
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics of the gas phase reaction of the Criegee intermediate CH2OO with SO2 as a function of temperature†
Lavinia
Onel
 a,
Rachel
Lade
a,
Jennifer
Mortiboy
a,
Mark A.
Blitz
ab,
Paul W.
Seakins
a,
Dwayne E.
Heard
a and
Daniel
Stone
*a
a,
Rachel
Lade
a,
Jennifer
Mortiboy
a,
Mark A.
Blitz
ab,
Paul W.
Seakins
a,
Dwayne E.
Heard
a and
Daniel
Stone
*a
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: d.stone@leeds.ac.uk; Tel: +44 113 343 6508
bNational Centre for Atmospheric Science, University of Leeds, Leeds, LS2 9JT, UK
First published on 25th August 2021
Abstract
The kinetics of the gas phase reaction of the Criegee intermediate CH2OO with SO2 have been studied as a function of temperature in the range 223–344 K at 85 Torr using flash photolysis of CH2I2/O2/SO2/N2 mixtures at 248 nm coupled to time-resolved broadband UV absorption spectroscopy. Measurements were performed under pseudo-first-order conditions with respect to SO2, revealing a negative temperature dependence. Analysis of experimental results using the Master Equation Solver for Multi-Energy well Reactions (MESMER) indicates that the observed temperature dependence, combined with the reported lack of a pressure dependence in the range 1.5–760 Torr, can be described by a reaction mechanism consisting of the formation of a pre-reaction complex leading to a cyclic secondary ozonide which subsequently decomposes to produce HCHO + SO3. The temperature dependence can be characterised by kCH2OO+SO2 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1. The observed negative temperature dependence for the title reaction in conjunction with the decrease in water dimer (the main competitor for the Criegee intermediate) concentration at lower temperatures means that Criegee intermediate chemistry can play an enhanced role in SO2 oxidation in the atmosphere at lower temperatures.
Introduction
Criegee intermediates are reactive species produced in the atmosphere following the ozonolysis of unsaturated volatile organic compounds (VOCs) in environments impacted by biogenic and/or anthropogenic emissions.1,2 The exothermicity associated with ozonolysis reactions leads to the formation of nascent Criegee intermediates with high internal energy, which may undergo either unimolecular decomposition or collisional energy transfer with surrounding gas molecules to form stabilised Criegee intermediates (SCIs). Bimolecular reactions of stabilised Criegee intermediates with atmospheric constituents such as water vapour, water vapour dimers, NO2, and SO2 can impact atmospheric budgets of NOx (NO + NO2), SO2, H2SO4, sulfate aerosol, and secondary organic aerosol, and thus influence air quality and climate.3–9 Welz et al.4 reported the first direct measurements of SCI kinetics using the photolysis of CH2I2 at 248 nm in the presence of O2 at low pressure (4 Torr) to generate the CH2OO Criegee intermediate ((R1)-(R2)), coupled with tunable VUV synchrotron photoionisation mass spectrometry (PIMS) to probe CH2OO:| CH2I2 + hν → CH2I + I | (R1) |
| CH2I + O2 → CH2OO + I | (R2) |
| CH2OO + SO2 → HCHO + SO3 | (R3) |
 at 298 K.17 However, the temperature dependence of the CH2OO + SO2 reaction has yet to be reported, with such an investigation necessary to improve the understanding of the mechanism and atmospheric impacts of the reaction between CH2OO and SO2.
at 298 K.17 However, the temperature dependence of the CH2OO + SO2 reaction has yet to be reported, with such an investigation necessary to improve the understanding of the mechanism and atmospheric impacts of the reaction between CH2OO and SO2.
| Reference | Technique | Photolysis wavelength/nm | Temperature/K | Pressure/Torr | Bath gas | k 3/10−11 cm3 molecule−1 s−1 |
|---|---|---|---|---|---|---|
| a LFP = laser flash photolysis of CH2I2 in the presence of O2. b PIMS = photoionisation mass spectrometry monitoring CH2OO. c UV absorption = time-resolved broadband ultraviolet absorption spectroscopy. d LIF = laser induced fluorescence. e CRDS = cavity ring down spectroscopy. | ||||||
| Welz et al. 20124 | LFPa/PIMSb | 248 | 298 | 4 | He | 3.9 ± 0.7 |
| Sheps 201310 | LFP/UV absc | 266 | 295 | 5 | He | 4.1 ± 0.3 |
| Stone et al. 201411 | LFP/LIFd HCHO | 355 | 295 | 50–450 | N2 | 3.42 ± 0.42 |
| LFP/PIMS | 248 | 295 | 1.5 | He | ||
| Liu et al. 201415 | LFP/LIF OH | 351 | 295 | 50–200 | Ar | 3.53 ± 0.29 |
| Chhantyal-Pun et al. 201512 | LFP/CRDSe | 355 | 295 | 30 | N2 | 3.80 ± 0.04 |
| Huang et al. 201513 | LFP/UV abs | 248 | 298 | 30–756 | N2 | 3.57 ± 0.28 |
| Liu et al. 201716 | LFP/LIF OH | 355 | 300 | 10 | Ar | 3.88 ± 0.13 |
| Howes et al. 20199 | LFP/PIMS | 248 | 295 | 2 | He | 3.74 ± 0.43 |
| LFP/UV abs | 248 | 295 | 50 | N2 | 3.87 ± 0.45 | |
| Qiu and Tonokura14 2019 | LFP/mid-IR abs | 266 | 295 | 10.4 | N2 | 3.6 ± 0.1 |
