices, such as exciton recombination before diffusion to the donor acceptor interface has occurred.4,5The cyanine dyes used in the OPV field comprise of a positively charged chromophore paired with a counterion; the chromophore generally consists of two heterocycles connected with a polymethine chain, the length of which can be altered to change the absorption profile of the dye. This has led to new applications for these dyes, such as in transparent devices, due to their high absorption in the near infrared and high transparency in the visible.6,7
Furthermore, the counterion can also be easily changed using ion exchange techniques,8 which allows for a wide variety of counterions to be employed including weakly coordinating anions (WCAs).9 While it is not possible for anions to be non-coordinating, WCAs closely approximate non-coordinating conditions10,11 and allow for preparation of ionic liquids virtually without ion pairing.12,13 Changing the counterion impacts upon the thin film properties as well as the device performance.14,15
Previous work regarding ultrafast processes in cyanine dyes has tended to focus on the fundamental photophysics of the cyanine chromophore in solution, such as the triplet state characteristics16 and the dynamics of higher lying excited states.17 The majority of studies pertaining to ultrafast processes in cyanine based OPVs have focused on cyanine:C60 bilayers. For example, time-resolved electroabsorption spectroscopy has been used to study the dissociation of charge transfer states in Cy3-P:C60 bilayers.18 Moving away from bilayers, transient absorption spectroscopy has been employed to study the impact of phase morphology in cyanine:PCBM blends, providing a better understanding of charge transfer dynamics in bulk heterojunction architecture OPVs.19 More recently, evidence was produced demonstrating that photocurrent generation can occur in a pristine cyanine thin film, despite the absence of a donor–acceptor interface.20 Furthermore, the generated charges exhibited a long carrier lifetime as a result of reduced recombination, providing potential material design concepts for future photodiodes. Cyanines have been utilised in several different photodiode designs, being employed to take advantage of their narrow band absorption, potential for high absorption in the near infrared,21 and ability to form J-aggregates.22–25
Intrinsic photogeneration has also been previously demonstrated in photodiodes utilising donor–acceptor oligomers,26 as well as in single layer SubNc, SubPc and squaraine dye-based devices. Here, the intrinsic charge generation was attributed to the high dielectric constant, resulting in efficient screening and therefore allowing free carrier generation.27,28 In comparison, cyanine dye-based photodiodes have shown substantially higher photon-to-electron quantum yields20 and could be a suitable and versatile alternative material to use in single layer photodiodes. The understanding of the symmetry-breaking charge separation mechanism, however, is still lacking and would spur the development of new materials design.
In this work, ultrafast transient absorption spectroscopy was used to unravel the photoinduced charge transfer mechanism in pristine pentamethine cyanine dyes, providing the first direct proof of high efficiency intrinsic charge generation in organic salt semiconductors. Furthermore, the impact of the counterion on the charge transfer process was assessed using four different counterions of varying sizes; the purpose of this was to vary the degree of interaction between the cyanine chromophores in the different dyes, and thus study its effect on the photogeneration of charges.
Finally, electroabsorption spectroscopy and time-of-flight (TOF) were used to determine the impact of counterion size on charge transport within the solid dye film.
Experimental section
Sample preparation
The cyanine dyes 2-[3-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)-propenyl]-1,3,3-trimethyl-3H-indolium chloride (Cy5-Cl) and its hexafluorophosphate analogue (Cy5-P) were purchased from FEW Chemicals, Germany. The tetrakis(nona-fluoro-tert-butoxy)aluminate (Al(pftb4)) and tetrakis(perfluoro-1-adamantoxy)aluminate (Al(pfad)4) WCA Cy5 salts were provided by the group of Prof. Ingo Krossing, and their synthesis is described in the ESI,† (S1).The Cy5-Cl and Cy5-P dyes were dissolved in acetonitrile (Sigma Aldrich), while the two Al WCA salts were dissolved in ethanol (Sigma Aldrich) in various concentrations. Thin films were subsequently fabricated by spin coating the solutions onto cleaned glass substrates inside a glovebox. All chemicals were used as supplied without further purification.
Samples for electroabsorption (EA) and time-resolved electroabsorption spectroscopy (TREAS) were prepared on glass patterned with fluorine-doped tin oxide (FTO). Spin coating aluminium sec-butoxide (0.25 M) in 2-methoxyethanol, calcination at 500 °C for 1 hour, and repeating 3 times yielded the 20 nm aluminium oxide insulating layer. The cyanine dye was then deposited as mentioned above, followed by thermal deposition of an aluminium top electrode.
Samples for TOF were also prepared on substrates with 6 layers of alumina atop the FTO patterned glass. The cyanine dyes were deposited via spin coating at 6000 rpm for 60 s.
Thin film thickness and the optical constants were determined using both ellipsometry (J. A. Woollam M2000-VI) and cross-sectional SEM (Zeiss Merlin).
