
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measurement of the conformational switching of azobenzenes from the macro- to attomolar scale in self-assembled 2D and 3D nanostructures†
Vanesa
Quintano
a,
Valentin
Diez-Cabanes
b,
Simone
Dell’Elce
c,
Lorenzo
Di Mario
d,
Stefano
Pelli Cresi
d,
Alessandra
Paladini
 d,
David
Beljonne
b,
Andrea
Liscio
*e and
Vincenzo
Palermo
*af
d,
David
Beljonne
b,
Andrea
Liscio
*e and
Vincenzo
Palermo
*af
aInstitute of Organic Synthesis and Photoreactivity (ISOF) – (CNR), via Gobetti 101, 40129 Bologna, Italy. E-mail: vincenzo.palermo@isof.cnr.it
bLaboratory for Chemistry of Novel Materials, University of Mons, Place du Parc 20, B-7000 Mons, Belgium
cGraphene-XT srl, via D’Azeglio 15, 40123 Bologna, Italy
dDivision of Ultrafast Processes in Materials (FLASHit), Institute of Structure of Matter (ISM) – CNR, via del Fosso del Cavaliere 100, 00133 Rome, Italy
eInstitute for Microelectronics and Microsystems (IMM), National Research Council of Italy (CNR), via del Fosso del Cavaliere 100, 00133 Rome, Italy. E-mail: andrea.liscio@artov.imm.cnr.it
fDepartment of Industrial and Materials Science, Chalmers University of Technology, Hörsalvägen 7, 41296 Gothenburg, Sweden
First published on 22nd April 2021
Abstract
It is important, but challenging, to measure the (photo)induced switching of molecules in different chemical environments, from solution through thin layers to solid bulk crystals. We compare the cis–trans conformational switching of commercial azobenzene molecules in different liquid and solid environments: polar solutions, liquid polymers, 2D nanostructures and 3D crystals. We achieve this goal by using complementary techniques: optical absorption spectroscopy, femtosecond transient absorption spectroscopy, Kelvin probe force microscopy and reflectance spectroscopy, supported by density functional theory calculations. We could observe the same molecule showing fast switching in a few picoseconds, when studied as an isolated molecule in water, or slow switching in tens of minutes, when assembled in 3D crystals. It is worth noting that we could also observe switching for small ensembles of molecules (a few attomoles), representing an intermediate case between single molecules and bulk structures. This was achieved using Kelvin probe force microscopy to monitor the change of surface potential of nanometric thin 2D islands containing ca. 106 molecules each, self-assembled on a substrate. This approach is not limited to azobenzenes, but can be used to observe molecular switching in isolated ensembles of molecules or other nano-objects and to study synergistic molecular processes at the nanoscale.
1. Introduction
Molecular switches are a class of molecules that are able to reversibly change their chemical structure under the effect of certain stimuli such as light irradiation or the change of temperature.1 They have been extensively studied for a wide range of applications, including solid-state electronics,2 molecular sensing,3 thermal fuel storage,4 and nanobiology,5 thus attracting more and more interest from the scientific community, as confirmed by the Nobel Prize in Chemistry 2016 awarded to Sauvage, Stoddart and Feringa “for the design and synthesis of molecular machines”.6Molecules with a central axis formed by a double bond are usually hindered rotors in the ground state, but they can also twist upon optical excitation, leading to cis–trans isomerization.7–9 In most cases, such conformational switching process occurs between two different well-defined states and is accompanied by the on/off switch of a certain property, which can be measured by a change in the electronic or optical absorption spectra, luminescence signals or nuclear magnetic resonance.10
Azobenzenes are one of the most well-known classes of molecular switches (Fig. 1). These molecules show photoisomerization from trans- to cis-conformation around the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond by absorbing UV light, whereas they return to trans-conformation when they are exposed to visible light or heat.
N bond by absorbing UV light, whereas they return to trans-conformation when they are exposed to visible light or heat.
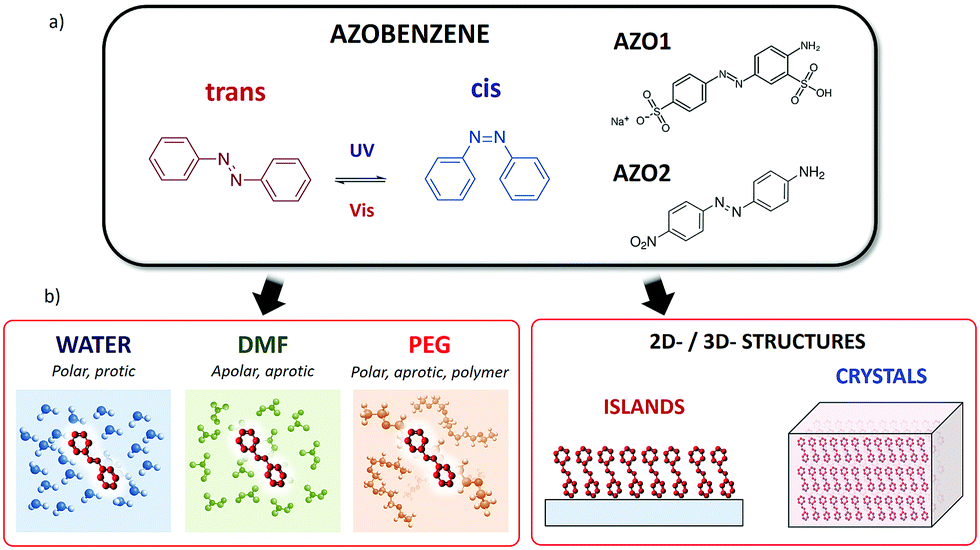 | ||
| Fig. 1 (a) Azobenzene: structure and different conformations. Molecules studied in this work: (AZO1) 2-amino-5-([4-sulfophenyl]azo)benzenesulfonic acid (acid yellow 9, CAS: 74543-21-8) and (AZO2) 4-(4-nitrophenylazo)aniline (CAS: 730-40-5). (b) Schematic representation of all the different solid and liquid environments where the cis → trans switching of a molecule was studied. | ||
This conformational switching can be easily observed in real time by using optical spectroscopy11 and it can be ideally exploited in order to obtain a light-driven operation in molecular machines or smart materials.12
In particular, dyes featuring the azo linkage (–NN–) are important for both fundamental studies and technological applications, due to their extensive use as dyes in the textile industry and colorant inks and more recently due to their new applications in optical switching and optical data storage techniques.13,14
The switching of azobenzenes has been vastly studied in solution, where macroscopic amounts of molecules can be studied using optical techniques.15,16 In an ideal case, molecules are free from steric hindrance, isolated from one another and dispersed in a uniform medium. Even in such an ideal situation, the switching mechanisms may occur by following different reaction pathways, including direct NN bond torsion, N center inversion or more complex coordinated movements of nitrogen and phenyl groups.17–20 In addition, the switching process in solution is influenced by the solvent polarity and proticity, while the solvent viscosity seems to have a lower impact on the switching dynamics (see ref. 21 and references therein).
While switching of azobenzenes in organic solvents can be easily studied for bulk solutions, most of their possible applications (e.g. in electronics) need to take place in solids or on surfaces, in a constrained environment, which is more challenging to study. In principle, azobenzene is able to isomerize even under the effect of strong constraints,22,23 such as the tight environments derived from solid matrices or from their inclusion in polymer chains.
As an example, some applications in electronics require the deposition of azobenzenes on a 2D substrate, possibly in ordered, self-assembled monolayers.24 In contrast, applications as industrial dyes require the dispersion of azobenzenes in a 3D matrix (typically a polymer).