Experimental studies employing LIF detection of HCHO produced in (R3)11 suggest that HCHO and thus SO3 are products of the reaction (R3), with the co-product SO3 directly observed by PIMS18 at 4 Torr and transient infrared absorption spectroscopy at 110 and 214 Torr.19 Theoretical studies also indicate that HCHO and SO3 are the dominant products under atmospheric conditions, formed via a five-membered secondary ozonide (SOZ) cycloadduct5,20–22 and thus the SO2-catalysed isomerisation of CH2OO to formic acid via SOZ evidenced by Aplincourt and Rui-López23 is a minor channel of the CH2OO + SO2 reaction. Experimental studies using PIMS18 and transient infrared absorption spectroscopy19 have not observed the reaction channel producing formic acid, also indicating that this channel is negligible. Vereecken et al.5 and Kuwata et al.22 have investigated the potential energy surface for the reaction between CH2OO and SO2, combined with Rice–Ramsperger–Kassel–Marcus (RRKM)/master equation simulations to predict product yields, but with contrasting results. Kuwata et al.22 predicted a yield for HCHO + SO3 of at least 97% at atmospheric pressure, while Vereecken et al.5 predicted a yield of 68% HCHO + SO3 at atmospheric pressure. Both studies report that the reaction is under the low-pressure regime, with negligible stabilisation of the SOZ up to pressures considerably above ambient conditions.5,22 However, the mechanisms for formation of the SOZ and for the subsequent production of HCHO and SO3 differ in the two theoretical studies. Vereecken et al.5 predicted barrierless formation of the SOZ in the initial CH2OO + SO2 encounter, with homolytic cleavage of the O–O bond in the SOZ yielding the biradical OCH2OS(O)O, which in turn predominantly leads to HCHO and SO3 by β-scission. The formation of formyl sulfinic acid, HC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OS(O)OH (15% at 1 atm) and bisoxy diradical, H2C(O˙)O˙, + SO2 (17% at 1 atm) by the decomposition of OCH2OS(O)O was also expected by Vereecken et al.5 In contrast, Kuwata et al.22 characterised the barrierless formation of a pre-reaction complex in the first reaction step, which then leads to the SOZ, with the reaction predominantly following a closed-shell pathway involving the direct formation of HCHO + SO3 from the SOZ by cycloreversion, in qualitative agreement with the earlier studies of Jiang et al.20 and Kurtén et al.21 Kuwata et al.22 employed a complex description of the potential energy surface, incorporating two distinct stereochemical pathways owing to the existence of diastereomeric endo and exo conformers of the SOZ. By combining the quantum chemical calculations with statistical rate theory models Kuwata et al.22 predicted that k3 = (3.68 ± 0.02) × 10−11 cm3 molecule−1 s−1 at 295 K with no statistically significant variation with pressure between 1–760 Torr, in good agreement with the results of the photolytic experiments.
O)OS(O)OH (15% at 1 atm) and bisoxy diradical, H2C(O˙)O˙, + SO2 (17% at 1 atm) by the decomposition of OCH2OS(O)O was also expected by Vereecken et al.5 In contrast, Kuwata et al.22 characterised the barrierless formation of a pre-reaction complex in the first reaction step, which then leads to the SOZ, with the reaction predominantly following a closed-shell pathway involving the direct formation of HCHO + SO3 from the SOZ by cycloreversion, in qualitative agreement with the earlier studies of Jiang et al.20 and Kurtén et al.21 Kuwata et al.22 employed a complex description of the potential energy surface, incorporating two distinct stereochemical pathways owing to the existence of diastereomeric endo and exo conformers of the SOZ. By combining the quantum chemical calculations with statistical rate theory models Kuwata et al.22 predicted that k3 = (3.68 ± 0.02) × 10−11 cm3 molecule−1 s−1 at 295 K with no statistically significant variation with pressure between 1–760 Torr, in good agreement with the results of the photolytic experiments.
The formation of SO3 by reaction (R3) indicates that the reaction is potentially significant in the production of H2SO4 and sulfate aerosol in the atmosphere. Atmospheric modelling studies have shown that a larger rate coefficient for CH2OO + SO2 has a significant impact on sulfate aerosol formation on a regional scale6 and on gas phase H2SO4 production in forested regions such as a boreal forest in Finland24 and tropical forests.25 However, the modelling results do depend on the competition between the reaction of CH2OO with SO2 and that with water vapour/water vapour dimers, with a number of recent studies indicating that the reaction of CH2OO with water dimers is more important than the CH2OO + water monomer reaction under atmospheric relevant conditions.26–29 IUPAC currently recommends a value of (6.4 ± 1.5) × 10−12 cm3 molecule−1 s−1 for the rate coefficient of the CH2OO + (H2O)2 reaction at 298 K, which is four orders of magnitude larger than the rate coefficient of the CH2OO + H2O reaction, (2.8 ± 1.1) × 10−16 cm3 molecule−1 s−1.30 Kinetic studies of the CH2OO + (H2O)2 reaction30 have led to the conclusion that the removal of CH2OO in the troposphere is dominated globally by the reaction with water dimer.2 However, the reaction of CH2OO with SO2 is fast enough to potentially compete with the CH2OO + water dimer reaction and impact on SO2 oxidation in SO2–rich environments and/or low absolute humidities. As the concentration of water dimer varies strongly with the saturated vapour pressure of water, the competition between SO2 and water dimers for CH2OO will be temperature dependent.
In the present work we report the kinetics of the CH2OO + SO2 reaction at 85 Torr in the temperature range of 223–344 K using 248 nm photolysis of CH2I2/O2/SO2/N2 mixtures under pseudo-first-order conditions with respect to SO2. Time-resolved broadband UV absorption spectroscopy was employed to monitor CH2OO directly during the reaction with SO2. This is the first study of the temperature dependence of the rate coefficient for the CH2OO + SO2 reaction.
