
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A computational investigation of the adsorption of small copper clusters on the CeO2(110) surface†
Rui
Zhang
 *a,
Arunabhiram
Chutia
*b,
Alexey A.
Sokol
c,
David
Chadwick
a and
C. Richard A.
Catlow
cd
*a,
Arunabhiram
Chutia
*b,
Alexey A.
Sokol
c,
David
Chadwick
a and
C. Richard A.
Catlow
cd
aDept of Chemical Engineering, Imperial College London, South Kensington Campus, London SW7 2AZ, UK. E-mail: rui.zhang13@imperial.ac.uk
bSchool of Chemistry, University of Lincoln, Lincoln LN6 7TS, UK. E-mail: achutia@lincoln.ac.uk
cDept of Chemistry, University College London, 20 Gordon St., London WC1H 0AJ, UK
dSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 1AT, UK
First published on 23rd August 2021
Abstract
We report a detailed density functional theory (DFT) study of the geometrical and electronic properties, and the growth mechanism of a Cun (n = 1–4) cluster on a stoichiometric, and especially on a defective CeO2(110) surface with one surface oxygen vacancy, without using pre-assumed gas-phase Cun cluster shapes. This gives new and valuable theoretical insight into experimental work regarding debatable active sites of promising CuOx/CeO2-nanorod catalysts in many reactions. We demonstrate that CeO2(110) is highly reducible upon Cun adsorption, with electron transfer from Cun clusters, and that a Cun cluster grows along the long bridge sites until Cu3, so that each Cu atom can interact strongly with surface oxygen ions at these sites, forming stable structures on both stoichiometric and defective CeO2(110) surface. Cu–Cu interactions are, however, limited, since Cu atoms are distant from each other, inhibiting the formation of Cu–Cu bonds. This monolayer then begins to grow into a bilayer as seen in the Cu3 to Cu4 transition, with long-bridge site Cu as anchoring sites. Our calculations on Cu4 adsorption reveal a Cu bilayer rich in Cu+ species at the Cu–O interface.
1. Introduction
Ceria-based catalysts have been widely studied in the past thirty years,1,2 stimulated by their successful applications, for example as a promoter in the automotive three-way catalysts (TWCs).3 Ceria (CeO2) crystals have a face-centred cubic fluorite structure, characterised by three low-index facets (100), (110), and (111). The material has a high oxygen storage capacity (OSC) as it can easily shift between Ce4+ and Ce3+, forming bulk and surface oxygen vacancies with consequent high reducibility,4 which is further enhanced in well-defined ceria nanostructures, such as nanoparticles, nanorods, and nanocubes which expose (111), (110)/(100), and (100) surfaces, respectively.5–7 Consequently, nanostructured ceria-based catalysts, such as CuOx/CeO2 catalysts, are active in many reactions, for example, the water gas shift (WGS) reaction8,9 and CO oxidation.10,11The structure and properties of CuOx/CeO2 catalysts have been widely studied. Chen et al. used high angle annular dark field scanning transmission electron microscope (HAADF-STEM) and in situ infra-red spectroscopy, as well as density functional theory (DFT) calculations to provide experimental and theoretical evidence of a Cu bilayer on a CeO2(111) surface.8 A top layer of Cu0 atoms were bonded with a bottom layer of mainly Cu+ ions, which in turn were bonded with surface oxygen vacancies (in a Cu+–Ov–Ce3+ form). This copper-ceria interfacial perimeter was identified as the active site for WGS. Kang et al. recently reported experimental and theoretical evidence of an active atomic [Cu(I)O2]3− site for CO oxidation which dynamically changed to/from [Cu(II)O4]6−via an electrophilic [Cu(II)O2(η2-O2)]4− intermediate on the CeO2(111) surface, both of which had a lower HOMO energy compared to Cu clusters on the surface.10
Besides these combined experimental and theoretical studies, there are several computational studies focusing mainly on the atomic and electronic structures of Cu/CeO2(111) (since CeO2(111) is the most stable surface12), employing density functional theory (DFT), commonly the DFT+U approach, in which an effective Hubbard Ueff parameter is used to consider on-site Coulomb repulsions. For example, Szabová et al. reported their most stable Cu/CeO2 structure with one oxidised Cu+ and one reduced surface Ce3+ furthest away from the Cu+, with the nearest neighbour surface oxygen ions bonding closely with the Cu+.13 For a Cu/CeO2−x system, the Cu atom sited above an oxygen vacancy was reduced to Cuδ−. Cu adsorption on surface oxygen vacancies was reported less stable than on a stoichiometric surface, suggesting that Cu nucleation was unlikely on the reduced CeO2(111) surface. Yang et al. calculated that small Cun (n = 1–4) clusters bonding with surface oxygen ions on a stoichiometric CeO2(111) surface, are positively charged and slightly polarised, showing shortened Cu–O distances.14 Cu2 and Cu3 adopted a planar shape, while the two-dimensional (2D) to 3D structural transition was predicted in a Cu4 cluster, because of the comparable strengths of Cu–Cu and Cu–O interactions. Paz-Borbon et al. calculated planar geometries of all Cun (n = 1–5) clusters on a stoichiometric CeO2(111) surface, due to strong Cu–O interactions and charge transfer effects.15 The number of surface Ce3+ ions increased with the Cu cluster size, with a maximum of three electrons transferred from a Cu5 cluster. Regarding ceria surface oxygen vacancies, Jerratsch et al. investigated Ce3+ localisation on a defective CeO2(111) surface with a single oxygen vacancy.16 They found at least one Ce3+ ion was not the nearest neighbour (NN) to the vacancy from both DFT calculations and scanning–tunnelling microscopy (STM).
Recently, Ning et al., based on a detailed H2-temperature programmed reduction (H2-TPR) and X-ray photoelectron spectra (XPS) study, reported that different ceria shapes (particles, rods, and cubes) significantly affected the dispersion and chemical properties of copper species of a CuO/CeO2 catalyst.11 They observed CuOx mono- and bilayer (using HAADF-STEM) as the dominant species, particularly on ceria nanorods, which were rich in Cu+ at the copper-ceria interface (Cu–[Ox]–Ce). The CuO/CeO2-nanorod catalyst had the highest concentration of surface Cu+ and oxygen vacancies, and thus showed a higher activity in CO oxidation, compared to catalysts with other shapes. Their experimental results strongly suggest that CuOx mono- and bilayer are likely to form on CeO2(110) and the copper-ceria interface may be rich in Cu+ and oxygen vacancies. These atomic and electronic features are important for CO oxidation,17 as well as many more reactions such as CO2 hydrogenation to methanol,18 N2O decomposition,6 WGS,9 and NO reduction.19
As CeO2(110) is less stable than CeO2(111), Cu/CeO2(110) is less studied. A number of computational studies are, however, reported. As with Cu adatom adsorption on CeO2(111), a DFT study of Nolan suggested a Cu+ ion and a Ce3+ ion on a CeO2(110) surface with significant local distortion.20 Cui et al. found a Cu+ or a Cu2+ ion on CeO2(110) when locating the Cu adatom at different adsorption sites.21 Recently, Chutia et al. studied in detail the geometric and electronic properties of a Cu adatom adsorbed at different sites on CeO2(110).22 They found the Otop initial structure (Cu on top of a surface O ion) led to the most stable optimised structure, where the Cu was at an O–Ce–O long bridge site, showing one electron transfer and strong Cu–O interactions. Ren et al. later studied the growth mechanism of a Cun (n = 1–5) cluster on CeO2(110).23 They observed a planar rhombus Cu4-p cluster transforming to a 3D tetrahedral Cu4-t cluster on the surface, and thus identified Cu3 as a critical size in Cu nucleation, which however was not favourable on CeO2(110). For a defective CeO2(110) surface, the modelling study of Kullgren et al. reported that the most stable structure had an asymmetrical bridge site, in which one nearest surface oxygen moved towards the vacancy, bridging two nearby surface Ce species, and the Ce3+ ions were localised at an NN and NNN (next-nearest neighbour) position, respectively.24
Considering debatable active sites of promising CuOx/CeO2-nanorod catalysts in many reactions,8,10,11,17,18 being it Cu species with different oxidation states, or the Cu-ceria interface, the understanding of atomic and electronic properties of small Cu clusters, a CuOx mono- and bilayer on CeO2(110), especially on a defective surface and at the copper-ceria interface, is thus of great interest and importance. However, there is a lack of comprehensive study of small Cu cluster morphologies and electronic interactions with CeO2(110) surface. Therefore, in this study, we have conducted systematic DFT calculations to investigate the atomic and electronic properties, and the growth mechanism of a Cun (n = 2–4) cluster on a stoichiometric, and especially on a defective CeO2(110) surface with one surface oxygen vacancy, growing from a Cun−1 cluster with an additional Cu atom placed at different adsorption sites. Our detailed investigation of small Cun (n = 1–4) cluster adsorption on CeO2(110), without using pre-assumed gas-phase Cun cluster shapes, provides fundamental understanding of highly reducible CeO2(110) surface upon Cun adsorption, and strong Cu–surface oxygen interactions with/out a surface oxygen vacancy, being the predominating factor in Cun (n = 1–4) cluster growth on CeO2(110), with relevance to experimental studies of CuOx/CeO2-nanorod catalysts. In the next section we present the theoretical methods employed, which we follow by the results and discussion first of Cun adsorption on a stoichiometric surface, and then on a defective surface. Our study leads to detailed and valuable understanding of structural and electronic properties of a Cun (n = 1–4) cluster adsorbed on stoichiometric and defective CeO2(110) surface, giving theoretical insights into the development of atomistic and electronic properties of a CuOx mono/bilayer at the Cu–O interface on CeO2(110).
