
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion⋯anion (MX3−)2 dimers (M = Zn, Cd, Hg; X = Cl, Br, I) in different environments†
Rafał
Wysokiński
 *a,
Wiktor
Zierkiewicz
*a,
Mariusz
Michalczyk
a and
Steve
Scheiner
*b
*a,
Wiktor
Zierkiewicz
*a,
Mariusz
Michalczyk
a and
Steve
Scheiner
*b
aFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: rafal.wysokinski@pwr.edu.pl; wiktor.zierkiewicz@pwr.edu.pl
bDepartment of Chemistry and Biochemistry, Utah State University Logan, Utah 84322-0300, USA. E-mail: steve.scheiner@usu.edu
First published on 16th June 2021
Abstract
The possibility that MX3− anions can interact with one another is assessed via ab initio calculations in gas phase as well as in aqueous and ethanol solution. A pair of such anions can engage in two different dimer types. In the bridged configuration, two X atoms engage with two M atoms in a rhomboid structure with four equal M–X bond lengths. The two monomers retain their identity in the stacked geometry which contains a pair of noncovalent M⋯X interactions. The relative stabilities of these two structures depend on the nature of the central M atom, the halogen substituent, and the presence of solvent. The interaction and binding energies are fairly small, generally no more than 10 kcal mol−1. The large electrostatic repulsion is balanced by a strong attractive polarization energy.
Introduction
The idea that ions of like charge can engage in stable complexes with one another has recently become a subject of intense scrutiny. Despite the obvious strong Coulombic repulsions which ought to hold these ions apart, there is an accumulating body of experimental data that supports the existence of such ion–ion complexes1–9 in the context of ionic liquids and crystal solids. The explanation that has been offered in some cases concerns the ability of H-bonds to act as a glue between the two anions if they are drawn close enough together. A balance is achieved between long and short-range interactions, as well as directional and non-directional contacts between ions in ionic liquids.2In comparison to cation-cation contacts, less data is available concerning the analogous anion–anion complexes. Such anion–anion interactions have been observed recently by Ganatra et al.10 in ionic liquids composed of mixtures of alkali halides/sodium acetate with sodium salt of butyric acid. The presence of anion–anion interactions was rationalized therein as the hydrophobic interaction between non-polar moieties (of high polarizability) which constitute the anions.10 Theoretical simulations of Miranda and co-workers treated the anion–anion interactions between aspartate dimers.11 Complexes studied there had positive interaction energies in the gas phase but these quantities turned negative in aqueous solution due to hydration of the monomers which considerably reduced the anion–anion repulsion. In the gas phase it was found that the high energy barrier prevented the dissociation of subunits.11 So far, similar results of metastable anion–anion complexes were also achieved by other researchers in the field of theoretical chemistry,12–15 including works published by our own group.16,17 Experimentally characterized structures containing anion–anion interactions have occurred in the literature for some time.18–30 As an example, the work of Chesman et al. noted five crystal structures of salts consisting of functionalized methanide anions. It was suggested that three of them are stabilized by π–π stacking interactions between anions while in the remaining two the anion–anion attraction results from the presence of hydrogen-bonding interactions.21
The possibility of stacking interactions between a pair of anions motivated us to search for analogous inorganic systems, containing transition metal, without the assistance of any hydrogen bonds. The Cambridge Structural Database (CSD)31 shows 8 hits for searching criteria comprising trihalogenated transition metals in subunits of charge −1, linked to each other by intermolecular contacts shorter than the sum of vdW radii. Surprisingly, within the abovementioned group of crystalline solids discovered in the CSD survey it is only mercury that fits this description20,32–38 where the Hg atom is engaged in unusual Hg⋯X (X = Cl, Br, I) interactions which stabilize anion–anion stacked polymeric chains. This particular type of interaction involving elements from the 12th group (Zn, Cd, Hg) with an electron-rich center has been introduced in literature very recently and baptized as spodium bond.39 It has been earlier shown that this noncovalent interaction has a complicated nature which incorporates both coulombic forces and some degree of covalency.40
The goal of the current work is a detailed investigation of the intriguing interanion stabilization in these crystal structures. We consider first whether such interactions between pairs of anions can be stabilizing, and how the nature and strength of each such interaction might be affected by the surrounding environment. To what specific forces can the attraction be attributed? How is the interaction affected by the identity of the central M atom and the three halogen ligands of the MX3− unit? The calculations confirm the experimental observation of two very different modes of binding of each pair of anions, and proceed to assess their relative stability, and their interconversion process.
