
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrostatic profiling of photosynthetic pigments: implications for directed spectral tuning†‡
Abhishek
Sirohiwal
 * and
Dimitrios A.
Pantazis
*
* and
Dimitrios A.
Pantazis
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: abhishek.sirohiwal@kofo.mpg.de; dimitrios.pantazis@kofo.mpg.de
First published on 8th October 2021
Abstract
Photosynthetic pigment–protein complexes harvest solar energy with a high quantum efficiency. Protein scaffolds are known to tune the spectral properties of embedded pigments principally through structured electrostatic environments. Although the physical nature of electrostatic tuning is straightforward, the precise spatial principles of electrostatic preorganization remain poorly explored for different protein matrices and incompletely characterized with respect to the intrinsic properties of different photosynthetic pigments. In this work, we study the electronic structure features associated with the lowest excited state of a series of eight naturally occurring (bacterio)chlorophylls and pheophytins to describe the precise topological differences in electrostatic potentials and hence determine intrinsic differences in the expected mode and impact of electrostatic tuning. The difference electrostatic potentials between the ground and first excited states are used as fingerprints. Both the spatial profile and the propensity for spectral tuning are found to be unique for each pigment, indicating spatially and directionally distinct modes of electrostatic tuning. The results define a specific partitioning of the protein matrix around each pigment as an aid to identify regions with a maximal impact on spectral tuning and have direct implications for dimensionality reduction in protein design and engineering. Thus, a quantum mechanical basis is provided for understanding, predicting, and ultimately designing sequence-modified or pigment-exchanged biological systems, as suggested for selected examples of pigment-reconstituted proteins.
1. Introduction
Photosynthesis is one of the most efficient natural processes that converts sunlight into chemical energy to power the biosphere of our planet.1 The primary tasks of any photosynthetic apparatus are the efficient collection of solar photons, excitation energy transfer, and charge separation.2–5 These tasks are performed by pigments embedded within protein scaffolds that are known to modulate and fine tune their spectral properties. Key pigments include bacteriochlorophylls and chlorophylls, whose lowest excitation energy (also known as “site energy” and typically associated with the “Qy” transition) forms the principal component in understanding the overall function of light harvesting complexes and photosynthetic reaction centers.5,6 The pigments are uniquely located within protein matrices and experience distinct electrostatic environments. As a result, chemically identical pigments can have distinct spectroscopic properties. Therefore, a key question is how does a protein tune the site energy of each pigment.Various factors are responsible for spectral tuning of pigments, such as long- and short-range protein electrostatics,7,8 hydrogen bonding,9 polarization, axial ligation, macrocyclic ring deformation,10–12 and out-of-plane motion of π-conjugated functional groups of the macrocyclic ring.13 In addition, excitonic coupling between closely placed pigments produces spectral shifts.14 Electrostatic effects are often found to be the leading factor.6–8,14–25 For example, a recent systematic study by Schapiro and co-workers8 on chlorophyll a based water soluble chlorophyll-binding proteins (WSCPs) demonstrated protein electrostatics to be dominant in modulating the site energy over ring deformations. In addition, the functioning of the reaction center of Photosystem II is also explicitly governed by protein electrostatics.7,18,22,26 The dominant effect of electrostatics in spectral tuning is not only limited to photosynthetic systems but is also prevalent in other photobiological systems.14,27–34 Moreover, modulation of site energies via protein electrostatics can be the basis of an engineered approach to achieve desired control of pigment properties via point mutations, and hence a holistic understanding of these leading effects in connection to the intrinsic properties of common pigments is essential.
A bottom-up understanding of the electrostatic tuning begins with the study of the intrinsic electronic structure of pigments in their ground and excited states. In this work, we study the intrinsic effects of electronic excitation as reflected on topological profiles of difference electrostatic potentials of several biologically relevant pigments to understand the respective spatial principles of matrix-induced electrostatic tuning. By directly comparing these pigments, we provide a basis for elucidating the distinct possibilities for spectral tuning in each pigment, which can be utilized in engineering protein scaffolds towards specific outcomes. The present results and comparisons are also essential for interpreting and analyzing spectroscopic observations from studies that involve the exchange of pigments. This can be physiological substitution of different chlorophylls, for example as part of a natural response of the photosynthetic apparatus to efficiently utilize far-red with red-shifted chlorophylls in place of Chl a,35–43 or artificial substitution of different chlorophylls (such as insertion of Chl d or f in natively Chl a systems) and even reconstitution of natural photosynthetic proteins with various non-native pigments.44–51 Our set includes the naturally occurring pigments chlorophylls a (Chl a), Chl b, Chl d, and Chl f, bacteriochlorophylls a (BChl a) and BChl b, bacteriopheophytin a (BPheo a), and pheophytin a (Pheo a). The key structural differences between the pigments are the substituents on the (bacterio)chlorin ring and the π-conjugation pattern (see Scheme 1).
 | ||
| Scheme 1 Structural definition of the various pigments under consideration in this work. (Bacterio)pheophytins are obtained by replacement of the central Mg2+ ion of the corresponding (bacterio)chlorophylls by two N-bound protons. The x and y axes correspond to the conventional representation of the transition dipole moment orientation in the macrocyclic ring plane. | ||
2. Methodology
The initial structures of the pigments under consideration were built upon the Chl a skeleton derived from the X-ray crystal structure of the Photosystem II (PDB ID: 3WU2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52). The phytyl chains of the pigments are not considered in these models and they were replaced with a methyl group (–CH3). The phytyl chain does not contribute to the lowest energy excitation and neglecting it reduces computational costs significantly.53–55 The pigments were optimized in vacuo using the B3LYP functional56 along with all-electron def2-TZVP57–59 basis sets on all atoms. Dispersion corrections were introduced using the D3(BJ) scheme.60,61 The resolution of the identity for the Coulomb integrals combined with the chain-of-spheres approximation to exchange integrals (RIJCOSX)62,63 was used along with matching auxiliary def2/J basis sets57 to speed up the computations. Very tight convergence criteria for the SCF and geometry optimizations were used throughout (VeryTightSCF and VeryTightOpt keywords in ORCA). Higher DFT integration grids were used, i.e. Grid6 and GridX7 in the ORCA nomenclature. No imaginary frequencies were found in the optimized structures.
