
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of surface contamination on the interfacial properties of CO2/water/calcite systems
Tran
Thi Bao Le
a,
Candice
Divine-Ayela
a,
Alberto
Striolo
 *a and
David R.
Cole
b
*a and
David R.
Cole
b
aDepartment of Chemical Engineering, University College London, London WC1E 6BT, UK. E-mail: a.striolo@ucl.ac.uk
bSchool of Earth Sciences, The Ohio State University, Columbus, Ohio 43210, USA
First published on 12th August 2021
Abstract
Understanding the wetting properties of reservoir rocks can be of great benefit for advanced applications such as the effective trapping and geological storage of CO2. Despite their importance, not all mechanisms responsible for wetting mineral surfaces in subsurface environments are well understood. Factors such as temperature, pressure and salinity are often studied, achieving results with little unanimity; other possible factors are left somewhat unexplored. One such factor is the effect of contamination. In the present study, the effects of adding a non-aqueous organic contaminant, ethanol, on the CO2–water interfacial tension (IFT) and the CO2/water/calcite contact angle were investigated using molecular dynamics simulations. Within the conditions studied, relatively small amounts of ethanol cause a significant decrease in the CO2–water IFTs, as well as a pronounced increase in the water-calcite-CO2 three phase contact angle. The latter result is due to the decrease of the IFT between CO2 and water and the strong adsorption of ethanol on the solid substrate. These findings could be helpful for explaining how impurities can affect experimental data and could lead to effective carbon sequestration strategies.
1. Introduction
Global emissions of one of the main greenhouse gas contributors, CO2, adds to worsening global warming effects, thus invoking the need for rapid action. Carbon sequestration (CS) or carbon capture and storage (CCS) – is a family of technologies developed to capture CO2 from industrial emitters, or directly from air, and sequester it, possibly in geological repositories. With the potential to trap 85–90% of industrial CO2 emissions,1 this method could have a major effect on managing climate change. Currently, geological storage in natural formations such as deep saline aquifers is considered to be the most likely option for the storage of large CO2 amounts.1–3 While the mineral composition varies greatly between different locations and reservoir types, calcite is present in many geological formations in the form of limestone and chalk rocks and it is readily available to use and model in laboratory studies.Among other risks associated with CCS, possible CO2 leakage – be it naturally occurring via faults and fractures in the rock, or via man-made structures such as faulty bore holes – could have negative consequences. Zwaan et al.4 suggested that CCS is essentially ineffective as a climate change mitigation option when the CO2 leakage rate is as high as 1% per year. Therefore, the success of containment strategies relies on the trapping mechanisms which are highly influenced by wettability characteristics of geologic minerals in the presence of aqueous brines and injected CO2. Wettability of a rock surface can be assessed by the three-phase (CO2–water–rock) contact angle, expressed by the Young's equation as:5
 | (1) |
In eqn (1), γS1 and γS2 are the surface tensions of the two fluids (water and CO2, sometimes at supercritical conditions, respectively) against the solid surface (calcite), while γ12 is the interfacial tension between the two fluids. For completeness, it should be noted that Makkonen recently suggested that interpreting the Young equation in terms of surface energies enables one to reconcile experimental observations such as the pinning of a contact line and the deformation of a solid substrate.6 Regarding the wettability of rock surfaces, several authors have reported that the contact angles can be controlled by factors such as surface roughness and/or chemical heterogeneity,7–9 pressure and temperature,10–14 and salinity.13,15 One additional parameter, commonly overlooked and frequently difficult to control, is the possible presence of impurities. Although CO2 is often processed before transport and storage to separate impurities, small amounts of other chemicals can remain.16,17 Many flue gas impurities can arise from industry and everyday life, such as N2, O2, Ar, NOx species (e.g., NO and NO2), sulphur species (e.g., H2S, SO2), alkali metal salts (e.g., KCl and K2SO4), and hydrocarbons (e.g., CH4 and C2H6).18,19 Understanding the impact of these impurities is of vital importance to the safety of operations as well as to control the cost of transport and storage of the captured CO2. Of particular relevance, it is known that various chemical species can affect both interfacial tension (IFT) and wettability.20–24 Using molecular dynamics (MD) simulations, Chen et al.20 studied the effects of adding CH4, Ar, and H2S to CO2 on the interfacial tension and wettability of the CO2/water/silica system at 20 MPa and 318 K. They found that Ar increases the CO2–water IFT, whereas H2S reduces it. Adding CH4 to CO2 has no significant effect on the IFT. On the other hand, the water contact angle increases when H2S is mixed with CO2, while Ar and CH4 decrease the water contact angle. Saraji et al.23 experimentally measured IFT and dynamic contact angles for CO2/brine/quartz systems at different pressures (13.79–27.58 MPa), temperatures (323–373 K), and salinities (0.2–5 M). They reported that increasing the SO2 amount in the system reduced the IFT between CO2 and brine, while quartz wettability was not affected.
Among other possible impurities, we concentrate here on ethanol, because this compound is expected to be surface active, and because CO2 from ethanol production facilities has increased significantly as more ethanol is utilised as a transportation fuel or mixed with gasoline.25,26 CO2 emitted from ethanol plants is of high purity (∼99% by volume), however, ethanol can remain as an impurity in the CO2 flue gas stream.25 The question we address is whether ethanol can affect interfacial properties of relevance to CCS. Because of computing power limitations, we consider small, yet notable ethanol concentrations in our systems. Although these concentrations could reflect the progressive accumulation of ethanol within a formation, it is recognised that the effects on both IFT and contact angle depend on ethanol concentration.
