
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking inversion symmetry by protonation: experimental and theoretical NEXAFS study of the diazynium ion, N2H+†
Rafael C.
Couto‡
 *a,
Weijie
Hua
*b,
Rebecka
Lindblad
cde,
Ludvig
Kjellsson
fg,
Stacey L.
Sorensen
c,
Markus
Kubin
d,
Christine
Bülow
d,
Martin
Timm
d,
Vicente
Zamudio-Bayer
d,
Bernd
von Issendorff
h,
Johan
Söderström
f,
J. Tobias
Lau
dh,
Jan-Erik
Rubensson
f,
Hans
Ågren
*fi and
Vincenzo
Carravetta
*j
*a,
Weijie
Hua
*b,
Rebecka
Lindblad
cde,
Ludvig
Kjellsson
fg,
Stacey L.
Sorensen
c,
Markus
Kubin
d,
Christine
Bülow
d,
Martin
Timm
d,
Vicente
Zamudio-Bayer
d,
Bernd
von Issendorff
h,
Johan
Söderström
f,
J. Tobias
Lau
dh,
Jan-Erik
Rubensson
f,
Hans
Ågren
*fi and
Vincenzo
Carravetta
*j
aDepartment of Theoretical Chemistry and Biology, School of Chemistry, Biotechnology and Health, Royal Institute of Technology, SE-106 91, Stockholm, Sweden. E-mail: rcc@kth.se
bMIIT Key Laboratory of Semiconductor Microstructure and Quantum Sensing, Department of Applied Physics, School of Science, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: wjhua@njust.edu.cn
cDepartment of Physics, Lund University, Box 118, S-22100 Lund, Sweden
dAbteilung für Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany
eInorganic Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, SE-75121 Uppsala, Sweden
fDepartment of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden. E-mail: hans.agren@physics.uu.se
gEuropean XFEL GmbH, Holzkoppel 4, 22869 Schenefeld, Germany
hPhysikalisches Institut, Albert-Ludwigs-Universität Freiburg, Hermann-Herder-Str. 3, 79104 Freiburg, Germany
iTomsk State University, 36 Lenin Avenue, Tomsk, Russia
jIPCF-CNR, via Moruzzi 1, 56124 Pisa, Italy. E-mail: carravetta@pi.ipf.cnr.it
First published on 29th July 2021
Abstract
As an example of symmetry breaking in NEXAFS spectra of protonated species we present a high resolution NEXAFS spectrum of protonated dinitrogen, the diazynium ion N2H+. By ab initio calculations we show that the spectrum consists of a superposition of two nitrogen 1s absorption spectra, each including a π* band, and a nitrogen 1s to H+ charge transfer band followed by a weak irregular progression of high energy excitations. Calculations also show that, as an effect of symmetry breaking by protonation, the π* transitions are separated by 0.23 eV, only slightly exceeding the difference in the corresponding dark (symmetry forbidden) and bright (symmetry allowed) core excitations of neutral N2. By DFT and calculations and vibrational analysis, the complex π* excitation band of N2H+ is understood as due to the superposition of the significantly different vibrational progressions of excitations from terminal and central nitrogen atoms, both leading to bent final state geometries. We also show computationally that the electronic structure of the charge transfer excitation smoothly depends on the nitrogen–proton distance and that there is a clear extension of the spectra going from infinity to close nitrogen–proton distance where fine structures show some, although not fully detailed, similarities. An interesting feature of partial localization of the nitrogen core orbitals, with a strong, non-monotonous, variation with nitrogen–proton distance could be highlighted. Specific effects could be unraveled when comparing molecular cation NEXAFS spectra, as represented by recently recorded spectra of N2+ and CO+, and spectra of protonated molecules as represented here by the N2H+ ion. Both types containing rich physical effects not represented in NEXAFS of neutral molecules because of the positive charge, whereas protonation also breaks the symmetry. The effect of the protonation on dinitrogen can be separated in charge, which extends the high-energy part of the spectrum, and symmetry-breaking, which is most clearly seen in the low-energy π* transition.
1 Introduction
Symmetry is exploited in a wide range of physical models. In molecular physics inversion-symmetric molecules have attracted particular interest as the concept of electron localization sometimes seems to be in conflict with the symmetry-imposed well-defined parity of the wave functions.1 In larger systems dynamic vibronic coupling leads to apparent loss of inversion symmetry,2 and in homonuclear diatomics symmetry breaking upon isotope substitution has been investigated.3 It has been shown that external fields influence dissociative photoionization of small diatomics, thereby breaking the symmetry.4 Also in the field-free case dissociative photoionization leads to symmetry breaking, a general phenomenon which can be understood in terms of entanglement between fragments of well-defined symmetry.5 It has been demonstrated that ultrafast core-hole localization in the N2 molecule is reflected in the angular distribution of Auger electrons,6 although a description in terms of a coherent superposition of symmetric states is also valid.The simplicity of near-edge X-ray absorption fine structure (NEXAFS) spectroscopy makes it ideal for a direct investigation of symmetry breaking. For inversion-symmetric molecules dipole selectivity demands parity change in the absorption process, i.e., an ungerade core-excited state must be reached from a gerade ground state. In the NEXAFS spectrum of the N2 molecule the population of the gerade π* orbital thus implies the opening of an ungerade core hole. In-line with the dipole selection rule a π* resonance that opens the forbidden gerade core hole is not been observed.
Here we explore the role of protonation on NEXAFS spectra by comparing neutral dinitrogen with the diazynium ion, N2H+. We make use of the new opportunities to measure NEXAFS spectra of molecular ions7–14 with high resolution to investigate symmetry breaking in N2 due to protonation. The N2H+ cation (diazynium) has attracted interest due to its astrophysical importance15,16 as one of the important components of molecular interstellar clouds, and its spectroscopic constants have been determined to high accuracy,17,18 while its neutral parent molecule has not been identified spectroscopically.
We focus here on the analysis of the N2H+ X-ray absorption spectrum in terms of valence transitions, Rydberg series and continuum resonance transitions, the role of core hole energy splitting and N2 to proton charge transfer transitions, all with respect to nitrogen–proton distance.
Through computational analysis of the energy separation, relative intensity and of different vibrational progressions for the two π* states of N2H+ the effect of the proton on the core-excited state of N2H+ in comparison to N2 could be unravelled. Our results indicate that the parity-forbidden gerade core-excitation channel of N2 opens up by protonation in N2H+. Due to a delicate balance between an electrostatic potential effect and the screening of both core hole and the proton by the electronic cloud, this π* splitting is limited to a small value, which, as shown by the analysis presented, still remains distinct from that of N2.
2 Experiment
NEXAFS in ion yield mode of the N2H+ molecular cation was measured at the UE52-PGM beamline at the BESSY II synchrotron radiation facility using the Ion Trap end-station.7,19–21 The core of this experimental station is a linear radio frequency ion trap cryogenically cooled to low temperatures. Compared to similar measurements obtained in a merged-beam setup,9,22 buffer-gas cooling in a cryogenic ion trap yields cold ions at increased target density and is more likely to give thermalized molecular ions in the electronic ground state.A mixture of N2 and H2 was ionized in a liquid-nitrogen cooled helium plasma created by a magnetron discharge ion source, where protonated N2H+ was formed.17,23 The beam of N2H+ molecular ions was selected by a quadrupole mass filter and further guided to the cryogenic ion trap, which was liquid-helium cooled and aligned with the X-ray beam. Following X-ray absorption, the core excited state relaxes mainly via Auger decay after which the molecular ion dissociates. The resulting product ions, N+ and NH+, were also stored in the ion trap, and detected with a reflectron time-of-flight mass spectrometer. Other product ions were not observed: Nq+(q ≥ 2) ions are likely to be quenched to N+ by collision with helium buffer gas because of the differences in ionization energies. Protons are not trapped at the operating frequencies of the ion trap. X-ray photodissociation action spectra of N2H+, assumed to be proportional to the X-ray absorption spectrum, were recorded by monitoring the N+ and NH+ product ion yield while scanning the photon energy. These different product ions gave very similar ion yield spectra.
The ion yield spectra were measured with different settings for the 400–404 eV photon energy region and for the photon energy region above 404 eV. These low (high) energy regions were measured with a step size of 10 meV (50 meV) and a beamline exit slit of 20 μm (50 μm) giving an energy resolution of 50 meV (75 meV). The π* resonance of neutral N2 was measured in a gas-cell with a step size of 8 meV and a beamline exit slit of 20 μm. The measurement of neutral N2 serves as a comparison to the nitrogen edge of N2H+, but also to cross-check the photon energy calibration24 that was performed on the neon 1s excitation in different reflection orders of the monochromator grating. The photon energy uncertainty at 400 eV is estimated as better than 50 meV.