52). The phytyl chains of the pigments are not considered in these models and they were replaced with a methyl group (–CH3). The phytyl chain does not contribute to the lowest energy excitation and neglecting it reduces computational costs significantly.53–55 The pigments were optimized in vacuo using the B3LYP functional56 along with all-electron def2-TZVP57–59 basis sets on all atoms. Dispersion corrections were introduced using the D3(BJ) scheme.60,61 The resolution of the identity for the Coulomb integrals combined with the chain-of-spheres approximation to exchange integrals (RIJCOSX)62,63 was used along with matching auxiliary def2/J basis sets57 to speed up the computations. Very tight convergence criteria for the SCF and geometry optimizations were used throughout (VeryTightSCF and VeryTightOpt keywords in ORCA). Higher DFT integration grids were used, i.e. Grid6 and GridX7 in the ORCA nomenclature. No imaginary frequencies were found in the optimized structures.
The excited states of the pigments were computed in the framework of time-dependent density functional theory (TD-DFT). We performed full TD-DFT (i.e. without the Tamm–Dancoff approximation) calculations using the range-separated hybrid ωB97X-V functional64,65 along with the def2-TZVP basis set. Compared to the original ωB97X range-separated functional of Chai and Head-Gordon,64 ωB97X-V was obtained by a ground-up reparameterization, which, crucially, incorporates non-local correlation via the VV10 scheme of Vydrov and Van Voorhis.66 The choice of this functional is motivated by extensive benchmarks against coupled-cluster level calculations.53 Our earlier investigation53 showed that ωB97X-V accurately predicts the energetics and ordering of the low-energy excited states of photosynthetic pigments. In addition, it accurately captures the spectral shifts originating from protein electrostatics.18 This functional has also been successfully employed in studies of the reaction center7 and the CP47 antenna complex of Photosystem II.19 A total of 10 roots were computed in all calculations. The RIJCOSX approximation was also used to speed up the TD-DFT computations. Similar to the geometry optimization protocol, very tight SCF criteria and dense integration grids were used. All the quantum chemical computations in this work are performed using the ORCA 4.2 program package.67,68 The difference electrostatic potentials between the ground and first excited states are based on the relaxed densities (ωB97X-V/def2-TZVP level of theory) computed using the Turbomole 7.5 program package.69
For determining the spectral shift of a pure protein electrostatic origin, we chose the ChlD1 and PheoD1 cavities derived from the reaction center of Photosystem II. In each case, the native pigments of the cavity and surrounding protein environment were optimized using a QM/MM approach, as described previously.7,18 In the next step, all pigments under consideration were created using the skeleton of the native pigments and re-optimized (B3LYP/def2-TZVP) in the gas phase. Using the optimized structures, excited state calculations (ωB97X-V/def2-TZVP) were performed in the gas phase and subsequently within the point-charge field (electrostatic embedding) to determine the protein matrix induced spectral shift.
3. Results and discussion
The electronic spectroscopy of these systems is often approached in the framework of the Gouterman model,70 which describes the low-energy absorption bands (Q and B) in terms of transitions between the four frontier molecular orbitals HOMO−1, HOMO, LUMO, and LUMO+1. The crucial lowest-energy excitation (from the ground S0 to the S1 state, typically identified as the Qy transition) is derived principally from the HOMO → LUMO excitation with secondary contribution from HOMO−1 → LUMO+1.53 All these orbitals have π character and are delocalized over the macrocyclic ring. As a result, small structural changes and replacement of substituent groups between the different pigments would alter the energetics of the frontier molecular orbitals, which would give rise to changes in the excitation energy as well as in the nature of the excitation itself. This is clearly reflected in our calculations of the vertical excitation energies of the eight pigments using the previously benchmarked7,18,53 range-separated ωB97X-V functional65 within the full time-dependent density functional theory framework (Table 1). We find that the pigments of the bacteriochlorin class (BChl a, BChl b and BPheo a) are significantly red-shifted compared to chlorin class pigments (Chl a, Chl b, Chl d, Chl f and Pheo a). The computed energies for the first excited state are in line with experimentally known trends71–74 and reflect the large impact on the S0 → S1 excitation energy of seemingly small differences in the π-conjugated system (e.g. C7–C8 bond) between the bacteriochlorin and chlorin ring types. Furthermore, significant differences in excitation energies are observed within each class, demonstrating the distinct role and impact of the different substituent groups.| Pigment | Energy (eV) | λ (nm) | f osc | Frontier molecular orbitals |
|---|---|---|---|---|
| BChl a | 1.519 | 816 | 0.39 | HOMO → LUMO (0.94) |
| HOMO−1 → LUMO+1 (0.03) | ||||
| BChl b | 1.471 | 843 | 0.38 | HOMO → LUMO (0.93) |
| HOMO−1 → LUMO+1 (0.03) | ||||
| BPheo a | 1.598 | 776 | 0.35 | HOMO → LUMO (0.90) |
| HOMO−1 → LUMO+1 (0.07) | ||||
| Chl a | 1.977 | 627 | 0.24 | HOMO → LUMO (0.82) |
| HOMO−1 → LUMO+1 (0.12) | ||||
| Chl b | 2.125 | 583 | 0.16 | HOMO → LUMO (0.73) |
| HOMO−1 → LUMO+1 (0.16) | ||||
| Chl d | 1.860 | 667 | 0.24 | HOMO → LUMO (0.84) |
| HOMO−1 → LUMO+1 (0.12) | ||||
| Chl f | 1.895 | 654 | 0.29 | HOMO → LUMO (0.85) |
| HOMO−1 → LUMO+1 (0.11) | ||||
| Pheo a | 1.959 | 633 | 0.19 | HOMO → LUMO (0.76) |
| HOMO−1 → LUMO+1 (0.16) |
Differences are also observed between the two classes in terms of the contribution to the excitation in the canonical orbital basis, which serves to rationalize the electronic origin of the differences. Specifically, the lowest excitation in the bacteriochlorin class has an almost exclusive contribution from the HOMO → LUMO transition compared to chlorins, which feature an enhanced secondary HOMO−1 → LUMO+1 component. The molecular orbitals themselves have largely the same character among all pigments (see Fig. 1 for a comparison between BChl a and Chl a frontier orbitals), therefore they do not readily reveal the detailed intrinsic differences between pigments.
