
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScreening of transition metal doped copper clusters for CO2 activation†
Máté
Szalay
a,
Dániel
Buzsáki
 a,
Júlia
Barabás
a,
Endre
Faragó
a,
Ewald
Janssens
b,
László
Nyulászi
ac and
Tibor
Höltzl
*acd
a,
Júlia
Barabás
a,
Endre
Faragó
a,
Ewald
Janssens
b,
László
Nyulászi
ac and
Tibor
Höltzl
*acd
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, H-1111 Budapest, Hungary. E-mail: tibor.holtzl@furukawaelectric.com
bQuantum Solid-State Physics, KU Leuven, Celestijnenlaan 200D, BE-3001 Leuven, Belgium
cMTA-BME Computation Driven Research Group, Budapest University of Technology and Economics, Szent Gellért tér 4, H-1111 Budapest, Hungary
dFurukawa Electric Institute of Technology, Nanomaterials Science Group, Késmárk utca 28/A, H-1158 Budapest, Hungary
First published on 22nd September 2021
Abstract
Activation of CO2 is the first step towards its reduction to more useful chemicals. Here we systematically investigate the CO2 activation mechanism on Cu3X (X is a first-row transition metal atom) using density functional theory computations. The CO2 adsorption energies and the activation mechanisms depend strongly on the selected dopant. The dopant electronegativity, the HOMO–LUMO gap and the overlap of the frontier molecular orbitals control the CO2 dissociation efficiency. Our calculations reveal that early transition metal-doped (Sc, Ti, V) clusters exhibit a high CO2 adsorption energy, a low activation barrier for its dissociation, and a facile regeneration of the clusters. Thus, early transition metal-doped copper clusters, particularly Cu3Sc, may be efficient catalysts for the carbon capture and utilization process.
Introduction
Due to the increased carbon dioxide emission from fossil fuel combustion, CO2 reduction is an urgent problem for our society. The rise of its atmospheric concentration leads to the severe problems of global warming and the decrease of the hydrosphere pH.1–8 To mitigate these and also solve the inevitable depletion of the fossil fuels, the capture of the CO2 and its storage or recycling to industrially applicable chemical compounds (e.g. methanol) is a possible solution.1,7,9–14 Although CO2 hydrogenation to methanol is thermodynamically feasible, in practice this reaction is hindered by the high kinetic barriers.15 Thus, a suitable catalyst is necessary to facilitate the reaction. Depending on the energy source, thermal,16 electrochemical,17,18 and photochemical19,20 catalysts are distinguished. It is well known from the seminal work of Hori et al. that copper surfaces catalyse the CO2 electroreduction to methane and various other small hydrocarbons.17 Methanol is industrially produced by thermal catalysis from synthesis gas, containing CO, CO2, and H2, using a Cu/ZnO/Al2O3 catalyst at high pressure (between 10 and 100 bar).21Since the late 1990s, nanocatalysis has also become a very actively researched field.22,23 In particular, it is well known since the work of Haruta et al., and subsequently from others, that small clusters can exhibit unexpectedly high catalytic activity.24,25 Specific reactivity at the nanoscale may provide special properties that are not achievable with standard macroscale materials (e.g. huge specific surface area, size dependent electronic structure, etc.).26
Due to their well-defined compositions and precise geometric structures, gas-phase clusters are excellent model systems to investigate the reactions at active sites of complex catalysts.27–29 With the help of gas-phase model systems it was possible to uncover reaction mechanisms and the role of cluster size,30,31 geometry,32,33 dopant atoms,34,35 and the charge state.27,36,37
Transition- and noble metal-based catalysts are among the most studied CO2 reduction catalysts, partly because of their relative stability under reaction conditions and the various synergistic effects that can be achieved by combining different metals. In particular, copper exhibits a promising activity,38–43 particularly in organic bond-coupling reactions44 and the hydrogenation of carbonate to methanol.45
Copper is one of the most promising metals for CO2 reduction. Copper catalysts of various sizes and types are widely used for the hydrogenation of CO2 to methanol and show exceptional activity and very good methanol selectivity for low temperature CO2 hydrogenation.46,47 Various copper surfaces (111, 211) and copper clusters Cun (n = 13, 15, 19, 55, 79) have also been studied, of which n = 19 was found to be the most active for CO2 dissociation.48 The mechanism of the metal-mediated reduction of CO2 to CO was investigated using DFT computations.49–52
Cationic copper clusters can, depending on their size, facilitate dissociative H2 adsorption, an important step in CO2 hydrogenation.53,54
The catalytic effect of copper nanoparticles and clusters deposited on the ZnO surface is outstanding.55 It was shown that the deposited clusters are much more active than the conventional industrial catalysts.56 Experiments and density functional theory (DFT) calculations showed that Cu4 clusters exhibit high activity and a low activation barrier for CO2 reduction to methanol57,58 and methane.59 Further tuning of the CO2 reduction activity of various catalysts is in the frontier of interest.
Several studies have investigated the effect of the cluster size and support on the reactivity towards CO2. Alumina surface deposited copper clusters exhibit non-monotonous size dependent activity (Cu4/Al2O3 > Cu20/Al2O3 > Cu3/Al2O3) for the CO2 reduction to methanol.60 Similarly, high methanol synthesis activity was observed for Cu4 clusters deposited on a Fe2O3 surface.61 It was shown that due to electron transfer from the catalyst to the CO2 molecule, Cux/TiO2 (x = 1–5) promotes CO2 activation in all cases compared to the pure TiO2 system.62,63 These results highlight the synergistic effects between clusters and their support. According to computational studies graphene-supported Cu4 clusters are promising for electrocatalysis,64 while experiments showed that carbon-supported copper clusters are highly active catalysts for CO2 reduction.65
For small (111)-like facets (Cu13, Cu55) the hydrocarbon production is favourable, while synergetic effect between (100)- and (111)-like facets (Cu38, Cu79) favours CO production.66 Cu3, Cu5, and Cu4, Cu6 exhibit similar CO2 reduction mechanisms. The rate-determining step is the cleavage of the adsorbed OH in the first case, while it is the proton–electron transfer to the adsorbed CO for the latter one.67 The redox behaviour of Cu5 and Cu20, and the effect of CO2 also has been investigated.68 CO2 adsorbs more strongly on Cu5 than on Cu20 and larger nanoparticles, and forms bicarbonate in solution. This explains the high electrochemical CO2 reduction activity of Cu5.
