
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective soft X-ray absorption as a tool to study protonation and electronic structure of gas-phase DNA
Xin
Wang
a,
Sivasudhan
Rathnachalam
 a,
Klaas
Bijlsma
a,
Wen
Li
a,
Ronnie
Hoekstra
a,
Markus
Kubin
b,
Martin
Timm
b,
Bernd
von Issendorff
c,
Vicente
Zamudio-Bayer
b,
J. Tobias
Lau
bc,
Shirin
Faraji
*a and
Thomas
Schlathölter
*a
a,
Klaas
Bijlsma
a,
Wen
Li
a,
Ronnie
Hoekstra
a,
Markus
Kubin
b,
Martin
Timm
b,
Bernd
von Issendorff
c,
Vicente
Zamudio-Bayer
b,
J. Tobias
Lau
bc,
Shirin
Faraji
*a and
Thomas
Schlathölter
*a
aZernike Institute for Advanced Materials, University of Groningen, Groningen, The Netherlands. E-mail: s.s.faraji@rug.nl; t.a.schlatholter@rug.nl
bAbteilung für Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin, Germany
cPhysikalisches Institut, Universität Freiburg, Freiburg, Germany
First published on 10th May 2021
Abstract
The conformation and the electronic structure of gas-phase oligonucleotides depends strongly on the protonation site. 5′-d(FUAG) can either be protonated at the A-N1 or at the G-N7 position. We have stored protonated 5′-d(FUAG) cations in a cryogenic ion trap held at about 20 K. To identify the protonation site and the corresponding electronic structure, we have employed soft X-ray absorption spectroscopy at the nitrogen K-edge. The obtained spectra were interpreted by comparison to time-dependent density functional theory calculations using a short-range exchange correlation functional. Despite the fact that guanine has a significantly higher proton affinity than adenine, the agreement between experiment and theory is better for the A-N1 protonated system. Furthermore, an inverse site sensitivity is observed in which the yield of the nucleobase fragments that contain the absorption site appears substantially reduced, which could be explained by non-statistical fragmentation processes, localized on the photoabsorbing nucleobase.
1 Introduction
Under most circumstances, DNA in biological systems is multiply negatively charged, because of the phosphate groups in its backbone. Under slightly acidic conditions, cytosine and adenine moieties become protonated and at even lower pH, also guanine is found protonated.1 Nucleobase protonation has important structural implications. It is for instance often key to formation of non-canonical DNA structures such as i-motifs, four-stranded DNA structures that form in cytosine(C)-rich sequences and that are stabilized by stacks of protonated C – neutral C base pairs. In solution, widespread techniques for investigating oligonucleotide structures (such as protonation sites) and structural transitions are UV absorption spectroscopy2 and circular dichroism spectroscopy.3 Due to unresolved issues concerning solvent interactions, however the exact correlation between measured spectral features and DNA structure is still under debate for many systems.A powerful approach for studying DNA structure in more detail and without the influence of solvent interactions are gas-phase studies. Electrospray ionization is an ideal tool to transfer intact biomolecular ions from solution into the gas phase, in particular when performed under “native” conditions, i.e. when for instance DNA tertiary structures are conserved in the process.4 Relatively straightforward mass-spectrometric tools such as collision induced dissociation can already deliver valuable information on the protonation site in gas-phase oligonucleotides.5 The much more structure-sensitive ion-mobility spectrometry has been used to determine the precise ground-state protonation site in gas-phase protonated nucleobases, and even different low-energy tautomeric forms could be distinguished.6 Infrared multiple photon dissociation action spectroscopy techniques in combination with quantum chemical calculations have also proven to be very useful for the determination of protonation sites in nucleosides and nucleotides.7