Methods
Full optimization of the (MX3−)2 dimers (M = Zn, Cd, Hg; X = Cl, Br, I) as well as the isolated MX3− monomers were performed at the MP2/aug-cc-pVDZ level of theory.41–43 The pseudopotential-containing aug-cc-pVDZ-PP basis set was used for metal and iodine atoms in order to incorporate relativistic effects.44,45 The accuracy of the energies was tested against calculations at a higher level, namely CCSD(T)/aug-cc-pVDZ(-PP)43–47 and MP2/aug-cc-pVQZ(-PP).43–45 Harmonic frequency analysis verified optimized geometries represent true minima on the potential energy surfaces with no imaginary frequencies. Calculations were carried out in the gas phase and solution. The chosen solvents water (ε = 78.4) and ethanol (ε = 25.3) were simulated by the PCM (polarizable continuum model).48 The interaction energy (Eint) of each complex was computed as the difference in total electronic energy between the fully optimized complex and its constituents in the geometries adopted within the complex, while the binding energy (Eb) takes as its reference the doubled value of the total electronic energy for the corresponding monomer in its fully optimized isolated state. The basis set superposition error (BSSE) was corrected via the counterpoise procedure introduced by Boys and Bernardi.49 As the Gaussian package does not allow the calculation of BSSE corrections within the context of solvent, the corrections were taken from the calculations in the gas phase for systems in their solvent geometry. Single-point calculations at the MP2/aug-cc-pVQZ and CCSD(T)/aug-cc-pVDZ levels were performed for the MP2/aug-cc-pVDZ optimized geometry. Calculations were carried out with the Gaussian 16, Rev. C.01 code.50 The MEP (molecular electrostatic potential) measures were assessed based on the values of its extrema on the 0.001 a.u. isodensity surface by the MultiWFN program.51,52 MEP maps were visualized by the VMD software.53 The AIM (atoms-in-molecules) method was engaged to elucidate bond paths and their topological properties by means of the AIMAll program.54 Decomposition of the interaction energies was carried out through the LMOEDA protocol55 realized in GAMESS-US (version 2020-R2) software.56 Energy barriers were quantified by the QST2 approach of the Synchronous Transit-Guided Quasi-Newton (STQN) method.57 The Cambridge Structural Database (CSD) supported the current study by providing X-ray structures similar to those studied in this work.31Results
Monomers
The MX3− anions are planar units with D3h symmetry in the gas phase, but with some small inequalities of the three M–X bond lengths in solution, in which the anions were not precisely planar. Optimized r(M–X) bond lengths are compiled in Table S1 (ESI†), where they may be seen to be between 2.2 and 2.8 Å. Not surprisingly these bond lengths are commensurate with the size of the atoms: Zn < Cd ≈ Hg and Cl < Br < I.Consideration of the topography of the molecular electrostatic potential (MEP) surrounding each monomer offers clues as to the preference for the way in which the two might approach one another. The MEP shown in Fig. 1 is illustrated for ZnCl3− as representative of all MX3− anions where the least and most negative regions are indicated by blue and red colors, respectively. The blue area immediately above the central Zn atom corresponds to what has come to be known as a π-hole, although since the species involved is an anion, the potential in this area is negative. The value of Vs,max, the potential at a maximum of the MEP on an isodensity surface, is listed in Table 1 not only in the gas phase, but in both ethanol and aqueous solvent. It might also be observed that the potential surrounding the Cl atoms is most negative in the ZnCl3 plane.
 | ||
| Fig. 1 MEP of ZnCl3− at the M06-2X/aug-cc-pVDZ level on a surface corresponding to 1.5 × vdW atomic radii from the (a) top and (b) side. Blue (−82 kcal mol−1) and red (−107 kcal mol−1) colors refer to least and most negative regions, respectively. | ||
| Isolated molecule | Water | Ethanol | Gas |
|---|---|---|---|
| ZnCl3− | −32.0 (−91.2) | −37.7 (−91.3) | −56.5 (−89.8) |
| ZnBr3− | −38.4 (−83.3) | −39.9 (−83.1) | −56.6 (−81.6) |
| ZnI3− | −35.5 (−72.1) | −40.0 (−72.2) | −56.7 (−71.6) |
| CdCl3− | −7.2 (−94.2) | −10.2 (−93.7) | −44.9 (−89.1) |
| CdBr3− | −7.3 (−84.7) | −9.1 (−84.2) | −45.9 (−80.9) |
| CdI3− | −3.9 (−75.2) | −14.4 (−72.6) | −47.8 (−70.9) |
| HgCl3− | −23.9 (−89.2) | −27.5 (−88.5) | −61.4 (−101.5) |
| HgBr3− | −32.7 (−81.5) | −31.5 (−74.6) | −61.5 (−78.4) |
| HgI3− | −27.8 (−67.4) | −36.6 (−68.9) | −61.9 (−68.8) |
There are several trends apparent in the data in Table 1. First considering the gas phase data, Vmax is least negative for Cd, followed by Zn and then by Hg with the most negative π-hole. The nature of the X halogen substituent has little effect on these quantities. Immersion of these anions in a polar medium makes each of these π-holes much less negative, particularly in water. Nonetheless, Cd retains its position as least negative π-hole followed now by Hg and then Zn. Within the context of solution, the nature of the X substituent plays a larger role, wherein Cl leaves the π-hole less negative than Br or I, although this pattern is not consistent throughout the set.
In addition to the π-holes above the M atoms, there are also secondary maxima near the X atoms, along the extensions of the M–X bonds. As shown by the parenthetical quantities in Table 1, these maxima are much more negative than the M π-hole, and so do not represent any sort of strong competition for a Lewis base. These secondary maxima, with σ-hole characteristics, are least negative for X = I and most negative for F.
Dimers
 | ||
| Fig. 2 Example of bridged structure of (Hg2Cl6)2−, refcode: BETPAR. | ||
| Atom | Cl | Br | I |
|---|---|---|---|
| Bridged | |||
| Zn | 51 | 11 | 2 |
| Cd | 8 | 7 | 39 |
| Hg | 91 | 39 | 101 |
| Stacked | |||
| Hg | 4 | 3 | 1 |
Another structural motif is less common, wherein each MX3− unit is stacked above another. As illustrated in Fig. 3 for (HgCl3)−2, this stacking places each Hg directly above a Cl atom of the neighboring unit, such that each dimer contains a pair of Hg⋯Cl direct interactions. This stacked arrangement occurs far less often with only 8 instances, all of which involve Hg, as indicated in Table 2.20,32–38 An essential difference between the stacked systems and bridged structures is the lack of a covalent bond between the two MX3 units in the former, leaving the central Hg atom in its quasi-D3h geometry. These stacked arrangement are also generally part of an extended polymeric geometry in a long (MX3)n chain.
