
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysisorption of bio oil nitrogen compounds onto montmorillonite†
Maisa
Vuorte
 a,
Susanna
Kuitunen
b and
Maria
Sammalkorpi
*ac
a,
Susanna
Kuitunen
b and
Maria
Sammalkorpi
*ac
aDepartment of Chemistry and Materials Science, School of Chemical Engineering, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland. E-mail: maria.sammalkorpi@aalto.fi
bNeste Engineering Solutions Oy, P.O. Box 310, FI-06101 Porvoo, Finland
cDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland
First published on 13th September 2021
Abstract
We assess computationally the adsorption of a series of nitrogen containing heterocycles and fatty acid amides from bio-oil on a model clay surface, Na-montmorillonite. The adsorption energies and conformations predicted by atomistic detail molecular dynamics (MD) simulations are compared against density functional theory (DFT) based molecular electrostatic potentials (MEP) and Hirshfeld, AIM, Merz-Singh-Kollman, and ChelpG charges. MD predicts systematically adsorption via cation bridging with adsorption strength of the heterocycles following purine > pyridine > imidazole > pyrrole > indole > quinoline. The fatty acid amides adsorption strength follows the steric availability and bulkiness of the head group. A comparison against the DFT calculations shows that MEP predicts adsorption geometries and the MD simulations reproduce the conformations for single adsorption site species. However, the DFT derived charge distibutions show that MD force-fields with non-polarizable fixed partial charge representations parametrized for aqueous environments cannot be used in apolar solvent environments without careful accuracy considerations. The overall trends in adsorption energies are reproduced by the Charmm GenFF employed in the MD simulations but the adsorption energies are systematically overestimated in this apolar solvent environment. The work has significance both for revealing nitrogen compound adsorption trends in technologically relevant bio oil environments but also as a methodological assessment revealing the limits of state of the art biomolecular force-fields and simulation protocols in apolar bioenvironments.
1 Introduction
Biofuels, that is, fuels derived from e.g. vegetable oils, animal fats, starch, waste biomass, or microbial biomass, are an environmentally more sustainable alternative to traditional fossil fuels. In comparison to fossil fuels, bio fuel alternatives result in a reduction of CO2, hydrocarbons, and particulate matter emissions. Additionally, bio fuels rely on energy from relatively recent carbon fixation, which further decreases net carbon emissions to the environment. Diversification of biofuel sources from traditional food agriculture commodities and efforts to reduce involved land usage has led to rising interest in biomass of marine origins.1,2 Especially algae biomass appears promising because of its high growth rate, efficient carbon dioxide fixation, high biomass production rate, and elevated oil yield.3–5The composition of biofuels depends strongly on the specific biomass used as feedstock and the chemical processes employed in conversion to biofuel.5–9 A particular feature of algae derived bio-oils is their elevated nitrogen compound content, 5–10 wt% of algae bio-oils procured through pyrolysis.1,4,10 These nitrogen compounds need to be removed from the oil for its use as a clean fuel, as well as to ensure optimal processing and shelf-life of the bio-oil. High concentrations of nitrogen containing compounds are problematic due to contamination of the catalyst, and the formation of NOx and N2O compounds during combustion of the bio-oil in fuel usage. These function as greenhouse gases, contribute to the depletion of the ozone layer, and have a role in the formation of acid rains and photochemical smog. Hence, denitrogenation is important in bio-oils refining. The nitrogen removal is commonly achieved via selective adsorption of nitrogen compounds from the rest of the oil onto the active sites of an adsorbent substrate, such as activated carbon,11,12 silica–alumina11 or metal-exchanged zeolites.11,13,14 The adsorbent may later be regenerated for reuse through solvent washing or thermal treatment.14
As selective adsorption of nitrogen compounds is in key role in their removal, it is enticing to pursue chemical detail level guidelines to understand and control the adsorption response. Presently, there are only a few targeted studies of nitrogen compound adsorption from algae oils, the most recent of these using phyllosilicate clays, e.g. montmorillonite,15,16 activated carbon,11,12 Cu–zeolites,11 silica gels,11 and aminated metal–organic frameworks.17 Computational approaches to surfactants in apolar environments have mostly focused on bulk assemblies, see e.g. ref. 18–25, or been limited to modelling adsorption of surfactants and other organic compounds at the apolar oil–solid interfaces, see e.g. ref. 26–33. Our previous work examines adsorption of vegetable oil minority species (fatty acids, monoglycerides, phospholipids) on silica via molecular dynamics simulation and links head group hydrogen bonding capability and charge to adsorption strength.34
Here, we pursue the adsorption response of a systematic series of nitrogen containing heterocycles, fatty acid amides, and water using atomistic detail molecular dynamics (MD) simulations. Specifically, physisorption of the compounds on a model clay (Na-montmorillonite) surface is probed in a triglyceride (triolein) solvent that describes a model bio-oil. Montmorillonite is chosen for the work as a common model adsorbent clay for drywashing bio-oils: montmorillonite-based adsorbents have formerly been used for both decolorization and deodorization of bio-oils as well as catalysts for bio fuel synthesis.15,16,35,36 Experimental studies of nitrogen compound adsorption on montmorillonite are mainly limited to chlorophyl, phospholipids, and sterols.15,16 We employ MD simulations with the umbrella sampling method to determine the adsorption conformations and energies of individual molecules on the Na-montmorillonite surface. As prior studies22,37–41 have raised concern on the performance accuracy of the aqueous environment force-field parametrizations used typically in atomistic detail MD simulations when taken to apolar solvent environments, we assess the modelling accuracy via density functional theory (DFT) electronic structure calculations with an implicit solvent model. The results enable an objective performance evaluation of the Charmm force field, in particular Charmm GenFF42 parameters, for examining simple organic molecules in low dielectric, apolar solvent environments. Besides the methodological assessment, the work has significance for revealing systematic nitrogen compound adsorption trends in technologically relevant bio oil environments. In particular, the work provides a mapping of adsorption preference order for the nitrogen compounds, which allows making predictions on the adsorption response based on the accessibility of the nitrogen but also bring out methodological pitfalls for modelling biobased surfactants in organic solvents.
2 Computational methods
2.1 Molecular dynamics simulations
The adsorption of six nitrogen containing, heterocyclic compounds, five fatty acid amides, and water from triglyceride (triolein) solvent onto Na-montmorillonite was studied in terms of adsorption conformation and free energy changes accompanying adsorption events via MD simulations. All MD simulations were carried out using the Gromacs 2016.5 simulation package.43 The montmorillonite surface was built based on the montmorillonite crystal structure (super cell) provided as part of the INTERFACE force field database,44 with Na+ counterions and optimized Al to Mg substitutions. This leads to chemical formula Na0.321[Si4O8][Al1.679Mg0.321O2(OH)2]. The chemical structures of the examined heterocyclic nitrogen compounds and fatty acid amides are presented in Fig. 1.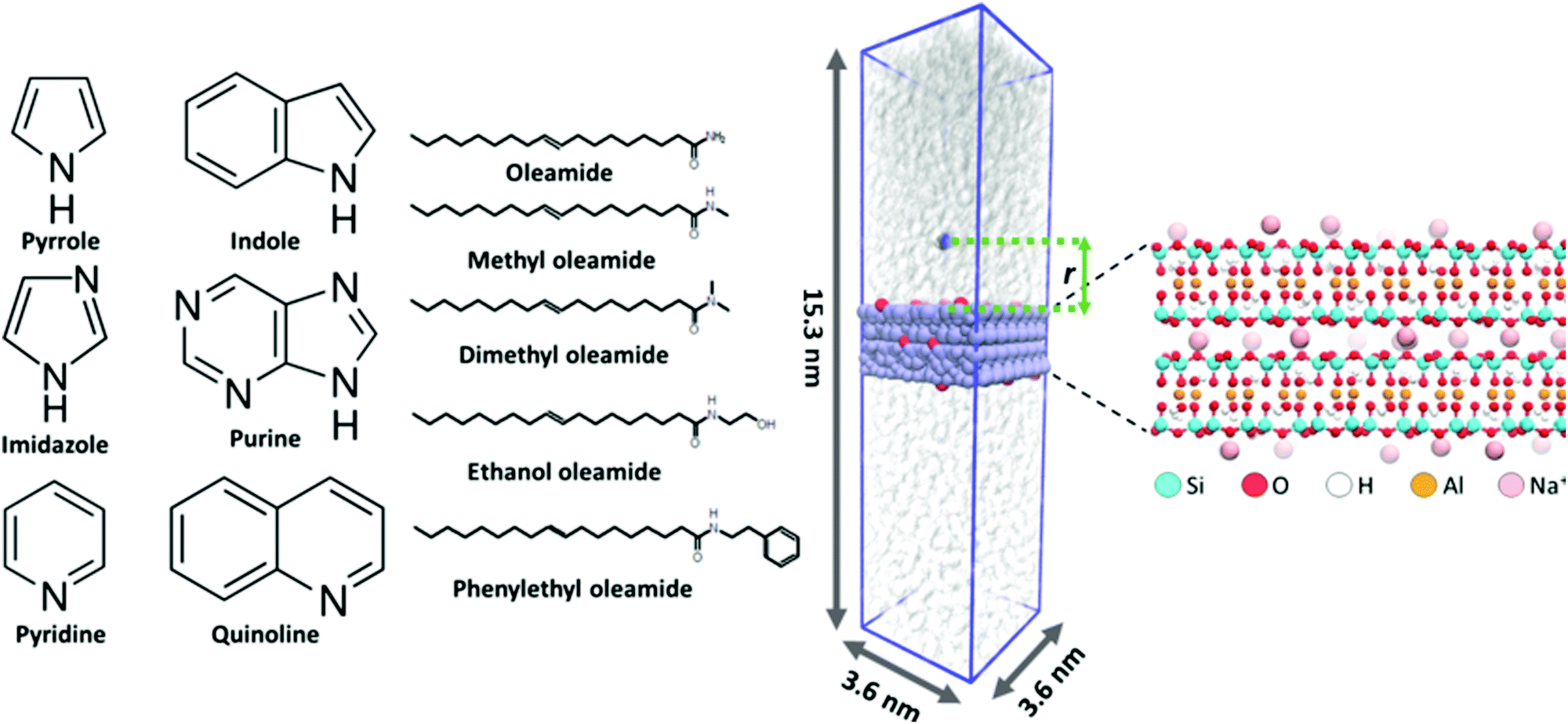 | ||
