
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural responses of model biomembranes to Mars-relevant salts†
Simon
Kriegler
a,
Marius
Herzog
a,
Rosario
Oliva
a,
Stewart
Gault
b,
Charles S.
Cockell
b and
Roland
Winter
 *a
*a
aPhysical Chemistry I - Biophysical Chemistry, Faculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Street 4a, 44227 Dortmund, Germany. E-mail: roland.winter@tu-dortmund.de
bUK Centre for Astrobiology, SUPA School of Physics and Astronomy, University of Edinburgh, James Clerk Maxwell Building, Peter Guthrie Tait Road, Edinburgh, EH9 3FD, Scotland
First published on 17th June 2021
Abstract
Lipid membranes are a key component of contemporary living systems and are thought to have been essential to the origin of life. Most research on membranes has focused on situations restricted to ambient physiological or benchtop conditions. However, the influence of more extreme conditions, such as the deep subsurface on Earth or extraterrestrial environments are less well understood. The deep subsurface environments of Mars, for instance, may harbor high concentrations of chaotropic salts in brines, yet we know little about how these conditions would influence the habitability of such environments for cellular life. Here, we investigated the combined effects of high concentrations of salts, including sodium and magnesium perchlorate and sulfate, and high hydrostatic pressure on the stability and structure of model biomembranes of varying complexity. To this end, a variety of biophysical techniques have been applied, which include calorimetry, fluorescence spectroscopies, small-angle X-ray scattering, dynamic light scattering, and microscopy techniques. We show that the structure and phase behavior of lipid membranes is sensitively dictated by the nature of the salt, in particular its anion and its concentration. We demonstrate that, with the exception of magnesium perchlorate, which can also induce cubic lipid arrangements, long-chain saturated lipid bilayer structures can still persist at high salt concentrations across a range of pressures. The lateral organization of complex heterogeneous raft-like membranes is affected by all salts. For simple, in particular bacterial membrane-type bilayer systems with unsaturated chains, vesicular structures are still stable at Martian brine conditions, also up to the kbar pressure range, demonstrating the potential compatibility of environments containing such ionic and pressure extremes to lipid-encapsulated life.
Introduction
An increasing number of organisms, so called extremophiles, have been found in extreme environments in recent years.1,2 They thrive in environments in which physical and chemical parameters, such as temperature, pressure, salinity and pH are extreme.1–11 Investigating and defining the physical and chemical limits of life and its associated macromolecules allows us to assess the habitability of extraterrestrial environments wih respect to known life and understand the conditions under which life might have originated.3Extremophilic organisms on Earth can tolerate and thrive under high ionic concentrations and pressures. In terms of ionic tolerance, the best characterised are the so-called halophiles (salt-loving microorganisms), found in all three domains of life, that can reproduce in NaCl concentration up to 35%.1,2 On Earth, pressure ranges from 1 bar to 1120 bar (0.1 to 112 MPa), with higher pressures observed at subduction zones (e.g., 9 kbar).1 Interestingly, piezophiles (pressure-loving microorganisms) are also found in all three domains of life,5–9 with several piezophilic and piezotolerant microorganisms having been isolated from deep-sea locations. The current record holder is Thermococcus piezophilus, a thermophilic archaeon able to survive up to 1250 bar.1 There have even been reports that life can survive brief exposures up to 20 kbar.12,13
Similar to Earth, the habitability of environments on other planetary bodies will be dictated by the interplay between chemical and physical environmental parameters. The subsurface represents one of the most important locations in the search for extinct and extant extraterrestrial life. On Earth alone, the subsurface is estimated to house 50–87% of the Earth's microorganisms.14,15 One of the requirements for life is the presence of liquid water. Over the past years, there has been increasing evidence of periods of surface water on Mars, and it is now thought that there may exist subsurface water in the form of lakes below the surface of Mars.3,16–18 Aqueous environments on Mars may take the form of deep groundwater or subglacial lakes, as observed beneath the Martian south pole. The Martian groundwater experiences hydrostatic pressures of ∼1 kbar at the base of the cryosphere if it reached a depth of 10 km.16 Similar harsh environments in subsurface lakes can be found on Earth, which have been shown to be capable of hosting life.2
Deep subsurface environments often harbor high concentrations of dissolved salts, yet we know little about how these highly concentrated ions, viz. such brines, shape the conditions for life, and even less about how combined salt-perssure effects limit the chances for cellular life. One particular ion is perchlorate, ClO4−, which is found in extreme environments on Earth and ubiquitously on Mars.19–21 Perchlorates are also known to act as chaotropes, perturbing the structure of water and its hydrogen bonding capacity,22 and might hence affect biomolecular hydration, structure and function. Perchlorate salts, particularly Mg(ClO4)2 and Ca(ClO4)2, exhibit deep eutectic temperatures allowing for the presence of liquid water at temperatures as low as −70 °C, therefore highlighting the case for their presence in subsurface Martian environments. In addition to perchlorate salts, sulfates are widely distributed across the Martian surface. On Earth, sodium chloride, NaCl, is the most well-studied salt relevant for biochemistry, but sulfate-containing environments are known and sulfates are an important component of many fluids in deep subsurface settings on Earth.1–3,23–27 To advance our understanding of the ability of subsurface environments to support life, we need to examine the combination of strong ionic effects imposed by these salts and high hydrostatic pressures (HHP).
Previous studies have demonstrated deleterious effects of destabilizing ions such as perchlorates on the activity of α-chymotrypsin (α-CT) and other enzymes at ambient conditions.20,21,28 Conversely, stabilizing salts, such as magnesium sulfate, MgSO4, have been shown to increase the activity and structural stability of α-CT.21 In recent work, we demonstrated that high pressures increase the enzymatic activity of α-CT even in the presence of high perchlorate concentrations, and the results suggested that high hydrostatic pressures may increase the habitability of environments under perchlorate stress. Therefore, deep subsurface environments that combine these stressors, potentially including the subsurface of Mars, may be more habitable than previously thought.21
The effects of these Martian salts on other biomolecular systems, such as the cellular membrane, is still unknown. Biological cells are surrounded by cytoplasmic membranes that function as barriers between the cytoplasm and the extracellular environment. The plasma membrane is essential for maintaining the internal conditions of the cells optimal for metabolism, energy transduction and signal processing. Membranes are complex structures,29 consisting of many different lipids, and organisms are able to adapt the properties of their cytoplasmic membranes in response to changes in the environment by changing the lipid composition.5–7,30–40 Bacterial and eukaryal lipids are mainly composed of two acyl chains, which are bound via an ester linkage to glycerol. These lipids are organized in a bilayer such that the polar head-groups stick into the water phases while the carbon chains are directed towards the inner side of the membrane. In contrast to bacterial and eukaryal lipids, archaeal lipids consist of two phytanyl chains, which are linked via an ether bond to glycerol or other alcohols like nonitol. In extremophilic archaea, ether lipids are found in which the phytanyl chains of two diether lipids are fused to form a C40 core. These tetraether lipids span the whole membrane.30–32
In this study we set out to investigate the combined effects of high pressure and high concentrations of Mars-relevant salts, including sodium and magnesium perchlorate and sulfate at the high concentrations anticipated, on the stability and structure of model biomembranes of varying complexity, ranging from one-component lipid bilayers to bacterial model biomembranes and ‘raft-like’ heterogeneous membranes mimicking eukaryotic cellular membranes. To this end, various biophysical techniques have been applied, which include calorimetry, fluorescence spectroscopies, small-angle X-ray scattering (SAXS), dynamic light scattering, and microscopy techniques. The results show, to our knowledge for the first time, the stability of lipid membranes under extreme salt and pressure conditions relevant to extraterrestrial environments and they reveal the distinct influence of these salts on the structure, fluidity, lateral organization, and morphology of lipid bilayer membranes, including the formation of cubic membrane structures at high magnesium perchlorate concentrations.
Results and discussion
To gain insights into the thermodynamic properties and phase transitions of the lipid bilayer membranes in the presence of the Mars-relevant salts, differential scanning calorimetry (DSC) measurements were carried out first. Depending on the lipid composition, temperature, pressure, and cosolute concentration, lipid bilayers can adopt various lamellar bilayer phases, namely the gel (Lβ′), ripple gel (Pβ′), and fluid (Lα, also referred to as liquid-crystalline) phases, given in the order of increasing temperature.29,37 The first-order main phase transition from the gel to the fluid phase occurs at a critical temperature Tm during the heating process. This phase transition is characterized by the melting of the ordering of the hydrocarbon chains in the interior of the phospholipid bilayer. It is important to note that a significant amount of fluidity, i.e. disordered phase, is needed for optimal physiological function.We first discuss the influence of the perchlorate and sulfate salts on the gel-to-fluid phase transition temperature of multilamellar vesicles (MLVs) consisting of pure dipalmitoylphosphatidylcholine (DPPC) bilayers, one of the best characterized simple model biomembrane systems. DPPC is a phospholipid consisting of two saturated C16 acyl chains attached to a zwitterionic phosphatidylcholine headgroup.
