
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicolour fluorescent molecular sensors for cations: design and experimental validation
Zoraida
Freixa
 *ab,
Iván
Rivilla
ac,
Francesc
Monrabal
ac,
Juan J.
Gómez-Cadenas
ac and
Fernando P.
Cossío
*cd
*ab,
Iván
Rivilla
ac,
Francesc
Monrabal
ac,
Juan J.
Gómez-Cadenas
ac and
Fernando P.
Cossío
*cd
aIkerbasque, Basque Foundation for Science, 48009 Bilbao, Spain. E-mail: zoraida_freixa@ehu.eus
bDepartment of Applied Chemistry, Faculty of Chemistry, University of the Basque Country (UPV/EHU), 20018 San Sebastián/Donostia, Spain
cDonostia International Physics Center (DIPC), 20018 San Sebastián/Donostia, Spain
dDepartment of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country (UPV/EHU), 20018 San Sebastián/Donostia, Spain
First published on 2nd July 2021
Abstract
Molecular entities whose fluorescence spectra are different when they bind metal cations are termed bicolour fluorescent molecular sensors. The basic design criteria of this kind of compound are presented and the different fluorescent responses are discussed in terms of their chemical behaviour and electronic features. These latter elements include intramolecular charge transfer (ICT), formation of intramolecular and intermolecular excimer/exciplex complexes and Förster resonance energy transfer (FRET). Changes in the electronic properties of the fluorophore based on the decoupling between its constitutive units upon metal binding are also discussed. The possibility of generating fluorescent bicolour indicators that can capture metal cations in the gas phase and at solid–gas interfaces is also discussed.
 Zoraida Freixa | Zoraida Freixa completed her PhD in the area of homogeneous catalysis at the Autonomous University of Barcelona. After a post-doctoral stage at Prof. van Leeuwen's group in Amsterdam, she enrolled as Ramón y Cajal Researcher at the Institute of Chemical Research of Catalonia (ICIQ) as a group manager of Prof. van Leeuwen's group. Since 2010, and after a short period as a Lecturer at the University of Barcelona, she has been holding an Ikerbasque Research Professor position at the University of the Basque Country (UPV/EHU) in San Sebastián. She leads a research group focused on the development of photoswitchable and/or chiral organometallic compounds with special emphasis on the study of their catalytic and luminescence properties. Since 2019, she has been involved in the development of bicolour sensors for barium and radium tagging in ββ0ν reactions. |
 Iván Rivilla | Iván Rivilla graduated in Chemistry at the University of Castilla-La Mancha. He obtained his PhD degree in Chemical Sciences in 2008. He did his first post-doctoral internship under the supervision of Prof. P. J. Pérez at the University of Huelva in 2009. In 2012, he moved to the University of the Basque Country (UPV/EHU). Since 2018, he has also been a visiting researcher at the La Sapienza University of Rome. Since 2020 he has been an Ikerbasque Research Associate at the Donostia International Physics Center (DIPC). His main research areas range from cycloaddition reactions to the design and synthesis of new chemical entities with biological activity. In addition, he is interested in the design and synthesis of new chemical sensors, for applications in particle physics or in the detection of pathogens. More recently, he has also begun to study reaction mechanisms by means of DFT calculations. |
 Francesc Monrabal | Francesc Monrabal graduated in Physics at the University of Valencia. He obtained his PhD at the same University developing the technology of gaseous xenon TPC for double beta decay search. In 2015 he started working with Prof. David R. Nygren at the University of Texas at Arlington where he was in charge of the design and construction of the Time Projection Chamber (TPC) for the NEXT (Neutrio Experiment with a Xenon Time Chamber)-White detector becoming a technical coordinator of the NEXT Collaboration the same year. Since 2019 he has been an Ikerbasque Research Fellow at the Donostia International Physics Center (DIPC), where he is leading the effort of the construction of the NEXT-100 detector. |
 Juan J. Gómez-Cadenas | Juan José Gómez Cadenas received both his MSc degree and his PhD from the University of Valencia. He has been a Staff member at the European Laboratory for Particle physics (CERN), a full professor at the University of Valencia and a full professor at CSIC. Since 2017, he has been an Ikerbasque Professor at DIPC. He is the co-spokesperson of NEXT, a leading experiment searching for neutrinoless double beta decay processes (ββ0ν). Together with the other authors of this paper and the members of the NEXT Collaboration, he is leading an interdisciplinary effort to develop a background-free ββ0ν experiment in xenon based on the tagging of the xenon daughter (Ba2+) in the double beta decay through the use of molecular sensors. |
 Fernando P. Cossío | Fernando P. Cossío received his MSc degree in chemistry from the University of Zaragoza and his PhD in organic chemistry from the University of the Basque Country (UPV/EHU). Since 2002, he has been a full professor in the UPV/EHU, where he served as the Dean of the Faculty of Chemistry and the vice chancellor for research and international relations. Since 2009, he has been the scientific director of Ikerbasque, the Basque foundation for the advance of science. His research interests are focused on experimental and computational aspects of cycloaddition chemistry and C–C bond forming reactions, with special emphasis in the design and chemical synthesis of new molecules with useful biological and photophysical properties in medicinal chemistry, molecular imaging and neutrinoless double beta decay reactions leading to Ba2+ cations. |
1. Introduction
Detection of metal cations by means of molecular sensors has become a very active research field, with important applications in analytical chemistry,1 materials2 and environmental sciences,3 and biological chemistry,4 among other scientific areas. Very recently, this technique has emerged as a promising tool in particle physics since luminescence sensing of cations is very well-suited for the detection of special events such as neutrinoless double beta decay nuclear processes.5Different techniques developed so far for the detection of metallic ions include atomic absorption spectroscopy, voltammetry, electrochemical analysis, inductively coupled plasma spectrometry, laser induced breakdown spectroscopy, neutron activation analysis and UV-Vis spectroscopy, among others.6,7 However, fluorescence8 is perhaps the most frequently used because of the relative simplicity of the associated devices, its very high sensitivity and its excellent spatial and temporal resolution.9–15 Since in many samples different metal cations can coexist, the corresponding sensors must exhibit high selectivity. In this respect, fluorescence sensing arrays have been developed as a useful alternative based on ensembles of different less selective sensors that exhibit characteristic responses for different analytes.16
Many different types of fluorescent sensors able to detect metal cations have been described, including nanoparticles,17 carbon-based quantum dots,18,19 and dendrimers,20 as well as metal–organic frameworks (MOFs),21 and biomolecules such as DNA22 and proteins.23 These clustered and macromolecular sensors are not included in the present Perspective Article, which is focused on discrete fluorescent molecular sensors.
The fluorescence emission24 spectrum of a given fluorophore is produced after excitation at a certain wavelength (normally in the UV region), followed by emission of photons at larger wavelengths. The Stokes shift expresses the difference between the maximum absorption wavelength λmaxabs and the maximum emitted wavelength λmaxem. If this difference is expressed in wavenumbers, the corresponding Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) Stokes is given by
Stokes is given by
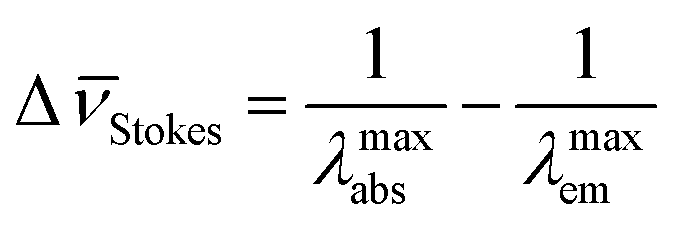
Since λmaxem > λmaxabs, ΔStokes > 0. The excitation process generates a transition from the ground state S0 of molecule A to the first singlet excited state S1(A*). The decay from S1(A*) to S0(A) is determined by the radiative kinetic constant kSr
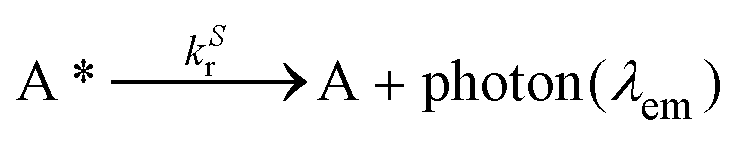
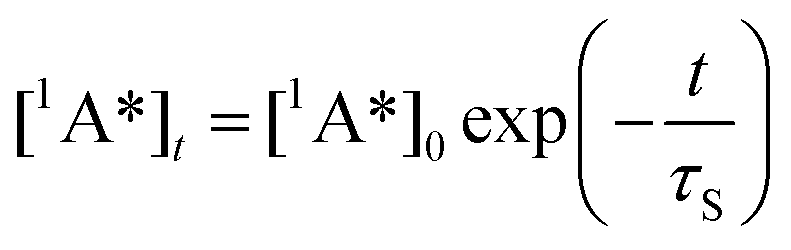
In the latter equation, [1A*]0 is the concentration of excited molecules at t = 0, which corresponds to the instant of the pulse light excitation, and τS is the lifetime of the excited state S1:
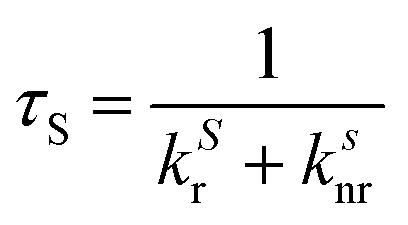
The time-dependent intensity of the fluorescence emission iF (t) after excitation at t = 0 is proportional to the concentration of molecule A in the singlet excited state and to the kinetic constant associated with the fluorescence emission:
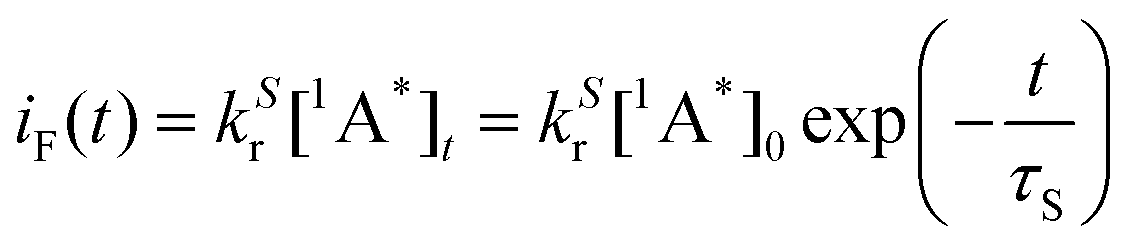
It is important to note that the measured fluorescence intensity IF is obtained from the total decay at different wavelengths and is expressed in arbitrary units (a.u., not to be confused with atomic units) that depend on the experimental conditions.
The fluorescence quantum yield for a given A* → A transition is the fraction of excited molecules S1(A*) that decay to the ground state S0(A) with emission of photons (vide supra):
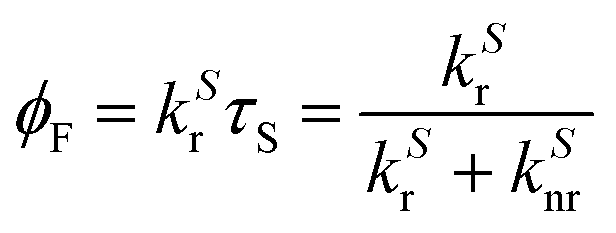
Thus, the quantum yield gives the fraction of emitted photons during the total decay with respect to the absorbed photons. This magnitude gives crucial information to measure the efficiency of fluorescence emission. Another relevant parameter is the brightness4,23 of the fluorescence emission BF, which is the product of the quantum yield and the molar extinction coefficient εabs at the excitation light wavelength.
| BF = ϕFεabs |
All these combined magnitudes characterize the photophysical behaviour of a given fluorophore.
The key point in fluorescence sensing of different metallic cations is the change of these magnitudes upon coordination or binding of the sensor with the analyte. In a fluorescent monocolour sensor the emitted photons are of identical wavelengths λem in the free state and upon coordination or binding with the metal cation. This can occur via photoinduced electron transfer1,25 (PET) phenomena via an electronic donation from one heteroatom of the sensor that quenches the fluorescence relaxation of the fluorophore in the free S1 state. Upon coordination, this donation is directed to the metal and the fluorescence decay takes place efficiently thus resulting in an off–on transition (turn-on enhancement) on going from the free to the metal-coordinated state. Another less frequently observed PET consists of the reverse on–off process (turn-off quenching). In most cases, the PET perturbations of the relaxation processes result in preferential emission of photons with λem values similar for free and bound states, the only difference being the very low and very high intensities in the off and on states, respectively. Therefore, the larger the difference between the intensities of the off and on states, the higher the sensitivity of the sensor. These monocolor probes require very high intensity-based separation factors between their ‘off’ and ‘on’ states; such separation factors are challenging to attain, given various analyte-independent factors which affect the probe intensity (e.g. fluctuations in the excitation source, the effect of the microenvironment, variations in the probe concentration, etc.).26,27 In this Perspective Article we rather focus on bicolor probes which rely on wavelength separation and are not affected by such factors.
Molecular bicolour sensors emit photons with different wavelengths in the free (λem) and bound 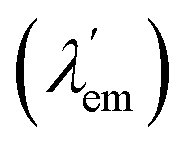 states. They can also be described as dual emission28 ratiometric probes with signal changes associated with two independent or correlated emitters.27 Depending on the magnitude of
states. They can also be described as dual emission28 ratiometric probes with signal changes associated with two independent or correlated emitters.27 Depending on the magnitude of 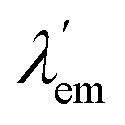 with respect to λem, blue (hypsochromic) or red (bathochromic) shifts will be observed. Thus, both free and bound bicolour sensors can be active fluorescent emitters within an on–on′ scheme. In this case, the sensitivity will be determined by the respective intensities and the corresponding photophysical parameters. Two relevant parameters are the wavelength difference
with respect to λem, blue (hypsochromic) or red (bathochromic) shifts will be observed. Thus, both free and bound bicolour sensors can be active fluorescent emitters within an on–on′ scheme. In this case, the sensitivity will be determined by the respective intensities and the corresponding photophysical parameters. Two relevant parameters are the wavelength difference 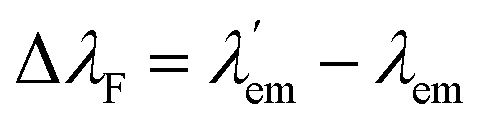 and the peak discrimination factor fλ, defined as
and the peak discrimination factor fλ, defined as
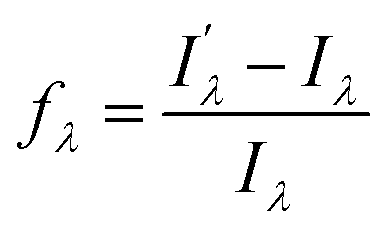
 and Iλ are the intensities of the signals characterized by a wavelength λ at the bound and free states, respectively. This factor is an indicator of the expected signal cleaning of the metal-bound sensor with respect to its free unbound state, especially when the concentration of the metallic cation is very low (vide infra). Thus, the greater the separation dictated by fλ, the lesser the overlap between both signals. Aside the intrinsic values of quantum yield, brightness and intensity, these last two factors will determine the sensitivity of the molecular sensor with respect to the background. This is particularly important in cation-tagging experiments with extremely low concentration of the metallic cation (eventually, only one!), in which the concentration of the free molecular bicolour sensor is therefore orders of magnitude more higher than that of the bound sensor.
and Iλ are the intensities of the signals characterized by a wavelength λ at the bound and free states, respectively. This factor is an indicator of the expected signal cleaning of the metal-bound sensor with respect to its free unbound state, especially when the concentration of the metallic cation is very low (vide infra). Thus, the greater the separation dictated by fλ, the lesser the overlap between both signals. Aside the intrinsic values of quantum yield, brightness and intensity, these last two factors will determine the sensitivity of the molecular sensor with respect to the background. This is particularly important in cation-tagging experiments with extremely low concentration of the metallic cation (eventually, only one!), in which the concentration of the free molecular bicolour sensor is therefore orders of magnitude more higher than that of the bound sensor.