2. Computational details
The Vienna Ab initio Simulation Package (VASP) was used to perform all the periodic spin-polarised DFT+U calculations.25–27 Blöchl's projector augmented wave (PAW) method was used to describe the core electrons of all atoms.28 The cut-off energy for the expansion of the plane-wave basis sets was set to 550 eV, with bulk energies converged to within 10−5 eV. A convergence criterion of 0.01 eV Å−1 was chosen for structural optimisation. The Perdew–Burke–Ernzerhof (PBE) version of the generalised gradient approximation (GGA) was used to carry out geometry optimisation and total energy calculations.29 The pristine CeO2(110) surface was modelled by a 3 × 3 supercell with 7 atomic layers in which the bottom four layers were fixed to mimic the bulk of the system. The slab was cut from the bulk CeO2 with a theoretical lattice constant of 5.492 Å, which is close to the experimental value of 5.411 Å. In the direction perpendicular to the surface, a vacuum gap of ∼18 Å was used. In all the calculations, Cu adsorption was only allowed on one of the two surfaces. Therefore, the dipole moment, due to Cun cluster adsorption, was corrected by using the methods proposed by Makov et al. and Neugebauer et al. as implemented in VASP.30,31 A 2 × 2 × 1 k-point sampling grid was employed in all slab calculations, using the Monkhorst–Pack scheme.32 A Hubbard parameter U33–35 in the Dudarev correction form35,36 was added to the energy functional, to correct the self-interaction error due to Ce localised 4f-orbital electrons. In this study, a Ueff value of 5.0 eV was employed for both Ce 4f-orbitals14,20,22,23 and Cu 3d-orbitals,22,37 which could correctly represent electron localisation in Ce 4f and Cu 3d orbitals, respectively. A single Cu atom and a Cu2 cluster in the gas phase were simulated using a 20 × 20 × 20 Å3 cubic cell.A two-stage optimisation procedure, originally proposed by Grau–Crespo38–40 was used to localise electrons in Ce 4f orbitals during CeO2(110) surface reduction, as the localisation is effected by lattice relaxation around the Ce3+ which is the response to the lower charge and larger radius of the Ce3+ compared with Ce4+. To generate this relaxation field Ce ions were replaced with larger La atoms. After geometry optimisation, the La atoms were then replaced by Ce atoms, which now have the appropriate surrounding relaxed configuration needed to localise an electron at the Ce site; the system is then fully geometry optimised.
Bader charges of different atoms were obtained by using the modified Bader charge analysis implemented by Tang et al.41 The Visualisation for Electronic and STructural Analysis (VESTA) package42 was employed to visualise different structures and spin densities.
The adsorption energy per Cu atom, Ead of any given Cun/CeO2(110) structure was calculated as follows,
 | (1) |
For the calculations involving reduced surfaces, the oxygen vacancy formation energy Ev was calculated as follows,
 | (2) |
Eqn (1) was also applied for the adsorption energy calculation of the Cun/CeO2(110)–Ov systems, where the energy of an optimised Cun/CeO2(110)–Ov structure and a relaxed/optimised defective CeO2(110)–Ov surface were used instead of the energy for the stoichiometric surface.
The charge density difference, ρdiff, was calculated by subtracting the sum of the charge densities of a Cun cluster (ρCun) and the ceria surface (ρceria) of the same geometry as the system from the total charge density of the system (ρsys), which is shown as follows.
| ρdiff = ρsys − (ρCun + ρceria) | (3) |
3. Results and discussion
3.1 Adsorption of Cu on CeO2(110) surface
We first reproduced the atomic and electronic investigation of a Cu adatom adsorbed on CeO2(110) at four different adsorption sites,22i.e. on top of a surface Ce atom (Cetop), a surface O atom (Otop), the middle of a surface four-fold hollow site (four-foldhollow) and the middle of a surface O–Ce short bridge site (O–Ceshort bdg). In the optimised structure having the most negative Cu adsorption energy of −3.258 eV, the Cu atom is close to the surface and bonded with two surface O ions on top of a second-layer Ce ion (named as an O–Ce–O long bridge site), which agrees with earlier work.21,22 Results and detailed discussion can be found in ESI,† Section S1.1.3.2 Adsorption of Cu2 on CeO2(110) surface
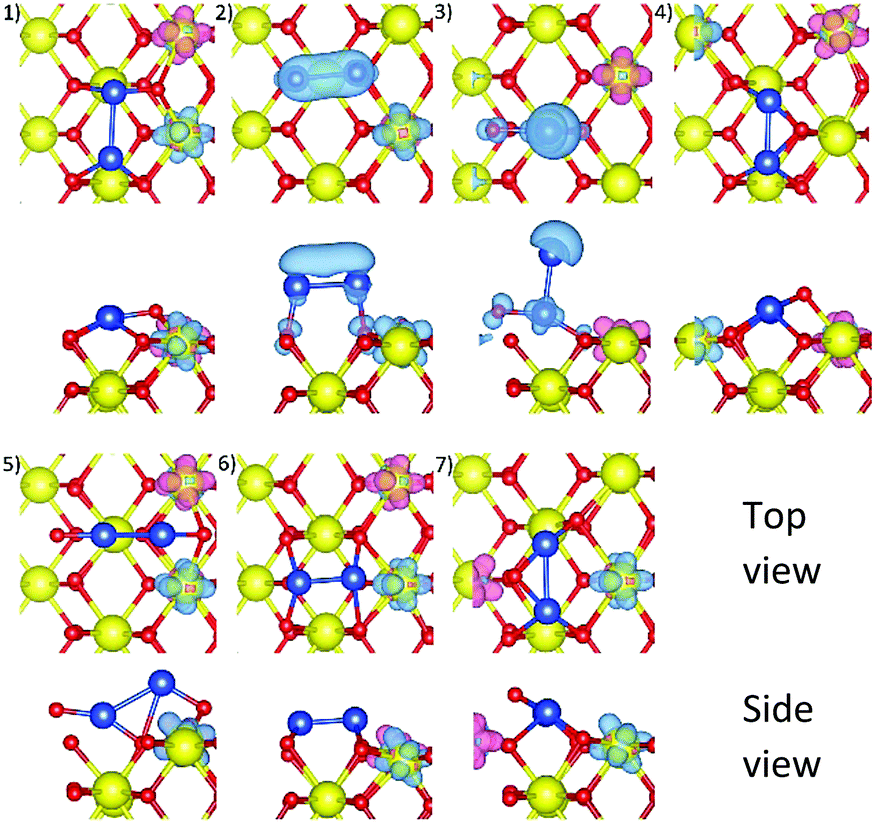
A Cu2 cluster with a Cu–Cu distance of 2.42 Å was placed either around an O–Ce–O long bridge site (Conf1–5) or on top of a second-layer four-fold hollow site (Conf6–7), parallel or perpendicular to the CeO2(110) surface, producing seven initial structures, illustrated in Fig. 1. The O–Ce–O long bridge site was the most stable adsorption site, as found in Section 4.1, therefore, this site and the associated second-layer four-fold hollow site were chosen. | ||
| Fig. 1 Top view and side view of seven initial configurations of a Cu2 cluster placed at different adsorption sites on the CeO2(110) surface, labelled as (1) to (7). Cerium, oxygen, and copper atoms are represented by red, yellow, and blue spheres respectively. | ||
Local surface distortion around the Cu2 cluster is observed in all optimised structures (see Fig. 2), also indicated by the average surface Ce–O bond lengths which are slightly larger than that of a pristine surface (2.342 Å), as listed in Table 1. Only Conf4 and Conf7 show significant structural changes from their corresponding initial structures. For Conf4, the two Cu atoms are bonded with nearby surface O ions at two long bridge sites, respectively, which were initially placed at one long bridge site. The optimised structure of Conf7 is essentially the same as that of Conf4 despite the Cu2 cluster being initially perpendicular to the surface, indicating that formation of a linear Cu2 cluster parallel and close to the surface is favoured. The optimised Conf7 has the most negative Cu adsorption energy, followed by Conf4, Conf1, Conf5, Conf3, Conf2 and Conf6. Therefore, only the most stable Conf7 and metastable Conf4 and 1 are discussed here.
