
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-consistent field modeling of mesomorphic phase changes of monoolein and phospholipids in response to additives†
N.
de Lange
 ,
J. M.
Kleijn
and
F. A. M.
Leermakers
*
,
J. M.
Kleijn
and
F. A. M.
Leermakers
*
Physical Chemistry & Soft Matter, Wageningen University and Research, Stippeneng 4, 6708 WE, Wageningen, The Netherlands. E-mail: frans.leermakers@wur.nl
First published on 17th June 2021
Abstract
Mapping the topological phase behaviour of lipids in aqueous solution is time consuming and finding the ideal lipid system for a desired application is often a matter of trial and error. Modelling techniques that can accurately predict the mesomorphic phase behaviour of lipid systems are therefore of paramount importance. Here, the self-consistent field theory of Scheutjens and Fleer (SF-SCF) in which a lattice refinement has been implemented, is used to scrutinize how various additives modify the self-assembled phase behaviour of monoolein (MO) and 1,2-dioleoyl-phosphatidylcholine (DOPC) lipids in water. The mesomorphic behaviour is inferred from trends in the mechanical properties of equilibrium lipid bilayers with increasing additive content. More specifically, we focus on the Helfrich parameters, that is, the mean and Gaussian bending rigidities (κ and ![[small kappa, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cb.gif) , respectively) supplemented with the spontaneous curvature of the monolayer (Jm0). We use previously established interaction parameters that position the unperturbed DOPC system in the lamellar Lα phase ( < 0, κ > 0 and Jm0 ≈ 0). Similar interaction parameters position the MO system firmly in a bicontinuous cubic phase ( > 0). In line with experimental data, a mixture of MO and DOPC tends to be in one of these two phases, depending on the mixing ratio. Moreover we find good correlations between predicted trends and experimental data concerning the phase changes of MO in response to a wide range of additives. These correlations give credibility to the use of SF-SCF modelling as a valuable tool to quickly explore the mesomorphic phase space of (phospho)lipid bilayer systems including additives.
, respectively) supplemented with the spontaneous curvature of the monolayer (Jm0). We use previously established interaction parameters that position the unperturbed DOPC system in the lamellar Lα phase ( < 0, κ > 0 and Jm0 ≈ 0). Similar interaction parameters position the MO system firmly in a bicontinuous cubic phase ( > 0). In line with experimental data, a mixture of MO and DOPC tends to be in one of these two phases, depending on the mixing ratio. Moreover we find good correlations between predicted trends and experimental data concerning the phase changes of MO in response to a wide range of additives. These correlations give credibility to the use of SF-SCF modelling as a valuable tool to quickly explore the mesomorphic phase space of (phospho)lipid bilayer systems including additives.
1 Introduction
The self-assembly of lipids in aqueous solutions yields a wide variety of structures of various shapes and morphologies. Relatively polar lipids can, for example, self-assemble into structures with a positive interfacial curvature, like spherical and cylindrical micelles. Phospholipids with their two hydrophobic tails typically form bilayers. Although this is not often emphasised, such bilayers can reside in various topological states. These may be lamellar, like the Lα phase, which dispersed in solution forms vesicles, or with saddle-shaped curvatures such as the L3 sponge phase, or cubic phases (QII) such as the primitive (P, Im3m), diamond (D, Pn3m) and gyroid (G, Ia3d) cubic phases. Lastly, very apolar lipids may exhibit large negative interfacial curvatures and form an inverted hexagonal phase (HII) or even inverted micelles. An overview of all types of lipid phases is readily available in literature.1,2The intriguing phenomenon of lipid self-assembly and the richness in resulting structures has garnered a lot of interest from academic researchers and companies alike, as the applications are vast. Lipid vesicles can be applied as drug delivery systems and bioreactors,3–6 whereas cubic mesophases can be used in other applications, such as biosensors and biofuel cells.7–9 Many different phases can also be found in nature. While the outer membrane of the cell and of various organelles exists in lamellar form for obvious reasons, the endoplasmic reticulum and inner membranes of mitochondria have been shown to contain cubic phase-like topologies as well.10 Additionally, saddle-shaped membranes arise during various cellular processes such as membrane fusion11 and occur in nuclear pores, the ‘holes’ in the double membrane of the nuclear envelope.12
While large protein complexes are associated with the formation of saddle-shaped membranes, for example for the formation of nuclear pores,13 the impact of the mechanical properties of the lipid bilayer itself is often overlooked. After all, if the Gaussian bending rigidity is strongly negative, the formation of bilayer handles could not occur. It is clear that the packing of lipids within the bilayer affects its mechanical properties, including its resistance to stretching and bending, and that these properties determine what shape the bilayer can adopt. In other words, each mesomorphic state of the bilayer, e.g. lamellar, sponge or cubic, has its own range of mechanical parameter values for which it is the stable configuration. Many questions such as what this range is, and what bilayer composition is required to obtain these specific properties still remain to be answered.
A theoretical model that is able to accurately model a lipid bilayer and predict the corresponding mechanical properties is greatly needed. Recently, we forwarded a quasi lattice-free Scheutjens–Fleer self-consistent field (SF-SCF) approach, which delivers this information for molecularly detailed models with unprecedented (numerical) accuracy.14 We applied this so-called lattice-refined method to model a phospholipid bilayer containing DOPC (dioleoyl phosphatidylcholine) in an aqueous solution and investigated the effects of several model parameter variations on the mechanical properties of the bilayer. We used the predicted trends to parametrize DOPC. The default parameter set that resulted from this study positions the DOPC bilayer to be, in accordance to experimental data, in a stable lamellar phase.
While the SF-SCF method is able to predict the mechanical properties of lipid bilayers, the theory continues to be under construction to better deliver on this promise and any validation of its predictions is welcomed to guide the development process. Therefore, predictions should be compared with experimental measurements.
In the current paper we follow how the mechanical parameters of model bilayers respond to the addition of a series of additives. In addition to DOPC, we model bilayers formed by the lipid monoolein (MO) because the response of the MO system to many additives is well documented in a recent review on this topic.15 Thus, we can correlate the obtained trends for the mechanical parameters directly to experimentally observed mesoscopic phase behaviour. The main idea behind this approach is not to just verify the obtained trends but to show that from these trends one can consistently predict the mesomorphic phase behaviour of lipids in response to different types of additives.
The self-assembly of MO is very different from that of DOPC. MO is one of the few lipids that readily forms QII mesophase bilayers, which makes this an ideal lipid in applications that utilize bicontinuous cubic phases such as membrane protein crystallization.16,17 In combination with additives, MO has shown a high versatility in the self-assembled topologies it can form.15 Addition of DOPC to MO, for example, causes the QII phase to swell after which it transitions into a lamellar bilayer phase.18 Apart from this lipid mixture, we have chosen to model both lipids in combination with a selection of additives. Criterion for this selection was that we could (as a first approximation) make use of the same parameter set so that no new uncalibrated parameters were needed. Fortunately we can do so and obtain additives that can direct the model systems (DOPC and MO) into different directions in phase space as we will see below. We have used models for additives aiming to represent ethanol, butanediol and t-butanol, three fatty acids (FA) with a C8, C12 and C16 tail, respectively, and a surfactant with a C12 tail and a polyethylene oxide headgroup (C12Em with m = 4, 5 or 10).
In this paper we will first review the membrane elasticity theory that links mechanical parameters to the corresponding topological state. This is followed by an introduction of the basic principles of the lattice-refined SF-SCF theory. For a detailed overview of the lattice-refined SF-SCF theory, we refer to our previous work. In the methods section, we sketch the routes used to determine the bending properties and structural properties of the bilayer and present the input specifications used in this work. These include the molecular architecture of the molecules and the default parameter set. In the results section we will first show a systematic survey into the effects of parameter variation for MO and the various additives to validate the chosen default parameters, as done previously for DOPC.14 Next, we will present results of the mechanical parameters of DOPC/MO mixed lipid bilayers, and of both DOPC and MO bilayers in combination with various additives. We will show how the results for MO correlate with the known phase behaviour of these lipids and subsequently discuss in turn how MO and the other additives influence the phase behaviour of DOPC. At the end of this article we discuss the direction future work may take us.
2 Linking mechanical parameters of bilayers to the corresponding topological state
The notion that mechanical parameters govern the physics of tensionless interfaces, such as membranes, stems from the work of Helfrich.19 He introduced the now famous Helfrich equation which expands the interfacial tension γ (grand potential per unit area) of a membrane as a function of the mean curvature and Gaussian curvature
and Gaussian curvature 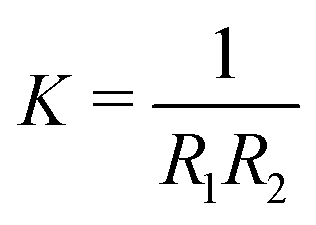 in a grand canonical ensemble. Here, R1 and R2 are the two principal curvatures that characterize the shape of the membrane.
in a grand canonical ensemble. Here, R1 and R2 are the two principal curvatures that characterize the shape of the membrane.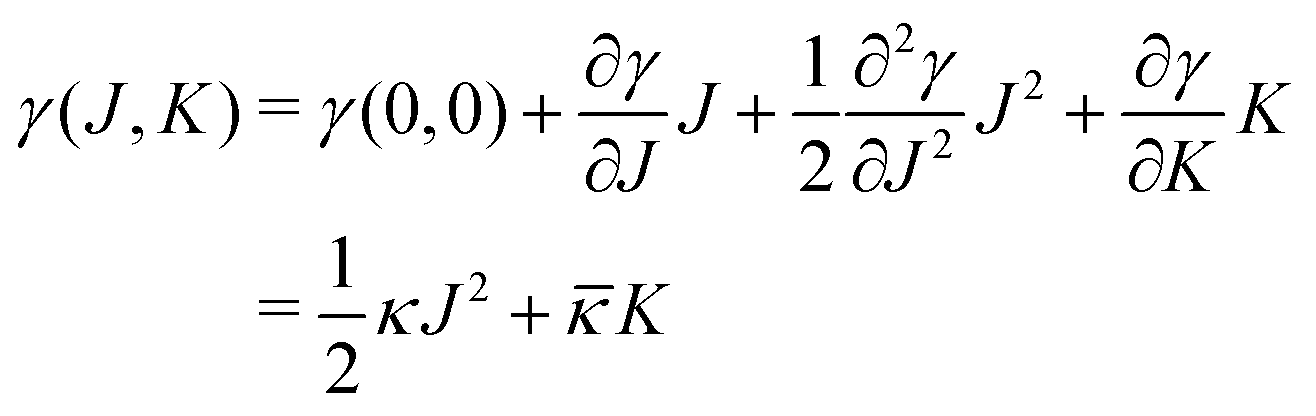 | (1) |
≡ ∂γ/∂K).