The DSC-thermogram of the pure DPPC shows a pretransition from the Lβ′ to the Pβ′ gel phase at 36.0 ± 0.5 °C, due to the rearrangement of lipid head groups, and the gel-to-fluid (Pβ′- to Lα-phase) main transition, due to the melting of hydrocarbon chains, at Tm = 41.6 ± 0.5 °C, (black thermograms in Fig. 1), in good agreement with literature data.29,37 As shown in Fig. 1a, addition of 0.25 M Mg(ClO4)2 leads to a decrease of the gel-to-fluid transition temperature by about 2 °C, i.e., the salt destabilizes the ordered gel phase of the lipid bilayer. The addition of 0.5 M of the salt largely abolishes the gel-to-fluid transition and a new DSC peak appears at high temperature (53.3 ± 0.2 °C). Upon addition of 1 M Mg(ClO4)2, a small endothermic peak appears at 59.6 ± 0.1 °C only, which indicates a high-temperature phase transition to another lipid phase (see below). Fig. 1b shows that the replacement of the Mg2+ by the Na+ cation destabilizes the gel-phase of DPPC as well. The exchange of the cation leads to a different scenario. Tm decreases by about 3 °C upon addition of 0.25 or 0.5 M NaClO4. The increase in the width of the DSC peak indicates a decrease of the cooperativity of the transition, viz. a broader phase separation region. The gel-to-gel (Lβ′–Pβ′) pretransition is not observed anymore, however, but no high-temperature DSC peak is recorded in this case.
 | ||
| Fig. 1 Comparative DSC measurements on DPPC multilamellar vesicles in the presence of different concentrations of (a) Mg(ClO4)2, (b) NaClO4, (c) MgSO4, and (d) Na2SO4 (the solubility limit is reached at ∼1 M concentration). For comparison, in each panel, the DSC thermogram of DPPC in pure buffer (black lines) is shown. | ||
A different scenario is observed in the presence of the corresponding sulfate salt, MgSO4 (Fig. 1c). In this case, addition of the sulfate leads to a shift of Tm to higher temperatures, 2 °C and 3 °C upon addition of 0.5 or 1 M MgSO4, respectively. This indicates that the sulfate is stabilizing the ordered gel phase of the lipid bilayer, probably through long-range electrostatic interactions of the highly charged sulfate with the choline ((CH3)3N+-) headgroup, and the stable lamellar bilayer structure persists up to the highest salt concentration measured. A similar behavior is observed if the Mg2+ cation is replaced by the monovalent Na+ (Fig. 1d). The difference between the results for the Mg2+ and Na+ perchlorate and sulfate salts indicates that the anions seem to control the change in phase behavior of the lipid bilayer.
To yield complementary information about the lipid order and fluidity of the membrane upon addition of the Mars-relevant salts, temperature-dependent Laurdan fluorescence spectroscopy measurements with membranes consisting of DPPC vesicles were performed. Laurdan, a solvatochromic membrane dye, presents a large excited state dipole moment, which results in its ability to report the extent of water penetration into the bilayer surface and upper chain region due to the dipolar relaxation effect. Because water penetration is correlated with lipid packing and hence membrane fluidity, Laurdan has been extensively used in membrane studies.41–44 The emission spectrum of Laurdan is centered at 490 nm when the lipids are in a disordered phase and is shifted to the blue (around 440 nm) when the lipids are in a more packed phase. The generalized polarization function, GP = (I440 − I490)/(I440 + I490), gives a convenient and quantitative way to measure the shift of the recorded emission intensities, I.
As shown in Fig. 2, the DPPC bilayer in pure buffer solution displays nearly constant GP-values of about 0.5 in the whole temperature range from 20 to 35 °C, which indicates that the membrane is in an ordered gel phase. In this state, the membrane has a high lipid order and the acyl chains mostly adopt the all-trans conformation. This state supports a less polar Laurdan environment in the upper chain region, resulting in an increased fluorescence emission at 440 nm. Above ∼41 °C, a relatively sharp transition from high to low GP-values is observed. At temperatures above the gel-to-fluid transition temperature, the membrane reaches an all-fluid (Lα) phase with a low lateral lipid order parameter owing to a high mobility and conformational disorder of the acyl chains, which results from an entropically favored large number of gauche conformers and kinks. In this state, water can penetrate the upper region of the bilayer, leading to an increase of the fluorescence intensity of Laurdan at 490 nm and a concomitant decrease of the GP-values.
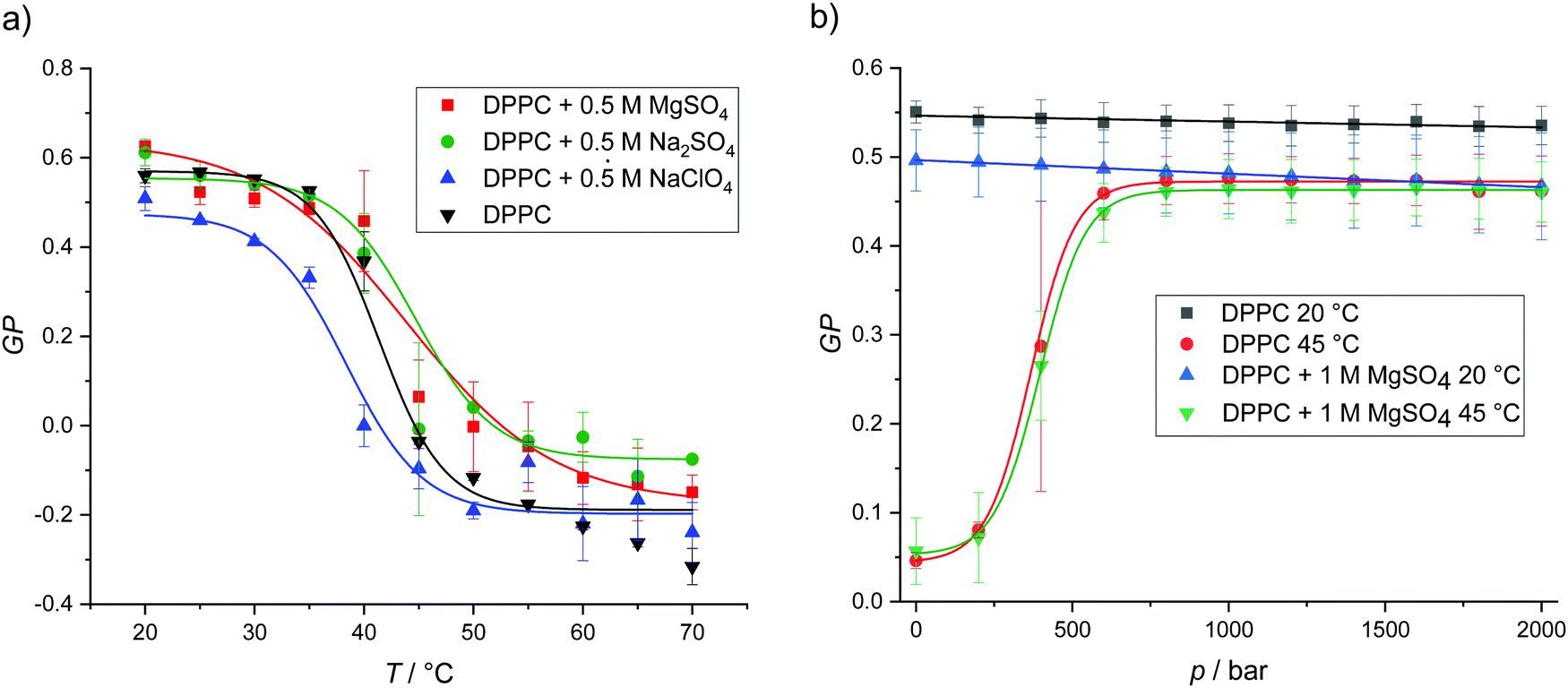 | ||
| Fig. 2 (a) Temperature-dependent GP-values of DPPC vesicles in pure buffer and in the presence of 0.5 M MgSO4, Na2SO4 and NaClO4. The transition temperatures were calculated from Boltzmann fits. (b) Pressure-dependent GP-values of DPPC vesicles at 20 and 45 °C in pure buffer and in the presence of 1 M MgSO4. Owing to the smaller partial molar volume of the lipids in the ordered gel phases, the gel state of the lipid bilayer is favored upon pressurization. According to literature data, Tm of DPPC bilayers increases with pressure at a rate of ∼20 °C kbar−1.37 | ||
The addition of 0.5 M MgSO4 and Na2SO4 leads to small increase of Tm and increases the order of the lipid chains in the fluid phase to some extent. Conversely, addition of 0.5 M NaClO4 decreases Tm, i.e. destabilizes the gel phase. Such behavior is consistent with the results obtained from the DSC measurements. These results are most likely due to an efficient binding of the chaotropic perchlorate anion to the lipid headgroup and penetration into the lipid chain region which prohibits dense packing of the lipid's acyl chains.45,46 The low charge density of the ClO4− anion is accompanied by a relatively high binding constant owing to the ease with which it can strip its hydration shell.45 In fact, the perchlorate anion has also been shown to interact even with apolar surface patches of proteins, i.e., the anion displays almost hydrophobic properties, which reduces the Gibbs energy of unfolding and therefore promotes destabilization of protein structures.47 The opposite effect is observed for strongly solvated highly charged anions, such as SO42−. GP-values of DPPC in Mg(ClO4)2 solution are not included here, as the addition of this salt leads to deterioration of the characteristic two-peak spectrum of Laurdan and a loss of the fluorescence signal at high temperatures. This can be explained by a lack of well defined incorporation into a stable lamellar bilayer structure and a loss of fluorescence intensity by degradation of the free fluorophore due to oxidation by perchlorate (Fig. SI1 shows the temperature dependence of the Laurdan emission spectra for the DPPC system in all salt solutions, ESI†).
