
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNa-ion mobility in P2-type Na0.5MgxNi0.17−xMn0.83O2 (0 ≤ x ≤ 0.07) from electrochemical and muon spin relaxation studies†
Le Anh
Ma
 *a,
Rasmus
Palm
b,
Elisabetta
Nocerino
b,
Ola Kenji
Forslund
b,
Nami
Matsubara
b,
Stephen
Cottrell
c,
Koji
Yokoyama
c,
Akihiro
Koda
d,
Jun
Sugiyama
ef,
Yasmine
Sassa
g,
Martin
Månsson
b and
Reza
Younesi
a
*a,
Rasmus
Palm
b,
Elisabetta
Nocerino
b,
Ola Kenji
Forslund
b,
Nami
Matsubara
b,
Stephen
Cottrell
c,
Koji
Yokoyama
c,
Akihiro
Koda
d,
Jun
Sugiyama
ef,
Yasmine
Sassa
g,
Martin
Månsson
b and
Reza
Younesi
a
aDepartment of Chemistry, Ångström Laboratory, Uppsala, Sweden. E-mail: le.anh.ma@kemi.uu.se; reza.younesi@kemi.uu.se
bDepartment of Applied Physics, KTH Royal Institute of Technology, SE-10691 Stockholm, Sweden
cISIS Pulsed Neutron and Muon Facility, STFC Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0QX, UK
dHigh Energy Accelerator Research Organization (KEK), Tokai, Ibaraki 319-1106, Japan
eNeutron Science and Technology Center, Comprehensive Research Organization for Science and Society (CROSS), Tokai, Ibaraki 319-1106, Japan
fAdvanced Science Research Center, Japan Atomic Energy Agency, Tokai, Ibaraki 319-1195, Japan
gDepartment of Physics, Chalmers University of Technology, 41296 Gothenburg, Sweden
First published on 5th October 2021
Abstract
Sodium transition metal oxides with a layered structure are one of the most widely studied cathode materials for Na+-ion batteries. Since the mobility of Na+ in such cathode materials is a key factor that governs the performance of material, electrochemical and muon spin rotation and relaxation techniques are here used to reveal the Na+-ion mobility in a P2-type Na0.5MgxNi0.17−xMn0.83O2 (x = 0, 0.02, 0.05 and 0.07) cathode material. Combining electrochemical techniques such as galvanostatic cycling, cyclic voltammetry, and the galvanostatic intermittent titration technique with μ+SR, we have successfully extracted both self-diffusion and chemical-diffusion under a potential gradient, which are essential to understand the electrode material from an atomic-scale viewpoint. The results indicate that a small amount of Mg substitution has strong effects on the cycling performance and the Na+ mobility. Amongst the tested cathode systems, it was found that the composition with a Mg content of x = 0.02 resulted in the best cycling stability and highest Na+ mobility based on electrochemical and μ+SR results. The current study clearly shows that for developing a new generation of sustainable energy-storage devices, it is crucial to study and understand both the structure as well as dynamics of ions in the material on an atomic level.
Introduction
With the increased demand in energy-storage technologies, rechargeable lithium-ion batteries (LIBs) have become a major component in the electronics industry as well as within the electrified transport sector. One main concern about LIBs is that Li resources are threatened to be exhausted in the future.1,2 Due to their cost effectiveness and abundance, sodium-ion batteries (SIBs) have become an interesting sustainable alternative. A major challenge in SIB research is to find suitable electrode materials with a good electrochemical performance, such as long-term cycling stability and high rate capability.3Amongst various cathode materials in SIBs,4–9 layered transition metal oxides of NaMO2-type (M = transition metals), especially manganese-based oxides, are the most widely studied materials due to the enhanced stability caused by the stable Mg ions.3,10 Here, layered oxides can be categorized into two main groups: O-type and P-type, depending on the Na+ coordination in the crystal structure with an appended number of unique oxide layer stacks consisting of edge-shared MO6-octahedra. The nomenclature of such layered oxides was first defined by Delmas et al.11 O-type oxides represent a structure with octahedral Na+ coordination, whereas P-type oxides have a prismatic coordination. If the appended number is 2, the metal oxide stacking in the unit cell along the c-axis is ABBA, and if the appended number is 3, the stacking in the unit cell is ABCABC. Hence, these are the two main types of sodium layered oxides: P2- and O3-type phases. During electrochemical cycling, both P2- and O3-types undergo phase transitions by MO2 slab-gliding. Materials belonging to the O3-type undergo different stages of phase transitions during desodiation, that is, O3–P3–O′3–P′3.12 In comparison with this, P2-phase materials undergo fewer phase transitions and only change into the O2-phase during desodiation. Even though O3-type NaxMO2 exhibits a higher capacity due to the higher Na content in the structure (x = 1), P2 structures have a better cyclability and rate performance due to the fewer phase transitions and higher Na diffusion.3,13
Manganese-based P2-NaxMnO2 (x < 1) is an attractive cathode material for SIBs, because of the low-cost and environmentally friendly properties of manganese.14 One drawback of some P2-structures is the P2–O2 phase transition above 4.0 V vs. Na+/Na, which leads to degradation and hence a shorter life time.15,16 Degradation at high voltages can be prevented using either a lower cut-off potential or by doping with different metals, such as Mg, Ni, Co and Fe.17–19 In 2001, P2-Na2/3Ni1/3Mn2/3O2 was first introduced by Dahn and Lu.20 Later studies have shown that by introducing magnesium into P2-type Na–Ni–Mn–O systems, a better rate capability and reversibility can be obtained. Compared with undoped P2-Na–Ni–Mn–O systems, Mg-doped systems undergo P2–OP4 phase transitions at high voltages, which are more reversible and stable than P2–O2 transitions.21–27
There have been a variety of studies focusing on the structural changes and electrochemistry of Mg-doped P2-Na–Ni–Mn–O cathode systems. For example, Bruce et al. studied the structural evolution of P2-Na2/3MgxNi1/3−xMn2/3O2 with different Mg contents (x = 0, 0.05, 0.1 and 0.2) at higher voltages and combined with ab initio studies, showing the stability and reversibility of the OP4 phase, which has an alternate stacking of octahedral and trigonal prismatic Na layers.27 Herein, we investigate different degrees of Mg doping in P2-Na0.5MgxNi0.17−xMn0.83O2 for a smaller range of Mg content (x = 0, 0.02, 0.05 and 0.07). With a variety of electrochemical techniques, such as galvanostatic cycling, cyclic voltammetry (CV) and the galvanostatic intermittent titration technique (GITT), we discuss the effects of Mg doping on the long-term cycle life and the diffusion coefficient and hence the Na-ion mobility.