Switching of self-assembled monolayers (SAMs) based on azobenzene molecules has been already studied at the macroscopic ensemble level by using spectroscopic techniques22,25–27 or at the molecular level by means of microscopic or computational techniques.21,27 The influence of the environment on the isomerization of azobenzenes in constrained confinements is still an important matter of debate in the community and requires careful consideration when comparing various methods.22
Here, we present a complete experimental study of the properties of model azobenzene compounds focusing on how the cis–trans switching behaviour is influenced by the environment, in particular when the molecule is solubilized in different solvents, dispersed in polymers, deposited in 2D layers on a substrate or arranged in 3D crystals (Fig. 1b). For this purpose, we had to overcome different limitations due to the solubility of the molecule, its processability and the complementary limitations of the different techniques used in this work. By combining spectroscopic studies in solution and on a surface with microscopic techniques, we observed the cis–trans switching of such azobenzene molecules both in bulk ensembles and in nanoscopic structures.
We chose two commercial azo compounds as target molecules: 2-amino-5-([4-sulfophenyl]azo)benzenesulfonic acid (acid yellow 9, CAS: 74543-21-8, Merck) named AZO1, and 4-(4-nitrophenylazo)aniline (CAS: 730-40-5, Merck) named AZO2.
AZO1 is an aminoazobenzene-class molecule commercialized as a biological stain and formerly used as a food dye (E105).28 One of the two azobenzene rings is decorated with one sulfonic acid (donor) and one amine (acceptor) group in the meta- and para-positions, respectively, while the other ring is substituted with a sulfonic acid sodium salt in the para-position.
Given its good solubility in a wide range of solvents due to the presence of two sulphonic groups, we studied the switching behaviour of AZO1 in polar solvents: protic (water) and aprotic (dimethylformamide, DMF).
Previous studies have suggested that solvent viscosity could have minimal influence on the switching rate of azobenzenes.21,29 Thus, besides small molecule solvents, we tested the switching behaviour of AZO1 also in a polymeric matrix environment, i.e., in polyethylene glycol with average Mn = 400 (PEG). This liquid polymer, widely used in academia and industry, also provides a polar, aprotic environment for solvated molecules. PEG has significant polarity and a viscosity of ≈90 mPa s, which is two orders of magnitude higher than those of water (0.89 mPa s) and DMF (0.8 mPa s).
AZO2 is a pseudostilbene-type molecule, a dye used in colouring natural and synthetic fabrics. It can be found in many clothing materials such as acetates, nylon, silk, wool, and cotton. This compound is a more conventional azobenzene bearing no charges, which possesses an electron-donating NH2 group on one phenyl ring and an electron-withdrawing NO2 group on the other, with dipole moment = 14.08 D. The donor–acceptor asymmetric functionalization provides a strong charge-transfer character to the π–π* transition (ca. 390 nm), which is then red-shifted and overlaps the n–π* transition (ca. 480 nm) (see Fig. S2, ESI,† for the calculated spectra), thus lowering the energy barrier between the cis and trans-forms and making the switching process extremely easy at room temperature. Such molecule is thus able to switch even in sterically hindered situations. Due to the lack of sulphonic groups, AZO2 is much less soluble than AZO1 and it cannot be solubilized in many solvents, including water. In this case, AZO2 could be solubilized in the DMF solvent, wherein it showed ultra-fast switching, similar to AZO1 in water.
2. Experimental
2.1 Materials
All the chemicals (solvents and azobenzene systems) were provided by Merck. The solvents used were HPLC water, DMF with a 99.8% purity, and PEG with average Mn = 400 (CAS number: 25322-68-3). The commercial azobenzene compounds used were 2-amino-5-([4-sulfophenyl]azo)benzenesulfonic acid (acid yellow 9, CAS number: 74543-21-8) named AZO1, and 4-(4-nitrophenylazo)aniline (CAS number: 730-40-5) named AZO2.2.2 Sample preparation
Silicon wafers were treated with ultrasonication in ethanol and acetone for twenty minutes each before being dried in an oven.2.3 Methods
The optical layout of the TA spectrometer consisted of a split-beam configuration in which 50% of the white light passes through the sample solution contained in a 1 mm static cell, while the remainder is used as a reference to account for pulse-to-pulse fluctuations in the white light generation. The pump pulse is focused (circular spot of diameter = 400 μm) onto the sample with an energy density of 280 μJ cm−2. The spot diameter of the probe pulse is approximately 150 μm, and its time delay with respect to the pump pulse is scanned in time by varying the length of its optical path.
The instrument response function (IRF) was measured to be approximately 50 fs. All measurements were performed in air at room temperature. More experimental details on the setup can be found in the previous studies.31,32
3. Results and discussion
3.1 AZO photoisomerization in liquid
We first used time-dependent density functional theory (DFT) calculations to estimate the relative stability and opto-electronic properties of the individual cis and trans conformers of AZO1 in each solvent in order to compare them with the experimental absorption spectra (Fig. 2).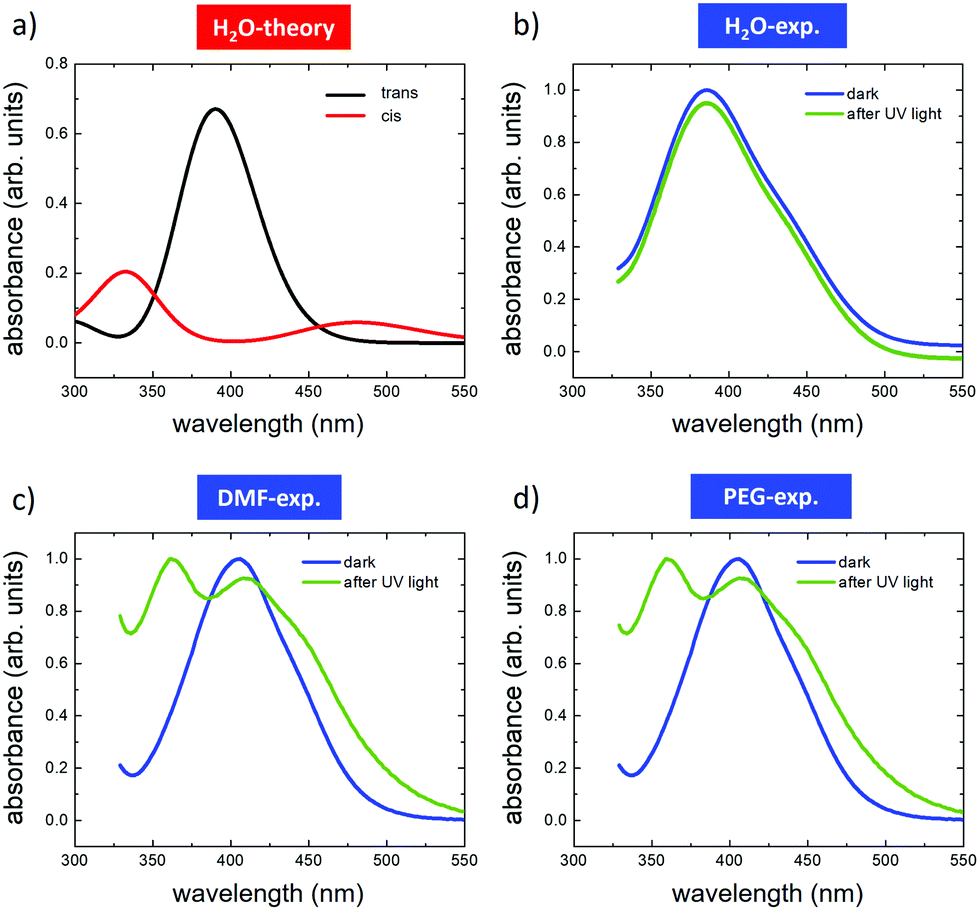 | ||
| Fig. 2 Optical absorption spectra of AZO1 in different solvents. (a) Calculated TD-DFT spectra of the trans (red) and cis (black) isomers in water. Spectra measured before (blue) and after (green) UV illumination in (b) water, (c) DMF and (d) PEG. In the case of water, the acquired curves have been Y-shifted to clearly distinguish them. The TD-DFT spectra for DMF and PEG are presented in Fig. S3 (ESI†). | ||
The complete set of spectra and full numerical details are available in Fig. S3 and Tables S1 and S2 in the ESI.†
In standard azobenzenes, the trans-form is energetically more stable than the cis form in the majority of cases. The photoisomerization of the cis form takes place upon UV light irradiation, while cis → trans thermal isomerization occurs spontaneously in the dark owing to the thermodynamic preference of the trans isomer. In addition to thermal isomerization, photoinduced cis → trans isomerization is also possible upon visible light irradiation.41
The DFT calculations predicted different absorption spectra for the cis and trans-forms, while only a minor effect of the solvent environment was observed. In all the studied cases, the absorption spectra of the trans form were dominated by an absorption peak centred between 300 and 400 nm due to the π–π* transition (see Tables S1 and S2, ESI†). Upon illumination, a wide absorption peak was predicted between 300 and 400 nm, which can be assigned to the π–π* transition, and another between 450 and 500 nm assigned to the n–π* transition (see Tables S1 and S2, ESI†). For the sake of simplicity, we depict only the simulation related to the water solvent (Fig. 2a).