Experimental
The kinetics of the CH2OO + SO2 reaction were studied as a function of temperature using laser flash photolysis of CH2I2/O2/SO2/N2 gas mixtures, coupled with time-resolved broadband UV absorption spectroscopy. The experimental apparatus has been described in detail previously31–33 therefore only a brief description is given here.Two reaction cells were used, both consisting of a 100 cm long glass tube of 3 cm inner diameter sealed with fused silica windows at both ends. The first cell was jacketed and used for experiments at T ≥ 296 K, in which the temperature was controlled by circulating thermofluid (HUBE6479 DW-therm oil) from a thermoregulator (Huber Unistat 360). The second reaction cell, used for temperatures at T < 296 K, was immersed in a cooling bath of 30% ethane-1,2-diol in methanol and surrounded by 3 cm thick polystyrene for insulation. The temperature for this cell was controlled using a refrigerated immersion chiller (LabPlant Refrigerated Immersion Probe, RP-100CD), with the probe immersed in the cooling mixture and magnetic stirring employed to achieve a constant temperature (within 98%) in the cooling mixture along the cell. Further details of temperature calibrations in the cell are given in the ESI.†
A dilute mixture of SO2 (Sigma-Aldrich, 99.9%) was prepared manometrically at a known concentration in nitrogen (BOC 99.998%) and stored in a glass bulb. Gases, i.e. SO2, O2 (BOC, 99.5%) and the carrier gas N2, were introduced through calibrated mass flow controllers (MFCs) in a glass mixing manifold. A few percent of the gas mixture was then passed through a bubbler containing liquid CH2I2 (Alfa Aesar, 99%) held at constant temperature in an ice bath, which was then mixed with the remaining fraction of the gas and delivered to the reaction cell. The initial concentration of CH2I2 in the cell was controlled by a needle valve at the entrance of the bubbler and determined by UV absorption measurements as the average of the concentration in the cell before and after each kinetic run.
The kinetic experiments were performed under pseudo-first-order conditions with respect to SO2. The initial concentrations were: [SO2] = (0.6–5.6) × 1013 molecule cm−3 (typical uncertainties of ∼5%), [CH2I2] = (1.0–7.4) × 1013 molecule cm−3, [O2] = 7.5 × 1017 molecule cm−3 and [CH2OO]0 = (0.4–2.7) × 1012 molecule cm−3.
The total flow rate was 18 slm, giving a residence time in the cell of ∼2 s. The total pressure in the cell was maintained at 85 Torr and measured by a capacitance manometer (MKS Instruments). The chemistry within the cell was initiated by pulsed excimer laser photolysis at 248 nm (KrF, Lambda-Physik CompEx 210) with a laser fluence of 2–6 × 1016 photon cm−2 (measured by a Molectron Powermax 500A power meter). The pulse repetition rate was 0.3 Hz to ensure a fresh gas mixture in the cell for each laser pulse.
Absorption of UV/visible radiation during the experiments was monitored using a laser-driven light source (LDLS, Energetiq EQ-99X), which provides ∼10 mW cm−2 of light between 200 nm and 800 nm. Approximately 10% of the probe light was aligned in a single pass arrangement along the length of the cell, which was used in the temperature calibrations (see ESI†) with the remaining light used for the kinetics measurements aligned in a 7 pass arrangement described previously.31–33 The total effective pathlength in the multi-pass arrangement, considering the total overlap between the photolysis and probe beams, was determined to be l = (471 ± 50) cm from comparison between the expected depletion of [CH2I2] owing to photolysis (Δ[CH2I2]), based on the measured concentration of CH2I2 and laser fluence in the cell, and Δ[CH2I2] × l derived from analysis of the post-photolysis absorption spectra. Further details are given in the ESI.† It should be noted that the value obtained for the total pathlength did not impact the determination of the rate coefficient of the CH2OO + SO2 reaction as the kinetic experiments were performed under pseudo-first-order conditions.
The probe beam exiting the reaction cell was passed through a sharp cut-on filter (248 nm RazorEdge ultrasteep long-pass edge filter, Semrock) to minimise the impacts of scattered excimer light and focused onto a fibre optic via a fibre launcher (Elliot Scientific). The output from the fibre optic was directed through a 25 μm slit onto a spectrograph equipped with a diffraction grating of 300 grooves per mm and imaged onto an integrated thermoelectrically cooled charge-coupled device (CCD) detector (FER-SCI-1024BRX, Princeton Instruments), giving spectral resolution of 1 nm (FWHM). The exposure time of the CCD to the light was varied from 10–100 μs. The spectral and temporal information were mapped spatially along the horizontal and vertical directions of the CCD, respectively, as described in detail in our previous work.33 The photocharge was shifted on the time axis (vertically) from an illuminated region to a storage region with a programmable rate in the range 5.6 μs per row to 35 μs per row. The rapid shift of photocharge on the CCD resulted in an instrument response function, which was included in the analysis of the measured concentration-time profiles as described in the Results section.
Wavelength calibrations were performed by recording the well-known Hg emission spectrum from a low pressure Hg pen-ray lamp (Oriel). Timings of the recording by the camera and the firing of the photolysis laser were controlled by a delay generator (SRS DG535). Intensity data recorded by the camera were typically averaged for 400–800 photolysis shots and transferred to a PC for analysis.