| Fig. 1 Left: Chemical structures of the nitrogen containing heterocyclic compounds and fatty acid amides examined in this work. Middle: Visualization of an example initial configuration and the simulation box are presented at right. The montmorillonite surface is shown in light purple, Na+ ions in red, and triolein solvent in translucent white. The PMF reaction coordinate r denotes the perpendicular distance between adsorbate center of mass and the plane defined by the topmost montmorillonite Si atoms. Right: Structure of the simulated two layer montmorillonite surface. | ||
The studied nitrogen compound set is selected because the chemical composition of algae derived bio oils can vary significantly depending on factors such as algae species and chemical processing methods used to process the biomass, such as lipid extraction or pyrolysis. During chemical processing, organic macromolecules, such as lipids and proteins, may depolymerize and recombine to form organic chemical species not present in the original biomass feedstock. Such species include for example fatty acid amides. Particularly, the combination of residual water in the feedstock and elevated temperature will rapidly hydrolyze proteins into amino acids. These then further decompose into ammonia and amines. Ammonia or amines that react with free fatty acids yield primary and secondary fatty acid amides, respectively7. Additionally, diverse nitrogen containing heterocycles are abundant in algae derived bio-oils.4,8,45 By covering the adsorption of heterocyclic compounds and fatty acid amides as a series that varies systematically in structure, see Fig. 1, we cover the species that can be expected to be present relatively well and also obtain a generalizable trend in the adsorption responses.
The montmorillonite surface and the nitrogen adsorbates (N-heterocycles and fatty acid amides) are described in fully atomistic detail level using the INTERFACE force field44 and Charmm GenFF 4.1,42 respectively. Notably, the INTERFACE force field enables fully dynamic montmorillonite slab and surface atoms. This allows for possible morphing of the surface structure. The triolein solvent is described by the Charmm c27 complementary united-atom description by Henin et al.46 Our previous work points toward Charmm c27 interactions being better balanced for apolar solvent environment simulations than the more recent Charmm parametrizations.37 The usage of a united atom description for the solvent leads to a significant speedup of the calculations. For water, the tip3p47,48 explicit water model is used. Notably, both Charmm and INTERFACE force fields were originally parametrized in aqueous environments and for fully hydrated biospecies. As the systems examined here contain charged species (montmorillonite Na+counter ions) in a low dielectric medium (triolein solvent), all results obtained for these systems require a careful and critical examination.
To prepare the simulation configurations, 100 triolein molecules were first inserted into a rectangular 3.6 nm × 3.6 nm × 25 nm simulation box. This leads to an excessively sparse, random mixture of triolein solvent. A montmorillonite surface 1.57 nm (2 repeat layers) in thickness was set to match the simulation box size in x- and y-dimensions as an infinite surface plane slab that spans the box in the x,y-plane (periodic boundary conditions). The simulation box was then gradually scaled smaller in the direction perpendicular to the surface (z-direction). Each scaling step was followed by a short 200 step steepest descent energy minimization. This was continued until appropriate density of the triolein solvent49,50 was reached. Next, a 10 ns NPT ensemble initial equilibration using a semi-isotropic Berendsen barostat51 with a reference pressure of 1 bar, compressibility of 4.5 × 10−5 bar−1, and a time constant of 2.0 ps was performed. In this, the average temperature was maintained at 343.15 K (70 °C) using the stochastic velocity rescale thermostat developed by Bussi et al.52 with a time constant of 0.5 ps. After this initial equilibration of the triglyceride solvent, a single adsorbate (N-heterocycle or fatty acid amide) molecule was placed 5.5 nm above the montmorillonite surface. During a 50 ps λ-coupling simulation, the interactions between the adsorbate and the rest of the system (triglyceride solvent and montmorillonite surface) were gradually turned on to avoid overlap of atoms. The system was then NPT equilibrated for 5 ns using the barostat and thermostat parameters specified above. Visualization of an example initial simulation configuration, the simulation box, and the montmorillonite slab is provided in Fig. 1. During simulation, the surface Na+ atom positions can fluctuate significantly, resulting in changes in local electrostatics. The effect of this is pronounced due to the low dielectric screening of the triglyceride solvent. To avoid the PMF convergence challenges rising from Na+ displacements at the surface, the Na+ ion positions were constrained using 500 kJ mol−1 nm−2 harmonic constraints after the NPT equilibration for all later simulation steps.
The binding energy of the adsorbate on the montmorillonite surface was assessed based on the free energy difference estimate of the adsorbate molecule being fully solvated by the triolein vs. adsorbed on the surface. This was estimated based on the potential of mean force (PMF) determined via umbrella sampling methodology. For the N-heterocycles, the perpendicular component (z-direction) of distance between the center of mass of adsorbed molecule and the plane defined by the topmost montmorillonite surface Si atoms was used as the PMF reaction coordinate, see Fig. 1. As the fatty amides are relatively large, the center of mass of the amide group connected to the head group (not the fatty acid tail) was used for the reaction coordinate.
Technical details for obtaining the PMF curves are as follows. To obtain the PMF curves, umbrella sampling windows were generated at 0.1 nm intervals along the reaction coordinate. Each of the simulated windows was run for a total of 200 ns using a 2 fs time step. The adsorbate was constrained in each window by a 1000–8000 kJ mol−1 nm−2 harmonic constraint. Similar to the simulation system preparation, the temperature of the system was controlled by the stochastic velocity rescale thermostat52 with a time constant of 0.5 ps and an average system temperature of 343.15 K. The average pressure was set to 1.0 bar using the Parrinello–Rahman semi-isotropic barostat with compressibility of 4.5 × 10−5 bar−1 and a time constant of 2.0 ps.53,54 The PMF curves were extracted from the trajectories of the simulated windows using the weighed histogram analysis method (WHAM) developed by Kumar et al.55. The presented PMF profiles represent the average profile of 300 bootstrapped profiles, with error estimate based on the standard deviation of the bootstrapped profiles. Fig. S1 of ESI,† presents an example set of these 300 bootstrapped profiles, calculated for imidazole, to enable assessment of PMF profile and adsorption energy convergence. The reported adsorption energies correspond to the difference between the PMF minimum, i.e. the adsorption site at the adsorbate surface, and the plateau value at large distance.
To assess the contributions of Lennard-Jones (LJ) and Coulombic non-bonded interactions to the overall PMF profiles, we performed a decomposition of the PMF data curve to LJ and Coulombic contributions for imidazole as the test case. The decomposed PMF is presented as Fig. S2 in the ESI.† The data supports the conclusion that the contribution of LJ-interactions to adsorption is much smaller than the Coulomb interaction contributions. The difference is so large that even taking into account the differences in molecule sizes, for species such as in this work the overall adsorption energy can be expected to scale with molecule point charges, particularly the adsorbing imidazole ring nitrogen. Imidazole is employed as the test case here due to its two ring nitrogens.