Another complementary and novel technique for studying the ion mobility in energy-related materials is muon spin rotation and relaxation (μ+SR). Here, the muon is an elementary particle with a spin (S = 1/2) that is very susceptible to magnetic fields due to the muon's exceptionally large gyromagnetic ratio, γμ = 135.5 MHz T−1. This makes μ+SR a unique and extremely sensitive (fractions of a Gauss) probe of both electronic as well as nuclear magnetic moments and fields.28 Traditionally, the μ+SR technique has been used extensively for studies of correlated electron physics, e.g., magnetism29–33 and superconductivity.34,35 However, lately, the scientific scope of this technique has broadened significantly. About a decade ago the targeted application for studies of ion diffusion in energy-related materials using μ+SR was initiated through a collaboration between academia and industry when the first systematic study of Li-ion diffusion in the archetypal battery cathode material LixCoO2 was presented.36 Since then, μ+SR has been utilized to study charge dynamics within a broad range of Li/Na/K-ion battery cathodes,37–40 anodes41,42 and solid electrolytes,43,44 as well as hydrogen diffusion/desorption in H-storage materials,45,46 along with ion movement in photovoltaic materials.47 Hence, μ+SR has become a novel and unique method in the area of sustainable energy materials.
Experimental methods
Polycrystalline Na0.5MgxNi0.17−xMn0.83O2 (x = 0, 0.02, 0.05 and 0.07) samples were prepared via solid-state reaction of Na2CO3, NiO, Mn2O3 and MgO in their respective ratios. The precursors were mixed using a planetary ball mill (PM 100 RETSCH) using two 20 mm zirconia balls (30 g) for 30 min. The mixture was then heated at 900 °C for 12 h in air and slowly cooled to room temperature. The structure and quality of the samples were verified using in-house X-ray diffraction (XRD) measurements (see ESI†). XRD data analysis and Rietveld refinements were performed using the software tools from the FullProf Suite.48Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed using an Avio 200 instrument. The powder samples were dissolved via acid digestion and diluted in ICP-grade water. A multi-standard solution (Multi-Element Calibration Standard 3, PerkinElmer, 5% HNO3) was used for calibration.
The electrodes were prepared by mixing the active material with conductive carbon (Super P) and polyvinyldene fluoride (PVdF Kynar Flex) as binder in a (weight or volume) ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, respectively. Drops of N-methyl-2-pyrrolidone (NMP; Sigma Aldrich) were added to the slurry mix, which was ball-milled at 600 rpm for 2 h. The slurry was cast onto a carbon-coated Al foil using the doctor-blade technique, dried at 80 °C and punched into 13-mm-diameter electrode discs. The discs were then introduced into an argon-filled glovebox (H2O <1 ppm and O2 <1 ppm) and dried under vacuum at 120 °C for 12 h to remove any residues of solvent and moisture. The average mass loading of the final electrode is 3.2 mg cm−2. Half-cells were assembled in polyethylene-coated aluminum pouch cells, in which Na metal (Sigma Aldrich) on Al foil was used as the counter electrode with Solupor® as the separator, Al as the current collector and 150 μL of 1 M NaPF6 (Stella) in PC (BASF) as the electrolyte. The cell assembly was carried out in an argon-filled glovebox (H2O <1 ppm and O2 <1 ppm). All cells were rested for 4 h before cycling.
:1:1, respectively. Drops of N-methyl-2-pyrrolidone (NMP; Sigma Aldrich) were added to the slurry mix, which was ball-milled at 600 rpm for 2 h. The slurry was cast onto a carbon-coated Al foil using the doctor-blade technique, dried at 80 °C and punched into 13-mm-diameter electrode discs. The discs were then introduced into an argon-filled glovebox (H2O <1 ppm and O2 <1 ppm) and dried under vacuum at 120 °C for 12 h to remove any residues of solvent and moisture. The average mass loading of the final electrode is 3.2 mg cm−2. Half-cells were assembled in polyethylene-coated aluminum pouch cells, in which Na metal (Sigma Aldrich) on Al foil was used as the counter electrode with Solupor® as the separator, Al as the current collector and 150 μL of 1 M NaPF6 (Stella) in PC (BASF) as the electrolyte. The cell assembly was carried out in an argon-filled glovebox (H2O <1 ppm and O2 <1 ppm). All cells were rested for 4 h before cycling.
Galvanostatic testing was performed using the Arbin cycling system (USA). As metallic Na was used as the counter electrode, all voltages used are vs. Na+/Na. Cycling tests were conducted in a voltage window of 2.0–4.5 V vs. Na+/Na for 8 cycles at 0.1C at room temperature. The 0.1C rate here is assumed for 10 h charge or discharge, i.e., desodiation or sodiation, respectively.
CV measurements were performed using a Bio-Logic MPG2 instrument. The cells were cycled for 10 cycles in a voltage window of 2.0–4.5 V vs. Na+/Na at different scan rates of 0.1 mV s−1, 0.2 mV s−1 and 0.5 mV s−1.
GITT measurements were conducted on a Bio-Logic MPG2 instrument by applying a current pulse of 2 μA mg−1 for 30 min, followed by a rest of 5 h. The rest periods were extended to 7 or 10 h, if the steady state potential was not reached.