Results
Fig. 1 shows an example of total absorbance recorded following photolysis, with contributions from the absorbance of CH2I2, CH2OO and IO. The absorbance, A(tot)λ,t, was observed as a function of wavelength (λ) and time (t) and analysed between 300 and 450 nm to determine the changes to concentrations of each species by fitting the reference absorption cross-sections for CH2OO,32 IO34 and CH2I235 to A(tot)λ,t at each time point (E1).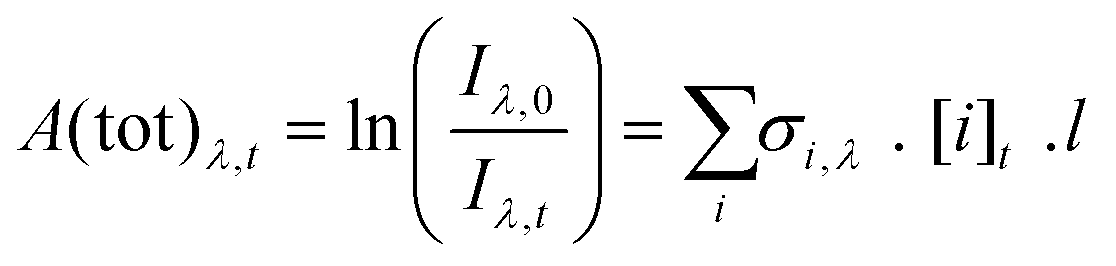 | (E1) |
 | ||
| Fig. 1 Observed absorbance (black), total fit (red), and the individual contributions from CH2OO (blue), IO (purple) and CH2I2 (orange) determined by fitting reference spectra to the measured absorbance using eqn (E1) at t = 1.0 ms following photolysis. For these data, T = 296 K, p = 85 Torr, [O2] = 7.5 × 1017 molecule cm−3, [CH2I2] = 8.0 × 1013 molecule cm−3, [CH2OO]0 = 1.8 × 1012 molecule cm−3 and [SO2] = 5.0 × 1012 molecule cm−3. | ||
Fig. 2 shows typical concentration–time profiles for CH2OO, which are given by a convolution of the ‘true’ kinetic decay with an instrument response function (IRF).33 The IRF is generated by the shift of the photocharge in the time direction of the CCD detector from the illuminated region to the storage region of the CCD (details in the Experimental). The rate coefficient for CH2I + O2 (1.7 × 10−12 cm3 molecule−1 s−1)36 and the O2 concentration (7.5 × 1017 molecule cm−3) employed in this work indicate a time scale of 1–2 μs for the CH2OO growth following photolysis. However, owing to the IRF, the observed [CH2OO] increases on a time scale of hundreds of μs before the subsequent decay which occurs on a time scale of 5–10 ms (Fig. 2). Typical concentration-time profiles for CH2I2 and IO are given in the ESI.†
 | ||
| Fig. 2 Examples of observations of CH2OO (open circles) and fit results (solid lines). For this experiment T = 316 K and p = 85 Torr. The initial concentration of the precursor was the same for all the kinetic decays, [CH2I2]0 = 4.2 × 1013 molecule cm−3, while the initial concentration of SO2 was varied from one measurement to another: no SO2 (pink circles), 0.5 × 1013 molecule cm−3 (green circles), 0.9 × 1013 molecule cm−3 (red circles), 1.5 × 1013 molecule cm−3 (black circles), 2.0 × 1013 molecule cm−3 (blue circles), 2.6 × 1013 molecule cm−3 (orange circles) and 3.1 × 1013 molecule cm−3 (purple circles). The global fit to the data using eqn (E4) results in the observed rate coefficients for the CH2OO reaction with SO2, kobs: (435 ± 5) s−1 (pink), (571 ± 5) s−1 (green), (783 ± 7) s−1 (red), (1002 ± 11) s−1 (black), (1261 ± 15) s−1 (blue), (1460 ± 19) s−1 (orange) and (1658 ± 23) s−1 (purple). For these data the IRF parameters are tc = (7.0 ± 0.1) × 10−5 s and w = (1.20 ± 0.02) × 10−4 s. Errors are given at 1σ level. | ||
As the kinetic experiments were performed under pseudo-first-order conditions with respect to SO2 the decay of [CH2OO] by reaction with SO2(R3) is described by an exponential decay (E2).
 | (E2) |
| kobs = kloss + k3[SO2] | (E3) |
The observed temporal behaviour of CH2OO can be described by a convolution of the exponential decay described by eqn (E2) with the IRF described by a Gaussian centred at time tc with width w(E4).33
 | (E4) |
Eqn (E4) was employed in the fit to the observed temporal profiles of CH2OO at each temperature to determine kobs and the Gaussian parameters tc and w. The parameters tc and w are dependent on the exposure time of the CCD detector to the light and the shift rate of the photocharge from the illuminated region to the storage region of the CCD. Concentration-time profiles obtained using the same exposure time and shift rate were fitted globally to determine the parameters tc and w. Fig. 2 shows an example of the global fit.
Under given conditions the decay rate coefficient of CH2OO in the absence of SO2, kloss is constant, hence a plot of kobs as a function of [SO2] is linear with gradient k3 (eqn (E3)). Typical kobsvs. [SO2] plots are shown in Fig. 3 at four different temperatures: 223 K, 270 K, 296 K and 344 K. It can be observed that k3 decreases with temperature. The plots of kobs against [SO2] at all temperatures employed are shown in the ESI.† The values of k3 determined at all temperatures are shown in Table 2. The temperature dependence of k3 is shown in Fig. 4.
 | ||
| Fig. 3 k obs vs. [SO2] plots at 85 Torr and four different temperatures. The linear fits to the data result in the second order rate coefficients k3 for each temperature: (6.4 ± 0.3) × 10−11 cm3 molecule−1 s−1 (223 K, dark red), (5.3 ± 0.3) × 10−11 cm3 molecule−1 s−1 (270 K, red), (4.0 ± 0.1) × 10−11 cm3 molecule−1 s−1 (296 K, green) and (3.3 ± 0.1) × 10−11 cm3 molecule−1 s−1 (344 K, blue). For each temperature [CH2I2] was constant: 3.4 × 1013 molecule cm−3 (dark red), 5.0 × 1013 molecule cm−3 (red), 4.8 × 1013 molecule cm−3 (green) and 4.5 × 1013 molecule cm−3 (blue). Laser fluence: 6.0 × 1016 photons cm−2 (dark red), 4.0 × 1016 photons cm−2 (red), 4.0 × 1016 photons cm−2 (green) and 3.0 × 1016 photons cm−2 (blue). Intercepts of the bimolecular plots are noted to decrease with increasing temperature, and are expected to be dominated by the reactions CH2OO + CH2OO and CH2OO + I.32 The rates of these reactions are expected to decrease with increasing temperature and are lower at the lower initial CH2OO and I concentrations at higher temperatures owing to lower laser fluence used in experiments at higher temperatures. | ||