2.2 Electronic structure calculations
Electronic structure calculations were carried out for the same set of N-heterocycles and fatty acid amides as the MD simulations, see Fig. 1. For the fatty acid amides, the hydrocarbon tail was truncated from 18 to 6 carbons and the double bond was omitted. Initial molecular geometries were constructed using Avogadro: an open-source molecular builder and visualization tool, version 1.1.1.56 The configurations were energy minimized in vacuo using the general Amber force field (GAFF)57 included in Avogadro. Gaussian g1658 was used for subsequent geometry optimization of the prepared molecular structures and single point calculations for population analysis to derive Hirshfeld,59 Merz-Singh-Kollman (MK),60,61 and ChelpG62 partial charges, as well as to calculate molecular electrostatic potential (MEP). The PBE0 hybrid functional63,64 and def2-TZVPP basis set65,66 were used. The solvent was modelled by an implicit CPCM solvent model67,68 with relative permittivity of 3.109 and refractive index of 1.477 based on literature values for triolein.69,70 Atoms in molecules (AIM) analysis71–74 was carried out to derive AIM75 partial charges based on the calculated electron densities. VMD was used for MEP visualizations.763 Results and discussion
To assess the adsorption energies of the nitrogen containing compounds, first the PMF curves corresponding to the free energy landscape observed by the nitrogen compound near the montmorillonite surface were calculated. For the heterocycles, the curves are presented in Fig. 2 and the corresponding adsorption energies ΔGads collected in Table 1. The data points toward adsorption strength of the examined N-heterocycles being purine > pyridine > imidazole > pyrrole > indole > quinoline.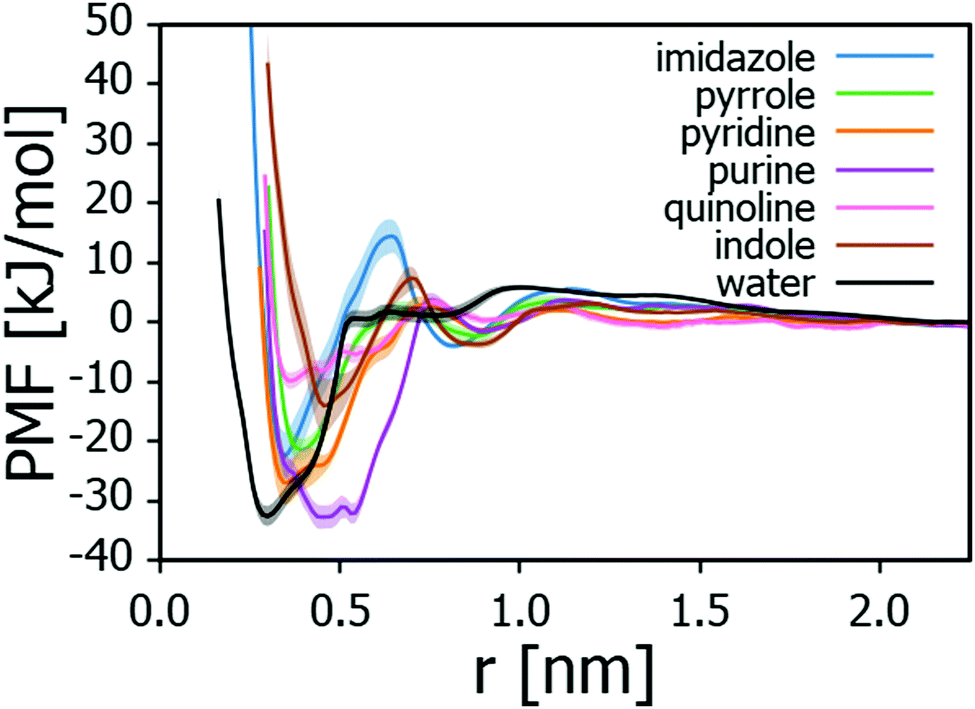 | ||
| Fig. 2 Potential of mean force (PMF) curves for N-heterocycles and water adsorbing onto Na-montmorillonite in triolein solvent. Distance r is the perpendicular distance between heterocycle center of mass and Na-montmorillonite surface topmost layer Si atoms. Shading indicates the PMF error estimate. | ||
| Adsorbate | ΔGads [kJ mol−1] |
|---|---|
| Quinoline | −9.8 ± 1.4 |
| Indole | −14.0 ± 4.7 |
| Pyrrole | −21.4 ± 1.7 |
| Imidazole | −22.4 ± 4.0 |
| Pyridine | −26.9 ± 3.6 |
| Purine | −32.7 ± 2.0 |
| Water | −32.5 ± 1.7 |
Let us next consider the binding conformations adopted by the heterocycles. Fig. 3 plots the radial distribution functions (RDFs) of N-heterocycle nitrogens with respect to montmorillonite surface Na+ ions. The RDFs were calculated based on the umbrella sampling windows corresponding to PMF minima in Fig. 2 over the last 30 ns of simulation time. The data shows that adsorption involves strong interaction of the adsorbate with the montmorillonite surface Na+ cations for all examined heterocycle species. Most examined heterocycles bind directly with the surface Na+ cations, as shown by the sharp localized short distance peak. For example, purine adsorbs on Na-montomorillonite in a parallel conformation with the surface, through interaction of a basic nitrogen with the surface Na+ ions. This adsorption conformation is supported by existing literature.77 However for indole and pyrrole, a more spread-out binding at larger separation from the surface cation is predicted. This presumably results from these heterocycles lacking a similar specific binding site for a cation. Both pyrrole78 and indole79,80 have been shown to bind cations through cation–π interactions. The use of static charges in a non-polarizable force field, such as Charmm GenFF, means this effect is not captured. Calculations using polarizable versions of both Charmm81–83 and OPLS84 force fields have shown that cation-benzene interaction in gas phase is greatly underestimated by non-polarizable force fields. Interestingly for this work, Turupcu et al. determined the interaction energy between K+ ion and benzene in water and THF using the cation–π interactions enabled OPLS/2020 force field as −7.5 kcal mol−1 (−31.4 kJ mol−1) and −6.7 kJ mol−1 (−28.0 kJ mol−1) respectively.84 In the approach, manual tuning of interactions is required to match ab initio and experimental target data.81,83,84 Furthermore, as hydrated Na+ ions rarely directly participate in cation–π interactions with benzene, this approach has not been pursued here. Nevertheless, the lack of competing cation-π interaction in the simulations steers adsorption to occur via an electronegative atom such as the ring nitrogen, instead of the π-conjugated rings.