The μ+SR measurements were performed using both the EMU instrument at the ISIS pulsed muon source in the UK as well as the S1 beamline of J-PARC MLF in Japan. The μ+SR time spectra were collected in the temperature range T = 50–400 K using a closed-cycle refrigerator (CCR). At each temperature, the μ+SR spectra were measured under the series of zero field (ZF), longitudinal field (LF = 5, 20 and 40 G) and transverse field (TF = 20 G), where LF (TF) is the external magnetic field applied parallel (perpendicular) to the initial muon spin polarization. This protocol was employed in order to conduct a robust global fitting procedure leading to an accurate determination of the Na+-ion hopping rate. For these experiments, approximately m = 1.5 g of pristine (non-cycled) powder sample was pressed into a pellet for each μ+SR measurement. Further detailed descriptions of the μ+SR experimental setup and procedures can be found in the ESI† as well as in the references.29,36,37
Results and discussion
The stoichiometry of all the samples was determined via ICP-OES (see Table S1 in the ESI†). The crystal structure and sample quality were verified using XRD analysis (the crystallographic structure and parameters are shown in Fig. 1 with detailed data and analysis presented in the ESI,† Fig. S1).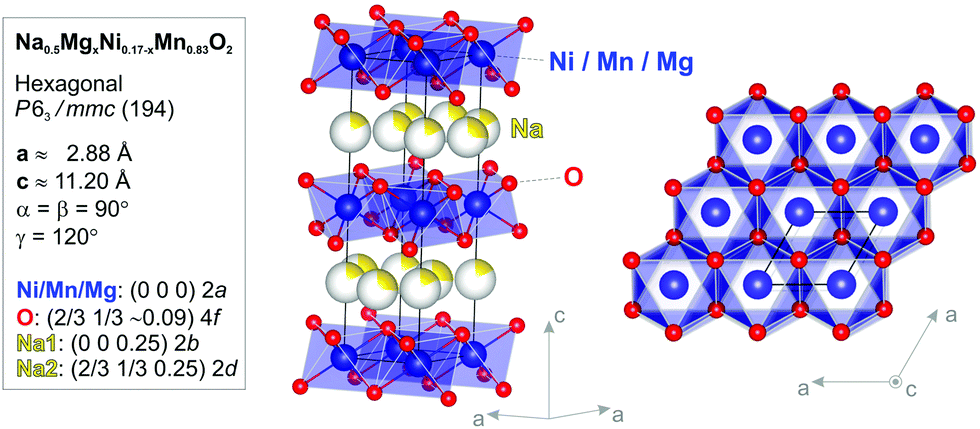 | ||
| Fig. 1 Atomic structure model (side- and top-view) as well as the crystallographic parameters and atomic positions for the Na0.5MgxNi0.17−xMn0.83O2 compound determined via Rietveld analysis of the RT-XRD pattern based on the x = 0 structure. The Mg atoms will be substituting Ni into the 2a lattice sites. The yellow partial shading of the Na atoms represents the fractional (2d) site occupancy. Also note the presence of two distinct Na1 (2b) and Na2 (2d) sites. | ||
Fig. 2 shows the galvanostatic performance of the different samples cycled at 0.1C in the voltage range of 2–4.5 V. The undoped sample shown in Fig. 2a, P2-Na0.5Ni0.17Mn0.87O2, exhibits three plateaus at 2.8 V, 3.7 V and 4.2 V, which have been reported before.16 The initial charge capacity was 150 mAh g−1, corresponding to 0.55 Na removal theoretically, which is more than the original Na content from the formula unit. There are different explanations for the additional capacity: first, at higher voltages, the material undergoes a phase transformation from P2 to the O2 phase, as shown in an earlier study,16 yielding more capacity; second, the extra capacity is caused by oxygen redox participation, as reported previously for similar cathode materials,22,49–52 and electrolyte decomposition at higher voltages, as reported before.22,50 With continued cycling, the plateau above 4.2 V disappeared, which indicates that processes at higher voltages decrease due to their irreversibility and instability. Fig. 2 shows the galvanostatic cycling of the cathode materials with different degrees of Mg doping, x = 0.02, 0.05 and 0.07. Previous reports have shown that doping with Mg enhances the sample's cycling stability by forming a more stable phase, the OP4 phase instead of the O2 phase at higher voltage ranges.22,53 The Mg-doped sample x = 0.02 had a lower initial charge capacity, ∼20 mAh g−1 less than in the subsequent cycles (Fig. 2b). This could be attributed to an insufficient amount of Na in the pristine sample, which then was more sodiated during discharge, demonstrating a higher discharge capacity. A similar influence of the Na deficiency can be seen to a smaller extent in the samples x = 0.05 and x = 0.07 in Fig. 2c and d, respectively.
 | ||
| Fig. 2 Galvanostatic measurements cycled at 0.1C in the range of 2–4.5 V for P2-Na0.5MgxNi0.17−xMn0.87O2 with different Mg contents (x = 0–0.07) using NaPF6 in PC as the electrolyte. Galvanostatic charge/discharge curves of (a) Na0.5Ni0.17Mn0.87O2, (b) Na0.5Mg0.02Ni0.15Mn0.87O2, (c) Na0.5Mg0.05Ni0.12Mn0.87O2 and (d) Na0.5Mg0.07Ni0.10Mn0.87O2. (e) Capacity vs. cycle number based on the measurements shown in (a–d) and capacity retention relative to cycle 2 vs. cycle number. The undoped sample exhibits a faster capacity fading compared with Mg-doped samples. | ||
Fig. 2e shows a comparison of the specific capacity amongst all four samples and their cyclability. Due to the Na deficiency in the initial cycle, the capacity retention is calculated relative to the capacity from the second cycle. Generally, all Mg-doped samples have a higher capacity retention than the undoped sample, which is due to the stable formation of the reversible OP4 phase at higher voltages compared with the irreversible O2 phase formation in the undoped sample.22,53 The sample x = 0.02 demonstrated the lowest capacity fading, while also exhibiting the highest specific capacity amongst the four samples.
From the CV measurements of the undoped sample, the current peak with the maximum current at 4.2 V decreased as a function of the number of cycles, whereas for Mg-doped samples the peak current remained at similar values (see ESI,† Fig. S2). This is also in agreement with the galvanostatic measurements in Fig. 2a, where for the undoped sample and Mg-doped sample with x = 0.05 and 0.07, the plateau above 4.2 V decreases with the cycle number, implying the disappearance of those high-voltage processes, and for the Mg-doped sample with x = 0.02, the plateau at 4.2 V remains, illustrating reversible processes at 4.2 V. The potential difference of the charge and discharge peak with the maximum current at around 4.2 V could give an indication of the reversibility of the corresponding redox processes. For ideal reversible diffusion-controlled processes, the peak potential difference Ediff is 0.059 V.54 If the peak potential difference is higher than 0.059 V, the process is quasi-reversible. The smaller the Ediff, the more reversible the redox processes. Fig. 3 shows the peak potential difference at ∼4.2 V plotted against the number of cycles for all four samples based on the CV measurements in the ESI† in Fig. S2. The Ediff values in the samples with x = 0.05 and x = 0.07 for all the cycles were relatively high compared with x = 0 and x = 0.02, indicating more irreversible processes at higher voltages for high-Mg-content compounds. During the initial cycles, x = 0.02 had the lowest Ediff values compared with x = 0, whereas after cycle 5 the Ediff values of x = 0 became slightly lower than for x = 0.02. When comparing these results with the results shown in Fig. 2, the CV results confirm the capacity-retention trend of the Mg-doped samples. However, the peak differences for x = 0 were not comparable to the Mg-doped samples, since its decrease in peak current is not taken into account for Ediff values. Future comparable studies of the atomic structure using XRD and Na+-ion dynamics using the μ+SR of cycled vs. pristine samples should therefore be of high interest.