 | ||
| Fig. 1 Frontier molecular orbitals of BChl a and Chl a associated with low-lying excitations. | ||
Electronic excitations are associated with redistribution of electron density within the macrocyclic ring. The interaction of the ground state (GS, S0) and first excited state (ES, S1) electron density with the protein environment determines the relative stability of the states, and hence affects the excitation energy. Qualitatively, a red-shift in the excitation energy can be achieved through stabilization of the ES or destabilization of the GS, while an opposite effect on the ES and GS would lead to a blue-shift. A systematic understanding of these effects can be achieved through the difference electrostatic potential (ESP), i.e. of the S1 minus the S0 state, originating from the electron density redistribution. We note that the difference ESP maps have been used successfully in previous studies75–79 to study and understand the electrostatic tuning of pigments within various light harvesting assemblies. Fig. 2 shows the difference ESP maps for the Qy excitation for all the pigments under consideration in this work. The complete original ωB97X-V results for the S0, S1, and S1–S0 ESPs are available in the form of cube files as an open-access data set.80 The regions with the negative (red color) and positive (blue color) ESPs indicate accumulation and depletion of electron density, respectively. To reduce the complexity in discussing the difference ESP maps and also to encompass the entire π-cloud, we adopt a geometric partitioning of the pigment and the space around it based on four “quadrants” Q1–Q4, which correspond approximately to the −y, +x, +y, and −x directions or the A, B, C/E, and D rings of the macrocycle.
 | ||
| Fig. 2 Difference electrostatic potential map (S1 minus S0, scale in a.u.) associated with the S0 → S1 (Qy) excitation for a variety of naturally occurring photosynthetic pigments. | ||
It is clear from Fig. 2 that BChl a, BChl b, and BPheo a show a larger redistribution of electron density upon excitation as compared to the other pigments (Chl a, Chl b, Chl d, Chl f and Pheo a). We note that the transition dipole moment can serve as a simple qualitative indicator for predicting the magnitude of spectral shifts. However, it does not provide insight into spatially highly resolved aspects of electrostatic tuning. In contrast, the difference ESP maps provide a more fine-grained description of the intrinsic topological differences among the pigments. This is particularly noticeable for the chlorophylls. In the case of BChl a and BPheo a, the electron density shifts from Q3 to Q1 upon excitation, whereas BChl b shows significant involvement of the Q2 region as well. In the case of Chl a, Chl b, and Chl d, the electron density shifts from Q2 towards Q1, whereas in the case of Chl f the electron density shifts from Q2 towards Q1 and Q3. Interestingly, Pheo a shows the largest redistribution of electron density (from Q3 and Q2 towards Q1) within the chlorin class. In addition, it becomes evident that the electron density redistribution upon excitation is strongly polarized along the y-axis in bacteriochlorin class pigments in stark contrast to chlorophylls, which reflects the different nature of the lowest excitation.
The specific interaction of the external (protein) electrostatic environment with the electron density redistribution region would lead to a significant spectral shift due to stabilization/destabilization of the excited/ground state. In terms of the plots of Fig. 2, a red-shift can be produced by (a) placing a positive charge near the negative ESP region, or (b) placing a negative charge near the positive ESP region. Similarly, a blue-shift can be produced by (a) placing a positive charge near the positive ESP region, or (b) placing a negative charge near the negative ESP region. Based on the obtained maps, it becomes clear that the bacteriochlorin class pigments present a higher propensity of spectral tuning compared to chlorin pigments due to a larger electron density redistribution leading to more pronounced difference ESP. In the case of BChl a and BPheo a, significant electrostatic induced spectral shifts can only be produced when the charges are placed in the Q1 and Q3 regions. In contrast, BChl b presents higher flexibility in spectral tuning as it also offers the possibility for the protein matrix to “use” the Q2 alongside the Q1 and Q3 quadrants. In the case of the chlorophylls and the pheophytin, maximal spectral tuning regions for Chl a, Chl f and Pheo a are Q1, Q2, and Q3. In contrast, a significant spectral shift in the Chl b and Chl d can be obtained when charges are located in the Q1 and Q2 regions. We found that the Q4 region is not strongly involved in the spectral tuning in any of the pigments. This is interesting and illuminating because this region of space is actually occupied by the phytyl chain. In other words, the side of the pigment that the protein matrix would be least able to electrostatically modulate simply because of steric considerations is also intrinsically the part of the pigment that is least responsive to electrostatic effects.