3 Theory
3.1 Electronic structure
The N2H+ is a linear molecule in its ground state;17,25,26 submillimeter-wave spectroscopy determines the equilibrium distances to RNa–Nb = 1.09369 Å and RNb–H = 1.03460 Å,18 where Na and Nb refer here, and in the following, to the terminal and the central nitrogen atom, respectively. The most accurate coupled cluster calculations (CCSD(T) with a TZ2P(f,d) basis set for N2H+ available in the literature27 confirm a linear geometry with RNa–Nb = 1.096 Å and RNb–H = 1.031 Å. Comparable values have been obtained by our present RASPT228 calculation with ANO-RCC-VQZP basis set29: RNa–Nb = 1.096 Å and RNb–H = 1.033 Å. Reported HF25 and our DFT calculations also reproduce the equilibrium geometry of N2H+ and show that the linear geometry is maintained when the Nb–H bond is stretched up to breaking. The HF dissociation energy is about 119 kcal mol−1, comparable to the CCSD(T) value of 123.7 kcal mol−1.27The electronic structure of the core excited states of N2H+ was studied by two ab initio methods with different characteristics: the static-exchange (STEX)30 independent particle approximation and the state-average restricted active space self-consistent field (RASSCF)31–33 method, followed by a multi-state second-order perturbation (MS-RASPT2),28 as implemented in the OpenMolcas software.34 In both cases the quasi-stationary core-excited states were optimized by imposing core-valence separation in order to avoid the variational collapse due to filling of the core orbital.35,36
The first method does not include electronic correlation but completely describes electronic relaxation and makes it possible to use an almost complete basis set, providing a description of the entire spectrum both below the ionization threshold and in the continuum. For the HF calculations, performed by using the DALTON code37, the cc-pVQZ basis set38,39 was employed, augmented by (25s, 25p, 10d) uncontracted gaussians, with even tempered exponents, on the excitation site in STEX calculations.
The second method (RASSCF) treats electron correlation explicitly but can be practically applied only up to the lowest Rydberg excitations. For these calculations the ANO-RCC-VQZP basis set29 along with an auxiliary (8s6p4d) Rydberg basis set was used. The Abelian point group C2v was used. Scalar relativistic effects have been included by using a second-order Douglas–Kroll–Hess Hamiltonian.40,41 In the RASPT2 step, an imaginary shift of 0.3 Hartree42 and the default ionization-potential electron-affinity (IPEA) shift of 0.25 Hartree43 were in use. The transition dipole moments in the velocity gauge were obtained by the RAS state-interaction approach.44,45 The results presented in Fig. 1 and Table 2 were obtained by considering an active space configuration which involves the use of all three RAS1, RAS2 and RAS3 spaces. The two nitrogen 1s orbitals were placed in the RAS1 space, where a maximum of one hole was allowed, and kept frozen in order to avoid variational collapse. The occupied 3σ, 4σ, 5σ and the two 1π orbitals were placed in the RAS2 space. A total of 20 unoccupied orbitals were included in the RAS3 space, where a maximum occupation of three electrons was allowed. This approach was successfully applied to other molecular ions,12,13 and extra details of the active space scheme and underlying analysis can be found in Couto et al.13
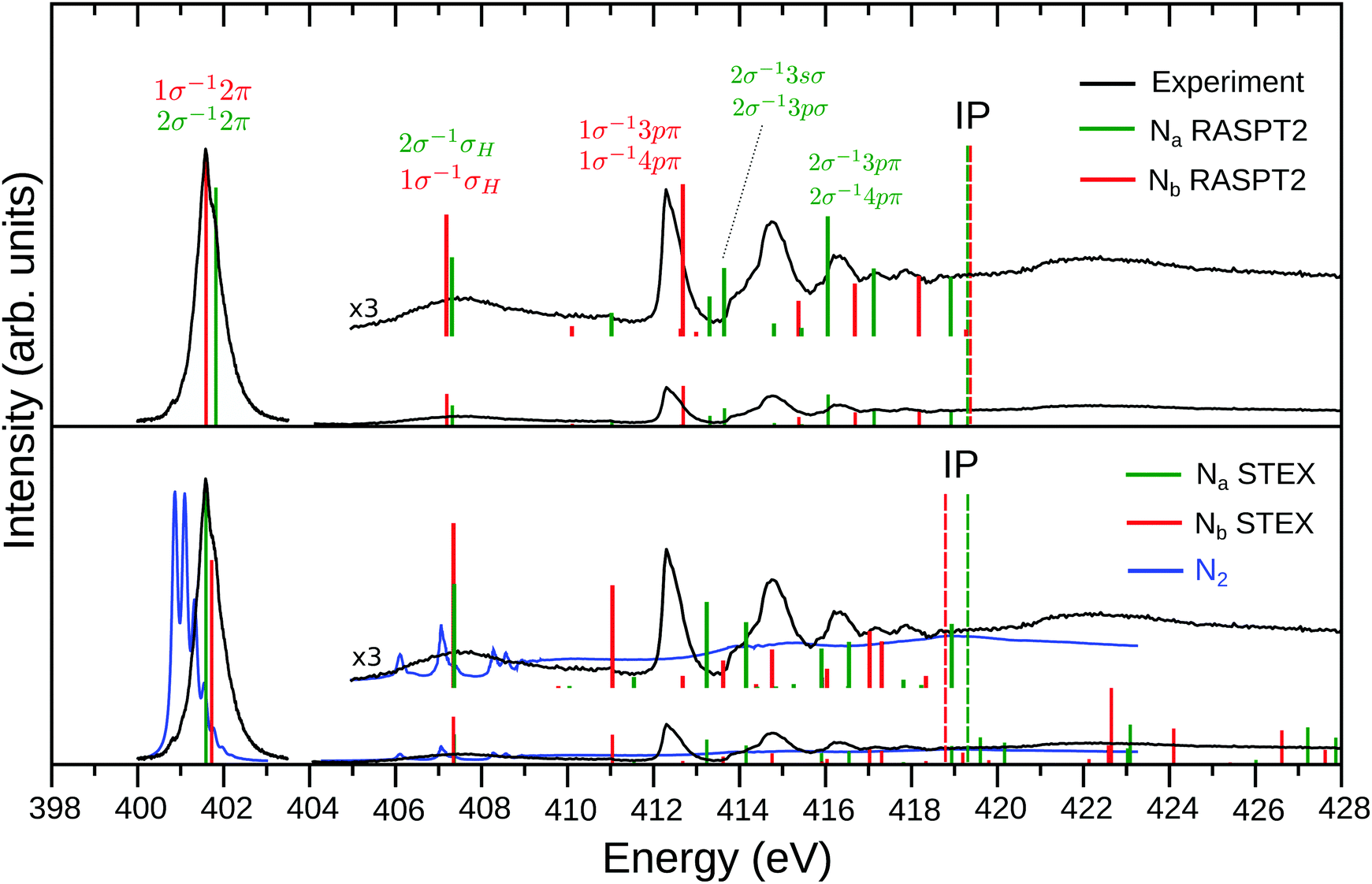 | ||
| Fig. 1 Experimental and theoretical NEXAFS spectrum of N2H+. The top panel shows the RASPT2 results while the STEX results are presented in the lower panel, together with the experimental spectrum (blue line) of neutral N2 (the region above 405 eV was taken from ref. 52). The Na (2σ excitations) and Nb (1σ excitations) contributions are represented by the green and red color bars, respectively. The experimental spectrum (black curve) is divided in two (402 eV band and region above 404 eV), as described in Section 2. In order to match the experimental 402 eV band, the RASPT2 spectrum was shifted by −2.2 eV, while the STEX one by −1.8 eV. The theoretical ionization potentials (IP) are represented by the dashed vertical lines. Labeling of the final states above 413 eV simply refers to the main single excitation component of the transition; see Table 2 for more details. | ||
3.2 Vibrational analysis
To further analyze the fine structure of the experimental nitrogen 1s → π* band of N2H+ around 402 eV (see Fig. 1), vibrationally resolved XAS spectrum was simulated using a locally modified version of the DynaVib package46 interfaced to the GAMESS-US47,48 package for electronic structure calculations. We chose the DFT method with the BLYP functional49 for the calculations. In the ground state, this cation has two degenerate π* orbitals πxz* and πyz*. Since they give equivalent contributions in a orientationally-averaged measurement, our vibronic analysis was only performed for one of these states, say, πxz*. A double-basis set technique50 was also adopted (Table S2, ESI†). The protocol was recently verified to produce accurate vibrationally-resolved X-ray photoelectron spectroscopy (XPS) at the C1s edge for a series of cyclic molecules.50 For the purpose of validation such simulation was first performed for N2 with various density functionals. The Franck–Condon (FC) factors were simulated by using the Duschinsky rotation (DR) method.51 N2 provides a good benchmark because the vibrationally-resolved band 1s → π* of this molecule is known with high experimental resolution. It turned out that BLYP density functional provides the best agreement with the experimental spectrum and thus such functional was adopted for studying N2H+ too.Special treatment was done for N2H+ whose ground state geometry is linear while there is no linear minimum for the 1s XCH state of either nitrogen. Only a linear saddle point (minima along the two stretching modes and the bending mode in the yz plane, maxima along the bending mode in the xz plane, see Fig. S4, ESI†) exists. We had scanned the potential energy surfaces (PESs) along the xz bending mode, which turned out to be of double-well shape, and solved the one-dimensional Shrödinger equation numerically to get the FC factors along this mode. More computational details for the vibrational fine structures are provided in the ESI.†
A Lorentzian line shape with half-width at half-maximum (HWHM) of ΓL = 0.0565 eV (in accord with the experimental lifetime of N2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12) was employed to convolute the stick spectrum of both N2 and N2H+ obtained by the vibrational energy levels and the corresponding Franck–Condon factors. For N2H+, an additional Gaussian broadening with HWHM of ΓG = 0.07 eV was applied in order to better compare with experimental profile (that is, a Voigt brodening). Finally, ad hoc uniform shifts of 1.10 and 1.23 eV were applied to the theoretical spectra of N2 and N2H+ spectra to better compare with corresponding experiments.