| Temperature/K | k 3/10−11 cm3 molecule−1 s−1 |
|---|---|
| 223 | 6.4 ± 0.3 |
| 259 | 5.5 ± 0.3 |
| 266 | 4.4 ± 0.3 |
| 270 | 5.3 ± 0.3 |
| 275 | 4.1 ± 0.5 |
| 281 | 4.1 ± 0.1 |
| 296 | 4.0 ± 0.1 |
| 316 | 4.2 ± 0.1 |
| 324 | 3.6 ± 0.2 |
| 331 | 3.4 ± 0.1 |
| 340 | 2.2 ± 0.1 |
| 344 | 3.3 ± 0.1 |
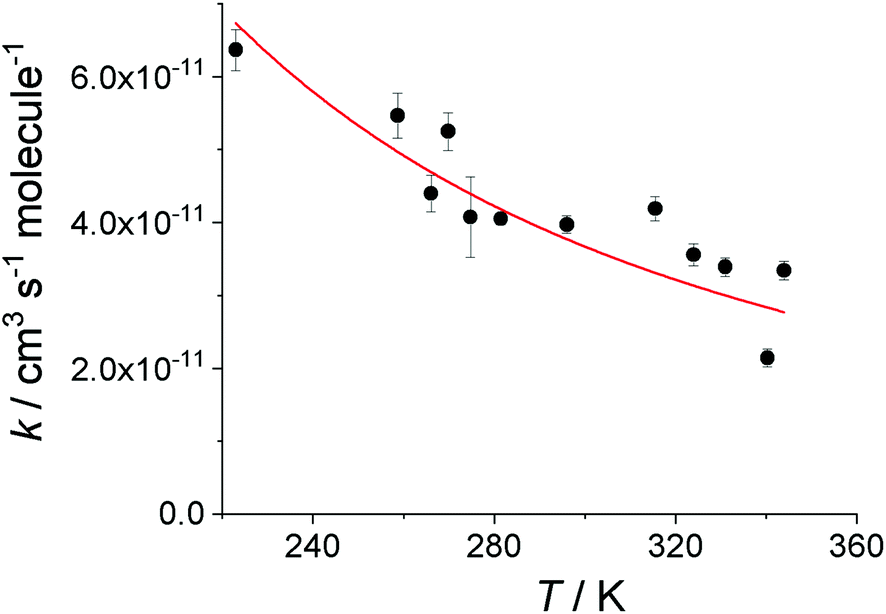 | ||
| Fig. 4 Temperature dependence of the rate coefficient for the CH2OO + SO2 reaction. The data are shown as black circles; error margins are statistical errors at 1σ level. The red line shows the parameterisation of the MESMER result, which gave k3 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1. | ||
Master equation calculations for the temperature dependence of the CH2OO + SO2 reaction using the MESMER program
A master equation analysis using the Master Equation Solver for Multi-Energy well Reactions (MESMER) was used to fit the potential energy surface (PES) for (R3) to the experimental rate coefficients. A full description of MESMER is given in previous work.38–41 MESMER solves the master equation and the subsequent eigenvalue–eigenvector analysis outputs the time profiles of species concentrations together with the phenomenological rate coefficients for the system. The most recent computational study of the CH2OO + SO2 by Kuwata et al.22 expanded the understanding of the reaction mechanism by characterising two distinct stereochemical pathways to provide a detailed description of the PES. The authors22 predicted that ∼90–91% of the reaction proceeds through a closed-shell transition structure to form HCHO + SO3, in qualitative agreement with previous studies,20,21 with a small contribution (6–7%) of the open-shell mechanism leading to HCHO + SO3 predicted by Vereecken et al. to be the dominant channel.5 The present calculations were based on a potential energy surface shown in Fig. 5, which was obtained by reducing the PES determined by Kuwata et al.22 to the dominant closed-shell mechanism. The reaction first proceeds by the formation of a pre-reaction complex leading to a cyclic secondary ozonide (SOZ). The SOZ subsequently decomposes, producing HCHO + SO3via two stereochemical pathways arising from the existence of endo and exo conformers of the pre-reaction complex (species 3a and 3b respectively in Fig. 5, using the same notation for the structures given by Kuwata et al.22) and of the SOZ (species 5a and 5b in Fig. 5). Simulations using MESMER showed that there was no significant loss of accuracy by the reduction in complexity of the PES to that shown in Fig. 5, and that the reduced mechanism dominates the fate of the CH2OO + SO2 under all conditions used in the MESMER calculations (T = 223–344 K and p = 2–760 Torr). | ||
| Fig. 5 Potential energy surface of the reaction CH2OO + SO2 showing the two stereo-chemical pathways proceeding through endo (light blue) and exo conformers (dark blue). Energies (kJ mol−1) of 3a, 3b, TS-4a and TS-4b were generated by MESMER calculations, while the energies of 5a, 5b, TS-7a, TS-7b and the products HCHO and SO3 were taken from the work of Kuwata et al.22 The energy levels calculated by Kuwata et al.22 for 3a and TS-4a are shown in light red and for 3b and TS-4b are shown in dark red. The notations for intermediates and transitions states are the same as in the work of Kuwata et al.19,22 Kuwata et al. describe the structures of the species.22 | ||
To characterise the observed negative temperature dependence and the pressure independence in the range 1.5–760 Torr11,13 of the CH2OO + SO2 reaction, the values of k3 obtained in this work for T = 220–350 K and p = 85 Torr and the 298 K IUPAC recommendation for p = 2–760 Torr, 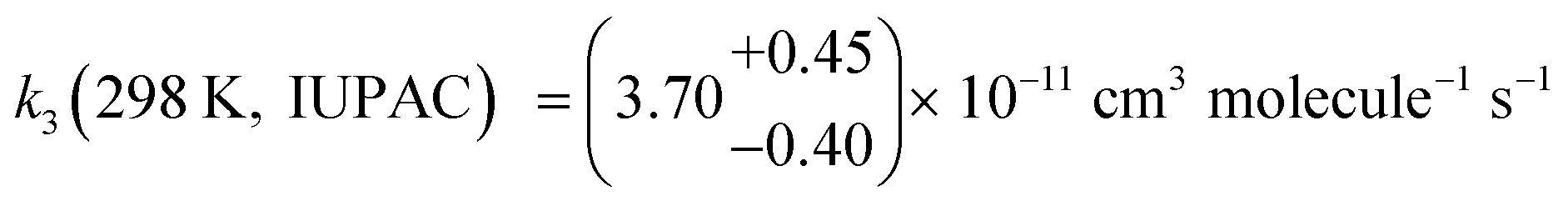 ,30 were fit in MESMER. The inverse Laplace transform (ILT) method was used to describe the barrierless formation of the pre-reaction complexes 3a and 3b, giving a canonical high pressure limiting rate coefficient k∞ = (5.5 ± 2.0) × 10−11 (T/298)−(1.0±0.4) cm3 molecule−1 s−1 for each stereo-chemical pathway. The calculations showed that the cycloaddition transition states TS-4a and TS-4b control the reaction rate and thus the relative energies of TS-4a and TS-4b were floated during the simulations. The energies of the pre-reaction complexes 3a and 3b and the transition states TS-4a and TS-4b were linked42 to maintain the same relative differences as those given by Kuwata et al.22 The values calculated by Kuwata et al.22 were used for the relative energies of 5a, 5b, TS-7a, TS-7b and the products HCHO and SO3. The fit results for k3 can be parameterised by k3 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1 and are shown in Fig. 4. The fit values for the energies of 3a, 3b, TS-4a and TS-4b (relative to CH2OO + SO2) are shown in Fig. 5, which also shows the values calculated by Kuwata et al.22 An agreement within 98% was obtained between the fit energies for 3a, 3b, TS-4a and TS-4b determined by MESMER and the energies predicted by Kuwata et al.,22i.e. the energies given by MESMER calculations are 0.9 kJ mol−1 more negative than the values of Kuwata et al. (Fig. 5). This small deviation is within the 2.5 kJ mol−1 errors of the MESMER calculations and the errors generated using the multi-coefficient Gaussian-3 quantum chemical method (MCG3) employed by Kuwata et al.22 to determine the energies of 3a, 3b, TS-4a and TS-4b (root-mean-squared error of 6.6 kJ mol−1).43