 | ||
| Fig. 3 Analysis from cyclic voltammetry (CV) results (see also ESI,† Fig. S2). Data plots taken from CV measurements at a scan rate of 0.1 mV s−1 for all samples. The potential difference Ediff values of the peak with the maximum current between the charge and discharge cycle, ∼4.2 V vs. Na+/Na, are plotted as a function of the number of cycles. The Ediff value can give insight into the reversibility of a redox process. The larger Ediff, the more irreversible the redox process is. | ||
One parameter that can affect the cycling performance is the effectiveness of its sodiation/desodiation, which is determined by the Na mobility and therefore the chemical diffusion coefficient. The cycling performance of all the samples was therefore further investigated using GITT to determine the chemical diffusion coefficient of Na. For the GITT measurements a current pulse of 2 μA mg−1 is applied for 30 min, followed by a resting time of 5 h. Fig. 4 shows the initial Na chemical diffusion coefficient (DCNa,in) determined after the first current pulse. The chemical diffusion coefficient DCNa is determined using the equation (eqn (S1)) in the ESI.† The initial mobility of the Na+ ions in sample x = 0.02 was the highest (DCNa = 5.4 × 10−10 cm2 s−1), followed by sample x = 0.07 (DC = 1.1 × 10−10 cm2 s−1) and the undoped sample (DCNa = 7.3 × 10−11 cm2 s−1). The lowest Na+-ion mobility was seen for the sample x = 0.05 (DCNa = 2.7 × 10−11 cm2 s−1).
 | ||
| Fig. 4 Initial chemical diffusion coefficient of Na+-ions determined after the first current pulse based on GITT measurements (at room-temperature) shown in the ESI,† Fig. S3 and analyzed using equation (eqn (S1) in the ESI†). | ||
As mentioned before, the μ+SR technique has recently been broadly utilized to study ion dynamics in energy-related materials. For layered battery cathode materials like the current title compounds, the principle behind this method is straightforward and simple. When the positive muons are implanted into the oxide material, they stop at the energetically favorable position and bind strongly to the negative oxygen atoms. At this position, the muon will mainly “feel” the nuclear magnetic moment of the alkali ion layers, along with a much weaker contribution from the very quickly vibrating paramagnetic (PM) electronic spins. If the alkali ions are static within the muon lifetime, the muon spin will mainly sense a static field distribution (Δ). On the other hand, if the alkali ions become mobile (jumping/diffusing), the muon spin will detect this as a dynamic contribution that is usually referred to as the field fluctuation rate (ν). For many battery materials, ν directly translates into the absolute value of the intrinsic ion-hopping rate, which makes this method unique. By studying the temperature dependence, ν(T), it is possible to extract the activation energy (Ea) for the self-diffusion process. Furthermore, if the jump paths in the crystallographic lattice are known (from, e.g., XRD) or assumed (from, e.g., computer modelling), it is also possible to use ν(T) to calculate the temperature dependence of the self-diffusion coefficient, DJion(T) [cm2 s−1]. The main limitation of using this method is that the ion (isotope) of interest needs to have a nuclear magnetic moment (μnuc), which the muon spin can detect. Hence, for battery materials both lithium [μnuc(7Li) = +3.256 (2) μN]38 and sodium [μnuc(23Na) = +2.217 (2) μN]37,44 are easily accessible while, e.g., potassium [μnuc(39K) = +0.3914(3) μN] is a bit more challenging (but still possible39). Most such studies have been performed on powder samples using so-called bulk μ+SR instruments. However, there is also the possibility of using a low-energy μ+SR (LEM) setup to perform depth-resolved investigations (∼5–300 nm) of thin films as well as across interfaces in multilayer samples.43
In this study, a systematic investigation of the Na+-ion dynamics has been performed on four samples of the P2-Na0.5MgxNi0.17−xMn0.83O2 (x = 0, 0.02, 0.05 and 0.07) compound using μ+SR. For each sample, temperature-dependent (T = 50–400 K) data were acquired and carefully analyzed, as described in great detail within the ESI.† The resulting Na+-ion-hopping rate ν(T,x) is shown below in Fig. 5a–d. For all compositions it is clear that the Na ions already start to become dynamic below room-temperature. The exponential increase with temperature is typical for a thermally activated ion-diffusion process and, hence, ν(T) can be well fitted to the Arrhenius-type equation:55
 | (1) |
 | ||
| Fig. 5 (a–d) Na+-ion-hopping rate ν(T,x) for the different samples extracted from fits to the μ+SR data (open symbols). Dashed blue lines are fits to the Arrhenius-type equation (eqn (1)). The extracted pre-exponential factor (A) and activation energy (Ea) are indicated for each Mg content (x). Dashed grey lines are a guide to the eye for comparing the hopping rates at room temperature. (e–h) Na+-ion-hopping paths and atomic distances along with Na-site occupancies as extracted from Rietveld refinement of the XRD data. | ||
For obtaining a robust extraction of both the A and Ea, it is reasonable and common practice to first investigate the temperature dependence of the ion-hopping rate in an Arrhenius-type plot, i.e., ln(ν) vs. 1000/T. In such a plot, the thermally activated (diffusive) behavior will be clearly visible as a linear dependence, which is evident for all samples shown in Fig. 6a. From a simple linear fit, Ea is robustly extracted from the derivative (slope) of the line. By conducting a second fitting to eqn (1) as shown in Fig. 5a–d, keeping the extracted Ea fixed, a robust determination of also A is possible. For the parent compound (x = 0) such a protocol yields Ea = 101.94 meV and A = 18.18 MHz. Considering the effect of Mg substitution (x), it is clear from both a simple visual inspection (Fig. 5a–d and Fig. 6a) as well as from the fits to eqn (1) (also summarized in Table 1 and Fig. 6b) that the μ+SR results are fully coherent with the GITT measurements (Fig. 4). The Na+-ion dynamics are increased by a small amount (x = 0.02) of Mg substitution, but then drastically decrease again, even below the parent compound (x = 0), for larger x = 0.05 and 0.07. From the Arrhenius fits this is seen as a strong decrease of Ea = 86.35 meV for the x = 0.02 sample. This is then followed by a drastic increase in Ea = 135.38/135.72 meV for the x = 0.05 and 0.07 compositions, respectively. It is noteworthy that both x = 0 and (especially) x = 0.02 samples display a similar or even lower Ea than for the archetypal LixCoO2 cathode material, where μ+SR experiments previously yielded Ea ≈ 100–150 meV.36,37 The x = 0.02 sample even displays a lower activation energy than the well-known Li-ion cathode material LiFePO4, which displayed Ea ≈ 100 meV in earlier μ+SR studies.38,40,56
 | ||
| Fig. 6 (a) Arrhenius-type plot of ln(ν) vs. 1000/T where the diffusive behavior of Na ions at higher temperatures is made visible as a linear dependence. A clear evolution with Mg content (x) is visible and a linear fit (solid lines) to the data allows us to extract the activation energy from the slope of the linear fit (∝ Ea). (b) Schematic view of energetically favorable values of x (green shaded area) based on the values of Ea (black squares/line) and hopping rate ν (red triangles/line) at T = 25 °C determined from the μ+SR experiments. See also Table 1 for detailed values of the parameters, including errors and goodness (χ2) of the fit. | ||
| x = 0 | x = 0.02 | x = 0.05 | x = 0.07 | |
