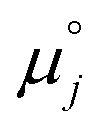Theory of the electrostatic surface potential and intrinsic dipole moments at the mixed ionic electronic conductor (MIEC)–gas interface
Received
15th April 2021
, Accepted 7th May 2021
First published on 7th May 2021
Abstract
The local activation overpotential describes the electrostatic potential shift away from equilibrium at an electrode/electrolyte interface. This electrostatic potential is not entirely satisfactory for describing the reaction kinetics of a mixed ionic–electronic conducting (MIEC) solid-oxide cell (SOC) electrode where charge transfer occurs at the electrode–gas interface. Using the theory of the electrostatic potential at the MIEC–gas interface as an electrochemical driving force, charge transfer at the ceria–gas interface has been modelled based on the intrinsic dipole potential of the adsorbate. This model gives a physically meaningful reason for the enhancement in electrochemical activity of a MIEC electrode as the steam and hydrogen pressure is increased in both fuel cell and electrolysis modes. This model was validated against operando XPS data from previous literature to accurately predict the outer work function shift of thin film Sm0.2Ce0.8O1.9 in a H2/H2O atmosphere as a function of overpotential.
1. Introduction
The solid oxide cell (SOC) is a highly efficient chemical-to-electrical and electrical-to-chemical energy conversion technology compatible with both existing fuel (i.e. natural gas) and future fuel (i.e. renewably sourced hydrogen) infrastructures. Fast kinetics at the nano-scale means that reaction mechanisms in intermediate temperature solid oxide cell electrodes (IT-SOC) can be difficult to fully understand. The electrical operation of the nickel/gadolinium doped ceria (Ni/CGO) electrode has been intensively studied for decades, however a unifying model for hydrogen electrooxidation or water electrolysis has not yet been agreed upon.1–12 As opposed to electronically insulating, pure oxide-ion conductors, the mixed ionic–electronic conductivity (MIEC) of doped ceria allows charge transfer with gas species to occur on the oxide surface, far from the triple phase boundary (TPB).13,14 The electroactive double phase boundary (2PB) is thought to enhance the rate of oxygen exchange due to the greatly increased area over which electrochemical reactions can take place.
The overall equilibrium between ceria and the gas-phase environment can be defined as:
| | 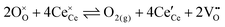 | (1) |
where
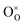
,
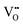
,
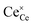
and
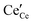
represent an oxide-ion at the anion site, a doubly charged oxygen vacancy, a cerium ion on the cation site, and small polaron (localised electron). Upon reduction of the lattice (
eqn (1) forward) additional highly mobile defects are created.
15,16 The increase in defect concentration induces a significant enhancement in bulk n-type conductivity of lattice, resulting in MIEC character. The bulk transport in doped ceria is relatively fast, and as such, the bulk-to-surface transport is not considered to be rate limiting for electrode processes.
17,18 The popular electrode bracket notation
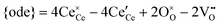
allows the chemical potential of MIEC defects to be considered as the difference between the electrochemical potential of electrons in the current collector (electron conducting phase),
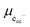
, and oxygen in the electrolyte (ionic conducting phase),
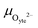
:
19,20| | 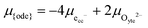 | (2) |
where,
Using this notation, the driving force for electrochemical reduction is determined by the position of the Fermi level of the metallic current collector or the oxygen chemical potential of the electrolyte phase. The electrochemical potential has been illustrated in two ways; the sum of the standard potential,
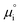
, activity potential,
kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
ln
ai and electric potential,
eϕi. A simplification can be made to express all the non-idealities within the excess chemical potential,
μexi. When an overpotential is applied, the electrochemical potential of electrons in the current collector shifts. The electrochemical potential of oxygen in the MIEC remains in equilibrium with the gas phase
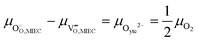
, and therefore the concentration of oxygen vacancies remains pinned.
21,22 Therefore, the local activation overpotential
η is given as:
| | 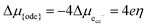 | (3) |
where the delta symbols refer to the difference between the working electrode and grounded electrode, which is in equilibrium with the gas phase.
17 The process of charge transfer at the MIEC electrode–gas interface comprises ambipolar exchange of ions and electronic species.
23 The result of such a process causes charge separation and an associated dipole moment at the electrode surface. This dipole moment is the origin of the electrostatic surface potential
χ:
where the electrode potential,
ϕode, is the potential for a MIEC phase, and
ϕad is the electrostatic potential at the location of the adsorbed species. By applying an overpotential to the MIEC electrode, an electrostatic surface potential shift may be established Δ
χ =
χ −
χeq where an effective double layer is formed between the electrode surface and the adsorbed species.
24 Although no net charge transfer occurs, this surface potential shift modifies the surface chemistry and is the driving force for the ambipolar exchange of ions and electronic species, such that the ratio of the forward,
Rf, and backward,
Rb, rates obeys the De Donder relation:
25| | 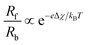 | (5) |
The surface electrostatic potential expressed in eqn (4) is defined as the difference in potential of the electrode and the adsorbed species. Under impedance spectroscopy, a small voltage perturbation (≈10 mV) of a MIEC electrode with a high concentration of electronic charge carriers will have a negligible shift in the concentration of electrons since the activity term is logarithmic. We can therefore redefine the surface potential in eqn (4) to be a function of overpotential when using near equilibrium electrochemical methods:18
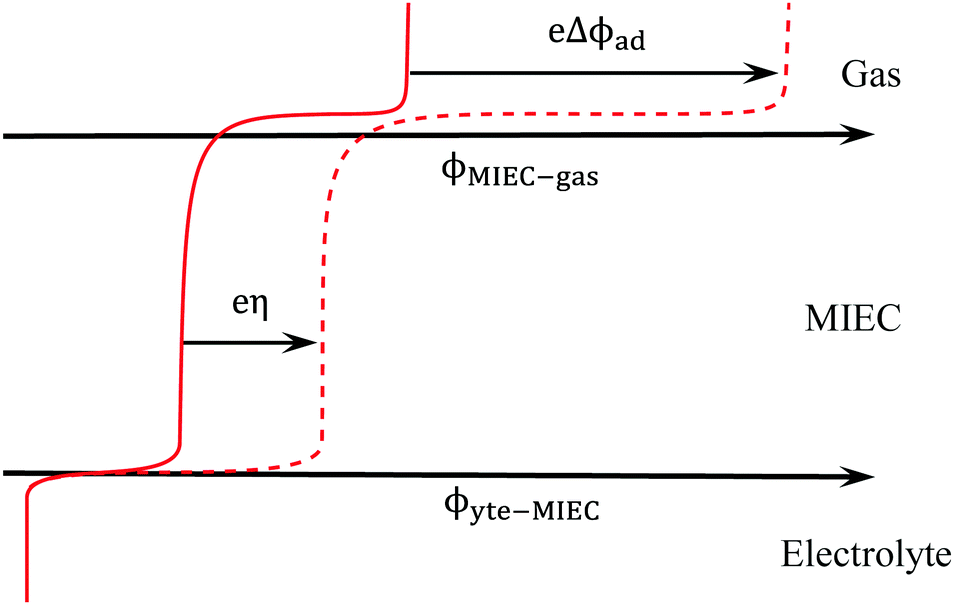
Fig. 1 illustrates the electrochemical driving force as described by eqn (6) where instead of a diffuse charge distribution, which would model the electrostatic potential step at a solid–liquid contact, the electrostatic potential step at the MIEC–gas interface is modelled as a step function since it is likely to be a single atomic layer in thickness. The ϕad will change upon an applied overpotential, but will not necessarily result in a non-zero Δχ. A change in surface chemistry then modifies the electrostatic potential of the adsorbate outside of equilibrium (i.e. modified bond orientation, polarisation, or coverage). This in turn results in a shift in magnitude of ϕad prompting a change in electrostatic potential at the surface. The thermodynamic relations between η and Δχ on the surface of a MIEC are regarded as being very complex.26
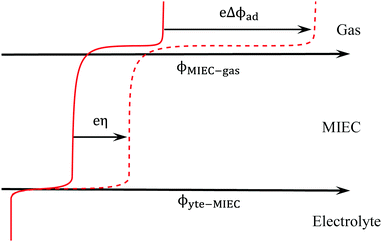 |
| | Fig. 1 Spatial variation of electrostatic potential in equilibrium (filled line) and under an applied overpotential (dashed line). | |
Studies on La0.6Sr0.4FeO3−δ suggest that the surface potential is not shifted upon an applied overpotential during the oxygen reduction reaction.27,28 However, analysing the shift in outer work function using in operando X-ray photoelectron spectroscopy (XPS) over an applied overpotential range, Chueh et al. observed that in a H2/H2O atmosphere, a significant electrostatic potential was established on the surface of Sm0.2Ce0.8O1.9 thin films.29,30 The adsorbate binding energy was invariant across the overpotential range, indicating that the origin of the electrostatic surface potential was associated with the intrinsic dipole of the adsorbate and thus independent of the potential of the MIEC electrode. When the electrode experienced anodic polarisation (+ve η) the absolute coverage of surface hydroxyls 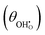 decreased, similarly, under cathodic polarisation (−ve η) the absolute coverage increased.
decreased, similarly, under cathodic polarisation (−ve η) the absolute coverage increased.
2. Thermodynamics of the ceria–gas interface
The chemistry at ceria–gas interfaces as a function of overpotential is complex and often overlooked. Recent developments in operando electrochemical analysis have allowed Chueh et al. to investigate the structure of the ceria–gas interface in a H2O/H2 atmosphere.29 To elucidate the thermodynamic driving force for the water electrolysis reaction, the global reaction 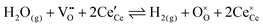 must be considered, with steps:
must be considered, with steps:
Under open circuit voltage (OCV) the water adsorption step R1 is fast and close to equilibrium even when η ≠ 0. R2 accounts for electron transfer coupled with hydrogen association, this step is considered rate limiting.22 Desorption of hydrogen gas in given by R3. To restore the defect composition of the MIEC after the hydrogen is produced, R4 and R5 are included to illustrate transport of oxygen vacancies from the electrolyte and electrons from the current collector, respectively. Most previous theoretical studies of ceria surfaces focused on the [111] termination which has the lowest surface energy.31–33 Here, the free energy of reduction of the subsurface oxygen site is lower than that of the surface site, while the most stable hydroxyl site is located at the surface site.32 We will therefore only consider adsorbate occupation of the surface site on the [111] plane. The (electro)chemical potential terms in R1 are given as:| | 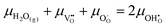 | (7) |
where the electrochemical potential for oxygen and oxygen vacancies are given as 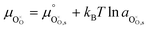 and
and 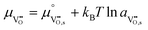 where we have cancelled the electrostatic potential terms by assuming the dilute solution approximation. The electrochemical potential of the adsorbate is defined as
where we have cancelled the electrostatic potential terms by assuming the dilute solution approximation. The electrochemical potential of the adsorbate is defined as 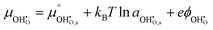 . Where the activity of a surface species, j, (aj) is given as the product of the activity coefficient, g, and the coverage, q, such that aj = γjθj, the hydroxyl coverage can be evaluated from:
. Where the activity of a surface species, j, (aj) is given as the product of the activity coefficient, g, and the coverage, q, such that aj = γjθj, the hydroxyl coverage can be evaluated from:| | 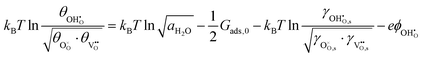 | (8) |
The adsorption energy for a dilute system where coverage is low and adsorbate–adsorbate interactions are negligible may be given as 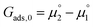 . For systems where the coverage is greater than a few per cent, competitive adsorbate considerations need to be included.34 The general competitive adsorption energy can be given as:
. For systems where the coverage is greater than a few per cent, competitive adsorbate considerations need to be included.34 The general competitive adsorption energy can be given as:
| | 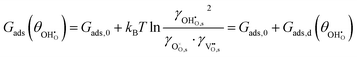 | (9) |
where
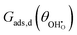
is a function which considers the coulombic dipole–dipole interactions as a function of coverage and accounts for all of the non-idealities of the adsorption step. For example, large adsorbates such as carbonates have greater adsorbate–adsorbate repulsion than smaller adsorbates like hydroxyls. Similarly, reducing the site density will push the adsorbates closer together and increase chemical potential, making adsorption less exothermic.
35,36 The free energy of adsorption is discussed in greater detail in the Section 3.