,30 were fit in MESMER. The inverse Laplace transform (ILT) method was used to describe the barrierless formation of the pre-reaction complexes 3a and 3b, giving a canonical high pressure limiting rate coefficient k∞ = (5.5 ± 2.0) × 10−11 (T/298)−(1.0±0.4) cm3 molecule−1 s−1 for each stereo-chemical pathway. The calculations showed that the cycloaddition transition states TS-4a and TS-4b control the reaction rate and thus the relative energies of TS-4a and TS-4b were floated during the simulations. The energies of the pre-reaction complexes 3a and 3b and the transition states TS-4a and TS-4b were linked42 to maintain the same relative differences as those given by Kuwata et al.22 The values calculated by Kuwata et al.22 were used for the relative energies of 5a, 5b, TS-7a, TS-7b and the products HCHO and SO3. The fit results for k3 can be parameterised by k3 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1 and are shown in Fig. 4. The fit values for the energies of 3a, 3b, TS-4a and TS-4b (relative to CH2OO + SO2) are shown in Fig. 5, which also shows the values calculated by Kuwata et al.22 An agreement within 98% was obtained between the fit energies for 3a, 3b, TS-4a and TS-4b determined by MESMER and the energies predicted by Kuwata et al.,22i.e. the energies given by MESMER calculations are 0.9 kJ mol−1 more negative than the values of Kuwata et al. (Fig. 5). This small deviation is within the 2.5 kJ mol−1 errors of the MESMER calculations and the errors generated using the multi-coefficient Gaussian-3 quantum chemical method (MCG3) employed by Kuwata et al.22 to determine the energies of 3a, 3b, TS-4a and TS-4b (root-mean-squared error of 6.6 kJ mol−1).43
A negative temperature dependence has also been observed for the reaction of the Criegee intermediate (CH3)2COO with SO2 between 283 and 303 K at 300 Torr following photolysis of (CH3)2CI2/O2/N2/SO2 mixtures at 248 nm coupled with time-resolved UV absorption at 340 nm.44 The kinetics of the reaction were characterised by k(CH3)2COO+SO2 = (1.36 ± 0.03) × 10−11 (T/298)(−6.45±0.64) cm3 molecule−1 s−1, and the dependence on temperature is thus more pronounced than that observed for CH2OO + SO2 in the current study, kCH2OO+SO2 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1.
Atmospheric implications
Fig. 6 compares the pseudo-first-order losses of CH2OO for a range of SO2 mixing ratios and relative humidities as a function of temperature. Losses to SO2 are given as k3[SO2], using the results for k3 determined in this work. Losses as a function of relative humidity are given as the product of the current IUPAC recommendation for the temperature dependence of the reaction between CH2OO and (H2O)217,28 and the water dimer concentration, which was determined from the saturated vapour pressure of water,45 relative humidity, and the equilibrium constant for 2H2O ⇌ (H2O)2.28,46 Although the reaction with water dimers dominates under most conditions, the reaction of CH2OO with SO2 is potentially important in regions with high SO2 concentrations and low humidity, particularly at low temperatures. Results for k3 obtained in this work may be useful for the interpretation of field measurements and data obtained in chamber experiments at temperatures away from 298 K. | ||
| Fig. 6 Comparison of pseudo-first-order losses for CH2OO for a range of SO2 mixing ratios and relative humidities as a function of temperature. Losses for CH2OO are shown as k[X], where X is SO2 or (H2O)2, and have been determined using results obtained in this work for k(T)[SO2], with k(T)[(H2O)2] determined from current IUPAC recommendations17 for CH2OO + (H2O)2 kinetics and water dimer concentrations determined from the saturated vapour pressure for water,45 relative humidity, and the equilibrium constant for 2H2O ⇌ (H2O)2.28,46 | ||
Conclusions
The kinetics for the reaction between the stabilised Criegee intermediate CH2OO and SO2 have been determined as a function of temperature using laser flash photolysis of CH2I2/O2/SO2/N2 mixtures at 248 nm coupled to time-resolved broadband UV absorption spectroscopy in the temperature range 223–344 K at a total pressure of 85 Torr. The reaction showed a negative temperature dependence which can be described by k3 = (3.72 ± 0.13) × 10−11 (T/298)(−2.05±0.38) cm3 molecule−1 s−1.Calculations in MESMER38 using a mechanism based on that proposed by Kuwata et al.22 show that the measured negative temperature dependence and the lack of pressure dependence between 1.5–760 Torr11,13,30 can be described by the barrierless formation of endo and exo pre-reaction complexes, which lead to endo and exo secondary cyclic ozonides through relatively low barriers. The secondary ozonides further decompose to HCHO + SO3. The energies of the transition states leading from the pre-reaction complexes to the SOZs were found to be 0.9 kJ mol−1 more negative than those reported in the computational study of Kuwata et al.22 In addition, this work reinforces the prediction that the reaction yield of HCHO + SO3 at atmospheric pressure is close to 100% and is in agreement with experimental work19 finding that HCHO + SO3 are the major products of the reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Natural Environment Research Council (NERC) for funding (grant references NE/L010798/1 and NE/P012876/1). We would also like to thank K. T. Kuwata for providing details of the calculations on the potential energy surface for CH2OO + SO2.References
- D. Johnson and G. Marston, The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 2008, 37(4), 699–716 RSC.