Of the introduced mechanical parameters, the spontaneous curvature of the monolayer and the Gaussian bending rigidity are of particular interest, as these control the topological (in)stability of the bilayer.14 For lipids to form bilayers, the value of Jm0, should be close to 0, i.e. indicating that the single leaflet of lipids does not have a strong preferred curvature. As such the interpretation of Jm0 conveniently follows the ideas behind the well-known (phenomenological) critical packing parameter P = v/la0 (with v the tail's volume per molecule, a0 the area of the lipid molecule at the core–corona interface and l the (average) length of the lipid tails).20 Clearly, Jm0 = 0 correlates with a packing parameter P = 1. In the limit where Jm0 ≈ 0, i.e. with the bilayer as the favourable state, governs the topology of the bilayers. More specifically, the phase transition between bilayers without saddle-shaped curvatures (vesicles, planar bilayers) to bilayers with saddle-shaped curvatures (sponge phases and cubic phases) are linked to the sign-switch of from negative to positive values.21,22
The mean bending modulus κ determines the membrane persistence length lm of the bilayer, which for example controls the size of thermodynamic stable vesicles or the inter-bilayer undulation repulsion, but has little to do with the topological state of the bilayer. Its value is necessarily positive, because negative values for this quantity would imply that the planar bilayer is at a maximum of the interfacial free energy.
As mentioned, using SCF theory it is possible to numerically accurately predict the mechanical properties of lipid bilayers, however for a correct parameterisation of the system, predictions should be compared and verified with existing experimental data. Unfortunately, while numerous experimental methods exist to determine κ,23–32 experimental methods to estimate or Jm0 are scarce. A quantitative indication of can only be obtained by carefully investigating membrane processes that involve topology changes, such as membrane fusion or fission.33 As such, only a handful experimental results11,34,35 are known to us and their accuracy is (debatable) uncertain, also because of a lack of molecularly detailed theoretical guidance. To date the only reliable experimental feedback to substantiate model predictions is the established topological phase behaviour of these lipids. The relation between theoretical predictions of mechanical properties of bilayers and their experimental phase behaviour is however only qualitative and consequently rather coarse.
3 Basic principles of lattice-refined SF-SCF theory
Computer simulations are the tool per excellence to find structural information on molecularly complex assemblies such as the lipid bilayer membrane. All we want to know on the nanometre length scale and the nanosecond time scale can be obtained from molecular dynamics (MD) simulations. However, to unravel lipid phase behaviour requires very long simulation times and extremely large system sizes and to date it seems not possible to execute a systematic MD study in this direction, even for the most coarse-grained MD approaches such as the MARTINI model.36 As usual in such situations one turns to approximate modelling methods. Invariably these routes make use of mean-field approximations. This precisely is the case for the method used in this paper, which is an extension of the self-consistent field theory of Scheutjens and Fleer.37,38 The key point of the mean-field route is that it is ‘free-energy’ based and as such it is delivering both structural and thermodynamic/mechanical parameters, and it is doing so at a (tiny) fraction of the MD computation time.The method of Scheutjens and Fleer extends mean-field approaches such as the regular solution and Flory–Huggins theory by allowing for concentration gradients. The molecules in the system are assumed to be composed of strings of equally sized segments. The free energy functional, which is written in terms of volume fraction (density) profiles and corresponding potential profiles for each segment type, is evaluated on a grid of lattice sites. The usually adopted chain model requires that the segments fit on the lattice sites and that neighbouring segments in the molecules occupy neighbouring sites on the lattice. In such a discretised world the account of conformational degrees of freedom for the chain molecules is facilitated. Accounting for fully self-avoiding chains would be ideal, but is computationally (still) extremely expensive. That is why a freely-jointed chain (FJC) model is adopted, which ignores the positional correlations along the chain that are more than two segments apart. The chain backfolding which in this approach is not excluded, is counteracted by an incompressibility constraint. Hence only on average backfolding is forbidden as each lattice site is on average filled exactly once. The ‘averaging’-range is implemented in layers of lattice sites.
The classical approach of Scheutjens and Fleer uses one length scale to discretise both the space and the molecules, that is, the segments of the molecules fit exactly on the lattice sites. Such an approach works well as long as the gradients in density are not too sharp. However, for a typical oil–water interface the interfacial width is comparable to the size of a water molecule and such coarse approach leads to so-called lattice artifacts. To alleviate these we recently implemented a lattice refinement,14 making the grid (lattice) size l smaller than the segment size b. The lattice-refinement value b/l takes integer values larger than 1. Typically the segment size is the unit length in the calculations and thus the lattice sites are half or a third, etc. of the size of a segment. The FJC model now can position neighbouring segments onto a larger set of nearby lattice sites and this leads of course to a model wherein the chains have a somewhat higher conformational entropy compared to the classical SF-SCF model.
Within the FJC model, even in the lattice-refined version, it is rather inexpensive to compute the statistical weight of all possible and allowed conformations of the chain molecules, but it requires the knowledge of so-called segment potentials uX(r) (where X is a segment type). Through these potentials the segments ‘feel’ each other: they represent the work per segment required to bring a segment X from the bulk to the coordinate r = {x,y,z}. Interactions in these potentials are parameterized by Flory–Huggins interaction parameters and contacts are evaluated using the Bragg–Williams mean-field approximation.39 The segment potentials serve as Boltzmann-like statistical weights GX(r). For a given conformation c we can add up all corresponding segment potentials and evaluate the statistical weight Gc. The sum of the statistical weights over all conformations are collected in so-called single molecule partition functions qi (i refers to a molecule type). This seemingly complex procedure is for the FJC model straightforwardly implemented in the propagator formalism.40 Knowing all statistical weights also allows the evaluation of relevant volume fraction profiles.
It turns out that one can compute the segment potentials only once the segment volume fractions are available. Hence, the segment potentials and the segment volume fractions mutually depend on each other. Fortunately this problem can be solved routinely using a numerical iteration scheme. The result of such a solution is referred to as the self-consistent field (SCF) result. The SCF solution is characterized by volume fraction profiles that both determine the potentials and follow from these and vice versa for the potentials.41
4 Methods
4.1 Data analysis
The calculation of both mechanical and structural parameters that characterise the bilayers is performed in the same way as in our previous article.14 In short, the lattice-refined SF-SCF machinery provides various types of outputs. First of all there are the volume fraction profiles per molecule type φi(r) as well as per segment type φX(r). Based on these volume fraction profiles, various structural properties can be calculated. Next to the volume fraction profiles, we can evaluate the mean-field free energy F, which can be expressed in terms of the set of volume fraction and segment potential profiles. Moreover, we can extract the so-called grand potential , where μi is the chemical potential of component i and ni is the number of molecules of type i in the system. Further, the grand potential can be written as a sum (integral) over the grand potential densities
, where μi is the chemical potential of component i and ni is the number of molecules of type i in the system. Further, the grand potential can be written as a sum (integral) over the grand potential densities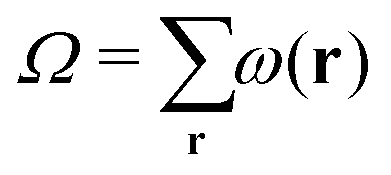 | (2) |
 | (3) |
 | (4) |
 | (5) |
Eqn (3) shows that the membrane tension follows from the zeroth moment over the (planar) grand potential density profile. In the SCF machinery the number of lipids per unit area will always be adjusted such that γ = 0 to a good approximation. Obviously, in order for the tension to vanish the grand potential density profile must have locally positive and negative contributions. Typically, negative contributions are connected to ‘stopping’ forces for membrane formation (chain stretching in the tail region and headgroup repulsion in the corona). The most important positive contribution, i.e. the main driving force for membrane self-assembly, is due to the ‘tension’ along the core–corona interface where the hydrophobic tails come in contact with the solvent. For phospholipid bilayers, this occurs in the region of the glycerol backbone of the lipid.
Eqn (4) encompasses the first moment over the grand potential density profile. The integral starts at the symmetry plane of the bilayer z = 0 and extends over just one of the leaflets and provides insight in the spontaneous curvature of the monolayer provided that the mean bending modulus κ = 2κm is known. Eqn (5) shows that the Gaussian bending rigidity is found by the second moment over the full grand potential density profile of the planar interface. When comparing eqn (4) and (5), the negative sign in eqn (4) and the fact that κ > 0 indicates that Jm0 tends to become negative when tends to become positive and vice versa. However, because of the different weighting (first moment vs. second moment) a sign switch of and Jm0 is not completely synchronized.
As is clear from eqn (4), the grand potential density profile does not give a direct evaluation of κ or Jm0. As such, we invariably have to involve the use of different geometries in order to compute these values. An important prerequisite for the comparison between different geometries is that the chemical potential of the components, i.e. lipids, water (and possible additives), are equal to that of the (tensionless) planar bilayer. In other words, comparing mechanical parameters within different geometries can only be done in the grand canonical ensemble. We can do so by evaluating the spherical vesicle. Here, the overall curvature energy of a spherical vesicle (Ωv) can be written as a function of κ and using the Helfrich equation (eqn (1)):
 | (6) |
as found by eqn (5) to extract the value for κ in eqn (6), and subsequently extract Jm0 with eqn (4).