 | ||
| Fig. 3 Example of stacked structure of (HgCl3−)2 (part of polymeric chain), refcode: TAGCAF. | ||
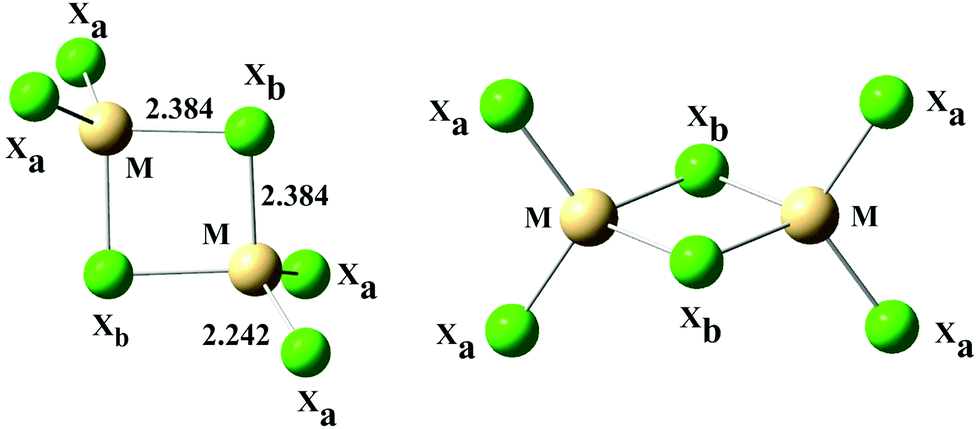 | ||
| Fig. 4 Structure of model bridged anions (M2X6)2− (M = Zn, Cd, Hg; X = Cl, Br, I) in two projections. Bond lengths for (Zn2Cl6)2− in water. | ||
| M–Xa | ∑Cov (%) | M–Xb | ∑Cov (%) | θ(Xa–M–Xa) | θ(Xb–M–Xb) | ∑X–M–Xa | |
|---|---|---|---|---|---|---|---|
| a Sum of θ(Xa–M–Xa) and two of θ(Xa–M–Xb) angles. | |||||||
| Vacuum | |||||||
| (Zn2Cl6)2− | 2.241 | 100 | 2.380 | 106 | 114.7 | 91.4 | 339 |
| (Zn2Br6)2− | 2.376 | 98 | 2.513 | 104 | 114.7 | 94.3 | 338 |
| (Zn2I6)2− | 2.576 | 99 | 2.709 | 104 | 114.9 | 95.2 | 338 |
| (Cd2Cl6)2− | 2.438 | 99 | 2.575 | 105 | 114.6 | 89.2 | 334 |
| (Cd2Br6)2− | 2.563 | 97 | 2.697 | 102 | 114.8 | 91.9 | 339 |
| (Cd2I6)2− | 2.747 | 97 | 2.878 | 102 | 114.9 | 94.4 | 339 |
| (Hg2Cl6)2− | 2.438 | 104 | 2.625 | 112 | 120.8 | 88.0 | 342 |
| (Hg2Br6)2− | 2.560 | 102 | 2.735 | 109 | 119.5 | 90.9 | 341 |
| (Hg2I6)2− | 2.737 | 101 | 2.892 | 107 | 118.0 | 93.9 | 339 |
| Ethanol | |||||||
| (Zn2Cl6)2− | 2.242 | 100 | 2.382 | 106 | 120.5 | 93.9 | 340 |
| (Zn2Br6)2− | 2.384 | 99 | 2.513 | 104 | 118.3 | 95.6 | 338 |
| (Zn2I6)2− | 2.579 | 99 | 2.713 | 104 | 117.5 | 98.1 | 337 |
| (Cd2Cl6)2− | 2.492 | 101 | 2.617 | 106 | 108.5 | 89.6 | 337 |
| (Cd2Br6)2− | 2.600 | 98 | 2.762 | 105 | 126.3 | 93.7 | 342 |
| (Cd2I6)2− | 2.785 | 98 | 2.947 | 104 | 125.4 | 93.8 | 342 |
| (Hg2Cl6)2− | 2.524 | 108 | 2.704 | 116 | 113.0 | 87.2 | 340 |
| (Hg2Br6)2− | 2.580 | 102 | 2.856 | 113 | 138.2 | 87.9 | 348 |
| (Hg2I6)2− | 2.788 | 103 | 2.993 | 110 | 120.4 | 96.3 | 339 |
| Water | |||||||
| (Zn2Cl6)2− | 2.242 | 100 | 2.384 | 106 | 121.1 | 93.9 | 340 |
| (Zn2Br6)2− | 2.378 | 98 | 2.518 | 104 | 119.3 | 95.3 | 336 |
| (Zn2I6)2− | 2.581 | 99 | 2.722 | 104 | 117.1 | 96.8 | 338 |
| (Cd2Cl6)2− | 2.496 | 101 | 2.636 | 107 | 120.6 | 89.4 | 342 |
| (Cd2Br6)2− | 2.660 | 101 | 2.761 | 105 | 119.5 | 92.5 | 340 |
| (Cd2I6)2− | 2.819 | 100 | 2.960 | 105 | 122.0 | 92.3 | 347 |
| (Hg2Cl6)2− | 2.464 | 105 | 2.822 | 121 | 135.8 | 86.8 | 347 |
| (Hg2Br6)2− | 2.622 | 104 | 2.860 | 113 | 126.8 | 87.2 | 344 |
| (Hg2I6)2− | 2.759 | 102 | 2.981 | 110 | 114.2 | 114.3 | 333 |
Analysis of the electron density topology of these bridged complexes by the AIM protocol provides some information about the internal bonding. The resulting AIM molecular diagrams are provided in Fig. S1 (ESI†). As delineated in Table S2 (ESI†), the electron densities at the M–X bond critical points lie in the range between 0.03 and 0.07 a.u. which would place them solidly in the noncovalent category, despite their short length. This characterization is supported by the positive Laplacians of roughly 0.01–0.02 a.u.