The denominator of the concentration term on the left-hand side of eqn (8) scales to the number of available dissociative adsorption sites. On the configurational entropy of the polarons and aliovalent dopants; due to their association with the oxygen vacancy defect in the surface prior to hydroxyl formation, their inclusion in eqn (8) would not have any effect on the equilibrium. Following the work of Wolverton et al., the enthalpy and entropy of reduction of bulk ceria was calculated to be 3.36 eV and 12kB, respectively,37 whereas at the surface the enthalpy and entropy of reduction was approximately 2.52 eV and 9kB, respectively.38 The lowering in enthalpy for the formation of defects at the surface can be attributed to fewer Ce–O bonds being broken, while the lowering in entropy is a result of less significant relaxation of atomic positions compared with the dense bulk. Enrichment of polarons at the ceria surface relative to the bulk has been observed by Haile et al. using XPS, where the polaron fraction was found to be between 60–80% of the total cerium sites.39
The driving force for the electrochemical water splitting reaction at the surface of a MIEC can be described by the difference in electrochemical potential of the adsorbate between equilibrium (η = 0) and quasi-equilibrium (η ≠ 0). This is understood as differences between a working electrode under current and a grounded electrode of the same material in equilibrium with the same gas atmosphere. The quasi-equilibrium state is held away from equilibrium by up to 1 V and is not to be confused with the near-equilibrium state which is a result of a perturbation on the scale of mV. R1 is fast and close to equilibrium, thus:
| | 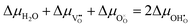 | (10) |
where the delta symbols refer to the difference between the working and reference electrodes.
17 When fast gas diffusion is assumed Δ
μH2O = 0, and the shift in electrochemical potential for oxygen and oxygen vacancies are given as
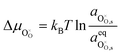
and
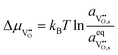
. The shift in adsorbate potential is given as:
| | 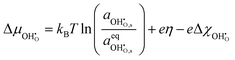 | (11) |
where the electrostatic potential of the adsorbate was expanded using eqn (6) by assuming the system is controlled by a small voltage perturbation. On expanding the electrochemical potential of the hydroxyl given in
eqn (11):
| | 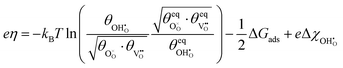 | (12) |
Eqn (12) may be used to solve for the coverage as a function of overpotential.
3. Hydroxyl dipole moment at the ceria–gas interface
While hydroxyls on the [111] surface of reduced and aliovalent doped ceria have been extensively studied using density functional theory (DFT), most work does not comment on the work function and intrinsic dipole moment of adsorbates.31–33,40 DFT calculations were carried out on 2 × 2 surfaces with a variety of hydroxyl-polaron (or aliovalent dopant) configurations (Fig. 2).
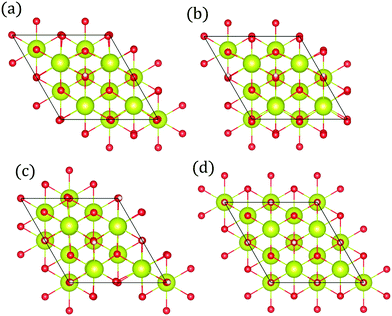 |
| | Fig. 2 Structure of lowest energy ceria after surface relaxation. (a) 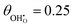 with a polaron positioned as the nearest neighbour (NN) with respect to the hydroxyl, (b) with a polaron positioned as the nearest neighbour (NN) with respect to the hydroxyl, (b) 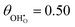 with polarons positioned NN with respect to the hydroxyls, (c) with polarons positioned NN with respect to the hydroxyls, (c) 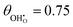 and (d) and (d) 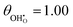 . Ce, O and H atoms are shown as yellow, red and white, respectively. . Ce, O and H atoms are shown as yellow, red and white, respectively. | |
3.1. Adsorbate-induced surface depolarisation
Attention must now be drawn to the electrostatic surface potential, which for a polar adsorbate is expressed in its dilute solution as 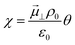 where
where ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) ⊥, ε0 and ρ0 represent the dipole moment normal to the surface, vacuum permittivity and the density of available adsorption sites. With respect to the system under inspection, hydroxyls on the ceria [111] surface align parallel to each other and normal to the surface (Fig. 2). Each dipole is exposed to the electrostatic field of all other dipoles, as are the charged defects on the ceria surface. The hydroxyl dipole moment is therefore a function of coverage ⊥(θ) = ⊥,0 − ⊥,d(θ) where ⊥,0 and ⊥,d represent the isolated dipole moment and depolarisation vector, respectively. The adsorbate-induced surface dipole moment as a function of coverage is thus given as:41
⊥, ε0 and ρ0 represent the dipole moment normal to the surface, vacuum permittivity and the density of available adsorption sites. With respect to the system under inspection, hydroxyls on the ceria [111] surface align parallel to each other and normal to the surface (Fig. 2). Each dipole is exposed to the electrostatic field of all other dipoles, as are the charged defects on the ceria surface. The hydroxyl dipole moment is therefore a function of coverage ⊥(θ) = ⊥,0 − ⊥,d(θ) where ⊥,0 and ⊥,d represent the isolated dipole moment and depolarisation vector, respectively. The adsorbate-induced surface dipole moment as a function of coverage is thus given as:41| | 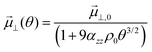 | (13) |
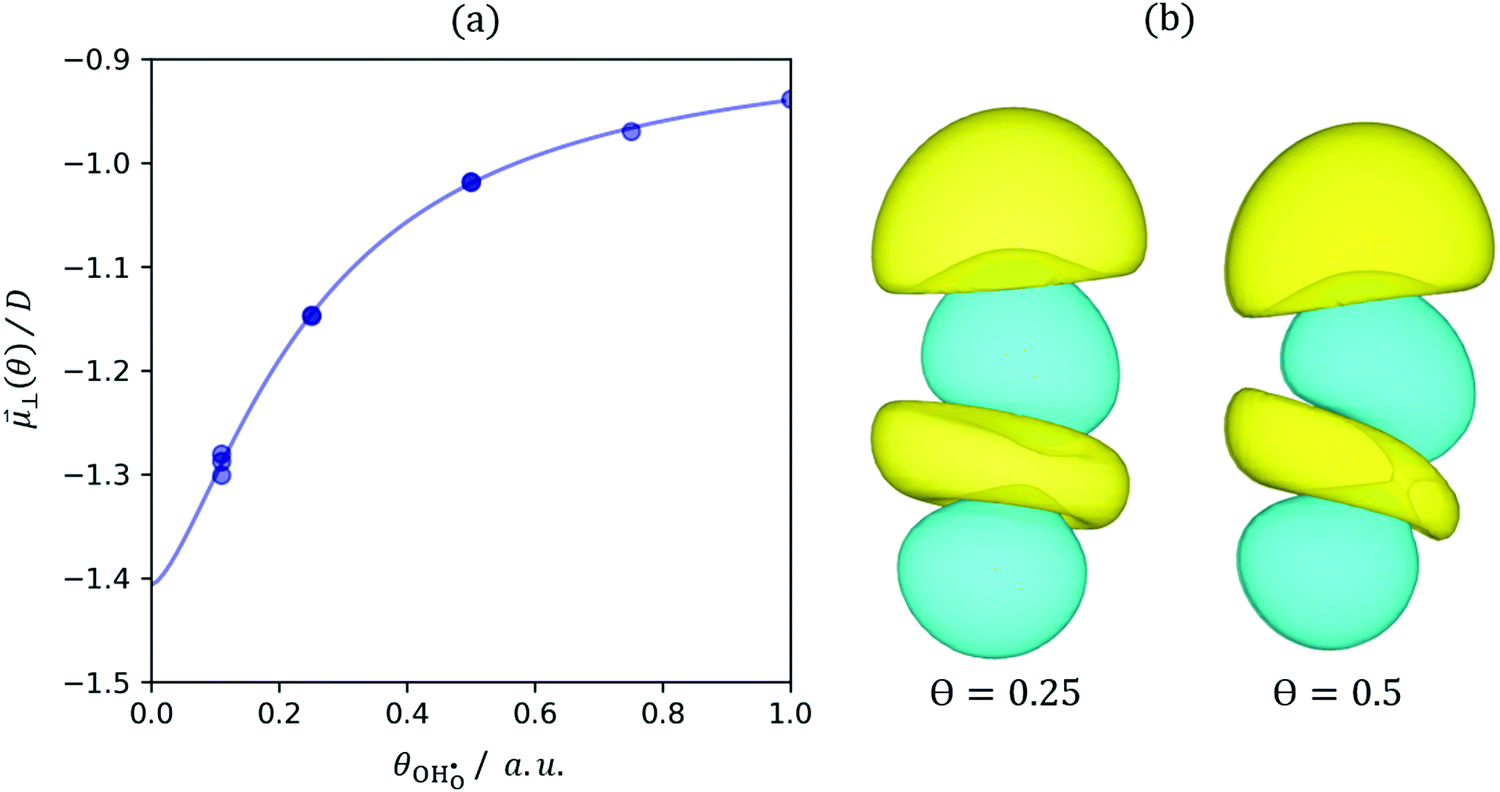
where αzz (units m3) is the O–H bond polarisability. The result of depolarisation is a shrinking in the hydroxyl bond length, thereby reducing the size of the electric field normal to the surface. Here we have shown how the surface composition influences the strength of the dipole (Fig. 3). The surfaces with coverages of 0.11, 0.25 and 0.5 allowed for arrangements in which the polaron/dopant was nearest neighbour, NN, or next nearest neighbour, NNN, with respect to the hydroxyl. However, the dipole moment was not significantly influenced by the defect arrangement, thereby supporting the theory that the essence of the electrostatic surface potential is the intrinsic dipole moment.29
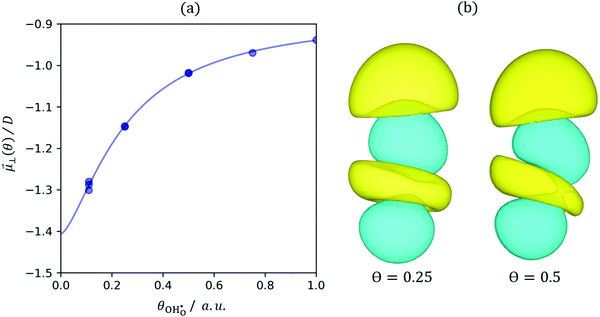 |
| | Fig. 3 (a) Calculated hydroxyl dipole moment normal to the ceria[111] termination as a function of coverage. The line fits to eqn (13). (b) Charge density difference isosurface (0.005 Bohr−3) of the hydroxyl on the ceria surface where negative and positive charge density is represented by blue and yellow isosurfaces, respectively. O site bottom and H site top. | |
3.2. Adsorption free energy
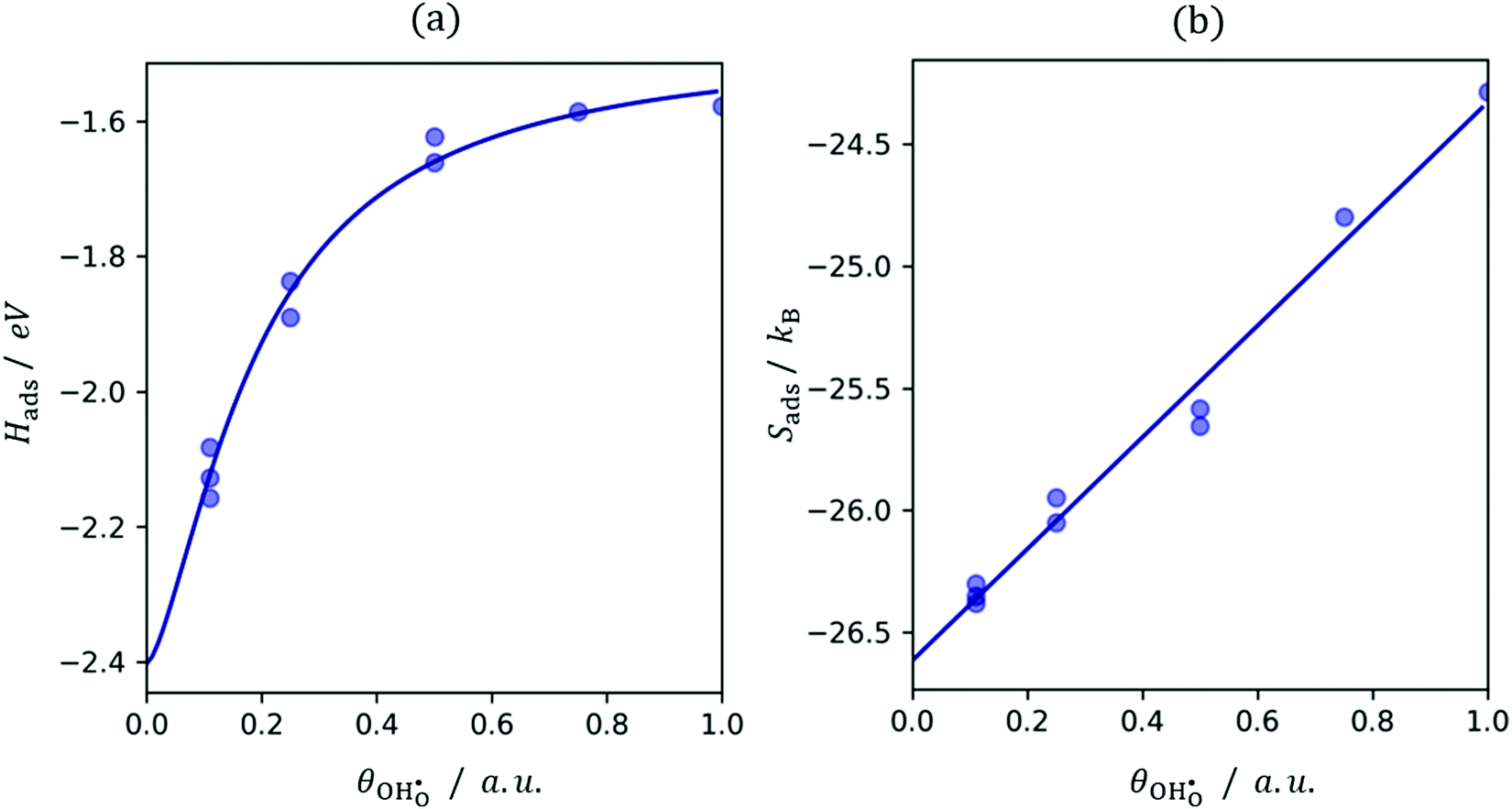
Due to the lateral interactions which provoke dipole–dipole depolarisation, the electrostatic field of all other dipoles destabilises the hydroxyl upon adsorption. This is demonstrated by the adsorption enthalpy Hads = Eelads + ZPEads as a function of coverage (Fig. 4a) where, Eel and ZPE represent the electronic energy and zero-point energy, respectively. The entropy of the adsorbate is calculated from the normal mode frequencies, vj, by the harmonic limit equation:35| | 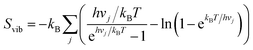 | (14) |
While the entropy of adsorption Sads is mostly dominated by the larger translational entropy of the gas phase species, the effect of the adsorbate–adsorbate interactions on Sads appears to be linear as a function of coverage (Fig. 4b). Lateral constraints on the adsorbate increase with coverage and reduce the energy of vibrational modes perpendicular to the surface. However, weakening of the ionic bond due to depolarisation enhances the energy of the vibrational mode of the displaced proton normal to the surface. The free energy of adsorption Gads = Hads − TSads can therefore be expressed as:| | 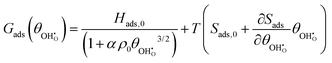 | (15) |
where α is an arbitrary constant. Fitting the energy of adsorption with the same coverage dependence as depolarisation, shows that it is the lateral dipole–dipole interactions which dominate the thermodynamic properties of competitive adsorption relative to defect interactions within the surface.