Another important aspect associated with the design of molecular fluorescent bicolour sensors is the necessity of developing sensors able to capture cations in the gas phase. This in turn implies the exploration of supramolecular chemistry on solid–gas interfaces, a research field much less developed than its in-solution analogues. The use of single cation identification has been proposed as a way to dramatically reduce the radioactive background29 in xenon detectors looking for neutrinoless double beta decay. The 136Xe isotope decays into barium, emitting two electrons (and two anti-neutrinos in the standard process). Identification of a coincidence signal of the two electrons with the appropriate energy with the appearance in the active volume of a Ba2+ ion will be an unequivocal signature of the neutrinoless process and, consequently, a demonstration that neutrinos are their own anti-particles (Majorana30 particles). Demonstrating this hypothesis would have deep consequences in particle physics and in cosmology.31,32 A promising “wet” barium tagging experiment has been reported as a proof of concept.33 In order to reproduce the required “dry” conditions imposed by these experiments, several interesting barium-tagging sensors have been reported so far,34,35 all of them based on PET off–on fluorescence emission. Our approach to barium-tagging is instead based on the development of suitable on–on’ bicolour fluorescent sensors, which benefit from a wavelength-shifting mechanism.
The many contributions to the development of ratiometric molecular fluorescent sensors for cations have been comprehensively described in leading reviews and book chapters in the area.9,26,27,36–42 In this Perspective Article, we shall present and discuss the main features and design criteria of molecular bicolour fluorescent sensors for metallic cations, intended for the detection of extremely low cation concentrations (eventually only one) on a surface covered by unbounded sensor molecules, in dry media. In the following sections, we will describe the main modes of sensor–cation interactions and the electronic phenomena responsible for the change in the fluorescence spectra upon metal coordination or after interaction with the cationic centre. We will also report on recent results obtained in our laboratories on barium tagging in dry media involving solid–gas phases.
Bicolour fluorescent probes can be considered a special type of the so-called single-fluorophore ratiometric probes with two analyte-sensitive emission signals, as they change the emission intensity and wavelength upon interaction with the analyte. They can also be described as dual emission materials composed of two independent emitters (free and bound) in equilibrium.28,43 In general, these molecules incorporate different components that include a synthetic ionophore (the cation recognition-binding domain), the fluorophore and, optionally, a spacer and a linker to anchor the sensor with suitable surfaces or biomolecules. The covalent connections among these components can be different, thus giving rise to different sensor architectures, the most usual ones being those shown in Fig. 1A.
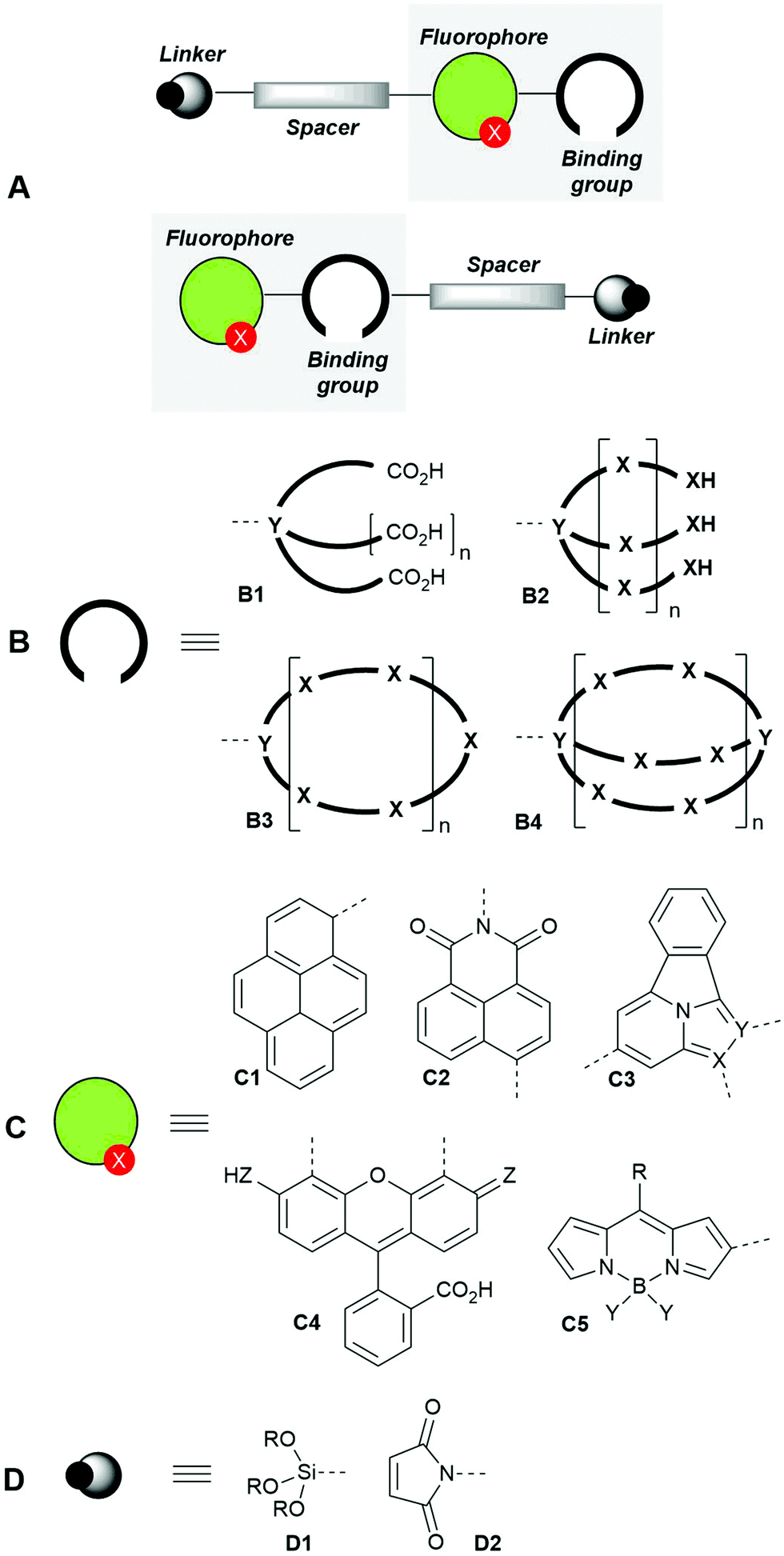 | ||
| Fig. 1 General design patterns of fluorescent molecular sensors for cations (A), showing their main components: binding groups (ionophores, B), fluorophores (C) and linkers (D). | ||
The binding groups (ionophores) are responsible for the efficient and preferably highly selective capture of the metal cation and can consist of chelating groups4 such as polycarboxylic acids B1, podands B2, coronands such as crown ethers44–46B3 and cryptands B4. Other ionophores47,48 such as calixarenes, cyclodextrins and pillararenes, not shown in Fig. 1B, have been described.
The structures of fluorophore components (Fig. 1C) are very varied4 and comprise aromatic hydrocarbons such as pyrenes C1, and polyheterocyclic aromatic moieties such as naphthylimides C2, tetracyclic structures based on imidazo[1,2(5)a]pyridines49,50C3, fluorescein derivatives C4 and BIODIPY-based structures C5, among others.
When required, the spacers can consist of aliphatic linear chains or polyethylene glycol (PEG) derivatives, which can intercalate with aromatic hydrocarbons or heterocycles. Finally, the linkers can be trialkoxysilyl derivatives D1 (very useful for anchoring the fluorescent molecular sensors to solid surfaces) or maleimides D2, which can bind – with low selectivity – cysteine-containing proteins or biothiols, thus giving rise to the corresponding conjugated derivatives.51
Molecular fluorescent bicolour sensors can change the intensity and emission wavelength of their fluorescence signals in different ways after interacting with metal cations (Fig. 2). When an ionophore is present, metal binding can modify the photophysical properties of the sensor via formation of monomeric and dimeric complexes (Fig. 2, mode A.1). In some cases, when the fluorophore itself is able to bind directly the metallic cation, the photophysical properties of the chelate can be different to those of the free fluorophore (Fig. 2, mode A.2), thus yielding a bicolour sensor. In many cases, a heteroatom X possessing a suitable lone-pair present in the fluorophore can also bind the metallic centre thus forming a complex in which both the synthetic ionophore and the heteroatom are bound to the cationic analyte (Fig. 2, note the dashed line in monomeric complex A.1). Finally, reactive functional groups decorating the fluorophore can react selectively with the metal to be transformed into a different fluorophore, thus giving rise to an ionophore-free fluorescent bicolour sensor (Fig. 2B). These considerations apply to not only single emitters from one excited electronic state, but also to dual emitters with a sole fluorophore possessing two emitting states and dual emitters with two independent or correlated emission centres.28
 | ||
| Fig. 2 Main types of bicolour sensors classified according to their structure and interaction with the metal. Different chelation modes (A, monomeric and dimeric chelating groups, as well as chelating fluorophores) are shown. Bicolour sensors in which the change in the fluorophore is generated by a chemical reaction with the metallic ion are also shown (mode B). Optional spacer and linker components are omitted. | ||
2. Mechanisms of bicolour sensing
2.1. Intramolecular charge transfer (ICT)
If changes in the electronic structure of the fluorescent bicolour sensor are considered, one possibility consists of modifying the polarization patterns between substituents of the fluorophore in the free and bound states. This process is named Photoinduced Charge Transfer (PCT), which almost in all cases takes place intramolecularly. Therefore, the latter process will be referred to as Intramolecular Charge Transfer (ICT). In these systems, an electron-releasing group (ERG), which acts as a donating group, transfers a certain electron density to an electron-withdrawing group (EWG), which is the acceptor of this charge transfer. This process involves complex electronic interactions. Within a simplified scheme involving delocalized frontier molecular orbitals, two situations can be distinguished, depending on the proximity of ERG and EWG moieties to the metallic cation. Of course, specific electronic interactions can be important, especially when there is a direct union between the fluorophore and the metal ion. When the ERG is close to the Mn+ centre bound to the ionophore, the two-electron ERG → Mn+ interaction will compete with the ERG → EWG donation thus increasing the frequency associated with the fluorescence S1 → S0 transition (Fig. 3A). Therefore, a blue shift will be observed, in many cases with a certain decrease in brightness. Alternatively, when the EWG is closer to the cation, the ERG → EWG donation will increase in magnitude and the EWG-Mn+ interaction will result in the lowering of the lowest unoccupied molecular orbital (LUMO) energy of the fluorophore, thus resulting in a lower
frequency associated with the fluorescence S1 → S0 transition (Fig. 3A). Therefore, a blue shift will be observed, in many cases with a certain decrease in brightness. Alternatively, when the EWG is closer to the cation, the ERG → EWG donation will increase in magnitude and the EWG-Mn+ interaction will result in the lowering of the lowest unoccupied molecular orbital (LUMO) energy of the fluorophore, thus resulting in a lower 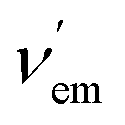 frequency and the corresponding red shift, usually associated with a brighter signal (Fig. 3B). In both cases, the sensitivity of the ICT sensor correlates with the respective differences in brightness, as well as with the peak discrimination factors (vide supra).
frequency and the corresponding red shift, usually associated with a brighter signal (Fig. 3B). In both cases, the sensitivity of the ICT sensor correlates with the respective differences in brightness, as well as with the peak discrimination factors (vide supra).
