
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-scan measurements of nuclear spin singlet order decay rates
Giulia
Melchiorre
a,
Ciara
Nelder
a,
Lynda J.
Brown
a,
Jean-Nicolas
Dumez
 b and
Giuseppe
Pileio
*a
b and
Giuseppe
Pileio
*a
aSchool of Chemistry, University of Southampton, SO17 1BJ, Southampton, UK. E-mail: g.pileio@soton.ac.uk
bCEISAM, CNRS, Université de Nantes, 44300 Nantes, France
First published on 26th March 2021
Abstract
Measurements of singlet spin order decay rates are time consuming due to the long-lived nature of this form of order and the typical pseudo-2D mode of acquisition. Additionally, this acquisition modality is not ideal for experiments run on hyperpolarized order because of the single-shot nature of hyperpolarization techniques. We present a methodology based on spatial encoding that not only significantly reduces the duration of these experiments but also confers compatibility using spin hyperpolarization techniques. The method condenses in a single shot the variable delay array used to measure decay rates in conventional pseudo-2D relaxation experiments. This results in a substantial time saving factor and, more importantly, makes the experiment compatible with hyperpolarization techniques since only a single hyperpolarized sample is required. Furthermore, the presented method, besides offering savings on time and costs, avoids reproducibility concerns associated with repetition in the hyperpolarization procedure. The method accelerates the measurement and characterization of singlet order decay times, and, when coupled with hyperpolarization techniques, can facilitate the quest for systems with very long decay times.
Introduction
NMR methodologies that exploit various aspects of long-lived nuclear spin singlet order – all groupable under the name singlet NMR – are under constant development.1–17 The three main properties of this form of spin order – NMR silent, accessible on demand and long-lived – have found a variety of applications including: quantification, with enhanced sensitivity, of ligand binding;18,19 measurements of very slow translational dynamics;20–25 and their exploitation as molecular tags that can preserve information over a long time.26–36 Long-lived spin order is exploited as a vehicle to preserve spin hyperpolarization gained through techniques such as PHIP,37 SABRE38 and dissolution–DNP.39 The quest for a biocompatible molecular marker that supports hyperpolarized long-lived spin order to be used for in vivo imaging applications is still open and has the potential to bring about important new possibilities in MRI.40At the core of all these developments is the measurement of the singlet order decay constant (TS) and its theoretical modelling.41 The intelligent design of molecules bearing singlet states has led to molecules displaying singlet order lifetimes of the order of hours.42,43 However, the procedure to interrogate whether a system displays such long lifetimes is quite tedious and costly in terms of money and time: a molecular system is devised on paper according to actual knowledge and numerical/analytical calculations; a synthetic route is designed (often involving multiple isotopic labeling) and the molecule synthesized before its relaxation properties are characterized. Understanding of the interplay of the different relaxation mechanisms in the molecule is then fed back into the molecule design and, often, the synthesis of a new derivative is required, which adds to time and costs. Such processes would be enormously facilitated (in terms of both time and costs) if one could measure the singlet lifetime on naturally abundant isotopic spin pairs by using hyperpolarization techniques to reveal the signal in such low-sensitivity species. However, although hyperpolarization is able to provide signals from singlet order in natural abundant systems, measuring their lifetime is not so straightforward.
The conventional method of measuring singlet decay constants is based on the following steps: (1) create longitudinal order either by waiting for the sample to return to equilibrium in a magnetic field or by hyperpolarizing the sample with one of the cited techniques (the duration of this step is indicated as τ0); (2) convert longitudinal order into singlet order8 (depending on the technique this is of the order of tens to hundreds of milliseconds, thus negligible in this context); (3) wait for an incremental variable time delay (τi) during which the singlet order gradually decays; (4) reconvert the remaining singlet order into transverse magnetization and acquire the signal (again of negligible duration in this context); (5) repeat all four previous steps nr-times (most typically 8 or 16) each time using a longer value for the variable delay τi. In the case of a low signal-to-noise ratio, steps 1 to 5 are also repeated ns times. The total duration of such an experiment is
 | (1) |
For example, to measure a TS estimated to be of the order of 60 min, one would wait for, at least, τ0 ≅ 3TS = 180 min and use a variable delay array going from 0 to 180 min in 7 spaced points (to make it simple), i.e. nr = 7 and τi = [0, 30, 60, 90, 120, 150, 180] min; under these conditions the total experimental time would result in  per scan (ns = 1). This time can be reduced to ∼11 h if the singlet order is destroyed6 at each of the nr repetitions such that τ0 could be kept of the order of 3T1 (T1 is assumed 30 s in this example) rather than 3TS. Experiments to measure TS in low-gamma singlet spin pairs can therefore take several days because of the combined need for taking many transients and the many minutes long typical TS values.
per scan (ns = 1). This time can be reduced to ∼11 h if the singlet order is destroyed6 at each of the nr repetitions such that τ0 could be kept of the order of 3T1 (T1 is assumed 30 s in this example) rather than 3TS. Experiments to measure TS in low-gamma singlet spin pairs can therefore take several days because of the combined need for taking many transients and the many minutes long typical TS values.