In our previous study,27 we simulated the CO2/water/calcite system comparing the contact angle predicted when different force fields were implemented. When the force field proposed by Xiao et al.28 was implemented to describe water–calcite interactions, the resultant water contact angle was very low, suggesting complete water wetting in the presence of CO2. On the other hand, when the force field parameters proposed by Raiteri et al.29 and Silvestri et al.30 were implemented, we found a contact angle of ∼46° for water surrounded by CO2 on the calcite surface, at 323 K and 20 MPa. The wide variation in the simulated results mirrors the variation reported experimentally. For example, Wang et al.13 reported a contact angle of ∼26.2° at 323 K and 20 MPa, while Arif et al.31 measured water contact angles as high as 80–90° at the same conditions. However, we note that it is generally expected that the calcite system is strongly water wet.32 Because of the large variation of contact angles observed experimentally, we could not conclude which, out of the two force fields tested for describing calcite, was more realistic. To further assess the reliability of the two force fields, Ali et al.33 investigated the structure of the hydration layer on calcite. Both force fields consistently yield a dense first hydration layer and predict similar distributions of aqueous electrolytes near the surface. Such predictions were found in agreement, within statistical uncertainty, with experimental data, suggesting that both force fields are acceptable for describing the calcite-water interface. However, it should be noted that, to facilitate the description of multi-component systems, Ali et al. implemented a parameterisation of the Raiteri et al. force field in which Buckingham potentials were fitted to reproduce Lennard-Jones potentials. This parameterisation, presented by Shen et al.,34 was found to reproduce hydration free energies of Ca2+ and CO32− ions, as well as the structure of interfacial water.
It is recognised that disagreement in the experimental data could be due to many effects, including the presence of impurities such as organics that might affect the contact angles, or the dissolution of calcite under acidic environment formed by the injection of CO2. Of note, the dissolution rates of calcite in CO2-saturated water were found to increase with temperature (323–373 K) and pressure (6.0–13.8 MPa).35 We decided not to include dissolution reactions, which could lead to changes in the calcite surface morphology. For computational reasons, we could not include the presence of common hydrated carbonates in the fluid system. While the effect of these approximations should be quantified in the future, the objective of this work is to test whether relatively small amounts of ethanol could lead to larger contact angles when the force field proposed by Xiao et al. is implemented to describe calcite. This force field was chosen because it yields complete water wetting at the conditions set for the simulations, a condition that follows expectations.32 Should it be confirmed that impurities could have a major effect on the measured contact angle for the CO2/water/calcite system, the need of synergistic experimental-computational studies for securing progress in this field would be reinforced, given the importance of wetting properties on designing carbon sequestration strategies. The remainder of this manuscript is organized as follows. In Section 2, we describe the simulation methods and algorithms implemented. In Section 3, we present the MD simulation results, starting from the prediction of IFT, followed by the wettability studies. We then summarize our main findings in the Conclusions, Section 4.
2. Simulaton methods and algorithms
2.1. Molecular models and force fields
The calcite slab was obtained from a calcite crystal terminated at the plane [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 4],36 following the protocol adopted in our previous study.27 We chose the force field developed by Xiao et al.28 to model the calcite surface. In our implementation, mimicking our prior study, calcium and carbon atoms in the solid substrate were tethered to their initial positions, whereas the oxygen atoms were allowed to move freely.
4],36 following the protocol adopted in our previous study.27 We chose the force field developed by Xiao et al.28 to model the calcite surface. In our implementation, mimicking our prior study, calcium and carbon atoms in the solid substrate were tethered to their initial positions, whereas the oxygen atoms were allowed to move freely.
The optimized potential for liquid simulation in the all atom form (OPLS-AA) was implemented to describe ethanol.37–39 To be consistent with our previous study,27 the simple point charge extended (SPC/E) model40 and the flexible version of the EPM2 model of Cygan et al.41 were used to describe water and CO2, respectively.
In all simulations, non-bonded dispersive interactions were described implementing the 12-6 Lennard-Jones (LJ) potential. The LJ parameters for all unlike interactions were determined by applying Lorentz–Berthelot mixing rules.42 The electrostatic interactions were modelled by the Coulombic potential, with the long-range corrections treated using the particle mesh Ewald method (PME).43 A cut-off distance of 12 Å was used for all interatomic interactions.
2.2. Simulation setup
The simulation box was periodic in the three directions for all simulations in this study.To predict the IFT, we performed NPT simulations of 3000 water molecules, 2000 CO2 molecules and various numbers of ethanol molecules (i.e., 100 and 200) placed in a XYZ box of size 48.57 × 50 × 100 Å3. The liquid water film was aligned parallel to the XY plane.
To study calcite wettability, we simulated systems in which a cylindrical water droplet, periodic along the X direction of the simulation box, was placed on the calcite surface and then surrounded by the other fluids (e.g., CO2 and ethanol). In our previous study,27 the contact angle for pure water droplet on calcite was found to be size-dependent for small amounts of water molecules. It converged to a certain value for large droplets. To generate droplets that are large enough, we first performed NVT simulations for 4000 water molecules placed on the calcite surface. Subsequent simulations were conducted in the NPT ensemble, wherein the pressure was controlled in the direction perpendicular to the calcite surface, to extract the values for the three-phase contact angles.