 | ||
| Fig. 5 Structure of model stacked anions (MX3−)2 (M = Zn, Cd, Hg; X = Cl, Br, I). Bond lengths for (ZnCl3)2− in water. | ||
| M–Xa | ∑Cov (%) | M–Xb | ∑Cov (%) | M⋯Xb | ∑Cov (%) | ∑vdW (%) | M-Xb⋯M | ∑X–M–X | |
|---|---|---|---|---|---|---|---|---|---|
| Ethanol | |||||||||
| (ZnCl3−)2 | 2.227 | 99 | 2.225 | 99 | 3.781 | 169 | 90 | 93.2 | 360 |
| (ZnBr3−)2 | 2.358 | 97 | 2.360 | 98 | 3.809 | 157 | 90 | 90.5 | 360 |
| (ZnI3−)2 | 2.559 | 98 | 2.564 | 98 | 4.093 | 157 | 92 | 87.8 | 360 |
| (CdCl3−)2 | 2.496 | 101 | 2.493 | 101 | 3.731 | 152 | 87 | 89.2 | 360 |
| (CdBr3−)2 | 2.613 | 99 | 2.606 | 99 | 3.850 | 146 | 89 | 96.0 | 360 |
| (CdI3−)2 | 2.784 | 98 | 2.787 | 98 | 4.012 | 142 | 89 | 85.2 | 360 |
| (HgCl3−)2 | 2.510 | 107 | 2.469 | 106 | 3.621 | 155 | 85 | 81.6 | 359 |
| (HgBr3−)2 | 2.573 | 102 | 2.535 | 101 | 3.689 | 146 | 86 | 90.1 | 359 |
| (HgI3−)2 | 2.792 | 103 | 2.782 | 103 | 3.944 | 146 | 88 | 79.2 | 359 |
| Water | |||||||||
| (ZnCl3−)2 | 2.231 | 100 | 2.226 | 99 | 3.782 | 169 | 90 | 93.5 | 360 |
| (ZnBr3−)2 | 2.362 | 98 | 2.362 | 98 | 3.815 | 158 | 90 | 90.6 | 360 |
| (ZnI3−)2 | 2.566 | 98 | 2.565 | 98 | 3.987 | 153 | 90 | 87.0 | 359 |
| (CdCl3−)2 | 2.503 | 102 | 2.502 | 102 | 3.737 | 152 | 87 | 87.9 | 360 |
| (CdBr3−)2 | 2.624 | 99 | 2.648 | 100 | 3.830 | 145 | 88 | 85.7 | 360 |
| (CdI3−)2 | 2.823 | 100 | 2.786 | 98 | 4.030 | 142 | 89 | 82.8 | 359 |
| (HgCl3−)2 | 2.524 | 108 | 2.474 | 106 | 3.616 | 155 | 85 | 80.2 | 359 |
| (HgBr3−)2 | 2.622 | 104 | 2.618 | 104 | 3.769 | 150 | 87 | 87.8 | 360 |
| (HgI3−)2 | 2.796 | 103 | 2.752 | 102 | 3.928 | 145 | 87 | 82.7 | 358 |
| Water | Ethanol | |
|---|---|---|
| a Gibbs free energy differences in parentheses. | ||
| (ZnCl3−)2 | 4.58 (3.37) | 5.52 (4.40) |
| (ZnBr3−)2 | 2.68 (3.63) | 4.00 (6.31) |
| (ZnI3−)2 | 0.42 (1.12) | 0.47 (1.11) |
| (CdCl3−)2 | 1.18 (0.54) | 2.56 (1.02) |
| (CdBr3−)2 | 0.91 (0.74) | 2.23 (3.47) |
| (CdI3−)2 | −6.59 (−8.16) | −0.07 (−1.60) |
| (HgCl3−)2 | −4.80 (−5.94) | −6.29 (−6.05) |
| (HgBr3−)2 | −4.00 (−5.53) | −4.90 (−5.12) |
| (HgI3−)2 | −6.26 (−4.36) | −8.08 (−7.44) |
As each MX3− unit retains its structural integrity in the stacked dimers, it is possible to evaluate both their interaction and binding energies, which are displayed in Table 6 at three different levels of theory. As a reminder, the interaction energy takes as its reference point the monomers in the geometry they adopt within the complex, so can be considered a pure measure of the interaction itself. In any case, there is little geometrical deformation of the subunits so Eint is only very slightly more exothermic than is Eb. The energetics of dimerization are more favourable in water than in ethanol, with all quantities negative in the former solvent. Dimerization is favoured with the larger X atoms I > Br > Cl, as well as for the larger M atoms Hg > Cd > Zn. The greater exothermicity of formation of stacked complexes with Hg is consistent with the CSD survey where it is only this M atom which engages in such arrangements (see Table 2). It is lastly important to point out the parallel nature of the MP2 (combined with aug-cc-pVDZ and aug-cc-pVQZ basis sets) energetic trends with those extracted via the more reliable CCSD(T) approach. When the BSSE correction is considered these trends become less coherent in the ethanol solvent. The reason for these discrepancies are due to the computational package limitation which does not give possibility of direct calculations of this correction in solvent (see details in the Method section).