Spectroscopy
Absorption spectra were measured using a PerkinElmer Lambda 950 UV/vis/NIR spectrophotometer. Emission spectra were measured using a Horiba Jobin-Yvon Fluorolog-3 instrument with a photomultiplier tube as a detector.Transient absorption (TA) spectra and electroabsorption (EA) spectra were measured using a femtosecond pump–probe spectrometer based on an amplified Ti:sapphire laser (Clark-MXR, CPA-2001) delivering 778 nm pulses of 150 fs at a 1 kHz repetition rate. In both cases, the pump beam was generated by passing a portion of the fundamental through a two-stage non-collinear optical parametric amplifier (NOPA-Plus, Clark-MXR) resulting in an excitation wavelength of 580 nm. The probe beam was constituted by a broadband white light continuum generated by passing part of the fundamental through a 5 mm-thick oscillating CaF2 plate (for TA, 450–1050 nm), or through a 4 mm sapphire window (for EA, 400–750 nm). The pump and probe beams, set at magic angle polarization, were spatially and temporally overlapped with respect to one another using a delay stage. For TA measurements, samples were measured in transmission mode while EA measurements were measured in reflection mode. The probe beam was split into a signal beam passing through the sample and a reference passing next to it in order to account for shot-to-shot fluctuations. The signal and reference beams were dispersed in two respective grating spectrographs (SpectraPro 2500i, Princeton Instruments or SR163, Andor Instruments) and detected shot-to-shot at 1 kHz by 512 × 58 pixel back-thinned charge-coupled device cameras (Hamamatsu S07030-0906). For TA measurements, a chopper set at 500 Hz modulated the frequency of the pump beam enabling for the absorption with and without the pump to be acquired. For EA spectroscopy, the electric field across the sample was modulated at half the amplifier frequency (500 Hz), using square pulses generated by a function generator (Tektronix AFG 2021, from −10 to 10 V, 100 μs pulse duration). The current responses across the samples were recorded using a 50 Ω series load with a 400 MHz bandpass oscilloscope (Tektronix TDS 3044B).
Time-of-flight
The excitation source was provided by a Nd:YAG Q-switched laser (Continuum Powerlite-7030) with a frequency-tripled output (355 nm) and a repetition rate of 0.2–20 Hz. This UV output was used to pump a broadband optical parametric oscillator (OPO-355, GWU) allowing for the wavelength of the excitation pulses (7 ns duration) to be tuned. A 24 V series battery was used to apply a voltage across the sample, and the samples only experienced a voltage during the single shot measurement. 10 shots were acquired to minimise the impact of oscillations, and between each measurement the voltage was switched off allowing the carriers to return to equilibrium. The photocurrent response from the samples was recorded using an oscilloscope (DPO 7104, Tektronix, 1 MΩ). A capacitor and a variable resistor box were placed in parallel to the battery in order to adjust the RC constant of the circuit.Results and discussion
Femtosecond pump–probe transient absorption (TA) spectroscopy measurements were first carried out on a solution and thin film sample of 1,3,3-trimethyl-2-[5-(1,3,3-trimethyl-1,3-dihydro-indol-2-ylidene)-penta-1,3-dienyl]-3H-indolium hexafluorophosphate, which will from now on be referred to as Cy5-P. The structure of the Cy5 chromophore can be seen in Fig. 1a and the normalized absorption spectra for the solution and thin film are shown in Fig. 1b. The sharp peak at around 640 nm in the solution spectrum corresponds to the Cy5 monomer (0, 0) transition, while the shoulder peaks at wavelengths ≤600 nm are assigned to vibronic transitions. The thin-film absorption spectrum is red shifted, as expected, due to increased intermolecular interactions in the solid state and is broadened with two main features appearing at 620 and 680 nm, respectively. The latter of these two features corresponds to the Cy5 monomer, while the former is assigned to the absorption of the H-dimer.29 | ||
| Fig. 1 (a) Structure of the Cy5 chromophore. (b) Absorption spectra of Cy5-P in acetonitrile and as a thin film. Normalized transient absorption spectra of Cy5-P solution (c) and thin film (d). Samples were excited at 580 nm. The transient absorption spectra were normalized to the 100 fs signal, and normalization was done for ease of comparison. | ||
Furthermore, the H-dimer peak exhibits broadening in the blue region due to higher order H-aggregates. The thin film spectrum corresponds largely to the absorption coefficient measured by ellipsometry (ESI,† Fig. S4.2) meaning that reflectance and interference features are only minor contributions to the absorbance spectrum.
Pulsed laser excitation at 580 nm yielded the transient absorption spectra of the solution and thin-film of Cy5-P (Fig. 1c and d). Four main features can be seen in the TA spectrum of the solution: two positive features at 450 and 500 nm, along with two negative features at 540–615 and 650 nm. The two positive peaks arise from the same process, as they display the same time dependence. They also exhibit no fluence (see ESI,† Fig. S2.1) or concentration dependence, and so can be assigned to S1–Sn transitions. Furthermore, this excited state absorption feature (ESA) is similar to those that have been seen previously.30 It is expected that a second positive feature, corresponding to trans–cis photoisomerization, would be seen if a longer time window were monitored.16 The negative band between 540 and 615 nm is assigned to ground state bleaching (GSB), due to its resemblance to the absorption spectrum of the monomer in the ground state. Finally, the negative feature at 650 nm corresponds to stimulated emission.
In comparison, the transient absorption spectrum for the Cy5-P film consists of three main features, of which the two negative features correspond to the GSB (620 nm) of the H-dimer and combined GSB and stimulated emission (680–720 nm). The major difference, however, between the solution and the film is the positive feature; unlike in the solution spectrum, the transient absorption spectrum of the film exhibits only a positive feature, which shifts towards higher wavelengths over time (from 470 nm at 0.1 ps to 495 nm at 1000 ps). The kinetics of this feature are also different to that of the ESA feature in the solution. Given that the proposed model for intrinsic charge generation in Cy5-P relies upon the generation of redox species,20 it was postulated that the single positive feature in the transient absorption spectrum could arise from the absorption of the oxidized Cy5 chromophore.
