
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV and VUV-induced fragmentation of tin-oxo cage ions†
Jarich
Haitjema
 a,
Lianjia
Wu
a,
Alexandre
Giuliani
bc,
Laurent
Nahon
c,
Sonia
Castellanos
a and
Albert M.
Brouwer
*ad
a,
Lianjia
Wu
a,
Alexandre
Giuliani
bc,
Laurent
Nahon
c,
Sonia
Castellanos
a and
Albert M.
Brouwer
*ad
aAdvanced Research Center for Nanolithography, P. O. Box 93019, 1090 BA Amsterdam, The Netherlands. E-mail: f.brouwer@arcnl.nl
bUAR 1008 Transfrom, INRAE, F-44316 Nantes, France
cSynchrotron SOLEIL, L’Orme des Merisiers, BP 48 Saint Aubin, 91192 Gif-sur-Yvette, France
dUniversity of Amsterdam, van’t Hoff Institute for Molecular Sciences, P. O. Box 94157, 1090 GD Amsterdam, The Netherlands
First published on 15th September 2021
Abstract
Photoresist materials are being optimized for the recently introduced Extreme Ultraviolet (EUV) photolithographic technology. Organometallic compounds are potential candidates for replacing the ubiquitous polymer-based chemically amplified resists. Tin (Sn) has a particularly large absorption cross section for EUV light (13.5 nm, 92 eV), which could lead to a lower required EUV dose for achieving the desired solubility change (improved sensitivity). However, the fundamental interaction between organometallic materials and higher energy photons is poorly understood. In this work, we exposed n-butyltin-oxo cage dications (M2+) in the gas phase to photons in the energy range 4–35 eV to explore their fundamental photoreactivity. Photoproducts were detected using mass spectrometry. Homolytic cleavage of tin–carbon bonds was observed for all photon energies above the onset of electronic absorption at ∼5 eV (∼250 nm), leading to photoproducts which have lost one or more of the attached butyl groups (Bu). Above 12 eV (<103 nm), dissociative photoionization occurred for the dication (M2+), competing with the neutral loss channels. The photoionization threshold is lowered by approximately 2 eV when one counterion (triflate, OTf− or tosylate, OTs−) is attached to the tin-oxo cage (MOTf+ and MOTs+). This threshold is expected to be even lower if each tin-oxo cage is attached to two counterions, as is the case in a solid film of tin-oxo cages. Addition of counterions also affected the fragmentation pathways; photoexcitation of (MX)+ (X = counterion, OTf or OTs) always led to formation of (MX-2Bu)+ rather than (MX-Bu)+. MOTs+ was much more reactive than MOTf+ in terms of reaction products per absorbed photon. A possible explanation for this is proposed, which involves the counterion reacting with the initially formed tin-based radical.
1 Introduction
The introduction of Extreme Ultraviolet (EUV) lithography into high-volume manufacturing comes with several challenges. In particular, the photoresist is a potential issue, since high resolution patterning places tight restrictions on its lithographic performance.1–5 The parameters resolution (minimum feature size that can be obtained), line edge roughness (average deviation of printed lines from a straight line), and sensitivity (light dose per unit of area that is required for pattern formation) are especially important. It is difficult to improve one of the resist parameters without simultaneously worsening at least one of the others, a problem that is known as the “RLS trade-off”.6 Chemically amplified resists, which are the workhorses of Ultraviolet photolithography, have only low absorption cross sections at the EUV wavelength of 13.5 nm. To achieve sufficient photon absorption in thin films, and potentially higher etch resistance, metal-containing photoresist materials have been proposed as alternatives.7–13An example of an organometallic photoresist material is the tin-oxo cage (see Fig. 1),14,15 first described by Puff and Reuter in 1989.16 Its potential use as an EUV photoresist was first investigated by Cardineau and coworkers in 2014.7 A film made of this compound has a strong absorption of EUV light, with an attenuation length of the order of 13 μm−1, compared to 5 μm−1 for conventional chemically amplified resists.17,18 The tin-oxo cage material is a negative tone resist, turning insoluble upon UV,19 EUV20 or e-beam irradiation.21,22 The photochemical mechanisms through which this material turns insoluble are not known in detail, although some have been proposed.19 It is generally accepted that cleavage of the tin–carbon bond is one of the first reaction steps, which leads to outgassing of volatile organic compounds and agglomeration of the remaining tin-oxo cores.7,23 This implies that shrinkage of the material occurs.24 A better knowledge of the mechanisms at work would aid the design of novel materials with improved resist properties.25
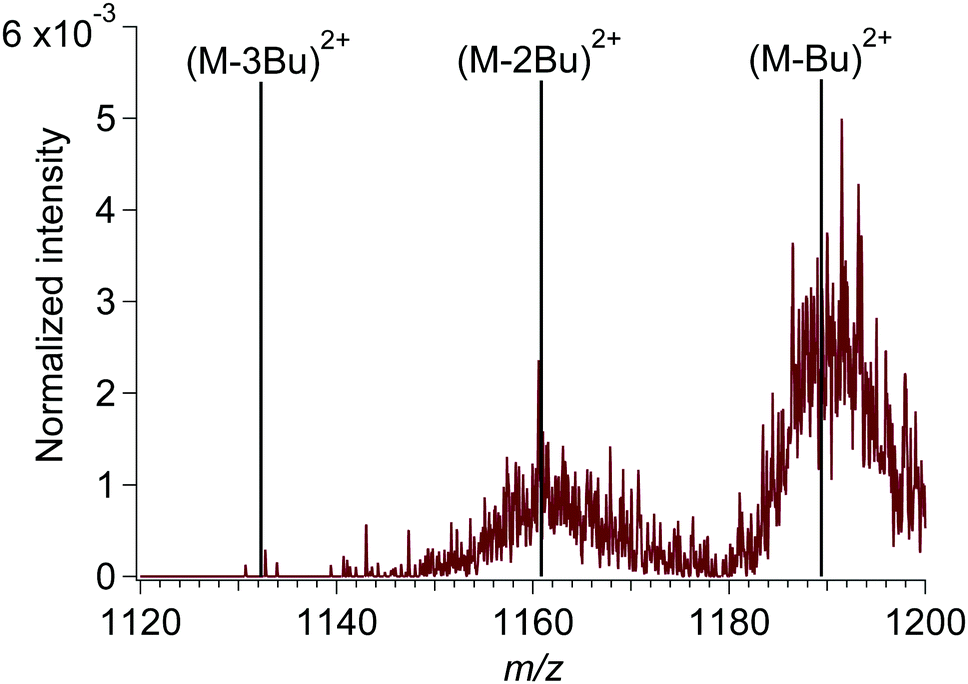 | ||
| Fig. 1 Mass spectrum of M2+ with 7 eV (177 nm) incident photon energy at m/z values just below the precursor ion peak (m/z 1218). Vertical lines indicate the m/z corresponding to loss of butyl (Bu): (M-3Bu)2+ (m/z 1132.5), (M-2Bu)2+ (m/z 1161) and (M-Bu)2+ (m/z 1189.5). Number of incident photons: 5.5 × 1011. The intensity was normalized to the intensity of the M2+ peak. | ||
