
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst hyperpolarizability of water at the air–vapor interface: a QM/MM study questions standard experimental approximations†
Guillaume
Le Breton
,
Oriane
Bonhomme
,
Pierre-François
Brevet
 ,
Emmanuel
Benichou
and
Claire
Loison
*
,
Emmanuel
Benichou
and
Claire
Loison
*
University of Lyon, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: claire.loison@univ-lyon1.fr
First published on 9th September 2021
Abstract
Surface Second-Harmonic Generation (S-SHG) experiments provide a unique approach to probe interfaces. One important issue for S-SHG is how to interpret the S-SHG intensities at the molecular level. Established frameworks commonly assume that each molecule emits light according to an average molecular hyperpolarizability tensor β(−2ω,ω,ω). However, for water molecules, this first hyperpolarizability is known to be extremely sensitive to their environment. We have investigated the molecular first hyperpolarizability of water molecules within the liquid–vapor interface, using a quantum description with explicit, inhomogeneous electrostatic embedding. The resulting average molecular first hyperpolarizability tensor depends on the distance relative to the interface, and it practically respects the Kleinman symmetry everywhere in the liquid. Within this numerical approach, based on the dipolar approximation, the water layer contributing to the Surface Second Harmonic Generation (S-SHG) intensity is less than a nanometer. The results reported here question standard interpretations based on a single, averaged hyperpolarizability for all molecules at the interface. Not only the molecular first hyperpolarizability tensor significantly depends on the distance relative to the interface, but it is also correlated to the molecular orientation. Such hyperpolarizability fluctuations may impact the S-SHG intensity emitted by an aqueous interface.
1 Introduction
Liquid water is ubiquitous on Earth; understanding the properties of its interfaces with other media is essential for many fields of physics, chemistry, and biology. Surface analytical tools have been developed to obtain information on the molecular organization at interfaces; in particular, non-linear optical techniques such as Surface Second Harmonic Generation (S-SHG) and Surface Sum Frequency Generation (S-SFG) are increasingly used to probe biological materials at interfaces,1–3 and to investigate the molecular structure of liquid–gas,4–9 liquid–liquid10,11 or liquid–solid12–16 interfaces. The SHG is an optical process whereby two photons at a fundamental frequency are converted into one photon at twice the fundamental frequency, i.e. the harmonic frequency. Coherent SHG techniques are surface-sensitive because of the cancellation of the SHG process within centrosymmetric media like bulk liquids17. Moreover, the SHG response is strongly dependent on the molecular arrangement and the molecular electrostatic environments,18,19 so that S-SHG has become a useful tool for probing molecular structure1,20,21 or surface electrostatics.2,8,9,12,14,22–24 However, linking the experimental S-SHG intensity to interface molecular structure often remains a challenge, for which different experimental characterizations,9,11 theoretical developments,17,25 and molecular modeling26–28 can benefit from each others.10,29–32The interpretation of the S-SHG signal of the water/air interface has been a subject of debate for decades.9,10,33 Efforts have been made to assess the contribution of the interface (local) contribution of “the Bonded Interface Layer” (BIL) and the one of bulk electric quadrupole and magnetic dipole contributions to the SHG signal appearing in the diffuse layer (DL).9,27
For a neat water/air interface, Dalstein et al.9 and Zhang et al.33 attribute the signal to the BIL, i.e. the layer where the inversion symmetry is broken by the interface-specific structure. Typically, under the electric dipole approximation, this contribution to the quadratic surface susceptibility tensor χ(2) is modeled as a sum of the molecular hyperpolarizability tensors β(−2ω,ω,ω)n, where n is not a power notation, but an index running over the molecules.10,33 To express χ(2) in the laboratory frame, one takes into account the orientation of the nth-molecule via the matrix Rn of its molecular coordinate system {a,b,c} in the laboratory frame  .10,33 To interpret the S-SHG signal of the water/air interface in terms of interface structure using this formalism, one therefore needs to know the hyperpolarizabilities of individual water molecules.33 Moreover, one considers that the signal can be interpreted in terms of molecular hyperpolarizability. The decomposition of the non linear optical response of a cluster into a sum of molecular properties is not obvious,36 since intermolecular interactions may strongly impact non linear optical properties (for a recent example see ref. 37). In the special case of water, Maroulis38 or Liang et al.39 compared the polarizabilities and/or hyperpolarizabilities of dimers to the sum of the properties of the individual molecules. Their results indicate that the single-molecule summation is a reasonable first step.
.10,33 To interpret the S-SHG signal of the water/air interface in terms of interface structure using this formalism, one therefore needs to know the hyperpolarizabilities of individual water molecules.33 Moreover, one considers that the signal can be interpreted in terms of molecular hyperpolarizability. The decomposition of the non linear optical response of a cluster into a sum of molecular properties is not obvious,36 since intermolecular interactions may strongly impact non linear optical properties (for a recent example see ref. 37). In the special case of water, Maroulis38 or Liang et al.39 compared the polarizabilities and/or hyperpolarizabilities of dimers to the sum of the properties of the individual molecules. Their results indicate that the single-molecule summation is a reasonable first step.
The molecular hyperpolarizability is commonly assumed to be the same for all the water molecules of a given phase, and respecting the molecular symmetry group C2v. These hypotheses permit to extract from the susceptibilities the mean molecular orientation – via the orientation matrices.4,20 However, it is known that solvation has a substantial effect on the measured hyperpolarizability of water molecules, and the liquid environment breaks the C2v symmetry.36,39–43 To reach quantitative interpretation of S-SHG signal of aqueous interfaces, the water hyperpolarizability must be known, precisely at these interfaces.