To confirm this, bromine vapor was used to oxidize a Cy5-P film in situ and the absorption spectrum was subsequently measured. As seen in Fig. 2a, there are two processes which take place when the film is exposed to bromine vapor. The first process is the disruption of H-aggregates within the film. This is evident from the broadened shoulder which is present in the spectrum of the neat film, but absent upon exposure to the bromine vapor. The broadened shoulder corresponds to higher order H-aggregates and so its absence points towards the oxidation of these large aggregates within the film. This process appears to occur more quickly since a single oxidation step is able to extinguish a large aggregate composed of several chromophores.
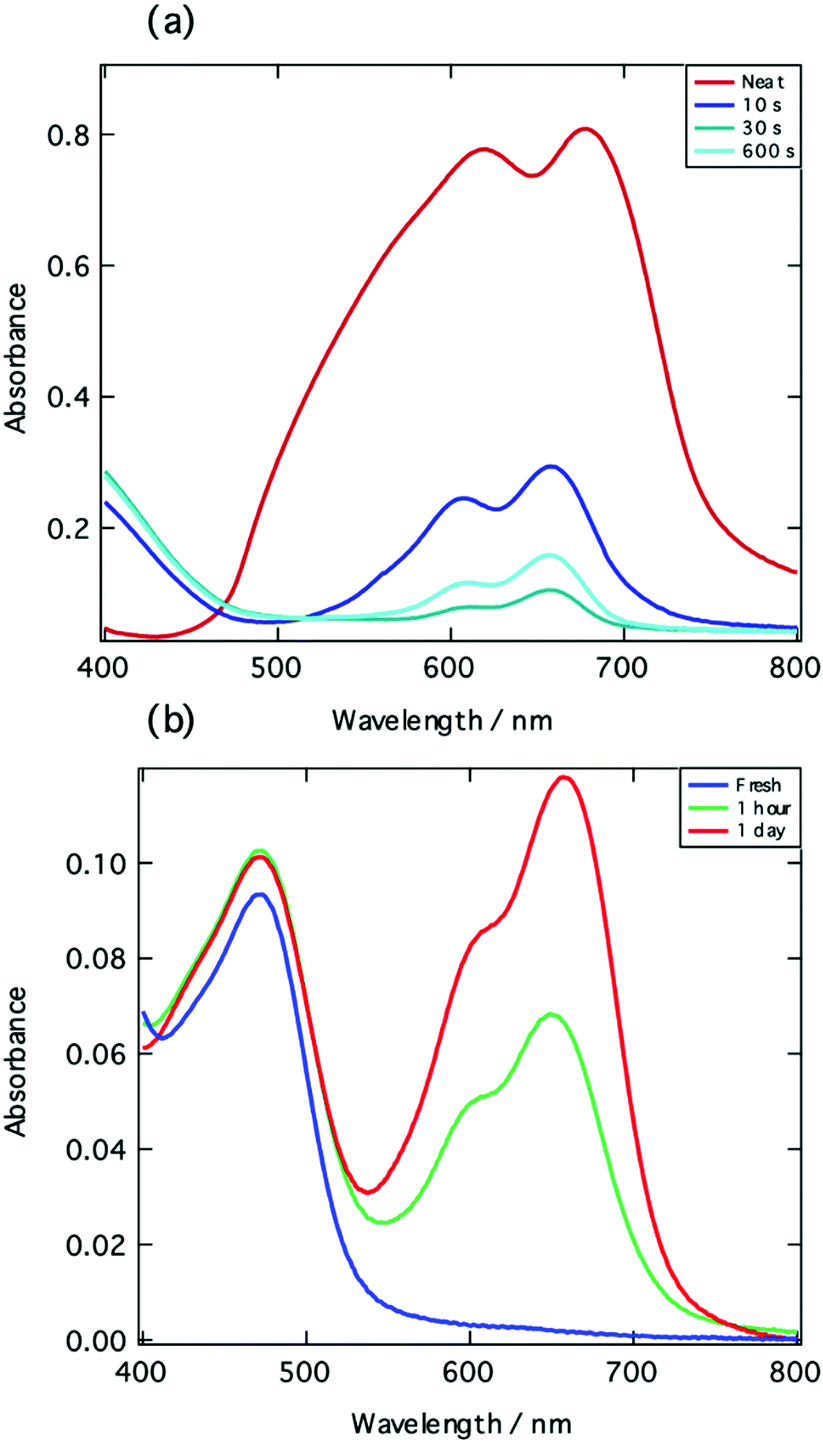 | ||
Fig. 2 (a) Absorption spectra of a thin film of Cy5-P before (red) and after (blue lines) exposure to bromine vapor for different amounts of time. (b) Absorption spectra of a thin film of Cy5-P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NOBF4 1:2.5 mol/mol blend ratio at different times after spin-coating. :NOBF4 1:2.5 mol/mol blend ratio at different times after spin-coating. | ||
The second process that takes place is then the oxidation of the cyanine chromophore in its monomeric state. Oxidation of the aggregated film, however, does not reveal the distinct peak at 470 nm seen in the transient absorption spectrum of Fig. 1d. A different behaviour is obtained when the chromophores are first oxidized in solution using NOBF4. Here, the oxidized film shows precisely the expected feature with a peak at 470 nm (Fig. 2b). After de-doping in air for several hours, the monomer spectrum reappears. Since the film was cast from a Cy5-P:NOBF4 1:2.5 mol/mol solution in acetonitrile, the large amount of salt probably inhibits H-aggregate formation. Also, from an electrostatic point of view, the cyanine di-cations are not likely to stack in close packed H-stacks.
Due to the close match of the absorption features measured in the transient absorption spectrum and the film oxidized with NOBF4, we are tempted to assign this positive feature to oxidized monomer species (Cy5+) present in the Cy5-P film.