Sensitivity can be greatly enhanced using hyperpolarized spin order. However, even in this case the measurements of singlet decay rates are complicated by the need to freshly prepare hyperpolarized order at each of the nr repetitions and this, depending on the hyperpolarization technique, is a costly and time-consuming procedure, often presenting issues of reproducibility. In dissolution–DNP, for example, the preparation of hyperpolarized order typically ranges between 10 min and 1 h, therefore at each nr repetition τ0 can be 10–60 min long. Methodology to circumvent these issues in hyperpolarized experiments has been published. The most basic method emulates the use of small-flip-angle pulses used in measuring T1 on hyperpolarized samples. For instance, a reduced efficiency singlet-to-magnetization step (such as, for example, spin-lock induced crossing (SLIC) or chemical shift scaling (CSS)) can be used to convert only a small amount of the hyperpolarized singlet pool into observable magnetization as demonstrated in long-lived hyperpolarization obtained with PHIP.44 However, because the spin order lost during each conversion step is not known, multiple experiments with variable repetition times are needed to access the value of TS. Also, only a fraction of the initial batch of hyperpolarized signal is used at each observation. In order to take advantage of the full hyperpolarized signal, a recycling scheme45 has been proposed. This scheme runs as follows: a batch of hyperpolarized longitudinal order is repeatedly converted into long-lived order, stored for a variable time, reconverted into transverse magnetization, read and, immediately after, re-stored in the form of long-lived order to be used later for the next point in the variable delay array. Both these methods solve costs and reproducibility issues at once and reduce the total experimental time by τ0(nr − 1) per scan since a single batch of hyperpolarized order is used. The disadvantage of the recycling procedure is that experimental losses are observed at each singlet-to-magnetization-to-singlet conversion and this appears as an extra contribution to the decay time that must be characterized and accounted for.
Spatial parallelization is a powerful approach to accelerate NMR experiments that require a stepwise incrementation of a delay. It has been applied, for example, to the evolution delay of 2D NMR experiments,46 the recovery delay in relaxation experiments,47 and the duration of the diffusion encoding gradient in diffusion experiments.48 Spatial parallelization is especially useful in combination with single-shot hyperpolarization methods and has notably been combined with dissolution dynamic nuclear polarisation.49–53
In this paper, we introduce a methodology that uses spatially selective long-lived spin order preparation techniques23 to reduce the experimental time required to measure TS to a duration equivalent to the longest point in the variable delay array:  . This procedure is valuable in further cutting down on experimental time (and associated costs) and, as the other existing methods, it would only require a single hyperpolarized sample thus reducing preparation time, costs and irreproducibility issues.
. This procedure is valuable in further cutting down on experimental time (and associated costs) and, as the other existing methods, it would only require a single hyperpolarized sample thus reducing preparation time, costs and irreproducibility issues.
Experimental
Proposed methodology
The methodology presented in this paper is based on the use of a space-selective magnetization to singlet conversion schemes (sM2S/sS2M)23 and can be implemented in two different ways which mix together either a single non-selective M2S and a train of sS2M (v1) or a train of sM2S and a single non-selective S2M (v2). Both variants achieve the same goal of shortening the measurement time, but only the first variant is compatible with hyperpolarized NMR. This is because in v2 the hyperpolarized spin order is stored as “short-lived”, longitudinal magnetization during the sM2S train. In v1 a non-selective M2S is run to convert thermal or hyperpolarized longitudinal order into singlet order across the whole sample. Then, a train of sS2M blocks followed by a relaxation delay and acquisition is run, repeatedly, to convert singlet order back to transverse order only in selected portions (slices) of the sample. At each run, the sS2M is instructed to address a different portion of the sample so that the singlet order from different portions is converted, and the associated signal acquired, at different times. Although the acquisition of a conventional FID is possible, we have here preferred to acquire the signal through a 1D spin echo imaging sequence so that the thickness and position of the slices addressed is clearer and under control. Using this methodology, singlet order is prepared in all slices simultaneously; however, the time elapsed from preparation to detection varies for the different slices, so the whole array of the variable delay time can be encoded in a single run. Singlet order is insensitive to gradient pulses, therefore, once this form of order is prepared in a given slice all successive gradients used to address a different slice have no deleterious effects. We have dubbed this general methodology as single-scan-TS (SSTS). The pulse sequence for its first implementation (SSTS-v1) is illustrated in Fig. 1A; its different steps can be summarized as follows: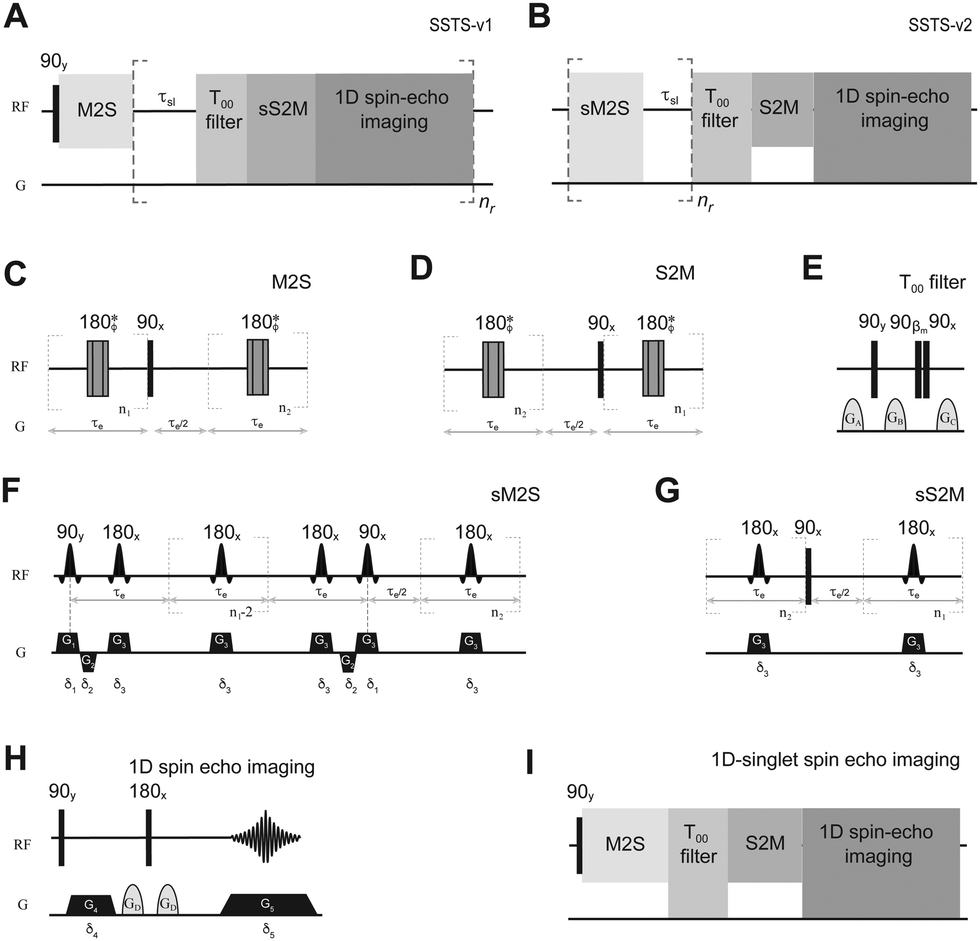 | ||
