Needles in a haystack: H-bonding in an optogenetic protein observed with isotope labeling and 2D-IR spectroscopy†
Received
5th March 2021
, Accepted 9th April 2021
First published on 19th April 2021
Abstract
Recently, re-purposing of cyanobacterial photoreceptors as optogentic actuators enabled light-regulated protein expression in different host systems. These new bi-stable optogenetic tools enable interesting new applications, but their light-driven working mechanism remains largely elusive on a molecular level. Here, we study the optogenetic cyanobacteriochrome Am1-c0023g2 with isotope labeling and two dimensional infrared (2D-IR) spectroscopy. Isotope labeling allows us to isolate two site-specific carbonyl marker modes from the overwhelming mid-IR signal of the peptide backbone vibrations. Unlike conventional difference-FTIR spectroscopy, 2D-IR is sensitive to homogeneous and inhomogeneous broadening mechanisms of these two vibrational probes in the different photostates of the protein. We analyse the 2D-IR line shapes in the context of available structural models and find that they reflect the hydrogen-bonding environment of these two marker groups.
I. Introduction
Cyanobacteriochromes (CBCRs) are photosensitive proteins that regulate diverse cellular responses to environmental light conditions in cyanobacteria.1–3 This class of proteins recently recieved increasing attention as building blocks for optogenetic tools because their modular domain structure allows to light-trigger cellular responses in different target organisms.4–7 One example is the light-regulated expression of target genes, which was recently achieved in yeast with a two-component optogenetic system8 based on the red/green CBCR Am1-c0023g2 (from here abbreviated AmI-g2) from the cyanobacterium Acarychloris marina.9 AmI-g2 is a member of the red/green lineage of CBCRs and can incorporate two different light-sensitive chromophores: phycocyanobilin (PCB) or biliverdin. Here, we investigate the PCB-assembled protein variant (Fig. 1) which can reversibly switch between a red- and green-light absorbing state (Pr and Pg, respectively).
 |
| | Fig. 1 Molecular properties of the red/green CBCR AmI-g2. (A) the protein adopts the typical GAF fold and covalently binds a PCB chromophore (cyan) to a conserved cysteine residue. Shown here is the crystal structure of the homologous GAF2 domain from the CBCR AnPixJ (PDB entry 3W2Z). (B) Structure of the PCB chromophore in the Pr state with the C15![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C16 double bond in Z configuration. (C) UV-vis absorption spectra of the Pr and Pg states. C16 double bond in Z configuration. (C) UV-vis absorption spectra of the Pr and Pg states. | |
All CBCRs share a common protein fold (called GAF fold) shown in Fig. 1A. They autocatalytically form a covalent thioether bond to the chromophore via addition of a conserved cysteine residue to tetrapyrrole ring A (Fig. 1B). Despite the fact that different CBCRs have been studied extensively with crystallographic,10–12 biochemical and spectroscopic methods, there is an ongoing debate about the role of structural heterogeneity. There are many evidences for the presence of several conformational sub-states with distinct properties in red/green CBCRs from nuclear magnetic resonance (NMR),13,14 resonance Raman15,16 or ultrafast optical spectroscopy.17–20 In these ultrafast spectroscopic studies, ground state structural heterogenieties were inferred from two observations in the past: (1) excitation wavelength dependence and (2) multiphasic behaviour of the excited state decay kinetics. While (2) can also be explained by solvation processes,21,22 the many other observations leave open questions: what exactly is heterogeneous in CBCRs and on which timescales does it equilibrate? Are the heterogenieties relevant for the function of CBCRs? There is some progress, especially from NMR and theoretical studies that ascribe these observations for example mixed conformations of tryptophan residues13–15 or the protonation of histidine residues.23 However NMR has intrinisically slow time resolution, and there are up to now no experimental studies that access the ultrafast dynamics of the electronic ground states. Here, we employ two dimensional infrared (2D-IR) spectroscopy in combination with isotope editing to isolate two vibrational modes that provide site-specific information about their direct environment within the protein in the Pr and Pg states. 2D-IR is a powerful tool to monitor the ultrafast dynamics of these modes and detect heterogenieties and their spectral diffusion with picosecond time-resolution.24 We observe various degrees of homogenous and inhomogenous broadening in the 2D-IR signals and relate them to differences in hydrogen-bonding.