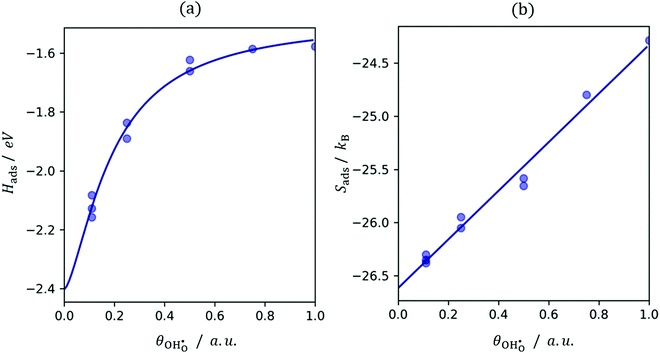 |
| | Fig. 4 Hydroxyl coverage dependence on (a) adsorption enthalpy fit to (15) and (b) adsorption entropy with linear fitting. | |
4. Analysis of model with previous experimental data
To elucidate the double-layer structure at the ceria–gas interface, the following section discusses the interpretation of previously reported operando XPS data from Sm0.2Ce0.8O1.9 thin films in a H2/H2O atmosphere using the equations discussed in the introduction.21,22,29,30
The work function, Φ, defines the energy required to remove an electron from the Fermi level, Ef, through the surface and into the vacuum level outside of the electrode, Evac. The work function can be split into two parts: the inner work function, Φin, and outer work function, Φout. The former correlates to the chemical potential of electrons in the bulk of the electrode, while the latter is an electrical potential term which corresponds to an electrostatic potential step at the electrode surface. For an electronically conductive material, a change in applied overpotential will result in a linear change in inner work function ΔΦin = −eη. The outer work function of a material will be influenced by an applied overpotential as long as the effective double layer at the electrode–gas interface has some overpotential dependence:
By considering the most stable ceria surface to be the [111] ceria termination, there are two oxygen sites; surface and subsurface with an equal site density of approximately 7.69 sites per nm
2.
32 Hydration of the surface site is considerably more thermodynamically stable so we do not consider occupation of the subsurface site.
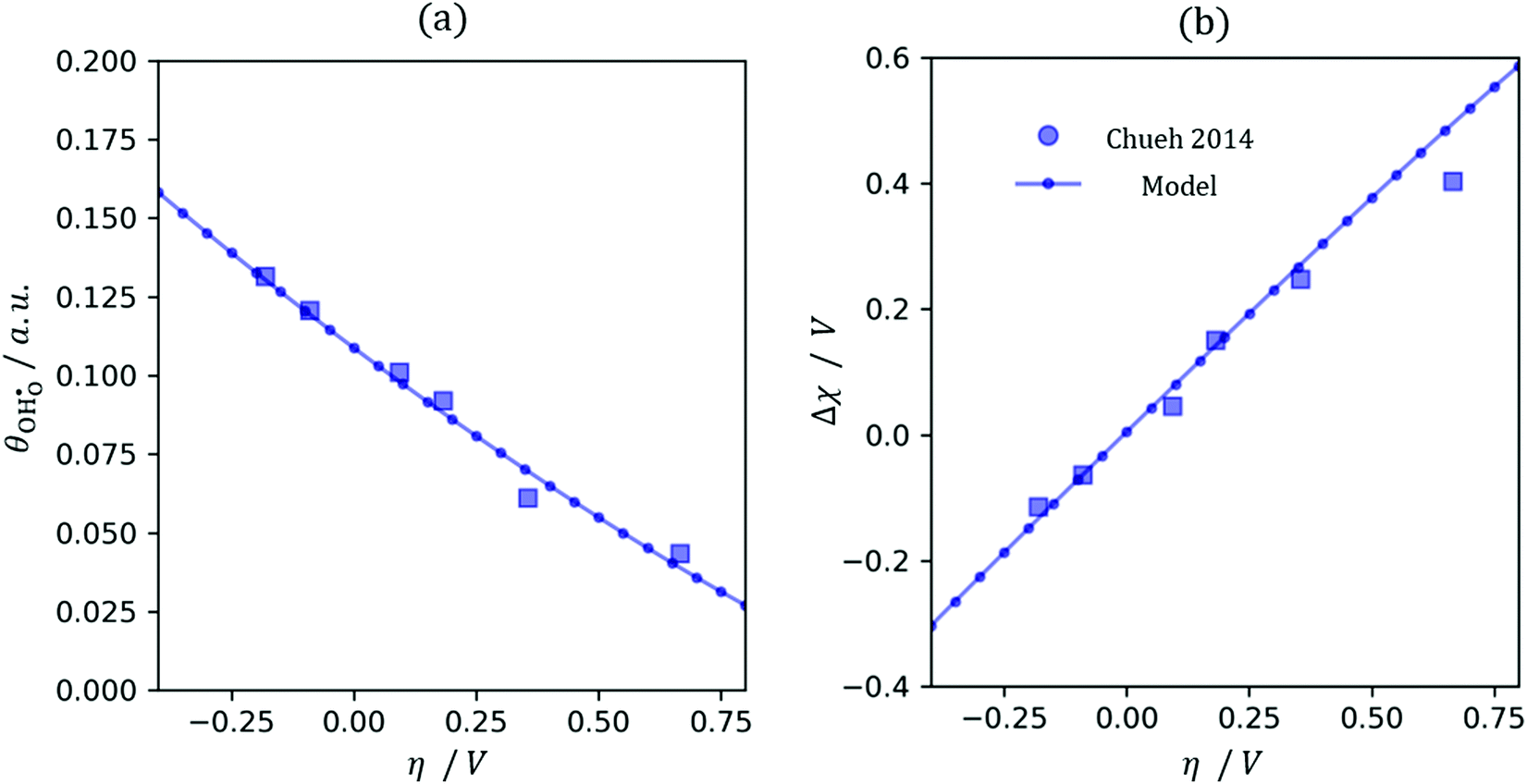
32Eqn (12) was used to solve for the coverage as a function of overpotential (
Fig. 5a). We have made the simplification in which all defects (
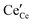
and
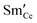
) have identical adsorption and electrostatic properties. In reality this is not the case, however more extensive multi-scale modelling would be required to evaluate the complex bimodal properties of the doped-reduced surface which is beyond the scope of the current study.
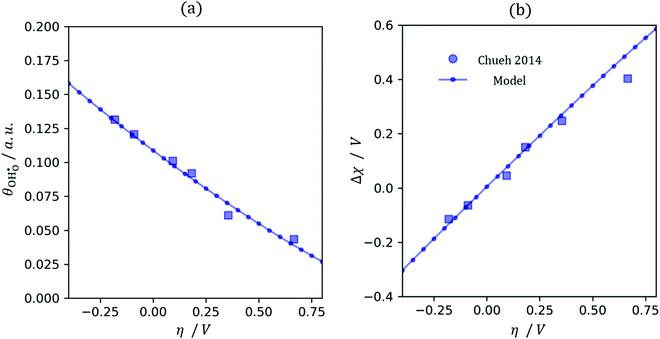 |
| | Fig. 5
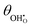 (a) and χ (b) on a Sm0.2Ce0.8O1.9–gas interface as a function of overpotential at 500 °C in 1:8:4 H2O:H2:Ar. Squares represent data collected by APXPS measurements (Chueh et al.).29 The dotted and filled lines represent the numerical and analytical solutions to (12), respectively. (a) and χ (b) on a Sm0.2Ce0.8O1.9–gas interface as a function of overpotential at 500 °C in 1:8:4 H2O:H2:Ar. Squares represent data collected by APXPS measurements (Chueh et al.).29 The dotted and filled lines represent the numerical and analytical solutions to (12), respectively. | |
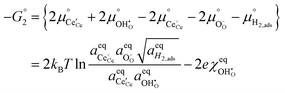
To determine 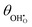 at open circuit voltage (OCV) we consider R1, R2 and R3 to be in equilibrium. Thus:
at open circuit voltage (OCV) we consider R1, R2 and R3 to be in equilibrium. Thus:
| | 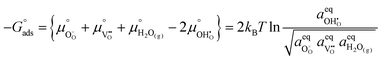 | (17) |
| |  | (18) |
| | 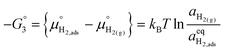 | (19) |
The thermodynamic constants when 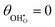 are given in Table 1. In Section 3.2 we demonstrated that dipole–dipole interactions increased enthalpy of adsorption in R1, thus making the adsorption reaction less exothermic as
are given in Table 1. In Section 3.2 we demonstrated that dipole–dipole interactions increased enthalpy of adsorption in R1, thus making the adsorption reaction less exothermic as 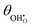 increases. The same effect can be observed with R2, whereby the enthalpy of electron transfer decreases as
increases. The same effect can be observed with R2, whereby the enthalpy of electron transfer decreases as 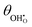 increases. This was demonstrated by simulating R2 using 2 × 2
increases. This was demonstrated by simulating R2 using 2 × 2 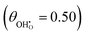 and 3 × 3
and 3 × 3 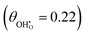 surfaces, where the change in electronic energy was found to be Eel2 = −2.39 eV and Eel2 = −2.86 eV, respectively. The adsorbed hydrogen dimer H2,ads is weakly bound to the ceria surface, resulting in a relatively small associated enthalpy for R3. However, the large entropy associated with the formation of a gas species means the free energy
surfaces, where the change in electronic energy was found to be Eel2 = −2.39 eV and Eel2 = −2.86 eV, respectively. The adsorbed hydrogen dimer H2,ads is weakly bound to the ceria surface, resulting in a relatively small associated enthalpy for R3. However, the large entropy associated with the formation of a gas species means the free energy 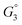 will be negative under operational conditions and the coverage of H2,ads is expected to be very low. Eqn (17)–(19) can be solved simultaneously to estimate the coverage of both adsorbates and defects (Fig. 5a).
will be negative under operational conditions and the coverage of H2,ads is expected to be very low. Eqn (17)–(19) can be solved simultaneously to estimate the coverage of both adsorbates and defects (Fig. 5a).