Over the last years, soft X-ray action spectroscopy techniques have evolved as novel tools for studying electronic structure and conformation of biomolecular ions in the gas phase, with some emphasis on protein folding.8, 9 By soft X-ray absorption, inner shell electrons are excited to unoccupied molecular orbitals (MOs) or ionized to the continuum. The soft X-ray excitation is element specific. Even more so, the absorption edge of a given element features resonances that reflect both, the chemical shift of the inner shell orbital and the unoccupied final MO. Tuning to a resonance thus allows to address a particular transition and electronically excite a molecule in a site-selective manner, for instance to induce scission of selected bonds or to probe the local electronic structure and conformation. Site selective bond scission induced by resonant soft X-ray absorption has been observed in photoion-photoelectron coincidence experiments on a selection of relatively small neutral molecules such as ClCH2Br.10 Also for N-methylacetamide, a molecule containing a peptide bond, excitation of the strongest N-1s resonance significantly increases breakage of that peptide bond.11 On the other hand, for 2Br-pyrimidine, a model halopyrimidine nucleobase analogue, fragmentation depends on the final electronic state of the molecule only, and not on the initial photoabsorption site.12 Very recently, Schwob and coworkers could demonstrate a site effect in the photofragmentation spectrum of the protonated S-containing peptide [Met5]-enkephalin for soft X-ray absorption at the sulfur L-edge.13
In this study we combine high resolution soft X-ray partial ion yield spectroscopy at cryogenic temperatures with time-dependent density functional theory (TDDFT) using a short-range exchange correlation functional (SRC) that is specifically tailored for inner-shell excitation processes.14, 15 The approach involves two different types of site-sensitivity: (i) the precise choice of soft X-ray photon energy not only allows to target a particular element by its inner shell absorption edge, but also to further specify the absorption site by selection of a suitable inner-shell excitation resonance. (ii) selection of the mass of the photoion under investigation defines a fragmentation channel, i.e. a particular bond scission that resulted from the initial absorption event. The two-fold site-sensitivity is employed to distinguish between two possible protonation sites in the gas-phase oligonucleotide 5′-d(FUAG) and to investigate energy dissipation in this molecule. The 5′-d(FUAG) model system contains the nucleobases adenine (A), guanine (G) and the nucleobase analog 5-fluorouracil (FU). The latter is employed because its high ionization potential and very low proton affinity render it a spectator for both, protonation and positive charge transport. Furthermore, it features a unique F atom that allows for selective soft X-ray targeting in future studies. The schematic structure is displayed in Fig. 1 with the conventional atom numbering. Note that atoms attached to a ring atom have the same number as the ring atom itself. 5′-d(FUAG) features a decrease in nucleobase vertical ionization potential from the 5′ to the 3'-terminal (FU: 9.46 eV,16A: 8.37 eV, G: 8.1 eV17) that favors hole migration to the 3′-terminal, i.e. charge transport. Proton affinities follow an opposite trend, being lowest for N atoms on FU and very similar but much higher for A-N1 (9.77 eV) and G-N7 (9.84 eV).18 By spectroscopically targeting the N K-edge around 400 eV, the focus of the study is set on the nucleobases (the only groups containing N atoms) and in particular on protonation of N atoms. N atoms in nucleobases have either amine (three single bonds, >N–) or imine (one double bond, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) character with the respective N-1s electrons being either weakly (imine) or strongly (amine) bound.19 In X-ray absorption spectra, this manifests in a transition-energy band π1 (imine-N 1s-π*) and a higher transition energy band π2 (amine-N 1s-π*), that has been observed for nucleotides in solution20 or for nucleobases in the gas-phase19, 21 but not for gas-phase DNA. In our model oligonucleotide, the ratio between amine and imine N atoms varies (amine: FU-N1,N3, A-N6,N9, G-N1,N2,N9; imine: A-N1,N3,N7, G-N3,N7). Protonation of either A-N1 or G-N7 (the most likely protonation sites) strongly increases the respective N-1s binding energy, as the N character changes from imine to an imine/amine mixture.
N–) character with the respective N-1s electrons being either weakly (imine) or strongly (amine) bound.19 In X-ray absorption spectra, this manifests in a transition-energy band π1 (imine-N 1s-π*) and a higher transition energy band π2 (amine-N 1s-π*), that has been observed for nucleotides in solution20 or for nucleobases in the gas-phase19, 21 but not for gas-phase DNA. In our model oligonucleotide, the ratio between amine and imine N atoms varies (amine: FU-N1,N3, A-N6,N9, G-N1,N2,N9; imine: A-N1,N3,N7, G-N3,N7). Protonation of either A-N1 or G-N7 (the most likely protonation sites) strongly increases the respective N-1s binding energy, as the N character changes from imine to an imine/amine mixture.