The composition dependence of the catalytic activity of doped clusters is less studied. It was shown that chalcogen doping of copper clusters lowers the activation barrier and enhances the selectivity for electrochemical CO2 reduction to methane.69 A combined spectroscopy and computational study of gas-phase MnxOy+ (x = 2–5, y ≥ x) showed linear binding of CO2 and only a small change in the CO2 electronic structure. Therefore, Mn-oxide does not seem to be a suitable material for CO2 activation.70 Experimental studies showed, that gas-phase Pt, Pd, Cu and Pd–Cu bimetallic hydride cluster anions can catalyse the formation of formate and formic acid from CO2.71–74 CO2 reduction on cationic titanium hydride complexes [LTiH]+ (L = cyclopentadyenil, O) showed ligand-dependent CO2 reduction pathways. These clearly indicate, that the product selectivity can be tuned by the composition.75 The study of CO2 hydrogenation to methanol and formate on Pd6M (M = Cu, Ni, Pt, Rh) showed that the dopants can reduce the activation barriers, thus enhancing the catalytic activity.76 The Ni dopant was found to be especially active. It was observed that doped clusters tend to be more selective toward formate than to CO, thus leading to methanol or methane formation. These clusters were found to be more active than the Cu(111) surface. CO2 adsorption and dissociation were studied on Ni4M clusters, with M = Ni, Mo, Sc, or Y. With the exception of the Mo dopant, the Ni4M clusters promote CO2 dissociation with the Sc-doped cluster being the most active.77 Another DFT study showed that Sc and other early 3d transition metal atoms are promising dopants for CO2 activation on [MMgO2]+/0/− clusters, while in contrast late 3d transition metals are favored for methane activation.78 It was found that the atomic d orbitals of the metals atoms play an important role in the CO2 activation, because the electron transfer takes place from the metal's d orbital to the unoccupied σ orbital of CO2.79 Gas-phase pure platinum clusters exhibited size-dependent CO2 activation.80 CO2 adsorption on gas-phase Cu4−xPtx (x = 0–4) was investigated using DFT calculations. High adsorption energies (from −78 to −158 kJ mol−1) and CO2 adsorption in a bent mode were found,81 and could be explained by charge transfer towards the cluster's LUMO.82 Based on the reversible structural transformation, the optimal strength of the interaction between the cluster and the adsorbent, and the low CO2 dissociation barrier, Cu3Pt is a promising catalyst.81 It was shown, that Ni- or Pd-doped Cu4 clusters exhibit higher adsorption energy and promote the CO2 activation, because the dopant enhances the charge transfer between the cluster and the adsorbent.83 Although none of these clusters decrease the CO2 dissociation barrier. Zr-doped copper clusters made the CO2 dissociation also possible and Cu3Zr was found to be especially active. The HOMO–LUMO gap and the charge of the adsorbed CO2 molecule were found to be the most important factors to determine the catalytic activity.84
While doping has been applied to fine-tune and improve the activity of metal clusters, a systematic investigation of the effect of the different dopants for CO2 activation is not yet available. The goal of this work is a comprehensive investigation of the effect of the first-row transition metal dopants (X) for the CO2 activation by small copper clusters. We selected the Cu3X clusters for this study because it was experimentally shown that Cu4/Al2O3 is effective for CO2 reduction to methanol57 and because computational studies indicated that the Pt-,81 Ni-,83 and Zr-84doped four-atomic copper clusters are effective for CO2 activation.
Computational methods
Density functional theory computations were carried out using the Q-Chem 5.3 program package.85 The structures were preoptimized using the TPSSh/def2-TZVP method,86,87 while the most stable structures were reoptimized using the TPSSh/def2-TZVP+D3 level of theory. This method was selected after a benchmarking analysis of the effect of the basis set size and the level of correlation (up to CCSD(T)/def2-QZVPPD reference) for the relative energies of different molecules and radicals bound to Cu4 (see Fig. S1 and S2 and Table S1 in ESI† for details). For each structure a stability analysis was performed to ensure that the SCF procedure converged to a minimum with respect to the molecular orbital coefficients. Subsequently, the harmonic vibrational frequencies were determined to confirm that the minima and transition structures have zero and one imaginary frequencies, respectively. Intrinsic reaction coordinates were computed starting from the transition structures.We selected seven initial structural isomers (rhombohedral, tetrahedral etc. see the ESI† for details of the most stable isomers) and performed geometry optimizations using possible multiplicities of 1 to 7 and 2 to 8 for clusters with an even and an odd number of electrons, respectively. After the identification of low energy bare metal clusters (up to ∼50 kJ mol−1 above the lowest energy structure), CO2 adsorption in different binding modes (O–Cu bond, O–X bond, C–Cu bond, C–X bond, ring, etc.) and structures with dissociated CO2 were systematically investigated to locate the intermediates and products of the CO2 activation reaction, as described in ref. 88. Subsequently, we explored the full reaction pathways.
Finally, we analysed the electronic structure using the Frontier Molecular Orbitals, Natural Charges,89,90 Wiberg bond indexes, and the Energy Decomposition Analysis based on the Absolutely Localized Molecular Orbitals (ALMO-EDA).91
Reaction energies are computed with reference to the lowest energy bare gas phase clusters and the CO2 molecule. Free energies were computed at a pressure of 1 atm and at various temperatures using the Tamkin library.92
Results
1. Geometry and electronic structure of bare clusters
The lowest energy isomers of the Cu3X clusters (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn) are depicted in Fig. 1. Except for Zn, their total atomization energies (the energy change during the dissociation of the clusters to separated atoms in their most stable spin states) are similar to that of the Cu4 (see Fig. S7 in the ESI†). As Al2O3-deposited Cu4 clusters were successfully applied to hydrogenate the CO2 to methanol,57 we expect that the doped clusters are also stable synthetic targets. | ||
| Fig. 1 Lowest energy structures of the bare Cu3X clusters, the CO2 entrance adducts/complexes, the intact CO2 adducts, and the lowest energy structures of Cu3X–CO2. Spin multiplicities of the clusters are indicated in the superscripts. Notations: oa – obtuse apex, aa – acute apex. | ||
The obtained lowest energy structures for Cu3Sc and Cu3V are consistent with previous results.93,94 All lowest energy Cu3X clusters have planar rhombus shapes. The frontier orbitals of the Cu3X clusters are consistent with the phenomenological shell model (PSM)95,96 as each copper atom contributes 1 valence electron, while the transition metal dopants contribute 2 to 4 itinerant electrons, resulting in 1s2p2–5 electronic structures. Consistently, the LUMOs of most clusters correspond to a p orbital in the PSM (Fig. 2). It is interesting to note that the HOMO and the LUMO indicate a partially filled D shell in the case of manganese doping. This is explained by the hybridization of the shell orbitals with the d atomic orbitals of manganese.97 A similar LUMO shape is observed for Cu3Zn.