Fig. 1 (A and B) SSTS pulse sequences proposed in this work with details of the M2S (C), S2M (D). T00 filter (E), sM2S (F) sS2M (G) and 1D spin echo imaging (H). The sequence for singlet-order-filtered 1D spin echo imaging is presented in (I). n1 = πJ/(2Δν) and n2 = n1/2.  . βm indicates the magic angle and * indicates a composite 180° pulse built as 90x180y90x and with overall phase ϕ cycled within each echo train as [x, x, . βm indicates the magic angle and * indicates a composite 180° pulse built as 90x180y90x and with overall phase ϕ cycled within each echo train as [x, x, ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) , , , x, x, , , , x, x, x, , , x]. , , , x, x, , , , x, x, x, , , x]. | ||
(1) A M2S54,55 block (Fig. 1C) creates long-lived singlet order at the same time across the whole sample.
(2) The singlet order is allowed to decay for a time τsl (kept constant or varied at each repetition).
(3) A T00 filter56 (Fig. 1E) is applied to filter out possible M2S by-products different from singlet order.
(4) A sS2M block (Fig. 1G) converts singlet order back to transverse magnetization only in a selected region of space (an axial slice perpendicular to the tube long axis in the examples below). The orientation, position and thickness of this slice depend on the axis along which the gradients G3 is applied, its intensity and the offset and bandwidth of the shaped 180 degrees pulses (details below).
(5) A 1D spin echo imaging sequence (Fig. 1H) is run to yield the sample profile along the direction selected by the gradients G4 and G5.
(6) Steps 2–5 are repeated nr times but the offset of the selective pulses in the sS2M block is changed so to address a different slice at each repetition.
Each spin-echo imaging acquisition is stored on a separate line of a Bruker 2D ser file. The file is then Fourier-transformed and processed in magnitude mode. The intensity of the images acquired at each step decays according to the singlet-order specific relaxation decay constant. For convenience in making comparisons, here we only report the positive projection rather than the full 2D spectrum. We point out that the spin-echo imaging acquisition can be replaced by a conventional NMR FID acquisition thus retaining spectral resolution and providing higher sensitivity, if necessary. However, since spectral resolution is unimportant in measuring TS and since molecules displaying very long-lived spin states are nearly equivalent spin-pairs (i.e. they generate a single peak on the spectrum) we have here preferred to use a spin-echo imaging modality that gives a clearer indication of the spatial region where the signal is coming from.
In its second implementation (SSTS-v2, Fig. 1B), a train of sM2S blocks followed by a relaxation delay is run, repeatedly, to convert longitudinal into singlet order only in selected portions of the sample. At each run, the sM2S is instructed to address a different portion of the sample so that different portions are addressed at distinct times. A successive non-selective S2M converts singlet order into transverse magnetization in all slices simultaneously which is then conveniently detected through a 1D spin echo imaging sequence. The singlet prepared in each slice starts decaying immediately after preparation but the time elapsed from preparation to detection is different for the different slices so the whole array of the variable delay time can be encoded in a single spectrum. The different steps involved in SSTS-v2 can be summarized as follows:
(1) A sM2S block (Fig. 1F) creates long-lived singlet order only in a selected region of space (an axial slice perpendicular to the tube long axis). The orientation, position and thickness of this slice depend on the axis along which the gradients G1, G2 and G3 are applied, their intensity and the offset and bandwidth of the shaped pulses (details below).
(2) During the time interval τsl the singlet order created in the current and all previously addressed slices starts decaying.
(3) The sM2S is repeated nr times but, at each repetition, the offset of the selective pulses is changed so as to address a different slice. After nr repetitions, singlet order will be created in all slices but at different times to each other.
(4) A T00 filter56 (Fig. 1E) is applied to filter out possible sM2S by-products distinct from the singlet order.
(5) A non-selective S2M54,55 (Fig. 1D) is run to convert the singlet order into transverse magnetization, simultaneously in all slices.
(6) A 1D spin echo imaging sequence is run to yield the sample profile along the z-axis.
The acquired echo signal is Fourier-transformed and processed in magnitude mode. The resulting spectrum contains nr peaks, each one directly reporting on the intensity of the singlet order evolved in one of the nr slices addressed in steps 1–3.