 | ||
| Fig. 1 A schematic of the 5′-d(FUAG) oligonucleotide showing the site-numbering convention as well as the vertical ionization potentials17 and the highest proton affinities18 for the isolated nucleobases. The two most likely protonation sites A-N1 and G-N7 are indicated in green. The bottom panel displays a high resolution N K-edge soft X-ray photofragmentation mass spectrum for [d(FUAG)+H]+ (EX = 407 eV, recorded at room temperature). IPv: vertical ionization potential, PA: proton affinity. | ||
2 Experiment
The bulk of the experiments was performed using the nanoclustertrap apparatus,22, 23, 24 which is a fixed endstation at the high-resolution soft X-ray beamline UE52-PGM of the BESSY II synchrotron radiation facility. The photon energy was calibrated using the Ne 1s line for the first 3 monochromator orders. For the N K-edge, the error bar of the photon energy was ±100 meV. Protonated [d(FUAG)+H]+ cations where produced by means of electrospray ionzation (ESI) from a 20 μM solution in methanol. The mass-selected cations where accumulated in a cryogenic (T ≈ 20 K) linear radiofrequency (RF) ion trap. An action-spectroscopy approach was employed for determination of the soft X-ray photoabsorption cross sections, i.e. photon energy EX-ray was ramped in 50 meV steps across the nitrogen K-edge (398–410 eV). For each EX-ray, the trapped protonated oligonucleotides were exposed to monochromatic (ΔE ≈ 100 meV) soft X-rays and the entire trap content including the photo-products were extracted into a time-of-flight (TOF) mass spectrometer. The relative ion yields for the different photofragments were determined from the TOF spectra and plotted against the X-ray photon energy to obtain N K-edge X-ray absorption spectra for the dominant fragments. Large trap volume and high soft X-ray resolution allowed for swift acquisition of high resolution spectra. Three independent scans of the X-ray energy were recorded and averaged. The variation between consecutively recorded spectra was small. Multi-photon absorption by the [d(FUAG)+H]+ precursor cations was negligible.The thermalization of the oligonucleotide ions with the cryogenic He buffer gas in the trap occurs on a several μs timescale. The relatively slow cooling process freezes out transient conformations but also reduces the number of fundamental conformers present in the trap. Possible effects of molecular conformation on the recorded soft X-ray spectra would therefore be minimized. Furthermore, the cryogenic buffer gas quenches fragmentation processes that occur on long (μs and more) timescales. This way, fragmentation processes that occur long after intramolecular vibrational redistribution are quenched.
3 Theory
For the theoretical part, the ground-state molecular structures were optimized in the gas phase using density functional theory (DFT) at the ωB97X-D/cc-pVDZ level of theory. As a reference, we first optimized the geometry of the neutral system. Subsequently, we optimized the geometry of the protonated oligonucleotide for two different positively charged systems being protonated on the A-N1 and on the G-N7 moiety, respectively (A-N1 and G-N7 are long known as the most likely protonation sites in isolated 2′-deoxyadenosine and 2′deoxyguanosine, respectively but also for nucleotides embedded in single stranded DNA.1). All minimized structures were confirmed by Hessian calculations and no imaginary frequency were found. A detailed exploration of the conformational space was beyond the scope of this study. The core-excitations were then computed using time-dependent DFT (TD-DFT) with Tamm–Dancoff approximation.25 Calculations were performed with restricted single excitation subspace that involved excitation only from the 1s orbital of a single N atom at a time. The short-range exchange correlation functional SRC2-R114, 15 with the parameters CSHF = 0.55, μSR = 0.69a0−1, CLHF = 0.08 and μLR = 1.02a0−1 in combintation with a cc-pVDZ basis set was employed. All computations were done using the Q-Chem 5.1 quantum chemistry package.264 Results and discussion
Fig. 2 displays the calculated oscillator strengths at the N K-edge for neutral d(FUAG) and for [d(FUAG)+H]+, protonated at A-N1 and G-N7 (top row, middle row, and bottom row, respectively). The columns in Fig. 2 show the data for the N sites belonging to each nucleobase, separately. The insets show isosurfaces of the canonical orbitals that dominate the final state character of the strongest transitions (isovalue: 0.05). It has been shown previously, that X-ray absorption spectra of nucleobases are generally only weakly affected by DNA secondary and tertiary structure21 and even their direct chemical environment27 and this is confirmed by the fact that all final states are localized on single nucleobases and have π* character. | ||
| Fig. 2 Oscillator strengths for [d(FUAG)+H] at the N K-edge. Top row: neutral molecule; middle row: protonation at A-N1; bottom row: protonation at G-N7. Initial N 1s site on FU (left column), A (middle column) or G (right column). Insets show isosurfaces (isovalue = 0.05) of the canonical orbitals that dominate the final state of the strongest transitions. The two blue arrows indicate the transition from A-N1, the two red arrows indicate transitions from G-N7. L + n stands for LUMO + n. | ||
For the neutral molecule (Fig. 2, top row) in the case of FU, the two strong lowest-energy transitions around 402.5 eV are from the FU-N1 and FU-N3 amine sites that both feature strongly bound 1s levels. In the case of A and G, the spectra in Fig. 2 are more complicated, as both nucleobases contain amine and imine nitrogens, with their different N-1s binding energies. For the neutral molecule and the case of A, transitions from the imine nitrogens A-N1,N3,N7 to the unoccupied π*-orbitals LUMO+1 and LUMO+3 all have transition energies around 400.2 eV. The transition from amine nitrogens A-N6,N9 to the LUMO+1 orbital are observed at 401.7 eV and 402.7 eV. A similar situation is found for G, where transitions from imine G-N7,N3 to the LUMO+5 orbital are found at 400.2 eV and 401.1 eV, whereas the transitions from amine G-N1,N2,N9 to the LUMO+4 orbital are found at 402.3 eV, 402.7 eV and 402.9 eV.