In particular, the origin of the failure of Kleinman symmetry observed experimentally for the neat air/water interface (χ(2)ZXX ≠ χ(2)XZX, for example) is debated.33,44,45 It was attributed by Zhang et al. to the asymmetry of the molecular hyperpolarizability, for instance to βaca ≠ βcaa or βbcb ≠ βcbb, where the c-axis is along the water dipole moment.33 A symmetry breaking of the hyperpolarizability tensor β indeed exists for water in the gas phase,10,33 but to our knowledge, it has been reported neither in the bulk liquid, nor at the interface.
In this article, we report theoretical chemistry calculations of the molecular hyperpolarizability of water molecules nearby the interface, and discuss the results in terms of the S-SHG measurements. Liquid water is a difficult system for computational modeling in general, and for the prediction of electric properties in particular because of the extended H-bond network.46,47 Modeling the molecular environment as homogeneous dielectric material is not sufficient to reproduce the sign change of water hyperpolarizability from the gas phase to the liquid phase.48 Here, we propose a sequential Quantum Mechanical/Molecular Mechanics (QM/MM) approach to provide new insights on the contribution of water molecular hyperpolarizability to the S-SHG signal of the liquid–vapor interface, see Fig. 1.
 | ||
| Fig. 1 Scheme of the numerical procedure (performed by the home-made software FROG). (a) Molecular dynamics snapshot of the water liquid–vapor simulation within a box of 5 × 5 × 40 nm3. (b) Zoom on the interface and on one water molecule at a given time step. (c) A single water molecule with its electrostatic environment, within a radius Rc, used for quantum mechanics calculation of individual hyperpolarizabilities. Average hyperpolarizability profiles are obtained as a function of the coordinate normal to the interface (Z) by selecting and analyzing molecules within 0.1 nm-thick slices. Pictures were prepared using VMD34 and Inkscape35 softwares. | ||
Such methods have been successful to describe the impact of solvation on the optical properties of NLO-dyes,28,49–54 and on other non-resonant molecules.39,41–43,55–57 First, a classical molecular description of the structure of the liquid–vapor water interface is obtained using Molecular Dynamics (MD) simulations. Then, the frequency dependent β(−2ω,ω,ω) of each water molecule at the interface is computed using a QM description within an electrostatic embedding framework58 to mimic the condensed phase environment.
In Section 2, we present the MD and QM methods and justify some important calculation parameters. In Section 3, we report our results concerning the interface structure, and the evolution of the hyperpolarizability components when the water molecule gets close to the liquid–vapor interface.
2 Method description and validation
2.1 Molecular mechanics simulations and analysis
LAMMPS59 V.11.08.2017 is used to perform the MD simulation along with the rigid TIP4P/2005 water force field.60 Using equilibrated bulk configurations, 9000 rigid TIP4P/2005 water molecules are placed in a simulation box (5 × 5 × 40 nm3) to form a water film, about 10 nm thick, see Fig. 1(a). Thanks to the sufficient film thickness, the middle part of the fluid film well mimics bulk water. Periodic boundary conditions were applied in all three directions, so that the simulation is actually that of a multi-film system, but the vacuum layer of 30 nm is thick enough to neglect interactions between the water interfaces. The target temperature is 300 K using a Nose–Hoover thermostat with the period of the temperature fluctuations set to τ = 0.4 ps. 3 ns of NVT equilibration is performed before the 20 ns NVT production run with a time step of 2 fs. The center of mass of the whole system is fixed every timestep. Both electrostatic and Lennard-Jones intermolecular interactions are computed using the long-range PPPM formalism.61,62 Neighbor lists were updated every timesteps with a radius of 1 nm.To compute the structural properties of the simulated system, such as the molecular orientations or the hyperpolarizabilities, a home-made Python software FROG is used, using the module MDAnalysis.63 Local composition and molecular orientation are computed along with the hydrogen bond (H-bond) network. The H-bond analysis uses the geometric criteria from Pezzotti et al., i.e. an H-bond is present if the distance O(–H)⋯O is smaller than 3.2 Å and the angle O–H⋯O is in the range of [140–220]°.27 For the bulk phase, we obtain 3.37 H-bonds per water molecule, to be compared to the value obtained by Pezzotti et al. of 3.4.27 The structural properties such as densities and H-bond numbers are averaged over time and space to produce profiles, as precised below.
2.2 Space averaging
In the system, two liquid–vapor interfaces are present, but we shall focus on a single interface here, see Fig. 1(b). The average position of the interface is supposed to be planar, parallel to the {X,Y} plane. To calculate profiles along the Z-direction, the various properties are averaged over the molecules belonging to 0.1 nm-thick slices normal to the Z direction. In the following, the profiles are plotted as function of the “altitude” ΔZ = Z − Z0 relative to the average position of the interface Z0. To calculate the error-bars, we have estimated space-correlations within each slice, see ESI,† for details (Section S1.3).2.3 Response properties within density functional theory
The QM/MM workflow was handled by our software FROG that reads the MD trajectories and writes input files for the QM calculations, to compute NLO properties within the PE framework.51 After the hyperpolarizability calculations have been performed, FROG analyses the individual QM outputs and computes the altitude-dependent distribution of the hyperpolarizability (see Section S1.1 of ESI†).Concerning the basis, large sets including polarization and diffuse functions are necessary to obtain reliable hyperpolarizabilities.67,68 The Dunning's correlation consistent (cc) basis sets,65,69,70 in particular d-aug-cc-VTZ basis set is commonly used for water.10,39,42,71 It can be considered as a very good compromise between efficiency and accuracy for the present QM/MM applications that has been used for water molecules in the gas and liquid bulk phases. For an isolated water molecule, Sections S1.2.1 and S1.2.2 of ESI,† show the convergence of DFT approaches relative to basis set.
