
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom high quality packing to disordered nucleation or phase separation in donor/acceptor interfaces: ClAlPc-C60 on Au(111)†
Esther
Barrena
 a,
Rogger
Palacios-Rivera
a,
José I.
Martínez
b and
Carmen
Ocal
*a
a,
Rogger
Palacios-Rivera
a,
José I.
Martínez
b and
Carmen
Ocal
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, Bellaterra, E-08193, Barcelona, Spain. E-mail: cocal@icmab.es
bDept. Nanostructures and Low-dimensional Materials, Institute of Material Science of Madrid (ICMM-CSIC), Campus UAM, C/Sor Juana Inés de la Cruz 3, E-28049 Madrid, Spain
First published on 8th June 2021
Abstract
The dramatic consequences that the orientation adopted by the molecular dipoles, in diverse arrays of chloroaluminum phthalocyanine (ClAlPc) on Au(111), have on the ulterior adsorption and growth of C60 are explored by means of an all scanning probe microscopy approach. The unidirectional downwards organization of the molecular dipoles at the first layer reduces charge transfer from the metal to C60. Imbalance between attractive and repulsive interactions of the fullerenes are crucial for their ordered supramolecular aggregation. The effect at the basis of such self-assembling seems to be released by the all upwards dipole orientation adopted on the ClAlPc second layer. The low electronic corrugation of the bilayer results in a higher mobility of the fullerenes which for similar coverages diffuse large distances to reach uncovered first layer regions. Density functional theory calculations corroborate the experimental observations indicating the relevance of charge transfer, potential energy surface corrugation, C60 on-surface diffusion barriers and screening. The structure of the co-adsorbed C60 and ClAlPc layers strongly depends on the deposition sequence. Phase-separation, where each molecule adopts the single-component assembly, occurs if C60 is deposited first. The present results contribute to understanding the influence of the dipolar nature of molecular layers on the electronic and structure of donor/acceptor heterojunctions, which is crucial for device design via engineering the energy level alignment at organic–organic and organic–metal interfaces.
Introduction
Understanding the fundamental processes taking place between organic molecules at the donor/acceptor interface, as well as the role of the metal surface, is of enormous importance in optimizing organic electronic devices, such as organic displays, organic field effect transistors, and organic photovoltaic cells. In particular, how competing intermolecular interactions at different scale lengths influence molecular assembly, affecting the interfacial structure, is relevant for practical applications because it controls the band alignment of the molecular systems. Investigating these issues implies, however, the complex task of considering several weak interactions, such as van der Waals, hydrogen bonds and electrostatic interactions. Moreover, supramolecular organization and self-assembly1,2 (the “bottom-up” fabrication method with the lowest energy consumption) on metals and charge transfer between metal surfaces and molecules3,4 (essential for charge injection/extraction in molecular electronics) depend on a delicate balance between inter and intramolecular interactions and molecule-surface interactions, which are highly system-dependent. Therefore, the relevance of describing the electronic processes at the molecule/molecule and molecule/metal interfaces, in connection with the structure of the interfaces themselves, is evident for application in devices that involve molecular species in contact with metallic electrodes.In the workbench of electronic interface design, sandwich structures, where one thin organic layer, acting as spacer, is intercalated between the metallic substrate and the electron acceptor material, have been routinely employed to tune surface work function and control charge transfer from the metal. Self-assembled monolayers covalently bonded to the metal are the most common dielectric spacers used to this end.5–8 Richer though much more complex scenarios are open at the donor/acceptor interface using small organic semiconductor molecules.
For years, C60 fullerenes have been considered as the archetypal electron acceptor materials due to their large electron affinity and good stability. Because of their practical relevance, a good deal of experimental and theoretical investigations have been devoted to both, electronic properties and structural details, for fullerenes on close-packed metals.9 Fewer exist concerning the growth of C60 on top of ordered organic monolayers on metals.10–13 In all cases, the subtle balance between molecule–molecule and molecule–substrate interactions is invoked. In particular, the observation of serpentine one dimensional chains or disordered phases have been interpreted in terms of repulsion between fullerene molecules that result charged to some extent after charge transfer from the metal to the fullerene through the intercalated layer.10 The possibility of modulating the weight of the contributing interactions by incorporating, under the acceptor molecules, layers made out of molecules with a permanent dipolar moment of diverse orientation emerges as an appealing benchmark. Wei et al. investigated how polymorphism of well-organized monolayers of the polar titanyl phtalocyanine (TiOPc)14 affects the interface built upon deposition of C60. Restructuring of the TiOPc monolayer, formation of co-crystalline networks and phase separation into domains with abrupt boundaries were reported. Interestingly, TiOPc molecules initially packed in a tilted (non-planar) configuration were displaced by the C60 deposited on top.15
In this context, the system formed by C60 and chloroaluminum phthalocyanine (ClAlPc) is attractive, because both are known to separately yield well-ordered monolayers on Au(111) which have been well electronically studied. Herein, the effect of the molecular dipoles within a layer intercalated between C60 and the metal is studied by depositing C60 molecules at room temperature (RT) on diverse ordered ClAlPc layers. To understand the influence of dipole orientation, three different scenarios are studied: all Cl-up monolayer, mixed Cl-up and Cl-down monolayer and a bilayer consisting of Cl-up and Cl-down molecules adopting a staggered stacking. Additionally, in an attempt to pursue the role of relative orientation between molecules, the reverse deposition order is also analyzed.
ClAlPc is a nonplanar molecule with permanent electric dipole perpendicular to its molecular π-plane that can adopt two distinct planar configurations (Cl-up and Cl-down) when adsorbed on metallic surfaces. Competing molecule–molecule and molecule-metal interactions make the adsorption of this dipolar molecule particularly complex. The ClAlPc/Au(111) system has been extensively studied16–19 and it is known that, after a moderate post-deposition annealing, the monolayer consists of a highly ordered array of all Cl-up molecules, where the interaction of the conjugated plane and the metal (π-dz2 coupling) overcomes the dipolar interaction between molecules. Molecules in the second layer adopt the Cl-down configuration and a fourfold adsorption that leads to a staggered stacking, optimizing the electrostatic interaction between first and second layers. It has also been reported that depending on the strain degree of the reconstructed topmost surface of the Au(111) substrate (that can vary with substrate preparation and post-growth annealing temperature) the first layer can be built up by the coexistence of Cl-down and Cl-up configurations, as reported for Au(111)/mica.16
The combination of scanning tunneling microscopy (STM) and frequency modulation atomic force microscopy (FM-AFM) with ab initio calculations provide a vast description of the created C60/ClAlPc interfaces. The consistency of the energetic and structural landscapes in this particular donor/acceptor system provides some general clues to envisage future fine-tuning of surface work function and design-controlled interfaces.