With the exception of Mg(ClO4)2, we have seen that the salts do not drastically affect the temperature dependent lipid phase behavior. As a representative example, Fig. 2b shows the effect of pressure up to 2000 bar on the lipid order and phase behavior of DPPC for two temperatures. As expected, in pure buffer at T = 20 °C, i.e. in the gel phase, pressure has no marked effect on the already densely packed phospholipid arrangement, i.e. the GP-values remain essentially constant up to high pressures. At 45 °C, i.e. ∼3 °C above the gel-to-fluid transition temperature of DPPC, we observe a pressure-induced transition from the fluid phase to a high-pressure gel phase around 500 bar. As the partial molar lipid volume in an ordered gel phase is smaller (∼3%) than in the fluid-like state, the densely packed gel phase is generally adopted at high pressures, the transition pressure strongly depending on the length and degree of unsaturation of the lipid's acyl chains.35,37–39,48,49 No significant changes are observed upon addition of 1 M MgSO4 at both temperatures, i.e., the lamellar bilayer structure is stable over the whole pressure range covered and the pressure dependent phase behavior is hardly affected by the sulfate salt.
To still be able to reveal the lipid order in the presence of the deteriorating Mg(ClO4)2 salt, we opted for another lipid packing-sensitive fluorophore, DPH (1,6-diphenyl-hexatriene), which is known to penetrate more deeply into the lipid bilayer structure. Hence, DPH fluorescence data provide information related to the central region of the membrane rather than reporting on the properties of the upper segments of the lipid acyl chains and the interfacial headgroup region. To this end, steady-state fluorescence anisotropy measurements were carried out. Anisotropy measurements are suitable for monitoring the molecular motions in membranes. Membrane fluidization increases the free volume for molecular rotations of the dye and decreases the intensity of the parallel component of the emitted light. Accordingly, the fluorescence anisotropy, r = (III − I⊥)/(III + 2I⊥), is reciprocally related to the membrane fluidity.39,41 More quantitatively, the measured end value and initial value in time-dependent fluorescence anisotropy experiments, recording r(t), with membrane systems are related to the average lipid order parameter S squared, i.e. r∞/r0 = S2 (with 0 ≤ S ≤ 1), which can also be approximated using steady-state fluorescence anisotropy measurements as the stead-state fluorescence anisotropy, rss, of DPH reflects mainly r∞.39,50
Fig. 3 shows the temperature dependence of the fluorescence anisotropy of DPPC vesicles in the absence and presence of Mg(ClO4)2. DPPC reveals its main phase transition around 41 °C, which is accompanied by a large decrease of the rss-value from about 0.35 to 0.12. The corresponding lipid order parameter, S, decreases from ∼0.93 in the gel phase to ∼0.37 in the fluid phase. In the presence of Mg(ClO4)2, the anisotropy values indicate disappearance of the gel-to-fluid chain melting transition and high fluidity over the whole temperature range from 5 to 75 °C. The endothermic peaks detected above 50 °C in our DSC experiments (Fig. 1a) are not distinctly reflected in the anisotropy experiments (despite possibly a change in slope around 50 °C), most likely as the conformational dynamics and hence fluidity of the acyl chains remain essentially unchanged at the endothermic phase transition.51
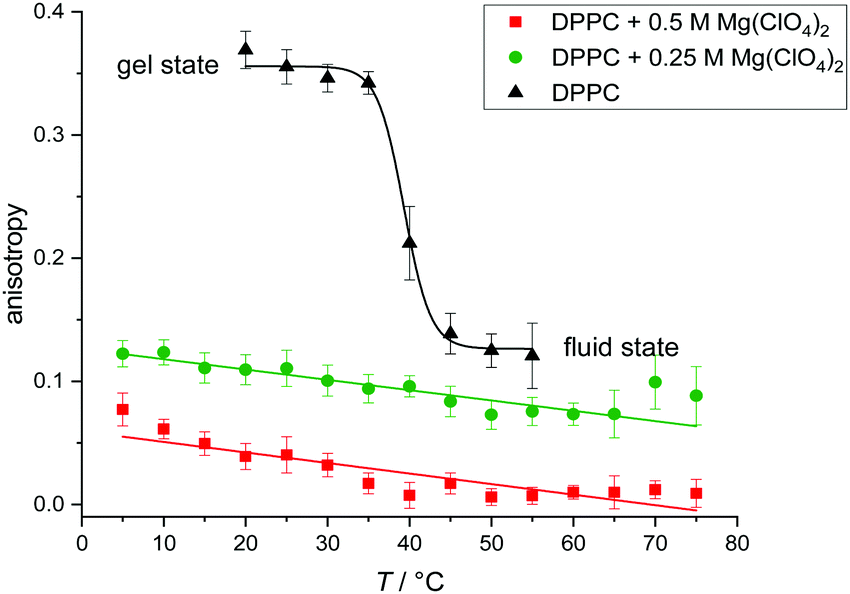 | ||
| Fig. 3 Temperature dependence of the steady-state fluorescence anisotropy, rss, for the fluorescence probe DPH embedded in DPPC vesicles in the absence and presence of 0.25 M and 0.5 M Mg(ClO4)2 salt. | ||
To reveal changes in the topology of the lipid phases, small-angle X-ray scattering (SAXS) and fluorescence microscopy studies were carried out. SAXS data allow for the determination of lamellar distance constants of multilamellar vesicles (MLVs) by the appearance of Bragg-peaks in the elastic scattering curve, I(Q). Q is the modulus of the wave vector transfer, given by Q = (4π/λ)sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, with 2θ being the scattering angle and λ the wavelength of the X-rays. The positions of the maxima were determined by the refinement of a Gaussian function to the shape of the Bragg-peaks after desmearing. The type of phase can be distinguished by the characteristic SAXS peak ratios in Q-space (Q1:Q2:Q3:…).37 For the lamellar lipid phase they are equidistant (1:2:3:…), for an inverse hexagonal phase (HII) they are
θ, with 2θ being the scattering angle and λ the wavelength of the X-rays. The positions of the maxima were determined by the refinement of a Gaussian function to the shape of the Bragg-peaks after desmearing. The type of phase can be distinguished by the characteristic SAXS peak ratios in Q-space (Q1:Q2:Q3:…).37 For the lamellar lipid phase they are equidistant (1:2:3:…), for an inverse hexagonal phase (HII) they are 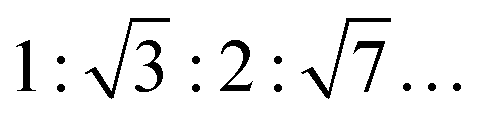 , for an inverse bicontinuous cubic phase Pn3m (QDII) they are:
, for an inverse bicontinuous cubic phase Pn3m (QDII) they are: 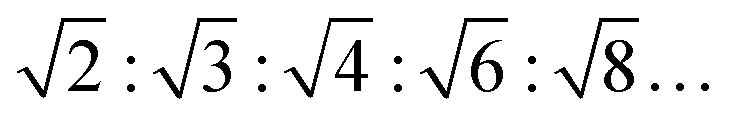 , and for an inverse bicontinuous cubic phase Im3m (QPII)
, and for an inverse bicontinuous cubic phase Im3m (QPII) 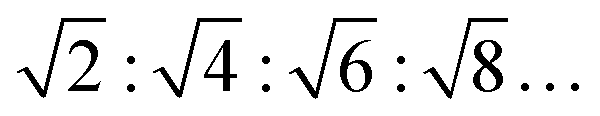 .52–55 The sponge phase (L3) is another bicontinuous phase; it has a 3D lipid bilayer surrounded by two different water channels similar to bicontinuous cubic phases, but it does not display long-range order and is more fluid compared to the cubic phases. According to Bragg's equation, the lattice parameter for the lamellar phase is given by dlam = 2π/Q1. The lamellar repeat distance, dlam, includes the thickness of the MLV's interlamellar water layer, dw, and the thickness of the lipid bilayer, dl, i.e., dlam = dw + dl.37
.52–55 The sponge phase (L3) is another bicontinuous phase; it has a 3D lipid bilayer surrounded by two different water channels similar to bicontinuous cubic phases, but it does not display long-range order and is more fluid compared to the cubic phases. According to Bragg's equation, the lattice parameter for the lamellar phase is given by dlam = 2π/Q1. The lamellar repeat distance, dlam, includes the thickness of the MLV's interlamellar water layer, dw, and the thickness of the lipid bilayer, dl, i.e., dlam = dw + dl.37
Fig. 4 presents selected SAXS patterns of 10 or 20 wt% DPPC and DOPC MLV suspensions in the absence and presence of 0.5 M MgSO4 or Mg(ClO4)2. The SAXS patterns of pure DPPC MLVs (in HBS buffer) at selected temperatures between 20 and 60 °C, i.e. at temperatures below and above Tm. The SAXS patterns show lamellar reflections of first, second and third order in the absence of salt. The lamellar spacings, dlam, amount to ∼6.5 nm at 30 °C (Lβ′ gel phase), ∼7.4 nm at 40 °C (Pβ′ gel phase), and decrease again after passing the chain-melting transition to ∼6.6 nm at 60 °C in the liquid-crystalline Lα phase. The addition of 0.5 M MgSO4 does not lead to a substantial change of the diffraction pattern; the dlam-values increase by ∼0.5 nm in the gel phase and by ∼1 nm in the fluid-like Lα phase. Such increase of dlam might be largely due to an increase of the interlamellar water layer (dw), but can also be due to an increase of lipid bilayer thickness (dl) as suggested by the measurements of the lipid order parameter (Fig. 2). A completely different scenario is observed upon addition of 0.5 M Mg(ClO4)2 to 10 wt% DPPC MLVs. The SAXS pattern changes completely: the broad Bragg reflections indicative of lamellar bilayer stacks vanish and Bragg reflections with spacings in the ratio of about 1:∼1.4 appear, which decrease in intensity with an increase of temperature. Such a diffraction pattern is indicative of a non-lamellar lipid phase, probably a bicontinuous inverse cubic lipid phase (QII) of space group Im3m (QPII), which should present spacings in the ratio 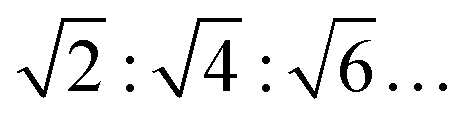 . The corresponding lattice constant,
. The corresponding lattice constant, 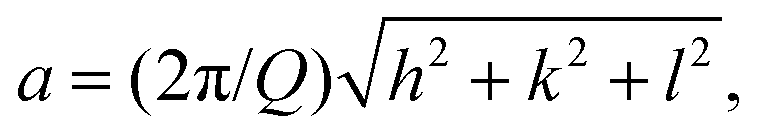 amounts to ∼8.9 nm. According to the geometrical representation shown at the bottom of Fig. 4, the cubic phase consists of a single contiguous fluid lipid bilayer folded into an infinite periodic minimal surface, which partitions the aqueous space into disjointed compartments.37,52–55 At a much higher lipid concentration, a 20 wt% DPPC dispersion in 0.5 M Mg(ClO4)2, the scattering pattern changes again. A particular structure (or a mixture of different topologies) cannot unambiguously be assigned for the scattering pattern at low temperatures. Above ∼50 °C, in agreement with the DSC data, a phase transition to a lamellar structure (Q-spacings 1:2:3) of 5.6 nm repeat distance is observed.