Table 1 Enthalpy and entropy for R1, R2 and R3 when 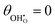 (T = 773 K)
(T = 773 K)
| Reaction, i |
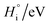
|
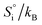
|
| R1 |
−2.39 |
−26.47 |
| R2 |
2.99 |
−0.567 |
| R3 |
0.08 |
11.11 |
Experimentally determining the coverage of an adsorbate on the polycrystalline samples used in ref. 29 is challenging and is limited by knowledge of the inelastic mean free path in the substrate material.42 The additional uncertainty from peak fitting and topological factors means that the error in determining the adsorbate coverage is substantially higher than calculating the shift in work function.43 The model also captures the generally linear 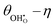 , demonstrating that the electrostatic component on the adsorbate electrochemical potential is dominant, something originally proposed by Fleig.18
, demonstrating that the electrostatic component on the adsorbate electrochemical potential is dominant, something originally proposed by Fleig.18
By combining eqn (12) and (15) the ΔΦout–η relation was numerically evaluated. The solution illustrates the expected linear-like character with respect to the gradient (Fig. 5b). Unlike measuring the hydroxyl coverage, the work function is considered to be more accurate and is not affected by topographical imperfections.42,43 The experimentally determined outer work function data is seen to give a modest validation of the model and constants used in this study. Importantly, Fig. 5b demonstrates that the complex Δχ–η relationship is approximately linear within the operational overpotential range of a SOC. As the overpotential is increased it is understood that the coverage of polar adsorbates increases with a linear-like tendency.
5. Outlook on electrode kinetics
In this section, an equation to describe the parameters discussed in Section 3 is derived to explain the current density resulting from the coupled electron transfer and hydrogen association step (R2) at the MIEC–gas interface.18,26,28 Thus, the general reaction rate per surface site for the forward and backward reactions is given as:| | 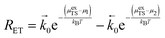 | (20) |
where μ1 and μ2 represents the electrochemical potential of the reactants and products, respectively, μexTS represents the excess chemical potential of the transition state, 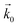 and
and 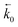 represent the forward and reverse rate constants, respectively. As a consequence of equilibrium
represent the forward and reverse rate constants, respectively. As a consequence of equilibrium 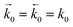 .25 The electrochemical potential of the transition state is defined following well-known Butler–Volmer kinetics:
.25 The electrochemical potential of the transition state is defined following well-known Butler–Volmer kinetics:| | 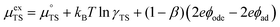 | (21) |
where β is defined as the symmetry factor 0 < β < 1.25 Under OCV the reactants and products of R2 are in equilibrium as shown in eqn (18). This expression is analogous to the Nernst equation which determines the voltage of an electrode–electrolyte interface in equilibrium.25 Using the definition for current density j = −neR, the equilibrium potentials are utilized to formulate the exchange current density, j0, by substituting (18) and (21) into (20):| | 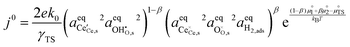 | (22) |
In this format the exchange current density appears to be a product of species activities. An additional simplification step yields the exchange current density as a function of electrostatic surface potential at equilibrium:
| | 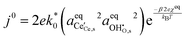 | (23) |
where
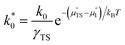
. The exchange current density is dependent not only on activity, as recognised in conventional Butler–Volmer kinetics, but also on the electrostatic potential at equilibrium,
χeq. Elucidating
γTS requires knowledge of the transition state, outlined by Bazant
et al.44 The hydroxyl coverage at equilibrium and the exchange current density as a function of partial pressure is displayed in
Fig. 6. On increasing
pH
2 the equilibrium hydroxyl coverage is increased as the R
2 reaction is driven backwards.
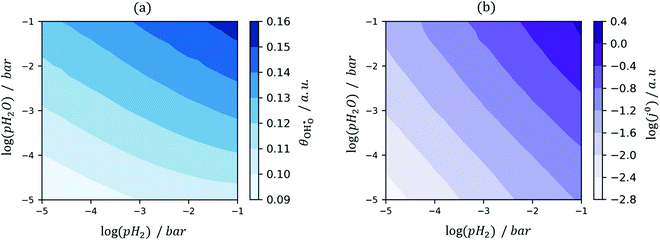 |
| | Fig. 6
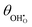 (a) and j0 (b) at the Sm0.2Ce0.8O1.9–gas interface as a function of steam and hydrogen partial pressure at 500 °C. (a) and j0 (b) at the Sm0.2Ce0.8O1.9–gas interface as a function of steam and hydrogen partial pressure at 500 °C. | |
On increasing pH2O the hydroxyl coverage constantly increases as expected, due to filling of extrinsic oxygen vacancies. Because increasing pH2O relative to pH2 hinders oxygen reduction, we observe a complex pH2O–j0 relationship, where the most important parameter is the exponentially scaled χeq. From a material selection point of view, one should try to maximise the χeq by tailoring the surface and cell operation to enhance the coverage of polar adsorbates. By extracting the equilibrium potentials to form the exchange current density, the current density can be given as:
| | 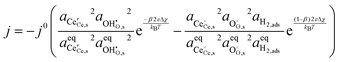 | (24) |
The overpotential relationship of
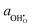
and Δ
χ are taken from
eqn (12). Due to the relatively large dipole moment of the hydroxyl on the ceria[111] surface, the change in adsorbate activity under an applied overpotential is linear and relatively small compared to the other exponential terms.
Eqn (24) may be experimentally evaluated through small electrical perturbations close to OCV which generates a linear response where all species activities remain close to equilibrium as Δ
χ → 0:
26| | 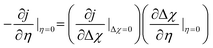 | (25) |
The first order partial derivative is separated into two relationships: j–Δχ and Δχ–η. The former term can be obtained by using a Maclaurin series expansion of eqn (23) to obtain the linear equation:
| | 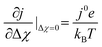 | (26) |
which is scaled with the exchange current density given in
eqn (23). While the latter term is projected as the gradient in
Fig. 7c and f where
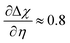
at OCV from the experimental data.
29 The gradient
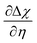
is dependent on the equilibrium potential.
45 Under low
pH
2O and
pH
2,
χeq is low and
χ experiences noticeable depletion as η is increased, relative to when the surface is “buffered” and
χ does not experience extensive loss under anodic polarization.
18,34,46
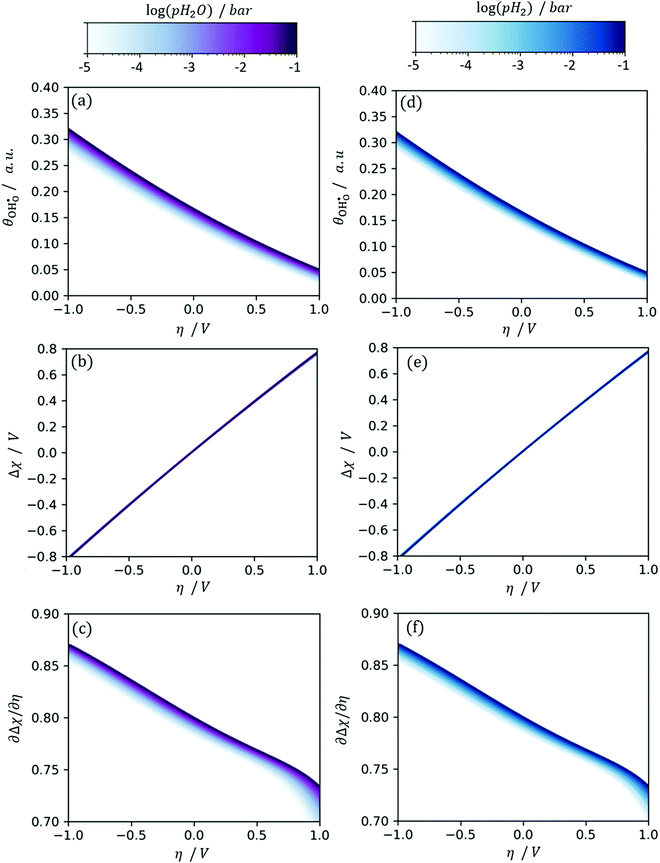 |
| | Fig. 7 (a) 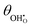 , (b) Δχ and (c) , (b) Δχ and (c) 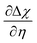 at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−5–10−1 bar and pH2 = 10−2 bar). (d) at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−5–10−1 bar and pH2 = 10−2 bar). (d) 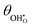 , (e) Δχ and (f) , (e) Δχ and (f) 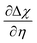 at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−2 bar and pH2 = 10−5–10−1 bar). at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−2 bar and pH2 = 10−5–10−1 bar). | |
6. Conclusion
Modelling of electrochemical double layers in thermodynamic non-equilibrium has been extensively studied at the electrolyte–electrode contact. The electrostatic surface potential at the MIEC–gas interface is relatively straightforward to model and appropriate in describing the driving force for charge exchange at the electrode surface. By adapting the electrochemical potential of molecular adsorbates to account for the intrinsic dipole moment, the adsorbate coverage, electrostatic surface potential and overpotential of a ceria–gas interface have been expressed in a parallel treatment. This theory was validated against in operando XPS data from previous reports collected from thin film Sm0.2Ce0.8O1.9 in a H2/H2O atmosphere, where the χ–η relationship was captured with the model developed.22,29,30 By virtue, the relatively strong intrinsic dipole moment of adsorbed hydroxyls was found to be the essence of the electrostatic surface potential and the linear-like 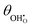 –η relationship. Butler–Volmer kinetic equations were then derived to account for the electrostatic potential at the ceria–gas interface, where the current density was shown to be extensively dependent on χeq. The MIEC–gas interface model is ubiquitous for all systems where the electrostatic potential at the electrode surface is not described as a diffuse charge layer. Moreover, by understanding the electronic structure of the adsorbate, electrodes and operational conditions could in future be adapted to maximise the charge transfer kinetics at the MIEC–gas interface.
–η relationship. Butler–Volmer kinetic equations were then derived to account for the electrostatic potential at the ceria–gas interface, where the current density was shown to be extensively dependent on χeq. The MIEC–gas interface model is ubiquitous for all systems where the electrostatic potential at the electrode surface is not described as a diffuse charge layer. Moreover, by understanding the electronic structure of the adsorbate, electrodes and operational conditions could in future be adapted to maximise the charge transfer kinetics at the MIEC–gas interface.
7. Computational methods
Spin-polarised density functional theory (DFT) calculations were carried out using the VASP code.47 The ionic cores were described by PAW potentials and the wavefunctions were expanded in plane waves with an energy cut off at 520 eV.48 The PBE-generalized gradient approximation (GGA) was used.49,50 To describe the Ce 4f electrons, DFT+U was implemented using the Dudarev treatment, where Ueff = 5 eV from previous work.51–53 The [111] surface was modelled as a symmetric O-terminated slab with a thickness of 12 atomic layers and 3 × 3 cell expansion in the lateral directions. The bottom three atomic layers were fixed during geometry optimisations. The periodic images of the slab were separated by a vacuum region of about 15 Å. The convergence parameters for electronic and ionic relaxation were set to 10−7 eV and 10−4 eV Å−1, respectively, to guarantee a sufficient accuracy of the calculated forces. The dipole correction was used to decouple the electrostatic interaction between the periodic images. The calculations were performed with a 4 × 4 × 1 Monkhorst Pack grid. For gases, electronic calculations were carried out in a 13 × 14 × 15 Å box. Vibrational frequencies at the gamma point were calculated using the ASE package with four finite difference displacements of 0.01 Å for each vibrational mode.54 Surface oxide sites and adsorbates were included in the calculation. Integration of the charge density difference in the x and y planes was carried out using the vaspkit code.55 Integration of the plane-averaged electron density difference was then carried out by fitting with a Fourier series followed by trapezoid integration.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix – nomenclature
| Symbol |
Description |
Unit |
|
a
e
|
Activity of species j |
1 |
|
c
j
|
Concentration of species j |
1 |
| cc |
Current collector, electron conducting phase |
— |
|
G
|
Gibbs free energy |
eV |
|
H
|
Enthalpy |
eV |
|
j
|
Current density |
A cm−2 |
|
j
0
|
Exchange current density |
A cm−2 |
|
k
0
|
Rate constant |
s−1 |
|
k
B
|
Boltzmann constant |
eV K−1 |
| MIEC |
Mixed ionic–electronic conducting phase |
— |
|
R
|
Rate of reaction |
s−1 |
|
S
|
Entropy |
k
B
|
| T |
Temperature |
K |
|
v
j
|
Normal vibrational mode |
Hz |
| yte |
Electrolyte, ion conducting phase |
— |
Greek symbols
| Symbol |
Description |
Unit |
|
α
zz
|
Bond polarizability normal to the surface |
m3 |
|
β
|
Electron transfer symmetry factor |
1 |
|
γ
j
|
Activity coefficient |
1 |
| Δ(j) |
Perturbation of quantity j relative to the equilibrium with the gas phase and grounded electrode |
1 |
|
ε
0
|
Vacuum permittivity |
e2 V−1 m−1 |
|
η
|
Overpotential |
V |
|
θ
j
|
Surface coverage of species j |
1 |
|
μ
j
|
Electrochemical potential of charged species j, or chemical potential of neutral species j |
eV |
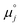
|
Standard potential of species j |
eV |
|
μ
exe
|
Excess chemical potential of species j |
eV |
|
⊥
|
Dipole moment |
D or C m |
|
ρ
0
|
Desnity of available adsorption sites |
m−2 |
|
ϕ
e
|
Electric potential of species j |
V |
|
Φ
|
Work function |
eV |
|
χ
|
Electrostatic surface potential |
V |
Acknowledgements
This work was supported by Ceres Power Ltd. All first principle calculations were performed using the Imperial College Research Computing Service (DOI: 10.14469/hpc/2232). We are also grateful for the discussion with Dr Alex Shard from the National Physical Laboratory, Teddington, UK, on analysing operando XPS data. We also acknowledge the support of the EPSRC through the award of a platform grant (EP/R002010/1) and through support for the Centre for Doctoral Training in the Advanced Characterisation of Materials (EP/L015277/1).