Methods
Experimental details
The Au(111) single crystal (Mateck GmbH, Germany) was prepared by repeated cycles of Ar+ sputtering (0.8 keV) plus annealing at 600 °C in a preparation chamber equipped with Low Energy Electron Diffraction (LEED) optics and diverse evaporators connected to the STM/AFM characterization chamber. After several cleaning cycles, the observation of a sharp pattern by LEED and the herringbone (HB) reconstruction by STM confirmed a well-ordered clean Au(111). The HB double rows of discommensuration lines (DL) along the 〈112〉 directions were used to establish the crystal azimuth orientation for the structural analysis throughout the present work. This in-situ reference is of relevance since the HB pattern, driven by tensile surface stress, can have a very different local appearance on diverse areas of the same surface.16,20,21 Extreme cases are the so called rosetta structure where HB domains converge in a complex star-like structure and the simplest pairwise parallel stripes with consequences on the characteristics of deposited molecular overlayers.20 It is worth mentioning that differences in Au(111) strain release have been shown to affect the orientation of the ClAlPc molecules within the monolayer leading to short-order and disordered mixed (Cl-up and Cl-down) monolayers.16 Conversely, the HB patterns have been reported to change upon adsorption of perylene and FePc due to an anisotropic stress induced by charge transfer at the Au interface.22The ClAlPc molecules (Sigma-Aldrich) were purified twice by gradient thermal annealing before being introduced in the ultra-high vacuum (UHV) chamber where they were degassed and sublimated employing a Knudsen cell at 345 °C. Well-ordered layers were obtained in samples annealed for 10 min at 150 °C immediately after deposition (post-growth annealing). The C60 molecules were also in situ deposited from the vapour phase using deposition rates of about 2–3 Å min−1 using a Knudsen cell (380 °C). The diverse molecular coverages (θ) at the surface have been estimated from topographic STM images.
The experiments were carried out at RT under a base pressure of 1 × 10−10 mbar, using a commercially combined STM/AFM-UHV system Aarhus SPM 150 with KolibriSensor™ probes (f0 ∼ 1 MHz, Q ∼ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and a Nanonis Control System (SPECS Surface Nano Analysis GmbH). The sharp metallic tip was cleaned in situ via Ar+ sputtering and, during STM measurements, was maintained oscillating at a constant amplitude (A = 200 pm) so that the tip–sample interaction was reflected in a frequency shift (Δf) from f0.23 Topographic STM was conducted in the constant current mode and the simultaneous Δf was recorded. Typical tunnelling parameters were: sample bias voltages of 1–2 V and currents of 100–200 pA. The in-plane lattice constant, in high resolution images, and step heights, in large-scale images of the in-situ cleaned Au(111) were employed for calibration of the STM piezo scanner. In the employed setup, the bias voltage is applied to the sample. The local contact potential difference (CPD) of the surfaces was determined from the parabolic dependence of the Δf versus bias voltage such that higher CPD corresponds to higher local work function. Details on CPD measurements are provided in the ESI.† All STM/AFM images were analyzed by using the WSxM freeware.24
000) and a Nanonis Control System (SPECS Surface Nano Analysis GmbH). The sharp metallic tip was cleaned in situ via Ar+ sputtering and, during STM measurements, was maintained oscillating at a constant amplitude (A = 200 pm) so that the tip–sample interaction was reflected in a frequency shift (Δf) from f0.23 Topographic STM was conducted in the constant current mode and the simultaneous Δf was recorded. Typical tunnelling parameters were: sample bias voltages of 1–2 V and currents of 100–200 pA. The in-plane lattice constant, in high resolution images, and step heights, in large-scale images of the in-situ cleaned Au(111) were employed for calibration of the STM piezo scanner. In the employed setup, the bias voltage is applied to the sample. The local contact potential difference (CPD) of the surfaces was determined from the parabolic dependence of the Δf versus bias voltage such that higher CPD corresponds to higher local work function. Details on CPD measurements are provided in the ESI.† All STM/AFM images were analyzed by using the WSxM freeware.24
Computational details
All ab initio calculations, for structural optimization, electronic structure properties and transition-state energy barriers, have been carried out by Density Functional Theory (DFT) as implemented in the plane-wave QUANTUM ESPRESSO simulation package.25 One-electron wave functions are expanded in a plane-waves basis with energy cut-offs of 450 and 550 eV for the kinetic energy and the electronic density, respectively. Exchange and Correlation (XC) have been computed in the revised version of the generalized gradient corrected approximation (GGA) of Perdew–Burke–Ernzerhof (rPBE).26 Rabe–Rappe–Kaxiras–Joannopoulos (RRKJ) ultrasoft pseudopotentials24–27 have been adopted to model the ion–electron interaction. In all the calculations, the Brillouin zone (BZ) has been sampled using optimal [2 × 2 × 1] Monkhorst–Pack grids.28 The van der Waals (vdW) interaction has been included via an empirical vdW R−6 correction (DFT+D3).29 Atomic positions have been optimized using a conjugate gradient minimization scheme until the maximum force on any atom was lower than 0.02 eV Å−1. The Fermi level was smeared out using the Methfessel–Paxton approach30 with a Gaussian width of 0.01 eV, and all energies were extrapolated to T = 0 K. Self-consistency in the electron density to a precision in the total energy better than 10−6 eV was achieved. Transition state geometries and energy barriers have been obtained with the Climbing-Image Nudged-Elastic Band method (CI-NEB),31 as implemented in QUANTUM ESPRESSO, using 12 intermediate images with a convergence threshold for the norm of the force orthogonal to the reaction path of 0.05 eV Å−1. To estimate the barriers for diffusion from the most stable configurations, we have computed minimum reaction paths (MEP) by the CI-NEB method taking as starting points these equilibrium configurations, stablishing a final state at around 4 Å from the starting point along the diverse directions indicated in each case and including an intermediate step at midway of 2 Å to boost the convergence.Structural models