Concerning the functional, CAM-B3LYP is a range-separated hybrid functional which adds long-range correction using the Coulomb-attenuating method and includes 19% of HF exchange at short-range and 65% at long-range.64 It is expected to provide reliable results for the non linear optical properties of medium-size molecules.72,73 While it is true that DFT/CAM-B3LYP is not very accurate to compute cross section at resonant frequency,74 the present work investigates SHG response for the fundamental frequency of 800 nm, i.e. in the non-resonant case, where no excited state is populated. For water, the excitation energies of the first states are around 7–10 eV, corresponding to a fundamental wavelength about 300 nm. Even if the excitation wavelength of interest in our article is 800 nm, these states should still contribute to the SHG response. CAM-B3LYP seems a reasonable choice, since it performs best to predict the cross section and the frequency compared to other DFT functionals.74 For single water molecules, the use of long-range-corrected functionals permits to obtain DFT hyperpolarizabilities in reasonable accordance with CCSD/d-aug-cc-pVTZ results: the typical absolute difference is 1.3 a.u., averaged over non-zero hyperpolarizability components (see Table S1, ESI† and ref. therein67,75). Finally, since the QM/MM approach involves an electrostatic environment, the second hyperpolarizability γ plays an important role. Besalú-Sala et al. have showed recently that CAM-B3LYP is among the best choices regarding the second hyperpolarizability of water.73
The first hyperpolarizability is extracted using the frequency-dependent quadratic response scheme.76 The hyperpolarizability values are given within the T convention,77 in atomic units. Frequency-dependent calculations were carried out using an incident wavelength λ of 800 nm, and compared to the results for a static field (λ → ∞). The vibrational contribution to the first hyperpolarizability is neglected. This contribution is typically of 10% for water in the gas phase.71
To characterize the effect of the MM embedding, we have calculated the electrostatic field generated by the point charges on the QM water molecule. More precisely, this electrostatic field is calculated at the position of the negative charge in the TIP4P/2005 force field −0.15 Å away from the oxygen atom, along the dipole vector.
| λ | Gas | Liquid | |||
|---|---|---|---|---|---|
| ∞a | 800a | ∞b | ∞a | 800a | |
a This work, using DFT-CAM-B3LYP/d-aug-pVTZ level within the PE-0 response scheme using the charges of the TIP4P-2005 force field. The values for the liquid are averages over 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 configurations obtained from 32 MD frames separated from 20 ps, with an electrostatic environment built up to Rc = 2 nm around the target molecule.
b Combined coupled cluster/molecular mechanics (CC/MM) method by Liang et al.39 000 configurations obtained from 32 MD frames separated from 20 ps, with an electrostatic environment built up to Rc = 2 nm around the target molecule.
b Combined coupled cluster/molecular mechanics (CC/MM) method by Liang et al.39
|
|||||
| β caa | −10.6 | −12.5 | −2.1 [1.1] | −1.9 [1.0] | −1.9 [1.1] |
| β aca | −10.6 | −12.4 | −2.1 [1.1] | −1.9 [1.0] | −2.0 [1.1] |
| β cbb | −4.2 | −5.0 | 1.6 [1.8] | 2.6 [1.7] | 2.7 [2.0] |
| β bcb | −4.2 | −7.4 | 1.6 [1.8] | 2.6 [1.7] | 2.3 [2.1] |
| β ccc | −12.1 | −15.3 | 3.0 [3.0] | 3.9 [2.8] | 4.0 [3.1] |
Based on these results, the rest of the work concerning the interface was done using the CAM-B3LYP functional and the d-aug-cc-VTZ basis set.
 | ||
| Fig. 2 Convergence of the averaged molecular first hyperpolarizability component 〈βccc〉 relative to the environment cutoff radius Rc, for molecules at the altitude ΔZ = −0.06 ± 0.05 nm. The percentage of the relative absolute error |〈βccc〉 − 〈βccc〉0|/〈βccc〉0 is plotted with a log. scale, where the reference value 〈βccc〉0 is obtained for Rc = 5 nm. The dashed line represents an error of 1%. See ESI,† Fig. S7 for other β components. | ||
A radius of about 2 nm is needed to reach a relative error below 1%, i.e. 0.1 atomic unit (dashed line on Fig. 2). The convergence is similar for molecules selected in the bulk phase (see Fig. S6 of ESI†). Therefore, in the following, the neighborhood is built up to a distance Rc = 2.3 nm. This result highlights that the hyperpolarizability is sensitive to the electrostatic environment up to several nanometers: for aqueous interfaces in general, the S-SHG signal of the BIL might be influenced indirectly by the structure and composition of a thicker interfacial layer. Indeed, special care should be granted to the choice of the electrostatic embedding description for QM/MM SHG studies.51,78
2.4 Time averaging of hyperpolarizability
The hyperpolarizability profiles are obtained from the analysis of Nf selected frames of the MD trajectory, separated from a time span Δt. This Section presents our methods to choose Δt and Nf.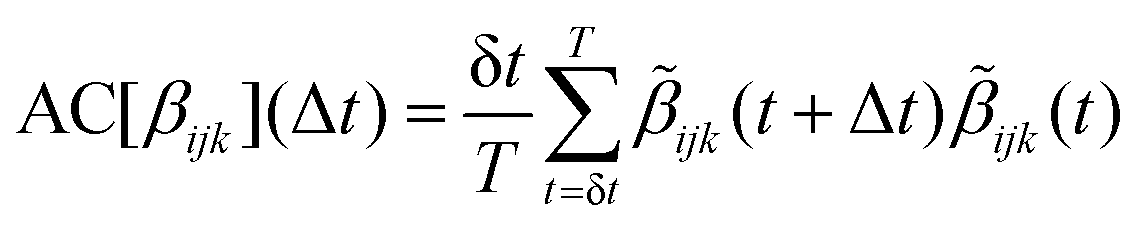 | (1) |