 | ||
| Fig. 2 Top view and side view of optimised Cu2/CeO2(110) structures with spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions, labelled as (1) to (7). Blue: up spin; pink: down spin. | ||
| System | M Cu cluster (µB) | M Ce (µB) | Number of Ce3+ reduced | Cu–O (Å) | Cu–Cu (Å) | (Ce–O)surf (Å) | E ad (eV) |
|---|---|---|---|---|---|---|---|
| Conf1 | 0 | 0.941/−0.969 | 2 | 1.846 | 2.601 | 2.360 | −2.810 |
| Conf2 | 0.329 | 0.966 | 1 | 1.834 | 2.268 | 2.349 | −1.725 |
| Conf3 | 0.326 | −0.968 | 1 | 1.781 | 2.342 | 2.352 | −2.054 |
| Conf4 | 0 | 0.966/−0.963 | 2 | 1.904 | 2.411 | 2.342 | −3.367 |
| Conf5 | 0 | 0.952/−0.952 | 2 | 1.936 | 2.512 | 2.367 | −2.163 |
| Conf6 | 0.022 | 0.859/−0.964 | 2 | 2.033 | 2.175 | 2.369 | −0.973 |
| Conf7 | 0 | 0.968/−0.966 | 2 | 1.908 | 2.478 | 2.345 | −3.492 |
Conf7, 4, and 1 have similar optimised structures, i.e. two Cu atoms bonded at two long bridge sites,23 in which the number of surface O ions available for Cu–O binding is maximised, showing short Cu–O distances in the range of 1.8–1.9 Å (see Table 1) and the most negative adsorption energies at −3.492, −3.367, and −2.810 eV, respectively.
In terms of electronic structures, Conf7, 4, and 1 have two electrons transferred from the Cu2 cluster to the surface, illustrated by the spin density isosurfaces of two reduced Ce3+ ions, Fig. 2, also their distinct magnetic moments in opposite spins (MCe, Table 1), and their Cu2 total magnetic moment being 0.
Different Ce3+ localisation has an impact on the Cu adsorption energy of Conf7, 4 and 1. In Conf7 and 4, the two Ce3+ ions are located at two different surface four-fold hollow sites, opposite to each other, which enables nearby oxygen ions to bind strongly with the Cu atoms, thus stabilising the structure. Differently, in Conf1, the two Ce3+ ions are on the same four-fold hollow site. Since a Ce3+ ion has a larger radius than a Ce4+ ion, the two Ce3+ ions move slightly away from each other, stretching Ce–O bonds and thus limiting movement of the bridging O ion towards its nearest Cu atom.
Besides, slightly different Cu–O interactions of Conf7 and 4 also affects their adsorption energies, though they have similar geometry and Ce3+ localisation. A detailed PDOS analysis was conducted, focusing on orbital interactions between one of the Cu atoms and its bonded three O ions (labelled as O1, O2, O3, in ESI,† Fig. S7). Conf7 and 4 show similar overall signatures (including Cu 4s, 3p and 3d, O 2s and 2p), Fig. 7a and b. They also demonstrate a noticeable overlap between Cu 3d and O 2p orbitals in the range of −6 to −5 eV, which is shown in more detail by PDOS plots of this Cu and its nearest O ion, ESI,† Fig. S7c and d. Additionally, a comparison of the 3d signatures of the Cu2 cluster before (ESI,† Fig. S8a) and after adsorption shows that they are broader in Conf7 as compared to Conf4. Further to this the number of states of O 2p signature in the range of −2 to 0 eV is larger in Conf7 than in Conf4, indicating stronger Cu–O interactions, which suggests why Conf7 has a slightly more negative adsorption energy. The strong Cu–O interactions are also confirmed by a deeper energy of Cu 3d and O 2p orbitals in both configurations than that of Cu 3d in a gaseous Cu2 cluster and O 2p on a pristine CeO2(110) surface (see ESI,† Fig. S8b).
For other configurations with less negative adsorption energies (see ESI,† Section S1.2), the observed weakening in adsorption energies of these configurations is seen to correlate with the decrease in Cu–Cu bond lengths, suggestive of a Coulomb repulsion between Cu atoms in sterically constrained structures. Partial oxidation of Cu2 to a top Cuδ− and a bottom Cu2+ species (Conf3) or two Cuδ+ (Conf2), and partial reduction of Ce4+ to Ce3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 (Conf6), also suggest electronic structures affecting Cu adsorption energy.
21 (Conf6), also suggest electronic structures affecting Cu adsorption energy.
Overall, we find the configuration with the most negative adsorption energy showing two Cu adsorbed at two adjacent long bridge sites, and intriguingly complex electronic structures with varied interactions between Cu species and between Cu and surface ions at different positions.
3.3 Adsorption of Cu3 on CeO2(110) surface
The two stable structures from the Cu2/CeO2(110) system with small adjustments were used to construct eight initial Cu3/CeO2(110) configurations. The third Cu was placed at different adsorption sites on the surface with respect to the Cu2 cluster at different heights from the surface (Fig. 3). | ||
| Fig. 3 Top and side view of eight initial configurations of a Cu3 cluster located at different adsorption sites on the CeO2(110) surface, labelled as (1) to (8). The Cu2 clusters are emphasised by the Cu–Cu bond. | ||
All optimised structures show surface distortion around the Cu atoms, as illustrated in Fig. 4, also shown by the average surface Ce–O bond lengths being larger than that of a pristine surface, as noted in Table 2. The optimised Conf3, 2, and 1 show a linear Cu3 structure23 and the most negative adsorption energies of −3.429 eV, −3.318 eV, and −3.307 eV, respectively. This is because the three Cu atoms are adsorbed at three adjacent long bridge sites, enabling them to bond strongly with at least two nearby surface O ions, showing short Cu–O distances in the range of 1.75–1.90 Å, Table 2, which agree with the calculated values reported by Chutia et al., yet smaller than their experimentally measured values of 1.9–2.4 Å.22 Cu–Cu interactions are weak since Cu atoms are far apart, hardly interacting, except in Conf3. This additional Cu–Cu bond (2.463 Å) thus leads to the most negative Cu adsorption energy of Conf3.
 | ||
| Fig. 4 Top view and side view of optimised Cu3/CeO2(110) structures with spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions, labelled as (1) to (8). | ||
| System | M Cu cluster (µB) | M Ce (µB) | Number of Ce3+ reduced | Cu–O (Å) | Cu–Cu (Å) | (Ce–O)surf (Å) | E ad (eV) |
|---|---|---|---|---|---|---|---|
| Conf1 | 0 | 0.969/0.972/−0.967 | 3 | 1.778 | — | 2.357 | −3.307 |
| Conf2 | 0 | 0.970/0.974/−0.964 | 3 | 1.781 | — | 2.455 | −3.318 |
| Conf3 | 0 | 0.960/0.966/−0.970 | 3 | 1.872 | 2.463 | 2.347 | −3.429 |
| Conf4 | 0 | 0.966/−0.967/−0.935 | 3 | 1.784 | 2.532 | 2.377 | −2.779 |
| Conf5 | 0 | 0.968 | 1 | 1.959 | 2.368 | 2.351 | −2.715 |
| Conf6 | 0 | 0.963/−0.959/−0.968 | 3 | 1.779 | 2.531 | 2.372 | −2.810 |
| Conf7 | 0 | 0.967 | 1 | 1.963 | 2.381 | 2.351 | −2.720 |
| Conf8 | 0 | 0.964 | 1 | 1.963 | 2.382 | 2.351 | −2.605 |
We note that Ren et al. also calculated the adsorption energy per Cu atom of a Cu3 linear cluster (−1.69 eV) on CeO2(110), which was greater than that of a Cu3 triangle cluster (−1.53 eV); yet with a difference of more than 1 eV in absolute values from ours, could that have resulted from different model parameters used, such as supercell size, cut-off energy, force convergence criteria, and k-point sampling.23
The most stable Conf3, and metastable Conf2 and 1, have three electrons transferred from Cu3 to the surface, as illustrated by the spin density isosurfaces around three reduced Ce3+ ions, Fig. 4.