Monolayer and bilayer ClAlPc/Au(111) interfacial models adopted here to theoretically analyze the different C60 adsorption geometries and energies, charge transfers and diffusion transition-state energy barriers, have already been introduced and described in detail in previous literature by our group.19 It is important to remark that, in order to eliminate the undesired perpendicular inter-cell interaction after the deposition of the C60 molecules on the different substrates, a minimum distance of 15 Å of vacuum is considered between neighboring cells along the axis perpendicular to the surface in our periodic modelling. A complete description of the substrate models to consider the adsorption of C60 molecules can be found in the ESI.†Results and discussion
Experimental results
Well-ordered monolayers of all Cl-up ClAlPc on Au(111) were prepared using a moderate post-growth annealing as described in the experimental section. The molecules self-arrange in the square lattice (aClAlPc = bClAlPc = 1.38 ± 0.02 nm) shown in Fig. 1 and full details are given in a previous work.19 In this monolayer, all molecules adopt the Cl-up configuration, that is, the layer consists of dipoles with unidirectional downwards orientation (μ↓) and their aromatic plane at about ∼3 Å from the surface and rotated 10° with respect to the square unit cell axes. | ||
| Fig. 1 (a) ClAlPc molecule, top and side view cartoons of the square lattice the Cl-up (downwards dipole) of the molecules within the monolayer. (b–d) STM topographic images for 1 ML of ClAlPc on Au(111); STM parameters: I = 130 pA and V = +1.25 V (b and c) and V = −1.04 V (d). | ||
As it can be seen in wide scan STM images (Fig. 1b and c), large domains of the HB pattern of the reconstructed gold surface are visible underneath and are used to determine the substrate main crystallographic directions. In general, the persistence of the HB is interpreted as an indication of molecular physisorption on a non-strained Au(111) surface. It is worth mentioning here that the HB reconstruction is a manifestation of strain relief due to an increased density (∼4%) of the topmost surface relative to a bulk-truncated (111) surface. Subtle differences in surface relaxation have been observed, for example, in gold films grown on mica, Au(111)/mica.16 Creation of highly mobile adatoms during the preparation procedure may also lead to small density variability and subsequent layer strain fluctuations manifested through HB domain distribution. Since the quality of the organic overlayer depends on it, we will come back to this fact later in this work.
On bare metal (111) surfaces, the C60 molecules are known to form close-packed hexagonal phases. In particular, on Au(111) it forms diverse well-ordered monolayers with commensurate structures among which the more frequently observed are: (2√3 × 2√3)R30° and (7 × 7)R0°,32 where R is the relative angle between the lattice orientation of the C60 overlayer and one nearest neighbour (NN) direction of the (111) substrate. Fig. 2 illustrates the result of depositing a submonolayer coverage (θ ∼ 0.3 ML) of C60 on a complete Cl-up monolayer. The particular surface region shown in Fig. 2a contains two terraces separated by a monoatomic step of Au(111). As expected from the symmetry relationship between the square adlayer and the hexagonal planes of the fcc crystal, the relative orientation of the square lattices of ClAlPc at each side of the step is 60°. The excellent quality of the C60 close-packed assemblies indicates the good mobility of these molecules at RT on highly ordered regions of the ClAlPc monolayer. The C60 islands have an apparent height of 0.5 ± 0.5 nm with respect to the underlying ClAlPc monolayer and display a remarkable flatness, with a corrugation of 65 ± 0.5 pm. The molecules exhibit a smooth hemispherical shape. The unit cell of aC60 = 0.98 ± 0.05 nm, oriented along the NN directions of the Au(111). The magnification images of Fig. 2b and c and their corresponding two-dimensional fast Fourier transform (2D-FFT) in Fig. 2d, indicate that the unidirectional epitaxial relationship of the two lattices is 5aClAlPc = 7aC60 along the common [01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ]. In contrast to the large well-ordered 2D islands, wrinkled chains, isolated molecules and small disordered nuclei of C60 are observed at the lower step edge and at less extended defects of the underlying monolayer on both terraces of Fig. 2a. Such defects are associated either to the presence of disordered regions of ClAlPc molecules close to the steps or to domain boundaries of the square lattice. This is the case shown in Fig. 2e. The existence of equivalent domains laterally displaced half unit cell, with the subsequent frontier along [2], is highlighted by the white dotted lines drawn in the Δf image of Fig. 2f.
]. In contrast to the large well-ordered 2D islands, wrinkled chains, isolated molecules and small disordered nuclei of C60 are observed at the lower step edge and at less extended defects of the underlying monolayer on both terraces of Fig. 2a. Such defects are associated either to the presence of disordered regions of ClAlPc molecules close to the steps or to domain boundaries of the square lattice. This is the case shown in Fig. 2e. The existence of equivalent domains laterally displaced half unit cell, with the subsequent frontier along [2], is highlighted by the white dotted lines drawn in the Δf image of Fig. 2f.