![[small beta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10e.gif) ijk(t) = βijk(t) − 〈βijk〉. In order to represent the contribution of all the components, a “total autocorrelation” (TAC) function is defined:
ijk(t) = βijk(t) − 〈βijk〉. In order to represent the contribution of all the components, a “total autocorrelation” (TAC) function is defined: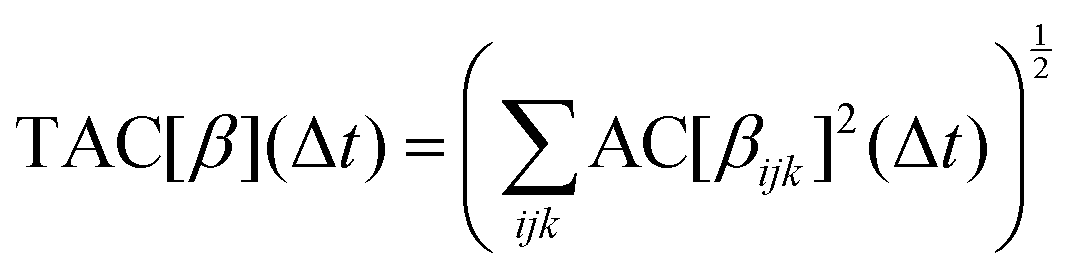 | (2) |
 | ||
| Fig. 3 Time auto-correlation (TAC) of the fluctuations of the beta tensor of a single molecule, either in the bulk phase (blue solid line), or at the interface (orange solid line), averaged over the components as defined by eqn (2). The ordinate axis follows a logarithmic scale. The dashed line represents a TAC of 5%. | ||
 | ||
| Fig. 4 Relative deviation of the average β components as a function of the number of QM calculations, for the typical altitude of ΔZ − Z0 = −0.06 ± 0.05 nm. The relative deviation (in %) is calculated with respect to the reference value obtained using the 4294 molecular configurations obtained for Nf = 80 selected MD frames. The dashed lines represent a deviation of 5%. | ||
Approximately 4200 configurations are needed to obtain relative deviation of the order of 2% at this typical altitude. These correspond to the Nf = 80 selected MD frames, which was chosen for the rest of the study, for all altitudes. Fig. S5 of ESI† reports the evolution for the other components, that converge towards zero, and the conclusions are the same.
For a fixed number of frames Nf, the number of QM calculations included in the hyperpolarizability average of a given slice is proportional to the density in this slice. Therefore, the hyperpolarizability averages are less precise for altitudes where the density is lower.
3 Results and discussion
3.1 Interface structure from classical molecular dynamics simulations
In a first step, the structure of the liquid–vapor interface has been obtained from the analysis of one interface in an MD simulation of a water film surrounded by vapor, see Fig. 1(a and b).We discuss here the density profile and the water dipole orientation profile displayed in Fig. 5(b). The density decreases progressively from the bulk value to zero with a profile close to a hyperbolic tangent shape.82 Its inflection point permits to define an absolute position Z0 for the interface, and the distance relative to it (ΔZ = Z − Z0) that is called “altitude” here.
 | ||
| Fig. 5 (a) Definition of the molecular frame {a,b,c} and of the angle θ = (c,Z) characterizing the orientation of the water dipole moment with respect to the interface normal Z-axis. (b) Water density profile (black circles) and 〈cosθ〉 profile (gray squares) as a function of the altitude. The solid black line is proportional to a hyperbolic tangent function fitted onto the density profile. The error bars are calculated using eqn (S3) of ESI.† | ||
The projection of the water dipole c-axis on the interface normal (Z), illustrated by Fig. 5(a), is particularly relevant for the interpretation of the S-SHG intensities. Indeed, the C∞ symmetry of the interface, associated to an expected C2v symmetry of the water hyperpolarizability tensor, yields surface susceptibility components expressed as linear combinations of 〈βijk(cosθ)l〉 where θ is the (c,Z) angle33,83 and l is 1 or 3. For example, if one focuses on the χ(2)ZZZ element (the dominant one, according to experimental observations on the water–air interface10):
| χ(2)ZZZ ∝ 〈β⊥cosθ〉 + 〈(βccc − β⊥)cos3θ〉, | (3) |
| β⊥ = (βcaa + βcbb + 2βaca+ 2βbcb)/2. | (4) |
The profile of 〈cosθ〉 is therefore plotted in Fig. 5(b) as a function of the altitude. The region where a net orientation appears, i.e. 〈cosθ〉 ≠ 0, spotlights the BIL and the thickness of the layer which should contribute to the S-SHG signal. Noticeably, the net water orientation appears few Angstroms further away from the interface than the density drop. The deviation for the H-bond network relative to the bulk occurs at a similar altitude (see Fig. S3 of ESI†).
Moreover, 〈cosθ〉 provides structural information: the negative values indicate that the dipole moment of the water molecule points on average with the hydrogens towards the bulk phase. But this propensity is weak, and the dipole moment of the water molecule in the most external water layers is preferentially close to parallel to the interface plane (see Fig. S4 of ESI†). These results agree with previous studies.10,47,84
To guide the interpretation, we have divided the system into 3 regions shown in Fig. 5: bulk (in purple), interface (in cyan) and vapor (in white). The limit between the bulk and interface is fixed at the altitude where 〈cosθ〉 significantly deviates from its bulk value. The edge of the vapor area is defined where the density reaches 5% of the bulk value. In practice, we have limited the vapor region at the point where accurate statistical averaging over the number of water molecules became difficult because of too small densities.
