
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light- and temperature-dependent dynamics of chromophore and protein structural changes in bathy phytochrome Agp2†
Galaan
Merga
a,
Maria Fernandez
Lopez
b,
Paul
Fischer
a,
Patrick
Piwowarski
a,
Żaneta
Nogacz
a,
Anastasia
Kraskov
b,
David
Buhrke‡
 b,
Francisco Velazquez
Escobar
b,
Norbert
Michael
b,
Friedrich
Siebert
c,
Patrick
Scheerer
d,
Franz
Bartl
a and
Peter
Hildebrandt
*b
b,
Francisco Velazquez
Escobar
b,
Norbert
Michael
b,
Friedrich
Siebert
c,
Patrick
Scheerer
d,
Franz
Bartl
a and
Peter
Hildebrandt
*b
aHumboldt Universität zu Berlin, Institut für Biologie, Biophysikalische Chemie, Invalidenstr. 42, D-10115 Berlin, Germany
bTechnische Universität Berlin, Institut für Chemie, Sekr. PC14, Straße des 17. Juni 135, D-10623 Berlin, Germany. E-mail: hildebrandt@chem.tu-berlin.de
cAlbert-Ludwigs-Universität Freiburg, Institut für Molekulare Medizin und Zellforschung, Sektion Biophysik, Hermann-Herderstr. 9, D-79104 Freiburg, Germany
dCharité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Institute of Medical Physics and Biophysics, Group Protein X-ray Crystallography and Signal Transduction, Charitéplatz 1, D-10117 Berlin, Germany
First published on 10th August 2021
Abstract
Bacterial phytochromes are sensoric photoreceptors that transform light absorbed by the photosensor core module (PCM) to protein structural changes that eventually lead to the activation of the enzymatic output module. The underlying photoinduced reaction cascade in the PCM starts with the isomerization of the tetrapyrrole chromophore, followed by conformational relaxations, proton transfer steps, and a secondary structure transition of a peptide segment (tongue) that is essential for communicating the signal to the output module. In this work, we employed various static and time-resolved IR and resonance Raman spectroscopic techniques to study the structural and reaction dynamics of the Meta-F intermediate of both the PCM and the full-length (PCM and output module) variant of the bathy phytochrome Agp2 from Agrobacterium fabrum. In both cases, this intermediate represents a branching point of the phototransformation, since it opens an unproductive reaction channel back to the initial state and a productive pathway to the final active state, including the functional protein structural changes. It is shown that the functional quantum yield, i.e. the events of tongue refolding per absorbed photons, is lower by a factor of ca. two than the quantum yield of the primary photochemical process. However, the kinetic data derived from the spectroscopic experiments imply an increased formation of the final active state upon increasing photon flux or elevated temperature under photostationary conditions. Accordingly, the branching mechanism does not only account for the phytochrome's function as a light intensity sensor but may also modulate its temperature sensitivity.
Introduction
Phytochromes are ubiquitous photoreceptors for controlling physiological processes in plants, bacteria, and fungi.1–3 They convert light energy, absorbed by a tetrapyrrole chromophore, into structural changes of the protein, which initiate the downstream signaling cascade. These structural changes may either lead to the formation of an interaction site for a reaction partner as suggested for plant phytochromes,2,4 or to the activation of an enzymatic output module, typically a histidine kinase, in prokaryotic phytochromes.1,3,5 The light-induced reaction sequence starts with the photoisomerisation of the chromophore to switch between a ZZZssa (Pr state) and ZZEssa configuration (Pfr state), followed by structural relaxations of the chromophore and the immediate protein environment up to the stage of the Meta intermediate states (Fig. 1). The subsequent protein structural changes that take place during the final step of the reaction cascade comprise a secondary structure transition of the tongue segment in the PHY domain between a largely α-helical and a β-sheet structure.6 This transition is then coupled to further extended secondary and/or tertiary structural changes and, in bacterial phytochromes, eventually communicated to the output module.6–8 | ||
| Fig. 1 Simplified reaction scheme of the photocycle of bathy phytochrome Agp2. The states in the dotted boxes on the left and right side include the chromophore in a ZZEssa and ZZZssa configuration, respectively. The states in the red and blue shaded regions possess an α-helical and β-sheet tongue segment, respectively. Thus, the reactions k1 and k2 reflect the α-helix → β-sheet secondary structure transition of the tongue and the thermal ZZZssa → ZZEssa chromophore isomerization, respectively. The thermal Pr → Pfr reversion k3 involves both processes. | ||
Detailed information about the molecular processes associated with the decay of the Meta states has been accumulated for the bathy phytochrome Agp2 (Agrobacterium fabrum) in which Pfr is the resting state (Fig. 1).3,9–14 Structural, spectroscopic, and theoretical studies have identified several molecular events in the chromophore binding pocket that are linked to the structural changes of the tongue, including the deprotonation of the ring C propionic side chain (propC) of the biliverdin (BV) chromophore and the repositioning of key amino acid side chains including Arg242, Arg211, Tyr205, His278, Tyr165, and Phe192.12,14 Of particular importance is the proton transfer from propC to His278,10 that is assisted by Arg211 and Tyr165.14 This proton translocation exerts two functions. First, it is essential for the structural transition of the tongue. Experimental and theoretical studies on Agp2 variants with site-specific amino acid substitutions and chemically modified BV chromophores indicate that the protonation-linked changes of the electrostatics in the chromophore binding pocket constitute the main determinant for inducing the protein structural changes.11,14 Second, proton transfer from propC to His278 promotes the thermal back conversion to the Pfr state (reaction step k3 in Fig. 1).
However, for the photosensor core module (PCM) of Agp2 also an alternative thermal back conversion route has been recently discovered.15 It starts with the formation of Meta-F and leads directly back to Pfr without undergoing the protein structural changes during the Meta-F to Pr transition (Fig. 1). A similar side reaction was found for the photoconversion of Pr of the prototypical phytochrome Agp1 also from A. fabrum suggesting that branching between a productive and unproductive route is a general phenomenon of phytochromes. However, so far, this reaction mechanism has only been detected for the truncated PCM variants while it remains to be shown for full-length phytochromes, which is the main objective of the present study. Furthermore, we ask for the parameters, which control switching between both reaction pathways. Employing resonance Raman (RR) and infrared (IR) spectroscopic techniques, it is shown that the branching mechanism substantially reduces the photoreceptor's yield of functional structural changes per absorbed photon. The consequences of these findings for the sensory functions of phytochrome are discussed.