At this stage, we cannot completely exclude the possibility that the feature at 470 nm arises from the absorption of reduced species of Cy5 (Cy5−), since absorption studies on electrochemically reduced pentamethine cyanine dyes revealed similar spectra for the neutral and the dicationic radicals.31 Also, neutral cyanine radicals are very unstable, promptly undergoing dimerization and are therefore inaccessible in steady state measurements.
Having established the presence of oxidized or reduced cyanine chromophores within the photoexcited film, the impact of the interchromophore distance upon the charge generation process was investigated. Poly(methylmethacrylate), PMMA, was added to Cy5-P in various molar ratios and thin films were fabricated. From ground state absorption measurements, it is evident that as the proportion of PMMA increases there is a decrease in the degree of aggregation, which is evidenced by the decrease in the H-dimer peak height (Fig. 3a and b). We also note that there is a decrease in the FWHM as the PMMA:Cy5-P ratio increases. While the precise composition of the film including monomers, H-dimers, larger H-aggregates, and perhaps J-aggregates, is very difficult to determine, the FWHM provides a useful indicator. From Fig. 3a, we see that a FWHM value of 90 nm is reached for well separated monomer species.
 | ||
| Fig. 3 (a) Normalized absorption spectra of varying ratios of PMMA:Cy5-P. (b) Ratio of H-dimer:monomer peak height as a function of the PMMA:Cy5-P blend ratio. | ||
From the TA spectra (Fig. 4a), it is evident that as the proportion of PMMA increases, resulting in the aforementioned decrease in aggregation, the positive feature transforms from one to two peaks which corresponds to the singlet excited state absorption as observed in solution (Fig. 1c).
 | ||
| Fig. 4 (a) Normalized TA spectra for 0:1, 1:1 and 12:1 (mol:mol) blends of PMMA:Cy5-P at a time of 1 ps. Samples were excited at 580 nm with a fluence of 30 μJ cm−2; (b)–(e) Global fits for different PMMA:Cy5-P blends (1:1, 4:1, 8:1 and 12:1); (f) Plot showing the rate of electron transfer as a function of PMMA:Cy5-P blend ratio. | ||
This confirms that the single positive peak occurs when the chromophores are closely packed allowing for a charge transfer process resulting in an oxidized and reduced species. The TA spectra were fitted with biexponentials, yielding time constants for the charge transfer processes occurring within the thin films, which can be found summarized in the ESI,† (Table S2). From there the corresponding spectra were extracted by a global fit (Fig. 4b–e). For more information on the global analysis and the global fitting procedure, the reader is directed to the ESI,† (S2) and the following literature.32–34
With increasing proportion of PMMA there is an increase in both of the time constants, and the rate constants for the different blends display a linear dependence with increasing PMMA ratio (Fig. 4f).
This helps us in confirming our hypothesis that the positive feature corresponds to photoinduced charge separation within the films, since the charge transfer kinetics are very sensitive to the intermolecular distance between the donor and the acceptor. Furthermore, Fig. 4b–e reveal that charge transfer occurs despite the fact that we radically change the degree of aggregation. Keeping the excitation wavelength at 580 nm, we change from exciting predominantly aggregates to monomers when the dye is diluted in PMMA.
These results allow us to propose two possible mechanisms for intrinsic charge generation in Cy5-P, both of which involve H-aggregates. The first reaction considers direct excitation of H-aggregates into higher energy exciton bands from where reductive quenching by the monomer species occurs (eqn (1)). On the other hand, charge transfer can proceed via electron transfer from the excited monomer species (eqn (2)). Both of these mechanisms generate the positive feature at 470 nm which is seen in the transient absorption spectrum.
| Cy5 + (H)Cy5 + hν → Cy5 + (H)Cy5* → Cy5+ + (H)Cy5− | (1) |
| Cy5 + (H)Cy5 + hν → (H)Cy5 + Cy5* → Cy5+ + (H)Cy5− | (2) |
The thermodynamic driving force has been proposed to arise from energetic disorder within the film.35 Apart from energy level shifts arising from electronic coupling between dye molecules, local electric fields may also substantially shift the HOMO and LUMO energy levels of the cyanine chromophores. When the cyanine cations form H-aggregate stacks, the anions are situated on the outside of the positively charged stack, which could give rise to the aforementioned local electric field, and thus lowering the energy levels of the H-aggregates with respect to those of the monomeric species.
Once the charges have undergone separation onto well separated sites, the positively charged hole is subsequently shielded from recombination by the negatively charged anions which surround the aggregates, which should result in an increase in the charge carrier lifetime, as found previously but not seen in these measurements.20 This difference can be explained by a much larger concentration of charge carriers produced at laser excitation intensity resulting in increased bimolecular charge carrier recombination and thus a much lower charge carrier lifetime than seen previously.
Impact of the counterion
Having established the presence of aggregate oxidized species within the photoexcited film, the impact of the counterion on the charge generation process was investigated. Alongside the hexafluorophosphate anion (Volume of anion determined by X-ray diffraction, V− = 0.093 nm3), three other counterions were studied in combination with the same chromophore: chloride (Cy5-Cl; V− = 0.029 nm3), tetrakis(nonafluoro-tert-butoxy)aluminate (Cy5-[Al(pftb)4]; V− = 0.707 nm3) and tetrakis(perfluoro-1-adamantoxy)aluminate (Cy5-[Al(pfad)4]; V− = 1.194 nm3). Thus, V− was changed by a factor of 41 if comparing Cy5-Cl with Cy5-[Al(pfad)4].The crystal structures of the PF6−, Al(pftb)4− and Al(pfad)4− salts were determined, and Hirshfeld analysis was conducted. An in-depth description of Hirshfeld surface analysis can be found here.36 From the Hirshfeld surfaces, fingerprint plots were produced (Fig. S3.2, ESI†), which provide a visual summary of the intermolecular contacts within the crystal structures.37 By examining the fingerprint plots of the H–H intermolecular contacts, it is evident that as the anion size increases the mean inter-chromophore distance increases. In addition, the dominating H–H intermolecular cation–cation contacts in Cy5-P (51.7% of the interactions) are replaced by others and the H–H cation–cation interactions only account for 13.6% of the contacts as a consequence of the now much larger counterion in Cy5-[Al(pfad)4].