In the classical approach, that is, in the SCF method before the lattice refinement implementation was available, discretisation artifacts prevented the evaluation of the grand potential density profiles for the tensionless planar bilayer. Instead, the only route available made use of the cylindrical geometry, despite the fact that lipids in cylindrical vesicles with a finite radius R do not have the same chemical potential as lipids in the corresponding planar or spherical bilayer.45,46 In retrospect the error that was introduced by using the cylindrical geometry was found to be sufficiently small so that results generated by the method of Pera et al.45 are indeed trustworthy. Older predictions however systematically overestimated the mean bending modulus and underestimated the Gaussian bending rigidity.47–49
 | (7) |
 | (8) |
 , i.e. the excess amount of segments of the lipid molecule per unit area (per leaflet) of the membrane, and N is the number of segments in the lipid molecule (measure for the molar volume).
, i.e. the excess amount of segments of the lipid molecule per unit area (per leaflet) of the membrane, and N is the number of segments in the lipid molecule (measure for the molar volume).
 | ||
| Fig. 1 Schematic overview of the molecular architectures used in this work. All specified united atoms (segments) have equal volume. We use two lipids: DOPC (without explicit charges) and MO. Both lipids have a hydrophobic tail of 18 carbons (lt = 18). We chose a single segment type for the phosphate group of DOPC and distinguish the carbon atoms surrounding the nitrogen, which are made more hydrophilic than the other carbon atoms. The segments of the glycerol group of MO are distinguished from those of DOPC and are made more hydrophilic. Apart from the two lipids, we use various additives such as solvents (ethanol, t-butanol and butanediol), fatty acids and C12Em surfactants. The main solvent in our system is always water, which in this model (and as in our previous study) consists of five equal monomers arranged in a configuration wherein one W is surrounded by four neighbouring W segments. For the additives we use the same χ values for all O groups, similar to CMO. We differentiate two χ values for the C groups of the additives. One for the tails, similar to C, and one representing the headgroups, comparable to CMO. | ||
4.2 Input parameters
The input required for SF-SCF modelling of lipid bilayers includes of course the molecular architectures of the lipids, the additives as well as the solvent(s). Secondly we need to specify all interaction parameters. Thirdly the method requires specification of the lattice geometry. In the lattice-refined version of course also a value for the discretisation b/l is needed.Calculations start with two-component systems where a lipid (by convention i = 1) in excess water (i = 0) forms a bilayer. Water normally forms an associative hydrogen-bonded network with other water molecules. This effectively prevents water to penetrate the bilayer core, which therefore can be regarded as ‘dry’. By modelling water as a small star-shaped cluster of five water segments, we can mimic this feature in first order. In our quest to understand how additives (i = 2) modify the bilayer's mechanical characteristics we introduce different molecules in the two-component systems: ethanol, t-butanol and butanediol; three fatty acids with different tail length (C8, C12 and C16); and C12Em surfactants with different amounts of ethylene oxide units (m = 4, 5 and 10). Apart from the longer tail carbon groups, we estimate the carbon and oxygen monomers of these additives to be relatively in the same order of hydrophilicity as the glycerol group of MO and have therefore modeled them with the same segments.
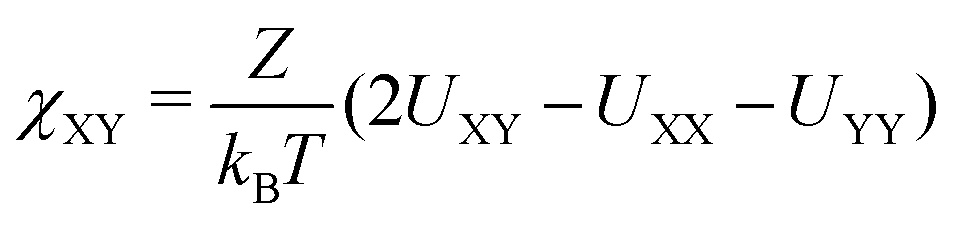 , where Z is the co-called lattice coordinate number. As soon as the average of the ‘like’-contacts (UXX + UYY)/2 is more favourable (more negative) than the unlike contacts UXY, we have a positive χ and this signals the tendency to cluster segments of the same type, leading to phase separation. Of course whether or not this happens depends also on the (mixing) entropy in the system. By definition the Flory–Huggins interaction parameter between similar segments is zero, i.e. χXX = 0. The χ between different segments in general may deviate from 0, i.e. have an effective repulsive potential (χXY > 0) or an attractive one (χXY < 0). An overview of the default interaction parameters used in this work can be found in Table 1.
, where Z is the co-called lattice coordinate number. As soon as the average of the ‘like’-contacts (UXX + UYY)/2 is more favourable (more negative) than the unlike contacts UXY, we have a positive χ and this signals the tendency to cluster segments of the same type, leading to phase separation. Of course whether or not this happens depends also on the (mixing) entropy in the system. By definition the Flory–Huggins interaction parameter between similar segments is zero, i.e. χXX = 0. The χ between different segments in general may deviate from 0, i.e. have an effective repulsive potential (χXY > 0) or an attractive one (χXY < 0). An overview of the default interaction parameters used in this work can be found in Table 1.
| W | OMO | CMO | CH | N | P | O | |
|---|---|---|---|---|---|---|---|
| C/CG | 1.2 | 2 | 0 | 0.5 | 2 | 2 | 2 |
| O | −0.2 | 0 | 2 | 1 | 0 | 0 | |
| P | −0.2 | 0 | 2 | 1 | −0.5 | ||
| N | −0.2 | 0 | 2 | 1 | |||
| CH | 0.6 | 1 | 0.5 | ||||
| CMO | 1.0 | 2 | |||||
| OMO | −0.5 |
The hydrophobicity of the lipid tails is represented by a higher interaction parameters of tail segments with water compared to glycerol and headgroup segments.
Previously14 the default interaction parameters were chosen such that SCF calculations on the DOPC–water system resulted in a bilayer with structural and mechanical characteristics in agreement with experimental data (e.g. a lamellar topology, and a relative flat headgroup orientation). We use an identical parameter set for DOPC in this paper and for a detailed explanation refer to our previous paper.14
One would expect that MO could simply be modelled with the same parameter set as for DOPC, since MO also combines a hydrophobic tail with a ‘glycerol’-like headgroup. However, it was found important to tune the parameters in such a way that the physics of the MO bilayer is in accordance with experimental data. A short survey how the mechanical parameters of the MO bilayer depends on some of the interaction parameters is given in the first part of the results section below. More specifically, we have varied the parameters for the glycerol moiety to find out how to provide the MO bilayer with a slightly positive Gaussian bending rigidity. The default parameters that we ended up with differ slightly from the interaction parameters used for DOPC. While the interaction parameter of the tails with water is kept the same (χC–W = 1.2), the glycerol segments are slightly more hydrophilic (χCMO–W = 1 and χOMO–W = −0.5, compared to (χCG–W = 1.2 and χO–W = −0.2 for DOPC. We can rationalize this as the glycerol backbone of MO contains two OH groups, and is therefore able to form hydrogen bonds with water.
All our ‘additives’ are molecules that combine C with O segments, each with their own specific interaction parameters. However, it is not practical to tune these for each of these additives individually. We thus decided to use the MO-parameterisation for all other additives. Hence long hydrocarbon stretches (e.g. the fatty acids tails) are taken as hydrophobic as the tail of MO (or the tails of DOPC) and the interaction parameters for hydrocarbons near O groups are as for the glycerol moiety of MO. For the oxygen groups of the additives we used the same interaction parameter values as for the glycerol groups of MO, i.e. χCMO–W = 1 and χOMO–W = −0.5.
As already mentioned, we can only compute all bending rigidities when using two different geometries, the planar bilayer and a spherical vesicle, for which we use one-gradient planar and spherical coordinate systems, respectively. In the planar coordinate system all quantities are evaluated per unit area (per lattice site). Here the r-coordinate as used in eqn (2) is implemented as a z-coordinate (normalised by the segment size, b): z = 1,2,…,Mz. The number of lattice sites (L) at each coordinate z for this system is independent of z. On both sides of the system we implement reflecting (mirror-like) boundary conditions. In the simulation volume there is just a single lipid layer near the lower boundary; this layer interacts with its mirror image forming a symmetric bilayer.50 We choose an appropriate number of molecules per unit area ni and initiate the iterations by introducing a guess for the bilayer density (or potentials) near the lower boundary. After the SCF solution is found we evaluate the interfacial tension from ω(z) using eqn (3).
In general this tension will not be zero and therefore we choose a new value for the number of (lipid) molecules per unit area n1 for the membrane constituents and a next loop of the iteration process is performed. The successive adjustment of the number of lipids per unit area continues until the membrane is free of tension (seven or more significant digits can routinely be reached). From this result the grand potential density profile ω0(z) is recorded (here the sub index 0 refers to the planar tensionless case). With this result we can evaluate eqn (4) and (5).