Spectroscopic studies on EUV-irradiated tin-oxo cages (ideally in situ) would be a valuable method towards improved understanding of their photochemistry. However, spectroscopic studies on the thin resist films (typical thickness 20–50 nm) are difficult because of the small amounts of material, which are below the detection limits for many highly informative methods such as NMR spectroscopy. An alternative is to perform gas-phase photoreaction experiments on individual ionic species, using mass spectrometry as a detection technique.26–29 This method was very recently used for studies on organometallic molecular hybrid materials that can potentially be used as photoresists.30,31 The tin-oxo cage dication can be isolated in the gas phase using a linear or quadrupole ion trap without any modification.32,33
While irradiation of the doubly charged tin-oxo cage (dication) gives inside into its fundamental reactivity, studies on mono–cation complexes are also of interest. In a tin-oxo cage film, the +2 charge of each cage is balanced by two counterions. For the gas phase dication, the electrons are more tightly bound as a result of Coulomb forces. Therefore, the value found for the ionization potential of the tin-oxo cage (∼12 eV)20 is higher than the ionization potential in solid tin-oxo cage films.34 The tin-oxo cage with two attached counterions has a net charge of zero, and therefore cannot be trapped. However, isolation of a tin-oxo cage complex with only one counterion (net charge +1) is very well possible. The counterions triflate (OTf−) and p-toluenesulfonate (tosylate, OTs−) were chosen for this purpose. These counterions interact with the tin-oxo cage dication through electrostatic and hydrogen bonding.35 Related structures, with the sulfonate group being attached either covalently or noncovalently, have been reported in the literature.36 Because the tosylate anion is a stronger base, its hydrogen bonding with the tin-oxo cage is expected to be stronger than that of the triflate.37 The counterion can have an influence on the reactivity; for example, the tin-oxo cage with trifluoroacetate counterions (TinF) was found to be less responsive to EUV light than the tin-oxo cage with acetate or hydroxide counterions, even though the fluorine atoms enhance EUV absorptivity.38, 39 The reason for this lower sensitivity is not known.
The aim of this paper is to study UV and VUV fragmentation and ionization pathways for the tin-oxo cage dication and monocation complexes (cage dication with one counter-anion). These pathways depend on the photon energy. At low energies, only excitation to bound electronic states (e.g. HOMO→LUMO) is possible, whereas at higher energies valence electrons can be ejected into the continuum (ionization). Tin 4d electrons could in principle also be ejected; their binding energy was reported to be about 24.4 eV for metallic tin40 and 29 eV for tin-oxo cage compounds.34 These energies are accessible with the used synchrotron source. While gas-phase experiments cannot directly be compared to exposure of photoresist films, the results give insight into the initial absorption and reaction events and therefore contribute to a fundamental understanding of the photochemistry.
2 Experimental
2.1 Sample preparation and measurements
Tin-oxo cage materials were prepared as described previously.7,20 The tin-oxo cage was synthesized with tosylate counterions (TinOTs), after which the material was converted to the hydroxide form (TinOH) by ion exchange using aqueous tetramethyl ammonium hydroxide.14 NMR spectra of TinOTs and TinOH (1H and 119Sn) corresponded to those reported in the literature.14,35,41 The tin-oxo cage in triflate form (TinOTf) was prepared by reacting TinOH with two molar equivalents of trifluoromethanesulfonic acid. The general procedure for this has been described previously.42 A characterization of TinOTf (1H and 19F NMR spectra) can be found in the ESI† (Fig. S1). UV absorption spectra of the compounds were recorded with a Shimadzu UV2700 spectrometer, using spectroscopic grade ethanol (EtOH) as a solvent.The tin-oxo cage compounds were dissolved in methanol (concentration ∼10 μM), filtered and electrosprayed for the gas phase VUV exposure experiments, using a syringe pump with a flow rate of about 5 μL min−1. Ions with m/z 1218 (M2+), 2585 (MOTf+) and 2607 (MOTs+) were trapped in a commercial linear ion trap (Thermo Finnigan LTQ XL) coupled to the DESIRS VUV beamline of the SOLEIL synchrotron.43,44 The trapping range was 30 m/z for all fragments; for example for the dication (m/z 1218) all ions with an m/z between 1203 (1218 − 15) and 1233 (1218 + 15) were trapped.
Photofragmentation was monitored for different photon energies, using 0.1 eV steps between 4.1 and 22.0 eV and 0.15 eV steps between 20.15 and 35.0 eV. The UV/VUV beam had an approximate spot size of 1 mm2 in the interaction region, a small energy bandwidth (typically 12 meV at 10 eV) and was free of high harmonics owing to the use of a Kr gas filter (7–14 eV) and a quartz window (4–7 eV). Above 14 eV the coatings of the gratings obviate the need for further filtering (4.5–22 eV: SiC, vanishing transmission above 28 eV (i.e. for first order light above 14 eV); >20 eV: Pt, vanishing transmission above 40 eV (for first order light above 20 eV). The photon flux was in the 1012–1013 photons per s range and was measured independently using an AXUV100 photodiode (International Radiation Detectors) under the measurement conditions. The light dose was kept low to limit (sequential) two-photon processes, aiming for ≤10% conversion of the primary ions to photoproducts. The dose was regulated by the exit slit and a mechanical shutter that controlled the irradiation time of the trapped ions between 65 and 2000 ms.