3.2 Water hyperpolarizabilities from quantum mechanics/molecular mechanics (QM/MM) calculations
The second step consists in computing, for each MD snapshot, the molecular hyperpolarizability β(2ω,ω,ω) of all water molecules. After justifying several calculation parameters (see Section 2.3), we discuss here the molecular hyperpolarizability components as a function of the altitude.First, the average hyperpolarizability tensor respects the C2v symmetry at the interface. Note that this was already true in the bulk (see Table S2 of ESI†). Indeed, the C2v-forbidden β components remain negligible (less than 0.3 a.u. in average) throughout the whole interface (see Fig. 6). All these values average to zero in the region where the density is large enough to obtain precise results, but their fluctuations are strong, as already noted by Liang et al.39 for the bulk phase. Concerning the non-vanishing hyperpolarizability components, Fig. 7 shows that they start to deviate from their bulk values roughly at the same altitude as the density, molecular orientation or H-bond network.
 | ||
| Fig. 6 Averages of the hyperpolarizability β(−2ω,ω,ω) as a function of the altitude, in the molecular frame {a,b,c}, for the C2v-forbidden components. The exciting wavelength is 800 nm. Errors bars are calculated using eqn (S3) of ESI.† | ||
 | ||
| Fig. 7 Averages of the components of the hyperpolarizability β(−2ω,ω,ω) as a function of the altitude, in the molecular frame {a,b,c}, for an exciting wavelength of 800 nm. βcaa and βaca are indistinguishable. β⊥ is defined by eqn (4). The errors bars, calculated using eqn (S3) of ESI,† are smaller than the symbols. | ||
Concerning the frequency dispersion, characterized for example by the difference between 〈βcbb〉 and 〈βbcb〉, it is much smaller in the water bulk phase (≃0.5 a.u.) than in the vacuum phase (≃2.5 a.u., see Table 1). In the interface region, the frequency dispersion remains weak, and the Kleinman symmetry is practically respected, i.e. 〈βcaa〉 = 〈βaca〉 and 〈βcbb〉 ≃ 〈βbcb〉. Therefore, within our methodology, the Kleinman symmetry breaking observed experimentally seems difficult to be explained solely by the individual dipolar molecular responses (see Section S4.3 of ESI†). Our calculations do not take into account the effective field (cavity field) effect, which is beyond the scope of this work. However, since the dispersion of the water polarizability between 800 and 400 nm is very small,10 it should not affect much our conclusion. More precise QM/MD calculations, local effective field effects, collective effects, or quadrupolar contributions should be further explored to explain this behavior.
The evolutions of individual components are relatively modest, and the hyperpolarizability tensor remains closer to the bulk one than to the gas one. Indeed, similarly to the structural properties depicted in Fig. 5, the electric field generated by the point-charge environment differs from the bulk one in the interface area. However, its projection along the molecular c-axis loses only about 30% of its bulk value. This can be seen on Fig. 8 that shows the electrostatic field generated by the point-charged environment on the central water molecule, projected along the c-molecular axis. We attribute this strong remaining field to the presence of close neighbors, and H-bonds, even for the most external layer of water molecules,47 see Fig. S3 of ESI.†
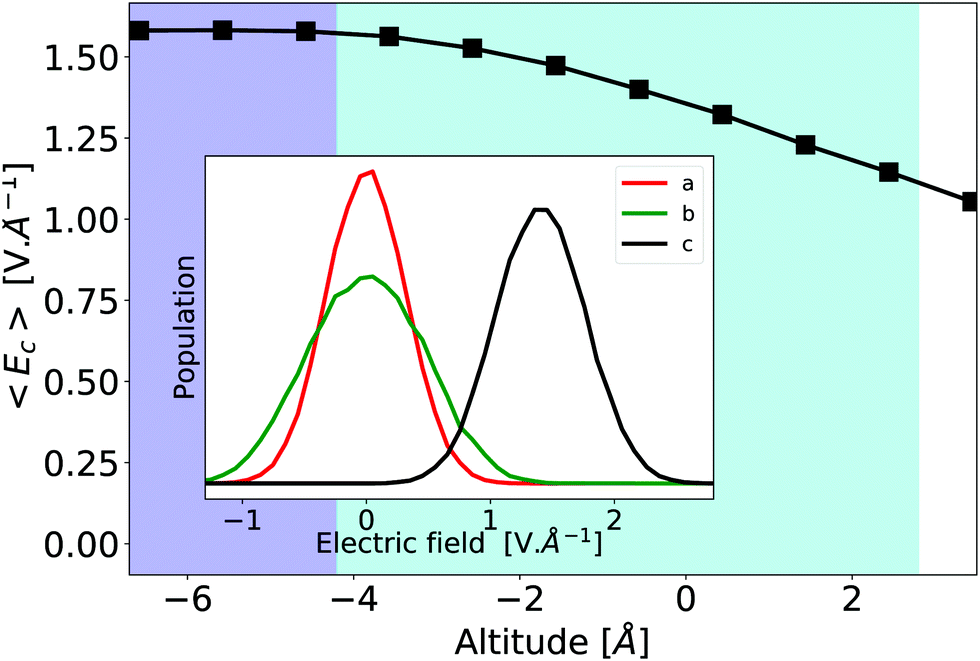 | ||
| Fig. 8 Electrostatic field generated by the environment along the molecular c-axis throughout the interface. In insert the electrostatic field distribution. The environment is built up to Rc = 2 nm. The average values of the electric field along the a and b-axis remain null across the interface – data not shown. | ||
3.3 Link with surface-second harmonic generation (S-SHG)
Even if the variations of individual molecular hyperpolarizability components could be considered as weak, the evolution of βccc and β⊥ is significant for the S-SHG interpretation. Whereas βccc is dominant in the bulk (4.0 vs. 0.5 a.u.), at the altitude of 0.04 nm, βccc and β⊥ have opposite values so that (βccc − β⊥) vanishes. The ratio of the two components of eqn (3) is strongly modulated by the altitude. This questions some frameworks used to analyze S-SHG signals, in which the ratio between βccc and β⊥ is typically considered as constant throughout the interface (see for example Section S4.4 of ESI†).Further, we tackle an even more basic question: can the β tensor distribution be replaced by a single, average value at a given altitude? In the dipolar approximation, one may write the susceptibility χ(2) as proportional to the average over molecular configurations of the product of the rotational matrix R with the β tensor. In practice, it is often assumed that all molecules can be described by a single hyperpolarizability tensor 〈βijk〉, and that the fluctuations of the hyperpolarizability tensor and the molecular orientation are uncorrelated, i.e. 〈βijkcoslθ〉 = 〈βijk〉〈coslθ〉. In our data, such equalities do not hold: Fig. 9 illustrates it for eqn (3), where the contributions of β⊥ and βccc to χ(2)ZZZ are calculated either taking into account the fluctuations, or using the average β tensor and average orientation at each altitude (see Section S4.2 of ESI,† for details). The difference between the two curves emphasizes correlations between the molecular orientation and molecular hyperpolarizabilities at the interface.