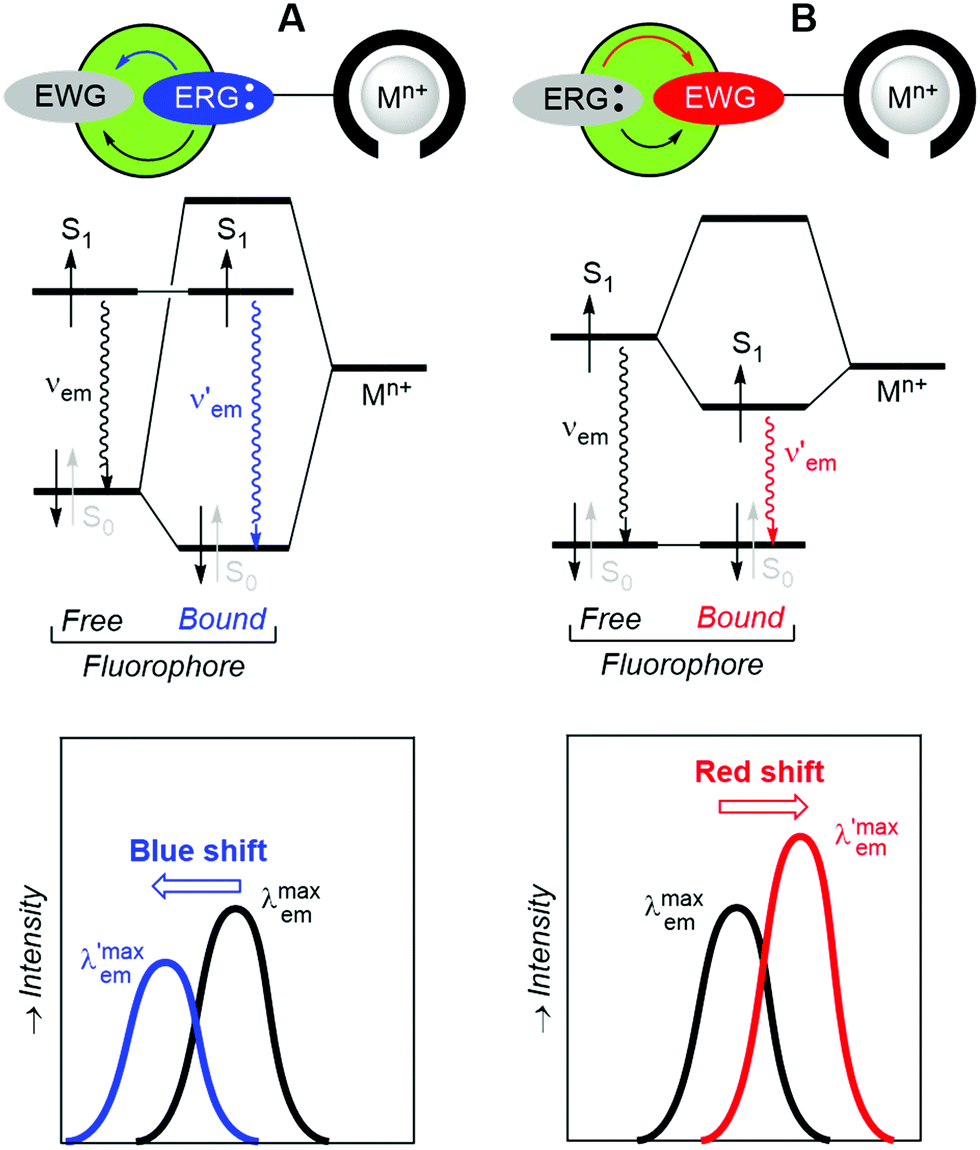 | ||
| Fig. 3 Schematic representation of intramolecular charge transfer processes (ICT) that modify the fluorescence emission of bicolour sensors in the presence of cations Mn+. (A) Blue shift effect of an electron-releasing group (ERC) close to the ionophore. (B) Red shift effect of an electron-withdrawing group (EWG) close to the ionophore. | ||
2.2. Excimers
Another way to obtain radiometric bicolour sensors consists of designing fluorophores that, upon binding the metallic cation through the adequate ionophore, form dimers (homo- or heterodimers) that are more stable in the S1 excited state. Thus, an excimer (excited dimer) originates from the interaction of an excited fluorophore with a nearby congener in the ground state. If the two interacting units are non-identical, the whole chemical entity is named an exciplex (excited complex). The two units can be connected by a spacer and incorporate their respective incomplete ionophore components that, upon coordination with the metallic cation, generate the optimal binding group (Fig. 4A). An analysis of the frontier molecular orbitals (FMOs) of the monomeric units of the sensor at the S0 and S1 (Fig. 4B) states shows that the energy levels of the FMOs of the excimer (or the exciplex) are modified upon dimerization and usually result in a shorter HOMO–LUMO gap (HOMO stands for the highest occupied molecular orbital).52 In turn, this shrinkage in energy levels generates a red shift that permits to discriminate the free and bound states of the sensor. The binding ability of the partial ionophores in the free monomeric state and the stability of the excimer/exciplex can result in a background signal that, depending upon the respective intensities, permits to establish an accurate measurement of the radiometric response (Fig. 4C). In other cases, the excimer/exciplex is relevant in the absence of the metallic cation, whose coordination results in the total or partial loss of fluorescent response associated with excimer inhibition promoted by cation coordination. | ||
| Fig. 4 (A) Cartoon showing the formation of excimers/exciplexes in the presence of a metallic cation. (B) Qualitative diagram showing the distortion of the FMO energy levels in the S1 state of the excimer with respect to those of the monomer. (C) Red shift of the emission spectra of monomers and excimers/exciplexes associated with the diagram shown in (B). | ||
2.3. FRET
A related phenomenon in which the emission involves an interaction between two fluorophores (Fig. 5A) is the Förster resonance energy transfer (FRET).53–55 This process consists of a non-radiative excitation transfer56 from the excited donating (D*) group to the acceptor group (A) in its ground state (Fig. 5B). The latter group relaxes via an A*(S1) → A(S0) process thus emitting with a different wavelength (Fig. 5C).57 Any FRET requires appropriate relative orbital energies associated with the FMOs of D and A. In the case of ratiometric bicolour sensors, the FRET is usually observed when the sensor is bound to the cationic centre. It is noteworthy that the distance R required for FRET is significantly larger than the average values of the combined van der Waals radii of the D and A groups. The Förster distance R0 is defined as the distance at which the FRET efficiency ϕFRET = [1 + (R/R0)6]1− is 0.5. For instance, if D = pyrene and A = coumarin, R0 = 3.9 nm.58 The FRET efficiency also depends on the relative orientation of D and A, as well as on the overlap between the emission spectrum of D and the absorption spectrum of A. | ||
| Fig. 5 (A) Schematic representation of a fluorescent sensor incorporating donor and acceptor units. R0 is the Förster radius (see the text). (B) Förster-resonance electron transfer (FRET) associated with the frontier molecular orbitals of the donor and the acceptor. (C) A representation of the fluorescence emission spectra of the FRET sensor in the free and bound states. | ||
2.4. Two-component fluorophores
In the previous sections, we have presented bicolour sensors based on fluorophores that act as indivisible photoactive ensembles. Another possibility consists of splitting a given fluorophore into two separate rotatable units connected by a single covalent bond (Fig. 6). In turn, this two-unit fluorophore 1 is covalently attached to an ionophore. When a metallic cation is captured by the latter binding moiety, the nature of the interaction is reinforced by additional coordination with a heteroatom of fluorophore 1, thus yielding an ICT-complex (Fig. 2, A.1 pattern) that in addition incorporates a π–cation interaction59 between the metallic centre Mn+ and the Ar2 group gathered in Fig. 25. This Ar2–Mn+ complex promotes a Ar2 → Mn+ charge transfer, as well as a rotation of Ar2 with respect to the polyheterocyclic HetAr1 unit. The outcome of these metal-induced changes is the decoupling between the HetAr1 and Ar2 groups. Therefore, the new HetAr1 fluorophore 2 thus formed (Fig. 25) is less conjugated than its predecessor fluorophore 1 in the unbound state and will emit photons with shorter wavelength. If an optical filter is installed in the detection system to hide the emission associated with fluorophore 1, this blue shift should permit an essentially background-free signal upon cation complexation if the peak discrimination factor fλ is large enough.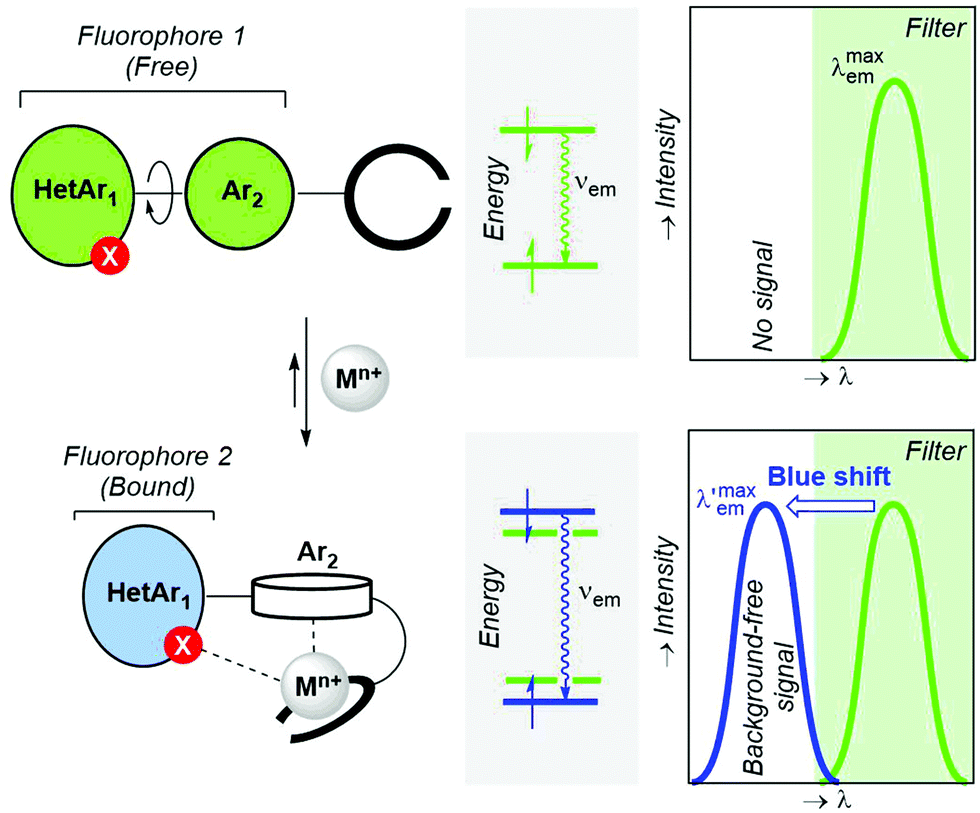 | ||
| Fig. 6 Design criteria of two-component fluorescent bicolour indicators (FBI) based on the decoupling of one component of the starting fluorophore upon metal coordination. The metal-induced hypsochromic shift and the recording of a background-free signal by means of an optical filter are also highlighted. HetAr and Ar denote polyheterocyclic aromatic and aromatic groups, respectively. | ||
3. Design and validation of bicolour probes
All of the above discussed bicolour sensors have been classified in terms of the chemical interactions involved (fluorophore–ionophore, direct chelation by the fluorophore and a chemical reaction with the cation) or the electronic effects that promote the change in the wavelength of the emitted photons (ICT, excimer/exciplex, FRET and related phenomena). In the following sections, an overview of selected examples of these strategies for the construction of bicolour fluorescent probes for cations is presented and discussed.3.1. ICT-Based sensors
Many review articles and books describe the photo-induced ICT phenomenon and the potential applications of ICT-based compounds.24,60 In polar solvents, rearrangement of the solvation shell around the ICT excited state produces its stabilization and the concomitant destabilization of the landing ground state, raising its energy level.41 Therefore, these compounds may show dual emission: a peak in the most energetic region of the emission spectra that originates at the locally excited state (LE), and a red-shifted signal from the solvent-stabilized ICT excited state. Additionally, if the donor and the acceptor are connected through a rotating π-electron bridge, a relaxation of the ICT excited state can occur through an intramolecular rotation, decoupling the orbitals of the donor and acceptor, forming a highly polar excited state. This phenomenon is referred to as “twisted intramolecular charge transfer” (TICT), and it is characterized by extra-large Stokes shifts in polar solvents.Both ICT- and TICT-based fluorophores are suitable scaffolds for the construction of bicolour probes for cations by integrating a cation receptor either on the ERG or on the EWG sites of the fluorophore (Fig. 3). The simplicity of the design makes ICT-based sensors the most widely used ratiometric probes for cations. Among the earliest examples are Indo-1 and Fura-2 (Fig. 7), developed in 1985 by Tsien and collaborators.61 Nowadays, Indo-1 and Fura-2 are the leading compounds of a series of commercially available fluorescent ratiometric probes for Ca2+ and other cations.62 They combine a stilbene skeletal fluorophore (reinforced by extra heterocyclic bonds) with the well-established BAPTA fragment as an ion-recognition site. These molecular probes are characterized by a strong fluorescent ratiometric response to Ca2+. Because of the inhibition of the ICT upon coordination of the Ca2+ to the aniline nitrogen of the BAPTA receptor, a blue-shifted fluorescenc emission is observed (Fig. 3A). One of the main limitations of these probes was the need for UV-light excitation. It was circumvented using two-photon absorption (TPA) microscopy, which permitted the study of cation distribution in live tissues.63
 | ||
| Fig. 7 Fura-2 and Indo-1 ICT-based sensors for Ca2+. | ||
More recent ICT-based sensors are based on fluorophores that can be excited in the visible region. In 2019 Woodruff and O’Halloran synthesized a family of BODIPY-based fluorophores containing an appended N,N,N′-tris(2-pyridylmethyl)-1,2-ethanediamine (trispicen) chelator as a Zn2+ receptor site.64 Among them, ZincBY-4 (Fig. 8), containing a pending acetamide on the 5-position of the BODIPY core outperformed other derivatives due to intramolecular hydrogen bonding between the amide hydrogen and the fluorine atoms of the BF2 centre, thus increasing the Zn2+ affinity. In polar solvents, the ICT-based fluorescence of the free probe is strongly quenched, whereas a blue-shifted emission is recovered upon Zn2+ chelation due to disruption of the ICT process.
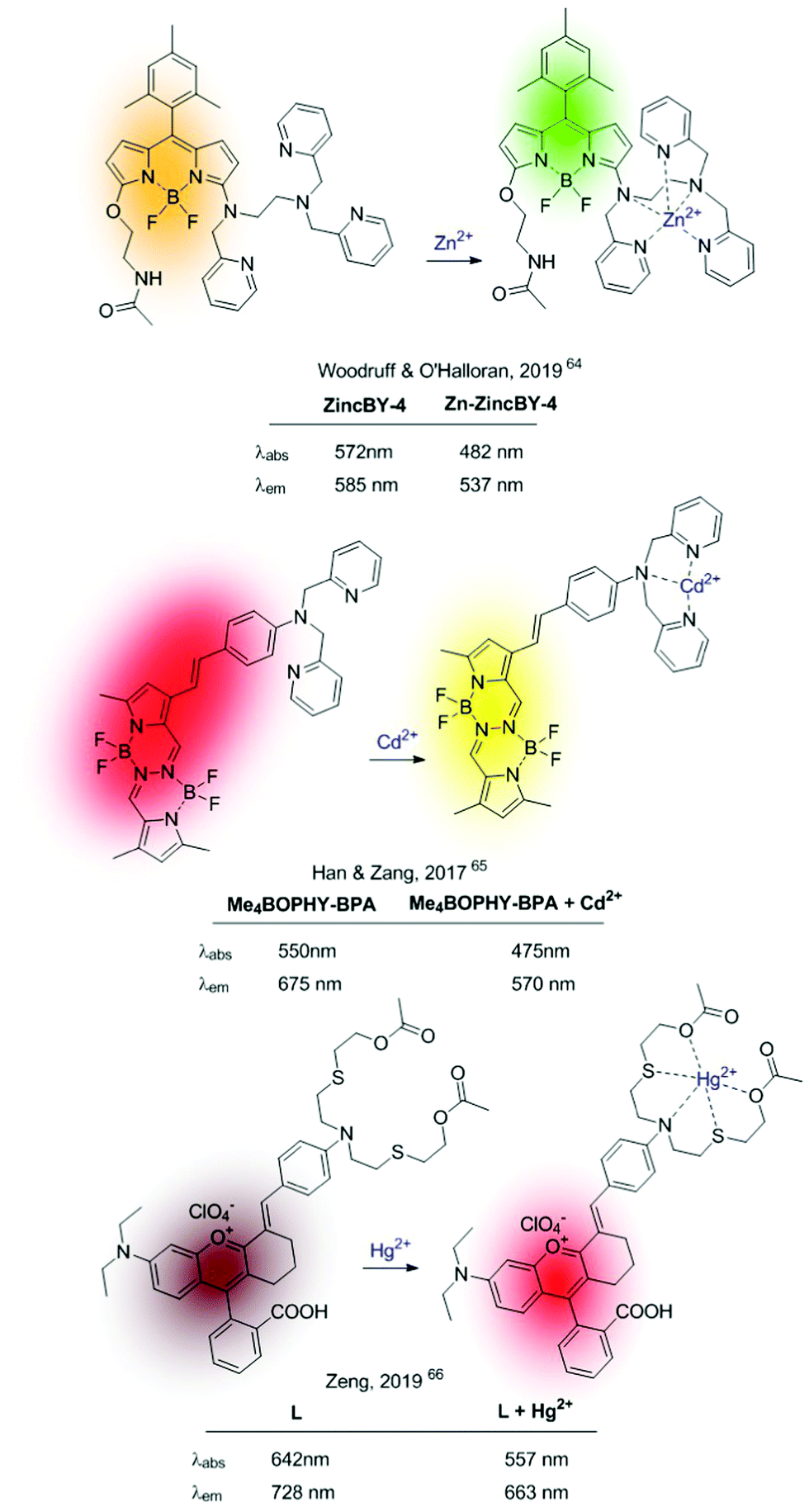 | ||
| Fig. 8 Recent examples of metal-induced ICT depletion probes. | ||
In 2017, Han, Zang and colleagues published ratiometric probes for Cd2+, also based on cation-induced ICT depletion, characterized by a long wavelength fluorescence emission (Me4BOPHY-BPA in Fig. 8).65 The sensor consisted of a tetramethylated BOPHY fluorophore conjugated to bis(pyridine-2-yl)amine (BPA) podand acting as a Cd2+ selective receptor. Cd2+–Chelation caused a clear hypsochromic shift of the fluorescence emission revealing the characteristic yellow emission of the Me4BOPHY dye. In 2019, Zeng et al. pushed this concept further developing a Hg2+-probe (L in Fig. 8) with NIR emission in both the holo and the apo form.66 This sensor consists of a chromenylium–cyanine dye containing a conjugated NS2O2 Hg2+–chelating unit. The strong ICT process between the N-atom of the cation receptor and the fluorescent dye, together with the NIR emission of the naked fluorophore, permitted the authors to construct this ratiometric NIR sensor operative under visible-light excitation.