It becomes evident from the present results that the spatial determinants of electrostatic spectral tuning are distinct for each pigment, even for pigments within the same class. To provide a proof of concept, we placed all the pigments in two genuine protein cavities to observe the protein electrostatic induced spectral shifts. These protein cavities are derived from the position of the ChlD1 (the native pigment is Chl a) and PheoD1 (the native pigment is Pheo a) pigments in the reaction center of Photosystem II, and have been refined through QM/MM simulations of a complete Photosystem II monomer.7,18 The ChlD1 and PheoD1 cavities are characterized by distinct electrostatic potentials (Fig. 3). Here, we report the spectral shifts of the pigments between the gas phase values and when placed in each of these two very different cavities. As shown in Fig. 3 and Table S1 (ESI‡), the spectral shift induced by each pocket to pigment is distinct, precisely due to the different effect of protein matrix electrostatics on the stability of the ground and excited states of the pigments as discussed above. Therefore, the difference ESP profiles in Fig. 2 serve as fingerprints to understand the role of the protein electrostatic preorganization in spectral tuning and can be useful not simply in understanding the matrix effect but potentially also in redesigning the cavities for tailored functionality.
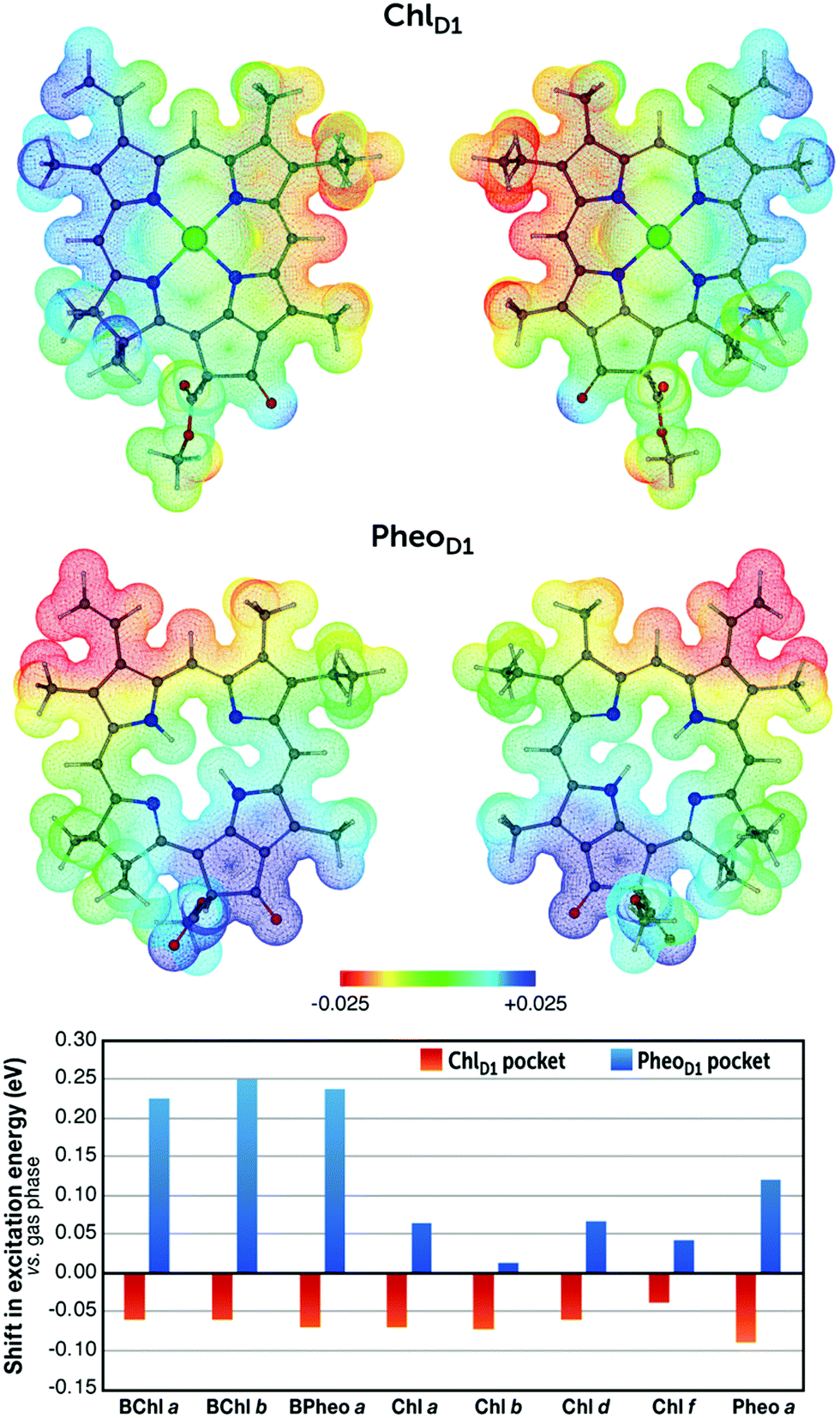 | ||
| Fig. 3 Top: Protein electrostatic potential at the ChlD1 and PheoD1 cavities of the reaction center of Photosystem II projected on the native ChlD1 (Chl a) and PheoD1 (Pheo a) pigments (scale in a.u.). Both “front” (left) and “back” (right) views are depicted. These plots were obtained by computing the difference between the electrostatic potential of the pigment with and without the protein matrix, represented as point-charges. The details of the associated molecular-mechanics model of Photosystem II are described elsewhere.7,18 Bottom: Spectral shift (cavity minus gas-phase) of the lowest excitation energy of the pigments considered in the present work, when placed in the corresponding protein cavities (see Table S1, ESI‡ for absolute values of excitation energies). | ||
Looking in greater detail at the cavity features (Fig. 3), it is seen that the electrostatic influence of the protein matrix on the ChlD1 cavity is mixed in nature. The protein exerts positive and negative ESP on the Q1 and Q2 region, respectively, and negative and positive ESP on the R12 and 13C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the Q3 region. Negative ESP on 10C–H is exerted, which is also found to be important for spectral tuning of all pigments in the present work. In the case of the PheoD1 pocket, the protein exerts a more directed influence compared to the ChlD1 cavity. Specifically, Q1 and Q3 experience a negative and positive ESP, respectively, whereas other regions experience rather neutral potential. Overall, only regions where the protein ESP influence is prominent can produce a significant spectral shift. Based on the complementarity of the identified regions for a spectral shift in the ChlD1 pocket and the difference ESP maps for various pigments, it can be qualitatively understood that the extent of red-shifting effects is higher than that of the blue-shifting effects, leading to a net small red-shift in the Qy excitation (see Table S2, ESI‡ for a more detailed analysis). On the other hand, in the case of the PheoD1 pocket, bacteriochlorin class pigments and Pheo a are expected to show large blue-shifts in Qy upon embedding, because of the clearly directional effect of the protein matrix, i.e. negative and positive ESPs in the Q1 and Q3 regions, and the near-perfect complementarity with the corresponding features of the intrinsic difference ESPs of these pigments (Fig. 2). By contrast, juxtaposition of Fig. 2 and 3 suggests that for chlorin-class pigments the extent of blue-shifting is expected to be slightly higher compared to the red-shifting effects in the PheoD1 pocket, leading to a net small blue-shift in the Qy excitation upon embedding (see Table S3, ESI‡). These deductions are qualitative or semi-quantitative in nature, but they are fully in line with the numerically precise, explicitly computed QM/MM shifts reported in Fig. 3 and Table S1 (ESI‡). On an altogether different level of analysis, a complete breakdown of the distinct protein matrix components that contribute to the total shifts can be very useful to pinpoint major contributors of electrostatic control. This arduous task has been demonstrated in the past for photosynthetic systems and can be achieved, for example, by systematically omitting the charges of specific amino acid residues or cofactors in an extended series of QM/MM calculations.7,22,26,81