| E int | E b | |||||
|---|---|---|---|---|---|---|
| I | II | III | I | II | III | |
| Water | ||||||
| (ZnCl3−)2 | −2.18 (−0.90) | −1.99 (−1.68) | −2.04 (−0.57) | −2.14 (−0.87) | −1.98 (−1.67) | −2.00 (−0.53) |
| (ZnBr3−)2 | −3.81 (−1.30) | −4.32 (−2.63) | −3.36 (−0.64) | −3.76 (−1.25) | −4.29 (−2.60) | −3.31 (−0.58) |
| (ZnI3−)2 | −5.35 (−2.35) | −7.16 (−4.47) | −4.42 (−1.23) | −5.34 (−2.34) | −6.96 (−4.27) | −4.36 (−1.07) |
| (CdCl3−)2 | −2.93 (−1.50) | −2.83 (−2.46) | −2.71 (−1.04) | −2.90 (−1.47) | −2.69 (−2.33) | −2.68 (−1.06) |
| (CdBr3−)2 | −4.74 (−1.95) | −5.14 (−3.68) | −4.17 (−1.13) | −4.70 (−1.91) | −5.10 (−3.65) | −4.12 (−1.08) |
| (CdI3−)2 | −6.25 (−2.91) | −8.36 (−5.58) | −5.34 (−2.05) | −6.02 (−2.68) | −7.23 (−4.45) | −5.28 (−1.99) |
| (HgCl3−)2 | −4.74 (−1.88) | −4.09 (−3.33) | −4.12 (−0.99) | −4.71 (−1.85) | −3.92 (−3.16) | −4.06 (−0.93) |
| (HgBr3−)2 | −6.11 (−2.00) | −6.13 (−4.08) | −5.11 (−0.75) | −6.05 (−1.94) | −6.13 (−4.08) | −5.08 (−0.72) |
| (HgI3−)2 | −8.44 (−3.49) | −10.00 (−6.43) | −6.99 (−1.69) | −8.22 (−3.27) | −9.85 (−6.28) | −6.26 (−0.96) |
| Ethanol | ||||||
| (ZnCl3−)2 | −0.46 (0.82) | −0.26 (0.05) | −0.33 (1.14) | −0.37 (0.90) | −0.29 (0.02) | −0.18 (1.29) |
| (ZnBr3−)2 | −2.11 (0.42) | −2.63 (−0.94) | −1.66 (1.08) | −2.04 (0.48) | −2.56 (−0.87) | −1.60 (1.15) |
| (ZnI3−)2 | −3.41 (−0.58) | −5.32 (−2.63) | −2.56 (0.55) | −3.35 (−0.52) | −5.27 (−2.58) | −2.49 (0.61) |
| (CdCl3−)2 | −1.25 (0.15) | −1.14 (−0.78) | −1.01 (0.60) | −1.18 (0.21) | −1.00 (−0.63) | −0.98 (0.63) |
| (CdBr3−)2 | −2.86 (−0.41) | −3.11 (−1.66) | −2.41 (0.25) | −2.83 (−0.37) | −3.03 (−1.57) | −2.39 (0.26) |
| (CdI3−)2 | −4.62 (−1.23) | −6.34 (−3.55) | −3.62 (0.10) | −4.52 (−1.13) | −6.19 (−3.40) | −3.55 (0.17) |
| (HgCl3−)2 | −2.98 (−0.10) | −2.27 (−1.50) | −2.25 (0.81) | −2.85 (−0.06) | −2.12 (−1.35) | −2.21 (0.84) |
| (HgBr3−)2 | −4.90 (−0.91) | −4.68 (−2.63) | −3.96 (0.27) | −4.81 (−0.82) | −4.54 (−2.49) | −3.91 (0.31) |
| (HgI3−)2 | −7.48 (−2.16) | −8.79 (−5.22) | −5.76 (−0.07) | −6.86 (−1.54) | −8.13 (−4.56) | −5.24 (0.44) |
One window into the fundamental nature of the interaction in the stacked dimers is opened with a LMOEDA decomposition of the total interaction energy. The data in Table 7 show first a strongly repulsive electrostatic term, not surprising in light of the close approach of the two anions. There is also a smaller complementary repulsion resulting from the overlap of the electron clouds of the two species. The polarization component furnishes the lion's share of the attractive force, with smaller increments arising from exchange and dispersion. Although of opposite sign, the magnitudes of Ees and Epol both diminish for larger X atoms and grow larger along with the size of the central M atom. Reducing the polarity of the solvent from water to ethanol reduces these quantities, but only by a relatively small amount.
| E es | E rep | E ex | E pol | E disp | E int | |
|---|---|---|---|---|---|---|
| Water | ||||||
| (ZnCl3−)2 | 178.66 | 6.90 | −4.55 | −178.86 | −3.11 | −0.95 |
| (ZnBr3−)2 | 165.37 | 11.50 | −7.47 | −166.19 | −4.43 | −1.23 |
| (ZnI3−)2 | 153.62 | 16.60 | −10.86 | −155.48 | −5.92 | −2.04 |
| (CdCl3−)2 | 213.66 | 7.93 | −4.96 | −214.22 | −3.78 | −1.36 |
| (CdBr3−)2 | 205.06 | 12.26 | −7.60 | −208.30 | −5.08 | −2.17 |
| (CdI3−)2 | 192.03 | 16.79 | −10.73 | −195.30 | −6.35 | −3.55 |
| (HgCl3−)2 | 225.07 | 13.11 | −8.07 | −226.73 | −5.37 | −1.99 |
| (HgBr3−)2 | 210.70 | 17.56 | −11.21 | −212.89 | −6.39 | −2.23 |
| (HgI3−)2 | 192.09 | 22.19 | −13.91 | −195.79 | −7.80 | −3.23 |
| Ethanol | ||||||
| (ZnCl3−)2 | 173.96 | 6.76 | −4.45 | −172.67 | −3.04 | 0.56 |
| (ZnBr3−)2 | 161.54 | 11.65 | −7.57 | −161.00 | −4.40 | 0.21 |
| (ZnI3−)2 | 152.85 | 15.44 | −10.40 | −152.76 | −5.75 | −0.62 |
| (CdCl3−)2 | 203.72 | 7.71 | −4.80 | −203.07 | −3.67 | −0.11 |
| (CdBr3−)2 | 187.79 | 10.53 | −6.54 | −187.89 | −4.34 | −0.44 |
| (CdI3−)2 | 183.33 | 18.45 | −11.84 | −184.80 | −6.63 | −1.49 |
| (HgCl3−)2 | 214.09 | 12.88 | −7.97 | −214.56 | −5.23 | −0.77 |
| (HgBr3−)2 | 197.04 | 15.24 | −9.51 | −197.94 | −5.86 | −1.04 |
| (HgI3−)2 | 190.15 | 25.08 | −16.00 | −192.69 | −8.74 | −2.20 |
The AIM diagrams of these stacked dimers in Fig. S2 (ESI†) show a pair of clear inter-unit bond M⋯X bond paths in each case. The values of the various properties of the bond critical point are compiled in Table S3 (ESI†), not only for the latter M⋯X bond, but also others of interest, e.g. the internal M–X bond. The density of the inter-anion M⋯X bond critical point is rather small, less than 0.01 a.u., much smaller than the internal M–X bonds. When viewed in concert with the positive Laplacian and the very small values of H, this interaction can be classified as noncovalent.