The clear advantages of TPA microscopy compared to conventional one-photon absorption (OPA) microscopy for the analysis of cation distribution in live cells, tissues, and organisms (i.e. improved penetration, reduced toxicity, reduced autofluorescence, and minimal light scattering) drove the development of molecular probes optimized for TPA.67–69 When considering ICT-based sensors, this has been pursued by binding the cation recognition site to the acceptor domain of an ICT–fluorophore. Therefore, upon metal binding, rather than ICT depletion (as observed in the examples mentioned above) the polarization of the ICT excited state will increase. This probe configuration should not only produce a bathochromic shift in the fluorescence emission of the cation-bound probe (Fig. 3B), but also enhance the TPA cross-section, and the brightness of the fluorescence emission.69
Based on this premise, in the last few years a series of ratiometric TPA probes appeared in the literature. Several Zn2+-selective sensors have been developed based on a BPA podand bound to different fluorophores. Representative examples are shown in Fig. 9.70–73MPVQ-series, initially developed by Meng's group,73 consisted of BPA-appended quinoline as the fluorescent skeleton with a conjugated donor on the 6-position, whereas chromis-1 contains 2-arylthiazole as a push–pull fluorophore. In all cases, a red-shifted fluorescence was observed in the presence of Zn2+, which was accompanied by an increased TPA cross-section.
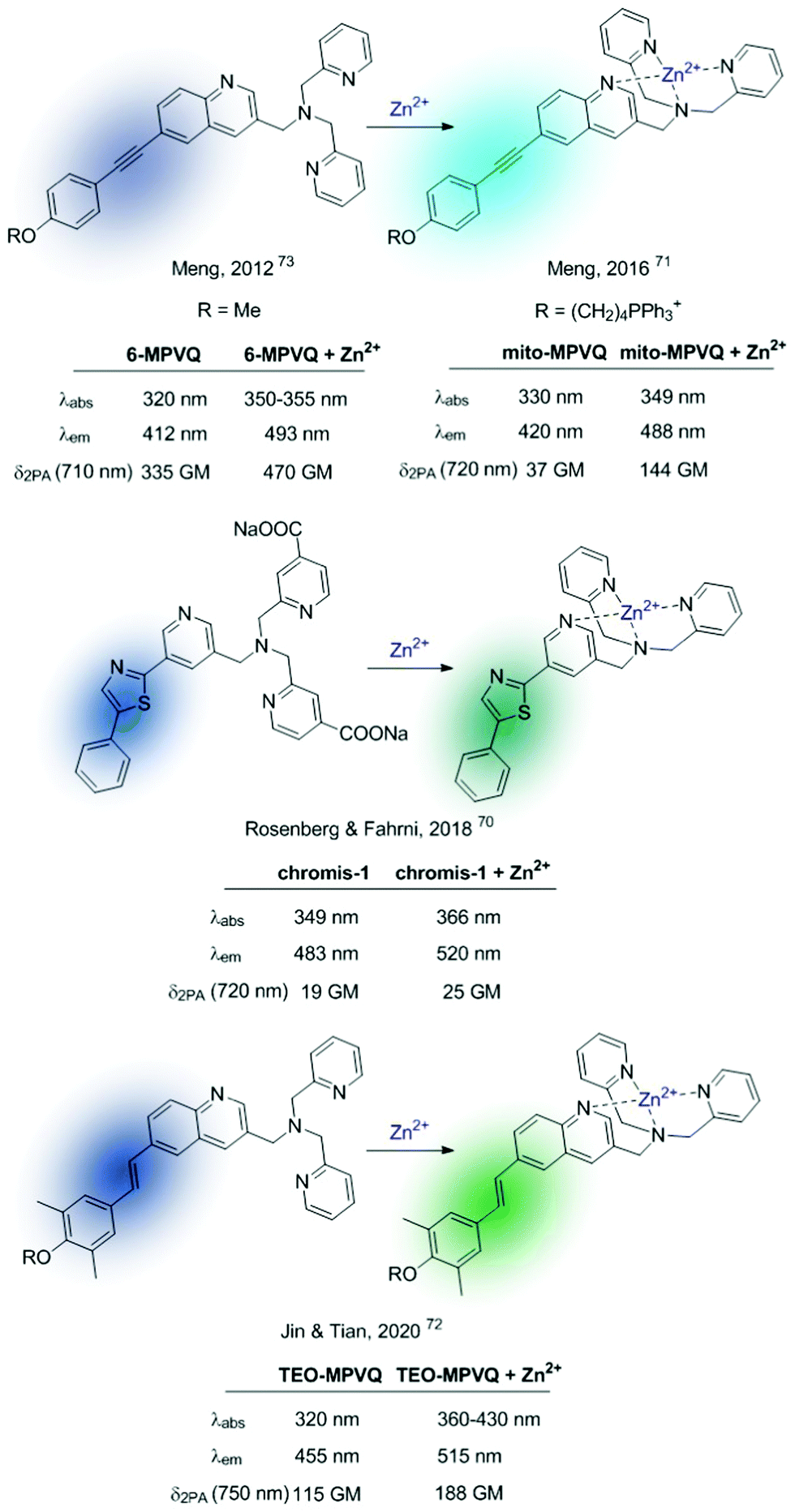 | ||
| Fig. 9 TPA ICT-based ratiometric probes using BPA as a selective Zn2+ receptor. | ||
More complex ratiometric sensors for cations, optimized for TPA, have been constructed using centrosymmetric donor–π–acceptor–π–donor quadrupolar configurations, in which the cation recognition site is integrated into the central acceptor unit, thus constituting a chelating fluorophore (Fig. 2, pattern A.2). The GBC probe (Fig. 10), developed in 2014,74 was a ground-breaking example of sensors based on this structural motif and inspired further developments.75,76 The cation recognition is achieved through a central bipyridine unit with two appended carbazole donors linked through vinylic π-electron bridges. GBC also contains oxyethylene side chains to increase its solubility in aqueous media and its cell permeability. The emission red shift and the enhanced TPA observed upon metal binding were initially attributed to ICT, because of the increased electron-withdrawing character of the Zn-coordinated bipyridine unit. The mechanism responsible for the large TPA characteristic of these systems has been further analysed from a theoretical perspective aiming at establishing structure–property relationships.77–79 These studies revealed that fluorescence emission under two-photon excitation originates from the S2 excited state rather than from S1. Additionally, coordination of the Zn2+ cation requires the rotation of the central bipyridine unit from the trans to the cis coplanar form. This conformational change results in an increase of the dipole and quadrupole moments of the molecule by a loss of the centre of inversion present in the most stable conformation of the unbound probe.77
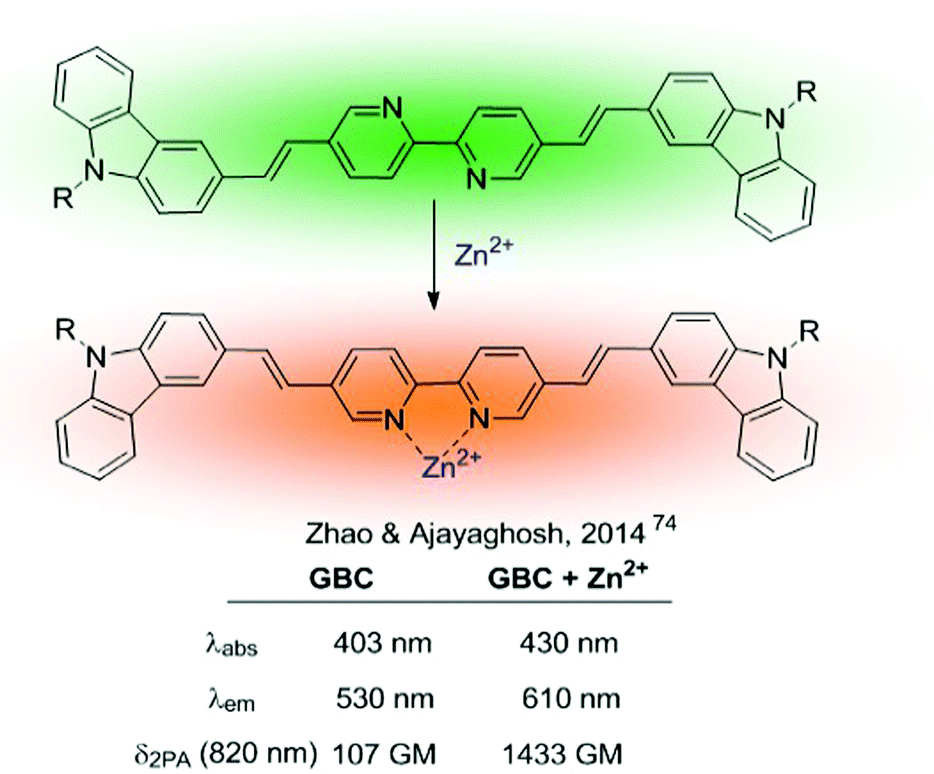 | ||
| Fig. 10 GBC ratiometric probe for Zn2+. | ||
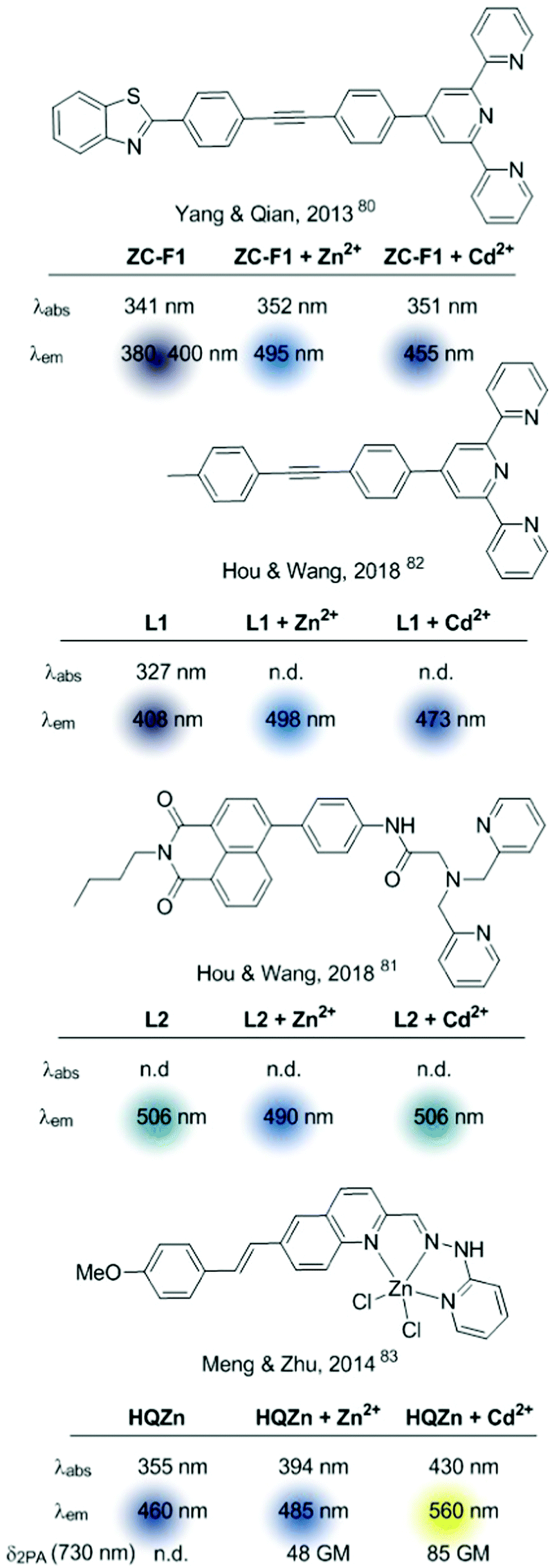 | ||
| Fig. 11 Multi-target sensors based on ICT. | ||
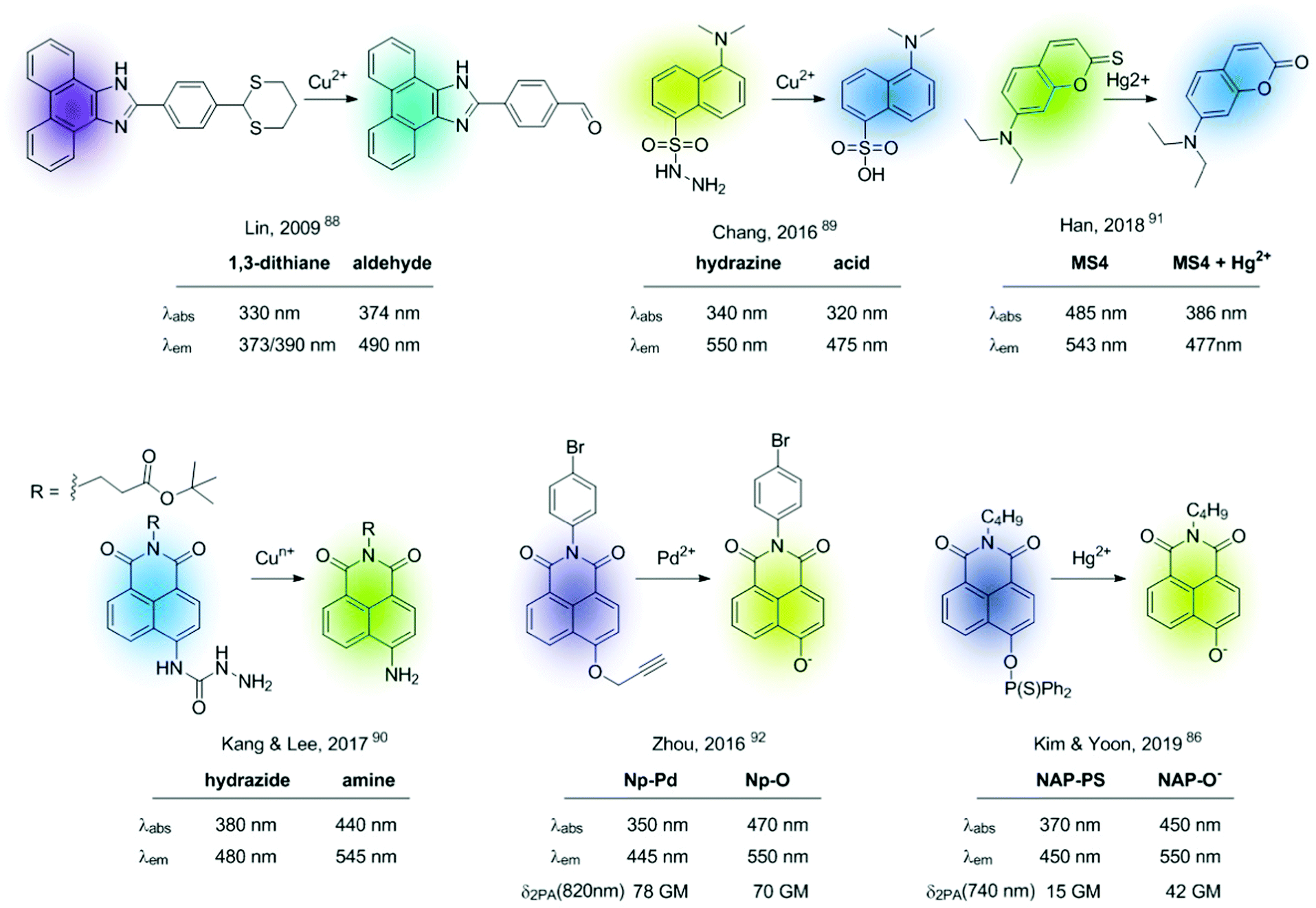 | ||
| Fig. 12 Representative examples of fluorescent chemodosimeters for cations. | ||
 | ||
| Fig. 13 NAP-PS chemodosimeter before (A) and after grinding with Hg(ClO4)2 (B). Reproduced from ref. 86 with permission from The Royal Society of Chemistry, copyright 2019. | ||
All chemodosimeters described above are based on metal-induced bond-cleavage reactions. Alternatively, they can also be constructed using the so-called “covalent assembly principle”. It refers to those probes in which the analyte triggers a cascade reaction on the probe, thus furnishing the conjugated backbone of a push–pull fluorescent dye.93 A coumarin-based probe (ATC-Hg), shown in Fig. 14, is a recent example of such a strategy.94 Hg2+ induced the deprotection of 1,3-dithiolane to a formyl group that condensates intramolecularly with the nearby amino group. This cascade reaction renders a heterocyclic aromatic donor–π–acceptor ICT fluorescent dye. This probe showed high selectivity to Hg2+ and MeHg+, with sensitivities of 27 nM and 5.8 μM, respectively.