The number of water molecules in the simulated systems was determined as a compromise between accuracy and computational cost. Although the formation of a hemi-cylindrical as opposed to hemi-spherical water droplet allows our system not to be affected by the line tension, prior studies show that simulated contact angles tend to change with the droplet size (especially for hemi-spherical droplets).27,44 However, the larger the system size, the more the computational cost. The system considered here (initial droplet radius ∼40 Å) is similar in size to the one considered in our previous work, allowing for reasonable comparison. The X, Y, and Z dimensions of the simulation box were 48.57, 270, and 120 Å, respectively. The solid substrate was parallel to the X and Y directions, while the solid–fluid interface was set perpendicularly to the Z direction. The Y and Z directions were chosen to be large enough to prevent interactions between periodic images of the simulated systems. The final hemi-cylindrical configuration of the droplet was then simulated in the presence of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CO2 and various numbers of ethanol molecules. In this set up, the fluid phase compositions for each system simulated are provided in Table 1. It is worth noting that the ratio between CO2 and H2O water molecules considered is expected to be consistent with realistic scenarios encountered near the wellbore during CO2 injection for CCS. There are recommended maximum levels of impurities in CO2 streams for the purpose of CO2 capture, transport and geological storage.45 Although it is possible that some of these impurities accumulate within a formation, it should be recognised that in our simulations, relatively high concentrations of ethanol in CO2 (∼4–9 wt%) were used, because of computational limitations. It is expected that the effects documented here will vary as the ethanol concentration changes.
000 CO2 and various numbers of ethanol molecules. In this set up, the fluid phase compositions for each system simulated are provided in Table 1. It is worth noting that the ratio between CO2 and H2O water molecules considered is expected to be consistent with realistic scenarios encountered near the wellbore during CO2 injection for CCS. There are recommended maximum levels of impurities in CO2 streams for the purpose of CO2 capture, transport and geological storage.45 Although it is possible that some of these impurities accumulate within a formation, it should be recognised that in our simulations, relatively high concentrations of ethanol in CO2 (∼4–9 wt%) were used, because of computational limitations. It is expected that the effects documented here will vary as the ethanol concentration changes.
| System | Number of molecules | ||
|---|---|---|---|
| H2O | CO2 | Ethanol | |
| S0 | 4000 | 14000 |
0 |
| S1 | 4000 | 14000 |
500 |
| S2 | 4000 | 14000 |
850 |
| S3 | 4000 | 14000 |
1200 |
2.3. Algorithms
All simulations were conducted by performing molecular dynamics (MD) simulations using the package GROMACS (version 5.1.4)46,47 in the NPT ensemble, where the number of particles (N), the pressure (P), and the temperature (T) are kept constant. The pressure of all systems was coupled in the Z direction using the Parrinello–Rahman barostat48 with a relaxation time of 1 ps. The temperature of all systems was maintained constant using Nosé–Hoover thermostats,49,50 as discussed below, with a relaxation time constant of 0.1 ps. The equations of motion were integrated by implementing the leapfrog algorithm51 with a time step of 1.0 fs.For contact angle simulations, NPT simulations were conducted at 323K and 20MPa. To maintain the kinetic energy distribution between solid and fluid,52 the temperature of calcite and that of the fluid were controlled separately using two Nosé–Hoover thermostats. The droplet shape did not change within a simulation time of 40 ns. Each simulation was repeated three times to assess the reliability of the results. Contact angles were extracted from two-dimensional density profiles of water following the procedure of de Ruijter et al.53 The contact angle was calculated every 2 ns during the last 6 ns of each simulation.
To complement the contact angle simulations, we quantified the effect of ethanol on the CO2/water interfacial tension. The simulation set up for these calculations is shown in Fig. 1. We performed the simulations of the CO2/water and (CO2 + ethanol)/water interfacial systems at selected P and T conditions representative of geological CS.54 A series of MD simulations were conducted at temperatures of 323 K over a pressure range of 5–50 MPa. All simulations were carried out for up to 24 ns at each pressure and temperature. The systems reached thermodynamic equilibrium within a simulated time of 20 ns. The trajectories of the last 4 ns of the simulations were used for further analysis presented in Section 3.1. Each simulation was repeated five times to assess the reliability of the results. After equilibration, two thermodynamically stable phases in the simulation box were separated by an interface perpendicular to the Z axis, as shown in Fig. 1. Therefore, the IFT between fluid phases was evaluated from the expression of pressure tensor as follows:55
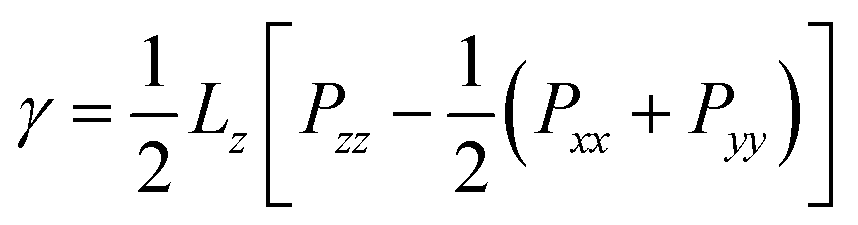 | (2) |
 | ||
| Fig. 1 Equilibrium interfacial tension models of (a) CO2/H2O, and (b) (CO2 + 100 ethanol)/H2O systems. The simulations were conducted at 323 K and 50 MPa. Water molecules are presented in yellow block, and cyan, red, and white spheres represent carbon, oxygen, and hydrogen atoms, respectively. | ||
In eqn (2), Lz is the box length in Z direction, Pzz is the interface-normal pressure component, Pxx and Pyy are the interface-parallel pressure components.