The gradients used for spatial selection (G1–G3) have trapezoidal shape and are applied along the z-axis (coinciding with the direction of the B0 field and the NMR tube long axis). All other gradients (GA–GD) have half-sine shape and are applied along directions perpendicular to z. The pulse shapes are chosen to provide a good selection profile for 180 degrees pulses in the pulse sequence, their duration is adjusted to reach the desired bandwidth (BW). The strength of the G1 gradient (g1) is adjusted so as to select the desired slice thickness TH according to
 | (2) |
 | (3) |
 | (4) |
1D spin-echo images along tube main axis
A 1D spin-echo image along the z-axis is acquired on sample S1 (see Materials and methods) in order to reveal the sample profile along the laboratory frame z-axis (aligned with the main field). This is achieved by using the pulse sequence in Fig. 1H with G4 and G5 applied along z and with g4 = g5 = 75 mT m−1 (5% of maximum). The resulting spectrum is shown in magnitude mode in Fig. 2 (grey line). The FWHM is ∼54 kHz corresponding to a sample extension along the z-axis of ∼16.9 mm (eqn (2)), slightly smaller than the coil dimension of 18 mm. The small discrepancy of dimension implies that B1 inhomogeneities at the edge of the coil may slightly compromise the image quality at the edges. There is, however, a roughly 32 kHz wide region where the spectrum intensity is high and flat, corresponding to a region of ∼10 mm where the coil performs at its best. | ||
| Fig. 2 1D image of sample S1 along the z-axis obtained using the pulse sequence in Fig. 1H (grey line); the pulse sequence in Fig. 1I where only the signal filtered through singlet order is acquired (red line); the pulse sequence in Fig. 1A with nr = 7 and τsl = 1 s (blue line – positive projection across all rows); the pulse sequence in Fig. 1A with nr = 7 and τsl = 60 s (black line – positive projection across all rows). A single transient was used to acquire each image and all other parameters are discussed in Materials and methods. | ||
The red line in Fig. 2 refers to the z-profile image of sample S1 acquired with the sequence in Fig. 1H where the 1D spin-echo imaging is run on the amount of magnetization which has been firstly converted into singlet order, filtered through the T00 filter and then converted back to transverse order. Theoretically, this filtering should reduce the signal by 1/3 but a more pronounced signal reduction of ∼2/3 is experimentally observed. However, the comparison between the two profiles is not straightforward since the solvent peaks also contribute to the image taken with the pulse sequence in Fig. 1H while their signal is suppressed by the T00 filter used in the sequence in Fig. 1I. The 1D spin-echo image of sample S1 produced by the SSTS-v1 pulse sequence of Fig. 1A (blue line in Fig. 2) run for nr = 7 and τsl = 1 s contains, in its positive projection across all rows, seven well-separated peaks, one for each of the nr slices selected. The intensity of these peaks is only slightly smaller than the singlet-filtered 1D sample profile meaning that there are no significative losses in running spatial-selective (sS2M) rather than non-selective S2M.
The 1D spin-echo image of sample S1 are still produced by the SSTS-v1 pulse sequence but nr = 7 and τsl = 60 s (black line in Fig. 2) also shows seven well-separated peaks whose intensity decays from right to left. In this experiment, the signal from the spins located in the slice corresponding to the rightmost peak has been acquired 60 s after the M2S was run; the second peak from the right has been acquired 120 s after the M2S was run, and so on for all other peaks. The areas underneath each peak can be plotted against time and fitted to retrieve the singlet relaxation constant. Unfortunately, this fitting gives a value of TS = 133 ± 15 s which is significantly shorter than the value of TS = 201 ± 1 s measured with a conventional pseudo-2D experiment. This is somewhat expected and is attributable to a combination of molecular diffusion and thermal convection currents. This issue is discussed and resolved below.
The effects of diffusion and thermal convection
Right from conception, we were well aware that thermal convection and Brownian diffusion would compromise the proposed methodology. The molecule used in this work (see Materials and methods), has an isotropic diffusion coefficient of 6.6 × 10−10 m2 s−1 as measured by a convection-compensated double-stimulated echo experiment. This means that it can travel an average distance of 0.25 mm per minute along one direction. On top of that, and much more effective, convection currents generated by temperature gradients in the sample can transport molecules across many millimeters per minute. The molecules which are in a specific location at the time the M2S is run, would be in a different position at the time the sS2M is run to retrieve the signal from that specific location; this would result in an apparent loss of signal which is attributable to the physical movement of the molecule rather than to a mere nuclear spin relaxation effect. This phenomenon can be directly tracked by using the SSTS-v2 pulse sequence which can visually report on molecular motions. In this version of the experiment, in fact, the molecules in each specific location are tagged as singlet order during the sM2S initial step. During the time needed for tagging all different slices, the singlet-tagged molecules move around the sample due to the motions discussed above. The final S2M followed by spin-echo imaging acquisition reveals the positions of these molecules wherever they are at the time of detection; if the molecules have moved out of the region where they were at the time of tagging, their final position will be revealed at the time of detection, i.e. the peaks in the image which correspond to the signal from the spins tagged in a given location will broaden proportionally to the extent of molecular motion. This is exactly what happened for sample S1 when the pulse sequence in Fig. 1B was run at nr = 7 and τsl = 1 s or 30 s as shown in Fig. 3. In this figure, the rightmost peak corresponds to the signal from the slice selected by the first occurrence of the sM2S, the second peak from the right corresponds to the signal from the slice selected by the second occurrence of the sM2S and so on for all other peaks. It becomes clear from these experiments that thermal convection is deleterious for the quantitative determination of TS in these experiments; even for τsl = 1 s, the peaks start to merge meaning that molecules have travelled the 0.5 mm left between two consecutive slices. This motion is almost entirely due to thermal convection since Brownian diffusion would only account for micrometric displacements in such a short time. For τsl = 30 s (Fig. 3B), the molecules originally tagged in a given slice have spread across the whole sample as indicated by the almost complete merging of all peaks. | ||
| Fig. 3 1D spin echo images of sample S1 obtained with the sequence in Fig. 1B for ns = 1, nr = 7 and τsl = 1 s (A) and τsl = 30 s (B). The intensity is normalized to the one of Fig. 2. | ||
Incidentally, the peaks produced by the pulse sequence SSTS-v1 remain of a fixed width irrespective of the value of τsl (compare blue and black lines in Fig. 2) since in this version of the experiment the time between the selective tagging of the molecules in a specific location occurs immediately before detection so that no displacement is actually visible. This does not mean that SSTS-v1 is actually immune to molecular motions since this is always going on in background, manifesting itself as a limiting factor on the accuracy on the TS measurements.