|---|---|---|---|---|
| E a [meV] | 101.94 ± 1.5 | 86.35 ± 1.1 | 135.38 ± 1.0 | 135.72 ± 1.0 |
| A [μs−1] | 18.18 ± 1.06 | 15.86 ± 0.63 | 24.26 ± 0.96 | 23.49 ± 0.80 |
| χ 2 (fit) | 0.00597 | 0.03734 | 0.00094 | 0.00129 |
| ν [μs−1] | 0.3438 | 0.5503 | 0.1249 | 0.1193 |
| μ D JNa [cm2 s−1] | 0.4726 × 10−10 | 0.7623 × 10−10 | 0.1720 × 10−10 | 0.1652 × 10−10 |
As already mentioned above, if the diffusion paths are known it is also possible to use the Na+-ion hopping rate (ν) to calculate the self-diffusion coefficient (μDJNa). This is conducted using the following well-known relationship,57 assuming a vacancy-diffusion mechanism:
 | (2) |
 | ||
| Fig. 7 Calculated Na+-ion self-diffusion coefficients (μDJNa) for different chemical compositions and temperatures as extracted from the μ+SR experiments. Detailed calculated values for μDJNa (x- and T-dependence) are listed in Table S3 of the ESI.† | ||
Comparing the x-dependencies of DCNa presented in Fig. 4 (GITT) and of μDJNa in Fig. 7 (μ+SR) it is evident that x = 0.02 is the most favorable chemical composition with the highest diffusion coefficients. It is also clear that samples with a higher Mg content display a strongly suppressed Na+-ion diffusion. This can be understood by the fact that substitution with the larger Mg will result in a larger disorder of Na within the crystallographic lattice, which has also been reported in a previous study.27 Such an explanation would fit rather well with the fact that Ea strongly increases for the higher Mg content (x). When comparing the absolute values for the diffusion coefficients obtained via GITT (Fig. 4) with those obtained via μ+SR (Fig. 7) it is clear that the latter method yield values that are approximately one order of magnitude lower at room temperature. This is very reasonable because DC = ΘDJ, where Θ is a thermodynamic factor. However, it is very important to point out that that μ+SR directly supplies the intrinsic material properties and the self-diffusion coefficient. On the other hand, the GITT measurements yield data from an actual battery (half) cell, i.e., they yield a characterization of the device (that includes the choice of electrolyte, area of the cathode, etc.) rather than a true intrinsic material property. Consequently, the relative comparison between different samples, temperatures, etc., is fully valid, while the absolute numbers cannot be directly associated without further input data and/or modeling. However, both intrinsic properties and processes in the presence of potential gradients are relevant in order to study the cathode systems critically and therefore highlight the importance of using complementary characterization techniques.58,59
Please note that for the calculation of the self-diffusion coefficients we have used the hopping rates extracted from the Arrhenius fits of the μ+SR data (listed in ESI,† Table S2) in order to achieve a more robust and systematic comparison between the different chemical compositions and temperatures. Furthermore, the jump paths, distances, and site occupancies are based on the XRD results acquired at room temperature. Here, we assume that there are no structural transitions within the temperature range where μDJNa is calculated. To verify the validity of such an assumption, careful temperature-dependent diffraction experiments would have to be conducted, cf. the previous studies of the closely related and well-known NaxCoO2 compound,37,60 which has a similar layered structure along with two distinct Na sites. This is, however, far beyond the scope of the current investigation and will have to be conducted within a future continuation study.
Conclusions
Our systematic and comprehensive study reveals that even a very small amount of Mg substitution has a strong effect on the overall performance of P2-type Na0.5MgxNi0.17−xMn0.83O2. Notably, the Mg substitution of x = 0.02 is the most favorable composition due to its highest capacity retention, the highest redox reversibility based on the peak potential differences, and the highest Na-ion mobility based on GITT and μ+SR measurements. Systematic results from GITT and μ+SR measurements are fully coherent and hence μ+SR is a powerful technique for electrode material studies. The μ+SR technique is a novel method that is clearly able to supply intrinsic material properties including the elusive self-diffusion coefficient. Moreover, it can provide information on Na-ion diffusion as a function of the Mg content and temperature. Compared with electrochemical testing, the results obtained using the μ+SR technique did not require the use of a half-cell system with metallic sodium and an electrolyte. On the other hand, the presence of the electrolyte involves complex reactions at the electrode surface interfaces, such as electrolyte degradation, which are temperature-dependent and could therefore influence the Na-ion diffusion, resulting in a different trend than with μ+SR measurements on the pristine powder.61 In our work, the electrochemical results at room temperature conform with the μ+SR results on the pristine powder. Therefore, our study has shown that μ+SR can be used as a complementary technique to electrochemical analysis to understand the ion mobility and the chemical kinetics of the material, which is crucial in order to optimize electrode materials for Na+-ion batteries.Author contributions
L. A. M. wrote the manuscript that was revised together with all authors. L. A. M. carried out electrochemical measurements and R.P. assisted the evaluation of the results. E. N., 0. K. F., N. M. and M. M. carried out the μ+SR and XRD measurements as well as the analysis. R.P., S. C., K. Y., A. K., J. S., Y. S. and M. M. contributed to the discussion of the μ+SR and XRD results. R. Y. and M. M. were involved in experimental planning and project discussions. All authors helped with overall data interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. A. M. and R. Y. acknowledge funding from the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) via grant 2016-01257 and STandUP for Energy. The KTH team acknowledge financial support from the Swedish Research Council, VR (Dnr. 2016-06955) as well as the Swedish Foundation for Strategic Research (SSF) within the Swedish national graduate school in neutron scattering (SwedNess). Furthermore, Y. S. is financed by the Swedish Research Council, VR (Dnr. 2017-05078) as well as the Chalmers Area of Advance Materials Science. J. S. was supported by the Japan Society for the Promotion Science (JSPS) KAKENHI Grant No. JP18H01863 and JP20K21149. We are grateful for allocation of the muon beamtimes at ISIS (RB1900049) as well as J-PARC (2019A0279) and highly appreciate the excellent technical support during the muon experiments from the staff of both facilities. The work of R. P. was supported by the Estonian Research Council Grant PUTJD957. Finally, the financial support from The High Energy Accelerator Research Organization (KEK) concerning local travelling and accommodation in Japan is acknowledged.References
- J.-M. Tarascon, Is lithium the new gold?, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- G. Alexander, R. E. Allen, A. Atala, W. P. Bowen, A. A. Coley, J. Goodenough, M. I. Katsnelson, E. V. Koonin, M. Krenn, L. S. Madsen, M. Månsson, N. P. Mauranyapin, A. I. Melvin, E. Rasel, L. E. Reichl, R. Yampolskiy, P. B. Yasskin, A. Zeilinger and S. Lidström, The sounds of science – A symphony for many instruments and voices, Phys. Scr., 2020, 95, 062501 CrossRef CAS.