A-N1 protonation (Fig. 2, middle row) significantly alters the molecular electronic structure. The additional positive charge leads to a general increase in binding energies in both, core and valence electrons. The overall appearance of the absorption spectra for FU and for G remains unchanged, even though protonation alters both, molecular geometry and energetic ordering of the final states. For the protonated nucleobase A itself, the situation is different. The LUMO is now localized on A. Protonation of the A-N1 site increases the binding energy of the respective 1s orbital from the lowest of all N 1s orbitals in the molecules to the highest. Accordingly, the respective transition changes from 400.1 eV (neutral) to 402.6 eV (protonated, see blue arrows in Fig. 2). The situation is very similar for G-N7 protonation (Fig. 2, bottom row). Despite a significant alteration in geometry and electronic structure, the overall appearance of the absorption spectra for FU and for A remains unchanged. However, the LUMO is now localized on G and protonation of the G-N7 site increases the (initially very low) binding energy of the corresponding 1s orbital to the highest N-1s binding energy in the molecule. As a consequence, the only low-energy transition on the G moiety shifts from 400.2 eV (neutral) up to 402.3 eV (protonated, see red arrows in Fig. 2). The final state of this transition is the LUMO.
How does this compare to the experimental data? Gas-phase protonated oligonucleotides are generally less stable with respect to soft X-ray absorption than their deprotonated counterparts. Gonzalez-Magaña et al. showed that for soft X-ray photoabsorption at the C, N and O K-edges, the doubly protonated 4-mer [d(GCAT)+2H]2+ is subject to extensive fragmentation, with glycosidic bond-cleavage (see Fig. 1) being a dominating process.28 The bottom panel in Fig. 1 shows a high-resolution photofragmentation mass spectrum for [d(FUAG)+H]+ at the N K-edge (Ephoton = 407 eV). The spectrum is again dominated by fragments due to glycosidic bond cleavage, in particular the nucleobase radical cations A+ and G+ and their protonated counterparts [A+H]+ and [G+H]+. The corresponding FU+ and [FU+H]+ ions are observed with much lower relative yields (the low-mass cutoff of the ion trap was around m/q= 110 amu).
Fig. 3 shows the sum of all 6 nucleobase ion ([G+H]+, [A+H]+, [FU+H]+, G+, A+ FU+) yields as a function of photon energy. For a direct comparison, the corresponding theoretical data from Fig. 2 both A-N1 and G-N7 protonated [d(FUAG)+H]+ are shown. For the theoretical spectra, the N atoms from all three nucleobases were combined and the calculated excitation energies and oscillator strengths were convoluted with Gaussian functions of 0.5 eV full width half maximum (FWHM), to account for the experimentally observed peak broadening. The energy scale for the theoretical data has been shifted by −0.65 eV with respect to the experimental data and the experimental data has been scaled to fit the theoretical data. For the π1 band, excellent agreement between experiment and theory is observed. For the π2 band, the agreement is markedly better for the A-N1 protonated system.