The extent of thermal convection depends on many factors including temperature difference across the sample, viscosity of the solvent, tube diameter and more,57 so its effects will manifest differently in different samples. Even if we could find a way to reduce convection to zero by, for example, a perfect control of the sample temperate, molecules would still move because of Brownian diffusion. Therefore, during the long τsl required to measure minutes long TS, molecules would still be excited while they are in a slice but are detected when they have moved to another one, thus compromising the whole experiment. The only way to make this experiment quantitative is to physically confine molecules within the selected slices. This is equivalent to the use of Shigemi tubes to confine all spins within the detecting coil in experiments where molecular motions can bring molecules outside of the coil at the time of detection.
Limiting molecular motions using compartmentation
To confine molecular motions in our experiments and thus make those quantitative, we propose to use a tube insert with a number of physically separated compartments (7 in our case) and set up the offsets of all selective pulses in the SSTS pulse sequences such that the center of each selected slice coincides with the center of these compartments. A prototype of this insert is discussed in Materials and methods and fits a conventional 10 mm medium-wall NMR tube. The insert has seven 1 mm thick compartments separated by 0.5 mm thick walls and is machined from a polyoxymethylene (POM) rod. The choice of the material is simply based on its chemical compatibility with most organic solvents and its mechanical properties in relation to the machining process. Since the insert OD is slightly smaller than the tube ID the compartments are easily filled as the solution is poured from the top making it quite compatible with dissolution–DNP experiments, for example. Air bubbles possibly trapped in the compartments are easily removed by a quick shake. For hyperpolarization experiments, where there is plenty of signal, the chambers in the insert can be fabricated thinner and arranged parallel to the tube main axis so as to make them easier to be filled with the sample coming out from the polariser. The 1D spin-echo image and the singlet-filtered 1D spin-echo image of a 0.5 M solution of EPM dissolved in acetone-d6 and contained in a LPV 10 mm OD medium-wall tube also containing the POM insert (sample S2) are shown in Fig. 4A and B. The signal from the molecules contained in each compartment appears in the form of well-separated peaks in the image. The small differences in the width and height of these peaks are due to imperfections in the machining which we can take into account as explained below. The small bumps at each side are due to the small amount of sample enclosed between the glass walls and the insert at the edges of the coil. Our coil is 18 mm long and therefore extends for about 4 mm each side of the compartmented region of the insert, which is just 10 mm long. | ||
| Fig. 4 1D spin-echo image of S2 obtained using the sequence in Fig. 1H (A); 1D singlet-filtered spin-echo image of S2 obtained using the sequence in Fig. 1I (B). Positive projections across all rows or the 2D spectrum resulting from the SSTS-v1 sequence in Fig. 1A run with nr = 7 and τsl = 1 s (C) and τsl = 60 s (D). The intensity is normalized across all panels such that the highest peak in panel A is 1. | ||
Fig. 4C displays the positive projection of the 2D spectrum resulting from the pulse sequence SSTS-v1 run with nr = 7 and τsl = 1 s on sample S2 where seven 1.2 mm thick slices are selected using g1 = −g2 = g3 = 420 mT m−1 and the offset of the selective pulses carefully chosen so as to center the selected slices with the center of the compartments in the POM insert – easily done using eqn (4). The thickness of these slices (1.2 mm) was deliberately chosen to be slightly bigger than the POM compartments (1 mm) to ensure a uniform excitation of the sample inside each compartment. The peaks are again well-separated, and the intensity pattern follows the one of the sample z-profiles in Fig. 4A. The spectrum in Fig. 4D is the positive projection of the 2D spectrum resulting from the pulse sequence SSTS-v1 run with nr = 7 and τsl = 60 s on sample S2.
The peaks remain well defined for the reasons discussed above but their intensities decay according to the time the corresponding slice has been selected, with the leftmost peak being selected at the first sS2M occurrence and the rightmost peak being selected at its last.