II. Materials and methods
Protein expression and purification
The apo AmI-g2 expressing cells were generated by transforming E. coli BL21 with a pET-30a(+) vector (kanamycin resistance) containing the sequence for AmI-g2 apo in the open reading frame (GenScript Biotech, Piscataway Township NJ, USA). The translated apo AmI-g2 includes a C-terminal His6-tag which was used to purify the protein under native conditions via Ni-affinity chromatography. Cells were grown at 37 °C and 155 rpm in Luria broth (LB) medium containing 35 μg ml−1 kanamycin to an OD600 of 0.6–0.8. M9 minimal medium containing 13C-glucose and 15N-ammoniumchloride was used for the expression of globally 13C15N-labeled protein. All cultures were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After incubation overnight at room temperature and 155 rpm, cells were harvested by centrifugation and stored at −20 °C. A shorter incubation time of 4 h was used for the isotope edited protein variant. The apo-protein was purified under native conditions via Ni-affinity chromatography and desalted into a reducing buffer based on PBS that also contained 5 mM EDTA and 5 mM β-mercaptoethanol using a Sephadex HiPrep 26/10 column (GE Healthcare Bio-Sciences, Uppsala, Sweden). 25 mM PCB in DMSO was added in 1.5× molar excess to the apoprotein and the in vitro assembly of AmI-g2-PCB was monitored using UV-vis spectroscopy. After ca. 30 min, no spectral changes were observed and the reaction mixture was desalted again into the final PBS buffer, thereby removing unbound PCB, EDTA and β-mercaptoethanol. For all IR experiments, the samples were prepared in D2O buffer by lyophilization as described earlier25 and deposited in CaF2 sandwich cells with 25 μm path length.
2D-IR spectroscopy
2D-IR experiments were performed with a previously described setup.26 Briefly, the output of a Yb-doped fiber laser/amplifier system (short-pulse Tangerine, Amplitude, France) with a repetition rate of 100 kHz was converted to the mid-IR with an OPA (Twin STARZZ, Fastlite, France) and subsequent frequency mixing. The IR pulses were split into pump, probe and reference pulses, and the pump pulses were sent through a pulse shaper (PhaseTech Spectroscopy) to generate a pulse pair with programmable delay times and phases. Working in a rotating frame, the delays were scanned in 50 fs time steps (spectral resolution of pump axis is 8 cm−1) and 4 phase cycles were used to suppress scattering. The delay between the pump pulse pair an the probe pulse (t2) was controlled with a conventional delay stage. Pump and probe pulses were focused and overlapped in the sample (spot size 100 μm), while the reference was slightly offset (ca. 500 μm). Probe and reference beams were transmitted through a spectrograph and detected with a 2 × 32-MCT detector array (spectral resolution ca. 4 cm−1) with custom electronics.27 The samples were prepared either in the Pr state by illumination with a green LED array (LIU525B, Thorlabs, Newton, MA, USA) or kept in the Pg state by illumination a red laser diode (HL6750MG) during the measurements. This illumination procedure resulted in a photostationary equilibrium with less than 20% residual Pr contribution in the Pg samples (see ESI†).