Material and methods
Materials
The full-length Agp2 (Agp2-FL) and the photosensor core module of Agp2 (Agp2-PCM) were expressed and purified as described previously.12,14 For spectroscopic experiments, the protein was dissolved in buffer (50 mM Tris/HCl, 5 mM EDTA, 300 mM NaCl, pH7.8) and concentrated using a Micropore 30 kD filter to a concentration of 50–90 mg mL−1.Spectroscopic measurements
Photoconversion of the sample was achieved by irradiation the Pfr (Pr) state at 785 nm (660 nm) using a light-emitting diode, providing a photon flux of ∼2 × 1021 (3 × 1020) photons per m2 per s. To establish a pure Pfr state as a starting point for the experiments, the sample was irradiated with 660 nm for 120 s at ambient temperature.For IR measurements the protein samples were placed in sample holders with a 6 μm PTFE-spacer. In cryogenic IR experiments, the sample was cooled to the desired temperature with an OptistatTNcryostat (Oxford Instruments) and the dark spectrum was recorded using a Bruker IFS66v/s spectrometer (2 cm−1 and 4 cm−1 spectral resolution at a time resolution of ca. 120 and 30 ms, respectively) equipped with a mercury cadmium telluride (MCT) detector (J15D series, EG&G Judson). The light-induced spectrum was recorded during irradiation for 0.5 or 120 s. The difference spectra calculated from the single-channel spectra before and after irradiation were preprocessed with the OPUS 7.5 software package (Bruker Optics, Karlsruhe, Germany).
Rapid scan IR measurements were performed with a Vertex 80v FTIR Spectrometer (Bruker Optics, Karlsruhe, Germany) equipped with a liquid N2 cooled MCT detector (Kolmar Technologies, Newburyport, MA, USA). Data acquisition rate and time resolution were 300 kHz and ca. 120 or 30 ms at 263 K. The single turnover illumination was performed with a 10 Hz pulsed Nd:YAG Powerlite 9010 LASER as pump source for a Horizon II optical parametric oscillator (Continuum, San Jose, CA, USA) set to 760 nm. After each illumination the protein was allowed to relax thermally to its dark adapted Pfr state. The presented data consists of an average of 80 single turnover measurements of the same sample. Difference spectra were calculated using averaged spectra recorded ca. 35 s before laser illumination as the reference. The IR data were analyzed via a SVD and rotation procedure as well as a subsequent global fit algorithm implemented in Octave 4.2.1.16,17 Further global fit analyses were performed with Origin 2019 (OriginLab Corporation, Northampton, MA, USA).
RR measurements were performed using a Bruker Fourier-transform Raman spectrometer RFS 100/S with 1064 nm excitation (Nd-YAG cw laser, line width 1 cm−1), equipped with a nitrogen-cooled cryostat from Resultec (Linkam). All spectra were recorded at 90 K with a laser power at the sample of 780 mW. In order to identify potential laser-induced damage of the phytochrome samples, spectra before and after a series of measurements were compared. In no case, changes between these control spectra were determined. For each spectrum the accumulation time was ca. 1 h. Photoconversion was achieved by irradiation with 785 nm for 1 min at the specified temperature. After irradiation and, in the case of freeze-quench RR experiments, a subsequent delay time in the dark, the sample was rapidly cooled to 90 K for RR measurements. The cooling rate was ca. 1 K s−1 such that within ca. 10 s the thermal processes of the protein were slowed down by a factor of 2–3 and essentially quenched after 30 s. RR spectra of the pure photoconversion products were obtained by subtracting residual contributions of the non-photolyzed Pfr. The spectra of individual states were analyzed by a band fitting procedure assuming Lorentzian band shapes. These Lorentzian functions obtained from the fits were then combined to yield the component spectrum of the respective state,17 which were used for a quantitative analysis of the spectra obtained from samples irradiated at different temperatures and different subsequent dark periods.
Results
Resonance Raman spectroscopy of cryogenically trapped photoconversion products
Intermediates of the Pfr photoconversion of Agp2 are obtained by irradiation of the dark state for 60 s at temperatures sufficient to block the thermal decay of the target intermediate. Immediately after irradiation, the sample is rapidly cooled to 90 K for RR measurements (Fig. 2). Mutual spectra subtraction of the unphotolyzed state and, if required, a global component analysis, yield the pure spectra of the respective intermediates.17,18 The same procedure was employed to obtain the spectrum of the Pr state whereas in this case the sample was irradiated at ambient temperature. In this study we are particularly interested in the Meta-F and Pr states which display very similar RR spectra.10 They mainly differ with respect to the position of the N–H in plane bending mode of the rings B and C which in Meta-F is observed at 1553 cm−1 but it is upshifted to 1571 cm−1 in Pr (Fig. 2). Further differences refer to bands of the enol tautomer in Pr (Fig. S1, ESI†).10 Irradiation of the sample at temperatures up to 253 K does not lead to the formation of Pr during the 60 s irradiation period as indicated by the lack of the 1571 cm−1 band (Fig. 2). Only at temperatures of 263 K and higher we note contributions of Pr but at T ≥ 273 K the Pfr contribution prevails. Similar results were obtained for Agp2-PCM, which displays essentially the same RR spectra of the intermediates and parent states as Agp2-FL.10,14 | ||