 | ||
| Fig. 9
χ
(2)
ZZZ
as a function of altitude calculated using the right hand side of eqn (3) in 0.1 nm-thick slices. Either the correlations between hyperpolarizability and orientation are considered (blue squares, using 〈βijk coslθ〉), or they are neglected (red circles, using 〈coslθ〉〈βijk〉). | ||
In Fig. 9, χ(2) is expected to vanish in the bulk region. The deviations relative to zero in this area indicate that the convergence relative to sampling is not completely achieved. Indeed, the configuration-convergence study presented in Fig. 4 is focused on the hyperpolarizability components β. Since the χ(2) elements also include molecular orientations, their convergence is more demanding. However, at the interface, the differences between the two ways of calculating χ(2) on our data exceed these errors.
We conclude that orientation/hyperpolarizability correlations may be relevant to interpret quantitatively S-SHG intensities. Such correlations have been mentioned by Champagne and co-workers in a more complex system, composed of an organic dye embedded in a lipid bilayer,85 and we show here that these may appear even at the neat liquid–vapor interface of pure water.
4 Conclusions
To summarize, a QM/MD approach was applied at the water liquid–vapor interface, to compute first hyperpolarizabilities of individual water molecules within the very specific environment where the S-SHG signal is generated. There is still a lot of room for improving the accuracy of the QM/MM approach (QM framework, electronic delocalization on several molecules, environment polarizability, long range electrostatic effects, local field factors,…), but qualitative results emerge. Within our approach, the dipolar contribution to the NLO response appears in a molecular layer of about half a nanometer thickness, where both the molecular orientation and the hyperpolarizability significantly differ from the bulk ones. The molecular first hyperpolarizabilities depend on the molecular environment up to radii larger than 2 nm, so that the S-SHG is very sensitive to the electrostatic properties of the interface. The hyperpolarizability tensor elements calculated in our PE approach almost respect the Kleinman symmetry, both in the bulk water phase and at the liquid–vapor interface, so that the Kleinman symmetry breaking observed experimentally may not originate in individual dipolar molecular response. The variations of the hyperpolarizability within the interface have an impact on the relative weights of its different components. Finally, we spotlight that the calculated molecular hyperpolarizabilities and molecular orientations are correlated: single average values for the molecular hyperpolarizability tensor and orientation are not sufficient to describe the entire distribution of molecular responses. The hyperpolarizability cannot be considered as constant for all water molecules at the liquid–vapor interface. More generally, this work indicates that one may need to consider different water populations at the interface to interpret the dipolar contributions to the surface second harmonic generation of aqueous interfaces. These hyperpolarizability fluctuations revealed at the neat air/water interface may also be relevant in more complex systems and could be more generally considered in quantitative interpretations of S-SHG experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thanks Zacharie Behel for his help during the writing of FROG. We gratefully acknowledge support from the PSMN (Pôle Scientifique de Modélisation Numérique) of the Ecole Normale Supérieure de Lyon for the computing resources.Notes and references
- E. Donohue, S. Khorsand, G. Mercado, K. M. Varney, P. T. Wilder, W. Yu, A. D. MacKerell, P. Alexander, Q. N. Van, B. Moree, A. G. Stephen, D. J. Weber, J. Salafsky and F. McCormick, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17290–17297 CrossRef CAS PubMed.
- A. C. McGeachy, E. R. Caudill, D. Liang, Q. Cui, J. A. Pedersen and F. Geiger, Chem. Sci., 2018, 9, 4285–4298 RSC.
- G. Licari, J. S. Beckwith, S. Soleimanpour, S. Matile and E. Vauthey, Phys. Chem. Chem. Phys., 2018, 20, 9328–9336 RSC.
- H.-T. Bian, R.-R. Feng, Y. Guo and H.-F. Wang, J. Chem. Phys., 2009, 130, 134709 CrossRef PubMed.
- H. C. Allen, N. N. Casillas-Ituarte, M. R. Sierra-Hernandez, X. Chen and C. Y. Tang, Phys. Chem. Chem. Phys., 2009, 11, 5538–5549 RSC.
- M. Sulpizi, M. Salanne, M. Sprik and M.-P. Gaigeot, J. Phys. Chem. Lett., 2012, 4, 83–87 CrossRef PubMed.
- Y. Chen, H. I. Okur, N. Gomopoulos, C. Macias-Romero, P. S. Cremer, P. B. Petersen, G. Tocci, D. M. Wilkins, C. Liang, M. Ceriotti and S. Roke, Sci. Adv., 2016, 2, 1–9 Search PubMed.
- P. E. Ohno, H. Chang, A. P. Spencer, Y. Liu, M. D. Boamah, H.-F. Wang and F. M. Geiger, J. Phys. Chem. Lett., 2019, 10, 2328–2334 CrossRef CAS PubMed.
- L. Dalstein, K.-Y. Chiang and Y.-C. Wen, J. Phys. Chem. Lett., 2019, 10, 5200–5205 CrossRef CAS PubMed.