 | ||
| Fig. 3 Comparison of integral N K-edge X-ray absorption spectrum for [d(FUAG)+H]+ fragments [G+H]+, [A+H]+, [FU+H]+, G+, A+ FU+ (grey symbols) and theoretical data from Fig. 2, convoluted with the experimentally observed resolution (0.5 eV) A-N1 (blue line) and G-N7 (red line) protonation. Note, that the axis of the theoretical data is shifted with respect to the experimental data by −0.65 eV. The non-resonant background has been subtracted from the experimental data and the spectrum has been scaled to the intensity of the theoretical spectrum. | ||
For a more detailed comparison, it is useful to utilize the final state information and plot the experimental data for each nucleobase fragment separately. The top panel in Fig. 4 displays the recorded soft X-ray spectra for the 3 hydrogenated nucleobase-related fragments [G+H]+, [A+H]+ and [FU+H]+. Note, that formation of these fragments requires glycosidic bond cleavage and hydrogen transfer. In addition, one (singly protonated nucleobase loss) or two (unprotonated nucleobase loss) H atoms need to have migrated towards the nucleobase.28 The panel below shows the results for the systematically less intense regular nucleobase fragments G+, A+ FU+. All 6 spectra are qualitatively very similar but differ in absolute and relative intensity. The spectra are dominated by the 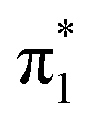 band centered at 399.5 eV and the
band centered at 399.5 eV and the  band around 401.7 eV. Above 403 eV, a broad continuum is observed for A and G that extends to higher photon energies. Whereas for
band around 401.7 eV. Above 403 eV, a broad continuum is observed for A and G that extends to higher photon energies. Whereas for  peak is found at identical energy for all fragments, the
peak is found at identical energy for all fragments, the  peak appears shifted by about 0.5 eV to lower energies for G+ and [G+H]+.
peak appears shifted by about 0.5 eV to lower energies for G+ and [G+H]+.
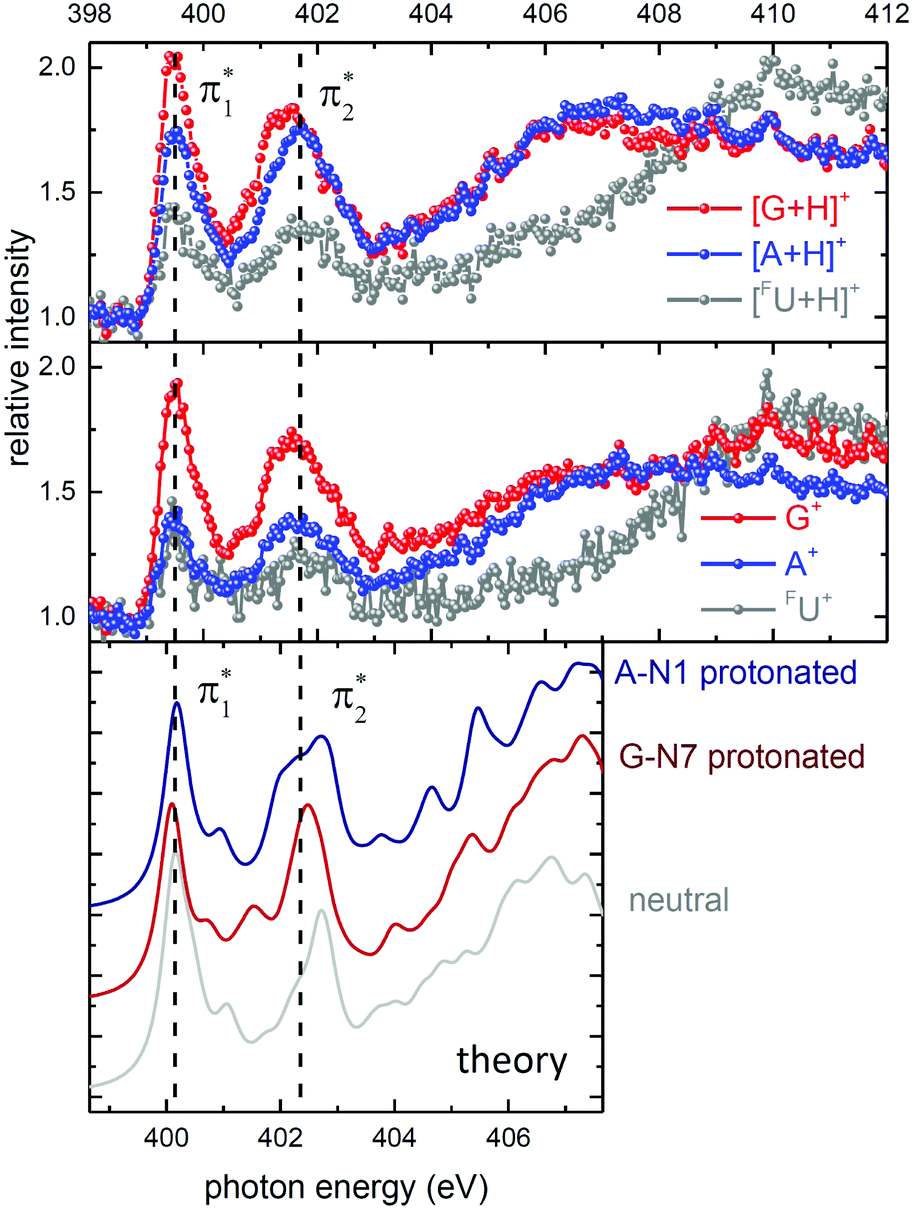 | ||
| Fig. 4 Top panel: Experimental N K-edge X-ray absorption spectra for [d(FUAG)+H]+ for the fragments [G+H]+, [A+H]+ and [FU+H]+ (top panel) and for G+, A+ FU+ (middle panel). The intensities are relative to the non-resonant intensity just below the N K-edge. Bottom panel: Theoretical data from Fig. 2, convoluted with the experimentally observed resolution (0.5 eV) for the neutral oligo as well as for A-N1 and G-N7 protonation. Note, that the axis of the theoretical data is shifted with respect to the experimental data by −0.65 eV. | ||
For comparison between experiment and theory, the bottom panel of Fig. 4 displays theoretical X-ray absorption spectra, obtained by plotting the FU, the A and the G data from Fig. 2 for the neutral oligo, the A-N1 protonated oligo and the G-N7 protonated oligo, respectively (the neutral data in only shown for comparison). The calculated excitation energies and oscillator strengths have again been convoluted with a 0.5 eV Gaussian and the energy scale of the theoretical data is shifted by −0.65 eV.