3. Simulation results
3.1. CO2/water interfacial tension
Fig. 2 shows the predicted interfacial tension for the systems considered in this study along with the experimental CO2/H2O data reported by Chiquet et al.54 and the MD results of Silvestri et al.44 In general, our predicted IFTs decrease rapidly with the increase of pressure and then remain approximately constant at pressures above ∼20 MPa. Our results for CO2–water IFT are in good agreement with those reported by Silvestri et al.44 However, we observe large deviations between experimental and simulation data at high pressures. This discrepancy could be ascribed to many factors, such as the difference between water models, long-range electrostatic interactions, and simulation parameters, e.g., LJ cutoff distance.56 Silvestri et al.44 recently reported that increasing LJ cut off from 10 to 24 Å does not improve significantly the CO2−water IFT predictions at 323 K and 1 MPa. Iglauer et al.10 suggested that the simulation models overpredict the experimental interfacial tension at high pressures due to the Lorentz–Berthelot combining rules. These rules ignore the non-additive contributions between CO2 and water, which are significant at high CO2 densities. Li et al.57 suggested that the deviation for the surface tension at high pressures could also be due to other simulation details, such as system size effects. Nevertheless, for pressures below 15 MPa, the agreement between simulated and experimental IFT seems satisfactory. We limit the remainder of this manuscript to the conditions at which the models implemented here yield reliable data. For completeness, it should be noted that Vlcek et al.58 optimized the unlike-pair interaction parameters for the SPC/E − rigid EPM2 models and was able to reproduce the mutual solubility of CO2 and water at conditions typical for carbon dioxide sequestration. | ||
| Fig. 2 Interfacial tension of the CO2/water and (CO2 + ethanol)/water systems as a function of pressure at 323 K. The green symbols represent the experimental interfacial tension of ethanol + water against the mole fraction of ethanol at 323 K, as reported by Vázquez et al.59 Error bars represent one standard deviation from the average. | ||
Fig. 2 also shows the IFT data upon the addition of ethanol at 323 K. Our results show that the CO2–water IFT decreases significantly with increasing ethanol content. This is consistent with the experimental data reported by Vázquez et al.,59 which show that the surface tension of aqueous solutions of ethanol decreased as the alcohol concentration increased at a given temperature (Fig. 2). We note that our simulations predict that the IFT does not change significantly at pressures above 20 MPs, as was the case for the water–CO2 IFT.
To further investigate the effects of ethanol on the IFT, we calculated the density profiles of all the compounds in the systems in the Z direction. Selected results are shown in Fig. 3. It can be seen that the density of ethanol increases significantly at the surface of water, compared to the bulk water, indicating its preferential adsorption at the water surface, which reduces the IFT. Our results are somewhat similar with observations reported by Chen et al.20 They found that the interfacial tension of the (CO2 + H2S)/water system is smaller than that of the CO2/water system due to a significant increase of H2S density at the water surface. We conclude that our results are qualitatively comparable to those of Chen et al. because both ethanol and H2S molecules are hydrophilic and tend to accumulate at the CO2–water interface.
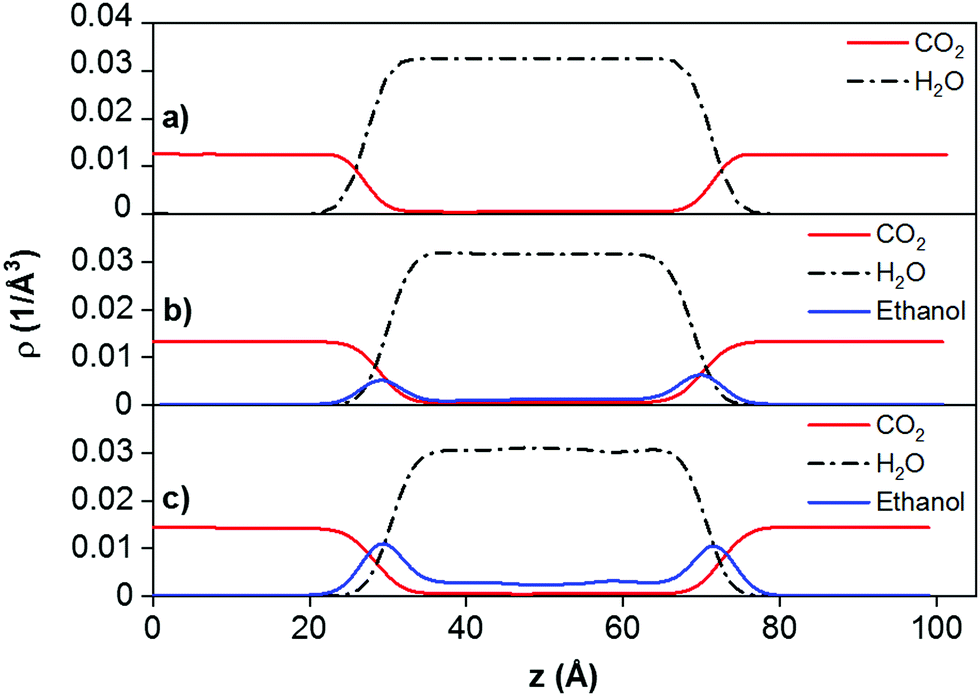 | ||
| Fig. 3 Density profiles of the fluid phase components obtained from (a) CO2/H2O, (b) (CO2 + 100 ethanol)/H2O, and (CO2 + 200 ethanol)/H2O systems. The simulations were conducted at T = 323 K, P = 40 MPa. In all panels we report the density profiles for the centre of mass of CO2, H2O, and ethanol molecules. | ||
3.2. Calcite wettability