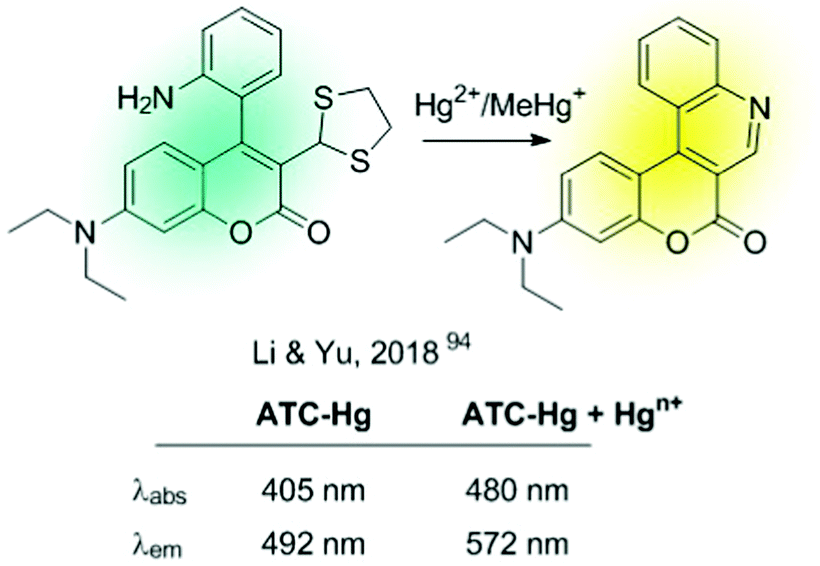 | ||
| Fig. 14 Chemodosimeter based on covalent assembly. | ||
Ratiometric sensors can also be constructed using metal-induced cascade reactions that reduce the conjugation on the probe. The group of Emrullahoğlu employed this strategy for the construction of BODIPY-based ratiometric probes for sensing Au3+ and Hg2+ (Fig. 15).95,96 Although strictly speaking these sensors cannot be considered ICT-based probes, it is convenient to include them here under the chemical pattern B shown in Fig. 2. In these examples, metal-mediated activation of the triple bond and subsequent intramolecular cyclisation and protonolysis generates the corresponding unconjugated BODIPY-furan fluorophores, as evidenced by the characteristic green emission after the cation-promoted reaction.
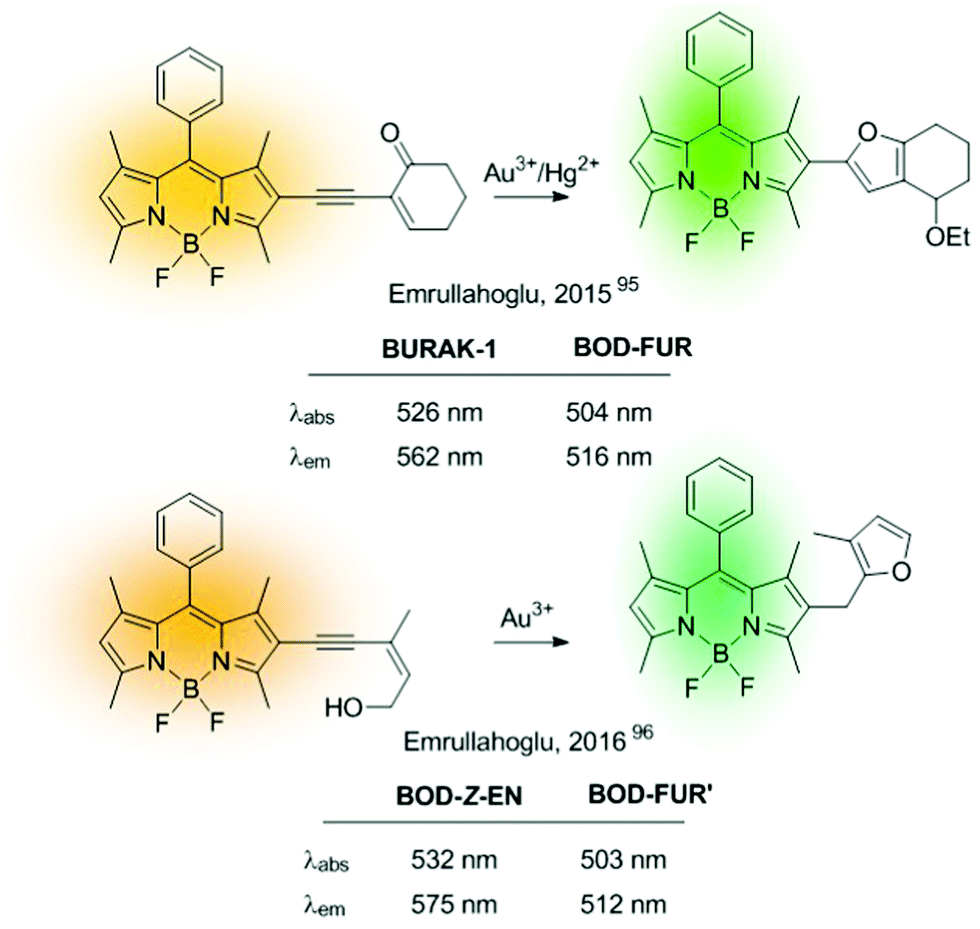 | ||
| Fig. 15 Example of ratiometric probes based on metal-induced cascade reactions. | ||
3.2. Excimer/exciplex-based sensors
An excimer was defined by Birks as “a dimer which is associated in an excited electronic state and which is dissociative (i.e. would dissociate in the absence of external restraints) in its ground electronic state”97 (Fig. 4). Therefore, excimer formation is an irradiation-induced phenomenon, strongly dependent on the fluorophore–fluorophore distance. In general, in this kind of bicolour sensor the fluorophore is a polyaromatic system, being pyrene the one most frequently used. Since excimer emission is often red-shifted compared to the fluorescence of the monomer, excimer-based sensors can be used for the construction of ratiometric probes. This type of molecular sensor is designed in such a manner that the interaction of the analyte with the recognition unit of the probe alters the fluorophore–fluorophore distance. Based on this premise, one can distinguish between two types of probes: (A) intermolecular excimers formed by cation-assembled dimers, and (B) intramolecular excimers based on probes containing two fluorophores whose conformation is altered by cation binding.98Some recent examples of sensors of type A are shown in Fig. 16.99–102 Within this configuration, the binding of two sensors to the cation places the fluorophores close enough to form an intramolecular excimer. Among the chosen examples, it is worth highlighting the molecular sensor developed by Misra et al. in 2015,99 which was modified and immobilized onto silica supports showing a ratiometric response to Hg2+ in the solid-state. Additionally, the interaction can be reversed by treatment of the silica-supported sensor with EDTA allowing their use as Hg2+ extractors.
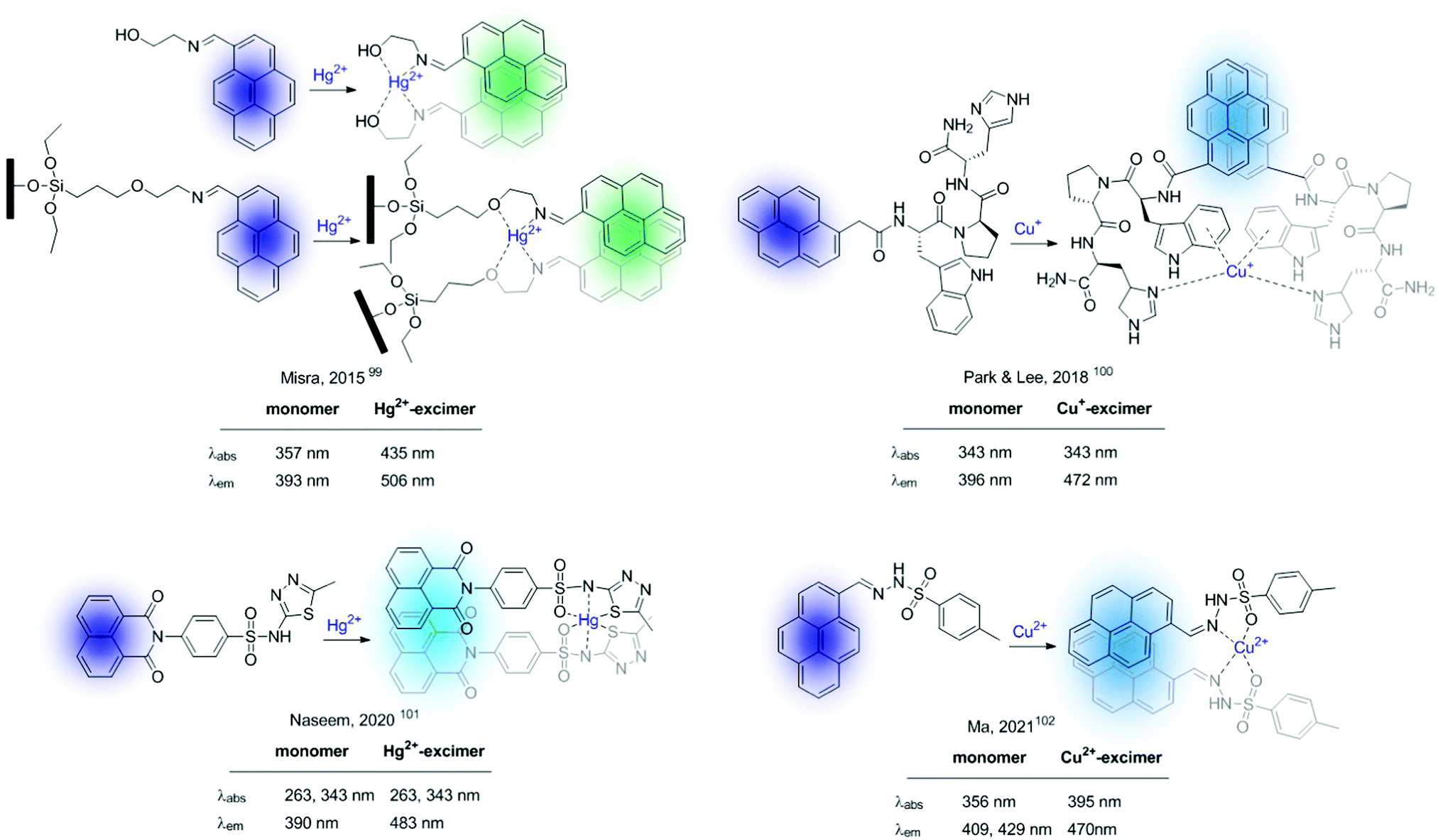 | ||
Fig. 16 Excimer-based probes for cations based on 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sensor/cation supramolecular complexes. :1 sensor/cation supramolecular complexes. | ||
An early example of intramolecular excimer-based sensors of type B is the Ba2+ and Sr2+ probe developed by Ushakov and Barigelletti in 1999 (Fig. 17).103 The probe consists of a bis(styryl) fluorophore containing two crown ether units. It relies on the formation of intramolecular sandwich complexes with the sensing ions (with radius larger than the cavity size of the crown ether), which facilitates excimer emission from the close-lying fluorophores (Fig. 17). Although in the example the fluorescence of the Ba2+-complexed probe presents a blue-shifted band compared to that of the unbound probe, a strong fluorescence attributed to excimer formation was also observed at ca. 600 nm.
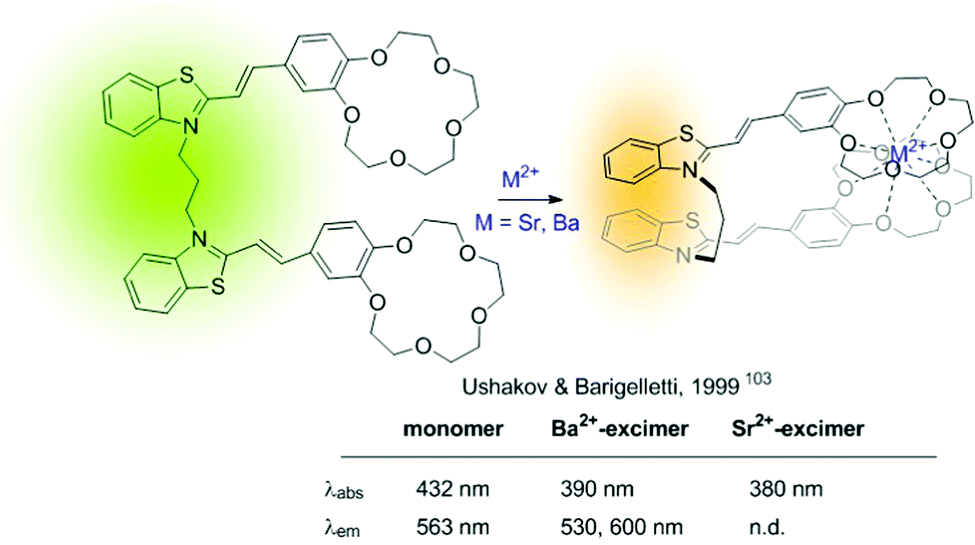 | ||
| Fig. 17 Intramolecular excimer-based probe for Ba2+. | ||
In 2017 Lee et al. developed a ratiometric sensor for Zn2+ based on an intramolecular excimer inhibition.104 It consists of a symmetric pyrene–peptidyl probe. In this example, the interaction of the probe with the cation modifies its conformation inhibiting pyrene–pyrene excimer formation (Fig. 18).