Singlet order decay constant quantification
To measure TS using the SSTS-v1 method we simply calculate the area under each peak in the spectrum with τsl = 60 s (Fig. 4D). Normalizing this to the area of the corresponding peak in the singlet-filtered 1D spin-echo image (Fig. 4B), the possible differences in volume between the compartments due to imprecise mechanical machining of the insert and/or differences in B1 homogeneity at the edges of the sample can be accounted for.The normalized area of the seven peaks is plotted versus time in Fig. 5 and fitted to a mono-exponential decay function to yield a TS = 134 ± 11 s which is in good agreement with a value of 142 ± 3 obtained using the conventional pseudo-2D method.
 | ||
| Fig. 5 Filled circles are the experimental area under the peaks in the spectrum of Fig. 4D. Each area has been normalized to the area of the corresponding peak in Fig. 4 and, successively, all points have been normalised so to have 1 at time 0. The time axis is obtained as an integer increment of τsl from 1 to 7. The grey line is the best fit to a mono-exponential decay. | ||
The total duration of the SSTS-v1 experiment in Fig. 5 was ∼nr × τsl = 420 s to be compared with the 1852 s needed by the conventional pseudo-2D measurements and as calculated using eqn (1) using the experimental value of T1 of 8.2 s and the same linear array of 7 points spanning from 60 to 420 s. This corresponds to a time saving factor of 4.4. The saving factor can change depending on the way the variable time array is arranged but the absolute time saving becomes more and more significant when the T1 and TS of the sample become longer and longer. Recalling the example in the Introduction, a molecule with an estimated TS of 60 min and a T1 of 30 s would require ∼11 h per scan to measure TS in a conventional pseudo-2D mode whereas our SSTS method would achieve the same goal in just 3 h per scan.
Materials and methods
Equipment
All experiments have been run on a 7.04 T wide bore magnet coupled to a Bruker Avance III console fitted with a 3-axis gradient system for micro-imaging experiments, able to deliver a maximum field gradient of 1.5 T m−1, and a Bruker MICWB40 microimaging probe equipped with a 1H/13C 10 mm resonator.Samples
All experiments have been run on samples containing a ∼0.5 M solution of ethyl-d5 (propyl-d7) maleate (EPM, Fig. 6A) dissolved in ethanol-d6 and degassed by bubbling oxygen-free N2 gas. | ||
| Fig. 6 Structure of molecule used and geometry of sample insets. (A) Molecular structure of ethyl-d5 (propyl-d7) maleate (EPM); (B) details of the POM insert used in sample S2. | ||
The molecule of EPM was synthetized in house according to reported procedures.43 For experiments referring to sample S1, the solution was contained in 10 mm medium-wall LPV NMR tube attached to a large J-Young valve that allows a 7.5 mm OD object to comfortably pass through its neck. For experiments referring to sample S2, the very same tube used for sample S1 was opened and a plastic structure inserted. This structure, machined from a polyoxymethylene rod with the dimensions specified in Fig. 6B, was designed in order to confine molecular motions within 7 parallel 1 mm-thick compartments spaced by six 0.5 mm-thick walls.
The T1 decay constant of samples S1 and S2 have been measured by a conventional saturation recovery experiment and found to be 8.2 ± 0.8 s. The TS for those two samples, measured with the conventional pseudo-2D mode, were found to be 201 ± 1 s for S1 and 143 ± 3 s for S2. The discrepancy between two samples is attributable to the likely higher level of O2 present in the sample S2 after the opening and placing of the plastic insert.
Pulse sequence parameters
Selective excitation and refocusing pulses occurring in the pulse sequences of Fig. 1 were designed using the Shinnar–Le Roux (SLR) algorithm using an equiripple filter and a time–bandwidth product of 6.58 These pulses were used with duration sp90 = 590 μs and sp180 = 635 μs for excitation and refocusing, respectively, corresponding to a bandwidth BW = 9 kHz. The strength of G4 and G5 gradients was kept constant at 75 mT m−1 (5% of maximum available) throughout all experiments, and their duration was 2.56 ms (matched to a SW of 100 kHz and 512 points in the time domain) and 1.28 ms, respectively. The absolute strengths g1, g2 and g3 were set to 180 mT m−1 (12% of maximum available). From eqn (2), these gradients excite a 1.2 mm thick slice. The optimized values of the echo delay and echo repetitions in the sM2S and S2M blocks were τe = 41.8 ms, n1 = 18 and n2 = 9. Seven slices were selected (nr = 7) and the offsets of the selective pulses to select the 7 slices were chosen so as to coincide with the center of the compartments in the POM insert.Conclusions
The methodology introduced in this paper allows single-scan measurements of the decay rate of singlet order. The method offers a significant saving factor in experimental time which translates into important and substantial absolute time saving when measuring samples with long T1 and TS values. Even more important is the fact that this is a single-scan method and therefore marries well with single-scan hyperpolarization techniques such as dissolution–DNP. Measuring TS lifetimes of hyperpolarized samples is otherwise very challenging, requiring multiple repetitions of the hyperpolarization procedures. Not only does this repetition have obvious implications on costs and time but, perhaps more importantly, causes irreproducibility associated with the difficulty of producing batches of identical polarization repeatedly.As in all other spatial-encoding based techniques, our method requires a trade-off between the number of points acquired simultaneously and the reduction in signal-to-noise due to the fact that the signal comes from a restricted portion of the sample. However, this should not be anything to worry about in the presence of hyperpolarization which is where we feel the technique expresses its best potential.
We believe that this methodology can play a fundamental role in the quest for singlet-bearing molecules displaying long lifetimes. Often in this field, we are required to design and synthesise doubly labelled molecules guided by scientific reasoning and/or simulations of the possible value of TS. This is a costly and time-consuming procedure and may well end up revealing that the molecule is not suitable for the desired purpose. The advances reported here provided by our new methodology in combination with a hyperpolarization method, could be used to measure TS in naturally abundant (or, at least, singly labelled) two-spin systems thus allowing evaluation of the suitability of a molecule, for any given application, before investing time and money on isotopically enriched syntheses. Investigation of hyperpolarized substrates will be carried out when the worldwide health emergency has passed, and interactions with other groups equipped with such technology becomes possible again.