O groups of the Q3 region. Negative ESP on 10C–H is exerted, which is also found to be important for spectral tuning of all pigments in the present work. In the case of the PheoD1 pocket, the protein exerts a more directed influence compared to the ChlD1 cavity. Specifically, Q1 and Q3 experience a negative and positive ESP, respectively, whereas other regions experience rather neutral potential. Overall, only regions where the protein ESP influence is prominent can produce a significant spectral shift. Based on the complementarity of the identified regions for a spectral shift in the ChlD1 pocket and the difference ESP maps for various pigments, it can be qualitatively understood that the extent of red-shifting effects is higher than that of the blue-shifting effects, leading to a net small red-shift in the Qy excitation (see Table S2, ESI‡ for a more detailed analysis). On the other hand, in the case of the PheoD1 pocket, bacteriochlorin class pigments and Pheo a are expected to show large blue-shifts in Qy upon embedding, because of the clearly directional effect of the protein matrix, i.e. negative and positive ESPs in the Q1 and Q3 regions, and the near-perfect complementarity with the corresponding features of the intrinsic difference ESPs of these pigments (Fig. 2). By contrast, juxtaposition of Fig. 2 and 3 suggests that for chlorin-class pigments the extent of blue-shifting is expected to be slightly higher compared to the red-shifting effects in the PheoD1 pocket, leading to a net small blue-shift in the Qy excitation upon embedding (see Table S3, ESI‡). These deductions are qualitative or semi-quantitative in nature, but they are fully in line with the numerically precise, explicitly computed QM/MM shifts reported in Fig. 3 and Table S1 (ESI‡). On an altogether different level of analysis, a complete breakdown of the distinct protein matrix components that contribute to the total shifts can be very useful to pinpoint major contributors of electrostatic control. This arduous task has been demonstrated in the past for photosynthetic systems and can be achieved, for example, by systematically omitting the charges of specific amino acid residues or cofactors in an extended series of QM/MM calculations.7,22,26,81
Hydrogen bonding is another important factor in spectral tuning of photosynthetic pigments9 that also has a dominant electrostatic component. From a structural point of view, each pigment can be a hydrogen bond acceptor through the 13CO group. In addition, bacteriochlorins can simultaneously hydrogen bond through the acetyl group (–COCH3), while among the chlorophylls only Chl b, Chl d and Chl f can form additional hydrogen bonds at the formyl group (–CHO). The nature of the spectral shift through hydrogen bonds can also be predicted qualitatively based on the difference ESP maps of Fig. 2. For example, a hydrogen bond to the 13C = O moiety is expected to result in a significant blue-shift in the case of BChl a, BPheo a and Pheo a, but only a slight blue-shift in the case of BChl b. Interestingly, a hydrogen bond to the acetyl group would result in a significant red-shift in the case of the bacteriochlorin pigments. The spectral shift due to a hydrogen bond to the 13CO moiety in the case of Chl a and Chl d would be negligible, whereas a red-shift can be expected in the case of Chl b and Chl f. A hydrogen bond to the formyl group in the case of Chl b would produce a blue-shift, whereas a strong and moderate red-shift is expected for Chl d and Chl f, respectively. We tested all these possibilities using explicit calculations with a water molecule playing the role of a hydrogen bond donor. The results, detailed in Table S4 (ESI‡), demonstrate that the effect of hydrogen bonding is overall smaller in magnitude compared to protein electrostatics, nevertheless the difference ESP maps of Fig. 2 are highly predictive also with regard to hydrogen bond induced spectral tuning.