- T. T. Pham, A. Jonchère, J. F. Dufrêche, P. F. Brevet and O. Diat, J. Chem. Phys., 2017, 146, 144701 CrossRef PubMed.
- L. B. Dreier, C. Bernhard, G. Gonella, E. H. Backus and M. Bonn, J. Phys. Chem. Lett., 2018, 9, 5685–5691 CrossRef CAS PubMed.
- R. K. Campen, A. K. Pymer, S. Nihonyanagi and E. Borguet, J. Phys. Chem. C, 2010, 114, 18465–18473 CrossRef CAS.
- S. Xu, S. Xing, S.-S. Pei, V. Ivanišev, R. Lynden-Bell and S. Baldelli, J. Phys. Chem. C, 2015, 119, 26009–26019 CrossRef CAS.
- A. Marchioro, M. Bischoff, C. Lütgebaucks, D. Biriukov, M. Předota and S. Roke, J. Phys. Chem. C, 2019, 123, 20393–20404 CrossRef CAS.
- J. H. Raberg, J. Vatamanu, S. J. Harris, C. H. van Oversteeg, A. Ramos, O. Borodin and T. Cuk, J. Phys. Chem. Lett., 2019, 10, 3381–3389 CrossRef CAS PubMed.
- S. Pezzotti, D. R. Galimberti and M.-P. Gaigeot, Phys. Chem. Chem. Phys., 2019, 21, 22188–22202 RSC.
- Y. Shen, Annu. Rev. Phys. Chem., 1989, 40, 327–350 CrossRef CAS.
- N. A. Murugan, R. Apostolov, Z. Rinkevicius, J. Kongsted, E. Lindahl and H. Ågren, J. Am. Chem. Soc., 2013, 135, 13590–13597 CrossRef CAS PubMed.
- G. C. Gschwend, M. Kazmierczak, A. J. Olaya, P.-F. Brevet and H. H. Girault, Chem. Sci., 2019, 10, 7633–7640 RSC.
- H.-T. Bian, R.-R. Feng, Y.-Y. Xu, Y. Guo and H.-F. Wang, Phys. Chem. Chem. Phys., 2008, 10, 4920–4931 RSC.
- Q. Wei, D. Zhou and H. Bian, Phys. Chem. Chem. Phys., 2018, 20, 11758–11767 RSC.
- S. Ong, X. Zhao and K. B. Eisenthal, Chem. Phys. Lett., 1992, 191, 327–335 CrossRef CAS.
- Y. Liu, E. C. Yan, X. Zhao and K. B. Eisenthal, Langmuir, 2001, 17, 2063–2066 CrossRef CAS.
- G. C. Gschwend, A. Olaya and H. H. Girault, Chem. Sci., 2020, 11, 10807–10813 RSC.
- A. G. F. de Beer and S. Roke, J. Chem. Phys., 2010, 132, 234702 CrossRef PubMed.
- G. Tocci, C. Liang, D. M. Wilkins, S. Roke and M. Ceriotti, J. Phys. Chem. Lett., 2016, 7, 4311–4316 CrossRef CAS PubMed.
- S. Pezzotti, D. R. Galimberti, Y. R. Shen and M.-P. Gaigeot, Phys. Chem. Chem. Phys., 2018, 20, 5190–5199 RSC.
- C. Bouquiaux, C. Tonnelé, F. Castet and B. Champagne, J. Phys. Chem. B, 2020, 124, 2101–2109 CrossRef CAS PubMed.
- S. Roke, ChemPhysChem, 2009, 10, 1380–1388 CrossRef CAS PubMed.
- M. N. Nasir, E. Benichou, C. Loison, I. Russier-Antoine, F. Besson and P.-F. Brevet, Phys. Chem. Chem. Phys., 2013, 15, 19919–19924 RSC.
- C. Loison, M. N. Nasir, E. Benichou, F. Besson and P.-F. Brevet, Phys. Chem. Chem. Phys., 2014, 16, 2136–2148 RSC.
- R. Hartkamp, A.-L. Biance, L. Fu, J.-F. Dufrêche, O. Bonhomme and L. Joly, Curr. Opin. Colloid Interface Sci., 2018, 37, 101–114 CrossRef CAS.
- W.-K. Zhang, D.-S. Zheng, Y.-Y. Xu, H.-T. Bian, Y. Guo and H.-F. Wang, J. Chem. Phys., 2005, 123, 224713 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- http://www.inkscape.org/ .
- I. Harczuk, O. Vahtras and H. Ågren, Phys. Chem. Chem. Phys., 2016, 18, 8710–8722 RSC.
- T. N. Ramos, F. Castet and B. Champagne, J. Phys. Chem. B, 2021, 125, 3386–3397 CrossRef CAS PubMed.
- G. Maroulis, J. Chem. Phys., 2000, 113, 1813–1820 CrossRef CAS.
- C. Liang, G. Tocci, D. M. Wilkins, A. Grisafi, S. Roke and M. Ceriotti, Phys. Rev. B, 2017, 96, 1–6 Search PubMed.
- P. Kaatz, E. A. Donley and D. P. Shelton, J. Chem. Phys., 1998, 108, 849–856 CrossRef CAS.
- T. D. Poulsen, P. R. Ogilby and K. V. Mikkelsen, J. Chem. Phys., 2001, 115, 7843–7851 CrossRef CAS.
- A. Osted, J. Kongsted, K. V. Mikkelsen, P. O. Åstrand and O. Christiansen, J. Chem. Phys., 2006, 124, 124503 CrossRef PubMed.
- C. B. Nielsen, O. Christiansen, K. V. Mikkelsen and J. Kongsted, J. Chem. Phys., 2007, 126, 154112 CrossRef PubMed.
- P. Guyot-Sionnest and Y. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 7985 CrossRef PubMed.
- C. A. Dailey, B. J. Burke and G. J. Simpson, Chem. Phys. Lett., 2004, 390, 8–13 CrossRef CAS.