From the theoretical data in Fig. 4 it is very clear that both energy and relative intensity of the ( ) band are very similar for both protonation states and even for the neutral oligo, whereas the
) band are very similar for both protonation states and even for the neutral oligo, whereas the 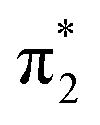 band is visibly influenced by protonation and protonation site. For A-N1 protonation, the energetic position and the width of the
band is visibly influenced by protonation and protonation site. For A-N1 protonation, the energetic position and the width of the  band resemble the experimental data rather closely, whereas for G-N7 protonation, the
band resemble the experimental data rather closely, whereas for G-N7 protonation, the  splitting is slightly too large and the
splitting is slightly too large and the  is significantly too narrow. This is in line with the fact that our calculations find the A-N1 protonated structure energetically favored by about 0.03 eV, in contrast to the energetic ordering of the proton affinities of the isolated nucleobases, which suggest G-N7 being favored (see Fig. 1).
is significantly too narrow. This is in line with the fact that our calculations find the A-N1 protonated structure energetically favored by about 0.03 eV, in contrast to the energetic ordering of the proton affinities of the isolated nucleobases, which suggest G-N7 being favored (see Fig. 1).
A deeper look into connection between photoabsorption site and fragmentation channel can be obtained by zooming into the π1 and π2 bands of the experimental data (Fig. 5) and by inspecting the actual transitions that contribute to the  bands. Fig. 2 shows that the
bands. Fig. 2 shows that the  band only has transitions from A-sites (G-N7 protonation) or from A and G-sites (A-N1 protonation). In the latter case, the A-contribution still is about twice the G-contribution, so theory shows that in any case
band only has transitions from A-sites (G-N7 protonation) or from A and G-sites (A-N1 protonation). In the latter case, the A-contribution still is about twice the G-contribution, so theory shows that in any case  -band excitations predominantly occur on the A moiety. However, experimentally the relative intensity of the
-band excitations predominantly occur on the A moiety. However, experimentally the relative intensity of the 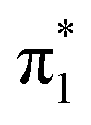 -band is highest for G+ and [G+H]+ fragments. (The yields for FU related fragments are generally smaller due to the high ionization potential of this nucleobase, but even though in the
-band is highest for G+ and [G+H]+ fragments. (The yields for FU related fragments are generally smaller due to the high ionization potential of this nucleobase, but even though in the  energy range, no N-1s absorption is possible on the FU moiety, both FU+ and [[FU+H]+] have comparable intensities for
energy range, no N-1s absorption is possible on the FU moiety, both FU+ and [[FU+H]+] have comparable intensities for  and
and  (see Fig. 4).) Fig. 5 also displays the difference [G+H]+–[A+H]+ (top) and G+−A+ (bottom). For the π1 band, the difference spectra both peak at 399.5 eV, as do the [G+H]+, [A+H]+, G+ and A+ peaks, i.e. the peaks merely differ in intensity. The same is true for the π2 band and the G+ and A+ peaks. It is however clear that in the [G+H]+ spectrum, the π2 band is markedly shifted to lower photon energies. A look at the difference spectrum shows that this is due to transitions at 401.1 eV, 0.6 eV below the 401.7 eV center of the π2 band.