Given the comparable energies of the stacked and bridged structures in solvent where both are present, it is natural to inquire about the process that would transform one to the other. This process was explored for the MCl3− systems in some depth and the energetics are displayed in Table 8.
| Complex | Bridged | TS | Stacked |
|---|---|---|---|
| Water | |||
| (ZnCl3−)2 | 0.00 | 14.75 | 4.58 |
| (CdCl3−)2 | 0.00 | 6.99 | 1.18 |
| (HgCl3−)2 | 0.00 | 1.86 | −4.80 |
| Ethanol | |||
| (ZnCl3−)2 | 0.00 | 11.92 | 5.52 |
| (CdCl3−)2 | 0.00 | 4.59 | 2.56 |
| (HgCl3−)2 | 0.00 | 4.73 | −6.29 |
Taking the transition from bridged to stacked (ZnCl3−)2 in water in the first row as an example, the stacked dimer is higher in energy than bridged by 4.58 kcal mol−1. The conversion must overcome an energy barrier of 14.75 kcal. This same transformation is exoergic for (HgCl3−)2 as the stacked dimer is more stable by 4.80 kcal mol−1. Nonetheless, there is a small energy barrier of 1.86 kcal mol−1 that the system must overcome for this transition to occur. In summary, as the M atom grows larger, the bridged → stacked conversion reverses from endoergic to exoergic, and the barrier rapidly diminishes. Replacement of the aqueous solvent by ethanol lowers the transition barrier for the two lighter metal atoms, but raises the barrier for (HgCl3−)2, even though the transition from bridged to stacked becomes more exoergic. These trends in the conversion process can be visualized more easily in Fig. 6 which also presents a schematic diagram of the geometry of the transition state. The transition state involves a stretch of one of the M–Clb bonds which is reduced again as the system settles into the stacked geometry. The magnitude of this stretching in the TS is provided in Table 9 which lists an interatomic distance between 4.2 and 4.5 Å.
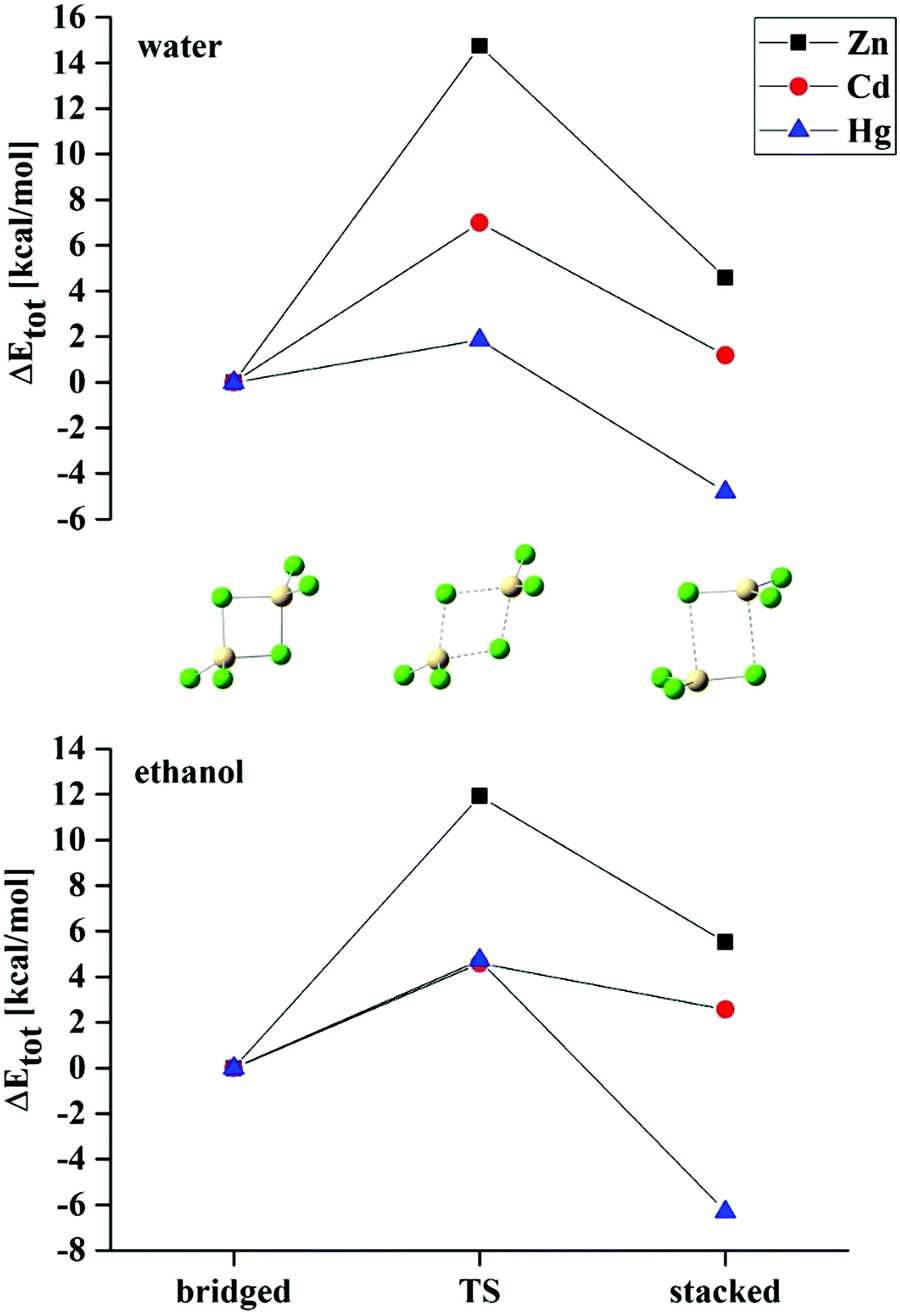 | ||
| Fig. 6 Energetics of conversion from bridged to stacked configurations of (MCl3−) dimers in water and ethanol solvents. | ||
| Complex | Bridged | TS | Stacked |
|---|---|---|---|
| Water | |||
| (ZnCl3−)2 | 2.384 | 4.253 | 3.782 |
| (CdCl3−)2 | 2.636 | 4.500 | 3.737 |
| (HgCl3−)2 | 2.822 | 4.170 | 3.616 |
| Ethanol | |||
| (ZnCl3−)2 | 2.382 | 4.350 | 3.781 |
| (CdCl3−)2 | 2.617 | 4.380 | 3.731 |
| (HgCl3−)2 | 2.704 | 4.516 | 3.621 |
Discussion
It might be surprising at first sight that anions can come together to form stable and metastable dimers, especially since the maximum in the MEP of the Lewis acid is clearly negative. But the systems discussed here fall into an ever enlarging category of anion⋯anion dimers which are either stable in their own right, or metastable in the sense that their dissociation requires surmounting an energy barrier. The largest subset of systems of this type are held together by H-bonds,15,58–72 but there have also been a number of halogen-bonded dianion complexes.12,24,73–78 Recent work has extended this growing list to pnicogen79 and triel bonds,80 as well as those involving metal atoms.16,17 The general finding is that such anion–anion complexes are stable in polar solvent but only metastable in the gas phase.In terms of overall stability, many of these complexes have fairly large binding energies. Considering water as a solvent, for example, the binding energy of CN− with the various MCl3− anions, where M is a Group 2 atom ranges up to 20 kcal mol−1 (ref. 17) and lies in a similar range for Group 12 atoms Zn, Cd, or Hg.16 Pnicogen bonds between anions are even larger in magnitude, more than 20 kcal mol−1 for the ZCl4− series, with Z = P, As or Sb.79 In the cases examined here, it is not a small and compact CN− base that is interacting with the Lewis acid, but rather a pair of MCl3− units with one another. Instead of a single strong N⋯CN bond, the interactions in these stacked dimers are dependent upon a pair of much weaker M⋯X interactions. It is therefore sensible to note the smaller total interaction energies here.