amounts to ∼8.9 nm. According to the geometrical representation shown at the bottom of Fig. 4, the cubic phase consists of a single contiguous fluid lipid bilayer folded into an infinite periodic minimal surface, which partitions the aqueous space into disjointed compartments.37,52–55 At a much higher lipid concentration, a 20 wt% DPPC dispersion in 0.5 M Mg(ClO4)2, the scattering pattern changes again. A particular structure (or a mixture of different topologies) cannot unambiguously be assigned for the scattering pattern at low temperatures. Above ∼50 °C, in agreement with the DSC data, a phase transition to a lamellar structure (Q-spacings 1:2:3) of 5.6 nm repeat distance is observed.
 | ||
| Fig. 4 SAXS patterns of 10 or 20 wt% MLV dispersions of DPPC, DOPC and DOPE:DOPG (70:30 molar ratio) bilayers at selected temperatures in the absence and presence of 0.5 M MgSO4 or Mg(ClO4)2. Bottom: Schematic representation of a multilamellar vesicle (MLV) and the inverted bicontinuous cubic phase (QPII) Im3m. | ||
In the case of the fluid C18:1cis-unsaturated phospholipid DOPC (Tm ≈ −22 °C37), broad lamellar Bragg reflections are visible in the case of pure DOPC and DOPC + 0.5 M MgSO4, with dlam-spacings of 6.5 ± 0.1 nm and 7.2 ± 0.1 nm at 20 °C, respectively, which shift to 5.4 ± 0.1 nm in the case of the 0.5 M perchlorate solution. Remarkably, the dlam-value in the latter case is much smaller, but multilamellar vesicles persist even in the presence of 0.5 M Mg(ClO4)2. The decrease in dlam-value is most likely due to a large decrease of the intermolecular water layer, dw, probably induced by the increase of the osmotic pressure imposed by the bulk salt solution of the MLV assembly.
To yield complementary information about the structure of a bacterial membrane-mimicking bilayer, SAXS measurements were also carried out on a DOPE:DOPG (70:30) lipid dispersion at 20 °C in the absence and presence of 0.5 M Mg(ClO4)2 (the d-spacings of all scattering data are shown in Table SI1, ESI†). The SAXS patterns indicate persistence of MLVs of lamellar spacings of about 5.5 ± 0.1 nm, which are similar to dlam-values of about 5.1 nm for pure DOPE bilayers.53 Owing to the large disorder of the MLVs of this lipid mixture, no higher-order reflections are visible. The multilamellarity disappeared largely for 0.5 M MgSO4, so that no pronounced Bragg reflections could be recorded. However, the existence of vesicular structures could be verified by complementary dynamic light scattering (DLS) experiments for this system as well (see Fig. SI 2, ESI†). With 5.6 ± 0.3 nm, the dlam-spacing in the presence of 0.5 M Mg(ClO4)2 is similar to that of pure DOPE:DOPG (70:30) bilayers, i.e. the bilayer structure of the bacterial model membrane is maintained in this highly concentrated magnesium perchlorate solution.
Finally, light microscopy and confocal fluorescence microscopy measurements were carried out to visualize morphological changes of the lipid vesicles (giant unilamellar vesicles, GUVs) on a larger (μm) length scale upon addition of the Mars-relevant salts. Fig. 5a shows light microscopy images of DPPC vesicles at ambient temperature (T = 20 °C), i.e. in its gel state, before and after the addition of MgSO4 and Mg(ClO4)2. As expected, the addition of salt at such high concentrations imposes distinct osmotic stress, which leads to deformation of the intact spherical GUVs. In case of the perchlorate salt, extreme shrinking and disappearance of vesicles occurs within minutes. Fig. 5b shows also corresponding data for DOPC vesicles at ambient temperature, i.e. a lipid vesicle in its fluid-like state (the gel phase of DOPC is reached below about −22 °C, only37). Here, a similar effect is observed, viz. deformation and budding of vesicles in case of MgSO4 and collapse and disintegration of the GUVs directly after injection of the Mg(ClO4)2 salt.
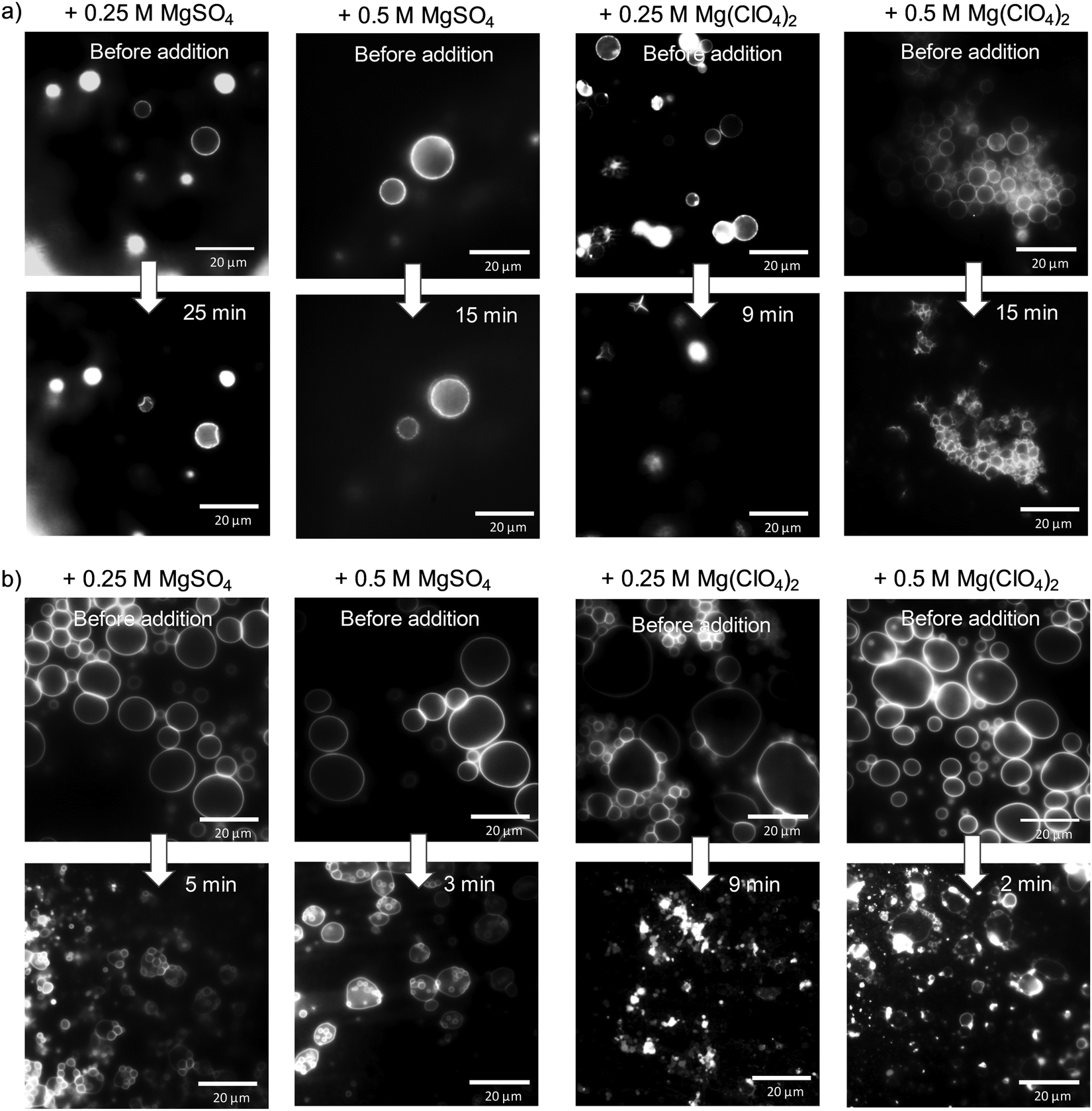 | ||
| Fig. 5 Light microscopy pictures of (a) DPPC and (b) DOPC giant unilamellar vesicles at T = 20 °C before and after addition of MgSO4 and Mg(ClO4)2 salt. | ||
Membranes of eukaryotic cells exhibit a considerably more complex picture of organization, including lateral heterogeneities arising from the tighter packing of cholesterol with saturated lipids in liquid-ordered (lo) domains and mostly unsaturated phospholipids in liquid-disordered (ld) domains. As a representative model membrane system, we present fluorescence microscopy data on a heterogeneous 5-component model membrane system (5-CS) in the absence and presence of salt. To this end, GUVs of the 5-CS, fluorescently labelled with N-Rh DHPE (ld-phase, red) and NBD-DHPE (lo-phase, green), were imaged before and after injection of the salt solutions (Fig. 6). At room temperature, before addition of the salts, the GUVs show the well-known distinct phase separation into liquid-disordered (ld, red) and cholesterol-rich liquid-ordered (lo, green) domains. Immediately after injection of the MgSO4 salt, the relative amount of both phases and the topology of vesicles changes. The amount of fluid ld phase decreases, and aggregation and budding of vesicles occurs. Upon addition of the corresponding perchlorate salt, disintegration and bursting of vesicles prevails. Interestingly, preparing the lipid vesicles in the Mg(ClO4)2 salt solution from the very beginning does not lead to formation of GUVs at all, whereas few and aggregated vesicles are still observed upon preparation in MgSO4 solution.