12) was employed to convolute the stick spectrum of both N2 and N2H+ obtained by the vibrational energy levels and the corresponding Franck–Condon factors. For N2H+, an additional Gaussian broadening with HWHM of ΓG = 0.07 eV was applied in order to better compare with experimental profile (that is, a Voigt brodening). Finally, ad hoc uniform shifts of 1.10 and 1.23 eV were applied to the theoretical spectra of N2 and N2H+ spectra to better compare with corresponding experiments.
4 Results
4.1 Overview
An overview of the experimental results for N2H+ is shown in Fig. 1 (black lines). The low energy region of the well-known NEXAFS spectrum of N2 (blue line in the lower panel of Fig. 1) is dominated by the excitation from the nitrogen core level to the lowest unoccupied orbital (LUMO), i.e., the 1σu−11πg excitation band centered at 401.7 eV commonly referred to as the π* resonance. At higher energies the Rydberg series converge to the ionization threshold around 410 eV, followed by double excitations and a shape resonance in the 412–420 eV range,52 see also Fig. 4. The extra proton potential in N2H+ extends the N2 spectrum roughly by a factor of two.The general appearance of the NEXAFS spectrum of the protonated molecule N2H+ shows similarity with the N2 spectrum; it is dominated by a π* band that is shifted by approx. 1 eV towards higher energies compared to N2. The fine-structure of this resonance is modified in the N2H+ spectrum as analyzed in detail below. The features at higher energies show a complex pattern below the ionization limits, which are theoretically estimated to be around 420 eV (see Fig. 1). The higher-energy features are more intense, relative to the π* resonance, than in the N2 spectrum, and the trend is that the sharp spectral structures shift to higher energies with the ionization limits. A salient difference between the N2H+ and N2 spectra, see also Fig. 4, is the appearance of an extra band in the former spectrum, in between the π* transition and Rydberg progressions. As firmly established below, this band originates in a charge transfer of a nitrogen core electron, terminal or central, to the proton, as was already predicted26 for N2H+ valence shell excitations.
As the N2+ ion has a vacancy in the 3σg orbital a dominating feature in the NEXAFS spectrum of N2+ is due to the 1σu → 3σg transition.12 No counterpart to this transition is found in the NEXAFS spectrum of N2H+ in Fig. 1; this experimental evidence suggests that there is no open 5σ orbital in N2H+.
4.2 Nitrogen–proton distance dependence of N2H+ electronic properties
Based on the previous observations, the HF description of the ground state may be considered valid, even when RNb–H varies over a large range. It is interesting to consider the variation of selected energetical (see Fig. 2) and structural (see Fig. 3) properties of the N2H+ system with the distance of the proton from the N2 fragment.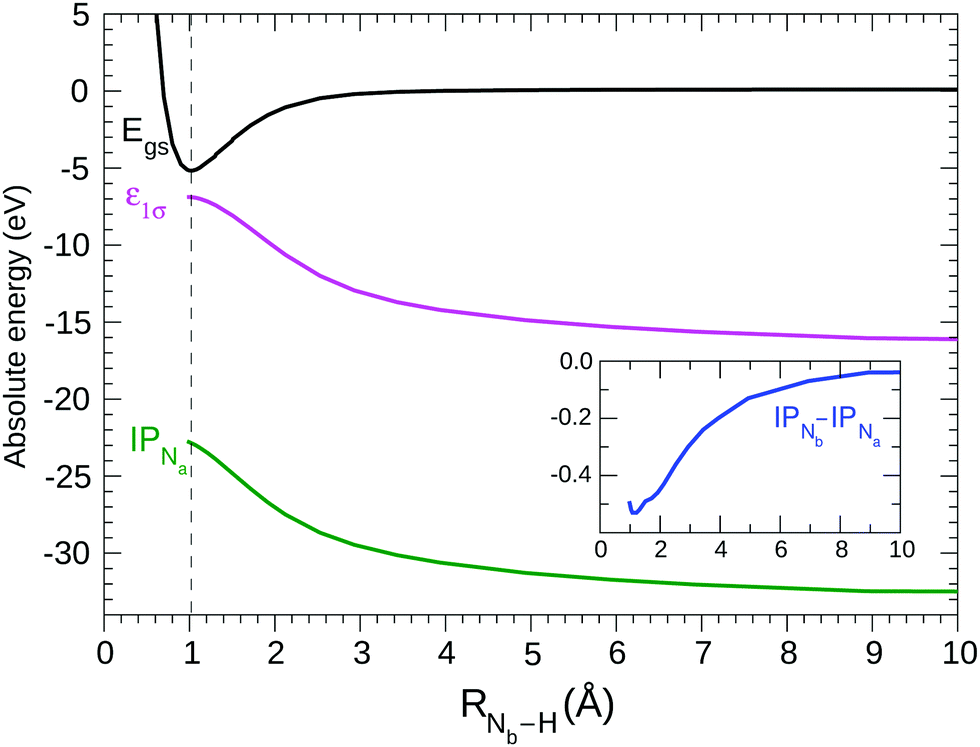 | ||
| Fig. 2 Energy properties of N2H+ vs the distance Nb–H computed at the HF level: ground state energy (black line); lowest frozen core ionization potential according to Koopmans' theorem (KT) (magenta line); relaxed core ionization potential IP of atom Na (green line); in the inset: difference between Nb and Na core ionization potentials; all the ionization potential values have been shifted by −444 eV, while the gs energy has been shifted by +2960 eV. The equilibrium value of RNb–H is 1.0 Å, indicated by the vertical dashed line. | ||
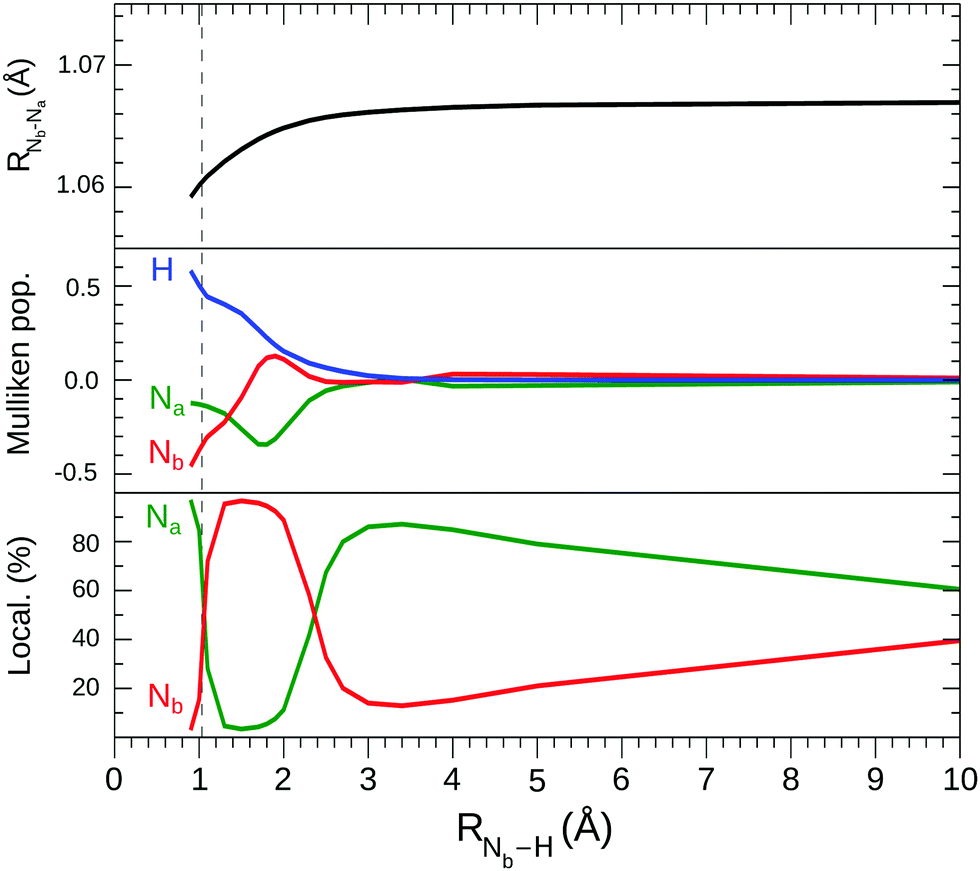 | ||
| Fig. 3 Structure properties of N2H+ vs the distance Nb–H computed at the HF level; top: N–N bond length; middle: variation of electron Mulliken population; bottom: localization of the lowest core orbital on the Na and Nb atoms. The equilibrium value of RNb–H is 1.0 Å, indicated by the vertical dashed line. | ||
By HF geometry optimization of the cation at different fixed RNb–H values the potential (black) curve in Fig. 2 is obtained; in agreement with the potential25 previously obtained keeping rigid the N2 fragment. Our HF calculations confirm,25 see Fig. 2, that the cation dissociates in the N2 and H+ fragments, as could be expected considering that the ionization potential of the N2 molecule (15.6 eV) is larger than that of H (13.6 eV). The contribution of the [N2+ + H] electronic configuration appears negligible at a great distance of H from N2, although it slightly contributes to the ground state wave function at the equilibrium geometry.