III. Results
Labeling and FTIR
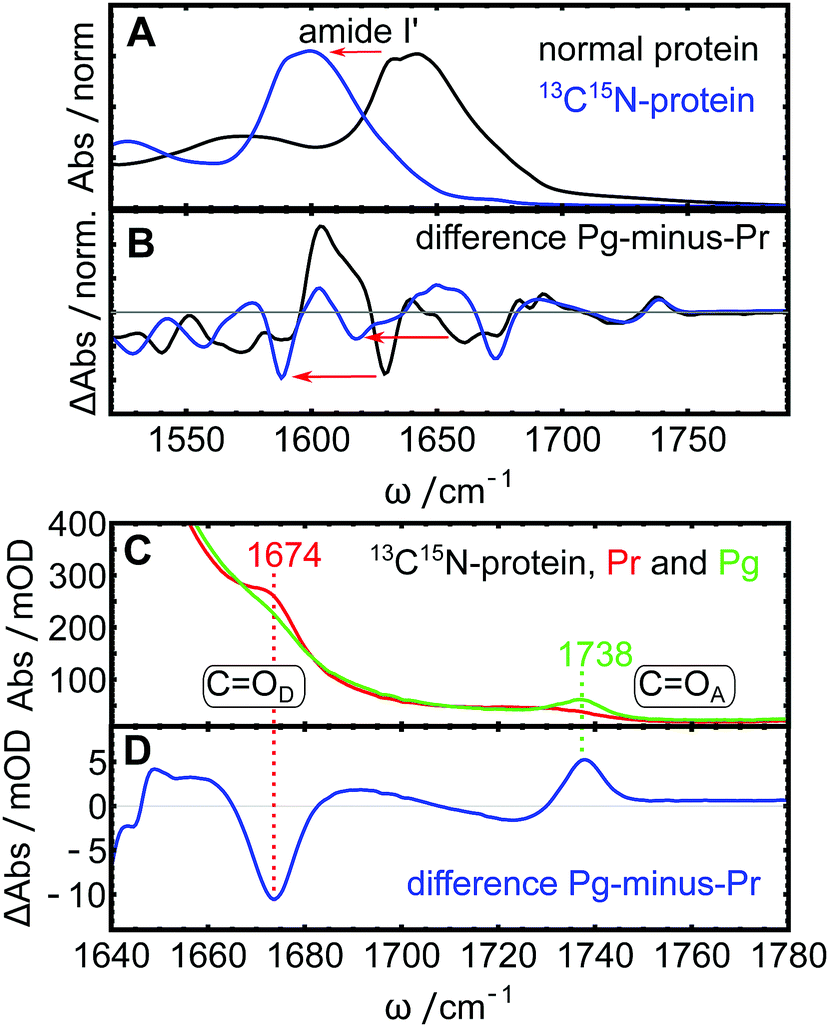
IR spectra of photoreceptor proteins in the region between 1600 and 1750 cm−1 contain overlapping contributions from different vibrational modes. Like in all other proteins, this region is dominated by the amide I vibrations from the protein backbone (termed I′ in deuterated samples), but also contains CO stretching vibrations from amino acid side chains28 and vibrational modes from the light-sensitive co-factor.16 These contributions can be distinguished from each other by isotope labeling parts of the system and comparing the IR spectra. Here, we 13C15N-labeled the entire AmI-g2 apo-protein and incooperated isotope-normal PCB. Fig. 2A shows Fourier transform IR (FTIR) absorption spectra of the labeled and normal samples with emphasis on the downshift of the amide I′ mode by ca. 50 cm−1 upon labeling (blue line). FTIR spectra of labeled and isotope normal AmI-g2 samples were recorded in the Pr and Pg states, and the corresponding “Pg-minus-Pr” difference spectra are shown in Fig. 2B. Some of the light-induced difference bands display the same shift pattern (downshifts by ca. 50 cm−1), and we can assign these to amide I′ difference bands that originate from structural changes of the protein backbone between the Pr and Pg states (indicated by red arrows). On the other hand, two difference-bands in the region between 1650 and 1760 cm−1 are completely insensitive to isotope labeling of the protein, and thus must originate from PCB. In the labeled sample, the collective tails of the downshifted amide I′ modes contribute to this spectral region only in the form of background absorption, and therefore both PCB bands are clearly visible in the absolute and the difference spectra (Fig. 2C and D). We assign these two bands to the carbonyl stretching vibrations of the pyrrole rings D (COD at 1674 cm−1) and A (COA at 1738 cm−1) based on the literature.29–31 Here, isotope labeling of individual carbon or oxygen atoms in PCB showed that the two carbonyl bands appear consistently at these position in several PCB-binding photoreceptors and model compounds. In PCB, pyrrole ring A is saturated, while all atoms in ring D are part of the conjugated system (see Fig. 1B), which accounts for major portion of the ca. 60 cm−1 frequency difference between the respective bands.29,30 Both bands show drastic changes in line shape depending on illumination. While COA is very broad and hardly distinguishable from the background in Pr, it shows up as a clear narrow peak at 1738 cm−1 in Pg. Interestingly, COD behaves opposite: it is quite narrow and well defined in Pr state at 1674 cm−1, and broadens in Pg. This observation also becomes very evident from the difference spectra (Fig. 2D), and reflects how the carbonyl groups sense their different environment in the Pr and Pg states. We now turn to 2D-IR spectroscopy to investigate these two bands in more depth.