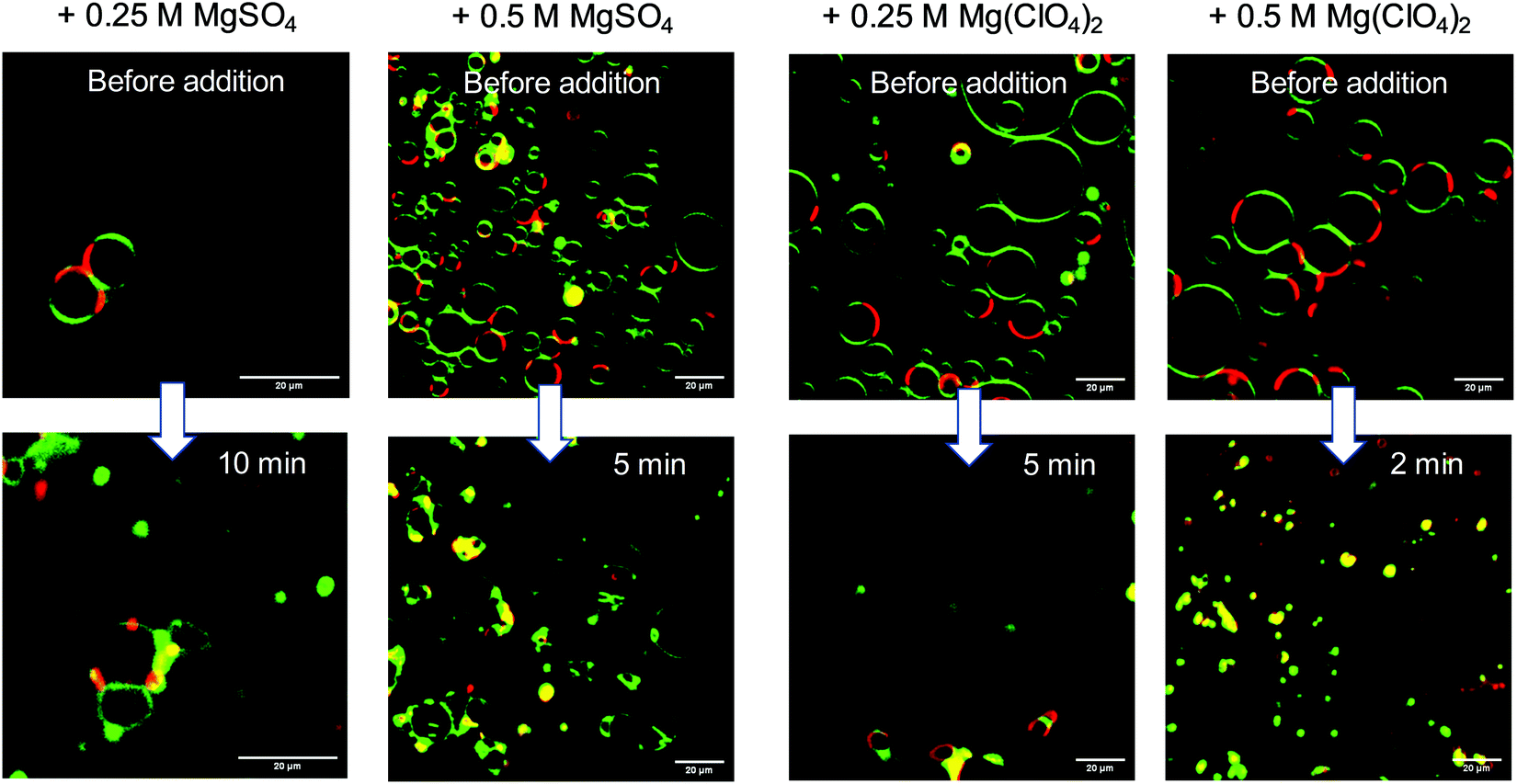 | ||
| Fig. 6 Selected time-dependent confocal fluorescence microscopy images (cross-sectional views) of GUVs composed of the anionic five-component (5-CS) heterogeneous (raft-like) membrane at 20 °C before and after injection of MgSO4 and Mg(ClO4)2 salt. Scale bar = 20 μm. The membrane fluorophores used were N-Rh-DHPE (red, labelling ld-phase) and NBD-DHPE (green, labelling lo-phase). | ||
Summary and conclusions
Generally, the binding mode of metal ions with a lipid membrane and their position in the bilayer depend upon their size, charge and bonding characteristics, and the salts across the Hofmeister series have a different impact.56–59 Using, for example, X-ray scattering and spectroscopies, such as fluorescence anisotropy, sum-frequency generation vibrational spectroscopy or isotope-edited (13C, 31P, 2H) NMR, salt-membrane interactions can be revealed. Highly charged ions, such as the divalent cations Ca2+ and Mg2+, are generally adsorbed on the lipid bilayer interface and ion-dipole interactions may cause conformational modifications in the headgroup and eventually also in the acyl tail region of the membrane.56–59 The binding strength follows the order Ca2+ > Mg2+, with binding constants to phosphatidylcholines such as DPPC of about 21 M−1 for Ca2+ and 2.5 M−1 for Mg2+.56 For lipid bilayers with unsaturated chains, such as DOPC, having larger cross-sectional areas, smaller affinities towards metal ions are observed, probably due to weaker interactions and as binding is connected with a significant loss of headgroup entropy. Ca2+ binds closer and stronger to the interfacial region than Mg2+ and induces partial dehydration and long-range interactions in the polar headgroup region, causing tightening of the acyl chain packing. Upon interaction with anionic lipids, even vesicle fusion is induced.57 Unlike for Ca2+, dehydration does not easily take place for Mg2+,56 which is rationalized by the tight hydration of Mg2+ and high free energy barrier of Mg2+ to bind to lipid moieties as compared to that of bulk water molecules. In fact, Mg2+ hydrates the liposomal surface by carrying water molecules. Hence, Mg2+ as prevailing divalent cation in Mars-relevant salts does not cause severe deleterious effects on the structure and fluidity of membranes.We show that the combined effects of high pressure and high concentrations of the Mars-relevant salts sodium and magnesium perchlorate and sulfate have diverse effects on the stability and structure of the model biomembranes. Concentrations below 0.5 M Mg(ClO4)2 lead to a decrease of the gel-to-fluid transition temperature of DPPC by a few degrees only, i.e. the perchlorate increases the fluidity of the membrane. At concentrations at and beyond 0.5 M of the perchlorate salt, the gel-to-fluid transition is abolished and a non-lamellar lipid phase, most likely of bicontinuous cubic structure, is formed from 10 wt% DPPC bilayer dispersions (other phases, such as fluid lamellar structures, are seen at high temperatures for very high phospholipid concentrations). Of note, such 3D bicontinuous structures could still serve as compartmentalization systems. For example, they are used as slow drug release systems in pharmacology.60,61 Remarkably, such structures have also been discovered in biological systems, such as in chloroplasts (thylakoid membranes) and the inner mitochondrial membrane, and planar biological membranes have been found to fold into cubic membranes in response to external stresses such as starvation.60,61 Interestingly, even in 1 M concentrations of sodium perchlorate salt lipid vesicles were still preserved with only some fluidizing effect. Vesicular structures are well preserved for the all-fluid phospholipid bilayer DOPC and the bacterial model membrane DOPE:DOPG (70:30) owing to the high conformational disorder of their cis-unsaturated acyl chains (e.g., Tm(1 bar) ≈ −22 °C, dTm/dp ≈ 10 °C kbar−1 for DOPC bilayers).37
A different scenario is observed in the presence of the corresponding sulfate salts, which have no drastic effect on the structure of the membranes even up to molar concentrations for both cations. They lead to a minor stabilization of the gel phase only owing to their strong electrostatic interaction of the sulfate group with the choline lipid head group. The differences between the results for the Mg2+ and Na+ perchlorate and sulfate salts indicate that the anions seem to be mainly responsible for the change in the structure and phase behavior of the lipid bilayer. An efficient binding of the chaotropic perchlorate anion to the lipid headgroup and penetration into the lipid chain region prohibits dense packing of the lipid's acyl chains,45,46 which might be supported by an increase of headgroup hydration upon binding of Mg2+. The low charge density of the ClO4− anion is accompanied by a relatively high binding constant owing to the ease with which it can strip its hydration shell. In fact, the perchlorate anion has also been shown to interact even with apolar environments such as surface patches of proteins.47 The opposite effect is observed for strongly solvated highly charged kosmotropic anions, such as SO42−, which support tighter packing of the lipid bilayer.