From Fig. 3 it can be seen (black line in the top graph) that the RNa–Nb bond length at the approach of the proton remains constant down to about RNb–H = 3 Å, then decreases until the equilibrium geometry where it takes on a value not very different from that of the N2 molecule. The presence of the proton has a larger effect on the electron density distribution,26 as shown in the middle graph of Fig. 3, which reports the variation of the electron charge around the proton and around the two N atoms estimated by the atomic Mulliken population. Starting from a zero value at a large distance, the proton begins to subtract electronic charge from N2 below 3 Å of distance. From this point towards the equilibrium geometry, the accumulation of electronic charge on H proceeds uniformly but with a strong polarization effect of the charge on the two nitrogen atoms. Initially it is the Na atom that preferably gives up electronic charge to H and also to the central N atom, while at a further approach to the equilibrium geometry it is the Nb atom that provides the greatest contribution. The protonation of N2 leads to the formation of a strong and peculiar N–H bond as the bonding electron charge is provided by the molecular fragment alone, in particular by the mixing of the 2σu orbital with the 1s hydrogen-like orbital. This polarization and then decrease of the electron charge in the N2 fragment is related to the symmetry breaking by protonation which becomes very evident in the electronic structure of the two core orbitals. The bottom graph of Fig. 3 shows the percentage localization of the “gerade like” core orbital around the two N nuclei vs the proton distance; this quantity is qualitatively estimated by the largest expansion coefficients of the core molecular orbital on the atomic basis functions of s type. Starting from a large RNb–H distance of >100 Å, where delocalization is complete, there is, decreasing the proton distance, an initial slow increase in localization followed by a quite peculiar variation, at short distances, due to the complex interaction of the σ orbitals (including the core orbitals) with the proton. Finally, at the equilibrium geometry, the 1σ and 2σ orbitals localize mostly on Na and Nb, respectively.
4.3 Nitrogen core orbitals
In light of these findings it is interesting to look at the N2H+ cation as formed by the two blocks N2 and H+ and consider, in comparison with the N2 molecule, the influence of the proton on the core electronic structure, and in particular to evaluate the relevance of the electrostatic interaction and the effect of the symmetry breaking from D∞h of N2 to C∞v of the fragment N2 in N2H+. In N2 the core orbitals are perfectly delocalized and have well-defined parity. They can be described, in terms of atomic orbitals, as 1σg = (+0.7071 1sNa + 0.7071 1sNb) and 1σu = (−0.7071 1sNa + 0.7071 1sNb). Inversion symmetry is broken in N2H+, the two sites become nonequivalent and localization is reflected in the magnitude of the expansion coefficients, see Fig. 3. In the ground state of N2H+ at the equilibrium geometry the two core orbitals tend to localize, although not perfectly: 1σ = (+0.8604 1sNa + 0.5094 1sNb), 2σ = (−0.5096 1sNa + 0.8609 1sNb). Via the sign of the expansion coefficients, the 1σ and 2σ orbitals of N2H+ are related to the parity constrained 1σg and 1σu orbitals of N2, respectively.The energy spacing of the core orbitals is 0.133 eV in N2H+, which can be compared to 0.102 eV in N2. In the KT approximation the two core ionization potentials have values IP1σ = 437.10 eV and IP2σ = 436.97 eV (see Table 1), while the lowest virtual orbitals 2π(π*) have energy −4.75 eV and can be related to the 1πg orbitals of N2. Neglecting electronic relaxation is a coarse approximation since the relaxation around the core hole is very rapid. However, if we introduce electronic relaxation without localizing the core orbitals, i.e. keeping the mixing observed in the ground state core orbitals, we obtain IP1σ = 430.03 eV and IP2σ = 429.88 eV with a difference of 0.15 eV very similar to that of the KT values but with absolute values still very far away from the ionization thresholds that can be guessed from the experimental spectrum in Fig. 1. Furthermore, using the two relaxed but not localized σ orbitals, we obtain (in parenthesis: excitation-energy (eV)/oscillator-strength): 1σ → 2π* (412.53/0.001) and 2σ → 2π* (412.41/0.092), i.e., the transition from 1σ, has around 0.12 eV higher energy than transitions from 2σ, and gets very little intensity. The situation is similar to the N2 case, where the excited state 1σg → 1πg is energy shifted relative to the excited state 1σu → 1πg by around 0.12 eV, and it is dipole forbidden due to parity.
| Atom | E exc (eV) | E ion (eV) | ||||
|---|---|---|---|---|---|---|
| Frozen | Relax-deloc | Relax-local | Frozen | Relax-deloc | Relax-local | |
| N | 402.43 | 410.16 | ||||
| Na | 432.35 | 412.53 | 403.38 | 437.10 | 430.03 | 421.07 |
| Nb | 432.22 | 412.41 | 403.52 | 436.97 | 429.88 | 420.56 |
By allowing complete electronic relaxation around the localized holes, the π* excitation energies are in much better agreement with the experiment; we obtain two “bright” core excited states: 1σ → π* (403.38/0.058) 2σ → π* (403.52/0.042) (see Table 1). The two transitions collect more or less the intensity of the allowed transition in the case of delocalized holes, with some increase due to the further contraction of the orbitals due to complete electronic relaxation. In this sense, symmetry breaking and core hole localization in N2H+ makes the “dark” 1σg → 1πg excited state of the “N2 fragment” visible by borrowing intensity from the “bright” 1σu → 1πg excited state. Finally we may observe that the trend for the ionization potentials (ΔIP line in the inset of Fig. 2), obtained by forcing the localization of the core holes, is quite regular and clearly shows how the presence of the proton separates the core IPs at the equilibrium geometry.
We add that all these features have been confirmed by RASSCF and DFT calculations, which differ from the HF prediction only in details. With respect to the energy separation of the two π* excitations, we indeed find differences between the theoretical approximations, but only in terms of variations of order of 0.1 eV, see summary Tables S1 and S2 in ESI.† Our best RASPT2 calculation, see Table 2, predicts a vertical energy difference of about 0.23 eV, while DFT calculations give a difference of about 0.3 eV (Table S3 of ESI†). This confirms a symmetry breaking as these values exceeds the corresponding difference between the bright and dark π* states of N2 (0.10 eV). Furthermore, the intensity ratio (fNa/fNb) of 0.71 from DFT, which is quite stable with the considered functionals, and of about 0.88 from RASPT2 (with more variation with the active space and basis set) is evidently quite different from zero.
| Ex. ener. (eV) | f | [CI weight (%)] assignment |
|---|---|---|
| 401.49 | 1.20 × 10−1 | [86] 1σ−12π |
| 401.72 | 1.06 × 10−1 | [55] 2σ−12π/[25] 2σ−11π−12π2 |
| 407.09 | 1.44 × 10−2 | [77] 1σ−1σH |
| 407.22 | 9.35 × 10−3 | [75] 2σ−1σH |
| 410.01 | 1.20 × 10−3 | [78] 1σ−13sσ |
| 410.48 | 6.99 × 10−5 | [73] 2σ−13sσ |
| 410.93 | 2.78 × 10−3 | [80] 2σ−11π−12π2 |
| 412.54 | 9.10 × 10−4 | [78] 1σ−15σ−12π2 |
| 412.59 | 1.80 × 10−2 | [61] 1σ−13pπ/[23] 1σ−14pπ |
| 412.9 | 5.30 × 10−4 | [71] 1σ−13pσ |
| 413.01 | 5.28 × 10−5 | [80] 1σ−11π−12π2 |
| 413.21 | 4.71 × 10−3 | [17] 2σ−13pπ/[17] 2σ−14pπ |
| [18] 2σ−11π−12π3pπ | ||
| [15] 2σ−11π−12π4pπ | ||
| 413.55 | 8.08 × 10−3 | [67] 2σ−13pσ/[12] 2σ−13sσ |
| 413.69 | 4.14 × 10−5 | [75] 2σ−11π−12π2 |
| 414.26 | 1.67 × 10−5 | [79] 1σ−14sσ |
| 414.71 | 1.52 × 10−3 | [80] 2σ−14sσ |
| 415.28 | 4.19 × 10−3 | [57] 1σ−15pπ/[18] 1σ−14pπ |
| 415.35 | 9.99 × 10−4 | [83] 2σ−15σ−12π |
| 415.96 | 1.42 × 10−2 | [21] 2σ−13pπ/[17] 2σ−14pπ |
| [20] 2σ−11π−12π3pπ | ||
| [18] 2σ−11π−12π4pπ | ||
| 416.59 | 6.24 × 10−3 | [45] 1σ−14pπ/[27] 1σ−15pπ |
| [13] 1σ−13pπ | ||
| 417.03 | 8.04 × 10−3 | [26] 2σ−11π−12π2/[11] 2σ−12π |
| 417.35 | 1.26 × 10−4 | [27] 2σ−15pπ |
| [27] 2σ−11π−12π5pπ | ||
| 417.96 | 3.93 × 10−5 | [74] 2σ−11π−1σH2π |
| 418.08 | 7.16 × 10−3 | [57] 1σ−11π−12π2 |
| 418.43 | 7.27 × 10−5 | [89] 1σ−11π−1σH2π |
| 418.82 | 7.01 × 10−3 | [38] 2σ−16pπ |
| [36] 2σ−11π−12π6pπ | ||
| 419.18 | 8.03 × 10−4 | [65] 1σ−15σ−1σH2π |
| 421.50 | — | 2σ−1 |
| 421.57 | — | 1σ−1 |
4.4 Computational analysis of the nitrogen–proton distance dependence of the N2H+ photoabsorption spectrum
To connect the N2H+ and N2 NEXAFS spectra, the transition from dinitrogen to diazynium was analyzed computationally.The NEXAFS spectrum was first calculated for various RNb–H using STEX. The results are shown in Fig. 4 and compared to the N2 spectrum computed by the same method.