 | ||
| Fig. 2 (a) STM topography (50 nm × 39 nm) of θ ∼ 0.3 ML of C60 on 1Ml of ClAlPc on Au(111) at RT. (b) and (c) are magnified images of the ClAlPc and C60 assemblies on the same terrace; STM parameters: I = 238 pA and V = +1.97 V. (d) Superimposed 2D FFT of ClAlPc (blue) and C60 (red) domains. Some crystallographic directions are indicated (see text). The cartoons at (g) and (h) illustrate the nucleation of the fullerenes over the monolayer and at defects. | ||
The similarity between the close-packed hexagonal phase, formed on the ordered ClAlPc monolayer (Fig. 2a and c), and those found for C60 layers on bare metal substrates is remarkable and manifestly contrasts with the quasi-one dimensional organization and low density disordered phases of C60 over zinc phtalocyanine (ZnPc), pentacene or α-sexithiophene (α-6T) layers on (111) oriented Au and/or Ag surfaces.10,12 The lack of 2D order in those systems has been attributed to the competitive weight of attractive forces and long-range repulsive interactions, regardless of the supramolecular structure of the underlying layers. It is clear that the presence of an intercalated layer between the metal and C60 may alter the C60 intermolecular interactions. For example, in the absence of an interlayer, the repulsive electrostatic interaction between charged molecules, due to charge transfer from the metal to the fullerenes, is effectively screened, therefore allowing molecular self-assembly. Even if charge transfer persists in the presence of an interlayer, the larger distance between the charged C60 and the metal results in diminished screening. For non-charged or weakly charged fullerenes, the hexagonal packing is favoured by strong van der Waals attractions between molecules but is also an indication of their facile diffusion favoured by weak intermolecular interactions with the underlying organic molecules. Indeed, as we will describe below, electronic corrugation influencing diffusion barriers also plays a role and, in the case of an interlayer made out of non-planar molecules with a permanent dipole, the scenario can get much more complicated.
As it is known, the work function (ϕ) of the metal's surface is modified by the adsorption of molecules though different processes among which, for example, molecule–substrate charge redistribution and “push-back” effect compete with permanent dipoles in the case of ClAlPc. Because the cases of the single layer (Cl-up) and the bilayer (Cl-down on Cl-up) have already been reported19 (see also Fig. S1 in the ESI†) we only briefly discuss here the consequences of the ClAlPc interlayer on the fullerene layer. Compared to the spatially averaged values obtained by UPS, CPD values obtained by local probes allow spatially resolved measurements on coexisting differentiated regions. Therefore, to get a picture of the energy level alignment, we performed systematic local CPDs on the similarly ordered layers of C60 on the bare and the ClAlPc monolayer. The result is presented in Fig. 3, where the clean Au(111) work function (ϕ = 5.35 eV) as measured separately by UPS is shown as a reference. On top of the well-ordered C60 islands directly grown on gold (see for example Fig. 5a), the work function is reduced by Δϕ = −100 meV. This reduction is lower than that reported by UPS for one monolayer.33 Otherwise, the effect of depositing an ordered ClAlPc monolayer on Au(111) is increasing the work function by Δϕ = +70 meV, a value that falls by Δϕ = −50 meV when C60 islands are grown on top. In summary, the intercalation of an ordered Cl-up ClAlPc monolayer between C60 and the metal surface slightly raises the work function above that of the clean Au(111). As we will discuss later, a direct consequence of this vacuum level offset is that electron transfer from the metal surface to C60 becomes unfavourable. Though in this particular system the absolute value of the changes are small, the results open new avenues for interface modification via surface engineering using dipolar molecular layers.
 | ||
| Fig. 3 Vacuum energy levels obtained from CPD measurements on the C60 islands, ClAlPc monolayer and C60/ClAlPc on Au(111). The work function of the substrate is used as a reference. Cartoons at the top illustrate the different cases. | ||
Despite the neat results described above for the all Cl-up monolayer formed over the highly ordered HB reconstruction of the relaxed Au(111) substrate, the situation becomes more complex if Cl-up and Cl-down configurations coexist on the very same molecular layer. In fact, in the absence of substrate influence, the dipolar interaction between ClAlPc neighbour molecules would favour assemblies with antiparallel orientation of the dipoles (Cl-up and Cl-down). Indeed, disordered or short-range order mixed 2D layers of ClAlPc have been reported for Au(111)/mica and ascribed to differences in the gold surface relaxation.16 Such a ClAlPc layer can be seen in Fig. 4a and b, for a coverage slightly over the monolayer, with the concurrence of the first layer and some individual and small aggregates of a second layer. Close inspection of the lower level reveals that, in addition to the rounded protuberances (bright in both topography and Δf) typical of molecules in the Cl-up configuration, molecules with Cl-down are also seen (flat four lobes appearance in topography and featureless aspect in Δf). The distribution of each dipole orientation is estimated to be ∼50% in the first layer. In the topographic image shown in Fig. 4c a bicolour scale has been used to clearly differentiate the deposited C60 (red) from the ClAlPc molecules (green). The contrast between the disordered C60 aggregates on the mixed layer monolayer and the well-packed hexagonal assemblies on the all Cl-up monolayer (Fig. 2a) is obvious. The lack of ordered packing of the fullerenes on the mixed surface is evidenced in the large-scale image of Fig. 4d. Molecules accumulate at extended defects and at small single layer patches, but no order takes place. Then, we can conclude that though the mobility of the molecules is considerably high so they meet and nucleate together, the repulsive interactions seem enough to impede optimal close packing.
 | ||
| Fig. 4 (a) STM topography and (b) simultaneously measured Δf for a mixed (Cl-up and Cl-down) monolayer of ClAlPc. Few second layer molecules are also seen. (c and d) STM topographic images of the same surface after deposition of θ ∼ 0.1ML of C60. STM parameters: I = 200 pA and V = +2.0 V. The bicolor scale of (c) highlights the disordered nucleation of C60 (red). | ||
After knowing that the misaligned disposition of the permanent dipoles in the first layer (top Fig. 5a) hinders organization of the fullerenes, we explore C60 deposition on an incomplete and defective bilayer. The staggered configuration of dipoles with opposite orientation within first and second layers is shown in Fig. 5a. Fig. 5c–f show different areas of the resulting surface after deposition of C60 (θ ∼ 0.2 ML). The most striking observation is that no matter how large the region is, there is neither adsorption nor nucleation on the second ClAlPc layer (Fig. 5c). In fact, and contrary to what happens in the case of the Cl-up monolayer, no nucleation is observed at punctual defects (vacancies) or extended defects (domain boundaries). The molecules diffuse long distances (several tens of unit cells) over this layer to reach empty first layer regions, where due to the Cl-up and Cl-up coexistence, they nucleate in a disordered manner (Fig. 5c and d) as schematically depicted in the cartoon of Fig. 5e. Remarkably, this process takes place even for relatively large amounts of deposited C60 (Fig. 5f). In the corresponding Δf map of Fig. 5g, the presence of two different domains and the downwards orientation of the ClAlPc molecules forming the second layer are clearly seen. A large number of fullerenes, individually recognized in the images, huddle together at the domain boundary. Outstandingly, at the tallest region of the C60 pile (circled at the Δf image centre) some short-range order is detected, likely indicating the vanishing influence of the layer underneath. The lack of C60 adsorption on the second ClAlPc layer, even for a sufficiently large coverage, is certainly a consequence of a high mobility and large diffusion path of the molecules on the upper level. However, the origin of such low energy barrier for surface diffusion is elusive. As commented above, the key will be found at the electronic corrugation of the ClAlPc layer.