The increase in inter-chromophore distance results in a decrease in the degree of aggregation within the film, as shown by a decrease in the ratio of the H-dimer:monomer peak height, as well as the reduced FWHM (Fig. 5a). We note that the same features are observed for the absorption coefficient measured by ellipsometry (Fig. S4.2, ESI†). The anion size also impacts upon the transient absorption spectra of the thin films (Fig. 5b).
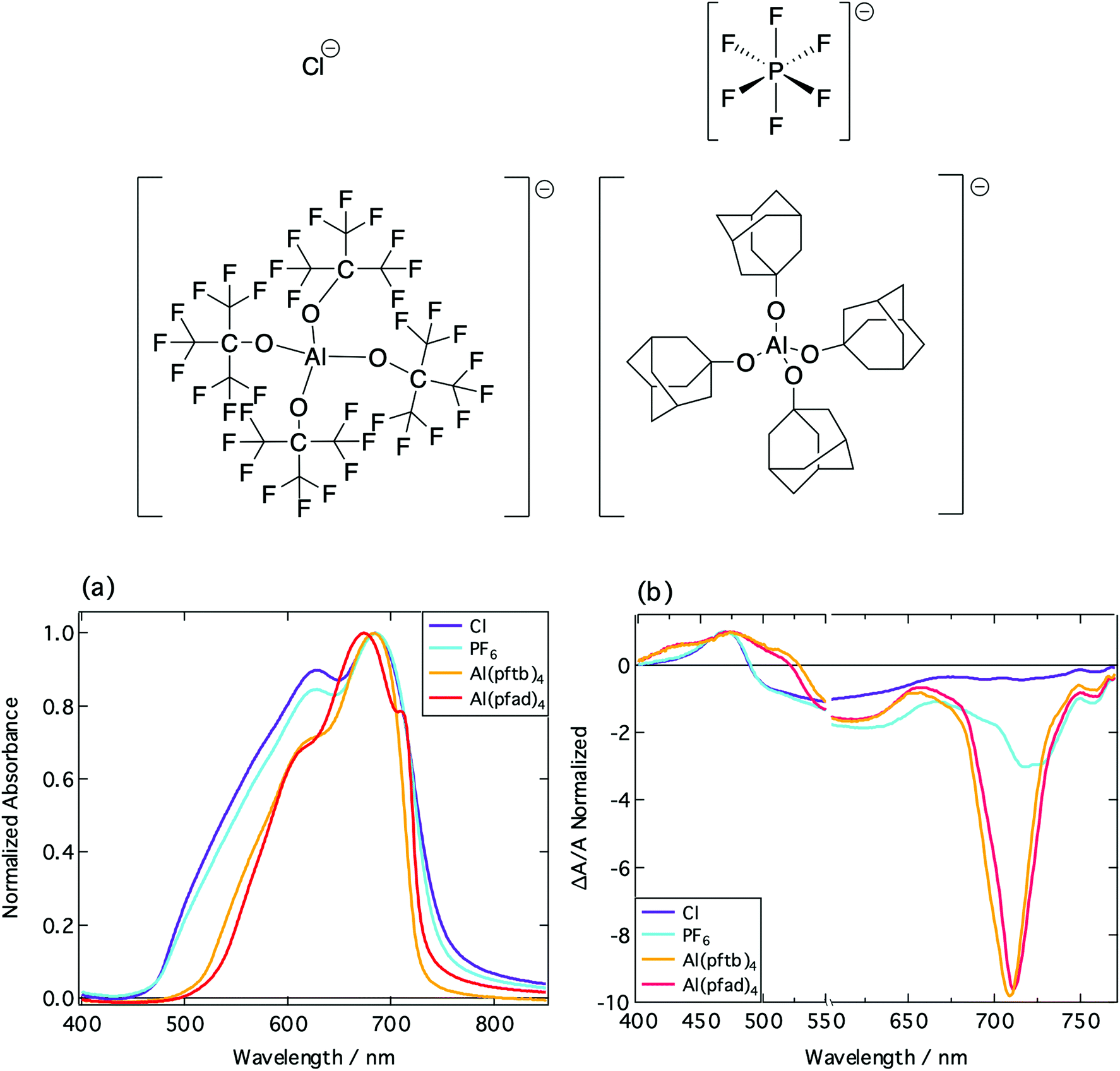 | ||
| Fig. 5 Structures of the different anion counterions: Cl−, PF6−, Al(pftb)4− (left) and Al(pfad)4− (right). Note that the fluorine atoms have been omitted from the Al(pfad)4− structure for clarity. (a) Normalized absorption spectra of Cy5 thin films incorporating different counterions: Cl− (purple), PF6− (cyano), Al(pftb)4− (orange) and Al(pfad)4− (red). (b) Normalized transient absorption spectra of different Cy5 thin films, taken at a time of 1 ps. Samples were excited at 580 nm. | ||
With increasing anion size, there is an increase in the relative intensity of the stimulated emission, arising due to a decrease in energetic disorder thus leading to a decrease in selfquenching.38 Furthermore, as the anion size increases, there is a slight shift in the positive feature in the TA spectra at 470 nm, and a second feature begins to be resolved. This reflects a diminished bleaching contribution of the H-aggregates, in accordance with the results obtained for Cy5 dispersed in PMMA. The thin films fabricated were reproducible, and the absorption spectra identical between batches. This allows us to conclude that the differences in the photophysics between the dyes incorporating different counterions are due to the difference in aggregation, and not due to structural defects occurring during film formation.