Other configurations have a Cu3 triangle adsorbed on the surface, thus resulting in weak copper–surface oxygen interactions, and weak electronic interactions (see ESI,† Section S1.3), and consequently less negative adsorption energies.
The Cu1–3/CeO2(110) configurations with the most negative Cu adsorption energies suggest that, with an increasing Cu loading, a Cu monolayer grows along the long bridge sites upon Cu adsorption, demonstrated by a Cu adatom growing to a Cu2, and a linear Cu3 cluster at the long bridge sites, agreeing with previous work.23 Our extensive examination of different initial configurations of Cun adsorbed at various adsorption sites, and associated detailed electronic structure investigation, provide insights into the impact of surface Cu–O and Cu–Cu interactions on optimised structures and adsorption energies.
3.4 Adsorption of Cu4 on CeO2(110) surface
Eight initial structures were constructed based on a stable linear Cu3 cluster and a triangular cluster from the Cu3/CeO2(110) system. The fourth Cu atom was placed at different adsorption sites with respect to the Cu3 cluster and at different heights above the surface, Fig. 5. | ||
| Fig. 5 Top view and side view of eight initial configurations of a Cu4 cluster located at different adsorption sites on the CeO2(110) surface, labelled as (1) to (8). The Cu3 clusters are emphasised by the Cu–Cu bonds which may not physically exist. | ||
All optimised structures show surface distortion around the Cu atoms, as illustrated in Fig. 6, also shown by the different values of average surface Ce–O bond length from that of a pristine surface, as listed in Table 3. Conf3, 4, and 1 have the most negative Cu adsorption energies at −2.971, −2.961, and −2.918 eV, respectively, followed by Conf2, Conf7, Conf8, Conf6 and Conf5, Table 3. Therefore, only the former three are discussed here (see ESI,† Section S1.4 for more details).
 | ||
| Fig. 6 Top view and side view of optimised Cu4/CeO2(110) structures with the spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions, labelled as (1) to (8). | ||
 | ||
| Fig. 7 Top view and side view of (0) an initial structure of CeO2(110)–Ov surface (the removed oxygen atom is highlighted using a light pink sphere); (1–3) three optimised CeO2(110) surface structures with spin density isosurfaces of 0.005 e Å−3 around reduced Ce3+ ions which are located at different positions on the surface. | ||
| System | M Cu cluster (µB) | M Ce-total (µB) | M Ce (µB) | Number of Ce3+ reduced | Cu–O (Å) | Cu–Cu (Å) | (Ce–O)surf (Å) | E ad (eV) |
|---|---|---|---|---|---|---|---|---|
| Conf1 | 0.006 | −0.009 | 0.958/0.952/−0.961/−0.958 | 4 | 1.790 | 2.516 | 2.346 | −2.918 |
| Conf2 | 0.577 | −0.954 | 0.967/−0.967/−0.954 | 3 | 1.894 | 2.428 | 2.352 | −2.859 |
| Conf3 | −0.007 | 1.925 | 0.945/0.966/0.965/−0.951 | 4 | 1.868 | 2.495 | 2.369 | −2.971 |
| Conf4 | 0 | 0.009 | 0.970/0.968/−0.964/−0.965 | 4 | 1.844 | 2.417 | 2.364 | −2.961 |
| Conf4-2 | 0.303 | −0.941 | 0.970/−0.949/−0.962 | 3 | 1.914 | 2.361 | 2.353 | −2.840 |
| Conf5 | 0 | 0 | 0.964/−0.968 | 2 | 1.785 | 2.441 | 2.338 | −2.470 |
| Conf6 | −0.041 | −0.020 | 0.949/−0.969 | 2 | 1.851 | 2.488 | 2.334 | −2.752 |
| Conf7 | −0.064 | 0.210 | −0.755/0.970/0.966/0.969 | 4 | 1.828 | 2.560 | 2.361 | −2.806 |
| Conf8 | 0 | 0.008 | 0.957/0.957/−0.942/−0.964 | 4 | 1.860 | 2.410 | 2.356 | −2.792 |
In Conf3 and 4, there is an isolated Cu bonded at a long bridge site, and a Cu3 cluster bonded at two adjacent long bridge sites. In Conf3, the fourth Cu is raised above the surface to bond with two Cu and one O ion, while in Conf4, the fourth Cu atom moves down slightly towards the surface, bonding with one second-layer and one surface O ion, and two nearest Cu atoms. In Conf1, the four Cu atoms are distributed at three adjacent long bridge sites, i.e. two isolated Cu at two long bridge sites, and a Cu2 cluster at one long bridge site. The fourth Cu atom moves from the Cetop site towards and bonds with the nearest surface O ion and one nearby Cu. The strong copper–surface interactions in Conf3, 4, and 1 thus contribute to their most negative adsorption energies.
From a Cu3 to a Cu4 cluster, the close competition between Cu–O and Cu–Cu interactions within a limited space leads to a Cu monolayer to bilayer transition along the adjacent long bridge sites. The small energy difference between Conf3, 4, 1, and 2 (maximum of 0.11 eV) and their different structures suggest that a Cu bilayer can start growing from several configurations. The stable and especially interesting optimised structure of Conf4, with one of the Cu atoms incorporated into the surface, is also observed experimentally.19,43
Conf2 has an adsorption energy very close to Con1, yet, it only has three electrons transferred, showing interesting electronic features in relevance to catalytic reactions. Its Cu 4s orbital PDOS plots, ESI,† Fig. S14a–c, suggest that the bottom three Cu atoms each donate one electron to the surface, becoming a Cu+ ion. Interestingly, the top Cu has two 4s electrons in opposite spins (a pair of distinct 4s signatures below EF), forming a Cuδ− species with a Bader charge of −0.437 e. This extra electron appearing in the top Cu 4s orbital originates from the bottom two Cu atoms with spin density isosurfaces, Fig. 6. In the plots of their 3d orbital PDOS, ESI,† Fig. S15, we note each has one unoccupied down-spin signature above EF, suggesting the electron in the top Cu is partially from these two bottom Cu 3d orbitals, though the contribution from the bottom Cu furthest away from the top is larger. Since this Cu is coordinated with three O ions, it can be easily stabilised as a Cu2+ ion, with a Bader charge of 0.724 e.21 As Conf2 and 3 have similar energies, electrons can easily exchange between Cu ions on CeO2(110), i.e. shift between Cu+ and Cu2+, which has been reported as providing active sites for many reactions.6,10,17
Paz-Borbón et al. showed a maximum of two electrons transferred from a Cu4 cluster to CeO2(111),15 whereas in our study, 2–4 electrons are transferred to CeO2(110). This significant difference in the number of electrons transferred suggests CeO2(110) could be more easily reduced after Cu cluster adsorption.
To investigate other additional possible electron transfers from Cu4 to CeO2(110), the initial structure of Conf4 was used to set up new structures, in which 1–3 pre-assumed Ce3+ ions were replaced with 1–3 La ions, respectively, for geometry optimisation. These La ions were then replaced by Ce ions for final optimisation. It was only possible to observe three electrons transferred, in one structure (labelled as Conf4–2, detailed discussion in ESI,† Section S1.4), whereas in the other two, four electrons were still transferred.
From the above discussion, we can conclude that CeO2(110) is highly reducible upon Cu4 adsorption, and competing Cu–Cu and Cu–O interactions are important in determining Cu4 shape and energetics, and electronic structure of Cu4/CeO2(110). Long-bridge site Cu atoms were the anchoring sites for Cu3 growth to Cu4.
3.5 Adsorption of Cu and Cu2 on CeO2(110) surface with one oxygen vacancy
An optimised defective CeO2(110) surface with one surface oxygen vacancy (CeO2(110)–Ov) is chosen as the new baseline for adsorption energy calculations when absorbing different Cu clusters on such a defective surface.
We thus first removed one oxygen from CeO2(110) (see Fig. 7(0)) and set up three configurations with different combinations of two pre-assumed Ce3+ sites around the oxygen vacancy, which are clearly illustrated in the optimised structures, Fig. 7(1–3). In Case1, the two surface Ce3+ ions are nearest neighbours (NN) of the vacancy. In Case2, one Ce3+ is a surface NN, while the other is a second-layer next nearest neighbour (NNN) of the vacancy. In Case3, one Ce3+ is a surface NN, while the other one is a surface NNN.