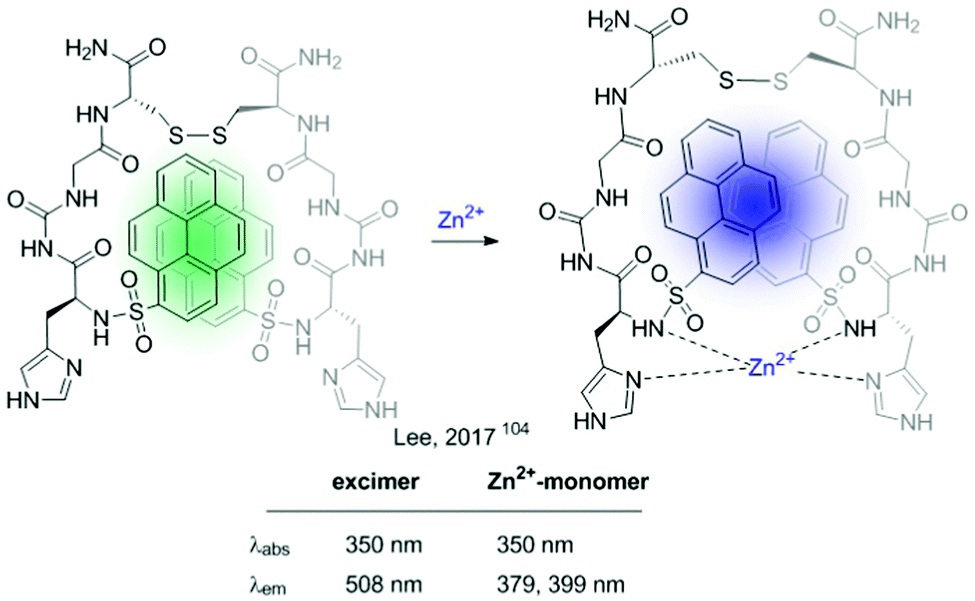 | ||
| Fig. 18 Intramolecular excimer inhibition-based probe for Zn2+. | ||
3.3. FRET-based sensors
Since the German scientist T. Förster described the FRET effect in 1948,105 many fluorescent sensors based on this effect have been developed,42,106 which have been widely applied in diverse fields, such as biology, physiology, medicine, and pharmacology. This type of sensor presents ideal radiometric sensing properties as it benefits from a simple structural design. This section describes some FRET-based fluorescent sensors and the combination of these with other mechanisms. As described in Fig. 5, these sensors are based on an energy donor/acceptor system. In many of them, rhodamine appears as a fundamental part of the fluorescent sensor (acceptor), and it is integrated in the sensor in the form of the non-fluorescent rhodamine spirolactam. The analyte's reaction promotes spiro opening, revealing rhodamine fluorescent dye, which results in a FRET-induced rhodamine-centered emission.In this context, Meng described in 2018 a two-photon radiometric fluorescent probe (RN3) for Pd2+ based on coumarin (donor) and rhodamine B (acceptor) connected by a diethylenetriamine linker (Fig. 19).107 The emission of a 7-substituted coumarin was determined in the range of 370–650 nm, which fully covered the rhodamine B's absorption (450–590 nm). RN3 showed high sensitivity and selectivity towards Pd2+ in aqueous solutions, with a clear ratiometric fluorescent response in a one- and two-photon excitation regime. Additionally, interactions with the cation boosted the TPA cross-section, which was attributed to ICT enhancement on the coumarin upon coordination to Pd2+. RN3 was tested for the detection of Pd2+ in deep tissues and living cells under TPA, showing low cytotoxicity.
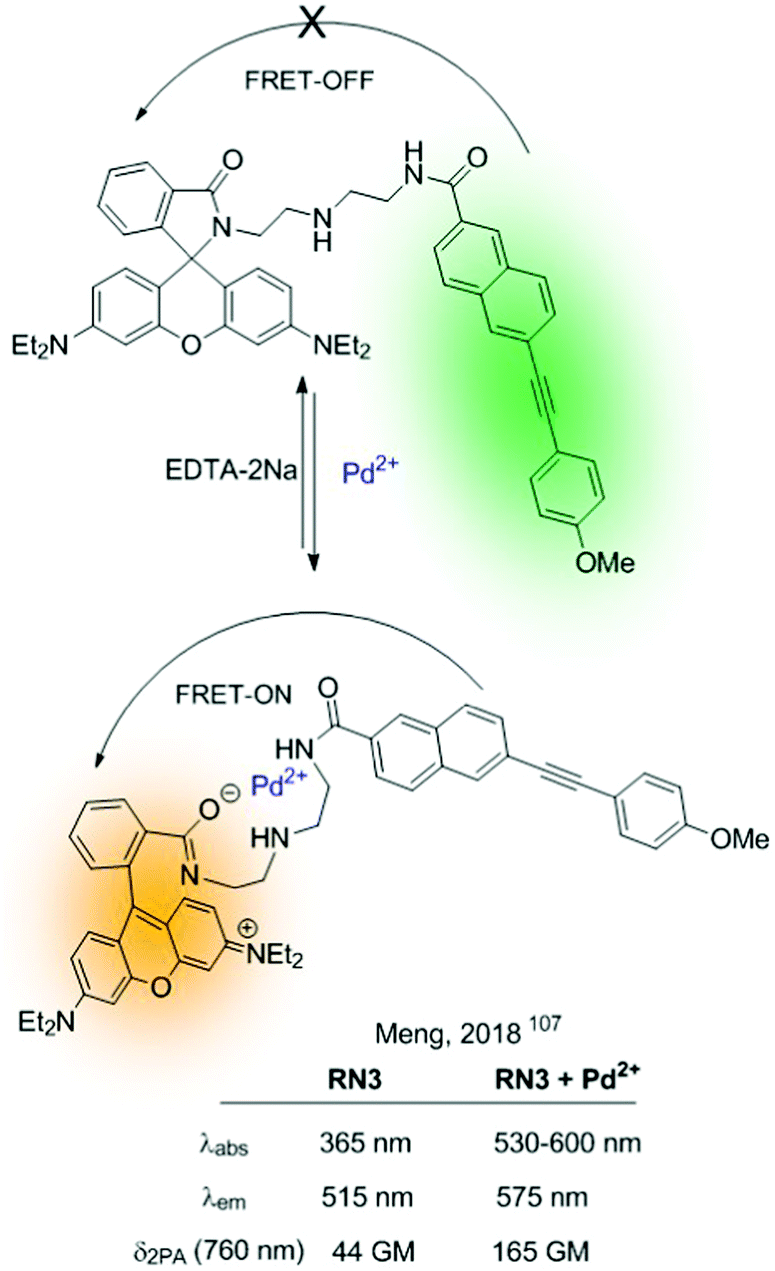 | ||
| Fig. 19 RN3 based on the FRET-effect to detect Pd2+ in aqueous media. | ||
Different rhodamine-based FRET-dyads have been used for the construction of ratiometric sensors (i.e. using BODIPY or quinoline-benzothiazole as a donor). They have been widely used for sensing highly polluting cations such as Hg2+108 or Cd2+. For the latter, in 2015 Goswami described RQBT, a rhodamine/quinoline-benzothiazole-based sensor (Fig. 20).109 The interaction of Cd2+ with the probe results in the cleavage of the spiro ring and formation of the Cd2+-chelated structure, as confirmed by single-crystal X-ray analysis. Probe RQTB also showed ratiometric fluorescence in dry media (silica-gel TLC plates), which was used for the construction of portable Cd2+ sensing kits (Fig. 21).
 | ||
| Fig. 20 Proposed FRET-based Cd2+-sensing mechanism of RQBT. | ||
 | ||
| Fig. 21 Photographs of TLC plates after immersion in an RQBT–MeOH solution (i) and after immersion in an RQBT–Cd2+ methanol solution (ii) taken in ambient light (a) and under hand-held UV light (b). Excitation wavelength of the UV light is 365 nm. Adapted from ref. 109 with permission from American Chemical Society, copyright 2015. | ||
Alternatively, FRET-probes for cations can be constructed by introducing an ICT sensor as part of a FRET dyad. Interaction of the cation with the ICT sensor will produce a shift in its absorption and/or emission properties, modifying the efficiency of the FRET process. A recent example of such a strategy is the CPBT probe for Zn2+developed by Chen, He, Guo and colleagues.110CPBT integrates a TPA donor coumarin derivative with an ICT fluorophore 4-amine-7-sulfamoylbenzo[c][1,2,5]-oxadiazole linked to the ionophore (Fig. 22). Within this type of construction, the interaction of the cation with the ICT fluorophore will cause a blue-shift in its emission, diminishing the spectral overlap between the donor and acceptor and weakening the FRET process. CPBT was tested for 2D and 3D ratiometric mapping of Zn2+ in living cells and in the head of zebrafish. Additionally, it showed TPA cross-section >40 GM, which makes it a suitable candidate to be used in two-photon excited fluorescence imaging.
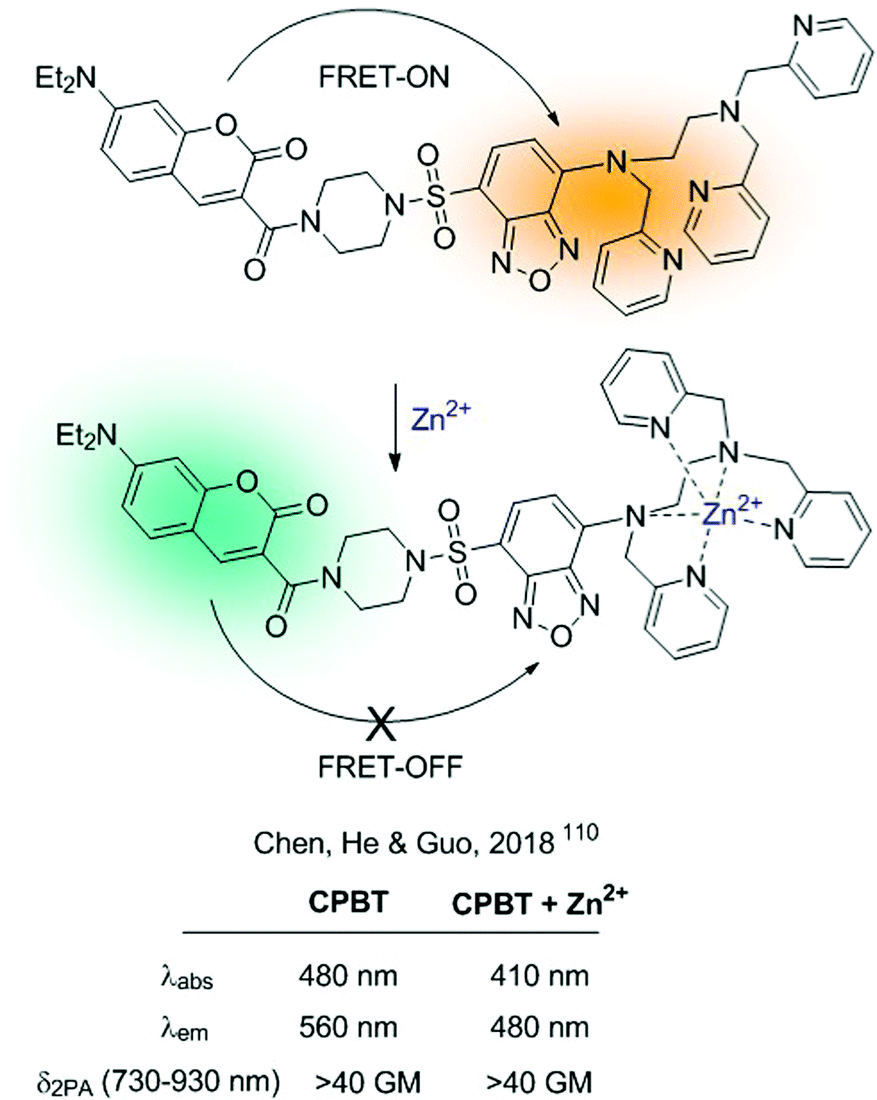 | ||
| Fig. 22 FRET-ICT probe for Zn2+, showing an FRET-ON/FRET-OFF mechanism. | ||
CPBT can be described as an FRET-on → FRET-off ratiometric probe. The reverse mechanism (FRET-off → FRET-on) can also be envisioned, and the QA sensor, developed by Mao et al., serves as a representative example (Fig. 23).111 This probe contains an anthracene unit as a donor and a quinoline ICT sensor as an acceptor. In the absence of the analyte (Zn2+), the probe shows the characteristic emission of anthracene. Interactions of Zn2+ with the quinolone sensor produce a red shift in its absorption band, together with a concomitant enhancement of the effective overlap with the anthracene emission, thus triggering the FRET process.