Focusing on the electrostatic component of the interactions here, Ees is large and positive, highly repulsive. It is the other components, chiefly polarization, which are responsible for the small cumulative attractions. This characteristic also differentiates these dual spodium bonded systems with some of the other anion–anion interactions. Ees is very small for the Group 2 complexes, and its sign depends on the specific central M atom.17 The electrostatic energy is quite attractive for the Group 12 analogues, in the 40–100 kcal mol−1 range16 and is further magnified to even larger negative quantities up to 111 kcal mol−1 for the pnicogen-bonded anion pairs.79 As a second issue leading to the large positive coulombic repulsions here, the minima surrounding the X atoms of MX3− are not directed at the M atom below. These minima instead lie in the MX3 plane, as is evident in Fig. 1, which would weaken any potential stabilizing interaction with the M π-hole.
Conclusions
MX3− ions can engage with one another in one of two different modes, despite their strong mutual coulombic repulsion. In the bridged structures, the bonding is largely covalent, with four equivalent short M–X bonds within a four-membered rhomboid ring. Each M atom is tetracoordinated and the bridging X atoms are covalently bonded to two M atoms. The bonding scenario is rather different in the stacked geometries where each MX3 unit largely retains its native D3h structure, which is held to the MX3 unit beneath it by a pair of noncovalent M⋯X bonds. The latter stacked structure does not represent a stable minimum in the gas phase, but occurs with an energy comparable to that of the bridged configuration in ethanol and aqueous solution. In fact, it is the noncovalently bonded stacked geometry that is preferred over bridged for the heavier HgCl3− dimers in either solvent. The overall energetics of formation of the noncovalently bonded stacked dimers are roughly thermoneutral. The binding energies are consistently exothermic in aqueous solution, albeit by only a few kcal mol−1, and Eb is smaller in magnitude in ethanol. There is a trend toward more exothermic stacking for heavier M atoms: Zn < Cd < Hg and for larger X atoms as well: Cl < Br < I.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology and by the US National Science Foundation under Grant No. 1954310. A generous allotment of computer time from the Wroclaw Supercomputer and Networking Center is acknowledged.References
- T. Niemann, P. Stange, A. Strate and R. Ludwig, ChemPhysChem, 2018, 19, 1691–1695 CrossRef CAS PubMed.
- T. Niemann, A. Strate, R. Ludwig, H. J. Zeng, F. S. Menges and M. A. Johnson, Angew. Chem., Int. Ed., 2018, 57, 15364–15368 CrossRef CAS PubMed.
- T. Niemann, D. Zaitsau, A. Strate, A. Villinger and R. Ludwig, Sci. Rep., 2018, 8, 14753 CrossRef PubMed.
- A. Strate, V. Overbeck, V. Lehde, J. Neumann, A. M. Bonsa, T. Niemann, D. Paschek, D. Michalik and R. Ludwig, Phys. Chem. Chem. Phys., 2018, 20, 5617–5625 RSC.
- I. A. Charushnikova, N. N. Krot and V. P. Perminov, Radiochemistry, 2020, 62, 9–17 CrossRef CAS.
- W.-Y. Wen, K. Miyajima and A. Otsuka, J. Phys. Chem., 1971, 75, 2148–2157 CrossRef CAS.
- W.-Y. Wen and K. Nara, J. Phys. Chem., 1967, 71, 3907–3914 CrossRef CAS.
- I. A. Charushnikova, M. S. Grigor'ev, A. M. Fedoseev, A. A. Bessonov and K. A. Lysenko, Radiochemistry, 2020, 62, 161–169 CrossRef CAS.
- R. Maurice, P. D. Dau, M. Hodee, E. Renault and J. K. Gibson, Eur. J. Inorg. Chem., 2020,(47), 4465–4476 CrossRef CAS.
- S. H. Ganatra, V. R. Shaikh, A. S. Ujjankar, S. D. Khobragade, P. A. Tomar and K. J. Patil, J. Mol. Liq., 2020, 315, 113654 CrossRef CAS.
- M. O. Miranda, D. J. R. Duarte and I. Alkorta, ChemPhysChem, 2020, 21, 1052–1059 CrossRef CAS PubMed.
- D. Quinonero, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2016, 18, 27939–27950 RSC.
- I. Iribarren, M. M. Montero-Campillo, I. Alkorta, J. Elguero and D. Quinonero, Phys. Chem. Chem. Phys., 2019, 21, 5796–5802 RSC.