In Case1, the nearest surface oxygen ion moved towards the vacancy on the surface plane. It bonds with two Ce3+ ions with equal Ce3+–O bond lengths (2.341 Å). There is no significant surface distortion, as indicated by an average surface Ce–O distance of 2.381 Å, closest to the value of a stoichiometric surface, unlike in the other two cases, possibly because of the hindrance to relaxation of two adjacent large Ce3+ ions locally.24 In Case2, the nearest oxygen ion moves towards the vacancy significantly. It is slightly raised from the surface, bridging one Ce3+ and one Ce4+ ion. Since a Ce3+ ion has a larger radius than a Ce4+ ion, the Ce3+–O bond is longer than the Ce4+–O bond, forming an asymmetric bridge site.24 A similar asymmetric bridge site is also observed in Case3.
Calculated oxygen vacancy formation energies are in the range of 0.98–1.43 eV, as reported in Table 4, which are slightly lower than those from previous work (1.54–2.69 eV),2,24,44–46 because of a more negative O2 binding energy of −9.863 eV (bond length 1.233 Å) used in our work24,47 (ESI,† Section S1.5). The well-known error of overbinding O2 using GGA/LDA DFT and PAW potentials,24,46 as well as different computational parameters used (e.g. supercell size, cut-off energy, U value, etc.) make it difficult to compare absolute values with earlier work; however the relative comparison between Case1 to 3 is not affected. Case3 shows the smallest oxygen vacancy formation energy, followed by Case1 and 2, which suggests it is energetically favourable to form surface rather than second-layer Ce3+ ions.24,46 An NN–NNN Ce3+ pair combination (Conf3) is more stable than a NN–NN combination (Conf1), suggesting it is favourable to coordinate a Ce3+ ion with Ce4+ ions rather than Ce3+ ions.24 Therefore, Case3 is chosen as the new baseline for Cu adsorption energy calculations.
| System | M Ce (µB) | Number of Ce3+ reduced | (Ce–O)surf (Å) | (Ce–O)sub (Å) | E v (eV) |
|---|---|---|---|---|---|
| Case1 | 0.969/−0.973 | 2 | 2.338 | 2.381 | 1.110 |
| Case2 | 0.962/−0.899 | 2 | 2.329 | 2.374 | 1.426 |
| Case3 | −0.966/0.946 | 2 | 2.333 | 2.373 | 0.978 |
Case1 and 2 show similar optimised structures, in which the Cu atom is located at a long bridge site, bonding with two surface O ions, as illustrated in Fig. 8. These two structures are similar to the optimised Otop structure, as shown in ESI,† Fig. S2.2. Because of the additional Cu–O interaction, the nearest surface O ion moved even closer to the vacancy and formed a Cu–O bond, compared to that in a defective CeO2(110) surface without Cu adsorption.
 | ||
| Fig. 8 Top view and side view of (0) the initial structure of the Otop configuration from the Cu/CeO2(110) system with one oxygen vacancy; (1–2) two optimised structures with spin density isosurfaces of 0.005 e Å−3 around reduced Ce3+ ions which are located at different positions on the surface. | ||
In both cases, there are three electrons trapped in three Ce3+ 4f orbitals, i.e. one from the Cu adatom, and two from the oxygen vacancy. However, their electronic structures are quite different. In Case1, there is one surface and one second-layer NN Ce3+ of the vacancy, and one surface NNN Ce3+. In Case2, there are two NN Ce3+ ions and one NNN Ce3+ ion, all of which are on the surface. Case2 has a slightly more negative adsorption energy at −3.690 eV, since it is energetically more favourable to form surface Ce3+ ions than second-layer Ce3+ ions. The shorter Cu–O distance of Case2 also contributes to its higher stability. Electron transfer is also confirmed by magnetic moments of these species, as listed in Table 5.
| System | M total (µB) | M Ce (µB) | Number of Ce3+ reduced | Cu–O (Å) | Cu–Cu (Å) | (Ce–O)surf (Å) | E ad (eV) |
|---|---|---|---|---|---|---|---|
| Case1 | 2.896 | 0.969/0.955/0.973 | 3 | 1.818 | — | 2.343 | −3.595 |
| Case2 | 0.974 | 0.969/0.972/−0.967 | 3 | 1.809 | — | 2.349 | −3.690 |
| Conf1v | −1.937 | −0.975/−0.971/0.967/−0.960 | 4 | 1.793 | — | 2.360 | −3.356 |
| Conf4v | −1.929 | −0.972/−0.971/−0.950/0.963 | 4 | 1.908 | 2.479 | 2.317 | −3.207 |
 | ||
| Fig. 9 Top view and side view of left (1) and (4): the initial structure of Conf1v and 4v, respectively, from the Cu2/CeO2(110) system with one oxygen vacancy; right (1) and (4) the corresponding optimised structures of Conf1v and 4v with spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions. | ||
In optimised Conf1v and 4v, shown in Fig. 9, the nearest surface oxygen ion moved very close to the vacancy, bonding to one of the Cu atoms. These two structures are very similar to the optimised Conf1 and 4 with a stoichiometric surface, although the Cu2 cluster bonds with the nearest oxygen ion of the vacancy instead of the oxygen ion originally at the vacancy.
Both Conf1v and 4v have four electrons localised at four Ce3+ ions. In Conf1v, all four Ce3+ ions are on the surface, i.e. two NNs and two NNNs of the vacancy. In Conf4v, there are two surface NNs, one second-layer NN, and one second-layer NNN, which introduces more structural perturbation to the surface, as suggested by a much smaller value of average surface Ce–O bond length (2.317 Å), compared to that of Conf1v (2.360 Å). As a result, Conf1v shows a more negative Cu adsorption energy of −3.356 eV than Conf4v (−3.207 eV). The stronger Cu–O bonding with a shorter Cu–O distance also contributes to the more negative adsorption energy of Conf1v. Electron transfer is also confirmed by the magnetic moments of these species, reported in Table 5.
3.6 Adsorption of Cu3 on CeO2(110) surface with one oxygen vacancy
The initial structure of Conf2, 3, 6 and 7 from the Cu3/CeO2(110) system were chosen to create one surface oxygen vacancy (see Fig. 10), whose optimised structures show stable linear Cu3 clusters and two types of unstable triangular Cu3 clusters, respectively. | ||
| Fig. 10 Top row: Top view and side view of initial structures of Conf1v, 2v, 6v and 7v, respectively, from the Cu3/CeO2(110) system with one oxygen vacancy. The Cu2 clusters are emphasised by the Cu–Cu bond. Bottom row: Top view and side view of the corresponding optimised structures of Conf1v, 2v, 6v and 7v, respectively, with spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions. | ||
Conf2v has the most negative Cu adsorption energy of −3.350 eV, because of strong Cu–O interactions. It is the only optimised structure showing a linear Cu3 cluster (see Fig. 10), similar to that of Conf2 with a stoichiometric surface. Surface oxygen ions on the same side as the vacancy are raised from the surface and bond closely with the Cu3 cluster, showing short Cu–O distances (Table 6), which include the nearest surface oxygen ion which moves close to the vacancy. Conf7v, 6v, and 3v have weaker Cu adsorption, showing one Cu far away from the surface, without Cu–surface O binding, as a result of weakened Cu–O interactions due to vacancy formation (ESI,† Section S1.6).
| System | M total (µB) | M Ce (µB) | Number of Ce3+ reduced | Cu–O (Å) | Cu–Cu (Å) | (Ce–O)surf (Å) | E ad (eV) |
|---|---|---|---|---|---|---|---|
| Cu3/CeO2(110)–Ov | |||||||
| Conf2v | 2.896 | 0.975/−0.975/0.969/0.964/0.964 | 5 | 1.795 | — | 2.351 | −3.350 |
| Conf3v | 2.898 | 0.971/0.969/0.959 | 3 | 1.796 | 2.258 | 2.345 | −2.420 |
| Conf6v | 0.948 | −0.972/0.969/0.952 | 3 | 1.910 | 2.355 | 2.347 | −2.557 |
| Conf7v | 2.888 | 0.971/0.969/0.950 | 3 | 1.911 | 2.360 | 2.342 | −2.575 |
| Cu4/CeO2(110)–Ov | |||||||
| Conf2v | −0.106 | −0.972/0.967/0.856/−0.956 | 4 | 1.860 | 2.353 | 2.342 | −2.495 |
| Conf3v | 0 | −0.968/0.967/0.952/−0.953 | 4 | 1.861 | 2.357 | 2.341 | −2.586 |
| Conf4v | −1.381 | −0.974/0.965/0.962/−0.964/−0.961 | 5 | 1.810 | 2.451 | 2.336 | −2.674 |
| Conf7v | 0 | 0.972/−0.967/0.963/−0.958 | 4 | 1.803 | 2.390 | 2.351 | −2.574 |
In Conf2v, there are five electrons trapped in Ce3+ 4f orbitals, whereas in Conf3v, 6v and 7v, only three electrons are trapped, and the other two electrons are found to locate in the Cu3 cluster (discussion in ESI,† Section S1.6).