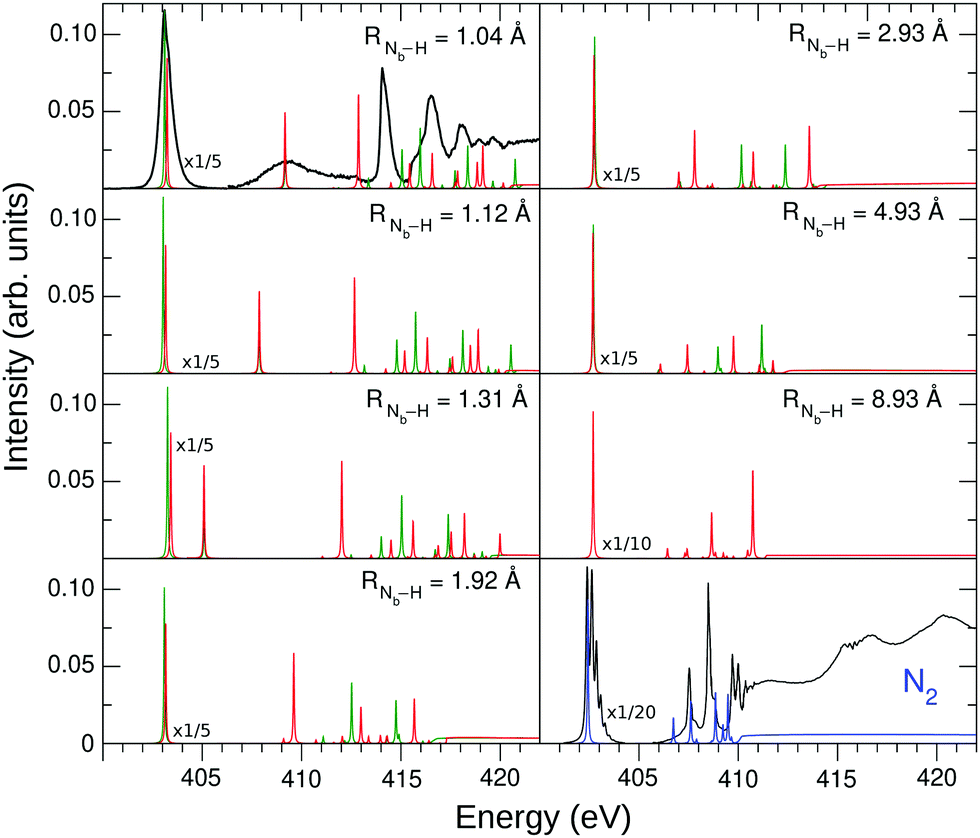 | ||
| Fig. 4 STEX N K-edge photoabsorption spectrum of N2H+ at different geometries, compared to that of N2. The green bars represent the Na contribution to the spectrum, while the Nb contribution is shown in red. The experimental spectra are represented by the black curves, which were arbitrary shifted to match the theoretical π* peak. The experimental π* N2 spectrum was obtained as described in Section 2, while the region above 405 eV was taken from ref. 52. | ||
As H+ approaches the fragment N2, two main effects can be noted. The first effect is that, while the values of the ionization thresholds (see IP curve in Fig. 2) increase as RNb–H decreases, the value of the STEX π* orbital energy (επ*) decreases instead (see also Fig. 5) and then the position of the lowest band shows a limited shift in N2H+ compared to N2. Such blue shift increases monotonously as the nitrogen-proton distance decreases, it is well appreciable at a distance well above the Nb–H bond length ad reaches a maximum value of about 1 eV at the equilibrium geometry, in good agreement with the experimental results in the first panel of Fig. 4. For values of RNb–H < 3 Å the effect of symmetry breaking induced by the presence of the proton becomes evident also in the separation of the two contributions (green for Na and red for Nb) to the first band. The second contribution, shifted by 0.13 eV at the equilibrium geometry and with lower intensity, appears as a robust shoulder of the first peak. These results suggested that the two bands could hardly be resolved experimentally since for N2H+ a rather “soft” bending mode is present with energy <0.1 eV,53 see further the vibrational analysis given in Section 4.6. The second effect is seen in the high energy region of the N2H+ spectrum, where the weak bands of N2 seem to acquire intensity by the interaction with the proton and some of them, especially that around 413 eV in the spectrum at the equilibrium geometry, show a striking energy variation versus RNb–H.
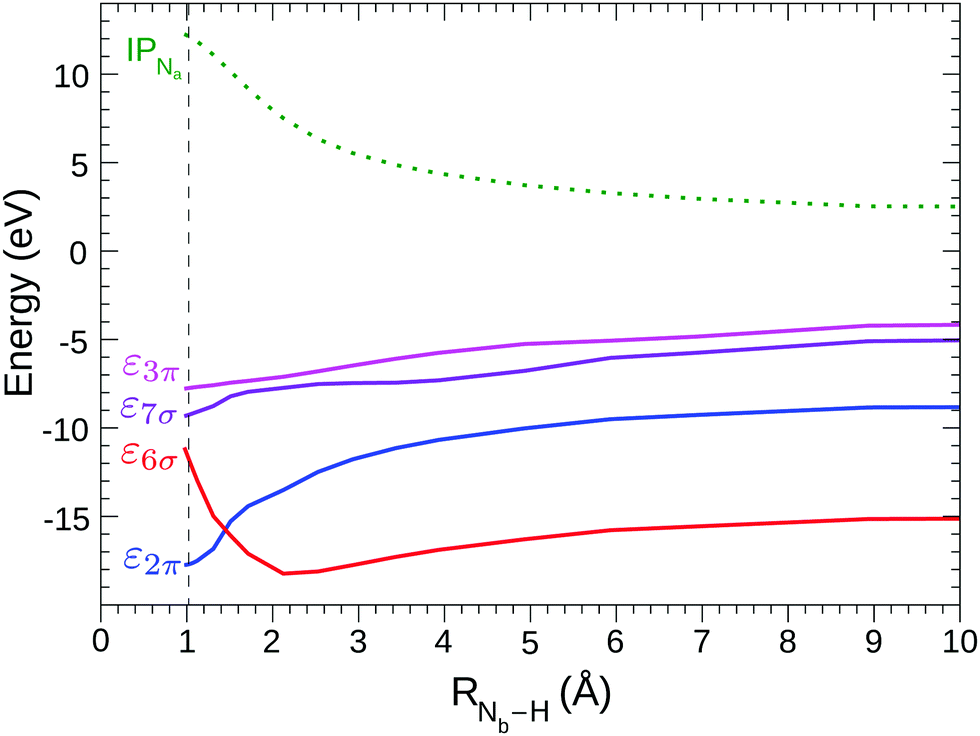 | ||
| Fig. 5 STEX energies of the four lowest virtual orbitals (1σ excitation channel) vs. the distance Nb–H; the label (H) of the lowest virtual orbital of σ symmetry (red line) underlines the localization of the orbital. The green dashed line shows the Na core ionization threshold shifted by −409 eV. The equilibrium value of RNb–H is 1.0 Å, indicated by the vertical dashed line. | ||
Fig. 5 shows the shifted core ionization potential (IP) and the lowest four (2π, 6σ, 7σ, 3π) STEX virtual orbital energies (ε), for the 1σ excitation channel, as a function of RNb–H. At equilibrium geometry the LUMO (blue line) corresponds to the orbital 2π (π*) related to the 1πg orbital of N2, while the LUMO+1 (6σ, red line) is the orbital with a clear localization around the proton and actually, both in energy and in form, it is similar to the hydrogenic orbital 1s. The increase of IP, as the proton approaches the N2 fragment, is due to the repulsive interaction between the charge of the proton and that of the core hole; conversely, the interaction between the proton and the electron in the π* orbital is attractive as shown by the blue curve in Fig. 5. It is somewhat surprising that, despite the different spatial distribution of the core hole and the π* orbital, the variation, in inverted sign, of IP and π* is very similar, so that, as already commented observing the spectra in Fig. 4, the position of the π* band varies little with RNb–H. The trend of ε6σ is completely different from that of επ* for small RNb–H values since, in this case, the approaching fragment H is not charged and the interaction with the charge distribution of the other fragment is more complex, also including a short-range repulsive component. As a consequence of the trend of the red curve (ε6σ) compared to the green dashed one (IP), the energy for the core excitation to such level, which is in fact a charge transfer excitation, is particularly mobile with RNb–H, as already observed in Fig. 4. The trend of the ε6σ curve, when compared to that of IPNa, also suggests that the 1σ → 6σ excited state, reached by vertical transition, has a decidedly dissociative character. RASPT2 calculations for the two lowest excited states confirm that, see Fig. S1 in the ESI.†
4.5 N2H+ photoabsorption spectrum at equilibrium geometry
Fig. 1 (lower panel) presents the STEX spectrum of N2H+ at the equilibrium geometry; the insert shows the high energy bands increased by a factor of 3. The RASPT2 results (excitation energies in eV, oscillator strengths, symmetry labels, and configurational expansion) for the bound excited states of N2H+ are collected in Fig. 1 (top panel) and Table 2.We recall here that while STEX represents the completion (or close to) of the 1-electron basis but with only a single configuration for excitations, RASPT2 is open ended towards full N-electronic excitations, but, in practice, with limitations of the 1-particle basis set. For details in the parametrization we refer to the Computational section. One can note that the RASPT2 results of the two core hole spectra are quite similar to the STEX predictions for the π* and charge transfer excitations, while they are more spread for the upper irregular progression between 412 and 420 eV. The configurational composition of the charge transfer state is for both Na and Nb transitions dominated by just one configuration, while one notes a difference for the π* states where a second, doubly excited component, appears for the excitation at Nb. Thus symmetry breaking appears to some extent also in the configurational representation of the two π* states. This type of difference is also reflected in the upper excitation region, 412–420 eV, where, similar to what was observed for CO+, where the localized 5σ orbital leads to a stronger splitting at the carbon center,13 the configurational mixing seems more pronounced for the Nb compared to the Na transitions. Also the charge transfer excitation can involve a concomitant extra excitation, but these transitions (which would be the counterpart of the semi-internal transitions in open-shell cationic spectra12) appear high up close to the IP and with small oscillator strengths.