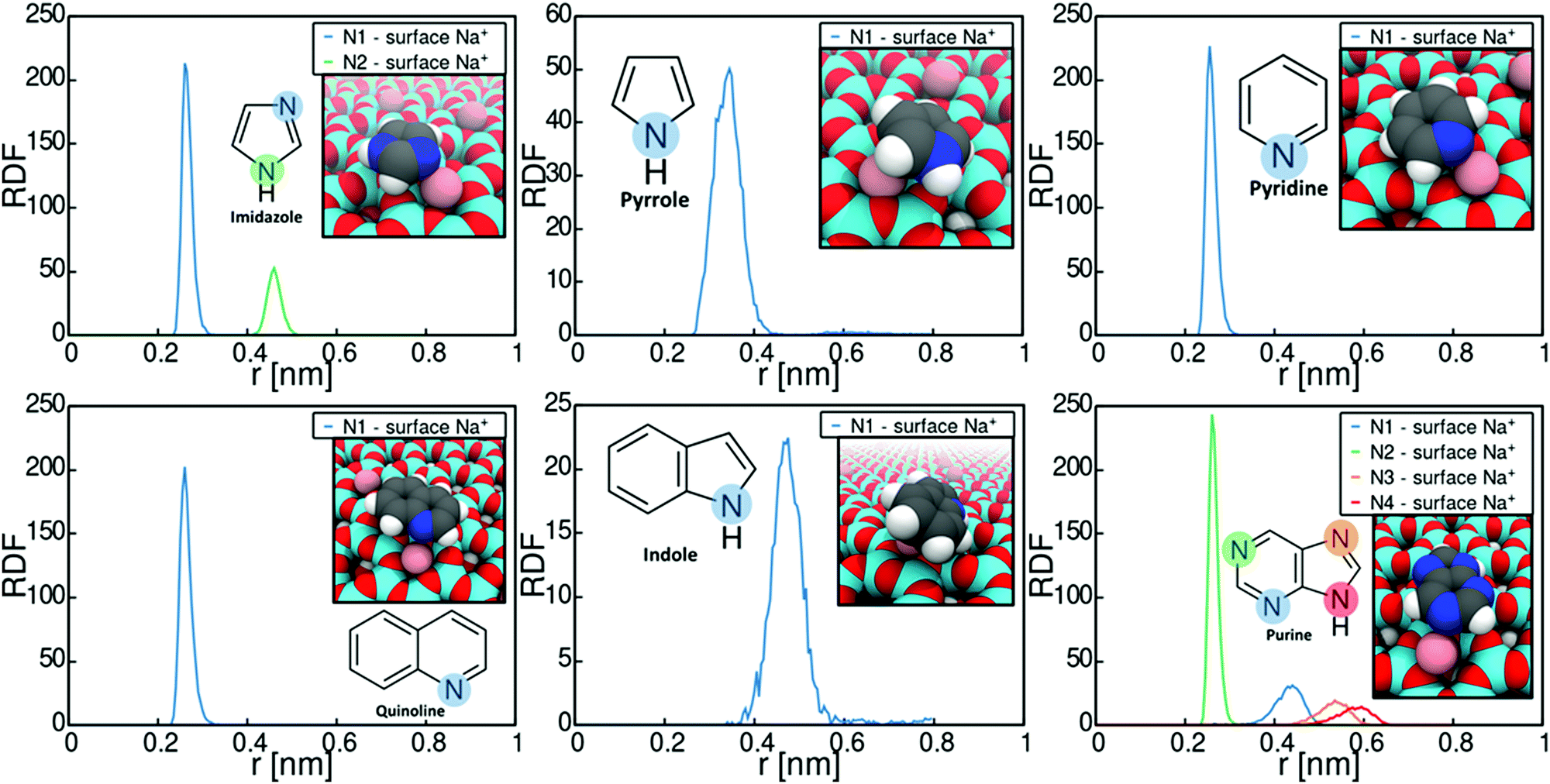 | ||
| Fig. 3 Radial distribution function (RDF) of N-heterocycle nitrogen with respect to montmorillonite surface Na+ ions. Insets are visualizations of molecule adsorption conformations at the PMF minimum. | ||
The significance of the strong surface cation correlation in the binding is that the type of cation present on the montmorillonite surface is likely to have a very strong influence on adsorption. Adsorption is driven mainly by Coulombic interactions while the Lennard-Jones contribution to the overall adsorption energy is minimal, see Fig. S2 in the ESI,† for decomposition of the imidazole adsorption PMF. This means adsorption energy can be expected to scale with the magnitude of the interacting point charges. Particularly, cation valency can be expected to be very significant in guiding adsorption, with higher valency leading to stronger adsorption. However, size of the surface cation will also affect steric availability of especially the amide carbonyl oxygen and divergence from the established adsorption preference can be expected.85–87 An indirect conclusion of the strong cation correlation of the adsorption is that also the presence of e.g. water on the montmorillonite surface can be expected to significantly affect adsorption strength. With water present on surface, adsorption may occur either via displacement of water by the adsorbate or via water bridging. For example, according to existing literature, the adsorption of e.g. nitroaromatic compounds in aqueous environments onto smectite clays involves the displacement on water at the clay surface by the adsorbate, followed by complexation with the clay surface cations.88
It is worth noting that periodic plane wave calculations derived adsorption energies of purine bases adenine (−84.5 kJ mol−1) and guanine (−115.4 kJ mol−1)77 on Na-montmorillonite in vacuo are higher compared to the adsorption energy of purine calculated here (−32.7 kJ mol−1), likely explained by the lack of charge transfer in the model. Similarly elevated in comparison to the here presented MD modelling derived values are the theoretical literature values for binding energies of pyrrole – Na+ (−107.9 kJ mol−1)78 and indole – Na+ (−131.8 to −136.5 kJ mol−1)79,80,89 complexes in gas phase. The elevated adsorption energy values are, however, calculated in vacuum or gas phase. Furthermore, evidence of chemisorption of, e.g. pyridine onto montmorillonite in aqueous environments exists.90 It should be noted that the MD model used here only accounts for physisorption of the adsorbate on the adsorbent. Finally, the adsorption energies reported in Table 1 correspond to single molecule adsorption on a surface and therefore cooperative and competitive adsorption effects (effect of other adsorbate molecules present on the surface) have not been assessed here.
The adsorption geometries inferred from Fig. 3 and the adsorption energies of Table 1 show that the accessibility of the non-protonated basic nitrogen (in pyridine and imidazole) leads to higher adsorption energies than those resulting from Na+ cation interaction with the protonated nitrogens (in pyrrole and indole). The nonprotonated nitrogens in pyridine and imidazole and their lone pairs are not involved in the formation of a delocalized π-orbital and thus do not contribute to the formation of resonance structures.91 This nitrogen lone pair explains the basicity of pyridine comparable to regular amines and imines.91
Table 2 summarises the adsorption energies of the examined fatty acid amides on Na-montmorillonite and the corresponding PMF profiles are presented in Fig. 4. The adsorption strengths follow the order: methyl oleamide > oleamide > dimethyl oleamide > phenylethyl oleamide > ethanol oleamide. In general, adsorption strength can be expected to be proportional to the steric availability of the headgroup for this type of linear, analogous molecules. Indeed, except for methyl oleamide, the results point toward introducing a bulkier headgroup to the amide significantly lowering the adsorption strength. This influence persists even if the substitute headgroup has some hydrogen bonding capability, such as ethanol oleamide. Methyl oleamide appears to be an outlier in the series by its significantly stronger adsorption than what predicted by its headgroup accessibility. Its adsorption strength is likely an artefact arising from the Charmm GenFF parametrization (imbalance of charge distribution vs. other interactions) and discussed below.
| Adsorbate | ΔGads [kJ mol−1] |
|---|---|
| Ethanol oleamide | −20.2 ± 1.3 |
| Phenylethyl oleamide | −20.3 ± 1.3 |
| Dimethyl oleamide | −23.0 ± 1.3 |
| Oleamide | −32.6 ± 0.9 |
| Methyl oleamide | −46.7 ± 1.8 |
| Water | −32.5 ± 1.7 |
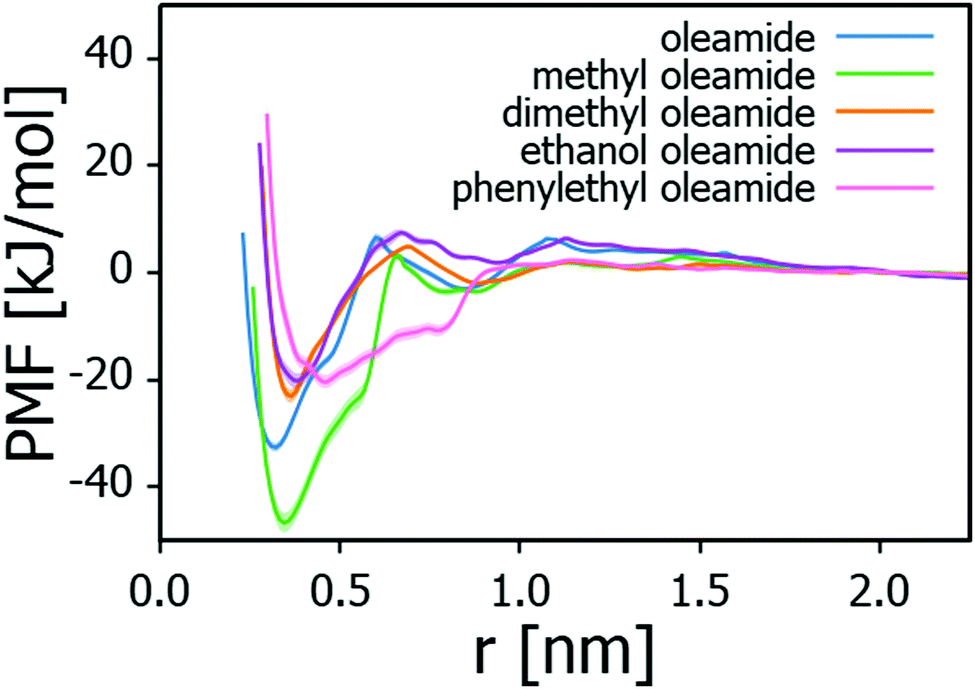 | ||
| Fig. 4 Potential of mean force (PMF) curves for fatty acid amides adsorbing onto Na-montmorillonite in triolein solvent. Distance r is the perpendicular distance between fatty acid amide head group center of mass and Na-montmorillonite surface topmost layer Si atoms. Shading indicates the PMF error estimate. | ||
Fig. 5 plots the radial distribution function of the carbonyl oxygen and amide nitrogen in relation to the montmorillonite surface Na+ cations, as well as visualizations of the binding. The data reveals the adsorption sites and conformations. Adsorption of all the examined amides occurs systematically via interaction of the carbonyl oxygen with the montmorillonite surface Na+ ions. This means that the adsorption strength can indeed be expected to be directly related to the steric availability of the carbonyl oxygen. Consequently, the adsorption response is directly subject to the partial negative charge assigned to the carbonyl oxygen atom in the force field parametrization.