In the spherical geometry case we have a radial coordinate r = 1,2,…,Mr (in units b) where each layer r has L(r) ∝r2 lattice sites. For a spherically shaped bilayer (vesicle) with a radius R (typically R = 100b is used) we fix the number of lipid molecules to n1 = 4πR2n0lipid, with n0lipid the number of lipid molecules per unit area in the planar bilayer. After an initial guess for the segment density or potential profile near r = R, the SCF iterations are resumed. Importantly, during these iterations the distribution of the additives is normalised using the bulk volume fractions φb2 identical to the ones found in the planar tensionless bilayer calculation. During the calculations the bilayer positions itself optimally in the spherical coordinate system such that for the converged SCF solution the grand potential 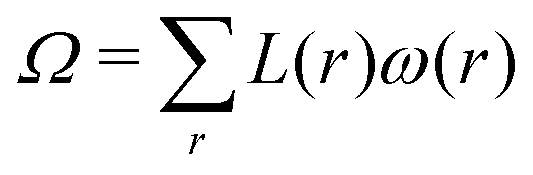 of the vesicle only contains curvature energy. This means that Ω = 4π(2κ + ).19,46 Using this result in combination with eqn (4) and (5) leads to the mechanical parameters. Importantly, the lipid component (i = 1) in the spherical vesicle system has the same bulk volume fraction as in the planar tensionless bilayer case. The same is true for the additive (enforced by the normalisation) and then necessarily the solvent also has a chemical potential that corresponds to the value found for the planar bilayer system (Gibbs–Duhem relation). Hence the vesicle system is exactly in the same thermodynamic state as the tensionless planar bilayer, that is, all components in the two systems have the same chemical potentials, and thus the two systems are in equilibrium.
of the vesicle only contains curvature energy. This means that Ω = 4π(2κ + ).19,46 Using this result in combination with eqn (4) and (5) leads to the mechanical parameters. Importantly, the lipid component (i = 1) in the spherical vesicle system has the same bulk volume fraction as in the planar tensionless bilayer case. The same is true for the additive (enforced by the normalisation) and then necessarily the solvent also has a chemical potential that corresponds to the value found for the planar bilayer system (Gibbs–Duhem relation). Hence the vesicle system is exactly in the same thermodynamic state as the tensionless planar bilayer, that is, all components in the two systems have the same chemical potentials, and thus the two systems are in equilibrium.
4.3 Limits to the additive-to-lipid ratio
There are limitations to the maximum amount of additives that can be included in the bilayers. In some cases these limitations are set by the system itself. For example, when hydrophobic additives are introduced in the MO bilayers, the system tends to go to inverted phases. It then may become impossible to find the tensionless state of the planar bilayer and this obviously frustrates the protocol to find the mechanical parameters. We therefore have set our own limits in such a way that the concentration of additives in the systems always is below their bulk binodal value. This does not pose major restrictions, because our primary interest is in predicting (initial) trends in how additives push a membrane system towards potential phase changes, rather than to pinpoint a critical composition for such a mesomorphic transition or a concentration at which the membrane falls apart (something which happens for example when large amounts of Triton are used). For surfactant-like additives their critical micelle concentration (CMC) is a threshold that we will not pass. In most cases we estimate the bulk binodal value of the additive from adsorption isotherms, that is the amount of additive that is absorbed in the membrane per unit area θσ as a function of its bulk concentration. The maximum amount of additives used does not depart far from the Henri regime, that is the initial linear part of the adsorption isotherm; see the ESI,† for more information. Important to note is that we do not take into account any lateral phase separation (i.e. lipid rafts) that could occur when working with lipid mixtures. However, as we keep the additive as a minority component in the membrane, we do not expect lateral phase separation to occur.5 Results and discussion
In the following we will first consider how the mechanical properties of the MO bilayer vary upon (small) changes in the parameters that characterise the way water interacts with the glycerol-like headgroup. This part of the work has been performed to obtain the default parameter set presented above. After this we will present, for illustration purposes, some structural properties of equilibrium DOPC and MO bilayers and a DOPC/MO mixed bilayer as these follow from using the default parameter setting. In the second part of the results section we will elaborate on how a mixed bilayer composed of DOPC and MO has some intermediate values for its mechanical parameters, κ and Jm0). In the third and final topic of the results section we will show how DOPC and MO bilayers respond to additives. Furthermore, we correlate the results of MO with published experimental phase behaviour, and discuss the biological relevance of the response of similar additives on DOPC bilayers.
5.1 Parametric study for MO bilayers
Our first priority was to properly model MO bilayers and find a suitable parameter set in line with its mesomorphic phase behaviour. Variation of the interaction parameters for the glycerol headgroup causes significant changes in both structural and mechanical parameters of MO bilayers, see Fig. 2. Fig. 2A and B show a decrease in bilayer (core) thickness (dOMOOMO) and an increase in the area per lipid (A0) with increasing hydrophilicity of the glycerol backbone (i.e. with more negative χCG–MO–W and χOMO–W). Such effects are in line with expectation and it is reassuring to see that the results are sensitive to these parameters.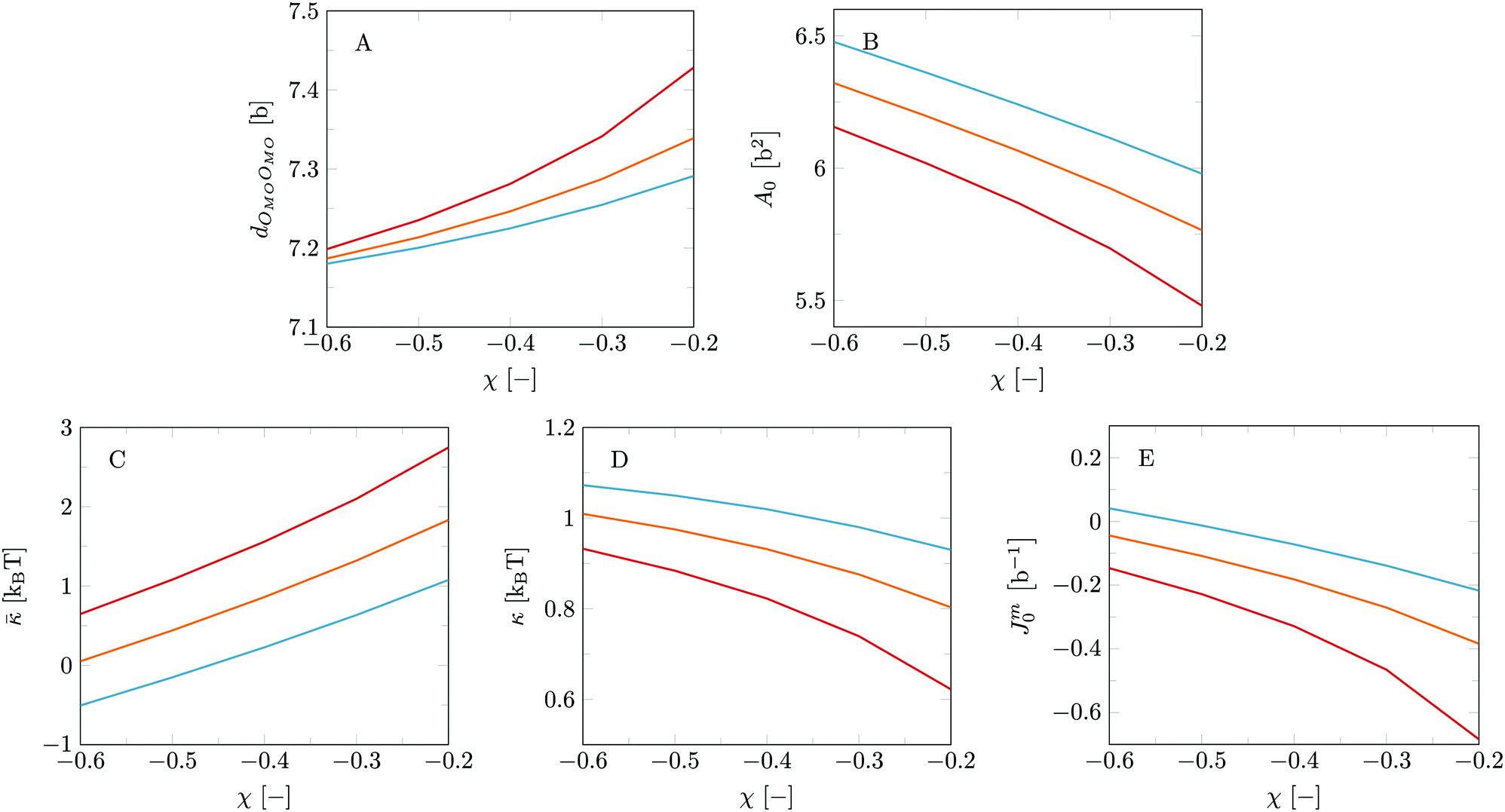 | ||
Fig. 2 Structural and mechanical properties as a function of χOMO–W for MO bilayers with different χCG–MO–W. (A) dOMOOMO, representing the bilayer core thickness; (B) A0, representing the area per lipid in the bilayer; (C) ; (D) κ; (E) Jm0. Red: χCG–MO–W = 1.2; orange: 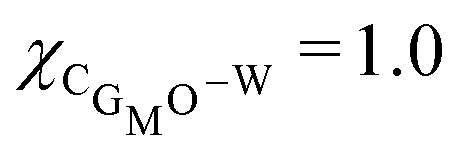 ; blue: χCG–MO–W = 0.8. ; blue: χCG–MO–W = 0.8. | ||
For the bending rigidities (Fig. 2C–E) we observe a decrease in and increases in κ and Jm0, respectively with increasing glycerol backbone hydrophilicity. These trends are relatively similar as found when changing the hydrophobicity of the glycerol backbone of DOPC.14 For an explanation of these trends we refer to this previous work; here, we focus on obtaining the interaction parameter set for MO that is in accordance experimental data.
As mentioned already it is well known that pure MO bilayers form cubic phases.15 This topological state limits the window for and Jm0. To be precise, the value of is expected to be positive, and the value of Jm0 slightly negative, but relatively close to zero as we obtain bilayers rather than (inverted) micelles. The value of κ is relatively insignificant for cubic phases as the mean curvature in these structures is not expected to be large (〈J〉 ≈ 0). Based on this information, we chose χCG–MO–W = 1.0 and χOMO–W = −0.5 as the interaction parameters for MO, as these values result into bilayers with and Jm0 in the expected range. As in this setting the Gaussian bending rigidity is not extremely far from zero, we may anticipate phase changes towards the lamellar phase in response to additives that induce a more positive preferential curvature of the leaflets of the bilayer.