| Fig. 2 RR spectra of Agp2-FL measured at 90 K immediately after irradiation the sample with 785 nm at different temperatures as indicated in the figure. The blue, green (top), and red traces (bottom) refer to the pure spectra of Pr, Meta-F, and Pfr, respectively. The same color code holds for the peak labels that mark the C–D stretching (1622, 1619, and 1600 cm−1 in Pr, Meta-F, and Pfr, respectively) and the N–H in-plane bending of rings B and C (1571, 1553, 1549 cm−1 in Pr, Meta-F, and Pfr, respectively). In addition, for Meta-F the weakly Raman-active B–C stretching mode is observed at 1585 cm−1. | ||
Freeze-quench resonance Raman spectroscopy
To probe the thermal decay processes of Meta-F, we have irradiated the Pfr state of Agp2-FL samples at 243 and 253 K to start with an essentially pure Meta-F state. In these experiments, the sample was irradiated for 60 s at a given temperature (irradiation temperature), followed by relaxation times in the dark from 0 to 11 minutes. Subsequently, the relaxation was quenched by rapid cooling to 90 K. At this temperature, the RR spectra were measured (Fig. 3 and Fig. S2, ESI†) to allow the direct comparison with the spectra of the pure states obtained at the same temperature (Fig. S1, ESI†). Note that the spectrum measured after freeze-quenching immediately following irradiation actually refers to a relaxation time of 60 s (corresponding to the irradiation period) since relaxation starts with the onset of the irradiation. | ||
| Fig. 3 RR spectra of Agp2-FL measured at 90 K after irradiation the sample with 785 nm at 243 K for 60 s (tirr) and keeping the sample in the dark for different times δ, corresponding to total relaxation times of tirr + δ (indicated in the figure). The red, green, and gray traces refer to the component spectra of Pfr, Meta-F, and the apoprotein, respectively. No contribution of the Pr spectrum (blue trace, top) was detected at any delay time. | ||
For the series of measurements with irradiation temperatures at 243 and 253 K the contribution of Pfr increases with the relaxation time as reflected by the growing in of the characteristic Pfr bands in the region between 1500 and 1700 cm−1 (Fig. 3 and Fig. S2 ESI†), which is most appropriate for distinguishing between the various (intermediates) states of phytochromes. Concomitantly, the bands related to Meta-F decrease but there is no indication of a (transient) formation of the Pr state, shown by the lack of RR intensity at 1571 cm−1 (Fig. 3). In fact, a global analysis of all experimental spectra was achieved with fidelity, solely on the basis of the component spectra of Pfr and Meta-F and the contribution of the apoprotein (Fig. 3 and Fig. S2, ESI†). The relative spectral contributions of Pfr and Meta-F (Irel,i) were then converted to relative concentrations (crel,i) by determining the relative RR cross sections (frr,i).19 These quantities were evaluated by relating the intensity of the strongest chromophore band to that of the phenylalanine breathing mode of the protein (1005 cm−1) as an internal standard in the spectra of the pure states (Fig. S1, ESI†). Accordingly, the RR cross section of Pfr was found to be ca. two times higher than that of Meta-F (equal to the keto form of Pr). Thus, the relative spectral contributions of all n species involved were normalized to 1.0 according to eqn (1)
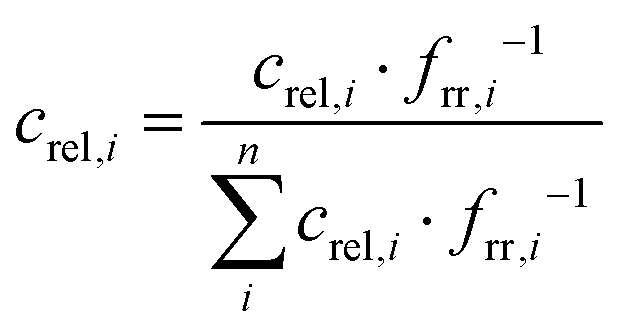 | (1) |
 | ||
| Fig. 4 Relative concentrations of Pfr of Agp2-FL as a function of the relaxation time after irradiation at 243 K (top, red symbols) and 253 K (bottom, blue symbols). The data were determined from the RR spectra in Fig. 3 and Fig. S2 (ESI†) as described in the text. The solid line represent simulations according to a kinetic model discussed in the text. | ||
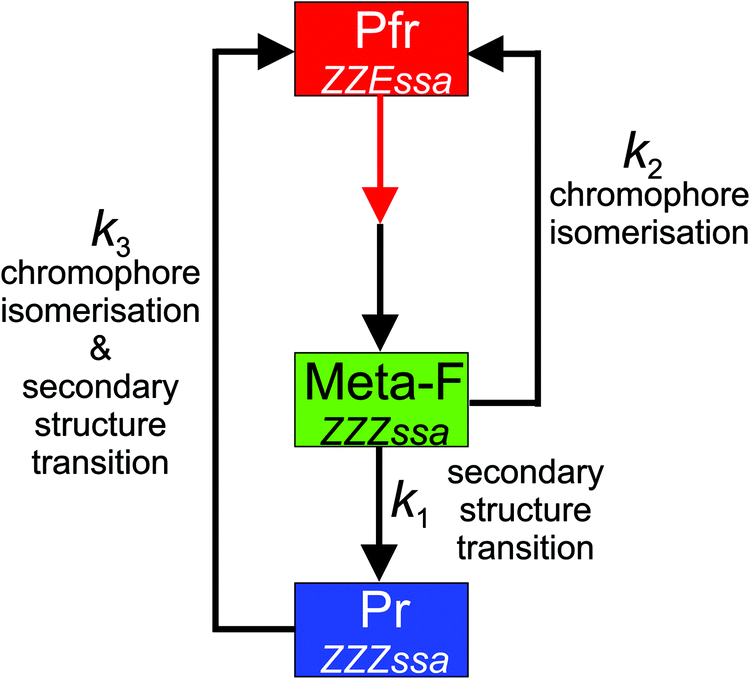 | ||
| Fig. 5 Kinetic model for the reactions of Meta-F. The black and red arrows indicate thermal and photochemical reactions. | ||
| T/K | k 1 | k 2 | k 3 | Method | |
|---|---|---|---|---|---|
| a IR, rapid-scan IR; FQ-RR; freeze-quench RR; UV, transient UV-vis absorption, data taken from ref. 14; TR-RR, pump–probe time-resolved RR, data taken from ref. 15. The average error associated with the rate constant determination was estimated to be ca. 10%. b Estimated from Arrhenius plots (Fig. S10, ESI). The average error of the activation energies was estimated to be between 15 and 30%, depending on the number of data points. | |||||
| Agp2-FL | 243 | 0.005 s−1 | 0.010 s−1 | 0.0009 s−1 | FQ-RR |
| 243 | <0.01 s−1 | 0.008 s−1 | — | IR | |
| 253 | 0.015 s−1 | 0.020 s−1 | 0.0018 s−1 | FQ-RR | |
| 253 | <0.02 s−1 | 0.022 s−1 | — | IR | |
| 263 | 0.15 s−1 | > 0.15 s−1 | 0.0027 s−1 | IR | |
| 283 | 1.1 s−1 | > 1.0 s−1 | 0.015 s−1 | IR | |
| 293 | — | — | 0.022 s−1 | UV | |
| Activation energyb | 80 kJ M−1 | 70 kJ M−1 | 40 kJ M−1 | ||
| Agp2-PCM | 243 | <0.005 s−1 | 0.0058 s−1 | — | IR |
| 253 | <0.015 s−1 | 0.013 s−1 | — | IR | |
| 263 | 0.13 s−1 | > 0.15 s−1 | 0.0036 s−1 | IR | |
| 283 | 0.87 s−1 | > 0.9 s−1 | 0.021 s−1 | IR | |
| 293 | 9.0 s−1 | 9.0 s−1 | 0.2 s−1 | TR-RR | |
| 293 | — | — | 0.12 s−1 | UV | |
| Activation energyb | 85 kJ M−1 | 90 kJ M−1 | 80 kJ M−1 | ||
Static IR difference spectroscopy
Irradiation-induced IR difference spectra of Agp2 were measured as a function of the temperature and irradiation time (Fig. 6). For the full-length protein, the difference spectrum at 248 K, corresponding to the Meta-F/Pfr difference spectrum, is similar to that at 183 K (Lumi-F/Pfr) inasmuch that no changes in the amide I band region are detected. The only difference between Lumi-F and Meta-F is a small 1 cm−1 shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the protonated propC. Increasing the temperature to 283 K leads to a decrease of the latter band concomitant to the growing in of positive and negative bands in the amide I band region. The amplitudes of the latter signals only slightly increase upon raising the temperature to 293 K. However, increasing the irradiation time from 0.5 to 120 s causes a substantial increase of the amide I signals such that one can clearly detect the formation of the β-hairpin and β-sheet structures (positive signals at 1641 and 1621 cm−1) in the Pr state at the expense of the coil and α-helical segments (negative signals at 1630 and 1656 cm−1) of the Pfr state. It is generally accepted that these spectral changes reflect the restructuring of the tongue segment in the PHY domain.10,20 Concomitantly, the CO stretching at 1756 cm−1 has disappeared indicating the deprotonation of the propC.