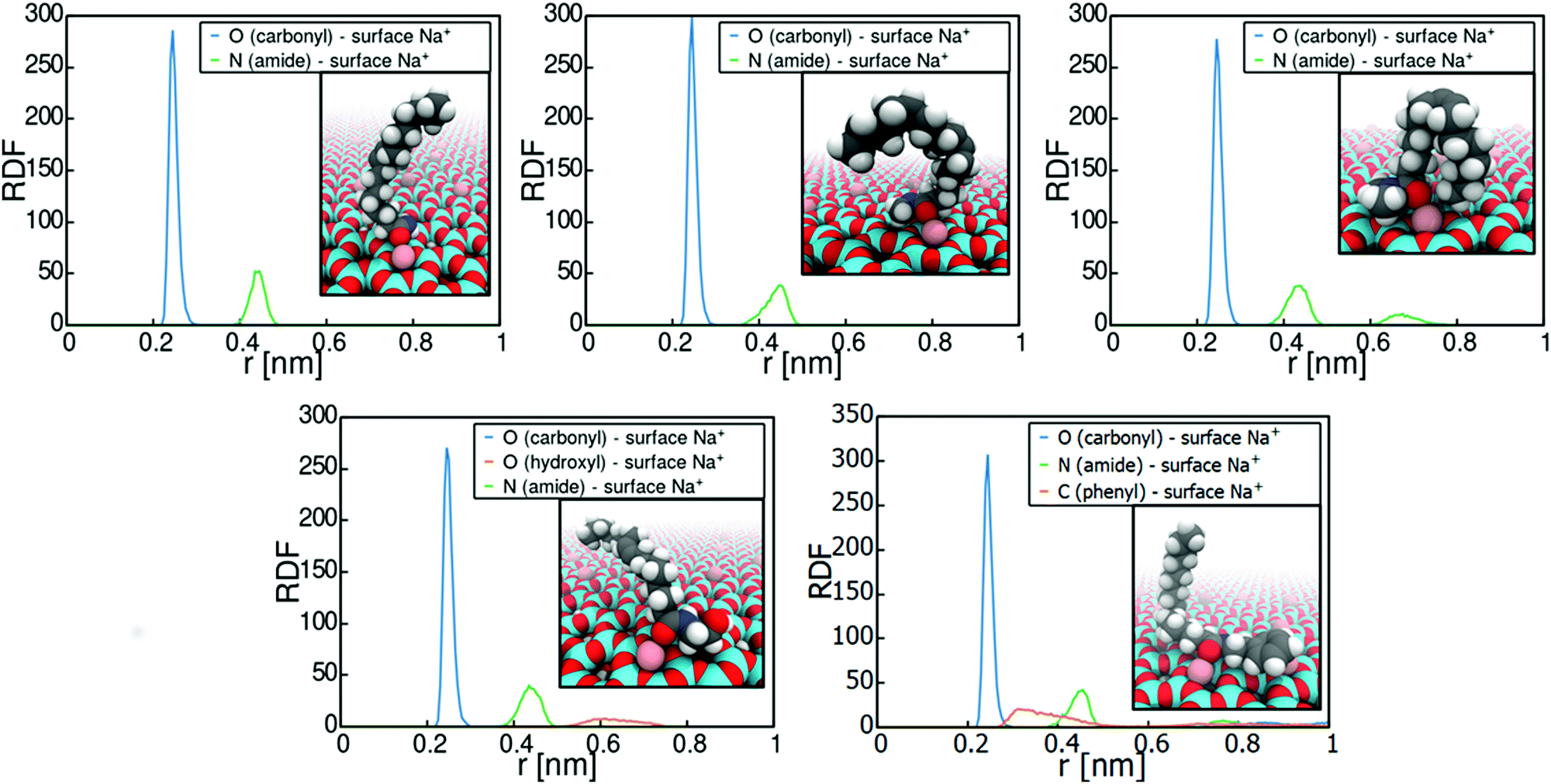 | ||
| Fig. 5 Radial distribution function (RDF) of fatty acid amide functional oxygens, amide nitrogens, and aromatic carbons (phenylethyl oleamide) with respect to montmorillonite surface Na+ ions. Insets are visualizations of molecule conformations at the PMF minimum. The hydrocarbon tail remains freely solvated in the triolein solvent and adopts a multitude of conformations. | ||
The assessment of adsorption of the nitrogen compounds in this work is on unhydrated Na-montmorillonite with no water present in the system. However, montmorillonite and other smectite clays are prone to surface hydration and significant swelling due to water in the interlayer space of the clay.92,93 Hence, we determined also the adsorption response and energy of water. Water adsorption energy is −32.5 kJ mol−1, see Fig. 2. This means that water at the montmorillonite surface, if present, will be bound at the surface by the Na+ ions. It may also influence the adsorption mechanism and adsorption strength of adsorbates. Notably, prior works have shown that adsorption related cation bridging primarily occurs at oil-wetted clay interfaces, while on hydrated clay surfaces cation bridging does not occur due to hydration of the ions.94 This is in agreement with deductions of the current work.
The here employed empirical force-fields, similar to most biomolecular force fields, such as Charmm, Amber, Gromos, and OPLS, are unpolarizable and rely on static partial charges of atoms.95,96 Furthermore, the parametrizations are optimized for aqueous environments and fully hydrated systems. Despite their shortcomings, empirical biomolecular force fields are successfully used to describe complex biochemical environments. However, the lack of polarization is known to become especially problematic for low dielectric environments, such as in apolar solvents,97,98 inside lipid membranes,99,100 and in hydrophobic protein core regions101. The parametrization procedure of e.g. the Charmm GenFF force field42 that is used in the MD simulations here involves matching of Hartree–Fock (HF) level interaction energies and distances for the compounds and water, along with additional matching to experimental properties. Because of the parametrization approach, the effect of electronic polarization and dielectric charge screening in aqueous environments are somewhat baked into the empirically derived interaction parameters of the force fields; however, the extent of this is unclear.
To assess the effect of this, we next compare the Charmm GenFF parameters, especially electrostatics in terms of partial charges, against DFT electronic structure calculations. The examination focuses on electrostatics because the adsorption to montmorillonite surface occurs here via cation bridging.
The molecular electrostatic potentials (MEPs) corresponding to the N-heterocycles and truncated fatty acid amides are presented in Fig. 6 and 7 respectively. The MEP describes the electrostatic potential felt by a positive test charge, i.e. proton, at a certain coordinate around a molecule, disregarding polarization of the molecule in response to the test charge. Typically, MEP is visualized as a molecular surface that is based on either the van der Waals radii of the atoms, solvent accessible surface, or a constant value of electron density. The latter, with an isovalue of 0.01e/a03, where e is unit charge and a0 the Bohr radius, was used here.
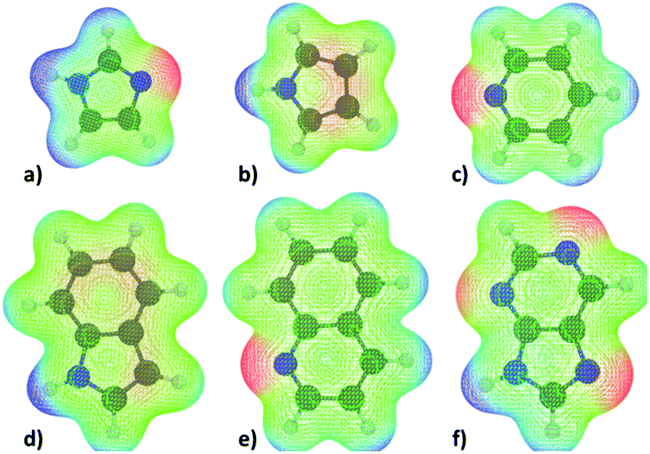 | ||
| Fig. 6 Visualizations of the molecular electrostatic potential (MEP) of (a) imidazole, (b) pyrrole, (c) pyridine, (d) indole, (e) quinoline, and (f) purine superimposed onto the molecular structure. Red corresponds to regions of negative potential and blue to regions of positive potential as experienced by a positive test charge (i.e. proton). | ||
 | ||
| Fig. 7 Visualizations of the molecular electrostatic potential (MEP) of (a) hexamide, (b) N-methylhexamide, (c) N-dimethylhexamide, (d) N-ethanolhexamide, and (e) N-phenylethylhexamide superimposed onto the molecular structure. Red corresponds to regions of negative potential and blue to regions of positive potential as experienced by a positive test charge (i.e. proton). | ||
The data of Fig. 6 and 7 shows that regions of negative electrostatic potential are concentrated at the nonprotonated basic nitrogens for N-heterocycles and at the carbonyl oxygens for the fatty acid amides. For N-ethanolhexamide, the alcohol hydroxyl oxygen introduces a second region of negative electrostatic potential. Comparison of the negative electrostatic potential regions with the adsorption geometries inferred from Fig. 3 and 5 reveals that MEP can be considered as a strong predictor for the site of adsorption via cation bridging for most of the examined adsorbent species. However for molecules with multiple possible adsorption sites, such as purine, discrepancy between the MEP prediction of most negative potential sites and the MD-simulation predicted adsorption sites can be observed.