Owing to the high salt concentrations and hence osmotic pressure, deformation, shrinking and budding of giant unilamellar vesicles was observed. The effect is most pronounced for the Mg(ClO4)2 salt, which induces immediate collapse and disintegration of the GUVs directly after injection of the salt solution. With regards to more complex heterogeneous anionic membranes, mimicking eukaryotic lipid bilayers that display lo–ld lateral phase segregation, we observed that the relative amount of both phases and the topology of vesicles changes after injection of the sulfate salt. The amount of fluid ld phase decreases, and aggregation and budding of vesicles occurs. Upon addition of the corresponding perchlorate salt, disintegration and bursting of vesicles occurs. Of note, preparing the lipid vesicles in the Mg(ClO4)2 salt solution from the very beginning does not lead to formation of GUVs at all, whereas few and aggregated vesicles are still observed upon preparation in MgSO4 solution.
Although water equilibrates readily across the lipid walls of GUVs, the passive permeation of solutes such as the Martian-like salts is strongly hindered. Owing to the large area expansion modulus (55–70 kBT nm−2), coupled with a low bending rigidity (10–20 kBT), lipid bilayer shells bend readily, but they tolerate only a limited area expansion (∼5%).62–68 Hence, GUVs experiencing high solute concentration differences across their lipid bilayer boundary adjust their volume, deforming in hypertonic media and swelling (via swell-burst lytic cycles) in hypotonic ones.67,68 Generally, the shape free-energy functional of lipid membranes has been found to depend sensitively on the vesicle's volume and area, the spontaneous curvature of the membrane surface, the bending rigidity, and the differential area between the outer and inner leaflet of the bilayer.62–64 In such hypertonic salt solutions as studied here, where osmotic pressures π ≈ RT(cout − cin) of more than 10 bar are reached at 0.5 M salt, dramatic shape transformations are seen which are driven by surface–area–volume changes and sustained by the flexible membrane interface and low compressibility of water. An increased surface–area–volume ratio upon efflux of water renders the vesicles flaccid and induces large-scale morphological changes leading to various topologies, viz. irregular shapes (such as prolate ellipsoids), budded and detached small daughter vesicles (which, according to the Laplace equation, can withstand higher pressure differences). In case of complex multicomponent vesicles, outside and inside budding and vesiculation is accompanied by changes of the lateral ld–lo phase segregation. Altered lateral tension imposed by osmotic pressure-induced shape changes is expected to change the line tension between domains of coexisting phases and hence the ld–lo phase separation propensity and the domain sizes.68,69
High concentrations of the chaotropic perchlorates affect other biomolecular systems as well, generally decreasing their stability. Recently, we investigated the combined effect of high perchlorate concentrations and high pressure on enzymatic activity. We showed that high pressures increase α-chymotrypsin enzyme activity even in the presence of high perchlorate concentrations.21 The results suggested that high pressures may increase the habitability of environments under perchlorate stress. In another study it has been shown that the driving force for liquid–liquid phase separation and condensate formation of proteins is strongly influenced by the type of Martian salt and its concentration.70 Interestingly, it has been found that confinement, mimicking cracks in subsurface saline water pools, can stabilize liquid phase droplets, leading to an increase in the temperature and pressure stability of protein droplet phases.70
To conclude, our data show that the structure and phase behavior of lipid membranes is sensitively dictated by the nature of the Mars-relevant salt (in particular, but not only, by its anion) and its concentration. Vesicular bilayer structures are largely deteriorated only for long-chain saturated phospholipids at magnesium perchlorate concentrations beyond 0.25 M and nonbilayer lipid phases may be formed. The lateral organization of complex heterogeneous raft-like membranes is affected by all salts, however. For simple, in particular bacterial membrane-type lipid bilayer systems with unsaturated chains, vesicular structures are still stable in the presence of Mars-relevant salts up to the kbar pressure range, opening up a window for organisms exposed to such environmental conditions to maintain the stability of their membranes. Adjustment of the membrane complexity would hence be necessary for membrane formation in salty solutions for any putative life at Mars-like conditions. These data reveal how high salt concentrations and pressure may influence the membrane organization and required adaptations in deep subsurface terrestrial life, for example in sulfate-rich continental crustal fluids.26 It remains to be seen whether membranes can exhibit the fluidity associated with biological complexity in the presence of perchlorate salts at the expected environmental temperatures of the Martian subsurface. Lipid bilayers consisting largely of (poly)unsaturated chains might still be able to sustain the required fluidity at subzero temperatures and high pressures.
Material and methods
Chemicals
The phospholipids, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phospho-(10-rac-glycerol) sodium salt (DPPG), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and 1,2-dioleoyl-sn-glycero-3-phospho-(10-rac-glycerol) sodium salt (DOPG) were purchased from Avanti Polar Lipids (Alabaster, USA). Cholesterol was obtained from Sigma-Aldrich (St. Louis, USA). The fluorescent dyes used in the confocal fluorescence microscopy measurements, N-(lissamine rhodamine B sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (N-Rh-DHPE) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamin (NBD-DHPE), were obtained from Molecular Probes (Invitrogen, California, USA). As a buffer in the fluorescence microscopy measurements, 10 mM HEPES (Sigma-Aldrich, St. Louis, USA) was chosen (pH 7.4). The fluorescence probes Laurdan (6-dodecanoyl-N,N-dimethyl-2-naphthylamine) and DPH (1,6-diphenyl-1,3,5-hexatrien) were obtained from Sigma-Aldrich (St. Louis, USA). Magnesium perchlorate was purchased from Thermo Fischer (Kandel, Germany), and magnesium sulfate from abcr (Karlsruhe, Germany). All other solvents and chemicals were obtained from Sigma-Aldrich (St. Louis, USA) and Merck (Darmstadt, Germany).Multilamellar vesicle (MLV) preparation
The lipid mixtures were prepared by mixing lipid stock solutions at a concentration of 10 or 20 mg mL−1 in chloroform. The magnesium salts were added before the vesicle preparation. For the fluorescence spectroscopy measurements, the desired amount of a 1 mM fluorophore solution was added. Then, the solvent from all samples was evaporated by a gentle stream of nitrogen gas to leave a thin lipid film behind. To remove the remaining solvent, the samples were freeze-dried for at least 12 h. Thereafter, 10 mM HEPES buffer of pH 7.4 was added, the solutions were mixed by vortexing with an additional 20 min ultrasonic treatment in a water bath at temperatures above the lipid's gel-to-liquid transition temperature. Subsequently, the solutions were subjected to a five-time freeze-and-thaw process using liquid nitrogen and a water bath at temperatures above the Tm to obtain fully hydrated multilamellar vesicles. The measurements were reproduced at least three times by use of separately prepared samples.Differential scanning calorimetry (DSC)
The prepared solutions containing multilamellar vesicles at a total lipid concentration of 10 or 20 wt% were transferred into Tzero Pans and closed with hermetic lids purchased from TA Instruments (New Castle, USA). The total volume of the sample and the reference buffer was 20 μL. A Q20 Differential Scanning Calorimeter (TA Instruments, New Castle, USA) was used for the DSC measurements. The heating rate was chosen to be 1 °C min−1 after a 5 min equilibration at the starting temperature.GUV preparation
The giant unilamellar vesicles (GUVs) were prepared by using the electroformation technique or the PVA (polyvinyl alcohol)-assisted method.43,44,71–73 In both cases, 100 μL of a 1 mg mL−1 concentrated lipid solution was prepared. The concentration of the fluorescently labeled lipids was 0.1 mol%, which corresponds to a ratio of labeled to unlabeled lipid of 1:500. Electroformation and PVA-assisted preparation were carried out as described before.71–73 For the electroformation, a constant frequency of 500 Hz and an alternating electric field of 0.14 V (5 min), 1.25 V (20 min) and 3.5 V (90 min) was applied for DPPC and the 5-CS at 75 °C. For GUVs consisting of DOPC, a constant frequency of 500 Hz was used with an alternating electric field of 0.1 V (10 min), 1 V (20 min) and 1.6 V (150 min) at 20 °C.