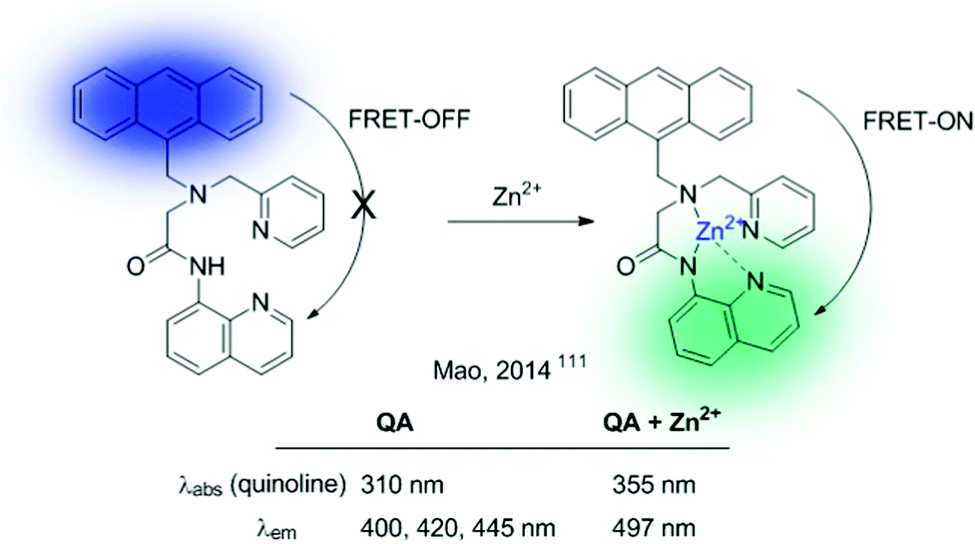 | ||
| Fig. 23 FRET-ICT probe for Zn2+, showing an FRET-OFF/FRET-ON mechanism. | ||
Closely related to FRET sensors, one can encounter the TBET-based probes (through bond energy transfer).112 They comprise sensors based on donor–acceptor dyads in which the donor and acceptor are connected through a conjugated bond. These probes can be used for the construction of sensors as long as the cation interaction alters the extent of the FRET process. The simplest strategy consists of revealing a “dormant” fluorophore (rhodamine), as described before for FRET probes. A representative example is Np-Rh, a TPA ratiometric TBET-based probe for Cu2+ shown in Fig. 24.113
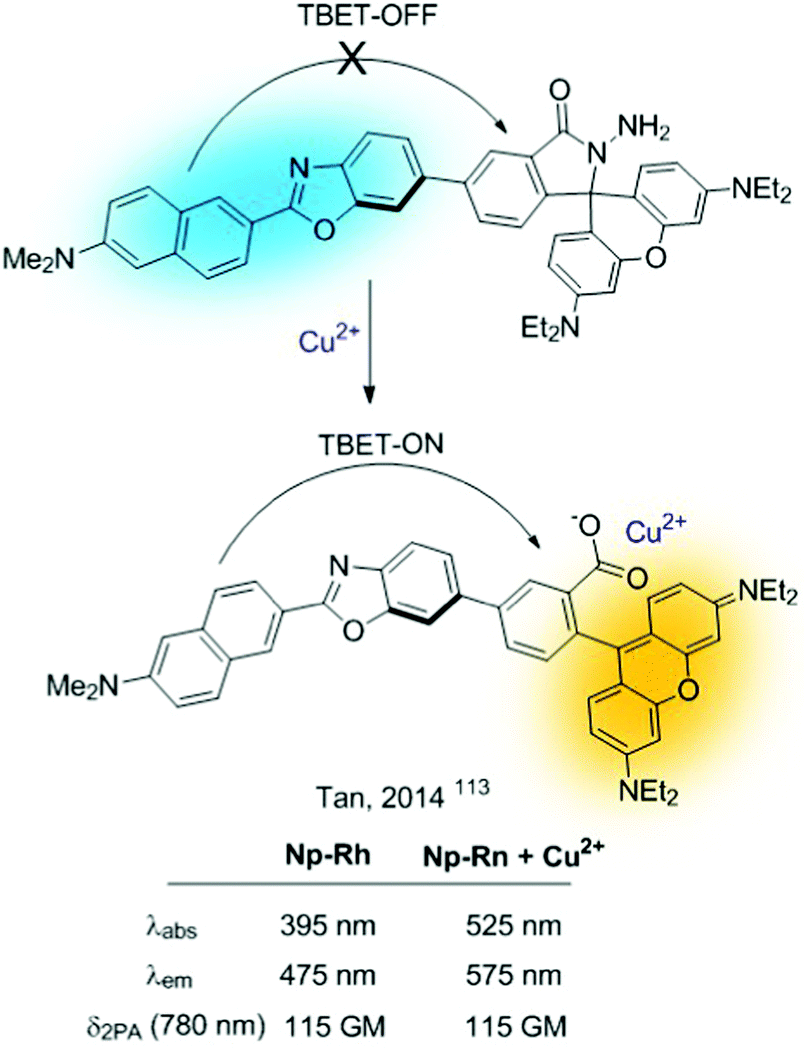 | ||
| Fig. 24 TBET-based probe for sensing Cu2+. | ||
3.4. Two-component fluorescent bicolour indicators
We applied the concept outlined in Section 2. 4 to the issue associated with the background-free detection of individual barium cations in ββ0ν experiments (vide supra). Application of indicators to this kind of problem requires a series of developments to enable its use. Although several molecular fluorescent sensors for Ba2+ capture and detection have been described in solution,114–120 the features of the Xe → Ba2+ + 2e experiment at a high pressure require the development of fluorescent sensors able to operate efficiently in the interface between a solid substrate and ultra-dry noble gas.The Xenon Chamber must incorporate “dry” molecular sensors capable of capturing the Ba2+ ion in a pressurised xenon environment. In addition, a very stable binding that prevents release of the barium cation before optical interrogation is required. As we have commented above, one possibility consists of developing monocolour ICT turn-on sensors.23,24 Our approach121 was based on the above-mentioned two-component design. Benzo[a]imidazo[5,1,2-cd]indolizine was chosen as a suitable fluorophore of type C3 (Fig. 1). To this HetAr1 group a para-phenylene group was added as an Ar2 component. Finally, a pentaoxa-16-azacyclooctadecane aza-crown ether unit was included as a B3 type coronand ionophore. This compound was named FBI, Fluorescent Bicolour Indicator (Fig. 25A). DFT calculations showed that the HetAr1–Ar2 group gives rise to the fluorophore 1 ensemble, in which the dihedral angle formed by both components was close to coplanarity.
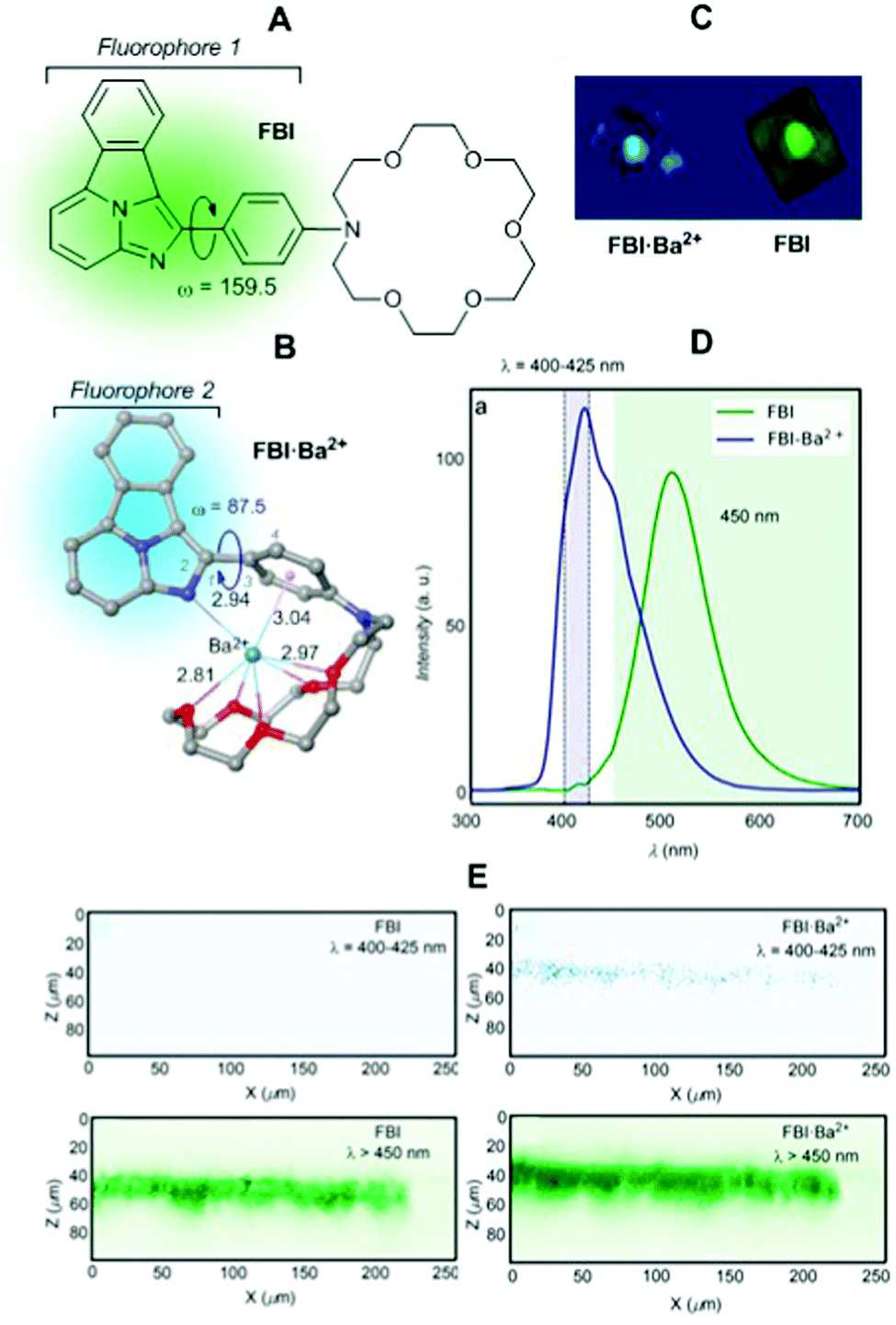 | ||
| Fig. 25 (A) Structure of the FBI molecule showing the approximate coplanarity of the two components of fluorophore 1. (B) DFT optimized structure of complex FBI·Ba2+. Bond angles and distances are given in deg and Å, respectively. (C) 3D tomography of FBI and FBI·Ba2+ obtained by means of a two-photon absorption (TPA) microscopy device. (D) Solid–gas phase emission spectra of FBI and FBI·Ba2+ adsorbed on silica. (E) Z–X profiles showing emission profiles (after TPA excitation) from both complexes with and without a λ > 450 nm filter. Adapted from ref. 121 with permission from Springer Nature, copyright 2020. | ||
Capture of Ba2+ by adding barium perchlorate in acetonitrile solution or in the gas phase led to a complex in which the Ba2+⋯Ph interaction disconnects both components of fluorophore 1 (Fig. 25B). This coordination pattern in which the barium cation, aside the cation–π interaction, complexes the five oxygen atoms of the aza-crown ether and one nitrogen atom of fluorophore 2 was elucidated by DFT calculations and NMR experiments.121 As expected, the FBI·Ba2+ complex shows a noticeable blue shift (Fig. 25C), with a peak discrimination factor fλ = 179.74 and a brightness value of Bλ = 8.06 M−1 cm−1. The fluorescence spectra in solution and in the gas phase show intense emission signals for FBI and FBI·Ba2+ (Fig. 25D) compatible with the FMO scheme shown in Fig. 25. Application of an optical filter for λ > 450 nm to both compounds shows a no signal pattern in the case of FBI. This separation of the chelated/unchelated photophysical properties results fundamental for ββ0ν experiments. In order to detect the Ba2+ cations produced by the decay, they will be driven by electric fields to the cathode where a region of a few centimetres will need to be scanned to look for a positive signal of the complexed molecule. This region will be fully covered, with a density of 105–106 molecules μm−2, by the bicolour indicator to guarantee a high efficiency to capture the cation. The discrimination factor between the response of the complexed indicator and the response of the residual background molecules on the surface must be large to allow separation of the single chelated molecule from the background emission produced by the surrounding molecules that will also be illuminated during the scan.
4. Conclusions and outlook
In this paper, we have highlighted significant advances in the design and experimental validation of fluorescent bicolour sensors. Most of these photo-indicators are based on binding ionophores and fluorophores connected by covalent bonds. If required, suitable spacer units and linkers are incorporated. In addition, some fluorescent bicolour sensors merge the ionophore and the fluorophore components, thus yielding chelating fluorophores whose photophysical properties change significantly upon cation binding. In other cases, the ionophore (most frequently a coronand) and the fluorophore work together upon cation coordination and induce a bathochromic or hypsochromic shift in their emission spectra. Finally, several fluorescent sensors generate their bicolour response by means of a chemical reaction promoted by the metallic cation that induces a change in the chemical structure of the fluorophore.These structural and chemical features result in a bicolour response as an attractive alternative to on–off or off–on changes via photoinduced electron transfer (PET) phenomena upon cation coordination. Instead, the bicolour response requires changes in the photophysical properties of the sensor, which is fluorescent in both the free and coordinated states within an on–on′ conceptual scheme. These dual patterns include intramolecular charge transfer processes (ICT), excimer/exciplex inter- or intramolecular complexes and Förster resonance energy transfer (FRET). As an additional design feature, the fluorophore can incorporate different rotatable units that can be decoupled in the presence of a suitable metal cation thus resulting in different fluorophores in the free and coordinated states.
It is expected that in the years to come these designs will permit the development of more sensitive and selective fluorescent bicolour sensors able to detect specifically metal cations in very low concentrations. This situation will permit further advances in fields ranging from cell biology to particle physics. In addition, the development of fluorescent sensors able to detect metallic cations in the gas phase and at solid–gas interfaces would permit the exploration of supramolecular chemistry in the absence of solvents, thus expanding the field from “wet” to “dry” chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basque Government (Grants IT-1346-19 and IT1180-19), by the Spanish Ministry of Science and Innovation (MICINN-FEDER, Grants PID2019-104772GB-I00, PID2019-111281GB-I00, RED2018-102387-T, and RED2018-102471-T), and by the European Research Council (ERC) under the European's Union Horizon 2020 research and innovation programme (Grant agreement ERC-2020-SyG 951281).References
- A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC.
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- N. De Acha, C. Elosua, J. M. Corres and F. J. Arregui, Sensors, 2019, 19, 599, DOI:10.3390/s19030599.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- J. J. Gómez-Cadenas, J. Martin-Albo, M. Mezzetto, F. Monrabal and M. Sorel, Riv. Nuovo Cimento Soc. Ital. Fis., 2012, 35, 29–98 Search PubMed.
- W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Analyst, 2016, 141, 4262–4271 RSC.
- L. Eddaif, A. Shaban and J. Telegdi, Int. J. Environ. Anal. Chem., 2019, 99, 824–853 CrossRef CAS.
- B. Valeur and M. N. Berberan-Santos, in Molecular Fluorescence, ed. B. Valeur and M. N. Berberan-Santos, 2012, pp. 409–478 Search PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- L. Yu, Y. Qiao, L. Miao, Y. He and Y. Zhou, Chin. Chem. Lett., 2018, 29, 1545–1559 CrossRef CAS.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- S. Chowdhury, B. Rooj, A. Dutta and U. Mandal, J. Fluoresc., 2018, 28, 999–1021 CrossRef CAS PubMed.
- N. S. Patil, R. B. Dhake, M. I. Ahamed and U. Fegade, J. Fluoresc., 2020, 30, 1295–1330 CrossRef CAS PubMed.
- L. Zhu, L. Zhang and A. H. Younes, Supramol. Chem., 2009, 21, 268–283 CrossRef CAS.
- J. Yin, Y. Hu and J. Yoon, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- D. G. Smith, I. L. Topolnicki, V. E. Zwicker, K. A. Jolliffe and E. J. New, Analyst, 2017, 142, 3549–3563 RSC.
- M. Ahmed, M. Faisal, A. Ihsan and M. M. Naseer, Analyst, 2019, 144, 2480–2497 RSC.
- D. Yoo, Y. Park, B. Cheon and M.-H. Park, Nanoscale Res. Lett., 2019, 14, 272 CrossRef PubMed.
- L. Wang, W. Li, L. Yin, Y. Liu, H. Guo, J. Lai, Y. Han, G. Li, M. Li, J. Zhang, R. Vajtai, P. M. Ajayan and M. Wu, Sci. Adv., 2020, 6, eabb6772 CrossRef CAS PubMed.
- D. S. a. R. B. I. Grabchev, Curr. Med. Chem., 2012, 19, 4976–4983 CrossRef PubMed.
- A. K. Jana and S. Natarajan, ChemPlusChem, 2017, 82, 1153–1163 CrossRef PubMed.
- S. S. Tan, S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 2664–2671 CrossRef CAS PubMed.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence-Principles and Applications, Wiley-VCH, Weinheim (Germany), 2nd edn, 2012 Search PubMed.
- J. A. Kemlo and T. M. Shepherd, Chem. Phys. Lett., 1977, 47, 158–162 CrossRef CAS.
- A. Bigdeli, F. Ghasemi, S. Abbasi-Moayed, M. Shahrajabian, N. Fahimi-Kashani, S. Jafarinejad, M. A. Farahmand Nejad and M. R. Hormozi-Nezhad, Anal. Chim. Acta, 2019, 1079, 30–58 CrossRef CAS PubMed.
- R. Gui, H. Jin, X. Bu, Y. Fu, Z. Wang and Q. Liu, Coord. Chem. Rev., 2019, 383, 82–103 CrossRef CAS.
- J. Gierschner, S. K. Behera and S. Y. Park, Angew. Chem., Int. Ed., 2021, 60 DOI:10.1002/anie.202009789.
- D. R. Nygren, J. Phys.: Conf. Ser., 2015, 650, 012002 CrossRef.
- E. Majorana, Il Nuovo Cimento (1924-1942), 2008, 14, 171 CrossRef.