 | ||
| Fig. 5 (a) Cartoons illustrating the side views of a mixed (disordered arrangement of Cl-up and Cl-down) monolayer (top) and an incomplete bilayer (bottom) of ClAlPc. (b) Top view of the ClAlPc bilayer. (c) and (d) STM topography of C60 deposited on a surface consisting of 70% of a bilayer of ClAlPc and 30% of a mixed monolayer. (f) STM topography and (g) simultaneously measured Δf for a nearly complete bilayer of ClAlPc. STM parameters: (c) I = 357 pA and V = +2.2 V, (d) I = 200 pA and V = +2.0 V, and (f and g) I = 150 pA and V = +1.8 V. | ||
In an attempt to rationalize from a theoretical perspective the different scenarios experimentally observed after the deposition of C60 molecules on: (i) well-ordered monolayers of all Cl-up ClAlPc on Au(111), and (ii) ClAlPc bilayer on Au(111), we have carried out a large and time-consuming battery of DFT-based calculations. A detailed structural description of the two types of ClAlPc adlayers on Au(111) can be found elsewhere.19 The factors employed for the choice of the possible configurations, for C60 on both adlayers as well as for the gas-phase interaction geometries, are described in the ESI.† Interestingly, in the gas phase, the calculation indicates that charge transfer and interaction energy strongly depend on the interaction geometry and relative orientation between the C60 and ClAlPc molecules (ESI,† Fig. S5). We have computed equilibrium geometries (keeping fixed the two bottommost Au(111) layers during the optimization process), adsorption energies and Bader charge transfers from the substrate for a C60 molecule (per unit cell) on a monolayer of Cl-up ClAlPc on Au(111) on three representative adsorption sites (for each analysing two cases: the interacting lowest part of the C60 being a C6-ring or a C2-bridge). The different adsorption sites and their corresponding optimized geometries obtained from the calculations are shown in Fig. 6. Most stable configurations are those with the C60 molecule lying on the terminal Cl atom (Fig. 6a and b), with adsorption energies per C60 of 1.31 and 1.14 eV for the interaction with a bottommost C6-ring or a C2-bridge, respectively. These values can be directly compared with 1.12 and 0.81 eV obtained for the gas-phase interaction between C60 and ClAlPc in the same interaction configurations (Fig. S5 in the ESI†). In the other on-surface configurations analysed (Fig. 6c–f) the C60 molecule lies on the molecular “wings” with residual lateral interactions with the Cl atoms, yielding adsorption energies ranging between 0.6 and 0.75 eV governed by vdW interactions, nonetheless sufficient to anchor the molecules to the surface.
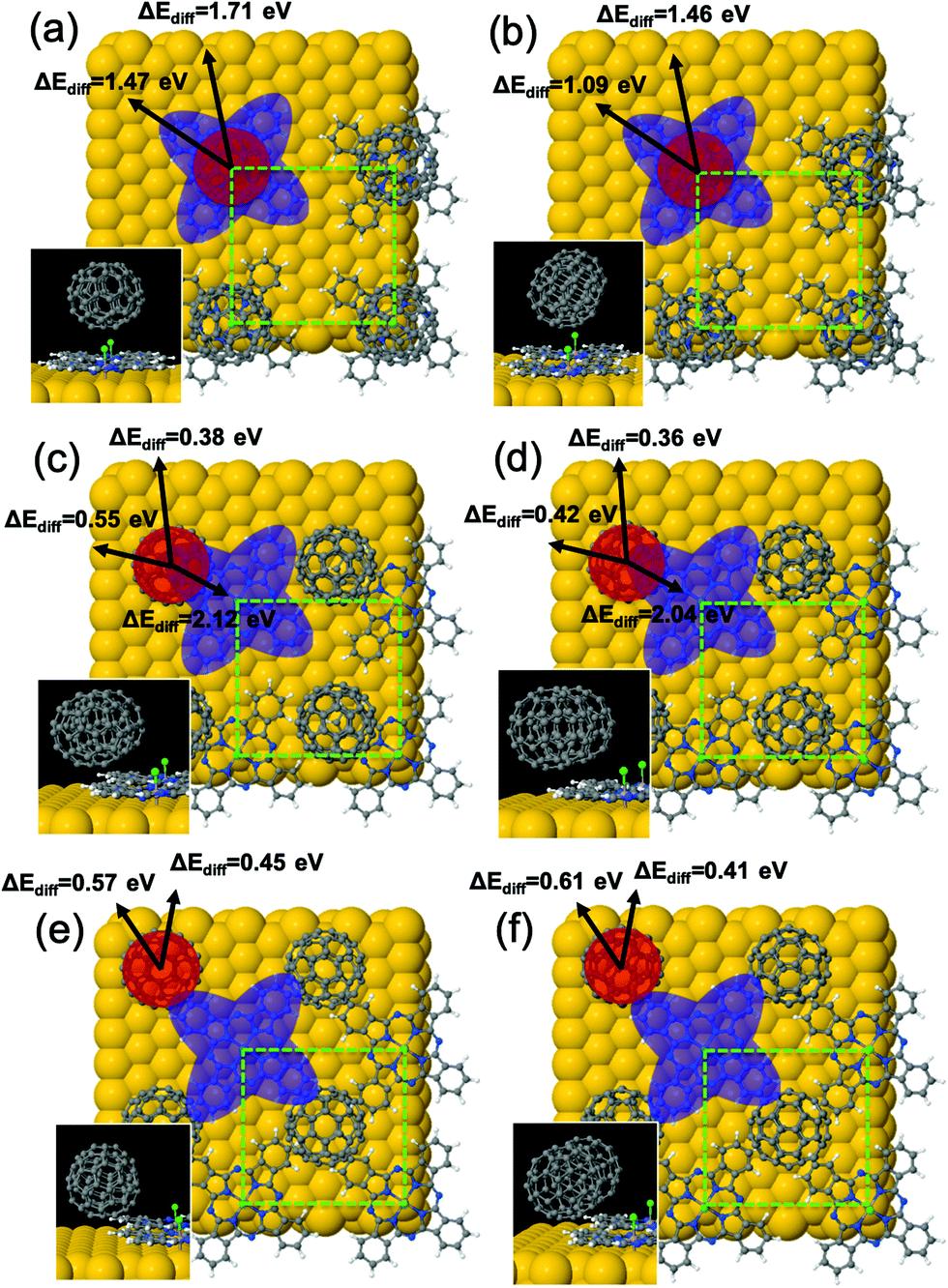 | ||
| Fig. 6 Top and side (insets) pictorial views of the DFT-optimized structural models for the adsorption of C60 molecules on an ordered monolayer of all Cl-up ClAlPc on Au(111) for six representative adsorption sites (a–f). The unit cell used in the calculations is shown as a light-green dashed-lined box. Blue and red shadowed regions represent the Cl-up ClAlPc and C60 molecules, respectively. Computed CI-NEB diffusion barriers (ΔEdiff; in eV) are indicated along specified directions (black arrows) for each case. More details are given in Fig. S2 of the ESI.† | ||