 |
| | Fig. 2 FTIR spectra of isotope-normal and 13C15N-labeled AmI-g2. (A) Downshift of the amide I′ mode (red arrows) in the labeled protein (blue line) compared to the isotope normal protein (black line). (B) Pg-minus-Pr difference spectra of the two variants. (C) Detailed view of the CO stretching region of the 13C15N protein in the Pr state (red line) and Pg state (green line), frequencies of the two CO bands indicated. (D) Detailed view of the difference spectrum in the same region. | |
2D-IR spectroscopy
2D-IR spectra in the region between 1640 and 1780 cm−1 were recorded for AmI-g2 in the Pr and Pg states at different population times t2 from 200 fs up to 3 ps (Fig. 3 shows the isotope labeled sample, isotope normal in ESI†). 2D-IR intensities scale with the square of the absorption cross section, while the linear absorbance scales linearly with the cross section.24 Therefore, the amide I′ background appears further suppressed in the 2D-IR spectra despite its high linear absorbance (compare to Fig. 2C). In the isotope labeled sample, both target CO bands can now be clearly detected in both states, and we do not observe any cross-peaks. In 2D-IR spectra, each signal consists of a negative peak on the diagonal axis (blue, ground state bleach and stimulated emission) and a positive counterpart (red, excited state absorption) shifted along the probe axis ωprobe due to anharmonicity.24 All 2D signals decay with increasing t2 due to vibrational population relaxation, and spectra collected at short t2 delays (e.g. 200 fs) have a higher signal-to-noise ratio than at long t2 delays. In Fig. 3, this decay was normalized out in the right panels, and spectra taken at 2 ps therefore appear more noisy. The vibrational lifetimes of the two modes in both states are around 1 ps, which is a typical value for carbonlys that usually decay between 0.8 and 5 ps.32
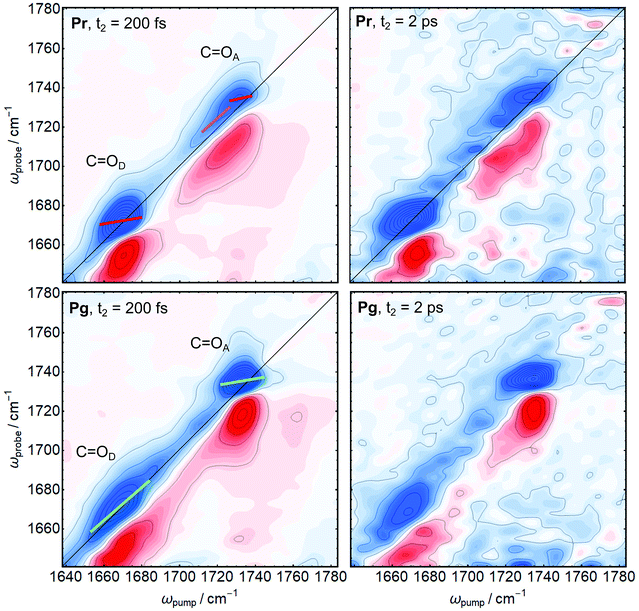 |
| | Fig. 3 2D-IR spectra of AmI-g2 in the Pr (top row) and Pg states (bottom row) at different population times. Spectra taken at t2 = 200 fs are shown on the left side, t2 = 2 ps on the right side. CLS of different bands are indicated as red, magenta and green lines. | |
The first important observable from 2D-IR spectroscopy is the 2D line shape, which allows to distinguish between homogeneous and inhomogeneous broadening mechanisms. A homogeneous band appears round, while an inhomogeneously broadened band that arises from many different sub-states with slightly different frequencies appears elongated along the diagonal.24 The two narrow bands that were identified in the FTIR spectra (COD in Pr COA in Pg) appear homogeneously broadened in the 2D-IR spectra, while the broad bands (COD in Pg and COA in Pr) are clearly inhomogeneously broadenend. The second important observable in 2D-IR is the t2-evolution of the center line slope (CLS), called spectral diffusion, which is a direct measure of equilibration on a ps time scale (CLS are indicated coloured bars in Fig. 3). While a