From the Cu1–3/CeO2(110)–Ov configurations with the most negative Cu adsorption energies, we could again conclude a Cu monolayer growth pattern along the long bridge sites after Cu adsorption, which is essentially the same as that on a stoichiometric surface. Both surface and second-layer Ce3+ ions are formed, but the latter are energetically less favoured.
3.7 Adsorption of Cu4 on CeO2(110) surface with one oxygen vacancy
The initial structures of Conf2, 3, 4, and 7 from the Cu4/CeO2(110) system were chosen to create one surface oxygen vacancy, Fig. 11, whose optimised structures demonstrate unique features and represent both stable and unstable configurations. | ||
| Fig. 11 Top row: Top view and side view of initial structures of Conf2v, 3v, 4v and 7v, respectively, from the Cu4/CeO2(110) system with one oxygen vacancy. The Cu3 clusters are emphasised by the Cu–Cu bonds which may not physically exist. Bottom row: Top view and side view of the corresponding optimised structures of Conf2v, 3v, 4v and 7v, respectively, with spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions. | ||
Conf4v has the most negative Cu adsorption energy of −2.674 eV, tightly followed by Conf3v, Conf7v, and Conf2v, Table 6, whose geometric and electronic structures discussed in detail in ESI,† Section S1.7. The optimised Conf4v is different from Conf4 with a stoichiometric surface, as a result of weakened Cu–O interactions. The fourth Cu in Conf4v moves away from the surface and bonds with two Cu and one O ion, whereas in Conf4, it moves down towards the surface and bonds with two Cu and both surface and second-layer O ions.
In Conf4v, five surface Ce4+ ions are reduced to Ce3+, whereas four Ce4+ ions are reduced on the surface in the other structures. Clearly, the most negative adsorption energy of Conf4v can be related to the greatest number of reduced Ce3+ ions on the surface. The Cu4 total magnetic moment is −0.409 µB, taking s, p and d orbitals into account. The two middle Cu atoms show a spin density isosurface around them, Fig. 11, and their 4s PDOS plots show two 4s signatures with similar magnitude below EF (see ESI,† Fig. S23). These observations suggest that three electrons are transferred from Cu4 to the surface, and one shared between the middle two Cu atoms, thus forming two Cu+ ions with Bader charges of 0.470 and 0.598 e, and two Cuδ+–Cuδ− species with Bader charges of 0.361 and −0.231 e. Similarly, in Conf2v, 3v, and 7v, three Cuδ+–Cu0 species and one Cu+ ion are formed on the surface (ESI,† Section S1.7).
Overall, we find that it is easier for a Cu4 cluster to retain and share one or more electrons between Cu atoms on a defective CeO2(110) surface than on a stoichiometric surface, forming Cu+ and Cuδ+–Cu0 species close to the vacancy, which has been proposed as active sites for reactions such as carbonate hydrogenation.48
3.8 Dispersion corrections
We note from previous studies that the inclusion of dispersion corrections in the DFT+U based calculations has a minimal effect on the local geometrical and electronic properties.47,49,50 Therefore, in this study we have only investigated the configurations which have the most negative Cu adsorption energies based on the DFT+U calculations, as listed in ESI,† Table S3, and compared the structures and energetics without (DFT+U) and with the van der Waals dispersion term (DFT+U+D3). We find that for each of the nine configurations investigated, the inclusion of the D3 term only makes the adsorption energies slightly more negative (maximum difference less than 0.23 eV) which agrees with the previous work.47,49,503.9 Discussion
For Cun (n = 1–4) adsorption on a stoichiometric CeO2(110) surface, a Cun cluster grows along the long bridge sites until Cu3, so that each Cu atom can strongly interact with surface oxygen ions at these sites, forming stable structures, as illustrated in Fig. 12(1–3), which, however, limits Cu–Cu interactions since they are distant from each other, hardly forming any Cu–Cu bonds. A linear Cu3 cluster represents a component of a Cu monolayer structure on the surface, where long bridge sites are first occupied upon Cu adsorption with an increasing Cu loading. This monolayer then grows into a bilayer in a way suggested by the Cu3 to Cu4 transition, with long-bridge site Cu as anchoring sites. The fourth Cu either rises up from the surface (Fig. 12(4.3)) or moves down towards the surface, as illustrated in Fig. 12(4.4), in between two adjacent long bridge sites, to bridge Cu atoms and bond with surface/subsurface oxygen ions from two adjacent long bridge sites. In this Cu monolayer to bilayer transition, Cu–Cu interactions gradually surpass in strength Cu–O interactions and become the dominant factor, resulting in Cu atoms at the top layer occupying the space in between long bridge sites and bonding with bottom-layer Cu atoms as well as surface oxygen ions; or some Cu atoms may be incorporated into the surface, as again seen in Fig. 12(4.4), and as is observed experimentally.19,43 This Cun cluster growth pattern is also demonstrated by the trend of adsorption energy per Cu atom versus Cun cluster size, given in Fig. 13. From Cu1 to Cu2, the adsorption energy per Cu becomes more negative by ∼0.2 eV to −3.492 eV, indicating a slightly more stable Cu2 cluster than a Cu adatom on the surface, due to additional Cu–Cu interactions besides surface Cu–O interactions. The value then becomes slightly less negative at −3.429 eV at Cu3, which then changes by ∼0.5 eV at Cu4. A similar Cun growth pattern is observed on a defective surface with one surface oxygen vacancy (CeO2(110)–Ov), as illustrated in Fig. 14, except we find that the Cu adatom on the defective surface has the most negative adsorption energy. However, Cu–O interactions are significantly weakened because of oxygen vacancy formation, thus showing a less negative adsorption energy per Cu than that with a stoichiometric surface, which becomes even more substantial in the Cu3 to Cu4 transition, where the adsorption energy per Cu becomes less negative by ∼0.7 eV. | ||
| Fig. 12 Top view and side view of the most stable Cun/CeO2(110) (n = 1–4) structures, labelled as (1) to (4); (4.3) and (4.4) are the optimised structures of Conf3 and 4, respectively, which have almost the same stability. | ||
 | ||
| Fig. 13 Adsorption energy per Cu atom as a function of the Cun cluster size on a stoichiometric and a defective CeO2(110) surface with one oxygen vacancy, respectively. | ||
 | ||
| Fig. 14 Top view and side view of the most stable Cun/CeO2(110)–Ov (n = 1–4) structures with the spin density isosurfaces of 0.005 e Å−3 around Cu and reduced Ce3+ ions, labelled as (1) to (4). | ||
Cun adsorption energy, shown in Fig. 13, suggests that growth of Cu4 on CeO2(110) with/out one surface oxygen vacancy is energetically less favoured, and Cu4 is likely to dissociate to Cu1–3. However, several experimental studies have reported Cu bilayers and large Cu particles on Cu/CeO2-nanorod catalysts, prepared in solutions by wet impregnation or deposition precipitation,8,11,51 suggesting that under kinetic conditions, for example, adsorption sites for single Cu atoms might become unavailable, or clustering of single Cu adsorbates may destabilise individual sites to some degree, formation of larger Cu clusters can become energetically preferable.