The experimental absorption spectra were acquired for each solution (Fig. 2b–d) in the dark (blue curves) and after illumination with UV light of λ = 365 nm (green curves). In the dark, all azobenzene molecules were in the trans configuration. In general, the calculations reproduced well the absorption spectra of the pristine trans-form, showing only a small effect due to the solvent environment. The spectra recorded for DMF were similar to those recorded for PEG, both showing the main peaks at ≈410 nm and a second one, not fully resolved, at ≈445 nm. Similar behaviour was observed in the case of water, where the main peak was shifted to ca. 390 nm, while the second one, not fully resolved, was located in the region between 430 and 450 nm. These differences were attributed to the small effect of the solvent on the HOMO–LUMO energies, which directly affected the band gap (see Tables S1 and S2, ESI†). All the measurements were performed in a saturation regime, i.e., there is no dependence of the trans–cis switching rate on the photon flux.
trans → cis isomerization was induced by light illumination at λ = 365 nm (power density = 3.2 mW cm−2) until no further spectral variation was observed, thus reaching the photostationary state (Fig. 3) given by a mixture of both cis- and trans-isomers and defined by the experiment-specific balance between the UV-induced trans–cis isomerization and the temperature-induced back (cis–trans) isomerization. Thereafter, we examined the backward (cis → trans) isomerization by turning off the 365 nm light source. The absorption spectra related to the back isomerization are reported in the ESI† (Fig. S3).
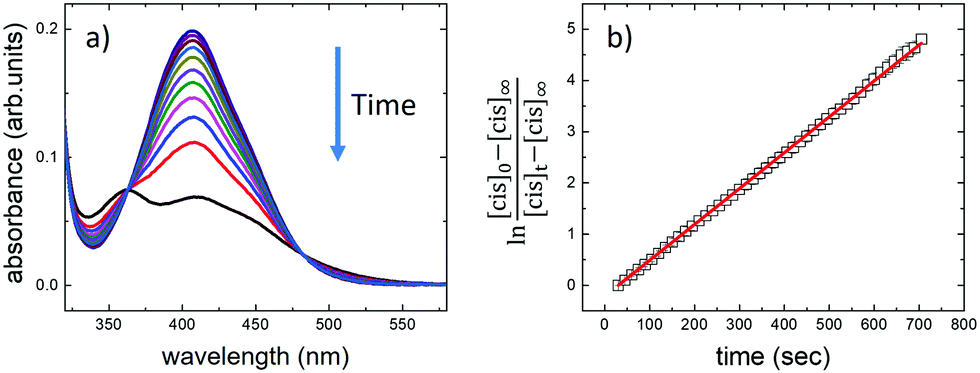 | ||
| Fig. 3 (a) Changes in the absorption spectra of AZO1 in DMF upon irradiation at 365 nm. The arrow indicates the changes over irradiation time. Different spectra were recorded at 30-second time steps. (b) Kinetics of isomerization. Absorbance at 410 nm (black squares) as a function of irradiation time. The red solid line shows a fit of the experimental data acquired during 5 cycles of irradiation under the same conditions. | ||
With the difference in the stationary absorption spectrum, the switching kinetics depended strongly on the polarity and proticity of the solvent used. In the case of DMF and PEG solvents, the formation of cis-AZO1 was clearly observed by the growth of a further peak at ca. 360 nm and the decrease of the relative intensity of the peak at 410 nm, thus indicating the presence of a mixture of cis- and trans-isomers. However, no variations in the absorption spectrum were observed for the AZO1 compound in water, where the optical absorption was stable upon prolonged illumination.
We used time-dependent measurements to estimate the kinetics of both the forward (trans → cis) and reverse (cis → trans) switching in the cases where the photoisomerization was observed, i.e., in DMF and PEG. The kinetic rate, k, of the forward (reverse) isomerization followed first-order kinetics (Fig. 3b), and it was estimated using the same procedure as that used in previous azobenzene studies42 by using the formula:
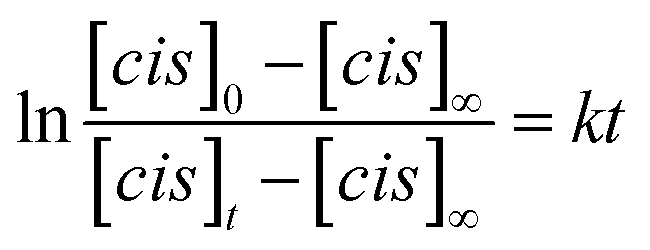 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) calculated using eqn (2)
calculated using eqn (2)
| k + (10−3 s−1) | k − (10−3 s−1) | K (k+/k−) | ε solvent | |
|---|---|---|---|---|
| PEG | 150 ± 20 | 45 ± 0 | 3 ± 1 | 12.4 |
| PEG:H2O (75%:25%) |
100 ± 50 | 40 ± 5 | 2.5 ± 0.6 | 27.8† |
| DMF | 22 ± 2 | 7.3 ± 0.2 | 3.0 ± 0.3 | 38.25 |
| PEG:H2O (50%:50%) |
No switching observed | No switching observed | — | 49.2† |
| PEG:H2O (25%:75%) |
No switching observed | No switching observed | — | 69.2† |
| H2O | No switching observed | No switching observed | — | 80.1 |
The kinetics of forward and reverse isomerization are intrinsically different. The isomerization rate constant of trans molecules (k+) consists solely of the UV photon-induced mechanism, whereas that for the reverse reaction of cis molecules (k−) consists of thermal relaxation mechanisms.
We observed that (i) in both solvents, as soon as the UV-light exposure stops, AZO1 still remains largely in its cis state, k+ > k−, and (ii) the photoisomerization mechanism takes place more slowly in DMF than in PEG. Considering the case of water in which the molecule does not photo-switch (no transition rate, kH2O ≅ 0), we could write the follow hierarchy: k+,PEG > k+,DMF > k+,H2O (k−,PEG > k−,DMF > k−,H2O). Taking into account that the rate of thermal-isomerization in non-polar solvents is faster than that in polar solvents,43 our finding indicates the role of solvent polarity in the photoisomerization kinetics of AZO1, since the polarity index hierarchy is PEG < DMF < H2O.
While the rate constant values are different, their ratio is instead a constant. The equilibrium constant K = k+/k− is the same for all the used solvents and amounts to 2.9 ± 0.1, clearly indicating that different solvents do not affect the electronic properties of azobenzenes.
This experimental result is confirmed by DFT calculations. The stabilization of AZO1 with the polarity of the solvent does not affect the frontier orbital (HOMO/LUMO) energies which are comparable for all three studied solvents (i.e. differences in energy between the isomers range within 20 meV; see Tables S1 and S2, ESI†). In particular, water is a solvent having the largest dielectric constant (εH2O = 80.1), thus being more effective than DMF (εDMF = 38.25) and PEG (εPEG = 12.4) for the stabilization of polar azobenzenes. However, this difference in stability has a minor influence on the relative energy difference between trans- and cis-forms, which is about 0.62–0.64 eV in all cases. We underline that such a theoretical description of the solvent is based on an explicit continuum model which is mainly dependent on the dielectric constant of the medium. Thus, no implicit effects (i.e. geometrical constraint) of the solvents are studied at this stage.