 | ||
| Fig. 2 (a) Total (black line) and partial (red and green lines for the s and d atomic orbital contributions, respectively) Density of States (DOS). Shaded areas show the occupied orbitals. For open shell clusters, the majority spin DOS is shown. (b) The frontier orbitals of Cu3X and the main Complementary occupied-virtual pairs (COVP). In the COVP pairs solid and transparent surfaces indicate the donor and acceptor part of each pair, respectively. | ||
Generally, the dopant atom prefers the high coordination, obtuse apex position. The only exception is the Zn, where the acute apex doped isomer is ∼10 kJ mol−1 more stable. Although in this case the CO2 dissociation is thermodynamically unfavoured. Because of this and for consistent comparison we only discuss the obtuse apex case. The Wiberg bond-indices (Table 1) show highly dopant-dependent partial covalent bonds between the dopant atom and the adjacent copper atoms. The Cu–X bonds are the strongest in the case of early transition metals (X = Sc, Ti or V) and the weakest in the case of X = Mn and Zn.
| X | Charge of X | Charge of Cuoa | Charge of Cuaa | Bond length of Cuoa–X [Å] | Bond length of Cuaa–X [Å] | Wiberg bond index of Cuoa–X | Wiberg bond index of Cuaa–X |
|---|---|---|---|---|---|---|---|
| a aa and oa refer to acute apex and obtuse apex positions, respectively (Fig. 1). | |||||||
| Sc | 0.50 | −0.17 | −0.16 | 2.55 | 2.48 | 0.82 | 0.77 |
| Ti | 0.39 | 0.04 | −0.22 | 2.53 | 2.57 | 0.42 | 0.51 |
| V | 0.26 | 0.14 | −0.20 | 2.51 | 2.57 | 0.39 | 0.46 |
| Cr | 0.40 | 0.18 | −0.29 | 2.43 | 2.58 | 0.42 | 0.27 |
| Mn | 0.46 | 0.04 | −0.21 | 2.51 | 2.53 | 0.15 | 0.31 |
| Fe | 0.34 | 0.19 | −0.27 | 2.34 | 2.45 | 0.50 | 0.36 |
| Co | 0.28 | 0.25 | −0.26 | 2.31 | 2.43 | 0.47 | 0.34 |
| Ni | 0.29 | 0.26 | −0.28 | 2.40 | 2.28 | 0.48 | 0.32 |
| Cu | 0.27 | 0.27 | −0.27 | 2.41 | 2.28 | 0.45 | 0.30 |
| Znoa,a | 0.42 | 0.02 | −0.22 | 2.42 | 2.41 | 0.14 | 0.28 |
| Znaa,a | 0.22 | −0.11 | 0.00 | 2.45 | 4.14 | 0.19 | 0.29 |
The variation of the Cu–X bond indices can be explained by the electron donation from copper 3d atomic orbitals to the partially filled valence 3d orbitals of the dopant, which was shown to lead to high coordination scandium-doped copper clusters.94 On the other hand, zinc has a fully occupied 3d shell and electron donation is only possible to the less favoured Zn 4p orbitals. This idea is consistent with the small difference between the stabilities of the low-coordinated zinc at the acute apex or the higher coordinated zinc at the obtuse apex position.
The partial atomic charges depend considerably on the dopant atom and at the obtuse apex dopant position they are generally in line with the dopant electronegativities. Thus, not only the electron donation to the unoccupied 3d atomic orbitals of the dopant, but also polarization effects determine the partial atomic charges. This is well reflected in the calculated density of states of the bare clusters (Fig. 2).
The variation of the magnetic moments of Cu3X follows similar trend to that of the free transition metal atoms: Cr and Mn dopants lead to the largest magnetic moments (see the ESI† for details).
2. CO2 adsorption and activation mechanism
Possible geometries of the different CO2 adducts were systematically investigated (lowest energy structures are depicted in Fig. 1; additional structures are available in the ESI†) and the CO2 dissociation reaction pathways are shown in Fig. 3.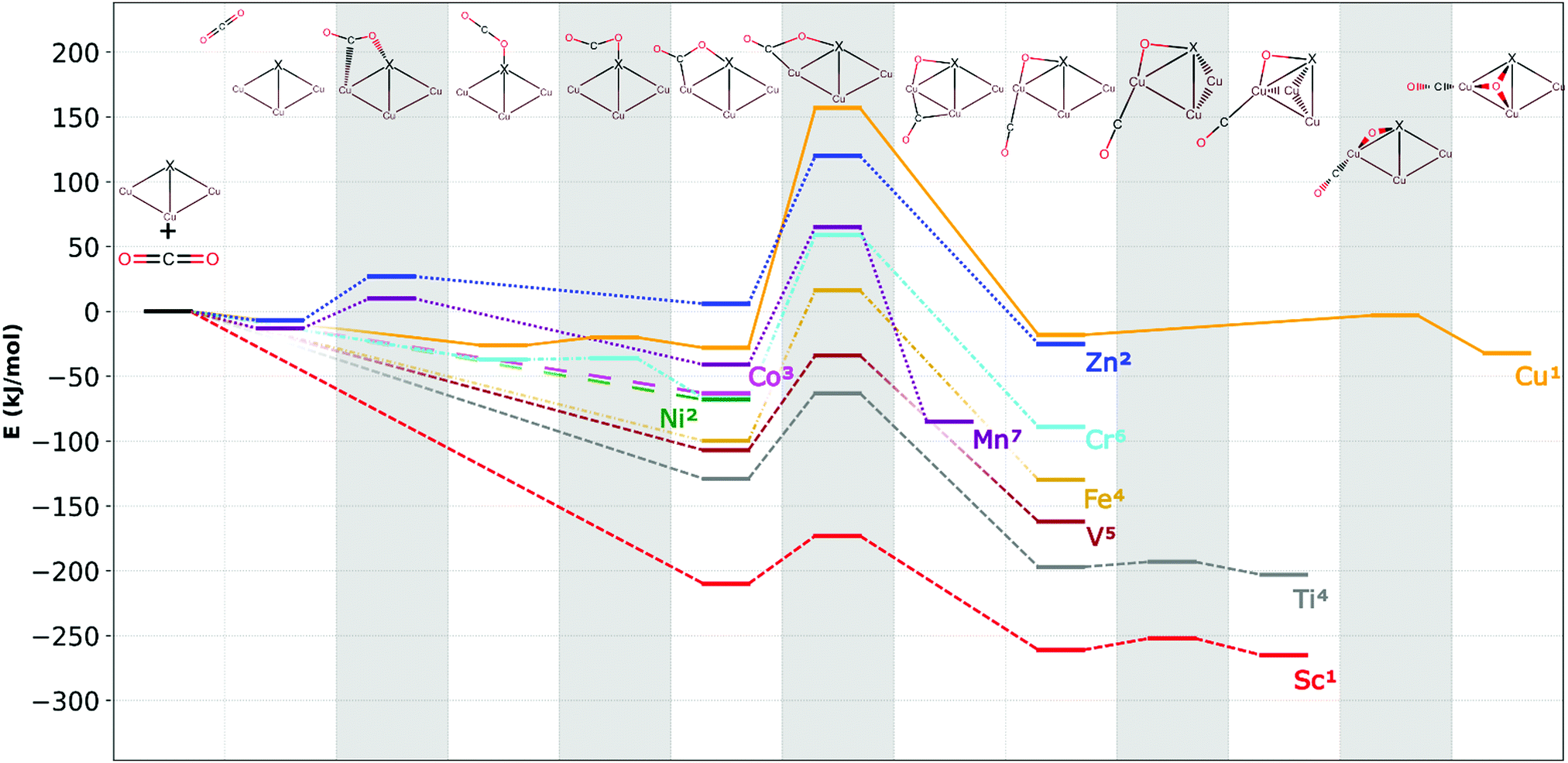 | ||
| Fig. 3 The reaction pathways for CO2 activation on Cu3X. Superscripts indicate the spin multiplicities of the different structures along the reaction paths. Shaded areas indicate the transition states. | ||
We analyse three classes of CO2 adducts: (i) entrance complexes that can be reached from the Cu3X and CO2 reactants without an energy barrier, (ii) intact CO2 adducts that contain non-dissociated CO2, and (iii) the lowest energy CO2 adducts are the thermodynamically most stable among the located Cu3X–CO2 isomers.