Experimental results obtained by Saga et al.46–48,51 on the reconstituted light-harvesting complex 2 of purple photosynthetic bacteria (where the native pigment is BChl a) with a variety of non-native pigments (BChl b and Chl a, b, d, and f) provide a solid example that connects directly to the fine-grained contrasts between the pigments discussed in the present work. Upon substitution with non-native pigments, Saga et al. observed a red-shift in the Qy band for all pigments (Qy band measured in acetone is taken as reference in their work), however they observed that the magnitude of this red-shift differs. For instance, BChl a and BChl b pigments were red-shifted by 32 nm (0.06 eV) and 37 nm (0.07 eV), respectively. In the case of chlorin class pigments, Chl d demonstrated the largest red-shift of 21 nm (0.05 eV) followed by Chl f (14 nm, 0.04 eV), Chl a (8 nm, 0.02 eV) and Chl b (4 nm, 0.01 eV). These trends show not only that bacteriochlorophylls possess a higher propensity for spectral tuning compared to the chlorophylls – which is to be expected – but also imply that the spatial determinants of electrostatic tuning are distinct for each pigment, in line with the distinct difference-ESP maps discussed here. Interestingly, it was found that hydrogen bonding interaction from the protein cavity (βArg20) to the 3-formyl group of the Chl d produces a larger red-shift (0.03 eV) in the Qy band when compared with the 2-formyl group containing the Chl f pigment.46 These results are also in line with the findings of Swainsbury et al.44 where mutation of the hydrogen bond donating residue with a hydrophobic residue caused a higher blue-shift in the Qy band of the Chl d (12 nm, 0.04 eV) compared to Chl f (4 nm, 0.01 eV). Similar observations are directly deducible from the current work, where simply based on the difference ESP maps a strong and moderate red-shift is predicted for Chl d and Chl f, respectively, due to hydrogen bonding interactions. A more detailed analysis of the topological determinants of spectral tuning in specific pigment-substituted systems will be presented in forthcoming work.
4. Conclusions and perspectives
In conclusion, we demonstrated the principal topological features of electrostatically induced spectral tuning in eight naturally occurring photosynthetic pigments by analyzing the profiles of S1–S0 difference electrostatic potentials that intrinsically characterize each pigment. The difference ESP maps reveal the spatial predisposition of the pigments to electrostatic modulation of their lowest excitation (or site energy in the case of protein-embedded pigments). These profiles are fundamentally distinct and show that each pigment is inherently amenable to spectral tuning through a different and unique distribution of matrix electrostatics. The utility of these profiles is demonstrated by QM/MM embedding of the different pigments in two well-defined but physically distinct cavities of Photosystem II, but they are also important analytical aids in the expanding field of pigment-substituted systems. The principles described here are not only useful in rationalizing the protein matrix control of site energies in physiological states of photosynthetic systems, but they also offer concrete and specific guidelines for protein engineering.The concept of spatially defined electrostatically induced spectral tuning provides a strong dimensionality reduction by reducing the protein search space in terms of the most important quadrants that differ intrinsically between different pigments, and by determining the electronic nature of a site mutation by correlating it topologically with the intrinsic S1–S0 difference ESP distribution. With such an approach, protein cavities and matrices may be redesigned with minimal intervention (site-directed mutagenesis82) to maximally control the impact of protein electrostatics on spectral tuning of a given pigment. In addition, the directions developed above can contribute to the chemical identification of specific pigments in cases where this is difficult from structural data alone (for example because of intrinsic limitations of methods such as cryo-electron microscopy83), but they can also aid in the selection of non-native – and possibly synthetic84 – pigments suitable for certain cavities in pigment-reconstituted proteins,44–48,50,51 for example with the aim of predictably expanding the solar absorption spectrum or otherwise tailoring photosynthetic efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Max Planck Society for financial support. Open Access funding provided by the Max Planck Society.References
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, John Wiley & Sons, 2014 Search PubMed.
- R. Croce and H. van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS PubMed.
- H. van Amerongen and R. Croce, Photosynth. Res., 2013, 116, 251–263 CrossRef CAS PubMed.
- R. Croce and H. van Amerongen, Science, 2020, 369, eaay2058 CrossRef CAS PubMed.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. Van Grondelle, Govindjee and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- C. Curutchet and B. Mennucci, Chem. Rev., 2017, 117, 294–343 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2020, 142, 18174–18190 CrossRef CAS PubMed.
- Y. Lahav, D. Noy and I. Schapiro, Phys. Chem. Chem. Phys., 2021, 23, 6544–6551 RSC.
- M. J. Llansola-Portoles, F. Li, P. Xu, S. Streckaite, C. Ilioaia, C. Yang, A. Gall, A. A. Pascal, R. Croce and B. Robert, Biochim. Biophys. Acta, Bioenerg., 2019, 1861, 148078 CrossRef PubMed.