O stretching of the protonated propC. Increasing the temperature to 283 K leads to a decrease of the latter band concomitant to the growing in of positive and negative bands in the amide I band region. The amplitudes of the latter signals only slightly increase upon raising the temperature to 293 K. However, increasing the irradiation time from 0.5 to 120 s causes a substantial increase of the amide I signals such that one can clearly detect the formation of the β-hairpin and β-sheet structures (positive signals at 1641 and 1621 cm−1) in the Pr state at the expense of the coil and α-helical segments (negative signals at 1630 and 1656 cm−1) of the Pfr state. It is generally accepted that these spectral changes reflect the restructuring of the tongue segment in the PHY domain.10,20 Concomitantly, the CO stretching at 1756 cm−1 has disappeared indicating the deprotonation of the propC.
 | ||
| Fig. 6 IR difference spectra of Agp2-FL (left) and Agp2-PCM (right) measured during irradiation with 785 nm and at different temperatures, as indicated in the figure. Gray traces refer to irradiation times of 0.5 s (both Agp2-FL and Agp2-PCM) whereas blue and red traces represent the spectra with 120 s irradiation time of Agp2-FL and Agp2-PCM, respectively. The right panel (bottom) includes a comparison of the Agp2-PCM (red) and Agp2-FL (blue) both measured during an irradiation time of 120 s at 293 K. The red- and blue-shaded regions highlight the signals referring to the formation of the β-sheet (positive signals) and the decay of the α-helix structure (negative signals) of the tongue, respectively. The green-shaded region marks the CO stretching of the protonated propC. | ||
The PCM variant of Agp2 shows a different behavior compared to the full-length protein (Fig. 6). Short irradiation times (0.5 s) at 273 and 283 K do not cause appreciable changes in the amide I band region and also propC remains protonated. Increasing the irradiation time to 120 s, however, substantially enhances the difference signals, particularly pronounced in the region of the CO stretching of ring D. Furthermore, distinct signals in the amide I band are now detectable and their amplitudes increase with increasing temperature to 293 K, accompanied by the disappearance of the positive propC signal. Finally, the difference spectra of Agp2-FL and Agp2-PCM at 293 K and 120 s irradiation time become similar albeit not identical: the band originating from the α-helical segment is hardly visible in the Agp2-PCM spectrum which, in addition, shows a weaker positive CO stretching of ring D, due to the lower contribution of the keto tautomer at pH 7.8.10
Rapid scan IR difference spectroscopy
To determine the dynamics of the protein structural changes, a set of time-resolved IR difference spectra of Agp2-FL were recorded at temperatures between 243 and 283 K and analyzed by SVD and subsequent global fitting. At 243 K (Fig. 7A) and 253 K (Fig. S4, ESI†) the two main component spectra are essentially identical and agree very well with the Meta-F/Pfr difference spectrum (cf.Fig. 6), characterized by the lack of signals in the amide I region and the shift of the propC signal from 1750 (in Pfr) to 1757 cm−1 (in Meta-F). In this sense, the distinction of two quasi-identical components has no physical meaning. This is also true for the time traces (Fig. 7B and Fig. S4, ESI†) which for each component can be described by a bi-exponential function with the same time constants but different amplitudes. Accordingly, the kinetic data of the rapid scan IR measurements exclusively refer to the decay of Meta-F directly back to Pfr. The respective rate constant k2 (Fig. 5) was determined by the amplitude-weighted kinetic traces of the two components (Table 1). | ||
| Fig. 7 Single value decomposition (SVD) and global fit analysis of the data sets of rapid scan IR difference spectra of Agp2-FL measured at 243 (A and B) and 283 K (C and D). The component spectra are shown in the left panel whereas the right panel presents the global fits to the temporal evolution of the first (red) and second component (blue). The red- and blue-shaded regions in the component spectra highlight the signals referring to the formation of the β-sheet (positive signals) and the decay of the α-helix structure (negative signals) of the tongue, respectively. The green-shaded region marks the CO stretching of the protonated propC. | ||
The rapid scan IR experiments at 263 and 283 K reveal a different behavior (Fig. 7C, D and Fig. S5, ESI†). Here, the SVD analysis yields two different component spectra corresponding to the Meta-F/Pfr (red) and the Pr/Pfr (blue) difference spectra. In principle, the decay of the Meta-F/Pfr component should reflect the transitions both from Meta-F to Pfr (k2) and to Pr (k1). The latter rate constant also determines the rise time of the Pr/Pfr difference spectrum. However, for both 263 and 283 K, the decay time of the Meta-F/Pfr and the rise time of the Pr/Pfr components were the same within the experimental and fitting accuracy. This implies that at these temperatures the direct decay from Meta-F to Pfr cannot be resolved in the present IR measurements and must be faster than the transition to Pr, consistent with the results obtained at lower temperatures (Table 1).