perfectly homogeneous band has a horizontal center line (CLS = 0), heterogeneous broadening leads to tilted center lines as the peaks are elongated along the diagonal (45° or CLS = 1). A decrease of the CLS with increasing t2 indicates an equilibration of the sub-states on this time scale.24 We observe a kink in the CLS of COA in the Pr state, indicating that this band consists of at least two distinguishable contributions with different CLS (red and magenta). This behaviour is highlighted in the diagonal cuts in Fig. 4. For COA(Pr), we observe a main peak at the same frequency as Pg (1736 cm−1), and a shoulder at 1720 cm−1. We attribute this shoulder to an additional sub-state of COA in the Pr state. We therefore treat COA(Pr) as two different bands centered at 1720 (magenta) and 1736 cm−1 (red). In line with the previous analysis, the homogeneous bands (COD(Pr) and COA(Pg)) already have lower CLS (ca. 0.4) compared to the broad COD(Pg) (ca. 0.8) after 200 fs. The low CLS of the narrow peaks decreases further with t2 (Fig. 4, bottom panels), which indicates that a comparably small heterogeneous component arises from fast fluctuations on the ps time scale. In contrast, the CLS of the the broad COD(Pg) band does not decay in the investigated time-window, indicating that the heterogeneous broadening arises from sub-states which equilibrate only on longer time scales that are not accessible due to the fast vibrational relaxation of all bands. Here, meaningful values for the CLS can only be obtained up to 2.5 ps due to the increasing noise level. The 1720 cm−1 component of COA(Pr) has a CLS of 0.7, while the 1736 cm−1 component is more homogeneous with a CLS of 0.4 and both do not decay in the accessible time window.
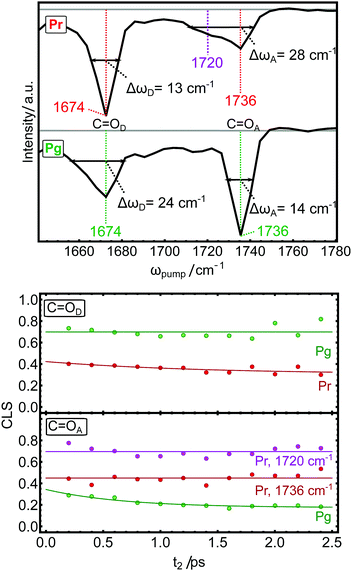 |
| | Fig. 4 Top panels: Diagonal cuts of the 2D-IR spectra in the Pr and Pg states. Bottom panels: The t2-evolution of the CLS of the carbonyl modes in the Pg state (green traces) and Pr state (red and magenta traces). | |
IV. Discussion
Spectral tuning of carbonyl frequencies
Three different factors are known to have major influence the frequency of CO vibrations: the chemical structure of the investigated compound, hydrogen bonding and electric fields.33 As already mentioned beforehand, the chemical structure (saturation of PCB) is the main reason why the two carbonyl stretches are detected at different frequencies. Furthermore, carbonyls are known to be very sensitive to hydrogen bond donation, where each oxygen can accept up to two H-bonds, and experience red shifts by ca. 15–25 cm−1 with each additional H-bond.33,34 In AmI-g2, we observe only neglible shifts of the peak positions for both investigated carbonyl groups upon photoconversion (Fig. 4), but predominately changes in line shapes. The line shapes are either homogeneous, narrow and symmetric around their maximum frequencies or show strong heterogeneous broadening, which we attribute to heterogeneous H-bonding environments sensed by the carbonyl groups.