Confocal fluorescence microscopy
For the confocal fluorescence microscopy detection, the laser combiner Oxxius Simply Light, L4Cc-CSB-130 (Lannion, France) or a CCD camera (Andor iXon Ultra, Acal BFi, Germany) in combination with a mercury-vapor lamp (Hg 100 W, Nikon, Tokyo, Japan) and the objective lens type CFI Plan Apochromat Lambda 100× Oil, NA 1.45, WD 0.13 (Nikon, Tokyo, Japan) were used.74,75 The 0.25 and 0.5 M concentrated salt-solutions were added after vesicle preparation using a syringe.Fluorescence spectroscopy
The fluorescence spectroscopy measurements were carried out on a K2 multifrequency phase modulation fluorometer (ISS Inc., Champaign, IL). The temperature dependent measurements were performed in a range of 5–75 °C, while the pressure-dependent measurements were carried out over a range of 1 bar to 2 kbar at 25 and 45 °C. The sample preparation was similar to that for the DSC experiments, apart from the total lipid concentration of 1 mg mL−1 and the addition of the fluorophores Laurdan or DPH at a ratio of 1 mol lipid to 0.000735 mol fluorophore. The excitation wavelength for the Laurdan measurements was chosen to be 390 nm and the emission intensity was collected in a range of 410 to 500 nm. Quantification of the spectral changes was achieved by use of the generalized polarization (GP) function to obtain information about the lateral order of the model membranes and their fluidity.41,42 The definition of the GP-function is GP = (IB − IR)/(IB + IR), with IR referring to the fluorescence intensity at 490 nm while IB indicates the fluorescence intensity at 440 nm. Emission at a wavelength of 490 nm is characteristic for a fluid (liquid-crystalline) phase, whereas an emission at 440 nm represents an ordered gel phase. Consequently, higher GP-values indicate a higher lateral order, while decreasing GP-values represent increasing disorder (fluidity) of the lipid acyl chains inside the membrane.The measurements containing the fluorophore DPH were carried out with an excitation wavelength of 350 nm in an emission range of 380–480 nm. The static anisotropy, rss, was monitored at the emission maximum of 420 nm to acquire information about the rotational dynamics and hence fluidity of the acyl chains inside the membrane. The anisotropy was calculated using rss = (IvvGIvh)/(Ivv + 2GIvh).39 The subscripts “v” and “h” indicate the vertical and horizontal positions of the excitation and emission polarizers, respectively. The instrumental correction factor G is defined as G = Ihv/Ihh and was calculated individually for each sample. For all experiments, the photon count was averaged over a time of 1 s.
Small-angle X-ray scattering (SAXS)
A SAXSess mc2 instrument from Anton-Paar (Graz, Austria) with a temperature control unit (TCS Control Unit, Anton-Paar, Graz, Austria) was used for the small-angle X-ray scattering measurements. An X-ray tube made of copper was used to generate the X-ray radiation, which was directed onto a quartz capillary (μ-cell, Anton-Paar, Graz, Austria) filled with 10 μL of a 10 wt% lipid solution. The sample preparation was identical to that for the DSC measurements. The scattering was detected in an evacuated chamber to reduce air scattering for every sample within 30 min via imaging plates in a temperature range of 20–60 °C for samples containing DPPC and at 20 °C for samples containing DOPC or DOPE:DOPG (70:30 molar ratio).
Dynamic light scattering
The DLS measurements were performed by means of a Zetasizer Nano-S from Malvern Instruments (Malvern, Worcs, UK) using macro-cuvettes (1 mm OS, Hellma GmbH & Co. KG, Müllheim, Germany). The total lipid concentration in the measured MLV solution was 0.5 mg mL−1. All measurements were carried out at 20 °C and the temperature was controlled through a thermostat. The Stokes-radius RS, respectively the particle diameter d, was calculated using the Stokes–Einstein equation, RS = (kBT)/(6πηD), for at least two separately prepared samples with a water viscosity, η, of 0.89 mPa s and the measured lateral diffusion coefficient, D.Author contributions
Conceptualization: R. W., C. S. C.; experiments and data analysis: S. K., M. H., R. O., discussion and writing: all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
R. W. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. C. S. C. acknowledges support from the Science and Technology Facilities Council (grant ST/V000586/1). S. G. was supported by an EPSRC studentship.References
- N. Merino, H. S. Aronson, D. P. Bojanova, J. Feyhl-Buska, M. L. Wong, S. Zhang and D. Giovannelli, Front. Microbiol., 2019, 10, 780 CrossRef.
- J. P. Harrison, N. Gheeraert, D. Tsigelnitskiy and C. S. Cockell, Trends Microbiol., 2013, 21, 204–212 CrossRef CAS PubMed.
- C. S. Cockell, Astrobiology: Understanding Life in the Universe, Blackwell Publ., Oxford, 2020 Search PubMed.
- A. Yayanos, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 9542–9546 CrossRef CAS PubMed.
- I. Daniel, P. Oger and R. Winter, Chem. Soc. Rev., 2006, 35, 858–875 RSC.
- M. Oger and M. Jebbar, Res. Microbiol., 2010, 161, 799–809 CrossRef.
- F. Meersmann, I. Daniel, D. H. Bartlett, R. Winter, R. Hazael and P. F. McMillan, Rev. Mineral. Geochem., 2013, 75, 607–648 CrossRef.
- H. Wang, Y. Zhang, D. H. Bartlett and X. Xiao, Microb. Ecol., 2021, 81, 617–629 CrossRef CAS.
- F. Inagaki, K.-U. Hinrichs, Y. Kubo, M. W. Bowles, V. B. Heuer, W.-L. Hong, T. Hoshino, A. Ijiri, H. Imachi, M. Ito, M. Kaneko, M.-A. Lever, Y.-S. Lin, B. A. Methé, S. Morita, Y. Morono, W. Tanikawa, M. Bihan, S. A. Bowden, M. Elvert, C. Glombitza, D. Gross, G. J. Harrington, T. Hori, K. Li, D. Limmer, C.-H. Liu, M. Murayama, N. Ohkouchi, S. Ono, Y.-S. Park, S. C. Phillips, X. Prieto-Mollar, M. Purkey, N. Riedinger, Y. Sanada, J. Sauvage, G. Snyder, R. Susilawati, Y. Takano, E. Tasumi, T. Terada, H. Tomaru, E. Trembath-Reichert, D. T. Wang and Y. Yamada, Science, 2015, 349, 420–424 CrossRef CAS.
- L. J. Rothschild and R. L. Mancinelli, Nature, 2001, 409, 1092–1101 CrossRef CAS.
- S. Magazù, F. Migliardo, M. A. Gonzalez, C. Mondelli, S. F. Parker and B. G. Vertessy, Life, 2012, 2, 364–376 CrossRef.
- A. Sharma, J. H. Scott, G. D. Cody, M. L. Fogel, R. M. Hazen, R. J. Hemley and W. T. Huntress, Science, 2002, 295, 1514–1516 CrossRef CAS PubMed.
- D. Vanlint, R. Mitchell, E. Bailey, F. Meersman, P. F. McMillan, C. W. Michiels and A. Aertsen, mBio, 2011, 2, e00130-10 CrossRef PubMed.
- J. Kallmeyer, R. Pockalny, R. R. Adhikari, D. C. Smith and S. D’Hondt, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16213–16216 CrossRef CAS PubMed.
- C. Magnabosco, L.-H. Lin, H. Dong, M. Bomberg, W. Ghiorse, H. Stan-Lotter, K. Pedersen, T. L. Kieft, E. van Heerden and T. C. Onstott, Nat. Geosci., 2018, 11, 707–717 CrossRef CAS.
- S. M. Clifford, J. Lasue, E. Heggy, J. Boisson, P. McGovern and M. D. Max, J. Geophys. Res., 2010, 115, E07001 CrossRef.
- R. Orosei, S. E. Lauro, E. Pettinelli, A. Cicchetti, M. Coradini, B. Cosciotti, F. Di Paolo, E. Flamini, E. Mattei, M. Pajola, F. Soldovieri, M. Cartacci, F. Cassenti, A. Frigeri, S. Giuppi, R. Martufi, A. Masdea, G. Mitri, C. Nenna, R. Noschese, M. Restano and R. Seu, Science, 2018, 361, 490–493 CAS.
- R. Orosei, C. Ding, W. Fa, A. Giannopoulos, A. Hérique, W. Kofman, S. E. Lauro, C. Li, E. Pettinelli, Y. Su, S. Xing and Y. Xu, Life, 2020, 10, 120 CrossRef PubMed.