Global fits of the transient spectra were obtained by fitting the spectra with three exponential functions, and these fits were then used to calculate the quantum yield for charge separation (S4 and S5, ESI†). The quantum yield of the charge separation process was found to be 83% and 86% for the Cy5-Cl and Cy5-P systems, respectively. This yield then decreased to 71% and 78% for the two aluminium-based analogues, Cy5-[Al(pftb)4] and Cy5-[Al(pfad)4].
The decrease in yield for the larger counterions further confirms that the interchromophore distance, and therefore the degree of aggregation, plays a key role in the charge transfer process. In the thin films of the cyanine dyes with the larger counterions (Al(pftb)4 and Al(pfad)4), the counterions are sufficiently large to ensure that higher order H-aggregates do not readily form, which minimises electron transfer from photoexcited monomer chromophores to the aggregates.
Charge transport
Having established a link between anion size and the mechanism of charge generation, the impact of the anion on charge transport was then investigated. For this, electroabsorption and time-resolved electroabsorption spectroscopy (TREAS) were used. These are spectroscopic techniques based upon the electric-field dependent optical response of a system (Stark effect).18Experimentally, the difference between electroabsorption measurements and TREAS is the addition of a pump beam in the latter. This photoexcitation of the sample generates charge pairs, which undergo separation under the applied electric field causing changes in the electroabsorption response of the material. By monitoring the time evolution, the charge carrier mobility can be determined. For more details on these techniques, the reader is directed to the following literature.18,39,40
While the electroabsorption spectra of the Cy5 dyes have similar shapes (Fig. S6.1, ESI†), there is a clear red shift in the positive feature as the anions increase in size (from 475 nm in Cy5-Cl to 550 nm in Cy5-[Al(pfad)4]). As the shape of the electroabsorption spectrum is related to the derivative of the absorption spectrum, this red shift arises from a decrease in higher order H-aggregates as the anion size increases. It is possible to determine the origin of certain features within the EA spectrum by comparing it to the derivatives of the steady state absorption spectrum. Furthermore, a linear combination of the zeroth, first and second derivatives can be fitted to the electroabsorption spectrum, with the accuracy of the fit determined by the number of transitions behind each absorption band.
The fits (Fig. S6.2, ESI†) for the Cy5-Cl and Cy5-P dyes are reasonably accurate; in contrast, however, the fits for the dyes with the two aluminium-based counterions are inaccurate. This implies that in these films there is inhomogeneity in the polarizability and the permanent dipole on a short length scale (tens of nm) and that the film is locally disordered. Furthermore, any aggregates that are present in these films do not exhibit uniform properties.41
The results from the TREAS experiments proved inconclusive. This was caused by the poor carrier mobility resulting in there being little time evolution in the electroabsorption spectra over the time-window of the experiment (approximately 1.6 ns). As the films of the cyanine dyes were already very thin (20–50 nm), decreasing the film thickness or increasing the applied voltage were not feasible solutions. As a result, time-of-flight (TOF) was instead used to estimate the impact of anion size on charge carrier mobility.
TOF is a technique whereby electron–hole pairs are generated in an electric field upon photoexcitation of the sample in the immediate vicinity of one electrode. The electric field then splits the electron–hole pair, where one charge is immediately collected by the nearby electrode, while the other traverses the sample to be collected at the counter electrode. The time taken for these charges to traverse the sample is called the transit time, and it can be used to calculate the charge mobility in the sample using the following equation:
 | (3) |
As well as giving the charge mobility in the sample, TOF can also give insight into the transport regime (dispersive vs. non-dispersive), by means of the appearance of the photocurrent curve. The mobility of the Cy5 dyes with the two smaller counterions (Cl− and PF6−) were investigated alongside [Al(pftb)4].
Samples were excited at 630 nm with an applied voltage of 8 V. An example of the resulting photocurrent curve is shown in the ESI† (Fig. S6.3). The transit time was estimated from the curve by constructing asymptotes with the intersection corresponding to the transit time.42 The thickness of the samples was determined by using cross-sectional scanning electron microscopy (Fig. 6 and Fig. S7, ESI†).
 | ||
| Fig. 6 Cross-sectional scanning electron microscopy (SEM) image of Cy5-[Al(pftb)4] time of flight sample. Sample comprises of glass/FTO/Al2O3/Cy5-[Al(pftb)4]/Al. | ||
As evident from Table 1, the mobility values are similar irrespective of the counterion size, and are comparable to those found in organic salt photovoltaics.43,44 This is an interesting result, as one would expect there to be a decrease in mobility as the anion size increases as a result of the chromophores being further apart, reducing the rate of charge hopping.
| Anion | Transit time/μs | Mobility/10−6 cm2 V−1 s−1 | Thickness/nm |
|---|---|---|---|
| Cl− | 2.47 ± 0.04 | 6.12 ± 0.09 | 110 |
| PF6− | 2.75 ± 0.13 | 3.69 ± 0.18 | 90 |
| Al(pftb)4− | 5.92 ± 0.30 | 5.75 ± 0.29 | 165 |
However, while the average separation of the Cy5 chromophores increases with increasing anion size and the percentage of interacting cation–cation surfaces is reduced by a factor of 4 (from 51.7 to 13.6%), the sections of the cations that mitigate charge transfer remain in contact. Therefore, even with the largest anions, it is not possible to break the interaction paths through the entire film.