For the interaction parameters chosen, we find κ to be close to 1kBT, dOMOOMO ≈ 7.2b and A0 ≈ 6.2b2, corresponding to 2.52 nm and 0.76 nm2 respectively. In our previous study on DOPC bilayers14 we found that the current model tends to underestimate the value for κ at least when we may trust corresponding experimental estimates. First of all it must be clear that the mean field predictions for the mean bending modulus are intrinsically on the low side because it underestimates the effects of excluded-volume correlations between densely packed neighbouring lipids, in particular when the systems are not too far from the gel-to-liquid phase transition. On top of this, the current lattice-refined approach further overestimates the chain flexibility, which leads to an additional softening of the bilayers compared to the classical predictions. One might think that also the neglect of electrostatic effects could have contributed to the fairly low values of the bending rigidity. However, in our previous paper14 we showed that at least for systems close to physiological conditions, this neglect was of minor importance. In line with these previous results we anticipate that κ found for the MO bilayers is also a lower estimate. In the same token we expect the membrane thickness of MO-bilayers, here estimated as twice the average position of the O's from the bilayer center dOMOOMO, to be an underestimation of the real value and correspondingly the area per lipid may be too high.51,52 The underestimation of the mean bending modulus κ is unfortunate. Typically we would like to argue that when the value is of order unity, regularly ordered bicontinuous cubic phases should give way to less ordered L3 phases. We still expect that when κ is sufficiently small that the L3 phase is preferred (that is when > 0) but that the threshold for such transition in the mean-field ‘world’ happens at values of κ lower than unity.
5.2 Structural and mechanical properties of DOPC and MO bilayers and mixtures thereof
With the parameter set in place a logical next step is to consider bilayers composed of more than one type of lipid. The phase behaviour of MO with added DOPC is well documented,18,53 which makes this lipid mixture an exciting starting point to verify our method to predict lipid phase behaviour. Here we present the bilayer properties as a function of the fraction MO (fMO). This fraction is calculated as a weight fraction assuming each monomer has equal mass: fMO = θσMO/(θσMO + θσDOPC). To provide some insight into the structures formed for pure DOPC, pure MO and a 50/50 wt% lipid mixture, we have plotted the volume fraction profiles in the planar geometry (Fig. 3). The center of the bilayers is set at z = 0. As expected the core of these bilayers is dry (ϕW = 0 at z = 0); water penetrates only up to the glycerol backbone. An in depth study on the properties of the DOPC bilayer is published in our previous article14 and many of the MO bilayer properties are surprisingly similar. Close inspection reveals that the MO bilayers are a little bit thinner than DOPC ones, which is traced to the fact that DOPC simply has a larger headgroup. The average position of the glycerol backbone is similar in the two membranes and also in the mixed bilayer (Fig. 3B). However, in the MO bilayer the glycerol has a wider distribution than in the DOPC bilayer. We can come up with two reasons for this. Firstly, the glycerol backbone of DOPC is oriented more parallel to the bilayer plane than that of MO, due to the extra tail attached to the sn2 position of the glycerol. Secondly, since the glycerol backbone of MO is more hydrophilic, we have a more gradual hydrophobic–hydrophilic transition.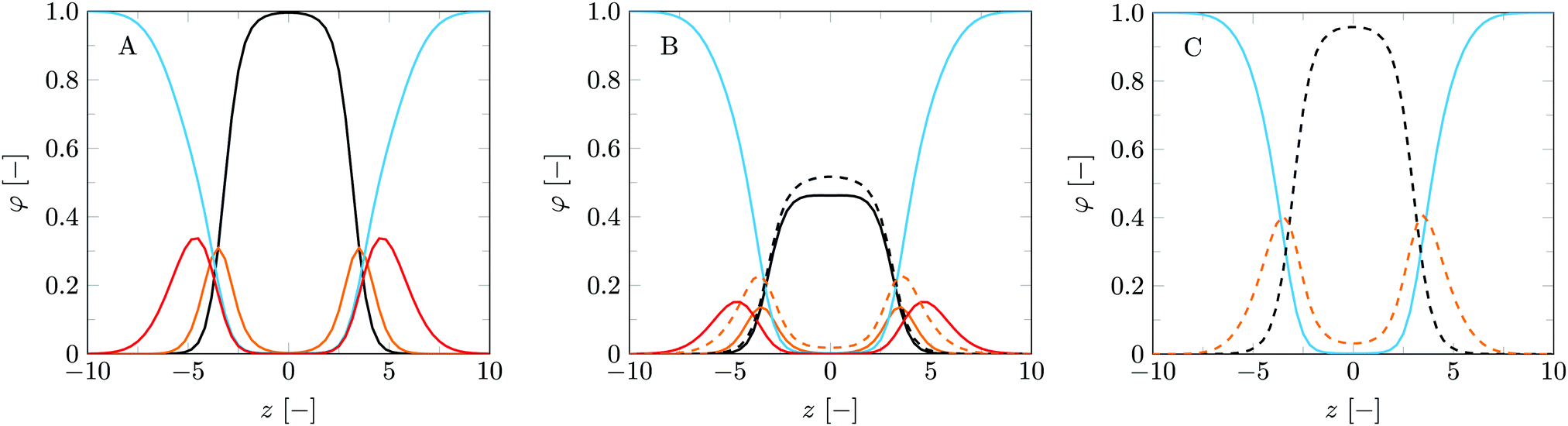 | ||
| Fig. 3 Volume fraction profiles for (A) a pure DOPC bilayer, (B) a mixed DOPC/MO bilayer (fMO = 0.5), and (C) a pure MO bilayer. Solid blue lines represent water, other solid lines represent DOPC and dashed lines represent MO. Black: lipid tails; orange: glycerol backbone of the lipids; red: lipid headgroups. | ||
The differences in structural properties between the MO and DOPC bilayers are reflected in their different natural topological states. As mentioned before, DOPC bilayers naturally occur in a lamellar topology while MO bilayers occur in cubic phases. The mechanical parameters reflect these states: is negative and Jm0 is very close to zero, even slightly positive, for DOPC, while these parameters are respectively positive and slightly negative for MO bilayers. See also Fig. 4 at fMO = 0 (pure DOPC bilayer) and at fMO = 1 (pure MO bilayer).
 | ||
| Fig. 4 Mechanical parameters , κ and Jm0 for DOPC bilayers as a function of fraction of MO with different χOMO–W. Blue: χOMO–W = −0.5; orange: χOMO–W = −0.55; red: χOMO–W = −0.6. | ||
Fig. 4 reports the mechanical parameters for the mixed DOPC/MO bilayers as a function of the fraction MO. The trends in these parameters can be translated into expected changes in the mesomorphic state of these bilayers with increasing fraction of MO. In particular we foresee a transition from a lamellar phase to a phase with saddle-shape topologies as signalled by the change of sign of . For the default χ-parameters chosen, we expect this to occur around 40 wt% (fMO ≈ 0.40): see the blue line in Fig. 4A. In reality this transition occurs around 50–55 wt%.18,53 This difference does not alarm us too much as the trend is in line with experimental data. Furthermore, small optimizations in the χ parameters can certainly improve these results as the sign-switch is highly dependent on the χ values chosen. This is showcased by the orange and red curves in Fig. 4, for which the interaction parameters for the MO glycerol moiety were chosen slightly different. In short, when the glycerol moiety of MO is made a bit more hydrophilic, decreases for the pure MO bilayers, and as a consequence, the transition point for which = 0 shifts to higher MO fractions in the mixed bilayer.
Next to the increase in , we observe a decrease in Jm0 with increasing fraction of MO. This trend seems to agree with the experimentally observed phase transition as well. Jm0 goes from slightly positive values, as expected for lamellar bilayer topologies, to slightly negative values, more expected for cubic phases. Like for , the sign-switch of Jm0 occurs at higher MO fractions as the glycerol moiety of MO is made more hydrophilic.
Small changes in the hydrophobicity of the glycerol moiety do not have an effect on κ, which remains above 1kBT for all DOPC/MO ratios. As κ thus remains relatively high, the modelling predicts a direct transition from lamellar to cubic without the L3 or sponge phase as an intermediate mesophase. This corresponds nicely to experimentally observed phase behaviour.18,53 Below we will argue that indirect transitions are expected for the addition of some solvents.
5.3 Effect of additives
Here we discuss the effects of adding various substances to pure MO or pure DOPC bilayers. We will not go into much detail regarding the structural parameters of the bilayer, but focus on (trends of) the mechanical parameters and aim to correlate these to known lipid phase behaviour in particular for the MO- but also for the DOPC system.The effect of these alcohols on the phase behaviour of DOPC bilayers is not documented. However, in several studies short alcoholic solvents have been added to lamellar bilayers.56–59 In some of these studies a few mechanical properties were measured, such as the mean bending modulus κ, using the micropipette aspiration technique.56,57 In general, it was observed that the addition of small alcohols largely lowers κ as the bilayers become much more flexible.
The effects of the alcohols on the mechanical properties of the lipid bilayers according to our SCF predictions are shown in Fig. 5.