However, the case of water is more complex because the role of hydrogen bonding has to be taken into account together with the permittivity. The absence of any change in the absorption spectrum indicates that either AZO1 is so stable that it cannot switch (kH2O ≅ 0) or that the kinetics is faster than the temporal response of the experimental setup (kH2O ≫ 0).
We faced such an issue when using two different approaches: (i) mixing solvents with different behaviors and (ii) using ultra-fast spectroscopic techniques.
In the first case, we mixed two solvents in which AZO1 showed opposite behaviors: PEG (observed switching) and H2O (not observed switching) corresponding to five different mediums: ranging from pure water to pure PEG together with mixtures composed of 25%, 50% and 75% PEG. Fig. 4b depicts the variations of the optical absorption measured on the five systems as a function of the time turning on/off the UV light (λ = 365 nm, irradiation time = 60 s).
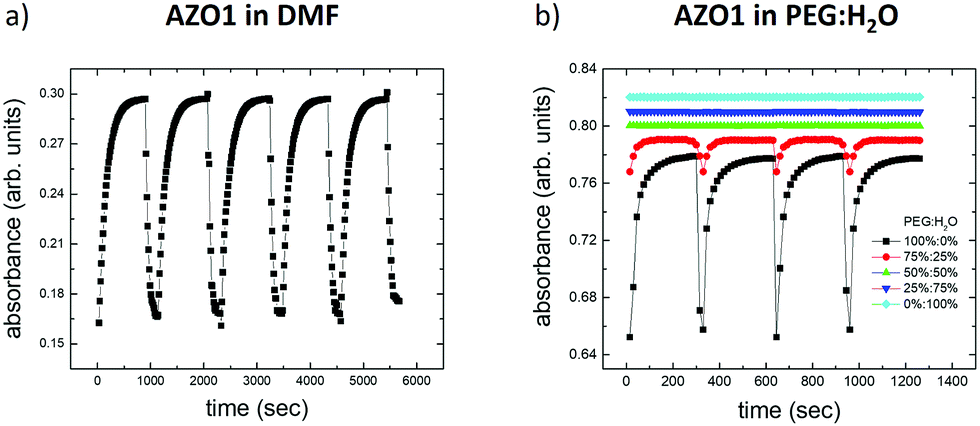 | ||
| Fig. 4 Variation of the optical absorption of AZO1 as a function of time in different solvents: (a) DMF and (b) five different mixtures of PEG:H2O, ranging from pure water (0%:100%) to pure PEG (100%:0%). | ||
Measurable switching was only observed with 75% PEG, even if the absorbance variation (abscis/abstrans ≈ 5%) was smaller than that in the case of pure PEG (abscis/abstrans ≈ 17%). Mixtures with a lower amount of PEG did not show any switching behavior. All the rate constants are reported in Table 1.
The dielectric properties of such mixtures have been studied previously.44 Due to the significant interaction between polyethylene glycols and water in the mixtures, the effective permittivity (εmix) is not linearly dependent on the volume fraction of PEG (VPEG). It follows instead a non-linear combination of the dielectric constants of the two solvents (εPEG and εH2O), described by the Bruggeman mixing formula45 (see also ref. 46 and the references therein for a comprehensive overview) and corrected by the structural rearrangement of the water molecules in the mixture, assuming the following form:
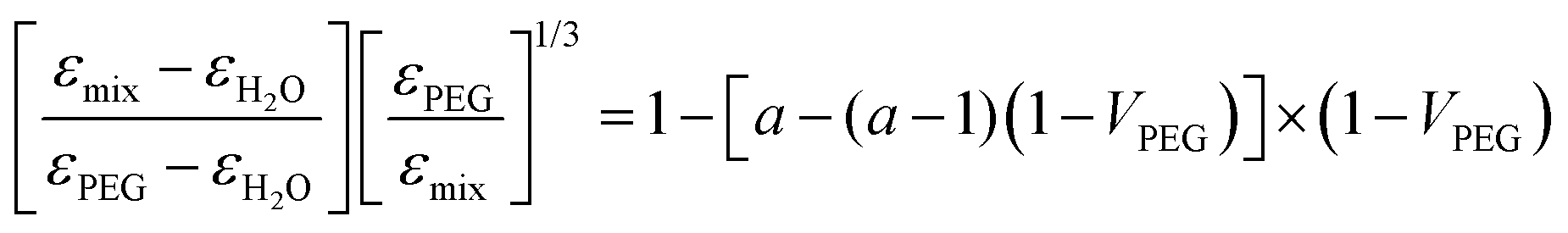 | (2) |
According to eqn (2), we calculated the dielectric permittivity values of the mixtures and compared them with those of pure solvents.47 All the values are reported in Table S1 (ESI†). We could note that the switching behavior caused by the UV light occurs when the dielectric constant of the solvent is observed only when εsolvent ≲ 40. Indeed, the effective dielectric constant of the mixture with 75% PEG (εmix = 27.8) is closer to the dielectric constant of PEG with respect to the one of water. As expected, the equilibrium constant, K, measured for the mixture agrees with the values calculated for DMF and PEG.
It is important to underline that the observed phenomenological relationship between isomerization and the effective dielectric constant of the solvent does not consider the role of OH, the effect of which is evinced in the case of isopropanol (εsolvent = 20) showing an intermediate behavior between that observed in water and that in DMF (Fig. S5, ESI†). Thus, the achieved relationship cannot be valid in the case of water due to the significant contribution of the hydrogen bonds. In general, the switching of sulfonic azobenzenes in an aqueous environment has been already demonstrated, meaning that the alcoholic groups of the azobenzenes do not interfere by inhibiting the switching in water.48
All these measurements were performed using conventional static UV-vis spectroscopy having a time resolution of a few seconds, which allowed us to observe phenomena with kinetic constants k± < 1 s−1. To overcome such an issue, we explored directly the fast isomerization phenomena by means of femtosecond transient absorption spectroscopy (FTAS).
We illuminated continuously a solution of 50 μM AZO1 in DMF with UV light (λ = 365 nm) and then irradiated with a pump pulse at 410 nm, which corresponds to the maximum of the static absorption of the trans-form (see Fig. 2b). In analogy with the TA spectra of other azobenzene molecules,49 the transient spectrum (see Fig. 5a) clearly showed features due to the trans-form of AZO1. In particular, we observed positive signals in the range of 450–750 nm (time decay of a few picoseconds) which could be assigned to the excited state absorption and a negative signal around 380 nm (hundreds of picoseconds time decay) due to the ground state bleaching. A further weak positive signal is observed at 345 nm corresponding to the photoinduced absorption from the vibration-excited cis-AZO1, which was produced by the pump irradiation of the sample. It is expected that this signal was cut on the high energy side by the edge of the white light which screened the actual intensity of the signal. Analogous experiments were performed on a solution of AZO1, this time in water (100 μM) (see Fig. 5b). Also, in this case, the transient measurements seem to indicate that the trans → cis isomerization did take place, even if the efficiency was lower than that in DMF, and the induced absorption signal of the cis product was less intense.