Similar results were obtained from rapid-scan IR experiments of Agp2-PCM as shown in Fig. S6–S9 (ESI†). We also re-analysed the previously published pump–probe time-resolved RR data in terms of the three reaction steps k1, k2, and k3 (Fig. S4, ESI†).15 These data as well as the kinetic data from time-resolved UV-vis absorption spectroscopy for the Pr → Pfr back reaction (k3) are listed in Table 1.
Discussion
Vibrational spectroscopies have been widely used to study the structural and reaction dynamics of proteins, covering a wide range from femtoseconds to seconds.21,22 However, application of time-resolved IR and RR spectroscopy to phytochromes is still not routine work but represents a considerable challenge,15,23–32 since the specific kinetic and spectral properties of the photoreceptor imposes severe constraints on the experiments.In this work we have employed different RR and IR spectroscopic techniques to analyse the decay processes of Meta-F for Agp2-PCM and Agp2-FL. Each technique has its specific advantages and drawbacks. Rapid scan IR spectroscopy is not able to distinguish between the reaction steps k1 and k2 which are both associated with the decay of the same component spectrum. Freeze-quench RR spectroscopy can overcome this obstacle but the time-resolution is too poor to extend the approach to temperatures higher than 260 K. Pump–probe time-resolved RR spectroscopic experiments require large amounts of sample such that they were restricted to Agp2-PCM at ambient temperature.15 Finally, transient UV-vis absorption spectroscopy has a sufficient time-resolution and low sample demands but suffers from the lack of spectral resolution, i.e. it is no able to distinguish between Meta-F and Pr. Thus, we have taken the results obtained by various techniques to compile a limited data set which, however, allows for an approximate quantitative kinetic analysis on the basis of the simplest reaction scheme shown in Fig. 5.
Branching mechanism in Agp2-FL and Agp2-PCM
The IR and RR spectroscopic analysis of full-length Agp2 revealed a direct transition of the Meta-F state to Pfr. Thus, as in Agp2-PCM,15 the decay of Meta-F proceeds via two routes in Agp2-FL, i.e., a decay directly to Pfr prior to the α-helix/β-sheet conversion of the tongue (k2) and a reaction via Pr that involves this secondary structure change in the first step (Meta-F → Pr; k1) and its reversal in the second step (Pr → Pfr; k3) (Fig. 5). The respective rate constants are comparable for Agp2-PCM and Agp2-FL. Only the Pr → Pfr back reaction (k3) is faster in Agp2-PCM, particularly at higher temperatures. This is also reflected by the activation energies EA which are – within the accuracy of the limited number of data – in general similar for Agp2-FL and Agp2-PCM except for the distinctly lower value for k3 in Agp2-FL (Table 1 and Fig. S10, ESI†). Taking into account that all three processes represent complex reaction sequences, in a first approximation the activation energies estimated in this work may be associated with the respective rate-limiting steps. Accordingly, the data for k1 and k2 point to similar activation energies for the secondary structure transition and chromophore isomerization (90–70 kJ mol−1). Indeed, values of this magnitude have been reported for peptide unfolding.33–35 For the thermal double bond isomerization of tetrapyrroles no reference data are available such that we compare the present results with calculations for retinal chromophores.36,37 In these computational studies, the authors underline the importance of protein–chromophore interactions to lower the energy barrier for double bond isomerization which otherwise are clearly overestimated. This argument most likely also holds for the protein-bound tetrapyrroles in phytochromes where thermal isomerization is accelerated due to the involvement of a keto–enol tautomerisation as discussed previously.10 In view of these considerations it is quite surprising that the rate constants and activation energies for the Pr → Pfr conversion (k3), including chromophore isomerization and tongue refolding, differ substantially between Agp2-FL and Agp2-PCM. It might be that the presence of the output module does not only affect the keto–enol equilibrium of the tetrapyrrole10 but also the isomerisation kinetics.Molecular events controlling of the branching reaction
The Meta-F state is the starting point for two competitive reactions. In this state, the chromophore adopts a relaxed ZZZssa geometry and is protonated at both the ring B and C pyrrole nitrogens and also propC carries a proton. It has been shown that deprotonation of propC is a prerequisite for the gross structural transition of the tongue that takes place during the decay from Meta-F to Pr (k1), presumably due to the substantial change of the local electric field.14 Furthermore, the proton transfer from propC to His278 transiently stabilizes a reactive enol tautomer that thus can initiate thermal isomerization of the C–D methine bridge to yield a ZZEssa chromophore.10Accordingly, we now ask if deprotonation of propC precedes the Meta-F to Pr decay (k1 – tongue refolding) or also the Meta-F to Pfr back reaction (k2 – chromophore isomerisation). The present kinetic data do not provide an unambiguous answer but in this context it is interesting to refer to previous results on Agp2-PCM variants which do not form the Pr state.14 In these variants, the only route for Pfr recovery goes via the direct reaction Meta-F to Pfr (k2) which is ca. 200–3000 times slower for the truncated Agp2-PCM variants Y165F, Y165F/F192Y, and Agp2-PAiRFP2 (which form a final photoproduct that is not Pr) than for wild-type Agp2-PCM. The Agp2-PCM variant R211A, however, shows only a slightly slower Pfr recovery (3 times). These differences can be related to specific structural properties. Whereas in the Meta-F state of R211A, propC is only partly deprotonated (pKa ≈ 7.5) and the chromophore shows a small enol population, the propC pKa of the other three variants is distinctly higher than 9 and no enol contribution can be detected at pH 7.8. These observations imply that the fast Meta-F to Pfr reaction is linked to the transient proton-coupled enol formation of the chromophore that is facilitated compared to the other variants with much slower Meta-F to Pfr reactions. Thus we conclude that deprotonation of propC is in general a prerequisite for the thermal isomerization of the chromophore in Meta-F of Agp2 and hence is the molecular event that precedes the branching point.