- A. D. Sakharov, Pis’ma Z. Eksp. Teor. Fiz., 1967, 5, 32–35 CAS.
- M. Fukugita and T. Yanagida, Phys. Lett. B, 1986, 174, 45–47 CrossRef CAS.
- A. D. McDonald, B. J. P. Jones, D. R. Nygren, C. Adams, V. Álvarez, C. D. R. Azevedo, J. M. Benlloch-Rodríguez, F. I. G. M. Borges, A. Botas, S. Cárcel, J. V. Carrión, S. Cebrián, C. A. N. Conde, J. Díaz, M. Diesburg, J. Escada, R. Esteve, R. Felkai, L. M. P. Fernandes, P. Ferrario, A. L. Ferreira, E. D. C. Freitas, A. Goldschmidt, J. J. Gómez-Cadenas, D. González-Díaz, R. M. Gutiérrez, R. Guenette, K. Hafidi, J. Hauptman, C. A. O. Henriques, A. I. Hernandez, J. A. Hernando Morata, V. Herrero, S. Johnston, L. Labarga, A. Laing, P. Lebrun, I. Liubarsky, N. López-March, M. Losada, J. Martín-Albo, G. Martínez-Lema, A. Martínez, F. Monrabal, C. M. B. Monteiro, F. J. Mora, L. M. Moutinho, J. Muñoz Vidal, M. Musti, M. Nebot-Guinot, P. Novella, B. Palmeiro, A. Para, J. Pérez, M. Querol, J. Repond, J. Renner, S. Riordan, L. Ripoll, J. Rodríguez, L. Rogers, F. P. Santos, J. M. F. dos Santos, A. Simón, C. Sofka, M. Sorel, T. Stiegler, J. F. Toledo, J. Torrent, Z. Tsamalaidze, J. F. C. A. Veloso, R. Webb, J. T. White and N. Yahlali, Phys. Rev. Lett., 2018, 120, 132504 CrossRef CAS PubMed.
- P. Thapa, I. Arnquist, N. Byrnes, A. A. Denisenko, F. W. Foss, B. J. P. Jones, A. D. McDonald, D. R. Nygren and K. Woodruff, Sci. Rep., 2019, 9, 15097 CrossRef CAS PubMed.
- P. Thapa, N. K. Byrnes, A. A. Denisenko, J. X. Mao, A. D. McDonald, C. A. Newhouse, T. T. Vuong, K. Woodruff, K. Nam, D. R. Nygren, B. J. P. Jones and F. W. Foss, Jr., ACS Sens., 2021, 6, 192–202 CrossRef CAS PubMed.
- S.-H. Park, N. Kwon, J.-H. Lee, J. Yoon and I. Shin, Chem. Soc. Rev., 2020, 49, 143–179 RSC.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS PubMed.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- X. Pei, Y. Pan, L. Zhang and Y. Lv, Appl. Spectrosc. Rev., 2020, 1–22, DOI:10.1080/05704928.2020.1793770.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- Y. Fu and N. S. Finney, RSC Adv., 2018, 8, 29051–29061 RSC.
- R. Zhang, F. Yan, Y. Huang, D. Kong, Q. Ye, J. Xu and L. Chen, RSC Adv., 2016, 6, 50732–50760 RSC.
- M. A. Haidekker and E. A. Theodorakis, J. Mater. Chem. C, 2016, 4, 2707–2718 RSC.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- S. Fery-Forgues, M. T. Le Bris, J. P. Guette and B. Valeur, J. Phys. Chem., 1988, 92, 6233–6237 CrossRef CAS.
- H. G. Loehr and F. Voegtle, Acc. Chem. Res., 1985, 18, 65–72 CrossRef CAS.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- J.-F. Chen, Q. Lin, Y.-M. Zhang, H. Yao and T.-B. Wei, Chem. Commun., 2017, 53, 13296–13311 RSC.
- M. Aginagalde, Y. Vara, A. Arrieta, R. Zangi, V. L. Cebolla, A. Delgado-Camón and F. P. Cossío, J. Org. Chem., 2010, 75, 2776–2784 CrossRef CAS PubMed.
- É. Lévesque, W. S. Bechara, L. Constantineau-Forget, G. Pelletier, N. M. Rachel, J. N. Pelletier and A. B. Charette, J. Org. Chem., 2017, 82, 5046–5067 CrossRef PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press Elsevier, London, UK, 2013 Search PubMed.
- J. Hoche, H.-C. Schmitt, A. Humeniuk, I. Fischer, R. Mitrić and M. I. S. Röhr, Phys. Chem. Chem. Phys., 2017, 19, 25002–25015 RSC.
- A. Kaur, P. Kaur and S. Ahuja, Anal. Methods, 2020, 12, 5532–5550 RSC.
- A. Kaur, P. Kaur and S. Ahuja, Anal. Methods, 2021, 13, 730 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3nd edn, 2006 Search PubMed.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2006, 128, 1188–1196 CrossRef CAS PubMed.
- P. G. Wu and L. Brand, Anal. Biochem., 1994, 218, 1–13 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- R. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer Theory and Applications, 2018 Search PubMed.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CrossRef CAS.
- S. Grisendi, Nat. Cell Biol., 2009, 11, S15–S15 CrossRef.
- H. Szmacinski, I. Gryczynski and J. R. Lakowicz, Photochem. Photobiol., 1993, 58, 341–345 CrossRef CAS PubMed.
- S. A. Garwin, M. S. Kelley, A. C. Sue, E. L. Que, G. C. Schatz, T. K. Woodruff and T. V. O’Halloran, J. Am. Chem. Soc., 2019, 141, 16696–16705 CrossRef CAS PubMed.
- D. Cheng, X. Liu, Y. Xie, H. Lv, Z. Wang, H. Yang, A. Han, X. Yang and L. Zang, Sensors, 2017, 17, 2517 CrossRef PubMed.
- X. Jiao, C. Liu, S. He, L. Zhao and X. Zeng, Dyes Pigm., 2019, 160, 86–92 CrossRef CAS.
- V. Juvekar, S. J. Park, J. Yoon and H. M. Kim, Coord. Chem. Rev., 2021, 427, 213574 CrossRef CAS.
- D. Kim, H. G. Ryu and K. H. Ahn, Org. Biomol. Chem., 2014, 12, 4550–4566 RSC.
- S. Sumalekshmy and C. J. Fahrni, Chem. Mater., 2011, 23, 483–500 CrossRef CAS PubMed.
- D. Bourassa, C. M. Elitt, A. M. McCallum, S. Sumalekshmy, R. L. McRae, M. T. Morgan, N. Siegel, J. W. Perry, P. A. Rosenberg and C. J. Fahrni, ACS Sens., 2018, 3, 458–467 CrossRef CAS PubMed.
- P. Ning, J. Jiang, L. Li, S. Wang, H. Yu, Y. Feng, M. Zhu, B. Zhang, H. Yin, Q. Guo and X. Meng, Biosens. Bioelectron., 2016, 77, 921–927 CrossRef CAS PubMed.
- W. Li, Z. Liu, B. Fang, M. Jin and Y. Tian, Biosens. Bioelectron., 2020, 148, 111666 CrossRef CAS PubMed.
- X. Meng, S. Wang, Y. Li, M. Zhu and Q. Guo, Chem. Commun., 2012, 48, 4196–4198 RSC.
- K. P. Divya, S. Sreejith, P. Ashokkumar, K. Yuzhan, Q. Peng, S. K. Maji, Y. Tong, H. Yu, Y. Zhao, P. Ramamurthy and A. Ajayaghosh, Chem. Sci., 2014, 5, 3469–3474 RSC.
- W. Li, B. Fang, M. Jin and Y. Tian, Anal. Chem., 2017, 89, 2553–2560 CrossRef CAS PubMed.
- L. Zhu, A. H. Younes, Z. Yuan and R. J. Clark, J. Photochem. Photobiol., A, 2015, 311, 1–15 CrossRef CAS PubMed.
- J. Bednarska, R. Zaleśny, N. Arul Murugan, W. Bartkowiak, H. Ågren and M. Odelius, J. Phys. Chem. B, 2016, 120, 9067–9075 CrossRef CAS PubMed.
- S. Huang, B.-Z. Yang and A.-M. Ren, J. Mol. Struct., 2016, 1114, 65–77 CrossRef CAS.
- D. Wang, A.-M. Ren, L.-Y. Zou, J.-F. Guo and S. Huang, J. Photochem. Photobiol., A, 2017, 341, 20–30 CrossRef CAS.
- Y. Tan, J. Gao, J. Yu, Z. Wang, Y. Cui, Y. Yang and G. Qian, Dalton Trans., 2013, 42, 11465–11470 RSC.
- Y. Zhang, X. Chen, J. Liu, G. Gao, X. Zhang, S. Hou and H. Wang, New J. Chem., 2018, 42, 19245–19251 RSC.
- Y.-Y. Zhang, X.-Z. Chen, X.-Y. Liu, M. Wang, J.-J. Liu, G. Gao, X.-Y. Zhang, R.-Z. Sun, S.-C. Hou and H.-M. Wang, Sens. Actuators, B, 2018, 273, 1077–1084 CrossRef CAS.
- W. Ye, S. Wang, X. Meng, Y. Feng, H. Sheng, Z. Shao, M. Zhu and Q. Guo, Dyes Pigm., 2014, 101, 30–37 CrossRef CAS.
- J. Young Choi, G.-H. Kim, Z. Guo, H. Yeon Lee, K. M. K. Swamy, J. Pai, S. Shin, I. Shin and J. Yoon, Biosens. Bioelectron., 2013, 49, 438–441 CrossRef CAS PubMed.
- Z. Li, Y. Xu, J. Fu, H. Zhu and Y. Qian, Chem. Commun., 2018, 54, 888–891 RSC.
- L. Chen, S. J. Park, D. Wu, H. M. Kim and J. Yoon, Chem. Commun., 2019, 55, 1766–1769 RSC.
- J. Hu, Z. Hu, S. Liu, Q. Zhang, H.-W. Gao and K. Uvdal, Sens. Actuators, B, 2016, 230, 639–644 CrossRef CAS.
- W. Lin, L. Yuan, W. Tan, J. Feng and L. Long, Chem. – Eur. J., 2009, 15, 1030–1035 CrossRef CAS PubMed.
- M. G. Choi, J. Kim, J. M. Hong, I. J. Chang, S. Ahn and S.-K. Chang, Tetrahedron Lett., 2016, 57, 975–978 CrossRef CAS.
- S. Y. Park, W. Kim, S.-H. Park, J. Han, J. Lee, C. Kang and M. H. Lee, Chem. Commun., 2017, 53, 4457–4460 RSC.
- S. Qin, B. Chen, J. Huang and Y. Han, New J. Chem., 2018, 42, 12766–12772 RSC.
- L. Zhou, S. Hu, H. Wang, H. Sun and X. Zhang, Spectrochim. Acta, Part A, 2016, 166, 25–30 CrossRef CAS PubMed.
- Z. Zhang, B. Zhang, X. Qian, Z. Li, Z. Xu and Y. Yang, Anal. Chem., 2014, 86, 11919–11924 CrossRef CAS PubMed.
- S.-L. Pan, K. Li, L.-L. Li, M.-Y. Li, L. Shi, Y.-H. Liu and X.-Q. Yu, Chem. Commun., 2018, 54, 4955–4958 RSC.
- M. Üçüncü, E. Karakuş and M. Emrullahoğlu, Chem. – Eur. J., 2015, 21, 13201–13205 CrossRef PubMed.
- M. Üçüncü, E. Karakuş and M. Emrullahoğlu, Chem. Commun., 2016, 52, 8247–8250 RSC.
- J. B. Birks, Rep. Prog. Phys., 1975, 38, 903–974 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- S. S. Razi, R. Ali, P. Srivastava and A. Misra, RSC Adv., 2015, 5, 79538–79547 RSC.
- P. K. Mehta, E.-T. Oh, H. J. Park and K.-H. Lee, Sens. Actuators, B, 2018, 256, 393–401 CrossRef CAS.
- B. Muzey and A. Naseem, J. Photochem. Photobiol., A, 2020, 391, 112354 CrossRef CAS.
- L.-J. Ma, Q. Liang, R. Feng, Z. Lv, F. Cui, L. Li, L. Yang, H. Liu and F. Sun, J. Photochem. Photobiol., A, 2021, 408, 113086 CrossRef CAS.
- E. N. Ushakov, S. P. Gromov, O. A. Fedorova, Y. V. Pershina, M. V. Alfimov, F. Barigelletti, L. Flamigni and V. Balzani, J. Phys. Chem. A, 1999, 103, 11188–11193 CrossRef CAS.
- P. K. Mehta, E.-T. Oh, H. J. Park and K.-H. Lee, Sens. Actuators, B, 2017, 245, 996–1003 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- L. Li, R. Guan, M. Guo, P. Ning, R. Shao and X. Meng, Sens. Actuators, B, 2018, 254, 949–955 CrossRef CAS.
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed.
- K. Aich, S. Goswami, S. Das, C. D. Mukhopadhyay, C. K. Quah and H.-K. Fun, Inorg. Chem., 2015, 54, 7309–7315 CrossRef CAS PubMed.
- H. Xu, C. Zhu, Y. Chen, Y. Bai, Z. Han, S. Yao, Y. Jiao, H. Yuan, W. He and Z. Guo, Chem. Sci., 2020, 11, 11037–11041 RSC.
- Y. Ma, H. Chen, F. Wang, S. Kambam, Y. Wang, C. Mao and X. Chen, Dyes Pigm., 2014, 102, 301–307 CrossRef CAS.
- D. Cao, L. Zhu, Z. Liu and W. Lin, J. Photochem. Photobiol., C, 2020, 44, 100371 CrossRef CAS.
- L. Zhou, X. Zhang, Q. Wang, Y. Lv, G. Mao, A. Luo, Y. Wu, Y. Wu, J. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 9838–9841 CrossRef CAS PubMed.
- Y. Nakahara, T. Kida, Y. Nakatsuji and M. Akashi, Chem. Commun., 2004, 224–225 RSC.
- F. Li, K. Zhong, S. Hou, L. Tang and Y. Bian, Tetrahedron Lett., 2020, 61, 151558 CrossRef CAS.
- K. Chaichana, N. Phutlaprungrueang, L. Chaicharoenwimolkul, M. Promkatkaew and S. Kongsriprapan, Spectrochim. Acta, Part A, 2019, 207, 118–122 CrossRef CAS PubMed.
- P. Saluja, N. Kaur, N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 6705–6708 CrossRef CAS.
- A. K. K. Bhasin, P. Chauhan and S. Chaudhary, Sens. Actuators, B, 2019, 294, 116–122 CrossRef CAS.
- P. N. Basa, A. Bhowmick, M. M. Schulz and A. G. Sykes, J. Org. Chem., 2011, 76, 7866–7871 CrossRef CAS PubMed.
- Y. Nakahara, T. Kida, Y. Nakatsuji and M. Akashi, Org. Biomol. Chem., 2005, 3, 1787–1794 RSC.
- I. Rivilla, B. Aparicio, J. M. Bueno, D. Casanova, C. Tonnelé, Z. Freixa, P. Herrero, C. Rogero, J. I. Miranda, R. M. Martínez-Ojeda, F. Monrabal, B. Olave, T. Schäfer, P. Artal, D. Nygren, F. P. Cossío and J. J. Gómez-Cadenas, Nature, 2020, 583, 48–54 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2021 |