- R. Prohens, A. Portell, M. Font-Bardia, A. Bauza and A. Frontera, Chem. Commun., 2018, 54, 1841–1844 RSC.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, Chem. – Eur. J., 2016, 22, 9226–9234 CrossRef CAS PubMed.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef CAS PubMed.
- W. Zierkiewicz, R. Wysokinski, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed.
- T. R. Griffiths and M. C. R. Symons, Trans. Faraday Soc., 1960, 56, 1752–1761 RSC.
- P. L. Goggin, P. King, D. M. Mcewan, G. E. Taylor, P. Woodward and M. Sandstrom, J. Chem. Soc., Dalton Trans., 1982, 875–882, 10.1039/dt9820000875.
- P. Biscarini, L. Fusina, G. Nivellini and G. Pelizzi, J. Chem. Soc., Dalton Trans., 1977, 664–668, 10.1039/dt9770000664.
- A. S. R. Chesman, J. L. Hodgson, E. I. Izgorodina, A. Urbatsch, D. R. Turner, G. B. Deacon and S. R. Batten, Cryst. Growth Des., 2014, 14, 1922–1932 CrossRef CAS.
- Y. H. Kim, S. Rhim, J. J. Park and J. Kang, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 317–323 CrossRef CAS.
- P. Kumar, M. K. Cabaj and P. M. Dominiak, Crystals, 2019, 9, 668 CrossRef CAS.
- T. Maxson, A. S. Jalilov, M. Zeller and S. V. Rosokha, Angew. Chem., Int. Ed., 2020, 59, 17197–17201 CrossRef CAS PubMed.
- Y. V. Nelyubina, M. Y. Antipin and K. A. Lyssenko, Russ. Chem. Rev., 2010, 79, 167–187 CrossRef CAS.
- R. Zangi, J. Chem. Phys., 2012, 136, 184501 CrossRef PubMed.
- F. Zapata, L. Gonzalez, A. Bastida, D. Bautista and A. Caballero, Chem. Commun., 2020, 56, 7084–7087 RSC.
- D. Braga and F. Grepioni, Chem. Commun., 1998, 911–912, 10.1039/a708438b.
- M. K. Cerreta and K. A. Berglund, J. Cryst. Growth, 1987, 84, 577–588 CrossRef CAS.
- Q. He, P. Y. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- L. A. Bengtsson, B. Noren and H. Stegemann, Acta Chem. Scand., 1995, 49, 391–398 CrossRef CAS.
- R. H. Fenn, Acta Crystallogr., 1966, 20, 20–23 CrossRef CAS.
- P. Ghosh and A. Chakravorty, Indian J. Chem. A, 2013, 52, 1247–1250 Search PubMed.
- D. Grdenic, M. Sikirica and I. Vickovic, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1977, 33, 1630–1632 CrossRef.
- V. V. Gritsenko, O. A. Dyachenko, G. V. Shilov, R. N. Lyubovskaya, T. V. Afanaseva, R. B. Lyubovskii and M. K. Makova, Russ. Chem. Bull., 1992, 41, 697–703 CrossRef.
- T. S. Lobana, S. A. Mbogo, W. R. Mcwhinnie, W. C. Patalinghug and A. H. White, J. Organomet. Chem., 1990, 390, 29–34 CrossRef CAS.
- T. G. Takhirov, O. A. Dyachenko, L. O. Atovmyan, E. I. Zhilyaeva and R. N. Lyubovskaya, J. Struct. Chem., 1990, 31, 712–719 CrossRef.
- A. Bauze, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef PubMed.
- T. Xia, D. Li and L. J. Cheng, Chem. Phys., 2020, 539, 110978 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- C. Moller and M. S. Plesset, Phys. Rev., 1934, 46, 0618–0622 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Phys. Chem., 1993, 98, 1358–1371 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Phys. Chem., 1987, 87, 5968–5975 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Headgordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- P. Su and H. Li, J. Phys. Chem., 2009, 131, 014102 CrossRef PubMed.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Włoch, P. Xu, F. Zahariev and M. S. Gordon, J. Phys. Chem., 2020, 152, 154102 CrossRef CAS.
- C. Peng and H. Bernhard Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- S. R. Kass, J. Am. Chem. Soc., 2005, 127, 13098–13099 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, ChemPhysChem, 2012, 13, 1421–1424 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. Phys. Lett., 2013, 555, 106–109 CrossRef CAS.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2014, 119, 183–194 CrossRef PubMed.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS.
- G. Frenking and G. F. Caramori, Angew. Chem., Int. Ed., 2015, 54, 2596–2599 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- F. Weinhold, Inorg. Chem., 2018, 57, 2035–2044 CrossRef CAS PubMed.
- R. Barbas, R. Prohens, A. Bauzá, A. Franconetti and A. Frontera, Chem. Commun., 2019, 55, 115–118 RSC.
- D. A. Cullen, M. G. Gardiner and N. G. White, Chem. Commun., 2019, 55, 12020–12023 RSC.
- N. G. White, CrystEngComm, 2019, 21, 4855–4858 RSC.
- W. Zhao, B. Qiao, J. Tropp, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2019, 141, 4980–4989 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, J. Phys. Chem. A, 2020, 124, 2207–2214 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, Phys. Chem. Chem. Phys., 2020, 22, 11348–11353 RSC.
- G. Wang, Z. Chen, Z. Xu, J. Wang, Y. Yang, T. Cai, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 610–620 CrossRef CAS PubMed.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481–487 CrossRef CAS PubMed.
- Z. Zhu, G. Wang, Z. Xu, Z. Chen, J. Wang, J. Shi and W. Zhu, Phys. Chem. Chem. Phys., 2019, 21, 15106–15119 RSC.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed.
- K. Ghosh, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 1429–1438 RSC.
- Y. Li, L. Meng and Y. Zeng, ChemPlusChem, 2021, 86, 232–240 CrossRef CAS.
- S. Scheiner, R. Wysokiński, M. Michalczyk and W. Zierkiewicz, J. Phys. Chem. A, 2020, 124, 4998–5006 CrossRef CAS PubMed.
- R. Wysokiński, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 4818–4828 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01502h |
| This journal is © the Owner Societies 2021 |