References
- M. C. Doppler, J. Fleig, M. Bram and A. K. Opitz, J. Power Sources, 2018, 380, 46–54 Search PubMed.
- K. Yamaji, N. Sakai, M. Ishikawa, H. Yokokawa and M. Dokiya, Ionics, 1997, 3, 67–74 Search PubMed.
- S. Primdahl and M. Mogensen, J. Electrochem. Soc., 1997, 144, 3409–3419 Search PubMed.
- J. H. Nam and D. H. Jeon, Electrochim. Acta, 2006, 51, 3446–3460 Search PubMed.
- H. Yokokawaa and T. Kawadab, Solid State Ionics, 1996, 2738, 1259–1266 Search PubMed.
- N. Sakai, K. Yamaji, T. Horita, H. Kishimoto, Y. P. Xiong and H. Yokokawa, Solid State Ionics, 2004, 175, 387–391 Search PubMed.
- M. Brown, S. Primdahl and M. Mogensen, J. Electrochem. Soc., 2000, 147, 475 Search PubMed.
- S. P. Jiang and S. P. S. Badwal, J. Electrochem. Soc., 1997, 144, 3777 Search PubMed.
- H. Yokokawa, T. Horita, N. Sakai, K. Yamaji, M. E. Brito, Y. P. Xiong and H. Kishimoto, Solid State Ionics, 2004, 174, 205–221 Search PubMed.
- A. Bieberle and L. J. Gauckler, Solid State Ionics, 2000, 135, 337–345 Search PubMed.
- T. Horita, H. Kishimoto, K. Yamaji, Y. Xiong, N. Sakai, M. E. Brito and H. Yokokawa, Solid State Ionics, 2006, 177, 1941–1948 Search PubMed.
- W. G. Bessler, S. Gewies and M. Vogler, Electrochim. Acta, 2007, 53, 1782–1800 Search PubMed.
- L. Holzer, B. Iwanschitz, T. Hocker, B. Munch, M. Prestat, D. Wiedenmann, U. Vogt, P. Holtappels, J. Sfeir, A. Mai and T. Graule, J. Power Sources, 2011, 196, 1279–1294 Search PubMed.
- M. Kishimoto, M. Lomberg, E. Ruiz-Trejo and N. P. Brandon, Electrochim. Acta, 2016, 190, 178–185 Search PubMed.
- L. Sun, X. Huang, L. Wang and A. Janotti, Phys. Rev. B, 2017, 95, 1–8 Search PubMed.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko and R. Schmitt, Chem. Soc. Rev., 2020, 49, 554–592 Search PubMed.
- A. Nenning, C. Bischof, J. Fleig, M. Bram and A. K. Opitz, Energies, 2020, 13, 1–30 Search PubMed.
- J. Fleig, Phys. Chem. Chem. Phys., 2005, 7, 2027–2037 Search PubMed.
- S. B. Adler, X. Y. Chen and J. R. Wilson, J. Catal., 2007, 245, 91–109 Search PubMed.
- Z. Guan, D. Chen and W. C. Chueh, Phys. Chem. Chem. Phys., 2017, 19, 23414–23424 Search PubMed.
- W. C. Chueh, A. H. Mcdaniel, M. E. Grass, Y. Hao, N. Jabeen, Z. Liu, S. M. Haile, K. F. Mccarty, H. Bluhm and F. El Gabaly, Chem. Mater., 2012, 24, 1876–1882 Search PubMed.
- Z. A. Feng, M. L. Machala and W. C. Chueh, Phys. Chem. Chem. Phys., 2015, 17, 12273–12281 Search PubMed.
- C. Zhang, Y. Yu, M. E. Grass, C. Dejoie, W. Ding, K. Gaskell, N. Jabeen, Y. P. Hong, A. Shavorskiy, H. Bluhm, W. X. Li, G. S. Jackson, Z. Hussain, Z. Liu and B. W. Eichhorn, J. Am. Chem. Soc., 2013, 135, 11572–11579 Search PubMed.
- C. Zhang, Y. Yu, M. E. Grass, C. Dejoie, W. Ding, K. Gaskell, N. Jabeen, Y. P. Hong, A. Shavorskiy, H. Bluhm, W. Li, G. S. Jackson, Z. Hussain, Z. Liu and B. W. Eichhorn, J. Am. Chem. Soc., 2013, 135, 11572–11579 Search PubMed.
- M. Z. Bazant, Acc. Chem. Res., 2013, 46, 1144–1160 Search PubMed.
- W. C. Chueh and S. M. Haile, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 313–341 Search PubMed.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Hävecker, A. Knop-Gericke, G. Rupprechter, B. Klötzer and J. Fleig, J. Phys. Chem. C, 2016, 120, 1461–1471 Search PubMed.
- A. Schmid and J. Fleig, J. Electrochem. Soc., 2019, 166, F831–F846 Search PubMed.
- Z. A. Feng, C. Balaji Gopal, X. Ye, Z. Guan, B. Jeong, E. Crumlin and W. C. Chueh, Chem. Mater., 2016, 28, 6233–6242 Search PubMed.
- Z. A. Feng, F. El Gabaly, X. Ye, Z. Shen and W. C. Chueh, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- A. R. Symington, M. Molinari, S. Moxon, J. M. Flitcroft, D. C. Sayle and S. C. Parker, J. Phys. Chem. C, 2020, 124, 3577–3588 Search PubMed.
- H. A. Hansen and C. Wolverton, J. Phys. Chem. C, 2014, 118, 27402–27414 Search PubMed.
- T. Wu, Q. Deng, H. A. Hansen and T. Vegge, J. Phys. Chem. C, 2019, 123, 5507–5517 Search PubMed.
- J. Fleig, R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2007, 9, 2713–2723 Search PubMed.
- J. Yang, J. M. Polfus, Z. Li, H. L. Tuller and B. Yildiz, Chem. Mater., 2020, 32, 5483–5492 Search PubMed.
- J. M. Polfus, B. Yildiz, H. L. Tuller and R. Bredesen, J. Phys. Chem. C, 2018, 122, 307–314 Search PubMed.
- S. Grieshammer and M. Martin, J. Mater. Chem. A, 2017, 5, 9241–9249 Search PubMed.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, J. Phys. Chem. C, 2016, 120, 16271–16289 Search PubMed.
- W. Yuan, Q. Ma, Y. Liang, C. Sun, K. V. L. V. Narayanachari, M. J. Bedzyk, I. Takeuchi and S. M. Haile, J. Mater. Chem. A, 2020, 8, 9850–9858 Search PubMed.
- X. Aparicio-Anglès, A. Roldan and N. H. De Leeuw, Chem. Mater., 2015, 27, 7910–7917 Search PubMed.
- O. L. A. Monti, J. Phys. Chem. Lett., 2012, 3, 2342–2351 Search PubMed.
- A. G. Shard, J. Vac. Sci. Technol., A, 2020, 38, 041201 Search PubMed.
- A. G. Shard, J. Wang and S. J. Spencer, Surf. Interface Anal., 2009, 41, 541–548 Search PubMed.
- D. Fraggedakis, M. McEldrew, R. B. Smith, Y. Krishnan, Y. Zhang, P. Bai, W. C. Chueh, Y. Shao-Horn and M. Z. Bazant, Electrochim. Acta, 2020, 367, 137432 Search PubMed.
- W. G. Bessler, J. Warnatz and D. G. Goodwin, Solid State Ionics, 2007, 177(39–40), 3371–3383 Search PubMed.
- J. Fleig and J. Jamnik, J. Electrochem. Soc., 2005, 152, E138 Search PubMed.
- M. Zhu, M. Yu, M. Xia, B. Li, P. Yu, S. Gao, Z. Qi, L. Liu, Y. Chen and H. Guan, Proc. ACM Symp. Appl. Comput., 2011, 554–559 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- C. W. M. Castleton, A. Lee and J. Kullgren, J. Phys. Chem. C, 2019, 123, 5164–5175 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- H. Zhou, D. Wang and X. Q. Gong, Phys. Chem. Chem. Phys., 2020, 22, 7738–7746 Search PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 Search PubMed.
- W. M. Temmerman, A. P. Sutton, S. L. Dudarev, G. A. Botton, S. Y. Savrasov and Z. Szotek, Phys. Status Solidi A, 1998, 166, 429–443 Search PubMed.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 1–30 Search PubMed.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W.-T. Geng, arXiv, 2019, 1–33 Search PubMed.
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ieuan D.
Seymour
a,
Robert T.
Leah
b,
Subhasish
Mukerjee
b,
Mark
Selby
b and
Stephen J.
Skinner
a,
Ieuan D.
Seymour
a,
Robert T.
Leah
b,
Subhasish
Mukerjee
b,
Mark
Selby
b and
Stephen J.