Photostationary conditions
Due to the interference with the branching reaction, static IR light-induced difference spectroscopy critically depends not only on the temperature but also on the irradiation time. For strictly sequential reaction cascades, variation of the irradiation time can solely affect the extent of product formation and thus the amplitude of the difference signals. In contrast, for the branched reaction mechanism in Agp2 with its rapid short-cut back reaction to the dark state (Fig. 5), variation of the irradiation time may cause changes of the photoproduct distribution as shown in Fig. 6 by the appearance of new difference signals and altered relative signal amplitudes comparing spectra with 120 s and 0.5 s irradiation time. These changes reflect the time-dependent development of the photostationary equilibrium. In fact, one can easily show that with increasing irradiation time the stationary concentration of Pr increases (Fig. S11, ESI†). In the same way, one can rationalize the deviations between the Pr/Pfr difference spectra of Agp2-FL and Agp2-PCM under identical irradiation conditions since then the stationary Pr concentrations depends on the thermal rate constants k1, k2, and k3, which differ between the full-length and the truncated PCM variant.More importantly, the branching mechanism has significant consequences for the function of phytochromes since it substantially reduces the efficiency by which light absorption leads to a physiological response. Assuming a photochemical quantum yield of the Pfr → Lumi-F conversion in Agp2 that is similar to Agp1,38 only 8% of the photoexcited Agp2 reaches the Meta-F state. With a k2/k1 ratio ≥1 for the rate constants of Agp2-FL at ambient temperature (cf.Table 1), it follows that only a fraction of less than 4% of the originally photoexcited molecules decays to Pr under secondary structure refolding of the tongue, a prerequisite for communicating the light signal to the output module and inducing a physiological response in bacterial phytochromes. Accordingly, one may readily understand the low yield of Pr formation (ca. 6%) in the bathy phytochrome PaBphP1 from Pseudomonas aeruginosa.39 Thus, the branching mechanism supports and modulates phytochrome's function as a light intensity sensor,40 implying that the probability for initiating the downstream physiological processes increases with the photon flux. This is illustrated by a simulation of the Pr formation for the limiting case of photostationary conditions in Fig. S11 (ESI†).
In addition, phytochromes have also suggested to serve as temperature sensors.41–43 The underlying molecular mechanism is not known. Early proposals claim temperature-dependent structural changes,41 but also the interplay between photoconversions and thermal reversions may provide a possible explanation.43 This is specifically true if the light-dependent distribution between the active and inactive parent states (Pr, Pfr) is modulated two temperature-dependent transitions as discovered in this work (Fig. S12, ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through CRC 1078 (B5 to F. B.; B6 to P. S. and P. H.). Further support was obtained from the Einstein Center of Catalysis EC2 (to P. H. and P. S.).Notes and references
- N. C. Rockwell, Y.-S. Su and J. C. Lagarias, Phytochrome Structure and Signaling Mechanisms, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed.
- H. Wang, Phytochrome signaling: Time to tighten up the loose ends, Mol. Plant, 2015, 8, 540–551 CrossRef CAS PubMed.
- T. Lamparter, N. Krauß and P. Scheerer, Phytochromes from Agrobacterium fabrum, Photochem. Photobiol., 2017, 93, 642–655 CrossRef CAS PubMed.
- S. Nagano, K. Guan, S. Shenkutie, C. Feiler, M. Weiss, A. Kraskov, D. Buhrke, P. Hildebrandt and J. Hughes, Structural insights into photoactivation and signalling in plant phytochromes, Nat. Plants, 2020, 6, 581–588 CrossRef CAS PubMed.
- B. Karniol and R. D. Vierstra, The pair of bacteriophytochromes from Agrobacterium tumefaciens are histidine kinases with opposing photobiological properties, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2807–2812 CrossRef CAS PubMed.
- H. Takala, A. Björling, O. Berntsson, H. Lehtivuori, S. Niebling, M. Hoernke, I. Kosheleva, R. Henning, A. Menzel, J. A. Ihalainen and S. Westenhoff, Signal amplification and transduction in phytochrome photosensors, Nature, 2014, 509, 245–248 CrossRef CAS PubMed.
- G. Gourinchas, S. Etzl, C. Göbl, U. Vide, T. Madl and A. Winkler, Long-range allosteric signaling in red light–regulated diguanylyl cyclases, Sci. Adv., 2017, 3, 1–12 Search PubMed.
- E. S. Burgie, J. Zhang and R. D. Vierstra, Crystal Structure of Deinococcus Phytochrome in the Photoactivated State Reveals a Cascade of Structural Rearrangements during Photoconversion, Structure, 2016, 24, 448–457 CrossRef CAS.