2.2 Data analysis
Results were obtained in the form of mass spectra for each photon energy, with intensity (I) obtained as a function of the mass-to-charge ratio (m/z). All mass spectra shown in this work were normalized to the precursor ion intensity.To quantify formation of fragments as a function of photon energy, action spectra were constructed. These spectra show the relative yield of fragments at each photon energy. To obtain meaningful results, this relative yield should be corrected for the number of incident photons (to correct for photon flux variations upon scanning) and for the total ion current TIC (to correct for fluctuations in the electrospray source). The relative intensity of fragment A can thus be calculated according to eqn (1).
 | (1) |
In eqn (1), IA is the relative intensity for fragment A, measured by the peak area. The integration range was chosen to be the predicted average mass-to-charge ratio plus or minus 7 (3+ ions), 14 (2+ ions) or 28 (1+ ions), rounded to the nearest whole number. TIC is the total ion current, measured by calculating the area of the complete mass spectrum. The incident number of photons Nphot is given by eqn (2):
 | (2) |
In eqn (2), Φq is the photon flux in s−1 measured in the reference scan with the photodiode, t is the irradiation time in s, wexp is the slit width during the experiment (in mm), and wref is the slit width during the reference scan (in mm). To account for small circumstantial differences between reference scans and measurement scans, a small scaling adjustment was applied to the photon flux curves as needed.
2.3 Computational
Quantum chemical calculations were performed using the B3LYP hybrid functional as implemented in Gaussian09 and Gaussian16.45 For geometry optimizations, the LANL2DZ basis set was used. Subsequent single point energy evaluations were performed with the higher-quality Def2TZVP basis set (B3LYP/Def2TZVP//LANL2DZ). Reaction energies are corrected for the change in zero-point vibrational energies (B3LYP/LANL2DZ). It should be noted that the conformational space of the molecule is large as a result of possible rotations about Sn–C and C–C bonds. Because exploration of this vast space for all species of interest was not possible within the scope of the present work, we have chosen one of the low-energy conformations of the butyltin oxo cage, and carried out all modifications of the structure starting from this initial structure. Thus, we cannot exclude that lower-energy rotational isomers can be found for the species discussed, which may slightly affect the reported bond energies. Considering the size of the system and the moderate level of theory that can be applied to it, the computational results should only be used as guidelines for interpretation of the experimental results. To reduce the size of the computational problem, the tosylate anion was replaced by methanesulfonate in the calculations. In preliminary explorations the butyl groups were replaced with methyl groups, but since the reaction energies were found to be different for the butyl-oxo cage system we carried out the reported calculations for the full n-butyltin-oxo cages. Full details of all relevant structures and energies are provided as ESI.†3 Results and discussion
3.1 Mass spectra of tin-oxo cage ions
The ESI-MS spectrum of the tin-oxo cage compound was reported earlier by Dakternieks and coworkers,32 who observed a main peak at m/z 1218, assigned to the dication [(nBuSn)12O14(OH)6]2+. This result was reproduced in our experiments (see ESI,† Fig. S2). The main peak is quite broad and shows an intricate structure, as a result of the 10 stable isotopes of tin. The high mass resolution of our instrumentation allowed us to resolve the isotope pattern. The pattern confirms the 2+ charge, because the spacing between the subpeaks is half an m/z unit.Depending on electrospray conditions, a peak with a lower m/z (1190) also appeared in the mass spectrum. This corresponds to a tin-oxo cage of which one of the butyl chains has been lost. By tuning the electrospray settings and applying a mass filter, we ensured that only [(nBuSn)12O14(OH)6]2+ (precursor ion, M2+) was present in the ion trap before irradiation.
Mass spectra of the isolated (non-irradiated) monocation complexes MOTf+ and MOTs+ clearly are in good agreement with predicted mass spectra (see ESI,† Fig. S3).
3.2 UV irradiation of tin-oxo cage ions
In an initial set of experiments, we exposed the tin-oxo cage dication to UV light ranging from 4 to 7 eV (310–177 nm). In this range the tin-oxo cage in solution shows a strong absorption centered around 220 nm.15 According to DFT calculations the Highest Occupied Molecular Orbital (HOMO) is primarily located on the six-coordinated Sn atoms and has Sn–C σ-bonding character. The LUMO, which is mostly located on the central belt of the cage, has significant Sn–C* character mixed with vacant tin-centered orbitals (5p, 5d).15,46The photofragmentation spectrum of the tin-oxo cage at a photon energy of 7.0 eV is shown in Fig. 1. It can be seen that the precursor ion (M2+) loses a butyl chain (Bu) upon photon absorption. This indicates cleavage of an Sn–C bond as the primary process in the electronically excited state. The isotope pattern shows that the fragments are doubly charged. Peaks near m/z 1160 correspond to the loss of two butyl chains, but loss of three or more butyl chains is negligible.
In Fig. 2, the action spectra of (M-Bu)2+ and (M-2Bu)2+ are shown. It can be seen that the yield of (M-Bu)2+ is low below 5 eV, peaks around 5.6 eV and increases again at higher energies. Although the 1175–1203 m/z peak area <5 eV is slightly above zero, a closer examination shows that this is caused by spectral background rather than by the presence of photochemically generated (M-Bu)2+ (see ESI,† Fig. S4).
 | ||
| Fig. 2 Comparison between absorption spectrum of TinOH in EtOH solution (red line) and action spectrum of (M-Bu)2+ and (M-2Bu)2+ in the gas phase (blue circles and green triangles, sum of both given by open circles). | ||
A similar excitation onset was found for (M-2Bu)2+. This fragment could be formed as a result of excess energy that remains in the system after cleavage of the first butyl group, which requires only about 2.2 eV.47 Cleavage of the second butyl group is expected to require even less energy, according to DFT calculations: at the B3LYP/Def2TZVP//LANL2DZ level the bond energies for the first and second butyl radical loss are 2.34 eV and 0.98 eV, respectively. For the first butyl loss, generating the radical dication of the tin cage, two isomers are conceivable, depending on whether the bond is broken of a 5-coordinated butyl-tin in the central “belt” of the molecule, or of a 6-coordinated butyl-tin unit in one of the two “caps” of the cage. It turns out that the latter is favored by 3.6 kcal mol−1 (B3LYP/Def2TZVP//LANL2DZ). The second butyl radical loss leads to a clearly preferred product, because the two radical sites can easily rearrange to a closed-shell structure when they arise on neighboring tin atoms in a cap, followed by a transfer of a bridging OH group to one of the two tin atoms. This yields one formal Sn(II) atom as illustrated in Scheme 2. This rearrangement occurs spontaneously in silico upon geometry optimization of the biradical species.