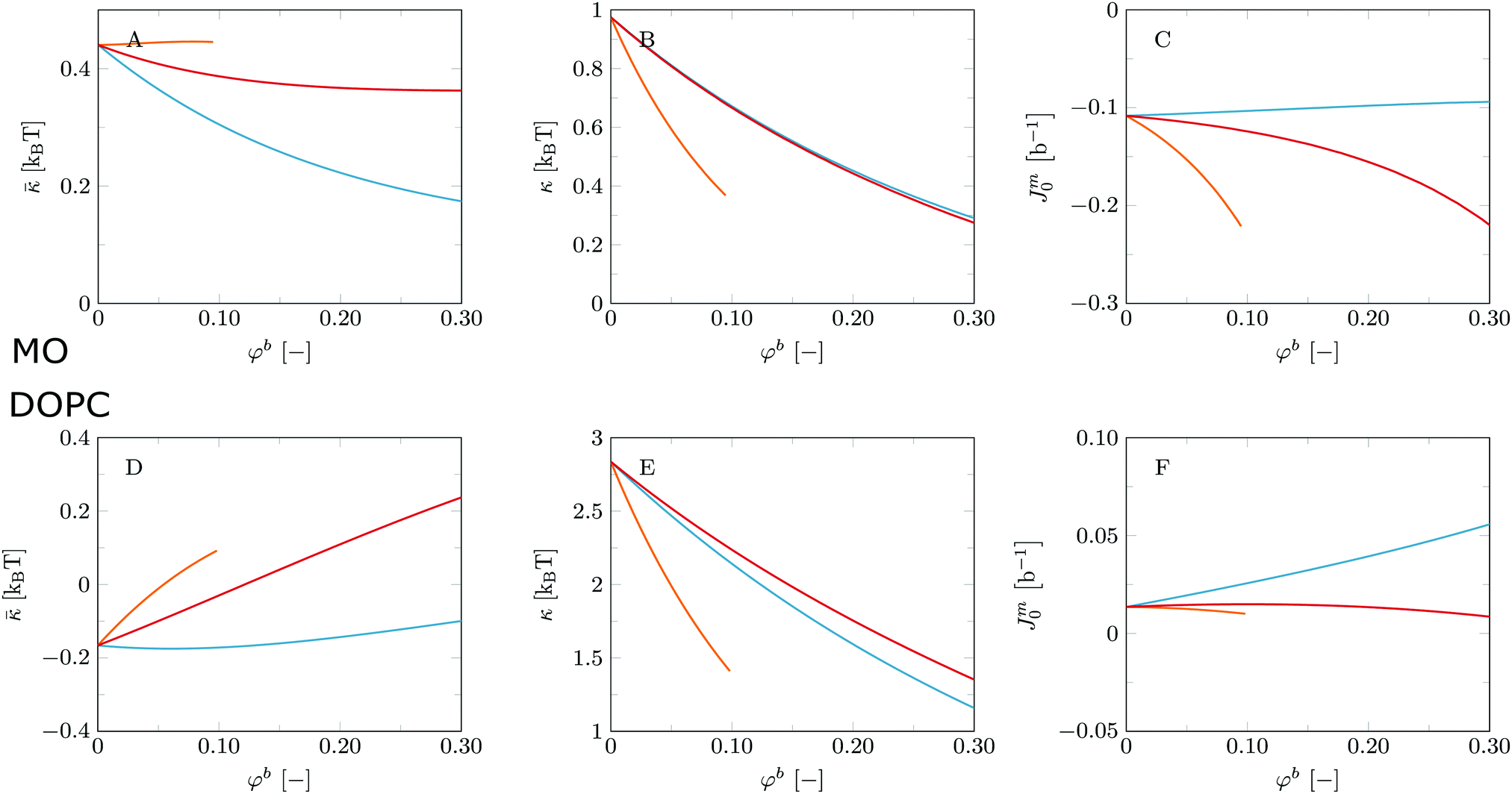 | ||
| Fig. 5 Mechanical parameters , κ and Jm0 as a function of bulk volume fraction φb of added ‘solvent’ for MO bilayers (top, A–C) and DOPC bilayers (bottom, D–F). Blue: added solvent is ethanol (C2O); orange: t-butanol (t-C4O); red: butanediol (C4O2). | ||
Let us first consider the MO system and discuss these results in light of the experimental results described above. In Fig. 5A we find that the Gaussian bending rigidity of the MO system decreases with the addition of ethanol and butanediol, albeit that the changes are modest. The response to t-butanol is very weak, so apparently there are competing effects that balance each other in this case. The spontaneous curvature of the monolayer (Fig. 5C) shows an opposite trend where addition of ethanol gives the smallest effect. The changes in and Jm0 upon addition of small amounts of ethanol, t-butanol or butanediol are not sufficient to point to phase changes. However, at high volume fractions of t-butanol inverted phases are likely, since for this case Jm0 is expected to be strongly negative. Fig. 5B shows that all three solvents tend to make the MO bilayers extremely flexible, since the mean bending modulus drops to sub-unity values rather dramatically. As explained above, this indicates the formation of less ordered phases such as the sponge or L3 phase. Indeed, as mentioned before this is what has been found experimentally.54
The corresponding results for the mechanical parameters for the DOPC bilayer system are presented in Fig. 5D–F. Fig. 5D shows that upon significant addition of ethanol as well as t-butanol and butanediol the Gaussian bending rigidity turns more positive and eventually changes sign. This suggests that this type of additive may destroy the lamellar topology of pure DOPC bilayers. The spontaneous curvature of the monolayer (Fig. 5F) increases both for addition of ethanol and butanediol, but decreases and may become negative for t-butanol. As the spontaneous curvature does not become extremely negative we expect that eventually at high volume fractions the alcohols may push DOPC bilayers towards bicontinuous cubic phases rather than the inverted hexagonal phase, although in experimental systems this transition may be hard to reach. Similar to the MO bilayer, the small alcohols make the DOPC bilayer more flexible (cf.Fig. 5E, but typically κ remains larger than unity and therefore transition into the sponge phase is very unlikely. The fact that small alcohols soften phospholipid bilayers has been reported in literature.56,57
There is much literature on the effect of fatty acids on lamellar (e.g. phospholipid) bilayers, yet on an experimental level almost all data concern long-chain fatty acids like oleic acids.61,62 From these, we only found one (very recent) article on fatty acid containing bilayers in the context of phase behaviour.61 It reported that the long chain fatty acids used, oleic acid (OA) and elaidic acid (EA), increased the mean bending rigidity of a DOPC bilayer. No topological phase transition was observed. In the few articles we found regarding the addition of medium-chain fatty acids (C6–C14) to lipid bilayers, interest was limited to general features, such as how much FA was incorporated63 or the effect on flip-flopping and transport across membranes.64–66
In our model calculations, above some threshold loading of FAs no longer tensionless bilayers could be found, indicating that inverted structures are dominating the phase space. The maximum amount of FA that could be incorporated was in the order of 20 wt% for MO bilayers and 30 wt% for DOPC bilayers. To provide a direct comparison for MO and DOPC bilayers we have chosen to show results up to 20 wt% FA (fFA = 0.2).
The effects of FA addition on the bending rigidities are shown in Fig. 6. From the figure we immediately observe that the effects of incorporating FA in MO bilayers or in DOPC bilayers are very similar. In general, we see increasing and both Jm0 and κ decreasing with addition of FA. The longer the tail of the FA, the more hydrophobic the FA is: this results into a stronger effect on and Jm0, but a weaker change in the mean bending modulus κ.
 | ||
| Fig. 6 Mechanical parameters , κ and Jm0 as a function of added fraction of fatty acid, for MO bilayers (top, A–C) and DOPC bilayers (bottom, D–F). Blue: C8O2; orange: C12O2; red: C16O2. | ||
Without additives the MO system is in a bicontinuous cubic phase. With increasing loading with FA the Gaussian bending modulus becomes more positive, implying that the system moves away even further from a lamellar state. In addition, the spontaneous curvature of the monolayer becomes significantly negative, implying that the system may well enter an inverted hexagonal phase (HII). This is especially expected for long-tail FA additives. The predictions are in this respect again in line with the experimental data. Experimental results that indicate the relative preference of inverted phases for long tail FA additives, were associated with a critical packing parameter P > 1.15 This is in accordance with our finding that Jm0 becomes strongly negative. Addition of FAs decreases the mean bending modulus a bit. The softening is much less than for the small molecular weight solvents discussed above. Hence the sponge phase is not expected in this case, especially not for the longer tail FA's. Short tail fatty acids (C3–C6) however may have a softening effect comparable to the small molecular weight solvents.
For DOPC bilayers switches sign as a function of FA loading (Fig. 6D) and we therefore anticipate that FAs will induce a transition from lamellar bilayers to saddle-shaped bilayers. The longer FA's are more effective in this respect than the shorter ones. As usual a strong positive response of the Gaussian bending modulus is mirrored by a strong negative response of the spontaneous curvature of the monolayer. As shown in Fig. 6F the value of the latter becomes significantly negative indicating that transition into an inverted hexagonal phase (HII) is a realistic scenario. In any case a sponge phase is not in sight because the mean bending modulus remains well above unity (Fig. 6E). This means that if a phase rich in saddle shapes is formed, it will be a bicontinuous cubic arrangement with long-range order.
We further note that the longer the FA tail, the smaller is the decrease in κ with fFA. This trend may suggest that a fatty acid with a tail length >16 could also increase the value of κ. This would be consistent with the experimental observation that the addition of oleic acid increases κ for DOPC bilayers.61
At high concentration, micelle-forming surfactants can even completely dissolve the bilayer into mixed micelles containing both lipids and surfactant molecules. A prime example is the addition of the non-ionic polyoxyethylene detergent Triton X-100 to phospholipid bilayers.67–69 This solubilization of lipid membranes is widely used to isolate, extract and characterize integral membrane proteins.70
In Triton X-100 the hydrophobic moiety features a benzene ring. In the modelling context this is not ideal because it definitely needs additional parameterisations. That is why we here chose to use C12Em surfactants to represent the class of surfactants. By varying the amount of ethylene oxide (E) units we can play with the size of the hydrophilic ‘head’ and thereby mimic both pure micelle-forming surfactants (C12E10) and surfactants for which additionally lamellar and even sponge phases have been observed (C12E4 and C12E5) depending on temperature and pressure.71,72 We further note that these surfactants can exhibit different lamellar phases such as the fluid lamellar (Lα) and liquid crystalline (LC) phases,73,74 between which we make no distinction in this work.