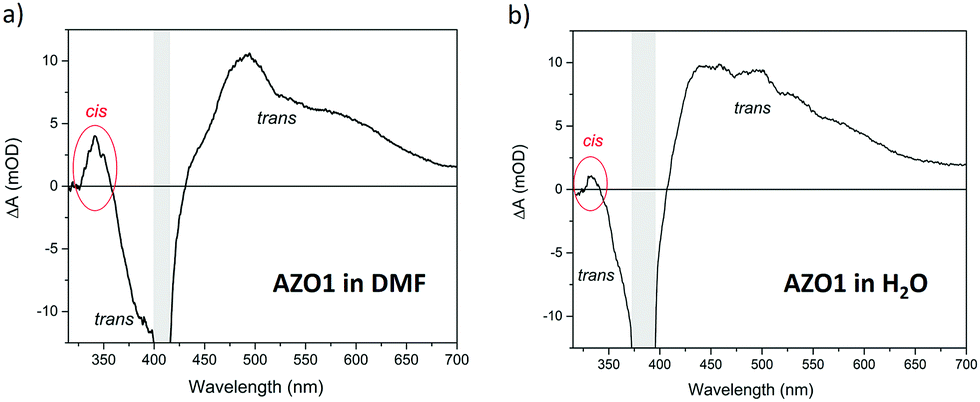 | ||
| Fig. 5 Transient spectra of AZO1 in (a) DMF and (b) H2O. The shaded areas show an intense negative feature due to the scattered pump radiation around 410 nm and 385 nm, respectively. The energy density is 280 μJ cm−2. The time delay between the pump and the probe is 0.65 ps. | ||
Although the kinetic rate was not directly measured (the estimated lower limit k± > 1 s−1), FTAS measurements clearly show the presence of both cis and trans isomers of AZO1 both in water and in DMF when illuminated with UV light confirming the photochromophore switching.
3.2 AZO photoisomerization in 2D nanostructures
In solution, molecules were isolated from each other and free to move (3 degrees of freedom of translational motion) and, as described above, the photoisomerization was mainly affected by the molecule–solvent interaction. Differently, when assembled in crystalline structures, azobenzenes were fixed (no translational motion) and the photoisomerization was often much less efficient due to the close packing of molecules that limits structural changes.50 We then explored an intermediate case where azobenzenes are constrained in 2D, forming demanding structures such as 2D assemblies created by depositing the molecules on a surface. In this case, molecules were not isolated and their possible switching was hindered due to the interplay of molecule–molecule and molecule–surface interactions.51We exploited thermodynamic effects to drive the self-assembly of azobenzene molecules to form micrometric islands, and we directly observed the photoswitching of the single aggregate by means of two scanning probe microscopy techniques: AFM and KPFM.
Techniques that allow the observation of molecular packing such as grazing-incidence small-angle X-ray scattering (GISAXS) and scanning tunneling microscopy (STM) show some limitations for the study of single micrometric molecular aggregates. In the case of the first technique, the measurement is the average of different islands since the measurement area is larger than that of the single object, while the second generally requires a conductive substrate.
Such issues can be overcome by using scanning probe microscopy. Although the AFM technique has no molecular resolution being typically <5 nm (>20 nm for KPFM), and therefore we cannot have direct information on the molecular packing, clear evidence on the arrangement could be provided by means of histogram analysis.35 Usually, a rough analysis of an AFM image is performed by studying a set of arbitrarily chosen profiles. Histogram analysis provided a complete description of the image in terms of 1D functions (i.e. frequency distribution of height, FDH) consisting of the sum of all traced profiles allowing us to calculate in a simple and direct way the thickness and roughness of azobenzene nano-aggregates.
We performed a comparative analysis of two azobenzene systems: AZO1 and AZO2 deposited on native silicon oxide under the same conditions (see Methods).
Since there were no thiol groups or moieties interacting with the substrate, we can neglect the interaction of azobenzenes with the superficial Si–OH sites of the substrate and point out the role of the molecule–molecule interaction. Moreover, due to the good wettability of the fresh cleaned substrates, molecules can easily move on the surface self-aggregating, without remarkable kinetic constraints, thanks to the slow evaporation rate of the solvent.
The KPFM technique has been successfully applied to study the in situ changes in the dipole moment in the monolayer,52–54 where the surface potential change is related to the dipole moment by the Helmholtz equation:55
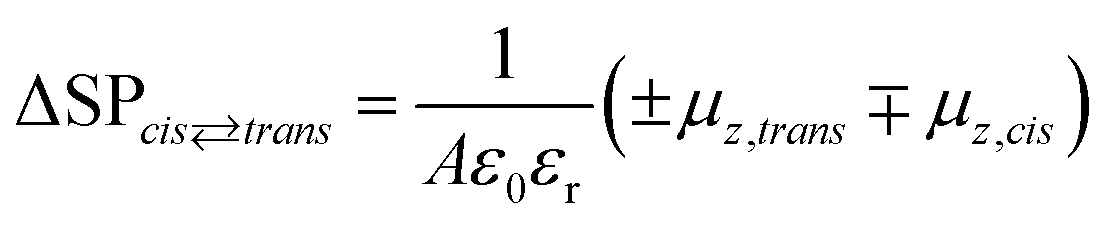 | (3) |
In the case of a disordered structure, eqn (3) has to be modified taking into account all the molecular orientations in the aggregate preventing a direct solution. In general, disordered arrays of molecules show an intrinsic value of the measured dipole moment lower than that of ordered structures due to the averaging over all orientations of the molecules.
Experimentally, the ΔSP value due to photoswitching was calculated as the difference in the surface potential measured by KPFM after UV irradiation at 365 nm for 60 min and after visible light irradiation to restore the trans-configuration:
| ΔSP = KPFMUV − KPFMvis. |
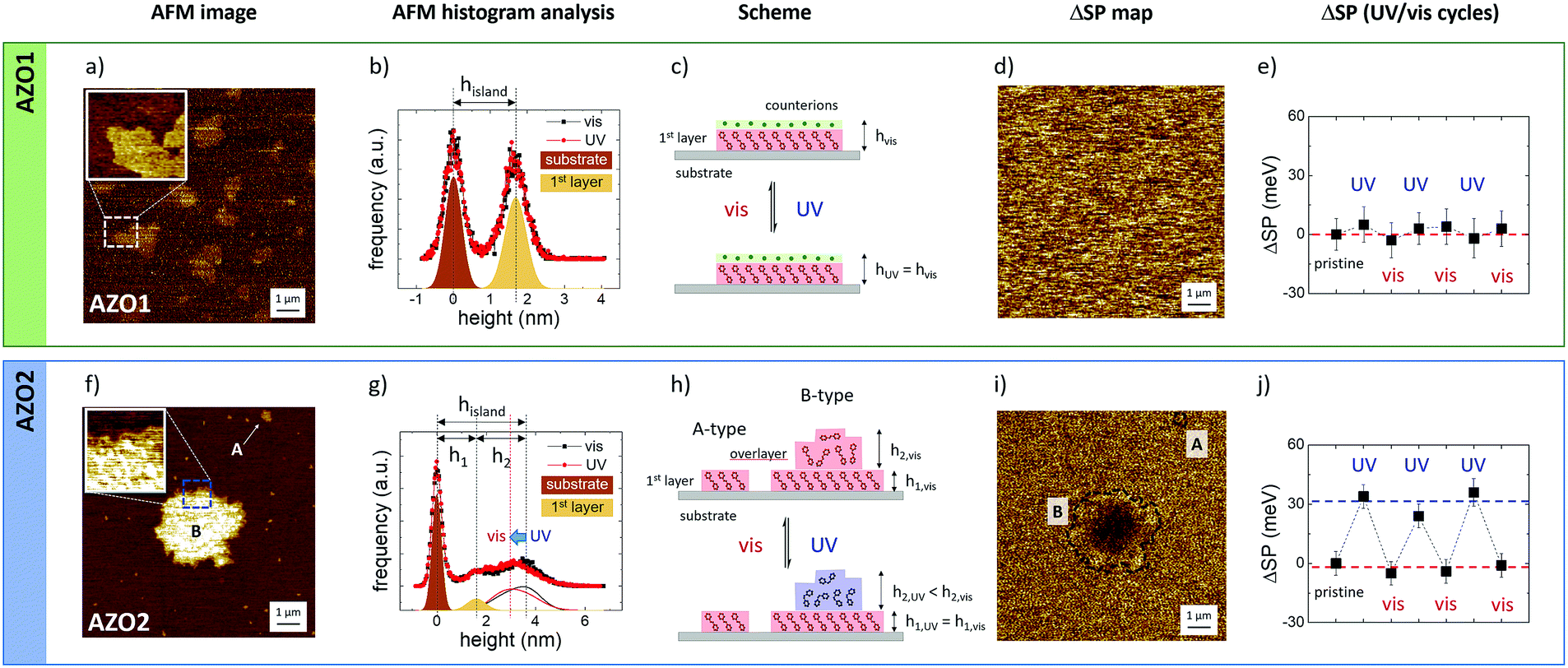 | ||
| Fig. 6 AFM morphology and the corresponding KPFM surface potential maps on (a and d) AZO1 and (f and i) AZO2 islands deposited on a silicon substrate. (b and g) Histogram analysis of the island edges reported in the two AFM image insets acquired under visible and UV-illumination. (c and h) Cartoon of the possible structure of the different islands before and after UV illumination. Time evolution of the surface potential, ΔSP, for (e) AZO1 and (i) AZO2, obtained by comparing the SP values measured in the central part of the islands, as acquired after UV irradiation at 365 nm for 60 min and after visible light illumination (436 nm) for further 60 minutes to restore the trans-configuration: ΔSP = KPFMUV − KPFMvis. Measurements were repeated for UV and visible light cycles. Z-Ranges: (a and f) AFM images = 6 nm and (d and i) ΔSP images = 30 mV. | ||
AZO1 is a polar salt, and the effective length of the azobenzene system is given by the molecular length (amounting to ≈ 1.4 nm in trans-conformation, as estimated by molecular mechanics) and the surrounding Na+ hydrated counter-ions with a diameter in solution of 0.72 nm (Fig. 6c). Thus, in analogy with previous work, we could assume that the uniform thickness is due to the arrangement of the AZO1 molecule in a tilted conformation surmounted by Na+ ions and a significant amount of water molecules, with the measurements being performed in air, as also suggested by the presence of the strip in the direction of the movement of the scanning tip in the AFM image.