Author contributions
G. P. and J. N. D. devised the research and ran experiments; G. M. and C. N. ran experiments and processed the data; L. J. B. synthetized the molecules. G. P. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Stuart J. Elliott, Ilya Kuprov and Ahmed J. M. Allami for fruitful discussions. This research was supported by EPSRC (UK) grant no. EP/P005187/1.Notes and references
- Long-lived Nuclear Spin Order: Theory and Applications, ed. G. Pileio, Royal Society of Chemistry, London, 2020 Search PubMed.
- C. Bengs, M. Sabba, A. Jerschow and M. H. Levitt, Phys. Chem. Chem. Phys., 2020, 22, 9703–9712 RSC.
- B. A. Rodin, C. Bengs, A. S. Kiryutin, K. F. Sheberstov, L. J. Brown, R. C. D. Brown, A. V. Yurkovskaya, K. L. Ivanov and M. H. Levitt, J. Chem. Phys., 2020, 152, 164201 CrossRef CAS PubMed.
- S. Mamone, N. Rezaei-Ghaleh, F. Opazo, C. Griesinger and S. Glöggler, Sci. Adv., 2020, 6, eaaz1955 CrossRef CAS PubMed.
- C. Bengs, L. Dagys and M. H. Levitt, J. Magn. Reson., 2020, 321, 106850 CrossRef CAS PubMed.
- B. A. Rodin, K. F. Sheberstov, A. S. Kiryutin, L. J. Brown, R. C. D. Brown, M. Sabba, M. H. Levitt, A. V. Yurkovskaya and K. L. Ivanov, J. Chem. Phys., 2019, 151, 234203 CrossRef PubMed.
- B. A. Rodin, A. S. Kiryutin, A. V. Yurkovskaya, K. L. Ivanov, S. Yamamoto, K. Sato and T. Takui, J. Magn. Reson., 2018, 291, 14–22 CrossRef CAS PubMed.
- G. Pileio, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 98–99, 1–19 CrossRef CAS PubMed.
- G. Stevanato, J. Eills, C. Bengs and G. Pileio, J. Magn. Res., 2017, 277, 169–178 CrossRef CAS PubMed.
- J. Eills, G. Stevanato, C. Bengs, S. Glöggler, S. J. Elliott, J. Alonso-Valdesueiro, G. Pileio and M. H. Levitt, J. Magn. Res., 2017, 274, 163–172 CrossRef CAS PubMed.
- A. S. Kiryutin, A. N. Pravdivtsev, A. V. Yurkovskaya, H. M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2016, 120, 11978–11986 CrossRef CAS PubMed.
- Z. Zhou, K. Claytor, W. S. Warren and T. Theis, J. Magn. Reson., 2016, 263, 108–115 CrossRef CAS PubMed.
- K. Claytor, T. Theis, Y. Feng and W. Warren, J. Magn. Reson., 2014, 239, 81–86 CrossRef CAS PubMed.
- K. Claytor, T. Theis, Y. Feng, J. Yu, D. Gooden and W. S. Warren, J. Am. Chem. Soc., 2014, 136, 15118–15121 CrossRef CAS PubMed.
- A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya, H.-M. Vieth and N. N. Lukzen, Phys. Chem. Chem. Phys., 2013, 15, 14248–14255 RSC.
- S. J. DeVience, R. L. Walsworth and M. S. Rosen, Phys. Rev. Lett., 2013, 111, 5 CrossRef PubMed.
- R. Sarkar, P. Ahuja, D. Moskau, P. R. Vasos and G. Bodenhausen, ChemPhysChem, 2007, 8, 2652–2656 CrossRef CAS PubMed.
- R. Buratto, D. Mammoli, E. Chiarparin, G. Williams and G. Bodenhausen, Angew. Chem., Int. Ed., 2014, 53, 11376–11380 CrossRef CAS PubMed.
- N. Salvi, R. Buratto, A. Bornet, S. Ulzega, I. Rentero Rebollo, A. Angelini, C. Heinis and G. Bodenhausen, J. Am. Chem. Soc., 2012, 134, 11076–11079 CrossRef CAS PubMed.
- M. C. Tourell, I.-A. Pop, L. J. Brown, R. C. D. Brown and G. Pileio, Phys. Chem. Chem. Phys., 2018, 20, 13705–13713 RSC.
- G. Pileio and S. Ostrowska, J. Magn. Reson., 2017, 285, 1–7 CrossRef CAS PubMed.
- G. Pileio, J.-N. Dumez, I.-A. Pop, J. T. Hill-Cousins and R. C. D. Brown, J. Magn. Reson., 2015, 252, 130–134 CrossRef CAS PubMed.
- J. N. Dumez, J. T. Hill-Cousins, R. C. D. Brown and G. Pileio, J. Magn. Reson., 2014, 246, 27–30 CrossRef CAS PubMed.
- P. Ahuja, R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Am. Chem. Soc., 2009, 131, 7498–7499 CrossRef CAS PubMed.
- S. Cavadini, J. Dittmer, S. Antonijevic and G. Bodenhausen, J. Am. Chem. Soc., 2005, 127, 15744–15748 CrossRef CAS PubMed.
- P. Saul, S. Mamone and S. Glöggler, Chem. Sci., 2019, 10, 413–417 RSC.