In particular, the phase behaviour of C12E5 is interesting as at high volume fractions at room temperature lamellar bilayers are observed, and at higher temperatures even the transition from lamellar to sponge phases is seen.75 This suggests that for pure C12E5 bilayers is close to zero at room temperature and positive at higher temperatures. This can be captured in the model by making the interaction parameters for the ethylene oxide part of the molecule temperature dependent76 such that the headgroup becomes less water soluble with increasing temperature. In the present paper we ignore this temperature dependence.
When we use the default parameters (Table 1) to model C12E5 bilayers, we find a Gaussian bending rigidity that is slightly negative = −0.52. Consistent with expectations, we observe an increase in when increasing χCD–W (not shown). For example, = 1.4 is obtained already for χCD–W = 1.2, which combined with a low mean bending modulus (κ = 0.34) indeed suggests the formation of the sponge phase at higher temperatures (i.e., for a more positive value for χCD–W).
Although the phase behavior of the pure surfactants is of interest, we here focus on the effect of surfactants as additives to MO and DOPC systems. We chose for this study two surfactants with a small headgroup (C12E5 and C12E4) and a surfactant with larger headgroup (C12E10).
The predicted mechanical parameters with increasing added amounts of C12Em surfactants to DOPC and MO bilayers are presented in Fig. 7. As with the addition of fatty acids, we observe similar trends for the DOPC and MO bilayers with the addition of C12Em surfactants. In line with expectation and the trends found for the fatty acids, i.e., an increase in and a decrease in Jm0 with increasing tail length, we now find the opposite effects with increasing surfactant headgroup size. We thus observe that with added fraction surfactant decreases while Jm0 increases and that these effects become more pronounced with increasing number of ethylene oxide units.
 | ||
| Fig. 7 Mechanical parameters , κ and Jm0 as a function of added fraction C12Em surfactants, for MO bilayers (top, A–C) and DOPC bilayers (bottom, D–F). Blue: C12E4; orange: C12E5; red: C12E10. | ||
For MO bilayers this means we expect transition of the bicontinuous cubic phase towards a lamellar phase. For the addition of C12E10 such a transition seems to occur already at about 10 wt% loading, as can be seen from Fig. 7A. As κ remains relatively close to unity, we do not expect the formation of an intermediate sponge phase. This phase behaviour corresponds closely to known phase behaviour of other MO/surfactant (or detergent) mixtures, which also shows a direct transition of the bicontinuous cubic phase to the lamellar Lα phase.15 The addition of C12E4 and C12E5 causes similar, but weaker, trends in and Jm0. The decrease in κ with C12E4,5 fraction seems to suggest that at high fractions the formation of a sponge phase, which requires a low value of κ, is possible. This is most likely for C12E4.
As expected, addition of these surfactants to DOPC bilayers induces the formation of micellar structures at high surfactant/lipid ratios. This follows mainly from the sharp increase in Jm0 to large positive values. The larger the polyethylene oxide headgroup the quicker, i.e. at lower molar fractions, this happens.
6 General discussion
In the previous sections we have shown that the lattice-refined SF-SCF theory can be used to estimate the mechanical parameters of MO and DOPC bilayer systems. We argue that we can use the trends in these parameters to consistently predict the mesomorphic phase behaviour of these lipids in response to a wide range of additives. More specifically, the phase transition from lamellar bilayer topologies to saddle-shaped topologies can be attributed to the sign-switch of. Additionally, the formation of micelles or inverted micelles follow from a large positive or large negative value of Jm0 respectively. The transitions between different saddle-shaped bilayer phases, i.e. various cubic phases and eventually the sponge phases, arguably are rather subtle. Invariably these phases can only be stable when the Gaussian bending modulus is positive. When the mean bending modulus is in addition much less than unity, we argue that the system loses its long-range order and forms a sponge phase. Bicontinuous triple periodic phases are likely to have a higher bending modulus compared with sponge phases. The higher the value for κ, the more the triple periodic cubic phase will go to a minimal surface, that is a surface on which the total curvature vanishes everywhere in the unit cell. This is the way the system can nullify the influence of the (relatively high) bending modulus. We cannot imagine that a strong preferential curvature of the monolayer, which typically is negative in this regime, is consistent with the bicontinuous cubic phase and this parameter should be responsible for the transitions towards the inverted hexagonal or inverted micelle systems. A strong criterion for this transition we do not yet have.
Now that we have established these rules, some general trends can be identified. In short, we observe that the inclusion of additives that are mainly hydrophobic and partition in the bilayer core, drive the bilayer towards a topological state of increased negative interfacial curvature and saddle shape configurations. That is, the DOPC system may lose the lamellar topology in favour of the inverted hexagonal phase or bicontinuous cubic phases and MO tends to go towards an inverted micellar phase. Micelle-forming surfactants do the opposite. The inclusion of small additives, both hydrophilic (i.e. ‘solvents’) or hydrophobic, generally decreases the mean bending modulus of the bilayers and thus makes bilayers more flexible. This may take a lipid system from a bicontinuous cubic phase closer to an L3-phase or decreases the persistence length of the bilayers and increase the undulation repulsion. These rules are highly complementary to the general rules for lipid self-assembly as outlined by van’t Hag et al.15
It should be stressed that small changes in the set of interaction parameters may have a relatively large impact on the results. This not necessarily is a bad property of the approach because in reality small variations in surfactant properties can also result in large shifts in phase behaviour. However, it means that the exact values of , κ and Jm0 as provided by the SF-SCF modelling should not be given too much weight. To arrive at our conclusions with respect to the effect of additives on the DOPC and MO bilayers mesomorphic phase behaviour, we only took into account the predicted trends in these mechanical quantities. It remains true that in order to provide more accurate estimates on the mechanical properties, a more detailed and structured parameterisation of each molecule used is required.
7 Conclusions
The self-assembly of lipids in aqueous solution yields a wide variety of structures of various shapes and morphologies. From an application perspective, it is of utmost importance to know the topological behaviour of the lipid system, in order to find relevant functionalities. With time, the experimental techniques used to follow this behaviour are becoming more accurate and more precise. However, mapping the topological phase behaviour can still be time consuming and finding the ideal lipid system for the desired application is a matter of trial and error. Modelling techniques that can accurately predict the mesomorphic phase behaviour of lipid systems are therefore in high demand. However when the computation time exceeds the experimental time scale, which still seems the case for all-atom molecular dynamics simulations, they are in this respect of little extra value. Only computationally inexpensive routes can map out a large parameter space needed to feed our intuition regarding the mesomorphism of lipids and lipid mixtures. Computationally inexpensive routes are approximate and we can only start trusting these routes if good correlations with experiments are produced.In this work, we used the lattice-refined self-consistent field theory of Scheutjens and Fleer (SF-SCF) which is computationally inexpensive. It can be fed with molecularly detailed models (on a united atom level) and come up with structural, thermodynamic and mechanical data that can be interpreted in terms of the preferred mesomorphic phase state of the system of interest. We have illustrated this by predicting the phase behaviour of MO and DOPC bilayers in response to a wide range of additives. For mixtures of MO and DOPC, and for the addition of various additives to MO bilayers, the obtained mechanical parameters clearly correlate with known phase behaviour. These correlations give credibility to the predicted phase behavior of DOPC in response to these additives. Importantly, this analysis can be readily extended to other lipid systems as well.
It must be understood that in the current SCF approach one can only predict the intrinsic values for the rigidities. These describe, e.g. the membrane fluctuations on a small, that is, on the lipid length scale. When the interest is in membrane fluctuations on a large length scale, one has to implement a renormalisation scheme for the rigidities, as derived (for example) by Leibler and coworkers.77
Furthermore, the lattice-refined SF-SCF approach as used in this work is not yet developed to the best of its possibilities. For instance, the chain model that is currently implemented is the freely-jointed chain. This chain model can be upgraded to the rotational isomeric state (RIS) scheme, implementing the chain flexibility more realistically.78 In addition the mean-field approximation is not suitable to account for thermotropic phase changes such as the gel-to-liquid phase transitions in bilayers. Solutions to overcome these shortcomings are in principle known79 but currently not implemented. Finally, and this is particularly important for lipid mixtures, we have not yet considered the possibility of lateral segregation of lipids in bilayer membrane (formation of rafts). Again at the expense of CPU time we can improve on this issue as well.80 We therefore conclude that the lattice-refined SF-SCF model has a great potential to predict mesomorphism of lipids and lipid mixtures and we can already use the approach to sharpen our intuition regarding the many possible phase changes that can occur.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Lewis and R. N. McElhaney, Struct. Biol. Membr., 1992, 2, 19–75 Search PubMed.
- M. Tate, E. Eikenberry, D. Turner, E. Shyamsunder and S. Gruner, Chem. Phys. Lipids, 1991, 57, 147–164 CrossRef CAS.
- G. V. Betageri and M. B. Yatvin, Liposome drug delivery, US Pat., 6761901, 2004 Search PubMed.
- E. Kisak, B. Coldren, C. Evans, C. Boyer and J. Zasadzinski, Curr. Med. Chem., 2004, 11, 199–219 CrossRef CAS.
- M. Michel, M. Winterhalter, L. Darbois, J. Hemmerle, J. C. Voegel, P. Schaaf and V. Ball, Langmuir, 2004, 20, 6127–6133 CrossRef CAS.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS.
- E. Nazaruk, R. Bilewicz, G. Lindblom and B. Lindholm-Sethson, Anal. Bioanal. Chem., 2008, 391, 1569 CrossRef CAS PubMed.
- E. Nazaruk, S. Smoliński, M. Swatko-Ossor, G. Ginalska, J. Fiedurek, J. Rogalski and R. Bilewicz, J. Power Sources, 2008, 183, 533–538 CrossRef CAS.
- J. J. Vallooran, S. Handschin, S. M. Pillai, B. N. Vetter, S. Rusch, H.-P. Beck and R. Mezzenga, Adv. Funct. Mater., 2016, 26, 181–190 CrossRef CAS.