CO2 adsorption quenches the magnetic moment of Sc-, Ti- and V-doped clusters and partially also for Mn, thus spin–flip should take place during the CO2 activation (see the ESI† for the details). However, the spin–orbit coupling is relatively weak in the case of the first-row transition metals, thus, we expect that the chemical reactions are much faster than the spin relaxation. Hence, the spin–flip is expected to take place in the reaction product and the reaction proceeds on the high-spin potential energy surface. Therefore, we computed the reaction paths using the initial spin state of the cluster. The only exception is the Cu3Sc, where the singlet and triplet states of the bare cluster are quasi-degenerate, but the all the singlet Cu3Sc–CO2 entrance adducts are significantly more stable. Therefore, we explored the reaction path on the singlet potential energy surface, while the most stable spin states are discussed in the ESI.†
Fig. 3 shows the dopant-dependent trends in the CO2 reactivities of Cu3X clusters. Generally, early transition metal Cu3X clusters yield thermodynamically more stable intermediates and products as well as lower transition state energies than the late transition metal Cu3X clusters. This opens the way towards fine-tuning the reactivity with an appropriate dopant.
The entrance complex geometries depend on the dopant type. There are three different CO2 adsorption modes in Fig. 3. In case of X = Sc, Ti, V, Fe, Co, or Ni the CO2 molecule binds to the clusters without an energy barrier in a di-σ mode, i.e., the molecule bends and forms formal oxygen–dopant and carbon–copper bonds.
In the case of Mn or Zn dopants, the adsorption is preceded by a van der Waals complex and the subsequent stabilization to a di-σ structure is hindered by an energy barrier. For X = Cr or Cu, the intact, linear CO2 binds to the cluster without an energy barrier, while very small barriers of 6 kJ mol−1 and 1 kJ mol−1, respectively lead the di-σ structures.
The barrier following the di-σ structure is the highest in all the reaction paths. This shows that the di-σ structure plays a central role in the reaction path of all the dopants.
The geometric structures of the most stable adducts depend considerably on the dopant atom type. In case of Cu3Co and Cu3Ni, the intact CO2 adduct is the most stable, while in all other cases CO2 dissociates to CO and a formal oxide ion. In the dissociated product the CO binds at the acute apex position in all doped clusters and oxygen is in a bridge position above the adjacent copper-dopant edge.
Based on the similarities of the reaction paths, the dopants can be classified into four categories:
Type I – scandium, titanium, and vanadium (short-dashed line in Fig. 3): the entrance complex is the lowest energy intact CO2 structure with a di-σ binding mode. This is formed from the reactants without an energy barrier. The cluster-CO2 adduct then isomerizes to a tetrahedral geometry. Here, the dopant atom locates at one of the apexes of the tetrahedral structure and not the oxide ion, as observed in Cu4. All the barriers are submerged, implying a facile CO2 dissociation reaction. The dissociated CO forms thermodynamically stable adduct with the cluster, thus its desorption and the cluster catalyst regeneration will be investigated in Section 4.
Type II – manganese and zinc (dotted line in Fig. 3): CO2 forms only weak van der Waals complexes in the case of Mn and Zn at the obtuse apex position. Subsequently, low barriers lead to the di-σ structure with intact CO2. This structure is unstable in the Zn-doped and metastable in the Mn-doped case that is energetically stabilized during the subsequent reaction step. The CO2 dissociation is hindered by energy barriers with the transition state energy being well above that of the reactants. Moreover, in the case of acute apex positioned Cu3Znaa the CO2 dissociation is thermodynamically unfavoured.
Type III – chromium and iron (dash-dotted line in Fig. 3): in case of Fe the initial CO2 adsorption is similar to the Type I and Type IV dopants, where the intact CO2 adduct formation is barrierless and the final product involves dissociated CO with a formal oxide ion in bridge position on rhombus shape cluster. In both the chromium and iron case, the rate determining step is the CO2 dissociation with a transition state lying somewhat higher in energy than the reactants. The preceding reaction barrier that exists in the Cr case is very small and does not make a qualitative difference between the chemical behaviour of Cr and Fe dopants.
Type IV – cobalt and nickel (long-dashed line in Fig. 3): in contrast to copper, no adsorption barrier was found for the clusters doped with Co and Ni and the most stable product is the di-σ structure with intact CO2. However, in contrast to pure Cu4, CO2 dissociation is thermodynamically unfavoured.