Skinner

 ,
,  ,
, 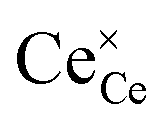 and
and 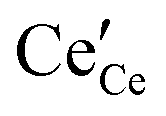 represent an oxide-ion at the anion site, a doubly charged oxygen vacancy, a cerium ion on the cation site, and small polaron (localised electron). Upon reduction of the lattice (eqn (1) forward) additional highly mobile defects are created.15,16 The increase in defect concentration induces a significant enhancement in bulk n-type conductivity of lattice, resulting in MIEC character. The bulk transport in doped ceria is relatively fast, and as such, the bulk-to-surface transport is not considered to be rate limiting for electrode processes.17,18 The popular electrode bracket notation
represent an oxide-ion at the anion site, a doubly charged oxygen vacancy, a cerium ion on the cation site, and small polaron (localised electron). Upon reduction of the lattice (eqn (1) forward) additional highly mobile defects are created.15,16 The increase in defect concentration induces a significant enhancement in bulk n-type conductivity of lattice, resulting in MIEC character. The bulk transport in doped ceria is relatively fast, and as such, the bulk-to-surface transport is not considered to be rate limiting for electrode processes.17,18 The popular electrode bracket notation  allows the chemical potential of MIEC defects to be considered as the difference between the electrochemical potential of electrons in the current collector (electron conducting phase),
allows the chemical potential of MIEC defects to be considered as the difference between the electrochemical potential of electrons in the current collector (electron conducting phase), 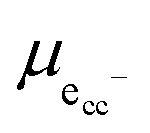 , and oxygen in the electrolyte (ionic conducting phase),
, and oxygen in the electrolyte (ionic conducting phase), 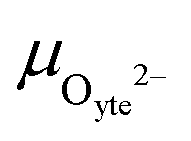 :19,20
:19,20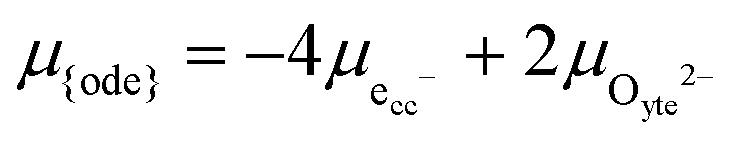
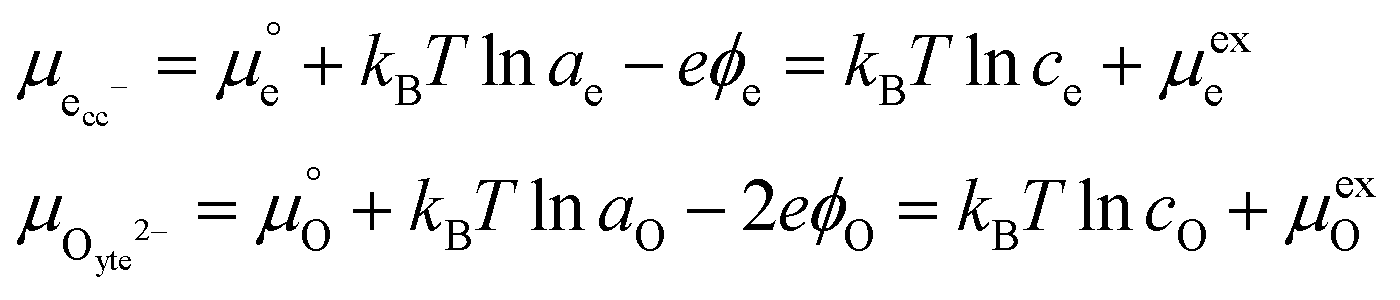
 , activity potential, kBT
, activity potential, kBT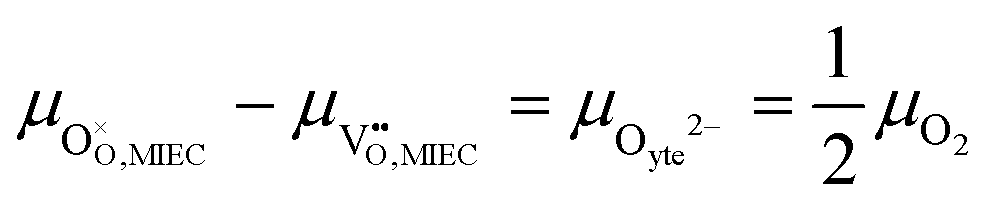 , and therefore the concentration of oxygen vacancies remains pinned.21,22 Therefore, the local activation overpotential η is given as:
, and therefore the concentration of oxygen vacancies remains pinned.21,22 Therefore, the local activation overpotential η is given as:

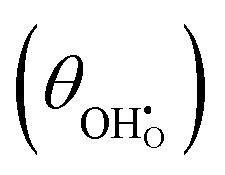 decreased, similarly, under cathodic polarisation (−ve η) the absolute coverage increased.
decreased, similarly, under cathodic polarisation (−ve η) the absolute coverage increased.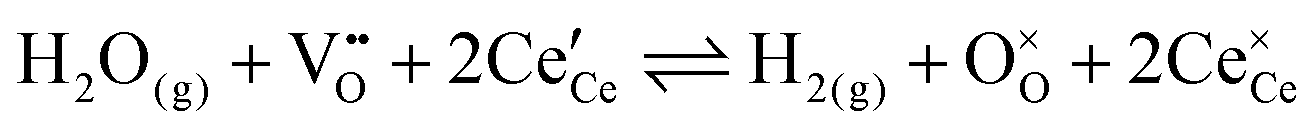 must be considered, with steps:
must be considered, with steps: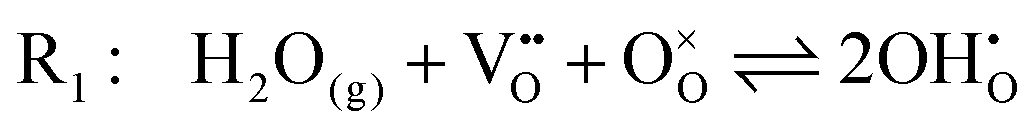
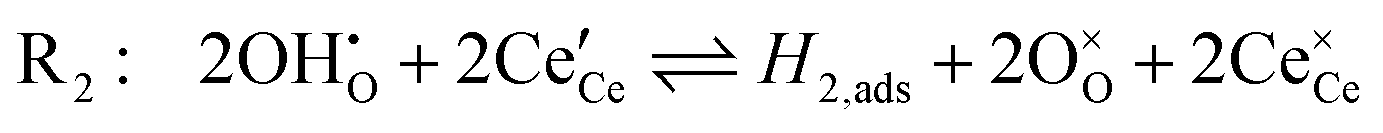
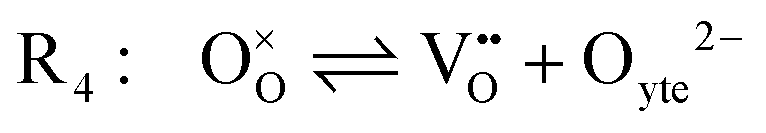
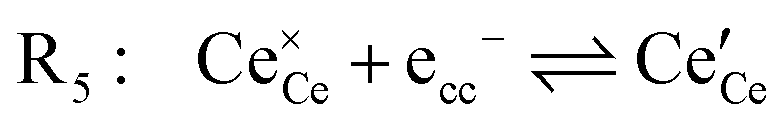
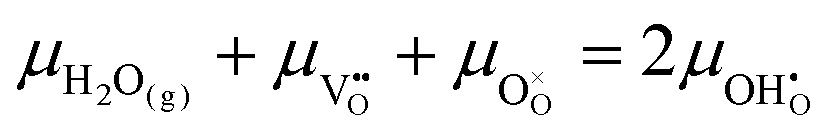
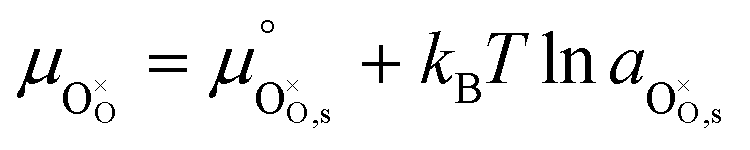 and
and 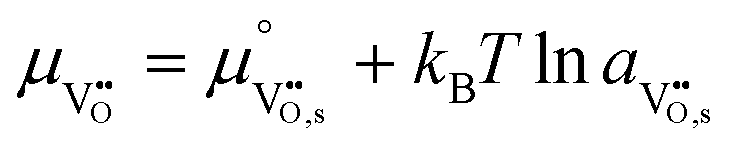 where we have cancelled the electrostatic potential terms by assuming the dilute solution approximation. The electrochemical potential of the adsorbate is defined as
where we have cancelled the electrostatic potential terms by assuming the dilute solution approximation. The electrochemical potential of the adsorbate is defined as 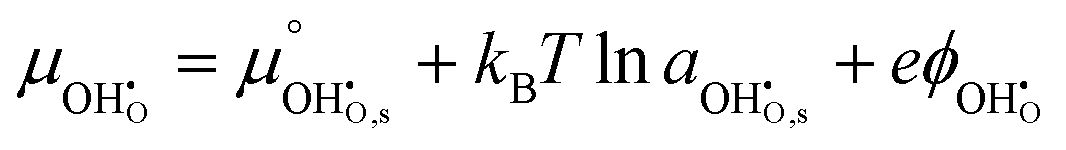 . Where the activity of a surface species, j, (aj) is given as the product of the activity coefficient, g, and the coverage, q, such that aj = γjθj, the hydroxyl coverage can be evaluated from:
. Where the activity of a surface species, j, (aj) is given as the product of the activity coefficient, g, and the coverage, q, such that aj = γjθj, the hydroxyl coverage can be evaluated from: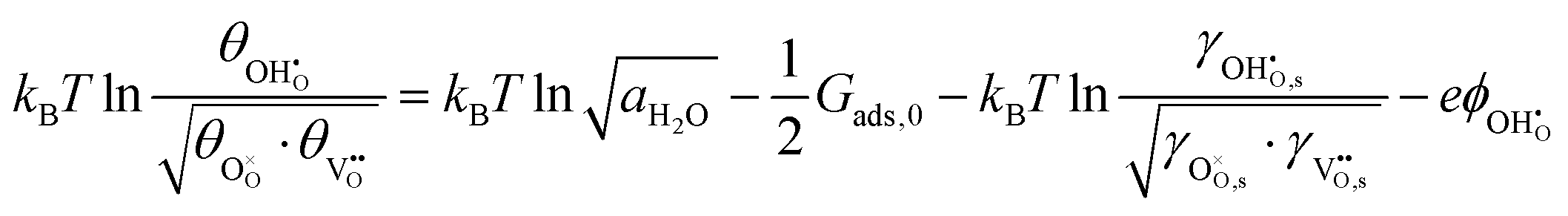
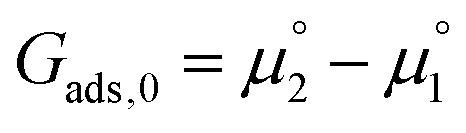 . For systems where the coverage is greater than a few per cent, competitive adsorbate considerations need to be included.34 The general competitive adsorption energy can be given as:
. For systems where the coverage is greater than a few per cent, competitive adsorbate considerations need to be included.34 The general competitive adsorption energy can be given as: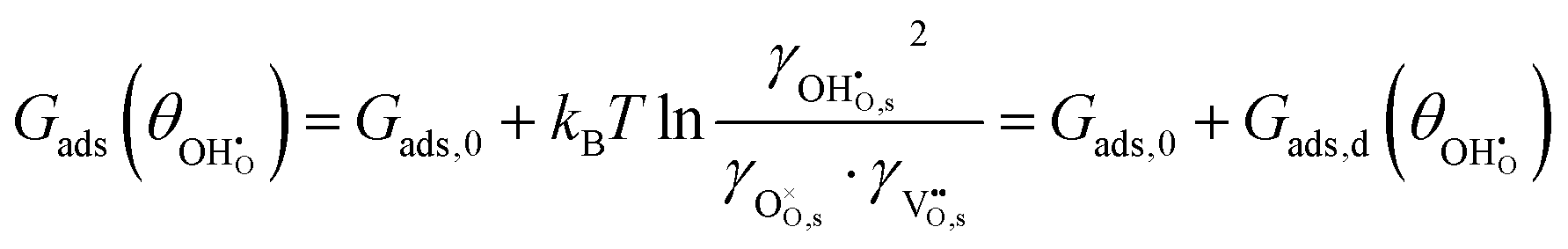
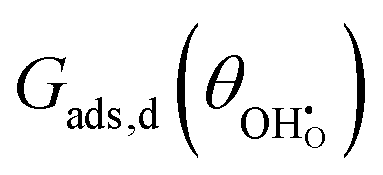 is a function which considers the coulombic dipole–dipole interactions as a function of coverage and accounts for all of the non-idealities of the adsorption step. For example, large adsorbates such as carbonates have greater adsorbate–adsorbate repulsion than smaller adsorbates like hydroxyls. Similarly, reducing the site density will push the adsorbates closer together and increase chemical potential, making adsorption less exothermic.35,36 The free energy of adsorption is discussed in greater detail in the Section 3.
is a function which considers the coulombic dipole–dipole interactions as a function of coverage and accounts for all of the non-idealities of the adsorption step. For example, large adsorbates such as carbonates have greater adsorbate–adsorbate repulsion than smaller adsorbates like hydroxyls. Similarly, reducing the site density will push the adsorbates closer together and increase chemical potential, making adsorption less exothermic.35,36 The free energy of adsorption is discussed in greater detail in the Section 3.