We have also computed the charge transfer processes (see the Methods section) occurring after the formation of the interface of C60 on the Cl-up ClAlPc/Au(111). We obtain charge transfers from the substrate to the C60 molecules of 0.22 and 0.16 e− for the configurations in Fig. 6a and b, respectively. Within the theoretical formalism employed in this work, the charge transfer from the Au(111) to the C60 in its most stable and fully relaxed (2√3 × 2√3)R30° periodic geometry34,35 is 0.28 e−. A value significantly lower than the 0.8 e− estimated by resonant photoemission for the C60/Au(111) interface.36 Although the computed values are of the same order with or without the ClAlPc monolayer, they are higher by 30% and 75% for C60/Au(111) than for the configurations in Fig. 6a and b. This can be considered as a non-negligible reduction of the charge transfer to C60 in the presence of the unidirectional dipole interlayer, and therefore diminishing the charge-induced repulsion between molecules. Besides, upon C60 adsorption the ClAlPc adlayer accumulates, on average per molecule, an excess of electronic charge of 0.19 e−, appreciably lower than without C60 molecules (0.32 e−).19 The consequences in molecular assembling are reinforced by the calculation of diffusion barriers from the most stable configurations in Fig. 6. The calculations yield on-surface diffusion barriers ranging between 0.36 eV and 1.71 eV, justifying only relative RT diffusion of the C60 molecules on the Cl-up ClAlPc monolayer. The low repulsion between C60 molecules, their moderate diffusion, summed to the intensity of the interaction with the underlying ClAlPc, favours the stabilization of the C60 molecules and leads to the formation of the robust close-packing of the fullerenes observed in Fig. 2a and c.
In a following step, we have computed equilibrium geometries, adsorption energies and Bader charge transfers from the substrate for a C60 molecule (per unit cell) on the ClAlPc bilayer on Au(111). Fig. 7 shows the 4 different adsorption sites studied (two with the interacting lowest part of the C60 being a C2-bridge and two being a C6-ring), and the corresponding optimized geometries. In this case the most stable configurations are those with the C60 molecule lying on the Al atom of the Cl-down ClAlPc molecules with a bottommost C2-bridge (Fig. 7a) and a C6-ring (Fig. 7b). The adsorption energies per C60 in each configuration are 0.48 and 0.56 eV, respectively, with charge transfers below 0.02 e−, in a clear physisorption regime. For the other two cases (Fig. 7c and d), adsorption energy and charge transfer are around 0.4 eV and <0.01 e−, respectively. The net charge accumulated in the bilayer of around 0.1 e− in all cases. The scenario has completely changed with respect to the adsorption on the Cl-up ClAlPc monolayer. Now, the nature of the interaction is merely vdW and adsorption energies are rather low. In order to evaluate the diffusion rate of the C60 molecules, we have computed the diffusion barriers, similarly to the previous case. The result of these calculations shows in all cases diffusion barriers <0.11 eV independently of the diffusion direction tested, which justifies a high diffusion capability of the molecules deposited on the bilayer. This effect is clearly driven by the low electronic corrugation of the bilayer, leaving the C60 molecules free to diffuse long distances on the surface until reaching highly-reactive on-surface defects or empty first layer regions, as experimentally evidenced.