3. Electronic structure analysis
The bent shape of the CO2 molecule and the atomic charges (Table 2) in the most stable intact adduct with di-σ structure (third column in Fig. 1) indicate its activation and significant charge transfer from the cluster to the molecule. Depending on the dopant, the carbon–oxygen bond orders decrease from 1.91 in the free CO2 molecule to 1.10–1.50 in the intactly bound CO2. The negative charge of the oxygen atoms imply a carboxyl group (Table 2). This negative charge is compensated mainly by the dopant atom.| Dopant | CO2 charge [e] | Dopant charge [e] | O–X bond index | Cu–C bond index | Cu–X bond index | Dissociating C–O bond index | CO2 dissociation barrier [kJ mol−1] |
|---|---|---|---|---|---|---|---|
| Sc | −1.00 | 1.04 | 0.60 | 0.25 | 0.47 | 1.10 | 37 |
| Ti | −0.84 | 0.58 | 0.49 | 0.35 | 0.33 | 1.20 | 67 |
| V | −0.81 | 0.50 | 0.42 | 0.36 | 0.33 | 1.29 | 73 |
| Cr | −0.71 | 0.55 | 0.28 | 0.38 | 0.25 | 1.42 | 126 |
| Mn | −0.89 | 0.96 | 0.25 | 0.42 | 0.05 | 1.21 | 107 |
| Fe | −0.77 | 0.64 | 0.27 | 0.29 | 0.36 | 1.39 | 117 |
| Co | −0.72 | 0.50 | 0.25 | 0.30 | 0.34 | 1.45 | — |
| Ni | −0.66 | 0.37 | 0.25 | 0.30 | 0.33 | 1.50 | — |
| Cu | −0.59 | 0.45 | 0.13 | 0.40 | 0.13 | 1.50 | 185 |
| Zn | −0.90 | 0.96 | 0.16 | 0.44 | 0.03 | 1.25 | 115 |
To investigate the electronic structure origin of the charge transfer between the cluster and the carbon-dioxide in the different cluster-CO2 adducts, the charge transfer energy was decomposed to contributions from Complementary Occupied Virtual Pairs (COVPs) (Fig. 2 and 4, see the ESI† for the details). In case of the intact adducts, the most significant COVP of the most stable intact CO2 adduct shows electron donation from the cluster HOMO to the CO2 LUMO (Fig. 2). As the CO2 LUMO is antibonding, electron donation to this orbital explains the weakened carbon–oxygen bond and the bent shape of the CO2 molecule. On the other hand, the back-donation from the CO2 HOMO to the cluster LUMO is generally small and a notable effect is observed only in the case of scandium doping (Fig. 4).
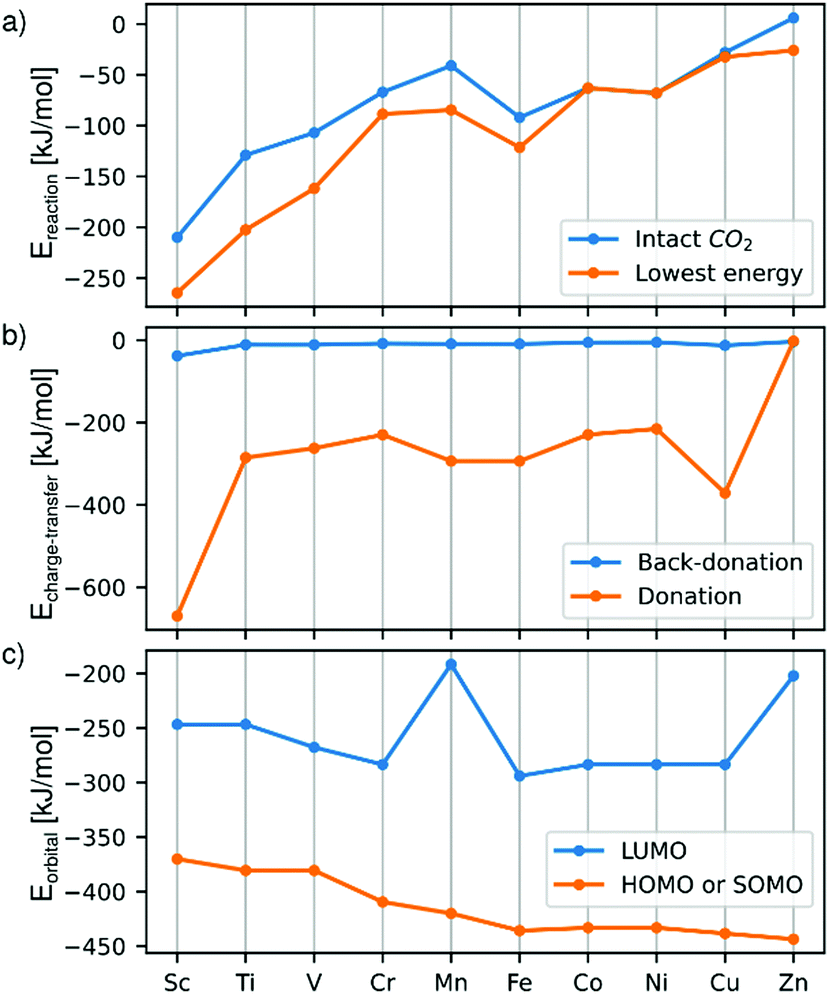 | ||
| Fig. 4 (a) Reaction energies of Cu3X-CO2 compared to the pure cluster and free CO2, (b) charge transfer energies due to electron donation from the cluster to the CO2 or back-donation in the opposite direction, and (c) frontier orbital energies of the most stable intact CO2–cluster adducts. | ||
This is in line with the observed negative charge of the adsorbed CO2. The dopant dependent contribution of valence s and d orbitals to the HOMOs/SOMOs (Fig. 2) are also reflected in the shape of the donor part of the COVPs. The SOMOs of Mn and Fe have small contributions from the valence s orbitals, thus the donor COVPs have clear, localized d characters, while in the early transition metal doped Cu3X with itinerant d electrons and high contributions from the valence s orbitals to the SOMOs, large, delocalized lobes on the donor part of the COVP clearly show the role of the hybridization of s and d atomic orbitals. Also, the HOMO/SOMO energies of the early transition metal-doped clusters are higher than those of clusters doped with the other transition metal dopants. This reduces the energy difference between the CO2 LUMO and the cluster HOMO. In addition to that, the shape of the LUMO (Fig. 2) clearly imply favourable overlap with the CO2 HOMO in case of early transition metals. This is in line with the slightly increased back-donation, especially in the case of Sc. These contribute to the higher binding energy in the case of early transition metals.
On the other hand, the overlap of the frontier orbitals of Cu3Mn and Cu3Zn with those of the CO2 molecule, and the increased energy gap between the cluster HOMO and CO2 LUMO explain the small energy barriers up to ∼30 kJ mol−1 in these cases from the entrance complex to the more stable di-σ structure.
The energies of the HOMOs/SOMOs decrease gradually from Sc to Zn, thus their energy differences to that of the CO2 LUMO increase. This is in line with the decreased stability of the intermediates and the products with the atomic number.