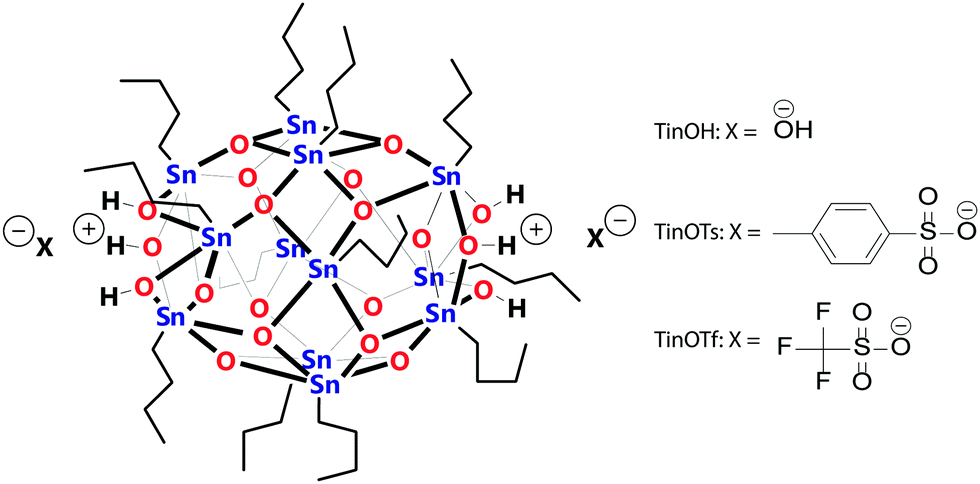 | ||
| Scheme 1 Tin-oxo cage compounds used in this work: TinOH (hydroxide counterions), TinOTs (tosylate counterions), and TinOTf (triflate counterions). TinOH was used as precursor of the dication (M2+), for the monocationic complexes (MOTf+, MOTs+) we used TinOTf and TinOTs, respectively. | ||
 | ||
| Scheme 2 Structural rearrangement upon subsequent loss of two Bu radicals from (M)2+ The drawing shows one “cap” of the tin oxo cage, with a formal charge of +1. Each of the Sn atoms has two additional bonds to oxygen atoms in the central “belt” unit, below the plane of the drawing. The view of the “cap” is from the top. | ||
The shape of both action spectra closely resembles that of the absorption spectrum of TinOH dissolved in EtOH (full red line), which indicates that photofragmentation in this energy regime scales linearly with the absorption coefficient. At photon energies >6.3 eV no reliable solution absorption spectrum could be recorded, because the solvent (EtOH) absorbs too strongly in this range. The gas phase experiment, however, shows a clear increase in absorption with increasing photon energy of M2+ above 6.5 eV.
Similarly to the study on tin-oxo cage dications, photofragmentation of the monocation complexes was studied. A mass spectrum of MOTf+, exposed to 7.0 eV photons, is shown in Fig. 3a. In contrast with the results on the bare tin-oxo cage dications, loss of a single butyl group is hardly observed. Rather, the major photoproduct at low photon energies is (MOTf –2Bu)+. For the monocation MOTs+ the same result was found (see Fig. 3b). The tosylate monocation (MOTs+) is more reactive than the triflate monocation (MOTf+), judging from the relative heights of the (M-2Bu)2+ peaks.
 | ||
| Fig. 3 Photofragmentation of MOTf+ (a) and MOTs+ (b) with incident light at 177 nm (7.0 eV), in the 1+ region of the spectrum. Vertical lines indicate the peaks corresponding to loss of multiple butyl groups. Number of incident photons: 8.3 × 1011. Normalization refers to the precursor ions. | ||
Computational results (with the methanesulfonate anion OMs as a computational surrogate for the OTs unit) indicate that breaking the first butyl-tin bonds in the cation complexes is slightly less endothermic (2.29 eV for OMs, 2.30 eV for OTf) than in the bare butyltin oxo cage (2.34 eV). For the second butyl loss, an additional reaction path opens, in which the sulfonate ion acts as a nucleophile and binds to the coordinatively unsaturated Sn atom (Scheme 3). The result resembles a bridging sulfonate group, a structural feature that was also observed by Prabusankar and coworkers.36 In this reaction, two hydrogen bonds with the bridging OH-groups are sacrificed to form an O–Sn bond, with a computed energy gain of 3.9 kcal mol−1 for OTf and 4.3 kcal mol−1 for OMs.‡ This result agrees with the observation that MOTs+ is more reactive than MOTf+, due to the stronger nucleophilicity of the tosylate anion. As a result of this rearrangement, the computed bond energy for the second butyl loss is 0.71 eV and 0.73 eV for the mesylate and triflate, respectively. This indirect weakening of the Sn–C bond may explain why in the complexes the (M-Bu)2+ species is hardly detected.
 | ||
| Scheme 3 Reaction mechanism for the loss of two butyl groups upon photo-excitation of MOTf+ (R = CF3) or MOTs+ (R = MePh), taking place at the positively charged cap of the tin-oxo cage (6-coordinated tin atoms; extra bonds are omitted for clarity). The view of the “cap” is from the side, as in Scheme 1. | ||
Similarly to the tin-oxo cage dication, the onset of photofragmentation corresponds to the onset of photo-absorption (see Fig. 4). This indicates that the initial excitation (leading to Sn–C cleavage) is not affected by the presence of counterions.
 | ||
| Fig. 4 Action spectrum of (MOTf-2Bu)+ (a) and (MOTs-2Bu)+ (b), blue circles for MOX-Bu and green triangles for MOX-2Bu, sum of both given by open circles. Red lines indicate absorption coefficients: TinOH (a) and MOTs+ (b, predicted, see Fig. S5, ESI†). | ||
A difference between the two counterions OTf and OTs is that OTf is mostly transparent at low photon energies (<6 eV), whereas OTs is absorbing (see ESI,† Fig. S5). This could have a minor influence on the reactivity at low photon energies, since the tosyl group could transfer its energy to the tin-oxo cage cation upon photoabsorption.