Selected snapshots for water droplets on the calcite surface after equilibration are shown in Fig. 4. To complement and better quantify the results from simulation snapshots, we calculated 2D density profiles of water within the plane perpendicular to the substrate and to the axis of the cylindrical water droplet. The results are shown in Fig. 5. It can be seen that in the presence of pure CO2, water completely spreads on the calcite surface, which is consistent with our previous study.27 Adding ethanol has a pronounced effect on the three-phase contact angle, and specifically the calcite surface becomes less water-wet. As the amount of ethanol increases, the water contact angle decreases. Analysis of simulation snapshots such as those shown in Fig. 4, supported by quantitative analysis of density profiles throughout the simulated systems reveal that adding ethanol causes water to ‘bead up’. Prior simulations for a variety of mineral–water interfaces suggested that, when the substrate strongly attracts water molecules, it is possible that a water monolayer forms on the mineral substrate, and then additional water yields a droplet on the water monolayer.60,61 This phenomenon is not observed for the system considered in the present manuscript. | ||
| Fig. 4 Snapshots of H2O droplets on the calcite surface in the presence of (a) pure CO2, (b) CO2 + 500 ethanol, and (c) CO2 + 850 ethanol, and (d) CO2 + 1200 ethanol. The simulations are conducted at 323 K and 20 MPa. Small orange dots represent CO2 molecules; cyan, white, red and green spheres represent carbon, hydrogen, oxygen atoms of water, and oxygen atoms of ethanol, respectively. | ||
 | ||
| Fig. 5 2D density distributions of water oxygen atoms averaged over the final 2 ns of simulations conducted at 323 K and 20 MPa for systems (a) S1, (b) S2, and (c) S3 on the calcite surface (see Table 1 for system composition). The colour bar shows density in the units of 1/Å3. | ||
In Fig. 6, we illustrate the procedure implemented to extract contact angle results from 2D atomic density profiles, using as example a water droplet in the presence of CO2. The predicted contact angles are summarized in Table 2. In addition to ethanol reducing the IFT between CO2 and water, which are described in Fig. 2, the strong adsorption of ethanol molecules onto the calcite surface also contributes to the simulated contact angle changes. Because the simulation snapshots of Fig. 4 suggest that ethanol preferentially adsorbs at the calcite–CO2 interface, both γS2 (between calcite and CO2) and γ12 (between water and CO2) decrease, but the effect on the former seems to be larger than that on the latter, leading to the changes in the three-phase contact angle. Although quantification of the solid–fluid surface energy is not attempted here, for completeness we refer to Grzelak and Errington,62,63 who showed how to implement the grand canonical matrix Monte Carlo approach for such investigations.
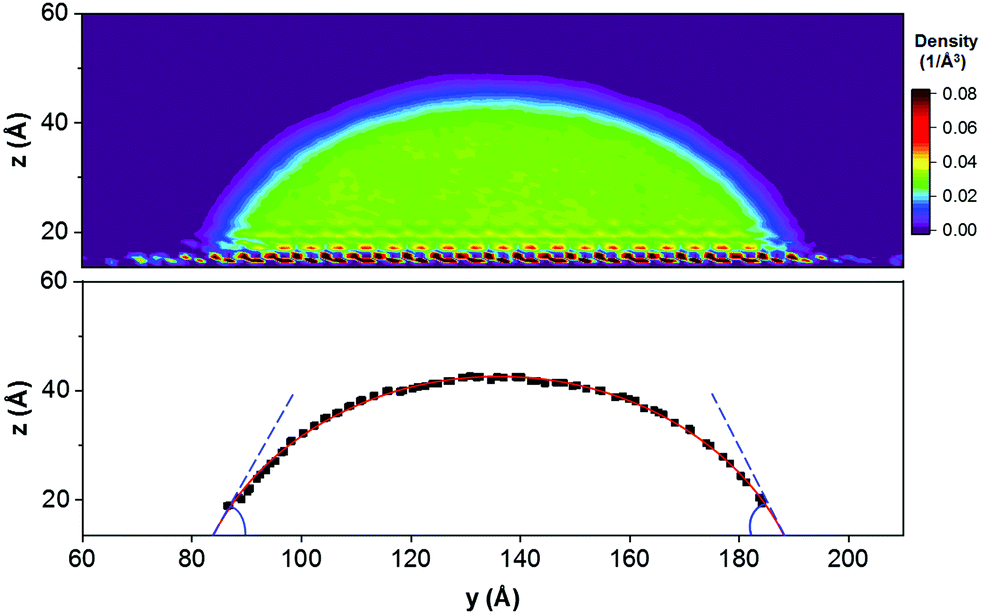 | ||
| Fig. 6 Top: 2D density distribution of water oxygen atoms averaged over the final 2 ns of simulation conducted for system S3 at 323 K and 20 MPa (see Table 1). Bottom: Illustration of the procedure implemented to extract the water contact angle. | ||
| System | Contact angle |
|---|---|
| S0 | ∼0° |
| S1 | 27.8° ± 2.2° |
| S2 | 41.2° ± 2.2° |
| S3 | 46.4° ± 2.7° |
To gain further insights into the adsorption behaviour of fluids near the calcite surface, we examined the density profiles of water and ethanol as a function of the vertical distance. The results are shown in Fig. 7. The water density profiles were calculated along the axis passing through the centre of the droplet. The results show that ethanol molecules formed a monolayer strongly adsorbed to the calcite surface at the calcite–water interface. This is supported by the planar density distributions obtained for the O atoms of ethanol in the plane perpendicular to the droplet, shown in Fig. 8. In Fig. 7, the hydrogen, oxygen, ethyl, and methyl peaks are formed at 1.87, 2.32, 3.37, and 4.57 Å from the calcium atoms in the surface, respectively. The atomic density profiles indicate that ethanol molecules orient their OH groups toward the surface, while they extend their methyl groups toward the CO2 phase. These results are consistent with experimental observations reported by Bovet et al.,64 who found that alcohols (e.g., methanol, ethanol, t-butanol and pentanol) form a compact, well-ordered monolayer on the calcite surface with their OH bonds oriented toward and the carbon chains pointing away from the surface.