Ring D carbonyl
There are three molecular structures available for red/green CBCRs in the Pr state that show strong similarities, namely AnPixJg2,11 NpR6012g413 and Slr1393g3.12 In all these structures, the ring D carbonyl is buried entirely inside the protein in H-bond distance with one conserved tyrosin residue. This conserved position is reflected in the 2D-IR spectra by the homogeneous band with a fast decaying, low CLS. While the different CBCRs of the red/green lineage have highly similar structures and spectral properties in the Pr state, their Pg states are much more diverse. Common to all, PCB is isomerised around the C15C16 bond, which leads to a rotation of ring D and consequently an entirely different protein environment compared to the Pr state. In the Pg structures of NpR6012g413 and Slr1393g3,12 COD moves into a solvent accessible position, which is occupied by a sodium ion in case of Slr1393g3. We observe a heterogeneous broadening of COD band with shoulders on both sides in Pg, while the center frequency does not shift. This is in line with a solvent accessible position in the Pg state, where the carbonyl can interact with zero to two interaction partners, i.e. water, ions, or other residues. On average, these interactions have the same effect on the carbonyl as the single H-bond to the Tyr residue in Pr, which accounts for the same central peak position. In the difference-FTIR spectra, this broadening in a heterogeneous environment is reflected by a sharp negative contribution at 1674 cm−1 on top of a broad positive background, which was also observed at similar frequencies in the difference FTIR spectra of AnpixJg2,35,36 NpR6012g437 and Slr1393g3.25
Ring A carbonyl
In contrast to the COD, the ring A carbonyl is H-bonded to a conserved tryptophan residue in the crystal structures of AnPixJg2 and Slr1393g3, but a heterogeneity is found in the NMR structure of NpR6012g4. Here, a two-state behaviour is observed, where the H-bond is sometimes broken and the tryptophan moves away from ring A. These two states were also observed in molecular dynamics simulations of AnPixJg2 independently by two different research groups, and the results of Rao et al. are shown in Fig. 5.14,23 Resonance Raman spectra of Slr1393g3 in the Pr state could be decomposed into the two sub-states, confirming the two-state nature for a third member of the red/green lineage and providing insight into the differences of PCB configuration and fluorescence properties of the two states.16 These two sub-states are observed in the 2D-IR spectra at 1720 (H-bond) and 1736 cm−1 (no H-bond). Interestingly, the H-bonded shoulder is heterogeneously broadened and maintains a high CLS of ca. 0.7 over the accessible time range, which reflects different H-bond strengths or distances between Trp and the ring A carbonyl. These states do not equilibrate on a ps timescale because they involve movements of the protein which expected to be much slower, but the transition can be observed on a ns time scale in the computer simulations. On the other hand, the higher frequency band is homogeneous with a constant CLS of 0.4, which agrees well with the assignment to the non H-bonded state. Interestingly, the indole group of the Trp residue forms a pi-stacking interaction with PCB ring D only in the Trp-on state, which apparently has negligible impact on the homogenous appearing COD. This result is in good agreement with a previous resonance Raman study of the related CBCR Slr-g3, where it was shown that the Trp sub-states are not sensed by other vibrational modes associated with ring D, but strongly by ring A.16 The published FTIR spectra of other red/green CBCRs differ quite substantially for COA, where the other proteins only show a weak bleach band in the FTIR difference spectra,25,35–37 and only AmI-g2 displays the sharp positive feature. In line with the assignment of the two COA bands in the Pr state, we conclude that in the Pg state of AmI-g2, the COA must be located in a hydrophobic environment that does not allow H-bonding. This transition apparently does not take place in other red/green CBCRs.
 |
| | Fig. 5 Tryptophan heterogeneity in the Pr state of the red/green CBCR AnPixJg2 observed by MD simulations.23 This figure was kindly provided by A. Rao and I. Schapiro. | |
V. Conclusions
While spectroscopic studies consistently showed that heterogeneity plays an important role in many CBCRs, the molecular basis for these observation is often unclear, mainly because most studies employ methods lacking spatial resolution (like transient vis spectroscopy). In this study, we gain site-specific information through isotope labeling and employ 2D-IR spectroscopy based on a novel 100 kHz Yb laser system, which provides a sufficient signal-to-noise ratio to detect only two localized carbonyl modes in a protein composed of 166 amino acids within a reasonable acquisition time. Remarkably, we find that the peak positions of both carbonyl marker modes do not shift upon photoconversions but instead change line shape. Shifts or broadenings cannot be distinguished by difference-FTIR spectroscopy, and we speculate that broadening might also be the main reason for CO difference signals in other bilin-photoreceptors, which are frequently interpreted in terms of frequency shifts. We observe that some modes are homogeneously broadened and quickly equilibrate to a large extent on a ps time scale, while others are inhomogenously broadened and do not equilibrate in the accessible time window. Analysis of our observations in the context of the available structural models for red/green CBCRs shows that spectral broadening correlates with heterogeneity in the hydrogen-bonding environment. Here, the COA vibration in the Pr state is an extreme case, where a two-state (H bond on/off) behaviour is reflected by two different carbonyl stretching frequencies with distinctive 2D line shapes.