- C. A. Taatjes, Criegee Intermediates: What Direct Production and Detection Can Teach Us About Reactions of Carbonyl Oxides, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2017, vol. 68, pp. 183–207 Search PubMed.
- L. Vereecken, Lifting the Veil on an Old Mystery, Science, 2013, 340(6129), 154–155 CrossRef CAS PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2, Science, 2012, 335(6065), 204–207 CrossRef CAS PubMed.
- L. Vereecken, H. Harder and A. Novelli, The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 2012, 14(42), 14682–14695 RSC.
- C. J. Percival, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, D. O. Topping, D. Lowe, S. R. Utembe, A. Bacak, G. McFiggans, M. C. Cooke, P. Xiao, A. T. Archibald, M. E. Jenkin, R. G. Derwent, I. Riipinen, D. W. K. Mok, E. P. F. Lee, J. M. Dyke, C. A. Taatjes and D. E. Shallcross, Regional and global impacts of Criegee intermediates on atmospheric sulphuric acid concentrations and first steps of aerosol formation, Faraday Discuss., 2013, 165, 45–73 RSC.
- R. Chhantyal-Pun, B. Rotavera, M. R. McGillen, M. A. H. Khan, A. J. Eskola, R. L. Caravan, L. Blacker, D. P. Tew, D. L. Osborn, C. J. Percival, C. A. Taatjes, D. E. Shallcross and A. J. Orr-Ewing, Criegee Intermediate Reactions with Carboxylic Acids: A Potential Source of Secondary Organic Aerosol in the Atmosphere. Acs Earth and Space, Chemistry, 2018, 2(8), 833–842 CAS.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Criegee intermediates and their impacts on the troposphere, Environ. Sci.: Processes Impacts, 2018, 20(3), 437–453 RSC.
- N. U. M. Howes, Z. S. Mir, M. A. Blitz, S. Hardman, T. R. Lewis, D. Stone and P. W. Seakins, Kinetic studies of C1 and C2 Criegee intermediates with SO2 using laser flash photolysis coupled with photoionization mass spectrometry and time resolved UV absorption spectroscopy, Phys. Chem. Chem. Phys., 2018, 20(34), 22218–22227 RSC.
- L. Sheps, Absolute Ultraviolet Absorption Spectrum of a Criegee Intermediate CH2OO. Journal of Physical, Chem. Lett., 2013, 4(24), 4201–4205 CAS.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 2014, 16(3), 1139–1149 RSC.
- R. Chhantyal-Pun, A. Davey, D. E. Shallcross, C. J. Percival and A. J. Orr-Ewing, A kinetic study of the CH2OO Criegee intermediate self-reaction, reaction with SO2 and unimolecular reaction using cavity ring-down spectroscopy, Phys. Chem. Chem. Phys., 2015, 17(5), 3617–3626 RSC.
- H. L. Huang, W. Chao and J. J. M. Lin, Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(35), 10857–10862 CrossRef CAS PubMed.
- J. Qiu and K. Tonokura, Detection of the simplest Criegee intermediate CH2OO in the ν4 band using a continuous wave quantum cascade laser and its kinetics with SO2 and NO2, Chem. Phys. Lett.: X, 2019, 2, 100019 CAS.
- Y. D. Liu, K. D. Bayes and S. P. Sander, Measuring Rate Constants for Reactions of the Simplest Criegee Intermediate (CH2OO) by Monitoring the OH Radical, J. Phys. Chem. A, 2014, 118(4), 741–747 CrossRef CAS PubMed.
- Y. Q. Liu, F. H. Liu, S. Y. Liu, D. X. Dai, W. R. Dong and X. M. Yang, A kinetic study of the CH2OO Criegee intermediate reaction with SO2, (H2O)2, CH2I2 and I atoms using OH laser induced fluorescence, Phys. Chem. Chem. Phys., 2017, 19(31), 20786–20794 RSC.
- R. A. Cox, M. Ammann, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII - Criegee intermediates, Atmos. Chem. Phys., 2020, 20(21), 13497–13519 CrossRef CAS.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 2013, 340(6129), 177–180 CrossRef CAS PubMed.
- Y. Y. Wang, M. R. Dash, C. Y. Chung and Y. P. Lee, Detection of transient infrared absorption of SO3 and 1,3,2-dioxathietane-2,2-dioxide cyc-(CH2)O(SO2)O in the reaction CH2OO+SO2, J. Chem. Phys., 2018, 148(6), 064301 CrossRef PubMed.
- L. Jiang, Y. S. Xu and A. Z. Ding, Reaction of Stabilized Criegee Intermediates from Ozonolysis of Limonene with Sulfur Dioxide Ab Initio and DFT Study, J. Phys. Chem. A, 2010, 114(47), 12452–12461 CrossRef CAS PubMed.
- T. Kurtén, J. R. Lane, S. Jorgensen and H. G. Kjaergaard, A Computational Study of the Oxidation of SO2 to SO3 by Gas-Phase Organic Oxidants, J. Phys. Chem. A, 2011, 115(31), 8669–8681 CrossRef PubMed.
- K. T. Kuwata, E. J. Guinn, M. R. Hermes, J. A. Fernandez, J. M. Mathison and K. Huang, A Computational Re-examination of the Criegee Intermediate-Sulfur Dioxide Reaction, J. Phys. Chem. A, 2015, 119(41), 10316–10335 CrossRef CAS PubMed.
- P. Aplincourt and M. F. Ruiz-López, Theoretical investigation of reaction mechanisms for carboxylic acid formation in the atmosphere, J. Am. Chem. Soc., 2000, 122(37), 8990–8997 CrossRef CAS.