Overall, the result is that the carrier mobility of the dyes with larger anions is similar to that of the dyes with smaller counterions, as the larger inter-chromophore distance is countered by the decreased degree of aggregation, and subsequent minimization of charge trapping by H-aggregates. Due to limitations with the sample preparation, it was difficult to get sufficiently thick samples to be able to conclusively determine the transport regime (dispersive vs. non-dispersive); furthermore, these limitations lead to a certain degree of uncertainty in regard to the mobility values found. However, while the absolute values may not be correct, we believe that the mobility values of the three dye materials tested are similar, and so the conclusions we have drawn are still valid.
Conclusions
In summary, we have shown using transient absorption spectroscopy that oxidized monomer species are photogenerated in pentamethine cyanine dye films incorporating small counterions, such as chloride or hexafluorophosphate. This has allowed us to confirm the charge generation mechanism in these materials, providing the first direct proof of high efficiency intrinsic charge generation in organic salt semiconductors. Upon photoexcitation, charge pairs are generated with the electron undergoing transfer from singlet excited states of monomeric species in the film to ground state aggregated chromophores. Alternatively, direct excitation of H-aggregates can occur, followed by reductive quenching by the monomer species. These charge transfer processes occur with a yield of up to 86%. The involvement of H-aggregates in the photoinduced charge separation process is essential as it ensures the necessary symmetry-breaking45,46 and the thermodynamic driving force to yield the charge separated pair.The impact of counterion size was then examined, with dyes containing two very large aluminium based counterions, Cy5-[Al(pftb)4] and Cy5-[Al(pfad)4], being studied alongside Cy5-Cl and Cy5-P. Transient absorption measurements demonstrated that monomer oxidized species were less present in the samples containing the two larger anions or samples where the cyanine dye was diluted in a polymer matrix. These results led us to conclude that in pentamethine cyanine dyes, where the anion is sufficiently large, higher order H-aggregates are not present in high concentration due to the chromophores being too spatially separated, thus diminishing the process of charge generation. However, despite an increase in spatial separation, time of flight measurements showed that the carrier mobility in Cy5-Al(pftb)4 was similar to that of the Cy5 dyes with the smaller counterions. This is interpreted as a counterbalancing effect for the charge carrier mobility, where the increased interchromophore distance in Cy5-[Al(pftb)4] films is compensated by the decrease in energetic disorder due to the suppression of H-aggregates.
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support by the Swiss National Science Foundation (SNF, Grant no. 200021_175729) and the National Center of Competence in Research “Molecular Ultrafast Science and Technology” (NCCR-MUST), a research instrument of the SNF, is gratefully acknowledged. The authors thank Linfeng Pan for cross-sectional SEM images, as well as Karen Strassel and Matthias Diethelm for their help in thin film preparation. Roland Hany is acknowledged for helpful discussions.References
- M. Bates and R. R. Lunt, Sustainable Energy Fuels, 2017, 1, 955–968 RSC.
- R. Hany, B. Fan, F. Araujo De Castro, J. Heier, W. Kylberg and F. Nüesch, Prog. Photovoltaics, 2011, 19, 851–857 CAS.
- E. Berner, T. Jäger, T. Lanz, F. Nüesch, J. N. Tisserant, G. Wicht, H. Zhang and R. Hany, Appl. Phys. Lett., 2013, 102, 98–102 CrossRef.
- B. Siegmund, M. T. Sajjad, J. Widmer, D. Ray, C. Koerner, M. Riede, K. Leo, I. D. W. Samuel and K. Vandewal, Adv. Mater., 2017, 29, 3–7 CrossRef PubMed.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- A. C. Véron, H. Zhang, A. Linden, F. Nüesch, J. Heier, R. Hany and T. Geiger, Org. Lett., 2014, 16, 1044–1047 CrossRef PubMed.
- H. Zhang, G. Wicht, C. Gretener, M. Nagel, F. Nüesch, Y. Romanyuk, J. N. Tisserant and R. Hany, Sol. Energy Mater. Sol. Cells, 2013, 118, 157–164 CrossRef CAS.
- D. Gesevičius, A. Neels, S. Jenatsch, E. Hack, L. Viani, S. Athanasopoulos, F. Nüesch and J. Heier, Adv. Sci., 2018, 5, 1700496 CrossRef PubMed.
- D. Gesevičius, A. Neels, S. Yakunin, E. Hack, M. V. Kovalenko, F. Nüesch and J. Heier, ChemPhysChem, 2018, 19, 3356–3363 CrossRef PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- A. Rupp, N. Roznyatovskaya, H. Scherer, W. Beichel, P. Klose, C. Sturm, A. Hoffmann, J. Tübke, T. Koslowski and I. Krossing, Chem. – Eur. J., 2014, 20, 9794–9804 CrossRef CAS PubMed.
- A. B. A. Rupp and I. Krossing, Acc. Chem. Res., 2015, 48, 2537–2546 CrossRef CAS PubMed.
- F. Nüesch, A. Faes, L. Zuppiroli, F. Meng, K. Chen and H. Tian, J. Mater. Sci., 2005, 40, 1353–1357 CrossRef.
- O. Malinkiewicz, T. Grancha, A. Molina-Ontoria, A. Soriano, H. Brine and H. J. Bolink, Adv. Energy Mater., 2013, 3, 472–477 CrossRef CAS.