Next, we determined based on the electronic structure calculations the Hirshfeld,59 AIM,75 Merz-Singh-Kollman (MK),60,61 and ChelpG62 partial charges corresponding to each atom site in the adsorbents. The partial atomic charge is not a quantum mechanical observable and therefore is not uniquely defined. Partial charge is highly method dependent, as demonstrated here by our comparison of the different partial charge determining methods. Fig. 8 presents a comparison of these DFT derived partial charges for the heterocycles to those of the Charmm GenFF. Exact values of the partial charges for the entire set of model compounds including the fatty acid amides are presented in Fig. S3–S5 of ESI.†Fig. 8 shows that both Hirshfeld and AIM charges differ significantly from Charmm GenFF, particularly for atoms at either extreme of the molecule charge distribution. The AIM partial charges are systematically larger and Hirshfeld charges smaller in magnitude in comparison to Charmm GenFF partial charges. Meanwhile, both MK and ChelpG result in values that match well with the Charmm GenFF partial charges. The data for the fatty acid amides is analogous, see ESI.†
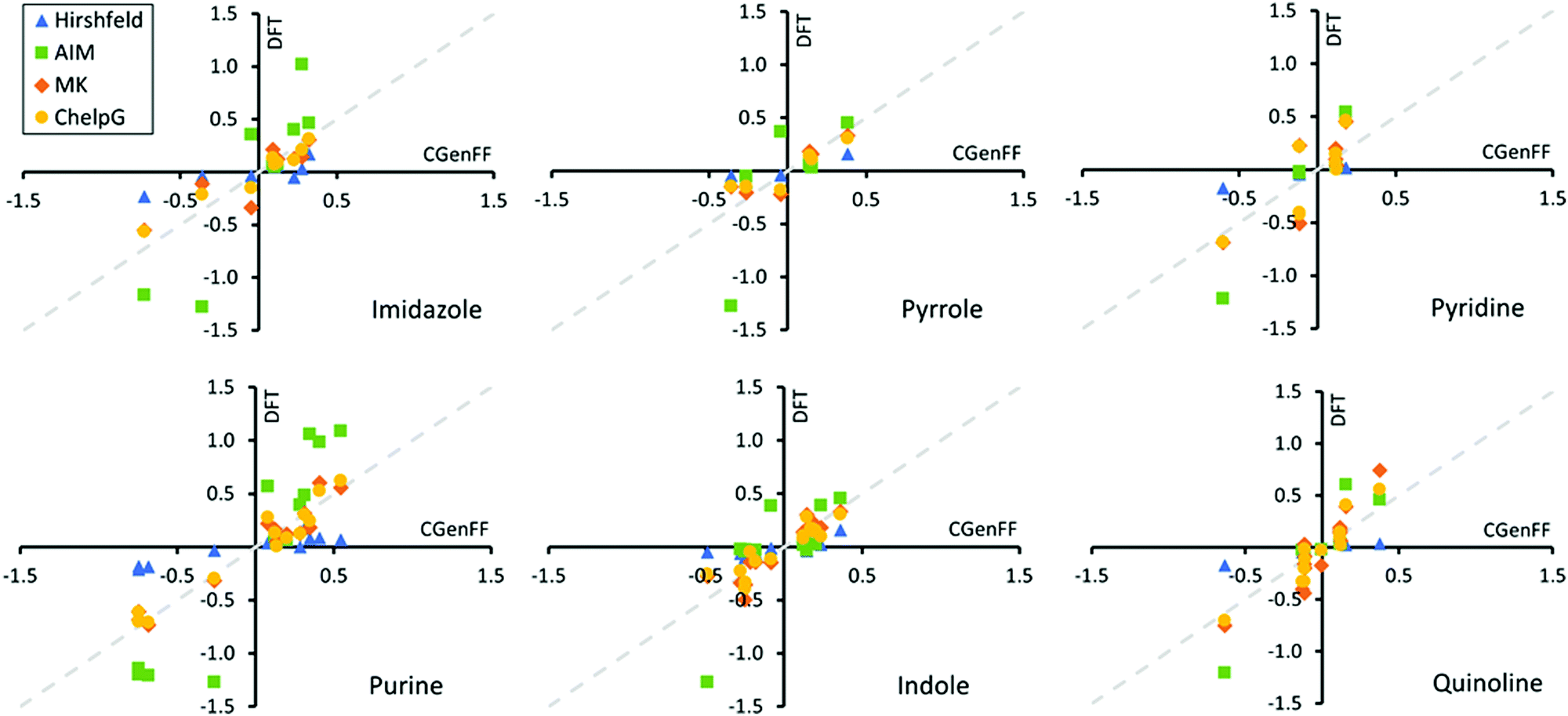 | ||
| Fig. 8 Comparison of the DFT calculations based Hirshfeld, AIM, Merz-Singh-Kollman (MK), and ChelpG charges with the Charmm GenFF partial charges for the studied N-heterocycles. Deviation from diagonal (dashed grey line) corresponds with a mismatch to the partial charge. | ||
To better understand the deviations, let us consider the charge determination schemes a bit more in detail. Hirshfeld population analysis involves partition of electron density in real space and assigned partial charge is dependent on the relative electron-withdrawing power of an atom relative to its environment.59,102 AIM charges are determined by dividing the molecular charge density into atomic contributions based on overall molecular topology through construction of zero-flux surfaces of the electronic density. MK and ChelpG partial charge schemes aim to replicate the MEP at various grid points around the molecule.61,62 The two methods differ in definition of these grid points. ChelpG uses a regularly spaced cubic lattice, while the MK scheme uses grid points on nested Connolly surfaces. Since the MK and ChelpG charges are fitted to reproduce MEP, atoms that lie deeply buried inside the molecule can vary in their charge which leads to uncertainty. On the other hand, the Charmm GenFF partial charges have been determined based on HF/6-31G(d) level theory interaction energies in aqueous environment, e.g., matching interactions with single water molecules placed at configurations that would arise via hydrogen bonding and/or lone pair interaction sites.42 The resulting partial charges of atoms are tuned until sufficient agreement with quantum mechanical calculations based water interaction distance and interaction energies. Commonly MK charges at the MP2/6-31G(d) theory level are used as initial estimates for partial charges.42 The Charmm GenFF parametrization compound set includes a wide variety of heterocycles, which often provide a scaffold for the majority of pharmaceuticals. This explains the good match between the MK, ChelpG and Charmm GenFF partial charges for the examined heterocycles. Mismatch between Charmm GenFF and the MK and ChelpG charges is greater for the fatty acid amides, see Fig. S5 of ESI,† for which parameter assignment was done mainly through analogy, accounting for neighbouring bonded atoms.103 Notably, atomic charges can depend strongly on the conformation of the molecule. This means the partial charges of a fixed point charge model, as in the case of most biomolecular force-fields, should be averaged over several molecular conformations for flexible molecules, such as the amides here. Here, partial charges were determined based only on a single molecular conformation. The provided DFT based charges are to indicate systematic deviations due to the apolar solvent environment. However, based on the good match with Charmm GenFF, the effect of this dielectric screening is minimal. Here the continuum solvent model with a relative permittivity of 3.109 is closely comparable to calculations in gas phase.
The good match between Charmm GenFF and MEP derived partial charges (MK and ChelpG) indicates that the Charmm GenFF based molecular dynamics simulations employ charge distributions that can be considered to accurately represent the equivalent of DFT simulations MEP even in this apolar solvent environment. Thus, the predictions of adsorption geometries and trends in adsorption energies for molecules with a single likely adsorption site can be considered reliable in comparison to quantum chemical calculations. This is supported by the good agreement with adsorption conformations from literature. However, subtle differences in electron density for molecules with multiple possible adsorption sites are clearly not captured by Charmm GenFF charge partition. For example, Charmm GenFF uses identical charges for the amide nitrogens and carbonyl oxygens of N-ethanolhexamide and N-phenylethylhexamide, despite the apparent differing chemical environments. Both MK and ChelpG predict more negative charges for both amide N and carbonyl O of N-phenylethylhexamide than those of Charmm GenFF. The persistence of this trend for other molecular configurations is unclear. Additionally for purine, Charmm GenFF assings a charge difference of 0.062e between the two nitrogens of the six-membered ring. This charge difference is not replicated by any of the charge calculation schemes. Together with the lack of polarization and proper charge screening in a low dielectric medium, such as the triolein solvent of this work, such charge distribution is likely to produce inaccurate adsorption geometries in the force-field based MD simulations. However, as the overall charge trends are in line with the various charge determination schemes, the qualitative response can be expected to persist.