- D. Bednarczyk, O. Dym, V. Prabahar, Y. Peleg, D. H. Pike and D. Noy, Angew. Chem., Int. Ed., 2016, 55, 6901–6905 CrossRef CAS PubMed.
- G. Zucchelli, D. Brogioli, A. P. Casazza, F. M. Garlaschi and R. C. Jennings, Biophys. J., 2007, 93, 2240–2254 CrossRef CAS PubMed.
- L. De Vico, A. Anda, V. A. Osipov, A. Ø. Madsen and T. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9051–E9057 CrossRef CAS PubMed.
- M. Ketelaars, J.-M. Segura, S. Oellerich, W. P. F. de Ruijter, G. Magis, T. J. Aartsma, M. Matsushita, J. Schmidt, R. J. Cogdell and J. Köhler, J. Phys. Chem. B, 2006, 110, 18710–18717 CrossRef CAS PubMed.
- B. M. Bold, M. Sokolov, S. Maity, M. Wanko, P. M. Dohmen, J. J. Kranz, U. Kleinekathöfer, S. Höfener and M. Elstner, Phys. Chem. Chem. Phys., 2020, 22, 10500–10518 RSC.
- K. Saito, K. Mitsuhashi and H. Ishikita, J. Photochem. Photobiol., A, 2020, 402, 112799 CrossRef CAS.
- H. Tamura, K. Saito and H. Ishikita, Chem. Sci., 2021, 12, 8131–8140 RSC.
- F. Segatta, L. Cupellini, M. Garavelli and B. Mennucci, Chem. Rev., 2019, 119, 9361–9380 CrossRef PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2021, 17, 1858–1873 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, Chem. Sci., 2021, 12, 4463–4476 RSC.
- L. Cupellini, D. Calvani, D. Jacquemin and B. Mennucci, Nat. Commun., 2020, 11, 662 CrossRef CAS PubMed.
- V. Macaluso, G. Salvadori, L. Cupellini and B. Mennucci, Chem. Sci., 2021, 12, 5555–5565 RSC.
- F. Müh, M. Plöckinger and T. Renger, J. Phys. Chem. Lett., 2017, 8, 850–858 CrossRef.
- M. Saggu, S. D. Fried and S. G. Boxer, J. Phys. Chem. B, 2019, 123, 1527–1536 CrossRef CAS PubMed.
- M. A. Steffen, K. Lao and S. G. Boxer, Science, 1994, 264, 810–816 CrossRef CAS PubMed.
- Y. Orozco-Gonzalez, M. P. Kabir and S. Gozem, J. Phys. Chem. B, 2019, 123, 4813–4824 CrossRef CAS PubMed.
- H. Tamura, K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16373–16382 CrossRef CAS PubMed.
- X. Zhou, D. Sundholm, T. A. Wesołowski and V. R. I. Kaila, J. Am. Chem. Soc., 2014, 136, 2723–2726 CrossRef CAS PubMed.
- V. R. I. Kaila, R. Send and D. Sundholm, Phys. Chem. Chem. Phys., 2013, 15, 4491–4495 RSC.
- F. Melaccio, N. Ferré and M. Olivucci, Phys. Chem. Chem. Phys., 2012, 14, 12485–12495 RSC.
- R. Liang, J. K. Yu, J. Meisner, F. Liu and T. J. Martinez, J. Am. Chem. Soc., 2021, 143, 5425–5437 CrossRef CAS PubMed.
- M. Wanko, M. Hoffmann, P. Strodel, A. Koslowski, W. Thiel, F. Neese, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2005, 109, 3606–3615 CrossRef CAS PubMed.
- M. Hoffmann, M. Wanko, P. Strodel, P. H. König, T. Frauenheim, K. Schulten, W. Thiel, E. Tajkhorshid and M. Elstner, J. Am. Chem. Soc., 2006, 128, 10808–10818 CrossRef CAS PubMed.
- F. Collette, T. Renger, F. Müh and M. Schmidt am Busch, J. Phys. Chem. B, 2018, 122, 4828–4837 CrossRef CAS PubMed.
- J. Neugebauer, ChemPhysChem, 2009, 10, 3148–3173 CrossRef CAS PubMed.
- D. J. Nürnberg, J. Morton, S. Santabarbara, A. Telfer, P. Joliot, L. A. Antonaru, A. V. Ruban, T. Cardona, E. Krausz, A. Boussac, A. Fantuzzi and A. W. Rutherford, Science, 2018, 360, 1210–1213 CrossRef PubMed.
- F. Gan, G. Shen and D. A. Bryant, Life, 2014, 5, 4–24 CrossRef PubMed.
- F. Gan, S. Zhang, N. C. Rockwell, S. S. Martin, J. C. Lagarias and D. A. Bryant, Science, 2014, 345, 1312–1317 CrossRef CAS PubMed.
- M. Tros, V. Mascoli, G. Shen, M.-Y. Ho, L. Bersanini, C. J. Gisriel, D. A. Bryant and R. Croce, Chem, 2021, 7, 155–173 CAS.
- V. Mascoli, L. Bersanini and R. Croce, Nat. Plants, 2020, 6, 1044–1053 CrossRef CAS PubMed.
- M.-Y. Ho, G. Shen, D. P. Canniffe, C. Zhao and D. A. Bryant, Science, 2016, 353, aaf9178 CrossRef PubMed.
- C. Gisriel, G. Shen, V. Kurashov, M.-Y. Ho, S. Zhang, D. Williams, J. H. Golbeck, P. Fromme and D. A. Bryant, Sci. Adv., 2020, 6, eaay6415 CrossRef CAS PubMed.