The inset figure in Fig. 6a presents a high-resolution image of the island edge, marked in the main image. The corresponding FDH (Fig. 6b) reveals two main peaks corresponding to the substrate and the island where the thickness of the latter (hisland) is given by the distance between the two peaks commonly fitted with Gaussian curves. Moreover, the peak width is the root mean square roughness (RRMS) of the corresponding nanostructure. The RRMS value measured for the substrate peak amounts to 0.36 nm, which is in excellent agreement with the values reported in the literature,35 proving the cleanliness of the substrate and the absence of experimental artifacts. In general, the variance of RRMS values was in the scale of 0.01 nm. For the sake of simplicity, the RRMS values were reported with two significant digits without any error.
AFM measurements were performed in situ and no measurable variation was observed between the FDHs measured on single islands upon illumination at 365 nm for 60 min (red dots and line) and after visible light illumination at 436 nm (black dots and line) to restore the trans-configuration. The RRMS value of the AZO1 island did not vary when illuminated and amounts to 0.4 nm (see the ESI† for more details), clearly indicating that the molecules arrange themselves in ordered structures that are not modified by a light stimulus.
Similarly, no changes of surface potential could be observed by KPFM at the nanoscale: ΔSP = 1 ± 3 mV (Fig. 6d) and the vis/UV cycles were repeated three times (Fig. 6e).
In summary, the lack of switching measured in the AZO1 island was attributed to different elements or to their combination: (i) the presence of counter-ions and charges that act as an electrical double layer, thus significantly shielding the variations of the dipole moment medium of the island due to switching, and (ii) tight packing of the molecules that could hinder isomerization.
Thus, we performed the same measurement on a different molecule, AZO2 (Fig. 6f and i), having a significant dipole moment (14.08 D) with respect to the one of AZO1 (2.5 D).
In this case, we observed two different structures of AZO2 with different lateral sizes: (A) small islands with a lateral size of up to 300 nm and a uniform height of h1 = 1.5 ± 0.2 nm, comparable to the AZO2 length, and (B) large micrometric structures (up to 10 μm) with jagged edges and a height of h1 = 1.6 ± 0.2 nm at the edges and a higher thickness of up to 5 nm, in the central part of the island, as depicted in the magnified AFM image inset and the corresponding histogram analysis (Fig. 6g). These values were determined on a set of 97 A-type and 14 B-type islands, corresponding to a total of about 1000 μm2 of scanned surface area.
The height of A-type islands suggests the presence of a single layer of azobenzene. Conversely, B-type islands show multi-layered structures with a first more ordered layer, visible at the islands’ edge, surmounted by more disordered overlayers.
The histogram analysis revealed that A-type islands and the first layer of B-type islands show the same root mean square roughness, RRMS = 0.2 nm, and, similarly to the case of AZO1, we did not observe any difference when the samples were illuminated with visible or UV light. Conversely, the overlayers behaved differently as evinced by the FDH measured during visible (black) or UV illumination (red). Focusing on the overlayer contribution (continuous lines), we clearly observed a change in the shape of the curves and a shift in the peak position of ca. 0.7 nm. Taking into account the variation of the shape (FDH is a weight function; see eqn (S2), ESI†), we observed that the average height of the overlayer drops by 0.4 nm when the sample is illuminated with UV light.
These findings confirmed the good structural order of the molecules forming the 1st layer, while the AZO2 molecules were supported quite randomly in the overlayer.
In general, AZO2 shows a more complex self-assembly behavior than AZO1 due to the presence of two moieties (–NH2 and –NO2) that can bind to the substrate surface and interact differently with the solvent. Only in the case of DMF we could observe the reversible cis–trans switching at the nanoscale due to the changes in the KPFM signal. Fig. 6h shows the ΔSP map acquired on single islands of AZO2. While no significant modification of the topography was observed upon irradiation, a significant, reversible change of the surface potential, ΔSP = 30 ± 10 mV, was observed on single B-type islands. Instead, the potential of A-type structures showed no remarkable variation. The KPFM images acquired before and after illumination are presented in Fig. S6 (ESI†). Moreover, the reversible change of the surface potential was observed on the B-type islands in correspondence with the wrinkled overlayer, while no change was observed on the well-ordered first layer (or A-type islands), suggesting that the structural disorder prevents steric constraints and allows the conformation switching of AZO2 molecules.
The role of the partial structural disorder was further confirmed by the analysis of different self-assembled AZO2 structures obtained by using different solvents. The switching behavior of AZO2 could not be observed on well-ordered islands; KPFM measurements performed on different, fiber-like structures with flat surfaces, RRMS = 0.3 nm (Fig. S7 (ESI†), obtained by deposition from THF, did not show any observable switching.
The ΔSP measured on the AZO2 isolated islands is in agreement with previous measurements performed on continuous SAM layers,53,54 which, however, did not reach the single-island lateral resolution obtained here. This fact remarkably demonstrates that it is possible to measure the switching of small ensembles of molecules in real time. Assuming a typical island of 1 × 1 μm2 and an average footprint of a typical azobenzene molecule of ≈0.5 nm2 obtained from STM measurements,15 we could observe a clear and reversible switching of a small yet finite amount of molecules (≈106 molecules or a few attomoles). This result confirms the great potential of KPFM to measure the molecular changes in mesoscopic ensembles of molecules in real time, previously demonstrated on more disordered systems such as polymer chains or nanocrystals.42,44,56
3.3 AZO photoisomerization in 3D crystals
Finally, we tried to monitor the cis–trans switching in 3D-ordered structures. In general, coordinated switching of molecules in bulk solids is much more challenging than in solution or in nanostructures, due to the much stronger steric constraints, and the significant rearrangements needed to accommodate the new molecular packing; see, for example, the studies of Feringa et al.,57 Koshima et al.58 and Li et al.59We were able to assemble 3D mesoscopic crystals for both AZO1 and AZO2 molecules on silicon substrates using standard solution processing, as depicted in Fig. 7a and b. Both molecules assembled in large elongated crystals with typical dimensions of a length of a few tens of microns, a width of a few hundreds of nanometers and a thickness ranging between 50 and 500 nm. The crystallinity of the structures was also supported by AFM measurements monitoring two features: the stepped surface and the roughness of the shelf of the step. In the first case, the crystal surface showed pits and steps with a thickness of ≈1.6 nm (inset of Fig. 7a), comparable to the thickness of the AZO1 islands. The second feature indicated that the crystal surface is flat having a roughness comparable to that of the support: RRMS = 0.3 ± 0.1 nm.