Author contributions
DB and JR expressed and purified the protein. JR and DB performed spectroscopic experiments. DB, JR and PH analysed the data. DB wrote the MS with contributions of all authors.
Data availability statement
The data are openly available on Zenodo at 10.5281/zenodo.4650475.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors would like to thank A. Rao and I. Schapiro for providing their results and Fig. 5 prior to publication. We thank Jan Helbing and Roland Zehnder for technical support and Ricardo Fernández-Terán for help with the CLS analysis. This work was supported by the Swiss National Science Foundation (SNF) through the NCCR MUST and Grant 200020B_188694/1.
References
- K. Anders and L. O. Essen, Curr. Opin. Struct. Biol., 2015, 35, 7–16 CrossRef CAS PubMed.
- K. Fushimi and R. Narikawa, Curr. Opin. Struct. Biol., 2019, 57, 39–46 CrossRef CAS PubMed.
- N. C. Rockwell and J. C. Lagarias, Curr. Opin. Plant Biol., 2017, 37, 87–93 CrossRef CAS PubMed.
- M. Blain-Hartung, N. C. Rockwell, M. V. Moreno, S. S. Martin, F. Gan, D. A. Bryant and J. C. Lagarias, J. Biol. Chem., 2018, 293, 8473–8483 CrossRef CAS PubMed.
- N. T. Ong and J. J. Tabor, ChemBioChem, 2018, 19, 1255–1258 CrossRef CAS PubMed.
- Z. Liu, J. Zhang, J. Jin, Z. Geng, Q. Qi and Q. Liang, Front. Microbiol., 2018, 9, 1–11 CrossRef PubMed.
- S. M. Castillo-Hair, E. A. Baerman, M. Fujita, O. A. Igoshin and J. J. Tabor, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- J. Jang, S. McDonald, M. Uppalapati and A. Woolley, bioRxiv, Synth. Biol., 2019, 1–23 Search PubMed.
- K. Fushimi, T. Nakajima, Y. Aono, T. Yamamoto, Ni-Ni. Win, M. Ikeuchi, M. Sato and R. Narikawa, Front. Microbiol., 2016, 7, 1–12 Search PubMed.
- E. S. Burgie, J. M. Walker, G. N. Phillips and R. D. Vierstra, Structure, 2013, 21, 88–97 CrossRef CAS PubMed.
- R. Narikawa, T. Ishizuka, N. Muraki, T. Shiba, G. Kurisu and M. Ikeuchi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 918–923 CrossRef CAS PubMed.
- X. Xu, A. Port, C. Wiebeler, K. H. Zhao, I. Schapiro and W. Gärtner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2432–2440 CrossRef CAS PubMed.
- S. Lim, Q. Yu, S. M. Gottlieb, C.-W. Chang, N. C. Rockwell, S. S. Martin, D. Madsen, J. C. Lagarias, D. S. Larsen and J. B. Ames, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4387–4392 CrossRef CAS PubMed.
- L. K. Scarbath-Evers, S. Jähnigen, H. Elgabarty, C. Song, R. Narikawa, J. Matysik and D. Sebastiani, Phys. Chem. Chem. Phys., 2017, 19, 13882–13894 RSC.
- F. Velázquez Escobar, T. Utesch, R. Narikawa, M. Ikeuchi, M.-A. Mroginski, W. Gärtner and P. Hildebrandt, Biochemistry, 2013, 52, 4871–4880 CrossRef PubMed.
- D. Buhrke, G. Battocchio, S. Wilkening, M. Blain-Hartung, T. Baumann, F. J. Schmitt, T. Friedrich, M. A. Mroginski and P. Hildebrandt, Biochemistry, 2020, 59, 509–519 CrossRef CAS PubMed.