(see Fig. 4).) Fig. 5 also displays the difference [G+H]+–[A+H]+ (top) and G+−A+ (bottom). For the π1 band, the difference spectra both peak at 399.5 eV, as do the [G+H]+, [A+H]+, G+ and A+ peaks, i.e. the peaks merely differ in intensity. The same is true for the π2 band and the G+ and A+ peaks. It is however clear that in the [G+H]+ spectrum, the π2 band is markedly shifted to lower photon energies. A look at the difference spectrum shows that this is due to transitions at 401.1 eV, 0.6 eV below the 401.7 eV center of the π2 band.
 | ||
| Fig. 5 Zoom into the region of the π1 and π2 bands from the two top panels in Fig. 4. Only the data [G+H]+ and [A+H]+ (top) and for G+ and A+ is shown. The black line shows the difference between the two spectra. | ||
Fig. 2 reveals that transitions on the A moiety contribute most in this energy range. Soft X-ray photofragmentation of protonated 5′-d(FUAG) cations thus shows a non-intuitive site-sensitivity: Yields of fragments directly related to the absorption site appear reduced. This could be explained in a model of competing fragmentation processes: glycosidic bond cleavage leading to the observed fragments necessarily is a relatively slow process, as it involves motion of a high-mass fragment as well as hydrogen transfer to the nucleobase. It is very likely that a large fraction of the photo-excitation energy has distributed over the entire molecule, before glycosidic bond cleavage is completed. However, in the photofragmentation spectra we also observe small peaks due to nucleobase fragmentation products. It is very likely that nucleobase fragmentation is a faster process that occurs before the energy has equilibrated. As a consequence, the yield of (intact) nucleobase fragments appears reduced for the X-ray absorbing nucleobase. This is in agreement with the thymidine XUV photoionization experiments of Mansson et al. who found lifetimes of about 200 fs for the glycosidic bond cleavage channel, but substantially shorter lifetimes of several 10 fs for the formation of small fragments.29
Only for EX > 407 eV, the yields for FU+ and [FU+H]+ are increasing to a similar relative intensity that is observed for A and G. In this energy range, direct N 1s ionization becomes the dominating channel, leading to removal of an additional electron. Coulomb repulsion of the charges increases the probability of finding the 3′-terminal FU positively charged.
5 Conclusion
In conclusion, we have shown that the combination of TDDFT calcuations using a short-range exchange correlation functional and high-resolution soft X-ray spectroscopy allows for a detailed understanding of the soft X-ray spectral features. Careful choice of the photon energy allows for well-localized photoexcitation, that appear to induce an increase of fast fragmentation in the absorbing nucleobase. Protonation leads to dramatic changes for the transisitions from the respective N atoms but the influence on the entire soft X-ray absorption spectrum is only moderate. As an outlook, the single F atom on the high IP terminal of the system will allow for localized F 1s excitation that can serve as a basis for soft X-ray pump–probe studies on charge migration and energy transport in oligonucleotides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank HZB for the allocation of synchrotron radiation beamtime. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. W. L. and X. W. acknowledge support by the Chinese Scholarship Council (CSC).The authors would like to acknowledge the contribution of the EU COST action MD-GAS (CA18212). The endstation at UE52-PGM is partially supported by BMBF grant 05K16VF1.Notes and references
- J. C. Gonzalez-Olvera, M. Durec, R. Marek, R. Fiala, M. d. R. J. Morales-Garcia, E. Gonzalez-Jasso and R. C. Pless, ChemBioChem, 2018, 19, 2088–2098 CrossRef CAS PubMed.
- J. Mergny, J. Li, L. Lacroix, S. Amrane and J. Chaires, Nucleic Acids Res., 2005, 33, e138 CrossRef PubMed.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- A. C. Leney and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef CAS PubMed.
- P. Wang, M. Bartlett and L. Martin, Rapid Commun. Mass Spectrom., 1997, 11, 846–856 CrossRef CAS.
- A. Anwar, J. Psutka, S. W. C. Walker, T. Dieckmann, J. S. Janizewski, J. L. Campbell and W. S. Hopkins, Int. J. Mass Spectrom., 2018, 429, 174–181 CrossRef CAS.
- R. R. Wu, B. Yang, G. Berden, J. Oomens and M. T. Rodgers, J. Phys. Chem. B, 2015, 119, 2795–2805 CrossRef CAS PubMed.
- A. R. Milosavljevic, C. Nicolas, M. L. J. Rankovic, F. Canon, C. Miron and A. Giuliani, J. Phys. Chem. Lett., 2015, 6, 3132–3138 CrossRef CAS.