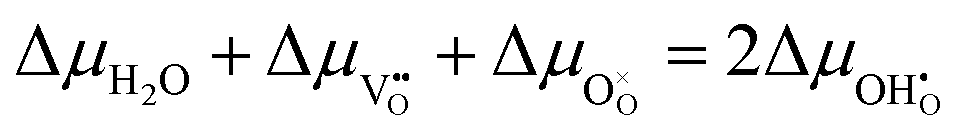
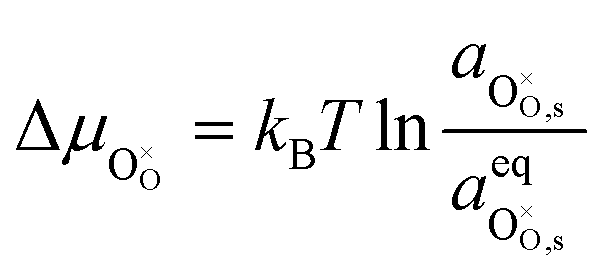 and
and 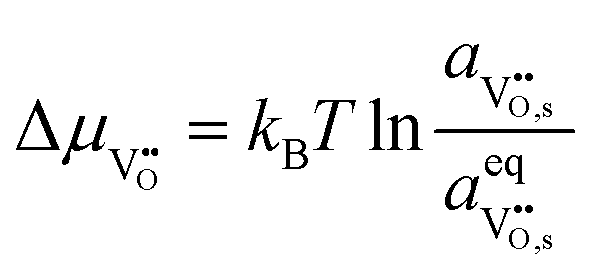 . The shift in adsorbate potential is given as:
. The shift in adsorbate potential is given as:
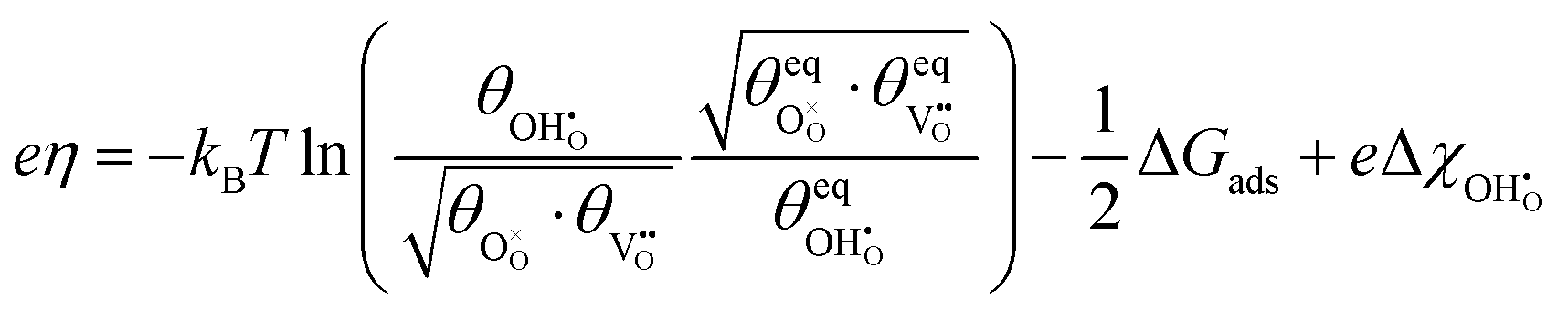

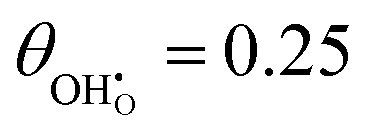 with a polaron positioned as the nearest neighbour (NN) with respect to the hydroxyl, (b)
with a polaron positioned as the nearest neighbour (NN) with respect to the hydroxyl, (b) 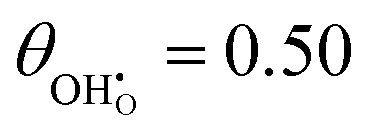 with polarons positioned NN with respect to the hydroxyls, (c)
with polarons positioned NN with respect to the hydroxyls, (c)  and (d)
and (d) 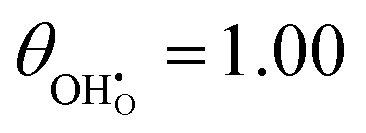 . Ce, O and H atoms are shown as yellow, red and white, respectively.
. Ce, O and H atoms are shown as yellow, red and white, respectively.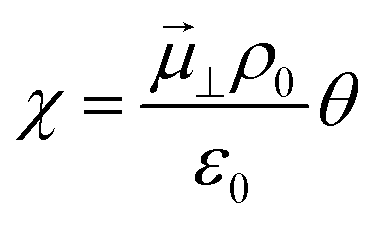 where
where 

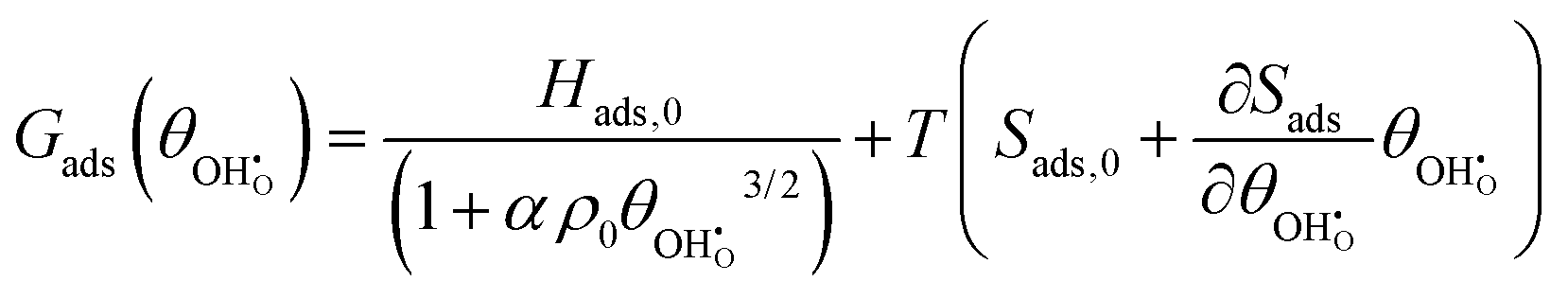
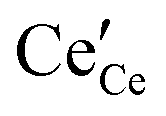 and
and 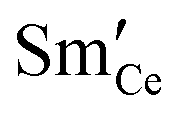 ) have identical adsorption and electrostatic properties. In reality this is not the case, however more extensive multi-scale modelling would be required to evaluate the complex bimodal properties of the doped-reduced surface which is beyond the scope of the current study.
) have identical adsorption and electrostatic properties. In reality this is not the case, however more extensive multi-scale modelling would be required to evaluate the complex bimodal properties of the doped-reduced surface which is beyond the scope of the current study.
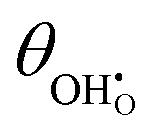 (a) and χ (b) on a Sm0.2Ce0.8O1.9–gas interface as a function of overpotential at 500 °C in 1
(a) and χ (b) on a Sm0.2Ce0.8O1.9–gas interface as a function of overpotential at 500 °C in 1 at open circuit voltage (OCV) we consider R1, R2 and R3 to be in equilibrium. Thus:
at open circuit voltage (OCV) we consider R1, R2 and R3 to be in equilibrium. Thus: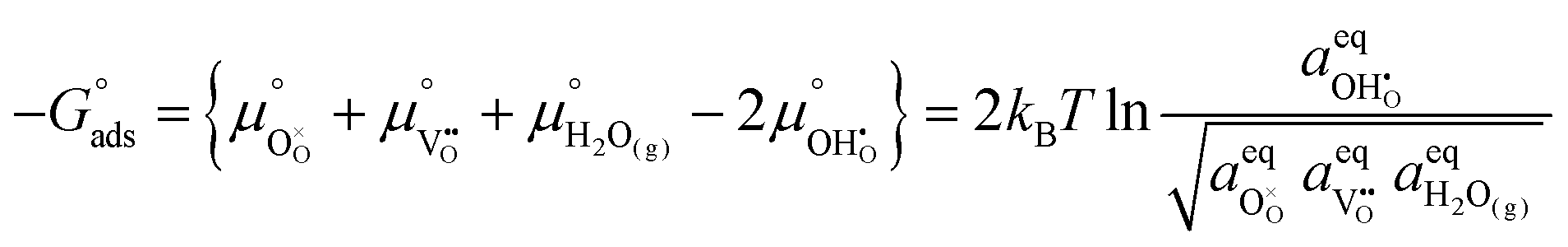
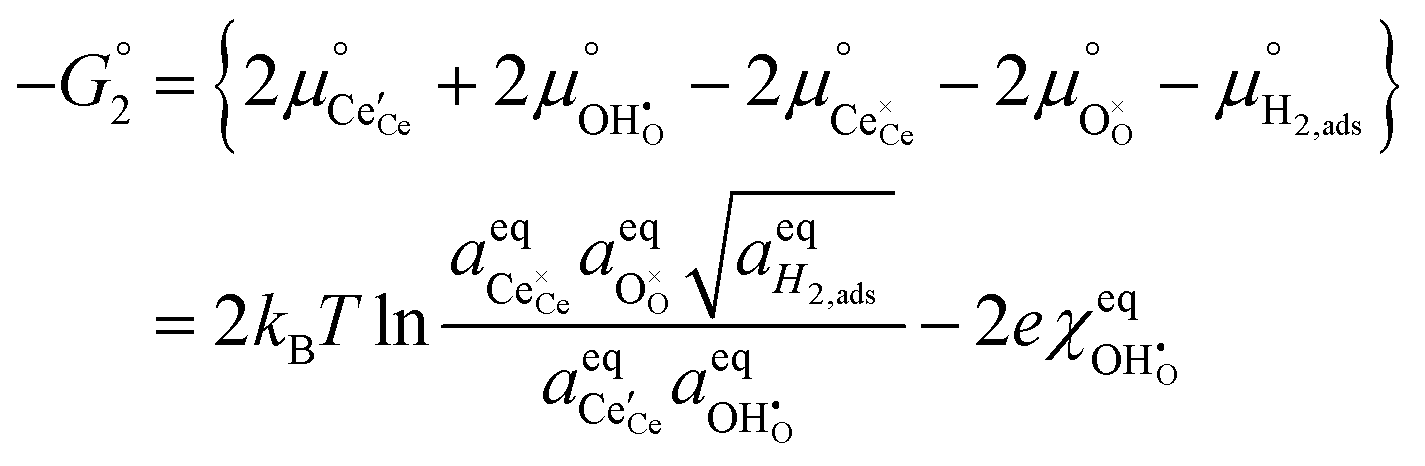
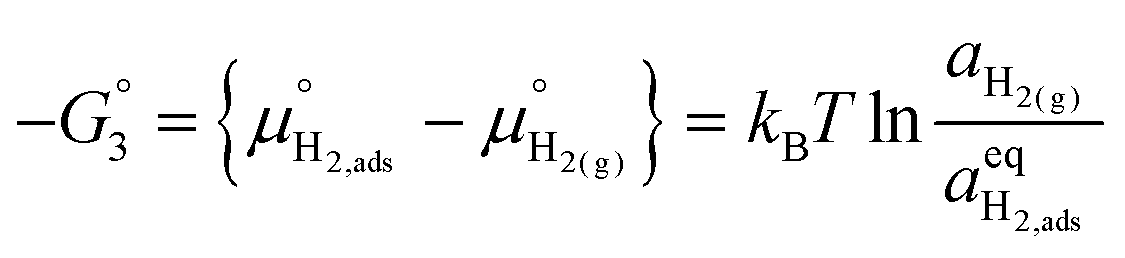
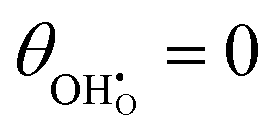 are given in Table 1. In Section 3.2 we demonstrated that dipole–dipole interactions increased enthalpy of adsorption in R1, thus making the adsorption reaction less exothermic as
are given in Table 1. In Section 3.2 we demonstrated that dipole–dipole interactions increased enthalpy of adsorption in R1, thus making the adsorption reaction less exothermic as 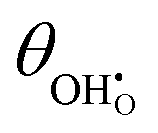 increases. The same effect can be observed with R2, whereby the enthalpy of electron transfer decreases as
increases. The same effect can be observed with R2, whereby the enthalpy of electron transfer decreases as 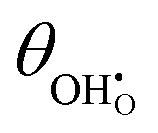 increases. This was demonstrated by simulating R2 using 2 × 2
increases. This was demonstrated by simulating R2 using 2 × 2  and 3 × 3
and 3 × 3 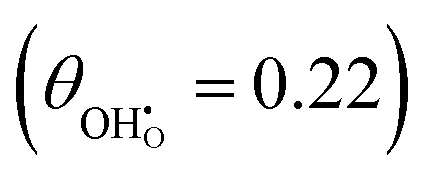 surfaces, where the change in electronic energy was found to be Eel2 = −2.39 eV and Eel2 = −2.86 eV, respectively. The adsorbed hydrogen dimer H2,ads is weakly bound to the ceria surface, resulting in a relatively small associated enthalpy for R3. However, the large entropy associated with the formation of a gas species means the free energy
surfaces, where the change in electronic energy was found to be Eel2 = −2.39 eV and Eel2 = −2.86 eV, respectively. The adsorbed hydrogen dimer H2,ads is weakly bound to the ceria surface, resulting in a relatively small associated enthalpy for R3. However, the large entropy associated with the formation of a gas species means the free energy 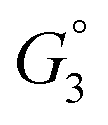 will be negative under operational conditions and the coverage of H2,ads is expected to be very low. Eqn (17)–(19) can be solved simultaneously to estimate the coverage of both adsorbates and defects (Fig. 5a).