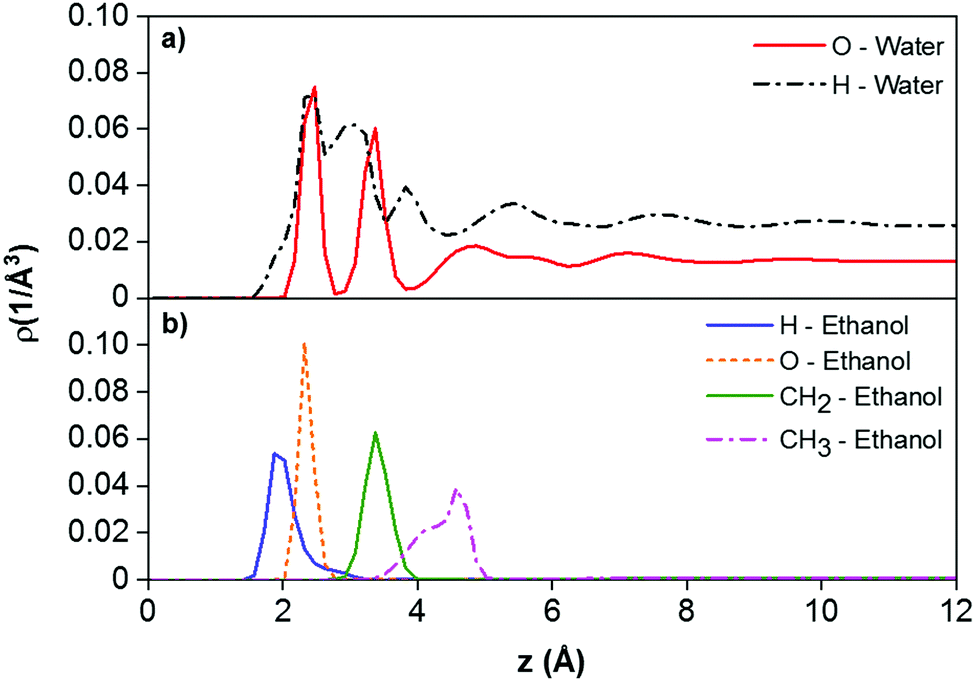 | ||
| Fig. 7 Z-Density profiles of (a) oxygen and hydrogen atoms of water molecules along the axis of symmetry of the droplet and normal to the surface, and (b) methyl (CH3), ethyl (CH2), oxygen, and hydrogen atoms of ethanol molecules along the surface normal. The simulation results were obtained from the system S3 at 323 K and 20 MPa. The reference (z = 0) corresponds the position of the plane of the surface Ca atoms. | ||
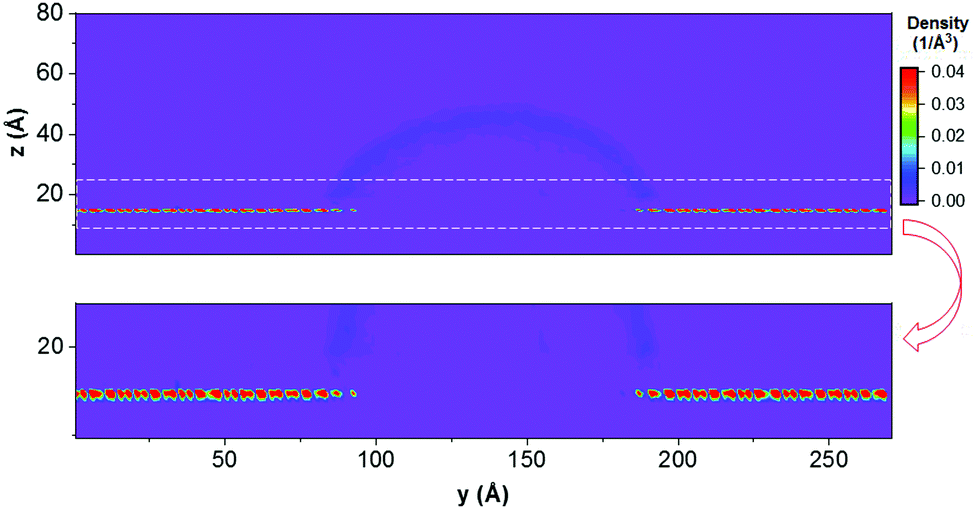 | ||
| Fig. 8 Top: 2D density profiles of O atoms of ethanol molecules surrounding the water droplet. The simulations were conducted for the system S3 and the last 2 ns of the simulations conducted at 323 K and 20 MPa are used for data analysis. Bottom: We report expanded view of the interfacial region. The colour bar shows density in the units of 1/Å3. | ||
3.3. Force field effects
To confirm that the results discussed above do not depend on the force field implemented to describe calcite, we repeated selected simulations for a system in which the calcite substrate was described following the parameterisation proposed by Shen et al.,34 which we had implemented in a prior study to quantify the structure of hydration water on calcite.33 The system composition is 4000 water molecules and 14000 CO2 molecules, the temperature is 323 K, and the pressure is 20 MPa. The contact angle for the water–calcite–CO2 system was compared with and without the ethanol molecules (see Fig. 9). Our results show that the contact angle increases from zero to ∼45° when 1200 ethanol molecules are added to the system. By comparison, when the Xiao et al. force field was implemented, the change observed in the simulations was from zero to ∼46°. Analysis of the simulation results also confirm that ethanol adsorbs predominantly at the calcite–CO2 interface, thereby supporting the observations discussed above.
 | ||
| Fig. 9 Snapshots of H2O droplets on calcite in the presence of (a) pure CO2, and (b) CO2 + 1200 ethanol molecules. The simulations are conducted at 323 K and 20 MPa, implementing the parameterisation proposed by Shen et al.34 to describe calcite. | ||
4. Conclusions
In the present study, the effects of impurities, and specifically that of ethanol on the contact angle of the CO2/water/calcite system were probed using molecular dynamics simulations.To support the direct simulation of the contact angle, which was interpreted based on the Young equation, we also simulated the interfacial tensions (IFT) for CO2/H2O and (CO2 + ethanol)/H2O systems at 323 K over a pressure range of 5–50 MPa. The results show that ethanol reduces the CO2/H2O IFT because it preferentially accumulates at the CO2/water interfaces. Our IFT results are in good agreement with previously reported MD simulations in the whole range of conditions tested, albeit the agreement with experiments is acceptable only up to 15–20 MPa.