- S. Bari, D. Egorov, T. L. C. Jansen, R. Boll, R. Hoekstra, S. Techert, V. Zamudio-Bayer, C. Buelow, R. Lindblad, G. Leistner, A. Lawicki, K. Hirsch, P. S. Miedema, B. von Issendorff, J. T. Lau and T. Schlathölter, Chem. – Eur. J., 2018, 24, 7631–7636 CrossRef CAS PubMed.
- C. Miron, M. Simon, N. Leclercq, D. Hansen and P. Morin, Phys. Rev. Lett., 1998, 81, 4104–4107 CrossRef CAS.
- P. Salen, M. Kaminska, R. J. Squibb, R. Richter, M. Alagia, S. Stranges, P. van der Meulen, J. H. D. Eland, R. Feifel and V. Zhaunerchyk, Phys. Chem. Chem. Phys., 2014, 16, 15231–15240 RSC.
- P. Bolognesi, J. A. Kettunen, A. Cartoni, R. Richter, S. Tosic, S. Maclot, P. Rousseau, R. Delaunay and L. Avaldi, Phys. Chem. Chem. Phys., 2015, 17, 24063–24069 RSC.
- L. Schwob, S. Dörner, K. Atak, K. Schubert, M. Timm, C. Bulow, V. Zamudio-Bayer, B. von Issendorff, J. T. Lau, S. Techert and S. Bari, J. Phys. Chem. Lett., 2020, 11, 1215–1221 CrossRef CAS PubMed.
- N. A. Besley, M. J. Peach and D. J. Tozer, Phys. Chem. Chem. Phys., 2009, 11, 10350–10358 RSC.
- N. A. Besley and F. A. Asmuruf, Phys. Chem. Chem. Phys., 2010, 12, 12024–12039 RSC.
- S. Wetmore, R. Boyd and L. Eriksson, Chem. Phys. Lett., 2001, 343, 151–158 CrossRef CAS.
- D. Roca-Sanjuan, M. Rubio, M. Merchan and L. Serrano-Andres, J. Chem. Phys., 2006, 125, 8 CrossRef PubMed.
- E. Hunter and S. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- O. Plekan, V. Feyer, R. Richter, M. Coreno, M. de Simone, K. C. Prince, A. B. Trofimov, E. V. Gromov, I. L. Zaytseva and J. Schirmer, Chem. Phys., 2008, 347, 360–375 CrossRef CAS.
- H. Shimada, T. Fukao, H. Minami, M. Ukai, K. Fujii, A. Yokoya, Y. Fukuda and Y. Saitoh, Chem. Phys. Lett., 2014, 591, 137–141 CrossRef CAS.
- W. Hua, H. Yamane, B. Gao, J. Jiang, S. Li, H. S. Kato, M. Kawai, T. Hatsui, Y. Luo, N. Kosugi and H. Agren, J. Phys. Chem. B, 2010, 114, 7016–7021 CrossRef CAS PubMed.
- K. Hirsch, J. T. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Moeller and B. von Issendorff, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154029 CrossRef.
- M. Niemeyer, K. Hirsch, V. Zamudio-Bayer, A. Langenberg, M. Vogel, M. Kossick, C. Ebrecht, K. Egashira, A. Terasaki, T. Moeller, B. V. Issendorff and J. T. Lau, Phys. Rev. Lett., 2012, 108, 057201 CrossRef CAS PubMed.
- A. Langenberg, K. Hirsch, A. Lawicki, V. Zamudio-Bayer, M. Niemeyer, P. Chmiela, B. Langbehn, A. Terasaki, B. V. Issendorff and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 184420 CrossRef.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kús, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. Distasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. Hanson-Heine, P. H. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. Oneill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y. C. Su, A. J. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- W. Hua, B. Gao, S. Li, H. Agren and Y. Luo, J. Phys. Chem. A, 2010, 114, 13214–13222 CrossRef CAS PubMed.
- O. Gonzalez-Magaña, M. Tiemens, G. Reitsma, L. Boschman, M. Door, S. Bari, P. O. Lahaie, J. R. Wagner, M. A. Huels, R. Hoekstra and T. Schlathölter, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 87, 032702 CrossRef.
- E. P. Mansson, S. De Camillis, M. C. Castrovilli, M. Galli, M. Nisoli, F. Calegari and J. B. Greenwood, Phys. Chem. Chem. Phys., 2017, 19, 19815–19821 RSC.
| This journal is © the Owner Societies 2021 |