- J. Kongsted, A. Osted, K. V. Mikkelsen and O. Christiansen, J. Chem. Phys., 2004, 120, 3787–3798 CrossRef CAS PubMed.
- S. Pezzotti, D. R. Galimberti and M. P. Gaigeot, J. Phys. Chem. Lett., 2017, 8, 3133–3141 CrossRef CAS PubMed.
- K. V. Mikkelsen, Y. Luo, H. Ågren and P. Jørgensen, J. Chem. Phys., 1995, 102, 9362–9367 CrossRef CAS.
- K. Garrett, X. Sosa Vazquez, S. B. Egri, J. Wilmer, L. E. Johnson, B. H. Robinson and C. M. Isborn, J. Chem. Theory Comput., 2014, 10, 3821–3831 CrossRef CAS PubMed.
- S. Osella, N. A. Murugan, N. K. Jena and S. Knippenberg, J. Chem. Theory Comput., 2016, 12, 6169–6181 CrossRef CAS PubMed.
- N. H. List, H. J. A. Jensen and J. Kongsted, Phys. Chem. Chem. Phys., 2016, 18, 10070–10080 RSC.
- G. Licari, L. Cwiklik, P. Jungwirth and E. Vauthey, Langmuir, 2017, 33, 3373–3383 CrossRef CAS PubMed.
- M. De Wergifosse, E. Botek, E. De Meulenaere, K. Clays and B. Champagne, J. Phys. Chem. B, 2018, 122, 4993–5005 CrossRef CAS PubMed.
- C. Tonnelé, B. Champagne, L. Muccioli and F. Castet, Chem. Mater., 2019, 31, 6759–6769 CrossRef.
- M. H. Cardenuto and B. Champagne, Phys. Chem. Chem. Phys., 2015, 17, 23634–23642 RSC.
- M. de Wergifosse, F. Castet and B. Champagne, J. Chem. Phys., 2015, 142, 194102 CrossRef PubMed.
- P. Beaujean and B. Champagne, Theor. Chem. Acc., 2018, 137, 50 Search PubMed.
- C. Steinmann, P. Reinholdt, M. S. Nørby, J. Kongsted and J. M. H. Olsen, Int. J. Quantum Chem., 2019, 119, e25717 CrossRef.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J. L. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- R. E. Isele-Holder, W. Mitchell and A. E. Ismail, J. Chem. Phys., 2012, 137, 174107 CrossRef PubMed.
- R. E. Isele-Holder, W. Mitchell, J. R. Hammond, A. Kohlmeyer and A. E. Ismail, J. Chem. Theory Comput., 2013, 9, 5412–5420 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2008, 393, 51–57 CrossRef.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani and P. Dahle, et al. , Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS.
- J. R. Hammond and K. Kowalski, J. Chem. Phys., 2009, 130, 194108 CrossRef PubMed.
- F. Castet, E. Bogdan, A. Plaquet, L. Ducasse, B. Champagne and V. Rodriguez, J. Chem. Phys., 2012, 136, 024506 CrossRef PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- P. Beaujean and B. Champagne, J. Chem. Phys., 2019, 151, 064303 CrossRef.
- F. Castet and B. Champagne, J. Chem. Theory Comput., 2012, 8, 2044–2052 CrossRef CAS PubMed.
- P. Besalú-Sala, S. P. Sitkiewicz, P. Salvador, E. Matito and J. M. Luis, Phys. Chem. Chem. Phys., 2020, 22, 11871–11880 RSC.
- M. J. Paterson, O. Christiansen, F. Pawłowski, P. Jørgensen, C. Hättig, T. Helgaker and P. Sałek, J. Chem. Phys., 2006, 124, 054322 CrossRef PubMed.
- P. Beaujean and B. Champagne, J. Chem. Phys., 2016, 145, 044311 CrossRef PubMed.
- P. Sałek, O. Vahtras, T. Helgaker and H. Ågren, J. Chem. Phys., 2002, 117, 9630–9645 CrossRef.
- A. Willetts, J. E. Rice, D. M. Burland and D. P. Shelton, J. Chem. Phys., 1992, 97, 7590–7599 CrossRef CAS.
- M. T. Beerepoot, A. H. Steindal, N. H. List, J. Kongsted and J. M. H. Olsen, J. Chem. Theory Comput., 2016, 12, 1684–1695 CrossRef CAS PubMed.
- E. R. Kjellgren, J. M. Haugaard Olsen and J. Kongsted, J. Chem. Theory Comput., 2018, 14, 4309–4319 CrossRef CAS PubMed.
- A. Marefat Khah, P. Reinholdt, J. M. H. Olsen, J. Kongsted and C. Hattig, J. Chem. Theory Comput., 2020, 16, 1373–1381 CrossRef CAS PubMed.
- G. Le Breton and L. Joly, J. Chem. Phys., 2020, 152, 241102 CrossRef CAS PubMed.
- W. Bu, D. Kim and D. Vaknin, J. Phys. Chem. C, 2014, 118, 12405–12409 CrossRef CAS.
- P.-F. Brevet, Surface Second Harmonic Generation, PPUR Presses Polytechniques, 1997 Search PubMed.
- W. Gan, D. Wu, Z. Zhang, R. R. Feng and H. F. Wang, J. Chem. Phys., 2006, 124, 114705 CrossRef PubMed.
- C. Bouquiaux, C. Tonnelé, F. Castet and B. Champagne, J. Phys. Chem. B, 2020, 124, 2101–2109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the QM/MD method and comparison with literature, MD structural analysis, molecular water hyperpolarizability analysis in the bulk and at the liquid/vapor interface, susceptibility expressions for the study of molecular orientation/β correlations. See DOI: 10.1039/d1cp02258j |
| This journal is © the Owner Societies 2021 |