- Y. Li, Y. Lu, C. Zhao, Y. S. Hu, M. M. Titirici, H. Li, X. Huang and L. Chen, Recent advances of electrode materials for low-cost sodium-ion batteries towards practical application for grid energy storage, Energy Storage Mater., 2017, 7, 130–151 CrossRef.
- W. R. Brant, R. Mogensen, S. Colbin, D. O. Ojwang, S. Schmid, L. Häggström, T. Ericsson, A. Jaworski, A. J. Pell and R. Younesi, Selective Control of Composition in Prussian White for Enhanced Material Properties, Chem. Mater., 2019, 31, 7203–7211 CrossRef CAS.
- V. A. Oltean, S. Renault, M. Valvo and D. Brandell, Sustainable materials for sustainable energy storage: Organic Na electrodes, Materials, 2016, 9, 142 CrossRef PubMed.
- M. Valvo, S. Doubaji, I. Saadoune and K. Edström, Pseudocapacitive charge storage properties of Na2/3Co2/3Mn2/9Ni1/9O2 in Na-ion batteries, Electrochim. Acta, 2018, 276, 142–152 CrossRef CAS.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Prussian blue: a new framework of electrode materials for sodium batteries, Chem. Commun., 2012, 48, 6544 RSC.
- S. Guo, H. Yu, Z. Jian, P. Liu, Y. Zhu, X. Guo, M. Chen, M. Ishida and H. Zhou, A high-capacity, low-cost layered sodium manganese oxide material as cathode for sodium-ion batteries, ChemSusChem, 2014, 7, 2115–2119 CrossRef CAS PubMed.
- Y.-S. Hu, L. Chen and H. Pan, Room-temperature stationary sodium-ion batteries for large-scale electric energy storage, Energy Environ. Sci., 2013, 6, 2338–2360 RSC.
- R. J. Clément, P. G. Bruce and C. P. Grey, Review-Manganese-based P2-type transition metal oxides as sodium-ion battery cathode materials, J. Electrochem. Soc., 2015, 162, A2589–A2604 CrossRef.
- C. Delmas, C. Fouassier and P. Hagenmuller, Structural classification and properties of the layered oxides, Phys. B + C, 1980, 99, 81–85 CrossRef CAS.
- C. Delmas, J. Braconnier, C. Fouassier and P. Hagenmuller, Electrochemical intercalation of sodium in NaxCoO2 bronzes, Solid State Ionics, 1981, 3–4, 165–169 CrossRef CAS.
- J. Su, Y. Pei, Z. Yang and X. Wang, First-principles investigation on crystal, electronic structures and Diffusion barriers of NaNi1/3Co1/3Mn1/3O2 for advanced rechargeable Na-ion batteries, Comput. Mater. Sci., 2015, 98, 304–310 CrossRef CAS.
- N. Ortiz-Vitoriano, N. E. Drewett, E. Gonzalo, T. T. Rojo, N. Ortiz Vitoriano, N. E. Drewett, E. Gonzalo and T. T. Rojo, High performance manganese-based layered oxide cathodes: Overcoming the challenges of sodium ion batteries, Energy Environ. Sci., 2017, 10, 1051–1074 RSC.
- Z. Lu and J. R. Dahn, In Situ X-Ray Diffraction Study of P2-Na[sub 2/3][Ni[sub 1/3]Mn[sub 2/3]]O[sub 2], J. Electrochem. Soc., 2001, 148, A1225 CrossRef CAS.
- D. H. Lee, J. Xu and Y. S. Meng, An advanced cathode for Na-ion batteries with high rate and excellent structural stability, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC.
- I. Hasa, S. Passerini and J. Hassoun, Toward high energy density cathode materials for sodium-ion batteries: investigating the beneficial effect of Aluminum doping on the P2-type structure, J. Mater. Chem. A, 2017, 5, 4467–4477 RSC.
- Z. Liu, J. Shen, S. Feng, Y. Huang, D. Wu, F. Li, Y. Zhu, M. Gu, Q. Liu, J. Liu and M. Zhu, Ultralow Volume Change of P2-type Layered Oxide Cathode with Controlled Phase Transition by Regulating Distribution of Na+, Angew. Chem., Int. Ed., 2021, 60, 20960–20969 CrossRef CAS PubMed.
- Z. Liu, X. Xu, S. Ji, L. Zeng, D. Zhang and J. Liu, Recent Progress of P2-Type Layered Transition-Metal Oxide Cathodes for Sodium-Ion Batteries, Chem. – Eur. J., 2020, 26, 7747–7766 CrossRef PubMed.
- Z. Lu and J. R. Dahn, In Situ X-Ray Diffraction Study of P2-Na 2/3[Ni 1/3Mn 2/3]O2, J. Electrochem. Soc., 2001, 148, 1225–1229 CrossRef.
- G. Singh, N. Tapia-Ruiz, J. M. Lopez del Amo, U. Maitra, J. W. Somerville, A. R. Armstrong, J. Martinez de Ilarduya, T. Rojo and P. G. Bruce, High Voltage Mg-Doped Na 0.67 Ni 0.3– x Mg x Mn 0.7 O 2 (x = 0.05, 0.1) Na-Ion Cathodes with Enhanced Stability and Rate Capability Supporting Information, Chem. Mater., 2016, 28, 5087–5094 CrossRef.
- L. A. Ma, F. Massel, A. J. Naylor, L. C. Duda and R. Younesi, Understanding charge compensation mechanisms in Na0.56Mg0.04Ni0.19Mn0.70O2, Commun. Chem., 2019, 2, 125 CrossRef.