The di-σ adducts with bent, intact CO2 are the key structures to interpret the reaction mechanism, as their properties directly correlate with the CO2 binding energy and the dissociation activation energy (Fig. 5). The oxygen-dopant bond index decreases with the mass of the dopant atom X, while the carbon-copper bond index follows the opposite trend. It is well visible that O–X bond order correlates both with the CO2 adsorption energy and also with the activation energy corresponding to its dissociation. It follows from the figure that the lower the O–X bond index, the less stable the intact CO2 adduct is and the higher the energy barrier for CO2 dissociation. As discussed above, the changes of the bond orders are due to electron donation from the cluster HOMO to the CO2 LUMO, but the correlation between the charge transfer energy and the activation energy is weak. On the other hand, the good correlation between the O–X bond orders and the activation energy of the CO2 dissociation clearly show that the strong bonds between the cluster and the CO2 enhance the activation of CO2.
 | ||
| Fig. 5 Correlation between the O–X and C–O bond orders and charge transfer energy corresponding to electron donation in the di-σ adduct with the reaction energies and dissociation barriers corresponding to CO2 dissociation. The orange lines are linear trendlines. Coefficients of determination are given in the top right corners. | ||
Similarly, to the Cu4 case, the atoms in the shorter diagonal bears a partially positive charge in all of the doped Cu3X clusters (Table 1). However, the electronegativity of the dopant atom plays an important role, as the nucleophilic attack will also take place more easily and the O–X bond will be stronger (Table 2) and structure with bent CO2 is greatly stabilized in the case of the lower electronegativity early transition metal dopants. The Wiberg indices show that the O–X bond is the weakest in the case of the Zn- or Mn-doped clusters (Table 2), so the dopant Mn and Zn atoms with relatively low electronegativity will be polarized not towards the oxygen but towards the adjacent copper atom. This effect is so strong that while the electronegativities of Sc and Ti are 1.36 and 1.54, the partial charges of the dopant atoms are 1.04 and 0.58, respectively. For Mn and Zn, which electronegativities are 1.55 and 1.65 and the partial charges equal to 0.96 in both cases (Table 2). Therefore, compared to other doped clusters, the bond strength between the more electron-rich Cu atom and C increases significantly and in return the C–X bond strength decreases (Table 2). This indicates a reduced donation interaction, which weakens the C–O bond to a lesser extent than in the Type I dopants. This leads to slightly higher dissociation barrier than for Sc, Ti, V, but still lower than in the Cu4 case.
In the Cu3Cr and Cu3Fe cases, large overlap is possible between the frontier orbitals, which is beneficial to strengthen the O–X bond. But, since the Cr and Fe have relatively high electronegativities (1.66 for Cr; 1.83 for Fe,), the dopant is less polarized, which has a weakening effect. Thus, intermediate strength O–X bond is formed between the dopant atoms and the CO2. In line, the C–O bond is weakened to a lesser extent than in the Type II case.
4. General considerations for CO2 reduction
Finally, we summarize the most important factors for the selection of the most suitable doped cluster catalyst.The very first of these is the relative stability and formation of the di-σ structure. For Zn, Mn, Cu and Cr dopants, an energy barrier precedes the formation of this structure. However, this barrier is relatively low, less than 35 kJ mol−1. The reaction energy of the intermediate plays an important role, since the released energy can be used to overcome the dissociation barrier, and the more stable our structure, the easier it can be realized in practice. This reaction energy is shown in Fig. 4. Based on these, Sc, Ti, and V are the most promising.
The second factor is the height of the dissociation barrier (Fig. 5). Sc, Ti and V have the lowest of these, while it is the highest in the case of the pure copper cluster. The third important factor is the relative stability of the final product, i.e. the reaction energy (Fig. 4). This is also favourable for the Sc-, V- and Ti-doped clusters.
Thus, the early transition metals of Type I are the most promising dopant atoms, as they lead to the smallest dissociation barriers and at the same time give the most stable structures. However, an over-stabilized product of the CO2 dissociation can also hinder the elimination of the product from the cluster surface, i.e. the cluster cannot be regenerated. For this reason, we must also study the CO desorption energy.
Our study has shown that in each of the cases discussed here, the CO desorption from the final structure proceeds without an energy barrier. Fig. 6 shows a relatively weak dopant dependence of the CO desorption energy. This could be interpreted by the fact that the CO binds to the opposite side of the cluster as the dopant. Among the different dopants, Type I leads to the lowest desorption energies and it is lowest in the case of the Ti dopant. On the other hand, the pure copper cluster has the highest CO desorption energy.
 | ||
| Fig. 6 CO desorption energy from the Cu3XO(CO) clusters. The oxide-cluster reduction energy is calculated based on the following reaction: Cu3XO + H2 → Cu3X + H2O stable dissociated CO2 product was not found for Co and Ni. | ||
Finally, the Cu3XO cluster must be reduced to close the catalytic cycle. While the exploration of the detailed, multistep reduction mechanism is beyond the scope of our present article, we studied the thermodynamic driving force according to Cu3XO + H2 → Cu3X + H2O. The reduction energy shows a strong dopant-dependence with most favoured reduction in Cu and Zn and least in Sc (Fig. 6).
In summary, early transition metals are favourable dopants to facilitate the CO2 dissociation, however the regeneration of the catalyst is difficult. Nevertheless, the energy gain from the CO2 dissociation is high, so it can cover large part of the regeneration energy. Thus, early transition metal-doped Cu3X clusters are promising catalysts in the gas phase.
On the other hand, CO2 dissociation is hindered with high energy barriers in late transition metal dopants, but the regeneration of the catalyst is facile. Transition metals from the middle of the row (Mn or Cr) represent a promising balance.
Finally, we investigated the effect of the finite temperature by computing the Gibbs-free energies from 300 K to 800 K for several intermediates and products of Cu3Sc (see the ESI† for details). As expected, due to the entropy effects, the relative Gibbs-free energies of the adducts increase with temperature. However, the adducts are all still highly stable thermodynamically, even at the highest temperature. On the other hand, the high temperature facilitates the regeneration of the catalyst, as the dissociation of the CO product and the reduction of the oxide are both favored. Only ∼100 kJ mol−1 Gibbs-free energy is necessary to regenerate the catalyst.
Conclusions
In summary, we found strongly dopant-dependent reaction energies, reaction mechanisms, dissociation barriers for CO2 and less dopant-dependent desorption energies for CO during the CO2 activation with first row transition metal doped Cu3X clusters. However, based on similar properties of some dopants, the reaction mechanisms of these clusters could be grouped into four categories.The low electronegativity dopants of Type I, i.e., early transition metals (Sc, Ti, V) lead to small HOMO–LUMO gaps and relatively high overlaps between the frontier molecular orbitals of the doped cluster and the molecule. This results in barrier-free CO2 adsorption in di-σ binding mode, low dissociation barriers, and notably high thermodynamic stability of the adducts. These make the early transition metals very promising dopants for CO2 activation.