- Z. Huang, D. Ji, S. Wang, A. Xia, F. Koberling, M. Patting and R. Erdmann, J. Phys. Chem. A, 2006, 110, 45–50 CrossRef CAS PubMed.
- C. A. Guarin, J. P. Villabona-Monsalve, R. López-Arteaga and J. Peon, J. Phys. Chem. B, 2013, 117, 7352–7362 CrossRef CAS PubMed.
- A. Devižis, A. Gelzinis, J. Chmeliov, M. Diethelm, L. Endriukaitis, D. Padula and R. Hany, Adv. Funct. Mater., 2021, 2102000, 1–8 Search PubMed.
- J. De Jonghe-Risse, J. Heier, F. Nüesch and J.-E. Moser, J. Mater. Chem. A, 2015, 3, 10935–10941 RSC.
- L. Wang, S. Jenatsch, B. Ruhstaller, C. Hinderling, D. Gesevičius, R. Hany and F. Nüesch, Adv. Funct. Mater., 2018, 28, 1–8 Search PubMed.
- M. Young, J. Suddard-Bangsund, T. J. Patrick, N. Pajares, C. J. Traverse, M. C. Barr, S. Y. Lunt and R. R. Lunt, Adv. Opt. Mater., 2016, 4, 1028–1033 CrossRef CAS.
- Y. Yang, S. Omi, R. Goto, M. Yahiro, M. Era, H. Watanabe and Y. Oki, Org. Electron., 2011, 12, 405–410 CrossRef CAS.
- T. P. Osedach, A. Iacchetti, R. R. Lunt, T. L. Andrew, P. R. Brown, G. M. Akselrod and V. Bulović, Appl. Phys. Lett., 2012, 101, 113303 CrossRef.
- Y. Yang, T. Nakamichi, H. Yoshioka, M. Yahiro, M. Era, H. Watanabe, Y. Cui, Y. Oki and G. Qian, J. Mater. Chem. C, 2013, 1, 1739–1744 RSC.
- S. B. Anantharaman, K. Strassel, M. Diethelm, A. Gubicza, E. Hack, R. Hany, F. A. Nüesch and J. Heier, J. Mater. Chem. C, 2019, 7, 14639–14650 RSC.
- A. L. Mannanov, P. S. Savchenko, Y. N. Luponosov, A. N. Solodukhin, S. A. Ponomarenko and D. Y. Paraschuk, Org. Electron., 2020, 78, 105588 CrossRef CAS.
- H. T. Chandran, T. W. Ng, Y. Foo, H. W. Li, J. Qing, X. K. Liu, C. Y. Chan, F. L. Wong, J. A. Zapien, S. W. Tsang, M. F. Lo and C. S. Lee, Adv. Mater., 2017, 29, 1606909 CrossRef PubMed.
- W. Li, D. Li, G. Dong, L. Duan, J. Sun, D. Zhang and L. Wang, Laser Photonics Rev., 2016, 10, 473–480 CrossRef CAS.
- E. N. Kaliteevskaya, V. P. Krutyakova, T. K. Razumova and A. A. Starovoitov, Opt. Spectrosc., 2016, 120, 482–491 CrossRef CAS.
- S. Chatterjee, B. Sauerwein, X. Yang, G. B. Schuster, P. D. Davis, P. Gottschalk and M. E. Kurz, J. Am. Chem. Soc., 1990, 112, 6329–6338 CrossRef CAS.
- J. R. Lenhard and A. D. Cameron, J. Phys. Chem., 1993, 97, 4916–4925 CrossRef CAS.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- R. Berera, R. van Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- M. E. F. Bouduban, A. Burgos-Caminal, R. Ossola, J. Teuscher and J.-E. Moser, Chem. Sci., 2017, 8, 4371–4380 RSC.
- U. B. Cappel, D. Moia, A. Bruno, V. Vaissier, S. A. Haque and P. R. F. Barnes, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- D. Oelkrug, H. J. Egelhaaf, J. Gierschner and A. Tompert, Synth. Met., 1996, 76, 249–253 CrossRef CAS.
- A. A. Paraecattil, J. De Jonghe-Risse, V. Pranculis, J. Teuscher and J.-E. Moser, J. Phys. Chem. C, 2016, 120, 19595–19602 CrossRef CAS.
- M. F. Bouduban, A. Burgos-Caminal, J. Teuscher and J.-E. Moser, Chimia, 2017, 71, 231–235 CrossRef CAS PubMed.
- A. Chowdhury, S. Wachsmann-Hogiu, P. R. Bangal, I. Raheem and L. A. Peteanu, J. Phys. Chem. B, 2001, 105, 12196–12201 CrossRef CAS.
- N. A. Mica, S. A. J. Thomson and I. D. W. Samuel, Org. Electron., 2018, 63, 415–420 CrossRef CAS.
- S. Jenatsch, R. Hany, A. Véron, M. Neukom, S. Zufle, A. Borgschulte, B. Ruhstaller and F. Nüesch, J. Phys. Chem. C, 2014, 118, 17036 CrossRef CAS.
- M. Bates and R. R. Lunt, Sustainable Energy Fuels, 2017, 1, 195 RSC.
- E. Vauthey, Chem. Phys. Chem., 2012, 13, 2001–2011 CrossRef CAS PubMed.
- C. E. Ramirez, S. Chen, N. E. Powers-Riggs, I. Schlesinger, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2020, 142, 18243–18250 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03251h |
| ‡ Current address: Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City Campus, London, W12 OBZ, UK. |
| This journal is © the Owner Societies 2021 |