 | ||
| Fig. 7 Top and side views of the DFT-optimized structural models for the adsorption of C60 on an ordered bilayer of Cl-up (bottom layer) and Cl-down (top layer) ClAlPc on Au(111). Four different fully DFT-optimized configurations are depicted resulting from the consideration of two representative adsorption sites and two different C60 orientations (interacting via a C6-ring or a C2-bridge). The most stable configurations are those with the C60 molecule lying on the Al atom of the Cl-down ClAlPc molecules with a bottommost C2-bridge (a) and a C6-ring (b). Terminal C2-bridges (left) and C6-rings (right) are represented by red rectangles and hexagons, respectively, in the insets. The unit cell used in the calculations is shown as a light-green dashed-lined box. Blue, green and red shadowed regions represent the Cl-up ClAlPc, Cl-down ClAlPc and C60 molecules, respectively. Computed CI-NEB diffusion barriers (ΔEdiff; in eV) are indicated along specified directions (black arrows) for each case. More details are given in Fig. S3 and S4 in the ESI.† | ||
With a last experimental case study, we will show that the structure of co-adsorbed C60 and ClAlPc layers strongly depends on the deposition sequence. As already known, if C60 is deposited first, islands of compact hexagonal packing are formed on the bare Au(111). For a coverage of θ ∼ 0.5 ML, the surface is made out of crystalline C60 patches surrounded by bare substrate (Fig. 8a–c). Some defects in the form of individual fullerenes with lower apparent height (darker in the image) is characteristic of C60 on Au(111).37 The result of the subsequent deposition of 1ML of ClAlPc on such a surface is shown in Fig. 8b. Remarkably, instead of covering the entire exposed surface (bare gold and C60), the impinging phtalocyanines do not adsorb on the fullerene layer but nucleate at the pristine substrate regions (Fig. 8d). Lateral phase-separation occurs, where each molecule adopts the well-ordered supramolecular order corresponding to the structures of the single-component cases. As the amount of ClAlPc (1ML) is about twice that needed to fill the bare Au(111) regions, the phtalocyanines pile up with the supramolecular packing of the bilayer (Fig. 5b).19 The segregation between molecules, both on the same terrace and on neighbouring terraces, can be seen in Fig. 8e. ClAlPc molecules in the first layer are in close contact with the C60 domains, while because the square lattice of the second layer is shifted a half unit cell in each direction, a thin empty rim exists at the frontier with C60 (see Fig. 8b and e). Though a very low ΔCPD ∼ 50 meV was measured between the two phases (see Fig. 3 and ref. 19) and conversely to abrupt boundaries with negligible dipole reported for phase separated C60-TiOPc films on Ag(111),15,38 disconnected boundaries between C60 and ClAlPc domains on Au(111) may influence the electronic coupling and electrical potential offset across the donor–acceptor interface of relevance for charge separation in OPV devices.
 | ||
| Fig. 8 STM topographic images of (a) θ ∼ 0.5 ML of C60 on Au(111). (b) The same surface after θ ∼ 1 ML of ClAlPc. (c) and (d) are cartoons illustrating the surface in (a) and (b), respectively. ClAlPc does not adsorb but diffuses on C60. (e) Larger area showing phase segregation of ClAlPc and C60. STM parameters: (a) I = 120 pA and V = +1.8 V, (b) I = 180 pA and V = +1.5 V, and (e) I = 200 pA and V = +2.0 V. | ||
Conclusions
We provide experimental evidence of the influence of an intercalated layer of dipolar ClAlPc molecules on the charge transfer between Au(111) and the electron acceptor C60. Both electronic and structural details of the ClAlPc layers become relevant for the organic–organic interface properties. The significance of the charge transfer and energy alignment across the intercalated ClAlPc film has consequences in the balance between attractive and repulsive interactions and, therefore, in the aggregation of the fullerenes. On top of the unidirectional monolayer of ClAlPc, with all molecules in the Cl-up configuration (downwards dipoles), C60 self-assembles in a robust close-packed hexagonal order. Conversely, C60 piles up disorderly when deposited on a monolayer with mixed orientation of the molecular dipoles (disordered coexistence of Cl-up and Cl-down). The overall compensation of dipoles in such mixed configuration would allow competition, between van der Waals intermolecular attraction and repulsion between charged C60 molecules, to manifest by a lack of order in their packing. Remarkably, despite the expected low electrostatic repulsion due to small charge transfer, C60 does not adsorb on the ClAlPc bilayer (topmost layer made out of all Cl-down, upwards dipoles) but nucleates at uncovered patches of the first layer. We ascribe this experimental evidence at the early stages of C60 growth to a lower adsorption energy and the lower diffusion barriers calculated over the bilayer stack with respect to the monolayer. The structure of co-adsorbed C60 and ClAlPc layers strongly depend on the deposition sequence. If fullerenes are deposited first, phase-separation occurs, where each molecule adopts the structure of the single component cases on clean Au(111).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Spanish Government under projects PID2019-110907GB-I00, MAT2016-77852-C2-1-R and MAT2017-85089-C2-1-R (AEI/FEDER, UE) and the “Severo Ochoa” Program for Centres of Excellence in R&D (CEX2019-000917-S). We also acknowledge the Generalitat de Catalunya grant 2017 SGR668. J. I. M. acknowledges Comunidad de Madrid via “Programa de Investigación Tecnologías 2018” (FOTOART-CM S2018/NMT-4367), and the EU Innovation Program under grant agreement 881603 (GrapheneCore3-Graphene-based disruptive technologies).References
- A. Kühnle, Self-assembly of organic molecules at metal surfaces, Curr. Opin. Colloid Interface Sci., 2009, 14, 157–168 CrossRef.
- C. A. Palma, M. Cecchini and P. Samori, Predicting self-assembly: From empirism to determinism, Chem. Soc. Rev., 2012, 41, 3713–3730 RSC.
- N. Koch, S. Duhm, J. P. Rabe, A. Vollmer and R. L. Johnson, Optimized hole injection with strong electron acceptors at organic–metal interfaces, Phys. Rev. Lett., 2005, 95, 237601 CrossRef.
- S. Braun, W. R. Salaneck and M. Fahlman, Energy-level alignment at organic/metal and organic/organic interfaces, Adv. Mater., 2009, 21, 1450–1472 CrossRef CAS.
- G. Witte and C. Wöll, Molecular beam deposition and characterization of thin organic films on metals for applications in organic electronics, Phys. Status Solidi, 2008, 205, 497–510 CrossRef CAS.
- B. de Boer, A. Hadipour, M. M. Mandoc, T. van Woundenbergh and P. W. M. Blom, Tuning of metal work functions with self-assembled monolayers, Adv. Mater., 2005, 17, 621–625 CrossRef CAS.
- J. Ch. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-assembled monolayers of thiolates on metals as a form of nanotechnology, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- M. Singh, N. Kaur and E. Comini, The role of self-assembled monolayers in electronic devices, J. Mater. Chem. C, 2020, 8, 3938–3955 RSC.
- X.-Q. Shi, M. A. Van Hove and R.-Q. Zhang, Survey of structural and electronic properties of C60 on close-packed metal surfaces, J. Mater. Sci., 2012, 47, 7341–7355 CrossRef CAS.