4.6 Vibrational analysis of the π* band
In order to characterize the profile of the first and most intense band of the experimental spectrum of N2H+ in Fig. 1, a detailed vibrational band analysis is called for. The band shows on the left side a rising slope, reaching maximum at 401.6 eV, and a more slowly descending slope on the right side, exhibiting a shoulder at 401.8 eV. We note that the low-energy side of the maximum shows an uncharacteristic rising slope. The question is if the band shape is simply due to the presence of two distinct π* excitations or it is also due to a vibrational structure more complex than that of N2. We have then applied a multidimensional Franck–Condon analysis to explore the origin of this band shape using DFT theory as described in Section 3.2 and in the ESI.†The validation of the method was performed by simulating the one-dimensional vibrational π* band of N2 and the best result, see Fig. 6, was obtained using the BLYP functional. It is here interesting to note that the vibrational profiles of the two π* excitations of N2H+ in Fig. 6, obtained by considering only the two stretching modes, are quite different; Na shows a more restricted progression, while Nb, as well as N2 shows a more extended one. The difference in these extensions reflects a difference in the excited state energy gradients, which also is a measure of symmetry breaking. The compound band shape (brown line in Fig. 6) is the superposition of the two vibrational structures based on the relative electronic transition energies and intensities. From Fig. 6 it appears clearly how the simulated vibrational profile for N2 is in quantitative agreement with the experimental one, while for N2H+ it is not possible to obtain even a qualitative agreement on the low-energy side of the band, for any relative alignment of the two calculated vibrational structures. This could be an indication of vibrational excitation in the initial state as was also observed for N2+,12 or/and to a broadening of the vibrational profile due to the bending mode.
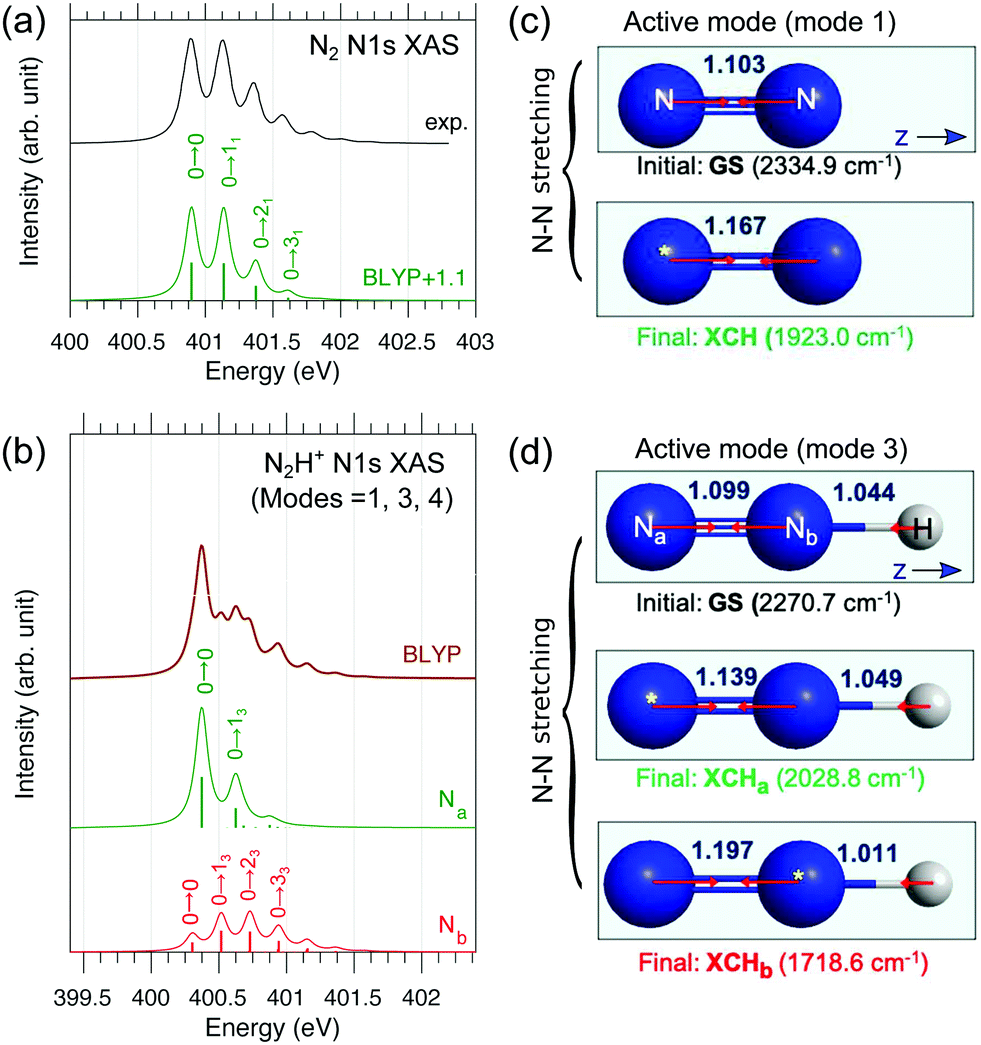 | ||
| Fig. 6 Theoretical simulation of the vibrationally-resolved N1s → π* XAS band of (a) N2 and (b) N2H+ computed by the BLYP and Duschinsky rotation method. For N2H+ only three modes (1, 3, 4; see Fig. S4, ESI†) are considered; dominant peaks were assigned. In panel b, atom-specific stick spectra are shown (already scaled with electronic oscillator strengths, see ESI†), and summation gives the total spectrum (brown). For N2, an ad hoc uniform shift of 1.1 eV was added to the theoretical spectrum to better compare with experiment. (c and d) Linear structures of the ground and XCH states (bond lengths are in Å; yellow stars denote the core holes) with active vibrational modes illustrated. For N2H+, the N–N stretching mode (mode 3) is dominantly active. | ||
The labels reported on the single peaks of the Na and Nb contributions (Fig. 6) show that the only mode activated by the excitation seems to be the N–N stretching assuming the linear geometry of the molecule. However in the initial and final states the bond angle changes from 180 to 120 degrees; this underlines how the anharmonicity of the bending motion must be explicitly considered. As discussed in Section 3 and in the ESI,† we have adopted a simplified 1D model for the description of the bending motion based on the decoupling of this mode from the other ones. This was accomplished by computing the potential energy curves for the core-excited state πxz* along the xz bending mode, which has a double-well shape (Fig. S2, ESI†). Thus, the total FC factors are given by
| Ωtotnlm = ΩbendnΩstrslm, | (1) |
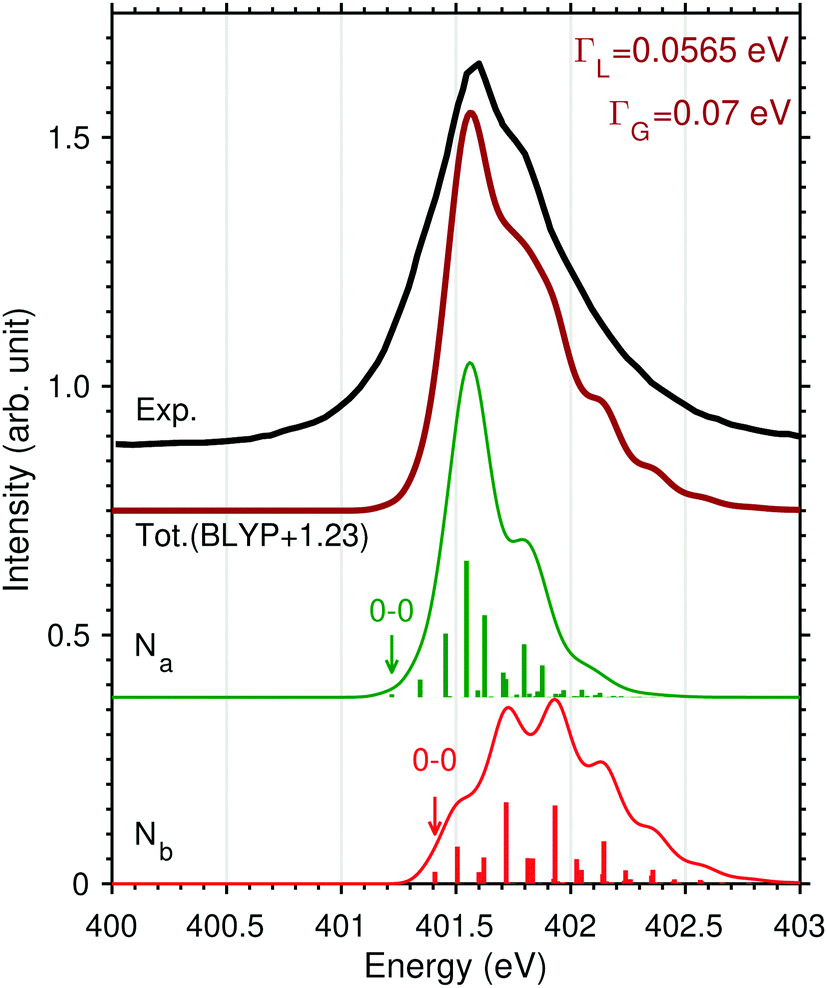 | ||
| Fig. 7 FC factors computed at the BLYP level for the N1s → π* excitations of N2H+ combing two stretching and one bending modes: Na (green), Nb (red). An uniform ad hoc shift of +1.23 eV was applied to the total theoretical profile (brown line) to better compare with experiment (black line). | ||
The superposition of the two vibrational progressions, obtained by using the experimental line broadening value given in the Experimental section, can offer, see Fig. 7, both an explanation of the higher energy slope including the shoulder at 401.8 eV and the slowly rising low-energy part of the band. It is also clear though that the superposition is delicately dependent on the electronic energy separation of the two π* excited states. All these effects are subtle and can probably only be resolved in simple protonated species as N2H+ studied here, whereas in XAS of protonated peptides or proteins55–58 these features would probably not be observed.
5 Summary