- P. W. Kim, L. H. Freer, N. C. Rockwell, S. S. Martin, J. C. Lagarias and D. S. Larsen, Biochemistry, 2012, 51, 608–618 CrossRef CAS PubMed.
- S. M. Gottlieb, P. W. Kim, C. W. Chang, S. J. Hanke, R. J. Hayer, N. C. Rockwell, S. S. Martin, J. C. Lagarias and D. S. Larsen, Biochemistry, 2015, 54, 1028–1042 CrossRef CAS PubMed.
- J. S. Kirpich, S. M. Gottlieb, C. W. Chang, P. W. Kim, S. S. Martin, J. C. Lagarias and D. S. Larsen, Biochemistry, 2019, 58, 2307–2317 CrossRef CAS PubMed.
- C. Slavov, X. Xu, K. H. Zhao, W. Gärtner and J. Wachtveitl, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1335–1344 CrossRef CAS PubMed.
- D. Wang, X. Li, S. Zhang, L. Wang, X. Yang and D. Zhong, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19731–19736 CrossRef CAS PubMed.
- D. Wang, X. Li, L. Wang, X. Yang and D. Zhong, J. Phys. Chem. Lett., 2020, 11, 8819–8824 CrossRef CAS PubMed.
- A. G. Rao, C. Wiebeler, S. Sen, D. S. Cerutti and I. Schapiro, Phys. Chem. Chem. Phys., 2021, 7–13 Search PubMed.
-
P. Hamm and M. T. Zanni, Concepts and Methods of 2D Infared Spectroscopy, Cambridge University Press, 2011 Search PubMed.
- D. Buhrke, K. T. Oppelt, P. J. Heckmeier, R. Fernández-Terán and P. Hamm, J. Chem. Phys., 2020, 153, 245101 CrossRef CAS PubMed.
- P. Hamm, J. Chem. Phys., 2021, 154, 1–6 CrossRef PubMed.
- K. M. Farrell, J. S. Ostrander, A. C. Jones, B. R. Yakami, S. S. Dicke, C. T. Middleton, P. Hamm and M. T. Zanni, Opt. Express, 2020, 28, 33584–33602 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–429 CrossRef CAS PubMed.
- F. Siebert, R. Grimm, W. Rüdiger, G. Schmidt and H. Scheer, Eur. J. Biochem., 1990, 194, 921–928 CrossRef CAS PubMed.
- H. Foerstendorf, C. Benda, W. Gärtner, M. Storf, H. Scheer and F. Siebert, Biochemistry, 2001, 40, 14952–14959 CrossRef CAS PubMed.
- J. J. van Thor, N. Fisher and P. R. Rich, J. Phys. Chem. B, 2005, 109, 20597–20604 CrossRef CAS PubMed.
- S. H. Schneider, H. T. Kratochvil, M. T. Zanni and S. G. Boxer, J. Phys. Chem. B, 2017, 121, 2331–2338 CrossRef CAS PubMed.
- V. A. Lorenz-Fonfria, Chem. Rev., 2020, 120, 3466–3576 CrossRef CAS PubMed.
- S. Woutersen, Y. Mu, G. Stock and P. Hamm, Chem. Phys., 2001, 266, 137–147 CrossRef CAS.
- Y. Fukushima, M. Iwaki, R. Narikawa, M. Ikeuchi, Y. Tomita and S. Itoh, Biochemistry, 2011, 50, 6328–6339 CrossRef CAS PubMed.
- C. Song, F. Velázquez Escobar, X. L. Xu, R. Narikawa, M. Ikeuchi, F. Siebert, W. Gärtner, J. Matysik and P. Hildebrandt, Biochemistry, 2015, 54, 5839–5848 CrossRef CAS PubMed.
- J. S. Kirpich, C. W. Chang, J. Franse, Q. Yu, F. V. Escobar, A. J. Jenkins, S. S. Martin, R. Narikawa, J. B. Ames, J. C. Lagarias and D. S. Larsen, Biochemistry, 2021, 60, 274–288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00996f |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Peter
Hamm
,
Peter
Hamm