Analysis of electronic structures of the configurations having the lowest adsorption energy clearly demonstrates electron transfer from Cu 4s to Ce 4f orbitals, readily reducing the CeO2(110) surface both with and without a surface oxygen vacancy. A maximum of four Ce3+ ions are found for a Cun/CeO2(110) (n = 1–4) system, and a maximum of five Ce3+ ions for a Cun/CeO2(110)–Ov (n = 1–4) system. Both surface and second-layer Ce3+ ions are formed, but the latter is energetically less favoured. Other metastable Cun/CeO2(110) structures also possess interesting electronic structures, in which either an electron pair with opposite spins or a single electron is observed on the Cun. For example, calculations of the metastable Cu4/CeO2(110)–Conf2 structure show coexistence of Cu+, Cu2+, and a topmost Cuδ− species, and intriguing Cu+/Cu2+ interchange at the Cu/CeO2 interface which has been reported as providing active sites for many reactions.6,10,17 In addition, surface oxygen vacancy formation makes it easier for a Cu4 cluster to retain and share one or more electrons between Cu atoms, forming mixed Cu+ and Cuδ+–Cu0 species close to the vacancy. The coexisting Cu+ and Cu0 species of a Cu bilayer at the Cu/CeO2 interface has been proposed as actives sites for reactions such as carbonate hydrogenation.48
By an extensive study of different possible Ce3+ electron spin arrangement of 38 configurations from both systems, we find that structures with an antiferromagnetic CeO2(110) or CeO2(110)–Ov surface are energetically favourable (see ESI,† Table S2) in most cases, with a maximum reduction in the adsorption energy of 0.18 eV, which strongly suggests that CeO2(110) in both systems does not show any ferromagnetic (FM) behaviour, as reported previously.52
A few previous studies of Cu and Cun adsorbed on other metal oxide surfaces, such as ZnO, MgO, TiO2, and SrTiO3, are also briefly discussed here and compared with our study. For non-reducible surfaces such as ZnO and MgO, Cu–surface metal cation interactions predominate. For example, on Zn terminated (0001) surface of ZnO, French et al.53 observed that neutrally charged Cu clusters were mainly attracted to Zn cations, and that charged Cu clusters had charges mostly localised on the anchoring Cu adatom, thus showing effectively charge neutral surface copper sites. They concluded that larger copper clusters were predominantly charge neutral, as electrostatic repulsion destabilised Cu+ ions. They54 later reported that copper atoms in the middle layer of planar and polyhedral clusters gained a small amount of charges from surface oxygen ions. For +2 charged Cu clusters, electron transfer from oxygens to the anchoring Cu facilitated interactions between second-layer Cu and surface Zn cations, thus promoting formation of polyhedral Cu clusters, with the formed Cux+ sites being the nucleation centres. Mora-Fonz et al.55 reported Cu adsorption energy on non-polar (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface of ZnO, in a range of 0.365–1.981 eV. On reconstructed polar Zn-terminated (0001) and O-terminated (000) surface, Higham et al.56 found that planar and 3D Cu cluster growth were favoured, respectively, because of strong attractive Cu–Zn and repulsive Cu–O interactions. On the O-rich Zn-terminated reconstructed surface, they also observed close interaction between Cu and surface oxygens, with electron transfer from coordinating Cu atoms to surface O ions.
0) surface of ZnO, in a range of 0.365–1.981 eV. On reconstructed polar Zn-terminated (0001) and O-terminated (000) surface, Higham et al.56 found that planar and 3D Cu cluster growth were favoured, respectively, because of strong attractive Cu–Zn and repulsive Cu–O interactions. On the O-rich Zn-terminated reconstructed surface, they also observed close interaction between Cu and surface oxygens, with electron transfer from coordinating Cu atoms to surface O ions.
Pacchioni and Rösch57 found that Cu–Cu interactions were stronger than Cu–surface interactions, in Cu4 adsorption on MgO(110). Cu and Cu4 were weakly oxidised by surface oxygens, showing a weak polar covalent bond with limited charge transfer from Cu 4s to surface O 2p, with adsorption energies of 0.34 and 0.36 eV, respectively. Geudtner et al.58 later revealed that Cu–Cu interactions were the dominating factor in larger Cun (n = 2–6) cluster formation on MgO(100), stronger than Cu–surface oxygen interactions, with reported adsorption energies of 1.91–2.31 eV.
For Cu adsorption on reducible surfaces such as TiO2, it was reported that Cu adatom bound strongly to TiO2(110) nearer to surface bridging O ions,59 and that a Cu7 cluster retained its pentagonal bipyramidal structure on TiO2 surface, because of strong Cu–O and weak Cu–Ti interactions.60,61 Natile et al.62 reported Cu2 adsorption on SrTiO3(100) with an adsorption energy of −1.74 eV, and observed strong interactions between Cu and surface oxygens.
Ceria is highly reducible, and electron transfer from Cun to surface Ce4+ is clearly observed upon Cun adsorption on CeO2(110), which, is very different from that on non-reducible surfaces such as ZnO and MgO, where Cu–surface metal cation interactions predominate, with a small amount of charge transfer either from Cu clusters to surface oxygens or vice versa, depending on the exact model studied. Yet, our detailed study of small Cun (n = 1–4) cluster adsorption on CeO2(110) agrees in general with the aforementioned studies of TiO2, that copper-metal oxide interactions are important in determining geometry and stability of Cu/metal oxide structures.60 The calculated adsorption energies of Cun on CeO2(110), absolute values of 2.971–3.492 eV, are generally higher than the abovementioned values for other surfaces, suggesting strong interactions between copper and ceria. In addition, experimental studies reported that nanostructured Cu/CeO2 catalysts had a copper particle size-activity dependence,51 which thus strongly stimulates further study of larger Cu cluster adsorption on CeO2(110).
4. Conclusions
The atomic and electronic structures of a Cun (n = 1–4) cluster adsorbed on either a stoichiometric CeO2(110) surface or a defective surface with one oxygen vacancy (CeO2–Ov) have been investigated by DFT calculations without using pre-assumed Cun cluster shapes. Both the stoichiometric and defective surface are readily reduced upon Cun adsorption, forming surface and second-layer Ce3+ ions, and do not show any FM behaviour. On both surfaces, Cu1 grows to Cu3 along the long bridge sites, forming strong Cu–O bonds at adjacent long bridge sites, which models a Cu monolayer growth mechanism. The Cu3 to Cu4 transition suggests that this monolayer then begins to grow into a bilayer, with long-bridge site Cu as anchoring sites, where top-layer Cu atoms11 occupy the space in between long bridge sites to bond strongly with bottom-layer Cu and surface oxygens; or some Cu atoms are incorporated into the CeO2(110) surface lattice, as observed experimentally.19,43 Surface oxygen vacancy formation however weakens Cu–O interactions at the surface, thus making Cu adsorption energy less negative.A Cu bilayer is rich in Cu+ species at the Cu–O interface (four Cu+ in Cu4/CeO2, two Cu+ and two Cuδ+ in Cu4/CeO2–Ov), agreeing with experimental results.11 In metastable structures, it also shows Cu2+ and Cuδ− species, and Cuδ+–Cu0 species on a stoichiometric and a defective surface, respectively. This interesting Cu2+/Cu+ and Cu+/Cu0 interplay observed in our work thus give a theoretical basis to many experimental studies where the Cu2+/Cu+ pair and the Cu+/Cu0 pair were proposed as active sites for CuOx/CeO2-nanorod catalysts in many reactions.6,10,17,48 In the future work we will explore the structures and energetics of larger Cu clusters adsorbed on the CeO2(110) surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Rui Zhang thanks the Department of Chemical Engineering, Imperial College London for the award of a Department Scholarship. This study was initiated under the EPSRC Low Carbon Fuels programme EP/N009533/1 & EP/N010531/1. We thank Dr Richardo Grau-Crespo for his helpful discussions on modelling the Ce4+ to Ce3+ reduction process. We also thank Dr David Mora-Fonz for his helpful discussions and suggestions. This work used the computing resources from the ARCHER UK National Super-computing Service http://www.archer.ac.uk/, the THOMAS and the YOUNG UK National Tier 2 Super-computing Service, via our membership of the UK's HEC Materials Chemistry Consortium (MCC), which is funded by EPSRC (EP/L000202). This work also used the UK Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/P020194 and EP/T022213). We also thank UCL for access to the KATHLEEN and Faraday computing facilities. We also acknowledge the use of HPC Midlands Plus (funded by the EPSRC (grant EP/P020232/1)) and Cirrus (funded by the University of Edinburgh and EPSRC(EP/PO20267/1)).References
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- J. Kaspar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef CAS.
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CrossRef CAS.
- W.-W. Wang, W.-Z. Yu, P.-P. Du, H. Xu, Z. Jin, R. Si, C. Ma, S. Shi, C.-J. Jia and C.-H. Yan, ACS Catal., 2017, 7, 1313–1329 CrossRef CAS.
- M. Zabilskiy, P. Djinović, E. Tchernychova, O. P. Tkachenko, L. M. Kustov and A. Pintar, ACS Catal., 2015, 5, 5357–5365 CrossRef CAS.