4 Conclusions
The adsorption of six nitrogen containing heterocyclic compounds and five fatty acid amides from model vegetable oil solvent to model phyllosilicate adsorbent (Na-montmorillonite) was examined via MD simulations and DFT electronic structure calculations. Adsorption energies and configurations corresponding to single molecule physisorption on dehydrated Na-montmorillonite were determined. We find that the adsorption of the examined heterocyclic compounds occurs through interaction of the ring nitrogen, particularly the basic, non-protonated nitrogen, with the montmorillonite surface Na+ cations. The examined fatty amides adsorb via interaction of the carbonyl oxygen with the montmorillonite Na+ ions. Adsorption of the heterocycles is dictated by the basicity of the ring nitrogen and, when applicable, the strength of cation–π interaction, while for the fatty acid amide series, the accessibility of the carbonyl group and the bulkiness of the head group govern the adsorption strength. Throughout, the adsorption takes place via a cation bridging mechanism, i.e. the montmorillonite surface Na+ ions are the adsorption site focii.The observed cation bridging adsorption mechanism means that the type and valency of the montmorillonite surface cations, but also the presence of water on the surface can be expected to greatly affect adsorption strength. Notably, when excess water is present in the system, hydration of the montmorillonite surface and the polar headgroups of the adsorbates can be expected. We assessed also water adsorption to the montmorillonite surface. If water is present in the system to wet the surface to hydrated montmorillonite, the results leave under debate whether the N-compounds on a hydrated montmorillonite surface adsorb via displacement of water from the surface to cationic sites or via water bridging.
The electronic structure calculations enabled determining the reliability and quality of the MD-simulation results. We conclude that the molecular electrostatic potential provides a good accuracy predictor for the site of adsorption for the here observed cation bridging type adsorption response. This good match with the MEP and the MD simulations results of the adsorption arises because for most of the examined molecules, the Charmm GenFF partial charges mirror the trends in molecular electron density. This was further analyzed via determining the Hirshfeld, AIM, MK and ChelpG partial charges. We noted that discrepancies between the DFT calculated electron density and Charmm GenFF arise for molecules with multiple possible adsorption sites and for molecules with poorer analogy in the force field parametrization set, such as the fatty acid amide head groups examined here. For such species, adsorption geometry predictions based on MD simulations remain unreliable. An additional source of error is the lack of polarization in the Charmm force field. This is especially apparent for interaction of ionic groups or ions in low dielectric mediums, such as the triolein solvent of this work. As a result, electrostatic interactions can be considered overestimated by a factor of 2.104 This consideration leads to a direct conclusion that the adsorption energies presented in this work for physisorption are significant overestimates in the apolar solvent environment considered here.
We presented here a systematic examination of nitrogen compound adsorption from bio oils via computational means. The obtained work demonstrates MD simulations can be used to extract reliable information about selective nitrogen adsorption but the findings also raise warranted attention to limitations of the molecular modelling approaches in quantitatively accurate adsorption modelling for apolar solvent systems. The significance of the work is that the adsorption findings provide a design guideline for adsorption response of related systems but the findings also provide means to assess accuracy of molecular level modelling approaches for similar apolar solvent systems. Overall, we conclude that molecular dynamics based modelling serves as a valuable tool in providing molecular level description and guidelines of physisorption responses in bio-oils.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Rasmus Kronberg and Prof. Kari Laasonen for assistance with the electronic structure calculations and for useful discussions. This work was partly financed by the Fortum and Neste Foundation project 20210130 (M. V.). The authors acknowledge the financial support of the Academy of Finland Grant No. 309324 (M. S.). We are grateful for the support by the FinnCERES Materials Bioeconomy Ecosystem. Computational resources by CSC IT Centre for Science, Finland, RAMI – RawMatTERS Finland Infrastructure, and Aalto Science-IT project are also gratefully acknowledged.Notes and references
- E. S. Shuba and D. Kifle, Renewable Sustainable Energy Rev., 2018, 81, 743–755 CrossRef CAS.
- K. Satyanarayana, A. Mariano and J. Vargas, Int. J. Energy Res., 2011, 35, 291–311 CrossRef.
- X. Miao, Q. Wu and C. Yang, J. Anal. Appl. Pyrolysis, 2004, 71, 855–863 CrossRef CAS.
- B. Maddi, S. Viamajala and S. Varanasi, Bioresour. Technol., 2011, 102, 11018–11026 CrossRef CAS PubMed.
- L. Sheng, X. Wang and X. Yang, Bioresour. Technol., 2018, 247, 14–20 CrossRef CAS PubMed.
- O. Palardy, C. Behnke and L. M. Laurens, Energy Fuels, 2017, 31, 8275–8282 CrossRef CAS.
- S. Chiaberge, I. Leonardis, T. Fiorani, G. Bianchi, P. Cesti, A. Bosetti, M. Crucianelli, S. Reale and F. De Angelis, Energy Fuels, 2013, 27, 5287–5297 CrossRef CAS.
- S. Wang, Q. Wang, X. Jiang, X. Han and H. Ji, Energy Convers. Manage., 2013, 68, 273–280 CrossRef CAS.
- A. Ross, J. Jones, M. Kubacki and T. Bridgeman, Bioresour. Technol., 2008, 99, 6494–6504 CrossRef CAS PubMed.
- N. H. Zainan, S. C. Srivatsa and S. Bhattacharya, Fuel, 2015, 161, 345–354 CrossRef CAS.
- F. Li, L. Katz and S. Qiu, Ind. Eng. Chem. Res., 2019, 58, 3959–3968 CrossRef CAS.
- F. Li, L. Katz and Z. Hu, ACS Sustainable Chem. Eng., 2019, 7, 16529–16538 CrossRef CAS.
- M. Hazrat, M. Rasul and M. M. K. Khan, Proc. Eng., 2015, 105, 865–876 CrossRef CAS.
- A. J. Hernández-Maldonado and R. T. Yang, Angew. Chem., 2004, 116, 1022–1024 CrossRef.
- P. Falaras, I. Kovanis, F. Lezou and G. Seiragakis, Clay Miner., 1999, 34, 221–232 CrossRef CAS.
- E. Santillan-Jimenez, R. Pace, S. Marques, T. Morgan, C. McKelphin, J. Mobley and M. Crocker, Fuel, 2016, 180, 668–678 CrossRef CAS.
- B. N. Bhadra and S. H. Jhung, Chem. Eng. J., 2020, 388, 124195 CrossRef CAS.
- P. A. Penttila, S. Vierros, K. Utriainen, N. Carl, L. Rautkari, M. Sammalkorpi and M. Österberg, Langmuir, 2019, 35, 8373–8382 CAS.
- O.-P. Lehtinen, R. W. N. Nugroho, T. Lehtimaa, S. Vierros, P. Hiekkataipale, J. Ruokolainen, M. Sammalkorpi and M. Österberg, Colloids Surf., B, 2017, 160, 355–363 CrossRef CAS PubMed.
- S. Abel and M. Marchi, J. Phys. Chem. B, 2020, 124, 11802–11818 CrossRef CAS PubMed.
- S. Abel, N. Galamba, E. Karakas, M. Marchi, W. H. Thompson and D. Laage, Langmuir, 2016, 32, 10610–10620 CrossRef CAS PubMed.
- R. de Souza, R. Ratochinski, M. Karttunen and L. Dias, J. Chem. Inf. Model., 2019, 60, 522–536 CrossRef PubMed.
- A. Khoshnood and A. Firoozabadi, Langmuir, 2015, 31, 5982–5991 CrossRef CAS PubMed.
- S. Vierros and M. Sammalkorpi, Phys. Chem. Chem. Phys., 2018, 20, 6287–6298 RSC.
- S. Vierros and M. Sammalkorpi, ACS Omega, 2019, 4, 15581–15592 CrossRef CAS PubMed.
- M. Doig and P. J. Camp, Phys. Chem. Chem. Phys., 2015, 17, 5248–5255 RSC.
- M. Doig, C. P. Warrens and P. J. Camp, Langmuir, 2013, 30, 186–195 CrossRef PubMed.
- J. L. Bradley-Shaw, P. J. Camp, P. J. Dowding and K. Lewtas, Langmuir, 2016, 32, 7707–7718 CrossRef CAS PubMed.