By comparison of diazynium to dinitrogen and the dinitrogen cation, we have explored the effect of symmetry breaking by protonation of homonuclear diatomic molecules on their NEXAFS spectra. Different from ionization, protonation of N2 changes the charge state without changing the number of valence electrons, but breaks inversion symmetry instead. As for the dinitrogen cation, the rich nitrogen K edge spectrum is extended by a factor of two for N2H+ over dinitrogen by the role of the positive charge in the initial state. Again similar to N2+, the role of electron correlation, as predicted by RASSCF calculations, is significant in the high-energy part of the spectrum of N2H+ as well.An instructive comparison of the appearance of cationic NEXAFS spectra can be made when the studied species have been charged by the addition of a proton versus the case when the molecule has been ionized through removal of an electron from the HOMO. Our recent studies of N2+12 and CO+13 indicated widely different and conspicuous features of the X-ray absorption spectra as compared to the corresponding neutral species. In particular, a three-fold division of the spectra could be made, none with correspondence in NEXAFS spectra of neutral molecules; at lowest transition energy, by an excitation to the valence orbital hole that was created in the ionization process, so creating a bare core hole state with correspondence to X-ray photoelectron spectroscopy but reached from a different initial state and following different selection rules; a second part showing spin-coupled split bands corresponding to core-valence transitions; and a remaining long and weak progression containing a manifold of peaks with irregular appearance manifesting highly excited states with a character of strong electron correlation. In case of protonated closed shell species, in place of the core → SOMO transition, we can have a charge transfer transition towards the proton forming a hydrogen, as we can conclude for N2H+ molecule at the equilibrium geometry.
Because inversion symmetry is broken in N2H+, there is no inversion-symmetry forbidden transition as in the dinitrogen molecule or molecular ion. This gives rise to two distinct 1sσ to π* transitions in the spectrum of N2H+, with substantial broadening because of complex vibrational excitation in the bent core excited state. Our detailed computational analysis links the transition in dinitrogen and N2H+, and shows that the presence of the proton in N2H+ opens the corresponding symmetry-forbidden excitation in N2. In order to analyze more in detail the complex shape of the experimental π* band, a multimode vibrational simulation was performed. It was predicted that the N–N stretch excitation in N2 is reduced for the terminal Na–π* excitation while being further prolonged for the Nb–π* excitation and that the bending mode causes a substantial broadening of the band.
We have also computationally traced the appearance of the NEXAFS spectrum for a proton approaching from infinity to the N2 molecule up to the equilibrium geometry of N2H+. The separation of the two π* peaks and the development of the electronic structure and of the charge transfer excitation show smooth dependence on the proton position. An interesting feature of partial localization of the nitrogen core orbitals with respect to proton distance could be unraveled. This localization shows a strong, non-monotonous, variation with proton distance qualitatively independent of the theory method applied.
In summary, as in the case of NEXAFS spectra of molecular cations obtained by ionization,12,13 our study on the N2H+ spectrum unravels new and unexplored effects also when the cation is obtained by molecular protonation. These spectra may also inherently contain rich information on chemistry in terms of electron localization, chemical bonding, and structural conformation when applied to larger molecular cations.
Author contributions
R. C. C., W. H. and V. C. performed all simulations and theoretical analysis, and prepared the text of manuscript and figures with J. T. L. and B. v. I; L. K., R. L., M. K., C. B., M. T., V. Z.-B., J. S., S. L. S., B. v. I., J. T. L., J.-E. R. suggested and planned the experiment, collected the data and carried out the data analysis; J. T. L., V. Z.-B., B. v. I. developed and built the experimental setup; H. Å., V. C. took main responsibility for the theoretical modelling, results discussion and the writing of the paper, in which all authors contributed. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The beamtime for this project was granted at BESSY II beamline UE52-PGM and Ion Trap endstation, operated by Helmholtz-Zentrum Berlin. The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC), through the project “Multiphysics Modeling of Molecular Materials” (SNIC 2019/2-41 and 2020/3-29), and the Shanghai Supercomputer Center. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 730872 and by the German Federal Ministry of Education and Research (BMBF) through Grant No. BMBF-05K16Vf2. R. L. and J.-E. R. acknowledge funding from the Swedish Research Council, contract 637-2014-6929, and 2014-04518, respectively. W. H. was supported by National Natural Science Foundation of China (11774174, 21703105).References
- P. S. Bagus and H. F. Schaefer, J. Chem. Phys., 1972, 56, 224–226 CrossRef CAS.
- F. Gel'mukhanov and H. Ågren, Phys. Rep., 1999, 312, 87–330 CrossRef.
- D. Rolles, M. Braune, S. Cvejanović, O. Geßner, R. Hentges, S. Korica, B. Langer, T. Lischke, G. Prümper and A. Reinköster, et al. , Nature, 2005, 437, 711–715 CrossRef CAS PubMed.
- M. F. Kling, C. Siedschlag, A. J. Verhoef, J. I. Khan, M. Schultze, T. Uphues, Y. Ni, M. Uiberacker, M. Drescher, F. Krausz and M. J. J. Vrakking, Science, 2006, 312, 246–248 CrossRef CAS PubMed.