- H. X. Mai, L. D. Sun, Y. W. Zhang, R. Si, W. Feng, H. P. Zhang, H. C. Liu and C. H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS.
- S. Y. Yao, W. Q. Xu, A. C. Johnston-Peck, F. Z. Zhao, Z. Y. Liu, S. Luo, S. D. Senanayake, A. Martínez-Arias, W. J. Liu and J. A. Rodriguez, Phys. Chem. Chem. Phys., 2014, 16, 17183–17195 RSC.
- L. Kang, B. Wang, Q. Bing, M. Zalibera, R. Buchel, R. Xu, Q. Wang, Y. Liu, D. Gianolio, C. C. Tang, E. K. Gibson, M. Danaie, C. Allen, K. Wu, S. Marlow, L. D. Sun, Q. He, S. Guan, A. Savitsky, J. J. Velasco-Velez, J. Callison, C. W. M. Kay, S. E. Pratsinis, W. Lubitz, J. Y. Liu and F. R. Wang, Nat. Commun., 2020, 11, 4008 CrossRef CAS PubMed.
- J. Ning, C. Dong, M. Li, Y. Zhou and W. Shen, J. Chem. Phys., 2020, 152, 094708 CrossRef CAS PubMed.
- J. C. Conesa, Surf. Sci., 1995, 339, 337–352 CrossRef CAS.
- L. Szabová, M. F. Camellone, M. Huang, V. Matolin and S. Fabris, J. Chem. Phys., 2010, 133, 234705 CrossRef PubMed.
- Z. Yang, L. Xie, D. Ma and G. Wang, J. Phys. Chem. C, 2011, 115, 6730–6740 CrossRef CAS.
- L. O. Paz-Borbon, F. Buendia, I. L. Garzon, A. Posada-Amarillas, F. Illas and J. Li, Phys. Chem. Chem. Phys., 2019, 21, 15286–15296 RSC.
- J. F. Jerratsch, X. Shao, N. Nilius, H. J. Freund, C. Popa, M. V. Ganduglia-Pirovano, A. M. Burow and J. Sauer, Phys. Rev. Lett., 2011, 106, 246801 CrossRef PubMed.
- S. Yao, K. Mudiyanselage, W. Xu, A. C. Johnston-Peck, J. C. Hanson, T. Wu, D. Stacchiola, J. A. Rodriguez, H. Zhao, K. A. Beyer, K. W. Chapman, P. J. Chupas, A. Martínez-Arias, R. Si, T. B. Bolin, W. Liu and S. D. Senanayake, ACS Catal., 2014, 4, 1650–1661 CrossRef CAS.
- L. Lin, S. Yao, Z. Liu, F. Zhang, N. Li, D. Vovchok, A. Martínez-Arias, R. Castañeda, J. Lin, S. D. Senanayake, D. Su, D. Ma and J. A. Rodriguez, J. Phys. Chem. C, 2018, 122, 12934–12943 CrossRef CAS.
- L. Liu, Z. Yao, Y. Deng, F. Gao, B. Liu and L. Dong, ChemCatChem, 2011, 3, 978–989 CrossRef CAS.
- M. Nolan, J. Chem. Phys., 2012, 136, 134703 CrossRef PubMed.
- L. Cui, Y. Tang, H. Zhang, L. G. Hector, Jr., C. Ouyang, S. Shi, H. Li and L. Chen, Phys. Chem. Chem. Phys., 2012, 14, 1923–1933 RSC.
- A. Chutia, E. K. Gibson, M. R. Farrow, P. P. Wells, D. O. Scanlon, N. Dimitratos, D. J. Willock and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2017, 19, 27191–27203 RSC.
- Z. Ren, N. Liu, B. Chen, J. Li and D. Mei, J. Phys. Chem. C, 2018, 122, 27402–27411 CrossRef CAS.
- J. Kullgren, K. Hermansson and C. Castleton, J. Chem. Phys., 2012, 137, 044705 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS PubMed.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- V. V. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed.
- V. V. Anisimov, I. I. Solovyev, M. A. Korotin, M. T. Czyzyk and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 16929–16934 CrossRef CAS PubMed.
- I. I. Solovyev, P. H. Dederichs and V. V. Anisimov, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 16861–16871 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- H. Raebiger, S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045209 CrossRef.
- E. L. Wilson, R. Grau-Crespo, C. L. Pang, G. Cabailh, Q. Chen, J. A. Purton, C. R. A. Catlow, W. A. Brown, N. H. de Leeuw and G. Thornton, J. Phys. Chem. C, 2008, 112, 10918–10922 CrossRef CAS.
- N. C. Hernández, R. Grau-Crespo, N. H. de Leeuw and J. F. Sanz, Phys. Chem. Chem. Phys., 2009, 11, 5246–5252 RSC.
- M. M. Branda, N. J. Castellani, R. Grau-Crespo, N. H. de Leeuw, N. C. Hernandez, J. F. Sanz, K. M. Neyman and F. Illas, J. Chem. Phys., 2009, 131, 094702 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- D. Gamarra, A. L. Cámara, M. Monte, S. B. Rasmussen, L. E. Chinchilla, A. B. Hungría, G. Munuera, N. Gyorffy, Z. Schay, V. C. Corberán, J. C. Conesa and A. Martínez-Arias, Appl. Catal., B, 2013, 130-131, 224–238 CrossRef CAS.
- Z. Yang, T. K. Woo, M. Baudin and K. Hermansson, J. Chem. Phys., 2004, 120, 7741–7749 CrossRef CAS PubMed.
- S. Fabris, G. Vicario, G. Balducci, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2005, 109, 22860–22867 CrossRef CAS PubMed.
- M. Nolan, D. G. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- A. Chutia, D. J. Willock and C. R. A. Catlow, Faraday Discuss., 2018, 208, 123–145 RSC.
- Y. Cui and W.-L. Dai, Catal. Sci. Technol., 2016, 6, 7752–7762 RSC.
- L. Chen, T. V. W. Janssens and H. Gronbeck, Phys. Chem. Chem. Phys., 2019, 21, 10923–10930 RSC.
- M. Delarmelina, M. G. Quesne and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2020, 22, 6660–6676 RSC.
- J. Zhu, Y. Su, J. Chai, V. Muravev, N. Kosinov and E. J. M. Hensen, ACS Catal., 2020, 10, 11532–11544 CrossRef CAS.
- P. R. Keating, D. O. Scanlon and G. W. Watson, J. Phys.: Condens. Matter, 2009, 21, 405502 CrossRef CAS PubMed.
- S. A. French, A. A. Sokol, S. T. Bromley, C. R. A. Catlow and P. Sherwood, Top. Catal., 2003, 24, 161–172 CrossRef CAS.
- S. A. French, A. A. Sokol, C. R. A. Catlow and P. Sherwood, J. Phys. Chem. C, 2008, 112, 7420–7430 CrossRef CAS.
- D. Mora-Fonz, T. Lazauskas, S. M. Woodley, S. T. Bromley, C. R. A. Catlow and A. A. Sokol, J. Phys. Chem. C, 2017, 121, 16831–16844 CrossRef CAS.
- M. D. Higham, D. Mora-Fonz, A. A. Sokol, S. M. Woodley and C. R. A. Catlow, J. Mater. Chem. A, 2020, 8, 22840–22857 RSC.
- G. Pacchioni and N. Rösch, J. Chem. Phys., 1996, 104, 7329 CrossRef CAS.
- G. Geudtner, K. Jug and A. M. Köster, Surf. Sci., 2000, 467, 98–106 CrossRef CAS.
- D. Pillay, Y. Wang and G. S. Hwang, Korean J. Chem. Eng., 2004, 21, 537–547 CrossRef CAS.
- S. M. Shearin, Mod. Concepts Mater. Sci., 2020, 2, 1–6 Search PubMed.
- Y. Lan, Y. Xie, J. Chen, Z. Hu and D. Cui, Chem. Commun., 2019, 55, 8068–8071 RSC.
- M. M. Natile, S. Carlotto, G. Bizzotto, A. Vittadini and A. Glisenti, Eur. J. Inorg. Chem., 2018, 3829–3834 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results and further discussion of some Cun/CeO2(110) structures with/out one surface oxygen vacancy. In addition, Cartesian coordinates of all the optimised structures of Cun (n = 1–4)/CeO2(110) and Cun (n = 1–4)/CeO2(110)–Ov from both DFT+U and DFT+U+D3 calculations can be found here (ZIP). See DOI: 10.1039/d1cp02973h |
| This journal is © the Owner Societies 2021 |