In the Type II electron-rich transition metal- (Mn, Zn) doped clusters, we observed very weak binding between the oxygen and the dopant due to a large HOMO–LUMO gap and small overlap between the frontier molecular orbitals of the doped cluster and the CO2. These lead to lower activation barriers for CO2 dissociation compared to the pure Cu4 cluster, but still higher than that in the Type I clusters.
The frontier orbitals of Type III metal (X = Cr, Fe) doped Cu3X clusters have high overlaps with those of the CO2, however due to their relatively high electronegativities the O–X bond is less polarized and thus CO2 adsorbs relatively weakly, and the dissociation barriers are high.
For Ni and Co doped clusters (Type IV), dissociation of CO2 is thermodynamically unfavourable, thus the intact CO2 structure is formed. Nevertheless, the bent structure of the molecule and its negative charge clearly shows that it is activated.
Our work has shown that early transition metal dopants facilitate the CO2 binding and can reduce the CO2 dissociation barrier, thereby promoting CO2 activation and reduction to CO. This highlights the potential of doping effects and motivates further studies to develop new CO2 reduction catalysts, to investigate further reduction to C1 or C2 product, and study the effect of the surface deposition on these doped copper clusters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the KU Leuven–Budapest University of Technology and Economics joint research funding (CELSA/18/032). This research has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 955650 (CATCHY).References
- W. Wang, Y. Himeda, J. Muckerman, G. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- P. Nejat, F. Jomehzadeh, M. Taheri, M. Gohari and M. Majid, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS.
- H. Arakawa, M. Aresta, J. Armor, M. Barteau, E. Beckman, A. Bell and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- N. Lewis and D. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- A. Appel, J. Bercaw, A. Bocarsly, H. Dobbek, D. DuBois, M. Dupuis, J. Ferry, E. Fujita, R. Hille, P. Kenis, C. Kerfeld, R. Morris, C. Peden, A. Portis, S. Ragsdale, T. Rauchfuss, J. Reek, L. Seefeldt, R. Thauer and G. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- A. Harriman, Philos. Trans. R. Soc., A, 2013, 371, 20110415 CrossRef PubMed.
- D. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- G. Olah, G. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- E. Kondratenko, G. Mul, J. Baltrusaitis, G. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- D. Talbi and E. Herbst, Astron. Astrophys., 2002, 386, 1139–1142 CrossRef CAS.
- M. Porosoff, B. Yan and J. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- R. Kortlever, J. Shen, K. Schouten, F. Calle-Vallejo and M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- E. Kondratenko, G. Mul, J. Baltrusaitis, G. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- S. Thomas, ChemCatChem, 2010, 2, 127–132 CrossRef CAS.
- D. Astruc, Nanopart. Catal., 2008, 16, 1–48 Search PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- G. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- K. Klabunde, Nanoscale materials in chemistry, Wiley, Hoboken, NJ, 2009 Search PubMed.
- T. Bligaard and J. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- J. Nørskov, T. Bligaard, J. Rossmeisl and C. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- J. Kleis, J. Greeley, N. Romero, V. Morozov, H. Falsig, A. Larsen, J. Lu, J. Mortensen, M. Dułak, K. Thygesen, J. Nørskov and K. Jacobsen, Catal. Lett., 2011, 141, 1067–1071 CrossRef CAS.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. House, E. Gottlieb, H. Kim, J. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- F. Schweinberger, M. Berr, M. Döblinger, C. Wolff, K. Sanwald, A. Crampton, C. Ridge, F. Jäckel, J. Feldmann, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2013, 135, 13262–13265 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. Song, S. House, S. Bernhard, J. Yang, G. Mpourmpakis and R. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W. Chun and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9810–9815 CrossRef CAS PubMed.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- H. Qian, D. Jiang, G. Li, C. Gayathri, A. Das, R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS PubMed.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- A. von Weber, E. Baxter, S. Proch, M. Kane, M. Rosenfelder, H. White and S. Anderson, Phys. Chem. Chem. Phys., 2015, 17, 17601–17610 RSC.
- S. Lang, T. Bernhardt, D. Kiawi, J. Bakker, R. Barnett and U. Landman, Angew. Chem., 2015, 127, 15328–15332 CrossRef.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- P. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- S. Vukojević, O. Trapp, J. Grunwaldt, C. Kiener and F. Schüth, Angew. Chem., Int. Ed., 2005, 44, 7978–7981 CrossRef PubMed.
- D. Bazin, D. Guillaume, C. Pichon, D. Uzio and S. Lopez, Oil Gas Sci. Technol., 2005, 60, 801–813 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617–8622 CrossRef CAS.
- J. Oliver-Messeguer, L. Liu, S. García-García, C. Canós-Giménez, I. Domínguez, R. Gavara, A. Doménech-Carbó, P. Concepción, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2015, 137, 3894–3900 CrossRef PubMed.
- M. Tamura, T. Kitanaka, Y. Nakagawa and K. Tomishige, ACS Catal., 2015, 6, 376–380 CrossRef.
- H. Arakawa, J. Dubois and K. Sayama, Energy Convers. Manage., 1992, 33, 521–528 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. Fischer, J. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- X. Zhang, J. Liu, B. Zijlstra, I. Filot, Z. Zhou, S. Sun and E. Hensen, Nano Energy, 2018, 43, 200–209 CrossRef CAS.
- H. Schwarz, Coord. Chem. Rev., 2017, 334, 112–123 CrossRef CAS.
- B. Szyja, D. Smykowski, J. Szczygieł, E. Hensen and E. Pidko, ChemCatChem, 2016, 8, 2500–2507 CrossRef CAS PubMed.
- G. Yan, Z. Gao, M. Zhao, K. Ma, Z. Ding, W. Yang and X. Ding, Mol. Catal., 2020, 497, 111205 CrossRef CAS.
- G. Yan, Z. Gao, M. Zhao, W. Yang and X. Ding, Appl. Surf. Sci., 2020, 517, 146200 CrossRef CAS.
- O. Lushchikova, D. Huitema, P. López-Tarifa, L. Visscher, Z. Jamshidi and J. Bakker, J. Phys. Chem. Lett., 2019, 10, 2151–2155 CrossRef CAS PubMed.
- O. Lushchikova, H. Tahmasbi, S. Reijmer, R. Platte, J. Meyer and J. Bakker, J. Phys. Chem. A, 2021, 125, 2836–2848 CrossRef CAS PubMed.
- A. Karelovic and P. Ruiz, Catal. Sci. Technol., 2015, 5, 869–881 RSC.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. Elkjær, J. Hummelshøj, S. Dahl, I. Chorkendorff and J. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS PubMed.