- Z. A. Almsherqi, S. D. Kohlwein and Y. Deng, J. Cell Biol., 2006, 173, 839–844 CrossRef CAS PubMed.
- D. P. Siegel, Biophys. J., 2008, 95, 5200–5215 CrossRef CAS PubMed.
- M. Torbati, T. P. Lele and A. Agrawal, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11094–11099 CrossRef CAS PubMed.
- S. R. Wente and M. P. Rout, Cold Spring Harbor Perspect. Biol., 2010, 2, a000562 CAS.
- N. de Lange, J. Kleijn and F. Leermakers, Phys. Chem. Chem. Phys., 2021, 23, 5152–5175 RSC.
- L. van’t Hag, S. L. Gras, C. E. Conn and C. J. Drummond, Chem. Soc. Rev., 2017, 46, 2705–2731 RSC.
- H. Qiu and M. Caffrey, Biomaterials, 2000, 21, 223–234 CrossRef CAS.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- V. Cherezov, J. Clogston, Y. Misquitta, W. Abdel-Gawad and M. Caffrey, Biophys. J., 2002, 83, 3393–3407 CrossRef CAS PubMed.
- W. Helfrich, Z. Naturforsch., C: J. Biosci., 1973, 28, 693–703 CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- D. P. Siegel and M. Kozlov, Biophys. J., 2004, 87, 366–374 CrossRef CAS PubMed.
- R. Varadharajan and F. A. M. Leermakers, Phys. Rev. Lett., 2018, 120, 028003 CrossRef CAS PubMed.
- J. Genova, V. Vitkova and I. Bivas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 022707 CrossRef PubMed.
- P. Méléard and T. Pott, Advances in planar lipid bilayers and liposomes, Elsevier, 2013, vol. 17, pp. 55–75 Search PubMed.
- A. F. Loftus, S. Noreng, V. L. Hsieh and R. Parthasarathy, Langmuir, 2013, 29, 14588–14594 CrossRef CAS PubMed.
- C. Monzel and K. Sengupta, J. Phys. D: Appl. Phys., 2016, 49, 243002 CrossRef.
- J. R. Henriksen and J. H. Ipsen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2004, 14, 149–167 CrossRef CAS PubMed.
- V. Heinrich and W. Rawicz, Langmuir, 2005, 21, 1962–1971 CrossRef CAS PubMed.
- T. Portet, S. E. Gordon and S. L. Keller, Biophys. J., 2012, 103, L35–L37 CrossRef CAS PubMed.
- W. Rawicz, K. C. Olbrich, T. McIntosh, D. Needham and E. Evans, Biophys. J., 2000, 79, 328–339 CrossRef CAS.
- J. Pan, S. Tristram-Nagle, N. Kučerka and J. F. Nagle, Biophys. J., 2008, 94, 117–124 CrossRef CAS PubMed.
- G. Pabst, N. Kučerka, M.-P. Nieh, M. Rheinstädter and J. Katsaras, Chem. Phys. Lipids, 2010, 163, 460–479 CrossRef CAS PubMed.
- M. Hu, J. J. Briguglio and M. Deserno, Biophys. J., 2012, 102, 1403–1410 CrossRef CAS PubMed.
- T. Baumgart, S. Das, W. W. Webb and J. T. Jenkins, Biophys. J., 2005, 89, 1067–1080 CrossRef CAS PubMed.
- S. Semrau, T. Idema, L. Holtzer, T. Schmidt and C. Storm, Phys. Rev. Lett., 2008, 100, 088101 CrossRef PubMed.
- S. J. Marrink and D. P. Tieleman, Chem. Soc. Rev., 2013, 42, 6801–6822 RSC.
- J. M. H. M. Scheutjens and G. J. Fleer, J. Phys. Chem., 1979, 83, 1619–1635 CrossRef CAS.
- J. M. H. M. Scheutjens and G. J. Fleer, J. Phys. Chem., 1980, 84, 178–190 CrossRef CAS.
- W. L. Bragg and E. J. Williams, Proc. R. Soc. London, Ser. A, 1934, 145, 699–730 CAS.
- G. Fleer, M. A. C. Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at interfaces, Springer Science & Business Media, 1993 Search PubMed.
- O. A. Evers, J. M. H. M. Scheutjens and G. J. Fleer, Macromolecules, 1990, 23, 5221–5233 CrossRef CAS.
- S. Oversteegen, P. Barneveld, F. Leermakers and J. Lyklema, Langmuir, 1999, 15, 8609–8617 CrossRef CAS.
- V. Markin, M. Kozlov and S. Leikin, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1149–1162 RSC.
- S. Safran, Statistical thermodynamics of surfaces, interfaces, and membranes, CRC Press, 2018 Search PubMed.
- H. Pera, J. Kleijn and F. Leermakers, J. Chem. Phys., 2014, 140, 02B606_1 CrossRef PubMed.
- F. A. M. Leermakers, J. Chem. Phys., 2013, 138, 04B610 Search PubMed.
- R. Kik, J. Kleijn and F. Leermakers, J. Phys. Chem. B, 2005, 109, 14251–14256 CrossRef CAS PubMed.
- R. A. Kik, F. A. M. Leermakers and J. M. Kleijn, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 021915 CrossRef PubMed.
- T. Umeda, Y. Suezaki, K. Takiguchi and H. Hotani, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 011913 CrossRef PubMed.
- F. A. M. Leermakers, J. M. H. M. Scheutjens and J. Lyklema, Biophys. Chem., 1983, 18, 353–360 CrossRef CAS.
- J. P. Dilger and R. Benz, J. Membr. Biol., 1985, 85, 181–189 CrossRef CAS PubMed.
- R. Fettiplace, D. Andrews and D. Haydon, J. Membr. Biol., 1971, 5, 277–296 CrossRef CAS PubMed.
- V. Chupin, J. Killian and B. de Kruijff, Biophys. J., 2003, 84, 2373–2381 CrossRef CAS.
- S. Engström, K. Alfons, M. Rasmusson and H. Ljusberg-Wahren, The Colloid Science of Lipids, Springer, 1998, pp. 93–98 Search PubMed.
- V. Cherezov, J. Clogston, M. Z. Papiz and M. Caffrey, J. Mol. Biol., 2006, 357, 1605–1618 CrossRef CAS PubMed.
- H. V. Ly and M. L. Longo, Biophys. J., 2004, 87, 1013–1033 CrossRef CAS PubMed.
- H. V. Ly, D. E. Block and M. L. Longo, Langmuir, 2002, 18, 8988–8995 CrossRef CAS.
- S.-Y. Chen, B. Yang, K. Jacobson and K. K. Sulik, Alcohol, 1996, 13, 589–595 CrossRef CAS PubMed.
- P. Westh, C. Trandum and Y. Koga, Biophys. Chem., 2001, 89, 53–63 CrossRef CAS.
- N. Tran, A. M. Hawley, J. Zhai, B. W. Muir, C. Fong, C. J. Drummond and X. Mulet, Langmuir, 2016, 32, 4509–4520 CrossRef CAS PubMed.
- A. I. Tyler, J. Greenfield, N. J. Brooks and J. M. Seddon, et al. , Front. Cell Dev. Biol., 2019, 7, 187 CrossRef PubMed.
- J. R. Simard, F. Kamp and J. A. Hamilton, Biophys. J., 2008, 94, 4493–4503 CrossRef CAS PubMed.
- M. Langner, T. Isac and S. Hui, Biochim. Biophys. Acta, Biomembr., 1995, 1236, 73–80 CrossRef.
- F. Kamp, D. Zakim, F. Zhang, N. Noy and J. A. Hamilton, Biochemistry, 1995, 34, 11928–11937 CrossRef CAS PubMed.
- J. A. Hamilton, J. Lipid Res., 1998, 39, 467–481 CrossRef CAS.
- F. Kamp and J. A. Hamilton, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2006, 75, 149–159 CrossRef CAS PubMed.
- R. J. Robson and E. A. Dennis, Biochim. Biophys. Acta, Lipids Lipid Metab., 1979, 573, 489–500 CrossRef CAS.
- E. London and D. A. Brown, Biochim. Biophys. Acta, Biomembr., 2000, 1508, 182–195 CrossRef CAS.
- H. Ahyayauch, B. Larijani, A. Alonso and F. M. Goñi, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 190–196 CrossRef CAS PubMed.
- A. Helenius and K. Simons, Biochim. Biophys. Acta, Rev. Biomembr., 1975, 415, 29–79 CrossRef CAS.
- D. J. Mitchell, G. J. Tiddy, L. Waring, T. Bostock and M. P. McDonald, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 975–1000 RSC.
- G. J. Tiddy, Phys. Rep., 1980, 57, 1–46 CrossRef CAS.
- B. Mädler, G. Klose, A. Möps, W. Richter and C. Tschierske, Chem. Phys. Lipids, 1994, 71, 1–12 CrossRef.
- H. Pfeiffer, G. Klose, K. Heremans and C. Glorieux, Chem. Phys. Lipids, 2006, 139, 54–67 CrossRef CAS PubMed.
- R. Strey, R. Schomäcker, D. Roux, F. Nallet and U. Olsson, J. Chem. Soc., Faraday Trans., 1990, 86, 2253–2261 RSC.
- B.-S. Lee, J. Mol. Liq., 2018, 262, 527–532 CrossRef CAS.
- S. Leibler, Stat. Mech. Membr. Surf., 2004, 45–103 Search PubMed.
- F. Leermakers and J. Scheutjens, J. Chem. Phys., 1988, 89, 3264–3274 CrossRef CAS.
- F. Leermakers and J. Scheutjens, J. Chem. Phys., 1988, 89, 6912–6924 CrossRef CAS.
- F. Leermakers, M. Ballauff and O. Borisov, Langmuir, 2007, 23, 3937–3946 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00697e |
| This journal is © the Owner Societies 2021 |