3.3 VUV irradiation of tin-oxo cage ions
At VUV energies >7 eV, additional 2+ fragments are observed, as exemplified in Fig. 5b (13.5 eV). (M-Bu)2+ is still the most abundant, although multiple butyl chain loss up to (M-5Bu)2+ is now detected as well. The subsequent fragmentation reactions are likely a result of the higher internal energy that remains in the various (M-nBu)2+ intermediates at higher photon energies.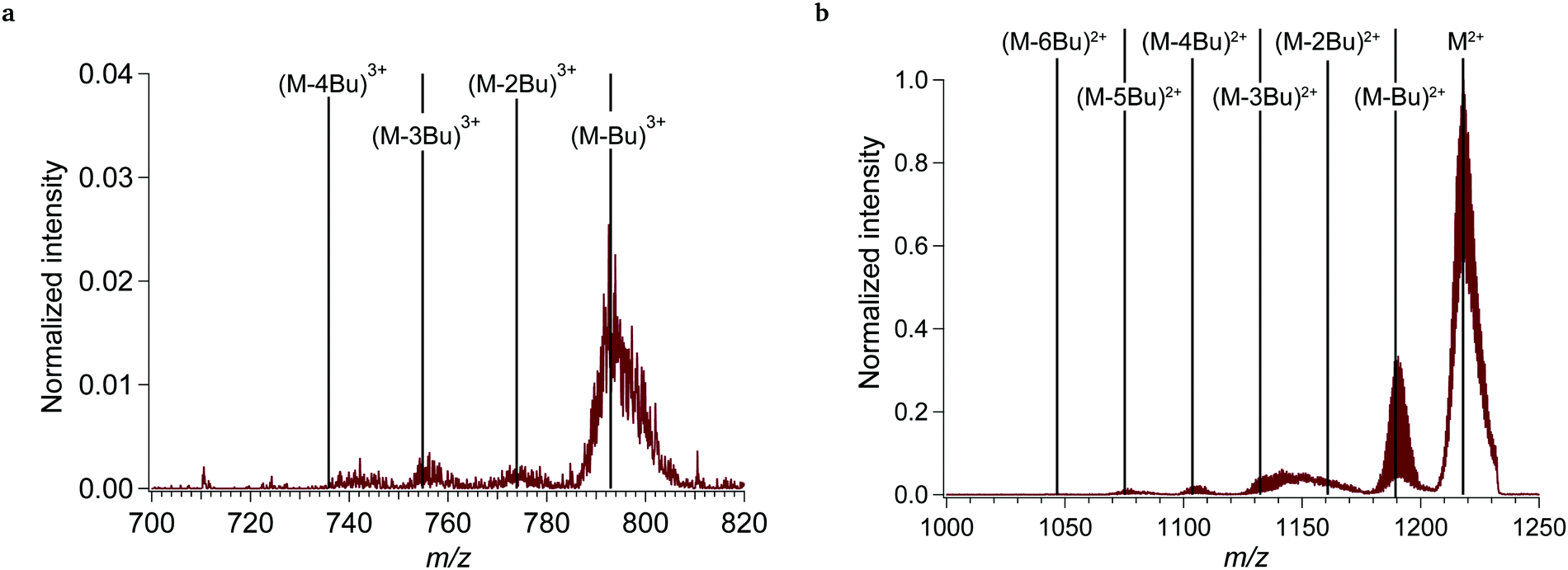 | ||
| Fig. 5 Mass spectrum of the tin-oxo cage dication with incident light of 92 nm (13.5 eV), showing (a) 3+ ions and (b) 2+ ions. Vertical lines indicate the main peak (m/z 1218) and peaks indicating various butyl chain losses. Number of incident photons: 1.7 × 1012. The intensity was normalized to the M2+ peak. | ||
Between (M-2Bu)2+ and (M-3Bu)2+, a broad feature is present in the mass spectrum, of which the origin is unclear. It could correspond to the loss of 2 butyl groups and one or multiple hydroxide(s).
As can be seen by comparing Fig. 5b with Fig. 1, fragmentation is much stronger at these higher photon energies. The relative intensity of the main fragment peak (M-Bu)2+ is about 100× larger at 13.5 eV, while the photon number at 13.5 eV is only about 3× larger. The fragmentation yield of (M-Bu)2+ increases gradually with photon energy (see Fig. 6) until it reaches a maximum around 13 eV, after which it gradually decreases. This decrease is likely due to competition with other fragmentation and ionization processes, although a decrease in total absorption cross section could also play a role.
 | ||
| Fig. 6 Action spectra of (M-Bu)2+, (M-2Bu)2+ and (M-Bu)3+ formation from the precursor ion M2+, between 5 and 20 eV. | ||
At photon energies >12.1 eV the dication can be ionized, leading to (M-Bu)3+ and smaller fragments (see Fig. 5a). The ionized precursor ion M3+ is not detected. The isotope pattern of the peaks unequivocally indicates a 3+ charge. The computed adiabatic ionization potential of 11.5 eV is in good agreement with the onset energy of ionization observed here. This ionization can either take place directly (short time scale) or by internal conversion to an auto-ionizing excited state, which contains sufficient internal energy to emit an electron (longer time scales). The structure of the M3+ species found by geometry optimization shows one Sn–C bond elongated from 213 pm to 245 pm (Fig. 7). Although the species is a minimum on the energy surface, it is rather weakly bound: the computed dissociation energy to (M-Bu)3+ + butyl radical is only 0.78 eV.15, 46
 | ||
| Fig. 7 Computed structure (B3LYP/LANL2DZ) and spin density (B3LYP/Def2TZVP//B3LYP/LANL2DZ) of M3+. | ||
The increasing yield of (M-Bu)3+ with increasing energy above 12 eV is accompanied by decreasing yields of (M-Bu)2+ and (M-2Bu)2+. This indicates that the ionization process is competitive with the excited–state bond cleavage leading to (M-Bu)2+. Because the (M-2Bu)2+ yield parallels the yield of (M-Bu)2+ it is likely that (M-2Bu)2+ is formed directly from (M-Bu)2+, rather than by loss of a butyl cation from (M-Bu)3+.
| M2+* → (M-Bu)2+˙ + Bu˙ → (M-2Bu)2+ + 2Bu˙ (>5 eV) | (a) |
| M2+** → [M3+˙] + e− → (M-Bu)3+ + Bu˙ + e− (>12 eV) | (b) |
| [M3+˙] → (M-Bu)2+˙ + Bu+ (>12 eV) | (c) |
In the above equation, M2+* is the tin-oxo cage dication in the first excited state, while M2+** represents a higher excited state.