- Z. Y. Li, R. Gao, J. Zhang, X. Zhang, Z. Hu and X. Liu, New insights into designing high-rate performance cathode materials for sodium ion batteries by enlarging the slab-spacing of the Na-ion diffusion layer, J. Mater. Chem. A, 2016, 4, 3453–3461 RSC.
- W. Zhao, H. Kirie, A. Tanaka, M. Unno, S. Yamamoto and H. Noguchi, Synthesis of metal ion substituted P2-Na2/3Ni 1/3Mn2/3O2 cathode material with enhanced performance for Na ion batteries, Mater. Lett., 2014, 135, 131–134 CrossRef CAS.
- K. Hemalatha, M. Jayakumar, P. Bera and A. S. Prakash, Improved electrochemical performance of Na 0.67 MnO 2 through Ni and Mg substitution, J. Mater. Chem. A, 2015, 3, 20908–20912 RSC.
- P. F. Wang, Y. You, Y. X. Yin, Y. S. Wang, L. J. Wan, L. Gu and Y. G. Guo, Suppressing the P2–O2 Phase Transition of Na0.67Mn0.67Ni0.33O2by Magnesium Substitution for Improved Sodium-Ion Batteries, Angew. Chem., Int. Ed., 2016, 55, 7445–7449 CrossRef CAS PubMed.
- N. Tapia-Ruiz, W. M. Dose, N. Sharma, H. Chen, J. Heath, J. W. Somerville, U. Maitra, M. S. Islam and P. G. Bruce, High voltage structural evolution and enhanced Na-ion diffusion in P2-Na2/3Ni1/3-: XMgxMn2/3O2 (0 ≤ x ≤ 0.2) cathodes from diffraction, electrochemical and ab initio studies, Energy Environ. Sci., 2018, 11, 1470–1479 RSC.
- A. Yaouanc and P. Dalmas De Reotier, Muon Spin Rotation, Relaxation, and Resonance Applications to Condensed Matter, Oxford University Press, Oxford, 2011 Search PubMed.
- Y. Ikedo, J. Sugiyama, O. Ofer, M. Månsson, H. Sakurai, E. Takayama-Muromachi, E. J. Ansaldo, J. H. Brewer and K. H. Chow, Comparative μ + SR study of the zigzag chain compounds NaMn2O4 & LiMn2O4, J. Phys.: Conf. Ser., 2010, 225, 012017 CrossRef.
- J. Sugiyama, H. Nozaki, M. Månsson, K. Pra, D. Andreica, A. Amato, M. Isobe and Y. Ueda, μ +SR study on ferromagnetic hollandite K 2Cr 8O 16 and Rb 2Cr 8O 16, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 214407 CrossRef.
- O. Ofer, Y. Ikedo, T. Goko, M. Månsson, J. Sugiyama, E. J. Ansaldo, J. H. Brewer, K. H. Chow and H. Sakurai, Magnetic structure of the zigzag chain family NaxCa 1-xV2O4 determined by muon-spin rotation, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094410 CrossRef.
- H. Nozaki, J. Sugiyama, M. Månsson, M. Harada, V. Pomjakushin, V. Sikolenko, A. Cervellino, B. Roessli and H. Sakurai, Incommensurate spin-density-wave order in quasi-one-dimensional metallic antiferromagnet NaV2O4, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 100410 CrossRef.
- O. K. Forslund, K. Papadopoulos, E. Nocerino, G. Morris, B. Hitti, D. Arseneau, V. Pomjakushin, N. Matsubara, J. C. Orain, P. Svedlindh, D. Andreica, S. Jana, J. Sugiyama, M. Månsson and Y. Sassa, Intertwined magnetic sublattices in the double perovskite compound LaSrNiReO6, Phys. Rev. B, 2020, 102, 144409 CrossRef CAS.
- J. Sugiyama, Y. Ikedo, M. Månsson, J. H. Brewer, S. L. Stubbs, E. J. Ansaldo, K. H. Chow, J. S. Lord, H. Ohta, C. Michioka and K. Yoshimura, Magnetic and superconducting nature of Na0.35CoO 2·yH2O and Na0.35CoO2· yD2O investigated by muon-spin spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 214505 CrossRef.
- Y. J. Uemura, G. M. Luke, B. J. Sternlieb, J. H. Brewer, J. F. Carolan, W. N. Hardy, R. Kadono, J. R. Kempton, R. F. Kiefl, S. R. Kreitzman, P. Mulhern, T. M. Riseman, D. L. Williams, B. X. Yang, S. Uchida, H. Takagi, J. Gopalakrishnan, A. W. Sleight, M. A. Subramanian, C. L. Chien, M. Z. Cieplak, G. Xiao, V. Y. Lee, B. W. Statt, C. E. Stronach, W. J. Kossler and X. H. Yu, Universal correlations between Tc and nsm* (Carrier density over effective mass) in High-Tc cuprate cuperconductors, Phys. Rev. Lett., 1989, 62, 2317–2320 CrossRef CAS PubMed.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Li diffusion in lixCoO2 probed by Muon-Spin spectroscopy, Phys. Rev. Lett., 2009, 103, 147601 CrossRef PubMed.
- M. Månsson and J. Sugiyama, Muon-spin relaxation study on Li- and Na-diffusion in solids, Phys. Scr., 2013, 88, 068509 CrossRef.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, Y. Ikedo, Y. Miyake, O. Ofer, M. Månsson, E. J. Ansaldo, K. H. Chow, G. Kobayashi and R. Kanno, Diffusive behavior in LiMPO 4 with M = Fe, Co, Ni probed by muon-spin relaxation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054111 CrossRef.
- N. Matsubara, E. Nocerino, O. K. Forslund, A. Zubayer, K. Papadopoulos, D. Andreica, J. Sugiyama, R. Palm, Z. Guguchia, S. P. Cottrell, T. Kamiyama, T. Saito, A. Kalaboukhov, Y. Sassa, T. Masese and M. Månsson, Magnetism and ion diffusion in honeycomb layered oxide K 2Ni 2TeO 6, Sci. Rep., 2020, 10, 18305 CrossRef CAS PubMed.
- P. Benedek, O. K. Forslund, E. Nocerino, N. Yazdani, N. Matsubara, Y. Sassa, F. Jurànyi, M. Medarde, M. Telling, M. Månsson and V. Wood, Quantifying Diffusion through Interfaces of Lithium-Ion Battery Active Materials, ACS Appl. Mater. Interfaces, 2020, 12, 16243–16249 CrossRef CAS PubMed.