- F. Martn, J. Fernández, T. Havermeier, L. Foucar, T. Weber, K. Kreidi, M. Schöffler, L. Schmidt, T. Jahnke, O. Jagutzki, A. Czasch, E. P. Benis, T. Osipov, A. L. Landers, A. Belkacem, M. H. Prior, H. Schmidt-Böcking, C. L. Cocke and R. Dörner, Science, 2007, 315, 629–633 CrossRef PubMed.
- M. S. Schöffler, J. Titze, N. Petridis, T. Jahnke, K. Cole, L. P. H. Schmidt, A. Czasch, D. Akoury, O. Jagutzki, J. B. Williams, N. A. Cherepkov, S. K. Semenov, C. W. McCurdy, T. N. Rescigno, C. L. Cocke, T. Osipov, S. Lee, M. H. Prior, A. Belkacem, A. L. Landers, H. Schmidt-Böcking, T. Weber and R. Dörner, Science, 2008, 320, 920–923 CrossRef PubMed.
- K. Hirsch, J. T. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. von Issendorff, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154029 CrossRef.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, A. aawicki, A. Terasaki, B. V. Issendorff and J. T. Lau, J. Chem. Phys., 2015, 143, 244318 CrossRef CAS PubMed.
- J.-P. Mosnier, E. T. Kennedy, P. van Kampen, D. Cubaynes, S. Guilbaud, N. Sisourat, A. Puglisi, S. Carniato and J.-M. Bizau, Phys. Rev. A, 2016, 93, 061401 CrossRef.
- S. Klumpp, A. A. Guda, K. Schubert, K. Mertens, J. Hellhund, A. Müller, S. Schippers, S. Bari and M. Martins, Phys. Rev. A, 2018, 97, 033401 CrossRef CAS.
- S. Bari, L. Inhester, K. Schubert, K. Mertens, J. O. Schunck, S. Dörner, S. Deinert, L. Schwob, S. Schippers, A. Müller, S. Klumpp and M. Martins, Phys. Chem. Chem. Phys., 2019, 21, 16505–16514 RSC.
- R. Lindblad, L. Kjellsson, R. C. Couto, M. Timm, C. Bülow, V. Zamudio-Bayer, M. Lundberg, B. von Issendorff, J. T. Lau, S. L. Sorensen, V. Carravetta, H. Ågren and J.-E. Rubensson, Phys. Rev. Lett., 2020, 124, 203001 CrossRef CAS PubMed.
- R. C. Couto, L. Kjellsson, H. Ågren, V. Carravetta, S. L. Sorensen, M. Kubin, C. Bülow, M. Timm, V. Zamudio-Bayer, B. von Issendorff, J. T. Lau, J. Söderström, J.-E. Rubensson and R. Lindblad, Phys. Chem. Chem. Phys., 2020, 22, 16215–16223 RSC.
- S. Carniato, J.-M. Bizau, D. Cubaynes, E. T. Kennedy, S. Guilbaud, E. Sokell, B. McLaughlin and J.-P. Mosnier, Atoms, 2020, 8, 67 CrossRef CAS.
- P. Caselli, P. C. Myers and P. Thaddeus, Astrophys. J., 1995, 455, L77–L80 CAS.
- W. D. Geppert and M. Larsson, Chem. Rev., 2013, 113, 8872–8905 CrossRef CAS PubMed.
- J. C. Owrutsky, C. S. Gudeman, C. C. Martner, L. M. Tack, N. H. Rosenbaum and R. J. Saykally, J. Chem. Phys., 1986, 84, 605–617 CrossRef CAS.
- T. Amano, T. Hirao and J. Takano, J. Mol. Spectrosc., 2005, 234, 170–175 CrossRef CAS.
- M. Niemeyer, K. Hirsch, V. Zamudio-Bayer, A. Langenberg, M. Vogel, M. Kossick, C. Ebrecht, K. Egashira, A. Terasaki, T. Möller, B. v. Issendorff and J. T. Lau, Phys. Rev. Lett., 2012, 108, 057201 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Niemeyer, M. Vogel, A. aawicki, A. Terasaki, J. T. Lau and B. von Issendorff, Angew. Chem., Int. Ed., 2015, 54, 4498–4501 CrossRef CAS PubMed.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, M. Kossick, A. aawicki, A. Terasaki, B. v. Issendorff and J. T. Lau, J. Chem. Phys., 2015, 142, 234301 CrossRef CAS PubMed.
- S. Schippers, A. L. D. Kilcoyne, R. A. Phaneuf and A. Müller, Contemp. Phys., 2016, 57, 215–229 CrossRef CAS.
- M. Meuwly, S. A. Nizkorodov, J. P. Maier and E. J. Bieske, J. Chem. Phys., 1996, 104, 3876–3885 CrossRef CAS.
- M. R. Weiss, R. Follath, K. J. S. Sawhney and T. Zeschke, Nucl. Instrum. Methods Phys. Res., 2001, 467–468, 482–484 CrossRef CAS.
- S. Forsen and B. Roos, Chem. Phys. Lett., 1970, 6, 128–132 CrossRef CAS.
- K. Vasudevan, S. Peyerimhoff and R. Buenker, Chem. Phys., 1974, 5, 149–165 CrossRef CAS.
- K. C. Sears, J. W. Ferguson, T. J. Dudley, R. S. Houk and M. S. Gordon, J. Phys. Chem. A, 2008, 112, 2610–2617 CrossRef CAS PubMed.
- P. Å. Malmqvist, K. Pierloot, A. R. M. Shahi, C. J. Cramer and L. Gagliardi, J. Chem. Phys., 2008, 128, 204109 CrossRef PubMed.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- H. Ågren, V. Carravetta, O. Vahtras and L. G. Pettersson, Theor. Chem. Acc., 1997, 97, 14–40 Search PubMed.
- H. J. A. Jensen, P. Jørgensen and H. Ågren, J. Chem. Phys., 1987, 87, 451–466 CrossRef CAS.
- H. Ågren, A. Flores-Riveros and H. Jensen, Phys. Scr., 1989, 40, 745–750 CrossRef.
- P. Å. Malmqvist, A. Rendell and B. O. Roos, J. Phys. Chem., 1990, 94, 5477–5482 CrossRef CAS.
- I. F. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov and R. K. Carlson, et al. , J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed.
- L. S. Cederbaum, W. Domcke and J. Schirmer, Phys. Rev. A, 1980, 22, 206–222 CrossRef CAS.
- M. G. Delcey, L. K. Sørensen, M. Vacher, R. C. Couto and M. Lundberg, J. Comput. Chem., 2019, 40, 1789–1799 CrossRef CAS PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertens, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjrgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutns, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A, 1986, 33, 3742–3748 CrossRef CAS.
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- P.-Å. Malmqvist and B. O. Roos, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- G. Tian, S. Duan, W. Hua and Y. Luo, DynaVib, version 1.0, Royal Institute of Technology, Sweden, 2012 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Advances in electronic structure theory: GAMESS a decade later, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, ch. 41, pp. 1167–1189 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- W. Hua, G. Tian and Y. Luo, Phys. Chem. Chem. Phys., 2020, 22, 20014–20026 RSC.
- F. Duschinsky, Acta Physicochim. URSS, 1937, 7, 551–566 Search PubMed.
- C. T. Chen, Y. Ma and F. Sette, Phys. Rev. A, 1989, 40, 6737–6740 CrossRef CAS PubMed.
- Y. Kabbadj, T. Huet, B. Rehfuss, C. Gabrys and T. Oka, J. Mol. Spectrosc., 1994, 163, 180–205 CrossRef CAS.
- J.-i. Adachi, N. Kosugi and A. Yagishita, J. Phys. B: At., Mol. Opt. Phys., 2005, 38, R127–R152 CrossRef CAS.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Phys. Chem. Chem. Phys., 2016, 18, 26213–26223 RSC.
- S. Bari, D. Egorov, T. L. C. Jansen, R. Boll, R. Hoekstra, S. Techert, V. Zamudio-Bayer, C. Bülow, R. Lindblad, G. Leistner, A. aawicki, K. Hirsch, P. S. Miedema, B. von Issendorff, J. T. Lau and T. Schlathölter, Chem. – Eur. J., 2018, 24, 7631–7636 CrossRef CAS PubMed.
- D. Egorov, S. Bari, R. Boll, S. Dörner, S. Deinert, S. Techert, R. Hoekstra, V. Zamudio-Bayer, R. Lindblad, C. Bülow, M. Timm, B. von Issendorff, J. T. Lau and T. Schlathölter, J. Am. Soc. Mass Spectrom., 2018, 29, 2138–2151 CrossRef CAS PubMed.
- S. Dörner, L. Schwob, K. Atak, K. Schubert, R. Boll, T. Schlathölter, M. Timm, C. Bülow, V. Zamudio-Bayer, B. von Issendorff, J. T. Lau, S. Techert and S. Bari, J. Am. Soc. Mass Spectrom., 2021, 32, 670–684 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information regarding the theoreticalresults and analysis. See DOI: 10.1039/d1cp02002a |
| ‡ Present address: Department of Physics, Stockholm University, Albanova University Center, SE-106 91 Stockholm, Sweden. |
| This journal is © the Owner Societies 2021 |