- A. Novelli, K. Hens, C. T. Ernest, M. Martinez, A. C. Nolscher, V. Sinha, P. Paasonen, T. Petaja, M. Sipila, T. Elste, C. Plass-Dulmer, G. J. Phillips, D. Kubistin, J. Williams, L. Vereecken, J. Lelieveld and H. Harder, Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 2017, 17(12), 7807–7826 CrossRef CAS.
- J. R. Pierce, M. J. Evans, C. E. Scott, S. D. D'Andrea, D. K. Farmer, E. Swietlicki and D. V. Spracklen, Weak global sensitivity of cloud condensation nuclei and the aerosol indirect effect to Criegee + SO2 chemistry, Atmos. Chem. Phys., 2013, 13(6), 3163–3176 CrossRef.
- W. Chao, J. T. Hsieh, C. H. Chang and J. J. M. Lin, Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 2015, 347(6223), 751–754 CrossRef CAS PubMed.
- T. R. Lewis, M. A. Blitz, D. E. Heard and P. W. Seakins, Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 2015, 17(7), 4859–4863 RSC.
- M. C. Smith, C. H. Chang, W. Chao, L. C. Lin, K. Takahashi, K. A. Boering and J. J. M. Lin, Strong Negative Temperature Dependence of the Simplest Criegee Intermediate CH2OO Reaction with Water Dimer, J. Phys. Chem. Lett., 2015, 6(14), 2708–2713 CrossRef CAS PubMed.
- L. C. Lin, H. T. Chang, C. H. Chang, W. Chao, M. C. Smith, C. H. Chang, J. J. M. Lin and K. Takahashi, Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 2016, 18(6), 4557–4568 RSC.
- IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation, http://iupac.pole-ether.fr. (accessed 18.06.2021).
- T. Lewis, D. E. Heard and M. A. Blitz, A novel multiplex absorption spectrometer for time-resolved studies, Rev. Sci. Instrum., 2018, 89(2), 024101 CrossRef PubMed.
- Z. S. Mir, T. R. Lewis, L. Onel, M. A. Blitz, P. W. Seakins and D. Stone, CH2OO Criegee intermediate UV absorption cross-sections and kinetics of CH2OO + CH2OO and CH2OO + I as a function of pressure, Phys. Chem. Chem. Phys., 2020, 22(17), 9448–9459 RSC.
- L. Onel, M. Blitz, P. Seakins, D. Heard and D. Stone, Kinetics of the Gas Phase Reactions of the Criegee Intermediate CH2OO with O3 and IO, J. Phys. Chem. A, 2020, 124(31), 6287–6293 CrossRef CAS PubMed.
- P. Spietz, J. C. G. Martin and J. P. Burrows, Spectroscopic studies of the I2/O3 photochemistry - Part 2. Improved spectra of iodine oxides and analysis of the IO absorption spectrum, J. Photochem. Photobiol., A, 2005, 176(1σ3), 50–67 CrossRef CAS.
- J. B. Burkholder, S. P. Sander, J. P. D. Abbatt, J. R. Barker, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, D. M. Wilmouth and P. H. Wine, Chemical kinetics and photochemical data for use in atmospheric studies - Evaluation No. 18. JPL Publication 15-10: available at: http://jpldataeval.jpl.nasa.gov/, last access: 25 May 2021, 2015.
- D. Stone, M. Blitz, L. Daubney, T. Ingham and P. Seakins, CH2OO Criegee biradical yields following photolysis of CH2I2 in O2, Phys. Chem. Chem. Phys., 2013, 15(44), 19119–19124 RSC.
- https://mathworld.wolfram.com/Erf.html. (accessed 18.06.2021).
- D. R. Glowacki, C. H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, MESMER: An Open-Source Master Equation Solver for Multi-Energy Well Reactions, J. Phys. Chem. A, 2012, 116(38), 9545–9560 CrossRef CAS PubMed.
- R. J. Shannon, R. L. Caravan, M. A. Blitz and D. E. Heard, A combined experimental and theoretical study of reactions between the hydroxyl radical and oxygenated hydrocarbons relevant to astrochemical environments, Phys. Chem. Chem. Phys., 2014, 16(8), 3466–3478 RSC.
- D. Stone, K. Au, S. Sime, D. J. Medeiros, M. Blitz, P. W. Seakins, Z. Decker and L. Sheps, Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO, Phys. Chem. Chem. Phys., 2018, 20(38), 24940–24954 RSC.
- C. A. Whelan, M. A. Blitz, R. Shannon, L. Onel, J. P. Lockhart, P. W. Seakins and D. Stone, Temperature and Pressure Dependent Kinetics of QOOH Decomposition and Reaction with O2: Experimental and Theoretical Investigations of QOOH Radicals Derived from Cl + (CH3)3COOH, J. Phys. Chem. A, 2019, 123(47), 10254–10262 CrossRef CAS PubMed.
- D. J. Medeiros, M. A. Blitz, L. James, T. H. Speak and P. W. Seakins, Kinetics of the Reaction of OH with Isoprene over a Wide Range of Temperature and Pressure Including Direct Observation of Equilibrium with the OH Adducts, J. Phys. Chem. A, 2018, 122(37), 7239–7255 CrossRef CAS PubMed.
- B. J. Lynch and D. G. Truhlar, Robust and affordable multicoefficient methods for thermochemistry and thermochemical kinetics: The MCCM/3 suite and SAC/3, J. Phys. Chem. A, 2003, 107(19), 3898–3906 CrossRef CAS.
- M. C. Smith, W. Chao, K. Takahashi, K. A. Boering and J. J. M. Lin, Unimolecular Decomposition Rate of the Criegee Intermediate (CH3)2COO Measured Directly with UV Absorption Spectroscopy, J. Phys. Chem. A, 2016, 120(27), 4789–4798 CrossRef CAS PubMed.
- NIST/SEMATECH e-Handbook of Statistical Methods. http://www.itl.nist.gov/div898/handbook/ (accessed 18.05.2021).
- B. Ruscic, Active Thermochemical Tables: Water and Water Dimer, J. Phys. Chem. A, 2013, 117(46), 11940–11953 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02932k |
| This journal is © the Owner Societies 2021 |