- J. Niederhausen, P. Amsalem, A. Wilke, R. Schlesinger, S. Winkler, A. Vollmer, J. P. Rabe and N. Koch, Doping of C60 (sub)monolayers by Fermi-level pinning induced electron transfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 081411 CrossRef.
- W. Jin, Q. Liu, D. B. Dougherty, W. G. Cullen, J. E. Reutt-Robey, J. Weeks and S. W. Robey, C60 chain phases on ZnPc/Ag(111) surfaces: Supramolecular organization driven by competing interactions, J. Chem. Phys., 2015, 142, 101910 CrossRef CAS PubMed.
- L. Chen, W. Chen, H. Huang, H. L. Zhang, J. Yuhara and A. T. S. Wee, Tunable arrays of C60 molecular chains, Adv. Mater., 2008, 20, 484–488 CrossRef CAS.
- H. L. Zhang, W. Chen, L. Chen, H. Huang, X. S. Wang, J. Yuhara and A. T. S. Wee, C60 molecular chains on a-sexithiophene nanostripes, Small, 2007, 3, 2015–2018 CrossRef CAS.
- X. Liu, Y. Wei, J. E. Reutt-Robey and S. W. Robey, Dipole–dipole interactions in TiOPc adlayers on Ag, J. Phys. Chem. C, 2014, 118(7), 3523–3532 CrossRef CAS.
- Y. Wei and J. E. Reutt-Robey, Molecular interface formation in titanyl phthalocyanine−C60 monolayer films, J. Phys. Chem. C, 2012, 116, 23773–23778 CrossRef CAS.
- T. Niu, Surface strain mediated dipole alignment of ClAlPc on Au(111), Appl. Phys. Lett., 2015, 106, 161601 CrossRef.
- Y. L. Huang, W. Chen, F. Bussolotti, T. C. Niu, A. T. S. Wee, N. Ueno and S. Kera, Impact of molecule-dipole orientation on energy level alignment at the submolecular scale, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 085205 CrossRef.
- Y. L. Huang, Y. Lu, T. C. Niu, H. Huang, S. Kera, N. Ueno, A. T. S. Wee and W. Chen, Reversible single-molecule switching in an ordered monolayer molecular dipole array, Small, 2012, 8, 1423–1428 CrossRef CAS PubMed.
- S. Matencio, R. Palacios-Rivera, J. I. Martínez, C. Ocal and E. Barrena, Chiral organization and charge redistribution in chloroaluminum phthalocyanine on Au(111) beyond the monolayer, J. Phys. Chem. C, 2018, 122, 16033–16041 CrossRef CAS.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Scanning tunneling microscopy observations on the reconstructed Au(111) surface: Atomic structure, long-range superstructure, rotational domains, and surface defects, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307–9318 CrossRef CAS PubMed.
- T. J. Roussel, E. Barrena, C. Ocal and J. Faraudo, Predicting supramolecular self-assembly on reconstructed metal surfaces, Nanoscale, 2014, 6, 7991–8001 RSC.
- J. T. Sun, L. Gao, X. B. He, Z. H. Cheng, Z. T. Deng, X. Lin, H. Hu, S. X. Du, F. Liu and H.-J. Gao, Surface reconstruction transition of metals induced by molecular adsorption, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115419 CrossRef.
- T. R. Albrecht, P. Grütter, D. Horne and D. Rugar, Frequency modulation detection using high-Q cantilevers for enhanced force microscope sensitivity, J. Appl. Phys., 1991, 69, 68–673 CrossRef.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, WSxM: A software for scanning probe microscopy and a tool for nanotechnology, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- P. Giannozzi, et al., QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Optimized pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227–1230 CrossRef PubMed.
- J. D. Pack and H. J. Monkhorst, “Special points for Brillouin-zone integrations”—a reply, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 16, 1748–1749 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, High-precision sampling for Brillouin-zone integration in metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- H. Shin, A. Schwarze, R. Diehl, K. Pussi, A. Colombier, É. Gaudry, J. Ledieu, G. M. McGuirk, L. N. Serkovic Loli, V. Fournée, L. L. Wang, G. Schull and R. Berndt, Structure and dynamics of C60 molecules on Au(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 245428 CrossRef.
- C.-T. Tzeng, W.-S. Lo, J.-Y. Yuh, R.-Y. Chu and K.-D. Tsuei, Photoemission, near-edge X-ray absorption spectroscopy, and low-energy electron-diffraction study of C60 on Au(111) surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 2263–2272 CrossRef CAS.
- E. Abad, J. Ortega and F. Flores, Metal/organic barrier formation for a C60/Au interface: From the molecular to the monolayer limit, Phys. Status Solidi A, 2012, 209, 636–646 CrossRef CAS.
- C. J. Villagómez, I. L. Garzón and L. Oliver Paz-Borbón, A first-principles DFT dispersion-corrected C60/Au(111) Raman study, Comput. Mater. Sci., 2020, 171, 109208 CrossRef.
- A. J. Britton, M. Weston, J. B. Taylor, A. Rienzo, L. C. Mayor and J. N. O’Shea, Charge transfer interactions of a Ru(II) dye complex and related ligand molecules adsorbed on Au(111), J. Chem. Phys., 2010, 133, 094705 CrossRef PubMed.
- J. A. Gardener, G. A. D. Briggs and M. R. Castell, Scanning tunneling microscopy studies of C60 monolayers on Au(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235434 CrossRef.
- K. M. Burson, Y. Wei, W. G. Cullen, M. S. Fuhrer and J. E. Reutt-Robey, Potential steps at C60–TiOPc–Ag(111) interfaces: Ultrahigh-vacuum-noncontact scanning probe metrology, Nano Lett., 2012, 12, 2859–2864 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on the CPD measurements and description of the choice of computed interaction configurtations. Figures showing topography and frequency shift data for the ClAlPc incomplete bilayer and providing the diverse structural models on the surface and gas-phase. See DOI: 10.1039/d1cp01312b |
| This journal is © the Owner Societies 2021 |