- X. Nie, M. Esopi, M. Janik and A. Asthagiri, Angew. Chem., 2013, 125, 2519–2522 CrossRef.
- A. Halder, C. Lenardi, J. Timoshenko, A. Mravak, B. Yang, L. Kolipaka, C. Piazzoni, S. Seifert, V. Bonačić-Koutecký, A. Frenkel, P. Milani and S. Vajda, ACS Catal., 2021, 6210–6224 CrossRef CAS.
- B. Yang, C. Liu, A. Halder, E. Tyo, A. Martinson, S. Seifert, P. Zapol, L. Curtiss and S. Vajda, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef CAS.
- B. Yang, X. Yu, A. Halder, X. Zhang, X. Zhou, G. Mannie, E. Tyo, M. Pellin, S. Seifert, D. Su and S. Vajda, ACS Sustainable Chem. Eng., 2019, 7, 14435–14442 CrossRef CAS.
- S. Iyemperumal and N. Deskins, Phys. Chem. Chem. Phys., 2017, 19, 28788–28807 RSC.
- P. López-Caballero, A. Hauser and M. Pilar de Lara-Castells, J. Phys. Chem. C, 2019, 123, 23064–23074 CrossRef PubMed.
- C. Liu, H. He, P. Zapol and L. Curtiss, Phys. Chem. Chem. Phys., 2014, 16, 26584–26599 RSC.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C. Sun, T. Li, J. Muntean, R. Winans, D. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Y. Bu, M. Zhao, G. Zhang, X. Zhang, W. Gao and Q. Jiang, ChemElectroChem, 2019, 6, 1831–1837 CrossRef CAS.
- R. Raju, P. Rodriguez and R. Johnston, J. Phys. Chem. C, 2019, 123, 14591–14609 CrossRef CAS.
- R. Passalacqua, S. Parathoner, G. Centi, A. Halder, E. Tyo, B. Yang, S. Seifert and S. Vajda, Catal. Sci. Technol., 2016, 6, 6977–6985 RSC.
- Q. Zhang, Q. Zhao, X. Liang, X. Wang, F. Ma, B. Suo, W. Zou, H. Han, Q. Song, Q. Wu, Y. Li and H. Zhu, Int. J. Hydrogen Energy, 2018, 43, 9935–9942 CrossRef CAS.
- N. Zimmermann, T. Bernhardt, J. Bakker, R. Barnett, U. Landman and S. Lang, J. Phys. Chem. A, 2020, 124, 1561–1566 CrossRef CAS PubMed.
- X. Zhang, G. Liu, K. Meiwes-Broer, G. Ganteför and K. Bowen, Angew. Chem., Int. Ed., 2016, 55, 9644–9647 CrossRef CAS PubMed.
- G. Liu, Z. Zhu, M. Marshall, M. Blankenhorn and K. Bowen, J. Phys. Chem. A, 2021, 125, 1747–1753 CrossRef CAS PubMed.
- Y. Liu, L. Jiang, X. Li, L. Wang, J. Chen and S. He, J. Phys. Chem. C, 2018, 122, 19379–19384 CrossRef CAS.
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. Alexandrova and K. Bowen, J. Am. Chem. Soc., 2020, 142, 7930–7936 CrossRef CAS PubMed.
- S. Tang, N. Rijs, J. Li, M. Schlangen and H. Schwarz, Chem. – Eur. J., 2015, 21, 8483–8490 CrossRef CAS PubMed.
- A. Saputro, A. Maulana, F. Fathurrahman, G. Shukri, M. Mahyuddin, M. Agusta, T. Wungu and H. Dipojono, Int. J. Hydrogen Energy, 2021, 46, 14418–14428 CrossRef CAS.
- A. Pangh, Catal. Commun., 2021, 149, 106245 CrossRef CAS.
- J. Li, P. González-Navarrete, M. Schlangen and H. Schwarz, Chem. – Eur. J., 2015, 21, 7780–7789 CrossRef CAS PubMed.
- Q. Tang, F. Shi, K. Li, W. Ji, J. Leszczynski, A. Russell, E. Eddings, Z. Shen and M. Fan, Fuel, 2020, 280, 118446 CrossRef CAS.
- A. Green, J. Justen, W. Schöllkopf, A. Gentleman, A. Fielicke and S. Mackenzie, Angew. Chem., Int. Ed., 2018, 57, 14822–14826 CrossRef CAS PubMed.
- L. Gálvez-González, J. Juárez-Sánchez, R. Pacheco-Contreras, I. Garzón, L. Paz-Borbón and A. Posada-Amarillas, Phys. Chem. Chem. Phys., 2018, 20, 17071–17080 RSC.
- Y. Wang, K. Wiberg and N. Werstiuk, J. Phys. Chem. A, 2007, 111, 3592–3601 CrossRef CAS PubMed.
- A. Alvarez-Garcia, E. Flórez, A. Moreno and C. Jimenez-Orozco, Mol. Catal., 2020, 484, 110733 CrossRef CAS.
- M. Sharma, K. Mondal, T. Ghanty and A. Banerjee, J. Phys. Chem. A, 2021, 125, 2558–2572 CrossRef PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. Gilbert, M. Wormit, J. Kussmann and V. Rassolov, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Liu, E. Proynov, J. Yu, T. Furlani and J. Kong, J. Chem. Phys., 2012, 137, 114104 CrossRef PubMed.
- G. Hou, E. Faragó, D. Buzsáki, L. Nyulászi, T. Höltzl and E. Janssens, Angew. Chem., Int. Ed., 2021, 60, 4756–4763 CrossRef CAS PubMed.
- A. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- E. Glendening, C. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- R. Khaliullin, E. Cobar, R. Lochan, A. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS PubMed.
- A. Ghysels, T. Verstraelen, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, J. Chem. Inf. Model., 2010, 50, 1736–1750 CrossRef CAS PubMed.
- D. Die, B. Zheng, L. Zhao, Q. Zhu and Z. Zhao, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- T. Höltzl, N. Veldeman, J. De Haeck, T. Veszprémi, P. Lievens and M. Nguyen, Chem. – Eur. J., 2009, 15, 3970–3982 CrossRef PubMed.
- W. Knight, K. Clemenger, W. de Heer, W. Saunders, M. Chou and M. Cohen, Phys. Rev. Lett., 1984, 52, 2141–2143 CrossRef CAS.
- E. Janssens, H. Tanaka, S. Neukermans, R. Silverans and P. Lievens, New J. Phys., 2003, 5, 46 CrossRef.
- T. Höltzl, P. Lievens, T. Veszprémi and M. Nguyen, J. Phys. Chem. C, 2009, 113, 21016–21018 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp02220b |
| This journal is © the Owner Societies 2021 |