According to the computations, separation of charge (eqn (c)) is thermodynamically favored over loss of a butyl radical (b), but apparently the latter process is faster. Loss of a second butyl group ((M-Bu)3+→(M-Bu)3+˙ + Bu˙) leads to formation of two radicals and is unfavorable (ΔE = 2.42 eV). Subsequent cleavage (M-2Bu)3+˙ → (M-3Bu)3+ + Bu˙ is again more facile(ΔE = 0.82 eV). Further loss of butyl groups is not observed in the 3+ ion series, but interestingly the yield of species (M-4Bu)2+ and (M-5Bu)2+ (which are formed via the electronic excitation pathway with Ephoton > 9 eV) continues to increase with increasing energy while the yield of (M-Bu)2+ and (M-2Bu)2+ levels off at Ephoton ≈ 12 eV. Another remarkable species is observed in Fig. 5a at m/z = 743. This corresponds with the composition (M-Bu-SnO2)3+. The peak-to-peak spacing confirms that this is a 3+ ion. A plausible structure is shown in Fig. S6 (ESI†). In this structure, the central belt has five instead of six O–Sn–O–Sn four-membered rings, and in each cap one of the Sn atoms is 5-coordinated instead of 6-coordinated.
The yield of (M-Bu)3+ increases with photon energy until it reaches a maximum around 19 eV. Above this energy, the yield of (M-Bu)3+ gradually decreases with increasing photon energy, with no evidence of ionization from an Sn 4d level (see Fig. 8). The absorption cross section of the tin-oxo cage dication, as predicted from the sum of the cross sections of all individual atoms from the CXRO database, is included in Fig. 8.48 It can be seen that the yield of (M-Bu)3+ drops faster than the predicted absorption coefficient. At higher photon energies, the “atomic approximation” becomes increasingly accurate,49 and a recent study shows that the data from the CXRO database correspond fairly well with the absorption spectrum of a thin film of tin-oxo cages between 25 eV and 40 eV.50 An alternative explanation could be that smaller fragments are formed at higher photon energies, which are not detected within the m/z range of the present experiment.
 | ||
| Fig. 8 Action spectra of (M-Bu)3+ (blue triangles) and (M-Bu)2+ (green circles) formation between 20 and 35 eV. The red line (right axis) shows the molar absorption coefficient of the tin-oxo cage dication (molecular formula: C48H114Sn12O20) calculated from tabulated values.48 | ||
At photon energies >10 eV, photoionization is observed for the monocationic complexes, similarly to the results of the tin-oxo cage dication; MOTf+ and MOTs+ are converted to doubly charged fragments (see Fig. 9). The photoionization is always dissociative, leading to the fragment (MOTf-Bu)2+ or (MOTs-Bu)2+. In both cases a small peak is observed just below 1220 m/z, which could correspond to the bare dication (m/z 1218) formed by transfer of an electron from the anion. Alternatively, this peak could result from transfer of a proton to the anion and loss of the acid. The monocation complexes do not seem to lose multiple butyl groups upon photoionization, contrary to the bare M2+ (Fig. 5b). The reason for this could be the rearrangement depicted in Scheme 4: after loss of a butyl group, the sulfonate anion attacks the vacant Sn-atom as a nucleophile, drastically reducing the charge separation. This is accompanied by an energy gain of ca. 1 eV.
 | ||
| Fig. 9 Photofragmentation of (a) MOTf+ and (b) MOTs+ with incident light at 92 nm (13.5 eV), in the 2+ region of the spectrum. Vertical lines indicate the peaks corresponding to loss of multiple butyl groups. Number of incident photons: 1.4 × 1011 for MOTf+, 3.3 × 1010 for MOTs+. | ||
 | ||
| Scheme 4 Rearrangement of (M-Bu)OR2+. | ||
The ionization threshold of the tin-oxo cage is approximately 10 eV when a counterion (OTs− or OTf−) is attached (see Fig. 10). This ionization threshold is found to be identical for the two counterions. MOTs+, however, appears to be more reactive towards fragmentation, since the (MOTs-Bu)2+ fragment has a higher relative intensity (∼3.5×). This could be related to the stronger nucleophilic nature of the tosylate, which favors formation of the complex shown in Scheme 4, or to a larger cross section of the larger tosylate anion. Another possibility is that MOTf2+ decays more through pathways that are not detected in our experiment.
 | ||
| Fig. 10 Fragmentation yield of (MOTs-Bu)2+ (black circles) and (MOTf-Bu)2+ (red triangles) upon irradiation of MOTf+ and MOTs+. Integration ranges: 1250–1278 m/z for MOTf+, 1261–1289 m/z for MOTs+. Note the lower relative intensity for MOTf (different y scale). | ||
It should be noted that both MOTf+ and MOTs+ appear to be more reactive than the bare cage (compare the intensities with Fig. 6). The quantum chemical calculations show similar structure changes after ionization for the complexes as for the dication, with increases of the length of one Sn–C bond to 247 pm.
The ionization threshold is ca. 2 eV lower than the 12 eV for the bare dication (compare with Fig. 6). As discussed before, this is due to electrostatic force; at higher charge, the electrons are more tightly bound. The computed adiabatic ionization potentials are 8.9 eV for MOTs+ (using MOMs+ as a model) and 9.1 eV for MOTf+. A recent XPS study on thin films of tin-oxo cages with different counterions reported valence electrons having a minimum binding energy of about 5.5 eV.34 This confirms that the ionization potential is lowered further in solid films, as a result of the two counterions (instead of one for MOX+) and additional stabilization by the polarizable medium.