- J. Salewski, F. V. Escobar, S. Kaminski, D. Von Stetten, A. Keidel, Y. Rippers, N. Michael, P. Scheerer, P. Piwowarski, F. Bartl, N. Frankenberg-Dinkel, S. Ringsdorf, W. Ga
![[r with combining umlaut]](https://www.rsc.org/images/entities/char_0072_0308.gif) tner, T. Lamparter, M. A. Mroginski and P. Hildebrandt, Structure of the biliverdin cofactor in the Pfr state of bathy and prototypical phytochromes, J. Biol. Chem., 2013, 288, 16800–16814 CrossRef CAS.
tner, T. Lamparter, M. A. Mroginski and P. Hildebrandt, Structure of the biliverdin cofactor in the Pfr state of bathy and prototypical phytochromes, J. Biol. Chem., 2013, 288, 16800–16814 CrossRef CAS. - F. Velazquez Escobar, P. Piwowarski, J. Salewski, N. Michael, M. Fernandez Lopez, A. Rupp, M. B. Qureshi, P. Scheerer, F. Bartl, N. Frankenberg-Dinkel, F. Siebert, M. A. Mroginski and P. Hildebrandt, A protonation-coupled feedback mechanism controls the signalling process in bathy phytochromes, Nat. Chem., 2015, 7, 423–430 CrossRef CAS PubMed.
- M. Fernandez Lopez, A. D. Nguyen, F. Velazquez Escobar, R. González, N. Michael, Ż. Nogacz, P. Piwowarski, F. Bartl, F. Siebert, I. Heise, P. Scheerer, W. Gärtner, M. A. Mroginski and P. Hildebrandt, Role of the Propionic Side Chains for the Photoconversion of Bacterial Phytochromes, Biochemistry, 2019, 58, 3504–3519 CrossRef CAS PubMed.
- A. Schmidt, L. Sauthof, M. Szczepek, M. F. Lopez, F. Velazquez Escobar, B. M. Qureshi, N. Michael, D. Buhrke, T. Stevens, D. Kwiatkowski, D. von Stetten, M. A. Mroginski, N. Krauß, T. Lamparter, P. Hildebrandt and P. Scheerer, Structural snapshot of a bacterial phytochrome in its functional intermediate state, Nat. Commun., 2018, 9, 1–13 CrossRef CAS.
- F. Velázquez Escobar, D. Buhrke, N. Michael, L. Sauthof, S. Wilkening, N. N. Tavraz, J. Salewski, N. Frankenberg-Dinkel, M. A. Mroginski, P. Scheerer, T. Friedrich, F. Siebert and P. Hildebrandt, Common Structural Elements in the Chromophore Binding Pocket of the Pfr State of Bathy Phytochromes, Photochem. Photobiol., 2017, 93, 724–732 CrossRef PubMed.
- A. Kraskov, A. D. Nguyen, J. Goerling, D. Buhrke, F. Velazquez Escobar, M. Fernandez Lopez, N. Michael, L. Sauthof, A. Schmidt, P. Piwowarski, Y. Yang, T. Stensitzki, S. Adam, F. Bartl, I. Schapiro, K. Heyne, F. Siebert, P. Scheerer, M. A. Mroginski and P. Hildebrandt, Intramolecular Proton Transfer Controls Protein Structural Changes in Phytochrome, Biochemistry, 2020, 59, 1023–1037 CrossRef CAS PubMed.
- D. Buhrke, U. Kuhlmann, N. Michael and P. Hildebrandt, The Photoconversion of Phytochrome Includes an Unproductive Shunt Reaction Pathway, ChemPhysChem, 2018, 19, 566–570 CrossRef CAS.
- E. Henry and J. Hofrichter, Singular value decomposition: Application to analysis of experimental data, Methods Enzymol., 1992, 210, 129–192 CAS.
- M. Elgeti, E. Ritter and F. J. Bartl, New Insights into Light-Induced Deactivation of Active Rhodopsin by SVD and Global Analysis of Time-Resolved UV/Vis- and FTIR-Data, Z. Phys. Chem., 2008, 222, 1117–1129 CrossRef CAS.
- F. Velazquez Escobar, C. Kneip, N. Michael, T. Hildebrandt, N. Tavraz, W. Gärtner, J. Hughes, T. Friedrich, P. Scheerer, M. A. Mroginski and P. Hildebrandt, The Lumi-R Intermediates of Prototypical Phytochromes, J. Phys. Chem. B, 2020, 124, 4044–4055 CrossRef CAS PubMed.
- B. Zienicke, I. Molina, R. Glenz, P. Singer, D. Ehmer, F. Velazquez Escobar, P. Hildebrandt, R. Diller and T. Lamparter, Unusual spectral properties of bacteriophytochrome Agp2 result from a deprotonation of the chromophore in the red-absorbing form Pr, J. Biol. Chem., 2013, 288, 31738–31751 CrossRef CAS.
- E. A. Stojković, K. C. Toh, M. T. A. Alexandre, M. Baclayon, K. Moffat and J. T. M. Kennis, FTIR spectroscopy revealing light-dependent refolding of the conserved tongue region of bacteriophytochrome, J. Phys. Chem. Lett., 2014, 5, 2512–2515 CrossRef PubMed.
- D. Buhrke and P. Hildebrandt, Probing Structure and Reaction Dynamics of Proteins Using Time-Resolved Resonance Raman Spectroscopy, Chem. Rev., 2020, 120, 3577–3630 CrossRef CAS PubMed.
- V. A. Lorenz-Fonfria, Infrared Difference Spectroscopy of Proteins: From Bands to Bonds, Chem. Rev., 2020, 120, 3466–3576 CrossRef CAS PubMed.
- K. C. Toh, E. A. Stojković, A. B. Rupenyan, I. H. M. Van Stokkum, M. Salumbides, M. L. Groot, K. Moffat and J. T. M. Kennis, Primary reactions of bacteriophytochrome observed with ultrafast mid-infrared spectroscopy, J. Phys. Chem. A, 2011, 115, 3778–3786 CrossRef CAS PubMed.
- Y. Yang, M. Linke, T. Von Haimberger, J. Hahn, R. Matute, L. González, P. Schmieder and K. Heyne, Real-time tracking of phytochrome's orientational changes during Pr photoisomerization, J. Am. Chem. Soc., 2012, 134, 1408–1411 CrossRef CAS PubMed.