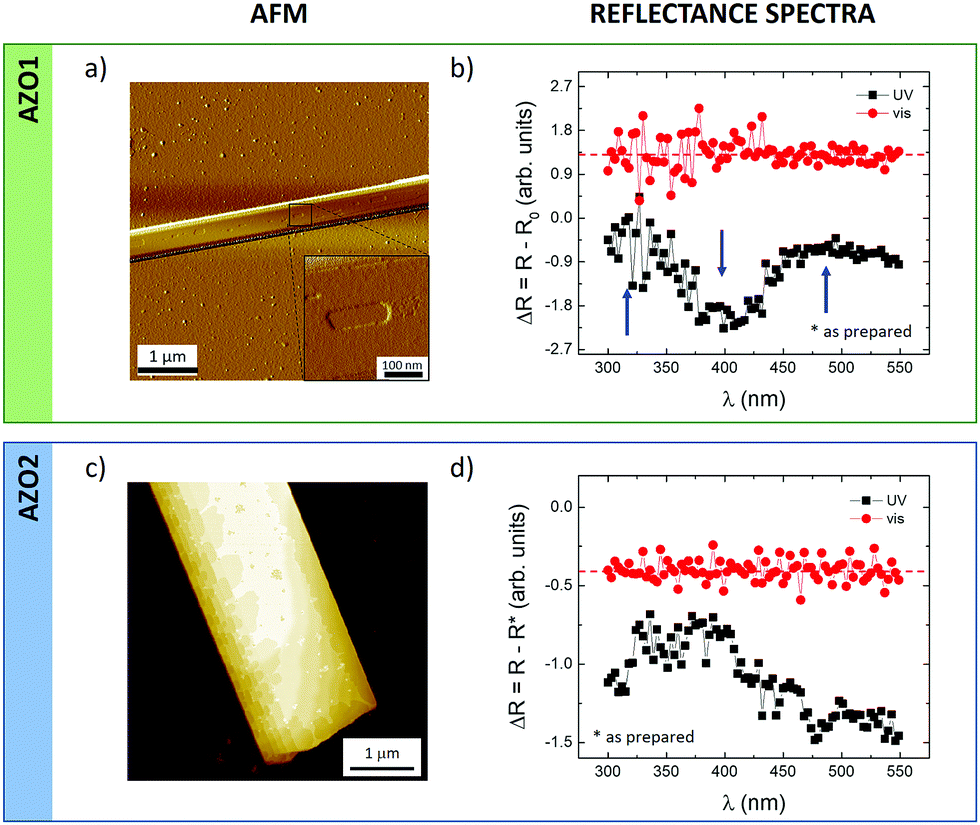 | ||
| Fig. 7 (a and c) AFM images of the corresponding (b and d) reflectance spectra acquired on mesoscopic AZO1 and AZO2 crystals self-assembled on silicon substrates (surface coverage <5%). (a) Phase AFM and (b) AFM morphology. Reflectance spectra, ΔR, given by the difference between the spectra measured after exposure to light and those obtained from non-irradiated samples (i.e. as prepared, R*); black curve, after 30 min exposure to UV light (λ = 365 nm, power density = 3.2 mW cm−2); red curve, after 30 min exposure to visible light (λ = 440 nm, power density = 0.56 mW cm−2). Z-Range: (a) arb. units and (c) 50 nm. | ||
KPFM measurements on the surface of the micrometric crystals were intrinsically better resolved than those in the case of nanostructures because of the lateral resolution. In particular, KPFM showed a small but significant change in the surface potential values measured before and after UV illumination: ΔSP was 5.8 ± 0.4 mV and 4.6 ± 0.4 mV for AZO1 and AZO2, respectively, confirming the low efficiency of cis–trans switching in 3D systems with respect to 2D islands or isolated molecules in solution. It is worth underlining that the measured ΔSP values agree with those achieved for 2D islands: AZO1 and the 1st layer of AZO2, where the potential resolution is ca. 10 meV.
To confirm the KPFM values, we also compared them with optical reflectometry measurements in which instead of measuring single islands the reflected light spectrum of the whole sample was measured. Fig. 7b and d present the reflectance spectra, ΔR, obtained after being irradiated with UV (black) and visible (red) light for both azobenzene molecules. All the spectra were obtained by subtracting the measurements collected from non-irradiated samples, which were previously stored in the dark (i.e. as prepared, R*), assuming that the film contained 100% trans azobenzene.
For both molecules, we observed a similar behavior in the overall reflectance signals measured after UV and light illumination. First, the samples were irradiated with UV light for 30 min and the reflectance spectra were recorded (black curve) showing a decrease close to 400 nm and an increase at ca. 325 and ca. 500 nm (marked with arrows for AZO1) corresponding to a partial conversion of trans- to cis isomerization, similarly to what has been observed previously.60,61 The following irradiation with visible light for 30 min revealed the opposite effect: the reflectance of the film (red line) became flat with irradiation time approaching the curve measured on non-irradiated samples, confirming the recovery of the cis-isomers to the trans-counterparts.
It should be noted that the reflectance spectra recorded on a reference film with no azobenzene showed no changes in the reflectance upon irradiation by UV or visible light (Fig. S8, ESI†), thus ruling out any artefact due to the optical changes of the substrate upon illumination.
Both microscopic KPFM and macroscopic reflectance measurements thus confirm that cis → trans switching in 3D crystals can be observed, but it is not very efficient in such 3D structures.
4. Conclusions
In this study, we measured the conformational switching of azobenzenes ranging from isolated molecules in liquid to attomolar-2D and macro-scale 3D self-assembled structures.Given the wide range of different environments, we used different complementary techniques. While optical steady-state spectroscopy is perfectly suited for conventional studies in solution, more refined techniques are needed to monitor switching, featuring very fast or very slow kinetics. Although the FTAS spectroscopy measurements fail to monitor switching, they are able to detect the presence of both the isomers also in the case of very fast cis → trans switching, confirming the photoisomerization of both the AZO molecules in all the tested liquid environments. Conversely, in the case of constrained 2D micro-systems, scanning probe microscopy techniques (AFM and KPFM) provide a unique way to observe collective switching for relatively small ensembles (a few attomoles, corresponding to ≈106 molecules) mapping the change of surface potential due to the dipole difference between cis- and trans-isomers.
When assembled in 2D and 3D aggregates, we evinced the role of structural disorder by means of a quantitative analysis of AFM images. Reversible photoswitching was observed only in the case of disordered aggregates, while ordered structures did not show remarkable variations.
This was confirmed by the measurements of ordered 3D structures where switching is hindered due to steric constraints, but could also be observed with some specific techniques, for example, macroscopic reflectometry measurements and, in best-case scenarios, microscopic techniques such as KPFM. The possibility to compare the switching ability and time scale in ensembles with tunable amounts of these molecules could allow us to better understand the effect of collective interactions to enhance and control their synergistic switching. In particular, the design of partially ordered molecular structures could allow a suitable way to better enhance their performance in, for example, solid-state devices and thin coatings for electronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the EC through the Marie Curie project ITN iSwitch (GA no. 642196), GrapheneCore2 (GA-785219), GrapheneCore 3 (GA-881603) projects – Graphene Flagship and the Swedish Research Council (under project Janus 2017-04456). Computational resources were provided by the Consortium des Équipements de Calcul Intensif (CÉCI) funded by the Belgian National Fund for Scientific Research (F. R. S.-FNRS) under Grant 2.5020.11. D.B. is a FNRS research director.Notes and references
- B. L. Feringa and W. R. Browne, Molecular switches, Wiley-VCH, Weinheim, Germany, 2nd completely rev. enl. edn, 2011 Search PubMed.