4 Conclusions
Tin-oxo cage ions were exposed to UV and VUV photons (4–35 eV) in the gas phase, and photoproducts were studied using mass spectrometry. The appearance of photoproducts indicated several photofragmentation pathways. Homolytic cleavage of tin–carbon bonds was observed for all photon energies above the onset of electronic absorption at ∼5 eV (∼250 nm), leading to photoproducts which have lost one or more of the attached butyl groups. Above 12 eV (<103 nm), dissociative photoionization occurred for the dication (M2+), competing with the regular fragmentation process. This ionization was always fully dissociative, leading to the (M-Bu)3+ fragment, the trication M3+ being unstable. When counterions (triflate or tosylate) were attached to the tin-oxo cage and the monocations (MOTf+ and MOTs+) were exposed to light, the ionization threshold was found to be lower (∼10 eV). The fundamental reactivity also changed, since loss of 2 rather than 1 butyl group was much more prominent upon electronic excitation. MOTs+ was found to be much more reactive than MOTf+. These two phenomena are tentatively explained by a reaction of the tin-centered radical with the sulfonate counterions. Such a reaction is likely to be relevant for the solubility-switching photochemistry in the solid state.No evidence was found for enhanced photofragmentation when the photon energies where above the binding energies of the 4d orbitals of the tin-oxo cages (29 eV in the solid state).34 This is because the cross section for the photoionization process near the threshold is small, and increases only gradually up to photon energies near 90 eV, corresponding to the delayed maximum of the 4d shape resonance.34
The photons used in this work (4–35 eV) have lower energy than those employed in EUV lithography (92 eV). Additionally, the experiments were performed on isolated cations rather than on solid-phase photoresist films. However, this also has the inherent advantage of being able to study initial key reactions that occur in the early stages of exposure, which is difficult to do for thin solid films. Furthermore, absorption of EUV photons inside resist films initiates Auger decay, associated shake processes and inelastic electron scattering after emission of primary electrons. These last processes generate secondary electrons that could have energies within the studied range of 4–35 eV.24 Lastly, this study shows that the counterion(s) can fundamentally alter the photoreactivity of the tin-oxo cage. This would mean that optimization of the counterion (X−) is a possible path towards improved resist parameters, while altering the attached organic group (butyl in this case) is another viable option.7 These various possibilities make the tin-oxo cage compound a versatile model compound that can be used in the search towards improved EUV photoresist materials.
Author contributions
All authors contributed to the experimental investigation; J. H. synthesized the compounds; A. M. B. performed the quantum-chemical calculations; A. G. and L. N. provided the instrumental facilities and software; J. H. analyzed the experimental data and wrote the first draft of the manuscript; all authors contributed to the review and editing; A. M. B. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was carried out at ARCNL, a public–private partnership of UvA, VU, NWO and ASML. The authors acknowledge Ed Zuidinga (UvA) for initial testing of the electrospray conditions and thank Niklas Ottosson, Thomas Schlathölter (RUG) and Ronnie Hoekstra (ARCNL/RUG) for performing preliminary experiments that initiated this research. We thank Mees Trouwborst for help with synthesis of tin-oxo cage compounds. We warmly thank the whole SOLEIL staff for running the facility under project 20161099.Notes and references
- A. Lio, Synchrotron Rad. News, 2019, 32, 9–14 CrossRef.
- E. van Setten, G. Bottiglieri, J. McNamara, J. van Schoot, K. Troost, J. Zekry, T. Fliervoet, S. Hsu, J. Zimmermann, M. Roesch, B. Bilski and P. Graeupner, Proc. SPIE, 2019, 10957, 1095709 Search PubMed.
- L. Li, X. Liu, S. Pal, S. Wang, C. K. Ober and E. P. Giannelis, Chem. Soc. Rev., 2017, 46, 4855–4866 RSC.
- T. Manouras and P. Argitis, Nanomaterials, 2020, 10, 1593 CrossRef CAS PubMed.
- C. Luo, C. Xu, L. Lv, H. Li, X. Huang and W. Liu, RSC Adv., 2020, 10, 8385–8395 RSC.
- J. Lee, J. Kim, S. Jeong, M. Lim, S. Koo, C.-M. Lim and Y.-S. Kim, Proc. SPIE, 2016, 9776, 977623 CrossRef.
- B. Cardineau, R. Del Re, M. Marnell, H. Al-Mashat, M. Vockenhuber, Y. Ekinci, C. Sarma, D. A. Freedman and R. L. Brainard, Microelectron. Eng., 2014, 127, 44–50 CrossRef CAS.
- L. Wu, J. Liu, M. Vockenhuber, Y. Ekinci and S. Castellanos, Eur. J. Inorg. Chem., 2019, 4136–4141 CrossRef CAS.
- N. Thakur, M. Vockenhuber, Y. Ekinci and S. Castellanos, Proc. SPIE, 2019, 10957, 109570D Search PubMed.
- M. C. Sharps, D. A. Marsh, L. N. Zakharov, J. E. Hutchison and D. W. Johnson, Cryst. Res. Technol., 2017, 52, 1700081 CrossRef.
- R. Frederick, T. Diulus, D. Hutchison, M. Nyman and G. S. Herman, ACS Appl. Mater. Interfaces, 2019, 11, 4514–4522 CrossRef CAS PubMed.
- J. T. Diulus, R. T. Frederick, M. Li, D. C. Hutchison, M. R. Olsen, I. Lyubinetsky, L. Árnadóttir, E. L. Garfunkel, M. Nyman, H. Ogasawara and G. S. Herman, ACS Appl. Mater. Interfaces, 2019, 11, 2526–2534 CrossRef CAS PubMed.
- Y. Zhu, M. R. Olsen, M. Nyman, L. Zhang and J. Zhang, Inorg. Chem., 2019, 58, 4534–4539 CrossRef CAS PubMed.
- C. Eychenne-Baron, F. Ribot and C. Sanchez, J. Organomet. Chem., 1998, 567, 137–142 CrossRef CAS.
- J. Haitjema, Y. Zhang, N. Ottosson and A. M. Brouwer, J. Photopolym. Sci. Technol., 2017, 30, 99–102 CrossRef CAS.
- H. Puff and H. Reuter, J. Organomet. Chem., 1989, 373, 173–184 CrossRef CAS.
- R. Fallica, J. K. Stowers, A. Grenville, A. Frommhold, A. P. G. Robinson and Y. Ekinci, J. Micro/Nanolithogr., MEMS, MOEMS, 2016, 15, 033506 CrossRef.
- R. Fallica, J. Haitjema, L. Wu, S. Castellanos, A. M. Brouwer and Y. Ekinci, J. Micro/Nanolithogr., MEMS, MOEMS, 2018, 17, 1 Search PubMed.