To compare against experimental observations, the contact angle simulations were conducted only at 323 K and 20 MPa. At these conditions, complete wetting behaviour of the calcite surface by water was observed in the presence of pure CO2. The addition of ethanol makes the calcite surface less water wet. The water contact angle increases up to ∼46° in the presence of 1200 ethanol molecules (which corresponds to 9 wt% with respect to CO2). Analysis of the simulation trajectories shows that ethanol does not adsorb at the water–calcite interface, while it accumulates at the CO2–calcite interface. This suggests that, even though ethanol reduces the CO2/water interfacial tension, the reduction of the CO2/calcite surface energy is much more pronounced, leading to an increase in the three-phase contact angle for the simulated system. When a different force field was implemented to describe calcite, the simulation results remained qualitatively consistent with the physical description just provided, although the simulation results also suggest that the force field implemented to describe interactions between calcite and fluid mixtures need to be further improved. Nevertheless, the results presented here suggest that the presence of impurities could explain, in part, the disagreement between experiments for the wetting properties of minerals in the presence of CO2 and brines. Our observations could aid the design of effective carbon sequestration strategies, as controlling the amount of impurities present in the system could allow practitioners to adjust the wetting properties as required to promote CO2 penetration within a geological formation as opposed to CO2 trapping, depending on the situation.
Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We acknowledge the financial support from the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-SC0006878 (Division of Chemical Sciences, Geosciences, and Biosciences), Geosciences Program. Additional financial support was provided by the A. P. Sloan Foundation via the Deep Carbon Observatory administered by the Carnegie Institution for Science. AS acknowledges financial support from the Science4CleanEnergy consortium (S4CE), which is supported by the Horizon 2020 R&D programme of the European Commission, via grant No. 764810. Generous allocations of computing time were provided by the National Energy Research Scientific Computing Center (NERSC) at Lawrence Berkeley National Laboratory, Berkeley, CA. NERSC is supported by the DOE Office of Science. We are also grateful to the University College London Research Computing Platforms Support (MYRAID and GRACE) and the UK Materials and Molecular Modelling Hub (THOMAS), for access to high-performance computing.References
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- M. A. Celia and J. M. Nordbotten, Ground Water, 2009, 47, 627–638 CrossRef CAS PubMed.
- Y. Fang, B. Baojun, T. Dazhen, S. Dunn-Norman and D. Wronkiewicz, Petrol Sci., 2010, 7, 83–92 CrossRef.
- B. van der Zwaan and K. Smekens, Environ. Model. Assess., 2009, 14, 135–148 CrossRef.
- T. Young, Philos. Trans. R. Soc. London, 1805, 95, 65–87 CrossRef.
- L. Makkonen, J. Phys.: Condens. Matter, 2016, 28, 135001 CrossRef PubMed.
- M. Andrew, B. Bijeljic and M. J. Blunt, Adv. Water Resour., 2014, 68, 24–31 CrossRef CAS.
- K. J. Kubiak, M. C. T. Wilson, T. G. Mathia and P. Carval, Wear, 2011, 271, 523–528 CrossRef CAS.
- S. B. Wang, Z. Y. Tao, S. M. Persily and A. F. Clarens, Environ. Sci. Technol., 2013, 47, 11858–11865 CrossRef CAS PubMed.
- S. Iglauer, M. S. Mathew and F. Bresme, J. Colloid Interface Sci., 2012, 386, 405–414 CrossRef CAS PubMed.
- D. Y. Yang, Y. G. Gu and P. Tontiwachwuthikul, Energy Fuels, 2008, 22, 504–509 CrossRef CAS.
- D. N. Espinoza and J. C. Santamarina, Water Resour. Res., 2010, 46, W07537 CrossRef.
- S. B. Wang, I. M. Edwards and A. F. Clarens, Environ. Sci. Technol., 2013, 47, 234–241 CrossRef CAS PubMed.
- S. Saraji, L. Goual, M. Piri and H. Plancher, Langmuir, 2013, 29, 6856–6866 CrossRef CAS PubMed.
- J. W. Jung and J. M. Wan, Energy Fuels, 2012, 26, 6053–6059 CrossRef CAS.
- C. Eickhoff, F. Neele, M. Hammer, M. DiBiagio, C. Hofstee, M. Koenen, S. Fischer, A. Isaenko, A. Brown and T. Kovacs, Energy Proc., 2014, 63, 7379–7388 CrossRef.
- F. Neele, J. Koornneef, J. P. Jakobsen, A. Brunsvold and C. Eickhoff, Energy Procedia, 2017, 114, 6536–6542 CrossRef CAS.
- R. T. J. Porter, M. Fairweather, M. Pourkashanian and R. M. Woolley, Int. J. Greenhouse Gas Control, 2015, 36, 161–174 CrossRef CAS.
- J. Y. Lee, T. C. Keener and Y. J. Yang, J. Air Waste Manage., 2009, 59, 725–732 CrossRef CAS PubMed.
- C. Chen, Z. Chai, W. J. Shen and W. Z. Li, Ind. Eng. Chem. Res., 2018, 57, 371–379 CrossRef CAS.
- Q. Y. Ren, G. J. Chen, W. Yan and T. M. Guo, J. Chem. Eng. Data, 2000, 45, 610–612 CrossRef CAS.