- E. Orgiu and P. Samori, Adv. Mater., 2014, 26, 1827–1845 CrossRef CAS PubMed.
- Y. Shiraishi, S. Sumiya and T. Hirai, Chem. Commun., 2011, 47, 4953–4955 RSC.
- B. Champagne, A. Plaquet, J. L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101–8103 CrossRef CAS PubMed.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- J.-P. Sauvage and V. Amendola, Molecular machines and motors, Springer, Berlin, New York, 2001 Search PubMed.
- H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175–182 CrossRef CAS.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS PubMed.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS PubMed.
- V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- M. Min, S. Seo, S. M. Lee and H. Lee, Adv. Mater., 2013, 25, 7045–7050 CrossRef CAS PubMed.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- I. K. Lednev, T. Q. Ye, P. Matousek, M. Towrie, P. Foggi, F. V. R. Neuwahl, S. Umapathy, R. E. Hester and J. N. Moore, Chem. Phys. Lett., 1998, 290, 68–74 CrossRef CAS.
- E. M. M. Tan, S. Amirjalayer, S. Smolarek, A. Vdovin, F. Zerbetto and W. J. Buma, Nat. Commun., 2015, 6, 5860 CrossRef PubMed.
- H. Dürr and H. Bouas-Laurent, Photochromism: molecules and systems, Elsevier, Amsterdam, Boston, Rev. edn, 2003 Search PubMed.
- H. Rau and E. Luddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 CrossRef CAS.
- T. Schultz, J. Quenneville, B. Levine, A. Toniolo, T. J. Martinez, S. Lochbrunner, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2003, 125, 8098–8099 CrossRef CAS PubMed.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, ChemPhysChem, 2010, 11, 1018–1028 CrossRef CAS PubMed.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS PubMed.
- T. Moldt, D. Przyrembel, M. Schulze, W. Bronsch, L. Boie, D. Brete, C. Gahl, R. Klajn, P. Tegeder and M. Weineltt, Langmuir, 2016, 32, 10795–10801 CrossRef CAS PubMed.
- H. Rau and Y. Q. Shen, J. Photochem. Photobiol., A, 1988, 42, 321–327 CrossRef CAS.
- H. T. Zhang, X. F. Guo, J. S. Hui, S. X. Hu, W. Xu and D. B. Zhu, Nano Lett., 2011, 11, 4939–4946 CrossRef CAS PubMed.
- V. Ferri, M. Elbing, G. Pace, M. D. Dickey, M. Zharnikov, P. Samori, M. Mayor and M. A. Rampi, Angew. Chem., Int. Ed., 2008, 47, 3407–3409 CrossRef CAS PubMed.
- D. Ishikawa, E. Ito, M. N. Han and M. Hara, Langmuir, 2013, 29, 4622–4631 CrossRef CAS PubMed.
- G. Pace, V. Ferri, C. Grave, M. Elbing, C. von Hanisch, M. Zharnikov, M. Mayor, M. A. Rampi and P. Samori, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9937–9942 CrossRef CAS PubMed.
- S. Das, P. V. Kamat, S. Padmaja, V. Au and S. A. Madison, J. Chem. Soc., Perkin Trans. 2, 1999, 1219–1223, 10.1039/a809720h.
- C. W. Chang, Y. C. Lu, T. T. Wang and E. W. G. Diau, J. Am. Chem. Soc., 2004, 126, 10109–10118 CrossRef CAS PubMed.
- C. Weber, T. Liebig, M. Gensler, A. Zykov, L. Pithan, J. P. Rabe, S. Hecht, D. Bleger and S. Kowarik, Sci. Rep., 2016, 6, 25605 CrossRef CAS PubMed.
- P. O'Keeffe, D. Catone, A. Paladini, F. Toschi, S. Turchini, L. Avaldi, F. Martelli, A. Agresti, S. Pesceteli, A. E. D. Castillo, F. Bonaccorso and A. Di Carlo, Nano Lett., 2019, 19, 684–691 CrossRef PubMed.
- F. Toschi, D. Catone, P. O’Keeffe, A. Paladini, S. Turchini, J. Dagar and T. M. Brown, Adv. Funct. Mater., 2018, 28, 1707126 CrossRef.
- A. Liscio, V. Palermo and P. Samori, Adv. Funct. Mater., 2008, 18, 907–914 CrossRef CAS.
- A. Liscio, V. Palermo and P. Samori, Acc. Chem. Res., 2010, 43, 541–550 CrossRef CAS PubMed.
- A. Liscio, ChemPhysChem, 2013, 14, 1283–1292 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- D. Bleger, J. Dokic, M. V. Peters, L. Grubert, P. Saalfrank and S. Hecht, J. Phys. Chem. B, 2011, 115, 9930–9940 CrossRef CAS PubMed.
- J. Dokic, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, GAUSSIAN 16 (Revision C.01), Gaussian Inc., Wallingford, CT, 2016.
- N. K. Joshi, M. Fuyuki and A. Wada, J. Phys. Chem. B, 2014, 118, 1891–1899 CrossRef CAS PubMed.
- T. Asano and T. Okada, J. Org. Chem., 1984, 49, 4387–4391 CrossRef CAS.
- L. Zhou, L. Chen, G. Ren, Z. Zhu, H. Zhao, H. Wang, W. Zhang and J. Han, Phys. Chem. Chem. Phys., 2018, 20, 27205–27213 RSC.
- C. S. Mali, S. D. Chavan, K. S. Kanse, A. C. Kumbharkhane and S. C. Mehrotra, Indian J. Pure Appl. Phys., 2007, 45, 476–481 CAS.
- D. A. G. Bruggeman, Ann. Phys., 1935, 416, 636–664 CrossRef.
- V. A. Markel, J. Opt. Soc. Am. A, 2016, 33, 1244–1256 CrossRef PubMed.
- P. W. Atkins and J. De Paula, Atkins' Physical Chemistry, Oxford University Press, Oxford, New York, 10th edn, 2014 Search PubMed.
- S. Loudwig and H. Bayley, J. Am. Chem. Soc., 2006, 128, 12404–12405 CrossRef CAS PubMed.
- J. Bahrenburg, K. Rottger, R. Siewertsen, F. Renth and F. Temps, Photochem. Photobiol. Sci., 2012, 11, 1210–1219 CrossRef CAS.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- V. Palermo, M. Palma and P. Samori, Adv. Mater., 2006, 18, 145–164 CrossRef CAS.
- B. Stiller, G. Knochenhauer, E. Markava, D. Gustina, I. Muzikante, P. Karageorgiev and L. Brehmer, Mater. Sci. Eng., C, 1999, 8–9, 385–389 CrossRef.
- S. Schuster, M. Füser, A. Asyuda, P. Cyganik, A. Terfort and M. Zharnikov, Phys. Chem. Chem. Phys., 2019, 21, 9098–9105 RSC.
- D. Schuhmann, J. Colloid Interface Sci., 1990, 134, 152–160 CrossRef CAS.
- A. Sihvola, Subsurf. Sens. Technol. Appl., 2000, 1, 393–415 CrossRef.
- J. W. Chen, F. K. C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS PubMed.
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS PubMed.
- Y. F. Wang, S. Chen, L. Qiu, K. Wang, H. T. Wang, G. P. Simon and D. Li, Adv. Funct. Mater., 2015, 25, 126–133 CrossRef CAS.
- M. Moniruzzaman, P. Christogianni and G. Kister, in Proceeding of 4th International Conference on Process Engineering and Advanced Materials, ed. M. A. Bustam, Z. Man, L. K. Keong, A. A. Hassankiadeh, Y. Y. Fong, M. Ayoub, M. Moniruzzaman and P. Mandal, 2016, vol. 148, pp. 114–121 Search PubMed.
- M. Moniruzzaman, C. J. Sabey and G. F. Fernando, Macromolecules, 2004, 37, 2572–2577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00740h |
| This journal is © the Owner Societies 2021 |