- Y. Zhang, J. Haitjema, X. Liu, F. Johansson, A. Lindblad, S. Castellanos, N. Ottosson and A. M. Brouwer, J. Micro/Nanolithogr., MEMS, MOEMS, 2017, 16, 023510 CrossRef.
- J. Haitjema, Y. Zhang, M. Vockenhuber, D. Kazazis, Y. Ekinci and A. M. Brouwer, J. Micro/Nanolithogr., MEMS, MOEMS, 2018, 16, 033510 Search PubMed.
- Y. Zhang, J. Haitjema, M. Baljozovic, M. Vockenhuber, D. Kazazis, T. A. Jung, Y. Ekinci and A. M. Brouwer, J. Photopolym. Sci. Technol., 2018, 31, 249–255 CrossRef CAS.
- M. C. Sharps, R. T. Frederick, M. L. Javitz, G. S. Herman, D. W. Johnson and J. E. Hutchison, Chem. Mater., 2019, 31, 4840–4850 CrossRef CAS.
- W. Hinsberg and S. Meyers, Proc. SPIE, 2017, 1014604 Search PubMed.
- I. Bespalov, Y. Zhang, J. Haitjema, R. Tromp, S. J. van de Molen, A. M. Brouwer, J. Jobst and S. Castellanos, ACS Appl. Mater. Interfaces, 2020, 12, 9881–9889 CrossRef CAS PubMed.
- S. Saha, D.-H. Park, D. C. Hutchison, M. R. Olsen, L. N. Zakharov, D. Marsh, S. Goberna-Ferrón, R. T. Frederick, J. T. Diulus, N. Kenane, G. S. Herman, D. W. Johnson, D. A. Keszler and M. Nyman, Angew. Chem., Int. Ed., 2017, 56, 10140–10144 CrossRef CAS PubMed.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Phys. Chem. Chem. Phys., 2016, 18, 26213–26223 RSC.
- A. Giuliani, A. R. Milosavljevic, F. Canon and L. Nahon, Mass Spectrom. Rev., 2014, 33, 424–441 CrossRef CAS PubMed.
- S. Daly, M. Krstic, A. Giuliani, R. Antoine, L. Nahon, A. Zavras, G. N. Khairallah, V. Bonačić-Koutecký, P. Dugourd and R. A. OHair, Phys. Chem. Chem. Phys., 2015, 17, 25772–25777 RSC.
- J. Zhen, S. R. Castillo, C. Joblin, G. Mulas, H. Sabbah, A. Giuliani, L. Nahon, S. Martin, J.-P. Champeaux and P. M. Mayer, Astrophys. J., 2016, 822, 1–8 CrossRef.
- L. Wu, M. Tiekink, A. Giuliani, L. Nahon and S. Castellanos, J. Mater. Chem. C, 2018, 7, 33–37 RSC.
- N. Thakur, A. Giuliani, L. Nahon and S. Castellanos, J. Photopolym. Sci. Technol., 2020, 33, 153–158 CrossRef CAS.
- D. Dakternieks, H. Zhu, E. R. T. Tiekink and R. Colton, J. Organomet. Chem., 1994, 476, 33–40 CrossRef CAS.
- J. Haitjema, L. Wu, A. Giuliani, L. Nahon, S. Castellanos and A. M. Brouwer, J. Photopolym. Sci. Technol., 2018, 31, 243–247 CrossRef CAS.
- Y. Zhang, J. Haitjema, S. Castellanos, O. Lugier, N. Sadegh, R. Ovsyannikov, E. Giangrisostomi, F. O. L. Johansson, E. Berggren, A. Lindblad and A. M. Brouwer, Appl. Phys. Lett., 2021, 118, 171903 CrossRef CAS.
- C. Eychenne-Baron, F. Ribot, N. Steunou, C. Sanchez, F. Fayon, M. Biesemans, J. C. Martins and R. Willem, Organometallics, 2000, 19, 1940–1949 CrossRef CAS.
- G. Prabusankar, B. Jousseaume, T. Toupance and H. Allouchi, Dalton Trans., 2007, 3121–3123 RSC.
- P. Thuéry, Y. Atoini and J. Harrowfield, Dalton Trans., 2019, 48, 8756–8772 RSC.
- Y. Zhang, PhD thesis, University of Amsterdam, 2019.
- O. Kostko, B. Xu, M. Ahmed, D. S. Slaughter, F. Ogletree, K. Closser, D. Prendergast, P. Naulleau, D. Olynick, P. Ashby, Y. Liu, W. Hinsberg and G. Wallraff, J. Chem. Phys., 2018, 149, 154305 CrossRef PubMed.
- J. C. Fuggle and N. Mårtensson, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 275–281 CrossRef CAS.
- F. Banse, P. Toledano, J. Maquet and C. Sanchez, Inorg. Chem., 1995, 34, 6371–6379 CrossRef CAS.
- L. Van Lokeren, R. Willem, D. van der Beek, P. Davidson, G. A. Morris and F. Ribot, J. Phys. Chem. C, 2010, 114, 16087–16091 CrossRef CAS.
- A. R. Milosavljević, C. Nicolas, J.-F. Gil, F. Canon, M. Réfrégiers, L. Nahon and A. Giuliani, J. Synchrotron Radiat., 2012, 19, 174–178 CrossRef PubMed.
- L. Nahon, N. de Oliveira, G. A. Garcia, J.-F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, 2016 Search PubMed.
- J. H. Ma, H. Wang, D. Prendergast, A. Neureuther and P. Naulleau, Proc. SPIE, 2020, 11323, 113231F Search PubMed.
- E. W. Abel, G. A. Stone and G. Wilkinson, Comprehensive Organometallic Chemistry II, Pergamon, 1995, p. 357 Search PubMed.
- B. L. Henke, E. M. Gullikson and J. C. Davis, At. Data Nucl. Data Tables, 1993, 54, 181–342 CrossRef CAS.
- R. Feng, G. Cooper and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 2002, 123, 199–209 CrossRef CAS.
- N. Sadegh, M. van der Geest, J. Haitjema, F. Campi, S. Castellanos, P. M. Kraus and A. M. Brouwer, J. Photopolym. Sci. Technol., 2020, 33, 145–151 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectra, details of computational results, computed structures. See DOI: 10.1039/d1cp03148a |
| ‡ Relative to the model in which the counterion is bound to the opposite side of the molecule. |
| This journal is © the Owner Societies 2021 |