- J. L. Bradley-Shaw, P. J. Camp, P. J. Dowding and K. Lewtas, Phys. Chem. Chem. Phys., 2018, 20, 17648–17657 RSC.
- S. J. Eder, A. Vernes and G. Betz, Langmuir, 2013, 29, 13760–13772 CrossRef CAS PubMed.
- Y. Xiong, T. Cao, Q. Chen, Z. Li, Y. Yang, S. Xu, S. Yuan, J. Sjöblom and Z. Xu, J. Phys. Chem. C, 2017, 121, 5020–5028 CrossRef CAS.
- J. Zhong, P. Wang, Y. Zhang, Y. Yan, S. Hu and J. Zhang, Energy, 2013, 59, 295–300 CrossRef CAS.
- Q. Liu, S. Yuan, H. Yan and X. Zhao, J. Phys. Chem. B, 2012, 116, 2867–2875 CrossRef CAS PubMed.
- M. Vuorte, S. Vierros, S. Kuitunen and M. Sammalkorpi, J. Colloid Interface Sci., 2020, 571, 55–65 CrossRef CAS PubMed.
- M. Munir, M. Ahmad, M. Rehan, M. Saeed, S. S. Lam, A. Nizami, A. Waseem, S. Sultana and M. Zafar, Environ. Res., 2021, 193, 110398 CrossRef CAS PubMed.
- M. Munir, M. Ahmad, M. Mubashir, S. Asif, A. Waseem, A. Mukhtar, S. Saqib, H. S. H. Munawaroh, M. K. Lam and K. S. Khoo, et al. , Bioresour. Technol., 2021, 328, 124859 CrossRef CAS PubMed.
- S. Vierros and M. Sammalkorpi, J. Chem. Phys., 2015, 142, 094902 CrossRef CAS PubMed.
- S. Vierros and M. Sammalkorpi, Phys. Chem. Chem. Phys., 2015, 17, 14951–14960 RSC.
- I. V. Kopanichuk, V. A. Novikov, A. A. Vanin and E. N. Brodskaya, J. Mol. Liq., 2019, 296, 111960 CrossRef CAS.
- D. P. Geerke and W. F. van Gunsteren, ChemPhysChem, 2006, 7, 671–678 CrossRef CAS PubMed.
- D. P. Geerke and W. F. van Gunsteren, J. Phys. Chem. B, 2007, 111, 6425–6436 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes and I. Vorobyov, et al. , J. Comput. Chem., 2010, 31, 671–690 CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- H. Heinz, T. J. Lin, R. K. Mishra and F. S. Emami, Langmuir, 2013, 29, 1754–1765 CrossRef CAS PubMed.
- Y. J. Bae, C. Ryu, J.-K. Jeon, J. Park, D. J. Suh, Y.-W. Suh, D. Chang and Y.-K. Park, Bioresour. Technol., 2011, 102, 3512–3520 CrossRef CAS PubMed.
- J. Henin, W. Shinoda and M. L. Klein, J. Phys. Chem. B, 2008, 112, 7008–7015 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, et al. , J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- M. J. E. O'Neil, The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 14th Revised edn, Elsevier Health Sciences, London, United Kingdom, 2006 Search PubMed.
- R. E. Weast, Handbook of Chemistry and Physics, 69th edn, CRC Press, Boca Raton, FL, 1988 Search PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- M. Parrinello, A. Rahman and P. Vashishta, Phys. Rev. Lett., 1983, 50, 1073–1076 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian’16 Revision B.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- B. H. Besler, K. M. Merz Jr and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- I. Gouw and I. Vlugter, Fette, Seifen, Anstrichm., 1967, 69, 223–226 CrossRef.
- L. A. Anderson, Chemical Stimulation of Lipid Production in Microalgae and Analysis by NMR Spectroscopy for Biofuel Applications, University of California, Davis, 2015 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- P. Mignon, P. Ugliengo and M. Sodupe, J. Phys. Chem. C, 2009, 113, 13741–13749 CrossRef CAS.
- A. Gapeev, C.-N. Yang, S. J. Klippenstein and R. C. Dunbar, J. Phys. Chem. A, 2000, 104, 3246–3256 CrossRef CAS.
- R. C. Dunbar, J. Phys. Chem. A, 1998, 102, 8946–8952 CrossRef CAS.
- C. Ruan, Z. Yang, N. Hallowita and M. Rodgers, J. Phys. Chem. A, 2005, 109, 11539–11550 CrossRef CAS PubMed.
- E. A. Orabi and G. Lamoureux, J. Chem. Theory Comput., 2012, 8, 182–193 CrossRef CAS PubMed.
- F.-Y. Lin and A. D. MacKerell Jr, J. Comput. Chem., 2020, 41, 439–448 CrossRef CAS PubMed.
- H. M. Khan, C. Grauffel, R. Broer, A. D. MacKerell Jr, R. W. Havenith and N. Reuter, J. Chem. Theory Comput., 2016, 12, 5585–5595 CrossRef CAS PubMed.
- A. Turupcu, J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2020, 16, 7184–7194 CrossRef CAS PubMed.
- R. Lin, A. Ladshaw, Y. Nan, J. Liu, S. Yiacoumi, C. Tsouris, D. W. DePaoli and L. L. Tavlarides, Ind. Eng. Chem. Res., 2015, 54, 10442–10448 CrossRef CAS.
- K. S. Walton, M. B. Abney and M. D. LeVan, Microporous Mesoporous Mater., 2006, 91, 78–84 CrossRef CAS.
- H. Yi, F. Jia, Y. Zhao, W. Wang, S. Song, H. Li and C. Liu, Appl. Surf. Sci., 2018, 459, 148–154 CrossRef CAS.
- H. Li, B. J. Teppen, C. T. Johnston and S. A. Boyd, Environ. Sci. Technol., 2004, 38, 5433–5442 CrossRef CAS PubMed.
- S. Mecozzi, A. P. West and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566–10571 CrossRef CAS PubMed.
- A. R. Swoboda and G. W. Kunze, Clays Clay Miner., 1964, 13, 277–288 CrossRef.
- V. Ram, A. Sethi, M. Nath and R. Pratap, The Chemistry of Heterocycles: Chemistry of Six to Eight Membered N,O, S, P and Se Heterocycles, Elsevier Science, 2019 Search PubMed.
- S. Karaborni, B. Smit, W. Heidug, J. Urai and E. van Oort, Science, 1996, 271, 1102–1104 CrossRef CAS.
- M. Segad, B. Jonsson, T. Åkesson and B. Cabane, Langmuir, 2010, 26, 5782–5790 CrossRef CAS PubMed.
- T. Underwood, V. Erastova, P. Cubillas and H. C. Greenwell, J. Phys. Chem. C, 2015, 119, 7282–7294 CrossRef CAS.
- A. D. MacKerell Jr, J. Comput. Chem., 2004, 25, 1584–1604 CrossRef PubMed.
- T. Fröhlking, M. Bernetti, N. Calonaci and G. Bussi, J. Chem. Phys., 2020, 152, 230902 CrossRef PubMed.
- I. Vorobyov, V. M. Anisimov, S. Greene, R. M. Venable, A. Moser, R. W. Pastor and A. D. MacKerell, J. Chem. Theory Comput., 2007, 3, 1120–1133 CrossRef CAS PubMed.
- I. V. Vorobyov, V. M. Anisimov and A. D. MacKerell, J. Phys. Chem. B, 2005, 109, 18988–18999 CrossRef CAS PubMed.
- G. Klesse, S. Rao, S. J. Tucker and M. S. Sansom, J. Am. Chem. Soc., 2020, 142, 9415–9427 CrossRef CAS PubMed.
- H. Li, J. Chowdhary, L. Huang, X. He, A. D. MacKerell Jr and B. Roux, J. Chem. Theory Comput., 2017, 13, 4535–4552 CrossRef CAS PubMed.
- T. Simonson and C. L. Brooks, J. Am. Chem. Soc., 1996, 118, 8452–8458 CrossRef CAS.
- S. Saha, R. K. Roy and P. W. Ayers, Int. J. Quantum Chem., 2009, 109, 1790–1806 CrossRef CAS.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- I. Leontyev and A. Stuchebrukhov, Phys. Chem. Chem. Phys., 2011, 13, 2613–2626 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Bootstrapped PMF profiles and decomposition of PMF into Coulombic and Lennard-Jones non-bonded contributions for imidazole adsorption on Na-montmorillonite, as well as partial charge decompositions of the N-heterocycles and fatty acid amides: Charmm GenFF, Hirshfeld, AIM, Merz-Singh-Kollman, and ChelpG charges. See DOI: 10.1039/d1cp01880a |
| This journal is © the Owner Societies 2021 |