- V. Kurashov, M.-Y. Ho, G. Shen, K. Piedl, T. N. Laremore, D. A. Bryant and J. H. Golbeck, Photosynth. Res., 2019, 141, 151–163 CrossRef CAS PubMed.
- K. Kato, T. Shinoda, R. Nagao, S. Akimoto, T. Suzuki, N. Dohmae, M. Chen, S. I. Allakhverdiev, J.-R. Shen, F. Akita, N. Miyazaki and T. Tomo, Nat. Commun., 2020, 11, 238 CrossRef CAS PubMed.
- D. J. K. Swainsbury, K. M. Faries, D. M. Niedzwiedzki, E. C. Martin, A. J. Flinders, D. P. Canniffe, G. Shen, D. A. Bryant, C. Kirmaier, D. Holten and C. N. Hunter, Biochim. Biophys. Acta, Bioenerg., 2019, 1860, 209–223 CrossRef CAS PubMed.
- E. Elias, N. Liguori, Y. Saga, J. Schäfers and R. Croce, Biomacromolecules, 2021, 22, 3313–3322 CrossRef CAS PubMed.
- Y. Saga, A. Tanaka, M. Yamashita, T. Shinoda, T. Tomo and Y. Kimura, Photochem. Photobiol., 2021 DOI:10.1111/php.13491.
- Y. Saga, M. Yamashita, M. Imanishi and Y. Kimura, Chem. Lett., 2018, 47, 1071–1074 CrossRef CAS.
- Y. Saga, M. Yamashita, M. Imanishi, Y. Kimura, Y. Masaoka, T. Hidaka and Y. Nagasawa, ACS Omega, 2020, 5, 6817–6825 CrossRef CAS PubMed.
- M. Germano, C. C. Gradinaru, A. Y. Shkuropatov, I. H. M. van Stokkum, V. A. Shuvalov, J. P. Dekker, R. van Grondelle and H. J. van Gorkom, Biophys. J., 2004, 86, 1664–1672 CrossRef CAS PubMed.
- M. Tros, L. Bersanini, G. Shen, M.-Y. Ho, I. H. M. van Stokkum, D. A. Bryant and R. Croce, Biochim. Biophys. Acta, Bioenerg., 2020, 1861, 148206 CrossRef CAS PubMed.
- Y. Saga, M. Yamashita, Y. Masaoka, T. Hidaka, M. Imanishi, Y. Kimura and Y. Nagasawa, J. Phys. Chem. B, 2021, 125, 2009–2017 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- A. Sirohiwal, R. Berraud-Pache, F. Neese, R. Izsák and D. A. Pantazis, J. Phys. Chem. B, 2020, 124, 8761–8771 CrossRef CAS PubMed.
- D. Sundholm, Chem. Phys. Lett., 2000, 317, 545–552 CrossRef CAS.
- D. Sundholm, Chem. Phys. Lett., 1999, 302, 480–484 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- O. A. Vydrov and T. V. Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- TURBOMOLE V7.5 2020, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.org.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- L. H. Andersen, E. Gruber, C. Kjær and S. Brøndsted Nielsen, Chem. – Eur. J, 2019, 25, 9153–9158 CrossRef PubMed.
- B. F. Milne, Y. Toker, A. Rubio and S. B. Nielsen, Angew. Chem., Int. Ed., 2015, 54, 2170–2173 CrossRef CAS PubMed.
- D. M. Niedzwiedzki, H. Liu, M. Chen and R. E. Blankenship, Photosynth. Res., 2014, 121, 25–34 CrossRef CAS PubMed.
- D. M. Niedzwiedzki and R. E. Blankenship, Photosynth. Res., 2010, 106, 227–238 CrossRef CAS PubMed.
- F. Müh, M. E.-A. Madjet, J. Adolphs, A. Abdurahman, B. Rabenstein, H. Ishikita, E.-W. Knapp and T. Renger, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16862–16867 CrossRef PubMed.
- J. Adolphs, F. Müh, M. E.-A. Madjet and T. Renger, Photosynth. Res., 2007, 95, 197 CrossRef PubMed.
- T. Renger and F. Müh, Phys. Chem. Chem. Phys., 2013, 15, 3348–3371 RSC.
- F. Müh, D. Lindorfer, M. Schmidt am Busch and T. Renger, Phys. Chem. Chem. Phys., 2014, 16, 11848–11863 RSC.
- F. Müh, M. Plöckinger, H. Ortmayer, M. Schmidt am Busch, D. Lindorfer, J. Adolphs and T. Renger, J. Photochem. Photobiol., B, 2015, 152, 286–300 CrossRef PubMed.
- A. Sirohiwal and D. A. Pantazis, Max Planck Society, 2021, DOI:10.17617/3.7f.
- F. C. Ramos, M. Nottoli, L. Cupellini and B. Mennucci, Chem. Sci., 2019, 10, 9650–9662 RSC.
- Y. Takegawa, M. Nakamura, S. Nakamura, T. Noguchi, J. Sellés, A. W. Rutherford, A. Boussac and M. Sugiura, Biochim. Biophys. Acta, Bioenerg., 2019, 1860, 297–309 CrossRef CAS PubMed.
- C. J. Gisriel, J. Wang, G. W. Brudvig and D. A. Bryant, Commun. Biol., 2020, 3, 408 CrossRef CAS PubMed.
- M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344–535 CrossRef CAS PubMed.
Footnotes |
| † Original ωB97X-V ESP data are provided as an open-access data set hosted by the Open Research Data Repository of the Max Planck Society at https://dx.doi.org/10.17617/3.7f. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02580e |
| This journal is © the Owner Societies 2021 |