- S. Yang, J. McCormick, S. Mamone, L. S. Bouchard and S. Glöggler, Angew. Chem., Int. Ed., 2019, 58, 2879–2883 CrossRef CAS PubMed.
- C. P. N. Tanner, J. R. Lindale, S. L. Eriksson, Z. Zhou, J. F. P. Colell, T. Theis and W. S. Warren, J. Chem. Phys., 2019, 151, 044201 CrossRef PubMed.
- S. Mamone and S. Glöggler, Phys. Chem. Chem. Phys., 2018, 20, 22463–22467 RSC.
- T. Theis, G. X. Ortiz, A. W. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev, Q. Wang and W. S. Warren, Sci. Adv., 2016, 2, e1501438 CrossRef PubMed.
- J.-N. Dumez, P. Håkansson, S. Mamone, B. Meier, G. Stevanato, J. T. Hill-Cousins, S. S. Roy, R. C. D. Brown, G. Pileio and M. H. Levitt, J. Chem. Phys., 2015, 142, 044506 CrossRef PubMed.
- Y. Feng, T. Theis, X. Liang, Q. Wang, P. Zhou and W. S. Warren, J. Am. Chem. Soc., 2013, 135, 9632–9635 CrossRef CAS PubMed.
- S. J. DeVience, R. L. Walsworth and M. S. Rosen, NMR Biomed., 2013, 26, 1204–1212 CrossRef CAS PubMed.
- Y. Zhang, X. Duan, P. C. Soon, V. Sychrovský, J. W. Canary and A. Jerschow, ChemPhysChem, 2016, 17, 2967–2971 CrossRef CAS PubMed.
- Y. Zhang, K. Basu, J. W. Canary and A. Jerschow, Phys. Chem. Chem. Phys., 2015, 17, 24370–24375 RSC.
- Y. N. Zhang, P. C. Soon, A. Jerschow and J. W. Canary, Angew. Chem., Int. Ed., 2014, 53, 3396–3399 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS PubMed.
- A. N. Pravdivtsev, F. D. Sönnichsen and J.-B. Hövener, PLoS One, 2020, 15, e0239982 CrossRef CAS PubMed.
- G. Pileio, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 217–231 CrossRef CAS PubMed.
- G. Stevanato, J. T. Hill-Cousins, P. Håkansson, S. S. Roy, L. J. Brown, R. C. D. Brown, G. Pileio and M. H. Levitt, Angew. Chem., Int. Ed., 2015, 54, 3740–3743 CrossRef CAS PubMed.
- L. J. Brown, in Long-lived nuclear spin order: theory and applications, ed. G. Pileio, Royal Society of Chemistry, London, 2020, ch. 4 Search PubMed.
- D. Graafen, M. B. Franzoni, L. M. Schreiber, H. W. Spiess and K. Münnemann, J. Magn. Res., 2016, 262, 68–72 CrossRef CAS PubMed.
- G. Pileio, S. Bowen, C. Laustsen, M. C. D. Tayler, J. T. Hill-Cousins, L. J. Brown, R. C. D. Brown, J. H. Ardenkjaer-Larsen and M. H. Levitt, J. Am. Chem. Soc., 2013, 135, 5084–5088 CrossRef CAS PubMed.
- L. Frydman, T. Scherf and A. Lupulescu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15858–15862 CrossRef CAS PubMed.
- N. M. Loening, M. J. Thrippleton, J. Keeler and R. G. Griffin, J. Magn. Reson., 2003, 164, 321–328 CrossRef CAS PubMed.
- M. J. Thrippleton, N. M. Loening and J. Keeler, Magn. Reson. Chem., 2003, 41, 441–447 CrossRef CAS.
- L. Frydman and D. Blazina, Nat. Phys., 2007, 3, 415–419 Search PubMed.
- J.-N. Dumez, J. Milani, B. Vuichoud, A. Bornet, J. Lalande-Martin, I. Tea, M. Yon, M. Maucourt, C. Deborde, A. Moing, L. Frydman, G. Bodenhausen, S. Jannin and P. Giraudeau, Analyst, 2015, 140, 5860–5863 RSC.
- L. Guduff, D. Kurzbach, C. van Heijenoort, D. Abergel and J.-N. Dumez, Chem. – Eur. J., 2017, 23, 16722–16727 CrossRef CAS PubMed.
- S. Ahola, V. V. Zhivonitko, O. Mankinen, G. Zhang, A. M. Kantola, H.-Y. Chen, C. Hilty, I. V. Koptyug and V.-V. Telkki, Nat. Commun., 2015, 6, 8363 CrossRef CAS PubMed.
- P. Giraudeau, Y. Shrot and L. Frydman, J. Am. Chem. Soc., 2009, 131, 13902–13903 CrossRef CAS PubMed.
- M. C. D. Tayler and M. H. Levitt, Phys. Chem. Chem. Phys., 2011, 13, 5556–5560 RSC.
- G. Pileio, M. Carravetta and M. H. Levitt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17135–17139 CrossRef CAS PubMed.
- M. C. D. Tayler, in Long-lived nuclear spin order: theory and applications, ed. G. Pileio, Royal Society of Chemistry, London, 2020, ch. 10 Search PubMed.
- I. Swan, M. Reid, P. W. Howe, M. A. Connell, M. Nilsson, M. A. Moore and G. A. Morris, J. Magn. Reson., 2015, 252, 120–129 CrossRef CAS PubMed.
- J. Pauly, P. Le Roux, D. Nishimura and A. Macovski, IEEE Trans. Med. Imag., 1991, 10, 53–65 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2021 |