- M. H. Hecht, R. C. Quinn, S. J. West, S. M. M. Young, D. W. Ming, D. C. Catling, B. C. Clark, W. V. Boynton, J. Hoffman, L. P. DeFlores, K. Gospodinova, J. Kapit and P. H. Smith, Science, 2009, 325, 64–67 CrossRef CAS PubMed.
- V. J. Laye and S. DasSarma, Astrobiology, 2018, 18, 412–418 CrossRef CAS.
- S. Gault, M. W. Jaworek, R. Winter and C. S. Cockell, Commun. Biol., 2020, 3, 550 CrossRef CAS PubMed.
- S. Lenton, N. H. Rhys, J. J. Towey, A. K. Soper and L. Dougan, Nat. Commun., 2017, 8, 1–5 CrossRef CAS PubMed.
- A. Gendrin, N. Mangold, J.-P. Bibring, Y. Langevin, B. Gondet, F. Poulet, G. Bonello, C. Quantin, J. Mustard, R. Arvidson and S. LeMouélic, Science, 2005, 307, 1587–1591 CrossRef CAS PubMed.
- A. Pontefract, T. F. Zhu, V. K. Walker, H. Hepburn, C. Lui, M. T. Zuber, G. Ruvkun and C. E. Carr, Front. Microbiol., 2017, 8, 1819 CrossRef PubMed.
- M. G. Fox-Powell and C. S. Cockell, Front. Microbiol., 2018, 9, 739 CrossRef PubMed.
- K. Bucher and I. Stober, Geofluids, 2010, 10, 241–253 CAS.
- J. D. Crisler, F. Chen, B. C. Clark and M. A. Schneegurt, Antonie van Leeuwenhoek, 2019, 112, 1105–1119 CrossRef CAS PubMed.
- S. Gault and C. S. Cockell, Astrobiology, 2021, 21, 405–412 CrossRef CAS PubMed.
- in Phospholipids Handbook, ed. G. Cevc, CRC Press, Boca Raton, Florida, 2018 Search PubMed.
- P. L.-G. Chong, Chem. Phys. Lipids, 2010, 163, 253–265 CrossRef CAS PubMed.
- W. N. Konings, S.-V. Albers, S. Koning and A. J. M. Driessen, Antonie van Leeuwenhoek, 2002, 81, 61–72 CrossRef CAS PubMed.
- P. L.-G. Chong, R. Ravindra, M. Khurana, V. English and R. Winter, Biophys. J., 2005, 89, 1841–1849 CrossRef CAS PubMed.
- P. L.-G. Chong, M. Sulc and R. Winter, Biophys. J., 2010, 99, 3319–3326 CrossRef CAS PubMed.
- R. Winter, Annu. Rev. Biophys., 2019, 48, 441–461 CrossRef CAS.
- R. Winter and W.-C. A. Pilgrim, Ber. Bunsenges. Phys. Chem., 1989, 93, 708–717 CrossRef CAS.
- M. Manisegaran, S. Bornemann, I. Kiesel and R. Winter, Phys. Chem. Chem. Phys., 2019, 21, 18533–18540 RSC.
- R. Winter and C. Jeworrek, Soft Matter, 2009, 5, 3157–3173 RSC.
- H. M. G. Barriga, R. V. Law, J. M. Seddon, O. Ces and N. J. Brooks, Phys. Chem. Chem. Phys., 2016, 18, 149–155 RSC.
- C. Bernsdorff, A. Wolf and R. Winter, Z. Phys. Chem., 1996, 193, 151–173 CrossRef CAS.
- M. Salvador-Castella, N. J. Brook, J. Peters and P. Oger, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183130 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edition, 2006 Search PubMed.
- L. A. Bagatolli, Fluorescent Methods to Study Biological Membranes, Springer Series on Fluorescence, Berlin, 2013, 13, pp. 3–36 Search PubMed.
- M. Herzog, T. Tiso, L. M. Blank and R. Winter, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183431 CrossRef CAS PubMed.
- S. Bornemann, M. Herzog, L. Roling, T. O. Paulisch, D. Brandis, S. Kriegler, H.-J. Galla, F. Glorius and R. Winter, Phys. Chem. Chem. Phys., 2020, 22, 9775–9788 RSC.
- J. R. Rydall and P. M. Macdonald, Biochemistry, 1992, 31, 1092–1099 CrossRef CAS PubMed.
- G. L. Jendrasiak, R. Smith and A. A. Ribeiro, Biochim. Biophys. Acta, 1993, 1145, 25–32 CrossRef CAS.
- J. W. Bye and R. J. Falconer, Protein Sci., 2013, 22, 1563–1570 CrossRef CAS PubMed.
- N. L. C. McCarthy, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Chem. Commun., 2015, 51, 8675–8678 RSC.
- D. L. Worcester and M. Weinrich, J. Phys. Chem. Lett., 2015, 6, 4417–4421 CrossRef CAS PubMed.
- D. Gmajner, P. A. Grabnar, M. T. Žnidarič, J. Štrus, M. Šentjurc and N. P. Ulrih, Biophys. Chem., 2011, 158, 150–156 CrossRef CAS PubMed.
- G. Basaiez, J. L. Nieva, E. Rivas, A. Alonso and F. M. Gonii, Biophys. J., 1996, 70, 2299–2306 CrossRef.
- J. M. Seddon and R. H. Templer, Philos. Trans. R. Soc., A, 1993, 344, 377–401 CrossRef CAS.
- J. Erbes, C. Czeslik, W. Hahn, G. Rapp and R. Winter, Ber. Bunsenges. Phys. Chem, 1994, 98, 1287–1293 CrossRef CAS.
- B. Tenchov and R. Koynova, Biochem. Biophys. Lipids, 2017, 208, 65–74 CAS.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahuallia, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- R. Kataoka, S. Aruga, S. Mitaku, Jr., K. Kinosita and A. Ikegami, Biophys. Chem., 1985, 21, 277–284 CrossRef CAS PubMed.
- C. P. Tilcock, P. R. Cullis and S. M. Gruner, Biochemistry, 1988, 27, 1415–1420 CrossRef CAS PubMed.
- R. Zorn and G. Nimtz, Ber. Bunsenges. Phys. Chem., 1990, 94, 573–578 CrossRef CAS.
- B. Tenchov, E. M. Vescio, G. D. Sprott, M. L. Zeidel and J. C. Mathai, J. Biol. Chem., 2006, 281, 10016–10023 CrossRef CAS PubMed.
- S. Hyde, S. Andersson, K. Larsson, Z. Blum, T. Landh, B. W. Ninham and S. Lidin, The Language of Shape, Elsevier, Amsterdam, 1997 Search PubMed.
- C. Cui, Y. Deg and L. Han, Sci. China: Mater., 2020, 63, 686–702 CrossRef.
- J. Käs and E. Sackmann, Biophys. J., 1991, 60, 825–844 CrossRef.
- U. Seifert, K. Berndl and R. Lipowsky, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 44, 1182–1202 CrossRef CAS PubMed.
- D. Boal, Mechanics of the Cell, Cambridge University Press, Cambridge, 2002 Search PubMed.
- R. Philipps, J. Kondev, J. Theriot and H. G. Garcia, Physical Biology of the Cell, Garland Science, London, 2013 Search PubMed.
- J. C. S. Ho, P. Rangamani, B. Liedberg and A. N. Parik, Langmuir, 2016, 32, 2151–2163 CrossRef CAS PubMed.
- K. Oglecka, P. Rangamani, B. Liedberg, R. S. Kraut and A. N. Parikh, eLife, 2014, 3, e03695 CrossRef PubMed.
- N. Wongsirojkul, N. Shimokawa, P. Opaprakasit, M. Takagi and T. Hamada, Langmuir, 2020, 36, 2937–2945 CrossRef CAS PubMed.
- R. D. Usery, T. A. Enoki, S. P. Wickramasinghe, M. D. Weiner, W.-C. Tsai, M. B. Kim, S. Wang, T. L. Torng, D. G. Ackerman, F. A. Heberle, J. Katsaras and G. W. Feigenson, Biophys. J., 2017, 112, 1431–1443 CrossRef CAS PubMed.
- Z. Fetahaj, L. Ostermeier, H. Cinar, R. Oliva and R. Winter, J. Am. Chem. Soc., 2021, 143, 5247–5259 CrossRef CAS PubMed.
- S. Bornemann, M. Herzog and R. Winter, Phys. Chem. Chem. Phys., 2019, 21, 5730–5743 RSC.
- A. Weinberger, F.-C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schröder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef CAS PubMed.
- H. Stein, S. Spindler, N. Bonakdar, C. Wang and V. Sandoghdar, Front. Physiol., 2017, 8, 63 Search PubMed.
- K. Weise, G. Triola, L. Brunsveld, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2009, 131, 1557–1564 CrossRef CAS PubMed.
- C. Nicolini, J. Baranski, S. Schlummer, J. Palomo, M. L. Burgues, M. Kahms, J. Kuhlmann, S. Sanchez, E. Gratton, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2006, 128, 192–201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and data. See DOI: 10.1039/d1cp02092g |
| This journal is © the Owner Societies 2021 |