- I. Umegaki, S. Kawauchi, H. Sawada, H. Nozaki, Y. Higuchi, K. Miwa, Y. Kondo, M. Månsson, M. Telling, F. C. Coomer, S. P. Cottrell, T. Sasaki, T. Kobayashi and J. Sugiyama, Li-ion diffusion in Li intercalated graphite C6Li and C12Li probed by μ + SR, Phys. Chem. Chem. Phys., 2017, 19, 19058–19066 RSC.
- I. Umegaki, Y. Higuchi, H. Nozaki, Y. Kondo, H. Oka, Y. Makimura, O. K. Forslund, M. Månsson, S. P. Cottrell and J. Sugiyama, Battery Materials Research with Muon Beam, JPS Conf. Proc., 2019, 25, 011009 Search PubMed.
- J. Sugiyama, H. Nozaki, I. Umegaki, K. Mukai, K. Miwa, S. Shiraki, T. Hitosugi, A. Suter, T. Prokscha, Z. Salman, J. S. Lord and M. Månsson, Li-ion diffusion in Li4Ti5 O12 and LiTi2 O4 battery materials detected by muon spin spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 014417 CrossRef.
- M. Månsson, I. Umegaki, H. Nozaki, Y. Higuchi, I. Kawasaki, I. Watanabe, H. Sakurai and J. Sugiyama, Na-ion dynamics in Quasi-1D compound NaV2O4, J. Phys.: Conf. Ser., 2014, 551, 012035 CrossRef.
- J. Sugiyama, Y. Ikedo, T. Noritake, K. Miwa, S. Towata, T. Goko, O. Ofer, M. Månsson, E. J. Ansaldo, J. H. Brewer and K. H. Chow, Microscopic indicator for thermodynamic stability of hydrogen storage materials provided by muon-spin spectroscopy, J. Phys.: Conf. Ser., 2010, 225, 012051 CrossRef.
- J. Sugiyama, I. Umegaki, M. Matsumoto, K. Miwa, H. Nozaki, Y. Higuchi, T. Noritake, O. K. Forslund, M. Månsson, S. P. Cottrell, A. Koda, E. J. Ansaldo and J. H. Brewer, Desorption reaction in MgH2 studied with in situ μ + SR, Sustain, Energy Fuels, 2019, 3, 956–964 CAS.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Partial cation substitution reduces iodide ion transport in lead iodide perovskite solar cells, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- J. Rodríguez-Carvajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192, 55–69 CrossRef.
- E. J. Kim, L. A. Ma, L. C. Duda, D. M. Pickup, A. V. Chadwick, R. Younesi, J. T. S. Irvine and A. Robert Armstrong, Oxygen Redox Activity through a Reductive Coupling Mechanism in the P3-Type Nickel-Doped Sodium Manganese Oxide, ACS Appl. Energy Mater., 2020, 3, 184–191 CrossRef CAS.
- C. Hakim, N. Sabi, L. A. Ma, M. Dahbi, D. Brandell, K. Edström, L. C. Duda, I. Saadoune and R. Younesi, Understanding the redox process upon electrochemical cycling of the P2-Na0.78Co1/2Mn1/3Ni1/6O2 electrode material for sodium-ion batteries, Commun. Chem., 2020, 3, 9 CrossRef CAS.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Oxygen redox chemistry without excess alkali-metal ions in Na 2/3 [Mg 0.28 Mn 0.72]O 2, Nat. Chem., 2018, 10, 288–295 CrossRef CAS PubMed.
- C. Ma, J. Alvarado, J. Xu, R. J. Clement, M. Kodur, W. Tong, C. P. Grey, Y. S. Meng, M. Kodur, W. Tong, C. P. Grey and Y. S. Meng, Exploring Oxygen Activity in the High Energy P2-Type Na0.78Ni0.23Mn0.69O2 Cathode Material for Na-Ion Batteries, J. Am. Chem. Soc., 2017, 139, 4835–4845 CrossRef CAS PubMed.
- G. Singh, N. Tapia-Ruiz, J. M. Lopez Del Amo, U. Maitra, J. W. Somerville, A. R. Armstrong, J. Martinez De Ilarduya, T. Rojo and P. G. Bruce, High Voltage Mg-Doped Na0.67Ni0.3-xMgxMn0.7O2(x = 0.05, 0.1) Na-Ion Cathodes with Enhanced Stability and Rate Capability, Chem. Mater., 2016, 28, 5087–5094 CrossRef.
- T. B. Reddy and D. Linden, Linden's Handbook of Batteries, Mc Graw-Hill, 4th edn, 2011 Search PubMed.
- K. J. Laidler, The development of the Arrhenius equation, J. Chem. Educ., 1984, 61, 494–498 CrossRef CAS.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, O. Ofer, M. Månsson, J. H. Brewer, E. J. Ansaldo, K. H. Chow, Y. Ikedo, Y. Miyake, K. Ohishi, I. Watanabe, G. Kobayashi and R. Kanno, Magnetic and diffusive nature of LiFePO4 investigated by muon spin rotation and relaxation, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 054430 CrossRef.
- R. J. Borg and G. J. Dienes, An Introduction to Solid State Diffusion, Elsevier, 1988 Search PubMed.
- L. A. Montoro and J. M. Rosolen, The role of structural and electronic alterations on the lithium diffusion in LixCo0.5Ni0.5O2, Electrochim. Acta, 2004, 49, 3243–3249 CrossRef CAS.
- A. Dunst, M. Sternad, V. Epp and M. Wilkening, Fast Li+ Self-Diffusion in Amorphous Li-Si Electrochemically Prepared from Semiconductor Grade, Monocrystalline Silicon: Insights from Spin-Locking Nuclear Magnetic Relaxometry, J. Phys. Chem. C, 2015, 119, 12183–12192 CrossRef CAS.
- M. Medarde, M. Mena, J. L. Gavilano, E. Pomjakushina, J. Sugiyama, K. Kamazawa, V. Y. Pomjakushin, D. Sheptyakov, B. Batlogg, H. R. Ott, M. Månsson and F. Juranyi, 1D to 2D Na+ ion diffusion inherently linked to structural transitions in Na0.7CoO2, Phys. Rev. Lett., 2013, 110, 266401 CrossRef CAS PubMed.
- F. Leng, C. M. Tan and M. Pecht, Effect of Temperature on the Aging rate of Li Ion Battery Operating above Room Temperature, Sci. Rep., 2015, 5, 12967 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03115e |
| This journal is © the Owner Societies 2021 |