- J. J. Van Thor, K. L. Ronayne and M. Towrie, Formation of the early photoproduct Lumi-R of cyanobacterial phytochrome Cph1 observed by ultrafast mid-infrared spectroscopy, J. Am. Chem. Soc., 2007, 129, 126–132 CrossRef CAS PubMed.
- J. Dasgupta, R. R. Frontiera, K. C. Taylor, J. C. Lagarias and R. A. Mathies, Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1784–1789 CrossRef CAS PubMed.
- P. Piwowarski, E. Ritter, K.-P. Hofmann, P. Hildebrandt, D. von Stetten, P. Scheerer, N. Michael, T. Lamparter and F. Bartl, Light-Induced Activation of Bacterial Phytochrome Agp1 Monitored by Static and Time-Resolved FTIR Spectroscopy, ChemPhysChem, 2010, 11, 1207–1214 CrossRef CAS PubMed.
- M. Kurttila, B. Stucki-Buchli, J. Rumfeldt, L. Schroeder, H. Häkkänen, A. Liukkonen, H. Takala, T. Kottke and J. A. Ihalainen, Site-by-site tracking of signal transduction in an azidophenylalanine-labeled bacteriophytochrome with step-scan FTIR spectroscopy, Phys. Chem. Chem. Phys., 2021, 23, 5615–5628 RSC.
- J. A. Ihalainen, E. Gustavsson, L. Schroeder, S. Donnini, H. Lehtivuori, L. Isaksson, C. Thöing, V. Modi, O. Berntsson, B. Stucki-Buchli, A. Liukkonen, H. Häkkänen, E. Kalenius, S. Westenhoff and T. Kottke, Chromophore-Protein Interplay during the Phytochrome Photocycle Revealed by Step-Scan FTIR Spectroscopy, J. Am. Chem. Soc., 2018, 140, 12396–12404 CrossRef CAS PubMed.
- N. Lenngren, P. Edlund, H. Takala, B. Stucki-Buchli, J. Rumfeldt, I. Peshev, H. Häkkänen, S. Westenhoff and J. A. Ihalainen, Coordination of the biliverdin D-ring in bacteriophytochromes, Phys. Chem. Chem. Phys., 2018, 20, 18216–18225 RSC.
- U. Choudry, D. J. Heyes, S. J. O. Hardman, M. Sakuma, I. V. Sazanovich, J. Woodhouse, E. De La Mora, M. N. Pedersen, M. Wulff, M. Weik, G. Schirò and N. S. Scrutton, Photochemical Mechanism of an Atypical Algal Phytochrome, ChemBioChem, 2018, 19, 1036–1043 CrossRef CAS.
- J. Kübel, M. Chenchiliyan, S. A. Ooi, E. Gustavsson, L. Isaksson, V. Kuznetsova, J. A. Ihalainen, S. Westenhoff and M. Maj, Transient IR spectroscopy identifies key interactions and unravels new intermediates in the photocycle of a bacterial phytochrome, Phys. Chem. Chem. Phys., 2020, 22, 9195–9203 RSC.
- I. K. Lednev, A. S. Karnoup, M. C. Sparrow and S. A. Asher, R -Helix Peptide Folding and Unfolding Activation Barriers: A Nanosecond UV Resonance Raman Study, J. Am. Chem. Soc., 1999, 121, 8074–8086 CrossRef CAS.
- C. Krejtschi and K. Hauser, Stability and folding dynamics of polyglutamic acid, Eur. Biophys. J., 2011, 40, 673–685 CrossRef CAS.
- D. R. Dee, Y. Horimoto and R. Y. Yada, Conserved Prosegment Residues Stabilize a Late- Stage Folding Transition State of Pepsin Independently of Ground States, PLoS One, 2014, 9, e101339 CrossRef PubMed.
- E. Tajkhorshid, B. Paizs and S. Suhai, Role of isomerization barriers in the pKa control of the retinal Schiff base: A density functional study, J. Phys. Chem. B, 1999, 103, 4518–4527 CrossRef CAS.
- S. Rinaldi, F. Melaccio, S. Gozem, F. Fanelli and M. Olivucci, Comparison of the isomerization mechanisms of human melanopsin and invertebrate and vertebrate rhodopsins, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1714–1719 CrossRef CAS PubMed.
- C. Schumann, R. Groß, M. M. N. Wolf, R. Diller, N. Michael and T. Lamparter, Subpicosecond midinfrared spectroscopy of the Pfr reaction of phytochrome Agp1 from Agrobacterium tumefaciens, Biophys. J., 2008, 94, 3189–3197 CrossRef CAS PubMed.
- E. Consiglieri, A. Gutt, W. Gärtner, L. Schubert, C. Viappiani, S. Abbruzzetti and A. Losi, Dynamics and efficiency of photoswitching in biliverdin-binding phytochromes, Photochem. Photobiol. Sci., 2019, 18, 2484–2496 CrossRef CAS PubMed.
- N. C. Rockwell, S. S. Martin and J. C. Lagarias, Red/green cyanobacteriochromes: Sensors of color and power, Biochemistry, 2012, 51, 9667–9677 CrossRef CAS PubMed.
- I. Njimona and T. Lamparter, Temperature effects on Agrobacterium phytochrome Agp1, PLoS One, 2011, 6(10), e25977 CrossRef CAS.
- Z. Yu, A. Ali, O. A. Igbalajobi, C. Streng, K. Leister, N. Krauß, T. Lamparter and R. Fischer, Two hybrid histidine kinases, TcsB and the phytochrome FphA, are involved in temperature sensing in Aspergillus nidulans, Mol. Microbiol., 2019, 112, 1814–1830 CrossRef CAS.
- C. Klose, F. Nagy and E. Schäfer, Thermal Reversion of Plant Phytochromes, Mol. Plant, 2020, 13, 386–397 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02494a |
| ‡ Present address: Department of Chemistry, University of Zürich, Winterthurerstr. 190, CH-8057 Zürich, Switzerland. |
| This journal is © the Owner Societies 2021 |