- V. Shah, D. Broseta, G. Mouronval and F. Montel, Int. J. Greenhouse Gas Control, 2008, 2, 594–604 CrossRef CAS.
- S. Saraji, M. Piri and L. Goual, Int. J. Greenhouse Gas Control, 2014, 28, 147–155 CrossRef CAS.
- A. Al-Yaseri, M. Sarmadivaleh, A. Saeedi, M. Lebedev, A. Barifcani and S. Iglauer, J. Pet. Sci. Eng., 2015, 129, 58–62 CrossRef CAS.
- Y. X. Xu, L. Isom and M. A. Hanna, Bioresource Technol., 2010, 101, 3311–3319 CrossRef CAS PubMed.
- M. E. D. De Oliveira, B. E. Vaughan and E. J. Rykiel, Bioscience, 2005, 55, 593–602 CrossRef.
- T. T. B. Le, A. Striolo and D. R. Cole, J. Phys. Chem. C, 2020, 124, 18532–18543 CrossRef CAS.
- S. J. Xiao, S. A. Edwards and F. Grater, J. Phys. Chem. C, 2011, 115, 20067–20075 CrossRef CAS.
- P. Raiteri, R. Demichelis and J. D. Gale, J. Phys. Chem. C, 2015, 119, 24447–24458 CrossRef CAS.
- A. Silvestri, A. Budi, E. Ataman, M. H. M. Olsson, M. P. Andersson, S. L. S. Stipp, J. D. Gale and P. Raiteri, J. Phys. Chem. C, 2017, 121, 24025–24035 CrossRef CAS.
- M. Arif, M. Lebedev, A. Barifcani and S. Iglauer, Int. J. Greenhouse Gas Control, 2017, 62, 113–121 CrossRef CAS.
- S. Iglauer, C. H. Pentland and A. Busch, Water Resour. Res., 2015, 51, 729–774 CrossRef CAS.
- A. Ali, T. T. B. Le, A. Striolo and D. R. Cole, J. Phys. Chem. C, 2020, 124, 24822–24836 CrossRef CAS.
- J. W. Shen, C. L. Li, N. F. A. van der Vegt and C. Peter, J. Phys. Chem. C, 2013, 117, 6904–6913 CrossRef CAS.
- C. Peng, J. P. Crawshaw, G. C. Maitland and J. P. M. Trusler, Chem. Geol., 2015, 403, 74–85 CrossRef CAS.
- S. Kerisit and S. C. Parker, J. Am. Chem. Soc., 2004, 126, 10152–10161 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2017, 121, 3864–3870 CrossRef CAS PubMed.
- L. S. Dodda, I. C. de Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- R. T. Cygan, V. N. Romanov and E. M. Myshakin, J. Phys. Chem. C, 2012, 116, 13079–13091 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer simulation of liquids, Oxford University Press, UK, 2004 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- A. Silvestri, E. Ataman, A. Budi, S. L. S. Stipp, J. D. Gale and P. Raiteri, Langmuir, 2019, 35, 16669–16678 CrossRef CAS PubMed.
- E. de Visser, C. Hendriks, M. Barrio, M. J. Molnvik, G. de Koeijer, S. Liljemark and Y. Le Gallo, Int. J. Greenhouse Gas Control, 2008, 2, 478–484 CrossRef CAS.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nose, Mol. Phys., 1984, 52, 255–268 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- R. W. Hockney, S. P. Goel and J. W. Eastwood, J. Comput. Phys., 1974, 14, 148–158 CrossRef.
- J. E. Basconi and M. R. Shirts, J. Chem. Theory Comput., 2013, 9, 2887–2899 CrossRef CAS PubMed.
- M. J. de Ruijter, T. D. Blake and J. De Coninck, Langmuir, 1999, 15, 7836–7847 CrossRef CAS.
- P. Chiquet, J. L. Daridon, D. Broseta and S. Thibeau, Energy Convers Manage., 2007, 48, 736–744 CrossRef CAS.
- A. R. van Buuren, S. J. Marrink and H. J. C. Berendsen, J. Phys. Chem., 1993, 97, 9206–9212 CrossRef CAS.
- P. K. Yuet and D. Blankschtein, J. Phys. Chem. B, 2010, 114, 13786–13795 CrossRef CAS PubMed.
- X. S. Li, D. A. Ross, J. P. M. Trusler, G. C. Maitland and E. S. Boek, J. Phys. Chem. B, 2013, 117, 5647–5652 CrossRef CAS PubMed.
- L. Vlcek, A. A. Chialvo and D. R. Cole, J. Phys. Chem. B, 2011, 115, 8775–8784 CrossRef CAS PubMed.
- G. Vazquez, E. Alvarez and J. M. Navaza, J. Chem. Eng. Data, 1995, 40, 611–614 CrossRef CAS.
- C. L. Wang, H. J. Lu, Z. G. Wang, P. Xiu, B. Zhou, G. H. Zuo, R. Z. Wan, J. Hu and H. P. Fang, Phys. Rev. Lett., 2009, 103, 137801 CrossRef PubMed.
- A. Phan, T. A. Ho, D. R. Cole and A. Striolo, J. Phys. Chem. C, 2012, 116, 15962–15973 CrossRef CAS.
- E. M. Grzelak and J. R. Errington, J. Chem. Phys., 2008, 128, 014710 CrossRef PubMed.
- E. M. Grzelak, V. K. Shen and J. R. Errington, Langmuir, 2010, 26, 8274–8281 CrossRef CAS PubMed.
- N. Bovet, M. Yang, M. S. Javadi and S. L. S. Stipp, Phys. Chem. Chem. Phys., 2015, 17, 3490–3496 RSC.
| This journal is © the Owner Societies 2021 |