will be negative under operational conditions and the coverage of H2,ads is expected to be very low. Eqn (17)–(19) can be solved simultaneously to estimate the coverage of both adsorbates and defects (Fig. 5a). (T = 773 K)
(T = 773 K)
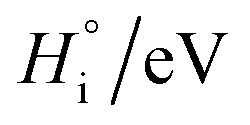

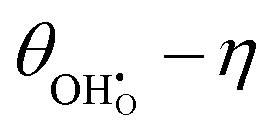 , demonstrating that the electrostatic component on the adsorbate electrochemical potential is dominant, something originally proposed by Fleig.18
, demonstrating that the electrostatic component on the adsorbate electrochemical potential is dominant, something originally proposed by Fleig.18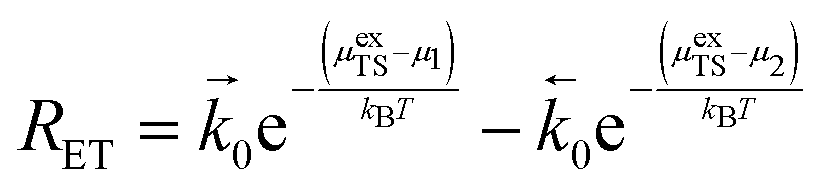
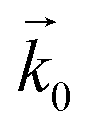 and
and 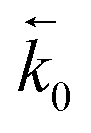 represent the forward and reverse rate constants, respectively. As a consequence of equilibrium
represent the forward and reverse rate constants, respectively. As a consequence of equilibrium 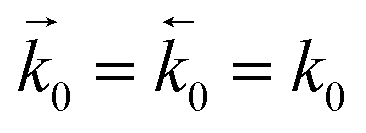 .25 The electrochemical potential of the transition state is defined following well-known Butler–Volmer kinetics:
.25 The electrochemical potential of the transition state is defined following well-known Butler–Volmer kinetics: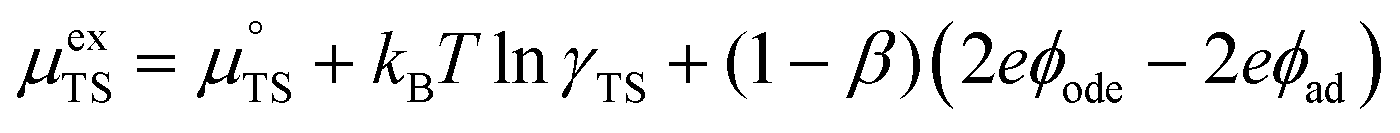

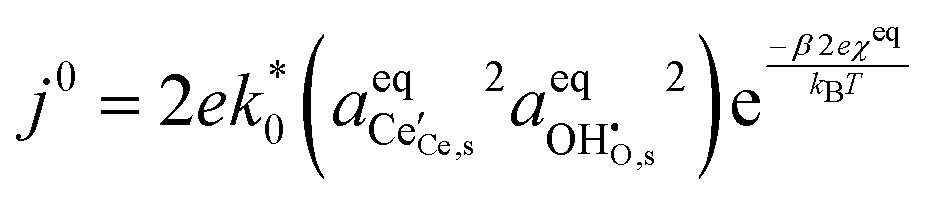
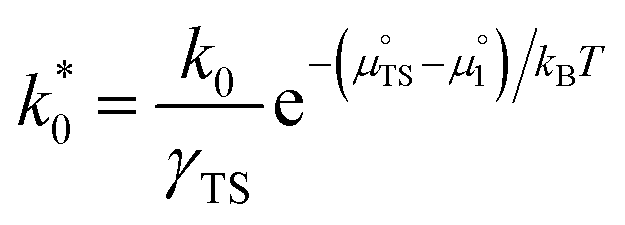 . The exchange current density is dependent not only on activity, as recognised in conventional Butler–Volmer kinetics, but also on the electrostatic potential at equilibrium, χeq. Elucidating γTS requires knowledge of the transition state, outlined by Bazant et al.44 The hydroxyl coverage at equilibrium and the exchange current density as a function of partial pressure is displayed in Fig. 6. On increasing pH2 the equilibrium hydroxyl coverage is increased as the R2 reaction is driven backwards.
. The exchange current density is dependent not only on activity, as recognised in conventional Butler–Volmer kinetics, but also on the electrostatic potential at equilibrium, χeq. Elucidating γTS requires knowledge of the transition state, outlined by Bazant et al.44 The hydroxyl coverage at equilibrium and the exchange current density as a function of partial pressure is displayed in Fig. 6. On increasing pH2 the equilibrium hydroxyl coverage is increased as the R2 reaction is driven backwards.
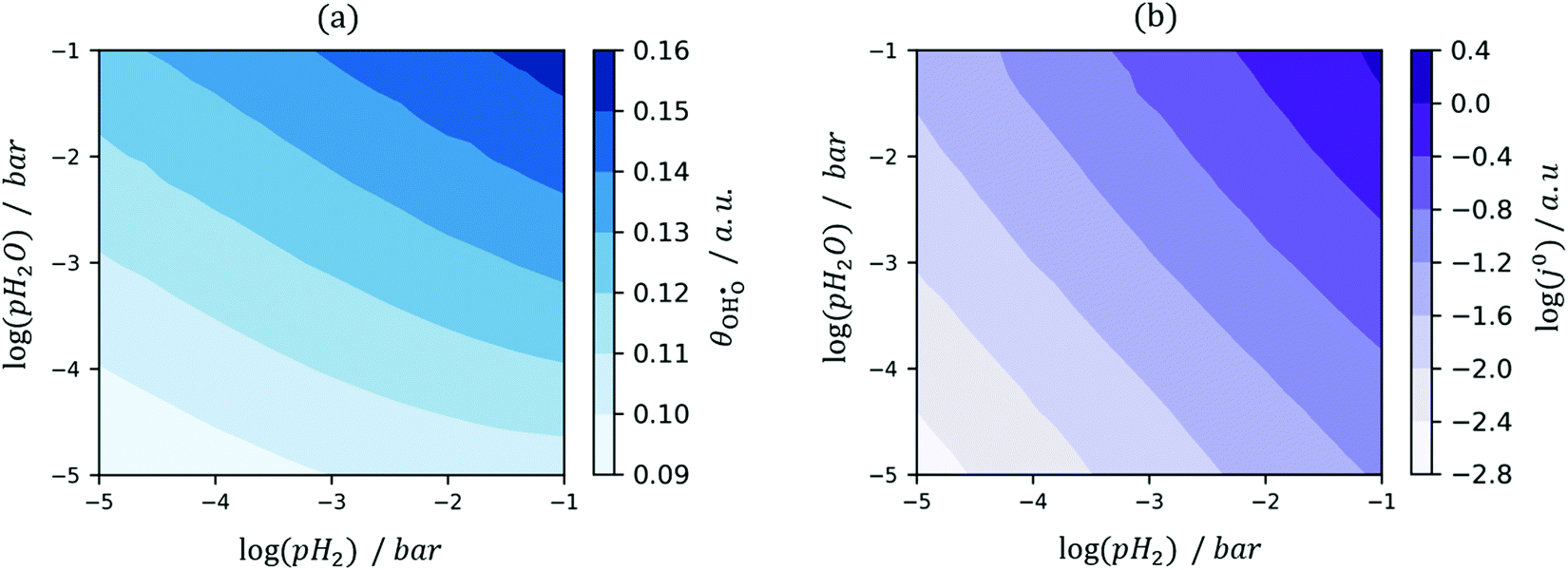
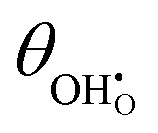 (a) and j0 (b) at the Sm0.2Ce0.8O1.9–gas interface as a function of steam and hydrogen partial pressure at 500 °C.
(a) and j0 (b) at the Sm0.2Ce0.8O1.9–gas interface as a function of steam and hydrogen partial pressure at 500 °C.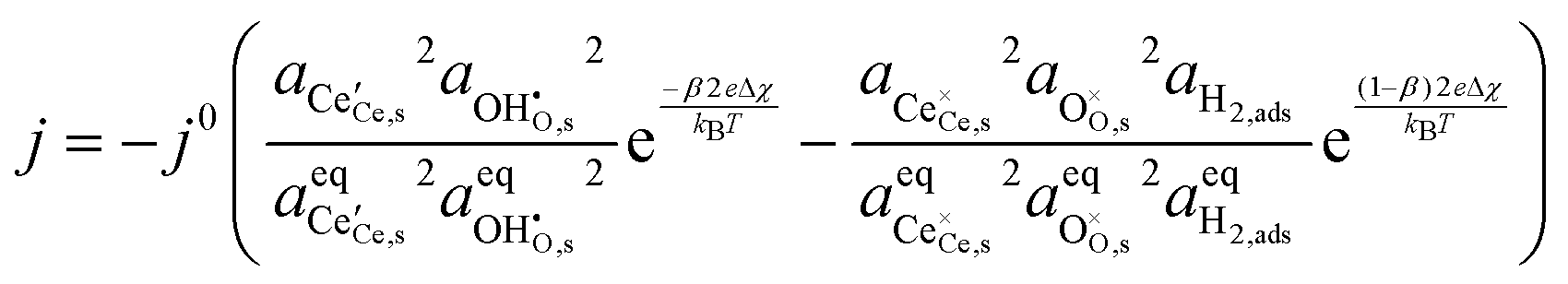
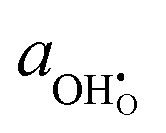 and Δχ are taken from eqn (12). Due to the relatively large dipole moment of the hydroxyl on the ceria[111] surface, the change in adsorbate activity under an applied overpotential is linear and relatively small compared to the other exponential terms. Eqn (24) may be experimentally evaluated through small electrical perturbations close to OCV which generates a linear response where all species activities remain close to equilibrium as Δχ → 0:26
and Δχ are taken from eqn (12). Due to the relatively large dipole moment of the hydroxyl on the ceria[111] surface, the change in adsorbate activity under an applied overpotential is linear and relatively small compared to the other exponential terms. Eqn (24) may be experimentally evaluated through small electrical perturbations close to OCV which generates a linear response where all species activities remain close to equilibrium as Δχ → 0:26
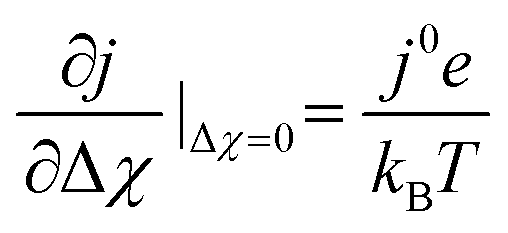
 at OCV from the experimental data.29 The gradient
at OCV from the experimental data.29 The gradient 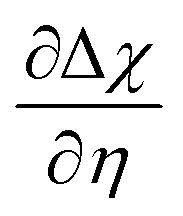 is dependent on the equilibrium potential.45 Under low pH2O and pH2, χeq is low and χ experiences noticeable depletion as η is increased, relative to when the surface is “buffered” and χ does not experience extensive loss under anodic polarization.18,34,46
is dependent on the equilibrium potential.45 Under low pH2O and pH2, χeq is low and χ experiences noticeable depletion as η is increased, relative to when the surface is “buffered” and χ does not experience extensive loss under anodic polarization.18,34,46

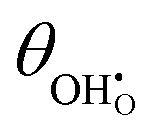 , (b) Δχ and (c)
, (b) Δχ and (c)  at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−5–10−1 bar and pH2 = 10−2 bar). (d)
at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−5–10−1 bar and pH2 = 10−2 bar). (d) 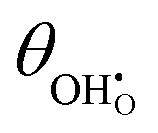 , (e) Δχ and (f)
, (e) Δχ and (f)  at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−2 bar and pH2 = 10−5–10−1 bar).
at the Sm0.2Ce0.8O1.9–gas interface as a function of η at 500 °C (pH2O = 10−2 bar and pH2 = 10−5–10−1 bar).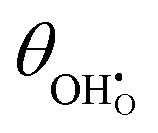 –η relationship. Butler–Volmer kinetic equations were then derived to account for the electrostatic potential at the ceria–gas interface, where the current density was shown to be extensively dependent on χeq. The MIEC–gas interface model is ubiquitous for all systems where the electrostatic potential at the electrode surface is not described as a diffuse charge layer. Moreover, by understanding the electronic structure of the adsorbate, electrodes and operational conditions could in future be adapted to maximise the charge transfer kinetics at the MIEC–gas interface.
–η relationship. Butler–Volmer kinetic equations were then derived to account for the electrostatic potential at the ceria–gas interface, where the current density was shown to be extensively dependent on χeq. The MIEC–gas interface model is ubiquitous for all systems where the electrostatic potential at the electrode surface is not described as a diffuse charge layer. Moreover, by understanding the electronic structure of the adsorbate, electrodes and operational conditions could in future be adapted to maximise the charge transfer kinetics at the MIEC–gas interface.